Attached files
| file | filename |
|---|---|
| EX-23.2 - EXHIBIT 23.2 - SPECTRUM PHARMACEUTICALS INC | consentex232.htm |
| EX-23.1 - EXHIBIT 23.1 - SPECTRUM PHARMACEUTICALS INC | consentex231.htm |
| EX-31.2 - EXHIBIT 31.2 - SPECTRUM PHARMACEUTICALS INC | sppi20151231ex312.htm |
| EX-31.1 - EXHIBIT 31.1 - SPECTRUM PHARMACEUTICALS INC | sppi20151231ex311.htm |
| EX-32.1 - EXHIBIT 32.1 - SPECTRUM PHARMACEUTICALS INC | sppi20151231ex321.htm |
| EX-32.2 - EXHIBIT 32.2 - SPECTRUM PHARMACEUTICALS INC | sppi20151231ex322.htm |
| EX-21.1 - EXHIBIT 21.1 - SPECTRUM PHARMACEUTICALS INC | listofsubsidiariesex211.htm |
| EX-10.56 - EXHIBIT 10.56 - SPECTRUM PHARMACEUTICALS INC | redactedsupplyagreementwit.htm |
| EX-10.55 - EXHIBIT 10.55 - SPECTRUM PHARMACEUTICALS INC | redactedlicenseassetpurcha.htm |
| EX-10.22 - EXHIBIT 10.22+* - SPECTRUM PHARMACEUTICALS INC | amendno1toseveranceagreeme.htm |
| EX-10.54 - EXHIBIT 10.54 - SPECTRUM PHARMACEUTICALS INC | redactedeaglepharmaco-prom.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
______________________________________________
Form 10-K
______________________________________________
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2015
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number: 001-35006

______________________________________________
SPECTRUM PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in its Charter)
______________________________________________
Delaware | 93-0979187 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
11500 South Eastern Avenue, Suite 240
Henderson, Nevada 89052
(Address of principal executive offices)
(702) 835-6300
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange on Which Registered | |
Common Stock, $0.001 par value Rights to Purchase Series B Junior Participating Preferred Stock | The NASDAQ Stock Market, LLC | |
Securities registered pursuant to Section 12(g) of the Act:
None
______________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 232.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer | ¨ | Accelerated filer | x | ||
Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | ||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of June 30, 2015, the aggregate market value of the voting and non-voting common equity held by non-affiliates of the Registrant was $459,959,918 (based upon the $6.84 per share closing sale price for shares of the Registrant’s Common Stock as reported by the NASDAQ Global Select Market on June 30, 2015, the last trading date of the Registrant’s most recently completed second fiscal quarter).
As of February 29, 2016, approximately 68,163,483 shares of the Registrant’s Common Stock, $0.001 par value, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for the registrant’s 2016 Annual Meeting of Stockholders, to be filed on or before April 29, 2016, are incorporated by reference into Part III, Items 10-14 of this Annual Report on Form 10-K.
TABLE OF CONTENTS
Page | |||
PART I | |||
PART II | |||
PART III | |||
PART IV | |||
Cautionary Note Concerning Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934 as amended (the “Exchange Act”), in reliance upon the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Forward-looking statements include, without limitation, statements regarding our future product development activities and costs, the revenue potential (licensing, royalty and sales) of our products and product candidates, the success, safety and efficacy of our drug products, revenues, development timelines, product acquisitions, liquidity and capital resources and trends, and other statements containing forward-looking words, such as, “believes,” “may,” “could,” “will,” “expects,” “intends,” “estimates,” “anticipates,” “plans,” “seeks,” “continues,” or the negative thereof or variation thereon or similar terminology (although not all forward-looking statements contain these words). Such forward-looking statements are based on the reasonable beliefs of our management as well as assumptions made by and information currently available to our management. Readers should not put undue reliance on these forward-looking statements. Forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified; therefore, our actual results may differ materially from those described in any forward-looking statements. Factors that might cause such a difference include, but are not limited to, those discussed elsewhere in this Annual Report on Form 10-K, and the following factors:
• | our ability to successfully develop, obtain regulatory approval for and market our products; |
• | our ability to continue to grow sales revenue of our marketed products; |
• | risks associated with doing business internationally; |
• | our ability to generate and maintain sufficient cash resources to fund our business; |
• | our ability to enter into strategic alliances with partners for manufacturing, development and commercialization; |
• | efforts of our development partners; |
• | the ability of our manufacturing partners to meet our timelines; |
• | the ability to timely deliver product supplies to our customers; |
• | our ability to identify new product candidates and to successfully integrate those product candidates into our operations; |
• | the timing and/or results of pending or future clinical trials, and our reliance on contract research organizations; |
• | our ability to protect our intellectual property rights; |
• | competition in the marketplace for our drugs; |
• | delay in approval of our products or new indications for our products by the U.S. Food and Drug Administration, or the “FDA”; |
• | actions by the FDA and other regulatory agencies, including international agencies; |
• | securing positive reimbursement for our products; |
• | the impact of any product liability, or other litigation to which we are, or may become a party; |
• | the impact of legislative or regulatory reform of the healthcare industry and the impact of recently enacted healthcare reform legislation; |
• | the availability and price of acceptable raw materials and components from third-party suppliers, and their ability to meet our demands; |
• | our ability, and that of our suppliers, development partners, and manufacturing partners, to comply with laws, regulations and standards, and the application and interpretation of those laws, regulations and standards, that govern or affect the pharmaceutical and biotechnology industries, the non-compliance with which may delay or prevent the development, manufacturing, regulatory approvals and sale of our products; |
• | defending against claims relating to improper handling, storage or disposal of hazardous chemical, radioactive or biological materials which could be time consuming and expensive; |
• | our ability to maintain the services of our key executives and technical and sales and marketing personnel; |
1
• | the difficulty in predicting the timing or outcome of product development efforts and regulatory approvals; and |
• | demand and market acceptance for our approved products. |
All subsequent written and oral forward-looking statements attributable to us or by persons acting on our behalf are expressly qualified in their entirety by these cautionary statements.
In addition, past financial or operating performance is not necessarily a reliable indicator of future performance, and you should not use our historical performance to anticipate results or future period trends. We can give no assurances that any of the events anticipated by the forward-looking statements will occur or, if any of them do, what impact they will have on our results of operations and financial condition. Except as required by law, we do not undertake to update any such forward-looking statements and expressly disclaim any duty to update the information contained in this Annual Report on Form 10-K.
Unless the context otherwise requires, all references in this Annual Report on Form 10-K to the “Company”, “we,” “us,” “our,” “Spectrum” and “Spectrum Pharmaceuticals” refer to Spectrum Pharmaceuticals, Inc. and its subsidiaries and other consolidated entities, as a consolidated entity. We primarily conduct our business activities as Spectrum Pharmaceuticals.
***
Spectrum Pharmaceuticals, Inc.®, FUSILEV®, FOLOTYN®, ZEVALIN®, MARQIBO®, BELEODAQ®, EVOMELA™, EOQUIN®, and RenaZorb® are registered trademarks of Spectrum Pharmaceuticals, Inc. and its subsidiaries. Redefining Cancer Care™, Turning Insights Into Hope™, RIT Oncology, LLC™, RIT™, RRZ™, and our logos are trademarks owned by Spectrum Pharmaceuticals, Inc. and its subsidiaries. All other trademarks and trade names are the property of their respective owners.
PART I
ITEM 1. BUSINESS
Company Overview and Business Strategy
Our primary strategy is comprised of acquiring, developing, and commercializing a broad and diverse pipeline of late-stage clinical and commercial products. In addition to an efficient in-house clinical development organization with regulatory and data management capabilities, we have established a commercial infrastructure for our marketed products. Currently, we have six approved oncology/hematology products that target different types of non-Hodgkin's lymphoma ("NHL"), advanced metastatic colorectal cancer, acute lymphoblastic leukemia ("ALL"), and multiple myeloma ("MM").
We also have two drugs in late stage development:
•SPI-2012, is being developed for chemotherapy-induced neutropenia in patients with breast cancer.
• | EOQUIN® (previously referred to as APAZIQUONE for intravesical instillation), is being developed for immediate intravesical instillation post-transurethral resection of bladder tumors in patients with non-muscle invasive bladder cancer. |
Our passion to identify, develop and deliver important options for patients suffering from cancer is behind every action we take. We are committed to excellence and strive to make a difference in the lives of patients every day.
Cancer Background and Market Size
Cancer is a group of diseases characterized by the uncontrolled growth and spread of abnormal cells, which can result in death. The development of cancer is multi-factorial and includes both external factors (tobacco, infectious organisms, chemicals, and radiation) and internal factors (inherited mutations, hormones, immune conditions, and mutations that occur from exposure to environmental factors or errors in making DNA (deoxyribonucleic acid) during normal cell division). These causal factors may act together or in sequence to initiate or promote the development of cancer. Ten or more years often pass between exposure to these factors and the development of detectable cancer. Cancer is treated through surgery, radiation, chemotherapy, hormone therapy, immune therapy, and/or targeted drug therapy.
According to the American Cancer Society’s publication Cancer Facts & Figures 2015, cancer is the second leading cause of death in the U.S. (only behind heart disease). In the U.S., approximately 1.7 million new cancer cases were expected to be diagnosed in 2015 and over 589,000 persons were expected to die from the disease. Anyone can develop cancer. Since the
2
risk of being diagnosed with cancer increases with age, most cases occur in adults who are middle aged or older. About 77% of all cancers are diagnosed in people 55 years of age and older. In the U.S., men have slightly less than a 1 in 2 lifetime risk of developing cancer; for women, the risk is a little more than 1 in 3. These probabilities are estimated based on the overall experience of the general population. Individuals within the population may have higher or lower risk because of differences in exposures (e.g., smoking), and/or genetic susceptibility. In addition, currently available treatments are variably effective in the different cancers and individual patients. Together these patients’ risks and the treatment limitations suggest a significant current and long-term demand for improved and novel cancer treatments.
Product Portfolio
We have a product portfolio consisting of both commercial stage and development stage products that address various cancer types (see “Research & Development” section below for our pipeline of cancer therapeutics that are in various development stages). We remain committed to growing the sales of our currently marketed products, as we strive to maintain a robust development pipeline.
Commercialized Products
Our commercialized drug products (or pending commercial launch in the case of EVOMELA), and their approved indications, are summarized in the following table:
FUSILEV
FUSILEV (levoleucovorin), a novel folate analog and the pharmacologically active isomer (the levo-isomer) of the racemic compound, calcium leucovorin. Leucovorin is a mixture of equal part of both isomers: the pharmacologically active levo-isomer and the inactive dextro-isomer. Preclinical studies have demonstrated that the inactive dextro-isomer may compete with the active levo-isomer for uptake at the cellular level. By removing the inactive dextro form, the dosage of FUSILEV is one-half that of leucovorin and patients are spared the administration of an inactive substance. FUSILEV is approved as a ready-to-use solution, and as freeze-dried powder. FUSILEV has the following indications for use:
• | in combination chemotherapy with 5-fluorouracil in the palliative treatment of patients with advanced metastatic colorectal cancer, or mCRC. |
• | for rescue after high-dose methotrexate therapy in osteosarcoma; and |
• | to diminish the toxicity and counteract the effects of impaired methotrexate elimination and of inadvertent over dosage of folic acid antagonists. |
FOLOTYN
FOLOTYN (pralatrexate injection), a folate analogue metabolic inhibitor, was discovered by Memorial Sloan-Kettering Cancer Center, SRI International and Southern Research Institute and developed by Allos Therapeutics, Inc. (“Allos”). In September 2009, the FDA granted accelerated approval for FOLOTYN for use as a single agent for the treatment of patients
3
with relapsed or refractory PTCL. FOLOTYN was the first chemotherapy approved by the FDA, under its accelerated approval program, for the treatment of relapsed or refractory PTCL and has been available to patients in the U.S. since October 2009.
According to the Lymphoma Research Foundation, lymphoma is the most common blood cancer. Hodgkin’s lymphoma and non-Hodgkin’s lymphoma (“NHL”) are the two main forms of lymphoma. Lymphoma occurs when lymphocytes, a type of white blood cell, grow abnormally and accumulate in one or more lymph nodes or lymphoid tissues. The body has two main types of lymphocytes that can develop into lymphomas: B-lymphocytes (B-cells) and T-lymphocytes (T-cells). PTCL comprises a group of rare and aggressive NHLs that develop from mature T-cells. PTCL accounts for approximately 5 to 15% of all NHL cases in the U.S and Europe.
Based on preclinical studies, we believe that FOLOTYN selectively enters cells expressing reduced folate carrier ("RFC"), a protein that is frequently over expressed on cancer cells compared to normal cells. Once inside cancer cells, FOLOTYN is efficiently polyglutamylated, which makes it less susceptible to efflux-based drug resistance and leads to high intracellular drug retention compared to other antifolates. Inside the cell, FOLOTYN targets the inhibition of dihydrofolate reductase ("DHFR"), an enzyme critical in the folate pathway, thereby interfering with DNA and RNA synthesis and triggering cancer cell death.
We are exploring additional settings for FOLOTYN where methotrexate (“MTX”), a drug in the same category as FOLOTYN, has been successfully used for decades in the treatment of breast cancer, bladder cancer, and lung cancer. We plan to test FOLOTYN’s benefits in these settings because FOLOTYN is designed to provide greater activity than MTX. In addition to its use alone as a single agent, we are evaluating FOLOTYN as part of different chemotherapy combinations.
ZEVALIN
ZEVALIN (ibritumomab tiuxetan) injection for intravenous use is a prescription medication that is part of a three step treatment regimen consisting of: two treatments of rituximab and one treatment of Yttrium-90 (Y-90) ZEVALIN. The National Cancer Institute (“NCI”) estimated 72,000 new cases of NHL in the U.S. in 2015. Rituximab is used to reduce the number of B-cells in the blood and Y-90 ZEVALIN is then given to treat NHL. It is currently approved in the U.S. and more than 40 countries outside the U.S. including countries in Europe, Latin America and Asia for (i) treatment of patients with recurring, low-grade or follicular B-cell NHL, after other anticancer drugs are no longer working, and (ii) newly diagnosed follicular NHL following a response to initial anticancer therapy.
MARQIBO
MARQIBO is a novel, sphingomyelin/cholesterol liposome-encapsulated formulation of the FDA-approved anticancer drug vincristine. MARQIBO’s approved indication is for the treatment of adult patients with ALL in second or greater relapse or whose disease has progressed following two or more lines of anti-leukemia therapy. In the U.S., approximately 6,000 patients per year are diagnosed with ALL, of which approximately 1,600 can be categorized as ALL in second or greater relapse.
MARQIBO is also currently being explored for the treatment of the broader ALL indication as well as in NHL in addition to its approved treatment for Philadelphia chromosome-negative ALL.
BELEODAQ
BELEODAQ (belinostat) is a histone deacytelase, (“HDAC”) inhibitor for the treatment of patients with relapsed or refractory PTCL. This indication was FDA approved in July 2014 under its accelerated approval program, based on tumor response rate and duration of response. BELEODAQ's anticancer effect is thought to be mediated through multiple mechanisms of action, including the inhibition of cell proliferation, induction of apoptosis (programmed cell death), inhibition of angiogenesis, induction of differentiation, and the activity in tumors that had become resistant to anticancer agents such as the platinums, taxanes and topoisomerase II inhibitors.
BELEODAQ is differentiated from other HDAC inhibitors that selectively inhibit a single class of HDAC enzymes because it inhibits all three classes of the zinc-dependent HDAC enzymes (Class I, Class II and Class IV); this leads to different alterations in histone and non-histone protein acetylation that, in turn, could importantly influence chromatin accessibility, gene transcription, and activity in different cancer patients, including those who develop drug resistant disease.
BELEODAQ has many attributes that separate it from other currently marketed HDACs, including efficacy when used alone and in combination, less toxicities (when compared to the reported rates of some adverse events with the other currently-
4
marketed HDACs), including less bone marrow toxicity, and a lack of other severe side effects, such as mucositis, that may enable full dose combinations of this drug with several other cytotoxic agents.
EVOMELA (previously referred to as Captisol-Enabled® MELPHALAN)
EVOMELA is intended for use as a conditioning treatment prior to autologous stem cell transplant for patients with MM. MM is a cancer of plasma cells, a type of white blood cell present mainly in the bone marrow that produces antibodies. In MM, a group of plasma cells (myeloma cells) become cancerous and multiply, raising the number of plasma cells to a higher-than-normal level, which can crowd out normal blood cells and lead to abnormally high proteins in the blood or urine. The NCI estimated 27,000 new cases of MM in the U.S. in 2015, with the incidence of new cases increasing by approximately 1.6% per year. The current intravenous Melphalan market is approximately $100 million annually, with predominant use in stem cell transplants.
The EVOMELA formulation avoids the use of propylene glycol (“PG”), which is required as a co-solvent in the currently-available formulation of this product. PG has been reported to cause adverse renal and cardiac side-effects that limit the ability to deliver higher quantities of intended therapeutic compounds. The use of Captisol technology to reformulate EVOMELA is anticipated to allow for longer administration durations and slower infusion rates, potentially enabling clinicians to avoid reductions, and safely achieve a higher dose intensity for pre-transplant chemotherapy.
EVOMELA was granted Orphan Drug status by the FDA for use as a high-dose conditioning regimen prior to hematopoietic progenitor (stem) cell transplantation. In December 2014, we filed our new drug application (“NDA”) for EVOMELA with the FDA. On October 23, 2015, we received a Complete Response Letter (“CRL”) from the FDA for this NDA that did not identify any clinical deficiencies, and we subsequently resubmitted our NDA. On March 10, 2016, the FDA communicated its approval of our NDA for EVOMELA as a high-dose conditioning treatment prior to hematopoietic progenitor (stem) cell transplantation in patients with MM, and for the palliative treatment of patients with MM for whom oral therapy is not appropriate. We plan to commercially launch EVOMELA as soon as possible.
Product Pipeline
SPI-2012
SPI-2012 is being investigated for the treatment of chemotherapy-induced neutropenia. In January 2012, we entered into a co-development and commercialization agreement for worldwide rights, except for Korea, China, and Japan, with Hanmi Pharmaceutical Co., Ltd. ("Hanmi"), for SPI-2012 based on Hanmi’s proprietary LAPSCOVERY Technology. Chemotherapy can cause myelosuppression and unacceptably low levels of white blood cells, making patients prone to infections, hospitalizations, and interruption of additional chemotherapy treatments.
Granulocyte colony-stimulating factor, or GCSF, stimulates the production of white blood cells by the bone marrow. A recombinant form of GCSF is used in appropriate cancer patients to accelerate recovery from neutropenia after chemotherapy, allowing higher-intensity treatment regimens to be given at full-dose and on schedule. We believe the worldwide annual market opportunity for GCSF-related drugs is over $6 billion.
In September 2014, we announced our decision to advance SPI-2012 to Phase 3 trials due to positive Phase 2 results in our collaboration program with Hanmi, and began discussions with the FDA and the European Medicines Agency (“EMA”) to discuss our Phase 3 design. In December 2015, we reached agreement with the FDA regarding our Phase 3 SPA for SPI-2012. We have designated more than 100 sites for this clinical trial, and initiated this Phase 3 clinical study beginning in January 2016. This trial will evaluate the safety and efficacy of SPI-2012 as a treatment for chemotherapy-induced neutropenia in patients with breast cancer, and will serve as the basis for our Biologics License Application ("BLA") filing.
POZIOTINIB
POZIOTINIB is a novel, oral pan-HER inhibitor that irreversibly blocks signaling through the Epidermal Growth Factor Receptor (EGFR, HER) Family of tyrosine-kinase receptors, including HER1 (erbB1; EGFR), HER2 (erbB2), HER4 (erbB4), and HER receptor mutations. This, in turn, leads to the inhibition of the proliferation of tumor cells that over-express these receptors. Mutations or over-expression/amplification of EGFR family receptors have been associated with a number of different cancers, including non-small cell lung cancer ("NSCLC"), breast cancer, and gastric cancer. POZIOTINIB has shown single agent activity in the treatment of various cancer types, including breast, gastric, colorectal and lung cancers. POZIOTINIB has shown promising early clinical activity in Phase 1 trials in patients who had failed multiple lines of treatment
5
including the HER2-directed therapies, trastuzumab and lapatinib. POZIOTINIB is currently being investigated by Hanmi in several mid-stage trials in different solid tumor indications including EGFR-mutant NSCLC, gastric cancer, head and neck cancer and HER2 positive breast cancer.
In November 2015, we submitted an Investigational New Drug ("IND") application with the FDA. In March 2016 we initiated our Phase 2 Breast Cancer Trial. The Phase 2 study is an open-label study that will enroll approximately 70 patients with HER-2 positive metastatic breast cancer, who have failed at least two HER-2 directed therapies. The dose and schedule of oral POZIOTINIB will be based on clinical experience from the studies in Korea, and in addition include the use of prophylactic therapies to help minimize known side-effects of HER-2 directed therapies.
In February 2015, we executed a global in-license agreement (excluding Korea and China) with Hanmi Pharmaceutical Co., Ltd for POZIOTINIB, a pan-HER inhibitor in Phase 2 clinical trials in return for our upfront payment and future regulatory and sales-dependent milestone payments. POZIOTINIB has shown single agent activity in the treatment of various cancer types, including breast, gastric, colorectal and lung cancers. In the Phase 1 study for this drug, 6 of 10 breast cancer patients demonstrated partial responses; we also believe the safety profile was consistent with similar drug classes, with four patients having a grade 3 diarrhea response.
EOQUIN (previously referred to as APAZIQUONE)
EOQUIN is a bio-reductive alkylating indoloquinone that is enzymatically activated by enzymes that are over expressed by bladder tumors that is being tested in non-muscle invasive bladder ("NMIBC").
The NCI estimates that the 2015 incidence and prevalence of bladder cancer in the U.S. was approximately 74,000 cases. The global presence of bladder cancer is estimated at 2.7 million cases. According to Botteman et al., (PharmacoEconomics 2003), bladder cancer is the most expensive cancer to treat on a lifetime basis. The overall cost of bladder cancer treatment in the U.S. is approximately $3.4 billion annually, most of which is related to the direct treatment of this disease.
The initial treatment of bladder cancer is to attempt a complete surgical removal of the tumor. However, bladder cancer is a highly recurrent disease with approximately 80% of patients recurring within five years, and a majority of patients recurring within two years. This high recurrence rate is attributed to:
• | the highly implantable nature of cancer cells that are dispersed during surgery; |
• | incomplete tumor resection; and |
• | tumors present in multiple locations in the bladder which may be missed or too small to visualize at the time of resection. |
Despite evidence in the published literature and guidance from the American and European Urology Associations, instillation of a chemotherapeutic agent immediately following surgery is not a standard clinical practice. Currently, there are no FDA approved drugs for this indication which may, in part, explain the difference between the literature and urology guidelines and actual clinical management of this disease. For more than 30 years, no new drugs have been introduced in the market for treatment of NMIBC. EOQUIN represents much needed therapy for patients and may provide a meaningful opportunity to reduce overall medical costs.
Pharmacokinetic studies have verified that EOQUIN is rarely detectable in the bloodstream of patients when it is administered either after surgical resection or as a part of a delayed multi-instillation protocol. EOQUIN is inactivated in the systemic circulation by the red blood cell fraction. The proposed dose therefore carries a minimal risk of systemic toxicity that could arise from absorption of a drug through the bladder wall into the bloodstream. These features of EOQUIN are distinct from other intravesical agents currently in use for the treatment of recurrent bladder cancer. An immediate instillation of EOQUIN may help by:
• | reducing tumor recurrence by destroying dispersed cancer cells that would otherwise re-implant onto the inner lining of the bladder; |
• | destroying remaining cancer cells at the site of tumor resection (also known as chemo-resection); and |
• | destroying tumors not observed during resection (also known as chemo-ablation). |
In August 2015, we reached agreement with the FDA on the Special Protocol Assessment ("SPA") of the planned Phase 3 clinical trial of EOQUIN. This trial commenced with its first patient dosing in October 2015, and is designed to evaluate the intravesical use of this drug for the treatment of patients with NMIBC as one or two instillations, immediately following
6
transurethral resection of bladder tumor ("TURBT"). In December 2015, we submitted our NDA for EOQUIN with the FDA, based on the pooled results of a previous Phase 3 program. In February 2016, the FDA accepted the EOQUIN NDA for review and indicated that it plans to hold an advisory committee meeting regarding the NDA and set a target decision date of December 11, 2016.
Manufacturing
We currently do not have internal manufacturing capabilities; therefore, all of our products are manufactured on a contract basis. We expect to continue to contract with third-party providers for manufacturing and packaging services, including active pharmaceutical ingredients (“API”) and finished-dosage products. We believe that our current agreements with third-party manufacturers provide for sufficient operating capacity to support the anticipated commercial demand and clinical requirements for our products. However, we are actively seeking multiple supplier sources for all our drug products in order to mitigate the risk of over-reliance on any one supplier. We attempt to prevent supply disruption through supply agreements, appropriate forecasting, and maintaining base stock levels. We believe that we could quickly enter into another supply or manufacturing agreement on substantially similar terms if we were required to do so.
Sales and Marketing
We presently market and sell our pharmaceutical products through a direct sales force in the U.S., and through distributors in Europe (and previously in Japan). Our U.S. sales team is divided between “corporate accounts” and “oncology accounts.” The primary decision makers for our products are oncologists and hematologists. As of December 31, 2015, our U.S. sales force (management, representatives, and direct support) numbered 100 employees.
Customers
Our product sales are concentrated to large pharmaceutical distributors (that ship and bill to hospitals and clinics). The customers that represent 10% or more of our total gross product sales in 2015, 2014 and 2013 are as follows:
Product Sales | ||||||||
2015 | 2014 | 2013 | ||||||
Oncology Supply, a division of ASD Specialty Healthcare, Inc., and its affiliates (excluding ICS) | 36.7 | % | 40.4 | % | 35.4 | % | ||
McKesson Corporation and its affiliates | 34.2 | % | 32.9 | % | 19.8 | % | ||
Cardinal Health, Inc. and its affiliates | 17.4 | % | * | * | ||||
Integrated Commercialization Solutions, Inc. (“ICS”) | * | * | 15.8 | % | ||||
* Less than 10%
We are exposed to credit risk associated with trade receivables that result from these product sales. We do not require collateral or deposits from our customers due to our assessment of their creditworthiness and our long-standing relationship with them. We maintain reserves for potential bad debt, though credit losses have historically been nominal and within management’s expectations. A summary of our customers that represent 10% or more of our accounts receivables, net, as of December 31, 2015 and 2014 are as follows:
Accounts Receivable, net | |||||
December 31, 2015 | December 31, 2014 | ||||
McKesson Corporation and its affiliates | 66.7 | % | 31.9 | % | |
Cardinal Health, Inc. and its affiliates | 23.8 | % | * | ||
Oncology Supply, a division of ASD Specialty Healthcare, Inc., and its affiliates (excluding ICS) | * | 51.1 | % | ||
ICS | * | 11.9 | % | ||
* Less than 10%
Competition
The pharmaceutical industry is characterized by rapidly-evolving biotechnology and intense competition, which we expect will continue. Many companies are engaged in research and development of compounds that are similar to ours – both commercialized and in development. In the event that one or more of our competitor’s programs are successful, the market for
7
some of our drug products could be reduced or eliminated. Any product for which we obtain FDA approval must also compete for market acceptance and market share.
Successful marketing of branded products depends primarily on the ability to communicate the effectiveness, safety, and value of the products to healthcare professionals in private practice, group practices, hospitals, academic institutions and managed care organizations. Competition for branded drugs is less driven by price and is more focused on innovation in treatment of disease, advanced drug delivery, and specific clinical benefits over competitive drug therapies. Unless our products are shown to be differentiated, i.e., have a better safety profile, efficacy, and cost-effectiveness, as compared to other alternatives, they may not gain acceptance by medical professionals and may therefore never be commercially successful.
Companies that have products on the market or in research and development that target the same indications as our products include, among others, Astra Zeneca PLC, Bayer AG, Endo International plc, Eli Lilly and Co., Novartis AG, Genentech, Inc. (Roche), Bristol-Myers Squibb Company, GlaxoSmithKline, Biogen-IDEC Pharmaceuticals, Inc., OSI Pharmaceuticals, Inc. (Astellas Pharma), Cephalon, Inc. (Teva Pharmaceuticals), Sanofi-Aventis, Inc., Pfizer, Inc., Genta Incorporated, Merck, Celgene Corporation, BiPar Sciences, Inc., Genzyme Corporation, Shire Pharmaceuticals, AbbVie, Poniard Pharmaceuticals, Inc., and Johnson & Johnson.
Each of the aforementioned companies may be more advanced in development of competing drug products. Many of these competitors are large and well-capitalized companies focusing on a wide range of cancers and drug indications, and have substantially greater resources and expertise than we do.
We believe that the current competitive landscape for each of our commercialized products is as follows:
(a) | FUSILEV is the levo-isomeric form of the racemic compound calcium, leucovorin, a product already approved for the same indication as FUSILEV. As there are currently two generic companies approved by the FDA to sell the leucovorin product, we are competing with a lower-cost alternative. In addition, as discussed in Section 1A, Risks Related to Our Business, FUSILEV faces competition from generic levo-leucovorin. |
(b) | ZEVALIN has two competitive products for its currently approved indications: |
• | Rituxan® (rituximab), marketed by Genentech and Biogen-IDEC Pharmaceuticals, is indicated for the treatment of patients with relapsed or refractory, low-grade or follicular, CD20-positive, B-cell NHL as a single agent; previously untreated follicular, CD20-positive, B-cell NHL in combination with CVP (cyclophosphamide, vincristine and prednisone combination) chemotherapy; and non-progressing (including stable disease), low-grade, CD20-positive B-cell NHL, as a single agent, after first-line CVP chemotherapy. Rituxan is administered as a part of various chemotherapy regimens and schedules, the vast majority of which, could be used in concert with other therapeutic agents, such as ZEVALIN, as part of a treatment plan. |
• | Treanda® (bendamustine hydrochloride) for Injection, for Intravenous Infusion, marketed by Cephalon, is indicated for the treatment of patients with indolent B-cell NHL that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. |
(c) | FOLOTYN, the first agent approved by the FDA for treatment of patients with relapsed or refractory PTCL, has two competitive products for its currently approved indications: |
• | Romidepsin, marketed by Celgene, Inc., was granted accelerated approval by the FDA in June 2011 for the treatment of patients with PTCL who have received at least one prior therapy. This was the second indication approved for romidepsin, which was initially approved by the FDA in November 2009 for the treatment of patients with CTCL who have received at least one prior systemic therapy. |
• | Brentuximab vedotin, marketed by Seattle Genetics, Inc., was also granted accelerated approval by the FDA in August 2011 for two indications, one of which was for the treatment of patients with systemic anaplastic large cell lymphoma (“ALCL”) after failure of at least one prior multi-agent chemotherapy regimen. ALCL is one of the subtypes of PTCL included in the labels of both FOLOTYN and romidepsin. |
We are aware of multiple investigational agents that are currently being studied in clinical trials for PTCL, including alisertib, which, if approved, may compete with FOLOTYN in the U.S. Because of the natural history of PTCL with repeated treatment failures, it is likely that many patients would receive treatment with more than one agent (e.g., BELEODAQ and
8
FOLOTYN). In addition, there are many existing approaches used in the treatment of relapsed or refractory PTCL, including combination chemotherapy and single agent regimens, which represent competition for FOLOTYN.
(d) | MARQIBO is a next generation liposomal form of standard vincristine. In its current indication, MARQIBO is approved for adult patients with relapsed or refractory Ph-ALL who have not responded or relapsed after two prior treatments. Currently, standard vincristine is not approved for the same indication as MARQIBO. |
(e) BELEODAQ is a histone deacetylase inhibitor indicated for the treatment of patients with relapsed or refractory PTCL. This indication is approved under accelerated approval based on tumor response rate and duration of response. An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trial.
Research and Development
New drug development is the process whereby drug product candidates are tested for the purpose of filing a BLA in the U.S. (or similar filing in other countries). Obtaining marketing approval from the FDA or similar regulatory authorities outside of the U.S. is an inherently uncertain, lengthy and expensive process that requires several phases of clinical trials to demonstrate to the satisfaction of the appropriate regulatory authorities that the products are both safe and effective for their respective indications. Our development focus is primarily based on acquiring and developing late-stage development drugs as compared to new drug discovery, which is particularly uncertain and lengthy.
Our in-development products are summarized below:

Our research and development expenses for drug development are comprised of our personnel expenses, contracted services with third parties, license fees and milestone payments to third parties, clinical trial costs, laboratory supplies, drug products, and certain allocations of corporate costs. The below table summarizes our research and development expenses by project in 2015, 2014 and 2013:
9
Research and Development Expenses for the Year Ended December 31, (in thousands) | |||||||||||
2015 | 2014 | 2013 | |||||||||
EOQUIN (previously APAZIQUONE) | $ | 4,147 | $ | 1,377 | $ | 1,078 | |||||
BELEODAQ | 1,320 | 20,911 | * | 6,733 | |||||||
FUSILEV | 885 | 442 | 4,517 | ||||||||
FOLOTYN | 2,650 | 4,927 | 2,992 | ||||||||
ZEVALIN | 3,025 | 6,950 | 8,572 | ||||||||
SPI-2012 | 1,133 | 4,141 | 1,403 | ||||||||
MARQIBO | 4,412 | 6,623 | 4,099 | ||||||||
EVOMELA (previously CE-MELPHALAN) | 8,568 | 5,966 | 3,400 | ||||||||
POZIOTINIB | 4,240 | — | — | ||||||||
Other in-development compounds and drugs | 633 | 1,967 | 3,632 | ||||||||
Total — Direct costs | 31,013 | 53,304 | 36,426 | ||||||||
Add: General research and development expenses (including personnel costs that correspond to more than one in-development project) | 21,571 | 21,073 | 13,335 | ||||||||
(Less): Reimbursements from development partners for SPI-2012 and BELEODAQ | (521 | ) | (2,758 | ) | (804 | ) | |||||
(Less): Incurred FOLOTYN study costs that reduce our drug development liability (see Note 16) | (1,297 | ) | (1,957 | ) | (2,287 | ) | |||||
Total research and development expenses | $ | 50,766 | $ | 69,662 | $ | 46,670 | |||||
* Inclusive of 2014 milestone payment of $17.8 million.
Patents and Proprietary Rights
Our Patents and Proprietary Rights
We in-license from third parties certain patent and related intellectual property rights related to our proprietary drug products. Under most of these license arrangements, we are generally responsible for all development, patent filing and maintenance costs, sales, marketing and liability insurance costs related to the drug products.
In addition, these licenses and agreements may require us to make royalty and other payments and to reasonably utilize the underlying technology of applicable patents. If we fail to comply with these and other terms in these licenses and agreements, we could lose the underlying rights to one or more of our potential products, which would adversely affect our product development and harm our business.
The protection, preservation and infringement-free commercial utilization of these patents and related intellectual property rights are very important to the successful execution of our strategy. However, the issuance of a patent is neither conclusive as to its validity nor as to the enforceable scope of the claims of the patent. Accordingly, our patents and the patents we have licensed may not prevent other companies from developing similar or functionally equivalent products or from successfully challenging the validity of our patents. If our patent applications are not allowed or, even if allowed and issued as patents, if such patents or the patents we have in-licensed are circumvented or not upheld by the courts, our ability to competitively utilize our patented products and technologies may be significantly reduced. Also, such patents may or may not provide competitive advantages for their respective products or they may be challenged or circumvented by competitors, in which case our ability to commercially sell these products may be diminished.
From time-to-time, we may need to obtain licenses to patents and other proprietary rights held by third parties to develop, manufacture and market our products. If we are unable to timely obtain these licenses on commercially reasonable terms, our ability to commercially exploit such products may be inhibited or prevented.
We believe that our patents and licenses are critical to operating our business, as summarized below by commercialized and in-development drug products.
FUSILEV: Fusilev had/has orphan drug exclusivity for two indications. These indications are for the treatment of (i) osteosarcoma (which expired on March 7, 2015) and (ii) metastatic colorectal cancer (which expires on April 29, 2018). FUSILEV also had/has a U.S. composition of matter patent that expires in March 2022.
10
In February 2015, the U.S. District Court for the District of Nevada found the asserted claims of the patent covering FUSILEV to be invalid, and on October 2, 2015, the Court of Appeals for the Federal Circuit affirmed that decision. On April 27, 2015, we filed suit in the U.S. District Court for the District of Columbia against the FDA seeking a temporary restraining order or preliminary injunction to suspend FDA approval of Sandoz Inc.'s ("Sandoz") Abbreviated New Drug Application ("ANDA"). The Company contends that Sandoz's ANDA should not have been approved until the expiry of our Orphan Drug Exclusivity on April 29, 2018. On April 29, 2015, the court denied the temporary restraining order and on May 27, 2015, the court entered summary judgment in favor of the FDA et al. On June 5, 2015, we filed our Notice of Appeal. Oral argument was held October 22, 2015. The ultimate outcome of this proceeding is uncertain.
ZEVALIN: We have sublicensed U.S. patents that cover the processes and tools for making monoclonal anti-bodies or MABs, in general, licensed U.S. patents that cover the CD-20 MAB in ZEVALIN as well as the use of ZEVALIN to treat NHL, and acquired patents covering the ZEVALIN compounding process (i.e., process of linking the CD-20 MAB to a radioactive isotope to make the patient-ready dosage form of ZEVALIN). These patents expire over a wide range of dates, and the licensed patents covering the CD-20 MAB began to expire in 2015. Additionally, we have U.S. patents covering the compounding process expiring in 2019, and we are considering filing new patent applications.
FOLOTYN: We have a composition of matter patent due to expire in 2022 following a five-year patent term extension in U.S. The composition of matter patent is due to expire in Europe in 2017 but is eligible for a similar patent term extension following regulatory approval in Europe. We also have patents covering the use of FOLOTYN for PTCL that will not expire until 2025. Additionally, we are considering filing new patent applications.
MARQIBO: We have U.S. and European patents covering the use of MARQIBO for leukemia, lymphoma and melanoma, and a U.S. patent covering the MARQIBO kit, all expiring in 2020. We have filed a patent cooperation treaty ("PCT") application claiming a method of encapsulating vincristine sulphate into liposomes. We are presently in the process of developing a “single vial” formulation of MARQIBO, and if we are successful, we believe our U.S. patent coverage could be extended to 2036.
BELEODAQ: The composition of matter patents that cover BELEODAQ and related compounds do not begin to expire until 2027. Currently, there are multiple U.S. and foreign patent applications pending that cover BELEODAQ formulations, uses and manufacturing and synthesis processes. We plan to file additional U.S. and foreign patent applications covering new formulations, uses, and manufacturing and synthesis processes.
EOQUIN: The U.S. formulation patent does not expire until 2022, and method of treatment of bladder cancer using a stabilized formulation that does not expire until 2024. Formulation patents outside the U.S. are due to expire in 2022. We have filed and plan to file additional U.S. and foreign patent applications covering new formulations and/or uses for this product.
SPI-2012: Composition of matter patents covering SPI-2012 are due to expire in 2025 in the U.S. and in 2024 outside the U.S. SPI-2012 is also covered by additional patents claiming various aspects of the technology that are due to expire between 2024 and 2030 and have filed patent applications for its formulation.
EVOMELA: This drug is covered by issued patents claiming improved Captisol ® technology that are due to expire between 2025 and 2029 in the U.S. Outside the U.S., we have issued patents that cover improved Captisol technology that are due to expire in 2025 and pending applications with anticipated expiry in 2029, if issued. We also have filed patent application covering Captisol-based formulation of EVOMELA in the U.S. and a number of other countries.
***
We are constantly evaluating our patent portfolio and are currently assessing and filing patent applications for our drug products and are considering new patent applications in order to maximize the life cycle of each of our products.
While the U.S. and the European Union (the "EU") are currently the largest potential markets for most of our products, we also have patents issued and patent applications pending outside of the U.S. and Europe. Limitations on patent protection in these countries, and the differences in what constitutes patentable subject matter in countries outside the U.S., may limit the protection we have on patents issued or licensed to us outside of the U.S. In addition, laws of foreign countries may not protect our intellectual property to the same extent as would laws in the U.S.
To minimize our costs and expenses and to maintain effective protection, we usually focus our patent and licensing activities within the U.S., the EU, Canada and Japan. In determining whether or not to seek a patent or to license any patent in a certain foreign country, we weigh the relevant costs and benefits, and consider, among other things, the market potential and profitability, the scope of patent protection afforded by the law of the jurisdiction and its enforceability, and the nature of terms
11
with any potential licensees. Failure to obtain adequate patent protection for our proprietary drugs and technology would impair our ability to be commercially competitive in these markets.
In conducting our business, we rely upon trade secrets, know-how, and licensing arrangements. We use customary practices for the protection of our confidential and proprietary information such as confidentiality agreements and trade secret protection measures. It is possible that these agreements will be breached or will not be enforceable in every instance, and that we will not have adequate remedies for any such breach. It is also possible that our trade secrets or know-how will otherwise become known or independently developed by competitors. The protection of know-how is particularly important because it is often necessary or useful information that allows us to practice the claims in the patents related to our proprietary drug products.
In addition to the specific intellectual property subjects discussed above, we have trademark protection in the U.S. for Spectrum Pharmaceuticals, Inc.®, FUSILEV®, MARQIBO®, BELEODAQ®, EOQUIN®, Spectrum Therapy Access Resources, STAR, ZEVALIN®, FOLOTYN flower design associated with FOLOTYN and RenaZorb®. We also have the FOLOTYN trademark and the associated flower design registered in Europe and other countries. Additionally, we have pending U.S. and ex-U.S. trademark applications for other potential marks.
The Patent Process
The U.S. Constitution provides Congress with the authority to provide inventors the exclusive right to their discoveries. Congress codified this right in U.S. Code Title 35, which gave the United States Patent and Trademark Office ("USPTO") the right to grant patents to inventors and defined the process for securing a U.S. patent. This process involves the filing of a patent application that instructs a person having ordinary skill in the respective art how to make and use the invention in clear and concise terms. The invention must be novel (i.e., not previously known) and non-obvious (i.e., not an obvious extension of what is already known). The patent application concludes with a series of claims that specifically describe the subject matter that the patent applicant considers his invention.
The USPTO undertakes an examination process that can take from one to seven years, or more, depending on the complexity of the patent and the problems encountered during examination.
In exchange for disclosing the invention to the public, for all U.S. patent applications filed after 1995, the successful patent applicant is currently provided a right to exclude others from making, using or selling the claimed invention for a period of 20 years from the effective filing date of the patent application.
Under certain circumstances, a patent term may be extended. Patent extensions are most frequently granted in the pharmaceutical and medical device industries under the Drug Price Competition and Pricing Term Restoration Act of 1984, or the Hatch-Waxman Act, to recover some of the time lost during the FDA regulatory process, subject to a number of limitations and exceptions. The patent term may be extended up to a maximum of five years; however, as a general rule, the average extension period granted for a new drug is approximately three years. Only one patent can be extended per FDA approved product, and a patent can only be extended once.
Product Exclusivity
Under the Hatch-Waxman Act, drug products are provided exclusivity whereby the FDA will not accept applications to market a generic form of an innovator reference listed drug product until the end of the prescribed period. A product is granted a five-year period of exclusivity if it contains a chemical entity never previously approved by the FDA either alone or in combination, although generic applications may be submitted after four years if they contain a certification of patent invalidity or non-infringement as further discussed below. A three-year period of exclusivity is granted to a previously approved product based on certain changes (e.g., in strength, dosage form, route of administration or conditions of use), where the application is supported by new clinical investigations that are essential to approval. In addition, in 1997, Congress amended the law to provide an additional six months of exclusivity as a reward for studying drugs in children. This pediatric exclusivity, which can be obtained during the approval process or after approval, effectively delays the approval of a generic application until six months after the expiration of any patent or other exclusivity that would otherwise delay approval, thus providing an additional six months free of generic competition. In order to qualify for pediatric exclusivity, the FDA must make a written request for pediatric studies, the application holder must agree to the request and complete the studies within the required timeframe, and the studies must be accepted by the FDA based on a determination that the studies fairly respond to the request. The provisions were enacted with a five-year sunset date, and have been reauthorized in 2002, 2007 and 2012.
Generic Approval and Patent Certification
12
The Hatch-Waxman Act also created the ANDA, approval process, which permits the approval of a generic version of a previously approved branded drug without the submission of a full NDA, and based in part on the FDA’s finding of safety and effectiveness for the reference listed drug. Applicants submitting an NDA are required to list patents associated with the drug product, which are published in the FDA Orange Book, and the timing of an ANDA approval depends in part on patent protection for the branded drug. When an ANDA is filed, the applicant must file a certification for each of the listed patents for the branded drug, stating one of the following: (1) that there is no patent information listed; (2) that such patent has expired; (3) that the patent will expire on a particular date (indicating that the ANDA may be approved on that date); or (4) that the drug for which approval is sought either does not infringe the patent or the patent is invalid, otherwise known as paragraph IV certification. If an ANDA applicant files a paragraph IV certification, it is required to provide the patent holder with notice of that certification. If the patent holder brings suit against the ANDA applicant for patent infringement within 45 days of receiving notice, the FDA may not approve the ANDA until the earlier of (i) 30 months from the patent holder’s receipt of the notice (the 30-month stay) or (ii) the issuance of a final, non-appealed, or non-appealable court decision finding the patent invalid, unenforceable or not infringed.
The Hatch-Waxman Act also provided an incentive for generic manufacturers to file paragraph IV certifications challenging patents that may be invalid, unenforceable, or not infringed, whereby the first company to successfully challenge a listed patent and receive ANDA approval is protected from competition from subsequent generic versions of the same drug product for up to 180 days after the earlier of (1) the date of the first commercial marketing of the first-filed ANDA applicant’s generic drug or (2) the date of a decision of a court in an action holding the relevant patent invalid, unenforceable, or not infringed. These 180-day exclusivity provisions have been the subject of litigation and administrative review, and the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or MMA, amended the provisions in several ways, including by providing that an ANDA applicant entitled to 180-day exclusivity may lose such exclusivity if any of the following events occur: (1) failure to market; (2) withdrawal of the ANDA; (3) change in patent certification; (4) failure to obtain tentative approval; (5) illegal settlement agreement; and (6) patent expiration.
With respect to the illegal settlement prong, the MMA amendments require that certain types of settlement agreements entered into between branded and generic pharmaceutical companies related to the manufacture, marketing and sale of generic versions of branded drugs are required to be filed with the Federal Trade Commission and the Department of Justice for review of potential anti-competitive practices. This requirement could affect the manner in which generic drug manufacturers resolve intellectual property litigation and other disputes with branded pharmaceutical companies, and could result generally in an increase in private-party litigation against pharmaceutical companies. The impact of this requirement, and the potential governmental investigations and private-party lawsuits associated with arrangements between brand name and generic drug manufacturers, remains uncertain. In addition, Congress has considered enacting legislation that would prohibit such settlements between brand name and generic drug manufacturers. Such a provision was considered as part of the Patient Protection and Affordable Care Act, or PPACA, signed into law on March 23, 2010. However, Congress removed the provision prior to passage. It is possible that Congress will again consider a ban on such settlements between brand name and generic drug manufacturers in the future.
The PPACA provides exclusivity protections for certain innovator biological products and a framework for FDA review and approval of biosimilar and interchangeable versions of innovator biologic products. The PPACA provides that no application for a biosimilar product may be approved until 12 years after the date on which the innovator product was first licensed, and no application may be submitted until four years after the date of first licensure. Products deemed interchangeable (as opposed to biosimilar) are also eligible for certain exclusivity.
Orphan Drug Designation
Some jurisdictions, including Europe and the U.S., may designate drugs for relatively small patient populations as “orphan” drugs. The FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the U.S., and a drug may also be considered an orphan even if the drug treats a disease or condition affecting more than 200,000 individuals in the U.S. Orphan drug designation does not necessarily convey any advantage in, or shorten the duration of, the regulatory review and process for marketing approval. If a product with an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to seven years of orphan drug exclusivity, during which time the FDA will not approve any other application to market the same drug for the same indication except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Also, competitors are not prohibited from receiving approval to market the same drug or biologic for a different indication than that which received orphan approval.
Under EU medicines laws, the criteria for designating an “orphan medicinal product” are similar in principle to those in the U.S. Criteria for orphan designation are set out in Article 3 of Regulation (EC) 141/2000 on the basis of two alternative
13
conditions. A medicinal product may be designated as orphan if it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the EU, when the application is made. This is commonly known as the “disease prevalence criterion” Alternatively, a product may be so designated if it is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the EU and if without incentives it is unlikely that the marketing of the product in the EU would generate sufficient return to justify the necessary investment. This is commonly known as the “insufficient return criterion.”
These two alternative criteria must cumulatively meet the second condition that there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the EU, or if such a method exists, the product will be of significant benefit to those affected by the condition. “Significant benefit” is defined in Regulation (EC) 847/2000 as a clinically relevant advantage or a major contribution to patient care.
Upon grant of a marketing authorization, orphan medicinal products are entitled to ten years of market exclusivity in respect of the approved therapeutic indication. Within the period of market exclusivity, no competent authority in the EU is permitted to accept an application for marketing authorization, a variation or a line-extension for the same approved therapeutic indication in respect of a similar medicinal product pursuant to Article 8.1 of Regulation 141/2000 unless one of the derogations set out in Article 8.3 of the same Regulation applies. In order to determine whether two products are considered similar, Regulation 847/2000 requires an assessment of the principal molecular structure and the underlying mode of action. Any minor variation or modification of the principal molecular structure would not ordinarily render the second product dissimilar to the first authorized product.
In order for the second applicant to break the market exclusivity granted to the first authorized similar medicinal product in respect of the same therapeutic indication, the second applicant would principally rely upon data to demonstrate that its product is safer, more efficacious or clinically superior to the first product pursuant to Article 8.3I of Regulation 141/2000. Ordinarily, such an assessment will require a head-to-head comparative clinical trial for the purpose of demonstrating clinical superiority.
The 10-year market exclusivity may be reduced to six years if at the end of the fifth year it is established that the product no longer meets the criteria for orphan designation on the basis of available evidence. To date, each of our five commercialized drugs continues to meet the orphan drug designation requirements.
FUSILEV had/has been granted orphan drug designations for its use in conjunction with high dose methotrexate in the treatment of osteosarcoma and for its use in combination chemotherapy with the approved agent 5-fluorouracil in the palliative treatment of metastatic adenocarcinoma of the colon and rectum (colorectal cancer). In addition, FOLOTYN has been granted an orphan drug designation for the treatment of T-cell lymphoma and BELEODAQ has been granted an orphan drug designation for PTCL. Lastly, MARQIBO has been granted orphan drug designations for its use in the treatment of adult patients with ALL in second or greater relapse or whose disease has progressed following two or more anti-leukemia therapies, and ZEVALIN has orphan drug designations for the treatment of patients with relapsed or refractory low-grade, follicular, or transformed B-cell NHL, including patients with Rituximab refractory follicular NHL.
Governmental Regulation
The development, production and marketing of our proprietary and generic drug and biologic products are subject to regulation for safety, efficacy and quality by numerous governmental authorities in the U.S. and other countries. In the U.S., drugs and biologics are subject to rigorous regulation. The Federal Food, Drug, and Cosmetic Act, as amended from time to time, and the regulations promulgated there under, as well as other federal and state statutes and regulations, govern, among other things, the development, approval, manufacture, safety, labeling, storage, record keeping, distribution, promotion, and advertising of our products. Product development and approval within this regulatory framework, including for drugs already at a clinical stage of development, can take many years and require the expenditure of substantial resources, and to obtain FDA approval, a product must satisfy mandatory quality, safety and efficacy requirements. In addition, each drug-manufacturing establishment must be registered with the FDA. Domestic manufacturing establishments must comply with the FDA’s current Good Manufacturing Practices (“cGMP”), regulations and are subject to inspections by the FDA. To supply drug ingredients or products for use in the U.S., foreign manufacturing establishments must also comply with cGMP and are subject to inspections by the FDA or by other regulatory authorities in certain countries under reciprocal agreements with the FDA.
General Information about the Drug Approval Process and Post-Marketing Requirements
The U.S. system of new drug and biologics approval is a rigorous process. Only a small percentage of compounds that enter the pre-clinical testing stage are ever approved for commercialization. Our strategy focuses on in-licensing clinical stage
14
drug products that are already in or about to enter human clinical trials. A late-stage focus helps us to effectively manage the high cost of drug development by focusing on compounds that have already passed the many hurdles in the pre-clinical and early clinical process.
The following general comments about the drug approval process are relevant to the development activities we are undertaking with our proprietary products.
Pre-clinical Testing: During the pre-clinical testing stage, laboratory and animal studies are conducted to show biological activity of a drug or biologic compound against the targeted disease. The compound is evaluated for safety. While all of our compounds are currently in clinical trials, it is possible that additional pre-clinical testing could be requested by a regulatory authority for any of our compounds.
Investigational New Drug Application: After certain pre-clinical studies are completed, an Investigational New Drug (“IND”) application is submitted to the FDA to request the ability to begin human testing of the drug or biologic. An IND becomes effective thirty days after the FDA receives the application (unless the FDA notifies the sponsor of a clinical hold), or upon prior notification by the FDA.
Phase 1 Clinical Trials: These trials typically involve small numbers of healthy volunteers or patients and usually define a drug candidate’s safety profile, including the safe dosage range.
Phase 2 Clinical Trials: In Phase 2 clinical trials, controlled studies of human patients with the targeted disease are conducted to assess the drug’s effectiveness. These studies are designed primarily to determine the appropriate dose levels, dose schedules and route(s) of administration, and to evaluate the effectiveness of the drug or biologic on humans, as well as to determine if there are any side effects on humans to expand the safety profile following Phase 1. These clinical trials, and Phase 3 trials discussed below, are designed to evaluate the product’s overall benefit-risk profile, and to provide information for physician labeling.
Phase 3 Clinical Trials: This phase usually involves a larger number of patients with the targeted disease. Investigators (typically physicians) monitor the patients to determine the drug candidate’s efficacy and to observe and report any adverse reactions that may result from long-term use of the drug on a large, more widespread, patient population. During the Phase 3 clinical trials, typically the drug candidate is compared to either a placebo or a standard treatment for the target disease.
New Drug Application or Biologic License Application: After completion of all three clinical trial Phases, if the data indicates that the drug is safe and effective, a NDA or BLA is filed with the FDA requesting FDA approval to market the new drug as a treatment for the target disease.
Fast Track and Priority Review: The FDA has established procedures for accelerating the approval of drugs to be marketed for serious or life threatening diseases for which the manufacturer can demonstrate the potential to address unmet medical needs.
Abbreviated New Drug Application: An ANDA is an abbreviated new drug application for generic drugs created by the Hatch-Waxman Act. When a company files an ANDA, it must make a patent certification regarding the patents covering the branded product listed in the FDA’s Orange Book. The ANDA drug development process generally takes less time than the NDA drug development process since the ANDA process usually does not require new clinical trials establishing the safety and efficacy of the drug product.
NDA/BLA and ANDA Approval: The FDA approves drugs and biologics that are subject to NDA and BLA review based on data in the application demonstrating the product is safe and effective in its proposed use(s) and that the product’s benefits outweigh its risks. The FDA will also review the NDA or BLA applicant’s manufacturing process and controls to ensure they are adequate to preserve the drug’s identity, strength, quality, and purity. Finally, the FDA will review and approve the product’s proposed labeling. As for the ANDA approval process, these “abbreviated” applications are generally not required to include preclinical or clinical data to establish safety and effectiveness. Rather, an ANDA must demonstrate both chemical equivalence and bio-equivalence (the rate and extent of absorption in the body) to the innovator drug — unless a bio-equivalence waiver is granted by the FDA.
Phase 4 Clinical Trials: After a drug has been approved by the FDA, Phase 4 studies may be conducted to explore additional patient populations, compare the drug to a competitor, or to further study the risks, benefits and optimal use of a drug. These studies may be a requirement as a condition of the initial approval of the NDA or BLA.
15
Post-Approval Studies Requirements under FDAAA: The Food and Drug Administration Amendments Act of 2007, or FDAAA, significantly added to the FDA’s authority to require post-approval studies. Under the FDAAA, if the FDA becomes aware of new safety information after approval of a product, they may require us to conduct further clinical trials to assess a known serious risk, signals of serious risk or to identify an unexpected serious risk. If required to conduct a post-approval study, periodic status reports must be submitted to the FDA. Failure to conduct such post-approval studies in a timely manner may result in administrative action being taken by FDA, including substantial civil fines.
Risk Evaluation and Mitigation Strategy Authority under FDAAA: The FDAAA also gave the FDA new authority to require the implementation of a Risk Evaluation and Mitigation Strategy, or REMS, for a product when necessary to minimize known and preventable safety risks associated with the product. The FDA may require the submission of a REMS before a product is approved, or after approval based on “new safety information,” including new analyses of existing safety information. A REMS may include a medication guide, patient package insert, a plan for communication with healthcare providers, or other elements as the FDA deems are necessary to assure safe use of the product, which could include imposing certain restrictions on distribution or use of a product. A REMS must include a timetable for submission of assessments of the strategy at specified time intervals. Failure to comply with a REMS, including the submission of a required assessment, may result in substantial civil or criminal penalties.
Other Issues Related to Product Safety: Adverse events that are reported after marketing approval also can result in additional limitations being placed on a product’s use and, potentially, withdrawal of the product from the market. In addition, under the FDAAA, the FDA has authority to mandate labeling changes to products at any point in a product’s lifecycle based on new safety information derived from clinical trials, post-approval studies, peer-reviewed medical literature, or post-market risk identification and analysis systems data.
FDA Enforcement
The development of drug and biologic products, as well as the marketing of approved drugs and biologics, is subject to substantial continuing regulation by the FDA, including regulation of adverse event reporting, manufacturing practices and the advertising and promotion of the product. Failure to comply with the FDA and other governmental regulations can result in fines, unanticipated compliance expenditures, recall or seizure of products, total or partial suspension of production and/or distribution, suspension of the FDA’s review of NDAs, BLAs, ANDAs or other product applications, enforcement actions, injunctions and criminal prosecution. Under certain circumstances, the FDA also has the authority to revoke previously granted drug approvals.
With respect specifically to information submitted to the FDA in support of marketing applications, the FDA, under its Fraud, Untrue Statements of Material Facts, Bribery and Illegal Gratuities Policy, can significantly delay the approval of a marketing application, or seek to withdraw an approved application where it identifies fraud or discrepancies in regulatory submissions. Such actions by the FDA may significantly delay or suspend substantive scientific review of a pending application during validity assessment or remove approved products from the market until the assessment is complete and questions regarding reliability of the data are resolved. In addition, the Generic Drug Enforcement Act of 1992 established penalties for wrongdoing in connection with the development or submission of an ANDA. Under this Act, the FDA has the authority to permanently or temporarily bar companies or individuals from submitting or assisting in the submission of an ANDA, and to temporarily deny approval and suspend applications to market generic drugs. The FDA may also suspend the distribution of all drugs approved or developed in connection with certain wrongful conduct and/or withdraw approval of an ANDA and seek civil penalties.
Healthcare Reform
Continuing studies of the proper utilization, safety and efficacy of pharmaceuticals and other health care products are being conducted by industry, government agencies and others. Such studies, which increasingly employ sophisticated methods and techniques, can call into question the utilization, safety and efficacy of previously marketed products and in some cases have resulted, and may in the future result, in the discontinuance of their marketing.
The Patient Centered Outcomes Research Institute (the "Institute"), a private, non-profit corporation created as a result of the PPACA, is tasked with assisting patients, clinicians, purchasers, and policy-makers in making informed health decisions. One of the Institute’s initiatives will be to conduct comparative clinical effectiveness research, which is defined as “research evaluating and comparing health outcomes and the clinical effectiveness, risks, and benefits of two or more medical treatments, services, and items.” It is important to note that the Institute would not be permitted to mandate coverage, reimbursement, or other policies for any public or private payer, however the outcome of the Institute's initiatives could influence prescriber behavior.
16
Foreign Regulation
Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country/region to country/region, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement also may vary, sometimes significantly, from country/region to country/region.
Under the EU regulatory systems, we may submit marketing authorization applications either under a centralized procedure or decentralized procedure or the mutual recognition procedure. The centralized procedure is mandatory for medicines produced by a biotechnological process. The procedure is also mandatory for new active substances which are indicated for treatment of several diseases or conditions, including cancer and orphan conditions. Companies may apply for centralized assessment if the product contains a new active substance or the product constitutes significant therapeutic, scientific or technical innovation or the granting of authorization under the centralized procedure is in the interests of the EU patients. A centralized marketing authorization is valid in all EU member states. This marketing authorization is issued in the form of a European Commission decision which is legally binding in its entirety to which it is addressed.
Directive 2004/27/EC introduced two parallel procedures to the centralized procedure to allow a product to be progressively authorized in each of the member states of the EU. They are the decentralized procedure and the mutual recognition procedure. The mutual recognition procedure applies where the product has already been authorized in a member state of the EU that will act as reference member state. The national marketing authorization granted by the reference member state forms the basis for mutual recognition in the member states chosen by the applicant. In the decentralized procedure, the product in question is not authorized in any one the EU member states. In such a situation, the applicant company will request a member state to act as the reference member state to lead the scientific assessment for the benefit/risk balance for agreement by the concerned member states. In both cases, the concerned member states have up to 90 days to accept or raise reasoned objections to the assessment made by the reference member state.
In addition, pricing and reimbursement is subject to negotiation and regulation in most countries outside the U.S. Increasingly, adoption of a new product for use in national health services is subject to health technology assessment under the national rules and regulations to establish the clinical effectiveness and cost-effectiveness of a new treatment. In some countries, in order to contain health care expenditures, reference price is introduced in order for the national healthcare providers to achieve a price comparable to the reference price in the same therapeutic category. We may therefore face the risk that the resulting prices would be insufficient to generate an acceptable return to us.
Third Party Reimbursement and Pricing Controls
In the U.S. and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payers, such as government and private insurance plans. Third-party payers are increasingly challenging the prices charged for medical products and services. It is time-consuming and expensive for us to go through the process of seeking reimbursement from Medicare and private payers. Our products may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis.
The PPACA enacted significant reforms, including revising the definition of “average manufacturer price” for reporting purposes, increasing Medicaid rebates, expanding the 340B drug discount program, and making changes to affect the Medicare Part D coverage gap, or “donut hole.” In the coming years, additional significant changes could be made to governmental healthcare programs, and the U.S. healthcare system as a whole, that may result in significantly increased rebates, decreased pricing flexibility, diminished negotiating flexibility, coverage and reimbursement limitations based upon comparative and cost-effectiveness reviews, and other measures that could significantly impact the success of our products.
In many foreign markets, including the countries in the EU, pricing of pharmaceutical products is subject to governmental control. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control.
Employees
As of December 31, 2015, we had 212 employees (as compared to 241 employees as of December 31, 2014), 10 of whom hold an M.D. degree, and 18 of whom hold a Ph.D. degree. We believe that the success of our business will depend, in part, on
17
our ability to attract and retain uniquely qualified personnel. Our employees are not part of any collective bargaining agreements, and we believe that we have good relations with our employees.
General Information
We are a Delaware corporation. We originally incorporated in Colorado in December 1987 as Americus Funding Corporation. We changed our corporate name in August 1996 to NeoTherapeutics, Inc., and reincorporated in Delaware in June 1997. We changed our corporate name in December 2002 to Spectrum Pharmaceuticals, Inc.
Our principal executive office is located at 11500 South Eastern Avenue, Suite 240, Henderson, Nevada 89052. Our telephone number is (702) 835-6300. Our website is located at www.sppirx.com. The information that can be accessed through our website is not incorporated by reference into this Annual Report on Form 10-K and should not be considered to be a part hereof.
We make our proxy statements, annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K (and related amendments to these reports, as applicable) available on our website free of charge as soon as practicable after filing or furnishing with the SEC.
All such reports are also available free of charge via EDGAR through the SEC website at www.sec.gov. In addition, the public may read and copy materials filed by us with the SEC at the SEC’s public reference room located at 100 F Street, NE, Washington, D.C., 20549. Information regarding operation of the SEC’s public reference room can be obtained by calling the SEC at 1-800-732-0330.
ITEM 1A. RISK FACTORS
Before deciding to invest in our company, or to maintain or increase your investment, you should carefully consider the risks described below, in addition to the other information contained in this Annual Report on Form 10-K and other reports we have filed with the SEC. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us, or that we currently deem immaterial, may also affect our business operations. If any of these risks are realized, our business, financial condition, or results of operations could be seriously harmed and in that event, the market price for our common stock could decline, and you may lose all or part of your investment.
These risk factors should be considered in connection with evaluating the forward-looking statements contained in this Annual Report on Form 10-K. These factors could cause actual results and conditions to differ materially from those projected in our forward-looking statements.
Risks Related to Our Business
Wholesaler actions could increase competitive and pricing pressures on pharmaceutical manufacturers, including us.
We sell certain of our products primarily through wholesalers, who in turn sell to end-users. These wholesalers comprise a significant part of the distribution network for pharmaceutical products in the U.S. A small number of large wholesalers control a significant share of the market, which can increase competitive and pricing pressures on pharmaceutical manufacturers, including us. In addition, wholesalers may apply pricing pressure through their fee-for-service arrangements.
We are aware of several competitors attempting to develop and market products competitive to our products, which may reduce or eliminate our commercial opportunities.
The pharmaceutical and biotechnology industries are intensely competitive and subject to rapid and significant technological changes, and a number of companies are pursuing the development of pharmaceuticals and products that target the same diseases and conditions that our products target, including products currently commercialized. We cannot predict with accuracy the timing or impact of the introduction of potentially competitive products or their possible effect on our sales. Certain potentially competitive products to our products are in various stages of development, some of which have pending applications for approval with the FDA or have been approved by regulatory authorities in other countries. Also, there are many ongoing studies with currently marketed products and other developmental products, which may yield new data that could adversely impact the use of our products in their current and potential future indications. The introduction of competitive products could significantly reduce our sales, which, in turn would adversely impact our financial and operating results.
18
We are a small company relative to our principal competitors, and our limited financial resources may limit our ability to develop and market our drug products.
Many companies, both public and private, including well-known pharmaceutical companies and smaller niche-focused companies, are developing products to treat many, if not all, of the diseases we are pursuing or are currently distributing drug products that directly compete with the drugs that we sell or that we intend to develop, market and distribute. Many of these companies have substantially greater financial, research and development, manufacturing, marketing and sales experience and resources than us. As a result, our competitors may be more successful than us in developing their products, obtaining regulatory approvals and marketing their products to consumers.
Competition for branded or proprietary drugs is less driven by price and is more focused on innovation in the treatment of disease, advanced drug delivery and specific clinical benefits over competitive drug therapies. We may not be successful in any or all of our current clinical studies; or if successful, and if one or more of our drug products is approved by the FDA, we may encounter direct competition from other companies who may be developing products for similar or the same indications as our drug products.
Companies that have products on the market or in research and development that target the same indications as our products target include, among others, Astra Zeneca PLC, Bayer AG, Endo International plc, Eli Lilly and Co., Novartis AG, Genentech, Inc. (Roche), Bristol-Myers Squibb Company, GlaxoSmithKline, Biogen-IDEC Pharmaceuticals, Inc., OSI Pharmaceuticals, Inc. (Astellas Pharma), Cephalon, Inc. (Teva Pharmaceuticals), Sanofi-Aventis, Inc., Pfizer, Inc., Genta Incorporated, Merck, Celgene Corporation, BiPar Sciences, Inc., Genzyme Corporation, Shire Pharmaceuticals, AbbVie, Poniard Pharmaceuticals, Inc., and Johnson & Johnson, who may be more advanced in the development of competing drug products or are more established.
Many of our competitors are large and well-capitalized companies focusing on a wide range of diseases and drug indications, and have substantially greater financial, research and development, marketing, human and other resources than we do. Furthermore, large pharmaceutical companies have significantly more experience than we do in pre-clinical testing, human clinical trials and regulatory approval procedures, among other things.
Our dependence on key executives, scientists and sales and marketing personnel could impact the development and management of our business.
We are highly dependent upon our ability to attract and retain qualified scientific, technical sales and marketing and managerial personnel. There is intense competition for qualified personnel in the pharmaceutical and biotechnology industries, and we cannot be sure that we will be able to continue to attract and retain the qualified personnel necessary, particularly as business prospects change, for the development and management of our business. Although we do not believe the loss of one individual would materially harm our business, our business might be harmed by the loss of the services of multiple existing personnel, as well as the failure to recruit additional key scientific, technical and managerial personnel in a timely manner. Much of the know-how we have developed resides in our scientific and technical personnel and is not readily transferable to other personnel. While we have an employment agreement with our Chief Executive Officer, we do not have employment agreements with any of our other key scientific, technical, or managerial employees.
If the distributors that we rely upon to sell our products fail to perform, our business may be adversely affected.
Our success depends on the continued customer support efforts of our network of distributors. In the U.S., we sell our products to a small number of distributors who in turn sell-through to patient health care providers. These distributors also provide multiple logistics services relating to the distribution of our products, including transportation, warehousing, cross-docking, inventory management, packaging and freight-forwarding. We do not promote products to these distributors and they do not set or determine demand for products. The use of distributors involves certain risks, including, but not limited to, risks that these distributors will:
• | not provide us with accurate or timely information regarding their inventories, the number of patients who are using our products or complaints about our products; |
• | not purchase sufficient inventory on hand to fulfill end user orders in a timely manner; |
• | be unable to satisfy financial obligations to us or others; and |
• | cease operations. |
19
Any such actions may result in decreased sales of our products, which would harm our business.
Because we have obtained accelerated approval to market FOLOTYN, BELEODAQ and MARQIBO, we are subject to ongoing regulatory obligations and review, including completion of the post-approval requirements.
FOLOTYN and BELEODAQ were approved for the treatment of patients with relapsed or refractory PTCL, and MARQIBO was approved for the treatment of adult patients with Philadelphia chromosome-negative acute lymphoblastic leukemia in second or greater relapse or whose disease has progressed following two or more anti-leukemia therapies, under the FDA’s accelerated approval regulations. These provisions allow the FDA to approve products for cancer or other serious or life threatening diseases based on initial positive data from clinical trials. Under these provisions, we are subject to certain post-approval requirements. Specifically, we are required to conduct Phase 1 dose escalating studies and a Phase 3 randomized study for FOLOTYN and BELEODAQ in patients with PTCL. The FDA also required that we conduct two Phase 1 trials to assess whether FOLOTYN poses a serious risk of altered drug levels resulting from organ impairment as well as additional post-marketing studies with BELEODAQ. For MARQIBO, we are required to conduct a randomized Phase 3 study in patients over 60 years of age with newly diagnosed ALL. Failure to complete the studies or adhere to the timelines established by the FDA could result in penalties, including fines or withdrawal of FOLOTYN, BELEODAQ, and/or MARQIBO from the market.
The FDA may also initiate proceedings to withdraw approval or request that we voluntarily withdraw these drugs from the market if our Phase 3 studies fail to confirm clinical benefit. Further, the FDA may require us to amend these drugs package insert, including by strengthening the warnings and precautions section or institute a REMS based on the results of these studies or clinical experience. We are also subject to additional, continuing post-approval regulatory obligations, including the possibility of additional clinical studies required by the FDA, safety reporting requirements and regulatory oversight of the promotion and marketing of these drugs.
We and our third-party contract manufacturers are subject to inspection by regulatory authorities.
We and our third-party manufacturers are required to adhere to cGMP. The cGMP regulations cover all aspects of the manufacturing, storage, testing, quality control and record keeping relating to our drugs.
We and or our third-party contract manufacturers are subject to periodic inspection by the FDA and foreign regulatory authorities to ensure compliance with cGMP or other applicable government regulations and corresponding foreign standards. We have limited control over a third-party manufacturer’s compliance with these regulations and standards. If we or our third-party manufacturers fail to comply with applicable regulatory requirements, we may be subject to fines, suspension, modification or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Our collaboration partner, Mundipharma, may not be successful in obtaining regulatory approval for FOLOTYN in a number of countries and FOLOTYN is subject to numerous complex regulatory requirements.
Our collaboration partner, Mundipharma, may not be successful in obtaining regulatory approval for FOLOTYN in a number of countries and FOLOTYN is subject to numerous complex regulatory requirements. Failure to comply with, or changes to, the regulatory requirements that are applicable to FOLOTYN outside the United States may result in a variety of consequences, including the following:
• | restrictions on FOLOTYN or our manufacturing processes; |
• | warning letters; |
• | withdrawal of FOLOTYN from the market; |
• | voluntary or mandatory recall of FOLOTYN; |
• | fines against us; |
• | suspension or withdrawal of regulatory approvals for FOLOTYN; |
• | suspension or termination of any of our ongoing clinical trials of FOLOTYN; |
• | refusal to permit import or export of FOLOTYN; |
• | refusal to approve pending applications or supplements to approved applications that we submit; |
20
• | denial of permission to file an application or supplement in a jurisdiction; |
• | product seizure; and/or |
• | injunctions, consent decrees, or the imposition of civil or criminal penalties against us. |
If actual future payments for allowances, discounts, returns, rebates and chargebacks exceed the estimates we made at the time of the sale of our products, including, without limitation, due to a change in the composition of our sales over time, our financial position, results of operations and cash flows may be materially and negatively impacted.
We recognize product revenue net of estimated allowances for discounts, returns, rebates and chargebacks. Such estimates require our most subjective and complex judgment due to the need to make estimates about matters that are inherently uncertain. Based on industry practice, pharmaceutical companies, including us, have liberal return policies. We are obligated to accept from customers the return of FUSILEV, MARQIBO, and BELEODAQ that have reached their expiration date, and up to six months thereafter. We authorize returns for damaged products and exchanges for expired products in accordance with our return goods policy and procedures. Also, like our competitors, we also give credits for chargebacks to wholesale customers that have contracts with us for their sales to hospitals, group purchasing organizations, pharmacies or other retail customers.
A chargeback is the difference between the price the wholesale customer (in our case, the GPOs) pays (wholesale acquisition cost) and the price that the GPO’s end-customer pays for a product (contracted customer). For instance, our products are subject to certain programs with federal government qualified entities whereby pricing on products is discounted to such entities and results in a chargeback claim to us. To the extent that our sales to discount purchasers, such as federal government qualified entities, increases, our chargebacks will also increase. There may be significant lag time between our original sale to the wholesaler and our receipt of the corresponding government chargeback claims from our wholesalers.
Our products are subject to state government-managed Medicaid programs, whereby rebates for purchases are issued to participating state governments. These rebates arise when the patient treated with our products is covered under Medicaid. Our calculations related to these Medicaid rebate accruals require us to estimate end-user and patient mix to determine which of our sales will likely be subject to these rebates. There is a significant time lag in us receiving these rebate notices (generally several months after our sale is made). Our estimates are based on our historical claims from participating state governments, as supplemented by management’s judgment.
Although we believe that we have sufficient allowances, actual results may differ significantly from our estimated allowances for discounts, returns, rebates and chargebacks. Changes in estimates and assumptions based upon actual results may have a material impact on our financial condition, results of operations and cash flows. Such changes to estimates will be made to the financial statements in the year in which the estimate is changed. In addition, our financial position, results of operations and cash flows may be materially and negatively impacted if actual future payments for allowances, discounts, returns, rebates and chargebacks exceed the estimates we made at the time of the sale of our products.
Reports of adverse events or safety concerns involving our products or similar agents, sold by us or our development partners and/or licensees, could delay or prevent us from obtaining or maintaining regulatory approval or negatively impact sales.
Certain of our products may cause serious adverse events. Discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, could interrupt, delay or halt clinical trials of such products, including the FDA-required post-approval studies, and could result in the FDA or other regulatory authorities denying or withdrawing approval of our products for any or all indications. The FDA, other regulatory authorities or we may suspend or terminate clinical trials at any time. We may also be required to update the package inserts based on reports of adverse events or safety concerns or implement a REMS, which could adversely affect such products acceptance in the market. In addition, the public perception of our products might be adversely affected, which could harm our business and results of operations and cause the market price of our common stock to decline, even if the concern relates to another company’s product or product candidate. Our planned trials to demonstrate efficacy in a variety of indications and to better manage side effect profiles of certain of our products may not be successful.
The marketing and sale of our products may be adversely affected by the marketing and sales efforts of third parties who sell our products or similar products outside of our territories.
21
We have only licensed the rights to develop and market our products in limited territories. Other companies market and sell the same products in other parts of the world. If, as a result of other companies’ actions, negative publicity is associated with our products or similar products, our own efforts to successfully market and sell our products in our markets may be adversely impacted.
Our sales depend on coverage and reimbursement from third-party payers and a reduction in the coverage and/or reimbursement for our products could have a material adverse effect on our product sales, business and results of operations.
Sales of our products are dependent on the availability and extent of coverage and reimbursement from third-party payers, including government programs and private insurance plans. Governments and private payers may regulate prices, reimbursement levels and/or access to our products to contain costs or to affect levels of use. We rely in large part on the reimbursement of our products through government programs such as Medicare and Medicaid in the United States, and a reduction in the coverage and/or reimbursement for our products could have a material adverse effect on our product sales, business and results of operations.
A substantial portion of our U.S. business relies on reimbursement from the U.S. federal government under Medicare Part B coverage. Most of our products furnished to Medicare beneficiaries in both a physician office setting and hospital outpatient setting are reimbursed under the Medicare Part B Average Sales Price, or ASP, payment methodology. ASP-based reimbursement of our products under Medicare may be below or could fall below the cost that some medical providers pay for such products, which could materially and adversely affect sales of our products. We also face risks relating to the reporting of pricing data that affect the U.S. reimbursement of and discounts for our products. ASP data are calculated by the manufacturer based on a formula defined by statute and regulation and are then submitted to the Centers for Medicare & Medicaid Services (the "CMS"), the agency responsible for administering the Medicare program, on a quarterly basis.
CMS uses those ASP data to determine the applicable reimbursement rates for our products under Medicare Part B. However, the statute, regulations and CMS guidance do not define specific methodologies for all aspects of the reporting of ASP data. For example, CMS has not provided specific guidance regarding administrative fees paid to group purchasing organizations, or GPOs, in the ASP calculation. CMS directs that manufacturers make “reasonable assumptions” in their calculation of ASP data in the absence of specific CMS guidance on a topic. As a result, we are required to apply our reasonable judgment to certain aspects of calculating ASP data. If our submitted ASP data are incorrect, we may become subject to substantial fines and penalties or other government enforcement actions, which could have a material adverse impact on our business and results of operations.
Servicing our debt requires a significant amount of cash, and we may not have sufficient cash flow from our business to pay our substantial debt.
As of December 31, 2015, we owed $120 million of principal, from our December 2013 issuance of convertible notes, which mature in December 2018. Any such indebtedness will require the dedication of a portion of our expected cash flow from operations to service our indebtedness, thereby reducing the amount of our cash flow available for other purposes. In addition, our ability to make scheduled payments of the principal, to pay interest on or to refinance our indebtedness, including our convertible notes, depends on our future performance, which is subject to regulatory, economic, financial, competitive and other factors beyond our control, and our ability to raise equity capital.
Our business may not continue to generate cash flow from operations in the future sufficient to service our debt and make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
Clinical trials may fail to demonstrate the safety and efficacy of our drug products, which could prevent or significantly delay obtaining regulatory approval.
Prior to receiving approval to commercialize any of our drug products, we must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA, and other regulatory authorities in the U.S. and other countries, that each of the products is both safe and effective. For each drug product, we will need to demonstrate its efficacy and monitor its safety throughout the process. If such development is unsuccessful, our business and reputation would be harmed and our stock price would be adversely affected.
22
All of our drug products are prone to the risks of failure inherent in drug development. Clinical trials of new drug products sufficient to obtain regulatory marketing approval are expensive, uncertain, and take years to complete. We may not be able to successfully complete clinical testing within the time frame we have planned, or at all. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our drug products. In addition, the results of pre-clinical studies and early-stage clinical trials of our drug products do not necessarily predict the results of later-stage clinical trials. Later-stage clinical trials may fail to demonstrate that a drug product is safe and effective despite having progressed through initial clinical testing. Even if we believe the data collected from clinical trials of our drug products is promising, data are susceptible to varying interpretations, and such data may not be sufficient to support approval by the FDA or any other U.S. or foreign regulatory approval. Pre-clinical and clinical data can be interpreted in different ways.
Accordingly, FDA officials could interpret such data in different ways than we or our partners do which could delay, limit or prevent regulatory approval. The FDA, other regulatory authorities, our institutional review boards, our contract research organizations, or we may suspend or terminate our clinical trials for our drug products. Any failure or significant delay in completing clinical trials for our drug products, or in receiving regulatory approval for the sale of any drugs resulting from our drug products, may severely harm our business and reputation. Even if we receive FDA and other regulatory approvals, our drug products may later exhibit adverse effects that may limit or prevent their widespread use, may cause the FDA to revoke, suspend or limit their approval, or may force us to withdraw products derived from those drug products from the market.
Moreover, the commencement and completion of clinical trials may be delayed by many factors that are beyond our control, including:
• | delays obtaining regulatory approval to commence a trial; |
• | reaching agreement on acceptable terms with contract research organizations, or CROs, and clinical trial sites; |
• | obtaining institutional review board, or IRB, approval at each site; |
• | slower than anticipated patient enrollment; |
• | scheduling conflicts with participating clinicians and clinical institutions; |
• | lack of funding; |
• | negative or inconclusive results; |
• | patient noncompliance with the protocol; |
• | adverse medical events or side effects among patients during the clinical trials; |
• | negative or problematic FDA inspections of our clinical operations or manufacturing operations; and |
• | real or perceived lack of effectiveness or safety. |
We could encounter delays if a clinical trial is suspended or terminated by us, the IRBs of the clinical trial sites in which such trials are being conducted, or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Our supply of active pharmaceutical ingredients, or APIs, and drug products will be dependent upon the production capabilities of contract manufacturing organizations, or CMOs, component and packaging supply sources, other third-party suppliers, and other providers of logistical services, some of whom are based overseas and, if these parties are not able to meet our demands and FDA scrutiny, we may be limited in our ability to meet demand for our products, ensure regulatory compliance or maximize profit on the sale of our products.
We have no internal manufacturing capacity for APIs or our drug products, and, therefore, we have entered into agreements with CMOs and other suppliers to supply us with APIs and our finished dose drug products. Success in the development and marketing of our drug products depends, in part, upon our ability to maintain, expand and enhance our existing relationships and establish new sources of supply. Some of the third-party manufacturing facilities used in the production of APIs and our drug products are located outside the U.S. The manufacture of APIs and finished drug products,
23
including the acquisition of compounds used in the manufacture of the finished drug product, may require considerable lead times. We have little or no control over the production processes of third-party manufacturers, CMOs or other suppliers.
Our ability to source APIs and drug products is also dependent on providers of logistical services who may be subject to disruptions that we cannot predict or sufficiently plan around. Accordingly, while we do not currently anticipate shortages of supply, circumstances could arise in which we will not have adequate supplies to timely meet our requirements or market demand for a particular drug product could outstrip the ability of our supply source to timely manufacture and deliver the product, thereby causing us to lose sales. In addition, our ability to make a profit on the sale of our drug products depends on our ability to obtain price arrangements that ensure a supply of product at favorable prices.
If problems arise during the production of a batch of our drug products, that batch of product may have to be discarded. This could, among other things, lead to increased costs, lost revenue, damage to customer relations, time and expense spent investigating the cause and, depending on the cause, similar losses with respect to other batches or products. If problems are not discovered before the product is released to the market, recall and product liability costs may also be incurred. To the extent that one of our suppliers experiences significant manufacturing problems, this could have a material adverse effect on our revenues and profitability.
Finally, reliance on CMOs entails risks to which we would not be subject if we manufactured products ourselves, including reliance on the third party for regulatory compliance and adherence to the cGMP, requirements, the possible breach of the manufacturing agreement by the CMO and the possibility of termination or non-renewal of the agreement by the CMO, based on its own business priorities, at a time that is costly or inconvenient for us. Before we can obtain marketing approval for our drug products, our CMO facilities must pass an FDA pre-approval inspection. In order to obtain approval, all of the facility’s manufacturing methods, equipment and processes must comply with cGMP requirements.
The cGMP requirements govern all areas of record keeping, production processes and controls, personnel and quality control. In addition, our CMOs will be subject to on-going periodic inspection by the FDA and corresponding state and foreign agencies for compliance with cGMP regulations, similar foreign regulations and other regulatory standards. We do not have control over our CMOs’ compliance with these regulations and standards. Any failure of our third party manufacturers or us to comply with applicable regulations, including an FDA pre-approval inspection and cGMP requirements, could result in sanctions being imposed on them or us, including warning letters, fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delay, suspension or withdrawal of approvals, license revocation, seizures or recalls of product, operation restrictions and criminal prosecutions, any of which could significantly and adversely affect our business.
Adverse economic conditions may have material adverse consequences on our business, results of operations and financial condition.
Unpredictable and unstable changes in economic conditions, including recession, inflation, increased government intervention, or other changes, may adversely affect our general business strategy. We rely upon our ability to generate positive cash flow from operations to fund our business. If we are not able to generate positive cash flow from operations, we may need to utilize sources of financing or other sources of cash. We may need to raise additional funds through public or private debt or equity financings in order to fund existing operations or to take advantage of opportunities, including acquisitions of complementary businesses or technologies. In addition, if our business deteriorates, we may not be able to maintain compliance with any covenants or representations and warranties in any such financings which could result in reduced availability of such financings, an event of default under such financings, or could make other sources of financing unavailable to us. Any such event would have a material adverse impact on our business, results of operations and financial condition.
While we believe we have adequate capital resources to meet current working capital and capital expenditure requirements, an economic downturn or an increase in our expenses could require additional financing on less than attractive rates or on terms that are excessively dilutive to existing stockholders. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans or plans to acquire additional technology.
Volatile economic conditions may not only limit our access to capital, but may also make it difficult for our customers and us to accurately forecast and plan future business activities, and they could cause businesses to slow spending on our products, which would delay and lengthen sales cycles. Furthermore, during challenging economic times, our customers may face issues gaining timely access to sufficient credit, which could result in an impairment of their ability to make timely
24
payments to us. In addition, adverse economic conditions could also adversely impact our suppliers’ ability to provide us with materials which would negatively impact on our business, financial condition and results of operations.
We may not be able to successfully integrate our acquisitions and any additional businesses we may acquire.
We regularly evaluate and, as appropriate, may make selective acquisitions of businesses and intellectual property that we believe complement or augment our existing business. Issues that could delay or prevent integration of the acquired business and/or intellectual property into our own include:
• | conforming standards, procedures and policies, business cultures and compensation structures; |
• | conforming information technology and accounting systems; |
• | consolidating corporate and administrative infrastructures; |
• | consolidating sales and marketing operations; |
• | retaining existing customers and attracting new customers; |
• | retaining key employees; |
• | identifying and eliminating redundant and under-performing operations and assets; |
• | minimizing the diversion of management’s attention from ongoing business concerns; |
• | coordinating geographically dispersed organizations; |
• | managing tax costs or inefficiencies associated with integrating operations; and |
• | making any necessary modifications to operating control standards to comply with the Sarbanes-Oxley Act of 2002 and the rules and regulations promulgated thereunder. |
If we are unable to successfully integrate our acquisitions with our existing business, we may not obtain the advantages that the acquisitions were intended to create, which may materially adversely affect our business, results of operations, financial condition and cash flows, our ability to develop and introduce new products and the market price of our stock. Actual costs and sales synergies, if achieved at all, may be lower than we expect and may take longer to achieve than we anticipate. Furthermore, the products of companies we acquire may overlap with our products or those of our customers, creating conflicts with existing relationships or with other commitments that are detrimental to the integrated businesses.
Our collaborations with outside scientists may be subject to change, which could limit our access to their expertise.
We work with scientific advisors and collaborators at research institutions. These scientists are not our employees and may have other commitments that would limit their availability to us. If a conflict of interest between their work for us and their work for another entity arises, we may lose their services, which could negatively impact our research and development activities.
We may rely on contract research organizations and other third parties to conduct clinical trials and, in such cases, we are unable to directly control the timing, conduct and expense of our clinical trials.
We may rely, in full or in part, on third parties to conduct our clinical trials. In such situations, we have less control over the conduct of our clinical trials, the timing and completion of the trials, the required reporting of adverse events and the management of data developed through the trial than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties may have staffing difficulties, may undergo changes in priorities or may become financially distressed, adversely affecting their willingness or ability to conduct our trials. We may experience unexpected cost increases that are beyond our control. Problems with the timeliness or quality of the work of a contract research organization may lead us to seek to terminate the relationship and use an alternative service provider. However, making this change may be costly and may delay our trials, and contractual restrictions may make such a change difficult or impossible. Additionally, it may be impossible to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost.
25
We may have conflicts with our third-party development partners that could delay or prevent the development or commercialization of our drug products.
We may have conflicts with our third-party development partners, such as conflicts concerning the interpretation of pre-clinical or clinical data, the achievement of milestones, the interpretation of contractual obligations, payments for services, development obligations or the ownership of intellectual property developed during our collaboration. If any conflicts arise with any of our third-party development partners, such partner may act in a manner that is adverse to our best interests. Any such disagreement could result in one or more of the following, each of which could delay or prevent the development or commercialization of our drug product, and in turn prevent us from generating revenues from such drug product:
• | unwillingness on the part of a third-party development partner to pay us milestone payments or royalties that we believe are due to us under a collaboration; |
• | uncertainty regarding ownership of intellectual property rights arising from our collaborative activities, which could prevent us from entering into additional collaborations; |
• | unwillingness to cooperate in the manufacture of the product, including providing us with product data or materials; |
• | unwillingness to keep us informed regarding the progress of its development and commercialization activities or to permit public disclosure of the results of those activities; |
• | initiation of litigation or alternative dispute resolution options by either party to resolve the dispute; |
• | attempts by either party to terminate the collaboration; |
• | our ability to maintain or defend our intellectual property rights may be compromised by our partner’s acts or omissions; |
• | a third-party development partner may utilize our intellectual property rights in such a way as to invite litigation that could jeopardize or invalidate our intellectual property rights or expose us to potential liability; |
• | a third-party development partner may change the focus of its development and commercialization efforts due to internal reorganizations, mergers, consolidations and otherwise; |
• | unwillingness to fully fund or commit sufficient resources to the testing, marketing, distribution or development of our products; |
• | unwillingness or ability to fulfill their obligations to us due to the pursuit of alternative products, conflicts of interest that arise or changes in business strategy or other business issues; and/or |
• | we may not be able to guarantee supplies of development or marketed products. |
Given these risks, it is possible that any collaborative arrangements which we have or may enter into may not be successful.
Our efforts to acquire or in-license and develop additional drug products may fail, which might limit our ability to grow our business.
To remain competitive and grow our business, our long-term strategy includes the acquisition or in-license of additional drug products. We are actively seeking to acquire, or in-license, additional commercial drug products as well as drug products that have demonstrated positive pre-clinical and/or clinical data. We have certain criteria that we are looking for in any drug product acquisition and in-license and we may not be successful in locating and acquiring, or in-licensing, additional desirable drug products on acceptable terms.
To accomplish our acquisition and in-license strategy, we intend to commit efforts, funds and other resources to research and development and business development. Even with acquired and in-licensed drug products, a high rate of failure is inherent in the development of such products. We must make ongoing substantial expenditures without any assurance that our efforts will be commercially successful. Failure can occur at any point in the process, including after significant funds have been invested. For example, promising new drug product candidates may fail to reach the market or may only have limited commercial success because of efficacy or safety concerns, failure to achieve positive clinical outcomes, inability to obtain necessary regulatory approvals, limited scope of approved uses, excessive costs to manufacture, the failure to establish or maintain intellectual property rights or infringement of the intellectual property rights of others.
In addition, many other large and small companies within the pharmaceutical and biotechnology industry seek to establish collaborative arrangements for product research and development, or otherwise acquire products in late-stage clinical
26
development, in competition with us. We face additional competition from public and private research organizations, academic institutions and governmental agencies in establishing collaborative arrangements for drug products in late-stage clinical development. Many of the companies and institutions that compete against us have substantially greater capital resources, research and development staffs and facilities than we have, and greater experience in conducting business development activities. These entities represent significant competition to us as we seek to expand our portfolio through the in-license or acquisition of compounds. Finally, while it is not feasible to predict the actual cost of acquiring and developing additional drug products, that cost could be substantial and we may need to raise additional financing for such purpose, which may further dilute existing stockholders.
From time to time we may need to license patents, intellectual property and proprietary technologies from third parties, which may be difficult or expensive to obtain.
We may need to obtain licenses to patents and other proprietary rights held by third parties to successfully develop, manufacture and market our drug products. As an example, it may be necessary to use a third party’s proprietary technology to reformulate one of our drug products in order to improve upon the capabilities of the drug product. If we are unable to timely obtain these licenses on reasonable terms, our ability to commercially exploit our drug products may be inhibited or prevented.
The potential size of the market for our drug products is uncertain.
We often provide estimates of the number of people who suffer from the diseases that our drugs are targeting. However, there is limited information available regarding the actual size of these patient populations. In addition, it is uncertain whether the results from previous or future clinical trials of drug products will be observed in broader patient populations, and the number of patients who may benefit from our drug products may be significantly smaller than the estimated patient populations.
Generic levo-leucovorin product competition could further adversely affect our FUSILEV revenues.
FUSILEV continues to face direct competition from generic levo-leucovorin products as a result of the FDA ANDA approval and competitive product launches by two companies in 2015. As a result, this generic competition is expected to continue to adversely impact our FUSILEV product value including (i) sales, demand and market share, (ii) the price we are able to charge, (iii) the inventory levels that wholesalers maintain, and (iv) product return rates. Additional companies are expected to launch their generic levo-leucovorin products in the future, which could further adversely impact FUSILEV revenues.
On April 27, 2015, we filed suit in the U.S. District Court for the District of Columbia against the FDA seeking a temporary restraining order or preliminary injunction to suspend FDA approval of Sandoz’s ANDA. We have contended that Sandoz’s ANDA should not have been approved until the expiry of our Orphan Drug Exclusivity on April 29, 2018. On April 29, 2015, the court denied the temporary restraining order and on May 27, 2015, the court entered summary judgment in favor of the FDA et al. On June 5, 2015, we filed our Notice of Appeal. Oral argument was held on October 22, 2015. The ultimate outcome of this proceeding and its impact on the market for FUSILEV is uncertain.
Our percentage of revenue and customer concentration for FUSILEV is significant. The loss of, or significant reduction or cancellation in sales to, any one of these customers could adversely affect our results of operations.
There is no assurance that FUSILEV sales will be sustainable, even at their current reduced levels due to generic competition. Our customer concentration of FUSILEV is high. A summary of our customers that represent 10% or more of our gross FUSILEV sales in 2015, 2014, and 2013 is as follows:
Product Sales | ||||||||
2015 | 2014 | 2013 | ||||||
Oncology Supply, a division of ASD Specialty Healthcare, Inc., and its affiliates (excluding ICS) | 28.3 | % | 36.2 | % | 32.0 | % | ||
McKesson Corporation and its affiliates | 18.1 | % | 25.7 | % | 19.8 | % | ||
Cardinal Health, Inc. and its affiliates | 11.4 | % | * | * | ||||
* Less than 10%
We expect significant customer concentration to continue for the foreseeable future. The loss of any large customer, a significant reduction in sales we make to them, any cancellation of orders they have made with us or any failure to pay for the products we have shipped to them could materially and adversely affect our results of operations, as it would likely take time for those sales to transition to another customer.
27
Changes in our effective income tax rate could adversely affect our profitability.
We are subject to federal and state income taxes in the U.S. and our tax liabilities are dependent upon the distribution of income among these different jurisdictions. Various factors may have favorable or unfavorable effects on our effective income tax rate. These factors include, but are not limited to:
• | interpretations of existing tax laws; |
• | the accounting for stock options and other share-based compensation; |
• | changes in tax laws and rates; |
• | future levels of research and development spending; |
• | changes in accounting standards; |
• | changes in the mix of earnings in the various tax jurisdictions in which we operate; |
• | the outcome of examinations by the Internal Revenue Service and other jurisdictions; |
• | the accuracy of our estimates for unrecognized tax benefits; |
• | realization of deferred tax assets; and |
• | changes in overall levels of pre-tax earnings. |
The impact on our income tax provision resulting from the above-mentioned factors may be significant and could have an impact on our profitability.
Earthquakes or other natural or man-made disasters and business interruptions could adversely affect our business.
Our operations are vulnerable to interruption by fire, power loss, floods, telecommunications failure and other events beyond our control. In addition, our operations are susceptible to disruption as a result of natural disasters such as earthquakes. So far we have never experienced any significant disruption of our operations as a result of earthquakes or other natural or man-made disasters. Although we have a contingency recovery plan, any significant business interruption could cause delays in our drug development and future sales and harm our business.
Risks Related to Our Industry
If we are unable to adequately protect our technology or enforce our patent rights, our business could suffer.
Our success with the drug products that we develop will depend, in part, on our ability and the ability of our licensors to obtain and maintain patent protection for these products. We currently have a number of U.S. and foreign patents issued and pending, however, we primarily rely on patent rights licensed from others. Our license agreements generally give us the right and/or obligation to maintain and enforce the subject patents. We may not receive patents for any of our pending patent applications or any patent applications we may file in the future. If our pending and future patent applications are not allowed or, if allowed and issued into patents, if such patents and the patents we have licensed are not upheld in a court of law, our ability to competitively exploit our drug products would be substantially harmed. Also, such patents may or may not provide competitive advantages for their respective products or they may be challenged or circumvented by our competitors, in which case our ability to commercially exploit these products may be diminished.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in pharmaceutical and biotechnology patents has emerged to date in the U.S. The laws of many countries may not protect intellectual property rights to the same extent as U.S. laws, and those countries may lack adequate rules and procedures for defending our intellectual property rights. Filing, prosecuting and defending patents on all our products or product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions and may not be covered by any of our patent claims or other intellectual property rights.
Changes in either patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. We do not know whether any of our patent applications will result in the issuance of any patents, and we cannot predict the breadth of claims that may be allowed in our patent applications or in the patent applications we license from others.
28
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
• | in certain jurisdictions, we or our licensors might not have been the first to make the inventions covered by each of our or our licensors’ pending patent applications and issued patents, and we may have to participate in expensive and protracted interference proceedings to determine priority of invention; |
• | we or our licensors might not have been the first to file patent applications for these inventions; |
• | others may independently develop similar or alternative product candidates or duplicate any of our or our licensors’ product candidates; |
• | our or our licensors’ pending patent applications may not result in issued patents; |
• | our or our licensors’ issued patents may not provide a basis for commercially viable products or may not provide us with any competitive advantages or may be challenged by third parties; |
• | others may design around our or our licensors’ patent claims to produce competitive products that fall outside the scope of our or our licensors’ patents; |
• | we may not develop or in-license additional patentable proprietary technologies related to our product candidates; or |
• | the patents of others may prevent us from marketing one or more of our product candidates for one or more indications that may be valuable to our business strategy. |
An issued patent does not guarantee us the right to practice the patented technology or commercialize the patented product. Third parties may have blocking patents that could be used to prevent us from commercializing our patented products and practicing our patented technology. Our issued patents and those that may be issued in the future may be challenged, invalidated or circumvented, which could limit our ability to prevent competitors from marketing related product candidates or could limit the length of the term of patent protection of our product candidates. Our competitors may independently develop similar technologies. In addition, because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We also rely on trade secret protection and contractual protections for our unpatented and proprietary drug compounds. Trade secrets are difficult to protect. While we enter into confidentiality agreements with our employees, consultants and others, these agreements may not successfully protect our trade secrets or other confidential and proprietary information. It is possible that these agreements will be breached, or that they will not be enforceable in every instance, and that we will not have adequate remedies for any such breach. Likewise, although we conduct periodic trade secret audits of certain partners, vendors and contract manufacturers, these trade secret audits may not protect our trade secrets or other confidential and proprietary information. It is possible that despite having certain trade secret audited security measures in place, trade secrets or other confidential and proprietary information may still be leaked or disclosed to a third party. It is also possible that our trade secrets will become known or independently developed by our competitors.
We also rely on trademarks to protect the names of our products. These trademarks may be challenged by others. If we enforce our trademarks against third parties, such enforcement proceedings may be expensive. Some of our trademarks, including ZEVALIN are owned by, or assignable to, our licensors and, upon expiration or termination of the applicable license agreements, we may no longer be able to use these trademarks. If we are unable to adequately protect our technology, trade secrets or proprietary know-how, or enforce our patents and trademarks, our business, financial condition and prospects could suffer.
Intellectual property rights are complex and uncertain and therefore may subject us to infringement claims.
The patent positions related to our drug products are inherently uncertain and involve complex legal and factual issues. We believe that there is significant litigation in the pharmaceutical and biotechnology industry regarding patent and other intellectual property rights. A patent does not provide the patent holder with freedom to operate in a way that infringes the patent rights of others. We may be accused of patent infringement at any time. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our products or methods do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity, in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents in the U.S.
29
Although we are not aware of any infringement by any of our drug products on the rights of any third party, there may be third party patents or other intellectual property rights, including trademarks and copyrights, relevant to our drug products of which we are not aware. Third parties may assert patent or other intellectual property infringement claims against us, or our licensors and collaborators, with products. Any claims that might be brought against us relating to infringement of patents may cause us to incur significant expenses and, if successfully asserted against us, may cause us to pay substantial damages and result in the loss of our use of the intellectual property that is critical to our business strategy.
In the event that we or our partners are found to infringe any valid claim of a patent held by a third party, we may, among other things, be required to:
• | pay damages, including up to treble damages and the other party’s attorneys’ fees, which may be substantial; |
• | cease the development, manufacture, use and sale of our products that infringe the patent rights of others through a court-imposed sanction such as an injunction; |
• | expend significant resources to redesign our products so they do not infringe others’ patent rights, which may not be possible; |
• | discontinue manufacturing or other processes incorporating infringing technology; or |
• | obtain licenses to the infringed intellectual property, which may not be available to us on acceptable terms, or at all. |
Rapid bio-technological advancement may render our drug products obsolete before we are able to recover expenses incurred in connection with their development. As a result, some of our drug products may never become profitable.
The pharmaceutical industry is characterized by rapidly evolving biotechnology. Biotechnologies under development by other pharmaceutical companies could result in treatments for diseases and disorders for which we are developing our own treatments. Several other companies are engaged in research and development of compounds that are similar to our research. A competitor could develop a new biotechnology, product or therapy that has better efficacy, a more favorable side-effect profile or is more cost-effective than one or more of our drug products and thereby cause our drug products to become commercially obsolete. Some of our drug products may become obsolete before we recover the expenses incurred in their development. As a result, such products may never become profitable.
Failure to obtain regulatory approval outside the U.S. will prevent us from marketing our product candidates abroad.
We intend to market certain of our existing and future product candidates in outside of the U.S. In order to market our existing and future product candidates in the EU and many other foreign jurisdictions, we must obtain separate regulatory approvals according to the applicable domestic laws and regulations. We have had limited interactions with foreign regulatory authorities, and the approval procedures vary among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA does not guarantee approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not necessarily ensure approval by regulatory authorities in other countries or by the FDA.
A failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval as well as other risks specific to the jurisdictions in which we may seek approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for foreign regulatory approvals and may not receive necessary approvals to commercialize our existing and future product candidates in any market.
Competition for patients in conducting clinical trials may prevent or delay product development and strain our limited financial resources.
Many pharmaceutical companies are conducting clinical trials involving patients with the disease indications that our drug products target. As a result, we must compete with them for clinical sites, physicians and the limited number of patients who fulfill the stringent requirements for participation in clinical trials. Also, due to the confidential nature of clinical trials, we do not know how many of the eligible patients may be enrolled in competing studies and who are consequently not available to us for our clinical trials. Our clinical trials may be delayed or terminated due to the inability to enroll enough patients. Patient enrollment depends on many factors, including the size of the patient population, the nature of the trial protocol, the proximity of patients to clinical sites and the eligibility criteria for the study. The delay or inability to meet planned patient enrollment
30
may result in increased costs and delays or termination of the trial, which could have a harmful effect on our ability to develop products.
Even after we receive regulatory approval to market our drug products, the market may not be receptive to our drug products upon their commercial introduction, which would negatively impact our ability to achieve profitability.
Our drug products may not gain market acceptance among physicians, patients, healthcare payers and the medical community. The degree of market acceptance of any approved drug products will depend on a number of factors, including:
• | the effectiveness of the drug product; |
• | the prevalence and severity of any side effects; |
• | potential advantages or disadvantages over alternative treatments; |
• | relative convenience and ease of administration; |
• | the strength of marketing and distribution support; |
• | the price of the drug product, both in absolute terms and relative to alternative treatments; and |
• | sufficient third-party coverage and reimbursement. |
If our drug products receive regulatory approval but do not achieve an adequate level of acceptance by physicians, healthcare payers and patients, we may not generate drug product revenues sufficient to attain profitability.
Guidelines and recommendations published by various organizations can reduce the use of our products.
Government agencies, such as the CMS, promulgate regulations, and issue guidelines, directly applicable to us and to our products. In addition, third parties such as professional societies, practice management groups, insurance carriers, physicians, private health/science foundations and organizations involved in various diseases from time to time may publish guidelines or recommendations to healthcare providers, administrators and payers, and patient communities. Recommendations may relate to such matters as utilization, dosage, route of administration and use of related therapies and coverage and reimbursement of our products by government and private payers. Third-party organizations like the above have in the past made recommendations about our products. Recommendations or guidelines that are followed by patients and healthcare providers could result in decreased utilization and/or dosage of our products, any of which could adversely affect our product sales and operating results materially.
The sale of our products is subject to regulatory approvals, and our business is subject to extensive regulatory requirements, and if we are unable to obtain regulatory approval for our product candidates, or if we fail to comply with governmental regulations we will be limited in our ability to commercialize our products and product candidates and/or subject us to penalties.
We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. Obtaining regulatory approval of a new drug is an uncertain, lengthy and expensive process, and success is never guaranteed. Despite the time, resources and effort expended, failure can occur at any stage. In order to receive approval from the FDA for each product candidate, we must demonstrate that the new drug product is safe and effective for its intended use and that the manufacturing processes for the product candidate comply with the FDA’s cGMPs. cGMPs include requirements related to production processes, quality control and assurance, and recordkeeping. The FDA has substantial discretion in the approval process for human medicines.
The FDA and comparable agencies in foreign countries impose many requirements related to the drug development process through lengthy and rigorous clinical testing and data collection procedures, and other costly and time consuming compliance procedures. While we believe that we are currently in compliance with applicable FDA regulations, if our partners, the contract research organizations or contract manufacturers with which we have relationships, or we fail to comply with the regulations applicable to our clinical testing, the FDA may delay, suspend or cancel our clinical trials, or the FDA might not accept the test results. The FDA, an institutional review board, third party investigators, any comparable regulatory agency in another country, or we, may suspend clinical trials at any time if the trials expose subjects participating in such trials to unacceptable health risks. Further, human clinical testing may not show any current or future drug product to be safe and effective to the satisfaction of the FDA or comparable regulatory agencies, or the data derived from the clinical tests may be unsuitable for submission to the FDA or other regulatory agencies. Once we submit an application seeking approval to market a
31
drug product, the FDA or other regulatory agencies may not issue their approvals on a timely basis, if at all. If we are delayed or fail to obtain these approvals, our business and prospects may be significantly damaged. In addition, any regulatory approvals that we receive for our future product candidates may also be subject to limitations on the indicated uses for which they may be marketed or contain requirements for potentially costly post-marketing follow-up studies and surveillance to monitor the safety and efficacy of the product.
If we obtain regulatory approval for our drug products, we, our partners, our manufacturers, and other contract entities will continue to be subject to extensive requirements by a number of national, foreign, state and local agencies. These regulations will impact many aspects of our operations, including testing, research and development, manufacturing, safety, effectiveness, labeling, storage, quality control, adverse event reporting, record keeping, approval, advertising and promotion of our future products. FDA and foreign regulatory authorities strictly regulate the promotional claims that may be made about prescription products and our product labeling, advertising and promotion is subject to continuing regulatory review. Physicians may nevertheless prescribe our product to their patients in a manner that is inconsistent with the approved label, or that is off-label. The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and if we are found to have improperly promoted off-label uses we may be subject to significant sanctions, civil and criminal fines and injunctions prohibiting us from engaging in specified promotional conduct. Moreover, our failure to comply with any applicable regulatory requirements could, among other things, result in:
• | warning letters; |
• | fines; |
• | changes in advertising; |
• | revocation or suspension of regulatory approvals of products; |
• | product recalls or seizures; |
• | delays, interruption, or suspension of product distribution, marketing and sales; |
• | civil or criminal sanctions; |
• | suspension or termination of ongoing clinical trials; |
• | imposition of restrictions on our operations; |
• | close the facilities of our contract manufacturers; and |
• | refusals to approve new products. |
The discovery of previously unknown safety risks with drug products approved to go to market may raise costs or prevent us from marketing such products or change the labeling of our products or take other potentially limiting or costly actions if we or others identify safety risks after our products are on the market.
The later discovery of previously unknown safety risks with our products may result in the imposition of restrictions on distribution or use of the drug product, including withdrawal from the market. The FDA may revisit and change its prior determinations with regard to the safety and efficacy of our products. If the FDA’s position changes, we may be required to change our labeling or to cease manufacture and marketing of the products at issue. Even prior to any formal regulatory action, we could voluntarily decide to cease the distribution and sale or recall any of our products if concerns about their safety or effectiveness develop.
The FDA has significant authority to take regulatory actions in the event previously unknown safety risks are identified or if data suggest that our products may present a risk to safety. For example, the FDA may:
• | require sponsors of marketed products to conduct post-approval clinical studies to assess a known serious risk, signals of serious risk or to identify an unexpected serious risk; |
• | mandate labeling changes to products, at any point in a product’s lifecycle, based on new safety information; and |
• | require sponsors to implement a REMS for a product which could include a medication guide, patient package insert, a communication plan to healthcare providers, or other elements as the FDA deems are necessary to assure safe use of the drug (either prior to approval or post-approval as necessary). |
32
Failure to comply with a REMS could result in significant civil monetary penalties or other administrative actions by the FDA. Further, regulatory agencies could change existing, or promulgate new, regulations at any time which may affect our ability to obtain or maintain approval of our existing or future products or require significant additional costs to obtain or maintain such approvals.
Legislative or regulatory reform of the healthcare system and pharmaceutical industry related to pricing, coverage or reimbursement may hurt our ability to sell our products profitably or at all.
Our ability to commercialize any products successfully will depend in part on the availability of coverage and reimbursement from third-party payers such as government authorities, private health insurers, health maintenance organizations including pharmacy benefit managers and other health care-related organizations, both in the U.S. and foreign markets. Even if we succeed in bringing one or more products to the market, the amount reimbursed for our products may be insufficient to allow us to compete effectively and could adversely affect our profitability. Coverage and reimbursement by governmental and other third-party payers may depend upon a number of factors, including a governmental or other third-party payer’s determination that use of a product is:
• | a covered benefit under its health plan; |
• | safe, effective and medically necessary; |
• | appropriate for the specific patient; |
• | cost-effective; and |
• | neither experimental nor investigational. |
Obtaining coverage and reimbursement approval for a product from each third-party and governmental payer is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payer. We may not be able to provide data sufficient to obtain coverage and adequate reimbursement.
In both the U.S. and certain foreign jurisdictions, there have been and may continue to be a number of legislative and regulatory proposals related to coverage and reimbursement that could impact our ability to sell our products profitably. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively referred to as the Healthcare Reform Law, was signed into law on March 30, 2010. The Healthcare Reform Law substantially changed the way healthcare is financed by both governmental and private insurers and significantly impacted the pharmaceutical industry. The Healthcare Reform Law included, among other things, an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, revisions to the definition of “average manufacturer price” for reporting purposes, increases in the amount of rebates owed by drug manufacturers under the Medicaid Drug Rebate Program, expansion of the 340B drug discount program that mandates discounts to certain hospitals, community centers and other qualifying providers, and changes to affect the Medicare Part D coverage gap, or “donut hole.” The full effects of these provisions will become apparent as these laws are implemented and the CMS and other agencies issue applicable regulations or guidance as required by the Healthcare Reform Law. Moreover, in the coming years, additional changes could be made to governmental healthcare programs that could significantly impact the success of our products.
The high cost of pharmaceuticals continues to generate substantial government interest. It is possible that proposals will be adopted, or existing regulations that affect the coverage and reimbursement of pharmaceutical and other medical products may change, that may impact our products currently on the market and any of our products approved for marketing in the future. Cost control initiatives could decrease the price that we receive for any of our products or product candidates. In addition, third-party payers are increasingly challenging the price and cost-effectiveness of medical products and services. Significant uncertainty exists as to the coverage and reimbursement status of newly-approved pharmaceutical products. Future developments may require us to decrease the price that we charge for our products, thereby negatively affecting our financial results.
In some foreign countries, particularly in the EU, prescription drug pricing is subject to governmental control. Drug pricing may be made against a reference price set by the healthcare providers as a measure for healthcare cost containment. Pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. If coverage and reimbursement of our products are unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels for the purpose of adoption of these products in the national health services in these jurisdictions, our profitability will likely be negatively affected.
33
If we market products in a manner that violates health care fraud and abuse laws, we may be subject to civil or criminal penalties, including exclusion from participation in government health care programs.
As a pharmaceutical company, even though we do not provide healthcare services or receive payments directly from or bill directly to Medicare, Medicaid or other third-party payers for our products, certain federal and state healthcare laws and regulations pertaining to fraud and abuse are and will be applicable to our business. We are subject to healthcare fraud and abuse laws by both the federal government and the states in which we conduct our business.
The laws that may affect our ability to operate include the federal health care program Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for purchasing, leasing, ordering, or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed health care programs. This statute applies to arrangements between pharmaceutical manufacturers and prescribers, purchasers and formulary managers. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common activities, the exceptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Pharmaceutical companies have been prosecuted under these laws for a variety of alleged promotional and marketing activities, such as providing free product to customers with the expectation that the customers would bill federal programs for the product; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion that caused claims to be submitted to Medicaid for non-covered off-label uses; and submitting inflated best price information to the Medicaid Drug Rebate Program.
The Health Insurance Portability and Accountability Act of 1996 also created prohibitions against health care fraud and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including private payers. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians. The Healthcare Reform Law imposed new requirements on manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and applicable manufacturers and group purchasing organizations to report annually to CMS ownership and investment interests held by physicians (as defined above) and their immediate family members and payments or other “transfers of value” to such physician owners and their immediate family members. Manufacturers were required to begin data collection on August 1, 2013 and to report such data to the government by March 31, 2014 and by the 90th calendar day of each year thereafter.
The majority of states also have statutes or regulations similar to these federal laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. In addition, some states have laws that require pharmaceutical companies to adopt comprehensive compliance programs. For example, under California law, pharmaceutical companies must comply with both the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and the PhRMA Code on Interactions with Healthcare Professionals, as amended. Certain states also mandate the tracking and reporting of gifts, compensation, and other remuneration paid by us to physicians and other health care providers. We have adopted and implemented a compliance program designed to comply with applicable federal, state and local requirements wherever we operate, including but not limited to the laws of the states of California and Nevada.
Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state laws may prove costly.
Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws. The Healthcare Reform Law also make several important changes to the federal Anti-Kickback Statute, false claims laws, and health care fraud statute by weakening the intent
34
requirement under the anti-kickback and health care fraud statutes that may make it easier for the government, or whistleblowers to charge such fraud and abuse violations. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the Health Care Reform Law provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. In addition, the Healthcare Reform Law increases penalties for fraud and abuse violations. If our past, present or future operations are found to be in violation of any of the laws described above or other similar governmental regulations to which we are subject, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and negatively impact our financial results.
We may be subject to product liability claims, and may not have sufficient product liability insurance to cover any such claims, which may expose us to substantial liabilities.
We may be held liable if any product we or our partners develop causes injury or is found otherwise unsuitable during product testing, manufacturing, clinical trials, marketing or sale. Regardless of merit or eventual outcome, product liability claims could result in decreased demand for our product candidates, injury to our reputation, withdrawal of patients from our clinical trials, substantial monetary awards to trial participants and the inability to commercialize any products that we may develop. These claims might be made directly by consumers, health care providers, pharmaceutical companies or others selling or testing our products. Although we currently carry product liability insurance in the amount of at least $15 million in the aggregate, it is possible that this coverage will be insufficient to protect us from future claims. Additionally, our insurance may not reimburse us or may not be sufficient to reimburse us for expenses or losses we may suffer. Moreover, if insurance coverage becomes more expensive, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. Failure to maintain sufficient insurance coverage could have a material adverse effect on our business, prospects and results of operations if claims are made that exceed our coverage.
On occasion, juries have awarded large judgments in class action lawsuits for claims based on drugs that had unanticipated side effects. In addition, the pharmaceutical and biotechnology industries, in general, have been subject to significant medical malpractice litigation. A successful product liability claim or series of claims brought against us could harm our reputation and business and financial condition.
We could be adversely affected by violations of the U.S. Foreign Corrupt Practices Act (“FCPA”) and other worldwide anti-bribery laws.
We are subject to the FCPA which prohibits companies and their intermediaries from making payments to non-U.S. government officials for purposes of obtaining or retaining business or securing any other improper advantage. We have policies and procedures in place to ensure that we comply with the FCPA and similar laws; however, there is no assurance that such policies and procedures will protect us against liability under the FCPA or related laws for actions taken by our employees and intermediaries with respect to our business. Failure to comply with the FCPA and related laws could disrupt our business and lead to criminal and civil penalties including fines, suspension of our ability to do business with the federal government and denial of government reimbursement of our products, which could result in a material adverse impact on our business, financial condition, results of operations and cash flows. We could also be adversely affected by any allegation that we violated such laws.
The use of hazardous materials, including radioactive and biological materials, in our research and development and commercial efforts imposes certain compliance costs on us and may subject us to liability for claims arising from the use or misuse of these materials.
Our research and development, manufacturing (including a radiolabeling step for ZEVALIN) and administration of our drugs involves the controlled use of hazardous materials, including chemicals, radioactive and biological materials, such as radioactive isotopes. We are subject to federal, state, local and foreign environmental laws and regulations governing, among other matters, the handling, storage, use and disposal of these materials and some waste products. We cannot completely eliminate the risk of contamination or injury from these materials and we could be held liable for any damages that result, which could exceed our financial resources. We currently maintain insurance coverage for injuries resulting from the hazardous materials we use; however, future claims may exceed the amount of our coverage. Also, we do not have insurance coverage for pollution cleanup and removal. Currently the costs of complying with such federal, state, local and foreign environmental regulations are not significant, and consist primarily of waste disposal expenses. However, they could become expensive, and current or future environmental laws or regulations may impair our research, development, production and commercialization efforts.
35
Risks Related to Our Common Stock
There are a substantial number of shares of our common stock eligible for future sale in the public market. The sale of these shares could cause the market price of our common stock to fall. Any future equity issuances by us may have dilutive and other effects on our existing stockholders.
As of December 31, 2015, there were approximately 68 million shares of our common stock outstanding. Security holders held outstanding options, restricted stock units, warrants, preferred stock and convertible notes which, if vested, exercised or converted, would obligate us to issue up to approximately 26 million additional shares of common stock. A substantial number of those shares, when we issue them upon vesting, conversion or exercise, will be available for immediate resale in the public market. In addition, we have reserved an aggregate of 13 million shares of our common stock for future issuance under our equity compensation plans. We may also sell additional shares of common stock or securities convertible or exercisable into common stock in public or private offerings, which would be available for resale in the market. Certain issuances by us of equity securities may be at or below the prevailing market price of our common stock and may have a dilutive impact on our existing stockholders. These issuances or other dilutive issuances would also cause our net income, if any, per share to decrease in future periods. The market price of our common stock could fall as a result of sales of any of these shares of common stock due to the increased number of shares available for sale in the market.
The convertible note hedge and warrant transactions that we entered into in December 2013 may affect the value of our common stock.
In connection with the pricing of our convertible notes in December 2013, we entered into convertible note hedge transactions and separate warrant transactions with RBC Capital Markets, LLC (“RBC”). The convertible note hedge transactions are expected generally to reduce the potential dilution upon any conversion of the notes and/or offset any cash payments we are required to make in excess of the principal amount of converted notes, as the case may be. The warrant transactions could separately have a dilutive effect to the extent that the market price per share of our common stock exceeds the strike price of the warrants. RBC and/or its affiliates may modify their hedge positions by entering into or unwinding various derivatives with respect to our common stock and/or purchasing or selling our common stock in secondary market transactions prior to the maturity of the convertible notes (and is likely to do so during any observation period related to a conversion of notes). This activity could cause or avoid an increase or a decrease in the market price of our common stock.
In addition, if the convertible note hedge and warrant transactions fail to become effective, through the failure of counterparties to perform or otherwise, RBC and/or its affiliates may unwind its hedge positions with respect to our common stock, which could adversely affect the value of our common stock. The potential effect, if any, of these transactions and activities on the market price of our common stock will depend in part on market conditions and cannot be ascertained at this time. Any of these activities could adversely affect the value of our common stock.
The market price and trading volume of our common stock fluctuate significantly and could result in substantial losses for individual investors.
The stock market from time to time experiences significant price and trading volume fluctuations that are unrelated to the operating performance of particular companies. These broad market fluctuations may cause the market price and trading volume of our common stock to decrease. In addition, the market price and trading volume of our common stock is often highly volatile.
Factors that may cause the market price and volume of our common stock to decrease include:
• | recognition on up-front licensing or other fees or revenues; |
• | payments of non-refundable up-front or license fees, or payment for cost-sharing expenses, to third parties; |
• | adverse results or delays in our clinical trials; |
• | fluctuations in our results of operations; |
• | timing and announcements of our technological innovations or new products or those of our competitors; |
• | developments concerning any strategic alliances or acquisitions we may enter into; |
36
• | announcements of FDA non-approval of our drug products, or delays in the FDA or other foreign regulatory review process or actions; |
• | changes in recommendations or guidelines of government agencies or other third parties regarding the use of our drug products; |
• | adverse actions taken by regulatory agencies with respect to our drug products, clinical trials, manufacturing processes or sales and marketing activities; |
• | concerns about our products being reimbursed; |
• | any lawsuit involving us or our drug products; |
• | developments with respect to our patents and proprietary rights; |
• | public concern as to the safety of products developed by us or others; |
• | regulatory developments in the U.S. and in foreign countries; |
• | changes in stock market analyst recommendations regarding our common stock or lack of analyst coverage; |
• | the pharmaceutical industry generally and general market conditions; |
• | failure of our results of operations to meet the expectations of stock market analysts and investors; |
• | sales of our common stock by our executive officers, directors and five percent stockholders or sales of substantial amounts of our common stock; |
• | hedging or arbitrage transactions by holders of our convertible notes; |
• | changes in accounting principles; and |
• | loss of any of our key scientific or management personnel. |
Also, certain dilutive securities such as warrants can be used as hedging tools which may increase volatility in our stock and cause a price decline. While a decrease in market price could result in direct economic loss for an individual investor, low trading volume could limit an individual investor’s ability to sell our common stock, which could result in substantial economic loss as well. From January 2, 2015 through February 29, 2016, the closing price of our common stock ranged between $4.28 and $7.66, and the daily trading volume was as high as 8.2 million shares and as low as 0.2 million shares.
Following periods of volatility in the market price of a company’s securities, a securities class action litigation may be instituted against that company. Regardless of their merit, these types of lawsuits generally result in substantial legal fees and management’s attention and resources being diverted from the operations of a business.
Provisions of our charter, bylaws and stockholder rights plan may make it more difficult for someone to acquire control of us or replace current management even if doing so would benefit our stockholders, which may lower the price an acquirer or investor would pay for our stock.
Provisions of our certificate of incorporation and bylaws, both as amended, may make it more difficult for someone to acquire control of us or replace our current management. These provisions include:
• | the ability of our board of directors to amend our bylaws without stockholder approval; |
• | the inability of stockholders to call special meetings; |
• | the ability of members of the board of directors to fill vacancies on the board of directors; |
• | the inability of stockholders to act by written consent, unless such consent is unanimous; and |
• | the establishment of advance notice requirements for nomination for election to our board of directors or for proposing matters that can be acted on by stockholders at stockholder meetings. |
These provisions may make it more difficult for stockholders to take certain corporate actions and could delay, discourage or prevent someone from acquiring our business or replacing our current management, even if doing so would benefit our stockholders. These provisions could limit the price that certain investors might be willing to pay for shares of our common stock.
37
We have a stockholder rights plan pursuant to which we distributed rights to purchase units of our Series B junior participating preferred stock. The rights become exercisable upon the earlier of ten days after a person or group of affiliated or associated persons has acquired 15% or more of the outstanding shares of our common stock or ten business days after a tender offer has commenced that would result in a person or group beneficially owning 15% or more of our outstanding common stock. These rights could delay or discourage someone from acquiring our business, even if doing so would benefit our stockholders. We currently have no stockholders who own 15% or more of the outstanding shares of our common stock.
Our failure to establish and maintain effective internal control over financial reporting could result in material misstatements in our financial statements, our failure to meet our reporting obligations and cause investors to lose confidence in our reported financial information, which in turn could cause the trading price of our common stock to decline.
Maintaining effective internal control over financial reporting is necessary for us to produce reliable financial statements. In connection with our assessment of the effectiveness of internal control over financial reporting and the preparation of our financial statements for the year ended December 31, 2013, we identified a material weakness related to ineffective design and operation of controls over our process of estimating the required period-end accruals for operating expenses, which resulted in net overstated operating expenses and accrued liabilities in multiple reporting periods in, and prior to, 2013. We have remediated this material weakness as of December 31, 2014.
Material weaknesses have adversely affected us in the past and could affect us in the future, and the results of our periodic management evaluations and annual auditor attestation reports regarding the effectiveness of our internal control over financial reporting required by Section 404 of the Sarbanes-Oxley Act of 2002. Any failure to maintain new and more precise monitoring controls and improved detection and communication of financial misstatements across all levels of the organization could result in (i) additional material weaknesses, (ii) material misstatements in our financial statements, requiring restatements of our previously-filed financial statements, and (iii) cause us to fail to meet our timely reporting obligations. These outcomes could cause us to lose public confidence, and could cause the trading price of our common stock to decline. For further information regarding our controls and procedures, see Item 9A, Controls and Procedures to this Annual Report on Form 10-K.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
We lease our principal executive office in Henderson, Nevada under a non-cancelable operating lease expiring April 30, 2019. We also lease our research and development facility in Irvine, California under a non-cancelable operating lease expiring May 31, 2019. In addition, we lease administrative space in Westlake Village, California; Westminster, Colorado; and Mumbai, India. We believe that these leased facilities are adequate to meet our current and planned business needs.
ITEM 3. LEGAL PROCEEDINGS
We are involved from time-to-time with various legal matters arising in the ordinary course of business. These claims and legal proceedings are of a nature we believe are normal and incidental to a pharmaceutical business, and may include product liability, intellectual property, employment matters, and other general claims.
We make provisions for liabilities when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Such provisions are assessed at least quarterly and adjusted to reflect the impact of any settlement negotiations, judicial and administrative rulings, advice of legal counsel, and other information and events pertaining to a particular case. Litigation is inherently unpredictable. Although the ultimate resolution of these various matters cannot be determined at this time, we do not believe that such matters, individually or in the aggregate, will have a material adverse effect on our consolidated results of operations, cash flows, or financial condition.
We are presently responding to ANDAs filed by companies seeking to market generic forms of FOLOTYN and FUSILEV. We are also responding to certain stockholder suits that purportedly stem from our March 12, 2013 press release, in which we announced anticipated changes in customer ordering patterns of FUSILEV. These stockholder complaints allege among other things that, as a result of this press release, our stock price declined.
38
FUSILEV ANDA Litigation
On January 20, 2012, March 2, 2012, June 18, 2014, January 23, 2015, July 17, 2015 and September 3, 2015 respectively, we filed suit against Sandoz Inc., Innopharma Inc., Ben Venue Laboratories, Inc., Amneal Pharmaceuticals, Inc., and Actavis LLC. respectively, following Paragraph IV certifications in connection with their filing separate ANDAs, to manufacture a generic version of FUSILEV. We filed the lawsuits in the U.S. District Court for the Districts of Nevada and Delaware seeking to enjoin the approval of their ANDAs plus recovery of our litigation fees and costs incurred in such matters. On December 9, 2013, three Mylan entities collaborating with Innopharma were joined to the Innopharma case. On November 24, 2014 the complaint in the Ben Venue case was amended to substitute the original defendant Ben Venue Laboratories, Inc. with successors West-Ward Pharmaceutical Corp. and Eurohealth International SARL.
A trial took place in the Sandoz case from January 12, 2015 through January 20, 2015 in the U.S. District Court for the District of Nevada and on February 20, 2015 the district court found certain of the asserted claims of the patent covering FUSILEV invalid. On February 27, 2015, we filed our Notice of Appeal. On August 4, 2015, the Delaware district court ordered that judgment be entered for Innopharma and Mylan due to the Nevada district court judgment in the Sandoz
action. On October 2, 2015, the U.S. Court of Appeals for the Federal Circuit affirmed the previously reported judgment from the U.S. District Court for the District of Nevada in favor of Sandoz.
On April 27, 2015, we filed suit in the U.S. District Court for the District of Columbia against the FDA seeking a temporary restraining order or preliminary injunction to suspend FDA approval of Sandoz’s ANDA. The Company contends that Sandoz’s ANDA should not have been approved until the expiry of our Orphan Drug Exclusivity on April 29, 2018. On April 29, 2015, the court denied the temporary restraining order and on May 27, 2015, the court entered summary judgment in favor of the FDA et al. On June 5, 2015, we filed our Notice of Appeal. Oral argument was held October 22, 2015. The ultimate
outcome of this proceeding is uncertain.
FOLOTYN ANDA Litigation
On June 19, 2014, we filed a lawsuit against five parties resulting from Paragraph IV certifications in connection with four separate ANDAs to manufacture a generic version of FOLOTYN: (1)Teva Pharmaceuticals USA, Inc., (2) Sandoz Inc., (3) Fresenius Kabi USA, LLC, (4) Dr. Reddy’s Laboratories, Ltd., and (5) Dr. Reddy’s Laboratories, Inc. We filed the lawsuit in the U.S. District Court for the District of Delaware seeking to enjoin the approval of their ANDAs plus recovery of our litigation fees and costs. The litigation is stayed with respect to the Dr. Reddy's entities pending resolution of the case against the other FOLOTYN ANDA filers. A trial date of September 12, 2016 has been set in the FOLOTYN lawsuit in the U.S. District Court for the District of Delaware. While we believe our patent rights are strong, the ultimate outcome of such action is uncertain.
Stockholder Litigation
John Perry v. Spectrum Pharmaceuticals, Inc. et al. (Filed March 14, 2013 in United States District Court, District of Nevada; Case Number 2:2013-cv-00433-LDG-CWH). This putative consolidated class action raises substantially identical claims and allegations against defendants Spectrum Pharmaceuticals, Inc., Dr. Rajesh C. Shrotriya, Brett L. Scott, and Joseph Kenneth Keller. The alleged class period is August 8, 2012 to March 12, 2013. The lawsuits allege a violation
Section 10(b) of the Securities Exchange Act of 1934 against all defendants and control person liability, as a violation of Section 20(b) of the Securities Exchange Act of 1934, against the individual defendants. The claims purportedly stem from the our March 12, 2013 press release, in which it announced that it anticipated a change in ordering patterns of FUSILEV. The complaints allege that, as a result of the March 12, 2013 press release, our stock price declined. The complaints further allege that during the putative class period certain defendants made misleadingly optimistic statements about FUSILEV sales, which inflated the trading price of our stock. The lawsuits seek relief in the form of monetary damages, costs and fees, and any other equitable or injunctive relief that the court deems appropriate. On March 21, 2014, the Court entered an order appointing Arkansas Teacher Retirement System as lead plaintiff. On May 20, 2014, Arkansas Teacher Retirement System filed a consolidated amended class action complaint. On July 18, 2014, we filed a motion to dismiss the consolidated amended class action complaint. On March 26, 2015, the court denied the motion to dismiss. On June 15, 2015, the Court ordered a stay of the
proceedings pending the outcome of mediation between the parties. On October 27, 2015, we reached a $7 million settlement in principle with the lead plaintiff (which involved our insurance carrier, as the reimbursing party in full), subject to preliminary and final court approval. We have included this settlement amount, along with $0.1 million of reimbursable legal expenses for this matter, on our accompanying Consolidated Balance Sheets as of December 31, 2015 within "other receivables" and "accounts payable and other accrued liabilities." On January 26, 2016, the Court preliminarily approved the settlement. The Court has scheduled a hearing on final approval of the settlement for June 13, 2016.
39
Timothy Fik v. Rajesh C. Shrotriya, et al. (Filed April 11, 2013 in United States District Court, District of Nevada; Case Number 2:2013-cv-00624-JCM-CWH); Christopher J. Watkins v. Rajesh C. Shrotriya, et al. (Filed April 22, 2013 in United States District Court, District of Nevada; Case Number 2:2013-cv-00684-JCM-VCF); and Stefan Muenchhagen v. Rajesh C. Shrotriya, et al. (Filed May 28, 2013; Case Number 2:2013-cv-00942-APG-PAL). These derivative complaints are brought by the respective purported stockholders on behalf of nominal plaintiff Spectrum against certain current and former directors and officers. The complaints generally allege breaches of fiduciary based on conduct relating to the events alleged in the consolidated Perry action. The complaints seek compensatory damages, corporate governance reforms, restitution and disgorgement of defendants’ alleged profits, and costs and fees. These actions are stayed pending resolution of the federal securities class action. Settlement discussions are ongoing, and accordingly, no agreement has yet been reached to resolve these derivative complaints. If a settlement were reached, we believe it would be reimbursable by our insurance carrier. However, the value of a potential settlement cannot be reasonably estimated given its highly uncertain nature.
Hardik Kakadia v. Rajesh C. Shrotriya, et al. (Filed April 23, 2013 in the Eighth Judicial District Court of the State of Nevada in and for Clark County; Case Number A-13-680643-B); and Joel Besner v. Rajesh C. Shrotriya, et al. (Filed May 31, 2013; Case Number A-13-682668-C) (collectively the “State Derivative Actions”). These consolidated State Derivative Actions are brought by the respective purported stockholders on behalf of nominal plaintiff Spectrum Pharmaceuticals, Inc. and are substantially similar to the consolidated federal derivative actions. These actions are stayed pending resolution of the federal securities class action. Settlement discussions are ongoing, and accordingly, no agreement has yet been reached to resolve these derivative complaints. If a settlement were reached, it would be reimbursable by our insurance carrier. However, the value of a potential settlement cannot be reasonably estimated given its highly uncertain nature.
Ira Gains v. Spectrum Pharmaceuticals, Inc. and Rajesh C. Shrotriya was filed in the United States District Court, District of Nevada on November 3, 2015. This putative class action was brought against defendants Spectrum Pharmaceuticals, Inc., and Dr. Rajesh C. Shrotriya. The alleged class period was May 7, 2015 to October 23, 2015. The complaint alleged a violation of Section 10(b) of the Securities Exchange Act of 1934 against all defendants and control person liability, as a violation of Section 20(a) of the Securities Exchange Act of 1934, against Dr. Rajesh C. Shrotriya. The claims purportedly stemmed from our October 23, 2015 press release in which we announced that the FDA issued a Complete Response Letter indicating that the FDA would not approve our NDA for EVOMELA in its present form. The complaint alleged that, as a result of the October 23, 2015 press release, our stock price declined. The complaint further alleged that during the putative class period defendants made misleadingly optimistic statements about the progress of the NDA for EVOMELA with the FDA, our expectations regarding FDA approval of the NDA, and EVOMELA’s potential as a future driver of our revenue, which inflated the trading price of our stock. The complaint sought relief in the form of monetary damages, costs and fees, and any other relief that the Court deemed appropriate. On December 11, 2015, plaintiff voluntarily dismissed the action without prejudice.
SEC Subpoena
On April 1, 2013, we received a subpoena from the SEC for documents pursuant to a formal order of investigation. The subpoena followed our March 12, 2013 announcement that we anticipated a change in customer ordering patterns of FUSILEV. We have cooperated with the SEC throughout its investigation, and on November 30, 2015, we received a notice that the SEC does not intend to recommend an enforcement action against our company.
Notice from HRSA
We received a notice on October 10, 2014 from the U.S. Health Resources and Services Administration, Office of Pharmacy Affairs (“HRSA”). In this notice HRSA asserted that, for at least one of our products with an “orphan drug” designation under section 526 of the Federal Food, Drug, and Cosmetic Act, we did not make the product(s) available for purchase by certain categories of providers at the applicable 340B price. The 340B price is a discounted price for covered outpatient drugs that manufacturers participating in Medicaid (which includes us) agree to make available to providers that participate in the 340B drug pricing program (“Covered Entities”). HRSA’s notice asserted that, by not selling our product(s) to certain categories of Covered Entities at 340B prices, we were overcharging them, and that we owed certain undefined refunds to those Covered Entities based on our previously made and reported product sales.
On October 14, 2015, the U.S. District Court for the District of Columbia issued a decision in the case of Pharmaceutical Research and Manufacturers of America v. United States Department of Health and Human Services, et al., invalidating HRSA’s July 21, 2014 “interpretive rule” relating to orphan drug pricing under the 340B drug pricing program. Because HRSA’s October 10, 2014 notice to us reflected the same interpretation of relevant orphan drug pricing statutes as the interpretation overturned by the District Court in the Pharmaceutical Research and Manufacturers of America decision, we believe the decision supports the propriety of our pricing to Covered Entities. Since we only make provisions for liabilities
40
when it is both probable that a liability has been incurred, and the amount can be reasonably estimated, we have not recorded a liability for this pending matter as of December 31, 2015.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is traded on the NASDAQ Global Market under the symbol “SPPI.” The high and low closing sale prices of our common stock reported by NASDAQ during each quarter ended in 2015 and 2014 were as follows:
High | Low | ||||||
Year Ended December 31, 2015: | |||||||
First Quarter | $ | 7.66 | $ | 5.95 | |||
Second Quarter | 7.37 | 5.65 | |||||
Third Quarter | 7.60 | 5.92 | |||||
Fourth Quarter | 6.93 | 5.07 | |||||
Year Ended December 31, 2014: | |||||||
First Quarter | $ | 10.24 | $ | 7.72 | |||
Second Quarter | 8.68 | 6.65 | |||||
Third Quarter | 8.90 | 6.89 | |||||
Fourth Quarter | 8.24 | 6.75 | |||||
On February 29, 2016, the closing price of our common stock on the NASDAQ Global Select Market was $4.52 per share, and there were 418 holders of record of our common stock.
During the year ended December 31, 2015, we repurchased an aggregate of 103,754 shares of common stock surrendered by our employees and members of our board of directors to satisfy their tax withholding obligations of restricted stock awards (at prices ranging from $5.15 to $7.41 per share). Such shares have been canceled by the transfer agent and returned to our authorized pool of common stock for issuance.
Stock Performance Graph (1)
The graph below compares the cumulative total stockholder return on $100 invested, assuming the reinvestment of all dividends, on December 31, 2010, the last trading day before our 2010 fiscal year, through the end of fiscal 2015 with the cumulative total return on $100 invested for the same period in the Russell 2000 index and our Peer Group.
41
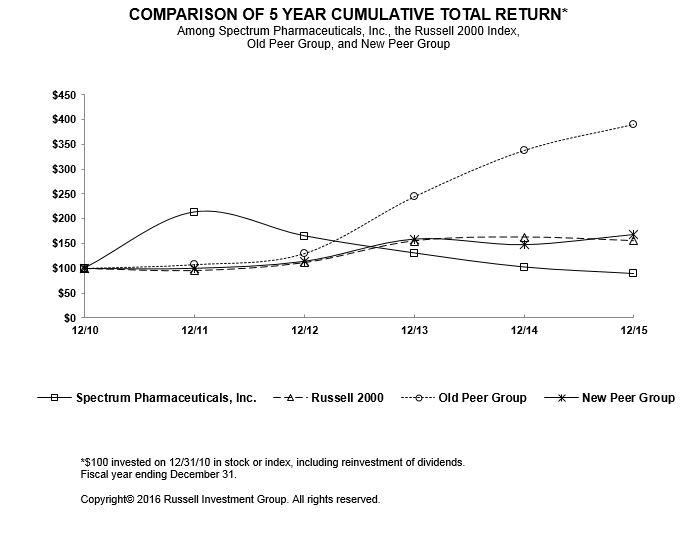
The New Peer Group (which we believe more closely reflects our operations and business characteristics than the Old Peer Group) consists of the following publicly-traded companies:
• | Acorda Therapeutics, Inc. |
• | Aegerion Pharmaceuticals, Inc. |
• | Auxilium Pharmaceuticals, Inc. |
• | Dendreon Corp. |
• | DepoMed Inc. |
• | Emergent BioSolutions, Inc. |
• | Genomic Health Inc. |
• | Hyperion Therapeutics, Inc. |
• | INSYS Therapeutics, Inc. |
• | Sagent Pharmaceuticals, Inc. |
• | SciClone Pharmaceuticals, Inc. |
• | Sucampo Pharmaceuticals, Inc. |
• | Supernus Pharmaceuticals, Inc. |
• | The Medicines Company |
• | VIVUS Inc. |
The Old Peer Group consisted of the following publicly-traded companies:
42
• | Acorda Therapeutics, Inc. |
• | Alkermes plc |
• | Amarin Corporation plc |
• | Auxilium Pharmaceuticals, Inc. |
• | BioMarin Pharmaceutical Inc. |
• | Dendreon Corporation |
• | Halozyme Therapeutics, Inc. |
• | Incyte Corporation |
• | Isis Pharmaceuticals, Inc. |
• | Jazz Pharmaceuticals plc |
• | The Medicines Company |
• | Momenta Pharmaceuticals, Inc. |
• | NPS Pharmaceuticals, Inc. |
• | Salix Pharmaceuticals, Ltd |
• | SciClone Pharmaceuticals, Inc. |
• | Theravance Biopharma, Inc. |
• | Vertex Pharmaceuticals Incorporated |
12/31/2010 | 12/31/2011 | 12/31/2012 | 12/31/2013 | 12/31/2014 | 12/31/2015 | ||||||||||||||||||
Spectrum Pharmaceuticals, Inc. | $ | 100 | $ | 213 | $ | 165 | $ | 131 | $ | 102 | $ | 89 | |||||||||||
Russell 2000 | $ | 100 | $ | 96 | $ | 111 | $ | 155 | $ | 162 | $ | 155 | |||||||||||
Old Peer Group | $ | 100 | $ | 108 | $ | 130 | $ | 244 | $ | 337 | $ | 390 | |||||||||||
New Peer Group | $ | 100 | $ | 100 | $ | 114 | $ | 159 | $ | 148 | $ | 168 | |||||||||||
(1) | The information in this section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any filing of the Company under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
Dividend Policy
We currently intend to retain all earnings, if any, for use in the expansion of our business and therefore do not anticipate paying any dividends in the foreseeable future. However, the payment of dividends, if any, will be at the discretion of the Board of Directors and subject to compliance at such time with any applicable restrictions contained in our various agreements.
ITEM 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data has been derived from our audited Consolidated Financial Statements. The audited Consolidated Financial Statements for the fiscal years ended December 31, 2015, 2014, and 2013 are included elsewhere in this Annual Report on Form 10-K. The information set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Item 7 and the Consolidated Financial Statements and Notes thereto in Item 8. The information set forth below is not necessarily indicative of our future financial condition or results of operations.
43
Year ended December 31, | |||||||||||||||||||
Selected Statement of Operations Data: | 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||
(In thousands, except per share data) | |||||||||||||||||||
Total revenues | $ | 162,556 | $ | 186,830 | $ | 155,854 | $ | 267,707 | $ | 192,963 | |||||||||
Operating costs and expenses: | |||||||||||||||||||
Cost of product sales (excludes amortization and impairment of intangible assets) | 27,689 | 27,037 | 28,580 | 46,633 | 33,838 | ||||||||||||||
Selling, general and administrative | 86,514 | 97,412 | 99,315 | 89,922 | 72,197 | ||||||||||||||
Research and development | 50,766 | 69,662 | 46,670 | 41,560 | 26,662 | ||||||||||||||
Amortization and impairment of intangible assets | 38,319 | 24,288 | 20,074 | 8,818 | 3,720 | ||||||||||||||
(Loss) Income from operations | (40,732 | ) | (31,569 | ) | (38,785 | ) | 80,774 | 56,546 | |||||||||||
Change in fair value of common stock warrant liability | — | — | — | — | (3,488 | ) | |||||||||||||
Change in fair value of contingent consideration related to acquisition | 676 | 987 | 2,871 | — | — | ||||||||||||||
Other (expense) income, net | (10,323 | ) | (12,951 | ) | (722 | ) | (844 | ) | 577 | ||||||||||
(Loss) income before provision for income taxes | (50,379 | ) | (43,533 | ) | (36,636 | ) | 79,930 | 53,635 | |||||||||||
(Provision) benefit for income taxes | (406 | ) | (2,186 | ) | (25,498 | ) | 14,271 | (3,704 | ) | ||||||||||
Net (loss) income | $ | (50,785 | ) | $ | (45,719 | ) | $ | (62,134 | ) | $ | 94,201 | $ | 49,931 | ||||||
Net (loss) income per share—basic | $ | (0.78 | ) | $ | (0.71 | ) | $ | (1.02 | ) | $ | 1.61 | $ | 0.94 | ||||||
Net (loss) income per share—diluted | $ | (0.78 | ) | $ | (0.71 | ) | $ | (1.02 | ) | $ | 1.46 | $ | 0.86 | ||||||
As of December 31, | |||||||||||||||||||
Selected Balance Sheet Data: | 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||
(In thousands) | |||||||||||||||||||
Working capital (current assets minus current liabilities) | $ | 114,981 | $ | 113,030 | $ | 145,206 | $ | 141,630 | $ | 151,443 | |||||||||
Total assets | $ | 421,220 | $ | 490,033 | $ | 499,155 | $ | 504,955 | $ | 280,780 | |||||||||
Long term obligations, less current portion | $ | 132,020 | $ | 126,040 | $ | 127,565 | $ | 93,031 | $ | 14,336 | |||||||||
Total stockholders’ equity | $ | 212,857 | $ | 254,554 | $ | 281,606 | $ | 288,681 | $ | 192,086 | |||||||||
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with “Selected Financial Data” and our consolidated financial statements and the related notes included in this Annual Report on Form 10-K. This discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of various factors including the risks we discuss in Item 1A of Part I, “Risk Factors” and elsewhere in this Annual Report on Form 10-K.
OVERVIEW
Our Business
Our primary strategy is comprised of acquiring, developing, and commercializing a broad and diverse pipeline of late-stage clinical and commercial products. In addition to an efficient in-house clinical development organization with regulatory and data management capabilities, we have established a commercial infrastructure for our marketed products. Currently, we have six oncology/hematology products approved that target different types of NHL, ALL, and MM.
We also have two drugs in late stage development:
•SPI-2012, is being developed for chemotherapy-induced neutropenia in patients with breast cancer.
• | EOQUIN® (previously referred to as APAZIQUONE for intravesical instillation), is being developed for immediate intravesical instillation post-transurethral resection of bladder tumors in patients with non-muscle invasive bladder cancer. |
44
Our passion to identify, develop and deliver important options for patients suffering from cancer is behind every action we take. We are committed to excellence and strive to make a difference in the lives of patients every day.
See Item 1, “Business,” for our discussion of:
• | Company Overview |
• | Cancer Background and Market Size |
• | Product Portfolio |
• | Manufacturing |
• | Sales and Marketing |
• | Customers |
• | Competition |
• | Research and Development |
Recent Highlights of Our Business, Product Development Initiatives, and Regulatory Approvals
During the year ended December 31, 2015 and through the filing date of this Annual Report on Form 10-K, we accomplished various critical business objectives, which included:
• | POZIOTINIB: In November 2015, we submitted an Investigational New Drug ("IND") application with the FDA. In March 2016 we initiated our Phase 2 Breast Cancer Trial. The Phase 2 study is an open-label study that will enroll approximately 70 patients with HER-2 positive metastatic breast cancer, who have failed at least two HER-2 directed therapies. The dose and schedule of oral POZIOTINIB will be based on clinical experience from the studies in Korea, and in addition include the use of prophylactic therapies to help minimize known side-effects of HER-2 directed therapies. |
In February 2015, we executed a global in-license agreement (excluding Korea and China) with Hanmi Pharmaceutical Co., Ltd for POZIOTINIB, a pan-HER inhibitor in Phase 2 clinical trials in return for our upfront payment and future regulatory and sales-dependent milestone payments. POZIOTINIB has shown single agent activity in the treatment of various cancer types, including breast, gastric, colorectal and lung cancers. In the Phase 1 study for this drug, 6 of 10 breast cancer patients demonstrated partial responses; we also believe the safety profile was consistent with similar drug classes, with four patients having a grade 3 diarrhea response.
• | EOQUIN (formerly referred to as APAZIQUONE): In August 2015, we reached agreement with the FDA on the SPA of the planned Phase 3 clinical trial of EOQUIN. This trial commenced with its first patient dosing in October 2015 and is designed to evaluate the intravesical use of this drug for the treatment of patients with non-muscle invasive bladder cancer (NMIBC) as one or two instillations, immediately following transurethral resection of bladder tumor (TURBT). Due to the high rate of recurrence for NMIBC, there is a significant unmet medical need and the overall cost of bladder cancer treatment in the U.S. is $3.4 billion annually, most of which is related to the direct treatment of this disease. Accordingly, this drug represents much-needed therapy for patients and provides a meaningful opportunity to reduce overall medical costs. In December 2015, we submitted our NDA for EOQUIN with the FDA, and in February 2016, the FDA communicated its acceptance of this NDA with a target decision date of December 11, 2016. |
• | EVOMELA (formerly referred to as Captisol-Enabled MELPHALAN): On October 23, 2015, we received a Complete Response Letter ("CRL") from the FDA for our EVOMELA NDA. A CRL is a standard communication from the FDA that informs companies that an application cannot be approved in its present form. Nonclinical deficiencies were identified, however, the FDA did not identify any clinical deficiencies for this drug in the CRL, and we subsequently resubmitted our NDA. On March 10, 2016, the FDA communicated its NDA approval for EVOMELA as a high-dose conditioning treatment prior to hematopoietic progenitor (stem) cell transplantation in patients with MM, and for the palliative treatment of patients with MM for whom oral therapy is not appropriate.. We plan to commercially launch EVOMELA as soon as possible. |
• | SPI 2012: In March 2015 positive Phase 2 data for this drug was presented at our Analyst Day, demonstrating non-inferiority and superiority to pegfilgrastim. In December 2015, we reached agreement with the FDA regarding our Phase 3 SPA for SPI-2012 and initiated the Phase 3 clinical study. We have designated more than 100 sites (with additional sites to be named) where patients are presently being dosed with SPI-2012 as part of this study. |
45
• | Sales Force Contracting Arrangement: On November 4, 2015, we executed a contract with Eagle Pharmaceuticals, Inc. ("Eagle") whereby designated members of our sales force will concurrently market (beginning January 2016) up to six of Eagle's pharmaceutical products, along with our products, in return for aggregate fixed proceeds of $12.8 million that will be paid over the 18-month service period. We are also eligible to receive variable, performance-based payments for sales of Eagle's products that exceed certain thresholds. |
• | ZEVALIN Ex-U.S. out-licenses: |
◦ | In November 2015, we entered into an out-license agreement with Mundipharma International Corporation Limited for their commercialization of ZEVALIN in Asia (excluding India and Greater China), Australia, New Zealand, Africa, the Middle East, and Latin America (including the Caribbean). In return, we received (i) $18 million (comprised of $15 million received in December 2015 and $3 million in January 2016), and (ii) unsecured notes aggregating $3.1 million. |
◦ | On January 8, 2016, we entered into a strategic partnership with Servier Canada, Inc. for the out-licenses of ZEVALIN, FOLOTYN, BELEODAQ, and MARQIBO. We will receive $6 million in upfront payments, development milestone payments if/when achieved, and a high single-digit royalty on their sales of these products. |
CHARACTERISTICS OF OUR REVENUE AND EXPENSES
The below summarizes the nature of our revenue and operating expense line items within our Consolidated Statements of Operations:
Revenue
The majority of our revenue is derived from sales of our drug products to large pharmaceutical wholesalers and distributors upon title transfer (which is typically at time of delivery), provided our other revenue recognition criteria have been met. To a lesser extent we also derive revenue from (i) license fees and royalties from out-licensing our drug products in named territories, and (ii) service revenue for our research and development activities conducted for the benefit of third parties.
Cost of Goods Sold (Excluding Amortization of Intangible Assets)
Cost of goods sold includes production and packaging materials, contract manufacturer fees, allocated personnel costs (excluding nominal stock-based compensation expense), shipping expenses, and royalty fees.
Selling, General and Administrative
Selling, general and administrative expenses primarily consist of compensation (including stock-based compensation) and benefits for our sales force and personnel that support our sales and marketing operations, and our general operations such as information technology, executive management, financial accounting, and human resources. It also includes costs attributable to marketing our products to our customers and prospective customers, patent and legal fees, financial statement audit fees, insurance coverage fees, bad debt expense, personnel recruiting fees, and other professional services.
Research and Development
Our research and development activities primarily relate to the development and testing of new drugs, and conducting studies in order to gain regulatory approval for the commercialization of our drug products. These expenses consist of compensation (including stock-based compensation) and benefits for research and development and clinical and regulatory personnel, materials and supplies, consultants, and regulatory and clinical payments related to studies. In addition, we include within research and development expense, technology transfer costs and manufacture qualification costs – prior to FDA approval of the product, its formulation, and/or its manufacturing sites.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
The preparation and presentation of financial statements in conformity with generally accepted accounting principles in the United States of America (“GAAP”) requires management to establish policies and make estimates and assumptions that affect (i) the amounts of assets and liabilities as of the date presented on the accompanying Consolidated Balance Sheets and (ii) the amounts of revenue and expenses for each year presented in the accompanying Consolidated Statements of Operations.
46
Our management believes its estimates and assumptions are supportable, reasonable, and consistently applied. Nonetheless, estimates are inherently uncertain. As a result, our financial position and operating results could materially differ from the amounts reported within the accompanying Consolidated Financial Statements if management’s estimates require prospective adjustment. Our critical accounting policies and estimates arise in conjunction with the following accounts:
• | Revenue recognition |
• | Inventories – lower of cost or market |
• | Fair value of acquired assets and assumed liabilities |
• | Goodwill and intangible assets – impairment evaluations |
• | Income taxes |
• | Stock-based compensation |
• | Litigation accruals (as required) |
Revenue Recognition
Product Sales: We recognize revenue from product sales when all of the following criteria are met:
(i) | Appropriate evidence of a binding arrangement exists with our customer; |
(ii) | Price is substantially fixed and determinable; |
(iii) | Collection from our customer is reasonably assured; |
(iv) | Our customer’s obligation to pay us is not contingent on resale of the product; |
(v) | We do not have significant obligations for future performance to directly bring about the resale of our product; and |
(vi) | We have a reasonable basis to estimate future returns. |
Our product sales are reduced by our gross-to-net (“GTN”) estimates, resulting in our reported “Product sales, net” in the accompanying Consolidated Statements of Operations. We defer revenue recognition in full if/when these estimates are not reasonably determinable at the time of sale.
Our GTN estimates reduce revenue in the same period that the related sale is recorded and include the following major categories:
(i) | Product Returns Allowances |
(ii) | Government Chargebacks |
(iii) | Discounts |
(iv) | Rebates |
(v) | Medicaid Rebates |
(vi) | Distribution and Data Fees |
Product Returns Allowances: Our FUSILEV, MARQIBO, and BELEODAQ customers are permitted to return purchased product beginning at its expiration date, and within six months thereafter. Returned product is generally not resold. Returns for expiry of ZEVALIN and FOLOTYN are not contractually, or customarily, allowed. We estimate potential returns based on historical rates of return.
Government Chargebacks: Our products are subject to pricing limits under certain federal government programs. Qualifying entities (end-users) purchase product from our wholesalers at their qualifying discounted price. The chargeback amount we incur represents the difference between our original sales price to the wholesaler, and the end-user’s applicable discounted purchase price. There may be significant lag time between our original sale to the wholesaler and our receipt of the corresponding government chargeback claims from our wholesalers.
47
Prompt Pay Discounts: Discounts for prompt payment are estimated at the time of sale, based on our eligible customers’ prompt payment history and the contractual discount percentage.
Commercial Rebates: Rebates are estimated based on our customers’ actual purchase level during the quarterly or annual rebate purchase period, and the corresponding contractual rebate tier we expect each customer to achieve.
Medicaid Rebates: Our products are subject to state government-managed Medicaid programs, whereby rebates for purchases are issued to participating state governments. These rebates arise when the patient treated with our products is covered under Medicaid. Our calculations related to these Medicaid rebate accruals require us to estimate end-user and patient mix to determine which of our sales will likely be subject to these rebates. There is a significant time lag in us receiving these rebate notices (generally several months after our sale is made). Our estimates are based on our historical claims, as supplemented by management’s judgment.
Distribution, Data, and GPO Administrative Fees: Distribution, data, and group purchasing organization (GPO) administrative fees are paid to authorized wholesalers of our products (except for U.S. sales of ZEVALIN) for various services, including: contract administration, inventory management, end-user sales data, and product returns processing. These fees are based on a contractually determined percentage of applicable sales.
License Fees: We recognize revenue for our licensing of intellectual property to third parties (i.e., out-licenses), based on the contractual terms of each agreement. This revenue may be associated with upfront license fees, milestone payments from our licensees’ sales or regulatory achievements, and royalties from our licensees’ sales in applicable territories.
Service Revenue: We receive fees under certain arrangements for research and development activities, sales and marketing activities, clinical trial management, and supply chain services. Payment may be triggered by the successful completion of a phase of development, results from a clinical trial, regulatory approval events, or completion of product or service delivery in our capacity as an agent or principal in such arrangement. We recognize revenue when the corresponding milestone is achieved, or the revenue is otherwise earned through our on-going activities.
Inventories – Lower of Cost or Market
We adjust our inventory value for estimated amounts of excess, obsolete, or unmarketable items. Such assumptions involve projections of future customer demand, as driven by economic and market conditions, and the product’s shelf life. If actual demand, or economic or market conditions are less favorable than those projected by us, incremental inventory write-downs may be required and could be significant.
Fair Value of Acquired Assets and Assumed Liabilities
The accounting for business combinations and asset acquisitions requires extensive use of estimates and judgments to measure the fair value of the identifiable tangible and intangible assets acquired, including in-process research and development, and liabilities assumed. Additionally, we must determine whether the acquisition meets the criteria for business combination accounting (rather than asset acquisition accounting), because in a business combination, the excess of the purchase price over the fair value of net assets acquired can only be recognized as “goodwill.”
The fair value of acquired tangible and identifiable intangible assets and liabilities assumed, are based on their estimated fair values at the acquisition date and requires extensive use of accounting estimates, judgments, and assumptions, including but not limited to: likelihood, timing, and costs to complete the in-process projects, probability of achieving regulatory approvals, cash flows to be derived from the acquired assets, and the application of appropriate discount rates. For each acquisition, we engage an independent third-party valuation specialist to assist management in determining the fair value of in-process research and development, identifiable intangible assets, and any contingent consideration.
In connection with certain of our acquisitions, we must record a contingent consideration liability for cash or stock payments upon the completion of certain future performance milestones. In these cases, a liability is recorded on the acquisition date for an estimate of the acquisition date fair value of the contingent consideration by applying the income approach utilizing variable inputs such as probability of achievement and risk-free adjusted discount rates. Any change in the fair value of the contingent consideration subsequent to the acquisition date is recognized in earnings.
Goodwill and Intangible Assets – Impairment Evaluations
48
Goodwill and other intangible assets with indefinite lives are not subject to amortization, but are evaluated for impairment annually as of October 1, or whenever events or changes in circumstance indicate that the asset might be impaired. We evaluate the possible impairment (i) if/when events or changes in circumstances occur that indicate that the carrying value of assets may not be recoverable; or (ii) in the case of goodwill and indefinite lived intangible assets, our annual impairment assessment date of October 1. These evaluations require significant judgment by our management in forecasting net cash flows to be derived by these intangible assets through our on-going operations. The discounted value of such cash flows (or our market capitalization in the case of goodwill) are compared to each asset’s carrying value to assess whether there is an indication of impairment and resulting charges to record.
Income Taxes
Our consolidated balance sheets reflect net deferred tax assets (net of a valuation allowance) that primarily represent the tax benefit of net operating loss and tax credit carryforwards, and credits and timing differences between book and tax. When it is more likely than not that all or some portion of deferred tax assets may not be realized, we establish a valuation allowance for the amount that may not be realized. Each quarter, we evaluate the need to retain all or a portion of the valuation allowance on our net deferred tax assets. Our evaluation considers historical earnings, estimated future taxable income and ongoing prudent and feasible tax planning strategies. Adjustments to the valuation allowance increase or decrease net income or loss in the period such adjustments are made. If our estimates require adjustments, it could have a significant impact on our consolidated financial statements.
Stock-Based Compensation
Stock-based compensation expense for equity awards granted to our employees and members of our board of directors is recognized on a straight-line basis over the award’s vesting period. Recognized compensation expense is net of an estimated forfeiture rate, which estimates those shares expected to be forfeited prior to vesting. We use the Black-Scholes option pricing model to determine the fair value of stock options (as of the date of grant) which carry service conditions for vesting. We use the Monte Carlo valuation model to value equity awards (as of the date of grant) which carry combined market conditions and service conditions for vesting. From time to time we issue stock warrants to non-employees. These awards are also valued using the Black-Scholes option pricing model, then are marked-to-market at each reporting period until fully vested.
Calculating stock-based compensation expense requires the input of highly subjective assumptions, including the pre-vesting forfeiture rate, expected term of the stock-based awards, stock price volatility, and risk-free interest rates. We estimate the expected term of options granted based on our employees’ historical exercise patterns, which we believe will be representative of their future behavior. We estimate the volatility of our common stock on the date of grant based on historical volatility of our common stock for a look-back period that corresponds with the expected term. We estimate the risk-free interest rate based upon the U.S. Treasury yields in effect at award grant, for a period equaling the stock option's expected term.
Litigation Accruals
From time-to-time, we are involved in various claims and legal proceedings of a nature considered normal and incidental to our business. These matters may include product liability, intellectual property, employment, and other general claims. We accrue for contingent liabilities when it is probable that a liability has been incurred and the amount can be reasonably estimated. The accruals are adjusted periodically as assessments change or as additional information becomes available.
RESULTS OF OPERATIONS
Operations Overview – 2015, 2014, and 2013
49
Year Ended December 31, | ||||||||||||||||||||
2015 | 2014 | 2013 | ||||||||||||||||||
($ in thousands) | ||||||||||||||||||||
Total revenues | $ | 162,556 | 100.0 | % | $ | 186,830 | 100.0 | % | $ | 155,854 | 100.0 | % | ||||||||
Operating costs and expenses: | ||||||||||||||||||||
Cost of product sales (excludes amortization and impairment of intangible assets) | 27,689 | 17.0 | % | 27,037 | 14.5 | % | 28,580 | 18.3 | % | |||||||||||
Selling, general and administrative | 86,514 | 53.2 | % | 97,412 | 52.1 | % | 99,315 | 63.7 | % | |||||||||||
Research and development | 50,766 | 31.2 | % | 69,662 | 37.4 | % | 46,670 | 29.9 | % | |||||||||||
Amortization and impairment of intangible assets | 38,319 | 23.6 | % | 24,288 | 13.0 | % | 20,074 | 12.9 | % | |||||||||||
Total operating costs and expenses | 203,288 | >100.0 | % | 218,399 | >100.0 | % | 194,639 | >100.0 | % | |||||||||||
Loss from operations | (40,732 | ) | (25.1 | )% | (31,569 | ) | (16.9 | )% | (38,785 | ) | (24.9 | )% | ||||||||
Interest expense, net | (9,074 | ) | (5.6 | )% | (8,584 | ) | (4.6 | )% | (2,192 | ) | (1.4 | )% | ||||||||
Change in fair value of contingent consideration related to acquisitions | 676 | 0.4 | % | 987 | 0.1 | % | 2,871 | 1.8 | % | |||||||||||
Other (expense) income, net | (1,249 | ) | (0.8 | )% | (4,367 | ) | (2.3 | )% | 1,470 | 0.1 | % | |||||||||
Loss before income tax | (50,379 | ) | (31.0 | )% | (43,533 | ) | (23.3 | )% | (36,636 | ) | (23.5 | )% | ||||||||
Provision for income taxes | (406 | ) | (0.2 | )% | (2,186 | ) | (1.1 | )% | (25,498 | ) | (16.4 | )% | ||||||||
Net loss | $ | (50,785 | ) | (31.2 | )% | $ | (45,719 | ) | (24.5 | )% | $ | (62,134 | ) | (39.9 | )% | |||||
YEAR ENDED DECEMBER 31, 2015 VERSUS DECEMBER 31, 2014
Total Revenues
Year Ended December 31, | ||||||||||||||
2015 | 2014 | $ Change | % Change | |||||||||||
($ in millions) | ||||||||||||||
Product sales, net: | ||||||||||||||
FUSILEV | $ | 60.7 | $ | 105.6 | $ | (44.9 | ) | (42.5 | )% | |||||
FOLOTYN | 40.6 | 47.6 | (7.0 | ) | (14.7 | )% | ||||||||
ZEVALIN | 17.5 | 22.1 | (4.6 | ) | (20.8 | )% | ||||||||
MARQIBO | 8.0 | 6.3 | 1.7 | 27.0 | % | |||||||||
BELEODAQ | 10.1 | 4.9 | 5.2 | >100.0 | % | |||||||||
136.9 | 186.5 | (49.6 | ) | (26.6 | )% | |||||||||
License fees and service revenue | 25.7 | 0.3 | 25.4 | >100.0 | % | |||||||||
Total revenues | $ | 162.6 | $ | 186.8 | $ | (24.2 | ) | (13.0 | )% | |||||
Product sales, net: To derive net product sales, gross product revenues in each period are reduced by management's latest estimated provisions for (i) product returns, (ii) government chargebacks, (iii) prompt pay discounts, (iv) commercial rebates, (v) Medicaid rebates, and (vi) distribution, data, and GPO administrative fees. Management considers various factors in the determination of these provisions, which are described in more detail within “Critical Accounting Policies and Estimates” above.
FUSILEV revenue decrease is primarily due to a significant decline in our unit sales to customers, as well as a decrease in our net average sales price per unit. This unit sales and price decline is due to the competitive launch in April 2015 of generic levo-leucovorin product (see Note 3(g)).
FOLOTYN revenue decrease is primarily due to moderate decline in units sold during the period, partially offset by a slight increase in our net average sales price per unit.
ZEVALIN revenue decrease is primarily due to moderate decline in units sold during the period in the U.S. and ex-U.S. territories, as well as a moderate decrease in our net average sales price per unit, specifically in ex-U.S. territories.
50
MARQIBO revenue increase is due to a moderate increase in units sold during the period, and a moderate increase in our net average sales price per unit.
BELEODAQ revenue increased as a result of units sold in the current period, in comparison to its commercial launch in the third quarter of 2014. The average net sales price per unit remained unchanged between the periods.
License fees and service revenue: In 2015, we recognized $25.7 million of license fees and service revenue which includes the following: (i) $9.7 million for the out-licenses of ZEVALIN, MARQIBO, and EVOMELA for the China territory (see Note 11), (ii) $15 million for our ZEVALIN out-license agreement with Mundipharma (see Note 12), and (iii) $0.8 million from our out-license royalties, all derived from FOLOTYN sales in Mundipharma’s (our co-development partner – see Note 16) territories.
Operating Expenses
Year Ended December 31, | ||||||||||||||
2015 | 2014 | $ Change | % Change | |||||||||||
($ in millions) | ||||||||||||||
Operating expenses: | ||||||||||||||
Cost of product sales (excludes amortization and impairment of intangible assets) | $ | 27.7 | $ | 27.0 | $ | 0.7 | 2.6 | % | ||||||
Selling, general and administrative | 86.5 | 97.4 | (10.9 | ) | (11.2 | )% | ||||||||
Research and development | 50.8 | 69.7 | (18.9 | ) | (27.1 | )% | ||||||||
Amortization and impairment of intangible assets | 38.3 | 24.3 | 14.0 | 57.6 | % | |||||||||
Total operating costs and expenses | $ | 203.3 | $ | 218.4 | $ | (15.1 | ) | (6.9 | )% | |||||
Cost of Product Sales. Cost of product sales increased, despite product revenue decline, primarily due to (i) $2.4 million for stability testing of ZEVALIN antibody for strategic supply stock in the current period, and (ii) recognition of $1.3 million of product cost associated with FUSILEV deferred revenue for shipments to our customers in the fourth quarter of 2015.
Selling, General and Administrative. Selling, general and administrative expenses decreased by $10.9 million, largely driven by our various 2015 operating expense reduction initiatives, as well as a $2.1 million reimbursement from our directors and officers insurance carrier, which was recognized as a reduction to current year expenses when these proceeds were received.
Research and Development. Research and development expenses decreased primarily due to the non-recurrence of our $17.8 million aggregate payment (in the form of cash and stock) in the first quarter of 2014, upon FDA milestone achievement associated with BELEODAQ. In addition, our clinical trial expenses in the current period have decreased, as we narrow our focus to certain drug development clinical studies. These reductions were partially offset by our upfront payment related to the POZIOTINIB in-license agreement (see Note 17 (b)(xii)) and technical transfer costs related to future ZEVALIN commercial production at a new contract manufacturer.
Amortization and Impairment of Intangible Assets. Amortization expense increased $14.0 million in the current year due to (i) $7.2 million impairment charge (non-cash) in the first quarter of 2015 for our FUSILEV distribution rights (See Note 3(g)), (ii) $3.7 million of accelerated amortization expense that was recorded in conjunction with the sale of certain ex-U.S. ZEVALIN rights to Mundipharma (see Note 12), and (iii) recognition of a full year of BELEODAQ amortization expense, which began in the third quarter of 2014 with its initial commercialization.
Total Other Expense
Year Ended December 31, | ||||||||||||||
2015 | 2014 | $ Change | % Change | |||||||||||
($ in millions) | ||||||||||||||
Total other expense | $ | (9.6 | ) | $ | (12.0 | ) | $ | 2.4 | 20.0 | % | ||||
51
Total other expenses decreased by $2.4 million due to multiple offsetting components. These items included (i) a decrease of $5.9 million related to foreign exchange loss adjustments on the value of intercompany loans, which are now recorded in "accumulated other comprehensive loss" in the Consolidated Balance Sheets, beginning April 1, 2015 (see Note 2 (ix)), (ii) $2.2 million related to a gain on the sale of certain stock holdings in 2014 that did not recur, (iii) $0.3 million increase in contingent consideration expense related to the acquisition of our MARQIBO and EVOMELA rights (see Note 10), (iv) a $0.5 million increase in interest expense on our 2018 Convertible Notes, and (v) $0.7 million increase in executive deferred compensation expense due to changes in the fair value of plan assets.
Provision for Income Taxes
Year Ended December 31, | ||||||||||||||
2015 | 2014 | $ Change | % Change | |||||||||||
($ in millions) | ||||||||||||||
Provision for income taxes | $ | (0.4 | ) | $ | (2.2 | ) | $ | 1.8 | 81.8 | % | ||||
In 2015, we recognized an income tax provision of $0.4 million, representing our minimum tax obligations and the impact of applied research and development tax credits in 2015. In 2014 our income tax provision primarily represented an adjustment to our prior year estimate of the benefit from the carryback of our 2013 federal net operating loss against 2012 taxes and an increase in the valuation allowance on deferred tax assets at January 1, 2014.
YEAR ENDED DECEMBER 31, 2014 VERSUS DECEMBER 31, 2013
Revenue
Year Ended December 31, | ||||||||||||||
2014 | 2013 | $ Change | % Change | |||||||||||
($ in millions) | ||||||||||||||
Product sales, net | ||||||||||||||
FUSILEV | $ | 105.6 | $ | 68.4 | $ | 37.2 | 54.4 | % | ||||||
FOLOTYN | 47.6 | 44.4 | 3.2 | 7.2 | % | |||||||||
ZEVALIN | 22.1 | 29.4 | (7.3 | ) | (24.8 | )% | ||||||||
MARQIBO | 6.3 | 1.3 | 5.0 | >100.0 | % | |||||||||
BELEODAQ | 4.9 | — | 4.9 | 100.0 | % | |||||||||
186.5 | 143.5 | 43.0 | 30.0 | % | ||||||||||
License fees and service revenue | 0.3 | 12.4 | (12.1 | ) | (97.6 | )% | ||||||||
Total revenues | $ | 186.8 | $ | 155.9 | $ | 30.9 | 19.8 | % | ||||||
Product sales, net: Gross product revenues are reduced by estimated provisions for product returns, sales discounts and rebates, distribution and data fees, and chargebacks established at the time revenues are recognized to arrive at product sales, net. Management considers various factors in the determination of such provisions, which are described in more detail within “Critical Accounting Policies and Estimates” above.
FUSILEV revenue increase was primarily due to (i) an increase in our average net price per unit as a result of certain non-recurring GTN adjustments that we experienced in the prior year period, and (ii) an increase in unit sales to our wholesalers during the current period, to satisfy end-user demand.
FOLOTYN revenue increase was due to an increase in unit sales to satisfy end-user demand, as well as a $1.0 million purchase by a new wholesaler in the first half of 2014; this resulted from the modification of our FOLOTYN distribution model and a bulk purchase by this wholesaler to fulfill anticipated end-user demand.
ZEVALIN revenue decrease was attributable to decreased end-user demand in the U.S. market, partially offset by an increase in our average net sales price per unit in 2014 versus 2013.
MARQIBO revenue increase was a result of our acquisition of Talon in July 2013 (and the product’s launch in late September 2013), as discussed in Note 10(a). In addition, during the third quarter of 2014, we modified the timing of our
52
revenue recognition model for this product from end-user receipt to wholesaler receipt, based on sufficient history of MARQIBO customer returns which provided a reasonable basis to estimate expected returns. This change had a one-time $0.4 million favorable impact in 2014 (i.e., representing units that had been shipped to our wholesalers in the second quarter of 2014 and earlier periods, but such revenue had been deferred through June 30, 2014).
BELEODAQ revenue was a result of its FDA approval and our subsequent commercial launch in the third quarter of 2014.
License fees and service revenue: In 2014, we recognized $0.3 million from our out-license royalties, all derived from FOLOTYN sales in Mundipharma’s (our co-development partner – see Note 16) territories. In 2013, we recognized $12.4 million from the amortization of deferred revenue that corresponded with our contracted research and development services. This revenue is associated with a $41.5 million upfront payment we received from Allergan in 2008, and an aggregate of $16.0 million in upfront payments we received from Nippon Kayaku and Handok in 2010.
Operating Expenses
Our operating expenses are summarized in the following table:
Year Ended December 31, | ||||||||||||||
2014 | 2013 | $ Change | % Change | |||||||||||
($ in millions) | ||||||||||||||
Operating expenses: | ||||||||||||||
Cost of product sales (excludes amortization and impairment of intangible assets) | $ | 27.0 | $ | 28.6 | $ | (1.6 | ) | (5.6 | )% | |||||
Selling, general and administrative | 97.4 | 99.3 | (1.9 | ) | (1.9 | )% | ||||||||
Research and development | 69.7 | 46.6 | 23.1 | 49.6 | % | |||||||||
Amortization and impairment of purchased intangible assets | 24.3 | 20.1 | 4.2 | 20.9 | % | |||||||||
Total operating costs and expenses | $ | 218.4 | $ | 194.6 | $ | 23.8 | 12.2 | % | ||||||
Cost of Product Sales. Despite our large increase in product sales, net, in 2014 as compared to 2013, our cost of product sales decreased 5.6%. This decrease was driven by a large inventory charge in 2013 for FUSILEV that did not recur in the current year, related to our program abandonment of a new vial size, as well as certain royalty adjustments from our in-license contract amendments in 2013 that did not recur in the current year.
Selling, General and Administrative. Selling, general and administrative expenses decreased due to the non-recurrence of $5.7 million of Talon acquisition expenses incurred in 2013, partially offset by increases in personnel expenses due to increased revenue, travel costs, and professional service fees as we continue to expand our sales and marketing activities.
Research and Development. Research and development expenses increased primarily due to the following: (i) an aggregate of $17.8 million from our cash payment and stock issuance to TopoTarget, upon the February 2014 contractual milestone achievement of the FDA acceptance of our new drug application for the PTCL indication of BELEODAQ; (ii) a $2.3 million increase in Phase 2 clinical study costs related to SPI-2012; (iii) a $1 million increase in new manufacturer technology transfer and qualification costs for our ZEVALIN product; and (iv) an increase in project consulting expenses of $1.2 million.
Amortization and Impairment of Intangible Assets. The amortization and impairment of intangible assets increased $4.2 million during 2014, primarily due to (i) the amortization of definite-lived intangible assets from the acquisition of Talon in July 2013 (through which we acquired MARQIBO distribution rights) and (ii) the commencement of the amortization of BELEODAQ distribution rights in August 2014.
Total Other Income (Expense)
Year Ended December 31, | |||||||||||||
2014 | 2013 | $ Change | % Change | ||||||||||
($ in millions) | |||||||||||||
Total other income (expense) | $ | (12.0 | ) | $ | 2.1 | $ | (14.1 | ) | >100.0% | ||||
53
Total other income (expenses) increased by $14.1 million primarily due to (i) a $6.4 million increase in interest expense attributable to our convertible senior notes issued in December 2013; (ii) a $8.0 million increase in unrealized loss from intercompany borrowings denominated in currencies other than the U.S. dollar; and (iii) a $1.9 million net increase in “acquisition-related contingent obligations” liability to the former stockholders of Talon and Ligand in the 2014 period, which resulted in an equal charge recognized within “change in fair value of contingent consideration related to acquisitions”. These amounts were partially offset by a $2.2 million gain on our sale of certain stock holdings during 2014.
Provision for Income Taxes
Year Ended December 31, | |||||||||||||
2014 | 2013 | $ Change | % Change | ||||||||||
($ in millions) | |||||||||||||
Provision for income taxes | $ | (2.2 | ) | $ | (25.5 | ) | $ | 23.3 | 91.4% | ||||
In 2014, we recognized an income tax provision of $2.2 million. This provision for income taxes primarily represents an adjustment to our prior year estimate of the benefit from the carryback of our 2013 federal net operating loss against 2012 taxes and an increase in the valuation allowance on deferred tax assets at January 1, 2014. In 2013, we recognized an income tax provision of $25.5 million. This provision was due to the recording of a full valuation allowance against our deferred tax assets.
LIQUIDITY AND CAPITAL RESOURCES
December 31, | |||||||
2015 | 2014 | ||||||
(in thousands, except financial metrics data) | |||||||
Cash and cash equivalents | $ | 139,741 | $ | 129,942 | |||
Marketable securities | $ | 245 | $ | 3,306 | |||
Accounts receivable, net | $ | 30,384 | $ | 70,758 | |||
Total current assets | $ | 191,324 | $ | 222,469 | |||
Total current liabilities | $ | 76,343 | $ | 109,439 | |||
Working capital surplus (a) | $ | 114,981 | $ | 113,030 | |||
Days sales outstanding (“DSO”) (b) | 56 | 126 | |||||
Current ratio (c) | 2.5 | 2.0 | |||||
(a) | Total current assets at period end minus total current liabilities at period end. |
(b) | Net accounts receivable at period end divided by revenue, net for the fourth quarter multiplied by 92 days. |
(c) | Total current assets at period end divided by total current liabilities at period end. |
Net Cash Provided By (Used In) Operating Activities
Cash provided by operating activities was $6.7 million in 2015, as compared to $3.6 million of cash used in the prior year period.
For the years ended December 31, 2015 and 2014, our cash collections from customers totaled $237.2 million and $242.5 million, respectively, representing 146% and 129% of reported net revenue for the same years.
For the years ended December 31, 2015 and 2014, cash payments to our employees and vendors for products, services, and rebates totaled $252.8 million and $279.5 million, respectively.
Net Cash Provided By (Used In) Investing Activities
Net cash provided by investing activities of $2.8 million is primarily due to $3.1 million from the redemption of mutual funds, partially offset by $0.2 million of purchases related to property, plant and equipment. This compares to cash used in investing activities of $21.8 million in the prior year, primarily related to the $25 million milestone payment due upon the FDA approval of BELEODAQ, partially offset by $4.1 million of proceeds from our sale of certain security holdings.
54
Net Cash Provided By Financing Activities
Net cash provided by financing activities of $1.5 million in 2015, as compared to $0.8 million in the prior year period. The cash provided by financing activities relates to (i) $1.5 million of proceeds from the issuance of common stock as a result of the exercise of employee stock options, and (ii) $0.6 million of proceeds from employee stock purchases under our employee stock purchase plan. These amounts were partially offset by our $0.6 million purchase and retirement of restricted stock at our employees’ election, in order to fund their corresponding minimum employee tax obligations at the time of vesting.
Convertible Senior Notes Due 2018
On December 17, 2013, we entered into an agreement for the sale of $120 million aggregate principal amount of 2.75% Convertible Senior Notes due December 2018 (the “2018 Convertible Notes”). The 2018 Convertible Notes are convertible into shares of our common stock at a conversion rate of 95 shares per $1,000 principal amount of the 2018 Convertible Notes, totaling 11.4 million common shares if fully converted. The in-the-money conversion price is equivalent to $10.53 per common share. The conversion rate and conversion price are subject to adjustment under certain limited circumstances. As of December 31, 2015, we may settle conversions of the 2018 Convertible Notes by paying or delivering, as the case may be, cash, shares of our common stock, or a combination of cash and shares, at our election.
The 2018 Convertible Notes bear interest at a rate of 2.75% per year, payable semiannually in arrears on June 15 and December 15 of each year, beginning on June 15, 2014. The 2018 Convertible Notes will mature and become payable on December 15, 2018, subject to earlier conversion into common stock at the holders’ option.
The sale of the 2018 Convertible Notes closed on December 23, 2013 and our net proceeds were $115.4 million, after deducting banker and professional fees of $4.6 million. We used a portion of these proceeds to simultaneously enter into “bought call” and “sold warrant” transactions with Royal Bank of Canada (collectively, the “Note Hedge”). We recorded the Note Hedge on a net cost basis of $13.1 million, as a reduction to “additional paid-in capital” in our accompanying Consolidated Balance Sheets. Under GAAP, the Note Hedge transaction is not expected to be marked-to-market through earnings or comprehensive income in future reporting periods.
Retired Credit Facility
On September 5, 2012, we entered into a credit agreement with Bank of America, N.A., as the administrative agent and an initial lender and Wells Fargo Bank, National Association, as an initial lender (the “Credit Agreement”). The Credit Agreement provided us with a committed $50 million revolving line of credit facility (the “Credit Facility”). The Credit Facility was to expire on September 5, 2014, but was repaid in full and canceled by us on December 20, 2013.
The Credit Facility bore interest, at our election, at a rate equal to the London Interbank Offer Rate (LIBOR), plus an applicable margin (2.75% to 4.25%, dependent on a defined liquidity ratio).
We recognized $1.2 million in 2013, within “interest expense” for this retired credit facility on the accompanying Consolidated Statement of Operations.
Future Capital Requirements
We believe that the future growth of our business will depend on our ability to successfully develop and acquire new drugs for the treatment of cancer and successfully bring these drugs to market.
The timing and amount of our future capital requirements will depend on many factors, including:
• | the need for additional capital to fund future development programs; |
• | the need for additional capital to fund strategic acquisitions; |
• | the need for additional capital to fund licensing arrangements; |
• | our requirement for additional information technology infrastructure and systems; and |
• | adverse outcomes from potential litigation and the cost to defend such litigation. |
We believe that our $140 million in aggregate cash and equivalents, and marketable securities as of December 31, 2015 will allow us to fund our current and planned operations for at least the next twelve months. However, we may seek additional capital through the sale of debt or equity securities, if necessary, especially in conjunction with opportunistic acquisitions or
55
licensing arrangements. We may be unable to obtain such additional capital when needed, or on terms favorable to us or our current stockholders and convertible senior note holders.
Contractual Obligations
The following table summarizes our contractual financial commitments as of December 31, 2015:
Total | Less than 1 Year | 1-3 Years | 3-5 Years | After 5 Years | |||||||||||||||
(in thousands) | |||||||||||||||||||
Operating lease obligations (1) | $ | 4,124 | $ | 1,283 | $ | 2,381 | $ | 460 | $ | — | |||||||||
Purchase obligations (2) | 39,421 | 32,916 | 6,345 | 118 | 42 | ||||||||||||||
Contingent milestone obligations (3) | 1,193,413 | 6,000 | 13,200 | 15,529 | 1,158,684 | ||||||||||||||
Drug development liability (4) | 14,685 | 259 | 735 | — | 13,691 | ||||||||||||||
Debt obligations (5) | 129,753 | 3,300 | 126,453 | — | — | ||||||||||||||
Total | $ | 1,381,396 | $ | 43,758 | $ | 149,114 | $ | 16,107 | $ | 1,172,417 | |||||||||
(1) | The operating lease obligations are primarily related to the facility lease for our corporate headquarters in Henderson, Nevada, expiring April 30, 2019; and our research and development and administrative facility in Irvine, California, expiring May 31, 2019. |
(2) | Purchase obligations represent the amount of open purchase orders and contractual commitments to vendors for products and services that have not been delivered, or rendered, as of December 31, 2015. |
(3) | Milestone obligations are payable contingent upon successfully reaching certain development and regulatory milestones. Given the unpredictability of the drug development process, and the impossibility of predicting the success of current and future clinical trials, these values assume that all development and regulatory milestones under all of our license agreements are successfully met, and represent our best estimate of each achievement date. In the event that the milestones are met, we believe it is likely that the increase in the potential value of the related drug product will exceed the amount of the milestone obligation. |
(4) | Research and development services under the Mundipharma Collaboration Agreement (see Note 16 to the accompanying Consolidated Financial Statements) over the period required to complete the jointly agreed-upon clinical development activities. |
(5) | Debt obligations represent amount due under our 2018 Convertible Notes issued in December 2013, inclusive of interest payments over its full term (see Note 15 to the accompanying Consolidated Financial Statements). |
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements (except for operating leases) that provide financing, liquidity, market or credit risk support, or involve derivatives. In addition, we have no arrangements that may expose us to liability that are not expressly reflected in the accompanying Consolidated Financial Statements and/or notes thereto.
As of December 31, 2015, we did not have any relationships with unconsolidated entities or financial partnerships, often referred to as “structured finance” or “special purpose entities,” established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. As such, we are not subject to any material financing, liquidity, market or credit risk that could arise if we had engaged in such relationships.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
In the normal course of business, our operations are exposed to risks associated with fluctuations in interest rates and foreign currency exchange rates.
The primary objective of our investment activities is to preserve principal, while at the same time maximizing yields without significantly increasing risk. We do not utilize hedging contracts or similar instruments. Because of our ability to generally redeem these investments at par at short notice and without penalty, changes in interest rates would have an immaterial effect on the fair value of these investments. If a 10% change in interest rates were to have occurred on
56
December 31, 2015, any decline in the fair value of our investments would not be material in the context of our accompanying Consolidated Financial Statements. In addition, we are exposed to certain market risks associated with credit ratings of corporations whose corporate bonds we may purchase from time to time. If these companies were to experience a significant detrimental change in their credit ratings, the fair market value of such corporate bonds may significantly decrease. If these companies were to default on these corporate bonds, we may lose part or all of our principal. We believe that we effectively manage this market risk by diversifying our investments, and investing in highly rated securities.
We are exposed to foreign currency exchange rate fluctuations relating to payments we make to vendors, suppliers and license partners using foreign currencies. In particular, some of our obligations are incurred in Euros. We mitigate such risk by maintaining a limited portion of our cash in Euros and other currencies.
57
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
Spectrum Pharmaceuticals, Inc. | ||
By: | /s/ RAJESH C. SHROTRIYA, M.D. | |
Rajesh C. Shrotriya, M.D. | ||
Chairman of the Board, Chief Executive Officer | ||
Date: March 14, 2016
POWER OF ATTORNEY
Each person whose signature appears below constitutes and appoints each of Rajesh C. Shrotriya and Kurt A. Gustafson as his attorney-in-fact, with full power of substitution, for him in any and all capacities, to sign any amendments to this Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each attorney-in-fact, or his substitute, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
Signature | Title | Dates |
/s/ RAJESH C. SHROTRIYA, M.D. | Chairman of the Board and Chief Executive Officer | March 14, 2016 |
Rajesh C. Shrotriya, M.D. | ||
/s/ KURT A. GUSTAFSON | Executive Vice President and Chief Financial Officer | March 14, 2016 |
Kurt A. Gustafson | ||
/s/ DOLOTRAI M. VYAS, PH.D. | Director | March 14, 2016 |
Dolatrai M. Vyas, Ph.D. | ||
/s/ LUIGI LENAZ, M.D. | Director | March 14, 2016 |
Luigi Lenaz, M.D. | ||
/s/ STUART M. KRASSNER, SC.D., PSY.D | Director | March 14, 2016 |
Stuart M. Krassner, Sc.D., Psy.D. | ||
/s/ ANTHONY E. MAIDA, III, M.A., M.B.A., PH.D. | Director | March 14, 2016 |
Anthony E. Maida, III, M.A., M.B.A., Ph.D. | ||
/s/ RAYMOND W. COHEN | Director | March 14, 2016 |
Raymond W. Cohen | ||
/s/ GILLES GAGNON | Director | March 14, 2016 |
Gilles Gagnon, M.Sc., M.B.A | ||
58
ITEM 8. CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
SPECTRUM PHARMACEUTICALS, INC.
FORM 10-K ANNUAL REPORT
For the Fiscal Year Ended December 31, 2015
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Page | ||
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Spectrum Pharmaceuticals, Inc.
Henderson, Nevada
We have audited the accompanying consolidated balance sheets of Spectrum Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2015 and 2014, and the related consolidated statements of operations, comprehensive loss, stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2015. Our audits also included the financial statement schedule listed in the Index at Item 15. These consolidated financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on the consolidated financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2015 and 2014, and the results of their operations and their cash flows for each of the two years in the period ended December 31, 2015, in conformity with accounting principles generally accepted in the United States of America. Also, in our opinion, such financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2015, based on the criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 14, 2016 expressed an unqualified opinion on the Company’s internal control over financial reporting.
/s/ Deloitte & Touche LLP
Costa Mesa, California
March 14, 2016
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
Spectrum Pharmaceuticals, Inc.
We have audited the accompanying consolidated statements of operations, comprehensive loss, stockholders’ equity, and cash flows for the year ended December 31, 2013. Our audit also included the financial statement schedule listed in the Index at Item 15(a). These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated results of operations and cash flows of Spectrum Pharmaceuticals, Inc. for the year ended December 31, 2013, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
/s/ Ernst & Young LLP
Irvine, California
March 12, 2014
F-3
SPECTRUM PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and par value amounts)
December 31, | |||||||
2015 | 2014 | ||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 139,741 | $ | 129,942 | |||
Marketable securities | 245 | 3,306 | |||||
Accounts receivable, net of allowance for doubtful accounts of $120 and $120, respectively | 30,384 | 70,758 | |||||
Other receivables | 12,572 | 5,489 | |||||
Inventories | 4,176 | 9,200 | |||||
Prepaid expenses and other assets | 4,206 | 3,774 | |||||
Total current assets | 191,324 | 222,469 | |||||
Property and equipment, net of accumulated depreciation | 918 | 1,405 | |||||
Intangible assets, net of accumulated amortization | 190,335 | 230,100 | |||||
Goodwill | 17,960 | 18,195 | |||||
Other assets | 20,683 | 17,864 | |||||
Total assets | $ | 421,220 | $ | 490,033 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable and other accrued liabilities | $ | 56,539 | $ | 84,994 | |||
Accrued payroll and benefits | 8,188 | 8,444 | |||||
Deferred revenue | 6,130 | 9,959 | |||||
Drug development liability | 259 | 1,141 | |||||
Acquisition-related contingent obligations | 5,227 | 4,901 | |||||
Total current liabilities | 76,343 | 109,439 | |||||
Drug development liability, less current portion | 14,427 | 14,644 | |||||
Deferred revenue, less current portion | 383 | — | |||||
Acquisition-related contingent obligations | 1,439 | 2,441 | |||||
Deferred tax liability | 6,779 | 6,569 | |||||
Other long-term liabilities | 7,444 | 6,088 | |||||
Convertible senior notes | 101,548 | 96,298 | |||||
Total liabilities | 208,363 | 235,479 | |||||
Commitments and contingencies | |||||||
Stockholders’ equity: | |||||||
Preferred stock, $0.001 par value; 5,000,000 shares authorized | |||||||
Series B Junior Participating Preferred Stock, $0.001 par value; 1,500,000 shares authorized: no shares issued and outstanding | — | — | |||||
Series E Convertible Voting Preferred Stock, $0.001 par value and $10,000 stated value; 2,000 shares authorized; 20 shares issued and outstanding at December 31, 2015 and 2014, respectively (convertible into 40,000 shares of common stock, with aggregate liquidation value of $240) | 123 | 123 | |||||
Common stock, $0.001 par value; 175,000,000 shares authorized; 68,228,935 and 65,969,699 issued and outstanding at December 31, 2015 and 2014, respectively | 68 | 66 | |||||
Additional paid-in capital | 552,108 | 538,553 | |||||
Accumulated other comprehensive loss | (5,319 | ) | (850 | ) | |||
Accumulated deficit | (334,123 | ) | (283,338 | ) | |||
Total stockholders’ equity | 212,857 | 254,554 | |||||
Total liabilities and stockholders’ equity | $ | 421,220 | $ | 490,033 | |||
See accompanying notes to these consolidated financial statements.
F-4
SPECTRUM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except share and per share amounts)
Year Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
Revenues: | |||||||||||
Product sales, net | $ | 136,851 | $ | 186,537 | $ | 143,475 | |||||
License fees and service revenue | 25,705 | 293 | 12,379 | ||||||||
Total revenues | 162,556 | 186,830 | 155,854 | ||||||||
Operating costs and expenses: | |||||||||||
Cost of product sales (excludes amortization and impairment of intangible assets) | 27,689 | 27,037 | 28,580 | ||||||||
Selling, general and administrative | 86,514 | 97,412 | 99,315 | ||||||||
Research and development | 50,766 | 69,662 | 46,670 | ||||||||
Amortization and impairment of intangible assets | 38,319 | 24,288 | 20,074 | ||||||||
Total operating costs and expenses | 203,288 | 218,399 | 194,639 | ||||||||
Loss from operations | (40,732 | ) | (31,569 | ) | (38,785 | ) | |||||
Other (expense) income: | |||||||||||
Interest expense, net | (9,074 | ) | (8,584 | ) | (2,192 | ) | |||||
Change in fair value of contingent consideration related to acquisitions | 676 | 987 | 2,871 | ||||||||
Other (expense) income, net | (1,249 | ) | (4,367 | ) | 1,470 | ||||||
Total other (expense) income | (9,647 | ) | (11,964 | ) | 2,149 | ||||||
Loss before income taxes | (50,379 | ) | (43,533 | ) | (36,636 | ) | |||||
Provision for income taxes | (406 | ) | (2,186 | ) | (25,498 | ) | |||||
Net loss | $ | (50,785 | ) | $ | (45,719 | ) | $ | (62,134 | ) | ||
Net loss per share: | |||||||||||
Basic | $ | (0.78 | ) | $ | (0.71 | ) | $ | (1.02 | ) | ||
Diluted | $ | (0.78 | ) | $ | (0.71 | ) | $ | (1.02 | ) | ||
Weighted average shares outstanding: | |||||||||||
Basic | 64,882,417 | 64,708,163 | 60,729,128 | ||||||||
Diluted | 64,882,417 | 64,708,163 | 60,729,128 | ||||||||
See accompanying notes to these consolidated financial statements.
F-5
SPECTRUM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands)
Year Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
Net loss | $ | (50,785 | ) | $ | (45,719 | ) | $ | (62,134 | ) | ||
Other comprehensive (loss) income, net of tax: | |||||||||||
Unrealized (loss) gain on available-for-sale securities | (1,429 | ) | (1,122 | ) | 690 | ||||||
Adjustment for realized gain on available-for-sale securities, and included in net income | — | (2,217 | ) | — | |||||||
Foreign currency translation adjustments | (3,040 | ) | 1,595 | (69 | ) | ||||||
Other comprehensive (loss) income, net | (4,469 | ) | (1,744 | ) | 621 | ||||||
Total comprehensive loss | $ | (55,254 | ) | $ | (47,463 | ) | $ | (61,513 | ) | ||
See accompanying notes to these consolidated financial statements.
F-6
SPECTRUM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share data)
Preferred Stock | Common Stock | Additional Paid-In Capital | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Treasury Stock | Total Stockholders' Equity | ||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||
Balance as of December 31, 2012 | 20 | $ | 123 | 60,026,675 | $ | 60 | $ | 463,710 | $ | 273 | $ | (175,485 | ) | — | $ | — | $ | 288,681 | ||||||||||||||||||
Net income | — | — | — | — | — | — | (62,134 | ) | — | — | (62,134 | ) | ||||||||||||||||||||||||
Other comprehensive income, net | — | — | — | — | — | 621 | — | — | — | 621 | ||||||||||||||||||||||||||
Issuance of common stock to 401(k) plan | — | — | 99,359 | — | 860 | — | — | — | — | 860 | ||||||||||||||||||||||||||
Issuance of common stock for ESPP | — | — | 74,925 | — | 495 | — | — | — | — | 495 | ||||||||||||||||||||||||||
Issuance of common stock upon exercise of stock options | — | — | 825,884 | 1 | 3,576 | — | — | — | — | 3,577 | ||||||||||||||||||||||||||
Share-based compensation expense and common stock issued (net of forfeitures) | — | — | 471,875 | — | 11,193 | — | — | — | — | 11,193 | ||||||||||||||||||||||||||
Repurchase of shares to satisfy employee tax withholding | — | — | (159,545 | ) | — | (1,509 | ) | — | — | — | — | (1,509 | ) | |||||||||||||||||||||||
Purchase of treasury stock | — | — | — | — | — | — | — | 235,000 | (1,651 | ) | (1,651 | ) | ||||||||||||||||||||||||
Retirement of treasury stock | — | — | (235,000 | ) | — | (1,651 | ) | — | — | (235,000 | ) | 1,651 | — | |||||||||||||||||||||||
Issuance of common stock for Talon acquisition | — | — | 3,000,000 | 3 | 26,307 | — | — | — | — | 26,310 | ||||||||||||||||||||||||||
Issuance of 2018 Convertible Notes | — | — | — | — | 14,443 | — | — | — | — | 14,443 | ||||||||||||||||||||||||||
Balance as of December 31, 2013 | 20 | $ | 123 | 64,104,173 | $ | 64 | $ | 518,144 | $ | 894 | $ | (237,619 | ) | — | $ | — | $ | 281,606 | ||||||||||||||||||
Net loss | — | — | — | — | — | — | (45,719 | ) | — | — | (45,719 | ) | ||||||||||||||||||||||||
Other comprehensive loss, net | — | — | — | — | — | (1,744 | ) | — | — | — | (1,744 | ) | ||||||||||||||||||||||||
Issuance of common stock to 401(k) plan | — | — | 133,734 | — | 1,028 | — | — | — | — | 1,028 | ||||||||||||||||||||||||||
Issuance of common stock for ESPP | — | — | 99,551 | — | 639 | — | — | — | — | 639 | ||||||||||||||||||||||||||
Issuance of common stock upon exercise of stock options | — | — | 485,260 | 1 | 1,905 | — | — | — | — | 1,906 | ||||||||||||||||||||||||||
Share-based compensation expense and common stock issued (net of forfeitures) | — | — | 396,083 | — | 10,781 | — | — | — | — | 10,781 | ||||||||||||||||||||||||||
Repurchase of shares to satisfy employee tax withholding | — | — | (249,102 | ) | (1,733 | ) | — | — | — | — | (1,733 | ) | ||||||||||||||||||||||||
Issuance of common stock to TopoTarget for milestone achievement | — | — | 1,000,000 | 1 | 7,789 | — | — | — | — | 7,790 | ||||||||||||||||||||||||||
Balance as of December 31, 2014 | 20 | $ | 123 | 65,969,699 | $ | 66 | $ | 538,553 | $ | (850 | ) | $ | (283,338 | ) | — | $ | — | $ | 254,554 | |||||||||||||||||
Net loss | — | — | — | — | — | — | (50,785 | ) | — | — | (50,785 | ) | ||||||||||||||||||||||||
Other comprehensive loss, net | — | — | — | — | — | (4,469 | ) | — | — | — | (4,469 | ) | ||||||||||||||||||||||||
Issuance of common stock to 401(k) plan | — | — | 179,865 | — | 1,124 | — | — | — | — | 1,124 | ||||||||||||||||||||||||||
Issuance of common stock for ESPP | — | — | 114,578 | — | 627 | — | — | — | — | 627 | ||||||||||||||||||||||||||
Issuance of common stock upon exercise of stock options | — | — | 456,082 | — | 1,482 | — | — | — | — | 1,482 | ||||||||||||||||||||||||||
Warrant modification | — | — | — | — | 568 | — | — | — | — | 568 | ||||||||||||||||||||||||||
Share-based compensation expense and common stock issued (net of forfeitures) | — | — | 1,613,553 | 2 | 10,392 | — | — | — | — | 10,394 | ||||||||||||||||||||||||||
Repurchase of shares to satisfy employee tax withholding | — | — | (104,842 | ) | (638 | ) | — | — | — | — | (638 | ) | ||||||||||||||||||||||||
Balance as of December 31, 2015 | 20 | $ | 123 | 68,228,935 | $ | 68 | $ | 552,108 | $ | (5,319 | ) | $ | (334,123 | ) | — | $ | — | $ | 212,857 | |||||||||||||||||
See accompanying notes to these consolidated financial statements.
F-7
SPECTRUM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands
Year Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
Cash Flows From Operating Activities: | |||||||||||
Net loss | $ | (50,785 | ) | $ | (45,719 | ) | $ | (62,134 | ) | ||
Adjustments to reconcile net loss to net cash (used in) provided by operating activities: | |||||||||||
Amortization of deferred revenue | — | — | (12,400 | ) | |||||||
Depreciation and amortization | 31,869 | 25,352 | 22,096 | ||||||||
Stock-based compensation | 12,084 | 11,809 | 12,423 | ||||||||
Change in fair value of common stock warrants issued to non-employees | — | — | 356 | ||||||||
Accretion of debt discount, recorded to interest expense on 2018 Convertible Notes (Note 15) | 5,250 | 4,818 | 43 | ||||||||
Amortization of deferred financing costs, recorded to interest expense on 2018 Convertible Notes (Note 15) | 662 | 599 | 101 | ||||||||
Bad debt (recovery) expense | — | (85 | ) | 127 | |||||||
Non-cash foreign currency exchange loss (income) | (157 | ) | 6,033 | 1,222 | |||||||
Impairment of intangible assets (Note 3(g)) | 7,160 | — | 1,023 | ||||||||
Change in fair value of contingent consideration related to Talon and EVOMELA acquisitions (Note 9) | (676 | ) | (987 | ) | (2,871 | ) | |||||
Change in fair value of Allos deferred development costs and deferred payment contingency (Note 16) | — | — | (2,869 | ) | |||||||
Research and development expense recognized for BELEODAQ in-license milestone achievement (Note 17(b)(x)) | — | 7,790 | — | ||||||||
Changes in operating assets and liabilities: | |||||||||||
Accounts receivable, net | 40,245 | (21,671 | ) | 42,559 | |||||||
Other receivables | (7,017 | ) | 2,070 | — | |||||||
Inventories | 1,863 | 4,253 | 1,570 | ||||||||
Prepaid expenses and current portion of other assets | (446 | ) | (718 | ) | (359 | ) | |||||
Deferred tax assets | — | 1,724 | 33,252 | ||||||||
Other assets | (1,731 | ) | (13,161 | ) | (8,989 | ) | |||||
Accounts payable and other accrued obligations | (28,298 | ) | 5,304 | (23,897 | ) | ||||||
Accrued payroll and benefits | (233 | ) | 1,594 | 425 | |||||||
Drug development liability | (1,100 | ) | (1,957 | ) | (5,917 | ) | |||||
Deferred revenue | (3,511 | ) | 9,803 | — | |||||||
Deferred tax liability | 210 | (602 | ) | — | |||||||
Other long-term liabilities | 1,355 | 124 | 2,153 | ||||||||
Net cash provided by (used in) operating activities | 6,744 | (3,627 | ) | (2,086 | ) | ||||||
Cash Flows From Investing Activities: | |||||||||||
Purchases of property and equipment | (223 | ) | (934 | ) | (161 | ) | |||||
Proceeds from sale of available-for-sale securities | 3,061 | 4,093 | — | ||||||||
Capitalized milestone payment upon FDA approval of BELEODAQ | — | (25,000 | ) | — | |||||||
Acquisition of EVOMELA (Note 10) | — | — | (3,000 | ) | |||||||
Acquisition of Talon, net of cash acquired (Note 10) | — | — | (11,169 | ) | |||||||
Net cash provided by (used in) investing activities | 2,838 | (21,841 | ) | (14,330 | ) | ||||||
Cash Flows From Financing Activities: | |||||||||||
Proceeds from Mundipharma related to FOLOTYN collaboration (Note 16) | — | — | 7,000 | ||||||||
Proceeds from exercise of stock options | 1,482 | 1,906 | 3,576 | ||||||||
Proceeds from sale of stock under employee stock purchase plan | 627 | 639 | 495 | ||||||||
Purchase of treasury stock | — | — | (1,651 | ) | |||||||
Purchase and retirement of restricted stock to satisfy employee tax liability at vesting | (638 | ) | (1,733 | ) | (1,509 | ) | |||||
Proceeds from revolving line of credit (Note 14) | — | — | 100,000 | ||||||||
Repayment of revolving line of credit (Note 14) | — | — | (175,000 | ) | |||||||
Proceeds from 2018 Convertible Notes (Note 15) | — | — | 120,000 | ||||||||
Deferred financing costs (Note 15) | — | — | (4,573 | ) | |||||||
Proceeds from sale of common stock warrants for 2018 Convertible Notes issuance (Note 15) | — | — | 12,612 | ||||||||
Purchase of common stock call options related to 2018 Convertible Notes issuance (Note 15) | — | — | (25,692 | ) | |||||||
Net cash provided by financing activities | 1,471 | 812 | 35,258 | ||||||||
Effect of exchange rates on cash and equivalents | (1,254 | ) | (1,708 | ) | (2,235 | ) | |||||
Net (decrease) increase in cash and equivalents | 9,799 | (26,364 | ) | 16,608 | |||||||
Cash and equivalents — beginning of year | 129,942 | 156,306 | 139,698 | ||||||||
Cash and equivalents — end of year | $ | 139,741 | $ | 129,942 | $ | 156,306 | |||||
Supplemental Disclosure of Cash Flow Information: | |||||||||||
Cash paid for income taxes | $ | 335 | $ | 329 | $ | — | |||||
Cash paid for interest | $ | 3,300 | $ | 3,227 | $ | 1,200 | |||||
Retirement of treasury shares | $ | — | $ | — | $ | 1,652 | |||||
Common stock issued for Talon acquisition | $ | — | $ | — | $ | 26,310 | |||||
See accompanying notes to these consolidated financial statements.
F-8
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
1. DESCRIPTION OF BUSINESS, BASIS OF PRESENTATION, AND OPERATING SEGMENT
(a) Description of Business
Spectrum Pharmaceuticals, Inc. (“Spectrum”, the “Company”, “we”, “our”, or “us”) is a biotechnology company, with a primary strategy comprised of acquiring, developing, and commercializing a broad and diverse pipeline of late-stage clinical and commercial products. In addition to an in-house clinical development organization with regulatory and data management capabilities, we have established a commercial infrastructure for our marketed products. Currently, we have six approved oncology/hematology products that target different types of non-Hodgkin's lymphoma ("NHL"), advanced metastatic colorectal cancer, acute lymphoblastic leukemia ("ALL"), and multiple myeloma ("MM").
We also have two drugs in late stage development:
•SPI-2012, is being developed for chemotherapy-induced neutropenia in patients with breast cancer.
• | EOQUIN® (previously referred to as APAZIQUONE for intravesical instillation), is being developed for immediate intravesical instillation post-transurethral resection of bladder tumors in patients with non-muscle invasive bladder cancer. |
(b) Basis of Presentation
Principles of Consolidation
The accompanying Consolidated Financial Statements in this Annual Report on Form 10-K have been prepared in accordance with generally accepted accounting principles in the United States of America (“GAAP”) and with the rules and regulations of the U.S. Securities and Exchange Commission (“SEC”). These financial statements include the financial position, results of operations, and cash flows of Spectrum and its subsidiaries, all of which are wholly-owned (except for SPC, as discussed below). All inter-company accounts and transactions among these legal entities have been eliminated in consolidation.
Variable Interest Entity
We own fifty-percent of Spectrum Pharma Canada (“SPC”), a legal entity organized in Quebec, Canada in January 2008. Certain of our drug clinical studies are conducted through this “variable interest entity” (as defined under applicable GAAP) and we fund all of SPC’s operating costs. Since we carry the full risks and rewards of SPC, we meet the applicable GAAP criteria as being its “primary beneficiary.” Accordingly, SPC’s balance sheets and statements of operations are included in our Consolidated Financial Statements as if it were a wholly-owned subsidiary for all periods presented.
(c) Operating Segment
We operate in one reportable operating segment that is focused exclusively on developing and commercializing oncology and hematology drug products. For the years ended December 31, 2015, 2014, and 2013, all of our revenue and related expenses were solely attributable to these activities. Substantially all of our assets (excluding our cash held in certain foreign bank accounts and our ZEVALIN distribution rights for the Ex-U.S. territory) are held in the U.S.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES AND USE OF ESTIMATES
The preparation of financial statements in conformity with GAAP requires our management to make informed estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses. However, actual values may materially differ, since estimates are inherently uncertain. On an on-going basis, our management evaluates its estimates and assumptions, including those related to (i) gross-to-net revenue adjustments; (ii) the timing of revenue recognition; (iii) the collectability of customer accounts; (iv) whether the cost of inventories can be recovered; (v) the fair value of goodwill and intangible assets; (vi) the realization of tax assets and estimates of tax liabilities; (vii) the likelihood of payment and value of contingent liabilities; (viii) the fair value of investments; (ix) the valuation of stock options and the periodic expense recognition of stock-based compensation; and (x) the potential outcome of ongoing or threatened litigation.
The estimates and assumptions that most significantly impact the presented amounts within these Consolidated Financial Statements are further described below:
F-9
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
(i) Revenue Recognition
(a) Product Sales: We sell our products to wholesalers/distributors (i.e., our customers), except for our U.S. sales of ZEVALIN in which case the end-user (i.e., clinic or hospital) is our customer. Our wholesalers distributors in turn sell our products directly to clinics, hospitals, and private oncology-based practices. Revenue from product sales is recognized when title and risk of loss have transferred to our customer, and the following additional criteria are met:
(1) | appropriate evidence of a binding arrangement exists with our customer; |
(2) | price is substantially fixed and determinable; |
(3) | collection from our customer is reasonably assured; |
(4) | our customer’s obligation to pay us is not contingent on resale of the product; |
(5) | we do not have significant continued performance obligations to our customer; and |
(6) | we have a reasonable basis to estimate returns. |
Our gross revenue is reduced by our gross-to-net (“GTN”) estimates each period, resulting in our reported “product sales, net” in the accompanying Consolidated Statements of Operations. We defer revenue recognition in full if these estimates are not reasonably determinable at the time of sale. These estimates are based upon information received from external sources (such as written and oral information obtained from our customers with respect to their period-end inventory levels and their sales to end-users during the period), in combination with management’s informed judgments. Due to the inherent uncertainty of estimates, the actual amount we incur may be materially different than our GTN estimates, and require prospective revenue adjustments in periods after the initial sale was recorded.
Our GTN estimates are comprised of the following categories:
Product Returns Allowances: Our FUSILEV, MARQIBO, and BELEODAQ customers are permitted to return purchased products beginning at its expiration date and within six months thereafter. Returned product is generally not resold. Returns for expiry of ZEVALIN and FOLOTYN are not contractually, or customarily, allowed. We estimate expected returns based on historical return rates.
Government Chargebacks: Our products are subject to pricing limits under certain federal government programs (e.g., Medicare and 340B Drug Pricing Program). Qualifying entities (i.e., end-users) purchase products from our customers at their qualifying discounted price. The chargeback amount we incur represents the difference between our contractual sales price to our customer, and the end-user’s applicable discounted purchase price under the government program. There may be significant lag time between our reported net product sales and our receipt of the corresponding government chargeback claims from our customers.
Prompt Pay Discounts: Discounts for prompt payment are estimated at the time of sale, based on our eligible customers’ prompt payment history and the contractual discount percentage.
Commercial Rebates: Commercial rebates are based on (i) our estimates of end-user purchases through a group purchasing organization ("GPO"), (ii) the corresponding contractual rebate percentage tier we expect each GPO to achieve, and (iii) our estimates of the impact of any prospective rebate program changes made by us.
Medicaid Rebates: Our products are subject to state government-managed Medicaid programs, whereby rebates are issued to participating state governments. These rebates arise when a patient treated with our product is covered under Medicaid, resulting in a discounted price for our product under the applicable Medicaid program. Our Medicaid rebate accrual calculations require us to project the magnitude of our sales, by state, that will be subject to these rebates. There is a significant time lag in us receiving rebate notices from each state (generally several months or longer after our sale is recognized). Our estimates are based on our historical claim levels by state, as supplemented by management’s judgment.
Distribution, Data, and GPO Administrative Fees: Distribution, data, and GPO administrative fees are paid to authorized wholesalers/distributors of our products (except for U.S. sales of ZEVALIN) for various commercial services, including: contract administration, inventory management, delivery of end-user sales data, and product returns processing. These fees are based on a contractually-determined percentage of our applicable sales.
F-10
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
(b) License Fees: We recognize revenue for our licensing of intellectual property to third-parties (out-licenses), based on the contractual terms of each agreement and our application of pertinent GAAP. This revenue may be associated with upfront license fees, milestone payments from our licensees’ sales or regulatory achievements, and royalties from our licensees’ sales in applicable territories.
(c) Service Revenue: We receive fees from third-parties under certain arrangements for our research and development activities, sales and marketing activities, clinical trial management, and supply chain services. Payment may be triggered by the successful completion of a phase of development, results from a clinical trial, regulatory approval events, or completion of product or service delivery in our capacity as an agent or principal in such arrangement. We recognize revenue when the corresponding milestone is achieved, or the revenue is otherwise earned and due to us through our on-going activities.
(d) New Revenue Recognition Standard: On April 1, 2015, the FASB voted for a one-year deferral of the effective date of the new revenue recognition standard, ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”). ASU 2014-09 is now effective for us beginning January 1, 2018, requiring revenue recognition in a manner that reasonably reflects the delivery of our goods or services to customers in return for expected consideration. To achieve this core principle, the guidance provides the following steps: (1) identify the contract(s) with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the entity satisfies a performance obligation.
We intend to apply the cumulative effect transition method of ASU 2014-09, and we continue to evaluate the impact of this new standard to our current revenue recognition models for product sales, license fees, and service revenue, as described above.
(ii) Cash and Cash Equivalents
Cash and cash equivalents consist of bank deposits and highly liquid investments with maturities of three months or less from the purchase date.
(iii) Marketable Securities
Our marketable securities consist of our holdings in mutual funds and bank certificates of deposit. Since we classify these securities as “available-for-sale” under applicable GAAP, any unrealized gains or losses from their change in value is reflected in “unrealized (loss) gain on securities” on the accompanying Consolidated Statements of Comprehensive Loss. Realized gains and losses on available-for-sale securities are included in “other (expense) income, net” on the accompanying Consolidated Statements of Operations.
(iv) Accounts Receivable
Our accounts receivables are derived from our product sales, license fees, and service revenue, and do not bear interest. The allowance for doubtful accounts is management’s best estimate of the amount of probable credit losses in existing accounts receivable. Account balances are charged off against the allowance after appropriate collection efforts are exhausted.
(v) Inventories
We value inventory at the lower of (i) the actual cost of its purchase or manufacture, or (ii) its current market value. Inventory cost is determined on the first-in, first-out method (FIFO). We regularly review our inventory quantities in process of manufacture and on hand. When appropriate, we record a provision for obsolete and excess inventory to derive its new cost basis, which takes into account our sales forecast by product and corresponding expiry dates.
Direct and indirect manufacturing costs related to the production of inventory prior to U.S. Food and Drug Administration ("FDA") approval are expensed through “research and development,” rather than being capitalized to inventory cost.
(vi) Property and Equipment
Our property and equipment is stated at historical cost, and is depreciated on a straight-line basis over an estimated useful life that corresponds with its designated asset category. We evaluate the recoverability of “long-lived assets” (which includes
F-11
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
property and equipment) whenever events or changes in circumstances in our business indicate that the asset’s carrying amount may not be recoverable through on-going operations.
(vii) Goodwill and Intangible Assets
Our goodwill represents the excess of our business acquisition cost over the estimated fair value of the net assets acquired in the corresponding transaction. Goodwill has an indefinite accounting life and is therefore not amortized. Instead, goodwill is evaluated for impairment on an annual basis (as of each October 1st), unless we identify impairment indicators that would require earlier testing.
We evaluate the recoverability of indefinite-lived intangible assets at least annually, or whenever events or changes in our business indicate that an intangible asset’s (whether indefinite or definite-lived) carrying amount may not be recoverable. Such circumstances could include, but are not limited to the following:
(a) a significant decrease in the market value of an asset;
(b) a significant adverse change in the extent or manner in which an asset is used; or
(c) an accumulation of costs significantly in excess of the amount originally expected for the acquisition of an asset.
Intangible assets with finite useful lives are amortized over their estimated useful lives on a straight-line basis. We review these assets for potential impairment if/when facts or circumstances suggest that the carrying value of these assets may not be recoverable.
(viii) Stock-Based Compensation
Stock-based compensation expense for equity awards granted to our employees and members of our board of directors is recognized on a straight-line basis over each award's vesting period. Recognized compensation expense is net of an estimated forfeiture rate, representing the percentage of awards that are expected to be forfeited (by termination of employment or service) prior to vesting. We use the Black-Scholes option pricing model to determine the fair value of stock options (as of the date of grant) which carry service conditions for vesting. We use the Monte Carlo valuation model to value equity awards (as of the date of grant) which carry combined market conditions and service conditions for vesting.
The calculation of the fair value of stock options and the recognition of stock-based compensation expense requires uncertain assumptions, including (a) the pre-vesting forfeiture rate of the award, (b) the expected term of the stock option, (c) the stock price volatility over the term of the stock option, and (d) the risk-free interest rate over the term of the stock option.
We estimate forfeiture rates based on our employees’ overall forfeiture history, which we believe will be representative of future results. We estimate the expected term of stock options granted based on our employees’ historical exercise patterns, which we believe will be representative of their future behavior. We estimate the volatility of our common stock on the date of grant based on historical volatility of our common stock for a look-back period that corresponds with the expected term. We estimate the risk-free interest rate based upon the U.S. Treasury yields in effect at award grant, for a period equaling the stock options’ expected term.
(ix) Foreign Currency Translation
We translate the assets and liabilities of our foreign subsidiaries that are stated in their functional currencies (i.e., local operating currencies), to U.S. dollars at the rates of exchange in effect at the reported balance sheet date. Revenues and expenses are translated using the monthly average exchange rates during the reported period. Unrealized gains and losses from the translation of our subsidiaries’ financial statements (that are initially denominated in the corresponding functional currency) are included as a separate component of “accumulated other comprehensive loss” in the Consolidated Balance Sheets.
We record foreign currency transactions, when initially denominated in a currency other than the respective functional currency of our subsidiary, at the prevailing exchange rate on the date of the transaction. Resulting unrealized foreign exchange gains and losses from transactions with third parties are included in “accumulated other comprehensive loss” in the Consolidated Balance Sheets.
Beginning April 1, 2015, all unrealized foreign exchange gains and losses associated with our intercompany loans were included in "accumulated other comprehensive loss" in the Consolidated Balance Sheets, as these loans with our foreign subsidiaries are no longer expected to be settled in the "foreseeable future." For the period January 1, 2015 through March 31, 2015, unrealized foreign exchange gains and losses associated with our intercompany loans were included in "accumulated
F-12
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
other comprehensive loss" in the Consolidated Balance Sheets and in "other expense (income), net" in the Consolidated Statements of Operations. In periods prior to January 1, 2015, all unrealized foreign exchange gains and losses associated with intercompany loans were included in “other expense (income), net” in the Consolidated Statements of Operations.
(x) Basic and Diluted Net (Loss) Income per Share
We calculate basic and diluted net (loss) income per share using the weighted average number of common shares outstanding during the periods presented. In periods of a net loss, basic and diluted loss per share are the same. For the diluted earnings per share calculation, we adjust the weighted average number of common shares outstanding to include only dilutive stock options, warrants, and other common stock equivalents outstanding during the period.
(xi) Income Taxes
Deferred tax assets and liabilities are recorded based on the estimated future tax effects of temporary differences between the tax basis of assets and liabilities and amounts reported in the financial statements, as well as operating losses and tax credit carry forwards using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Realization of deferred tax assets is dependent upon future earnings, the timing and amount of which are uncertain.
We have recorded a valuation allowance to reduce our net deferred tax assets, because we believe that, based upon a weighting of positive and negative factors, it is more likely than not that these deferred tax assets will not be realized. If/when we were to determine that our deferred tax assets are realizable, an adjustment to the corresponding valuation allowance would increase our net income in the period that such determination was made.
In the event that we are assessed interest and/or penalties from taxing authorities that have not been previously accrued, such amounts would be included in “Provision for income taxes” within the Consolidated Statements of Operations in the period the notice was received.
(xii) Research and Development Costs
Research and development costs are expensed as incurred, or as certain milestone payments become due, generally triggered by contractual clinical or regulatory events.
(xiii) Fair Value Measurements
We determine measurement-date fair value based on the proceeds that would be received through the sale of the asset, or that we would pay to settle or transfer the liability, in an orderly transaction between market participants. We utilize valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible. Fair value measurements are based on a three-tier hierarchy that prioritizes the inputs used to measure fair value. These tiers include the following:
Level 1: Quoted prices (unadjusted) in active markets for identical assets or liabilities that are publicly accessible at the measurement date.
Level 2: Observable prices that are based on inputs not quoted on active markets, but that are corroborated by market data. These inputs may include quoted prices for similar assets or liabilities or quoted market prices in markets that are not active to the general public.
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
3. BALANCE SHEET ACCOUNT DETAIL
The composition of selected financial statement captions that comprise the accompanying Consolidated Balance Sheets are summarized below:
(a) Cash and Cash Equivalents and Marketable Securities
F-13
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
As of December 31, 2015 and December 31, 2014, our holdings included in “cash and cash equivalents” and “marketable securities” were at major financial institutions.
Our investment policy requires that investments in marketable securities be in only highly-rated instruments, which are primarily U.S. treasury bills or U.S. treasury-backed securities, and limited investments in securities of any single issuer. We maintain cash balances in excess of federally insured limits with reputable financial institutions. To a limited degree, the Federal Deposit Insurance Corporation (FDIC) and other third parties insure these investments. However, these investments are not insured against the possibility of a complete loss of earnings or principal and are inherently subject to the credit risk related to the continued credit worthiness of the underlying issuer and general credit market risks. We manage such risks on our portfolio by investing in highly liquid, highly rated instruments, and limit investing in long-term maturity instruments.
The carrying amount of our equity securities, money market funds, bank certificate of deposits (“Bank CDs”), and mutual funds approximates their fair value (utilizing Level 1 or Level 2 inputs – see Note 2 (xiii)) because of our ability to immediately convert these instruments into cash with minimal expected change in value.
The following is a summary of our presented “cash and cash equivalents” and “marketable securities”:
Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated fair Value | Cash and equivalents | Marketable Securities | ||||||||||||||||||||||
Current | Long Term | ||||||||||||||||||||||||||
December 31, 2015 | |||||||||||||||||||||||||||
Bank deposits | $ | 59,625 | $ | — | $ | — | $ | 59,625 | $ | 59,625 | $ | — | $ | — | |||||||||||||
Money market funds | 80,116 | — | — | 80,116 | 80,116 | — | — | ||||||||||||||||||||
Bank certificate of deposits | 245 | — | — | 245 | — | 245 | — | ||||||||||||||||||||
Total cash and equivalents and marketable securities | $ | 139,986 | $ | — | $ | — | $ | 139,986 | $ | 139,741 | $ | 245 | $ | — | |||||||||||||
December 31, 2014 | |||||||||||||||||||||||||||
Bank deposits | $ | 62,997 | $ | — | $ | — | $ | 62,997 | $ | 62,997 | $ | — | $ | — | |||||||||||||
Money market funds | 66,945 | — | — | 66,945 | 66,945 | — | — | ||||||||||||||||||||
Bank certificate of deposits | 244 | — | — | 244 | — | 244 | — | ||||||||||||||||||||
Mutual funds | 3,062 | — | — | 3,062 | — | 3,062 | — | ||||||||||||||||||||
Total cash and equivalents and marketable securities | $ | 133,248 | $ | — | $ | — | $ | 133,248 | $ | 129,942 | $ | 3,306 | $ | — | |||||||||||||
As of December 31, 2015, none of these securities had been in a continuous unrealized loss position longer than one year.
(b) Property and Equipment
“Property and equipment, net of accumulated depreciation” consist of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Computers hardware and software | $ | 3,785 | $ | 3,616 | |||
Laboratory equipment | 608 | 643 | |||||
Office furniture | 355 | 344 | |||||
Leasehold improvements | 2,872 | 2,847 | |||||
Property and equipment, at cost | 7,620 | 7,450 | |||||
(Less): Accumulated depreciation | (6,702 | ) | (6,045 | ) | |||
Property and equipment, net of accumulated depreciation | $ | 918 | $ | 1,405 | |||
F-14
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Depreciation expense (included within “total operating costs and expenses” in the accompanying Consolidated Statements of Operations) for the years ended December 31, 2015, 2014, and 2013 was $0.7 million, $1.1 million, and $1.2 million, respectively.
(c) Inventories
“Inventories” consist of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Raw materials | $ | 1,606 | $ | 1,507 | |||
Work in process* | 4,228 | 3,979 | |||||
Finished goods | 1,498 | 3,714 | |||||
(Less:) Non-current portion of inventories included within "other assets" ** | $ | (3,156 | ) | $ | — | ||
Inventories | $ | 4,176 | $ | 9,200 | |||
*Our work-in-process inventory at December 31, 2015 includes $0.8 million of packaged but unlabeled ZEVALIN vials with expiry in December 2017 (with possible extensions thereafter). In addition to the amounts presented at December 31, 2015, we received $3.4 million of ZEVALIN antibody materials in January 2016 for its future manufacture (representing strategic long-term supply). We expect to sell our existing and committed ZEVALIN inventory over the next few years. However, if our production strategy changes in our transition to a new contract manufacturer, it could result in a charge in that period to “cost of product sales (excludes amortization of intangible assets)” within the Consolidated Statements of Operations.
** The non-current portion of inventories included within "other assets" at December 31, 2015 of $3.2 million, represents inventories we don't expect to sell within the next twelve months based on current estimates.
(d) Accounts receivables, net of allowance for doubtful accounts
“Accounts receivables, net” consists of trade receivables from our customers. We are exposed to credit risk associated with trade receivables that result from these product sales. We do not require collateral or deposits from our customers due to our assessment of their creditworthiness and our long-standing relationship with them. We maintain reserves for potential bad debt, though credit losses have historically been nominal and within management’s expectations. A summary of our customers that represent 10% or more of our accounts receivables as of December 31, 2015 and 2014 are as follows:
Year Ended December 31, | |||||||||||||
2015 | 2014 | ||||||||||||
McKesson Corporation and its affiliates | $ | 20,281 | 66.7 | % | $ | 22,534 | 31.9 | % | |||||
Cardinal Health, Inc. and its affiliates | 7,241 | 23.8 | % | 8,432 | 11.9 | % | |||||||
Oncology Supply, a division of ASD Specialty Healthcare, Inc., and its affiliates (excluding ICS) | * | — | % | 36,154 | 51.1 | % | |||||||
All other customers | 2,862 | 9.4 | % | 3,638 | 5.1 | % | |||||||
Total Accounts Receivables, net | $ | 30,384 | 100.0 | % | $ | 70,758 | 100.0 | % | |||||
* | Less than 10% |
(e) Prepaid expenses and other assets
“Prepaid expenses and other assets” consist of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Prepaid operating expenses | $ | 3,507 | $ | 3,112 | |||
2018 Convertible Notes issuance costs (current portion) | 699 | 662 | |||||
Prepaid expenses and other assets | $ | 4,206 | $ | 3,774 | |||
F-15
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
(f) Other receivables
“Other receivables” consist of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Income tax receivable | $ | 1,301 | $ | 1,387 | |||
Insurance receivable | 7,100 | — | |||||
Mundipharma promissory note | 2,215 | — | |||||
Research and development expenses - reimbursements due | 1,699 | 4,102 | |||||
Other miscellaneous receivables | 257 | — | |||||
Other receivables | $ | 12,572 | $ | 5,489 | |||
(g) Intangible Assets and Goodwill
“Intangible assets, net of accumulated amortization” consist of the following:
December 31, 2015 | |||||||||||||||||||||||
Historical Cost | Accumulated Amortization | Foreign Currency Translation | Impairment | Net Amount | Full Amortization Period (months) | Remaining Amortization Period (months) | |||||||||||||||||
MARQIBO IPR&D (NHL indication) | $ | 17,600 | $ | — | $ | — | $ | — | $ | 17,600 | n/a | n/a | |||||||||||
EVOMELA IPR&D | 7,700 | — | — | — | 7,700 | n/a | n/a | ||||||||||||||||
BELEODAQ distribution rights | 25,000 | (2,812 | ) | — | — | 22,188 | 160 | 142 | |||||||||||||||
MARQIBO distribution rights | 26,900 | (8,544 | ) | — | — | 18,356 | 81 | 51 | |||||||||||||||
FOLOTYN distribution rights | 118,400 | (29,474 | ) | — | — | 88,926 | 152 | 113 | |||||||||||||||
ZEVALIN distribution rights – U.S. | 41,900 | (30,608 | ) | — | — | 11,292 | 123 | 39 | |||||||||||||||
ZEVALIN distribution rights – Ex-U.S. | 23,490 | (12,632 | ) | *** | (4,353 | ) | — | 6,505 | 96 | 51 | |||||||||||||
FUSILEV distribution rights* | 16,778 | (9,618 | ) | — | (7,160 | ) | — | 56 | 0 | ||||||||||||||
FOLOTYN out-license** | 27,900 | (9,109 | ) | — | (1,023 | ) | 17,768 | 110 | 79 | ||||||||||||||
Total intangible assets | $ | 305,668 | $ | (102,797 | ) | $ | (4,353 | ) | $ | (8,183 | ) | $ | 190,335 | ||||||||||
* | On February 20, 2015, the U.S. District Court for the District of Nevada found the patent covering FUSILEV to be invalid, which was upheld on appeal. On April 24, 2015, Sandoz began to commercialize a generic version of FUSILEV. This represented a “triggering event” under applicable GAAP in evaluating the value of our FUSILEV distribution rights as of March 31, 2015, resulting in a $7.2 million impairment charge (non-cash) in the first quarter of 2015. We accelerated amortization expense recognition for the remaining net book value of FUSILEV distribution rights. |
** | On May 29, 2013, we amended our FOLOTYN collaboration agreement with Mundipharma. As a result of the amendment, Europe and Turkey were excluded from Mundipharma’s commercialization territory, and their royalty rates and milestone payments to us were modified. This constituted a change under which we originally valued the FOLOTYN out-license as part of business combination accounting, resulting in an impairment charge (non-cash) of $1.0 million resulted from this amendment. |
*** | This value is inclusive of $3.7 million of accelerated amortization expense, recorded in the fourth quarter of 2015, which specifically relates to our allocation of the historical cost of certain ZEVALIN ex-U.S. rights out-licensed to Mundipharma (see Note 12) at that time. |
Our annual impairment evaluation (as of October 1st) of our indefinite-lived intangible assets was completed by our management, with no resulting impairment. The assets under review included MARQIBO IPR&D and EVOMELA IPR&D, which we carry on our accompanying Consolidated Balance Sheet as of December 31, 2015, at $17.6 million and $7.7 million, respectively.
F-16
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
December 31, 2014 | |||||||||||||||||||
Historical Cost | Accumulated Amortization | Foreign Currency Translation | Impairment | Net Amount | |||||||||||||||
MARQIBO IPR&D (NHL indications) | $ | 17,600 | $ | — | $ | — | $ | — | $ | 17,600 | |||||||||
EVOMELA IPR&D | 7,700 | — | — | — | 7,700 | ||||||||||||||
BELEODAQ distribution rights | 25,000 | (937 | ) | — | — | 24,063 | |||||||||||||
MARQIBO distribution rights | 26,900 | (4,225 | ) | — | — | 22,675 | |||||||||||||
FOLOTYN distribution rights | 118,400 | (20,030 | ) | — | — | 98,370 | |||||||||||||
ZEVALIN distribution rights – U.S. | 41,900 | (27,134 | ) | — | — | 14,766 | |||||||||||||
ZEVALIN distribution rights – Ex-U.S. | 23,490 | (7,402 | ) | (2,162 | ) | — | 13,926 | ||||||||||||
FUSILEV distribution rights | 16,778 | (6,270 | ) | — | — | 10,508 | |||||||||||||
FOLOTYN out-license | 27,900 | (6,385 | ) | — | (1,023 | ) | 20,492 | ||||||||||||
Total intangible assets | $ | 305,668 | $ | (72,383 | ) | $ | (2,162 | ) | $ | (1,023 | ) | $ | 230,100 | ||||||
Intangible asset amortization and impairment expense recognized in 2015, 2014, and 2013 was $38.3 million, $24.3 million, and $21.2 million, respectively.
Estimated intangible asset amortization expense (excluding incremental amortization from the reclassification of IPR&D to developed technology) for the five succeeding years and thereafter is as follows:
Years Ending December 31 | |||
2016 | $ | 23,360 | |
2017 | 23,368 | ||
2018 | 23,368 | ||
2019 | 20,762 | ||
2020 | 15,505 | ||
2021 and thereafter | 58,672 | ||
$ | 165,035 | ||
“Goodwill” is comprised of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Acquisition of Talon | $ | 10,526 | $ | 10,526 | |||
Acquisition of ZEVALIN Ex-U.S. distribution rights | 2,525 | 2,525 | |||||
Acquisition of Allos | 5,346 | 5,346 | |||||
Foreign currency exchange translation effects | (437 | ) | (202 | ) | |||
Goodwill | $ | 17,960 | $ | 18,195 | |||
(h) Other assets
“Other assets” are comprised of the following:
F-17
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
December 31, | |||||||
2015 | 2014 | ||||||
Equity securities and secured promissory note (see Note 11)* | $ | 6,689 | $ | 8,501 | |||
Supplies and deposits | 185 | 234 | |||||
2018 Convertible Notes issuance costs (excluding current portion)** | 1,472 | 2,171 | |||||
Executive officer life insurance – cash surrender value | 9,181 | 6,958 | |||||
Inventories - non-current portion | $ | 3,156 | $ | — | |||
Other assets | $ | 20,683 | $ | 17,864 | |||
* These equity securities were excluded from “marketable securities” (see Note 3(a)) due to our intent to hold these securities for at least one year beyond December 31, 2015, as discussed in Note 11. Unrealized losses from these equity securities were recognized through “unrealized (loss) gain on available-for-sale securities" within the Consolidated Statements of Comprehensive Loss, and were $1.4 million for the year ended December 31, 2015.
** In April 2015, the FASB issued Accounting Standards Update 2015-03, Simplifying the Presentation of Debt Issuance Costs (“ASU 2015-03”). ASU 2015-03 requires that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability. However, ASU 2015-03 does not impact the recognition and measurement guidance for debt issuance costs. ASU 2015-03 is effective for our annual and interim reporting periods beginning January 1, 2016. Accordingly, we will record a reclassification of our 2018 Convertible Notes issuance costs, from “other assets” to “convertible senior notes” within our Consolidated Balance Sheets, beginning January 1, 2016.
(i) Accounts payable and other accrued obligations
Accounts payable and other accrued obligations are comprised of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Trade accounts payable and other accrueds | $ | 26,684 | $ | 24,571 | |||
Accrued rebates | 18,166 | 41,782 | |||||
Accrued product royalty | 4,908 | 5,182 | |||||
Allowance for returns | 1,394 | 1,135 | |||||
Accrued data and distribution fees | 1,830 | 3,952 | |||||
Accrued GPO administrative fees | 1,058 | 3,222 | |||||
Inventory management fee | 498 | 1,110 | |||||
Allowance for chargebacks | 2,001 | 4,040 | |||||
Accounts payable and other accrueds | $ | 56,539 | $ | 84,994 | |||
Amounts presented within “accounts payable and other accrued liabilities” in the accompanying Consolidated Balance Sheets for GTN estimates (see Note 2(i)) were as follows:
Description | Rebates and Chargebacks | Data and Distribution, GPO Fees, and Inventory Management Fees | Returns | ||||||||
Balance as of December 31, 2013 | $ | 33,967 | $ | 5,373 | $ | 2,900 | |||||
Add: provisions (recovery) | 76,636 | 21,330 | (78 | ) | |||||||
(Less): credits or actual allowances | (64,781 | ) | (18,419 | ) | (1,687 | ) | |||||
Balance as of December 31, 2014 | 45,822 | 8,284 | 1,135 | ||||||||
Add: provisions | 75,498 | 15,928 | 1,486 | ||||||||
(Less): credits or actual allowances | (101,153 | ) | (20,826 | ) | (1,227 | ) | |||||
Balance as of December 31, 2015 | $ | 20,167 | $ | 3,386 | $ | 1,394 | |||||
F-18
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
(j) Deferred revenue
Deferred revenue (including current and long-term) is comprised of the following:
December 31, | |||||||
2015 | 2014 | ||||||
CASI out-license (see Note 11) | $ | — | $ | 9,959 | |||
FUSILEV deferred revenue* | 6,083 | — | |||||
Dr. Reddy's out-license (see Note 17(b)(iii)) | 430 | — | |||||
Deferred revenue | $ | 6,513 | $ | 9,959 | |||
* During the third quarter 2015, we deferred revenue recognition related to certain FUSILEV product shipments that did not meet our revenue recognition criteria (see Note 2(i)(a)), aggregating $9.9 million. Specifically, this deferral is a result of our inability to estimate future rebate values (with requisite precision) offered to our customers in order to compete with generic products. During the fourth quarter of 2015, we recognized $3.8 million for these third quarter shipments within "Product sales, net." As of December 31, 2015, $6.1 million of these third quarter 2015 shipments remains deferred.
(k) Other long-term liabilities
Other long-term liabilities are comprised of the following:
December 31, | |||||||
2015 | 2014 | ||||||
Accrued executive deferred compensation | $ | 6,458 | $ | 4,694 | |||
Deferred rent (non-current portion) | 248 | 364 | |||||
Business acquisition liability | — | 300 | |||||
Other tax liabilities | 738 | 730 | |||||
Other long-term liabilities | $ | 7,444 | $ | 6,088 | |||
4. GROSS-TO-NET PRODUCT SALES
The below table presents a GTN product sales reconciliation for the accompanying Consolidated Statement of Operations:
2015 | 2014 | 2013 | |||||||||
Gross product sales | $ | 215,136 | $ | 284,685 | $ | 224,301 | |||||
Commercial rebates and government chargebacks | (61,283 | ) | (76,636 | ) | (63,610 | ) | |||||
Distribution, data, and GPO administrative fees | (15,613 | ) | (21,330 | ) | (19,067 | ) | |||||
Prompt pay discounts | (16 | ) | (260 | ) | (183 | ) | |||||
Product returns allowances | (1,373 | ) | 78 | 2,034 | |||||||
Product sales, net | $ | 136,851 | $ | 186,537 | $ | 143,475 | |||||
5. NET PRODUCT SALES BY GEOGRAPHIC REGION, PRODUCT LINE, AND GROSS PRODUCT SALES BY SIGNIFICANT CUSTOMERS
The below table presents our net product sales by geography for the years ended December 31, 2015, 2014, and 2013:
Net Product Sales by Geographic Region
F-19
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Year Ended December 31, | ||||||||||||||||||||
2015 | 2014 | 2013 | ||||||||||||||||||
United States | $ | 130,432 | 95.3 | % | $ | 177,979 | 95.4 | % | $ | 133,462 | 93.0 | % | ||||||||
International: | ||||||||||||||||||||
Europe (ZEVALIN and FOLOTYN only) | 2,234 | 1.6 | % | 3,357 | 1.8 | % | 3,953 | 2.8 | % | |||||||||||
Asia Pacific (ZEVALIN only) | 4,185 | 3.1 | % | 5,201 | 2.8 | % | 6,060 | 4.2 | % | |||||||||||
Total International | 6,419 | 4.7 | % | 8,558 | 4.6 | % | 10,013 | 7.0 | % | |||||||||||
Net product sales | $ | 136,851 | 100 | % | $ | 186,537 | 100.0 | % | $ | 143,475 | 100.0 | % | ||||||||
Net Sales by Product
The below table presents our net product sales by product for the years ended December 31, 2015, 2014, and 2013:
Year Ended December 31, | ||||||||||||||||||||
2015 | 2014 | 2013 | ||||||||||||||||||
FUSILEV | $ | 60,710 | 44.4 | % | $ | 105,608 | 56.6 | % | $ | 68,397 | 47.7 | % | ||||||||
FOLOTYN | 40,606 | 29.7 | % | 47,556 | 25.5 | % | 44,370 | 30.9 | % | |||||||||||
ZEVALIN | 17,457 | 12.8 | % | 22,169 | 11.9 | % | 29,393 | 20.5 | % | |||||||||||
MARQIBO | 8,006 | 5.9 | % | 6,328 | 3.4 | % | 1,315 | 0.9 | % | |||||||||||
BELEODAQ | 10,072 | 7.4 | % | 4,876 | 2.6 | % | — | — | % | |||||||||||
Net product sales | $ | 136,851 | 100.0 | % | $ | 186,537 | 100.0 | % | $ | 143,475 | 100.0 | % | ||||||||
Gross Product Sales by Customer
The below table presents the customers that represent 10% or more of our gross product sales in 2015, 2014, and 2013:
Year Ended December 31, | ||||||||||||||||||||
2015 | 2014 | 2013 | ||||||||||||||||||
Oncology Supply, a division of ASD Specialty Healthcare, Inc., and its affiliates (excluding ICS) | $ | 78,989 | 36.7 | % | $ | 115,079 | 40.4 | % | $ | 79,497 | 35.4 | % | ||||||||
McKesson Corporation and its affiliates | 73,577 | 34.2 | % | 93,656 | 32.9 | % | 44,350 | 19.8 | % | |||||||||||
Integrated Commercialization Solutions, Inc. (“ICS”) | * | — | % | * | — | % | 35,548 | 15.8 | % | |||||||||||
Cardinal Health, Inc. and its affiliates | 37,414 | 17.4 | % | * | — | % | * | — | % | |||||||||||
All Other Customers | 25,156 | 11.7 | % | 75,950 | 26.7 | % | 64,906 | 29.0 | % | |||||||||||
Gross product sales | $ | 215,136 | 100.0 | % | $ | 284,685 | 100.0 | % | $ | 224,301 | 100.0 | % | ||||||||
* | Less than 10% |
6. STOCK-BASED COMPENSATION
2009 Stock Incentive Plan Overview
We have one active stockholder-approved stock-based compensation plan, the 2009 Incentive Award Plan (the “2009 Plan”), which replaced our former stockholder-approved plans. We may grant incentive stock options, non-qualified options, restricted stock awards, and stock appreciation rights under the 2009 Plan.
The maximum number of our common stock available for issuance under the 2009 Plan at inception was 10 million shares. Beginning on January 1, 2010, and each January 1st thereafter, the number of shares of common stock available for issuance under the 2009 Plan automatically increases by the greater of (i) 2.5 million shares or (ii) a number of shares such that the total number of shares of common stock available for issuance under the 2009 Plan shall equal 30% of the then number of shares of common stock issued and outstanding. As of December 31, 2015, 8.6 million shares were available for grant. It is our
F-20
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
policy that before stock is issued through the exercise of stock options, we must first receive all required cash payment for such shares (whether through an upfront cash exercise or net-settlement exercise).
Stock-based awards are governed by agreements between us and the recipients. Incentive stock options and nonqualified stock options may be granted under the 2009 Plan at an exercise price of not less than 100% of the closing fair market value of our common stock on the respective date of grant. The grant date is generally the date the award is approved by the Compensation Committee of the Board of Directors, though for aggregate awards of 50,000 or less in each quarter, the grant date is the date the award is approved by our Chief Executive Officer.
Stock-based awards generally vest 25% on the first anniversary of the date of grant, or for new hires, the first anniversary of their initial date of employment. Awards generally vest monthly thereafter on a straight-line basis over three years. Stock options must be exercised, if at all, no later than 10 years from the date of grant. Upon termination of employment, vested stock options may be exercised within 90 days from the last date of employment. In the event of an optionee’s death, disability, or retirement, the exercise period is 365 days from the last date of employment.
Employee Stock Purchase Plan
Under the terms of our 2009 Employee Stock Purchase Plan (the “ESPP”), eligible employees can purchase common stock through payroll deductions. The purchase price is equal to the closing price of our common stock on the first or last day of the offering period (whichever is less), minus a 15% discount. We use the Black-Scholes option-pricing model, in combination with the discounted employee price, in determining the value of ESPP expense to be recognized during each offering period. A participant may purchase a maximum of 50,000 shares of common stock during a six-month offering period, not to exceed $25,000 worth of stock on the offering date during each plan year.
A total of 4.3 million shares of common stock are authorized for issuance under the ESPP. Beginning on January 1, 2010, and each January 1st thereafter, the number of shares of common stock available for issuance under the ESPP shall automatically increase by an amount equal to the lesser of (i) one million shares or (ii) an amount determined by the ESPP administrator. However, in no event shall the number of shares of common stock available for future sale under the ESPP exceed 10 million shares, subject to capitalization adjustments occurring due to dividends, splits, dissolution, liquidation, mergers, or changes in control.
Stock-Based Compensation Expense Summary
We classify our stock-based compensation expense (inclusive of our incentive stock plan, employee stock purchase plan, and 401(k) contribution matching program) in the accompanying Consolidated Statements of Operations, based on the department to which the recipient belongs. Stock-based compensation expense included within “Total operating costs and expenses” for years ended December 31, 2015, 2014 and 2013 was as follows:
Year Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
Cost of product sales | $ | 62 | $ | — | $ | — | |||||
Selling, general and administrative | 10,049 | 10,053 | 10,762 | ||||||||
Research and development | 1,973 | 1,756 | 2,017 | ||||||||
Total | $ | 12,084 | $ | 11,809 | $ | 12,779 | |||||
Employee stock-based compensation expense for the years ended December 31, 2015, 2014 and 2013 was recognized (reduced for estimated forfeitures) on a straight-line basis over the vesting period. Forfeitures are estimated at the time of grant and prospectively revised if actual forfeitures differ from those estimates. We estimate forfeitures of stock options using the historical exercise behavior of our employees. For purposes of this estimate, we have applied an estimated forfeiture rate of 11%, 8%, and 8% for the years ended December 31, 2015, 2014 and 2013.
Valuation Assumptions – Restricted Stock and Stock Options
F-21
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
The grant-date fair value per share for restricted stock awards was based upon the closing market price of our common stock on the award grant-date.
The fair value of stock options granted was estimated at the date of grant using the Black-Scholes option-pricing model. The following assumptions were used to determine fair value for the stock awards granted in the applicable year:
Year Ended December 31, | |||||
2015 | 2014 | 2013 | |||
Expected option life (in years) (a) | 5.43 | 4.95 | 4.95 | ||
Risk-free interest rate (b) | 1.25% - 1.68% | 0.58% - 1.52% | 0.35% - 0.78% | ||
Volatility (c) | 48.0% - 50.2% | 48.9% - 62.1% | 58.3% -71.5% | ||
Dividend yield (d) | —% | —% | —% | ||
Weighted-average grant-date fair value per stock option | $2.85 | $3.49 | $4.66 | ||
(a) | Determined by the historical stock option exercise behavior of our employees (maximum term is 10 years). |
(b) | Based upon the U.S. Treasury yields in effect during the period which the options were granted (for a period equaling the stock options’ expected term). |
(c) | Measured using our historical stock price for a period equal to stock options’ expected term. |
(d) | We do not expect to declare any cash dividends in the foreseeable future. |
Stock Option Activity
Stock option activity during the years ended December 31, 2015, 2014 and 2013 is as follows:
Number of Shares | Weighted- Average Exercise Price/Share | Weighted- Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | ||||||||||||
Outstanding — December 31, 2012 | 10,399,265 | $ | 6.57 | ||||||||||||
Granted | 2,041,300 | 8.92 | |||||||||||||
Exercised | (825,884 | ) | 4.40 | $ | 3,435 | (1 | ) | ||||||||
Forfeited | (202,882 | ) | 8.22 | ||||||||||||
Expired | (82,581 | ) | 8.91 | ||||||||||||
Outstanding — December 31, 2013 | 11,329,218 | 7.10 | |||||||||||||
Granted | 2,576,292 | 7.60 | |||||||||||||
Exercised | (485,260 | ) | 4.77 | $ | 1,629 | (1 | ) | ||||||||
Forfeited | (557,109 | ) | 9.65 | ||||||||||||
Expired | (214,039 | ) | 10.70 | ||||||||||||
Outstanding — December 31, 2014 | 12,649,102 | 7.12 | |||||||||||||
Granted | 2,219,587 | 6.04 | |||||||||||||
Exercised | (456,082 | ) | 4.45 | $ | 977 | (1 | ) | ||||||||
Forfeited | (296,162 | ) | 8.06 | ||||||||||||
Expired | (279,594 | ) | 9.05 | ||||||||||||
Outstanding — December 31, 2015 | 13,836,851 | $ | 6.97 | 6.34 | $ | 7,516 | (2 | ) | |||||||
Vested (exercisable) — December 31, 2015 | 10,235,721 | $ | 6.94 | 5.40 | $ | 7,159 | (2 | ) | |||||||
Unvested (unexercisable) — December 31, 2015 | 3,601,130 | $ | 7.06 | 9.00 | $ | 357 | (2 | ) | |||||||
(1) | Represents the total difference between our closing stock price at the time of exercise and the stock option exercise price, multiplied by the number of options exercised. |
F-22
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
(2) | Represents the total difference between our closing stock price on the last trading day of 2015 and the stock option exercise price, multiplied by the number of in-the-money options as of December 31, 2015. The amount of intrinsic value will change based on the fair market value of our stock. |
The following table summarizes information with respect to stock option grants as of December 31, 2015:
Outstanding | Exercisable | ||||||||||||||
Exercise Price | Granted Stock Options Outstanding | Weighted- Average Remaining Contractual Life (Years) | Weighted- Average Exercise Price | Granted Stock Options Exercisable | Weighted- Average Exercise Price | ||||||||||
$0.92 - 3.15 | 1,090,385 | 2.49 | $ | 2.18 | 1,090,385 | $ | 2.18 | ||||||||
$3.16 - 4.95 | 1,378,444 | 4.29 | 4.24 | 1,378,444 | 4.24 | ||||||||||
$4.96 - 6.9 | 4,394,294 | 6.14 | 6.08 | 2,999,511 | 6.22 | ||||||||||
$6.91 - 8.99 | 4,500,895 | 7.61 | 7.78 | 2,595,943 | 7.95 | ||||||||||
$9.00 - 16.32 | 2,472,833 | 7.21 | 10.71 | 2,171,438 | 10.83 | ||||||||||
13,836,851 | 6.34 | $ | 6.97 | 10,235,721 | $ | 6.94 | |||||||||
As of December 31, 2015, there was unrecognized compensation expense of $8.8 million related to unvested stock options, which we expect to recognize over a weighted average period of 2.41 years.
Restricted Stock Award Activity
A summary of restricted stock award activity is as follows:
Number of Restricted Stock Awards | Weighted Average Fair Value per Share at Grant Date | |||||
Unvested — December 31, 2012 | 1,034,604 | $ | 11.00 | |||
Granted | 523,800 | 8.74 | ||||
Vested | (501,660 | ) | 9.72 | |||
Forfeited | (49,625 | ) | 10.60 | |||
Unvested — December 31, 2013 | 1,007,119 | 10.09 | ||||
Granted | 581,194 | 7.52 | ||||
Vested | (578,985 | ) | 10.24 | |||
Forfeited | (185,111 | ) | 9.88 | |||
Unvested — December 31, 2014 | 824,217 | 8.22 | ||||
Granted | 1,948,585 | 6.32 | ||||
Vested | (364,507 | ) | 8.47 | |||
Forfeited | (234,313 | ) | 7.32 | |||
Unvested — December 31, 2015 | 2,173,982 | $ | 6.58 | |||
2015 | 2014 | 2013 | |||||||||
Restricted stock expense | $ | 4,006 | $ | 3,830 | $ | 4,202 | |||||
As of December 31, 2015, there was approximately $9.8 million of unrecorded expense related to issued restricted stock that will be recognized over an estimated weighted average period of 2.29 years. These unvested shares are included in our issued and outstanding common stock as of December 31, 2015.
401(k) Plan – Stock Matching Contribution
F-23
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
We issued shares of common stock to our employees in connection with our 401(k) program, partially matching our employees’ annual 401(k) contributions, as summarized below:
2015 | 2014 | 2013 | |||||||||
Shares of common stock issued | 179,865 | 133,734 | 99,359 | ||||||||
Match contribution value* | $ | 1,124 | $ | 1,028 | $ | 860 | |||||
* | Represents our stock price on the date of the common stock issuance multiplied by the number of shares of common stock issued. |
7. STOCKHOLDERS’ EQUITY
Authorized Stock
On December 13, 2010, we filed the Certificate of Designation of Rights, Preferences and Privileges of Series B Junior Participating Preferred Stock with the Delaware Secretary of State which authorized 1.5 million shares to be designated as Series B Junior Participating Preferred Stock. On June 13, 2011, our stockholders approved an amendment to our Certificate of Incorporation to increase the authorized number of shares of our common stock from 100 million shares to 175 million shares. The amendment was filed with the Delaware Secretary of State on June 24, 2011. As of December 31, 2015, we also had five million shares of preferred stock authorized, of which 1.5 million shares were designated as Series B Junior Participating Preferred Stock and 2,000 shares were designated as Series E Convertible Voting Preferred Stock.
Stockholder Rights Agreement
On November 29, 2010, our Board of Directors approved a replacement rights agreement, effective December 13, 2010, that replaced the stockholder rights agreement which was originally adopted in 2000. This replacement rights agreement will extend until December 13, 2020. A stockholder rights agreement is designed to deter coercive, unfair, or inadequate takeovers and other abusive tactics that might be used in an attempt to gain control of our company. A stockholder rights agreement will not prevent takeovers at a full and fair price, but rather is designed to deter coercive takeover tactics and to encourage anyone attempting to acquire our company to first negotiate with our Board of Directors.
Under the terms of the new Stockholder Rights Agreement, the rights become exercisable upon the earlier of 10 days after a person or group of affiliated or associated persons has acquired 15% or more of the outstanding shares of our common stock or 10 business days after a tender offer has commenced that would result in a person or group beneficially owning 15% or more of our outstanding common stock. These rights could delay or discourage someone from acquiring our company, even if doing so would potentially benefit our stockholders.
We currently have no stockholders who own 15% or more of the outstanding shares of our common stock. five days after the rights become exercisable, each right, other than rights held by the person or group of affiliated persons whose acquisition of more than 15% of our outstanding common stock caused the rights to become exercisable, will entitle its holder to buy, in lieu of shares of Series B Preferred Stock, a number of shares of our common stock having a market value of two times the exercise price of the rights. After the rights become exercisable, if we are a party to certain merger or business combination transactions or transfers 50% or more of our assets or earnings power (as defined in the Stockholder Rights Agreement), each right will entitle its holder to buy a number of shares of common stock of the acquiring or surviving entity having a market value of twice the exercise price of the right.
Series E Preferred Stock
In September 2003, we received gross cash proceeds of $20 million in exchange for the issuance of 2,000 shares of our Series E Convertible Voting Preferred Stock, convertible into four million shares of common stock. As of December 31, 2015 and 2014, 20 shares of Series E Preferred Stock were outstanding. No dividends are payable on the Series E Preferred Stock. Pursuant to certain provisions of the Certificate of Designation, Rights and Preferences of the Series E Preferred Stock, we have the option to redeem all of the unconverted Series E Preferred Stock outstanding at the end of a 20-day trading period if, among other things, in that period our common stock trades above $12.00 per share.
In the event we are the subject of any voluntary or involuntary liquidation, dissolution or winding up, before any distribution of our assets shall be made to the common stockholders, the holders of the Series E Preferred Stock shall be entitled to receive a liquidation preference in an amount equal to 120% of the stated value per share plus any declared and unpaid dividends thereon.
F-24
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Common Stock Issuable
As of December 31, 2015, 26 million shares of our common stock were issuable upon conversion, or exercise of rights granted (regardless of whether in or out-of-the-money), as summarized below:
Conversion of Series E Preferred Stock | 40,000 | |
2018 Convertible Notes | 11,401,284 | |
Exercise of issued employee stock options | 13,836,851 | |
Exercise of issued warrants | 445,000 | |
Management incentive plan restricted stock units | 260,000 | |
Total common shares | 25,983,135 | |
Warrant Activity
We typically issue warrants to purchase shares of our common stock to investors as part of a financing transaction or in connection with services rendered by placement agents or consultants. Our outstanding warrants expire on varying dates through December 2020. A summary of warrant activity is as follows:
Number of Shares | Weighted Average Exercise Price | |||||
Outstanding — December 31, 2012 | 395,000 | $ | 5.45 | |||
Granted | 50,000 | 7.51 | ||||
Outstanding — December 31, 2013 | 445,000 | $ | 6.39 | |||
Granted | — | — | ||||
Outstanding — December 31, 2014 | 445,000 | $ | 6.39 | |||
Granted | — | — | ||||
Outstanding — December 31, 2015 | 445,000 | $ | 6.78 | |||
Exercisable — December 31, 2015 | 445,000 | $ | 6.78 | |||
8. NET LOSS PER SHARE
Net loss per share was computed by dividing net loss by the weighted average number of common shares outstanding for the years ended December 31, 2015, 2014, and 2013:
Year Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
Net loss | $ | (50,785 | ) | $ | (45,719 | ) | $ | (62,134 | ) | ||
Weighted average shares—basic | 64,882,417 | 64,708,163 | 60,729,128 | ||||||||
Net loss per share—basic | $ | (0.78 | ) | $ | (0.71 | ) | $ | (1.02 | ) | ||
Weighted average shares—diluted | 64,882,417 | 64,708,163 | 60,729,128 | ||||||||
Net loss income per share—diluted | $ | (0.78 | ) | $ | (0.71 | ) | $ | (1.02 | ) | ||
F-25
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
The below outstanding securities were excluded from the above calculation of net loss per share because their impact would have been anti-dilutive due to net loss per share, for the years ended, as summarized below:
Year Ended December 31, | ||||||||
2015 | 2014 | 2013 | ||||||
2018 Convertible Notes | 11,401,284 | 11,401,284 | 343,600 | |||||
Common stock options | 1,441,086 | 2,173,916 | 2,934,625 | |||||
Restricted stock awards | 2,173,615 | 824,217 | 1,007,119 | |||||
Common stock warrants | 9,357 | 120,702 | 160,816 | |||||
Preferred stock | 40,000 | 40,000 | 40,000 | |||||
Total | 15,065,342 | 14,560,119 | 4,486,160 | |||||
9. FAIR VALUE MEASUREMENTS
The table below summarizes certain asset and liability fair values that are included within our accompanying Consolidated Balance Sheets, and their designations among three fair value measurement categories:
December 31, 2015 Fair Value Measurements | |||||||||||||||
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
Assets: | |||||||||||||||
Bank certificates of deposits | $ | — | $ | 245 | $ | — | $ | 245 | |||||||
Money market currency funds | — | 80,116 | — | 80,116 | |||||||||||
Equity securities | 5,189 | — | — | 5,189 | |||||||||||
Mutual funds | — | — | — | — | |||||||||||
Deferred compensation investments, including life insurance cash surrender value | — | 9,181 | — | 9,181 | |||||||||||
$ | 5,189 | $ | 89,542 | $ | — | $ | 94,731 | ||||||||
Liabilities: | |||||||||||||||
Deferred executive compensation liability | — | 6,458 | — | 6,458 | |||||||||||
Deferred drug development liability | — | — | 14,686 | 14,686 | |||||||||||
Ligand Contingent Consideration | — | — | 5,227 | 5,227 | |||||||||||
Talon CVR | — | — | 1,377 | 1,377 | |||||||||||
Corixa Liability | — | — | 62 | 62 | |||||||||||
$ | — | $ | 6,458 | $ | 21,352 | $ | 27,810 | ||||||||
F-26
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
December 31, 2014 Fair Value Measurements | |||||||||||||||
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
Assets: | |||||||||||||||
Bank certificates of deposits | $ | — | $ | 244 | $ | — | $ | 244 | |||||||
Money market currency funds | — | 66,945 | — | 66,945 | |||||||||||
Equity securities | 7,191 | — | — | 7,191 | |||||||||||
Mutual funds | — | 3,062 | — | 3,062 | |||||||||||
Deferred compensation investments, including life insurance cash surrender value | — | 6,958 | — | 6,958 | |||||||||||
$ | 7,191 | $ | 77,209 | $ | — | $ | 84,400 | ||||||||
Liabilities: | |||||||||||||||
Deferred executive compensation liability | — | 4,694 | — | 4,694 | |||||||||||
Deferred development liability | — | — | 15,785 | 15,785 | |||||||||||
Ligand Contingent Consideration | — | — | 4,901 | 4,901 | |||||||||||
Talon CVR | — | — | 2,379 | 2,379 | |||||||||||
Corixa Liability | — | — | 62 | 62 | |||||||||||
$ | — | $ | 4,694 | $ | 23,127 | $ | 27,821 | ||||||||
We did not have any transfers between Levels 1 and 2 for all periods presented. The following presents a roll forward of our liabilities for which we utilize Level 3 inputs in determining period-end value. These liabilities are included on our Consolidated Balance Sheets within “acquisition related contingent obligations” and “drug development liability”. The basis of the various Level 3 valuation inputs are discussed in the Notes to these accompanying Consolidated Financial Statements.
Our carrying amounts of financial instruments such as cash equivalents, accounts receivable, prepaid expenses, accounts payable, and accrued liabilities, excluding acquisition related contingent consideration liabilities, approximate their related fair values due to their short-term nature.
Fair Value Measurements of Unobservable Inputs (Level 3) | |||
Balance at December 31, 2013 | $ | 26,071 | |
Deferred development costs (see Note 16) | (1,957 | ) | |
Ligand Contingent Consideration (see Note 10(b)) | 901 | ||
Talon CVR (see Note 10(a)) | (1,950 | ) | |
Corixa Liability (see Note 17(b)(i)) | 62 | ||
Balance at December 31, 2014 | $ | 23,127 | |
Deferred development costs (see Note 16) | (1,099 | ) | |
Ligand Contingent Consideration (see Note 10(b)) | 326 | ||
Talon CVR (see Note 10(a)) | (1,002 | ) | |
Corixa Liability (see Note 17(b)(i)) | — | ||
Balance at December 31, 2015 | $ | 21,352 | |
10. BUSINESS COMBINATIONS AND CONTINGENT CONSIDERATION
(a) Acquisition of Talon Therapeutics, Inc.
Talon Acquisition Overview
F-27
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
On July 17, 2013, we purchased all of the outstanding shares of common stock of Talon Therapeutics, Inc. (“Talon”). Through the acquisition of Talon, we gained worldwide rights to MARQIBO. The Talon purchase consideration comprised of (i) an aggregate upfront cash amount of $11.3 million, (ii) issuance of 3.0 million shares of our common stock, then equivalent to $26.3 million (based on a closing price of $8.77 per share on July 17, 2013), and (iii) the issuance of contingent value rights (“CVR”) initially valued at $6.5 million.
The CVR was valued using a valuation model that probability-weights expected outcomes (ranging from 50% to 100%) and discounts those amounts to their present value, using an appropriate discount rate (these represent unobservable inputs and are therefore classified as Level 3 inputs – see Note 2 (xiii)). The CVR has a maximum payout of $195.0 million if all sales and regulatory approval milestones are achieved, as summarized below:
• | $5.0 million upon the achievement of net sales of MARQIBO in excess of $30.0 million in any calendar year |
• | $10.0 million upon the achievement of net sales of MARQIBO in excess of $60.0 million in any calendar year |
• | $25.0 million upon the achievement of net sales of MARQIBO in excess of $100.0 million in any calendar year |
• | $50.0 million upon the achievement of net sales of MARQIBO in excess of $200.0 million in any calendar year |
• | $100.0 million upon the achievement of net sales of MARQIBO in excess of $400.0 million in any calendar year |
• | $5.0 million upon receipt of marketing authorization from the FDA regarding Menadione Topical Lotion |
Talon CVR Fair Value as of December 31, 2015 and December 31, 2014
The CVR fair value will continue to be evaluated on a quarterly basis. Current and future changes in its fair value results from the likelihood and timing of milestone achievement and/or the corresponding discount rate applied thereon. Adjustments to CVR fair value are recognized within “change in fair value of contingent consideration related to acquisitions” in the accompanying Consolidated Statements of Operations.
Fair Value of Talon CVR | |||
December 31, 2014 | $ | 2,379 | |
Fair value adjustment for the year ended December 31, 2015 | (1,002 | ) | |
December 31, 2015 | $ | 1,377 | |
(b) Acquisition of Rights to EVOMELA and Related Contingent Consideration
Overview of Acquisition of Rights to EVOMELA
In March 2013, we completed the acquisition of exclusive global development and commercialization rights to Captisol-enabled®, propylene glycol-free MELPHALAN (which we recently branded as “EVOMELA”) for use as a conditioning treatment prior to autologous stem cell transplant for patients with MM. We acquired these rights from CyDex Pharmaceuticals, Inc. a wholly-owned subsidiary of Ligand Pharmaceuticals Incorporated (“Ligand”) for an initial license fee of $3.0 million.
We accounted for this transaction as a business combination, which requires that assets acquired and liabilities assumed be recognized on the balance sheet at their fair values as of the transaction date.
We are required to pay Ligand additional amounts up to an aggregate $66.0 million, upon the achievement of certain regulatory milestones and net sales thresholds, and we also assumed full financial responsibility for its ongoing clinical and regulatory development program. We also must pay royalties of 20% on our future net sales of EVOMELA in all territories.
Consideration Transferred
The acquisition-date fair value of the consideration transferred consisted of the following:
F-28
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Cash consideration | $ | 3,000 | |
Ligand Contingent Consideration | 4,700 | ||
Total purchase consideration | $ | 7,700 | |
Fair Value Estimate of Asset Acquired and Liability Assumed
The total purchase consideration is allocated to the acquisition of the net tangible and intangible assets based on their estimated fair values as of the closing date. The allocation of the total purchase price to the net assets acquired is as follows:
IPR&D EVOMELA rights | $ | 7,700 | |
We estimated the fair value of the in-process research and development using the income approach. The income approach uses valuation techniques to convert future amounts to a single present amount (discounted). Our measurement is based on the value indicated by current market expectations about those future amounts. The fair value estimate took into account our estimates of future incremental earnings that may be achieved upon regulatory approval, promotion, and distribution associated with the rights, and included estimated cash flows of approximately 10 years and a discount rate of approximately 25%.
The fair value of the contingent consideration liability assumed was determined using the probability of success and the discounted cash flow method of the income approach (representing unobservable inputs and therefore represent Level 3 values - see Note 2(xiii), which assumed that FDA approval of EVOMELA will occur on or about May 9, 2016 (on March 10, 2016, the FDA communicated its approval of this drug; however, under GAAP this event does not affect our reported liability estimate as of December 31, 2015). Upon receipt of FDA approval, we are contractually obligated to make a $6.0 million milestone payment to Ligand within 30 days ("Ligand Contingent Consideration"); accordingly, this payment is due no later than April 9, 2016.
Ligand Contingent Consideration Fair Value as of December 31, 2015 and December 31, 2014
The Ligand Contingent Consideration fair value will continue to be evaluated on a quarterly basis. Any changes in its fair value results from the likelihood and timing of milestone achievement and/or the corresponding discount rate applied thereon. Adjustments to Ligand Contingent Consideration fair value are recognized within “change in fair value of the contingent consideration related to acquisitions” in the accompanying Consolidated Statements of Operations.
Fair Value of Ligand Contingent Consideration | |||
December 31, 2014 | $ | 4,901 | |
Fair value adjustment for year ended December 31, 2015 | 326 | ||
December 31, 2015 | $ | 5,227 | |
(c) Allos Acquisition
We acquired Allos Therapeutics, Inc. (“Allos”) on September 5, 2012, which was accounted for as a business combination. Our total cash consideration for this acquisition was $205.2 million, through which we acquired FOLOTYN distribution rights. We have no contingent consideration obligations as part of this transaction.
11. OUT-LICENSE OF MARQIBO, ZEVALIN, & EVOMELA IN CHINA TERRITORY
Overview of CASI Out-License
On September 17, 2014, we executed three product out-license agreements with a perpetual term (collectively, the “CASI Out-License”) with CASI Pharmaceuticals, Inc. (“CASI”), a publicly-traded biopharmaceutical company (NASDAQ: CASI) with a primary focus on the China market. Under the CASI Out-License, we granted CASI the exclusive rights to distribute two of our commercialized oncology drugs, ZEVALIN and MARQIBO, and our Phase 3 drug candidate, EVOMELA (“CASI Out-Licensed Products”) in greater China (which includes Taiwan, Hong Kong and Macau). In return, we received CASI equity for the rights related to ZEVALIN and EVOMELA and a secured promissory note for the rights related to MARQIBO.
F-29
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Additionally, under certain conditions which generally expire on September 17, 2019, we have a right to receive additional CASI common stock in order to maintain our post-investment ownership percentage if CASI issues additional securities.
CASI will be responsible for the development and commercialization of these three drugs, including the submission of import drug registration applications to regulatory authorities and conducting any confirmatory clinical studies in greater China. We will provide CASI with future commercial supply of the CASI Out-Licensed Products under typical market terms.
Proceeds Received
The proceeds we received, and its fair value on the CASI Out-License execution date, consisted of the following:
CASI common stock (5.4 million shares) | $ | 8,649 | (a) | |
CASI secured promissory note due March 17, 2016 (since extended to March 17, 2017), net of fair value discount ($1.5 million face value and 0.5% annual coupon) | 1,310 | (b) | ||
Total consideration received, net of fair value discount | $ | 9,959 | (c) | |
(a) | Value determined based on the September 17, 2014 closing price of 5.4 million shares of CASI common stock on the NASDAQ Capital Market of $1.60 per share. Our current intention is to hold these securities on a long-term basis. Accordingly, we have presented its value of $5.2 million as of December 31, 2015 within “other assets” (rather than “marketable securities”) on our accompanying Consolidated Balance Sheets. The change in the value of these securities is reported within “unrealized (loss) gain on available-for-sale securities” on the accompanying Consolidated Statement of Comprehensive Loss. |
(b) | Value estimated using the terms of the $1.5 million promissory note, and the application of a synthetic debt rating based on CASI’s publicly-available financial information, and the prevailing interest yields on similar public debt securities as of September 17, 2014. The face value of the promissory note as of December 31, 2015 is included within "other assets" on the accompanying Consolidated Balance Sheets. |
(c) | Presented within “license fees and service revenue” in the accompanying Consolidated Statement of Operations as of December 31, 2015. |
In addition, CASI will be responsible for paying any royalties or milestones that we are obligated to pay to our third-party licensors resulting from the achievement of certain milestones and/or sales of CASI Out-Licensed Products, but only to the extent of the greater China portion of such royalties or milestones.
Recognition of Proceeds – License Fee Revenue in 2015
The $9.7 million value (undiscounted, and net of certain foreign exchange adjustments) of the upfront proceeds that we received from CASI were recognized in the second quarter of 2015 within “license fees and service revenue” through our Consolidated Statements of Operations. The timing of this revenue recognition corresponds with the execution of supply agreements with CASI for ZEVALIN, MARQIBO, and EVOMELA. These agreements allow CASI to procure CASI Out-Licensed Products directly from approved third parties, and in such case, do not require our future involvement for their supply.
12. OUT-LICENSE OF ZEVALIN IN CERTAIN EX-U.S. TERRITORIES
Out-License of ZEVALIN to Mundipharma
On November 16, 2015, we entered into an out-license agreement with Mundipharma International Corporation Limited for their commercialization of ZEVALIN in Asia (excluding India and Greater China), Australia, New Zealand, Africa, the Middle East, and Latin America (including the Caribbean). In return, we received $18 million (comprised of $15 million received in December 2015 and $3 million received in January 2016), of which $15 million is included within "license fees and service revenue" on our accompanying Consolidated Statement of Operations. Revenue for the $3 million payment from January 2016 will be deferred and recognized within "license fees and service revenue" on a per unit basis with Mundipharma's future sales of ZEVALIN kits. We are also eligible to receive an additional $2 million upon Mundipharma's achievement of a specified sales milestone, that if/when achieved, will also be reported within "license fees and service revenue".
F-30
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
In connection with this out-license, on November 16, 2015, we concurrently sold to Mundipharma K.K., all common stock of Spectrum Pharmaceuticals GK (the legal entity through which we previously sold ZEVALIN in Japan) for $2.2 million (in the form of an unsecured note, payable no later than May 2016), representing its net asset value (excluding inventory) as of November 16, 2015.
13. CO-PROMOTION ARRANGEMENT WITH EAGLE PHARMACEUTICALS, INC.
On November 4, 2015, we executed an agreement with Eagle Pharmaceuticals, Inc. ("Eagle") whereby designated members of our sales force will concurrently market up to six of Eagle's pharmaceutical products along with our products, in return for fixed monthly payments over the initial 18 month contract term through June 30, 2017, aggregating $12.8 million (the "Eagle Agreement"). We are also eligible to receive milestone payments of up to $5 million for sales made in 2016 that exceed certain thresholds, and up to $4 million for sales made in the first half 2017 that exceed certain thresholds. In addition, for performance above such sales thresholds in 2016, and in the first half of 2017, we are eligible to receive certain further payments if specified targets for annual net sales of Eagle's products are exceeded. The fixed payments to us, as well as costs for certain marketing activities that we coordinate with third parties, will be recognized within "license fees and service revenue" on our accompanying Consolidated Statement of Operations, beginning January 2016 (variable payments to us will be recognized in the period earned). An allocation of our corresponding sales personnel costs, and reimbursable costs for marketing activities will be recognized within a new caption on our Consolidated Statement of Operations, "cost of service revenue."
Eagle may extend the initial term of this agreement by six months to December 31, 2017 at its sole election. Any extensions after December 31, 2017 require mutual consent and will be for six months per extension. The Eagle Agreement may be terminated by either party for uncured material breaches and certain other events following a change of control or insolvency of either party, and solely by Eagle for convenience with 60 days written notice, subject to an established termination fee, as calculated within the Eagle Agreement.
14. REVOLVING LINE OF CREDIT
We entered into a credit agreement on September 5, 2012 with Bank of America, N.A, as the administrative agent and Wells Fargo Bank, N.A, as an initial lender (the “Credit Agreement”). The Credit Agreement provided us with a committed $50 million revolving line of credit facility (the “Credit Facility”). The Credit Facility was repaid in full, then immediately terminated, on December 20, 2013 in connection with the sale and issuance of our 2018 Convertible Senior Notes (see Note 15). We recognized $1.2 million within “interest expense, net” in 2013, for this retired Credit Facility on the accompanying Consolidated Statement of Operations.
15. CONVERTIBLE SENIOR NOTES
Overview
On December 17, 2013, we entered into an agreement for the sale of $120 million aggregate principal amount of 2.75% Convertible Senior Notes due December 2018 (the “2018 Convertible Notes”). The 2018 Convertible Notes are convertible into shares of our common stock at a conversion rate of 95 shares per $1,000 principal amount of the 2018 Convertible Notes, equating to 11.4 million common shares if fully converted. The in-the-money conversion price is equivalent to $10.53 per common share. The conversion rate and conversion price is subject to adjustment under certain limited circumstances. The 2018 Convertible Notes bear interest at a rate of 2.75% per year, payable semiannually in arrears on June 15 and December 15 of each year. The 2018 Convertible Notes will mature and become payable on December 15, 2018, subject to earlier conversion into common stock at the holders’ option.
The sale of the 2018 Convertible Notes closed on December 23, 2013 and our net proceeds were $115.4 million, after deducting banker and professional fees of $4.6 million. We used a portion of these net proceeds to simultaneously enter into “bought call” and “sold warrant” transactions with Royal Bank of Canada (collectively, the “Note Hedge”). We recorded the Note Hedge on a net cost basis of $13.1 million, as a reduction to “additional paid-in capital” in our accompanying Consolidated Balance Sheets. Under applicable GAAP, the Note Hedge transaction is not expected to be marked-to-market through earnings or comprehensive income in future reported periods.
Conversion Hedge
F-31
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
We entered into Note Hedge transactions to reduce the potential dilution to our stockholders and/or offset any cash payments that we are required to make in excess of the principal amount, upon conversion of the 2018 Convertible Notes (in the event that the market price of our common stock is greater than the conversion price). The strike price of the “bought call” is equal to the conversion price and conversion rate of the 2018 Convertible Notes, matching the 11.4 million common shares the 2018 Convertible Notes may be converted into. The strike price of our “sold warrant” is $14.03 per share of our common stock, and is also for 11.4 million common shares.
Conversion Events
On and after June 15, 2018, and until the close of business on the second scheduled trading day immediately preceding the maturity date, holders may convert all or a portion of their 2018 Convertible Notes. Prior to June 15, 2018, holders may convert all or a portion of their 2018 Convertible Notes only under any of the following circumstances: (1) during any fiscal quarter (and only during such fiscal quarter), if, for at least 20 trading days (whether or not consecutive) during the 30 consecutive trading day period ending on the last trading day of the immediately preceding fiscal quarter, the last reported sale price of our common stock on such trading day is greater than or equal to 130% of the applicable conversion price on such trading day; (2) during the five consecutive business day period immediately following any five consecutive trading day period in which, for each trading day of that measurement period, the trading price per $1,000 principal amount of 2018 Convertible Notes for such trading day was less than 98% of the product of (i) the last reported sale price of our common stock on such trading day and (ii) the applicable conversion rate on such trading day; (3) upon the occurrence of certain corporate transactions; and (4) at any time prior to our stockholders’ approval to settle the 2018 Convertible Notes in our common shares and/or cash.
As of December 31, 2015, the 2018 Convertible Notes are not eligible to be converted into common as none of the above elements (1) through (4) were met. Our stockholders’ approval of "flexible settlement" occurred at our Annual Meeting of Stockholders on June 29, 2015. As a result, we may (at our election) settle any future conversions of the 2018 Convertible Notes by paying or delivering cash, shares of common stock, or a combination of cash and shares of common stock. However, if the holders of the Convertible Notes do not elect any conversion into our common stock, our December 2018 obligation to repay the principal amount of $120 million in cash, plus any accrued and unpaid interest, is unchanged.
Carrying Value and Fair Value
The carrying value of the 2018 Convertible Notes as of December 31, 2015 is summarized as follows:
Principal amount | $ | 120,000 | |
(Less): Unamortized debt discount (amortized through December 2018) | (18,452 | ) | |
December 31, 2015 | $ | 101,548 | |
As of December 31, 2015 and December 31, 2014, the estimated aggregate fair value of the 2018 Notes is $105.1 million and $113.2 million, respectively. These fair value estimates are less than the principal amount of $120 million, largely since the conversion feature of the 2018 Notes was, and remains, out-of-the money. These estimated fair values represent a Level 2 measurement (see Note 2(xiii)), based upon the 2018 Convertible Notes' quoted bid price at each date in a thinly-traded market.
Components of Interest Expense on 2018 Convertible Notes
The following table sets forth the components of interest expense recognized in the accompanying Consolidated Statements of Operations for the 2018 Convertible Notes for the years ended December 31, 2015 and 2014:
Year Ended December 31, | |||||||
2015 | 2014 | ||||||
Contractual coupon interest expense | $ | 3,300 | $ | 3,300 | |||
Amortization of debt issuance costs | 662 | 599 | |||||
Accretion of debt discount | 5,250 | 4,818 | |||||
Total | $ | 9,212 | $ | 8,717 | |||
Effective interest rate | 8.66 | % | 8.66 | % | |||
F-32
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
16. MUNDIPHARMA AGREEMENT
As the result of our acquisition of Allos Therapeutics, Inc. on September 5, 2012 (through which we obtained distribution rights for FOLOTYN), we assumed its obligations under an active strategic collaboration agreement with a third-party, Mundipharma (the “Mundipharma Collaboration Agreement”). Under the Mundipharma Collaboration Agreement, we retained full commercialization rights for FOLOTYN in the U.S. and Canada, with Mundipharma having exclusive rights to commercialize FOLOTYN in all other countries in the world (the “Mundipharma Territories”).
On May 29, 2013, the Mundipharma Collaboration Agreement was amended and restated (the “Amended Mundipharma Collaboration Agreement”), in order to modify: (i) the scope of the licensed territory, (ii) milestone payments, (iii) royalty rates, and (iv) drug development obligations. In connection with the Amended Mundipharma Collaboration Agreement, we received a one-time $7.0 million payment from Mundipharma for certain research and development activities to be performed by us.
As a result of the Amended Mundipharma Collaboration Agreement, (a) Europe and Turkey were excluded from Mundipharma’s commercialization territory, (b) we may receive regulatory milestone payments of up to $16.0 million, and commercial progress and sales-dependent milestone payments of up to $107.0 million, (c) we will receive tiered double-digit royalties based on net sales of FOLOTYN within Mundipharma’s licensed territories, and (d) we and Mundipharma will bear our own FOLOTYN development costs.
On May 29, 2015 and effective as of May 1, 2015, we entered into an amendment to the Amended Mundipharma Collaboration Agreement (the “Amendment”). Pursuant to the Amendment, among other things, the parties revised the conditions to our exercise of the option to gain commercialization rights in Switzerland from Mundipharma, and also revised tiered double digit royalties payable by Mundipharma on net sales in Switzerland.
The fair value of this liability is included in the current and long-term portions of “drug development liability” within the accompanying Consolidated Balance Sheets, and it includes our assumptions about personnel needed to perform these research and development activities, third party costs for projected clinical trial enrollment, and patient treatment-related follow up through approximately 2031.
The fair value of our “drug development liability” within our accompanying Consolidated Balance Sheets was estimated using the discounted income approach model. The unobservable inputs (i.e., Level 3 inputs - see Note 2(xiii)) in this valuation model that have the most significant effect on these liabilities include (i) estimates of research and development personnel costs needed to perform the research and development services, (ii) estimates of expected cash outflows to third parties for services and supplies during the expected period of performance through 2031, and (iii) an appropriate discount rate for these expenditures. These inputs are reviewed by management on a quarterly basis for continued applicability.
We assess this liability at each reporting date and record its adjustment through “research and development” expense in our accompanying Consolidated Statements of Operations.
Year Ended December 31, | |||||||||||
Drug Development Liability, Current – FOLOTYN | Drug Development Liability, Long Term – FOLOTYN | Total Drug Development Liability – FOLOTYN | |||||||||
Balance at December 31, 2014 | $ | 1,141 | $ | 14,644 | $ | 15,785 | |||||
Transfer from long term to current in 2015 | 217 | (217 | ) | — | |||||||
(Less): Expenses incurred in 2015 | (1,099 | ) | — | (1,099 | ) | ||||||
Balance at December 31, 2015 | $ | 259 | $ | 14,427 | $ | 14,686 | |||||
17. COMMITMENTS AND CONTINGENCIES
(a) Facility and Equipment Leases
We lease our principal executive office in Henderson, Nevada under a non-cancelable operating lease expiring April 30, 2019. We also lease our research and development and administrative facility in Irvine, California under a non-cancelable operating lease expiring May 31, 2019, in addition to several other administrative office leases. Each lease agreement contains
F-33
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
scheduled rent increases which are accounted for on a straight-line basis. Our total rental expense in 2015, 2014, and 2013 was $1.7 million, $1.9 million, and $1.2 million, respectively.
Our future minimum lease payments are as follows:
Year ending December 31, | Operating Lease Minimum Payments | ||
2016 | $ | 1,283 | |
2017 | 1,172 | ||
2018 | 1,209 | ||
2019 | 460 | ||
2020 | — | ||
$ | 4,124 | ||
(b) Licensing Agreements, Co-Development Agreements and Milestone Payments
Our drug candidates are being developed pursuant to license agreements that provide us with territory-specific rights to its manufacture, sublicense, and sale. We are generally responsible for all development costs, patent filings and maintenance costs, sales and marketing costs, and liability insurance costs. We are also obligated to make certain milestone payments to third parties upon the achievement of regulatory and sales milestones that are specified in these license agreements. We estimate and present a corresponding liability on our Consolidated Balance Sheets when amounts are probable and reasonably estimable. In addition, we are obligated to pay royalties based on our current and future net sales of in-licensed products.
Our most significant of these agreements are listed and summarized below:
(i) ZEVALIN U.S.: In-Licensing and development in the U.S.
In December 2008, we acquired rights to commercialize and develop ZEVALIN in the U.S. as the result of a transaction with Cell Therapeutics, Inc. (“CTI”) through our wholly-owned subsidiary, RIT Oncology LLC (“RIT”). We assumed certain agreements with various third parties related to ZEVALIN intellectual property for its manufacture, use, and sale in the U.S.
In accordance with the terms of assumed contracts, we are required to meet specified payment obligations, including a milestone payment to Corixa Corporation of $5.0 million based on ZEVALIN sales in the U.S. (the “Corixa Liability”). This milestone has not yet been met, and $0.1 million for this potential milestone achievement is included within “acquisition-related contingent obligations” in our accompanying Consolidated Balance Sheet as of December 31, 2015 and December 31, 2014, respectively. Our U.S. net sales-based royalties are in the low to mid-single digits to Genentech, Inc. and mid-teens to Biogen.
(ii) ZEVALIN Ex-U.S.: In-License and Asset Purchase Agreement with Bayer Pharma
In April 2012, through our wholly-owned subsidiary, Spectrum Pharmaceuticals Cayman, L.P., we completed the acquisition of licensing rights to market ZEVALIN outside of the U.S. from Bayer Pharma AG (“Bayer”). ZEVALIN is currently approved in approximately 40 countries outside the U.S. for the treatment of B-cell non-Hodgkin lymphoma, including countries in Europe, Latin America and Asia.
In consideration for the rights granted under the agreement, concurrent with the closing, we paid Bayer a one-time fee of €19.0 million. Our ex-U.S. net sales-based royalty to Bayer ranges between the single digits to mid-teens. Unless earlier terminated, the term of the agreement continues until the expiration of the last-to-expire patent covering the sale of a licensed product in the relevant country, or 15 years from the date of first commercial sale of the licensed product in such country, whichever is longer.
(iii) ZEVALIN Ex-U.S.: Out-License Agreement with Dr. Reddy’s
Effective June 27, 2014, we executed an exclusive License Agreement with Dr. Reddy’s Laboratories Ltd. (“Dr. Reddy’s”), for the distribution rights of ZEVALIN within India. The agreement term is 15 years from the receipt of pending approval of ZEVALIN from the Drug Controller General of India. On December 17, 2014, upon the execution of a supply agreement, an upfront and non-refundable payment of $0.5 million was triggered and was paid to us in February 2015. This recognition of this upfront payment is reported on a straight line basis, within “license fee and service revenue” on the Consolidated Statement of Operations over a 10-year term through December 2024. Additionally, sales and regulatory
F-34
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
milestones (aggregating $3 million) will become payable to us when achieved by Dr. Reddy’s, as well as a 20% royalty on net sales of ZEVALIN in India.
(iv) FUSILEV: Amended and Restated In-License Agreement with Merck & Cie AG
In May 2006, we amended and restated a license agreement with Merck & Cie AG (“Merck”), which we assumed in connection with our March 2006 acquisition of the assets of Targent, Inc. Pursuant to the license agreement with Merck, we obtained the exclusive license to use regulatory filings related to FUSILEV and a non-exclusive license under certain patents and know-how to develop, manufacture, use, and sell FUSILEV in the field of oncology in North America in return for a royalty percentage (in the mid-single digits) of net sales. Merck is eligible to receive a $0.2 million payment from us upon the achievement of a FDA approval of an oral form of FUSILEV. This milestone has not yet been met, and no amounts have been accrued in our accompanying Consolidated Balance Sheets for its potential achievement.
(v) FOLOTYN: In-License Agreement with Sloan-Kettering Institute, SRI International and Southern Research Institute
In December 2002, Allos entered into the FOLOTYN License Agreement with Sloan-Kettering Institute for Cancer Research, SRI International, and Southern Research Institute. As a result of Allos becoming our wholly owned subsidiary in September 2012, we are bound by the FOLOTYN License Agreement under which we obtained exclusive worldwide rights to a portfolio of patents and patent applications related to FOLOTYN and its uses. Under the terms of the FOLOTYN License Agreement, we are required to fund all development programs and have sole responsibility for all commercialization activities. In addition, we pay graduated royalties to our licensors based on our (including sub licensees) worldwide annual net sales of FOLOTYN. Royalties are 8% of annual worldwide net sales up to $150 million; 9% of annual worldwide net sales of $150 million through $300 million; and 11% of annual worldwide net sales in excess of $300 million.
(vi) EVOMELA: In-License Agreement with Cydex Pharmaceuticals, Inc.
In March 2013, we completed the acquisition of exclusive global development and commercialization rights to EVOMELA from Ligand (see Note 10(b)). We filed an NDA with the FDA in December 2015 for its use as a conditioning treatment prior to autologous stem cell transplant for patients with MM. On March 10, 2016, the FDA communicated its approval of our NDA for EVOMELA. Upon receipt of FDA approval, we are contractually obligated to make a $6.0 million milestone payment to Ligand within 30 days ("Ligand Contingent Consideration"); accordingly, this payment is due no later than April 9, 2016.
We are required to pay Ligand amounts of up to $66 million (inclusive of the $6.0 million milestone for FDA approval), upon achievement of certain regulatory milestones and net sales thresholds, which we have valued at $5.2 million and $4.9 million within “acquisition-related contingent obligations” in our accompanying Consolidated Balance Sheets as of December 31, 2015 and December 31, 2014, respectively. We will also pay royalties of 20% on our net sales of licensed products in all territories.
(vii) MARQIBO: Contingent Consideration Agreement with Talon Therapeutics, Inc.
In July 2013, we completed the acquisition of Talon, through which we obtained exclusive global development and commercialization rights to MARQIBO (see Note 10(a)). As part of this acquisition, we issued the former Talon stockholders contingent value rights (“CVR”) that we have valued and presented on our accompanying Consolidated Balance Sheets as a $1.4 million and $2.4 million liability within “acquisition-related contingent obligations” as of December 31, 2015 and December 31, 2014, respectively. The CVR has a maximum payout value of $195 million if all sales and regulatory approval milestones are achieved.
(viii) EOQUIN (previously referred to as APAZIQUONE): License Agreement with Allergan, Inc. and NDDO Research Foundation
In October 2008, we entered into an exclusive development and commercialization collaboration agreement with Allergan for EOQUIN. Pursuant to the terms of the agreement, Allergan paid us an up-front non-refundable fee of $41.5 million at closing (which we have amortized through revenue within “license fees and service revenue” in full as of December 31, 2013). In October 2008, pursuant to a letter agreement with NDDO Research Foundation (“NDDO”), we agreed to pay NDDO the following in relation to EOQUIN milestones: (a) upon FDA acceptance of the NDA, the issuance of 25,000 of our common shares and (b) upon FDA approval of the drug (its target decision date is set for December 11, 2016), a one-time payment of $0.3 million.
F-35
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
In January 2013, we entered into a second amendment to the license, development, supply and distribution agreement with Allergan to amend the agreement and reacquire the rights originally licensed to Allergan in the U.S., Europe, and other territories in exchange for a tiered single-digit royalty on net sales of certain products containing EOQUIN, and relieved Allergan of its development and commercialization obligations.
(ix) EOQUIN: Collaboration Agreement with Nippon Kayaku Co. LTD.
In November 2009, we entered into a collaboration agreement with Nippon Kayaku Co., LTD. (“Nippon Kayaku”) for the development and commercialization of EOQUIN in Asia, except North and South Korea (the “Nippon Kayaku Territory”). In addition, Nippon Kayaku received exclusive rights to EOQUIN for the treatment of non-muscle invasive bladder cancer in Asia (other than North and South Korea), including Japan and China. Nippon Kayaku will conduct EOQUIN clinical trials in the Nippon Kayaku Territory pursuant to a development plan. Further, Nippon Kayaku will be responsible for all expenses relating to the development and commercialization of EOQUIN in the Nippon Kayaku Territory.
Under the terms of this agreement, Nippon Kayaku paid us an upfront fee of $15 million (which we have amortized through revenue within “license fees and service revenue” in full as of December 31, 2013). Nippon Kayaku is also obligated to make additional payments to us based on the achievement of certain development, regulatory and commercialization milestones. Under the terms of the agreement, we are entitled to payment of $10 million and $126 million upon achievement of certain regulatory and commercialization milestones, respectively. Also, Nippon Kayaku has agreed to pay us royalties based on a percentage of net sales of the subject products in the defined territory in the mid-teen digits.
(x) BELEODAQ: In-Licensing and Collaboration Agreement with TopoTarget
In February 2010, we entered into a licensing and collaboration agreement with TopoTarget A/S (now Onxeo SA) (“TopoTarget”), as amended in October 2013, for the development and commercialization of BELEODAQ. The agreement provides that we have the exclusive right to manufacture, develop, and commercialize BELEODAQ in North America and India, with an option for China. Pursuant to the terms of this agreement, we paid TopoTarget an upfront fee of $30 million in 2010.
Under continuing terms, all development, including studies, will be conducted under a joint development plan, which we will fund 70% of such costs, and TopoTarget will fund 30%. We have final decision-making authority for all developmental activities in North America and India (and China upon exercise of our option). TopoTarget has final decision-making authority for all developmental activities in all other jurisdictions. In February 2014, upon FDA acceptance of our new drug application, we issued one million shares of our common stock, and made a $10 million milestone payment to TopoTarget. The aggregate payout value of this first milestone at achievement was $17.8 million, and is recognized within “research and development” of the accompanying Consolidated Statement of Operations during the first quarter of 2014.
In July 2014, we received approval from the FDA for BELEODAQ’s use for injection and treatment of relapsed or refractory PTCL. As a result, we paid a second milestone payment to TopoTarget of $25 million in November 2014, which we capitalized as an amortizable intangible asset. Other potential milestone payments due upon BELEODAQ regulatory achievements and sales thresholds (aggregating $278 million) are not included within “total liabilities” in our accompanying Consolidated Balance Sheets.
We will pay TopoTarget future royalties in the mid-teen digits based on our net sales of BELEODAQ. The agreement will continue until the expiration of the last royalty payment period in the last country in the defined territory with certain provisions surviving, unless earlier terminated in accordance with its terms.
(xi) SPI-2012: Co-Development and Commercialization Agreement with Hanmi Pharmaceutical Company
In January 2012 (and as amended in March 2014 and October 2014), we entered into a License, Development and Supply Agreement with Hanmi Pharmaceutical Co., Ltd. (“Hanmi”), for SPI-2012, formerly known as “LAPS-GCSF”, a drug based on Hanmi’s proprietary LAPSCOVERY™ technology for the treatment of chemotherapy induced neutropenia. Under the terms of the agreement, as amended, we have primary financial responsibility for the SPI-2012 development plan. We have worldwide rights for SPI-2012, except for Korea, China, and Japan. We will also be responsible for milestone payments related to SPI-2012 Phase 3 clinical trial commencement (resulting in a triggered $3 million milestone payment, based on initial patient dosing in January 2016; we intend to settle this liability through the issuance of our stock to Hanmi by March 2016), regulatory approvals, and sales thresholds (aggregating $238 million), which are not included within “total liabilities” in our accompanying Consolidated Balance Sheets. We will pay Hanmi royalties in the mid-teen digits on our net sales of SPI-2012.
F-36
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
(xii) POZIOTINIB: In-License Agreement with Hanmi
In February 2015, we executed an in-license agreement with Hanmi Pharmaceutical Co., Ltd for POZIOTINIB, a pan-HER inhibitor in Phase 2 clinical trials, requiring our upfront payment for these rights. This drug has shown single agent activity in the treatment of various cancer types during Phase I studies, including breast, gastric, colorectal and lung cancers.
Under the terms of this agreement, we received the exclusive rights to commercialize POZIOTINIB, excluding Korea and China. Hanmi, and its development partners, will bear full responsibility for the on-going Phase 2 trials in Korea. We will bear full financial responsibility for all other clinical studies. We will pay Hanmi future regulatory and sales-dependent milestones (aggregating $358 million), which are not included within “total liabilities” in our accompanying Consolidated Balance Sheets. We will pay Hanmi royalties in the low to mid-teen digits on our net sales of POZIOTINIB.
(c) Service Agreements
In connection with the research and development of our drug products, we have entered into contracts with numerous third party service providers, such as radio-pharmacies, distributors, clinical trial centers, clinical research organizations, data monitoring centers, and with drug formulation, development and testing laboratories. The financial terms of these agreements are varied and generally obligate us to pay in stages, depending on achievement of certain events specified in the agreements, such as contract execution, reservation of service or production capacity, actual performance of service, or the successful accrual and dosing of patients.
At each period end, we accrue for all services received, with such accruals based on factors such as estimates of work performed, patient enrollment, completion of patient studies and other events. Should we decide to discontinue and/or slow-down the work on any project, the associated costs for those projects would be limited to the extent of the work completed. Generally, we are able to terminate these contracts due to the discontinuance of the related project(s) and thus avoid paying for the services that have not yet been rendered.
(d) Supply Agreements
We have entered into certain supply agreements, or have issued purchase orders, which require us to make minimum purchases from vendors for the manufacture of our products. These commitments do not exceed our planned commercial requirements, and the contracted prices do not exceed their fair market value.
(e) Employment Agreements
We have entered into an employment agreement with our Chief Executive Officer under which cash compensation and benefits would become payable in the event of termination by us for any reason other than cause, his resignation for good reason, or upon a change in control of our Company.
(f) Deferred Compensation Plan
The Spectrum Pharmaceuticals, Inc. Deferred Compensation Plan (the “DC Plan”) is administered by the Compensation Committee of our Board of Directors and is intended to comply with the requirements of Section 409A of the Internal Revenue Code of 1986, as amended.
The DC Plan is maintained to provide deferred compensation benefits for a select group of our employees (the “DC Participants”). Under the DC Plan, we provide the DC Participants with the opportunity to make annual elections to defer up to a specified amount or percentage of their eligible cash compensation, and we have the option to make discretionary contributions. At December 31, 2015 and December 31, 2014, this DC Plan liability was $6.5 million and $4.7 million, respectively, and is included within “other long-term liabilities” in the accompanying Consolidated Balance Sheets.
(g) Litigation
We are involved from time-to-time with various legal matters arising in the ordinary course of business. These claims and legal proceedings are of a nature we believe are normal and incidental to a pharmaceutical business, and may include product liability, intellectual property, employment matters, and other general claims.
We make provisions for liabilities when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Such provisions are assessed at least quarterly and adjusted to reflect the impact of any settlement
F-37
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
negotiations, judicial and administrative rulings, advice of legal counsel, and other information and events pertaining to a particular case. Litigation is inherently unpredictable. Although the ultimate resolution of these various matters cannot be determined at this time, we do not believe that such matters, individually or in the aggregate, will have a material adverse effect on our consolidated results of operations, cash flows, or financial condition.
We are presently responding to ANDAs filed by companies seeking to market generic forms of FOLOTYN. We are also responding to certain stockholder suits that purportedly stem from our March 12, 2013 press release, in which we announced anticipated changes in customer ordering patterns of FUSILEV. These complaints allege that, as a result of this press release, our stock price declined.
FUSILEV ANDA Litigation
On January 20, 2012, March 2, 2012, June 18, 2014, January 23, 2015, July 17, 2015 and September 3, 2015 respectively, we filed suit against Sandoz Inc., Innopharma Inc., Ben Venue Laboratories, Inc., Amneal Pharmaceuticals, Inc., and Actavis LLC. respectively, following Paragraph IV certifications in connection with their filing separate ANDAs, to manufacture a generic version of FUSILEV. We filed the lawsuits in the U.S. District Court for the Districts of Nevada and Delaware seeking to enjoin the approval of their ANDAs plus recovery of our litigation fees and costs incurred in such matters. On December 9, 2013, three Mylan entities collaborating with Innopharma were joined to Innopharma case. On November 24, 2014, the complaint in the Ben Venue case was amended to substitute the original defendant Ben Venue Laboratories, Inc. with successors West-Ward Pharmaceutical Corp. and Eurohealth International SARL.
A trial took place in the Sandoz case from January 12, 2015 through January 20, 2015 in the U.S. District Court for the District of Nevada and on February 20, 2015 the district court found certain of the asserted claims of the patent covering FUSILEV invalid. On February 27, 2015, we filed our Notice of Appeal. On August 4, 2015 the Delaware district court ordered that judgment be entered for Innopharma and Mylan due to the Nevada district court judgment in the Sandoz action. On October 2, 2015, the U.S. Court of Appeals for the Federal Circuit affirmed the previously reported judgment from the U.S. District Court for the District of Nevada in favor of Sandoz Inc.
On April 27, 2015, we filed suit in the U.S. District Court for the District of Columbia against the FDA seeking a temporary restraining order or preliminary injunction to suspend FDA approval of Sandoz’s ANDA. The Company contends that Sandoz’s ANDA should not have been approved until the expiry of the Company’s Orphan Drug Exclusivity on April 29, 2018. On April 29, 2015, the court denied the temporary restraining order and on May 27, 2015, the court entered summary judgment in favor of the FDA et al. On June 5, 2015, we filed our Notice of Appeal. Oral argument was held October 22, 2015. The ultimate outcome of this proceeding is uncertain.
FOLOTYN ANDA Litigation
On June 19, 2014, we filed a lawsuit against five parties resulting from Paragraph IV certifications in connection with four separate ANDAs to manufacture a generic version of FOLOTYN: (1)Teva Pharmaceuticals USA, Inc., (2) Sandoz Inc., (3) Fresenius Kabi USA, LLC, (4) Dr. Reddy’s Laboratories, Ltd., and (5) Dr. Reddy’s Laboratories, Inc. We filed the lawsuit in the U.S. District Court for the District of Delaware seeking to enjoin the approval of their ANDAs plus recovery of our litigation fees and costs. The litigation is stayed with respect to the Dr. Reddy's entities pending resolution of the case against the other FOLOTYN ANDA filers. A trial date of September 12, 2016 has been set in the FOLOTYN lawsuit in the U.S. District Court for the District of Delaware. While we believe our patent rights are strong, the ultimate outcome of such action is uncertain.
Stockholder Litigation
John Perry v. Spectrum Pharmaceuticals, Inc. et al. (Filed March 14, 2013 in United States District Court, District of Nevada; Case Number 2:2013-cv-00433-LDG-CWH). This putative consolidated class action raises substantially identical claims and allegations against defendants Spectrum Pharmaceuticals, Inc., Dr. Rajesh C. Shrotriya, Brett L. Scott, and Joseph Kenneth Keller. The alleged class period is August 8, 2012 to March 12, 2013. The lawsuits allege a violation of Section 10(b) of the Securities Exchange Act of 1934 against all defendants and control person liability, as a violation of Section 20(b) of the Securities Exchange Act of 1934, against the individual defendants. The claims purportedly stem from the Company’s March 12, 2013 press release, in which it announced that it anticipated a change in ordering patterns of FUSILEV. The complaints allege that, as a result of the March 12, 2013 press release, the Company’s stock price declined. The complaints further allege that during the putative class period certain defendants made misleadingly optimistic statements about FUSILEV sales, which inflated the trading price of Company stock. The lawsuits seek relief in the form of monetary damages, costs and fees, and any other equitable or injunctive relief that the court deems appropriate. On March 21, 2014, the Court entered an order appointing Arkansas Teacher Retirement System as lead plaintiff. On May 20, 2014, Arkansas Teacher Retirement System filed a
F-38
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
consolidated amended class action complaint. On July 18, 2014, we filed a motion to dismiss the consolidated amended class action complaint. On March 26, 2015, the court denied the motion to dismiss. On June 15, 2015, the Court ordered a stay of the proceedings pending the outcome of mediation between the parties. On October 27, 2015, we reached a $7 million settlement in principle with the lead plaintiff (which involved our insurance carrier, as the reimbursing party in full), subject to preliminary and final court approval. We have included this settlement amount, along with $0.1 million of reimbursable legal expenses for this matter, on our accompanying Consolidated Balance Sheets as of December 31, 2015 within "other receivables" and "accounts payable and other accrued liabilities." On January 26, 2016, the Court preliminarily approved the settlement. The Court has scheduled a hearing on final approval of the settlement for June 13, 2016.
Timothy Fik v. Rajesh C. Shrotriya, et al. (Filed April 11, 2013 in United States District Court, District of Nevada; Case Number 2:2013-cv-00624-JCM-CWH); Christopher J. Watkins v. Rajesh C. Shrotriya, et al. (Filed April 22, 2013 in United States District Court, District of Nevada; Case Number 2:2013-cv-00684-JCM-VCF); and Stefan Muenchhagen v. Rajesh C. Shrotriya, et al. (Filed May 28, 2013; Case Number 2:2013-cv-00942-APG-PAL). These derivative complaints are brought by the respective purported stockholders on behalf of nominal plaintiff Spectrum against certain current and former directors and officers. The complaints generally allege breaches of fiduciary based on conduct relating to the events alleged in the consolidated Perry action. The complaints seek compensatory damages, corporate governance reforms, restitution and disgorgement of defendants’ alleged profits, and costs and fees. These actions are stayed pending resolution of the federal securities class action. Settlement discussions are ongoing, and accordingly, no agreement has yet been reached to resolve these derivative complaints. If a settlement were reached, we believe it would be reimbursable by our insurance carrier. However, the value of a potential settlement cannot be reasonably estimated given its highly uncertain nature.
Hardik Kakadia v. Rajesh C. Shrotriya, et al. (Filed April 23, 2013 in the Eighth Judicial District Court of the State of Nevada in and for Clark County; Case Number A-13-680643-B); and Joel Besner v. Rajesh C. Shrotriya, et al. (Filed May 31, 2013; Case Number A-13-682668-C) (collectively the “State Derivative Actions”). These consolidated State Derivative Actions are brought by the respective purported stockholders on behalf of nominal plaintiff Spectrum Pharmaceuticals, Inc. and are substantially similar to the consolidated federal derivative actions. These actions are stayed pending resolution of the federal securities class action. Settlement discussions are ongoing, and accordingly, no agreement has yet been reached to resolve these derivative complaints. If a settlement were reached, it would be reimbursable by our insurance carrier. However, the value of a potential settlement cannot be reasonably estimated given its highly uncertain nature.
Ira Gains v. Spectrum Pharmaceuticals, Inc. and Rajesh C. Shrotriya was filed in the United States District Court, District of Nevada on November 3, 2015. This putative class action was brought against defendants Spectrum Pharmaceuticals, Inc., and Dr. Rajesh C. Shrotriya. The alleged class period was May 7, 2015 to October 23, 2015. The complaint alleged a violation of Section 10(b) of the Securities Exchange Act of 1934 against all defendants and control person liability, as a violation of Section 20(a) of the Securities Exchange Act of 1934, against Dr. Rajesh C. Shrotriya. The claims purportedly stemmed from our October 23, 2015 press release in which we announced that the FDA issued a Complete Response Letter indicating that the FDA would not approve our NDA for EVOMELA in its present form. The complaint alleged that, as a result of the October 23, 2015 press release, our stock price declined. The complaint further alleged that during the putative class period defendants made misleadingly optimistic statements about the progress of the NDA for EVOMELA with the FDA, our expectations regarding FDA approval of the NDA, and EVOMELA’s potential as a future driver of our revenue, which inflated the trading price of our stock. The complaint sought relief in the form of monetary damages, costs and fees, and any other relief that the Court deemed appropriate. On December 11, 2015, plaintiff voluntarily dismissed the action without prejudice.
(h) SEC Subpoena
On April 1, 2013, we received a subpoena from the SEC for documents pursuant to a formal order of investigation. The subpoena followed our March 12, 2013 announcement that we anticipated a change in customer ordering patterns of FUSILEV. We have cooperated with the SEC throughout its investigation, and on November 30, 2015, we received a notice from that the SEC does not intend to recommend an enforcement action against our company.
(i) Notice from HRSA
We received a notice on October 10, 2014 from the U.S. Health Resources and Services Administration, Office of Pharmacy Affairs (“HRSA”). In this notice HRSA asserted that, for at least one of our products with an “orphan drug” designation under section 526 of the Federal Food, Drug, and Cosmetic Act, we did not make the product(s) available for purchase by certain categories of providers at the applicable 340B price. The 340B price is a discounted price for covered outpatient drugs that manufacturers participating in Medicaid (which includes us) agree to make available to providers that participate in the 340B drug pricing program (“Covered Entities”). HRSA’s notice asserted that, by not selling our product(s) to
F-39
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
certain categories of Covered Entities at 340B prices, we were overcharging them, and that we owed certain undefined refunds to those Covered Entities based on our previously made and reported product sales.
On October 14, 2015, the U.S. District Court for the District of Columbia issued a decision in the case of Pharmaceutical Research and Manufacturers of America v. United States Department of Health and Human Services, et al., invalidating HRSA’s July 21, 2014 “interpretive rule” relating to orphan drug pricing under the 340B drug pricing program. Because HRSA’s October 10, 2014 notice to us reflected the same interpretation of relevant orphan drug pricing statutes as the interpretation overturned by the District Court in the Pharmaceutical Research and Manufacturers of America decision, we believe the decision supports the propriety of our pricing to Covered Entities. Since we only make provisions for liabilities when it is both probable that a liability has been incurred, and the amount can be reasonably estimated, we have not recorded a liability for this pending matter as of December 31, 2015 or any earlier period.
18. INCOME TAXES
The components of loss before provision for income taxes are as follows:
For the Years Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
United States | $ | (56,554 | ) | $ | (37,327 | ) | $ | (30,437 | ) | ||
Foreign | 6,175 | (6,205 | ) | (6,199 | ) | ||||||
Total | $ | (50,379 | ) | $ | (43,532 | ) | $ | (36,636 | ) | ||
The provision for income taxes consist of the following:
For the Years Ended December 31, | |||||||||||
2015 | 2014 | 2013 | |||||||||
Current: | |||||||||||
Federal | $ | 113 | $ | 1,529 | $ | (8,357 | ) | ||||
State | 5 | 126 | (691 | ) | |||||||
Foreign | 148 | 29 | — | ||||||||
$ | 266 | $ | 1,684 | $ | (9,048 | ) | |||||
Deferred: | |||||||||||
Federal | 114 | 495 | 36,183 | ||||||||
State | 26 | 7 | (1,637 | ) | |||||||
Foreign | — | — | — | ||||||||
140 | 502 | 34,546 | |||||||||
Total income tax provision | $ | 406 | $ | 2,186 | $ | 25,498 | |||||
The 2014 income tax provision includes $1.5 million related to the correction of our prior year estimates of carryback of federal net operating losses, book tax differences on acquisition-related liabilities, and credits ineligible for offset against federal income taxes. Management has evaluated the materiality of these adjustments quantitatively and qualitatively, and has concluded that the corrections are immaterial to the accompanying Consolidated Financial Statements, taken as a whole.
The income tax provision differs from that computed using the federal statutory rate applied to income before taxes as follows:
F-40
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
2015 | 2014 | 2013 | |||||||||
Tax provision computed at the federal statutory rate | $ | (17,619 | ) | $ | (15,236 | ) | $ | (12,822 | ) | ||
State tax, net of federal benefit | 232 | 66 | (246 | ) | |||||||
Research credits | (2,974 | ) | (2,134 | ) | (2,254 | ) | |||||
FIN 48 uncertain tax positions | — | — | — | ||||||||
Change in tax credit carryforwards | (4,965 | ) | — | — | |||||||
Transaction costs | — | (11 | ) | 880 | |||||||
Officers compensation | 1,577 | 1,895 | 2,178 | ||||||||
Stock based compensation | 535 | 299 | 501 | ||||||||
Permanent items and other | (487 | ) | 21,742 | (1,080 | ) | ||||||
Domestic manufacturing deduction | — | (630 | ) | 767 | |||||||
Tax differential on foreign earnings | 1,435 | 1,570 | 1,123 | ||||||||
Change in tax rate | (903 | ) | (519 | ) | (283 | ) | |||||
Valuation allowance | 23,575 | (4,856 | ) | 36,734 | |||||||
Income tax provision | $ | 406 | $ | 2,186 | $ | 25,498 | |||||
The Protecting Americans from Tax Hikes Act of 2015, which President Obama signed into law on December 18, 2015, reinstated the research and development credit for 2015 and included a permanent extension of the research credit under Section 41. We recorded a $0.4 million benefit, before impact of valuation allowance, related to the research and development credit in 2015.
Significant components of our deferred tax assets as of December 31, 2015 and 2014 are presented below. A valuation allowance has been recognized to offset the net deferred tax assets as realization of such deferred tax assets no longer meets the “more-likely-than-not” threshold under GAAP.
2015 | 2014 | ||||||
Deferred tax assets: | |||||||
Net operating loss carry forwards | $ | 41,251 | $ | 40,505 | |||
Research credits | 22,082 | 9,045 | |||||
Stock based compensation | 4,828 | 3,703 | |||||
Deferred revenue | 2,280 | 1,893 | |||||
Development costs | 5,504 | 5,950 | |||||
Returns and allowances | 1,363 | 4,161 | |||||
Other, net | 9,887 | 9,082 | |||||
Total deferred tax assets before valuation allowance | 87,195 | 74,339 | |||||
Valuation allowance | (71,815 | ) | (45,983 | ) | |||
Total deferred tax assets | 15,380 | 28,356 | |||||
Deferred tax liabilities: | |||||||
Basis difference in debt | (713 | ) | (907 | ) | |||
Depreciation and amortization differences | (21,446 | ) | (34,088 | ) | |||
Net deferred tax liability | $ | (6,779 | ) | $ | (6,639 | ) | |
At December 31, 2015 and 2014, we recorded a valuation allowance of $71.8 million and $46.0 million, respectively. The valuation allowance increased by $25.8 million and decreased by $3.6 million, during 2015 and 2014, respectively. The increase in the valuation allowance in 2015 was due to increased research & development and orphan drug credits and the decrease in deferred tax liabilities. The decrease in valuation allowance in 2014 was due to a $17.2 million adjustment to write off deferred tax assets acquired from Talon that were determined not to be realizable.
At December 31, 2015, we had federal and state net operating loss carryforwards of approximately $111.5 million and $92.7 million, respectively. We have approximately $8.9 million of foreign loss carryforwards that will begin to expire in 2022. The federal and state loss carry forwards will begin to expire in 2018 and 2016, respectively, unless previously utilized. At
F-41
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
December 31, 2015, we had federal and state tax credits of approximately $22.9 million and $3.0 million, respectively. The federal tax credit carryovers begin to expire in 2027 unless previously utilized. The state research and development credit carryforwards have an indefinite carryover period.
As a result of the prior ownership changes, the utilization of certain net operating loss and research and development tax credit carryforwards including those acquired in connection with the acquisition of Allos and Talon are subject to annual limitations under Sections 382 and 383 of the Internal Revenue Code of 1986 and similar state provisions. Any net operating losses or credits that would expire unutilized as a result of Section 382 and 383 limitations have been removed from the table of deferred tax assets and the accompanying disclosures of net operating loss and research and development carryforwards.
Accounting guidance clarifies the accounting for uncertain tax positions and prescribes a recognition threshold and measurement process for recording in the financial statements uncertain tax positions taken or expected to be taken in a tax return. Additionally, the authoritative guidance addresses the de-recognition, classification, accounting in interim periods and disclosure requirements for uncertain tax positions. Only tax positions that meet the more-likely-than-not recognition threshold at the effective date may be recognized.
In November 2015, the FASB issued ASU 2015-17, Balance Sheet Classification of Deferred Taxes, which requires companies to classify all deferred tax assets and liabilities as noncurrent on the balance sheet instead of separating deferred taxes into current and noncurrent amounts by jurisdiction. For public business entities, the guidance is effective for financial statements issued for annual periods beginning after December 15, 2016. We decided to early adopt these provision on a prospective basis as of December 31, 2015, and the adoption did not have a material impact on the accompanying Consolidated Financial Statements.
The following tabular reconciliation summarizes activity related to unrecognized tax benefits:
2015 | 2014 | 2013 | |||||||||
Balance at beginning of year | $ | 1,944 | $ | 2,212 | $ | 5,482 | |||||
Adjustments related to prior year tax positions | 1,318 | (915 | ) | (200 | ) | ||||||
Increases related to current year tax positions | 1,236 | 647 | 648 | ||||||||
Decreases due to settlements | — | — | (1,227 | ) | |||||||
Decreases related to prior year tax positions | — | — | (2,491 | ) | |||||||
Balance at end of year | $ | 4,498 | $ | 1,944 | $ | 2,212 | |||||
We continue to believe that our tax positions meet the more-likely-than-not standard required under the recognition phase of the authoritative guidance. However, we consider the amounts and probabilities of the outcomes that can be realized upon ultimate settlement with the tax authorities and determined unrecognized tax benefits primarily related to credits should be established as noted in the summary rollforward above.
Approximately $0.7 million, $0.7 million, and $0.3 million of the total unrecognized tax benefits as of December 31, 2015, 2014, and 2013, respectively, would reduce our annual effective tax rate if recognized. Additional amounts in the summary rollforward could impact our effective tax rate if we did not maintain a full valuation allowance on our net deferred tax assets.
We do not expect our unrecognized tax benefits to change significantly over the next 12 months. With a few exceptions, we are no longer subject to U.S. federal, state and local income tax examinations for years before 2009. Our policy is to recognize interest and/or penalties related to unrecognized tax benefits in income tax expense in the consolidated statements of operations.
19. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
Selected quarterly financial data (unaudited) for the year ended December 31, 2015 and 2014 is presented below:
F-42
Notes to Consolidated Financial Statements
(all tabular amounts presented in thousands, except share, per share, per unit, and number of years)
Quarter Ended (Unaudited) | |||||||||||||||
March 31 | June 30 | September 30 | December 31 | ||||||||||||
2015 | |||||||||||||||
Total revenues | $ | 38,618 | $ | 44,982 | $ | 28,627 | $ | 50,329 | |||||||
Operating loss | $ | (21,661 | ) | $ | (34 | ) | $ | (16,074 | ) | $ | (2,963 | ) | |||
Net loss | $ | (25,562 | ) | $ | (2,346 | ) | $ | (18,724 | ) | $ | (4,153 | ) | |||
Net loss per share, basic | $ | (0.39 | ) | $ | (0.04 | ) | $ | (0.28 | ) | $ | (0.07 | ) | |||
Net loss per share, diluted | $ | (0.39 | ) | $ | (0.04 | ) | $ | (0.28 | ) | $ | (0.07 | ) | |||
2014 | |||||||||||||||
Total revenues | $ | 40,124 | $ | 46,855 | $ | 47,990 | $ | 51,861 | |||||||
Operating loss | $ | (24,414 | ) | $ | (1,396 | ) | $ | (4,127 | ) | $ | (1,632 | ) | |||
Net loss | $ | (27,641 | ) | $ | (3,563 | ) | $ | (11,539 | ) | $ | (2,976 | ) | |||
Net loss per share, basic | $ | (0.44 | ) | $ | (0.06 | ) | $ | (0.18 | ) | $ | (0.05 | ) | |||
Net loss per share, diluted | $ | (0.44 | ) | $ | (0.06 | ) | $ | (0.18 | ) | $ | (0.05 | ) | |||
Net loss per basic and diluted shares are computed independently for each of the quarters presented based on basic and diluted shares outstanding per quarter and, therefore, may not sum to the totals for the year.
20. SUBSEQUENT EVENTS
Out-License of ZEVALIN, FOLOTYN, BELEODAQ, and MARQIBO in Canada
On January 8, 2016, we entered into a strategic partnership with Servier Canada, Inc. for the out-licenses of ZEVALIN, FOLOTYN, BELEODAQ, and MARQIBO. We will receive $6 million in upfront payments, development milestone payments if/when achieved, and a high single-digit royalty on their sales of these products.
FDA Approval of EVOMELA
On March 10, 2016, the FDA communicated its approval of our NDA for EVOMELA. Upon receipt of FDA approval, we are contractually obligated to make a $6.0 million milestone payment to Ligand Pharmaceuticals Incorporated within 30 days (see Note 10(b)); accordingly, this payment is due no later than April 9, 2016.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None
Item 9A. Controls and Procedures
Our principal executive officer and principal financial officer have provided certifications filed as Exhibits 31.1 and 32.1, and 31.2, and 32.2, respectively. Such certifications should be read in conjunction with the information contained in this Item 9A for a more complete understanding of the matters covered by those certifications.
(a) Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rule 13a-15(f) of the Securities Exchange Act of 1934 (the “Exchange Act”). Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of the financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles. This process includes those policies and procedures (i) that pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) that receipts and expenditures are being made only in accordance with authorizations of our management and directors; (iii) that provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on
F-43
our financial statements; and (iv) that provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of the internal control over financial reporting to future periods are subject to risk that the internal control may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate. Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2015. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 framework) (“2013 COSO”).
Based on our management’s assessment, we have concluded that as of December 31, 2015, our internal control over financial reporting was effective, as evaluated under the 2013 COSO criteria. Our independent registered public accounting firm, Deloitte & Touche LLP, has issued a report on our internal control over financial reporting. Deloitte & Touche LLP’s report appears within Item 9A in this Annual Report on Form 10-K and expresses an unqualified opinion on the effectiveness of our internal control over financial reporting.
(b) Disclosure Controls and Procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures as of December 31, 2015, pursuant to Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures, as of such date, were effective.
(c) Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting during the fiscal fourth quarter of the year ended December 31, 2015 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
F-44
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Spectrum Pharmaceuticals, Inc.
Henderson, Nevada
We have audited the internal control over financial reporting of Spectrum Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2015, based on the criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed by, or under the supervision of, the company’s principal executive and principal financial officers, or persons performing similar functions, and effected by the company’s board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on the criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements and financial statement schedule as of and for the year ended December 31, 2015 of the Company and our report dated March 14, 2016 expressed an unqualified opinion on those consolidated financial statements and financial statement schedule.
/s/ Deloitte & Touche LLP
Costa Mesa, California
March 14, 2016
F-45
Item 9B. Other Information
None.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required under this item is incorporated by reference from our definitive proxy statement related to our 2016 Annual Meeting of Stockholders, or the Proxy Statement, to be filed pursuant to Regulation 14A, on or before April 29, 2016.
Item 11. Executive Compensation
The information required under this item is incorporated herein by reference from the Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required under this item is incorporated herein by reference from the Proxy Statement.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required under this item is incorporated herein by reference from the Proxy Statement.
Item 14. Principal Accountant Fees and Services
The information required under this item is incorporated herein by reference from the Proxy Statement.
F-46
Part IV
Item 15. Exhibits and Financial Statement Schedules
(a) | Financial Statements and Schedules |
The following financial statements and schedules listed below are included in this Annual Report on Form 10-K:
Financial Statements
Reports of Independent Registered Public Accounting Firms
Consolidated Balance Sheets as of December 31, 2015 and 2014
Consolidated Statements of Operations for the years ended December 31, 2015, 2014, and 2013
Consolidated Statements of Comprehensive Loss for the years ended December 31, 2015, 2014, and 2013
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2015, 2014, and 2013
Consolidated Statements of Cash Flows for the years ended December 31, 2015, 2014, and 2013
Notes to the Consolidated Financial Statements
Schedule II – Valuation and Qualifying Accounts for the years ended December 31, 2015, 2014, and 2013
(All other schedules are omitted, as required information is either not applicable or the information is presented in the consolidated financial statements).
F-47
SCHEDULE II – VALUATION AND QUALIFYING ACCOUNTS
Years Ended December 31, 2015, 2014, and 2013
Additions (Reductions) | |||||||||||||||||||
Description | Balance at Beginning of Period | Additions (Recovery) to Bad Debt Expense | Charged to Other Accounts | Deductions (1) | Balance at End of Period | ||||||||||||||
(in thousands) | |||||||||||||||||||
December 31, 2015 | |||||||||||||||||||
Allowance for doubtful accounts | $ | 120 | $ | — | $ | — | $ | — | $ | 120 | |||||||||
December 31, 2014 | |||||||||||||||||||
Allowance for doubtful accounts | $ | 206 | $ | (85 | ) | $ | — | $ | (1 | ) | $ | 120 | |||||||
December 31, 2013 | |||||||||||||||||||
Allowance for doubtful accounts | $ | 228 | $ | 11 | $ | — | $ | (33 | ) | $ | 206 | ||||||||
(1) | Deductions represent the actual write-off of accounts receivable balances. |
(b) Exhibits
The following is a list of exhibits required by Item 601 of Regulation S-K filed as part of this Annual Report on Form 10-K. For exhibits that previously have been filed, the Company incorporates those exhibits herein by reference. The exhibit table below includes the Form Type and Filing Date of the previous filing and the original exhibit number in the previous filing which is being incorporated by reference herein.
Exhibit No. | Description | |
2.1 | Asset Purchase Agreement, dated August 15, 2007, by and between Cell Therapeutics, Inc. and Biogen Idec Inc. (Filed as Exhibit 10.1 to Cell Therapeutics, Inc.’s Form 8-K, No. 001-12465, as filed with the Securities and Exchange Commission on August 21, 2007, and incorporated herein by reference.) | |
2.2 | First Amendment to Asset Purchase Agreement, dated December 9, 2008, by and between Cell Therapeutics, Inc. and Biogen Idec Inc. (Filed as Exhibit 10.48 to Cell Therapeutics, Inc.’s Form 10K, No. 001-12465, as filed with the Securities and Exchange Commission on March 16, 2009, and incorporated herein by reference.) | |
2.3# | License and Asset Purchase Agreement, dated January 23, 2012, by and between Spectrum Pharmaceuticals Cayman, L.P. and Bayer Pharma AG. (Filed as Exhibit 2.1 to Form 10-Q, No. 001-35006, as filed with the Securities and Exchange Commission on April 27, 2012, and incorporated herein by reference.) | |
2.4 | Agreement and Plan of Merger, dated as of April 4, 2012, by and among Spectrum Pharmaceuticals, Inc., Sapphire Acquisition Sub, Inc. and Allos Therapeutics, Inc., including a Form of Contingent Value Rights Agreement and a Form of Tender and Voting Agreement. (Filed as Exhibits 2.1, 2.2 and 2.3, respectively, to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on April 5, 2012, and incorporated herein by reference.) | |
2.5 | Securities Purchase Agreement, dated July 16, 2013, by and among Spectrum Pharmaceuticals, Inc., Eagle Acquisition Merger Sub, Inc., certain entities affiliated with Warburg Pincus & Co. and certain entities affiliated with Deerfield Management, LLC. (Filed as Exhibit 2.1 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on July 19, 2013, and incorporated herein by reference.) | |
2.6 | Stock Purchase Agreement, dated July 16, 2013, by and among Spectrum Pharmaceuticals, Inc., Eagle Acquisition Merger Sub, Inc. and Talon Therapeutics, Inc. (Filed as Exhibit 2.2 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on July 19, 2013, and incorporated herein by reference.) | |
2.7 | Contingent Value Rights Agreement, dated July 16, 2013, by and among Spectrum Pharmaceuticals, Inc., Talon Therapeutics, Inc. and Corporate Stock Transfer Inc. as rights agent. (Filed as Exhibit 2.3 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on July 19, 2013, and incorporated herein by reference.) | |
F-48
2.8 | Exchange Agreement, dated July 16, 2013, by and among Talon Therapeutics, Inc. and certain entities affiliated with Deerfield Management, LLC, including the Registration Rights Agreement by and among Spectrum Pharmaceuticals, Inc. and certain entities affiliated with Deerfield Management, LLC, as Exhibit A thereto. (Filed as Exhibit 2.4 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on July 19, 2013, and incorporated herein by reference.) | |
3.1 | Certificate of Incorporation, as amended through June 24, 2011. (Filed as Exhibit 3.1 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 2, 2012, and incorporated herein by reference.) | |
3.2 | Second Amended and Restated Bylaws of Spectrum Pharmaceuticals, Inc. (Filed as Exhibit 3.2 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on August 8, 2012, and incorporated herein by reference.) | |
4.1 | Rights Agreement, dated as of December 13, 2010, between Spectrum Pharmaceuticals, Inc. and ComputerShare Trust Company, N.A. (formerly U.S. Stock Transfer Corporation), as Rights Agent, which includes as Exhibit A thereto the form of Certificate of Designation for the Series B Junior Participating Preferred Stock, as Exhibit B thereto the Form of Rights Certificate and as Exhibit C thereto a Summary of Rights of Stockholder Rights Plan. (Filed as Exhibit 4.1 to Form 8-K, No. 000-28782, as filed with the Securities and Exchange Commission on December 13, 2010, and incorporated herein by reference.) | |
4.2 | Registration Rights and Stockholder Agreement, dated as of February 2, 2010, by and between Spectrum Pharmaceuticals, Inc. and Topotarget A/S. (Filed as Exhibit 4.2 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 12, 2014, and incorporated herein by reference.) | |
4.3 | Indenture, dated as of December 23, 2013, by and between Spectrum Pharmaceuticals, Inc. and Wilmington Trust, National Association, dated as of December 23, 2013. (Filed as Exhibit 4.1 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
4.4 | Form of Note for Spectrum Pharmaceuticals, Inc.’s 2.75% Convertible Senior Notes due 2018. (Filed as Exhibit 4.2 to Form 8-K, No. 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
10.1 | Sublease Agreement, dated as of December 2, 2010, between Spectrum Pharmaceuticals, Inc. and Del Webb Corporation. (Filed as Exhibit 10.1 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 10, 2011, and incorporated herein by reference.) | |
10.2 | First Amendment to Sublease Agreement, dated November 16, 2011, between Spectrum Pharmaceuticals, Inc. and Del Webb Corporation. (Filed as Exhibit 10.2 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 2, 2012, and incorporated herein by reference.) | |
10.3 | Second Amendment to Sublease Agreement, dated November 12, 2012, between Spectrum Pharmaceuticals, Inc. and Del Webb Corporation. (Filed as Exhibit 10.10 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on February 28, 2013, and incorporated herein by reference.) | |
10.4 | Industrial Lease Agreement, dated as of January 16, 1997, between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.11 to Form 10-KSB, No. 000-28782, as filed with the Securities and Exchange Commission on March 31, 1997, and incorporated herein by reference.) | |
10.5 | First Amendment, dated March 25, 2004, to Industrial Lease Agreement dated as of January 16, 1997 by and between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.1 to Form 10-Q, No. 000-28782, as filed with the Securities and Exchange Commission on May 17, 2004, and incorporated herein by reference.) | |
10.6 | Second Amendment, dated March 7, 2006, to Industrial Lease Agreement dated as of January 16, 1997 by and between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.6 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 12, 2014, and incorporated herein by reference.) | |
10.7 | Third Amendment, dated February 12, 2006, to Industrial Lease Agreement dated as of January 16, 1997 by and between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.7 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 12, 2014, and incorporated herein by reference.) | |
10.8 | Fourth Amendment, dated July 29, 2009, to Industrial Lease Agreement dated as of January 16, 1997 by and between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.29 to Form 10-K, 000-28782, as filed with the Securities and Exchange Commission on April 5, 2010, and incorporated herein by reference.) | |
F-49
10.9 | Fifth Amendment, dated November 21, 2013, to Industrial Lease Agreement dated as of January 16, 1997 by and between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.9 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 12, 2014, and incorporated herein by reference.) | |
10.10 | Sixth Amendment, dated January 31, 2014, to Industrial Lease Agreement dated as of January 16, 1997 by and between Spectrum Pharmaceuticals, Inc. and the Irvine Company. (Filed as Exhibit 10.10 to Form 10-K, No. 001-35006, as filed with the Securities and Exchange Commission on March 12, 2014, and incorporated herein by reference.) | |
10.11 | Lease Agreement, dated April 7, 2014, by and between Spectrum Pharmaceuticals, Inc. and 11500 South Eastern Avenue, LLC. (Filed as Exhibit 10.1 to Form 10-Q, No. 001-35006, as filed with the Securities and Exchange Commission on August 8, 2014 and incorporated herein by reference.) | |
10.12 | Preferred Stock and Warrant Purchase Agreement, dated as of September 26, 2003, by and among Spectrum Pharmaceuticals, Inc. and the purchasers listed on Schedule 1 attached thereto. (Filed as Exhibit 10.1 to Form 8-K, 000-28782, as filed with the Securities and Exchange Commission on September 30, 2003, and incorporated herein by reference.) | |
10.13* | Form of Stock Option Agreement under the 2003 Amended and Restated Incentive Award Plan. (Filed as Exhibit 10.1 to Form 8-K, 000-28782, as filed with the Securities and Exchange Commission on December 17, 2004, and incorporated herein by reference.) | |
10.14* | Form of Non-Employee Director Stock Option Agreement under the 2003 Amended and Restated Incentive Award Plan. (Filed as Exhibit 10.5 to Form 10-Q, 000-28782, as filed with the Securities and Exchange Commission on May 10, 2005, and incorporated herein by reference.) | |
10.15* | 2003 Amended and Restated Incentive Award Plan. (Filed as Exhibit 10.1 to Form 8-K, 000-28782, as filed with the Securities and Exchange Commission on July 2, 2009, and incorporated herein by reference.) | |
10.16* | Amendment No. 1 to 2003 Amended and Restated Incentive Award Plan. (Filed as Exhibit 10.1 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 6, 2015, and incorporated herein by reference.) | |
10.17* | Long-Term Retention and Management Incentive Plan. (Filed as Exhibit 10.36 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on August 4, 2011, and incorporated herein by reference.) | |
10.18* | Deferred Compensation Plan (Filed as Exhibit 4.1 to Form S-8, Reg. No. 333-176681, as filed with the Securities and Exchange Commission on September 6, 2011, and incorporated herein by reference). | |
10.19* | Executive Employment Agreement by and between Spectrum Pharmaceuticals, Inc. and Rajesh C. Shrotriya, M.D., entered into June 20, 2008 and effective as of January 2, 2008. (Filed as Exhibit 10.1 to Form 8-K, 000-28782, as filed with the Securities and Exchange Commission on June 26, 2008, and incorporated herein by reference.) | |
10.20* | First Amendment to Executive Employment Agreement, dated as of April 17, 2014, by and between Spectrum Pharmaceuticals, Inc. and Dr. Rajesh C. Shrotriya. (Filed as Exhibit 10.2 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on August 8, 2014 and incorporated herein by reference. | |
10.21* | Form of Change in Control Severance Agreement. (Filed as Exhibit 10.1 to Form 8-K, 001-35006 as filed with the Securities and Exchange Commission on March 31, 2014, and incorporated herein by reference.) | |
10.22+* | First Amendment to Change in Control Severance Agreement, dated February 18, 2015, by and between Spectrum Pharmaceuticals, Inc. and Joseph W. Turgeon. | |
10.23* | Second Amendment to Change in Control Severance Agreement, dated August 6, 2015, by and between Spectrum Pharmaceuticals, Inc. and Joseph W. Turgeon. (Filed as Exhibit 10.2 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on August 7, 2015, and incorporated herein by reference.) | |
10.24* | Form of Indemnity Agreement of Spectrum Pharmaceuticals, Inc. (Filed as Exhibit 10.32 to Form 10-K, 000-28782, as filed with the Securities and Exchange Commission on March 31, 2009, and incorporated herein by reference.) | |
10.25* | 2009 Employee Stock Purchase Plan. (Filed as Exhibit 99.1 to Form S-8, Reg. No. 333-160312, as filed with the Securities and Exchange Commission on June 29, 2009, and incorporated herein by reference.) | |
10.26* | 2009 Incentive Award Plan. (Filed as Exhibit 99.2 to Form S-8, Reg. No. 333-160312, as filed with the Securities and Exchange Commission on June 29, 2009, and incorporated herein by reference.) | |
F-50
10.27* | Term Sheet for 2009 Incentive Award Plan Stock Option Award. (Filed as Exhibit 10.8 to Form 10-Q, 000-28782, as filed with the Securities and Exchange Commission on August 13, 2009, and incorporated herein by reference.) | |
10.28* | Term Sheet for 2009 Incentive Award Plan, Nonqualified Stock Option Award Awarded to Non-Employee Directors (Revised July 2012). (Filed as Exhibit 10.2 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.29* | Term Sheet for 2009 Incentive Award Plan, Restricted Stock Award. (Filed as Exhibit 10.10 to Form 10-Q, 000-28782, as filed with the Securities and Exchange Commission on August 13, 2009, and incorporated herein by reference.) | |
10.30* | Amendment No. 1 to 2009 Incentive Award Plan. (Filed as Exhibit 10.2 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 6, 2015, and incorporated herein by reference.) | |
10.31# | License and Collaboration Agreement, dated February 2, 2010, by and between Spectrum Pharmaceuticals, Inc. and Topotarget A/S. (Filed as Exhibit 10.37 to Form 10-K, 000-28782, as filed with the Securities and Exchange Commission on April 5, 2010, and incorporated herein by reference.) | |
10.32# | Amendment to License and Collaboration Agreement, dated October 3, 2013, by and between Spectrum Pharmaceuticals, Inc. and Topotarget A/S. (Filed as Exhibit 99.1 to Form 8-K/A, 001-35006, as filed with the Securities and Exchange Commission on November 18, 2013, and incorporated herein by reference.) | |
10.33# | License Agreement for 10-Propargyl-10-Deazaaminopterin “PDX” dated December 23, 2002 and amended May 9, 2006 between Allos Therapeutics, Inc. and SRI International, Sloan-Kettering Institute for Cancer Research and Southern Research Institute. (Filed as Exhibit 10.1 to Allos Therapeutics, Inc.’s Form 10-Q/A, File No. 000-29815, as filed with the Securities and Exchange Commission on August 17, 2012, and incorporated herein by reference.) | |
10.34# | Second Amendment to License Agreement for 10-Propargyl-10-Deazaaminopterin “PDX” dated November 6, 2007 between Allos Therapeutics, Inc. and SRI International, Sloan-Kettering Institute for Cancer Research and Southern Research Institute. (Filed as Exhibit 10.13.1 to Allos Therapeutics, Inc.’s Form 10-K, File No. 000-29815, as filed with the Securities and Exchange Commission on March 1, 2010, and incorporated herein by reference.) | |
10.35# | Third Amendment to License Agreement for 10-Propargyl-10-Deazaaminopterin “PDX” dated May 10, 2011 between Allos Therapeutics, Inc. and SRI International, Sloan-Kettering Institute for Cancer Research and Southern Research Institute. (Filed as Exhibit 10.3 to Allos Therapeutics, Inc.’s Form 10-Q, File No. 000-29815, as filed with the Securities and Exchange Commission on August 4, 2011, and incorporated herein by reference.) | |
10.36# | Amended and Restated License, Development and Commercialization Agreement, dated May 29, 2013, by and between Allos Therapeutics, Inc. and Mundipharma International Corporation Limited (Filed as Exhibit 10.1 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on August 9, 2013, and incorporated herein by reference.) | |
10.37# | First Amendment to Amended and Restated License, Development and Commercialization Agreement, dated May 29, 2015, by and between Allos Therapeutics, Inc. and Mundipharma International Corporation Limited. (Filed as Exhibit 10.1 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on August 7, 2015, and incorporated herein by reference.) | |
10.38# | Amended and Restated Supply Agreement, dated May 29, 2013, by and between Allos Therapeutics, Inc. and Mundipharma Medical Company (Filed as Exhibit 10.2 to Form 10-Q, File No. 001-35006, as filed with the Securities and Exchange Commission on August 9, 2013, and incorporated herein by reference.) | |
10.39 | License Agreement, dated December 21, 2007, by and between Biogen Idec Inc. and Cell Therapeutics, Inc. (Filed as Exhibit 10.8 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.40 | License-Back Agreement, dated December 21, 2007, by and between Biogen Idec Inc. and Cell Therapeutics, Inc. (Filed as Exhibit 10.9 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.41# | Sublicense Agreement, dated December 21, 2007, by and among Cell Therapeutics, Inc., Biogen Idec Inc., SmithKline Beecham Corporation d/b/a GlaxoSmithKline and Glaxo Group Limited. (Filed as Exhibit 10.11 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
F-51
10.42# | Sublicense Agreement, dated December 21, 2007, by and among Cell Therapeutics, Inc., Biogen Idec Inc., Corixa Corporation, Coulter Pharmaceutical, Inc., The Regents of the University of Michigan and SmithKline Beecham Corporation d/b/a GlaxoSmithKline. (Filed as Exhibit 10.12 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.43 | Security Agreement, dated December 15, 2008, by and between RIT Oncology, LLC and Biogen Idec Inc. (Filed as Exhibit 10.35 to Form 10-K, 001-35006, as filed with the Securities and Exchange Commission on March 10, 2011, and incorporated herein by reference.) | |
10.44# | Omnibus Amendment to Zevalin Supply Arrangements, dated October 1, 2012, by and between Biogen Idec US Corporation and RIT Oncology, LLC, a wholly-owned subsidiary of Spectrum Pharmaceuticals, Inc.. (Filed as Exhibit 10.14 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.45 | License Agreement, dated May 23, 2006, by and between Merck Eprova AG and Spectrum Pharmaceuticals, Inc. (Filed as Exhibit 10.16 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.46 | First Amendment to License Agreement, dated as of June 20, 2014, by and between Spectrum Pharmaceuticals, Inc. and Merck Eprova AG. (Filed as Exhibit 99.1 to Form 8-K, 001-35006, as filed with the Securities and Exchange Commission on June 26, 2014, and incorporated herein by reference). | |
10.47 | Manufacturing and Supply Agreement, dated May 23, 2006, by and between Merck Eprova AG and Spectrum Pharmaceuticals, Inc. (Filed as Exhibit 10.17 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on November 9, 2012, and incorporated herein by reference.) | |
10.48# | License Agreement, dated March 8, 2013, by and between Spectrum Pharmaceuticals, Inc. and CyDex Pharmaceuticals, Inc. (Filed as Exhibit 10.1 to Form 10-Q, 001-35006, as filed with the Securities and Exchange Commission on May 9, 2013, and incorporated herein by reference.) | |
10.49 | Purchase Agreement, dated as of December 17, 2013, by and among Spectrum Pharmaceuticals, Inc., Jefferies LLC and RBC Capital Markets, LLC. (Filed as Exhibit 10.1 to Form 8-K, 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
10.50 | Base Call Option Transaction Confirmation, dated as of December 17, 2013, by and between Spectrum Pharmaceuticals, Inc. and Royal Bank of Canada. (Filed as Exhibit 10.2 to Form 8-K, 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
10.51 | Base Warrant Transaction Confirmation, dated as of December 17, 2013, by and between Spectrum Pharmaceuticals, Inc. and Royal Bank of Canada. (Filed as Exhibit 10.3 to Form 8-K, 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
10.52 | Additional Call Option Transaction Confirmation, dated as of December 20, 2013, by and between Spectrum Pharmaceuticals, Inc. and Royal Bank of Canada. (Filed as Exhibit 10.4 to Form 8-K, 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
10.53 | Additional Warrant Transaction Confirmation, dated as of December 20, 2013, by and between Spectrum Pharmaceuticals, Inc. and Royal Bank of Canada. (Filed as Exhibit 10.5 to Form 8-K, 001-35006, as filed with the Securities and Exchange Commission on December 23, 2013, and incorporated herein by reference.) | |
10.54+# | Co-Promotion Agreement between Eagle Pharmaceuticals, Inc. and Spectrum Pharmaceuticals, Inc., dated November 4, 2015. | |
10.55+# | License and Asset Purchase Agreement by and between Spectrum Pharmaceuticals Cayman, L.P. and Mundipharma International Corporation Limited, dated November 16, 2015. | |
10.56+# | Supply Agreement by and between Spectrum Pharmaceuticals Cayman, L.P. and Mundipharma Medical Company, dated November 16, 2015. | |
10.57 | At Market Issuance Sales Agreement dated as of December 23, 2015, by and among Spectrum Pharmaceuticals, Inc., FBR Capital Markets & Co., MLV & Co. LLC and H.C. Wainwright & Co., LLC. (Filed as Exhibit 1.2 to Form S-3, Reg. No. 333-208760, as filed with the Securities and Exchange Commission on December 23, 2015, and incorporated herein by reference.) | |
21.1+ | Subsidiaries of Registrant. | |
23.1+ | Consent of Independent Registered Public Accounting Firm (Deloitte & Touche LLP). | |
23.2+ | Consent of Independent Registered Public Accounting Firm (Ernst & Young LLP). | |
24.1 | Power of Attorney (included in the signature page.) | |
F-52
31.1+ | Certification of Principal Executive Officer, pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934. | |
31.2+ | Certification of Principal Financial Officer, pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934. | |
32.1+ | Certification of Principal Executive Officer, pursuant to Rule 13a-14(b)/15d-14(b) of the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. | |
32.2+ | Certification of Principal Financial Officer, pursuant to Rule 13a-14(b)/15d-14(b) of the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. | |
101.INS | XBRL Instance Document | |
101.SCH | XBRL Taxonomy Extension Schema Document | |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |
* Indicates a management contract or compensatory plan or arrangement.
# Confidential portions omitted and filed separately with the U.S. Securities and Exchange Commission pursuant to Rule 24b-2 promulgated under the Securities Exchange Act of 1934, as amended.
+ Filed herewith.
F-53