UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended
December 31, 2010
|
|
|
|
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
Commission File Number:
001-35006
SPECTRUM PHARMACEUTICALS,
INC.®
(Exact Name of Registrant as
Specified in its Charter)
| |
|
|
|
Delaware
|
|
93-0979187
|
(State or other jurisdiction
of
incorporation or organization)
|
|
(I.R.S. Employer
Identification No.)
|
|
|
11500 South Eastern Avenue, Suite 240
Henderson, Nevada 89052
(Address of principal executive
offices)
|
(702) 260-7421
(Registrant’s telephone
number, including area code)
Securities
registered pursuant to Section 12(b) of the Act:
| |
|
|
|
Title of Each Class
|
|
Name of Each Exchange on Which
Registered
|
|
Common Stock, $0.001 par value
|
|
The NASDAQ Stock Market, LLC
|
|
Common Stock Purchase Warrants
|
|
|
|
Rights to Purchase Series B Junior Participating Preferred
Stock
|
|
|
Securities registered pursuant
to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
(§ 229.405 of this chapter) during the preceding
12 months (or for such shorter period that the registrant
was required to submit and post such
files). Yes o No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K
o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in
Rule 12b-2
of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large accelerated
filer o
|
|
Accelerated
filer þ
|
|
Non-accelerated
filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting
company o
|
Indicate by check mark whether the Registrant is a shell company
(as defined in
Rule 12b-2
of the Exchange
Act). Yes o No þ
The aggregate market value of the voting and non-voting common
equity held by non-affiliates of the registrant as of
June 30, 2010 was $189,068,452 based on the closing sale
price of such common equity on such date.
As of February 25, 2011 there were 52,003,514 shares
of the registrant’s common stock were outstanding.
DOCUMENTS
INCORPORATED BY REFERENCE
None
FORWARD-LOOKING
STATEMENTS
Spectrum Pharmaceuticals, Inc.’s Annual Report on
Form 10-K
contains certain forward-looking statements. These
forward-looking statements involve a number of risks and
uncertainties. These forward-looking statements can generally be
identified as such because the context of the statement will
include certain words, including but not limited to,
“believes,” “may,” “will,”
“expects,” “intends,” “estimates,”
“anticipates,” “plans,” “seeks,”
“continues,” “predicts,”
“potential,” “likely,” or
“opportunity,” and also contains predictions,
estimates and other forward-looking statements within the
meaning of Section 27A of the Securities Act of 1933, as
amended, and Section 21E of the Securities Exchange Act of
1934, as amended, and in reliance upon the safe harbor
provisions of the Private Securities Litigation Reform Act of
1995. Such forward-looking statements are based on the current
beliefs of the Company’s management, as well as assumptions
made by and information currently available to the
Company’s management. Readers of this Annual Report on
Form 10-K
should not put undue reliance on these forward-looking
statements, which speak only as of the time this Annual Report
on
Form 10-K
was filed with the Securities and Exchange Commission, or SEC.
Reference is made in particular to forward-looking statements
regarding the success, safety and efficacy of our drug products,
product approvals, product sales, revenues, development
timelines, product acquisitions, liquidity and capital resources
and trends. Forward-looking statements are inherently subject to
risks and uncertainties, some of which cannot be predicted or
quantified. Spectrum Pharmaceuticals, Inc.’s actual results
may differ materially from the results projected in the
forward-looking statements. Factors that might cause such a
difference include, but are not limited to, those discussed in
this Report, including the “Risk Factors” in
“Item 1A — Risk Factors”, and in
“Item 7 — Management’s Discussion and
Analysis of Financial Condition and Results of Operations”
included in Part II. In addition, past financial or
operating performance is not necessarily a reliable indicator of
future performance, and you should not use our historical
performance to anticipate results or future period trends. We
can give no assurances that any of the events anticipated by the
forward-looking statements will occur or, if any of them do,
what impact they will have on our results of operations and
financial condition. Except as required by law, we do not
undertake to update any such forward-looking statements and
expressly disclaim any duty to update the information contained
in this Annual Report on
Form 10-K.
Unless the context otherwise requires, all references in this
Annual Report on
Form 10-K
to the “Company”, “we,” “us,”
“our,” “Spectrum” and “Spectrum
Pharmaceuticals” refer to Spectrum Pharmaceuticals, Inc.
and its subsidiaries and other consolidated entities, as a
consolidated entity. We primarily conduct all our activities as
Spectrum Pharmaceuticals.
Spectrum Pharmaceuticals,
Inc.®,
Fusilev®,
Zevalin®
and
RenaZorb®
are registered trademarks of Spectrum Pharmaceuticals, Inc. and
its subsidiaries. Redefining Cancer
Caretm,
Turning Insights Into
Hopetm,
RIT Oncology,
LLCtm,
RITtm,
and our logos are trademarks owned by Spectrum Pharmaceuticals,
Inc. and its subsidiaries.
EOquin®
is a registered trademark of Allergan, Inc. All other trademarks
and trade names are the property of their respective owners.
3
PART I
Overview
We are a biotechnology company with fully integrated commercial
and drug development operations with a primary focus in
oncology. Our strategy is comprised of acquiring, developing and
commercializing a broad and diverse pipeline of late-stage
clinical and commercial products. We market two oncology drugs,
ZEVALIN®
and
FUSILEV®
and have two drugs, apaziquone and belinostat, in late stage
development along with a diversified pipeline of novel drug
candidates. We have assembled an integrated in-house scientific
team, including formulation development, clinical development,
medical research, regulatory affairs, biostatistics and data
management, and have established a commercial infrastructure for
the marketing of our drug products. We also leverage the
expertise of our worldwide partners to assist in the execution
of our strategy. Apaziquone is presently being studied in two
large Phase 3 clinical trials for non-muscle invasive bladder
cancer, or NMIBC, under strategic collaborations with Allergan,
Inc., or Allergan, Nippon Kayaku Co. Ltd., orNippon Kayaku, and
Handok Pharmaceuticals Co. Ltd., or Handok. Belinostat, is being
studied in multiple indications including a Phase 2
registrational trial for relapsed or refractory peripheral
T-cell lymphoma, or PTCL, under a strategic collaboration with
TopoTarget A/S or TopoTarget.
Our business strategy is comprised of the following initiatives:
|
|
|
| |
•
|
Maximizing the growth potential of our marketed drugs,
Zevalin and Fusilev. Our near-term outlook
largely depends on sales and marketing successes for our two
marketed drugs. For Zevalin, we stabilized sales in 2009,
increased sales in 2010 and believe we can continue to grow
sales in 2011 and beyond. For Fusilev, which we launched in
August 2008, we were able to benefit from broad utilization in
community clinics and hospitals and recognized a dramatic
increase in sales during 2010 due to a shortage of generic
leucovorin. While we cannot predict how long the shortage may
continue, our focus now is to obtain approval for Fusilev in
advanced metastatic colorectal cancer. As part of its review of
our supplemental new drug application (sNDA) for metastatic
colorectal cancer, the FDA requested additional data to which we
submitted a response on October 29, 2010. The FDA formally
accepted the submission and established a decision date (PDUFA)
of April 29, 2011.
|
For both Zevalin and Fusilev, we initiated and continue to stage
appropriate infrastructure expansions and additional initiatives
to facilitate broad customer reach and to address other market
requirements, as appropriate. We have formed a dedicated
commercial organization comprised of highly experienced and
motivated sales representatives, account managers, and a
complement of other support marketing personnel to manage the
sales and marketing of these drugs. In addition our scientific
department supports field activities through various MDs, PhDs
and other medical science liaison personnel.
|
|
|
| |
•
|
Optimizing our development portfolio and maximizing the asset
values of its components. While over the recent
few years, we have evolved from a development-stage to a
commercial-stage pharmaceutical company, we have maintained a
highly focused development portfolio. Our strategy with regard
to our development portfolio is to focus on late-stage drugs and
to develop them rapidly to the point of regulatory approval. We
plan to develop some of these drugs ourselves or with our
subsidiaries and affiliates, or secure collaborations such that
we are able to suitably monetize these assets.
|
We have assembled a drug development infrastructure that is
comprised of highly experienced and motivated MDs, PhDs,
clinical research associates and a complement of other support
personnel to rapidly develop these drugs. During 2009, this team
achieved our goal of completing enrollment in the two Phase 3
apaziquone trials (with more than 1,600 patients enrolled).
We expect to continue to maximize the value of apaziquone
through further developmental efforts and initiation of
additional trials.
We have several other exciting compounds in earlier stages of
development in our portfolio. Based upon a criteria-based
portfolio review, we are in the process of streamlining our
pipeline drugs, allowing for greater focus and integration of
our development and commercial goals.
4
|
|
|
| |
•
|
Expanding our pipeline of late stage and commercial drugs
through licensing and business development. It is
our goal to identify new strategic opportunities that will
create strong synergies with our currently marketed drugs and
identify and pursue partnerships for out-licensing certain of
our drugs in development. To this end, we will continue to
explore strategic collaborations as these relate to drugs that
are either in advanced clinical trials or are currently on the
market. We believe that such opportunistic collaborations will
provide synergies with respect to how we deploy our internal
resources. In this regard, we intend to identify and secure
drugs that have significant growth potential either through
enhanced marketing and sales efforts or through pursuit of
additional clinical development. We believe our in-licensing of
belinostat, a novel histone deacetylase (HDAC) inhibitor, is
demonstrative of such licensing and business development efforts
outlined above.
|
| |
| |
•
|
Managing our financial resources
effectively. We remain committed to fiscal
discipline, a policy which has allowed us to become well
capitalized among our peers, despite a very challenging capital
markets environment during 2009 and continuing through 2010.
This policy includes the pursuit of non-dilutive funding
options, prudent expense management, and the achievement of
critical synergies within our operations in order to maintain a
reasonable burn rate. Even with the continued
build-up in
operational infrastructure to facilitate the marketing of our
two commercial drugs, we intend to be fiscally prudent in any
expansion we undertake. In terms of revenue generation, we plan
to become more reliant on sales from currently marketed drugs
and intend to pursue out-licensing of select pipeline drugs in
select territories, as discussed above. When appropriate, we may
pursue other sources of financing, including non-dilutive
financing alternatives. While we are currently focused on
advancing our key drug development programs, we anticipate that
we will make regular determinations as to which other programs,
if any, to pursue and how much funding to direct to each program
on an ongoing basis, based on clinical success and commercial
potential, including termination of our existing development
programs, especially if we do not expect value being driven from
continued development. We ended 2010 with over $100 million
in cash, cash equivalents and investments which was net of the
$30 million license fee paid for belinostat in early 2010
offset by cash received from out-license of apaziquone of
approximately $17.5 million.
|
| |
| |
•
|
Further enhancing the organizational structure to meet our
corporate objectives. We have highly experienced
staff in pharmaceutical operations, clinical development,
regulatory and commercial functions who previously held
positions at both small to mid-size biotech companies, as well
as large pharmaceutical companies. We have strengthened the
ranks of our management team, and will continue to pursue talent
on an opportunistic basis. Finally, we remain committed to
running a lean and efficient organization, while effectively
leveraging our critical resources.
|
Recent
Developments
In 2010 and early 2011, we have continued to execute on our
business strategy described above. We discuss below the key
developments during that period.
We recorded approximately $61 million in sales of our
products for the year 2010. We successfully increased Zevalin
sales to approximately $29 million in 2010 as compared to
approximately $16 million in 2009. We also recorded Fusilev
sales of $32 million in 2010, compared to approximately
$13 million in 2009. We believe that in 2011 and beyond,
revenues from these products have the potential to grow. However
future growth in Fusilev revenue will depend largely on its
approval by the FDA for metatstatic colorectal cancer indication
(expected on April 29, 2011) and or a continuing
shortage of leucovorin.
In September 2009, Zevalin received FDA approval in first-line
setting; expanding its label for the treatment of patients with
previously untreated follicular NHL who achieve a partial or
complete response to first-line chemotherapy. This new and
expanded indication supplements the 2002 FDA approval of Zevalin
as treatment for patients with relapsed or refractory, low-grade
or follicular B-cell NHL. Additionally, in November 2009, the
Centers for Medicare and Medicaid Services (CMS) decided that
Zevalin should be reimbursed under an Average Sales Price (ASP)
methodology in the Hospital Outpatient Prospective Payment
System (HOPPS) and issued a corresponding proposed rule, which
became effective on January 1, 2010. The ASP methodology is
widely used for
5
injectable chemotherapy drugs and creates a consistent
reimbursement standard in the hospital setting. Effective
January 1, 2011 Zevalin reimbursement rate increased from
ASP plus 4% to ASP plus 5%.
In December 2010 at the Annual Meeting of the American Society
of Hematology in Orlando, Florida, a total of 12 scientific
papers on Zevalin were presented. Of these, 6 papers were
selected by the program committee of ASH for oral presentations.
These included a five year update on the progression free
survival in the “FIT” (First-Line Indolent Trial),
Zevalin’s use in bone marrow conditioning regimen and the
first results from an international Phase 2 clinical trial using
Zevalin as a first line monotherapy treatment in certain newly
diagnosed patients with follicular lymphoma.
We have been working with the FDA to remove the requirement for
a bioscan prior to Zevalin administration and on
January 20, 2011 we submitted a Post Approval Supplement
(sBLA) with the FDA containing data supporting the removal of
the bioscan. The FDA has up to sixty (60) days to accept or
reject the submission.
In October 2009, the FDA issued a Complete Response letter
regarding the sNDA for Fusilev in metastatic colorectal cancer
indication. In the Complete Response letter, the FDA recommended
that we meet with them to discuss options for continuing to seek
approval of Fusilev in advanced metastatic colorectal cancer. We
promptly requested such a meeting, which occurred in January
2010. In that meeting, the FDA requested additional data which
we submitted on October 29, 2010. The FDA formally accepted
the submission and we expect a decision by the FDA (PDUFA date)
on April 29, 2011. In addition, on January 4, 2011, we
submitted a sNDA with the FDA for a
Ready-to-Use
formulation of Fusilev for Injection. This submission is in
support of Fusilev’s use in colorectal cancer and the FDA
has up to 60 days to formally accept or reject the
submission.
As for apaziquone, in November 2009, we entered into a
collaboration agreement with Nippon Kayaku Co. Ltd. for the
development and commercialization of apaziquone in Asia, with
the exception of North and South Korea. In exchange, Nippon
Kayaku paid Spectrum an up-front payment of $15 million and
agreed to make additional payments of up to $136.0 million
based on the achievement of certain regulatory and
commercialization milestones contained in the collaboration
agreement, as well as royalties on net sales. Nippon Kayaku
received exclusive rights to apaziquone for the treatment of
NMIBC in Asia, including Japan and China. Under the terms of the
Nippon Kayaku collaboration agreement, Nippon Kayaku will
conduct the apaziquone clinical trials pursuant to a development
plan, and will be responsible for all expenses relating to the
development and commercialization of apaziquone in the Nippon
Kayaku territory. As for South Korea, or the Republic of Korea,
and North Korea, or the Democratic People’s Republic of
Korea, (collectively, “Korea”), we entered into a
collaboration agreement with Handok Pharmaceuticals Co. Ltd. for
the development and commercialization of apaziquone for the
treatment of NMIBC. Under the terms of the Handok collaboration
agreement, Handok paid us an up-front payment of
$1.0 million and there are potential milestone payments of
approximately $19 million, as well as royalties on net
sales. The potential milestones will be based on the achievement
of certain regulatory and commercialization milestones.
Additionally, Handok will conduct the apaziquone clinical trials
pursuant to a development plan and will be responsible for all
expenses relating to the development and commercialization of
apaziquone in North and South Korea.
In the fourth quarter of 2009, we completed enrollment of two
Phase 3 pivotal clinical trials for apaziquone. The two trials
enrolled more than 1,600 patients with non-muscle invasive
bladder cancer and we expect top-line data in 2012. Per the
terms of the collaboration agreement, we received a
$1.5 million milestone payment in January 2010 from
Allergan for the completion of patient accrual in these clinical
trials.
In February 2010, we entered into a licensing and collaboration
agreement with TopoTarget, for the development and
commercialization of belinostat, a drug being studied in
multiple indications, including a Phase 2 registrational
trial for patients with relapsed/refractory PTCL. The licensing
and collaboration agreement provides that we have the exclusive
right to make, develop and commercialize belinostat in North
America and India, with an option for China. In consideration
for the rights granted under the licensing and collaboration
agreement, we paid TopoTarget an up-front fee of
$30 million. In addition, we will pay up to
$313 million and one million shares of Spectrum common
stock based on the achievement of certain development,
regulatory and sales milestones, as well as double-digit
royalties on net sales of belinostat.
6
In January 2011 we signed a letter of agreement with Viropro,
Inc., for the development of a biosimilar version of the
monoclonal antibody drug rituximab. Biosimilars, or follow-on
biologics, are terms used to describe officially-approved
subsequent versions of innovator biopharmaceutical products made
by a different sponsor following patent and exclusivity expiry.
We continued our efforts to build a global pharmaceutical
organization in 2010. For two of our non-US business entities,
Spectrum Pharma Canada, Inc., a Canadian affiliate headquartered
in the Province of Quebec, Canada, and OncoRx Pharma Private
Ltd., a wholly-owned Indian subsidiary headquartered in Mumbai,
India, we continued to grow and establish these entities in an
effort to facilitate the opening of clinical trials sites in
these countries to continue the clinical development of our
products at a reduced cost.
Product
Portfolio
We have a product portfolio consisting of both commercial stage
and development stage products. While we are committed to
growing the sales of our marketed products, we strive to
maintain a robust pipeline of products under development to
bring to the market.
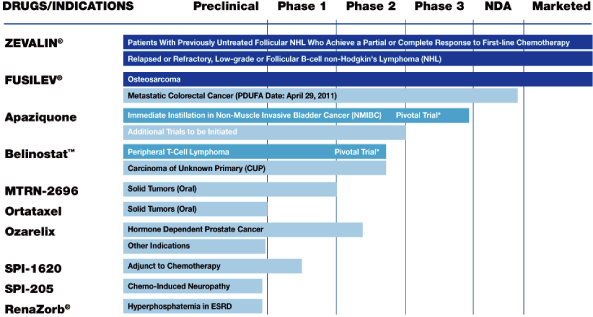
Our drug products, their approved
and/or
target indications, and status of development are summarized in
the following table, and discussed below in further detail:
|
|
|
|
*
|
|
Pivotal Trial under Special
Protocol Assessment (SPA) |
Some of our drugs may prove to be beneficial in additional
disease indications as we continue their study and development.
In addition, we have intellectual property rights to neurology
compounds that we may out-license to third parties for further
development.
Overview
of Cancer
According to the American Cancer Society’s publication
Cancer Facts & Figures 2010, cancer is the
second leading cause of death in the United States, accounting
for approximately 25% of all deaths. In the United States,
approximately 1.5 million new cancer cases were expected to
be diagnosed in 2010 and over 569,000 persons were
7
expected to die from the disease in 2010. Accordingly, there is
significant demand for improved and novel cancer treatments.
Cancer develops when cells in a part of the body begin to grow
out of control. Although there are many kinds of cancer, they
all start because of
out-of-control
growth of abnormal cells. Normal body cells grow, divide, and
die in an orderly fashion. During the early years of a
person’s life, normal cells divide more rapidly until the
person becomes an adult. After that, cells in most parts of the
body divide only to replace worn-out or dying cells and to
repair injuries. Because cancer cells continue to grow and
divide, they are different from normal cells. Instead of dying,
they outlive normal cells and continue to form new abnormal
cells.
Cancer cells develop because of damage to DNA. Most of
the time, when DNA becomes damaged, the body is able to repair
it. In cancer cells, the damaged DNA is not repaired. People can
inherit damaged DNA, which accounts for inherited cancers. More
often, however, a person’s DNA becomes damaged by exposure
to something in the environment, such as smoking.
Cancer usually forms as a tumor. Some cancers, like
leukemia, do not form tumors. Instead, these cancer cells
involve the blood and blood-forming organs and circulate through
other tissues where they grow. Often, cancer cells travel to
other parts of the body where they begin to grow and replace
normal tissue. This process is called metastasis. Regardless of
where a cancer may spread, however, it is always named for the
place it began. For instance, breast cancer that spreads to the
liver is still called breast cancer, not liver cancer.
Different types of cancer can behave very differently.
For example, lung cancer and breast cancer are very different
diseases. They grow at different rates and respond to different
treatments. That is why people with cancer need treatment that
is aimed at their particular kind of cancer. Cancer is currently
treated by surgery, chemotherapy, radiation therapy, hormonal
therapy, biological therapy and immunotherapy. Cancer is
referred to as refractory when it has not responded, or is no
longer responding, to a treatment.
We are seeking novel drugs that address cancer or cancer related
indications with significant unmet medical need, that:
|
|
|
| |
•
|
are already approved for sale or have demonstrated initial
safety and efficacy in clinical trials
and/or we
believe have a higher probability of regulatory approval than
that of a typical compound at a similar stage of development;
|
| |
| |
•
|
target cancer indications with significant unmet medical need,
where current treatments either do not exist or are not deemed
to be effective; and
|
| |
| |
•
|
we believe we can acquire at a fair value based on our judgment
of clinical success and commercial potential.
|
Our drug
products
Zevalin ([90Y]-ibritumomab
tiuxetan): In December 2008, we acquired
rights to commercialize and develop Zevalin in the United
States, as the result of a transaction with Cell Therapeutics,
Inc., orCTI as further described below.
Zevalin is a prescribed form of cancer therapy called
radioimmunotherapy. Radioimmunotherapy combines a source of
radiation, called a radioisotope, with an antibody. As part of
the Zevalin therapeutic regimen, the Y-90 radioisotope is
combined with a monoclonal antibody (CD20 MAB) that specifically
recognizes a particular part of a B-cell (the cells of the
immune system that make antibodies to invading pathogens) called
the CD20 antigen. The CD20 antigen is found on malignant and
normal B-cells. As the patient is infused with Y-90 Zevalin and
it enters the bloodstream, the antibody portion recognizes and
attaches to the CD20 antigen on tumor cells, allowing the
radiation energy emitted from the Y-90 radioisotope
(i.e., beta emission) to penetrate and damage the
malignant B-cells as well as nearby neighboring cells, many of
which are also lymphoma cells.
Zevalin was approved by the FDA in February of 2002 as the first
radioimmunotherapeutic agent for the treatment of NHL. Zevalin
was approved as part of a Zevalin therapeutic regimen for
treatment of relapsed or refractory, low-grade or follicular
B-cell NHL, including patients with rituximab-refractory
follicular NHL. For reference, the term refractory refers to
lymphoma that does not respond to a particular therapy. The term
relapsed
8
refers to lymphoma that returns after initially responding to
therapy. The terms low-grade and follicular refer to types of
lymphoma cells as determined by laboratory tests, which have an
indolent (slow growing) clinical course. Rituximab is a
monoclonal antibody that specifically recognizes a particular
part of a B-cell also called the CD 20 antigen, and is used as
monotherapy or in combination with other agents for the
treatment of B-cell NHL.
The current Zevalin therapeutic regimen also requires a bioscan
(also known as an “imaging study”) of the prospective
patient prior to treatment with Y-90 Zevalin. For the bioscan,
the patient is infused with In-111 Zevalin, the In-111
radioisotope combined with the CD20 MAB. In-111 Zevalin produces
a kind of radiation called gamma emission, which is very similar
to the kind of radiation used to produce x-rays. Once infused
with In-111, the prospective patient goes through a bioscan. The
bioscan allows a physician to follow In-111 Zevalin as it
travels within the prospective patient’s body. Based upon
the distribution of In-111 Zevalin (whether the In-111 Zevalin
goes to certain unintended areas of the body), the physician may
elect to not infuse the patient with Y-90 Zevalin. Many
healthcare providers throughout the world who provide Zevalin
therapy do not believe that the In-111 bioscan is a necessary
part of the Zevalin therapeutic regimen. In the EU, most
countries do not perform the In-111 bioscan prior to the Y-90
Zevalin infusion. On January 20, 2011, we submitted a Post
Approval Supplement with the FDA containing data supporting the
removal of the bioscan. The FDA has up to sixty (60) days
to accept or reject the submission.
NHL is caused by the abnormal proliferation of white blood cells
and normally spreads through the lymphatic system, a system of
vessels that drains fluid from the body. There are many
different types of NHL which can be divided into aggressive NHL,
a rapidly spreading acute form of the disease, and indolent NHL,
which progresses more slowly, and can be classified as either
B-cell or T-cell NHL. According to the National Cancer
Institute’s SEER database there were nearly
400,000 people in the U.S. with NHL in 2004. The
American Cancer Society estimated that in the United States
65,540 people were expected to be newly diagnosed with NHL
in 2010. Additionally, approximately 20,210 were expected to die
from this disease in 2010.
In December 2008, the FDA accepted for filing and review, and
granted priority review status for RIT Oncology, LLC’s or
RIT’s, supplemental biologics license application (sBLA)
for the use of Zevalin as first-line therapy for patients with a
previously untreated follicular NHL who achieve a partial or
complete response of first-line chemotherapy.
The sBLA was based upon data from the multinational, randomized
Phase 3 First-line Indolent Trial (FIT) which evaluated the
efficacy and safety of a single infusion of Zevalin in
414 patients with CD20-positive follicular NHL who had
achieved a partial response or a complete response after
receiving one of the standard first-line chemotherapy regimens.
The FIT trial demonstrated that when used as a first-line
consolidation therapy for patients with follicular NHL, Zevalin
significantly improved the median progression-free survival time
from 18 months (control arm) to 38 months (Zevalin
arm) (p<0.0001).
The primary investigators of the study concluded that Zevalin
consolidation of first remission in advanced stage follicular
NHL is highly effective, resulting in a total complete response
(CR + CRu) rate of 87 percent and prolongation of median
progression-free survival by almost two years, with a toxicity
profile comparable to that seen with Zevalin’s use in
relapsed or refractory indications. In September 2009, we
received FDA approval for the sBLA.
Additionally, in November 2009, the CMS decided that Zevalin
should be reimbursed under an ASP methodology in the HOPPS and
issued a corresponding proposed rule, which went into effect on
January 1, 2010. The ASP methodology is widely used for
injectable chemotherapy drugs and creates a consistent
reimbursement standard in the hospital setting.
In December 2010 at the Annual Meeting of the American Society
of Hematology in Orlando, Florida, a total of 12 scientific
papers on Zevalin were presented. Of these, 6 papers were
selected by the program committee of ASH for oral presentations.
These included a five year update on the progression free
survival in the “FIT” (First-Line Indolent Trial),
Zevalin’s use in bone marrow conditioning regimen and the
first results from an international Phase 2 clinical trial using
Zevalin as a first line monotherapy treatment in certain newly
diagnosed patients with follicular lymphoma.
9
The following describes the principal commercial terms relating
to Zevalin licensing and development:
|
|
|
| |
•
|
On December 15, 2008, we closed a transaction to enter into
a
50/50
owned joint venture called RIT, with CTI. CTI previously
acquired the U.S. rights to develop, market and sell
Zevalin from Biogen Idec, Inc. or Biogen on December 21,
2007.
|
| |
| |
•
|
Upon entering into the joint venture arrangement, CTI
contributed the Zevalin product assets to RIT in exchange for a
50% membership interest in RIT and the cash payments to CTI
noted below. CTI received an initial cash payment of
$7.5 million at the closing of the joint venture
transaction on December 15, 2008, and received an
additional $7.5 million cash payment in early January 2009.
CTI also had the option to sell its remaining 50% membership
interest in RIT to us, subject to adjustment for any amounts
owed between RIT and CTI at the time of sale. CTI exercised this
“Put” option in February 2009. On March 15, 2009,
we entered into an agreement with CTI to complete such sale for
an aggregate amount of $16.5 million subject to certain
adjustments for, among other things, payables determined to be
owed between CTI and RIT. CTI disputed the adjustments, but in a
May 2009 arbitration proceeding, we were awarded approximately
$4.3 million. As a result of the sale, we own 100% of RIT
and are its sole member and therefore, we have, through
licenses, all of the U.S. rights to Zevalin.
|
| |
| |
•
|
In connection with obtaining the required consent of Biogen to
the foregoing joint venture arrangement, we entered into certain
agreements with Biogen. Such agreements included:
|
|
|
|
| |
•
|
an amendment to the original asset purchase agreement between
CTI and Biogen (CTI/Biogen Agreement), modifying future
milestone payments, to provide that (i) concurrently with
the execution of the amendment CTI was required to pay Biogen
$0.2 million (which was reimbursed to CTI by RIT from the
initial capital contributions made by CTI and us),
(ii) upon the December 2008 closing of the joint venture
transaction, CTI was required to pay Biogen an additional
$2.0 million (which was paid by RIT as successor to CTI
under the amendment), (iii) upon the achievement of the
specified FDA approval milestone, RIT (as successor to CTI) was
required to pay Biogen an additional amount of $5.5 million
if the milestone event occurred in 2009 (provided that RIT may
elect to defer any such payment until January 1, 2010, but
upon such election the required payment will increase to
$6.0 million), $7.0 million if the milestone event
occurs in 2010, $9.0 million if the milestone event occurs
in 2011, or $10.0 million if the milestone event occurs in
2012 or later. As disclosed above, we received FDA approval for
the treatment of patients with previously untreated follicular
NHL who achieve a partial or complete response to first-line
chemotherapy and in accordance with the amendment, we paid
Biogen $5.5 million. No other material terms of the
CTI/Biogen Agreement were modified. CTI’s rights and
obligations, including its payment obligations to Biogen,
including royalties on net sales of Zevalin and an additional
regulatory milestone payment, under both the CTI/Biogen
Agreement and the amendment were assigned to and assumed by RIT
in connection with the closing of the joint venture transaction.
|
| |
| |
•
|
an amendment to the original supply agreement between Biogen and
CTI (CTI/Biogen Supply Agreement), modifying certain of the
pricing and manufacturing technology transfer terms contained in
the CTI/Biogen Supply Agreement and also providing that the term
of the agreement may be shortened in some instances in the event
of a mid-term manufacturing technology transfer. CTI’s
rights and obligations, including its payment obligations to
Biogen, under both the CTI/Biogen Supply Agreement and the
amendment were assigned to and assumed by RIT in connection with
the closing of the joint venture transaction.
|
| |
| |
•
|
a security agreement, by and between RIT and Biogen whereby RIT
granted to Biogen a first priority security interest in all of
RIT’s assets, including the assets contributed to RIT by
CTI in connection with the closing of the joint venture
transaction, to secure certain payment, indemnification and
other obligations of RIT to Biogen.
|
| |
| |
•
|
a guarantee, by us for the benefit of Biogen whereby we have,
among other things, guaranteed the payment and performance all
of RIT’s obligations to Biogen (including its obligations
as assignee of CTI under all contractual arrangements between
CTI and Biogen that were assigned to and assumed by RIT in
connection with the closing of the joint venture transaction).
|
10
|
|
|
| |
•
|
pursuant to the transfer of Zevalin assets from CTI to RIT in
December 2008, RIT assumed certain license and sublicense
agreements with various third parties related to Zevalin
intellectual property under which RIT is required to make
certain payment obligations including milestone payments and
royalties.
|
Fusilev®
(levoleucovorin) for injection: On
March 7, 2008, our new drug application (NDA) for our
proprietary drug Fusilev was approved by the FDA. We
commercially launched Fusilev in August 2008, with an in-house
sales force and commercialization team. Subsequent to the
launch, in November 2008, we received a unique J-code for
Fusilev from CMS, which went into effect on January 1,
2009. The J-code is a unique, product-specific billing code that
assists providers (e.g., physicians that prescribe
Fusilev) in obtaining reimbursement for Fusilev.
Fusilev is a novel folate analog formulation and the
pharmacologically active isomer (the levo-isomer) of the
racemic compound, calcium leucovorin. Isomers are compounds with
the same molecular formula, but “mirror image” atomic
structures. Leucovorin is a mixture of equal parts of both
isomers: the pharmacologically active levo-isomer and the
inactive dextro-isomer. Preclinical studies have
demonstrated that the inactive dextro-isomer may compete
with the active levo-isomer for uptake at the cellular
level. By removing the inactive dextro form, the dosage
of Fusilev is one-half that of leucovorin and patients are
spared the administration of an inactive substance.
Fusilev rescue is indicated after high-dose methotrexate therapy
in patients with osteosarcoma, and to diminish the toxicity and
counteract the effects of impaired methotrexate elimination or
inadvertent overdose of folic acid antagonists. Fusilev has been
designated as an orphan drug for its approved indications.
Methotrexate is a widely used anti-cancer drug. It is a
therapeutic option in the treatment of solid tumors and
hematological malignancies, such as NHL. In addition,
methotrexate is also used to treat autoimmune diseases such as
rheumatoid arthritis, psoriasis and some rare opportunistic
infections.
Leucovorin is currently a standard combination agent with 5-FU
in various colorectal cancer treatment regimens. Leucovorin
potentiates the effects of 5-FU and its derivatives by
stabilizing the binding of the drug’s metabolite to its
target enzyme, thus prolonging drug activity. There are
peer-reviewed publications wherein Fusilev is used in place of
the leucovorin in combination with 5-FU containing regimens for
adjuvant and advanced colorectal cancer and in combination with
oxaliplatin
and/or
irinotecan for advanced disease. The National Comprehensive
Cancer Network Clinical Practice Guidelines in
Oncologytm
in colon cancer and rectal cancer have been updated to reflect
that Fusilev is available in the United States. Additionally, in
the fourth quarter of 2008, Fusilev was listed and continues to
be listed in the NCCN Drugs and Biologic Compendium for use in
combination with high-dose methotrexate for the treatment of
bone cancer (osteosarcoma and de-differentiated
chrondrosarcoma). The NCCN Drugs and Biologics Compendium is an
important reference that has been recognized by United
HealthCare as a formal guidance for coverage policy. In
addition, CMS announced in June 2008 that it would recognize the
NCCN Drugs & Biologics Compendium as a source of
information to determine which drugs may be covered under
Medicare Part B.
The following describes the principal commercial terms relating
to Fusilev licensing and development.
|
|
|
| |
•
|
In April 2006, we acquired all of the oncology drug product
assets of Targent, Inc. Targent is eligible to receive payments,
in the form of our common stock
and/or cash,
upon achievement of certain regulatory and sales milestones. At
our option, any amounts due in cash under the purchase agreement
may be paid by issuing shares of our common stock having a
value, determined as provided in the purchase agreement, equal
to the cash payment amount.
|
| |
| |
•
|
In May 2006, we amended and restated a license agreement with
Merck & Cie AG, a Swiss corporation, that we assumed
in connection with the acquisition of the assets of Targent.
Pursuant to the license agreement with Merck & Cie, we
obtained the exclusive license to use regulatory filings related
to Fusilev and a non-exclusive license under certain patents and
know-how related to Fusilev to develop, make, have made, use,
sell and have sold Fusilev in the field of oncology in North
America. In addition, we have the right of first opportunity to
negotiate an exclusive license to manufacture, have
manufactured, use and sell Fusilev products outside the field of
oncology in North America. Also, under the terms of the license
agreement, we paid Merck & Cie $100,000 for the
achievement of FDA approval of Fusilev. Eprova is also eligible
to receive a payment upon achievement of another regulatory
milestone, in addition to royalties on net sales. The term of
the license agreement is determined on a
product-by-product
and
country-by-country
basis until
|
11
|
|
|
| |
|
royalties are no longer owed under the license agreement. The
license agreement expires in its entirety after the date that we
no longer owe any royalties to Merck & Cie. We have
the unilateral right to terminate the license agreement, in its
entirety or on a
product-by-product
or
country-by-country
basis, at any time for any reason and either party may terminate
the license agreement due to material breach of the terms of the
license agreement by or insolvency of the other party.
|
Apaziquone: Apaziquone is an
anti-cancer agent that becomes activated by certain enzymes
often present in higher amounts in cancer cells than in normal
cells. It is currently being investigated for the treatment of
NMIBC, which is a cancer that is only in the innermost layer of
the bladder and has not spread to deeper layers of the bladder.
The American Cancer Society estimated that the 2010 incidence
and prevalence of bladder cancer in the United States would be
approximately 70,530 and over 500,000 respectively. According to
Botteman et al., (PharmacoEconomics 2003), bladder cancer is the
most expensive cancer to treat on a lifetime basis.
The initial treatment of this cancer is complete surgical
removal of the tumor. However, bladder cancer is a highly
recurrent disease with approximately 75% of patients recurring
within 5 years, and a majority of patients recurring within
2 years. This high recurrence rate is attributed to:
1) the highly implantable nature of cancer cells that are
dispersed during surgery, 2) incomplete tumor resection,
and 3) tumors present in multiple locations in the bladder
which may be missed or too small to visualize at the time of
resection. Despite evidence in the published literature and
guidance from the American and European Urology Associations,
instillation of a chemotherapeutic agent immediately following
surgery is not a standard clinical practice. Currently, there
are no approved drugs for this indication which may, in part,
explain the difference between the literature and urology
guidelines and actual clinical management of this disease. For
more than 30 years, no new drugs have been introduced in
the market for treatment of NMIBC. An immediate instillation of
apaziquone may help by 1) reducing tumor recurrence by
destroying dispersed cancer cells that would otherwise
re-implant onto the inner lining of the bladder, 2) by
destroying remaining cancer cells at the site of tumor resection
(also known as chemo-resection), and 3) by destroying
tumors not observed during resection (also known as
chemo-ablation).
Apaziquone is a bio-reductive alkylating indoloquinone that is
enzymatically activated by enzymes that are over expressed by
bladder tumors. Pharmacokinetic studies have verified that
apaziquone is not detectable in the bloodstream of patients when
it is administered either after surgical resection or as a part
of a delayed multi-instillation protocol. The proposed dose
therefore carries a minimal risk of systemic toxicity which
could arise from absorption of a drug through the bladder wall
into the bloodstream. Additionally, the current proposed dose is
a fraction of the systemic toxic dose. These features of
apaziquone are distinct from other intravesical agents currently
in use for the treatment of recurrent bladder cancer.
A Phase 1 dose-escalation marker lesion (tumor) study
demonstrated that apaziquone had no systemic toxicity, and was
well tolerated at the dose level being used in the Phase 3
trials. Apaziquone also demonstrated anti-tumor activity against
NMIBC, as evidenced by eight of twelve patients showing a
complete response, defined as the complete disappearance of the
marker lesion as confirmed by biopsy, after receiving six
treatments with apaziquone over a period of six weeks.
Phase 2 data has confirmed anti-tumor activity in patients with
multiple, recurrent NMIBC, as evidenced by 31 of
46 patients (67%) showing a complete response after
receiving six weekly treatments with 4 mg of apaziquone
instilled into the urinary bladder in this marker lesion study.
Apaziquone was well-tolerated, with no significant systemic
toxicity, and local toxicity limited to temporary chemical
cystitis (inflammation of the urinary bladder) resulting in
increased urinary frequency, dysuria (painful urination) and
hematuria (blood in the urine) in a few patients. At the
two-year follow up, eighteen patients (38%) were disease free.
In September 2005, we initiated an open label, multi-center
clinical study in Europe in high-risk NMIBC in 53 patients.
Patients with high-risk NMIBC usually have more aggressive
bladder cancer with higher incidence of recurrence
and/or
progression to a more invasive stage, where the cancer invades
the muscle wall of the bladder, which may require total surgical
removal of the bladder. Apaziquone was well-tolerated over
multiple instillations in this study of patients with high-risk
superficial bladder cancer. At 18 months follow up 55% of
the patients were recurrence free.
12
In 2006, we performed a 20 patient pilot safety study in
low-grade NMIBC. In this study, apaziquone was found to be well
tolerated when a single 4 mg dose is given to patients
immediately following surgery. In addition, there was no adverse
effect on wound healing and apaziquone was not detected in the
bloodstream.
In March 2007, we received concurrence from the FDA for the
design of a Phase 3 study protocol for the treatment of
non-invasive bladder cancer under a special protocol assessment
procedure. The development plan for apaziquone is two
randomized, double-blind, placebo-controlled Phase 3 clinical
trials, each with 562 evaluable patients with
TaG1-G2
(low-grade) NMIBC. Patients are being randomized in a
one-to-one
ratio to apaziquone or placebo. Under the protocol, the patients
are given a single 4 mg dose following surgical removal of
the tumors. The primary endpoint is a statistically significant
difference (p < 0.05) in the rate of tumor recurrence at
year two between the apaziquone patient group and the placebo
group. The first study began during the second quarter of 2007,
and the second study began during the third quarter of 2007. In
2008, we received scientific advice from the European Medicines
Agency (EMEA) whereby the EMEA agreed that the two Phase 3
studies as designed should be sufficient for a regulatory
decision regarding European registration. In December 2009, we
achieved our goal of completing enrollment for both Phase 3
clinical trials and we expect top-line data in 2012.
The following describes the principal commercial terms relating
to apaziquone licensing and development.
|
|
|
| |
•
|
In October 2008, we terminated our 2001 license agreement for
apaziquone with INC
Research®,
formerly NDDO Research
Foundation®
(INC) in the Netherlands, as the patents underlying the
agreement were all about to expire. Pursuant to the termination,
INC assigned to us all rights it had in the know-how or
intellectual property licensed under the agreement and all
rights in may have had in any know-how or intellectual property
created during the term of the agreement. In exchange, we paid
INC a nominal amount of cash and issued them a nominal number of
shares of our common stock. In addition, INC is entitled to up
to 25,000 additional shares of our common stock and an
additional payment of $300,000 upon achievement of certain
regulatory milestones.
|
| |
| |
•
|
In October, 2008, we entered into a license, development, supply
and distribution agreement with Allergan pursuant to which we
and Allergan agreed to a collaboration for the development and
commercialization of a formulation of apaziquone suitable for
use in treating cancer or precancerous conditions via
instillation. The agreement with Allergan also provides that
Allergan has the exclusive right to make, develop and
commercialize apaziquone for the treatment of bladder cancer, or
pre-bladder cancer conditions worldwide except for Asia (as is
defined in the agreement). We also entered into a co-promotion
agreement with Allergan providing for the joint
commercialization of apaziquone in the United States, whereby we
and Allergan will share equally all profits and
commercialization expenses. We also have the right, in our sole
discretion, to opt-out of the co-promotion agreement before
January 1, 2012. If we elect to opt-out of the co-promotion
agreement, our share of any future development costs shall be
significantly reduced. Part of the aggregate development costs
and marketing expenses incurred by us since January 1, 2009
shall be reimbursed by Allergan in the form of a one-time
payment. In addition, if we opt-out of the co-promotion
agreement, the co-promotion agreement will terminate and instead
of a sharing of profit and expenses, Allergan will pay us
royalties on a percentage of net sales of the apaziquone in the
United States that are slightly greater than the royalties paid
on net sales outside the United States. In addition, Allergan
will pay us up to $245 million in additional milestones
based upon the achievement of certain sales milestones in the
United States.
|
| |
| |
•
|
In consideration for the rights granted under our license,
development, supply and distribution agreements with Allergan,
Allergan paid us an up-front fee of $41.5 million. In
addition, Allergan will pay us up to $304.0 million based
on the achievement of certain development, regulatory and sales
milestones. For example, for completing enrollment of both
aforementioned Phase III trials by year-end 2009, Allergan
paid us a $1.5 million milestone payment. Also, Allergan
has agreed to pay us tiered royalties starting in the mid-teens
based on a percentage of net sales of the apaziquone outside of
the United States.
|
| |
| |
•
|
We will continue to conduct the current Phase 3 clinical trials
as well as certain future planned clinical trials pursuant to a
joint development plan, of which Allergan will fund 65% of the
development costs. In November 2009, we entered into a
collaboration agreement with the Nippon Kayaku Co., LTD. for the
development and commercialization of apaziquone in Asia, except
North and South Korea (the “Nippon
|
13
|
|
|
| |
|
Kayaku Territory”). In exchange, Nippon Kayaku paid
Spectrum an up-front payment of $15 million and agreed to
make additional payments of up to $136.0 million based on
the achievement of certain regulatory and commercialization
milestones contained in the agreement. In addition, Nippon
Kayaku received exclusive rights to apaziquone for the treatment
of NMIBC in Asia (other than North and South Korea), including
Japan and China. Nippon Kayaku will conduct apaziquone clinical
trials in the Nippon Kayaku Territory pursuant to a development
plan. In addition, Nippon Kayaku will be responsible for all
expenses relating to the development and commercialization of
apaziquone in the Nippon Kayaku Territory. In January 2011
Nippon Kayaku initiated a Phase 1 study with the first patient
being dosed in Japan. The Phase 1 study is required by the local
regulatory authorities and is designed to enroll up to
6 patients.
|
|
|
|
| |
•
|
Also in November 2009, we entered into a collaboration agreement
with Handok Pharmaceuticals for the development and
commercialization of apaziquone in North and South Korea. Under
the terms of the Handok collaboration agreement, Handok paid us
an up-front payment of $1.0 million and potential milestone
payments totaling approximately $19 million. The potential
milestone payments will be based on the achievement of certain
regulatory and commercialization milestones. Handok received
rights to apaziquone for the treatment of NMIBC in North and
South Korea. Additionally, Handok will conduct the apaziquone
clinical trials in North and South Korea pursuant to a
development plan and will be responsible for all expenses
relating to the development and commercialization of apaziquone
in North and South Korea.
|
Belinostat: Belinostat is a histone
deacytelase or HDAC inhibitor that is being studied in multiple
clinical trials, both as a single drug and in combination with
chemotherapeutic drugs for the treatment of various
hematological and solid tumors. HDACs catalyze the removal of
chemical groups known as acetyl groups from certain portions of
human DNA, and thus regulate gene expression. By inhibiting this
enzyme, belinostat induces cell cycle arrest, and leads to
inhibition of cancer cell proliferation and induction of
apoptosis, or cell death. Additional mechanisms of action
thought to be responsible for belinostat’s anti-cancer
effect include inhibition of angiogenesis, or blood vessel
growth, and the resensitization of cells that have overcome drug
resistance to anticancer drugs, such as platinums and taxanes.
Belinostat is currently the only HDAC inhibitor in clinical
development with multiple potential routes of administration,
including intravenous administration, continuous intravenous
infusion and oral administration, which we believe may afford
belinostat with a significant competitive advantage.
Belinostat is currently in a registrational trial, under a
special protocol assessment, as a monotherapy for
relapsed/refractory Peripheral T-Cell Lymphoma or PTCL an
indication which has been granted Orphan Drug and Fast Track
designation by the FDA. The registrational trial is an
open-label, multicenter, single arm efficacy and safety study,
in which we plan to enroll approximately 120 patients with
relapsed or refractory peripheral T-cell lymphoma, who have
failed at least one prior systemic therapy. We expect to file an
NDA for belinostat in PTCL in 2011/2012.
Belinostat is also currently in a randomized Phase 2 trial for
carcinoma of unknown primary or CUP, in combination with
carboplatin and paclitaxel being conducted by our collaborator,
Topotarget. Target enrollment was reached in December 2010 and
Topotarget expects top-line results of progression free survival
and response rate in the third quarter of 2011. There are
currently no approved therapies or drugs for treatment of CUP,
which is an indication with a large patient population. The NCI
estimated that for 2008, approximately 2 to 4% of all cancers
are CUP.
Based on the data from past and ongoing studies, we believe
there are many potential attributes associated with belinostat
that separate it from other currently marketed HDACs, including
efficacy when used alone and in combination, less toxicities
(when compared to other currently-marketed HDACs), including
less bone marrow toxicity, and a lack of other severe side
effects, such as mucositis, that may enable full dose
combinations of this drug with several other cytotoxic agents.
Hence, belinostat is currently being investigated in multiple
indications, both as monotherapy and in combination with other
treatment regimens. Numerous studies have been conducted, and
are ongoing, through the NCI and other well-known oncologic
academic institutions. Additionally, we plan on a comprehensive
development program for belinostat, which includes both
hematologic indications, such as PTCL, and solid tumor
indications, such as ovarian cancer, colorectal cancer and CUP.
Based upon the foregoing, we
14
believe belinostat potentially has broad applicability and
hence, commercial potential beyond that of currently marketed
HDACs.
The following describes the principal commercial terms relating
to belinostat licensing and development.
|
|
|
| |
•
|
In February 2010, we entered into a licensing and collaboration
agreement with TopoTarget, for the development and
commercialization of belinostat, pursuant to which TopoTarget
and we agreed to a collaboration for the development and
commercialization of belinostat. The agreement provides that we
have the exclusive right to make, develop and commercialize
belinostat in North America and India, with an option for China.
The agreement also grants TopoTarget a co-promote option if and
only if we do not maintain a minimum number (subject to
adjustment for certain events outside of our control) of field
personnel (as defined in the agreement) for a certain number of
years post-approval of the PTCL indication.
|
| |
| |
•
|
In consideration for the rights granted to us under the license
and collaboration agreement with TopoTarget, we paid TopoTarget
an up-front fee of $30.0 million. In addition, we will pay
up to $313 million and one million shares of Spectrum
common stock based on the achievement of certain development,
regulatory and sales milestones. as well as certain royalties on
net sales of belinostat.
|
| |
| |
•
|
Under the terms of the agreement, all development, including
studies, will be conducted under a joint development plan and in
accordance with a mutually agreed upon target product profile
provided that we have final decision-making authority for all
developmental activities in North America and India (and China
upon exercise of the option for China) and TopoTarget has final
decision-making authority for all developmental activities in
all other jurisdictions, We will assume all responsibility for
and future costs of the ongoing registrational PTCL trial while
TopoTarget will assume all responsibility for and future costs
of the ongoing Phase 2 CUP trial. We and TopoTarget will conduct
future planned clinical trials pursuant to the joint development
plan, of which we will fund 70% of the development costs
and TopoTarget will fund 30% of the development costs.
|
| |
| |
•
|
We and TopoTarget will each pay 50% of the costs for chemical,
pharmaceutical and other process development related to the
manufacturing of the product that are incurred with a mutually
agreed upon budget in the joint development plan. TopoTarget is
responsible for supplying us with both clinical and commercial
product.
|
Ozarelix: Ozarelix is a Luteinizing
Hormone Releasing Hormone, or LHRH, antagonist (a substance that
blocks the effects of a natural hormone found in the body).
Mechanistically, LHRH antagonists exert rapid inhibition of
luteinizing hormone and follicle stimulating hormone with an
accompanying rapid decrease in sex hormones and would therefore
be expected to be effective in a variety of hormonally dependent
disease states including ovarian cancer, prostate cancer, BPH,
infertility, uterine myoma and endometriosis.
In January 2010, based upon the mixed results of our earlier
Phase 2 study of ozarelix for the treatment of BPH and the
recently announced failure of Aeterna Zentaris’s large,
Phase 3, registrational trial of cetrorelix (another LHRH
antagonist), we discontinued development of ozarelix in BPH. We
estimate that this discontinuation will result in substantial
reduction in future clinical development expenses. Currently, we
are conducting a phase II clinical trial of ozarelix in
prostate cancer patients.
The following describes the principal commercial terms relating
to ozarelix licensing and development.
|
|
|
| |
•
|
In 2004, we entered into a license agreement with a subsidiary
of Aeterna Zentaris, Inc., Aeterna Zentaris GmbH, whereby we
acquired an exclusive license to develop and commercialize
ozarelix in North America (including Canada and Mexico) and
India. In addition, we have a 50% financial interest in any
income Aeterna Zentaris derives from ozarelix in Japan. We are
contingently obligated to pay amounts based upon achievement of
milestones and a royalty based on any future net sales. In
November 2010, we amended the terms of the agreement to expand
the territory covered by the exclusive license.
|
| |
| |
•
|
The term of the license agreement expires ten years after the
first commercial sale of a product in any country within the
territory or as long as any product is covered by a patent in
any country in the territory, and where there is no generic
competition in such country of the territory, whichever term is
longer, although some obligations survive termination. In
addition, the agreement may be terminated earlier by either
party (in
|
15
|
|
|
| |
|
some cases either in whole or on a
product-by-product
and/or
country-by-country
and/or
indication-by-indication
basis), based upon material breach or the commencement of
bankruptcy or insolvency proceedings involving the other, or by
us upon sixty days’ notice to Aeterna Zentaris.
|
Ortataxel: In July 2007, we entered
into an exclusive worldwide license agreement for ortataxel, a
third-generation taxane with Indena S.p.A. In clinical studies,
ortataxel has been shown to be bioavailable when administered
orally to patients with solid tumors. In addition, it belongs to
a new generation of taxanes with the potential to be active
against tumors resistant to paclitaxel (Bristol-Myers
Squibb’s
Taxol®)
and docetaxel (Sanofi-Aventis’
Taxotere®).
Phase 1 and 2 studies in more than 350 patients with solid
tumors have shown activity in patients that were refractory to
treatment with the available taxane drugs. The safety profile of
ortataxel is comparable to that of paclitaxel and docetaxel.
While optimizing the oral formulation for better
bioavailability, we will consider future studies with the oral
formulation.
The following describes the principal commercial terms relating
to ortataxel licensing and development.
|
|
|
| |
•
|
Under the terms of the license agreement with Indena, we are
obligated to make payments based on the achievement of certain
development, regulatory filing and sales milestones. We will
also pay Indena certain royalties on worldwide sales of
ortataxel, if and when the product is approved. On
October 11, 2010, we amended the agreement to extend
payments of certain development and regulatory milestones.
|
| |
| |
•
|
Also, we are obligated to purchase all of our requirements of
ortataxel active pharmaceutical ingredient from Indena.
|
Lucanthone: Lucanthone is an
orally administered small-molecule which inhibits
Topoisomerase II and AP endonuclease. In preclinical tests,
lucanthone was shown to enhance the sensitivity of animals to an
anticancer agent in a time dependent and reversible manner.
Lucanthone was originally used as an antiparasitic agent for the
treatment of schistosomiasis in the 1950s and 1960s, and has a
demonstrated safety profile. It was later discontinued because
better anti-parasitic medications became available. We are
currently working on the development plan for lucanthone.
The following describes the principal commercial terms relating
to lucanthone licensing and development.
|
|
|
| |
•
|
We entered into a license agreement with Dr. Robert E.
Bases, the inventor of a method of treating cancer of the
central nervous system through the administration of lucanthone
and radiation, whereby we acquired worldwide exclusive rights to
develop and commercialize a product based upon his invention in
May 2005. Under the terms of the license agreement, we made a
small up-front payment and are obligated to make additional
periodic payments, a payment upon achievement of a certain
regulatory milestone and royalties on potential net sales, if
any.
|
SPI-1620: SPI-1620 is a highly
selective peptide agonist of endothelin B receptors, which can
stimulate receptors on endothelial cells, the innermost layer of
cells lining the blood vessels. This technology takes advantage
of the fact that the blood supply to tumors is different than
the blood supply to healthy organs. Blood vessels in the growing
part of tumors are relatively devoid of smooth muscle covering
and are rich in endothelial cells. Therefore, by stimulating the
endothelial B receptors present on the endothelial cells,
SPI-1620 should selectively increase tumor blood flow while
sparing healthy tissue.
Chemotherapy is one of the mainstays of therapy for solid
carcinomas, including breast, lung, and prostate. Chemotherapy
uses drugs called cytotoxic agents that are poisonous to cells
and kill cancer cells. Chemotherapy often fails because adequate
and uniform distribution of the cytotoxic agents is not achieved
in the tumor, and serious side effects can result from toxicity
to normal cells. Consequently, any means to increase the
delivery of a cytotoxic agent selectively to tumors, while
minimizing its concentration in normal tissues may be beneficial.
SPI-1620 is being developed as an adjunct to chemotherapy. In
pre-clinical studies, when anti-cancer drugs, such as
paclitaxel, are administered shortly after SPI-1620, the
anti-cancer drug concentration in the tumor is increased several
fold. This results in increased anti-tumor efficacy at a given
dose of a cytotoxic agent, and might
16
allow physicians to maximize efficacy with reduced cytotoxic
agent doses with resultant decreased toxicity to the normal
organs.
In the first quarter of 2008, we initiated an open label,
dose-escalation Phase 1 study assessing the safety,
tolerability, pharmacokinetics and pharmacodynamics of SPI-1620
in patients with recurrent or progressive carcinoma. We enrolled
the first patient in this study in February 2008, and are
continuing to enroll patients in this study.
The following describes the principal commercial terms relating
to SPI-1620 licensing and development.
|
|
|
| |
•
|
We acquired an exclusive worldwide license to develop and
commercialize SPI-1620 for the prevention and treatment of
cancer from Chicago Labs, Inc. in February 2005. We paid Chicago
Labs a small up-front fee and are obligated to make future
payments contingent upon the successful achievement of certain
development and regulatory milestones. In addition, we will pay
royalties and sales milestones on net sales, after marketing
approval is obtained.
|
RenaZorb: RenaZorb, a
second-generation lanthanum-based nanoparticle phosphate binding
agent, has the potential to treat hyperphosphatemia, (high
phosphate levels in blood), in patients with stage 5 chronic
kidney disease (end-stage renal disease). Hyperphosphatemia
affects patients with chronic kidney disease, especially end-
stage kidney disease patients on dialysis. It can lead to
significant bone disease (including pain and fractures) and
cardiovascular disease, and is independently associated with
increased mortality.
According to The United States Renal Data System in 2010, there
will be an estimated 600,000 patients with end-stage renal
disease in the United States. Treatment of hyperphosphatemia is
aimed at lowering blood phosphate levels by:
(1) restricting dietary phosphorus intake; and
(2) using, on a daily basis, and with each meal, oral
phosphate binding drugs that facilitate fecal elimination of
dietary phosphate before its absorption from the
gastrointestinal tract into the bloodstream. Restricting dietary
phosphorus intake has historically not been a successful means
of serum phosphate control, therefore phosphate binders are the
mainstay of hyperphosphatemia management.
Currently marketed therapies for treating hyperphosphatemia
include polymer-based and lanthanum-based phosphate binders,
aluminum-based phosphate binders, and calcium-based phosphate
binders. Under the National Kidney Foundation K/DOQI guidelines,
both calcium-based phosphate binders and non-calcium,
non-aluminum, non-magnesium phosphate binders are recommended as
first line or long-term therapy for the management of
hyperphosphatemia. However, the current therapies require use of
a large number of pills or large pills to be chewed or swallowed
along with each meal, leading to problems with patient
compliance with the treatment regimen.
We believe that RenaZorb has the opportunity, because of its
potentially higher capacity for binding phosphate on an equal
weight basis, to significantly improve patient compliance by
offering the
lowest-in-class
dosage to achieve the same therapeutic benefit as other
phosphate binders. We continue to perform preclinical
development work on RenaZorb.
The following describes the principal commercial terms relating
to RenaZorb licensing and development.
|
|
|
| |
•
|
We entered into a license agreement with Altair Nanomaterials,
Inc. and its parent Altair Nanotechnologies, Inc., whereby we
acquired an exclusive worldwide right to develop and
commercialize RenaZorb for all human therapeutic and diagnostic
uses in January 2005. Under the terms of the license agreement,
we made up-front and milestone payments and are obligated to
make additional payments upon achievement of certain clinical
development and regulatory and sales milestones, in addition to
royalties on potential net sales.
|
| |
| |
•
|
In August 2009, we entered into an acquisition agreement with
Altair, in which we acquired 100% of the rights to Renazorb and
all of Altair’s life science technology. Our acquisition of
RenaZorb expands upon our prior license agreement with Altair,
pursuant to which Altair granted us human uses. Our acquisition
of RenaZorb provides us with access to all uses of and
intellectual property for RenaZorb. In consideration for the
acquisition, we paid Altair a total of $750,000 in the form of
restricted shares of our common stock.
|
17
Manufacturing
We currently do not have internal manufacturing capabilities;
therefore, all of our products are manufactured on a contract
basis. We expect to continue to contract with third party
providers for manufacturing services, including active
pharmaceutical ingredient, or API, finished-dosage product, as
well as packaging operations. We believe that our current
agreements with third party manufacturers provide for sufficient
operating capacity to support the anticipated commercial demand
for our products. However, we have only one approved contract
manufacturer for each aspect of the manufacturing process for
Zevalin and are in the process of qualifying additional contract
manufacturers for Fusilev. If we are unable to obtain a
sufficient supply of our required products, or if we should
encounter delays or difficulties in our relationships with our
manufacturers, we may lose potential sales.
We attempt to prevent disruption of supplies through supply
agreements, appropriate forecasting, maintaining stock levels
and other strategies. We believe that the market for such
manufacturers and suppliers is such that we could quickly enter
into another supply or manufacturing agreement, on substantially
similar terms, if we were required to do so. However, in the
event we are unable to manufacture our products, either directly
or indirectly through others or on commercially acceptable
terms, if at all, we may not be able to commercialize our
products as planned. Although we are taking these actions to
avoid a disruption in supply, we cannot provide assurance that
we may not experience a disruption in the future.
Sales,
Marketing and Distribution
We have built, and continue to build, a sales and marketing
infrastructure as part of our commercialization efforts for
Fusilev and Zevalin. While we maintain a relatively small sales
force, we believe that the size of our sales force is
appropriate to effectively reach our target audience for our two
commercial products.
For Fusilev, we utilize a third-party logistics company to store
and distribute this drug product. The same third party logistics
company also stores and ships Zevalin kits containing the CD20
MAB.
For Zevalin, we changed the supply and distribution model in
2009. Previously, we sold Zevalin kits containing the CD20 MAB
to radiopharmacies, who then in turn ordered the radioactive
isotope (Y-90 or In-111) separately and radiolabeled (or
attached) the radioactive isotope to the CD20 MAB. The
radiopharmacy then sold the end user product to the consumer.
Under the current model we do not sell the Zevalin kits
containing the CD20 MAB to the radiopharmacies, but instead
contract with them, as a
fee-for-service,
to radiolabel the individual components of the CD20 MAB to the
radioactive isotope, and then, also under a
fee-for-service
arrangement, have them distribute the end use product to the end
user, which are clinics, hospitals or other medical settings. In
this regard, we now sell the CD20 MAB together with the
radioactive isotope as the end user product directly to the
healthcare service provider.
Customers
Our product sales are concentrated in a limited number of
customers. Sales to Customer A for the years ended
December 31, 2010, 2009 and 2008 were 45.7%, 21.7 and 35.7%
respectively, of our total consolidated gross product sales. No
other single customer generates over 10% of our consolidated
gross product sales.
We are exposed to risks associated with extending credit to our
customers related to the sale of products. We do not require
collateral or other security to support credit sales, however,
we maintain reserves for potential bad debt and to date, credit
losses have been within management’s expectations. Customer
A owed us 56.1% of net receivables as of December 31, 2010
and no customer owed us more than 10% of net receivables as of
December 31, 2009. All sales were to customers in the
United States.
Competition
The pharmaceutical industry is characterized by rapidly evolving
biotechnology and intense competition. We expect
biotechnological developments and improvements in the fields of
our business to continue to occur at a rapid rate and, as a
result, expect competition to remain intense. Many companies are
engaged in research and development of compounds that are
similar to our research. Biotechnologies under development by
these and other pharmaceutical companies could result in
treatments for the diseases and disorders for which we are
18
developing our own treatments. In the event that one or more of
those programs are successful, the market for some of our drug
products could be reduced or eliminated. Any product for which
we obtain FDA approval must also compete for market acceptance
and market share.
Competing in the branded product business requires us to
identify and quickly bring to market new products embodying
therapeutic innovations. Successful marketing of branded
products depends primarily on the ability to communicate the
effectiveness, safety and value of the products to healthcare
professionals in private practice, group practices, hospitals
and academic institutions, and managed care organizations.
Competition for branded drugs is less driven by price and is
more focused on innovation in treatment of disease, advanced
drug delivery and specific clinical benefits over competitive
drug therapies. Unless our products are shown to have a better
safety profile, efficacy and cost-effectiveness as compared to
other alternatives, they may not gain acceptance by medical
professionals and may therefore never be successful commercially.
Companies that have products on the market or in research and
development that target the same indications as our products
target include, among others, Abraxis Bioscience, Inc., Astra
Zeneca LP, Bayer AG, Endo Pharmaceuticals, Eli Lilly and Co.,
Novartis Pharmaceuticals, Corporation, Genentech, Inc.,
Bristol-Myers Squibb Company, GlaxoSmithKline, Biogen-IDEC
Pharmaceuticals, Inc., OSI Pharmaceuticals, Inc., Cephalon,
Inc., Sanofi-Aventis, Inc., Pfizer, Inc., Genta Incorporated,
Merck, Celgene Corporation, Allos Therapeutics, Inc., BiPar
Sciences, Inc., Genzyme Corporation, Shire Pharmaceuticals,
Abbott Laboratories, Poniard Pharmaceuticals, Inc., Roche
Pharmaceuticals and Johnson & Johnson who may be more
advanced in development of competing drug products or are more
established and are currently marketing products for the
treatment of various indications that our drug products target.
Many of our competitors are large and well-capitalized companies
focusing on a wide range of diseases and drug indications, and
have substantially greater financial, research and development,
marketing, human and other resources than we do. Furthermore,
large pharmaceutical companies have significantly more
experience than we do in pre-clinical testing, human clinical
trials and regulatory approval procedures, among other things.
As noted above, we launched our proprietary product, Fusilev, in
August 2008. Fusilev is the levo-isomeric form of the racemic
compound calcium leucovorin, a product already approved for the
same indications our product is approved for. Leucovorin has
been sold as a generic product on the market for a number of
years. There are two generic companies currently selling the
leucovorin product and therefore we are competing against a low
cost alternative. Also, Fusilev will be offered as part of a
treatment regimen, and that regimen may change to exclude
Fusilev. For these reasons, we may not recognize the full
potential value of our investment in the product.
Regarding Zevalin, there are three products which are potential
competitors for the indications it is currently approved for.
Treanda®
(bendamustine hydrochloride) for Injection, for Intravenous
Infusion, marketed by Cephalon, is indicated for the treatment
of patients with indolent B-cell NHL that has progressed during
or within six months of treatment with rituximab or a
rituximab-containing regimen.
Also, the
Bexxar®
therapeutic regimen (Tositumomab and Iodine I 131 Tositumomab),
a radiopharmaceutical marketed by GlaxoSmithKline, is indicated
for the treatment of patients with CD20 antigen-expressing
relapsed or refractory, low-grade, follicular, or transformed
NHL, including patients with Rituximab-refractory NHL.
Finally,
Rituxan®
(rituximab), marketed by Genentech and Biogen, is indicated for
the treatment of patients with relapsed or refractory, low-grade
or follicular, CD20-positive, B-cell NHL as a single agent;
previously untreated follicular, CD20-positive, B-cell NHL in
combination with CVP (cyclophosphamide, vincristine and
prednisone combination) chemotherapy; and non-progressing
(including stable disease), low-grade, CD20-positive B-cell NHL,
as a single agent, after first-line CVP chemotherapy. Rituxan is
administered as a part of various chemotherapy regimens and
schedules, the vast majority of which, could be used in concert
with other therapeutic agents, such as Zevalin, as part of a
treatment plan.
For more information regarding competition to our products,
please also read our discussion of competition matters in
Item 1A “Risk Factors” of this report.
19
Research
and Development
New drug development, which is the process whereby drug product
candidates are tested for the purpose of filing a NDA or BLA (or
similar filing in other countries) and eventually obtaining
marketing approval from the FDA or a similar marketing
authorization from other regulatory authorities outside of the
United States, is an inherently uncertain, lengthy and expensive
process that requires several phases of clinical trials to
demonstrate to the satisfaction of the appropriate regulatory
authorities that the products are both safe and effective for
their respective indications. Our development focus is primarily
based on acquiring and developing late-stage development drugs
as compared to new drug discovery, which is very uncertain and
lengthy.
Research and development expenses for such drug development are
comprised of the following types of costs incurred in performing
research and development activities: personnel expenses,
facility costs, contract services, license fees and milestone
payments, costs of clinical trials, laboratory supplies and drug
products, and allocations of corporate costs. Research and
development expenditures, including related stock-based charges
but not including amortization of intangibles or expensing of
in-process research and development costs, are expensed as we
incur them and were approximately $57.3 million,
$21.1 million and $26.7 million, respectively, in
2010, 2009 and 2008 broken out by product as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Apaziquone
|
|
$
|
6,165
|
|
|
$
|
10,915
|
|
|
$
|
5,477
|
|
|
Ozarelix
|
|
|
1,916
|
|
|
|
1,168
|
|
|
|
2,435
|
|
|
Ortataxel
|
|
|
716
|
|
|
|
311
|
|
|
|
150
|
|
|
Fusilev
|
|
|
1,281
|
|
|
|
1,125
|
|
|
|
2,096
|
|
|
Zevalin
|
|
|
421
|
|
|
|
563
|
|
|
|
151
|
|
|
Belinostat
|
|
|
36,045
|
|
|
|
—
|
|
|
|
—
|
|
|
Other development drugs
|
|
|
3,469
|
|
|
|
1,535
|
|
|
|
1,304
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total — Direct Costs
|
|
|
50,013
|
|
|
|
15,617
|
|
|
|
11,613
|
|
|
Indirect Costs (including non-cash share-based compensation of
$2.4 million, $3.2 million and $4.0 million,
respectively)
|
|
|
14,838
|
|
|
|
16,652
|
|
|
|
15,070
|
|
|
Partner Reimbursement
|
|
|
(7,550
|
)
|
|
|
(11,211
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total Research & Development
|
|
$
|
57,301
|
|
|
$
|
21,058
|
|
|
$
|
26,683
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Patents
and Proprietary Rights
Our
Patents and Proprietary Rights
We in-license from third parties certain patent and related
intellectual property rights related to our proprietary
products. In particular, we have licensed patent rights with
respect to Fusilev, Zevalin, ozarelix, ortataxel, lucanthone,
belinostat and SPI-1620, in each case for the remaining life of
the applicable patents. Except for Zevalin, Fusilev, belinostat
and ozarelix, our agreements generally provide us with exclusive
worldwide rights to, among other things, develop, sublicense,
and commercialize the drug products. Under most of these license
arrangements, we are generally responsible for all development,
patent filing and maintenance costs, sales, marketing and
liability insurance costs related to the drug products. In
addition, these licenses and agreements may require us to make
royalty and other payments and to reasonably exploit the
underlying technology of applicable patents. If we fail to
comply with these and other terms in these licenses and
agreements, we could lose the underlying rights to one or more
of our potential products, which would adversely affect our
product development and harm our business. In addition, with
regard to Zevalin, apaziquone and RenaZorb, we own patent and
related intellectual property rights related to these products.
The protection, preservation and infringement-free commercial
exploitation of these patents and related intellectual property
rights is very important to the successful execution of our
strategy. However, the issuance of a patent is neither
conclusive as to its validity nor as to the enforceable scope of
the claims of the patent. Accordingly,
20
our patents and the patents we have may not prevent other
companies from developing similar or functionally equivalent
products or from successfully challenging the validity of our
patents. If our patent applications are not allowed or, even if
allowed and issued as patents, if such patents or the patents we
have in-licensed, are circumvented or not upheld by the courts,
our ability to competitively exploit our patented products and
technologies may be significantly reduced. Also, such patents
may or may not provide competitive advantages for their
respective products or they may be challenged or circumvented by
competitors, in which case our ability to commercially exploit
these products may be diminished.
From time to time, we may need to obtain licenses to patents and
other proprietary rights held by third parties to develop,
manufacture and market our products. If we are unable to timely
obtain these licenses on commercially reasonable terms, our
ability to commercially exploit such products may be inhibited
or prevented.
As mentioned above, we own and in-license from third parties
certain patent rights related to our products. We believe that
our patents and licenses are important to our business, but that
with the exception of the United States and European patents
discussed in this paragraph, no one patent or license is
currently of material importance to our business. For Fusilev,
we have one United States formulation patent that covers Fusilev
that expires in 2019. For Zevalin, we have sublicensed United
States patents that cover the processes and tools for making
monoclonal anti-bodies or MABs, in general, licensed United
States patents that cover the CD-20 MAB in Zevalin as well as
the use of Zevalin to treat NHL, and acquired patent
applications covering the Zevalin compounding process
(i.e., process of linking the CD20 MAB to a radioactive
isotope to make the patient-ready dosage form of Zevalin). These
patents expire over a wide range of dates beginning in 2009, but
the licensed patents covering the CD-20 MAB itself do not begin
to expire until 2015. Additionally, we have pending United
States patent applications covering the compounding process, and
will consider filing more patent applications, if the
opportunity arises. For belinostat, there are composition of
matter patents that cover belinostat and related compounds that
do not begin to expire until 2021. Currently, there are multiple
United States and foreign patent applications pending that cover
belinostat formulations, uses and manufacturing and synthesis
processes. We plan to file additional United States and foreign
patent applications covering new formulations, uses and
manufacturing and synthesis processes, where appropriate. For
apaziquone, there is a United States formulation patent that
does not expire until 2022. We have filed and plan to file
additional United States and foreign patent applications
covering new formulations
and/or uses
for this product. For ozarelix, there is a United States
composition patent that will expire in 2020, and method of use
and formulation patent applications on file in the United
States. For ortataxel, there are two United States composition
patents that will expire in 2013 and multiple manufacturing and
synthesis patents that do not begin to expire till 2021, and the
corresponding European patents will expire in 2014. We
anticipate filing new method of use and formulation patent
applications for the ortataxel product in the future. There is
one United States patent covering satraplatin, a method of use
patent expired in 2010. For lucanthone, there is a United States
method of use patent that expires in 2019. For RenaZorb, there
is one method of use patent that expires in 2024 and pending
United States and foreign patent applications covering
compositions of matter directed to treating hyperphosphatemia.
For SPI-1620, we have filed method of use patent applications in
the United States and Europe. We also have multiple United
States method of use patents that expire in 2021 and 2022, and
there is ongoing prosecution for their European counterparts. We
have also filed another method of use patent application in the
United States and Europe and anticipate filing future patent
applications pending the continued development of new methods of
use and new formulations. We are constantly evaluating our
patent portfolio and are currently prosecuting patent
applications for our drug products and are considering new
patent applications in order to maximize the life cycle of each
of our products.
While the United States and the European Union are currently the
largest potential markets for most of our products, we also have
patents issued and patent applications pending outside of the
United States and Europe. Limitations on patent protection in
these countries, and the differences in what constitutes
patentable subject matter in countries outside the United
States, may limit the protection we have on patents issued or
licensed to us outside of the United States. In addition, laws
of foreign countries may not protect our intellectual property
to the same extent as would laws in the United States. To
minimize our costs and expenses and to maintain effective
protection, we usually focus our patent and licensing activities
within the United States, the European Union, Canada and Japan.
In determining whether or not to seek a patent or to license any
patent in a certain foreign country, we weigh the relevant costs
and benefits, and consider, among other things, the market
potential and profitability, the scope of patent protection
afforded by the law of the jurisdiction and its enforceability,
and the nature of terms with any
21
potential licensees. Failure to obtain adequate patent
protection for our proprietary drugs and technology would impair
our ability to be commercially competitive in these markets.
In addition to the specific intellectual property subjects
discussed above, we have trademark protection in the United
States for Spectrum Pharmaceuticals,
Inc®,
Fusilev®,
Turning Insights Into
Hopetm,
Zevalin®
and
RenaZorb®.
Additionally, for some other of these and other works related to
our business, we have pending United States and
ex-United
States trademark applications.
EOquin®
is a registered trademark of Allergan.
In conducting our business generally, we rely upon trade
secrets, know-how, and licensing arrangements and use customary
practices for the protection of our confidential and proprietary
information such as confidentiality agreements and trade secret
protection measures, such as periodic internal and external
trade secret audits. It is possible that these agreements will
be breached or will not be enforceable in every instance, and
that we will not have adequate remedies for any such breach. It
is also possible that our trade secrets or know-how will
otherwise become known or independently developed by
competitors. The protection of know-how is particularly
important because the know-how is often the necessary or useful
information that allows us to practice the claims in the patents
related to our proprietary drug products.
We may find it necessary to initiate litigation to enforce our
patent rights, to protect our trade secrets or know-how or to
determine the scope and validity of the proprietary rights of
others. Litigation concerning patents, trademarks, copyrights
and proprietary technologies can often be protracted and
expensive and, as with litigation generally, the outcome is
inherently uncertain. See Item 1A “Risk Factors”
for more information.
The
Patent Process
The United States Constitution provides Congress with the
authority to provide inventors the exclusive right to their
discoveries. Congress codified this right in United States Code
Title 35, which gave the U.S. Patent and Trademark
Office, or USPTO, the right to grant patents to inventors and
defined the process for securing a United States patent. This
process involves the filing of a patent application that teaches
a person having ordinary skill in the respective art how to make
and use the invention in clear and concise terms. The invention
must be novel (not previously known) and non-obvious (not an
obvious extension of what is already known). The patent
application concludes with a series of claims that specifically
describe the subject matter that the patent applicant considers
his invention.
The USPTO undertakes an examination process that can take from
one to seven years, or more, depending on the complexity of the
patent and the problems encountered during examination.
In exchange for disclosing the invention to the public, for all
United States patent applications filed after 1995, the
successful patent applicant is currently provided a right to
exclude others from making, using or selling the claimed
invention for a period of 20 years from the effective
filing date of the patent application.
Under certain circumstances, a patent term may be extended.
Patent extensions are most frequently granted in the
pharmaceutical and medical device industries under the Drug
Price Competition and Pricing Term Restoration Act of 1984, or
Hatch 1984, or Hatch-Waxman Act, to recover some of the time
lost during the FDA regulatory process, subject to a number of
limitations and exceptions. The patent term may be extended up
to a maximum of five years; however, as a general rule, the
average extension period granted for a new drug is approximately
three years. Only one patent can be extended per FDA approved
product, and a patent can only be extended once.
Product
Exclusivity
Under the Hatch-Waxman Act, drug products are provided
exclusivity whereby the FDA will not accept applications to
market a generic form of an innovator reference listed drug
product until the end of the prescribed period. A product is
granted a five-year period of exclusivity if it contains a
chemical entity never previously approved by the FDA either
alone or in combination, although generic applications may be
submitted after four years if they contain a certification of
patent invalidity or non-infringement as further discussed
below. A three-year period of exclusivity is granted to a
previously approved product based on certain changes, e.g.,
in strength, dosage form, route of administration or
conditions of use, where the application is supported by new
clinical investigations that are essential to approval. In
addition, in 1997 Congress amended the law to provide an
additional six months of
22
exclusivity as a reward for studying drugs in children. This
pediatric exclusivity, which can be obtained during the approval
process or after approval, effectively delays the approval of a
generic application until six months after the expiration of any
patent or other exclusivity that would otherwise delay approval,
thus providing an additional six months free of generic
competition. In order to qualify for pediatric exclusivity, the
FDA must make a written request for pediatric studies, the
application holder must agree to the request and complete the
studies with required timeframe, and the studies must be
accepted by the FDA based on a determination that the studies
fairly respond to the request. The provisions were enacted with
a five-year sunset date, and have been reauthorized in 2002 and
2007. The current provisions are set to expire in October 2012,
and Congress is likely to consider reauthorizing the statute
again.
Generic
Approval and Patent Certification
The Hatch-Waxman Act also created the abbreviated new drug
application, or ANDA, approval process, which permits the
approval of a generic version of a previously approved branded
drug without the submission of a full new drug application, or
NDA, and based in part on the FDA’s finding of safety and
effectiveness for the reference listed drug. Applicants
submitting an NDA are required to list patents associated with
the drug product, which are published in the FDA Orange Book,
and the timing of an ANDA approval depends in part on patent
protection for the branded drug. When an ANDA is filed, the
applicant must file a certification for each of the listed
patents for the branded drug, stating one of the following:
(1) that there is no patent information listed;
(2) that such patent has expired; (3) that the patent
will expire on a particular date (indicating that the ANDA may
be approved on that date); or (4) that the drug for which
approval is sought either does not infringe the patent or the
patent is invalid, otherwise known as paragraph IV
certification. If an ANDA applicant files a paragraph 4
certification, it is required to provide the patent holder with
notice of that certification. If the patent holder brings suit
against the ANDA applicant for patent infringement within
45 days of receiving notice, the FDA may not approve the
ANDA until the earlier of (i) 30 months from the
patent holder’s receipt of the notice (the
30-month
stay) or (ii) the issuance of a final, non-appealed, or
non-appealable court decision finding the patent invalid,
unenforceable or not infringed.
The Hatch-Waxman Act also provided an incentive for generic
manufacturers to file paragraph 4 certifications
challenging patents that may be invalid unenforceable, or not
infringed, whereby the first company to successfully challenge a
listed patent and receive ANDA approval is protected from
competition from subsequent generic versions of the same drug
product for 180 days after the earlier of (1) the date
of the first commercial marketing of the first-filed ANDA
applicant’s generic drug or (2) the date of a decision
of a court in an action holding the relevant patent invalid,
unenforceable, or not infringed. These
180-day
exclusivity provisions have been the subject of litigation and
administrative review, and the Medicare Prescription Drug,
Improvement, and Modernization Act of 2003, or MMA, amended the
provisions in several ways, including by providing that an ANDA
applicant entitled to
180-day
exclusivity may lose such exclusivity if any of the following
events occur: (1) failure to market; (2) withdrawal of
the ANDA; (3) change in patent certification;
(4) failure to obtain tentative approval; (5) illegal
settlement agreement; and (6) patent expiration.
With respect to the illegal settlement prong, the MMA amendments
require that certain types of settlement agreements entered into
between branded and generic pharmaceutical companies related to
the manufacture, marketing and sale of generic versions of
branded drugs are required to be filed with the Federal Trade
Commission and the Department of Justice for review of potential
anti-competitive practices. This requirement could affect the
manner in which generic drug manufacturers resolve intellectual
property litigation and other disputes with branded
pharmaceutical companies, and could result generally in an
increase in private-party litigation against pharmaceutical
companies. The impact of this requirement, and the potential
governmental investigations and private-party lawsuits
associated with arrangements between brand name and generic drug
manufacturers, remains uncertain and could adversely affect our
business. In addition, Congress has considered enacting
legislation that would prohibit such settlements between brand
name and generic drug manufacturers. Such a provision was
considered as part of the recently enacted healthcare reform,
the Patient Protection and Affordable Care Act or PPACA signed
into law on March 23, 2010. However, Congress removed the
provision prior to passage. It is possible that Congress will
again consider a ban on such settlements between brand name and
generic drug manufacturers in the future.
With the passage of the PPACA, there are now exclusivity
protections for certain innovator biological products and a
framework for FDA review and approval of biosimilar and
interchangeable versions of innovator biologic
23
products. The PPACA provides that no application for a
biosimilar product may be approved until 12 years after the
date on which the innovator product was first licensed, and no
application may be submitted until four years after the date of
first licensure. Products deemed interchangeable (as opposed to
biosimilar) are also eligible for certain exclusivity.
Please also read our discussion of patent and intellectual
property matters in Item 1A “Risk Factors”
section of this report.
Orphan
Drug Designation
Some jurisdictions, including Europe and the United States, may
designate drugs for relatively small patient populations as
“orphan” drugs. The FDA may grant orphan drug
designation to a drug intended to treat a rare disease or
condition that affects fewer than 200,000 individuals in the
United States, and a drug may also be considered an orphan even
if the drug treats a disease or condition affecting more than
200,000 individuals in the United States where the drug has no
expected profitability. Orphan drug designation does not
necessarily convey any advantage in, or shorten the duration of,
the regulatory review and process for marketing approval. If a
product with an orphan drug designation subsequently receives
the first FDA approval for the indication for which it has such
designation, the product is entitled to seven years of orphan
drug exclusivity, during which time FDA will not approve any
other application to market the same drug for the same
indication except in limited circumstances, such as a showing of
clinical superiority to the product with orphan exclusivity.
Also, competitors are not prohibited from receiving approval to
market the same drug or biologic for a different indication than
that which received orphan approval.
Under European Union medicines laws, the criteria for
designating an “orphan medicinal product” are similar
in principle to those in the United States. Criteria for orphan
designation are set out in Article 3 of Regulation (EC)
141/2000 on the basis of two alternative conditions. A medicinal
product may be designated as orphan if it is intended for the
diagnosis, prevention or treatment of a life-threatening or
chronically debilitating condition affecting not more than five
in 10 thousand persons in the European Union, or EU, when the
application is made. This is commonly known as the “disease
prevalence criterion” Alternatively, a product may be so
designated if it is intended for the diagnosis, prevention or
treatment of a life-threatening, seriously debilitating or
serious and chronic condition in the EU and if without
incentives it is unlikely that the marketing of the product in
the EU would generate sufficient return to justify the necessary
investment. This is commonly known as the “insufficient
return criterion.”
These two alternative criteria must cumulatively meet the second
condition that there exists no satisfactory method of diagnosis,
prevention or treatment of the condition in question that has
been authorized in the EU, or if such a method exists, the
product will be of significant benefit to those affected by the
condition. “Significant benefit” is defined in
Regulation (EC) 847/2000 as a clinically relevant advantage or a
major contribution to patient care.
Upon grant of a marketing authorization, orphan medicinal
products are entitled to ten years of market exclusivity in
respect of the approved therapeutic indication. Within the
period of market exclusivity, no competent authority in the EU
is permitted to accept an application for marketing
authorization, a variation or a line-extension for the same
approved therapeutic indication in respect of a similar
medicinal product pursuant to Article 8.1 of
Regulation 141/2000 unless one of derogations set out in
Article 8.3 of the same Regulation applies. In order to
determine whether two products are considered similar,
Regulation 847/2000 requires an assessment of the principal
molecular structure and the underlying mode of action. Any minor
variation or modification of the principal molecular structure
would not ordinarily render the second product dissimilar to the
first authorized product.
In order for the second applicant to break the market
exclusivity granted to the first authorized similar medicinal
product in respect of the same therapeutic indication, the
second applicant would principally rely upon data to demonstrate
that his product is safer, more efficacious or clinically
superior to the first product pursuant to Article 8.3I of
Regulation 141/2000. Ordinarily, such an assessment will
require a
head-to-head
comparative clinical trial for the purpose of demonstrating
clinical superiority.
24
The 10-year
market exclusivity may be reduced to 6 years if at the end
of the fifth year it is established that the product no longer
meets the criteria for orphan designation on the basis of
available evidence.
Fusilev has been granted orphan drug designations for its use in
conjunction with high dose methotrexate in the treatment of
osteosarcoma and for its use in combination chemotherapy with
the approved agent 5-fluorouracil in the palliative treatment of
metastatic adenocarcinoma of the colon and rectum (colorectal
cancer). In addition, belinostat has been granted an orphan drug
designation for PTCL. As discussed above, a drug with orphan
designation status may obtain orphan exclusivity upon marketing
approval under specified conditions set out in the applicable
laws and regulations.
Governmental
Regulation
The development, production and marketing of our proprietary and
generic drug products are subject to regulation for safety,
efficacy and quality by numerous governmental authorities in the
United States and other countries. In the United States, drugs
are subject to rigorous regulation. The Federal Food, Drug, and
Cosmetic Act, as amended from time to time, and the regulations
promulgated there under, as well as other federal and state
statutes and regulations, govern, among other things, the
development, approval, manufacture, safety, labeling, storage,
record keeping, distribution, promotion, and advertising of our
products. Product development and approval within this
regulatory framework, including for drugs already at a clinical
stage of development, can take many years and require the
expenditure of substantial resources, and to obtain FDA
approval, a product must satisfy mandatory procedures and safety
and efficacy requirements. In addition, each drug-manufacturing
establishment must be registered with the FDA. Domestic
manufacturing establishments must comply with the FDA’s
current good manufacturing practice, orcGMP, regulations and are
subject to inspections by the FDA. To supply drug ingredients or
products for use in the United States, foreign manufacturing
establishments must also comply with cGMP and are subject to
inspections by the FDA or by other regulatory authorities in
certain countries under reciprocal agreements with the FDA.
General
Information about the Drug Approval Process and Post-Marketing
Requirements
The United States system of new drug approval is one of the most
rigorous in the world. Only a small percentage of compounds that
enter the pre-clinical testing stage are ever approved for
commercialization. Our strategy focuses on in-licensing clinical
stage drug products that are already in or about to enter human
clinical trials. A late-stage focus helps us to effectively
manage the high cost of drug development by focusing on
compounds that have already passed the many hurdles in the
pre-clinical and early clinical process.
The following general comments about the drug approval process
are relevant to the development activities we are undertaking
with our proprietary drugs.
Pre-clinical Testing: During the pre-clinical
testing stage, laboratory and animal studies are conducted to
show biological activity of a drug compound against the targeted
disease and the compound is evaluated for safety.
Investigational New Drug Application: After
pre-clinical testing, an Investigational New Drug, or IND,
Application is submitted to the FDA to request the ability to
begin human testing of the drug. An IND becomes effective thirty
days after the FDA receives the application (unless the FDA
notifies the sponsor of a clinical hold), or upon prior
notification by the FDA.
Phase 1 Clinical Trials: These trials,
typically involving small numbers of healthy volunteers or
patients, usually define a drug candidate’s safety profile,
including the safe dosage range.
Phase 2 Clinical Trials: In phase 2 clinical
trials, controlled studies of human patients with the targeted
disease are conducted to assess the drug’s effectiveness.
These studies are designed primarily to determine the
appropriate dose levels, dose schedules and route(s) of
administration, and to evaluate the effectiveness of the drug on
humans, as well as to determine if there are any side effects on
humans to expand the safety profile following phase 1. These
clinical trials, and phase 3 trials discussed below, are
designed to evaluate the drug’s overall benefit-risk
profile, and to provide information to inform physician labeling.
25
Phase 3 Clinical Trials: This phase usually
involves larger number of patients with the targeted disease.
Investigators (typically physicians) monitor the patients to
determine the drug candidate’s efficacy and to observe and
report any adverse reactions that may result from long-term use
of the drug on a large, more widespread, patient population.
During the phase 3 clinical trials, typically the drug candidate
is compared to either a placebo or a standard treatment for the
target disease.
New Drug Application: After completion of all
three clinical trial phases, if the data indicates that the drug
is safe and effective, a NDA is filed with the FDA requesting
FDA approval to market the new drug as a treatment for the
target disease.
Fast Track and Priority Review: The FDA has
established procedures for accelerating the approval of drugs to
be marketed for serious or life threatening diseases for which
the manufacturer can demonstrate the potential to address unmet
medical needs.
Abbreviated New Drug Application: An ANDA is
the abbreviated review and approval process for generic drugs
created by the Hatch-Waxman Act. When a company files an ANDA,
it must make a patent certification regarding the patents
covering the branded product listed in the FDA’s Orange
Book. The ANDA drug development and approval process generally
takes less time than the NDA drug development and approval
process since the ANDA process usually does not require new
clinical trials establishing the safety and efficacy of the drug
product.
NDA and ANDA Approval: The FDA approves drugs
that are subject to NDA review based on data in the application
demonstrating the drug is safe and effective in its proposed
use(s) and that the drug’s benefits outweigh its risks. FDA
will also review the NDA applicant’s manufacturing process
and controls to ensure they are adequate to preserve the
drug’s identity, strength, quality, and purity, and FDA
will review and approve the drug’s proposed labeling. As
for the ANDA approval process, these “abbreviated”
applications are generally not required to include preclinical
or clinical data to establish safety and effectiveness. Rather,
an ANDA must demonstrate both chemical equivalence and
bio-equivalence (the rate and extent of absorption in the body)
to the innovator drug — unless a bio-equivalence
waiver is granted by the FDA.
Phase 4 Clinical Trials: After a drug has been
approved by the FDA, phase 4 studies may be conducted to explore
additional patient populations, compare the drug to a
competitor, or to further study the risks, benefits and optimal
use of a drug. These studies may be a requirement as a condition
of the initial approval of the NDA.
Post-Approval Studies Requirements under
FDAAA: The Food and Drug Administration
Amendments Act of 2007, or FDAAA, which was signed into law in
September 2007, significantly added to the FDA’s authority
to require post-approval studies. Under the FDAAA, if the FDA
becomes aware of new safety information after approval of a
product, they may require us to conduct further clinical trials
to assess a known serious risk, signals of serious risk or to
identify an unexpected serious risk. If required to conduct a
post-approval study, periodic status reports must be submitted
to the FDA. Failure to conduct such post-approval studies in a
timely manner may result in administrative action being taken by
FDA, including substantial civil fines.
Risk Evaluation and Mitigation Strategy Authority under
FDAAA: The FDAAA also gave the FDA new authority
to require the implementation of a Risk Evaluation and
Mitigation Strategy, or REMS, for a product when necessary to
minimize known and preventable safety risks associated with the
product. The FDA may require the submission of a REMS before a
product is approved, or after approval based on “new safety
information,” including new analyses of existing safety
information. A REMS may include a medication guide, patient
package insert, a plan for communication with healthcare
providers, or other elements as the FDA deems are necessary to
assure safe use of the product, which could include imposing
certain restrictions on distribution or use of a product. A REMS
must include a timetable for submission of assessments of the
strategy at specified time intervals. Failure to comply with a
REMS, including the submission of a required assessment, may
result in substantial civil or criminal penalties.
Other Issues Related to Product
Safety: Adverse events that are reported after
marketing approval also can result in additional limitations
being placed on a product’s use and, potentially,
withdrawal of the product from the market. In addition, under
the FDAAA, the FDA has authority to mandate labeling changes to
products at any point in a product’s lifecycle based on new
safety information derived from clinical trials, post-approval
studies, peer-reviewed medical literature, or post-market risk
identification and analysis systems data.
26
FDA
Enforcement
The development of drug products, as well as the marketing of
approved drugs, is subject to substantial continuing regulation
by the FDA, including regulation of adverse event reporting,
manufacturing practices and the advertising and promotion of the
drug. Failure to comply with the FDA and other governmental
regulations can result in fines, unanticipated compliance
expenditures, recall or seizure of products, total or partial
suspension of production
and/or
distribution, suspension of the FDA’s review of NDAs, ANDAs
or other product applications, enforcement actions, injunctions
and criminal prosecution. Under certain circumstances, the FDA
also has the authority to revoke previously granted drug
approvals. Although we have internal compliance programs, if
these programs do not meet regulatory agency standards or if our
compliance is deemed deficient in any significant way, it could
have a material adverse effect on our business. See Item 1A
“Risks Factors — Our failure to comply with
governmental regulation may delay or prevent approval of our
products
and/or
subject us to penalties.”
With respect specifically to information submitted to FDA in
support of marketing applications, the FDA, under its Fraud,
Untrue Statements of Material Facts, Bribery and Illegal
Gratuities Policy, can significantly delay the approval of a
marketing application, or seek to withdraw an approved
application where it identifies fraud or discrepancies in
regulatory submissions. Such actions by the FDA may
significantly delay or suspend substantive scientific review of
a pending application during validity assessment or remove
approved products from the market until the assessment is
complete and questions regarding reliability of the data are
resolved. In addition, the Generic Drug Enforcement Act of 1992
established penalties for wrongdoing in connection with the
development or submission of an ANDA. Under this Act, the FDA
has the authority to permanently or temporarily bar companies or
individuals from submitting or assisting in the submission of an
ANDA, and to temporarily deny approval and suspend applications
to market generic drugs. The FDA may also suspend the
distribution of all drugs approved or developed in connection
with certain wrongful conduct
and/or
withdraw approval of an ANDA and seek civil penalties.
Healthcare
Reform
Continuing studies of the proper utilization, safety and
efficacy of pharmaceuticals and other health care products are
being conducted by industry, government agencies and others.
Such studies, which increasingly employ sophisticated methods
and techniques, can call into question the utilization, safety
and efficacy of previously marketed products and in some cases
have resulted, and may in the future result, in the
discontinuance of their marketing.
The Patient Centered Outcomes Research Institute, a private,
non-profit corporation created as a result of the PPACA, is
tasked with assisting patients, clinician, purchasers, and
policy-makers in making informed health decisions. One of the
Institute’s initiatives will be to conduct comparative
clinical effectiveness research, which is defined as
“research evaluating and comparing health outcomes and the
clinical effectiveness, risks, and benefits of 2 or more medical
treatments, services, and items.” It is important to note
that the Institute would not be permitted to mandate coverage,
reimbursement, or other policies for any public or private
payer, however the outcome of the Institute’s initiatives
could influence prescriber behavior.
Foreign
Regulation
Whether or not we obtain FDA approval for a product, we must
obtain approval of a product by the comparable regulatory
authorities of foreign countries before we can commence clinical
trials or marketing of the product in those countries. The
approval process varies from country/region to country/region,
and the time may be longer or shorter than that required for FDA
approval. The requirements governing the conduct of clinical
trials, product licensing, pricing and reimbursement also may
vary, sometimes significantly, from country/region to
country/region.
Under the EU regulatory systems, we may submit marketing
authorization applications either under a centralized procedure
or decentralized procedure or the mutual recognition procedure.
The centralized procedure is mandatory for medicines produced by
a biotechnological process. The procedure is also mandatory for
new active substances which are indicated for treatment of
several diseases or conditions, including cancer and orphan
conditions. Companies may apply for centralized assessment if
the product contains a new active substance or the
27
product constitutes significant therapeutic, scientific or
technical innovation or the granting of authorization under the
centralized procedure is in the interests of the EU patients. A
centralized marketing authorization is valid in all European
Union member states. This marketing authorization is issued in
the form of a Commission decision which is legally binding in
its entirety to which it is addressed.
Directive 2004/27/EC introduced two parallel procedures to the
centralized procedure to allow a product to be progressively
authorized in each of the member states of the EU. They are the
decentralized procedure and the mutual recognition procedure.
The mutual recognition procedure applies where the product has
already been authorized in a member state of the EU that will
act as reference member state. The national marketing
authorization granted by the reference member state forms the
basis for mutual recognition in the member states chosen by the
applicant. In the decentralized procedure, the product in
question is not authorized in any one the EU member states. In
such a situation, the applicant company will request a member
state to act as the reference member state to lead the
scientific assessment for the benefit/risk balance for agreement
by the concerned member states. In both cases, the concerned
member states have up to 90 days to accept or raise
reasoned objections to the assessment made by the reference
member state.
In addition, pricing and reimbursement is subject to negotiation
and regulation in most countries outside the United States.
Increasingly, adoption of a new product for use in national
health services is subject to health technology assessment under
the national rules and regulations to establish the clinical
effectiveness and cost-effectiveness of a new treatment. In some
countries, in order to contain health care expenditures,
reference price is introduced in order for the national
healthcare providers to achieve a price comparable to the
reference price in the same therapeutic category. We may
therefore face the risk that the resulting prices would be
insufficient to generate an acceptable return to us.
Third
Party Reimbursement and Pricing Controls
In the United States and elsewhere, sales of pharmaceutical
products depend in significant part on the availability of
reimbursement to the consumer from third-party payers, such as
government and private insurance plans. Third-party payers are
increasingly challenging the prices charged for medical products
and services. It is time-consuming and expensive for us to go
through the process of seeking reimbursement from Medicare and
private payers. Our products may not be considered cost
effective, and coverage and reimbursement may not be available
or sufficient to allow us to sell our products on a competitive
and profitable basis.
The PPACA enacted significant reforms, including revising the
definition of “average manufacturer price” for
reporting purposes, increasing Medicaid rebates, expanding the
340B drug discount program, and making changes to affect the
Medicare Part D coverage gap, or “donut hole.” In
the coming years, additional significant changes could be made
to governmental healthcare programs, and the United States
healthcare system as a whole, that may result in significantly
increased rebates, decreased pricing flexibility, diminished
negotiating flexibility, coverage and reimbursement limitations
based upon comparative and cost-effectiveness reviews, and other
measures that could significantly impact the success of our
products.
In many foreign markets, including the countries in the EU,
pricing of pharmaceutical products is subject to governmental
control. In the United States, there have been, and we expect
that there will continue to be, a number of federal and state
proposals to implement similar governmental pricing control.
While we cannot predict whether such legislative or regulatory
proposals will be adopted, the adoption of such proposals could
have a material adverse effect on our business, financial
condition and profitability.
Employees
The efforts of our employees are critical to our success. We
believe that we have assembled a strong management team with the
experience and expertise needed to execute our business
strategy. We anticipate hiring additional personnel as needs
dictate to implement our growth strategy. As of
December 31, 2010, we had 139 employees, of which 10
held a M.D. degree and 8 held a Ph.D. degree. We cannot be sure
that we will be able to attract and retain qualified personnel
in sufficient numbers to meet our needs. Our employees are not
subject to any collective bargaining agreements, and we regard
our relations with our employees to be good.
28
Corporate
Background and Available Information
We are a Delaware corporation that was originally incorporated
in Colorado as Americus Funding Corporation in December 1987,
became NeoTherapeutics, Inc. in August 1996, was reincorporated
in Delaware in June 1997, and was renamed Spectrum
Pharmaceuticals, Inc. in December 2002.
We also maintain websites located at
http://www.sppirx.com
and
http://www.spectrumpharm.com,
and electronic copies of our periodic and current reports, proxy
statements for our annual stockholder’s meetings, and any
amendments to those reports, are available, free of charge,
under the “Investor Relations” link on our website as
soon as practicable after such material is filed with, or
furnished to, the SEC.
For financial information regarding our business activities,
please see “Item 8 — Financial Statements
and Supplementary Data.”
In addition to other information included in this Annual Report
on
Form 10-K,
the following factors, among others, could cause actual results
to differ materially from those contained in forward-looking
statements contained in this Annual Report on
Form 10-K,
and thus should be considered carefully in evaluating our
business and future prospects. The following risk factors are
not an exhaustive list of the risks associated with our
business. New factors may emerge or changes to these risks could
occur that could materially affect our business.
Risks
Related to Our Business
Like
other early-stage biotech companies, we have a history of
operating losses and our losses may continue to increase as we
expand our commercialization and development efforts, and our
efforts may never result in profitability.
Our cumulative losses since our inception in 1987 through
December 31, 2010 were approximately $310.4 million.
Our net losses in 2010, 2009 and 2008 were approximately
$48.8 million, $19.0 million and $14.2 million,
respectively, after recording approximately $2.7 million,
$8.1 million and $1.3 million, respectively, of
warrant based income. We expect to continue to incur additional
losses as we implement our growth strategy of commercializing
our approved drug products and developing our pipeline products
for at least the next few years. We may never achieve
significant revenues from sales of products or become
profitable. Even if we eventually generate significant revenues
from sales, we will likely continue to incur losses over the
next several years.
Our
business does not generate sufficient cash to finance our
ongoing operations and therefore, we will likely need to
continue to raise additional capital.
Our current commercial operations do not generate sufficient
operating cash to finance the clinical development of all our
drug products, to commercialize our approved drug products and
to capitalize on growth opportunities. While we have been
successful recently in generating funds through the licensing
and sale of our assets, we have historically relied primarily on
raising capital through the sale of our securities and
out-licensing our drug products to meet our financial needs.
Although we began selling products in 2008, we believe that in
the near-term we will likely need to continue to raise funds in
order to continue drug product commercialization, development
and acquisition.
We may not be able to raise additional capital on favorable
terms, if at all, particularly with the current volatile
financial market conditions. Accordingly, we may be forced to
significantly change our business plans and restructure our
operations to conserve cash, which would likely involve
out-licensing or selling some or all of our intellectual,
technological and tangible property not presently contemplated
and at terms that we believe would not be favorable to us,
and/or
reducing the scope and nature of our currently planned drug
development and commercialization activities. An inability to
raise additional capital would also materially impact our
ability to expand operations.
29
Our
drug product Fusilev may not be more cost-effective than
competing drugs and otherwise may not have any competitive
advantage, which could hinder our ability to successfully
commercialize it.
Fusilev is a novel folate analog formulation and the
pharmacologically active isomer (the levo-isomer) of the racemic
compound calcium leucovorin, a product already approved for the
same indications our product is approved for. Leucovorin has
been sold as a generic product on the market for a number of
years. There are generic companies currently selling the product
and therefore, Fusilev competes against a low-cost alternative.
Also, Fusilev will be offered as part of a treatment regimen,
and that regimen may change to exclude Fusilev. Accordingly, it
may not gain acceptance by the medical field or become
commercially successful.
Our
revenue from Fusilev sales may not be sustainable and our
customer concentration is significant
Most of the Fusilev revenue the Company recorded in 2010
resulted from the leucovorin drug shortage, the depth of which
we cannot predict. Even with approval for colorectal cancer a
possibility in 2011, there is not surety that Fusilev sales will
be sustainable. Our customer concentration of Fusilev is high.
Sales to Customer A for the years ended December 31, 2010,
2009 and 2008 were 45.7%, 27.1%, and 35.7% respectively, of our
total consolidated gross product sales. If our relationship with
our top distributors is impaired our sales of Fusilev would be
negatively impacted.
If we
are unable to expand the approved usage of Fusilev, the
product’s operating results may be harmed, which could
adversely affect our financial and operating
results.
We have filed a supplemental new drug application for Fusilev
for use in combination with 5-FU-containing regimens in the
treatment of colorectal cancer. The greatest potential use of
this product is in this indication. If we are not able to obtain
approval for this indication, we may not recognize the full
anticipated value of our investment in the product and our
financial and operating results could be adversely affected.
Adverse
economic conditions may have material adverse consequences on
our business, results of operations and financial
condition.
Unpredictable and unstable changes in economic conditions,
including recession, inflation, increased government
intervention, or other changes, may adversely affect our general
business strategy. If the current equity and credit markets
further deteriorate, or do not continue to improve, it may make
any necessary debt or equity financing more difficult, more
costly, and more dilutive. While we believe we have adequate
capital resources to meet current working capital and capital
expenditure requirements, a radical economic downturn, a
double-dip recession, or an increase in our expenses could
require additional financing on less than attractive rates or on
terms that are excessively dilutive to existing stockholders.
Failure to secure any necessary financing in a timely manner and
on favorable terms could have a material adverse effect on our
growth strategy, financial performance and stock price and could
require us to delay or abandon clinical development plans or
plans to acquire additional technology.
These economic conditions not only limit our access to capital,
but also make it difficult for our customers and us to
accurately forecast and plan future business activities, and
they could cause businesses to slow spending on our products,
which would delay and lengthen sales cycles. Furthermore, during
challenging economic times, our customers may face issues
gaining timely access to sufficient credit, which could result
in an impairment of their ability to make timely payments to us.
In addition, the recent economic crisis could also adversely
impact our suppliers’ ability to provide us with materials
which would negatively impact on our business, financial
condition and results of operations.
Clinical
trials may fail to demonstrate the safety and efficacy of our
drug products, which could prevent or significantly delay
obtaining regulatory approval.
Prior to receiving approval to commercialize any of our drug
products, we must demonstrate with substantial evidence from
well-controlled clinical trials, and to the satisfaction of the
FDA, and other regulatory authorities in the United States and
other countries, that each of the products is both safe and
effective. For each drug product, we will need to demonstrate
its efficacy and monitor its safety throughout the process. If
such development is unsuccessful, our business and reputation
would be harmed and our stock price would be adversely affected.
30
All of our drug products are prone to the risks of failure
inherent in drug development. Clinical trials of new drug
products sufficient to obtain regulatory marketing approval are
expensive and take years to complete. We may not be able to
successfully complete clinical testing within the time frame we
have planned, or at all. We may experience numerous unforeseen
events during, or as a result of, the clinical trial process
that could delay or prevent us from receiving regulatory
approval or commercializing our drug products. In addition, the
results of pre-clinical studies and early-stage clinical trials
of our drug products do not necessarily predict the results of
later-stage clinical trials. Later-stage clinical trials may
fail to demonstrate that a drug product is safe and effective
despite having progressed through initial clinical testing. Even
if we believe the data collected from clinical trials of our
drug products is promising, such data may not be sufficient to
support approval by the FDA or any other United States or
foreign regulatory approval. Pre-clinical and clinical data can
be interpreted in different ways.
Accordingly, FDA officials could interpret such data in
different ways than we or our partners do which could delay,
limit or prevent regulatory approval. The FDA, other regulatory
authorities, our institutional review boards, our contract
research organizations, or we may suspend or terminate our
clinical trials for our drug products. Any failure or
significant delay in completing clinical trials for our drug
products, or in receiving regulatory approval for the sale of
any drugs resulting from our drug products, may severely harm
our business and reputation. Even if we receive FDA and other
regulatory approvals, our drug products may later exhibit
adverse effects that may limit or prevent their widespread use,
may cause the FDA to revoke, suspend or limit their approval, or
may force us to withdraw products derived from those drug
products from the market.
If we
are unable to maintain or obtain improved reimbursement rates
for Zevalin, the product’s sales may be harmed, which could
adversely affect our financial and operating
results.
Effective January 1, 2010, the Centers for
Medicare & Medicaid Services finalized a policy to
allow reimbursement for Zevalin in the Hospital Outpatient
Prospective Payment System, or HOPPS, based on the average sales
price methodology applicable to other injectable drugs and
biologicals. If we are not able to maintain this reimbursement
methodology in the HOPPS setting, we could face significant
difficulty in getting health care providers to prescribe
Zevalin, which will have an adverse impact on the product’s
expected sales, and in turn adversely impact our financial and
operating results.
We may
face difficulties in achieving broader market acceptance of
Zevalin if we do not invest significantly in our sales and
marketing infrastructure.
Sales of Zevalin in the United States declined over the several
years prior to our acquisition of the Zevalin assets. We believe
that an enhanced sales and marketing strategy for Zevalin, in
conjunction with efforts to obtain approval by the FDA for
expanded uses of Zevalin, has significant potential to increase
sales of and revenue from Zevalin in the future. However,
implementation of the sales and marketing strategy for Zevalin,
and the efforts to expand approved usage of Zevalin, will
require a continued significant investment of financial and
other resources by us for the foreseeable future and may not
ultimately increase Zevalin sales or allow us to realize the
anticipated benefits from our investment in the product.
Additionally, our efforts to establish an effective commercial
team for Zevalin will require significant commitments of both
financial and management resources by us, and may not ultimately
be successful due a variety of factors, including industry
competition for effective commercial personnel or the inability
of us to dedicate the necessary resources to those efforts.
We are
aware of several competitors attempting to develop and market
products competitive to Zevalin, which may reduce or eliminate
our commercial opportunity.
The pharmaceutical and biotechnology industries are intensely
competitive and subject to rapid and significant technological
changes, and a number of companies are pursuing the development
of pharmaceuticals and products that target the same diseases
and conditions that Zevalin targets. We cannot predict with
accuracy the timing or impact of the introduction of potentially
competitive products or their possible effect on our sales.
Certain potentially competitive products to Zevalin are in
various stages of development, some of which have been filed for
approval with the FDA or have been approved by regulatory
authorities in other countries. Also, there are many ongoing
studies with currently marketed products including
Rituxan®,
Treanda®
and other developmental products,
31
which may yield new data that could adversely impact the use of
Zevalin in specific states for which it has obtained FDA
approval. The introduction of competitive products to Zevalin
could significantly reduce the sales of Zevalin, which, in turn
would adversely impact our financial and operating results.
The
intellectual property and assets owned by our subsidiary, RIT,
are subject to a security agreement with Biogen that secures the
entity’s payment and other obligations to Biogen, and we
have guaranteed all of those obligations.
In connection with the formation of RIT, it entered into a
security agreement with Biogen pursuant to which RIT granted to
Biogen a first priority security interest in all of its assets,
which primarily consist of the Zevalin-related intellectual
property. The security agreement secures certain payment,
indemnification and other obligations of RIT to Biogen related
to Zevalin. If RIT were to default on certain of its obligations
to Biogen or generally become bankrupt or insolvent, Biogen
could seek to foreclose on the collateral under the security
agreement to satisfy RIT’s obligations. If RIT were to
default on its obligations to Biogen, and Biogen were to
foreclose on the collateral under the security agreement,
RIT’s business could be materially and adversely impacted,
which could in turn materially and adversely impact our
investment in RIT and our financial condition and results of
operations.
Additionally, we are a guarantor on all of RIT’s
obligations to Biogen. If RIT were to default on its obligations
to Biogen, Biogen could require us alone to satisfy all of those
obligations under our guarantee. The financial and other
obligations that we would incur as a result of any such default
could have a material and adverse effect on our financial
condition and results of operations.
The
marketing and sale of Fusilev and Zevalin may be adversely
affected by the marketing and sales efforts of third parties who
sell these products outside of our territories.
We have only licensed the rights to develop, market and sell
Fusilev in North America, and have licensed the rights to
develop, market and sell Zevalin in the United States. Other
companies market and sell the same products in other parts of
the world. If, as a result of other companies’ actions,
negative publicity is associated with either product, our own
efforts to successfully market and sell such products in our
markets may be adversely impacted.
Our
supply of active pharmaceutical ingredients, or APIs, and drug
products will be dependent upon the production capabilities of
contract manufacturing organizations, or CMOs, component and
packaging supply sources, other third-party suppliers, and other
providers of logistical services, some of whom are based
overseas and, if these parties are not able to meet our demands
and FDA scrutiny, we may be limited in our ability to meet
demand for our products, ensure regulatory compliance or
maximize profit on the sale of our products.
We have no internal manufacturing capacity for APIs or our drug
products, and, therefore, we have entered into agreements with
CMOs and other suppliers to supply us with APIs and our finished
dose drug products. Success in the development and marketing of
our drug products depends, in part, upon our ability to
maintain, expand and enhance our existing relationships and
establish new sources of supply. Some of the third-party
manufacturing facilities used in the production of APIs and our
drug products are located outside the United States. The
manufacture of APIs and finished drug products, including the
acquisition of compounds used in the manufacture of the finished
drug product, may require considerable lead times. We have
little or no control over the production processes of
third-party manufacturers, CMOs or other suppliers. Our ability
to source APIs and drug products is also dependent on providers
of logistical services who may be subject to disruptions that we
cannot predict or sufficiently plan around. Accordingly, while
we do not currently anticipate shortages of supply,
circumstances could arise in which we will not have adequate
supplies to timely meet our requirements or market demand for a
particular drug product could outstrip the ability of our supply
source to timely manufacture and deliver the product, thereby
causing us to lose sales. In addition, our ability to make a
profit on the sale of our drug products depends on our ability
to obtain price arrangements that ensure a supply of product at
favorable prices.
Additionally, all of our regular suppliers are sole-source
suppliers, including for Zevalin and Fusilev, and no currently
qualified alternative suppliers exist. Our efforts to qualify
additional suppliers of Fusilev may take longer than anticipated
and the timing for securing necessary regulatory approvals is
uncertain. If problems arise during the
32
production of a batch of our drug products, that batch of
product may have to be discarded. This could, among other
things, lead to increased costs, lost revenue, damage to
customer relations, time and expense spent investigating the
cause and, depending on the cause, similar losses with respect
to other batches or products. If problems are not discovered
before the product is released to the market, recall and product
liability costs may also be incurred. To the extent that one of
our suppliers experiences significant manufacturing problems,
this could have a material adverse effect on our revenues and
profitability.
Finally, reliance on CMOs entails risks to which we would not be
subject if we manufactured products ourselves, including
reliance on the third party for regulatory compliance and
adherence to the FDA’s current Good Manufacturing Practice,
or cGMP, requirements, the possible breach of the manufacturing
agreement by the CMO and the possibility of termination or
non-renewal of the agreement by the CMO, based on its own
business priorities, at a time that is costly or inconvenient
for us. Before we can obtain marketing approval for our drug
products, our CMO facilities must pass an FDA pre-approval
inspection. In order to obtain approval, all of the
facility’s manufacturing methods, equipment and processes
must comply with cGMP requirements. The cGMP requirements govern
all areas of record keeping, production processes and controls,
personnel and quality control. In addition, our CMOs will be
subject to on-going periodic inspection by the FDA and
corresponding state and foreign agencies for compliance with
cGMP regulations, similar foreign regulations and other
regulatory standards. We do not have control over our CMOs’
compliance with these regulations and standards. Any failure of
our third party manufacturers or us to comply with applicable
regulations, including an FDA pre-approval inspection and cGMP
requirements, could result in sanctions being imposed on them or
us, including warning letters, fines, injunctions, civil
penalties, failure of regulatory authorities to grant marketing
approval of our products, delay, suspension or withdrawal of
approvals, license revocation, seizures or recalls of product,
operation restrictions and criminal prosecutions, any of which
could significantly and adversely affect our business.
The
development of our drug product, apaziquone, may be adversely
affected if the development efforts of Allergan, who retained
certain rights to the product, are not successful.
In 2008, we entered into a co-development and license agreement
with Allergan, Inc., or Allergan, for the worldwide development
and commercialization of our drug product, apaziquone. Allergan
has agreed to partially fund development and commercialization
expenses for apaziquone. We do not fully control the drug
development process under the license agreement and may have
disagreements with our partner. In addition, if we do not
achieve certain milestones under the license agreement and it
has been determined that failure to achieve these milestones was
a result of our actions or inactions, Allergan is entitled to
assume additional control over the development process. As a
result, success of this product could depend, in part, upon the
efforts of Allergan. Allergan may not be successful in the
clinical development of the drug, obtaining approval of the
product by regulatory authorities, or the eventual
commercialization of apaziquone.
The
development of our drug product, belinostat, may be adversely
affected if the development efforts of TopoTarget, who retained
certain rights to the product, are not successful.
TopoTarget licensed to us the rights to develop and market
belinostat in the United States, Canada, Mexico and India.
TopoTarget is currently fully funding and overseeing one
clinical study underway with belinostat. In addition, TopoTarget
has agreed to partially fund other development expenses for
belinostat. We do not fully control the drug development process
under our agreement with TopoTarget and may have disagreements
with our partner. TopoTarget, or its partners, may conduct their
own clinical trials on belinostat for regulatory approval in all
other parts of the world. We will not have control over such
development activities and our ability to attain regulatory
approvals for belinostat may be adversely impacted if
TopoTarget’s efforts are not successful.
The
development of our drug product, ozarelix, may be adversely
affected if the development efforts of Aeterna Zentaris, who
retained certain rights to the product, are not
successful.
Aeterna Zentaris licensed to us the rights to develop and market
ozarelix worldwide except Japan, Korea, Indonesia, Malaysia, the
Philippines, and Singapore. Aeterna Zentaris, or its partners,
may conduct their own clinical trials on ozarelix for regulatory
approval in all these countries. We will not have control over
such
33
development activities and our ability to attain regulatory
approvals for ozarelix may be adversely impacted if Aeterna
Zentaris’ efforts are not successful.
Our
dependence on key executives, scientists and sales and marketing
personnel could impact the development and management of our
business.
We are highly dependent upon our ability to attract and retain
qualified scientific, technical sales and marketing and
managerial personnel. There is intense competition for qualified
personnel in the pharmaceutical and biotechnology industries,
and we cannot be sure that we will be able to continue to
attract and retain the qualified personnel necessary for the
development and management of our business. Although we do not
believe the loss of one individual would materially harm our
business, our business might be harmed by the loss of the
services of multiple existing personnel, as well as the failure
to recruit additional key scientific, technical and managerial
personnel in a timely manner. Much of the know-how we have
developed resides in our scientific and technical personnel and
is not readily transferable to other personnel. While we have an
employment agreement with our Chief Executive Officer, we do not
have employment agreements with any of our other key scientific,
technical and managerial employees.
As we
evolve from a company primarily involved in development to a
company also involved in commercialization, we may encounter
difficulties in managing our growth and expanding our operations
successfully.
We only recently began commercial sales of our products and have
had to increase our personnel accordingly, including
establishing a direct sales force and complete commercial team.
In addition, as we advance our drug products through clinical
trials, we will need to expand our development, regulatory,
manufacturing, marketing and sales capabilities or contract with
third parties to provide these capabilities for us. As our
operations expand, we expect that we will need to manage
additional relationships with such third parties, as well as
additional collaborators and suppliers. Maintaining these
relationships and managing our future growth will impose
significant added responsibilities on members of our management.
We must be able to: manage our development efforts effectively;
manage our clinical trials effectively; hire, train and
integrate additional management, development, administrative and
sales and marketing personnel; improve our managerial,
development, operational and finance systems and expand our
facilities, all of which may impose a strain on our
administrative and operational infrastructure. If we are not
able to effectively manage our growth, our product sales and
resulting revenues will be negatively impacted.
If we
acquire additional businesses, we may not be able to
successfully integrate their operations.
We regularly evaluate and, as appropriate, may make selective
acquisitions of businesses that we believe complement or augment
our existing business. We may not be able to integrate any
acquired business successfully or operate any acquired business
profitably. Issues that could delay or prevent integration of
the acquired business into our own include:
|
|
|
| |
•
|
conforming standards, controls, procedures and policies,
business cultures and compensation structures;
|
| |
| |
•
|
conforming information technology and accounting systems;
|
| |
| |
•
|
consolidating corporate and administrative infrastructures;
|
| |
| |
•
|
consolidating sales and marketing operations;
|
| |
| |
•
|
retaining existing customers and attracting new customers;
|
| |
| |
•
|
retaining key employees;
|
| |
| |
•
|
identifying and eliminating redundant and underperforming
operations and assets;
|
| |
| |
•
|
minimizing the diversion of management’s attention from
ongoing business concerns;
|
| |
| |
•
|
coordinating geographically dispersed organizations;
|
| |
| |
•
|
managing tax costs or inefficiencies associated with integrating
operations; and
|
34
|
|
|
| |
•
|
making any necessary modifications to operating control
standards to comply with the Sarbanes-Oxley Act of 2002 and the
rules and regulations promulgated thereunder.
|
If we are unable to successfully integrate our acquisitions with
our existing business, we may not obtain the advantages that the
acquisitions were intended to create, which may materially
adversely affect our business, results of operations, financial
condition and cash flows, our ability to develop and introduce
new products and the market price of our stock. Actual costs and
sales synergies, if achieved at all, may be lower than we expect
and may take longer to achieve than we anticipate. Furthermore,
the products of companies we acquire may overlap with our
products or those of our customers, creating conflicts with
existing relationships or with other commitments that are
detrimental to the integrated businesses.
Our
collaborations with outside scientists may be subject to change,
which could limit our access to their expertise.
We work with scientific advisors and collaborators at research
institutions. These scientists are not our employees and may
have other commitments that would limit their availability to
us. If a conflict of interest between their work for us and
their work for another entity arises, we may lose their
services, which could negatively impact our research and
development activities.
We may
rely on contract research organizations and other third parties
to conduct clinical trials and, in such cases, we are unable to
directly control the timing, conduct and expense of our clinical
trials.
We may rely, in full or in part, on third parties to conduct our
clinical trials. In such situations, we have less control over
the conduct of our clinical trials, the timing and completion of
the trials, the required reporting of adverse events and the
management of data developed through the trial than would be the
case if we were relying entirely upon our own staff.
Communicating with outside parties can also be challenging,
potentially leading to mistakes as well as difficulties in
coordinating activities. Outside parties may have staffing
difficulties, may undergo changes in priorities or may become
financially distressed, adversely affecting their willingness or
ability to conduct our trials. We may experience unexpected cost
increases that are beyond our control. Problems with the
timeliness or quality of the work of a contract research
organization may lead us to seek to terminate the relationship
and use an alternative service provider. However, making this
change may be costly and may delay our trials, and contractual
restrictions may make such a change difficult or impossible.
Additionally, it may be impossible to find a replacement
organization that can conduct our trials in an acceptable manner
and at an acceptable cost.
We are
subject to risks associated with doing business
internationally.
Since we conduct clinical trials and manufacture our drug
products internationally, our business is subject to certain
risks inherent in international business, many of which are
beyond our control. These risks include, among other things:
|
|
|
| |
•
|
maintaining compliance with foreign legal requirements,
including employment law;
|
| |
| |
•
|
unexpected changes in foreign regulatory requirements, including
quality standards and other certification requirements;
|
| |
| |
•
|
tariffs, customs, duties and other trade barriers;
|
| |
| |
•
|
changing economic conditions in countries where our products are
manufactured;
|
| |
| |
•
|
exchange rate risks;
|
| |
| |
•
|
product liability, intellectual property and other claims;
|
| |
| |
•
|
political instability;
|
| |
| |
•
|
new export license requirements; and
|
| |
| |
•
|
difficulties in coordinating and managing foreign operations.
|
Any of these factors could have an adverse effect on our
business, financial condition and results of operations.
35
We may
have conflicts with our partners that could delay or prevent the
development or commercialization of our drug
products.
We may have conflicts with our partners, such as conflicts
concerning the interpretation of preclinical or clinical data,
the achievement of milestones, the interpretation of contractual
obligations, payments for services, development obligations or
the ownership of intellectual property developed during our
collaboration. If any conflicts arise with any of our partners,
such partner may act in a manner that is adverse to our best
interests. Any such disagreement could result in one or more of
the following, each of which could delay or prevent the
development or commercialization of our drug product, and in
turn prevent us from generating revenues:
|
|
|
| |
•
|
unwillingness on the part of a partner to pay us milestone
payments or royalties that we believe are due to us under a
collaboration;
|
| |
| |
•
|
uncertainty regarding ownership of intellectual property rights
arising from our collaborative activities, which could prevent
us from entering into additional collaborations;
|
| |
| |
•
|
unwillingness by the partner to cooperate in the development or
manufacture of the product, including providing us with product
data or materials;
|
| |
| |
•
|
unwillingness on the part of a partner to keep us informed
regarding the progress of its development and commercialization
activities or to permit public disclosure of the results of
those activities;
|
| |
| |
•
|
initiation of litigation or alternative dispute resolution
options by either party to resolve the dispute;
|
| |
| |
•
|
attempts by either party to terminate the collaboration;
|
| |
| |
•
|
our ability to maintain or defend our intellectual property
rights may be compromised by our partner’s acts or
omissions;
|
| |
| |
•
|
a partner may utilize our intellectual property rights in such a
way as to invite litigation that could jeopardize or invalidate
our intellectual property rights or expose us to potential
liability;
|
| |
| |
•
|
a partner may change the focus of its development and
commercialization efforts due to internal reorganizations,
mergers, consolidations and otherwise;
|
| |
| |
•
|
unwillingness of a partner to fully fund or commit sufficient
resources to the testing, marketing, distribution or development
of our products;
|
| |
| |
•
|
unwillingness or ability of a partner to fulfill their
obligations to us due to the pursuit of alternative products,
conflicts of interest that arise or changes in business strategy
or other business issues; and/or
|
| |
| |
•
|
we may not be able to guarantee supplies of development or
marketed products.
|
Given these risks, it is possible that any collaborative
arrangements which we have or may enter into may not be
successful.
Our
efforts to acquire or in-license and develop additional drug
products may fail, which might limit our ability to grow our
business.
To remain competitive and grow our business, our long-term
strategy includes the acquisition or in-license of additional
drug products. We are actively seeking to acquire, or
in-license, additional commercial drug products as well as drug
products that have demonstrated positive pre-clinical
and/or
clinical data. We have certain criteria that we are looking for
in any drug product acquisition and in-license and we may not be
successful in locating and acquiring, or in-licensing,
additional desirable drug products on acceptable terms.
To accomplish our acquisition and in-license strategy, we intend
to commit efforts, funds and other resources to research and
development and business development. Even with acquired and
in-licensed drug products, a high rate of failure is inherent in
the development of such products. We must make ongoing
substantial expenditures without any assurance that our efforts
will be commercially successful. Failure can occur at any point
in the process, including after significant funds have been
invested. For example, promising new drug product candidates may
fail to reach the market or may only have limited commercial
success because of efficacy or safety concerns, failure to
36
achieve positive clinical outcomes, inability to obtain
necessary regulatory approvals, limited scope of approved uses,
excessive costs to manufacture, the failure to establish or
maintain intellectual property rights or infringement of the
intellectual property rights of others.
In addition, many other large and small companies within the
pharmaceutical and biotechnology industry seek to establish
collaborative arrangements for product research and development,
or otherwise acquire products in late-stage clinical
development, in competition with us. We face additional
competition from public and private research organizations,
academic institutions and governmental agencies in establishing
collaborative arrangements for drug products in late-stage
clinical development. Many of the companies and institutions
that compete against us have substantially greater capital
resources, research and development staffs and facilities than
we have, and greater experience in conducting business
development activities. These entities represent significant
competition to us as we seek to expand our portfolio through the
in-license or acquisition of compounds. Finally, while it is not
feasible to predict the actual cost of acquiring and developing
additional drug products, that cost could be substantial and we
may need to raise additional financing for such purpose, which
may further dilute existing stockholders.
From
time to time we may need to license patents, intellectual
property and proprietary technologies from third parties, which
may be difficult or expensive to obtain.
We may need to obtain licenses to patents and other proprietary
rights held by third parties to successfully develop,
manufacture and market our drug products. As an example, it may
be necessary to use a third party’s proprietary technology
to reformulate one of our drug products in order to improve upon
the capabilities of the drug product. If we are unable to timely
obtain these licenses on reasonable terms, our ability to
commercially exploit our drug products may be inhibited or
prevented.
We are
a small company relative to our principal competitors, and our
limited financial resources may limit our ability to develop and
market our drug products.
Many companies, both public and private, including well-known
pharmaceutical companies and smaller niche-focused companies,
are developing products to treat many, if not all, of the
diseases we are pursuing or are currently distributing drug
products that directly compete with the drugs that we sell or
that we intend to develop, market and distribute. Many of these
companies have substantially greater financial, research and
development, manufacturing, marketing and sales experience and
resources than us. As a result, our competitors may be more
successful than us in developing their products, obtaining
regulatory approvals and marketing their products to consumers.
Competition for branded or proprietary drugs is less driven by
price and is more focused on innovation in the treatment of
disease, advanced drug delivery and specific clinical benefits
over competitive drug therapies. We may not be successful in any
or all of our current clinical studies; or if successful, and if
one or more of our drug products is approved by the FDA, we may
encounter direct competition from other companies who may be
developing products for similar or the same indications as our
drug products. Companies that have products on the market or in
research and development that target the same indications as our
products target include, among others, Abraxis Bioscience, Inc.,
Astra Zeneca LP, Bayer AG, Endo Pharmaceuticals, Eli Lilly and
Co., Novartis Pharmaceuticals Corporation, Genentech, Inc.,
Bristol-Myers Squibb Company, GlaxoSmithKline, Biogen-IDEC
Pharmaceuticals, Inc., OSI Pharmaceuticals, Inc., Cephalon,
Inc., Sanofi-aventis, Inc., Pfizer, Inc., Genta Incorporated,
Merck, Celgene Corporate, Allos Therapeutics, Inc., BiPar
Sciences, Inc., Genzyme Corporation, Shire Pharmaceuticals,
Abbott Laboratories, Poniard Pharmaceuticals, Inc., Roche
Pharmaceuticals and Johnson & Johnson who may be more
advanced in the development of competing drug products or are
more established. Many of our competitors are large and
well-capitalized companies focusing on a wide range of diseases
and drug indications, and have substantially greater financial,
research and development, marketing, human and other resources
than we do. Furthermore, large pharmaceutical companies have
significantly more experience than we do in pre-clinical
testing, human clinical trials and regulatory approval
procedures, among other things.
37
Our
drug products may not be more effective, safer or more
cost-efficient than a competing drug and otherwise may not have
any competitive advantage, which could hinder our ability to
successfully commercialize our drug products.
Any drug product for which we obtain FDA approval must compete
for market acceptance and market share. Drugs produced by other
companies are currently on the market for each disease type we
are pursuing. Even if one or more of our drug development
products ultimately receives FDA approval, our drug products may
not have better efficacy in treating the target indication than
a competing drug, may not have a more favorable side-effect
profile than a competing drug, may not be more cost-efficient to
manufacture or apply, or otherwise may not demonstrate a
competitive advantage over competing therapies. Accordingly,
even if FDA approval is obtained for one or more of our drug
development products, they may not gain acceptance by the
medical field or become commercially successful.
The
size of the market for our potential products is
uncertain.
We often provide estimates of the number of people who suffer
from the diseases that our drugs are targeting. However, there
is limited information available regarding the actual size of
these patient populations. In addition, it is uncertain whether
the results from previous or future clinical trials of drug
products will be observed in broader patient populations, and
the number of patients who may benefit from our drug products
may be significantly smaller than the estimated patient
populations.
If
actual future payments for allowances, discounts, returns,
rebates and chargebacks exceed the estimates we made at the time
of the sale of our products, our financial position, results of
operations and cash flows may be materially and negatively
impacted.
We recognize product revenue net of estimated allowances for
discounts, returns, rebates and chargebacks. Such estimates
require our most subjective and complex judgment due to the need
to make estimates about matters that are inherently uncertain.
Based on industry practice, pharmaceutical companies, including
us, have liberal return policies. Generally, we are obligated to
accept from customers the return of pharmaceuticals that have
reached their expiration date up to twelve months after their
expiration. We authorize returns for damaged products and
exchanges for expired products in accordance with our return
goods policy and procedures. In addition, like our competitors,
we also give credits for chargebacks to wholesale customers that
have contracts with us for their sales to hospitals, group
purchasing organizations, pharmacies or other retail customers.
A chargeback is the difference between the price the wholesale
customer (in our case, the GPOs) pays (wholesale acquisition
cost) and the price that the GPO’s end-customer pays for a
product (contracted customer). Since we have only recently begun
commercial distribution of our products, we do not have
historical data on returns and allowances. Although we believe
that we have estimated the allowances very conservatively,
actual results may differ significantly from our estimated
allowances for discounts, returns, rebates and chargebacks.
Changes in estimates and assumptions based upon actual results
may have a material impact on our results of operations
and/or
financial condition. Such changes to estimates will be made to
the financial statements in the year in which the estimate is
charged. In addition, our financial position, results of
operations and cash flows may be materially and negatively
impacted if actual future payments for allowances, discounts,
returns, rebates and chargebacks exceed the estimates we made at
the time of the sale of our products.
Earthquakes
or other natural or man-made disasters and business
interruptions could adversely affect our business.
Our operations are vulnerable to interruption by fire, power
loss, floods, telecommunications failure and other events beyond
our control. In addition, our operations are susceptible to
disruption as a result of natural disasters such as earthquakes.
So far we have never experienced any significant disruption of
our operations as a result of earthquakes or other natural
disasters. Although we have a contingency recovery plan, any
significant business interruption could cause delays in our drug
development and future sales and harm our business.
38
Risks
Related to Our Industry
If
third-party payors do not adequately reimburse providers for any
of our products, if approved for marketing, we may not be
successful in selling them.
Our ability to commercialize any products successfully will
depend in part on the extent to which reimbursement will be
available from governmental and other third-party payors, both
in the United States and in foreign markets. Even if we succeed
in bringing one or more products to the market, the amount
reimbursed for our products may be insufficient to allow us to
compete effectively and could adversely affect our profitability.
Reimbursement by a governmental and other third-party payors may
depend upon a number of factors, including a governmental or
other third-party payor’s determination that use of a
product is:
|
|
|
| |
•
|
a covered benefit under its health plan;
|
| |
| |
•
|
safe, effective and medically necessary;
|
| |
| |
•
|
appropriate for the specific patient;
|
| |
| |
•
|
cost-effective; and
|
| |
| |
•
|
neither experimental nor investigational.
|
Obtaining reimbursement approval for a product from each
third-party and governmental payor is a time-consuming and
costly process that could require us to provide supporting
scientific, clinical and cost-effectiveness data for the use of
our products to each payor. We may not be able to provide data
sufficient to obtain reimbursement.
In the United States, there have been, and we expect there will
continue to be, a number of state and federal proposals that
limit the amount that private insurance plans may pay to
reimburse the cost of drugs, including our products. We believe
the increasing emphasis on managed care in the United States has
and will continue to put pressure on the price and usage of our
products, which may also impact sales of our products. In
addition, current third-party reimbursement policies for our
products may change at any time. Negative changes in
reimbursement or our failure to obtain reimbursement for our
products may reduce the demand for, or the price of, products,
which could result in lower sales of our products, thereby
weakening our competitive position and negatively impacting our
results of operations.
Eligibility for coverage does not imply that any drug product
will be reimbursed in all cases or at a rate that allows us to
make a profit. Interim payments for new products, if applicable,
may also not be sufficient to cover our costs and may not become
permanent. Reimbursement rates may vary according to the use of
the drug and the clinical setting in which it is used, may be
based on payments allowed for lower-cost drugs that are already
reimbursed, may be incorporated into existing payments for other
products or services, and may reflect budgetary constraints
and/or
Medicare or Medicaid data used to calculate these rates. Net
prices for products also may be reduced by mandatory discounts
or rebates required by government health care programs or by any
future relaxation of laws that restrict imports of certain
medical products from countries where they may be sold at lower
prices than in the United States.
Wholesaler
actions could increase competitive and pricing pressures on
pharmaceutical manufacturers, including us.
We sell Fusilev primarily through
wholesalers. These wholesale customers comprise a
significant part of the distribution network for pharmaceutical
products in the United States. A small number of large wholesale
distributors control a significant share of the market, which
can increase competitive and pricing pressures on pharmaceutical
manufacturers, including us. In addition, wholesalers may apply
pricing pressure through
fee-for-service
arrangements, and their purchases may exceed customer demand,
resulting in reduced wholesaler purchases in later quarters. We
cannot assure you that we can manage these pressures or that
wholesaler purchases will not decrease as a result of this
potential excess buying.
39
Rapid
bio-technological advancement may render our drug products
obsolete before we are able to recover expenses incurred in
connection with their development. As a result, our drug
products may never become profitable.
The pharmaceutical industry is characterized by rapidly evolving
biotechnology. Biotechnologies under development by other
pharmaceutical companies could result in treatments for diseases
and disorders for which we are developing our own treatments.
Several other companies are engaged in research and development
of compounds that are similar to our research. A competitor
could develop a new biotechnology, product or therapy that has
better efficacy, a more favorable side-effect profile or is more
cost-effective than one or more of our drug products and thereby
cause our drug products to become commercially obsolete. Some of
our drug products may become obsolete before we recover the
expenses incurred in their development. As a result, such
products may never become profitable.
Competition
for patients in conducting clinical trials may prevent or delay
product development and strain our limited financial
resources.
Many pharmaceutical companies are conducting clinical trials in
patients with the disease indications that our drug products
target. As a result, we must compete with them for clinical
sites, physicians and the limited number of patients who fulfill
the stringent requirements for participation in clinical trials.
Also, due to the confidential nature of clinical trials, we do
not know how many of the eligible patients may be enrolled in
competing studies and who are consequently not available to us
for our clinical trials. Our clinical trials may be delayed or
terminated due to the inability to enroll enough patients.
Patient enrollment depends on many factors, including the size
of the patient population, the nature of the trial protocol, the
proximity of patients to clinical sites and the eligibility
criteria for the study. The delay or inability to meet planned
patient enrollment may result in increased costs and delays or
termination of the trial, which could have a harmful effect on
our ability to develop products.
Failure
to obtain regulatory approval outside the United States will
prevent us from marketing our product candidates
abroad.
We intend to market certain of our existing and future product
candidates in outside of the United States. In order to market
our existing and future product candidates in the European Union
and many other foreign jurisdictions, we must obtain separate
regulatory approvals according to the applicable domestic laws
and regulations. We have had limited interactions with foreign
regulatory authorities, and the approval procedures vary among
countries and can involve additional testing, and the time
required to obtain approval may differ from that required to
obtain FDA approval. Approval by the FDA does not guarantee
approval by regulatory authorities in other countries, and
approval by one or more foreign regulatory authorities does not
necessarily ensure approval by regulatory authorities in other
countries or by the FDA. The foreign regulatory approval process
may include all of the risks associated with obtaining FDA
approval as well as other risks specific to the jurisdictions in
which we may seek approval. We may not obtain foreign regulatory
approvals on a timely basis, if at all. We may not be able to
file for foreign regulatory approvals and may not receive
necessary approvals to commercialize our existing and future
product candidates in any market.
Even
after we receive regulatory approval to market our drug
products, the market may not be receptive to our drug products
upon their commercial introduction, which would negatively
impact our ability to achieve profitability.
Our drug products may not gain market acceptance among
physicians, patients, healthcare payors and the medical
community. The degree of market acceptance of any approved drug
products will depend on a number of factors, including:
|
|
|
| |
•
|
the effectiveness of the drug product;
|
| |
| |
•
|
the prevalence and severity of any side effects;
|
| |
| |
•
|
potential advantages or disadvantages over alternative
treatments;
|
| |
| |
•
|
relative convenience and ease of administration;
|
40
|
|
|
| |
•
|
the strength of marketing and distribution support;
|
| |
| |
•
|
the price of the drug product, both in absolute terms and
relative to alternative treatments; and
|
| |
| |
•
|
sufficient third-party coverage or reimbursement.
|
If our drug products receive regulatory approval but do not
achieve an adequate level of acceptance by physicians,
healthcare payors and patients, we may not generate drug product
revenues sufficient to attain profitability.
Guidelines
and recommendations published by various organizations can
reduce the use of our products.
Government agencies such as the Centers for Medicare &
Medicaid Services promulgate regulations, and issue guidelines,
directly applicable to us and to our products. In addition,
third parties such as professional societies, practice
management groups, insurance carriers, physicians, private
health/science foundations and organizations involved in various
diseases from time to time may publish guidelines or
recommendations to healthcare providers, administrators and
payers, and patient communities. Recommendations may relate to
such matters as usage, dosage, route of administration and use
of related therapies and reimbursement of our products by
government and private payers. Third-party organizations like
the above have in the past made recommendations about our
products. Recommendations or guidelines that are followed by
patients and healthcare providers could result in decreased use
and/or
dosage of our products. Any recommendations or guidelines that
result in decreased use, dosage or reimbursement of our products
could adversely affect our product sales and operating results
materially.
Our
failure to comply with governmental regulations may delay or
prevent approval of our drug products and/or subject us to
penalties.
The FDA and comparable agencies in foreign countries impose many
requirements related to the drug development process through
lengthy and rigorous clinical testing and data collection
procedures, and other costly and time consuming compliance
procedures. While we believe that we are currently in compliance
with applicable FDA regulations, if our partners, the contract
research organizations or contract manufacturers with which we
have relationships, or we fail to comply with the regulations
applicable to our clinical testing, the FDA may delay, suspend
or cancel our clinical trials, or the FDA might not accept the
test results. The FDA, an institutional review board, third
party investigators, any comparable regulatory agency in another
country, or we, may suspend clinical trials at any time if the
trials expose subjects participating in such trials to
unacceptable health risks. Further, human clinical testing may
not show any current or future drug product to be safe and
effective to the satisfaction of the FDA or comparable
regulatory agencies, or the data derived from the clinical tests
may be unsuitable for submission to the FDA or other regulatory
agencies. Once we submit an application seeking approval to
market a drug product, the FDA or other regulatory agencies may
not issue their approvals on a timely basis, if at all. If we
are delayed or fail to obtain these approvals, our business and
prospects may be significantly damaged.
If we obtain regulatory approval for our drug products, we, our
partners, our manufacturers, and other contract entities will
continue to be subject to extensive requirements by a number of
national, foreign, state and local agencies. These regulations
will impact many aspects of our operations, including testing,
research and development, manufacturing, safety, effectiveness,
labeling, storage, quality control, adverse event reporting,
record keeping, approval, advertising and promotion of our
future products. Failure to comply with applicable regulatory
requirements could, among other things, result in:
|
|
|
| |
•
|
warning letters;
|
| |
| |
•
|
fines;
|
| |
| |
•
|
changes in advertising;
|
| |
| |
•
|
revocation or suspension of regulatory approvals of products;
|
| |
| |
•
|
product recalls or seizures;
|
| |
| |
•
|
delays, interruption, or suspension of product distribution,
marketing and sales;
|
41
|
|
|
| |
•
|
civil or criminal sanctions;
|
| |
| |
•
|
suspension or termination of ongoing clinical trials;
|
| |
| |
•
|
imposition of restrictions on our operations;
|
| |
| |
•
|
close the facilities of our contract manufacturers; and
|
| |
| |
•
|
refusals to approve new products.
|
The
discovery of previously unknown safety risks with drug products
approved to go to market may raise costs or prevent us from
marketing such products or change the labeling of our products
or take other potentially limiting or costly actions if we or
others identify safety risks after our products are on the
market.
The later discovery of previously unknown safety risks with our
products may result in the imposition of restrictions on
distribution or use of the drug product, including withdrawal
from the market. The FDA may revisit and change its prior
determinations with regard to the safety and efficacy of our
products. If the FDA’s position changes, we may be required
to change our labeling or to cease manufacture and marketing of
the products at issue. Even prior to any formal regulatory
action, we could voluntarily decide to cease the distribution
and sale or recall any of our products if concerns about their
safety or effectiveness develop.
The Food and Drug Administration Amendments Act of 2007
significantly added to the FDA’s authority, including
allowing the FDA to:
|
|
|
| |
•
|
require sponsors of marketed products to conduct
post-approval clinical studies to assess a known serious risk,
signals of serious risk or to identify an unexpected serious
risk;
|
| |
| |
•
|
mandate labeling changes to products, at any point in a
product’s lifecycle, based on new safety
information; and
|
| |
| |
•
|
require sponsors to implement a Risk Evaluation and Mitigation
Strategy, or REMS, for a product which could include a
medication guide, patient package insert, a communication plan
to healthcare providers, or other elements as the FDA deems are
necessary to assure safe use of the drug (either prior to
approval or post-approval as necessary).
|
Failure to comply with a REMS could result in significant civil
monetary penalties or other administrative actions by FDA.
Further, regulatory agencies could change existing, or
promulgate new, regulations at any time which may affect our
ability to obtain or maintain approval of our existing or future
products or require significant additional costs to obtain or
maintain such approvals.
Our
failure to comply with FDA (and related) regulations applicable
to our business may subject us to sanctions, which could damage
our reputation and adversely affect our business
condition.
In the United States, the FDA, and comparable state regulatory
agencies and enforcement authorities, impose requirements on us
as a manufacturer and marketer of prescription drug products.
Drug manufacturers are required to register with FDA, and are
required to comply with various regulatory requirements
regarding drug research, manufacturing, distribution, reporting
and recordkeeping. Most drug products must be approved by the
FDA prior to marketing, and companies are required to comply
with numerous post-marketing requirements.
Further, drug manufacturers are required to comply with FDA
requirements for labeling and advertising, as well as other
Federal and state requirements for advertising. This includes a
prohibition on promotion for unapproved or “off-label”
uses, e.g., promotion of products for uses that are not
described in the product’s FDA-approved labeling. While a
physician may prescribe a medication for off-label uses where
appropriate, companies may not generally promote drug products
for off-label uses.
If FDA or other Federal and state agencies believe that a
company is not in compliance with applicable regulations, they
have various enforcement authorities to address violations. FDA
can issue a warning letter and seek voluntary compliance from a
company in the form of remedial or corrective action. FDA may
also impose civil money penalties by administrative action, and
through judicial enforcement seek actions including injunctions,
42
seizures, and criminal penalties. FDA or other federal and state
authorities may also seek operating restrictions on a company in
order to achieve compliance, including termination or suspension
of company activities. Such agencies and enforcement authorities
may also disseminate information to the public about their
enforcement actions.
If we were to become subject to any FDA or similar enforcement
action related to any of our drug products, our business
condition could be adversely affected, and the public release of
such information could be damaging to our reputation.
Legislative
or regulatory reform of the healthcare system and pharmaceutical
industry related to pricing or reimbursement may hurt our
ability to sell our products profitably or at all.
In both the United States and certain foreign jurisdictions,
there have been and may continue to be a number of legislative
and regulatory proposals related to pricing and reimbursement
that could impact our ability to sell our products profitably.
The Patient Protection and Affordable Care Act and the Health
Care and Education Reconciliation Act of 2010 were signed into
law on March 23, 2010 and March 30, 2010,
respectively, and are referred to collectively as the Healthcare
Reform Acts. The Healthcare Reform Acts enacted provisions
including a revision to the definition of “average
manufacturer price” for reporting purposes, increasing
Medicaid rebates, expanding the 340B drug discount program, and
making changes to affect the Medicare Part D coverage gap,
or “donut hole.” These reforms will significantly
impact the pharmaceutical industry. The full effects of these
provisions will become apparent as these laws are implemented
and the Centers for Medicare & Medicaid Services and
other agencies issue applicable regulations or guidance as
required by the Acts. Moreover, in the coming years, additional
changes could be made to governmental healthcare programs that
could significantly impact the success of our products.
The sales of our products depend in part on the availability of
reimbursement from third-party payors such as government health
administration authorities, private health insurers, health
maintenance organizations including pharmacy benefit managers
and other health care-related organizations. Both the federal
and state governments in the United States and foreign
governments continue to propose and pass new legislation and
regulations designed to contain or reduce the cost of health
care. Such legislation and regulations may result in decreased
reimbursement for prescription drugs, which may further
exacerbate industry-wide pressure to reduce the prices charged
for prescription drugs. This could harm our ability to market
our products and generate revenues.
It is possible that proposals will be adopted, or existing
regulations that affect the coverage or pricing of
pharmaceutical and other medical products may change, before any
of our products are approved for marketing. Cost control
initiatives could decrease the price that we receive for any of
our products that we are developing. In addition, third-party
payors are increasingly challenging the price and
cost-effectiveness of medical products and services. Significant
uncertainty exists as to the reimbursement status of
newly-approved pharmaceutical products.
The high cost of pharmaceuticals continues to generate
substantial government interest. Various governmental entities
may focus on pharmaceutical prices by holding hearings or
launching investigations regarding the pricing for drugs by
pharmaceutical companies such as ours and the ability of
patients to obtain drugs. In December 2009, the Government
Accounting Office released its report on the growing cost of
brand-name prescription drugs. In addition, in July 2008, the
Joint Economic Committee of Congress held hearings on the
pricing of drugs for rare conditions. Future developments may
require us to decrease the price that we charge for our
products, thereby negatively affecting our financial results.
In some foreign countries, particularly in the European Union,
prescription drug pricing is subject to governmental control.
Drug pricing may be made against a reference price set by the
healthcare providers as a measure for healthcare cost
containment. Pricing negotiations with governmental authorities
can take considerable time after the receipt of marketing
approval for a product. To obtain reimbursement or pricing
approval in some countries, we may be required to conduct a
clinical trial that seeks to address the clinical effectiveness
and cost-effectiveness of our product candidate as compared with
other available therapies as part of the health technology
assessment. If reimbursement of our products is unavailable or
limited in scope or amount, or if pricing is set at
unsatisfactory levels for the purpose of adoption of these
products in the national health services in these jurisdictions,
our profitability will likely be negatively affected.
43
If we
market products in a manner that violates health care
anti-kickback or other anti-fraud and anti-abuse laws, we may be
subject to civil or criminal penalties, including exclusions
from participation in federal health care
programs.
The federal health care program anti-kickback statute prohibits,
among other things, knowingly and willfully offering, paying,
soliciting, or receiving remuneration to induce or in return for
purchasing, leasing, ordering, or arranging for the purchase,
lease or order of any health care item or service reimbursable
under Medicare, Medicaid or other federally financed health care
programs. This statute applies to arrangements between
pharmaceutical manufacturers and prescribers, purchasers and
formulary managers. Although there are a number of statutory
exemptions and regulatory safe harbors protecting certain common
activities, the exemptions and safe harbors are drawn narrowly,
and practices that involve remuneration intended to induce
prescribing, purchases or recommendations may be subject to
scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly
presenting, or causing to be presented, a false claim for
payment to the federal government, or knowingly making, or
causing to be made, a false statement to get a false claim paid.
Pharmaceutical companies have been prosecuted under these laws
for a variety of alleged promotional and marketing activities,
such as providing free product to customers with the expectation
that the customers would bill federal programs for the product;
reporting to pricing services inflated average wholesale prices
that were then used by federal programs to set reimbursement
rates; engaging in off-label promotion that caused claims to be
submitted to Medicaid for non-covered off-label uses; and
submitting inflated best price information to the Medicaid
Rebate Program.
The Health Insurance Portability and Accountability Act of 1996
also created prohibitions against health care fraud and false
statements relating to health care matters. The health care
fraud statute prohibits knowingly and willfully executing a
scheme to defraud any health care benefit program, including
private payors. The false statements statute prohibits knowingly
and willfully falsifying, concealing or covering up a material
fact or making any materially false, fictitious or fraudulent
statement in connection with the delivery of or payment for
health care benefits, items or services.
The majority of states also have statutes or regulations similar
to these federal laws, which apply to items and services
reimbursed under Medicaid and other state programs, or, in
several states, apply regardless of the payor. In addition, some
states have laws that require pharmaceutical companies to adopt
comprehensive compliance programs. For example, under California
law, pharmaceutical companies must comply with both the April
2003 Office of Inspector General Compliance Program Guidance for
Pharmaceutical Manufacturers and the PhRMA Code on Interactions
with Healthcare Professionals, as amended. We have adopted and
implemented a compliance program which we believe satisfies the
applicable requirements of California law.
Sanctions under these federal and state laws may include civil
monetary penalties, exclusion of a manufacturer’s products
from reimbursement under government programs, criminal fines and
imprisonment. Because of the breadth of these laws and the
narrowness of the safe harbors, it is possible that some of our
business activities could be subject to challenge under one or
more of such laws. The Healthcare Reform Acts make several
important changes to the federal anti-kickback statute, false
claims laws, and health care fraud statute for example, by
weakening the intent requirement under the anti-kickback and
health care fraud statutes that may make it easier for the
government, or whistleblowers to charge such fraud and abuse
violations. In addition, the Healthcare Reform Acts increase
penalties for fraud and abuse violations. In addition, the
Healthcare Reform Acts increases penalties for fraud and abuse
violations. If our past, present or future operations are found
to be in violation of any of the laws described above or other
similar governmental regulations to which we are subject, we may
be subject to the applicable penalty associated with the
violation which could adversely affect our ability to operate
our business and negatively impact our financial results.
If we
are unable to adequately protect our technology or enforce our
patent rights, our business could suffer.
Our success with the drug products that we develop will depend,
in part, on our ability and the ability of our licensors to
obtain and maintain patent protection for these products. We
currently have a number of United States and foreign patents
issued and pending, however, we primarily rely on patent rights
licensed from others. Our license agreements generally give us
the right
and/or
obligation to maintain and enforce the subject patents. We may
44
not receive patents for any of our pending patent applications
or any patent applications we may file in the future. If our
pending and future patent applications are not allowed or, if
allowed and issued into patents, if such patents and the patents
we have licensed are not upheld in a court of law, our ability
to competitively exploit our drug products would be
substantially harmed. Also, such patents may or may not provide
competitive advantages for their respective products or they may
be challenged or circumvented by our competitors, in which case
our ability to commercially exploit these products may be
diminished.
The patent positions of pharmaceutical and biotechnology
companies can be highly uncertain and involve complex legal and
factual questions. No consistent policy regarding the breadth of
claims allowed in pharmaceutical and biotechnology patents has
emerged to date in the United States. The laws of many countries
may not protect intellectual property rights to the same extent
as United States laws, and those countries may lack adequate
rules and procedures for defending our intellectual property
rights. Filing, prosecuting and defending patents on all our
products or product candidates throughout the world would be
prohibitively expensive. Competitors may use our technologies in
jurisdictions and may not be covered by any of our patent claims
or other intellectual property rights.
Changes in either patent laws or in interpretations of patent
laws in the United States and other countries may diminish the
value of our intellectual property. We do not know whether any
of our patent applications will result in the issuance of any
patents, and we cannot predict the breadth of claims that may be
allowed in our patent applications or in the patent applications
we license from others.
The degree of future protection for our proprietary rights is
uncertain because legal means afford only limited protection and
may not adequately protect our rights or permit us to gain or
keep our competitive advantage. For example:
|
|
|
| |
•
|
in certain jurisdictions, we or our licensors might not have
been the first to make the inventions covered by each of our or
our licensors’ pending patent applications and issued
patents, and we may have to participate in expensive and
protracted interference proceedings to determine priority of
invention;
|
| |
| |
•
|
we or our licensors might not have been the first to file patent
applications for these inventions;
|
| |
| |
•
|
others may independently develop similar or alternative product
candidates or duplicate any of our or our licensors’
product candidates;
|
| |
| |
•
|
our or our licensors’ pending patent applications may not
result in issued patents;
|
| |
| |
•
|
our or our licensors’ issued patents may not provide a
basis for commercially viable products or may not provide us
with any competitive advantages or may be challenged by third
parties;
|
| |
| |
•
|
others may design around our or our licensors’ patent
claims to produce competitive products that fall outside the
scope of our or our licensors’ patents;
|
| |
| |
•
|
we may not develop or in-license additional patentable
proprietary technologies related to our product
candidates; or
|
| |
| |
•
|
the patents of others may prevent us from marketing one or more
of our product candidates for one or more indications that may
be valuable to our business strategy.
|
Moreover, an issued patent does not guarantee us the right to
practice the patented technology or commercialize the patented
product. Third parties may have blocking patents that could be
used to prevent us from commercializing our patented products
and practicing our patented technology. Our issued patents and
those that may be issued in the future may be challenged,
invalidated or circumvented, which could limit our ability to
prevent competitors from marketing related product candidates or
could limit the length of the term of patent protection of our
product candidates. In addition, our competitors may
independently develop similar technologies. Moreover, because of
the extensive time required for development, testing and
regulatory review of a potential product, it is possible that,
before any of our product candidates can be commercialized, any
related patent may expire or remain in force for only a short
period following commercialization, thereby reducing any
advantage of the patent.
45
We also rely on trade secret protection and contractual
protections for our unpatented, confidential and proprietary
technology. Trade secrets are difficult to protect. While we
enter into confidentiality agreements with our employees,
consultants and others, these agreements may not successfully
protect our trade secrets or other confidential and proprietary
information. It is possible that these agreements will be
breached, or that they will not be enforceable in every
instance, and that we will not have adequate remedies for any
such breach. Likewise, although we conduct periodic trade secret
audits of certain partners, vendors and contract manufacturers,
these trade secret audits may not protect our trade secrets or
other confidential and proprietary information. It is possible
that despite having certain trade secret audited security
measures in place, trade secrets or other confidential and
proprietary information may still be leaked or disclosed to a
third party. It is also possible that our trade secrets will
become known or independently developed by our competitors.
We also rely on trademarks to protect the names of our products.
These trademarks may be challenged by others. If we enforce our
trademarks against third parties, such enforcement proceedings
may be expensive. Some of our trademarks, including Zevalin are
owned by, or assignable to, our licensors and, upon expiration
or termination of the applicable license agreements, we may no
longer be able to use these trademarks.
If we are unable to adequately protect our technology, trade
secrets or proprietary know-how, or enforce our patents and
trademarks, our business, financial condition and prospects
could suffer.
Intellectual
property rights are complex and uncertain and therefore may
subject us to infringement claims.
The patent positions related to our drug products are inherently
uncertain and involve complex legal and factual issues. We
believe that there is significant litigation in the
pharmaceutical and biotechnology industry regarding patent and
other intellectual property rights. A patent does not provide
the patent holder with freedom to operate in a way that
infringes the patent rights of others. We may be accused of
patent infringement at any time. The coverage of patents is
subject to interpretation by the courts, and the interpretation
is not always uniform. If we are sued for patent infringement,
we would need to demonstrate that our products or methods do not
infringe the patent claims of the relevant patent
and/or that
the patent claims are invalid or unenforceable, and we may not
be able to do this. Proving invalidity, in particular, is
difficult since it requires a showing of clear and convincing
evidence to overcome the presumption of validity enjoyed by
issued patents in the United States.
Although we are not aware of any infringement by any of our drug
products on the rights of any third party, there may be third
party patents or other intellectual property rights, including
trademarks and copyrights, relevant to our drug products of
which we are not aware. Third parties may assert patent or other
intellectual property infringement claims against us, or our
licensors and collaborators, with products. Any claims that
might be brought against us relating to infringement of patents
may cause us to incur significant expenses and, if successfully
asserted against us, may cause us to pay substantial damages and
result in the loss of our use of the intellectual property that
is critical to our business strategy.
In the event that we or our partners are found to infringe any
valid claim of a patent held by a third party, we may, among
other things, be required to:
|
|
|
| |
•
|
pay damages, including up to treble damages and the other
party’s attorneys’ fees, which may be substantial;
|
| |
| |
•
|
cease the development, manufacture, use and sale of our products
that infringe the patent rights of others through a
court-imposed sanction such as an injunction;
|
| |
| |
•
|
expend significant resources to redesign our products so they do
not infringe others’ patent rights, which may not be
possible;
|
| |
| |
•
|
discontinue manufacturing or other processes incorporating
infringing technology; or
|
| |
| |
•
|
obtain licenses to the infringed intellectual property, which
may not be available to us on acceptable terms, or at all.
|
46
Intellectual
property litigation is increasingly common and increasingly
expensive and may result in restrictions on our business and
substantial costs, even if we prevail.
Patent and other intellectual property litigation is becoming
more common in the pharmaceutical industry. The pharmaceutical
field is characterized by a large number of patent filings
involving complex legal and factual questions, and, therefore,
we cannot predict with certainty whether our licensed patents
will be enforceable. Competitors may have filed applications for
or have been issued patents and may obtain additional patents
and proprietary rights related to products or processes that
compete with or are similar to ours. We may not be aware of all
of the patents potentially adverse to our interests that may
have been issued to others. Litigation is sometimes necessary to
defend against or assert claims of infringement, to enforce our
patent rights, including those we have licensed from others, to
protect trade secrets or to determine the scope and validity of
proprietary rights of third parties. We have not conducted an
extensive search of patents issued to other parties and such
patents which contain claims relating to our technology and
products may exist, may have been filed, or could be issued. If
such patents do exist, we may be infringing upon a third
party’s patent rights or other intellectual property, and
litigation asserting such claims might be initiated in which we
would not prevail, or we would not be able to obtain the
necessary licenses on reasonable terms, if at all. All such
litigation, whether meritorious or not, as well as litigation
initiated by us against third parties, is time-consuming and
very expensive to defend or prosecute and to resolve and we
cannot be certain that we will have the required resources to
pursue litigation or otherwise to protect our proprietary
rights. In addition, if we infringe the intellectual property
rights of others, we could lose our right to develop,
manufacture or sell our products or could be required to pay
monetary damages or royalties to license proprietary rights from
third parties. An adverse determination in a judicial or
administrative proceeding or a failure to obtain necessary
licenses could prevent us from manufacturing or selling our
products, which could harm our business, financial condition and
prospects.
If our competitors prepare and file patent applications in the
United States or Europe that claim technology we also claim, we
may have to participate in interference proceedings required by
the USPTO to determine priority of invention or opposition
proceedings in Europe, both of which could result in substantial
costs, even if we ultimately prevail. Results of interference
and opposition proceedings are highly unpredictable and may
result in us having to try to obtain licenses which may not be
available on commercially reasonable terms, or at all, in order
to continue to develop or market certain of our products. If we
need but cannot obtain a license, we may be prevented from
marketing the affected product.
We may
be subject to damages resulting from claims that we, or our
employees, have wrongfully used or disclosed alleged trade
secrets of our employees’ former employers.
Many of our employees were previously employed at universities
or biotechnology or pharmaceutical companies, including our
competitors or potential competitors. Although we have not
received any claim to date, we may be subject to claims that
these employees through their employment inadvertently or
otherwise used or disclosed trade secrets or other proprietary
information of their former employers. Litigation may be
necessary to defend against these claims. If we fail in
defending such claims, in addition to paying monetary damages,
we may lose valuable intellectual property rights or personnel.
We may
be subject to product liability claims, and may not have
sufficient product liability insurance to cover any such claims,
which may expose us to substantial liabilities.
We may be held liable if any product we or our partners develop
causes injury or is found otherwise unsuitable during product
testing, manufacturing, clinical trials, marketing or sale.
Regardless of merit or eventual outcome, product liability
claims could result in decreased demand for our product
candidates, injury to our reputation, withdrawal of patients
from our clinical trials, substantial monetary awards to trial
participants and the inability to commercialize any products
that we may develop. These claims might be made directly by
consumers, health care providers, pharmaceutical companies or
others selling or testing our products. Although we currently
carry product liability insurance in the amount of at least
$15.0 million in the aggregate, it is possible that this
coverage will be insufficient to protect us from future claims.
Additionally, our insurance may not reimburse us or may not be
sufficient to reimburse us for expenses or losses we may suffer.
Moreover, if insurance coverage becomes more expensive, we may
not be able to maintain insurance coverage at a reasonable cost
or in sufficient amounts to
47
protect us against losses due to liability. Failure to maintain
sufficient insurance coverage could have a material adverse
effect on our business, prospects and results of operations if
claims are made that exceed our coverage.
On occasion, juries have awarded large judgments in class action
lawsuits for claims based on drugs that had unanticipated side
effects. In addition, the pharmaceutical and biotechnology
industries, in general, have been subject to significant medical
malpractice litigation. A successful product liability claim or
series of claims brought against us could harm our reputation
and business and financial condition.
The
use of hazardous materials, including radioactive and biological
materials, in our research and development and commercial
efforts imposes certain compliance costs on us and may subject
us to liability for claims arising from the use or misuse of
these materials.
Our research and development, manufacturing (including a
radiolabeling step for Zevalin) and administration of our drugs
involves the controlled use of hazardous materials, including
chemicals, radioactive and biological materials, such as
radioactive isotopes. However, we do not physically handle these
radioactive isotopes or such hazardous materials. We are subject
to federal, state and local laws and regulations governing the
storage, use and disposal of these materials and some waste
products. We believe that our safety procedures for the storage,
use and disposal of these materials comply with the standards
prescribed by federal, state and local regulations. However, we
cannot completely eliminate the risk of accidental contamination
or injury from these materials. If there were to be an accident,
we could be held liable for any damages that result, which could
exceed our financial resources. We currently maintain insurance
coverage for injuries resulting from the hazardous materials we
use; however, future claims may exceed the amount of our
coverage. Also, we do not have insurance coverage for pollution
cleanup and removal. Currently the costs of complying with
federal, state and local regulations are not significant, and
consist primarily of waste disposal expenses, however, they
could become expensive, and current or future environmental
regulations may impair our research, development, production and
commercialization efforts.
Risks
Related to Our Common Stock
There
are a substantial number of shares of our common stock eligible
for future sale in the public market. The sale of these shares
could cause the market price of our common stock to fall. Any
future equity issuances by us may have dilutive and other
effects on our existing stockholders.
As of December 31, 2010, there were approximately
51,459,284 shares of our common stock outstanding, and in
addition, security holders held options, warrants and preferred
stock which, if vested, exercised or converted, would obligate
us to issue up to approximately 12.6 million additional
shares of common stock. However, we would receive over
$62 million from the issuance of shares of common stock
upon the exercise of all of the options and warrants. A
substantial number of those shares, when we issue them upon
vesting, conversion or exercise, will be available for immediate
resale in the public market. In addition, we may sell additional
shares of common stock or securities convertible or exercisable
into common stock in public or private offerings, which would be
available for resale in the market. The market price of our
common stock could fall as a result of sales of any of these
shares of common stock due to the increased number of shares
available for sale in the market.
We have primarily financed our operations, and we anticipate
that we will have to finance a large portion of our operating
cash requirements, by issuing and selling our common stock or
securities convertible into or exercisable for shares of our
common stock. Any issuances by us of equity securities may be at
or below the prevailing market price of our common stock and may
have a dilutive impact on our existing stockholders. These
issuances or other dilutive issuances would also cause our net
income, if any, per share to decrease in future periods. As a
result, the market price of our common stock could drop.
The
market price and trading volume of our common stock fluctuate
significantly and could result in substantial losses for
individual investors.
The stock market from time to time experiences significant price
and trading volume fluctuations that are unrelated to the
operating performance of particular companies. These broad
market fluctuations may cause the market price and trading
volume of our common stock to decrease. In addition, the market
price and trading volume of our common stock is often highly
volatile.
48
Factors that may cause the market price and volume of our common
stock to decrease include:
|
|
|
| |
•
|
recognition on up-front licensing or other fees or revenues;
|
| |
| |
•
|
payments of non-refundable up-front or license fees, or payment
for cost-sharing expenses, to third parties;
|
| |
| |
•
|
adverse results or delays in our clinical trials;
|
| |
| |
•
|
fluctuations in our results of operations;
|
| |
| |
•
|
timing and announcements of our technological innovations or new
products or those of our competitors;
|
| |
| |
•
|
developments concerning any strategic alliances or acquisitions
we may enter into;
|
| |
| |
•
|
announcements of FDA non-approval of our drug products, or
delays in the FDA or other foreign regulatory review process or
actions;
|
| |
| |
•
|
changes in recommendations or guidelines of government agencies
or other third parties regarding the use of our drug products;
|
| |
| |
•
|
adverse actions taken by regulatory agencies with respect to our
drug products, clinical trials, manufacturing processes or sales
and marketing activities;
|
| |
| |
•
|
concerns about our products being reimbursed;
|
| |
| |
•
|
any lawsuit involving us or our drug products;
|
| |
| |
•
|
developments with respect to our patents and proprietary rights;
|
| |
| |
•
|
public concern as to the safety of products developed by us or
others;
|
| |
| |
•
|
regulatory developments in the United States and in foreign
countries;
|
| |
| |
•
|
changes in stock market analyst recommendations regarding our
common stock or lack of analyst coverage;
|
| |
| |
•
|
the pharmaceutical industry generally and general market
conditions;
|
| |
| |
•
|
failure of our results of operations to meet the expectations of
stock market analysts and investors;
|
| |
| |
•
|
sales of our common stock by our executive officers, directors
and five percent stockholders or sales of substantial amounts of
our common stock;
|
| |
| |
•
|
changes in accounting principles; and
|
| |
| |
•
|
loss of any of our key scientific or management personnel.
|
Also, certain dilutive securities such as warrants can be used
as hedging tools which may increase volatility in our stock and
cause a price decline. While a decrease in market price could
result in direct economic loss for an individual investor, low
trading volume could limit an individual investor’s ability
to sell our common stock, which could result in substantial
economic loss as well. Since January 1, 2010 through
February 25, 2011, the price of our common stock ranged
between $7.10 and $3.70, and the daily trading volume was as
high as 5,960,800 shares and as low as 83,900 shares.
In addition, due in large part to the current global economic
crisis many institutional investors that historically had
invested in specialty pharmaceutical companies have ceased
operations or further investment in these companies, which has
had negatively impacted trading volume for our stock.
Following periods of volatility in the market price of a
company’s securities, securities class action litigation
may be instituted against that company. Regardless of their
merit, these types of lawsuits generally result in substantial
legal fees and management’s attention and resources being
diverted from the operations of a business.
49
Provisions
of our charter, bylaws and stockholder rights plan may make it
more difficult for someone to acquire control of us or replace
current management even if doing so would benefit our
stockholders, which may lower the price an acquirer or investor
would pay for our stock.
Provisions of our certificate of incorporation and bylaws, both
as amended, may make it more difficult for someone to acquire
control of us or replace our current management. These
provisions include:
|
|
|
| |
•
|
the ability of our board of directors to amend our bylaws
without stockholder approval;
|
| |
| |
•
|
the inability of stockholders to call special meetings;
|
| |
| |
•
|
the ability of members of the board of directors to fill
vacancies on the board of directors;
|
| |
| |
•
|
the inability of stockholders to act by written consent, unless
such consent is unanimous; and
|
| |
| |
•
|
the establishment of advance notice requirements for nomination
for election to our board of directors or for proposing matters
that can be acted on by stockholders at stockholder meetings.
|
These provisions may make it more difficult for stockholders to
take certain corporate actions and could delay, discourage or
prevent someone from acquiring our business or replacing our
current management, even if doing so would benefit our
stockholders. These provisions could limit the price that
certain investors might be willing to pay for shares of our
common stock.
We have a stockholder rights plan pursuant to which we
distributed rights to purchase units of our series B junior
participating preferred stock. The rights become exercisable
upon the earlier of ten days after a person or group of
affiliated or associated persons has acquired 15% or more of the
outstanding shares of our common stock or ten business days
after a tender offer has commenced that would result in a person
or group beneficially owning 15% or more of our outstanding
common stock. These rights could delay or discourage someone
from acquiring our business, even if doing so would benefit our
stockholders. We currently have no stockholders who own 15% or
more of the outstanding shares of our common stock.
If we
fail to maintain an effective system of internal control over
financial reporting, we may not be able to accurately report our
financial results, and current and potential stockholders may
lose confidence in our financial reporting.
We are required by the SEC to establish and maintain adequate
internal control over financial reporting that provides
reasonable assurance regarding the reliability of our financial
reporting and the preparation of financial statements in
accordance with generally accepted accounting principles. We are
likewise required, on a quarterly basis, to evaluate the
effectiveness of our internal controls and to disclose any
changes and material weaknesses in those internal controls.
As described in our Annual Report on
Form 10-K
for the year ended December 31, 2009, we identified a
material weakness with regard to accounting for warrant
instruments in our internal control over financial reporting for
such period. Given this material weakness with regard to
warrants, management was unable to conclude that we maintained
effective internal control over financial reporting as of
December 31, 2009. Since the determination regarding this
material weakness, we devoted significant effort and resources
to the remediation and improvement of our internal control over
financial reporting. As described in Item 9A of this Annual
Report on
Form 10-K
for the year ended December 31, 2010, no new or existing
material weaknesses were identified and we determined that our
internal control over financial reporting was effective as of
December 31, 2010.
Any failure to maintain such internal controls in the future
could adversely impact our ability to report our financial
results on a timely and accurate basis. If our financial
statements are not accurate, investors may not have a complete
understanding of our operations. Likewise, if our financial
statements are not filed on a timely basis as required by the
SEC and NASDAQ, we could face severe consequences from those
authorities. In either case, there could result a material
adverse affect on our business. Inferior internal controls could
also cause investors to lose confidence in our reported
financial information, which could have a negative effect on the
trading price of our stock.
50
Our
publicly-filed SEC reports are reviewed by the SEC from time to
time and any significant changes required as a result of any
such review may result in material liability to us and have a
material adverse impact on the trading price of our common
stock.
The reports of publicly-traded companies are subject to review
by the SEC from time to time for the purpose of assisting
companies in complying with applicable disclosure requirements
and to enhance the overall effectiveness of companies’
public filings, and reviews of such reports are now required at
least every three years under the Sarbanes-Oxley Act of 2002.
SEC reviews may be initiated at any time, and we could be
required to modify or reformulate information contained in prior
filings as a result of an SEC review. Any modification or
reformulation of information contained in such reports could be
significant and could result in material liability to us and
have a material adverse impact on the trading price of our
common stock.
Changes
in our effective income tax rate could adversely affect our
results of operations.
We are subject to federal and state income taxes in the United
States and our tax liabilities are dependent upon the
distribution of income among these different jurisdictions.
Various factors may have favorable or unfavorable effects on our
effective income tax rate. These factors include, but are not
limited to:
|
|
|
| |
•
|
interpretations of existing tax laws,
|
| |
| |
•
|
the accounting for stock options and other share-based
compensation,
|
| |
| |
•
|
changes in tax laws and rates,
|
| |
| |
•
|
future levels of research and development spending,
|
| |
| |
•
|
changes in accounting standards,
|
| |
| |
•
|
changes in the mix of earnings in the various tax jurisdictions
in which we operate,
|
| |
| |
•
|
the outcome of examinations by the Internal Revenue Service and
other jurisdictions,
|
| |
| |
•
|
the accuracy of our estimates for unrecognized tax benefits,
|
| |
| |
•
|
realization of deferred tax assets, and
|
| |
| |
•
|
changes in overall levels of pre-tax earnings.
|
The impact on our income tax provision resulting from the
above-mentioned factors may be significant and could have an
impact on our results of operations.
We do
not anticipate declaring any cash dividends on our common
stock.
We have never declared or paid cash dividends on our common
stock and do not plan to pay any cash dividends on our common
stock in the foreseeable future. Our current policy is to retain
all funds and any earnings for use in the operation and
expansion of our business. If we do not pay dividends, our stock
may be less valuable to investors because a return on their
investment will only occur if our stock price appreciates.
|
|
|
Item 1B.
|
Unresolved
Staff Comments
|
None.
We sublease our principal executive office in Henderson, Nevada
under a non cancelable operating lease expiring April 30,
2014. We lease our research and development facility in Irvine,
California under a non cancelable operating lease expiring
June 30, 2016. We also lease small administrative offices
in Zurich, Switzerland, Montreal, Canada, and Mumbai, India on
an expense-sharing basis. The financial and other terms of these
lease arrangements are not material to our business. We believe
that our leased facilities are adequate to meet our needs at
this time.
51
|
|
|
Item 3.
|
Legal
Proceedings
|
We are involved with various legal matters arising from the
ordinary course of business. Although the ultimate resolution of
these various matters cannot be determined at this time, we do
not believe that such matters, individually or in the aggregate,
will have a material adverse effect on our future consolidated
results of operations, cash flows or financial condition.
52
PART II
|
|
|
Item 5.
|
Market
for Registrant’s Common Equity Related Stockholder Matters
and Issuer Purchases of Equity Securities
|
Common
Stock
As of February 25, 2011 there were 52,003,514 shares
of common stock outstanding and 382 stockholders of record. On
February 25, 2011, the closing sale price of our common
stock was $6.80 per share.
Market
for Securities
Our common stock is traded on the NASDAQ Global Market under the
symbol “SPPI.” The high and low sale prices of our
common stock reported by NASDAQ during each quarter ended in
2010 and 2009 were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
High
|
|
Low
|
|
|
|
Year 2010:
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
5.48
|
|
|
$
|
4.28
|
|
|
Second Quarter
|
|
$
|
5.24
|
|
|
$
|
3.79
|
|
|
Third Quarter
|
|
$
|
4.66
|
|
|
$
|
3.67
|
|
|
Fourth Quarter
|
|
$
|
7.08
|
|
|
$
|
4.05
|
|
|
Year 2009
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
2.10
|
|
|
$
|
1.39
|
|
|
Second Quarter
|
|
$
|
8.15
|
|
|
$
|
1.75
|
|
|
Third Quarter
|
|
$
|
10.00
|
|
|
$
|
4.76
|
|
|
Fourth Quarter
|
|
$
|
6.74
|
|
|
$
|
3.97
|
|
53
Stock
Performance Graph (1)
The graph below compares the cumulative total stockholder return
on $100 invested, assuming the reinvestment of all dividends, on
December 31, 2005, the last trading day before our 2005
fiscal year, through the end of fiscal 2010 with the cumulative
total return on $100 invested for the same period in the Russell
2000 index and a Peer Group.
COMPARISON
OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among
Spectrum Pharmaceuticals, Inc., the Russell 2000 Index,
and a Peer Group
|
|
|
|
* |
|
$100 invested on 12/31/05 in stock or index, including
reinvestment of dividends. Fiscal year ending December 31. |
The Peer Group included the following companies:
| |
|
|
|
|
|
|
|
•
|
|
Affymax Inc
|
|
•
|
|
Immunomedics Inc
|
|
•
|
|
Allos Therapeutics Inc
|
|
•
|
|
Intermune Inc
|
|
•
|
|
Amicus Therapeutics Inc
|
|
•
|
|
Isis Pharmaceuticals Inc
|
|
•
|
|
Arena Pharmaceuticals Inc
|
|
•
|
|
Mannkind Corp
|
|
•
|
|
Biocryst Pharmaceuticals Inc
|
|
•
|
|
Medivation Inc
|
|
•
|
|
Biomarin Pharmaceutical Inc
|
|
•
|
|
Onyx Pharmaceuticals Inc
|
|
•
|
|
Cell Therapeutics Inc
|
|
•
|
|
Sangamo Biosciences Inc
|
|
•
|
|
Cytokinetics Inc
|
|
•
|
|
Seattle Genetics Inc
|
|
•
|
|
Dendreon Corp
|
|
•
|
|
Supergen Inc
|
|
•
|
|
Enzon Pharmaceuticals Inc
|
|
•
|
|
Theravance Inc
|
|
•
|
|
Exelixis Inc
|
|
•
|
|
Vertex Pharmaceuticals Inc
|
54
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
12/31/05
|
|
|
12/31/06
|
|
|
12/31/07
|
|
|
12/31/08
|
|
|
12/31/09
|
|
|
12/31/10
|
|
|
|
|
Spectrum Pharmaceuticals, Inc.
|
|
|
100.00
|
|
|
|
130.73
|
|
|
|
64.30
|
|
|
|
35.22
|
|
|
|
104.96
|
|
|
|
162.41
|
|
|
Russell 2000
|
|
|
100.00
|
|
|
|
118.37
|
|
|
|
116.51
|
|
|
|
77.15
|
|
|
|
98.11
|
|
|
|
124.46
|
|
|
Peer Group
|
|
|
100.00
|
|
|
|
123.99
|
|
|
|
126.64
|
|
|
|
92.09
|
|
|
|
128.95
|
|
|
|
134.43
|
|
|
|
|
|
(1) |
|
The information in this section is not “soliciting
material,” is not deemed “filed” with the SEC and
is not to be incorporated by reference in any filing of the
Company under the Securities Act of 1933, as amended, or the
Securities Exchange Act of 1934, as amended, whether made before
or after the date hereof and irrespective of any general
incorporation language in any such filing. |
Unregistered
Equity Issuances
We did not issue any unregistered securities during the year
ended December 31, 2010 that were not otherwise disclosed
in a previously filed Quarterly Report on Form 10-Q or a
Current Report on Form 8-K.
Equity
Repurchases
We did not repurchase any of our securities during the quarter
ended December 31, 2010.
Dividends
We have never paid cash dividends on our common stock and we do
not intend to pay cash dividends of our common stock in the
foreseeable future. We currently intend to retain our earnings,
if any, to finance future growth.
|
|
|
Item 6.
|
Selected
Financial Data
|
The following table presents selected historical financial data.
We derived the selected statements of operations data for the
years ended December 31, 2010, 2009, and 2008 and balance
sheet data as of December 31, 2010 and 2009 from our
audited consolidated financial statements and notes thereto that
are included elsewhere in this annual report. We derived the
selected statements of operations data for the years ended
December 31, 2007 and 2006 and the balance sheet data as of
December 31, 2008, 2007 and 2006 from our audited
consolidated financial statements that do not appear in this
annual report.
You should read the following financial information together
with the information under “Management’s Discussion
and Analysis of Financial Condition and Results of
Operations” and our consolidated financial statements and
related notes included elsewhere in this annual report. The
information set forth below is not necessarily indicative of our
future financial condition or results of operations.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years ended December 31,
|
|
|
Statement of Operations Data:
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
Total revenues
|
|
$
|
74,113
|
|
|
$
|
38,025
|
|
|
$
|
28,725
|
|
|
$
|
7,672
|
|
|
$
|
5,673
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of product sales (excludes amortization of purchased
intangible assets)
|
|
|
17,439
|
|
|
|
8,148
|
|
|
|
1,193
|
|
|
|
—
|
|
|
|
97
|
|
|
Selling, general and administrative
|
|
|
48,550
|
|
|
|
33,607
|
|
|
|
15,156
|
|
|
|
11,577
|
|
|
|
7,736
|
|
|
Research and development
|
|
|
57,301
|
|
|
|
21,058
|
|
|
|
26,683
|
|
|
|
33,285
|
|
|
|
23,728
|
|
|
Amortization of purchased intangibles
|
|
|
3,720
|
|
|
|
3,720
|
|
|
|
158
|
|
|
|
—
|
|
|
|
—
|
|
|
Acquired in-process research and development
|
|
|
—
|
|
|
|
—
|
|
|
|
4,700
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(52,897
|
)
|
|
|
(28,508
|
)
|
|
|
(19,165
|
)
|
|
|
(37,190
|
)
|
|
|
(25,888
|
)
|
|
Change in fair value of common stock warrant liability
|
|
|
2,731
|
|
|
|
8,075
|
|
|
|
1,271
|
|
|
|
12,055
|
|
|
|
(2,485
|
)
|
|
Other income, net
|
|
|
1,279
|
|
|
|
662
|
|
|
|
1,165
|
|
|
|
3,139
|
|
|
|
2,606
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss before provision for income taxes
|
|
|
(48,887
|
)
|
|
|
(19,771
|
)
|
|
|
(16,729
|
)
|
|
|
(21,996
|
)
|
|
|
(25,767
|
)
|
|
Benefit (Provision) for income taxes
|
|
|
43
|
|
|
|
(421
|
)
|
|
|
(5
|
)
|
|
|
(5
|
)
|
|
|
(5
|
)
|
|
Net loss attributable to non-controlling interest
|
|
|
—
|
|
|
|
1,146
|
|
|
|
2,538
|
|
|
|
20
|
|
|
|
3
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to Spectrum Pharmaceuticals, Inc.
stockholders
|
|
$
|
(48,844
|
)
|
|
$
|
(19,046
|
)
|
|
$
|
(14,196
|
)
|
|
$
|
(21,981
|
)
|
|
$
|
(25,769
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share — basic and diluted
|
|
$
|
(0.99
|
)
|
|
$
|
(0.48
|
)
|
|
$
|
(0.45
|
)
|
|
$
|
(0.76
|
)
|
|
$
|
(1.06
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
55
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
Balance Sheet Data:
|
|
2010
|
|
2009
|
|
2008
|
|
2007
|
|
2006
|
|
|
|
(In thousands)
|
|
|
|
Cash, cash equivalents and investments
|
|
$
|
104,243
|
|
|
$
|
113,341
|
|
|
$
|
75,938
|
|
|
$
|
55,659
|
|
|
$
|
50,967
|
|
|
Working capital
|
|
|
58,543
|
|
|
|
86,758
|
|
|
|
54,677
|
|
|
|
48,813
|
|
|
|
46,054
|
|
|
Total assets
|
|
|
163,631
|
|
|
|
173,133
|
|
|
|
129,509
|
|
|
|
57,540
|
|
|
|
53,117
|
|
|
Common stock warrant liability (at fair value)
|
|
|
3,904
|
|
|
|
6,635
|
|
|
|
765
|
|
|
|
2,035
|
|
|
|
14,090
|
|
|
Long term obligations, less current portion
|
|
|
25,833
|
|
|
|
25,310
|
|
|
|
42,822
|
|
|
|
992
|
|
|
|
1,035
|
|
|
Total stockholders’ equity (including non-controlling
interest)
|
|
|
74,476
|
|
|
|
108,324
|
|
|
|
53,116
|
|
|
|
46,714
|
|
|
|
31,759
|
|
|
|
|
Item 7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
This Annual Report on
Form 10-K
contains forward-looking statements within the meaning of the
federal securities laws. These statements are subject to risks
and uncertainties that could cause actual results and events to
differ materially from those expressed or implied by such
forward-looking statements. For a detailed discussion of these
risks and uncertainties, see the “Risk Factors”
section in Item 1A of Part I of this
Form 10-K.
We caution the reader not to place undue reliance on these
forward-looking statements, which reflect management’s
analysis only as of the date of this
Form 10-K.
We undertake no obligation to update forward-looking statements
to reflect events or circumstances occurring after the date of
this
Form 10-K.
Overview
We are a biotechnology company with fully integrated commercial
and drug development operations with a primary focus in
oncology. Our strategy is comprised of acquiring, developing and
commercializing a broad and diverse pipeline of late-stage
clinical and commercial products. We market two oncology drugs,
ZEVALIN®
and
FUSILEV®
and have two drugs, apaziquone and belinostat, in late stage
development along with a diversified pipeline of novel drug
candidates. We have assembled an integrated in-house scientific
team, including formulation development, clinical development,
medical research, regulatory affairs, biostatistics and data
management, and have established a commercial infrastructure for
the marketing of our drug products. We also leverage the
expertise of our worldwide partners to assist in the execution
of our strategy. Apaziquone is presently being studied in two
large Phase 3 clinical trials for non-muscle invasive bladder
cancer, or NMIBC, under strategic collaborations with Allergan,
Inc., (“Allergan”), Nippon Kayaku Co. Ltd.,
(“Nippon Kayaku”), and Handok Pharmaceuticals Co.
Ltd., (“Handok”). Belinostat, is being studied in
multiple indications including a Phase 2 registrational trial
for relapsed or refractory peripheral T-cell lymphoma, or PTCL,
under a strategic collaboration with TopoTarget A/S or
TopoTarget.
Our business strategy is comprised of the following initiatives:
|
|
|
| |
•
|
Maximizing the growth potential of our marketed drugs,
Zevalin and Fusilev. Our near-term outlook
largely depends on sales and marketing successes for our two
marketed drugs. For Zevalin, we stabilized sales in 2009,
increased sales in 2010 and believe we can continue to grow
sales in 2011 and beyond. For Fusilev, which we launched in
August 2008, we were able to benefit from broad utilization in
community clinics and hospitals and recognized a dramatic
increase in sales during 2010 due to a shortage of generic
leucovorin. While we cannot predict how long the shortage may
continue, our focus now is to obtain approval for Fusilev in
advanced metastatic colorectal cancer. As part of its review of
our supplemental new drug application, or sNDA, for metastatic
colorectal cancer, the FDA requested additional data to which we
submitted a response on October 29, 2010. The FDA formally
accepted the submission and established a decision date, or
PDUFA, of April 29, 2011.
|
For both Zevalin and Fusilev, we initiated and continue to stage
appropriate infrastructure expansions and additional initiatives
to facilitate broad customer reach and to address other market
requirements, as appropriate. We have formed a dedicated
commercial organization comprised of highly experienced and
motivated sales representatives, account managers, and a
complement of other support marketing personnel
56
to manage the sales and marketing of these drugs. In addition
our scientific department supports field activities through
various MDs, PhDs and other medical science liaison personnel.
|
|
|
| |
•
|
Optimizing our development portfolio and maximizing the
asset values of its components. While over
the recent few years, we have evolved from a development-stage
to a commercial-stage pharmaceutical company, we have maintained
a highly focused development portfolio. Our strategy with regard
to our development portfolio is to focus on late-stage drugs and
to develop them rapidly to the point of regulatory approval. We
plan to develop some of these drugs ourselves or with our
subsidiaries and affiliates, or secure collaborations such that
we are able to suitably monetize these assets.
|
We have assembled a drug development infrastructure that is
comprised of highly experienced and motivated MDs, PhDs,
clinical research associates and a complement of other support
personnel to rapidly develop these drugs. During 2009, this team
achieved our goal of completing enrollment in the two Phase 3
apaziquone trials (with more than 1,600 patients enrolled).
We expect to continue to maximize the value of apaziquone
through further developmental efforts and initiation of
additional trials.
We have several other exciting compounds in earlier stages of
development in our portfolio. Based upon a criteria-based
portfolio review, we are in the process of streamlining our
pipeline drugs, allowing for greater focus and integration of
our development and commercial goals.
|
|
|
| |
•
|
Expanding our pipeline of late stage and commercial drugs
through licensing and business
development. It is our goal to identify new
strategic opportunities that will create strong synergies with
our currently marketed drugs and identify and pursue
partnerships for out-licensing certain of our drugs in
development. To this end, we will continue to explore strategic
collaborations as these relate to drugs that are either in
advanced clinical trials or are currently on the market. We
believe that such opportunistic collaborations will provide
synergies with respect to how we deploy our internal resources.
In this regard, we intend to identify and secure drugs that have
significant growth potential either through enhanced marketing
and sales efforts or through pursuit of additional clinical
development. We believe our in-licensing of belinostat, a novel
histone deacetylase, or HDAC, inhibitor, is demonstrative of
such licensing and business development efforts outlined above.
|
| |
| |
•
|
Managing our financial resources
effectively. We remain committed to fiscal
discipline, a policy which has allowed us to become well
capitalized among our peers, despite a very challenging capital
markets environment during 2009 and continuing through 2010.
This policy includes the pursuit of non-dilutive funding
options, prudent expense management, and the achievement of
critical synergies within our operations in order to maintain a
reasonable burn rate. Even with the continued
build-up in
operational infrastructure to facilitate the marketing of our
two commercial drugs, we intend to be fiscally prudent in any
expansion we undertake. In terms of revenue generation, we plan
to become more reliant on sales from currently marketed drugs
and intend to pursue out-licensing of select pipeline drugs in
select territories, as discussed above. When appropriate, we may
pursue other sources of financing, including non-dilutive
financing alternatives. While we are currently focused on
advancing our key drug development programs, we anticipate that
we will make regular determinations as to which other programs,
if any, to pursue and how much funding to direct to each program
on an ongoing basis, based on clinical success and commercial
potential, including termination of our existing development
programs, especially if we do not expect value being driven from
continued development. We ended 2010 with over $100 million
in cash, cash equivalents and investments which was net of the
$30 million license fee paid for belinostat in early 2010
offset by cash received from an out-license of apaziquone of
approximately $17.5 million.
|
| |
| |
•
|
Further enhancing the organizational structure to meet our
corporate objectives. We have highly
experienced staff in pharmaceutical operations, clinical
development, regulatory and commercial functions who previously
held positions at both small to mid-size biotech companies, as
well as large pharmaceutical companies. We have strengthened the
ranks of our management team, and will continue to pursue talent
on an opportunistic basis. Finally, we remain committed to
running a lean and efficient organization, while effectively
leveraging our critical resources.
|
57
Financial
Condition
Liquidity
and Capital Resources
Our cumulative losses, since inception in 1987 through
December 31, 2010, are approximately $310.4 million.
We expect to continue to incur additional losses for at least
the next few years, as we implement our growth strategy of
commercializing marketed drugs, while continuing to develop our
portfolio of late-stage drug products. Our long-term strategy is
to generate profits from the sale and licensing of our drug
products. Accordingly, in the next several years, we expect to
supplement our cash position with sales of Zevalin and Fusilev
and generate licensing revenue from out-licensing our other drug
products.
While we believe that the approximately $104 million in
cash, cash equivalents and investments, which includes long term
marketable securities, we had available on December 31,
2010 will allow us to fund our current planned operations for at
least the next twelve to eighteen months, we may, however, seek
to obtain additional capital through the sale of debt or equity
securities, if necessary, especially in conjunction with
opportunistic acquisitions or license of drugs. We may be unable
to obtain such additional capital when needed, or on terms
favorable to us or our stockholders, if at all. If we raise
additional funds by issuing equity securities, the percentage
ownership of our stockholders will be reduced, stockholders may
experience additional dilution or such equity securities may
provide for rights, preferences or privileges senior to those of
the holders of our common stock. If additional funds are raised
through the issuance of debt securities, the terms of such
securities may place restrictions on our ability to operate our
business. If and when appropriate, just as we have done in the
past, we may pursue non-dilutive financing alternatives as well.
Zevalin sales growth is largely dependent on the successful
launch of Zevalin for use as part of first-line therapy for
follicular NHL, continued use in its initial indication, and
establishing a consistent and accurate reimbursement standard.
As noted above, we recently obtained a CMS decision for a
reimbursement standard based on ASP methodology in the HOPPS
setting. As discussed earlier, during 2010 our sales of Fusilev
grew considerably over prior years because of a shortage of
generic leucovorin. We are unable to predict how long this
current shortage may last and we believe that the growth of
future Fusilev sales largely depends upon obtaining FDA approval
for use of Fusilev in combination with 5-FU containing regimens
for the treatment of colorectal cancer and favorable
reimbursement. The FDA stated in their October 2009 Complete
Response letter that the submission did not demonstrate that
Fusilev is non-inferior to leucovorin; and at our January 2010
meeting the FDA requested additional data which we submitted in
late 2010. The FDA formally accepted the submission and
established a decision date (PDUFA) of April 29, 2011. We
are unable to reasonably estimate when, if ever, we will realize
sustainable net profit from sales of these two products or any
of our other products, if they are approved by the FDA.
Our expenditures for research and development (R&D”)
consist of direct product specific costs (such as up-front
license fees, milestone payments, active pharmaceutical
ingredients, clinical trials, patent related legal costs, and
product liability insurance, among others) and non-product
specific, or indirect, costs (such as personnel costs, rent, and
utilities, among others). The following summarizes our research
and development expenses for the periods indicated and include
related stock-based charges but not amortization of intangibles
or expensing of in-process research and development costs. We
charge all research and development expenses to operations as
incurred.
58
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Apaziquone
|
|
$
|
6,165
|
|
|
$
|
10,915
|
|
|
$
|
5,477
|
|
|
Ozarelix
|
|
|
1,916
|
|
|
|
1,168
|
|
|
|
2,435
|
|
|
Ortataxel
|
|
|
716
|
|
|
|
311
|
|
|
|
150
|
|
|
Fusilev
|
|
|
1,281
|
|
|
|
1,125
|
|
|
|
2,096
|
|
|
Zevalin
|
|
|
421
|
|
|
|
563
|
|
|
|
151
|
|
|
Belinostat
|
|
|
36,045
|
|
|
|
—
|
|
|
|
—
|
|
|
Other development drugs
|
|
|
3,469
|
|
|
|
1,535
|
|
|
|
1,304
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total — Direct Costs
|
|
|
50,013
|
|
|
|
15,617
|
|
|
|
11,613
|
|
|
Indirect Costs (including non-cash share-based compensation of
$2.4 million, $3.2 million and $4.0 million,
respectively)
|
|
|
14,838
|
|
|
|
16,652
|
|
|
|
15,070
|
|
|
Partner Reimbursement
|
|
|
(7,550
|
)
|
|
|
(11,211
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total Research & Development
|
|
$
|
57,301
|
|
|
$
|
21,058
|
|
|
$
|
26,683
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Our primary focus areas for the foreseeable future, and the
programs that are expected to represent a significant part of
our R&D expenditures, are the on-going registrational
clinical trials of apaziquone and belinostat and additional
clinical studies in supporting the expanded utilization of our
FDA products (ZEVALIN and FUSILEV). While we are currently
focused on advancing these key product development programs, we
continually evaluate our R&D programs of other pipeline
products in response to the scientific and clinical success of
each product candidate, as well as an ongoing assessment as to
the product candidate’s commercial potential. Our
anticipated net use of cash for R&D in the fiscal year
ending December 31, 2011, excluding the cost of
in-licensing or acquisitions of additional drugs, if any, is
expected to range between approximately $30 and $40 million.
Under our various existing licensing agreements, we are
contingently obligated to make various regulatory and business
milestone payments. In connection with the development of
certain in-licensed drug products, we anticipate the occurrence
of certain of these milestones during 2011. Upon successful
achievement of these milestones, we will likely become obligated
to pay up to approximately $5.0 million during 2011,
payable in cash or stock at our discretion.
Further, while we do not receive any funding from third parties
for research and development that we conduct, co-development and
out-licensing agreements with other companies for any of our
drug products may reduce our expenses. In this regard, we
entered into a collaboration agreement with Allergan whereby,
commencing January 1, 2009, Allergan has borne 65% of the
development costs of apaziquone. Additionally, we entered into a
collaboration agreement with TopoTarget, whereby, commencing
February 2, 2010, TopoTarget bears, for belinostat, 100% of
the CUP trial costs and 30% of other development costs unrelated
to the PTCL study.
In addition to our present portfolio of drug product candidates,
we continually evaluate proprietary products for acquisition. If
we are successful in acquiring rights to additional products, we
may pay up-front licensing fees in cash
and/or
common stock and our research and development expenditures would
likely increase.
Net
Cash used in Operating Activities
Net cash used in operating activities was $22.5 million for
2010 which includes the non-recurring $30.0 million upfront
payment for in licensing a novel drug belinostat in late stage
pivotal clinical trials. The principal components of such cash
usage was a net loss in the period of $48.8 million
adjusted for net non-cash credits of $1.3 million, offset
by changes in working capital of $25.0 million during the
period.
59
Net
Cash used for Investing Activities
Net cash used in investing activities of $9.8 million in
2010 was primarily due to the $8.3 million net purchase of
marketable securities and a $1.6 million increase in
property and equipment acquisitions, of which $1.0 million
relates to landlord contributions to tenant improvements.
Net
Cash provided by Financing Activities
Net cash provided by financing activities of $3.6 million
in 2010, primarily relates to proceeds from the issuance of
common stock as a result of the exercise of stock options and
purchases of shares under our Employee Stock Purchase Plan.
Results
of Operations
Results
of Operations for Fiscal 2010 Compared to Fiscal
2009
Net Revenues. Net revenues increased
$36.1 million, or 94.9%, to $74.1 million in 2010 (net
of estimates for promotional, price and other adjustments
including distributor chargebacks, rebates and returns reserves)
from $38.0 million in 2009. We recorded approximately
$60.9 million of revenue from the sales of Zevalin and
Fusilev as compared to approximately $28.2 million in 2009.
Zevalin and Fusilev revenues in 2010 were approximately $28.9
and $32.0 million respectively, compared to approximately
$15.7 million and $12.5 million, respectively in 2009.
The increase in Zevalin revenues included both an increase in
unit sales and average selling prices. Revenues from the sales
of Fusilev have fluctuated in 2009 and 2010. During the first
half of 2009, Fusilev sales were higher due to a supply
disruption of leucovorin. The disruption in supply abated in the
second quarter of 2009, and subsequent Fusilev sales were
significantly lower than experienced in the first half of 2009.
Commencing late in the second quarter of 2010, a similar
disruption emerged and accordingly, in the second, third and
fourth quarters of 2010, sales of Fusilev grew significantly.
Looking forward we are unable to predict how long this
disruption may continue and cannot predict our ability to
manufacture sufficient quantities to meet fluctuating commercial
demand.
We also recorded $13.2 million in 2010 and
$9.8 million in 2009 of licensing revenues from the
amortization of the upfront payments received from Allergan in
2008 and from Nippon Kayaku and Handok payments received in
2010. In January 2007, we received approximately
$0.9 million, representing our 50% share of an economic
interest that Aeterna Zentaris had from an arrangement with
Nippon Kayaku for certain rights to ozarelix in Japan and
recognized the amount as deferred revenue. In early 2010 we
reevaluated the basis for deferral having determined that there
are no further ongoing obligations and recorded the
approximately $0.9 million as license revenue during 2010.
Cost of Product Sales. As a result of
increased product revenues and a reserve for expiring inventory
of $50,000, the cost of product sales increased
$9.3 million to $17.4 million in 2010 from
$8.1 million in 2009. As a percentage of revenue, the cost
of product sales increased from 21.4% in 2009 to 23.5% in 2010.
Selling, General and Administrative. Selling,
general and administrative expenses increased
$15.0 million, or 44.5%, to $48.6 million in 2010,
from $33.6 million in 2009. The increase is primarily due
to approximately:
|
|
|
| |
•
|
$13.6 million increase attributable to sales and marketing
expenses, including payroll costs, incurred with the sales of
Zevalin and Fusilev. We expect sales and marketing expenses
related to Zevalin and Fusilev to increase in 2011
|
| |
| |
•
|
$1.8 million increase in non-cash compensation expenses.
|
Research and Development. Total research and
development expenses increased $36.2 million, or 172.1%, to
$57.3 million in 2010, from $21.1 million in 2009. The
increase is primarily due to the $30.0 million upfront
payment for the licensing of belinostat, and a one-time charge
of $3.1 million, representing the fair value of
751,956 shares of our common stock issued as consideration
for the acquisition and licensing of compounds. We anticipate
research and development expenses in 2011 to be higher than
2010, before any payments for drug licensing, primarily due to
continued development of belinostat through our collaboration
with TopoTarget.
60
Change in Fair Value of Common Stock Warrant
Liability. We recorded income of
$2.7 million for the change in the fair value of the
warrant obligations during 2010 compared to income of
$8.1 million in the same period of 2009. The decrease from
2009 is due to the expiration of 6,931,607 common stock warrants
issued in 2009, which expired unexercised and a decrease in the
fair value of 3,747,312 warrants expiring in September 2011.
Other Net Income. The principal components of
other income of $1.3 million and $662,000 during 2010 and
2009, respectively, consisted of $977,000 related to grants
under the Qualifying Therapeutic Discovery Project Program
administered under section 48D of the Internal Revenue Code
as well as currency gains and losses and net interest income. We
have lower investment yields due to the shift in our investment
strategy to more conservative US Treasury investments. We expect
similar yields going forward until such time the credit markets
improve.
Results
of Operations for Fiscal 2009 Compared to Fiscal
2008
In 2009, we incurred a net loss of approximately
$19.0 million as compared to a net loss of
$14.2 million in 2008. The principal components of the
year-to-year
changes in line items are discussed below.
Net Revenues. We recognized revenue of
approximately $38.0 million in 2009 as compared to
$28.7 million in 2008. During 2009, we recorded
approximately $28.2 million of revenue from the sales of
Zevalin and Fusilev as compared to approximately
$8.0 million in 2008. Zevalin and Fusilev revenues in 2009
were approximately $15.7 and $12.5 million respectively,
compared to approximately $0.3 million and
$7.7 million, respectively in 2008. While shipments of
Fusilev for the period ended December 31, 2008 were
approximately $10.8 million (net of estimates for
promotional, price and other adjustments), based on our revenue
recognition policy, we had deferred the recognition of
approximately $3.1 million of such revenue until we had
more experience with product returns. We also recognized
approximately $0.3 million net sales of Zevalin from the
consolidation of RIT Oncology, LLC (RIT) effective
December 15, 2008. During 2009, we also recognized
$8.3 million of licensing revenues from the amortization of
the $41.5 million up-front payment we received from
Allergan in 2008. We also recognized a milestone payment from
Allergan of $1.5 million on the completion of enrollment of
our two pivotal clinical trials for apaziquone. No similar
revenues were recognized in 2008. During 2008, we recognized
revenue from: (i) an agreement with Par Pharmaceutical, our
former marketing partner for sumatriptan injection, pursuant to
which we received a non-refundable $20 million cash payment
from Par for the transfer of our share of the profits from the
commercialization of sumatriptan injection; and (ii) the
transfer of rights to certain of our ANDAs to Sagent
Pharmaceuticals for $660,000. No similar revenues were generated
during 2009.
Selling, General and Administrative. Selling,
general and administrative expenses increased by approximately
$18.4 million, from approximately $15.2 million in
2008 to approximately $33.6 million in 2009, primarily due
to approximately:
|
|
|
| |
•
|
$10.6 million increase attributable to sales and marketing
expenses, including payroll costs, incurred with the launch of
Zevalin and Fusilev.
|
| |
| |
•
|
$3.3 million increase in general and administrative costs
due to increased activities, including payroll costs and higher
professional costs due to business development activities
|
| |
| |
•
|
$1.6 million increase in non-cash compensation expenses.
|
Research and Development. Research and
development expenses decreased by approximately
$5.7 million, from approximately $26.7 million in 2008
to approximately $21 million in 2009, which included
non-cash amortization and write off of Zevalin related
intangibles and in-process research, and development charges of
approximately $3.7 million and $4.9 million in 2009
and 2008, respectively. Research and development expenses also
reduced primarily due to sharing of apaziquone related
development costs by our development partner, Allergan, of
approximately $11.2 million. In addition, we incurred
reduced development expense in other development products,
including ozarelix. During 2009, in line with the strategy
outlined at the start of the year, and in response to the global
financial crisis we focused on executing a successful launch of
Zevalin and Fusilev and prioritized our research and development
efforts to complete the rapid enrollment in the apaziquone
clinical studies.
As reported above, we recorded approximately $3.7 million
of expense from amortization of Zevalin related intangibles for
the year ended December 31, 2009 as compared to
$0.2 million during the same period in 2008. This
61
was a full year’s amortization expense as compared to
15 day’s prorated amortization during 2008, since
Zevalin was acquired in December 2008. During the year 2008, we
recorded expense of $4.7 million for in-process research
and development, or IPRD, on the Zevalin related intangibles; no
similar expense was recorded in 2009.
Change in Fair Value of Common Stock Warrant
Liability. We recorded approximately
$8.1 million of income from warrant obligations in 2009 as
compared to $1.3 million in 2008, in connection with the
restatement of our previously reported financial statements.
Other Net Income. Other income consisted of
net interest income of approximately $0.7 million and
$1.2 million for the years ended December 31, 2009 and
2008, respectively and in 2008 included approximately $200,000
realized investment gains. The decrease in interest income was
primarily due to lower investment yields in 2009 due to the
shift in our investment strategy to more conservative US
Treasury investments.
Nature of
each accrual that reduces gross revenue to net revenue
Provisions for product returns, sales discounts and rebates,
distribution and data fees, and estimates for chargebacks are
established as a reduction of product sales revenue at the time
revenues are recognized. Management considers various factors in
determination of such provisions, which are described more in
detail below. Such estimated amounts are deducted from our gross
sales to determine our net revenues. Provisions for bad and
doubtful accounts are deducted from gross receivables to
determine net receivables. Changes in our estimates, if any,
would be recorded in the statement of operations in the period
the change is determined. If we materially over or under
estimate the amount, there could be a material impact on our
consolidated financial statements.
For the periods ended December 31, 2010 and 2009, the
following is a roll forward of the provisions for product
returns, discounts and rebates, data and distribution fees and
chargeback allowances and estimated doubtful account allowances:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Chargebacks
|
|
|
|
|
|
|
|
|
Data and
|
|
|
|
|
|
|
|
|
|
|
and
|
|
|
|
|
|
|
|
|
Distribution
|
|
|
Doubtful
|
|
|
|
|
|
|
|
Discounts
|
|
|
Rebates
|
|
|
Returns
|
|
|
Fees
|
|
|
accounts
|
|
|
Total
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Period ended December 31, 2010:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at beginning of the period
|
|
$
|
860
|
|
|
|
388
|
|
|
$
|
1,176
|
|
|
$
|
213
|
|
|
$
|
150
|
|
|
$
|
2,787
|
|
|
Add provisions:
|
|
|
1,750
|
|
|
|
14,721
|
|
|
|
3,540
|
|
|
|
3,029
|
|
|
|
359
|
|
|
|
23,389
|
|
|
Less: Credits or actual allowances:
|
|
|
(1,935
|
)
|
|
|
(635
|
)
|
|
|
(2,716
|
)
|
|
|
(1,368
|
)
|
|
|
(170
|
)
|
|
|
(6,824
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at the close of the period
|
|
$
|
675
|
|
|
$
|
14,474
|
|
|
$
|
2,000
|
|
|
$
|
1,874
|
|
|
$
|
339
|
|
|
$
|
19,352
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period ended December 31, 2009:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at beginning of period
|
|
$
|
1,631
|
|
|
$
|
—
|
|
|
$
|
3,144
|
|
|
$
|
—
|
|
|
$
|
150
|
|
|
$
|
4,925
|
|
|
Add provisions:
|
|
|
3,760
|
|
|
|
469
|
|
|
|
95
|
|
|
|
1,212
|
|
|
|
—
|
|
|
|
5,536
|
|
|
Less: Credits or actual allowances:
|
|
|
(4,531
|
)
|
|
|
(81
|
)
|
|
|
(2,063
|
)
|
|
|
(999
|
)
|
|
|
—
|
|
|
|
(7,674
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at the close of the period
|
|
$
|
860
|
|
|
$
|
388
|
|
|
$
|
1,176
|
|
|
$
|
213
|
|
|
$
|
150
|
|
|
$
|
2,787
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amounts recorded as allowances on our consolidated balance
sheets for 2010 and 2009 are reflected in the table above. The
basis and methods of estimating these allowances, used by
management, are described below.
Chargebacks,
discounts and rebates
Chargebacks represent a provision against gross accounts
receivable and related reduction to gross revenue. A chargeback
is the difference between the price the wholesale customer, in
our case the wholesaler or distributor, pays (the wholesale
acquisition cost, or WAC) and the price (contracted price) that
a contracted customer (e.g., a Group Purchasing Organization, or
GPO, member) pays for a product. We accrue for chargebacks in
the relevant
62
period on the presumption that all units of product sold to
members of the GPOs will be charged back. We estimate
chargebacks at the time of sale of our products to the members
of the GPOs based on:
(1) volume of all products sold via distributors to members
of the GPOs and the applicable chargeback rates for the relevant
period;
(2) applicable WAC and the contract prices agreed with the
GPOs; and
(3) the information of inventories remaining on hand at the
wholesalers and distributors at the end of the period, actual
chargeback reports received from our wholesalers and
distributors as well as the chargebacks not yet billed (product
shipped less the chargebacks already billed back) in the
calculation and validation of our chargeback estimates and
reserves.
Discounts (generally prompt payment discounts) are accrued at
the end of every reporting period based on the gross sales made
to the customers during the period and based on their terms of
trade for a product. We generally review the terms of the
contracts, specifically price and discount structures, payment
terms in the contracts between the customer and the Company to
estimate the discount accrual.
Customer rebates are estimated at every period end, based on
direct purchases, depending on whether any rebates have been
offered. The rebates are recognized when products are purchased
and a periodic credit is given. Medicaid rebates are based on
the data we receive from the public sector benefit providers,
which is based on the final dispensing of our product by a
pharmacy to a benefit plan participant.
We record Medicaid and Medicare rebates based on estimates for
such expense. However, such amount have not been material to the
financial statements.
Product
returns allowances
Customers are typically permitted to return products within
thirty days after shipment, if incorrectly shipped or not
ordered, and within a window of time six months before and
twelve months after the expiration of product dating, subject to
certain restocking fees and preauthorization requirements, as
applicable. The returned product is destroyed if it is damaged,
quality is compromised or past its expiration date. Based on our
returns policy, we refund the sales price to the customer as a
credit and record the credit against receivables. In general,
returned product is not resold. As of each balance sheet date,
we estimate potential returns, based on several factors,
including: inventory held by distributors, sell through data of
distributor sales to end users, customer and end-user ordering
and re-ordering patterns, aging of accounts receivables, rates
of returns for directly substitutable products and
pharmaceutical products for the treatment of therapeutic areas
similar to indications served by our products, shelf life of our
products and based on experience of our management with selling
similar oncology products. We record an allowance for future
returns by debiting revenue, thereby reducing gross revenues and
crediting a reserve for returns to other accrued liabilities.
Distribution
and Data Fees
Distribution and data fees are paid to authorized wholesalers
and specialty distributors of Fusilev as a percentage of WAC for
products sold. The services provided include contract
administration, inventory management, product sales reporting by
customer, returns for clinics and hospitals. We accrue
distribution and data fees based on a percentage of Fusilev
revenues that are set and governed by distribution agreements.
Doubtful
Accounts
An allowance for doubtful accounts is estimated based on the
customer payment history and a review by management of the aging
of the accounts receivables as of the balance sheet date. We
accrue for doubtful accounts by recording an expense and
creating an allowance for such accounts. If we are privy to
information on the solvency of a customer or observe a payment
history change, we estimate the accrual for such doubtful
receivables or write the receivable off.
63
Off-Balance
Sheet Arrangements
We do not have any off balance sheet arrangements.
Contractual
and Commercial Obligations
The following table summarizes our contractual and other
commitments, including obligations under a facility lease and
equipment leases, as of December 31, 2010, approximately:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Less than
|
|
|
|
|
|
|
|
|
After
|
|
|
($ in ’000’s)
|
|
Total
|
|
|
1 Year
|
|
|
2-3 Years
|
|
|
4-5 Years
|
|
|
5 Years
|
|
|
|
|
Contractual Obligations(1)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Capital Lease Obligations(2)
|
|
|
90
|
|
|
|
45
|
|
|
|
45
|
|
|
|
—
|
|
|
|
—
|
|
|
Operating Lease Obligations(3)
|
|
|
3,245
|
|
|
|
568
|
|
|
|
1,233
|
|
|
|
1,152
|
|
|
|
292
|
|
|
Purchase Obligations(4)
|
|
|
6,323
|
|
|
|
5,931
|
|
|
|
392
|
|
|
|
—
|
|
|
|
—
|
|
|
Contingent Milestone Obligations(5)
|
|
|
204,043
|
|
|
|
6,089
|
|
|
|
69,058
|
|
|
|
37,926
|
|
|
|
90,970
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
213,701
|
|
|
|
12,633
|
|
|
|
70,728
|
|
|
|
39,078
|
|
|
|
91,262
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
The table of contractual and commercial obligations excludes
contingent payments that we may become obligated to pay upon the
occurrence of future events whose outcome is not readily
determinable. Such significant contingent obligations are
described below under “Employment Agreement.” |
| |
|
(2) |
|
The capital lease obligations are related to leased office
equipment. |
| |
|
(3) |
|
The operating lease obligations are primarily related to the
facility lease for our principal executive office in Henderson,
Nevada expiring April 30, 2014; and for our research and
development facility in Irvine, California expiring
June 30, 2016 |
| |
|
(4) |
|
Purchase obligations represent the amount of open purchase
orders and contractual commitments to vendors for products and
services that have not been delivered, or rendered, as of
December 31, 2010. Approximately 90% of the purchase
obligations consist of expenses associated with clinical trials
and related costs for apaziquone and ozarelix for each of the
periods presented. Please see “Service Agreements”
below for further information. |
| |
|
(5) |
|
Milestone obligations are payable contingent upon successfully
reaching certain development and regulatory milestones as
further described below under “Licensing Agreements.”
While the amounts included in the table above represent all of
our potential cash development and regulatory milestone
obligations as of December 31, 2010, given the
unpredictability of the drug development process, and the
impossibility of predicting the success of current and future
clinical trials, the timelines estimated above do not represent
a forecast of when payment milestones will actually be reached,
if at all. Rather, they assume that all development and
regulatory milestones under all of our license agreements are
successfully met, and represent our best estimates of the
timelines. In the event that the milestones are met, we believe
it is likely that the increase in the potential value of the
related drug product will exceed the amount of the milestone
obligation. |
Licensing
Agreements
Almost all of our drug candidates are being developed pursuant
to license agreements that provide us with rights to certain
territories to, among other things, develop, sublicense, and
sell the drugs. We are required to use commercially reasonable
efforts to develop the drugs, are generally responsible for all
development, patent filing and maintenance costs, sales,
marketing and liability insurance costs, and are generally
contingently obligated to make milestone payments to the
licensors if we successfully reach development and regulatory
milestones specified in the agreements. In addition, we are
obligated to pay royalties and, in some cases, milestone
payments based on net sales, if any, after marketing approval is
obtained from regulatory authorities.
The potential contingent development and regulatory milestone
obligations under all our licensing agreements are generally
tied to progress through the FDA approval process, which
approval significantly depends on positive clinical trial
results. The following list is typical of milestone events
relevant for us: conclusion of Phase 2 or commencement of Phase
3 clinical trials; filing of new drug applications in each of
the United States, Europe and Japan; and approvals from each of
the regulatory agencies in those jurisdictions.
64
Service
Agreements
In connection with the research and development of our drug
products, we have entered into contracts with numerous third
party service providers, such as clinical trial centers,
clinical research organizations, data monitoring centers, and
with drug formulation, development and testing laboratories. The
financial terms of these agreements are varied and generally
obligate us to pay in stages, depending on achievement of
certain events specified in the agreements, such as contract
execution, reservation of service or production capacity, actual
performance of service, or the successful accrual and dosing of
patients.
At each period end, we accrue for all costs of goods and
services received, with such accruals based on factors such as
estimates of work performed, patient enrollment, completion of
patient studies and other events. We are in a position to
accelerate, slow-down or discontinue any or all of the projects
that we are working on at any given point in time. Should we
decide to discontinue
and/or
slow-down the work on any project, the associated costs for
those projects would get limited to the extent of the work
completed. Generally, we are able to terminate these contracts
due to the discontinuance of the related project(s) and thus
avoid paying for the services that have not yet been rendered
and our future purchase obligations would reduce accordingly.
Employment
Agreement
We have entered into an employment agreement with
Dr. Shrotriya, our President and Chief Executive Officer,
which expires January 2, 2012. The employment agreement
automatically renews for a one-year calendar term unless either
party gives written notice of such party’s intent not to
renew the agreement at least ninety days prior to the
commencement of the next year. The employment agreement requires
Dr. Shrotriya to devote his full working time and effort to
the business and affairs of the Company during the term of the
agreement. The employment agreement provides for a minimum
annual base salary with annual increases, periodic bonuses and
option grants as determined by the Compensation Committee of the
Board of Directors.
Dr. Shrotriya’s employment may be terminated due to
non-renewal of his employment agreement by us, mutual agreement,
death or disability, or by us for cause (as that term is defined
in the employment agreement) or without cause, or by
Dr. Shrotriya for no reason, good reason (as defined in the
agreement) or non-renewal. The employment agreement provides for
various guaranteed severance payments and benefits if:
(i) the agreement is not renewed by us,
(ii) Dr. Shrotriya’s employment is terminated
without cause, (iii) Dr. Shrotriya resigns for good
reason, (iv) the agreement is terminated due to death or
disability of Dr. Shrotriya, (v) if Dr. Shrotriya
voluntarily resigns his employment for no reason or (vi) if
Dr. Shrotriya’s employment is terminated (other than
by Dr. Shrotriya) without cause within twelve months after
a change in control, or Dr. Shrotriya is adversely affected
in connection with a change in control and resigns within twelve
months. If the agreement is terminated due to mutual agreement,
Dr. Shrotriya’s non-renewal of the agreement, or by us
for cause, Dr. Shrotriya shall not be entitled to any
severance.
If any payment or distribution by us to or for the benefit of
Dr. Shrotriya is subject to the excise tax imposed by
Section 4999 of the Internal Revenue Code, or IRC, or any
interest or penalties are incurred by Dr. Shrotriya with
respect to such excise tax, then Dr. Shrotriya shall be
entitled to receive an additional payment in an amount such that
after payment by Dr. Shrotriya of all taxes (including any
interest and penalties imposed with respect thereto) and excise
tax imposed upon such payment, Dr. Shrotriya retains an
amount of the payment equal to the excise tax imposed upon the
payment.
If we determine that any payments to Dr. Shrotriya under
the agreement fail to satisfy the distribution requirement of
Section 409A(a)(2)(A) of the IRC, the payment schedule of
that benefit shall be revised to the extent necessary so that
the benefit is not subject to the provisions of
Section 409A(a)(1) of the IRC. We may attach conditions to
or adjust the amounts so paid to preserve, as closely as
possible, the economic consequences that would have applied in
the absence of this adjustment; provided, however, that no such
condition or adjustment shall result in the payments being
subject to Section 409A(a)(1) of the IRC.
65
Critical
Accounting Policies, Estimates and Assumptions
Our discussion and analysis of our consolidated financial
condition and results of operations are based upon our
consolidated financial statements, which have been prepared in
conformity with accounting principles generally accepted in the
United States, or GAAP. The preparation of these financial
statements requires us to make estimates and assumptions that
affect the reported amounts of assets, liabilities, revenues and
expenses, and related disclosure of contingent assets and
liabilities reported in our consolidated financial statements.
The estimation process requires assumptions to be made about
future events and conditions, and is consequently inherently
subjective and uncertain. Actual results could differ materially
from our estimates. We regularly evaluate our estimates,
including cash requirements, by assessing: planned research and
development activities and general and administrative
requirements; required clinical trial activity; market need for
our drug candidates; and other major business assumptions.
The SEC defines critical accounting policies as those that are,
in management’s view, most important to the portrayal of
our financial condition and results of operations and most
demanding of our judgment. We consider the following policies to
be critical to an understanding of our consolidated financial
statements and the uncertainties associated with the complex
judgments made by us that could impact our results of
operations, financial position and cash flows.
Revenue
Recognition
We sell our products to wholesalers and distributors of oncology
products and directly to the end user, directly or through GPOs
(e.g., certain hospitals or hospital systems and clinics with
whom we have entered into a direct purchase agreement). Our
wholesalers and distributors purchase our products and sell the
products directly to the end users, which include, but are not
limited to, hospitals, clinics, medical facilities, managed care
facilities and private oncology based practices etc. Revenue
from product sales is recognized upon shipment of product when
title and risk of loss have transferred to the customer, and the
following additional criteria specified by ASC
No. 605-15,
“Revenue Recognition: Products” are met:
(i) the price is substantially fixed and determinable;
(ii) our customer has economic substance apart from that
provided by us;
(iii) our customer’s obligation to pay us is not
contingent on resale of the product; and
(iv) we do not have significant obligations for future
performance to directly bring about the resale of our
product; and
(v) we have a reasonable basis to estimate future returns.
Generally, revenue is recognized when all four of the following
criteria are met:
(i) persuasive evidence that an arrangement exists;
(ii) delivery of the products has occurred, or services
have been rendered;
(iii) the selling price is both fixed and
determinable; and
(iv) collectibility is reasonably assured.
Provisions for estimated product returns, sales discounts,
rebates and charge backs are established as a reduction of gross
product sales at the time such revenues are recognized. Thus,
revenue is recorded, net of such estimated provisions. Our
estimates for product returns are based our review of inventory
in the channels and review of historical rates of actual returns.
Consistent with industry practice, our product return policy
permits our customers to return products within thirty days
after shipment, if incorrectly shipped or not ordered, and
within a window of time six months before and twelve months
after the expiration of product dating, subject to certain
restocking fees and preauthorization requirements, as
applicable. Currently, our returns policy does not allow for
replacement of product. The returned product is destroyed if it
is damaged, its quality is compromised or it is past its
expiration date. Based on our returns
66
policy, we refund the sales price to the customer as a credit
and record the credit against receivables. In general returned
product is not resold. We generally reserve the right to decline
granting a return and to decide on product destruction. As of
each balance sheet date, we estimate potential returns, based on
several factors, including: inventory held by distributors, sell
through data of distributor sales to end users, customer and
end-user ordering and re-ordering patterns, aging of accounts
receivables, rates of returns for directly substitutable
products and other pharmaceutical products for the treatment of
therapeutic areas similar to indications served by our products,
shelf life of our products and the extensive experience of our
management with selling the same and similar oncology products.
We record an allowance for future returns by debiting revenue,
thereby reducing gross revenues and crediting a reserve for
returns to reduce gross receivables. If allowances exceed the
related accounts receivables, we reclassify such allowances to
accrued obligations.
We also state the related accounts receivable at net realizable
value, with any allowance for doubtful accounts charged to
general operating expenses. If revenue from sales is not
reasonably determinable due to provisions for estimates,
promotional adjustments, price adjustments, returns or any other
potential adjustments, we defer the revenue and recognize
revenue when the estimates are reasonably determinable, even if
the monies for the gross sales have been received.
Up-front fees representing non-refundable payments received upon
the execution of licensing or other agreements are recognized as
revenue upon execution of the agreements where we have no
significant future performance obligations and collectibility of
the fees is reasonably assured. Milestone payments, which are
generally based on developmental or regulatory events, are
recognized as revenue when the milestones are achieved,
collectibility is reasonably assured, and we have no significant
future performance obligations in connection with the milestone.
In those instances where we have collected fees or milestone
payments but have significant future performance obligations
related to the development of the drug product, we record
deferred revenue and recognize it over the period of our future
obligations.
Fair
Value of Acquired Assets
The fair value of acquired tangible and identifiable intangible
assets and liabilities assumed based on their estimated fair
values at the acquisition date requires extensive accounting
estimates and judgments, including in process research and
development. For each acquisition, we engaged an independent
third-party valuation firm to assist in determining the fair
value of in-process research and development and identifiable
intangible assets. Such a valuation requires significant
estimates and assumptions including but not limited to:
determining the timing and expected costs to complete the
in-process projects, projecting regulatory approvals, estimating
future cash flows from product sales resulting from in-process
projects, and developing appropriate discount rates and
probability rates by project. We believe the fair values
assigned to the assets acquired and liabilities assumed are
based on reasonable assumptions. However, these assumptions may
be inaccurate, and unanticipated events and circumstances may
occur. Additionally, we must determine whether an acquired
entity considered to be a business or a set of net assets
because a portion of the purchase price can only be allocated to
goodwill in a business combination.
Research
and Development
Research and development expenses include salaries and benefits,
clinical trial and related manufacturing costs, contract and
other outside service fees, and facilities and overhead costs
related to our research and development efforts. Research and
development expenses also consist of costs incurred for
proprietary and collaboration research and development and
include activities such as product registries and
investigator-sponsored trials. Research and development costs
are expensed as incurred. In certain instances we enter into
agreements with third parties for research and development
activities, where we may prepay fees for services at the
initiation of the contract. We record such prepayment as a
prepaid asset and charge research and development expense over
the period of time the contracted research and development
services are performed. In connection with the October 2008
co-development agreement, Allergan bears 65% of the development
costs incurred for apaziquone in NMIBC, commencing
January 1, 2009. During the year ended December 31,
2010 and December 31, 2009, approximately $7.5 million
$11.2 million, respectively, of development costs were
reimbursed by Allergan, and credited against total related
research and development expense.
67
As of each balance sheet date, we review purchase commitments
and accrue drug development expenses based on factors such as
estimates of work performed, patient enrollment, completion of
patient studies and other events. Accrued clinical study costs
are subject to revisions as trials progress to completion.
Revisions are recorded in the period in which the facts that
give rise to the revision become known.
Amortization
and impairment of intangible assets
Identifiable intangible assets with definite lives are amortized
on a straight-line basis over their estimated useful lives,
ranging from 1 to 10 years.
We evaluate the recoverability of intangible assets whenever
events or changes in circumstances indicate that an intangible
asset’s carrying amount may not be recoverable. Such
circumstances could include, but are not limited to the
following:
(i) a significant decrease in the market value of an asset;
(ii) a significant adverse change in the extent or manner
in which an asset is used; or
(iii) an accumulation of costs significantly in excess of
the amount originally expected for the acquisition of an asset.
We measure the carrying amount of the asset against the
estimated undiscounted future cash flows associated with it.
Should the sum of the expected future net cash flows be less
than the carrying value of the asset being evaluated, an
impairment loss would be recognized. The impairment loss would
be calculated as the amount by which the carrying value of the
asset exceeds its fair value.
Share-Based
Compensation
We recognize compensation expenses for all share-based awards to
employees and directors. In estimating the fair value of
share-based compensation, we use the quoted closing market
price, based on the date prior to our grant date, of our common
stock for stock awards and the Black-Scholes option pricing
model for stock options and warrants. We estimate future
volatility based on historical volatility of our common stock,
and we estimate the expected life of options based on several
criteria, including the vesting period of the grant and the
expected volatility.
Share based compensation is recognized only for those awards
that are ultimately expected to vest, and we have applied or
estimated forfeiture rate to unvested awards for purposes of
calculating compensation costs. These estimates will be revised
in future periods if actual forfeitures differ from the
estimates. Changes in forfeiture estimates impact compensation
cost in the period in which the change in estimate occurs.
We account for registered common stock warrants pursuant to
applicable accounting guidance on the understanding that in
compliance with applicable securities laws, the registered
warrants require the issuance of registered securities upon
exercise and do not sufficiently preclude an implied right to
net cash settlement. We classify registered warrants on the
consolidated balance sheet as a current liability which is
revalued at each balance sheet date subsequent to the initial
issuance. Determining the appropriate fair-value model and
calculating the fair value of registered warrants requires
considerable judgment, including estimating stock price
volatility and expected warrant life. We develop our estimates
based on historical data. A small change in the estimates used
may have a relatively large change in the estimated valuation.
We use the Black-Scholes pricing model to value the registered
warrants. Changes in the fair market value of the warrants are
reflected in the consolidated statement of operations as
“Change in fair value of common stock warrant
liability.”
New
Accounting Pronouncements
See Note 2: Recent Accounting Pronouncements of our
accompanying consolidated financial statements for a description
of recent accounting pronouncements that have a potentially
significant impact on our financial reporting and our
expectations of their impact on our results of operations and
financial condition.
68
|
|
|
Item 7A.
|
Quantitive
and Qualitative Disclosures About Market Risk
|
The primary objective of our investment activities is to
preserve principal, while at the same time maximizing yields
without significantly increasing risk. We do not utilize hedging
contracts or similar instruments.
We are exposed to certain market risks. Our primary exposures
relate to (1) interest rate risk on our investment
portfolio, (2) credit risk of the companies’ bonds in
which we invest, (3) general credit market risks as have
existed since late 2007 and (4) the financial viability of
the institutions which hold our capital and through which we
have invested our funds. We manage such risks on our investment
portfolio by investing in highly liquid, highly rated
instruments and not investing in long-term maturity instruments.
In response to the dislocation in the credit markets since the
latter part of 2007, in early 2008 we converted substantially
all of our investments, including all of our market auction debt
securities, into highly liquid and safe instruments. Our
investments, as of December 31, 2010 and December 31,
2009, were primarily in money market accounts, short-term
corporate bonds, certificate of deposits, U.S. Treasury
bills and U.S. Treasury-backed securities. We believe the
financial institutions through which we have invested our funds
are strong, well capitalized and our instruments are held in
accounts segregated from the assets of the institutions.
However, due to the current extremely volatile financial and
credit markets and liquidity crunch faced by most banking
institutions, the financial viability of these institutions, and
the safety and liquidity of our funds is being constantly
monitored.
Because of our ability to generally redeem these investments at
par at short notice and without penalty, changes in interest
rates would have an immaterial effect on the fair value of these
investments. If a 10% change in interest rates were to have
occurred on December 31, 2010 or December 31, 2009,
any decline in the fair value of our investments would not be
material in the context of our consolidated financial
statements. In addition, we are exposed to certain market risks
associated with credit ratings of corporations whose corporate
bonds we may purchase from time to time. If these companies were
to experience a significant detrimental change in their credit
ratings, the fair market value of such corporate bonds may
significantly decrease. If these companies were to default on
these corporate bonds, we may lose part or all of our principal.
We believe that we effectively manage this market risk by
diversifying our investments, and investing in highly rated
securities.
In addition, we are exposed to foreign currency exchange rate
fluctuations relating to payments we make to vendors, suppliers
and license partners using foreign currencies. In particular,
some of our obligations are incurred in Euros. We mitigate such
risk by maintaining a limited portion of our cash in Euros and
other currencies.
|
|
|
Item 8.
|
Financial
Statements and Supplementary Data
|
Our annual consolidated financial statements are included in
Item 15 of this report.
|
|
|
Item 9.
|
Changes
in and Disagreements with Accoutants on Accounting and Financial
Disclosure
|
None
|
|
|
Item 9A.
|
Controls
and Procedures
|
Our principal executive officer and principal financial officer
have provided certifications filed as Exhibits 31.1 and
32.1, and 31.2 and 32.2, respectively. Such certifications
should be read in conjunction with the information contained in
this Item 9A for a more complete understanding of the
matters covered by such certifications.
|
|
|
(i)
|
Disclosure
Controls and Procedures
|
We have established disclosure controls and procedures (as such
terms are defined in
Rules 13a-15(e)
and
15d-15(e)
under the Exchange Act, that are designed to ensure that
information required to be disclosed in our Exchange Act reports
is recorded, processed, summarized and reported within the time
periods specified in the SEC’s rules and forms, and that
such information is accumulated and communicated to our
management, including our Chief Executive Officer (our principal
executive officer) and Acting Chief Financial Officer (our
principal financial officer), as appropriate, to allow for
timely decisions regarding required disclosure. In designing and
evaluating the disclosure controls and procedures, our
management is required to apply its judgment in evaluating
69
the cost-benefit relationship of possible controls and
procedures. Our disclosure controls and procedures are designed
to provide a reasonable level of assurance of reaching our
desired disclosure control objectives.
As required by SEC
Rule 13a-15(b),
we carried out an evaluation, under the supervision and with the
participation of our management, including our Chief Executive
Officer and our Acting Chief Financial Officer, of the
effectiveness of the design and operation of our disclosure
controls and procedures as of December 31, 2010, the end of
the period covered by this report (the “Evaluation
Date”). Based on the foregoing, our Chief Executive Officer
and Acting Chief Financial Officer concluded that our disclosure
controls and procedures, as of the end of the period covered by
this report, were effective in timely alerting them to material
information relating to the Company required to be included in
our periodic SEC filings.
|
|
|
(ii)
|
Internal
Control Over Financial Reporting
|
|
|
|
(a)
|
Management’s
annual report on internal control over financial
reporting
|
Our management is responsible for establishing and maintaining
adequate internal control over financial reporting, as such term
is defined in Exchange Act
Rules 13a-15(f)
and
15d-15(f).
Our internal control system was designed to provide reasonable
assurance to our management and board of directors regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. Due to the small size
of our company and the limited number of employees, it is not
possible for us to fully segregate duties associated with the
financial reporting process; accordingly, we rely on mitigating
controls to reduce the risks from such lack of segregation of
duties. Further, all internal control systems, no matter how
well designed, have inherent limitations. Therefore, even those
systems determined to be effective can provide only reasonable
assurance with respect to financial statement preparation and
presentation. Because of such inherent limitations, internal
control over financial reporting may not prevent or detect
misstatements. Projections of any evaluation of effectiveness to
future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our
management, including our principal executive officer and
principal financial officer, we conducted an evaluation of the
effectiveness of our internal control over financial reporting
based on the framework in Internal Control —
Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission, or COSO. Based on our
evaluation under the framework in COSO, our management concluded
that our internal control over financial reporting was effective
as of the Evaluation Date.
Our independent registered public accounting firm,
Ernst & Young LLP, has issued a report on our internal
control over financial reporting. Ernst & Young
LLP’s report appears below under Item 9A(ii)(b) and
expresses an unqualified opinion on the effectiveness of our
internal control over financial reporting.
|
|
|
(b)
|
Changes
in internal control over financial reporting
|
During the quarter ended December 31, 2010, there were no
changes in our internal control over financial reporting that
have materially affected, or are reasonably likely to materially
affect, our internal control over financial reporting.
70
Report of
Ernst & Young LLP, Independent Registered Public
Accounting Firm
The Board of Directors and
Stockholders of Spectrum Pharmaceuticals, Inc.
We have audited Spectrum Pharmaceuticals, Inc. and
Subsidiaries’ internal control over financial reporting as
of December 31, 2010, based on criteria established in
Internal Control — Integrated Framework issued by the
Committee of Sponsoring Organizations of the Treadway Commission
(the COSO criteria). Spectrum Pharmaceuticals, Inc. and
Subsidiaries’ management is responsible for maintaining
effective internal control over financial reporting, and for its
assessment of the effectiveness of internal control over
financial reporting included in the accompanying
Management’s Annual Report on Internal Control over
Financial Reporting. Our responsibility is to express an opinion
on the company’s internal control over financial reporting
based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control
over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of
internal control over financial reporting, assessing the risk
that a material weakness exists, testing and evaluating the
design and operating effectiveness of internal control based on
the assessed risk, and performing such other procedures as we
considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s
internal control over financial reporting includes those
policies and procedures that (1) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the
company; (2) provide reasonable assurance that transactions
are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting
principles, and that receipts and expenditures of the company
are being made only in accordance with authorizations of
management and directors of the company; and (3) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
In our opinion, Spectrum Pharmaceuticals, Inc. and Subsidiaries
maintained, in all material respects, effective internal control
over financial reporting as of December 31, 2010 based on
the COSO criteria.
We also have audited in accordance with standards of the Public
Company Accounting Oversight Board (United States), the
consolidated balance sheets of Spectrum Pharmaceuticals Inc. and
Subsidiaries as of December 31, 2010 and 2009, and the
related consolidated statements of operations,
stockholders’ equity and cash flows for each of the two
years in the period ended December 31, 2010 of Spectrum
Pharmaceuticals, Inc. and Subsidiaries and our report dated
March 9, 2011 expressed an unqualified opinion thereon.
Irvine, California
March 9, 2011
71
|
|
|
Item 9B.
|
Other
Information
|
None.
PART III
|
|
|
Item 10.
|
Directors,
Executive Officers and Corporate Governance
|
Directors
Our board of directors consists of six directors, each of whom
serves for a one-year term expiring at the following annual
meeting and until his successor has been duly elected and
qualified. The names of our directors and certain biographical
information about them are set forth below:
| |
|
|
Krishan K. Arora, Ph.D.

|
|
Dr. Arora, 70, has been a director of Spectrum since June
2010. Prior to his election, Dr. Arora had been providing
consulting services to Spectrum since February 2010.
Dr. Arora is a business executive with global experience in
driving strategic thinking, management and implementation of
operations for drug development worldwide. Dr. Arora has
provided consulting services to senior management at several
pharmaceutical companies, including Astellas Pharma Global
Development, Inc., for global drug development, from May 2008 to
June 2009, and UCB, Inc., for applications in global regulatory
affairs, electronic document management, pharmacovigilance and
worldwide quality assurance and compliance, from November 2003
to February 2006. Prior to that, Dr. Arora held senior
management positions with several pharmaceutical companies,
including Vice President of R&D Global Regulatory Affairs
for Management Information at Pfizer Inc. from 1998 to 2003,
Senior Director of Regulatory Affairs at Novartis AG from 1993
to 1998 and Group Director of Biometrics Operations at
Sanofi-Aventis. In addition, from 1994 to 2003, Dr. Arora
served as Chairman of the Electronic Regulatory Submissions
Working Group at PhRMA, which consisted of business and
information technology experts from the FDA and 20
biopharmaceutical companies and was the PhRMA lead at ICH on
establishing electronic standards for submission of marketing
applications to regulatory authorities. Dr. Arora received
a B.Sc. in Mathematics, Physics and Chemistry from Lucknow
University in India, a B.Sc. (Honors) in Agriculture and a M.Sc.
in Animal Genetics from G. B. Pant University of Agriculture
& Technology in India and a Ph.D. in Population Genetics
from Iowa State University.
|
|
|
|
Dr. Arora has significant experience in the pharmaceutical
industry, which includes 11 years’ experience in
global regulatory affairs and 18 years’ experience in
biometrics operations, including statistics, clinical data
systems, clinical data management and medical writing.
Furthermore, Dr. Arora has operational management
experience, a keen understanding of the regulatory environment
in which pharmaceutical companies operate and extensive
knowledge in drug development operations and regulatory
submissions and approvals. As a result, Dr. Arora is well
qualified to serve on our board of directors.
|
72
| |
|
|
Stuart M. Krassner,
Sc.D., Psy.D

|
|
Dr. Krassner, 75, has been a director of Spectrum since
December 2004 and was previously a member of our Scientific
Advisory Board from 1996 to 2001. Dr. Krassner’s
career spans four decades of experience in various positions at
the University of California, Irvine, or UCI, most recently as
Professor Emeritus of Developmental and Cell Biology at the
School of Biological Sciences. While at UCI, he developed and
reinforced FDA and NIH compliance procedures for UCI-sponsored
human clinical trials, established UCI’s first
Institutional Review Board, and at one time headed all contract
and grant activities. Dr. Krassner has also been retained
by a number of public and private pharmaceutical, medical device
and other companies to provide scientific and regulatory
advisory services, including FDA compliance.
Dr. Krassner’s work has been published in numerous
peer-reviewed U.S. journals. Dr. Krassner has been awarded
grants from the National Institute of Health, the National
Science Foundation and the World Health Organization.
Dr. Krassner has been a member of the American Society of
Protozoology, the American Society of Tropical Medicine and
Hygiene, the Corporation of the Marine Biological Laboratories,
Woods Hole, MA, and Sigma Xi, among others. Dr. Krassner
received a B.S. in Biology from Brooklyn College and an Sc.D.
from the Bloomberg School of Public Health at Johns Hopkins
University.
|
|
|
|
Dr. Krassner’s extensive and distinctive experience in
business and academia brings valuable perspective to our board.
He has a strong background in research in the area of
developmental and cell biology and his work in the area has been
published in numerous peer-reviewed U.S. journals. Moreover, his
expertise in scientific and regulatory advisory services,
including FDA compliance, makes him well qualified to serve on
our board of directors.
|
|
|
|
|
Luigi Lenaz, M.D.

|
|
Dr. Lenaz, 70, has been a director of Spectrum since June
2010. Dr. Lenaz served as Spectrum’s Chief Scientific
Officer from February 2005 to June 2008 and as President of
Spectrum’s Oncology Division from 2000 to 2005. Since
retiring as Spectrum’s Chief Scientific Officer in June
2008, Dr. Lenaz provided consulting services to Spectrum
from June 2008 to June 2010. From 1997 to 2000, Dr. Lenaz
served as Senior Vice President of Clinical Research, Medical
Affairs at SuperGen, Inc., a NASDAQ listed pharmaceutical
company dedicated to cancer drug development. From 1978 to 1997,
Dr. Lenaz held several senior management positions with
Bristol- Myers Squibb, a NYSE-listed pharmaceutical company,
including Senior Vice President of Oncology Franchise Management
from 1990 to 1997 and Director of Scientific Affairs,
Anti-Cancer from 1985 to 1990. Dr. Lenaz is also a
prominent researcher, having conducted research in the areas of
pharmacology, experimental chemotherapy, histology, general
physiology, and experimental therapeutics at various
institutions for cancer research, including Roswell Park
Memorial Institute, Memorial Sloan-Kettering Cancer Center and
the National Cancer Institute in Milan. He is a member of
several scientific societies, including the American Association
for Cancer Research, American Association for Clinical Oncology,
European Society for Medical Oncology, and International
Association for the Study of Lung Cancer. Dr. Lenaz has
served as a director of Pharmaco-Kinesis Corporation, a
privately held medical device company, since January 2009.
Dr. Lenaz is a graduate of Liceo Scientifico A. Righi in
Bologna, Italy and he received a medical degree from the
University of Bologna Medical School in 1966.
|
73
| |
|
|
|
|
|
Dr. Lenaz is a renowned and accomplished oncologist who
will bring to the board of directors over 35 years’
experience in the pharmaceutical industry and a wealth of
knowledge in the field of cancer drug development.
Dr. Lenaz’s qualifications to serve on the board of
directors include his expertise in the development of cancer
drugs, his tenure as our Chief Scientific Officer, as well as
his subsequent consulting services for our company, his
significant management experience with Bristol-Myers Squibb, and
his prominent research in the field of oncology. As a result,
Dr. Lenaz is well qualified to serve on our board of
directors.
|
|
|
|
|
Anthony E. Maida, III,
M.A., M.B.A., Ph.D.

|
|
Dr. Maida, 59, has been a director of Spectrum since
December 2003. Dr. Maida is currently Vice President of
Clinical Research and General Manager, Oncology, world-wide for
PharmaNet, Inc. Dr. Maida has been the acting Chairman of
Dendri Therapeutics, Inc., a startup company focused on the
clinical development of therapeutic vaccines for patients with
cancer, since 2003. Dr. Maida has been serving as Chairman,
Founder and Director of BioConsul Drug Development Corporation
and Principal of Anthony Maida Consulting International since
1999, providing consulting services to large and small
biopharmaceutical firms in the clinical development of oncology
products and product acquisitions and to venture capital firms
evaluating life science investment opportunities. Additionally,
Dr. Maida formerly served as a member of the board of
directors of Sirion Therapeutics, Inc., a privately held
ophthalmic-focused company, and GlycoMetrix, Inc., a startup
company focused on the development of tests to identify
carbohydrates that can indicate cancer. Dr. Maida served as
the President and Chief Executive Officer of Replicon
NeuroTherapeutics, Inc., a biopharmaceutical company focused on
the therapy of patients with tumors (both primary and
metastatic) of the central nervous system, where he successfully
raised financing from both venture capital and strategic
investors and was responsible for all financial and operational
aspects of the company, from June 2001 to July 2003. From 1999
to 2001, Dr. Maida held positions as Interim Chief
Executive Officer for Trellis Bioscience, Inc., a privately held
biotechnology company that addresses high clinical stage failure
rates in pharmaceutical development, and President of CancerVax
Corporation, a biotechnology company dedicated to the treatment
of cancer. From 1992 until 1999, Dr. Maida served as
President and CEO of Jenner Biotherapies, Inc., a
biopharmaceutical company. From 1980 to 1992, Dr. Maida
held senior management positions with various companies
including Vice President Finance and Chief Financial Officer of
Data Plan, Inc., a wholly owned subsidiary of Lockheed
Corporation. Dr. Maida serves on the Advisory Boards of
EndPoint BioCapital and Sdn Bhd (Kuala Lumpur, Malaysia) and
serves or has served as a consultant and technical analyst for
several investment firms, including CMX Capital, LLC, Sagamore
Bioventures, Roaring Fork Capital, North Sound Capital, The
Bonnie J. Addario Lung Cancer Foundation and Pediaric
BioScience, Inc. Additionally, Dr. Maida has been retained
by Abraxis BioScience, Inc., Northwest Biotherapeutics, Inc.,
Takeda Chemical Industries, Ltd. (Osaka, Japan), and Toucan
Capital to conduct corporate and technical due diligence on
investment opportunities. Dr. Maida is a speaker at
industry conferences and is a member of the American Society of
Clinical Oncology, the American Association for Cancer Research,
the Society of Neuro-Oncology, the International Society for
Biological Therapy of Cancer, the American Association of
Immunologists and the American Chemical Society. Dr. Maida
received a B.A. in History from Santa Clara University in
1975, a B.A. in Biology from San Jose State University in
1977, an M.B.A. from Santa Clara University in 1978, an
M.A. in Toxicology from San Jose State University in 1986
and a Ph.D. in Immunology from the University of California in
2010.
|
74
| |
|
|
|
|
|
Dr. Maida’s qualifications to serve on the board of
directors include the extensive experience he has gained holding
senior management positions, including chairman, president,
chief financial officer and chief executive officer, at various
biotechnology and biopharmaceutical companies. He has
successfully raised financing from venture capital and strategic
investors for biopharmaceutical companies and he currently
provides consulting services to hedge funds, venture capital
firms interested in biopharmaceutical firms. Furthermore,
Dr. Maida’s vast knowledge in the area of clinical
development of oncology products and product acquisitions, in
addition to his continuous research in the field of oncology,
provides unique and valuable insight to our board of directors.
As a result, Dr. Maida is well qualified to serve on our
board of directors.
|
|
|
|
|
Dilip J. Mehta, M.D., Ph.D.

|
|
Dr. Mehta, 78, has been a director of Spectrum since June
2010. Dr. Mehta served on Spectrum’s board of
directors from June 2003 to July 2007. Dr. Mehta has been
self-employed as a pharmaceutical consultant since 1998 and
provided consulting services to Spectrum from July 2007 to June
2010. Dr. Mehta is a venture partner at Radius Ventures,
LLC in New York. From 1982 until his retirement in 1997,
Dr. Mehta held several senior management positions with
Pfizer Inc., including Senior Vice President, U.S. Clinical
Research, with responsibility for clinical research (Phases 1, 2
and 3) including data processing and statistical analysis for
Pfizer’s drugs in the U.S., as well as supervised
submissions of new drug applications for Cardura, Norvasc,
Zoloft, Zithromax, Diflucan, Unasyn, Trovan, Viagra, Geodon, and
a number of other drugs/supplements. Dr. Mehta served as
Chairman of the board of directors of Quintiles Spectral (India)
Limited (Ahmedabad, India) from 1998 to 2001 and as a member of
the board of directors of Bharat Serums & Vaccines Limited
(Mumbai, India) from 2006 to 2008 and Targanta Therapeutics
Corporation, a NASDAQ-listed biopharmaceutical company acquired
by The Medicines Company in February 2009, from 2005 to 2009.
From 1993 to 1997, Dr. Mehta served as Chair, Efficacy
Section for the Pharmaceutical Research and Manufacturers of
America, or PhRMA, in the International Conference on
Harmonization and was a PhRMA topic leader for one of the Expert
Working Group in Efficacy. From 1966 to 1982, Dr. Mehta
held the position of Group Director, Clinical Research in the
U.S. for Hoechst AG with supervision of Internal Medicine,
Metabolic and Infectious Diseases and Cardiovascular groups.
Dr. Mehta received an M.D., an M.B.B.S. (Bachelor of
Medicine and Bachelor of Surgery — equivalent to an
M.D. degree in the U.S.) and a Ph.D. from the University of
Bombay. Dr. Mehta was a Research Fellow in Clinical
Pharmacology at Cornell University Medical College.
|
|
|
|
Dr. Mehta brings to the board of directors over
28 years’ experience in the pharmaceutical industry
and a wealth of knowledge in the field of clinical research and
drug development. Dr. Mehta’s qualifications to serve
on the board of directors include his expertise in clinical
research, drug development and FDA matters, his prior service on
Spectrum’s board of directors, as well as his service on
the boards of directors of other publicly traded and privately
held biopharmaceutical companies and his significant management
experience with Pfizer. As a result, Dr. Mehta is well
qualified to serve on our board of directors.
|
75
| |
|
|
Rajesh C. Shrotriya, M.D.

|
|
Dr. Shrotriya, 66, has been Chairman of the Board, Chief
Executive Officer and President since August 2002 and a director
of Spectrum since June 2001. From September 2000 to August 2002,
Dr. Shrotriya served as President and Chief Operating
Officer of Spectrum. Dr. Shrotriya also serves as a member
of the board of directors of Antares Pharma, Inc., an
AMEX-listed drug delivery systems company. Prior to joining
Spectrum, Dr. Shrotriya held the position of Executive Vice
President and Chief Scientific Officer from November 1996 until
August 2000, and as Senior Vice President and Special Assistant
to the President from November 1996 until May 1997, for
SuperGen, Inc., a publicly-held pharmaceutical company focused
on drugs for life-threatening diseases, particularly cancer.
From August 1994 to October 1996, Dr. Shrotriya held the
positions of Vice President, Medical Affairs and Vice President,
Chief Medical Officer of MGI Pharma, Inc., an oncology-focused
biopharmaceutical company. Dr. Shrotriya spent
18 years at Bristol-Myers Squibb Company in a variety of
positions, most recently as Executive Director, Worldwide CNS
Clinical Research. Previously, Dr. Shrotriya held various
positions at Hoechst Pharmaceuticals, most recently as Medical
Advisor. Dr. Shrotriya was an attending physician and held
a courtesy appointment at St. Joseph Hospital in Stamford,
Connecticut. In addition, he received a certificate for Advanced
Biomedical Research Management from Harvard University.
Dr. Shrotriya received an M.D. from Grant Medical College,
Bombay, India, in 1974; a D.T.C.D. (Post Graduate Diploma in
Chest Diseases) from Delhi University, V.P. Chest Institute,
Delhi, India, in 1971; an M.B.B.S. (Bachelor of Medicine and
Bachelor of Surgery — equivalent to an M.D. degree in
the U.S.) from the Armed Forces Medical College, Poona, India,
in 1967; and a B.S. in Chemistry from Agra University, Aligarh,
India, in 1962.
|
|
|
|
Dr. Shrotriya is a demonstrated leader in the
biopharmaceutical industry. His significant leadership
experience includes 8 years of serving as our Chairman and
Chief Executive Officer as well as his service on the board of
directors of Antares Pharma, Inc. Dr. Shrotriya has held
prior leadership roles in the biopharmaceutical industry
including his positions as our President and Chief Operating
Officer, as the executive vice president and chief scientific
officer for a publicly-held pharmaceutical company, and
18 years of experience in various positions he held in
Bristol- Myers Squibb. Dr. Shrotriya’s significant
leadership experience in the biopharmaceutical sector, along
with his experience as a physician and his expertise in drug
development, position him well to serve on our board of
directors.
|
76
Executive
Officers
Each of our executive officers serves at the discretion of the
board of directors. The names of our executive officers and
certain biographical information about them are set forth below:
| |
|
|
|
Name and Age
|
|
|
|
|
|
Rajesh C. Shrotriya, M.D. (66) Chairman of the
Board, Chief Executive Officer and President
|
|
Information regarding Dr. Shrotriya is provided above.
|
|
George Tidmarsh, M.D, Ph.D. (51) Senior Vice
President, Chief Scientific Officer & Head of
Research & Development Operations
|
|
Dr. Tidmarsh has served as Senior Vice President, Chief
Scientific Officer and Head of Research & Development at
Spectrum since July 2010. Before joining Spectrum,
Dr. Tidmarsh served as the Chief Executive Officer of
Metronome Therapeutics, a privately held biopharmaceutical
company focused on novel cancer drug development from 2006 until
2010. From 2005 through 2008, he was the Founder and Chief
Executive Officer of Horizon Therapeutics, Inc., a venture
funded private company, where he successfully completed four
Phase 1 and two large Phase 3 trials, and authored all patent
applications. Dr. Tidmarsh also has published over twenty
articles. He earned his Bachelor of Science, M.D., and
Ph.D. from Stanford University and is currently an Associate
Professor of Pediatrics and Neonatology at Stanford University.
|
|
James E. Shields (59)
Senior Vice President, Chief Commercial Officer
|
|
Mr. Shields has served as Senior Vice president, Chief
Commercial Officer since May 2010. Previously Mr. Shields
served as Area Business Director for a Division of TEVA
Pharmaceutical Industries Limited from September 2007 through
April 2010 and Regional Business Director and National Director
of Sales for Commercial Divisions of Altana AG from March 2001
until the US Commercial Division was dissolved in December
2006. Mr. Shields also held positions of increasing
responsibility with several pharmaceutical companies, including
Centocor, Bristol-Myers Squibb, and ICI Stuart Pharmaceuticals.
Mr. Shields earned his Bachelor’s Degree from the
University of Kentucky.
|
|
Shyam Kumaria (61)
Senior Vice President of Finance
|
|
Mr. Kumaria has served as Vice President of Finance since
December 2003. From 1996 to 2003, he provided financial and
management consulting services to private companies. From 1984
to 1996, he served in senior executive and management positions
for several companies including Deloitte & Touche. Mr.
Kumaria became a Chartered Accountant in London, England in 1973
and a Certified Public Accountant in 1978. He received an
Executive M.B.A. from Columbia University in 1984.
|
|
Brett L. Scott (60)
Senior Vice President and Acting Chief Financial Officer
|
|
Mr. Scott has served as Senior Vice President and Acting Chief
Financial Officer since October 2010. Previously Mr. Scott
served as Chief Financial Officer at Biolase Technology a
Southern California-based medical device company. Prior to
Biolase, Mr. Scott was Executive Vice President and Chief
Financial Officer of North American Scientific, Inc., a Southern
California-based medical device company. In March 2009, North
American Scientific sought protection under Chapter 11 of the
U.S. Bankruptcy Code, and as part of an orderly plan to sell its
assets, during the following two months successfully completed
the sale of its prostate and breast cancer businesses to Best
Theratronics, Ltd. and Portola Medical Inc. respectively. Prior
to North American Scientific, Mr. Scott was Chief Financial
Officer of Irvine, California-based Alsius Corporation from
January 2006 to August 2008. Mr. Scott is a Certified Public
Accountant and received a bachelor of science degree in business
administration from the University of Southern California.
|
77
There are no
family relationships between any director or executive officer
and any other director or executive officer.
Section 16(A)
Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our executive
officers and directors, and persons who beneficially own more
than ten percent of our common stock, to file initial reports of
ownership and reports of changes in ownership with the SEC and
NASDAQ. Executive officers, directors and persons who
beneficially own more than ten percent of our common stock are
required by SEC regulations to furnish us with copies of all
Section 16(a) forms they file.
Based solely upon our review of the copies of reporting forms
furnished to us, and written representations that no other
reports were required, we believe that all filing requirements
under Section 16(a) of the Exchange Act applicable to our
directors, officers and any persons holding 10% or more of our
common stock with respect to our fiscal year ended
December 31, 2010 were satisfied on a timely basis with the
exception of Mr. George Tidmarsh, our Chief Scientific
Officer, who filed a Form 5 on February 14, 2011 to
report an earlier acquisition of shares of the Company’s
common stock in connection with an asset purchase transaction
that closed in July 2010.
Code of
Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics that
applies to all of our directors, officers and employees,
including the principal executive officer, principal financial
officer, principal accounting officer, controller or persons
performing similar functions. A copy of the Code of Business
Conduct and Ethics will be provided to any person, without
charge, upon oral request to
(949) 788-6700
or upon written request to Investor Relations, Spectrum
Pharmaceuticals, Inc., 157 Technology Drive, Irvine, CA 92618.
Amendments to the Code of Business Conduct and Ethics that apply
to our principal executive officer, principal financial officer,
principal accounting officer, controller or persons performing
similar functions, if any, will be posted on our website at
www.sppirx.com. We will disclose any waivers of provisions of
our Code of Business Conduct and Ethics that apply to our
directors and principal executive, financial and accounting
officers by disclosing such information on
Form 8-K.
Audit
Committee
We have a standing Audit Committee that is currently comprised
of Drs. Maida (Chair), Arora and Krassner, each of whom
satisfies the NASDAQ and SEC rules for Audit Committee
membership. Our board of directors has determined that
Drs. Maida, Arora and Krassner are each Audit Committee
financial experts within the meaning of SEC rules and that all
three are “independent” within the meaning of the
NASDAQ director independence standards and SEC
Rule 10A-3.
|
|
|
Item 11.
|
Executive
Compensation
|
Compensation
Discussion and Analysis
Executive
summary
In 2010, the company established aggressive plans for future
growth, including strategic business milestones —
maximizing the growth potential of our marketed drugs,
Zevalin®
and
Fusilev®,
managing multiple large, late-stage clinical trials for
apaziquone and belinostat, as well as developing a pipeline of
anti-cancer drugs. In addition, we targeted to grow our Zevalin
revenues by 25% to approximately $20 million and continue
to create shareholder value.
Our results in 2010 reflect achievement of certain key strategic
and financial objectives:
|
|
|
| |
•
|
Record product revenues of both of our marketed, proprietary
anticancer drugs
ZEVALIN®
and
FUSILEV®
|
| |
| |
•
|
Overall revenue for the company grew by almost 95% to
$74 million from $38 million in 2009
|
| |
| |
•
|
Closing the year with $104 million in cash, cash
equivalents and investments during difficult economic times and
market conditions,
|
78
|
|
|
| |
•
|
Strengthening the leadership and streamlining operations at each
of the Company’s functions,
|
| |
| |
•
|
Growing the Company’s market capitalization by
approximately 40% to levels which greatly exceeded the
Board’s expectations and peer company performance.
|
As a result, the Company is transforming from a small drug
development company to a biotechnology company with fully
integrated commercial and drug development operations.
These accomplishments are testaments to our Chairman and Chief
Executive Officer, Dr. Rajesh Shrotriya’s consistently
demonstrated clear vision and leadership in directing the
affairs of the Company through the past eight years. In
alignment with the its compensation philosophy, the Compensation
Committee determined that Dr. Shrotriya’s reward for
this performance would be in the form of an increased equity
incentive opportunity, the details of which are provided within
the Compensation Discussion and Analysis, or CD&A, and the
accompanying compensation table disclosures.
In addition, our transformation necessitated that we add three
newly appointed executive officers to our leadership team in
2010 to lead our sales, research & development, and
finance functions in the future. The specific compensation
programs that we established for each of these executive
officers are provided within the CD&A and accompanying
compensation table disclosures.
Compensation
Philosophy and Objectives
The Compensation Committee’s executive compensation
philosophy is to attract and retain professionals of the highest
caliber, capable of leading us to fulfillment of our ambitious
business objectives, by offering competitive compensation
opportunities that reward executives for their individual
contributions towards both our long-term and short-term goals.
Competition for attracting the best talent in the pharmaceutical
industry is very intense, and such competition is national in
scope. Accordingly, in light of the intense competition for
highly qualified executives, our executive officers are eligible
for competitive salary adjustments, cash bonuses and equity
compensation based upon periodic evaluations of individual and
company performance, relative to goals established at the start
of the year.
The Compensation Committee believes that three principal
compensation elements — base salary, annual bonus, and
equity incentive awards — in combination effectively
support the company’s overall compensation objectives of
attracting top talent for executive positions, incentivizing
such executive officers, rewarding them for achievement of
individual and company goals, and aligning the interests of
executive officers with those of our stockholders. The
Compensation Committee has not established specific competitive
market levels to target for each element, but has strived to
position the total compensation delivered by the three elements
as follows:
|
|
|
| |
•
|
Chairman & CEO; between the 75th and 90th percentile
of the peer group and industry in general
|
| |
| |
•
|
Other Named Executive Officers: between the median and 75th
percentile of the peer group and industry in general
|
The different positioning strategies reflect the contributions
made since 2002 by our current Chairman and CEO, as well as his
significant impact on our current and future business success.
In contrast, three of the four other named executive officers
were hired into their current roles in 2010. Finally, executive
officer total compensation is delivered primarily through the
annual bonus and equity incentive awards, hence actual
achievement of our desired compensation positioning vs. market
is largely dependent upon
pay-for-performance.
The Compensation Committee believes that its compensation
philosophy aligns the interests of our executive officers with
those of our stockholders, and is necessary to incentivize
individual executives to peak performance in advancing our
short-term and long-term business objectives. It is designed to
reward hard work, dedication and the achievement of both
individual and company goals.
Named
Executive Officers
For 2010, our named executive officers were as follows:
| |
|
|
|
Executive Name
|
|
Title
|
|
|
|
Rajesh Shrotriya
|
|
Chairman and Chief Executive Officer
|
79
| |
|
|
|
Executive Name
|
|
Title
|
|
|
|
George Tidmarsh
|
|
Chief Scientific Officer
|
|
Jim Shields
|
|
Chief Commercial Officer
|
|
Brett Scott
|
|
Acting Chief Financial Officer
|
|
Shyam Kumaria
|
|
Senior Vice President, Finance
|
Determining
Competitive Practices
Peer
Group for Compensation Benchmarking
In 2010, as part of our business transformation, we worked with
Grant Thornton LLP to develop a revised peer group consisting of
22 companies used for evaluating competitive total
compensation levels. This peer group represents a mix of
companies in which we would likely compete for business and
talent, with revenues and market capitalization similar to that
of Spectrum. Specifically, for 2009 (the most recent year in
which compensation benchmarking data is available) the peer
group companies had median revenue of approximately
$63 million and a market capitalization of
$564 million compared to Spectrum’s revenue of
$74 million and market capitalization of approximately
$354 million as of December 31, 2010. We used this
peer group specifically to review the level of base salaries,
mix and size of annual and long-term incentives, and other
benefits and perquisites provided to executives of similar-sized
companies.
The compensation peer group included the following companies:
| |
|
|
|
|
|
|
|
•
|
|
Affymax Inc
|
|
•
|
|
Immunomedics Inc
|
|
•
|
|
Allos Therapeutics Inc
|
|
•
|
|
Intermune Inc
|
|
•
|
|
Amicus Therapeutics Inc
|
|
•
|
|
Isis Pharmaceuticals Inc
|
|
•
|
|
Arena Pharmaceuticals Inc
|
|
•
|
|
Mannkind Corp
|
|
•
|
|
Biocryst Pharmaceuticals Inc
|
|
•
|
|
Medivation Inc
|
|
•
|
|
Biomarin Pharmaceutical Inc
|
|
•
|
|
Onyx Pharmaceuticals Inc
|
|
•
|
|
Cell Therapeutics Inc
|
|
•
|
|
Sangamo Biosciences Inc
|
|
•
|
|
Cytokinetics Inc
|
|
•
|
|
Seattle Genetics Inc
|
|
•
|
|
Dendreon Corp
|
|
•
|
|
Supergen Inc
|
|
•
|
|
Enzon Pharmaceuticals Inc
|
|
•
|
|
Theravance Inc
|
|
•
|
|
Exelixis Inc
|
|
•
|
|
Vertex Pharmaceuticals Inc
|
Other
Competitive Benchmarks
To supplement compensation data gathered from our peer group
companies, compensation for our named executive officers is also
compared to published survey data from the Radford Life Sciences
Survey. This survey includes data from various companies within
the same industry and of similar size to Spectrum.
Key
Elements of Executive Compensation
The principal elements of compensation for our executive
officers are:
|
|
|
| |
•
|
Base salary;
|
| |
| |
•
|
Annual bonuses; and
|
| |
| |
•
|
Equity incentive awards.
|
Base Salary. The base salaries
of our executive officers are established as part of an annual
compensation adjustment cycle. Base salaries for the year are
established either at the end of the prior year or the beginning
of the current year, i.e. the base salary for 2010 was set at
the end of 2009. In establishing those salaries, the
Compensation Committee considers the executive’s level of
responsibility, experience and individual performance, impact on
company results, and company performance, as well as information
regarding salary levels paid to executives with comparable
duties in companies at a similar stage as ours.
80
Annual Bonuses. The Compensation
Committee typically awards cash bonuses to executives as part of
their annual overall compensation at the beginning of the next
year for prior year performance, i.e. the cash bonus for 2010
performance was set in early 2011. Such cash bonuses are a
reward for company results, individual achievement of goals, as
well as achievement of key milestones. We have not established
formal bonus target levels for our executive officers; however,
the actual bonus awarded is determined based upon the
Committee’s evaluation of each executive’s performance
along with a review of bonus opportunities and actual bonuses
paid to executives with similar duties and impact on results
working in comparable companies.
Equity Incentive Awards. Cash
compensation is viewed as a reward for annual performance vs.
goals, Equity incentive awards are important long-term
compensation tools for employee retention as well as
incentivizing future performance. In addition, equity incentive
awards are an important compensation tool to utilize in
attracting and retaining high caliber executive talent. The
Compensation Committee believes that granting equity incentive
awards, including stock options and restricted stock, to our
executive officers is very beneficial to stockholders because it
aligns management’s interests in the enhancement of
stockholder value. An executive officer receives value from
these grants only if he or she remains employed by us during the
vesting period, and, with regard to stock options, only if our
common stock appreciates from the fair market value of our
common stock on the date of the grant. In determining the number
of shares subject to an equity incentive award, the Compensation
Committee takes into account the executive officer’s
position and level of responsibility, the executive
officer’s past performance and potential future
contribution, the executive officer’s existing stock and
unvested restricted stock holdings, the competitiveness of the
executive officer’s total compensation, including equity
awards, and with reference to equity award levels of executives
with comparable duties in comparable companies.
Typically, the Compensation Committee grants a lesser grant
value of restricted stock than stock options because when
restricted stock vests, it provides immediate value to the
recipient, with less risk than stock options. In deciding
whether to grant stock options or restricted stock, the
Compensation Committee will review market factors such as our
stock price, the different benefits offered by each type of
award, past equity grants and the desired balance between
performance and retention. Typically, the Compensation Committee
grants equity incentive awards once annually; however, it may
make additional grants based upon a number of factors, including
company results and individual achievements.
Benefits
and Perquisites
The named executive officers are eligible for benefits that are
generally available to all Spectrum employees and are subject to
favorable tax treatment by the IRS under the current tax code.
Such benefits include health insurance, life insurance,
vacation, and 401(k) retirement savings. Named executive
officers and employees are required to contribute to offset a
portion of the cost of certain plans.
We maintain a 401(k) Plan and an Employee Stock Purchase Plan,
each available to all employees, to encourage employees to save
for retirement and to provide incentives for our employees to
exert maximum effort for our success. The 401(k) Plan provides
matching employee contributions in shares of our common stock
and the Employee Stock Purchase Plan provides employees with the
opportunity to purchase common stock through accumulated payroll
deductions, each in order to, among other things, align
employees’ interests with our stockholders.
Fiscal
2010 Compensation
The company’s achievement of the strategic, financial and
share value objectives in 2010 shaped the Compensation
Committee’s decisions with respect to 2010 base salary
changes, annual bonuses and equity incentive awards.
Base
Salaries
We provide base salaries to compensate executives for the
services rendered during the fiscal year. In setting base
salaries for the CEO and other named executive officers, the
Compensation Committee considers various factors including
competitive market data, the experience, skills and impact of
the individual, the compensation of
81
the individual relative to other members of the executive team;
and performance of the executive and the company in the prior
year.
Based on these factors, the Compensation Committee determined
that the base salaries for selected named executive officers in
2009 and 2010 were as follows:
| |
|
|
|
|
|
|
|
|
|
Executive
|
|
2009
|
|
2010
|
|
|
|
Rajesh Shrotriya
|
|
$
|
600,000
|
|
|
$
|
650,000
|
|
|
George Tidmarsh
|
|
|
—
|
|
|
$
|
400,000
|
|
|
Jim Shields
|
|
|
—
|
|
|
$
|
240,000
|
|
|
Brett Scott
|
|
|
—
|
|
|
$
|
225,000
|
|
|
Shyam Kumaria
|
|
$
|
275,000
|
|
|
$
|
290,000
|
|
Annual
Bonus
The Board of Directors set stretch goals for the Company at the
start of 2010, which constitute the performance objectives for
the Chief Executive Officer and the Company’s other
executive officers. In January 2011, the Compensation Committee
determined that the Company’s executive officers met or
exceeded each of the above-listed business objectives, as
follows:
| |
|
|
|
|
|
|
|
Goals For 2010
|
|
|
|
Results For 2010
|
|
|
|
Marketed Products
|
|
|
|
|
|
•
|
|
ZEVALIN: Grow sales 25% over 2009 sales of $15.7M; Submit data
to FDA for bioscan removal; Arrange alternate back-up supplier
for Yttrium; Reduce cost of goods; Secure reimbursement in HOPPs
and community setting.
|
|
•
|
|
All of the Zevalin goals have been accomplished, including Sales
of $29 million representing an 84% increase, compared to a
target growth of 25%, for a drug that had experienced a
declining sales trend prior to our acquisition of rights to the
drug in 2009. Zevalin cost of goods was reduced significantly in
2010.
|
|
•
|
|
FUSILEV: Maximize FUSILEV sales, with a goal of $4 million for
2010; while advancing an sNDA for its use in the treatment of
colorectal cancer.
|
|
•
|
|
All of the Fusilev goals have been accomplished: Sales of $32
million represent an increase of over 150% compared 2009 sales;
and the FDA has established a PDUFA date of April 29, 2011 for a
decision on our sNDA for colorectal cancer.
|
|
|
|
|
|
•
|
|
Achieved profitability in the fourth quarter of 2010
|
|
|
|
|
|
•
|
|
Fourth quarter 2010 product revenues in excess of $30 million,
up 500% as compared to approximately $5 million in the fourth
quarter of 2009
|
|
|
|
|
|
•
|
|
Fiscal year 2010 product revenues in excess of $60 million, up
114% as compared to approximately $28 million in fiscal year
2009.
|
|
Development pipeline
|
|
|
|
|
|
|
|
Acquire a late stage (in Phase II or III/pivotal trial) or
marketed drug
|
|
•
|
|
We in-licensed rights to belinostat, a HDAC inhibitor from
TopoTarget Inc. and have accelerated the clinical trial timeline
in a difficult to enroll study, with the objective of filing an
NDA in 2011, or early 2012.
|
|
|
|
Continue active monitoring of apaziquone clinical trials
|
|
•
|
|
The apaziquone clinical trials are on track for NDA submission
in 2012.
|
|
|
|
Active management of alliances with drug development partners
|
|
•
|
|
We have continued to maintain excellent relations with our
alliance partners.
|
82
| |
|
|
|
|
|
|
|
Goals For 2010
|
|
|
|
Results For 2010
|
|
|
|
Financial Resources
|
|
|
|
|
|
•
|
|
Maintain tight control over the Company’s expenses.
|
|
•
|
|
We continued to exercise tight control over cash used in
operations. In spite of the approximately $30 million paid for
the licensing of belinostat, we closed the year with $104
million cash,cash equivalents and investments, compared to $125
million at the start of the year; a net decrease of $21 million.
|
|
•
|
|
Continue to manage expenses and capital such that we close 2010
with at least $50M in the bank (if we do acquire an asset for
about $30 million cash)
|
|
•
|
|
During the fourth quarter cash, cash equivalents and investments
increased from $92 million as of September 30, 2010 to
approximately $104 million as of December 31, 2010, reflecting a
net increase of approximately $12 million.
|
|
Human Resources
|
|
|
|
|
|
•
|
|
Enhance leadership capabilities of personnel.
|
|
•
|
|
Maintained the companywide training and employee development to
enhance corporate
|
|
•
|
|
Hire appropriate personnel to support ZEVALIN sales growth; and
personnel to support development of belinostat and other
pipeline drugs.
|
|
|
|
communication and teamwork; and also one-on-one training for key individuals in leadership roles to enable desired levels of performance to advance corporate objectives
During 2010 we reviewed our staffing and strengthened the leadership in all functions — Commercial, Development, Legal and compliance, Finance and Business Development; at the same time reduced headcount by approximately 20 personnel compared to the start of the year.
|
|
Investor Relations
|
|
|
|
|
|
•
|
|
Continue to present at strategic healthcare and partnership
conferences.
|
|
•
|
|
The Company presented at several strategic healthcare and
partnership conferences and continued to build on its investor
base.
|
|
•
|
|
Continue to build a base of strategic investors while
maintaining relationships with NASDAQ and current investors.
|
|
•
|
|
During 2010, the Company experienced an increase in its market
capitalization of approximately 40%.
|
Based upon results versus established goals, for fiscal 2010,
the Compensation Committee determined that the individual
performance of Dr. Shrotriya, the Company’s Chairman,
President and Chief Executive Officer, exceeded expectations.
In making its determination regarding Dr. Shrotriya’s
total compensation in 2010, the Compensation Committee took into
account the fact that Dr. Shrotriya had the principal
authority and executive decision-making ability required to
execute, or to oversee the execution of, the Company’s
goals and objectives and to lead the Company’s
transformation from a small drug development company to its
current status as a biotechnology company with fully integrated
commercial and drug development operations by, among other
things, building the Company’s research and development,
sales and marketing and managerial infrastructure, attracting
and retaining key management talent. In addition, the
Compensation Committee acknowledged Dr. Shrotriya’s
success in continuing to build a base of strategic investors,
who have expressed interest and desire to invest additional
capital to spur the Company’s future growth. For these
accomplishments, the Compensation Committee reasoned, and the
Board of Directors endorsed, that the level of
Dr. Shrotriya’s achievements was far in excess of the
achievements of principal executive officers of peer companies
and thus merited a superior bonus commensurate with preceding
years. Accordingly, the Committee awarded Dr. Shrotriya a
cash bonus of $950,000.
83
In making decisions related to the annual bonus for the other
named executive officers, the Compensation Committee, based
primarily on Dr. Shrotriya’s recommendations,
concluded as follows:
Shyam Kumaria — Mr. Kumaria’s 2010 goals
were aligned with Spectrum’s overall objectives, with an
emphasis on supporting attainment of the financial objectives.
Based upon results achieved, Dr. Shrotriya determined that
Mr. Kumaria’s individual performance exceeded
expectations as follows: financial management and reporting
achievements, with a focus on cost containment; completing
value-added corporate projects such as successfully obtaining
approximately $1 million in Government grant funds, gross
margin improvements on Zevalin, and optimizing tax planning.
Based on Dr. Shrotriya’s recommendations, the
Compensation Committee determined that Mr. Kumaria’s
contributions to the achievement of the Company’s goals
merited a cash bonus of $100,000 based on 2010 performance.
George Tidmarsh — Mr. Tidmarsh joined us in July
2010 and, in the short period that he has been with the Company,
has made contributions to achievement of Spectrum’s
strategic milestones. The Compensation Committee determined that
Mr. Tidmarsh’s contributions to the achievement of the
Company’s goals merited a cash bonus of $50,000 based on
2010 performance.
Jim Shields — Mr. Shields joined u s in May
2010. As Chief Commercial officer, he participates in
Spectrum’s established sales incentive plan; and earned a
commission of $51,975 from such participation. In addition, the
Compensation Committee determined that Mr. Shields’s
contributions to the achievement of the Company’s goals
merited a cash bonus of $35,000 based on 2010 performance.
Brett Scott — Mr. Scott joined us in October
2010. In view of the limited service period in 2010,
the Compensation Committee determined that Mr. Scott would
not receive a cash bonus for 2010.
Equity
Incentive Opportunities
Spectrum reviews the mix of the equity-based awards, which
primarily consist of stock options and restricted stock awards,
each year to ensure that the optimal balance on performance and
retention is met for each executive officer. The Compensation
Committee considers the performance of Spectrum and each
executive in determining the appropriate level of equity
incentive opportunity granted to each executive officer in 2010,
which is summarized in the Grants of Plan-based Awards table on
page 87.
In addition, based on the factors previously discussed, the
Compensation Committee approved stock option and restricted
awards to each named executive officer on January 3, 2011
as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock Options
|
|
Restricted Stock
|
|
Executive
|
|
# Granted
|
|
Exercise price
|
|
# Granted
|
|
Fair Market Value
|
|
|
|
Rajesh Shrotriya
|
|
|
1,000,000
|
|
|
$
|
6.87
|
|
|
|
250,000
|
|
|
$
|
6.87
|
|
|
George Tidmarsh
|
|
|
50,000
|
|
|
$
|
6.87
|
|
|
|
20,000
|
|
|
$
|
6.87
|
|
|
Jim Shields
|
|
|
50,000
|
|
|
$
|
6.87
|
|
|
|
20,000
|
|
|
$
|
6.87
|
|
|
Brett Scott(1)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Shyam Kumaria
|
|
|
50,000
|
|
|
$
|
6.87
|
|
|
|
20,000
|
|
|
$
|
6.87
|
|
|
|
|
|
(1) |
|
Mr. Scott was not awarded stock options or restricted Stock
due to his limited tenure with the Company. |
Payments
upon Termination of Employment or
Change-in-Control
Dr. Shrotriya has an employment agreement that provides for
certain payments and benefits upon separation from the Company.
The payments and benefits as provided within the agreement are
designed to achieve the following objectives: protect earned
benefits in the case that Dr. Shrotriya is terminated
without cause or as may result from a change in control of
Spectrum, and to act in the best interests of the stockholders
at all times. The level of benefits provided under
Dr. Shrotriya’s agreement reflects the Compensation
Committee’s assessment of market conditions to provide a
competitive level of compensation if he is impacted by a
termination without cause or a change of control of Spectrum as
well as a recognition of the results achieved by
Dr. Shrotriya over his time of service to us.
84
Dr. Shrotriya would receive certain severance benefits if
he is terminated by Spectrum at the expiration of the term of
the agreement (if not renewed), if his employment is terminated
by us without cause, if his employment is terminated as a result
of a change in control or his position is adversely affected due
to a change in control and he resigns, or if he voluntarily
terminates from Spectrum. The benefits are described in greater
detail in this annual report under the heading “Executive
Employment Agreements, Termination of Employment and
Change-in-Control
Arrangements.”
We currently do not provide any severance benefits to the other
named executive officers, other than would be provided under
general policies that apply to all of our other employees.
Impact
of Accounting and Tax Considerations on
Compensation
Stock —
based compensation
The fair value of stock-based compensation, which includes
equity incentives such as stock options and restricted stock, is
measured in accordance with authoritative guidance. We measure
compensation cost for all stock-based awards at fair value on
the date of grant and recognize compensation expense over the
service period over which the awards are expected to vest.
162(m)
Section 162(m) of the Internal Revenue Code limits the
Company’s ability to deduct compensation paid in any given
year to a named executive officer in excess of
$1.0 million. Performance-based compensation plans are not
subject to this restriction. In the event the proposed
compensation for any of the Company’s named executive
officers is expected to exceed the $1.0 million limitation,
the Compensation Committee will, in making a decision, balance
the benefits of tax deductibility with its responsibility to
hire, retain and motivate executive officers with competitive
compensation programs. In light of our significant net operating
losses, Section 162(m) is not considered a significant
factor in executive officer compensation decisions at this time.
280G and
4999
Sections 280G and 4999 of the Internal Revenue Codes relate
to a 20% excise tax that may be levied on a payment made to an
executive as a result of a
change-in-control
if the payment exceeds 2.99 times the executive’s base
earnings (as defined by the code section). Our agreement with
Dr. Shrotriya provides that we will compensate him for any
excise taxes as might arise upon a
change-in-control
of Spectrum. The decision to provide Dr. Shrotriya with
this tax
gross-up
reflects the relatively low cash compensation Dr, Shrotriya
received in prior years (which increases his potential 280G tax
liability should a change in control occur), and to encourage
Dr. Shrotriya to hold his stock options awarded in prior
years for an extended period (which he has done). We do not
currently provide similar tax protection to its other named
executive officers, should a
change-in-control
occur.
Risk
Assessment of Compensation Policies and Practices
Our Compensation Committee has considered the concept of risk as
it relates to the Company’s compensation program, and based
on such consideration, the Compensation Committee does not
believe our compensation program encourages excessive or
inappropriate risk-taking for the following reasons:
We structure our pay to consist of both fixed and variable
compensation. The fixed (or salary) portion of compensation is
designed, in part, to provide a steady income regardless of
stock price performance so that executives do not feel pressured
to focus exclusively on stock price performance to the detriment
of other important business metrics. The variable (cash bonus
and equity) portions of compensation are designed to reward both
short and long-term corporate performance. For short-term
performance, a cash bonus is awarded based on the achievement of
the performance criteria established for the Company
and/or each
executive officer based on performance criteria that the
Compensation Committee believes to be challenging, yet does not
encourage risk-taking, such as the achievement of product
development and regulatory milestones, the acquisition of new
products and the continuation of strategic corporate
collaborations. For long-term performance, our stock option
awards generally vest over four years and are only valuable if
the Company’s stock price increases over time, which
further
85
aligns the interests of our executives with those of our
stockholders. The Compensation Committee believes that these
variable elements of overall compensation are a sufficient
percentage of overall compensation to motivate executive
officers to produce superior short and long-term corporate
results, while the fixed element is also sufficiently high such
that the executive officers are not encouraged to take
unnecessary or excessive risks. In addition, our internal
controls and ethics code also help mitigate risks associated
with our compensation program.
Based on the foregoing, the Compensation Committee concluded
that risks arising from the Company’s compensation program
are not reasonably likely to have a material adverse effect on
the Company and do not encourage or incentivize excessive or
inappropriate risk-taking by the Company’s employees.
Role of
Executives in Setting Compensation
The Company’s CEO makes recommendations on changes to
compensation to the Compensation Committee for all executive
officers except himself. Executives are not involved in
decisions regarding their own compensation. The Compensation
Committee has overall responsibility for the compensation
programs for the CEO and other named executive officers.
Role of
the Compensation Consultant
In 2010, management engaged Grant Thornton LLP to develop our
compensation peer group, perform total compensation benchmarking
analysis and make recommendations on potential annual and equity
incentive opportunity levels for each named executive officer.
Grant Thornton also assisted the Company in complying with the
SEC proxy disclosure requirements as it relates to the
preparation of the CD&A and related tabular calculations.
Grant Thornton is independent and all work performed by Grant
Thornton is subject to review and approval of the Compensation
Committee.
Compensation
of Directors
The following table shows fiscal 2010 compensation for our
non-employee directors.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fees Earned or
|
|
Option
|
|
|
|
|
|
Paid in Cash
|
|
Awards
|
|
|
|
Name
|
|
(1)($)
|
|
(2)($)
|
|
Total ($)
|
|
|
|
Krishan K. Arora(5)
|
|
|
25,000
|
|
|
|
69,000
|
|
|
|
94,000
|
|
|
Stuart M. Krassner(3)
|
|
|
85,000
|
|
|
|
157,200
|
|
|
|
242,200
|
|
|
Luigi Lenaz(4)(6)
|
|
|
50,000
|
|
|
|
69,000
|
|
|
|
119,000
|
|
|
Anthony E. Maida(3)
|
|
|
85,000
|
|
|
|
157,200
|
|
|
|
242,200
|
|
|
Dilip J. Mehta(3)(7)
|
|
|
60,000
|
|
|
|
69,000
|
|
|
|
129,000
|
|
|
|
|
|
1. |
|
This column reports the dollar amount of cash compensation paid
in 2011 for board and committee service. Effective as of
June 26, 2009, each non-employee director received annual
retainers for the period of service from the date of election
(or reelection) as a director to the subsequent annual
stockholder meeting, each retainer being payable on annually or
semi-annually at the election of the director as follows:
$25,000 director retainer, $25,000 retainer in lieu of
meeting fees of the board and committees of the board, and
$10,000 each to the chairs of the Audit and Compensation
Committees. Our directors are also reimbursed for certain
out-of-pocket
expenses incurred in connection with attendance at board
meetings. Directors who are also our employees receive no
compensation for service as directors. |
| |
|
2. |
|
The amounts reflect the aggregate grant date fair value of the
following option awards made to such non-employee director. On
April 20, 2010, each Board member of record on that date
received a stock option to purchase up to 30,000 shares of
our common stock at $5.05 per share; with 100% of the shares
vesting on the last day of service as a Board member for the
2009-2010
period. On July 1, 2010, upon confirmation of election as a
Board member for the
2010-2011
period, each elected non-employee director received an option to
purchase up to 30,000 shares of our common stock at $3.92
per share; 25% of the shares vested on the date of grant and the
remaining shares vested equally in three annual increments from
the date of grant; and on. For |
86
|
|
|
|
|
|
additional information, refer to note 12 of our financial
statements that are included elsewhere in this annual report. |
| |
|
3. |
|
Includes $30,000 related to Board, Committee and Committee Chair
service from January 1, 2011 until June 30, 2011, or
date of Annual Stockholder Meeting. |
| |
|
4. |
|
Includes $25,000 related to Board and Committee service for the
period from January 1, 2011 until June 30, 2011, or
date of Annual Stockholder Meeting. |
| |
|
5. |
|
Excludes $119,250 compensation as consultant prior to election
as director. Such consulting arrangement terminated effective
July 1, 2010. |
| |
|
6. |
|
Excludes $127,399 compensation as consultant prior to election
as director. While his consulting agreement expired
December 31, 2010. Dr. Lenaz’s consulting
arrangements terminated effective July 1, 2010. |
| |
|
7. |
|
Excludes $3,000 compensation as consultant prior to election as
director. Such consulting arrangement terminated effective
July 1, 2010. |
COMPENSATION
COMMITTEE INTERLOCKS AND INSIDER PARTICIPATION
The Compensation Committee is currently comprised of
Drs. Krassner, Maida and Arora. None of the members of our
Compensation Committee is or has been an officer or employee of
Spectrum. None of our executive officers has served as a
director or member or the compensation committee of any other
entity, any of whose executive officers served on our Board of
Directors or our Compensation Committee.
REPORT OF
THE COMPENSATION COMMITTEE
The following Compensation Committee Report does not
constitute soliciting material and should not be deemed filed or
incorporated by reference into any other company filing under
the Securities Act of 1933 or the Exchange Act, except to the
extent we specifically incorporate it by reference therein.
The Compensation Committee has reviewed and discussed the
CD&A with management. Based on its review and discussions
with management, the Compensation Committee recommended to the
Board of Directors that the CD&A be included in our 2010
Annual Report on
Form 10-K.
Stuart M. Krassner, Sc.D., Psy.D, Chair.
Anthony E. Maida, III, M.A., M.B.A., Ph.D
Krishan K. Arora, Ph.D.
Summary
Compensation Table
The following information sets forth summary information
concerning the compensation we awarded or accrued during 2010,
2009 and 2008 to our Chief Executive Officer, Chief Scientific
Officer, Senior Vice President Of Finance, Chief Commercial
Officer and Acting Chief Financial Officer. In 2008 and 2009, we
had no other named executive officers.
87
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock
|
|
Option
|
|
All Other
|
|
|
|
|
|
|
|
|
|
|
|
Awards
|
|
Awards
|
|
Compensation
|
|
|
|
Name and Principal Position
|
|
Year
|
|
Salary ($)
|
|
Bonus ($)
|
|
($)(1)
|
|
($)(1)
|
|
($)
|
|
Total ($)
|
|
|
|
Rajesh Shrotriya
|
|
|
2010
|
|
|
|
650,000
|
|
|
|
950,000
|
|
|
|
930,000
|
|
|
|
2,530,000
|
|
|
|
454,195
|
(2,4)
|
|
|
5,514,195
|
|
|
Chairman, Chief Executive
|
|
|
2009
|
|
|
|
600,000
|
|
|
|
1,000,000
|
|
|
|
—
|
|
|
|
2,159,000
|
|
|
|
33,956
|
(2)
|
|
|
3,792,956
|
|
|
Officer and President
|
|
|
2008
|
|
|
|
600,000
|
|
|
|
1,000,000
|
|
|
|
388,000
|
|
|
|
839,500
|
|
|
|
34,694
|
(2)
|
|
|
2,862,194
|
|
|
George Tidmarsh
|
|
|
2010
|
|
|
|
181,258
|
|
|
|
50,000
|
|
|
|
60,300
|
|
|
|
472,000
|
|
|
|
6,276
|
|
|
|
769,835
|
|
|
Chief Scientific Officer
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shyam Kumaria
|
|
|
2010
|
|
|
|
290,000
|
|
|
|
100,000
|
|
|
|
46,500
|
|
|
|
394,000
|
|
|
|
97,524
|
(3,4)
|
|
|
928,024
|
|
|
Senior Vice President,
|
|
|
2009
|
|
|
|
275,000
|
|
|
|
60,000
|
|
|
|
—
|
|
|
|
429,900
|
|
|
|
21,449
|
(3)
|
|
|
786,349
|
|
|
Finance
|
|
|
2008
|
|
|
|
250,000
|
|
|
|
50,000
|
|
|
|
65,850
|
|
|
|
113,000
|
|
|
|
22,113
|
(3)
|
|
|
500,963
|
|
|
James Shields
|
|
|
2010
|
|
|
|
138,433
|
|
|
|
86,975
|
|
|
|
—
|
|
|
|
288,234
|
|
|
|
17,566
|
|
|
|
531,208
|
|
|
Chief Commercial Officer
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Brett Scott
|
|
|
2010
|
|
|
|
49,959
|
|
|
|
—
|
|
|
|
—
|
|
|
|
247,000
|
|
|
|
2,374
|
|
|
|
299,333
|
|
|
Acting Chief Financial Officer
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
The amounts reflect the aggregate grant date fair value of
awards made to such named executive officers. For additional
information, refer to note 12 of our financial statements
that are included elsewhere in this annual report. |
| |
|
(2) |
|
Amounts include: (a) annual 401(k) matching contribution
made by us in shares of our common stock and healthcare
premiums, which is a benefit offered to all our employees,
(b) premiums paid on life insurance policies covering his
life and having as beneficiary his estate or other
beneficiaries, (c) amounts related to the personal use of a
leased company car, gas and repairs, and (d) legal fees
related to negotiations of his employment agreement. No
individual component of this amount exceeds $25,000. |
| |
|
(3) |
|
Amounts include annual 401(k) matching contribution made by us
in shares of our common stock, and premiums paid on healthcare
and life insurance policies,which are benefits that are offered
to all of our employees. |
Grants
of Plan Based Awards in 2010
The following table provides information about equity awards
granted to the named executive officers in 2010. There can be no
assurance that the grant date fair value of stock and option
awards will be realized by the named executive officers.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
All Other
|
|
All Other
|
|
|
|
|
|
|
|
|
|
Option
|
|
Stock
|
|
|
|
|
|
|
|
|
|
Awards:
|
|
Awards:
|
|
|
|
Grant Date
|
|
|
|
|
|
Number of
|
|
Number of
|
|
Exercise or
|
|
Fair Value
|
|
|
|
|
|
Securities
|
|
Shares of
|
|
Base Price
|
|
of Stock
|
|
|
|
|
|
Underlying
|
|
Stock or
|
|
of Option
|
|
and Option
|
|
Name
|
|
Grant Date
|
|
Options (#)
|
|
Units (#)
|
|
Awards ($)
|
|
Awards ($)(1)
|
|
|
|
Rajesh Shrotriya
|
|
|
1/8/2010
|
|
|
|
500,000
|
|
|
|
|
|
|
|
4.65
|
|
|
$
|
1,380,000
|
|
|
|
|
|
7/1/2010
|
|
|
|
500,000
|
|
|
|
|
|
|
|
3.92
|
|
|
$
|
1,150,000
|
|
|
|
|
|
1/8/2010
|
|
|
|
|
|
|
|
200,000
|
|
|
|
|
|
|
$
|
930,000
|
|
|
George Tidmarsh
|
|
|
7/15/2010
|
|
|
|
200,000
|
|
|
|
|
|
|
|
4.02
|
|
|
$
|
472,000
|
|
|
|
|
|
7/15/2010
|
|
|
|
|
|
|
|
15,000
|
|
|
|
|
|
|
$
|
60,300
|
|
|
Shyam Kumaria
|
|
|
1/8/2010
|
|
|
|
50,000
|
|
|
|
|
|
|
|
4.65
|
|
|
$
|
138,000
|
|
|
|
|
|
2/5/2010
|
|
|
|
10,000
|
|
|
|
|
|
|
|
4.39
|
|
|
$
|
26,000
|
|
|
|
|
|
7/1/2010
|
|
|
|
100,000
|
|
|
|
|
|
|
|
3.92
|
|
|
$
|
230,000
|
|
|
|
|
|
1/8/2010
|
|
|
|
|
|
|
|
10,000
|
|
|
|
|
|
|
$
|
46,500
|
|
|
James Shields
|
|
|
5/10/2010
|
|
|
|
100,000
|
|
|
|
|
|
|
|
4.65
|
|
|
$
|
275,000
|
|
|
|
|
|
6/30/2010
|
|
|
|
1,080
|
|
|
|
|
|
|
|
3.9
|
|
|
$
|
2,473
|
|
|
|
|
|
7/1/2010
|
|
|
|
1,000
|
|
|
|
|
|
|
|
3.92
|
|
|
$
|
2,300
|
|
|
|
|
|
9/30/2010
|
|
|
|
3,570
|
|
|
|
|
|
|
|
4.09
|
|
|
$
|
8,461
|
|
|
Brett L. Scott
|
|
|
10/11/2010
|
|
|
|
100,000
|
|
|
|
|
|
|
|
4.28
|
|
|
$
|
247,000
|
|
88
The exercise price of all the option awards listed above is
equal to the fair market value on the dates of grant in
accordance with the terms of our equity incentive plans. All the
awards listed above vest annually in equal 25% increments with
25% immediately vested on the date of grant.
|
|
|
|
(1) |
|
The amounts reflect the grant date fair value dollar amount for
financial statement reporting purposes. Fair value assumptions
can be found in Note 11 to our financial statements. |
Outstanding
Equity Awards at Fiscal Year-End 2010
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Option Awards
|
|
Stock Awards
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity
|
|
|
|
Number of
|
|
Number of
|
|
|
|
|
|
Equity
|
|
Incentive
|
|
|
|
Securities
|
|
Securities
|
|
|
|
|
|
Incentive
|
|
Plan Awards:
|
|
|
|
Underlying
|
|
Underlying
|
|
|
|
|
|
Plan Awards:
|
|
Market Value of
|
|
|
|
Unexercised
|
|
Unexercised
|
|
Option
|
|
Option
|
|
Number of
|
|
Shares of
|
|
|
|
Options (#)
|
|
Options (#)
|
|
Exercise
|
|
Expiration
|
|
Shares of
|
|
Stock Not Vested
|
|
Name
|
|
Exercisable
|
|
Unexercisable
|
|
Price
|
|
Date
|
|
Stock Not Vested (#)
|
|
($)(7)
|
|
|
|
Rajesh Shrotriya
|
|
|
12,000
|
|
|
|
—
|
|
|
|
4.75
|
|
|
|
06/17/12
|
|
|
|
|
|
|
|
|
|
|
|
|
|
75,000
|
|
|
|
—
|
|
|
|
1.06
|
|
|
|
09/25/12
|
|
|
|
|
|
|
|
|
|
|
|
|
|
225,000
|
|
|
|
—
|
|
|
|
1.99
|
|
|
|
09/05/13
|
|
|
|
|
|
|
|
|
|
|
|
|
|
215,000
|
|
|
|
—
|
|
|
|
4.90
|
|
|
|
09/12/13
|
|
|
|
|
|
|
|
|
|
|
|
|
|
200,000
|
|
|
|
—
|
|
|
|
4.23
|
|
|
|
01/01/16
|
|
|
|
|
|
|
|
|
|
|
|
|
|
150,000
|
|
|
|
—
|
|
|
|
5.08
|
|
|
|
09/26/16
|
|
|
|
|
|
|
|
|
|
|
|
|
|
350,000
|
|
|
|
—
|
|
|
|
5.53
|
|
|
|
01/01/17
|
|
|
|
|
|
|
|
|
|
|
|
|
|
100,000
|
|
|
|
—
|
|
|
|
3.15
|
|
|
|
12/06/17
|
|
|
|
|
|
|
|
|
|
|
|
|
|
500,000
|
|
|
|
—
|
|
|
|
2.55
|
|
|
|
03/25/18
|
|
|
|
|
|
|
|
|
|
|
|
|
|
112,500
|
|
|
|
37,500
|
(1)
|
|
|
1.43
|
|
|
|
12/06/18
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
50,000
|
(4)
|
|
|
343,500
|
|
|
|
|
|
175,000
|
|
|
|
175,000
|
(1)
|
|
|
1.47
|
|
|
|
01/16/19
|
|
|
|
|
|
|
|
|
|
|
|
|
|
250,000
|
|
|
|
250,000
|
(1)
|
|
|
6.09
|
|
|
|
06/26/19
|
|
|
|
|
|
|
|
|
|
|
|
|
|
125,000
|
|
|
|
375,000
|
(1)
|
|
|
4.65
|
|
|
|
01/08/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
125,000
|
|
|
|
375,000
|
(1)
|
|
|
3.92
|
|
|
|
07/01/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
150,000
|
(5)
|
|
|
1,030,500
|
|
|
George Tidmarsh
|
|
|
—
|
|
|
|
200,000
|
(3)
|
|
|
4.02
|
|
|
|
07/15/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
15,000
|
(6)
|
|
|
103,050
|
|
|
Shyam Kumaria
|
|
|
40,000
|
|
|
|
—
|
|
|
|
4.26
|
|
|
|
12/06/15
|
|
|
|
|
|
|
|
|
|
|
|
|
|
20,000
|
|
|
|
—
|
|
|
|
3.15
|
|
|
|
12/06/17
|
|
|
|
|
|
|
|
|
|
|
|
|
|
50,000
|
|
|
|
—
|
|
|
|
2.55
|
|
|
|
03/25/18
|
|
|
|
|
|
|
|
|
|
|
|
|
|
37,500
|
|
|
|
12,500
|
(1)
|
|
|
1.43
|
|
|
|
12/06/18
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7,500
|
(4)
|
|
|
51,525
|
|
|
|
|
|
25,000
|
|
|
|
25,000
|
(1)
|
|
|
1.47
|
|
|
|
01/16/19
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,500
|
|
|
|
2,500
|
(1)
|
|
|
4.89
|
|
|
|
05/21/19
|
|
|
|
|
|
|
|
|
|
|
|
|
|
50,000
|
|
|
|
50,000
|
(1)
|
|
|
6.09
|
|
|
|
06/26/19
|
|
|
|
|
|
|
|
|
|
|
|
|
|
12,500
|
|
|
|
37,500
|
(1)
|
|
|
4.65
|
|
|
|
01/08/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10,000
|
|
|
|
—
|
(2)
|
|
|
4.39
|
|
|
|
02/05/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
25,000
|
|
|
|
75,000
|
(1)
|
|
|
3.92
|
|
|
|
07/01/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7,500
|
(5)
|
|
|
51,525
|
|
|
James Shields
|
|
|
—
|
|
|
|
100,000
|
(3)
|
|
|
4.65
|
|
|
|
05/10/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
250
|
|
|
|
750
|
(1)
|
|
|
3.92
|
|
|
|
07/01/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1,080
|
|
|
|
—
|
(2)
|
|
|
3.90
|
|
|
|
06/30/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,570
|
|
|
|
—
|
(2)
|
|
|
4.09
|
|
|
|
09/30/20
|
|
|
|
|
|
|
|
|
|
|
Brett Scott
|
|
|
—
|
|
|
|
100,000
|
(3)
|
|
|
4.28
|
|
|
|
10/11/20
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Option shares vest annually in equal 25% increments, with 25%
immediately vested on the grant date. |
| |
|
(2) |
|
Option shares vest immediately. |
| |
|
(3) |
|
Option shares vest 25% on anniversary date, and in 36 equal
monthly increments thereafter. |
89
|
|
|
|
(4) |
|
Shares granted on December 6, 2008 with 25% vesting on the
grant date, and continuing to vest in equal 25% increments every
December 6th thereafter. |
| |
|
(5) |
|
Shares granted on January 8, 2010 with 25% vesting on the
grant date, and continuing to vest in equal 25% increments every
January 8th thereafter. |
| |
|
(6) |
|
Shares granted on July 15, 2010, and vesting in equal 25%
increments every July 15th thereafter. |
| |
|
(7) |
|
Calculation based on the closing price of the common stock on
December 31, 2010 of $6.87 per share. |
Stock
Vested Table in Fiscal Year 2010
The following table provides information regarding the number of
shares acquired upon exercise
and/or
vesting in 2010 and the value realized by the named executive
officers.
| |
|
|
|
|
|
|
|
|
|
|
|
Stock Awards
|
|
|
|
Number of
|
|
|
|
|
|
Shares
|
|
Value realized
|
|
|
|
acquired on
|
|
on Vesting
|
|
Name
|
|
Vesting (#)
|
|
($)(1)
|
|
|
|
Rajesh Shrotriya
|
|
|
125,000
|
|
|
|
602,250
|
|
|
Shyam Kumaria
|
|
|
15,000
|
|
|
|
71,975
|
|
|
|
|
|
(1) |
|
The value realized on vesting in the above table was calculated
based on the price of the common stock on the vesting date. |
Executive
Employment Agreements, Termination of Employment and
Change-in-Control
Arrangements
On June 20, 2008, we entered into an employment agreement
with Dr. Shrotriya, our President and Chief Executive
Officer, which became effective as of January 2, 2008 and
replaced his previous employment agreement. The employment
agreement expires on January 2, 2012, unless terminated
earlier, and automatically renews for a one-year term unless
either party gives written notice of such party’s intent
not to renew the agreement at least 90 days prior to the
commencement of the next year. The employment agreement requires
Dr. Shrotriya to devote his full working time and effort to
our business and affairs of us during the term of the agreement.
The employment agreement provides for a minimum annual base
salary with annual increases, periodic bonuses and option grants
as determined by the Compensation Committee.
Compensation
and Benefits
Dr. Shrotriya shall receive an annual base salary of
$600,000, as adjusted annually based upon the performance of
Dr. Shrotriya and Spectrum, as determined by the
Compensation Committee.
Dr. Shrotriya shall also be paid an annual performance
bonus in cash
and/or
equity based awards, no later than January 31 of the year
following, in an amount to be determined by the Compensation
Committee according to Dr. Shrotriya’s achievement of
annual performance objectives mutually agreed upon by
Dr. Shrotriya and our Board of Directors.
Under the agreement, Dr. Shrotriya is entitled to receive
additional employment benefits, including the right to
participate in any incentive plans and to receive life, medical,
dental, paid vacation, estate planning services, a leased
vehicle and reimbursements for automobile related expenses, and
other benefits.
Termination
Dr. Shrotriya’s employment may be terminated due to
non-renewal of the agreement by us, by mutual agreement of the
parties, by us for cause (as that term is defined in the
agreement) or without cause, on grounds of disability or death
of Dr. Shrotriya, by Dr. Shrotriya for no reason or
for good reason (as those terms are defined in the agreement),
or by Dr. Shrotriya’s non-renewal of the agreement.
If (i) the agreement is not renewed by us,
(ii) Dr. Shrotriya is terminated without cause, or
(iii) Dr. Shrotriya resigns for good reason, then
Dr. Shrotriya’s guaranteed severance payments include
the right to receive (a) a lump
90
sum payment equivalent to the aggregate of two years’ cash
compensation; (b) company-paid continued coverage for
Dr. Shrotriya and his eligible dependents under our
existing health and benefit plans for two years; and
(c) immediate vesting of all options, restricted stock and
other equity based awards granted to Dr. Shrotriya.
Dr. Shrotriya shall have three years to exercise all vested
equity based awards. Since options issued to Dr. Shrotriya
pursuant to our 1997 Stock Incentive Plan can only be exercised
for ninety days after termination, a replacement option shall be
granted to Dr. Shrotriya at termination to allow for three
years’ of exercisability.
In the event Dr. Shrotriya voluntarily resigns for good
reason, or is terminated by us without cause, we will pay or
reimburse Dr. Shrotriya for reasonable relocation expenses
up to a certain amount.
If Dr. Shrotriya’s employment is terminated without
cause prior to the end of a calendar year, then our Board of
Directors shall determine the amount of any bonus that would
have been paid to Dr. Shrotriya had his employment
continued through the end of the calendar year and we shall pay
Dr. Shrotriya the pro rata amount of the bonus.
If the agreement is terminated due to death or disability of
Dr. Shrotriya, a lump sum equal to three months of base
salary, at the time of his termination, shall be paid to
Dr. Shrotriya, his legal representative or estate, as
applicable. All equity based awards, such as options and
restricted stock, shall immediately vest and shall remain
exercisable in accordance with the terms of the respective
equity plan and individual agreement(s) governing such options
and as otherwise set forth in the agreement.
If Dr. Shrotriya voluntarily resigns his employment for no
reason, any stock options or other equity based awards (except
for restricted stock) shall immediately become fully vested upon
the effective date of Dr. Shrotriya’s resignation, and
he shall have three years to exercise all such vested equity
based awards. Dr. Shrotriya shall receive the same benefits
for any unexpired options issued pursuant to our 1997 Plan as if
he had been terminated without cause by us.
If during the term of the agreement, Dr. Shrotriya resigns
for good reason (as defined in the agreement) other than
pursuant to the circumstances of a change in control and the
board has not cured the condition(s) that constitute good
reason, then Dr. Shrotriya shall receive all of the
severance benefits he would receive if he had been terminated
without cause by us. Upon a change of control of Spectrum, if
(i) Dr. Shrotriya’s employment is terminated
(other than by Dr. Shrotriya) without cause within twelve
months thereafter; or (ii) Dr. Shrotriya is adversely
affected in certain terms outlined in the agreement, and
Dr. Shrotriya, within twelve months after an event
constituting a change of control, elects to resign his
employment with us, then in either case, Dr. Shrotriya
shall be provided with company-paid senior executive
outplacement and shall receive the same severance benefits as he
would receive if he was terminated by us without cause. However,
instead of two years’ cash compensation, Dr. Shrotriya
shall receive three years cash compensation. In addition, upon a
change of control, we shall pay Dr. Shrotriya a one-time
payment of $600,000.
If the agreement is terminated due to mutual agreement,
Dr. Shrotriya’s non-renewal of the agreement, or by us
for cause, he shall not be entitled to any severance.
Other
If any payment or distribution by us to or for the benefit of
Dr. Shrotriya is subject to the excise tax imposed by
Section 4999 of the IRC or any interest or penalties are
incurred by the Dr. Shrotriya with respect to such excise
tax, then Dr. Shrotriya shall be entitled to receive an
additional payment in an amount such that after payment by
Dr. Shrotriya of all taxes (including any interest and
penalties imposed with respect thereto) and excise tax imposed
upon such payment, Dr. Shrotriya retains an amount of the
payment equal to the excise tax imposed upon the payment.
If we determine that any payments to Dr. Shrotriya under
the agreement fail to satisfy the distribution requirement of
Section 409A(a)(2)(A) of the IRC, the payment schedule of
that benefit shall be revised to the extent necessary so that
the benefit is not subject to the provisions of
Section 409A(a)(1) of the IRC. We may attach conditions to
or adjust the amounts so paid to preserve, as closely as
possible, the economic consequences that would have applied in
the absence of this adjustment; provided, however, that no such
condition or adjustment shall result in the payments being
subject to Section 409A(a)(1) of the IRC.
91
Potential
Payments Upon Termination or Following a Change in
Control
The tables below reflect the amount of compensation to each of
our named executive officers in the event of termination of such
executive’s employment or following a change in control of
our Company. The amount of compensation payable to each named
executive officer upon voluntary termination without cause,
retirement, involuntary termination without cause, involuntary
termination for cause or termination following a change of
control, in the event of disability or death of the executive,
and following a change in control of our Company is shown below.
Where applicable, the amounts shown assume that termination was
effective as of December 31, 2010 and use the closing price
of our common stock as of December 31, 2010 ($6.87), and
are estimates of the amounts which would be paid out to the
executives upon their termination. In the case of payments upon
termination, the actual amounts to be paid out can only be
determined at the time of such executive’s separation from
our Company.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Voluntary
|
|
|
|
|
|
|
|
|
|
|
|
Involuntary
|
|
|
|
|
|
Change in
|
|
|
|
|
|
|
|
Termination
|
|
|
|
|
|
|
|
|
|
|
|
Termination
|
|
|
Involuntary
|
|
|
Control
|
|
|
|
|
|
|
|
Without
|
|
|
|
|
|
|
|
|
|
|
|
Without
|
|
|
Termination
|
|
|
(Qualifying
|
|
|
Change in
|
|
|
|
|
Cause ($)
|
|
|
Retirement
|
|
|
Death ($)
|
|
|
Disability ($)
|
|
|
Cause ($)
|
|
|
For Cause
|
|
|
Termination) ($)
|
|
|
Control ($)
|
|
|
|
|
Rajesh Shrotriya
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Severance payments
|
|
|
—
|
|
|
|
—
|
|
|
|
162,500
|
|
|
|
162,500
|
|
|
|
3,200,000
|
|
|
|
—
|
|
|
|
4,800,000
|
|
|
|
—
|
|
|
Cash payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
600,000
|
|
|
Benefit payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
173,690
|
|
|
|
—
|
|
|
|
263,036
|
|
|
|
—
|
|
|
Vesting Acceleration — Options
|
|
|
3,282,750
|
|
|
|
—
|
|
|
|
3,282,750
|
|
|
|
3,282,750
|
|
|
|
3,282,750
|
|
|
|
—
|
|
|
|
3,282,750
|
|
|
|
—
|
|
|
Vesting Acceleration— Restricted stock
|
|
|
—
|
|
|
|
—
|
|
|
|
1,374,000
|
|
|
|
1,374,000
|
|
|
|
1,374,000
|
|
|
|
—
|
|
|
|
1,374,000
|
|
|
|
—
|
|
|
280 G gross up
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
2,251,286
|
|
|
|
—
|
|
|
Total
|
|
|
3,282,750
|
|
|
|
0
|
|
|
|
4,819,250
|
|
|
|
4,819,250
|
|
|
|
8,030,440
|
|
|
|
—
|
|
|
|
11,971,072
|
|
|
|
600,000
|
|
|
George Tidmarsh
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Severance payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Benefits payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Vesting Acceleration — Options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
570,000
|
|
|
|
—
|
|
|
Vesting Acceleration — Restricted stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
103,050
|
|
|
|
—
|
|
|
Total
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
673,050
|
|
|
|
—
|
|
|
Shyam Kumaria
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Severance payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Benefits payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Vesting Acceleration — Options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
551,450
|
|
|
|
—
|
|
|
Vesting Acceleration — Restricted stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
103,050
|
|
|
|
—
|
|
|
Total
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
654,500
|
|
|
|
—
|
|
|
James Shields
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Severance payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Benefits payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Vesting Acceleration — Options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
224,213
|
|
|
|
—
|
|
|
Vesting Acceleration — Restricted stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Total
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
224,213
|
|
|
|
—
|
|
|
Brett Scott
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Severance payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Benefits payments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Vesting Acceleration — Options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
259,000
|
|
|
|
—
|
|
|
Vesting Acceleration — Restricted stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
0
|
|
|
|
—
|
|
|
Total
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
259,000
|
|
|
|
—
|
|
Cash severance payments: Includes base salary,
bonus and auto allowance payable, pursuant to terms of the
employment agreement described above, for two years.
92
Cash payments: Consists of a one-time payment
upon a change in control of our Company pursuant to terms of the
employment agreement described above.
Benefit payments: Includes COBRA insurance
payments for healthcare insurance premiums payable, pursuant to
terms of the employment agreements described above, for two
years unless the lump-sum option is elected. Under the
“Change in Control” scenario, an estimated cost for
outplacement services is also included, pursuant to terms of the
employment agreement described above.
Vesting Acceleration —
Options: Includes the aggregate intrinsic value
of those stock options whose vesting is accelerated upon
termination, either pursuant to terms of the employment
agreements described above, or pursuant to terms of our equity
incentive plans. The calculation of such fair value is based on
the difference between the last closing price of our common
stock, on or before December 31, 2010, and the exercise
price of the options.
Vesting Acceleration — Restricted
stock: Includes the aggregate fair market value
of restricted stock whose vesting is accelerated upon
termination pursuant to terms of our equity incentive plans. The
calculation of such fair value is based on the last closing
price of our common stock, on or before December 31, 2010.
|
|
|
Item 12.
|
Security
Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters.
|
Based on information publicly filed and provided to us by
certain holders, the following table shows the amount of our
Series E Preferred Stock and common stock beneficially
owned on February 25, 2011 (unless otherwise indicated) by
holders of more than 5% of the outstanding shares of any class
of our voting securities, other than with respect to
Dr. Rajesh C. Shrotriya (our Chairman, Chief Executive
Officer and President) whose ownership is included in the second
table below. Beneficial ownership is determined in accordance
with the rules of the SEC and generally includes voting
and/or
investment power with respect to our voting securities, unless
footnoted to the contrary. For purposes of the following tables,
the percentage ownership is based upon 20 shares of our
Series E Preferred Stock, and 52,003,514 shares of our
common stock, outstanding as of February 25, 2011. Unless
otherwise indicated, the business address of each stockholder is
c/o Spectrum
Pharmaceuticals, Inc., 11500 S. Eastern Avenue,
Suite 240, Henderson, Nevada 89052.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common
|
|
|
|
|
|
|
|
|
|
|
|
Shares and
|
|
|
|
Percent of
|
|
|
|
Preferred
|
|
Percent of
|
|
Common
|
|
Percent of
|
|
Shares Eligible
|
|
|
|
Shares
|
|
Preferred
|
|
Equivalents
|
|
Common
|
|
to Vote on
|
Name and Address
|
|
Beneficially
|
|
Stock
|
|
Beneficially
|
|
Shares
|
|
February 25,
|
|
of Beneficial Owner
|
|
Owned(1)
|
|
Outstanding(2)
|
|
Owned(3)
|
|
Outstanding(3)
|
|
2011(4)
|
|
|
|
BlackRock, Inc.(5)
|
|
|
—
|
|
|
|
—
|
|
|
|
3,161,135
|
|
|
|
6.08
|
%
|
|
|
6.08
|
%
|
|
40 East 52nd Street
New York, NY 10022
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Eastern Capital Limited(6)
|
|
|
—
|
|
|
|
—
|
|
|
|
4,737,307
|
|
|
|
9.11
|
%
|
|
|
9.11
|
%
|
|
P.O. Box 31363/P.O. Box 31300 Grand Cayman,
KY1-1206, Cayman Islands
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sands Brothers Venture Capital Funds 1-IV, LLC(7)
|
|
|
20
|
|
|
|
100.00
|
%
|
|
|
40,000
|
|
|
|
|
*
|
|
|
|
*
|
|
90 Park Avenue, 31st Floor New York, NY 10016
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
|
Less than 1% |
| |
|
(1) |
|
The amount relates to the shares of our Series E Preferred
Stock owned by the entity as of February 25, 2011. There
are no outstanding shares of any other series of our preferred
stock. |
| |
|
(2) |
|
Represents the percentage ownership of the total number of our
outstanding shares of Series E Preferred Stock. |
| |
|
(3) |
|
Shares of common stock owned as of February 25, 2011 and
shares of common stock subject to preferred stock and warrants
currently convertible or exercisable, or convertible or
exercisable within 60 days of February 25, 2011, are
deemed beneficially owned and outstanding for computing the
percentage of the person holding such securities, but are not
considered outstanding for computing the percentage of any other
person. |
93
|
|
|
|
(4) |
|
Reflects actual voting percentage. Each holder of Series E
Preferred Stock shall be entitled to the number of votes equal
to the number of shares of common stock into which such shares
of Series E Preferred Stock could be converted on the
record date at the then current conversion value as determined
pursuant to the Certificates of Designations. At the current
conversion value, each share of Series E Preferred Stock is
entitled to 2,000 votes on each matter at the annual meeting.
Consequently, the holders of our Series E Preferred Stock
shall have a total of 40,000 votes on each matter at the annual
meeting. |
| |
|
(5) |
|
The information set forth herein is based solely on information
contained in a Schedule 13G filed with the SEC on
February 8, 2011 by BlackRock, Inc.
(“BlackRock”). According to the Schedule 13G,
BlackRock has sole voting and dispositive power over
3,161,135 shares of our common stock. |
| |
|
(6) |
|
The information set forth herein is based solely on information
contained in a Schedule 13G filed with the SEC on
February 14, 2011 by Eastern Capital Limited. Eastern
Capital Limited is a direct wholly-owned subsidiary of Portfolio
Services Ltd. Kenneth B. Dart is the beneficial owner of all of
the outstanding shares of Portfolio Services Ltd., which in
turns owns all the outstanding shares of Eastern Capital
Limited. As of the date of the Schedule 13G filing, Eastern
Capital Limited and Mr. Dart beneficially own in the
aggregate 4,737,307 shares of our common stock. Eastern
Capital Limited and Mr. Dart have shared voting and
dispositive powers with respect to 4,737,307 shares of our
common stock. |
| |
|
(7) |
|
Based upon the information provided to us by the holder, SB
Venture Capital Management I-IV, LLCs are the Investment
Advisors to Sands Brothers Venture Capital LLC
(“SBV”), Sands Brothers Venture Capital II LLC
(“SBV II”), Sands Brothers Venture Capital LLC III
(“SBV III”) and Sands Brothers Venture Capital IV
LLC (“SBV IV”) (collectively, the “Funds”).
The Funds’ beneficial ownership includes the effect of
converting the 20 shares of Series E Preferred Stock
into 40,000 shares of common stock. Martin S. Sands and
Steven B. Sands are co-Member Managers of SB Venture Capital
Management LLC, SB Venture Capital Management II LLC, SB
Venture Capital Management III LLC, and SB Venture Capital
Management IV LLC, each a New York limited liability
company and each the member-manager of SBV, SBV-II, SBV-III and
SBV-IV, respectively, and are the natural persons exercising
voting and investment control over securities beneficially owned
by the Funds. |
94
The following table sets forth certain information regarding the
beneficial ownership of our common stock as of February 25,
2011 (unless otherwise noted) by: (i) each of our directors
and director nominees, (ii) our named executive officers,
and (iii) all of our directors, director nominees and
executive officers as a group. Shares of common stock owned as
of February 25, 2011 and shares of common stock subject to
options or warrants currently exercisable or exercisable within
60 days of February 25, 2011, are deemed beneficially
owned and outstanding for computing the percentage of the person
holding such securities, but are not considered outstanding for
computing the percentage of any other person. Unless otherwise
noted, each person listed below has sole voting power and sole
investment power with respect to shares shown as owned by him.
Information as to beneficial ownership is based upon statements
furnished to us or filed with the SEC by such persons. Unless
otherwise indicated, the business address of each stockholder is
c/o Spectrum
Pharmaceuticals, Inc., 11500 S. Eastern Avenue,
Suite 240, Henderson, Nevada 89052.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Percent of
|
|
|
|
|
|
|
|
Total
|
|
Shares
|
|
Name of Beneficial Owner
|
|
Options
|
|
Shares(1)
|
|
Owned
|
|
Outstanding
|
|
|
|
Named Executive Officers
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shrotriya, Rajesh(2)
|
|
|
3,077,000
|
|
|
|
1,423,466
|
|
|
|
4,500,466
|
|
|
|
8.2
|
%
|
|
Kumaria, Shyam(3)
|
|
|
310,000
|
|
|
|
221,798
|
|
|
|
531,798
|
|
|
|
1.0
|
%
|
|
Tidmarsh, George(4)
|
|
|
12,500
|
|
|
|
42,818
|
|
|
|
55,318
|
|
|
|
|
*
|
|
Shields, James(5)
|
|
|
17,400
|
|
|
|
26,269
|
|
|
|
43,669
|
|
|
|
|
*
|
|
Scott, Brett
|
|
|
—
|
|
|
|
4,000
|
|
|
|
4,000
|
|
|
|
|
*
|
|
Directors/Directors Nominees
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Krassner, Stuart
|
|
|
152,500
|
|
|
|
10,750
|
|
|
|
163,250
|
|
|
|
|
*
|
|
Maida, Anthony
|
|
|
174,500
|
|
|
|
2,250
|
|
|
|
176,750
|
|
|
|
|
*
|
|
Lenaz, Luigi
|
|
|
52,500
|
|
|
|
21,877
|
|
|
|
74,377
|
|
|
|
|
*
|
|
Arora, Krishan
|
|
|
7,500
|
|
|
|
—
|
|
|
|
7,500
|
|
|
|
|
*
|
|
Mehta, Dilip
|
|
|
7,500
|
|
|
|
32,000
|
|
|
|
39,500
|
|
|
|
|
*
|
|
All Executive Offices and Director/Director Nominees as a group
(10 persons)(6)
|
|
|
3,811,400
|
|
|
|
1,785,228
|
|
|
|
5,596,628
|
|
|
|
10.0
|
%
|
|
|
|
|
* |
|
less than 1% |
| |
|
(1) |
|
The holders of restricted stock are entitled to vote and receive
dividends, if declared, on the shares of common stock covered by
the restricted stock grant. |
| |
|
(2) |
|
The number of shares includes 337,500 unvested restricted shares
of our common stock subject to future vesting within
60 days of February 25, 2011. |
| |
|
(3) |
|
The number of shares includes 27,500 unvested restricted shares
of our common stock subject to future vesting within
60 days of February 25, 2011. |
| |
|
(4) |
|
The number of shares includes 30,000 unvested restricted shares
of our common stock subject to future vesting within
60 days of February 25, 2011. |
| |
|
(5) |
|
The number of shares includes 15,000 unvested restricted shares
of our common stock subject to future vesting within
60 days of February 25, 2011. |
| |
|
(6) |
|
The number of shares includes 410,000 unvested restricted shares
of our common stock held as a group subject to future vesting
within 60 days of February 25, 2011. |
We are not aware of any arrangements that may at a subsequent
date result in a change of control of the Company.
95
Equity
Compensation Plan Information
The following table summarizes all equity compensation plans
including those approved by security holders and those not
approved by security holders, as of December 31, 2010.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of
|
|
|
|
|
|
Number of Securities
|
|
|
|
|
Securities to
|
|
|
|
|
|
Remaining Available
|
|
|
|
|
be Issued
|
|
|
|
|
|
for Future Issuance
|
|
|
|
|
Upon Exercise
|
|
|
Weighted-average
|
|
|
Under Equity
|
|
|
|
|
of Outstanding
|
|
|
Exercise Price of
|
|
|
Compensation Plans
|
|
|
|
|
Options,
|
|
|
Warrants
|
|
|
(excluding securities
|
|
|
Plan Category
|
|
Warrants or Rights
|
|
|
and Rights
|
|
|
reflected in column (a))
|
|
|
|
|
Equity compensation plans approved by security holders(1)
|
|
|
8,756,594
|
|
|
|
4.16
|
|
|
|
10,816,185
|
|
|
Equity compensation plans not approved by security holders(2)
|
|
|
445,000
|
|
|
|
5.04
|
|
|
|
—
|
|
|
Employee Stock Purchase Plan approved by security holders
|
|
|
N/A
|
|
|
|
N/A
|
|
|
|
4,766,002
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
9,201,594
|
|
|
|
4.20
|
|
|
|
15,582,187
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
We have three stock incentive plans: the 1997 Stock Incentive
Plan, or the 1997 Plan, the 2003 Incentive Award Plan, or the
2003 Plan, and the 2009 Plan collectively as the Plans. We are
not granting any more options pursuant to the 1997 Plan or the
2003 Plan. The 2009 Plan authorizes annual increases in the
number of shares of our common stock available for issuance
under the 2009 Plan by an amount equal to the greater of
(i) 2,500,000 and (ii) a number of shares such that
the total number of shares available for issuance equals 30% of
the then number of shares of our common stock issued and
outstanding. Thus, the authorized and available shares may
fluctuate over time. |
| |
|
(2) |
|
The number represents 445,000 shares of common stock
issuable upon exercise of warrants issued to our non-employees
under plans approved by our board of directors that we believe
are not required to be approved by our stockholders pursuant to
the rules of the NASDAQ Stock Market. We issued these warrants
in circumstances that enable us to adequately compensate,
without the payment in cash, for outside consultant services, in
order to conserve our cash for operating activities. The number
of securities remaining available for future issuance under
these types of equity compensation plans is zero; however, our
Board of Directors may approve additional issuances of warrants
under circumstances that it decides are appropriate. |
The above table does not include warrants issued to investors in
connection with financing transactions. As of December 31,
2010, there were outstanding investor warrants to purchase up to
an aggregate of 3,747,312 shares of our common stock, with
a weighted average exercise price of $6.62 per share, which are
scheduled to expire on September 15, 2011, if not exercised.
Further details regarding warrants issued by us are included in
note 11 to our audited consolidated financial statements
included elsewhere in this annual report.
|
|
|
Item 13.
|
Certain
Relationships and Related Transactions, and Director
Independence.
|
Transactions with Related Parties
Dr. Lenaz
Consulting Arrangement
On April 28, 2008, we entered into a consulting agreement
with Dr. Lenaz, our former Chief Scientific Officer, which
provided for Dr. Lenaz to provide part-time consulting
services from June 30, 2008, the date of his retirement as
our Chief Scientific Officer, through December 31, 2010. On
July 1, 2010, effective with his election as a director of
the Company, Dr. Lenaz’s consulting services were
terminated. Under the terms of the consulting agreement, as
amended from time to time, Dr. Lenaz was paid $127,399
during 2010 for consulting services provided through
June 30, 2010.
96
Policy
on the Review, Approval or Ratification of Transactions with
Related Persons
We have adopted a written policy for approval or ratification of
all transactions with related parties that are required to be
reported under Item 404(a) or
Regulation S-K.
The policy provides that the Audit Committee of the board of
directors shall review the material facts of all transactions
and either approve or disapprove of the entry into the
transaction. If advance Audit Committee approval of a
transaction is not feasible, then the transaction shall be
considered by the Audit Committee chair and, if the Audit
Committee determines it to be appropriate, ratified by the Audit
Committee.
The Audit Committee may establish that certain transactions may
be pre-approved by the Audit Committee. However the Audit
Committee has not established any such transactions.
No director shall participate in any approval of a transaction
for which he or she is a related party. The director shall
provide all material information concerning the transaction to
the Audit Committee.
Director
Independence
In determining whether members of our Board of Directors are
independent, the Board reviews a summary of the relationships of
each director with the Company and other facts relevant to the
analysis of whether the directors qualify as independent
director under the NASDAQ Global Market listing standards.
All members of the Board, expect for Dr. Rajesh C.
Shrotriya, President and Chief Executive Officer of the Company,
and Dr. Luigi Lenaz, Board Member, are independent pursuant
to the listing standards of the NASDAQ Global Market. All
members of the Audit, Compensation and Nominating and Corporate
Governance Committees are independent pursuant to the listing
standards of the NASDAQ Global Market and the rules promulgated
by the SEC for the Audit Committee.
|
|
|
Item 14.
|
Principal
Accountant Fees and Services
|
The following summarizes aggregate fees billed to us by our
independent registered public accounting firms, E&Y and
K&C, for the fiscal years ended December 31, 2010 and
2009:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ernst &
|
|
|
Ernst &
|
|
|
|
|
|
|
|
Young LLP
|
|
|
Young LLP
|
|
|
Kelley & Co.
|
|
|
|
|
2010($)
|
|
|
2009($)
|
|
|
2009($)(1)
|
|
|
|
|
Audit Fees
|
|
|
416,000
|
|
|
|
210,000
|
|
|
|
85,280
|
|
|
Audit-related Fees
|
|
|
5,200
|
|
|
|
—
|
|
|
|
27,210
|
|
|
Tax Fees
|
|
|
253,825
|
|
|
|
—
|
|
|
|
24,800
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
675,025
|
|
|
|
210,000
|
|
|
|
137,290
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Represents fees billed to us by K&C as our independent
registered public accounting firm through December 3, 2009. |
The fees billed to us by E&Y and K&C during or related
to our 2010 and 2009 fiscal years consist solely of audit fees,
audit-related fees and tax fees, as follows:
Audit Fees. Represents the aggregate fees
billed to us for professional services rendered for the audit of
our annual consolidated financial statements and our internal
controls over financial reporting, for the reviews of our
consolidated financial statements included in our
Form 10-Q
filings for each fiscal quarter, and the preparation of comfort
letters and consents with respect to registration statements.
Audit-related Fees. Represents the aggregate
fees billed to us for assurance and related services that are
reasonably related to the performance of the audit and review of
our consolidated financial statements that are not already
reported in Audit Fees. These services include accounting
consultations and attestation services that are not required by
statute.
Tax Fees. Represents the aggregate fees billed
to us for professional services rendered for tax returns,
compliance and tax advice.
97
Policy on
Audit Committee Pre-approval of Audit and Permissible Non-audit
Services of Independent Auditor
All audit and permissible non-audit services by our independent
registered public accounting firms were pre-approved by our
Audit Committee. Pursuant to its charter, the Audit Committee
may establish pre-approval policies and procedures, subject to
SEC and NASDAQ rules and regulations, to approve audit and
permissible non-audit services, however, it has not yet done so.
PART IV
|
|
|
Item 15.
|
Exhibits and
Financial Statement Schedules
|
(a)(1) Consolidated Financial Statements:
| |
|
|
|
|
|
|
|
Page
|
|
|
|
|
|
|
F-2
|
|
|
|
|
|
F-4
|
|
|
|
|
|
F-5
|
|
|
|
|
|
F-6
|
|
|
|
|
|
F-7
|
|
|
|
|
|
F-8
|
|
(a)(2) Financial Statement Schedules: All financial
statement schedules are omitted because they are not applicable
or the required information is included in the Consolidated
Financial Statements or notes thereto.
(a)(3) Exhibits.
98
Index to
Exhibits
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
2
|
.1
|
|
Asset Purchase Agreement by and between the Registrant, Targent
Inc. and Certain Stockholders of Targent, Inc., dated March 17
2006. (Filed as Exhibit 2.1 to
Form 10-K/A,
Amendment No. 1, as filed with the Securities and Exchange
Commission on May 1, 2006, and incorporated herein by
reference.)
|
|
|
2
|
.2
|
|
Asset Purchase Agreement by and between the Registrant and Par
Pharmaceutical, Inc., dated as of May 6, 2008. (Filed as
Exhibit 2.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 11, 2008, and incorporated herein by reference.)
|
|
|
2
|
.3#
|
|
Purchase and Formation Agreement, dated as of November 26,
2008, by and among the Registrant, Cell Therapeutics, Inc. and
RIT Oncology, LLC. (Filed as Exhibit 2.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 19, 2008, and incorporated herein by reference.)
|
|
|
2
|
.4#
|
|
Limited Liability Company Interest Assignment Agreement, dated
as of March 15, 2009, by and between the Registrant and
Cell Therapeutics, Inc. (Filed as Exhibit 2.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 15, 2009, and incorporated herein by reference.)
|
|
|
3
|
.1+
|
|
Amended Certificate of Incorporation, as filed.
|
|
|
3
|
.2
|
|
Form of Amended and Restated Bylaws of the Registrant. (Filed as
Exhibit 3.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 16, 2004, and incorporated herein by reference.)
|
|
|
4
|
.1
|
|
Rights Agreement, dated as of December 13, 2010, between
the Registrant and ComputerShare Trust Company, N.A.
(formerly U.S. Stock Transfer Corporation), as Rights Agent,
which includes as Exhibit A thereto the form of Certificate
of Designation for the Series B Junior Participating
Preferred Stock, as Exhibit B thereto the Form of Rights
Certificate and as Exhibit C thereto a Summary of Rights of
Stockholder Rights Plan. (Filed as Exhibit 4.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 13, 2010, and incorporated herein by reference.)
|
|
|
4
|
.2
|
|
Registration Rights Agreement, dated as of September 26,
2003, by and among the Registrant and the persons listed on
Schedule 1 attached thereto. (Filed as Exhibit 4.4 to
Form 8-K,
as filed with the Securities and Exchange Commission on
September 30, 2003, and incorporated herein by reference.)
|
|
|
4
|
.3
|
|
Investor Rights Agreement, dated as of April 20, 2004, by
and among the Registrant and the persons listed on
Schedule 1 attached thereto. (Filed as Exhibit 4.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
April 23, 2004, and incorporated herein by reference.)
|
|
|
4
|
.4
|
|
Form of Warrant, dated September 15, 2005. (Filed as
Exhibit 4.35 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 15, 2006, and incorporated herein by reference.)
|
|
|
4
|
.5
|
|
Registration Rights Agreement, dated as of April 20, 2006,
by and among the Registrant and Targent, Inc. (Filed as
Exhibit 4.2 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 8, 2006, and incorporated herein by reference.)
|
|
|
10
|
.1+
|
|
Sublease Agreement, dated as of December 2, 2010, between
the Registrant and Del Webb Corporation.
|
|
|
10
|
.2
|
|
Industrial Lease Agreement, dated as of January 16, 1997,
between the Registrant and the Irvine Company. (Filed as
Exhibit 10.11 to
Form 10-KSB,
as filed with the Securities and Exchange Commission on
March 31, 1997, and incorporated herein by reference.)
|
|
|
10
|
.3
|
|
Preferred Stock and Warrant Purchase Agreement, dated as of
September 26, 2003, by and among the Registrant and the
purchasers listed on Schedule 1 attached thereto. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
September 30, 2003, and incorporated herein by reference.)
|
|
|
10
|
.4
|
|
First Amendment, dated March 25, 2004, to Industrial Lease
Agreement dated as of January 16, 1997 by and between the
Registrant and the Irvine Company. (Filed as Exhibit 10.1
to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 17, 2004, and incorporated herein by reference.)
|
|
|
10
|
.5
|
|
Common Stock and Warrant Purchase Agreement, dated as of
April 20, 2004, by and among the Registrant and the
purchasers listed on Schedule 1 attached thereto. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
April 23, 2004, and incorporated herein by reference.)
|
|
|
10
|
.6#+
|
|
Amended and Restated License and Collaboration Agreement by and
between the Registrant and Aeterna Zentaris GmbH (formerly
Zentaris GmbH), dated as of November 5, 2010.
|
99
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
10
|
.7*
|
|
Form of Stock Option Agreement under the 2003 Amended and
Restated Incentive Award Plan. (Filed as Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 17, 2004, and incorporated herein by reference.)
|
|
|
10
|
.8#
|
|
License Agreement by and between the Registrant and Chicago
Labs, Inc. (Filed as Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
February 25, 2005, and incorporated herein by reference.)
|
|
|
10
|
.9*
|
|
Form of Non-Employee Director Stock Option Agreement under the
2003 Amended and Restated Incentive Award Plan. (Filed as
Exhibit 10.5 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 10, 2005, and incorporated herein by reference.)
|
|
|
10
|
.10*
|
|
Restricted Stock Award Grant Notice and Restricted Stock Award
Agreement under the 2003 Amended and Restated Incentive Award
Plan. (Filed as Exhibit 10.44 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 15, 2006, and incorporated herein by reference.)
|
|
|
10
|
.11#
|
|
License Agreement between the Registrant and Merck Eprova AG,
dated May 23, 2006. (Filed as Exhibit 10.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 8, 2006, and incorporated herein by reference.)
|
|
|
10
|
.12*
|
|
Third Amended and Restated 1997 Stock Incentive Plan. (Filed as
Exhibit 10.2 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
November 3, 2006, and incorporated herein by reference.)
|
|
|
10
|
.13#
|
|
Agreement by and between the Registrant and Glaxo Group Limited
(d/b/a GlaxoSmithKline), dated November 10, 2006. (Filed as
Exhibit 10.38 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 14, 2007, and incorporated herein by reference.)
|
|
|
10
|
.14*
|
|
2003 Amended and Restated Incentive Award Plan. (Filed as
Exhibit 10.3 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 9, 2007, and incorporated herein by reference.)
|
|
|
10
|
.15#
|
|
License Agreement by and between the Registrant and Indena,
S.p.A., dated July 17, 2007. (Filed as Exhibit 10.4 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
November 9, 2007, and incorporated herein by reference.)
|
|
|
10
|
.16*
|
|
Executive Employment Agreement by and between the Registrant and
Rajesh C. Shrotriya, M.D., entered into June 20, 2008
and effective as of January 2, 2008. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
June 26, 2008, and incorporated herein by reference.)
|
|
|
10
|
.17*
|
|
Consulting Agreement by and between the Registrant and Luigi
Lenaz, M.D., effective as of July 1, 2008. (Filed as
Exhibit 10.2 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 11, 2008, and incorporated herein by reference.)
|
|
|
10
|
.18*+
|
|
Amendment Number One to Consulting Agreement by and between the
Registrant and Luigi Lenaz, M.D., effective as of
March 16, 2010.
|
|
|
10
|
.19*+
|
|
Amendment Number Two to Consulting Agreement by and between the
Registrant and Luigi Lenaz, M.D., effective as of
July 2, 2010.
|
|
|
10
|
.20*
|
|
Form of Indemnity Agreement of the Registrant. (Filed as
Exhibit 10.32 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 31, 2009, and incorporated herein by reference.)
|
|
|
10
|
.21#
|
|
License, Development, Supply and Distribution Agreement, dated
October 28, 2008, by and among the Registrant, Allergan
Sales, LLC, Allergan USA, Inc. and Allergan, Inc. (Filed as
Exhibit 10.33 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 31, 2009, and incorporated herein by reference.)
|
|
|
10
|
.22*
|
|
2009 Employee Stock Purchase Plan. (Filed as Exhibit 99.1
to
Form S-8,
as filed with the Securities and Exchange Commission on
June 29, 2009, and incorporated herein by reference.)
|
|
|
10
|
.23*
|
|
2009 Incentive Award Plan. (Filed as Exhibit 99.2 to
Form S-8,
as filed with the Securities and Exchange Commission on
June 29, 2009, and incorporated herein by reference.)
|
|
|
10
|
.24*
|
|
2003 Amended and Restated Incentive Award Plan. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
July 2, 2009, and incorporated herein by reference.)
|
|
|
10
|
.25
|
|
Fourth Amendment, dated July 29, 2009, to Industrial Lease
Agreement dated as of January 16, 1997 by and between the
Registrant and the Irvine Company. (Filed as Exhibit 10.29
to
Form 10-K,
as filed with the Securities and Exchange Commission on
April 5, 2010, and incorporated herein by reference.)
|
100
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
10
|
.26*
|
|
Term Sheet for 2009 Incentive Award Plan Stock Option Award.
(Filed as Exhibit 10.8 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 13, 2009, and incorporated herein by reference.)
|
|
|
10
|
.27*
|
|
Term Sheet for 2009 Incentive Award Plan, Nonqualified Stock
Option Award Awarded to Non-Employee Directors. (Filed as
Exhibit 10.9 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 13, 2009, and incorporated herein by reference.)
|
|
|
10
|
.28*
|
|
Term Sheet for 2009 Incentive Award Plan, Restricted Stock
Award. (Filed as Exhibit 10.10 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 13, 2009, and incorporated herein by reference.)
|
|
|
10
|
.29#
|
|
License Agreement, dated November 6, 2009, by and between
the Registrant and Nippon Kayaku Co., Ltd. (Filed as
Exhibit 10.36 to
Form 10-K,
as filed with the Securities and Exchange Commission on
April 5, 2010, and incorporated herein by reference.)
|
|
|
10
|
.30#
|
|
License and Collaboration Agreement, dated February 2,
2010, by and between the Registrant and TopoTarget A/S. (Filed
as Exhibit 10.37 to
Form 10-K,
as filed with the Securities and Exchange Commission on
April 5, 2010, and incorporated herein by reference.)
|
|
|
10
|
.31
|
|
Asset Purchase Agreement, dated August 15, 2007, by and between
Cell Therapeutics, Inc. and Biogen Idec Inc. (Filed as Exhibit
10.1 to Cell Therapeutics, Inc.’s Form 8-K, No. 001-12465,
as filed with the Securities and Exchange Commission on August
21, 2007, and incorporated herein by reference.)
|
|
|
10
|
.32
|
|
First Amendment to Asset Purchase Agreement, dated December 9,
2008, by and between Cell Therapeutics, Inc. and Biogen Idec
Inc. (Filed as Exhibit 10.48 to Cell Therapeutics, Inc.’s
Form 10K, No. 001-12465, as filed with the Securities and
Exchange Commission on March 16, 2009, and incorporated herein
by reference.)
|
|
|
10
|
.33
|
|
Supply Agreement, dated December 21, 2007, by and between Cell
Therapeutics, Inc. and Biogen Idec Inc. (Filed as Exhibit 10.2
to Cell Therapeutics, Inc.’s Form 8-K, No. 001-12465, as
filed with the Securities and Exchange Commission on December
31, 2007, and incorporated herein by reference.)
|
|
|
10
|
.34#+
|
|
First Amendment to Supply Agreement, dated December 15, 2008, by
and between Cell Therapeutics, Inc. and Biogen Idec Inc.
|
|
|
10
|
.35+
|
|
Security Agreement, dated December 15, 2008, by and between RIT
Oncology, LLC and Biogen Idec Inc.
|
|
|
16
|
.1
|
|
Letter from Kelly and Company to the Securities and Exchange
Commission, dated December 3, 2009. (Filed as
Exhibit 16.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 8, 2009, and incorporated herein by reference.)
|
|
|
21
|
+
|
|
Subsidiaries of Registrant.
|
|
|
23
|
.1+
|
|
Consent of Ernst & Young LLP, Independent Registered
Public Accounting Firm.
|
|
|
23
|
.2+
|
|
Consent of Kelly & Company, Independent Registered
Public Accounting Firm.
|
|
|
24
|
.1
|
|
Power of Attorney (included in the signature page.)
|
|
|
31
|
.1+
|
|
Certification of Principal Executive Officer, pursuant to
Rule 13a-14(a)/15d-14(a)
of the Securities Exchange Act of 1934.
|
|
|
31
|
.2+
|
|
Certification of Principal Financial Officer, pursuant to
Rule 13a-14(a)/15d-14(a)
of the Securities Exchange Act of 1934.
|
|
|
32
|
.1+
|
|
Certification of Principal Executive Officer, pursuant to
Rule 13a-14(b)/15d-14(b)
of the Securities Exchange Act of 1934 and 18 U.S.C.
Section 1350.
|
|
|
32
|
.2+
|
|
Certification of Principal Financial Officer, pursuant to
Rule 13a-14(b)/15d-14(b)
of the Securities Exchange Act of 1934 and 18 U.S.C.
Section 1350.
|
|
|
|
|
* |
|
Indicates a management contract or compensatory plan or
arrangement. |
| |
|
# |
|
Confidential portions omitted and filed separately with the U.S.
Securities and Exchange Commission pursuant to
Rule 24b-2
promulgated under the Securities Exchange Act of 1934, as
amended. |
| |
|
+ |
|
Filed herewith. |
101
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the registrant has duly caused
this Annual Report on
Form 10-K
to be signed on its behalf by the undersigned, thereunto duly
authorized.
Spectrum Pharmaceuticals, Inc.
|
|
|
| |
By:
|
/s/ Rajesh
C. Shrotriya, M.D.
|
Rajesh C. Shrotriya, M.D.
Chief Executive Officer and President
Date: March 9, 2011
POWER OF
ATTORNEY
Each person whose signature appears below constitutes and
appoints each of Rajesh C. Shrotriya and Brett L. Scott Kumaria
as his attorney-in-fact, with full power of substitution, for
him in any and all capacities, to sign any amendments to this
Form 10-K,
and to file the same, with all exhibits thereto and other
documents in connection therewith, with the Securities and
Exchange Commission, hereby ratifying and confirming all that
each attorney-in-fact, or his substitute, may do or cause to be
done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of
1934, this Annual Report on
Form 10-K
has been signed below by the following persons on behalf of the
Registrant and in the capacities and on the dates indicated:
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Rajesh
C. Shrotriya, M.D.
Rajesh
C. Shrotriya, M.D.
|
|
Chairman of the Board, Chief Executive
Officer, and President
(Principal Executive Officer)
|
|
March 9, 2011
|
|
|
|
|
|
|
|
/s/ Brett
L. Scott
Brett
L. Scott
|
|
Senior Vice President and Acting Chief
Financial Officer
(Principal Financial and Accounting Officer)
|
|
March 9, 2011
|
|
|
|
|
|
|
|
/s/ Krishan
K. Arora, Ph.D.
Krishan
K. Arora, Ph.D.
|
|
Director
|
|
March 9, 2011
|
|
|
|
|
|
|
|
/s/ Luigi
Lenaz, M.D.
Luigi
Lenaz, M.D.
|
|
Director
|
|
March 9, 2011
|
|
|
|
|
|
|
|
/s/ Stuart
M. Krassner, Sc.D., Psy.D.
Stuart
M. Krassner, Sc.D., Psy.D.
|
|
Director
|
|
March 9, 2011
|
|
|
|
|
|
|
|
/s/ Anthony
E. Maida, III, M.A., M.B.A., Ph.D
Anthony
E. Maida, III, M.A., M.B.A., Ph.D
|
|
Director
|
|
March 9, 2011
|
|
|
|
|
|
|
|
/s/ Dilip
J. Mehta, M.D., Ph.D.
Dilip
J. Mehta, M.D., Ph.D.
|
|
Director
|
|
March 9, 2011
|
102
Spectrum Pharmaceuticals, Inc. and Subsidiaries
Consolidated Financial Statements
As of December 31, 2010 and 2009 and
For Each of the Three Years Ended December 31, 2010
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Consolidated
Financial Statements
INDEX TO
FINANCIAL STATEMENTS
| |
|
|
|
|
|
|
|
Page
|
|
|
|
|
|
|
F-2
|
|
|
|
|
|
F-4
|
|
|
|
|
|
F-5
|
|
|
|
|
|
F-6
|
|
|
|
|
|
F-7
|
|
|
|
|
|
F-8
|
|
F-1
Report of
Ernst & Young LLP, Independent Registered Public
Accounting Firm
The Board of Directors and
Stockholders of Spectrum Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of
Spectrum Pharmaceuticals, Inc. and Subsidiaries as of
December 31, 2010 and 2009, and the related consolidated
statements of operations, stockholders’ equity and cash
flows for each of the two years in the period ended
December 31, 2010. These financial statements are the
responsibility of the Company’s management. Our
responsibility is to express an opinion on these financial
statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes examining, on a
test basis, evidence supporting the amounts and disclosures in
the financial statements. An audit also includes assessing the
accounting principles used and significant estimates made by
management, as well as evaluating the overall financial
statement presentation. We believe that our audits provide a
reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred
to above present fairly, in all material respects, the
consolidated financial position of Spectrum Pharmaceuticals,
Inc. and Subsidiaries at December 31, 2010 and 2009, and
the consolidated results of its operations and its cash flows
for each of the two years in the period ended December 31,
2010, in conformity with U.S. generally accepted accounting
principles.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States),
Spectrum Pharmaceuticals, Inc. and Subsidiaries internal control
over financial reporting as of December 31, 2010, based on
criteria established in Internal Control-Integrated Framework
issued by the Committee of Sponsoring Organizations of the
Treadway Commission and our report dated March 9, 2011
expressed an unqualified opinion thereon.
Irvine, California
March 9, 2011
F-2
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
Spectrum Pharmaceuticals, Inc. and Subsidiaries.
We have audited the accompanying consolidated statements of
operations, stockholders’ equity and cash flows for the
year ended December 31, 2008 of Spectrum Pharmaceuticals,
Inc. and Subsidiaries. These consolidated financial statements
are the responsibility of the Company’s management. Our
responsibility is to express an opinion on these consolidated
financial statements based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes examining, on a
test basis, evidence supporting the amounts and disclosures in
the financial statements. An audit also includes assessing the
accounting principles used and significant estimates made by
management, as well as evaluating the overall financial
statement presentation. We believe that our audit provides a
reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred
to above present fairly, in all material respects, the results
of operations and cash flows of Spectrum Pharmaceuticals, Inc.
and Subsidiaries for the year ended December 31, 2008 in
conformity with U.S. generally accepted accounting
principles.
Kelly & Company
Costa Mesa, California
March 31, 2009
F-3
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
(In thousands, except share and per share data)
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
ASSETS
|
|
Current Assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
53,557
|
|
|
$
|
82,336
|
|
|
Marketable securities
|
|
|
42,117
|
|
|
|
31,005
|
|
|
Accounts receivable, net of allowance for doubtful accounts of
$339, and $150, respectively
|
|
|
21,051
|
|
|
|
8,658
|
|
|
Inventories
|
|
|
4,234
|
|
|
|
3,230
|
|
|
Prepaid expenses and other current assets
|
|
|
906
|
|
|
|
1,028
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
121,865
|
|
|
|
126,257
|
|
|
Bank certificates of deposit
|
|
|
8,569
|
|
|
|
11,438
|
|
|
Property and equipment, net
|
|
|
3,158
|
|
|
|
1,928
|
|
|
Zevalin related intangible assets, net of accumulated
amortization of $12,295 and $8,575, respectively
|
|
|
29,605
|
|
|
|
33,325
|
|
|
Other assets
|
|
|
434
|
|
|
|
185
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
163,631
|
|
|
$
|
173,133
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current Liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable and other accrued obligations
|
|
$
|
38,704
|
|
|
$
|
16,606
|
|
|
Accrued compensation and related expenses
|
|
|
3,313
|
|
|
|
3,360
|
|
|
Deferred revenue
|
|
|
12,300
|
|
|
|
8,300
|
|
|
Common stock warrant liability
|
|
|
3,904
|
|
|
|
6,635
|
|
|
Accrued drug development costs
|
|
|
5,101
|
|
|
|
4,598
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
63,322
|
|
|
|
39,499
|
|
|
Capital lease obligations
|
|
|
40
|
|
|
|
69
|
|
|
Deferred revenue and other credits — less current
portion
|
|
|
25,495
|
|
|
|
24,943
|
|
|
Zevalin related contingent obligations
|
|
|
298
|
|
|
|
298
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities
|
|
|
89,155
|
|
|
|
64,809
|
|
|
Commitments and contingencies
|
|
|
|
|
|
|
|
|
|
Stockholders’ Equity:
|
|
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value; 5,000,000 shares
authorized of which 1,000,000 shares have been designated
as Series B junior participating preferred stock, no shares
issued and outstanding
|
|
|
—
|
|
|
|
—
|
|
|
Series E convertible voting preferred stock —
$10,000 par value; 2,000 shares authorized; 26 and
68 shares issued and outstanding at December 31, 2010
and 2009, respectively, (aggregate liquidation value of $312)
|
|
|
160
|
|
|
|
419
|
|
|
Common stock, $0.001 par value —
100,000,000 shares authorized; 51,459,284 and 48,926,314
issued and outstanding at December 31, 2010 and 2009,
respectively
|
|
|
51
|
|
|
|
49
|
|
|
Additional paid-in capital
|
|
|
384,757
|
|
|
|
369,482
|
|
|
Accumulated other comprehensive loss
|
|
|
(92
|
)
|
|
|
(70
|
)
|
|
Accumulated deficit
|
|
|
(310,400
|
)
|
|
|
(261,556
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
74,476
|
|
|
|
108,324
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
163,631
|
|
|
$
|
173,133
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to consolidated financial statements.
F-4
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
(In thousands, except share and per share data)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Revenues:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales, net
|
|
$
|
60,921
|
|
|
$
|
28,225
|
|
|
$
|
8,049
|
|
|
License and contract revenue
|
|
|
13,192
|
|
|
|
9,800
|
|
|
|
20,676
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
$
|
74,113
|
|
|
$
|
38,025
|
|
|
$
|
28,725
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating costs and expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of product sales (excludes amortization of purchased
intangible assets)
|
|
|
17,439
|
|
|
|
8,148
|
|
|
|
1,193
|
|
|
Selling general and administrative
|
|
|
48,550
|
|
|
|
33,607
|
|
|
|
15,156
|
|
|
Research and development
|
|
|
57,301
|
|
|
|
21,058
|
|
|
|
26,683
|
|
|
Amortization of purchased intangible assets
|
|
|
3,720
|
|
|
|
3,720
|
|
|
|
158
|
|
|
Acquired in-process research and development
|
|
|
—
|
|
|
|
—
|
|
|
|
4,700
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating costs and expenses
|
|
|
127,010
|
|
|
|
66,533
|
|
|
|
47,890
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(52,897
|
)
|
|
|
(28,508
|
)
|
|
|
(19,165
|
)
|
|
Non-operating income:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Change in fair value of common stock warrant liability
|
|
|
2,731
|
|
|
|
8,075
|
|
|
|
1,271
|
|
|
Other income, net
|
|
|
1,279
|
|
|
|
662
|
|
|
|
1,165
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss before provision for income taxes
|
|
|
(48,887
|
)
|
|
|
(19,771
|
)
|
|
|
(16,729
|
)
|
|
Benefit (provision) for income taxes
|
|
|
43
|
|
|
|
(421
|
)
|
|
|
(5
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
(48,844
|
)
|
|
|
(20,192
|
)
|
|
|
(16,734
|
)
|
|
Net loss attributable to non-controlling interest
|
|
|
—
|
|
|
|
1,146
|
|
|
|
2,538
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to Spectrum Pharmaceuticals, Inc.
stockholders
|
|
$
|
(48,844
|
)
|
|
$
|
(19,046
|
)
|
|
$
|
(14,196
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share:
|
|
$
|
(0.99
|
)
|
|
$
|
(0.48
|
)
|
|
$
|
(0.45
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted weighted average shares outstanding:
|
|
|
49,502,854
|
|
|
|
39,273,905
|
|
|
|
31,551,152
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to consolidated financial statements.
F-5
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
(In thousands, except share data)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other
|
|
|
|
|
|
Total
|
|
|
Non-
|
|
|
|
|
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Additional
|
|
|
Comprehensive
|
|
|
Accumulated
|
|
|
Stockholders
|
|
|
Controlling
|
|
|
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Paid-In Capital
|
|
|
Income (Loss)
|
|
|
Deficit
|
|
|
Equity
|
|
|
Interest
|
|
|
Total
|
|
|
|
|
Balance at December 2007
|
|
|
170
|
|
|
$
|
1,048
|
|
|
|
31,233,798
|
|
|
$
|
31
|
|
|
$
|
273,455
|
|
|
$
|
493
|
|
|
$
|
(228,313
|
)
|
|
$
|
46,713
|
|
|
$
|
—
|
|
|
$
|
46,713
|
|
|
Net Loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(14,196
|
)
|
|
|
(14,196
|
)
|
|
|
(2,538
|
)
|
|
|
(16,734
|
)
|
|
Realized gains on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(493
|
)
|
|
|
—
|
|
|
|
(493
|
)
|
|
|
—
|
|
|
|
(493
|
)
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(146
|
)
|
|
|
—
|
|
|
|
(146
|
)
|
|
|
—
|
|
|
|
(146
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total comprehensive loss, net
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(639
|
)
|
|
|
(14,196
|
)
|
|
|
(14,835
|
)
|
|
|
(2,538
|
)
|
|
|
(17,373
|
)
|
|
Contributions by non-controlling interest
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
16,800
|
|
|
|
16,800
|
|
|
Conversion of Series E Preferred Stock into Common Stock
|
|
|
(102
|
)
|
|
|
(629
|
)
|
|
|
204,000
|
|
|
|
—
|
|
|
|
629
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Fair value of common stock issued to Targent, Inc. for NDA
Approval
|
|
|
—
|
|
|
|
—
|
|
|
|
125,000
|
|
|
|
—
|
|
|
|
305
|
|
|
|
—
|
|
|
|
—
|
|
|
|
305
|
|
|
|
—
|
|
|
|
305
|
|
|
Fair value of common stock issued to NDDO, University of
Bradford et al
|
|
|
—
|
|
|
|
—
|
|
|
|
75,000
|
|
|
|
—
|
|
|
|
74
|
|
|
|
—
|
|
|
|
—
|
|
|
|
74
|
|
|
|
—
|
|
|
|
74
|
|
|
Share-based compensation expense and common stock issued (net of
forfeitures)
|
|
|
—
|
|
|
|
—
|
|
|
|
362,088
|
|
|
|
1
|
|
|
|
6,322
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,323
|
|
|
|
—
|
|
|
|
6,323
|
|
|
Issuance of common stock to 401(k) plan
|
|
|
—
|
|
|
|
—
|
|
|
|
166,430
|
|
|
|
—
|
|
|
|
274
|
|
|
|
—
|
|
|
|
—
|
|
|
|
274
|
|
|
|
—
|
|
|
|
274
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
68
|
|
|
$
|
419
|
|
|
|
32,166,316
|
|
|
$
|
32
|
|
|
$
|
281,059
|
|
|
$
|
(146
|
)
|
|
$
|
(242,510
|
)
|
|
$
|
38,854
|
|
|
$
|
14,262
|
|
|
$
|
53,116
|
|
|
Net Loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(19,046
|
)
|
|
|
(19,046
|
)
|
|
|
(1,146
|
)
|
|
|
(20,192
|
)
|
|
Realized gains on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
101
|
|
|
|
—
|
|
|
|
101
|
|
|
|
—
|
|
|
|
101
|
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(25
|
)
|
|
|
—
|
|
|
|
(25
|
)
|
|
|
—
|
|
|
|
(25
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total comprehensive loss, net
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
76
|
|
|
|
(19,046
|
)
|
|
|
(18,970
|
)
|
|
|
(1,146
|
)
|
|
|
(20,116
|
)
|
|
Issuance of common stock and warrants for cash, net of issuance
costs
|
|
|
—
|
|
|
|
—
|
|
|
|
15,187,715
|
|
|
|
15
|
|
|
|
81,779
|
|
|
|
—
|
|
|
|
—
|
|
|
|
81,794
|
|
|
|
—
|
|
|
|
81,794
|
|
|
Contributions by non-controlling interest
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,067
|
|
|
|
2,067
|
|
|
Purchase of non controlling interest
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,798
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,798
|
)
|
|
|
(15,183
|
)
|
|
|
(16,981
|
)
|
|
Issuance of common stock to employees — shelf takedown
|
|
|
—
|
|
|
|
—
|
|
|
|
432,200
|
|
|
|
1
|
|
|
|
1,166
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,167
|
|
|
|
—
|
|
|
|
1,167
|
|
|
Stock Options tender offer
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,520
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,520
|
)
|
|
|
—
|
|
|
|
(2,520
|
)
|
|
Issuance of common stock to 401(k) plan
|
|
|
—
|
|
|
|
—
|
|
|
|
139,795
|
|
|
|
—
|
|
|
|
448
|
|
|
|
—
|
|
|
|
—
|
|
|
|
448
|
|
|
|
—
|
|
|
|
448
|
|
|
Issuance of common stock for ESPP
|
|
|
—
|
|
|
|
—
|
|
|
|
65,715
|
|
|
|
—
|
|
|
|
292
|
|
|
|
—
|
|
|
|
—
|
|
|
|
292
|
|
|
|
—
|
|
|
|
292
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
488,750
|
|
|
|
1
|
|
|
|
1,261
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,262
|
|
|
|
—
|
|
|
|
1,262
|
|
|
Share-based compensation expense and common stock issued (net of
forfeitures)
|
|
|
—
|
|
|
|
—
|
|
|
|
207,014
|
|
|
|
—
|
|
|
|
6,860
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,860
|
|
|
|
—
|
|
|
|
6,860
|
|
|
Fair value of common stock issued to Targent, Inc. for NDA
Approval
|
|
|
—
|
|
|
|
—
|
|
|
|
125,000
|
|
|
|
—
|
|
|
|
185
|
|
|
|
—
|
|
|
|
—
|
|
|
|
185
|
|
|
|
—
|
|
|
|
185
|
|
|
Fair value of common stock issued to Altair Inc. for Renazorb
rights
|
|
|
—
|
|
|
|
—
|
|
|
|
113,809
|
|
|
|
—
|
|
|
|
750
|
|
|
|
—
|
|
|
|
—
|
|
|
|
750
|
|
|
|
—
|
|
|
|
750
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
68
|
|
|
$
|
419
|
|
|
|
48,926,314
|
|
|
$
|
49
|
|
|
$
|
369,482
|
|
|
$
|
(70
|
)
|
|
$
|
(261,556
|
)
|
|
$
|
108,324
|
|
|
$
|
—
|
|
|
$
|
108,324
|
|
|
Net Loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(48,844
|
)
|
|
|
(48,844
|
)
|
|
|
—
|
|
|
|
(48,844
|
)
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(22
|
)
|
|
|
—
|
|
|
|
(22
|
)
|
|
|
—
|
|
|
|
(22
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total comprehensive loss, net
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(22
|
)
|
|
|
(48,844
|
)
|
|
|
(48,866
|
)
|
|
|
—
|
|
|
|
48,866
|
|
|
Conversion of Series E preferred stock to common stock
|
|
|
(42
|
)
|
|
|
(259
|
)
|
|
|
84,000
|
|
|
|
—
|
|
|
|
259
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock to 401(k) plan
|
|
|
—
|
|
|
|
—
|
|
|
|
136,121
|
|
|
|
—
|
|
|
|
598
|
|
|
|
—
|
|
|
|
—
|
|
|
|
598
|
|
|
|
—
|
|
|
|
598
|
|
|
Issuance of common stock for ESPP
|
|
|
—
|
|
|
|
—
|
|
|
|
168,283
|
|
|
|
—
|
|
|
|
554
|
|
|
|
—
|
|
|
|
—
|
|
|
|
554
|
|
|
|
—
|
|
|
|
554
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
1,135,340
|
|
|
|
1
|
|
|
|
3,073
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,074
|
|
|
|
—
|
|
|
|
3,074
|
|
|
Share-based compensation expense and common stock issued (net of
forfeitures)
|
|
|
—
|
|
|
|
—
|
|
|
|
257,270
|
|
|
|
—
|
|
|
|
7,687
|
|
|
|
—
|
|
|
|
—
|
|
|
|
7,687
|
|
|
|
—
|
|
|
|
7,687
|
|
|
Fair value of common stock issued in connection with drug license
|
|
|
—
|
|
|
|
—
|
|
|
|
751,956
|
|
|
|
1
|
|
|
|
3,104
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,105
|
|
|
|
—
|
|
|
|
3,105
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2010
|
|
|
26
|
|
|
$
|
160
|
|
|
|
51,459,284
|
|
|
$
|
51
|
|
|
$
|
384,757
|
|
|
$
|
(92
|
)
|
|
$
|
(310,400
|
)
|
|
$
|
74,476
|
|
|
$
|
—
|
|
|
$
|
74,476
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to consolidated financial statements.
F-6
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
(In thousands)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Cash Flows From Operating Activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(48,844
|
)
|
|
$
|
(20,192
|
)
|
|
$
|
(16,734
|
)
|
|
Adjustments to reconcile net loss to net cash used in operating
activities:
|
|
Amortization of deferred revenue
|
|
|
(12,300
|
)
|
|
|
(9,186
|
)
|
|
|
—
|
|
|
Depreciation and amortization
|
|
|
4,506
|
|
|
|
4,244
|
|
|
|
610
|
|
|
Stock-based compensation
|
|
|
8,285
|
|
|
|
7,423
|
|
|
|
6,537
|
|
|
Acquired in-process research and development
|
|
|
—
|
|
|
|
—
|
|
|
|
4,700
|
|
|
Fair value of common stock issued for drug license
|
|
|
3,105
|
|
|
|
935
|
|
|
|
379
|
|
|
Change in fair value of common stock warrants
|
|
|
(2,731
|
)
|
|
|
(8,075
|
)
|
|
|
(1,271
|
)
|
|
Provision for bad debt
|
|
|
359
|
|
|
|
—
|
|
|
|
—
|
|
|
Provision for expiring inventory
|
|
|
50
|
|
|
|
—
|
|
|
|
—
|
|
|
Loss on disposal of fixed assets
|
|
|
11
|
|
|
|
—
|
|
|
|
—
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable
|
|
|
(12,752
|
)
|
|
|
1,118
|
|
|
|
(4,811
|
)
|
|
Inventories
|
|
|
(1,054
|
)
|
|
|
(1,389
|
)
|
|
|
(1,841
|
)
|
|
Prepaid expenses and other assets
|
|
|
(134
|
)
|
|
|
(250
|
)
|
|
|
101
|
|
|
Accounts payable and other accrued obligations
|
|
|
21,546
|
|
|
|
7,097
|
|
|
|
2,387
|
|
|
Accrued compensation and related expenses
|
|
|
(47
|
)
|
|
|
404
|
|
|
|
1,845
|
|
|
Accrued drug development costs
|
|
|
503
|
|
|
|
1,149
|
|
|
|
—
|
|
|
Landlord contributions to tenant improvements
|
|
|
995
|
|
|
|
—
|
|
|
|
—
|
|
|
Deferred revenue and other credits
|
|
|
15,958
|
|
|
|
(912
|
)
|
|
|
93
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(22,544
|
)
|
|
|
(17,634
|
)
|
|
|
(8,005
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Flows From Investing Activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Maturities of marketable securities
|
|
|
32,391
|
|
|
|
25,783
|
|
|
|
—
|
|
|
Purchases of marketable securities
|
|
|
(40,649
|
)
|
|
|
—
|
|
|
|
(13,056
|
)
|
|
Investment in Zevalin acquisition
|
|
|
—
|
|
|
|
(30,940
|
)
|
|
|
(10,202
|
)
|
|
Purchases of property and equipment
|
|
|
(1,576
|
)
|
|
|
(673
|
)
|
|
|
(1,518
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in investing activities
|
|
|
(9,834
|
)
|
|
|
(5,830
|
)
|
|
|
(24,776
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash Flows From Financing Activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock and warrants, net of
related offering costs and expenses
|
|
|
—
|
|
|
|
95,810
|
|
|
|
—
|
|
|
Proceeds from issuance of common stock option exercises
|
|
|
3,074
|
|
|
|
1,262
|
|
|
|
—
|
|
|
Proceeds from issuance of common stock to employees —
shelf takedown
|
|
|
—
|
|
|
|
1,167
|
|
|
|
—
|
|
|
Proceeds from issuance of common stock under the ESPP plan
|
|
|
554
|
|
|
|
292
|
|
|
|
—
|
|
|
Proceeds from Allergan, Inc. collaboration
|
|
|
—
|
|
|
|
—
|
|
|
|
41,500
|
|
|
Repurchase of warrants
|
|
|
—
|
|
|
|
(71
|
)
|
|
|
—
|
|
|
Repurchase of stock options pursuant to tender offer
|
|
|
—
|
|
|
|
(2,520
|
)
|
|
|
—
|
|
|
Repayment of capital leases
|
|
|
(29
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
3,599
|
|
|
|
95,940
|
|
|
|
41,500
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net (decrease) increase in cash and cash equivalents
|
|
|
(28,779
|
)
|
|
|
72,476
|
|
|
|
8,719
|
|
|
Cash and cash equivalents — beginning of year
|
|
|
82,336
|
|
|
|
9,860
|
|
|
|
1,141
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents — end of year
|
|
$
|
53,557
|
|
|
$
|
82,336
|
|
|
$
|
9,860
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental Disclosure of Cash Flow Information:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for income taxes
|
|
$
|
27
|
|
|
$
|
45
|
|
|
$
|
5
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest
|
|
$
|
17
|
|
|
$
|
28
|
|
|
$
|
36
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Financed portion of leasehold improvements
|
|
$
|
451
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of preferred stock to common stock
|
|
$
|
259
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to consolidated financial statements.
F-7
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Spectrum Pharmaceuticals, Inc. is a biotechnology company with
fully integrated commercial and drug development operations,
with a primary focus in oncology. We are a Delaware corporation
that was originally incorporated in Colorado as Americus Funding
Corporation in December 1987, became NeoTherapeutics, Inc. in
August 1996, was reincorporated in Delaware in June 1997, and
was renamed Spectrum Pharmaceuticals, Inc. in December 2002.
Our strategy is comprised of acquiring, developing and
commercializing a broad and diverse pipeline of late-stage
clinical and commercial products. We market two oncology drugs,
ZEVALIN®
and
FUSILEV®
and have two drugs, apaziquone and belinostat, in late stage
development along with a diversified pipeline of novel drug
candidates. We have assembled an integrated in-house scientific
team, including formulation development, clinical development,
medical research, regulatory affairs, biostatistics and data
management, and have established a commercial infrastructure for
the marketing of our drug products. We also leverage the
expertise of our worldwide partners to assist in the execution
of our strategy. Apaziquone is presently being studied in two
large Phase 3 clinical trials for non-muscle invasive bladder
cancer, or NMIBC, under strategic collaborations with Allergan,
Inc., (“Allergan”), Nippon Kayaku Co. Ltd.,
(“Nippon Kayaku”), and Handok Pharmaceuticals Co.
Ltd., (“Handok”). Belinostat, is being studied in
multiple indications including a Phase 2 registrational trial
for relapsed or refractory peripheral T-cell lymphoma,
(“PTCL”), under a strategic collaboration with
TopoTarget A/S (“TopoTarget”).
|
|
|
2.
|
Summary
of Significant Accounting Policies
|
Basis
of Presentation
The accompanying consolidated financial statements have been
prepared in accordance with accounting principles generally
accepted in the United States of America.
Principles
of Consolidation
The consolidated financial statements include the accounts of
Spectrum Pharmaceuticals, Inc., our wholly-owned subsidiaries,
and joint ventures the Company controls, or of which it is
determined to be the primary beneficiary. We evaluate the need
to consolidate joint ventures in accordance with authoritative
guidance. Investments by outside parties in our consolidated
entities are recorded as non-controlling interest in our
consolidated financial statements, and stated net after
allocation of income and losses in the entity.
As of December 31, 2010, we had three consolidated
subsidiaries: OncoRx Pharma Private Limited
(“OncoRx”), 100% owned, organized in Mumbai, India in
2008; Spectrum Pharmaceuticals GmbH, wholly-owned inactive
subsidiary, incorporated in Switzerland in April 1997; RIT
Oncology, LLC (“RIT”), 100% owned since March 15,
2009, organized in Delaware in October 2008; and a consolidated
joint venture, Spectrum Pharma Canada, organized in Quebec,
Canada in January 2008. We have eliminated all significant
intercompany balances and transactions among the consolidated
entities from the consolidated financial statements.
Use of
Estimates
The preparation of financial statements in conformity with
accounting principles generally accepted in the United States
requires management to make estimates and assumptions that
affect the reported amounts of assets, liabilities, revenues and
expenses and disclosure of contingent obligations in the
consolidated financial statements and accompanying notes. The
estimation process requires assumptions to be made by management
about future events and conditions, and as such, is inherently
subjective and uncertain. Actual results could differ materially
from those estimates.
F-8
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Segment
and Geographic Information
We operate in one reportable segment: acquiring, developing and
commercializing prescription drug products. Accordingly, we
report the accompanying consolidated financial statements in the
aggregate, including all of our activities in one reportable
segment. Foreign operations were not significant for any of the
periods presented herein.
Cash,
Cash Equivalents and Marketable Securities
Cash, cash equivalents and marketable securities primarily
consist of bank checking deposits, short-term treasury
securities, institutional money market funds, corporate debt and
equity, municipal obligations, government agency notes, and
certificates of deposit. We classify highly liquid short-term
investments, with insignificant interest rate risk and
maturities of 90 days or less at the time of acquisition,
as cash and cash equivalents. Other investments, which do not
meet the above definition of cash equivalents, are classified as
either
“held-to-maturity”
or
“available-for-sale”
marketable securities. Investments that lack immediate
liquidity, or which we intend to hold for more than one year are
classified as long-term investments, and included in other
assets. All of our “available for sale securities” are
classified as current assets based on our intent and ability to
use any and all of these securities as necessary to satisfy our
cash needs as they arise, by redeeming them at par with short
notice and without penalty.
As of December 31, 2010, we held substantially all of our
cash, cash equivalents and marketable securities at major
financial institutions, which must invest our funds in
accordance with our investment policy with the principal
objectives of such policy being preservation of capital,
fulfillment of liquidity needs and above market returns
commensurate with preservation of capital. Our investment policy
also requires that investments in marketable securities be in
only highly rated instruments, which are primarily US treasury
bills or US treasury backed securities, with limitations on
investing in securities of any single issuer. We maintain cash
balances in excess of federally insured limits in reputable
financial institutions. To a limited degree, the Federal Deposit
Insurance Corporation and third parties insure these
investments. However, these investments are not insured against
the possibility of a complete loss of earnings or principal and
are inherently subject to the credit risk related to the
continued credit worthiness of the underlying issuer and general
credit market risks. We manage such risks on our portfolio by
investing in highly liquid, highly rated instruments and limit
investing in long-term maturity instruments.
“Available-for-sale”
marketable securities are carried at fair value, with any
unrealized gains and losses included as a component of
accumulated other comprehensive income (loss) in
stockholders’ equity. Realized gains and losses and
declines in value judged to be
other-than-temporary,
as well as interest income and dividends on investments, are
included in other income and expense. We have classified
$8.6 million of our investments with maturity dates over
1 year from December 31, 2010 as long term based on
our intention to hold to maturity.
F-9
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Fair
Value Measurements
The carrying values of our cash and cash equivalents, marketable
securities, other securities and common stock warrants, carried
at fair value as of December 31, 2010 and 2009, are
classified in the table below in one of the three categories
described below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair Value Measurements
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Assets:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
53,557
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
53,557
|
|
|
FDIC insured bank CDs
|
|
|
—
|
|
|
|
29,985
|
|
|
|
—
|
|
|
|
29,985
|
|
|
Money market currency funds
|
|
|
—
|
|
|
|
15,488
|
|
|
|
—
|
|
|
|
15,488
|
|
|
U.S. Government securities
|
|
|
—
|
|
|
|
2,909
|
|
|
|
—
|
|
|
|
2,909
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
2,304
|
|
|
|
—
|
|
|
|
2,304
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents and marketable securities
|
|
|
53,557
|
|
|
|
50,686
|
|
|
|
—
|
|
|
|
104,243
|
|
|
Other securities
|
|
|
26
|
|
|
|
—
|
|
|
|
—
|
|
|
|
26
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
53,583
|
|
|
$
|
50,686
|
|
|
$
|
—
|
|
|
$
|
104,269
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock warrant liability
|
|
|
—
|
|
|
|
—
|
|
|
|
3,904
|
|
|
|
3,904
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
3,904
|
|
|
$
|
3,904
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Assets:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
82,336
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
82,336
|
|
|
FDIC insured bank CDs
|
|
|
—
|
|
|
|
20,948
|
|
|
|
—
|
|
|
|
20,948
|
|
|
Money market currency funds
|
|
|
—
|
|
|
|
4,800
|
|
|
|
—
|
|
|
|
4,800
|
|
|
U.S. Government securities
|
|
|
—
|
|
|
|
16,542
|
|
|
|
—
|
|
|
|
16,542
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
153
|
|
|
|
—
|
|
|
|
153
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents and marketable securities
|
|
|
82,336
|
|
|
|
42,443
|
|
|
|
—
|
|
|
|
124,779
|
|
|
Other securities
|
|
|
35
|
|
|
|
—
|
|
|
|
—
|
|
|
|
35
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
82,371
|
|
|
$
|
42,443
|
|
|
$
|
—
|
|
|
$
|
124,814
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock warrant liability
|
|
|
—
|
|
|
|
—
|
|
|
|
6,635
|
|
|
|
6,635
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
6,635
|
|
|
$
|
6,635
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
We measure fair value based on the prices that would be received
to sell an asset or paid to transfer a liability in an orderly
transaction between market participants at the measurement date.
Fair value measurements are based on a three-tier hierarchy that
prioritizes the inputs used to measure fair value. These tiers
include the following:
Level 1: Quoted prices (unadjusted) in
active markets for identical assets or liabilities that are
accessible at the measurement date. The fair value hierarchy
gives the highest priority to Level 1 inputs.
Level 2: Observable prices that are based
on inputs not quoted on active markets, but corroborated by
market data. These inputs include quoted prices for similar
assets or liabilities; quoted market prices in markets that are
not
F-10
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
active; or other inputs that are observable or can be
corroborated by observable market data for substantially the
full term of the assets or liabilities.
Level 3: Unobservable inputs are used
when little or no market data is available. The fair value
hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, we utilize valuation techniques that
maximize the use of observable inputs and minimize the use of
unobservable inputs to the extent possible, as well as consider
counterparty credit risk in the assessment of fair value. Cash
equivalents consist of certificates of deposit and are valued at
cost, which approximates fair value due to the short-term
maturities of these instruments. Marketable securities consist
of certificates of deposit, US Government Treasury bills, US
treasury-backed securities and corporate deposits, which are
stated at fair market value, based on values provided us by the
financial institutions where we invest our funds.
We had classified all of our marketable securities as
Level 1 measurements as of December 31, 2009. Based on
the recent guidance on disclosures for fair value measurements
and in order to be consistent with industry practice, as of
December 31, 2010 and 2009, we have reclassified all of our
marketable securities under Level 2 measurements. There
were no other transfers in and out of Level 2 during the
year ended December 31, 2010.
The following summarizes the activity of Level 3 inputs
measured on a recurring basis for the years ended
December 31, 2010 and 2009:
| |
|
|
|
|
|
|
|
Fair Value Measurements of
|
|
|
|
|
Common Stock Warrants
|
|
|
|
|
Using Significant
|
|
|
|
|
Unobservable Inputs (Level 3)
|
|
|
|
|
($ in 000’s)
|
|
|
|
|
Balance at December 31, 2008
|
|
$
|
765
|
|
|
Transfers in / (out) of Level 3
|
|
|
—
|
|
|
Issuance of common stock warrants
|
|
|
14,016
|
|
|
Repurchases or forfeitures
|
|
|
(394
|
)
|
|
Gain on repurchase recognized in earnings
|
|
|
323
|
|
|
Adjustments resulting from change in value of warrants
recognized in earnings
|
|
|
(8,075
|
)
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
6,635
|
|
|
Transfers in / (out) of Level 3
|
|
|
—
|
|
|
Forfeitures
|
|
|
(788
|
)
|
|
Adjustments resulting from change in value of warrants
recognized in earnings
|
|
|
(1,943
|
)
|
|
|
|
|
|
|
|
Balance at December 31, 2010
|
|
$
|
3,904
|
|
|
|
|
|
|
|
The fair value of common stock warrants are measured on their
respective origination dates and at the end of each reporting
period using Level 3 inputs. The significant assumptions we
use in the calculations under the Black-Scholes Option Pricing
Model as of December 31, 2010 and 2009, included an
expected term based on the remaining contractual life of the
warrants, a risk-free interest rate based upon observed interest
rates appropriate for the expected term of the instruments,
volatility based on the historical volatility of our common
stock, and a zero dividend rate based on our past, current and
expected practices of granting dividends on common stock. These
warrants will expire on September 14, 2011 if not exercised.
We did not elect the fair value option, as allowed, for our
financial assets and liabilities that were not previously
carried at fair value. Therefore, material financial assets and
liabilities that are not carried at fair value, such as trade
accounts receivable and payable, are still reported at their
historical carrying values, which approximates fair value due to
their short term nature.
F-11
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Concentration
of credit risk
Our cash investments are subject to concentration of credit
risk, which is managed by diversification of the investment
portfolio and by the purchase of investment-grade securities.
Our product sales are concentrated in a limited number of
customers. Sales to Customer A for the years ended
December 31, 2010, 2009 and 2008 were 45.7%, 27.1%, and
35.7% respectively, of our total consolidated gross product
sales. No other single customer generated over 10% of our
consolidated gross product sales.
We are exposed to risks associated with extending credit to our
customers related to the sale of products. We do not require
collateral or other security to support credit sales, however,
we maintain reserves for potential bad debt and to date, credit
losses have been within management’s expectations. Customer
A owed us 56.1% of net receivables as of December 31, 2010
and no single customer owed us more than 10% at
December 31, 2009. All sales were to customers in the
United States.
We have single source suppliers for raw materials and the
manufacturing of finished product of Zevalin and Fusilev and
have begun the process of qualifying additional contract
manufacturers for Fusilev. We are exposed to loss of revenue
from the sale of these products if the supplier cannot fulfill
demand. We also have single source suppliers for raw materials,
and manufactured finished product for our development drug
candidates. If we are unable to obtain sufficient quantities of
such product, our research and development activities may be
adversely affected.
Inventories
Inventories are valued at the lower of cost
(first-in,
first-out method) or market. The lower of cost or market is
determined based on net estimated realizable value after
appropriate consideration is given to obsolescence, excessive
levels, deterioration, and other factors.
We continually review product inventories on hand, evaluating
inventory levels relative to product demand, remaining shelf
life, future marketing plans and other factors. We adjust our
inventory to reflect situations in which the cost of inventory
is not expected to be recovered. We record a reserve to adjust
inventory to its net realizable value if (i) a product is
close to expiration and not expected to be sold, (ii) when
a project has reached its expiration date or (iii) when a
product is not expected to be saleable. In determining reserves
for these products, we consider factors such as the amount of
inventory on hand and its remaining shelf life, and current and
expected market conditions, including management forecasts and
levels of competition. We have evaluated the current level of
inventory considering historical trends and other factors, and
based on our evaluation, we have recorded adjustments to reflect
inventory at its net realizable value. These adjustments are
estimates, which could vary significantly from actual results if
future economic conditions, customer demand, competition or
other relevant factors differ from expectations. These estimates
require us to make assessments about the future demand for our
products in order to categorize the status of such inventory
items as slow-moving, obsolete or in
excess-of-need.
These future estimates are subject to the ongoing accuracy of
our forecasts of market conditions, industry trends, competition
and other factors. Differences between our estimated reserves
and actual inventory adjustments have not been significant, and
are accounted for in the current period as a change in estimate.
During 2010, expiring inventory reserves of $50,000 were
recorded against cost of goods sold and the total reserve was
$50,000 and $89,000 at December 31, 2010 and 2009,
respectively.
Property
and Equipment
Property and equipment are recorded at cost less accumulated
depreciation. Maintenance and repairs are expensed as incurred.
Upon sale or disposition of assets, any gain or loss is included
in the statements of operations.
F-12
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
The cost of property, plant and equipment is depreciated using
the straight-line method over the following estimated useful
lives of the respective assets. Leasehold improvements are
amortized over the lesser of the estimated useful lives of the
respective assets or the related lease terms.
| |
|
|
|
|
|
Computers and software
|
|
|
3 to 5 years
|
|
|
Office furniture and equipment
|
|
|
5 to 7 years
|
|
|
Lab and media equipment
|
|
|
2 to 7 years
|
|
All long-lived assets, including property and equipment, are
reviewed for impairment whenever events or changes in business
circumstances indicate that the carrying amount of the assets
may not be fully recoverable. If impairment is indicated, we
reduce the carrying value of the asset to fair value. Fair value
would be determined by the use of appraisals, discounted cash
flow analyses or comparable fair values of similar assets.
Patents
and Licenses
We expense all licensing and patent application costs as they
are incurred.
Intangible
Assets
As described in note 3 below, we acquired 50% of the rights
in RIT in December 2008 and the remaining 50% in March 2009.
The purchase price for the acquisition of Zevalin rights was
allocated to identifiable intangible assets acquired and
liabilities assumed based on their estimated fair values at the
acquisition date. Such a valuation requires significant
estimates and assumptions including but not limited to:
determining the timing and expected costs to complete the
in-process projects, projecting regulatory approvals, estimating
future cash flows from product sales resulting from in-process
projects, and developing appropriate discount rates and
probability rates by project. We believe the fair values
assigned to the assets acquired and liabilities assumed are
based on reasonable assumptions.
Identifiable intangible assets with definite lives are amortized
on a straight-line basis over their estimated useful lives,
ranging from 1 to 10 years.
We evaluate the recoverability of indefinite and definite
intangible assets whenever events or changes in circumstances
indicate that an intangible asset’s carrying amount may not
be recoverable. Such circumstances could include, but are not
limited to the following:
(i) a significant decrease in the market value of an asset;
(ii) a significant adverse change in the extent or manner
in which an asset is used; or
(iii) an accumulation of costs significantly in excess of
the amount originally expected for the acquisition of an asset.
We measure the carrying amount of the asset against the
estimated undiscounted future cash flows associated with it.
Should the sum of the expected future net cash flows be less
than the carrying value of the asset being evaluated, an
impairment loss would be recognized. The impairment loss would
be calculated as the amount by which the carrying value of the
asset exceeds its fair value. No impairment loss was recorded
during the years 2010, 2009, or 2008.
Revenue
Recognition
Revenue from product sales is recognized upon shipment of
product when title and risk of loss have transferred to the
customer. We sell our products to wholesalers and distributors
of oncology products and directly to the end user, directly or
through GPOs (e.g., certain hospitals or hospital systems and
clinics with whom we have entered into a direct purchase
agreement). Our wholesalers and distributors purchase our
products and sell the products
F-13
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
directly to end users, which include, but are not limited to,
hospitals, clinics, medical facilities, managed care facilities
and private oncology based practices. Revenue from product sales
is recognized upon shipment of product when title and risk of
loss have transferred to the customer, and the following
additional criteria are met:
(i) the price is substantially fixed and determinable;
(ii) our customer has economic substance apart from that
provided by us;
(iii) our customer’s obligation to pay us is not
contingent on resale of the product;
(iv) we do not have significant obligations for future
performance to directly bring about the resale of our
product; and
(v) we have a reasonable basis to estimate future returns.
Generally, revenue is recognized when all four of the following
criteria are met:
(i) persuasive evidence that an arrangement exists;
(ii) delivery of the products has occurred, or services
have been rendered;
(iii) the selling price is both fixed and
determinable; and
(iv) collectibility is reasonably assured.
Provision for estimated product returns, sales discounts,
rebates, chargebacks and distribution and data fees are
established as a reduction of gross product sales at the time
such revenues are recognized. Thus, revenue is recorded, net of
such estimated provisions.
Shipments of Fusilev for the year ended December 31, 2008
were approximately $10.8 million (net of estimates for
promotional, price and other adjustments). We deferred the
recognition of approximately $3.1 million of such revenue
to allow for potential sales returns. In 2010 and 2009, based on
our evaluation of return history to date combined with inventory
held by distributors, sell through data of distributor sales to
end users, customer and end-user ordering and re-ordering
patterns, aging of accounts receivables, rates of returns for
directly substitutable products and other pharmaceutical
products for the treatment of therapeutic areas similar to
indications served by our products, shelf life of our products
and the extensive experience of our management with selling the
same and similar oncology products, we reserved approximately
$2.0 million and $1.2 million as of December 31,
2010 and 2009, respectively. No returns reserve is recorded for
Zevalin since we invoice our end user customers and recognize
revenues only when a patient is treated with Zevalin.
We also state the related accounts receivable at net realizable
value, with any allowance for doubtful accounts charged to
general operating expenses. If revenue from sales is not
reasonably determinable due to provisions for estimates,
promotional adjustments, price adjustments, returns or any other
potential adjustments, we defer the revenue and recognize
revenue when the estimates are reasonably determinable, even if
the monies for the gross sales have been received.
We utilize a third-party logistics company to store and
distribute Fusilev. The same third party logistics company also
stores and ships Zevalin kits containing the CD20 MAB.
During 2009, we changed the supply and distribution model for
Zevalin. Previously, we sold Zevalin kits containing the CD20
MAB to radiopharmacies, who in turn ordered the radioactive
isotope (Y-90 or In-111) separately and radiolabeled (or
attached) the radioactive isotope to the CD20 MAB. The
radiopharmacy then sold the end user product to the consumer.
Under the current model we do not sell the Zevalin kits
containing the CD20 MAB to the radiopharmacies, but instead
contract with them, as a
fee-for-service,
to radiolabel the individual components of the CD20 MAB to the
radioactive isotope, and then, also under a
fee-for-service
arrangement, have them distribute the end use product to the end
user; the clinics, hospitals or other medical settings. In this
regard, we now sell the CD20 MAB together with the radioactive
isotope as the end user product.
F-14
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Consistent with industry practice, our product return policy
permits our customers to return products within 30 days
after shipment, if incorrectly shipped or not ordered, and
within a window of time 6 months before and 12 months
after the expiration of product dating, subject to certain
restocking fees and preauthorization requirements, as
applicable. The returned product is destroyed if it is damaged,
it’s quality is compromised or it is past its expiration
date. Based on our returns policy, we refund the sales price to
the customer as a credit and record the credit against
receivables. In general, returned product is not resold. We
generally reserve the right to decline granting a return and to
decide on product destruction. As of each balance sheet date, we
estimate potential returns, based on several factors, including:
inventory held by distributors, sell through data of distributor
sales to end users, customer and end-user ordering and
re-ordering patterns, aging of accounts receivables, rates of
returns for directly substitutable products and other
pharmaceutical products for the treatment of therapeutic areas
similar to indications served by our products, shelf life of our
products and the extensive experience of our management with
selling the same and similar oncology products. We record an
allowance for future returns by reducing gross revenues and
increasing the allowance for returns based upon the
Company’s historical patterns of product returns matched
against sales, and management’s evaluation of specific
factors that may influence the risk of product returns. If
allowances exceed the related accounts receivables, we
reclassify such allowances to accrued obligations. Historical
allowances for product returns have been within estimated
amounts reserved or accrued.
Up-front fees representing non-refundable payments received upon
the execution of licensing or other agreements are recognized as
revenue upon execution of the agreements where we have no
significant future performance obligations and collectibility of
the fees is reasonably assured. Milestone payments, which are
generally based on developmental or regulatory events, are
recognized as revenue when the milestones are achieved,
collectibility is reasonably assured, and we have no significant
future performance obligations in connection with the milestone.
In those instances where we have collected fees or milestone
payments but have significant future performance obligations
related to the development of the drug product, we record
deferred revenue and recognize it over the period of our future
obligations.
Research
and Development
Research and development expenses include salaries and benefits,
clinical trial and related manufacturing costs, contract and
other outside service fees, and facilities and overhead costs
related to our research and development efforts. Research and
development expenses also consist of costs incurred for
proprietary and collaborative research and development and
include activities such as product registries and
investigator-sponsored trials. Research and development costs
are expensed as incurred. In certain instances, we enter into
agreements with third parties for research and development
activities, where we may prepay fees for services at the
initiation of the contract. We record such prepayment as a
prepaid asset and charge research and development expense over
the period of time the contracted research and development
services are performed. Other types of arrangements with third
parties may be fixed fee or fee for service, and may include
monthly payments or payments upon the completion of milestones
or receipt of deliverables.
As of each balance sheet date, we review purchase commitments
and accrue drug development expenses based on factors such as
estimates of work performed, patient enrollment, completion of
patient studies and other events. Accrued clinical study costs
are subject to revisions as trials progress to completion.
Revisions are recorded in the period in which the facts that
give rise to the revision become known.
Basic
and Diluted Net (Loss) Per Share
We calculate basic and diluted net income (loss) per share using
the weighted average number of common shares outstanding during
the periods presented, and adjust the amount of net income
(loss) used in this calculation for preferred stock dividends
(if any) declared during the period. In periods of a net loss
position, basic and diluted weighted average shares are the
same. For the diluted earnings per share calculation, we adjust
the weighted average
F-15
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
number of common shares outstanding to include dilutive stock
options, warrants and other common stock equivalents outstanding
during the period.
The following shows the amounts used in computing basic and
diluted loss per share for each of the three years in the period
ended December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
($ in ‘000’s except per share data)
|
|
|
|
|
Net loss — attributable to Spectrum Pharmaceuticals,
Inc. stockholders
|
|
$
|
(48,844
|
)
|
|
$
|
(19,046
|
)
|
|
$
|
(14,196
|
)
|
|
Less:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Preferred dividends paid in cash or stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss attributable to Spectrum stockholders
|
|
$
|
(48,844
|
)
|
|
$
|
(19,046
|
)
|
|
$
|
(14,196
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares issued and outstanding
|
|
|
49,502,854
|
|
|
|
39,273,905
|
|
|
|
31,551,152
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(0.99
|
)
|
|
|
(0.48
|
)
|
|
|
(0.45
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following table sets forth the number of shares excluded
from the computation of diluted earnings per share, as their
inclusion would have been anti-dilutive:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Series E Preferred Shares
|
|
|
52,000
|
|
|
|
136,000
|
|
|
|
136,000
|
|
|
Stock Options
|
|
|
5,157,935
|
|
|
|
4,451,733
|
|
|
|
5,097,835
|
|
|
Warrants
|
|
|
4,142,312
|
|
|
|
8,379,912
|
|
|
|
5,444,555
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9,352,247
|
|
|
|
12,967,645
|
|
|
|
10,678,390
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounting
for Employee Share-Based Compensation
We measure compensation cost for all share-based awards at fair
value on the date of grant and recognize compensation expense in
our consolidated statements of operations over the service
period that the awards are expected to vest. We have elected to
recognize compensation expense for all options with graded
vesting on a straight-line basis over the vesting period of the
entire option.
The fair value of share-based compensation is estimated based on
the closing market price of our common stock on the day prior to
the award grants for stock awards, and the Black-Scholes Option
Pricing Model for stock options and warrants. We estimate
volatility based on historical volatility of our common stock,
and estimate the expected term based on several criteria,
including the vesting period of the grant and the term of the
award.
We recorded share-based employee compensation as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Research and development expense
|
|
$
|
2,484
|
|
|
$
|
3,192
|
|
|
$
|
3,925
|
|
|
Selling, general and administrative expense
|
|
|
5,801
|
|
|
|
4,231
|
|
|
|
2,612
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
8,285
|
|
|
$
|
7,423
|
|
|
$
|
6,537
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-16
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Warrant
accounting
We account for common stock warrants pursuant to the applicable
guidance on accounting for derivative financial instruments
indexed to, and potentially settled in, a company’s own
stock, on the understanding that in compliance with applicable
securities laws, registered warrants require the issuance of
registered securities upon exercise and do not sufficiently
preclude an implied right to net cash settlement. We classify
registered warrants on the consolidated balance sheet as a
current liability, which is revalued at each balance sheet date
subsequent to the initial issuance. Determining the appropriate
fair-value model and calculating the fair value of registered
warrants requires considerable judgment, including estimating
stock price volatility and expected warrant life. We develop our
estimates based on historical data. A small change in the
estimates used may have a relatively large change in the
estimated valuation. We use the Black-Scholes pricing model to
value the registered warrants. Changes in the fair market value
of the warrants are reflected in the consolidated statement of
operations as “Change in the fair value of common stock
warrant liability.”
Income
Taxes
Deferred tax assets and liabilities are recognized for the
future tax consequences attributable to differences between the
financial statement carrying amounts of existing assets and
liabilities and their respective tax bases. Deferred tax assets
and liabilities are measured using enacted tax rates expected to
apply to taxable income in the years in which those temporary
differences are expected to be recovered or settled. The effect
on the deferred tax assets and liabilities of a change in tax
rates is recognized in income in the period that includes the
enactment date.
The Company has determined that the net deferred tax asset does
not meet the “more likely than not” to be realized
criteria and, accordingly, a valuation allowance has been
recorded to reduce the net deferred tax asset to zero.
Acquisitions
and Collaborations
For all in-licensed products, pursuant authoritative guidance,
we perform an analysis to determine whether we hold a variable
interest or interests that give us a controlling financial
interest in a variable interest entity. On the basis of our
interpretations and conclusions, we determine whether the
acquisition falls under the purview of variable interest entity
accounting and if so, consider the necessity to consolidate the
acquisition.
We also perform an analysis to determine if the inputs
and/or
processes acquired in an acquisition qualify as a business. On
the basis of our interpretations and conclusions, we determine
if the in-licensed products qualify as a business and whether to
account for such products as a business combination or an asset
acquisition.
Comprehensive
Income (loss)
Comprehensive income (loss) is calculated in accordance with
authoritative guidance which requires the disclosure of all
components of comprehensive income, including net income (loss)
and changes in equity during a period from transactions and
other events and circumstances generated from non-owner sources.
Our accumulated other comprehensive loss at December 31,
2010 and 2009, respectively consisted primarily of net
unrealized gains/losses on investments in marketable securities
as of that date.
Recent
Accounting Pronouncements
In June 2009, the FASB issued authoritative guidance that
requires companies to perform an analysis to determine whether
such companies’ variable interest or interests give it a
controlling financial interest in a variable interest entity.
This analysis identifies the primary beneficiary of a variable
interest entity as the enterprise that has both the power to
direct the activities of a variable interest entity that most
significantly impact the entity’s economic performance, and
the obligation to absorb losses or the right to receive benefits
of the entity that could potentially be significant to the
variable interest entity. This guidance also requires ongoing
reassessments of
F-17
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
whether an enterprise is the primary beneficiary of a variable
interest entity and eliminates the quantitative approach
previously required for determining the primary beneficiary. We
adopted the provisions of this guidance in the first quarter of
2010, and determined that none of the unconsolidated entities
with which we currently conduct business or collaborations are
variable interest entities to be consolidated.
New
Accounting Standards Not Yet Adopted
In December 2010, the FASB issued an accounting standards update
that provides guidance on the recognition and classification of
the annual fee imposed by the Patient Act and Affordable Care
Act as amended by the Health Care and Education Reconciliation
Act on pharmaceutical companies that manufacture or import
branded prescription drugs. Under this guidance, the annual fee
should be estimated and recognized in full as a liability upon
the first qualifying sale with a corresponding deferred cost
that is amortized to operating expense using a straight-line
method of allocation unless another method better allocates the
fee over the calendar year in which it is payable. The annual
fee ranges from $2.5 billion to $4.1 billion for all
affected entities in total, a portion of which will be allocated
to us on the basis of the amount of our branded prescription
drug sales for the preceding year as a percentage of the
industry’s branded prescription drug sales for the same
period. The annual fee is not deductible for federal income tax
purposes. This guidance will be effective for calendar years
beginning after December 31, 2010, which will be the
Company’s fiscal year 2011. We are currently evaluating the
potential impact of adopting this guidance on our consolidated
financial statements.
In April 2010, the FASB issued an accounting standards update
that provides guidance on the milestone method of revenue
recognition for research and development arrangements. This
guidance allows an entity to make an accounting policy election
to recognize revenue that is contingent upon the achievement of
a substantive milestone in its entirety in the period in which
the milestone is achieved. This guidance is effective for fiscal
years beginning on or after June 15, 2010, which will be
our 2011 fiscal year, and may be applied prospectively to
milestones achieved after the adoption date or retrospectively
for all periods presented, with earlier application permitted.
The Company does not expect that the adoption of the guidance
will have a material impact on the Company’s consolidated
financial statements.
In October 2009, the FASB issued an accounting standards update
that requires an entity to allocate arrangement consideration at
the inception of an arrangement to all of its deliverables based
on their relative selling prices, eliminates the use of the
residual method of allocation, and requires the
relative-selling-price method in all circumstances in which an
entity recognizes revenue of an arrangement with multiple
deliverables. This guidance is effective for revenue
arrangements entered into or materially modified in fiscal years
beginning on or after June 15, 2010, which will be our 2011
fiscal year, with earlier application permitted. We are
currently evaluating the potential impact of adopting this
guidance on our consolidated financial statements.
|
|
|
3.
|
Commercial
and Drug Development Drug Products
|
We currently market two oncology products in the United States,
Zevalin and Fusilev. In addition, we have several products in
clinical development, primarily including apaziquone which has
completed patient enrollment for two Phase 3 clinical trials for
bladder cancer and belinostat is being studied under a Special
Protocol Assessment, or SPA, in a Phase 2 trial for
relapsed or refractory PTCL. The following is a brief
description of our key products as of December 31, 2010.
Zevalin: Zevalin is a prescribed form
of cancer therapy called radioimmunotherapy which combines a
source of radiation, called a radioisotope, with an antibody.
During the year ended December 31, 2010, 2009 and 2008, we
recorded net revenues of $29.0 million, $15.7 million
and $0.3 million, respectively from sales of Zevalin.
In December 2008, we partnered with Cell Therapeutics, Inc., or
CTI, to form a
50-50 owned
joint venture, RIT Oncology, LLC, or RIT, to commercialize and
develop Zevalin, a CD20-directed radiotherapeutic antibody, in
F-18
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
the United States. We paid $15 million for our 50% interest
in RIT. Pursuant to provisions of the 2008 joint-venture
agreement, in March 2009, we acquired the remaining 50%
ownership of RIT for $16.5 million, resulting in RIT
becoming our wholly-owned subsidiary. In April 2009, we disputed
payment of an installment of $3.5 million of the
$16.5 million, on the grounds that CTI’s unpaid
liabilities pertaining to Zevalin, and CTI’s share of joint
venture expenses equaled or exceeded the installment amount. In
May 2009, we received an arbitration award of approximately
$4.3 million. The entire $3.5 million was released to
us and CTI additionally paid us approximately $0.8 million.
The award was final, binding and non-appealable by either party.
The assets contributed by CTI to RIT were all of its interests
in the Zevalin business, which included the U.S. development,
sales and marketing rights to Zevalin as well as other assets
acquired in the December 2007 agreement with Biogen. The assets
acquired included the Zevalin FDA registration, FDA dossier,
U.S. trademark, trade name and trade dress, customer list,
certain patents and the assignment of numerous contracts. There
was no continuity of physical facilities or personnel from the
December 2007 transaction. The assets contributed by CTI to RIT
also included the assets acquired in the June 2008 Access
Agreement with Bayer Schering Pharma AG, which holds the rights
to Zevalin outside of the United States. Under the Access
Agreement, Bayer gave CTI access to data from Bayer’s
Phase 3 first-line indolent trial of Zevalin. Finally, the
assets contributed by CTI to RIT included CTI’s
September 30, 2008 submission of the Zevalin supplemental
Biologics License Application, or sBLA, for use in first-line
consolidation therapy for patients with B-cell follicular NHL.
The joint venture also assumed obligations of $2.2 million
in current liabilities and certain contingent obligations.
The allocation of the initial capitalization of the joint
venture, detailed below, was based on the relative fair values
of the intangible assets acquired, as determined by an
independent valuation consultant, and the obligations assumed by
the joint venture.
| |
|
|
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
|
|
|
Developed technology
|
|
|
|
|
|
$
|
23,100
|
|
|
Core technology
|
|
|
|
|
|
|
14,100
|
|
|
Acquired in-process research and development
|
|
|
|
|
|
|
4,700
|
|
|
Assumed obligation to pay Biogen
|
|
|
|
|
|
|
(2,200
|
)
|
|
Acquisition transaction costs
|
|
|
|
|
|
|
(902
|
)
|
|
Fair value of assumed contingent obligations
|
|
$
|
12,500
|
|
|
|
|
|
|
Less: Limitation based on excess of values of intangibles
acquired over initial capitalization
|
|
|
(1,898
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Maximum amount available
|
|
$
|
10,602
|
|
|
|
|
|
|
Contingent obligations, restricted out of $10,602 as recorded
|
|
|
|
|
|
|
(8,798
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total initial capitalization of joint venture
|
|
|
|
|
|
$
|
30,000
|
|
|
|
|
|
|
|
|
|
|
|
The total fair value of developed and core assets equals
$37.2 million. The developed technology asset relates to
intellectual property and rights thereon related to Zevalin as
approved by the FDA for relapsed or refractory, low-grade or
follicular B-cell NHL. The core technology asset represents the
value of the intellectual property and rights therein expected
to be leveraged in the development of label expansions for
Zevalin. Developed and core technologies are amortized over the
term of the patents related to such technologies. Identifiable
intangible assets with definite lives are amortized on a
straight-line basis over their estimated useful lives. The
developed and core technology assets will be amortized over
10 years, or approximately $3.7 million annually
through 2018. In addition, during 2008 an amount of
$4.7 million of in process research and development, or
IPR&D, for a medical indication still awaiting approval by
the FDA was recorded to operating expenses. Such amount was
completely written off during the year ended December 31,
2008. Amortization expense expected to be recorded over the next
five years is approximately $18.5 million. IPR&D for
RIT was evaluated utilizing the present value of the estimated
after- tax cash flows expected to be generated by purchased
undeveloped technology related to the Zevalin business
F-19
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
or label expansions for indications that have not been approved
by the FDA. Since, at the effective time of the transaction
establishing RIT, the IPR&D had not reached technological
feasibility, such amount was charged to operations for the year
ended December 31, 2008 as of the formation date of RIT.
The March 2009 50% acquisition of the non-controlling interest
in RIT included a premium of $1.8 million, including
certain acquisition related costs, and was charged to additional
paid in capital.
The RIT transaction involved contingent consideration, therefore
we recognized $8.8 million as a Zevalin related contingent
obligation on the balance sheet, which is equal to the excess of
the fair value of the intangible assets over the initial
capitalization, and is less than the approximately
$12.5 million fair value of the contingent consideration,
as determined by the independent valuation consultant. Certain
contingencies were resolved during 2009 and $8.5 million of
the contingent consideration payable was charged to the recorded
amount, which reduced contingent liabilities to approximately
$298,000 at December 31, 2010 and 2009.
In December 2008, the FDA had accepted for filing and review,
and granted priority review status for a sBLA for the use of
Zevalin as part of a first-line therapy for patients with
previously untreated follicular non-Hodgkin’s lymphoma, or
NHL. The sBLA application was approved by the FDA on
September 3, 2009, which now allows the use of Zevalin for
a substantially larger patient population. Zevalin is now FDA
approved and marketed by Spectrum for treatment of patients with
previously untreated follicular NHL who achieve a partial or
complete response to chemotherapy and with relapsed or
refractory, low-grade or follicular B-cell NHL, including
patients who have rituximab-refractory follicular NHL. In
connection with the FDA approval, we paid $8.5 million in
milestone payments. In November 2009, the Centers for
Medicare & Medicaid Services, or CMS finalized a
policy to allow reimbursement for
Zevalin®,
in the Hospital Outpatient Prospective Payment System, based on
the Average Sales Price methodology applicable to other
injectable drugs and biologicals. This reimbursement methodology
went into effect on January 1, 2010.
Fusilev for Injection: Fusilev is the
only commercially available drug containing only the pure active
L-isomer of racemic (L and R forms) leucovorin. Fusilev is
currently indicated after high-dose methotrexate therapy in
patients with osteosarcoma, and to diminish the toxicity and
counteract the effects of impaired methotrexate elimination or
inadvertent overdose of folic acid antagonists.
We commercially launched Fusilev in August 2008 and recorded net
revenues of approximately $32.0 million, $12.5 million
and $7.7 million from Fusilev sales for the years ended
December 31, 2010, 2009 and 2008 respectively.
In April 2006, we acquired all of the oncology drug assets of
Targent, Inc. The principal asset in the transaction was a
license agreement to market Fusilev in the field of oncology in
North America. We paid an up-front fee in common stock, with a
fair market value of approximately $2.7 million, and are
contingently obligated to pay additional amounts based upon
achievement of milestones. At our option, cash payments for
milestones specified in the agreement may be paid in shares of
the Company’s common stock having a value determined as
provided in the asset purchase agreement, equal to the cash
payment amount. In 2009 and 2008, we recorded stock-based
research and development charges of $185,000 and $305,000,
respectively, which represents the fair market value of
125,000 shares of our common stock issued at each of March
2008 and 2009 as milestone payments to Targent, LLC.
Apaziquone: Apaziquone, a synthetic
drug which is activated by certain enzymes present in higher
amounts in cancer cells than in normal tissues, is currently
being developed for non-muscle invasive bladder cancer.
In October 2008, we signed an exclusive development and
commercialization collaboration agreement with Allergan for
apaziquone. Under the terms of the agreement, Allergan paid us
an up-front non-refundable $41.5 million at closing and
will make additional payments of up to $304 million based
on the achievement of certain development, regulatory and
commercialization milestones. We retained exclusive rights to
apaziquone in Asia, including Japan and China. Allergan received
exclusive rights to apaziquone for the treatment of bladder
cancer in the rest of the world, including the United States,
Canada and Europe.
F-20
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
In the United States, we will co-promote apaziquone with
Allergan and share equally in its profits and expenses. Allergan
will also pay us royalties on all of its apaziquone sales
outside of the United States. Under the terms of the agreement,
we will continue to conduct the development program, including
the manufacture of clinical supplies and two Phase 3
registrational trials, and will be jointly responsible for
obtaining regulatory approval for the product. Both parties
share development expenses with Allergan bearing 65% of the
cost. Pursuant to our revenue recognition policy, we will
recognize the up-front payment of $41.5 million over the
period of the development work, estimated at 4 to 5 years.
As of December 31, 2010, and 2009, we have classified
$8.3 million of such amount recorded on the consolidated
balance sheet as current portion of deferred revenue.
In December 2009, we completed enrollment of our two Phase 3
pivotal clinical trials enrolling more than 1,600 patients
with non-muscle invasive bladder cancer. As per the
collaboration agreement with Allergan, Spectrum recorded a
$1.5 million milestone payment from Allergan. Such amount
was received in January 2010.
We also have the right, in our sole discretion, to opt-out of
the co-promotion agreement before January 1, 2012. If we do
so, our share of any future development costs shall be
significantly reduced. Part of the aggregate development costs
and marketing expenses incurred by us since January 1, 2009
shall be reimbursed by Allergan in the form of a one-time
payment. The co-promotion agreement will terminate and instead
of a sharing of profit and expenses, Allergan will pay us
royalties on a percentage of net sales of the apaziquone in the
United States that are slightly greater than the royalties paid
on net sales outside the United States. In addition, Allergan
will pay us up to $245 million in additional milestones
based upon the achievement of certain sales milestones in the
United States.
In October 2008, we terminated our 2001 license agreement for
apaziquone with INC
Research®,
formerly NDDO Research Foundation, or INC, in the Netherlands,
as the patents underlying the agreement were all about to
expire. Pursuant to the termination, INC assigned to us all
rights it had in the know-how or intellectual property licensed
under the agreement and all rights in may have had in any
know-how or intellectual property created during the term of the
agreement. In exchange we paid INC a nominal amount of cash and
issued them a nominal number of shares of our common stock. In
addition, INC is entitled to up to 25,000 additional shares of
our common stock and an additional payment of $300,000 upon
achievement of certain regulatory milestones.
In November 2009, we entered into a collaboration agreement with
Nippon Kayaku Co., LTD. for the development and
commercialization of apaziquone in Asia, except North and South
Korea (the “Nippon Kayaku Territory”). In exchange,
Nippon Kayaku paid Spectrum an up-front payment of
$15.0 million, which was received in January 2010, and
agreed to make additional payments of up to $136.0 million
based on the achievement of certain regulatory and
commercialization milestones. Nippon Kayaku received exclusive
rights to apaziquone for the treatment of non-muscle invasive
bladder cancer in Asia (other than North and South Korea),
including Japan and China. Under the terms of the Nippon Kayaku
collaboration agreement, Nippon Kayaku will conduct the
apaziquone clinical trials pursuant to a development plan. In
addition, Nippon Kayaku will be responsible for all expenses
relating to the development and commercialization of apaziquone
in the Nippon Kayaku Territory.
Also in November 2009, we entered into collaboration agreement
with Handok Pharmaceuticals of Korea for the development and
commercialization of apaziquone for the treatment of non-muscle
invasive bladder cancer in North and South Korea. Under the
terms of the Handok collaboration agreement, Handok paid us an
up-front payment of $1.0 million, which was received in
January 2010, and potential milestone payments totaling
approximately $19 million. The potential milestones will be
based on the achievement of certain regulatory and
commercialization milestones. Additionally, Handok will conduct
the apaziquone clinical trials pursuant to a development plan
and will be responsible for all expenses relating to the
development and commercialization of apaziquone in North and
South Korea.
Belinostat: Belinostat is a histone
deacytelase, or HDAC, inhibitor that is being studied in
multiple clinical trials, both as a single drug and in
combination with chemotherapeutic drugs for the treatment of
various hematological and solid tumors.
The following describes the principal commercial terms relating
to belinostat licensing and development.
F-21
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
In February 2010, we entered into a licensing and collaboration
agreement with TopoTarget, for the development and
commercialization of belinostat, pursuant to which TopoTarget
and the Company agreed to a collaboration for the development
and commercialization of belinostat. The agreement provides that
we have the exclusive right to make, develop and commercialize
belinostat in North America and India, with an option for China.
The agreement also grants TopoTarget a co-promote option if and
only if we do not maintain a minimum number (subject to
adjustment for certain events outside of our control) of field
personnel (as defined in the agreement) for a certain number of
years post-approval of the PTCL indication.
In consideration for the rights granted to us under the license
and collaboration agreement with TopoTarget, we paid TopoTarget
an up-front fee of $30.0 million which is recorded as
research and development expense in the accompanying
consolidated financial statements. In addition, we will pay up
to $313 million and one million shares of Spectrum common
stock based on the achievement of certain development,
regulatory and sales milestones, as well as certain royalties on
net sales of belinostat.
Under the terms of the agreement, all development, including
studies, will be conducted under a joint development plan and in
accordance with a mutually agreed upon target product profile
provided that we have final decision-making authority for all
developmental activities in North America and India (and China
upon exercise of the option for China) and TopoTarget has final
decision-making authority for all developmental activities in
all other jurisdictions. We will assume all responsibility for
and future costs of the ongoing registrational PTCL trial while
TopoTarget will assume all responsibility for and future costs
of the ongoing Phase 2 CUP trial. We and TopoTarget will conduct
future planned clinical trials pursuant to the joint development
plan, of which we will fund 70% of the development costs
and TopoTarget will fund 30% of the development costs.
The Company and TopoTarget will each pay 50% of the costs for
chemical, pharmaceutical and other process development related
to the manufacturing of the product that are incurred with a
mutually agreed upon budget in the joint development plan.
TopoTarget is responsible for supplying us with both clinical
and commercial product.
|
|
|
4.
|
Cash,
Cash Equivalents and Investments
|
Cash, cash equivalents and investments in marketable securities,
including long term bank certificates of deposits, totaled
$104.2 million and $124.8 million as of
December 31, 2010 and 2009, respectively. Long term bank
certificates of deposit include a $500,000 restricted
certificate of deposit that collateralizes tenant
F-22
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
improvement obligations to the lessor of our principal offices.
The following is a summary of such investments (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross
|
|
|
Gross
|
|
|
Estimated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amortized
|
|
|
Unrealized
|
|
|
Unrealized
|
|
|
Fair
|
|
|
|
|
|
Marketable Security
|
|
|
|
|
Cost
|
|
|
Gains
|
|
|
Losses
|
|
|
Value
|
|
|
Cash
|
|
|
Current
|
|
|
Long Term
|
|
|
|
|
December 31, 2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
53,557
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
53,557
|
|
|
$
|
53,557
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Bank CDs (including restricted certificate of deposit of $500)
|
|
|
29,985
|
|
|
|
—
|
|
|
|
—
|
|
|
|
29,985
|
|
|
|
—
|
|
|
|
21,416
|
|
|
|
8,569
|
|
|
Money market currency funds
|
|
|
15,488
|
|
|
|
—
|
|
|
|
—
|
|
|
|
15,488
|
|
|
|
—
|
|
|
|
15,488
|
|
|
|
—
|
|
|
U.S. Government securities
|
|
|
2,909
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,909
|
|
|
|
—
|
|
|
|
2,909
|
|
|
|
—
|
|
|
Corporate debt securities
|
|
|
2,304
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,304
|
|
|
|
—
|
|
|
|
2,304
|
|
|
|
—
|
|
|
Other securities (included in other assets)
|
|
|
35
|
|
|
|
—
|
|
|
|
9
|
|
|
|
26
|
|
|
|
—
|
|
|
|
—
|
|
|
|
26
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total investments
|
|
$
|
104,278
|
|
|
$
|
—
|
|
|
$
|
9
|
|
|
$
|
104,269
|
|
|
$
|
53,557
|
|
|
$
|
42,117
|
|
|
$
|
8,595
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
82,336
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
82,336
|
|
|
$
|
82,336
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Bank CDs
|
|
|
20,948
|
|
|
|
—
|
|
|
|
—
|
|
|
|
20,948
|
|
|
|
—
|
|
|
|
12,260
|
|
|
|
8,688
|
|
|
Money market currency funds
|
|
|
4,800
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,800
|
|
|
|
—
|
|
|
|
4,800
|
|
|
|
—
|
|
|
U.S. Government securities
|
|
|
16,542
|
|
|
|
—
|
|
|
|
—
|
|
|
|
16,542
|
|
|
|
—
|
|
|
|
13,792
|
|
|
|
2,750
|
|
|
Corporate debt securities
|
|
|
153
|
|
|
|
—
|
|
|
|
—
|
|
|
|
153
|
|
|
|
—
|
|
|
|
153
|
|
|
|
—
|
|
|
Other securities (included in other assets)
|
|
|
47
|
|
|
|
—
|
|
|
|
12
|
|
|
|
35
|
|
|
|
—
|
|
|
|
—
|
|
|
|
35
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total investments
|
|
$
|
124,826
|
|
|
$
|
—
|
|
|
$
|
12
|
|
|
$
|
124,814
|
|
|
$
|
82,336
|
|
|
$
|
31,005
|
|
|
$
|
11,473
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inventories consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Raw materials
|
|
$
|
962
|
|
|
$
|
280
|
|
|
Finished goods
|
|
|
3,272
|
|
|
|
2,950
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
4,234
|
|
|
$
|
3,230
|
|
|
|
|
|
|
|
|
|
|
|
F-23
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
|
|
|
6.
|
Property
and Equipment
|
Property and equipment consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Computers and software
|
|
$
|
1,299
|
|
|
$
|
1,195
|
|
|
Lab and media equipment
|
|
|
892
|
|
|
|
724
|
|
|
Office furniture and equipment
|
|
|
889
|
|
|
|
697
|
|
|
Leasehold improvements
|
|
|
2,742
|
|
|
|
1,255
|
|
|
Assets held under capital lease obligations
|
|
|
146
|
|
|
|
146
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5,968
|
|
|
|
4,017
|
|
|
Less accumulated depreciation and amortization
|
|
|
(2,810
|
)
|
|
|
(2,089
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
3,158
|
|
|
$
|
1,928
|
|
|
|
|
|
|
|
|
|
|
|
Included in accumulated depreciation and amortization is $88,000
and $29,000 of amortization related to assets held under capital
lease obligations at December 31, 2010 and 2009,
respectively.
|
|
|
7.
|
Accounts
Payable and Other Accrued Obligations
|
Accounts payable and other accrued obligations consisted of the
following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Trade payables
|
|
$
|
8,734
|
|
|
$
|
5,611
|
|
|
Allowance for rebates
|
|
|
14,474
|
|
|
|
388
|
|
|
Accrued product royalty
|
|
|
4,026
|
|
|
|
1,911
|
|
|
Allowance for returns
|
|
|
2,000
|
|
|
|
1,176
|
|
|
Accrued data and distribution fees
|
|
|
1,874
|
|
|
|
213
|
|
|
Allowance for chargebacks
|
|
|
350
|
|
|
|
851
|
|
|
Other accrued obligations
|
|
|
7,246
|
|
|
|
6,456
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
38,704
|
|
|
$
|
16,606
|
|
|
|
|
|
|
|
|
|
|
|
F-24
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
For the periods ended December 31, 2010 and 2009, the
following is a roll forward of the provisions for product
returns, rebates, data and distribution fees and chargeback
allowances (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Data and
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Distribution
|
|
|
|
|
Chargebacks
|
|
|
Rebates
|
|
|
Returns
|
|
|
Fees
|
|
|
|
|
Period ended December 31, 2010:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at beginning of the period
|
|
$
|
851
|
|
|
|
388
|
|
|
$
|
1,176
|
|
|
$
|
213
|
|
|
Add provisions:
|
|
|
862
|
|
|
|
14,721
|
|
|
|
3,540
|
|
|
|
3,029
|
|
|
Less: Credits or actual allowances:
|
|
|
(1,363
|
)
|
|
|
(635
|
)
|
|
|
(2,716
|
)
|
|
|
(1,368
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at the close of the period
|
|
$
|
350
|
|
|
$
|
14,474
|
|
|
$
|
2,000
|
|
|
$
|
1,874
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period ended December 31, 2009:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at beginning of period
|
|
$
|
1,439
|
|
|
$
|
—
|
|
|
$
|
3,144
|
|
|
$
|
—
|
|
|
Add provisions:
|
|
|
3,454
|
|
|
|
469
|
|
|
|
95
|
|
|
|
1,212
|
|
|
Less: Credits or actual allowances:
|
|
|
(4,042
|
)
|
|
|
(81
|
)
|
|
|
(2,063
|
)
|
|
|
(999
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances at the close of the period
|
|
$
|
851
|
|
|
$
|
388
|
|
|
$
|
1,176
|
|
|
$
|
213
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Significant components of the provision for income taxes consist
of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Current:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal
|
|
$
|
(128
|
)
|
|
$
|
78
|
|
|
|
—
|
|
|
State
|
|
|
82
|
|
|
|
343
|
|
|
$
|
5
|
|
|
Foreign
|
|
|
3
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
(43
|
)
|
|
$
|
421
|
|
|
$
|
5
|
|
|
Deferred:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
State
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Foreign
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
(43
|
)
|
|
$
|
421
|
|
|
$
|
5
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-25
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
The income tax provision differs from that computed using the
federal statutory rate applied to income before taxes as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Tax benefit computed at the federal statutory rate
|
|
$
|
(16,658
|
)
|
|
$
|
(6,697
|
)
|
|
$
|
(6,086
|
)
|
|
State tax, net of federal benefit
|
|
|
(2,082
|
)
|
|
|
(981
|
)
|
|
|
(1,039
|
)
|
|
Expired tax attributes
|
|
|
32,236
|
|
|
|
8,097
|
|
|
|
—
|
|
|
Credits
|
|
|
(406
|
)
|
|
|
(1,644
|
)
|
|
|
—
|
|
|
Common stock warrant liability
|
|
|
(928
|
)
|
|
|
(2,745
|
)
|
|
|
(432
|
)
|
|
Stock based compensation
|
|
|
1,397
|
|
|
|
1,533
|
|
|
|
—
|
|
|
Permanent items and other
|
|
|
1,548
|
|
|
|
(737
|
)
|
|
|
—
|
|
|
Valuation allowance
|
|
|
(15,150
|
)
|
|
|
3,595
|
|
|
|
7,562
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income tax provision
|
|
$
|
(43
|
)
|
|
$
|
421
|
|
|
$
|
5
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Significant components of the Company’s deferred tax assets
as of December 31, 2010 and 2009 are shown below. A
valuation allowance has been recognized to offset the net
deferred tax assets as realization of such deferred tax assets
has not met the more likely than not threshold.
| |
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Deferred tax assets:
|
|
|
|
|
|
|
|
|
|
Net operating loss carry forwards
|
|
$
|
36,279
|
|
|
$
|
58,597
|
|
|
Research credits
|
|
|
7,044
|
|
|
|
10,230
|
|
|
Stock based compensation
|
|
|
2,535
|
|
|
|
2,641
|
|
|
Deferred revenue
|
|
|
9,611
|
|
|
|
12,839
|
|
|
Depreciation and amortization differences
|
|
|
14,451
|
|
|
|
1,466
|
|
|
Other, net
|
|
|
1,914
|
|
|
|
1,211
|
|
|
Valuation allowance
|
|
|
(71,834
|
)
|
|
|
(86,984
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
At December 31, 2010, the Company has federal and state net
operating loss carry forwards of approximately $92 million
and $81 million, respectively. The Company has
approximately $1.9 million of foreign loss carry forwards
that begin to expire in 2011. The federal and state loss carry
forwards begin to expire in 2018 and 2016, respectively, unless
previously utilized. At December 31, 2010, the Company has
federal and state research and development tax credits of
approximately $6.7 million and $1.5 million,
respectively. The federal research tax credit begins to expire
in 2023 unless previously utilized. The state research and
development credits have an indefinite carryover period.
The utilization of the net operating loss and research and
development tax credit carry forwards is subject to an annual
limitation under Sections 382 and 383 of the Internal
Revenue Code of 1986, and similar state tax provisions due to
the amount of the net operating loss and research and
development tax credits carry forwards and other deferred tax
assets that can be utilized to offset future taxable income and
tax, respectively.
The Company completed its most recent Section 382 study in
December of 2010. As a result of the ownership changes from that
study, we removed approximately $27.9 million of deferred
tax assets relating to net operating losses and approximately
$4.4 million of deferred tax assets related to research and
development credits from the table of deferred taxes presented
above as these deferred tax assets are expected to expire
unutilized. The Company
F-26
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
will continue to monitor for additional ownership changes as
another change could result in additional net operating losses
and credits expiring unutilized in the future.
In July 2006, the Financial Accounting Standards Board, or FASB,
issued authoritative guidance for the accounting for uncertainty
in income taxes, which prescribes a recognition threshold and
measurement process for recording in the financial statements
uncertain tax positions taken or expected to be taken in a tax
return. Additionally, the authoritative guidance addresses the
derecognition, classification, accounting in interim periods and
disclosure requirements for uncertain tax positions. Only tax
positions that meet the more likely than not recognition
threshold at the effective date may be recognized.
The following table summarizes activity related to our gross
unrecognized tax benefits:
| |
|
|
|
|
|
|
|
2010
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Balance at Beginning of year
|
|
|
—
|
|
|
Adjustments related to prior year tax positions
|
|
|
1,310
|
|
|
Increases related to current year tax positions
|
|
|
387
|
|
|
Decreases due to settlements
|
|
|
—
|
|
|
Decreases due to IRC Section 382 limitation
|
|
|
—
|
|
|
|
|
|
|
|
|
Balance at end of year
|
|
|
1,697
|
|
|
|
|
|
|
|
There were no unrecognized tax benefits as of December 31,
2009 and 2008.
During 2010, the Company continues to believe that its tax
positions meet the more likely than not standard required under
the recognition phase of the authoritative guidance. However, it
considers the amounts and probabilities of the outcomes that can
be realized upon ultimate settlement with the tax authorities
and determined unrecognized tax benefits primarily related to
credits should be established as noted in the summary
rollforward above.
Approximately $42,000 of the total unrecognized tax benefits as
of December 31, 2010, would reduce our annual effective tax
rate if recognized. Additional amounts in the summary
rollforward could impact our effective tax rate if we did not
maintain a full valuation allowance on our net deferred tax
assets.
We do not expect our unrecognized tax benefits to change
significantly over the next 12 months. With a few
exceptions, the Company is no longer subject to
U.S. federal, state and local income tax examinations for
years before 2006. The Company’s policy is to recognize
interest
and/or
penalties related to unrecognized tax benefits in income tax
expense in the consolidated statements of operations.
On November 1, 2010 we received notice that we were awarded
grants totaling $978,000 under the Qualifying Therapeutic
Discovery Project, or QTDP, Program administered under
section 48D of the Internal Revenue Code, of which we
recorded $978,000 in December 2010 as other income in the
consolidated financial statements. The QTDP tax credit is
provided under new section 48D of the Internal Revenue
Code, enacted as part of the Patient Protection and Affordable
Care Act of 2010 (P.L.
111-148).
|
|
|
9.
|
Commitments
and Contingencies
|
Facility
and Equipment Leases
We sublease our principal executive office in Henderson, Nevada
under a non cancelable operating lease expiring April 30,
2014. We also lease our research and development facility in
Irvine, California under a non cancelable operating lease
expiring June 30, 2016. The lease agreement contains
certain scheduled rent increases which are accounted for on a
straight-line basis.
As part of our Irvine facility lease renewal in 2009, the
landlord agreed to contribute up to approximately
$1.5 million toward the cost of tenant improvements. The
tenant improvements were completed in the second
F-27
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
quarter of 2010 at an aggregate cost of approximately
$1.4 million, of which, $451,000 is being financed. This
landlord contribution is being amortized on a straight-line
basis over the term of the lease as a reduction to rent expense.
The Company has acquired certain furniture, fixtures and
equipment under capital leases that are payable in various
scheduled monthly installments through December 2012.
Future minimum lease payments are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
Operating
|
|
|
Capital
|
|
|
|
|
Leases
|
|
|
Leases
|
|
|
|
|
($ in ‘000’s)
|
|
|
|
|
Year ending December 31,
|
|
|
|
|
|
|
|
|
|
2011
|
|
$
|
568
|
|
|
$
|
45
|
|
|
2012
|
|
|
601
|
|
|
|
45
|
|
|
2013
|
|
|
632
|
|
|
|
—
|
|
|
2014
|
|
|
582
|
|
|
|
—
|
|
|
2015
|
|
|
570
|
|
|
|
—
|
|
|
Thereafter
|
|
|
292
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
3,245
|
|
|
$
|
90
|
|
|
|
|
|
|
|
|
|
|
|
Rent expense for the years ended December 31, 2010, 2009
and 2008 was approximately $640,000, $593,000, and $583,000,
respectively.
Licensing
Agreements
Almost all of our drug candidates are being developed pursuant
to license agreements that provide us with rights to certain
territories to, among other things, develop, sublicense, and
sell the drugs. We are required to use commercially reasonable
efforts to develop the drugs, are generally responsible for all
development, patent filing and maintenance costs, sales,
marketing and liability insurance costs, and are generally
contingently obligated to make milestone payments to the
licensors if we successfully reach development and regulatory
milestones specified in the agreements. In addition, we are
obligated to pay royalties and, in some cases, milestone
payments based on net sales, if any, after marketing approval is
obtained from regulatory authorities.
The potential contingent development and regulatory milestone
obligations under all our licensing agreements are generally
tied to progress through the FDA approval process, which
approval significantly depends on positive clinical trial
results. The following represents typical milestone events for
the Company: conclusion of Phase 2 or commencement of Phase 3
clinical trials; filing of new drug applications in each of the
United States, Europe and Asia; and approvals from each of the
regulatory agencies in those jurisdictions.
Given the uncertainty of the drug development and regulatory
approval process, we are unable to predict with any certainty
when any of the milestones will occur, if at all. Accordingly,
the milestone payments represent contingent obligations that
will be recorded as expense when the milestone is achieved.
While it is difficult to predict when milestones will be
achieved, we estimate that if all of our contingent milestones
are successfully achieved within our anticipated timelines, our
potential contingent cash development and regulatory milestone
obligations, are $204 million as of December 31, 2010.
In connection with the development of certain in-licensed drug
products, we anticipate the occurrence of certain of these
milestones during 2011. Upon successful achievement of these
milestones, we will likely become obligated to pay up to
approximately $6.1 million during 2011, payable in cash or
stock at our discretion.
F-28
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Service
Agreements
In connection with the research and development of our drug
products, we have entered into contracts with numerous third
party service providers, such as radio-pharmacies, distributors,
clinical trial centers, clinical research organizations, data
monitoring centers, and with drug formulation, development and
testing laboratories. The financial terms of these agreements
are varied and generally obligate us to pay in stages, depending
on achievement of certain events specified in the agreements,
such as contract execution, reservation of service or production
capacity, actual performance of service, or the successful
accrual and dosing of patients.
At each period end, we accrue for all costs of goods and
services received, with such accruals based on factors such as
estimates of work performed, patient enrollment, completion of
patient studies and other events. We are in a position to
accelerate, slow-down or discontinue any or all of the projects
that we are working on at any given point in time. Should we
decide to discontinue
and/or
slow-down the work on any project, the associated costs for
those projects would be limited to the extent of the work
completed. Generally, we are able to terminate these contracts
due to the discontinuance of the related project(s) and thus
avoid paying for the services that have not yet been rendered
and our future purchase obligations would reduce accordingly.
Supply
Agreements
In connection with our acquisition of Zevalin, RIT assumed a
supply agreement with Biogen Idec Inc., or Biogen, to
manufacture Zevalin for sale in the United States pursuant to
which we would purchase from Biogen, and Biogen would provide to
us, kits to make Zevalin doses for sale to end-users in the
United States at a “cost plus” manufacturing price.
In 2010, we have a single source API supplier as well as a
single source finished product manufacturer for Fusilev. In
2011, we began qualifying additional contract manufacturers.
Employment
Agreement
We have entered into an employment agreement with
Dr. Rajesh C. Shrotriya, our President and Chief Executive
Officer, which expires January 2, 2012. The employment
agreement automatically renews for subsequent one-year calendar
term unless either party gives written notice of such
party’s intent not to renew the agreement at least
90 days prior to the commencement of the new term. The
employment agreement requires Dr. Shrotriya to devote his
full working time and effort to our business and affairs during
the term of the agreement. The employment agreement provides for
a minimum annual base salary with annual increases, periodic
bonuses and option grants as determined by the Compensation
Committee of our Board of Directors.
Litigation
We are involved with various legal matters arising in the
ordinary course of our business. Although the ultimate
resolution of these various matters cannot be determined at this
time, we do not believe that such matters, individually or in
the aggregate, will have a material adverse effect on our
consolidated results of operations, cash flows or financial
condition.
Authorized
Stock
On July 6, 2006, our stockholders approved an amendment to
our Certificate of Incorporation to increase the authorized
number of shares of our common stock from 50,000,000 shares
to 100,000,000 shares. The amendment was filed with the
Delaware Secretary of State on July 7, 2006. Further, on
July 7, 2006, we amended the Certificate of Designation of
Rights, Preferences and Privileges of Series B Junior
Participating Preferred Stock filed with the
F-29
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Delaware Secretary of State on December 18, 2000 to
increase the authorized number of Series B Junior
Participating Preferred Stock from 200,000 shares to
1,000,000 shares.
Preferred
Stock
Stockholder
Rights Agreement
In December 2000, we adopted a stockholder rights agreement
pursuant to which we distributed rights to purchase units of our
Series B Junior Participating Preferred Stock
(“Series B Preferred Stock”). On
November 29, 2010 our Board of Directors approved a
replacement rights agreement, effective December 13, 2010,
that replaced the stockholder rights agreement which was
originally was adopted in 2000 and expired on December 13,
2010. The new replacement rights agreement will extend until
December 13, 2020 the framework of the expired rights plan.
A stockholder rights agreement is designed to deter coercive,
unfair, or inadequate takeovers and other abusive tactics that
might be used in an attempt to gain control of Spectrum
Pharmaceuticals without paying all stockholders a fair price for
their shares. A stockholder rights agreement will not prevent
takeovers at a full and fair price, but rather is designed to
deter coercive takeover tactics and to encourage anyone
attempting to acquire Spectrum Pharmaceuticals to first
negotiate with the Board of Directors.
Under the terms of the new stockholder rights plan, the rights
become exercisable upon the earlier of ten days after a person
or group of affiliated or associated persons has acquired 15% or
more of the outstanding shares of our common stock or ten
business days after a tender offer has commenced that would
result in a person or group beneficially owning 15% or more of
our outstanding common stock. These rights could delay or
discourage someone from acquiring our business, even if doing so
would benefit our stockholders. We currently have no
stockholders who own 15% or more of the outstanding shares of
our common stock. Five days after the rights become exercisable,
each right, other than rights held by the person or group of
affiliated persons whose acquisition of more than 15% of our
outstanding common stock caused the rights to become
exercisable, will entitle its holder to buy, in lieu of shares
of Series B Preferred Stock, a number of shares of our
common stock having a market value of twice the exercise price
of the rights. After the rights become exercisable, if we are a
party to certain merger or business combination transactions or
transfers 50% or more of our assets or earnings power (as
defined), each right will entitle its holder to buy a number of
shares of common stock of the acquiring or surviving entity
having a market value of twice the exercise price of the right.
Series E
Preferred Stock
In September 2003, we received gross cash proceeds of
$20,000,000 in exchange for the issuance of 2,000 shares of
our Series E Convertible Voting Preferred Stock, or the
Series E Preferred Stock, convertible into
4,000,000 shares of common stock, and warrants, exercisable
for five years, to purchase up to a total of
2,800,000 shares of our common stock at an exercise price
of $6.50 per share. As of December 31, 2010 and 2009, 26
and 68 shares of Series E Preferred Stock, convertible
into 52,000 and 136,000 shares of common stock,
respectively, were outstanding. No dividends are payable on the
Series E Preferred Stock. Pursuant to certain provisions of
the Certificate of Designation, Rights and Preferences of the
Series E Preferred Stock, we have the option to redeem all
of the unconverted Series E Preferred Stock outstanding at
the end of a
20-day
trading period if, among other things, in that period the common
stock of the Company trades above $12.00 per share.
In the event of any voluntary or involuntary liquidation,
dissolution or winding up of the Corporation, before any
distribution of assets of the Corporation shall be made to the
common stockholders, the holders of the Series E Preferred
Stock shall be entitled to receive a liquidation preference in
an amount equal to 120% of the stated value per share plus any
declared and unpaid dividends thereon.
F-30
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Common
Stock Issuances for Cash
On May 6, 2009, we sold an aggregate of 432,200 shares
of common stock to certain of our employees at a purchase price
of $2.70 per share, which was the closing price of our common
stock on May 6, 2009. This offering resulted in gross
proceeds to us of approximately $1.2 million. The investors
in this offering included Dr. Rajesh Shrotriya, M.D.,
our Chairman, President and Chief Executive Officer, and Shyam
Kumaria, our Vice President of Finance. Dr. Shrotriya
purchased 290,000 shares of common stock and
Mr. Kumaria purchased 85,000 shares of common stock.
We decided to conduct this offering with certain of our
employees to allow such employees to invest their personal cash
directly into the Company at the current fair market value of
our stock. The purchase agreements include provisions
prohibiting the investors from disposing of the shares of common
stock purchased in the offering for ninety days. The offering
was approved by the Placement Committee of the Board of
Directors. In addition, the Audit Committee of the Board of
Directors approved the offering pursuant to our Related Party
Transaction Policies and Procedures.
On May 26, 2009, we sold 3,913,895 shares of our
common stock at a purchase price of $5.11 per share for net cash
proceeds of approximately $19 million, after placement
agent fees and other offering costs of approximately
$1 million. In connection with this offering, 1,956,947
common stock warrants exercisable at $5.11 between November 27,
2009 and February 25, 2010, were issued to the investors.
All warrants expired unexercised as of February 25, 2010.
On June 15, 2009, we sold 1,715,266 shares of our
common stock at a purchase price of $5.83 per share for net cash
proceeds of approximately $9.5 million, after placement
agent fees and other offering costs of approximately
$0.5 million. In connection with this offering, 857,633
common stock warrants exercisable at $5.83 between
December 15, 2009 and March 15, 2010, were issued to
the investors. All warrants expired unexercised as of
March 15, 2010.
On June 30, 2009, we sold 2,936,037 shares of our
common stock at a purchase price of $7.15 per share for net cash
proceeds of approximately $20 million, after placement
agent fees and other offering costs of approximately
$1 million. In connection with this offering, 1,468,020
common stock warrants exercisable at $7.10 between
December 30, 2009 and March 30, 2010, were issued to
the investors. All warrants expired unexercised as of
March 30, 2010.
On September 18, 2009, we sold 6,622,517 shares of our
common stock at a purchase price of $7.55 per share for net cash
proceeds of approximately $47.5 million, after placement
agent fees and other offering costs of approximately
$2.5 million. In connection with this offering, 2,649,007
common stock warrants exercisable at $7.55 between
March 22, 2010 and June 21, 2010, were issued to the
investors. All warrants expired unexercised as of June 21,
2010.
Other
Equity Transactions
Pursuant to the terms of the April 2006 asset purchase agreement
with Targent, LLC, upon achievement of certain regulatory and
sales milestones Targent is eligible to receive payments in the
form of shares of the Company’s common stock
and/or cash.
At our option, cash payments specified in the agreement may be
paid in shares of the Company’s common stock having a value
determined as provided in the asset purchase agreement, equal to
the cash payment amount. During the three years ended
December 31, 2010, we issued shares of common stock, for
achievement of certain regulatory milestones, as follows. The
fair value of the issued stock was recorded as stock-based
research and development expense for the period in which the
milestone was achieved:
|
|
|
| |
•
|
March 2008: 125,000 shares with a fair value of $305,000.
|
| |
| |
•
|
March 2009: 125,000 shares with a fair value of $185,000.
|
F-31
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
In October 2008, we issued 75,000 shares of the
Company’s common stock in connection with the assignment to
us of certain intellectual property rights related to apaziqone.
The fair value of the stock, $74,000, was recorded as a
stock-based research and development expense for the year ended
December 31, 2008.
In August 2009, we acquired 100% of the rights to
RenaZorb®
and
Renalin®,
lanthanum-based nanotechnology compounds with potent and
selective phosphate binding properties, for all uses pursuant to
an amended and restated agreement that we entered into with
Altair Nanomaterials, Inc. and Altair Nanotechnologies. In 2005,
the Company had acquired the worldwide license from Altair to
develop and commercialize Altair’s lanthanum-based
nanotechnology compounds and related technology for all human
therapeutic uses. The August 2009 acquisition expanded the
worldwide, exclusive license to include all uses. In conjunction
with the expanded license, Altair assigned all intellectual
property associated with
RenaZorb®
(associated with human uses),
Renalin®
(associated with animal or veterinarian use), its
lanthanum-based nanotechnology and all of its other life
sciences research and development to us. In consideration, we
issued 113,809 shares of our common stock, with a then fair
value of approximately $750,000.
In July 2010, we issued 425,000 shares of our common stock
in exchange for certain intellectual property and drug assets.
The fair market value of the stock, approximately
$1.7 million, was charged to research and development
during the third quarter of 2010. In December 2010, we issued
326,956 shares of our common stock in exchange for
intellectual property and drug assets. The fair market value of
the stock, approximately $1.4 million, was charged to
research and development during the fourth quarter of 2010.
Common
Stock Reserved for Future Issuance
As of December 31, 2010, approximately 12.6 million
shares of common stock were issuable upon conversion or exercise
of rights granted under prior financing arrangements, stock
options and warrants, as follows:
| |
|
|
|
|
|
Conversion of Series E preferred shares
|
|
|
52,000
|
|
|
Exercise of stock options
|
|
|
8,397,094
|
|
|
Exercise of warrants
|
|
|
4,192,312
|
|
|
|
|
|
|
|
|
Total shares of common stock reserved for future issuances
|
|
|
12,641,406
|
|
|
|
|
|
|
|
Of the warrants outstanding at December 31, 2010, 3,747,312
warrants relate to 2005 issuances which expire in September 2011
if not exercised.
F-32
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Warrant
Activity
We typically issue warrants to purchase shares of our common
stock to investors as part of a financing transaction or in
connection with services rendered by placement agents and
consultants. Our outstanding warrants expire on varying dates
through December 2015. A summary of warrant activity is as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
Number of
|
|
|
Average
|
|
|
|
|
Shares
|
|
|
Exercise Price
|
|
|
|
|
Outstanding — December 31, 2007
|
|
|
9,652,051
|
|
|
$
|
6.51
|
|
|
Issued
|
|
|
50,000
|
|
|
|
1.79
|
|
|
Forfeited
|
|
|
(157,450
|
)
|
|
|
6.62
|
|
|
Expired
|
|
|
(4,100,046
|
)
|
|
|
5.43
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2008
|
|
|
5,444,555
|
|
|
|
7.28
|
|
|
Issued
|
|
|
6,931,607
|
|
|
|
6.55
|
|
|
Repurchased
|
|
|
(95,238
|
)
|
|
|
6.62
|
|
|
Expired
|
|
|
(1,252,005
|
)
|
|
|
10.03
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2009
|
|
|
11,028,919
|
|
|
|
6.52
|
|
|
Issued
|
|
|
275,000
|
|
|
|
5.59
|
|
|
Forfeited
|
|
|
(180,000
|
)
|
|
|
5.16
|
|
|
Expired
|
|
|
(6,931,607
|
)
|
|
|
6.55
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2010
|
|
|
4,192,312
|
|
|
$
|
6.45
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable — December 31, 2010
|
|
|
4,142,312
|
|
|
$
|
6.48
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes information with respect to
warrants outstanding and exercisable at December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
Weighted-
|
|
|
|
|
|
Weighted-
|
|
|
|
|
Number
|
|
|
Contractual
|
|
|
Average
|
|
|
Number
|
|
|
Average
|
|
|
Exercise Price
|
|
Outstanding
|
|
|
Life (Years)
|
|
|
Exercise Price
|
|
|
Exercisable
|
|
|
Exercise Price
|
|
|
|
|
$1.00 - 2.50
|
|
|
50,000
|
|
|
|
2.3
|
|
|
$
|
1.79
|
|
|
|
50,000
|
|
|
$
|
1.79
|
|
|
$2.51 - 5.00
|
|
|
75,000
|
|
|
|
4.5
|
|
|
$
|
3.82
|
|
|
|
25,000
|
|
|
$
|
3.82
|
|
|
$5.01 - 6.00
|
|
|
120,000
|
|
|
|
2.7
|
|
|
$
|
5.13
|
|
|
|
120,000
|
|
|
$
|
5.13
|
|
|
$6.01 - 7.00
|
|
|
3,947,312
|
|
|
|
0.9
|
|
|
$
|
6.60
|
|
|
|
3,947,312
|
|
|
$
|
6.60
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,192,312
|
|
|
|
|
|
|
|
|
|
|
|
4,142,312
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Of the warrants outstanding at December 31, 2010, 3,747,312
warrants relate to 2005 issuances which expire in September 2011
if not exercised.
|
|
|
11.
|
Share-Based
Compensation
|
Stock
Options
We have three stock incentive plans: the 1997 Stock Incentive
Plan, or the 1997 Plan, the 2003 Amended and Restated Incentive
Award Plan, or the 2003 Plan and the 2009 Incentive Award Plan,
or the 2009 Plan which was approved by our stockholders in June
2009 (collectively, the “Plans”). The 2003 Plan
authorizes the grant of incentive awards, including stock
options, for the purchase of up to a total of
10,000,000 shares. Subsequent to the adoption of the 2009
Plan, no new options have been granted pursuant the 2003 Plan or
1997 Plan. The Board and
F-33
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
the stockholders approved 10,000,000 shares of common stock
available for issuance under the 2009 Plan. Beginning on
January 1, 2010, and each January 1st thereafter,
the number of shares of common stock available for issuance
under the 2009 Plan shall increase by the greater of
(i) 2,500,000 and (ii) a number of shares such that
the total number of shares of common stock available for
issuance under the Plan shall equal 30% of the then number of
shares of common stock issued and outstanding. As of
December 31, 2010, approximately 10.8 million
incentive awards were available for grant under the 2009 Plan.
During each of the three years in the period ended
December 31, 2010, we granted stock options at exercise
prices equal to or greater than the quoted market price of our
common stock on the grant date. The fair value of each option
grant was estimated on the date of grant using the Black-Scholes
option pricing model with the following weighted average
assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
Expected life (years)
|
|
4.93
|
|
|
5.0
|
|
|
|
5.0
|
|
|
Risk-free interest rate
|
|
1.04% - 2.75%
|
|
|
2.27
|
%
|
|
|
2.66
|
%
|
|
Volatility
|
|
70.7%
|
|
|
72.4
|
%
|
|
|
65.9
|
%
|
|
Dividend yield
|
|
0%
|
|
|
0
|
%
|
|
|
0
|
%
|
The expected option life assumption is estimated based on actual
historical exercise activity and assumptions regarding future
exercise activity of unexercised, outstanding options. The
risk-free interest rate assumption is based upon observed
interest rates appropriate for the expected term of the
Company’s employee stock options. The expected volatility
is based on the historical volatility of the Company’s
stock. The Company has not paid any dividends on common stock
since its inception and does not anticipate paying dividends on
its common stock in the foreseeable future.
The Company recognizes shared-based compensation cost over the
vesting period using the straight-line single option method.
Share-based compensation expense is recognized only for those
awards that are ultimately expected to vest. An estimated
forfeiture rate has been applied to unvested awards for the
purpose of calculating compensation cost. Forfeitures were
estimated based on historical experience. These estimates are
revised, if necessary, in future periods if actual forfeitures
differ from the estimates. Changes in forfeiture estimates
impact compensation cost in the period in which the change in
estimate occurs.
F-34
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
A summary of stock option activity is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
Remaining
|
|
|
Aggregate
|
|
|
|
|
Number of
|
|
|
Exercise
|
|
|
Contractual
|
|
|
Intrinsic
|
|
|
|
|
Shares
|
|
|
Price
|
|
|
Term (Years)
|
|
|
Value
|
|
|
|
|
Outstanding — December 31, 2007
|
|
|
6,482,260
|
|
|
$
|
5.91
|
|
|
|
|
|
|
|
|
|
|
Granted (weighted-average fair value of $1.19 per share)
|
|
|
2,148,000
|
|
|
|
2.10
|
|
|
|
|
|
|
|
|
|
|
Forfeited
|
|
|
(294,521
|
)
|
|
|
4.38
|
|
|
|
|
|
|
|
|
|
|
Expired
|
|
|
(1,219,967
|
)
|
|
|
6.08
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2008
|
|
|
7,115,772
|
|
|
|
4.80
|
|
|
|
|
|
|
|
|
|
|
Granted (weighted-average fair value of $2.87 per share)
|
|
|
4,141,000
|
|
|
|
4.70
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
(488,750
|
)
|
|
|
2.58
|
|
|
|
|
|
|
|
|
|
|
Forfeited
|
|
|
(551,130
|
)
|
|
|
5.35
|
|
|
|
|
|
|
|
|
|
|
Cancelled
|
|
|
(2,165,372
|
)
|
|
|
7.75
|
|
|
|
|
|
|
|
|
|
|
Expired
|
|
|
(106,275
|
)
|
|
|
4.62
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2009
|
|
|
7,945,245
|
|
|
|
4.04
|
|
|
|
|
|
|
|
|
|
|
Granted (weighted-average fair value of $2.65 per share)
|
|
|
3,350,070
|
|
|
|
4.26
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
(1,135,340
|
)
|
|
|
3.21
|
|
|
|
|
|
|
|
|
|
|
Forfeited
|
|
|
(1,347,786
|
)
|
|
|
4.04
|
|
|
|
|
|
|
|
|
|
|
Expired
|
|
|
(415,095
|
)
|
|
|
5.45
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding — December 31, 2010
|
|
|
8,397,094
|
|
|
$
|
4.17
|
|
|
|
7.4
|
|
|
$
|
22,747,151
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested (exercisable) — December 31, 2010
|
|
|
5,157,935
|
|
|
$
|
4.12
|
|
|
|
6.6
|
|
|
$
|
14,250,135
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unvested (unexercisable) — December 31, 2010
|
|
|
3,239,159
|
|
|
$
|
4.26
|
|
|
|
8.7
|
|
|
$
|
8,492,016
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes information with respect to stock
options outstanding and exercisable at December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
Weighted-
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
Remaining
|
|
|
Average
|
|
|
|
|
|
Average
|
|
|
|
|
Number
|
|
|
Contractual
|
|
|
Exercise
|
|
|
Number
|
|
|
Exercise
|
|
|
Exercise Price
|
|
Outstanding
|
|
|
Life (Years)
|
|
|
Price
|
|
|
Exercisable
|
|
|
Price
|
|
|
|
|
$0.92 - 1.40
|
|
|
110,000
|
|
|
|
2.6
|
|
|
$
|
1.09
|
|
|
|
104,176
|
|
|
$
|
1.08
|
|
|
$1.43 - 2.47
|
|
|
1,180,665
|
|
|
|
6.7
|
|
|
$
|
1.61
|
|
|
|
810,750
|
|
|
$
|
1.64
|
|
|
$2.49 - 3.77
|
|
|
1,019,542
|
|
|
|
7.2
|
|
|
$
|
2.70
|
|
|
|
929,224
|
|
|
$
|
2.65
|
|
|
$3.82 - 5.79
|
|
|
4,353,942
|
|
|
|
7.8
|
|
|
$
|
4.45
|
|
|
|
2,206,612
|
|
|
$
|
4.67
|
|
|
$5.80 - 8.33
|
|
|
1,732,945
|
|
|
|
7.4
|
|
|
$
|
6.28
|
|
|
|
1,107,173
|
|
|
$
|
6.35
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8,397,094
|
|
|
|
|
|
|
|
|
|
|
|
5,157,935
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Due to our rapid growth over the past few years and personnel
turnover rate, in early 2009, we had a limited number of shares
available for future grant under the 2003 Plan. Primarily in
order to increase the pool of shares available for future grant
under such plan, we conducted a tender offer to eligible
employees to acquire options
F-35
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
granted to certain employees of the Company pursuant to the 1997
Plan and 2003 Plan, and which were outstanding at March 23,
2009. Eligible employees were employees of Spectrum or its
subsidiaries who held options with exercise prices in excess of
$5.00. The cash amount offered to those employees was $0.01 for
options with an exercise price over $10.00 and $1.15 for the
options with an exercise price between $5.00 and $9.99.
On April 23, 2009, a total of 2,165,372 shares
underlying eligible options were tendered by eligible employees
and were accepted by us, representing 73% of the shares
underlying eligible options that were eligible to be tendered in
the offer. We made a cash payment in the aggregate of
approximately $2.5 million to the eligible employees
participating in the offer.
As of December 31, 2010, there was unrecognized
compensation expense of $6.9 million related to unvested
stock options, which we expect to recognize over a weighted
average period of 2.4 years. The aggregate intrinsic value
of the options outstanding and options exercisable as of
December 31, 2010 was $22.7 million and
$14.3 million, respectively. The weighted average grant
date fair value of stock options granted during the year ended
December 31, 2010 was $2.65.
Restricted
Stock
A summary of restricted stock activity is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
Number of
|
|
|
Weighted Average
|
|
|
|
|
Restricted
|
|
|
Grant Date
|
|
|
|
|
Shares
|
|
|
Fair Value
|
|
|
|
|
Non-vested — December 31, 2007
|
|
|
277,500
|
|
|
$
|
5.03
|
|
|
Granted
|
|
|
372,500
|
|
|
|
1.65
|
|
|
Vested
|
|
|
(272,500
|
)
|
|
|
3.17
|
|
|
Forfeited
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested — December 31, 2008
|
|
|
377,500
|
|
|
|
3.04
|
|
|
Granted
|
|
|
262,500
|
|
|
|
1.86
|
|
|
Vested
|
|
|
(284,375
|
)
|
|
|
2.82
|
|
|
Forfeited
|
|
|
(2,500
|
)
|
|
|
5.45
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested — December 31, 2009
|
|
|
353,125
|
|
|
|
2.32
|
|
|
Granted
|
|
|
390,000
|
|
|
|
4.48
|
|
|
Vested
|
|
|
(261,500
|
)
|
|
|
3.58
|
|
|
Forfeited
|
|
|
(122,125
|
)
|
|
|
1.87
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested — December 31, 2010
|
|
|
359,500
|
|
|
$
|
3.96
|
|
|
|
|
|
|
|
|
|
|
|
The fair value of restricted stock awards is the quoted market
price of our stock on the grant date, and is charged to expense
over the period of vesting. These awards are subject to
forfeiture to the extent that the recipient’s service is
terminated prior to the shares becoming vested.
During the years ended December 31, 2010, 2009 and 2008,
the stock-based charge in connection with the expensing of
restricted stock awards was approximately $974,000, $665,000 and
$862,000, respectively. As of December 31, 2010, there was
approximately $1.1 million of unrecognized stock-based
compensation cost related to non-vested restricted stock awards,
which is expected to be recognized over a weighted average
period of 2.0 years.
F-36
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
401(k)
Plan Matching Contribution
During the years ended December 31, 2010, 2009 and 2008 we
issued 136,121, 139,795, and 166,430 shares of common stock
as the Company’s match of approximately $598,000, $448,000,
and $274,000 on the 401(k) contributions of our employees during
those periods.
Employee
Stock Purchase Plan
Effective July 2009, we adopted the 2009 Employee Stock Purchase
Plan, or Purchase Plan. The Purchase Plan provides our eligible
employees with an incentive by providing a method whereby they
may voluntarily purchase shares of our common stock upon terms
described in the Purchase Plan. The Purchase Plan is designed to
be operated on the basis of six consecutive month offering
periods commencing January 1 and July 1 of each year. The
Purchase Plan provides that eligible employees may authorize
payroll deductions to purchase shares of our common stock at 85%
of the fair market value of common stock on the first or last
day of the applicable purchase period. A participant may
purchase a maximum of 50,000 shares of common stock during
a 6-month
offering period, not to exceed $25,000 worth of stock on the
offering date during each plan year. The Purchase Plan
terminates in 2019.
A total of 5,000,000 shares of common stock are authorized
for issuance under the Purchase Plan, and as of
December 31, 2010, 233,998 shares have been issued
under the Purchase Plan. Beginning on January 1, 2010, and
each January 1st thereafter, the number of shares of
common stock available for issuance under the Purchase Plan
shall increase by an amount equal to the lesser of
(i) 1,000,000 shares or (ii) an amount determined
by the Purchase Plan Administrator. However, in no event shall
the number of shares of common stock available for future sale
under the Purchase Plan exceed 10,000,000 shares, subject
to capitalization adjustments occurring due to dividends,
splits, dissolution, liquidation, mergers, or changes in control.
The Purchase Plan replaces our 2001 Employee Stock Purchase
Program, which was terminated by the Board effective
June 26, 2009. Total Purchase Plan stock based compensation
expense for the years ended December 31, 2010 and 2009 was
$230,000 and $44,000, respectively.
|
|
|
12.
|
Quarterly
Financial Data (Unaudited)
|
A summary of quarterly financial data (unaudited) is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended
|
|
|
|
March 31
|
|
June 30
|
|
September 30
|
|
December 31
|
|
|
|
Year ended December 31, 2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
$
|
11,089
|
|
|
$
|
12,343
|
|
|
$
|
16,735
|
|
|
$
|
33,946
|
|
|
Operating (loss) income
|
|
$
|
(40,492
|
)
|
|
$
|
(12,266
|
)
|
|
$
|
(6,880
|
)
|
|
$
|
6,742
|
|
|
Net (loss) income
|
|
$
|
(39,014
|
)
|
|
$
|
(9,676
|
)
|
|
$
|
(4,594
|
)
|
|
$
|
4,440
|
|
|
Net (loss) income per share, basic
|
|
$
|
(0.80
|
)
|
|
$
|
(0.20
|
)
|
|
$
|
(0.09
|
)
|
|
$
|
0.09
|
|
|
Net (loss) income, diluted
|
|
$
|
(0.80
|
)
|
|
$
|
(0.20
|
)
|
|
$
|
(0.09
|
)
|
|
$
|
0.08
|
|
|
Year ended December 31, 2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
$
|
14,163
|
|
|
$
|
8,141
|
|
|
$
|
7,101
|
|
|
$
|
8,620
|
|
|
Operating (loss)
|
|
$
|
(626
|
)
|
|
$
|
(9,831
|
)
|
|
$
|
(8,761
|
)
|
|
$
|
(9,290
|
)
|
|
Net loss attributable to non-controlling interest
|
|
$
|
1,146
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Net income (loss)
|
|
$
|
115
|
|
|
$
|
(29,819
|
)
|
|
$
|
474
|
|
|
$
|
10,184
|
|
|
Net income (loss) per share — basic
|
|
$
|
0.00
|
|
|
$
|
(0.87
|
)
|
|
$
|
0.01
|
|
|
$
|
0.21
|
|
|
Net income (loss) per share — diluted
|
|
$
|
0.00
|
|
|
$
|
(0.87
|
)
|
|
$
|
(0.07
|
)
|
|
$
|
0.20
|
|
F-37
Spectrum
Pharmaceuticals, Inc. and Subsidiaries
Notes to
Consolidated Financial
Statements — (Continued)
Earnings per basic and diluted shares are computed independently
for each of the quarters presented based on basic and diluted
shares outstanding per quarter and, therefore, may not sum to
the totals for the year.
In January 2011, we signed a letter of agreement with Viropro,
Inc., for the development of a biosimilar version of the
monoclonal antibody drug rituximab. Biosimilars, or follow-on
biologis, are terms used to describe officially-approved
subsequent versions of innovator biopharmaceutical products made
by a different sponsor following patent and exclusivity expiry.
Terms of the agreement call for a nominal upfront payment and
additional payments based on certain development and regulatory
milestones should the Company elect to continue development
efforts.
F-38
Index to
Exhibits
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
2
|
.1
|
|
Asset Purchase Agreement by and between the Registrant, Targent
Inc. and Certain Stockholders of Targent, Inc., dated March 17
2006. (Filed as Exhibit 2.1 to
Form 10-K/A,
Amendment No. 1, as filed with the Securities and Exchange
Commission on May 1, 2006, and incorporated herein by
reference.)
|
|
|
2
|
.2
|
|
Asset Purchase Agreement by and between the Registrant and Par
Pharmaceutical, Inc., dated as of May 6, 2008. (Filed as
Exhibit 2.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 11, 2008, and incorporated herein by reference.)
|
|
|
2
|
.3#
|
|
Purchase and Formation Agreement, dated as of November 26,
2008, by and among the Registrant, Cell Therapeutics, Inc. and
RIT Oncology, LLC. (Filed as Exhibit 2.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 19, 2008, and incorporated herein by reference.)
|
|
|
2
|
.4#
|
|
Limited Liability Company Interest Assignment Agreement, dated
as of March 15, 2009, by and between the Registrant and
Cell Therapeutics, Inc. (Filed as Exhibit 2.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 15, 2009, and incorporated herein by reference.)
|
|
|
3
|
.1+
|
|
Amended Certificate of Incorporation, as filed.
|
|
|
3
|
.2
|
|
Form of Amended and Restated Bylaws of the Registrant. (Filed as
Exhibit 3.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 16, 2004, and incorporated herein by reference.)
|
|
|
4
|
.1
|
|
Rights Agreement, dated as of December 13, 2010, between
the Registrant and ComputerShare Trust Company, N.A.
(formerly U.S. Stock Transfer Corporation), as Rights Agent,
which includes as Exhibit A thereto the form of Certificate
of Designation for the Series B Junior Participating
Preferred Stock, as Exhibit B thereto the Form of Rights
Certificate and as Exhibit C thereto a Summary of Rights of
Stockholder Rights Plan. (Filed as Exhibit 4.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 13, 2010, and incorporated herein by reference.)
|
|
|
4
|
.2
|
|
Registration Rights Agreement, dated as of September 26,
2003, by and among the Registrant and the persons listed on
Schedule 1 attached thereto. (Filed as Exhibit 4.4 to
Form 8-K,
as filed with the Securities and Exchange Commission on
September 30, 2003, and incorporated herein by reference.)
|
|
|
4
|
.3
|
|
Investor Rights Agreement, dated as of April 20, 2004, by
and among the Registrant and the persons listed on
Schedule 1 attached thereto. (Filed as Exhibit 4.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
April 23, 2004, and incorporated herein by reference.)
|
|
|
4
|
.4
|
|
Form of Warrant, dated September 15, 2005. (Filed as
Exhibit 4.35 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 15, 2006, and incorporated herein by reference.)
|
|
|
4
|
.5
|
|
Registration Rights Agreement, dated as of April 20, 2006,
by and among the Registrant and Targent, Inc. (Filed as
Exhibit 4.2 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 8, 2006, and incorporated herein by reference.)
|
|
|
10
|
.1+
|
|
Sublease Agreement, dated as of December 2, 2010, between
the Registrant and Del Webb Corporation.
|
|
|
10
|
.2
|
|
Industrial Lease Agreement, dated as of January 16, 1997,
between the Registrant and the Irvine Company. (Filed as
Exhibit 10.11 to
Form 10-KSB,
as filed with the Securities and Exchange Commission on
March 31, 1997, and incorporated herein by reference.)
|
|
|
10
|
.3
|
|
Preferred Stock and Warrant Purchase Agreement, dated as of
September 26, 2003, by and among the Registrant and the
purchasers listed on Schedule 1 attached thereto. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
September 30, 2003, and incorporated herein by reference.)
|
|
|
10
|
.4
|
|
First Amendment, dated March 25, 2004, to Industrial Lease
Agreement dated as of January 16, 1997 by and between the
Registrant and the Irvine Company. (Filed as Exhibit 10.1
to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 17, 2004, and incorporated herein by reference.)
|
|
|
10
|
.5
|
|
Common Stock and Warrant Purchase Agreement, dated as of
April 20, 2004, by and among the Registrant and the
purchasers listed on Schedule 1 attached thereto. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
April 23, 2004, and incorporated herein by reference.)
|
|
|
10
|
.6#+
|
|
Amended and Restated License and Collaboration Agreement by and
between the Registrant and Aeterna Zentaris GmbH (formerly
Zentaris GmbH), dated as of November 5, 2010.
|
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
10
|
.7*
|
|
Form of Stock Option Agreement under the 2003 Amended and
Restated Incentive Award Plan. (Filed as Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 17, 2004, and incorporated herein by reference.)
|
|
|
10
|
.8#
|
|
License Agreement by and between the Registrant and Chicago
Labs, Inc. (Filed as Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
February 25, 2005, and incorporated herein by reference.)
|
|
|
10
|
.9*
|
|
Form of Non-Employee Director Stock Option Agreement under the
2003 Amended and Restated Incentive Award Plan. (Filed as
Exhibit 10.5 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
May 10, 2005, and incorporated herein by reference.)
|
|
|
10
|
.10*
|
|
Restricted Stock Award Grant Notice and Restricted Stock Award
Agreement under the 2003 Amended and Restated Incentive Award
Plan. (Filed as Exhibit 10.44 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 15, 2006, and incorporated herein by reference.)
|
|
|
10
|
.11#
|
|
License Agreement between the Registrant and Merck Eprova AG,
dated May 23, 2006. (Filed as Exhibit 10.1 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 8, 2006, and incorporated herein by reference.)
|
|
|
10
|
.12*
|
|
Third Amended and Restated 1997 Stock Incentive Plan. (Filed as
Exhibit 10.2 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
November 3, 2006, and incorporated herein by reference.)
|
|
|
10
|
.13#
|
|
Agreement by and between the Registrant and Glaxo Group Limited
(d/b/a GlaxoSmithKline), dated November 10, 2006. (Filed as
Exhibit 10.38 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 14, 2007, and incorporated herein by reference.)
|
|
|
10
|
.14*
|
|
2003 Amended and Restated Incentive Award Plan. (Filed as
Exhibit 10.3 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 9, 2007, and incorporated herein by reference.)
|
|
|
10
|
.15#
|
|
License Agreement by and between the Registrant and Indena,
S.p.A., dated July 17, 2007. (Filed as Exhibit 10.4 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
November 9, 2007, and incorporated herein by reference.)
|
|
|
10
|
.16*
|
|
Executive Employment Agreement by and between the Registrant and
Rajesh C. Shrotriya, M.D., entered into June 20, 2008
and effective as of January 2, 2008. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
June 26, 2008, and incorporated herein by reference.)
|
|
|
10
|
.17*
|
|
Consulting Agreement by and between the Registrant and Luigi
Lenaz, M.D., effective as of July 1, 2008. (Filed as
Exhibit 10.2 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 11, 2008, and incorporated herein by reference.)
|
|
|
10
|
.18*+
|
|
Amendment Number One to Consulting Agreement by and between the
Registrant and Luigi Lenaz, M.D., effective as of
March 16, 2010.
|
|
|
10
|
.19*+
|
|
Amendment Number Two to Consulting Agreement by and between the
Registrant and Luigi Lenaz, M.D., effective as of
July 2, 2010.
|
|
|
10
|
.20*
|
|
Form of Indemnity Agreement of the Registrant. (Filed as
Exhibit 10.32 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 31, 2009, and incorporated herein by reference.)
|
|
|
10
|
.21#
|
|
License, Development, Supply and Distribution Agreement, dated
October 28, 2008, by and among the Registrant, Allergan
Sales, LLC, Allergan USA, Inc. and Allergan, Inc. (Filed as
Exhibit 10.33 to
Form 10-K,
as filed with the Securities and Exchange Commission on
March 31, 2009, and incorporated herein by reference.)
|
|
|
10
|
.22*
|
|
2009 Employee Stock Purchase Plan. (Filed as Exhibit 99.1
to
Form S-8,
as filed with the Securities and Exchange Commission on
June 29, 2009, and incorporated herein by reference.)
|
|
|
10
|
.23*
|
|
2009 Incentive Award Plan. (Filed as Exhibit 99.2 to
Form S-8,
as filed with the Securities and Exchange Commission on
June 29, 2009, and incorporated herein by reference.)
|
|
|
10
|
.24*
|
|
2003 Amended and Restated Incentive Award Plan. (Filed as
Exhibit 10.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
July 2, 2009, and incorporated herein by reference.)
|
|
|
10
|
.25
|
|
Fourth Amendment, dated July 29, 2009, to Industrial Lease
Agreement dated as of January 16, 1997 by and between the
Registrant and the Irvine Company. (Filed as Exhibit 10.29
to
Form 10-K,
as filed with the Securities and Exchange Commission on
April 5, 2010, and incorporated herein by reference.)
|
| |
|
|
|
|
Exhibit
|
|
|
|
No.
|
|
Description
|
|
|
|
|
10
|
.26*
|
|
Term Sheet for 2009 Incentive Award Plan Stock Option Award.
(Filed as Exhibit 10.8 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 13, 2009, and incorporated herein by reference.)
|
|
|
10
|
.27*
|
|
Term Sheet for 2009 Incentive Award Plan, Nonqualified Stock
Option Award Awarded to Non-Employee Directors. (Filed as
Exhibit 10.9 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 13, 2009, and incorporated herein by reference.)
|
|
|
10
|
.28*
|
|
Term Sheet for 2009 Incentive Award Plan, Restricted Stock
Award. (Filed as Exhibit 10.10 to
Form 10-Q,
as filed with the Securities and Exchange Commission on
August 13, 2009, and incorporated herein by reference.)
|
|
|
10
|
.29#
|
|
License Agreement, dated November 6, 2009, by and between
the Registrant and Nippon Kayaku Co., Ltd. (Filed as
Exhibit 10.36 to
Form 10-K,
as filed with the Securities and Exchange Commission on
April 5, 2010, and incorporated herein by reference.)
|
|
|
10
|
.30#
|
|
License and Collaboration Agreement, dated February 2,
2010, by and between the Registrant and TopoTarget A/S. (Filed
as Exhibit 10.37 to
Form 10-K,
as filed with the Securities and Exchange Commission on
April 5, 2010, and incorporated herein by reference.)
|
|
|
10
|
.31
|
|
Asset Purchase Agreement, dated August 15, 2007, by and between
Cell Therapeutics, Inc. and Biogen Idec Inc. (Filed as Exhibit
10.1 to Cell Therapeutics, Inc.’s Form 8-K, No. 001-12465,
as filed with the Securities and Exchange Commission on August
21, 2007, and incorporated herein by reference.)
|
|
|
10
|
.32
|
|
First Amendment to Asset Purchase Agreement, dated December 9,
2008, by and between Cell Therapeutics, Inc. and Biogen Idec
Inc. (Filed as Exhibit 10.48 to Cell Therapeutics, Inc.’s
Form 10K, No. 001-12465, as filed with the Securities and
Exchange Commission on March 16, 2009, and incorporated herein
by reference.)
|
|
|
10
|
.33
|
|
Supply Agreement, dated December 21, 2007, by and between Cell
Therapeutics, Inc. and Biogen Idec Inc. (Filed as Exhibit 10.2
to Cell Therapeutics, Inc.’s Form 8-K, No. 001-12465, as
filed with the Securities and Exchange Commission on December
31, 2007, and incorporated herein by reference.)
|
|
|
10
|
.34#+
|
|
First Amendment to Supply Agreement, dated December 15, 2008, by
and between Cell Therapeutics, Inc. and Biogen Idec Inc.
|
|
|
10
|
.35+
|
|
Security Agreement, dated December 15, 2008, by and between RIT
Oncology, LLC and Biogen Idec Inc.
|
|
|
16
|
.1
|
|
Letter from Kelly and Company to the Securities and Exchange
Commission, dated December 3, 2009. (Filed as
Exhibit 16.1 to
Form 8-K,
as filed with the Securities and Exchange Commission on
December 8, 2009, and incorporated herein by reference.)
|
|
|
21
|
+
|
|
Subsidiaries of Registrant.
|
|
|
23
|
.1+
|
|
Consent of Ernst & Young LLP, Independent Registered
Public Accounting Firm.
|
|
|
23
|
.2+
|
|
Consent of Kelly & Company, Independent Registered
Public Accounting Firm.
|
|
|
24
|
.1
|
|
Power of Attorney (included in the signature page.)
|
|
|
31
|
.1+
|
|
Certification of Principal Executive Officer, pursuant to
Rule 13a-14(a)/15d-14(a)
of the Securities Exchange Act of 1934.
|
|
|
31
|
.2+
|
|
Certification of Principal Financial Officer, pursuant to
Rule 13a-14(a)/15d-14(a)
of the Securities Exchange Act of 1934.
|
|
|
32
|
.1+
|
|
Certification of Principal Executive Officer, pursuant to
Rule 13a-14(b)/15d-14(b)
of the Securities Exchange Act of 1934 and 18 U.S.C.
Section 1350.
|
|
|
32
|
.2+
|
|
Certification of Principal Financial Officer, pursuant to
Rule 13a-14(b)/15d-14(b)
of the Securities Exchange Act of 1934 and 18 U.S.C.
Section 1350.
|
|
|
|
|
* |
|
Indicates a management contract or compensatory plan or
arrangement. |
| |
|
# |
|
Confidential portions omitted and filed separately with the U.S.
Securities and Exchange Commission pursuant to
Rule 24b-2
promulgated under the Securities Exchange Act of 1934, as
amended. |
| |
|
+ |
|
Filed herewith. |