Attached files
| file | filename |
|---|---|
| 8-K - 8-K - Innoviva, Inc. | a10-18069_18k.htm |
| EX-99.1 - EX-99.1 - Innoviva, Inc. | a10-18069_1ex99d1.htm |
| EX-99.6 - EX-99.6 - Innoviva, Inc. | a10-18069_1ex99d6.htm |
| EX-99.5 - EX-99.5 - Innoviva, Inc. | a10-18069_1ex99d5.htm |
| EX-99.2 - EX-99.2 - Innoviva, Inc. | a10-18069_1ex99d2.htm |
| EX-99.3 - EX-99.3 - Innoviva, Inc. | a10-18069_1ex99d3.htm |
Exhibit 99.4
|
POSTER P1168 |
Fluticasone furoate (FF), an inhaled
corticosteroid (ICS), demonstrates efficacy |
|
Busse W(1), Bleecker ER(2), Bateman ED(3), Lötvall J(4), Woodcock A(5), Forth R(6), Davis AM(6), Jacques L(7), Haumann B(7)
(1)Department of Medicine, University of Wisconsin, Madison, USA; (2)Translational Sciences, Wake Forest University Health Sciences Winston-Salem, USA (3)Department of Medicine, University of Cape Town, Cape Town, South Africa; (4)Krefting Research Centre, Gothenburg, Sweden; (5)School of Translational Medicine, University Hospital of Manchester Manchester, UK, (6)Respiratory Medicine Development Center, GlaxoSmithKline, North Carolina, USA; (7)Respiratory Medicine Development Centre, GlaxoSmithKline, Uxbridge, UK
UPDATED ABSTRACT
Introduction: FF (GW685698X) is a novel ICS still active at 24h, in development as a once-daily treatment in combination with the long-acting beta2 agonist (LABA) vilanterol trifenatate (VI; GW642444M) for asthma and chronic obstructive pulmonary disease.
Objectives: To evaluate the efficacy and safety of FF administered using a novel single-step activation dry powder inhaler (DPI) in patients >12 years uncontrolled on moderate doses of ICS (fluticasone propionate [FP] 500mcg/day or equivalent). FP was used as an active control.
Methods: In this randomised, double-blind, placebo-controlled, double-dummy, parallel group study, 627 patients were randomised to FF (200, 400, 600 or 800mcg) once daily, FP 500mcg twice daily or placebo for 8 weeks. Primary endpoint: change in trough (pre-dose) forced expiratory volume in 1 second (FEV1) at Week 8.
Results: Primary: the test of linear trend in FF dose response relative to placebo for trough FEV1 at Week 8 was significant (p<0.001). However, no dose response between FF doses was observed. Secondary: peak expiratory flow (PEF), symptom-free and rescue medication-free 24h periods, and withdrawals due to lack of efficacy supported the efficacy of FF 200–800mcg doses and FP. Incidence of oral candidiasis was highest for FF 800mcg (12%). 24h urinary cortisol excretion ratios (Week 8/baseline) for FF were similar to placebo except for FF 800mcg, which was significantly lower.
Conclusion: This study supports the use of FF as a once-daily treatment for patients with asthma uncontrolled on moderate ICS doses.
INTRODUCTION
· ICS are the most effective anti-inflammatory treatment for persistent asthma(1)–(3) and play a critical role in first-line treatment.
· There is a need for improved treatment options for patients with uncontrolled disease; most ICS are dosed twice daily, but once-daily dosing can improve treatment adherence,(4) and may improve outcomes for patients.(5),(6)
· FF a novel ICS still active at 24h, is currently in development as the ICS component of a new ICS/LABA once daily inhaled combination treatment for asthma.
OBJECTIVE
· Evaluate the efficacy and safety of four doses of FF dosed once daily in the evening for 8 weeks, using a novel single-step activation DPI, in patients >12 years whose symptoms were uncontrolled on moderate doses of ICS (FP 500mcg/day or equivalent).
METHODS
· A phase IIb, randomised, double-blind, double-dummy, placebo-controlled, parallel-group, multicentre, dose-ranging study in adolescent and adult patients with persistent asthma (Figure 1).
· Eligible patients (>12 years old) had persistent asthma,(2) with an evening pre-dose % predicted FEV1 of 40–90% of their predicted normal value, and >12% and >200mL reversibility of FEV1 following albuterol/salbutamol inhalation. They had been using an ICS for >8 weeks prior to their first visit (at a stable dose for >4 weeks).
· All patients replaced their short-acting beta2 agonists with albuterol/salbutamol inhalation aerosol at visit 1, for use as needed during the study.
· Following a 28-day run-in (to assess baseline asthma status, daily diary compliance, and for safety evaluations) during which patients continued their prior ICS therapy at a fixed dose, they were randomly assigned to receive
· FF (200, 400, 600 or 800mcg once daily [evening; PM]) via novel DPI plus placebo twice daily (morning [AM] and PM) via DISKUS™/ACCUHALER™
· FP (500mcg twice daily AM and PM) via DISKUS™/ACCUHALER™ plus placebo once daily (PM) via novel DPI
· placebo once daily (PM) via novel DPI plus placebo twice daily (AM and PM) via DISKUS™/ACCUHALER™.
· All medications were blinded to investigators and patients, and each patient received two devices at each visit (novel DPI and DISKUS™/ACCUHALER™).
· Patients stopped their maintenance ICS therapy for the duration of treatment (visit 3–8), and received study medication for 56 days (8 weeks). A follow-up clinic visit or phone contact was conducted 1 week after completing treatment.
Efficacy and safety measures
· The primary endpoint was the mean change from baseline in PM trough FEV1 (pre-dose and pre-rescue bronchodilator) at Week 8
· the primary comparison was the test for linear dose response in FEV1 across the four doses of FF and placebo (intent-to-treat [ITT] population; analysis of covariance [ANCOVA] with last observation carried forward [LOCF]).
· Secondary endpoints
· mean change from baseline in daily PM trough (pre-dose and pre-rescue bronchodilator) PEF and daily AM PEF, averaged over the treatment period
· mean change from baseline in % symptom-free and % rescue medication-free 24h periods
· withdrawals due to lack of efficacy.
Figure 1. Study design.

OD = once daily; BD = twice daily
· Safety assessments included
· incidence of adverse events (AEs; categorised using the MedDRA coding dictionary, Version 11)
· oropharyngeal examination for oral candidiasis
· laboratory tests (haematology, clinical chemistry, urinalysis parameters [pre- and post-treatment]) and vital signs
· 24h urinary cortisol excretion (pre- and post-treatment).
RESULTS
· 627 patients were randomised to FF (200, 400, 600 or 800mcg) once daily PM, FP 500mcg twice daily or placebo for 8 weeks; 622 patients received one or more doses (the ITT population).
· 82–92% of the FF groups and 88% of the FP group completed the study, vs 63% of the placebo group (Table 1).
Table 1. Patient disposition.
|
|
|
|
|
FF dose |
|
FP dose |
|
||||||
|
|
|
Placebo |
|
200mcg OD |
|
400mcg OD |
|
600mcg OD |
|
800mcg OD |
|
500mcg BD |
|
|
|
|
n (%) |
|
n (%) |
|
n (%) |
|
n (%) |
|
n (%) |
|
n (%) |
|
|
Received study medication |
|
103 |
|
99 |
|
101 |
|
107 |
|
102 |
|
110 |
|
|
Completed |
|
65 (63) |
|
81 (82) |
|
93 (92) |
|
94 (88) |
|
85 (83) |
|
97 (88) |
|
|
Discontinued |
|
38 (37) |
|
18 (18) |
|
8 (8) |
|
13 (12) |
|
17 (17) |
|
13 (12) |
|
|
Lack of efficacy |
|
34 (33) |
|
11 (11) |
|
6 (6) |
|
11 (10) |
|
12 (12) |
|
8 (7) |
|
|
AE |
|
2 (2) |
|
3 (3) |
|
0 |
|
1 (<1) |
|
0 |
|
4 (4) |
|
|
Withdrew consent |
|
1 (<1) |
|
0 |
|
0 |
|
1 (<1) |
|
3 (3) |
|
1 (<1) |
|
|
Protocol deviation |
|
0 |
|
3 (3) |
|
1 (<1) |
|
0 |
|
1 (<1) |
|
0 |
|
|
Investigator discretion |
|
1 (<1) |
|
0 |
|
0 |
|
0 |
|
1 (<1) |
|
0 |
|
|
Lost to follow-up |
|
0 |
|
1 (1) |
|
1 (<1) |
|
0 |
|
0 |
|
0 |
|
· Demographic characteristics were evenly matched between treatment groups, as were clinical characteristics and measures of pulmonary function at screening, indicating a similar severity of illness between groups (Table 2).
Table 2. Demographic and clinical characteristics
(ITT population).
|
|
|
|
|
FF dose |
|
FP dose |
|
||||||
|
|
|
Placebo |
|
200mcg OD |
|
400mcg OD |
|
600mcg OD |
|
800mcg OD |
|
500mcg BD |
|
|
|
|
(n=103) |
|
(n=99) |
|
(n=101) |
|
(n=107) |
|
(n=102) |
|
(n=110) |
|
|
Age, years (range)* |
|
47.2±14.03 |
|
45.7±15.02 |
|
47.2±14.39 |
|
45.7±14.38 |
|
46.6±14.09 |
|
46.1±13.86 |
|
|
|
|
(16–78) |
|
(12–77) |
|
(13–70) |
|
(13–75) |
|
(12–76) |
|
(12–74) |
|
|
Gender, n (%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Females |
|
64 (62) |
|
63 (64) |
|
62 (61) |
|
67 (63) |
|
63 (62) |
|
68 (62) |
|
|
Race, n (%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
White |
|
83 (81) |
|
74 (75) |
|
80 (79) |
|
77 (72) |
|
80 (78) |
|
83 (76) |
|
|
Asian |
|
6 (6) |
|
9 (9) |
|
7 (7) |
|
11 (10) |
|
7 (7) |
|
7 (6) |
|
|
Other |
|
14 (14) |
|
16 (16) |
|
14 (14) |
|
19 (18) |
|
15 (15) |
|
19 (17) |
|
|
Lung function at screening* |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pre-bronchodilator FEV1 (L) |
|
2.043±0.6022 |
|
2.046±0.6387 |
|
2.066±0.6358 |
|
2.043±0.6076 |
|
2.057±0.5865 |
|
2.064±0.5644 |
|
|
% predicted FEV1 (%) |
|
64.12±11.133 |
|
65.08±11.684 |
|
66.59±12.771 |
|
64.33±11.983 |
|
66.00±12.118 |
|
65.43±12.353 |
|
|
Post-bronchodilator FEV1 |
|
2.557±0.7454 |
|
2.603±0.7591 |
|
2.566±0.7522 |
|
2.597±0.7176 |
|
2.639±0.7355 |
|
2.592±0.6413 |
|
|
Reversibility of FEV1 (%) |
|
26.14±14.448 |
|
28.93±17.305 |
|
25.53±13.892 |
|
28.55±14.005 |
|
29.32±15.409 |
|
27.02±15.013 |
|
|
Reversibility of FEV1 (mL) |
|
513.4±278.54 |
|
556.1±305.62 |
|
500.9±258.55 |
|
553.5±260.75 |
|
582.4±316.95 |
|
528.5±253.20 |
|
*Values are mean ± standard deviation
Efficacy
Primary outcome
· At baseline, PM trough FEV1 was similar between the six treatment groups; between baseline and Week 8 there was a reduction in trough FEV1 with placebo, but an increase with all FF once daily PM doses, and FP twice daily (Figure 2).
· The results of a test of linear trend in dose response, excluding the placebo group showed no significant relationship between response and dose of FF (p=0.306; 95% CI: –0.229 to 0.072). The estimate of slope was –0.078mL/mcg. The results of the test for linear trend in dose response, including placebo were significant (p<0.001; 95% CI: 0.112 to 0.333). The estimate of the slope was 0.223mL/mcg.
· Results for the per-protocol population were consistent with those for the ITT population.
Figure 2. Adjusted treatment differences of change from baseline in trough FEV1 (LOCF) at Week 8 (ITT population).

PEF
· All FF once daily PM doses and FP twice daily had greater improvements in PM and AM trough PEF (LSM change between baseline and Weeks 1–8) than placebo (p<0.001; Figure 3).
Figure 3. PEF over Weeks 1–8
a) PM and b) AM.

Symptom-free and rescue medication-free periods
· Differences in the percentage of symptom-free 24h periods (LSM change between baseline and Weeks 1–8) were greater for FF 200mcg and 400mcg (p<0.001), 600mcg (p=0.022), 800mcg (p=0.002) once daily PM and FP twice daily (p=0.017) than for placebo (Figure 4a). Differences in the percentage of rescue medication-free 24h periods (LSM change between baseline and Weeks 1–8) were greater for all FF once daily PM doses and FP twice daily than for placebo (p<0.001; Figure 4b).
Figure 4. Symptom-free (a) and rescue-free (b) 24h periods over Weeks 1–8.

Treatment discontinuation
· More patients on placebo (33%) than on active treatment (6–12%) discontinued treatment due to lack of efficacy (p<0.001, Figure 5).
Figure 5. Treatment discontinuation due to lack of efficacy (cumulative incidence).

Note: patients are represented from their date of randomisation to their date of withdrawal due to lack of efficacy
Safety
· A low incidence (<1–6%) of asthma exacerbations occurred during active treatment, vs 16% with placebo; most were due to lack of efficacy
· 8% of patients receiving placebo took oral corticosteroid for exacerbations, vs 2%, 0%, 0% and <1% with FF 200, 400, 600 and 800mcg respectively, and 3% with FP.
· FF was generally well tolerated; Table 3 lists the most commonly reported AEs in each treatment group.
· Other than for oral candidiasis (higher incidence at highest dose level), the incidence of drug-related AEs was similar between FF dose groups.
· No serious AEs were related to study medication.
Table 3. Most common on-treatment AEs.
|
|
|
|
|
FF dose |
|
FP dose |
|
||||||
|
|
|
Placebo |
|
200mcg OD |
|
400mcg OD |
|
600mcg OD |
|
800mcg OD |
|
500mcg BD |
|
|
|
|
(n=103) |
|
(n=99) |
|
(n=101) |
|
(n=107) |
|
(n=102) |
|
(n=110) |
|
|
Patients with any on- treatment AE, n (%) |
|
23 (22) |
|
31 (31) |
|
34 (34) |
|
37 (35) |
|
36 (35) |
|
39 (35) |
|
|
Headache |
|
10 (10) |
|
3 (3) |
|
10 (10) |
|
12 (11) |
|
10 (10) |
|
10 (9) |
|
|
Nasopharyngitis |
|
4 (4) |
|
3 (3) |
|
5 (5) |
|
2 (2) |
|
7 (7) |
|
4 (4) |
|
|
Oropharyngeal candidiasis* |
|
1 (<1) |
|
4 (4) |
|
4 (4) |
|
1 (<1) |
|
4 (4) |
|
4 (4) |
|
|
Dysphonia |
|
1 (<1) |
|
4 (4) |
|
5 (5) |
|
1 (<1) |
|
4 (4) |
|
2 (2) |
|
|
Oral candidiasis* |
|
0 |
|
2 (2) |
|
2 (2) |
|
1 (<1) |
|
7 (7) |
|
0 |
|
|
Pharyngolaryngeal pain |
|
1 (<1) |
|
2 (2) |
|
0 |
|
3 (3) |
|
1 (<1) |
|
4 (4) |
|
|
URTI |
|
1 (<1) |
|
2 (2) |
|
0 |
|
3 (3) |
|
1 (<1) |
|
3 (3) |
|
|
Back pain |
|
1 (<1) |
|
1 (1) |
|
1 (<1) |
|
4 (4) |
|
2 (2) |
|
0 |
|
|
Influenza |
|
1 (<1) |
|
1 (1) |
|
2 (2) |
|
3 (3) |
|
0 |
|
0 |
|
|
Nausea |
|
0 |
|
0 |
|
3 (3) |
|
0 |
|
2 (2) |
|
0 |
|
|
Pain in extremity |
|
0 |
|
0 |
|
0 |
|
3 (3) |
|
1 (<1) |
|
0 |
|
*One additional patient (<1%), in the FF 800mcg OD arm, was diagnosed as having oral candidiasis, coded as ‘candidiasis’ URTI = upper respiratory tract infection
· In general, 24h urinary cortisol excretion ratios (Week 8/baseline) for FF were similar to placebo (the ratio for FF 800mcg was significantly lower than for placebo [p<0.001; Figure 6]).
· No other safety concerns were raised.
Figure 6. Adjusted 24h urinary cortisol excretion ratios.*
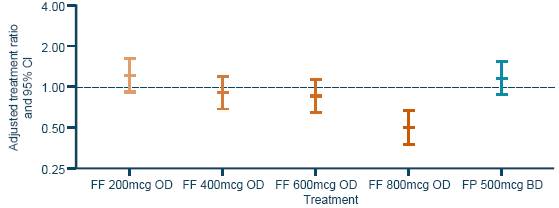
Note: analysis performed using ANCOVA with covariates of country, sex, age, treatment and the log of the baseline values; *The urinary cortisol population consisted of 414 (67%) patients in the ITT population who had urine samples with no factors that would confound the analysis of urinary cortisol
CONCLUSIONS
· The current findings support the use of FF as a once-daily treatment for patients with asthma, uncontrolled on moderate ICS doses
· the primary efficacy analysis was statistically significant, with all FF doses providing greater improvements in PM trough FEV1 than placebo; the lack of FF dose dependency is typical of ICS studies in patients symptomatic on moderate doses of ICS
· secondary efficacy endpoints consistently confirmed the findings of the primary endpoint
· FF once daily appears to be generally well tolerated with AEs equivalent to FP (1000mcg total daily dose) except for a higher incidence of oral candidiasis and a lower urinary cortisol excretion ratio at the highest dose (800mcg once daily PM) overall, the incidence of AEs was low, with no evidence of dose dependency for the most commonly reported AEs.
· On the basis of these findings, FF 200–600mcg appears to be an appropriate once-daily dose for patients with moderate-to-severe persistent asthma.
REFERENCES
(1) Global Initiative for Asthma (GINA). Global Strategy for Asthma Management and Prevention. 2007. Available at: www.ginasthma.org.
(2) National Institutes of Health (NIH). Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Full Report 2007. NIH Publication No. 07-4051. Available at: http://www.nhlbi.nih.gov.
(3) British Thoracic Society. Thorax 1997;52:S1–2.
(4) Price D, et al. BMC Pulm Med 2010;10:1.
(5) Smith LA, et al. Pediatrics 2008;122:760–9.
(6) Stanford RH, et al. J Asthma 2010;47:257–62.
ACKNOWLEDGEMENTS
· This study was sponsored by GlaxoSmithKline (ClinicalTrials.gov: NCT00603746; protocol number FFA109684).
· Editorial support (in the form of writing assistance, assembling tables and figures, collating author comments, grammatical editing and referencing) was provided by Geoff Weller at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline.
|
|
|
Presented at the Annual Conference of the European Respiratory Society (ERS), Barcelona, Spain, 18–22 September 2010 |
