Attached files
| file | filename |
|---|---|
| EX-32.1 - CERTIFICATION OF PRINCIPAL EXECUTIVE AND PRINCIPAL FINANCIAL OFFICERS - LEXICON PHARMACEUTICALS, INC. | exh321certificationofprinc.htm |
| EX-31.2 - CERTIFICATION OF PRINCIPAL FINANCIAL OFFICER - LEXICON PHARMACEUTICALS, INC. | exh312certificationofprinc.htm |
| EX-31.1 - CERTIFICATION OF PRINCIPAL EXECUTIVE OFFICER - LEXICON PHARMACEUTICALS, INC. | exh311certificationofprinc.htm |
| EX-23.1 - CONSENT OF ERNST & YOUNG LLP - LEXICON PHARMACEUTICALS, INC. | exh231ernst_youngconsent10.htm |
| EX-10.26 - LOAN AND SECURITY AGREEMENT - LEXICON PHARMACEUTICALS, INC. | exh1026loanandsecurityagre.htm |
| EX-10.13 - STOCK OPTION NOTICE TO DIRECTORS - LEXICON PHARMACEUTICALS, INC. | exh1013stockoptionnoticeto.htm |
| EX-10.12 - OFFICER RESTRICTED STOCK UNIT AGREEMENT - LEXICON PHARMACEUTICALS, INC. | exh1012officerrestrictedst.htm |
| EX-10.11 - OFFICER STOCK OPTION AGREEMENT - LEXICON PHARMACEUTICALS, INC. | exh1011officerstockoptiona.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One) | |
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2017 | |
or
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period from _____________ to _____________ | |
Commission File Number: 000-30111
Lexicon Pharmaceuticals, Inc.
(Exact Name of Registrant as Specified in its Charter)
Delaware | 76-0474169 | |||
(State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification Number) | |||
8800 Technology Forest Place The Woodlands, Texas 77381 (Address of Principal Executive Offices and Zip Code) | (281) 863-3000 (Registrant’s Telephone Number, Including Area Code) | |||
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange on which Registered | |||
Common Stock, par value $0.001 per share | Nasdaq Global Select Market | |||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act of 1933. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Securities Exchange Act of 1934. (check one): Large accelerated filer þ Accelerated filer o Non-accelerated filer o Smaller reporting company o Emerging growth company o
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Securities Exchange Act of 1934. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes o No þ
The aggregate market value of voting stock held by non-affiliates of the registrant as of the last day of the registrant’s most recently completed second quarter was approximately $715.3 million, based on the closing price of the common stock on the Nasdaq Global Select Market on June 30, 2017 of $16.45 per share. For purposes of the preceding sentence only, our directors, executive officers and controlling stockholders are assumed to be affiliates. As of February 26, 2018, 105,591,828 shares of common stock were outstanding.
Documents Incorporated by Reference
Certain sections of the registrant’s definitive proxy statement relating to the registrant’s 2018 annual meeting of stockholders, which proxy statement will be filed under the Securities Exchange Act of 1934 within 120 days of the end of the registrant’s fiscal year ended December 31, 2017, are incorporated by reference into Part III of this annual report on Form 10-K.
Lexicon Pharmaceuticals, Inc.
Table of Contents
Item | ||
PART I | ||
1. | ||
1A. | ||
1B. | ||
2. | ||
3. | ||
4. | ||
PART II | ||
5. | ||
6. | ||
7. | ||
7A. | ||
8. | ||
9. | ||
9A. | ||
9B. | ||
PART III | ||
10. | ||
11. | ||
12. | ||
13. | ||
14. | ||
PART IV | ||
15. | ||
16. | ||
The Lexicon name and logo and XERMELO® are registered trademarks of Lexicon Pharmaceuticals, Inc.
_____________________________________________________
In this annual report on Form 10-K, “Lexicon Pharmaceuticals,” “Lexicon,” “we,” “us” and “our” refer to Lexicon Pharmaceuticals, Inc. and its subsidiaries.
_____________________________________________________
This annual report on Form 10-K contains forward-looking statements. These statements relate to future events or our future financial performance. We have attempted to identify forward-looking statements by terminology including “anticipate,” “believe,” “can,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “should” or “will” or the negative of these terms or other comparable terminology. These statements are only predictions and involve known and unknown risks, uncertainties and other factors, including the risks outlined under “Item 1A. Risk Factors,” that may cause our or our industry’s actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements.
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. We are not under any duty to update any of the forward-looking statements after the date of this annual report on Form 10-K to conform these statements to actual results, unless required by law.
PART I
Item 1. Business
Overview
Lexicon Pharmaceuticals is a biopharmaceutical company focused on the development and commercialization of breakthrough treatments for human disease. We are presently devoting most of our resources to the commercialization or development of our four most advanced drug programs:
• | We have obtained approval from the U.S. Food and Drug Administration, or FDA, to sell our first commercial product, XERMELO® (telotristat ethyl), an orally-delivered small molecule drug for the treatment of carcinoid syndrome diarrhea in combination with somatostatin analog, or SSA, therapy in adults inadequately controlled by SSA therapy. We have commenced sales and marketing of XERMELO, and it is commercially available to patients in the United States. We have granted Ipsen Pharma SAS, or Ipsen, an exclusive, royalty-bearing right to commercialize XERMELO outside of the United States and Japan, and Ipsen has obtained approval from the European Commission to market XERMELO in the member states of the European Union, Norway and Iceland. Ipsen has commenced sales and marketing of XERMELO, and it is commercially available to patients in the United Kingdom, Germany and certain other European Union member states. |
• | We are developing sotagliflozin, an orally-delivered small molecule drug candidate, as a treatment for type 1 and type 2 diabetes. We have reported positive top-line data from two pivotal Phase 3 clinical trials and a third Phase 3 clinical trial of sotagliflozin in type 1 diabetes patients. We have granted Sanofi-Aventis Deutschland GmbH, or Sanofi, an exclusive, worldwide (excluding Japan), royalty-bearing right to develop, manufacture and commercialize sotagliflozin. We and Sanofi are presently preparing applications for regulatory approval to market sotagliflozin for type 1 diabetes in the United States and the European Union, and Sanofi is presently conducting Phase 3 development of sotagliflozin in type 2 diabetes. |
• | We are developing LX2761, an orally-delivered small molecule drug candidate, as a treatment for diabetes. We are presently conducting Phase 1 clinical development of LX2761. We have granted Sanofi certain rights of first negotiation with respect to the future development and commercialization of LX2761. |
• | We are developing LX9211, an orally-delivered small molecule drug candidate, as a treatment for neuropathic pain. We are presently conducting Phase 1 clinical development of LX9211. |
Compounds from our most advanced drug programs, as well as compounds from a number of additional drug discovery and development programs that we have advanced into various stages of clinical and preclinical development, originated from our own internal drug discovery efforts. These efforts were driven by a systematic, target biology-driven approach in which we used gene knockout technologies and an integrated platform of advanced medical technologies to systematically study the physiological and behavioral functions of almost 5,000 genes in mice and assessed the utility of the proteins encoded by the corresponding human genes as potential drug targets. We have identified and validated in living animals, or in vivo, more than 100 targets with promising profiles for drug discovery.
We are working both independently and through strategic collaborations and alliances with third parties to capitalize on our drug target discoveries and drug discovery and development programs. We seek to retain exclusive or co-exclusive rights to the benefits of certain drug discovery and development programs by developing and commercializing drug candidates from those programs internally, particularly in the United States for indications treated by specialist physicians. We seek to collaborate with other pharmaceutical and biotechnology companies, such as Ipsen and Sanofi, with respect to drug discovery or the development and commercialization of certain of our drug candidates, particularly with respect to commercialization in territories outside the United States, commercialization in the United States for indications treated by primary care physicians, or when the collaboration may otherwise provide us with access to expertise and resources that we do not possess internally or are complementary to our own.
Lexicon Pharmaceuticals was incorporated in Delaware in July 1995, and commenced operations in September 1995. Our corporate headquarters are located at 8800 Technology Forest Place, The Woodlands, Texas 77381, and our telephone number is (281) 863-3000.
Our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 are made available free of charge on our corporate website located at www.lexpharma.com as soon as reasonably practicable after the filing of
1
those reports with the Securities and Exchange Commission. Information found on our website should not be considered part of this annual report on Form 10-K.
Drug Programs
We are presently devoting most of our resources to the commercialization or development of our four most advanced drug programs: XERMELO (telotristat ethyl) for carcinoid syndrome diarrhea, sotagliflozin for type 1 and type 2 diabetes, LX2761 for diabetes and LX9211 for neuropathic pain. We have also advanced a number of additional compounds into various stages of clinical and preclinical development.
XERMELO (telotristat ethyl)
We commercially launched XERMELO, an orally-delivered small molecule compound, following regulatory approval in the United States in February 2017 for the treatment of carcinoid syndrome diarrhea in combination with SSA therapy in adults inadequately controlled by SSA therapy. XERMELO was internally generated by our scientists and inhibits tryptophan hydroxylase, or TPH, the rate-limiting enzyme for serotonin production found primarily in enterochromaffin cells of the gastrointestinal tract. Carcinoid syndrome is characterized by frequent and debilitating diarrhea and can result when these cells become cancerous and metastisize to the liver or other organs, where they overproduce serotonin. The recommended dose of XERMELO is 250mg three times daily, and the full prescribing information for XERMELO includes certain warnings and precautions relating to constipation.
We have entered into a license and collaboration agreement under which we granted Ipsen an exclusive, royalty-bearing right and license to commercialize XERMELO outside of the United States and Japan. In September 2017, Ipsen received approval from the European Commission for the marketing of telotristat ethyl for the treatment of carcinoid syndrome diarrhea in combination with SSA therapy in adults inadequately controlled by SSA therapy in all member states of the European Union, Norway and Iceland. Ipsen has commenced sales and marketing of XERMELO, and it is now commercially available to patients in the United Kingdom, Germany and certain other European Union member states.
Our pivotal TELESTAR Phase 3 clinical trial assessed the safety and efficacy of XERMELO and served as the primary basis for the regulatory approval of XERMELO in the United States and the European Union. Data from the study showed that patients who added XERMELO to SSA therapy experienced a statistically significant reduction from baseline compared to placebo in the average number of daily bowel movements over the 12-week study period (p<0.001), meeting the study’s primary endpoint. Thirty three percent of patients who added XERMELO to SSA therapy at the approved 250mg dose experienced a reduction in overall bowel movements from baseline of at least two per day, as compared to four percent with placebo. The proportion of patients with treatment-emergent adverse events, serious adverse events and discontinuation due to adverse events were generally similar in all three treatment arms, with the tolerability profile of the approved 250mg dose appearing similar to placebo and somewhat better than the 500mg dose with respect to gastrointestinal discomfort and mood.
We are presently preparing to submit Investigational New Drug applications, or INDs, and commence clinical development of XERMELO in cholangiocarcinoma and neuroendocrine tumors as part of our life cycle management of the program.
Sotagliflozin
Sotagliflozin is an orally-delivered small molecule compound that we and Sanofi are developing for the treatment of type 1 and type 2 diabetes mellitus. Sotagliflozin was internally generated by our scientists and inhibits both sodium-glucose cotransporter type 2, or SGLT2, a transporter responsible for most of the glucose reabsorption performed by the kidney, and sodium-glucose cotransporter type 1, or SGLT1, a transporter responsible for glucose and galactose absorption in the gastrointestinal tract. Our scientists identified mice lacking SGLT1, SGLT2 or both as having potent anti-diabetic phenotypes across multiple measures of glucose control and metabolism, and found that compounds inhibiting both targets had a favorable preclinical profile relative to compounds selective for SGLT2.
We have entered into a collaboration and license agreement with Sanofi under which we granted Sanofi an exclusive, worldwide (excluding Japan), royalty-bearing right and license to develop, manufacture and commercialize sotagliflozin. Under the alliance, we are responsible for conducting all clinical development activities relating to type 1 diabetes and Sanofi is responsible for conducting all clinical development activities relating to type 2 diabetes.
2
Type 1 Diabetes.
We reported top-line primary efficacy endpoint data in September 2016 and additional data in May 2017 from our pivotal inTandem1 Phase 3 clinical trial evaluating the safety and tolerability of sotagliflozin and its effects on glycemic parameters associated with type 1 diabetes. The trial enrolled 793 patients with type 1 diabetes in the United States and Canada in a randomized, double-blind, placebo-controlled study of 200mg and 400mg once daily doses of sotagliflozin over a 24-week treatment period, followed by a 28-week extension. Insulin therapy was optimized in patients over a 6-week period prior to dosing. The primary efficacy endpoint under evaluation in the trial was the reduction of hemoglobin A1c, or A1C, versus placebo on optimized insulin treatment at 24 weeks, with secondary endpoints including percentage of patients achieving A1C levels of less than 7% without experiencing an event of severe hypoglycemia or diabetic ketoacidosis, or DKA, change in meal-time, or bolus, insulin use, body weight, fasting plasma glucose and patient-reported assessments. Data from the study showed that patients treated with sotagliflozin experienced statistically significant reductions in A1C from baseline of 0.43% for the 200mg dose (p<0.001) and 0.48% for the 400mg dose (p<0.001), as compared to a reduction of 0.07% on placebo after 24 weeks of treatment, meeting the study’s primary efficacy endpoint at both dose levels. The A1C benefit achieved with sotagliflozin was sustained with statistically significant results over the full 52-week duration of the study for both the 200mg and 400mg doses. Benefits in all secondary efficacy endpoints were observed in both the 200mg and 400mg dose arms compared to placebo, with statistically significant improvements in all secondary efficacy endpoints observed in the 400mg dose arm and in the percentage of patients achieving A1C levels of less than 7% without any severe hypoglycemia or DKA events and weight loss observed in the 200mg dose arm and statistically significant improvements in all secondary efficacy endpoints observed in the 400mg dose arm. Over the full 52-week treatment period, the incidences of treatment-emergent adverse events in the placebo, 200mg and 400mg dose arms were 80.6%, 81.7% and 79.8%, respectively; the incidences of serious adverse events were 7.5%, 10.3% and 11.1%, respectively; and the incidences of discontinuation due to adverse events were 4.1%, 4.9% and 6.5%, respectively. Potential cases of severe hypoglycemia and DKA were reviewed by a blinded adjudication panel, which determined whether such cases met pre-established diagnostic criteria. The number of patients with positively adjudicated severe hypoglycemic events during the full 52-week treatment period was 26 (9.7%), 17 (6.5%) and 17 (6.5%) in the placebo, 200mg and 400mg dose arms, respectively. The number of patients with positively adjudicated DKA events during the full 52-week treatment period was 1 (0.4%), 9 (3.4%) and 11 (4.2%) in the placebo, 200mg and 400mg dose arms, respectively.
We reported top-line primary efficacy endpoint data in December 2016 and additional data in August 2017 from our pivotal inTandem2 Phase 3 clinical trial evaluating the safety and tolerability of sotagliflozin and its effects on glycemic parameters associated with type 1 diabetes. The trial enrolled 782 patients with type 1 diabetes in Europe and Israel in a randomized, double-blind, placebo-controlled study of 200mg and 400mg once daily doses of sotagliflozin over a 24-week treatment period, followed by a 28-week extension. Insulin therapy was optimized in patients over a 6-week period prior to dosing. As with inTandem1, the primary efficacy endpoint under evaluation in the trial was the reduction of A1C versus placebo on optimized insulin treatment at 24 weeks, with secondary endpoints including percentage of patients achieving A1C levels of less than 7% without experiencing a severe hypoglycemia or DKA event, change in bolus insulin use, body weight, fasting plasma glucose and patient-reported assessments. Data from the study showed that patients treated with sotagliflozin experienced statistically significant reductions in A1C from baseline of 0.39% for the 200mg dose (p<0.001) and 0.37% for the 400mg dose (p<0.001), as compared to a reduction of 0.02% on placebo after 24 weeks of treatment, meeting the study’s primary efficacy endpoint at both dose levels. The A1C benefit achieved with sotagliflozin was sustained with statistically significant results over the full 52-week duration of the study for both the 200mg and 400mg doses. Statistically significant improvements in all secondary efficacy endpoints were observed in both the 200mg and 400mg dose arms compared to placebo. Over the full 52-week treatment period, the incidences of treatment-emergent adverse events in the placebo, 200mg and 400mg dose arms were 61.2%, 68.2% and 68.8%, respectively; the incidences of serious adverse events were 6.6%, 10.0% and 8.0%, respectively; and the incidences of discontinuation due to adverse events were 3.5%, 3.8% and 6.8%, respectively. Potential cases of severe hypoglycemia and DKA were reviewed by a blinded adjudication panel, which determined whether such cases met pre-established diagnostic criteria. The number of patients with positively adjudicated severe hypoglycemic events during the full 52-week treatment period was 13 (5.0%), 13 (5.0%) and 6 (2.3%) in the placebo, 200mg and 400mg dose arms, respectively. The number of patients with positively adjudicated DKA events during the full 52-week treatment period was 0 (0.0%), 6 (2.3%) and 9 (3.4%) in the placebo, 200mg and 400mg dose arms, respectively.
We reported pooled continuous glucose monitoring, or CGM, data in September 2017 from the inTandem1 and inTandem2 clinical trials. The percentage of time during the initial 24-week treatment period spent inside the target range for CGM glucose (70-180 mg/dL) increased from 52.2% to 57.8% in patients treated with 200mg of sotagliflozin and from 50.7% to 64.1% in patients treated with 400mg of sotagliflozin, with no relevant change observed in patients receiving placebo. The differences from placebo were clinically significant for both the 200mg and 400mg dose groups (p=0.026 and p<0.001, respectively). The increase in time spent in range by both sotagliflozin dose groups was a result of significantly reduced time spent above 180 mg/dL, while the time spent below 70 mg/dL was not increased. These results translate into an additional 1.41
3
hours and 3.02 hours that a patient would spend within the 70-180 mg/dL target range in a 24-hour period, for the 200mg and 400mg dose groups respectively.
We reported top-line data in June 2017 from our inTandem3 Phase 3 clinical trial evaluating the safety and tolerability of sotagliflozin and its effects on glycemic parameters associated with type 1 diabetes. The trial enrolled 1,405 patients with type 1 diabetes in the United States and Europe in a randomized, double-blind, placebo-controlled study of a 400mg once daily dose of sotagliflozin over a 24-week treatment period. Insulin therapy was not optimized in patients and eligibility criteria included any background insulin therapy. The primary efficacy endpoint under evaluation in the trial was the proportion of patients achieving A1C levels of less than 7% at 24 weeks without experiencing a severe hypoglycemic or DKA event, with secondary endpoints including the change from baseline in A1C, body weight, systolic blood pressure and bolus insulin use. Data from the study showed statistically significant superiority of sotagliflozin (28.6%) compared to placebo (15.2%) in the proportion of patients achieving A1C levels of less than 7% without experiencing a severe hypoglycemic or DKA event (p<0.001), meeting the study’s primary endpoint. Patients treated with sotagliflozin also experienced statistically significant improvements in all secondary efficacy endpoints compared to placebo. The incidences of treatment-emergent adverse events in the placebo and 400mg dose arms were 52.5% and 55.1%, respectively; the incidences of serious adverse events were 3.3% and 6.9%, respectively; and the incidences of discontinuation due to adverse events were 2.3% and 6.3%, respectively. Potential cases of severe hypoglycemia and DKA were reviewed by a blinded adjudication panel, which determined whether such cases met pre-established diagnostic criteria. The number of patients with positively adjudicated severe hypoglycemic events during the 24-week treatment period was 17 (2.4%) and 21 (3.0%) in the placebo and 400mg dose arms, respectively. The number of patients with positively adjudicated DKA events during the 24-week treatment period was 4 (0.6%) and 21 (3.0%) in the placebo and 400mg dose arms, respectively. Results from the inTandem3 trial were published in the New England Journal of Medicine in September 2017.
We and Sanofi are presently preparing for the submission of applications for regulatory approval to market sotagliflozin for the treatment of type 1 diabetes in the United States and the European Union.
Type 2 Diabetes.
Sanofi is presently conducting a comprehensive Phase 3 development program for sotagliflozin in type 2 diabetes patients, including the following randomized, double-blind, placebo-controlled studies:
• | 200mg and 400mg once daily doses of sotagliflozin as monotherapy in approximately 400 patients over a 26-week treatment period; |
• | 400mg once daily dose of sotagliflozin in approximately 500 patients on background metformin therapy over a 26-week treatment period, followed by a 52-week extension; |
• | 400mg once daily dose of sotagliflozin in approximately 500 patients added to sulfonylurea alone or in combination with metformin over a 26-week treatment period, followed by a 52-week extension; |
• | 200mg or 400mg once daily dose of sotagliflozin in approximately 10,500 patients with cardiovascular risk factors and moderately impaired renal function over a treatment period to be determined by cardiovascular outcome events, currently expected to be approximately four years; |
• | 200mg and 400mg once daily doses of sotagliflozin in approximately 780 patients with moderate renal impairment over a 52-week treatment period; |
• | 200mg and 400mg once daily doses of sotagliflozin in approximately 276 patients with severe renal impairment over a 52-week treatment period; |
• | 200mg and 400mg once daily doses of sotagliflozin in approximately 560 patients on background basal insulin alone or in addition to other oral antidiabetic drug therapies over an 18-week treatment period, followed by a 34-week extension; |
• | 200mg and 400mg once daily doses of sotagliflozin in approximately 700 patients on dipeptidyl peptidase-4, or DPP-4, inhibitors, with or without metformin, compared to 25mg dose of empagliflozin over a 26-week treatment period; |
• | 200mg and 400mg once daily doses of sotagliflozin in approximately 930 patients on background metformin therapy compared to 2-6mg dose of glimepiride over a 52-week treatment period; and |
4
• | 200mg and 400mg once daily doses of sotagliflozin in approximately 360 patients aged 55 years or older, with or without any stable anti-diabetes therapy, evaluating efficacy and bone safety over a 26-week treatment period, followed by a 78-week extension. |
We previously completed two Phase 2 clinical trials evaluating the safety and tolerability of sotagliflozin and its effects on glycemic parameters associated with type 2 diabetes.
The Phase 2b clinical trial enrolled 299 patients with type 2 diabetes who were not adequately controlled on metformin monotherapy in a double-blind, randomized, placebo-controlled study of 75mg once daily, 200mg once daily, 200mg twice daily and 400mg once daily doses of sotagliflozin, each administered in combination with standard metformin therapy over a 12‑week treatment period. The primary efficacy endpoint under evaluation in the trial was the change in A1C from baseline to week 12. Secondary efficacy endpoints included percentage of patients achieving A1C levels of less than 7%, as well as changes in fasting plasma glucose levels, weight, blood pressure and triglyceride levels. Data from the study showed that treatment with sotagliflozin demonstrated statistically significant benefits in the primary and multiple secondary endpoints. Patients in each of the 75mg once daily, 200mg once daily, 200mg twice daily and 400mg once daily sotagliflozin treatment arms had mean A1C reductions from baseline of 0.43, 0.52, 0.79 and 0.92 percent, respectively (p<0.001 for all treatment arms), while in patients randomized to placebo, A1C decreased by 0.09 percent. We also observed that patients treated with sotagliflozin showed significant reductions in body weight and blood pressure. Sotagliflozin was well tolerated and adverse events were generally mild to moderate, with the overall incidence of adverse events with sotagliflozin being similar to placebo.
The Phase 2a clinical trial enrolled 36 patients with non-insulin dependent type 2 diabetes in a double-blind, randomized, placebo-controlled study of 150mg and 300mg doses of sotagliflozin, each administered once daily over a four-week treatment period. The efficacy endpoints under evaluation in the trial included urinary glucose excretion, fasting plasma glucose, response to oral glucose tolerance testing, and change in A1C. Data from the study showed that treatment with 150mg and 300mg of sotagliflozin provided improvements in glycemic control and demonstrated statistically significant benefits in the primary and multiple secondary efficacy endpoints. A marked and statistically significant decrease in fasting plasma glucose was observed at each measurement point throughout the treatment period in both treatment arms relative to placebo. After four weeks of dosing, patients in both dose groups exhibited statistically significant reductions in A1C as compared to patients receiving placebo (p=0.001 and p<0.001 for the 150mg and 300mg treatment arms, respectively). Patients in both treatment arms also exhibited statistically significant improvements in glucose tolerance in response to oral glucose tolerance testing (p<0.001 for both treatment arms). Consistent with the mechanism of action of sotgliflozin, there was also a significant, dose-dependent increase in 24-hour urinary glucose excretion in both treatment arms at each measurement point throughout the study period relative to placebo (p<0.001 at all time points measured). Patients in both treatment arms also showed positive trends in broader metabolic and cardiovascular parameters, including weight reduction, decreased blood pressure and lower triglyceride levels. Sotagliflozin was well tolerated in the trial, with no dose-limiting toxicities observed and adverse events being generally mild and equally distributed across all treatment groups, including the placebo group.
LX2761
LX2761 is an orally-delivered small molecule compound that we are developing for the treatment of diabetes. LX2761 was internally generated by our scientists and is designed to inhibit SGLT1 locally in the gastrointestinal tract without any significant inhibition of SGLT2 in the kidney. We are presently conducting Phase 1 clinical development of LX2761.
We have granted Sanofi certain rights of first negotiation with respect to the future development and commercialization of LX2761.
LX9211
LX9211 is an orally-delivered small molecule compound that we are developing for the treatment of neuropathic pain. LX9211 was jointly generated by our and Bristol-Myers Squibb’s scientists as part of our drug discovery alliance with Bristol-Myers Squibb and inhibits adaptor associated kinase 1, or AAK1, in the central nervous system. Our scientists identified mice lacking AAK1 as having increased resistance to induced neuropathic pain in preclinical models. We are presently conducting Phase 1 clinical development of LX9211.
We have obtained exclusive research, development and commercialization rights to LX9211 and additional compounds acting through AAK1 from Bristol-Myers Squibb.
Drug Target Discoveries
5
Our internal drug discovery efforts were driven by a systematic, target biology-driven approach in which we used gene knockout technologies and an integrated platform of advanced medical technologies to systematically study the physiological and behavioral functions of almost 5,000 genes in mice and assessed the utility of the proteins encoded by the corresponding human genes as potential drug targets. We have identified and validated in living animals, or in vivo, more than 100 targets with promising profiles for drug discovery.
Collaborations
We are working both independently and through strategic collaborations and alliances with third parties to capitalize on our drug target discoveries and drug discovery and development programs. Consistent with this approach, we seek to retain exclusive rights to the benefits of certain drug discovery and development programs by developing and commercializing drug candidates from those programs internally, particularly in the United States for indications treated by specialist physicians, as we have with XERMELO in the United States. We seek to collaborate with other pharmaceutical and biotechnology companies, such as Ipsen and Sanofi, with respect to drug discovery or the development and commercialization of certain of our drug candidates, particularly with respect to commercialization in territories outside the United States, commercialization in the United States for indications treated by primary care physicians, or when the collaboration may provide us with access to expertise and resources that we do not possess internally or are complementary to our own. We also seek to collaborate with other pharmaceutical and biotechnology companies, research institutes and academic institutions to capitalize on our drug target discoveries.
Strategic Collaborations
Sanofi. We entered into a collaboration and license agreement with Sanofi in November 2015 under which we granted Sanofi an exclusive, worldwide, royalty-bearing right and license to develop, manufacture and commercialize sotagliflozin. In December 2016, Sanofi terminated its rights under the agreement with respect to Japan. We received a $300 million upfront payment under the agreement and we are eligible to receive up to $210 million upon the achievement of specified clinical development milestones, up to $220 million upon the achievement of specified regulatory milestones and up to $990 million upon the achievement of specified commercial milestones. We are also entitled to tiered, escalating royalties ranging from low double digit percentages to 40 percent of net sales of sotagliflozin, based on indication and territory, with royalties for the higher band of such range attributable to net sales for type 1 diabetes in the United States, and subject in each case to customary royalty reduction provisions.
We are responsible for all clinical development activities relating to type 1 diabetes and have exercised an exclusive option to co-promote and have a significant role, in collaboration with Sanofi, in the commercialization of sotagliflozin for the treatment of type 1 diabetes in the United States. Under the terms of the exercised co-promotion option, we will fund 40 percent of the commercialization costs relating to such co-promotion activities. Sanofi is responsible for all clinical development and commercialization of sotagliflozin for the treatment of type 2 diabetes worldwide and is solely responsible for the commercialization of sotagliflozin for the treatment of type 1 diabetes outside the United States. We share in the funding of a portion of the planned type 2 diabetes development costs over the first three years of the collaboration, up to an aggregate of $100 million. Sanofi will book sales worldwide in all indications.
Ipsen. We entered into a license and collaboration agreement with Ipsen in October 2014 under which we granted Ipsen an exclusive, royalty-bearing right and license to commercialize telotristat ethyl outside of the United States, Canada and Japan. The collaboration was expanded in March 2015 to include Canada. We have received $24.5 million in upfront payments and $19.2 million in regulatory and commercial launch milestones under the agreement. In addition, we are eligible to receive up to an additional $13.1 million upon the achievement of additional specified regulatory and commercial launch milestones and up to €72 million upon the achievement of specified sales milestones. We are also entitled to tiered, escalating royalties ranging from low twenties to mid-thirties percentages of net sales of telotristat ethyl in the licensed territory, subject to a credit for Ipsen’s payments to us for the manufacture and supply of such units of telotristat ethyl and customary royalty reduction provisions.
Bristol-Myers Squibb. We established a drug discovery alliance with Bristol-Myers Squibb Company in December 2003 to discover, develop and commercialize small molecule drugs in the neuroscience field. Bristol-Myers Squibb extended the target discovery term of the alliance in May 2006. We initiated the alliance with a number of neuroscience drug discovery programs at various stages of development and used our gene knockout technologies to identify additional drug targets with promise in the neuroscience field. For those targets that were selected for the alliance, we and Bristol-Myers Squibb worked together, on an exclusive basis, to identify, characterize and carry out the preclinical development of small molecule drugs. Bristol-Myers Squibb has the first option to assume full responsibility for clinical development and commercialization of any
6
drugs resulting from the alliance which enter clinical trials, other than LX9211 and additional compounds acting through AAK1. We received $86 million in upfront payments and research funding under the agreement during the target discovery portion of the alliance, which expired in October 2009. In addition, we are entitled to receive clinical and regulatory milestone payments ranging, depending on the timing and extent of our efforts in the alliance, up to $76 million for each drug developed by Bristol-Myers Squibb under the alliance. We will also earn royalties on sales of drugs commercialized by Bristol-Myers Squibb under the alliance.
We jointly developed LX9211 with Bristol-Myers Squibb as part of the alliance, and separately obtained from Bristol-Myers Squibb exclusive research, development and commercialization rights to LX9211 and additional compounds acting through AAK1. We have agreed to pay Bristol-Myers Squibb up to $34.5 million in clinical and regulatory milestones for the first indication and up to $16 million in clinical and regulatory milestones for each of the second and third indications, if applicable. We have also agreed to pay single digit royalties on worldwide net sales and up to $40 million in commercial milestones.
Genentech. We established a drug discovery alliance with Genentech, Inc. in December 2002 to discover novel therapeutic proteins and antibody targets. We and Genentech expanded the alliance in November 2005 for the advanced research, development and commercialization of new biotherapeutic drugs. Under the original alliance agreement, we used our target validation technologies to discover the functions of secreted proteins and potential antibody targets identified through Genentech’s internal drug discovery research. In the expanded alliance, we conducted additional, advanced research on a broad subset of those proteins and targets. We have exclusive rights to develop and commercialize biotherapeutic drugs for two of these targets, while Genentech has exclusive rights to develop and commercialize biotherapeutic drugs for the other targets. We retain certain other rights to discoveries made in the alliance, including non-exclusive rights, along with Genentech, for the development and commercialization of small molecule drugs addressing the targets included in the alliance. We received $58 million in upfront payments, research funding and research milestone payments under the agreement during the research collaboration term, which expired in November 2008. In addition, we are entitled to receive clinical and regulatory milestone payments ranging, depending on the extent of our efforts in the alliance, up to $25 million for each drug target for which Genentech develops a biotherapeutic drug under the alliance. We will also earn royalties on sales of biotherapeutic drugs commercialized by Genentech under the alliance. Genentech is entitled to receive milestone payments and royalties on sales of biotherapeutic drugs which we develop or commercialize under the alliance.
Other Collaborations
We have established collaborations with a number of pharmaceutical and biotechnology companies, research institutes and academic institutions under which we have received fees in exchange for generating knockout mice for genes requested by the collaborator, providing phenotypic data with respect to such knockout mice or otherwise granting access to some of our technologies and discoveries. In some cases, we remain eligible to receive milestone or royalty payments on the sale of mice and phenotypic data or on products that our collaborators discover or develop using our technology.
Manufacturing and Product Supply
We do not own or operate manufacturing or distribution facilities or resources for clinical or commercial production and distribution of XERMELO or any of our drug candidates. Instead, we have multiple contractual agreements in place with third-party contract manufacturing organizations, or CMOs, who, on our behalf, manufacture clinical and commercial supplies of XERMELO and clinical supplies of our drug candidates, and will continue to do so for the foreseeable future. Sanofi is responsible for the manufacture of all clinical and commercial supplies of sotagliflozin under the terms of our collaboration. We have selected well-established and reputable global CMOs for our active pharmaceutical ingredient, or API, and drug product manufacturing that have good regulatory standing, large manufacturing capacities, and multiple manufacturing sites within their business footprint. We employ highly skilled personnel with both technical and manufacturing experience to diligently manage the activities at our CMOs. Our quality department audits these suppliers on a periodic basis. Our commercial suppliers are subject to routine inspections by regulatory agencies. We work closely with our third-party manufacturers to ensure compliance with current good manufacturing practices, or cGMP, and other stringent regulatory requirements enforced by the FDA or foreign regulatory agencies in other territories, as applicable.
Raw materials that are used to manufacture our API are sourced from multiple third-party suppliers in Asia and Europe. Third-party API contract manufacturers in Asia and Europe stock sufficient quantities of these materials to ensure they can manufacture adequate API quantities per our requirements, for both clinical and commercial purposes. We store API at third-party facilities, and provide appropriate amounts to third-party drug product contract manufacturers in Asia and North America who then manufacture, package and label our specified quantities of finished goods for XERMELO and our drug candidates. We rely on sole source third-party drug product contract manufacturers in the United States to manufacture,
7
package and label finished drug product for commercial distribution of XERMELO. We also rely on a single third-party logistics provider, with two distribution locations, to provide shipping and warehousing services for our commercial supply of XERMELO in the United States. Our third-party contract manufacturers also need to obtain materials such as excipients, components and reagents to manufacture our API and finished drug products.
Within our supply chain, we have established safety stock amounts for both our API and drug products, and store those quantities for XERMELO in multiple locations. The quantities that we store are based on our business needs and take into account scenarios for market and clinical demand, production lead times, potential supply interruptions and shelf life for our API and drug products. In parallel, for business continuity reasons, we are in the process of evaluating and expect to establish additional or backup suppliers for our API and drug product manufacturers in the near future. We believe that our current manufacturing network has the appropriate capacity to produce sufficient commercial quantities of XERMELO for both our and Ipsen’s commercialization efforts in support of the current approved indication of carcinoid syndrome diarrhea, as well as potential indications of cholangiocarcinoma and neuroendocrine tumors, if those indications prove to be successful and gain regulatory approval in the future.
Marketing, Sales and Distribution
We have a fully integrated commercial team consisting of sales, marketing, market access, and commercial operations functions. Our specialized sales team promotes XERMELO in the United States, concentrating their efforts on oncologists, oncology nurses and pharmacists. We have also built an internal medical affairs function with responsibility for responding to external inquiries regarding the appropriate use of XERMELO with regularly updated and well-substantiated scientific and medical information. We have contracted with two independent specialty pharmacies to dispense XERMELO and provide specialty pharmacy services in fulfillment of prescriptions in the United States, allowing for efficient delivery of XERMELO by mail directly to patients. We rely on Ipsen for the commercialization and distribution of XERMELO in territories outside of the United States.
To help ensure that all eligible patients in the United States have appropriate access to XERMELO, we have established a comprehensive reimbursement and support program called LexCares. Through LexCares, we provide co-pay assistance to qualified, commercially insured patients to help minimize out-of-pocket costs and provide free drug to uninsured or under-insured patients who meet certain clinical and financial criteria. In addition, LexCares is designed to provide comprehensive reimbursement support services, such as prior authorization support, benefits investigation and, if needed, appeals support.
Competition
The biotechnology and pharmaceutical industries are highly competitive and characterized by rapid technological change. We face significant competition in each of the aspects of our business from other pharmaceutical and biotechnology companies, as well as academic research institutions, clinical reference laboratories and governmental agencies that are pursuing research or development activities similar to ours. Many of our competitors have substantially greater research, development and commercialization capabilities and financial, scientific, marketing and human resources than we do. As a result, our competitors may succeed in developing products earlier than we do, obtaining approvals from the FDA or other regulatory agencies for those products more rapidly than we do, developing products that are more effective than those we develop or commercializing products more effectively and profitably than we do. Similarly, our collaborators face similar competition from other competitors who may succeed in developing products more quickly, developing products that are more effective than those developed by our collaborators or commercialize products more effectively and profitably than our collaborators.
The competition for our products and drug candidates includes both marketed products and drug candidates that are being developed by others, including pharmaceutical products that are currently in a more advanced stage of clinical development or commercialization than are our own drug candidates. These competitive marketed products and drug candidates include compounds that employ different mechanisms of action in addressing diseases and conditions for which we are developing our own drug candidates and, in some cases such as sotagliflozin, that employ the same or similar mechanisms of action.
We believe that our ability to successfully compete with these potentially competitive drug candidates and other competitive products currently on the market will depend on, among other things:
• | the efficacy, safety and reliability of our products; |
8
• | our ability, and the ability of our collaborators, to complete preclinical and clinical development and obtain regulatory approvals for our drug candidates; |
• | the timing and scope of regulatory approvals of our products; |
• | our ability, and the ability of our collaborators, to obtain product acceptance by physicians and other health care providers and secure coverage and adequate reimbursement for product use in approved indications; |
• | our ability, and the ability of our collaborators, to manufacture and sell commercial quantities of our products; |
• | the skills of our employees and our ability to recruit and retain skilled employees; |
• | protection of our intellectual property; and |
• | the availability of substantial capital resources to fund development and commercialization activities. |
Our principal competition for XERMELO includes the use, above their maximum labeled dose, of the established SSA therapies octreotide and lanreotide, injectable products currently marketed by Novartis and Ipsen, respectively. In addition, we also expect that XERMELO will experience competition from lutetium Lu 177 dotatate, a radiopharmaceutical product currently marketed for the treatment of gastroenteropancreatic neuroendocrine tumors by Advanced Accelerator Applications (a subsidiary of Novartis).
If approved for the treatment of type 1 diabetes, we expect that our principal competition for sotagliflozin will include established insulin therapies, as well as selective SGLT2 inhibitors which may gain regulatory approval for the treatment of type 1 diabetes, such as dapagliflozin, empagliflozin and canagliflozin, currently marketed for the treatment of type 2 diabetes by AstraZeneca, Boehringer Ingelheim and Eli Lilly, and Janssen (a subsidiary of Johnson & Johnson), respectively. If approved for the treatment of type 2 diabetes, we expect that our principal competition for sotagliflozin will include such selective SGLT2 inhibitors, as well as DPP-4 inhibitors such as sitagliptin, currently marketed for the treatment of type 2 diabetes by Merck.
Government Regulation
The development, manufacture and sale of pharmaceutical products are subject to extensive regulation by United States and foreign governmental authorities, including federal, state and local authorities. In the United States, new drugs are subject to regulation under the Federal Food, Drug and Cosmetic Act and the regulations promulgated thereunder, or the FDC Act. The FDA and comparable governmental authorities regulate, among other things, research and development activities and the testing, manufacture, quality control, safety, efficacy, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, export and import of pharmaceutical products.
The standard process required by the FDA before a drug candidate may be marketed in the United States generally includes the following:
• | preclinical laboratory and animal tests performed under current good laboratory practices, or cGLP; |
• | submission of an IND, which must become effective before human clinical trials may commence; |
• | adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug candidate for its intended use; |
• | submission of a New Drug Application, or NDA, for approval of commercial marketing and sale, or of an NDA supplement, or sNDA, for approval of a new indication if the product is already approved for another indication; |
• | pre-approval inspection of manufacturing facilities and selected clinical investigators for their compliance with cGMP and current good clinical practices, or cGCP; |
• | if FDA convenes an advisory committee, satisfactory completion of the advisory committee review; and |
• | FDA approval of the NDA or sNDA. |
9
This process for the testing and approval of drug candidates requires substantial time, effort and financial resources. Preclinical development of a drug candidate can take from one to several years to complete, with no guarantee that an IND based on those studies will become effective to even permit clinical testing to begin. Before commencing the first clinical trial of a drug candidate in the United States, we must submit an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial. In such a case, we and the FDA must resolve any outstanding concerns before the clinical trial may begin. Submission of an IND may not result in FDA authorization to commence a clinical trial. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development, and the FDA must grant permission for each clinical trial to start and continue. Further, an independent institutional review board for each medical center proposing to participate in the clinical trial must review and approve the plan for any clinical trial before it commences at that center. Regulatory authorities or an institutional review board or we may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
For purposes of NDA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
• | Phase 1 clinical trials are conducted in a limited number of healthy human volunteers or, in some cases, patients, to evaluate the safety, dosage tolerance, absorption, metabolism, distribution and excretion of the drug candidate; |
• | Phase 2 clinical trials are conducted in groups of patients afflicted with a specified disease or condition to obtain preliminary data regarding efficacy as well as to further evaluate safety and optimize dosing of the drug candidate. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials; and |
• | Phase 3 clinical trials are conducted in larger patient populations at multiple clinical trial sites to obtain statistically significant evidence of the efficacy of the drug candidate for its intended use and to further test for safety in an expanded patient population. |
In addition, the FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so-called Phase 4 studies may be made a condition to be satisfied after a drug receives approval. Failure to satisfy such post-marketing commitments can result in FDA enforcement action, up and to including withdrawal of NDA approval. The results of phase 4 studies can confirm the effectiveness of a drug candidate and can provide important safety information to augment the FDA’s adverse drug reaction reporting system.
After completion of clinical trials, FDA approval of an NDA must be obtained before a new drug may be marketed in the United States. The submission of an NDA requires payment of a substantial user fee to the FDA. An NDA must contain, among other things, information on chemistry, manufacturing controls and potency and purity, non-clinical pharmacology and toxicology, human pharmacokinetics and bioavailability and clinical data. There can be no assurance that the FDA will accept an NDA for filing and, even if accepted for filing, that approval will be granted. The FDA may convene an advisory committee to provide clinical insight on NDA review questions. Although the FDA is not required to follow the recommendations of an advisory committee, the agency typically does so. Among other things, the FDA reviews an NDA to determine whether a product is safe and effective for its intended use and whether the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may deny approval of an NDA by way of a Complete Response letter if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or an additional pivotal Phase 3 clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. An NDA may be approved with significant restrictions on its labeling, marketing and distribution under a Risk Evaluation and Mitigation Strategy or otherwise that could restrict the commercial applications of a product or impose costly procedures in connection with the commercialization or use of the product. Once issued, the FDA may withdraw product approval if ongoing regulatory standards are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
In addition to obtaining FDA approval for each product, each drug manufacturing establishment must be inspected and approved by the FDA. All manufacturing establishments are subject to inspections by the FDA and by other federal, state and local agencies and must comply with current Good Manufacturing Practices requirements. Non-compliance with these requirements can result in, among other things, total or partial suspension of production, failure of the government to grant approval for marketing and withdrawal, suspension or revocation of marketing approvals.
10
Satisfaction of FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes many years, with the actual time required varying substantially based on, among other things, the nature, novelty and complexity of the drug candidate and of the disease or condition. Government regulation may delay or prevent marketing of drug candidates or new diseases for a considerable period of time and impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approvals for new indications for our product candidates on a timely basis, if at all. Success in earlier-stage clinical trials does not ensure success in later-stage clinical trials. Targets and pathways identified in vitro may be determined to be less relevant in clinical studies and results in animal model studies may not be predictive of human clinical results. Furthermore, data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. Even if a drug candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
Once the FDA approves a product, a manufacturer must provide certain updated safety and efficacy information. Product changes as well as certain changes in a manufacturing process or facility would necessitate additional FDA review and approval. Other post-approval changes may also necessitate further FDA review and approval. Additionally, a manufacturer must meet other requirements including those related to adverse event reporting and record keeping.
Products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, which impose certain procedural and documentation requirements upon us and our third-party manufacturers.
The FDA closely regulates the marketing and promotion of drugs, including restricting the promotion of uses for which a drug is not approved by the agency. Not only must a company have appropriate substantiation to support claims made about a drug, under the FDA’s current interpretation of relevant laws, a company can make only those claims relating to safety and efficacy that are for indications for which FDA has approved the drug and are otherwise consistent with the FDA-approved label for the drug. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may, in their independent medical judgment, prescribe legally available drugs for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturers’ communications on the subject of off-label use. Additionally, a significant number of pharmaceutical companies have been the target of inquiries and investigations by various United States federal and state regulatory, investigative, prosecutorial and administrative entities in connection with the promotion of products for off-label uses and other sales practices. These investigations have alleged violations of various United States federal and state laws and regulations, including claims asserting antitrust violations, violations of the FDC Act, false claims laws, the Prescription Drug Marketing Act, anti-kickback laws, and other alleged violations in connection with the promotion of products for unapproved uses, pricing and Medicare and/or Medicaid reimbursement.
The United States Orphan Drug Act is intended to incentivize the development of products for rare diseases or conditions that affect fewer than 200,000 people in the United States. If a drug is being developed for a rare disease or condition, to be eligible for designation as an orphan drug, the FDA must not have previously approved a drug considered the “same drug” for the same orphan indication. If the FDA has previously approved another same drug for the same indication, the sponsor of the subsequent drug would be required to provide a plausible hypotheses of clinical superiority over the previously approved drug to obtain an orphan designation. Upon FDA receipt of orphan drug designation, the sponsor is eligible for tax credits of up to 25% for qualified clinical trial expenses, the ability to apply for annual grant funding and waiver of PDUFA application fee. In addition, upon marketing approval, an orphan-designated drug could be eligible for seven years of market exclusivity for the approved orphan-designated indication. Such orphan drug exclusivity, if awarded, would only block the approval of any drug considered the same drug for the same orphan indication. Moreover, a subsequent same drug could break a previously approved drug’s orphan exclusivity through a demonstration of clinical superiority over the previously approved drug.
The FDA has various programs, including Fast Track, priority review and accelerated approval, which are intended to expedite or simplify the process for developing and reviewing promising drugs, or to provide for the approval of a drug on the basis of a surrogate endpoint. Generally, drugs that are eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of drugs to
11
treat serious or life-threatening diseases or conditions and fill unmet medical needs. Priority review is designed to give drugs that treat serious conditions and offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months of NDA filing as compared to a standard review time of 10 months from NDA filing. Certain other types of drug applications are also eligible for priority review. Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track-designated drug and expedite review of the application for a drug designated for priority review. Accelerated approval provides for an earlier approval for a new drug that is intended to treat a serious or life-threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint. As a condition of approval, the FDA may require that a sponsor of a product candidate receiving accelerated approval perform post-marketing clinical trials to confirm the clinically meaningful outcome as predicted by the surrogate marker trial. In addition to the Fast Track, accelerated approval and priority review programs, the FDA also designates Breakthrough Therapy status to drugs that are intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. The FDA will seek to ensure the sponsor of a breakthrough therapy product candidate receives intensive guidance on an efficient drug development program, intensive involvement of senior managers and experienced staff on a proactive, collaborative and cross-disciplinary review and rolling review.
Additional programs intended to expedite the development of drug products were included in the 21st Century Cures Act, or the Cures Act. The Cures Act includes various provisions to accelerate the development and delivery of new treatments, such as those intended to expand the types of evidence manufacturers may bring to the FDA to support drug approval, to encourage patient-centered drug development, to liberalize the communication of healthcare economic information to payers, and to create greater transparency with regard to manufacturer expanded access programs. Central to the Cures Act are provisions that enhance and accelerate the FDA’s processes for reviewing and approving new drugs and supplements to approved NDAs, including provisions that:
• | require the FDA to establish a program to evaluate the potential use of real world evidence to help support the approval of a new indication for an approved drug and to help support or satisfy post-approval study requirements; |
• | provide that the FDA may rely upon qualified data summaries to support the approval of a supplemental application with respect to a qualified indication for an already approved drug; |
• | require the FDA to issue guidance for purposes of assisting sponsors in incorporating complex adaptive and other novel trial designs into proposed clinical protocols and applications for new drugs; and |
• | require the FDA to establish a process for the qualification of drug development tools for use in supporting or obtaining FDA approval for or investigational use of a drug. |
The Cures Act amends Section 114 of the Food and Drug Administration Modernization Act of 1997 to help clarify and facilitate the dissemination of healthcare economic information, including by broadening the definition of healthcare economic information, expressly extending the dissemination of healthcare economic information to payors, and clarifying that healthcare economic information must only relate to an FDA-approved indication rather than directly relate to the indication.
Regulation Outside of the United States
In addition to regulations in the United States, we are subject to the regulations of other countries governing clinical trials and the manufacturing, commercial sales and distribution of our products outside of the United States. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of countries outside of the United States before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European Union regulatory systems, a company may submit marketing authorization applications, or MAAs, either under a centralized or decentralized procedure. Under the centralized procedure, MAAs are submitted to the European Medicines Agency, or EMA, whose Committee for Medicinal Products for Human Use reviews the application and issues an opinion on it. The opinion is considered by the European Commission which is responsible for deciding applications. If the application is approved, the European Commission grants a single marketing authorization that is valid for all European Union member states as well as Iceland, Liechtenstein and Norway, or the EEA. The national authorization procedures, the
12
decentralized and mutual recognition procedures, as well as national applications, are available for products for which the centralized procedure is not compulsory. The mutual recognition procedure provides for the European Union member states selected by the applicant to mutually recognize a national marketing authorization that has already been granted by the competent authority of another member state, referred to as the Reference Member State, or RMS. The decentralized procedure is used when the product in question has yet to be granted a marketing authorization in any member state. Under this procedure the applicant can select the member state that will act as the RMS. In both the mutual recognition and decentralized procedures, the RMS reviews the application and submits its assessment of the application to the member states where marketing authorizations are being sought, referred to as Concerned Member States or CMS. Within 90 days of receiving the application and assessment report, each CMS must decide whether to recognize the RMS assessment. If a member state does not agree with the assessment, and the disputed points cannot be resolved the matter is eventually referred to the European Commission, whose decision is binding on all member states. If the application is successful national marketing authorizations will be granted by the competent authorities in each of the member states chosen by the applicant.
Conditional marketing authorizations may be granted for a limited number of medicinal products for human use referenced in European Union law applicable to conditional marketing authorizations where the clinical dataset is not comprehensive, if the risk-benefit balance of the product is positive, it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, unmet medical needs will be fulfilled and the benefit to public health of the immediate availability on the market of the medicinal product outweighs the risk inherent in the fact that additional data are still required. Specific obligations, such as the completion of ongoing or new studies and obligations relating to the collection of pharmacovigilance data, may be amongst the conditions stipulated in the marketing authorization.
As in the United States, we may apply for designation of a product as an Orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. In the European Union, orphan designation is available for products in development which are either intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union, or intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the community and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the medicinal product. Additionally, the sponsor of an application for orphan drug designation must establish that there exists no satisfactory authorized method of diagnosis, prevention, or treatment of the condition or even if such treatment exists, the product will be of significant benefit to those affected by that condition.
Orphan drugs in the European Union enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan-designated product. The period of market exclusivity may be reduced to six years if at the end of the fifth year it is established that the criteria for orphan designation are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Healthcare Regulation
Federal and state healthcare laws, including fraud and abuse and health information privacy and security laws, also apply to our business. If we fail to comply with those laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected. The laws that may affect our ability to operate include, but are not limited to: the federal Anti-Kickback Statute, which prohibits. among other things, soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; and federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent. Additionally, we are subject to state law equivalents of each of the above federal laws, which may be broader in scope and apply regardless of whether the payer is a federal healthcare program, and many of which differ from each other in significant ways and may not have the same effect, further complicate compliance efforts.
Numerous federal and state laws, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use and disclosure of personal information. Other countries also have, or are developing, laws governing the collection, use and transmission of personal information. In addition, most healthcare providers who are expected to prescribe our products and from whom we obtain patient health information, are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology and Clinical Health Act, or HIPAA. Although we are not directly subject to HIPAA, we could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a HIPAA-covered entity, including healthcare providers, in a manner that is not authorized or permitted by HIPAA. The
13
legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues with the potential to affect our business, including recently enacted laws in a majority of states requiring security breach notification. These laws could create liability for us or increase our cost of doing business. International laws, such as the EU Data Privacy Directive and Swiss Federal Act on Data Protection, regulate the processing of personal data within the European Union and between countries in the European Union and countries outside of the European Union, including the United States. Failure to provide adequate privacy protections and maintain compliance with safe harbor mechanisms could jeopardize business transactions across borders and result in significant penalties.
In addition, the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act, or the PPACA, created a federal requirement under the federal Open Payments program, that requires certain manufacturers to track and report to the Centers for Medicare and Medicaid Services, or CMS, annually certain payments and other transfers of value provided to physicians and teaching hospitals made in the previous calendar year. In addition, there are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities.
For those marketed products which are covered in the United States by the Medicaid program, we have various obligations, including government price reporting and rebate requirements, which generally require products be offered at substantial rebates/discounts to Medicaid and certain purchasers. We are also required to discount such products to authorized users of the Federal Supply Schedule of the General Services Administration, under which additional laws and requirements apply. These programs require submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations, and the guidance governing such calculations is not always clear. Compliance with such requirements can require significant investment in personnel, systems and resources, but failure to properly calculate our prices, or offer required discounts or rebates could subject us to substantial penalties.
Other Regulations
In addition to the foregoing, our business is subject to regulation under various state and federal environmental laws, including the Occupational Safety and Health Act, the Resource Conservation and Recovery Act and the Toxic Substances Control Act. These and other laws govern our use, handling and disposal of various biological, chemical and radioactive substances used in and wastes generated by our operations. We believe that we are in material compliance with applicable environmental laws and that our continued compliance with these laws will not have a material adverse effect on our business. We cannot predict, however, whether new regulatory restrictions will be imposed by state or federal regulators and agencies or whether existing laws and regulations will adversely affect us in the future.
Patents and Proprietary Rights
We are able to protect our proprietary rights from unauthorized use by third parties only to the extent that those rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. Accordingly, patents and other proprietary rights are an essential element of our business. We own or exclusively license patents and/or patent applications throughout the world that claim our approved drug, XERMELO, and our drug candidates, including:
• | issued patents and pending patent applications in Europe, the United States, and other countries throughout the world, including Australia, Argentina, Brazil, Canada, China, Europe, India, Israel, Japan, Mexico, New Zealand, South Africa, and South Korea, that claim telotristat ethyl and associated crystalline forms, pharmaceutical compositions comprising telotristat ethyl, and methods of its manufacture and use; |
• | issued patents and pending patent applications in Europe, the United States, and other countries throughout the world, including Australia, Argentina, Brazil, Canada, China, Europe, India, Israel, Japan, Mexico, New Zealand, South Africa, and South Korea, that claim sotagliflozin and associated crystalline forms, pharmaceutical compositions comprising sotagliflozin, and methods of its manufacture and use; |
• | pending patent applications in Europe, the United States, and other countries throughout the world, including Australia, Argentina, Brazil, Canada, China, Europe, India, Israel, Japan, Mexico, New Zealand, South Africa, and South Korea, that disclose and/or claim LX2761, pharmaceutical compositions comprising LX2761, and methods of its use; and |
14
• | pending patent applications in Europe, the United States, and other countries throughout the world, including Australia, Argentina, Brazil, Canada, China, Europe, India, Israel, Japan, Mexico, New Zealand, South Africa, and South Korea, that disclose and/or claim LX9211, pharmaceutical compositions comprising LX9211, and methods of its use. |
Additionally, we hold rights to a number of patents and patent applications under license agreements with third parties. Many of these licenses are nonexclusive, although some are exclusive in specified fields. Most of the licenses have terms that extend for the life of the licensed patents.
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country. We have filed patent applications and hold issued patents covering our approved drug, XERMELO, and each of our drug candidates. None of our United States patents that claim XERMELO or one of our drug candidates has a normal expiration date earlier than 2026.
All of our employees, consultants and advisors are required to execute a proprietary information agreement upon the commencement of employment or consultation. In general, the agreement provides that all inventions conceived by the employee or consultant, and all confidential information developed or made known to the individual during the term of the agreement, shall be our exclusive property and shall be kept confidential, with disclosure to third parties allowed only in specified circumstances. We cannot assure you, however, that these agreements will provide useful protection of our proprietary information in the event of unauthorized use or disclosure of such information.
Our patent and intellectual property rights are subject to certain rights and uncertainties. See “Risks Related to Our Intellectual Property” under “Item 1A. Risk Factors.”
Executive Officers
Our executive officers and their ages and positions are listed below.
Name | Age | Position with the Company |
Lonnel Coats | 53 | President and Chief Executive Officer and Director |
Pablo Lapuerta, M.D. | 54 | Executive Vice President and Chief Medical Officer |
Alan J. Main, Ph.D. | 64 | Executive Vice President, Commercial Supply Operations |
Alexander A. Santini | 59 | Executive Vice President and Chief Commercial Officer |
Praveen Tyle, Ph.D. | 57 | Executive Vice President, Research and Development |
Jeffrey L. Wade | 53 | Executive Vice President, Corporate and Administrative Affairs and Chief Financial Officer |
James F. Tessmer | 58 | Vice President, Finance and Accounting |
Lonnel Coats has been our president and chief executive officer and a director since July 2014. Mr. Coats previously served in a series of executive leadership positions at Eisai Inc. and Eisai Corporation of North America, where he worked for 18 years before joining our company, most recently as chief executive officer from 2010 to 2014 and president and chief operating officer from 2004 to 2010. Prior to joining Eisai, Mr. Coats spent eight years with Janssen Pharmaceuticals, Inc., a division of Johnson & Johnson, where he held a variety of management and sales positions. Mr. Coats serves as a director of Blueprint Medicines Corporation and holds a B.S. from Oakland University.
Pablo Lapuerta, M.D. has been our executive vice president and chief medical officer since February 2015 and previously served in a series of medical and clinical leadership positions since joining our company in 2011. Dr. Lapuerta was formerly vice president at Bristol-Myers Squibb Company with responsibility for global development of an Alzheimer’s disease drug candidate, and prior to that served as senior vice president, clinical strategy and chief medical officer of Cogentus Pharmaceuticals, Inc. and in a variety of clinical development leadership roles at Bristol-Myers Squibb, where he worked for 11 years before joining Cogentus. He holds a B.A. in biology from Harvard College and an M.D. from Harvard Medical School.
Alan J. Main, Ph.D. has been our executive vice president, commercial supply operations since May 2017 and previously served in a series of manufacturing and scientific leadership positions since joining our company in 2001. Dr. Main was president and chief executive officer of Coelacanth Corporation, a leader in using proprietary chemistry technologies to
15
rapidly discover new chemical entities for drug development, until our acquisition of Coelacanth in 2001. Dr. Main was formerly senior vice president, U.S. Research at Novartis Pharmaceuticals Corporation, where he worked for 20 years before joining Coelacanth. Dr. Main holds a B.S. from the University of Aberdeen, Scotland and a Ph.D. in organic chemistry from the University of Liverpool, England and completed postdoctoral studies at the Woodward Research Institute.
Alexander A. Santini has been our executive vice president and chief commercial officer since November 2016 and previously served in a series of commercial leadership positions since joining our company in April 2015. Mr. Santini was formerly vice president of market access and an executive member at Bayer Healthcare Pharmaceuticals, where he had executive responsibility for market access, pricing, trade and channel management and payer account management, and prior to that served in a variety of commercial leadership roles of increasing responsibility during eight years of service at Bayer and 22 years of service at Berlex Laboratories. Mr. Santini served as a non-commissioned officer in the United States Air Force, where he completed the Radiologic Technology Program at the United States Air Force School of Health Care Science and an AAS in business marketing from Westchester Community College.
Praveen Tyle, Ph.D. has been our executive vice president of research and development since May 2016. Dr. Tyle was previously a member of the executive management team at Osmotica Pharmaceutical Corp., serving as president and chief executive officer from January 2013 through April 2016 and prior to that as executive vice president and chief scientific officer. Prior to his service at Osmotica, Dr. Tyle held a series of scientific leadership positions within the pharmaceutical industry, including executive vice president and chief science officer for the United States Pharmacopeia, senior vice president and global head of research and development and business development and licensing at Novartis OTC, corporate senior vice president of global research and development and chief scientific officer at Bausch & Lomb Incorporated and vice president and global head of pharmaceutical sciences at Pharmacia Corporation. Dr. Tyle serves as director of Eyegate Pharmaceuticals, Inc. and Orient Europharma Ltd. Dr. Tyle received his B.Pharm. from the Indian Institute of Technology, Banaras Hindu University and his Ph.D. in pharmaceutics and pharmaceutical chemistry from the Ohio State University.
Jeffrey L. Wade has been our executive vice president, corporate and administrative affairs and chief financial officer since February 2015 and previously served in a series of finance and legal leadership positions since joining our company in 1999. Mr. Wade was previously a corporate securities and finance attorney for ten years with the law firm of Andrews & Kurth L.L.P., where he represented companies in the biotechnology, information technology and energy industries. Mr. Wade is a member of the board of directors of the Texas Healthcare and Bioscience Institute. He received his B.A. and J.D. from the University of Texas.
James F. Tessmer has been our vice president, finance and accounting since November 2007 and previously served in a series of finance and accounting leadership positions since joining our company in 2001. Mr. Tessmer was previously assistant controller for Mariner Health Network, Inc. and prior to that served in a variety of financial and accounting management positions for HWC Distribution Corp. and American General Corporation. Mr. Tessmer is a certified public accountant and received his B.B.A. from the University of Wisconsin – Milwaukee and his M.B.A. from the University of Houston.
Employees
As of February 26, 2018, we employed 174 persons, of whom 33 hold M.D. or Ph.D. degrees and another 41 hold other advanced degrees. All of our employees are located in the United States. None of our employees are represented by a labor union and we believe that our relationship with our employees is good.
Research and Development Expenses
In 2017, 2016 and 2015, respectively, we incurred expenses of $156.8 million, $178.2 million and $95.2 million in company-sponsored as well as collaborative research and development activities, including $4.9 million, $3.9 million and $3.7 million of stock-based compensation expense in 2017, 2016 and 2015, respectively.
16
Item 1A. Risk Factors
The following risks and uncertainties are important factors that could cause actual results or events to differ materially from those indicated by forward-looking statements. The factors described below are not the only ones we face and additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations.
Risks Related to Our Business and Industry
We depend heavily on the commercial success of XERMELO. If we do not achieve commercial success with XERMELO, our business will suffer and our stock price will likely decline.
We expect that a significant portion of our total revenues for the next several years will be attributable to sales of XERMELO in the United States, but we cannot be certain that XERMELO will be commercially successful. Our future sales of XERMELO will depend on numerous factors, including:
• | the number of patients with carcinoid syndrome diarrhea who are inadequately controlled by SSA therapy, as well as the number of newly diagnosed carcinoid syndrome diarrhea patients; |
• | competition from SSA therapies, radiopharmaceutical products and any additional products for the treatment of carcinoid syndrome diarrhea that may be approved by the FDA in the future; |
• | the safety profile of XERMELO, including whether previously unknown side effects or increased incidence or severity of known side effects as compared to those seen during development are identified with the increased use of XERMELO after approval; |
• | the effectiveness of our commercial strategy for marketing XERMELO and our execution of that strategy, including our pricing strategy and the effectiveness of our efforts to obtain adequate third-party reimbursement; |
• | the acceptance of XERMELO by patients, the medical community and third-party payers; and |
• | our ability to meet the demand for commercial supplies of XERMELO and to maintain and successfully monitor commercial manufacturing arrangements for XERMELO with third-party manufacturers to ensure they meet our standards and those of the FDA, which extensively regulates and monitors pharmaceutical manufacturing facilities. |
While we believe that XERMELO has a competitive commercial profile, our current estimates of the revenues that XERMELO could generate in future periods may change based upon the above factors, and could prove to be incorrect. If our revenues, market share or other indicators of market acceptance of XERMELO fail to meet the expectations of investors or public market analysts, the market price of our common stock could decline. In addition, if one or more of the factors above negatively affects XERMELO sales, our business and financial condition could be materially harmed and we may be more heavily dependent on the success of our other drug programs.
We depend heavily on our and Sanofi’s ability to obtain regulatory approval in the United States and the European Union for sotagliflozin in type 1 diabetes. If we and Sanofi fail to obtain such regulatory approval or fail to successfully commercialize sotagliflozin for type 1 diabetes upon regulatory approval, our business will suffer and our stock price will likely decline.
We and Sanofi are presently preparing for the submission of applications for regulatory approval to market sotagliflozin for the treatment of type 1 diabetes in the United States and the European Union. We cannot offer any assurances or predict with any certainty that the FDA and/or EMA will accept such applications for filing or grant marketing approval for sotagliflozin, in either case on the expected timelines. Furthermore, regulatory approvals for sotagliflozin, even if obtained, may limit the type of patients in which sotagliflozin may be used or otherwise require specific warning or labeling language, each of which may reduce the commercial potential of sotagliflozin. Even if approved, we and Sanofi might not be successful in commercializing sotagliflozin for type 1 diabetes. Should we and Sanofi fail to obtain regulatory approval for sotagliflozin in type 1 diabetes or fail to successfully commercialize sotagliflozin upon such regulatory approval, our business and financial condition could be materially harmed and we may be more heavily dependent on the success of our other drug programs.
17
Clinical testing of our drug candidates in humans is an inherently risky and time-consuming process that may fail to demonstrate safety and efficacy, which could result in the delay, limitation or prevention of regulatory approval.
In order to obtain regulatory approvals for the commercial sale of any products that we or our collaborators may develop in addition to XERMELO, we or our collaborators are required to complete extensive clinical trials in humans to demonstrate the safety and efficacy of our drug candidates. We or our collaborators may not be able to obtain authority from the FDA, or other equivalent foreign regulatory agencies to initiate or complete any clinical trials. In addition, we have limited internal resources for making regulatory filings and interacting with regulatory authorities.
Clinical trials are inherently risky and the results from nonclinical testing of a drug candidate that is under development may not be predictive of results that will be obtained in human clinical trials. In addition, the results of early human clinical trials may not be predictive of results that will be obtained in larger-scale, advanced stage clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after achieving positive results in earlier trials. Although Phase 2 proof-of-concept clinical trials of sotagliflozin in type 2 diabetes patients were positive, we cannot assure you that the Phase 3 clinical development program for sotagliflozin being conducted by Sanofi in type 2 diabetes patients will yield positive results. Negative or inconclusive results from a nonclinical study or a clinical trial could cause us, our collaborators or the FDA or other equivalent foreign regulatory agencies to terminate a nonclinical study or clinical trial or require that we or our collaborators repeat or modify it. Furthermore, we, one of our collaborators or a regulatory agency with jurisdiction over the trials may suspend clinical trials at any time if the subjects or patients participating in such trials are being exposed to unacceptable health risks or for other reasons.
Any nonclinical or clinical test may fail to produce results satisfactory to the FDA or foreign regulatory authorities. Nonclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval. The FDA or institutional review boards at the medical institutions and healthcare facilities where we or our collaborators sponsor clinical trials may suspend any trial indefinitely if they find deficiencies in the conduct of these trials. Clinical trials must be conducted in accordance with the FDA’s current Good Clinical Practices. The FDA and these institutional review boards have authority to oversee our and our collaborators’ clinical trials, and the FDA may require large numbers of subjects or patients. In addition, we or our collaborators must manufacture, or contract for the manufacture of, the drug candidates that we use in our clinical trials under the FDA’s current Good Manufacturing Practices.
The rate of completion of clinical trials is dependent, in part, upon the rate of enrollment of patients. Patient accrual is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the study, the nature of the study, the existence of competitive clinical trials and the availability of alternative treatments. Delays in planned patient enrollment may result in increased costs and prolonged clinical development, which in turn could allow our competitors to bring products to market before we do and impair our ability to commercialize our products or potential products.
We or our collaborators may not be able to successfully complete any clinical trial of a drug candidate within any specified time period. In some cases, we or our collaborators may not be able to complete the trial at all. Moreover, clinical trials may not show our drug candidates to be both safe and effective. Thus, the FDA and other regulatory authorities may not approve any additional drug candidates that we develop for any indication or may limit the approved indications or impose other conditions.
Our drug candidates are subject to a lengthy and uncertain regulatory process that may not result in the necessary regulatory approvals, which could adversely affect our and our collaborators’ ability to commercialize products.
Our drug candidates, as well as the activities associated with their research, development and commercialization, are subject to extensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain regulatory approval for any drug candidate would prevent us from commercializing that drug candidate. Other than XERMELO, we and our collaborators have not received regulatory approval to market any of our drug candidates in any jurisdiction. The process of obtaining regulatory approvals is expensive, and often takes many years, if approval is obtained at all, and can vary substantially based upon the type, complexity and novelty of the drug candidates involved. Before a new drug application can be filed with the FDA, the drug candidate must undergo extensive clinical trials, which can take many years and may require substantial expenditures. Any clinical trial may fail to produce results satisfactory to the FDA. For example, the FDA could determine that the design of a clinical trial is inadequate to produce reliable results. Furthermore, prior to approving a new drug, the FDA typically requires that the efficacy of the drug be demonstrated in two double-blind, controlled studies. The regulatory process also requires nonclinical testing, and data obtained from nonclinical and clinical activities are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. In addition, delays or rejections may be encountered based upon changes in regulatory policy for product approval
18
during the period of product development and regulatory agency review. Changes in regulatory approval policy, regulations or statutes or the process for regulatory review during the development or approval periods of our drug candidates may cause delays in the approval or rejection of an application. Even if the FDA or a comparable authority in another country approves a drug candidate, the approval may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product and may impose ongoing requirements for post-approval studies, including additional research and development and clinical trials. These agencies also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval.
The commercial success of XERMELO and any other products that we or our collaborators may develop will depend upon the degree of market acceptance among physicians, patients, health care payers and the medical community.
Our ability to commercialize XERMELO and our or our collaborators’ ability to commercialize any other products that we or they may develop will be highly dependent upon the extent to which XERMELO and such other products gain market acceptance among physicians, patients, health care payers, such as commercial health insurers, Medicare and Medicaid, and the medical community. If XERMELO and such other products do not achieve an adequate level of acceptance, we may not generate adequate product revenues and we may not become profitable. The degree of market acceptance of XERMELO and such other products will depend upon a number of factors, including:
• | the effectiveness, or perceived effectiveness, of our products in comparison to competing products; |
• | the existence of any significant side effects, as well as their severity in comparison to any competing products; |
• | potential advantages or disadvantages in relation to alternative treatments; |
• | current and future indications for which our products may be approved; |
• | the ability to offer our products for sale at competitive prices; |
• | relative convenience and ease of administration; |
• | the strength of marketing and distribution support; and |
• | sufficient third-party coverage or reimbursement. |
If we are unable to implement and maintain an effective and specialized sales force, marketing infrastructure and distribution capabilities, we will not be able to successfully commercialize XERMELO or any other products that we or our collaborators may develop.
In order to successfully commercialize XERMELO, we have built a marketing organization and a specialized sales force for XERMELO and established distribution capabilities in the United States. However, we had no prior experience in building and maintaining such a commercialization infrastructure. Factors that may hinder our efforts to effectively manage and maintain such infrastructure for XERMELO or establish, manage and maintain such infrastructure for other products that we or our collaborators may develop include:
• | inability to recruit, retain and effectively manage adequate numbers of effective sales and marketing personnel; |
• | inability to maintain relationships with third-party logistics providers, specialty pharmacies, third-party manufacturers and other third parties instrumental in the commercial manufacture and distribution of XERMELO and any other products; |
• | inability to establish or implement internal controls and procedures required in connection with sales of pharmaceutical products; |
• | inability of sales personnel to obtain access to or convince adequate numbers of physicians to prescribe XERMELO or any other products; and |
• | lack of complementary products to be offered by our sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines. |
19
If we are unable to implement and sustain our sales force, marketing infrastructure and distribution capability for XERMELO or any other products, we may not be able to generate any product revenue, may generate increased expenses and may never become profitable.
We will need to continue to expend significant time and resources to train our XERMELO sales force to be credible, persuasive and compliant in discussing XERMELO with the specialists treating the patients indicated under the label. We will also need to continue to train our sales force to ensure that a consistent and appropriate message about XERMELO is being delivered to our potential customers. If we are unable to effectively train our sales force and equip them with effective materials, including medical and sales literature to help them inform and educate potential customers about the benefits and risks of XERMELO and its proper administration, our ability to successfully commercialize XERMELO could be diminished, which could have a material adverse effect on our financial condition, stock price and operations.
If we are unable to obtain adequate coverage and reimbursement from third-party payers for XERMELO and any other products that we or our collaborators may develop, our revenues and prospects for profitability will suffer.
Our ability to successfully commercialize XERMELO and any other products that we or our collaborators may develop will be highly dependent on the extent to which coverage and reimbursement for such products will be available from third-party payers, including governmental payers, such as Medicare and Medicaid, and private health insurers, including managed care organizations and group purchasing organizations. Many patients will not be capable of paying themselves for XERMELO and some or all of the other products that we or our collaborators may develop, and will rely on third-party payers to pay for, or subsidize, their medical needs. If third-party payers do not provide coverage or reimbursement for XERMELO or any products that we or our collaborators may develop, our revenues and prospects for profitability will suffer. In addition, even if third-party payers provide some coverage or reimbursement for such products, the availability of such coverage or reimbursement for prescription drugs under private health insurance and managed care plans often varies based on the type of contract or plan purchased.
In addition, in some foreign countries, particularly the countries in the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, price negotiations with governmental authorities can take six to 12 months or longer after the receipt of regulatory marketing approval for a product. To obtain reimbursement and/or pricing approval in some countries, we or our collaborators may be required to conduct a clinical trial that compares the cost effectiveness of our drug candidates or products to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in the commercialization of our drug candidates. Third-party payers are challenging the prices charged for medical products and services, and many third-party payers limit reimbursement for newly approved health care products. In particular, third-party payers may limit the indications for which they will reimburse patients who use any products that we or our collaborators may develop. Cost-control initiatives could decrease prices we or our collaborators might establish for products that may be developed, which would result in lower product revenues to us.
We may not be able to manufacture XERMELO and any other products that we or our collaborators may develop in commercial quantities, which would impair our ability to commercialize such products.
Other than XERMELO, our drug candidates have been manufactured in relatively small quantities for nonclinical and clinical trials. If any of these drug candidates are approved by the FDA or other regulatory agencies for commercial sale, we or our collaborators will need to manufacture them in larger quantities. We may not be able to successfully increase the manufacturing capacity, whether in collaboration with third-party manufacturers or on our own, for any of such drug candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we or our collaborators are unable to successfully increase the manufacturing capacity for a drug candidate, the regulatory approval or commercial launch of that drug candidate may be delayed or there may be a shortage in supply. Our drug candidates require precise, high-quality manufacturing. The failure to achieve and maintain these high manufacturing standards, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously hurt our business.
We and our collaborators are subject to extensive and rigorous ongoing regulation relating to XERMELO and any other products that we or our collaborators may develop.
We are subject to extensive and rigorous ongoing domestic and foreign government regulation of, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing of XERMELO and any other products which receive regulatory approvals from the FDA or foreign regulatory authorities. The failure to comply with these requirements or the identification of safety problems during commercial marketing could lead to the need
20
for product marketing restrictions, product withdrawal or recall or other voluntary or regulatory action, which could delay further marketing until the product is brought into compliance. The failure to comply with these requirements may also subject us or our collaborators to stringent penalties.
We are subject to certain healthcare laws, regulation and enforcement; our failure to comply with those laws could have a material adverse effect on our results of operations and financial condition.
We are subject to certain healthcare laws and regulations and enforcement by the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include, without limitation:
• | the federal Anti-Kickback Law, which constrains our business activities, which includes our marketing practices, educational programs, pricing policies, and relationships with healthcare providers or other entities, by prohibiting, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
• | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent; |
• | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
• | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; |
• | the Foreign Corrupt Practices Act, a United States law which regulates certain financial relationships with foreign government officials (which could include, for example, certain medical professionals); |
• | federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
• | state and federal government price reporting laws that require us to calculate and report complex pricing metrics to government programs, where such reported price may be used in the calculation of reimbursement and/or discounts on our marketed drugs (participation in these programs and compliance with the applicable requirements may subject us to potentially significant discounts on our products, increased infrastructure costs, and potentially limit our ability to offer certain marketplace discounts); and |
• | state and federal marketing expenditure tracking and reporting laws, which generally require certain types of expenditures in the United States to be tracked and reported. Compliance with such requirements may require investment in infrastructure to ensure that tracking is performed properly, and some of these laws result in the public disclosure of various types of payments and relationships, which could potentially have a negative effect on our business and/or increase enforcement scrutiny of our activities. |
In addition, certain marketing practices, including off-label promotion, may also violate certain federal and state health regulatory fraud and abuse laws as well as false claims laws, including the civil False Claims Act. Suits filed under the civil False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals, commonly known as “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. The filing of qui tam actions has caused a number of pharmaceutical, medical device and other healthcare companies to defend a civil False Claims Act action. When an entity is determined to have violated the civil False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we, or our officers or employees, may be subject to penalties, including administrative civil and criminal penalties, damages, fines, withdrawal of regulatory approval, the curtailment or restructuring of our operations, the exclusion
21
from participation in Medicare, Medicaid and other federal and state healthcare programs, individual imprisonment, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, any of which could adversely affect our ability to sell our products or operate our business and also adversely affect our financial results. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
Numerous federal and state laws, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use and disclosure of personal information. Other countries also have, or are developing, laws governing the collection, use and transmission of personal information. In addition, most healthcare providers who may be expected to prescribe our products and from whom we may obtain patient health information are subject to privacy and security requirements under the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA. Although we are not directly subject to HIPAA, we could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues with the potential to affect our business, including recently enacted laws in a majority of states requiring security breach notification. These laws could create liability for us or increase our cost of doing business. International laws, such as the EU Data Privacy Directive and Swiss Federal Act on Data Protection, regulate the processing of personal data within Europe and between European countries and the United States. Failure to provide adequate privacy protections and maintain compliance with safe harbor mechanisms could jeopardize business transactions across borders and result in significant penalties.
Current healthcare laws and regulations and future legislative or regulatory reforms to the healthcare system may negatively affect our revenues and prospects for profitability.
A primary trend in the United States and some foreign countries is toward reform and cost containment in the health care industry. The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals that may have the effect of reducing the prices that we are able to charge for XERMELO and other products we or our collaborators may develop. Healthcare reform measures which may be adopted in the future in the United States and foreign jurisdictions may result in more rigorous coverage criteria and significant downward pressure on the prices drug manufacturers may charge. As a result, our revenues and prospects for profitability could be significantly harmed.
As a result of the overall trend towards cost-effectiveness criteria and managed healthcare in the United States, third-party payers are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs. They may use tiered reimbursement and may adversely affect demand for XERMELO and other products we or our collaborators may develop by placing them in an expensive tier. They may also refuse to provide any coverage of uses of approved products for medical indications other than those for which the FDA has granted market approvals. As a result, significant uncertainty exists as to whether and how much third-party payers will reimburse for newly approved drugs, which in turn will put pressure on the pricing of drugs. Further, we do not have experience in ensuring approval by applicable third-party payers outside of the United States for coverage and reimbursement of pharmaceutical products. We also anticipate pricing pressures in connection with the sale of XERMELO and other products we or our collaborators may develop due to the increasing influence of health maintenance organizations and additional legislative proposals.
Pricing for pharmaceutical products has come under increasing scrutiny by governments, legislative bodies and enforcement agencies. These activities may result in actions that have the effect of reducing our revenue or harming our business or reputation.
Many companies in our industry have received a governmental request for documents and information relating to drug pricing and patient support programs. We may become subject to similar requests, which would require us to incur significant expense and result in distraction for our management team. Additionally, to the extent there are findings, or even allegations, of improper conduct on the part of our company, such findings could further harm our business, reputation and/or prospects. It is possible that such inquiries could result in negative publicity or other negative actions that could harm our reputation, changes in our product pricing and distribution strategies, reduced demand for our approved products and/or reduced reimbursement of approved products, including by federal health care programs such as Medicare and Medicaid and state health care programs.
22
Our competitors may develop products that impair the value of XERMELO or any other products that we or our collaborators may develop.
The pharmaceutical and biotechnology industries are highly diversified and are characterized by rapid technological change. We and our collaborators face, and will continue to face, intense competition from biotechnology and pharmaceutical companies, as well as academic research institutions, clinical reference laboratories and government agencies that are pursuing research and development activities similar to ours. In addition, significant delays in the development of our drug candidates could allow our competitors to bring products to market before us, which would impair our or our collaborators’ ability to commercialize our drug candidates. XERMELO and any other products that we or our collaborators develop will compete in highly competitive markets. Further, our competitors may be more effective at using their technologies to develop commercial products. Many of the organizations competing with us have greater capital resources, larger research and development staff and facilities, more experience in obtaining regulatory approvals and more extensive product manufacturing and marketing capabilities. As a result, our competitors may be able to more easily develop products that would render XERMELO and any other products that we or our collaborators develop obsolete and noncompetitive. For example, dapagliflozin, empagliflozin and canagliflozin are currently being marketed by AstraZeneca, Boehringer Ingelheim and Eli Lilly, and Janssen (a subsidiary of Johnson & Johnson), respectively, for the treatment of type 2 diabetes. Each of those products act through SGLT2, one of the targets of sotagliflozin. In addition, there may be drug candidates of which we are not aware at an earlier stage of development that may compete with our drug candidates.
Risks Related to Our Capital Requirements and Financial Results
We will need additional capital in the future and, if it is unavailable, we will be forced to delay, reduce or eliminate our commercialization efforts or product development programs. If additional capital is not available on reasonable terms, we will be forced to obtain funds, if at all, by entering into financing agreements on unattractive terms.
As of December 31, 2017, we had $310.8 million in cash, cash equivalents and investments. We anticipate that our existing capital resources and the cash and revenues we expect to derive from product revenues, collaborations and other sources will enable us to fund our currently planned operations for at least the next 12 months. However, we caution you that we may generate less cash and revenues or incur expenses more rapidly than we currently anticipate. Our currently planned operations for the next twelve months include the continued commercialization of XERMELO in the United States; preparations with Sanofi for the submission of regulatory applications to market sotagliflozin for type 1 diabetes in the United States and the European Union; preparations for the commercial launch of sotagliflozin for type 1 diabetes in the United States, if approved; the initiation of clinical development of XERMELO for cholangiocarcinoma and neuroendocrine tumors; the continued clinical development of LX2761 for diabetes; and the continued clinical development of LX9211 for neuropathic pain.
Although difficult to accurately predict, the amount of our future capital requirements will be substantial and will depend on many factors, including:
• | the success of our sales, marketing, distribution and other commercialization activities for XERMELO in the United States and the revenues we generate from that approved product; |
• | the success of Ipsen’s sales, marketing, distribution and other commercialization activities for XERMELO outside of the United States and Japan; |
• | our and Sanofi’s ability to obtain regulatory approval for the marketing and sale of sotagliflozin for type 1 diabetes; |
• | if approved, our and Sanofi’s ability to successfully commercialize sotagliflozin for type 1 diabetes in the United States and Sanofi’s ability to successfully commercialize sotagliflozin for type 1 diabetes outside of the United States and Japan; |
• | the progress and scope of Sanofi’s development activities with respect to sotagliflozin in type 2 diabetes patients; |
• | the timing, progress and results of our clinical trials of XERMELO, LX2761 and LX9211; |
• | the amount and timing of payments, if any, under our existing collaboration agreements with Sanofi, Ipsen and other entities and any future collaboration agreements; |
• | the amount and timing of our research, development and commercialization expenditures; |
• | future results from clinical trials of our other drug candidates; |
23
• | the cost and timing of regulatory approvals and commercialization of additional drug candidates that we successfully develop; |
• | the market acceptance and commercial success of additional products that we successfully develop and commercially launch; |
• | the effect of competing programs and products, and of technological and market developments; |
• | the filing, maintenance, prosecution, defense and enforcement of patent claims and other intellectual property rights; and |
• | the cost and timing of establishing or contracting for commercialization capabilities of any other approved drug candidate. |
Our capital requirements have and will continue to be substantial as we market XERMELO in the United States, prepare for the submission of regulatory applications to market sotagliflozin for type 1 diabetes in the United States and the European Union, prepare for the commercial launch of sotagliflozin for type 1 diabetes in the United States, continue to share in the funding of the type 2 diabetes development costs for sotagliflozin; initiate clinical trials of XERMELO for cholangiocarcinoma and neuroendocrine tumors, continue to conduct early stage clinical trials of LX2761 and LX9211 and advance new drug candidates into clinical development. Our capital requirements will also be affected by any expenditures we make in connection with license agreements and acquisitions of and investments in complementary products and technologies. For all of these reasons, our future capital requirements cannot easily be quantified.
If our capital resources are insufficient to meet future capital requirements, we will need to raise additional funds to continue our currently planned operations. Our ability to raise additional capital is dependent on a number of factors, including the market demand for our securities, which itself is subject to a number of pharmaceutical development and business risks and uncertainties, as well as uncertainty that we would be able to raise such additional capital at a price or on terms that are favorable to us. If we raise additional capital by issuing equity securities, our then-existing stockholders will experience dilution and the terms of any new equity securities may have preferences over our common stock. The affirmative and restrictive covenants and the pledge of substantially all of our assets as collateral under our existing term loan with BioPharma Credit PLC and BioPharma Credit Investments IV Sub LP, or the BioPharma Term Loan, restrict our ability to raise additional capital by issuing debt securities. We cannot be certain that additional financing, whether debt or equity, will be available in amounts or on terms acceptable to us, if at all. We may be unable to raise sufficient additional capital on reasonable terms, and if so, we will be forced to delay, reduce or eliminate our clinical development programs or commercialization efforts or obtain funds, if at all, by entering into financing agreements on unattractive terms.
We have a history of net losses, and we expect to continue to incur net losses and may not achieve or maintain profitability.
We have incurred net losses since our inception, including net losses of $129.1 million for the year ended December 31, 2017, $141.4 million for the year ended December 31, 2016 and $4.7 million for the year ended December 31, 2015. As of December 31, 2017, we had an accumulated deficit of $1.4 billion. Because of the numerous risks and uncertainties associated with successfully developing and commercializing drugs, we are unable to predict the extent of any future losses or whether or when we will become profitable, if at all. The size of our net losses will depend, in part, on the rate of decline or growth in our revenues and on the amount of our expenses. We expect to continue to incur significant expenses over the next several years as we expect to make significant investments in the commercialization of XERMELO in the United States, the commercialization of sotagliflozin for type 1 diabetes in the United States, if approved, and the ongoing clinical development of XERMELO, sotagliflozin and our other drug candidates.
We commercially launched XERMELO following regulatory approval in February 2017 for the treatment of carcinoid syndrome diarrhea in combination with SSA therapy in adults inadequately controlled by SSA therapy in the United States. Prior to the launch of XERMELO, we derived substantially all of our revenues from strategic collaborations and other research and development collaborations and technology licenses.
Future revenues from our commercialization of XERMELO are uncertain because they depend on a number of factors, including market acceptance of XERMELO, the success of our sales, marketing, distribution and other commercialization activities and the cost and availability of reimbursement for XERMELO.
Future revenues from our existing collaborations are uncertain because they depend, to a large degree, on the achievement of milestones and payment of royalties we earn from any future products developed under the collaborations. Our ability to secure future revenue-generating agreements will depend upon our ability to address the needs of our potential future
24
collaborators and licensees, and to negotiate agreements that we believe are in our long-term best interests. We may determine, as we have with certain of our drug candidates, including XERMELO in the United States and Japan, that our interests are better served by retaining rights to our discoveries and advancing our therapeutic programs to a later stage, which could limit our near-term revenues and increase expenses. Because of these and other factors, our operating results have fluctuated in the past and are likely to do so in the future, and we do not believe that period-to-period comparisons of our operating results are a good indication of our future performance.
We expect to spend significant amounts to fund our commercialization activities with respect to XERMELO in the United States, our preparations for the commercial launch of sotagliflozin for type 1 diabetes in the United States and our nonclinical and clinical development activities, including the conduct of ongoing and planned clinical trials for XERMELO, sotagliflozin, LX2761 and LX9211. As a result, we will need to generate substantial additional revenues to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
Our operating results have been and likely will continue to fluctuate, and we believe that period-to-period comparisons of our operating results are not a good indication of our future performance.
Our operating results have fluctuated in the past and are likely to fluctuate in the future. A number of factors, many of which we cannot control, could subject our operating results to volatility, including:
• | our ability to successfully commercialize XERMELO in the United States and the amount of revenues generated from such commercialization efforts; |
• | our and Sanofi’s ability to obtain regulatory approval for the marketing and sale of sotagliflozin for type 1 diabetes; |
• | the amount and timing of payments, if any, under our existing collaboration agreements with Sanofi, Ipsen and other entities; |
• | the success of our ongoing preclinical and clinical development efforts; |
• | the timing and amount of expenses incurred with respect to our preclinical and clinical development and commercialization efforts; |
• | our success in establishing new collaborations and technology licenses, and the timing of such arrangements; |
• | the success rate of our development efforts leading to opportunities for new collaborations and licenses, as well as milestone payments and royalties; |
• | the timing and willingness of our collaborators to commercialize pharmaceutical products that would result in milestone payments and royalties; |
• | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our products and technologies; |
• | general and industry-specific economic conditions, which may affect our and our collaborators’ research and development expenditures. |
Because of these and other factors, including the risks and uncertainties described in this section, our operating results have fluctuated in the past and are likely to do so in the future. Due to the likelihood of fluctuations in our revenues and expenses, we believe that period-to-period comparisons of our operating results are not a good indication of our future performance.
We have substantial indebtedness that may limit cash flow available to invest in the ongoing needs of our business.
We have incurred $245.7 million of indebtedness. Although the affirmative and restrictive covenants and the pledge of substantially all of our assets as collateral under the BioPharma Term Loan restrict our ability to obtain additional debt financing, we could in the future incur additional indebtedness beyond such amount. Our substantial debt combined with our other financial obligations and contractual commitments could have significant adverse consequences, including:
25
• | requiring us to dedicate a substantial portion of cash flow from operations to the payment of interest on, and principal of, our debt, which will reduce the amounts available to fund working capital, capital expenditures, product commercialization and development efforts and other general corporate purposes; |
• | increasing our vulnerability to adverse changes in general economic, industry and market conditions; |
• | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we compete; and |
• | placing us at a competitive disadvantage compared to our competitors that have less debt or better debt servicing options. |
We intend to satisfy our current and future debt service obligations with our existing cash and cash equivalents and marketable securities and funds from external sources. However, we may not have sufficient funds or may be unable to arrange for additional financing to pay the amounts due under our existing debt. Funds from external sources may not be available on acceptable terms, if at all. In addition, a failure to comply with the covenants under our existing debt instruments could result in an event of default under those instruments. In the event of an acceleration of amounts due under our debt instruments as a result of an event of default, including upon the occurrence of an event that would reasonably be expected to have a material adverse effect on our business, operations, properties, assets or condition or a failure to pay any amount due, we may not have sufficient funds or may be unable to arrange for additional financing to repay our indebtedness or to make any accelerated payments, and the lenders could seek to enforce their security interests in the collateral securing such indebtedness.
If we do not effectively manage our affirmative and restrictive covenants under the BioPharma Term Loan, our financial condition and results of operations could be adversely affected. In addition, we may not achieve the amount of XERMELO net sales required for us to access the second tranche available under the BioPharma Term Loan.
Our obligations under the BioPharma Term Loan are secured by a first lien security interest in substantially all of our assets. In addition, the BioPharma Term Loan requires that we comply with certain affirmative and restrictive covenants, including among other things, covenants restricting dispositions, fundamental changes in our business, mergers or acquisitions, indebtedness, encumbrances, distributions, investments, transactions with affiliates and subordinated debt, any of which could restrict our business and operations, particularly our ability to respond to changes in our business or to take specified actions to take advantage of certain business opportunities that may be presented to us. Our failure to comply with any of these covenants could result in a default under the BioPharma Term Loan, which could permit the lenders to declare all or part of any outstanding borrowings to be immediately due and payable. If we are unable to repay those amounts, the lenders could enforce the security interest granted to them to secure that debt, which would seriously harm our business.
Moreover, the second $50.0 million tranche is only available for draw by March 2019 if XERMELO net sales are greater than $25 million in the preceding quarter. We may be unable to achieve such amount of XERMELO net sales, in which case our liquidity could be negatively affected.
Risks Related to Our Relationships with Third Parties
We are significantly dependent upon our collaborations with Ipsen, Sanofi and other pharmaceutical and biotechnology companies. If pharmaceutical products are not successfully and timely developed and commercialized under our collaborations, our opportunities to generate revenues from milestones and royalties will be greatly reduced.
We have entered into collaboration agreements with Ipsen for the commercialization of XERMELO outside of the United States and Japan and with Sanofi for the worldwide (excluding Japan) development and commercialization of sotagliflozin. We have also established collaborative arrangements with other pharmaceutical and biotechnology companies with respect to the research, development and commercialization of drug candidates from other programs. We have derived a substantial majority of our revenues to date from these strategic collaborations and other research and development collaborations and technology licenses. Future revenues from our existing collaborations depend upon the achievement of milestones and payment of royalties we earn from any future products developed under the collaborations. If our relationship terminates with any of our collaborators, particularly Ipsen and Sanofi, our reputation in the business and scientific community may suffer and revenues will be negatively impacted to the extent such losses are not offset by additional collaboration agreements. If milestones are not achieved under our collaborations or our collaborators are unable to successfully develop and commercialize products from which milestones and royalties are payable, we will not earn the revenues contemplated by those collaborations.
26
We have limited or no control over the resources that any collaborator may devote to the development and commercialization of products under our alliances. For example, Sanofi is responsible for all clinical development activities relating to sotagliflozin for the treatment of type 2 diabetes and we have limited influence on the manner in which Sanofi may conduct such clinical development. Any of our present or future collaborators may not perform their obligations as expected. These collaborators may breach or terminate their agreements with us or otherwise fail to conduct research, development or commercialization activities successfully or in a timely manner. Further, our collaborators may elect not to develop pharmaceutical products arising out of our collaborative arrangements or may not devote sufficient resources to the development, regulatory approval, manufacture, marketing or sale of these products. If any of these events occurs, we may not receive collaboration revenue or otherwise realize anticipated benefits from such collaborations, our product development efforts may be delayed and our business, operating results and financial condition could be adversely affected.
Conflicts with our collaborators could jeopardize the success of our collaborative agreements and harm our product development efforts.
We may pursue opportunities in specific disease and therapeutic modality fields that could result in conflicts with our collaborators, if any of our collaborators takes the position that our internal activities overlap with those activities that are exclusive to our collaboration. Moreover, disagreements could arise with our collaborators over rights to our intellectual property or our rights to share in any of the future revenues of compounds or therapeutic approaches developed by our collaborators. Any conflict with or among our collaborators could result in the termination of our collaborative agreements, delay collaborative research or development activities, impair our ability to renew or obtain future collaborative agreements or lead to costly and time consuming litigation. Conflicts with our collaborators could also have a negative impact on our relationship with existing collaborators, materially impairing our business and revenues. Some of our collaborators are also potential competitors or may become competitors in the future. Our collaborators could develop competing products, preclude us from entering into collaborations with their competitors or terminate their agreements with us prematurely. Any of these events could harm our product development efforts.
We depend on third-party manufacturers, including sole source suppliers, to manufacture commercial quantities of XERMELO. We may not be able to maintain these relationships and could experience supply disruptions outside of our control.
We rely on a network of third-party manufacturers to manufacture and supply XERMELO for commercial sale. As a result of our reliance on these third-party manufacturers and suppliers, including sole source suppliers for certain steps in the manufacture of XERMELO, we could be subject to significant supply disruptions. Our supply chain for sourcing raw materials and manufacturing drug product ready for distribution is a multi-step endeavor. Third-party contract manufacturers procure raw materials, convert these raw materials into API, and then convert the API into final dosage form. Establishing and managing this supply chain requires a significant financial commitment and the creation and maintenance of numerous third party contractual relationships. Although we attempt to effectively manage the business relationships with companies in our supply chain, we do not have control over their operations.
We require our own commercial supply of XERMELO for sale in the United States, and are required under our collaboration agreement to supply Ipsen’s commercial requirements of XERMELO in the European Union and other territories outside of the United States and Japan once approved in such jurisdictions. We currently rely, and expect to continue to rely, on sole source third-party manufacturers to produce final drug product and package and label XERMELO. While we have identified and expect to qualify and engage back-up third-party manufacturers as additional or alternative suppliers for the production of final drug product and packaging and labeling of XERMELO, we currently do not have such arrangements in place. Moreover, some of these alternative manufacturers will need to be approved by the FDA before we can use them for manufacturing XERMELO. It is also possible that supplies of materials that cannot be second-sourced can be managed with inventory planning. There can be no assurance, however, that failure of any of our sole source third-party manufacturers to meet our and Ipsen’s commercial demands for XERMELO in a timely manner, or our failure to engage qualified additional or back-up suppliers for the production of final drug product and packaging and labeling of XERMELO, would not have a material adverse effect on commercialization of XERMELO and our business.
Supply disruptions may result from a number of factors, including shortages in product raw materials, labor or technical difficulties, regulatory inspections or restrictions, shipping or customs delays or any other performance failure by any third-party manufacturer on which we rely. Any supply disruptions could disrupt sales of XERMELO, which could have a material adverse impact on our business.
We rely on a single third-party logistics provider and two independent specialty pharmacies for distribution of XERMELO in the United States, and their failure to distribute XERMELO effectively would adversely affect sales of XERMELO.
27
We rely on a single third-party logistics provider for shipping and warehousing of our commercial supply of XERMELO and two independent specialty pharmacies for dispensation of XERMELO to patients in fulfillment of prescriptions in the United States. Although our third-party logistics provider stores our commercial supply of XERMELO at two separate warehouses, the use of a single third-party logistics provider increases the risk that a fire or damage from another type of disaster at either of the warehouses may result in a disruption of our commercialization efforts. A specialty pharmacy is a pharmacy that specializes in the dispensing of medications for complex or chronic conditions, which often require a high level of patient education and ongoing management. The use of specialty pharmacies involves certain additional risks, including, but not limited to, risks that these specialty pharmacies will:
• | not provide us accurate or timely information regarding their inventories, the number of patients who are using XERMELO or complaints about XERMELO; |
• | reduce or discontinue their efforts to sell or support or otherwise not effectively sell or support XERMELO; |
• | not devote the resources necessary to sell XERMELO in the volumes and within the time frames that we expect; |
• | be unable to satisfy their financial obligations to us; or |
• | cease operations. |
If our third-party logistics provider or either or both of our specialty pharmacies do not fulfill their contractual obligations to us, or refuse or fail to adequately distribute XERMELO and serve patients, or the agreements are terminated without adequate notice, shipments of XERMELO, and associated revenues, would be adversely affected. In addition, we expect that it may take a significant amount of time if we were required to change our third-party logistics provider or either of our specialty pharmacies.
We rely on third parties to carry out drug development activities.
We rely on clinical research organizations and other third-party contractors to carry out many of our drug development activities, including the performance of nonclinical laboratory and animal tests under the FDA’s current Good Laboratory Practices regulations and the conduct of clinical trials of our drug candidates in accordance with protocols we establish. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, our drug development activities may be delayed, suspended or terminated. Such a failure by these third parties could significantly impair our ability to develop and commercialize the affected drug candidates.
We lack the capability to manufacture materials for nonclinical studies, clinical trials or commercial sales and rely on third parties to manufacture our drug candidates, which may harm or delay our product development and commercialization efforts.
We currently do not have the manufacturing capabilities or experience necessary to produce materials for nonclinical studies, clinical trials or commercial sales and intend in the future to continue to rely on collaborators and third-party contractors to produce such materials. We will rely on selected manufacturers to deliver materials on a timely basis and to comply with applicable regulatory requirements, including the current Good Manufacturing Practices of the FDA, which relate to manufacturing and quality control activities. These manufacturers may not be able to produce material on a timely basis or manufacture material at the quality level or in the quantity required to meet our development timelines and applicable regulatory requirements. In addition, there are a limited number of manufacturers that operate under the FDA’s current Good Manufacturing Practices and that are capable of producing such materials, and we may experience difficulty finding manufacturers with adequate capacity for our needs. If we are unable to contract for the production of sufficient quantity and quality of materials on acceptable terms, our product development and commercialization efforts may be delayed. Moreover, noncompliance with the FDA’s current Good Manufacturing Practices can result in, among other things, fines, injunctions, civil and criminal penalties, product recalls or seizures, suspension of production, failure to obtain marketing approval and withdrawal, suspension or revocation of marketing approvals.
Risks Related to Our Intellectual Property
If we are unable to adequately protect our intellectual property, third parties may be able to use our products and technologies, which could adversely affect our ability to compete in the market.
Our success will depend in part upon our ability to obtain patents and maintain adequate protection of the intellectual property related to our products and technologies. The patent positions of biotechnology and pharmaceutical companies,
28
including our patent position, are generally uncertain and involve complex legal and factual questions. We will be able to protect our intellectual property rights from unauthorized use by third parties only to the extent that our products and technologies are covered by valid and enforceable patents or are effectively maintained as trade secrets. We will continue to apply for patents covering our products and technologies as, where and when we deem appropriate. However, pending patent applications do not provide protection against competitors because they are not enforceable until they issue as patents. Further, the disclosures contained in our current and future patent applications may not be sufficient to meet statutory requirements for patentability and our applications may fail to result in issued patents. Once issued, patents still may not provide commercially meaningful protection. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from developing competing products and technologies. Furthermore, others may independently develop similar or alternative products or technologies or design around our patents. If anyone infringes upon our or our collaborators’ patent rights, enforcing these rights may be difficult, costly and time-consuming and, as a result, it may not be cost-effective or otherwise expedient to pursue litigation to enforce those patent rights.
Our patents may be challenged by third parties as invalid or unenforceable under U.S. or foreign laws, or they may be infringed by third parties. As a result, we may be involved in the defense and enforcement of our patent or other intellectual property rights in a court of law, U.S. Patent and Trademark Office inter partes review or reexamination proceeding, foreign opposition proceeding or related legal and administrative proceeding in the United States and elsewhere. The costs of defending our patents or enforcing our proprietary rights in post-issuance administrative proceedings and litigation may be substantial and the outcome can be uncertain. An adverse outcome may allow third parties to use our intellectual property without a license and negatively impact our business.
In addition, because patent applications can take many years to issue, third parties may have pending applications, unknown to us, which may later result in issued patents that cover the production, manufacture, commercialization or use of our products and drug candidates. If any such patents are issued to other entities, we will be unable to obtain patent protection for the same or similar discoveries that we make relating to our products and drug candidates. Moreover, we may be blocked from using our drug targets or drug candidates or developing or commercializing our products and other drug candidates, or may be required to obtain a license that may not be available on reasonable terms, if at all. Further, others may discover uses for our drug targets and drug candidates other than those covered in our issued or pending patents, and these other uses may be separately patentable. Even if we have a patent claim on a particular technology or product, the holder of a patent covering the use of that technology or product could exclude us from selling a product that is based on the same use of that product.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (for example, if the patent owner has failed to “work” the invention in that country or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life-saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our products and drug candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patent and other intellectual property protection, which makes it difficult to stop infringement.
We rely on trade secret protection for some of our confidential and proprietary information. We have taken security measures to protect our proprietary information and trade secrets, but these measures may not provide adequate protection. While we seek to protect our proprietary information by entering into confidentiality agreements with employees, collaborators and consultants, we cannot assure you that our proprietary information will not be disclosed, or that we can meaningfully protect our trade secrets. In addition, our competitors may independently develop substantially equivalent proprietary information or may otherwise gain access to our trade secrets.
We may be involved in patent litigation and other disputes regarding intellectual property rights and may require licenses from third parties for our planned nonclinical and clinical development and commercialization activities. We may not prevail in any such litigation or other dispute or be able to obtain required licenses.
Our products and those of our collaborators, as well as our nonclinical and clinical development efforts, may give rise to claims that they infringe the patents of others. We are aware that other companies and institutions are developing products acting through the same drug targets through which some of our drug candidates currently in clinical development act, have conducted research on many of the same targets that we have identified and have filed patent applications potentially covering drug targets that we have identified and certain therapeutic products addressing such targets. In some cases, patents have issued from these applications. In addition, many companies and institutions have well-established patent portfolios directed to
29
common techniques, methods and means of developing, producing and manufacturing pharmaceutical products. These or other companies or institutions could bring legal actions against us or our collaborators for damages or to stop us or our collaborators from engaging in certain nonclinical or clinical development activities or from manufacturing and marketing therapeutic products that allegedly infringe their patent rights. If any of these actions are successful, in addition to our potential liability for damages, these entities would likely require us or our collaborators to obtain a license in order to continue engaging in the infringing activities or to manufacture or market the infringing therapeutic products or may force us to terminate such activities or manufacturing and marketing efforts.
We may deem it advisable to pursue litigation against others to enforce our patents and intellectual property rights and may be the subject of litigation brought by third parties to enforce their patent and intellectual property rights. In addition, we may become involved in litigation based on intellectual property indemnification undertakings that we have given to certain of our collaborators. Patent litigation is expensive and requires substantial amounts of management attention. The eventual outcome of any such litigation is uncertain and involves substantial risks.
We believe that there will continue to be significant litigation in our industry regarding patent and other intellectual property rights. We have expended and many of our competitors have expended and are continuing to expend significant amounts of time, money and management resources on intellectual property litigation. If we become involved in future intellectual property litigation, it could consume a substantial portion of our resources and could negatively affect our results of operations.
Data breaches and cyber-attacks could compromise our intellectual property or other sensitive information and cause significant damage to our business and reputation.
In the ordinary course of our business, we collect, maintain and transmit sensitive data on our networks and systems, including our intellectual property and proprietary or confidential business information (such as research data and personal information) and confidential information with respect to our customers, clinical trial patients and our business partners. We have also outsourced significant elements of our information technology infrastructure and, as a result, third parties may or could have access to our confidential information. The secure maintenance of this information is critical to our business and reputation. We believe that companies have been increasingly subject to a wide variety of security incidents, cyber-attacks and other attempts to gain unauthorized access. These threats can come from a variety of sources, ranging in sophistication from an individual hacker to a state-sponsored attack and motive (including corporate espionage). Cyber threats may be generic, or they may be custom-crafted against our information systems. Our network and storage applications and those of our vendors may be subject to unauthorized access by hackers or breached due to operator error, malfeasance or other system disruptions. It is often difficult to anticipate or immediately detect such incidents and the damage caused by such incidents. These data breaches and any unauthorized access or disclosure of our information or intellectual property could compromise our intellectual property and expose sensitive business information. A data security breach could also lead to public exposure of personal information of our clinical trial patients, customers and others. Cyber-attacks could cause us to incur significant remediation costs, result in product development delays, disrupt key business operations and divert attention of management and key information technology resources. Our network security and data recovery measures and those of our vendors may not be adequate to protect against such security breaches and disruptions. These incidents could also subject us to liability, expose us to significant expense and cause significant harm to our reputation and business.
We may be subject to damages resulting from claims that we, our employees or independent contractors have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees and independent contractors were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, independent contractors or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and divert management’s attention. If we fail in defending such claims, in addition to paying money claims, we may lose valuable intellectual property rights or personnel. A loss of key research personnel and/or their work product could hamper or prevent our ability to commercialize certain drug candidates, which could severely harm our business.
Risks Related to Employees and Facilities Operations
If we are unable to manage our growth, our business, financial condition, results of operations and prospects may be adversely affected.
30
We have experienced and expect to continue to experience growth in the number of our employees and in the scope of our operations. This growth places significant demands on our management, operational and financial resources, and our current and planned personnel, systems, procedures and controls may not be adequate to support our growth. To effectively manage our growth, we must continue to improve existing, and implement new, operational and financial systems, procedures and controls and must expand, train and manage our growing employee base, and there can be no assurance that we will effectively manage our growth without experiencing operating inefficiencies or control deficiencies. We expect that we may need to increase our medical, clinical, commercial and other personnel, and recruiting and retaining qualified individuals is difficult. If we are unable to manage our growth effectively, or are unsuccessful in recruiting qualified personnel when advisable, our business, financial condition, results of operations and prospects may be adversely affected.
The loss of key personnel or the inability to attract and retain additional personnel could impair our ability to operate and expand our operations.
We are highly dependent upon the principal members of our management, as well as medical, clinical and commercial staff, the loss of whose services might adversely impact the achievement of our objectives. Retaining and, where advisable, recruiting qualified medical, clinical and commercial personnel will be critical to support activities related to successfully executing on our commercial plan for XERMELO and advancing our nonclinical and clinical development programs for sotagliflozin and our other drug programs. Competition is intense for experienced medical, clinical and commercial personnel, and we may be unable to retain or recruit such personnel with the expertise or experience necessary to allow us to successfully develop and commercialize our products. Further, all of our employees are employed “at will” and, therefore, may leave our employment at any time.
Facility security breaches may disrupt our operations, subject us to liability and harm our operating results.
Any break-in or trespass of our facilities that results in the misappropriation, theft, sabotage or any other type of security breach with respect to our proprietary and confidential information, including research or clinical data, or that results in damage to our equipment and assets could subject us to liability and have a material adverse impact on our business, operating results and financial condition.
Our facilities are located near coastal zones, and the occurrence of a hurricane or other disaster could damage our facilities and equipment, which could harm our operations.
Our facilities are located in The Woodlands, Texas and Basking Ridge, New Jersey, and therefore our facilities are vulnerable to damage from hurricanes. We are also vulnerable to damage from other types of disasters, including fire, floods, power loss, communications failures, terrorism and similar events and any insurance we may maintain may not be adequate to cover our losses. If any disaster were to occur, our ability to operate our business at our facilities could be seriously, or potentially completely, impaired.
Risks Related to Environmental and Product Liability
We have used hazardous chemicals and radioactive and biological materials in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research and development processes have historically involved the controlled use of hazardous materials, including chemicals and radioactive and biological materials. Our operations have produced hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from these materials. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. We may face liability for any injury or contamination that results from our use or the use by third parties of these materials, and such liability may exceed our insurance coverage and our total assets. Compliance with environmental laws and regulations may be expensive, and current or future environmental regulations may impair our research, development and production efforts.
In addition, our collaborators may use hazardous materials in connection with our collaborative efforts. In the event of a lawsuit or investigation, we could be held responsible for any injury caused to persons or property by exposure to, or release of, these hazardous materials used by these parties. Further, we may be required to indemnify our collaborators against all damages and other liabilities arising out of our development activities or products produced in connection with these collaborations.
31
Our business has a substantial risk of product liability and we face potential product liability exposure far in excess of our limited insurance coverage.
We may be held liable if XERMELO or any other product that we or our collaborators develop or commercialize, or any product that is made with the use or incorporation of any of our technologies, causes injury or is found otherwise unsuitable during product testing, manufacturing, marketing or sale. Regardless of merit or eventual outcome, product liability claims could result in decreased demand for our products and product candidates, injury to our reputation, withdrawal of patients from our clinical trials, product recall, substantial monetary awards to third parties and the inability to commercialize any products that we may develop. These claims might be made directly by consumers, health care providers, pharmaceutical companies or others selling or testing our products. We have obtained limited product liability insurance coverage for our clinical trials and commercial activities. However, our insurance may not reimburse us or may not be sufficient to reimburse us for expenses or losses we may suffer. Moreover, if insurance coverage becomes more expensive, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. On occasion, juries have awarded large judgments in class action lawsuits for claims based on drugs that had unanticipated side effects. In addition, the pharmaceutical and biotechnology industries, in general, have been subject to significant medical malpractice litigation. A successful product liability claim or series of claims brought against us could harm our reputation and business.
Risks Related to Our Common Stock
Invus, L.P., Invus C.V. and their affiliates own a controlling interest in our outstanding common stock and may have interests which conflict with those of our other stockholders.
Invus, L.P. and Invus C.V., which we collectively refer to as Invus, and their affiliates currently own approximately 59.3% of the outstanding shares of our common stock and are thereby able to control the election and removal of our directors and determine our corporate and management policies, including potential mergers or acquisitions, asset sales, the amendment of our articles of incorporation or bylaws and other significant corporate transactions. This concentration of ownership may delay or deter possible changes in control of our company, which may reduce the value of an investment in our common stock. The interests of Invus and its affiliates may not be aligned with the interests of other holders of our common stock.
Invus has additional rights under our stockholders’ agreement with Invus, L.P. relating to the membership of our board of directors, which provides Invus with substantial influence over significant corporate matters.
Under our stockholders’ agreement with Invus, L.P., Invus has the right to designate a number of directors equal to the percentage of all the outstanding shares of our common stock owned by Invus and its affiliates, rounded up to the nearest whole number of directors. Invus has designated three of the nine current members of our board of directors. While Invus has not presently exercised its director designation rights in full, it may exercise them at any time in the future in its sole discretion. To facilitate the exercise of such rights, we have agreed, upon written request from Invus, to take all necessary steps in accordance with our obligations under the stockholders’ agreement to (1) increase the number of directors to the number specified by Invus (which number shall be no greater than reasonably necessary for the exercise of Invus’ director designation rights under the stockholders’ agreement) and (2) cause the appointment to the newly created directorships of directors so designated by Invus pursuant to its rights under the stockholders’ agreement.
Invus also has the right to require proportionate representation of Invus-appointed directors on the audit, compensation and corporate governance committees of our board of directors, subject to certain restrictions. Invus-designated directors currently serve as one of the three members of each of the compensation committee and the corporate governance committee of our board of directors. No Invus-designated directors currently serve on the audit committee of our board of directors.
The provisions of the stockholders’ agreement relating to Invus’ rights to designate members of our board of directors and its audit, compensation and corporate governance committees will terminate if the percentage of all the outstanding shares of our common stock owned by Invus and its affiliates falls below 10%. Invus also has the right to terminate these provisions at any time in its discretion.
Our stock price may be extremely volatile.
The trading price of our common stock has been highly volatile, and we believe the trading price of our common stock will remain highly volatile and may fluctuate substantially due to factors such as the following, many of which we cannot control:
32
• | the commercial success of XERMELO and the revenues we generate from sales of XERMELO; |
• | adverse results or delays in our or our collaborators’ clinical trials; |
• | the timing and achievement of milestones under our collaboration agreements; |
• | the announcement of FDA approval or non-approval, or delays in the FDA review process, of our or our collaborators’ drug candidates or those of our competitors or actions taken by regulatory agencies with respect to our, our collaborators’ or our competitors’ clinical trials; |
• | actions taken by regulatory agencies with respect to XERMELO, sotagliflozin and our other drug candidates; |
• | the announcement of new products by our competitors; |
• | quarterly variations in our or our competitors’ results of operations; |
• | developments in our relationships with our collaborators, including conflicts, litigation or the termination or modification of our agreements; |
• | the announcement of an in-licensed drug candidate or strategic acquisition; |
• | litigation, including intellectual property infringement and product liability lawsuits, involving us; |
• | failure to achieve operating results projected by securities analysts; |
• | changes in earnings estimates or recommendations by securities analysts; |
• | the satisfaction of outstanding debt obligations or entry into new financing arrangements; |
• | developments in the biotechnology or pharmaceutical industry; |
• | sales of large blocks of our common stock or sales of our common stock by our executive officers, directors and significant stockholders; |
• | departures of key personnel or board members; |
• | FDA or international regulatory actions; |
• | third-party coverage and reimbursement policies; |
• | disposition of any of our drug programs or other technologies; and |
• | other factors, including general market, economic and political conditions and other factors unrelated to our operating performance or the operating performance of our competitors. |
These factors may materially adversely affect the market price of our common stock. In addition, the stock markets in general, and the markets for biotechnology and pharmaceutical stocks in particular, have historically experienced significant volatility that has often been unrelated or disproportionate to the operating performance of particular companies. For example, negative publicity regarding drug pricing and price increases by pharmaceutical companies has negatively impacted, and may continue to negatively impact, the markets for biotechnology and pharmaceutical stocks. Likewise, the broader financial markets could experience significant volatility that could also negatively impact the markets for biotechnology and pharmaceutical stocks. These broad market fluctuations have adversely affected and may in the future adversely affect the trading price of our common stock. Excessive volatility may continue for an extended period of time.
In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted. A securities class action suit against us could result in substantial costs and divert management’s attention and resources, which could have a material and adverse effect on our business.
33
Future sales of our common stock, or the perception that such sales may occur, may depress our stock price.
A substantial number of shares of our common stock is reserved for issuance upon conversion of notes evidencing our current indebtedness, upon the exercise of stock options and upon vesting of restricted stock units. If our stockholders sell substantial amounts of our common stock (including shares issued upon the conversion of notes, exercise of stock options or vesting of restricted stock units) in the public market, or if the market perceives that such sales may occur, the market price of our common stock could fall and it may become more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem appropriate. For example, following an acquisition, a significant number of shares of our common stock held by new stockholders may become freely tradable or holders of registration rights could cause us to register their shares for resale. Sales of these shares of common stock held by existing stockholders could cause the market price of our common stock to decline.
Conversion of our 5.25% Convertible Senior Notes due 2021 may dilute the ownership interest of our existing stockholders, including holders who had previously converted their notes, or may otherwise depress the price of our common stock.
The conversion of some or all of our 5.25% Convertible Senior Notes due 2021 will dilute the ownership interests of existing stockholders to the extent we deliver shares upon conversion of any of the notes. Any sales in the public market of the common stock issuable upon such conversion could adversely affect prevailing market prices of our common stock. In addition, the existence of the notes may encourage short selling by market participants because the conversion of the notes could be used to satisfy short positions, or anticipated conversion of the notes into shares of our common stock could depress the price of our common stock.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our common stock could decrease, which might cause our stock price and trading volume to decline.
We may engage in future acquisitions, which may be expensive and time consuming and from which we may not realize anticipated benefits.
We may acquire additional businesses, technologies and products if we determine that these businesses, technologies and products complement our existing technology or otherwise serve our strategic goals. If we do undertake any transactions of this sort, the process of integrating an acquired business, technology or product may result in operating difficulties and expenditures and may not be achieved in a timely and non-disruptive manner, if at all, and may absorb significant management attention that would otherwise be available for ongoing development of our business. If we fail to integrate acquired businesses, technologies or products effectively or if key employees of an acquired business leave, the anticipated benefits of the acquisition would be jeopardized. Moreover, we may never realize the anticipated benefits of any acquisition, such as increased revenues and earnings or enhanced business synergies. Future acquisitions could result in potentially dilutive issuances of our equity securities, the incurrence of debt and contingent liabilities and amortization expenses related to intangible assets, which could materially impair our results of operations and financial condition.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We currently own approximately 260,000 square feet of space for our corporate offices and laboratories in buildings located in The Woodlands, Texas, a suburb of Houston, Texas, and lease approximately 25,000 square feet of office space in Basking Ridge, New Jersey.
In April 2004, we obtained a $34.0 million mortgage on our facilities in The Woodlands, Texas. The mortgage loan originally had a ten-year term with a 20-year amortization and a fixed rate of 8.23%. The mortgage was amended in September 2013 to extend the maturity date from April 2014 to April 2017 and again in April 2017 to extend the maturity date to April 2018, in each case with the mortgage loan’s monthly payment amount and fixed interest rate remaining unchanged. The mortgage had a principal balance outstanding of $14.1 million as of December 31, 2017. The entire principal balance is
34
recorded as current portion of long-term debt in the accompanying consolidated balance sheet as of December 31, 2017 as there is a balloon payment due in April 2018. We intend to refinance the mortgage prior to the balloon payment becoming due.
In March 2015, our subsidiary Lexicon Pharmaceuticals (New Jersey), Inc. leased a 25,000 square-foot office space in Basking Ridge, New Jersey. The term of the lease extends from June 1, 2015 through December 31, 2022, and provides for escalating yearly base rent payments starting at $482,000 and increasing to $646,000 in the final year of the lease.
We believe that our facilities are well-maintained, in good operating condition and acceptable for our current operations.
Item 3. Legal Proceedings
We are from time to time party to claims and legal proceedings that arise in the normal course of our business and that we believe will not have, individually or in the aggregate, a material adverse effect on our results of operations, financial condition or liquidity.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is quoted on The Nasdaq Global Select Market under the symbol “LXRX.” The following table sets forth, for the periods indicated, the high and low sales prices for our common stock as reported on The Nasdaq Global Select Market.
High | Low | ||||||
2016 | |||||||
First Quarter | $ | 13.45 | $ | 7.65 | |||
Second Quarter | $ | 15.17 | $ | 11.52 | |||
Third Quarter | $ | 19.62 | $ | 13.73 | |||
Fourth Quarter | $ | 19.50 | $ | 13.71 | |||
2017 | |||||||
First Quarter | $ | 17.48 | $ | 13.41 | |||
Second Quarter | $ | 18.00 | $ | 13.48 | |||
Third Quarter | $ | 17.29 | $ | 11.80 | |||
Fourth Quarter | $ | 12.38 | $ | 8.07 | |||
As of February 26, 2018, there were approximately 224 holders of record of our common stock.
We have never paid cash dividends on our common stock. We anticipate that we will retain all of our future earnings, if any, for use in the expansion and operation of our business and do not anticipate paying cash dividends in the foreseeable future.
Performance Graph
The following performance graph compares the performance of our common stock to the Nasdaq Composite Index and the Nasdaq Biotechnology Index for the period beginning December 31, 2012 and ending December 31, 2017. The graph assumes that the value of the investment in our common stock and each index was $100 at December 31, 2012, and that all dividends were reinvested.
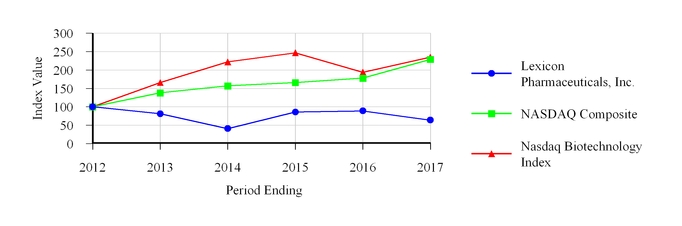
36
December 31, | |||||||||||||||||
2012 | 2013 | 2014 | 2015 | 2016 | 2017 | ||||||||||||
Lexicon Pharmaceuticals, Inc. | 100 | 81 | 41 | 86 | 89 | 64 | |||||||||||
Nasdaq Composite Index | 100 | 138 | 157 | 166 | 178 | 229 | |||||||||||
Nasdaq Biotechnology Index | 100 | 166 | 222 | 247 | 194 | 235 | |||||||||||
The foregoing stock price performance comparisons shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, or otherwise subject to the liabilities of that section, nor shall it be deemed incorporated by reference by any general statement incorporating by reference this annual report on Form 10-K into any filing under the Securities Act of 1933 or under the Securities Exchange Act of 1934, except to the extent that we specifically incorporate such comparisons by reference.
37
Item 6. Selected Financial Data
The statements of comprehensive loss data for the years ended December 31, 2017, 2016 and 2015 and the balance sheet data as of December 31, 2017 and 2016 have been derived from our audited financial statements included elsewhere in this annual report on Form 10-K. The statements of comprehensive loss data for the years ended December 31, 2014 and 2013, and the balance sheet data as of December 31, 2015, 2014 and 2013 have been derived from our audited financial statements not included in this annual report on Form 10-K. Our historical results are not necessarily indicative of results to be expected for any future period. The data presented below has been derived from financial statements that have been prepared in accordance with accounting principles generally accepted in the United States and should be read with our financial statements, including the notes, and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this annual report on Form 10-K.
Year Ended December 31, | |||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
Statements of Comprehensive Loss Data: | (in thousands, except per share data) | ||||||||||||||||||
Revenues | $ | 90,335 | $ | 83,337 | $ | 130,014 | $ | 22,854 | $ | 2,222 | |||||||||
Operating expenses: | |||||||||||||||||||
Cost of sales (including finite-lived intangible asset amortization) | 1,899 | — | — | — | — | ||||||||||||||
Research and development, including stock-based compensation of $4,905 in 2017, $3,938 in 2016, $3,693 in 2015, $4,020 in 2014 and $4,376 in 2013 | 156,813 | 178,151 | 95,187 | 89,279 | 89,682 | ||||||||||||||
Increase (decrease) in fair value of Symphony Icon, Inc. purchase liability | 2,101 | (703 | ) | 5,927 | 1,428 | (2,210 | ) | ||||||||||||
Selling, general and administrative, including stock-based compensation of $4,567 in 2017, $3,514 in 2016, $3,150 in 2015, $3,061 in 2014 and $3,045 in 2013 | 66,203 | 43,044 | 23,835 | 19,411 | 17,121 | ||||||||||||||
Impairment loss on buildings | — | — | 3,597 | 13,102 | — | ||||||||||||||
Total operating expenses | 227,016 | 220,492 | 128,546 | 123,220 | 104,593 | ||||||||||||||
Income (loss) from operations | (136,681 | ) | (137,155 | ) | 1,468 | (100,366 | ) | (102,371 | ) | ||||||||||
Interest and other income (expense), net | (5,030 | ) | (4,274 | ) | (6,150 | ) | 2 | (1,755 | ) | ||||||||||
Consolidated net loss before taxes | (141,711 | ) | (141,429 | ) | (4,682 | ) | (100,364 | ) | (104,126 | ) | |||||||||
Income tax benefit | 12,661 | — | — | 70 | — | ||||||||||||||
Consolidated net loss | $ | (129,050 | ) | $ | (141,429 | ) | $ | (4,682 | ) | $ | (100,294 | ) | $ | (104,126 | ) | ||||
Consolidated net loss per common share, basic and diluted | $ | (1.23 | ) | $ | (1.36 | ) | $ | (0.05 | ) | $ | (1.31 | ) | $ | (1.42 | ) | ||||
Shares used in computing consolidated net loss per common share, basic and diluted | 105,237 | 103,863 | 103,591 | 76,347 | 73,302 | ||||||||||||||
As of December 31, | |||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
Balance Sheet Data: | (in thousands) | ||||||||||||||||||
Cash, cash equivalents and short-term investments, including restricted cash and investments | $ | 310,788 | $ | 346,504 | $ | 521,352 | $ | 339,339 | $ | 129,128 | |||||||||
Working capital | 197,868 | 193,231 | 409,443 | 324,018 | 115,260 | ||||||||||||||
Total assets | 436,539 | 475,625 | 651,960 | 471,376 | 274,160 | ||||||||||||||
Long-term debt, net of current portion | 231,576 | 85,167 | 100,960 | 87,500 | 20,167 | ||||||||||||||
Accumulated deficit | (1,381,404 | ) | (1,250,363 | ) | (1,108,934 | ) | (1,104,252 | ) | (1,003,958 | ) | |||||||||
Lexicon Pharmaceuticals, Inc. stockholders’ equity | 52,102 | 157,401 | 285,850 | 284,018 | 170,163 | ||||||||||||||
38
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis should be read with “Selected Financial Data” and our financial statements and notes included elsewhere in this annual report on Form 10-K.
Overview
We are a biopharmaceutical company focused on the development and commercialization of breakthrough treatments for human disease. We are presently devoting most of our resources to the commercialization or development of our four most advanced drug programs:
• | We have obtained approval from the FDA to sell our first commercial product, XERMELO (telotristat ethyl), an orally-delivered small molecule drug for the treatment of carcinoid syndrome diarrhea in combination with SSA therapy in adults inadequately controlled by SSA therapy. We have commenced sales and marketing of XERMELO, and it is now commercially available to patients in the United States. We have granted Ipsen an exclusive, royalty-bearing right to commercialize XERMELO outside of the United States and Japan, and Ipsen has obtained approval from the European Commission to market XERMELO in the member states of the European Union, Norway and Iceland. Ipsen has commenced sales and marketing of XERMELO, and it is commercially available to patients in the United Kingdom, Germany and certain other European Union member states. |
• | We are developing sotagliflozin, an orally-delivered small molecule drug candidate, as a treatment for type 1 and type 2 diabetes. We have reported positive top-line data from two pivotal Phase 3 clinical trials and a third Phase 3 clinical trial of sotagliflozin in type 1 diabetes patients. We have granted Sanofi an exclusive, worldwide (excluding Japan), royalty-bearing right to develop, manufacture and commercialize sotagliflozin. We and Sanofi are presently preparing applications for regulatory approval to market sotagliflozin for type 1 diabetes in the United States and the European Union, and Sanofi is presently conducting Phase 3 development of sotagliflozin in type 2 diabetes. |
• | We are developing LX2761, an orally-delivered small molecule drug candidate, as a treatment for diabetes. We are presently conducting Phase 1 clinical development of LX2761. We have granted Sanofi certain rights of first negotiation with respect to the future development and commercialization of LX2761. |
• | We are developing LX9211, an orally-delivered small molecule drug candidate, as a treatment for neuropathic pain. We are presently conducting Phase 1 clinical development of LX9211. |
Compounds from our most advanced drug programs, as well as compounds from a number of additional drug discovery and development programs that we have advanced into various stages of clinical and preclinical development, originated from our own internal drug discovery efforts. These efforts were driven by a systematic, target biology-driven approach in which we used gene knockout technologies and an integrated platform of advanced medical technologies to systematically study the physiological and behavioral functions of almost 5,000 genes in mice and assessed the utility of the proteins encoded by the corresponding human genes as potential drug targets. We have identified and validated in living animals, or in vivo, more than 100 targets with promising profiles for drug discovery.
We are working both independently and through strategic collaborations and alliances with third parties to capitalize on our drug target discoveries and drug discovery and development programs. We seek to retain exclusive or co-exclusive rights to the benefits of certain drug discovery and development programs by developing and commercializing drug candidates from those programs internally, particularly in the United States for indications treated by specialist physicians. We seek to collaborate with other pharmaceutical and biotechnology companies, such as Ipsen and Sanofi, with respect to drug discovery or the development and commercialization of certain of our drug candidates, particularly with respect to commercialization in territories outside the United States, commercialization in the United States for indications treated by primary care physicians, or when the collaboration may otherwise provide us with access to expertise and resources that we do not possess internally or are complementary to our own.
We commercially launched XERMELO following regulatory approval in February 2017 for the treatment of carcinoid syndrome diarrhea in combination with SSA therapy in adults inadequately controlled by SSA therapy in the United States. Prior to the launch of XERMELO, we derived substantially all of our revenues from strategic collaborations and other research and development collaborations and technology licenses. To date, we have generated a substantial portion of our revenues from a limited number of sources.
39
Our operating results and, in particular, our ability to generate additional revenues are dependent on many factors, including our ability to successfully commercialize XERMELO in the United States and the amount of revenues generated from such commercialization efforts; our and Sanofi’s ability to obtain regulatory approval for the marketing and sale of sotagliflozin for type 1 diabetes; the amount and timing of payments, if any, under our existing collaboration agreements with Sanofi, Ipsen and other entities; the success of our ongoing preclinical and clinical development efforts and ability to obtain necessary regulatory approvals; our success in establishing new collaborations and licenses; the timing and willingness of such new collaborators to commercialize products that would result in milestone payments and royalties and their success in such efforts; and general and industry-specific economic conditions which may affect research and development expenditures.
Future revenues from our commercialization of XERMELO are uncertain because they depend on a number of factors, including market acceptance of XERMELO, the success of our sales, marketing, medical affairs, distribution and other commercialization activities and the cost and availability of reimbursement for XERMELO.
Future revenues from our existing collaborations are uncertain because they depend, to a large degree, on the achievement of milestones and payment of royalties we earn from any future products developed under the collaborations. Our ability to secure future revenue-generating agreements will depend upon our ability to address the needs of our potential future collaborators and licensees, and to negotiate agreements that we believe are in our long-term best interests. We may determine, as we have with certain of our drug candidates, including XERMELO in the United States and Japan, that our interests are better served by retaining rights to our discoveries and advancing our therapeutic programs to a later stage, which could limit our near-term revenues and increase expenses. Because of these and other factors, our operating results have fluctuated in the past and are likely to do so in the future, and we do not believe that period-to-period comparisons of our operating results are a good indication of our future performance.
Since our inception, we have incurred significant losses and, as of December 31, 2017, we had an accumulated deficit of $1.4 billion. Our losses have resulted principally from costs incurred in research and development, selling, general and administrative costs associated with our operations, and non-cash stock-based compensation expenses associated with stock options and restricted stock granted to employees and consultants. Research and development expenses consist primarily of salaries and related personnel costs, external research costs related to our nonclinical and clinical efforts, material costs, facility costs, depreciation on property and equipment, and other expenses related to our drug discovery and development programs. Selling, general and administrative expenses consist primarily of salaries and related expenses for executive, sales and marketing, and administrative personnel, professional fees and other corporate expenses, including information technology, facilities costs and general legal activities. We expect to continue to incur significant research and development costs in connection with the continuing development of our drug candidates. As a result, we will need to generate significantly higher revenues to achieve profitability.
Critical Accounting Policies
Revenue Recognition
We recognize revenues when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed or determinable and collectibility is reasonably assured.
Product Revenues
Product revenues consist of commercial sales of XERMELO in the United States and sales of bulk tablets of XERMELO to Ipsen. Product revenues are recognized once we meet all four revenue recognition criteria described above. In March 2017, we began shipping XERMELO to our customers in the United States. We recognize revenue for product sales of XERMELO at the time product is received by our specialty pharmacy customers, net of allowances for customer credits, including estimated rebates, chargebacks, discounts, returns, distribution service fees, and government rebates, such as Medicare Part D coverage gap reimbursements in the United States. Product shipping and handling costs are included in cost of sales.
Customer Credits: Our specialty pharmacy customers are offered various forms of consideration, including allowances, service fees and prompt payment discounts. We expect that the specialty pharmacies will earn prompt payment discounts. As a result, we deduct the full amount of those discounts from total product sales when revenues are recognized. Service fees are also deducted from product sales as they are earned.
Rebates: Allowances for rebates include mandated discounts under the Medicaid Drug Rebate Program. Rebate amounts are based upon contractual agreements or legal requirements with public sector (e.g. Medicaid) benefit providers.
40
Rebates are amounts owed after the final dispensing of the product to a benefit plan participant and are based upon contractual agreements or legal requirements with public sector benefit providers. The allowance for rebates is based on statutory discount rates and expected utilization. Our estimates for expected utilization of rebates are based on third party market research data and data received from the specialty pharmacies. Rebates are generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known unpaid rebates from the prior quarter. If actual future rebates vary from estimates, we may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Chargebacks: Chargebacks are discounts that occur when contracted customers purchase directly from a specialty pharmacy. Contracted customers, which currently consist primarily of Public Health Service institutions, non-profit clinics, and federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The specialty pharmacies, in turn, charge back us the difference between the price initially paid by the specialty pharmacies and the discounted price paid to the specialty pharmacies by the customer. The allowance for chargebacks is based on known sales to contracted customers.
Medicare Part D Coverage Gap: The Medicare Part D prescription drug benefit mandates manufacturers to fund 50% of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Our estimates for the expected Medicare Part D coverage gap are based on data received from the specialty pharmacies. Funding of the coverage gap is generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters. If actual future funding varies from estimates, we may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Co-payment assistance: Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. We accrue a liability for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators.
Collaborative Agreements
Revenues under collaborative agreements include both license revenue and contract research revenue. Activities under collaborative agreements are evaluated to determine if they represent a multiple element revenue agreement. We identify the deliverables included within the agreement and evaluate which deliverables represent separate units of accounting. We account for those components as separate units of accounting if the following two criteria are met:
• | The delivered item or items have value to the customer on a stand-alone basis; and |
• | If there is a general right of return relative to the delivered items, delivery or performance of the undelivered items is considered probable and within our control. |
Factors considered in this determination include, among other things, whether any other vendors sell the items separately and if the licensee could use the delivered item for its intended purpose without the receipt of the remaining deliverables. If multiple deliverables included in an arrangement are separable into different units of accounting, we allocate the arrangement consideration to those units of accounting. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. Arrangement consideration is allocated at the inception of the arrangement to the identified units of accounting based on their relative estimated selling price. Revenue is recognized for each unit of accounting when the appropriate revenue recognition criteria are met.
Future milestone payments that are contingent upon the achievement of a substantive milestone are recognized in their entirety in the period in which the milestone is achieved. A milestone is substantive if:
• | The consideration payable to us is commensurate with our performance necessary to achieve the milestone or the increase in value to the collaboration resulting from our performance; |
• | The milestone relates solely to our past performance; and |
• | The milestone is reasonable relative to all of the other deliverables and payments within the arrangement. |
Commercial milestones will be accounted for as royalties and recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria were met. Subscription and license fees are recognized as other revenue upon the grant of the technology license when performance is complete and there is no continuing involvement. Royalty revenues are recognized as earned in accordance with the contract terms at the time the royalty amount is fixed and determinable based on information received from the sublicensees and at the time collectibility is reasonably assured.
41
A change in our revenue recognition policy or changes in the terms of contracts under which we recognize revenues could have an impact on the amount and timing of our recognition of revenues.
Research and Development Expenses
Research and development expenses consist of costs incurred for research and development activities solely sponsored by us as well as collaborative research and development activities. These costs include direct and research-related overhead expenses and are expensed as incurred. Technology license fees for technologies that are utilized in research and development and have no alternative future use are expensed when incurred.
We are presently devoting most of our resources to the commercialization or development of our four most advanced drug programs:
• | XERMELO, an orally-delivered small molecule drug approved by the FDA for the treatment of carcinoid syndrome diarrhea in combination with SSA therapy in adults inadequately controlled by SSA therapy; |
• | Sotagliflozin, an orally-delivered small molecule drug candidate that we are developing as a treatment for type 1 and type 2 diabetes; |
• | LX2761, an orally-delivered small molecule drug candidate, that we are developing as a treatment for diabetes; and |
• | LX9211, an orally-delivered small molecule drug candidate, that we are developing as a treatment for neuropathic pain. |
Compounds from our most advanced drug programs, as well as compounds from a number of additional drug discovery and development programs that we have advanced into various stages of clinical and preclinical development, originated from our own internal drug discovery efforts. These efforts were driven by a systematic, target biology-driven approach in which we used gene knockout technologies and an integrated platform of advanced medical technologies to systematically study the physiological and behavioral functions of almost 5,000 genes in mice and assessed the utility of the proteins encoded by the corresponding human genes as potential drug targets. We have identified and validated in living animals, or in vivo, more than 100 targets with promising profiles for drug discovery.
The drug development process takes many years to complete. The cost and length of time varies due to many factors including the type, complexity and intended use of the drug candidate. We estimate that drug development activities are typically completed over the following periods:
Phase | Estimated Completion Period | |
Preclinical development | 1-2 years | |
Phase 1 clinical trials | 1-2 years | |
Phase 2 clinical trials | 1-2 years | |
Phase 3 clinical trials | 2-4 years | |
We expect research and development costs to remain substantial in the future as we continue to fund our share of type 2 diabetes development expenses for sotagliflozin and our clinical trials of XERMELO, LX2761 and LX9211 and advance new drug candidates into clinical development. Due to the variability in the length of time necessary for drug development, the uncertainties related to the cost of these activities and ultimate ability to obtain governmental approval for commercialization, accurate and meaningful estimates of the ultimate costs to bring our potential drug candidates to market are not available.
We record significant accrued liabilities related to unbilled expenses for products or services that we have received from service providers, specifically related to ongoing nonclinical studies and clinical trials. These costs primarily relate to clinical study management, monitoring, laboratory and analysis costs, drug supplies, toxicology studies and investigator grants. We have multiple drugs in concurrent nonclinical studies and clinical trials at clinical sites throughout the world. In order to ensure that we have adequately provided for ongoing nonclinical and clinical development costs during the period in which we incur such costs, we maintain accruals to cover these expenses. Substantial portions of our nonclinical studies and clinical trials are performed by third-party laboratories, medical centers, contract research organizations and other vendors. For nonclinical studies, we accrue expenses based upon estimated percentage of work completed and the contract milestones remaining. For clinical studies, expenses are accrued based upon the number of patients enrolled and the duration of the study. We monitor patient enrollment, the progress of clinical studies and related activities to the extent possible through internal reviews of data reported to us by the vendors and clinical site visits. Our estimates depend on the timeliness and accuracy of
42
the data provided by our vendors regarding the status of each program and total program spending. We periodically evaluate the estimates to determine if adjustments are necessary or appropriate based on information we receive. Although we use consistent milestones or subject or patient enrollment to drive expense recognition, the assessment of these costs is a subjective process that requires judgment. Upon settlement, these costs may differ materially from the amounts accrued in our consolidated financial statements.
We record our research and development costs by type or category, rather than by project. Significant categories of costs include personnel, facilities and equipment costs and third-party and other services. In addition, a significant portion of our research and development expenses is not tracked by project as it benefits multiple projects. Consequently, fully-loaded research and development cost summaries by project are not available.
Stock-based Compensation Expense
We recognize compensation expense in our statements of comprehensive loss for share-based payments, including stock options and restricted stock units issued to employees, based on their fair values on the date of the grant, with the compensation expense recognized over the period in which an employee is required to provide service in exchange for the stock award. Stock-based compensation expense for awards without performance conditions is recognized on a straight-line basis. Stock-based compensation expense for awards with performance conditions is recognized over the period from the date the performance condition is determined to be probable of occurring through the time the applicable condition is met. We had stock-based compensation expense of $9.5 million for the year ended December 31, 2017, or $0.09 per share. As of December 31, 2017, stock-based compensation cost for all outstanding unvested options and restricted stock units was $18.6 million, which is expected to be recognized over a weighted-average vesting period of 1.3 years.
The fair value of stock options is estimated at the date of grant using the Black-Scholes option-pricing model. For purposes of determining the fair value of stock options, we segregate our options into two homogeneous groups, based on exercise and post-vesting employment termination behaviors, resulting in a change in the assumptions used for expected option lives and forfeitures. Expected volatility is based on the historical volatility in our stock price. The following weighted-average assumptions were used for options granted in the years ended December 31, 2017, 2016 and 2015, respectively:
Expected Volatility | Risk-free Interest Rate | Expected Term | Dividend Rate | |||||||
December 31, 2017: | ||||||||||
Employees | 61 | % | 1.7 | % | 4 | 0 | % | |||
Officers and non-employee directors | 70 | % | 2.2 | % | 8 | 0 | % | |||
December 31, 2016: | ||||||||||
Employees | 63 | % | 1.1 | % | 4 | 0 | % | |||
Officers and non-employee directors | 83 | % | 1.6 | % | 8 | 0 | % | |||
December 31, 2015: | ||||||||||
Employees | 64 | % | 1.2 | % | 4 | 0 | % | |||
Officers and non-employee directors | 81 | % | 1.8 | % | 8 | 0 | % | |||
Impairment of Long-Lived Assets
Our long-lived assets include property, plant and equipment, intangible assets and goodwill. We regularly review long-lived assets for impairment. The recoverability of long-lived assets, other than goodwill, is measured by comparing the assets carrying amount to the expected undiscounted future cash flows that the asset is expected to generate. Determining whether an impairment has occurred typically requires various estimates and assumptions, including determining which cash flows are directly related to the potentially impaired asset, the useful life over which cash flows will occur, their amount, and the asset's residual value, if any. We use internal cash flow estimates, quoted market prices when available and independent appraisals as appropriate to determine fair value. We derive the required cash flow estimates from our historical experience and our internal business plans and apply an appropriate discount rate. During 2015, we determined that our buildings were impaired and therefore recorded an impairment loss of $3.6 million, which was recorded in impairment loss on buildings in the accompanying consolidated statements of comprehensive loss. There were no significant impairments of long-lived assets in 2017 or 2016.
Indefinite-lived intangible assets, composed primarily of in-process research and development, or IPR&D, projects acquired in business combinations which have not reached technological feasibility, are reviewed annually for impairment and
43
whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. Estimating future cash flows of an IPR&D product candidate for purposes of an impairment analysis requires us to make significant estimates and assumptions regarding the amount and timing of costs to complete the project and the amount, timing and probability of achieving revenues from the completed product similar to how the acquisition date fair value of the project was determined.
Goodwill is not amortized, but is tested at least annually for impairment at the reporting unit level. We have determined that the reporting unit is the single operating segment disclosed in our current financial statements. Impairment is the condition that exists when the carrying amount of goodwill exceeds its implied fair value. The first step in the impairment process is to determine the fair value of the reporting unit and then compare it to the carrying value, including goodwill. We determined that the market capitalization approach is the most appropriate method of measuring fair value of the reporting unit. Under this approach, fair value is calculated as the average closing price of our common stock for the 30 days preceding the date that the annual impairment test is performed, multiplied by the number of outstanding shares on that date. A control premium, which is representative of premiums paid in the marketplace to acquire a controlling interest in a company, is then added to the market capitalization to determine the fair value of the reporting unit. If the fair value exceeds the carrying value, no further action is required and no impairment loss is recognized. Additional impairment assessments may be performed on an interim basis if we encounter events or changes in circumstances that would indicate that, more likely than not, the carrying value of goodwill has been impaired. There was no impairment of goodwill in 2017, 2016 and 2015.
Business Combinations
We allocate the purchase price of acquired businesses to the tangible and intangible assets acquired and liabilities assumed based upon their estimated fair values on the acquisition date. The purchase price allocation process requires management to make significant estimates and assumptions, especially at acquisition date with respect to intangible assets and in-process research and development.
These assumptions are based in part on historical experience and are inherently uncertain. Examples of critical estimates in valuing certain of the intangible assets we have acquired or may acquire in the future include but are not limited to: the feasibility and timing of achievement of development, regulatory and commercial milestones; expected costs to develop the in-process research and development into commercially viable products; and future expected cash flows from product sales.
In connection with the purchase price allocations for acquisitions, we estimate the fair value of the contingent payments. The estimated fair value of any contingent payments is determined utilizing a probability-based income approach inclusive of an estimated discount rate.
Unanticipated events and circumstances may occur which may affect the accuracy or validity of such assumptions, estimates or actual results.
Recent Accounting Pronouncements
See Note 3, Recent Accounting Pronouncements, of the Notes to Consolidated Financial Statements, for a discussion of the impact of new accounting standards on our consolidated financial statements.
Results of Operations – Comparison of Years Ended December 31, 2017, 2016 and 2015
Revenues
Total revenues and dollar and percentage changes as compared to the prior year are as follows (dollar amounts are presented in millions):
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Total revenues | $ | 90.3 | $ | 83.3 | $ | 130.0 | |||||
Dollar increase (decrease) | $ | 7.0 | $ | (46.7 | ) | ||||||
Percentage increase (decrease) | 8 | % | (36 | )% | |||||||
Years Ended December 31, 2017 and 2016
44
• | Net product revenues – Net product revenue was $15.9 million in 2017, representing revenues recognized from the sale of XERMELO in the United States and sales of bulk tablets of XERMELO to Ipsen. Product revenues are recorded net of estimated product returns, pricing discounts including rebates offered pursuant to mandatory federal and state government programs and chargebacks, prompt pay discounts and distribution fees and co-pay assistance. Revenue recognition policies require estimates of the aforementioned sales allowances each period. |
• | Collaborative agreements – Revenue from collaborative agreements decreased 11% in 2017 to $74.3 million, primarily due to a decrease in revenues recognized from the collaboration and license agreement with Sanofi, partially offset by increases in milestone revenues recognized in 2017 from the license and collaboration agreement with Ipsen. Revenues under the Sanofi agreement in 2017 and 2016 were primarily attributable to the development activities we performed relating to type 1 diabetes, together with funding of our share of type 2 diabetes development expenses. |
• | Royalties and other revenue – Revenues from royalties and other revenue increased 15% in 2017 to $0.2 million. |
Years Ended December 31, 2016 and 2015
• | Collaborative agreements – Revenue from collaborative agreements decreased 36% in 2016 to $83.2 million, primarily due to a decrease in revenues recognized from the collaboration and license agreement with Sanofi. Revenues under the Sanofi agreement in 2016 were primarily attributable to the development activities we performed relating to type 1 diabetes, together with funding of our share of type 2 diabetes development expenses. Revenues under the Sanofi agreement in 2015 were primarily attributable to the license portion of the upfront payment made by Sanofi in connection with the agreement. |
• | Royalties and other revenue – Revenues from royalties and other revenue decreased 46% in 2016 to $0.2 million. |
In 2017, Sanofi and Ipsen represented 64% and 18% of revenues, respectively. In 2016, Sanofi and Ipsen represented 90% and 9% of revenues, respectively. In 2015, Ipsen represented 98% of revenues.
Cost of Sales
Cost of sales for 2017 was $1.9 million. We began capitalizing inventory in 2017 following FDA approval of XERMELO, as the related costs were expected to be recoverable through the commercialization of the product. Costs incurred prior to FDA approval were recorded as research and development expenses in the consolidated statements of comprehensive loss. Cost of sales consists of third-party manufacturing costs, freight and indirect overhead costs associated with sales of XERMELO. The pre-commercialized inventory is expected to be sold over approximately the next two years. As a result, cost of sales will reflect a lower average per unit cost of materials. Cost of sales in 2017 includes $1.5 million of amortization of intangible assets related to XERMELO.
Research and Development Expenses
Research and development expenses and dollar and percentage changes as compared to the prior year are as follows (dollar amounts are presented in millions):
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Total research and development expense | $ | 156.8 | $ | 178.2 | $ | 95.2 | |||||
Dollar increase (decrease) | $ | (21.3 | ) | $ | 83.0 | ||||||
Percentage increase (decrease) | (12 | )% | 87 | % | |||||||
Research and development expenses consist primarily of third-party and other services principally related to nonclinical and clinical development activities, salaries and other personnel-related expenses, facility and equipment costs and stock-based compensation.
Years Ended December 31, 2017 and 2016
• | Third-party and other services – Third-party and other services decreased 17% in 2017 to $121.0 million, primarily due to decreases in our external clinical development costs relating to sotagliflozin. Third-party and other services |
45
relate principally to our clinical trial and related development activities, such as nonclinical and clinical studies and contract manufacturing.
• | Personnel – Personnel costs increased 18% in 2017 to $22.2 million, primarily due to increases in personnel. Salaries, bonuses, employee benefits, payroll taxes, recruiting and relocation costs are included in personnel costs. |
• | Stock-based compensation – Stock-based compensation expense increased 25% in 2017 to $4.9 million. |
• | Facilities and equipment – Facilities and equipment costs decreased 11% in 2017 to $3.0 million. |
• | Other – Other costs increased 3% in 2017 to $5.8 million. |
Years Ended December 31, 2016 and 2015
• | Third-party and other services – Third-party and other services increased 110% in 2016 to $146.5 million, primarily due to increases in our external clinical development costs relating to sotagliflozin. Third-party and other services relate principally to our clinical trial and related development activities, such as nonclinical and clinical studies and contract manufacturing. |
• | Personnel – Personnel costs increased 27% in 2016 to $18.8 million, primarily due to increases in personnel, including increases in medical affairs personnel, in preparation for commercialization of XERMELO. |
• | Stock-based compensation – Stock-based compensation expense increased 7% in 2016 to $3.9 million. |
• | Facilities and equipment – Facilities and equipment costs increased 7% in 2016 to $3.3 million. |
• | Other – Other costs increased 52% in 2016 to $5.6 million, primarily due to increases in travel and sponsorships. |
Increase (Decrease) in Fair Value of Symphony Icon Liability
The fair value of the Symphony Icon purchase liability increased by $2.1 million in the year ended December 31, 2017, decreased by $0.7 million in the year ended December 31, 2016, and increased by $5.9 million for the year ended December 31, 2015, respectively (see Note 10, Arrangements with Symphony Icon, Inc., of the Notes to Consolidated Financial Statements, for more information).
Selling, General and Administrative Expenses
Selling, general and administrative expenses and dollar and percentage changes as compared to the prior year are as follows (dollar amounts are presented in millions):
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Total selling, general and administrative expense | $ | 66.2 | $ | 43.0 | $ | 23.8 | |||||
Dollar increase | $ | 23.2 | $ | 19.2 | |||||||
Percentage increase | 54 | % | 81 | % | |||||||
Selling, general and administrative expenses consist primarily of personnel costs to support the commercialization of XERMELO and our research and development activities, professional and consulting fees, stock-based compensation expense, and facility and equipment costs.
Years Ended December 31, 2017 and 2016
• | Personnel – Personnel costs increased 86% in 2017 to $30.1 million, primarily due to increases in personnel, including increases in sales and marketing personnel, in connection with commercialization of XERMELO. Salaries, bonuses, employee benefits, payroll taxes, recruiting and relocation costs are included in personnel costs. |
46
• | Professional and consulting fees – Professional and consulting fees increased 13% in 2017 to $20.8 million, primarily due to increases in legal and patent fees, as well as marketing and consulting costs in connection with commercialization of XERMELO. |
• | Stock-based compensation – Stock-based compensation expense increased 30% in 2017 to $4.6 million, primarily due to awards granted to sales and marketing personnel. |
• | Facilities and equipment – Facilities and equipment costs increased 28% in 2017 to $2.0 million. |
• | Other – Other costs increased 161% in 2017 to $8.7 million, primarily due to increases in travel and contributions to charitable foundations. |
Years Ended December 31, 2016 and 2015
• | Professional and consulting fees – Professional and consulting fees increased 149% in 2016 to $18.5 million, primarily due to increased consulting costs in preparation for commercialization of XERMELO. |
• | Personnel – Personnel costs increased 58% in 2016 to $16.2 million, primarily due to increases in personnel, including increases in sales and marketing personnel, in preparation for commercialization of XERMELO. |
• | Stock-based compensation – Stock-based compensation expense increased 12% in 2016 to $3.5 million. |
• | Facilities and equipment – Facilities and equipment costs increased 59% in 2016 to $1.5 million, primarily due to increases in depreciation expense and property taxes. |
• | Other – Other costs increased 65% in 2016 to $3.3 million, primarily due to travel and training expenses. |
Impairment Loss on Buildings
In 2015, we recognized an impairment loss on our buildings of $3.6 million for the year ended December 31, 2015, as a result of writing down the buildings to the estimated net selling price.
Interest Expense and Interest and Other Income (Expense), Net
Interest Expense. Interest expense increased 6% in 2017 to $7.0 million from $6.6 million in 2016, due to a new loan agreement completed in December 2017. Interest expense decreased 2% in 2016 from $6.7 million in 2015.
Interest and Other Income (Expense), Net. Interest and other income, net was $2.0 million, $2.3 million, and $0.6 million in the years ended December 31, 2017, 2016, and 2015, respectively.
Income Tax Benefit
The income tax benefit for the year ended December 31, 2017 was $12.7 million (see Note 7, Income Taxes of the Notes to Consolidated Financial Statements, for more information). There was no income tax expense or benefit in 2016 or 2015.
Consolidated Net Loss and Consolidated Net Loss per Common Share
Consolidated net loss decreased to $129.1 million in 2017 from $141.4 million in 2016 and $4.7 million in 2015. Net loss per common share was $1.23 in 2017, $1.36 in 2016, and $0.05 in 2015.
Liquidity and Capital Resources
We have financed our operations from inception primarily through sales of common and preferred stock, contract and milestone payments we received under our strategic and other collaborations, target validation, database subscription and technology license agreements, product sales, government grants and contracts, and financing under debt and lease arrangements. We have also financed certain of our research and development activities under our agreements with Symphony Icon, Inc. From our inception through December 31, 2017, we had received net proceeds of $1.4 billion from issuances of common and preferred stock and convertible debt. In December 2017, we received $145.9 million in net proceeds from the
47
first tranche of a loan agreement with BioPharma Credit PLC and BioPharma Credit Investments IV Sub LP, as discussed below. In addition, from our inception through December 31, 2017, we received $828.7 million in cash payments from strategic and other collaborations, target validation, database subscription and technology license agreements, sales of compound libraries and reagents, product sales, and government grants and contracts, of which $771.3 million had been recognized as revenues through December 31, 2017.
As of December 31, 2017, we had $310.8 million in cash, cash equivalents and short-term investments. As of December 31, 2016, we had $346.5 million in cash, cash equivalents and short-term investments. We used cash of $185.4 million in operations in 2017. This consisted primarily of the consolidated net loss for the year of $129.1 million and a net decrease in other operating liabilities net of assets of $59.3 million and the deferred tax benefit of $12.7 million, partially offset by non-cash charges of $9.5 million related to stock-based compensation expense, $3.4 million related to depreciation and amortization expense and $2.1 million related to the increase in fair value of the Symphony Icon purchase liability. Investing activities provided cash of $50.5 million in 2017, primarily due to net maturities of investments of $50.8 million. Financing activities provided cash of $149.9 million, primarily due to net proceeds of $145.9 million from a new loan agreement completed in December 2017 and $8.0 million from issuance of common stock, partially offset by repayment of debt borrowings of $2.3 million and repurchases of common stock of $1.7 million.
Symphony Drug Development Financing Agreements. In June 2007, we entered into a series of related agreements providing for the financing of the clinical development of certain drug programs, including XERMELO, along with any other pharmaceutical compositions modulating the same targets as those drug candidates. Under the financing arrangement, we licensed to Symphony Icon, Inc., a then wholly-owned subsidiary of Symphony Icon Holdings LLC, our intellectual property rights related to the programs and Holdings contributed $45 million to Symphony Icon in order to fund the clinical development of the programs. We also issued and sold to Holdings shares of our common stock in exchange for $15 million and received an exclusive option to acquire all of the equity of Symphony Icon, thereby allowing us to reacquire the programs.
Upon the recommendation of Symphony Icon’s development committee, which was comprised of an equal number of representatives from us and Symphony Icon, Symphony Icon’s board of directors had the right to require us to pay Symphony Icon up to $15 million for Symphony Icon’s use in the development of the programs in accordance with a specified development plan and related development budget. Symphony Icon’s board of directors requested that we pay Symphony Icon $9.3 million under the agreement, all of which was paid prior to the exercise of the purchase option in July 2010.
In July 2010, we entered into an amended and restated purchase option agreement with Symphony Icon and Holdings and simultaneously exercised our purchase option. Pursuant to the amended terms of the purchase option, we paid Holdings $10 million in July 2010 and issued 1,891,074 shares of common stock to designees of Holdings in July 2012 in satisfaction of an additional $35 million base payment obligation. We also agreed to make up to $45 million in additional contingent payments upon the occurrence of certain specified events. In December 2014, we paid $5.8 million in cash and issued 666,111 shares of common stock to designees of Holdings in satisfaction of a $11.5 million contingent payment obligation as a result of receiving an upfront payment pursuant to our license and collaboration agreement with Ipsen. In April 2015, we paid $0.75 million in cash to Holdings in satisfaction of our contingent payment obligation as a result of receiving an additional upfront payment from Ipsen in March 2015. In September 2016, we paid $3.2 million in cash to Holdings in satisfaction of our contingent payment obligation as a result of receiving a milestone payment from Ipsen in August 2016 (see Note 15, Collaboration and License Agreements, of the Notes to Consolidated Financial Statements, for more information).
In September 2016, we entered into an amendment to the amended and restated purchase option agreement pursuant to which we agreed to pay Holdings $21.0 million upon our receipt of regulatory approval in the United States for the marketing and sale of XERMELO, such buyout amount to be in lieu of any remaining payments due to Holdings under the amended and restated purchase option agreement. In March 2017, we paid $10.5 million in cash and issued 659,905 shares of common stock to designees of Holdings in satisfaction of our remaining contingent payment obligation as a result of receiving regulatory approval in the United States for the marketing and sale of XERMELO.
Loan Agreement. In December 2017, we entered into a loan agreement that provides up to $200.0 million in borrowing capacity available in two tranches, each maturing in December 2022 and bearing interest at 9.0% per year. The first $150.0 million tranche was funded in December 2017 and we plan to use the net proceeds of $145.9 million to fund working capital and other general corporate purposes. The second $50.0 million tranche is available for draw at our discretion by March 2019 if net sales of XERMELO are greater than $25 million in the preceding quarter.
Texas Institute for Genomic Medicine. In July 2005, we received an award from the Texas Enterprise Fund for the creation of a knockout mouse embryonic stem cell library containing 350,000 cell lines for the Texas Institute for Genomic Medicine, or TIGM, using our proprietary gene trapping technology, which we completed in 2007. We also equipped TIGM
48
with the bioinformatics software required for the management and analysis of data relating to the library. The Texas Enterprise Fund made an additional award to the Texas A&M University System for the creation of facilities and infrastructure to house the library.
Under the terms of our award, we are responsible for the creation of a specified number of jobs beginning in 2012, reaching an aggregate of 1,616 new jobs in Texas by December 31, 2016. We will receive credits against those job obligations based on funding received by TIGM and certain related parties from sources other than the State of Texas. We will also receive credits against those job obligations for any surplus jobs we create. Subject to these credits, the State may require us to repay $2,415 for each job we fall short beginning in 2013. Our maximum aggregate exposure for such payments, if we fail to create any new jobs, is approximately $14.2 million, without giving effect to any credits to which we may be entitled.
Facilities. In April 2004, we obtained a $34.0 million mortgage on our facilities in The Woodlands, Texas. The mortgage loan originally had a ten-year term with a 20-year amortization and a fixed interest rate of 8.23%. The mortgage was amended in September 2013 to extend the maturity date from April 2014 to April 2017, with the mortgage loan’s monthly payment amount and fixed interest rate each remaining unchanged. In April 2017, the mortgage was amended to extend the maturity date to April 2018, with the mortgage loan’s monthly payment amount and fixed interest rate each remaining unchanged. The mortgage had a principal balance outstanding of $14.1 million as of December 31, 2017. The entire principal balance is recorded as current portion of long-term debt in the accompanying consolidated balance sheet as of December 31, 2017 as there is a balloon payment due in April 2018. We intend to refinance the mortgage prior to the balloon payment becoming due.
In March 2015, our subsidiary Lexicon Pharmaceuticals (New Jersey), Inc. leased a 25,000 square-foot office space in Basking Ridge, New Jersey. The term of the lease extends from June 1, 2015 through December 31, 2022, and provides for escalating yearly base rent payments starting at $482,000 and increasing to $646,000 in the final year of the lease.
Including the lease and debt obligations described above, we had incurred the following contractual obligations as of December 31, 2017:
Payments due by period (in millions) | |||||||||||||||||||
Contractual Obligations | Total | Less than 1 year | 2-3 years | 4-5 years | More than 5 years | ||||||||||||||
Debt | $ | 251.6 | $ | 14.1 | $ | — | $ | 237.5 | $ | — | |||||||||
Interest payment obligations | 86.3 | 18.7 | 36.6 | 31.0 | — | ||||||||||||||
Operating leases | 3.2 | 0.7 | 1.2 | 1.3 | — | ||||||||||||||
Total | $ | 341.1 | $ | 33.5 | $ | 37.8 | $ | 269.8 | $ | — | |||||||||
Our future capital requirements will be substantial and will depend on many factors, including the success of our sales, marketing, distribution and other commercialization activities for XERMELO in the United States and the revenues we generate from that approved product; the success of Ipsen’s sales, marketing, distribution and other commercialization activities for XERMELO outside of the United States and Japan; our and Sanofi’s ability to obtain regulatory approval for the marketing and sale of sotagliflozin for type 1 diabetes; if approved, our and Sanofi’s ability to successfully commercialize sotagliflozin for type 1 diabetes in the United States and Sanofi’s ability to successfully commercialize sotagliflozin for type 1 diabetes outside of the United States and Japan; the progress and scope of Sanofi’s development activities with respect to sotagliflozin in type 2 diabetes patients; the timing, progress and results of clinical trials of XERMELO, LX2761 and LX9211; the amount and timing of payments, if any, under our existing collaboration agreements with Sanofi, Ipsen and other entities; the amount and timing of our research, development and commercialization expenditures; the resources we devote to developing and supporting our products and other factors. Our capital requirements will also be affected by any expenditures we make in connection with license agreements and acquisitions of and investments in complementary technologies and businesses. We expect to devote substantial capital resources to commercialize XERMELO; to seek regulatory approval and prepare for commercialization in the United States for sotagliflozin in type 1 diabetes; to our clinical development efforts with respect to XERMELO, LX2761 and LX9211; and for other general corporate activities. We believe that our current unrestricted cash and investment balances and cash and revenues we expect to derive from strategic and other collaborations and other sources will be sufficient to fund our operations for at least the next 12 months. During or after this period, if cash generated by operations is insufficient to satisfy our liquidity requirements, we will need to sell additional equity or debt securities or obtain additional credit arrangements. Additional financing may not be available on terms acceptable to us or at all. The sale of additional equity or convertible debt securities may result in additional dilution to our stockholders.
49
Disclosure about Market Risk
We are exposed to limited market and credit risk on our cash equivalents which have maturities of three months or less at the time of purchase. We maintain a short-term investment portfolio which consists of U.S. Treasury bills and corporate debt securities that mature three to 12 months from the time of purchase, which we believe are subject to limited market and credit risk. We currently do not hedge interest rate exposure or hold any derivative financial instruments in our investment portfolio.
We had approximately $310.8 million in cash and cash equivalents and short-term investments as of December 31, 2017, which included $145.9 million in net proceeds from Tranche A borrowings. We believe that the working capital available to us will be sufficient to meet our cash requirements for at least the next 12 months.
We have operated primarily in the United States and substantially all sales to date have been made in U.S. dollars. Accordingly, we have not had any material exposure to foreign currency rate fluctuations.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
See “Disclosure about Market Risk” under “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” for quantitative and qualitative disclosures about market risk.
Item 8. Financial Statements and Supplementary Data
The financial statements required by this Item are incorporated under Item 15 in Part IV of this report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures (as defined in rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934) are effective to ensure that the information required to be disclosed by us in the reports we file under the Securities Exchange Act is gathered, analyzed and disclosed with adequate timeliness, accuracy and completeness, based on an evaluation of such controls and procedures as of the end of the period covered by this report.
Subsequent to our evaluation, there were no significant changes in internal controls or other factors that could significantly affect internal controls, including any corrective actions with regard to significant deficiencies and material weaknesses.
Management Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act).
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 Framework).
Based on such assessment using those criteria, management believes that, as of December 31, 2017, our internal control over financial reporting is effective.
Our independent auditors have also audited our internal control over financial reporting as of December 31, 2017 as stated in the audit report which appears on page F-2 and is incorporated under Item 15 in Part IV of this report.
50
Item 9B. Other Information
None.
51
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this Item is hereby incorporated by reference from (a) the information appearing under the captions “Election of Directors,” “Stock Ownership of Certain Beneficial Owners and Management,” “Corporate Governance” and “Executive and Director Compensation” in our definitive proxy statement which involves the election of directors and is to be filed with the Securities and Exchange Commission pursuant to the Securities Exchange Act of 1934 within 120 days of the end of our fiscal year on December 31, 2017 and (b) the information appearing under Item 1 in Part I of this report.
Item 11. Executive Compensation
The information required by this Item is hereby incorporated by reference from the information appearing under the captions “Corporate Governance” and “Executive and Director Compensation” in our definitive proxy statement which involves the election of directors and is to be filed with the Commission pursuant to the Securities Exchange Act of 1934 within 120 days of the end of our fiscal year on December 31, 2017. Notwithstanding the foregoing, in accordance with the instructions to Item 407(e)(5) of Regulation S-K, the information contained in our proxy statement under the sub-heading “Compensation Committee Report” shall not be deemed to be filed as part of or incorporated by reference into this annual report on Form 10-K.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item is hereby incorporated by reference from the information appearing under the captions “Stock Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in our definitive proxy statement which involves the election of directors and is to be filed with the Commission pursuant to the Securities Exchange Act of 1934 within 120 days of the end of our fiscal year on December 31, 2017.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is hereby incorporated by reference from the information appearing under the captions “Corporate Governance” and “Transactions with Related Persons” in our definitive proxy statement which involves the election of directors and is to be filed with the Commission pursuant to the Securities Exchange Act of 1934 within 120 days of the end of our fiscal year on December 31, 2017.
Item 14. Principal Accounting Fees and Services
The information required by this Item as to the fees we pay our principal accountant is hereby incorporated by reference from the information appearing under the caption “Ratification and Approval of Independent Auditors” in our definitive proxy statement which involves the election of directors and is to be filed with the Commission pursuant to the Securities Exchange Act of 1934 within 120 days of the end of our fiscal year on December 31, 2017.
52
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) | Documents filed as a part of this report: |
1. | Consolidated Financial Statements |
Page | |
2. | Financial Statement Schedules |
All other financial statement schedules are omitted because they are not applicable or not required, or because the required information is included in the financial statements or notes thereto.
3. | Exhibits |
Exhibit No. | Description | ||
3.1 | — | Amended and Restated Certificate of Incorporation (filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K dated April 26, 2012 and incorporated by reference herein). | |
3.2 | — | Certificate of Amendment to Amended and Restated Certificate of Incorporation (filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K dated May 20, 2015 and incorporated by reference herein). | |
3.3 | — | Second Amended and Restated Bylaws (filed as Exhibit 3.2 to the Company’s Current Report on Form 8‑K dated April 26, 2012 and incorporated by reference herein). | |
4.1 | — | Securities Purchase Agreement, dated June 17, 2007, with Invus, L.P. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated June 17, 2007 and incorporated by reference herein). | |
4.2 | — | Amendment, dated October 7, 2009, to Securities Purchase Agreement, dated June 17, 2007, with Invus, L.P. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated October 7, 2009 and incorporated by reference herein). | |
4.3 | — | Registration Rights Agreement, dated June 17, 2007, with Invus, L.P. (filed as Exhibit 10.3 to the Company’s Current Report on Form 8-K dated June 17, 2007 and incorporated by reference herein). | |
4.4 | — | Stockholders’ Agreement, dated June 17, 2007, with Invus, L.P. (filed as Exhibit 10.4 to the Company’s Current Report on Form 8-K dated June 17, 2007 and incorporated by reference herein). | |
4.5 | — | Supplement to Transaction Agreements, dated March 15, 2010, with Invus, L.P. and Invus C.V. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated March 15, 2010 and incorporated by reference herein). | |
4.6 | — | Supplement No. 2 to Transaction Agreements, dated February 23, 2012, with Invus, L.P. and Invus C.V. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated February 23, 2012 and incorporated by reference herein). | |
4.7 | — | Indenture related to the 5.25% Convertible Senior Notes due 2021, dated as of November 26, 2014, with Wells Fargo Bank, N.A. (filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K dated November 26, 2014 and incorporated by reference herein). | |
53
Exhibit No. | Description | ||
4.8 | — | Form of 5.25% Convertible Senior Notes due 2021 (filed as Exhibit A to Exhibit 4.1 to the Company’s Current Report on Form 8-K dated November 26, 2014 and incorporated by reference herein). | |
10.1 | — | Offer Letter, dated July 1, 2014, with Lonnel Coats (filed as Exhibit 10.3 to the Company’s Current Report on Form 8-K dated July 7, 2014 and incorporated by reference herein). | |
10.2 | — | Offer Letter, dated March 10, 2011, with Pablo Lapuerta, M.D. (filed as Exhibit 10.5 to the Company’s Annual Report on Form 10-K for the period ended December 31, 2011 and incorporated by reference herein). | |
10.3 | — | Offer Letter, dated March 23, 2016, with Praveen Tyle, Ph.D. (filed as Exhibit 10.4 to the Company’s Annual Report on Form 10-K for the period ended December 31, 2016 and incorporated by reference herein). | |
10.4 | — | Employment Agreement with Jeffrey L. Wade, J.D. (filed as Exhibit 10.3 to the Company’s Registration Statement on Form S-1 (Registration No. 333-96469) and incorporated by reference herein). | |
10.5 | — | Consulting Agreement with Alan S. Nies, M.D. dated February 19, 2003, as amended (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the period ended September 30, 2010 and incorporated by reference herein). | |
10.6 | — | Consulting Agreement with Robert J. Lefkowitz, M.D. dated March 31, 2003 (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the period ended March 31, 2003 and incorporated by reference herein). | |
10.7 | — | Form of Indemnification Agreement with Officers and Directors (filed as Exhibit 10.7 to the Company’s Registration Statement on Form S-1 (Registration No. 333-96469) and incorporated by reference herein). | |
10.8 | — | Summary of Non-Employee Director Compensation (filed as Exhibit 10.3 to the Company’s Current Report on Form 8-K dated April 27, 2017 and incorporated by reference herein). | |
10.9 | — | 2017 Equity Incentive Plan (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated April 27, 2017 and incorporated by reference herein). | |
10.10 | — | 2017 Non-Employee Directors’ Equity Incentive Plan (filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K dated April 27, 2017 and incorporated by reference herein). | |
*10.11 | — | Form of Stock Option Agreement with Officers under the 2017 Equity Incentive Plan. | |
*10.12 | — | Form of Restricted Stock Unit Agreement with Officers under the 2017 Equity Incentive Plan. | |
*10.13 | — | Form of Notice of Stock Option Grant to Directors under the 2017 Non-Employee Directors’ Equity Incentive Plan. | |
†10.14 | — | Collaboration and License Agreement, dated November 5, 2015, with Sanofi (filed as Exhibit 10.14 to the Company’s Annual Report on Form 10-K/A for the period ended December 31, 2015 and incorporated by reference herein). | |
†10.15 | — | Amendment No. 1, dated July 1, 2017, to Collaboration and License Agreement, dated November 5, 2015, with Sanofi (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the period ended September 30, 2017 and incorporated by reference herein). | |
†10.16 | — | License and Collaboration Agreement, dated October 21, 2014, with Ipsen Pharma SAS (filed as Exhibit 10.1 to the amendment to the Company’s Quarterly Report on Form 10-Q/A for the period ended September 30, 2014, as filed on December 23, 2014, and incorporated by reference herein). | |
†10.17 | — | First Amendment, dated March 17, 2015, to License and Collaboration Agreement, dated October 21, 2014, with Ipsen Pharma SAS (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the period ended March 31, 2015 and incorporated by reference herein). | |
10.18 | — | Collaboration and License Agreement, dated December 17, 2003, with Bristol-Myers Squibb Company (filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the period ended March 31, 2015 and incorporated by reference herein). | |
54
Exhibit No. | Description | |
10.19 | — | First Amendment, dated May 30, 2006, to Collaboration and License Agreement, dated December 17, 2003, with Bristol-Myers Squibb Company (filed as Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the period ended March 31, 2015 and incorporated by reference herein). |
†10.20 | — | Second Amendment, dated November 2, 2016, to Collaboration and License Agreement, dated December 17, 2003, with Bristol-Myers Squibb Company (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated November 2, 2016 and incorporated by reference herein). |
†10.21 | — | Second Amended and Restated Collaboration and License Agreement, dated November 30, 2005, with Genentech, Inc. (filed as Exhibit 10.22 to the Company’s Annual Report on Form 10-K for the period ended December 31, 2005 and incorporated by reference herein). |
10.22 | — | Amendment, dated June 8, 2009, to Second Amended and Restated Collaboration and License Agreement, dated November 30, 2005, with Genentech, Inc. (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K/A dated June 8, 2009 and incorporated by reference herein). |
†10.23 | — | Commercial Supply Agreement, dated June 6, 2016, with Catalent CTS, LLC (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q/A for the period ended March 31, 2017 and incorporated by reference herein). |
10.24 | — | Economic Development Agreement, dated July 15, 2005, with the State of Texas and the Texas A&M University System (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the period ended September 30, 2005 and incorporated by reference herein). |
10.25 | — | Amendment, dated April 30, 2008, to Economic Development Agreement, dated July 15, 2005, with the State of Texas and the Texas A&M University System (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated April 30, 2008 and incorporated by reference herein). |
†*10.26 | — | |
†10.27 | — | Loan Agreement, dated December 4, 2017, with BioPharma Credit PLC and BioPharma Credit Investments IV Sub LP (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated December 4, 2017 and incorporated by reference herein). |
21.1 | — | Subsidiaries (filed as Exhibit 21.1 to the Company’s Annual Report on Form 10-K for the period ended December 31, 2010 and incorporated by reference herein). |
*23.1 | — | |
*24.1 | — | Power of Attorney (contained in signature page). |
*31.1 | — | |
*31.2 | — | |
*32.1 | — | |
*101.INS | — | XBRL Instance Document. |
*101.SCH | — | XBRL Taxonomy Extension Schema Document. |
*101.CAL | — | XBRL Taxonomy Extension Calculation Linkbase Document. |
*101.DEF | — | XBRL Taxonomy Extension Definition Linkbase Document. |
*101.LAB | — | XBRL Taxonomy Extension Label Linkbase Document. |
*101.PRE | — | XBRL Taxonomy Extension Presentation Linkbase Document. |
_____________________
* | Filed herewith. |
† | Confidential treatment has been requested for a portion of this exhibit. The confidential portions of this exhibit have been omitted and filed separately with the Securities and Exchange Commission. |
Item 16. Form 10-K Summary
Not applicable.
55
Signatures
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
Lexicon Pharmaceuticals, Inc. | |||
Date: | March 1, 2018 | By: | /s/ LONNEL COATS |
Lonnel Coats | |||
President and Chief Executive Officer | |||
Date: | March 1, 2018 | By: | /s/ JEFFREY L. WADE |
Jeffrey L. Wade | |||
Executive Vice President, Corporate and Administrative Affairs and Chief Financial Officer | |||
Power of Attorney
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Lonnel Coats and Jeffrey L. Wade, or either of them, each with the power of substitution, his or her attorney-in-fact, to sign any amendments to this Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, here ratifying and confirming all that each of said attorneys-in-fact, or his or her substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | |
/s/ LONNEL COATS | President, Chief Executive Officer and Director (Principal Executive Officer) | March 1, 2018 | |
Lonnel Coats | |||
/s/ JEFFREY L. WADE | Executive Vice President, Corporate and Administrative Affairs and Chief Financial Officer (Principal Financial Officer) | March 1, 2018 | |
Jeffrey L. Wade | |||
/s/ JAMES F. TESSMER | Vice President, Finance and Accounting (Principal Accounting Officer) | March 1, 2018 | |
James F. Tessmer | |||
/s/ RAYMOND DEBBANE | Chairman of the Board of Directors | March 1, 2018 | |
Raymond Debbane | |||
/s/ PHILIPPE J. AMOUYAL | Director | March 1, 2018 | |
Philippe J. Amouyal | |||
/s/ SAMUEL L. BARKER | Director | March 1, 2018 | |
Samuel L. Barker, Ph.D. | |||
/s/ ROBERT J. LEFKOWITZ | Director | March 1, 2018 | |
Robert J. Lefkowitz, M.D. | |||
/s/ ALAN S. NIES | Director | March 1, 2018 | |
Alan S. Nies, M.D. | |||
/s/ FRANK P. PALANTONI | Director | March 1, 2018 | |
Frank P. Palantoni | |||
/s/ CHRISTOPHER J. SOBECKI | Director | March 1, 2018 | |
Christopher J. Sobecki | |||
/s/ JUDITH L. SWAIN | Director | March 1, 2018 | |
Judith L. Swain, M.D. | |||
56
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Lexicon Pharmaceuticals, Inc.:
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Lexicon Pharmaceuticals, Inc. as of December 31, 2017 and 2016, the related consolidated statements of comprehensive loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the consolidated financial position of the Company at December 31, 2017 and 2016, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 1, 2018 expressed an unqualified opinion thereon.
Basis of Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2002.
Houston, Texas
March 1, 2018
F-1
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Lexicon Pharmaceuticals, Inc.:
Opinion on Internal Control over Financial Reporting
We have audited Lexicon Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Lexicon Pharmaceuticals, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the accompanying consolidated balance sheets of Lexicon Pharmaceuticals, Inc. as of December 31, 2017 and 2016, the related consolidated statements of comprehensive loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”) of the Company and our report dated March 1, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Houston, Texas
March 1, 2018
F-2
Lexicon Pharmaceuticals, Inc.
Consolidated Balance Sheets
(In thousands, except par value)
As of December 31, | |||||||
2017 | 2016 | ||||||
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 61,661 | $ | 46,600 | |||
Short-term investments | 249,127 | 299,904 | |||||
Accounts receivable, net of allowances of $4 | 4,825 | 7,492 | |||||
Inventory | 1,948 | — | |||||
Prepaid expenses and other current assets | 4,434 | 3,878 | |||||
Total current assets | 321,995 | 357,874 | |||||
Property and equipment, net of accumulated depreciation and amortization of $58,623 and $59,875, respectively | 17,687 | 19,390 | |||||
Goodwill | 44,543 | 44,543 | |||||
Other intangible assets | 51,885 | 53,357 | |||||
Other assets | 429 | 461 | |||||
Total assets | $ | 436,539 | $ | 475,625 | |||
Liabilities and Equity | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 57,652 | $ | 52,877 | |||
Accrued liabilities | 12,282 | 32,114 | |||||
Current portion of deferred revenue | 40,099 | 63,372 | |||||
Current portion of long-term debt, net of deferred financing costs | 14,094 | 16,280 | |||||
Total current liabilities | 124,127 | 164,643 | |||||
Deferred revenue, net of current portion | 22,428 | 48,934 | |||||
Long-term debt, net of deferred financing costs | 231,576 | 85,167 | |||||
Deferred tax liabilities | 6,014 | 18,675 | |||||
Other long-term liabilities | 292 | 805 | |||||
Total liabilities | 384,437 | 318,224 | |||||
Commitments and contingencies | |||||||
Equity: | |||||||
Preferred stock, $.01 par value; 5,000 shares authorized; no shares issued and outstanding | — | — | |||||
Common stock, $.001 par value; 225,000 shares authorized; 105,711 and 104,582 shares issued, respectively | 106 | 105 | |||||
Additional paid-in capital | 1,435,526 | 1,411,222 | |||||
Accumulated deficit | (1,381,404 | ) | (1,250,363 | ) | |||
Accumulated other comprehensive loss | (222 | ) | (195 | ) | |||
Treasury stock, at cost, 122 and 306 shares, respectively | (1,904 | ) | (3,368 | ) | |||
Total equity | 52,102 | 157,401 | |||||
Total liabilities and equity | $ | 436,539 | $ | 475,625 | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Lexicon Pharmaceuticals, Inc.
Consolidated Statements of Comprehensive Loss
(In thousands, except per share amounts)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Revenues: | |||||||||||
Net product revenue | $ | 15,890 | $ | — | $ | — | |||||
Collaborative agreements | 74,267 | 83,182 | 129,728 | ||||||||
Royalties and other revenue | 178 | 155 | 286 | ||||||||
Total revenues | 90,335 | 83,337 | 130,014 | ||||||||
Operating expenses: | |||||||||||
Cost of sales (including finite-lived intangible asset amortization) | 1,899 | — | — | ||||||||
Research and development, including stock-based compensation of $4,905, $3,938 and $3,693, respectively | 156,813 | 178,151 | 95,187 | ||||||||
Increase (decrease) in fair value of Symphony Icon, Inc. purchase liability | 2,101 | (703 | ) | 5,927 | |||||||
Selling, general and administrative, including stock-based compensation of $4,567, $3,514 and $3,150, respectively | 66,203 | 43,044 | 23,835 | ||||||||
Impairment loss on buildings | — | — | 3,597 | ||||||||
Total operating expenses | 227,016 | 220,492 | 128,546 | ||||||||
Income (loss) from operations | (136,681 | ) | (137,155 | ) | 1,468 | ||||||
Interest expense | (6,984 | ) | (6,567 | ) | (6,722 | ) | |||||
Interest and other income, net | 1,954 | 2,293 | 572 | ||||||||
Consolidated net loss before taxes | (141,711 | ) | (141,429 | ) | (4,682 | ) | |||||
Income tax benefit | 12,661 | — | — | ||||||||
Consolidated net loss | $ | (129,050 | ) | $ | (141,429 | ) | $ | (4,682 | ) | ||
Consolidated net loss per common share, basic and diluted | $ | (1.23 | ) | $ | (1.36 | ) | $ | (0.05 | ) | ||
Shares used in computing consolidated net loss per common share, basic and diluted | 105,237 | 103,863 | 103,591 | ||||||||
Other comprehensive loss: | |||||||||||
Unrealized gain (loss) on investments | (27 | ) | 24 | (156 | ) | ||||||
Comprehensive loss | $ | (129,077 | ) | $ | (141,405 | ) | $ | (4,838 | ) | ||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
Lexicon Pharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
(In thousands)
Accumulated | ||||||||||||||||||||||||||
Additional | Other | |||||||||||||||||||||||||
Common Stock | Paid-In | Accumulated | Comprehensive | Treasury | ||||||||||||||||||||||
Shares | Par Value | Capital | Deficit | Gain (Loss) | Stock | Total | ||||||||||||||||||||
Balance at December 31, 2014 | 103,663 | $ | 104 | $ | 1,390,619 | $ | (1,104,252 | ) | $ | (63 | ) | $ | (2,390 | ) | $ | 284,018 | ||||||||||
Stock-based compensation | — | — | 6,843 | — | — | — | 6,843 | |||||||||||||||||||
Issuance of common stock under Equity Incentive Plans | 197 | — | 114 | — | — | — | 114 | |||||||||||||||||||
Repurchase of common stock | — | — | — | — | — | (357 | ) | (357 | ) | |||||||||||||||||
Consolidated net loss | — | — | — | (4,682 | ) | — | — | (4,682 | ) | |||||||||||||||||
Unrealized loss on investments | — | — | — | — | (156 | ) | — | (156 | ) | |||||||||||||||||
Other | — | — | 70 | — | — | — | 70 | |||||||||||||||||||
Balance at December 31, 2015 | 103,860 | 104 | 1,397,646 | (1,108,934 | ) | (219 | ) | (2,747 | ) | 285,850 | ||||||||||||||||
Stock-based compensation | — | — | 7,452 | — | — | — | 7,452 | |||||||||||||||||||
Issuance of common stock under Equity Incentive Plans | 722 | 1 | 6,124 | — | — | — | 6,125 | |||||||||||||||||||
Repurchase of common stock | — | — | — | — | — | (621 | ) | (621 | ) | |||||||||||||||||
Consolidated net loss | — | — | — | (141,429 | ) | — | — | (141,429 | ) | |||||||||||||||||
Unrealized gain on investments | — | — | — | — | 24 | — | 24 | |||||||||||||||||||
Balance at December 31, 2016 | 104,582 | 105 | 1,411,222 | (1,250,363 | ) | (195 | ) | (3,368 | ) | 157,401 | ||||||||||||||||
Cumulative effect of change in accounting principle | — | — | 1,991 | (1,991 | ) | — | — | — | ||||||||||||||||||
Stock-based compensation | — | — | 9,472 | — | — | — | 9,472 | |||||||||||||||||||
Issuance of common stock to designees of Symphony Icon Holdings LLC | 660 | — | 10,499 | — | — | — | 10,499 | |||||||||||||||||||
Issuance of common stock under Equity Incentive Plans | 469 | 1 | 5,485 | — | — | — | 5,486 | |||||||||||||||||||
Issuance of treasury stock | — | — | (3,143 | ) | — | — | 3,143 | — | ||||||||||||||||||
Repurchase of common stock | — | — | — | — | — | (1,679 | ) | (1,679 | ) | |||||||||||||||||
Consolidated net loss | — | — | — | (129,050 | ) | — | — | (129,050 | ) | |||||||||||||||||
Unrealized loss on investments | — | — | — | — | (27 | ) | — | (27 | ) | |||||||||||||||||
Balance at December 31, 2017 | 105,711 | $ | 106 | $ | 1,435,526 | $ | (1,381,404 | ) | $ | (222 | ) | $ | (1,904 | ) | $ | 52,102 | ||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Lexicon Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(In thousands)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Cash flows from operating activities: | |||||||||||
Consolidated net loss | $ | (129,050 | ) | $ | (141,429 | ) | $ | (4,682 | ) | ||
Adjustments to reconcile consolidated net loss to net cash provided by (used in) operating activities: | |||||||||||
Depreciation and amortization | 3,399 | 2,056 | 727 | ||||||||
Impairment of assets | — | — | 3,597 | ||||||||
Increase (decrease) in fair value of Symphony Icon, Inc. purchase liability | 2,101 | (703 | ) | 5,927 | |||||||
Stock-based compensation | 9,472 | 7,452 | 6,843 | ||||||||
(Gain) loss on disposal of property and equipment | 3 | 16 | (47 | ) | |||||||
Amortization of debt issuance costs | 599 | 527 | 520 | ||||||||
Deferred tax benefit | (12,661 | ) | — | — | |||||||
Changes in operating assets and liabilities: | |||||||||||
(Increase) decrease in accounts receivable | 166 | (4,080 | ) | 124 | |||||||
Increase in inventory | (1,948 | ) | — | — | |||||||
(Increase) decrease in prepaid expenses and other current assets | (557 | ) | 6,259 | (5,373 | ) | ||||||
(Increase) decrease in other assets | 33 | (32 | ) | (416 | ) | ||||||
Increase (decrease) in accounts payable and other liabilities | (7,172 | ) | 27,650 | 6,203 | |||||||
Increase (decrease) in deferred revenue | (49,779 | ) | (73,344 | ) | 171,353 | ||||||
Net cash provided by (used in) operating activities | (185,394 | ) | (175,628 | ) | 184,776 | ||||||
Cash flows from investing activities: | |||||||||||
Purchases of property and equipment | (228 | ) | (231 | ) | (910 | ) | |||||
Proceeds from disposal of property and equipment | — | — | 335 | ||||||||
Purchases of investments | (267,873 | ) | (425,673 | ) | (326,446 | ) | |||||
Maturities of investments | 318,623 | 444,156 | 210,000 | ||||||||
Net cash provided by (used in) investing activities | 50,522 | 18,252 | (117,021 | ) | |||||||
Cash flows from financing activities: | |||||||||||
Proceeds from issuance of common stock, net of fees | 7,987 | 3,624 | 114 | ||||||||
Repurchase of common stock | (1,679 | ) | (621 | ) | (357 | ) | |||||
Proceeds from debt borrowings, net of fees | 145,905 | — | — | ||||||||
Repayment of debt borrowings | (2,280 | ) | (2,016 | ) | (1,859 | ) | |||||
Other financing activities | — | — | 70 | ||||||||
Net cash provided by (used in) financing activities | 149,933 | 987 | (2,032 | ) | |||||||
Net increase (decrease) in cash and cash equivalents | 15,061 | (156,389 | ) | 65,723 | |||||||
Cash and cash equivalents at beginning of year | 46,600 | 202,989 | 137,266 | ||||||||
Cash and cash equivalents at end of year | $ | 61,661 | $ | 46,600 | $ | 202,989 | |||||
Supplemental disclosure of cash flow information: | |||||||||||
Cash paid for interest | $ | 5,870 | $ | 6,050 | $ | 6,270 | |||||
Supplemental disclosure of noncash investing and financing activities: | |||||||||||
Common stock issued in satisfaction of Symphony Icon payment obligation | $ | 10,499 | $ | — | $ | — | |||||
Unrealized gain(loss) on investments | $ | (27 | ) | $ | 24 | $ | (156 | ) | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
Lexicon Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
December 31, 2017
1. Organization and Operations
Lexicon Pharmaceuticals, Inc. (“Lexicon” or the “Company”) is a Delaware corporation incorporated on July 7, 1995. Lexicon was organized to discover the functions and pharmaceutical utility of genes and use those gene function discoveries in the discovery and development of pharmaceutical products for the treatment of human disease.
Lexicon has financed its operations from inception primarily through sales of common and preferred stock, contract and milestone payments to it under strategic collaborations and other research and development collaborations, target validation, database subscription and technology license agreements, product sales, government grants and contracts and financing under debt and lease arrangements. The Company’s future success is dependent upon many factors, including, but not limited to, its ability to successfully commercialize XERMELO (telotristat ethyl) and any other products which gain regulatory approval, develop and obtain regulatory approval for its other drug candidates, achieve milestones under its collaboration agreements, establish new collaboration and license agreements, obtain and enforce patents and other proprietary rights in its discoveries, comply with federal and state regulations, and maintain sufficient capital to fund its activities. As a result of the aforementioned factors and the related uncertainties, there can be no assurance of the Company’s future success.
2. Summary of Significant Accounting Policies
Basis of Presentation: The accompanying consolidated financial statements include the accounts of Lexicon and its wholly-owned subsidiaries. Intercompany transactions and balances are eliminated in consolidation.
Use of Estimates: The preparation of financial statements in conformity with U. S. generally accepted accounting principles (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the period. Actual results could differ from those estimates.
Cash, Cash Equivalents and Short-Term Investments: Lexicon considers all highly-liquid investments with original maturities of three months or less to be cash equivalents. As of December 31, 2017 and December 31, 2016, short-term investments consist of U.S. treasury bills and corporate debt securities. The Company’s short-term investments are classified as available-for-sale securities and are carried at fair value, based on quoted market prices of the securities. The Company views its available-for-sale securities as available for use in current operations regardless of the stated maturity date of the security. Unrealized gains and losses on such securities are reported as a separate component of stockholders’ equity. Net realized gains and losses, interest and dividends are included in interest income. The cost of securities sold is based on the specific identification method.
Accounts Receivable: Lexicon records trade accounts receivable in the normal course of business related to the sale of products or services. The allowance for doubtful accounts takes into consideration such factors as historical write-offs, the economic climate and other factors that could affect collectibility. Write-offs are evaluated on a case by case basis.
Inventory: Inventories are determined at the lower of cost or market value with cost determined under the specific identification method and may consist of raw materials, work in process and finished goods. The Company began capitalizing inventory during 2017 after the approval of XERMELO by the FDA, as the related costs were expected to be recoverable through the commercialization of the product. Costs incurred prior to approval of XERMELO were recorded as research and development expense in the consolidated statements of comprehensive loss. As a result, cost of sales for approximately the next two years will reflect a lower average per unit cost of materials. Inventory consisted of the following as of December 31, 2017 (in thousands):
Raw materials | $ | 616 | ||
Work-in-process | 149 | |||
Finished goods | 1,183 | |||
Total inventory | $ | 1,948 | ||
F-7
Concentration of Credit Risk: Lexicon’s cash equivalents, investments and accounts receivable represent potential concentrations of credit risk. The Company attempts to minimize potential concentrations of risk in cash equivalents and investments by placing investments in high-quality financial instruments. The Company’s accounts receivable are unsecured and are concentrated in pharmaceutical and biotechnology companies located in Europe and the United States. The Company has not experienced any significant credit losses to date. In 2017, customers in France and the United States represented 82% and 18% of revenue, respectively. In 2016, customers in France and the United States represented 99% and 1%, respectively. In 2015, customers in France and the United States represented 99% and 1% of revenue, respectively. At December 31, 2017, management believes that the Company has no significant concentrations of credit risk.
Segment Information and Significant Customers: Lexicon operates in one business segment, which primarily focuses on the discovery, development and commercialization of pharmaceutical products for the treatment of human disease. Substantially all of the Company’s revenues have been derived from drug discovery alliances, target validation collaborations for the development and, in some cases, analysis of the physiological effects of genes altered in knockout mice, technology licenses, subscriptions to its databases, product sales, government grants and contracts and compound library sales. In 2017, Sanofi and Ipsen Pharma SAS (“Ipsen”) represented 64% and 18% of revenues, respectively. In 2016, Sanofi and Ipsen represented 90% and 9% of revenues, respectively. In 2015, Sanofi represented 98% of revenues.
Other Intangible Assets: Other intangible assets, net consist of in-process research and development acquired in business combinations, which are reported at fair value, less accumulated amortization. Intangible assets with finite lives are amortized using the straight-line method over their estimated useful lives. During 2017, intangible assets relating to XERMELO of $24.7 million were reclassified from indefinite-lived to finite-lived assets following the approval of XERMELO by the FDA. The Company recorded $1.5 million in amortization expense related to this asset, which is recorded as cost of sales in the accompanying consolidated statements of comprehensive loss for the year ended December 31, 2017.
Estimated future amortization expense for intangible assets as of December 31, 2017 is as follows:
For the Year Ending December 31 | |||
(in thousands) | |||
2018 | $ | 1,766 | |
2019 | 1,766 | ||
2020 | 1,766 | ||
2021 | 1,766 | ||
2022 | 1,766 | ||
Thereafter | 14,417 | ||
$ | 23,247 | ||
Property and Equipment: Property and equipment that is held and used is carried at cost and depreciated using the straight-line method over the estimated useful life of the assets which ranges from three to 40 years. Maintenance, repairs and minor replacements are charged to expense as incurred. Leasehold improvements are amortized over the shorter of the estimated useful life or the remaining lease term. Significant renewals and betterments are capitalized.
Impairment of Long-Lived Assets: Long-lived assets and certain identifiable intangible assets to be held and used are reviewed for impairment when events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Recoverability of assets to be held and used is measured by comparison of the carrying amount of an asset to future net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount that the carrying amount of the assets exceeds the fair value of the assets. Assets to be disposed of are reported at the lower of the carrying amount or fair value less costs to sell. During 2015, the Company determined that its buildings were impaired and therefore recorded an impairment loss of $3.6 million, which was recorded in impairment loss on buildings in the accompanying consolidated statements of comprehensive loss. There were no impairments of long-lived assets, including finite-lived intangible assets, in 2017 or 2016.
Indefinite lived intangible assets are also tested annually for impairment and whenever indicators of impairment are present. When performing the impairment assessment, the Company first assesses qualitative factors to determine whether it is
F-8
necessary to recalculate the fair value of its intangible assets. If management believes, as a result of the qualitative assessment, that it is more likely than not that the fair value of the intangible assets is less than its carrying amount, the Company calculates the asset’s fair value. If the carrying value of the asset exceeds its fair value, then the intangible asset is written down to its fair value. There were no impairments of indefinite lived intangible assets in 2017, 2016 or 2015.
Goodwill Impairment: Goodwill is not amortized, but is tested at least annually for impairment at the reporting unit level. The Company has determined that the reporting unit is the single operating segment disclosed in its current financial statements. Impairment is the condition that exists when the carrying amount of goodwill exceeds its implied fair value. The first step in the impairment process is to determine the fair value of the reporting unit and then compare it to the carrying value, including goodwill. If the fair value exceeds the carrying value, no further action is required and no impairment loss is recognized. Additional impairment assessments may be performed on an interim basis if the Company encounters events or changes in circumstances that would indicate that, more likely than not, the carrying value of goodwill has been impaired. There was no impairment of goodwill in 2017, 2016 or 2015.
Revenue Recognition: Revenues are recognized when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed or determinable and collectibility is reasonably assured.
Product Revenues
Product revenues consist of commercial sales of XERMELO in the United States and sales of bulk tablets of XERMELO to Ipsen. Product revenues are recognized once the Company meets all four revenue recognition criteria described above. In March 2017, Lexicon began shipping XERMELO to its customers in the United States. The Company recognizes revenue for product sales of XERMELO at the time the product is received by its specialty pharmacy customers, net of allowances for customer credits, including estimated rebates, chargebacks, discounts, returns, distribution service fees, and government rebates, such as Medicare Part D coverage gap reimbursements in the United States. Product shipping and handling costs are included in cost of sales.
Customer Credits: The Company’s specialty pharmacy customers are offered various forms of consideration, including allowances, service fees and prompt payment discounts. The Company expects the specialty pharmacies will earn prompt payment discounts and, therefore, the Company deducts the full amount of these discounts from total product sales when revenues are recognized. Service fees are also deducted from product sales as they are earned.
Rebates: Allowances for rebates include mandated discounts under the Medicaid Drug Rebate Program. Rebate amounts are based upon contractual agreements or legal requirements with public sector (e.g. Medicaid) benefit providers. Rebates are amounts owed after the final dispensing of the product to a benefit plan participant and are based upon contractual agreements or legal requirements with public sector benefit providers. The allowance for rebates is based on statutory discount rates and expected utilization. The Company’s estimates for expected utilization of rebates are based in part on third party market research data, and data received from the specialty pharmacies. Rebates are generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarter’s unpaid rebates. If actual future rebates vary from estimates, the Company may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Chargebacks: Chargebacks are discounts that occur when contracted customers purchase directly from a specialty pharmacy. Contracted customers, which currently consist primarily of Public Health Service institutions, non-profit clinics, and Federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The specialty pharmacy, in turn, charges back to Lexicon the difference between the price initially paid by the specialty pharmacy and the discounted price paid to the specialty pharmacy by the customer. The allowance for chargebacks is based on known sales to contracted customers.
Medicare Part D Coverage Gap: Medicare Part D prescription drug benefit mandates manufacturers to fund 50% of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. The Company’s estimates for the expected Medicare Part D coverage gap are based on data received from the specialty pharmacies. Funding of the coverage gap is generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters. If actual future funding varies from estimates, the Company may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Co-payment assistance: Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. The Company accrues a liability for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators.
F-9
Collaborative Agreements
Revenues under collaborative agreements include both license revenue and contract research revenue. Activities under collaborative agreements are evaluated to determine if they represent a multiple element revenue agreement. The Company identifies the deliverables included within the agreement and evaluates which deliverables represent separate units of accounting. The Company accounts for those components as separate units of accounting if the following two criteria are met:
• | The delivered item or items have value to the customer on a stand-alone basis; and |
• | If there is a general right of return relative to the delivered items, delivery or performance of the undelivered items is considered probable and within the Company’s control. |
Factors considered in this determination include, among other things, whether any other vendors sell the items separately and if the licensee could use the delivered item for its intended purpose without the receipt of the remaining deliverables. If multiple deliverables included in an arrangement are separable into different units of accounting, the Company allocates the arrangement consideration to those units of accounting. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. Arrangement consideration is allocated at the inception of the arrangement to the identified units of accounting based on their relative estimated selling price. Revenue is recognized for each unit of accounting when the appropriate revenue recognition criteria are met.
Future milestone payments that are contingent upon the achievement of a substantive milestone are recognized in their entirety in the period in which the milestone is achieved. A milestone is substantive if:
• | The consideration payable to the Company is commensurate with the Company’s performance necessary to achieve the milestone or the increase in value to the collaboration resulting from the Company’s performance; |
• | The milestone relates solely to the Company’s past performance; and |
• | The milestone is reasonable relative to all of the other deliverables and payments within the arrangement. |
Commercial milestones will be accounted for as royalties and recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria are met. Subscription and license fees are recognized as other revenue upon the grant of the technology license when performance is complete and there is no continuing involvement. Royalty revenues are recognized as earned in accordance with the contract terms at the time the royalty amount is fixed and determinable based on information received from the sublicensees and at the time collectibility is reasonably assured.
Cost of Sales: Cost of sales consists of third-party manufacturing costs, freight and indirect overhead costs associated with sales of XERMELO. The Company began capitalizing inventory during 2017 following approval of XERMELO by the FDA, as the related costs were expected to be recoverable through the commercialization of the product. Costs incurred prior to approval of XERMELO have been recorded as research and development expense in the consolidated statements of comprehensive loss. As a result, cost of sales for approximately the next two years will reflect a lower average per unit cost of materials. Product shipping and handling costs are included in cost of sales. Cost of sales also includes the amortization of the in-process research and development intangible asset for XERMELO using the straight-line method over the estimated useful life of 14 years.
Research and Development Expenses: Research and development expenses consist of costs incurred for company-sponsored as well as collaborative research and development activities. These costs include direct and research-related overhead expenses and are expensed as incurred. Technology license fees for technologies that are utilized in research and development and have no alternative future use are expensed when incurred. Substantial portions of the Company’s preclinical and clinical trials are performed by third-party laboratories, medical centers, contract research organizations and other vendors. For preclinical studies, the Company accrues expenses based upon estimated percentage of work completed and the contract milestones remaining. For clinical studies, expenses are accrued based upon the number of patients enrolled and the duration of the study. The Company monitors patient enrollment, the progress of clinical studies and related activities to the extent possible through internal reviews of data reported to the Company by the vendors and clinical site visits. The Company’s estimates depend on the timeliness and accuracy of the data provided by the vendors regarding the status of each program and total program spending. The Company periodically evaluates the estimates to determine if adjustments are necessary or appropriate based on information it receives.
Stock-Based Compensation: The Company recognizes compensation expense in its statements of comprehensive loss for share-based payments, including stock options and restricted stock units issued to employees, based on their fair values on the date of the grant, with the compensation expense recognized over the period in which an employee is required to provide
F-10
service in exchange for the stock award. Stock-based compensation expense for awards without performance conditions is recognized on a straight-line basis. Stock-based compensation expense for awards with performance conditions is recognized over the period from the date the performance condition is determined to be probable of occurring through the time the applicable condition is met. As of December 31, 2017, stock-based compensation cost for all outstanding unvested options and restricted stock units was $18.6 million, which is expected to be recognized over a weighted-average period of 1.3 years.
The fair value of stock options is estimated at the date of grant using the Black-Scholes method. The Black-Scholes option-pricing model requires the input of subjective assumptions. Because the Company’s employee stock options have characteristics significantly different from those of traded options, and because changes in the subjective input assumptions can materially affect the fair value estimate, in management’s opinion, the existing models do not necessarily provide a reliable single measure of the fair value of its employee stock options. For purposes of determining the fair value of stock options, the Company segregates its options into two homogeneous groups, based on exercise and post-vesting employment termination behaviors, resulting in a change in the assumptions used for expected option lives and forfeitures. Expected volatility is based on the historical volatility in the Company’s stock price. The following weighted-average assumptions were used for options granted in the years ended December 31, 2017, 2016 and 2015, respectively:
Expected Volatility | Risk-free Interest Rate | Expected Term | Dividend Rate | |||||
December 31, 2017: | ||||||||
Employees | 61% | 1.7% | 4 | 0 | % | |||
Officers and non-employee directors | 70% | 2.2% | 8 | 0 | % | |||
December 31, 2016: | ||||||||
Employees | 63% | 1.1% | 4 | 0 | % | |||
Officers and non-employee directors | 83% | 1.6% | 8 | 0 | % | |||
December 31, 2015: | ||||||||
Employees | 64% | 1.2% | 4 | 0 | % | |||
Officers and non-employee directors | 81% | 1.8% | 8 | 0 | % | |||
Net Loss per Common Share: Net loss per common share is computed using the weighted average number of shares of common stock outstanding. Shares associated with convertible debt, stock options and restricted stock units are not included because they are antidilutive.
3. Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09, “Revenue from Contracts with Customers”, which amends FASB ASC Topic 606. ASU 2014-09 provides a single, comprehensive revenue recognition model for all contracts with customers. This standard contains principles for the determination of the measurement of revenue and the timing of when such revenue is recognized. Revenue recognition will reflect the transfer of goods or services to customers at an amount that is expected to be earned in exchange for those goods or services. In August 2015, the FASB issued ASU No. 2015-14, “Revenue from Contracts with Customers: Deferral of Effective Date”, which defers the effective date of ASU 2014-09 by one year. ASU 2014-09 is now effective for annual periods after December 15, 2017 including interim periods within that reporting period. Early application is permitted only for annual periods beginning after December 15, 2016, including interim periods within that reporting period. In 2016, the FASB issued four additional ASUs related to Topic 606: ASU Nos. 2016-08, 2016-10, 2016-12 and 2016-20. These ASUs clarify various aspects of the new revenue guidance, including principal versus agent considerations, identifying performance obligations and licensing, and they include other improvements and practical expedients. Two adoption methods are permitted; retrospectively to all prior reporting periods presented, with certain practical expedients; or the modified retrospective method with the cumulative effect of initially adopting the ASU recognized at the date of initial application. The Company has elected to adopt this new standard effective January 1, 2018, using the modified retrospective transition method.
To date, the Company has assessed that ASU 2014-09 will not have a material impact on revenue recognition from product revenue. The Company’s only source of product revenue has been sales of XERMELO, which the Company received FDA approval for in February 2017, and subsequently, entered into a limited number of arrangements with specialty pharmacies (“SPs”) in the U.S. (collectively, the “customers”), under which the Company began shipping to its customers in March 2017. Under current GAAP, the Company recognizes revenue on its product sales when the customer obtains control of
F-11
the product, which occurs upon delivery. Product revenue is recorded net of applicable reserves for variable consideration, including discounts and allowances. These estimates are based on the most likely amount method for relevant factors such as current contractual and statutory requirements, industry data and forecasted customer buying and payment patterns. The Company’s net product revenues reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the respective underlying contracts. Under ASU 2014-09, the Company expects to be able to utilize a process and controls approach consistent with its historical process and based on the nature of its current contracts with customers, does not anticipate a significant amount of variable consideration subject to constraint. As a result, the Company does not believe that the adoption of this ASU will have a material impact on the timing or amount of revenues recognized related to its contracts with customers for the sale of product.
The Company expects the accounting for contingent milestone payments to be the most significant change in the accounting for its license and collaboration agreements. Topic 605 provides guidance specific to the accounting for milestone payments, including the ability to defer the recognition of any milestones until received and, if certain criteria are met, the ability to recognize milestone payments as revenue when received. Under the Company’s current accounting policy, Lexicon recognizes contingent or milestone payments as revenue in the period that the payment-triggering event occurs or is achieved. However, under the new revenue standard, it is possible to recognize contingent or milestone payments before the payment-triggering event is completely achieved, subject to management’s assessment of the probability of achievement of the milestone and the likelihood of a significant reversal of such milestone revenue at each reporting date. This assessment may result in recognizing milestone revenue before the milestone event has been achieved. The Company expects to evaluate estimates and timing of milestone achievement and related variable consideration based on the most likely amount method in its application of this ASU to collaborative agreements. Estimating variable consideration and the related constraint will require the use of significant management judgment.
To date, the Company’s primary sources of collaboration revenue have been license and collaboration agreements with three separate third-party licensees: Texas Institute for Genomic Medicine (“TIGM”), Sanofi and Ipsen. The Company has performed an evaluation of the expected effect of adoption in its accounting for license and collaboration agreements as discussed further below.
With respect to its contract with TIGM, the Company evaluated the variable consideration related to the remaining milestone in the adoption of this ASU and determined based on the most likely amount method that it was not probable that a significant reversal would occur and therefore, no constraint was required. As a result, under the modified retrospective method, the Company will record a $14.2 million cumulative-effect adjustment to its accumulated deficit on the date of adoption.
With respect to its collaboration agreements with Sanofi and Ipsen, the Company evaluated the variable consideration relating to future milestone payments and determined, based on the most likely amount method, that the estimated amounts could be considered as part of the transaction price. The Company then evaluated the variable constraint and determined that the variable consideration amounts are constrained, primarily by future events that are not within the control of the Company. The future events primarily related to receipt of positive results from studies, approval from regulatory agencies, and upon achieving sales in certain locations. As a result, the Company has determined that there is no cumulative adjustment necessary for these agreements on the date of adoption.
The adoption of the ASU will have no significant impact to the provision for income taxes and will have no impact to the net cash provided by or used in operating, investing or financing activities on the Company’s consolidated statements of cash flows. Estimated impacts from adoption of this ASU may differ upon the final adoption and implementation in the first quarter of 2018. As the Company completes its analysis of the accounting for the collaboration agreements under the new revenue standard, management is assessing the required changes to its accounting policies, systems and internal control over financial reporting.
In January 2016, the FASB issued ASU No. 2016-01, “Recognition and Measurement of Financial Assets and Financial Liabilities.” ASU 2016-01 requires that most equity investments be measured at fair value, with subsequent changes in fair value recognized in net income. The pronouncement also impacts financial liabilities under the fair value option and the presentation and disclosure requirements for financial instruments. This pronouncement is effective for fiscal years, and interim periods within those years, beginning after December 15, 2017, and early adoption is not permitted. The adoption of this ASU on January 1, 2018 is not expected to have a material impact on Lexicon’s consolidated financial statements.
In February 2016, the FASB issued ASU No. 2016-02, “Leases.” ASU 2016-02 requires companies that lease assets to recognize a right-of-use asset and a lease liability, initially measured at the present value of the lease payments, in its balance sheet. The pronouncement will also require additional disclosures about the amount, timing and uncertainty of cash flows
F-12
arising from leases. This pronouncement is effective for fiscal years, and interim periods within those years, beginning after December 15, 2018, and early adoption is permitted. This ASU is required to be adopted using a modified retrospective approach. Management plans to adopt ASU 2016-02 on January 1, 2019, and anticipates that most of its operating leases will result in the recognition of additional assets and corresponding liabilities on the consolidated balance sheet. The Company does not expect that the implementation of the ASU will have a material impact on its financial position. The actual impact will depend on the Company’s lease portfolio at the time of adoption. The Company continues to assess all implications of the standard and related financial disclosures.
In March 2016, the FASB issued ASU No. 2016-09, “Stock Compensation,” which is intended to simplify several aspects of the accounting for share-based payment award transactions. The Company adopted this pronouncement effective January 1, 2017. Upon adoption, the Company recognized approximately $6.1 million of accumulated excess tax benefits as deferred tax assets that under the previous guidance could not be recognized until the benefits were realized through a reduction in cash taxes paid. This part of the guidance is applied using a modified retrospective method with a cumulative-effect adjustment to the accumulated deficit for the excess tax benefits not previously recognized. However, given the full valuation allowance placed on the additional $6.1 million of deferred tax assets, the recognition of this provision of ASU 2016-09 had no impact to the Company’s accumulated deficit as of January 1, 2017. Additionally, the Company recorded an adjustment to accumulated deficit of $2.0 million as a result of making an entity-wide accounting policy election to account for forfeitures of share-based payment awards as they occur instead of estimating the number of awards that are expected to vest.
4. Cash and Cash Equivalents and Investments
The fair value of cash and cash equivalents and investments held at December 31, 2017 and 2016 are as follows:
As of December 31, 2017 | |||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | ||||||||||||
(in thousands) | |||||||||||||||
Cash and cash equivalents | $ | 61,661 | $ | — | $ | — | $ | 61,661 | |||||||
Securities maturing within one year: | |||||||||||||||
U.S. treasury securities | 222,316 | — | (168 | ) | 222,148 | ||||||||||
Corporate debt securities | 27,033 | — | (54 | ) | 26,979 | ||||||||||
Total short-term investments | $ | 249,349 | $ | — | $ | (222 | ) | $ | 249,127 | ||||||
Total cash and cash equivalents and investments | $ | 311,010 | $ | — | $ | (222 | ) | $ | 310,788 | ||||||
As of December 31, 2016 | |||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | ||||||||||||
(in thousands) | |||||||||||||||
Cash and cash equivalents | $ | 46,600 | $ | — | $ | — | $ | 46,600 | |||||||
Securities maturing within one year: | |||||||||||||||
U.S. treasury securities | 227,911 | 1 | (107 | ) | 227,805 | ||||||||||
Corporate debt securities | 72,188 | 1 | (90 | ) | 72,099 | ||||||||||
Total short-term investments | $ | 300,099 | $ | 2 | $ | (197 | ) | $ | 299,904 | ||||||
Total cash and cash equivalents and investments | $ | 346,699 | $ | 2 | $ | (197 | ) | $ | 346,504 | ||||||
There were $7,000 in realized losses for the year ended December 31, 2017. There were no realized gains or losses for the years ended December 31, 2016 and 2015.
5. Fair Value Measurements
The Company uses various inputs in determining the fair value of its investments and measures these assets on a recurring basis. Assets and liabilities recorded at fair value in the consolidated balance sheets are categorized by the level of
F-13
objectivity associated with the inputs used to measure their fair value. The following levels are directly related to the amount of subjectivity associated with the inputs to fair valuation of these assets and liabilities:
• | Level 1 – quoted prices in active markets for identical assets, which include U.S. treasury securities |
• | Level 2 – other significant observable inputs (including quoted prices for similar investments, market corroborated inputs, etc.), which include corporate debt securities |
• | Level 3 – significant unobservable inputs (including the Company’s own assumptions in determining the fair value of the Symphony Icon purchase consideration liability) |
The inputs or methodology used for valuing securities are not necessarily an indication of the credit risk associated with investing in those securities. The following tables provide the fair value measurements of applicable Company assets and liabilities that are measured at fair value on a recurring basis according to the fair value levels defined above as of December 31, 2017 and 2016.
Assets and Liabilities at Fair Value | |||||||||||||||
As of December 31, 2017 | |||||||||||||||
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
(in thousands) | |||||||||||||||
Assets | |||||||||||||||
Cash and cash equivalents | $ | 61,661 | $ | — | $ | — | $ | 61,661 | |||||||
Short-term investments | 222,148 | 26,979 | — | 249,127 | |||||||||||
Total cash and cash equivalents and investments | $ | 283,809 | $ | 26,979 | $ | — | $ | 310,788 | |||||||
Assets and Liabilities at Fair Value | |||||||||||||||
As of December 31, 2016 | |||||||||||||||
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
(in thousands) | |||||||||||||||
Assets | |||||||||||||||
Cash and cash equivalents | $ | 45,093 | $ | 1,507 | $ | — | $ | 46,600 | |||||||
Short-term investments | 227,805 | 72,099 | — | 299,904 | |||||||||||
Total cash and cash equivalents and investments | $ | 272,898 | $ | 73,606 | $ | — | $ | 346,504 | |||||||
Liabilities | |||||||||||||||
Accrued liabilities | $ | — | $ | — | $ | 18,912 | $ | 18,912 | |||||||
Total liabilities | $ | — | $ | — | $ | 18,912 | $ | 18,912 | |||||||
The Company did not have any Level 3 assets during the years ended December 31, 2017, 2016 and 2015. Transfers between levels are recognized at the actual date of circumstance that caused the transfer. In 2016, the Company’s Level 3 liabilities represented the contingent purchase consideration payable to Symphony Icon, and was estimated using a probability-based income approach utilizing an appropriate discount rate. Subsequent changes in the fair value of the Symphony Icon (“Symphony Icon”) purchase consideration liability are recorded as an increase or decrease in Symphony Icon purchase liability in the accompanying consolidated statements of comprehensive loss. The fair value of the Symphony Icon purchase consideration liability increased by $2.1 million during the year ended December 31, 2017, decreased by $0.7 million during the year ended December 31, 2016, and increased by $5.9 million during the year ended December 31, 2015. The following table summarizes the change in consolidated balance sheet carrying value associated with Level 3 liabilities for the years ended December 31, 2015, 2016 and 2017.
F-14
Other Long-term Liabilities | |||
(in thousands) | |||
Balance at January 1, 2015 | $ | 17,638 | |
Change in valuation of purchase consideration payable to former Symphony Icon stockholders | 5,927 | ||
Payment of base payment obligation with common stock and cash | (750 | ) | |
Balance at December 31, 2015 | 22,815 | ||
Change in valuation of purchase consideration payable to former Symphony Icon stockholders | (703 | ) | |
Payment of contingent payment obligation with cash | (3,200 | ) | |
Balance at December 31, 2016 | 18,912 | ||
Change in valuation of purchase consideration payable to former Symphony Icon stockholders | 2,101 | ||
Payment of contingent payment obligation with common stock and cash | (21,013 | ) | |
Balance at December 31, 2017 | $ | — | |
The Company also has assets that under certain conditions are subject to measurement at fair value on a non-recurring basis. These assets include goodwill associated with the acquisitions of Coelacanth Corporation in 2001 and Symphony Icon in 2010 and intangible assets associated with the acquisition of Symphony Icon in 2010. For these assets, measurement at fair value in periods subsequent to their initial recognition is applicable if one or more is determined to be impaired.
6. Property and Equipment
Property and equipment at December 31, 2017 and 2016 are as follows:
Estimated Useful Lives | As of December 31, | |||||||||||
In Years | 2017 | 2016 | ||||||||||
(in thousands) | ||||||||||||
Computers and software | 3-5 | $ | 4,605 | $ | 7,667 | |||||||
Furniture and fixtures | 5-7 | 6,006 | 6,003 | |||||||||
Laboratory equipment | 3-7 | 3,423 | 3,423 | |||||||||
Leasehold improvements | 7-10 | 400 | 296 | |||||||||
Buildings | 15-40 | 59,212 | 59,212 | |||||||||
Land | — | 2,664 | 2,664 | |||||||||
Total property and equipment | 76,310 | 79,265 | ||||||||||
Less: Accumulated depreciation and amortization | (58,623 | ) | (59,875 | ) | ||||||||
Net property and equipment | $ | 17,687 | $ | 19,390 | ||||||||
7. Income Taxes
The Tax Cuts and Jobs Act (the “2017 Tax Act”) was enacted on December 22, 2017. The 2017 Tax Act significantly changes U.S. corporate income tax laws, including reducing the U.S. corporate income tax rate from 35 percent to 21 percent beginning in 2018. At December 31, 2017, Lexicon has not completed the accounting for the tax effects of the 2017 Tax Act; however, an estimate of the effects on the existing deferred tax balances has been made, as further discussed below.
Lexicon recognizes deferred tax liabilities and assets for the expected future tax consequences of events that have been recognized differently in the financial statements and tax returns. Under this method, deferred tax liabilities and assets are determined based on the difference between the financial statement carrying amounts and tax bases of liabilities and assets using enacted tax rates and laws in effect in the years in which the differences are expected to reverse. Accordingly, Lexicon remeasured certain deferred tax assets and liabilities based on the newly enacted U.S. corporate income tax rate, which resulted in a decrease of $171.4 million. Lexicon will continue to make and refine calculations and estimates, which could potentially affect the measurement of the deferred tax balances or give rise to new deferred tax amounts. Where the Company has not yet been able to make reasonable estimates of the impact of certain elements, the Company has not recorded any amounts related to
F-15
those elements and has continued accounting for them in accordance with ASC 740 on the basis of the tax laws in effect immediately prior to the enactment of the 2017 Tax Act. Deferred tax assets are evaluated for realization based on a more-likely-than-not criteria in determining if a valuation allowance should be provided.
The components of Lexicon’s deferred tax assets (liabilities) at December 31, 2017 and 2016 are as follows:
As of December 31, | |||||||
2017 | 2016 | ||||||
(in thousands) | |||||||
Deferred tax assets: | |||||||
Net operating loss carryforwards | $ | 186,967 | $ | 258,405 | |||
Research and development tax credits | 46,682 | 44,111 | |||||
Orphan drug credits | 26,524 | 24,233 | |||||
Capitalized research and development | 69,561 | 86,845 | |||||
Stock-based compensation | 3,923 | 7,060 | |||||
Deferred revenue | 12,950 | 39,307 | |||||
Other | 5,579 | 8,432 | |||||
Total deferred tax assets | 352,186 | 468,393 | |||||
Deferred tax liabilities: | |||||||
Deferred tax liability related to acquisition of Symphony Icon | (10,896 | ) | (18,675 | ) | |||
Other | (1 | ) | — | ||||
Total deferred tax liabilities | (10,897 | ) | (18,675 | ) | |||
Less: valuation allowance | (347,303 | ) | (468,393 | ) | |||
Net deferred tax liabilities | $ | (6,014 | ) | $ | (18,675 | ) | |
The $10.9 million deferred tax liability relates to the tax impact of future amortization or possible impairments associated with intangible assets acquired with Symphony Icon, which are not deductible for tax purposes. During 2017, after XERMELO was approved by the FDA, the intangible asset related to XERMELO became finite-lived and as a result $8.7 million of the related deferred tax liability could be considered as a source of taxable income. Lexicon does not believe it can estimate the reversal of the temporary difference related to the remaining assets acquired with sufficient certainty such that $6.0 million of the deferred tax liability is not considered as a source of taxable income in assessing the Company’s need for a valuation allowance in accordance with ASC 740 on the basis of the tax laws in effect immediately prior to the enactment of the 2017 Tax Act.
At December 31, 2017, Lexicon had both federal and state NOL carryforwards of approximately $851.4 million and $382.0 million, respectively. The federal and state NOL carryforwards will begin to expire in 2018. The Company’s R&D tax credit carryforwards of approximately $46.7 million began to expire in 2018. The orphan drug credit relates to a credit that is calculated as a percentage of expenditures for development of XERMELO, which has received Orphan Drug designation from the FDA. Utilization of the NOL, R&D credit and orphan drug credit carryforwards may be subject to a significant annual limitation due to ownership changes that have occurred previously or could occur in the future provided by Section 382 of the Internal Revenue Code. Based on the federal tax law limits and the Company’s cumulative loss position, Lexicon concluded it was appropriate to establish a full valuation allowance for its net deferred tax assets, excluding the deferred tax liability relating to the XERMELO finite-lived asset, until an appropriate level of profitability is sustained. During the year ended December 31, 2017, the valuation allowance decreased $121.1 million, primarily due to the effect of the new U.S. federal corporate tax rate. Lexicon recorded income tax benefits of $12.7 million in the year ended December 31, 2017. Of the $12.7 million tax benefits, $8.7 million is the release of a valuation allowance as a result of the ability to estimate the reversal of the deferred tax liability related to the intangible associated with XERMELO, as discussed above. The remaining $4.0 million was recorded to remeasure the deferred tax liability associated with the remaining indefinite-lived intangible asset associated with Symphony Icon at the newly enacted U.S. corporate income tax rate. There were no income tax benefits in the years ended December 31, 2016 and 2015, respectively. As of December 31, 2017 and 2016, the Company did not have any unrecognized tax benefits.
The Company is primarily subject to U.S. federal and New Jersey and Texas state income taxes. The tax years 1995 to current remain open to examination by U.S. federal authorities and 2004 to current remain open to examination by state
F-16
authorities. The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. As of December 31, 2017 and 2016, the Company had no accruals for interest or penalties related to income tax matters.
8. Goodwill
On July 12, 2001, Lexicon completed the acquisition of Coelacanth Corporation in a merger. Coelacanth, now Lexicon Pharmaceuticals (New Jersey), Inc., formed the core of the Company’s division responsible for small molecule compound discovery. The results of Lexicon Pharmaceuticals (New Jersey), Inc. are included in the Company’s results of operations for the period subsequent to the acquisition. Goodwill associated with the acquisition of $25.8 million, which represents the excess of the $36.0 million purchase price over the fair value of the underlying net identifiable assets, was assigned to the consolidated entity, Lexicon.
On July 30, 2010, Lexicon exercised its Purchase Option (as defined in Note 10) and completed the acquisition of Symphony Icon, Inc. Goodwill associated with the acquisition of $18.7 million, which represents the assets recognized in connection with the deferred tax liability acquired and did not result from excess purchase price, was assigned to the consolidated entity, Lexicon.
Goodwill is not subject to amortization, but is tested at least annually for impairment at the reporting unit level, which is the Company’s single operating segment. The Company performed an impairment test of goodwill on its annual impairment assessment date. This test did not result in an impairment of goodwill.
9. Debt Obligations
Convertible Notes. In November 2014, Lexicon completed an offering of $87.5 million in aggregate principal amount of its 5.25% Convertible Senior Notes due 2021 (the “Convertible Notes”). The conversion feature did not meet the criteria for bifurcation as required by generally accepted accounting principles and the entire principal amount was recorded as long-term debt on the Company’s consolidated balance sheets.
The Convertible Notes are governed by an indenture (the “Indenture”), dated as of November 26, 2014, between the Company and Wells Fargo Bank, N.A., as trustee. The Convertible Notes bear interest at a rate of 5.25% per year, payable semiannually in arrears on June 1 and December 1 of each year, beginning on June 1, 2015. The Convertible Notes mature on December 1, 2021. The Company may not redeem the Convertible Notes prior to the maturity date, and no sinking fund is provided for the Convertible Notes.
Holders of the Convertible Notes may convert their Convertible Notes at their option at any time prior to the close of business on the business day immediately preceding the maturity date. Upon conversion, the Company will deliver for each $1,000 principal amount of converted Convertible Notes a number of shares of its common stock equal to the conversion rate, as described in the Indenture. The conversion rate is initially 118.4553 shares of common stock per $1,000 principal amount of Convertible Notes (equivalent to an initial conversion price of $8.442 per share of common stock). The conversion rate is subject to adjustment in some events but will not be adjusted for any accrued and unpaid interest. In addition, following certain corporate events that occur prior to the maturity date, the Company will increase the conversion rate for a holder who elects to convert its Convertible Notes in connection with such a corporate event in certain circumstances.
If the Company undergoes a fundamental change, holders may require the Company to repurchase for cash all or any portion of their Convertible Notes at a fundamental change repurchase price equal to 100% of the principal amount of the Convertible Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date.
In connection with the issuance of the Convertible Notes, the Company incurred $3.4 million of debt issuance costs, which offsets long-term debt on the consolidated balance sheets. The debt issuance costs are amortized as interest expense over the expected life of the Convertible Notes using the effective interest method. The Company determined the expected life of the debt was equal to the seven-year term of the Convertible Notes. As of December 31, 2017, the balance of unamortized debt issuance costs was $1.9 million, which offsets long-term debt on the consolidated balance sheets.
The fair value of the Convertible Notes was $127.3 million as of December 31, 2017 and was determined using Level 2 inputs based on the indicative pricing published by certain investment banks or trading levels of the Convertible Notes, which are not listed on any securities exchange or quoted on an inter-dealer automated quotation system.
Mortgage Loan. In April 2004, Lexicon purchased its existing laboratory and office buildings and animal facilities in The Woodlands, Texas with proceeds from a $34.0 million third-party mortgage financing and $20.8 million in cash. The mortgage loan originally had a ten-year term with a 20-year amortization and a fixed interest rate of 8.23%. The mortgage was amended in September 2013 to extend the maturity date from April 2014 to April 2017, with the mortgage loan’s monthly
F-17
payment amount and fixed interest rate each remaining unchanged. In April 2017, the mortgage was amended to extend the maturity date to April 2018, with the mortgage loan’s monthly payment amount and fixed interest rate each remaining unchanged. The mortgage had a principal balance of $14.1 million as of December 31, 2017. This entire balance is recorded as current portion of long-term debt in the accompanying consolidated balance sheet as of December 31, 2017 as there is a balloon payment due in April 2018. Lexicon intends to refinance this debt prior to paying the balloon payment. The buildings and land that serve as collateral for the mortgage loan are included in property and equipment at $59.2 million and $2.7 million, respectively, before accumulated depreciation, as of December 31, 2017. The fair value of Lexicon’s mortgage loan approximates its carrying value. The fair value of Lexicon’s mortgage loan was determined using Level 2 inputs using discounted cash flow analysis, based on the Company’s estimated current incremental borrowing rate.
BioPharma Term Loan. In December 2017, Lexicon entered into a loan agreement with BioPharma Credit PLC and BioPharma Credit Investments IV Sub LP that provides up to $200 million borrowing capacity (the “BioPharma Term Loan”) available in two tranches, each maturing in December 2022. The BioPharma Term Loan bears interest at 9% per year, subject to additional interest if an event of default occurs and is continuing, and is payable quarterly. The first $150 million tranche was funded in December 2017. The second $50 million tranche is available for draw by March 2019 at Lexicon’s option if net XERMELO sales are greater than $25 million in the preceding quarter.
The BioPharma Term Loan is subject to mandatory prepayment provisions that require prepayment upon a change of control or receipt of proceeds from certain non-ordinary course transfers of assets. The Company may prepay the BioPharma Term Loan in whole at its option at any time. Any prepayment of the BioPharma Term Loan is subject to customary make-whole premiums and prepayment premiums.
The Company’s obligations under the BioPharma Term Loan are secured by a first lien security interest in substantially all of the assets of the Company and certain of its subsidiaries. The loan agreement contains certain customary representations and warranties, affirmative and negative covenants and events of default applicable to the Company and certain of its subsidiaries, including among other things, covenants restricting dispositions, fundamental changes in our business, mergers or acquisitions, indebtedness, encumbrances, distributions, investments, transactions with affiliates and subordinated debt. If an event of default occurs and is continuing, all amounts outstanding under the BioPharma Term Loan may be declared immediately due and payable.
In connection with the BioPharma Term Loan, the Company incurred $4.1 million of debt issuance costs, which offsets long-term debt on the consolidated balance sheets. The debt issuance costs are amortized as interest expense over the expected life of the BioPharma Term Loan using the effective interest method. The Company determined the expected life of the debt was equal to the five-year term of the BioPharma Term Loan. The fair value of the BioPharma Term Loan approximates its carrying value. The fair value of the BioPharma Term Loan was determined using Level 2 inputs using discounted cash flow analysis, based on the Company’s estimated current incremental borrowing rate.
The following table includes the aggregate scheduled future principal payments of the Company’s long-term debt as of December 31, 2017:
For the Year Ending December 31 | |||
(in thousands) | |||
2018 | $ | 14,094 | |
2019 | — | ||
2020 | — | ||
2021 | 87,500 | ||
2022 | 150,000 | ||
Thereafter | — | ||
Total debt | 251,594 | ||
Less deferred financing costs | (5,924 | ) | |
Less current portion | (14,094 | ) | |
Total long-term debt | $ | 231,576 | |
F-18
10. Arrangements with Symphony Icon, Inc.
On June 15, 2007, Lexicon entered into a series of related agreements providing for the financing of the clinical development of certain of its drug candidates, including XERMELO, along with any other pharmaceutical compositions modulating the same targets as those drug candidates (the “Programs”). The agreements included a Novated and Restated Technology License Agreement pursuant to which the Company licensed to Symphony Icon, a then wholly-owned subsidiary of Symphony Icon Holdings LLC (“Holdings”), the Company’s intellectual property rights related to the Programs. Holdings contributed $45 million to Symphony Icon in order to fund the clinical development of the Programs.
Under a Share Purchase Agreement, dated June 15, 2007, between the Company and Holdings, the Company issued and sold to Holdings 1,092,946 shares of its common stock on June 15, 2007 in exchange for $15 million and an exclusive purchase option (the “Purchase Option”) that gave the Company the right to acquire all of the equity of Symphony Icon, thereby allowing the Company to reacquire all of the Programs. On July 30, 2010, Lexicon entered into an Amended and Restated Purchase Option Agreement with Symphony Icon and Holdings and simultaneously exercised the Purchase Option, thereby reacquiring the Programs. Pursuant to the amended terms of the Purchase Option, Lexicon paid Holdings $10 million on July 30, 2010 and issued 1,891,074 shares of common stock to designees of Holdings on July 30, 2012 in satisfaction of an additional $35 million base payment obligation.
Lexicon also agreed to make up to $45 million in additional contingent payments, which would consist of 50% of any consideration Lexicon received pursuant to any licensing transaction (a “Licensing Transaction”) under which Lexicon grants a third party rights to commercialize XERMELO or other pharmaceutical compositions modulating the same target as XERMELO (the “LG103 Programs”), subject to certain exceptions. The contingent payments would be due if and when Lexicon received such consideration from a Licensing Transaction. In the event Lexicon received regulatory approval in the United States for the marketing and sale of any product resulting from the LG103 Programs prior to entering into a Licensing Transaction for the commercialization of such product in the United States, in lieu of any contingent payment from such a Licensing Transaction, Lexicon would pay Holdings the sum of $15 million and the amount of certain expenses Lexicon incurred after its exercise of the Purchase Option which were attributable to the development of such product, reduced by up to 50% of such sum on account of any contingent payments paid prior to such United States regulatory approval attributable to any such Licensing Transaction outside of the United States with respect to such product. In the event Lexicon made any such payment upon United States regulatory approval, Lexicon would have no obligation to make subsequent contingent payments attributable to any such Licensing Transactions for the commercialization of such product outside the United States until the proceeds of such Licensing Transactions exceed 50% of the payment made as a result of such United States regulatory approval. The contingent payments were payable in cash or a combination of cash and common stock, in Lexicon’s discretion, provided that no more than 50% of any contingent payment would be paid in common stock. In December 2014, Lexicon paid $5.8 million in cash and issued 666,111 shares of common stock to designees of Holdings in satisfaction of a $11.5 million contingent payment obligation as a result of receiving an upfront payment pursuant to Lexicon’s license and collaboration agreement with Ipsen. In April 2015, Lexicon paid $0.75 million in cash to Holdings in satisfaction of its contingent payment obligation as a result of receiving an additional upfront payment from Ipsen in March 2015. In September 2016, Lexicon paid $3.2 million in cash to Holdings in satisfaction of its contingent payment obligation as a result of receiving a milestone payment from Ipsen in August 2016 (see Note 15, Collaboration and License Agreements).
In September 2016, Lexicon entered into an amendment (the “Amendment”) to the Purchase Option Agreement with Holdings and Symphony Icon pursuant to which Lexicon agreed to pay Holdings $21.0 million upon Lexicon’s receipt of regulatory approval in the United States for the marketing and sale of XERMELO, such buyout amount to be in lieu of any remaining payments which may be or become payable to Holdings under the Purchase Option Agreement. In March 2017, Lexicon paid $10.5 million in cash and issued 659,905 shares of common stock to designees of Holdings in satisfaction of its remaining contingent payment obligation as a result of receiving regulatory approval in the United states for the marketing and sale of XERMELO.
Lexicon accounted for the exercise of the Purchase Option and acquisition of Symphony Icon as a business combination. In connection with its acquisition of Symphony Icon, Lexicon paid $10.0 million in cash, and has also agreed to pay Holdings additional base and contingent payments as discussed above. The fair value of the base and contingent consideration payments was $45.6 million and was estimated by applying a probability-based income approach utilizing an appropriate discount rate. This estimation was based on significant inputs that are not observable in the market, referred to as Level 3 inputs. Key assumptions include: (1) a discount rate of 14% for the base payments; (2) a discount rate of 18% for the contingent payments; and (3) a probability adjusted contingency. No discount rate was used in the valuation of the contingent consideration liability as of December 31, 2016 as the expected buyout was short-term in nature. Subsequent changes in the fair value of the Symphony Icon purchase consideration liability were recorded as increase or decrease in fair value of Symphony Icon purchase liability expense in the accompanying consolidated statements of comprehensive loss. The fair value
F-19
of the Symphony Icon purchase consideration liability increased by $2.1 million during the year ended December 31, 2017, decreased by $0.7 million during the year ended December 31, 2016, and increased by $5.9 million during the year ended December 31, 2015.
11. Commitments and Contingencies
Operating Lease Obligations: A Lexicon subsidiary leases office space in Basking Ridge, New Jersey under a lease agreement, the term of which began in June 2015 and terminates in December 2022. Rent expense is recognized on a straight-line basis over the lease term. Additionally, Lexicon leases certain equipment under operating leases.
Rent expense for all operating leases was approximately $0.6 million, $0.5 million and $0.1 million for the years ended December 31, 2017, 2016 and 2015, respectively. The following table includes non-cancelable, escalating future lease payments for the facility in New Jersey:
For the Year Ending December 31 | |||
(in thousands) | |||
2018 | $ | 625 | |
2019 | 614 | ||
2020 | 626 | ||
2021 | 639 | ||
2022 | 651 | ||
Thereafter | — | ||
Total | $ | 3,155 | |
Employment Arrangements: Lexicon has entered into employment arrangements with certain of its corporate officers. Under the arrangements, each officer receives a base salary, subject to adjustment, with an annual discretionary bonus based upon specific objectives to be determined by the compensation committee. The employment arrangements are at-will and some contain non-competition agreements. Some of the arrangements also provide for certain severance payments for either six or 12 months and, in some cases, payment of a specified portion of the officer’s bonus target for such year, in the event of a specified termination of the officer’s employment.
Legal Proceedings: Lexicon is from time to time party to claims and legal proceedings that arise in the normal course of its business and that it believes will not have, individually or in the aggregate, a material adverse effect on its results of operations, financial condition or liquidity.
12. Other Capital Stock Agreements
Reverse Stock Split: Effective May 20, 2015, Lexicon completed a one-for-seven reverse split of its common stock. All references to shares of common stock and per-share data for all periods presented in this report have been adjusted to give effect to this reverse stock split. Proportional adjustments were also made to all shares of common stock issuable under Lexicon’s equity incentive plans and upon conversion of Lexicon’s Notes. Concurrent with the reverse stock split, the authorized shares of common stock were reduced from 900 million (prior to the reverse stock split) to 225 million. As no change was made to the par value of the common shares, common stock and additional paid-in capital were adjusted on a retroactive basis to give effect to the reverse stock split. No fractional shares were issued in connection with the reverse stock split. Any fractional share of common stock that would otherwise have resulted from the reverse stock split were converted into cash payments equal to such fraction multiplied by the closing sales price of the common stock as last reported on the last trading day immediately preceding the effective date of the reverse stock split.
F-20
13. Equity Incentive Awards
Equity Incentive Plans
2017 Equity Incentive Plan: In September 1995, Lexicon adopted the 1995 Stock Option Plan, which was subsequently amended and restated in February 2000, April 2009, April 2012, April 2015 and April 2017 and renamed the 2017 Equity Incentive Plan (the “Equity Incentive Plan”).
The Equity Incentive Plan provides for the grant of incentive stock options to employees and nonstatutory stock options to employees, directors and consultants of the Company. The plan also permits the grant of stock bonus awards, restricted stock awards, restricted stock unit awards, stock appreciation rights and performance stock awards. Incentive and nonstatutory stock options have an exercise price of 100% or more of the fair market value of the Company’s common stock on the date of grant. Most stock options granted under the Equity Incentive Plan become vested and exercisable over a period of four years; however some have been granted with different vesting schedules. Stock options granted under the Equity Incentive Plan have a term of ten years from the date of grant.
The total number of shares of common stock that may be issued pursuant to stock awards under the Equity Incentive Plan shall not exceed in the aggregate 15,000,000 shares. As of December 31, 2017, an aggregate of 15,000,000 shares of common stock had been reserved for issuance, options to purchase 4,773,915 shares and 945,723 restricted stock units were outstanding, 1,812,584 shares had been issued upon the exercise of stock options, 1,118,151 shares had been issued pursuant to restricted stock units and 113,940 shares had been issued pursuant to stock bonus awards or restricted stock awards granted under the Equity Incentive Plan.
2017 Non-Employee Directors’ Equity Incentive Plan: In February 2000, Lexicon adopted the 2000 Non-Employee Directors’ Stock Option Plan, which was subsequently amended and restated in April 2009, April 2012, April 2015 and April 2017 and renamed the 2017 Non-Employee Directors’ Equity Incentive Plan (the “Directors’ Plan”). Under the Directors’ Plan, non-employee directors may be granted awards under the plan with an aggregate grant date fair value of more than $500,000 during any calender year, taken together with any cash fees paid to such non-employee director in compensation for service on Lexicon’s board of directors during such calender year. Stock options granted under the Directors’ Plan have an exercise price equal to the fair market value of the Company’s common stock on the date of grant and a term of ten years from the date of grant.
The total number of shares of common stock that may be issued pursuant to stock awards under the Directors’ Plan shall not exceed in the aggregate 600,000 shares. As of December 31, 2017, an aggregate of 600,000 shares of common stock had been reserved for issuance, stock options to purchase 187,119 shares were outstanding, none had been issued upon the exercise of stock options and 82,696 shares had been issued pursuant to restricted stock awards granted under the Directors’ Plan.
Stock Option Activity: The following is a summary of stock option activity under Lexicon’s equity incentive plans:
2017 | 2016 | 2015 | |||||||||||||||||||
(in thousands, except exercise price data) | Options | Weighted Average Exercise Price | Options | Weighted Average Exercise Price | Options | Weighted Average Exercise Price | |||||||||||||||
Outstanding at beginning of year | 4,834 | $ | 11.24 | 4,217 | $ | 12.35 | 3,371 | $ | 14.98 | ||||||||||||
Granted | 892 | 14.31 | 1,370 | 10.40 | 1,207 | 6.83 | |||||||||||||||
Exercised | (458 | ) | 11.97 | (495 | ) | 12.17 | (19 | ) | 11.14 | ||||||||||||
Expired | (157 | ) | 26.42 | (195 | ) | 27.33 | (187 | ) | 27.29 | ||||||||||||
Forfeited | (150 | ) | 13.84 | (63 | ) | 10.45 | (155 | ) | 8.51 | ||||||||||||
Outstanding at end of year | 4,961 | 11.17 | 4,834 | 11.24 | 4,217 | 12.35 | |||||||||||||||
Exercisable at end of year | 3,077 | $ | 10.95 | 2,727 | $ | 12.55 | 2,686 | $ | 14.53 | ||||||||||||
The weighted average estimated grant date fair value of stock options granted during the years ended December 31, 2017, 2016 and 2015 were $8.59, $6.43 and $4.58, respectively. The total intrinsic value of stock options exercised during the years ended December 31, 2017, 2016 and 2015 were $2.0 million, $1.7 million and $35,000, respectively. The weighted average remaining contractual term of stock options outstanding and exercisable was 6.4 and 5.2 years, respectively, as of
F-21
December 31, 2017. At December 31, 2017, the aggregate intrinsic value of the outstanding stock options and the exercisable stock options was $4.6 million and $2.8 million, respectively.
Stock Bonus and Restricted Stock Unit Activity:
During the years ended December 31, 2017, 2016 and 2015, Lexicon granted its non-employee directors 10,248, 11,456 and 21,360 shares, respectively, of restricted stock awards. The restricted stock awards had weighted average grant date fair values of $15.61, $13.96 and $7.49 per share, respectively, and vested immediately.
During the years ended December 31, 2017, 2016 and 2015, Lexicon granted its employees restricted stock units in lieu of or in addition to annual stock option awards. These restricted stock units vest in four annual installments. The following is a summary of restricted stock units activity under Lexicon’s stock-based compensation plans for the year ended December 31, 2017:
Shares | Weighted Average Grant Date Fair Value | ||||||
(in thousands) | |||||||
Outstanding at December 31, 2016 | 875 | $ | 8.13 | ||||
Granted | 418 | 14.44 | |||||
Vested | (286 | ) | 8.78 | ||||
Forfeited | (61 | ) | 11.57 | ||||
Outstanding at December 31, 2017 | 946 | $ | 10.50 | ||||
Aggregate Shares Reserved for Issuance
As of December 31, 2017, an aggregate of 5,906,757 shares of common stock were reserved for issuance upon exercise of outstanding stock options and vesting of outstanding restricted stock units and 6,565,872 additional shares were available for future grants under Lexicon’s equity incentive plans. The Company has a policy of using either authorized and unissued shares or treasury shares, including shares acquired by purchase in the open market or in private transactions, to satisfy equity award exercises.
14. Benefit Plan
Lexicon maintains a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code. The plan covers substantially all full-time employees. Participating employees may defer a portion of their pretax earnings, up to the Internal Revenue Service annual contribution limit. Beginning in 2000, the Company was required to match employee contributions according to a specified formula. The matching contributions totaled $1,033,000, $733,000 and $332,000 in the years ended December 31, 2017, 2016 and 2015, respectively. Company contributions are vested based on the employee’s years of service, with full vesting after four years of service.
15. Collaboration and License Agreements
Lexicon has derived substantially all of its revenues from drug discovery and development alliances, target validation collaborations for the development and, in some cases, analysis of the physiological effects of genes altered in knockout mice, product sales, government grants and contracts, technology licenses, subscriptions to its databases and compound library sales.
Sanofi. In November 2015, Lexicon entered into a Collaboration and License Agreement, which was subsequently amended in July 2017 (collectively, the “Sanofi Agreement”), with Sanofi for the worldwide development of Lexicon’s diabetes drug candidate sotagliflozin. In December 2016, Sanofi terminated its rights under the Sanofi Agreement with respect to Japan.
Under the Sanofi Agreement, Lexicon has granted Sanofi an exclusive, worldwide (excluding Japan), royalty-bearing right and license under its patent rights and know-how to develop, manufacture and commercialize sotagliflozin. Subject to specified exceptions, neither party may (a) perform clinical development activities relating to any other compound which inhibits sodium-glucose cotransporters type 1 or type 2 or (b) commercialize any such compounds in the United States, countries of the European Union and certain other specified countries, in each case during the royalty terms applicable in such
F-22
countries. Among the specified exceptions is a right Lexicon retained to pursue the development of its LX2761 drug candidate, with respect to which Lexicon granted Sanofi certain rights of first negotiation specified in the Sanofi Agreement.
Under the Sanofi Agreement, Sanofi paid Lexicon an upfront payment of $300 million. In addition, Lexicon is eligible to receive from Sanofi (a) up to an aggregate of $110 million upon the achievement of four development milestones relating to the results of certain Phase 3 clinical trials of sotagliflozin in type 2 diabetes patients, (b) up to an aggregate of $220 million upon the achievement of four regulatory milestones relating to the first commercial sale following regulatory approval of sotagliflozin for type 1 and type 2 diabetes, respectively, in each of the United States and Europe, of which two milestones representing the substantial majority of such aggregate amount relate to type 2 diabetes and the remaining two milestones relate to type 1 diabetes, (c) $100 million upon the achievement of a milestone based on the results of either of two outcomes studies in type 2 diabetes patients, the completion of which would likely occur after initial regulatory approval of sotagliflozin in type 2 diabetes, and (d) up to an aggregate of $990 million upon the achievement of six commercial milestones that will be achieved upon reaching specified levels of sales. The Company believes that each of the development and regulatory milestones under the Sanofi Agreement is substantive. Due to the uncertainty surrounding the achievement of the future development and regulatory milestones, these payments will not be recognized as revenue unless and until they are earned, as the Company is not able to reasonably predict if and when the milestones will be achieved. Commercial milestones, which are not encompassed within the definition of milestones under generally accepted accounting principles, will be accounted for as royalties and recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria were met. Lexicon is also entitled to tiered, escalating royalties ranging from low double digit percentages to forty percent of net sales of sotagliflozin, based on indication and territory, with royalties for the higher band of such range attributable to net sales for type 1 diabetes in the United States, and subject in each case to customary royalty reduction provisions.
Lexicon will continue to be responsible for all clinical development activities relating to type 1 diabetes and has exercised an exclusive option to co-promote and have a significant role, in collaboration with Sanofi, in the commercialization of sotagliflozin for the treatment of type 1 diabetes in the United States. Under the terms of its co-promotion option, Lexicon will fund forty percent of the commercialization costs relating to such co-promotion activities. Sanofi will be responsible for all clinical development and commercialization of sotagliflozin for the treatment of type 2 diabetes worldwide and will be solely responsible for the commercialization of sotagliflozin for the treatment of type 1 diabetes outside the United States. Lexicon will share in the funding of a portion of the planned type 2 diabetes development costs over the first three years of the collaboration, up to an aggregate of $100 million. Sanofi will book sales worldwide in all indications.
The parties are responsible for using commercially reasonable efforts to perform their development and commercialization obligations pursuant to mutually approved development and commercialization plans.
The parties’ activities under the Sanofi Agreement are governed by a joint steering committee and certain other governance committees which reflect equal or other appropriate representation from both parties. If the applicable governance committee is not able to make a decision by consensus and the parties are not able to resolve the issue through escalation to specified senior executive officers of the parties, then Sanofi will have final decision-making authority, subject to limitations specified in the Sanofi Agreement.
The Sanofi Agreement will expire upon the expiration of all applicable royalty terms for all licensed products in all countries. The royalty term for each licensed product in each country is the period commencing on the effective date of the Sanofi Agreement and ending on the latest of expiration of specified patent coverage, expiration of specified regulatory exclusivity and 10 years following the first commercial sale in the applicable country. Either party may terminate the Sanofi Agreement in the event of an uncured material breach by the other party. Prior to completion of the core development activities for type 2 diabetes specified in the development plan, Sanofi may terminate the Sanofi Agreement on a country-by-country and licensed product-by-licensed product basis, in the event of (a) notification of a material safety issue relating to the licensed product or the class of sodium-glucose cotransporters type 1 or type 2 inhibitors resulting in a recommendation or requirement that Lexicon or Sanofi cease development, (b) failure to achieve positive results with respect to certain clinical trial results, (c) the occurrence of specified fundamental adverse events or (d) the exploitation of the licensed product infringing third party intellectual property rights in specified major markets and Sanofi is unable to obtain a license to such third party intellectual property rights.
The Company considered the following deliverables with respect to the revenue recognition of the $300 million upfront payment:
•The exclusive worldwide license granted to Sanofi to develop and commercialize sotagliflozin;
•The development services Lexicon is performing for sotagliflozin relating to type 1 diabetes; and
•The funding Lexicon will provide for development relating to type 2 diabetes.
F-23
The Company determined that the license had stand-alone value because it is an exclusive license that gives Sanofi the right to develop and commercialize sotagliflozin or to sublicense its rights. In addition, sotagliflozin is currently in development and it is possible that Sanofi or another third party could conduct clinical trials without assistance from Lexicon. As a result, the Company considers the license and the development services under the Sanofi Agreement to be separate units of accounting. The Company recognized the portion of the consideration allocated to the license immediately because Lexicon delivered the license and earned the revenue at the inception of the arrangement. The Company is recognizing as revenue the amount allocated to the development services for type 1 diabetes and the obligation to provide funding for development services for type 2 diabetes over the period of time Lexicon performs services or provides funding, currently expected to be through 2020.
The Company determined that the initial allocable arrangement consideration was the $300 million upfront payment because it was the only payment that was fixed and determinable at the inception of the arrangement. There was considerable uncertainty at the date of the agreement as to whether Lexicon would earn milestone payments or royalty payments. As such, the Company did not include those payments in the allocable consideration. The Company allocated the allocable consideration based on the relative best estimate of selling price of each unit of accounting. The Company estimated the selling price of the license deliverable by applying a probability-based income approach utilizing an appropriate discount rate. The significant inputs the Company used to determine the projected income of the license included: exercising the option to co-promote, estimated future product sales, estimated cost of goods sold, estimated operating expenses, income taxes, and an appropriate discount rate. The Company estimated the selling price of the development services for type 1 diabetes by using internal estimates of the cost to hire third parties to perform the services over the expected period to perform the development. The Company estimated the obligation to provide funding for type 2 diabetes by using internal estimates of the expected cash flows and timing for $100 million in funding.
As a result of the allocation, the Company recognized $126.8 million of the $300 million upfront payment for the license in 2015. The Company is recognizing the $113.8 million allocated to the development services deliverable and the $59.4 million allocated to the funding deliverable over the estimated period of performance as the development and funding occurs. Revenue recognized under the Sanofi Agreement was $56.3 million, $75.4 million and $126.8 million for the years ended December 31, 2017, 2016 and 2015, respectively. Revenue for the years ended December 31, 2017 and 2016 includes $1.9 million and $6.3 million, respectively, of sales of clinical trial materials to Sanofi.
Ipsen. In October 2014, Lexicon entered into a License and Collaboration Agreement, which was subsequently amended in March 2015 (collectively, the “Ipsen Agreement”), with Ipsen for the development and commercialization of XERMELO outside of the United States and Japan (the “Licensed Territory”).
Under the Ipsen Agreement, Lexicon granted Ipsen an exclusive, royalty-bearing right and license under its patent rights and know-how to commercialize XERMELO in the Licensed Territory. Ipsen is responsible for using diligent efforts to commercialize XERMELO in the Licensed Territory pursuant to a mutually approved commercialization plan. Subject to certain exceptions, Lexicon was responsible for conducting clinical trials required to obtain regulatory approval for XERMELO for carcinoid syndrome in the European Union, including those contemplated by a mutually approved initial development plan, and has the first right to conduct most other clinical trials of XERMELO. Lexicon was responsible for the costs of all clinical trials contemplated by the initial development plan. The costs of additional clinical trials will be allocated between the parties based on the nature of such clinical trials. Under the Ipsen Agreement, Ipsen has paid Lexicon an aggregate of $43.7 million through December 31, 2017, consisting of $24.5 million in upfront payments, a $6.4 million milestone upon the acceptance of the filing submitted by Ipsen to the European Medicines Agency for XERMELO as an adjunct to somatostatin analog therapy for the long-term treatment of carcinoid syndrome, a $5.1 million milestone upon Ipsen’s receipt of approval from the European Commission for the marketing of XERMELO in all member states of the European Union, Norway and Iceland, a $3.84 million milestone upon Ipsen’s first commercial sale in Germany, and a $3.84 million milestone upon Ipsen’s first commercial sale in the United Kingdom. In addition, Lexicon is eligible to receive from Ipsen (a) up to an aggregate of approximately $13.1 million upon the achievement of specified regulatory and commercial launch milestones and (b) up to an aggregate of €72 million upon the achievement of specified sales milestones. Due to the uncertainty surrounding the achievement of the future regulatory and sales milestones, these payments will not be recognized as revenue unless and until they are earned as the Company is not able to reasonably predict if and when the milestones will be achieved. Lexicon is also entitled to tiered, escalating royalties ranging from low twenties to mid-thirties percentages of net sales of XERMELO in the Licensed Territory, subject to a credit for amounts previously paid to Lexicon by Ipsen for the manufacture and supply of such units of XERMELO. Lexicon and Ipsen have entered into a commercial supply agreement pursuant to which Lexicon supplies Ipsen’s commercial requirements of XERMELO, and Ipsen pays an agreed upon transfer price for such commercial supply.
F-24
The Company considered the following deliverables with respect to the revenue recognition of the $24.5 million upfront payment:
•The exclusive license granted to Ipsen to develop and commercialize XERMELO in the Licensed
Territory;
•The development services Lexicon is performing for XERMELO;
•The obligation to participate in committees which govern the development of XERMELO
until commercialization; and
•The obligation to supply commercial supply of XERMELO, under a commercial supply agreement.
The Company determined that the license had stand-alone value because it is an exclusive license that grants Ipsen the right to develop and commercialize XERMELO or to sublicense its rights. In addition, at the time of the agreement, it would have been possible for Ipsen or another third party to conduct clinical trials without assistance from Lexicon. As a result, the Company considers the license and the development services under the Agreement to be separate units of accounting. The Company recognized the portion of the consideration allocated to the license immediately because Lexicon delivered the license and earned the revenue at the inception of the arrangement. The Company is recognizing as revenue the amount allocated to the development services and the obligation to participate in committees over the period of time Lexicon performs services, currently expected to be through mid-2018.
The Company determined that the commercial supply agreement is a contingent deliverable at the onset of the Agreement. There was inherent uncertainty in obtaining regulatory approval at the time of the agreement, thus, making the applicability of the commercial supply agreement outside the control of Lexicon and Ipsen. As a result, the Company has determined the commercial supply agreement does not meet the definition of a deliverable that needs to be accounted for at the inception of the arrangement. The Company has also determined that there is no significant and incremental discount related to the commercial supply agreement that should be accounted for at the inception of the arrangement.
The Company determined that the initial allocable arrangement consideration was the $24.5 million upfront payments because they were the only payments that were fixed and determinable at the inception of the arrangement. There was considerable uncertainty at the date of the agreement as to whether Lexicon would earn milestone payments, royalty payments or payments for finished drug product. As such, the Company did not include those payments in the allocable consideration. The Company allocated the allocable consideration based on the relative best estimate of selling price of each unit of accounting. The Company estimated the selling price of the license deliverable by applying a probability-based income approach utilizing an appropriate discount rate. The significant inputs the Company used to determine the projected income of the license included: estimated future product sales, estimated cost of goods sold, estimated operating expenses, income taxes, and an appropriate discount rate. The Company estimated the selling price of the development services by using internal estimates of the cost to hire third parties to perform the services over the expected period to perform the development. The Company estimated the selling price of the obligation to participate in committees by using internal estimates of the number of internal hours and salary and benefits costs to perform these services.
As a result of the allocation, the Company recognized $21.2 million of the $24.5 million upfront payment for the license in 2014, and an additional $1.4 million in 2015 upon entering into the amendment. The Company is recognizing the $1.7 million allocated to the development services deliverable over the estimated period of performance as development occurs, and is recognizing the $0.1 million allocated to the committee participation deliverable ratably over the estimated period of performance. Milestone payments that are contingent upon the achievement of a substantive milestone are recognized in their entirety in the period in which the milestone is achieved. Revenue recognized under the Agreement was $16.1 million, $7.2 million and $2.3 million for the years ended December 31, 2017, 2016 and 2015, respectively. Revenue for the year ended December 31, 2017 includes $0.8 million from sales of bulk tablets of XERMELO to Ipsen.
Texas Institute for Genomic Medicine. In July 2005, Lexicon received a $35.0 million award from the Texas Enterprise Fund for the creation of a knockout mouse embryonic stem cell library containing 350,000 cell lines for the Texas Institute for Genomic Medicine (“TIGM”) using Lexicon’s proprietary gene trapping technology, which Lexicon completed in 2007. Lexicon also equipped TIGM with the bioinformatics software required for the management and analysis of data relating to the library. The Texas Enterprise Fund made an additional award of $15.0 million to the Texas A&M University System for the creation of facilities and infrastructure to house the library.
Under the terms of the award, Lexicon is responsible for the creation of a specified number of jobs beginning in 2012, reaching an aggregate of 1,616 new jobs in Texas by December 31, 2016. Lexicon will receive credits against those job obligations based on funding received by TIGM and certain related parties from sources other than the State of Texas. Lexicon will also receive credits against those job obligations for any surplus jobs that Lexicon created. Subject to these credits, the
F-25
state may require Lexicon to repay $2,415 for each job Lexicon falls short beginning in 2013. Lexicon’s maximum aggregate exposure for such payments, if Lexicon fails to create any new jobs, is approximately $14.2 million, without giving effect to any credits to which Lexicon may be entitled. Lexicon has recorded this obligation as deferred revenue in the accompanying consolidated balance sheets.
16. Selected Quarterly Financial Data (Unaudited)
The table below sets forth certain unaudited statements of comprehensive loss data, and net loss per common share data, for each quarter of 2017 and 2016:
(in thousands, except per share data)
Quarter Ended | |||||||||||||||
March 31 | June 30 | September 30 | December 31 | ||||||||||||
(Unaudited) | |||||||||||||||
2017 | |||||||||||||||
Revenues | $ | 18,293 | $ | 12,053 | $ | 26,942 | $ | 33,047 | |||||||
Loss from operations | $ | (42,485 | ) | $ | (33,893 | ) | $ | (29,518 | ) | $ | (30,785 | ) | |||
Consolidated net loss | $ | (34,891 | ) | $ | (35,059 | ) | $ | (30,722 | ) | $ | (28,378 | ) | |||
Consolidated net loss per common share, basic and diluted | $ | (0.33 | ) | $ | (0.33 | ) | $ | (0.29 | ) | $ | (0.27 | ) | |||
Shares used in computing consolidated net loss per common share, basic and diluted | 104,461 | 105,300 | 105,582 | 105,588 | |||||||||||
2016 | |||||||||||||||
Revenues | $ | 12,494 | $ | 20,089 | $ | 27,717 | $ | 23,037 | |||||||
Loss from operations | $ | (33,871 | ) | $ | (37,021 | ) | $ | (34,933 | ) | $ | (31,330 | ) | |||
Consolidated net loss | $ | (34,883 | ) | $ | (38,112 | ) | $ | (36,015 | ) | $ | (32,419 | ) | |||
Consolidated net loss per common share, basic and diluted | $ | (0.34 | ) | $ | (0.37 | ) | $ | (0.35 | ) | $ | (0.31 | ) | |||
Shares used in computing consolidated net loss per common share, basic and diluted | 103,682 | 103,830 | 103,885 | 104,052 | |||||||||||
For all periods presented, the weighted average number of shares outstanding are the same for both basic and diluted consolidated net loss per common share. For these periods, shares associated with convertible debt, stock options and restricted stock units are not included in the weighted average number of shares of common stock outstanding because they are antidilutive.
F-26