Attached files
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
10-K
(Mark
One)
|
|
þ
|
ANNUAL REPORT PURSUANT TO
SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
|
For
the Fiscal Year Ended December 31, 2009
or
|
|
q
|
TRANSITION REPORT PURSUANT TO
SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
|
For
the Transition Period from _____________ to _____________
Commission
File Number: 000-30111
Lexicon
Pharmaceuticals, Inc.
(Exact
Name of Registrant as Specified in its Charter)
|
Delaware
|
76-0474169
|
|
(State
or Other Jurisdiction of Incorporation
or Organization)
|
(I.R.S.
Employer Identification
Number)
|
|
8800
Technology Forest Place
The
Woodlands, Texas 77381
(Address
of Principal Executive Offices and Zip Code)
|
(281)
863-3000
(Registrant’s Telephone
Number,
Including
Area Code)
|
Securities
registered pursuant to Section 12(b) of the Act:
|
Title
of Each Class
|
Name
of Each Exchange on which Registered
|
||||
|
Common
Stock, par value $0.001 per share
|
Nasdaq
Global Market
|
||||
Securities registered pursuant to
Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act of 1933. Yes o No þ
Indicate
by check mark if the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Securities Exchange Act of
1934. Yes o No þ
Indicate
by check mark whether the registrant (1) has filed all reports required to be
filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the
preceding 12 months (or for such shorter period that the registrant was required
to file such reports) and (2) has been subject to such filing requirements for
the past 90 days. Yes þ No o
Indicate
by check mark whether the registrant has submitted electronically and posted on
its corporate Web site, if any, every Interactive Data File required to be
submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding
12 months (or for such shorter period that the registrant was required to submit
and post such files). Yes o No o
Indicate
by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or information statements
incorporated by reference in Part III of this Form 10-K or any amendment to this
Form 10-K. þ
Indicate
by check mark whether the registrant is a large accelerated filer, an
accelerated filer, a non-accelerated filer or a smaller reporting
company. See definitions of “large accelerated filer,” “accelerated
filer” and “smaller reporting company” in Rule 12b-2 of the Securities Exchange
Act of 1934. (check one): Large accelerated filer o Accelerated
filer þ Non-accelerated
filer o Smaller
reporting company o
Indicate
by check mark whether the registrant is a shell company (as defined in Rule
12b-2 of the Securities Exchange Act of 1934). Yes o No þ
The
aggregate market value of voting stock held by non-affiliates of the registrant
as of the last day of the registrant’s most recently completed second quarter
was approximately $99.1 million, based on the closing price of the common stock
on the Nasdaq Global Market on June 30, 2009 of $1.24 per
share. For purposes of the preceding sentence only, all directors,
executive officers and beneficial owners of ten percent or more of the
registrant’s common stock are assumed to be affiliates. As of
March 1, 2010, 175,633,988 shares of common stock were
outstanding.
Documents
Incorporated by Reference
Certain
sections of the registrant’s definitive proxy statement relating to the
registrant’s 2010 annual meeting of stockholders, which proxy statement will be
filed under the Securities Exchange Act of 1934 within 120 days of the end of
the registrant’s fiscal year ended December 31, 2009, are incorporated by
reference into Part III of this annual report on
Form
10-K.
Lexicon
Pharmaceuticals, Inc.
|
Item
|
|||
|
PART
I
|
|||
|
1.
|
1
|
||
|
1A.
|
15
|
||
|
1B.
|
30
|
||
|
2.
|
30
|
||
|
3.
|
30
|
||
|
4.
|
30
|
||
|
PART
II
|
|||
|
5.
|
31
|
||
|
6.
|
33
|
||
|
7.
|
34
|
||
|
7A.
|
44
|
||
|
8.
|
44
|
||
|
9.
|
44
|
||
|
9A.
|
44
|
||
|
9B.
|
44
|
||
|
PART
III
|
|||
|
10.
|
45
|
||
|
11.
|
45
|
||
|
12.
|
45
|
||
|
13.
|
45
|
||
|
14.
|
45
|
||
|
PART
IV
|
|||
|
15.
|
46
|
||
|
50
|
|||
The
Lexicon name and logo, OmniBank® and
LexVision® are
registered trademarks and Genome5000™ is a
trademark of Lexicon Pharmaceuticals, Inc.
In this
annual report on Form 10-K, “Lexicon Pharmaceuticals,” “Lexicon,” “we,” “us” and
“our” refer to Lexicon Pharmaceuticals, Inc.
Factors Affecting Forward Looking Statements
This
annual report on Form 10-K contains forward-looking statements. These
statements relate to future events or our future financial
performance. We have attempted to identify forward-looking statements
by terminology including “anticipate,” “believe,” “can,” “continue,” “could,”
“estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “should”
or “will” or the negative of these terms or other comparable terminology. These
statements are only predictions and involve known and unknown risks,
uncertainties and other factors, including the risks outlined under “Item
1A. Risk Factors,” that may cause our or our industry’s actual
results, levels of activity, performance or achievements to be materially
different from any future results, levels of activity, performance or
achievements expressed or implied by these forward-looking
statements.
Although
we believe that the expectations reflected in the forward-looking statements are
reasonable, we cannot guarantee future results, levels of activity, performance
or achievements. We are not under any duty to update any of the forward-looking
statements after the date of this annual report on Form 10-K to conform these
statements to actual results, unless required by law.
PART
I
Item
1. Business
Overview
Lexicon
Pharmaceuticals is a biopharmaceutical company focused on the discovery and
development of breakthrough treatments for human disease. We have
used our proprietary gene knockout technologies and an integrated platform of
advanced medical technologies to systematically study the physiological and
behavioral functions of almost 5,000 genes in mice and assessed the utility of
the proteins encoded by the corresponding human genes as potential drug
targets. We have identified and validated in living animals, or in vivo, more than 100
targets with promising profiles for drug discovery. For targets that
we believe have high pharmaceutical value, we engage in programs for the
discovery and development of potential new drugs, focusing in the core
therapeutic areas of immunology, metabolism, cardiology and
ophthalmology.
We have
announced positive results from Phase 2 clinical trials of each of our two most
advanced drug candidates: LX1031, an orally-delivered small molecule
compound that we are developing as a potential treatment for irritable bowel
syndrome and other gastrointestinal disorders and LX4211, an orally-delivered
small molecule compound that we are developing as a potential treatment for type
2 diabetes. We are presently conducting Phase 2 clinical trials of
two other drug candidates: LX2931, an orally-delivered small molecule
compound that we are developing as a potential treatment for rheumatoid
arthritis and other autoimmune diseases and LX1032, an orally-delivered small
molecule compound that we are developing as a potential treatment for the
symptoms associated with carcinoid syndrome. We have advanced one
other drug candidate into preclinical development: LX7101, a topically-delivered
small molecule compound that we are developing as a potential treatment for
glaucoma. We have small molecule compounds from a number of
additional drug discovery programs in various stages of preclinical research and
believe that our systematic, target biology-driven approach to drug discovery
will enable us to continue to expand our clinical pipeline.
We are
working both independently and through strategic collaborations and alliances to
capitalize on our technology, drug target discoveries and drug discovery and
development programs. Consistent with this approach, we seek to
retain exclusive rights to the benefits of certain of our small molecule drug
programs by developing drug candidates from those programs internally and to
collaborate with third parties with respect to the discovery, development and
commercialization of small molecule and biotherapeutic drug candidates for other
targets, particularly when the collaboration provides us with access to
expertise and resources that we do not possess internally or are complementary
to our own. We have established drug discovery and development
collaborations with a number of leading pharmaceutical and biotechnology
companies which have enabled us to generate near-term cash while offering us the
potential to retain economic participation in products our collaborators develop
through the collaboration. In addition, we have established
collaborations and license agreements with other leading pharmaceutical and
biotechnology companies, research institutes and academic institutions under
which we received fees and, in some cases, are eligible to receive milestone and
royalty payments, in return for granting access to some of our technologies and
discoveries.
Lexicon
Pharmaceuticals was incorporated in Delaware in July 1995, and commenced
operations in September 1995. Our corporate headquarters are located
at 8800 Technology Forest Place, The Woodlands, Texas 77381, and our telephone
number is (281) 863-3000.
Our
annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on
Form 8-K, and amendments to those reports filed or furnished pursuant to Section
13(a) or 15(d) of the Securities Exchange Act of 1934 are made available free of
charge on our corporate website located at www.lexpharma.com as soon as
reasonably practicable after the filing of those reports with the Securities and
Exchange Commission. Information found on our website should not be
considered part of this annual report on Form 10-K.
Our
Drug Development Pipeline
Human
clinical trials are currently underway for four of our drug candidates, with one
additional drug candidate in preclinical development and compounds from a number
of additional programs in various stages of preclinical research:
|
Drug Program
|
|
Potential Indication
|
|
Stage of
Development
|
||||||||||||||
|
Preclinical
Research
|
Preclinical
Development
|
IND
|
Phase
1
|
Phase
2
|
Phase
3
|
|||||||||||||
|
LX1031
|
Irritable
Bowel Syndrome
|
|||||||||||||||||
|
LX4211
|
Type
2 Diabetes
|
|||||||||||||||||
|
LX2931
|
Rheumatoid
Arthritis
|
|||||||||||||||||
|
LX1032
|
Carcinoid
Syndrome
|
|||||||||||||||||
|
LX7101
|
Glaucoma
|
|||||||||||||||||
LX1031
LX1031 is
an orally-delivered small molecule compound that we are developing for the
potential treatment of irritable bowel syndrome and other gastrointestinal
disorders. We reported top-line data in November 2009 from a Phase 2
clinical trial evaluating the safety and tolerability of LX1031 and its effects
on symptoms associated with irritable bowel syndrome. The Phase 2
clinical trial, which began in December 2008, enrolled 155 patients suffering
from either diarrhea-predominant or mixed irritable bowel syndrome in a
randomized, double-blind, placebo-controlled study of 250mg and 1,000mg doses of
LX1031, each administered four times daily over a four-week treatment
period. The efficacy endpoints under evaluation in the trial included
a global assessment of adequate relief, number of bowel movements, symptom
severity evaluation (bloating, urgency and pain), and stool
form. Top-line data from the study showed that treatment with 1,000mg
of LX1031 four times daily produced a statistically significant improvement in
the global assessment of relief of irritable bowel syndrome pain and discomfort
over the four-week treatment period compared to placebo. Improvements
in the global assessment were observed in the first week of treatment and were
maintained in each of the four weeks of the study, achieving statistical
significance relative to placebo at the end of the first and second week and
showing an improved trend relative to placebo that did not reach statistical
significance at the end of the third and fourth weeks. Improvements
in the global assessment of adequate relief corresponded with statistically
significant improvements in stool consistency in the same dose
group. Increased clinical response correlated with a greater
reduction in serotonin synthesis as reflected by measures of urinary 5-HIAA, the
primary metabolite of serotonin and a biomarker for serotonin production. LX1031
was well tolerated with no notable differences in adverse events observed
between placebo and either treatment group. We are presently seeking
to develop an improved formulation of LX1031in preparation for use in
future clinical trials.
We
previously completed a Phase 1a single ascending-dose study and two Phase 1b
multiple ascending-dose studies exploring safety and tolerability and the
effects of LX1031 on serotonin synthesis. In Phase 1 clinical trials,
all dose levels were well tolerated, no dose-limiting toxicities were observed,
and LX1031 was shown to reduce levels of urinary 5-HIAA.
We
designed LX1031 to reduce production of serotonin in the gastrointestinal tract
and therefore reduce the serotonin available for receptor activation without
affecting serotonin levels in the brain. LX1031 was internally
generated by our medicinal chemists as an inhibitor of tryptophan hydroxylase,
or TPH, the rate-limiting enzyme for serotonin production found primarily in
enterochromaffin, or EC, cells of the gastrointestinal tract. Our
scientists found that mice lacking the non-neuronal form of this enzyme, TPH1,
have virtually no serotonin in the gastrointestinal tract, but maintain normal
levels of serotonin in the brain. In preclinical studies, LX1031
demonstrated a dose-dependent reduction of serotonin levels in the
gastrointestinal tract of multiple species without affecting brain serotonin
levels.
Clinical
development of LX1031 is being funded through our product development
collaboration with Symphony Icon Holdings LLC, or Holdings, pursuant to which we
have licensed to a wholly-owned subsidiary of Holdings, Symphony Icon, Inc. or
Symphony Icon, the intellectual property rights related to LX1031 and hold
an exclusive option to acquire all the equity of Symphony Icon, thereby allowing
us to reacquire LX1031. See “—Our Commercialization Strategy—Drug
Development Financing Collaborations—Symphony Icon.”
LX4211
LX4211 is
an orally-delivered small molecule compound that we are developing for the
potential treatment of type 2 diabetes mellitus. We reported top-line
data in January 2010 from a Phase 2 clinical trial evaluating the safety and
tolerability of LX4211and its effects on biomarkers associated with type 2
diabetes. The Phase 2 trial, which began in September 2009, enrolled
36 patients with non-insulin dependent type 2 diabetes in a double-blind,
randomized, placebo-controlled study of 150mg and 300mg doses of LX4211, each
administered once daily over a four-week treatment period. The
efficacy endpoints under evaluation in the trial included urinary glucose
excretion, fasting plasma glucose, response to oral glucose tolerance testing,
and hemoglobin A1c, also known as HbA1c or A1c, a measure of blood glucose
levels over time. Top-line data from the study showed that treatment
with 150mg and 300mg of LX4211 provided improvements in glycemic control and
demonstrated statistically significant benefits in the primary and multiple
secondary efficacy endpoints. A marked and statistically significant
decrease in fasting plasma glucose was observed throughout the treatment period
in both dose groups relative to placebo. After four weeks of dosing,
patients in both dose groups exhibited statistically significant reductions in
HbA1c as compared to patients receiving placebo. Patients in both
dose groups also exhibited statistically significant improvements in glucose
tolerance in response to oral glucose tolerance testing. Consistent
with the mechanism of action of LX4211, there was also a significant,
dose-dependent increase in 24-hour urinary glucose excretion in both dose groups
throughout the study period relative to placebo. Patients in both
dose groups showed positive trends that did not reach statistical significance
in broader metabolic and cardiovascular parameters, including weight reduction,
decreased blood pressure and lower triglyceride levels. LX4211
demonstrated a favorable safety profile in the trial, with no dose-limiting
toxicities observed. Adverse events were generally mild and equally
distributed across all groups, including the placebo group. We are
presently completing a 13-week preclinical toxicology study of LX4211 to permit
longer-term clinical trials following which we intend to conduct a
bioavailability study of a solid oral dose formulation of LX4211 in 2010
before initiating a Phase 2 dose-ranging study.
We
previously completed a combined Phase 1 single ascending-dose and multiple
ascending-dose study of LX4211. In the Phase 1 clinical trial, LX4211
was well tolerated at all dose levels and produced a dose-dependent increase in
urinary glucose excretion.
LX4211
was internally generated by our medicinal chemists to target sodium-glucose
cotransporter type 2, or SGLT2, a transporter responsible for most of the
glucose reabsorption performed by the kidney. Our scientists
discovered that mice lacking SGLT2 have improved glucose tolerance and increased
urinary glucose excretion. LX4211 also inhibits sodium-glucose
cotransporter type 1, or SGLT1, a transporter responsible for glucose and
galactose absorption in the gastrointestinal tract, and to a lesser extent than
SGLT2, glucose reabsorption in the kidney. In preclinical studies,
animals treated with LX4211 demonstrated increased urinary glucose excretion and
decreased HbA1c levels, with urinary glucose excretion returning to baseline
after treatment was discontinued.
LX2931
LX2931 is
an orally-delivered small molecule compound that we are developing for the
potential treatment of autoimmune diseases such as rheumatoid
arthritis. We initiated a Phase 2 clinical trial in August 2009 to
evaluate the safety and tolerability of LX2931 and its effects on symptoms and
signs associated with rheumatoid arthritis. The Phase 2 trial is
expected to enroll up to 200 patients with rheumatoid arthritis who are also
taking methotrexate, a standard therapy, in a double-blind, randomized,
placebo-controlled study of three dose levels of LX2931 against placebo over a
12-week treatment period. The efficacy endpoints under evaluation in the trial
include ACR20 at 12 weeks and ACR20/50/70 and DAS28 at four, eight and 12
weeks. Top-line data from this trial are expected to be available in
late 2010 or shortly thereafter.
We
previously completed a drug-drug interaction study of LX2931 in rheumatoid
arthritis patients taking methotrexate. We also completed two Phase
1a single ascending-dose studies, a Phase 1b multiple ascending-dose study and a
multiple dose study assessing the pharmacokinetics of a solid dose form of
LX2931. In the Phase 1 clinical trials, LX2931 demonstrated a
dose-dependent reduction in circulating lymphocytes similar to those associated
with a beneficial response observed in animal arthritis models after treatment
with LX2931 and produced a dose-dependent decrease in absolute lymphocyte
counts, with systemic exposure plateauing at doses of 100 to 125
mg. An episode of acute abdominal pain resolving within 24 hours was
observed in two out of 24 subjects in the single ascending-dose trials who
received doses above 175 mg, potentially representing dose-limiting
tolerability. All other doses were well tolerated with mild to
moderate adverse events equally distributed across all groups, including the
placebo group.
LX2931
was internally generated by our medicinal chemists to target
sphingosine-1-phosphate lyase, or S1P lyase, an enzyme in the sphingosine-1
phosphate (S1P) pathway associated with the activity of
lymphocytes. Lymphocytes are a cellular component and key driver of
the immune system, and are involved in a number of autoimmune and inflammatory
disorders. Our scientists discovered that mice lacking this enzyme
have increased retention of immune cells in the thymus and spleen with a
corresponding reduction in the deployment of T-cells and B-cells into the
circulating blood. In preclinical studies, LX2931 produced a
consistent reduction in circulating lymphocyte counts in multiple species, and
reduced joint inflammation and prevented arthritic destruction of joints in
mouse and rat models of arthritis.
LX1032
LX1032 is
an orally-delivered small molecule compound that we are developing for the
potential treatment of symptoms associated with carcinoid
syndrome. We initiated a Phase 2 clinical trial in July 2009 to
evaluate the safety and tolerability of LX1032 and its effects on symptoms
associated with carcinoid syndrome. The Phase 2 trial is expected to
enroll up to 28 patients with symptomatic carcinoid syndrome refractory to
octreotide therapy in a double-blind, randomized, placebo-controlled study
assessing a series of escalating doses of LX1032 against placebo over a 28-day
treatment period, followed by cohort expansion at optimal dose. The efficacy
endpoints under evaluation in the trial include the number of daily bowel
movements, stool form, urgency, a global assessment of symptoms associated with
carcinoid syndrome, flushing episodes and an assessment of pain and discomfort.
Top-line data from this trial are expected to be available in the second half of
2010. We also intend to initiate a complementary open-label clinical trial
of LX1032 in the first half of 2010, which is expected to enroll up to 16
additional patients.
We
previously completed a Phase 1a single ascending-dose study and a Phase 1b
multiple ascending-dose study of LX1032. In Phase 1 clinical trials,
LX1032 was generally well tolerated at all dose levels, and results demonstrated
a potent dose-dependent reduction in both blood serotonin levels and urinary
5-HIAA which was consistent with the reductions observed in preclinical animal
models.
LX1032
was internally generated by our medicinal chemists as an inhibitor of TPH, the
same target as LX1031, but LX1032 is chemically distinct and, unlike LX1031, was
specifically designed to achieve enhanced systemic exposure to address disorders
such as carcinoid syndrome that require regulation of serotonin levels beyond
the enterochromaffin cells in the gastrointestinal tract without impacting brain
serotonin production. In preclinical studies, LX1032 was able to
reduce peripheral serotonin levels in several different species without
affecting serotonin levels in the brain. LX1032 has received Fast
Track status from the United States Food and Drug Administration, or FDA, which
provides for an expedited review process that may shorten FDA approval
times.
Clinical
development of LX1032 is being funded through our product development
collaboration with Holdings, pursuant to which we have licensed to Symphony
Icon the intellectual property rights related to LX1032 and hold an
exclusive option to acquire all the equity of Symphony Icon, thereby allowing us
to reacquire LX1032. See “—Our Commercialization Strategy—Drug
Development Financing Collaborations—Symphony Icon.”
LX7101
LX7101 is
a topically-delivered small molecule compound that we are developing for the
potential treatment of glaucoma. We have commenced preclinical
studies of LX7101 and certain associated back-up molecules.
LX7101
was internally generated by our medicinal chemists to target a kinase
responsible for regulating intraocular pressure and is designed to lower
intraocular pressure by enhancing the fluid outflow facility of the
eye. Our scientists discovered that mice lacking the gene encoding
the target of LX7101 exhibited lower intraocular pressure compared to normal
mice. In preclinical studies, LX7101 significantly reduced
intraocular pressure in an animal model of ocular hypertension.
Discovery
Programs
We have
advanced a number of additional drug discovery programs into various stages of
preclinical research in preparation for formal preclinical development
studies. Through the end of 2009, we had identified and validated,
in vivo, more than 100
targets with promising profiles for drug discovery.
Our
Drug Discovery and Development Process
Our drug
discovery and development process began with our Genome5000 program, in which we
used our gene knockout and medical evaluative technologies to discover the
putative physiological and behavioral functions of almost 5,000 human genes
through analysis of the corresponding mouse knockout models. In our
Genome5000 program, we used our patented gene knockout technologies to generate
knockout mice – mice whose DNA has been modified to disrupt, or knock out, the
function of the altered gene – by altering the DNA of genes in a special variety
of mouse cells, called embryonic stem cells, which were then cloned and used to
generate mice with the altered gene. We then studied the physiology
and behavior of the knockout mice using a comprehensive battery of advanced
medical technologies, each of which was adapted specifically for the analysis of
mouse physiology. This systematic use of these evaluative
technologies allowed us to discover, in vivo, the physiological
and behavioral functions of the genes we knocked out and assess the prospective
pharmaceutical utility of the potential drug targets encoded by the
corresponding human genes. The study of the effects of knocking out
genes in mice has historically proven to be a powerful tool for understanding
human genes because of the close similarity of gene function and physiology
between mice and humans, with approximately 99% of all human genes having a
counterpart in the mouse genome.
We engage
in programs for the discovery of potential small molecule drugs for those in vivo-validated drug
targets that we consider to have high pharmaceutical value. We have
established extensive internal small molecule drug discovery capabilities, in
which we use our own sophisticated libraries of drug-like chemical compounds in
high-throughput screening assays to identify “hits,” or chemical compounds
demonstrating activity, against these targets. We then employ
medicinal chemistry efforts to optimize the potency and selectivity of these
hits and to identify lead compounds for potential development. We
have established extensive internal capabilities to characterize the absorption,
distribution, metabolism and excretion of our potential drug candidates and
otherwise evaluate their safety in mammalian models in preparation for
preclinical and clinical development. In all of our drug discovery
programs, we use a parallel physiological analysis technology platform that we
used in the discovery of gene function to analyze the in vivo activity and safety
profiles of drug candidates in mice as part of our preclinical research
efforts.
Once we
identify a potential drug candidate, we initiate formal preclinical development
studies in preparation for regulatory filings for the commencement of human
clinical trials. We have established internal expertise in each of
the critical areas of preclinical and clinical development, including clinical
trial design, study implementation and oversight, and regulatory affairs, with
demonstrated experience by members of our clinical development team in the
successful implementation of Phase 1, 2 and 3 clinical trials and regulatory
approval for the commercialization of therapeutic products.
We
believe that our systematic, biology-driven approach and our underlying
technology platform provide us with substantial advantages over alternative
approaches to drug target discovery. In particular, we believe that
the comprehensive nature of our approach has allowed us to identify potential
drug targets within the context of mammalian physiology that might have been
missed by more narrowly focused efforts. We also believe our approach
is more likely to reveal those side effects that may be a direct result of
inhibiting or otherwise modulating the drug target and may limit the utility of
potential therapeutics directed at the drug target. We believe these
advantages have contributed to better target selection and, therefore, to a
greater likelihood of success for our drug discovery and development
efforts.
Our
Technology
The core
elements of our technology platform include our patented technologies for the
generation of knockout mice, our integrated platform of advanced medical
technologies for the systematic and comprehensive biological analysis of in vivo behavior and
physiology, and our specialized approach to medicinal chemistry and the
generation of high-quality, drug-like compound libraries.
Gene
Knockout Technologies
Our gene
targeting technology, which we have licensed from third parties for the life of
the licensed patents, enables us to generate highly-specific alterations in
targeted genes. The technology replaces DNA of a gene in a mouse
embryonic stem cell through a process known as homologous recombination to
disrupt the function of the targeted gene, permitting the generation of knockout
mice. By using this technology in combination with one or more
additional technologies, we are able to generate alterations that selectively
disrupt, or conditionally regulate, the function of the targeted gene for the
analysis of the gene’s function in selected tissues, at selected stages in the
animal’s development or at selected times in the animal’s life. We
can also use this technology to replace the targeted gene with its corresponding
human gene for use for preclinical research in our drug programs.
Our gene
trapping technology, which we invented and is covered by issued patents that we
own, is a high-throughput method of generating knockout mouse
clones. The technology uses genetically engineered retroviruses that
infect mouse embryonic stem cells in vitro, integrate into the
chromosome of the cell and disrupt the function of the gene into which it
integrates, permitting the generation of knockout mice. This process
also allows us to identify and catalogue each embryonic stem cell clone by DNA
sequence from the trapped gene and to select embryonic stem cell clones by DNA
sequence for the generation of knockout mice. We have used our gene
trapping technology in an automated process to create our OmniBank library of
more than 270,000 frozen gene knockout embryonic stem cell clones, each
identified by DNA sequence in a relational database.
Physiological
Analysis Technologies
We have
employed an integrated platform of advanced analytical technologies to rapidly
and systematically discover the physiological and behavioral effects resulting
from loss of gene function in the knockout mice we have generated using our gene
knockout technologies and catalogued those effects in our comprehensive and
relational LexVision database. These analyses include many of the
most sophisticated diagnostic technologies and tests currently available, many
of which might be found in a major medical center. Each of these
technologies was adapted specifically for the analysis of mouse
physiology. This state-of-the-art technology platform has enabled us
to assess the consequences of loss of gene function in a living mammal across a
wide variety of parameters relevant to human disease.
We employ
portions of this same physiological analysis technology to analyze the in vivo efficacy and safety
profiles of drug candidates in mice. We believe that this approach
will allow us, at an early stage, to identify and optimize drug candidates for
further preclinical and clinical development that demonstrate in vivo efficacy and to
distinguish side effects caused by a specific compound from the target-related
side effects that we defined using the same comprehensive series of
tests.
Chemistry
Technologies
We have
used solution-phase chemistry to generate our own diverse libraries of optically
pure compounds that are targeted against the same pharmaceutically-relevant gene
families that we addressed in our Genome5000 program. These libraries
have been built using highly robust and scalable organic reactions that allow us
to generate compound collections of great diversity and to specially tailor the
compound collections to address various therapeutic target
families. We designed these libraries by analyzing the chemical
structures of drugs that have been proven safe and effective against human
disease and using that knowledge in the design of scaffolds and chemical
building blocks for the generation of large numbers of new drug-like
compounds. When we identify a hit against one of our in vivo-validated targets, we
can rapidly reassemble these building blocks to create hundreds or thousands of
variations around the structure of the initial compound, enabling us to
accelerate our medicinal chemistry efforts. We have supplemented our
internally-generated compound libraries with collections of compounds acquired
from third parties. We have also established an alliance with
Nuevolution A/S providing us with access to Nuevolution’s Chemetics® platform
chemistry technology, allowing us to screen our targets against ultra-large
libraries of fragment-based chemical compounds.
Our
Commercialization Strategy
We are
working both independently and through strategic collaborations and alliances
with leading pharmaceutical and biotechnology companies, research institutes and
academic institutions to capitalize on our technology, drug target discoveries
and drug discovery and development programs. Consistent with this
approach, we seek to retain exclusive rights to the benefits of certain of our
small molecule programs by developing drug candidates from those programs
internally and to collaborate with third parties with respect to the discovery,
development and commercialization of small molecule and biotherapeutic drug
candidates for other targets, particularly when the collaboration provides us
with access to expertise and resources that we do not possess internally or are
complementary to our own.
Drug
Discovery and Development Collaborations
Bristol-Myers
Squibb. We established a drug discovery alliance with
Bristol-Myers Squibb Company in December 2003 to discover, develop and
commercialize small molecule drugs in the neuroscience
field. Bristol-Myers Squibb extended the target discovery term of the
alliance in May 2006. We initiated the alliance with a number of
neuroscience drug discovery programs at various stages of development and have
used our gene knockout technologies to identify additional drug targets with
promise in the neuroscience field. For those targets that were
selected for the alliance, we and Bristol-Myers Squibb are working together, on
an exclusive basis, to identify, characterize and carry out the preclinical
development of small molecule drugs, and share equally both in the costs and in
the work attributable to those efforts. As drugs resulting from the
alliance enter clinical trials, Bristol-Myers Squibb will have the first option
to assume full responsibility for clinical development and
commercialization.
We
received $86 million in upfront payments and research funding under the
agreement during the target discovery portion of the alliance, which expired in
October 2009. In addition, we are entitled to receive clinical and
regulatory milestone payments ranging, depending on the timing and extent of our
efforts in the alliance, up to $76 million for each drug developed by
Bristol-Myers Squibb under the alliance. We will also earn royalties
on sales of drugs commercialized by Bristol-Myers Squibb under the
alliance.
Genentech. We
established a drug discovery alliance with Genentech, Inc. in December 2002 to
discover novel therapeutic proteins and antibody targets. We and
Genentech expanded the alliance in November 2005 for the advanced research,
development and commercialization of new biotherapeutic drugs. Under
the original alliance agreement, we used our target validation technologies to
discover the functions of secreted proteins and potential antibody targets
identified through Genentech’s internal drug discovery research. In
the expanded alliance, we conducted additional, advanced research on a broad
subset of those proteins and targets. We have exclusive rights to
develop and commercialize biotherapeutic drugs for two of these targets, while
Genentech has exclusive rights to develop and commercialize biotherapeutic drugs
for the other targets. We retain certain other rights to discoveries
made in the alliance, including non-exclusive rights, along with Genentech, for
the development and commercialization of small molecule drugs addressing the
targets included in the alliance.
We
received $58 million in upfront payments, research funding and research
milestone payments under the agreement during the research collaboration term,
which expired in November 2008. In addition, we are entitled to
receive clinical and regulatory milestone payments ranging, depending on the
extent of our efforts in the alliance, up to $25 million for each drug target
for which Genentech develops a biotherapeutic drug under the
alliance. We will also earn royalties on sales of biotherapeutic
drugs commercialized by Genentech under the alliance. Genentech is
entitled to receive milestone payments and royalties on sales of biotherapeutic
drugs which we develop or commercialize under the alliance.
Schering-Plough/Organon. We
established a drug discovery alliance with N.V. Organon in May 2005 to discover,
develop and commercialize novel biotherapeutic drugs. In the
collaboration, we created and analyzed knockout mice for 300 genes selected by
the parties that encode secreted proteins or potential antibody targets,
including two of our preexisting drug discovery programs. We and
Organon agreed to equally share costs of and responsibility for research,
preclinical and clinical activities, jointly determine the manner in which
collaboration products would be commercialized, and equally benefit from product
revenue. Organon, formerly a subsidiary of Akzo Nobel N.V., was
acquired by Schering-Plough Corporation in November 2007, which subsequently
merged with Merck & Co., Inc. in November 2009. In February 2010,
we entered into a revised collaboration and license agreement with Organon and
Schering Corporation, acting through its Schering-Plough Research Institute
division, amending the terms of the alliance to provide that Schering-Plough
will assume the full cost of research activities conducted by either party in
the alliance, and will assume the full cost of and responsibility for
preclinical, clinical and commercialization activities with respect to
biotherapeutic drugs resulting from the alliance.
We
received $52.5 million in upfront payments and research funding under the
agreement during the target discovery portion of the alliance, which expired in
December 2009, and may receive additional research funding for any future work
we do under the alliance. In addition, we are entitled to receive clinical
and regulatory milestone payments of up to $39 million for each drug target for
which Schering-Plough develops a biotherapeutic drug under the
alliance. We will also earn royalties on sales of biotherapeutic
drugs commercialized by Schering-Plough under the alliance.
Takeda. We
established a drug discovery alliance with Takeda Pharmaceutical Company Limited
in July 2004 to discover new drugs for the treatment of high blood
pressure. In the collaboration, we used our gene knockout
technologies to identify drug targets that control blood
pressure. Takeda is responsible for the screening, medicinal
chemistry, preclinical and clinical development and commercialization of drugs
directed against targets selected for the alliance, and bears all related
costs.
We
received $18.5 million in upfront payments and research milestone payments under
the agreement during the target discovery portion of the alliance, which expired
in July 2007. In addition, we are entitled to receive clinical
development and product launch milestone payments of up to $29 million for each
drug developed by Takeda under the alliance. We will also earn
royalties on sales of drugs commercialized by Takeda under the
alliance.
Drug
Development Financing Collaborations
Symphony Icon. In
June 2007, we entered into a series of related agreements providing for the
financing of the clinical development of certain drug programs, including LX1031
and LX1032, along with any other pharmaceutical compositions modulating the same
targets as those drug candidates. Under the financing arrangement, we
licensed to Symphony Icon, a wholly-owned subsidiary of Holdings, our
intellectual property rights related to the programs and Holdings contributed
$45 million to Symphony Icon in order to fund the clinical development of the
programs. We also entered into a share purchase agreement with
Holdings under which we issued and sold to Holdings shares of our common stock
in exchange for $15 million and we received an exclusive option to acquire all
of the equity of Symphony Icon, thereby allowing us to reacquire the
programs. The purchase option is exercisable by us at any time, in
our sole discretion, until June 15, 2011 at an exercise price of (a) $81
million, if the purchase option is exercised before June 15, 2010 and (b) $90
million, if the purchase option is exercised on or after June 15, 2010 and
before June 15, 2011. The purchase option exercise price may be paid
in cash or a combination of cash and common stock, at our sole discretion,
provided that the common stock portion may not exceed 40% of the purchase option
exercise price.
We and
Symphony Icon are developing the programs in accordance with a specified
development plan and related development budget. We are the party
primarily responsible for the development of the programs. Our development
activities are supervised by Symphony Icon’s development committee, which is
comprised of an equal number of representatives from us and Symphony Icon. The
development committee reports to Symphony Icon’s board of directors, which is
currently comprised of five members, including one member we designated, two
members designated by Holdings, and two independent directors whom we and
Holdings selected mutually.
Upon the
recommendation of Symphony Icon’s development committee, Symphony Icon’s board
of directors may require us to pay Symphony Icon up to $15 million for Symphony
Icon’s use in the development of the programs in accordance with the specified
development plan and related development budget. The development
committee’s right to recommend that Symphony Icon’s board of directors submit
any further funding requirements to us will terminate on the one-year
anniversary of the expiration of the purchase option, subject to limited
exceptions. Through December 31, 2009, Symphony Icon’s board of
directors has requested us to pay Symphony Icon $4.3 million under the
agreement, all of which has been paid, and we expect that additional funding
will be needed in the future.
Other
Collaborations and Licenses
Texas Institute for Genomic
Medicine. In July 2005, we received an award from the Texas
Enterprise Fund for the creation of a knockout mouse embryonic stem cell library
containing 350,000 cell lines for the Texas Institute for Genomic Medicine, or
TIGM, using our proprietary gene trapping technology, which we completed in
2007. We also equipped TIGM with the bioinformatics software required
for the management and analysis of data relating to the library. The
Texas Enterprise Fund made an additional award to the Texas A&M University
System for the creation of facilities and infrastructure to house the
library. Under the terms of our award, we are responsible for the
creation of a specified number of jobs beginning in 2012, but will receive
credits against those job obligations based on funding received by TIGM and
certain related parties. We may be required to repay the state a
portion of the award if we fail to meet those job obligations.
Taconic Farms. We
established a collaboration with Taconic Farms, Inc. in November 2005 for the
marketing, distribution and licensing of certain lines of our knockout mice and
entered into an expanded collaboration with Taconic in July
2009. Taconic is an industry leader in the breeding, housing, quality
control and global marketing and distribution of rodent models for medical
research and drug discovery. Under the terms of the collaboration, we
are presently making available through Taconic more than 3,600 distinct lines of
knockout mice, and in some cases, phenotypic data relating to such lines of
knockout mice, for use by pharmaceutical and biotechnology companies, academic
and non-profit institutions and other researchers. We receive license
fees and royalties from payments received by Taconic from customers obtaining
access to knockout mice and any related phenotypic data.
Research
Collaborations. We have established research collaborations
with a number of leading pharmaceutical and biotechnology companies, research
institutes and academic institutions under which we received fees in exchange
for generating knockout mice for genes requested by the collaborator, providing
phenotypic data with respect to such knockout mice or otherwise granting access
to some of our technologies and discoveries. In some cases, we are
also eligible to receive milestone and royalty payments on products that our
collaborators discover or develop using our technology.
Technology
Licenses. We have granted non-exclusive, internal research-use
sublicenses under certain of our gene targeting patent rights to a number of
leading pharmaceutical and biotechnology companies. Many of these
agreements extend for the life of the patents. Others have terms of
one to three years, in some cases with provisions for subsequent
renewals. We have typically received up-front license fees and, in
some cases, received additional license fees or milestone payments on products
that the sublicensee discovers or developed using our technology.
Our
Executive Officers
Our
executive officers and their ages and positions are listed below.
|
Name
|
Age
|
Position with the Company
|
|
Arthur
T. Sands, M.D., Ph.D.
|
48
|
President
and Chief Executive Officer and Director
|
|
Alan
J. Main, Ph.D.
|
56
|
Executive
Vice President of Pharmaceutical Research
|
|
Jeffrey
L. Wade, J.D.
|
45
|
Executive
Vice President and General Counsel
|
|
Brian
P. Zambrowicz, Ph.D.
|
47
|
Executive
Vice President and Chief Scientific Officer
|
|
Philip
M. Brown, M.D., J.D.
|
48
|
Senior
Vice President of Clinical Development
|
|
Steven
A. Tragash
|
63
|
Senior
Vice President of Corporate Affairs
|
|
James
F. Tessmer
|
50
|
Vice
President, Finance and Accounting
|
Arthur T. Sands, M.D., Ph.D.
co-founded our company and has been our president and chief executive officer
and a director since September 1995. At Lexicon, Dr. Sands pioneered the
development of large-scale gene knockout technology for use in drug
discovery. Before founding our company, Dr. Sands served as an
American Cancer Society postdoctoral fellow in the Department of Human and
Molecular Genetics at Baylor College of Medicine. Dr. Sands received
his B.A. in economics and political science from Yale University and his M.D.
and Ph.D. from Baylor College of Medicine.
Alan J. Main, Ph.D. has been
our executive vice president of pharmaceutical research since February 2007 and
served as our senior vice president, Lexicon Pharmaceuticals from July 2001
until February 2007. Dr. Main was president and chief executive
officer of Coelacanth Corporation, a leader in using proprietary chemistry
technologies to rapidly discover new chemical entities for drug development,
from January 2000 until our acquisition of Coelacanth in
July 2001. Dr. Main was formerly senior vice president,
U.S. Research at Novartis Pharmaceuticals Corporation, where he worked for
20 years before joining Coelacanth. Dr. Main holds a B.S.
from the University of Aberdeen, Scotland and a Ph.D. in organic chemistry from
the University of Liverpool, England and completed postdoctoral studies at the
Woodward Research Institute.
Jeffrey L. Wade, J.D. has
been our executive vice president and general counsel since February 2000
and was our senior vice president and chief financial officer from
January 1999 to February 2000. From 1988 through
December 1998, Mr. Wade was a corporate securities and finance
attorney with the law firm of Andrews & Kurth L.L.P., for the last
two years as a partner, where he represented companies in the biotechnology,
information technology and energy industries. Mr. Wade is a
member of the boards of directors of the Texas Healthcare and Bioscience
Institute and the Texas Life Science Center for Innovation and
Commercialization. He received his B.A. and J.D. from the University
of Texas.
Brian P. Zambrowicz, Ph.D.
co-founded our company and has been our executive vice president and chief
scientific officer since February 2007. Dr. Zambrowicz served as our
executive vice president of research from August 2002 until February 2007,
senior vice president of genomics from February 2000 to August 2002,
vice president of research from January 1998 to February 2000 and
senior scientist from April 1996 to January 1998. From 1993
to April 1996, Dr. Zambrowicz served as a National Institutes of
Health postdoctoral fellow at the Fred Hutchinson Cancer Center in Seattle,
Washington, where he studied gene trapping and gene targeting
technology. Dr. Zambrowicz received his B.S. in biochemistry
from the University of Wisconsin. He received his Ph.D. from the
University of Washington, where he studied tissue-specific gene regulation using
transgenic mice.
Philip M. Brown, M.D., J.D.
has been our senior vice president of clinical development since February 2008
and was our vice president of clinical development from April 2003 to
February 2008. Dr. Brown served as vice president of clinical
development for Encysive Pharmaceuticals Inc. (formerly Texas Biotechnology
Corporation), a biopharmaceutical company, from June 2000 until
April 2003, and was senior medical director within the organization from
December 1998 until June 2000. From July 1994 to
December 1998, Dr. Brown served as associate vice president of medical
affairs for Pharmaceutical Research Associates, a clinical research
organization. He has conducted numerous clinical trials as an investigator in a
variety of therapeutic areas, as well as managed programs from IND through NDA
and product commercialization. He is a fellow of the American College of Legal
Medicine and serves as an adjunct faculty member at the Massachusetts General
Hospital, Institute of Health Professions in Boston. He received his B.A. from
Hendrix College, his M.D. from Texas Tech University School of Medicine, and his
J.D. from the University of Texas.
Steven A. Tragash has been
our senior vice president of corporate affairs since February 2010 and
previously served as our vice president of corporate affairs from February 2009
to February 2010 and executive director of corporate affairs from March 2008 to
February 2009. Mr. Tragash was vice president and director of Guidant
Corporation from February 2001 to July 2007 and previously served as president
and chief executive officer of Strategica Management Consulting and executive
vice president, corporate relations and marketing of Glendale Federal Savings
Bank. Mr. Tragash received his B.S. from Bowling Green
University.
James F. Tessmer has been our
vice president, finance and accounting since November 2007 and previously served
as our senior director of finance from February 2004 to November 2007 and
director of finance from April 2001 to February 2004. From January
1997 to April 2001, Mr. Tessmer was assistant controller for Mariner Health
Network, Inc. and prior to that served in a variety of financial and accounting
management positions for HWC Distribution Corp. and American General
Corporation. Mr. Tessmer is a certified public accountant and
received his B.B.A. from the University of Wisconsin – Milwaukee and his M.B.A.
from the University of Houston.
Patents
and Proprietary Rights
We will
be able to protect our proprietary rights from unauthorized use by third parties
only to the extent that those rights are covered by valid and enforceable
patents or are effectively maintained as trade secrets. Accordingly, patents and
other proprietary rights are an essential element of our business. We
seek patent protection for all chemical compounds, antibodies and other
potential therapeutic agents that we discover and their use in treating human
diseases and conditions. We own patent applications, and in some
cases issued patents, covering each of our drug candidates in clinical and
preclinical development, including:
|
|
·
|
patent
applications pending worldwide that claim LX1031 and associated
crystalline forms, pharmaceutical compositions, and methods of manufacture
and use, which we have exclusively licensed to Symphony Icon pursuant to
our product development collaboration with Symphony
Icon.
|
|
|
·
|
patent
applications pending worldwide that claim LX4211 and associated
crystalline forms, pharmaceutical compositions, and methods of manufacture
and use.
|
|
|
·
|
patent
applications pending worldwide that claim LX2931 and associated
crystalline forms, pharmaceutical compositions, and methods of manufacture
and use, from which one U.S. patent has issued to
date.
|
|
|
·
|
patent
applications pending worldwide that claim LX1032 and associated
crystalline forms, pharmaceutical compositions, and methods of manufacture
and use, from which one U.S. patent has issued to date, which we have
exclusively licensed to Symphony Icon pursuant to our product development
collaboration with Symphony Icon.
|
|
|
·
|
patent
applications pending worldwide that claim LX7101 and associated
pharmaceutical compositions, and methods of manufacture and
use.
|
We
additionally seek patent protection for:
|
|
·
|
the
sequences of genes that we believe to be novel, the proteins they encode
and their predicted utility as a drug target or therapeutic
protein;
|
|
|
·
|
the
utility of genes and the drug targets or proteins they encode based on our
discoveries of their biological functions using knockout
mice;
|
|
|
·
|
drug
discovery assays for our in vivo-validated
targets; and
|
|
|
·
|
various
enabling technologies in the fields of mutagenesis, embryonic stem cell
manipulation and transgenic or knockout
mice.
|
Additionally,
we hold rights to a number of patents and patent applications under license
agreements with third parties. Many of these licenses are
nonexclusive, although some are exclusive in specified fields. Most
of the licenses have terms that extend for the life of the licensed
patents.
Patents
extend for varying periods according to the date of patent filing or grant and
the legal term of patents in the various countries where patent protection is
obtained. The actual protection afforded by a patent, which can vary from
country to country, depends on the type of patent, the scope of its coverage and
the availability of legal remedies in the country. We have filed
patent applications and, in some cases, hold issued patents covering each of our
drug candidates in clinical and preclinical development. While the
patents issued under such applications have or will have a variety of expiration
dates, depending on, among other things, the date of filing and possible patent
term extension, no U.S. patent that has issued or may issue based on a patent
application we have filed relating to one of our drug candidates in clinical or
preclinical development has a normal expiration date earlier than
2026.
All of
our employees, consultants and advisors are required to execute a proprietary
information agreement upon the commencement of employment or consultation. In
general, the agreement provides that all inventions conceived by the employee or
consultant, and all confidential information developed or made known to the
individual during the term of the agreement, shall be our exclusive property and
shall be kept confidential, with disclosure to third parties allowed only in
specified circumstances. We cannot assure you, however, that these agreements
will provide useful protection of our proprietary information in the event of
unauthorized use or disclosure of such information.
Competition
The
biotechnology and pharmaceutical industries are highly competitive and
characterized by rapid technological change. We face significant
competition in each of the aspects of our business from other pharmaceutical and
biotechnology companies. In addition, a large number of universities
and other not-for-profit institutions, many of which are funded by the U.S. and
foreign governments, are also conducting research to discover genes and their
functions and to identify potential therapeutic products. Many of our
competitors have substantially greater research and product development
capabilities and financial, scientific, marketing and human resources than we
do. As a result, our competitors may succeed in developing products
earlier than we do, obtaining approvals from the FDA or other regulatory
agencies for those products more rapidly than we do, or developing products that
are more effective than those we propose to develop. Similarly, our
collaborators face similar competition from other competitors who may succeed in
developing products more quickly, or developing products that are more
effective, than those developed by our collaborators. Any products
that we may develop or discover are likely to be in highly competitive
markets.
The
competition for our drug candidates includes both marketed products and drug
candidates that are being developed by others, including drug candidates that
are currently in a more advanced stage of clinical development than are our own
drug candidates. These competitive marketed products and drug
candidates include compounds that employ different mechanisms of action in
addressing diseases and conditions for which we are developing our own drug
candidates and, in some cases such as LX4211, that employ the same or similar
mechanisms of action.
We
believe that our ability to successfully compete with these potentially
competitive drug candidates and other competitive products currently on the
market will depend on, among other things:
|
|
·
|
the
efficacy, safety and reliability of our drug
candidates;
|
|
|
·
|
our
ability, and the ability of our collaborators, to complete preclinical
testing and clinical development and obtain regulatory approvals for our
drug candidates;
|
|
|
·
|
the
timing and scope of regulatory approvals for our drug
candidates;
|
|
|
·
|
our
ability, and the ability of our collaborators, to obtain product
acceptance by physicians and other health care providers and reimbursement
for product use in approved
indications;
|
|
|
·
|
our
ability, and the ability of our collaborators, to manufacture and sell
commercial quantities of our
products;
|
|
|
·
|
the
skills of our employees and our ability to recruit and retain skilled
employees;
|
|
|
·
|
protection
of our intellectual property; and
|
|
|
·
|
the
availability of substantial capital resources to fund development and
commercialization activities.
|
Government
Regulation
Regulation
of Pharmaceutical Products
The
development, manufacture and sale of any drug or biologic products developed by
us or our collaborators will be subject to extensive regulation by United States
and foreign governmental authorities, including federal, state and local
authorities. In the United States, new drugs are subject to
regulation under the Federal Food, Drug and Cosmetic Act and the regulations
promulgated thereunder, or the FDC Act, and biologic products are subject to
regulation both under certain provisions of the FDC Act and under the Public
Health Services Act and the regulations promulgated thereunder, or the PHS
Act. The FDA regulates, among other things, the development,
preclinical and clinical testing, manufacture, safety, efficacy, record keeping,
reporting, labeling, storage, approval, advertising, promotion, sale,
distribution and export of small molecule and biotherapeutic drugs.
The
standard process required by the FDA before a drug candidate may be marketed in
the United States includes:
|
|
·
|
preclinical
laboratory and animal tests performed under the FDA’s current Good
Laboratory Practices regulations;
|
|
|
·
|
submission
to the FDA of an Investigational New Drug application, or IND, which must
become effective before human clinical trials may
commence;
|
|
|
·
|
adequate
and well-controlled human clinical trials to establish the safety and
efficacy of the drug candidate for its intended
use;
|
|
|
·
|
for
drug candidates regulated as small molecule drugs, submission of a New
Drug Application, or NDA, and, for drug candidates regulated as
biotherapeutic drugs, submission of a Biologic License Application, or
BLA, with the FDA; and
|
|
|
·
|
FDA
approval of the NDA or BLA prior to any commercial sale or shipment of the
product.
|
This
process for the testing and approval of drug candidates requires substantial
time, effort and financial resources. Preclinical development of a
drug candidate can take from one to several years to complete, with no guarantee
that an IND based on those studies will become effective to even permit clinical
testing to begin. Before commencing the first clinical trial of a
drug candidate in the United States, we must submit an IND to the
FDA. The IND automatically becomes effective 30 days after receipt by
the FDA, unless the FDA, within the 30-day time period, raises concerns or
questions about the conduct of the clinical trial. In such a case, we
and the FDA must resolve any outstanding concerns before the clinical trial may
begin. A separate submission to the existing IND must be made for
each successive clinical trial conducted during product development, and the FDA
must grant permission for each clinical trial to start and
continue. Further, an independent institutional review board for each
medical center proposing to participate in the clinical trial must review and
approve the plan for any clinical trial before it commences at that
center. Regulatory authorities or an institutional review board or
the sponsor may suspend a clinical trial at any time on various grounds,
including a finding that the subjects or patients are being exposed to an
unacceptable health risk.
For
purposes of NDA or BLA approval, human clinical trials are typically conducted
in three sequential phases that may overlap.
|
|
·
|
Phase
1 clinical trials are conducted in a limited number of healthy human
volunteers or, in some cases, patients, to evaluate the safety, dosage
tolerance, absorption, metabolism, distribution and excretion of the drug
candidate;
|
|
|
·
|
Phase
2 clinical trials are conducted in groups of patients afflicted with a
specified disease or condition to obtain preliminary data regarding
efficacy as well as to further evaluate safety and optimize dosing of the
drug candidate; and
|
|
|
·
|
Phase
3 clinical trials are conducted in larger patient populations at multiple
clinical trial sites to obtain statistically significant evidence of the
efficacy of the drug candidate for its intended use and to further test
for safety in an expanded patient
population.
|
In
addition, the FDA may require, or companies may pursue, additional clinical
trials after a product is approved. These so-called Phase 4 studies
may be made a condition to be satisfied after a drug receives
approval.
Completion
of the clinical trials necessary for an NDA or BLA submission typically takes
many years, with the actual time required varying substantially based on, among
other things, the nature and complexity of the drug candidate and of the disease
or condition. Success in earlier-stage clinical trials does not
ensure success in later-stage clinical trials. Furthermore, data
obtained from clinical activities is not always conclusive and may be
susceptible to varying interpretations, which could delay, limit or prevent
proceeding with further clinical trials, filing or acceptance of an NDA or BLA,
or obtaining marketing approval.
After
completion of clinical trials, FDA approval of an NDA or BLA must be obtained
before a new drug may be marketed in the United States. An NDA or
BLA, depending on the submission, must contain, among other things, information
on chemistry, manufacturing controls and potency and purity, non-clinical
pharmacology and toxicology, human pharmacokinetics and bioavailability and
clinical data. There can be no assurance that the FDA will accept an
NDA or BLA for filing and, even if filed, that approval will be
granted. Among other things, the FDA reviews an NDA to determine
whether a product is safe and effective for its intended use and a BLA to
determine whether a product is safe, pure and potent and the facility in which
it is manufactured, processed, packed, or held meets standards designed to
assure the product’s continued safety, purity and potency. The FDA
may deny approval of an NDA or BLA if the applicable regulatory criteria are not
satisfied, or it may require additional clinical data or an additional pivotal
Phase 3 clinical trial. Even if such data are submitted, the FDA may
ultimately decide that the NDA or BLA does not satisfy the criteria for
approval. Once issued, the FDA may withdraw product approval if
ongoing regulatory standards are not met or if safety problems occur after the
product reaches the market. In addition, the FDA may require testing
and surveillance programs to monitor the effect of approved products which have
been commercialized, and the FDA has the power to prevent or limit further
marketing of a product based on the results of these post-marketing
programs. Limited indications for use or other conditions could also
be placed on any approvals that could restrict the commercial applications of a
product or impose costly procedures in connection with the commercialization or
use of the product.
In
addition to obtaining FDA approval for each product, each drug manufacturing
establishment must be inspected and approved by the FDA. All
manufacturing establishments are subject to inspections by the FDA and by other
federal, state and local agencies and must comply with current Good
Manufacturing Practices requirements. Non-compliance with these
requirements can result in, among other things, total or partial suspension of
production, failure of the government to grant approval for marketing and
withdrawal, suspension or revocation of marketing approvals.
Once the
FDA approves a product, a manufacturer must provide certain updated safety and
efficacy information. Product changes as well as certain changes in a
manufacturing process or facility would necessitate additional FDA review and
approval. Other post-approval changes may also necessitate further
FDA review and approval. Additionally, a manufacturer must meet other
requirements including those related to adverse event reporting and record
keeping.
The FDA
closely regulates the marketing and promotion of drugs. A company can
make only those claims relating to safety and efficacy that are approved by the
FDA. Failure to comply with these requirements can result in adverse
publicity, warning letters, corrective advertising and potential civil and
criminal penalties.
Violations
of the FDC Act, the PHS Act or regulatory requirements may result in agency
enforcement action, including voluntary or mandatory recall, license suspension
or revocation, product seizure, fines, injunctions and civil or criminal
penalties.
In
addition to regulatory approvals that must be obtained in the United States,
drugs are also subject to regulatory approval in other countries in which they
are marketed. The conduct of clinical trials of drugs in countries
other than the United States is likewise subject to regulatory oversight in such
countries. The requirements governing the conduct of clinical trials,
product licensing, pricing, and reimbursement vary widely from country to
country. No action can be taken to market any drug in a country until
the regulatory authorities in that country have approved an appropriate
application. FDA approval does not assure approval by other
regulatory authorities. The current approval process varies from
country to country, and the time spent in gaining approval varies from that
required for FDA approval. In some countries, the sale price of a
drug or biologic product must also be approved. The pricing review
period often begins after marketing approval is granted. Even if a
foreign regulatory authority approves a drug, it may not approve satisfactory
prices for the product.
Other
Regulations
In
addition to the foregoing, our business is and will be subject to regulation
under various state and federal environmental laws, including the Occupational
Safety and Health Act, the Resource Conservation and Recovery Act and the Toxic
Substances Control Act. These and other laws govern our use, handling
and disposal of various biological, chemical and radioactive substances used in
and wastes generated by our operations. We believe that we are in
material compliance with applicable environmental laws and that our continued
compliance with these laws will not have a material adverse effect on our
business. We cannot predict, however, whether new regulatory
restrictions will be imposed by state or federal regulators and agencies or
whether existing laws and regulations will adversely affect us in the
future.
Research
and Development Expenses
In 2009,
2008 and 2007, respectively, we incurred expenses of $81.2 million,
$107.2 million and $103.2 million in company-sponsored as well as
collaborative research and development activities, including $3.0 million,
$3.9 million and $5.2 million of stock-based compensation expense in
2009, 2008 and 2007, respectively.
Employees
and Consultants
We
believe that our success will be based on, among other things, achieving and
retaining scientific and technological superiority and identifying and retaining
capable management. We have assembled a highly qualified team of
scientists as well as executives with extensive experience in the biotechnology
industry.
As of
February 28, 2010, we employed 345 persons, of whom 91 hold M.D., Ph.D. or
D.V.M. degrees and another 51 hold other advanced degrees. We believe
that our relationship with our employees is good.
Item
1A. Risk
Factors
The
following risks and uncertainties are important factors that could cause actual
results or events to differ materially from those indicated by forward-looking
statements. The factors described below are not the only ones we face
and additional risks and uncertainties not presently known to us or that we
currently deem immaterial also may impair our business operations.
Risks
Related to Our Need for Additional Financing and Our Financial
Results
We
will need additional capital in the future and, if it is unavailable, we will be
forced to significantly curtail or cease our operations. If it is not
available on reasonable terms, we will be forced to obtain funds by entering
into financing agreements on unattractive terms.
As of
December 31, 2009, we had $157.1 million in cash, cash equivalents and
investments, including $56.0 million of auction rate securities and related
rights, and $5.4 million in investments held by Symphony
Icon. We anticipate that our existing capital resources and the cash
and revenues we expect to derive from collaborations, technology licenses and
other sources will enable us to fund our currently planned operations for at
least the next 12 months. Our currently planned operations for that
time period consist of the completion of our ongoing clinical trials, the
initiation and conduct of additional clinical trials and the continuation of our
small molecule drug discovery and preclinical research
efforts. However, we caution you that we may generate less cash and
revenues or incur expenses more rapidly than we currently
anticipate.
Although
difficult to accurately predict, the amount of our future capital requirements
will be substantial and will depend on many factors, including:
|
|
·
|
our
ability to obtain additional funds from collaborations, technology
licenses and other sources;
|
|
|
·
|
the
amount and timing of payments under such
agreements;
|
|
|
·
|
the
level and timing of our research and development
expenditures;
|
|
|
·
|
the
timing and progress of the clinical development of our drug candidates
LX1031, LX4211, LX2931 and LX1032;
|
|
|
·
|
our
election whether to exercise our exclusive option to acquire all of the
equity of Symphony Icon, thereby allowing us to reacquire LX1031 and
LX1032;
|
|
|
·
|
future
results from clinical trials of our drug
candidates;
|
|
|
·
|
the
cost and timing of regulatory approvals of drug candidates that we
successfully develop;
|
|
|
·
|
market
acceptance of products that we successfully develop and commercially
launch;
|
|
|
·
|
the
effect of competing programs and products, and of technological and market
developments;
|
|
|
·
|
the
filing, maintenance, prosecution, defense and enforcement of patent claims
and other intellectual property rights;
and
|
|
|
·
|
the
cost and timing of establishing or contracting for sales, marketing and
distribution capabilities.
|
Our
capital requirements will increase substantially as we advance our drug
candidates into more advanced stage clinical development. Our capital
requirements will also be affected by any expenditures we make in connection
with license agreements and acquisitions of and investments in complementary
products and technologies. For all of these reasons, our future
capital requirements cannot easily be quantified.
If our
capital resources are insufficient to meet future capital requirements, we will
need to raise additional funds to continue our currently planned
operations. If we raise additional capital by issuing equity
securities, our then-existing stockholders will experience dilution and the
terms of any new equity securities may have preferences over our common
stock. We cannot be certain that additional financing, whether debt
or equity, will be available in amounts or on terms acceptable to us, if at
all. We may be unable to raise sufficient additional capital on
reasonable terms, and if so, we will be forced to significantly curtail or cease
our operations or obtain funds by entering into financing agreements on
unattractive terms.
Invus,
L.P., our largest stockholder, may decline to grant its consent which is
required for us to conduct additional equity offerings at prices less than $4.50
per share. In addition, we can provide no assurance that Invus will
exercise its rights to require us to initiate up to two pro rata rights
offerings in which it would be obligated to purchase its pro rata portion of the
offering.
In June
2007, we entered into a securities purchase agreement with Invus, L.P., under
which Invus made an initial investment of $205.4 million to purchase 50,824,986
shares of our common stock in August 2007 and has the right to require us to
initiate up to two pro rata rights offerings to our stockholders, which would
provide all stockholders with non-transferable rights to acquire shares of our
common stock, in an aggregate amount of up to $344.5 million, less the
proceeds of any “qualified offerings” that we may complete in the interim
involving the sale of our common stock at prices above $4.50 per
share. We have not completed any such qualified
offering. Invus may exercise its right to require us to conduct the
first rights offering by giving us notice within a period of one year beginning
on November 28, 2009 (which we refer to as the first rights offering
trigger date). Invus may exercise its right to require us to conduct
a second rights offering by giving us notice within a period of one year
beginning on the date that is 90 days after Invus’ exercise of its right to
require us to conduct the first rights offering or, if Invus does not exercise
its right to require us to conduct the first rights offering, within a period of
one year beginning on the first anniversary of the first rights offering trigger
date. If Invus elects to exercise its right to require us to initiate
a rights offering, Invus would be required to purchase its pro rata portion of
the offering.
Under the
securities purchase agreement, until the later of the completion of the second
rights offering or the expiration of the period following the second rights
offering trigger date during which Invus may require us to initiate the second
rights offering, we have agreed not to issue any of our common stock for a per
share price of less than $4.50 without the prior written consent of Invus,
except pursuant to an employee or director stock option, incentive compensation
or similar plan or to persons involved in the pharmaceutical industry in
connection with simultaneous strategic transactions involving such persons in
the ordinary course. In addition, if we notify Invus of a proposed
public offering for an offering above $4.50 per share during the period in which
Invus may initiate a rights offering, Invus will have a period of 10 business
days in which to exercise its right to require us to conduct a rights offering,
in which case we would be required to forego the proposed public offering and
proceed with the rights offering. If we are not able to issue common
stock at prices equal to or greater than $4.50 per share in the future, due to
market conditions or otherwise, this obligation will limit our ability to raise
capital by issuing additional equity securities without the consent of
Invus. In the event Invus declines to grant such consent and, in
addition, elects not to exercise its right to require us to initiate the first
rights offering, or elects to limit the size of the first rights offering, our
ability during this period to satisfy our future capital requirements by issuing
equity securities will be limited if we are unable to do so by issuing common
stock at prices equal to or greater than $4.50 per share.
We
have a history of net losses, and we expect to continue to incur net losses and
may not achieve or maintain profitability.
We have
incurred net losses since our inception, including net losses of
$82.8 million for the year ended December 31, 2009, $76.9 million for
the year ended December 31, 2008 and $58.8 million for the year ended
December 31, 2007. As of December 31, 2009, we had an
accumulated deficit of $570.2 million. We are unsure when we
will become profitable, if ever. The size of our net losses will
depend, in part, on the rate of decline or growth in our revenues and on the
level of our expenses.
We derive
substantially all of our revenues from drug discovery and development
collaborations and other collaborations and technology licenses, and will
continue to do so for the foreseeable future. Our future revenues
from collaborations and technology licenses are uncertain because our existing
agreements have fixed terms or relate to specific projects of limited duration
and future revenues from such agreements, if any, depend on the achievement of
milestones and payment of royalties we earn from any future products developed
under the collaborations. As a result, we depend, in part, on
securing new collaboration and license agreements. Our ability to
secure future revenue-generating agreements will depend upon our ability to
address the needs of our potential future collaborators and licensees, and to
negotiate agreements that we believe are in our long-term best
interests. We may determine, as we have to date with respect to our
four clinical drug candidates, that our interests are better served by retaining
rights to our discoveries and advancing our therapeutic programs to a later
stage, which could limit our near-term revenues. Given the
early-stage nature of our operations, we do not currently derive any revenues
from sales of pharmaceutical products.
A large
portion of our expenses is fixed, including expenses related to facilities,
equipment and personnel. In addition, we expect to spend significant
amounts to fund our research and development activities, including the conduct
of clinical trials and the advancement of additional potential therapeutics into
clinical development. As a result, we expect that our operating
expenses will continue to increase significantly as our drug programs progress
into and through human clinical trials and, consequently, we will need to
generate significant additional revenues to achieve
profitability. Even if we do achieve profitability, we may not be
able to sustain or increase profitability on a quarterly or annual
basis.
We
have licensed the intellectual property, including commercialization rights, to
our drug candidates LX1031 and LX1032 to Symphony Icon and will not receive any
future royalties or revenues with respect to these drug candidates unless we
exercise our option to purchase Symphony Icon.
Our
option to purchase all of the equity of Symphony Icon, thereby allowing us to
reacquire these drug candidates, is exercisable by us at any time, in our sole
discretion, until June 15, 2011 at an exercise price of (a) $81
million, if the purchase option is exercised before June 15, 2010 and (b) $90
million, if the purchase option is exercised on or after June 15, 2010 and
before June 15, 2011. The purchase option exercise price may be paid
in cash or a combination of cash and common stock, at our sole discretion,
provided that the common stock portion may not exceed 40% of the purchase option
exercise price. Any such issuance of common stock may also be subject
to Invus providing its consent to such issuance as required by our securities
purchase agreement with Invus.
If we
elect to exercise the purchase option, we will be required to make a substantial
cash payment or to make a lesser but still substantial cash payment and issue a
substantial number of shares of our common stock, which may in turn require us
to enter into a financing arrangement or license arrangement with one or more
third parties. The amount of any such cash payment would reduce our
capital resources. Payment in shares of our common stock could result
in dilution to our stockholders at that time. Other financing or
licensing alternatives may be expensive or impossible to obtain. If
we do not exercise the purchase option prior to its expiration, our rights to
purchase all of the equity in Symphony Icon and to reacquire LX1031 and LX1032
will terminate. We may not have the financial resources to exercise
the option, which may result in our loss of these
rights. Additionally, we may not receive clinical data from future
clinical trials of LX1031 before the expiration of our option on June 15,
2011 or the clinical data available to us may otherwise be insufficient for us
to make a determination of whether we should exercise the option prior to June
15, 2010 or June 15, 2011.
At
December 31, 2009, we held $56.2 million (par value), with an
estimated fair value of $46.3 million, of auction rate securities for which
auctions have failed and, as a result, we may not be able to access at least a
portion of these funds without a loss of principal.
At
December 31, 2009, we held $56.2 million (par value), with an estimated
fair value of $46.3 million, of investments with an auction interest rate
reset feature, known as auction rate securities. Until February 2008,
the market for our auction rate securities was highly
liquid. However, starting in February 2008, a substantial number of
auctions “failed,” meaning that there was not enough demand to sell all of the
securities that holders desired to sell at auction.
In
November 2008, we accepted an offer from UBS AG, the investment bank that sold
us our auction rate securities, providing us with certain rights related to our
auction rate securities. The rights permit us to require UBS to
purchase our auction rate securities from us at par value during the period from
June 30, 2010 through July 2, 2012. Conversely, UBS has the
right, in its discretion, to purchase or sell the securities at any time by
paying us the par value of the securities. In connection with our
acceptance of UBS’s offer, in January 2009, we entered into a credit line
agreement with UBS Bank USA that provides, as of December 31, 2009, up to
an aggregate amount of $37.5 million in the form of an uncommitted, demand,
revolving line of credit. The credit line is secured only by the
auction rate securities and advances under the credit line will be made on a “no
net cost” basis, meaning that the interest paid by us on advances will not
exceed the interest or dividends paid to us by the issuer of the auction rate
securities. As of December 31, 2009, we had $37.4 million
outstanding under this credit line.
Although
we have accessed substantially the maximum amount permitted under the credit
line and expect to exercise the rights and sell our auction rate securities to
UBS on June 30, 2010, the earliest date allowable under the rights, we will
have no means to access approximately $18.7 million (par value), as of
December 31, 2009, invested in auction rate securities before such date
without a loss of principal. Further, UBS and its affiliates may not
be able to maintain the financial resources necessary to perform its obligations
under the rights or credit line. UBS and the Swiss government are
currently engaged in discussions with the United States government regarding the
disclosure, pursuant to an August 2009 settlement between UBS and the United
States government, of the identities of certain UBS customers subject to an
ongoing investigation of tax fraud, the outcome of which could also impact UBS’
ability to perform its obligations under the rights or credit
line. As a result, we cannot provide any assurance that we will be
able to access the funds invested in auction rate securities without a loss of
principal, unless a future auction on these investments is successful or the
issuer redeems the securities.
Our
operating results have been and likely will continue to fluctuate, and we
believe that period-to-period comparisons of our operating results are not a
good indication of our future performance.
Our
operating results and, in particular, our ability to generate additional
revenues are dependent on many factors, including:
|
|
·
|
our
ability to establish new collaborations and technology licenses, and the
timing of such arrangements;
|
|
|
·
|
the
expiration or other termination of collaborations and technology licenses,
which may not be renewed or
replaced;
|
|
|
·
|
the
success rate of our discovery and development efforts leading to
opportunities for new collaborations and licenses, as well as milestone
payments and royalties;
|
|
|
·
|
the
timing and willingness of our collaborators to commercialize
pharmaceutical products that would result in milestone payments and
royalties; and
|
|
|
·
|
general
and industry-specific economic conditions, which may affect our and our
collaborators’ research and development
expenditures.
|
Because
of these and other factors, including the risks and uncertainties described in
this section, our operating results have fluctuated in the past and are likely
to do so in the future. Due to the likelihood of fluctuations in our
revenues and expenses, we believe that period-to-period comparisons of our
operating results are not a good indication of our future
performance.
Risks
Related to Discovery and Development of Our Drug Candidates
We
are an early-stage company, and have not proven our ability to successfully
develop and commercialize drug candidates based on our drug target
discoveries.
Our
business strategy of using our discovery of the functions of genes using
knockout mice to select promising drug targets and developing and
commercializing drug candidates based on our target discoveries, in significant
part through collaborations, is unproven. Our success will depend
upon our ability to successfully generate, select and develop drug candidates
for targets we consider to have pharmaceutical value, whether on our own or
through collaborations, and to select an appropriate commercialization strategy
for each potential therapeutic we choose to pursue.
We have
not proven our ability to develop or commercialize drug candidates based on our
drug target discoveries. The generation and selection of potential
drug candidates for a target is a difficult, expensive and time-consuming
process that is subject to substantial technical and scientific challenges and
uncertainties, without any assurance of ever identifying a drug candidate
warranting clinical testing. The process involves the optimization of
a wide variety of variables, including among many other things potency against
the target, selectivity for the intended target relative to other proteins,
absorption, metabolism, distribution and excretion characteristics, activity in
animal models of disease and the results of other preclinical research, and
feasibility and cost of manufacture, each of which may affect one or more of the
others in ways that conflict with the desired profile.
Furthermore,
we do not know that any pharmaceutical products based on our drug target
discoveries can be successfully developed or commercialized. Our
strategy is focused principally on the discovery and development of drug
candidates for targets that have not been clinically validated in humans by
drugs or drug candidates generated by others. As a result, the drug
candidates we develop are subject to uncertainties as to the effects of
modulating the human drug target as well as to those relating to the
characteristics and activity of the particular compound. For example, we
are presently seeking to develop an improved formulation of LX1031 in
preparation for use in future clinical trials and cannot provide assurance that
we will be able to develop a commercially viable formulation.
In
addition, we may experience unforeseen technical complications in the processes
we use to identify potential drug targets or discover and develop potential drug
candidates. These complications could materially delay or limit the
use of our resources, substantially increase the anticipated cost of conducting
our drug target or drug candidate discovery efforts or prevent us from
implementing our processes at appropriate quality and throughput
levels.
Clinical
testing of our drug candidates in humans is an inherently risky and
time-consuming process that may fail to demonstrate safety and efficacy, which
could result in the delay, limitation or prevention of regulatory
approval.
In order
to obtain regulatory approvals for the commercial sale of any products that we
may develop, we will be required to complete extensive clinical trials in humans
to demonstrate the safety and efficacy of our drug candidates. We or
our collaborators may not be able to obtain authority from the FDA, or other
equivalent foreign regulatory agencies to initiate or complete any clinical
trials. In addition, we have limited internal resources for making
regulatory filings and interacting with regulatory authorities.
Clinical
trials are inherently risky and the results from preclinical testing of a drug
candidate that is under development may not be predictive of results that will
be obtained in human clinical trials. In addition, the results of
early human clinical trials may not be predictive of results that will be
obtained in larger-scale, advanced stage clinical trials. A number of
companies in the pharmaceutical industry have suffered significant setbacks in
advanced clinical trials, even after achieving positive results in earlier
trials. Negative or inconclusive results from a preclinical study or
a clinical trial could cause us, one of our collaborators or the FDA to
terminate a preclinical study or clinical trial or require that we repeat
it. Furthermore, we, one of our collaborators or a regulatory agency
with jurisdiction over the trials may suspend clinical trials at any time if the
subjects or patients participating in such trials are being exposed to
unacceptable health risks or for other reasons.
Any
preclinical or clinical test may fail to produce results satisfactory to the FDA
or foreign regulatory authorities. Preclinical and clinical data can
be interpreted in different ways, which could delay, limit or prevent regulatory
approval. The FDA or institutional review boards at the medical
institutions and healthcare facilities where we sponsor clinical trials may
suspend any trial indefinitely if they find deficiencies in the conduct of these
trials. Clinical trials must be conducted in accordance with the
FDA’s current Good Clinical Practices. The FDA and these
institutional review boards have authority to oversee our clinical trials, and
the FDA may require large numbers of subjects or patients. In
addition, we must manufacture, or contract for the manufacture of, the drug
candidates that we use in our clinical trials under the FDA’s current Good
Manufacturing Practices.
The rate
of completion of clinical trials is dependent, in part, upon the rate of
enrollment of patients. Patient accrual is a function of many
factors, including the size of the patient population, the proximity of patients
to clinical sites, the eligibility criteria for the study, the nature of the
study, the existence of competitive clinical trials and the availability of
alternative treatments. Delays in planned patient enrollment may
result in increased costs and prolonged clinical development, which in turn
could allow our competitors to bring products to market before we do and impair
our ability to commercialize our products or potential products.
We or our
collaborators may not be able to successfully complete any clinical trial of a
potential product within any specified time period. In some cases, we
or our collaborators may not be able to complete the trial at all. Moreover,
clinical trials may not show our potential products to be both safe and
effective. Thus, the FDA and other regulatory authorities may not
approve any products that we develop for any indication or may limit the
approved indications or impose other conditions.
Risks
Related to Regulatory Approval of Our Drug Candidates
Our
drug candidates are subject to a lengthy and uncertain regulatory process that
may not result in the necessary regulatory approvals, which could adversely
affect our ability to commercialize products.
Our drug
candidates, as well as the activities associated with their research,
development and commercialization, are subject to extensive regulation by the
FDA and other regulatory agencies in the United States and by comparable
authorities in other countries. Failure to obtain regulatory approval
for a drug candidate would prevent us from commercializing that drug
candidate. We have not received regulatory approval to market any of
our drug candidates in any jurisdiction and have only limited experience in
preparing and filing the applications necessary to gain regulatory
approvals. The process of obtaining regulatory approvals is
expensive, and often takes many years, if approval is obtained at all, and can
vary substantially based upon the type, complexity and novelty of the drug
candidates involved. Before a new drug application can be filed with
the FDA, the drug candidate must undergo extensive clinical trials, which can
take many years and may require substantial expenditures. Any
clinical trial may fail to produce results satisfactory to the
FDA. For example, the FDA could determine that the design of a
clinical trial is inadequate to produce reliable results. The
regulatory process also requires preclinical testing, and data obtained from
preclinical and clinical activities are susceptible to varying interpretations,
which could delay, limit or prevent regulatory approval. In addition,
delays or rejections may be encountered based upon changes in regulatory policy
for product approval during the period of product development and regulatory
agency review. Changes in regulatory approval policy, regulations or
statutes or the process for regulatory review during the development or approval
periods of our drug candidates may cause delays in the approval or rejection of
an application. Even if the FDA or a comparable authority in another
country approves a drug candidate, the approval may impose significant
restrictions on the indicated uses, conditions for use, labeling, advertising,
promotion, marketing and/or production of such product and may impose ongoing
requirements for post-approval studies, including additional research and
development and clinical trials. These agencies also may impose
various civil or criminal sanctions for failure to comply with regulatory
requirements, including withdrawal of product approval.
If
our potential products receive regulatory approval, we or our collaborators will
remain subject to extensive and rigorous ongoing regulation.
If we or
our collaborators obtain initial regulatory approvals from the FDA or foreign
regulatory authorities for any products that we may develop, we or our
collaborators will be subject to extensive and rigorous ongoing domestic and
foreign government regulation of, among other things, the research, development,
testing, manufacture, labeling, promotion, advertising, distribution and
marketing of our products and drug candidates. The failure to comply
with these requirements or the identification of safety problems during
commercial marketing could lead to the need for product marketing restrictions,
product withdrawal or recall or other voluntary or regulatory action, which
could delay further marketing until the product is brought into
compliance. The failure to comply with these requirements may also
subject us or our collaborators to stringent penalties.
Risks
Related to Commercialization of Products
The
commercial success of any products that we may develop will depend upon the
degree of market acceptance of our products among physicians, patients, health
care payors, private health insurers and the medical community.
Even if
approved by the relevant regulatory authority, our ability to commercialize any
products that we may develop will be highly dependent upon the extent to which
these products gain market acceptance among physicians, patients, health care
payors, such as Medicare and Medicaid, private health insurers, including
managed care organizations and group purchasing organizations, and the medical
community. If these products do not achieve an adequate level of
acceptance, we may not generate adequate product revenues, if at all, and we may
not become profitable. The degree of market acceptance of our drug
candidates, if approved for commercial sale, will depend upon a number of
factors, including:
|
|
·
|
the
effectiveness, or perceived effectiveness, of our products in comparison
to competing products;
|
|
|
·
|
the
existence of any significant side effects, as well as their severity in
comparison to any competing
products;
|
|
|
·
|
potential
advantages over alternative
treatments;
|
|
|
·
|
the
ability to offer our products for sale at competitive
prices;
|
|
|
·
|
relative
convenience and ease of
administration;
|
|
|
·
|
the
strength of marketing and distribution support;
and
|
|
|
·
|
sufficient
third-party coverage or
reimbursement.
|
If
we are unable to establish sales and marketing capabilities or enter into
agreements with third parties to market and sell our drug candidates, we may be
unable to generate product revenues.
We have
no experience as a company in the sales, marketing and distribution of
pharmaceutical products and do not currently have a sales and marketing
organization. Developing a sales and marketing force would be
expensive and time-consuming, could delay any product launch, and we may never
be able to develop this capacity. To the extent that we enter into
arrangements with third parties to provide sales, marketing and distribution
services, our product revenues are likely to be lower than if we market and sell
any products that we develop ourselves. If we are unable to establish
adequate sales, marketing and distribution capabilities, independently or with
others, we may not be able to generate product revenues.
If
we are unable to obtain adequate coverage and reimbursement from third-party
payors for any products that we may develop, our revenues and prospects for
profitability will suffer.
Our
ability to commercialize any products that we may develop will be highly
dependent on the extent to which coverage and reimbursement for our products
will be available from third-party payors, including governmental payors, such
as Medicare and Medicaid, and private health insurers, including managed care
organizations and group purchasing organizations. Many patients will
not be capable of paying themselves for some or all of the products that we may
develop and will rely on third-party payors to pay for, or subsidize, their
medical needs. If third-party payors do not provide coverage or
reimbursement for any products that we may develop, our revenues and prospects
for profitability will suffer. In addition, even if third-party
payors provide some coverage or reimbursement for our products, the availability
of such coverage or reimbursement for prescription drugs under private health
insurance and managed care plans often varies based on the type of contract or
plan purchased.
A primary
trend in the United States health care industry is toward reform and cost
containment. Current and future prescription drug benefit programs,
including any programs that may become effective as a result of such trend, may
have the effect of reducing the prices that we are able to charge for products
we develop and sell through plans under the programs. These
prescription drug programs may also cause third-party payors other than the
federal government, including the states under the Medicaid program, to
discontinue coverage for products we develop or to lower the price that they
will pay.
Proponents
of drug reimportation may attempt to pass legislation, which would allow direct
reimportation under certain circumstances. If legislation or
regulations were passed allowing the reimportation of drugs, it could decrease
the price we receive for any products that we may develop, thereby negatively
affecting our revenues and prospects for profitability.
In
addition, in some foreign countries, particularly the countries in the European
Union, the pricing of prescription pharmaceuticals is subject to governmental
control. In these countries, price negotiations with governmental
authorities can take six to 12 months or longer after the receipt of regulatory
marketing approval for a product. To obtain reimbursement and/or
pricing approval in some countries, we may be required to conduct a clinical
trial that compares the cost effectiveness of our drug candidates or products to
other available therapies. The conduct of such a clinical trial could
be expensive and result in delays in the commercialization of our drug
candidates. Third-party payors are challenging the prices charged for
medical products and services, and many third-party payors limit reimbursement
for newly approved health care products. In particular, third-party
payors may limit the indications for which they will reimburse patients who use
any products that we may develop. Cost-control initiatives could
decrease the price we might establish for products that we may develop, which
would result in lower product revenues to us.
Our
competitors may develop products that make our products obsolete.
The
biotechnology industry is highly fragmented and is characterized by rapid
technological change. We face, and will continue to face, intense
competition from biotechnology and pharmaceutical companies, as well as academic
research institutions, clinical reference laboratories and government agencies
that are pursuing research and development activities similar to
ours. In addition, significant delays in the development of our drug
candidates could allow our competitors to bring products to market before us,
which would impair our ability to commercialize our drug
candidates. Any products that we develop will compete in highly
competitive markets. Further, our competitors may be more effective
at using their technologies to develop commercial products. Many of
the organizations competing with us have greater capital resources, larger
research and development staff and facilities, more experience in obtaining
regulatory approvals and more extensive product manufacturing and marketing
capabilities. As a result, our competitors may be able to more easily
develop products that would render our products, and those of our collaborators,
obsolete and noncompetitive. For example, drug candidates are
currently being developed by other pharmaceutical companies for the treatment of
type 2 diabetes that act through SGLT2, one of the targets of LX4211, which
are in more advanced stages of development than LX4211. In addition, there
may be drug candidates of which we are not aware at an earlier stage of
development that may compete with our drug candidates.
We
may not be able to manufacture our drug candidates in commercial quantities,
which would prevent us from commercializing our drug candidates.
To date,
our drug candidates have been manufactured in small quantities for preclinical
and clinical trials. If any of these drug candidates are approved by
the FDA or other regulatory agencies for commercial sale, we will need to
manufacture them in larger quantities. We may not be able to
successfully increase the manufacturing capacity, whether in collaboration with
third-party manufacturers or on our own, for any of our drug candidates in a
timely or economic manner, or at all. Significant scale-up of
manufacturing may require additional validation studies, which the FDA must
review and approve. If we are unable to successfully increase the
manufacturing capacity for a drug candidate, the regulatory approval or
commercial launch of that drug candidate may be delayed or there may be a
shortage in supply. Our drug candidates require precise, high-quality
manufacturing. The failure to achieve and maintain these high
manufacturing standards, including the incidence of manufacturing errors, could
result in patient injury or death, product recalls or withdrawals, delays or
failures in product testing or delivery, cost overruns or other problems that
could seriously hurt our business.
Risks
Related to Our Relationships with Third Parties
Disagreements
with Symphony Icon regarding the development of our drug candidates LX1031 or
LX1032 could negatively affect or delay their development.
While we
are the party primarily responsible for development of our drug candidates
LX1031 and LX1032 in accordance with a specified development plan and related
development budget, our development activities are supervised by Symphony Icon’s
development committee, which is comprised of an equal number of representatives
from us and Symphony Icon. Any disagreements between us and Symphony
Icon regarding a development decision may cause delays in the development and
commercialization of those drug candidates or lead to development decisions that
do not reflect our interests. Any such delays or development
decisions not in our interest could negatively affect the value of LX1031 or
LX1032.
We
are dependent in many ways upon our collaborations with major pharmaceutical
companies. The revenues we receive under our existing collaboration
agreements have been decreasing in recent periods and are likely to continue to
decrease in the future. If we are unable to achieve milestones under
our collaborations or if our collaborators’ efforts fail to yield pharmaceutical
products on a timely basis, our opportunities to generate revenues and earn
royalties will be reduced.
We have
derived a substantial majority of our revenues to date from collaborative drug
discovery and development alliances with a limited number of major
pharmaceutical companies. Revenues from our drug discovery and
development alliances depend upon continuation of the collaborations, the
achievement of milestones and payment of royalties we earn from any future
products developed under the collaborations. If our relationship
terminates with any of our collaborators, our reputation in the business and
scientific community may suffer and revenues will be negatively impacted to the
extent such losses are not offset by additional collaboration
agreements. If we are unable to achieve milestones or our
collaborators are unable to successfully develop products from which royalties
are payable, we will not earn the revenues contemplated by those drug discovery
and development collaborations. In addition, some of our alliances
are exclusive and preclude us from entering into additional collaborative
arrangements with other parties in the field of exclusivity.
We have
limited or no control over the resources that any collaborator may devote to the
development and commercialization of products under our
alliances. Any of our present or future collaborators may not perform
their obligations as expected. These collaborators may breach or
terminate their agreements with us or otherwise fail to conduct discovery,
development or commercialization activities successfully or in a timely
manner. Further, our collaborators may elect not to develop
pharmaceutical products arising out of our collaborative arrangements or may not
devote sufficient resources to the development, approval, manufacture, marketing
or sale of these products. If any of these events occurs, we may not
be able to develop or commercialize potential pharmaceutical
products.
Conflicts
with our collaborators could jeopardize the success of our collaborative
agreements and harm our product development efforts.
We may
pursue opportunities in specific disease and therapeutic modality fields that
could result in conflicts with our collaborators, if any of our collaborators
takes the position that our internal activities overlap with those activities
that are exclusive to our collaboration. Moreover, disagreements
could arise with our collaborators over rights to our intellectual property or
our rights to share in any of the future revenues of compounds or therapeutic
approaches developed by our collaborators. Any conflict with or among
our collaborators could result in the termination of our collaborative
agreements, delay collaborative research or development activities, impair our
ability to renew or obtain future collaborative agreements or lead to costly and
time consuming litigation. Conflicts with our collaborators could
also have a negative impact on our relationship with existing collaborators,
materially impairing our business and revenues. Some of our
collaborators are also potential competitors or may become competitors in the
future. Our collaborators could develop competing products, preclude
us from entering into collaborations with their competitors or terminate their
agreements with us prematurely. Any of these events could harm our
product development efforts.
We
rely on third parties to carry out drug development activities.
We rely
on clinical research organizations and other third party contractors to carry
out many of our drug development activities, including the performance of
preclinical laboratory and animal tests under the FDA’s current Good Laboratory
Practices regulations and the conduct of clinical trials of our drug candidates
in accordance with protocols we establish. If these third parties do
not successfully carry out their contractual duties or regulatory obligations or
meet expected deadlines, our drug development activities may be delayed,
suspended or terminated. Such a failure by these third parties could
significantly impair our ability to develop and commercialize the affected drug
candidates.
We
lack the capability to manufacture materials for preclinical studies, clinical
trials or commercial sales and rely on third parties to manufacture our drug
candidates, which may harm or delay our product development and
commercialization efforts.
We
currently do not have the manufacturing capabilities or experience necessary to
produce materials for preclinical studies, clinical trials or commercial sales
and intend in the future to continue to rely on collaborators and third-party
contractors to produce such materials. We will rely on selected
manufacturers to deliver materials on a timely basis and to comply with
applicable regulatory requirements, including the current Good Manufacturing
Practices of the FDA, which relate to manufacturing and quality control
activities. These manufacturers may not be able to produce material
on a timely basis or manufacture material at the quality level or in the
quantity required to meet our development timelines and applicable regulatory
requirements. In addition, there are a limited number of
manufacturers that operate under the FDA’s current Good Manufacturing Practices
and that are capable of producing such materials, and we may experience
difficulty finding manufacturers with adequate capacity for our
needs. If we are unable to contract for the production of sufficient
quantity and quality of materials on acceptable terms, our product development
and commercialization efforts may be delayed. Moreover, noncompliance
with the FDA’s current Good Manufacturing Practices can result in, among other
things, fines, injunctions, civil and criminal penalties, product recalls or
seizures, suspension of production, failure to obtain marketing approval and
withdrawal, suspension or revocation of marketing approvals.
Risks
Related to Our Intellectual Property
If
we are unable to adequately protect our intellectual property, third parties may
be able to use our technology, which could adversely affect our ability to
compete in the market.
Our
success will depend in part upon our ability to obtain patents and maintain
adequate protection of the intellectual property related to our technologies and
products. The patent positions of biotechnology companies, including
our patent position, are generally uncertain and involve complex legal and
factual questions. We will be able to protect our intellectual
property rights from unauthorized use by third parties only to the extent that
our technologies are covered by valid and enforceable patents or are effectively
maintained as trade secrets. We will continue to apply for patents
covering our technologies and products as and when we deem
appropriate. Pending patent applications do not provide protection
against competitors because they are not enforceable until they issue as
patents. Further, the disclosures contained in our current and future
patent applications may not be sufficient to meet statutory requirements for
patentability. Once issued, patents still may not provide
commercially meaningful protection. Our existing patents and any
future patents we obtain may not be sufficiently broad to prevent others from
practicing our technologies or from developing competing
products. Furthermore, others may independently develop similar or
alternative technologies or design around our patents. If anyone
infringes upon our or our collaborators’ patent rights, enforcing these rights
may be difficult, costly and time-consuming and, as a result, it may not be
cost-effective or otherwise expedient to pursue litigation to enforce those
patent rights. In addition, our patents may be challenged or
invalidated or may fail to provide us with any competitive advantages, if, for
example, others were the first to invent or to file patent applications for
these inventions.
Because
patent applications can take many years to issue, there may be currently pending
applications, unknown to us, which may later result in issued patents that cover
the production, manufacture, commercialization or use of our technologies, drug
targets or drug candidates. If any such patents are issued to
other entities, we will be unable to obtain patent protection for the same or
similar discoveries that we make. Moreover, we may be blocked from
using or developing some of our existing or proposed technologies and products,
or may be required to obtain a license that may not be available on reasonable
terms, if at all. Further, others may discover uses for our
technologies or products other than those covered in our issued or pending
patents, and these other uses may be separately patentable. Even if
we have a patent claim on a particular technology or product, the holder of a
patent covering the use of that technology or product could exclude us from
selling a product that is based on the same use of that product.
The laws
of some foreign countries do not protect intellectual property rights to the
same extent as the laws of the United States, and many companies have
encountered significant problems in protecting and defending such rights in
foreign jurisdictions. Many countries, including certain countries in
Europe, have compulsory licensing laws under which a patent owner may be
compelled to grant licenses to third parties (for example, if the patent owner
has failed to “work” the invention in that country or the third party has
patented improvements). In addition, many countries limit the
enforceability of patents against government agencies or government
contractors. In these countries, the patent owner may have limited
remedies, which could materially diminish the value of the
patent. Compulsory licensing of life-saving drugs is also becoming
increasingly popular in developing countries either through direct legislation
or international initiatives. Such compulsory licenses could be
extended to include some of our drug candidates, which could limit our potential
revenue opportunities. Moreover, the legal systems of certain
countries, particularly certain developing countries, do not favor the
aggressive enforcement of patent and other intellectual property protection,
which makes it difficult to stop infringement.
We rely
on trade secret protection for our confidential and proprietary
information. We have taken security measures to protect our
proprietary information and trade secrets, but these measures may not provide
adequate protection. While we seek to protect our proprietary
information by entering into confidentiality agreements with employees,
collaborators and consultants, we cannot assure you that our proprietary
information will not be disclosed, or that we can meaningfully protect our trade
secrets. In addition, our competitors may independently develop
substantially equivalent proprietary information or may otherwise gain access to
our trade secrets.
We
may be involved in patent litigation and other disputes regarding intellectual
property rights and may require licenses from third parties for our discovery
and development and planned commercialization activities. We may not
prevail in any such litigation or other dispute or be able to obtain required
licenses.
Our
discovery and development efforts as well as our potential products and those of
our collaborators may give rise to claims that they infringe the patents of
others. We are aware that other companies and institutions are
developing products acting through the same drug targets through which some of
our drug candidates currently in clinical development act, have conducted
research on many of the same targets that we have identified and have filed
patent applications potentially covering many of the genes and encoded drug
targets that are the focus of our drug discovery programs. In some
cases, patents have issued from these applications. In addition, many
companies and institutions have well-established patent portfolios directed to
common techniques, methods and means of developing, producing and manufacturing
pharmaceutical products. Other companies or institutions could bring
legal actions against us or our collaborators for damages or to stop us or our
collaborators from engaging in certain discovery or development activities or
from manufacturing and marketing any resulting therapeutic
products. If any of these actions are successful, in addition to our
potential liability for damages, these entities would likely require us or our
collaborators to obtain a license in order to continue engaging in the
infringing activities or to manufacture or market the resulting therapeutic
products or may force us to terminate such activities or manufacturing and
marketing efforts.
We may
need to pursue litigation against others to enforce our patents and intellectual
property rights and may be the subject of litigation brought by third parties to
enforce their patent and intellectual property rights. In addition,
we may become involved in litigation based on intellectual property
indemnification undertakings that we have given to certain of our
collaborators. Patent litigation is expensive and requires
substantial amounts of management attention. The eventual outcome of
any such litigation is uncertain and involves substantial risks.
We
believe that there will continue to be significant litigation in our industry
regarding patent and other intellectual property rights. We have
expended and many of our competitors have expended and are continuing to expend
significant amounts of time, money and management resources on intellectual
property litigation. If we become involved in future intellectual
property litigation, it could consume a substantial portion of our resources and
could negatively affect our results of operations.
We
use intellectual property that we license from third parties. If we
do not comply with these licenses, we could lose our rights under
them.
We rely,
in part, on licenses to use certain technologies that are important to our
business, and we do not own the patents that underlie these
licenses. Most of these licenses, however, have terms that extend for
the life of the licensed patents. Our rights to use these
technologies and practice the inventions claimed in the licensed patents are
subject to our abiding by the terms of those licenses and the licensors not
terminating them. We believe we are currently in material compliance
with all requirements of these licenses. In many cases, we do not
control the filing, prosecution or maintenance of the patent rights to which we
hold licenses and rely upon our licensors to prosecute infringement of those
rights. The scope of our rights under our licenses may be subject to
dispute by our licensors or third parties.
We
have not sought patent protection outside of the United States for some of our
inventions, and some of our licensed patents only provide coverage in the United
States. As a result, our international competitors could be granted
foreign patent protection with respect to our discoveries.
We have
decided not to pursue patent protection with respect to some of our inventions
outside the United States, both because we do not believe it is cost-effective
and because of confidentiality concerns. Accordingly, our
international competitors could develop, and receive foreign patent protection
for, genes or gene sequences, uses of those genes or gene sequences, gene
products and drug targets, assays for identifying potential therapeutic
products, potential therapeutic products and methods of treatment for which we
are seeking United States patent protection.
We
may be subject to damages resulting from claims that we, our employees or
independent contractors have wrongfully used or disclosed alleged trade secrets
of their former employers.
Many of
our employees and independent contractors were previously employed at
universities, other biotechnology or pharmaceutical companies, including our
competitors or potential competitors. We may be subject to claims
that these employees, independent contractors or we have inadvertently or
otherwise used or disclosed trade secrets or other proprietary information of
their former employers. Litigation may be necessary to defend against
these claims. Even if we are successful in defending against these
claims, litigation could result in substantial costs and divert management’s
attention. If we fail in defending such claims, in addition to paying
money claims, we may lose valuable intellectual property rights or
personnel. A loss of key research personnel and/or their work product
could hamper or prevent our ability to commercialize certain drug candidates,
which could severely harm our business.
Risks
Related to Employees, Growth and Facilities Operations
The
loss of key personnel or the inability to attract and retain additional
personnel could impair our ability to expand our operations.
We are
highly dependent upon the principal members of our management and scientific
staff, the loss of whose services might adversely impact the achievement of our
objectives and the continuation of existing
collaborations. Recruiting and retaining qualified clinical and
scientific personnel will be critical to support activities related to advancing
our clinical and preclinical development programs, and supporting our
collaborative arrangements and our internal proprietary research and development
efforts. Competition is intense for experienced clinical personnel,
in particular, and we may be unable to retain or recruit clinical personnel with
the expertise or experience necessary to allow us to pursue collaborations,
develop our products or expand our operations to the extent otherwise
possible. Further, all of our employees are employed “at will” and,
therefore, may leave our employment at any time.
Our
collaborations with outside scientists may be subject to restriction and
change.
We work
with scientific and clinical advisors and collaborators at academic and other
institutions that assist us in our research and development
efforts. These advisors and collaborators are not our employees and
may have other commitments that limit their availability to
us. Although these advisors and collaborators generally agree not to
perform competing work, if a conflict of interest between their work for us and
their work for another entity arises, we may lose their services. In
such a circumstance, our development efforts with respect to the matters on
which they were working maybe significantly delayed or otherwise adversely
affected. In addition, although our advisors and collaborators sign
agreements not to disclose our confidential information, it is possible that
valuable proprietary knowledge may become publicly known through
them.
Security
breaches may disrupt our operations and harm our operating results.
Our
network security and data recovery measures may not be adequate to protect
against computer viruses, break-ins, and similar disruptions from unauthorized
tampering with our computer systems. The misappropriation, theft,
sabotage or any other type of security breach with respect to any of our
proprietary and confidential information that is electronically stored,
including research or clinical data, could have a material adverse impact on our
business, operating results and financial condition. Additionally,
any break-in or trespass of our facilities that results in the misappropriation,
theft, sabotage or any other type of security breach with respect to our
proprietary and confidential information, including research or clinical data,
or that results in damage to our research and development equipment and assets
could have a material adverse impact on our business, operating results and
financial condition.
Because
most of our operations are located at a single facility, the occurrence of a
disaster could significantly disrupt our business.
Most of
our operations are conducted at our facility in The Woodlands,
Texas. While we have developed redundant and emergency backup systems
to protect our resources and the facilities in which they are stored, they may
be insufficient in the event of a severe fire, flood, hurricane, tornado,
mechanical failure or similar disaster. If such a disaster
significantly damages or destroys the facility in which our resources are
maintained, our business could be disrupted until we could regenerate the
affected resources. Our business interruption insurance may not be
sufficient to compensate us in the event of a major interruption due to such a
disaster.
Risks
Related to Environmental and Product Liability
We
use hazardous chemicals and radioactive and biological materials in our
business. Any claims relating to improper handling, storage or disposal of these
materials could be time consuming and costly.
Our
research and development processes involve the controlled use of hazardous
materials, including chemicals and radioactive and biological
materials. Our operations produce hazardous waste
products. We cannot eliminate the risk of accidental contamination or
discharge and any resultant injury from these materials. Federal,
state and local laws and regulations govern the use, manufacture, storage,
handling and disposal of hazardous materials. We may face liability
for any injury or contamination that results from our use or the use by third
parties of these materials, and such liability may exceed our insurance coverage
and our total assets. Compliance with environmental laws and
regulations may be expensive, and current or future environmental regulations
may impair our research, development and production efforts.
In
addition, our collaborators may use hazardous materials in connection with our
collaborative efforts. In the event of a lawsuit or investigation, we could be
held responsible for any injury caused to persons or property by exposure to, or
release of, these hazardous materials used by these parties. Further,
we may be required to indemnify our collaborators against all damages and other
liabilities arising out of our development activities or products produced in
connection with these collaborations.
We
may be sued for product liability.
We or our
collaborators may be held liable if any product that we or our collaborators
develop, or any product that is made with the use or incorporation of any of our
technologies, causes injury or is found otherwise unsuitable during product
testing, manufacturing, marketing or sale. Although we currently have
and intend to maintain product liability insurance, this insurance may become
prohibitively expensive or may not fully cover our potential
liabilities. Our inability to obtain sufficient insurance coverage at
an acceptable cost or otherwise to protect against potential product liability
claims could prevent or inhibit the commercialization of products developed by
us or our collaborators. If we are sued for any injury caused by our
or our collaborators’ products, our liability could exceed our total
assets.
Risks
Related to Our Common Stock
Our
stock price may be extremely volatile.
The
trading price of our common stock has been highly volatile, and we believe the
trading price of our common stock will remain highly volatile and may fluctuate
substantially due to factors such as the following:
|
|
·
|
adverse
results or delays in clinical
trials;
|
|
|
·
|
announcement
of FDA approval or non-approval, or delays in the FDA review process, of
our or our collaborators’ product candidates or those of our competitors
or actions taken by regulatory agencies with respect to our, our
collaborators’ or our competitors’ clinical
trials;
|
|
|
·
|
the
announcement of new products by us or our
competitors;
|
|
|
·
|
quarterly
variations in our or our competitors’ results of
operations;
|
|
|
·
|
conflicts
or litigation with our
collaborators;
|
|
|
·
|
litigation,
including intellectual property infringement and product liability
lawsuits, involving us;
|
|
|
·
|
failure
to achieve operating results projected by securities
analysts;
|
|
|
·
|
changes
in earnings estimates or recommendations by securities
analysts;
|
|
|
·
|
financing
transactions;
|
|
|
·
|
developments
in the biotechnology or pharmaceutical
industry;
|
|
|
·
|
sales
of large blocks of our common stock or sales of our common stock by our
executive officers, directors and significant
stockholders;
|
|
|
·
|
departures
of key personnel or board members;
|
|
|
·
|
developments
concerning current or future
collaborations;
|
|
|
·
|
FDA
or international regulatory
actions;
|
|
|
·
|
third-party
reimbursement policies;
|
|
|
·
|
acquisitions
of other companies or technologies;
|
|
|
·
|
disposition
of any of our subsidiaries, drug programs or other technologies;
and
|
|
|
·
|
other
factors, including factors unrelated to our operating performance or the
operating
performance of our
competitors.
|
These
factors, as well as general economic, political and market conditions, may
materially adversely affect the market price of our common stock.
In the
past, following periods of volatility in the market price of a company’s
securities, securities class action litigation has often been
instituted. A securities class action suit against us could result in
substantial costs and divert management’s attention and resources, which could
have a material and adverse effect on our business.
Invus’
ownership of our common stock and its other rights under the stockholders’
agreement we entered into in connection with Invus’ $205.4 million initial
investment in our common stock provide Invus with substantial influence over
matters requiring stockholder approval, including the election of directors and
approval of significant corporate transactions, as well as other corporate
matters.
Under the
stockholders’ agreement we entered into in connection with Invus’ $205.4 million
initial investment in our common stock, Invus currently has the right to
designate the greater of three members or 30% (or the percentage of all the
outstanding shares of our common stock owned by Invus and its affiliates, if
less than 30%) of all members of our board of directors, rounded up to the
nearest whole number of directors, pursuant to which Invus has designated
Raymond Debbane, president and chief executive officer of The Invus Group, LLC,
an affiliate of Invus, and Philippe J. Amouyal and Christopher J. Sobecki, each
of whom are managing directors of The Invus Group, LLC. In the event
that the number of shares of our common stock owned by Invus and its affiliates
ever exceeds 50% of the total number of shares of our common stock then
outstanding (not counting for such purpose any shares acquired by Invus from
third parties in excess of 40% (or, if higher, its then pro rata amount) of the
total number of outstanding shares of common stock, as permitted by the
standstill provisions of the stockholders’ agreement), from and after that time,
Invus will have the right to designate a number of directors equal to the
percentage of all the outstanding shares of our common stock owned by Invus and
its affiliates (not counting for such purpose any shares acquired by Invus from
third parties in excess of 40% (or, if higher, its then pro rata amount) of the
total number of outstanding shares of common stock, as permitted by the
standstill provisions of the stockholders’ agreement), rounded up to the nearest
whole number of directors. The directors appointed by Invus have
proportionate representation on the compensation and corporate governance
committees of our board of directors.
Invus’
rights with respect to the designation of members of our board of directors and
its compensation and corporate governance committees will terminate if the
percentage of all the outstanding shares of our common stock owned by Invus and
its affiliates falls below 10%. Invus will also have the right to
terminate these provisions at any time following the date on which the
percentage of all the outstanding shares of our common stock owned by Invus and
its affiliates exceeds 50% (not counting for such purpose any shares acquired by
Invus and its affiliates from third parties in excess of 40% (or, if higher, its
then pro rata amount) of the total number of outstanding shares of our common
stock, as permitted by the standstill provisions of the stockholders’
agreement).
Invus has
preemptive rights under the stockholders’ agreement to participate in future
equity issuances by us (including any qualified offering), subject to certain
exceptions, so as to maintain its then-current percentage ownership of our
capital stock. Subject to certain limitations, Invus will be required
to exercise its preemptive rights in advance with respect to certain marketed
offerings, in which case it will be obligated to buy its pro rata share of the
number of shares being offered in such marketed offering, including any
overallotment (or such lesser amount specified in its exercise of such rights),
so long as the sale of the shares were priced within a range
within 10% above or below the market price on the date we notified Invus of the
offering and we met certain other conditions.
The
provisions of the stockholders’ agreement relating to preemptive rights will
terminate on the earlier to occur of August 28, 2017 and the date on which the
percentage of all the outstanding shares of our common stock owned by Invus and
its affiliates falls below ten percent.
Invus is
subject to standstill provisions restricting its ability to purchase or
otherwise acquire additional shares of common stock from third parties to an
amount that would result in its ownership of our common stock not exceeding 49%
of the total number of shares outstanding. These standstill
provisions will not apply to the acquisitions of securities by way of stock
splits, stock dividends, reclassifications, recapitalizations, or other
distributions by us, acquisitions contemplated by the securities purchase
agreement and the stockholders’ agreement, including in the rights offerings and
upon Invus’ exercise of preemptive rights under the stockholders’
agreement.
Except
for acquisitions pursuant to the provisions described above, and subject to
certain exceptions, Invus has agreed that it will not, and will cause its
affiliates not to, without the approval of our unaffiliated board, directly or
indirectly:
|
|
·
|
solicit
proxies to vote any of our voting securities or any voting securities of
our subsidiaries;
|
|
|
·
|
submit
to our board of directors a written proposal for any merger,
recapitalization, reorganization, business combination or other
extraordinary transaction involving an acquisition of us or any of our
subsidiaries or any of our or our subsidiaries’ securities or assets by
Invus and its affiliates;
|
|
|
·
|
enter
into discussions, negotiations, arrangements or understandings with any
third party with respect to any of the foregoing;
or
|
|
|
·
|
request
us or any of our representatives, directly or indirectly, to amend or
waive any of these standstill
provisions.
|
The
standstill provisions of the stockholders’ agreement will terminate on the
earliest to occur of (a) August 28, 2017, (b) the date on which the percentage
of all the outstanding shares of our common stock owned by Invus and its
affiliates falls below 10%, (c) the date on which the percentage of all of the
outstanding shares of our common stock owned by Invus and its affiliates exceeds
50% (not counting for such purpose any shares acquired by Invus from third
parties in excess of 40% (or, if higher, its then pro rata amount) of the total
number of outstanding shares of common stock, as permitted by the standstill
provisions of the stockholders’ agreement), (d) the date on which any third
party makes a public proposal to acquire (by purchase, exchange, merger or
otherwise) assets or business constituting 50% or more of our revenues, net
income or assets or 50% of any class of our equity securities our board of
directors recommends or approves, or proposes to recommend or approve, any such
transaction or (e) the date on which any third party acquires beneficial
ownership (by purchase, exchange, merger or otherwise) of assets or business
constituting 20% or more of our revenues, net income or assets or 20% of any
class of our equity securities or our board of directors recommends or approves,
or proposes to recommend or approve, any such transaction.
Subject
to certain exceptions, Invus has agreed that neither it nor its affiliates will
sell any shares of common stock to third parties that are not affiliated with
Invus if, to Invus’ knowledge, such transfer would result in any such third
party (or any person or group including such third party) owning more than 14.9%
of the total number of outstanding shares of our common stock.
The
provisions of the stockholders’ agreement relating to sales to third parties
will terminate on the earliest to occur of (a) August 28, 2017,
(b) the date on which the percentage of all the outstanding shares of our
common stock owned by Invus and its affiliates falls below 10%, and (c) the
date on which the percentage of all the outstanding shares of our common stock
owned by Invus and its affiliates exceeds 50% (not counting for such purpose any
shares acquired by Invus and its affiliates from third parties in excess of 40%
(or, if higher, its then pro rata amount) of the total number of outstanding
shares of our common stock, as permitted by the standstill provisions of the
stockholders’ agreement).
In any
election of persons to serve on our board of directors, Invus will be obligated
to vote all of the shares of common stock held by it and its affiliates in favor
of the directors nominated by our board of directors, as long as we have
complied with our obligation with respect to the designation of members of our
board of directors described above and the individuals designated by Invus for
election to our board of directors have been nominated, and, if applicable, are
serving on our board of directors. With respect to all other matters
submitted to a vote of the holders of our common stock, Invus will be obligated
to vote any shares that it acquired from third parties in excess of 40% (or, if
higher, its then pro rata amount) of the total number of outstanding shares of
common stock, as permitted by the standstill provisions of the stockholders’
agreement, in the same proportion as all the votes cast by other holders of our
common stock, unless Invus and we (acting with the approval of the unaffiliated
board) agree otherwise. Invus may vote all other shares of our common
stock held by it in its sole discretion.
The
provisions of the stockholders’ agreement relating to voting will terminate on
the earliest to occur of (a) August 28, 2017, (b) the date on which
the percentage of all the outstanding shares of our common stock held by Invus
and its affiliates falls below 10%, (c) the date on which the percentage of
all outstanding shares of our common stock owned by Invus and its affiliates
exceeds 50% (not counting for such purpose any shares acquired by Invus from
third parties in excess of 40% (or, if higher, its then pro rata amount) of the
total number of outstanding shares of our common stock, as permitted by the
provisions of the stockholders’ agreement), and (d) the termination of the
standstill provisions in accordance with the stockholders’
agreement.
Invus is
entitled to certain minority protections, including consent rights over (a) the
creation or issuance of any new class or series of shares of our capital stock
(or securities convertible into or exercisable for shares of our capital stock)
having rights, preferences or privileges senior to or on parity with our common
stock, (b) any amendment to our certificate of incorporation or bylaws, or
amendment to the certificate of incorporation or bylaws of any of our
subsidiaries, in a manner adversely affecting Invus’ rights under the securities
purchase agreement and the related agreements, (c) the repurchase, retirement,
redemption or other acquisition of our or our subsidiaries’ capital stock (or
securities convertible into or exercisable for shares of our or our
subsidiaries’ capital stock), (d) any increase in the size of our board of
directors to more than 12 members and (e) the adoption or proposed adoption of
any stockholders’ rights plan, “poison pill” or other similar plan or agreement,
unless Invus is exempt from the provisions of such plan or
agreement.
The
provisions of the stockholders’ agreement relating to minority protections will
terminate on the earlier to occur of August 28, 2017 and the date on which Invus
and its affiliates hold less than 15% of the total number of outstanding shares
of our common stock.
We
may engage in future acquisitions, which may be expensive and time consuming and
from which we may not realize anticipated benefits.
We may
acquire additional businesses, technologies and products if we determine that
these businesses, technologies and products complement our existing technology
or otherwise serve our strategic goals. If we do undertake any
transactions of this sort, the process of integrating an acquired business,
technology or product may result in operating difficulties and expenditures and
may not be achieved in a timely and non-disruptive manner, if at all, and may
absorb significant management attention that would otherwise be available for
ongoing development of our business. If we fail to integrate acquired
businesses, technologies or products effectively or if key employees of an
acquired business leave, the anticipated benefits of the acquisition would be
jeopardized. Moreover, we may never realize the anticipated benefits
of any acquisition, such as increased revenues and earnings or enhanced business
synergies. Future acquisitions could result in potentially dilutive
issuances of our equity securities, the incurrence of debt and contingent
liabilities and amortization expenses related to intangible assets, which could
materially impair our results of operations and financial
condition.
Future
sales of our common stock may depress our stock price.
If our
stockholders sell substantial amounts of our common stock (including shares
issued upon the exercise of options) in the public market, the market price of
our common stock could fall. These sales also might make it more
difficult for us to sell equity or equity-related securities in the future at a
time and price that we deem appropriate. For example, following an
acquisition, a significant number of shares of our common stock held by new
stockholders may become freely tradable or holders of registration rights could
cause us to register their shares for resale. Sales of these shares
of common stock held by existing stockholders could cause the market price of
our common stock to decline.
If
we are unable to meet Nasdaq continued listing requirements, Nasdaq may take
action to delist our common stock.
Our
common stock trades on The Nasdaq Global Market, which has qualitative and
quantitative listing criteria, including operating results, net assets,
corporate governance, minimum trading price and minimums for public float, which
is the amount of stock not held by our affiliates. If we are unable
to meet Nasdaq continued listing requirements, Nasdaq may take action to delist
our common stock. A delisting of our common stock could negatively impact us and
our shareholders by reducing the liquidity and market price of our common stock
and potentially reducing the number of investors willing to hold or acquire our
common stock.
Item
1B. Unresolved Staff Comments
None.
Item
2.
Properties
We
currently own approximately 300,000 square feet of space for our corporate
offices and laboratories in buildings located in The Woodlands, Texas, a suburb
of Houston, Texas, and lease approximately 76,000 square feet of space for
offices and laboratories near Princeton, New Jersey.
In April
2004, we purchased our facilities in The Woodlands, Texas from the lessor under
our previous synthetic lease agreement. In connection with such
purchase, we obtained a $34.0 million mortgage which has a ten-year term with a
20-year amortization and bears interest at a fixed rate of 8.23%. The
mortgage had a principal balance outstanding of $29.5 million as of December 31,
2009.
In May
2002, our subsidiary Lexicon Pharmaceuticals (New Jersey), Inc. entered into a
lease for our facility in Hopewell, New Jersey. The term of the lease
extends until June 30, 2013. The lease provides for an escalating
yearly base rent payment of $1.3 million in the first year, $2.1 million in
years two and three, $2.2 million in years four to six, $2.3 million in
years seven to nine and $2.4 million in years ten and eleven. We are
the guarantor of the obligations of our subsidiary under the lease.
We
believe that our facilities are well-maintained, in good operating condition and
acceptable for our current operations.
Item
3. Legal
Proceedings
We are
from time to time party to claims and legal proceedings that arise in the normal
course of our business and that we believe will not have, individually or in the
aggregate, a material adverse effect on our results of operations, financial
condition or liquidity.
Item
4. [Reserved]
PART
II
|
Item
5.
|
Market for
Registrant’s Common Equity, Related Stockholder Matters and Issuer
Purchases of Equity
Securities
|
Our
common stock is quoted on The Nasdaq Global Market under the symbol
“LXRX.” The following table sets forth, for the periods indicated,
the high and low sales prices for our common stock as reported on The Nasdaq
Global Market.
|
High
|
Low
|
|||||||
|
2008
|
||||||||
|
First
Quarter
|
$ | 3.07 | $ | 1.27 | ||||
|
Second
Quarter
|
$ | 2.44 | $ | 1.57 | ||||
|
Third
Quarter
|
$ | 2.60 | $ | 1.51 | ||||
|
Fourth
Quarter
|
$ | 1.90 | $ | 0.74 | ||||
|
2009
|
||||||||
|
First
Quarter
|
$ | 1.75 | $ | 0.81 | ||||
|
Second
Quarter
|
$ | 1.63 | $ | 0.94 | ||||
|
Third
Quarter
|
$ | 3.78 | $ | 1.11 | ||||
|
Fourth
Quarter
|
$ | 2.13 | $ | 1.30 | ||||
As of
February 28, 2010, there were approximately 228 holders of record of our common
stock.
We have
never paid cash dividends on our common stock. We anticipate that we will retain
all of our future earnings, if any, for use in the expansion and operation of
our business and do not anticipate paying cash dividends in the foreseeable
future.
Performance
Graph
The
following performance graph compares the performance of our common stock to the
Nasdaq Composite Index and the Nasdaq Biotechnology Index for the period
beginning December 31, 2004 and ending December 31, 2009. The graph assumes that
the value of the investment in our common stock and each index was $100 at
December 31, 2004, and that all dividends were reinvested.
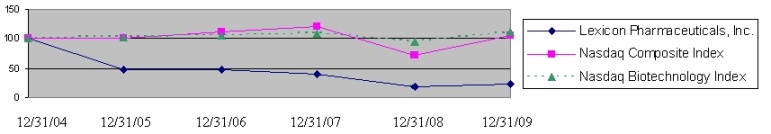
|
December
31,
|
||||||||||||
|
2004
|
2005
|
2006
|
2007
|
2008
|
2009
|
|||||||
|
Lexicon
Pharmaceuticals, Inc.
|
100
|
47
|
47
|
39
|
18
|
22
|
||||||
|
Nasdaq
Composite Index
|
100
|
101
|
111
|
122
|
72
|
104
|
||||||
|
Nasdaq
Biotechnology Index
|
100
|
103
|
104
|
109
|
95
|
110
|
||||||
The
foregoing stock price performance comparisons shall not be deemed “filed” for
purposes of Section 18 of the Securities Exchange Act of 1934, or otherwise
subject to the liabilities of that section, nor shall it be deemed incorporated
by reference by any general statement incorporating by reference this annual
report on Form 10-K into any filing under the Securities Act of 1933 or under
the Securities Exchange Act of 1934, except to the extent that we specifically
incorporate such comparisons by reference.
Purchases
of Equity Securities
The
following table provides information about our purchases of shares of our common
stock during the six months ended December 31, 2009:
|
Period
|
Total
Number of Shares Purchased
|
Average
Price Paid Per Share
|
Total
Number of Shares Purchased as Part of a Publicly Announced
Program
|
Maximum
Number of Shares that May Yet be Purchased Under the Program (1)
|
||||||||||||
|
July
1 – 31, 2009
|
— | $ | — | — | $ | — | ||||||||||
|
August 1 – 31,
2009
|
67,891 | (2) | $ | 1.30 | (3) | — | $ | — | ||||||||
|
September 1 – 30,
2009
|
— | $ | — | — | $ | — | ||||||||||
|
October 1 – 31,
2009
|
— | $ | — | — | $ | — | ||||||||||
|
November 1 – 30,
2009
|
— | $ | — | — | $ | — | ||||||||||
|
December 1 – 31,
2009
|
— | $ | — | — | $ | — | ||||||||||
|
|
|
|
(1)
|
In
connection with the vesting of restricted stock bonus awards granted under
our Equity Incentive Plan, which was adopted in February 2009 as an
amendment and restatement of our 2000 Equity Incentive Plan and expires in
February 2019, certain recipients of such restricted stock bonus awards
elected to satisfy their withholding tax obligations with respect to such
vesting event by having us retain a portion of the vested
shares. In the future, we may grant additional restricted
equity securities under our Equity Incentive Plan for which the
recipient’s tax withholding obligations may be satisfied by our retention
of a portion of such securities. Further, the number of such
restricted equity securities which may vest will be dependent on the
continued employment of such recipients or other performance-based
conditions. As a result, we cannot predict with any certainty
either the total amount of restricted equity securities or the approximate
dollar value of such securities that we may purchase in future years as
those securities vest.
|
|
|
(2)
|
Represents
shares retained by us at the election of certain recipients of restricted
stock bonus awards granted under our Equity Incentive Plan in satisfaction
of their withholding tax obligations with respect to the vesting of those
awards.
|
|
|
(3)
|
Represents
the market price of our common stock on the date of vesting, calculated in
accordance with the process for determination of fair market value under
our Equity Incentive Plan.
|
Item
6. Selected Financial
Data
The
statement of operations data for the years ended December 31, 2009, 2008
and 2007 and the balance sheet data as of December 31, 2009 and 2008 have
been derived from our audited financial statements included elsewhere in this
annual report on Form 10-K. The statements of operations data
for the years ended December 31, 2006 and 2005, and the balance sheet data as of
December 31, 2007, 2006 and 2005 have been derived from our audited financial
statements not included in this annual report on Form 10-K. Our
historical results are not necessarily indicative of results to be expected for
any future period. The data presented below has been derived from
financial statements that have been prepared in accordance with accounting
principles generally accepted in the United States and should be read with our
financial statements, including the notes, and with “Management’s Discussion and
Analysis of Financial Condition and Results of Operations” included elsewhere in
this annual report on Form 10-K.
|
Year
Ended December 31,
|
||||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
||||||||||||||||
|
Statements
of Operations Data:
|
(in
thousands, except per share data)
|
|||||||||||||||||||
|
Revenues
|
$
|
10,700
|
$
|
32,321
|
$
|
50,118
|
$
|
72,798
|
$
|
75,680
|
||||||||||
|
Operating
expenses:
|
||||||||||||||||||||
|
Research and
development, including stock-based compensation of $3,022 in
2009, $3,941 in 2008, $5,150 in 2007, $4,394 in 2006 and
($21) in 2005
|
81,238
|
107,232
|
103,237
|
105,494
|
92,539
|
|||||||||||||||
|
General and
administrative, including stock-based compensation of $2,252 in 2009,
$2,559 in 2008, $2,776 in 2007, $2,636 in 2006 and $0 in
2005
|
19,418
|
21,624
|
21,835
|
22,535
|
19,260
|
|||||||||||||||
|
Total
operating expenses
|
100,656
|
128,856
|
125,072
|
128,029
|
111,799
|
|||||||||||||||
|
Loss
from operations
|
(89,956
|
)
|
(96,535
|
)
|
(74,954
|
)
|
(55,231
|
)
|
(36,119
|
)
|
||||||||||
|
Interest
and other income (expense), net
|
(3,463
|
)
|
(349
|
)
|
3,721
|
801
|
(77
|
)
|
||||||||||||
|
Consolidated
net loss before taxes
|
(93,419
|
)
|
(96,884
|
)
|
(71,233
|
)
|
(54,430
|
)
|
(36,196
|
)
|
||||||||||
|
Income
tax benefit (provision)
|
102
|
—
|
—
|
119
|
(119
|
)
|
||||||||||||||
|
Consolidated
net loss
|
(93,317
|
)
|
(96,884
|
)
|
(71,233
|
)
|
(54,311
|
)
|
(36,315
|
)
|
||||||||||
|
Less:
net loss attributable to noncontrolling interest in Symphony Icon,
Inc.
|
10,537
|
20,024
|
12,439
|
—
|
—
|
|||||||||||||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc.
|
$
|
(82,780
|
)
|
$
|
(76,860
|
)
|
$
|
(58,794
|
)
|
$
|
(54,311
|
)
|
$
|
(36,315
|
)
|
|||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc. per common share, basic
and diluted
|
$
|
(0.57
|
)
|
$
|
(0.56
|
)
|
$
|
(0.59
|
)
|
$
|
(0.81
|
)
|
$
|
(0.57
|
)
|
|||||
|
Shares
used in computing net loss attributable to Lexicon Pharmaceuticals, Inc.
per common share, basic and diluted
|
145,465
|
136,797
|
99,798
|
66,876
|
63,962
|
|||||||||||||||
|
As
of December 31,
|
||||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
||||||||||||||||
|
Balance
Sheet Data:
|
(in
thousands)
|
|||||||||||||||||||
|
Cash,
cash equivalents and short-term investments, including restricted cash and
investments of $430
|
$
|
157,096
|
$
|
86,502
|
$
|
222,109
|
$
|
79,999
|
$
|
99,695
|
||||||||||
|
Short-term
investments held by Symphony Icon, Inc.
|
5,417
|
16,610
|
36,666
|
—
|
—
|
|||||||||||||||
|
Long-term
investments
|
—
|
55,686
|
—
|
—
|
—
|
|||||||||||||||
|
Working
capital
|
118,730
|
87,991
|
229,303
|
39,586
|
48,584
|
|||||||||||||||
|
Total
assets
|
257,761
|
261,508
|
369,296
|
190,266
|
218,714
|
|||||||||||||||
|
Long-term
debt, net of current portion
|
28,482
|
29,529
|
30,493
|
31,372
|
32,189
|
|||||||||||||||
|
Accumulated
deficit
|
(570,175
|
)
|
(487,395
|
)
|
(410,535
|
)
|
(351,741
|
)
|
(297,430
|
)
|
||||||||||
|
Lexicon
Pharmaceuticals, Inc. stockholders’ equity
|
163,787
|
185,580
|
256,300
|
85,501
|
85,802
|
|||||||||||||||
Item
7.
Management’s Discussion and
Analysis of Financial Condition and Results of Operations
The
following discussion and analysis should be read with “Selected Financial Data”
and our financial statements and notes included elsewhere in this annual report
on Form 10-K.
Overview
We are a
biopharmaceutical company focused on the discovery and development of
breakthrough treatments for human disease. We have used our
proprietary gene knockout technologies and an integrated platform of advanced
medical technologies to identify and validate, in vivo, more than 100
targets with promising profiles for drug discovery. For targets that
we believe have high pharmaceutical value, we engage in programs for the
discovery and development of potential new drugs, focusing in the core
therapeutic areas of immunology, metabolism, cardiology and
ophthalmology. Human clinical trials are currently underway for four
of our drug candidates, with one additional drug candidate in preclinical
development and compounds from a number of additional programs in various stages
of preclinical research.
We are
working both independently and through strategic collaborations and alliances to
capitalize on our technology, drug target discoveries and drug discovery and
development programs. Consistent with this approach, we seek to
retain exclusive rights to the benefits of certain of our small molecule drug
programs by developing drug candidates from such programs internally and to
collaborate with third parties with respect to the discovery, development and
commercialization of small molecule and biotherapeutic drug candidates for other
targets, particularly when the collaboration provides us with access to
expertise and resources that we do not possess internally or are complementary
to our own. We have established drug discovery and development
collaborations with a number of leading pharmaceutical and biotechnology
companies which have enabled us to generate near-term cash while offering us the
potential to retain economic participation in products our collaborators develop
through the collaboration. In addition, we have established
collaborations and license agreements with other leading pharmaceutical and
biotechnology companies, research institutes and academic institutions under
which we received fees and, in some cases, are eligible to receive milestone and
royalty payments, in return for granting access to some of our technologies and
discoveries.
We derive
substantially all of our revenues from drug discovery and development
collaborations and other collaborations and technology licenses. To
date, we have generated a substantial portion of our revenues from a limited
number of sources.
Our
operating results and, in particular, our ability to generate additional
revenues are dependent on many factors, including our success in establishing
new collaborations and technology licenses, expirations of our existing
collaborations and alliances, the success rate of our discovery and development
efforts leading to opportunities for new collaborations and licenses, as well as
milestone payments and royalties, the timing and willingness of collaborators to
commercialize products that would result in milestone payments and royalties,
and general and industry-specific economic conditions which may affect research
and development expenditures. Our future revenues from collaborations
and technology licenses are uncertain because our existing agreements have fixed
terms or relate to specific projects of limited duration and we depend, in part,
on securing new agreements. Our ability to secure future
revenue-generating agreements will depend upon our ability to address the needs
of our potential future collaborators and licensees, and to negotiate agreements
that we believe are in our long-term best interests. We may
determine, as we have with our four clinical drug candidates, that our interests
are better served by retaining rights to our discoveries and advancing our
therapeutic programs to a later stage, which could limit our near-term
revenues. Because of these and other factors, our operating results
have fluctuated in the past and are likely to do so in the future, and we do not
believe that period-to-period comparisons of our operating results are a good
indication of our future performance.
Since our
inception, we have incurred significant losses and, as of December 31, 2009, we
had an accumulated deficit of $570.2 million. Our losses have resulted
principally from costs incurred in research and development, general and
administrative costs associated with our operations, and non-cash stock-based
compensation expenses associated with stock options granted to employees and
consultants. Research and development expenses consist primarily of
salaries and related personnel costs, external research costs related to our
preclinical and clinical efforts, material costs, facility costs, depreciation
on property and equipment, and other expenses related to our drug discovery and
development programs. General and administrative expenses consist primarily of
salaries and related expenses for executive and administrative personnel,
professional fees and other corporate expenses, including information
technology, facilities costs and general legal activities. In
connection with the continued expansion of our drug discovery and development
programs, we expect to continue to incur significant research and development
costs. As a result, we will need to generate significantly higher revenues to
achieve profitability.
Critical
Accounting Policies
Revenue
Recognition
We
recognize revenues when persuasive evidence of an arrangement exists, delivery
has occurred or services have been rendered, the price is fixed or determinable,
and collectibility is reasonably assured. Payments received in
advance under these arrangements are recorded as deferred revenue until
earned.
Upfront
fees under our drug discovery and development alliances are recognized as
revenue on a straight-line basis over the estimated period of service, generally
the contractual research term, as this period is our best estimate of the period
over which the services will be rendered, to the extent they are
non-refundable. We have determined that the level of effort we
perform to meet our obligations is fairly constant throughout the estimated
periods of service. As a result, we have determined that it is
appropriate to recognize revenue from such agreements on a straight-line basis,
as we believe this reflects how the research is provided during the initial
period of the agreement. When it becomes probable that a collaborator
will extend the research period, we adjust the revenue recognition method as
necessary based on the level of effort required under the agreement for the
extension period.
Research
funding under these alliances is recognized as services are performed to the
extent they are non-refundable, either on a straight-line basis over the
estimated service period, generally the contractual research term; or as
contract research costs are incurred. Milestone-based fees are
recognized upon completion of specified milestones according to contract
terms. Payments received under target validation collaborations and
government grants and contracts are recognized as revenue as we perform our
obligations related to such research to the extent such fees are
non-refundable. Non-refundable technology license fees are recognized
as revenue upon the grant of the license, when performance is complete and there
is no continuing involvement.
Revenues
recognized from multiple element contracts are allocated to each element of the
arrangement based on the relative fair value of the elements. An
element of a contract can be accounted for separately if the delivered elements
have standalone value to the collaborator and the fair value of any undelivered
elements is determinable through objective and reliable evidence. If
an element is considered to have standalone value but the fair value of any of
the undelivered items cannot be determined, all elements of the arrangement are
recognized as revenue over the period of performance for such undelivered items
or services.
A change
in our revenue recognition policy or changes in the terms of contracts under
which we recognize revenues could have an impact on the amount and timing of our
recognition of revenues.
Research
and Development Expenses
Research
and development expenses consist of costs incurred for research and development
activities solely sponsored by us as well as collaborative research and
development activities. These costs include direct and
research-related overhead expenses and are expensed as
incurred. Technology license fees for technologies that are utilized
in research and development and have no alternative future use are expensed when
incurred.
We have
announced positive top-line results from Phase 2 clinical trials of each of our
two most advanced drug candidates: LX1031, an orally-delivered small
molecule compound that we are developing as a potential treatment for irritable
bowel syndrome and other gastrointestinal disorders and LX4211, an
orally-delivered small molecule compound that we are developing as a potential
treatment for type 2 diabetes. We are presently conducting Phase 2
clinical trials of two other drug candidates: LX2931, an
orally-delivered small molecule compound that we are developing as a potential
treatment for rheumatoid arthritis and other autoimmune diseases and LX1032, an
orally-delivered small molecule compound that we are developing as a potential
treatment for the symptoms associated with carcinoid syndrome. We
have advanced one other drug candidate into preclinical development: LX7101, a
topically-delivered small molecule compound that we are developing as a
potential treatment for glaucoma. We have small molecule compounds
from a number of additional drug discovery programs in various stages of
preclinical research and believe that our systematic, target biology-driven
approach to drug discovery will enable us to continue to expand our clinical
pipeline. The drug development process takes many years to
complete. The cost and length of time varies due to many factors
including the type, complexity and intended use of the drug
candidate. We estimate that drug development activities are typically
completed over the following periods:
|
Phase
|
Estimated
Completion Period
|
|
|
Preclinical
development
|
1-2
years
|
|
|
Phase
1 clinical trials
|
1-2
years
|
|
|
Phase
2 clinical trials
|
1-2
years
|
|
|
Phase
3 clinical trials
|
2-4
years
|
We expect
research and development costs to increase in the future as our existing
clinical drug programs advance to later stage clinical trials and new drug
programs enter preclinical and clinical development. Due to the
variability in the length of time necessary for drug development, the
uncertainties related to the cost of these activities and ultimate ability to
obtain governmental approval for commercialization, accurate and meaningful
estimates of the ultimate costs to bring our potential drug candidates to market
are not available.
We record
significant accrued liabilities related to unbilled expenses for products or
services that we have received from service providers, specifically related to
ongoing preclinical studies and clinical trials. These costs
primarily relate to clinical study management, monitoring, laboratory and
analysis costs, drug supplies, toxicology studies and investigator
grants. We have multiple drugs in concurrent preclinical studies and
clinical trials at clinical sites throughout the world. In order to
ensure that we have adequately provided for ongoing preclinical and clinical
development costs during the period in which we incur such costs, we maintain
accruals to cover these expenses. We update our estimates for these
accruals on a monthly basis. Although we use consistent milestones or
subject or patient enrollment to drive expense recognition, the assessment of
these costs is a subjective process that requires judgment. Upon
settlement, these costs may differ materially from the amounts accrued in our
consolidated financial statements.
We record
our research and development costs by type or category, rather than by
project. Significant categories of costs include personnel,
facilities and equipment costs, laboratory supplies and third-party and other
services. In addition, a significant portion of our research and
development expenses is not tracked by project as it benefits multiple
projects. Consequently, fully-loaded research and development cost
summaries by project are not available.
Consolidation
of Variable Interest Entity
We
consolidate the financial condition and results of operations of Symphony
Icon. While we have determined Symphony Icon is a variable interest
entity for which we are the primary beneficiary, Symphony Icon is wholly-owned
by the noncontrolling interest holders. Therefore, we reduce the
amount of our reported net loss in our consolidated statements of operations by
the loss attributed to the noncontrolling interest and we also reduce the
noncontrolling interest holders’ ownership interest in the consolidated balance
sheets by Symphony Icon’s losses.
As
discussed in Note 3, Recent Accounting Pronouncements, of the Notes to
Consolidated Financial Statements, we have determined that upon adoption of
SFAS 167, Amendments to FASB Interpretation No. 46(R), on
January 1, 2010, we are not the primary beneficiary of Symphony Icon, and
therefore, we will no longer include the financial condition and results of
operations of Symphony Icon in our consolidated financial
statements.
Stock-based
Compensation Expense
We
recognize compensation expense in our statements of operations for share-based
payments, including stock options issued to employees, based on their fair
values on the date of the grant, with the compensation expense recognized over
the period in which an employee is required to provide service in exchange for
the stock award. Stock-based compensation expense is recognized on a
straight-line basis. We had stock-based compensation expense of
$5.3 million for the year ended December 31, 2009, or $0.04 per
share. Stock-based compensation expense has no impact on cash flows
from operating activities or financing activities. As of December 31,
2009, stock-based compensation cost for all outstanding unvested options was
$6.8 million, which is expected to be recognized over a weighted-average
vesting period of 1.2 years.
The fair
value of stock options is estimated at the date of grant using the Black-Scholes
option-pricing model. For purposes of determining the fair value of
stock options, we segregate our options into two homogeneous groups, based on
exercise and post-vesting employment termination behaviors, resulting in a
change in the assumptions used for expected option lives and
forfeitures. Expected volatility is based on the historical
volatility in our stock price. The following weighted-average
assumptions were used for options granted in the years ended December 31, 2009,
2008 and 2007, respectively:
|
Expected
Volatility
|
Risk-free
Interest Rate
|
Expected
Term
|
Estimated
Forfeitures
|
Dividend
Rate
|
||||||||||||||||
|
December
31, 2009:
|
||||||||||||||||||||
|
Employees
|
78 | % | 1.9 | % | 5 | 24 | % | 0 | % | |||||||||||
|
Officers
and non-employee directors
|
77 | % | 2.7 | % | 8 | 7 | % | 0 | % | |||||||||||
|
December
31, 2008:
|
||||||||||||||||||||
|
Employees
|
66 | % | 2.9 | % | 6 | 22 | % | 0 | % | |||||||||||
|
Officers
and non-employee directors
|
66 | % | 3.8 | % | 9 | 6 | % | 0 | % | |||||||||||
|
December
31, 2007:
|
||||||||||||||||||||
|
Employees
|
66 | % | 4.5 | % | 6 | 21 | % | 0 | % | |||||||||||
|
Officers
and non-employee directors
|
67 | % | 4.6 | % | 9 | 4 | % | 0 | % | |||||||||||
Goodwill
Impairment
Goodwill
is not amortized, but is tested at least annually for impairment at the
reporting unit level. We have determined that the reporting unit is
the single operating segment disclosed in our current financial
statements. Impairment is the condition that exists when the carrying
amount of goodwill exceeds its implied fair value. The first step in
the impairment process is to determine the fair value of the reporting unit and
then compare it to the carrying value, including goodwill. We
determined that the market capitalization approach is the most appropriate
method of measuring fair value of the reporting unit. Under this
approach, fair value is calculated as the average closing price of our common
stock for the 30 days preceding the date that the annual impairment test is
performed, multiplied by the number of outstanding shares on that
date. A control premium, which is representative of premiums paid in
the marketplace to acquire a controlling interest in a company, is then added to
the market capitalization to determine the fair value of the reporting
unit. If the fair value exceeds the carrying value, no further action
is required and no impairment loss is recognized. Additional
impairment assessments may be performed on an interim basis if we encounter
events or changes in circumstances that would indicate that, more likely than
not, the carrying value of goodwill has been impaired. There was no
impairment of goodwill in 2009.
Valuation
of Investments that Do Not Have Active Markets
At
December 31, 2009, we held $56.2 million (par value) of investments with an
auction interest rate reset feature, known as auction rate
securities. The securities have historically traded at par and are
redeemable at par plus accrued interest at the option of the
issuer. Until February 2008, the carrying value of our auction rate
securities approximated fair value. With the liquidity issues
experienced in the global credit and capital markets, our auction rate
securities have experienced multiple failed auctions and the estimated market
value of these securities is less than cost.
We
estimated the fair value of these auction rate securities using a discounted
cash flow analysis that considered the following key inputs: (a) the
underlying structure of each security; (b) the present value of the future
principal and interest payments discounted at rates considered to reflect
current market conditions and the relevant risk associated with each security;
and (c) consideration of the time horizon that the market value of each
security could return to its cost. We also considered secondary
market trading date in estimating the fair value of these auction rate
securities. We estimate that the fair value of these securities at
December 31, 2009 was $46.3 million.
In
November 2008, we accepted an offer from UBS AG, the investment bank that sold
us our auction rate securities, providing us with certain rights related to our
auction rate securities. These rights permit us to require UBS to
purchase our $56.2 million (par value) of auction rate securities at par
value during the period from June 30, 2010 through July 2,
2012. Conversely, UBS has the right, in its discretion, to purchase
or sell the securities at any time by paying us the par value of the
securities. We expect to exercise these rights and sell our auction
rate securities back to UBS on June 30, 2010, the earliest date allowable
under the rights. We estimate that the fair value of these rights at
December 31, 2009 was $9.7 million.
The
enforceability of the rights results in a separate asset that is measured at its
fair value. We elected to measure the rights under a fair value
option, which permits entities to choose, at certain election dates, to measure
eligible items at fair value. As a result of accepting the rights, we
elected in 2008 to classify the rights and reclassify our investments in auction
rate securities as trading securities from available-for-sale
securities. As a result, we will assess the fair value of these two
individual assets and record changes each period until the rights are exercised
and the auction rate securities are redeemed. We expect that
subsequent changes in the value of the rights will largely offset the subsequent
fair value movements of the auction rate securities, subject to the continued
expected performance by UBS of its obligations under the agreement.
The fair
value of the auction rate securities and the associated rights could further
change significantly in the future and we may be required to record additional
other-than-temporary impairment charges related to the auction rate securities
and gains related to the rights if there are further reductions in fair value of
the auction rate securities in future periods.
Recent
Accounting Pronouncements
See Note
3, Recent Accounting Pronouncements, of the Notes to Consolidated Financial
Statements, for a discussion of the impact of new accounting standards on our
consolidated financial statements.
Results
of Operations – Comparison of Years Ended December 31, 2009, 2008 and 2007
Revenues
Total
revenues and dollar and percentage changes as compared to the prior year are as
follows (dollar amounts are presented in millions):
|
Year
Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Total
revenues
|
$
|
10.7
|
$
|
32.3
|
$
|
50.1
|
||||||
|
Dollar
decrease
|
$
|
(21.6
|
)
|
$
|
(17.8
|
)
|
||||||
|
Percentage
decrease
|
(67%
|
)
|
(36%
|
)
|
||||||||
Years
Ended December 31, 2009 and 2008
|
|
·
|
Collaborative research
– Revenue from collaborative research decreased 66% to $9.3 million,
primarily due to reduced revenues under our alliances with Bristol-Myers
Squibb and N.V. Organon due to the completion in 2009 of the target
discovery portion of these alliances, and completion in 2008 of the target
discovery portion of our alliance with Genentech, partially offset by
increases in revenue from our collaboration with
Taconic.
|
|
|
·
|
Subscription and license
fees – Revenue from subscriptions and license fees decreased 73% to
$1.4 million, primarily due to a decrease in technology license
fees.
|
Years
Ended December 31, 2008 and 2007
|
|
·
|
Collaborative research
– Revenue from collaborative research decreased 43% to $27.2 million,
primarily due to the completion in 2007 of the knockout mouse embryonic
stem cell library project funded by our award from the Texas Enterprise
Fund, reduced revenues under our alliance with N.V. Organon due to our
progress towards completion of the target discovery portion of the
alliance, and the completion in 2007 of the target discovery portion of
our alliance with Takeda Pharmaceutical
Limited.
|
|
|
·
|
Subscription and license
fees – Revenue from subscriptions and license fees increased 152%
to $5.1 million, primarily due to an increase in technology license
fees.
|
In 2009,
Bristol-Myers Squibb, Organon and Taconic represented 31%, 23% and 21% of
revenues, respectively. In 2008, Bristol-Myers Squibb, Organon and
Genentech represented 32%, 29% and 13% of revenues, respectively. In
2007, Organon, Bristol-Myers Squibb and the Texas Enterprise Fund represented
27%, 23% and 22% of revenues, respectively.
Research
and Development Expenses
Research
and development expenses and dollar and percentage changes as compared to the
prior year are as follows (dollar amounts are presented in
millions):
|
Year
Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Total
research and development expense
|
$
|
81.2
|
$
|
107.2
|
$
|
103.2
|
||||||
|
Dollar
increase (decrease)
|
$
|
(26.0
|
)
|
$
|
4.0
|
|||||||
|
Percentage
increase (decrease)
|
(24%
|
)
|
4%
|
|
||||||||
Research
and development expenses consist primarily of salaries and other
personnel-related expenses, third-party and other services principally related
to preclinical and clinical development activities, facility and equipment
costs, laboratory supplies and stock-based compensation expenses.
Years
Ended December 31, 2009 and 2008
|
|
·
|
Personnel – Personnel
costs decreased 21% in 2009 to $32.7 million, primarily due to
reductions in our personnel in May 2008 and January
2009. Salaries, bonuses, employee benefits, payroll taxes,
recruiting and relocation costs are included in personnel
costs.
|
|
|
·
|
Third-party and other services
– Third-party and other services decreased 33% in 2009 to
$20.2 million, primarily due to a decrease in external preclinical
research and development costs. Third-party and other services
include third-party research services, technology licenses and
subscriptions to third-party
databases.
|
|
|
·
|
Facilities and equipment –
Facilities and equipment costs decreased 17% in 2009 to
$15.3 million, primarily due to decreases in depreciation expense and
utilities expense.
|
|
|
·
|
Laboratory supplies –
Laboratory supplies expense decreased 28% in 2009 to
$6.2 million, primarily due to reductions in our genetics research
activities.
|
|
|
·
|
Stock-based compensation
– Stock-based compensation expense decreased 23% in 2009 to
$3.0 million, primarily as a result of the reduction in our
personnel.
|
|
|
·
|
Other – Other costs
decreased 22% in 2009 to $3.8 million, primarily due to a decrease in
computer software expense.
|
Years
Ended December 31, 2008 and 2007
|
|
·
|
Personnel – Personnel
costs decreased 7% to $41.4 million, primarily due to a reduction in our
personnel in May 2008.
|
|
|
·
|
Third-party and other
services – Third-party and other services increased 72% to
$29.9 million, primarily due to an increase in external preclinical
and clinical research and development
costs.
|
|
|
·
|
Facilities and
equipment – Facilities and equipment costs decreased 8% to
$18.5 million, primarily due to a decrease in depreciation
expense.
|
|
|
·
|
Laboratory supplies –
Laboratory supplies expense decreased 25% to $8.6 million, primarily
due to the reduction in personnel in May
2008.
|
|
|
·
|
Stock-based
compensation – Stock-based compensation expense decreased 23% to
$3.9 million, primarily due to the reduction in our personnel in May
2008.
|
|
|
·
|
Other – Other costs
increased by 2% to
$4.9 million.
|
General
and Administrative Expenses
General
and administrative expenses and dollar and percentage changes as compared to the
prior year are as follows (dollar amounts are presented in
millions):
|
Year
Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Total
general and administrative expense
|
$
|
19.4
|
$
|
21.6
|
$
|
21.8
|
||||||
|
Dollar
decrease
|
$
|
(2.2
|
)
|
$
|
(0.2
|
)
|
||||||
|
Percentage
decrease
|
(10%
|
)
|
(1%
|
)
|
||||||||
General
and administrative expenses consist primarily of personnel costs to support our
research and development activities, professional fees such as legal fees,
facility and equipment costs, and stock-based compensation
expenses.
Years
Ended December 31, 2009 and 2008
|
|
·
|
Personnel – Personnel
costs decreased 13% in 2009 to $9.0 million, primarily due to
reductions in our personnel in May 2008 and January
2009. Salaries, bonuses, employee benefits, payroll taxes,
recruiting and relocation costs are included in personnel
costs.
|
|
|
·
|
Professional fees –
Professional fees decreased 7% in 2009 to $4.0 million, primarily due
to decreased consulting fees.
|
|
|
·
|
Facilities and
equipment – Facilities and equipment costs were $2.5 million,
consistent with the prior year.
|
|
|
·
|
Stock-based
compensation – Stock-based compensation expense decreased 12% in
2009 to $2.3 million, primarily as a result of the reduction in our
personnel.
|
|
|
·
|
Other – Other costs
decreased 16% in 2009 to
$1.6 million.
|
Years
Ended December 31, 2008 and 2007
|
|
·
|
Personnel – Personnel
costs decreased 3% to $10.4 million, primarily due to lower bonus and
benefit costs, offset in part by severance costs associated with
reductions in personnel.
|
|
|
·
|
Professional fees –
Professional fees increased 20% to $4.3 million, primarily due to
increased market research and other consulting
costs.
|
|
|
·
|
Facilities and
equipment – Facilities and equipment costs were $2.5 million,
consistent with the prior year.
|
|
|
·
|
Stock-based
compensation – Stock-based compensation expense decreased 8% to
$2.6 million.
|
|
|
·
|
Other – Other costs
decreased 16% to $1.9 million.
|
Gain
(Loss) on Investments, Net, Interest Income, Interest Expense and Other
(Expense) Income, Net
Gain (Loss) on Investments,
Net. Gain on investments was $3.5 million for the year
ended December 31, 2009, representing the increase in fair value of our
student loan auction rate securities. This gain was partially offset
by a loss on investments of $2.3 million for the year ended December 31,
2009, representing the decline in fair value of the rights obtained from
UBS. Loss on investments was $13.4 million for the year ended
December 31, 2008, representing the decline in fair value of our student
loan auction rate securities. This loss was partially offset by a
gain on investments of $12.1 million for the year ended December 31,
2008, representing the fair value of the rights.
Interest Income. Interest income decreased
85% in 2009 to $0.9 million from $5.8 million in 2008 primarily due to
lower average cash and investment balances as well as lower yields on our
investments. Interest income decreased 21% in 2008 from
$7.3 million in 2007, primarily due to lower average cash and investment
balances as well as lower yields on our investments.
Interest Expense. Interest expense
increased 10% in 2009 to $3.0 million from $2.7 million in 2008 and
decreased 3% in 2008 from $2.8 million in 2007.
Other (Expense) Income,
Net. Other expense, net increased 21% in 2009 to
$2.6 million from 2008 primarily due to an impairment of surplus equipment
as a result of our reduction of personnel in January 2009. Other
expense, net increased 165% in 2008 to $2.1 million from 2007 primarily due
to the increase in amortization of the asset related to the option to purchase
the equity of Symphony Icon. We have recorded the value of the
purchase option as an asset, and we are amortizing this asset over the four-year
option period (see Note 10, Arrangements with Symphony Icon, Inc., of the
Notes to Consolidated Financial Statements, for more information).
Income
Tax Benefit
The
income tax benefit for the year ended December 31, 2009 was
$102,000.
Noncontrolling
Interest in Symphony Icon, Inc.
The loss
attributed to the noncontrolling interest holders of Symphony Icon decreased 47%
to $10.5 million from $20.0 million in 2008 and increased 61% in 2008
from $12.4 million in 2007, due to the timing of expenditures related to
clinical development of the drug candidates licensed to Symphony
Icon.
Net
Loss Attributable to Lexicon Pharmaceuticals, Inc. and Net Loss Attributable to
Lexicon Pharmaceuticals, Inc. per Common Share
Net loss
attributable to Lexicon Pharmaceuticals, Inc. increased to $82.8 million in
2009 from $76.9 million in 2008 and $58.8 million in
2007. Net loss attributable to Lexicon Pharmaceuticals, Inc. per
common share increased to $0.57 in 2009 from $0.56 in 2008 and decreased from
$0.59 in 2007.
Liquidity
and Capital Resources
We have
financed our operations from inception primarily through sales of common and
preferred stock, contract and milestone payments to us under our drug discovery
and development collaborations, target validation, database subscription and
technology license agreements, government grants and contracts and financing
under debt and lease arrangements. We have also financed certain of
our research and development activities under our agreements with Symphony Icon,
Inc. From our inception through December 31, 2009, we had received
net proceeds of $605.4 million from issuances of common and preferred
stock. In addition, from our inception through December 31, 2009, we
received $448.7 million in cash payments from drug discovery and
development collaborations, target validation, database subscription and
technology license agreements, sales of compound libraries and reagents and
government grants and contracts, of which $434.7 million had been
recognized as revenues through December 31, 2009.
As of
December 31, 2009, we had $157.1 million in cash, cash equivalents and
investments, including $56.0 million in auction rate securities and related
rights as discussed below under “Disclosure about Market Risk,” and $5.4 million
in investments held by Symphony Icon. As of December 31, 2008, we had
$142.2 million in cash, cash equivalents and investments, including $55.7
million of auction rate securities and related rights, and $16.6 million in
investments held by Symphony Icon. We used cash of $89.0 million in
operations in 2009. This consisted primarily of the consolidated net loss for
the period of $93.3 million, a $4.7 million decrease in deferred revenue, a net
decrease in other operating liabilities net of assets of $3.8 million, and a net
gain on investments and auction rate security rights of $1.2 million, partially
offset by non-cash charges of $6.2 million related to depreciation expense, $5.3
million related to stock-based compensation expense and $2.1 million related to
the amortization of the Symphony Icon purchase option. Investing
activities provided cash of $11.9 million in 2009, primarily due to maturities
of investments of $76.3 million, partially offset by purchases of investments of
$64.2 million. Financing activities provided cash of $91.8 million
due to proceeds from issuance of common stock of $55.4 million, proceeds
from debt borrowings of $38.6 million, partially offset by repayment of debt
borrowings of $2.1 million.
UBS Credit
Line. In January 2009, we entered into a credit line agreement
with UBS Bank USA that provides, as of December 31, 2009, up to an
aggregate amount of $37.5 million in the form of an uncommitted, demand,
revolving line of credit. We entered into the credit line in
connection with our acceptance of an offer from UBS AG, the investment bank that
sold us our auction rate securities, providing us with rights to require UBS to
purchase our $56.2 million (par value) of auction rate securities at par value
during the period from June 30, 2010 through July 2,
2012. The credit line is secured only by these auction rate
securities and advances under the credit line will be made on a “no net cost”
basis, meaning that the interest paid by us on advances will not exceed the
interest or dividends paid to us by the issuer of the auction rate
securities. As of December 31, 2009 we had $37.4 million
outstanding under the credit line.
Invus Securities Purchase
Agreement. In June 2007, we entered into a securities purchase
agreement with Invus, L.P., under which Invus made an initial investment of
$205.4 million to purchase 50,824,986 shares of our common stock in August 2007
and has the right to require us to initiate up to two pro rata rights offerings
to our stockholders, which would provide all stockholders with non-transferable
rights to acquire shares of our common stock, in an aggregate amount of up to
$344.5 million, less the proceeds of any “qualified offerings” that we may
complete in the interim involving the sale of our common stock at prices above
$4.50 per share. We have not completed any such qualified
offering. Invus may exercise its right to require us to conduct the
first rights offering by giving us notice within a period of one year beginning
on November 28, 2009 (which we refer to as the first rights offering
trigger date). Invus may exercise its right to require us to conduct
a second rights offering by giving us notice within a period of one year
beginning on the date that is 90 days after Invus’ exercise of its right to
require us to conduct the first rights offering or, if Invus does not exercise
its right to require us to conduct the first rights offering, within a period of
one year beginning on the first anniversary of the first rights offering trigger
date. If Invus elects to exercise its right to require us to initiate
a rights offering, Invus would be required to purchase its pro rata portion of
the offering.
In
connection with the securities purchase agreement, we entered into a
stockholders’ agreement with Invus under which Invus (a) has specified rights
with respect to designation of directors and participation in future equity
issuances by us, (b) is subject to certain standstill restrictions, as well as
restrictions on transfer and the voting of the shares of common stock held by it
and its affiliates, and (c), as long as Invus holds at least 15% of the total
number of outstanding shares of our common stock, is entitled to certain
minority protections.
Symphony Drug Development Financing
Agreement. In June 2007, we entered into a series of related
agreements providing for the financing of the clinical development of certain
drug programs, including LX1031 and LX1032, along with any other pharmaceutical
compositions modulating the same targets as those drug
candidates. Under the financing arrangement, we licensed to Symphony
Icon, a wholly-owned subsidiary of Symphony Icon Holdings LLC, our intellectual
property rights related to the programs and Holdings contributed $45 million to
Symphony Icon in order to fund the clinical development of the
programs. We also entered into a share purchase agreement with
Holdings under which we issued and sold to Holdings 7,650,622 shares of our
common stock in exchange for $15 million and an exclusive option to acquire
all of the equity of Symphony Icon, thereby allowing us to reacquire the
programs. The purchase option is exercisable by us at any time, in
our sole discretion, until June 15, 2011 at an exercise price of
(a) $81 million, if the purchase option is exercised before
June 15, 2010 and (b) $90 million, if the purchase option is
exercised on or after June 15, 2010 and before June 15,
2011. The purchase option exercise price may be paid in cash or a
combination of cash and common stock, at our sole discretion, provided that the
common stock portion may not exceed 40% of the purchase option exercise
price.
Upon the
recommendation of Symphony Icon’s development committee, which is comprised of
an equal number of representatives from us and Symphony Icon, Symphony Icon’s
board of directors may require us to pay Symphony Icon up to $15 million for
Symphony Icon’s use in the development of the programs in accordance with the
specified development plan and related development budget. The
development committee’s right to recommend that Symphony Icon’s board of
directors submit such funding requirement to us will terminate on the one-year
anniversary of the expiration of the purchase option, subject to limited
exceptions. Through December 31, 2009, Symphony Icon’s board of
directors has requested us to pay Symphony Icon $4.3 million under the
agreement, all of which has been paid, and we expect that additional funding
will be needed in the future.
Facilities. In
April 2004, we obtained a $34.0 million mortgage on our facilities in The
Woodlands, Texas. The mortgage loan has a ten-year term with a
20-year amortization and bears interest at a fixed rate of 8.23%. In
May 2002, our subsidiary Lexicon Pharmaceuticals (New Jersey), Inc. signed a
ten-year lease for its facility in Hopewell, New Jersey extending the term until
June 30, 2013. The lease provides for an escalating yearly base
rent payment of $1.3 million in the first year, $2.1 million in years two
and three, $2.2 million in years four to six, $2.3 million in years
seven to nine and $2.4 million in years ten and eleven. We are
the guarantor of the obligations of our subsidiary under the lease.
Including
the lease and debt obligations described above, we had incurred the following
contractual obligations as of December 31, 2009:
|
Payments
due by period (in millions)
|
||||||||||||||||||||
|
Contractual
Obligations
|
Total
|
Less
than 1 year
|
1-3
years
|
3-5
years
|
More
than 5 years
|
|||||||||||||||
|
Debt
|
$
|
67.0
|
$
|
38.5
|
$
|
2.4
|
$
|
26.1
|
$
|
—
|
||||||||||
|
Interest
payment obligations
|
9.8
|
2.4
|
4.6
|
2.8
|
—
|
|||||||||||||||
|
Operating
leases
|
9.0
|
2.5
|
5.1
|
1.4
|
—
|
|||||||||||||||
|
Total
|
$
|
85.8
|
$
|
43.4
|
$
|
12.1
|
$
|
30.3
|
$
|
—
|
||||||||||
Our
future capital requirements will be substantial and will depend on many factors,
including our ability to obtain drug discovery and development collaborations
and other collaborations and technology license agreements, the amount and
timing of payments under such agreements, the level and timing of our research
and development expenditures, market acceptance of our products, the resources
we devote to developing and supporting our products and other
factors. Our capital requirements will also be affected by any
expenditures we make in connection with license agreements and acquisitions of
and investments in complementary technologies and businesses. We
expect to devote substantial capital resources to continue our research and
development efforts, to expand our support and product development activities,
and for other general corporate activities. We believe that our
current unrestricted cash and investment balances and cash and revenues we
expect to derive from drug discovery and development collaborations, other
collaborations and technology licenses and other sources will be sufficient to
fund our operations for at least the next 12 months. During or after
this period, if cash generated by operations is insufficient to satisfy our
liquidity requirements, we will need to sell additional equity or debt
securities or obtain additional credit arrangements. Additional
financing may not be available on terms acceptable to us or at all. The sale of
additional equity or convertible debt securities may result in additional
dilution to our stockholders.
Disclosure
about Market Risk
We are
exposed to limited market and credit risk on our cash equivalents which have
maturities of three months or less at the time of purchase. We
maintain a short-term investment portfolio which consists of U.S. Treasury
bills, money market accounts, corporate debt securities and certificates of
deposit that mature three to 12 months from the time of purchase and a long-term
investment portfolio which consists of auction rate securities that mature
greater than 12 months from the time of purchase, which we believe are subject
to limited market and credit risk, other than as discussed below. We
currently do not hedge interest rate exposure or hold any derivative financial
instruments in our investment portfolio.
At
December 31, 2009, we held $56.2 million (par value), with an estimated fair
value of $46.3 million, of investments with an auction interest rate reset
feature, known as auction rate securities. These notes are issued by
various state agencies for the purpose of financing student
loans. The securities have historically traded at par and are
redeemable at par plus accrued interest at the option of the
issuer. Interest is typically paid at the end of each auction period
or semiannually. Until February 2008, the market for our auction rate
securities was highly liquid. However, starting in February 2008, a
substantial number of auctions “failed,” meaning that there was not enough
demand to sell all of the securities that holders desired to sell at
auction. The immediate effect of a failed auction is that such
holders cannot sell the securities at auction and the interest rate on the
security generally resets to a maximum interest rate. In the case of
funds invested by us in auction rate securities which are the subject of a
failed auction, we may not be able to access the funds without a loss of
principal, unless a future auction on these investments is successful or the
issuer redeems the security. We have modified our current investment
strategy to reallocate our investments more into U.S. treasury securities and
U.S. treasury-backed money market investments.
At
December 31, 2009, observable auction rate securities market information
was not available to determine the fair value of our investments. We have
estimated the fair value of these securities at $46.3 million as of
December 31, 2009 using models of the expected future cash flows related to
the securities and taking into account assumptions about the cash flows of the
underlying student loans, as well as secondary market data. The
assumptions used in preparing the discounted cash flow model include estimates
of interest rates, timing and amount of cash flows, liquidity premiums and
expected holding periods of the auction rate securities, based on data available
as of December 31, 2009. The underlying sources of these
assumptions are volatile and the assumptions are subject to change as those
sources and market conditions change. If the current market conditions
deteriorate further, or a recovery in market values does not occur, we may be
required to record additional unrealized or realized losses in future
quarters.
In
November 2008, we accepted an offer from UBS AG, the investment bank that sold
us our auction rate securities, providing us with certain rights related to our
auction rate securities. The rights permit us to require UBS to
purchase our $56.2 million (par value) of auction rate securities at par value
during the period from June 30, 2010 through July 2,
2012. Conversely, UBS has the right, in its discretion, to purchase
or sell the securities at any time by paying us the par value of such
securities. We expect to exercise the rights and sell our auction
rate securities back to UBS on June 30, 2010, the earliest date allowable
under the rights.
The
enforceability of the rights results in a separate asset that is measured at its
fair value. We elected to measure the rights under a fair value
option, which permits entities to choose, at certain election dates, to measure
eligible items at fair value. As a result of accepting the rights, we
have elected to classify the rights and reclassify our investments in auction
rate securities as trading securities from available-for-sale
securities. As a result, we will assess the fair value of these two
individual assets and record changes each period until the rights are exercised
and the auction rate securities are redeemed. We expect that
subsequent changes in the value of the rights will largely offset the subsequent
fair value movements of the auction rate securities, subject to the continued
expected performance by the investment bank of its obligations under the
agreement.
Excluding
auction rate securities and the related rights, at December 31, 2009, we
had approximately $106.5 million in cash and cash equivalents and
short-term investments, including $5.4 million in investments held by
Symphony Icon. We believe that the working capital available to us
excluding the funds held in auction rate securities will be sufficient to meet
our cash requirements for at least the next 12 months.
We have
operated primarily in the United States and substantially all sales to date have
been made in U.S. dollars. Accordingly, we have not had any material exposure to
foreign currency rate fluctuations.
Item 7A. Quantitative and Qualitative
Disclosures About Market Risk
See
“Disclosure about Market Risk” under “Item 7. Management’s Discussion and
Analysis of
Financial Condition and Results of Operations” for quantitative and qualitative
disclosures about market risk.
Item
8. Financial Statements and
Supplementary Data
The
financial statements required by this Item are incorporated under Item 15 in
Part IV of this report.
Item
9. Changes in and Disagreements with
Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and
Procedures
Our
principal executive officer and principal financial officer have concluded that
our disclosure controls and procedures (as defined in rules 13a-15(e) and
15d-15(e) under the Securities Exchange Act of 1934) are effective to ensure
that the information required to be disclosed by us in the reports we file under
the Securities Exchange Act is gathered, analyzed and disclosed with adequate
timeliness, accuracy and completeness, based on an evaluation of such controls
and procedures as of the end of the period covered by this report.
Subsequent
to our evaluation, there were no significant changes in internal controls or
other factors that could significantly affect internal controls, including any
corrective actions with regard to significant deficiencies and material
weaknesses.
Management
Report on Internal Control over Financial Reporting
Our
management is responsible for establishing and maintaining adequate internal
control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f)
under the Securities Exchange Act).
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements. Projections of any evaluation of
effectiveness to future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree of compliance
with the policies or procedures may deteriorate.
Our
management assessed the effectiveness of our internal control over financial
reporting as of December 31, 2009. In making this assessment,
management used the criteria set forth by the Committee of Sponsoring
Organizations of the Treadway Commission in Internal Control-Integrated
Framework.
Based on
such assessment using those criteria, management believes that, as of December
31, 2009, our internal control over financial reporting is
effective.
Our
independent auditors have also audited our internal control over financial
reporting as of December 31, 2009 as stated in the audit report which
appears on page F-2 and is incorporated under Item 15 in Part IV of
this report.
Item 9B. Other
Information
None.
PART
III
Item
10. Directors, Executive Officers and
Corporate Governance
The
information required by this Item is hereby incorporated by reference from (a)
the information appearing under the captions “Election of Directors,” “Stock
Ownership of Certain Beneficial Owners and Management,” “Corporate Governance”
and “Executive and Director Compensation” in our definitive proxy statement
which involves the election of directors and is to be filed with the Securities
and Exchange Commission pursuant to the Securities Exchange Act of 1934 within
120 days of the end of our fiscal year on December 31, 2009 and (b) the
information appearing under Item 1 in Part I of this
report.
Item 11. Executive
Compensation
The
information required by this Item is hereby incorporated by reference from the
information appearing under the captions “Corporate Governance” and “Executive
and Director Compensation” in our definitive proxy statement which involves the
election of directors and is to be filed with the Commission pursuant to the
Securities Exchange Act of 1934 within 120 days of the end of our fiscal year on
December 31, 2009. Notwithstanding the foregoing, in accordance with the
instructions to Item 407(e)(5) of Regulation S-K, the information contained in
our proxy statement under the sub-heading “Compensation Committee Report” shall
not be deemed to be filed as part of or incorporated by reference into this
annual report on Form 10-K.
Item
12. Security Ownership of Certain
Beneficial Owners and Management and Related Stockholder
Matters
The
information required by this Item is hereby incorporated by reference from the
information appearing under the captions “Stock Ownership of Certain Beneficial
Owners and Management” and “Equity Compensation Plan Information” in our
definitive proxy statement which involves the election of directors and is to be
filed with the Commission pursuant to the Securities Exchange Act of 1934 within
120 days of the end of our fiscal year on December 31, 2009.
Item
13. Certain Relationships and Related
Transactions, and Director Independence
The
information required by this Item is hereby incorporated by reference from the
information appearing under the captions “Corporate Governance” and
“Transactions with Related Persons” in our definitive proxy statement which
involves the election of directors and is to be filed with the Commission
pursuant to the Securities Exchange Act of 1934 within 120 days of the end of
our fiscal year on December 31, 2009.
Item
14. Principal Accounting Fees and
Services
The
information required by this Item as to the fees we pay our principal accountant
is hereby incorporated by reference from the information appearing under the
caption “Ratification and Approval of Independent Auditors” in our definitive
proxy statement which involves the election of directors and is to be filed with
the Commission pursuant to the Securities Exchange Act of 1934 within 120 days
of the end of our fiscal year on December 31, 2009.
PART
IV
Item
15. Exhibits and Financial Statement
Schedules
|
|
(a)
|
Documents
filed as a part of this report:
|
1. Consolidated
Financial Statements
|
Page
|
|
|
Report
of Independent Registered Public Accounting Firm
|
F-1
|
|
Report
of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated
Balance Sheets
|
F-3
|
|
Consolidated
Statements of Operations
|
F-4
|
|
Consolidated
Statements of Stockholders’ Equity
|
F-5
|
|
Consolidated
Statements of Cash Flows
|
F-6
|
|
Notes
to Consolidated Financial Statements
|
F-7
|
2. Financial
Statement Schedules
All other
financial statement schedules are omitted because they are not applicable or not
required, or because the required information is included in the financial
statements or notes thereto.
3. Exhibits
|
Exhibit No.
|
Description
|
|
|
3.1
|
—
|
Restated
Certificate of Incorporation (filed as Exhibit 3.1 to the Company’s
Registration Statement on Form S-1 (Registration No. 333-96469) and
incorporated by reference herein).
|
|
3.2
|
—
|
First
Certificate of Amendment to Restated Certificate of Incorporation (filed
as Exhibit 3.2 to the Company’s Annual Report on Form 10-K for the period
ended December 31, 2007 and incorporated by reference
herein).
|
|
3.3
|
—
|
Second
Certificate of Amendment to Restated Certificate of Incorporation (filed
as Exhibit 3.3 to the Company’s Annual Report on Form 10-K for the period
ended December 31, 2007 and incorporated by reference
herein).
|
|
3.4
|
—
|
Third
Certificate of Amendment to Restated Certificate of Incorporation (filed
as Exhibit 3.1 to the Company’s Quarterly Report on Form 10-Q for the
period ended June 30, 2009 and incorporated by reference
herein).
|
|
3.5
|
—
|
Amended
and Restated Bylaws (filed as Exhibit 3.1 to the Company’s Current Report
on Form 8-K dated October 24, 2007 and incorporated by reference
herein).
|
|
4.1
|
—
|
Securities
Purchase Agreement, dated June 17, 2007, with Invus, L.P. (filed as
Exhibit 10.1 to the Company’s Current Report on Form 8-K dated June 17,
2007 and incorporated by reference herein).
|
|
4.2
|
—
|
Amendment,
dated October 7, 2009, to Securities Purchase Agreement, dated
June 17, 2007, with Invus, L.P. (filed as Exhibit 10.1 to the
Company’s Current Report on Form 8-K dated October 7, 2009 and
incorporated by reference herein).
|
|
4.3
|
—
|
Registration
Rights Agreement, dated June 17, 2007, with Invus, L.P. (filed as Exhibit
10.3 to the Company’s Current Report on Form 8-K dated June 17, 2007 and
incorporated by reference herein).
|
|
4.4
|
—
|
Stockholders’
Agreement, dated June 17, 2007, with Invus, L.P. (filed as Exhibit 10.4 to
the Company’s Current Report on Form 8-K dated June 17, 2007 and
incorporated by reference herein).
|
|
10.1
|
—
|
Restated
Employment Agreement with Arthur T. Sands, M.D., Ph.D. (filed as Exhibit
10.1 to the Company’s Annual Report on Form 10-K for the period ended
December 31, 2005 and incorporated by reference
herein).
|
| Exhibit No. | Description | |
|
10.2
|
—
|
Employment
Agreement with Alan Main, Ph.D. (filed as Exhibit 10.1 to the Company’s
Quarterly Report on Form 10-Q for the period ended September 30, 2001 and
incorporated by reference herein).
|
|
10.3
|
—
|
Employment
Agreement with Jeffrey L. Wade, J.D. (filed as Exhibit 10.3 to the
Company’s Registration Statement on Form S-1 (Registration No. 333-96469)
and incorporated by reference herein).
|
|
10.4
|
—
|
Employment
Agreement with Brian P. Zambrowicz, Ph.D. (filed as Exhibit 10.4 to the
Company’s Registration Statement on Form S-1 (Registration No. 333-96469)
and incorporated by reference herein).
|
|
10.5
|
—
|
Offer
Letter, dated May 4, 2009, with Ajay Bansal (filed as Exhibit 10.1 to the
Company’s Current Report on Form 8-K dated May 4, 2009 and incorporated by
reference herein).
|
|
10.6
|
—
|
Consulting
Agreement with Alan S. Nies, M.D. dated February 19, 2003, as amended
(filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for
the period ended March 31, 2004 and incorporated by reference
herein).
|
|
10.7
|
—
|
Consulting
Agreement with Robert J. Lefkowitz, M.D. dated March 31, 2003 (filed as
Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the period
ended March 31, 2003 and incorporated by reference
herein).
|
|
10.8
|
—
|
Form
of Indemnification Agreement with Officers and Directors (filed as Exhibit
10.7 to the Company’s Registration Statement on Form S-1 (Registration No.
333-96469) and incorporated by reference herein).
|
|
10.9
|
—
|
Summary
of Non-Employee Director Compensation (filed as Exhibit 10.3 to the
Company’s Current Report on Form 8-K dated April 23, 2009 and incorporated
by reference herein).
|
|
10.10
|
—
|
Summary
of 2010 Named Executive Officer Cash Compensation (filed as Exhibit 10.1
to the Company’s Current Report on Form 8-K dated February 15, 2010 and
incorporated by reference herein).
|
|
10.11
|
—
|
Equity
Incentive Plan (filed as Exhibit 10.1 to the Company’s Current Report on
Form 8-K dated April 23, 2009 and incorporated by reference
herein).
|
|
10.12
|
—
|
Non-Employee
Directors’ Stock Option Plan (filed as Exhibit 10.2 to the Company’s
Current Report on Form 8-K dated April 23, 2009 and incorporated by
reference herein).
|
|
10.13
|
—
|
Coelacanth
Corporation 1999 Stock Option Plan (filed as Exhibit 99.1 to the Company’s
Registration Statement on Form S-8 (Registration No. 333-66380) and
incorporated by reference herein).
|
|
10.14
|
—
|
Form
of Stock Option Agreement with Chairman of Board of Directors under the
Equity Incentive Plan (filed as Exhibit 10.17 to the Company’s Annual
Report on Form 10-K for the period ended December 31, 2005 and
incorporated by reference herein).
|
|
*10.15
|
—
|
Form
of Stock Option Agreement with Directors under the Non-Employee Directors’
Stock Option Plan.
|
|
*10.16
|
—
|
Form
of Stock Option Agreement with Officers under the Equity Incentive
Plan.
|
|
10.17
|
—
|
Form
of Restricted Stock Bonus Agreement with Officers under the Equity
Incentive Plan (filed as Exhibit 10.2 to the Company’s Current Report on
Form 8-K dated February 12, 2009 and incorporated by reference
herein).
|
|
10.18
|
—
|
Form
of Restricted Stock Unit Agreement with Officers under the Equity
Incentive Plan (filed as Exhibit 10.2 to the Company’s Current Report
on Form 8-K dated February 15, 2010 and incorporated by reference
herein).
|
|
†10.19
|
—
|
Collaboration
and License Agreement, dated December 17, 2003, with Bristol-Myers
Squibb Company (filed as Exhibit 10.15 to the amendment to the Company’s
Annual Report on Form 10-K/A for the period ended December 31, 2003, as
filed on July 16, 2004, and incorporated by reference
herein).
|
| Exhibit No. | Description | |
|
†10.20
|
—
|
First
Amendment, dated May 30, 2006, to Collaboration and License Agreement,
dated December 17, 2003, with Bristol-Myers Squibb Company (filed as
Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the period
ended June 30, 2006, and incorporated by reference
herein).
|
|
†10.21
|
—
|
Collaboration
Agreement, dated July 27, 2004, with Takeda Pharmaceutical Company Limited
(filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for
the period ended September 30, 2004 and incorporated by reference
herein).
|
|
†10.22
|
—
|
Collaboration
and License Agreement, dated May 16, 2005, with N.V. Organon and (only
with respect to Section 9.4 thereof) Intervet Inc. (filed as Exhibit 10.1
to the Company’s Quarterly Report on Form 10-Q for the period ended June
30, 2005 and incorporated by reference herein).
|
|
†10.23
|
—
|
Second
Amended and Restated Collaboration and License Agreement, dated November
30, 2005, with Genentech, Inc. (filed as Exhibit 10.22 to the Company’s
Annual Report on Form 10-K for the period ended December 31, 2005 and
incorporated by reference herein).
|
|
10.24
|
—
|
Amendment,
dated June 8, 2009, to Second Amended and Restated Collaboration and
License Agreement, dated November 30, 2005, with Genentech, Inc. (filed as
Exhibit 10.1 to the Company’s Current Report on Form 8-K/A dated June 8,
2009 and incorporated by reference herein).
|
|
10.25
|
—
|
Economic
Development Agreement dated July 15, 2005, with the State of Texas and the
Texas A&M University System (filed as Exhibit 10.1 to the Company’s
Quarterly Report on Form 10-Q for the period ended September 30, 2005 and
incorporated by reference herein).
|
|
10.26
|
—
|
Amendment,
dated April 30, 2008, to Economic Development Agreement, dated July 15,
2005, with the State of Texas and the Texas A&M University System
(filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated
April 30, 2008 and incorporated by reference herein).
|
|
†10.27
|
—
|
Novated
and Restated Technology License Agreement, dated June 15, 2007, with
Symphony Icon Holdings LLC and Symphony Icon, Inc. (filed as Exhibit 10.1
to the Company’s Quarterly Report on Form 10-Q for the period ended June
30, 2007 and incorporated by reference herein).
|
|
†10.28
|
—
|
Amended
and Restated Research and Development Agreement, dated June 15, 2007, with
Symphony Icon Holdings LLC and Symphony Icon, Inc. (filed as Exhibit 10.2
to the Company’s Quarterly Report on Form 10-Q for the period ended June
30, 2007 and incorporated by reference herein).
|
|
†10.29
|
—
|
Purchase
Option Agreement, dated June 15, 2007, with Symphony Icon Holdings LLC and
Symphony Icon, Inc. (filed as Exhibit 10.3 to the Company’s Quarterly
Report on Form 10-Q for the period ended June 30, 2007 and incorporated by
reference herein).
|
|
†10.30
|
—
|
Research
Cost Sharing, Payment and Extension Agreement, dated June 15, 2007, with
Symphony Icon Holdings LLC and Symphony Icon, Inc. (filed as Exhibit 10.4
to the Company’s Quarterly Report on Form 10-Q for the period ended June
30, 2007 and incorporated by reference herein).
|
|
10.31
|
—
|
Credit
Line Agreement, dated January 27, 2009, with UBS Bank USA (filed as
Exhibit 10.1 to the Company’s Current Report on Form 8-K dated January 27,
2009 and incorporated by reference herein).
|
|
10.32
|
—
|
Loan
and Security Agreement, dated April 21, 2004, between Lex-Gen Woodlands,
L.P. and iStar Financial Inc. (filed as Exhibit 10.18 to the Company’s
Annual Report on Form 10-K for the period ended December 31, 2004 and
incorporated by reference herein).
|
| Exhibit No. | Description | |
|
10.33
|
—
|
Lease
Agreement, dated May 23, 2002, between Lexicon Pharmaceuticals (New
Jersey), Inc. and Townsend Property Trust Limited Partnership (filed as
Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the
period ended June 30, 2002 and incorporated by reference
herein).
|
|
21.1
|
—
|
Subsidiaries
(filed as Exhibit 21.1 to the Company’s Annual Report on Form 10-K for the
year ended December 31, 2004 and incorporated by reference
herein).
|
|
*23.1
|
—
|
Consent
of Independent Registered Public Accounting Firm
|
|
*24.1
|
—
|
Power
of Attorney (contained in signature page)
|
|
*31.1
|
—
|
Certification
of Principal Executive Officer Pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
*31.2
|
—
|
Certification
of Principal Financial Officer Pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002
|
|
*32.1
|
—
|
Certification
of Principal Executive and Principal Financial Officers Pursuant to
Section 906 of the Sarbanes-Oxley Act of
2002
|
_____________________
* Filed
herewith.
|
|
†
|
Confidential
treatment has been requested for a portion of this exhibit. The
confidential portions of this exhibit have been omitted and filed
separately with the Securities and Exchange
Commission.
|
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of
1934, the registrant has duly caused this report to be signed on its behalf by
the undersigned thereunto duly authorized.
|
Lexicon
Pharmaceuticals, Inc.
|
|||
|
Date: March
5, 2010
|
By:
|
/s/
Arthur T.
Sands
|
|
|
Arthur
T. Sands, M.D., Ph.D.
|
|||
|
President
and Chief Executive Officer
|
|||
|
Date: March
5, 2010
|
By:
|
/s/
James F.
Tessmer
|
|
|
James
F. Tessmer
|
|||
|
Vice
President, Finance and Accounting
|
|||
Power
of Attorney
KNOW ALL
PERSONS BY THESE PRESENTS, that each person whose signature appears below
constitutes and appoints Jeffrey L. Wade and James F. Tessmer, or either of
them, each with the power of substitution, his or her attorney-in-fact, to sign
any amendments to this Form 10-K, and to file the same, with exhibits thereto
and other documents in connection therewith, with the Securities and Exchange
Commission, here ratifying and confirming all that each of said
attorneys-in-fact, or his or her substitute or substitutes, may do or cause to
be done by virtue hereof.
Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the registrant and in the
capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
|
|
/s/
Arthur T. Sands
|
President
and Chief Executive Officer
(Principal
Executive Officer)
|
March
5, 2010
|
|
|
Arthur
T. Sands, M.D., Ph.D.
|
|||
|
/s/
James F. Tessmer
|
Vice
President, Finance and Accounting
(Principal
Financial and Accounting Officer)
|
March
5, 2010
|
|
|
James
F. Tessmer
|
|||
|
/s/
Samuel L. Barker
|
Chairman
of the Board of Directors
|
March
5, 2010
|
|
|
Samuel
L. Barker, Ph.D.
|
|||
|
/s/
Philippe J. Amouyal
|
Director
|
March
5, 2010
|
|
|
Philippe
J. Amouyal
|
|||
|
/s/
Raymond Debbane
|
Director
|
March
5, 2010
|
|
|
Raymond
Debbane
|
|||
|
/s/
Robert J. Lefkowitz
|
Director
|
March
5, 2010
|
|
|
Robert
J. Lefkowitz, M.D.
|
|||
|
/s/
Alan S. Nies
|
Director
|
March
5, 2010
|
|
|
Alan
S. Nies, M.D.
|
|||
|
/s/
Frank P. Palantoni
|
Director
|
March
5, 2010
|
|
|
Frank
P. Palantoni
|
|||
|
/s/
Christopher J. Sobecki
|
Director
|
March
5, 2010
|
|
|
Christopher
J. Sobecki
|
|||
|
/s/
Judith L. Swain
|
Director
|
March
5, 2010
|
|
|
Judith
L. Swain,
M.D.
|
|||
Report
of Independent
Registered
Public Accounting Firm
The Board
of Directors and Stockholders
of
Lexicon Pharmaceuticals, Inc.:
We have
audited the accompanying consolidated balance sheets of Lexicon Pharmaceuticals,
Inc. and subsidiaries as of December 31, 2009 and 2008, and the related
consolidated statements of operations, stockholders’ equity, and cash flows for
each of the three years in the period ended December 31, 2009. These
financial statements are the responsibility of the Company’s
management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We
conducted our audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we plan
and perform the audit to obtain reasonable assurance about whether the financial
statements are free of material misstatement. An audit includes examining, on a
test basis, evidence supporting the amounts and disclosures in the financial
statements. An audit also includes assessing the accounting principles used and
significant estimates made by management, as well as evaluating the overall
financial statement presentation. We believe that our audits provide a
reasonable basis for our opinion.
In our
opinion, the financial statements referred to above present fairly, in all
material respects, the consolidated financial position of Lexicon
Pharmaceuticals, Inc. and subsidiaries as of December 31, 2009 and 2008,
and the consolidated results of their operations and their cash flows for each
of the three years in the period ended December 31, 2009, in conformity
with U.S. generally accepted accounting principles.
We also
have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), Lexicon Pharmaceuticals, Inc.’s internal
control over financial reporting as of December 31, 2009, based on criteria
established in Internal Control—Integrated Framework issued by the Committee of
Sponsoring Organizations of the Treadway Commission and our report dated
March 5, 2010 expressed an unqualified opinion thereon.
/s/ Ernst
& Young LLP
Houston,
Texas
March 5,
2010
Report
of Independent
Registered
Public Accounting Firm
The Board
of Directors and Stockholders
of
Lexicon Pharmaceuticals, Inc.:
We have
audited Lexicon Pharmaceuticals, Inc.’s internal control over
financial reporting as of December 31, 2009, based on criteria established
in Internal Control—Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission (the COSO criteria). Lexicon
Pharmaceuticals, Inc.’s management is responsible for maintaining effective
internal control over financial reporting, and for its assessment of the
effectiveness of internal control over financial reporting included in the
accompanying Management Report on Internal Control over Financial Reporting. Our
responsibility is to express an opinion on the company’s internal control over
financial reporting based on our audit.
We
conducted our audit in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we plan
and perform the audit to obtain reasonable assurance about whether effective
internal control over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of internal control over
financial reporting, assessing the risk that a material weakness exists, testing
and evaluating the design and operating effectiveness of internal control based
on the assessed risk, and performing such other procedures as we considered
necessary in the circumstances. We believe that our audit provides a reasonable
basis for our opinion.
A
company’s internal control over financial reporting is a process designed to
provide reasonable assurance regarding the reliability of financial reporting
and the preparation of financial statements for external purposes in accordance
with generally accepted accounting principles. A company’s internal control over
financial reporting includes those policies and procedures that (1) pertain
to the maintenance of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the company;
(2) provide reasonable assurance that transactions are recorded as
necessary to permit preparation of financial statements in accordance with
generally accepted accounting principles, and that receipts and expenditures of
the company are being made only in accordance with authorizations of management
and directors of the company; and (3) provide reasonable assurance
regarding prevention or timely detection of unauthorized acquisition, use or
disposition of the company’s assets that could have a material effect on the
financial statements.
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements. Also, projections of any evaluation
of effectiveness to future periods are subject to the risk that controls may
become inadequate because of changes in conditions, or that the degree of
compliance with the policies or procedures may deteriorate.
In our
opinion, Lexicon Pharmaceuticals, Inc. maintained, in all material respects,
effective internal control over financial reporting as of December 31,
2009, based on the COSO criteria.
We also
have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the consolidated balance sheets of Lexicon
Pharmaceuticals, Inc. and subsidiaries as of December 31, 2009 and 2008,
and the related consolidated statements of operations, stockholders’ equity, and
cash flows for each of the three years in the period ended December 31,
2009 and our report dated March 5, 2010 expressed an unqualified opinion
thereon.
/s/ Ernst
& Young LLP
Houston,
Texas
March 5,
2010
Lexicon
Pharmaceuticals, Inc.
Consolidated
Balance Sheets
(In
thousands, except par value)
|
As
of December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
Assets
|
||||||||
|
Current
assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
100,554
|
$
|
85,873
|
||||
|
Short-term investments,
including restricted investments of $430
|
56,542
|
629
|
||||||
|
Short-term investments held by
Symphony Icon, Inc.
|
5,417
|
16,610
|
||||||
|
Accounts receivable, net of
allowances of $35
|
815
|
568
|
||||||
|
Prepaid expenses and other
current assets
|
6,356
|
5,487
|
||||||
|
Total current assets
|
169,684
|
109,167
|
||||||
|
Long-term
investments
|
—
|
55,686
|
||||||
|
Property
and equipment, net of accumulated depreciation and amortization of $75,795
and $71,102, respectively
|
58,754
|
65,087
|
||||||
|
Goodwill
|
25,798
|
25,798
|
||||||
|
Other
assets
|
3,525
|
5,770
|
||||||
|
Total assets
|
$
|
257,761
|
$
|
261,508
|
||||
|
Liabilities
and Equity
|
||||||||
|
Current
liabilities:
|
||||||||
|
Accounts payable
|
$
|
5,919
|
$
|
7,926
|
||||
|
Accrued liabilities
|
5,611
|
6,615
|
||||||
|
Current portion of deferred
revenue
|
942
|
5,672
|
||||||
|
Current portion of long-term
debt
|
38,482
|
963
|
||||||
|
Total current liabilities
|
50,954
|
21,176
|
||||||
|
Deferred
revenue, net of current portion
|
14,212
|
14,212
|
||||||
|
Long-term
debt
|
28,482
|
29,529
|
||||||
|
Other
long-term liabilities
|
616
|
764
|
||||||
|
Total liabilities
|
94,264
|
65,681
|
||||||
|
Commitments
and contingencies
|
||||||||
|
Equity:
|
||||||||
|
Lexicon Pharmaceuticals, Inc.
stockholders’ equity:
|
||||||||
|
Preferred stock, $.01 par
value; 5,000 shares authorized; no shares issued and
outstanding
|
—
|
—
|
||||||
|
Common stock, $.001 par value;
900,000 and 300,000 shares authorized, respectively; 175,785 and 136,797
shares issued, respectively
|
176
|
137
|
||||||
|
Additional paid-in capital
|
733,874
|
672,838
|
||||||
|
Accumulated deficit
|
(570,175
|
)
|
(487,395
|
)
|
||||
|
Treasury stock, at cost, 80
and no shares, respectively
|
(88
|
)
|
—
|
|||||
|
Total Lexicon Pharmaceuticals,
Inc. stockholders’ equity
|
163,787
|
185,580
|
||||||
|
Noncontrolling interest in
Symphony Icon, Inc.
|
(290
|
)
|
10,247
|
|||||
|
Total equity
|
163,497
|
195,827
|
||||||
|
Total liabilities and equity
|
$
|
257,761
|
$
|
261,508
|
||||
The
accompanying notes are an integral part of these consolidated financial
statements.
Lexicon
Pharmaceuticals, Inc.
Consolidated
Statements of Operations
(In
thousands, except per share amounts)
|
Year
Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Revenues:
|
||||||||||||
|
Collaborative research
|
$
|
9,334
|
$
|
27,177
|
$
|
48,080
|
||||||
|
Subscription and license fees
|
1,366
|
5,144
|
2,038
|
|||||||||
|
Total revenues
|
10,700
|
32,321
|
50,118
|
|||||||||
|
Operating
expenses:
|
||||||||||||
|
Research and development,
including stock-based compensation of $3,022, $3,941 and $5,150,
respectively
|
81,238
|
107,232
|
103,237
|
|||||||||
|
General and administrative,
including stock-based compensation of $2,252, $2,559 and $2,776,
respectively
|
19,418
|
21,624
|
21,835
|
|||||||||
|
Total operating
expenses
|
100,656
|
128,856
|
125,072
|
|||||||||
|
Loss
from operations
|
(89,956
|
)
|
(96,535
|
)
|
(74,954
|
)
|
||||||
|
Gain
(loss) on investments, net
|
1,173
|
(1,314
|
)
|
—
|
||||||||
|
Interest
income
|
880
|
5,762
|
7,286
|
|||||||||
|
Interest
expense
|
(2,966
|
)
|
(2,691
|
)
|
(2,771
|
)
|
||||||
|
Other
expense, net
|
(2,550
|
)
|
(2,106
|
)
|
(794
|
)
|
||||||
|
Consolidated
net loss before taxes
|
(93,419
|
)
|
(96,884
|
)
|
(71,233
|
)
|
||||||
|
Income
tax benefit
|
102
|
—
|
—
|
|||||||||
|
Consolidated
net loss
|
(93,317
|
)
|
(96,884
|
)
|
(71,233
|
)
|
||||||
|
Less:
net loss attributable to Symphony Icon, Inc.
|
10,537
|
20,024
|
12,439
|
|||||||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc.
|
$
|
(82,780
|
)
|
$
|
(76,860
|
)
|
$
|
(58,794
|
)
|
|||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc. per common share, basic
and diluted
|
$
|
(0.57
|
)
|
$
|
(0.56
|
)
|
$
|
(0.59
|
)
|
|||
|
Shares
used in computing net loss attributable to Lexicon Pharmaceuticals, Inc.
per common share, basic and diluted
|
145,465
|
136,797
|
99,798
|
|||||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
Lexicon
Pharmaceuticals, Inc.
Consolidated
Statements of Stockholders’ Equity
(In
thousands)
|
Lexicon
Pharmaceuticals, Inc. Stockholders
|
||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||
|
Additional
|
Other
|
|||||||||||||||||||||||||||||||||||
|
Common
Stock
|
Paid-In
|
Accumulated
|
Comprehensive
|
Treasury
|
Noncontrolling
|
Total
|
||||||||||||||||||||||||||||||
|
Shares
|
Par
Value
|
Capital
|
Deficit
|
Loss
|
Stock
|
Total
|
Interest
|
Equity
|
||||||||||||||||||||||||||||
|
Balance
at December 31, 2006
|
77,804 | $ | 78 | $ | 437,180 | $ | (351,741 | ) | $ | (16 | ) | $ | — | $ | 85,501 | $ | — | $ | 85,501 | |||||||||||||||||
|
Stock-based
compensation
|
— | — | 7,926 | — | — | — | 7,926 | — | 7,926 | |||||||||||||||||||||||||||
|
Issuance
of common stock to Invus, L.P., net of fees
|
50,825 | 51 | 197,911 | — | — | — | 197,962 | — | 197,962 | |||||||||||||||||||||||||||
|
Issuance
of common stock to Symphony Holdings, LLC, net of fees
|
7,651 | 8 | 22,793 | — | — | — | 22,801 | — | 22,801 | |||||||||||||||||||||||||||
|
Purchase
of noncontrolling interest by preferred shareholders of Symphony Icon,
Inc.
|
— | — | — | — | — | — | — | 42,710 | 42,710 | |||||||||||||||||||||||||||
|
Issuance
of common stock
|
516 | — | 892 | — | — | — | 892 | — | 892 | |||||||||||||||||||||||||||
|
Net
loss
|
— | — | — | (58,794 | ) | — | — | (58,794 | ) | (12,439 | ) | (71,233 | ) | |||||||||||||||||||||||
|
Unrealized
gain on investments
|
— | — | — | — | 12 | — | 12 | — | 12 | |||||||||||||||||||||||||||
|
Comprehensive
loss
|
(58,782 | ) | (71,221 | ) | ||||||||||||||||||||||||||||||||
|
Balance
at December 31, 2007
|
136,796 | 137 | 666,702 | (410,535 | ) | (4 | ) | — | 256,300 | 30,271 | 286,571 | |||||||||||||||||||||||||
|
Stock-based
compensation
|
— | — | 6,135 | — | — | — | 6,135 | — | 6,135 | |||||||||||||||||||||||||||
|
Exercise
of common stock options
|
1 | — | 1 | — | — | — | 1 | — | 1 | |||||||||||||||||||||||||||
|
Net
loss
|
— | — | — | (76,860 | ) | — | — | (76,860 | ) | (20,024 | ) | (96,884 | ) | |||||||||||||||||||||||
|
Unrealized
gain on investments
|
— | — | — | — | 4 | — | 4 | — | 4 | |||||||||||||||||||||||||||
|
Comprehensive
loss
|
(76,856 | ) | (96,880 | ) | ||||||||||||||||||||||||||||||||
|
Balance
at December 31, 2008
|
136,797 | 137 | 672,838 | (487,395 | ) | — | — | 185,580 | 10,247 | 195,827 | ||||||||||||||||||||||||||
|
Stock-based
compensation
|
— | — | 5,639 | — | — | — | 5,639 | — | 5,639 | |||||||||||||||||||||||||||
|
Grant
of restricted stock
|
534 | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
|
Issuance
of common stock, net of fees
|
38,333 | 39 | 55,133 | — | — | — | 55,172 | — | 55,172 | |||||||||||||||||||||||||||
|
Exercise
of common stock options
|
121 | — | 264 | — | — | — | 264 | — | 264 | |||||||||||||||||||||||||||
|
Repurchase
of common stock
|
— | — | — | — | — | (88 | ) | (88 | ) | — | (88 | ) | ||||||||||||||||||||||||
|
Net
loss
|
— | — | — | (82,780 | ) | — | — | (82,780 | ) | (10,537 | ) | (93,317 | ) | |||||||||||||||||||||||
|
Balance
at December 31, 2009
|
175,785 | $ | 176 | $ | 733,874 | $ | (570,175 | ) | $ | — | $ | (88 | ) | $ | 163,787 | $ | (290 | ) | $ | 163,497 | ||||||||||||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
Lexicon
Pharmaceuticals, Inc.
Consolidated
Statements of Cash Flows
(In
thousands)
|
Year
Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Cash
flows from operating activities:
|
||||||||||||
|
Consolidated net loss
|
$
|
(93,317
|
)
|
$
|
(96,884
|
)
|
$
|
(71,233
|
)
|
|||
|
Adjustments to reconcile net
loss to net cash used in operating activities:
|
||||||||||||
|
Depreciation
|
6,159
|
7,929
|
9,262
|
|||||||||
|
Impairment of fixed assets
|
436
|
—
|
—
|
|||||||||
|
Amortization of Symphony Icon
purchase option
|
2,141
|
2,141
|
1,160
|
|||||||||
|
Stock-based compensation
|
5,274
|
6,500
|
7,926
|
|||||||||
|
(Gain) loss on auction rate
securities (“ARS”)
|
(3,508
|
)
|
13,374
|
—
|
||||||||
|
(Gain) loss on ARS Rights
|
2,335
|
(12,060
|
)
|
—
|
||||||||
|
Changes in operating assets and
liabilities:
|
||||||||||||
|
(Increase) decrease in
receivables
|
(247
|
)
|
1,195
|
(577
|
)
|
|||||||
|
(Increase) decrease in prepaid
expenses and other current assets
|
(869
|
)
|
(1,375
|
)
|
255
|
|||||||
|
Decrease in other assets
|
104
|
108
|
109
|
|||||||||
|
Increase (decrease) in accounts
payable and other liabilities
|
(2,794
|
)
|
(2,256
|
)
|
2,619
|
|||||||
|
Decrease in deferred
revenue
|
(4,730
|
)
|
(14,272
|
)
|
(23,844
|
)
|
||||||
|
Net cash used in operating
activities
|
(89,016
|
)
|
(95,600
|
)
|
(74,323
|
)
|
||||||
|
Cash
flows from investing activities:
|
||||||||||||
|
Purchases of property and
equipment
|
(369
|
)
|
(2,187
|
)
|
(1,900
|
)
|
||||||
|
Proceeds from disposal of
property and equipment
|
107
|
—
|
1
|
|||||||||
|
Purchases of investments held
by Symphony Icon, Inc.
|
(4,250
|
)
|
—
|
(44,991
|
)
|
|||||||
|
Maturities of investments held
by Symphony Icon, Inc.
|
15,443
|
20,056
|
8,325
|
|||||||||
|
Purchase of short-term
investments
|
(59,955
|
)
|
(39,847
|
)
|
(260,739
|
)
|
||||||
|
Maturities of short-term
investments
|
60,901
|
181,393
|
111,353
|
|||||||||
|
Net cash provided by (used in)
investing activities
|
11,877
|
159,415
|
(187,951
|
)
|
||||||||
|
Cash
flows from financing activities:
|
||||||||||||
|
Proceeds from issuance of
common stock to Invus, L.P., net of fees
|
—
|
—
|
197,962
|
|||||||||
|
Proceeds from issuance of
common stock to Symphony Holdings, LLC, net of fees
|
—
|
—
|
14,237
|
|||||||||
|
Proceeds from issuance of
common stock, net of fees
|
55,436
|
1
|
892
|
|||||||||
|
Repurchase of common stock
|
(88
|
)
|
—
|
—
|
||||||||
|
Proceeds from debt
borrowings
|
38,592
|
—
|
—
|
|||||||||
|
Repayment of debt
borrowings
|
(2,120
|
)
|
(881
|
)
|
(815
|
)
|
||||||
|
Proceeds from purchase of
noncontrolling interest by preferred shareholders of Symphony Icon,
Inc.
|
—
|
—
|
42,710
|
|||||||||
|
Net cash provided by (used in)
financing activities
|
91,820
|
(880
|
)
|
254,986
|
||||||||
|
Net
increase (decrease) in cash and cash equivalents
|
14,681
|
62,935
|
(7,288
|
)
|
||||||||
|
Cash
and cash equivalents at beginning of
year
|
85,873
|
22,938
|
30,226
|
|||||||||
|
Cash
and cash equivalents at end of
year
|
$
|
100,554
|
$
|
85,873
|
$
|
22,938
|
||||||
|
Supplemental
disclosure of cash flow information:
|
||||||||||||
|
Cash paid for interest
|
$
|
2,519
|
$
|
2,599
|
$
|
2,665
|
||||||
|
Cash received related to income
taxes
|
$
|
102
|
$
|
—
|
$
|
—
|
||||||
|
Supplemental
disclosure of noncash investing and financing activities:
|
||||||||||||
|
Common
stock issued for purchase option in conjunction with Symphony Icon
financing
|
$
|
—
|
$
|
—
|
$
|
8,564
|
||||||
|
Unrealized
gain on investments
|
$
|
—
|
$
|
4
|
$
|
12
|
||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
Lexicon
Pharmaceuticals, Inc.
Notes
to Consolidated Financial Statements
December
31, 2009
1. Organization
and Operations
Lexicon
Pharmaceuticals, Inc. (“Lexicon” or the “Company”) is a Delaware corporation
incorporated on July 7, 1995. Lexicon was organized to discover the
functions and pharmaceutical utility of genes and use those gene function
discoveries in the discovery and development of pharmaceutical products for the
treatment of human disease.
Lexicon
has financed its operations from inception primarily through sales of common and
preferred stock, payments received under collaboration and alliance agreements,
database subscription agreements, government grants and contracts and technology
licenses, and financing obtained under debt and lease arrangements. The
Company’s future success is dependent upon many factors, including, but not
limited to, its ability to discover and develop pharmaceutical products for the
treatment of human disease, establish additional collaboration and license
agreements, achieve milestones under such agreements, obtain and enforce patents
and other proprietary rights in its discoveries, comply with federal and state
regulations, and maintain sufficient capital to fund its
activities. As a result of the aforementioned factors and the related
uncertainties, there can be no assurance of the Company’s future
success.
2. Summary
of Significant Accounting Policies
Basis of Presentation: The
accompanying consolidated financial statements include the accounts of Lexicon
and its wholly-owned subsidiaries, as well as one variable interest entity,
Symphony Icon, Inc. (“Symphony Icon”), of which the Company is the primary
beneficiary. The Company has therefore consolidated the financial
condition and results of operations of Symphony Icon. Intercompany
transactions and balances are eliminated in consolidation.
Certain
amounts in the prior year’s financial statements have been reclassified to
conform to the current year presentation. These include the
reclassification of $1.3 million and $1.1 million of patent-related
legal costs from research and development expense to general and administrative
expense on the consolidated statements of operations for the years ended
December 31, 2008 and 2007, respectively.
Use of Estimates: The
preparation of financial statements in conformity with U. S. generally accepted
accounting principles requires management to make estimates and assumptions that
affect the reported amounts of assets and liabilities and disclosure of
contingent assets and liabilities at the date of the financial statements and
the reported amounts of revenues and expenses during the period. Actual results
could differ from those estimates.
Cash, Cash Equivalents and
Short-Term Investments: Lexicon considers all highly-liquid investments
with original maturities of three months or less to be cash
equivalents. As of December 31, 2009, short-term investments
consist of certificates of deposit, auction rate securities and auction rate
security rights (“ARS Rights”) obtained from UBS AG, the investment bank that
sold Lexicon the auction rate securities it currently holds (see
Note 4). As of December 31, 2008, short-term investments
consisted of certificates of deposit. The certificates of deposits
are classified as available-for-sale securities and are carried at fair value,
based on quoted market prices of the securities. The Company views
its available-for-sale securities as available for use in current operations
regardless of the stated maturity date of the security. Unrealized
gains and losses on such securities are reported as a separate component of
stockholders’ equity. Net realized gains and losses, interest and
dividends are included in interest income. Lexicon has elected to
classify its auction rate securities and ARS Rights as trading securities, which
requires recording these securities at fair value. The cost of
securities sold is based on the specific identification method.
Restricted Cash and
Investments: Lexicon is required to maintain restricted cash
or investments to collateralize standby letters of credit for the lease on its
office and laboratory facilities in Hopewell, New Jersey (see
Note 11). As of December 31, 2009 and 2008, restricted cash
and investments were $0.4 million.
Long-Term
Investments: Lexicon classifies its investments as either
current or long-term based upon the investments' contractual maturities and
Lexicon’s intent and ability to convert such instruments to cash within one
year. As of December 31, 2008, long-term investments consisted
of auction rate securities and ARS Rights. Lexicon has elected to
classify its long-term investments as trading securities, which requires
recording these securities at fair value.
Accounts
Receivable: Lexicon records trade accounts receivable in the
normal course of business related to the sale of products or
services. The allowance for doubtful accounts takes into
consideration such factors as historical write-offs, the economic climate and
other factors that could affect collectibility. Write-offs are
evaluated on a case by case basis.
Concentration of Credit Risk:
Lexicon’s cash equivalents, investments and accounts receivable represent
potential concentrations of credit risk. The Company attempts to minimize
potential concentrations of risk in cash equivalents and investments by placing
investments in high-quality financial instruments. The Company’s accounts
receivable are unsecured and are concentrated in pharmaceutical and
biotechnology companies located in the United States, Europe and
Japan. The Company has not experienced any significant credit losses
to date. In 2009, customers in the United States and Europe
represented 75% and 25% of revenue, respectively. In 2008, customers
in the United States and Europe represented 68% and 32% of revenue,
respectively. In 2007, customers in the United States, Europe and
Japan represented 66%, 29% and 5% of revenue, respectively. At December 31,
2009, management believes that the Company has no significant concentrations of
credit risk.
Segment Information and Significant
Customers: Lexicon operates in one business segment, which primarily
focuses on the discovery of the functions and pharmaceutical utility of genes
and the use of those gene function discoveries in the discovery and development
of pharmaceutical products for the treatment of human disease. Substantially all
of the Company’s revenues have been derived from drug discovery alliances,
target validation collaborations for the development and, in some cases,
analysis of the physiological effects of genes altered in knockout mice,
technology licenses, subscriptions to its databases, government grants and
contracts and compound library sales. In 2009, Bristol-Myers Squibb
Company, N.V. Organon and Taconic Farms, Inc. represented 31%, 23% and 21% of
revenues, respectively. In 2008, Bristol-Myers Squibb, Organon and
Genentech, Inc. represented 32%, 29% and 13% of revenues,
respectively. In 2007, Organon, Bristol-Myers Squibb and the
Texas Enterprise Fund represented 27%, 23% and 22% of revenues,
respectively.
Property and Equipment:
Property and equipment are carried at cost and depreciated using the
straight-line method over the estimated useful life of the assets which ranges
from three to 40 years. Maintenance, repairs and minor replacements
are charged to expense as incurred. Leasehold improvements are
amortized over the shorter of the estimated useful life or the remaining lease
term. Significant renewals and betterments are
capitalized.
Impairment of Long-Lived
Assets: Long-lived assets and certain identifiable intangible
assets to be held and used are reviewed for impairment when events or changes in
circumstances indicate that the carrying amount of such assets may not be
recoverable. Determination of recoverability is based on an estimate
of undiscounted future cash flows resulting from the use of the asset and its
eventual disposition. In the event that such cash flows are not
expected to be sufficient to recover the carrying amount of the assets, the
assets are written down to their estimated fair values.
Goodwill
Impairment: Goodwill is not amortized, but is tested at least
annually for impairment at the reporting unit level. The Company has
determined that the reporting unit is the single operating segment disclosed in
its current financial statements. Impairment is the condition that
exists when the carrying amount of goodwill exceeds its implied fair
value. The first step in the impairment process is to determine the
fair value of the reporting unit and then compare it to the carrying value,
including goodwill. If the fair value exceeds the carrying value, no
further action is required and no impairment loss is
recognized. Additional impairment assessments may be performed on an
interim basis if the Company encounters events or changes in circumstances that
would indicate that, more likely than not, the carrying value of goodwill has
been impaired. There was no impairment of goodwill in 2009, 2008 or
2007.
Revenue Recognition: Revenues
are recognized when persuasive evidence of an arrangement exists, delivery has
occurred or services have been rendered, the price is fixed or determinable and
collectibility is reasonably assured. Payments received in advance
under these arrangements are recorded as deferred revenue until
earned. Revenues are earned from drug discovery and development
collaborations, target validation collaborations, database subscriptions,
technology licenses, and government grants and contracts. Revenues
generated from third parties under collaborative arrangements are recorded on a
gross basis on the consolidated statements of operations as Lexicon is the
principal participant for these transactions for the purpose of accounting for
these arrangements.
Upfront
fees under drug discovery and development collaborations are recognized as
revenue on a straight-line basis over the estimated period of service, generally
the contractual research term, as this period is Lexicon’s best estimate of the
period over which the services will be rendered, to the extent they are
non-refundable. Lexicon has determined that the level of effort it
performs to meet its obligations is fairly constant throughout the estimated
periods of service. As a result, Lexicon has determined that it is
appropriate to recognize revenue from such agreements on a straight-line basis,
as management believes this reflects how the research is provided during the
initial period of the agreement. When it becomes probable that a
collaborator will extend the research period, Lexicon adjusts the revenue
recognition method as necessary based on the level of effort required under the
agreement for the extension period.
Research
funding under these alliances is recognized as services are performed to the
extent they are non-refundable, either on a straight-line basis over the
estimated service period, generally the contractual research term, or as
contract research costs are incurred. Milestone-based fees are
recognized upon completion of specified milestones according to contract
terms. Payments received under target validation collaborations and
government grants and contracts are recognized as revenue as Lexicon performs
its obligations related to such research to the extent such fees are
non-refundable. Non-refundable technology license fees are recognized
as revenue upon the grant of the license when performance is complete and there
is no continuing involvement.
The
Company analyzes its multiple element arrangements to determine whether the
elements can be separated and accounted for individually as separate units of
accounting. An element of a contract can be accounted for separately
if the delivered elements have standalone value to the collaborator and the fair
value of any undelivered elements is determinable through objective and reliable
evidence. If an element is considered to have standalone value but the fair
value of any of the undelivered items cannot be determined, all elements of the
arrangement are recognized as revenue over the period of performance for such
undelivered items or services.
Research and Development Expenses:
Research and development expenses consist of costs incurred for
company-sponsored as well as collaborative research and development activities.
These costs include direct and research-related overhead expenses and are
expensed as incurred. Technology license fees for technologies that
are utilized in research and development and have no alternative future use are
expensed when incurred.
Stock-Based
Compensation: The Company recognizes compensation expense in
its statement of operations for share-based payments, including stock options
issued to employees, based on their fair values on the date of the grant, with
the compensation expense recognized over the period in which an employee is
required to provide service in exchange for the stock
award. Stock-based compensation expense is recognized on a
straight-line basis. As of December 31, 2009, stock-based
compensation cost for all outstanding unvested options was $6.8 million,
which is expected to be recognized over a weighted-average period of
1.2 years.
The fair
value of stock options is estimated at the date of grant using the Black-Scholes
method. The Black-Scholes option-pricing model requires the input of
subjective assumptions. Because the Company’s employee stock options
have characteristics significantly different from those of traded options, and
because changes in the subjective input assumptions can materially affect the
fair value estimate, in management’s opinion, the existing models do not
necessarily provide a reliable single measure of the fair value of its employee
stock options. For purposes of determining the fair value of stock
options, the Company segregates its options into two homogeneous groups, based
on exercise and post-vesting employment termination behaviors, resulting in a
change in the assumptions used for expected option lives and
forfeitures. Expected volatility is based on the historical
volatility in the Company’s stock price. The following
weighted-average assumptions were used for options granted in the years ended
December 31, 2009, 2008 and 2007, respectively:
|
Expected
Volatility
|
Risk-free
Interest Rate
|
Expected
Term
|
Estimated
Forfeitures
|
Dividend
Rate
|
|||||
|
December
31, 2009:
|
|||||||||
|
Employees
|
78%
|
1.9%
|
5
|
24%
|
0%
|
||||
|
Officers
and non-employee directors
|
77%
|
2.7%
|
8
|
7%
|
0%
|
||||
|
December
31, 2008:
|
|||||||||
|
Employees
|
66%
|
2.9%
|
6
|
22%
|
0%
|
||||
|
Officers
and non-employee directors
|
66%
|
3.8%
|
9
|
6%
|
0%
|
||||
|
December
31, 2007:
|
|||||||||
|
Employees
|
66%
|
4.5%
|
6
|
21%
|
0%
|
||||
|
Officers
and non-employee directors
|
67%
|
4.6%
|
9
|
4%
|
0%
|
Net Loss per Common Share:
Net loss per common share is computed using the weighted average number
of shares of common stock outstanding. Shares associated with stock options and
warrants are not included because they are antidilutive.
Comprehensive
Loss: Comprehensive loss is comprised of net loss and
unrealized gains and losses on available-for-sale
securities. Comprehensive loss is reflected in the consolidated
statements of stockholders’ equity. Comprehensive loss equals net
loss for the year ended December 31, 2009. There were no
unrealized gains for the year ended December 31, 2009. There
were unrealized gains of $4,000 and $12,000 in the years ended December 31, 2008
and 2007, respectively.
3. Recent
Accounting Pronouncements
In
September 2006, the Financial Accounting Standards Board (“FASB”) issued a new
accounting pronouncement regarding fair value measurements (formerly Statement
of Financial Standards (“SFAS”) No. 157, “Fair Value
Measurements”). The pronouncement, found under FASB Accounting
Standards Codification (“ASC”) Topic 820, defines fair value, establishes a
framework for measuring fair value in generally accepted accounting principles,
and expands disclosures about fair value measurements. This
pronouncement applies under other accounting pronouncements that require or
permit fair value measurements, the FASB having previously concluded in those
accounting pronouncements that fair value is the relevant measurement
attribute. Accordingly, this pronouncement does not require any new
fair value measurements. More specifically, this pronouncement
emphasizes that fair value is a market-based measurement, not an entity-specific
measurement, and sets out a fair value hierarchy, which ranks the quality and
reliability of the information used to determine fair values. This
pronouncement was effective January 1, 2008 for financial assets and liabilities
and January 1, 2009 for non-financial assets and liabilities. The
adoption of this pronouncement did not have an effect on the Company’s financial
position or results of operations.
In
December 2007, the FASB issued a new accounting pronouncement regarding business
combinations (formerly SFAS No. 141(Revised), “Business Combinations”), which
requires an acquirer to recognize the assets acquired, the liabilities assumed,
and any non-controlling interest in the acquiree at the acquisition date,
measured at their fair values as of that date, with limited exceptions. This
pronouncement, found under FASB ASC Topic 805, also requires the acquirer in a
business combination achieved in stages to recognize the identifiable assets and
liabilities, as well as the non-controlling interest in the acquiree, at the
full amounts of their fair values. This pronouncement makes various other
amendments to authoritative literature intended to provide additional guidance
or to confirm the guidance in that literature to that provided in this
pronouncement. This pronouncement applies prospectively to business combinations
for which the acquisition date is on or after the beginning of the first annual
reporting period beginning on or after December 15, 2008. The adoption of this
pronouncement did not have an effect on the Company’s financial position or
results of operations.
In
December 2007, the FASB issued a new pronouncement regarding noncontrolling
interests (formerly SFAS No. 160, “Noncontrolling Interests in Consolidated
Financial Statements”) to improve the relevance, comparability, and transparency
of the financial information that a reporting entity provides in its
consolidated financial statements. This pronouncement, found under FASB ASC
Topic 810, establishes accounting and reporting standards that require the
ownership interests in subsidiaries not held by the parent to be clearly
identified, labeled and presented in the consolidated statement of financial
position within equity, but separate from the parent’s equity. This
pronouncement also requires the amount of consolidated net income attributable
to the parent and to the non-controlling interest to be clearly identified and
presented on the face of the consolidated statement of income. Changes in a
parent’s ownership interest while the parent retains its controlling financial
interest must be accounted for consistently, and when a subsidiary is
deconsolidated, any retained non-controlling equity investment in the former
subsidiary must be initially measured at fair value. The gain or loss on the
deconsolidation of the subsidiary is measured using the fair value of any
non-controlling equity investment. The pronouncement also requires entities to
provide sufficient disclosures that clearly identify and distinguish between the
interests of the parent and the interests of the non-controlling owners. This
pronouncement applies prospectively to all entities that prepare consolidated
financial statements and applies prospectively for fiscal years, and interim
periods within those fiscal years, beginning on or after December 15, 2008. The
Company’s adoption of this pronouncement on January 1, 2009 did not materially
affect its financial position or results of operations, other than reclassifying
the noncontrolling interest in Symphony Icon to equity for all periods
presented.
In
December 2007, the FASB ratified a new pronouncement regarding collaborative
arrangements (formerly Emerging Issues Task Force Issue No. 07-01, “Accounting
for Collaborative Arrangements”), which provides guidance on how the parties to
a collaborative agreement should account for costs incurred and revenue
generated on sales to third parties, how sharing payments pursuant to a
collaboration agreement should be presented in the income statement and certain
related disclosure requirements. The adoption of this pronouncement,
found under FASB ASC Topic 808, did not have an effect on the Company's
financial position or results of operations, other than requiring additional
disclosures.
In May
2009, the FASB issued a new accounting pronouncement regarding subsequent events
(formerly SFAS No. 165, “Subsequent Events”), which provides guidance to
establish general standards of accounting for, and disclosures of, events that
occur after the balance sheet date but before financial statements are issued or
are available to be issued. This pronouncement, found under FASB ASC
Topic 855, is effective for interim or fiscal periods ending after June 15,
2009. The Company's adoption of this pronouncement did not have an
effect on its financial position or results of operations.
In June
2009, the FASB issued a new accounting pronouncement regarding variable interest
entities (formerly SFAS No. 167, “Amendments to FASB Interpretation No. 46(R)),”
which changes how a company determines when an entity that is insufficiently
capitalized or is not controlled through voting (or similar rights) should be
consolidated. The determination of whether a company is required to
consolidate an entity is based on, among other things, an entity’s purpose and
design and a company’s ability to direct the activities that most significantly
impacts the entity’s economic performance. The impact of the adoption
of this pronouncement may be applied retrospectively with a cumulative-effect
adjustment to retained earnings as of the beginning of the first year restated,
or through a cumulative-effect adjustment on the date of
adoption. This pronouncement, found under FASB ASC Topic 810, is
effective for fiscal years, and interim periods within those fiscal years,
beginning on or after November 15, 2009. The Company has determined
that upon adoption of this pronouncement on January 1, 2010, Lexicon will
no longer be the primary beneficiary of Symphony Icon, and therefore will no
longer include the financial condition and results of operations of Symphony
Icon in its consolidated financial statements. As of
December 31, 2009, Symphony Icon had $6.2 million in current assets,
$5.4 million of which was short-term investments, and $4.2 million in
current liabilities. On January 1, 2010, Lexicon will record a
cumulative-effect adjustment to retained earnings (accumulated deficit) as a
result of adopting this pronouncement, which will increase the accumulated
deficit balance by $1.5 million.
In
October 2009, the FASB issued Accounting Standards Update (“ASU”)
No. 2009-13, “Multiple-Deliverable Revenue Arrangements”, which amends FASB
ASC Topic 605. ASU No. 2009-13 addresses how to determine
whether an arrangement involving multiple deliverables contain more than one
unit of accounting and how to allocate consideration to each unit of accounting
in the arrangement. This pronouncement replaces all references to
fair value as the measurement criteria with the term selling price and
establishes a hierarchy for determining the selling price of a
deliverable. The pronouncement also eliminates the use of the
residual value method for determining the allocation of arrangement
consideration, and requires additional disclosures. This
pronouncement should be applied prospectively for revenue arrangements entered
into or materially modified in fiscal years beginning on or after June 15,
2010, with early adoption permitted. This pronouncement’s impact on
accounting for revenue arrangements is dependent upon arrangements entered into
on or after that time.
4. Cash
and Cash Equivalents and Investments
The fair
value of cash and cash equivalents and investments held at December 31,
2009 and 2008 are as follows:
|
As
of December 31, 2009
|
||||||||||||||||
|
Amortized
Cost
|
Gross
Unrealized Gains
|
Gross
Unrealized Losses
|
Estimated
Fair Value
|
|||||||||||||
|
(in
thousands)
|
||||||||||||||||
|
Cash
and cash equivalents
|
$
|
100,554
|
$
|
—
|
$
|
—
|
$
|
100,554
|
||||||||
|
Securities
maturing within one year:
|
||||||||||||||||
|
Certificates of deposit
|
508
|
—
|
—
|
508
|
||||||||||||
|
ARS rights
|
—
|
9,725
|
—
|
9,725
|
||||||||||||
|
Securities
maturing after ten years:
|
||||||||||||||||
|
Auction rate securities
|
56,175
|
—
|
(9,866
|
)
|
46,309
|
|||||||||||
|
Total
short-term investments
|
$
|
56,683
|
$
|
9,725
|
$
|
(9,866
|
)
|
$
|
56,542
|
|||||||
|
Short-term
investments held by Symphony Icon, Inc.:
|
||||||||||||||||
|
Cash and cash equivalents
|
5,417
|
—
|
—
|
5,417
|
||||||||||||
|
Total
short-term investments held by Symphony Icon, Inc.
|
$
|
5,417
|
$
|
—
|
$
|
—
|
$
|
5,417
|
||||||||
|
Total
cash and cash equivalents and investments
|
$
|
162,654
|
$
|
9,725
|
$
|
(9,866
|
)
|
$
|
162,513
|
|||||||
|
As
of December 31, 2008
|
||||||||||||||||
|
Amortized
Cost
|
Gross
Unrealized Gains
|
Gross
Unrealized Losses
|
Estimated
Fair Value
|
|||||||||||||
|
(in
thousands)
|
||||||||||||||||
|
Cash
and cash equivalents
|
$
|
85,873
|
$
|
—
|
$
|
—
|
$
|
85,873
|
||||||||
|
Securities
maturing within one year:
|
||||||||||||||||
|
Certificates of deposit
|
629
|
—
|
—
|
629
|
||||||||||||
|
Total short-term investments
|
$
|
629
|
$
|
—
|
$
|
—
|
$
|
629
|
||||||||
|
Securities
maturing after one year through five years:
|
||||||||||||||||
|
ARS rights
|
—
|
12,060
|
—
|
12,060
|
||||||||||||
|
Securities
maturing after ten years:
|
||||||||||||||||
|
Auction rate securities
|
57,000
|
—
|
(13,374
|
)
|
43,626
|
|||||||||||
|
Total
long-term investments
|
$
|
57,000
|
$
|
12,060
|
$
|
(13,374
|
)
|
$
|
55,686
|
|||||||
|
Short-term
investments held by Symphony Icon, Inc.:
|
||||||||||||||||
|
Cash and cash equivalents
|
16,610
|
—
|
—
|
16,610
|
||||||||||||
|
Total
short-term investments held by Symphony Icon, Inc.
|
$
|
16,610
|
$
|
—
|
$
|
—
|
$
|
16,610
|
||||||||
|
Total
cash and cash equivalents and investments
|
$
|
160,112
|
$
|
12,060
|
$
|
(13,374
|
)
|
$
|
158,798
|
|||||||
There
were no realized gains or losses for the year ended December 31,
2009. There were realized gains of $123,000 for the year ended
December 31, 2008. There were no realized gains or losses for
the year ended December 31, 2007.
At
December 31, 2009, Lexicon held $56.2 million (par value), with an
estimated fair value of $46.3 million, of investments with an auction interest
rate reset feature, known as auction rate securities. These notes are
issued by various state agencies for the purpose of financing student
loans. The securities have historically traded at par and are
redeemable at par plus accrued interest at the option of the issuer. Interest is
typically paid at the end of each auction period or
semiannually. Until February 2008, the market for Lexicon’s auction
rate securities was highly liquid. However, starting in February
2008, a substantial number of auctions “failed,” meaning that there was not
enough demand to sell all of the securities that holders desired to sell at
auction. The immediate effect of a failed auction is that such holders cannot
sell the securities at auction and the interest rate on the security generally
resets to a maximum interest rate. In the case of funds invested by Lexicon in
auction rate securities which are the subject of a failed auction, Lexicon may
not be able to access the funds without a loss of principal, unless a future
auction on these investments is successful or the issuer redeems the
security. Lexicon has modified its current investment strategy to
reallocate its investments more into U.S. treasury securities and U.S.
treasury-backed money market investments.
At
December 31, 2009 and 2008, observable auction rate securities market
information was not available to determine the fair value of Lexicon’s
investments. Lexicon has estimated the fair value of these securities at
$46.3 million and $43.6 million as of December 31, 2009 and 2008,
respectively, using models of the expected future cash flows related to the
securities and taking into account assumptions about the cash flows of the
underlying student loans, as well as secondary market trading
data. The assumptions used in preparing the discounted cash flow
model include estimates of interest rates, timing and amount of cash flows,
liquidity premiums and expected holding periods of the auction rate securities,
based on data available as of December 31, 2009 and 2008. The
underlying sources of these assumptions are volatile and the assumptions are
subject to change as those sources and market conditions change. If
the current market conditions deteriorate further, or a recovery in market
values does not occur, Lexicon may be required to record additional unrealized
or realized losses in future quarters.
In
November 2008, Lexicon accepted an offer from UBS AG, the investment bank that
sold Lexicon the auction rate securities, providing Lexicon with rights related
to its auction rate securities (“ARS Rights”). The ARS Rights permit
Lexicon to require UBS to purchase its $56.2 million (par value) of auction
rate securities at par value during the period from June 30, 2010 through
July 2, 2012. Conversely, UBS has the right, in its discretion,
to purchase or sell the securities at any time by paying Lexicon the par value
of such securities. Management expects to exercise the ARS Rights and
sell Lexicon’s auction rate securities back to UBS on June 30, 2010, the
earliest date allowable under the ARS Rights. Lexicon is also
eligible to borrow from UBS Bank USA, an affiliate of UBS, at no net cost up to
75% of the market value of the securities, as determined by UBS Bank USA, which
loans would become payable upon the investment bank’s purchase or sale of the
securities (see Note 9).
The
enforceability of the ARS Rights results in a separate asset that is measured at
its fair value. Lexicon elected to measure the ARS Rights under a
fair value option, which permits entities to choose, at certain election dates,
to measure eligible items at fair value. As a result of accepting the
ARS Rights, Lexicon has elected to classify the ARS Rights and reclassify its
investments in auction rate securities as trading securities from
available-for-sale securities. As a result, Lexicon will be required
to assess the fair value of these two individual assets and record changes each
period until the ARS Rights are exercised and the auction rate securities are
redeemed. Lexicon expects that subsequent changes in the value of the
ARS Rights will largely offset the subsequent fair value movements of the
auction rate securities, subject to the continued expected performance by the
investment bank of its obligations under the agreement.
5. Fair
Value Measurements
The
Company uses various inputs in determining the fair value of its investments and
measures these assets on a recurring basis. Financial assets recorded
at fair value in the consolidated balance sheet are categorized by the level of
objectivity associated with the inputs used to measure their fair
value. The following levels are directly related to the amount of
subjectivity associated with the inputs to fair valuation of these financial
assets:
|
·
|
Level
1 – quoted prices in active markets for identical
investments
|
|
·
|
Level
2 – other significant observable inputs (including quoted prices for
similar investments, market corroborated inputs,
etc.)
|
|
·
|
Level
3 – significant unobservable inputs (including the Company’s own
assumptions in determining the fair value of
investments)
|
The
inputs or methodology used for valuing securities are not necessarily an
indication of the credit risk associated with investing in those
securities. Based on market conditions and the unavailability of
Level 1 inputs, during the year ended December 31, 2008, the Company
adopted a valuation methodology that involves discounted cash flow analysis and
secondary market data for its auction rate securities. Accordingly,
the investments in auction rate securities changed from Level 1 to
Level 3 within the fair value levels described above. In
addition, the Company obtained ARS Rights from UBS, and the ARS Rights have been
recorded at fair value as determined using a discounted cash flow valuation
methodology. The following tables provide the fair value measurements
of applicable Company financial assets according to the fair value levels
defined above as of December 31, 2009 and 2008.
|
Financial
Assets at Fair Value
As
of December 31, 2009
|
||||||||||||||||
|
Level
1
|
Level
2
|
Level
3
|
Total
|
|||||||||||||
|
(in
thousands)
|
||||||||||||||||
|
Cash
and cash equivalents
|
$
|
100,554
|
$
|
—
|
$
|
—
|
$
|
100,554
|
||||||||
|
Short-term
investments
|
508
|
—
|
56,034
|
56,542
|
||||||||||||
|
Short-term
investments held by Symphony Icon, Inc.
|
5,417
|
—
|
—
|
5,417
|
||||||||||||
|
Total cash and cash equivalents
and investments
|
$
|
106,479
|
$
|
—
|
$
|
56,034
|
$
|
162,513
|
||||||||
|
Financial
Assets at Fair Value
As
of December 31, 2008
|
||||||||||||||||
|
Level
1
|
Level
2
|
Level
3
|
Total
|
|||||||||||||
|
(in
thousands)
|
||||||||||||||||
|
Cash
and cash equivalents
|
$
|
85,873
|
$
|
—
|
$
|
—
|
$
|
85,873
|
||||||||
|
Short-term
investments
|
629
|
—
|
—
|
629
|
||||||||||||
|
Short-term
investments held by Symphony Icon, Inc.
|
16,610
|
—
|
—
|
16,610
|
||||||||||||
|
Long-term
investments
|
—
|
—
|
55,686
|
55,686
|
||||||||||||
|
Total cash and cash equivalents
and investments
|
$
|
103,112
|
$
|
—
|
$
|
55,686
|
$
|
158,798
|
||||||||
The table
presented below summarizes the change in consolidated balance sheet carrying
value associated with Level 3 financial assets for the years ended
December 31, 2008 and 2009.
|
Short-term
Investments
|
Long-term
Investments
|
Total
|
||||||||||
|
(in
thousands)
|
||||||||||||
|
Balance
at December 31, 2007
|
$
|
—
|
$
|
—
|
$
|
—
|
||||||
|
Unrealized
losses included in earnings as loss on investments
|
—
|
(13,374
|
)
|
(13,374
|
)
|
|||||||
|
Unrealized
gains included in earnings as gain on investments
|
—
|
12,060
|
12,060
|
|||||||||
|
Net
sales and settlements
|
—
|
(21,050
|
)
|
(21,050
|
)
|
|||||||
|
Transfers
into Level 3
|
—
|
78,050
|
78,050
|
|||||||||
|
Balance
at December 31, 2008
|
—
|
55,686
|
55,686
|
|||||||||
|
Unrealized
gains included in earnings as gain on investments
|
350
|
823
|
1,173
|
|||||||||
|
Net
sales and settlements
|
(725
|
)
|
(100
|
)
|
(825
|
)
|
||||||
|
Reclassification
from long-term to short-term investments
|
56,409
|
(56,409
|
)
|
—
|
||||||||
|
Balance
at December 31, 2009
|
$
|
56,034
|
$
|
—
|
$
|
56,034
|
||||||
The
Company also has assets that under certain conditions are subject to measurement
at fair value on a non-recurring basis. These assets include goodwill
associated with the acquisition of Coelacanth Corporation in
2001. For these assets, measurement at fair value in periods
subsequent to their initial recognition is applicable if one or more is
determined to be impaired.
6. Property
and Equipment
Property
and equipment at December 31, 2009 and 2008 are as follows:
|
Estimated
Useful Lives
|
As
of December 31,
|
|||||||||||
|
In Years
|
2009
|
2008
|
||||||||||
|
(in
thousands)
|
||||||||||||
|
Computers
and software
|
3-5
|
$
|
10,986
|
$
|
12,328
|
|||||||
|
Furniture
and fixtures
|
5-7
|
7,634
|
7,648
|
|||||||||
|
Laboratory
equipment
|
3-7
|
39,047
|
39,385
|
|||||||||
|
Leasehold
improvements
|
7-10
|
9,786
|
9,756
|
|||||||||
|
Buildings
|
15-40
|
63,532
|
63,508
|
|||||||||
|
Land
|
—
|
3,564
|
3,564
|
|||||||||
|
Total
property and equipment
|
134,549
|
136,189
|
||||||||||
|
Less:
Accumulated depreciation and amortization
|
(75,795
|
)
|
(71,102
|
)
|
||||||||
|
Net
property and equipment
|
$
|
58,754
|
$
|
65,087
|
||||||||
7. Income
Taxes
Lexicon
recognizes deferred tax liabilities and assets for the expected future tax
consequences of events that have been recognized differently in the financial
statements and tax returns. Under this method, deferred tax liabilities and
assets are determined based on the difference between the financial statement
carrying amounts and tax bases of liabilities and assets using enacted tax rates
and laws in effect in the years in which the differences are expected to
reverse. Deferred tax assets are evaluated for realization based on a
more-likely-than-not criteria in determining if a valuation allowance should be
provided.
The
components of Lexicon’s deferred tax assets (liabilities) at December 31, 2009
and 2008 are as follows:
|
As
of December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
(in
thousands)
|
||||||||
|
Deferred
tax assets:
|
||||||||
|
Net
operating loss carryforwards
|
$
|
161,020
|
$
|
159,917
|
||||
|
Research and development tax
credits
|
29,440
|
25,000
|
||||||
|
Capitalized research and
development
|
29,870
|
—
|
||||||
|
Stock-based compensation
|
9,778
|
8,815
|
||||||
|
Deferred revenue
|
5,296
|
6,368
|
||||||
|
Other
|
1,699
|
902
|
||||||
|
Total deferred tax assets
|
237,103
|
201,002
|
||||||
|
Deferred
tax liabilities:
|
||||||||
|
Other
|
(413
|
)
|
(397
|
)
|
||||
|
Total deferred tax liabilities
|
(413
|
)
|
(397
|
)
|
||||
|
Less: valuation allowance
|
(236,690
|
)
|
(200,605
|
)
|
||||
|
Net deferred tax assets
|
$
|
—
|
$
|
—
|
||||
At
December 31, 2009, Lexicon had both federal and state NOL carryforwards of
approximately $443.3 million and $110.8 million,
respectively. The federal and state NOL carryforwards begin to expire
in 2011. The Company has R&D tax credit carryforwards of
approximately $29.4 million expiring beginning in
2011. Utilization of the NOL and R&D credit carryforwards may be
subject to a significant annual limitation due to ownership changes that have
occurred previously or could occur in the future provided by Section 382 of
the Internal Revenue Code. Based on the federal tax law limits and
the Company’s cumulative loss position, Lexicon concluded it was appropriate to
establish a full valuation allowance for its net deferred tax assets until an
appropriate level of profitability is sustained. During the year
ended December 31, 2009, the valuation allowance increased
$36.1 million, primarily due to the Company’s current year net
loss. Lexicon recorded an income tax benefit of $102,000 in the year
ended December 31, 2009. As of December 31, 2008 and 2007,
the Company did not have any unrecognized tax benefits.
The
Company is primarily subject to U.S. federal and New Jersey and Texas state
income taxes. The tax years 1995 to current remain open to
examination by U.S. federal authorities and 2004 to current remain open to
examination by state authorities. The Company’s policy is to
recognize interest and penalties related to income tax matters in income tax
expense. As of December 31, 2009 and 2008, the Company had no
accruals for interest or penalties related to income tax matters.
8. Goodwill
On July
12, 2001, Lexicon completed the acquisition of Coelacanth Corporation in a
merger. Coelacanth, now Lexicon Pharmaceuticals (New Jersey), Inc., forms the
core of the Company’s division responsible for small molecule compound
discovery. The results of Lexicon Pharmaceuticals (New Jersey), Inc.
are included in the Company’s results of operations for the period subsequent to
the acquisition.
Goodwill
associated with the acquisition of $25.8 million, which represents the
excess of the $36.0 million purchase price over the fair value of the
underlying net identifiable assets, was assigned to the consolidated entity,
Lexicon. There was no change in the carrying amount of goodwill for
the year ended December 31, 2009. Goodwill is not subject to
amortization, but is tested at least annually for impairment at the reporting
unit level, which is the Company’s single operating segment. The
Company performed an impairment test of goodwill on its annual impairment
assessment date. This test did not result in an impairment of
goodwill.
9. Debt
Obligations
Mortgage Loan: In April 2004,
Lexicon purchased its existing laboratory and office buildings and animal
facilities in The Woodlands, Texas with proceeds from a $34.0 million
third-party mortgage financing and $20.8 million in cash. The
mortgage loan has a ten-year term with a 20-year amortization and bears interest
at a fixed rate of 8.23%. The buildings and land that serve as
collateral for the mortgage loan are included in property and equipment at
$63.5 million and $3.6 million, respectively, before accumulated
depreciation.
The fair
value of Lexicon’s mortgage loan approximates its carrying value. The
fair value of Lexicon’s mortgage loan is estimated using discounted cash flow
analysis, based on the Company’s estimated current incremental borrowing
rate.
UBS Credit
Line: In January 2009, Lexicon entered into a credit line
agreement with UBS Bank USA that provides, as of December 31, 2009, up to
an aggregate amount of $37.5 million in the form of an uncommitted, demand,
revolving line of credit. Lexicon entered into the credit line in
connection with its acceptance of an offer from UBS AG, providing Lexicon with
rights to require UBS to purchase its $56.2 million (par value) of auction
rate securities at par value during the period from June 30, 2010 through
July 2, 2012. The credit line is secured only by these auction
rate securities and advances under the credit line will be made on a “no net
cost” basis, meaning that the interest paid by Lexicon on advances will not
exceed the interest or dividends paid to Lexicon by the issuer of the auction
rate securities. The interest rate paid on the line of credit is less
than Lexicon’s current incremental borrowing rate. As of
December 31, 2009, Lexicon had $37.4 million outstanding under this
credit line.
The
following table includes the aggregate future principal payments of the
Company’s long-term debt as of December 31, 2009:
|
For
the Year Ending December 31
|
||||
|
(in
thousands)
|
||||
|
2010
|
$
|
38,482
|
||
|
2011
|
1,138
|
|||
|
2012
|
1,230
|
|||
|
2013
|
1,343
|
|||
|
2014
|
24,771
|
|||
|
66,964
|
||||
|
Less
current portion
|
(38,482
|
)
|
||
|
Total
long-term debt
|
$
|
28,482
|
||
10. Arrangements
with Symphony Icon, Inc.
On June
15, 2007, Lexicon entered into a series of related agreements providing for the
financing of the clinical development of certain of its drug candidates,
including LX1031 and LX1032, along with any other pharmaceutical compositions
modulating the same targets as those drug candidates (the
“Programs”). The agreements include a Novated and Restated Technology
License Agreement pursuant to which the Company licensed to Symphony Icon, a
wholly-owned subsidiary of Symphony Icon Holdings LLC (“Holdings”), the
Company’s intellectual property rights related to the
Programs. Holdings contributed $45 million to Symphony Icon in
order to fund the clinical development of the Programs.
Under a
Share Purchase Agreement, dated June 15, 2007, between the Company and Holdings,
the Company issued and sold to Holdings 7,650,622 shares of its common stock on
June 15, 2007 in exchange for $15 million and the Purchase Option (as defined
below).
Under a
Purchase Option Agreement, dated June 15, 2007, among the Company, Symphony Icon
and Holdings, the Company has received from Holdings an exclusive purchase
option (the “Purchase Option”) that gives the Company the right to acquire all
of the equity of Symphony Icon, thereby allowing the Company to reacquire all of
the Programs. The Purchase Option is exercisable by the Company at
any time, in its sole discretion, until June 15, 2011 at an exercise price
of (a) $81 million, if the Purchase Option is exercised before
June 15, 2010 and (b) $90 million, if the Purchase Option is exercised
on or after June 15, 2010 and before June 15, 2011. The
Purchase Option exercise price may be paid in cash or a combination of cash and
common stock, at the Company’s sole discretion, provided that the common stock
portion may not exceed 40% of the Purchase Option exercise
price. Lexicon has calculated the value of the Purchase Option as the
difference between the fair value of the common stock issued to Holdings of
$23.6 million and the $15.0 million in cash received from Holdings for
the issuance of the common stock. Lexicon has recorded the value of
the Purchase Option as an asset, and is amortizing this asset over the four-year
option period. The unamortized balance of $3.1 million and
$5.3 million is recorded in other assets in the accompanying consolidated
balance sheets as of December 31, 2009 and 2008, respectively, and the
amortization expense of $2.1 million is recorded in other expense, net in
the accompanying consolidated statements of operations for each of the years
ended December 31, 2009 and 2008.
Under an
Amended and Restated Research and Development Agreement, dated June 15, 2007,
among the Company, Symphony Icon and Holdings (the “R&D Agreement”),
Symphony Icon and the Company are developing the Programs in accordance with a
specified development plan and related development budget. The
R&D Agreement provides that the Company will continue to be primarily
responsible for the development of the Programs. The Company’s
development activities are supervised by Symphony Icon’s development committee,
which is comprised of an equal number of representatives from the Company and
Symphony Icon. The development committee will report to Symphony
Icon’s board of directors, which is currently comprised of five members,
including one member designated by the Company and two independent
directors.
Under a
Research Cost Sharing, Payment and Extension Agreement, dated June 15, 2007,
among the Company, Symphony Icon and Holdings, upon the recommendation of the
development committee, Symphony Icon’s board of directors may require the
Company to pay Symphony Icon up to $15 million for Symphony Icon’s use in
the development of the Programs in accordance with the specified development
plan and related development budget. The development committee’s
right to recommend that Symphony Icon’s board of directors submit such funding
requirement to the Company will terminate on the one-year anniversary of the
expiration of the Purchase Option, subject to limited
exceptions. Through December 2009, Symphony Icon’s board of directors
has requested the Company to pay Symphony Icon $4.3 million under this
agreement, all of which has been paid through December 31, 2009, and
management expects that additional funding will be needed in the
future.
Lexicon
has determined that Symphony Icon is a variable interest entity for which it is
the primary beneficiary. This determination was based on Holdings’
lack of controlling rights with respect to Symphony Icon’s activities and the
limitation on the amount of expected residual returns Holdings may expect from
Symphony Icon if Lexicon exercises its Purchase Option. Lexicon has
determined it is a variable interest holder of Symphony Icon due to its
contribution of the intellectual property relating to the Programs and its
issuance of shares of its common stock in exchange for the Purchase Option,
which Lexicon intends to exercise if the development of the Programs is
successful. Lexicon has determined that it is a primary beneficiary
as a result of certain factors, including its primary responsibility for the
development of the Programs and its contribution of the intellectual property
relating to the Programs. As a result, Lexicon has included the
financial condition and results of operations of Symphony Icon in its
consolidated financial statements. Symphony Icon’s cash and cash
equivalents have been recorded on Lexicon’s consolidated financial statements as
short-term investments held by Symphony Icon. The noncontrolling
interest in Symphony Icon on Lexicon’s consolidated balance sheet initially
reflected the $45 million proceeds contributed into Symphony Icon less
$2.3 million of structuring and legal fees. As the collaboration
progressed, this line item was reduced by Symphony Icon’s losses, which were
$10.5 million, $20.0 million and $12.4 million in the years ended
December 31, 2009, 2008 and 2007, respectively. The reductions to the
noncontrolling interest in Symphony Icon have been reflected in Lexicon’s
consolidated statements of operations using a similar caption and has reduced
the amount of Lexicon’s reported net loss. Through December 31,
2009, Lexicon has not charged any license fees and has not recorded any revenue
from Symphony Icon, and does not expect to do so based on the current agreements
with Symphony Icon and Holdings.
As
discussed in Note 3, Recent Accounting Pronouncements, Lexicon has
determined that upon adoption of SFAS 167 on January 1, 2010, Lexicon
will no longer be the primary beneficiary of Symphony Icon, and therefore will
no longer include the financial condition and results of operations of Symphony
Icon in its consolidated financial statements.
11. Commitments
and Contingencies
Operating Lease
Obligations: A Lexicon subsidiary leases laboratory and office
space in Hopewell, New Jersey under an agreement that expires in June
2013. The lease provides for two five-year renewal options at 95% of
the fair market rent and includes escalating lease payments. Rent
expense is recognized on a straight-line basis over the original lease
term. Lexicon is the guarantor of the obligation of its subsidiary
under this lease. The Company is required to maintain restricted
investments to collateralize a standby letter of credit for this
lease. The Company had $0.4 million in restricted investments as
collateral as of December 31, 2009
and 2008. Additionally, Lexicon leases certain equipment
under operating leases.
Rent
expense for all operating leases was approximately $2.4 million,
$2.5 million and $2.5 million for the years ended December 31, 2009,
2008 and 2007, respectively. The following table includes
non-cancelable, escalating future lease payments for the facility in New
Jersey:
|
For
the Year Ending December 31
|
||||
|
(in
thousands)
|
||||
|
2010
|
$
|
2,502
|
||
|
2011
|
2,539
|
|||
|
2012
|
2,593
|
|||
|
2013
|
1,296
|
|||
|
Total
|
$
|
8,930
|
||
Employment Agreements:
Lexicon has entered into employment agreements with certain of its
corporate officers. Under the agreements, each officer receives a base salary,
subject to adjustment, with an annual discretionary bonus based upon specific
objectives to be determined by the compensation committee. The employment
agreements are at-will and contain non-competition agreements. The agreements
also provide for a termination clause, which requires either a six or 12-month
payment based on the officer’s salary and payment of a specified portion of the
officer’s bonus target for such year, in the event of termination.
Legal
Proceedings: Lexicon is from time to time party to claims and
legal proceedings that arise in the normal course of its business and that it
believes will not have, individually or in the aggregate, a material adverse
effect on its results of operations, financial condition or
liquidity.
12. Agreements
with Invus, L.P.
On June
17, 2007, Lexicon entered into a series of agreements with Invus, L.P. (“Invus”)
under which Invus made an investment in the Company’s common stock and has
certain other rights described below.
Lexicon
entered into a Securities Purchase Agreement (the “Securities Purchase
Agreement”) with Invus under which the Company issued and sold to Invus
50,824,986 shares in an initial investment (the “Initial Investment”) and
permitted Invus to require, subject to specific conditions, that the Company
conduct certain rights offerings (the “Rights Offerings”). In
connection with the Securities Purchase Agreement, Lexicon also entered into a
Warrant Agreement with Invus under which the Company issued to Invus warrants
(the “Warrants”) to purchase 16,498,353 shares of its common stock at an
exercise price of $3.0915 per share. The Warrant Agreement provided
that, to the extent not previously exercised, the Warrants would terminate
concurrently with the closing of the Initial Investment.
Initial
Investment: In the Initial Investment, which closed on August
28, 2007, Invus purchased 50,824,986 shares of Lexicon’s common stock for a
total of approximately $205.4 million, resulting in net proceeds of
$198.0 million after deducting fees and expenses of approximately
$7.5 million. Simultaneously with the closing of the Initial
Investment, all Warrants issued under the Warrant Agreement terminated
unexercised according to their terms. This purchase resulted in
Invus’ ownership of 40% of the post-transaction outstanding shares of Lexicon’s
common stock.
Rights
Offerings: For a period of one year following
November 28, 2009 (the “First Rights Offering Trigger Date”), Invus will
have the right to require Lexicon to make a pro rata offering of
non-transferable rights to acquire common stock to all of its stockholders (the
“First Rights Offering”) in an aggregate amount to be designated by Invus not to
exceed $172.3 million, minus the aggregate net proceeds received in all
Qualified Offerings (as defined below), if any, completed prior to the First
Rights Offering Trigger Date. The price per share of the First Rights
Offering would be designated by Invus in a range between $4.50 and a
then-current average market price of the Company’s common stock. All
stockholders would have oversubscription rights with respect to the First Rights
Offering, and Invus would be required to purchase its pro rata portion of the
First Rights Offering.
For a
period of one year following the date (the “Second Rights Offering Trigger
Date”) which is 90 days after (a) Invus’ exercise of its right to
require us to conduct the First Rights Offering or (b) if Invus does not
exercise its right to require Lexicon to conduct the First Rights Offering, the
First Rights Offering Trigger Date, Invus would have the right to require the
Company to make a pro rata offering of non-transferable rights to acquire common
stock to all of its stockholders (the “Second Rights Offering” and, together
with the First Rights Offering, the “Rights Offerings”) in an aggregate amount
to be designated by Invus not to exceed an amount equal to $344.5 million, minus
the amount of the First Rights Offering, minus the aggregate net proceeds
received in all Qualified Offerings, if any, completed prior to the Second
Rights Offering Trigger Date. The price per share of the Second
Rights Offering would be designated by Invus in a range between $4.50 and a
then-current average market price of the Company’s common stock. All
stockholders would have oversubscription rights with respect to the Second
Rights Offering, and Invus would be required to purchase its pro rata portion of
the Second Rights Offering. Lexicon has determined that the First
Rights Offering and the Second Rights Offering should be treated as equity
instruments, and accordingly has not recorded a liability for the future
settlement of any rights offerings.
A
“Qualified Offering” consists of a bona fide financing transaction comprised of
Lexicon’s issuance of shares of its common stock at a price greater than $4.50
per share, which transaction is not entered into in connection with the
Company’s entry into any other transaction (including, a collaboration or
license for the discovery, development or commercialization of pharmaceutical
products) involving the purchaser of such common stock. Lexicon has
not completed any such Qualified Offering. Until the later of the
completion of the Second Rights Offering or the expiration of the 90-day period
following the Second Rights Offering Trigger Date, Lexicon will not, without
Invus’ prior consent, issue any shares of its common stock at a price below
$4.50 per share, subject to certain exceptions.
In
connection with the Securities Purchase Agreement, Lexicon entered into a
Stockholders’ Agreement with Invus under which Invus (a) has specified rights
with respect to designation of directors and to participate in future equity
issuances by the Company, (b) is subject to certain standstill restrictions, as
well as restrictions on transfer and the voting of the shares of common stock
held by it and its affiliates, and (c), as long as Invus holds at least 15% of
the total number of outstanding shares of the Company’s common stock, is
entitled to certain minority protections.
13. Other
Capital Stock Agreements
Common Stock: In October
2009, Lexicon completed the public offering and sale of 38,333,332 shares of its
common stock at a price of $1.50 per share, resulting in net proceeds of
$55.2 million, after deducting underwriting discounts and commissions of
$1.9 million and offering expenses of $0.4 million. Invus
purchased 15,455,145 of these shares. All of the net proceeds of this
offering are reflected as issuance of common stock in the accompanying financial
statements.
14. Stock
Options and Warrants
Stock
Option Plans
Equity Incentive
Plan: In September 1995, Lexicon adopted the 1995 Stock Option
Plan, which was subsequently amended and restated in February 2000 as the 2000
Equity Incentive Plan, and later amended and restated in April 2009 as the
Equity Incentive Plan (the “Equity Incentive Plan”).
The
Equity Incentive Plan provides for the grant of incentive stock options to
employees and nonstatutory stock options to employees, directors and consultants
of the Company. The plan also permits the grant of stock bonus awards,
restricted stock awards, phantom stock awards and stock appreciation rights.
Incentive and nonstatutory stock options have an exercise price of 100% or more
of the fair market value of our common stock on the date of
grant. The purchase price of restricted stock awards may not be less
than 85% of fair market value. However, the plan administrator may
award stock bonus awards in consideration of past services or phantom stock
awards without a purchase payment. Shares may be subject to a repurchase option
in the discretion of the plan administrator. Most options granted
under the Equity Incentive Plan become vested and exercisable over a period of
four years; however some have been granted with different vesting
schedules. Options granted under the Equity Incentive Plan have a
term of ten years from the date of grant.
The total
number of shares of common stock that may be issued pursuant to stock awards
under the Equity Incentive Plan shall not exceed in the aggregate 35,000,000
shares. No more than 3,500,000 shares may be issued pursuant to
awards other than stock options and stock appreciation rights. As of
December 31, 2009, an aggregate of 35,000,000 shares of common stock had been
reserved for issuance, options to purchase 16,739,448 shares were outstanding,
4,310,864 shares had been issued upon the exercise of stock options and 552,073
shares had been issued pursuant to stock bonus awards or restricted stock awards
granted under the Equity Incentive Plan.
Non-Employee Directors’ Stock Option
Plan: In February 2000, Lexicon adopted the 2000 Non-Employee
Directors’ Stock Option Plan, which was subsequently amended and restated in
April 2009 as the Non-Employee Directors’ Stock Option Plan, (the “Directors’
Plan”) to provide for the automatic grant of nonstatutory stock options to
non-employee directors of the Company. Under the Directors’ Plan,
non-employee directors receive an initial option to purchase 30,000 shares of
common stock. In addition, on the day following each of the Company’s
annual meetings of stockholders, each non-employee director who has been a
director for at least six months is automatically granted an option to purchase
10,000 shares of common stock, and the non-employee chairman of the board of
directors is automatically granted an option to purchase 20,000 shares of common
stock. Initial option grants become vested and exercisable over a
period of five years and annual option grants become vested over a period of
12 months from the date of grant. Options granted under the
Directors’ Plan have an exercise price equal to the fair market value of the
Company’s common stock on the date of grant and a term of ten years from the
date of grant.
The total
number of shares of common stock that may be issued pursuant to stock awards
under the Directors’ Plan shall not exceed in the aggregate 1,200,000
shares. As of December 31, 2009, an aggregate of 1,200,000 shares of
common stock had been reserved for issuance, options to purchase 604,000 shares
were outstanding, and none had been exercised under the Directors’
Plan.
Coelacanth Corporation 1999 Stock
Option Plan: Lexicon assumed the Coelacanth Corporation 1999
Stock Option Plan (the “Coelacanth Plan”) and the outstanding stock options
under the plan in connection with its July 2001 acquisition of Coelacanth
Corporation. The Company will not grant any further options under the
plan. As outstanding options under the plan expire or terminate, the
number of shares authorized for issuance under the plan will be correspondingly
reduced.
The
purpose of the plan was to provide an opportunity for employees, directors and
consultants of Coelacanth to acquire a proprietary interest, or otherwise
increase their proprietary interest, in Coelacanth as an incentive to continue
their employment or service. Both incentive and nonstatutory options
are outstanding under the plan. Most outstanding options vest over
time and expire ten years from the date of grant. The exercise price
of options awarded under the plan was determined by the plan administrator at
the time of grant. In general, incentive stock options have an
exercise price of 100% or more of the fair market value of Coelacanth common
stock on the date of grant and nonstatutory stock options have an exercise price
as low as 85% of fair market value on the date of grant.
As of
December 31, 2009, an aggregate of 30,142 shares of common stock had been
reserved for issuance, options to purchase 2,225 shares of common stock were
outstanding and 27,917 shares of common stock had been issued upon the exercise
of stock options issued under the Coelacanth Plan.
Stock Option
Activity: The following is a summary of option activity under
Lexicon’s stock option plans:
|
2009
|
2008
|
2007
|
||||||||||||||||||||||
|
(in
thousands, except exercise price data)
|
Options
|
Weighted
Average Exercise Price
|
Options
|
Weighted
Average Exercise Price
|
Options
|
Weighted
Average Exercise Price
|
||||||||||||||||||
|
Outstanding
at beginning of year
|
16,898
|
$
|
5.13
|
16,351
|
$
|
5.65
|
15,815
|
$
|
5.99
|
|||||||||||||||
|
Granted
|
4,864
|
1.44
|
4,077
|
2.08
|
2,952
|
3.85
|
||||||||||||||||||
|
Exercised
|
(121
|
)
|
2.18
|
(1
|
)
|
1.89
|
(516
|
)
|
1.80
|
|||||||||||||||
|
Expired
|
(3,372
|
)
|
5.66
|
(2,663
|
)
|
4.32
|
(1,137
|
)
|
8.11
|
|||||||||||||||
|
Forfeited
|
(923
|
)
|
2.46
|
(866
|
)
|
3.03
|
(763
|
)
|
4.68
|
|||||||||||||||
|
Outstanding
at end of year
|
17,346
|
4.16
|
16,898
|
5.13
|
16,351
|
5.65
|
||||||||||||||||||
|
Exercisable
at end of year
|
10,462
|
$
|
5.69
|
11,410
|
$
|
6.28
|
11,946
|
$
|
6.21
|
|||||||||||||||
The
weighted average estimated grant date fair value of options granted during the
years ended December 31, 2009, 2008 and 2007 were $1.04, $1.43
and $2.71, respectively. The total intrinsic value of
options exercised during the years ended December 31, 2009, 2008 and 2007
were $600, $300 and $982,000, respectively. The weighted average
remaining contractual term of options outstanding and exercisable was 6.1 and
4.4 years, respectively, as of December 31, 2009. At December
31, 2009, the aggregate intrinsic value of the outstanding options and the
exercisable options was $1,344,000 and $51,000, respectively.
The
following is a summary of the nonvested options as of December 31, 2009,
and changes during the year then ended, under Lexicon’s stock option
plans:
|
Options
|
Weighted
Average Grant Date Fair Value
|
|||||||
|
(in
thousands)
|
||||||||
|
Nonvested
at beginning of year
|
5,488
|
$
|
1.91
|
|||||
|
Granted
|
4,864
|
1.04
|
||||||
|
Vested
|
(2,546
|
)
|
1.99
|
|||||
|
Canceled
|
(923
|
)
|
1.71
|
|||||
|
Nonvested
at end of year
|
6,883
|
$
|
1.30
|
|||||
Restricted
Stock Activity:
During
the year ended December 31, 2009, Lexicon granted its officers restricted
stock bonus awards under the Equity Incentive Plan in lieu of cash bonus
awards. The shares subject to the awards vested in two installments
over the one-year period following the date of grant. The following
is a summary of restricted stock activity under Lexicon’s stock option plans for
the year ended December 31, 2009:
|
Shares
|
Weighted
Average Grant Date Fair Value
|
|||||||
|
(in
thousands)
|
||||||||
|
Outstanding
at December 31, 2008
|
—
|
$
|
—
|
|||||
|
Granted
|
534
|
1.45
|
||||||
|
Vested
|
(267
|
)
|
1.45
|
|||||
|
Forfeited
|
(12
|
)
|
1.45
|
|||||
|
Nonvested
at December 31, 2009
|
255
|
$
|
1.45
|
|||||
Warrants
In
connection with the acquisition of Coelacanth in July 2001, Lexicon assumed
Coelacanth’s outstanding warrants to purchase 25,169 shares of common
stock. The warrants expired on March 31, 2009. The
fair value of the warrants was included in the total purchase price for the
acquisition.
Aggregate
Shares Reserved for Issuance
As of
December 31, 2009, an aggregate of 17,345,673 shares of common stock were
reserved for issuance upon exercise of outstanding stock options and 14,005,665
additional shares were available for future grants under Lexicon’s stock option
plans. The Company has a policy of using either authorized and
unissued shares or treasury shares, including shares acquired by purchase in the
open market or in private transactions, to satisfy equity award
exercises.
15. Benefit
Plans
Lexicon
has established an Annual Profit Sharing Incentive Plan (the “Profit Sharing
Plan”). The purpose of the Profit Sharing Plan is to provide for the payment of
incentive compensation out of the profits of the Company to certain of its
employees. Participants in the Profit Sharing Plan are entitled to an annual
cash bonus equal to their proportionate share (based on salary) of 15 percent of
the Company’s annual pretax income, if any.
Lexicon
maintains a defined-contribution savings plan under Section 401(k) of the
Internal Revenue Code. The plan covers substantially all full-time
employees. Participating employees may defer a portion of their
pretax earnings, up to the Internal Revenue Service annual contribution
limit. Beginning in 2000, the Company was required to match employee
contributions according to a specified formula. The matching
contributions totaled $614,000, $828,000 and $862,000 in the years ended
December 31, 2009, 2008 and 2007, respectively. Company
contributions are vested based on the employee’s years of service, with full
vesting after four years of service.
16. Collaboration
and License Agreements
Lexicon
has derived substantially all of its revenues from drug discovery and
development alliances, target validation collaborations for the development and,
in some cases, analysis of the physiological effects of genes altered in
knockout mice, government grants and contracts, technology licenses,
subscriptions to its databases and compound library sales.
Drug
Discovery and Development Alliances
Bristol-Myers Squibb. Lexicon
established an alliance with Bristol-Myers Squibb Company in December 2003 to
discover, develop and commercialize small molecule drugs in the neuroscience
field. Lexicon initiated the alliance with a number of drug discovery
programs at various stages of development and has used its gene knockout
technology to identify additional drug targets with promise in the neuroscience
field. For those targets that were selected for the alliance, Lexicon
and Bristol-Myers Squibb are working together, on an exclusive basis, to
identify, characterize and carry out the preclinical development of small
molecule drugs, and share equally both in the costs and in the work attributable
to those efforts. As drugs resulting from the collaboration enter
clinical trials, Bristol-Myers Squibb will have the first option to assume full
responsibility for clinical development and commercialization.
Lexicon
received an upfront payment of $36.0 million and research funding of $30.0
million in the initial three years of the agreement, or the target function
discovery term. This funding was in consideration for access to
Lexicon’s technology and infrastructure and for Lexicon’s production and
specified phenotypic analysis of knockout mice in support of the target function
discovery portion of the alliance. Bristol-Myers Squibb extended the
target discovery term of the alliance in May 2006 for an additional two years in
exchange for $20.0 million in additional research funding over the two year
extension, which commenced in January 2007. This additional funding
was in consideration for additional research and phenotypic analysis of knockout
mice which supplemented the phenotypic analysis conducted in the initial target
function discovery term. Lexicon will also receive clinical and
regulatory milestone payments ranging, depending on the timing and extent of its
efforts in the alliance, up to $76 million for each drug developed by
Bristol-Myers Squibb under the alliance. Lexicon will earn royalties
on sales of drugs commercialized by Bristol-Myers Squibb. The party
with responsibility for the clinical development and commercialization of drugs
resulting from the alliance will bear the costs of those efforts. The
target discovery portion of the alliance expired in October 2009. The
original upfront payment of $36.0 million and research funding of
$30.0 million was recognized over the initial estimated period of service
of three years. The additional research funding of $20.0 million
was recognized over the estimated performance period of two and one-half
additional years subject to the extension, beginning in January
2007. Lexicon recorded a change in estimate that increased net loss
and net loss per share by $1.7 million and $0.01 per share, respectively,
in the year ended December 31, 2008 due to an increase in estimated
performance period of this extension.
The
upfront payment of $36.0 million was not related to a deliverable with
standalone value at inception, and Lexicon accounted for the entire agreement
with Bristol-Myers Squibb as a single unit of accounting. Milestone
payments received are in consideration for additional
performance. Therefore, Lexicon recognizes revenue from such
milestone payments upon achievement of the milestones.
Revenue
recognized under this agreement was $1.7 million, $9.3 million and
$10.0 million for the years ended December 31, 2009, 2008 and 2007,
respectively.
Genentech. Lexicon
established an alliance with Genentech, Inc. in December 2002 to discover novel
therapeutic proteins and antibody targets. Lexicon used its target
validation technologies to discover the functions of secreted proteins and
potential antibody targets identified through Genentech’s internal drug
discovery research. Lexicon received an upfront payment of $9.0
million and funding under a $4.0 million loan in 2002. In addition,
Lexicon received $24.0 million in performance payments for its work in the
collaboration as it was completed. The upfront payment of
$9.0 million was recognized over the initial estimated period of service of
three years, which was subsequently extended to three and one-half
years.
In
November 2005, Lexicon and Genentech expanded the alliance to include additional
research, as well as the development and commercialization of new biotherapeutic
drugs. Lexicon received a total of $25.0 million in upfront and
milestone payments and research funding for the three-year advanced research
portion of the expanded alliance. In the expanded alliance, Lexicon
conducted advanced research on a broad subset of targets validated in the
original collaboration using Lexicon’s proprietary gene knockout
technology. The upfront payment under the new agreement was
recognized over the estimated period of service of three years.
Lexicon
has exclusive rights to develop and commercialize biotherapeutic drugs for two
of the targets included in the alliance, while Genentech has exclusive rights to
develop and commercialize biotherapeutic drugs for the other
targets. Lexicon retains certain other rights to discoveries made in
the alliance, including non-exclusive rights, along with Genentech, for the
development and commercialization of small molecule drugs addressing the targets
included in the alliance. Lexicon will receive clinical and
regulatory milestone payments ranging, depending on the extent of Lexicon’s
efforts in the alliance, up to $25 million for each drug target for which
Genentech develops a biotherapeutic drug under the alliance. Lexicon
will also earn royalties on sales of biotherapeutic drugs commercialized by
Genentech under the alliance. Genentech is entitled to receive
milestone payments and royalties on sales of biotherapeutic drugs which Lexicon
develops or commercializes under the alliance. The research
collaboration term under the agreement expired in November 2008.
The
upfront payment was not related to a deliverable with standalone value at
inception and Lexicon accounted for the entire agreement with Genentech as a
single unit of accounting. Milestone payments received are in
consideration for additional performance. Therefore, Lexicon
recognizes revenue from such milestone payments upon achievement of the
milestones.
Revenue
recognized under this agreement was $4.0 million and $4.3 million for
the years ended December 31, 2008 and 2007, respectively.
Schering-Plough/Organon. Lexicon
established a drug discovery alliance with N.V. Organon in May 2005 to discover,
develop and commercialize novel biotherapeutic drugs. In the
alliance, Lexicon created and analyzed knockout mice for 300 genes selected by
the parties that encode secreted proteins or potential antibody targets,
including two of Lexicon’s preexisting drug discovery
programs. Lexicon and Organon agreed to equally share costs of and
responsibility for research, preclinical and clinical activities, jointly
determine the manner in which collaboration products would be commercialized,
and equally benefit from product revenue. Organon, formerly a
subsidiary of Akzo Nobel N.V., was acquired by Schering-Plough Corporation in
November 2007, which subsequently merged with Merck & Co., Inc. in November
2009. In February 2010, Lexicon entered into a revised collaboration
and license agreement with Organon and Schering Corporation, acting through its
Schering-Plough Research Institute division, amending the terms of the alliance
to provide that Schering-Plough will assume the full cost of research activities
conducted by either party in the alliance, and will assume the full cost of and
responsibility for preclinical, clinical and commercialization activities with
respect to biotherapeutic drugs resulting from the alliance. Lexicon
is entitled to receive clinical and regulatory milestone payments of up to $39
million for each drug target for which Schering-Plough develops a biotherapeutic
drug under the alliance. Lexicon will also earn royalties on sales of
biotherapeutic drugs commercialized by Schering-Plough under the
alliance.
Lexicon
received an upfront payment of $22.5 million from Organon in exchange for access
to Lexicon’s drug target discovery capabilities and the exclusive right to
co-develop biotherapeutic drugs for the 300 genes selected for the
alliance. Organon has also provided Lexicon with annual research
funding totaling $30.0 million for its 50% share of the alliance’s costs during
this same period. The target discovery portion of the alliance
expired in December 2009.
The
upfront payment of $22.5 million was not related to a deliverable with
standalone value at inception, and Lexicon accounted for the entire agreement
with Organon as a single unit of accounting. Revenue from the upfront
payment is recognized on a straight-line basis over the four-year period that
Lexicon expects to perform its obligations under the target function discovery
portion of the alliance. Revenue from the research funding fees is
recognized as Lexicon performs its obligations under the target function
discovery portion of the alliance, reflecting the gross amount billed to Organon
on the basis of shared costs during the period. Milestone payments
received are in consideration for additional performance. Therefore,
Lexicon recognizes revenue from such milestone payments upon achievement of the
milestones.
Revenue
recognized under this agreement was $2.3 million, $9.2 million and
$13.5 million for the years ended December 31, 2009, 2008 and 2007,
respectively.
Takeda. Lexicon
established an alliance with Takeda Pharmaceutical Company Limited in July 2004
to discover new drugs for the treatment of high blood pressure. In
the collaboration, Lexicon used its gene knockout technologies to identify drug
targets that control blood pressure. Takeda is responsible for the
screening, medicinal chemistry, preclinical and clinical development and
commercialization of drugs directed against targets selected for the alliance,
and bears all related costs. Lexicon received an upfront payment of
$12.0 million from Takeda for the initial, three-year term of the
agreement. This payment was in consideration for access to Lexicon’s technology
and infrastructure during the target discovery portion of the
alliance. In addition, Lexicon received $6.5 million in research
milestone payments for targets selected for therapeutic
development. Lexicon is entitled to receive clinical development and
product launch milestone payments of up to $29 million for each product
commercialized from the collaboration. Lexicon will also earn
royalties on sales of drugs commercialized by Takeda. The target
discovery portion of the alliance, which ended in 2007, had a term of three
years.
The
upfront payment of $12.0 million was not related to a deliverable with
standalone value at inception, and Lexicon accounted for the entire agreement
with Takeda as a single unit of accounting. Revenue was recognized
from the upfront payment on a straight-line basis over the three-year period
Lexicon expected to perform its obligations under the
agreement. Milestone payments received are in consideration for
additional performance. Therefore, Lexicon recognizes revenue from
such milestone payments upon achievement of the milestones.
Revenue
recognized under this agreement was $2.3 million for the year ended
December 31, 2007.
Other
Collaborations and Licenses
Texas Institute for Genomic
Medicine. In July 2005, Lexicon received a
$35.0 million award from the Texas Enterprise Fund for the creation of a
knockout mouse embryonic stem cell library containing 350,000 cell lines for the
Texas Institute for Genomic Medicine (“TIGM”) using Lexicon’s proprietary gene
trapping technology, which Lexicon completed in 2007. Lexicon also
equipped TIGM with the bioinformatics software required for the management and
analysis of data relating to the library. The Texas Enterprise Fund
also awarded $15.0 million to the Texas A&M University System for the
creation of facilities and infrastructure to house the
library. Revenue recognized under this agreement was
$0.1 million and $10.6 million for the years ended December 31,
2008 and 2007, respectively. Lexicon recorded a change in estimate
that increased revenue and therefore decreased net loss and net loss per share
by $3.7 million and $0.04 per share, respectively, in the year ended
December 31, 2007 due to a reduction in the estimated performance period of
this agreement.
Under the
terms of the award, Lexicon is responsible for the creation of a specified
number of jobs beginning in 2012, reaching an aggregate of 1,616 new jobs in
Texas by December 31, 2016. Lexicon will obtain credits based on
funding received by TIGM and certain related parties from sources other than the
State of Texas that it may offset against its potential liability for any job
creation shortfalls. Lexicon will also obtain credits against future
jobs commitment liabilities for any surplus jobs it creates. Subject
to these credits, if Lexicon fails to create the specified number of jobs, the
state may require Lexicon to repay $2,415 for each job Lexicon falls
short. Lexicon’s maximum aggregate exposure for such payments, if
Lexicon fails to create any new jobs, is approximately $14.2 million,
without giving effect to any credits to which Lexicon may be
entitled. Lexicon has recorded this obligation as deferred revenue in
the accompanying consolidated balance sheets. The Texas A&M
University System, together with TIGM, has independent job creation obligations
and is obligated for an additional period to maintain an aggregate of 5,000
jobs, inclusive of those Lexicon creates.
Taconic
Farms. Lexicon established a collaboration with Taconic Farms,
Inc. in November 2005 for the marketing, distribution and licensing of certain
lines of knockout mice and entered into an expanded collaboration with Taconic
in July 2009. Under the terms of the collaboration, Lexicon is
presently making available through Taconic more than 3,600 distinct lines of
knockout mice, and in some cases, phenotypic data relating to such lines of
knockout mice, for use by pharmaceutical and biotechnology companies, academic
and non-profit institutions and other researchers. Lexicon receives
license fees and royalties from payments received by Taconic from customers
obtaining access to knockout mice and any related phenotypic
data. The Company received payments totaling $2.8 million
through December 31, 2009. Revenue recognized under these agreements
was $2.2 million, $747,000 and $558,000 for the years ended December 31, 2009,
2008 and 2007, respectively.
Bristol-Myers
Squibb. Lexicon entered into drug target validation agreements
with Bristol-Myers Squibb Company in December 2004, January 2006, October
2006, November 2007 and February 2009, under which Lexicon is developing mice
and phenotypic data for certain genes requested by Bristol-Myers Squibb under
those agreements. The collaboration term under each of these
agreements will expire after the final phenotypic data set has been delivered by
Lexicon under that agreement. The Company received payments totaling
$10.6 million under these agreements through December 31,
2009. Revenue recognized under these agreements was
$1.1 million, $1.1 million and $1.7 million for the years ended
December 31, 2009, 2008 and 2007, respectively.
Genentech. Lexicon
entered into a drug target validation agreement with Genentech, Inc. in February
2007. Under this agreement, Lexicon developed mice with mutations
requested by Genentech. The collaboration term under the agreement
has expired as the final delivery of the selected mice has been performed by
Lexicon. The Company received payments totaling $1.1 million
under the agreement through December 31, 2009. Revenue recognized
under this agreement was $26,000, $0.1 million and
$0.9 million for the years ended December 31, 2009, 2008 and 2007,
respectively.
17. Selected
Quarterly Financial Data
The table
below sets forth certain unaudited statements of operations data, and net loss
per common share data, for each quarter of 2009 and 2008:
(in
thousands, except per share data)
|
Quarter
Ended
|
||||||||||||||||
|
March
31
|
June
30
|
September
30
|
December
31
|
|||||||||||||
|
(Unaudited)
|
||||||||||||||||
|
2009
|
||||||||||||||||
|
Revenues
|
$
|
4,168
|
$
|
2,989
|
$
|
2,131
|
$
|
1,412
|
||||||||
|
Loss
from operations
|
$
|
(23,570
|
)
|
$
|
(22,782
|
)
|
$
|
(21,757
|
)
|
$
|
(21,847
|
)
|
||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc.
|
$
|
(21,560
|
)
|
$
|
(20,073
|
)
|
$
|
(19,142
|
)
|
$
|
(22,005
|
)
|
||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc. per common share, basic
and diluted
|
$
|
(0.16
|
)
|
$
|
(0.15
|
)
|
$
|
(0.14
|
)
|
$
|
(0.13
|
)
|
||||
|
Shares
used in computing net loss per common share
|
137,075
|
137,331
|
137,313
|
169,872
|
||||||||||||
|
2008
|
||||||||||||||||
|
Revenues
|
$
|
8,893
|
$
|
9,566
|
$
|
7,512
|
$
|
6,350
|
||||||||
|
Loss
from operations
|
$
|
(24,438
|
)
|
$
|
(26,386
|
)
|
$
|
(24,822
|
)
|
$
|
(20,889
|
)
|
||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc.
|
$
|
(17,950
|
)
|
$
|
(20,034
|
)
|
$
|
(23,459
|
)
|
$
|
(15,417
|
)
|
||||
|
Net
loss attributable to Lexicon Pharmaceuticals, Inc. per common share, basic
and diluted
|
$
|
(0.13
|
)
|
$
|
(0.15
|
)
|
$
|
(0.17
|
)
|
$
|
(0.11
|
)
|
||||
|
Shares
used in computing net loss per common share
|
136,795
|
136,796
|
136,796
|
136,797
|
||||||||||||
F-25