Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10–K
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2015
Commission file number: 000-28508
FLAMEL TECHNOLOGIES S.A.
(Exact name of registrant as specified in its charter)
| Republic of France | 43-1050617 | |
| State or other jurisdiction of incorporation or organization | (I.R.S. Employer Identification No.) | |
Parc Club du Moulin à Vent 33, avenue du Docteur Georges Levy Vénissieux France |
69200 | |
| (Address of principal executive offices) | (Zip Code) | |
| Registrant's telephone number, including area code: +33 472 78 34 34 | ||
Securities registered pursuant to Section 12(b) of the Act:
American Depositary Shares* Ordinary Shares** |
NASDAQ Global Market | |
| Title of each class | Name of exchange on which registered | |
| * | American Depositary Shares evidenced by American Depository Receipts. Each American Depositary Share represents one (1) Ordinary Share. |
| ** | Nominal value 0.122 Euros per share. Not for trading, but only in connection with the listing of American Depositary Shares. |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
Large accelerated filer x Accelerated filer ¨
Non-accelerated filer ¨ Smaller reporting company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
Aggregate market value of the voting stock held by non-affiliates of the registrant:
| $845,720,744 | June 30, 2015 | |
| Value | Date of Valuation |
The calculation of the aggregate market value of voting stock excludes 587,450 ordinary shares of the registrant that are understood to be held by employees, directors and shareholders that the registrant concluded were affiliates of the registrant on that date. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
Number of the registrant's American Depositary Shares, 0.122 Euro per share par value, outstanding as of March 7, 2016 was 40,234,506.
The following documents are incorporated by reference in the Parts of this Form 10-K indicated below:
| Documents Incorporated by Reference | Parts of Form 10-K into which Incorporated | |
| None | N/A | |
TABLE OF CONTENTS
| -2- |
Cautionary Disclosure Regarding Forward-Looking Statements
This annual report on Form 10-K contains forward-looking statements. We may make additional written or oral forward-looking statements from time to time in filings with the Securities and Exchange Commission or otherwise. The words “will,” “may,” “believe,” “expect,” “anticipate,” “estimate,” “project” and the negative of these and similar expressions identify forward-looking statements, which speak only as of the date the statement is made. Such forward-looking statements are within the meaning of that term in Section 27A of the Securities Act of 1933 as amended (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Although we believe that our forward-looking statements are based on reasonable assumptions within the bounds of our knowledge of our business and operations, our business is subject to significant risks and there can be no assurance that actual results of our research, development and commercialization activities and our results of operations will not differ materially from our expectations. Factors that could cause actual results to differ from expectations in our forward-looking statements include, among others, those specified in “Risk Factors” in this Part I, Item 1A, including:
| · | we depend on a small number of products and customers for the majority of our revenues and the loss of any one of these products or customers could reduce our revenues significantly. |
| · | our Bloxiverz® and Vazculep® products are not patent protected and could face substantial competition resulting in a loss of market share or forcing us to reduce the prices we charge for those products, which would have a material adverse effect on our revenues and results of operation. |
| · | we could fail to successfully complete the research and development for the two pipeline products we are evaluating for potential application to the FDA pursuant to our UMD strategy, or our competitors could complete the development of such products and apply for FDA approval of such products before us, which would have a material adverse effect on our future business opportunities. |
| · | we may depend on partnership arrangements or strategic alliances for the commercialization of some of our products, and the failure of any third party to fulfill its duties under such an arrangement or alliance could have a material adverse effect on our financial condition and results of operation. |
| · | our products may not gain market acceptance, and lack of such market acceptance would limit our ability to generate revenue which would have a material adverse effect on our business. |
| · | our products may not reach the commercial market for a number of reasons, which would adversely affect our future revenues. |
| · | we must invest substantial sums in research and development (“R&D”) in order to remain competitive, and we may not fully recover these investments. |
| · | the development of several of our drug delivery platforms and products depends on the services of a single provider and any interruption of such provider’s operations could significantly delay or have a material adverse effect on our product pipeline. |
| · | we depend upon a limited number of suppliers to manufacture our products and to deliver certain raw materials used in our products and the failure of any such supplier to timely deliver sufficient quantities of products or raw materials could have a material adverse effect on our business. |
| · | if our competitors develop and market technologies or products that are more effective or safer than ours, or obtain regulatory approval and market such technologies or products before we do, our commercial opportunity will be diminished or eliminated. |
| · | Our newly acquired pediatric products could fail to generate enough physician interest to make them successful products. |
| · | if third party payors choose not to reimburse our pediatric products, or to reimburse them with a greater burden on the patient, our business could suffer, irrespective of a physician’s preference for using our product.. Since some of our pediatric competitors enjoy OTC status, this could cause our revenues to suffer. |
| · | We could fail to successfully or efficiently integrate the FSC business into our existing business. |
| · | Our acquisition of FSC, and its larger employee base, could increase our exposure to additional regulatory risks associated with the promotion of our products. |
| · | if we cannot adequately protect our drug delivery platforms and proprietary information, we may be unable to sustain a competitive advantage. |
| · | we depend on key personnel to execute our business plan and the loss of any one or more of these key personnel may limit our ability to effectively pursue our business plan. |
| · | we ceased to qualify as a foreign private issuer, which will increase the costs and expenses we incur to comply with U.S. Securities Laws. |
Forward-looking statements are subject to inherent risks and uncertainties, some of which cannot be predicted or quantified. Future events and actual results could differ materially from those set forth in, contemplated by or underlying the forward-looking statements. We undertake no obligation to update these forward-looking statements as a result of new information, future events or otherwise. You should not place undue reliance on these forward-looking statements. Statements in this annual report on Form 10-K including those set forth above and in “Risk Factors” section of this annual report on Form 10-K, describe factors, among others, that could contribute to or cause such differences.
| -3- |
| Item 1. | Business |
General Overview
Flamel Technologies S.A. (“Flamel,” the “Company,” “we” or “us”) is a specialty pharmaceutical company utilizing core competencies in drug delivery and formulation development to create safer and more efficacious pharmaceutical products to address unmet medical needs and/or reduce overall healthcare costs. The Company has a balanced business model consisting of:
| (i) | an Unapproved Marketed Drugs (“UMDs”) business with two approved products in the USA, Bloxiverz® (neostigmine methylsulfate injection) and Vazculep® (phenylephrine hydrochloride injection) that are currently marketed, and a third product currently being reviewed by the FDA, all obtained through the acquisition of Éclat Pharmaceuticals, LLC’s (or “Éclat”) portfolio on March 13, 2012, |
| (ii) | a branded pediatric business, acquired through the acquisition of FSC Laboratories and FSC Pediatrics (“FSC”) on February 8, 2016; and |
| (iii) | a branded business, focusing on the development of products utilizing Flamel’s proprietary drug delivery platforms. |
The branded products that are based on Flamel’s proprietary drug delivery platforms target high-value solid oral and alternative dosage forms using 505(b)(2) and Biosimilar pathways where the Company can develop strong intellectual property positions and deliver meaningful patient benefits. Flamel is headquartered in Lyon, France and has operations in St. Louis, Missouri and Charlotte, NC, USA, and Dublin, Ireland.
Corporate Information
The Company was incorporated as a société anonyme (or SA), a form of corporation under the laws of the Republic of France, in August 1990 as Flamel Technologies S.A. and its shares, represented by American Depositary Shares, began to be quoted on the NASDAQ National Market in 1996 and are now quoted on the NASDAQ Global Market. As per the Company’s by-laws, its legal existence expires in 2099, unless extended. Flamel’s principal place of business is located at Parc Club du Moulin à Vent, 33, avenue du Docteur Georges Lévy, 69200 Venissieux, France (a suburb of Lyon); phone number +33 472 78 34 34, fax number +33 472 78 34 35. Its website is www.flamel.com.
The Company currently has two direct wholly owned operating subsidiaries: Flamel US Holdings, Inc., and Flamel Irish Holdings, Ltd. Flamel US Holdings, Inc. is a Delaware corporation, created for the acquisition of Éclat in March 2012 and is the acquiring entity of FSC Holdings, LLC (“FSC Holdings”). Éclat Pharmaceuticals, LLC, a Delaware limited liability company (“Éclat), is a wholly owned subsidiary of Flamel US Holdings, Inc. Talec Pharma, LLC, a Delaware limited liability company (“Talec”), is a wholly owned subsidiary of Éclat. FSC Therapeutics, LLC, a Delaware limited liability company (“FSC Therapeutics”) is a wholly owned subsidiary of FSC Holdings, LLC and owns the intangible property acquired in the acquisition of FSC Holdings on February 8, 2016. FSC Laboratories, Inc. is a Delaware corporation and a wholly owned subsidiary of FSC Holdings. FSC Pediatrics, Inc. is a Delaware corporation and is a wholly owned subsidiary of FSC Laboratories. Flamel Irish Holdings, Ltd is a corporation organized under the laws of Ireland. Its wholly owned subsidiary, Flamel Ireland, Ltd., a corporation organized under the laws of Ireland, is where all intangible property was relocated on December 16, 2014. A complete list of the Company’s subsidiaries can be found in Exhibit 21.1 to this annual report on form 10-K.
Our Business Model
Since the acquisitions of Éclat and FSC, we have implemented a balanced business model allowing Flamel to (i) commercialize niche branded (Bloxiverz and Vazculep) and generic pharmaceutical products in the U.S., (ii) commercialize branded products and devices in the pediatric therapeutic area (Karbinal™ER ,Aciphex® Sprinkle™, Cefaclor and Flexichamber® (for more details, see “– Lead Products” in this Part I, Item 1), and (iii) blend novel, high-value internally developed products with our drug delivery and capabilities (for more details, see “– Other Products Under Development” in this Part I, Item 1 of this Annual Report on Form 10-K).
Flamel’s business model allows us to select, develop, and then license or acquire niche branded products mainly in the U.S. for FDA approval and commercialization. By adopting this strategy, the Company makes itself less dependent on the often changing strategies of partners in the future. Nevertheless, Flamel still explores development, supply and licensing opportunities for either its drug delivery platforms (Micropump® oral sustained release platform, and its derivatives LiquiTime® and Trigger Lock™, and the long acting injectable platform Medusa™; see “– Flamel’s Drug Delivery Platforms Overview” in this Part I, Item 1 for details) or its proprietary products (as the case may be; see “– Other Products Under Development” in this Part I, Item 1 of this Annual Report on Form 10-K for details) with carefully selected third parties, but, unlike our historical operations, will not be dependent completely on those partnerships to create revenue and profit opportunities.
Business Strengths and Strategies
Éclat, which has focused on pursuing U.S. Food and Drug Administration (“FDA”) approvals through the 505(b)(2) regulatory pathway (see “– Patent Restoration and Exclusivity” in this Part I, Item 1 of this Annual Report on Form 10-K), and FSC, which has focused on the commercialization of pediatric products and devices, add to our Company’s marketing and licensing knowledge of the commercial and regulatory process in the U.S, which we believe enhances the ability of the Company to identify potential product candidates for development, leverage new opportunities for the application of our drug delivery platforms, and license and market products in both the U.S. and EU.
| -4- |
We anticipate this commercialization capability will allow us to retain a greater portion of the economic benefits associated with sales not only of our proprietary products, but additional Unapproved Marketed Drug products as well (see “– Other Products Under Development – Additional Products using the Unapproved Marketed Drug (or UMD) Strategy”) in this Part I, Item 1 of this Annual Report on Form 10-K). In addition, we intend to pursue FDA approval of new products developed using our drug delivery platforms (see “– Other Products Under Development – Proprietary pipeline to deliver several regulatory filings (US and/or EU) through 2018” in this Part I, Item 1 of this Annual Report on Form 10-K).
Our versatile, proprietary drug delivery platforms (Micropump®, LiquiTime®, Trigger Lock™, Medusa™) allow us to select unique product development opportunities, representing either “life cycle” opportunities for marketed chemical and biological drugs (via 505(b)(2) or ANDA regulatory paths), or innovative formulation opportunities for NCEs or NBEs (via NDA regulatory path). Our drug delivery platforms allow us to generate competitive differentiated product profiles. These product development opportunities offer the ability to grow market share and to protect market position, through patent protection and/or product differentiation in multiple marketplaces. As part of our new business model, several products formulated using our proprietary drug delivery platforms are currently under various stages of development at Flamel. These products will be marketed either by the Company and/or by partners via licensing/distribution agreements (see “– Other Products Under Development – Proprietary pipeline to deliver several regulatory filings (US and/or EU) through 2018”) in this Part I, Item 1 of this Annual Report on Form 10-K).
The key elements of our strategy that enable us to build upon our strengths are:
| · | Maximizing the commercial potential of our Unapproved Marketed Drug and pediatric products; |
| · | Continuing to build commercially successful products utilizing Micropump; |
| · | Identifying and optimizing time-to-market for our (not yet approved) drug delivery platforms, i.e. LiquiTime, Trigger Lock and Medusa. |
| · | Maximizing the technical potential of our existing drug delivery platforms for developing new and proprietary products; |
| · | Developing and validating additional drug delivery platforms utilizing our current drug delivery platforms; and |
| · | Leveraging the capabilities of our existing (and future) proprietary products and/or drug delivery platforms with pharmaceutical and biotechnology partners. |
Developments in 2015 and 2016
On March 27, 2015, we announced positive results of first-in-man (“FIM”) clinical trial with LiquiTime guaifenesin. Three different twice-daily formulations of LiquiTime guaifenesin were evaluated against immediate release guaifenesin tablets dosed every four hours in a four-way crossover pharmacokinetic study with 16 healthy volunteers (see “- Proprietary Pipeline Products in this Part I, Item 1 of this Annual Report on Form 10-K).
On April 7, 2015, we announced the departure of Mr. Steve Lisi, Senior Vice President of Business and Corporate Development at Flamel. Simultaneously, Flamel also reaffirmed its product revenue guidance for 2015 of $170 to $185 million for combined sales of Bloxiverz® and Vazculep®.
On June 26, 2015, we announced the appointment of Ms. Sandra Hatten as Senior Vice President of Quality and Regulatory Affairs and Mr. Gregory Davis as Vice President of Business and Corporate Development.
On June 29, 2015, we announced positive results from two pilot pharmacokinetic (PK) studies in healthy volunteers of FT227, an abuse-deterrent, extended-release, oral hydromorphone product using its proprietary Trigger Lock drug delivery platform. The PK studies were intended to provide sufficient data for the Company to select a preferred prototype formulation to move forward into pivotal studies (see “- Proprietary Pipeline Products” in this Part I, Item 1 of this Annual Report on Form 10-K).
On September 9, 2015, we announced that we had received a Prescription Drug User Fee Act (PDUFA) date of April 30, 2016 from the U.S. Food and Drug Administration (FDA) for our third Unapproved Marketed Drug product (see “- Additional Unapproved Marketed Drug Products in this Part I, Item 1 of this Annual Report on Form 10-K).
On October 5, 2015, we announced that our Irish subsidiary, Flamel Ireland Limited, had licensed exclusive U.S. rights to the LiquiTime drug delivery platform to Perrigo Company plc’s Irish subsidiary, Elan Pharma International Limited, for the U.S. Over-the-Counter (OTC) drug market. Ibuprofen and guaifenesin were included along with five potential new products (for more details see “– Products In Development With Partners” in this Part I, Item 1 of this Annual Report on Form 10-K).
On November 23, 2015, we announced the appointment of Mr. Michael Kanan as Senior Vice President and Chief Financial Officer.
On December 22, 2015, we announced positive interim results from a Phase 1a clinical trial in healthy volunteers of a once weekly subcutaneous injection formulation of exenatide using our Medusa technology (FT228). The study achieved all safety and PK assessment objectives throughout ascending single dose administrations of FT228. The safety profile of FT228 was favorable in healthy volunteers up to a dose of 140 mcg, with an extremely low incidence of the gastrointestinal side effects commonly related to twice daily subcutaneous administration of exenatide (nausea, vomiting, abdominal discomfort, loss of appetite), in addition to a low incidence of mild injection site reactions. We plan to initiate a Phase 1b study of FT228 in Type 2 Diabetes Mellitus patients in the first quarter of 2016. One dose per week of FT228 will be administered over a one month period in order to assess safety in patients, the steady-state PK profile and the product’s potential effect on surrogate biomarkers for the disease.
| -5- |
On January 8, 2016, we provided an update on the Company’s corporate progress as it related to a number of initiatives, including ongoing clinical programs and 2016 revenue guidance. We announced that we would be submitting a Special Protocol Assessment (SPA) for once-nightly Micropump sodium oxybate in the first quarter of 2016 and upon approval would commence patient recruitment likely in the second quarter. We also announced that we had requested a meeting with FDA in the first quarter of 2016 to discuss continued development of FT227 related to Trigger Lock and expect to initiate licensing discussions for the technology in early 2016. Additionally, we announced our plans to move the first two LiquiTime products, ibuprofen and guaifenesin, into pivotal testing in 2016. Finally, we provided product revenue guidance for 2016 in the range of $100 to $120 million.
On February 8, 2016, we announced the acquisition of FSC Holdings, LLC, together with its wholly owned subsidiaries FSC Pediatrics, Inc., FSC Therapeutics, LLC, and FSC Laboratories, Inc. from Deerfield CSF, LLC, an affiliate of Deerfield Management, one of the Company’s major shareholders. (see Note 19: Subsequent Events to the consolidated financial statements within Part II, Item 8 of this Annual Report on Form 10-K.). FSC is a Charlotte, NC-based specialty pharmaceutical company that markets three pediatric pharmaceutical products indicated for infection, allergy, gastroesophageal disease (GERD), and a medical device for the administration of aerosolized medication using pressurized Metered Dose Inhalers (pMDIs) for the treatment of asthma. Under the terms of the acquisition, Flamel will pay $20.25 million over a five year period to Deerfield for all of its equity interests in FSC Holdings. Specifically, Flamel will pay $1.05 million annually for five years and will make a final payment in January 2021 of $15.0 million. Flamel will also pay Deerfield a 15% royalty per annum on net sales of the current FSC products, up to $12.5 million for a period not exceeding ten years.
Lead Products
Bloxiverz® (neostigmine methylsulfate injection), Flamel’s first NDA approval. Bloxiverz’s NDA was filed on July 31, 2012. Bloxiverz was approved by the FDA on May 31, 2013 and was launched in July 2013. Bloxiverz is a drug used intravenously in the operating room for the reversal of the effects of non-depolarizing neuromuscular blocking agents after surgery. Bloxiverz was the first FDA-approved version of neostigmine methylsulfate. Today, neostigmine is the most frequently used product for the reversal of the effects of other agents used for neuromuscular blocks. There are approximately 5 million vials sold annually in the U.S. On January 8, 2015 and December 28, 2015, the FDA approved Fresenius Kabi USA (“Fresenius”)’s NDA for neostigmine methylsulfate (for both 0.5 mg/1mL and 1 mg/1mL strengths) and an ANDA submitted by Eurohealth International, an affiliate of West-Ward Pharmaceuticals Corp., neostigmine methylsulfate (for both 0.5 mg/1mL and 1 mg/1mL strengths), respectively. In 2014, we recognized a total amount of $10.2 million as revenues from product sales and $150.3 million in 2015 (for more details, see “– Results of Operations” in Part II, Item 7 of this Annual Report on Form 10-K). In the future, the volume of sales of Bloxiverz is dependent upon the competitive market dynamics between Flamel, APP, West-Ward and any subsequent ANDA approvals that may occur. Additionally, an alternative product marketed by Merck, Bridion (sugammadex), was approved in early 2016 and will likely reduce the size of the neostigmine market.
Vazculep® (phenylephrine hydrochloride injection), Flamel’s second NDA approval. On June 28, 2013, the Company filed an NDA for a second product developed by Éclat, Vazculep (phenylephrine hydrochloride injection). The product was approved by the FDA on June 27, 2014. Flamel’s subsidiary Éclat Pharmaceuticals started shipping Vazculep™ (in 1mL single use vials, and 5mL and 10mL pharmacy bulk package vials) to wholesalers in October 2014. There are approximately 7 million vials sold annually in the U.S. Vazculep is the only FDA-approved version of phenylephrine hydrochloride to be available in all three vial sizes. West-Ward Pharmaceuticals Corp. (“West Ward”) commercializes the 1mL single-dose vial, as an approved product in the U.S. The volume of sales of Vazculep is dependent upon the competitive landscape in the marketplace.
Karbinal™ER (carbinoxamine maleate extended-release oral suspension). Karbinal ER is an H1 receptor antagonist (antihistamine) indicated for children two years of age and older, is the only first generation extended release oral suspension antihistamine available in U.S. Karbinal ER provides physicians with a new, effective and easy to use treatment option for children with seasonal and perennial allergic rhinitis that need symptomatic relief for runny nose, sneezing, itchy nose or throat and itchy and watery eyes. Karbinal ER was launched in 2015 and is exclusively licensed from Tris Pharma.
AcipHex® Sprinkle™ (rabeprazole sodium). AcipHex Sprinkle is a delayed-release capsule, in dosages of 5 mg and 10 mg, indicated for the treatment of GERD in children 1 to 11 years of age for up to 12 weeks. AcipHex Sprinkle can be sprinkled on a small amount of soft food (e.g., applesauce, fruit or vegetable based baby food, or yogurt) or the capsule granules can be emptied into a small amount of liquid (e.g., infant formula, apple juice, or pediatric electrolyte solution). The U.S. marketing rights for this product were acquired from Eisai Inc. and the product was launched in 2015.
Cefaclor for Oral Suspension, 125 mg/5 mL, 250 mg/5 mL and 375 mg/5 mL. Cefaclor is indicated for the treatment of otitis media, lower respiratory infections, pharyngitis and tonsillitis, urinary tract infections, and skin and skin structure infections, caused by susceptible organisms. It is a second generation cephalosporin antibiotic used to treat certain infections, caused by susceptible bacteria. Cefaclor was launched in 2015.
Flexichamber ™. Flexichamber is a collapsible holding chamber for use by patients under the care or treatment of a licensed healthcare professional to administer aerosolized medication from most pressurized Metered Dose Inhalers (MDI), is a prescription medical device. Flexichamber is comprised of antistatic materials to help improve delivery of medication from MDIs to the patient, while minimizing the adherence of the medication to the walls of the chamber. Flexichamber can be used with or without a mask. FSC received FDA 510(k) clearance for Flexichamber in October 2014 and the product is expected to be launched in 2016.
| -6- |
Coreg CR®, the Micropump-based marketed product. Coreg CR is an extended-release formulation (once-a-day) of Coreg (i.e. carvedilol phosphate), a non-selective antagonist of Beta 1, Beta 2 adrenergic receptors and a selective antagonist of Alpha 1 adrenergic receptors. Coreg and Coreg CR are the only beta blockers indicated for the treatment of moderate to severe heart failure and left ventricular dysfunction following myocardial infarction. Coreg CR was developed in partnership with GlaxoSmithKline (“GSK”) and is approved, marketed and sold in the U.S since 2007. To date, we have generated (i) $23 million in milestone payments and (ii) $62.5 million in royalty revenue from Coreg CR. Until December 1, 2014, we received royalty revenue of $6.3 million and a total amount of $6.7 million from product sales. In December 2014, as part of the divestiture of our development and manufacturing facility (“Pessac Facility”), the Company transferred the Supply Agreement for Coreg CR and, transferred and assigned to Recipharm all rights, titles and interests in the royalties of the License Agreements by and between Flamel and GSK (for more details, see “– Strategic Alliances” in this Part I, Item 1, and Note 17: Discontinued Operations to the consolidated financial statements in Part II, Item 8 of this Annual Report on Form 10-K).
Other Products Under Development
Additional Unapproved Marketed Drug Products. The Company still intends to develop and seek NDA or ANDA approval for select products that are currently marketed in the U.S. but are currently not approved by the FDA. One of the Company’s principle criteria is previously, well established efficacy. This strategy should create opportunities, to have the only approved version of products in niche markets, potentially enjoying a period of defacto exclusivity through the 505(b)(2) approval pathway. However, this strategy has a limited number of opportunities where a meaningful return on investment is possible. Through Éclat, Flamel has acquired additional opportunities beyond Bloxiverz and Vazculep:
| · | Flamel filed one of these products in mid-2015 and received a Prescription Drug User Fee Act (PDUFA) date of April 30, 2016 from the U.S. Food and Drug Administration (FDA). Based on IMS and other third-party data, the Company estimates that current U.S. market sales of the unapproved versions of this drug are in the range of $100 million per year. |
| · | Another UMD Product is currently being reviewed internally and the Company anticipates its development in 2016. |
Proprietary Pipeline Products. Five product development opportunities (i.e. four using Micropump or LiquiTime or Trigger Lock, and one using Medusa) have been selected for internal development. After setting differentiated targeted product profiles and establishing development plans, pharmaceutical development activities have been initiated.
| · | Sodium Oxybate, a Micropump based formulation for one single dose before bedtime for patients suffering from narcolepsy, eliminating the need for a second dose. The results of Flamel’s FIM clinical study in healthy volunteers, published in April 2014, demonstrated elimination of the need for a second, middle of the night dose, as is required with JAZZ’s Xyrem® product. This was further confirmed by a second clinical study performed at higher doses, published in December 2014. The elimination of the second dose for narcolepsy patients not only provides more convenience, but could also benefit patients by eliminating or reducing the current disruption in nighttime sleep. The potential for additional benefits of Micropump Sodium Oxybate, including improved safety, will be examined in future clinical studies. Prior to commencement of clinical studies, the Company plans to submit to the FDA, a Special Protocol Assessment (SPA) for once-nightly Micropump sodium oxybate in the first quarter of 2016. SPAs serve as a way to reduce the risks associated with clinical studies, as the acceptance represents general agreement by the FDA of a pivotal trial’s design, endpoints and analyses. Upon acceptance and approval of the SPA by the FDA, we plan to commence patient recruitment. The clinical trial is expected to run through early to mid-2017. It is expected to be a placebo-controlled efficacy study of approximately 200-300 patients and will be conducted at between 50 and 60 clinical sites in North America and Europe. In conjunction with the SPA, we expect to file for Investigational Medicinal Product Dossier (IMPD) approval in the EU. Any additional studies, such as pivotal pharmacokinetic (PK) studies, needed for a New Drug Application (NDA) approval will be run simultaneously, and the target for trial completion is mid-2017. Ultimately, we believe we will be able to demonstrate improved efficacy, improved safety and improved patient satisfaction over the standard of care, JAZZ’s Xyrem®, a twice nightly sodium oxybate formulation, which generated approximately $955 million in revenues in 2015. |
| · | Ibuprofen, LiquiTime-based formulation for twice-daily dosing for the treatment of pain. Flamel’s pharmacokinetic results, published in September 2014, demonstrated equivalent exposure (or “AUC”) to immediate-release ibuprofen, similar onset and similar blood levels at 12 hours, with no safety or tolerability issues. In October 2015, we entered into a license agreement with Elan Pharma International Limited (“Elan”) for the U.S. OTC rights to LiquiTime including our ibuprofen product (see “– Products In Development With Partners in this Part I, Item 1 of this Annual Report on Form 10-K). In consultation with Elan, Flamel anticipates a US regulatory filing in 2017. Additionally, Flamel may follow such filing with a later filing in the European Union, either by itself or with another licensing partner. LiquiTime ibuprofen opens the door to the OTC cough, cold and allergy markets, where LiquiTime delivery platform can provide significant benefit through combination products containing frequently used together active ingredients with tailored and extended release profiles. |
| -7- |
| · | Guaifenesin, LiquiTime-based formulation for twice-daily dosing for the treatment of chest congestion associated with various indications (common cold, infections, or allergies). Flamel’s pharmacokinetic results, published in March 2015, clearly met the intention of the study, i.e. to allow selecting the best formulation prototype, which satisfied most of the criteria necessary for proving bioequivalence of AUC to the immediate release guaifenesin tablets, for further optimization and scale up. Our guaifenesin product was also part of the license agreement with Elan. We plan, in consultation with Elan, to begin pivotal testing in 2016. This second product will expand our twice-daily oral suspension offerings for the OTC market in the near future. |
| · | Exenatide, a once a week Medusa-based injectable formulation of exenatide (FT228), a glucagon-like peptide-1 (“GLP-1”) agonist for the treatment of type 2 diabetes. Flamel’s preclinical results in minipigs, published in June 2014, demonstrated improved bioavailability versus commercially available once-a-week exenatide, similar release profiles for two successive injections, no adverse clinical signs and an excellent local tolerability. The pharmacokinetics profile of Medusa-exenatide was compatible with a release over one week in human. In 2015, we completed a Phase 1a clinical trial in healthy volunteers of a once weekly subcutaneous injection formulation of exenatide. The trial was conducted in two periods. The first, a 2-arm study of 20 healthy volunteers in each arm, evaluated the safety and PK profiles of FT228 versus Byetta®, a twice daily injection of exenatide marketed by Astra Zeneca, at a total dose of 10 mcg. The second, a 2-arm study of 20 healthy volunteers in each arm, evaluated the safety and PK profiles of FT228 at total doses of 70 mcg and 140 mcg. The safety profile of FT228 was favorable in healthy volunteers up to a dose of 140 mcg, with an extremely low incidence of the gastrointestinal side effects commonly related to twice daily subcutaneous administration of exenatide (nausea, vomiting, abdominal discomfort, loss of appetite), in addition to a low incidence of mild injection site reactions. Two subjects dropped out of the study prior to completion of the 140 mcg dosing for reasons unrelated to the clinical trial. The study achieved all safety and pharmacokinetic (PK) assessment objectives throughout ascending single dose administrations of FT228. We initiated a Phase 1b study of FT228 in Type 2 Diabetes Mellitus patients in the first quarter of 2016. One dose per week of FT228 is being administered for a one month period at two different doses in order to assess safety in patients, the steady-state PK profile and the product’s potential effect on surrogate biomarkers for the disease. |
| · | Hydromorphone. Flamel also has a Trigger Lock-based abuse-deterrent, extended-release, oral hydromorphone product (FT227) in development. Hydromorphone is used for relief of moderate to severe pain in patients requiring continuous around-the-clock opioid treatment for an extended period of time. We announced in June 2015, positive results from two pilot pharmacokinetic (PK) studies in healthy volunteers of FT227. The PK studies were intended to provide sufficient data for the Company to select a preferred prototype formulation to move forward into pivotal studies. The studies compared three FT227 prototypes to the comparator product Jurnista© (sold as Exalgo© in the United States) in both fasted and fed conditions at a dose of 32mg. Under fasted conditions, comparing the AUC and the Cmax of FT227 to Jurnista in 16 subjects, the results identified a FT227 formulation that met the bioequivalence criteria for both parameters. Under fed conditions (14 subjects), the same formulation was bioequivalent in terms of AUC to Jurnista but outside of the Cmax bioequivalence criterion at the lower confidence interval level. Comparing the effect of food on the PK parameters of the FT227 prototypes across the two studies, no notable difference is seen in either AUC or Cmax in fed and fasted conditions. This suggests that administration of FT227 will not be subject to a clinically relevant food effect. In both studies FT227 was well tolerated and no serious adverse events were reported. In addition, Flamel has generated substantial in vitro data comparing the abuse deterrence properties of FT227 compared to other marketed abuse-deterrent opioid products. The Company is confident that Trigger Lock is a robust platform for opioids that will set a high standard in terms of abuse deterrence. Further in vitro data have been generated on FT227 by an independent contract research organization which confirmed the excellent abuse deterrent properties of the product. FT227 is designed to be filed as a 505(b)(2) New Drug Application (NDA). We recently requested a meeting with the FDA to discuss the further development of FT227 and expect to initiate licensing discussions for the technology in early 2016. |
These proprietary pipeline products will be marketed either by Flamel (and/or its subsidiary Éclat) or by partners via licensing/distribution agreements.
Products in development with partners.
Following the rationalization of the Company’s products pipeline, as described below under the caption “Proprietary Intellectual Property.”, the four partnerships that remained in effect in 2013, were either terminated in 2014 or transferred to Recipharm AB, as part the divestiture of our Pessac Facility. As a term of this divestiture, Recipharm will be allowed to use Micropump platform for the continued manufacturing of microparticles for Coreg CR®, marketed by GlaxoSmithKline in the USA (for more details see above “– Lead Products” in this Part I, Item 1, and below “– Strategic Alliances” in this Part I, Item 1 and “– Manufacturing” in this Part I, Item 1 of this Annual Report on Form 10-K).
We entered into an Exclusive License Agreement on September 30, 2015, with Elan Pharma International Limited, a subsidiary of Perrigo Company plc, for the U.S. rights to our LiquiTime drug delivery platform for the U.S. (OTC) drug market. Under the multi-product license agreement, we received an upfront payment of $6.0 million and will be eligible for at least $50 million in approval and launch milestones. In addition, we will receive mid-single digit royalties on net sales of the products. Flamel and Elan believe there is a large market opportunity for other OTC extended release liquid drug formulations, including products containing active ingredient combinations for the US cough/cold market, which analysts have estimated between $6 billion to $8 billion annually.
| -8- |
Proprietary Product Pipeline.
The status of Flamel’s proprietary product pipelines is detailed in the followings table:
| Proprietary Product Pipeline |
| Development Strategy/Platform |
Drug/Product | Indication | Stage | Sales Forces | ||||
| UMD[1] | Neostigmine Methylsulfate Injection/Bloxiverz® | Anesthesia | Marketed in the U.S. | Flamel (via Éclat) | ||||
| Phenylephrine Hydrochloride Injection/Vazculep® | Anesthesia | Marketed in the U.S. | ||||||
| UMD#3 (Undisclosed or “UD”)[2] | UD | NDA filing in 2015 with PDUFA date of April 30, 2016 | ||||||
| UMD #4 (UD) | UD | Development beginning in 2016 with anticipated filing in 2017 | ||||||
| Pediatrics | Karbinal ER | Allergy/Antihistamine | Marketed in the U.S. | FSC Pediatrics | ||||
| AchipHex Sprinkle | GERD | Marketed in the U.S. | ||||||
| Cefaclor Oral Suspension | Infections | Marketed in the U.S. | ||||||
| Flexichamber [4] | Asthma | Marketed in the U.S. | ||||||
| Micropump® | Sodium oxybate | CNS (narcolepsy) | 2 clinical studies completed SPA to be submitted in early 2016 (pivotal clinical study initiation expected as early as 2016) |
To be determined[3] | ||||
| LiquiTime® | Ibuprofen | Pain |
FIM clinical study completed (pivotal clinical study initiation expected in 2016) |
LiquiTime®-based product(s) out-licensed | ||||
| Guaifenesin | Respiratory | Proof of concept FIM clinical study completed (pivotal clinical study initiation expected in 2016) |
||||||
| Trigger Lock™ | Hydromorphone | Pain | PK study completed in 2015 (pivotal clinical study initiation expected in 2016)
|
To be determined[3] | ||||
| Medusa™ | Exenatide (once-a-week) | Diabetes |
Phase 1(a) study completed in 2015 (Phase 1(b) study initiated Q1 2016) |
| [1] | Company’s Unapproved Marketed Drug Products. |
| [2] | For competitive reasons, Flamel has decided not to identify those products for the time being, but intends to provide additional information upon the achievement of pre-clinical, clinical and regulatory milestones. |
| [3] | Those products are expected to be licensed to partners for completion of development and commercialization. |
| [4] | Medical device |
| -9- |
Competition and Market Opportunities
Competition
Competition in the pharmaceutical and biotechnology industry is intense and is expected to increase. We compete with academic laboratories, research institutions, universities, joint ventures, and other pharmaceutical and biotechnology companies, including other companies developing niche brand or generic specialty pharmaceutical products or drug delivery platforms. Some of these competitors may also be our business partners. There can be no assurance that our competitors will not obtain patent protection or other intellectual property rights that would make it difficult or impossible for us to compete with their products. Furthermore, major technological changes can happen quickly in the pharmaceutical and biotechnology industries. Such rapid technological change, or the development by our competitors of technologically improved or differentiated products, could render our drug delivery platforms obsolete or noncompetitive.
The drug delivery industry landscape has dramatically changed over the past decade and even more so during the past five years, largely as a function of the growing importance of generic drugs. The growth of generics (typically small molecules) and of large molecules (biosimilars) has been accelerated by the demand for less expensive pharmaceutical products. As a result, the pricing power of pharmaceutical companies will be more tightly controlled in the future.
In addition, the overall landscape of the Pharma/Biotech industry has changed, as consolidation has reduced our pool of potential partners and further accelerated the competition among drug delivery and specialty pharmaceutical companies. Over the past ten years, numerous stand-alone drug delivery companies have been acquired (partly or entirely) by pharmaceutical, biotech, generic or other drug delivery companies. By acquiring drug delivery platforms, those companies are internalizing their previously outsourced R&D efforts while potentially preventing competitors from accessing the acquired technologies. In the meantime, certain drug delivery companies have consolidated their existing positioning or have entered new markets via M&A transactions and/or restructuring.
Just as Flamel has undertaken a strategy of developing and commercializing its own products, few of Flamel’s “historical” competitors still pursue a sole drug delivery business model as many others have moved or are moving to the Specialty Pharma model. A few examples include Alkermes, Depomed, Ethypharm and Octoplus.
Our drug delivery platforms primarily compete with technologies from companies such as:
| Flamel’s
Drug Delivery Platforms |
Competition category* | Selected Competitive Companies* | ||
| Micropump® (oral) | Solid sustained release | Alkermes plc; COSMO Pharmaceuticals SpA; Depomed, Inc.; Durect Corp.; Supernus Pharmaceuticals, Inc.; Veloxis Pharmaceuticals A/S (formerly LifeCycle Pharma) | ||
| LiquiTime® (oral) | Liquid sustained release | Neos Therapeutics, Inc. (“Neos”); Tris Pharma, Inc. (“Tris”) | ||
| Trigger-Lock™ (oral) | Abuse resistance | Acura Pharmaceuticals, Inc.; Altus Formulation, Cima (Cephalon); Collegium Pharmaceutical, Inc.; Durect Corp.; Egalet Corporation; Elite Pharmaceuticals, Inc.; Ethypharm; Grünenthal Group; Intellipharmaceutics International, Inc.; QRx Pharma, Ltd.; KemPharm, Inc. | ||
| Medusa™ (injectable) | Depot
(PLA/PLGA microspheres, liposomes and other technologies) |
Alkermes plc.; Biodel Inc.; Debiopharm Group; Durect Corp.; LG Life Sciences; InnoCore Pharmaceuticals; Marina Biotech, Inc. (Novosom AG technology); MedinCell SA; Octoplus N.V. (subsidiary of Dr. Reddy’s); Onxeo (formerly BioAlliance Pharma); Pacira Pharmaceuticals, Inc.; Q Chip Ltd. (Midatech); REcoly N.V.; Soligenix, Inc. (formerly DOR BioPharma Inc); Surmodics, Inc.; Xenetic Biosciences plc. (formerly Lipoxen plc) |
* From companies’ web site and/or press releases.
| -10- |
Flamel’s Specialty Pharma model (focusing on optimized re-formulations development capabilities) competes with a number of companies, based upon the product being developed. Examples of companies with whom we or future partners would compete, given our current pipeline, include Jazz Pharmaceuticals, Purdue Pharma, Tris Pharma and others. Flamel as a specialty pharmaceutical company has various capabilities, including the use of the 505(b)(2) regulatory pathway, the life cycle management of drugs, and direct commercialization of drugs.
Market Opportunities
Drug delivery platforms are of particular interest for managing the life cycle of pharmaceutical products, as they offer many advantages:
| · | improvements in bioavailability |
| · | pharmacokinetic improvements |
| · | enhanced efficacy |
| · | reduction of adverse events |
| · | improved patient compliance |
Drug delivery capabilities also provide companies the ability to protect the improved drugs by allowing patent protection to the differentiated drugs the technologies provide. Market exclusivity can also be granted for improvements to existing drugs. BCC Research estimated the global drug delivery market to worth an estimated $188 billion in 2014 and that the market grew to $194 billion in 2015. The increased number of geriatric patients and the demand for convenient drug delivery options offer major opportunities for the development of innovative and easy-to-use drug delivery platforms. In 2015, FDA’s Center for Drug Evaluation and Research (“CDER”) approved 41 novel new drugs, as new molecular entities (“NMEs”) under New Drug Applications (NDAs) or as new therapeutic biologics under Biologics License Applications (“BLAs”) (FDA, Novel New Drugs 2014, Summary, January 2015). Additionally, the FDA approved 82 “first time generic drugs” (FDA, ANDA (Generic) Drug Approvals in 2015, www.fda.gov).
Market opportunities for its current proprietary pipeline are estimated by Flamel to be worth at least several hundred million dollars each. For example, Xyrem® (sodium oxybate) recorded $955 million in sales in the U.S. for 2015 (source: Jazz press release Full Year And Fourth Quarter 2015 Financial Results, February 23, 2016); OTC ibuprofen products recorded sales in the U.S. beyond $480 million including combination products in 2015 (source: IMS). The U.S. cough, cold, pain and allergy markets targeted by our LiquiTime-based products, is estimated between $6 billion and $8 billion annually – (source: Nielsen Data Trend). Sales of GLP-1 drugs exceeded $3 billion in 2015 according to IMS and the U.S. market for prescription painkillers exceeded $7 billion in 2015 (source: IMS).
The industry faces many challenges. There are four main forces currently affecting all pharmaceutical and drug delivery companies and forcing the industry to adapt and to change: (i) the rise of generics; (ii) the rise in costs for new product development; (iii) the commoditization and acquisition of drug delivery technologies; (iv) the integration of the drug delivery-based formulation development occurs at much earlier stage in the overall pharmaceutical development; and (v) the higher regulatory and reimbursement hurdles.
These forces have affected the small molecule space to a greater extent, as biologics enjoy higher barriers to entry and have been sheltered as a consequence. But they are at work in the biologics space as well. In particular, in today’s environment, a drug has to demonstrate significant therapeutic efficacy advantage over current standard of care in order to successfully solicit third party payer coverage. Alternatively, changes in the delivery of a drug must create a demonstrable reduction in costs. Dosing convenience, by itself, is no longer sufficient to gain reimbursement acceptance. It has a serious impact on drug delivery companies as they have to now demonstrate, through costly Phase 3 trials, therapeutic efficacy of their new formulations. The FDA has actually encouraged drug companies developing enhanced formulations to use an abbreviated regulatory pathway: the 505(b)(2) NDA. Most drug delivery companies today are using this approach or the supplemental NDA pathway (“sNDA”). An NDA or sNDA is necessary to market an already approved drug for a new indication, or in a different dosage form or formulation. However, the sNDA approach requires cross-referencing the originator’s drug dossier, and eventually an alliance with the originator’s company for commercialization.
Because the drug delivery industry is highly competitive, participants must find ways to lessen the pressure and increase profitability. Flamel, resulting from the combination of its existing proprietary drug delivery platforms with the established commercial capability of Éclat, has evolved into a Specialty Pharma company focusing on re-formulations and requiring shorter product development cycles by using a “fast track” NDA mechanism (505(b)(2)). The pharmaceutical and biotechnology sectors, with an impending “patent cliff”, are forcing Big Pharma/Biotech to reorganize and creating niche opportunities for Specialty Pharma companies like the “new” Flamel.
Flamel’s Drug Delivery Platforms
Overview
Flamel owns and develops drug delivery platforms that address key formulation challenges, leading to the development of differentiated drug products for administration in various forms (e.g. capsules, tablets, sachets or liquid suspensions for oral use; or injectables for subcutaneous administration) and can be applied to a broad range of drugs (novel, already-marketed, or off-patent):
| -11- |
| · | Micropump® is a microparticulate system that allows the development and marketing of modified and/or controlled release of solid, oral dosage formulations of drugs (Micropump®-carvedilol and Micropump®-aspirin formulations have been approved in the U.S. and in the E.U., respectively. |
| · | LiquiTime® allows development of modified/controlled release oral products in a liquid suspension formulation particularly suited to children or for patients having issues swallowing tablets or capsules. |
| · | Trigger Lock™ allows development of abuse-resistant modified/controlled release formulations of narcotic/opioid analgesics and other drugs susceptible to abuse. |
We believe the versatility of Micropump which permits us to develop differentiated product profiles (modified/controlled release formulations) under various dosage forms including capsules, tablets, sachets and liquid suspensions (LiquiTime) for oral use, is a competitive advantage. With Trigger Lock potentially addressing the issue of narcotic/opioid analgesics abuse, we have broad and versatile presentations to serve most markets from pediatric to geriatric.
| · | Medusa™ allows the development of extended/modified release of injectable dosage formulations of drugs (e.g. peptides, polypeptides, proteins, and small molecules). |
We believe also that the Medusa platform provides a competitive advantage for developing differentiated injectable product profiles. Medusa-based formulations permit drugs’ full activity to be preserved in an extended release format with other potential advantages being, improved solubility, stability, and resistance to aggregation. Overall, Medusa™ can improve the patient experience through a change in the route of administration (e.g. switching from intravenous to subcutaneous injection) and may improve compliance through reduction of administration frequency (e.g. from once-a-day to once-a-week).
The Company will continue to selectively partner its proprietary formulations capabilities and will either commercialize products based on its drug delivery platforms on its own or partner them.
Micropump®: Delivery Platform for the Modified and/or Controlled Release of Solid, Oral Dosage Formulations of Drugs
Flamel’s Micropump platform permits either extended or delayed delivery of small molecule drugs via the oral route. Micropump consists of a multiple-particulate system containing 5,000 to 10,000 microparticles per capsule or tablet. The 200-500 microns diameter-sized microparticles are released in the stomach and pass into the small intestine, where each microparticle, operating as a miniature delivery system, releases the drug at an adjustable rate and over an extended period of time. The design of the Micropump microparticles allows an extended release in the Gastro-Intestinal (“GI”) tract allowing mean plasma residence times to be extended for up to 24 hours. The microparticles’ design can be adapted to each drug’s specific characteristics by modifying the coating composition and thickness as well as the composition of the excipients encapsulated with the drug. The resultant formulations can potentially offer improved efficacy (by extending therapeutic coverage), reduced toxicity and/or side effects (by reducing Cmax or peak drug concentration in the plasma, or by reducing intra- and inter-patient variability), and improved patient compliance (by reducing frequency of administration). The platform is applicable to poorly soluble (< 0.01mg/L) as well as highly soluble (> 500g/L) and to low dose (e.g. 4 mg) or high dose (e.g. 1,000 mg) drugs, while providing excellent mouth feel and taste masking properties. Micropump allows the achievement of extremely precise pharmacokinetic profiles extended (and/or delayed) release of single or combination of drugs, in a variety of formats (such as tablets, capsules, sachet, or liquids (LiquiTime), while preserving the targeted release rate over the shelf-life of the product.
Considering R&D costs for reformulating a drug are typically substantially lower than for developing NCEs, “reformulation approvals” provide an opportunity to extend the exclusivity period of already marketed drugs or create new market exclusivity for off-patent drug. The Micropump platform has successfully transitioned to commercial stage with Coreg CR® (see “– Lead Products”) in this Part I, Item 1 of this Annual Report on Form 10-K. Flamel currently has additional Micropump-based internal products in development including sodium oxybate for narcolepsy, which has been successfully tested in two Phase 1 clinical studies; see “– Proprietary Product Pipeline” in this Part I, Item 1 of this Annual Report on Form 10-K).
Micropump (and related products) is patent protected (see “– Proprietary Intellectual Property”). Coreg CR® Micropump-based microparticles are now being manufactured for GSK by Recipharm (see “– Strategic Alliances” in this Part I, Item 1 and “– Manufacturing” in this Part I, Item 1 of this Annual Report on Form 10-K).
LiquiTime®: Delivery Platform for the Modified/Controlled Release of Liquid, Oral Dosage Formulations of Drugs
The U.S. sales of drugs (Rx and OTC) in liquid form for oral administration exceeded $5.8 billion for the year 2015 (source: IMS). Amongst marketed “extended release” (twice-a-day or once-daily) liquid products are Tussionex® (hydrocodone polistirex and chlorpheniramine polistirex) and its branded and generic alternatives, Delsym® and Delsym Children® (dextromethorphan polistirex developed and sold by Reckitt Benckiser plc.) and Quillivant XR marketed by Pfizer. These products totaled $249 million sales in 2015 (source: IMS).
| -12- |
Flamel’s LiquiTime platform uses Micropump’s competitive advantages to allow us to develop modified/controlled release (e.g. zero-order kinetics) in liquid suspension formulations. The LiquiTime products are particularly suitable for dosing to children and for use by patients having issues swallowing tablets or capsules. Unlike the other product examples described in the previous paragraph, which are all based on ion exchange resin technology, LiquiTime does not have the limitation of having to work solely with ionic drugs and therefore has applicability to a much broader range of drug molecules. As with Micropump, LiquiTime can be applied to the development of combination products and it is readily able to be scaled-up to commercial quantities. We believe that LiquiTime, designed to provide a controlled, extended release of oral liquids principally for pediatric and geriatric patients will be also effective for certain prescription products. The increasing number of geriatric patients and the demand for convenient drug delivery options for children offer striking opportunities for the development of LiquiTime-based formulations. Flamel has described two LiquiTime-based products in development: ibuprofen LiquiTime and guaifenesin LiquiTime both successfully tested in FIM clinical studies. The rights to these products along with the LiquiTime technology has been licensed to Elan Pharm International Limited for U.S. OTC rights. The first pivotal clinical study could be expected to be initiated as early as in the end of 2016 with respect to these products (see “Proprietary Product Pipeline” in this Part I, Item 1 of this Annual Report on Form 10-K).
LiquiTime (and related products) is patent protected (see “– Proprietary Intellectual Property” in this Part I, Item 1 of this Annual Report on Form 10-K).
Trigger Lock™: Delivery Platform for Abuse-Resistant Modified/Controlled Release Formulations of Narcotic/Opioid Analgesics
A major problem faced by the industry is the growing abuse and misuse of opioids by drug abusers, who attempt to extract the opioids from the drug products for the purposes of injection or otherwise achieve the immediate release of the large doses contained in extended release products. The proportion of narcotic/opioid analgesics abuse associated with emergency room admissions has more than tripled in ten years, from 6.8% in 1998 to 26.5% in 2008 (TEDS report, July 15, 2010). Narcotic/opioid analgesics abuse continues to increase as current products remain easy to abuse. In 2010, enough prescription painkillers were prescribed to medicate every American adult every 4 hours for one month (PBS 2013). The number of prescription medicine abusers in 2010 was 8.76 million, 5.1 million of whom abused painkillers (drugabuse.com 2013). The market for opioid drugs, used to treat patients suffering from severe and chronic pain in the seven major markets (USA, Japan, and five European countries) was estimated to exceed $7.4 billion in 2010, dominated by oxycodone. In 2015, the U.S. sales of oral opioid drugs (hydrocodone, hydromorphone, morphine, oxycodone and oxymorphone, including combination products) exceeded $6.4 billion (source: IMS).
Flamel’s Trigger Lock platform utilizes Micropump’s competitive advantages to allow the development of abuse-resistant modified/controlled release formulations of narcotics and other drugs susceptible to abuse:
| · | Micropump particles are extremely difficult to crush to extract the narcotic/opioid analgesics; |
| · | Additional formulation modifications are made to prevent other less publicized methods of abusing controlled release technologies are available; and, |
| · | Trigger Lock can provide products that are either bioequivalent to or have improved pharmacokinetics over marketed narcotic/opioid analgesics. |
| · | The FDA’s moves to restrict the prescribing of extended-release opioid analgesics should benefit abuse-resistant formulations, such as Trigger Lock. The FDA issued a “Draft Guidance for Abuse Deterrent Opioids” on January 9, 2013. |
| · | We believe that Trigger Lock has the potential to satisfy the FDA Draft Guidance for Abuse Deterrent Opioids: |
| · | Laboratory-based in vitro manipulation and extraction studies (Category 1) – Success with Trigger Lock |
| · | Pharmacokinetic studies (Category 2) – Success with Trigger Lock |
| · | Clinical abuse potential studies (Category 3) – To be performed prior marketing |
| · | Analysis of post marketing data to assess the impact of an abuse-resistant formulation on actual abuse in a community setting (Category 4) – To be performed post marketing |
| · | Flamel has one Trigger Lock-based internal product, hydromorphone, under development for which certain PK and independent in-vitro abuse resistance data was gathered in 2015; a pivotal clinical study is expected to be initiated in 2016 (see “– Proprietary Product Pipeline” in this Part I, Item 1 of this Annual Report on Form 10-K). |
| · | Trigger Lock (and related products) is patent protected (see “– Proprietary Intellectual Property” in this Part I, Item 1 of this Annual Report on Form 10-K). |
Medusa™ Delivery Platform for the Modified/Controlled Release of Injectable Dosage Formulations of Drugs
U.S. sales for injectable products in 2015 were $132 billion, including nearly $16 billion for “long-acting” products (source: IMS). Conversely, global sales of biologics were between $190 to $222 billion in 2014 according to various analysts, and are expected to exceed $386 billion by 2019 (source: BCC Research).
Flamel’s Medusa, a hydrogel depot formulation approach that does not alter the drug substance, enables the modified/controlled delivery from one day up to one week of drugs. Medusa is particularly suited to the development of subcutaneously administered formulations.
The Medusa platform consists of proprietary and versatile drug carrier polymers that form hydrogel depots after injection. Medusa polymers are made of glutamic acid, a naturally occurring aminoacid, and alpha tocopherol (Vitamin E). These polymers are amphiphilic and spontaneously form stable hydrogels in water. These hydrogels contain hydrophobic nanodomains rich in Vitamin E and hydrophilic polyglutamate that are exposed to water. The hydrogels are robust over a wide range of pH values and can be stored, in particular as a stable freeze-dry form, that can be easily reconstituted in water for Injection. Those polymers have been proven to be safe and biodegradable. A comprehensive ADME and regulatory toxicology package for the key Medusa polymer was completed in 2014 in order to update the Type IV Drug Master File (“DMF”) filed with the FDA in February 2011.
| -13- |
The drug is loaded in the hydrogel (nano- or micro-gel) via non-covalent, hydrophobic and electrostatic, bonds. Once in the body, the hydrogel releases the drugs in a controlled manner with no initial burst effect, lower Cmax and uniform plasma concentration, over an extended period of time. Both drug loading (in fully aqueous solution, and usually, under solvent- and surfactant-free conditions) and release (essentially by displacement of the loaded drug by circulating endogenous proteins) are non-denaturing, which preserves structural integrity - and hence activity - of the drug. The transient, non-covalent interactions dictate the pharmacokinetic parameters (Cmax and bioavailability in particular) of the released drugs.
Flamel is focusing on Medusa-based “biobetter” development opportunities, which can be summarized as follows:
| · | Proven biologic drugs with established markets and proven clinical development approaches; |
| · | Product differentiation e.g. improvement of pharmacokinetic (and potentially pharmacodynamics) parameters; |
| · | Protection of market position through product differentiation and/or patent extension; and, |
| · | Ability to grow market share and resist price competition. |
Flamel has one Medusa-based internal product in development (see “– Proprietary Product Pipeline” in this Part I, Item 1 of this Annual Report on Form 10-K). Flamel completed a Phase 1(a) study of its once-a-week Medusa exenatide product in 2015 and initiated a Phase 1(b) study in Q1 2016.
Medusa is patent protected until June 2031 in the United States (see “– Proprietary Intellectual Property” in this Part I, Item 1 of this Annual Report on Form 10-K).
Proprietary Intellectual Property
Patents and other proprietary rights are essential to our business. Our proprietary product pipeline and our strategic alliances are dependent on our drug delivery platforms and related products (formulation, process, etc.) being patent protected. As a matter of policy, we seek patent protection of our inventions and trademarks and also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to maintain and develop our competitive position.
On a case-by-case basis, an invention developed jointly by Flamel and a partner may be assigned to and prosecuted by the partner. The information provided in this section herein, does not refer to such patent applications.
In 2014, Flamel engaged in a rationalization process of its patent portfolio to focus on key patent families that protect our core drug delivery platforms. As a result of this process, the total number of patents is significantly lower than in previous years. As of December 31, 2015, we owned the following patent and patent applications:
| US | EUROPE | ROW* | TOTAL | |||||||||||||
| Granted patents | 15 | 170 | 99 | 284 | ||||||||||||
| Pending patent applications | 12 | 14 | 42 | 68 | ||||||||||||
| Patents granted in 2015 | 1 | 2 | 16 | 19 | ||||||||||||
| Patent applications filed in 2015 | 5 | 1 | 9 | 15 | ||||||||||||
* ROW: Rest of the World
| -14- |
The Company’s granted patents protecting its drug delivery platforms have the following latest dates of expiration by technology platform:
| Drug Delivery | Date of expiration of granted patents | |||
| Platforms | U.S. | Europe | ||
| Micropump® | July 2027 | May 2030 | ||
| LiquiTime® | September 2025 | April 2023 | ||
| Trigger Lock™ | April 2027 | May 2026 | ||
| Medusa™ | June 2031 | November 2024 | ||
Flamel’s key patents include protection for the following:
| · | Micropump® platform is patented under multiple granted patents. Among them is Flamel’s Micropump®-related key patent, WO 2003/030878, which discloses an efficacious coating formulation for providing delayed and sustained release of an active ingredient with absorption limited to the upper part of intestinal tract. It is granted in the U.S. as US Patent 8,101,209 and will expire on October 2025. It covers Coreg CR® formulation and, as such, has been listed at the FDA Orange Book by our partner GSK on February 23, 2012. Equivalent patents are granted in China, Hong Kong, Israel, India, Singapore, Japan, South Korea, Canada, South Africa, Mexico (expiry date: October 2022) and in France (expiry date: October 2021). Patent applications are pending in Brazil and Europe; and, would expire on October 2022. |
| · | LiquiTime® platform is protected by a Flamel’s patent granted in the U.S. (US 7,906,145; expiry date: September 2025) and in South Korea, Canada, Israel, Japan, Australia, China, Austria, Belgium, Switzerland, Liechtenstein, Germany, Spain, France, United Kingdom, Italy, Ireland, Luxembourg, Netherlands, Portugal, Sweden, Turkey, India, Mexico, South Africa that expire on April 2023. A patent application is pending in Brazil and 3 continuation application are pending in the U.S. |
| · | Trigger Lock™ platform is protected by 7 (seven) Flamel’s patent application families. Within these patent families, 12 (twelve) patents are granted in the U.S., Europe and Japan; and, 20 (twenty) patent applications are pending including other countries and will expire between November 2025 and December 2033. |
| · | Medusa™ platform is patented under Flamel’s key patent WO 2003/104303 granted in the U.S. and which will expire in July 2023. Equivalent patents to WO 2003/104303 are granted in China, Israel, Mexico, Australia, Japan, South Korea, Canada, Europe, India and South Africa. A patent application is pending in Brazil. These patents will expire in June 2023. |
| - | Medusa™-based nanogels are protected by issued patents from WO 2005/051416’ family in the U.S., Australia, China, Israel, Japan, South Korea, Mexico, South Africa, India, Canada and Europe expiring on November 2024. Corresponding patent application is pending in Brazil. |
| - | Medusa™-based microgels are protected by granted patents from WO 2007/141344’ patents family in the U.S., Australia, Japan, Canada, China, Israel, South Korea, Mexico and South Africa. Patent applications are pending in Europe, India and Brazil. This patents family will expire on June 2027. |
Strategic Alliances
The four partnerships left in 2013, after the rationalization of the Company’s products pipeline initiated in 2012, have, in 2014, either been terminated or transferred to Recipharm, as part the divestiture of our Pessac Facility, as follows:
| · | In May 2014: Effective termination of pilot (feasibility) study agreement, including an option for a license to be exercised prior engaging IND-/IMPD-enabling studies, with an undisclosed large international pharmaceutical company for the development of a Medusa™-enabled formulation of a partner’s controlled compound for cardiovascular indication (confidential); |
| · | In August 2014: Effective termination of the license and development agreement with an undisclosed specialty pharmaceutical company for the development of a Micropump-based, once-daily formulation of a central nervous system medication that is currently being marketed by that partner. Before the termination, we recognized $2.3 million in development and license fees in 2014; |
| · | In December 2014: The multi-year development partnership agreement with an undisclosed, large international pharmaceutical company was transferred to Recipharm, as part the divestiture of Pessac Facility. Before this divestiture, we received $2.0 million in development fees in 2014 classified as Discontinued Operations; and, |
| -15- |
| · | In December 2014: As part the divestiture of our development and manufacturing facility, the royalty payment under the license agreement and the supply agreement with GSK were, respectively, delegated and transferred to Recipharm. |
| · | Before the divestiture, we received royalty revenue of $6.3 million and a total amount of $6.7 million as revenues from product sales in 2014 (see “Note 17: Discontinued Operations to the consolidated financial statements in Part II, Item 8 of this Annual Report on Form 10-K). As a result of this divestiture, Flamel and Recipharm have entered into a five year services and manufacturing agreement to support the development of Flamel’s proprietary portfolio. Additionally, Recipharm has an option, for a certain period of time, to negotiate with Flamel for the European rights to product that Flamel plans to license for sale in the European market. In addition, Recipharm and Flamel have agreed to enter into a license agreement whereby Recipharm will be allowed to selectively offer Flamel’s drug delivery platforms in its CDMO business, further enhancing the economic benefits to both companies. |
In 2014 about 41% of our revenues came from partnerships. Today, however, the Company’s business is financially less dependent on its need to work with partners to develop products using our drug delivery platforms. By design, we now have revenue generating marketed products and products in late stage development that are not dependent on these partnerships (see “– Lead Products” in this Part I, Item 1 and “– Other Products Under Development” in this Part I, Item 1 of this Annual Report on Form 10-K).
Nonetheless, we are still open to entering into new partnerships, in particular, with pharmaceutical and biotechnology companies providing new formulation development opportunities (especially, based on partners’ proprietary or controlled therapeutic compounds), but also for certain of our proprietary products that Flamel and/or its US marketing unit, Éclat, will not market itself (such as the LiquiTime-based OTC products in late stage developments) and access to complementary expertise (regulatory, medical and commercial). Under such partnership agreements, our partners typically assume responsibility for all formulation development, manufacturing, polymer supply, clinical, regulatory and marketing costs and make payments to us at the time the agreement is signed and upon the achievement of significant technical, pre-clinical, clinical and regulatory milestones. We also typically are entitled to receive royalty payments on the sales of products that incorporate our drug delivery platforms.
Manufacturing
The manufacturing facilities for our drug delivery platforms were located in Pessac, France, near Bordeaux (hereinafter referred as “Pessac Facility”). This Pessac Facility provided us with two commercial scale production lines for the manufacture of Coreg CR® microparticles, and another production line used for other Micropump, and LiquiTime/Trigger Lock-based formulations (i.e. the production of certain pharmaceutical products, including commercial scale quantities of our intermediate formulated products). During 2014, our commercial manufacturing capacity utilization ranged from 50% to 65% of total capacity.
On December 1, 2014, the Pessac Facility was divested to Recipharm. This divestiture agreement allows Flamel to retain access to the development and manufacturing capabilities of Pessac Facility for all its drug delivery platforms. In particular, this facility can support, like any CDMO, certain of our needs for scale-up activities and clinical batch manufacturing for our Micropump, LiquiTime and Trigger Lock platforms, as well as for the synthesis of Medusa’s polymers and technical batch manufacturing for non-clinical studies pertaining to our Medusa-based formulations. In addition, this agreement permits us to utilize other Recipharm’s manufacturing facilities for the development and/or manufacture of our proprietary pipeline if needed.
The Pessac Facility was never used for the production of finished products commercialized by our US operations. Indeed, the manufacture of the UMDs products marketed by the Company’s US operations is outsourced to cGMP compliant and FDA-audited CDMOs in accordance with supply agreements.
Flamel intends to continue to outsource to third party contract manufacturing companies like Recipharm when appropriate. For example, in 2014, Flamel has transferred the scale up of certain of its own proprietary products to CDMOs in the U.S. This will be beneficial to the Company for products that will ultimately be submitted and sold in the United States.
Government Regulation
The design, testing, manufacturing and marketing of certain new or substantially modified drugs, biological products or medical devices must be approved, cleared or certified by regulatory agencies, regulatory authorities and Notified Bodies under applicable laws and regulations, the requirements of which may vary from country to country. This regulatory process is lengthy, expensive and uncertain. In the United States, the FDA regulates such products under various federal statutes, including the Federal Food, Drug, and Cosmetic Act (“FDCA”) and the Public Health Service Act. Similar requirements exist in the Member States of the European Union and are imposed by the European Commission and the competent authorities of EU Member States. There can be no assurance that we or our collaborative partners will be able to obtain such regulatory approvals or clearances or certification of conformity on a timely basis, if at all, for any products under development. Delays in receipt or failure to receive such approvals, clearances, or certifications of conformity, the revocation of previously received approvals or clearances, or certifications of conformity, or failure to comply with existing or future regulatory requirements could have a material adverse effect on our business, financial condition and results of operations.
| -16- |
We believe our delivery platforms, when used in conjunction with therapeutic pharmaceuticals, and development products acquired from Éclat, are subject to drug and biological product approval or marketing authorization requirements. In the United States and the European Union, biological products, such as therapeutic proteins and peptides, generally are subject to the same FDA and EU regulatory requirements as other drugs, although some differences exist. For example, a biologic license application (BLA) is submitted for approval for commercialization of some biological products instead of the New Drug Application (“NDA”) or Abbreviated New Drug Application (“ANDA”) used for other drugs. Also, unlike other drug products, some biological products are subject to FDA lot-by-lot release requirements and those approved under a BLA currently cannot be the subject of ANDAs. However, the FDA is working on a variety of issues pertaining to the possible development of biosimilars and there can be no assurance that this type of submission will continue to be unavailable for biological products. Additionally, our delivery platforms likely will be regulated by the FDA as ‘combination products’ if they are used together with a biologic or medical device. In order to facilitate pre-market review of combination products, the FDA designates one of its centers to have primary jurisdiction for the pre-market review and regulation of both components. In the European Union, applications for marketing authorization of innovative drugs, which are essentially products that are neither generics nor biosimilars, are addressed on a case-by-case basis by the European Medicines Agency (“EMA”), followed by a decision of the European Commission, or by the competent authorities of the EU Member States.
New Drug and Biological Product Development and Approval Process
United States and European Union
Regulation by governmental authorities in the United States and other countries has a significant impact on the development, manufacture, and marketing of biological and drug products and on ongoing research and product development activities. The products of all of our pharmaceutical and biotechnology partners as well as our own products will require regulatory approval by governmental agencies and regulatory authorities prior to commercialization. In particular, these products are subject to manufacturing according to stringent cGMP quality principles, and rigorous, pre-clinical and clinical testing and other pre-market approval requirements by the FDA, the European Commission and regulatory authorities in other countries. In the United States and the European Union, various statutes and regulations also govern, or influence the manufacturing, safety, labeling, storage, record keeping and marketing of pharmaceutical and biological products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources.
The FDA and European Union’s statutes, regulations, or policies may change and additional statutes or government regulations may be enacted which could prevent or delay regulatory approvals of biological or drug products. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
Regulatory approval, when and if obtained, may be limited in scope. In particular, regulatory approvals will restrict the marketing of a product to specific uses. Approved biological and other drugs, as well as their manufacturers, are subject to ongoing review (including requirements and restrictions related to record keeping and reporting, FDA, European Commission and EU Member States competent authorities’ approval of certain changes in manufacturing processes or product labeling, product promotion and advertising, and pharmacovigilance, which includes monitoring and reporting adverse reactions, maintaining safety measures, and conducting dossier reviews for marketing authorization renewal). Discovery of previously unknown problems with these products may result in restrictions on their manufacture, sale or use, or in their withdrawal from the market. Failure to comply with regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other actions affecting the commercial prospects of our pharmaceutical and biotechnology partners’ potential products or uses or products that incorporate our technologies. Any failure by our pharmaceutical and biotechnology partners to comply with current or new and changing regulatory obligations, and any failure to obtain and maintain, or any delay in obtaining, regulatory approvals, could materially adversely affect our business.
The process for new drug and biological product development and approval has many steps, including:
Chemical and Formulation Development
Pharmaceutical formulation taking into account the chemistry and physical characteristics of the drug or biological substance is the beginning of a new product. If initial laboratory experiments reveal that the concept for a new drug or biological product looks promising, then a variety of further development steps and tests complying with internationally recognized guidance documents will have to be continued, in order to provide for a product ready for testing in animals and, after sufficient animal test results, also in humans.
Concurrent with pre-clinical studies and clinical trials, companies must continue to develop information about the properties of the drug product and finalize a process for manufacturing the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product, and the manufacturer must develop and validate methods for testing the quality, purity and potency of the final products. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life.
Pre-Clinical Testing
Once a biological or drug candidate is identified for development, the candidate enters the pre-clinical testing stage. This includes laboratory evaluation of product chemistry and formulation, as well as animal studies of pharmacology (mechanism of action, pharmacokinetics) and toxicology which may have to be conducted over lengthy periods of time, to assess the potential safety and efficacy of the product as formulated. Pre-clinical tests must be conducted in compliance with good laboratory practice regulations, the Animal Welfare Act and its regulations in the US and the Clinical Trials Directive and related national laws and guidelines in the EU Member States. Violations of these laws and regulations can, in some cases, lead to invalidation of the studies, then requiring such studies to be replicated. In some cases, long-term pre-clinical studies are conducted while clinical studies are ongoing.
| -17- |
Investigational New Drug Application
USA: The entire body of chemical or biochemical, pharmaceutical and pre-clinical development work necessary to administer investigational drugs to human volunteers or patients is summarized in an Investigational New Drug (“IND”) application to the FDA. The IND becomes effective if not rejected by the FDA within thirty (30) days after filing. There is no assurance that the submission of an IND will eventually allow a company to commence clinical trials. All clinical trials must be conducted under the supervision of a qualified investigator in accordance with good clinical practice regulations to ensure the quality and integrity of clinical trial results and data. These regulations include the requirement that, with limited exceptions, all subjects provide informed consent. In addition, an institutional review board (“IRB”), composed primarily of physicians and other qualified experts at the hospital or clinic where the proposed studies will be conducted, must review and approve each human study. The IRB also continues to monitor the study and must be kept aware of the study’s progress, particularly as to adverse events and changes in the research. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if adverse events occur. Failure to adhere to good clinical practices and the protocols, and failure to obtain IRB approval and informed consent, may result in FDA rejection of clinical trial results and data, and may delay or prevent the FDA from approving the drug for commercial use.
European Union: The European equivalent to the IND is the Investigational Medicinal Product Dossier (“IMPD”) which likewise must contain pharmaceutical, pre-clinical and, if existing, previous clinical information on the drug substance and product. An overall risk-benefit assessment critically analyzing the non-clinical and clinical data in relation to the potential risks and benefits of the proposed trial must also be included. The intended clinical trial must be submitted for authorization by the regulatory authority(ies) of each EU Member States in which the trial is intended to be conducted prior to its commencement. The trial must be conducted on the basis of the protocol as approved by an Ethics Committee(s) in each EU Member State (EU equivalent to IRBs) before the trial commences. Before submitting an application to the competent authority, the sponsor must register the trial in the EudraCT database where it will be provided with a unique EudraCT number.
Clinical Trials
Typically, clinical testing involves the administration of the drug or biological product first to healthy human volunteers and then to patients with conditions needing treatment under the supervision of a qualified principal investigator, usually a physician, pursuant to a ‘protocol’ or clinical plan reviewed by the FDA and the competent authorities of the EU Member States along with the IRB or Ethics Committee (via the IND or IMPD submission). The protocol details matters such as a description of the condition to be treated, the objectives of the study, a description of the patient population eligible for the study and the parameters to be used to monitor safety and efficacy.
Clinical trials are time-consuming and costly, and typically are conducted in three sequential phases, which sometimes may overlap. Phase I trials consist of testing the product in a small number of patients or normal volunteers, primarily for safety, in one or more dosages, as well as characterization of a drug’s pharmacokinetic and/or pharmacodynamic profile. In Phase II, in addition to safety, the product is studied in a patient population to evaluate the product’s efficacy for the specific, targeted indications and to determine dosage tolerance and optimal dosage. Phase III trials typically involve additional testing for safety and clinical efficacy in an expanded patient population at geographically dispersed sites. With limited exceptions, all patients involved in a clinical trial must provide informed consent prior to their participation. Meeting clinical endpoints in early stage clinical trials does not assure success in later stage clinical trials. Phase I, II, and III testing may not be completed successfully within any specified time period, if at all.
The FDA and the competent authorities of EU Member States monitor the progress of each clinical trial phase conducted under an IND or IMPD and may, at their discretion, reevaluate, alter, suspend or terminate clinical trials at any point in this process for various reasons, including a finding that patients are being exposed to an unacceptable health risk or a determination that it is unethical to continue the study. The FDA, the European Commission and the competent authorities of EU Member States can also request that additional clinical trials be conducted as a condition to product approval. The IRB, the Ethics Committee, and sponsor also may order the temporary or permanent discontinuance of a clinical trial at any time for a variety of reasons, particularly if safety concerns arise. Such holds can cause substantial delay and in some cases may require abandonment of product development. These clinical studies must be conducted in conformance with the FDA’s bioresearch monitoring regulations, the Clinical Trials Directive and/or internationally recognized guidance (such as “ICH”, or “International Conference on Harmonization”).
New Drug Application or Biological License Application
After the completion of the clinical trial phases of development, if the sponsor concludes that there is substantial evidence that the drug or biological candidate is effective and that the drug is safe for its intended use, an NDA or “BLA” (“Biological License Application”) may be submitted to the FDA. The application must contain all of the information on the drug or biological candidate gathered to that date, including data from the pre-clinical and clinical trials, information pertaining to the preparation of the drug or biologic, analytical methods, product formulation, details on the manufacture of finished products, proposed product packaging, labeling and stability (shelf-life). NDAs and BLAs are often over 100,000 pages in length. If FDA determines that a Risk Evaluation And Mitigation Strategy (“REMS”) is necessary to ensure that the benefits of the drug outweigh the risks, a sponsor may be required to include as part of the application a proposed REMS, including a package insert directed to patients, a plan for communication with healthcare providers, restrictions on a drug’s distribution, or a medication guide to provide better information to consumers about the drug’s risks and benefits. Submission of an NDA or BLA does not assure FDA approval for marketing.
| -18- |
The FDA reviews all submitted NDAs and BLAs before it accepts them for filing (the U.S. prerequisite for dossier review). It may refuse to file the application and request additional information rather than accepting an application for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA or BLA to determine, among other things, whether a product is safe and effective for its intended use. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation. There is a strong presumption for advisory committee review for any drug containing an active ingredient not previously approved. The FDA is not bound by the recommendation of an advisory committee. Under the Prescription Drug User Fee Act (“PDUFA”), submission of an NDA or BLA with clinical data requires payment of a fee. In return, the FDA assigns an action date of 10 months from acceptance of the application to return of a first ‘complete response,’ in which the FDA may approve the product or request additional information. (Although PDUFA also provides for a six-month “priority review” process, we do not anticipate it applying to any of our products or our partners’ products.) There can be no assurance that an application will be approved within the performance goal timeframe established under PDUFA, if at all. If the FDA’s evaluation of the NDA or BLA is not favorable, the FDA usually will outline the deficiencies in the submission and request additional testing or information. Notwithstanding the submission of any requested additional information, or even in lieu of asking for additional information, the FDA may decide that the marketing application does not satisfy the regulatory criteria for approval and issue a complete response letter, communicating the agency’s decision not to approve the application.
FDA approval of an NDA or BLA will be based, among other factors, on the agency’s review of the pre-clinical and clinical data submitted, a risk/benefit analysis of the product, and an evaluation of the manufacturing processes and facilities. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its NDA or BLA. For instance, FDA may require us to provide data from additional preclinical studies or clinical trials to support approval of certain development products acquired from Éclat. Among the conditions for NDA or BLA approval is the requirement that each prospective manufacturer’s quality control and manufacturing procedures conform to cGMP standards and requirements. Manufacturing establishments often are subject to inspections prior to NDA or BLA approval to assure compliance with cGMPs and with manufacturing commitments made in the relevant marketing application.
Patent Restoration and Exclusivity
The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, establishes two abbreviated approval pathways for drug products that are in some way follow-on versions of already approved products.
Generic Drugs. A generic version of an approved drug is approved by means of an Abbreviated New Drug Application, or ANDA, by which the sponsor demonstrates that the proposed product is the same as the approved, brand-name drug, which is referred to as the “Reference Listed Drug,” or “RLD”. Generally, an ANDA must contain data and information showing that the proposed generic product and RLD (1) have the same active ingredient, in the same strength and dosage form, to be delivered via the same route of administration, (2) are intended for the same uses, and (3) are bioequivalent. This is instead of independently demonstrating the proposed product’s safety and effectiveness, which are inferred from the fact that the product is the same as the RLD, which the FDA previously found to be safe and effective.
505(b)(2) NDAs. If a product is similar, but not identical, to an already approved product, it may be submitted for approval via an NDA under Section 505(b)(2) of the Act. Unlike an ANDA, this does not excuse the sponsor from demonstrating the proposed product’s safety and effectiveness. Rather, the sponsor is permitted to rely to some degree on published scientific literature and the FDA’s finding that the RLD is safe and effective, and must submit its own data of safety and effectiveness to an extent necessary because of the differences between the products. With regard to certain UMD products, we intend to submit 505(b)(2) NDAs, relying solely on published scientific literature. We do not plan to conduct additional preclinical studies or clinical trials for these 505(b)(2) NDAs; and, if we were required to do so, would review the continued value of the product.
RLD Patents. An NDA sponsor must advise the FDA about patents that claim the drug substance or drug product or a method of using the drug. When the drug is approved, those patents are among the information about the product that is listed in the FDA publication, Approved Drug Products with Therapeutic Equivalence Evaluations, which is referred to as the Orange Book. The sponsor of an ANDA or 505(b)(2) application seeking to rely on an approved product as the RLD must make one of several certifications regarding each listed patent. A “Paragraph III” certification is the sponsor’s statement that it will wait for the patent to expire before obtaining approval for its product. A “Paragraph IV” certification is a challenge to the patent; it is an assertion that the patent does not block approval of the later product, either because the patent is invalid or unenforceable or because the patent, even if valid, is not infringed by the new product.
Once the FDA accepts for filing an ANDA or 505(b)(2) application containing a Paragraph IV certification, the applicant must within 20 days provide notice to the RLD NDA holder and patent owner that the application with patent challenge has been submitted, and provide the factual and legal basis for the applicant’s assertion that the patent is invalid or not infringed. If the NDA holder or patent owner file suit against the ANDA or 505(b)(2) applicant for patent infringement within 45 days of receiving the Paragraph IV notice, FDA is prohibited from approving the ANDA or 505(b)(2) application for a period of 30 months from the date of receipt of the notice. If the RLD has NCE exclusivity and the notice is given and suit filed during the fifth year of exclusivity, the 30-month stay does not begin until five years after the RLD approval. The FDA may approve the proposed product before the expiration of the 30-month stay if a court finds the patent invalid or not infringed or if the court shortens the period because the parties have failed to cooperate in expediting the litigation.
| -19- |
Regulatory Exclusivities. The Hatch-Waxman Act may provide periods of regulatory exclusivity for products that would serve as RLDs. If a product is a “new chemical entity,” or NCE, – generally meaning that the active moiety has never before been approved in any drug – there may be a period of five years from the product’s approval during which the FDA may not accept for filing any ANDA or 505(b)(2) application for a drug with the same active moiety. An ANDA or 505(b)(2) application may be submitted after four years, however, if the sponsor makes a Paragraph IV certification challenging a listed patent. Because it takes time for the FDA to review and approve an application once it has been accepted for filing, five-year NCE exclusivity usually effectively means the ANDA or 505(b)(2) application is not approved for a period well beyond five years from approval of the RLD.
A product that is not an NCE may qualify for a three-year period of exclusivity, if the NDA contains clinical data that were necessary for approval. In that instance, the exclusivity period does not preclude filing or review of the ANDA or 505(b)(2) application; rather, the FDA is precluded from granting final approval to the ANDA or 505(b)(2) application until three years after approval of the RLD. Additionally, the exclusivity applies only to the conditions of approval that required submission of the clinical data. For example, if an NDA is submitted for a product that is not an NCE, but that seeks approval for a new indication, and clinical data were required to demonstrate the safety or effectiveness of the product for that use, the FDA could not approve an ANDA or 505(b)(2) application for another product with that active moiety for that use. For example, Coreg CR received three-year exclusivity for the clinical trials that demonstrated the safety and efficacy of the new, controlled-release dosage form; that exclusivity, which has expired, blocked other controlled-release products.
Patent Term Restoration. Under the Hatch-Waxman Act, a portion of the patent term lost during product development and FDA review of an NDA or 505(b)(2) application is restored if approval of the application is the first permitted commercial marketing of a drug containing the active ingredient. The patent term restoration period is generally one-half the time between the effective date of the IND and the date of submission of the NDA, plus the time between the date of submission of the NDA and the date of FDA approval of the product. The maximum period of restoration is five years, and the patent cannot be extended to more than 14 years from the date of FDA approval of the product. Only one patent claiming each approved product is eligible for restoration and the patent holder must apply for restoration within 60 days of approval. The United States Patent and Trademark Office, or PTO, in consultation with the FDA, reviews and approves the application for patent term restoration. When any of our products is approved, we intend to seek patent term restoration for an applicable patent when it is appropriate.
Other Countries
Whether or not FDA approval has been obtained, approval of a pharmaceutical product by regulatory authorities must be obtained in any other country prior to the commencement of marketing of the product in that country. The approval procedure may vary from country to country, can involve additional testing, and the time required may differ from that required for FDA approval. Under European Union legislation, product authorization is granted for an initial period of five years. The authorization may subsequently be renewed for an unlimited period on the basis of a re-evaluation of the risk-benefit balance by the competent authorizing authority. In the EU, marketing authorization of drugs is according to either a centralized, decentralized or mutual recognition procedure, generally depending on the nature and type of drug. Certain designated drugs may be authorized only in accordance with the centralized procedure by the European Commission following an opinion by the European Medicines Agency (“EMA”). The centralized procedure is mandatory for pharmaceutical products developed by means of biotechnological processes (recombinant DNA, controlled expression of genes coding, hybridoma and monoclonal antibody methods), products containing new actives substances indicated for the treatment of AIDS, cancer, diabetes and neuro-degenerative diseases, orphan designated medicinal products and advanced therapy products. Other pharmaceutical products may be authorized in accordance with the centralized procedure where it is demonstrated that they contain new active substances or are demonstrated to have a significant therapeutic benefit, or where they constitute a scientific or technical innovation, or are in the interest of patients at Community level. Where authorization is in accordance with the decentralized or mutual recognition procedures, approval is either by “mutual recognition,” whereby the authorization granted by the competent authorities of one EU Member States are recognized by the authorities of other EU Member States, or where the competent authorities of each EU Member State authorize a product on the basis of an identical dossier, with one national authority taking care of the dossier intensively and coordinating activities. To the extent possible, clinical trials of our products are designed to develop a regulatory package sufficient for the grant of marketing authorization in the EU approval according to the Community Code on medicinal products.
Regulatory approval of prices for certain drugs is required in France and in many other countries outside the United States. In particular, many EU Member States make the reimbursement of a product within the national social security system conditional on the agreement by the seller not to sell the product above a fixed price in that country. Also common is the unilateral establishment of a reimbursement price by the national authorities, often accompanied by the inclusion of the product on a list of reimbursable products. Related pricing discussions and ultimate governmental approvals can take several months to years. Some countries require periodic pricing updates and renewals at intervals ranging from two to five years. Some countries also impose price freezes or obligatory price reductions. We cannot assure you that, if regulatory authorities establish lower prices for any product incorporating our technology in any one EU Member State, this will not have the practical effect of requiring our collaborative partner correspondingly to reduce its prices in other EU Member States. We can offer no assurance that the resulting prices would be sufficient to generate an acceptable return on our investment in our products.
| -20- |
Regulation of Combination Drugs
Medical products containing a combination of drugs or biological products may be regulated as ‘combination products’ in the United States. A combination product generally is defined as a product comprising components from two or more regulatory categories (e.g., drug/device, device/biologic, drug/biologic). Each component of a combination product is subject to the requirements established by the FDA for that type of component, whether a drug, biologic or device.
To determine which FDA center or centers will review a combination product submission, companies may submit a request for assignment to the FDA. Those requests may be handled formally or informally. In some cases, jurisdiction may be determined informally based on FDA experience with similar products. However, informal jurisdictional determinations are not binding on the FDA. Companies also may submit a formal Request for Designation to the FDA Office of Combination Products. The Office of Combination Products will review the request and make its jurisdictional determination within 60 days of receiving a Request for Designation.
In order to facilitate pre-market review of combination products, the FDA designates one of its centers to have primary jurisdiction for the pre-market review and regulation of both components. The determination whether a product is a combination product or two separate products is made by the FDA on a case-by-case basis. It is possible that our delivery platforms, when coupled with a drug, biologic or medical device component, could be considered and regulated by the FDA as a combination product.
If the primary mode of action is determined to be a drug, the product will be reviewed by the Center for Drug Evaluation and Research (“CDER”) either in consultation with another center or independently. If the primary mode of action is determined to be a medical device, the product would be reviewed by Center for Devices and Radiological Health (“CDRH”) either in consultation with another center, such as CDER, or independently. In addition, FDA could determine that the product is a biologic and subject to the jurisdiction of the Center for Biologic Evaluation and Research (“CBER”), although it is also possible that a biological product will be regulated by CDER.
In the European Union, drug combinations, that is, drug products containing two or more drug substances each of which has to contribute a proven advantage of therapy (e.g., synergism, less adverse reactions), are subject to drug regulations like all others. Products combining drug substances or drugs with a device may be subject to device and/or drug regulations, or may be classified as medical devices, depending on the individual case.
Marketing Approval and Reporting Requirements
If the FDA approves an NDA or BLA, the product becomes available for physicians to prescribe. The FDA may require post-marketing studies, also known as Phase IV studies, as a condition of approval to develop additional information regarding the safety of a product. These studies may involve continued testing of a product and development of data, including clinical data, about the product’s effects in various populations and any side effects associated with long-term use. After approval, the FDA may require post-marketing studies or clinical trials, as well as periodic status reports, if new safety information develops. These post-marketing studies may include clinical trials to investigate known serious risks or signals of serious risks or identify unexpected serious risks. Failure to conduct these studies in a timely manner may result in substantial civil fines and can result in withdrawal of approval.
In addition, the FDA may require distribution to patients of a medication guide such as a REMS for prescription products that the agency determines pose a serious and significant health concern in order to provide information necessary to patients’ safe and effective use of such products.
In the European Union, the marketing authorization of a medicinal product may be made conditional on the conduct of Phase IV post-marketing studies. Failure to conduct these studies in relation to centrally authorized products can lead to the imposition of substantial fines. Moreover, Phase IV studies are often conducted by companies in order to obtain further information on product efficacy and positioning on the market in view of competitors and to assist in application for pricing and reimbursement.
Post-Marketing Obligations
Any products manufactured and/or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping requirements, reporting of adverse experiences with the product, submitting other periodic reports, drug sampling and distribution requirements, notifying the FDA and gaining its approval of certain manufacturing or labeling changes, complying with certain electronic records and signature requirements, submitting periodic reports to the FDA, maintaining and providing updated safety and efficacy information to the FDA, and complying with FDA promotion and advertising requirements. For example, with respect to the Éclat product Vazculep, the FDA has required the Company to conduct post-marketing clinical and non-clinical studies to be completed between 2016 and 2019.
Drug and biologics manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and to list their products with the FDA. The FDA periodically inspects manufacturing facilities in the United States and abroad in order to assure compliance with the applicable cGMP regulations and other requirements. Facilities also are subject to inspections by other federal, foreign, state or local agencies. In complying with the cGMP regulations, manufacturers must continue to expend time, money and effort in recordkeeping and quality control to assure that the product meets applicable specifications and other post-marketing requirements. Failure of the Company or our licensees to comply with FDA’s cGMP regulations or other requirements could have a significant adverse effect on the Company’s business, financial condition and results of operations.
| -21- |
Also, newly discovered or developed safety or efficacy data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, additional pre-clinical or clinical studies, or even in some instances, revocation or withdrawal of the approval. Violations of regulatory requirements at any stage, including after approval, may result in various adverse consequences, including the FDA’s delay in approving or refusal to approve a product, withdrawal or recall of an approved product from the market, other voluntary or FDA-initiated action that could delay or restrict further marketing, and the imposition of civil fines and criminal penalties against the manufacturer and NDA or BLA holder. In addition, later discovery of previously unknown problems may result in restrictions on the product, manufacturer or NDA or BLA holder, including withdrawal of the product from the market. Furthermore, new government requirements may be established that could delay or prevent regulatory approval of our products under development, or affect the conditions under which approved products are marketed.
The Food and Drug Administration Amendments Act of 2007 provides the FDA with expanded authority over drug products after approval. This legislation enhances the FDA’s authority with respect to post-marketing safety surveillance, including, among other things, the authority to require additional post-marketing studies or clinical trials, labeling changes as a result of safety findings, registering clinical trials, and making clinical trial results publicly available.
In the European Union, stringent pharmacovigilance regulations oblige companies to appoint a suitably qualified and experienced Qualified Person resident in the European Economic Area, to prepare and submit to the competent authorities adverse event reports within specific time lines, prepare Periodic Safety Update Reports (PSURs) and provide other supplementary information, report to authorities at regular intervals and take adequate safety measures agreed with regulatory agencies as necessary. Failure to undertake these obligations can lead to the imposition of substantial fines.
Biologics Price Competition and Innovation Act of 2009
The Hatch-Waxman construct applies only to conventional chemical drug compounds, sometimes referred to as small molecule compounds approved under an NDA. On March 23, 2010, however, the “Biologics Price Competition and Innovation Act” of 2009, or “BPCIA”, was signed into law. It creates an abbreviated approval pathway for biological products that are “biosimilar” to a previously approved biological product, which is called the “reference product.” This abbreviated approval pathway is intended to permit a biosimilar product to come to market more quickly and less expensively than if a “full” BLA were submitted, by relying to some extent on FDA’s previous review and approval of the reference product to which the proposed product is similar. If a proposed biosimilar product meets the statutory standards for approval (which include demonstrating that it is highly similar to the reference product and there are no clinically meaningful differences in safety, purity or potency between the products), the proposed biosimilar may be approved on the basis of an application that is different than the standard BLA. In addition, a biosimilar product may be approved as interchangeable with the reference product if the proposed product application meets standards intended to ensure that the biosimilar product can be expected to produce the same clinical result as the reference product.
Other Regulation
Controlled Substances Act. Our Trigger Lock delivery platform is designed to control the release of narcotics and other active ingredients subject to abuse. Narcotics are “controlled substances” under the Controlled Substances Act. The federal “Controlled Substances Act” (“CSA”), Title II of the Comprehensive Drug Abuse Prevention and Control Act of 1970, regulates the manufacture and distribution of narcotics and other controlled substances, including stimulants, depressants and hallucinogens. The CSA is administered by the “Drug Enforcement Administration” (“DEA”), a division of the U.S. Department of Justice, and is intended to prevent the abuse or diversion of controlled substances into illicit channels of commerce.
Any person or firm that manufactures, distributes, dispenses, imports, or exports any controlled substance (or proposes to do so) must register with the DEA. The applicant must register for a specific business activity related to controlled substances, including manufacturing or distributing, and may engage in only the activity or activities for which it is registered. The DEA conducts periodic inspections of registered establishments that handle controlled substances and allots quotas of controlled drugs to manufacturers and marketers’ failure to comply with relevant DEA regulations, particularly as manifested in the loss or diversion of controlled substances, can result in regulatory action including civil penalties, refusal to renew necessary registrations, or proceedings to revoke those registrations. In certain circumstances, violations can lead to criminal prosecution. In addition to these federal statutory and regulatory obligations, there may be state and local laws and regulations relevant to the handling of controlled substances or listed chemicals.
cGMP. Current Good Manufacturing Practices rules apply to the manufacturing of drugs and medical devices. Our manufacturing facilities and laboratories are subject to inspection and regulation by French regulatory authorities in accordance with applicable EU provisions governing cGMP and may also be subject to the United States’ and other countries’ regulatory agencies. Mutual recognition agreements for government inspections exist between the United States, the EU, Canada, Australia and New Zealand.
In addition to regulations enforced by the FDA, we are also subject to French, U.S. and other countries’ rules and regulations governing permissible laboratory activities, waste disposal, handling of toxic, dangerous or radioactive materials and other matters. Our R&D involves the controlled use of hazardous materials, chemicals, viruses and various radioactive compounds. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by French, EU, U.S. and other foreign rules and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated.
| -22- |
Health Care Fraud and Abuse. We are subject to a number of federal and state laws pertaining to health care “fraud and abuse,” such as anti-kickback and false claims laws. Under anti-kickback laws, it is illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance via regulations and that there are few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third-party payors (such as the Medicare and Medicaid programs) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our sales and marketing activities relating to our products could be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal health care programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. In addition, similar sanctions and penalties can be imposed upon executive officers and employees, including criminal sanctions against executive officers. As a result of the potential penalties that can be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the government were to allege or convict us or our executive officers of violating these laws, our business could be harmed. In addition, private individuals have the ability to bring similar actions. In addition to the reasons noted above, our activities could be subject to challenge due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. There also are an increasing number of federal and state laws that require manufacturers to make reports to states on pricing, marketing information, and payments and other transfers of value to healthcare providers. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent authorities.
Healthcare Reimbursement
In both U.S. and foreign markets, sales of our potential products as well as products of pharmaceutical and biotechnology companies that incorporate our technology into their products, if any, will depend in part on the availability of reimbursement by third-party payers, such as government health administration authorities, private health insurers and other organizations. The U.S. market for pharmaceutical products is increasingly being shaped by managed care organizations, pharmacy benefit managers, cooperative buying organizations and large drugstore chains. Third-party payers are challenging the price and cost effectiveness of medical products and services. Uncertainty particularly exists as to the reimbursement status of newly approved healthcare products. There can be no assurance reimbursement will be available to enable us to maintain price levels sufficient to realize an appropriate return on our product development investment. Legislation and regulations affecting the pricing of pharmaceuticals may change before our proposed products are approved for marketing and any such changes could further limit reimbursement for medical products and services.
| Item 1A. | Risk Factors |
Our business faces many risks. The risks described below may not be the only risks we face. Additional risks that we do not yet know of or that we currently believe are immaterial may also impair our business operations. If any of the events or circumstances described in the following risks actually occur, our business, financial condition or results of operations could suffer, and the trading price of our securities could decline. As a result, you should consider all of the following risks, together with all of the other information in this annual report on Form 10-K, before making an investment decision regarding our securities.
Risks Relating to Our Business and Industry
We depend on a small number of products and customers for the majority of our revenues and the loss of any one of these products or customers could reduce our revenues significantly.
We derive a majority of our revenues from sales of two products, Bloxiverz and Vazculep. Additionally, we depend on a small number of customers for the majority of our revenues from sales of these two drug products. Three customers, AmeriSource Bergen, Cardinal and McKesson accounted for approximately 95% of revenues from sales of these products in 2015. These customers comprise a significant portion of the distribution network for pharmaceutical products in the U.S. This distribution network is continuing to undergo consolidation marked by mergers and acquisitions among wholesale distributors and retail drug store chains. As a result, a small number of large wholesale distributors and large chain drug stores control a significant share of the market. We expect that continuing consolidation will increase pricing and other competitive pressures on pharmaceutical companies. The loss of any one of these products or the termination of our relationship with any of these customers or our failure to broaden our customer base could cause our revenues to decrease significantly and result in losses from our operations. Further, we may be unable to negotiate favorable business terms with customers that represent a significant portion of our revenues, and any such inability could have a material adverse effect on our business, results of operations, financial condition and prospects.
| -23- |
We may depend on partnership arrangements or strategic alliances for the commercialization of some of our products in development, in particular those incorporating our drug delivery platforms.
The commercialization of some of our drug delivery platforms-based products in development, such as Trigger Lock based-hydromorphone and Medusa based-exenatide, will require resources and expertise that we currently do not have. Therefore, we will need to seek partners, and/or enter into strategic alliances, licenses or other arrangements to successfully commercialize these products, as we did with respect to the license to Elan for the OTC rights for LiquiTime (see “– Products In Development With Partners” in this Part I, Item 1 of this Annual Report on Form 10-K).
Our products may not gain market acceptance.
Even if we and/or our partners obtain the necessary regulatory approval to market products, such products, technologies and product candidates may not gain market acceptance among physicians, patients, healthcare payers and medical communities. The degree of market acceptance of any product, technology or product candidate will depend on a number of factors, including:
| · | the scope of regulatory approvals, including limitations or warnings in a product’s regulatory-approved labeling; |
| · | demonstration of the clinical safety and efficacy of the product or technology; |
| · | the absence of evidence of undesirable side effects of the product or technology that delay or extend trials; |
| · | the lack of regulatory delays or other regulatory actions; |
| · | its cost-effectiveness; |
| · | its potential advantage over alternative treatment methods; |
| · | the availability of third-party reimbursement; and |
| · | the marketing and distribution support it receives. |
If any of our products or drug delivery platforms fail to achieve market acceptance, our ability to generate additional revenue will be limited, which would have a material adverse effect on our business. In addition, even if we gain regulatory approval and market acceptance, further delays due to, for example, the FDA not removing unapproved products from the market in a timely manner, may affect our ability to generate revenue quickly after market acceptance.
Our products may not reach the commercial market for a number of reasons.
Drug development is an inherently uncertain process with a high risk of failure at every stage of development. Successful research and development (“R&D”) of pharmaceutical products is difficult, expensive and time consuming. Many product candidates fail to reach the market. Our success will depend on the development and the successful commercialization of previously Unapproved Marketed Drugs (“UMDs”) products, development of products that utilize our drug delivery platforms, and the continued development and marketing of the products we obtained in the FSC acquisition in February 2016. If any of the UMDs products, products incorporating our drug delivery platforms, or FSC products fail to reach the commercial market, our future revenues would be adversely affected.
Even if our products and current drug delivery platforms appear promising during development, there may not be successful commercial applications developed for them for a number of reasons, including:
| · | the FDA, the European Medicines Agency (“EMA”), the competent authority of an EU Member State or an Institutional Review Board (“IRB”), or an Ethics Committee (EU equivalent to IRB), or our partners may delay or halt applicable clinical trials; |
| · | we or our partners may face slower than expected rate of patient recruitment and enrollment in clinical trials, or may devote insufficient funding to the clinical trials; |
| · | our current drug delivery platforms and drug products may be found to be ineffective or cause harmful side effects, or may fail during any stage of pre-clinical testing or clinical trials; |
| · | we or our partners may find certain products cannot be manufactured on a commercial scale and, therefore, may not be economical to produce; |
| · | managed care providers may be unwilling or unable to reimburse patients at an economically attractive level for products under development; or |
| · | our products could fail to obtain regulatory approval or, if approved, fail to achieve market acceptance, fail to be included within the pricing and reimbursement schemes of the U.S. or EU Member States, or be precluded from commercialization by proprietary rights of third parties. |
We must invest substantial sums in R&D in order to remain competitive, and we may not fully recover these investments.
To be successful in the highly competitive pharmaceutical industry, we must commit substantial resources each year to R&D in order to develop new products and enhance our technologies. In 2015, we spent $25.6 million on R&D. Our ongoing investments in R&D for future products could result in higher costs without a proportionate increase, or any increase, in revenues. The R&D process is lengthy and carries a substantial risk of failure. If our R&D does not yield sufficient products that achieve commercial success, our future operating results will be adversely affected.
| -24- |
The development of several of our drug delivery platforms and products depend on the services of a single provider and any interruption of operations of such provider could significantly delay or have a material adverse effect on our product pipeline.
As part of the divestiture of our development and manufacturing facility (“Pessac Facility”) to Recipharm AB (“Recipharm”), we entered into certain agreements with Recipharm for the development, supply of clinical materials and potentially the supply of commercial batches for several of our products incorporating our drug delivery platforms, as well as our Medusa™ polymer(s); for details see “Business – Information on the Company” in this Part I, Item 1 of this Annual Report on Form 10-K. Any disruption in the operations of Recipharm or if Recipharm fails to supply acceptable quantity and quality materials or services to us for any reason, such disruption or failure could delay our product development and could have a material adverse effect on our business, financial condition and results of operations. In case of a disruption, we may need to establish alternative manufacturing sources for our drug delivery products, and this would likely lead to substantial production delays as we build or locate replacement facilities and seek to satisfy necessary regulatory obligations.
We depend on a limited number of suppliers for the manufacturing of our products and certain raw materials used in of our products and any failure of such suppliers to deliver sufficient quantities of supplies of product or these raw materials could have a material adverse effect on our business.
Currently, we depend on a single manufacturer for both all of our drug products. Additionally, we purchase certain raw materials used in our products from a limited number of suppliers, including a single supplier for certain key ingredients. If the supplies of these products or materials were interrupted for any reason, our manufacturing and marketing of certain products could be delayed. These delays could be extensive and expensive, especially in situations where a substitution was not readily available or required variations of existing regulatory approvals and certifications or additional regulatory approval. For example, an alternative supplier may be required to pass an inspection by the FDA, EMA or the competent authorities of EU Member States for compliance with current Good Manufacturing Practices (“cGMP”) requirements before supplying us with product or before we may incorporate that supplier’s ingredients into the manufacturing of our products by our contract, development, and manufacturing organizations (“CDMOs”). Failure to obtain adequate supplies in a timely manner could have a material adverse effect on our business, financial condition and results of operations.
If our competitors develop and market technologies or products that are safer or more effective than ours, or obtain regulatory approval and market such technologies or products before we do, our commercial opportunity will be diminished or eliminated.
Competition in the pharmaceutical and biotechnology industry is intense and is expected to increase. We compete with academic laboratories, research institutions, universities, joint ventures and other pharmaceutical and biotechnology companies, including other companies developing drug delivery platforms or niche brand or generic specialty pharmaceutical products. Some of these competitors may be also our business partners.
Our drug delivery platforms compete with technologies provided by several other companies (for details see “Business – Competition and Market Opportunities” in this Part I, Item 1 of this Annual Report on Form 10-K). In particular, New Biological or Chemical Entities (“NBEs” or “NCEs”) could be developed that, if successful, could compete against our drug delivery platforms or products. Among the many experimental therapies being tested in the U.S. and in the EU, there may be some that we do not now know of that may compete with our drug delivery platforms or products in the future. These new biological or chemical products may be safer or may work better than our products.
With respect to our UMD drug products, the FDA could approve generic versions or previously filed NDAs of our marketed products, as was the case with the approval of APP’s (a division of Fresenius Kabi USA, LLC) and Eurohealth International’s (an affiliate of West-Ward Pharmaceuticals Corp.) neostigmine methylsulfate products, competitive products to Bloxiverz in January and December 2015 respectively.
With respect to our pediatric products acquired from FSC, we have competitors selling products in both the OTC and the prescription markets. Many of these competitors have greater market shares for their products and have leverage in the physician’s office and in the retail distribution channel. As a result of these dynamics, we could have competitors with a greater share of voice in the market, which would impede our ability to grow the FSC products.
Many of these competitors have substantially greater financial, technological, manufacturing, marketing, managerial and R&D resources and experience than we do. Furthermore, acquisitions of competing drug delivery companies by large pharmaceutical companies could enhance our competitors’ resources. Accordingly, our competitors may succeed in developing competing technologies and products, obtaining regulatory approval and gaining market share for these products more rapidly than we do.
If third party payors choose not to reimburse the use of our pediatric products our sales and profitability could suffer.
Because several of the categories in which we participate are available on an over-the-counter basis (OTC) some insurance programs may drive consumers to those products by requiring large co-pays for our products. In some cases this could require a patient failure with OTCs before our products are allowed to be used. Additionally, some health plans may prefer generic alternatives in our therapeutic categories, which is manifested by requiring higher copays for our products. Other health plans could omit coverage for our products altogether. Any of these types of dynamics could negatively impact the sale of our products.
| -25- |
If we cannot keep pace with the rapid technological change in our industry, we may lose business, and our drug delivery platforms could become obsolete or noncompetitive.
Our success also depends, in part, on maintaining a competitive position in the development of products and technologies in a rapidly evolving field. Major technological changes can happen quickly in the biotechnology and pharmaceutical industries. If we cannot maintain competitive products and technologies, our competitors may succeed in developing competing technologies or obtaining regulatory approval for products before us, and the products of our competitors may gain market acceptance more rapidly than our products. Such rapid technological change, or the development by our competitors of technologically improved or different products, could render our drug delivery platforms obsolete or noncompetitive.
We may fail to effectively pursue our business strategy.
Our business strategy is to obtain FDA approval and commercialize certain UMD product candidates, continue to develop and commercialize our drug delivery platforms, develop and market the FSC products and identify and acquire additional businesses or new product opportunities. There can be no assurance that we will be successful in any of these objectives; and a failure in any of these objectives could negatively impact our business and operating results.
In particular, we may be unable to successfully identify attractive acquisition candidates or complete any acquisitions, or successfully integrate any acquired business, product or technology or retain any key employees of acquired businesses. Integrating any business, product or technology we acquire could be expensive and time consuming, and could disrupt our ongoing business and distract our management. If we were to be unable to complete these acquisitions or to successfully integrate any acquired businesses, products or technologies effectively, our business would suffer. In addition, any amortization or charges resulting from the costs of acquisitions could negatively impact our operating results.
The impact of the acquisition of FSC on our financial results may be worse than the assumptions we have used.
Even if the integration of the FSC business is successful, we have made certain assumptions relating to the impact on our financial results in respect of the acquisition. These assumptions relate to numerous matters, including:
| · | the amount of intangible assets that will result from the acquisition; |
| · | the impact of fair value adjustments to contingent acquisition consideration payable as a result of changes in estimated probability and timing of achieving the targets; |
| · | acquisition costs, including transaction and integration costs; |
| · | the impact of impairment and other charges if the FSC products are unsuccessful; and |
| · | other financial and strategic risks of the acquisition. |
Irrespective of our assumptions, we may incur higher than expected operating, transaction and integration costs, and we may encounter general economic and business conditions that adversely affect us following the acquisition. If one or more of these assumptions are incorrect, it could have an adverse effect on our business and operating results, and the perceived benefits from the acquisition may not be realized.
If we cannot adequately protect our intellectual property and proprietary information, we may be unable to sustain a competitive advantage.
Our success depends, in part, on our ability to obtain and enforce patents for our products, processes and drug delivery platforms and to preserve our trade secrets and other proprietary information. If we cannot do so, our competitors may exploit our inventions and deprive us of the ability to realize revenues and profits from our products and technologies.
Any patent applications that we have made or may make relating to our potential products, processes and technologies may not result in patents being issued. Patent law relating to the scope of claims in the pharmaceutical and biotechnology fields in which we operate is continually evolving and can be the subject of some uncertainty. The laws providing patent protection may change in a way that would limit protection. Our current patents may not be exclusive, valid or enforceable. They may not protect us against competitors that challenge our patents, such as companies that submit drug marketing applications to the FDA, the EMA, or the competent authorities of EU Member States that rely, at least in part, on safety and efficacy data from our products or our business partners’ products, or competitors may obtain patents that may have an adverse effect on our ability to conduct business or discover ways to circumvent our patents. The scope of any patent protection may not be sufficiently broad to cover our products or to exclude competing products. Our partnerships with third parties expose us to risks that they will claim intellectual property rights on our inventions or fail to keep our unpatented technology or processes confidential.
Further, patent protection once obtained is limited in time, after which competitors may use the covered product or technology without obtaining a license from us. Because of the time required to obtain regulatory marketing approval, the period of effective patent protection for a marketed product is frequently substantially shortened.
| -26- |
We also rely on trademarks, copyrights, trade secrets and know-how to develop, maintain and strengthen our competitive position. To protect our trade secrets and proprietary technologies and processes, we rely, in part, on confidentiality agreements with our employees, consultants, advisors and partners. These agreements may not provide adequate protection for our trade secrets and other proprietary information in the event of any unauthorized use or disclosure, or if others lawfully develop the information. If these agreements are breached, we cannot be certain that we will have adequate remedies. Further, we cannot guarantee that third parties will not know, discover or independently develop equivalent proprietary information or technologies or processes, or that they will not gain access to our trade secrets or disclose our trade secrets to the public. Therefore, we cannot guarantee that we can maintain and protect unpatented proprietary information and trade secrets. Misappropriation or other loss of our intellectual property would adversely affect our competitive position and may cause us to incur substantial litigation or other costs.
The implementation of the Leahy-Smith America Invents Act of 2011 may adversely affect our business.
The Leahy-Smith America Invents Act of 2011 (“AIA”), changes the current U.S. “first-to-invent” system to a system that awards a patent to the “first-inventor-to-file” for an application for a patentable invention. This change alters the pool of available materials that can be used to challenge patents in the U.S. and eliminates the ability to rely on prior research to lay claim to patent rights. Disputes will be resolved through new derivation proceedings and the AIA creates mechanisms to allow challenges to newly issued patents in reexamination proceedings. New bases and procedures may make it easier for competitors to challenge our patents, which could result in increased competition and have a material adverse effect on our business and results of operations. The AIA may also make it harder to challenge third-party patents and place greater importance on being the first inventor to file a patent application on an invention. The AIA amendments to patent filing and litigation procedures in the U.S. may result in litigation being more complex and expensive and divert the efforts of our technical and management personnel.
Third parties may claim that our products infringe their rights, and we may incur significant costs resolving these claims.
Third parties may claim, that the manufacture, use, import, offer for sale or sale of our drug delivery platforms or our other products infringes on their patent rights. In response to such claims, we may have to seek licenses, defend infringement actions or challenge the validity of those patent rights in court. If we cannot obtain required licenses, are found liable for infringement or are not able to have such patent rights declared invalid, we may be liable for significant monetary damages, encounter significant delays in bringing products to market or be precluded from the manufacture, use, import, offer for sale or sale of products or methods of drug delivery covered by the patents of others. We may not have identified, or be able to identify in the future, U.S. or foreign patents that pose a risk of potential infringement claims.
Any claims that our products or drug delivery platforms infringe proprietary rights of third parties, with or without merit, could be time-consuming, result in costly litigation or divert the efforts of our technical and management personnel, any of which could disrupt our relationships with our partners and could significantly harm our operating results.
If we or our partners are required to obtain licenses from third parties, our revenues and royalties on any commercialized products could be reduced.
The development of some of our drug delivery platforms-based products may require the use of raw materials (e.g. proprietary excipient), active ingredients or drugs (e.g., proprietary proteins), technologies/processes, etc. developed by third parties. The extent to which efforts by other researchers have resulted or will result in patents and the extent to which we or our partners are forced to obtain licenses from others, if available, on commercially reasonable terms is currently unknown. If we or our partners must obtain licenses from third parties, fees must be paid for such licenses, which could reduce the revenues and royalties we may receive on commercialized products that incorporate our drug delivery platforms.
Security breaches and other disruptions could compromise confidential information and expose us to liability and cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store proprietary data, including intellectual property, as well as our proprietary business information and that of our customers, suppliers and business partners, on our networks. The secure maintenance and transmission of this information is critical to our operations and business strategy. Despite our security measures, our information systems and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, investigations by regulatory authorities in the U.S. and EU Member States, disruption to our operations and damage to our reputation, any of which could adversely affect our business.
Failure to comply with domestic and international privacy and security laws could result in the imposition of significant civil and criminal penalties.
The costs of compliance with privacy and security laws, including protecting electronically stored information from cyber-attacks, and potential liability associated with failure to do so could adversely affect our business, financial condition and results of operations. We are subject to various domestic and international privacy and security regulations, including but not limited to The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”). HIPAA mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. In addition, many states have enacted comparable laws addressing the privacy and security of health information, some of which are more stringent than HIPAA.
| -27- |
Fluctuations in foreign currency exchange rates may cause fluctuations in our financial results.
For the year ended December 31, 2015, we derived 100% of our total revenues from continuing operations from transactions in U.S. dollars, but have 24% of our cash and cash equivalents, and 28% of our marketable securities, and the majority of our expenses denominated in Euros. Our functional currency is the Euro and our reporting currency is the U.S. Dollar. As a result, both our actual and reported financial results could be significantly affected by fluctuations of the Euro relative to the U.S. dollar. We do not currently engage in substantial hedging activities with respect to the risk of exchange rate fluctuations, but we expect to implement hedging activities to manage exchange rate risk in the future.
Uncertainty remains about the ability of certain EU Member States to continue to service their sovereign debt obligations. This debt crisis and the related financial restructuring efforts may cause the value of the Euro to deteriorate, reducing the value of the Euro relative to the U.S. Dollar. Any strengthening in the U.S. Dollar relative to the Euro would have a negative effect on our balance sheet while a weakening in the U.S. Dollar relative to the Euro would have a positive effect. If global economic and market conditions, or economic conditions in the European Union, the U.S. or other key markets, remain uncertain, persist or deteriorate further, our business, financial condition, results of operations and cash flows may be adversely affected.
We may not maintain an effective system of internal control over financial reporting, which could harm our business and financial results.
During its evaluation of the effectiveness of internal control over financial reporting as of December 31, 2015, our management identified material weaknesses related to: lack of sufficient personnel, resulting in, among other things, a failure to implement a proper segregation of duties; ineffective controls over the revenue, income tax, and financial close processes; ineffective controls over information technology and key spreadsheets used in preparing financial statements; and ineffective monitoring of our internal control systems. See Item 9A. Disclosure Controls and Procedures. While we have begun to implement measures that we believe are necessary and appropriate to address these material weaknesses, we cannot assure you that our efforts will prove wholly successful in remediating these material weaknesses. While we have not incurred and do not expect to incur material expenses specifically related to the remediation of these material weaknesses, actual expenses may exceed our current estimates and may be material. In addition, we cannot assure you that we have identified all these material weaknesses, or that we will not identify other these material weaknesses in the future. If we are unable to successfully identify and remediate any material weakness that may exist in our internal control over financial reporting, the accuracy and timing of our financial reporting may be adversely affected, we may be unable to maintain compliance with securities law requirements and applicable stock exchange listing requirements regarding timely filing of periodic reports, our stock price may decline, and we could be subject to shareholder litigation.
Our effective tax rate could be highly volatile and could adversely affect our operating results.
Our future effective tax rate may be adversely affected by a number of factors, many of which are outside of our control, including:
| · | the jurisdictions in which profits are determined to be earned and taxed; |
| · | adjustments to estimated taxes upon finalization of various tax returns; |
| · | increases in expenses not deductible for tax purposes, including write-offs of acquired in-process R&D and impairment of goodwill in connection with acquisitions; |
| · | changes in available tax credits; |
| · | changes in share-based compensation expense; |
| · | changes in the valuation of our deferred tax assets and liabilities; |
| · | changes in domestic or international tax laws or the interpretation of such tax laws; |
| · | the resolution of issues arising from tax audits with various tax authorities; |
| · | the tax effects of purchase accounting for acquisitions that may cause fluctuations between reporting periods; and |
| · | taxes that may be incurred upon a repatriation of cash from foreign operations. |
| · | Any significant increase in our future effective tax rates could impact our results of operations for future periods adversely. |
We depend upon consultants, advisors and outside contractors extensively in important roles within our Company.
We outsource many key functions of our business and therefore rely on a substantial number of consultants, advisors and outside contractors. If we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our development activities may be extended, delayed or terminated which would have an adverse effect on our development program and our business.
We depend on key personnel to execute our business plan. If we cannot attract and retain key personnel, we may not be able to successfully implement our business plan.
Our success depends in large part upon our ability to attract and retain highly qualified personnel. During our operating history, we have assigned many key responsibilities within our Company to a relatively small number of individuals, each of whom has played key roles in executing various important components of our business. We do not maintain material key person life insurance for any of our key personnel. If we lose the services of Mr. Anderson, our Chief Executive Officer, or other members of our senior executive team, we may have difficulty executing our business plan in the manner we currently anticipate. Further, because each of our key personnel is involved in numerous roles in various components of our business, the loss of any one or more of such individuals could have an adverse effect on our business.
| -28- |
Risks Relating to Regulatory and Legal Matters
Products that incorporate our drug delivery platforms and other products we may develop are subject to regulatory approval. If we or our pharmaceutical and biotechnology company partners do not obtain such approvals, or if such approvals are delayed, our revenues may be adversely affected.
Although products that incorporate our drug delivery platforms and other products we may develop may appear promising, in particular at their early stages of development and in clinical trials, none of these potential platforms or products may gain regulatory approval and reach the commercial market for a variety of reasons.
In the U.S., federal, state and local government agencies, primarily the FDA, regulate all pharmaceutical products, including existing products and those under development. We cannot control, and our pharmaceutical and biotechnology partners cannot control, the timing of regulatory approval for any of these products, or if approval is obtained at all. We, or our partners, may experience significant delays in expected product releases while attempting to obtain regulatory approval for products incorporating our technologies. If we, or our partners, are not successful, our revenues and profitability may decline.
Applicants for FDA approval often must submit to the FDA extensive clinical and pre-clinical data, as well as information about product manufacturing processes and facilities and other supporting information. Varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of a drug product. The FDA also may require us, or our partners, to conduct additional pre-clinical studies or clinical trials. For instance, the FDA may require additional toxicology tests and clinical trials to confirm the safety and effectiveness of Medusa-based product candidates, which would impact development plans for product candidates. In addition, although Flamel has submitted a Drug Master File (“DMF”) for its lead Medusa polymer, the FDA may require additional information prior to the conduct of clinical trials or for commercialization of any product that uses our Medusa polymer and cross-references our DMF.
Similarly, although we anticipate submitting applications for approval for our development products that rely on existing data to demonstrate safety and effectiveness, FDA may determine that additional studies particular to our products are necessary. If FDA requires such additional data, it would impact development plans for those products.
Changes in FDA approval policy during the development period, or changes in regulatory review for each submitted new product application, also may delay an approval or result in rejection of an application. For instance, under the Food and Drug Administration Amendments Act of 2007 (“FDAAA”), we or our partners may be required to develop Risk Evaluations and Mitigation Strategies (“REMS”), to ensure the safe use of product candidates. If the FDA disagrees with our or our partners’ REMS proposals, it may be more difficult and costly for us, or our partners, to obtain regulatory approval for product candidates. Similarly, FDAAA provisions may make it more likely that the FDA will refer a marketing application for a new product to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. This review may add to the wait time for approval, and, although the FDA is not bound by the recommendation of an advisory committee, objections or concerns expressed by an advisory committee may cause the FDA to delay or deny approval.
The FDA has substantial discretion in the approval process and may disagree with our or our partners’ interpretations of data and information submitted in an application, which also could cause delays of an approval or rejection of an application. Even if the FDA approves a product, the approval may limit the uses or indications for which a product may be marketed, restrict distribution of the product or require further studies. With respect to Vazculep, the FDA has required the Company to conduct post-marketing non-clinical and clinical studies to be completed between 2016 and 2019.
The FDA may also withdraw product clearances and approvals for failure to comply with regulatory requirements or if problems follow initial marketing. In the same way, medicinal products for supply on the EU market are subject to marketing authorization by either the European Commission, following an opinion by the EMA, or by the competent authorities of EU Member States. Applicants for marketing authorization must submit extensive technical and clinical data essentially in the form of the ICH Common Technical Document. The data is subject to extensive review by the competent authorities and may be considered inappropriate or insufficient. If applications for marketing authorization by pharmaceutical and biotechnology company partners are delayed, or rejected, if the therapeutic indications for which the product is approved are limited, or if conditional marketing authorization imposing post-marketing clinical trials or surveillance is imposed, our revenues may decline and earnings may be negatively impacted.
Commercial products are subject to continuing regulation, and we on our own, and in conjunction with our pharmaceutical and biotechnology partners, may be subject to adverse consequences if we or they fail to comply with applicable regulations.
We on our own and in conjunction with our pharmaceutical and biotechnology partners will be subject to extensive regulatory requirements for our and the co-developed products and product candidates that incorporate our drug delivery platforms, even if the products receive regulatory approval. These regulations are wide-ranging and govern, among other things:
| -29- |
| · | adverse drug experiences and other reporting requirements; |
| · | product promotion and marketing; |
| · | active pharmaceutical ingredients and/or product manufacturing, including cGMP compliance; |
| · | record keeping; |
| · | distribution of drug samples; |
| · | required clinical trials and/or post-marketing studies; |
| · | authorization renewal procedures; |
| · | authorization variation procedures; |
| · | compliance with any required REMS; |
| · | updating safety and efficacy information; |
| · | processing of personal data; |
| · | use of electronic records and signatures; and |
| · | changes to product manufacturing or labeling. |
If we or our partners, including any CDMOs that we use, fail to comply with these laws and regulations, the FDA, the European Commission, competent authorities of EU Member States, or other regulatory organizations, may take actions that could significantly restrict or prohibit commercial distribution of our products and products that incorporate our technologies. If the FDA, the European Commission or competent authorities of EU Member States determine that we are not in compliance with these laws and regulations, they could, among other things:
| · | issue warning letters; |
| · | impose fines; |
| · | seize products or request or order recalls; |
| · | issue injunctions to stop future sales of products; |
| · | refuse to permit products to be imported into, or exported out of, the United States or the European Union; |
| · | suspend or limit our production; |
| · | withdraw or vary approval of marketing applications; |
| · | order the competent authorities of EU Member States to withdraw or vary national authorization; and |
| · | initiate criminal prosecutions. |
We are subject to U.S. federal and state laws prohibiting “kickbacks” and false claims that, if violated, could subject us to substantial penalties, and any challenges to or investigation into our practices under these laws could cause adverse publicity and be costly to respond to, and thus could harm our business.
We are subject to extensive and complex U.S. federal and state and international laws and regulations, including but not limited to, health-care “fraud and abuse” laws, such as anti-kickback and false claims laws and regulations pertaining to government benefit program reimbursement, price reporting and regulations, and sales and marketing practices. These laws and regulations are broad in scope and subject to evolving interpretations, which could require us to incur substantial costs associated with compliance or to alter one or more of our sales or marketing practices. In addition, violations of these laws, or allegations of such violations, could disrupt our business and result in a material adverse effect on our revenues, profitability, and financial condition. In the current environment, there appears to be a greater risk of investigations of possible violations of these laws and regulations. This is reflected by recent enforcement activity and pronouncements by the US Office of Inspector General of the Department of Health and Human Services that it intends to continue to vigorously pursue fraud and abuse violations by pharmaceutical companies, including through the potential to impose criminal penalties on pharmaceutical company executives. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Healthcare reform and restrictions on reimbursements may limit our financial returns.
Our ability to successfully commercialize our products and technologies may depend on the extent to which the government health administration authorities, the health insurance funds in the EU Member States, private health insurers and other third party payers in the U.S. will reimburse consumers for the cost of these products, which would affect the volume of drug products sold by pharmaceutical and biotechnology companies that incorporate our technology into their products. Third party payers are increasingly challenging both the need for, and the price of, novel therapeutic drugs and uncertainty exists as to the reimbursement status of newly approved therapeutics. The commercial success of our products depends in part on the conditions under which products incorporating our technology are reimbursed. Adequate third party reimbursement may not be available for such drug products to enable us to maintain price levels sufficient to realize an appropriate return on our investments in research and product development, which could materially and adversely affect our business. We cannot predict the effect that changes in the healthcare system, especially cost containment efforts, may have on our business. In particular, it is difficult to predict the effect of health care reform legislation enacted in the U.S. in 2010, certain provisions of which are still subject to regulatory implementation, further legislative change and ongoing judicial review. Any such changes or changes due to future legislation governing the pricing and reimbursement of healthcare products in the EU Member States may adversely affect our business.
| -30- |
Regulatory reforms may adversely affect our ability to sell our products profitably.
From time to time, the US Congress, the Council of the European Union and the European Parliament, as well as the legislators of the EU Member States, adopt changes to the statutes that the FDA, the European Commission and the competent authorities of the EU Member States enforce in ways that could significantly affect our business. In addition, the FDA, the European Commission and the competent authorities of the EU Member States often issue new regulations or guidance, or revise or reinterpret their current regulations and guidance in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or FDA, EU or EU Member State’s regulations, guidance or interpretations changed, and what the impact of any such changes may be.
Any such changes could have a significant impact on the path to approval of products incorporating our drug delivery platforms, our products or of competing products, and on our obligations and those of our pharmaceutical and biotechnology company partners.
We and companies to which we have licensed, or will license our products or drug delivery platforms and subcontractors we engage for services related to the development and manufacturing of our products are subject to extensive regulation by the FDA and other regulatory authorities. Our and their failure to meet strict regulatory requirements could adversely affect our business.
We, and companies to which we license our products or drug delivery platforms, as well as companies acting as subcontractors for our product developments, including but not limited to non-clinical, pre-clinical and clinical studies, and manufacturing, are subject to extensive regulation by the FDA, other domestic regulatory authorities and equivalent foreign regulatory authorities, particularly the European Commission and the competent authorities of EU Member States. Those regulatory authorities may conduct periodic audits or inspections of the applicable facilities to monitor compliance with regulatory standards and we remain responsible for the compliance of our subcontractors. If the FDA or another regulatory authority finds failure to comply with applicable regulations, the authority may institute a wide variety of enforcement actions, including:
| · | warning letters or untitled letters; |
| · | fines and civil penalties; |
| · | delays in clearing or approving, or refusal to clear or approve, products; |
| · | withdrawal, suspension or variation of approval of products; product recall or seizure; |
| · | orders to the competent authorities of EU Member States to withdraw or vary national authorization; |
| · | orders for physician notification or device repair, replacement or refund; |
| · | interruption of production; |
| · | operating restrictions; |
| · | injunctions; and |
| · | criminal prosecution. |
Any adverse action by a competent regulatory agency could lead to unanticipated expenditures to address or defend such action and may impair the ability to produce and market applicable products, which could significantly impact our revenues and royalties that we receive from our customers.
We may face product liability claims related to clinical trials for our products or their misuse.
The testing, including through clinical trials, manufacturing and marketing, and the use of our products may expose us to potential product liability and other claims. If any such claims against us are successful, we may be required to make significant compensation payments. Any indemnification that we have obtained, or may obtain, from Contract Research Organizations (“CROs”) or pharmaceutical and biotechnology companies or hospitals conducting human clinical trials on our behalf may not protect us from product liability claims or from the costs of related litigation. Insurance coverage is expensive and difficult to obtain, and we may be unable to obtain coverage in the future on acceptable terms, if at all. We currently maintain general liability insurance with a limit of €10 million and product liability and recall insurance with a limit of €10 million. We cannot be certain that the coverage limits of our insurance policies or those of our strategic partners will be adequate. If we are unable to obtain sufficient insurance at an acceptable cost, a product liability claim or recall could adversely affect our financial condition. Similarly, any indemnification we have obtained, or may obtain, from pharmaceutical and biotechnology companies with whom we are developing, or will develop, our products may not protect us from product liability claims from the consumers of those products or from the costs of related litigation.
If we use hazardous biological and/or chemical materials in a manner that causes injury, we may be liable for significant damages.
Our R&D activities involve the controlled use of potentially harmful biological and/or chemical materials, and are subject to U.S., state, EU, national and local laws and regulations governing the use, storage, handling and disposal of those materials and specified waste products. We cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling or disposal of these materials, including fires and/or explosions, storage tank leaks and ruptures and discharges or releases of toxic or hazardous substances. These operating risks can cause personal injury, property damage and environmental contamination, and may result in the shutdown of affected facilities and the imposition of civil or criminal penalties. The occurrence of any of these events may significantly reduce the productivity and profitability of a particular manufacturing facility and adversely affect our operating results.
| -31- |
We currently maintain property, business interruption and casualty insurance with aggregate maximum limits of €60 million, which are limits that we believe to be commercially reasonable, but may be inadequate to cover any actual liability or damages.
Risks Relating to Ownership of Our Securities
Our share price has been volatile and may continue to be volatile.
The trading price of our shares has been, and is likely to continue to be, highly volatile. The market value of an investment in our shares may fall sharply at any time due to this volatility. During the year ended December 31, 2015, the closing sale price of our ADSs as reported on the NASDAQ National Market ranged from $11.50 to $25.69. During the year ended December 31, 2014, the closing sale price of our ADSs as reported on the NASDAQ National Market ranged from $8.15 to $18.89. The market prices for securities of drug delivery, specialty pharma, biotechnology and pharmaceutical companies historically have been highly volatile. Factors that could adversely affect our share price include, among others:
| · | fluctuations in our operating results; |
| · | announcements of technological partnerships, innovations or new products by us or our competitors; |
| · | actions with respect to the acquisition of new or complementary businesses; |
| · | governmental regulations; |
| · | developments in patent or other proprietary rights owned by us or others; |
| · | public concern as to the safety of drug delivery platforms developed by us or drugs developed by others using our platform; |
| · | the results of pre-clinical testing and clinical studies or trials by us or our competitors; |
| · | adverse events related to our products or products developed by pharmaceutical and biotechnology company partners that use our drug delivery platforms; |
| · | lack of efficacy of our products; |
| · | litigation; |
| · | decisions by our pharmaceutical and biotechnology company partners relating to the products incorporating our technologies; |
| · | actions by the FDA, the EMA or national authorities of EU Member States in connection with submissions related to the products incorporating our technologies; |
| · | the perception by the market of biotechnology and high technology companies generally; and |
| · | general market conditions, including the impact of the current financial environment. |
If we are not profitable in the future, the value of our shares may fall.
Prior to 2015, we had a history of operating losses and have accumulated aggregate net loss from our inception of approximately $281.5 million through December 31, 2015. If we are unable to earn a profit in future periods, the market price of our stock may fall. The costs for R&D of our products and drug delivery platforms and general and administrative expenses have been the principal causes of our net losses in recent years. Our ability to operate profitably depends upon a number of factors, many of which are beyond our direct control. These factors include:
| · | the demand for our drug delivery platforms and products; |
| · | the level of product and price competition; |
| · | our ability to develop new partnerships and additional commercial applications for our products; |
| · | our ability to control our costs; |
| · | our ability to broaden our customer base; |
| · | the effectiveness of our marketing strategy; |
| · | the effectiveness of our partners’ marketing strategy for products that use our technology; and |
| · | general economic conditions. |
We may require additional financing, which may not be available on favorable terms or at all, and which may result in dilution of our shareholders’ equity interest.
We may require additional financing to fund the development and possible acquisition of new products and businesses. We may consume available resources more rapidly than currently anticipated, resulting in the need for additional funding. If we cannot obtain financing when needed, or obtain it on favorable terms, we may be required to curtail our plans to continue to develop drug delivery platforms, develop new products, or acquire additional products and businesses. Other factors that will affect future capital requirements and may require us to seek additional financing include:
| · | the development and acquisition of new products and drug delivery platforms; |
| · | the progress of our research and product development programs; |
| -32- |
| · | results of our partnership efforts with potential pharmaceutical and biotechnology company partners; and |
| · | the timing of, and amounts received from, future product sales, product development fees and licensing revenue and royalties. |
If adequate funds are not available, we may be required to significantly reduce or refocus our product development efforts, resulting in loss of sales, increased costs and reduced revenues. Alternatively, to obtain needed funds for acquisitions or operations, we may choose to issue shares of our common stock or preferred stock, either through public or private financings. Additional funds may not be available on terms that are favorable to us and, in the case of such equity financings, may result in dilution to our stockholders.
We currently do not intend to pay dividends and cannot assure shareholders that we will make dividend payments in the future.
We have never declared or paid a cash dividend on any of our capital stock and do not anticipate declaring cash dividends in the foreseeable future. Declaration of dividends on our shares will depend upon, among other things, future earnings, if any, the operating and financial condition of our business, our capital requirements, general business conditions and such other factors as our Board of Directors deems relevant.
Judgments of United States courts, including those predicated on the civil liability provisions of the federal securities laws of the United States, may not be enforceable in French courts.
An investor in the U.S. may find it difficult to:
| · | effect service of process within the U.S. against us and our non-U.S. resident directors and officers; |
| · | enforce United States court judgments based upon the civil liability provisions of the United States federal securities laws against us and our non-U.S. resident directors and officers in France; or |
| · | bring an original action in a French court to enforce liabilities based upon the U.S. federal securities laws against us and our non-U.S. resident directors and officers. |
Holders of ADSs have fewer rights than shareholders and have to act through the Depositary to exercise those rights.
Holders of ADSs do not have the same rights as shareholders and, accordingly, cannot exercise rights of shareholders against us. The Bank of New York Mellon, as depositary, or the “Depositary”, is the registered shareholder of the deposited shares underlying the ADSs. Therefore, holders of ADSs will generally have to exercise the rights attached to those shares through the Depositary. We will use reasonable efforts to request that the Depositary notify the holders of ADSs of upcoming votes and ask for voting instructions from them. If a holder fails to return a voting instruction card to the Depositary by the date established by the Depositary for receipt of such voting instructions, or if the Depositary receives an improperly completed or blank voting instruction card, or if the voting instructions included in the voting instruction card are illegible or unclear, then such holder will be deemed to have instructed the Depositary to vote its shares, and the Depositary shall vote such shares in favor of any resolution proposed or approved by our Board of Directors and against any resolution not so proposed or approved.
Preferential subscription rights may not be available for U.S. persons.
Under French law, shareholders have preferential rights to subscribe for cash issuances of new shares or other securities giving rights to acquire additional shares on a pro rata basis. U.S. holders of our securities (which might not be shares but ADSs) may not be able to exercise preferential subscription rights for their securities unless a registration statement under the Securities Act is effective with respect to such rights or an exemption from the registration requirements imposed by the Securities Act is available. We may, from time to time, issue new shares or other securities giving rights to acquire additional shares (such as warrants) at a time when no registration statement is in effect and no Securities Act exemption is available. If so, United States holders of our securities will be unable to exercise any preferential rights and their interests will be diluted. We are under no obligation to file any registration statement in connection with any issuance of new shares or other securities.
For holders of our shares in the form of ADSs, the Depositary may make these rights or other distributions available to holders in the United States if we instruct it to do so. If we fail to issue such instruction and the Depositary determines that it is impracticable to sell the rights, it may allow these rights to lapse. In that case, the holders will receive no value for them.
Our largest shareholders own a significant percentage of the share capital and voting rights of the Company.
As of February 16, 2016, Broadfin Capital and certain of its affiliates beneficially owned approximately 10.85% of our outstanding shares (in the form of ADRs), Janus Capital Management, LLC and certain of its affiliates beneficially owned 10.72% of our outstanding shares (in the form of ADRs) and Deerfield Capital and certain of its affiliates beneficially owned approximately 10.06% of our outstanding shares (in the form of ADRs). To the extent these shareholders continue to hold a large percentage of our share capital and voting rights, they will remain in a position to exert heightened influence in the election of the directors of the Company and in other corporate actions that require shareholder approval, including change of control transactions.
| Item 1B. | Unresolved Staff Comments |
None.
| -33- |
| Item 2. | Properties |
Our corporate headquarters and the research center are located in Venissieux, France (a suburb of Lyon) in four adjacent leased facilities totaling approximately 58,000 square feet. One building of approximately 12,800 square feet houses administrative offices and analytical research laboratories. The lease on this facility expires in 2016. A second facility comprising approximately 12,800 square feet houses equipment dedicated to our Micropump, LiquiTime and Trigger Lock platforms has a lease which expired in 2015 and is expected to be renewed. The third facility of approximately 6,800 square feet houses analytical laboratories and the lease expires in 2016. The fourth facility of approximately 26,000 square feet houses research and biochemistry (Medusa) laboratories and quality/regulatory affairs.
We previously owned manufacturing facilities, of approximately 103,900 square feet, located in Pessac, France (“Pessac Facility”), which included (i) approximately 6,800 square feet used for the manufacture of Coreg CR® microparticles for GSK as well as other Micropump, and LiquiTime/Trigger Lock-based formulations (up to commercial scale; altogether the “Micropump Pilot Development facilities”) and housed two suites of equipment, as well as a dedicated warehouse, analytical control laboratory and a technical area with air compressor units, refrigeration units for solvents, and a heat boiler. This facility was divested to Recipharm on December 1, 2014 (for more detail, see Note 17: Discontinued Operations to the consolidated financial statements in Item II, Part 8 of this Annual Report on Form 10-K ).
We have commercial and administrative activities located in Chesterfield, Missouri. In November 2015, we relocated to new office space in Chesterfield, Missouri. The office space consists of 12,100 square feet, and the lease expires in 2020. We still maintain the lease on our former office space which expires in 2018. Additionally, we have commercial and administrative activities located in Charlotte, North Carolina. This office space consists of 6,300 square feet, and the lease expires in 2020
We have intellectual property, clinical, quality, regulatory, and supply chain activities located in Dublin, Ireland. The office space consists of 5,059 square feet and the lease expires in 2025.
During 2015, we expended $1.6 million on property and equipment essentially limited to maintenance and investment in a global ERP.
See “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II, Item 7 of this Annual Report on Form 10-K for more information regarding our investment activities and principal capital expenditures over the last three years.
| Item 3. | Legal Proceedings |
While we may be engaged in various claims and legal proceedings in the ordinary course of business, we are not involved (whether as a defendant or otherwise) in, and, we have no knowledge of any threat of, any litigation, arbitration or administrative or other proceeding that management believes will have a material adverse effect on our consolidated financial position or results of operations.
| Item 4. | Mine Safety Disclosures |
Not applicable.
| -34- |
| Item 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Common Stock Data (per share) (Unaudited):
The principal trading market for the Company’s securities in ADSs is the NASDAQ Global Market. There is no foreign trading market for the Company’s ordinary shares, ADSs or any other equity security issued by the Company. Each ADS represents one ordinary share, nominal value 0.122 Euros. Each ADS is evidenced by an ADR. The Bank of New York Mellon is the Depositary for the ADRs.
As of March 7, 2016, there were 40,234,506 ADSs outstanding, and the closing stock price of the Company was $8.67 per share.
The following table reports the high and low trading prices of the ADSs on the NASDAQ Market for the periods indicated.
| 2015 Price Range | 2014 Price Range | |||||||||||||||
| High | Low | High | Low | |||||||||||||
| First Quarter | $ | 18.47 | $ | 11.50 | $ | 14.70 | $ | 8.15 | ||||||||
| Second Quarter | 22.32 | 13.88 | 15.17 | 9.94 | ||||||||||||
| Third Quarter | 25.69 | 15.37 | 15.78 | 13.21 | ||||||||||||
| Fourth Quarter | 19.27 | 12.21 | 18.89 | 11.76 | ||||||||||||
Holders
As of March 7, 2016 there were 30 holders of record of our Ordinary Shares or ADSs. Because almost all of our ordinary shares are held by brokers, nominees and other institutions on behalf of shareholders, we are unable to estimate the total number of individual shareholders represented by these record holders.
Dividends
The Company has never declared or paid a cash dividend on any of its capital stock and does not anticipate declaring cash dividends in the foreseeable future.
Securities authorized for issuance under equity compensation plans
Information regarding our equity compensation plans is presented below as of December 31, 2015 (in thousands, except per share data).
| Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted-average exercise price per share of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | ||||||||||
| (a) | (b) | (c) | ||||||||||
| Stock options | 2,326 | $ | 13.84 | 97 | ||||||||
| Warrants | 668 | $ | 16.97 | 45 | ||||||||
| Free share awards | 226 | n/a | 250 | |||||||||
| -35- |
Stock performance graph
The following graph compares the cumulative 5-year return provided to stockholders of Flamel’s ADSs relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. We believe these indices are the most appropriate indices against which the total shareholder return of Flamel should be measured. The NASDAQ Biotechnology Index has been selected because it is an index of U.S. quoted biotechnology and pharmaceutical companies. An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our ADSs and in each of the indexes on January 1, 2011 and its relative performance is tracked through December 31, 2015. The comparisons shown in the graph are based upon historical data and we caution that the stock price performance shown in the graph is not indicative of, nor intended to forecast, the potential future performance of our stock.
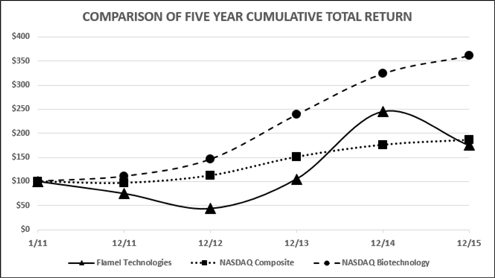
Notwithstanding anything to the contrary set forth in any of our filings under the Securities Act of 1933, or the Exchange Act that might incorporate future filings, including this annual report on Form 10-K, in whole or, in part, the performance graph presented above shall not be incorporated by reference into any such filings.
| -36- |
| Item 6. | Selected Financial Data (in thousands, except per share amounts) |
Annual Financial Data:
The following selected financial data should be read in conjunction with our consolidated financial statements and related notes and Item 7—Management's Discussion and Analysis of Financial Condition and Results of Operations in Part II of this Report.
| 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||||
| Statement of Income Data: | ||||||||||||||||||||
| Revenues | $ | 173,209 | $ | 14,775 | $ | 4,179 | $ | 7,534 | $ | 9,238 | ||||||||||
| Gross profit (a) | 162,288 | 11,392 | 3,617 | 20,241 | 26,316 | |||||||||||||||
| Operating income (loss) | 71,447 | (93,857 | ) | (53,700 | ) | (8,363 | ) | (9,583 | ) | |||||||||||
| Net income (loss) from continuing operations | 40,659 | (88,924 | ) | (46,509 | ) | (4,740 | ) | (11,449 | ) | |||||||||||
| Net income (loss) from discontinued operations | - | 4,018 | 3,584 | 1,512 | 2,675 | |||||||||||||||
| Net income (loss) | 40,659 | (84,906 | ) | (42,925 | ) | (3,228 | ) | (8,774 | ) | |||||||||||
| Earnings (loss) per share - Basic: | ||||||||||||||||||||
| Continuing operations | 1.00 | (2.45 | ) | (1.83 | ) | (0.19 | ) | (0.46 | ) | |||||||||||
| Discontinued operations | - | 0.11 | 0.14 | 0.06 | 0.11 | |||||||||||||||
| Net income (loss) | 1.00 | (2.34 | ) | (1.69 | ) | (0.13 | ) | (0.36 | ) | |||||||||||
| Earnings (loss) per share - Diluted: | ||||||||||||||||||||
| Continuing operations | 0.93 | (2.45 | ) | (1.83 | ) | (0.19 | ) | (0.46 | ) | |||||||||||
| Discontinued operations | - | 0.11 | 0.14 | 0.06 | 0.11 | |||||||||||||||
| Net income (loss) | 0.93 | (2.34 | ) | (1.69 | ) | (0.13 | ) | (0.36 | ) | |||||||||||
(a) Gross margin is computed by subtracting cost of products and services sold from total revenues.
| 2015 | 2014 | 2013 | 2012 | 2011 | ||||||||||||||||
| Balance Sheet Data: | ||||||||||||||||||||
| Cash and cash equivalents | $ | 65,064 | $ | 39,760 | $ | 6,636 | $ | 2,742 | $ | 3,456 | ||||||||||
| Marketable securities | 79,738 | 53,074 | 401 | 6,413 | 21,035 | |||||||||||||||
| Goodwill | 18,491 | 18,491 | 18,491 | 18,491 | - | |||||||||||||||
| Intangible assets, net | 15,825 | 28,389 | 40,139 | 41,589 | - | |||||||||||||||
| Total assets | 214,977 | 174,205 | 116,252 | 117,311 | 69,402 | |||||||||||||||
| Long-term debt (incl. current portion) | 1,118 | 3,717 | 30,249 | 10,409 | 3,715 | |||||||||||||||
| Long-term contingent consideration payable (incl. current portion) | 122,693 | 114,750 | 55,265 | 26,220 | - | |||||||||||||||
| -37- |
Quarterly Financial Data (Unaudited):
The following tables present certain unaudited consolidated quarterly financial information for each quarter of 2015 and 2014. Year-to-date Earnings (loss) per share amounts may be different than the sum of the applicable quarters due to differences in weighted average shares outstanding for the respective periods.
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| 2015: | ||||||||||||||||
| Revenues | $ | 32,726 | $ | 49,795 | $ | 47,320 | $ | 43,368 | ||||||||
| Gross profit (a) | 29,096 | 47,039 | 45,233 | 40,920 | ||||||||||||
| Operating income (loss) | 10,214 | (1,177 | ) | (14,479 | ) | 76,889 | ||||||||||
| Net income (loss) | 11,647 | (17,400 | ) | (29,685 | ) | 76,097 | ||||||||||
| Net income (loss) per share - Basic | 0.29 | (0.43 | ) | (0.73 | ) | 1.85 | ||||||||||
| Net income (loss) per share - Diluted | 0.27 | (0.43 | ) | (0.73 | ) | 1.75 | ||||||||||
| 2014: | ||||||||||||||||
| Revenues | $ | 4,578 | $ | 4,318 | $ | 2,913 | $ | 2,966 | ||||||||
| Gross profit (a) | 3,805 | 3,811 | 2,206 | 1,570 | ||||||||||||
| Operating loss | (24,231 | ) | (19,399 | ) | (16,739 | ) | (33,488 | ) | ||||||||
| Net loss from continuing operations | (26,861 | ) | (20,200 | ) | (9,980 | ) | (31,883 | ) | ||||||||
| Net income (loss) from discontinued operations | 223 | (873 | ) | (66 | ) | 4,734 | ||||||||||
| Net loss | (26,638 | ) | (21,073 | ) | (10,046 | ) | (27,149 | ) | ||||||||
| Earnings (loss) per share - Basic: | ||||||||||||||||
| Continuing operations | (0.95 | ) | (0.53 | ) | (0.26 | ) | (0.81 | ) | ||||||||
| Discontinued operations | 0.01 | (0.02 | ) | - | 0.12 | |||||||||||
| Net income (loss) | (0.94 | ) | (0.55 | ) | (0.26 | ) | (0.69 | ) | ||||||||
| Earnings (loss) per share - Diluted: | ||||||||||||||||
| Continuing operations | (0.95 | ) | (0.53 | ) | (0.26 | ) | (0.81 | ) | ||||||||
| Discontinued operations | 0.01 | (0.02 | ) | - | 0.12 | |||||||||||
| Net income (loss) | (0.94 | ) | (0.55 | ) | (0.26 | ) | (0.69 | ) | ||||||||
(a) Gross margin is computed by subtracting cost of products and services sold from total revenues.
The quarter ended September 30, 2015 includes the unfavorable correction to revenue of $1,193, which should have been recorded in the second quarter of 2015, and is due to the fact that the Company incorrectly recorded revenue in the second quarter of 2015 as a result of improper reconciliation to revenue data communicated by service providers.
The quarter ended December 31, 2015 includes the unfavorable correction to revenue of $1,200 with respect to the Company’s rebate calculations which should have been recorded in the third quarter of 2015.
The quarter ended December 31, 2015 includes the favorable correction to net income (loss) of $1,969 with respect to tax benefits from stock-based compensation which should have been recorded in prior periods ($359 in 2012, $333 in 2013, $(692) in the first quarter of 2014, and $618, $814 and $537 in the first, second and third quarter of 2015 respectively).
The impact of these errors was not material to any prior periods.
In addition, the cumulative impact of the corrections was not material to the Company's consolidated financial statements for
the quarter ended September 30, 2015, the quarter ended December 31, 2015 or the year ended December 31, 2015.
| -38- |
| Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations. |
Management's Discussion And Analysis
(In Thousands, Except Per Share Data)
You should read the discussion and analysis of our financial condition and results of operations set forth in this Item 7 together with our consolidated financial statements and the related notes appearing elsewhere in this annual report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this annual report on Form 10-K, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties, and reference is made to the “Cautionary Disclosure Regarding Forward-Looking Statements” set forth immediately following the Table of Content of this annual report on Form 10-K for further information on the forward looking statements herein. In addition, you should read the “Risk Factors” section of this annual report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis and elsewhere in this annual report on Form 10-K.
Overview
We are a specialty pharmaceutical company utilizing core competencies in drug delivery and formulation to develop safer and more efficacious pharmaceutical products to address unmet medical needs and/or reduce overall healthcare costs. Flamel has a balanced business model consisting of a successful previously Unapproved Marketed Drugs (“UMDs”) business with two marketed products in the USA, Bloxiverz and Vazculep, and a branded development business, focusing on the development of products utilizing Flamel’s proprietary drug delivery platforms. The branded products are based on proprietary drug delivery platforms and target high-value solid oral and alternative dosage forms using 505(b)(2) and Biosimilar pathways where the Company can develop strong intellectual property positions and deliver meaningful patient benefits. Flamel’s business model allows the Company to select, develop, seek approval for, and commercialize niche branded and generic products, initially targeted for the U.S. market. The Company is able to self-fund the development of most product development opportunities.
Strategy
The Company's business strategy is designed to drive overall sales and earnings growth while maintaining a return on invested capital at an appropriate premium above the Company's cost of capital. Our key areas of focus address the most significant opportunities and challenges facing the Company, including:
| · | Unapproved Marketed Drug Development: The Company now derives cash flow and profitability from the sales of two of its UMD products. During 2015 the Company generated $172,488 of sales from the UMD products compared to $11,993 in 2014. The first UMD product, Bloxiverz, which had sales of $150,283 in 2015 was approved by the FDA on May 31, 2013, and is currently being marketed in the U.S. The second UMD product, Vazculep, which had sales of $20,251 in 2015, was approved by the FDA on June 27, 2014 and launched in October, 2014 in the USA. Both products are commercialized in the USA by Flamel’s subsidiary Éclat. These sales were derived from the acquisition of Éclat, which has focused on pursuing FDA approvals through the 505(b)(2) regulatory pathway. Through our acquisition of Éclat we obtained marketing and licensing knowledge of the commercial and regulatory process in the U.S. and EU. We believe this knowledge has enhanced our ability to identify product candidates for development, leverage new opportunities for the application of our drug delivery platforms, and license and market products in the U.S and EU. The revenues from these UMD products are now generating cash flow which we can use to fund our second strategy, the development and commercialization of our drug delivery products. |
| · | Development and Commercialization of the Company’s Drug Delivery Pipeline Products: In addition to the UMD strategy, the Company is continuing to advance the commercialization of its innovative drug delivery platforms. We have now enhanced our ability to identify new product candidates and to pursue commercial opportunities associated with our drug delivery platforms. The Company’s drug delivery platforms allow the creation of competitive and differentiated drug product profiles (e.g., with improved pharmacokinetics, efficacy and/or safety). Flamel owns and develops drug delivery platforms that address key formulation challenges, leading to the development of differentiated drug products for administration in various forms (e.g., capsules, tablets, sachets or liquid suspensions for oral use; or injectables for subcutaneous administration) and can be applied to a broad range of drugs (novel, already-marketed, or off-patent). These product development opportunities allow us to protect our products through patent protection and product differentiation. As a result of developing its own drug delivery platforms the Company’s business is now less dependent on the development activities performed by partners, and relies more on the development of its own, self-funded, products. Our proprietary drug delivery platforms include: |
| o | Micropump® is a microparticulate system that allows the development and marketing of modified and/or controlled release of solid, oral dosage formulations of drugs (Micropump®-carvedilol and Micropump®-aspirin formulations have been approved in the U.S. and in the E.U., respectively. |
| o | LiquiTime® allows development of modified/controlled release oral products in a liquid suspension formulation particularly suited to children or for patients having issues swallowing tablets or capsules. |
| -39- |
| o | Trigger Lock™ allows development of abuse-resistant modified/controlled release formulations of narcotic/opioid analgesics and other drugs susceptible to abuse. |
| o | Medusa™ allows the development of extended/modified release of injectable dosage formulations of drugs (e.g., peptides, polypeptides, proteins, and small molecules). |
Several products formulated using our proprietary drug delivery platforms are currently under various stages of development for possible marketing either by the Company and/or by partners via licensing/distribution agreements:
The key elements of our pipeline strategy include:
| o | Continuing to build commercially successful products utilizing Micropump; |
| o | Identifying and optimizing time-to-market for our (not yet approved) drug delivery platforms, i.e., LiquiTime, Trigger Lock and Medusa; |
| o | Maximizing the technical potential of our existing drug delivery platforms for developing new and proprietary products; |
| o | Developing and validating additional drug delivery platforms utilizing our current drug delivery platforms |
| · | Inorganic growth through Acquisitions and/or Partnerships: The Company maintains a strong balance sheet with substantial liquidity and little long term debt. As part of its overall enterprise strategy, the company expects to explore and pursue appropriate inorganic growth opportunities that complement its drug delivery platforms or acquire proprietary products that enhance profitability and cash flow. This was evidenced in early 2016 with the acquisition of FSC Holdings, LLC, a Charlotte, NC-based specialty pharmaceutical company, dedicated to providing innovative solutions to unmet medical needs for pediatric patients. Additionally, the Company will leverage the capabilities of its existing and future proprietary products and/or drug delivery platforms with pharmaceutical and biotechnology partnerships or licensing transactions. In 2015 the Company completed a licensing transaction for its LiquiTime technology based-OTC products which was licensed to Elan Pharma International Limited. |
Key Business Trends and Highlights
In operating our business and monitoring our performance, we consider a number of performance measures, as well as trends affecting our industry as a whole, which include the following:
| · | Healthcare and Regulatory reform: Various health care reform laws in the United States may impact our ability to successfully commercialize our products and technologies. The success of our commercialization efforts may depend on the extent to which the government health administration authorities, the health insurance funds in the EU Member States, private health insurers and other third party payers in the U.S. will reimburse consumers for the cost of healthcare products and services. |
| · | Pricing Environment for Pharmaceuticals: The pricing environment continues to be in the spotlight of many regulators. As a result the need to obtain and maintain appropriate pricing and reimbursement for our products may become more challenging due to, among other things, the attention being paid to healthcare cost containment and other austerity measures in the U.S. and worldwide. |
| · | Generics playing a larger role in healthcare: Generic pharmaceutical products will continue to play a large role in the US healthcare system. Specifically the Company has seen additional generic competition to its products and continues to expect generic competition in the future. |
| · | Access to and Cost of Capital: The recent tightening of credit in the US may create challenges for the Company if it were to have the need to raise capital. Currently the Company has no needs to raise capital. |
Highlights of our consolidated results for the year ended December 31, 2015 are as follows:
| · | Revenue was $173,209 for the year ended December 31, 2015 compared to $14,775 in the same period last year. This substantial increase was the result of product launches in 2014 which had a full year’s worth of revenues in 2015. |
| · | Operating income was $71,447 compared to an operating loss of ($93,857) in 2014. |
| · | Net Income was $40,659 compared to a net loss of ($84,906) in 2014. |
| · | Diluted net income per share was $0.93, compared to diluted net loss of ($2.34) in 2014. |
| · | Cash and marketable securities increased $51,968 to $144,802 from $92,834 at December 31, 2014. |
Critical Accounting Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to use judgment in making estimates and assumptions that affect the reported amounts of assets and liabilities, disclosures of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of sales and expenses during the periods presented. Actual results could differ from those estimates under different assumptions or conditions.
The following accounting policies are based on, among other things, judgments and assumptions made by management that include inherent risks and uncertainties. Management's estimates are based on the relevant information available at the end of each period.
| -40- |
Revenue. Revenue includes sales of pharmaceutical products, upfront licensing fees, milestone payments for R&D achievements, and compensation for the execution of R&D activities.
Product Sales and Services
Revenue is generally realized or realizable and earned when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the seller’s price to the buyer is fixed or determinable, and collectability is reasonably assured. The Company records revenue from product sales when title and risk of ownership have been transferred to the customer, which is typically upon delivery to the customer and when the selling price is determinable. As is customary in the pharmaceutical industry, the Company’s gross product sales are subject to a variety of deductions in arriving at reported net product sales. When the Company recognizes revenue from the sale of its products, an estimate of provision for sales return and allowances is recorded which reduces product sales. These adjustments include estimates for product returns, chargebacks, payment discounts and other sales allowances and rebates. The estimate for chargebacks is determined when product is shipped from the wholesalers to their customers. The return allowance, when estimable, is based on an analysis of the historical returns of the product or similar products.
For generic products and branded products sold in mature and stable markets where changes in selling price are rare, the Company recognizes revenues upon shipment. For products where market conditions remain volatile and selling price is subject to changes, which is the Company’s situation in 2015, 2014 and 2013, the Company delays revenue recognition until the wholesaler sells the product to its customers. For new product launches the Company recognizes revenue once sufficient data is available to determine product acceptance in the marketplace such that product returns may be estimated based on historical data and there is evidence of reorders and consideration is made of wholesaler inventory levels. Net product sales of wholesalers to their customers are determined using sales data from an independent, renowned wholesaler inventory tracking service. Net sales of wholesalers to their customers are calculated by deducting estimates for returns for wholesaler customers, chargebacks, payment discounts and other sales or discounts offered from the applicable gross sales value. Estimates for product returns are adjusted periodically based upon historical rates of returns, inventory levels in the distribution channel and other related factors.
License and Research Revenue
Where agreements have more than one deliverable, a determination is made as to whether the license and R&D elements should be recognized separately or combined into a single unit of account in accordance with ASU 2009-13, Revenue with Multiple Deliverables.
The Company uses a Multiple Attribution Model, referred to as the milestone-based method:
| · | As milestones relate to discrete development steps (i.e., can be used by the partners to decide whether to continue the development under the agreement), the Company views that milestone events have substance and represent the achievement of defined goals worthy of the payments. Therefore, milestone payments based on performance are recognized when the performance criteria are met and there are no further performance obligations. |
| · | Non-refundable technology access fees received from collaboration agreements that require the Company's continuing involvement in the form of development efforts are recognized as revenue ratably over the development period. |
| · | R&D work is compensated at a non-refundable hourly rate for a projected number of hours. Revenue on such agreements is recognized at the hourly rate for the number of hours worked as the R&D work is performed. Costs incurred under these contracts are considered costs in the period incurred. Payments received in advance of performance are recorded as deferred revenue and recognized in revenue as services are rendered. |
When Flamel receives revenue under signed feasibility study agreements, revenue is then recognized over the term of the agreement as services are performed.
R&D. Research and development expenses consist primarily of costs related to clinical studies and outside services, personnel expenses, and other research and development costs. Clinical study and outside services costs relate primarily to services performed by clinical research organizations and related clinical or development manufacturing costs, materials and supplies, filing fees, regulatory support, and other third party fees. Personnel expenses relate primarily to salaries, benefits and stock-based compensation. Other research and development expenses primarily include overhead allocations consisting of various support and facilities-related costs. R&D expenditures are charged to operations as incurred.
The Company recognizes R&D tax credits received from the French government for spending on innovative R&D as an offset of R&D expenses.
The Company does not track fully-burdened research and development expenses on a project-by-project basis. We manage our R&D expense by identifying the research and development activities we anticipate will be performed during a given period and then prioritizing efforts based on scientific data, probability of successful development, market potential, available human and capital resources and other similar considerations. We continually review our pipeline and the development status of product candidates and, as necessary, reallocate resources among the research and development portfolio that we believe will best support the future growth of our business.
Long-Lived Assets. Long-lived assets include fixed assets and intangible assets. Intangible assets consist primarily of purchased licenses and intangible assets corresponding to acquired, in progress R&D recognized as part of the Éclat acquisition purchase price allocation. Acquired IPR&D has an indefinite life and is not amortized until completion and development of the project, at which time the IPR&D becomes an amortizable asset. Amortization of acquired IPR&D is computed using the straight-line method over estimated useful life of the assets.
| -41- |
Long-lived assets are reviewed for impairment whenever conditions indicate that the carrying value of the assets may not be fully recoverable. Such impairment tests are based on a comparison of the pretax undiscounted cash flows expected to be generated by the asset to the recorded value of the asset. If impairment is indicated, the asset value is written down to its market value if readily determinable or its estimated fair value based on discounted cash flows. Any significant unanticipated changes in business or market conditions that vary from current expectations could have an impact on the fair value of these assets and any potential associated impairment. There were no indications of impairment as of December 31, 2015 or 2014.
Contingent Consideration. The acquisition-related contingent consideration payable arising from the acquisition of Éclat Pharmaceuticals are accounted at fair-value (see Note 8 : Long-Term Contingent Consideration Payable). The fair value of warrant consideration is estimated on a quarterly basis using a Black-Scholes option pricing model, and the fair value of earn-out payment consideration is estimated on a quarterly basis using a discounted cash flow model based on probability-adjusted annual gross profit of the specified Éclat Pharmaceuticals products at an appropriate discount rate. Changes in fair value are recorded in the consolidated statements of operations within operating expenses as changes in fair value of related party acquisition-related contingent consideration.
The Company elected the fair value option for the measurement of the financing-related contingent consideration payable associated with the Deerfield and Broadfin Royalty Agreements (see Note 8 : Long-Term Contingent Consideration Payable). The fair value of royalty agreement consideration is estimated on a quarterly basis using a discounted cash flow model based on probability-adjusted annual revenues of the specified Éclat Pharmaceuticals products at an appropriate discount rate. Changes in fair value are recorded in the consolidated statements of operations as Interest expense - changes in fair value of related party financing-related contingent consideration.
Discontinued Operations. The Company followed the guidance in Financial Accounting Standards Board Accounting Standards Codification (ASC) Topic 205 Presentation of Financial Statements (ASC 205), Topic 360 Property, Plant and Equipment (ASC 360) and Accounting Standards Update (ASU 2014-08), Reporting of Discontinued Operations and Disclosures of Disposals of Components of an Entity in determining the accounting for the divestiture of the Pessac facility. In 2014, the Company opted to early adopt the provisions of ASU 2014-08 as management believed that all criteria for presenting the disposal of Pessac Facility and its business as a discontinued operation were met, and that presenting the disposal as a discontinued operation would better reflect the ongoing operations of the entity.
The divestiture of the Pessac facility represented a strategic shift that had and will have a major effect on the Company’s operations and financial results. Since 2012, the Company’s business model of combining novel, high-value internally developed products with its leading drug delivery capabilities and commercializing niche branded and general pharmaceutical products. Previously, the Company’s focus was to develop and license its proprietary drug delivery platforms (Micropump®, LiquiTime®, Trigger Lock™ and Medusa™) with pharmaceutical companies and biotechnology partners (e.g. the licensing of Micropump® to GSK to develop Coreg CR® with GSK bringing and commercializing the product to market). The divestiture of Pessac Facility to Recipharm and the transfer to Recipharm of the GSK’s Supply Agreement and royalty income relating to Coreg CR ® is an implementation of this revised strategy. The Company is reducing its sole reliance on products developed with partners, explaining the transfer of its rights and obligations pertaining to Coreg CR®, including the Pessac Facility. Flamel sold over 50% of its historical revenues as a result of this transaction which has a major impact on the Company’s operations and results.
The divestiture of the Pessac facility was accomplished in a single transaction and the assets, contracts and liabilities referred to in the Asset Purchase Agreement signed between Flamel and Recipharm were determined to represent a disposal group. This disposal group was considered to be a component of the Company. While the Pessac Facility and its related business were not identified as reportable segment or operating segment, as the Company operates in only one segment, the Pessac Facility and its related business is considered to be an asset group as the transferred assets, liabilities and contracts represent the lowest level for which identifiable cash flows are largely independent of the cash flows of other group of assets and liabilities. The Company transferred all future cash outflows and inflows relating to the Pessac Facility that can be clearly distinguished operationally and for financial reporting purposes.
The results of discontinued operations, less income taxes, have been reported as a separate component of income in the consolidated statements of operations. The assets and liabilities of the discontinued operation have been reported separately in the asset and liability sections of the consolidated statements of financial position for the periods presented in the statement. Note 17: Discontinued Operations contains a description of the facts and circumstances related to the disposal, the gain and loss on disposal and the specific line items included in the consolidated statements of operations, financial position and cash flows relative to the disposal group.
Foreign Currency Translation. The reporting currency of the Company and its wholly-owned subsidiaries is the U.S. dollar. Each of the Company's non-U.S. subsidiaries and the parent entity use local currency as their functional currency. Subsidiaries and entities that do not use the U.S. Dollar as their functional currency translate 1) profit and loss accounts at the weighted average exchange rates during the reporting period, 2) assets and liabilities at period end exchange rates and 3) shareholders' equity accounts at historical rates. Resulting translation gains and losses are included as a separate component of stockholders' equity in AOCI. Assets and liabilities denominated in a currency other than the subsidiary's functional currency are translated to the subsidiary's functional currency at period end exchange rates. Resulting gains and losses are recognized in the consolidated statements of income.
| -42- |
Results of Operations
The following is a summary of our financial results (in thousands, except per share amounts):
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Product sales and services | $ | 172,488 | $ | 11,993 | $ | 1,153 | $ | 160,495 | 1,338.2 | % | $ | 10,840 | 940.2 | % | ||||||||||||||
| License and research revenue | 721 | 2,782 | 3,026 | (2,061 | ) | (74.1 | )% | (244 | ) | (8.1 | )% | |||||||||||||||||
| Total revenues | 173,209 | 14,775 | 4,179 | 158,434 | 1,072.3 | % | 10,596 | 253.6 | % | |||||||||||||||||||
| Cost of products and services sold | 10,921 | 3,383 | 562 | 7,538 | 222.8 | % | 2,821 | 502.0 | % | |||||||||||||||||||
| Research and development expenses | 25,608 | 17,298 | 15,966 | 8,310 | 48.0 | % | 1,332 | 8.3 | % | |||||||||||||||||||
| Selling, general and administrative expenses | 21,712 | 15,698 | 13,216 | 6,014 | 38.3 | % | 2,482 | 18.8 | % | |||||||||||||||||||
| Intangible asset amortization | 12,564 | 11,749 | - | 815 | 6.9 | % | 11,749 | n/a | ||||||||||||||||||||
| Changes in fair value of acquisition-related contingent consideration | 30,957 | 57,491 | 28,135 | (26,534 | ) | (46.2 | )% | 29,356 | 104.3 | % | ||||||||||||||||||
| Loss on early repayment of related party acquisition-related note | - | 3,013 | - | (3,013 | ) | (100.0 | )% | 3,013 | n/a | |||||||||||||||||||
| Total operating expenses | 101,762 | 108,632 | 57,879 | (6,870 | ) | (6.3 | )% | 50,753 | 87.7 | % | ||||||||||||||||||
| Operating income (loss) | 71,447 | (93,857 | ) | (53,700 | ) | 165,304 | 176.1 | % | (40,157 | ) | (74.8 | )% | ||||||||||||||||
| Investment Income | 2,651 | 963 | 254 | 1,688 | 175.3 | % | 709 | 279.1 | % | |||||||||||||||||||
| Interest Expense | (1,415 | ) | (5,747 | ) | (2,602 | ) | 4,332 | 75.4 | % | (3,145 | ) | (120.9 | )% | |||||||||||||||
| Interest Expense - changes in fair value of financing-related contingent consideration | (4,883 | ) | (3,525 | ) | (1,990 | ) | (1,358 | ) | (38.5 | )% | (1,535 | ) | (77.1 | )% | ||||||||||||||
| Foreign exchange gain (loss) | 10,594 | 11,871 | (288 | ) | (1,277 | ) | (10.8 | )% | 12,159 | 4,221.9 | % | |||||||||||||||||
| Income (loss) before income taxes | 78,394 | (90,295 | ) | (58,326 | ) | 168,689 | 186.8 | % | (31,969 | ) | (54.8 | )% | ||||||||||||||||
| Income tax provision (benefit) | 37,735 | (1,407 | ) | (11,244 | ) | 39,142 | 2,781.9 | % | 9,837 | 87.5 | % | |||||||||||||||||
| Net income (loss) from continuing operations | 40,659 | (88,888 | ) | (47,082 | ) | 129,547 | 145.7 | % | (41,806 | ) | (88.8 | )% | ||||||||||||||||
| Net income from discontinued operations | - | 4,018 | 3,584 | (4,018 | ) | (100.0 | )% | 434 | 12.1 | % | ||||||||||||||||||
| Net income (loss) | $ | 40,659 | $ | (84,870 | ) | $ | (43,498 | ) | $ | 125,529 | 147.9 | % | $ | (41,372 | ) | (95.1 | )% | |||||||||||
| Earnings (loss) per share - Diluted | $ | 0.93 | $ | (2.34 | ) | $ | (1.69 | ) | $ | 3.28 | 139.8 | % | $ | (0.66 | ) | (39.0 | )% | |||||||||||
Revenues
The revenues for each of the Company's significant products were as follows:
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Bloxiverz | $ | 150,283 | $ | 10,211 | $ | - | $ | 140,072 | 1,371.8 | % | $ | 10,211 | n/a | |||||||||||||||
| Vazculep | 20,151 | - | - | 20,151 | n/a | - | - | |||||||||||||||||||||
| Dextroamphetamine | 1,946 | 1,643 | - | 303 | 18.4 | % | 1,643 | n/a | ||||||||||||||||||||
| Other | 108 | 139 | 1,153 | (31 | ) | (22.3 | )% | (1,014 | ) | (87.9 | )% | |||||||||||||||||
| Total product sales and services | 172,488 | 11,993 | 1,153 | 160,495 | 1,338.2 | % | 10,840 | 940.2 | % | |||||||||||||||||||
| License and research revenue | 721 | 2,782 | 3,026 | (2,061 | ) | (74.1 | )% | (244 | ) | (8.1 | )% | |||||||||||||||||
| Total revenues | $ | 173,209 | $ | 14,775 | $ | 4,179 | $ | 158,434 | 1,072.3 | % | $ | 10,596 | 253.6 | % | ||||||||||||||
2015 Compared to 2014
Product sales and services revenues were $172,488 for the year ended December 31, 2015, compared to $11,993 for the prior year. This represents a $160,495 increase in 2015 from 2014. The primary driver of growth was higher sales of Bloxiverz® of $140,072 resulting from volume increases due to market share gains and, to a lesser degree, higher net selling prices. Bloxiverz® was launched in 2014. Additional sales volume resulting from the 2015 launch of Vazculep® also contributed to the sales increase and generated $20,151 of higher sales when compared to the same period last year.
License and research revenues were $721 for the year ended December 31, 2015, compared to $2,782 for the prior year. This represents a $2,061 decrease in 2015 from 2014, largely driven by the termination of remaining development partnership contracts as the Company continues to pursue its strategy of depending less on the development activities performed by partners, and more on the development of its own, self-funded, products.
| -43- |
2014 Compared to 2013
Product sales and services revenues were $11,993 for the year ended December 31, 2014, compared to $1,153 for the prior year. This represents a $10,840 increase in 2014 from 2013. The primary driver of growth was additional sales volume of $10,211 resulting from the 2014 launch of Bloxiverz®.
License and research revenues were fairly consistent between the years ended December 31, 2014 and 2013.
Cost of Products and Services Sold
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Cost of products and services sold | $ | 10,921 | $ | 3,383 | $ | 562 | $ | 7,538 | 222.8 | % | $ | 2,821 | 502.0 | % | ||||||||||||||
| Percentage of sales | 6.3 | % | 22.9 | % | 13.4 | % | ||||||||||||||||||||||
Cost of products and services sold increased $7,538 during the year ended December 31, 2015 as compared to the same period in 2014 primarily due to increases in respective product sales and services. As a percentage of sales, cost of products sold decreased to 6.3% in 2015 compared to 22.9% in 2014 due primarily to higher sales volumes and a favorable change in product mix.
Cost of products and services sold increased $2,821 during the year ended December 31, 2014 as compared to the same period in 2013 primarily due to increases in respective product sales and services. As a percentage of sales, cost of products sold increased to 22.9% in 2014 compared to 13.4% in 2013 primarily due to an unfavorable change in product mix.
Research and Development Expenses
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Research and development expenses | $ | 25,608 | $ | 17,298 | $ | 15,966 | $ | 8,310 | 48.0 | % | $ | 1,332 | 8.3 | % | ||||||||||||||
| Percentage of sales | 14.8 | % | 117.1 | % | 382.1 | % | ||||||||||||||||||||||
The following table provides a breakout of our research and development expenses by major categories of expense:
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Salaries and employee benefits | $ | 9,466 | $ | 12,420 | $ | 8,395 | $ | (2,954 | ) | (23.8 | )% | $ | 4,025 | 47.9 | % | |||||||||||||
| Clinical studies and outside services | 17,196 | 6,844 | 9,211 | 10,352 | 151.3 | % | (2,367 | ) | (25.7 | )% | ||||||||||||||||||
| Other | 4,258 | 4,049 | 4,591 | 209 | 5.2 | % | (542 | ) | (11.8 | )% | ||||||||||||||||||
| Government grants and R&D tax credit | (5,312 | ) | (6,015 | ) | (6,231 | ) | 703 | 11.7 | % | 216 | 3.5 | % | ||||||||||||||||
| Total research and development expenses | $ | 25,608 | $ | 17,298 | $ | 15,966 | $ | 8,310 | 48.0 | % | $ | 1,332 | 8.3 | % | ||||||||||||||
Research and development expenses increased $8,310 or 48.0% during the year ended December 31, 2015 as compared to the same period in 2014 primarily due to higher clinical studies and outside services costs including a $2,300 filing fee for the third Éclat product and the Company’s overall continued investment in its pipeline products. These costs were partially offset by a decrease in salaries and employee benefits which were driven largely by changes in foreign exchange rates and a decrease in certain employee benefits in 2015 relative to 2014.
Research and development expenses increased $1,332 or 8.3% during the year ended December 31, 2014 as compared to the same period in 2013 primarily due primarily to increases in certain employee benefits, partially offset by approximately $2,000 for Vazculep filing fees made in 2013 that did not repeat in 2014.
Selling, General and Administrative Expenses
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Selling, general and administrative expenses | $ | 21,712 | $ | 15,698 | $ | 13,216 | $ | 6,014 | 38.3 | % | $ | 2,482 | 18.8 | % | ||||||||||||||
| Percentage of sales | 12.5 | % | 106.2 | % | 316.2 | % | ||||||||||||||||||||||
Selling, general and administrative expenses increased $6,014 or 38.3% during the year ended December 31, 2015 as compared to the same period in 2014 primarily due to higher stock-based compensation expenses of $4,356 and additional employee recruitment costs associated with the Company’s efforts to reinforce its management team.
| -44- |
Selling, general and administrative expenses increased $2,482 or 18.8% during the year ended December 31, 2014 as compared to the same period in 2013 primarily due to increased legal costs, post marketing studies requested by the FDA for Bloxiverz®, FDA product fees and advisory costs related to the Pessac facility divestiture and transfer of the Company’s intellectual property from our French entity to our Irish-based entity.
Intangible Asset Amortization
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Intangible asset amortization | $ | 12,564 | $ | 11,749 | $ | - | $ | 815 | 6.9 | % | $ | 11,749 | n/a | |||||||||||||||
| Percentage of sales | 7.3 | % | 79.5 | % | - | |||||||||||||||||||||||
Intangible asset amortization expense increased $815 or 6.9% during the year ended December 31, 2015 as compared to the same period in 2014 due to the commencement of amortization related to the acquired In-Process R&D (“IPR&D”) Vazculep intangible asset upon the product’s launch in 2015.
Intangible asset amortization expense increased $11,749 during the year ended December 31, 2014 as compared to the same period in 2013 due to the commencement of amortization related to the acquired IPR&D Bloxiverz intangible asset upon the product’s launch in 2014.
Changes in Fair Value of Related Party Acquisition-Related Contingent Consideration
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Changes in fair value of related party acquisition-related contingent consideration | $ | 30,957 | $ | 57,491 | $ | 28,135 | $ | (26,534 | ) | (46.2 | )% | $ | 29,356 | 104.3 | % | |||||||||||||
| Percentage of sales | 17.9 | % | 389.1 | % | 673.2 | % | ||||||||||||||||||||||
Changes in fair value of related party acquisition-related contingent consideration decreased $26,534 or 46.2% during the year ended December 31, 2015 as compared to the same period in 2014 primarily due to decreases in the changes in fair value of warrants of $37,970 driven by a reduction in the Company’s stock price, which were partially offset by increases in the changes in fair value of the earn-out payments liability of $11,436 due to changes in the associated long-term product sales forecasts.
Changes in fair value of related party acquisition-related contingent consideration increased $29,356 or 104.3% during the year ended December 31, 2014 as compared to the same period in 2013 primarily due to increases in the changes in fair value of the earn-out payments liability of $18,642 driven by changes in the associated long-term product sales forecasts and increases in the fair value of warrants of $15,075 driven largely by an increase in the Company’s stock price.
Income Taxes
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Income tax provision (benefit) | $ | 37,735 | $ | (1,407 | ) | $ | (11,244 | ) | $ | 39,142 | 2,781.9 | % | $ | 9,837 | 87.5 | % | ||||||||||||
| Percentage of income (loss) before income taxes | 48.1 | % | 1.6 | % | 19.5 | % | ||||||||||||||||||||||
| -45- |
The items accounting for the difference between the income tax provision (benefit) computed at the French statutory rate and the Company's effective tax rate are as follows for the years ended December 31:
| 2015 | 2014 | 2013 | ||||||||||
| Statutory tax rate | 33.3 | % | 33.3 | % | 33.3 | % | ||||||
| Non-deductible changes in fair value of contingent consideration | 13.8 | % | (24.7) | % | (16.6) | % | ||||||
| Change in valuation allowance | (9.5) | % | 5.2 | % | (4.5) | % | ||||||
| Income tax deferred charge | - | (16.9 | )% | - | ||||||||
| International tax rates differential | 10.9 | % | 6.7 | % | 6.2 | % | ||||||
| Other | (0.4) | % | (2.0) | % | 1.1 | % | ||||||
| Effective income tax rate | 48.1 | % | 1.6 | % | 19.5 | % | ||||||
| Income tax provision (benefit) - at Statutory tax rate | $ | 26,105 | $ | (30,080 | ) | $ | (19,232 | ) | ||||
| Non-deductible changes in fair value of contingent consideration | 10,835 | 22,326 | 9,592 | |||||||||
| Change in valuation allowance | (7,425 | ) | (4,732 | ) | 2,616 | |||||||
| Income tax deferred charge | - | 15,273 | - | |||||||||
| International tax rates differential | 8,559 | (6,026 | ) | (3,609 | ) | |||||||
| Other | (339 | ) | 1,832 | (611 | ) | |||||||
| Income tax provision (benefit) - at Effective income tax rate | $ | 37,735 | $ | (1,407 | ) | $ | (11,244 | ) | ||||
The Company's effective tax rates for the years ended December 31, 2015, 2014 and 2013 were 48.1%, 1.6% and 19.5%, respectively. Increased expenses reported during 2015, 2014 and 2013 due to changes in fair value of contingent consideration are not deductible for income tax purposes, and therefore unfavorably impact the Company’s effective income tax rate. In 2013, valuation allowances were placed against the deferred tax assets generated by pre-tax losses in France. In 2014, the Company entered into an intra-entity transaction to transfer intellectual property from France to Ireland which resulted in a charge of $15,273 after utilization of net operating loss carryforwards that were previously fully reserved with a valuation allowance. In 2015 certain valuation allowances were reversed with respect to pre-tax income of our U.S. and French operations as the Company had sufficient taxable income to utilize net operating losses. These rates also reflect the additional tax impacts of the Company's significant U.S. operations at a higher statutory tax rate than the French statutory tax rate, and in 2015 are additionally unfavorably impacted by losses incurred at our Irish entity at a lower statutory tax rate.
Net Income From Discontinued Operations
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Net income from discontinued operations | $ | - | $ | 4,018 | $ | 3,584 | $ | (4,018 | ) | (100.0 | )% | $ | 434 | 12.1 | % | |||||||||||||
On December 1, 2014, Flamel divested its Pessac Facility to Recipharm AB (“Recipharm”). Under the divestiture agreement, Recipharm paid the Company $13,200. This divestiture agreement allowed Flamel to use the development and manufacturing capabilities of the acquired Pessac Facility and to use Recipharm’s other facilities for the development or manufacture of its proprietary pipeline if needed. As part of the divestiture agreement, as of December 1, 2014 the Company transferred to Recipharm the Supply Agreement and the associated royalties pertaining to Coreg CR® under the License agreement with GSK to Recipharm. The divestiture of the Pessac Facility has been classified as Discontinued Operations for the twelve month periods ended December 31, 2014, 2013 and 2012 (see Note 17 – Discontinued Operations to the Consolidated Financial Statements in “Item 8. Financial Statements”).
The divestiture of the Pessac Facility has been classified as Discontinued Operations for the twelve months ended December 31, 2015, 2014 and 2013, with net income attributable to such Discontinued Operations of $0, $4018 and $3,584, respectively. The gain on sale of the Pessac Facility in 2014 amounted to $5,007.
The summary statement of operations of the Discontinued Operations for each of the last three years is as follows:
| 2015 | 2014 | 2013 | ||||||||||
| Revenues | $ | - | $ | 14,967 | $ | 18,265 | ||||||
| Operating income (loss) | - | (875 | ) | 3,667 | ||||||||
| Gain on disposal | - | 5,007 | - | |||||||||
| Interest Expense | - | (4 | ) | (9 | ) | |||||||
| Income tax provision (benefit) | - | 110 | 74 | |||||||||
| Net income from discontinued operations | $ | - | $ | 4,018 | $ | 3,584 | ||||||
| -46- |
Liquidity and Capital Resources
The Company's cash flows from operating, investing and financing activities, as reflected in the Consolidated Statements of Cash Flows, are summarized in the following table:
| Increase / (Decrease) | ||||||||||||||||||||||||||||
| Years Ended December 31, | 2015 vs. 2014 | 2014 vs. 2013 | ||||||||||||||||||||||||||
| 2015 | 2014 | 2013 | $ | % | $ | % | ||||||||||||||||||||||
| Net cash provided by (used in): | ||||||||||||||||||||||||||||
| Operating activities | $ | 84,293 | $ | (10,618 | ) | $ | (20,676 | ) | $ | 94,911 | 893.9 | % | $ | 10,058 | 48.6 | % | ||||||||||||
| Investing activities | (31,730 | ) | (43,083 | ) | 6,045 | 11,353 | 26.4 | % | (49,128 | ) | (812.7 | )% | ||||||||||||||||
| Financing activites | (23,751 | ) | 95,995 | 18,274 | (119,746 | ) | (124.7 | )% | 77,721 | 425.3 | % | |||||||||||||||||
Operating Activities
Net cash provided by operating activities of $84,293 for the year ended December 31, 2015 increased $94,911 from the same prior year period. The primary driver of this growth was a $96,574 increase in net income after adjusting for non-cash changes in fair value of contingent consideration and depreciation and amortization expenses.
Net cash used in operating activities of $10,618 for the year ended December 31, 2014 decreased $10,058 from the same period in 2013. The primary driver of this for this decrease in the use of cash related to proceeds in 2014 of $13,210 from the French Government for R&D credits for the reimbursement of a portion of R&D expenses.
Investing Activities
Cash used in investing activities of $31,730 for the year ended December 31, 2015 decreased $11,353 compared to the same prior year period. This decrease was primarily driven by lower use of cash for net purchases of marketable securities of $24,496, which was partially offset by $13,242 of proceeds from the sale of the Pessac facility in 2014 that did not repeat in 2015.
Cash used in investing activities of $43,038 for the year ended December 31, 2014 increased $49,128 compared to the same period in 2013. This increase was primarily driven by higher net purchases of marketable securities of $60,664 after implementing a revised investment strategy to leverage cash received from the Company’s 2014 capital raise. These outflows were partially offset by $13,242 of proceeds from the sale of the Pessac facility in 2014.
Financing Activities
Cash used in financing activities of $23,751 for the year ended December 31, 2015 increased $119,746 compared to $95,995 of cash provided by financing activities in the same period of 2014. This increase was primarily driven by significant 2014 activity that did not repeat in 2015, consisting of $132,260 of proceeds from the 2014 capital raise that were partially offset by $34,392 of 2014 debt repayments, each of which are specifically addressed in the below description of 2014 activity. The increase in cash used in financing activities during 2015 was further increased by $23,169 of additional earn-out payments paid to Deerfield driven by higher 2015 gross profit earned on sales of the related Éclat products.
Cash provided by financing activities of $95,995 for the year ended December 31, 2014 increased $77,721 compared to the same period in 2013. This increase was primarily driven by $132,260 of proceeds from the 2014 capital raise, comprised of the issuance of ordinary shares and warrants primarily in connection with the underwriter public offering fulfilled in March 2014 and issuance of shares to Recipharm in December 2014. These inflows were partially offset by $34,392 of debt repayments, which primarily consisted of:
| · | $12,000 to Deerfield, a related party, to repay an acquisition liability note originally incurred as part of the consideration for the Company’s acquisition of Éclat Pharmaceuticals, LLC on March 13, 2012, |
| · | $15,000 to Deerfield, a related party, to repay a facility agreement originally borrowed as part of a February 2013 debt financing transaction, and |
| · | $5,000 to Broadfin, a related party, to repay a facility agreement originally borrowed as part of a December 2013 debt financing transaction. |
The increase in cash provided by financing activities in 2014 compared to 2013 was also partially offset by $20,000 of loan proceeds in 2013 related to the above $15,000 and $5,000 from Deerfield and Broadfin, respectively, which did not repeat in 2014.
Liquidity and Risk Management
We believe that our existing cash and marketable securities balances and cash we expect to generate from operations will be sufficient to fund our operations and to meet our existing obligations for the foreseeable future. The adequacy of our cash resources depends on many assumptions, including primarily our assumptions with respect to product revenues and expenses, as well as the other factors set forth in “Risk Factors.” To continue to grow our business, we will need to commit substantial resources, which could result in future losses or otherwise limit our opportunities or affect our ability to operate our business. Our assumptions may prove to be wrong or other factors may adversely affect our business, and as a result we could exhaust or significantly decrease our available cash and marketable securities balances which could, among other things, force us to raise additional funds and/or force us to reduce our expenses, either of which could have a material adverse effect on our business.
| -47- |
To continue to grow our business over the longer term, we plan to commit substantial resources to product development and clinical trials of product candidates. In this regard, we have evaluated and expect to continue to evaluate a variety of strategic transactions as part of our strategy to acquire or in-license and develop additional products and product candidates. Acquisition opportunities that we pursue could materially affect our liquidity and capital resources and may require us to incur indebtedness, seek equity capital or both. Raising additional capital could be accomplished through one or more public or private debt or equity financings, collaborations or partnering arrangements. Any equity financing would be dilutive to our shareholders.
Other Matters
Litigation
The Company is subject to potential liabilities generally incidental to its business arising out of present and future lawsuits and claims related to product liability, personal injury, contract, commercial, intellectual property, tax, employment, compliance and other matters that arise in the ordinary course of business. The Company accrues for potential liabilities when it is probable that future costs (including legal fees and expenses) will be incurred and such costs can be reasonably estimated. At December 31, 2015, there were no contingent liabilities with respect to any threat of litigation, arbitration or administrative or other proceeding that are reasonably likely to have a material adverse effect on the Company’s consolidated financial position, results of operations, cash flows or liquidity.
Material Commitments
The Company has commitments to purchase services from Recipharm Pessac for a total of $22,500 for a five-year period commencing January 1, 2015 (disclosed in Note 17- Discontinued Operations) and to purchase one batch per year for the next five years of the generic pharmaceutical product it markets for $46 per year. During the year ended December 31, 2015, the Company recorded $4,089 of research and development expenses, and cash outflows of $5,679, related to this commitment to Recipharm
The Company and its subsidiaries lease office facilities under noncancelable operating leases expiring at various dates. Rent expense was $752, $844 and $759 in 2015, 2014, and 2013, respectively. Minimum rental commitments for non-cancelable leases in effect at December 31, 2015 are as follows:
| 2016 | $ | 784 | ||
| 2017 | 472 | |||
| 2018 | 463 | |||
| 2019 | 389 | |||
| 2020 | 310 | |||
| Thereafter | - | |||
| Total | $ | 2,418 |
Other than the above commitments to Recipharm and for operating leases, there were no other material commitments outside of the normal course of business. Material commitments in the normal course of business include long-term debt, long-term contingent consideration payable, and post-retirement benefit plan obligations which are disclosed in Note 7 - Long-Term Debt , Note 8 – Long-term Contingent Consideration Payable, and Note 10 – Post-Retirement Benefit Plans, respectively.
Aggregate Contractual Obligations
The following table presents contractual obligations of the Company at December 31, 2015:
| Payments due by period | ||||||||||||||||||||
| Total | Less
than 1 Year | 1
to 3 Years | 3
to 5 Years | More
than 5 Years | ||||||||||||||||
| Statement of Income Data: | ||||||||||||||||||||
| Long-term debt | $ | 1,118 | $ | 434 | $ | 539 | $ | 145 | $ | - | ||||||||||
| Long-term contingent consideration payable (undiscounted) | 155,155 | 20,981 | 41,011 | 28,849 | 64,314 | |||||||||||||||
| Recipharm purchase commitment | 18,220 | 4,250 | 9,110 | 4,860 | - | |||||||||||||||
| Operating leases | 2,418 | 784 | 935 | 699 | - | |||||||||||||||
| Other | 184 | 46 | 92 | 46 | - | |||||||||||||||
| Total contractual cash obligations | $ | 177,095 | $ | 26,495 | $ | 51,687 | $ | 34,599 | $ | 64,314 | ||||||||||
| -48- |
See Note 7 : Long-Term Debt and Note 8 : Long-Term Contingent Consideration Payable to the Company's consolidated financial statements contained in Item 8 – Financial Statements for obligations with respect to the respective items within the above table. Obligations relative to the Deerfield warrant-based contingent consideration of $20,617 are not included within the above table. The Company’s long-term debt does not bear interest and therefore no interest is included in the above table.
See Note 17 – Discontinued Operations to the Company's consolidated financial statements contained in Item 8 – Financial Statements for obligations with respect to the Company's Recipharm and other obligations within the above table.
See Note 10 : Post-Retirement Benefit Plans to the Company's consolidated financial statements contained in Item 8 – Financial Statements for obligations with respect to the Company's post-retirement benefit plans. Obligations of $2,170 related to the post-retirement benefit plans are not included within the above table.
| -49- |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
Interest Rate Risk
The Company is subject to interest rate risk as a result of its portfolio of marketable securities. The primary objectives of our investment policy are as follows: safety and preservation of principal and diversification of risk; liquidity of investments sufficient to meet cash flow requirements; and competitive yield. Although our investments are subject to market risk, our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure from any single issue, issuer or certain types of investment. Our investment policy allows us to maintain a portfolio of cash equivalents and marketable securities in a variety of instruments, including U.S. federal government and federal agency securities, European Government bonds, corporate bonds or commercial paper issued by U.S. or European corporations, money market instruments, certain qualifying money market mutual funds, certain repurchase agreements, tax-exempt obligations of states, agencies, and municipalities in the U.S and Europe, and common stocks.
Foreign Exchange Risk
We have significant operations in Europe as well as in the U.S. The functional currency of each foreign subsidiary is generally the local currency. We are exposed to foreign currency exchange risk as the functional currency financial statements of foreign subsidiaries are translated to U.S. dollars. The assets and liabilities of our foreign subsidiaries having a functional currency other than the U.S. dollar are translated into U.S. dollars at the exchange rate prevailing at the balance sheet date, and at the average exchange rate for the reporting period for revenue and expense accounts. The cumulative foreign currency translation adjustment is recorded as a component of accumulated other comprehensive loss in shareholders’ equity. The reported results of our foreign subsidiaries will be influenced by their translation into U.S. dollars by currency movements against the U.S. dollar. Our primary currency translation exposure is related to our subsidiaries that have functional currencies denominated in Euro. A 10% strengthening/(weakening) in the rates used to translate the results of our foreign subsidiaries that have functional currencies denominated in the euro would have increased/(decreased) net income for the year ended December 31, 2015 by approximately $9,500.
Transactional exposure arises where transactions occur in currencies other than the functional currency. Transactions in foreign currencies are recorded at the exchange rate prevailing at the date of the transaction. The resulting monetary assets and liabilities are translated into the appropriate functional currency at exchange rates prevailing at the balance sheet date and the resulting gains and losses are reported in foreign currency gain (loss) in the consolidated statements of income. As of December 31, 2015, our primary exposure to transaction risk related to euro net monetary liabilities held by subsidiaries with a U.S. dollar functional currency. Realized and unrealized foreign exchange gains resulting from transactional exposure were $10,594 for the year ended December 31, 2015.
| -50- |
| Item 8. | Financial Statements and Supplementary Data. |
Flamel Technologies S.A.
Consolidated Statements of Income (Loss)
(In Thousands, Except Per Share Data)
| Years ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Revenues: | ||||||||||||
| Product sales and services | $ | 172,488 | $ | 11,993 | $ | 1,153 | ||||||
| License and research revenue | 721 | 2,782 | 3,026 | |||||||||
| Total | 173,209 | 14,775 | 4,179 | |||||||||
| Operating expenses: | ||||||||||||
| Cost of products and services sold | 10,921 | 3,383 | 562 | |||||||||
| Research and development expenses | 25,608 | 17,298 | 15,966 | |||||||||
| Selling, general and administrative expenses | 21,712 | 15,698 | 13,216 | |||||||||
| Intangible asset amortization | 12,564 | 11,749 | - | |||||||||
| Changes in fair value of related party acquisition-related contingent consideration | 30,957 | 57,491 | 28,135 | |||||||||
| Loss on early repayment of related party acquisition-related note | - | 3,013 | - | |||||||||
| Total | 101,762 | 108,632 | 57,879 | |||||||||
| Operating income (loss) | 71,447 | (93,857 | ) | (53,700 | ) | |||||||
| Investment Income | 2,651 | 963 | 254 | |||||||||
| Interest Expense | (1,415 | ) | (5,747 | ) | (2,602 | ) | ||||||
| Interest Expense - changes in fair value of related party financing-related contingent consideration | (4,883 | ) | (3,525 | ) | (1,990 | ) | ||||||
| Foreign exchange gain (loss) | 10,594 | 11,871 | (288 | ) | ||||||||
| Other income (expense) | - | (36 | ) | 573 | ||||||||
| Income (loss) before income taxes | 78,394 | (90,331 | ) | (57,753 | ) | |||||||
| Income tax provision (benefit) | 37,735 | (1,407 | ) | (11,244 | ) | |||||||
| Net income (loss) from continuing operations | 40,659 | (88,924 | ) | (46,509 | ) | |||||||
| Net income from discontinued operations | - | 4,018 | 3,584 | |||||||||
| Net income (loss) | $ | 40,659 | $ | (84,906 | ) | $ | (42,925 | ) | ||||
| Earnings (loss) per share - Basic: | ||||||||||||
| Continuing operations | $ | 1.00 | $ | (2.45 | ) | $ | (1.83 | ) | ||||
| Discontinued operations | - | 0.11 | 0.14 | |||||||||
| Net income (loss) | $ | 1.00 | $ | (2.34 | ) | $ | (1.69 | ) | ||||
| Earnings (loss) per share - Diluted: | ||||||||||||
| Continuing operations | $ | 0.93 | $ | (2.45 | ) | $ | (1.83 | ) | ||||
| Discontinued operations | - | 0.11 | 0.14 | |||||||||
| Net income (loss) | $ | 0.93 | $ | (2.34 | ) | $ | (1.69 | ) | ||||
| Weighted average number of shares outstanding - Basic | 40,580 | 36,214 | 25,450 | |||||||||
| Weighted average number of shares outstanding - Diluted | 43,619 | 36,214 | 25,450 | |||||||||
See accompanying notes to consolidated financial statements.
| -51- |
Flamel Technologies S.A.
Consolidated Statements of Comprehensive Income (Loss)
(In Thousands)
| Years ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Net income/(loss) | $ | 40,659 | $ | (84,906 | ) | $ | (42,925 | ) | ||||
| Other comprehensive income/(loss), net of tax: | ||||||||||||
| Foreign currency translation gain/(loss), net | (15,087 | ) | (18,040 | ) | 562 | |||||||
| Unrealized gain/(loss) on marketable securities | (147 | ) | (198 | ) | - | |||||||
| Total other comprehensive income/(loss), net of tax | (15,234 | ) | (18,238 | ) | 562 | |||||||
| Total comprehensive income/(loss) | $ | 25,425 | $ | (103,144 | ) | $ | (42,363 | ) | ||||
See accompanying notes to consolidated financial statements.
| -52- |
Flamel Technologies S.A.
Consolidated Balance Sheets
(In Thousands, Except Per Share Data)
| December 31, | ||||||||
| 2015 | 2014 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 65,064 | $ | 39,760 | ||||
| Marketable securities | 79,738 | 53,074 | ||||||
| Accounts receivable (net of allowance of $35 and $127 at December 31, 2015 and 2014 respectively) | 6,978 | 1,679 | ||||||
| Inventories | 4,155 | 6,729 | ||||||
| Research and development tax credit receivable - current portion | 2,382 | 5,932 | ||||||
| Prepaid expenses and other current assets | 7,989 | 4,418 | ||||||
| Current assets held for sale | - | 730 | ||||||
| Total current assets | 166,306 | 112,322 | ||||||
| Property and equipment, net | 2,616 | 1,776 | ||||||
| Goodwill | 18,491 | 18,491 | ||||||
| Intangible assets, net | 15,825 | 28,389 | ||||||
| Income tax deferred charge | 11,581 | 13,102 | ||||||
| Other | 158 | 125 | ||||||
| Total assets | $ | 214,977 | $ | 174,205 | ||||
| LIABILITIES AND SHAREHOLDERS' EQUITY | ||||||||
| Current liabilities: | ||||||||
| Current portion of long-term debt | $ | 434 | $ | 2,440 | ||||
| Current portion of long-term related party contingent consideration payable | 28,614 | 39,892 | ||||||
| Accounts payable | 10,565 | 8,024 | ||||||
| Deferred revenue | 5,121 | 1,336 | ||||||
| Accrued expenses | 3,598 | 5,667 | ||||||
| Income taxes | 323 | 7,643 | ||||||
| Other | 133 | 5,672 | ||||||
| Current liabilities held for sale | - | 168 | ||||||
| Total current liabilities | 48,788 | 70,842 | ||||||
| Long-term debt, less current portion | 684 | 1,277 | ||||||
| Long-term related party contingent consideration payable, less current portion | 94,079 | 74,858 | ||||||
| Deferred taxes | 1,351 | - | ||||||
| Other | 2,210 | 2,333 | ||||||
| Total liabilities | 147,112 | 149,310 | ||||||
| Shareholders' equity: | ||||||||
| Ordinary shares, nominal value of 0.122 euro per share; 53,198 shares authorized; 41,241 and 40,191 issued and outstanding at December 31, 2015 and 2014, respectively | 6,331 | 6,188 | ||||||
| Additional paid-in capital | 363,984 | 346,582 | ||||||
| Accumulated deficit | (279,793 | ) | (320,452 | ) | ||||
| Accumulated other comprehensive loss | (22,657 | ) | (7,423 | ) | ||||
| Total shareholders' equity | 67,865 | 24,895 | ||||||
| Total liabilities and shareholders' equity | $ | 214,977 | $ | 174,205 | ||||
See accompanying notes to consolidated financial statements.
| -53- |
Flamel Technologies S.A.
Consolidated Statements of Shareholders' Equity
(In Thousands)
| Ordinary shares | Additional | Accumulated | Accumulated other comprehensive | Total shareholders' | ||||||||||||||||||||
| Shares | Amount | paid-in capital | deficit | loss | equity | |||||||||||||||||||
| Balance, December 31 2012 | 25,416 | $ | 3,714 | $ | 209,158 | $ | (192,621 | ) | $ | 10,253 | $ | 30,504 | ||||||||||||
| Net loss | - | - | - | (42,925 | ) | - | (42,925 | ) | ||||||||||||||||
| Other comprehensive income | - | - | - | - | 562 | 562 | ||||||||||||||||||
| Subscription of warrants | - | - | (27 | ) | - | - | (27 | ) | ||||||||||||||||
| Exercise of stock options or warrants | 50 | 8 | 391 | - | - | 399 | ||||||||||||||||||
| Vesting of free shares | 147 | 24 | (24 | ) | - | - | - | |||||||||||||||||
| Stock-based compensation expense | - | - | 1,975 | - | - | 1,975 | ||||||||||||||||||
| Balance, December 31 2013 | 25,613 | $ | 3,746 | $ | 211,473 | $ | (235,546 | ) | $ | 10,815 | $ | (9,512 | ) | |||||||||||
| Net loss | - | - | - | (84,906 | ) | - | (84,906 | ) | ||||||||||||||||
| Other comprehensive loss | - | - | - | - | (18,238 | ) | (18,238 | ) | ||||||||||||||||
| Subscription of warrants | - | - | 351 | - | - | 351 | ||||||||||||||||||
| Exercise of stock options or warrants | 1,001 | 164 | 5,861 | - | - | 6,025 | ||||||||||||||||||
| Vesting of free shares | 151 | 24 | (24 | ) | - | - | - | |||||||||||||||||
| Stock-based compensation expense | - | - | 2,894 | - | - | 2,894 | ||||||||||||||||||
| Public offering | 12,400 | 2,099 | 113,133 | - | - | 115,232 | ||||||||||||||||||
| Shares granted to Recipharm AB | 1,026 | 155 | 12,894 | - | - | 13,049 | ||||||||||||||||||
| Balance, December 31 2014 | 40,191 | $ | 6,188 | $ | 346,582 | $ | (320,452 | ) | $ | (7,423 | ) | $ | 24,895 | |||||||||||
| Net income | - | - | - | 40,659 | - | 40,659 | ||||||||||||||||||
| Other comprehensive income | - | - | - | - | (15,234 | ) | (15,234 | ) | ||||||||||||||||
| Subscription of warrants | - | - | 601 | - | - | 601 | ||||||||||||||||||
| Exercise of stock options or warrants | 899 | 123 | 6,266 | - | - | 6,389 | ||||||||||||||||||
| Vesting of free shares | 151 | 20 | (20 | ) | - | - | - | |||||||||||||||||
| Stock-based compensation expense | - | - | 7,741 | - | - | 7,741 | ||||||||||||||||||
| Excess tax benefit from stock-based compensation | - | - | 2,814 | - | - | 2,814 | ||||||||||||||||||
| Balance, December 31 2015 | 41,241 | $ | 6,331 | $ | 363,984 | $ | (279,793 | ) | $ | (22,657 | ) | $ | 67,865 | |||||||||||
See accompanying notes to consolidated financial statements.
| -54- |
Flamel Technologies S.A.
Consolidated Statements of Cash Flows
(In Thousands)
| Years ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net income (loss) | $ | 40,659 | $ | (84,906 | ) | $ | (42,925 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
| Depreciation and amortization | 13,132 | 14,141 | 3,062 | |||||||||
| Loss (gain) on disposal of property and equipment | - | (4,952 | ) | 14 | ||||||||
| Loss (gain) on sale of marketable securities | 779 | - | - | |||||||||
| Unrealized exchange gain | (8,969 | ) | (6,252 | ) | - | |||||||
| Grants recognized in research and development expenses and operating income | (1,498 | ) | (589 | ) | (676 | ) | ||||||
| Remeasurement of related party acquisition-related contingent consideration | 30,957 | 60,503 | 28,135 | |||||||||
| Remeasurement of related party financing-related contingent consideration | 4,883 | 3,319 | 1,990 | |||||||||
| Imputed interest on long-term debt | - | - | 712 | |||||||||
| Change in deferred tax and income tax deferred charge | 1,420 | (2,806 | ) | (11,320 | ) | |||||||
| Stock-based compensation expense | 7,741 | 2,690 | 2,029 | |||||||||
| Increase (decrease) in cash from: | ||||||||||||
| Accounts receivable | (8,108 | ) | 3,426 | (511 | ) | |||||||
| Inventories | 2,547 | (3,112 | ) | (2,186 | ) | |||||||
| Prepaid expenses and other current assets | (610 | ) | (2,330 | ) | 315 | |||||||
| Research and development tax credit receivable | 2,975 | 13,210 | 665 | |||||||||
| Accounts payable & other current liabilities | (3,887 | ) | 8,090 | 800 | ||||||||
| Deferred revenue | 3,815 | (55 | ) | (14 | ) | |||||||
| Accrued expenses | (1,617 | ) | (265 | ) | 1,211 | |||||||
| Other long-term assets and liabilities | 74 | (10,730 | ) | (1,977 | ) | |||||||
| Net cash provided by (used in) operating activities | 84,293 | (10,618 | ) | (20,676 | ) | |||||||
| Cash flows from investing activities: | ||||||||||||
| Purchases of property and equipment | (1,629 | ) | (1,728 | ) | (1,029 | ) | ||||||
| Proceeds from disposal of property and equipment | - | 13,242 | 1,007 | |||||||||
| Proceeds from sales of marketable securities | 20,490 | 13,678 | 7,152 | |||||||||
| Purchase of marketable securities | (50,591 | ) | (68,275 | ) | (1,085 | ) | ||||||
| Net cash provided by (used in) investing activities | (31,730 | ) | (43,083 | ) | 6,045 | |||||||
| Cash flows from financing activities: | ||||||||||||
| Reimbursment of loans | (4,911 | ) | (34,186 | ) | - | |||||||
| Reimbursment of conditional R&D grants | (747 | ) | (355 | ) | (475 | ) | ||||||
| Proceeds from loans or conditional R&D grants | - | - | 19,333 | |||||||||
| Principal payments on capital lease obligations | - | (161 | ) | (77 | ) | |||||||
| Earn-out payments for related party acquisition-related contingent consideration | (24,526 | ) | (1,357 | ) | (907 | ) | ||||||
| Royalty payments for related party financing-related contingent consideration | (3,371 | ) | (206 | ) | - | |||||||
| Excess tax benefit from stock-based compensation | 2,814 | - | - | |||||||||
| Cash proceeds from issuance of ordinary shares and warrants | 6,990 | 132,260 | 400 | |||||||||
| Net cash provided by (used in) financing activities | (23,751 | ) | 95,995 | 18,274 | ||||||||
| Effect of exchange rate changes on cash and cash equivalents | (3,508 | ) | (9,170 | ) | 251 | |||||||
| Net increase in cash and cash equivalents | 25,304 | 33,124 | 3,894 | |||||||||
| Cash and cash equivalents at January 1 | 39,760 | 6,636 | 2,742 | |||||||||
| Cash and cash equivalents at December 31 | $ | 65,064 | $ | 39,760 | $ | 6,636 | ||||||
| Supplemental disclosures of cash flow information: | ||||||||||||
| Income tax paid | $ | 42,121 | $ | 403 | $ | 153 | ||||||
| Interest paid | $ | 4,738 | $ | 4,431 | $ | 1,701 | ||||||
| Supplemental schedule of non-cash investing and financing activities: | ||||||||||||
| Income tax paid Grants recognized in research and development expenses and operating income | $ | (1,498 | ) | $ | - | $ | - | |||||
See accompanying notes to consolidated financial statements.
| -55- |
Flamel Technologies S.A.
Notes to Consolidated Financial Statements
(In Thousands, Except Per Share Data)
NOTE 1 : Summary of Significant Accounting Policies
Nature of Operations. Flamel Technologies, S.A. (“Flamel” or the "Company") is organized as a Société Anonyme, a form of corporation under the laws of The Republic of France. The Company was founded in 1990. Flamel is a specialty pharmaceutical company utilizing core competencies in drug delivery and formulation development to create safer and more efficacious pharmaceutical products to address unmet medical needs and/or reduce overall healthcare costs. The Company is headquartered in Lyon, France and has operations in St. Louis, Missouri, and Charlotte, NC, USA, and Dublin, Ireland.
Basis of Presentation. These Consolidated Financial Statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP). The consolidated financial statements include the accounts of the Company and all majority-owned subsidiaries. All significant intercompany accounts and transactions have been eliminated.
Reclassifications. The accompanying consolidated financial statements for prior years contain certain reclassifications to conform to the presentation used in 2015.
Revenue. Revenue includes sales of pharmaceutical products, upfront licensing fees, milestone payments for R&D achievements, and compensation for the execution of R&D activities.
Product Sales and Services
Revenue is generally realized or realizable and earned when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the seller’s price to the buyer is fixed or determinable, and collectability is reasonably assured. The Company records revenue from product sales when title and risk of ownership have been transferred to the customer, which is typically upon delivery to the customer and when the selling price is determinable. As is customary in the pharmaceutical industry, the Company’s gross product sales are subject to a variety of deductions in arriving at reported net product sales. When the Company recognizes revenue from the sale of its products, an estimate of provision for sales return and allowances is recorded which reduces product sales. These adjustments include estimates for product returns, chargebacks, payment discounts and other sales allowances and rebates. The estimate for chargebacks is determined when product is shipped from the wholesalers to their customers. The return allowance, when estimable, is based on an analysis of the historical returns of the product or similar products.
For generic products and branded products sold in mature and stable markets where changes in selling price are rare, the Company recognizes revenues upon shipment. For products where market conditions remain volatile and selling price is subject to changes, which is the Company’s situation in 2015, 2014 and 2013, the Company delays revenue recognition until the wholesaler sells the product to its customers. For new product launches the Company recognizes revenue once sufficient data is available to determine product acceptance in the marketplace such that product returns may be estimated based on historical data and there is evidence of reorders and consideration is made of wholesaler inventory levels. Net product sales of wholesalers to their customers are determined using sales data from an independent, renowned wholesaler inventory tracking service. Net sales of wholesalers to their customers are calculated by deducting estimates for returns for wholesaler customers, chargebacks, payment discounts and other sales or discounts offered from the applicable gross sales value. Estimates for product returns are adjusted periodically based upon historical rates of returns, inventory levels in the distribution channel and other related factors.
License and Research Revenue
Where agreements have more than one deliverable, a determination is made as to whether the license and R&D elements should be recognized separately or combined into a single unit of account in accordance with ASU 2009-13, Revenue with Multiple Deliverables.
The Company uses a Multiple Attribution Model, referred to as the milestone-based method:
| · | As milestones relate to discrete development steps (i.e., can be used by the partners to decide whether to continue the development under the agreement), the Company views that milestone events have substance and represent the achievement of defined goals worthy of the payments. Therefore, milestone payments based on performance are recognized when the performance criteria are met and there are no further performance obligations. |
| · | Non-refundable technology access fees received from collaboration agreements that require the Company's continuing involvement in the form of development efforts are recognized as revenue ratably over the development period. |
| · | R&D work is compensated at a non-refundable hourly rate for a projected number of hours. Revenue on such agreements is recognized at the hourly rate for the number of hours worked as the R&D work is performed. Costs incurred under these contracts are considered costs in the period incurred. Payments received in advance of performance are recorded as deferred revenue and recognized in revenue as services are rendered. |
| -56- |
When Flamel receives revenue under signed feasibility study agreements, revenue is then recognized over the term of the agreement as services are performed.
Government Grants. The Company receives financial support for various research or investment projects from governmental agencies.
The Company receives funds to finance R&D projects. These funds are repayable on commercial success of the project. In the absence of commercial success, the Company is released of its obligation to repay the funds and as such the funds are recognized in the Income Statement as an offset to R&D expense. The absence of commercial success must be formally confirmed by the granting authority. Should the Company wish to discontinue the R&D to which the funding is associated, the granting authorities must be informed.
R&D. Research and development expenses consist primarily of costs related to clinical studies and outside services, personnel expenses, and other research and development costs. Clinical study and outside services costs relate primarily to services performed by clinical research organizations and related clinical or development manufacturing costs, materials and supplies, filing fees, regulatory support, and other third party fees. Personnel expenses relate primarily to salaries, benefits and stock-based compensation. Other research and development expenses primarily include overhead allocations consisting of various support and facilities-related costs. R&D expenditures are charged to operations as incurred.
The Company recognizes R&D tax credits received from the French government for spending on innovative R&D as an offset of R&D expenses.
Stock-based Compensation. The Company accounts for Stock-based compensation based on grant-date fair value estimated in accordance with ASC 718. The fair value of stock options and warrants is estimated using Black-Scholes option-pricing valuation models (“Black-Scholes model”). The term used within the Black-Sholes valuation models is determined using a simplified method, as historical data was considered insufficient and irrelevant relative to the grant of stock-options and warrants to a limited population. The Company recognizes compensation cost, net of an estimated forfeiture rate, using the accelerated method over the requisite service period of the award.
Income Taxes. The provision for income taxes is based on pretax income reported in the consolidated statements of income and currently enacted tax rates for each jurisdiction. Deferred tax assets are determined based on the difference between the financial reporting and tax basis of assets and liabilities, applying enacted statutory tax rates in effect for the year in which the tax differences are expected to reverse. Deferred tax assets and liabilities are adjusted for the effects of changes in the tax laws and rates on the date of enactment. The Company regularly reviews its deferred tax assets for recoverability and establishes a valuation allowance when it believes it is more likely than not that such assets may not be recovered, taking into consideration historical operating results, expectations of future earnings, changes in its operations and the expected timing of the reversals of existing temporary differences.
Discontinued Operations. The Company followed the guidance in Financial Accounting Standards Board Accounting Standards Codification (ASC) Topic 205 Presentation of Financial Statements (ASC 205), Topic 360 Property, Plant and Equipment (ASC 360) and Accounting Standards Update (ASU 2014-08), Reporting of Discontinued Operations and Disclosures of Disposals of Components of an Entity in determining the accounting for the divestiture of the Pessac facility. In 2014, the Company opted to early adopt the provisions of ASU 2014-08 as management believed that all criteria for presenting the disposal of Pessac Facility and its business as a discontinued operation were met, and that presenting the disposal as a discontinued operation would better reflect the ongoing operations of the entity.
The divestiture of the Pessac facility represented a strategic shift that had and will have a major effect on the Company’s operations and financial results. Since 2012, the Company’s business model of combining novel, high-value internally developed products with its leading drug delivery capabilities and commercializing niche branded and general pharmaceutical products. Previously, the Company’s focus was to develop and license its proprietary drug delivery platforms (Micropump®, LiquiTime®, Trigger Lock™ and Medusa™) with pharmaceutical companies and biotechnology partners (e.g. the licensing of Micropump® to GSK to develop Coreg CR® with GSK bringing and commercializing the product to market). The divestiture of Pessac Facility to Recipharm and the transfer to Recipharm of the GSK’s Supply Agreement and royalty income relating to Coreg CR ® is an implementation of this revised strategy. The Company is reducing its sole reliance on products developed with partners, explaining the transfer of its rights and obligations pertaining to Coreg CR®, including the Pessac Facility. Flamel sold over 50% of its historical revenues as a result of this transaction which has a major impact on the Company’s operations and results.
The divestiture of the Pessac facility was accomplished in a single transaction and the assets, contracts and liabilities referred to in the Asset Purchase Agreement signed between Flamel and Recipharm were determined to represent a disposal group. This disposal group was considered to be a component of the Company. While the Pessac Facility and its related business were not identified as reportable segment or operating segment, as the Company operates in only one segment, the Pessac Facility and its related business is considered to be an asset group as the transferred assets, liabilities and contracts represent the lowest level for which identifiable cash flows are largely independent of the cash flows of other group of assets and liabilities. The Company transferred all future cash outflows and inflows relating to the Pessac Facility that can be clearly distinguished operationally and for financial reporting purposes.
The results of discontinued operations, less income taxes, have been reported as a separate component of income in the consolidated statements of operations. The assets and liabilities of the discontinued operation have been reported separately in the asset and liability sections of the consolidated statements of financial position for the periods presented in the statement. Note 17: Discontinued Operations contains a description of the facts and circumstances related to the disposal, the gain and loss on disposal and the specific line items included in the consolidated statements of operations, financial position and cash flows relative to the disposal group.
| -57- |
Cash and Cash Equivalents. Cash and cash equivalents consist of cash on hand, cash on deposit and fixed term deposits which are highly liquid investments with original maturities of less than three months.
Marketable Securities. Marketable securities consist of investments in available-for-sale marketable equity securities and are recorded at fair value.
Accounts Receivable. Accounts receivable are stated at amounts invoiced net of allowances for doubtful accounts. The Company makes judgments as to its ability to collect outstanding receivables and provides allowances for the portion of receivables deemed uncollectible. Provision is made based upon a specific review of all significant outstanding invoices. A majority of accounts receivable is due from three significant customers which are disclosed in Note 16: Company Operations by Product, Customer and Geographic Area.
Inventories. Inventories consist of raw materials and finished products, which are stated at lower of cost or market determined under the first-in, first-out ("FIFO") method. Raw materials used in the production of pre-clinical and clinical products are expensed as R&D costs when consumed. The Company establishes reserves for inventory estimated to be obsolete, unmarketable or slow-moving on a case by case basis.
Property and Equipment. Property and equipment is stated at historical cost less accumulated depreciation. Depreciation and amortization are computed using the straight-line method over the following estimated useful lives:
| Land and buildings | 20 years |
| Laboratory equipment | 4-8 years |
| Office and computer equipment | 3 years |
| Furniture, fixtures and fittings | 5-10 years |
Goodwill. Goodwill represents the excess of the acquisition consideration over the fair value of assets acquired and liabilities assumed. The Company has determined that it operates in a single segment and has a single reporting unit associated with the development and commercialization of pharmaceutical products. The annual test for goodwill impairment is a two-step process. The first step is a comparison of the fair value of the reporting unit with its carrying amount, including goodwill. If this step indicates impairment, then, in the second step, the loss is measured as the excess of recorded goodwill over the implied fair value of the goodwill. Implied fair value of goodwill is the excess of the fair value of the reporting unit as a whole over the fair value of all separately identified assets and liabilities within the reporting unit. The Company tests goodwill for impairment annually and when events or changes in circumstances indicate that the carrying value may not be recoverable. The Company relies upon projections of future discounted cash flows and takes into account assumptions regarding the evolution of the market and its ability to successfully develop and commercialize its products. Changes in market conditions could have a major impact on the valuation of these assets and could result in potential associated impairment. The Company has determined that no impairment of goodwill existed at December 31, 2015 or 2014.
Long-Lived Assets. Long-lived assets include fixed assets and intangible assets. Intangible assets consist primarily of purchased licenses and intangible assets corresponding to acquired, in progress R&D recognized as part of the Éclat acquisition purchase price allocation. Acquired IPR&D has an indefinite life and is not amortized until completion and development of the project, at which time the IPR&D becomes an amortizable asset. Amortization of acquired IPR&D is computed using the straight-line method over estimated useful life of the assets.
Long-lived assets are reviewed for impairment whenever conditions indicate that the carrying value of the assets may not be fully recoverable. Such impairment tests are based on a comparison of the pretax undiscounted cash flows expected to be generated by the asset to the recorded value of the asset. If impairment is indicated, the asset value is written down to its market value if readily determinable or its estimated fair value based on discounted cash flows. Any significant unanticipated changes in business or market conditions that vary from current expectations could have an impact on the fair value of these assets and any potential associated impairment. The Company has determined that no indications of impairment existed at December 31, 2015 or 2014.
Contingent Consideration. The acquisition-related contingent consideration payable arising from the acquisition of Éclat Pharmaceuticals are accounted at fair-value (see Note 8 : Long-Term Contingent Consideration Payable). The fair value of warrant consideration is estimated on a quarterly basis using a Black-Scholes option pricing model, and the fair value of earn-out payment consideration is estimated on a quarterly basis using a discounted cash flow model based on probability-adjusted annual gross profit of the specified Éclat Pharmaceuticals products at an appropriate discount rate. Changes in fair value are recorded in the consolidated statements of operations within operating expenses as changes in fair value of related party acquisition-related contingent consideration.
The Company elected the fair value option for the measurement of the financing-related contingent consideration payable associated with the Deerfield and Broadfin Royalty Agreements, both of whom are related parties (see Note 8: Long-Term Contingent Consideration Payable). The fair value of royalty agreement consideration is estimated on a quarterly basis using a discounted cash flow model based on probability-adjusted annual revenues of the specified Éclat Pharmaceuticals products at an appropriate discount rate. Changes in fair value are recorded in the consolidated statements of operations as interest expense - changes in fair value of related party financing-related contingent consideration.
Fair Value Measurement. The Company is often required to measure certain assets and liabilities at fair value, either upon initial recognition or for subsequent accounting or reporting. For example, we use fair value extensively when accounting for and reporting of certain financial instruments, when measuring certain contingent consideration liabilities and in the initial recognition of net assets acquired in a business combination.
| -58- |
ASC 820, Fair Value Measurements and Disclosures defines fair value as a market-based measurement that should be determined based on the assumptions that marketplace participants would use in pricing an asset or liability. When estimating fair value, depending on the nature and complexity of the asset or liability, we may generally use one or each of the following techniques:
| · | Income approach, which is based on the present value of a future stream of net cash flows. |
| · | Market approach, which is based on market prices and other information from market transactions involving identical or comparable assets or liabilities. |
As a basis for considering the assumptions used in these techniques, the standard establishes a three-tier fair value hierarchy which prioritizes the inputs used in measuring fair value as follows:
| · | Quoted prices for identical assets or liabilities in active markets (Level 1 inputs). |
| · | Quoted prices for similar assets or liabilities in active markets, or quoted prices for identical or similar assets or liabilities in markets that are not active, or inputs other than quoted prices that are directly or indirectly observable, or inputs that are derived principally from, or corroborated by, observable market data by correlation or other means (Level 2 inputs). |
| · | Unobservable inputs that reflect estimates and assumptions (Level 3 inputs). |
Foreign Currency Translation. The reporting currency of the Company and its wholly-owned subsidiaries is the U.S. dollar. Each of the Company's non-U.S. subsidiaries and the parent entity uses local currency as its functional currency. Subsidiaries and entities that do not use the U.S. Dollar as their functional currency translate 1) profit and loss accounts at the weighted average exchange rates during the reporting period, 2) assets and liabilities at period end exchange rates and 3) shareholders' equity accounts at historical rates. Resulting translation gains and losses are included as a separate component of stockholders' equity in Accumulated Other Comprehensive Income. Assets and liabilities denominated in a currency other than the subsidiary's functional currency are translated to the subsidiary's functional currency at period end exchange rates. Resulting gains and losses are recognized in the consolidated statements of income.
Use of Estimates. The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of sales and expenses during the periods presented. Actual results could differ from those estimates under different assumptions or conditions.
NOTE 2 : Effect of New Accounting Standards
In July 2015, the FASB issued ASU 2015-14 Revenue from Contracts with Customers: Deferral of the Effective Date (ASU 2015-14) which deferred the effective date for ASU No. 2014-09, Revenue from Contracts with Customers (ASU 2014-09), by one year. ASU 2014-09 will supersede the revenue recognition requirements in Revenue Recognition (Topic 605) and requires entities to recognize revenue in a way that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 is now effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period, which for the Company is January 1, 2018. Early adoption is permitted only as of annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period.. The new standard can be applied retrospectively to each prior reporting period presented or retrospectively with the cumulative effect of the change recognized at the date of the initial application in retained earnings. The Company is currently evaluating the potential impact the adoption of ASU 2014-09 will have on its consolidated financial statements and has not yet selected a transition method.
In July 2015, the FASB issued ASU No. 2015-11, Simplifying the Measurement of Inventory (ASU 2015-11), which requires an entity to measure inventory within the scope of this ASU at the lower of cost and net realizable value. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. The effective date for the standard is for fiscal years beginning after December 15, 2016, which for the Company is January 1, 2017. Early adoption is permitted. The new standard is to be applied prospectively. The Company does not expect ASU 2015-11 to have a material impact on its consolidated financial statements.
In September 2015, the FASB issued ASU No. 2015-16, Simplifying the Accounting for Measurement-Period Adjustments (ASU 2015-16). The amended guidance requires that an acquirer recognize adjustments to provisional amounts that are identified during the measurement period in the reporting period in which the adjustment amounts are determined. The amendments are effective prospectively for the fiscal years, and the interim reporting periods within those years, beginning on or after December 15, 2015 and early adoption is permitted. The Company has elected not to early adopt ASU 2015-16.
In November 2015, the FASB issued ASU No. 2015-17, Balance Sheet Classification of Deferred Taxes (ASU 2015-17), which requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. ASU is effective for the Company in its first quarter of fiscal 2017, with early application permitted and, upon adoption, may be applied either prospectively or retrospectively. The Company early adopted the provisions of ASU 2015-17 for the year ended December 31, 2015 on a prospective basis.
| -59- |
In February 2016, the FASB issued ASU No. 2016-02, Leases (ASU 2016-02), which supersedes ASC 840 “Leases” and creates a new topic, ASC 842 "Leases." This update requires lessees to recognize on their balance sheet a lease liability and a lease asset for all leases, including operating leases, with a term greater than 12 months. The update also expands the required quantitative and qualitative disclosures surrounding leases. This update is effective for fiscal years beginning after December 15, 2018 and interim periods within those fiscal years, with earlier application permitted. This update will be applied using a modified retrospective transition approach for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. The Company is currently evaluating the effect of this update on its consolidated financial statements.
NOTE 3 : Marketable Securities
The Company has investments in available-for-sale marketable equity securities which are recorded at fair market value and measured using quoted prices in their respective active market, thus representing a level 1 fair value measurement as defined in ASC 820. Unrealized gains and losses are recorded as other comprehensive income (loss) in shareholders’ equity, net of income tax effects.
The value at cost, gross unrealized holding gains or losses, and fair value of the Company’s available-for-sale marketable securities are summarized below as of December 31:
| 2015 | 2014 | |||||||
| Value at cost | $ | 79,885 | $ | 53,272 | ||||
| Gross unrealized holding gains | - | - | ||||||
| Gross unrealized holding losses | (147 | ) | (198 | ) | ||||
| Fair value | $ | 79,738 | $ | 53,074 | ||||
NOTE 4 : Inventories
The principal categories of inventories at December 31, 2015 and 2014 are as follows:
| 2015 | 2014 | |||||||
| Finished Goods | $ | 2,545 | $ | 5,068 | ||||
| Raw Materials | 1,610 | 1,661 | ||||||
| Total | $ | 4,155 | $ | 6,729 | ||||
NOTE 5 : Property and Equipment, net
The principal categories of property and equipment, net at December 31, 2015 and 2014 are as follows:
| 2015 | 2014 | |||||||
| Laboratory equipment | $ | 9,963 | $ | 9,801 | ||||
| Office and computer equipment | 2,968 | 3,288 | ||||||
| Furniture, fixtures and fittings | 4,315 | 4,544 | ||||||
| Less - accumulated depreciation | (14,630 | ) | (15,857 | ) | ||||
| Total | $ | 2,616 | $ | 1,776 | ||||
Depreciation expense for the years ended December 31, 2015, 2014 and 2013 was $568, $681 and $774, respectively.
| -60- |
NOTE 6 : Goodwill and Intangible Assets
The Company's amortizable and unamortizable intangible assets at December 31, 2015 and 2014 are as follows:
| 2015 | 2014 | |||||||||||||||||||||||
| Gross Value | Accumulated Amortization | Net
Book Value | Gross Value | Accumulated Amortization | Net
Book Value | |||||||||||||||||||
| Amortizable intangible assets: | ||||||||||||||||||||||||
| Acquired IPR&D - Bloxiverz | $ | 35,248 | $ | (23,498 | ) | $ | 11,750 | $ | 35,248 | $ | (11,749 | ) | $ | 23,499 | ||||||||||
| Acquired IPR&D - Vazculep | 12,061 | (7,986 | ) | 4,075 | 12,061 | (7,171 | ) | 4,890 | ||||||||||||||||
| Total amortizable intangible assets | $ | 47,309 | $ | (31,484 | ) | $ | 15,825 | $ | 47,309 | $ | (18,920 | ) | $ | 28,389 | ||||||||||
| Unamortizable intangible assets: | ||||||||||||||||||||||||
| Goodwill | 19,035 | (544 | ) | 18,491 | 19,035 | (544 | ) | 18,491 | ||||||||||||||||
| Total unamortizable intangible assets | $ | 19,035 | $ | (544 | ) | $ | 18,491 | $ | 19,035 | $ | (544 | ) | $ | 18,491 | ||||||||||
The Company recorded amortization expense related to amortizable intangible assets of $12,564, $11,749 and $0 for the years ended December 31, 2015, 2014 and 2013, respectively. Accumulated amortization in the above tables includes an impairment charge of $7,171 which was recognized during the year ended December 31, 2012 relative to the Acquired IPR&D – Vazculep intangible asset.
Amortizable intangible assets are amortized over their estimated useful lives, which range from three to six years, using the straight-line method. Total future amortization of intangible assets for the next five years is as follows:
| Years ending December 31, | Estimated Amortization Expense | |||
| 2016 | $ | 12,565 | ||
| 2017 | 815 | |||
| 2018 | 815 | |||
| 2019 | 815 | |||
| 2020 | 815 | |||
| Total | $ | 15,825 | ||
During the years ended December 31, 2015 and 2014, the Company did not acquire any intangible assets or goodwill.
NOTE 7 : Long-Term Debt
French government agencies provide financing to French companies for research and development. At December 31, 2015 and 2014, the Company had outstanding loans of $1,118 and $3,717, respectively for various programs. These loans do not bear interest and are repayable only in the event the research project is technically or commercially successful. Potential repayment is scheduled to occur from 2015 through 2019.
During the years ended December 31, 2015, 2014 and 2013, the Company repaid $747, $355 and $475, of loans associated with specific research projects, respectively. In addition, during 2015 the Company received waivers of repayment for the remaining portion of certain loans of $1,498 on the basis of limited commercial and technical success. Amounts waived are reported as reductions to R&D expenses in the Company’s consolidated statements of income. No such waivers were received during 2014 or 2013.
| -61- |
NOTE 8 : Long-Term Contingent Consideration Payable
Long-term contingent consideration payable and related activity are reported at fair value and consist of the following at December 31, 2015 and 2014:
| Activity During Year Ended December 31, 2015 | ||||||||||||||||||||
| Changes in
Fair Value of Contingent Consideration | ||||||||||||||||||||
Balance, December 31, 2014 | Payments to
Related Parties | Operating
Expense; Acquisition- Related | Interest Expense; Financing- Related | Balance, December 31, 2015 | ||||||||||||||||
| Acquisition-related: | ||||||||||||||||||||
| Warrants - Éclat Pharmaceuticals (a) | $ | 34,542 | $ | - | $ | (13,925 | ) | $ | - | $ | 20,617 | |||||||||
| Earn-out payments - Éclat Pharmaceuticals (b) | 70,112 | (24,526 | ) | 44,882 | - | 90,468 | ||||||||||||||
| Financing-related: | ||||||||||||||||||||
| Royalty agreement - Deerfield (c) | 6,837 | (2,282 | ) | - | 3,307 | 7,862 | ||||||||||||||
| Royalty agreement - Broadfin (d) | 3,259 | (1,089 | ) | - | 1,576 | 3,746 | ||||||||||||||
| Total contingent consideration | $ | 114,750 | $ | (27,897 | ) | $ | 30,957 | $ | 4,883 | $ | 122,693 | |||||||||
| Less: Current portion | (39,892 | ) | (28,614 | ) | ||||||||||||||||
| Total long-term contingent consideration payable | $ | 74,858 | $ | 94,079 | ||||||||||||||||
Each of the above items is associated with related parties as further described in Footnote 18 – Related Parties.
| (a) | As part of the consideration for the Company’s acquisition of Éclat Pharmaceuticals, LLC on March 13, 2012, the Company issued two warrants with a six-year term which allow for the purchase of a combined total of 3,300 ordinary shares of Flamel. One warrant is exercisable for 2,200 shares at an exercise price of $7.44 per share, and the other warrant is exercisable for 1,100 shares at an exercise price of $11.00 per share. |
The fair value of the warrants is estimated on a quarterly basis using a Black-Scholes option pricing model with the following assumptions as of December 31, 2015 and 2014:
| 2015 | 2014 | |||||||
| Weighted average exercise price per share | $ | 8.63 | $ | 8.63 | ||||
| Expected term (years) | 2.63 | 3.63 | ||||||
| Expected volatility | 64.54 | % | 54.80 | % | ||||
| Risk-free interest rate | 0.93 | % | 1.25 | % | ||||
| Expected dividend yield | - | - | ||||||
These Black-Scholes fair value measurements are based on significant inputs not observable in the market and thus represent a level 3 measurement as defined in ASC 820. The fair value of the warrant consideration is most sensitive to movement in the Company’s share price and expected volatility at the balance sheet date.
Expected term: The expected term of the options or warrants represents the period of time between the grant date and the time the options or warrants are either exercised or forfeited, including an estimate of future forfeitures for outstanding options or warrants. Given the limited historical data and the grant of stock options and warrants to a limited population, the simplified method has been used to calculate the expected life.
Expected volatility: The expected volatility is calculated based on an average of the historical volatility of the Company's stock price for a period approximating the expected term.
Risk-free interest rate: The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant and a maturity that approximates the expected term.
Expected dividend yield: The expected dividend yield is based on the Company's authorized periodic dividend and the Company's expectation for dividend yields over the expected term. The Company has not distributed any dividends since its inception, and has no plan to distribute dividends in the foreseeable future.
| -62- |
At the closing date of the 2012 Éclat acquisition, it was uncertain as to whether the Company would ultimately fulfill its obligation under these warrants using Company shares and therefore would be required to settle these warrants by transferring cash. Accordingly, pursuant to the guidance of ASC 480, the Company determined that these warrants should be classified as a long-term liability. This classification as a long-term liability was further supported by the Company’s determination, pursuant to the guidance of ASC 815-40-15-7(i), that these warrants could also not be considered as being indexed to the Company’s own stock, on the basis that the exercise price for the warrants is determined in U.S. dollars, although the functional currency of the Company at the closing date of the Éclat acquisition was the Euro.
| (b) | As part of the consideration for the Company’s acquisition of Éclat Pharmaceuticals, LLC in March 2012, the Company committed to provide quarterly earn-out payments equal to 20% of any gross profit generated by certain Éclat Pharmaceuticals products. These payments will continue in perpetuity. |
| (c) | As part of a February 2013 debt financing transaction conducted with Deerfield Management, a related party and current shareholder, the Company received cash of $2,600 in exchange for entering into a royalty agreement whereby the Company shall pay quarterly a 1.75% royalty on the net sales of certain Éclat Pharmaceuticals products until December 31, 2024. |
| (d) | As part of a December 2013 debt financing transaction conducted with Broadfin Healthcare Master Fund, a related party and current shareholder, the Company received cash of $2,200 in exchange for entering into a royalty agreement whereby the Company shall pay quarterly a 0.834% royalty on the net sales of certain Éclat Pharmaceuticals products until December 31, 2024. |
The fair value of each contingent consideration item in (b), (c) and (d) above was estimated separately on a quarterly basis by using a discounted cash flow model based on probability-adjusted annual net sales or gross profit, as appropriate, of each of the specified Éclat Pharmaceuticals products at an appropriate discount rate. These fair value measurements are based on significant inputs not observable in the market and thus represent a level 3 measurement as defined in ASC 820. Subsequent changes in the fair value of the acquisition-related and financing-related contingent consideration payable, resulting primarily from management’s revision of key assumptions, will be recorded in the changes in fair value of related party acquisition-related contingent consideration and interest expense – changes in fair value of related party financing-related contingent consideration lines, respectively, within the Company’s Consolidated Statements of Income.
NOTE 9 : Income Taxes
The components of income (loss) before income taxes consist of the following for the years ended December 31:
| 2015 | 2014 | 2013 | ||||||||||
| United States | $ | 101,241 | $ | (89,939 | ) | $ | (53,864 | ) | ||||
| France | 6,622 | (392 | ) | (3,889 | ) | |||||||
| Ireland | (29,469 | ) | - | - | ||||||||
| Total income (loss) before income taxes | $ | 78,394 | $ | (90,331 | ) | $ | (57,753 | ) | ||||
The income tax provision (benefit) consists of the following for the years ended December 31,:
| 2015 | 2014 | 2013 | ||||||||||
| Current: | ||||||||||||
| United States - Federal | $ | 33,748 | $ | - | $ | - | ||||||
| United States - State | 980 | - | - | |||||||||
| France | 1,657 | 1,400 | 76 | |||||||||
| Total current | 36,385 | 1,400 | 76 | |||||||||
| Deferred: | ||||||||||||
| United States - Federal | 1,830 | (2,457 | ) | (9,905 | ) | |||||||
| United States - State | 1,267 | (350 | ) | (1,415 | ) | |||||||
| France | (1,747 | ) | - | - | ||||||||
| Total deferred | 1,350 | (2,807 | ) | (11,320 | ) | |||||||
| Income tax provision (benefit) | $ | 37,735 | $ | (1,407 | ) | $ | (11,244 | ) | ||||
| -63- |
The items accounting for the difference between the income tax provision (benefit) computed at the French statutory rate and the Company's effective tax rate are as follows for the years ended December 31,:
| 2015 | 2014 | 2013 | ||||||||||
| Statutory tax rate | 33.3 | % | 33.3 | % | 33.3 | % | ||||||
| Non-deductible changes in fair value of contingent consideration | 13.8 | % | (24.7) | % | (16.6) | % | ||||||
| Change in valuation allowance | (9.5) | % | 5.2 | % | (4.5) | % | ||||||
| Income tax deferred charge | - | (16.9 | )% | - | ||||||||
| International tax rates differential | 10.9 | % | 6.7 | % | 6.2 | % | ||||||
| Other | (0.4) | % | (2.0) | % | 1.1 | % | ||||||
| Effective income tax rate | 48.1 | % | 1.6 | % | 19.5 | % | ||||||
| Income tax provision (benefit) - at Statutory tax rate | $ | 26,105 | $ | (30,080 | ) | $ | (19,232 | ) | ||||
| Non-deductible changes in fair value of contingent consideration | 10,835 | 22,326 | 9,592 | |||||||||
| Change in valuation allowance | (7,425 | ) | (4,732 | ) | 2,616 | |||||||
| Income tax deferred charge | - | 15,273 | - | |||||||||
| International tax rates differential | 8,559 | (6,026 | ) | (3,609 | ) | |||||||
| Other | (339 | ) | 1,832 | (611 | ) | |||||||
| Income tax provision (benefit) - at Effective income tax rate | $ | 37,735 | $ | (1,407 | ) | $ | (11,244 | ) | ||||
The Company's effective tax rates for the years ended December 31, 2015, 2014 and 2013 were 48.1%, 1.6% and 19.5%, respectively. Increased expenses reported during 2015, 2014 and 2013 due to changes in fair value of contingent consideration are not deductible for income tax purposes, and therefore unfavorably impact the Company’s effective income tax rate. In 2013, valuation allowances were placed against the deferred tax assets generated by pre-tax losses in France. In 2014, the Company entered into an intra-entity transaction to transfer intellectual property from France to Ireland which resulted in a charge of $15,273 after utilization of net operating loss carryforwards that were previously fully reserved with a valuation allowance. In 2015 certain valuation allowances were reversed with respect to pre-tax income of our U.S. and French operations as the Company had sufficient taxable income to utilize net operating losses. These rates also reflect the additional tax impacts of the Company's significant U.S. operations at a higher statutory tax rate than the French statutory tax rate, and in 2015 are additionally unfavorably impacted by losses incurred at our Irish entity at a lower statutory tax rate.
Deferred Tax Assets (Liabilities)
Deferred income tax provisions reflect the effect of temporary differences between consolidated financial statement and tax reporting of income and expense items. The net deferred tax assets/liabilities at December 31, 2015 and 2014 resulted from the following temporary differences:
| 2015 | 2014 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 44,587 | $ | 66,167 | ||||
| Stock based compensation | 1,767 | - | ||||||
| Fair value royalty agreements | 2,435 | 2,130 | ||||||
| Other | 1,025 | 1,039 | ||||||
| Total deferred tax assets | 49,814 | 69,336 | ||||||
| Valuation allowances | (45,516 | ) | (57,980 | ) | ||||
| Net deferred tax assets | 4,298 | 11,356 | ||||||
| Deferred tax liabilities: | ||||||||
| Amortization of IPR&D | (5,649 | ) | (11,356 | ) | ||||
| Total deferred tax liabilities | (5,649 | ) | (11,356 | ) | ||||
| Net deferred tax assets (liabilities) | $ | (1,351 | ) | $ | - | |||
The net operating loss carryforwards at December 31, 2015 relate primarily to Irish and French operations. While these net operating losses have no expiration in France or Ireland, in France they are limited to annual utilization of €1,000 plus fifty per cent (50%) of any taxable income in excess of €1,000. The company continually reviews the adequacy of the valuation allowance. A valuation allowance is recorded if, based on the weight of available evidence, it is more likely than not that a deferred tax asset will not be realized. This assessment is based on an evaluation of the level of historical taxable income and projections for future taxable income. The Company has provided valuation allowances covering 100% of these deferred tax assets as of December 31, 2015.
At December 31, 2015, the Company has no unremitted earnings outside of France as measured on a US GAAP basis. Whereas the measure of earnings for purposes of taxation of a distribution may be different for tax purposes, these earnings, which are considered to be invested indefinitely, would become subject to income tax if they were remitted as dividends or if the Company were to sell its stock in the subsidiaries. It is not practicable to estimate the amount of deferred tax liability on such earnings.
| -64- |
The Company and its subsidiaries file income tax returns in France, Ireland and U.S. federal and various state jurisdictions. The Company is no longer subject to, with limited exceptions, French income tax examinations by tax authorities for years prior to 2013. Any other tax period or jurisdiction in which the Company files remains subject to potential tax examination by the respective tax authorities.
Certain vested and exercised employee equity compensation awards have resulted in tax deductions in excess of previously recorded tax benefits based on the value of such equity compensation awards at the time of grant, and have been recorded as additional paid-in capital. The excess tax benefit from stock-based compensation recognized in 2015 is $2,814.
Research and Development Tax Credits Receivable
The French government provides tax credits to companies for spending on innovative R&D. These credits are recorded as an offset of R&D expenses and are credited against income taxes payable in each of the four years after being incurred or, if not so utilized, are recoverable in cash. As of December 31, 2015, the Company’s net Research tax credit receivable amounts to $2,382 and represents a gross research tax credit of $3,720, partially offset by current income tax payable of $1,338.
Income Tax Deferred Charge
On December 16, 2014, the Company transferred all of its intangible intellectual property from its French entity to its Irish entity as a part of a global reorganization. The intellectual property includes patents on drug delivery platforms, clinical data sets and other intangible assets related to the pipeline of proprietary products in development. This intra-entity transaction resulted in a charge of $14,088 of related taxes to the French government in December 2014. As this represents an intra-entity transaction, no deferred tax asset has been recognized, but rather was originally recorded as $986 of prepaid expenses and $13,102 of a long-term Income tax deferred charge asset in accordance with ASC 740-10-25-3 (e). This income tax deferred charge asset is amortized over the tax life of the asset at a rate of 7% per year and will result in tax relief in Ireland of $8.5 million from 2016 to 2029. At December 31, 2015, the balance of these respective accounts was classified as prepaid expenses of $842 and Income tax deferred charge asset of $11,581. This deferred charge asset will not be changed by future events other than the sale or amortization, including market write-down or impairment measured on a pretax basis, of the related asset in Ireland. No impairment has been identified in connection with the impairment test performed as of December 31, 2015.
NOTE 10 : Post-Retirement Benefit Plans
Post-Retirement Benefit Contributions to French Government Agencies
The Company is required by French law to deduct specific monthly payroll amounts to support post-retirement benefit programs sponsored by the relevant government agencies in France. As the ultimate obligation is maintained by the French government agencies, there is no additional liability recorded by the Company in connection with these plans. Expenses recognized for these plans were $573 in 2015, $719 in 2014, and $701 in 2013.
Retirement Indemnity Obligation – France
French law requires the Company to provide for the payment of a lump sum retirement indemnity to French employees based upon years of service and compensation at retirement. The retirement indemnity has been actuarially calculated on the assumption of voluntary retirement at a government-defined retirement age. Benefits do not vest prior to retirement. Any actuarial gains or losses are recognized in the Company’s consolidated statements of income in the periods in which they occur.
The benefit obligation is calculated as the present value of estimated future benefits to be paid, using the following assumptions:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Compensation rate increase | 3.00 | % | 3.00 | % | 3.00 | % | ||||||
| Discount rate | 2.03 | % | 1.49 | % | 3.25 | % | ||||||
| Employee turn-over | ------------ Actuarial standard and average of the last 5 years ----------- | |||||||||||
| Average age of retirement | ----- 60 to 65 years actuarial standard based on age and professional status ------ | |||||||||||
Certain actuarial assumptions, such as discount rate, have a significant effect on the amounts reported for net periodic benefit cost and accrued retirement indemnity benefit obligation amounts. The discount rate is determined annually by benchmarking a published long-term bond index using the iBoxx € Corporates AA 10+ index.
| -65- |
Changes in the funded status of the retirement indemnity benefit plans were as follows:
| 2015 | 2014 | |||||||
| Retirement indemnity benefit obligation, beginning of year | $ | 2,350 | $ | 2,142 | ||||
| Service cost | 117 | 99 | ||||||
| Interest cost | 20 | 36 | ||||||
| Plan amendments | - | - | ||||||
| Benefits paids | (46 | ) | (87 | ) | ||||
| Actuarial loss (gain) | (27 | ) | 460 | |||||
| Exchange rate changes | (244 | ) | (300 | ) | ||||
| Retirement indemnity benefit obligation, end of year | $ | 2,170 | $ | 2,350 | ||||
The lump sum retirement indemnity is accrued on the Company’s Consolidated Balance Sheets within non-current other liabilities, excluding the current portion. As these are not funded benefit plans, there are no respective assets recorded.
The future expected benefits to be paid over the next five years and for the five years thereafter is as follows:
Retirement indemnity benefit obligation | ||||
| Expected benefit payments for year ending December 31,: | ||||
| 2016 | $ | - | ||
| 2017 | - | |||
| 2018 | - | |||
| 2019 | 11 | |||
| 2020 | - | |||
| Next five years | 221 | |||
NOTE 11 : Other Assets and Liabilities
Various other assets and liabilities are summarized as follows for the year ending December 31,:
| 2015 | 2014 | |||||||
| Prepaid expenses and other current assets | ||||||||
| Valued-added tax recoverable | $ | 1,099 | $ | 1,077 | ||||
| Prepaid expenses | 2,846 | 3,225 | ||||||
| Advance to suppliers and other current assets | 518 | 116 | ||||||
| Income tax receivable | 3,526 | - | ||||||
| Total | $ | 7,989 | $ | 4,418 | ||||
| 2015 | 2014 | |||||||
| Accrued expenses | ||||||||
| Accrued compensation | $ | 1,888 | $ | 3,792 | ||||
| Accrued social charges | 1,710 | 1,875 | ||||||
| Total | $ | 3,598 | $ | 5,667 | ||||
| 2015 | 2014 | |||||||
| Other current liabilities | ||||||||
| R&D credit tax financing short term | $ | - | $ | 5,382 | ||||
| Provision for retirement indemnity short term | - | 55 | ||||||
| Other | 133 | 235 | ||||||
| Total | $ | 133 | $ | 5,672 | ||||
| -66- |
| 2015 | 2014 | |||||||
| Other non-current liabilities | ||||||||
| Provision for retirement indemnity | $ | 2,170 | $ | 2,295 | ||||
| Other | 40 | 38 | ||||||
| Total | $ | 2,210 | $ | 2,333 | ||||
NOTE 12 : Contingent Liabilities and Commitments
Litigation
The Company is subject to potential liabilities generally incidental to its business arising out of present and future lawsuits and claims related to product liability, personal injury, contract, commercial, intellectual property, tax, employment, compliance and other matters that arise in the ordinary course of business. The Company accrues for potential liabilities when it is probable that future costs (including legal fees and expenses) will be incurred and such costs can be reasonably estimated. At December 31, 2015 and 2014, there were no contingent liabilities with respect to any threat of litigation, arbitration or administrative or other proceeding that are reasonably likely to have a material adverse effect on the Company’s consolidated financial position, results of operations, cash flows or liquidity.
Material Commitments
The Company has commitments to purchase services from Recipharm Pessac for a total of $22,500 for a five-year period commencing January 1, 2015 (disclosed in Note 17- Discontinued Operations) and to purchase one batch per year for the next five years of the generic pharmaceutical product it markets for $46 per year.
The Company and its subsidiaries lease office facilities under noncancelable operating leases expiring at various dates. Rent expense was $752, $844 and $759 in 2015, 2014, and 2013, respectively. Minimum rental commitments for non-cancelable leases in effect at December 31, 2015 are as follows:
| 2016 | $ | 784 | ||
| 2017 | 472 | |||
| 2018 | 463 | |||
| 2019 | 389 | |||
| 2020 | 310 | |||
| Thereafter | - | |||
| Total | $ | 2,418 |
Other than the above commitments to Recipharm and for operating leases, there were no other material commitments outside of the normal course of business. Material commitments in the normal course of business include long-term debt, long-term contingent consideration payable, and post-retirement benefit plan obligations which are disclosed in Note 7 - Long-Term Debt , Note 8 – Long-term Contingent Consideration Payable, and Note 10 – Post-Retirement Benefit Plans, respectively.
NOTE 13 : Equity Instruments and Stock Based Compensation
Compensation expense included in the Company’s Consolidated Statements of Income for all stock-based compensation arrangements was as follows:
| 2015 | 2014 | 2013 | ||||||||||
| Cost of products and services sold | $ | - | $ | 38 | $ | 20 | ||||||
| Research and development expenses | 1,587 | 1,027 | 779 | |||||||||
| Selling, general and administrative expenses | 6,154 | 1,798 | 1,230 | |||||||||
| Total stock-based compensation expense | $ | 7,741 | $ | 2,863 | $ | 2,029 | ||||||
As of December 31, 2015, the Company expects $13,297 of unrecognized expense related to granted, but nonvested stock-based compensation arrangements to be incurred in future periods. This expense is expected to be recognized over a weighted average period of 1.7 years.
The Company recognized a tax benefit related to stock-based compensation of $1,767 for the year ended December 31, 2015.
Upon exercise of stock options or warrants, or upon the issuance of free share awards, the Company issues new shares.
| -67- |
Determining the Fair Value of Stock Options and Warrants
The Company measures the total fair value of stock options and warrants on the grant date using the Black-Scholes option-pricing model and recognizes each grant's fair value as compensation cost over the period that the option or warrant vests. Options are granted to employees of the Company and generally become exercisable within four years following the grant date and expire ten years after the grant date. Warrants are typically issued to the Company’s Board of Directors as compensation for services rendered and generally become exercisable within one year following the grant date, and expire four years after the grant date.
The weighted-average assumptions under the Black-Scholes option-pricing model for stock option and warrant grants are as follows:
| 2015 | 2014 | 2013 | ||||||||||
| Stock Option grants: | ||||||||||||
| Expected term (years) | 6.25 | 6.25 | 6.25 | |||||||||
| Expected volatility | 58.59 | % | 59.00 | % | 58.15 | % | ||||||
| Risk-free interest rate | 1.89 | % | 1.79 | % | 1.66 | % | ||||||
| Expected dividend yield | - | - | - | |||||||||
| Warrant grants: | ||||||||||||
| Expected term (years) | 2.50 | 2.50 | 2.50 | |||||||||
| Expected volatility | 55.00 | % | 58.00 | % | 54.00 | % | ||||||
| Risk-free interest rate | 0.89 | % | 0.75 | % | 0.47 | % | ||||||
| Expected dividend yield | - | - | - | |||||||||
Expected term: The expected term of the options or warrants represents the period of time between the grant date and the time the options or warrants are either exercised or forfeited, including an estimate of future forfeitures for outstanding options or warrants. Given the limited historical data and the grant of stock options and warrants to a limited population, the simplified method has been used to calculate the expected life.
Expected volatility: The expected volatility is calculated based on an average of the historical volatility of the Company's stock price for a period approximating the expected term.
Risk-free interest rate: The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant and a maturity that approximates the expected term.
Expected dividend yield: The expected dividend yield is based on the Company's authorized periodic dividend and the Company's expectation for dividend yields over the expected term. The Company has not distributed any dividends since its inception, and has no plan to distribute dividends in the foreseeable future.
Stock Options
A summary of the combined stock option activity and other data for the Company's stock option plans for the year ended December 31, 2015 is as follows:
| Number of Stock Options | Wtd. Avg. Exercise Price per Share | Wtd. Avg. Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||
| Stock options outstanding, January 1, 2015 | 2,498 | $ | 11.45 | |||||||||||
| Granted | 934 | 16.71 | ||||||||||||
| Exercised | (708 | ) | 8.75 | |||||||||||
| Forfeited | (398 | ) | 14.59 | |||||||||||
| Stock options outstanding, December 31, 2015 | 2,326 | $ | 13.84 | 8.26 years | $ | 6,426 | ||||||||
| Stock options exercisable, December 31, 2015 | 798 | $ | 10.80 | 6.12 years | $ | 4,880 | ||||||||
The aggregate intrinsic value of options exercised during the years ended December 31, 2015, 2014 and 2013 was $10,063 and $3,789, respectively. There were no options exercised in 2013.
The weighted average grant date fair value of options granted during the years ended December 31, 2015, 2014 and 2013 was $9.38, $9.19 and $3.36 per share, respectively.
| -68- |
At December 31, 2015, there were 97 shares authorized for stock option grants in subsequent periods.
Warrants
A summary of the combined warrant activity and other data for the year ended December 31, 2015 is as follows:
| Number of Warrants | Wtd. Avg. Exercise Price per Share | Wtd. Avg. Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||
| Warrants outstanding, January 1, 2015 | 553 | $ | 10.47 | |||||||||||
| Granted | 305 | 21.67 | ||||||||||||
| Exercised | (190 | ) | 5.57 | |||||||||||
| Forfeited | - | - | ||||||||||||
| Warrants outstanding, December 31, 2015 | 668 | $ | 16.97 | 2.90 years | $ | 687 | ||||||||
| Warrants exercisable, December 31, 2015 | 363 | $ | 13.04 | 2.41 years | $ | 687 | ||||||||
The aggregate intrinsic value of warrants exercised during the years ended December 31, 2015, 2014 and 2013 was $2,698, $3,107 and $2, respectively.
The weighted average grant date fair value of warrants granted during the years ended December 31, 2015, 2014 and 2013 was $5.92, $3.90 and $1.50 per share, respectively.
At December 31, 2015, an additional 3,300 warrants were outstanding and exercisable relative to consideration paid for the Company’s acquisition of Éclat Pharmaceuticals, LLC on March 13, 2012. These warrants are not considered stock-based compensation and are therefore excluded from the above tables, and instead are addressed within Note 8 - Long-Term Contingent Consideration Payable.
At December 31, 2015, there were 45 shares authorized for warrant grants in subsequent periods.
Free Share Awards
Free share awards represent Company shares issued free of charge to employees of the Company as compensation for services rendered. The Company measures the total fair value of free share awards on the grant date using the Company's stock price at the time of the grant. Free share awards generally cliff vest at the end of a four year vesting period, and are expensed over this vesting period.
A summary of the Company's free share awards as of December 31, 2015, and changes during the year then ended, is reflected in the table below.
| Number of Free Share Awards | Wtd. Avg. Grant Date Fair Value | |||||||
| Nonvested free share awards outstanding, January 1, 2015 | 402 | $ | 11.29 | |||||
| Granted | - | - | ||||||
| Vested | (151 | ) | 7.36 | |||||
| Forfeited | (25 | ) | 10.95 | |||||
| Nonvested free shares awards outstanding, December 31, 2015 | 226 | $ | 13.95 | |||||
The weighted average grant date fair value of free share awards granted during the years ended December 31, 2014 and 2013 was $16.30 and $7.36, respectively. There were no free share awards granted in 2015.
At December 31, 2015, there were 250 shares authorized for free share award grants in subsequent periods.
NOTE 14 : Earnings (Loss) Per Share
Basic earnings (loss) per share is calculated using the weighted average number of shares outstanding during each period. The diluted earnings (loss) per share calculation includes the impact of dilutive equity compensation awards and contingent consideration warrants.
A reconciliation of basic and diluted earnings (loss) per share, together with the related shares outstanding in thousands for the years ended December 31, is as follows:
| -69- |
| 2015 | 2014 | 2013 | ||||||||||
| Net income (loss) from continuing operations | $ | 40,659 | $ | (88,924 | ) | $ | (46,509 | ) | ||||
| Net income (loss) from discontinued operations | - | 4,018 | 3,584 | |||||||||
| Net income (loss) | $ | 40,659 | $ | (84,906 | ) | $ | (42,925 | ) | ||||
| Weighted average shares: | ||||||||||||
| Basic shares | 40,580 | 36,214 | 25,450 | |||||||||
| Effect of dilutive securities—options and warrants outstanding | 3,039 | - | - | |||||||||
| Diluted shares | 43,619 | 36,214 | 25,450 | |||||||||
| Earnings (loss) per share - Basic: | ||||||||||||
| Continuing operations | $ | 1.00 | $ | (2.45 | ) | $ | (1.83 | ) | ||||
| Discontinued operations | - | 0.11 | 0.14 | |||||||||
| Net income (loss) | $ | 1.00 | $ | (2.34 | ) | $ | (1.69 | ) | ||||
| Earnings (loss) per share - Diluted: | ||||||||||||
| Continuing operations | $ | 0.93 | $ | (2.45 | ) | $ | (1.83 | ) | ||||
| Discontinued operations | - | 0.11 | 0.14 | |||||||||
| Net income (loss) | $ | 0.93 | $ | (2.34 | ) | $ | (1.69 | ) | ||||
Potential common shares of 635, 6,753, and 7,753 were excluded from the calculation of weighted average shares for the years ended December 31, 2015, 2014 and 2013, because their effect was considered to be anti-dilutive. For the years ended December 31, 2014 and 2013, the effects of dilutive securities were entirely excluded from the calculation of earnings per share as a net loss was reported in these periods.
NOTE 15 : Comprehensive Income (Loss)
The following table shows the components of accumulated other comprehensive income (loss) for the twelve months ended December 31, 2015, 2014, and 2013 net of immaterial tax effects:
| Foreign Currency Translation Adjustment Income (Loss), Net | Unrealized Gain (Loss) on Marketable Securities, Net | Total | ||||||||||
| Balance - January 1, 2013 | $ | 10,253 | $ | - | $ | 10,253 | ||||||
| Other comprehensive income | 562 | - | 562 | |||||||||
| Balance - December 31, 2013 | 10,815 | - | 10,815 | |||||||||
| Other comprehensive loss | (18,040 | ) | (198 | ) | (18,238 | ) | ||||||
| Balance - December 31, 2014 | (7,225 | ) | (198 | ) | (7,423 | ) | ||||||
| Other comprehensive loss | (15,087 | ) | (147 | ) | (15,234 | ) | ||||||
| Balance - December 31, 2015 | $ | (22,312 | ) | $ | (345 | ) | $ | (22,657 | ) | |||
The effect on the Company’s consolidated financial statements of amounts reclassified out of Accumulated other comprehensive income was immaterial for all years presented.
NOTE 16 : Company Operations by Product, Customer and Geographic Area
The Company has determined that it operates in one segment, the development and commercialization of pharmaceutical products, including controlled-release therapeutic products based on its proprietary polymer based technology. The Company's Chief Operating Decision Maker is the CEO. The CEO and the Board review profit and loss information on a consolidated basis to assess performance and make overall operating decisions as well as resource allocations. All products are included in one segment because the majority of our products have similar economic and other characteristics, including the nature of the products and production processes, type of customers, distribution methods and regulatory environment.
| -70- |
The following table presents a summary of total revenues by these products:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Bloxiverz | $ | 150,283 | $ | 10,211 | $ | - | ||||||
| Vazculep | 20,151 | - | - | |||||||||
| Dextroamphetamine | 1,946 | 1,643 | - | |||||||||
| Other | 108 | 139 | 1,153 | |||||||||
| Total product sales and services | 172,488 | 11,993 | 1,153 | |||||||||
| License and research revenue | 721 | 2,782 | 3,026 | |||||||||
| Total revenues | $ | 173,209 | $ | 14,775 | $ | 4,179 | ||||||
The following table presents a summary of total revenues by significant wholesaler customer:
| Years Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
| Customer A | $ | 60,620 | $ | 3,659 | $ | 354 | ||||||
| Customer B | 53,988 | 3,937 | 541 | |||||||||
| Customer C | 43,434 | 3,563 | (124 | ) | ||||||||
| Other | 14,446 | 834 | 382 | |||||||||
| Total product sales and services | 172,488 | 11,993 | 1,153 | |||||||||
| License and research revenue | 721 | 2,782 | 3,026 | |||||||||
| Total revenues | $ | 173,209 | $ | 14,775 | $ | 4,179 | ||||||
The following table summarizes revenues and non-monetary long-lived assets by geographic region. Non-monetary long-lived assets primarily consist of property and equipment, goodwill and intangible assets:
| 2015 | 2014 | 2013 | ||||||||||
| Revenues, for the years ended December 31, | ||||||||||||
| United States | $ | 172,379 | $ | 14,302 | $ | 3,368 | ||||||
| France | 89 | 473 | 811 | |||||||||
| Ireland | 741 | - | - | |||||||||
| Total | $ | 173,209 | $ | 14,775 | $ | 4,179 | ||||||
| Long-lived assets, as of December 31, | ||||||||||||
| United States | $ | 34,515 | $ | 47,077 | $ | 58,868 | ||||||
| France | 2,317 | 1,704 | 2,307 | |||||||||
| Ireland | 258 | - | - | |||||||||
| Total | $ | 37,090 | $ | 48,781 | $ | 61,175 | ||||||
NOTE 17 : Discontinued Operations
On December 1, 2014, the Company signed an Asset Purchase Agreement with Recipharm AB (“Recipharm”) to divest its development and manufacturing facility and associated business located in Pessac, France. The assets included in the divestiture were tangible equipment, furniture and fixtures, inventories and all intellectual property rights relating to the operation and technological know-how necessary in manufacturing the products that are produced in the facility, as well as the assignment to Recipharm of all employees, customer contracts and liabilities which primarily relate to agreements of the Company with GlaxoSmithKline (“GSK”) for the manufacture and sale of Coreg CR®, which was Flamel’s lead product at the time, using its Micropump drug delivery platform and manufactured in the Pessac Facility.
The aggregate consideration received for the divested assets and business was $13,200, plus the value of divested inventory as determined using inventory valuation methodology as defined by the two parties. All cash and receivables pertaining to the Pessac Facility business prior to the sale were retained by the Company. A contribution of $700 was made by the Company to finance potential future retirement indemnities payable on transferred employees. The business was accounted for as a discontinued operation in the fourth quarter of 2014 and, therefore, the operating results of our Pessac Facility business were included in Discontinued Operations in the Company’s consolidated financial statements for all applicable years presented. The Company recognized a $5,007 gain on disposal, which was included in our income from Discontinued Operations, in fiscal year 2014. Concurrently with the above, Recipharm made an investment of $13,000 in newly issued Flamel shares, the purchase price of which was based on the average of the trailing 20 days’ trading prices of the Company’s shares prior to the closing date.
| -71- |
In connection with the Asset Purchase Agreement, the Company also entered into a number of other agreements with Recipharm:
Master Agreement on Supply and Services of Products (“MSA”)
Recipharm will provide various services in the domain of R&D and manufacture of pharmaceutical products for an initial non-cancellable period of five years.
Over the initial term, any services to be provided to shall include internal and external costs incurred by Recipharm plus 20%, which has been determined to be fair value for such services. The minimum amount of services per year, for a cumulative total of $22,500 as follows:
| Year 1 | $ | 4,250 | ||
| Year 2 | $ | 4,250 | ||
| Year 3 | $ | 4,250 | ||
| Year 4 & 5 (each) | $ | 4,860 |
During the year ended December 31, 2015, the Company recorded $4,089 of research and development expenses, and cash outflows of $5,679, related to this commitment to Recipharm.
Option Agreement
Recipharm has a first option (right of first refusal) to discuss and negotiate licenses of the Company’s intellectual property rights for the sale of certain products in Europe. Upon exercise of the option, Recipharm and the Company shall agree in good faith on terms and conditions of the related license agreement within forty-five (45) days from the exercise of the option. The term of the Option Agreement is from the signing of the agreement through December 31, 2017. The Company received no compensation related to the option agreement.
Summary results of operations for the divested Pessac business were as follows for the years ended December 31,:
| 2015 | 2014 | 2013 | ||||||||||
| Revenues | $ | - | $ | 14,967 | $ | 18,265 | ||||||
| Operating income (loss) | - | (875 | ) | 3,667 | ||||||||
| Gain on disposal | - | 5,007 | - | |||||||||
| Interest Expense | - | (4 | ) | (9 | ) | |||||||
| Income tax provision (benefit) | - | 110 | 74 | |||||||||
| Net income from discontinued operations | $ | - | $ | 4,018 | $ | 3,584 | ||||||
Carrying amounts of major classes of assets and liabilities classified as held for sale in the Consolidated balance sheets are as follows as of December 31,:
| 2015 | 2014 | |||||||
| Accounts receivable, net | $ | - | $ | 730 | ||||
| Total assets of the disposal group classified as held for sale | - | 730 | ||||||
| Accounts payable | - | 168 | ||||||
| Total liabilities of the disposal group classified as held for sale | $ | - | $ | 168 | ||||
The major cash flows related to Discontinued Operations as included in the Consolidated statements of cash flows are as follows for the years ended December 31,:
| 2015 | 2014 | 2013 | ||||||||||
| Capital expenditures | $ | - | $ | 1,271 | $ | 872 | ||||||
| Depreciation and amortization | - | 1,709 | 1,751 | |||||||||
| Operating and investing non-cash elements | - | (740 | ) | (676 | ) | |||||||
| -72- |
NOTE 18 : Related Party Transactions
In March 2012, the Company acquired all of the membership interests of Éclat from Breaking Stick Holdings, L.L.C. (“Breaking Stick”, formerly Éclat Holdings), an affiliate of Deerfield Capital L.P (“Deerfield”), a significant shareholder of the Company. As of December 31, 2015 and 2014, the remaining consideration obligations for this transaction consisted of two warrants to purchase a total of 3,300 shares of Flamel and commitments to make earnout payments to Breaking Stick of 20% of any gross profit generated by certain Éclat products (the “Products”). Breaking Stick is majority owned by Deerfield, with a minority interest owned by Mr. Michael Anderson, the Company’s CEO, and certain other current and former employees. The original consideration for the acquisition of Éclat also included a $12 million senior note payable to the majority owners of Breaking Stick, which was fully repaid in March 2014 using the net proceeds from the Company’s public offering of ADS’s.
As part of a February 2013 debt financing transaction conducted with Deerfield Management, Éclat entered into a Royalty Agreement with Horizon Santé FLML, Sarl and Deerfield Private Design Fund II, L.P., both affiliates of the Deerfield Entities (together, “Deerfield PDF/Horizon”). The Royalty Agreement provides for Éclat to pay Deerfield PDF/Horizon 1.75% of the net sales of the Products sold by the Company and any of its affiliates until December 31, 2024, with royalty payments accruing daily and paid in arrears for each calendar quarter during the term of the Royalty Agreement. The Company has also entered into a Security Agreement dated February 4, 2013 with Deerfield PDF/Horizon, whereby Deerfield PDF/Horizon was granted a security interest in the intellectual property and regulatory rights related to the Products to secure the obligations of Éclat and Flamel US, including the full and prompt payment of royalties to Deerfield PDF/Horizon under the Royalty Agreement. This original Deerfield debt financing transaction also included a $15 million facility agreement which was repaid in full in March 2014 using the net proceeds from the Company’s public offering of ADS’s.
As part of a December 2013 debt financing transaction conducted with Broadfin Healthcare Master Fund (“Broadfin”), the Company entered into a Royalty Agreement with Broadfin, a significant shareholder of the Company, dated as of December 3, 2013 (the “Broadfin Royalty Agreement”). Pursuant to the Broadfin Royalty Agreement, the Company is required to pay a royalty of 0.834% on the net sales of certain products sold by the Company and any of its affiliates until December 31, 2024. This original Broadfin debt financing transaction also included a $5 million facility agreement which was repaid in full in March 2014 using the net proceeds from the Company’s public offering of ADS’s.
NOTE 19 : Subsequent Event
On February 8, 2016, the Company entered into an agreement to acquire FSC Holdings, LLC (“FSC”), a Charlotte, NC-based specialty pharmaceutical company dedicated to providing innovative solutions to unmet medical needs for pediatric patients, from Deerfield CSF, LLC, a Deerfield Management company (“Deerfield”), a related party. Under the terms of the acquisition, which was completed on February 8, 2016, the Company will pay $1,050 annually for five years with a final payment in January 2021 of $15,000 for a total of $20,250 to Deerfield for all of the equity interests in FSC. The Company will also pay Deerfield a 15% royalty per annum on net sales of the current FSC products, up to $12,500 for a period not exceeding ten years. The Company is in the initial stages of allocating the purchase price to the acquired net assets of FSC.
| -73- |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Flamel Technologies SA
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of income/(loss), of comprehensive income/(loss), of shareholders’ equity and of cash flows present fairly, in all material respects, the financial position of Flamel Technologies SA and its subsidiaries at December 31, 2015 and December 31, 2014, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2015 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the index appearing under Item 15(a)(2) presents fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company did not maintain, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) because material weaknesses in internal control over financial reporting existed as of that date.
The material weaknesses related to 1) an insufficient complement of personnel with an appropriate level of knowledge, experience and training commensurate with the Company’s financial reporting requirements, 2) a lack of effective controls over segregation of duties and restricted access for processes, including an assessment of incompatible management responsibilities at its US subsidiary relating to a) journal entries as the Company has not designed or maintained effective controls over the review, approval and documentation related to journal entries recorded, b) third party vendors and third party payments as the Company has not designed or maintained effective controls over the review, approval, ongoing maintenance of third party vendor contracts and vendor payments, c) cash payments as the Company does not have an adequate process to ensure the safeguarding of the Company’s assets, d) payroll and related accounts as the Company has not designed or maintained effective controls over the payroll process, 3) revenue as the Company has not designed or maintained effective controls over product pricing and rebate arrangements and the use of service providers in the revenue process, 4) income taxes as the Company has not designed or maintained effective controls related to the completeness, accuracy and valuation of income taxes process, 5) information technology general controls and key spreadsheets as the Company has not designed or maintained effective controls over information technology general controls and key spreadsheets, 6) financial close process as the Company has not designed or maintained effective controls over the financial close process, 7) monitoring controls as the Company did not maintain an internal audit function sufficient to monitor control activities.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. The material weaknesses referred to above are described in Management's Report on Internal Control Over Financial Reporting, appearing under Item 9a. We considered these material weaknesses in determining the nature, timing, and extent of audit tests applied in our audit of the December 31, 2015 consolidated financial statements, and our opinion regarding the effectiveness of the Company’s internal control over financial reporting does not affect our opinion on those consolidated financial statements. The Company's management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in management's report referred to above. Our responsibility is to express opinions on these financial statements, on the financial statement schedule, and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
As discussed in Note 2 to the consolidated financial statements, the Company changed the manner in which it accounts for the classification of deferred income tax balances in 2015.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Lyon, France,
March 15, 2016
PricewaterhouseCoopers Audit
| Represented by | |
| /s/ Frédéric Charcosset | |
| Frédéric Charcosset |
| -74- |
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. |
None.
| Item 9A. | Controls and Procedures. |
Disclosure Controls and Procedures
Management of the Company, with the participation of its Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of the Company’s disclosure controls and procedures as of December 31, 2015, the end of the period covered by this annual report on Form 10-K. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (“Exchange Act”), means controls and other procedures of a company that are designed to provide reasonable assurance that information required to be disclosed by the Company in the reports it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures are also designed to provide reasonable assurance that such information is accumulated and communicated to the Company’s management, including its Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosure. Based on their evaluation, as of the end of the period covered by this Form 10-K, the Company’s Chief Executive Officer and Chief Financial Officer have concluded that the Company’s disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) were not effective at the reasonable assurance level because of the material weaknesses in our internal control over financial reporting described below.
Management, including our Chief Executive Officer and Chief Financial Officer, believes the consolidated financial statements included in this annual report on Form 10-K fairly present in all material respects our financial condition, results of operations and cash flows at the end of and for the periods presented in accordance with U.S. GAAP.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed by, or under the supervision of, our CEO and CFO, or persons performing similar functions, and effected by the Company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. In addition, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management of the Company has assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2015. In making its assessment of internal control over financial reporting, management used the criteria described in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis.
Management has identified the following control deficiencies that constituted material weaknesses in our internal control over financial reporting as of December 31, 2015:
| · | Lack of sufficient personnel: We did not maintain a sufficient number of personnel with an appropriate level of knowledge, experience and training in internal control over financial reporting commensurate with our financial reporting requirements. |
| · | Segregation of duties: As a consequence of the above weakness we have not designed or maintained effective controls over segregation of duties and restricted access for processes, including an assessment of incompatible management responsibilities relating to the following: |
| o | Journal entries: We have not designed or maintained effective controls over the review, approval and documentation related to journal entries recorded in the US. |
| o | Third party vendors and third party payments: We have not designed or maintained effective controls in the US over the review, approval, ongoing maintenance of third party vendor contracts and vendor payments. |
| o | Cash payment process: We have not designed or maintained effective controls over the cash payment process in the US. We do not have an adequate process to ensure the safeguarding of the Company’s assets to support the proper functioning of internal controls. |
| o | Payroll and related accounts: We have not designed or maintained effective controls over the US payroll process. |
| -75- |
| · | Revenue: We have not designed or maintained effective controls over the revenue process. Specifically, we have not designed or maintained effective controls over the review and approval of product prices and subsequent changes to customer prices, the authorization, review and accounting for rebate arrangements included in customer contracts and the company’s use of service providers in the revenue process. |
| · | Income taxes: We have not designed or maintained effective controls over the income tax process. Specifically, we have not designed or maintained effective controls related to the income tax process on a quarterly or year-end basis, including deferred tax reconciliations, valuation allowances, review of state income taxes and related apportionment, review of tax returns and return to provision differences, review of information provided to and received from the outsourced tax provider, review of effective income tax rates and any uncertain tax positions. |
| · | Information technology general controls and key spreadsheets: We have not designed or maintained effective controls over information technology general controls and key spreadsheets used within certain processes and management’s controls to support account balances or in the preparation of the financial statements. |
| · | Financial close process: We have not designed or maintained effective controls over the financial close process. |
| · | Monitoring controls: We did not design and maintain effective monitoring controls related to the design and operational effectiveness of our internal controls. Specifically, we did not maintain an internal audit function sufficient to monitor control activities. |
These control deficiencies could result in misstatements that could be pervasive to the financial statements and disclosures which would result in a material misstatement of the consolidated financial statements that would not be prevented or detected. Accordingly, our management has determined that these control deficiencies constitute material weaknesses.
Because of these material weaknesses, management concluded that the Company did not maintain effective internal control over financial reporting as of December 31, 2015, based on criteria in Internal Control-Integrated Framework (2013) issued by the COSO.
The effectiveness of our internal control over financial reporting as of December 31, 2015 has been audited by PricewaterhouseCoopers Audit, our independent registered public accounting firm. Their report appears in Item 8. Financial Statements and Supplementary Data of this annual report on Form 10-K.
Remediation Plan and Status
Management believes that the rapid growth of the size and complexity of the Company’s business during 2015, without a commensurate growth in the size and expertise of our finance and accounting organizations contributed to the weaknesses described above.
We have commenced and continue to identify and implement actions to improve the effectiveness of our internal control over financial reporting and disclosure controls and procedures. These plans include but are not limited to, adding to our finance staff and enhancing our training with respect to financial reporting and disclosure responsibilities. Recently, we hired a Chief Financial Officer and Chief Accounting officer, and expect to hire a tax professional, all of whom have the appropriate experience, certification, education, and training in financial reporting and accounting. Additionally, to assist in mitigating some of the segregation of duties weaknesses, we expect to complete the implementation of a new information technology system in 2016 that will employ a more robust and effective set of general computer and access controls. We will review our remediation plans with our audit committee during 2016 and periodically update them with our progress. Leading this remediation process is our Senior Vice President and Chief Financial Officer, who was hired in November 2015, with the assistance of our Chief Accounting Officer, who was hired in January 2016. Further, our Board of Directors recognizes the critical importance of a sound internal control structure and has directed senior management to ensure that a proper, consistent tone is communicated throughout the organization, which emphasizes the expectation that these deficiencies will be rectified through implementation of more effective processes and controls. To assist management in this regard, the Company has engaged a nationally recognized consulting firm to enhance existing documentation and implement improvements or changes to the existing internal control structure.
While our remediation actions described above represent significant progress to enhance our internal control over financial reporting relating to the identified material weaknesses, we continue to implement and test the effectiveness of these actions and procedures and additional time is required to complete implementation and to assess and ensure the sustainability of the resulting enhancements. We believe the above actions, together with any modifications thereto which we may determine to be appropriate and such further additional remedial steps we may identify during 2016, will ultimately be effective in remediating the material weaknesses described above, and we will continue to devote significant time and attention to these remedial efforts. However, the material weaknesses cannot be considered remediated until the applicable remedial enhancements operate for a sufficient period of time and management has concluded, through testing, that our internal controls are operating effectively.
| -76- |
Changes in Internal Control Over Financial Reporting
As described above under "Remediation Plan and Status”, there were changes to our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) that occurred during the most recent fiscal quarter and annual period that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information. |
None.
| -77- |
Certain information required by Part III is omitted from this annual report on Form 10-K because we intend to file our definitive proxy statement for our 2016 annual general meeting of shareholders pursuant to Regulation 14A of the Securities Exchange Act of 1934 (our “Definitive 2016 Proxy Statement”), not later than 120 days after the end of the fiscal year covered by this annual report on Form 10-K, and certain information to be included in our Definitive 2016 Proxy Statement is incorporated herein by reference.
| Item 10. | Directors, Executive Officers and Corporate Governance. |
Information regarding Directors, Executive Officers and Corporate Governance is hereby incorporated by reference to our Definitive 2016 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2015.
| Item 11. | Executive Compensation. |
Information regarding Executive Compensation is hereby incorporated by reference to our Definitive 2016 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2015.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
Information regarding Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters is hereby incorporated by reference to our Definitive 2016 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2015.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
Information regarding Certain Relationships and Related Transactions, and Director Independence is hereby incorporated by reference to our Definitive 2016 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2015.
| Item 14. | Principal Accountant Fees and Services. |
Information regarding Principal Accountant Fees and Services is hereby incorporated by reference to our Definitive 2016 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2015..
| -78- |
| Item 15. | Exhibits and Financial Statement Schedules |
| (a) | Documents Filed as Part of this Report: |
| 1. | Financial Statements See Item 8 - Financial Statements and Supplementary Data of Part II of this Report |
| 2. | Financial Statement Schedules See below for Schedule II: Valuation and Qualifying Accounts. All other schedules are omitted as they are not applicable, not required or the information is included in the consolidated financial statements or related notes to the consolidated financial statements |
Schedule II
Valuation and Qualifying Accounts
(in thousands)
| Balance
at beginning of period | Additions: Charges to expense (a) | Deductions
(b) | Other
changes (c) | Balance
at end of period | ||||||||||||||||
| Deferred tax asset valuation allowance: | ||||||||||||||||||||
| 2015 | $ | 57,980 | $ | 4,312 | $ | (11,737 | ) | $ | (5,039 | ) | $ | 45,516 | ||||||||
| 2014 | 69,939 | 8,453 | (13,185 | ) | (7,227 | ) | 57,980 | |||||||||||||
| 2013 | 64,356 | 2,616 | - | 2,967 | 69,939 | |||||||||||||||
| a) | Additions to the deferred tax asset valuation allowance relate to movements on certain French, Irish and U.S. deferred tax assets where we continue to maintain a valuation allowance until sufficient positive evidence exists to support reversal. |
| b) | Deductions to the deferred tax asset valuation allowance include movements relating to utilization of NOLs and tax credit carryforwards, release in valuation allowance and other movements including adjustments following finalization of tax returns. |
| c) | Other changes to the deferred tax asset valuation allowance relate primarily to currency translation adjustments recorded directly in equity. |
| 3. | Exhibits The exhibits listed on the following Index to Exhibits are filed as part of this annual report on Form 10-K. |
| -79- |
Index to Exhibits
| Number | Description | |
| 3.1 | Revised Statuts or ByLaws of the Company (1) | |
| 4.1 | Guaranty of Note made by Flamel Technologies S.A. in favor of Éclat Holdings, LLC, dated March 13, 2012 (2) | |
| 4.2 | Warrant to purchase 2,200,000 American Depositary Shares, each representing one Ordinary Share of Flamel Technologies S.A. (2) | |
| 4.3 | Warrant to purchase 1,100,000 American Depositary Shares, each representing one Ordinary Share of Flamel Technologies S.A. (2) | |
| 10.1 | Amended and Restated Deposit Agreement among Flamel, The Bank of New York, as Depositary, and holders from time to time of American Depositary Shares issued thereunder (including as an exhibit the form of American Depositary Receipt) (3) | |
| 10.2* | Note Agreement among Flamel Technologies S.A., Flamel US Holdings, Inc. and Éclat Holdings, LLC, dated March 13, 2012 (2) | |
| 10.3 | Registration Rights Agreement between Flamel Technologies S.A. and Éclat Holdings, LLC, dated March 13, 2012 (2) | |
| 10.4 | Facility Agreement among Flamel US Holdings, Deerfield Private Design Fund II, L.P. and Deerfield Private Design International II, L.P dated December 31, 2012 (4) | |
| 10.5* | Royalty Agreement among Eclat Pharmaceuticals LLC, Horizon Santé FLML, Sarl and Deerfield Private Design Fund II, L.P dated December 31, 2012 (4) | |
| 10.6* | Security Agreement between Éclat Pharamaceuticals, LLC and Deerfield Private Design Fund II, L.P. and Horizon Santé FLML, Sarl, dated February 4, 2013 (4) | |
| 10.7 | Broadfin Facility Agreement, effective as of December 3, 2013 (5) | |
| 10.8* | Broadfin Royalty Agreement, dated as of December 3, 2013 (5) | |
| 10.9 | Asset Purchase Agreement by and among Flamel Technologies and Recipharm Pessac, dated November 26, 2014 (1) | |
| 10.10 | Master Agreement on Supply of Services and Products by and between Flamel Technologies and Recipharm Pessac, dated December 1, 2014 (1) | |
| 10.11 | Service Agreement by and between Flamel Technologies and Recipharm Pessac, dated December 1, 2014 (1) | |
| 10.12 | Supply Agreement by and between Flamel Technologies and Recipharm Pessac, dated December 1, 2014 (1) | |
| 10.13* | Membership Interest Purchase Agreement by and among Éclat Holdings LLC, Éclat Pharmaceuticals LLC, Flamel Technologies S.A. and Flamel US Holdings Inc., dated March 13, 2012 (1) | |
| 10.14* | License Agreement by and between Elan Pharma International Limited and Flamel Ireland Limited, dated September 30, 2015 (Filed herewith) | |
| 10.15 | Lease Agreement by and between Nine East, LLC and Eclat Pharmaceuticals LLC, dated July 23, 2013 (Filed herewith) | |
| 10.16 | Lease Agreement by and between Grove II LLC and Eclat Pharmaceuticals LLC, dated October 5, 2015 (Filed herewith) | |
| 10.17 | Lease Agreement by and between Channor Limited, Blanchardstown Corporate Park Management Limited, Flamel Ireland Limited, and Flamel Technologies S.A., dated July 3, 2015 (Filed herewith) | |
| -80- |
| 10.18‡ | Employment Agreement by and between Flamel Technologies S.A. and Sandra Hatten, dated July 8th, 2015 (Filed herewith) | |
| 10.19‡ | Employment Agreement by and between Flamel Technologies S.A. and Phillandas T. Thompson, dated July 7th, 2015 (Filed herewith) | |
| 10.20 | Membership Interest Purchase Agreement dated as of February 5, 2016, by and among James Flynn, Peter Steelman, Deerfield CSF, LLC, FSC Holding Company, LLC, FSC Therapeutics, LLC, FSC Laboratories, Inc., Flamel Technologies SA, and Flamel US Holdings, Inc. (Filed herewith) | |
| 10.21‡ | Rules Governing the Free Share Plan – December 2014 (Filed herewith) | |
| 10.22‡ | Rules Governing the Free Share Plan – December 2014 (Filed herewith) | |
| 10.23‡ | June 2015 Stock Warrant Rules (Filed herewith) | |
| 10.24‡ | Subscription Form of Stock Warrant (Filed herewith) | |
| 10.25‡ | December 2015 Stock Option Rules (Filed herewith) | |
| 10.26‡ | Form of Stock Option Grant Letter (Filed herewith) | |
| 14.1 | Code of Ethics for CEO (Directeur Général), Delegated Managing Directors (Directeurs Généraux Délégués) and Senior Financial Officers (6) | |
| 21.1 | List of Subsidiaries (Filed herewith) | |
| 23.1 | Consent of PricewaterhouseCoopers Audit (Filed herewith) | |
| 31.1 | Certification of the Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Filed herewith) | |
| 31.2 | Certification of the Principal Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Filed herewith) | |
| 32.1 | Certification of the Chief Executive Officer pursuant to USC Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (Furnished herewith) | |
| 32.2 | Certification of the Principal Financial Officer pursuant to USC Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (Furnished herewith) | |
| 101.INS | XBRL Instant Document | |
| 101.SCH | XBRL Taxonomy Extension Schema Document | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document |
| (1) | Incorporated by reference to the Company’s Annual Report on Form 20-F for the year ended December 31, 2014, filed on April 30, 2015. |
| (2) | Incorporated by reference to the Company’s Current Report on Form 6-K, filed March 21, 2012. |
| (3) | Incorporated by reference to the Company’s registration statement on Form F-6 filed February 12, 2014, as amended (No. 333-193892). |
| (4) | Incorporated by reference to the Company’s Annual Report on Form 20-F for the year ended December 31, 2012, filed on April 30, 2013. |
| (5) | Incorporated by reference to the Company’s Annual Report on Form 20-F for the year ended December 31, 2013, filed on April 30, 2014. |
| (6) | Incorporated by reference to the Company’s Annual Report on Form 20-F for the year ended December 31, 2003, filed on April 26, 2004. |
| * | Confidential treatment has been requested for the redacted portions of this agreement. A complete copy of the agreement, including the redacted portions, has been filed separately with the Securities and Exchange Commission. |
| ‡ | Management contract or compensatory plan or arrangement filed pursuant to Item 15(b) of Form 10-K. |
| -81- |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| FLAMEL TECHNOLOGIES S.A. | ||
| Dated: March 15, 2016 | By: | /s/ Michael S. Anderson |
| Name: Michael S. Anderson | ||
| Title: Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each of each of Craig R. Stapleton, Guillaume Cerutti, Francis J.T. Fildes, Benoit Van Assche and Christophe Navarre, by their respective signatures below, irrevocably constitutes and appoints Michael S. Anderson and Phillandas T. Thompson, and each of them individually acting alone without the other, his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either of them, or their or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
| Signature | Title | Date | ||
| /s/ Michael S. Anderson | Chief Executive Office (Principal Executive Officer) and Director | March 15, 2016 | ||
| Michael S. Anderson | ||||
| /s/ Michael F. Kanan | Chief Financial Officer (Principal Financial Officer) | March 15, 2016 | ||
| Michael F. Kanan | ||||
| /s/ David P. Gusky | Corporate Controller (Principal Accounting Officer) | March 15, 2016 | ||
| David P. Gusky | ||||
| /s/ Craig R. Stapleton | Non-Executive Chairman of the Board and Director | March 15, 2016 | ||
| Craig R. Stapleton | ||||
| /s/ Guillaume Cerutti | Director | March 15, 2016 | ||
| Guillaume Cerutti | ||||
| /s/ Francis J.T. Fildes | Director | March 15, 2016 | ||
| Francis J.T. Fildes | ||||
| /s/ Benoit Van Assche | Director | March 15, 2016 | ||
| Benoit Van Assche | ||||
| /s/ Christophe Navarre | Director | March 15, 2016 | ||
| Christophe Navarre |
| -82- |