Attached files
| file | filename |
|---|---|
| EX-10.40 - EXHIBIT 10.40 - AVADEL PHARMACEUTICALS PLC | a123119ex-1040.htm |
| EX-32.2 - EXHIBIT 32.2 - AVADEL PHARMACEUTICALS PLC | a123119ex-322.htm |
| EX-32.1 - EXHIBIT 32.1 - AVADEL PHARMACEUTICALS PLC | a123119ex-321.htm |
| EX-31.2 - EXHIBIT 31.2 - AVADEL PHARMACEUTICALS PLC | a123119ex-312.htm |
| EX-31.1 - EXHIBIT 31.1 - AVADEL PHARMACEUTICALS PLC | a123119ex-311.htm |
| EX-23.1 - EXHIBIT 23.1 - AVADEL PHARMACEUTICALS PLC | a123119ex-231.htm |
| EX-21.1 - EXHIBIT 21.1 - AVADEL PHARMACEUTICALS PLC | a123119ex-211.htm |
| EX-4.6 - EXHIBIT 4.6 - AVADEL PHARMACEUTICALS PLC | a123119ex-46.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2019
Commission file number: 000-28508
AVADEL PHARMACEUTICALS PLC
(Exact name of registrant as specified in its charter)
Ireland | 98-1341933 | |
State or other jurisdiction of incorporation or organization | (I.R.S. Employer Identification No.) | |
10 Earlsfort Terrace Dublin 2, Ireland D02 T380 | Not Applicable | |
(Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: +011-1-485-1200 | ||
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol (s) | Name of exchange on which registered |
American Depositary Shares* | AVDL | The Nasdaq Global Market |
Ordinary Shares, nominal value $0.01 per share** | AVDL | The Nasdaq Global Market |
* | American Depositary Shares may be evidenced by American Depository Receipts. Each American Depositary Share represents one (1) Ordinary Share. | |
** | Not for trading, but only in connection with the listing of American Depositary Shares. on The Nasdaq Global Market. | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically, every Interactive Data File required to be submitted and pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ý No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer ý
Non-accelerated filer ¨ Smaller reporting company ¨
Emerging growth company ¨
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No ý
The aggregate market value of voting stock held by non-affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter was $97,947,088 based on the closing sale price of the registrant’s American Depositary Shares as reported by the Nasdaq Global Market on June 28, 2019. Such market value excludes 3,464,274 ordinary shares, $0.01 per share nominal value, held by each officer and director and by shareholders that the registrant concluded were affiliates of the registrant on that date. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
The number of the registrant’s ordinary shares, $0.01 per share nominal value, outstanding as of March 10, 2020 was 46,404,432.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of either (a) a definitive proxy statement involving the election of directors or (b) an amendment to this Form 10-K, either of which will be filed within 120 days after December 31, 2019, are incorporated by reference into Part III of this Form 10-K.
-1-
TABLE OF CONTENTS
Page # | ||
Item 1. | ||
Item 1A. | ||
Item 1B. | ||
Item 2. | ||
Item 3. | ||
Item 4. | ||
Item 5. | ||
Item 6. | ||
Item 7. | ||
Item 7A. | ||
Item 8. | ||
Item 9. | ||
Item 9A. | ||
Item 9B. | ||
Item 10. | ||
Item 11. | ||
Item 12. | ||
Item 13. | ||
Item 14. | ||
Item 15. | ||
-2-
Cautionary Disclosure Regarding Forward-Looking Statements
This Annual Report on Form 10-K includes “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Any statements about our expectations, beliefs, plans, objectives, assumptions or future events or performance are not historical facts and may be forward-looking. These statements are often, but are not always, made through the use of words or phrases such as “may,” “will,” “could,” “should,” “expects,” “intends,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “projects,” “potential,” “continue,” and similar expressions, or the negative of these terms, or similar expressions. Accordingly, these statements involve estimates, assumptions, risks and uncertainties which could cause actual results to differ materially from those expressed in them. Any forward-looking statements are qualified in their entirety by reference to the factors discussed throughout this prospectus, and in particular those factors referenced in the section “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K.
This Annual Report on Form 10-K contains forward-looking statements that are based on our management’s belief and assumptions and on information currently available to our management. These statements relate to future events or our future financial performance, and involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
• | Our reliance on a single product candidate, FT218, the expected completion of the Phase III clinical trial for FT218 and our ability to obtain regulatory approval of and successfully commercialize FT218; |
• | Our plans and expectations regarding the effectiveness of our restructuring plan announced in February 2019, including our ability to achieve the desired cost savings; |
• | Any further restructuring actions that may be required and our ability to obtain any required consents (including any consents required pursuant to the Indenture governing our exchange notes due 2023, or the 2023 Notes); |
• | Our reliance on a small number of products to generate all or substantially all of our revenue and the competitive pressures that these products face; |
• | The lack of patent protection for three of our approved products, Bloxiverz, Vazculep and Akovaz; |
• | Our ability to successfully launch Nouress in the United States; |
• | Our ability to develop and obtain U.S. Food and Drug Administration (“FDA”), approval for any future potential “unapproved marketed drug” product candidates in the future; |
• | Our ability to continue to service the 2023 Notes, including making the ongoing interest payments on the 2023 Notes, settling exchanges of the 2023 Notes in cash or completing any required repurchases of the 2023 Notes; |
• | The ability of our product candidates and products to gain market acceptance; |
• | Our ability to enter into strategic partnerships for the development, commercialization, manufacturing and distribution of our products and product candidates; |
• | Our dependence on a limited number of suppliers for the manufacturing of our products and certain raw materials in our products and any failure of such suppliers to deliver sufficient quantities of these raw materials, which could have a material adverse effect on our business; |
• | Our ability to finance our operations on acceptable terms, either through the raising of capital, the incurrence of convertible or other indebtedness or through strategic financing or commercialization partnerships; |
• | Our expectations about the potential market sizes and market participation potential for our approved or proposed products; |
• | Our ability to retain members of our management team and our employees; and |
• | Competition existing today or that will likely arise in the future. |
These forward-looking statements are neither promises nor guarantees of future performance due to a variety of risks and uncertainties and other factors more fully discussed in the “Risk Factors” section in Part I, Item 1A of this Annual Report on Form 10-K and the risk factors and cautionary statements described in other documents that we file from time to time with the SEC. Given these uncertainties, readers should not place undue reliance on our forward-looking statements. These forward-looking statements speak only as of the date on which the statements were made and are not guarantees of future performance. Except as may be required by applicable law, we do not undertake to update any forward-looking statements after the date of this Annual Report or the respective dates of documents incorporated by reference herein or therein that include forward-looking statements.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to revise any forward-looking statements to reflect events or developments occurring after the date of this Annual Report, even if new information becomes available in the future.
-3-
NOTE REGARDING TRADEMARKS
The Company is the owner of the Avadel, Akovaz, Bloxiverz, Vazculep, Nouress, Micropump, Liquitime, and Medusa trademarks, as well as certain other trademarks, including design versions of some of these trademarks. The symbols ™ and ® may not be used in connection with the presentation of these trademarks in this report, but the absence of such symbols does not indicate a lack of trademark rights. Certain other trademarks used in this report are the property of third-party trademark owners and may similarly be presented with or without trademark references.
-4-
PART I
Item 1. Business.
(Dollar amounts in thousands, except per-share amounts and as otherwise noted)
General Overview
Avadel Pharmaceuticals plc (Nasdaq: AVDL) (“Avadel,” the “Company,” “we,” “our,” or “us”) is an emerging biopharmaceutical company. Our lead product candidate, FT218, is an investigational once-nightly formulation of sodium oxybate for the treatment of excessive daytime sleepiness (“EDS”), and cataplexy in narcolepsy patients. FT218, which uses our Micropump drug-delivery technology, is in a Phase 3 clinical trial for the treatment of narcolepsy patients suffering from EDS and cataplexy. In addition, we have three approved commercial products developed under our “unapproved marketed drug,” or UMD, program, Akovaz, Bloxiverz and Vazculep, and a fourth approved product, Nouress, which are sterile injectable drugs used in the hospital setting.
We are primarily focused on the development and potential U.S. Food and Drug Administration (“FDA”) approval of FT218. In addition, we continue to market and distribute our current approved hospital products portfolio and, pending resolution of the existing patent infringement claim (as described below), we plan to commercialize Nouress. Outside of our product candidate and our existing commercial products, we continue to evaluate opportunities to expand our product portfolio.
FT218 (Micropump sodium oxybate)
FT218 is a once-nightly formulation of sodium oxybate that uses our Micropump controlled release drug-delivery technology for the treatment of EDS and cataplexy in patients suffering from narcolepsy. Sodium oxybate is the sodium salt of gamma hydroxybutyrate, an endogenous compound and metabolite of the neurotransmitter gamma-aminobutyric acid. Sodium oxybate is approved in Europe and the United States (“U.S.”) as a twice-nightly formulation indicated for the treatment of EDS and cataplexy in patients with narcolepsy.
In December 2019, we completed patient enrollment of our Phase 3 REST-ON clinical trial of FT218 to assess the safety and efficacy of a once-nightly formulation of FT218 for the treatment of EDS and cataplexy in patients suffering from narcolepsy. The REST-ON trial is a randomized, double-blind, placebo-controlled study that has enrolled 212 patients and is being conducted in clinical sites in the U.S., Canada, Western Europe and Australia. Top line data from the REST-ON trial is currently expected in the second quarter of 2020.
In January 2018, the FDA granted FT218 Orphan Drug Designation, which makes the drug eligible for certain development and commercial incentives, including a potential U.S. market exclusivity for up to seven years. Additionally, in April 2019, our first FT218 patent was issued, providing intellectual property protection into 2037. There are additional patent applications currently in development and/or pending at the U.S. Patent and Trademark Office (“USPTO”), as well as foreign patent offices.
We believe FT218 has the potential to demonstrate improved dosing compliance, safety and patient satisfaction over the current standard of care for EDS and cataplexy in patients with narcolepsy, which is a twice-nightly sodium oxybate formulation. If approved, we believe FT218 has the potential to take a significant share of the sodium oxybate market. The current market size for the twice-nightly administration of sodium oxybate is estimated at an annualized revenue run rate of $1.7 billion.
Micropump Drug-Delivery Technology
Our Micropump drug-delivery technology allows for the delayed delivery of small molecule drugs taken orally, which has the potential to improve dosing compliance, reduce toxicity and improve patient compliance. Beyond FT218, we believe there could be other product development opportunities for our Micropump drug delivery technology, representing either “life cycle” opportunities, whereby additional intellectual property can be added to a pharmaceutical product to extend the commercial viability of a currently marketed product, or innovative formulation opportunities for new chemical entities.
Unapproved Marketed Drugs Program
The FDA allows certain unapproved prescription drugs to be marketed if (i) they are relied on by health care professionals to treat serious medical conditions, and (ii) there is no FDA-approved drug to treat such condition or insufficient supply of FDA-approved drugs. In most cases, these prescription drugs pre-date the establishment of the FDA. Although these products are typically not protected by patents or similar intellectual property, FDA guidance states that, if it approves an NDA for any such products via a 505(b)(2) process, the FDA is more likely to seek enforcement action, such as seizure or injunction, against remaining unapproved drugs of the same type, potentially after a grace period provided by the FDA.
-5-
Existing Commercial Products
To date, we have received FDA approvals for three previously unapproved prescription drugs:
• | Bloxiverz (neostigmine methylsulfate injection) - Bloxiverz was approved by the FDA in May 2013 and was launched in July 2013. Bloxiverz is a drug used intravenously in the operating room to reverse the effects of non-depolarizing neuromuscular blocking agents after surgery. Bloxiverz was the first FDA-approved version of neostigmine methylsulfate. Today, neostigmine is one of the two most frequently used products for the reversal of the effects of other agents used for neuromuscular blocks. There are approximately 2,500 vials of neostigmine sold annually in the U.S. |
• | Vazculep (phenylephrine hydrochloride injection) - Vazculep was approved by the FDA in June 2014 and was launched in October 2014. Vazculep is indicated for the treatment of clinically important hypotension occurring in the setting of anesthesia. There are approximately 7,100 vials of Vazculep sold annually in the U.S. |
• | Akovaz (ephedrine sulfate injection). Akovaz, was approved by the FDA in April 2016 and was launched in August 2016. Akovaz was the first FDA approved formulation of ephedrine sulfate, an alpha- and beta- adrenergic agonist and a norepinephrine-releasing agent that is indicated for the treatment of clinically important hypotension occurring in the setting of anesthesia. There are approximately 7,500 vials of Akovaz sold annually in the U.S. |
Nouress
In December 2019, we received FDA approval for Nouress (cysteine hydrochloride injection), a sterile injectable product for use in the hospital setting, and currently have two patents covering that product. Several additional patent applications for Nouress are pending with the USPTO. In light of the recently filed patent suit by Exela Pharma Sciences, LLC, (“Exela”) we are currently evaluating the timing and process for a commercial launch of Nouress in the U.S. See Note 16: Contingent Liabilities and Commitments in Part II of this Form 10-K for a discussion on the filed patent suit by Exela.
We use the revenue from our UMD products to fund the research and development of FT218. In addition, we believe evaluating opportunities to commercialize other unapproved drugs in markets with a limited number of competitors may provide us with near-term revenue growth and potentially provide cash flows that can also be used to fund research and development initiatives for the development of FT218 and other potential product candidates.
Corporate Information
We were incorporated on December 1, 2015 as an Irish private limited company, and re-registered as an Irish public limited company (“plc”), on November 21, 2016. Our principal place of business is located at 10 Earlsfort Terrace, Dublin 2, Ireland and our phone number is 00 353 1 920 1000. We file annual, quarterly and current reports, proxy statements and other documents with the U.S. Securities and Exchange Commission (“SEC”) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Our website is www.avadel.com, where we make available free of charge our reports (and any amendments thereto) on Forms 10-K, 10-Q and 8-K as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. These filings are also available to the public at www.sec.gov. We do not incorporate the information on or accessible through our website into this Annual Report on Form 10-K.
We currently have five direct wholly-owned subsidiaries: (a) Avadel US Holdings, Inc., (b) Flamel Ireland Limited, which conducts business under the name Avadel Ireland, (c) Avadel Investment Company Limited, (d) Avadel Finance Ireland Designated Activity Company and (e) Avadel France Holding SAS. Avadel US Holdings, Inc., a Delaware corporation, is the holding entity of (i) Avadel Specialty Pharmaceuticals, LLC (currently the subject of a voluntary Chapter 11 bankruptcy proceeding), (ii) Avadel Legacy Pharmaceuticals, LLC, (iii) Avadel Management Corporation, (iv) FSC Holding Company, (v) Avadel Operations Company, Inc. and (vi) Avadel CNS Pharmaceuticals LLC. Avadel Finance Ireland Designated Activity Company is the holding entity of Avadel Finance Cayman Limited. Flamel Ireland Limited (operating under the trade name Avadel Ireland) is an Irish corporation. Avadel France Holding SAS, a French société par actions simplifiée, is the holding entity of Avadel Research SAS through which Avadel conducts substantially all of its R&D activities. A complete list of our subsidiaries can be found in Exhibit 21.1 to this Annual Report on Form 10-K.
-6-
Recent Developments
Shelf Registration Statement on Form S-3
In February 2020, we filed with the SEC a new shelf registration statement on Form S-3 (the 2020 Shelf Registration Statement) (File No. 333-236258) that allows issuance and sale by us, from time to time, of:
(a) | up to $250,000 in aggregate of ordinary shares, nominal value US$0.01 per share (the “Ordinary Shares”), each of which may be represented by ADSs, preferred shares, nominal value US$0.01 per share (the “Preferred Shares”), debt securities (the “Debt Securities”), warrants to purchase Ordinary Shares, ADSs, Preferred Shares and/or Debt Securities (the “Warrants”), and/or units consisting of Ordinary Shares, ADSs, Preferred Shares, one or more Debt Securities or Warrants in one or more series, in any combination, pursuant to the terms of the 2020 Shelf Registration Statement, the base prospectus contained in the 2020 Shelf Registration Statement (the “Base Prospectus”), and any amendments or supplements thereto (together, the “Securities”); including |
(b) | up to $50,000 of ADSs that may be issued and sold from time to time pursuant to the terms of an Open Market Sale AgreementSM (“the Sales Agreement”), entered into with Jefferies LLC on February 4, 2020 (the “Sales Agreement”), the 2020 Shelf Registration Statement, the Base Prospectus and the terms of the sales agreement prospectus contained in the 2020 Shelf Registration Statement. |
Securities Purchase Agreement
On February 21, 2020, we announced that we have entered into a definitive agreement for the sale of our ADSs and Series A Non-Voting Convertible Preferred Shares (“Series A Preferred”) in a private placement to a group of institutional accredited investors. The private placement has resulted in gross proceeds of approximately $65,000 before deducting placement agent and other offering expenses, and resulting in net proceeds of approximately $61,000.
Pursuant to the terms of the private placement, we issued 8,680 ADSs and 488 shares of Series A Preferred at a price of $7.09 per share, priced at-the-market under Nasdaq rules. Each share of non-voting Series A Preferred is convertible into one ADS, provided that conversion will be prohibited if, as a result, the holder and its affiliates would own more than 9.99% of the total number of Avadel ADSs outstanding. The closing of the private placement occurred on February 25, 2020. Proceeds from the private placement will be used to fund continued clinical and program development of FT218, including an open-label extension study for REST-ON, a switch study to evaluate patients switching from twice-nightly sodium oxybate to once-nightly FT218, as well as for general corporate purposes.
Competition and Market Opportunities
Competition
Competition in the pharmaceutical and biotechnology industry is intense and is expected to increase. We compete with academic laboratories, research institutions, universities, joint ventures, and other pharmaceutical and biotechnology companies, including other companies developing brand or generic specialty pharmaceutical products or drug delivery platforms. Some of these competitors may also be our business partners. There can be no assurance that our competitors will not obtain patent protection or other intellectual property rights that would make it difficult or impossible for us to compete with their products. Furthermore, major technological changes can happen quickly in the pharmaceutical and biotechnology industries. Such rapid technological change, or the development by our competitors of technologically improved or differentiated products, could render our products, including our drug delivery technologies, obsolete or noncompetitive.
The pharmaceutical industry has dramatically changed in recent years, largely as a function of the growing importance of generic drugs. The growth of generics (typically small molecules) and of large molecules (biosimilars) has been accelerated by the demand for less expensive pharmaceutical products. As a result, the pricing power of pharmaceutical companies will be more tightly controlled in the future.
In addition, the overall landscape of the Pharma/Biotech industry has changed, and consolidation has reduced our pool of potential partners and acquisition opportunities within the biopharmaceutical space.
We complete with a number of companies based upon our current marketed products and those in development. Examples of companies with whom Avadel or any of our future partners would compete, given our current products and pipeline, include Jazz Pharmaceuticals, Fresenius Kabi, Par Pharmaceuticals, Hikma Pharmaceuticals and Ferring.
-7-
Potential competition for FT218
If FT218 receives FDA approval, it will compete with the current approved twice-nightly sodium oxybate formulation, as well as a number of daytime stimulants including lisdexamfetamine, modafinil, armodafinil, solriamfetol and pitolisant, which are widely prescribed, or prescribed concomitantly with sodium oxybate. Sodium oxybate is currently the only product approved for both EDS and cataplexy. In addition, we anticipate that our FT218 product may face competition from manufacturers of generic twice-nightly sodium oxybate formulations, who have reached settlement agreements with the current marketer for entry by 2023. In addition, there are other products in development that may be approved in the future that could have an impact on the sodium oxybate market prior to FT218’s potential FDA approval, including low or no sodium versions of sodium oxybate and reboxetine.
Market Opportunities
Because the pharmaceutical industry is highly competitive, participants seek ways to increase profitability by reducing competition through patent protection. Avadel, combining its existing proprietary drug delivery technologies with the established commercial capability of our unapproved to approved product strategy has evolved into a biopharmaceutical company focusing on re-formulations and requiring shorter product development cycles by using an abbreviated NDA mechanism (505(b)(2)).
In particular, in today’s environment, a drug has to demonstrate significant therapeutic improvements over the current standard of care in order to obtain third party payer coverage. Alternatively, changes in the delivery of a drug must create a demonstrable reduction in costs. Dosing convenience, by itself, is no longer sufficient to gain reimbursement acceptance. Biopharmaceutical companies must now demonstrate, through costly Phase 3 trials, therapeutic efficacy of their new formulations. The FDA has encouraged drug companies developing enhanced formulations to use an abbreviated regulatory pathway: the 505(b)(2) NDA. Many biopharmaceutical companies today are using this approach or the supplemental NDA pathway (“sNDA”). An NDA or sNDA is necessary to market an already approved drug for a new indication, or in a different dosage form or formulation. However, the sNDA approach requires cross-referencing the originator’s drug dossier, and eventually an alliance with the originator for commercialization.
Avadel’s Drug Delivery Technologies
We own drug delivery technologies that address key formulation challenges, potentially allowing the development of differentiated drug products for administration in various forms (e.g., capsules, tablets, sachets or liquid suspensions for oral use; or injectables for subcutaneous administration) that could be applied to a broad range of drugs (novel, already-marketed, or off-patent).
A brief discussion of each of our drug delivery technologies is set forth below.
• | Micropump® Technology. Our Micropump allows for the development of modified and/or controlled release solid, oral dosage formulations of drugs. Micropump-carvedilol and Micropump-aspirin formulations have been approved in the U.S. Further, Micropump technology is being employed in our investigational FT218 product. |
• | LiquiTime®. Our LiquiTime technology allows for development of modified/controlled release oral products in a liquid suspension formulation, which may make such formulations particularly well suited for children and/or patients having issues swallowing tablets or capsules. Although we own this technology, we are currently not pursuing any commercial pharmaceutical drug development opportunities using it. |
• | Medusa™. Our Medusa technology allows for the development of extended-/modified-release injectable dosage formulations of drugs (e.g., peptides, polypeptides, proteins, and small molecules). Although we own this technology, we are currently not pursuing any commercial pharmaceutical drug development opportunities using it. |
Proprietary Intellectual Property
Parts of our product pipeline and strategic alliances utilize our drug delivery platforms and related products of which certain features are the subject of patents or patent applications. As a matter of policy, we seek patent protection of our inventions and also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to maintain and develop competitive positions.
• | FT218 Patents. We were awarded our first FT218-related U.S. patent on April 30, 2019, which is directed to modified release formulations of gamma-hydroxybutyrate (“GHB”), and which expires in 2037. We have a number of additional, FT218-related patent applications pending at the USPTO as well as at non-U.S. patent offices. |
-8-
• | Nouress Patents. We have two issued U.S. patents directed to cysteine solutions and methods of using cysteine solutions, both of which are listed in FDA’s Orange Book for Nouress, and both of which expire in 2039. Further, we have several Nouress-related patent applications pending at the USPTO. |
• | Drug Delivery Technology Patents. Our drug delivery technologies are the subject of certain patents, including: (i) for Micropump, patents relating to coating technologies that provide for delayed and sustained release of an active ingredient and that tend to allow for absorption in the upper part of the intestinal tract (expiring in 2025 in the U.S. and 2022 in non-U.S. jurisdictions); (ii) for LiquiTime, patents relating to film-coated microcapsules and a method comprising orally administering such microcapsules to a patient (expiring in 2023); and (iii) for Medusa, patents relating to an aqueous colloidal suspension of low viscosity based on submicronic particles of water-soluble biodegradable polymer PO (polyolefin) carrying hydrophobic groups (expiring in 2023). |
The patent positions of biopharmaceutical companies like us are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and patent scope can be reinterpreted by the courts after issuance. Moreover, many jurisdictions permit third parties to challenge issued patents in administrative proceedings, which may result in further narrowing or even cancellation of patent claims. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any of our licensed or owned patents will provide sufficient protection from competitors. Any of our licensed or owned patents may be challenged, circumvented, or invalidated by third parties. For more information, please see the information set forth under the caption “– Risks Related to Our Intellectual Property – If we cannot adequately protect our intellectual property and proprietary information, we may be unable to sustain a competitive advantage” in the “Risk Factors” included in Part I, Item 1A of this Annual Report on Form 10-K.
Supplies and Manufacturing
We attempt to maintain multiple suppliers in order to mitigate the risk of shortfall and inability to supply market demand. Nevertheless, for most of our products, we rely on a limited number of suppliers, and in certain cases only one supplier, for sourcing active pharmaceutical ingredients (“APIs”).
The manufacture of our sterile hospital injectable products marketed by Avadel in the U.S. is outsourced to current good manufacturing practices (“cGMP”) -compliant and FDA-audited contract manufacturing organizations (“CMOs”) pursuant to supply agreements. We will continue to outsource to third-party CMOs, and have no present plans to acquire manufacturing facilities. We believe this outsourcing policy is beneficial to us for products to be marketed in the U.S.
Government Regulation
The design, testing, manufacturing and marketing of certain new or substantially modified drugs, biological products or medical devices must be approved, cleared or certified by regulatory agencies, regulatory authorities and notified bodies under applicable laws and regulations, the requirements of which may vary from country to country. This regulatory process is lengthy, expensive and uncertain. In the U.S., the FDA regulates such products under various federal statutes, including the Federal Food, Drug, and Cosmetic Act (“FDCA”) and the Public Health Service Act.
New Drug Product Development and Approval Process
Regulation by governmental authorities in the U.S. and other countries has a significant impact on the development, manufacture, and marketing of drug products and on ongoing research and product development activities. The products of Avadel’s pharmaceutical partners as well as its own products require regulatory approval by governmental agencies and regulatory authorities prior to commercialization. In particular, these products are subject to manufacturing according to stringent requirements known as cGMP which are promulgated by the FDA in the U.S. and by other authorities in other jurisdictions, and rigorous, pre-clinical and clinical testing and other pre-market approval requirements by the FDA, the European Commission and regulatory authorities in other countries. In the U.S. and the European Union, various statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of pharmaceutical products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources.
Regulatory approval, when and if obtained, may be limited in scope. In particular, regulatory approvals may restrict the marketing of a product to specific uses. Approved drugs, as well as their manufacturers, are subject to ongoing review (including requirements and restrictions related to record keeping and reporting, FDA, European Commission and EU Member States competent authorities’ approval of certain changes in manufacturing processes or product labeling, product promotion and advertising, and
-9-
pharmacovigilance, which includes monitoring and reporting adverse reactions, maintaining safety measures, and conducting dossier reviews for marketing authorization renewal). Discovery of previously unknown problems with these products may result in restrictions on their manufacture, sale or use, or in their withdrawal from the market. Failure to comply with regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other actions affecting Avadel’s potential products and commercial prospects or the potential products and commercial prospects of Avadel’s pharmaceutical partners who may utilize Avadel’s technologies. Any failure by Avadel or our pharmaceutical partners to comply with current or new and changing regulatory obligations, and any failure to obtain and maintain, or any delay in obtaining, regulatory approvals, could materially adversely affect our business.
The process for new drug product development and approval has many steps, including:
Chemical and Formulation Development. Pharmaceutical formulation taking into account the chemistry and physical characteristics of the drug or biological substance, is the beginning of a new product. If initial laboratory experiments reveal that the concept for a new drug product looks promising, then a variety of further development steps and tests complying with internationally recognized guidance documents will have to be continued, in order to provide for a product ready for testing in animals and, after sufficient animal test results, also in humans.
Concurrent with pre-clinical studies and clinical trials, companies must continue to develop information about the properties of the drug product and finalize a process for manufacturing the product in accordance with cGMP. The manufacturing process must be capable of consistently producing quality batches of the product, and the manufacturer must develop and validate methods for testing the quality, purity and potency of the final products. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life.
Pre-Clinical Testing. Once a drug candidate is identified for development, the candidate enters the pre-clinical testing stage. This includes laboratory evaluation of product chemistry and formulation, as well as animal studies of pharmacology (mechanism of action, pharmacokinetics) and toxicology which may have to be conducted over lengthy periods of time, to assess the potential safety and efficacy of the product as formulated. Pre-clinical tests must be conducted in compliance with good laboratory practice regulations, the Animal Welfare Act and its regulations in the U.S. and the Clinical Trials Directive and related national laws and guidelines in the EU Member States. Violations of these laws and regulations can, in some cases, lead to invalidation of the studies, then requiring such studies to be replicated. In some cases, long-term pre-clinical studies are conducted while clinical studies are ongoing.
Investigational New Drug Application.
U.S. The entire body of chemical or biochemical, pharmaceutical and pre-clinical development work necessary to administer investigational drugs to human volunteers or patients is summarized in an Investigational New Drug (“IND”) application to the FDA. The IND becomes effective, if not rejected by the FDA within thirty (30) days after filing. There is no assurance that the submission of an IND will eventually allow a company to commence clinical trials. All clinical trials must be conducted under the supervision of a qualified investigator in accordance with good clinical practice regulations to ensure the quality and integrity of clinical trial results and data. These regulations include the requirement that, with limited exceptions, all subjects provide informed consent. In addition, an institutional review board (“IRB”), composed primarily of physicians and other qualified experts at the hospital or clinic where the proposed studies will be conducted, must review and approve each human study. The IRB also continues to monitor the study and must be kept aware of the study’s progress, particularly as to adverse events and changes in the research. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if adverse events occur. Failure to adhere to good clinical practices and the protocols, and failure to obtain IRB approval and informed consent, may result in FDA rejection of clinical trial results and data, and may delay or prevent the FDA from approving the drug for commercial use.
European Union. The European equivalent to the IND is the Investigational Medicinal Product Dossier (“IMPD”) which likewise must contain pharmaceutical, pre-clinical and, if existing, previous clinical information on the drug substance and product. An overall risk-benefit assessment critically analyzing the non-clinical and clinical data in relation to the potential risks and benefits of the proposed trial must also be included. The intended clinical trial must be submitted for authorization by the regulatory authority(ies) of each EU Member States in which the trial is intended to be conducted prior to its commencement. The trial must be conducted in accordance with the protocol as approved by an Ethics Committee(s) in each EU Member State (EU equivalent to IRBs). Before submitting an application to the competent authority, the sponsor must register the trial in the EudraCT database where in the U.S. it will be provided with a unique EudraCT number.
Clinical Trials. Typically, clinical testing involves the administration of the drug product first to healthy human volunteers and then to patients with conditions needing treatment under the supervision of a qualified principal investigator, usually a physician,
-10-
pursuant to a ‘protocol’ or clinical plan reviewed by the FDA and the competent authorities of the EU Member States along with the IRB or Ethics Committee (via the IND or IMPD submission). The protocol details matter such as a description of the condition to be treated, the objectives of the study, a description of the patient population eligible for the study, and the parameters to be used to monitor safety and efficacy.
Clinical trials are time-consuming and costly, and typically are conducted in three sequential phases, which sometimes may overlap. Phase I trials consist of testing the product in a small number of patients or normal volunteers, primarily for safety, in one or more dosages, as well as characterization of a drug’s pharmacokinetic and/or pharmacodynamic profile. In Phase 2, in addition to safety, the product is studied in a patient population to evaluate the product’s efficacy for the specific, targeted indications and to determine dosage tolerance and optimal dosage. Phase 3 trials typically involve additional testing for safety and clinical efficacy in an expanded patient population at geographically dispersed sites. With limited exceptions, all patients involved in a clinical trial must provide informed consent prior to their participation. Meeting clinical endpoints in early stage clinical trials does not assure success in later stage clinical trials. Phase 1, 2, and 3 testing may not be completed successfully within any specified time period, if at all.
The FDA and the competent authorities of EU Member States monitor the progress of each clinical trial phase conducted under an IND or IMPD and may, at their discretion, reevaluate, alter, suspend or terminate clinical trials at any point in this process for various reasons, including a finding that patients are being exposed to an unacceptable health risk or a determination that it is unethical to continue the study. The FDA, the European Commission and the competent authorities of EU Member States can also request that additional clinical trials be conducted as a condition to product approval. The IRB, the Ethics Committee, and sponsor also may order the temporary or permanent discontinuance of a clinical trial at any time for a variety of reasons, particularly if safety concerns arise. Such holds can cause substantial delay and, in some cases, may require abandonment of product development. These clinical studies must be conducted in conformance with the FDA’s bioresearch monitoring regulations, the EU Clinical Trials Directive and/or internationally recognized guidance such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (“ICH”).
New Drug Application. After the completion of the clinical trial phases of development, if the sponsor concludes substantial evidence exists that the drug candidate is effective and that the drug is safe for its intended use, a new Drug Application (“NDA”) may be submitted to the FDA. The application must contain all of the information on the drug candidate gathered to that date, including data from the pre-clinical and clinical trials, information pertaining to the preparation of the drug, analytical methods, product formulation, details on the manufacture of finished products, proposed product packaging, labeling and stability (shelf-life). NDAs are often over 100,000 pages in length. If FDA determines that a Risk Evaluation and Mitigation Strategy (“REMS”) is necessary to ensure that the benefits of the drug outweigh the risks, a sponsor may be required to include as part of the application a proposed REMS, including a package insert directed to patients, a plan for communication with healthcare providers, restrictions on a drug’s distribution, or a medication guide to provide better information to consumers about the drug’s risks and benefits. Submission of an NDA does not assure FDA approval for marketing.
The FDA reviews all submitted NDAs before it accepts them for filing (the U.S. prerequisite for dossier review). The FDA may refuse to file the application and request additional information rather than accepting an application for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA to determine, among other things, whether a product is safe and effective for its intended use. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation. There is a strong presumption for advisory committee review for any drug containing an active ingredient not previously approved. The FDA is not bound by the recommendation of an advisory committee. Under the Prescription Drug User Fee Act (“PDUFA”), submission of an NDA with clinical data requires payment of a fee. In return, the FDA assigns an action date of 10 months from acceptance of the application to return of a first ‘complete response,’ in which the FDA may approve the product or request additional information (PDUFA also provides for an expedited, six-month, “priority review” process. There can be no assurance an application will be approved within the performance goal timeframe established under PDUFA, if at all. If the FDA’s evaluation of the NDA is not favorable, the FDA usually will outline the deficiencies in the submission and request additional testing or information. Notwithstanding the submission of any requested additional information, or even in lieu of asking for additional information, the FDA may decide the marketing application does not satisfy the regulatory criteria for approval and issue a complete response letter, communicating the Agency’s decision not to approve the application.
FDA approval of an NDA will be based, among other factors, on the Agency’s review of the pre-clinical and clinical data submitted, a risk/benefit analysis of the product, and an evaluation of the manufacturing processes and facilities. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its NDA. For instance, FDA may require Avadel to provide data from additional preclinical studies or clinical trials to support approval of certain development steps or the NDA itself. Among the conditions for NDA approval is the requirement
-11-
that each prospective manufacturer’s quality control and manufacturing procedures conform to cGMP standards and requirements. Manufacturing establishments often are subject to Pre-Approval Inspections prior to NDA approval to assure compliance with cGMP manufacturing commitments made in the relevant marketing application.
Patent Restoration and Exclusivity. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, establishes two abbreviated approval pathways for drug products that are in some way follow-on versions of already approved products.
Generic Drugs. A generic version of an approved drug is approved by means of an Abbreviated New Drug Application (“ANDA”), by which the sponsor demonstrates the proposed product is the same as the approved, brand-name drug, which is referred to as the “Reference Listed Drug” (“RLD”). Generally, an ANDA must contain data and information showing the proposed generic product and RLD (1) have the same active ingredient, in the same strength and dosage form, to be delivered via the same route of administration, (2) are intended for the same uses, and (3) are bioequivalent. This is instead of independently demonstrating the proposed product’s safety and effectiveness, which are inferred from the product being the same as the RLD, which the FDA previously found to be safe and effective.
505(b)(2) NDAs. If a product is similar, but not identical, to an already approved product, it may be submitted for approval via an NDA under Section 505(b)(2) of the Act. Unlike an ANDA, this does not excuse the sponsor from demonstrating the proposed product’s safety and effectiveness. Rather, the sponsor is permitted to rely to some degree on published scientific literature and the FDA’s finding that the RLD is safe and effective, and must submit its own data of safety and effectiveness to an extent necessary because of the differences between the products.
RLD Patents. An NDA sponsor must advise the FDA about patents that claim the drug substance or drug product or a method of using the drug. When the drug is approved, those patents are among the information about the product that is listed in the FDA publication, Approved Drug Products with Therapeutic Equivalence Evaluations, which is referred to as the “Orange Book”. The sponsor of an ANDA or 505(b)(2) application seeking to rely on an approved product as the RLD must make one of several certifications regarding each listed patent. A “Paragraph III” certification is the sponsor’s statement that it will wait for the patent to expire before obtaining approval for its product. A “Paragraph IV” certification is a challenge to the patent; it is an assertion that the patent does not block approval of the later product, because the patent is invalid, unenforceable, and/or not infringed by the new product.
Once the FDA accepts for filing an ANDA or 505(b)(2) application containing a Paragraph IV certification, the applicant must within 20 days provide notice to the RLD NDA holder and patent owner that the application with patent challenge has been submitted, and provide the factual and legal basis for the applicant’s assertion that the patent is invalid, unenforceable, or not infringed. If the NDA holder or patent owner file suit against the ANDA or 505(b)(2) applicant for patent infringement within 45 days of receiving the Paragraph IV notice, the FDA is prohibited from approving the ANDA or 505(b)(2) application for a period of 30 months from the date of receipt of the notice. If the RLD has new chemical entity (“NCE”) exclusivity and the notice is given and suit filed during the fifth year of exclusivity, the 30-month stay does not begin until five years after the RLD approval. The FDA may approve the proposed product before the expiration of the 30-month stay i) if a court finds the patent invalid, unenforceable, or not infringed, ii) if the court shortens the period because the parties have failed to cooperate in expediting the litigation, or iii) if the parties reach a settlement agreement and notify the FDA of same.
As an alternative to Paragraph IV certification, and only with respect to method of use patents, an applicant can ‘carve around’ a “Patent Use Code” associated with a particular patent in the FDA’s Orange Book. A Patent Use Code is supposed to describe an indication/use of the RLD that is i) set forth in the RLD label and ii) covered by the corresponding method of use patent. As such, in lieu of a Paragraph IV certification, an applicant can demonstrate to the FDA that its proposed label does not include the method of use described by the RLD’s Patent Use Code for the corresponding method of use patent and, thus, ‘carve around’ that Patent Use Code. If a ‘carve around’ is successful, the notice requirement and 30-month stay on FDA approval of the application described above with respect to a Paragraph IV certification for that particular method of use patent does not apply.
Regulatory Exclusivities. The Hatch-Waxman Act may provide periods of regulatory exclusivity for products that would serve as RLDs. If a product is a “new chemical entity,” or NCE, - generally meaning that the active moiety has never before been approved in any drug - there may be a period of five years from the product’s approval during which the FDA may not accept for filing any ANDA or 505(b)(2) application for a drug with the same active moiety. An ANDA or 505(b)(2) application may be submitted after four years, however, if the sponsor makes a Paragraph IV certification challenging a listed patent.
An RLD that is not an NCE may qualify for a three-year period of exclusivity, if the NDA contains clinical data necessary for approval. In that instance, the exclusivity period does not preclude filing or review of the ANDA or 505(b)(2) application; rather, the FDA is precluded from granting final approval to the ANDA or 505(b)(2) application until three years after approval of the
-12-
RLD. Additionally, the exclusivity applies only to the conditions of approval that required submission of the clinical data. For example, if an NDA is submitted for an RLD that is not an NCE, but that seeks approval for a new indication, and clinical data were required to demonstrate the safety or effectiveness of the RLD for that use, the FDA could not approve an ANDA or 505(b)(2) application for another product with that active moiety for that use. For example, Coreg CRTM received three-year exclusivity for the clinical trials that demonstrated the safety and efficacy of the new, controlled-release dosage form; that exclusivity, which has expired, blocked other controlled-release products.
For a brief discussion of potential marketing exclusivity that could be available under certain conditions with respect to Avadel’s product candidate FT218, please see the information set forth under the caption “Risks Related to Regulatory and Legal Matters – If FT218 is approved by the FDA, we may not obtain orphan drug marketing exclusivity” in the “Risk Factors” included in Part I, Item 1A of this Annual Report on Form 10-K.
Patent Term Restoration. Under the Hatch-Waxman Act, a portion of the patent term lost during product development and FDA review of an NDA or 505(b)(2) application is restored if approval of the application is the first permitted commercial marketing of a drug containing the active ingredient. The patent term restoration period is generally one-half the time between the effective date of the IND and the date of submission of the NDA, plus the time between the date of submission of the NDA and the date of FDA approval of the product. The maximum period of restoration is five years, and the patent cannot be extended to more than 14 years from the date of FDA approval of the product. Only one patent claiming each approved product is eligible for restoration and the patent holder must apply for restoration within 60 days of approval. The USPTO, in consultation with the FDA, reviews and approves the application for patent term restoration. In the event that Avadel applies for patent term extensions on patents covering Avadel’s products, the FDA and the USPTO may not agree with Avadel’s assessment of whether such extensions are available, and may refuse to grant extensions to Avadel’s patents, or may grant more limited extensions than Avadel requests. Moreover, Avadel may not receive an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements.
Regulation of Combination Drugs. Medical products containing a combination of drugs, biologic, or device products may be regulated as ‘combination products’ in the U.S. A combination product generally is defined as a product comprising components from two or more regulatory categories (e.g., drug/device, device/biologic, drug/biologic). Each component of a combination product is subject to the requirements established by the FDA for that type of component, whether a drug, biologic or device.
To determine which FDA center or centers will review a combination product submission, companies may submit a request for assignment to the FDA. Those requests may be handled formally or informally. In some cases, jurisdiction may be determined informally based on FDA experience with similar products. However, informal jurisdictional determinations are not binding on the FDA. Companies also may submit a formal Request for Designation to the FDA Office of Combination Products. The Office of Combination Products will review the request and make its jurisdictional determination within 60 days of receiving a Request for Designation.
In order to facilitate pre-market review of combination products, the FDA designates one of its centers to have primary jurisdiction for the pre-market review and regulation of both components. The determination whether a product is a combination product or two separate products is made by the FDA on a case-by-case basis. It is possible that Avadel’s delivery platforms, when coupled with a drug or medical device component, could be considered and regulated by the FDA as a combination product.
If the primary mode of action is determined to be a drug, the product will be reviewed by the Center for Drug Evaluation and Research (“CDER”) either in consultation with another center or independently. If the primary mode of action is determined to be a medical device, the product would be reviewed by Center for Devices and Radiological Health (“CDRH”) either in consultation with another center, such as CDER, or independently. In addition, FDA could determine that the product is a biologic and subject to the jurisdiction of the Center for Biologic Evaluation and Research (“CBER”), although it is also possible that a biological product will be regulated by CDER.
Marketing Approval and Reporting Requirements. If the FDA approves an NDA, the product becomes available for physicians to prescribe. The FDA may require post-marketing studies, also known as Phase IV studies, as a condition of approval to develop additional information regarding the safety of a product. These studies may involve continued testing of a product and development of data, including clinical data, about the product’s effects in various populations and any side effects associated with long-term use. After approval, the FDA may require post-marketing studies or clinical trials, as well as periodic status reports, if new safety information develops. These post-marketing studies may include clinical trials to investigate known serious risks or signals of serious risks or identify unexpected serious risks. Failure to conduct these studies in a timely manner may result in substantial civil fines and can result in withdrawal of approval. Avadel has several Phase IV obligations with its current approvals.
-13-
In addition, the FDA may require distribution to patients of a medication guide such as a Risk Evaluation and Mitigation Strategies (“REMS”) for prescription products that the agency determines pose a serious and significant health concern in order to provide information necessary to patients’ safe and effective use of such products. We expect our FT218 product, if approved by the FDA will be subject to a REMS program.
In the European Union, the marketing authorization of a medicinal product may be made conditional on the conduct of Phase IV post-marketing studies. Failure to conduct these studies in relation to centrally authorized products can lead to the imposition of substantial fines. Moreover, Phase IV studies are often conducted by companies in order to obtain further information on product efficacy and positioning on the market in view of competitors and to assist in application for pricing and reimbursement.
Other Post-Marketing Obligations. Any products manufactured and/or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping requirements, reporting of adverse experiences with the product, submitting other periodic reports, drug sampling and distribution requirements, notifying the FDA and gaining its approval of certain manufacturing or labeling changes, complying with certain electronic records and signature requirements, submitting periodic reports to the FDA, maintaining and providing updated safety and efficacy information to the FDA, and complying with FDA promotion and advertising requirements. For example, the FDA has required Avadel to conduct post-marketing clinical and non-clinical studies for several of its products to be completed between 2016 and 2019.
Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and to list their products with the FDA. The FDA periodically inspects manufacturing facilities in the U.S. and elsewhere in order to assure compliance with the applicable cGMP regulations and other requirements. Facilities also are subject to inspections by other U.S. federal, foreign, state or local agencies. In complying with the cGMP regulations, manufacturers must continue to expend time, money and effort in recordkeeping and quality control to assure that the product meets applicable specifications and other post-marketing requirements. Failure of Avadel or its licensees to comply with FDA’s cGMP regulations or other requirements could have a significant adverse effect on Avadel’s business, financial condition and results of operations.
Also, newly discovered or developed safety or efficacy data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, additional pre-clinical or clinical studies, or even in some instances, revocation or withdrawal of the approval. Violations of regulatory requirements at any stage, including after approval, may result in various adverse consequences, including the FDA’s delay in approving or refusal to approve a product, withdrawal or recall of an approved product from the market, other voluntary or FDA-initiated action that could delay or restrict further marketing, and the imposition of civil fines and criminal penalties against the manufacturer and NDA holder. In addition, later discovery of previously unknown problems may result in restrictions on the product, manufacturer or NDA holder, including withdrawal of the product from the market. Furthermore, new government requirements may be established that could delay or prevent regulatory approval of Avadel’s products under development, or affect the conditions under which approved products are marketed.
The Food and Drug Administration Amendments Act of 2007 (“FDAAA”) provides the FDA with expanded authority over drug products after approval. This legislation enhances the FDA’s authority with respect to post-marketing safety surveillance, including, among other things, the authority to require additional post-marketing studies or clinical trials, labeling changes as a result of safety findings, registering clinical trials, and making clinical trial results publicly available.
In the European Union, stringent pharmacovigilance regulations oblige companies to appoint a suitably qualified and experienced Qualified Person resident in the European Economic Area, to prepare and submit to the competent authorities adverse event reports within specific time lines, prepare Periodic Safety Update Reports (“PSURs”) and provide other supplementary information, report to authorities at regular intervals and take adequate safety measures agreed with regulatory agencies as necessary. Failure to undertake these obligations can lead to the imposition of substantial fines.
Other Regulation
Controlled Substances Act. Narcotics and other APIs, such as sodium oxybate and ephedrine sulfate are “controlled substances” under the Controlled Substances Act. The U.S. federal “Controlled Substances Act” (“CSA”), Title II of the Comprehensive Drug Abuse Prevention and Control Act of 1970, regulates the manufacture and distribution of narcotics and other controlled substances, including stimulants, depressants and hallucinogens in the U.S. The CSA is administered by the “Drug Enforcement Administration” (“DEA”), a division of the U.S. Department of Justice, and is intended to prevent the abuse or diversion of controlled substances into illicit channels of commerce. Avadel has several products marketed under this Act and has at least one product under development.
Any person or firm that manufactures, distributes, dispenses, imports, or exports any controlled substance (or proposes to do so) must register with the DEA. The applicant must register for a specific business activity related to controlled substances, including
-14-
manufacturing or distributing, and may engage in only the activity or activities for which it is registered. The DEA conducts periodic inspections of registered establishments that handle controlled substances and allots quotas of controlled drugs to manufacturers and marketers’ failure to comply with relevant DEA regulations, particularly as manifested in the loss or diversion of controlled substances, can result in regulatory action including civil penalties, refusal to renew necessary registrations, or proceedings to revoke those registrations. In certain circumstances, violations can lead to criminal prosecution. In addition to these federal statutory and regulatory obligations, there may be state and local laws and regulations relevant to the handling of controlled substances or listed chemicals.
cGMP. Current Good Manufacturing Practices rules apply to the manufacturing of drugs and medical devices. In addition to regulations enforced by the FDA, Avadel is also subject to French, U.S. and other countries’ rules and regulations governing permissible laboratory activities, waste disposal, handling of toxic, dangerous or radioactive materials and other matters. Avadel’s R&D involves the controlled use of hazardous materials, chemicals, viruses and various radioactive compounds. Although Avadel believes that its safety procedures for handling and disposing of such materials comply with the standards prescribed by French, EU, U.S. and other foreign rules and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated.
Health Care Fraud and Abuse. Avadel is subject to a number of federal and state laws pertaining to health care “fraud and abuse,” such as anti-kickback and false claims laws. Under anti-kickback laws, it is illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance via regulations and that there are few court decisions addressing industry practices, it is possible that Avadel’s practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third-party payors (such as the Medicare and Medicaid programs) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Avadel’s sales and marketing activities relating to its products could be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal health care programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. In addition, similar sanctions and penalties can be imposed upon executive officers and employees, including criminal sanctions against executive officers. As a result of the potential penalties that can be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the U.S. government were to allege or convict Avadel or its executive officers of violating these laws, Avadel’s business could be harmed. In addition, private individuals have the ability to bring similar actions. In addition to the reasons noted above, Avadel’s activities could be subject to challenge due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. There also are an increasing number of U.S. federal and state laws that require manufacturers to make reports to states on pricing, marketing information, and payments and other transfers of value to healthcare providers. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, Avadel’s reporting actions could be subject to the penalty provisions of the pertinent authorities.
Healthcare Privacy and Security Laws. Avadel may be subject to, or its marketing activities may be limited by the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), as amended by the Health Information Technology and Clinical Health Act and their respective implementing regulations, which established uniform standards for certain “covered entities” (healthcare providers, health plans and healthcare clearinghouses) governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of protected health information. Among other things, HIPAA’s privacy and security standards are directly applicable to “business associates” – independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. In addition to possible civil and criminal penalties for violations, state attorney generals are authorized to file civil actions for damages or injunctions in U.S. federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. In the EU/EEA, Directive 95/46/EEC (as amended) or its successor applies to identified or identifiable personal data processed by automated means (e.g., a computer database of customers) and data contained in, or intended to be part of, non-automated filing systems (traditional paper files) as well as transfer of such data to a country outside of the EU/EEA.
“Sunshine” and Marketing Disclosure Laws. There are an increasing number of U.S. federal and state “sunshine” laws that require pharmaceutical manufacturers to make reports to states on pricing and marketing information. Several U.S. states have enacted legislation requiring pharmaceutical companies to, among other things, establish marketing compliance programs, file periodic reports with the state, and make periodic public disclosures on sales and marketing activities, and prohibiting certain other sales
-15-
and marketing practices. In addition, a similar recently implemented federal requirement requires manufacturers, including pharmaceutical manufacturers, to track and report to the federal government certain payments and other transfers of value made to physicians and other healthcare professionals and teaching hospitals and ownership or investment interests held by physicians and their immediate family members. The U.S. federal government began disclosing the reported information on a publicly available website in 2014. These laws may adversely affect Avadel’s sales, marketing, and other activities with respect to its medicines in the U.S. by imposing administrative and compliance burdens on us. If Avadel fails to track and report as required by these laws or otherwise comply with these laws, it could be subject to the penalty provisions of the pertinent U.S. state and federal authorities.
Government Price Reporting. For those marketed medicines which are covered in the U.S. by the Medicaid programs, Avadel has various obligations, including government price reporting and rebate requirements, which generally require medicines be offered at substantial rebates/discounts to Medicaid and certain purchasers (including “covered entities” purchasing under the 340B Drug Discount Program). Avadel is also required to discount such medicines to authorized users of the Federal Supply Schedule of the General Services Administration, under which additional laws and requirements apply. These programs require submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations, and the guidance governing such calculations is not always clear. Compliance with such requirements can require significant investment in personnel, systems and resources, but failure to properly calculate Avadel’s prices, or offer required discounts or rebates could subject it to substantial penalties. One component of the rebate and discount calculations under the Medicaid and 340B programs, respectively, is the “additional rebate”, a complex calculation which is based, in part, on the rate at which a branded drug price increases over time more than the rate of inflation (based on the CPI-U). This comparison is based on the baseline pricing data for the first full quarter of sales associated with a branded drug’s NDA, and baseline data cannot generally be reset, even on transfer of the NDA to another manufacturer. This “additional rebate” calculation can, in some cases where price increases have been relatively high versus the first quarter of sales of the NDA, result in Medicaid rebates up to 100 percent of a drug’s “average manufacturer price” and 340B prices of one penny.
Healthcare Reimbursement
In both U.S. and non-U.S. markets, sales of Avadel’s potential products as well as products of pharmaceutical and biotechnology companies that incorporate Avadel’s technology into their products, if any, will depend in part on the availability of reimbursement by third-party payers, such as government health administration authorities, private health insurers and other organizations. The U.S. market for pharmaceutical products is increasingly being shaped by managed care organizations, pharmacy benefit managers, cooperative buying organizations and large drugstore chains. Third-party payers are challenging the price and cost effectiveness of medical products and services. Uncertainty particularly exists as to the reimbursement status of newly approved healthcare products. There can be no assurance reimbursement will be available to enable Avadel to maintain price levels sufficient to realize an appropriate return on our product development investment. Legislation and regulations affecting the pricing of pharmaceuticals may change before Avadel’s proposed products are approved for marketing and any such changes could further limit reimbursement for medical products and services.
Employees
As of December 31, 2019, we had approximately 50 employees, all of which were full-time. Except for employees at our French subsidiaries, none of our other employees is subject to a union or other collective bargaining agreement. Additionally, employees at our French subsidiaries (approximately 19 employees) are represented by a works’ council in which employee representatives have the right to be consulted as to certain matters affecting our French subsidiaries. We believe that our relations with our employees are satisfactory.
-16-
Item 1A. Risk Factors.
An investment in Avadel involves a high degree of risk. You should carefully consider the risks described below, as well as the other information included or incorporated by reference in this Annual Report on Form 10-K, before making an investment decision. Avadel’s business, financial condition, results of operations and cash flows could be materially adversely affected by any of these risks. The market or trading price of Avadel’s securities could decline due to any of these risks. In addition, please read “Cautionary Disclosure Regarding Forward-Looking Statements” in this Annual Report on Form 10-K, where we describe additional uncertainties associated with Avadel’s business and with the forward-looking statements included or incorporated by reference in this Annual Report on Form 10-K. Please note that additional risks not presently known to us or that we currently deem immaterial may also impair Avadel’s business and operations.
Risks Relating to Our 2019 Net Loss and 2019 Restructuring Plan
Our net loss and use of cash from operating activities may limit our ability to fully pursue our business strategy.
We reported a net loss of $33.2 million in 2019 and a loss from operating activities of $38.3 million. As a result, our cash and marketable securities as of December 31, 2019 totaled $64.2 million. Our business strategy is to primarily focus on the development and potential FDA approval for FT218 which is in a Phase 3 clinical trial for the treatment of narcolepsy patients suffering from EDS and cataplexy. In addition, we will continue to maximize the value of our current approved hospital products portfolio, including the potential commercialization of our fourth product, Nouress (cysteine hydrochloride injection). The successful pursuit of all components of our strategy will require substantial financial resources, and there can be no assurance that our existing cash and marketable securities assets and the cash generated by our operations will be adequate for these purposes. Failure to implement any component of our strategy may prevent us from achieving profitability in the future or may otherwise have a material adverse effect on our financial condition and results of operation.
Our restructuring plan announced in February 2019 may not be as effective as anticipated, and we may fail to fully realize the expected cost savings or may experience unintended negative impacts from the restructuring.
In February 2019, we announced a restructuring plan intended to achieve future cost savings through, among other actions, a reduction of our overall workforce by approximately 50 percent. In conjunction with the restructuring plan, we also announced the voluntary Chapter 11 bankruptcy filing by our subsidiary, Avadel Specialty Pharmaceuticals, LLC (“Specialty Pharma”), which was responsible solely for the sales, marketing and distribution of our Noctiva product. We refer to the restructuring plan and the Chapter 11 bankruptcy of Specialty Pharma as the 2019 Restructuring. We implemented the restructuring plan in light of disappointing results from the commercial launch of Noctiva, and in order to focus our resources on other product development activities, in particular the ongoing Phase 3 clinical trial of FT218 for the treatment of excessive daytime sleepiness (“EDS”), and cataplexy in narcolepsy patients. The 2019 Restructuring requires the devotion of management attention as well as significant resources, including pre-tax cash charges which were approximately $12.0 million in 2019, and may pose significant risks. The 2019 Restructuring may not be as effective as we anticipated and may not fully produce the expected cost savings or the effective re-focusing of our efforts toward completing the development of FT218. In addition, the restructuring plan may result in greater implementation costs than we have estimated or may result in unintended negative consequences. For example, because of the speed and magnitude of the workforce reduction that occurred during the 2019 restructuring, it may be difficult in the future to retain certain remaining employees who are critical to our ability to successfully pursue our business plan.
If we need to take further restructuring actions, necessary third-party consents may not be granted.
Our management may determine we need to take further restructuring actions to achieve additional cost savings, to generate additional capital needed for our business strategy, or for other purposes. Certain restructuring scenarios that management consider could require obtaining the consent of third parties, such as holders of our Exchangeable Senior Notes, or the 2023 Notes. For example, the voluntary bankruptcy filing by Specialty Pharma required the consent of holders of a majority in principal amount of our 2023 Notes in order to avoid a default under the Indenture governing such 2023 Notes. While we were successful in obtaining that consent, there can be no assurance we will be successful in obtaining additional consents in the future from such holders or from other third parties whose consents may be required. Failure to obtain these third-party consents would prevent us from taking additional restructuring actions, which could have a material adverse effect on our cash flow, financial resources and ability to successfully pursue our business strategy.
The Chapter 11 bankruptcy filing by Specialty Pharma may have unexpected adverse results.
As part of the 2019 Restructuring, Avadel US Holdings Inc., Specialty Pharma’s immediate parent and our wholly-owned subsidiary, agreed to provide debtor-in-possession financing to Specialty Pharma of up to $2.7 million. We could face challenges in having
-17-
the liquidation plan for Specialty Pharma approved and costs from the restructuring may exceed the amount of financing Avadel US Holdings Inc. has committed to provide. Adverse or unexpected results from the bankruptcy proceeding could impair the success of the 2019 Restructuring.
A management-directed third-party evaluation of our FT218 development program could result in changes that increase the cost of the program and further delay its completion.
We recently announced completion of enrollment in the clinical trial for our FT218 product and an estimated completion date for that clinical trial. Management, in conjunction with pharmaceutical industry consulting firms, continues its evaluation of the FT218 program. The results of this on-going evaluation, in addition to the results of our FT218 development program, including our pivotal phase III REST ON study and all other components of an NDA submission, could cause us to modify our development plan with respect to FT218 in ways that could materially increase the ultimate cost of such development or further delay its completion, or could identify unknown risks or problems with the product. Any such cost increases, added delays, risks or problems could have a material adverse effect on our financial condition and results of operation.
Risks Relating to Our Business and Industry
We derive a substantial majority of our revenues from a small number of products facing competitive pressures, and from a small number of customers, and the loss of any one of these products or customers could reduce Avadel’s revenues significantly.
In 2019, we derived our $59.2 million in revenues from sales of our three hospital products, Bloxiverz, Vazculep and Akovaz. Product sales of these three products declined in the aggregate from 2018 to 2019 by $38.3 million, or 39.2%, from $97.5 million to $59.2 million. Our Noctiva product failed to achieve anticipated revenue levels despite a substantial investment of resources toward its commercialization, and these disappointing results led to the voluntary Chapter 11 bankruptcy filing by Specialty Pharma in February 2019. In addition, we depend on a small number of customers for the majority of our revenues from our three hospital products. Three customers accounted for approximately 71% of total revenues from sales of these products in 2019. These three customers comprise a significant portion of the distribution network for pharmaceutical products in the U.S.
Competition for our hospital products in 2019 caused significant downward pricing pressure and loss of market share by us resulting in lower aggregate revenues for these products. Further competition in the future could cause further reductions in prices and market share. The distribution network for pharmaceutical products is continuing to undergo consolidation marked by mergers and acquisitions among wholesale distributors and retail drug store chains. As a result, a small number of large wholesale distributors and large chain drug stores control a significant share of the market. We expect that continuing consolidation in the industry may cause competitive pressures on pharmaceutical companies. The loss of any one of our three hospital products, the termination of our relationship with any of our customers or our failure to broaden our customer base could cause our revenues to further decrease significantly and result in further losses from our operations. Further, we may be unable to negotiate favorable business terms with customers that represent a significant portion of our revenues, and any such inability could have a material adverse effect on our business, results of operations, financial condition and prospects.
Further, as part of our 2013 debt financing transaction with Deerfield, we granted Deerfield a security interest in the intellectual property and product registration rights of certain legacy products. If we default on the terms of the loan agreement with Deerfield, Deerfield could enforce its security interest, which could further impact our revenues.
We must invest substantial sums in research and development (“R&D”) in order to remain competitive, and we may not fully recover these investments.
To be successful in the highly-competitive pharmaceutical industry, we must commit substantial resources each year to R&D in order to develop new products and enhance our technologies. In 2019, we spent $32.9 million on R&D, including on our FT218 product candidate and on Nouress, which was approved by the FDA in December 2019. Our ongoing investments in R&D for FT218 as well as possible future products could result in higher costs without a proportionate increase, or any increase, in revenues. The R&D process is lengthy and carries a substantial risk of failure. If our R&D does not yield sufficient products that achieve commercial success, our future operating results will be adversely affected.
Our products may not reach the commercial market for a number of reasons.
Drug development is an inherently uncertain process with a high risk of failure at every stage of development. Successful R&D of pharmaceutical products is difficult, expensive and time consuming. Many product candidates fail to reach the market. Our success will depend on the development and the successful commercialization of new drugs, including additional previously unapproved marketed drugs (“UMD”) products, and products that utilize our drug delivery technologies. If any of our additional
-18-
UMD products or products incorporating our drug delivery technologies fails to reach the commercial market, our future revenues would be adversely affected.
Even if our products and current drug delivery technologies appear promising during development, there may not be successful commercial applications developed for them for a number of reasons, including:
• | the FDA, the European Medicines Agency (“EMA”), the competent authority of an EU Member State or an Institutional Review Board (“IRB”), or an Ethics Committee (EU equivalent to IRB), or our partners may delay or halt applicable clinical trials; |
• | we or our partners may face slower than expected rate of patient recruitment and enrollment in clinical trials, or may devote insufficient funding to the clinical trials; |
• | our drug delivery technologies and drug products may be found to be ineffective or to cause harmful side effects, or may fail during any stage of pre-clinical testing or clinical trials; |
• | we or our partners may find that certain products cannot be manufactured on a commercial scale and, therefore, may not be economical or feasible to produce; |
• | we or our partners may face delays in completing our clinical trials due to circumstances outside of our control, including natural disasters, labor or civil unrest, global health concerns or pandemics or acts of war or terrorism; or |
• | our products could fail to obtain regulatory approval or, if approved, could fail to achieve market acceptance, could fail to be included within the pricing and reimbursement schemes of the U.S. or EU Member States, or could be precluded from commercialization by proprietary rights of third parties. |
We may rely on collaborations with third parties to commercialize certain of our products in development, particularly products using our drug delivery technologies, and such strategy involves risks that could impair our prospects for realizing profits from such products.
We expect that the commercialization of some of our products in development, which utilize our drug delivery technologies, may require collaboration with third-party partners involving strategic alliances, licenses, product divestitures or other arrangements. We may not be successful in entering into such collaborations on favorable terms, if at all, or our collaboration partners may not adequately perform under such arrangements, and as a result our ability to commercialize these products will be negatively affected and our prospects will be impaired.
Our products may not gain market acceptance.
Our products and technologies may not gain market acceptance among physicians, patients, healthcare payor and medical communities. The degree of market acceptance of any product or technology will depend on a number of factors, including, but not limited to:
• | the scope of regulatory approvals, including limitations or warnings in a product’s regulatory-approved labeling; or other restrictions under a FDA Risk Evaluations and Mitigations Strategies (“REMS”), program; |
• | in the case of any new UMD product we may successfully pursue, whether and the extent to which the FDA removes competing products from the market; |
• | in the case of our product candidates that are controlled substances the U.S. Drug Enforcement Administration (“DEA”), scheduling classification; |
• | demonstration of the clinical safety and efficacy of the product or technology; |
• | the absence of evidence of undesirable side effects of the product or technology that delay or extend trials; |
• | the lack of regulatory delays or other regulatory actions; |
• | its cost-effectiveness and related access to payor coverage; |
• | its potential advantage over alternative treatment methods; |
• | the availability of third-party reimbursement; and |
• | the marketing and distribution support it receives. |
If any of our products or technologies fails to achieve market acceptance, our ability to generate additional revenue will be limited, which would have a material adverse effect on our business.
The development of several of our drug delivery technologies and products depends on the services of a single provider and any interruption of operations of such provider could significantly delay or have a material adverse effect on our product pipeline.
Currently, we use a single source provider for the development, supply of clinical materials and potentially the supply of commercial
-19-
batches for several of our products incorporating our drug delivery technologies. Any disruption in the operations of this provider or if this provider fails to supply acceptable quantity and quality materials or services to us for any reason, such disruption or failure could delay our product development and could have a material adverse effect on our business, financial condition and results of operations. In case of a disruption, we may need to establish alternative manufacturing sources for our drug delivery products, and this would likely lead to substantial production delays as we build or locate replacement facilities and seek to satisfy necessary regulatory requirements.
We depend on a limited number of suppliers for the manufacturing of our products and certain raw materials used in our products and any failure of such suppliers to manufacture or supply sufficient quantities of product or these raw materials could have a material adverse effect on our business.
Currently, we depend on a limited number of contract manufacturing organizations (“CMOs”), for three products, Bloxiverz, Vazculep and Akovaz, from which we derive our revenues. Additionally, we purchase certain raw materials used in our products from a limited number of suppliers, including a single supplier for certain key ingredients. If the supplies of these products or materials were interrupted for any reason, including but not limited to, natural disasters, labor or civil unrest, global health concerns or pandemics or acts of war or terrorism, the manufacturing and supply of certain products could be delayed. If the supplies of these products or materials were interrupted for any reason, our manufacturing and marketing of certain products could be delayed. These delays could be extensive and expensive, especially in situations where a substitution was not readily available or required variations of existing regulatory approvals and certifications or additional regulatory approval. For example, an alternative supplier may be required to pass an inspection by the FDA, EMA or the competent authorities of EU Member States for compliance with current cGMP, requirements before supplying us with product or before we may incorporate that supplier’s ingredients into the manufacturing of our products by our contract, development, and manufacturing organizations (“CDMOs”). Failure to obtain adequate supplies in a timely manner could have a material adverse effect on our business, financial condition and results of operations.
If our competitors develop and market technologies or products that are safer or more effective than ours, or obtain regulatory approval and market such technologies or products before we do, our commercial opportunity will be diminished or eliminated.
Competition in the pharmaceutical and biotechnology industry is intense and is expected to increase. We compete with academic laboratories, research institutions, universities, joint ventures and other pharmaceutical and biotechnology companies, including other companies developing drug delivery technologies or niche brand or generic specialty pharmaceutical products. Some of these competitors may also be our business partners.
Our drug delivery technologies compete with technologies provided by several other companies. In particular, delivery technologies and products, could be developed that, if successful, could compete against our drug delivery technologies or products. Among the many experimental therapies being tested in the U.S. and in the EU, there may be some that we do not now know of that may compete with our drug delivery technologies or products in the future. These new biological or chemical products may be safer or may work better than our products.
With respect to our UMD products, the FDA has approved generic versions or previously filed NDAs of our marketed products and may approve additional generic versions in the future.
Many of our competitors have substantially greater financial, technological, manufacturing, marketing, managerial and R&D resources and experience than we do. Furthermore, acquisitions of competing drug delivery companies by large pharmaceutical companies could enhance our competitors’ resources. Accordingly, our competitors may succeed in developing competing technologies and products, obtaining regulatory approval and gaining market share for their products more rapidly than we do.
Our revenues may be negatively affected by healthcare reforms and increasing pricing pressures.
Future prices for our pharmaceutical products and medical devices will be substantially affected by reimbursement policies of third-party payors such as government healthcare programs, private insurance plans and managed care organizations; by our contracts with the drug wholesalers who distribute our products; and by competitive market forces generally. In recent years, third-party payors have been exerting downward pressure on prices at which products will be reimbursed, and the drug wholesale industry has been undergoing consolidation which gives greater market power to the remaining, larger drug wholesalers. In the U.S., the Trump administration has made public and social media statements causing uncertainty as to future federal U.S. government policies regulating drug prices. Further, the trend toward increased availability of generic products has contributed to overall pricing pressures in the pharmaceutical industry. Additionally, on December 18, 2019, President Trump, the U.S. Department of Health and Human Services, and the FDA issued a notice of proposed rulemaking that, if finalized, would allow for the importation of certain prescription drugs from Canada. FDA also issued a Draft Guidance document outlining a potential
-20-
pathway for manufacturers to obtain an additional National Drug Code (“NDC”), for an FDA-approved drug that was originally intended to be marketed in a foreign country and that was authorized for sale in that foreign country. The regulatory and market implications of the notice of proposed rulemaking and Draft Guidance are unknown at this time, but legislation, regulations or policies allowing the reimportation of drugs, if enacted and implemented, could decrease the price we receive for our products and adversely affect our future revenues and prospects for profitability. Similarly, any future changes in laws, regulations, practices or policies, in the drug wholesale industry, or in the prevalence of generic products, may adversely affect our financial condition and results of operations.
If we cannot keep pace with the rapid technological change in our industry, we may lose business, and our products and technologies could become obsolete or noncompetitive.
Our success also depends, in part, on maintaining a competitive position in the development of products and technologies in a rapidly evolving field. Major technological changes can happen quickly in the biotechnology and pharmaceutical industries. If we cannot maintain competitive products and technologies, our competitors may succeed in developing competing technologies or obtaining regulatory approval for products before us, and the products of our competitors may gain market acceptance more rapidly than our products. Such rapid technological change, or the development by our competitors of technologically improved or different products, could render our products or technologies obsolete or noncompetitive.
We may fail to effectively execute our business strategy.
Our business strategy is to continue our UMD program by commercializing our fourth UMD product, Nouress, as well as potentially seek FDA approval for and commercialize additional future UMD product candidates, and to continue to seek FDA approval for FT218, which completed enrollment for its Phase 3 clinical trial and for which we expect topline data in the second quarter of 2020. There can be no assurance that we will be successful in any of these objectives; and a failure in any of these objectives could negatively impact our business and operating results.
Failure to comply with domestic and international privacy and security laws could result in the imposition of significant civil and criminal penalties.
The costs of compliance with privacy and security laws, including protecting electronically stored information from cyber-attacks, and potential liability associated with any compliance failures could adversely affect our business, financial condition and results of operations. We are subject to various domestic and international privacy and security regulations, including but not limited to HIPAA and the General Data Protection Regulation (“GDPR”), (Regulation EU 2016/679). HIPAA mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. In addition, many U.S. states have enacted comparable laws addressing the privacy and security of health information, some of which are more stringent than HIPAA. GDPR requires Avadel to ensure personal data collected by Avadel is gathered legally and under strict conditions and to protect such personal data from misuse and exploitation. If Avadel fails to comply with GDPR, we will face significant fines and penalties that could adversely affect our business, financial condition and results of operations.
Our effective tax rate could be highly volatile and could adversely affect our operating results.
Our future effective tax rate may be adversely affected by a number of factors, many of which are outside of our control, including:
• | the jurisdictions in which profits are determined to be earned and taxed; |
• | changes in the valuation of our deferred tax assets and liabilities; |
• | the resolution of issues arising from tax audits with various tax authorities; |
• | changes in share-based compensation expense; |
• | changes in domestic or international tax laws or the interpretation of such tax laws; |
• | changes in available tax credits; |
• | increases in expenses not deductible for tax purposes, including increases in the fair value of related party payables, write-offs of acquired in-process R&D and impairment of goodwill in connection with acquisitions; |
• | adjustments to estimated taxes upon finalization of various tax returns; |
• | the tax effects of purchase accounting for acquisitions that may cause fluctuations between reporting periods. |
Any significant increase in our future effective tax rates could impact our results of operations for future periods adversely.
-21-
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of December 31, 2019, we had U.S. federal net operating loss carryforwards of approximately $75.5 million due to prior period losses, some of which, if not utilized, will expire in 2034 and 2035 for federal tax purposes. Approximately $10.4 million of these net operating loss carryforwards could expire unused and be unavailable to offset future income tax liabilities, which could adversely affect our profitability. The $10.4 million of U.S. federal net operating loss carryforwards are subject to an annual limitation as a result of the FSC acquisition under Internal Revenue Code Section 382 and may not be fully utilized before they expire.
Under U.S. federal tax legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act (“Tax Act”), U.S. federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such U.S. federal net operating losses is limited. Under Sections 382 and 383 of the U.S. Internal Revenue Code of 1986 (the “Code”) if a corporation undergoes an “ownership change” (generally defined as a greater than 50 percentage-point cumulative change (by value) in the equity ownership of certain shareholders over a rolling three-year period), the corporation’s ability to use its pre-change net operating losses and other pre-change tax attributes to offset its post-change taxable income or taxes may be limited. We may also experience ownership changes as a result of this offering or future issuances of our stock or as a result of subsequent shifts in our stock ownership, some of which are outside our control. We have completed an analysis to determine that no events have been triggered in the past. If any ownership changes are determined to be triggered in the future, our ability to use our current net operating losses to offset post-change taxable income or taxes would be subject to limitation. We will be unable to use our net operating losses if we do not attain profitability sufficient to offset our available net operating losses prior to their expiration.
As of December 31, 2019, we had approximately $95.8 million of net operating losses in Ireland that do not have an expiration date. While these losses do not have an expiration date, substantial changes in the activities performed in these jurisdictions could have an impact on our ability to utilize these tax attributes in the future.
We outsource important activities to consultants, advisors and outside contractors.
We outsource many key functions of our business and therefore rely on a substantial number of consultants, advisors and outside contractors. If we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by such third parties is compromised for any reason, our development activities may be extended, delayed or terminated which would have an adverse effect on our development program and our business.
We depend on key personnel to execute our business plan. If we cannot attract and retain key personnel, we may not be able to successfully implement our business plan.
Our success depends in large part upon our ability to attract and retain highly qualified personnel. During our operating history, we have assigned many key responsibilities within our Company to a relatively small number of individuals, each of whom has played key roles in executing various important components of our business. We do not maintain material key person life insurance for any of our key personnel. If we lose the services of Greg Divis, our Chief Executive Officer, or other members of our senior executive team, we may have difficulty executing our business plan in the manner we currently anticipate. Further, because each of our key personnel is involved in numerous roles in various components of our business, the loss of any one or more of such individuals could have an adverse effect on our business.
Risks Related to Our Intellectual Property
If we cannot adequately protect our intellectual property and proprietary information, we may be unable to effectively compete.
Our success depends, in part, on our ability to obtain and enforce patents and other intellectual property rights for our products and technology, including our drug delivery technologies, and to preserve our trade secrets and other proprietary information. If we cannot do so, our competitors may exploit our technologies and deprive us of the ability to realize revenues and profits from our products and technologies.
To the extent any of our products may benefit from protections afforded by patents, we face the risk that patent law relating to the scope of claims in the pharmaceutical and biotechnology fields is continually evolving and can be the subject of uncertainty and may change in a way that would limit protection. Our patents may not be exclusive, valid or enforceable. For example, our patents may not protect us against challenges by companies that submit drug marketing applications to the FDA, or the competent authorities of EU Member States or other jurisdictions in which we may attempt to compete, in particular where such applications rely, at least in part, on safety and efficacy data from our products or our business partners’ products. In addition, competitors may obtain patents that may have an adverse effect on our ability to conduct business, or they may discover ways to circumvent our patents. The scope of any patent protection may not be sufficiently broad to cover our products or to exclude competing products. Any
-22-
patent applications we have made or may make relating to our potential products or technologies may not result in patents being issued. Even after issuance, our patents may be challenged in the courts or patent offices in the U.S. and abroad. Such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical product candidates, or limit the duration of the patent protection of our product candidates. Further, patent protection once obtained is limited in time, after which competitors may use the covered product or technology without obtaining a license from us. Because of the time required to obtain regulatory marketing approval, the remaining period of effective patent protection for a marketed product is frequently substantially shorter than the full duration of the patent. While a patent term extension can be requested under certain circumstances, the grant of such a request is not guaranteed.
Our partnerships with third parties expose us to risks that they will claim intellectual property rights on our inventions or fail to keep our unpatented products or technology confidential.
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be materially adversely affected and our business would be harmed.
We also rely on trademarks, copyrights, trade secrets and know-how to develop, maintain and strengthen our competitive position.
To protect our products, trade secrets and proprietary technologies, we rely, in part, on confidentiality agreements with our employees, suppliers, consultants, advisors and partners. These agreements may not provide adequate protection for our trade secrets and other proprietary information in the event of any unauthorized use or disclosure, or if others lawfully develop the information. If these agreements are breached, we cannot be certain we will have adequate remedies. Further, we cannot guarantee that third parties will not know, discover or independently develop equivalent proprietary information or technologies, or that they will not gain access to our trade secrets or disclose our trade secrets to the public. Therefore, we cannot guarantee we can maintain and protect unpatented proprietary information and trade secrets. Misappropriation or other loss of our intellectual property would adversely affect our competitive position and may cause us to incur substantial litigation or other costs.
Changes in U.S. or ex-U.S. patent laws could diminish the value of patents in general, thereby impairing our ability to protect our products.
Changes in either the patent laws or interpretation thereof in the U.S. or in ex-U.S. jurisdictions could increase uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. For example, the Leahy-Smith America Invents Act of 2011 (“AIA”), changed the previous U.S. “first-to-invent” system to the current system that awards a patent to the “first-inventor-to-file” for an application for a patentable invention. This change alters the pool of available materials that can be used to challenge patents in the U.S. and limits the ability to rely on prior research to lay claim to patent rights. Under the current system, disputes are resolved through new derivation proceedings, and the AIA includes mechanisms to allow challenges to issued patents in reexamination, inter partes review and post grant proceedings. The AIA also includes bases and procedures that may make it easier for competitors to challenge our patents, which could result in increased competition and have a material adverse effect on our business and results of operations. The AIA may also make it harder to challenge third-party patents and place greater importance on being the first inventor to file a patent application on an invention. The AIA amendments to patent filing and litigation procedures in the U.S. may result in litigation being more complex and expensive and divert the efforts of our technical and management personnel.
In addition, the patent positions of companies in the development and commercialization of pharmaceuticals may be particularly uncertain. Depending on future actions by the U.S. Congress, the U.S. federal courts, and the USPTO, or by similarly legislative, judicial, and regulatory authorities in other jurisdictions, the laws and regulations governing patents could change in unpredictable ways that could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property in the future.
Third parties may claim that our products infringe their rights, and we may incur significant costs resolving these claims.
Third parties may claim infringement of their patents and other intellectual property rights by the manufacture, use, import, offer for sale or sale of our drug delivery technologies or our other products. For example, an Orange Book patent exists related to Exela’s currently marketed cysteine hydrochloride injection product. In this regard, Exela filed a complaint against us and our subsidiary, Avadel Legacy Pharmaceuticals, LLC (“Avadel Legacy”), in the United States District for the District of Delaware in January of 2020 alleging infringement of that Orange Book patent by our recently-approved Nouress product. As another example, approximately 14 Orange Book patents exist related to Jazz Pharmaceuticals’ currently marketed sodium oxybate product, and, in connection with us seeking regulatory approval for FT218, Jazz may allege that FT218 infringes its patents or other intellectual property rights and file suit to prevent us from commercializing FT218. In response to any claim of infringement, we may choose
-23-
or be forced to seek licenses, defend infringement actions or challenge the validity or enforceability of those patent rights in court or administrative proceedings. If we cannot obtain required licenses on commercially reasonably terms, or at all, are found liable for infringement or are not able to have such patent rights declared invalid or unenforceable, our business could be materially harmed. We may be subject to claims (and even held liable) for significant monetary damages (including enhanced damages and/or attorneys’ fees), encounter significant delays in bringing products to market or be precluded from the manufacture, use, import, offer for sale or sale of products or methods of drug delivery covered by the patents of others. Even if a license is available, it may not be available on commercially reasonable terms or may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. We may not have identified, or be able to identify in the future, U.S. or foreign patents that pose a risk of potential infringement claims.
Parties making claims against us may be able to sustain the costs of patent litigation more effectively than we can because they have substantially greater resources. In addition, any claims, with or without merit, that our products or drug delivery technologies infringe proprietary rights of third parties could be time-consuming, result in costly litigation or divert the efforts of our technical and management personnel, any of which could disrupt our relationships with our partners and could significantly harm our financial positions and operating results.
If we or our partners are required to obtain licenses from third parties, our revenues and royalties on any commercialized products could be reduced.
The development of certain products based on our drug delivery technologies may require the use of raw materials (e.g., proprietary excipient), active ingredients, drugs (e.g., proprietary proteins) or technologies developed by third parties. The extent to which efforts by other researchers have resulted or will result in patents and the extent to which we or our partners are forced to obtain licenses from others, if available, on commercially reasonable terms is currently unknown. If we or our partners must obtain licenses from third parties, fees may be required for such licenses, which could reduce the net revenues and royalties we receive on commercialized products that incorporate our drug delivery technologies.
Security breaches and other disruptions could compromise confidential information and expose us to liability and cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store on our networks various intellectual property including our proprietary business information and that of our customers, suppliers and business partners. The secure maintenance and transmission of this information is critical to our operations and business strategy. Despite our security measures, our information systems and infrastructure may be vulnerable to disruptions such as computer hacking, phishing attacks, ransomware, dissemination of computer viruses, worms and other destructive or disruptive software, attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, investigations by regulatory authorities in the U.S. and EU Member States, disruption to our operations and damage to our reputation, any of which could adversely affect our business.
We could suffer financial loss or the loss of valuable confidential information. Although we develop and maintain systems and controls designed to prevent these events from occurring and we have a process to identify and mitigate threats, the development and maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become increasingly sophisticated. Moreover, despite our efforts, the possibility of these events occurring cannot be eliminated entirely and there can be no assurance that any measures we take will prevent cyber-attacks or security breaches that could adversely affect our business.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the U.S., if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products, including generics or biosimilars. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
-24-
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or other intellectual property. If we were to initiate legal proceedings against a third party to enforce a patent covering our product(s) or product candidate(s), the defendant could counterclaim that the patent is invalid and/or unenforceable. In patent litigation in the U.S., defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, written description or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. There is risk that a court could rule in favor of the defendant with respect to such a counterclaim of patent invalidity and/or unenforceability.
Interference or derivation proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Our defense of litigation or interference or derivation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties, or enter into development partnerships that would help us bring our product candidates to market.
Because of the substantial amount of discovery that can occur in connection with intellectual property-related litigation and/or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation/proceeding. There could also be public announcements of the results of hearings, motions, or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
We employ or may employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we endeavor to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying any awarded monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and/or be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the U.S. in several stages over the lifetime of the patents and/or applications. We rely on our outside counsel to coordinate payment of these fees due to patent agencies. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our product candidates in all countries throughout the world would be prohibitively
-25-
expensive, and intellectual property rights in some countries outside the U.S. can be less extensive than those in the U.S. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the U.S., or from selling or importing products made using our inventions in and into the U.S. or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export infringing products to territories where we have patent protection, but enforcement is not as strong as that in the U.S. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in non-U.S. jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property we develop or license.
Risks Related to Regulatory and Legal Matters
Our products will generally be subject to regulatory approval. If we or our pharmaceutical and biotechnology company partners do not obtain such approvals, or if such approvals are delayed, our revenues may be adversely affected.
Our FT218 product, as well as products we may wish to market in the future, may not gain regulatory approval and reach the commercial market for a variety of reasons.
In the U.S., federal, state and local government agencies, primarily the FDA, regulate all pharmaceutical products, including existing products and those under development. Neither we nor our pharmaceutical and biotechnology partners can control whether we obtain regulatory approval for any of these products or, if obtained, the timing thereof. There may be significant delays in expected product releases while attempting to obtain regulatory approval for products incorporating our technologies. If we or our partners are not successful in timely obtaining such approvals, our revenues and profitability may decline.
Applicants for FDA approval often must submit to the FDA extensive clinical and pre-clinical data, as well as information about product manufacturing processes and facilities and other supporting information. Varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of a drug product. The FDA also may require us, or our partners to conduct additional pre-clinical studies or clinical trials.
Similarly, although we anticipate submitting applications for approval for our development products that rely on existing data to demonstrate safety and effectiveness, the FDA may determine that additional studies particular to our products are necessary. If the FDA requires such additional studies, it would impact development plans for those products.
Changes in FDA approval policy during the development period, or changes in regulatory review for each submitted new product application, also may delay an approval or result in rejection of an application. For instance, under the FDAAA, we or our partners may be required to develop REMS to ensure the safe use of product candidates. If the FDA disagrees with such REMS proposals, it may be more difficult and costly to obtain regulatory approval for our product candidates. Similarly, FDAAA provisions may make it more likely that the FDA will refer a marketing application for a new product to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. This review may add to the time for approval, and, although the FDA is not bound by the recommendation of an advisory committee, objections or concerns expressed by an advisory committee may cause the FDA to delay or deny approval.
The FDA has substantial discretion in the approval process and may disagree with our or our partners’ interpretations of data and information submitted in an application, which also could cause delays of an approval or rejection of an application. Even if the FDA approves a product, the approval may limit the uses or indications for which the product may be marketed, restrict distribution of the product or require further studies.
The FDA may also withdraw product approvals for failure to comply with regulatory requirements or if problems follow initial marketing. In the same way, medicinal products for supply on the EU market are subject to marketing authorization by either the European Commission, following an opinion by the EMA, or by the competent authorities of EU Member States. Applicants for
-26-
marketing authorization must submit extensive technical and clinical data essentially in the form of the ICH Common Technical Document. The data is subject to extensive review by the competent authorities, and after such review the data may be considered inappropriate or insufficient. If applications for marketing authorization by pharmaceutical and biotechnology company partners are delayed or rejected, if the therapeutic indications for which the product is approved are limited, or if conditional marketing authorization imposing post-marketing clinical trials or surveillance is imposed, our revenues, operating results and liquidity may decline and earnings may be negatively impacted.
Our products are subject to continuing regulation, and we on our own, and in conjunction with our pharmaceutical partners, may be subject to adverse consequences if we or they fail to comply with applicable regulations.
We, on our own, and in conjunction with our pharmaceutical partners will be subject to extensive regulatory requirements for our and the co-developed products and product candidates, even if the products receive regulatory approval. These regulations are wide-ranging and govern, among other things:
• | adverse drug experiences and other reporting requirements; |
• | product promotion and marketing; |
• | APIs and/or product manufacturing, including cGMP compliance; |
• | record keeping; |
• | distribution of drug samples; |
• | required clinical trials and/or post-marketing studies; |
• | authorization renewal procedures; |
• | authorization variation procedures; |
• | compliance with any required REMS; |
• | updating safety and efficacy information; |
• | processing of personal data; |
• | use of electronic records and signatures; and |
• | changes to product manufacturing or labeling. |
Clinical development of drugs is costly and time-consuming, and the outcomes are uncertain. A failure to prove that our product candidates are safe and effective in clinical trials could materially and adversely affect our business, financial condition, results of operations and growth prospects.
We have made significant investments in our REST-ON Phase 3 clinical trial of FT218. Clinical trials are expensive and can take many years to complete, and the outcome is uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of potential medicine candidates may not be predictive of the results of later-stage clinical trials. Drug candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical testing. Any failure of our REST-ON Phase 3 clinical trial would prevent or delay the potential approval and commercialization of our sodium oxybate product, which would materially and adversely affect our business, financial condition, results of operations and growth prospects.
In addition to issues relating to the results generated in clinical trials, clinical trials can be delayed or halted for a variety of reasons, including:
• | failure in obtaining regulatory approval to commence a trial; |
• | failure in reaching agreement on acceptable terms with prospective contract research organizations (“CROs”) and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
• | failure in obtaining institutional review board or ethics committee approval at each site; |
• | failure in recruiting suitable patients to participate in a trial; |
• | failure in having patients complete a trial or return for post-treatment follow-up; |
• | failure in clinical sites dropping out of a trial; |
• | failure in adding new sites; or |
• | failure in manufacturing sufficient quantities of medicine candidates for use in clinical trials. |
We rely and expect to rely on CROs and clinical trial sites to ensure the proper and timely conduct of our future clinical trials and while we have and intend to have agreements governing their committed activities, we will have limited influence over their actual performance.
-27-
We rely on third parties to conduct our clinical trials, and if they do not properly and successfully perform their contractual, legal and regulatory duties, we may not be able to obtain regulatory approvals for or commercialize our drug product candidates.
We rely on CROs and other third parties to assist us in designing, managing, monitoring and otherwise carrying out our clinical trials, including with respect to site selection, contract negotiation and data management. We do not control these third parties and, as a result, they may not treat our clinical studies as a high priority, which could result in delays. We are responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol, as well as the FDA’s and non-U.S. regulatory agencies’ requirements, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to ensure that the data and results are credible and accurate and that the trial participants are adequately protected. The FDA and non-U.S. regulatory agencies enforce good clinical practices through periodic inspections of trial sponsors, principal investigators and trial sites. If we, CROs or other third parties assisting us or our study sites fail to comply with applicable good clinical practices, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or its non-U.S. counterparts may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA or non-U.S. regulatory agencies will determine that any of our clinical trials comply with good clinical practices. In addition, our clinical trials must be conducted with product produced under the FDA’s cGMP regulations and similar regulations outside of the U.S. Our failure, or the failure of our product suppliers, to comply with these regulations may require us to repeat or redesign clinical trials, which would delay the regulatory approval process.
If third parties do not successfully carry out their duties under their agreements with us, if the quality or accuracy of the data they obtain is compromised due to failure to adhere to our clinical protocols, including dosing requirements, or regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, our clinical trials may not meet regulatory requirements. If our clinical trials do not meet regulatory requirements or if these third parties need to be replaced, our clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates or succeed in our efforts to create approved line extensions for certain of our existing products or generate additional useful clinical data in support of these products.
If we or our partners, including any CDMOs that we use, fail to comply with these laws and regulations, the FDA, the European Commission, competent authorities of EU Member States, or other regulatory organizations, may take actions that could significantly restrict or prohibit commercial distribution of our products and products that incorporate our technologies. If the FDA, the European Commission or competent authorities of EU Member States determine that we are not in compliance with these laws and regulations, they could, among other things:
• | issue warning letters; |
• | impose fines; |
• | seize products or request or order recalls; |
• | issue injunctions to stop future sales of products; |
• | refuse to permit products to be imported into, or exported out of, the U.S. or the E.U.; |
• | suspend or limit our production; |
• | withdraw or vary approval of marketing applications; |
• | order the competent authorities of EU Member States to withdraw or vary national authorization; and |
• | initiate criminal prosecutions. |
If FT218 is approved by the FDA, we may not obtain orphan drug marketing exclusivity.
Orphan drug status may be granted by the FDA to certain products intended to treat diseases and conditions that affect fewer than 200,000 individuals in the U.S. or, if they affect more than 200,000 individuals in the U.S., there is no reasonable expectation of recovering the cost of developing and making the product available in the U.S. for the applicable disease or condition.
Our proposed product FT218 obtained orphan drug designation for the treatment of narcolepsy from the FDA in January 2018. Generally, a product with orphan drug designation that subsequently receives the first FDA approval for the disease or condition for which it has such designation will be entitled to certain U.S. marketing exclusivity for a period of seven years. FT218 would not be the first sodium oxybate product with such FDA approval. However, if the FDA concludes that FT218 is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care, the FDA could award FT218 with such marketing exclusivity. A designated orphan drug may not receive orphan drug exclusivity. Among other factors, the FDA will consider the results of our FT218 Phase 3 clinical trial with respect to the efficacy and safety of the previously approved sodium oxybate product. Thus, there can be no assurance that FT218 will receive orphan drug status exclusivity, if approved. In addition, even if such orphan drug marketing exclusivity rights were granted by the FDA, such exclusivity rights may be lost if the FDA later determines that our request for such designation was materially defective or if the manufacturer is unable to assure sufficient
-28-
quantity of the drug to meet the needs of patients with the rare disease or condition to be treated with the product. Further, even with respect to the indications for which we have received orphan designation, we may not be the first to obtain marketing approval for any particular orphan indication due to the uncertainties associated with developing pharmaceutical products, and thus, for example, approval of our product candidates could be blocked for seven years if another company previously obtained approval and orphan drug exclusivity in the U.S. for the same drug and same condition.
We are subject to U.S. federal and state and international laws and regulations prohibiting “kickbacks” and false claims that, if violated, could subject us to substantial penalties, and any challenges to or investigation into our practices under these laws could cause adverse publicity and be costly to respond to, and thus could harm our business.
We are subject to extensive and complex U.S. federal and state and international laws and regulations, including but not limited to, health-care “fraud and abuse” laws, such as anti-kickback and false claims laws and regulations pertaining to government benefit program reimbursement, price reporting and regulations, and sales and marketing practices. These laws and regulations are broad in scope and subject to evolving interpretations, which could require us to incur substantial costs associated with compliance or to alter one or more of our sales or marketing practices. In addition, violations of these laws, or allegations of such violations, could disrupt our business and result in a material adverse effect on our revenues, profitability, and financial condition. In the current environment, there appears to be a greater risk of investigations of possible violations of these laws and regulations. This increased risk is reflected by recent enforcement activity and pronouncements by the US Office of Inspector General of the Department of Health and Human Services that it intends to continue to vigorously pursue fraud and abuse violations by pharmaceutical companies, including through the potential to impose criminal penalties on pharmaceutical company executives. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Healthcare reform and restrictions on reimbursements may limit our financial returns.
Our ability to successfully commercialize our products and technologies may depend on the extent to which the government health administration authorities, the health insurance funds in the EU Member States, private health insurers and other third-party payor in the U.S. will reimburse consumers for the cost of these products, which would affect the volume of drug products sold by pharmaceutical and biotechnology companies that incorporate our technology into their products. Third party payor are increasingly challenging both the need for, and the price of, novel therapeutic drugs and uncertainty exists as to the reimbursement status of newly approved therapeutics. The commercial success of our products depends in part on the conditions under which products incorporating our technology are reimbursed. Adequate third-party reimbursement may not be available for such drug products to enable us to maintain price levels sufficient to realize an appropriate return on our investments in research and product development, which could materially and adversely affect our business. We cannot predict the effect that changes in the healthcare system, especially cost containment efforts, may have on our business. In particular, it is difficult to predict the effect of health care reform legislation enacted in the U.S. in 2010, certain provisions of which are still subject to regulatory implementation, further legislative change and ongoing judicial review. Any such changes or changes due to future legislation governing the pricing and reimbursement of healthcare products in the EU Member States may adversely affect our business.
Regulatory reforms may adversely affect our ability to sell our products profitably.
From time to time, the U.S. Congress, the Council of the European Union and the European Parliament, as well as the legislators of the EU Member States, adopt changes to the statutes that the FDA, the European Commission and the competent authorities of the EU Member States enforce in ways that could significantly affect our business. In addition, the FDA, the European Commission and the competent authorities of the EU Member States often issue new regulations or guidance, or revise or reinterpret their current regulations and guidance in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or FDA, EU or EU Member State’s regulations, guidance or interpretations changed, and what the impact of any such changes may be. Any such changes could have a significant impact on the path to approval of our proposed products or of competing products, and on our obligations and those of our pharmaceutical industry partners.
We and companies to which we have licensed, or will license our products or drug delivery technologies and subcontractors we engage for services related to the development and manufacturing of our products are subject to extensive regulation by the FDA and other regulatory authorities. Our and their failure to meet strict regulatory requirements could adversely affect our business.
We, and companies to which we license our products or drug delivery technologies, as well as companies acting as subcontractors for our product developments, including but not limited to non-clinical, pre-clinical and clinical studies, and manufacturing, are subject to extensive regulation by the FDA, other domestic regulatory authorities and equivalent foreign regulatory authorities,
-29-
particularly the European Commission and the competent authorities of EU Member States. Those regulatory authorities may conduct periodic audits or inspections of the applicable facilities to monitor compliance with regulatory standards and we remain responsible for the compliance of our subcontractors. If the FDA or another regulatory authority finds failure to comply with applicable regulations, the authority may institute a wide variety of enforcement actions, including:
• | warning letters or untitled letters; |
• | fines and civil penalties; |
• | delays in clearing or approving, or refusal to clear or approve, products; |
• | withdrawal, suspension or variation of approval of products; product recall or seizure; |
• | orders to the competent authorities of EU Member States to withdraw or vary national authorization; |
• | orders for physician notification or device repair, replacement or refund; |
• | interruption of production; |
• | operating restrictions; |
• | injunctions; and |
• | criminal prosecution. |
Any adverse action by a competent regulatory agency could lead to unanticipated expenditures to address or defend such action and may impair our ability to produce and market applicable products, which could significantly impact our revenues and royalties that we receive from our customers.
We may face product liability claims related to clinical trials for our products or their misuse.
The testing, including through clinical trials, manufacturing and marketing, and the use of our products may expose us to potential product liability and other claims. If any such claims against us are successful, we may be required to make significant compensation payments. Any indemnification that we have obtained, or may obtain, from CROs or pharmaceutical and biotechnology companies or hospitals conducting human clinical trials on our behalf may not protect us from product liability claims or from the costs of related litigation. Insurance coverage is expensive and difficult to obtain, and we may be unable to obtain coverage in the future on acceptable terms, if at all. We currently maintain general liability insurance and product liability and recall insurance. We cannot be certain that the coverage limits of our insurance policies or those of our strategic partners will be adequate. If we are unable to obtain sufficient insurance at an acceptable cost, a product liability claim or recall could adversely affect our financial condition.
Similarly, any indemnification we have obtained, or may obtain, from pharmaceutical and biotechnology companies with whom we are developing, or will develop, our products may not protect us from product liability claims from the consumers of those products or from the costs of related litigation.
If we use hazardous biological and/or chemical materials in a manner that causes injury, we may be liable for significant damages.
Our R&D activities involve the controlled use of potentially harmful biological and/or chemical materials, and are subject to U.S., state, EU, national and local laws and regulations governing the use, storage, handling and disposal of those materials and specified waste products. We cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling or disposal of these materials, including fires and/or explosions, storage tank leaks and ruptures and discharges or releases of toxic or hazardous substances. These operating risks can cause personal injury, property damage and environmental contamination, and may result in the shutdown of affected facilities and the imposition of civil or criminal penalties. The occurrence of any of these events may significantly reduce the productivity and profitability of a particular manufacturing facility and adversely affect our operating results.
We currently maintain property, business interruption and casualty insurance with limits that we believe to be commercially reasonable, but may be inadequate to cover any actual liability or damages.
Risks Related to Ownership of Our Securities
The price of ADSs representing our ordinary shares has been volatile and may continue to be volatile.
The trading price of American Depositary Shares representing our ordinary shares, or ADSs, has been, and is likely to continue to be, highly volatile. The market value of an investment in ADSs may fall sharply at any time due to this volatility. During the year ended December 31, 2019, the closing sale price of ADSs as reported on the Nasdaq Global Market ranged from $1.09 to $7.70. During the year ended December 31, 2018, the closing sale price of ADSs as reported on the Nasdaq Global Market ranged
-30-
from $1.74 to $11.70. The market prices for securities of drug delivery, specialty pharma, biotechnology and pharmaceutical companies historically have been highly volatile. Factors that could adversely affect our share price include, among others:
• | fluctuations in our operating results; |
• | announcements of technological partnerships, innovations or new products by us or our competitors; |
• | actions with respect to the acquisition of new or complementary businesses; |
• | governmental regulations; |
• | developments in patent or other proprietary rights owned by us or others; |
• | public concern as to the safety of drug delivery technologies developed by us or drugs developed by others using our platform; |
• | the results of pre-clinical testing and clinical studies or trials by us or our competitors; |
• | adverse events related to our products or products developed by pharmaceutical and biotechnology company partners that use our drug delivery technologies; |
• | lack of efficacy of our products; |
• | litigation; |
• | decisions by our pharmaceutical and biotechnology company partners relating to the products incorporating our technologies; |
• | the perception by the market of specialty pharma, biotechnology, and high technology companies generally; |
• | general market conditions, including the impact of the current financial environment; and |
• | the dilutive impact of any new equity or convertible debt securities we may issue or have issued. |
We incurred a net loss in 2019 and we will likely incur a net loss in 2020, and if we are not able to regain profitability in the future, the value of our shares may fall.
We reported a net loss of $33.2 million and $95.3 million for the years ended December 31, 2019 and 2018, respectively. In addition, in part because we expect sales of our hospital products to suffer further substantial declines during 2020 and we will incur substantial expenses to develop our products, we will likely incur a net loss in 2020 as well, the amount of which is not known to us at this time. We cannot predict if we will be able to regain profitability. If we are unable to earn a profit in future periods, the market price of our shares may fall. Our ability to operate profitably depends upon a number of factors, many of which are beyond our direct control. These factors include:
• | the demand for our drug delivery technologies and products; |
• | the level of product and price competition; |
• | our ability to develop new partnerships and additional commercial applications for our products; |
• | our ability to control our costs; |
• | our ability to broaden our customer base; |
• | the effectiveness of our marketing strategy; |
• | our effective tax rate; |
• | the effectiveness of our partners’ marketing strategy for products that use our technology; and |
• | general economic conditions. |
We may require additional financing, which may not be available on favorable terms or at all, and which may result in dilution of the equity interest of the holders of ADSs.
We may require additional financing to fund the development and possible acquisition of new products and businesses. We may consume available resources more rapidly than currently anticipated, resulting in the need for additional funding. If we cannot obtain financing when needed, or obtain it on favorable terms, we may be required to curtail our plans to continue to develop drug delivery technologies, develop new products, or acquire additional products and businesses. Other factors that will affect future capital requirements and may require us to seek additional financing include:
• | the development and acquisition of new products and drug delivery technologies; |
• | the progress of our research and product development programs; and |
• | the timing of, and amounts received from, future product sales, product development fees and licensing revenue and royalties. |
If adequate funds are not available, we may be required to significantly reduce or refocus our product development efforts, resulting in loss of sales, increased costs and reduced revenues. Alternatively, to obtain needed funds for acquisitions or operations, we may choose to issue additional ADSs representing our ordinary shares, or issue equity-linked debt, or we may choose to issue preferred shares, in either case through public or private financings. Additional funds may not be available on terms that are favorable to
-31-
us and, in the case of such equity financings, may result in dilution to the holders of ADSs. See also the discussion elsewhere in these Risk Factors under the caption “Our net loss and the resulting decrease in our available liquid assets may limit our ability to fully pursue our business strategy.”
We have broad discretion in the use of our cash and may not use it effectively.
Our management has broad discretion in the use of our cash, and may not apply our cash in ways that ultimately increases the value of any investment in our securities. We currently intend to use our cash to fund marketing activities for our commercialized products, to fund certain clinical trials for product candidates, to fund R&D activities for potential new product candidates, and for working capital, capital expenditures and general corporate purposes. As in the past we expect to invest our excess cash in available-for-sale marketable securities, including corporate bonds, U.S. government securities, other fixed income securities and equities; and these investments may not yield a favorable return. If we do not invest or apply our cash effectively, our financial position and the price of ADSs may decline.
We currently do not intend to pay dividends and cannot assure the holders of our ADSs that we will make dividend payments in the future.
We have never declared or paid a cash dividend on any of our ordinary shares or ADSs and do not anticipate declaring cash dividends in the foreseeable future. Declaration of dividends will depend upon, among other things, future earnings, if any, the operating and financial condition of our business, our capital requirements, general business conditions and such other factors as our Board of Directors deems relevant.
Provisions of our articles of association could delay or prevent a third-party’s effort to acquire us.
Our articles of association could delay, defer or prevent a third-party from acquiring us, even where such a transaction would be beneficial to the holders of ADSs, or could otherwise adversely affect the price of ADSs. For example, certain provisions of our articles of association:
• | permit our board of directors to issue preferred shares with such rights and preferences as they may designate, subject to applicable law; |
• | impose advance notice requirements for shareholder proposals and director nominations to be considered at annual shareholder meetings; and |
• | require the approval of a supermajority of the voting power of our shares entitled to vote at a general meeting of shareholders to amend or repeal any provisions of our articles of association. |
We believe these provisions, if implemented in compliance with applicable law, may provide some protection to holders of ADSs from coercive or otherwise unfair takeover tactics. These provisions are not intended to make us immune from takeovers. They will, however, apply even if some holders of ADSs consider an offer to be beneficial and could delay or prevent an acquisition that our Board of Directors determines is in the best interest of the holders of ADSs. Certain of these provisions may also prevent or discourage attempts to remove and replace incumbent directors.
In addition, mandatory provisions of Irish law could prevent or delay an acquisition of the Company by a third party. For example, Irish law does not permit shareholders of an Irish public limited company to take action by written consent with less than unanimous consent. In addition, an effort to acquire us may be subject to various provisions of Irish law relating to mandatory bids, voluntary bids, requirements to make a cash offer and minimum price requirements, as well as substantial acquisition rules and rules requiring the disclosure of interests in ADSs in certain circumstances.
These provisions may discourage potential takeover attempts or bids for our ordinary shares at a premium over the market price or they may adversely affect the market price of, and the voting and other rights of the holders of, ADSs. These provisions could also discourage proxy contests and make it more difficult for holders of ADSs to elect directors other than the candidates nominated by our board of directors, and could depress affect the market price of ADSs.
Irish law differs from the laws in effect in the U.S. and might afford less protection to the holders of ADSs.
Holders of ADSs could have more difficulty protecting their interests than would the shareholders of a U.S. corporation. As an Irish company, we are governed by Irish law, including the Irish Companies Act 2014 and the Irish Takeover Rules, which differs in some significant, and possibly material, respects from provisions set forth in various U.S. state laws applicable to U.S. corporations and their shareholders, including provisions relating to interested directors, mergers and acquisitions, takeovers, shareholder lawsuits and indemnification of directors.
-32-
The duties of directors and officers of an Irish company are generally owed to the company only. Therefore, under Irish law shareholders of Irish companies do not generally have a right to commence a legal action against directors or officers, and may only do so in limited circumstances. Directors of an Irish company must act with due care and skill, honestly and in good faith with a view to the best interests of the company. Directors must not put themselves in a position in which their duties to the company and their personal interests conflict and must disclose any personal interest in any contract or arrangement with the company or any of our subsidiaries. A director or officer can be held personally liable to the company in respect of a breach of duty to the company.
Judgments of U.S. courts, including those predicated on the civil liability provisions of the federal securities laws of the U.S., may not be enforceable in Irish courts.
An investor in the U.S. may find it difficult to:
• | effect service of process within the U.S. against us and our non-U.S. resident directors and officers; |
• | enforce U.S. court judgments based upon the civil liability provisions of the U.S. federal securities laws against us and our non-U.S. resident directors and officers in Ireland; or |
• | bring an original action in an Irish court to enforce liabilities based upon the U.S. federal securities laws against us and our non-U.S. resident directors and officers. |
Judgments of U.S. courts, including those predicated on the civil liability provisions of the federal securities laws of the United States, may not be enforceable in Cayman Islands courts.
We have been advised by our Cayman Islands legal counsel, Maples and Calder, that the courts of the Cayman Islands are unlikely (i) to recognize or enforce against us or Avadel judgments of courts of the U.S. predicated upon the civil liability provisions of the securities laws of the U.S. or any State; and (ii) in original actions brought in the Cayman Islands, to impose liabilities against us or Avadel predicated upon the civil liability provisions of the securities laws of the U.S. or any State, so far as the liabilities imposed by those provisions are penal in nature. In those circumstances, although there is no statutory enforcement in the Cayman Islands of judgments obtained in the U.S., the courts of the Cayman Islands will recognize and enforce a foreign money judgment of a foreign court of competent jurisdiction without retrial on the merits based on the principle that a judgment of a competent foreign court imposes upon the judgment debtor an obligation to pay the sum for which judgment has been given provided certain conditions are met. For a foreign judgment to be enforced in the Cayman Islands, such judgment must be final and conclusive and for a liquidated sum, and must not be in respect of taxes or a fine or penalty, inconsistent with a Cayman Islands judgment in respect of the same matter, impeachable on the grounds of fraud or obtained in a manner, and or be of a kind the enforcement of which is, contrary to natural justice or the public policy of the Cayman Islands (awards of punitive or multiple damages may well be held to be contrary to public policy). A Cayman Islands Court may stay enforcement proceedings if concurrent proceedings are being brought elsewhere.
Holders of ADSs have fewer rights than shareholders and have to act through the Depositary to exercise those rights.
Holders of ADSs do not have the same rights as shareholders and, accordingly, cannot exercise rights of shareholders against us. The Bank of New York Mellon, as depositary, or the “Depositary”, is the registered shareholder of the deposited shares underlying the ADSs. Therefore, holders of ADSs will generally have to exercise the rights attached to those shares through the Depositary. We will use reasonable efforts to request that the Depositary notify the holders of ADSs of upcoming votes and ask for voting instructions from them. If a holder fails to return a voting instruction card to the Depositary by the date established by the Depositary for receipt of such voting instructions, or if the Depositary receives an improperly completed or blank voting instruction card, or if the voting instructions included in the voting instruction card are illegible or unclear, then such holder will be deemed to have instructed the Depositary to vote its shares, and the Depositary shall vote such shares in favor of any resolution proposed or approved by our Board of Directors and against any resolution not so proposed or approved.
Our largest shareholders own a significant percentage of the share capital and voting rights of the Company.
As of February 25, 2020, RTW Investments LP. owned approximately 10.0% of Avadel’s outstanding shares (in the form of ADSs), Avoro Capital Advisors LLC owned approximately 8.4% of our outstanding shares (in the form of ADSs) and Vivo Opportunity, LLC and certain of its affiliates owned approximately 7.6% of our outstanding shares (in the form of ADSs). To the extent these shareholders continue to hold a large percentage of our share capital and voting rights, they will remain in a position to exert heightened influence in the election of the directors of the Company and in other corporate actions that require shareholder approval, including change of control transactions.
-33-
U.S. Holders of ordinary shares or ADSs may suffer adverse U.S. tax consequences if we are classified as a passive foreign investment company.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties that are received from unrelated parties in connection with the active conduct of a trade or business. Our status as a PFIC depends on the composition of our income and the composition and value of our assets (for which purpose the total value of our assets may be determined in part by the market value of the ordinary shares or ADSs, which are subject to change) from time to time. If we are characterized as a PFIC, U.S. Holders (as defined below under “Material U.S. Federal Income Tax Considerations for U.S. Holders”) of ordinary shares or ADSs may suffer materially adverse tax consequences, including having gains realized on the sale of ordinary shares or ADSs treated as ordinary income, rather than capital gain, the loss of the preferential rate applicable to dividends received on ordinary shares or ADSs by individuals who are U.S. Holders, and having interest charges apply to distributions by us and the proceeds of sales of ordinary shares or ADSs. See “Material U.S. Federal Income Tax Considerations for U.S. Holders—PFIC rules.”
We believe that we were not a PFIC for the taxable year ending December 31, 2019 and, based on the expected value of our assets, including any goodwill, and the expected nature and composition of our income and assets, we do not anticipate that we will be a PFIC for our current taxable year. However, our status as a PFIC is a fact-intensive determination subject to various uncertainties, and we cannot provide any assurances regarding our PFIC status for the current, prior or future taxable years.
Certain U.S. Holders that own 10 percent or more of the vote or value of ordinary shares or ADSs may suffer adverse U.S. tax consequences because our non-U.S. subsidiaries are expected to be classified as controlled foreign corporations.
Each ‘‘Ten Percent Shareholder’’ (as defined below) in a non-U.S. corporation that is classified as a ‘‘controlled foreign corporation,’’ or a CFC, for U.S. federal income tax purposes generally is required to include in income for U.S. federal tax purposes such Ten Percent Shareholder’s pro rata share of the CFC’s ‘‘Subpart F income’’ and investment of earnings in U.S. property, even if the CFC has made no distributions to its shareholders. Subpart F income generally includes dividends, interest, rents, royalties, ‘‘global intangible low-taxed income,’’ gains from the sale of securities and income from certain transactions with related parties. In addition, a Ten Percent Shareholder that realizes gain from the sale or exchange of shares in a CFC may be required to classify a portion of such gain as dividend income rather than capital gain. A non-U.S. corporation generally will be classified as a CFC for U.S. federal income tax purposes if Ten Percent Shareholders own, directly or indirectly, more than 50% of either the total combined voting power of all classes of stock of such corporation entitled to vote or of the total value of the stock of such corporation. A ‘‘Ten Percent Shareholder’’ is a U.S. person (as defined by the Code) who owns or is considered to own 10% or more of the total combined voting power of all classes of stock entitled to vote or 10% or more of the total value of all classes of stock of such corporation.
We believe that we were not a CFC in the 2019 taxable year, but that our non-U.S. subsidiaries were CFCs in the 2019 taxable year. We anticipate that our non-U.S. subsidiaries will remain CFCs in the 2020 taxable year, and it is possible that we may become a CFC in the 2020 taxable year or in a subsequent taxable year. The determination of CFC status is complex and includes attribution rules, the application of which is not entirely certain. U.S. Holders should consult their own tax advisors with respect to the potential adverse U.S. tax consequences of becoming a Ten Percent Shareholder in a CFC, including the possibility and consequences of becoming a Ten Percent Shareholder in one or more of our non-U.S. subsidiaries that are anticipated to be treated as CFCs. If we are classified as both a CFC and a PFIC, we generally will not be treated as a PFIC with respect to those U.S. Holders that meet the definition of a Ten Percent Shareholder during the period in which we are a CFC, subject to certain exceptions.
A transfer of ordinary shares may be subject to Irish stamp duty.
Transfers of ordinary shares (as opposed to ADSs) could be subject to Irish stamp duty (currently at the rate of 1% of the higher of the price paid or the market value of the shares acquired). Payment of Irish stamp duty is generally a legal obligation of the transferee. Although transfers of ADSs are not subject to Irish stamp duty, the potential for stamp duty to arise on transfers of ordinary shares could adversely affect the price of our ordinary shares or ADSs.
-34-
Risks Related to the 2023 Notes
Servicing our 2023 Notes may require a significant amount of cash, and we may not have sufficient cash or the ability to raise the funds necessary to settle exchanges of the 2023 Notes in cash, repay the Notes at maturity, or repurchase the 2023 Notes as required following a fundamental change.
In February 2018 we issued $143.75 million aggregate principal amount of our Senior Exchangeable Notes. Prior to February 2023, the 2023 Notes are convertible at the option of the holders only under certain conditions or upon the occurrence of certain events. If holders of the 2023 Notes elect to exchange their 2023 Notes, unless we elect to deliver solely our ADSs to settle such exchanges, we will be required to make cash payments in respect of the 2023 Notes being exchanged. Holders of the 2023 Notes also have the right to require us to repurchase all or a portion of their 2023 Notes upon the occurrence of a fundamental change (as defined in the applicable indenture governing the 2023 Notes) at a repurchase price equal to 100% of the principal amount of the 2023 Notes to be repurchased, plus accrued and unpaid interest. If the 2023 Notes have not previously been exchanged or repurchased, we will be required to repay the 2023 Notes in cash at maturity. Our ability to make cash payments in connection with exchanges of the 2023 Notes, repurchase the 2023 Notes in the event of a fundamental change, or to repay or refinance the 2023 Notes at maturity will depend on market conditions and our future performance, which is subject to economic, financial, competitive, and other factors many of which are beyond our control. We incurred significant net losses in 2019 and we may continue to incur significant losses. As a result, we may not have enough available cash or be able to obtain financing at the time we are required to repurchase or repay the 2023 Notes or in the event we elect to pay cash with respect to 2023 Notes being exchanged.
The conditional exchange feature of the 2023 Notes, if triggered, may adversely affect our financial condition and operating results.
In the event the conditional exchange feature of the 2023 Notes is triggered, holders of 2023 Notes will be entitled to exchange the 2023 Notes at any time during specified periods at their option (see the discussion under the caption “Recent Developments -- Issuance of Exchangeable Notes” in Item 1 of this Annual Report on Form 10-K). If one or more holders elect to exchange their 2023 Notes, unless we elect to satisfy our exchange obligation by causing to be delivered solely ADSs (other than paying cash in lieu of any fractional ADSs), we would be required to settle a portion or all of our exchange obligation through the payment of cash, which could adversely affect our liquidity. In addition, even if holders do not elect to exchange their 2023 Notes, we could be required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the 2023 Notes as a current rather than long-term liability, which would result in a material reduction of our net working capital.
The accounting method for convertible and exchangeable debt securities that may be settled in cash, such as the 2023 Notes, could have a material effect on our reported financial results.
Under Accounting Standards Codification 470-20, Debt with Conversion and Other Options, which we refer to as ASC 470-20, an entity must separately account for the liability and equity components of the convertible or exchangeable debt instruments (such as the 2023 Notes) that may be settled entirely or partially in cash upon conversion or exchange in a manner that reflects the issuer’s economic interest cost. However, entities must first consider the guidance in ASC 815-15, Embedded Derivatives (“ASC 815-15”), to determine if an instrument contains an embedded feature that should be separately accounted for as a derivative. ASC 815 provides for an exception to this rule when convertible notes, as host instruments, are deemed to be conventional, as defined by ASC 815-40. Should this exception apply, the effect of ASC 470-20 on the accounting for the 2023 Notes is that the equity component would be required to be included in the additional paid-in capital section of shareholders’ equity on Avadel’s consolidated balance sheet, and the value of the equity component would be treated as original issue discount for purposes of accounting for the debt component of the 2023 Notes. As a result, Avadel would be required to record a greater amount of non-cash interest expense in current periods presented as a result of the amortization of the discounted carrying value of the 2023 Notes to their face amount over the term of the 2023 Notes. Avadel would report lower net income in its financial results because ASC 470-20 would require interest to include both the current period’s amortization of the debt discount and the instrument’s coupon interest, which could adversely affect Avadel’s reported or future financial results, the trading price of the ADSs and the trading price of the 2023 Notes.
In addition, under certain circumstances, convertible or exchangeable debt instruments (such as the 2023 Notes) that may be settled entirely or partly in cash are currently accounted for utilizing the treasury stock method, the effect of which is that the ADSs deliverable upon exchange of the 2023 Notes are not included in the calculation of diluted earnings per share except to the extent that the exchange value of the 2023 Notes exceeds their principal amount. Under the treasury stock method, for diluted earnings per share purposes, the transaction is accounted for as if the number of ADSs that would be necessary to settle such excess, if we elected to settle such excess in ADSs, are issued. We cannot be sure that the accounting standards in the future will continue to
-35-
permit the use of the treasury stock method. If Avadel is unable to use the treasury stock method in accounting for the ADSs deliverable upon exchange of the 2023 Notes, then Avadel’s diluted earnings per share would be adversely affected.
Exchanges of the 2023 Notes will dilute the ownership interest of Avadel’s existing shareholders and holders of the ADSs, including holders who had previously exchanged their 2023 Notes and received ADSs upon exchange, to the extent our exchange obligation includes ADSs.
The exchange of some or all of the 2023 Notes will dilute the ownership interests of Avadel’s existing shareholders and holders of the ADSs to the extent our exchange obligation includes ADSs. Any sales in the public market of the ADSs issuable upon such exchange of the 2023 Notes could adversely affect prevailing market prices of the ADSs and, in turn, the price of the 2023 Notes. In addition, the existence of the 2023 Notes may encourage short selling by market participants because the exchange of the 2023 Notes could depress the price of the ADSs.
The fundamental change repurchase feature of the 2023 Notes may delay or prevent an otherwise beneficial takeover attempt of Avadel.
The indenture governing the 2023 Notes will require us to repurchase the 2023 Notes for cash upon the occurrence of a fundamental change and, in certain circumstances, to increase the exchange rate for a holder that exchanges its 2023 Notes in connection with a make-whole fundamental change. A takeover of Avadel may trigger the requirement that we repurchase the 2023 Notes and/or increase the exchange rate, which could make it more costly for a potential acquirer to engage in a combinatory transaction with us or Avadel. Such additional costs may have the effect of delaying or preventing a takeover of Avadel that would otherwise be beneficial to investors.
Dividends paid by the Parent may be subject to Irish dividend withholding tax
In certain circumstances, as an Irish tax resident company, Avadel will be required to deduct Irish dividend withholding tax (currently at the rate of 20%) from dividends paid to its shareholders. Shareholders that are resident in the U.S., EU countries (other than Ireland) or other countries with which Ireland has signed a tax treaty (whether the treaty has been ratified or not) generally should not be subject to Irish withholding tax so long as the shareholder has provided its broker, for onward transmission to Avadel’s qualifying intermediary or other designated agent (in the case of shares held beneficially), or Avadel or its transfer agent (in the case of shares held directly), with all the necessary documentation by the appropriate due date prior to payment of the dividend. However, some shareholders may be subject to withholding tax, which could adversely affect the price of ordinary shares and the value of their 2023 Notes.
Item 1B. Unresolved Staff Comments.
Not applicable.
Item 2. Properties.
(Amounts in thousands, except square foot amounts)
We have commercial and administrative activities located in Chesterfield, Missouri. Our current office space consists of 24,236 square feet, and the lease expires in 2025. Additionally, we still maintain the lease on the former headquarters of FSC Laboratories, Inc. located in Charlotte, North Carolina. This office space consists of 6,300 square feet, and the lease expires in 2020.
Avadel Research SAS, our prior research center, was located in Venissieux, France (a suburb of Lyon) in two adjacent leased facilities totaling approximately 25,600 square feet. One building of approximately 12,800 square feet housed the administrative offices and analytical research laboratories. A second facility comprising approximately 12,800 square feet housed equipment dedicated to our Micropump and LiquiTime platforms. Both leases were terminated during the three months ended December 31, 2019 as part of the 2019 French Restructuring plan. See Note 18: Restructuring Costs in Part II, item 8 of this Annual Report on Form 10-K for more information.
We had intellectual property, clinical, quality, regulatory, and supply chain activities located in Dublin, Ireland. The office space consisted of 5,059 square feet and the lease expired in 2025. This lease was surrendered during the three months ended December 31, 2019 as part of the 2019 Corporate Restructuring plan. See Note 18: Restructuring Costs in Part II, item 8 of this Annual Report on Form 10-K for more information.
See “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II, Item 7 of this Annual
-36-
Report on Form 10-K for more information regarding our investment activities and principal capital expenditures over the last three years.
Item 3. Legal Proceedings.
Voluntary Chapter 11 Bankruptcy Filing by Specialty Pharma update
Note 3: Subsidiary Bankruptcy and Deconsolidation on Part II, item 8 of this annual report on From 10-K briefly describes the Chapter 11 bankruptcy case which our subsidiary Specialty Pharma commenced on February 6, 2019, and which on April 26, 2019 resulted in the bankruptcy court-approved sale of all of Specialty Pharma’s intangible assets and inventory to an unaffiliated third party. As a result of such sale, Specialty Pharma has completed its divestment of the assets of the Noctiva business. During the pendency of the bankruptcy case, all pending litigation against Specialty Pharma is automatically stayed and any new litigation against Specialty Pharma is precluded unless the bankruptcy court orders otherwise. Below are descriptions of a litigation to which Specialty Pharma is a party and a contract dispute involving Specialty Pharma, both of which matters are subject to the automatic stay during the bankruptcy case.
Ferring Litigation. Some of the patents covering the Noctiva product (the “Noctiva Patents”) are the subject of litigation initiated by Ferring Pharmaceuticals Inc. and two of its foreign affiliates, who manufacture a competing product known as Nocdurna. Nocdurna was approved by the FDA in June 2018 and commercially launched in the U.S. in November 2018. In this litigation, filed in the United States District Court for the Southern District of New York, Ferring seeks to invalidate and disputes the inventorship of the Noctiva Patents, seeks damages for various alleged breaches of contractual and common law duties, and seeks damages for alleged infringement by Noctiva of Ferring’s “Nocdurna” trademark. Specialty Pharma and certain other parties including Serenity Pharmaceuticals, LLC (“Serenity”) (the licensor of the Noctiva Patents) have defended this litigation, and have made counterclaims against Ferring, including for infringement of the Noctiva Patents and a declaratory judgment of noninfringement with respect to Ferring’s “Nocdurna” trademark. The court dismissed Ferring’s inventorship claim and its claims for alleged breaches of contractual and common law duties, although these dismissals may be appealed by Ferring. On February 15, 2019, Specialty Pharma and its co-defendants moved to stay the litigation pending completion of the bankruptcy proceeding of Specialty Pharma. On May 15, 2019, that motion was denied due to a pending settlement of the litigation with respect to just Ferring and Specialty Pharma. On February 25, 2020, Ferring and Specialty Pharma jointly moved for bankruptcy court approval of a settlement agreement with respect to the claims alleged in the litigation. In accordance with the terms of the settlement agreement, promptly following bankruptcy court approval of the settlement agreement, the parties would dismiss with prejudice their respective claims against each other in the litigation. On March 13th, 2020, the bankruptcy court entered an order approving the settlement with Ferring. Pursuant to the terms of the settlement, the parties are to dismiss their respective claims against each other in the District Court litigation in the Southern District of New York, with such dismissals to be effective concurrently. That joint dismissal has not yet been filed with District Court for the Southern District of New York.
Contract Dispute. On January 21, 2019, Serenity gave notice to Specialty Pharma of an alleged breach of the parties’ Noctiva license agreement. Serenity alleges that Specialty Pharma breached its contractual obligation to devote commercially reasonable efforts to the commercialization of Noctiva and seeks unspecified damages. On January 27, 2019, Specialty Pharma notified Serenity of a claim for $1.7 million in damages as a result of Serenity’s breach of its contractual obligation to pay the costs of the Ferring Litigation. Serenity’s notice to Specialty Pharma invoked the dispute resolution provisions of the Noctiva license agreement, which culminate in arbitration, but neither party has yet initiated an arbitration proceeding or filed suit.
Exela Litigation. On January 7, 2020, Exela filed a complaint against us and our subsidiary, Avadel Legacy in the United States District for the District of Delaware. The complaint alleges infringement of a certain Exela patent related to its cysteine hydrochloride product. Exela is most notably seeking i) a declaratory judgment that the Nouress product infringes its patent, ii) an injunction (both preliminary and permanent) precluding the launch of Nouress, and iii) monetary damages (including enhanced damages, prejudgment interest and attorneys’ fees) in the event Nouress is commercially launched and found to infringe Exela’s patent. We have not yet been served with the complaint.
Item 4. Mine Safety Disclosures.
Not applicable.
-37-
PART II
Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Common Stock Data (per share):
The principal trading market for our securities in ADSs is the Nasdaq Global Market under the symbol “AVDL”. There is no foreign trading market for our ordinary shares, ADSs or any other equity security issued by us. Each ADS represents one ordinary share, nominal value $0.01. Each ADS is evidenced by an ADR. The Bank of New York Mellon is the Depositary for the ADRs.
As of March 10, 2020, there were 46,404,432 ordinary shares outstanding, and our closing stock price was $8.57 per share.
The following table reports the high and low trading prices of the ADSs on the Nasdaq Global Market for the periods indicated:
2019 Price Range | 2018 Price Range | ||||||||||||||
High | Low | High | Low | ||||||||||||
First quarter | $ | 3.29 | $ | 1.44 | $ | 11.70 | $ | 6.76 | |||||||
Second quarter | 3.19 | 1.09 | 7.78 | 5.89 | |||||||||||
Third quarter | 4.47 | 1.92 | 7.14 | 4.08 | |||||||||||
Fourth quarter | 7.70 | 3.34 | 4.66 | 1.74 | |||||||||||
Holders
As of March 10, 2020, there were 79 holders of record of our ordinary shares and 42 accounts registered with The Bank of New York Mellon, the depositary of our ADS program, as holders of ADSs, one of which ADS accounts is registered to the Depositary Trust Corporation (DTC). Because our ADSs are generally held of record by brokers, nominees and other institutions as participants in DTC on behalf of the beneficial owners of such ADSs, we are unable to estimate the total number of beneficial owners of the ADSs held by these record holders.
Dividends
We have never declared or paid a cash dividend on any of our shares and do not anticipate declaring cash dividends in the foreseeable future.
Equity Compensation Plan
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report on Form 10-K.
Issuer Purchases of Equity Securities
We did not repurchase any of our equity securities during the year ended December 31, 2019.
-38-
Share Performance Graph
The following graph compares the cumulative 5-year return provided to shareholders of Avadel’s ADSs relative to the cumulative total returns of the Nasdaq Composite Index and the Nasdaq Biotechnology Index. We believe these indices are the most appropriate indices against which the total shareholder return of Avadel should be measured. The Nasdaq Biotechnology Index has been selected because it is an index of U.S. quoted biotechnology and pharmaceutical companies. An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our ADSs and in each of the indexes on January 1, 2013 and our relative performance is tracked through December 31, 2019. The comparisons shown in the graph are based upon historical data and we caution that the stock price performance shown in the graph is not indicative of, or intended to forecast, the potential future performance of our stock.
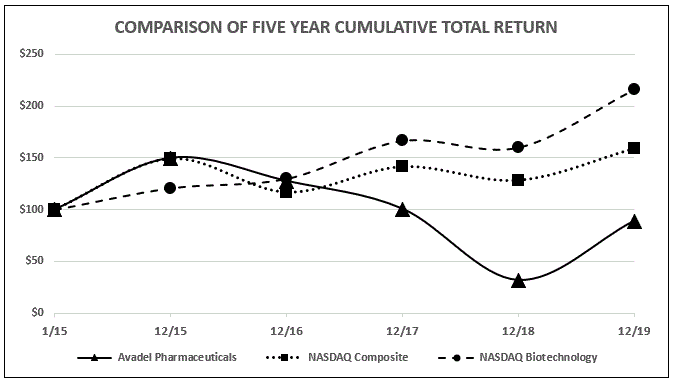
This performance graph shall not be deemed “filed” for purposes of Section 18 of the Exchange Act. Notwithstanding any statement to the contrary set forth in any of our filings under the Securities Act of 1933 or the Exchange Act that might incorporate future filings, including this Annual Report on Form 10-K, in whole or in part, this performance graph shall not be incorporated by reference into any such filings except as may be expressly set forth by specific reference in any such filing.
-39-
Item 6. Selected Financial Data (in thousands, except per share amounts).
Annual Financial Data:
The consolidated statements of (loss) income data for fiscal 2019, 2018 and 2017, and the consolidated balance sheet data as of December 31, 2019 and 2018 were derived from our consolidated financial statements and accompanying notes included elsewhere in this Annual Report on Form 10-K. The consolidated statements of (loss) income for fiscal 2016 and 2015 and the consolidated balance sheet data as of December 31, 2017, 2016 and 2015 were derived from our audited consolidated financial statements that are not included in this Annual Report on Form 10-K.
The following selected financial data should be read in conjunction with our consolidated financial statements and related notes appearing in Item 8 “Financial Statements and Supplementary Data” and Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II of this Annual Report on Form 10-K. Our historical results are not necessarily indicative of the results to be expected in any future period.
Statement of (Loss) Income Data: | 2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
Total revenues | $ | 59,215 | $ | 103,269 | $ | 173,245 | $ | 150,246 | $ | 173,009 | ||||||||||
Gross profit (a) | 47,090 | 85,753 | 156,944 | 136,998 | 161,599 | |||||||||||||||
Operating (loss) income (b) | (24,112 | ) | (104,926 | ) | 89,505 | (4,965 | ) | 70,758 | ||||||||||||
Net (loss) income | (33,226 | ) | (95,304 | ) | 68,271 | (41,276 | ) | 41,798 | ||||||||||||
Net (loss) income per share - basic | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.69 | $ | (1.00 | ) | $ | 1.03 | |||||||
Net (loss) income per share - diluted | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.63 | $ | (1.00 | ) | $ | 0.96 | |||||||
Balance Sheet Data: | 2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
Cash and cash equivalents | $ | 9,774 | $ | 9,325 | $ | 16,564 | $ | 39,215 | $ | 65,064 | ||||||||||
Marketable securities | 54,384 | 90,590 | 77,511 | 114,980 | 79,738 | |||||||||||||||
Goodwill | 18,491 | 18,491 | 18,491 | 18,491 | 18,491 | |||||||||||||||
Intangible assets, net | 813 | 1,629 | 92,289 | 22,837 | 15,825 | |||||||||||||||
Total assets | 151,436 | 190,300 | 253,277 | 245,482 | 215,081 | |||||||||||||||
Long-term debt (incl. current portion) | 121,686 | 115,840 | 267 | 815 | 1,118 | |||||||||||||||
Long-term related party payable (incl. current portion) | 17,327 | 28,840 | 98,925 | 169,347 | 122,693 | |||||||||||||||
(a) Gross profit is computed by subtracting cost of products from total revenues.
(b) We recorded an impairment charge of $66,087 during the year ended December 31, 2018.
-40-
Quarterly Financial Data (Unaudited):
The following tables present certain unaudited consolidated quarterly financial information for each quarter of 2019 and 2018. Year-to-date net income (loss) per share amounts may be different than the sum of the applicable quarters due to differences in weighted average shares outstanding for the respective periods.
2019: | March 31 | June 30 | September 30 | December 31 | |||||||||||
Revenues | $ | 16,437 | $ | 17,554 | $ | 14,229 | $ | 10,995 | |||||||
Gross profit (a) | 13,171 | 13,932 | 11,406 | 8,581 | |||||||||||
Operating loss | (8,167 | ) | (4,451 | ) | (4,147 | ) | (7,347 | ) | |||||||
Net loss | (13,018 | ) | (8,605 | ) | (8,864 | ) | (2,739 | ) | |||||||
Net loss per share - basic | (0.35 | ) | (0.23 | ) | (0.24 | ) | (0.07 | ) | |||||||
Net loss per share - diluted | (0.35 | ) | (0.23 | ) | (0.24 | ) | (0.07 | ) | |||||||
2018: | March 31 | June 30 | September 30 | December 31 | |||||||||||
Revenues | $ | 33,293 | $ | 29,230 | $ | 19,826 | $ | 20,920 | |||||||
Gross profit (a) | 26,701 | 25,718 | 16,706 | 16,628 | |||||||||||
Operating loss (b) | (12,625 | ) | (2,785 | ) | (14,095 | ) | (75,421 | ) | |||||||
Net loss | (12,236 | ) | (3,438 | ) | (15,771 | ) | (63,859 | ) | |||||||
Net loss per share - basic | (0.32 | ) | (0.09 | ) | (0.43 | ) | (1.72 | ) | |||||||
Net loss per share - diluted | (0.32 | ) | (0.09 | ) | (0.43 | ) | (1.72 | ) | |||||||
(a) Gross profit is computed by subtracting cost of products from total revenues.
(b) We recorded an impairment charge of $66,087 during the three months ended December 31, 2018.
-41-
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
MANAGEMENT’S DISCUSSION AND ANALYSIS
(In thousands, except per share data)
You should read the discussion and analysis of our financial condition and results of operations set forth in this Item 7 together with our consolidated financial statements and the related notes appearing elsewhere in this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report on Form 10-K, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties, and reference is made to the “Cautionary Disclosure Regarding Forward-Looking Statements” set forth immediately following the Table of Content of this Annual Report on Form 10-K for further information on the forward looking statements herein. In addition, you should read the “Risk Factors” section of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis and elsewhere in this Annual Report on Form 10-K.
Information pertaining to fiscal year 2017 was included in the Company’s Annual Report on Form 10-K for the year-ended December 31, 2018, on pages 42 through 58, under Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” which was filed with the SEC on March 15, 2019.
Overview
Nature of Operations
Avadel Pharmaceuticals plc (Nasdaq: AVDL) (“Avadel,” the “Company,” “we,” “our,” or “us”) is an emerging biopharmaceutical company. Our lead product candidate, FT218, is an investigational once-nightly formulation of sodium oxybate for the treatment of excessive daytime sleepiness (“EDS”), and cataplexy in narcolepsy patients. FT218, which uses our Micropump drug-delivery technology, is in a Phase 3 clinical trial for the treatment of narcolepsy patients suffering from EDS and cataplexy. In addition, we have three approved commercial products developed under our “unapproved marketed drug,” or UMD, program, Akovaz, Bloxiverz and Vazculep, and a fourth approved product, Nouress, which are sterile injectable drugs used in the hospital setting.
We are primarily focused on the development and potential U.S. Food and Drug Administration (“FDA”) approval of FT218. In addition, we continue to market and distribute our current approved hospital products portfolio and, pending resolution of the existing patent infringement claim (as described below), we plan to commercialize Nouress. Outside of our product candidate and our existing commercial products, we continue to evaluate opportunities to expand our product portfolio.
FT218 (Micropump sodium oxybate)
FT218 is a once-nightly formulation of sodium oxybate that uses our Micropump controlled release drug-delivery technology for the treatment of EDS and cataplexy in patients suffering from narcolepsy. Sodium oxybate is the sodium salt of gamma hydroxybutyrate, an endogenous compound and metabolite of the neurotransmitter gamma-aminobutyric acid. Sodium oxybate is approved in Europe and the United States (“U.S.”) as a twice-nightly formulation indicated for the treatment of EDS and cataplexy in patients with narcolepsy.
In December 2019, we completed patient enrollment of our Phase 3 REST-ON clinical trial of FT218 to assess the safety and efficacy of a once-nightly formulation of FT218 for the treatment of EDS and cataplexy in patients suffering from narcolepsy. The REST-ON trial is a randomized, double-blind, placebo-controlled study that has enrolled 212 patients and is being conducted in clinical sites in the U.S., Canada, Western Europe and Australia. Top line data from the REST-ON trial is currently expected in the second quarter of 2020.
In January 2018, the FDA granted FT218 Orphan Drug Designation, which makes the drug eligible for certain development and commercial incentives, including a potential U.S. market exclusivity for up to seven years. Additionally, in April 2019, our first FT218 patent was issued, providing intellectual property protection into 2037. There are additional patent applications currently in development and/or pending at the USPTO, as well as foreign patent offices.
We believe FT218 has the potential to demonstrate improved dosing compliance, safety and patient satisfaction over the current standard of care for EDS and cataplexy in patients with narcolepsy, which is a twice-nightly sodium oxybate formulation. If
-42-
approved, we believe FT218 has the potential to take a significant share of the sodium oxybate market. The current market size for the twice-nightly administration of sodium oxybate is estimated at an annualized revenue run rate of $1.7 billion.
Micropump Drug-Delivery Technology
Our Micropump drug-delivery technology allows for the delayed delivery of small molecule drugs taken orally, which has the potential to improve dosing compliance, reduce toxicity and improve patient compliance. Beyond FT218, we believe there could be other product development opportunities for our Micropump drug delivery technology, representing either “life cycle” opportunities, whereby additional intellectual property can be added to a pharmaceutical product to extend the commercial viability of a currently marketed product, or innovative formulation opportunities for new chemical entities.
Unapproved Marketed Drugs Program
The FDA allows certain unapproved prescription drugs to be marketed if (i) they are relied on by health care professionals to treat serious medical conditions, and (ii) there is no FDA-approved drug to treat such condition or insufficient supply of FDA-approved drugs. In most cases, these prescription drugs pre-date the establishment of the FDA. Although these products are typically not protected by patents or similar intellectual property, FDA guidance states that, if it approves an NDA for any such products via a 505(b)(2) process, the FDA is more likely to seek enforcement action, such as seizure or injunction, against remaining unapproved drugs of the same type, potentially after a grace period provided by the FDA.
Existing Commercial Products
To date, we have received FDA approvals for three previously unapproved prescription drugs:
• | Bloxiverz (neostigmine methylsulfate injection) - Bloxiverz was approved by the FDA in May 2013 and was launched in July 2013. Bloxiverz is a drug used intravenously in the operating room to reverse the effects of non-depolarizing neuromuscular blocking agents after surgery. Bloxiverz was the first FDA-approved version of neostigmine methylsulfate. Today, neostigmine is one of the two most frequently used products for the reversal of the effects of other agents used for neuromuscular blocks. |
• | Vazculep (phenylephrine hydrochloride injection) - Vazculep was approved by the FDA in June 2014 and was launched in October 2014. Vazculep is indicated for the treatment of clinically important hypotension occurring in the setting of anesthesia. |
• | Akovaz (ephedrine sulfate injection). Akovaz, was approved by the FDA in April 2016 and was launched in August 2016. Akovaz was the first FDA approved formulation of ephedrine sulfate, an alpha- and beta- adrenergic agonist and a norepinephrine-releasing agent that is indicated for the treatment of clinically important hypotension occurring in the setting of anesthesia. |
Nouress
In December 2019, we received FDA approval for Nouress (cysteine hydrochloride injection), a sterile injectable product for use in the hospital setting, and currently have two patents covering that product. Several additional patent applications for Nouress are pending with the USPTO. In light of the recently filed patent suit by Exela Pharma Sciences, LLC, (“Exela”) we are currently evaluating the timing and process for a commercial launch of Nouress in the U.S. See Note 16: Contingent Liabilities and Commitments for a discussion on the filed patent suit by Exela Pharma Sciences, LLC.
We use the revenue from our UMD products to fund the research and development of FT218. In addition, we believe evaluating opportunities to commercialize other unapproved drugs in markets with a limited number of competitors may provide us with near-term revenue growth and potentially provide cash flows that can also be used to fund research and development initiatives for the development of FT218 and other potential product candidates.
Corporate Information
We were incorporated on December 1, 2015 as an Irish private limited company, and re-registered as an Irish public limited company (“plc”), on November 21, 2016. Our principal place of business is located at 10 Earlsfort Terrace, Dublin 2, Ireland and our phone number is 00 353 1 920 1000. We file annual, quarterly and current reports, proxy statements and other documents with the U.S. Securities and Exchange Commission (“SEC”) under the Securities Exchange Act of 1934, as amended (the “Exchange
-43-
Act”). Our website is www.avadel.com, where we make available free of charge our reports (and any amendments thereto) on Forms 10-K, 10-Q and 8-K as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. These filings are also available to the public at www.sec.gov.
We currently have five direct wholly-owned subsidiaries: (a) Avadel US Holdings, Inc., (b) Flamel Ireland Limited, which conducts business under the name Avadel Ireland, (c) Avadel Investment Company Limited, (d) Avadel Finance Ireland Designated Activity Company and (e) Avadel France Holding SAS. Avadel US Holdings, Inc., a Delaware corporation, is the holding entity of (i) Avadel Specialty Pharmaceuticals, LLC (currently the subject of a voluntary Chapter 11 bankruptcy proceeding), (ii) Avadel Legacy Pharmaceuticals, LLC, (iii) Avadel Management Corporation, (iv) FSC Holding Company, (v) Avadel Operations Company, Inc. and (vi) Avadel CNS Pharmaceuticals LLC. Avadel Finance Ireland Designated Activity Company is the holding entity of Avadel Finance Cayman Limited. Flamel Ireland Limited (operating under the trade name Avadel Ireland) is an Irish corporation. Avadel France Holding SAS, a French société par actions simplifiée, is the holding entity of Avadel Research SAS through which Avadel conducts substantially all of its R&D activities. A complete list of our subsidiaries can be found in Exhibit 21.1 to this Annual Report on Form 10-K.
References in these consolidated financial statements and the notes thereto to “Avadel,” the “Company,” “we,” “our,” “us,” and similar terms shall be deemed to be references to Flamel prior to the completion of the Merger, unless the context otherwise requires.
Key Business Trends and Highlights
In operating our business and monitoring our performance, we consider a number of performance measures, as well as trends affecting our industry as a whole, which include the following:
• | Healthcare and Regulatory Reform: Various health care reform laws in the U.S. may impact our ability to successfully commercialize our products and technologies. The success of our commercialization efforts may depend on the extent to which the government health administration authorities, the health insurance funds in the E.U. Member States, private health insurers and other third-party payers in the U.S. will reimburse consumers for the cost of healthcare products and services. |
• | Competition and Technological Change: Competition in the pharmaceutical and biotechnology industry continues to be intense and is expected to increase. We compete with academic laboratories, research institutions, universities, joint ventures, and other pharmaceutical and biotechnology companies, including other companies developing niche branded or generic specialty pharmaceutical products or drug delivery platforms. Furthermore, major technological changes can happen quickly in the pharmaceutical and biotechnology industries. Such rapid technological change, or the development by our competitors of technologically improved or differentiated products, could render our drug delivery platforms obsolete or noncompetitive. |
• | Pricing Environment for Pharmaceuticals: The pricing environment continues to be in the political spotlight in the U.S. As a result, the need to obtain and maintain appropriate pricing for our products may become more challenging due to, among other things, the attention being paid to healthcare cost containment and other austerity measures in the U.S. and worldwide. |
• | Generics Playing a Larger Role in Healthcare: Generic pharmaceutical products will continue to play a large role in the U.S. healthcare system. Specifically, we have seen, or likely will see, additional generic competition to our current and future products and we continue to expect generic competition in the future. |
• | Access to and Cost of Capital: The process of raising capital and associated cost of such capital for a company of our financial profile can be difficult and potentially expensive. If the need were to arise to raise additional capital, access to that capital may be difficult and/or expensive and, as a result, could create liquidity challenges for us. |
• | Possible Net Loss from Operations in 2020: In part because we expect sales of our hospital products to significantly decline from 2019 and we will incur substantial expenses to further the clinical development of FT218, we likely will incur a net loss in 2020. |
Recent Developments
Shelf Registration Statement on Form S-3
-44-
In February 2020, we filed with the SEC a new shelf registration statement on Form S-3 (the 2020 Shelf Registration Statement) (File No. 333-236258) that allows issuance and sale by us, from time to time, of:
(a) | up to $250,000 in aggregate of ordinary shares, nominal value US$0.01 per share (the “Ordinary Shares”), each of which may be represented by ADSs, preferred shares, nominal value US$0.01 per share (the “Preferred Shares”), debt securities (the “Debt Securities”), warrants to purchase Ordinary Shares, ADSs, Preferred Shares and/or Debt Securities (the “Warrants”), and/or units consisting of Ordinary Shares, ADSs, Preferred Shares, one or more Debt Securities or Warrants in one or more series, in any combination, pursuant to the terms of the 2020 Shelf Registration Statement, the base prospectus contained in the 2020 Shelf Registration Statement (the “Base Prospectus”), and any amendments or supplements thereto (together, the “Securities”); including |
(b) | up to $50,000 of ADSs that may be issued and sold from time to time pursuant to the terms of an Open Market Sale AgreementSM (“the Sales Agreement”), entered into with Jefferies LLC on February 4, 2020 (the “Sales Agreement”), the 2020 Shelf Registration Statement, the Base Prospectus and the terms of the sales agreement prospectus contained in the 2020 Shelf Registration Statement. |
Securities Purchase Agreement
On February 21, 2020, we announced that we have entered into a definitive agreement for the sale of our ADSs and Series A Non-Voting Convertible Preferred Shares (“Series A Preferred”) in a private placement to a group of institutional accredited investors. The private placement has resulted in gross proceeds of approximately $65,000 before deducting placement agent and other offering expenses, and resulting in net proceeds of approximately $61,000.
Pursuant to the terms of the private placement, we issued 8,680 ADSs and 488 shares of Series A Preferred at a price of $7.09 per share, priced at-the-market under Nasdaq rules. Each share of non-voting Series A Preferred is convertible into one ADS, provided that conversion will be prohibited if, as a result, the holder and its affiliates would own more than 9.99% of the total number of Avadel ADSs outstanding. The closing of the private placement occurred on February 25, 2020. Proceeds from the private placement will be used to fund continued clinical and program development of FT218, including an open-label extension study for REST-ON, a switch study to evaluate patients switching from twice-nightly sodium oxybate to once-nightly FT218, as well as for general corporate purposes.
Financial Highlights
Highlights of our consolidated results for the year ended December 31, 2019 are as follows:
• | Revenue was $59,215 for the year ended December 31, 2019 compared to $103,269 in the same period last year. This year over year decrease was primarily the result of increased competition driving lower prices as noted above in our discussion of Key Business Trends and Highlights. We experienced price and unit volume declines across all our hospital products due to additional competition. |
• | Operating loss was $24,112 for the year ended December 31, 2019 compared to an operating loss of $104,926 for the year ended December 31, 2018. The primary reasons for the decrease in operating loss was due to the 2018 impairment of the Noctiva intangible asset of $66,087 and lower SG&A and R&D expenses in the current period of $70,176 and $6,412, respectively, primarily driven by the exit of Noctiva. These decreases were partially offset by lower total revenues of $44,054 as discussed above and a decrease of $23,576 in the gain in fair value of related party contingent consideration recognized during the year ended December 31, 2018 of $22,731 compared to expense of $845 recognized during the current year. |
• | Net loss was $33,226 for the year ended December 31, 2019 compared to net loss of $95,304 in the same period last year. |
• | Diluted net loss per share was $0.89 for the year ended December 31, 2019 compared to diluted net loss per share of $2.55 in the same period last year. |
• | Cash and marketable securities decreased $35,757 to $64,158 at December 31, 2019 from $99,915 at December 31, 2018. This decrease was largely driven from $38,325 use of cash in operations. |
-45-
Critical Accounting Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to use judgment in making estimates and assumptions that affect the reported amounts of assets and liabilities, disclosures of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the periods presented. Actual results could differ from those estimates under different assumptions or conditions.
The following accounting policies are based on, among other things, judgments and assumptions made by management that include inherent risks and uncertainties. Management’s estimates are based on the relevant information available at the end of each period.
Revenue. Revenue includes sales of pharmaceutical products, licensing fees, and, if any, milestone payments for research and development (“R&D”) achievements.
Product Sales
We sell products primarily through wholesalers and considers these wholesalers to be its customers. Under ASC 606, revenue from product sales is recognized when the customer obtains control of our product, which occurs typically upon receipt by the customer. Our gross product sales are subject to a variety of price adjustments in arriving at reported net product sales. These adjustments include estimates of product returns, chargebacks, payment discounts, rebates, and other sales allowances and are estimated based on analysis of historical data for the product or comparable products, future expectations for such products and other judgments and analysis.
License Revenue
From time to time we may enter into out-licensing agreements which are within the scope of ASC 606 under which it licenses to third parties certain rights to its products or intellectual property. The terms of these arrangements typically include payment to us of one or more of the following: non-refundable, upfront license fees; development, regulatory, and commercial milestone payments; and sales-based royalty payments. Each of these payments results in license revenue. During the years ended December 31, 2019 and 2018, we recognized $0 and $1,846 of revenue from license agreements, respectively.
Research and Development (“R&D”). R&D expenses consist primarily of costs related to clinical studies and outside services, personnel expenses, and other R&D expenses. Clinical studies and outside services costs relate primarily to services performed by clinical research organizations and related clinical or development manufacturing costs, materials and supplies, filing fees, regulatory support, and other third-party fees. Personnel expenses relate primarily to salaries, benefits and share-based compensation. Other R&D expenses primarily include overhead allocations consisting of various support and facilities-related costs. R&D expenditures are charged to operations as incurred.
We recognize R&D tax credits received from the French and Irish government for spending on innovative R&D as an offset of R&D expenses.
Share-based Compensation. We account for share-based compensation based on the estimated grant-date fair value. The fair value of stock options and warrants is estimated using Black-Scholes option-pricing valuation models (“Black-Scholes model”). As required by the Black-Sholes model, estimates are made of the underlying volatility of AVDL stock, a risk-free rate and an expected term of the option or warrant. We estimated the expected term using a simplified method, as we do not have enough historical exercise data for a majority of such options and warrants upon which to estimate an expected term. We recognize compensation cost, net of an estimated forfeiture rate, using the accelerated method over the requisite service period of the award.
Income Taxes. Our income tax (benefit) provision, deferred tax assets and liabilities, and liabilities for unrecognized tax benefits reflect management’s best estimate of current and future taxes to be paid. We are subject to income taxes in Ireland, France and the U.S. Significant judgments and estimates are required in the determination of the consolidated income tax (benefit) provision.
Deferred income taxes arise from temporary differences between the tax basis of assets and liabilities and their reported amounts in the financial statements, which will result in taxable or deductible amounts in the future. In evaluating our ability to recover our deferred tax assets in the jurisdiction from which they arise, we consider all available positive and negative evidence, including scheduled reversals of deferred tax liabilities, projected future taxable income or loss, tax-planning strategies, and results of recent operations. The assumptions about future taxable income or loss require the use of significant judgment and are consistent with the plans and estimates we are using to manage the underlying businesses.
-46-
The calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws and regulations in a multitude of jurisdictions across our global operations. A tax benefit from an uncertain tax position may be recognized when it is more likely than not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, on the basis of the technical merits.
We record unrecognized tax benefits as liabilities and adjust these liabilities when our judgment changes as a result of the evaluation of new information not previously available. Because of the complexity of some of these uncertainties, the ultimate resolution may result in a payment that is materially different from our current estimate of the unrecognized tax benefit liabilities. These differences will be reflected as increases or decreases to income tax expense in the period in which new information is available.
We have not recorded a deferred tax liability for any income or withholding taxes that may arise as the result of the distribution of unremitted earnings within our Company. At December 31, 2019, we had unremitted earnings of $3,961 outside of Ireland as measured on a U.S. GAAP basis. Based on our estimates that future domestic cash generation will be sufficient to meet future domestic cash needs along with our specific plans for reinvestment, we have not recorded a deferred tax liability for any income or withholding taxes that may arise from a distribution that would qualify as a dividend for tax purposes. It is not practicable to estimate the amount of deferred tax liability on such remittances, if any.
Goodwill. Goodwill represents the excess of the acquisition consideration over the fair value of assets acquired and liabilities assumed. We have determined that we operate in a single segment and has a single reporting unit associated with the development and commercialization of pharmaceutical products. Prior to November 2019, we tested for goodwill impairment using a two-step process. The first step is a comparison of the fair value of the reporting unit with its carrying amount, including goodwill. If this step indicates impairment, then, in the second step, the loss is measured as the excess of recorded goodwill over the implied fair value of the goodwill. Implied fair value of goodwill is the excess of the fair value of the reporting unit as a whole over the fair value of all separately identified assets and liabilities within the reporting unit. As discussed in Note 2: Effect of New Accounting Standards, we elected to early adopt Accounting Standards Update (“ASU”) 2017-04 in November 2019, which eliminated step 2 from the goodwill impairment test. We test goodwill for impairment annually and when events or changes in circumstances indicate that the carrying value may not be recoverable. We performed our required impairment test of goodwill and have determined that no impairment of goodwill existed at December 31, 2019 and 2018.
Long-Lived Assets. Long-lived assets include fixed assets and intangible assets. Intangible assets consist primarily of purchased licenses and intangible assets recognized as part of the Éclat Pharmaceuticals acquisition. Acquired IPR&D has an indefinite life and is not amortized until completion and development of the project, at which time the IPR&D becomes an amortizable asset, for which amortization of such intangible assets is computed using the straight-line method over the estimated useful life of the assets.
Long-lived assets are reviewed for impairment whenever conditions indicate that the carrying value of the assets may not be fully recoverable. Such impairment tests are based on a comparison of the pretax undiscounted cash flows expected to be generated by the asset to the recorded value of the asset or other market based value approaches. If impairment is indicated, the asset value is written down to its market value if readily determinable or its estimated fair value based on discounted cash flows. Any significant changes in business or market conditions that vary from current expectations could have an impact on the fair value of these assets and any potential associated impairment. During the fourth quarter of 2018, we recorded a $66,087 impairment charge to the entire acquired developed technology related to Noctiva (see Note 9: Goodwill and Intangible Assets). We determined that no impairment existed at December 31, 2019.
Acquisition-related Contingent Consideration. The acquisition-related contingent consideration payables arising from the acquisition of Éclat Pharmaceuticals (i.e., our hospital products) and FSC (our pediatrics products), which was assumed by the buyer as part of the disposition of the pediatrics products on February 16, 2018, are accounted for at fair-value (see Note 12: Long-Term Related Party Payable and Note 17: Divestiture of the Pediatric Assets). The fair value of the warrants issued in connection with the Éclat acquisition were estimated using a Black-Scholes model. A portion of these warrants were exercised on February 23, 2018 and the remaining warrants expired on March 12, 2018. See Note 12: Long-Term Related Party Payable. The fair value of acquisition-related contingent consideration payable is estimated using a discounted cash flow model based on the long-term sales or gross profit forecasts of the specified hospital or pediatric products using an appropriate discount rate. There are a number of estimates used when determining the fair value of these earn-out payments. These estimates include, but are not limited to, the long-term pricing environment, market size, market share the related products are forecast to achieve, the cost of goods related to such products and an appropriate discount rate to use when present valuing the related cash flows. These estimates can and often do change based on changes in current market conditions, competition, management judgment and other factors. Changes to these estimates can have and have had a material impact on our consolidated statements of (loss) income and balance sheets. Changes in fair value of these liabilities are recorded in the consolidated statements of (loss) income within operating expenses as changes in fair value of related party contingent consideration.
-47-
Financing-related Royalty Agreements. We also entered into two royalty agreements with related parties in connection with certain financing arrangements. We elected the fair value option for the measurement of the financing-related contingent consideration payable associated with the royalty agreements with certain Deerfield and Broadfin entities, both of whom are related parties (see Note 12: Long-Term Related Party Payable). The fair value of financing-related royalty agreements is estimated using the same components used to determine the fair value of the acquisition-related contingent consideration noted above, with the exception of cost of products sold. Changes to these components can also have a material impact on our consolidated statements of (loss) income and balance sheets. Changes in the fair value of this liability are recorded in the consolidated statements of (loss) income as other (expense) income - changes in fair value of related party payable.
Results of Operations
The following is a summary of our financial results (in thousands, except per share amounts):
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Comparative Statements of (Loss) Income: | 2019 | 2018 | $ | % | |||||||||||
Product sales | $ | 59,215 | $ | 101,423 | $ | (42,208 | ) | (41.6 | )% | ||||||
License revenue | — | 1,846 | (1,846 | ) | (100.0 | )% | |||||||||
Total revenues | 59,215 | 103,269 | (44,054 | ) | (42.7 | )% | |||||||||
Operating expenses: | |||||||||||||||
Cost of products | 12,125 | 17,516 | (5,391 | ) | (30.8 | )% | |||||||||
Research and development expenses | 32,917 | 39,329 | (6,412 | ) | (16.3 | )% | |||||||||
Selling, general and administrative expenses | 30,183 | 100,359 | (70,176 | ) | (69.9 | )% | |||||||||
Intangible asset amortization | 816 | 6,619 | (5,803 | ) | (87.7 | )% | |||||||||
Changes in fair value of related party contingent consideration | 845 | (22,731 | ) | 23,576 | 103.7 | % | |||||||||
Impairment of intangible asset | — | 66,087 | (66,087 | ) | (100.0 | )% | |||||||||
Restructuring costs | 6,441 | 1,016 | 5,425 | 534.0 | % | ||||||||||
Total operating expenses | 83,327 | 208,195 | (124,868 | ) | (60.0 | )% | |||||||||
Operating (loss) income | (24,112 | ) | (104,926 | ) | 80,814 | 77.0 | % | ||||||||
Investment and other income, net | 1,069 | 452 | 617 | 136.5 | % | ||||||||||
Interest expense | (12,483 | ) | (10,622 | ) | (1,861 | ) | (17.5 | )% | |||||||
Loss on deconsolidation of subsidiary | (2,678 | ) | — | (2,678 | ) | (100.0 | )% | ||||||||
Other (expense) income - changes in fair value of related party payable | (378 | ) | 1,899 | (2,277 | ) | (119.9 | )% | ||||||||
(Loss) income before income taxes | (38,582 | ) | (113,197 | ) | 74,615 | 65.9 | % | ||||||||
Income tax (benefit) provision | (5,356 | ) | (17,893 | ) | 12,537 | 70.1 | % | ||||||||
Net (loss) income | $ | (33,226 | ) | $ | (95,304 | ) | $ | 62,078 | 65.1 | % | |||||
Net (loss) income per share - diluted | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.66 | 65.1 | % | |||||
-48-
The revenues for each of our significant products were as follows:
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Revenues | 2019 | 2018 | $ | % | |||||||||||
Bloxiverz | $ | 7,479 | $ | 20,850 | $ | (13,371 | ) | (64.1 | )% | ||||||
Vazculep | 33,152 | 42,916 | (9,764 | ) | (22.8 | )% | |||||||||
Akovaz | 18,642 | 33,759 | (15,117 | ) | (44.8 | )% | |||||||||
Other | (58 | ) | 3,898 | (3,956 | ) | (101.5 | )% | ||||||||
Product sales | 59,215 | 101,423 | (42,208 | ) | (41.6 | )% | |||||||||
License revenue | — | 1,846 | (1,846 | ) | (100.0 | )% | |||||||||
Total revenues | $ | 59,215 | $ | 103,269 | $ | (44,054 | ) | (42.7 | )% | ||||||
Total product sales were $59,215 for the year ended December 31, 2019, compared to $101,423 for the same prior year period. Bloxiverz’s revenue declined $13,371 when compared to the same period last year, primarily due to lower net selling prices driven largely by new competitors that entered the market in 2018 and 2019 and continued market penetration from an alternative molecule to neostigmine. Vazculep’s revenue decreased by $9,764 primarily due to a decrease in unit volumes and lower realized net selling prices when compared to the prior year. Akovaz’s revenue decreased $15,117 driven by lower unit volumes and net selling prices due largely to new competitors that entered the market in 2018 and 2019. Other revenues, which includes Noctiva, which was deconsolidated in February 2019, and the pediatric products, which were divested in February 2018, declined when compared to the prior year due to the divestiture of those products as well as an increased returns reserve related to these products during the year ended December 31, 2019.
License and research revenue was $0 for the year ended December 31, 2019 compared to $1,846 in the same period last year. In December 2018, we reached an agreement to exit a contract and our remaining performance obligations and recognized the remaining $1,600 of deferred revenue during the year ended December 31, 2018, which represented the unsatisfied performance obligations associated with a license agreement.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Cost of Products | 2019 | 2018 | $ | % | |||||||||||
Cost of products | $ | 12,125 | $ | 17,516 | $ | (5,391 | ) | (30.8 | )% | ||||||
Percentage of sales | 20.5 | % | 17.0 | % | |||||||||||
Cost of products decreased $5,391, or 30.8% during the year ended December 31, 2019 compared to the prior year due to lower sales volumes. As a percentage of total revenue, cost of products sold was higher than the prior year period due to lower net selling prices of our hospital products.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Research and Development Expenses | 2019 | 2018 | $ | % | |||||||||||
Research and development expenses | $ | 32,917 | $ | 39,329 | $ | (6,412 | ) | (16.3 | )% | ||||||
Percentage of sales | 55.6 | % | 38.1 | % | |||||||||||
R&D expenses decreased $6,412 or 16.3% during the year ended December 31, 2019 as compared to the same period in 2018. This decline was a result of $2,800 of lower spending associated with the exit of Noctiva, lower payroll, benefits and share-based compensation of $2,900 related to the 2019 Corporate and French restructuring plans and approximately $900 of cost reductions at our Lyon, France R&D center. We continue to invest a substantial portion of R&D in our FT218 development program.
-49-
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Selling, General and Administrative Expenses | 2019 | 2018 | $ | % | |||||||||||
Selling, general and administrative expenses | $ | 30,183 | $ | 100,359 | $ | (70,176 | ) | (69.9 | )% | ||||||
Percentage of sales | 51.0 | % | 97.2 | % | |||||||||||
Selling, general and administrative (“SG&A”) expenses decreased $70,176 or 69.9% during the year ended December 31, 2019 as compared to the prior year. This decrease was primarily due to a decrease of $60,100 of sales and marketing costs related to the exit of Noctiva during the first quarter 2019 as well as $2,700 of decreased costs due to the 2018 divestiture of the pediatric products. Also contributing to the decrease is lower overall payroll and share-based compensation of $7,200 due to reduced headcount as a result of the 2019 Corporate and French restructuring plans.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Intangibles Asset Amortization | 2019 | 2018 | $ | % | |||||||||||
Intangible asset amortization | $ | 816 | $ | 6,619 | $ | (5,803 | ) | (87.7 | )% | ||||||
Percentage of sales | 1.4 | % | 6.4 | % | |||||||||||
Intangible asset amortization expense decreased $5,803 or 87.7% during the year ended December 31, 2019 as compared to the prior year driven by the impairment of the intangible asset related to Noctiva at December 31, 2018.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Changes in Fair Value of Related Party Contingent Consideration | 2019 | 2018 | $ | % | |||||||||||
Changes in fair value of related party contingent consideration | $ | 845 | $ | (22,731 | ) | $ | 23,576 | 103.7 | % | ||||||
Percentage of sales | 1.4 | % | (22.0 | )% | |||||||||||
We compute the fair value of the related party contingent consideration using several significant assumptions and when these assumptions change, due to underlying market conditions, the fair value of these liabilities change as well. Each of the underlying assumptions used to determine the fair values of these contingent liabilities can, and often do, change based on adjustments in current market conditions, competition and other factors. These changes can have a material impact on our consolidated statements of loss (income) and balance sheets.
As a result of changes in the underlying assumptions used to determine the estimated fair values of our acquisition-related contingent consideration earn-out payments - Éclat, we recorded expense of $845 and a gain of $22,731 and increased/lowered the fair value of the acquisition-related contingent consideration earn-out payments - Éclat for the years ended December 31, 2019 and 2018, respectively. In Item 7 Critical Accounting Estimates, there are numerous assumptions and estimates we use when determining the fair value of the acquisition-related earn-out payments - Éclat. These assumptions include estimates of pricing, market size, the market share the related products are forecast to achieve, the cost of goods related to such products and an appropriate discount rate to use when determining the present value of the related cash flows.
For the year ended December 31, 2019, as a result of changes to these estimates when compared to the same estimates at December 31, 2018, we recorded an increase in the fair value of our contingent consideration liabilities. The increase was due to the timing of anticipated market competition when compared to the original estimates.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Impairment of Intangible Asset | 2019 | 2018 | $ | % | |||||||||||
Impairment of intangible asset | $ | — | $ | 66,087 | $ | (66,087 | ) | (100.0 | )% | ||||||
Percentage of sales | — | % | 64.0 | % | |||||||||||
During the fourth quarter of 2018, an impairment charge of $66,087 was recorded to write-off the remaining carrying value of the acquired developed technology intangible asset related to Noctiva. During the fourth quarter 2018, certain conditions came to light, largely the lack of a meaningful increase in Noctiva prescriptions despite the substantial investment of resources, which indicated that the carrying value of the asset, may not be fully recoverable. As such, we performed an impairment test based on
-50-
a comparison of the pretax discounted cash flows expected to be generated by the asset, which is a Level 3 fair value estimate, to the recorded value of the asset and concluded that the associated cash flows did not support any of the carrying value of the intangible asset and we recorded a full impairment charge. The Chapter 11 bankruptcy filing of Avadel Specialty Pharmaceuticals, LLC (“Specialty Pharma”) commenced on February 6, 2019, the subsidiary which marketed, sold and distributed Noctiva, confirmed management’s conclusion on the impairment. There were no such impairment charges during the year ended December 31, 2019.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Restructuring Costs | 2019 | 2018 | $ | % | |||||||||||
Restructuring costs | $ | 6,441 | $ | 1,016 | $ | 5,425 | 534.0 | % | |||||||
Percentage of sales | 10.9 | % | 1.0 | % | |||||||||||
Restructuring costs increased $5,425 during the year ended December 31, 2019 as compared to the same prior year period driven by the 2019 French Restructuring and 2019 Corporate Restructuring actions and included severance and legal costs, impairment of certain assets, the reversal of certain retirement indemnity obligations, share-based compensation and the termination payments for vacating the Irish and French office leases during 2019. See Note 18: Restructuring Costs.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Investment and Other Income, net | 2019 | 2018 | $ | % | |||||||||||
Investment and other income, net | $ | 1,069 | $ | 452 | $ | 617 | 136.5 | % | |||||||
Percentage of sales | 1.8 | % | 0.4 | % | |||||||||||
Investment and other income, net increased $617 during the year ended December 31, 2019 as compared to the same prior year period driven by higher realized and unrealized gains on our marketable securities during the current period when compared to realized and unrealized losses on our marketable securities the prior year period, partially offset by a legal settlement of $1,750, of which a majority of the settlement is due to a former employee and the remainder to the former employee’s legal counsel. See Note 16: Contingent Liabilities and Commitments.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Interest Expense | 2019 | 2018 | $ | % | |||||||||||
Interest expense | $ | 12,483 | $ | 10,622 | $ | 1,861 | 17.5 | % | |||||||
Percentage of sales | (21.1 | )% | (10.3 | )% | |||||||||||
Interest expense increased $1,861 for the year ended December 31, 2019 when compared to the year ended December 31, 2018 as a result of twelve months of interest recorded in 2019 versus 10.5 months of interest recorded in 2018 due to the 2023 Notes issued in February 2018.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Loss on Deconsolidation of Subsidiary | 2019 | 2018 | $ | % | |||||||||||
Loss on deconsolidation of subsidiary | $ | (2,678 | ) | $ | — | $ | (2,678 | ) | (100.0 | )% | |||||
Percentage of sales | (4.5 | )% | — | % | |||||||||||
As a result of Specialty Pharma’s bankruptcy filing on February 6, 2019, we concluded that we no longer controls its operations and accordingly deconsolidated this subsidiary. We recorded a loss on the deconsolidation as a result of removing the net assets and certain liabilities of this subsidiary from our consolidated financial statements. See Note 3: Subsidiary Bankruptcy and Deconsolidation for more discussion.
-51-
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Other (Expense) Income - Changes in Fair Value of Related Party Payable | 2019 | 2018 | $ | % | |||||||||||
Other (expense) income - changes in fair value of related party payable | $ | (378 | ) | $ | 1,899 | $ | (2,277 | ) | (119.9 | )% | |||||
Percentage of sales | (0.6 | )% | 1.8 | % | |||||||||||
We recorded expense of $378 to increase the fair value of these liabilities during the year ended December 31, 2019 and income of $1,899 to reduce the fair value of these liabilities during the year ended December 31, 2018, due to the same reasons associated with the Éclat product sales forecasts as described in the section “Changes in Fair Value of Related Party Contingent Consideration” for these periods. As noted in Item 7 Critical Accounting Estimates, there are a number of assumptions and estimates we use when determining the fair value of the related party payable payments. These estimates include pricing, market size, the market share the related products are forecast to achieve and an appropriate discount rate to use when determining the present value of the related cash flows. These estimates often do change based on changes in current market conditions, competition and other factors.
Years Ended December 31, | Increase / (Decrease) | ||||||||||||||
Income Taxes | 2019 | 2018 | $ | % | |||||||||||
Income tax (benefit) provision | $ | (5,356 | ) | $ | (17,893 | ) | $ | 12,537 | 70.1 | % | |||||
Percentage of (loss) income before income taxes | 13.9 | % | 15.8 | % | |||||||||||
In 2019, the income tax benefit decreased by $12,537 when compared to the same period in 2018. The decrease in the income tax benefit in 2019 was primarily driven by the impairment of the Noctiva intangible asset in 2018, which did not recur in 2019. In addition to the non-recurring impairment, an increase in the valuation allowance in 2019, when compared to the same period in 2018 also contributed to the decrease in tax benefit recorded in 2019. As a part of a corporate reorganization, the Company entered into an internal sale transaction in December 2019. The internal sale transaction included transfer of intangible assets from an Irish entity to a U.S. entity. The internal sale transaction resulted in a decrease of $5,536 to Irish deferred tax asset with corresponding decrease of $5,536 to valuation allowance, an increase of $8,190 to U.S. deferred tax asset associated with amortization of intangible assets, and a $8,190 deferred tax benefit.
-52-
Liquidity and Capital Resources
Our cash flows from operating, investing and financing activities, as reflected in the consolidated statements of cash flows, are summarized in the following table:
Years Ended December 31, | Increase / (Decrease) | |||||||||||||
Net Cash (Used In) Provided By | 2019 | 2018 | $ | % | ||||||||||
Operating activities | $ | (38,325 | ) | $ | (82,716 | ) | $ | 44,391 | 53.7 | % | ||||
Investing activities | 38,723 | (36,981 | ) | 75,704 | 204.7 | % | ||||||||
Financing activities | (27 | ) | 112,659 | (112,686 | ) | (100.0 | )% | |||||||
Operating Activities
Net cash used in operating activities of $38,325 for the year ended December 31, 2019 decreased from net cash used in operating activities of $82,716 in the prior year. This decrease in cash used in operating activities is driven by lower cash loss (net loss adjusted for non-cash credits and charges) of $20,858 when compared to the same period last year. This decrease is driven by less cash used in SG&A and R&D expenses in the current year driven by the Specialty Pharma bankruptcy and deconsolidation, partially offset by lower revenue in the current year when compared to the prior year. The decrease in cash used in operating activities was also due to the increase in accounts payable and accrued expenses of $17,670 and a decrease in the earn-out and royalty payments for related party contingent payable of $9,570 during the year ended December 31, 2019 compared to the prior year.
Investing Activities
Cash provided by investing activities was $38,723 for the year ended December 31, 2019 compared to cash used in investing activities of $36,981 in the same prior year period. In 2019, we had net proceeds of $38,598 for the sale of marketable securities compared to our use of $16,803 cash for the purchase of marketable securities during the year ended December 31, 2018. Additionally, we also had a $20,000 Noctiva related milestone payment as part of the Exclusive License and Assignment Agreement with Serenity Pharmaceuticals, LLC (“Serenity”) during the year ended December 31, 2018.
Financing Activities
Cash used in financing activities of $27 for the year ended December 31, 2019 decreased $112,686 compared to cash provided by financing activities of $112,659 for the same prior year period. During the year ended December 31, 2018, $143,750 of cash was provided by financing activities through the issuance of the 2023 Notes. A portion of the proceeds from the offering of the 2023 Notes was used for share repurchases totaling $27,637 and to pay direct expenses associated with the issuance of the 2023 Notes of $6,190 during the first half of 2018.
Liquidity and Risk Management
The adequacy of our cash resources depends on the outcome of certain business conditions including the cost of our FT218 clinical development plan, our cost structure, our hospital products revenue stream and other factors set forth in “Risk Factors” within Part I, Item 1A of this Annual Report on Form 10-K. To complete the FT218 clinical development plan and to ensure an adequate and robust NDA for filing with the FDA we will need to commit substantial resources, which could result in future losses or otherwise limit our opportunities or affect our ability to operate our business. Our assumptions concerning the outcome of certain business conditions may prove to be wrong or other factors may adversely affect our business, and as a result we could exhaust or significantly decrease our available cash and marketable securities balances which could, among other things, force us to raise additional funds and/or force us to reduce our expenses, either of which could have a material adverse effect on our business. If available to us raising additional capital may be accomplished through one or more public or private debt or equity financings, collaborations or partnering arrangements. Any equity financing would be dilutive to our shareholders.
On February 21, 2020, we announced that we have entered into a definitive agreement for the sale of our ADSs and Series A Non-Voting Convertible Preferred Shares (“Series A Preferred”) in a private placement to a group of institutional accredited investors. The private placement has resulted in gross proceeds of approximately $65,000 before deducting placement agent and other offering expenses, and resulting in net proceeds of approximately $61,000.
-53-
Borrowings
In February 2018, we issued the 2023 Notes. We received net proceeds of approximately $137,560 from the sale of the 2023 Notes, after deducting fees and expenses of $6,190.
Share Repurchase Programs
We fully completed our authorized share buyback program during the year ended December 31, 2018. No share purchases were made during the year ended December 31, 2019.
Other Matters
Litigation
We are subject to potential liabilities generally incidental to our business arising out of present and future lawsuits and claims related to product liability, personal injury, contract, commercial, intellectual property, tax, employment, compliance and other matters that arise in the ordinary course of business. We accrue for potential liabilities when it is probable that future costs (including legal fees and expenses) will be incurred and such costs can be reasonably estimated. At December 31, 2019 and December 31, 2018, there were no contingent liabilities with respect to any litigation, arbitration or administrative or other proceeding that are reasonably likely to have a material adverse effect on our consolidated financial position, results of operations, cash flows or liquidity.
Note 3: Subsidiary Bankruptcy and Deconsolidation briefly describes the Chapter 11 bankruptcy case which our subsidiary Specialty Pharma commenced on February 6, 2019, and which on April 26, 2019 resulted in the bankruptcy court-approved sale of all of Specialty Pharma’s intangible assets and inventory to an unaffiliated third party. As a result of such sale, Specialty Pharma has completed its divestment of the assets of the Noctiva business. During the pendency of the bankruptcy case, all pending litigation against Specialty Pharma is automatically stayed and any new litigation against Specialty Pharma is precluded unless the bankruptcy court orders otherwise. Below are descriptions of a litigation to which Specialty Pharma is a party and a contract dispute involving Specialty Pharma, both of which matters are subject to the automatic stay during the bankruptcy case.
Ferring Litigation. Some of the patents covering the Noctiva product (the “Noctiva Patents”) are the subject of litigation initiated by Ferring Pharmaceuticals Inc. and two of its foreign affiliates, who manufacture a competing product known as Nocdurna. Nocdurna was approved by the FDA in June 2018 and commercially launched in the U.S. in November 2018. In this litigation, filed in the United States District Court for the Southern District of New York, Ferring seeks to invalidate and disputes the inventorship of the Noctiva Patents, seeks damages for various alleged breaches of contractual and common law duties, and seeks damages for alleged infringement by Noctiva of Ferring’s “Nocdurna” trademark. Specialty Pharma and certain other parties including Serenity Pharmaceuticals, LLC (“Serenity”) (the licensor of the Noctiva Patents) have defended this litigation, and have made counterclaims against Ferring, including for infringement of the Noctiva Patents and a declaratory judgment of noninfringement with respect to Ferring’s “Nocdurna” trademark. The court dismissed Ferring’s inventorship claim and its claims for alleged breaches of contractual and common law duties, although these dismissals may be appealed by Ferring. On February 15, 2019, Specialty Pharma and its co-defendants moved to stay the litigation pending completion of the bankruptcy proceeding of Specialty Pharma. On May 15, 2019, that motion was denied due to a pending settlement of the litigation with respect to just Ferring and Specialty Pharma. On February 25, 2020, Ferring and Specialty Pharma jointly moved for bankruptcy court approval of a settlement agreement with respect to the claims alleged in the litigation. In accordance with the terms of the settlement agreement, promptly following bankruptcy court approval of the settlement agreement, the parties would dismiss with prejudice their respective claims against each other in the litigation. On March 13th, 2020, the bankruptcy court entered an order approving the settlement with Ferring. Pursuant to the terms of the settlement, the parties are to dismiss their respective claims against each other in the District Court litigation in the Southern District of New York, with such dismissals to be effective concurrently. That joint dismissal has not yet been filed with District Court for the Southern District of New York.
Contract Dispute. On January 21, 2019, Serenity gave notice to Specialty Pharma of an alleged breach of the parties’ Noctiva license agreement. Serenity alleges that Specialty Pharma breached its contractual obligation to devote commercially reasonable efforts to the commercialization of Noctiva and seeks unspecified damages. On January 27, 2019, Specialty Pharma notified Serenity of a claim for $1.7 million in damages as a result of Serenity’s breach of its contractual obligation to pay the costs of the Ferring Litigation. Serenity’s notice to Specialty Pharma invoked the dispute resolution provisions of the Noctiva license agreement, which culminate in arbitration, but neither party has yet initiated an arbitration proceeding or filed suit.
Exela Litigation. On January 7, 2020, Exela filed a complaint against us and our subsidiary, Avadel Legacy in the United States District for the District of Delaware. The complaint alleges infringement of a certain Exela patent related to its cysteine hydrochloride product. Exela is most notably seeking i) a declaratory judgment that the Nouress product infringes its patent, ii)
-54-
an injunction (both preliminary and permanent) precluding the launch of Nouress, and iii) monetary damages (including enhanced damages, prejudgment interest and attorneys’ fees) in the event Nouress is commercially launched and found to infringe Exela’s patent. We have not yet been served with the complaint.
Former employee dispute. On January 10, 2020, we settled a dispute with a former employee for $1,750.
Material Commitments
At December 31, 2019,we had various commitments to purchase finished product from customers. Commitments for these arrangements, at maximum quantities and at contractual prices over the remaining life of the contract, and excluding any waived commitments, are as follows for the years ended December 31:
Purchase Commitments: | Balance | |||
2020 | $ | 1,434 | ||
2021 | 1,430 | |||
2022 | 1,430 | |||
2023 | 1,430 | |||
2024 | — | |||
Thereafter | — | |||
Total | $ | 5,724 | ||
We also had a commitment with a contract manufacturer related to the construction and preparation of a production suite at the contract manufacturer’s facility, which is substantially complete at December 31, 2019. Subsequent to the initial build and preparation of the production suite, this commitment also includes annual fees which would commence at the time of FDA approval of the product and continue thereafter for five years. These amounts are not included in the table above, as the start date has not been determined.
We and our subsidiaries lease office facilities under non-cancellable operating leases expiring at various dates. See Note 10: Leases for disclosure of these.
Other than the above commitments, there were no other material commitments outside of the normal course of business. Material commitments in the normal course of business include long-term debt and long-term related party payable, which are disclosed in Item 8. Financial Statements and Supplementary Data, Note 11: Long-Term Debt and Note 12: Long-Term Related Party Payable, respectively.
Aggregate Contractual Obligations
The following table presents our contractual obligations at December 31, 2019:
Payments Due by Period | ||||||||||||||||||||
Contractual Obligations: | Total | Less than 1 Year | 1 to 3 Years | 3 to 5 Years | More than 5 Years | |||||||||||||||
Long-term debt and interest | $ | 166,391 | $ | 6,469 | $ | 12,938 | $ | 146,984 | $ | — | ||||||||||
Long-term related party payable (undiscounted) | 29,847 | 5,554 | 5,181 | 4,262 | 14,850 | |||||||||||||||
Purchase commitments | 5,724 | 1,434 | 2,860 | 1,430 | — | |||||||||||||||
Operating leases | 3,368 | 779 | 1,167 | 1,216 | 206 | |||||||||||||||
Total contractual cash obligations | $ | 205,330 | $ | 14,236 | $ | 22,146 | $ | 153,892 | $ | 15,056 | ||||||||||
See Note 11: Long-Term Debt and Note 12: Long-Term Related Party Payable to our consolidated financial statements contained in Item 8 – Financial Statements for obligations with respect to the respective items within the above table.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Interest Rate Risk
-55-
We are subject to interest rate risk as a result of our portfolio of marketable securities. The primary objectives of our investment policy are as follows: safety and preservation of principal and diversification of risk; liquidity of investments sufficient to meet cash flow requirements; and competitive yield. Although our investments are subject to market risk, our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure from any single issue, issuer or certain types of investment. Our investment policy allows us to maintain a portfolio of cash equivalents and marketable securities in a variety of instruments, including U.S. federal government and federal agency securities, European Government bonds, corporate bonds or commercial paper issued by U.S. or European corporations, money market instruments, certain qualifying money market mutual funds, certain repurchase agreements, tax-exempt obligations of states, agencies, and municipalities in the U.S and Europe, and equities.
Foreign Exchange Risk
We are exposed to foreign currency exchange risk as the functional currency financial statements of a non-U.S. subsidiary is translated to U.S. dollars. The assets and liabilities of this non-U.S. subsidiary having a functional currency other than the U.S. dollar is translated into U.S. dollars at the exchange rate prevailing at the balance sheet date, and at the average exchange rate for the reporting period for revenue and expense accounts. The cumulative foreign currency translation adjustment is recorded as a component of accumulated other comprehensive loss in shareholders’ equity. The reported results of this non-U.S. subsidiary will be influenced by their translation into U.S. dollars by currency movements against the U.S. dollar. Our primary currency translation exposure is related to one subsidiary that has functional currencies denominated in euro. A 10% strengthening/weakening in the rates used to translate the results of our non-U.S. subsidiaries that have functional currencies denominated in euro as of December 31, 2019 would have had an immaterial impact on net loss for the year ended December 31, 2019.
Transactional exposure arises where transactions occur in currencies other than the functional currency. Transactions in foreign currencies are recorded at the exchange rate prevailing at the date of the transaction. The resulting monetary assets and liabilities are translated into the appropriate functional currency at exchange rates prevailing at the balance sheet date and the resulting gains and losses are reported in foreign exchange gain (loss) in the consolidated statements of (loss) income. As of December 31, 2019, our primary exposure is to transaction risk related to Euro net monetary assets and liabilities held by subsidiaries with a U.S. dollar functional currency. Realized and unrealized foreign exchange gains resulting from transactional exposure were immaterial for the year ended December 31, 2019.
-56-
Item 8. Financial Statements and Supplementary Data.
AVADEL PHARMACEUTICALS PLC
CONSOLIDATED STATEMENTS OF (LOSS) INCOME
(In thousands, except per share data)
Years ended December 31, | ||||||||||||
2019 | 2018 | 2017 | ||||||||||
Revenues: | ||||||||||||
Product sales | $ | 59,215 | $ | 101,423 | $ | 172,841 | ||||||
License revenue | — | 1,846 | 404 | |||||||||
Total revenues | 59,215 | 103,269 | 173,245 | |||||||||
Operating expenses: | ||||||||||||
Cost of products | 12,125 | 17,516 | 16,301 | |||||||||
Research and development expenses | 32,917 | 39,329 | 33,418 | |||||||||
Selling, general and administrative expenses | 30,183 | 100,359 | 58,860 | |||||||||
Intangible asset amortization | 816 | 6,619 | 3,659 | |||||||||
Changes in fair value of related party contingent consideration | 845 | (22,731 | ) | (31,040 | ) | |||||||
Impairment of intangible asset | — | 66,087 | — | |||||||||
Restructuring costs | 6,441 | 1,016 | 2,542 | |||||||||
Total operating expenses | 83,327 | 208,195 | 83,740 | |||||||||
Operating (loss) income | (24,112 | ) | (104,926 | ) | 89,505 | |||||||
Investment and other income, net | 1,069 | 452 | 2,136 | |||||||||
Interest expense | (12,483 | ) | (10,622 | ) | (1,052 | ) | ||||||
Loss on deconsolidation of subsidiary | (2,678 | ) | — | — | ||||||||
Other (expense) income - changes in fair value of related party payable | (378 | ) | 1,899 | 2,071 | ||||||||
(Loss) income before income taxes | (38,582 | ) | (113,197 | ) | 92,660 | |||||||
Income tax (benefit) provision | (5,356 | ) | (17,893 | ) | 24,389 | |||||||
Net (loss) income | $ | (33,226 | ) | $ | (95,304 | ) | $ | 68,271 | ||||
Net (loss) income per share - basic | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.69 | ||||
Net (loss) income per share - diluted | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.63 | ||||
Weighted average number of shares outstanding - basic | 37,403 | 37,325 | 40,465 | |||||||||
Weighted average number of shares outstanding - diluted | 37,403 | 37,325 | 41,765 | |||||||||
See accompanying notes to consolidated financial statements.
-57-
AVADEL PHARMACEUTICALS PLC
CONSOLIDATED STATEMENTS OF COMPREHENSIVE (LOSS) INCOME
(In thousands)
Years ended December 31, | ||||||||||||
2019 | 2018 | 2017 | ||||||||||
Net (loss) income | $ | (33,226 | ) | $ | (95,304 | ) | $ | 68,271 | ||||
Other comprehensive income (loss), net of tax: | ||||||||||||
Foreign currency translation (loss) gain | (117 | ) | (419 | ) | 134 | |||||||
Net other comprehensive income, net of ($43), ($18), $28 tax, respectively | 727 | 269 | 165 | |||||||||
Total other comprehensive income (loss), net of tax | 610 | (150 | ) | 299 | ||||||||
Total comprehensive (loss) income | $ | (32,616 | ) | $ | (95,454 | ) | $ | 68,570 | ||||
See accompanying notes to consolidated financial statements.
-58-
AVADEL PHARMACEUTICALS PLC
CONSOLIDATED BALANCE SHEETS
(In thousands, except per share data)
December 31, | ||||||||
2019 | 2018 | |||||||
ASSETS | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 9,774 | $ | 9,325 | ||||
Marketable securities | 54,384 | 90,590 | ||||||
Accounts receivable | 8,281 | 11,330 | ||||||
Inventories, net | 3,570 | 4,770 | ||||||
Research and development tax credit receivable | 2,107 | 283 | ||||||
Prepaid expenses and other current assets | 4,264 | 8,553 | ||||||
Total current assets | 82,380 | 124,851 | ||||||
Property and equipment, net | 544 | 1,911 | ||||||
Operating lease right-of-use assets | 3,612 | — | ||||||
Goodwill | 18,491 | 18,491 | ||||||
Intangible assets, net | 813 | 1,629 | ||||||
Research and development tax credit receivable | 6,322 | 7,272 | ||||||
Other non-current assets | 39,274 | 36,146 | ||||||
Total assets | $ | 151,436 | $ | 190,300 | ||||
LIABILITIES AND SHAREHOLDERS’ (DEFICIT) EQUITY | ||||||||
Current liabilities: | ||||||||
Current portion of long-term debt | $ | — | $ | 106 | ||||
Current portion of long-term related party payable | 5,554 | 9,439 | ||||||
Current portion of operating lease liability | 645 | — | ||||||
Accounts payable | 6,100 | 3,503 | ||||||
Accrued expenses | 19,810 | 21,695 | ||||||
Other current liabilities | 3,875 | 3,640 | ||||||
Total current liabilities | 35,984 | 38,383 | ||||||
Long-term debt, less current portion | 121,686 | 115,734 | ||||||
Long-term related party payable, less current portion | 11,773 | 19,401 | ||||||
Long-term operating lease liability | 2,319 | — | ||||||
Other non-current liabilities | 8,873 | 14,002 | ||||||
Total liabilities | 180,635 | 187,520 | ||||||
Shareholders’ (deficit) equity: | ||||||||
Preferred shares, nominal value of $0.01 per share; 50,000 shares authorized; none issued or outstanding at December 31, 2019 and December 31, 2018, respectively | — | — | ||||||
Ordinary shares, nominal value of $0.01 per share; 500,000 shares authorized; 42,927 issued and 37,520 outstanding at December 31, 2019, and 42,720 issued and 37,313 outstanding at December 31, 2018 | 429 | 427 | ||||||
Treasury shares, at cost, 5,407 shares held at December 31, 2019 and December 31, 2018, respectively | (49,998 | ) | (49,998 | ) | ||||
Additional paid-in capital | 434,391 | 433,756 | ||||||
Accumulated deficit | (391,215 | ) | (357,989 | ) | ||||
Accumulated other comprehensive loss | (22,806 | ) | (23,416 | ) | ||||
Total shareholders’ (deficit) equity | (29,199 | ) | 2,780 | |||||
Total liabilities and shareholders’ (deficit) equity | $ | 151,436 | $ | 190,300 | ||||
See accompanying notes to consolidated financial statements.
-59-
AVADEL PHARMACEUTICALS PLC
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ (DEFICIT) EQUITY
(In thousands)
Ordinary shares | Additional | Accumulated | Accumulated other comprehensive | Treasury Shares | Total shareholders’ | |||||||||||||||||||||||||
Shares | Amount | paid-in capital | deficit | loss | Shares | Amount | (deficit) equity | |||||||||||||||||||||||
Balance, December 31, 2016 | 41,371 | $ | 414 | $ | 385,020 | $ | (319,800 | ) | $ | (23,565 | ) | — | $ | — | $ | 42,069 | ||||||||||||||
Net income | — | — | — | 68,271 | — | — | — | 68,271 | ||||||||||||||||||||||
Other comprehensive income | — | — | — | — | 299 | — | — | 299 | ||||||||||||||||||||||
Exercise of stock options | 69 | — | 396 | — | — | — | — | 396 | ||||||||||||||||||||||
Vesting of restricted shares | 23 | — | — | — | — | — | — | — | ||||||||||||||||||||||
Share-based compensation expense | — | — | 8,062 | — | — | — | — | 8,062 | ||||||||||||||||||||||
Share repurchases | — | — | — | — | — | 2,117 | (22,361 | ) | (22,361 | ) | ||||||||||||||||||||
Adjustment to accumulated deficit (see Note 13: Income Taxes) | — | — | — | (11,156 | ) | — | — | — | (11,156 | ) | ||||||||||||||||||||
Balance, December 31, 2017 | 41,463 | 414 | 393,478 | (262,685 | ) | (23,266 | ) | 2,117 | (22,361 | ) | 85,580 | |||||||||||||||||||
Net loss | — | — | — | (95,304 | ) | — | — | — | (95,304 | ) | ||||||||||||||||||||
Other comprehensive loss | — | — | — | — | (150 | ) | — | — | (150 | ) | ||||||||||||||||||||
Exercise of stock options | 82 | 1 | 534 | — | — | — | — | 535 | ||||||||||||||||||||||
Exercise of warrants | 603 | 6 | 2,905 | — | — | — | — | 2,911 | ||||||||||||||||||||||
Expiration of warrants | — | — | 2,167 | — | — | — | — | 2,167 | ||||||||||||||||||||||
Vesting of restricted shares | 547 | 6 | (6 | ) | — | — | — | — | — | |||||||||||||||||||||
Employee share purchase plan share issuance | 25 | — | 127 | — | — | — | — | 127 | ||||||||||||||||||||||
Share-based compensation expense | — | — | 7,852 | — | — | — | — | 7,852 | ||||||||||||||||||||||
Equity component of 2023 Notes | — | — | 26,699 | — | — | — | — | 26,699 | ||||||||||||||||||||||
Share repurchases | — | — | — | — | — | 3,290 | (27,637 | ) | (27,637 | ) | ||||||||||||||||||||
Balance, December 31, 2018 | 42,720 | 427 | 433,756 | (357,989 | ) | (23,416 | ) | 5,407 | (49,998 | ) | 2,780 | |||||||||||||||||||
Net loss | — | — | — | (33,226 | ) | — | — | — | (33,226 | ) | ||||||||||||||||||||
Other comprehensive income | — | — | — | — | 610 | — | — | 610 | ||||||||||||||||||||||
Vesting of restricted shares | 153 | 2 | (2 | ) | — | — | — | — | — | |||||||||||||||||||||
Employee share purchase plan share issuance | 54 | — | 118 | — | — | — | — | 118 | ||||||||||||||||||||||
Share-based compensation expense | — | — | 519 | — | — | — | — | 519 | ||||||||||||||||||||||
Balance, December 31, 2019 | 42,927 | $ | 429 | $ | 434,391 | $ | (391,215 | ) | $ | (22,806 | ) | 5,407 | $ | (49,998 | ) | $ | (29,199 | ) | ||||||||||||
See accompanying notes to consolidated financial statements.
-60-
AVADEL PHARMACEUTICALS PLC
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Years ended December 31, | ||||||||||||
2019 | 2018 | 2017 | ||||||||||
Cash flows from operating activities: | ||||||||||||
Net (loss) income | $ | (33,226 | ) | $ | (95,304 | ) | $ | 68,271 | ||||
Adjustments to reconcile net (loss) income to net cash (used in) provided by operating activities: | ||||||||||||
Depreciation and amortization | 2,486 | 7,430 | 4,883 | |||||||||
Impairment of intangible asset | — | 66,087 | — | |||||||||
Amortization of premiums on marketable securities | 41 | 2,823 | 732 | |||||||||
Remeasurement of related party acquisition-related contingent consideration | 845 | (22,731 | ) | (31,040 | ) | |||||||
Remeasurement of related party financing-related contingent consideration | 378 | (1,899 | ) | (2,071 | ) | |||||||
Amortization of debt discount and debt issuance costs | 5,995 | 4,830 | — | |||||||||
Changes in deferred tax | (6,334 | ) | (19,152 | ) | 3,556 | |||||||
Share-based compensation expense | 519 | 7,852 | 8,072 | |||||||||
Loss on deconsolidation of subsidiary | 1,750 | — | — | |||||||||
Other adjustments | (295 | ) | 1,365 | (968 | ) | |||||||
Net changes in assets and liabilities | ||||||||||||
Accounts receivable | 2,471 | 3,452 | 3,054 | |||||||||
Inventories, net | 1,155 | 711 | (2,899 | ) | ||||||||
Prepaid expenses and other current assets | (1,187 | ) | 3,577 | (3,741 | ) | |||||||
Research and development tax credit receivable | (1,014 | ) | (2,545 | ) | (3,141 | ) | ||||||
Accounts payable & other current liabilities | 4,641 | (2,032 | ) | 595 | ||||||||
Deferred revenue | (114 | ) | (1,892 | ) | (216 | ) | ||||||
Accrued expenses | 357 | (10,640 | ) | 13,187 | ||||||||
Accrued income taxes | (30 | ) | (341 | ) | (786 | ) | ||||||
Earn-out payments for related party contingent consideration in excess of acquisition-date fair value | (10,988 | ) | (19,468 | ) | (31,636 | ) | ||||||
Royalty payments for related party payable in excess of original fair value | (1,748 | ) | (2,838 | ) | (4,429 | ) | ||||||
Other assets and liabilities | (4,027 | ) | (2,001 | ) | (4,761 | ) | ||||||
Net cash (used in) provided by operating activities | (38,325 | ) | (82,716 | ) | 16,662 | |||||||
Cash flows from investing activities: | ||||||||||||
Purchases of property and equipment | (29 | ) | (178 | ) | (591 | ) | ||||||
Proceeds from disposal of property and equipment | 154 | — | — | |||||||||
Purchase of intangible assets | — | (20,000 | ) | (53,111 | ) | |||||||
Proceeds from sales of marketable securities | 63,246 | 359,507 | 189,009 | |||||||||
Purchases of marketable securities | (24,648 | ) | (376,310 | ) | (151,005 | ) | ||||||
Net cash provided by (used in) investing activities | 38,723 | (36,981 | ) | (15,698 | ) | |||||||
Cash flows from financing activities: | ||||||||||||
Proceeds from debt issuance | — | 143,750 | — | |||||||||
Payments for debt issuance costs | — | (6,190 | ) | — | ||||||||
Earn-out payments for related party contingent consideration | — | (645 | ) | (1,246 | ) | |||||||
Exercise of warrants | — | 2,911 | — | |||||||||
Proceeds from issuance of ordinary shares | 118 | 577 | 404 | |||||||||
Share repurchases | — | (27,637 | ) | (22,361 | ) | |||||||
Other financing activities, net | (145 | ) | (107 | ) | (115 | ) | ||||||
Net cash (used in) provided by financing activities | (27 | ) | 112,659 | (23,318 | ) | |||||||
Effect of foreign currency exchange rate changes on cash and cash equivalents | 78 | (201 | ) | (297 | ) | |||||||
Net change in cash and cash equivalents | 449 | (7,239 | ) | (22,651 | ) | |||||||
Cash and cash equivalents at January 1 | 9,325 | 16,564 | 39,215 | |||||||||
Cash and cash equivalents at December 31 | $ | 9,774 | $ | 9,325 | $ | 16,564 | ||||||
Supplemental disclosures of cash flow information: | ||||||||||||
Income tax paid | $ | 140 | $ | 776 | $ | 19,143 | ||||||
Interest paid | 6,469 | 3,359 | 1,050 | |||||||||
See accompanying notes to consolidated financial statements.
-61-
AVADEL PHARMACEUTICALS PLC
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(In thousands, except per share data)
NOTE 1: Summary of Significant Accounting Policies
Nature of Operations. Avadel Pharmaceuticals plc (Nasdaq: AVDL) (“Avadel,” the “Company,” “we,” “our,” or “us”) is an emerging biopharmaceutical company. Our lead product candidate, FT218, is an investigational once-nightly formulation of sodium oxybate for the treatment of excessive daytime sleepiness (“EDS”), and cataplexy in narcolepsy patients. FT218, which uses our Micropump drug-delivery technology, is in a Phase 3 clinical trial for the treatment of narcolepsy patients suffering from EDS and cataplexy. In addition, we have three approved commercial products developed under our “unapproved marketed drug,” or UMD, program, Akovaz, Bloxiverz and Vazculep, and a fourth approved product, Nouress, which are sterile injectable drugs used in the hospital setting.
We are primarily focused on the development and potential U.S. Food and Drug Administration (“FDA”) approval of FT218. In addition, we continue to market and distribute our current approved hospital products portfolio and, pending resolution of the existing patent infringement claim (as described below), we plan to commercialize Nouress. Outside of our product candidate and our existing commercial products, we continue to evaluate opportunities to expand our product portfolio.
Current marketed products
• | Bloxiverz (neostigmine methylsulfate injection) - Bloxiverz was approved by the FDA in May 2013 and was launched in July 2013. Bloxiverz is a drug used intravenously in the operating room to reverse the effects of non-depolarizing neuromuscular blocking agents after surgery. Bloxiverz was the first FDA-approved version of neostigmine methylsulfate. Today, neostigmine is one of the two most frequently used products for the reversal of the effects of other agents used for neuromuscular blocks. |
• | Vazculep (phenylephrine hydrochloride injection) - Vazculep was approved by the FDA in June 2014 and was launched in October 2014. Vazculep is indicated for the treatment of clinically important hypotension occurring in the setting of anesthesia. |
• | Akovaz (ephedrine sulfate injection). Akovaz, was approved by the FDA in April 2016 and was launched in August 2016. Akovaz was the first FDA approved formulation of ephedrine sulfate, an alpha- and beta- adrenergic agonist and a norepinephrine-releasing agent that is indicated for the treatment of clinically important hypotension occurring in the setting of anesthesia. |
Nouress
In December 2019, we received FDA approval for Nouress (cysteine hydrochloride injection), a sterile injectable product for use in the hospital setting, and currently have two patents covering that product. Several additional patent applications for Nouress are pending with the USPTO. In light of the recently filed patent suit by Exela Pharma Sciences, LLC, we are currently evaluating the timing and process for a commercial launch of Nouress in the U.S. See Note 16: Contingent Liabilities and Commitments.
The Company was incorporated in Ireland on December 1, 2015 as a private limited company, and re-registered as an Irish public limited company on November 21, 2016. Our headquarters are in Dublin, Ireland and we have operations in St. Louis, Missouri, U.S., and Lyon, France.
Basis of Presentation. These consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the U.S. (U.S. GAAP). The consolidated financial statements include the accounts of the Company and all subsidiaries. All intercompany accounts and transactions have been eliminated.
On February 6, 2019, the Company’s indirect wholly-owned subsidiary, Avadel Specialty Pharmaceuticals, LLC (“Specialty Pharma”), filed a voluntary petition for reorganization under Chapter 11 of the U.S. Code (the “Bankruptcy Code”). in the U.S. District Bankruptcy Court for the District of Delaware (the “Bankruptcy Court”), Case No. 19-10248. Specialty Pharma is operating and managing its business as “debtors-in-possession” under the jurisdiction of the Bankruptcy Court and in accordance with the applicable provisions of the Bankruptcy Code and order of the Bankruptcy Court. As a result of Specialty Pharma’s voluntary bankruptcy filing on February 6, 2019, we no longer controlled the operations of Specialty Pharma; therefore, we deconsolidated Specialty Pharma effective with the bankruptcy filing and the Company recorded its investment in Specialty Pharma under the cost method. See Note 3: Subsidiary Bankruptcy and Deconsolidation. Our results of operations for the period January 1, 2019
-62-
through February 6, 2019 include the results of Specialty Pharma prior to its February 6, 2019 voluntary petition for reorganization under Chapter 11 of the U.S. Bankruptcy Code.
Our results of operations for the period January 1, 2018 through February 16, 2018 and for the year ended December 31, 2017 include the results of FSC Therapeutics and FSC Laboratories, Inc., (collectively “FSC”), prior to its February 16, 2018 disposition date. See Note 17: Divestiture of the Pediatric Assets, for additional information.
Revenue. Revenue includes sales of pharmaceutical products, licensing fees, and, if any, milestone payments for research and development (“R&D”) achievements.
Effective January 1, 2018, the Company adopted Accounting Standards Codification (“ASC”) Topic 606, “Revenue from Contracts with Customers” (“ASC 606”) using the modified retrospective transition method applied to all open contracts as at December 31, 2017. The adoption of the new standard did not have a material effect on the overall timing or amount of revenue recognized when compared to prior accounting standards. See Note 4: Revenue Recognition.
ASC 606 applies to all contracts with customers, except for contracts that are within the scope of other standards, such as leases, insurance, collaboration arrangements and financial instruments. Under ASC 606, an entity recognizes revenue when the performance obligations to the customer have been satisfied through the transfer of control of the goods or services. To determine the appropriate revenue recognition for arrangements that the Company believes are within the scope of ASC 606, we perform the following five steps: (i) Identify the contract(s) with a customer; (ii) Identify the performance obligations in the contract; (iii) Determine the transaction price; (iv) Allocate the transaction price to the performance obligations in the contract; and (v) Recognize revenue when (or as) the entity satisfies a performance obligation. The Company applies the five-step model to contracts only when the Company and its customer’s rights and obligations under the contract can be determined, the contract has commercial substance, and it is probable that the Company will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. For contracts that are determined to be within the scope of ASC 606, the Company identifies the promised goods or services in the contract to determine if they are separate performance obligations or if they should be bundled with other goods and services into a single performance obligation. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
Product Sales
We sell products primarily through wholesalers and considers these wholesalers to be its customers. Under ASC 606, revenue from product sales is recognized when the customer obtains control of our product, which occurs typically upon receipt by the customer. Our gross product sales are subject to a variety of price adjustments in arriving at reported net product sales. These adjustments include estimates of product returns, chargebacks, payment discounts, rebates, and other sales allowances and are estimated based on analysis of historical data for the product or comparable products, future expectations for such products and other judgments and analysis.
License Revenue
From time to time we may enter into out-licensing agreements which are within the scope of ASC 606 under which it licenses to third parties certain rights to its products or intellectual property. The terms of these arrangements typically include payment to us of one or more of the following: non-refundable, upfront license fees; development, regulatory, and commercial milestone payments; and sales-based royalty payments. Each of these payments results in license revenue.
For a complete discussion of the accounting for net product revenue and license revenues, see Note 4: Revenue Recognition.
Research and Development (“R&D”). R&D expenses consist primarily of costs related to clinical studies and outside services, personnel expenses, and other R&D expenses. Clinical studies and outside services costs relate primarily to services performed by clinical research organizations and related clinical or development manufacturing costs, materials and supplies, filing fees, regulatory support, and other third-party fees. Personnel expenses relate primarily to salaries, benefits and share-based compensation. Other R&D expenses primarily include overhead allocations consisting of various support and facilities-related costs. R&D expenditures are charged to operations as incurred.
We recognize R&D tax credits received from the French and Irish government for spending on innovative R&D as an offset of R&D expenses.
-63-
Advertising Expenses. We expense the costs of advertising as incurred. Advertising expenses were $372, $17,562 and $2,214 for the years ended December 31, 2019, 2018 and 2017, respectively. The decrease in advertising for the year ended December 31, 2019 is due to Specialty Pharma’s bankruptcy and deconsolidation. See Note 3: Subsidiary Bankruptcy and Deconsolidation.
Share-based Compensation. We account for share-based compensation based on the estimated grant-date fair value. The fair value of stock options and warrants is estimated using Black-Scholes option-pricing valuation models (“Black-Scholes model”). As required by the Black-Sholes model, estimates are made of the underlying volatility of AVDL stock, a risk-free rate and an expected term of the option or warrant. We estimated the expected term using a simplified method, as we do not have enough historical exercise data for a majority of such options and warrants upon which to estimate an expected term. We recognize compensation cost, net of an estimated forfeiture rate, using the accelerated method over the requisite service period of the award.
Income Taxes. We account for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, we determine deferred tax assets and liabilities on the basis of the differences between the financial statement and tax bases of assets and liabilities by using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.
We recognize deferred tax assets to the extent that we believe that these assets are more likely than not to be realized. In making such a determination, we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax-planning strategies, and results of recent operations. If we determine that we would be able to realize our deferred tax assets in the future in excess of their net recorded amount, we would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes.
We record uncertain tax positions in accordance with ASC 740 on the basis of a two-step process in which (1) we determine whether it is more likely than not that the tax positions will be sustained on the basis of the technical merits of the position and (2) for those tax positions that meet the more-likely-than-not recognition threshold, we recognize the largest amount of tax benefit that is more than 50 percent likely to be realized upon ultimate settlement with the related tax authority.
We recognize interest and penalties related to unrecognized tax benefits in the income tax expense line in the accompanying consolidated statements of (loss) income. Accrued interest and penalties are included on the related tax liability line in the consolidated balance sheets.
Cash and Cash Equivalents. Cash and cash equivalents consist of cash on hand, cash on deposit and fixed term deposits which are highly liquid investments with original maturities of less than three months.
Marketable Securities. The Company’s marketable securities are considered to be available for sale and are carried at fair value, with unrealized gains and losses, net of taxes, reported as a component of accumulated other comprehensive (loss) income (“AOCI”) in shareholders’ equity, with the exception of unrealized gains and losses on equity instruments and unrealized losses believed to be other-than-temporary, if any, which are reported in earnings in the current period. The cost of securities sold is based upon the specific identification method.
Accounts Receivable. Accounts receivable are stated at amounts invoiced net of allowances for doubtful accounts and certain other gross to net variable consideration deductions. The Company makes judgments as to our ability to collect outstanding receivables and provides allowances for the portion of receivables deemed uncollectible. Provision is made based upon a specific review of all significant outstanding invoices. A majority of accounts receivable is due from four significant customers.
Inventories. Inventories consist of raw materials and finished products, which are stated at lower of cost or net realizable value, using the first-in, first-out (“FIFO”) method. Raw materials used in the production of pre-clinical and clinical products are expensed as R&D costs when consumed. The Company establishes reserves for inventory estimated to be obsolete, unmarketable or slow-moving on a case by case basis.
Property and Equipment. Property and equipment is stated at historical cost less accumulated depreciation. Depreciation and amortization are computed using the straight-line method over the following estimated useful lives:
Laboratory equipment | 4-8 years |
Software, office and computer equipment | 3 years |
Leasehold improvements, furniture, fixtures and fittings | 5-10 years |
-64-
Goodwill. Goodwill represents the excess of the acquisition consideration over the fair value of assets acquired and liabilities assumed. We have determined that we operate in a single segment and has a single reporting unit associated with the development and commercialization of pharmaceutical products. Prior to November 2019, we tested for goodwill impairment using a two-step process. The first step is a comparison of the fair value of the reporting unit with its carrying amount, including goodwill. If this step indicates impairment, then, in the second step, the loss is measured as the excess of recorded goodwill over the implied fair value of the goodwill. Implied fair value of goodwill is the excess of the fair value of the reporting unit as a whole over the fair value of all separately identified assets and liabilities within the reporting unit. As discussed in Note 2: Effect of New Accounting Standards, we elected to early adopt Accounting Standards Update (“ASU”) 2017-04 in November 2019, which eliminated step 2 from the goodwill impairment test. We test goodwill for impairment annually and when events or changes in circumstances indicate that the carrying value may not be recoverable. We performed our required impairment test of goodwill and have determined that no impairment of goodwill existed at December 31, 2019 and 2018.
Long-Lived Assets. Long-lived assets include fixed assets and intangible assets. Intangible assets consist primarily of purchased licenses and intangible assets recognized as part of the Éclat Pharmaceuticals acquisition. Acquired IPR&D has an indefinite life and is not amortized until completion and development of the project, at which time the IPR&D becomes an amortizable asset, for which amortization of such intangible assets is computed using the straight-line method over the estimated useful life of the assets.
Long-lived assets are reviewed for impairment whenever conditions indicate that the carrying value of the assets may not be fully recoverable. Such impairment tests are based on a comparison of the pretax undiscounted cash flows expected to be generated by the asset to the recorded value of the asset or other market based value approaches. If impairment is indicated, the asset value is written down to its market value if readily determinable or its estimated fair value based on discounted cash flows. Any significant changes in business or market conditions that vary from current expectations could have an impact on the fair value of these assets and any potential associated impairment. During the fourth quarter of 2018, we recorded a $66,087 impairment charge to the entire acquired developed technology related to Noctiva (see Note 9: Goodwill and Intangible Assets). We determined that no impairment existed at December 31, 2019.
Lease Obligations. On January 1, 2019, the Company adopted ASU 2016-02, “Leases” which supersedes ASC 840 “Leases” and creates a new topic, ASC 842 “Leases”. The Company adopted ASU 2016-02 using the modified retrospective transition approach and elected the transition option to recognize the adjustment in the period of adoption rather than in the earliest period presented. Results and disclosure requirements for reporting periods beginning after January 1, 2019 are presented under Topic 842, while prior period amounts have not been adjusted and continue to be reported in accordance with our historical accounting under Topic 840. Upon adoption, we recognized total ROU assets of $5,046, with corresponding lease liabilities of $5,131 on the consolidated balance sheets. The adoption did not impact our beginning retained earnings, or our prior year consolidated statements of income and statements of cash flows.
The Company elected the package of practical expedients permitted under the transition guidance, which allowed us to carryforward our historical lease classification, our assessment on whether a contract was or contains a lease, and our initial direct costs for any leases that existed prior to January 1, 2019. The Company also elected to combine our lease and non-lease components and to keep leases with an initial term of 12 months or less off the balance sheet and recognize the associated lease payments in the consolidated statements of (loss) income on a straight-line basis over the lease term.
Under ASU 2016-02, the Company determines if a contract is a lease at the inception of the arrangement. Right-of-use assets and operating lease liabilities are recognized at commencement date based on the present value of remaining lease payments over the lease term. For this purpose, the Company considers only payments that are fixed and determinable at the time of commencement. The Company reviews all options to extend, terminate, or purchase its right-of-use assets at the inception of the lease and will include these options in the lease term when they are reasonably certain of being exercised. Nearly all of the Company’s lease contracts do not provide a readily determinable implicit rate. For these contracts, the Company’s estimated incremental borrowing rate is based on information available at the inception of the lease. Our lease agreements may contain variable costs such as common area maintenance, insurance, real estate taxes or other costs. Variable lease costs are expensed as incurred on the consolidated statements of income.
Acquisition-related Contingent Consideration. The acquisition-related contingent consideration payables arising from the acquisition of Éclat Pharmaceuticals (i.e., our hospital products) and FSC (our pediatrics products), which was assumed by the buyer as part of the disposition of the pediatrics products on February 16, 2018, are accounted for at fair-value (see Note 12: Long-Term Related Party Payable and Note 17: Divestiture of the Pediatric Assets). The fair value of the warrants issued in connection with the Éclat acquisition were estimated using a Black-Scholes model. A portion of these warrants were exercised on February 23, 2018 and the remaining warrants expired on March 12, 2018. See Note 12: Long-Term Related Party Payable. The fair value
-65-
of acquisition-related contingent consideration payable is estimated using a discounted cash flow model based on the long-term sales or gross profit forecasts of the specified hospital or pediatric products using an appropriate discount rate. There are a number of estimates used when determining the fair value of these earn-out payments. These estimates include, but are not limited to, the long-term pricing environment, market size, market share the related products are forecast to achieve, the cost of goods related to such products and an appropriate discount rate to use when present valuing the related cash flows. These estimates can and often do change based on changes in current market conditions, competition, management judgment and other factors. Changes to these estimates can have and have had a material impact on our consolidated statements of (loss) income and balance sheets. Changes in fair value of these liabilities are recorded in the consolidated statements of (loss) income within operating expenses as changes in fair value of related party contingent consideration.
Financing-related Royalty Agreements. We also entered into two royalty agreements with related parties in connection with certain financing arrangements. We elected the fair value option for the measurement of the financing-related contingent consideration payable associated with the royalty agreements with certain Deerfield and Broadfin entities, both of whom are related parties (see Note 12: Long-Term Related Party Payable). The fair value of financing-related royalty agreements is estimated using the same components used to determine the fair value of the acquisition-related contingent consideration noted above, with the exception of cost of products sold. Changes to these components can also have a material impact on our consolidated statements of (loss) income and balance sheets. Changes in the fair value of this liability are recorded in the consolidated statements of (loss) income as other (expense) income - changes in fair value of related party payable.
Use of Estimates. The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, including marketable securities and contingent liabilities at the date of the consolidated financial statements and the reported amounts of sales and expenses during the periods presented. These estimates and assumptions are based on the best information available to management at the balance sheet dates and depending on the nature of the estimate can require significant judgments. Changes to these estimates and judgments can have and have had a material impact on our consolidated statements of (loss) income and balance sheets. Actual results could differ from those estimates under different assumptions or conditions.
NOTE 2: Newly Issued Accounting Standards
Recently Adopted Accounting Guidance
In February 2016, the Financial Accounting Standards Board (“FASB”) issued ASU 2016-02, “Leases” which supersedes ASC 840 “Leases” and creates a new topic, ASC 842 “Leases.” This update requires lessees to recognize on their balance sheet a lease liability and a lease asset for all leases, including operating leases, with a term greater than 12 months. The update also expands the required quantitative and qualitative disclosures surrounding leases. This update is effective for fiscal years beginning after December 15, 2018 and interim periods within those fiscal years, with earlier application permitted. In July 2018, the FASB issued ASU 2018-11 “Targeted Improvements”, amending certain aspects of the new leasing standard. The amendment allows an additional optional transition method whereby an entity records a cumulative effect adjustment to opening retained earnings in the year of adoption without restating prior periods, which the Company has elected. The Company adopted this ASU during the first quarter of 2019. See Note 10: Leases for more information.
In January 2017, the FASB issued ASU 2017-04, “Intangibles - Goodwill and Other: Simplifying the Test for Goodwill Impairment.” This update eliminates step 2 from the goodwill impairment test, and requires the goodwill impairment test to be performed by comparing the fair value of a reporting unit with its carrying amount. An impairment charge should be recognized for the amount by which the carrying amount exceeds the reporting unit’s fair value; however, the loss recognized should not exceed the total amount of goodwill allocated to that reporting unit. This guidance is effective for the Company in the first quarter of 2020. Early adoption is permitted for interim or annual goodwill impairment tests performed on testing dates after January 1, 2017. In November 2019, the Company elected to early adopt ASU 2017-04, and the adoption had no impact on our consolidated financial statements.
Recent Accounting Guidance Not Yet Adopted
In December 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, as part of its overall simplification initiative to reduce costs and complexity of applying accounting standards while maintaining or improving the usefulness of the information provided to users of financial statements. The FASB’s amendments primarily impact ASC 740, Income Taxes, and may impact both interim and annual reporting periods. ASU 2019-12 will be effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years and early adoption is permitted. We are currently evaluating the impact of adopting ASU 2019-12.
-66-
In August 2018, the FASB issued ASU 2018-13, “Fair Value Measurement (Topic 820): Disclosure Framework— Changes to the Disclosure Requirement for Fair Value Measurement” which amends certain disclosure requirements over Level 1, Level 2 and Level 3 fair value measurements. The amendments in ASU 2018-13 are effective for fiscal years beginning after December 15, 2019, with early adoption permitted. The Company is currently evaluating the impact of adopting ASU 2018-13 and does not believe the standard will have a material impact on the consolidated financial statements.
In June 2016, the FASB issued ASU No. 2016-13, “Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”).” This standard requires entities to measure all expected credit losses for financial assets held at the reporting date based on historical experience, current conditions and reasonable and supportable forecasts. ASU 2016-13 will be effective for the Company for fiscal years beginning on or after January 1, 2020, including interim periods within those annual reporting periods and early adoption is permitted. We are currently evaluating the impact of our adoption of ASU 2016-13 on our condensed consolidated financial statements and currently does not believe the standard will have a material impact on the consolidated financial statements.
NOTE 3: Subsidiary Bankruptcy and Deconsolidation
Bankruptcy Filing and Deconsolidation
As a result of Specialty Pharma’s bankruptcy filing on February 6, 2019, Avadel has ceded authority for managing the business to the Bankruptcy Court, and Avadel management cannot carry on Specialty Pharma’s activities in the ordinary course of business without Bankruptcy Court approval. Avadel manages the day-to-day operations of Specialty Pharma, but does not have discretion to make significant capital or operating budgetary changes or decisions and purchase or sell significant assets, as Specialty Pharma’s material decisions are subject to review by the Bankruptcy Court. For these reasons, we have concluded that Avadel has lost control of Specialty Pharma, and no longer has significant influence over Specialty Pharma during the pendency of the bankruptcy. Therefore, we deconsolidated Specialty Pharma effective with the filing of the Chapter 11 bankruptcy in February 2019.
In order to deconsolidate Specialty Pharma, the carrying values of the assets and certain liabilities of Specialty Pharma were removed from our consolidated balance sheet as of February 5, 2019, and we recorded our investment in Specialty Pharma at its estimated fair value of $0. As the estimated fair value of our investment in Specialty Pharma was lower than its net book value immediately prior to the deconsolidation, we recorded a non-cash charge of approximately $2,678 for the year ended December 31, 2019 associated with the deconsolidation of Specialty Pharma. Subsequent to the deconsolidation of Specialty Pharma, we are accounting for our investment in Specialty Pharma using the cost method of accounting because Avadel does not exercise significant influence over the operations of Specialty Pharma due to the Chapter 11 filing.
On April 26, 2019, Specialty Pharma sold its intangible assets and remaining inventory to an unaffiliated third party in exchange for aggregate cash proceeds of approximately $250, pursuant to an order approving such sale which was issued by the Bankruptcy Court on April 15, 2019. As a result of such sale, Specialty Pharma has completed its divestment of the assets of the Noctiva business.
On July 2, 2019, Specialty Pharma was made aware of a $50,695 claim made by the Internal Revenue Service (IRS) as part of the bankruptcy claims process against Specialty Pharma. On October 2, 2019 the IRS amended the original claim filed in July, reducing the claim to $9,302. Specialty Pharma files its U.S. federal tax return as a member of the Company’s consolidated U.S. tax group. As such, the IRS claim was filed against Specialty Pharma in the bankruptcy proceedings due to IRS tax law requirements for joint and several liability of all members in a consolidated U.S. tax group. Both Specialty Pharma and the Company disagree with the merits of the amended IRS claim and intend to defend their positions vigorously.
Debtor in Possession (“DIP”) Financing – Related Party Relationship
In connection with the bankruptcy filing, Specialty Pharma entered into a DIP Credit and Security Agreement with Avadel US Holdings (“DIP Credit Agreement”) dated as of February 8, 2019, in an aggregate amount of up to $2,700, of which the funds are to be used by Specialty Pharma solely to fund operations through February 6, 2020. As of December 31, 2019, the Company had funded $407 under the DIP Credit Agreement. As we have assessed that it is unlikely that Specialty Pharma will pay back the loan to Avadel, the $407 has been recorded as part of the loss on deconsolidation of subsidiary within the consolidated statements of (loss) income. At December 31, 2019, the fair value of the remaining commitment under the DIP Credit Agreement is not material.
NOTE 4: Revenue Recognition
The Company generates revenue primarily from the sale of pharmaceutical products to customers. From time to time the Company also generates revenue from licensing arrangements whereby the Company provides access to certain of its intellectual property.
-67-
Periods prior to January 1, 2018
Product Sales and Services
Revenue was generally realized or realizable and earned when persuasive evidence of an arrangement existed, delivery had occurred or services had been rendered, the seller’s price to the buyer was fixed or determinable, and collectability was reasonably assured. The Company recorded revenue from product sales when title and risk of ownership transferred to the customer, which was typically upon delivery to the customer and when the selling price was determinable.
Licensing Revenues
From time to time, the Company enters into licensing agreements for the license of technology used for developing modified controlled release of oral pharmaceutical products. Non-refundable fees where the Company had continuing performance obligations were deferred and recognized ratably over the projected performance period. Milestone payments, which were typically related to regulatory, commercial or other achievements by the Company or their licensees and distributors, were recognized as revenues when the milestone was accomplished and collection was reasonably assured.
Periods commencing January 1, 2018
Product Sales and Services
Effective January 1, 2018, the Company implemented ASC 606, Revenue From Contracts With Customers. The Company sells products primarily through wholesalers and considers these wholesalers to be its customers. Under ASC 606, revenue from product sales is recognized when the customer obtains control of the Company’s product and the Company’s performance obligations are met, which occurs typically upon receipt of delivery to the customer. As is customary in the pharmaceutical industry, the Company’s gross product sales are subject to a variety of price deductions in arriving at reported net product sales. These adjustments include estimates for product returns, chargebacks, payment discounts, rebates, and other sales allowances and are estimated when the product is delivered based on analysis of historical data for the product or comparable products, as well as future expectations for such products.
Reserves to reduce Gross Revenues to Net Revenues
Revenues from product sales are recorded at the net selling price, which includes estimated reserves to reduce gross product sales to net product sales resulting from product returns, chargebacks, payment discounts, rebates, and other sales allowances that are offered within contracts between the Company and its customers and end users. These reserves are based on the amounts earned or to be claimed on the related sales and are classified as reductions of accounts receivable if the amount is payable to the customer, except in the case of the estimated reserve for future expired product returns, which are classified as a liability. The reserves are classified as a liability if the amount is payable to a party other than a customer. Where appropriate, these estimated reserves take into consideration relevant factors such as the Company’s historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, these reserves reflect the Company’s best estimates to reduce gross selling price to net selling price to which it expects to be entitled based on the terms of its contracts. The actual net selling price ultimately received may differ from the Company’s estimates. If actual results in the future vary from the Company’s estimates, the Company adjusts these estimates, which would affect net product revenue and earnings in the period such variances become known.
Product Returns
Consistent with industry practice the Company maintains a returns policy that generally offers customers a right of return for product that has been purchased from the Company. The Company estimates the amount of product returns and records this estimate as a reduction of revenue in the period the related product revenue is recognized. The Company currently estimates product return liabilities based on analysis of historical data for the product or comparable products, as well as future expectations for such products and other judgments and analysis.
Chargebacks, Discounts and Rebates
Chargebacks, discounts and rebates represent the estimated obligations resulting from contractual commitments to sell products to its customers or end users at prices lower than the list prices charged to our wholesale customers. Customers charge the Company for the difference between the gross selling price they pay for the product and the ultimate contractual price agreed to between
-68-
the Company and these end users. These reserves are established in the same period that the related revenue is recognized, resulting in a reduction of product revenue and accounts receivable. Chargebacks, discounts and rebates are estimated at the time of sale to the customer.
Revenue from licensing arrangements
The terms of the Company’s licensing agreements may contain multiple performance obligations, including certain R&D activities. The terms of these arrangements typically include payment to the Company of one or more of the following: non-refundable, up-front license fees; development, regulatory and commercial milestone payments. Each of these payments are recorded as license revenues. The Company did not have any license revenue during the year ended December 31, 2019.
License of Intellectual Property
If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from non-refundable, up-front fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are bundled with other promises, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, up-front fees. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition.
Disaggregation of revenue
The Company’s primary source of revenue is from the sale of pharmaceutical products, which are equally affected by the same economic factors as it relates to the nature, amount, timing, and uncertainty of revenue and cash flows. For further detail about the Company’s revenues by product, see Note 22: Company Operations by Product, Customer and Geography.
Contract Balances
The Company does not recognize revenue in advance of invoicing its customers and therefore has no related contract assets.
A receivable is recognized in the period the Company sells its products and when the Company’s right to consideration is unconditional.
There were no material deferred contract costs at December 31, 2019 and 2018.
Transaction Price Allocated to the Remaining Performance Obligation
For product sales, the Company generally satisfies its performance obligations within the same period the product is delivered. Product sales recognized in 2019 from performance obligations satisfied (or partially satisfied) in previous periods were immaterial.
For certain licenses of intellectual property, specifically those with performance obligations satisfied over time, the Company allocates a portion of the transaction price to that performance obligation and recognizes revenue using an appropriate measure of progress towards development of the product. In December 2018, we reached an agreement to exit a contract and our remaining performance obligations and recognized the remaining $1,600 of deferred revenue, which represented the unsatisfied performance obligations associated with a license agreement. At December 31, 2019, the deferred revenue balance related to this obligation is $0.
We have elected certain of the practical expedients from the disclosure requirement for remaining performance obligations for specific situations in which an entity need not estimate variable consideration to recognize revenue. Accordingly, the Company applies the practical expedient in ASC 606 to its stand-alone contracts and does not disclose information about variable consideration from remaining performance obligations for which we recognize revenue.
NOTE 5: Fair Value Measurements
The Company is required to measure certain assets and liabilities at fair value, either upon initial recognition or for subsequent accounting or reporting. For example, we use fair value extensively when accounting for and reporting certain financial instruments, when measuring certain contingent consideration liabilities and in the initial recognition of net assets acquired in a business combination. Fair value is estimated by applying the hierarchy described below, which prioritizes the inputs used to measure fair
-69-
value into three levels and bases the categorization within the hierarchy upon the lowest level of input that is available and significant to the fair value measurement.
ASC 820, “Fair Value Measurements and Disclosures,” defines fair value as a market-based measurement that should be determined based on the assumptions that marketplace participants would use in pricing an asset or liability. When estimating fair value, depending on the nature and complexity of the asset or liability, we may generally use one or each of the following techniques:
• | Income approach, which is based on the present value of a future stream of net cash flows. |
• | Market approach, which is based on market prices and other information from market transactions involving identical or comparable assets or liabilities. |
As a basis for considering the assumptions used in these techniques, the standard establishes a three-tier fair value hierarchy which prioritizes the inputs used in measuring fair value as follows:
• | Level 1 - Quoted prices for identical assets or liabilities in active markets. |
• | Level 2 - Quoted prices for similar assets or liabilities in active markets, or quoted prices for identical or similar assets or liabilities in markets that are not active, or inputs other than quoted prices that are directly or indirectly observable, or inputs that are derived principally from, or corroborated by, observable market data by correlation or other means. |
• | Level 3 - Unobservable inputs that reflect estimates and assumptions. |
The following table summarizes the financial instruments measured at fair value on a recurring basis classified in the fair value hierarchy (Level 1, 2 or 3) based on the inputs used for valuation in the accompanying consolidated balance sheets:
As of December 31, 2019 | As of December 31, 2018 | |||||||||||||||||||||||
Fair Value Measurements: | Level 1 | Level 2 | Level 3 | Level 1 | Level 2 | Level 3 | ||||||||||||||||||
Marketable securities (see Note 6) | ||||||||||||||||||||||||
Equity securities | $ | 4,404 | $ | — | $ | — | $ | 9,145 | $ | — | $ | — | ||||||||||||
Money market funds | 38,799 | — | — | 52,996 | — | — | ||||||||||||||||||
Corporate bonds | — | 4,098 | — | — | 6,339 | — | ||||||||||||||||||
Government securities - U.S. | — | 5,446 | — | — | 12,701 | — | ||||||||||||||||||
Other fixed-income securities | — | 1,637 | — | — | 9,409 | — | ||||||||||||||||||
Total assets | $ | 43,203 | $ | 11,181 | $ | — | $ | 62,141 | $ | 28,449 | $ | — | ||||||||||||
Related party payable (see Note 12) | — | — | 17,327 | — | — | 28,840 | ||||||||||||||||||
Total liabilities | $ | — | $ | — | $ | 17,327 | $ | — | $ | — | $ | 28,840 | ||||||||||||
A review of fair value hierarchy classifications is conducted on a quarterly basis. Changes in the observability of valuation inputs may result in a reclassification for certain financial assets or liabilities. During the fiscal year ended December 31, 2019, there were no transfers in and out of Level 1, 2, or 3. During the twelve months ended December 31, 2019, 2018 and 2017, we did not recognize any other-than-temporary impairment loss.
Some of the Company’s financial instruments, such as cash and cash equivalents, accounts receivable and accounts payable, are reflected in the balance sheet at carrying value, which approximates fair value due to their short-term nature.
Debt
We estimate the fair value of our $143,750 aggregate principal amount of 4.50% exchangeable senior notes due 2023 (the “2023 Notes”), a Level 2 input, based on interest rates that would be currently available to the Company for issuance of similar types of debt instruments with similar terms and remaining maturities or recent trading prices obtained from brokers. The estimated fair value of the 2023 Notes at December 31, 2019 is $107,692 compared to a book value of $121,686.
See Note 11: Long-Term Debt for additional information regarding our debt obligations.
-70-
NOTE 6: Marketable Securities
We have investments in available-for-sale marketable securities which are recorded at fair market value. Prior to January 1, 2018, unrealized gains and losses on all securities are recorded as other comprehensive income (loss) in shareholders’ equity, net of income tax effects.
On January 1, 2018, the Company adopted ASU 2016-01, which requires the change in the fair value of available-for-sale equity investments to be recognized in our consolidated statements of (loss) income rather than as a component of our consolidated statement of comprehensive (loss) income. For the years ended December 31, 2019 and 2018, the net unrealized loss on our available-for-sale equity investments, recorded as a component of investment income in the accompanying consolidated statements of (loss) income, was income of $170 and a loss of $956, respectively. The net unrealized gain on our available-for-sale equity investments was immaterial for the year ended December 31, 2017 and was recorded as other comprehensive income in shareholders’ equity, net of income tax effects.
The following tables show the Company’s available-for-sale securities’ adjusted cost, gross unrealized gains, gross unrealized losses and fair value by significant investment category as of December 31, 2019 and 2018, respectively:
2019 | ||||||||||||||||
Marketable Securities: | Adjusted Cost | Unrealized Gains | Unrealized Losses | Fair Value | ||||||||||||
Equity securities | $ | 4,234 | $ | 170 | $ | — | $ | 4,404 | ||||||||
Money market funds | 38,028 | 771 | — | 38,799 | ||||||||||||
Corporate bonds | 4,021 | 77 | — | 4,098 | ||||||||||||
Government securities - U.S. | 5,341 | 110 | (5 | ) | 5,446 | |||||||||||
Other fixed-income securities | 1,614 | 23 | — | 1,637 | ||||||||||||
Total | $ | 53,238 | $ | 1,151 | $ | (5 | ) | $ | 54,384 | |||||||
2018 | ||||||||||||||||
Marketable Securities: | Adjusted Cost | Unrealized Gains | Unrealized Losses | Fair Value | ||||||||||||
Equity securities | $ | 10,101 | $ | — | $ | (956 | ) | $ | 9,145 | |||||||
Money market funds | 52,733 | 316 | (53 | ) | 52,996 | |||||||||||
Corporate bonds | 6,411 | 7 | (79 | ) | 6,339 | |||||||||||
Government securities - U.S. | 12,714 | 66 | (79 | ) | 12,701 | |||||||||||
Other fixed-income securities | 9,400 | 22 | (13 | ) | 9,409 | |||||||||||
Total | $ | 91,359 | $ | 411 | $ | (1,180 | ) | $ | 90,590 | |||||||
We determine realized gains or losses on the sale of marketable securities on a specific identification method. We recognized gross realized gains of $483, $317, and $1,677 for the twelve months ended December 31, 2019, 2018, and 2017, respectively. These realized gains were offset by realized losses of $151, $565, and $1,390 for the twelve-months ended December 31, 2019, 2018, and 2017, respectively. We reflect these gains and losses as a component of investment income in the accompanying consolidated statements of (loss) income.
The following table summarizes the estimated fair value of our investments in marketable debt securities, accounted for as available-for-sale securities and classified by the contractual maturity date of the securities as of December 31, 2019:
Maturities | ||||||||||||||||||||
Marketable Debt Securities: | Less than 1 Year | 1-5 Years | 5-10 Years | Greater than 10 Years | Total | |||||||||||||||
Corporate bonds | $ | 293 | $ | 3,464 | $ | 341 | $ | — | $ | 4,098 | ||||||||||
Government securities - U.S. | — | 4,744 | 315 | 387 | 5,446 | |||||||||||||||
Other fixed-income securities | — | 1,637 | — | — | 1,637 | |||||||||||||||
Total | $ | 293 | $ | 9,845 | $ | 656 | $ | 387 | $ | 11,181 | ||||||||||
-71-
We have classified our investment in available-for-sale marketable securities as current assets in the consolidated balance sheets as the securities need to be available for use, if required, to fund current operations. There are no restrictions on the sale of any securities in our investment portfolio.
NOTE 7: Inventories
The principal categories of inventories, net of reserves of $914 and $4,757 at December 31, 2019 and 2018, respectively, are comprised of the following:
Inventory: | 2019 | 2018 | ||||||
Finished goods | $ | 3,020 | $ | 4,270 | ||||
Raw materials | 550 | 500 | ||||||
Total | $ | 3,570 | $ | 4,770 | ||||
Total net reserves decreased by $3,843 during the year ended December 31, 2019 driven largely by the deconsolidation of Specialty Pharma.
NOTE 8: Property and Equipment, net
The principal categories of property and equipment, net at December 31, 2019 and 2018, respectively, are as follows:
Property and Equipment, net: | 2019 | 2018 | ||||||
Laboratory equipment | $ | — | $ | 8,864 | ||||
Software, office and computer equipment | 1,258 | 2,487 | ||||||
Furniture, fixtures and fittings | 300 | 3,715 | ||||||
Less - accumulated depreciation | (1,014 | ) | (13,155 | ) | ||||
Total | $ | 544 | $ | 1,911 | ||||
The decrease in the principal categories of property and equipment, net during the year ended December 31, 2019, is a result of the restructuring efforts discussed in Note 18: Restructuring Costs.
Depreciation expense for the years ended December 31, 2019, 2018 and 2017 was $459, $811 and $1,224, respectively.
NOTE 9: Goodwill and Intangible Assets
The Company’s amortizable and unamortizable intangible assets at December 31, 2019 and 2018, respectively, are as follows:
2019 | 2018 | |||||||||||||||||||||||||||
Goodwill and Intangible Assets: | Gross Value | Accumulated Amortization | Net Carrying Amount | Gross Value | Accumulated Amortization | Impairment | Net Carrying Amount | |||||||||||||||||||||
Amortizable intangible assets: | ||||||||||||||||||||||||||||
Acquired developed technology - Noctiva | $ | — | $ | — | $ | — | $ | 73,111 | $ | (7,024 | ) | $ | (66,087 | ) | $ | — | ||||||||||||
Acquired developed technology - Vazculep | 1,629 | (816 | ) | 813 | 12,061 | (10,432 | ) | — | 1,629 | |||||||||||||||||||
Total amortizable intangible assets | $ | 1,629 | $ | (816 | ) | $ | 813 | $ | 85,172 | $ | (17,456 | ) | $ | (66,087 | ) | $ | 1,629 | |||||||||||
Unamortizable intangible assets - Goodwill | $ | 18,491 | $ | — | $ | 18,491 | $ | 18,491 | $ | — | $ | — | $ | 18,491 | ||||||||||||||
-72-
The Company recorded amortization expense related to amortizable intangible assets of $816, $6,619 and $3,659 for the years ended December 31, 2019, 2018 and 2017, respectively.
No impairment loss related to goodwill or intangible assets was recognized during the year ended December 31, 2019.
During the fourth quarter 2018, certain conditions came to light, largely the lack of a meaningful increase in Noctiva prescriptions despite the substantial investment of resources, which indicated that the carrying value of the asset, may not be fully recoverable. As such, we performed an impairment test based on a comparison of the pretax discounted cash flows expected to be generated by the asset, which is a Level 3 fair value estimate, to the recorded value of the asset and concluded that the associated cash flows did not support any of the carrying value of the intangible asset and we recorded a full impairment charge of $66,087 at December 31, 2018 related to the acquired developed technology associated with Noctiva. The Chapter 11 bankruptcy filing of Specialty Pharma commenced on February 6, 2019, the subsidiary which marketed, sold and distributed Noctiva, confirmed management’s conclusion on the impairment. This impairment charge is included in the line “Impairment of intangible asset” in the consolidated statements of (loss) income.
Amortizable intangible assets are amortized over their estimated useful lives, which range from three to fifteen years, using the straight-line method. At December 31, 2019, total amortization of intangible assets for the year ended December 31, 2020 is $813. There is no estimated amortization during the years ended December 31, 2021-2024 as the acquired developed technology - Vazculep will be fully amortized at December 31, 2020.
NOTE 10: Leases
On January 1, 2019, the Company adopted the ASU 842 using the modified retrospective transition approach and elected the transition option to recognize the adjustment in the period of adoption rather than in the earliest period presented. The adoption resulted in the initial recognition of operating lease right-of-use assets of $5,046 and operating lease liabilities of $5,131. At December 31, 2019, the balances of the operating lease right-of-use asset and total operating lease liability were $3,612 and $2,964, respectively, of which $645 of the operating lease liability is classified as a current liability.
The Company leases certain office facilities, comprising approximately 81% of the total lease population at December 31, 2019. All leased facilities are classified as operating leases with remaining lease terms between one and five years. The Company determines if a contract is a lease at the inception of the arrangement. The Company reviews all options to extend, terminate, or purchase its right-of-use assets at the inception of the lease and will include these options in the lease term when they are reasonably certain of being exercised. For all of the Company’s leases, lease and non-lease components are accounted for as a single lease component, as all non-lease components are immaterial to break out separately.
The components of lease costs, which are included in selling, general and administrative expenses in the consolidated statements of (loss) income of year ended December 31, 2019 were as follows:
Lease cost: | 2019 | |||
Operating lease costs (1) | $ | 1,515 | ||
Sublease income (2) | (276 | ) | ||
Total lease cost | $ | 1,239 | ||
(1) Variable lease costs were immaterial for the year ended December 31, 2019.
(2) Represents sublease income received for the vacated office facility in Charlotte, North Carolina, which was acquired with the FSC acquisition in February 2016. The lease and sublease agreements terminate in December 2020. The Company also vacated portions of its office facility in St. Louis, Missouri during May 2019 and August 2019 and started receiving sublease income starting in May 2019 and August 2019 from two different tenants. The lease agreement ends in April 2025 and the sublease agreement that started in May 2019 ends in May 2021 and the sublease agreement that started in August 2019 ends in July 2020 and can continue thereafter on a month-to-month basis.
During the year ended December 31, 2019, the Company reduced its operating lease liabilities by $1,480 for cash paid. The remainder of the decrease is related to the exit of certain leases discussed below. In addition, during the year ended December 31, 2019, one new operating lease commenced resulting in the recognition of an operating lease right-of-use asset and liability of $1,000 and $0, respectively, as the entire lease payment was paid on March 31, 2019. As of December 31, 2019, the Company is aware of one additional potential embedded lease that has not yet commenced and will not commence until certain conditions are
-73-
met. If these conditions are met and the start date is determined, annual fees would commence and at that time an operating lease right-of-use asset and corresponding operating lease liability will be recorded.
In connection with the 2019 Corporate Restructuring plan discussed in Note 18: Restructuring Costs, during the year ended December 31, 2019, the Company came to an agreement with the landlord of the leased office space in Ireland, to surrender the lease by December 31, 2019 with an early termination payment of approximately $288. This amount was recorded as a restructuring cost in the consolidated statements of (loss) income.
In connection with the 2019 French Restructuring plan discussed in Note 18: Restructuring Costs, during the year ended December 31, 2019, the Company came to an agreement with the landlord of the leased office space in France, to surrender the lease by December 31, 2019 with an early termination payment of approximately $820. The Company accounted for this change in the lease term as a modification of the original lease. As a result of this modification, the right-of-use asset and liability related to the French office lease was remeasured, and the asset was subsequently tested for impairment as the fair value was less than the book value. Since the fair value was determined to be less than the book value, the Company recorded an impairment to the right of use asset of $826, which is recorded as a restructuring cost in the consolidated statements of loss.
As of December 31, 2019, our operating leases have a weighted-average remaining lease term of 5.0 years and a weighted-average discount rate of 5.3%. Nearly all of Avadel’s lease contracts do not provide a readily determinable implicit rate. For these contracts, Avadel’s estimated incremental borrowing rate is based on information available at the inception of the lease.
Maturities of the Company’s operating lease liabilities were as follows:
Maturities: | Operating Leases | |||
2020 | $ | 779 | ||
2021 | 578 | |||
2022 | 590 | |||
2023 | 602 | |||
2024 | 614 | |||
Thereafter | 201 | |||
Total lease payments | 3,364 | |||
Less: interest | 400 | |||
Present value of lease liabilities | $ | 2,964 | ||
Under the prior lease guidance, minimum rental commitments for non-cancelable leases as of December 31, 2019 were:
Lease Commitment: | Operating Leases | |||
2020 | $ | 779 | ||
2021 | 578 | |||
2022 | 590 | |||
2023 | 602 | |||
2024 | 614 | |||
Thereafter | 201 | |||
Total minimum lease payments | $ | 3,364 | ||
-74-
NOTE 11: Long-Term Debt
Long-Term debt is summarized as follows:
December 31, 2019 | December 31, 2018 | |||||||
Principal amount of 4.50% exchangeable senior notes due 2023 | $ | 143,750 | $ | 143,750 | ||||
Less: debt discount and issuance costs, net | (22,064 | ) | (28,059 | ) | ||||
Net carrying amount of liability component | 121,686 | 115,691 | ||||||
Other debt | — | 149 | ||||||
Subtotal | 121,686 | 115,840 | ||||||
Less: current maturities | — | (106 | ) | |||||
Long-term debt | $ | 121,686 | $ | 115,734 | ||||
Equity component: | ||||||||
Equity component of exchangeable notes, net of issuance costs | $ | (26,699 | ) | $ | (26,699 | ) | ||
Issuance of Debt Securities
On February 16, 2018, Avadel Finance Cayman Limited, a Cayman Islands exempted company (the “Issuer”) and an indirect wholly-owned subsidiary of the Company, issued $125,000 aggregate principal amount of 4.50% exchangeable senior notes due 2023 (the “2023 Notes”) in a private placement (the “Offering”) to qualified institutional buyers pursuant to Rule 144A under the Securities Act. In connection with the Offering, the Issuer granted the initial purchasers of the 2023 Notes a 30-day option to purchase up to an additional $18,750 aggregate principal amount of the 2023 Notes, which was fully exercised on February 16, 2018. Net proceeds received by the Company, after issuance costs and discounts, were approximately $137,560.
The Company pays 4.50% cash interest per year on the principal amount of the 2023 Notes, payable semi-annually in arrears on February 1 and August 1 of each year, beginning on August 1, 2018, to holders of record at the close of business on the preceding January 15 or July 15, respectively. Interest accrues on the principal amount of the 2023 Notes from and including the date the 2023 Notes were issued or from, and including, the last date in respect of which interest has been paid or provided for, as the case may be, to, but excluding, the next interest payment date. The 2023 Notes are general, unsecured obligations of the Issuer, and are fully and unconditionally guaranteed by the Company on a senior unsecured basis. There are no financial debt covenants associated with the 2023 Notes.
The 2023 Notes are the Company’s senior unsecured obligations and rank equally in right of payment with all of the Company’s existing and future senior unsecured indebtedness and effectively junior to any of the Company’s existing and future secured indebtedness, to the extent of the value of the assets securing such indebtedness.
The 2023 Notes will be exchangeable at the option of the holders at an initial exchange rate of 92.6956 ADSs per $1 principal amount of 2023 Notes, which is equivalent to an initial exchange price of approximately $10.79 per ADS. Such initial exchange price represents a premium of approximately 20% to the $8.99 per ADS closing price on The Nasdaq Global Market on February 13, 2018. Upon the exchange of any 2023 Notes, the Issuer will pay or cause to be delivered, as the case may be, cash, ADSs or a combination of cash and ADSs, at the Issuer’s election. Holders of the 2023 Notes may convert their 2023 Notes, at their option, only under the following circumstances prior to the close of business on the business day immediately preceding August 1, 2022, under the circumstances and during the periods set forth below and regardless of the conditions described below, on or after August 1, 2022 and prior to the close of business on the business day immediately preceding the maturity date:
• | Prior to the close of business on the business day immediately preceding August 1, 2022, a holder of the 2023 Notes may surrender all or any portion of its 2023 Notes for exchange at any time during the five business day period immediately after any five consecutive trading day period (the “Measurement Period”) in which the trading price per $1 principal amount of 2023 Notes, as determined following a request by a holder of the 2023 Notes, for each trading day of the measurement period was less than 98% of the product of the last reported sale price of the ADSs and the exchange rate on each such trading day. |
• | If a transaction or event that constitutes a fundamental change or a make-whole fundamental change occurs prior to the close of business on the business day immediately preceding August 1, 2022, regardless of whether a holder of the 2023 Notes has the right to require the Company to repurchase the 2023 Notes, or if Avadel is a party to a merger event that |
-75-
occurs prior to the close of business on the business day immediately preceding August 1, 2022, all or any portion of a the holder’s 2023 Notes may be surrendered for exchange at any time from or after the date that is 95 scheduled trading days prior to the anticipated effective date of the transaction (or, if later, the earlier of (x) the business day after the Company gives notice of such transaction and (y) the actual effective date of such transaction) until 35 trading days after the actual effective date of such transaction or, if such transaction also constitutes a fundamental change, until the related fundamental change repurchase date.
• | Prior to the close of business on the business day immediately preceding August 1, 2022, a holder of the 2023 Notes may surrender all or any portion of its 2023 Notes for exchange at any time during any calendar quarter commencing after the calendar quarter ending on June 30, 2018 (and only during such calendar quarter), if the last reported sale price of the ADSs for at least 20 trading days (whether or not consecutive) during the period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the exchange price on each applicable trading day. |
• | If the Company calls the 2023 Notes for redemption pursuant to Article 16 to the Indenture prior to the close of business on the business day immediately preceding August 1, 2022, then a holder of the 2023 Notes may surrender all or any portion of its 2023 Notes for exchange at any time prior to the close of business on the second business day prior to the redemption date, even if the 2023 Notes are not otherwise exchangeable at such time. After that time, the right to exchange shall expire, unless the Company defaults in the payment of the redemption price, in which case a holder of the 2023 Notes may exchange its 2023 Notes until the redemption price has been paid or duly provided for. |
We considered the guidance in ASC 815-15, Embedded Derivatives, to determine if this instrument contains an embedded feature that should be separately accounted for as a derivative. ASC 815 provides for an exception to this rule when convertible notes, as host instruments, are deemed to be conventional, as defined by ASC 815-40. We determined that this exception applies due, in part, to our ability to settle the 2023 Notes in cash, ADSs or a combination of cash and ADSs, at our option. We have therefore applied the guidance provided by ASC 470-20, Debt with Conversion and Other Options which requires that the 2023 Notes be separated into debt and equity components at issuance and a value be assigned to each. The carrying amount of the liability component was calculated by measuring the fair value of a similar liability that does not have an associated convertible feature. The allocation was performed in a manner that reflected our non-convertible debt borrowing rate for similar debt. The equity component of the 2023 Notes was recognized as a debt discount and represents the difference between the proceeds from the issuance of the 2023 Notes and the fair value of the liability of the 2023 Notes on its issuance date. The excess of the principal amount of the liability component over its carrying amount (the “Debt Discount”) is amortized to interest expense using the effective interest method over the term of the 2023 Notes. The equity component is not remeasured as long as it continues to meet the conditions for equity classification.
In connection with the issuance of the 2023 Notes, we incurred approximately $6,190 of debt issuance costs, which primarily consisted of underwriting, legal and other professional fees, and allocated these costs to the liability and equity components based on the allocation of the proceeds. Of the total $6,190 of debt issuance costs, $1,201 were allocated to the equity component and recorded as a reduction to additional paid-in capital and $4,989 were allocated to the liability component and recorded as a reduction to debt on our consolidated balance sheets. The portion allocated to the liability component is amortized to interest expense using the effective interest method over the same five-year term as the related 2023 Notes.
-76-
NOTE 12: Long-Term Related Party Payable
Long-term related party payable and related activity are reported at fair value and consist of the following at December 31, 2019 and 2018, respectively:
Activity during the Twelve Months Ended December 31, 2019 | ||||||||||||||||||||
Changes in Fair Value of Related Party Payable | ||||||||||||||||||||
Balance, December 31, 2018 | Payments to Related Parties | Operating Expense | Other Expense | Balance, December 31, 2019 | ||||||||||||||||
Acquisition-related contingent consideration: | ||||||||||||||||||||
Earn-out payments - Éclat Pharmaceuticals (a) | $ | 25,615 | $ | (10,988 | ) | $ | 845 | $ | — | $ | 15,472 | |||||||||
Financing-related: | ||||||||||||||||||||
Royalty agreement - Deerfield (b) | 2,184 | (1,183 | ) | — | 250 | 1,251 | ||||||||||||||
Royalty agreement - Broadfin (c) | 1,041 | (565 | ) | — | 128 | 604 | ||||||||||||||
Total related party payable | 28,840 | $ | (12,736 | ) | $ | 845 | $ | 378 | 17,327 | |||||||||||
Less: Current portion | (9,439 | ) | (5,554 | ) | ||||||||||||||||
Total long-term related party payable | $ | 19,401 | $ | 11,773 | ||||||||||||||||
Each of the above items is associated with related parties as further described in Note 23: Related Party Transactions.
(a) | In March 2012, the Company acquired all of the membership interests of Éclat from Breaking Stick Holdings, L.L.C. (“Breaking Stick”, formerly Éclat Holdings), an affiliate of Deerfield. Breaking Stick is majority owned by Deerfield, with a minority interest owned by certain current and former employees. As part of the consideration, the Company committed to provide quarterly earn-out payments equal to 20% of any gross profit generated by certain Éclat products. These payments will continue in perpetuity, to the extent gross profit of the related products also continue in perpetuity. |
(b) | As part of a February 2013 debt financing transaction conducted with Deerfield, the Company received cash of $2,600 in exchange for entering into a royalty agreement whereby the Company shall pay quarterly a 1.75% royalty on the net sales of certain Éclat products until December 31, 2024. In connection with such debt financing transaction, the Company granted Deerfield a security interest in the product registration rights of the Éclat Pharmaceuticals products. |
(c) | As part of a December 2013 debt financing transaction conducted with Broadfin Healthcare Master Fund, a related party and current shareholder, the Company received cash of $2,200 in exchange for entering into a royalty agreement whereby the Company shall pay quarterly a 0.834% royalty on the net sales of certain Éclat products until December 31, 2024. |
At December 31, 2019, the fair value of each related party payable listed in (a), (b) and (c) above was estimated using a discounted cash flow model based on estimated and projected annual net revenues or gross profit, as appropriate, of each of the specified Éclat products using an appropriate risk-adjusted discount rate of 14%. These fair value measurements are based on significant inputs not observable in the market and thus represent a level 3 measurement as defined in ASC 820. Subsequent changes in the fair value of the acquisition-related related party payables, resulting primarily from management’s revision of key assumptions, will be recorded in the consolidated statements of (loss) income in the line items entitled “Changes in fair value of related party contingent consideration” for items noted in (b) above and in “Other (expense) income - changes in fair value of related party payable” for items (b) and (c) above. See Note 1: Summary of Significant Accounting Policies under the caption Acquisition-related Contingent Consideration and Financing-related Royalty Agreements for more information on key assumptions used to determine the fair value of these liabilities.
We have chosen to make a fair value election pursuant to ASC 825, “Financial Instruments” for its royalty agreements detailed in items (b) and (c) above. These financing-related liabilities are recorded at fair market value on the consolidated balance sheets and the periodic change in fair market value is recorded as a component of “Other (expense) income – changes in fair value of related party payable” on the consolidated statements of (loss) income.
-77-
The following table summarizes changes to the related party payables, a recurring Level 3 measurement, for the twelve-month periods ended December 31, 2019, 2018 and 2017:
Related Party Payable: | Balance | |||
Balance at December 31, 2016 | $ | 169,347 | ||
Payments of related party payable | (37,311 | ) | ||
Fair value adjustments (1) | (33,111 | ) | ||
Balance at December 31, 2017 | 98,925 | |||
Payments of related party payable | (22,951 | ) | ||
Fair value adjustments (1) | (24,630 | ) | ||
Expiration of warrants | (2,167 | ) | ||
Disposition of the pediatrics assets | (20,337 | ) | ||
Balance at December 31, 2018 | 28,840 | |||
Payments of related party payable | (12,736 | ) | ||
Fair value adjustments (1) | 1,223 | |||
Balance at December 31, 2019 | $ | 17,327 | ||
(1) Fair value adjustments are reported as “Changes in fair value of related party contingent consideration” and “Other (expense) income - changes in fair value of related party payable” in the consolidated statements of (loss) income.
NOTE 13: Income Taxes
The components of (loss) income before income taxes for the years ended twelve months ended December 31, are as follows:
(Loss) Income Before Income Taxes: | 2019 | 2018 | 2017 | |||||||||
Ireland | $ | (50,134 | ) | $ | (42,604 | ) | $ | (3,123 | ) | |||
U.S. | 10,401 | (70,340 | ) | 92,754 | ||||||||
France | 1,151 | (253 | ) | 3,029 | ||||||||
Total (loss) income before income taxes | $ | (38,582 | ) | $ | (113,197 | ) | $ | 92,660 | ||||
The income tax provision consists of the following for the years ended December 31:
Income Tax (Benefit) Provision: | 2019 | 2018 | 2017 | |||||||||
Current: | ||||||||||||
U.S. - Federal | $ | — | $ | — | $ | 18,064 | ||||||
U.S. - State | 97 | 330 | 331 | |||||||||
France | — | — | 265 | |||||||||
Total current | 97 | 330 | 18,660 | |||||||||
Deferred: | ||||||||||||
Ireland | (1,256 | ) | — | — | ||||||||
U.S. - Federal | (4,093 | ) | (19,503 | ) | 4,686 | |||||||
U.S. - State | (104 | ) | 1,280 | 1,043 | ||||||||
Total deferred | (5,453 | ) | (18,223 | ) | 5,729 | |||||||
Income tax (benefit) provision | $ | (5,356 | ) | $ | (17,893 | ) | $ | 24,389 | ||||
-78-
The reconciliation between income taxes at the statutory rate and the Company’s (benefit) provision for income taxes is as follows for the years ended December 31:
Reconciliation to Effective Income Tax Rate: | 2019 | 2018 | 2017 | |||||||||
Statutory tax rate | 12.5 | % | 12.5 | % | 12.5 | % | ||||||
Differences in international tax rates | 3.2 | % | 8.0 | % | 22.2 | % | ||||||
Nondeductible changes in fair value of contingent consideration | (0.3 | )% | 4.0 | % | (11.6 | )% | ||||||
Intercompany asset transfer | 21.2 | % | — | % | — | % | ||||||
Change in valuation allowances | (19.1 | )% | (5.3 | )% | (0.7 | )% | ||||||
Nondeductible stock-based compensation | (2.7 | )% | (1.3 | )% | (0.4 | )% | ||||||
Cross border merger | — | % | — | % | 0.3 | % | ||||||
Unrealized tax benefits | 0.7 | % | (1.3 | )% | 1.4 | % | ||||||
State and local taxes (net of federal) | — | % | (0.3 | )% | 0.3 | % | ||||||
Change in U.S. tax law | — | % | (0.2 | )% | 3.8 | % | ||||||
Nondeductible interest expense | (2.5 | )% | (1.1 | )% | — | % | ||||||
Other | 0.9 | % | 0.7 | % | (1.5 | )% | ||||||
Effective income tax rate | 13.9 | % | 15.7 | % | 26.3 | % | ||||||
Income tax (benefit) provision - at statutory tax rate | $ | (4,823 | ) | $ | (14,149 | ) | $ | 11,582 | ||||
Differences in international tax rates | (1,218 | ) | (9,039 | ) | 20,557 | |||||||
Nondeductible changes in fair value of contingent consideration | 121 | (4,559 | ) | (10,779 | ) | |||||||
Intercompany asset transfer | (8,190 | ) | — | — | ||||||||
Change in valuation allowances | 7,379 | 5,998 | (610 | ) | ||||||||
Nondeductible stock-based compensation | 1,039 | 1,499 | (375 | ) | ||||||||
Cross-border merger | — | — | 265 | |||||||||
Unrecognized tax benefits | (261 | ) | 1,440 | 1,296 | ||||||||
State and local taxes (net of federal) | (7 | ) | 299 | 252 | ||||||||
Change in U.S. tax law | — | 274 | 3,513 | |||||||||
Nondeductible interest expense | 982 | 1,269 | — | |||||||||
Other | (378 | ) | (925 | ) | (1,312 | ) | ||||||
Income tax (benefit) provision - at effective income tax rate | $ | (5,356 | ) | $ | (17,893 | ) | $ | 24,389 | ||||
In 2019, the income tax benefit decreased by $12,537 when compared to the same period in 2018. The decrease in the income tax benefit in 2019 was primarily driven by the impairment of the Noctiva intangible asset in 2018, which did not recur in 2019. In addition to the non-recurring impairment, an increase in the valuation allowance in 2019, when compared to the same period in 2018 also contributed to the decrease in tax benefit recorded in 2019. As a part of a corporate reorganization, the Company entered into an internal sale transaction in December 2019. The internal sale transaction included transfer of intangible assets from an Irish entity to a U.S. entity. The internal sale transaction resulted in a decrease of $5,536 to Irish deferred tax asset with corresponding decrease of $5,536 to valuation allowance, an increase of $8,190 to U.S. deferred tax asset associated with amortization of intangible assets, and a $8,190 deferred tax benefit.
In 2018, the income tax provision decreased by $42,282 when compared to the same period in 2017, resulting in an income tax benefit. The decrease in the income tax provision was primarily driven by a significant reduction in the amount of taxable income recorded in the U.S. and Ireland in 2018, when compared to 2017. There was also a significant increase in valuation allowance in 2018, when compared to the same period in 2017 as a result of this. In 2018, there was a significant decrease in amounts related to change in U.S. tax law due to the 2017 U.S. Tax Cuts and Jobs Act.
Unrecognized Tax Benefits
The Company or one of its subsidiaries files income tax returns in Ireland, France, U.S. and various states. The Company is no longer subject to Irish, French, U.S. Federal, and state and local examinations for years before 2015. The Internal Revenue Service (IRS) commenced an examination of the Company's U.S. income tax return for 2015 in the 4th quarter of 2016. In the second
-79-
quarter of 2019, the IRS added the 2016 and 2017 U.S. income tax return to the ongoing 2015 audit. The French tax authority commenced an examination of the Company's French tax return for 2017 in the first quarter of 2019.
The following table summarizes the activity related to the Company’s unrecognized tax benefits for the twelve months ended December 31:
Unrecognized Tax Benefit Activity | 2019 | 2018 | 2017 | |||||||||
Balance at January 1: | $ | 5,315 | $ | 3,954 | $ | 1,686 | ||||||
Additions based on tax positions related to the current year | — | 1,087 | 2,268 | |||||||||
Increases (decreases) for tax positions of prior years | 2,416 | 274 | — | |||||||||
Statute of limitations expiration | (1,266 | ) | — | — | ||||||||
Balance at December 31: | $ | 6,465 | $ | 5,315 | $ | 3,954 | ||||||
We expect that within the next twelve months the unrecognized tax benefits could decrease by $2,000 - $3,000. Additionally, interest could increase by up to $400.
At December 31, 2019, 2018, and 2017, there are $3,806, $4,597, and $3,349 of unrecognized tax benefits that if recognized would affect the annual effective tax rate.
We recognize interest and penalties accrued related to unrecognized tax benefits in income tax expense. During the years ended December 31, 2019, 2018, and 2017, we recognized approximately $555, $725, and $304 in interest and penalties. We had approximately $1,612, and $1,057 for the payment of interest and penalties accrued at December 31, 2019, and 2018, respectively.
Deferred Tax Assets (Liabilities)
Deferred income tax provisions reflect the effect of temporary differences between consolidated financial statement and tax reporting of income and expense items. The net deferred tax assets/liabilities at December 31, 2019 and 2018 resulted from the following temporary differences:
Net Deferred Tax Assets and Liabilities: | 2019 | 2018 | ||||||
Deferred tax assets: | ||||||||
Net operating loss carryforwards | $ | 30,275 | $ | 19,510 | ||||
Amortization | 11,602 | 6,830 | ||||||
Stock based compensation | 3,577 | 4,587 | ||||||
Accounts receivable | 53 | — | ||||||
Fair value contingent consideration | 264 | 384 | ||||||
Restructuring costs (Noctiva) | — | 13,812 | ||||||
Other | 901 | 479 | ||||||
Gross deferred tax assets | 46,672 | 45,602 | ||||||
Deferred tax liabilities: | ||||||||
Amortization | (172 | ) | (308 | ) | ||||
Accounts receivable | — | (661 | ) | |||||
Prepaid expenses | (35 | ) | (405 | ) | ||||
Gross deferred tax liabilities | (207 | ) | (1,374 | ) | ||||
Less: valuation allowances | (17,038 | ) | (21,199 | ) | ||||
Net deferred tax assets | $ | 29,427 | $ | 23,029 | ||||
At December 31, 2019, we had $95,771 of net operating losses in Ireland that do not have an expiration date and $75,537 of net operating loss in the U.S. Of the $75,537 of net operating loss in the U.S., $10,365 were acquired due to the acquisition of FSC and $65,172 is due to the losses at US Holdings. The portion due to the acquisition of FSC will expire in 2034 through 2035. A valuation allowance is recorded if, based on the weight of available evidence, it is more likely than not that a deferred tax asset
-80-
will not be realized. This assessment is based on an evaluation of the level of historical taxable income and projections for future taxable income. For the year ended December 31, 2019 we recorded $13,320 of valuation allowances related to Irish net operating losses and $309 of valuation allowances related to the U.S. net operating losses. The U.S. net operating losses are subject to an annual limitation as a result of the FSC acquisition under Internal Revenue Code Section 382 and will not be fully utilized before they expire.
We recorded a valuation allowance against all of our net operating losses in Ireland as of December 31, 2019, and all of our net operating losses in Ireland and France as of December 31, 2018. We intend to continue maintaining a full valuation allowance on the Irish net operating losses until there is sufficient evidence to support the reversal of all or some portion of these allowances. In 2019, we removed $3,259 of French net operating losses and the corresponding valuation allowance as a result of the 2019 restructuring activities in France. See Note 18: Restructuring Costs.
While we believe it is more likely than not that it will be able to realize the deferred tax assets in the U.S., we continue to monitor changes in the U.S. hospital products market as unfavorable changes could ultimately impact our assessment of the realizability of our U.S. deferred tax assets. If we experience an ownership change under Internal Revenue Code Section 382, the U.S. net operating losses could also be limited in their utilization.
At December 31, 2019, we have unremitted earnings of $3,961 outside of Ireland as measured on a U.S. GAAP basis. Whereas the measure of earnings for purposes of taxation of a distribution may be different for tax purposes, these earnings, which are considered to be invested indefinitely, would become subject to income tax if they were remitted as dividends or if we were to sell our stock in the subsidiaries, net of any prior income taxes paid. It is not practicable to estimate the amount of deferred tax liability on such earnings, if any.
Research and Development Tax Credits Receivable
The French and Irish governments provide tax credits to companies for spending on innovative R&D. These credits are recorded as an offset of R&D expenses and are credited against income taxes payable in years after being incurred or, if not so utilized, are recoverable in cash after a specified period of time, which may differ depending on the tax credit regime. As of December 31, 2019, our net research tax credit receivable amounts to $8,429 and represents a French gross research tax credit of $7,608 and an Irish gross research tax credit of $821. As of December 31, 2018, our net Research tax credit receivable amounts to $7,555 and represents a French gross research tax credit of $6,922 and an Irish gross research tax credit of $633.
Income Tax Deferred Charge
On December 16, 2014, we transferred all of our intangible intellectual property from our French entity to our Irish entity as part of a global reorganization. The intellectual property includes patents on drug delivery platforms, clinical data sets and other intangible assets related to the pipeline of proprietary products in development. This intra-entity transaction resulted in a charge of $14,088 of related taxes to the French government in December 2014. As this represents an intra-entity transaction, no deferred tax asset was originally recognized, but rather was recorded as $986 of prepaid expenses and $13,102 of a long-term income tax deferred charge asset in accordance with ASC 740-10-25-3 (e). This income tax deferred charge asset is amortized over the tax life of the asset at a rate of 7% per year and will result in tax relief in Ireland of $8,500 from 2016 to 2029, subject to the ability to realize tax benefits for additional deductions. At December 31, 2016, the balance of these respective accounts was classified as prepaid expenses of $814 and income tax deferred charge asset of $10,342. In 2017, we adopted the provisions of ASU 2016-16, related to Intra-Entity Transfers of Assets Other Than Inventory. Adoption of ASU 2016-16 eliminated the $11,156 income tax deferred charge recorded within the consolidated balance sheet as of December 31, 2016. In addition to the elimination of the income tax deferred charge, we recorded a deferred tax asset of $7,954 related to the remaining unamortized tax basis of the intangible intellectual property. A full valuation allowance was recorded against the deferred tax asset as sufficient evidence does not exist at this time that we will be able to utilize these benefits.
2017 Tax Cuts and Jobs Act
On December 22, 2017, the U.S. government enacted the Tax Cuts and Jobs Act (the “Tax Act”). The Tax Act includes significant changes to the U.S. corporate income tax system including: a federal corporate rate reduction from 35% to 21%; limitations on the deductibility of interest expense and executive compensation; creation of the base erosion anti-abuse tax (“BEAT”) and a new minimum tax. As a result of the Act being signed into law, we recognized a provisional charge of $274 and $3,513 in 2018 and 2017, respectively, related to the re-measurement of its U.S. net deferred tax assets and certain unrecognized tax benefits at the lower enacted corporate tax rates. A majority of the provisions in the Tax Act were effective January 1, 2018.
-81-
NOTE 14: Post-Retirement Benefit Plans
Post-Retirement Benefit Contributions to French Government Agencies
The Company is required by French law for our French employees to deduct specific monthly payroll amounts to support post-retirement benefit programs sponsored by the relevant government agencies in France. As the ultimate obligation is maintained by the French government agencies, there is no additional liability recorded by the Company in connection with this plan. Expenses recognized for this plan were $288 in 2019, $356 in 2018, and $606 in 2017.
Retirement Indemnity Obligation – France
French law requires the Company to provide for the payment of a lump sum retirement indemnity to French employees based upon years of service and compensation at retirement. The retirement indemnity has been actuarially calculated on the assumption of voluntary retirement at a government-defined retirement age. Benefits do not vest prior to retirement. Any actuarial gains or losses are recognized in the Company’s consolidated statements of (loss) income in the periods in which they occur.
During the second quarter of 2019, the Company initiated a plan to substantially reduce all of its workforce at its Vénissieux, France site (“2019 French Restructuring”). As a result of this decision, the Company reversed the French retirement indemnity obligation during the year ended December 31, 2019. See Note 18: Restructuring Costs.
The benefit obligation is calculated as the present value of estimated future benefits to be paid, using the following assumptions for the years ended December 31:
Retirement Benefit Obligation Assumptions: | 2018 | 2017 | ||||
Compensation rate increase | 2.75 | % | 3.00 | % | ||
Discount rate | 1.50 | % | 1.25 | % | ||
Employee turn-over | Actuarial standard and average of the last 5 years | |||||
Average age of retirement | 60 to 65 years actuarial standard based on age and professional status | |||||
Certain actuarial assumptions, such as discount rate, have a significant effect on the amounts reported for net periodic benefit cost and accrued retirement indemnity benefit obligation amounts. The discount rate is determined annually by benchmarking a published long-term bond index using the iBoxx € Corporates AA 10+ index.
Changes in the funded status of the retirement indemnity benefit plans were as follows for the years ended December 31:
Retirement Benefit Obligation Activity: | 2019 | 2018 | ||||||
Retirement indemnity benefit obligation, beginning of year | $ | 1,024 | $ | 1,303 | ||||
Service cost | — | 93 | ||||||
Interest cost | — | 17 | ||||||
Plan amendment | — | — | ||||||
Benefits paid | — | (12 | ) | |||||
Curtailment gain | (1,000 | ) | (148 | ) | ||||
Actuarial loss | — | (178 | ) | |||||
Exchange rate changes | (24 | ) | (51 | ) | ||||
Retirement indemnity benefit obligation, end of year | $ | — | $ | 1,024 | ||||
The lump sum retirement indemnity was accrued at December 31, 2018 on the Company’s consolidated balance sheets within non-current other liabilities, excluding the current portion. As these are not funded benefit plans, there are no respective assets recorded.
Due to the 2019 French Restructuring plan, at December 31, 2019 there are no future expected retirement indemnity benefits to be paid. See Note 18: Restructuring Costs.
-82-
NOTE 15: Other Assets and Liabilities
Various other assets and liabilities are summarized for the years ended December 31, as follows:
Prepaid Expenses and Other Current Assets: | 2019 | 2018 | ||||||
Valued-added tax recoverable | $ | 1,051 | $ | 1,378 | ||||
Prepaid and other expenses | 2,116 | 2,145 | ||||||
Guarantee from Armistice (see Note 17) | 454 | 534 | ||||||
Income tax receivable | 536 | 921 | ||||||
Short-term deposit | — | 3,350 | ||||||
Other | 107 | 225 | ||||||
Total | $ | 4,264 | $ | 8,553 | ||||
Other Non-Current Assets: | 2019 | 2018 | ||||||
Deferred tax assets | $ | 29,427 | $ | 23,029 | ||||
Long-term deposits | 1,477 | 1,477 | ||||||
Guarantee from Armistice (see Note 17) | 1,367 | 5,697 | ||||||
Right of use assets at contract manufacturing organizations | 6,428 | 5,894 | ||||||
Other | 575 | 49 | ||||||
Total | $ | 39,274 | $ | 36,146 | ||||
Accrued Expenses: | 2019 | 2018 | ||||||
Accrued compensation | $ | 3,944 | $ | 3,971 | ||||
Accrued social charges | 592 | 1,009 | ||||||
Accrued restructuring (see Note 18) | 2,949 | 879 | ||||||
Customer allowances | 6,470 | 6,541 | ||||||
Accrued contract research organization charges | 2,098 | 1,000 | ||||||
Accrued contract manufacturing organization costs | 735 | 2,028 | ||||||
Accrued contract sales organization and marketing costs | — | 3,469 | ||||||
Other | 3,022 | 2,798 | ||||||
Total | $ | 19,810 | $ | 21,695 | ||||
Other Non-Current Liabilities: | 2019 | 2018 | ||||||
Provision for retirement indemnity | $ | — | $ | 1,024 | ||||
Customer allowances | 981 | 1,352 | ||||||
Unrecognized tax benefits | 6,465 | 5,315 | ||||||
Guarantee to Deerfield (see Note 17) | 1,372 | 5,717 | ||||||
Other | 55 | 594 | ||||||
Total | $ | 8,873 | $ | 14,002 | ||||
NOTE 16: Contingent Liabilities and Commitments
Litigation
The Company is subject to potential liabilities generally incidental to our business arising out of present and future lawsuits and claims related to product liability, personal injury, contract, commercial, intellectual property, tax, employment, compliance and other matters that arise in the ordinary course of business. The Company accrues for potential liabilities when it is probable that future costs (including legal fees and expenses) will be incurred and such costs can be reasonably estimated. At December 31, 2019 and December 31, 2018, there were no contingent liabilities with respect to any litigation, arbitration or administrative or
-83-
other proceeding that are reasonably likely to have a material adverse effect on the Company’s consolidated financial position, results of operations, cash flows or liquidity.
Note 3: Subsidiary Bankruptcy and Deconsolidation briefly describes the Chapter 11 bankruptcy case which our subsidiary Specialty Pharma commenced on February 6, 2019, and which on April 26, 2019 resulted in the bankruptcy court-approved sale of all of Specialty Pharma’s intangible assets and inventory to an unaffiliated third party. As a result of such sale, Specialty Pharma has completed its divestment of the assets of the Noctiva business. During the pendency of the bankruptcy case, all pending litigation against Specialty Pharma is automatically stayed and any new litigation against Specialty Pharma is precluded unless the bankruptcy court orders otherwise. Below are descriptions of a litigation to which Specialty Pharma is a party and a contract dispute involving Specialty Pharma, both of which matters are subject to the automatic stay during the bankruptcy case.
Ferring Litigation. Some of the patents covering the Noctiva product (the “Noctiva Patents”) are the subject of litigation initiated by Ferring Pharmaceuticals Inc. and two of its foreign affiliates, who manufacture a competing product known as Nocdurna. Nocdurna was approved by the FDA in June 2018 and commercially launched in the U.S. in November 2018. In this litigation, filed in the United States District Court for the Southern District of New York, Ferring seeks to invalidate and disputes the inventorship of the Noctiva Patents, seeks damages for various alleged breaches of contractual and common law duties, and seeks damages for alleged infringement by Noctiva of Ferring’s “Nocdurna” trademark. Specialty Pharma and certain other parties including Serenity Pharmaceuticals, LLC (“Serenity”) (the licensor of the Noctiva Patents) have defended this litigation, and have made counterclaims against Ferring, including for infringement of the Noctiva Patents and a declaratory judgment of noninfringement with respect to Ferring’s “Nocdurna” trademark. The court dismissed Ferring’s inventorship claim and its claims for alleged breaches of contractual and common law duties, although these dismissals may be appealed by Ferring. On February 15, 2019, Specialty Pharma and its co-defendants moved to stay the litigation pending completion of the bankruptcy proceeding of Specialty Pharma. On May 15, 2019, that motion was denied due to a pending settlement of the litigation with respect to just Ferring and Specialty Pharma. On February 25, 2020, Ferring and Specialty Pharma jointly moved for bankruptcy court approval of a settlement agreement with respect to the claims alleged in the litigation. In accordance with the terms of the settlement agreement, promptly following bankruptcy court approval of the settlement agreement, the parties would dismiss with prejudice their respective claims against each other in the litigation. On March 13th, 2020, the bankruptcy court entered an order approving the settlement with Ferring. Pursuant to the terms of the settlement, the parties are to dismiss their respective claims against each other in the District Court litigation in the Southern District of New York, with such dismissals to be effective concurrently. That joint dismissal has not yet been filed with District Court for the Southern District of New York.
Contract Dispute. On January 21, 2019, Serenity gave notice to Specialty Pharma of an alleged breach of the parties’ Noctiva license agreement. Serenity alleges that Specialty Pharma breached its contractual obligation to devote commercially reasonable efforts to the commercialization of Noctiva and seeks unspecified damages. On January 27, 2019, Specialty Pharma notified Serenity of a claim for $1.7 million in damages as a result of Serenity’s breach of its contractual obligation to pay the costs of the Ferring Litigation. Serenity’s notice to Specialty Pharma invoked the dispute resolution provisions of the Noctiva license agreement, which culminate in arbitration, but neither party has yet initiated an arbitration proceeding or filed suit.
Exela Litigation. On January 7, 2020, Exela filed a complaint against us and our subsidiary, Avadel Legacy in the United States District for the District of Delaware. The complaint alleges infringement of a certain Exela patent related to its cysteine hydrochloride product. Exela is most notably seeking i) a declaratory judgment that the Nouress product infringes its patent, ii) an injunction (both preliminary and permanent) precluding the launch of Nouress, and iii) monetary damages (including enhanced damages, prejudgment interest and attorneys’ fees) in the event Nouress is commercially launched and found to infringe Exela’s patent. We have not yet been served with the complaint.
Former employee dispute. On January 10, 2020, we settled a dispute with a former employee for $1,750.
-84-
Material Commitments
At December 31, 2019, we have various commitments to purchase finished product from customers. For the year ended December 31, 2019, the Company paid $7,400 related to the below purchase commitments. Commitments for these arrangements, at maximum quantities and at contractual prices over the remaining life of the contract, and excluding any waived commitments, are as follows for the years ended December 31:
Purchase Commitments: | Balance | |||
2020 | $ | 1,434 | ||
2021 | 1,430 | |||
2022 | 1,430 | |||
2023 | 1,430 | |||
2024 | — | |||
Thereafter | — | |||
Total | $ | 5,724 | ||
The Company also has a commitment with a contract manufacturer related to the construction and preparation of a production suite at the contract manufacturer’s facility, which is substantially complete at December 31, 2019. Subsequent to the initial build and preparation of the production suite, this commitment also includes annual fees which would commence at the time of FDA approval of the product and continue thereafter for five years. These amounts are not included in the table above, as the start date has not been determined.
Leases
The Company and our subsidiaries lease office facilities under non-cancellable operating leases expiring at various dates. See Note 10: Leases for disclosure of these.
Other than the above commitments, there were no other material commitments outside of the normal course of business. Material commitments in the normal course of business include long-term debt and long-term related party payable, which are disclosed in Note 11: Long-Term Debt and Note 12: Long-Term Related Party Payable, respectively.
Contractual Obligations
The following table presents contractual obligations of the Company at December 31, 2019:
Payments Due by Period | ||||||||||||||||||||
Contractual Obligations: | Total | Less than 1 Year | 1 to 3 Years | 3 to 5 Years | More than 5 Years | |||||||||||||||
Long-term debt and interest | $ | 166,391 | $ | 6,469 | $ | 12,938 | $ | 146,984 | $ | — | ||||||||||
Long-term related party payable (undiscounted) | 29,847 | 5,554 | 5,181 | 4,262 | 14,850 | |||||||||||||||
Purchase commitments | 5,724 | 1,434 | 2,860 | 1,430 | — | |||||||||||||||
Operating leases | 3,368 | 779 | 1,167 | 1,216 | 206 | |||||||||||||||
Total contractual cash obligations | $ | 205,330 | $ | 14,236 | $ | 22,146 | $ | 153,892 | $ | 15,056 | ||||||||||
NOTE 17: Divestiture of the Pediatric Assets
On February 12, 2018, the Company, together with its subsidiaries Avadel Pharmaceuticals (USA), Inc., Avadel Pediatrics, Inc., FSC Therapeutics, LLC (“FSC Therapeutics”), and Avadel US Holdings, Inc. (“Holdings”), as the “Sellers,” entered into an asset purchase agreement (the “Purchase Agreement”) with Cerecor, Inc. (“Cerecor”). The transaction closed on February 16, 2018 wherein Cerecor purchased from the Sellers four pediatric commercial stage assets – Karbinal™ ER, Cefaclor, Flexichamber™ and AcipHex® Sprinkle™, together with certain associated business assets – which were held by FSC. The Company acquired FSC in February 2016 from Deerfield and certain of its affiliates. Pursuant to the Purchase Agreement, Cerecor assumed the Company’s remaining payment obligations to Deerfield under the Membership Interest Purchase Agreement, dated as of February 5, 2016, between Holdings, Flamel Technologies SA (the predecessor of the Company) and Deerfield and certain of its affiliates, which payment obligations consisted of the following (collectively, the “Assumed Obligations”): (i) a quarterly payment
-85-
of $263 beginning in July 2018 and ending in October 2020, amounting to an aggregate payment obligation of $2,625; (ii) a payment in January 2021 of $15,263; and (iii) a quarterly royalty payment of 15% on net sales of the FSC products through February 5, 2026 (“FSC Product Royalties”), in an aggregate amount of up to approximately $10,300. Cerecor also assumed certain contracts and other obligations related to the acquired assets, and in that connection Holdings agreed to pay Cerecor certain make-whole payments associated with obligations Cerecor is assuming related to a certain supply contract related to Karbinal™ ER.
In conjunction with the divestiture, the Company also entered into the following arrangements:
License and Development Agreement
Also, in connection with the closing under the Purchase Agreement, Flamel Ireland Limited, an Irish private limited company operating under the trade name of Avadel Ireland (“Avadel Ireland”) and a wholly-owned subsidiary of the Company, and Cerecor entered into a license and development agreement (the “License and Development Agreement”) pursuant to which, among other things:
• | Avadel Ireland will provide Cerecor with four product formulations utilizing Avadel Ireland’s LiquiTime™ technology, and will complete pilot bioequivalence studies for such product formulations within 18 months; |
• | Cerecor will reimburse Avadel Ireland for development costs of the four LiquiTime™ products in excess of $1,000 in the aggregate; |
• | Upon transfer of the four product formulations, Cerecor will assume all remaining development costs and responsibilities for the product development, clinical studies, NDA applications and associated filing fees; and |
• | Upon regulatory approval and commercial launch of any LiquiTime™ products, Cerecor will pay Avadel Ireland quarterly royalties based on a percentage of net sales of any such products in the mid-single digit range. |
Effective October 25, 2019, Cerecor and Avadel Ireland agreed to terminate the License and Development Agreement.
Deerfield Guarantee
In connection with the closing under the Purchase Agreement, the Company and Holdings provided their guarantee (the “Deerfield Guarantee”) in favor of Deerfield. Under the Deerfield Guarantee, the Company and Holdings guaranteed to Deerfield the payment by Cerecor of the Assumed Obligations under the Membership Interest Purchase Agreement between the Company and Deerfield dated February 5, 2016. The Assumed Obligations include (i) a quarterly payment of $263 beginning in July 2018 and ending in October 2020, amounting to an aggregate payment obligation of $2,625; (ii) a payment in January 2021 of $15,263; and (iii) a quarterly royalty payment of 15% on net sales of the FSC products through February 6, 2026 (“FSC Product Royalties”), in an aggregate amount of up to approximately $10,300. In addition, under the Deerfield Guarantee, the Company and Holdings guaranteed that Deerfield would receive certain minimum annual FSC Product Royalties through February 6, 2026 (the “Minimum Royalties”). Given the Company’s explicit guarantee to Deerfield, the Company recorded the guarantee in accordance with ASC 460. A valuation was performed, which was based largely on an analysis of the potential timing of each possible cash outflow described above and the likelihood of Cerecor’s default on such payments assuming an S&P credit rating of CCC+. The result of this valuation identified a guarantee liability of $6,643. This liability is being amortized proportionately based on undiscounted cash outflows through the remainder of the contract with Deerfield.
On October 10, 2019, Cerecor entered into a purchase and sale agreement with Aytu BioScience, Inc (“Buyer”) pursuant to which the Buyer will purchase certain assets from Cerecor and assume certain of Cerecor’s liabilities, including all of Cerecor’s liabilities assumed as part of the Purchase Agreement noted above. As part of this transaction, on November 1, 2019, Armistice has agreed to deposit $15,000 in an escrow account governed by an escrow agreement between Armistice and Deerfield having the purpose of securing the $15,000 balloon payment due January 2021 as part of the Membership Interest Purchase Agreement. As part of the Cerecor transaction with the buyer, Deerfield contractually acknowledges and agrees that it will seek payment from the escrow funds before requesting payment from the Company pursuant to the Deerfield Guarantee discussed above. Due to the change in circumstances, a new valuation was performed based on an analysis of the possible timing of the updated possible cash flow which excludes the $15,000 that Armistice has deposited in an escrow account. The updated valuation identified an updated guarantee liability of $1,827 at December 31, 2019, which will be amortized proportionately based on undiscounted cash outflows through the remainder of the contract with Deerfield.
-86-
Armistice Guarantee
In connection with the closing under the Purchase Agreement, Armistice Capital Master Fund, Ltd., the majority shareholder of Cerecor, guaranteed to Holdings the payment by Cerecor of the Assumed Obligations, including the Minimum Royalties. A valuation of the guarantee asset was performed in accordance with ASC 460 “Guarantees” and a guarantee asset of $6,620 was recorded. This asset is being amortized proportionately based on undiscounted cash outflows through the remainder of the contract with Deerfield noted above.
As discussed above, based on the purchase and asset sale between Cerecor and the Buyer, an updated valuation was performed and identified an updated guarantee asset of $1,821 at December 31, 2019.
The fair values of the Avadel guarantee to Deerfield and the guarantee received by Avadel from Armistice largely offset and when combined are not material.
Based on management’s review of ASU 2014-08, “Reporting Discontinued Operations and Disclosures of Disposals of Components of an Entity”, the disposition of our pediatric assets and related liabilities did not qualify for discontinued operations reporting. Our results of operations for the period January 1, 2018 through February 16, 2018 and for the years ended December 31, 2018 and 2017 include the results of FSC, prior to its February 16, 2018 disposition date.
The net impact of this transaction was not material to the consolidated statements of (loss) income.
NOTE 18: Restructuring Costs
2019 French Restructuring
During the second quarter of 2019, the Company initiated a plan to substantially reduce all of its workforce at its Vénissieux, France site (“2019 French Restructuring”). This reduction is an effort to align the Company’s cost structure with our ongoing and future planned projects. The reduction in workforce is substantially complete at December 31, 2019. Restructuring charges associated with this plan of $4,855 were recognized during the year ended December 31, 2019. Included in the 2019 French Restructuring charges of $4,855 were charges for employee severance, benefits and other costs of $4,339, charges related to fixed asset impairment of $629, charges related to the early termination penalty related to the office and copier lease terminations of $887 (see Note 10: Leases), partially offset by a benefit of $1,000 related to the reversal of the French retirement indemnity obligation. The following table sets forth activities for the Company’s cost reduction plan obligations for the year ended December 31, 2019:
2019 French Restructuring Obligation: | 2019 | |||
Balance of restructuring accrual at January 1, | $ | — | ||
Charges for employee severance, benefits and other costs | 4,339 | |||
Payments | (2,441 | ) | ||
Foreign currency impact | 24 | |||
Balance of restructuring accrual at December 31, | $ | 1,922 | ||
The 2019 French Restructuring liabilities of $1,849, $42 and $31 are included in the consolidated balance sheet in accrued expenses, other long-term liabilities and accounts payable at December 31, 2019, respectively.
2019 Corporate Restructuring
During the first quarter of 2019, the Company announced a plan to reduce its Corporate workforce by more than 50% (the “2019 Corporate Restructuring”). The reduction in workforce is primarily a result of the exit of Noctiva during the first quarter of 2019 (see Note 3: Subsidiary Bankruptcy and Deconsolidation), as well as an effort to better align the Company’s remaining cost structure at our U.S. and Ireland locations with our ongoing and future planned projects. The reduction in workforce is substantially complete at December 31, 2019. The restructuring charges associated with this plan of $1,755 were recognized during the year ended December 31, 2019, respectively. Included in the 2019 Corporate Restructuring charges of $1,755 for the year ended December 31, 2019, were charges for employee severance, benefit and other costs of $3,406, charges related to the early termination penalty related to the office lease termination of $288, the write-off of $125 of property, plant and equipment, net,
-87-
partially offset by a benefit of $2,064 related to share based compensation forfeitures related to the employees affected by the global reduction in workforce.
The following table sets forth activities for the Company’s cost reduction plan obligations for the year ended December 31, 2019:
2019 Corporate Restructuring Obligation: | 2019 | |||
Balance of restructuring accrual at January 1, | $ | — | ||
Charges for employee severance, benefits and other costs | 3,406 | |||
Payments | (2,326 | ) | ||
Balance of restructuring accrual at December 31, | $ | 1,080 | ||
The restructuring accrual at December 31, 2019 is included in the consolidated balance sheet in accrued expenses.
2017 French Restructuring
During the first quarter of 2017, the Company announced a plan to reduce its workforce at our Venissieux, France site by approximately 50% (the “2017 French Restructuring”). This reduction was an effort to align the Company’s cost structure with our ongoing and future planned projects. In July 2017, the Company completed negotiations with the works council for our French operations and received approval from the French Labor Commission (DIRECCTE) to implement the plan. The reduction was substantially complete at December 31, 2019. The 2017 French Restructuring benefit for the year ended December 31, 2019 was $169 and the 2017 French Restructuring expense for the year ended December 31, 2018 was $1,164.
The following table sets forth activities for the Company’s cost reduction plan obligations for the years ended December 31, 2019 and 2018:
2017 French Restructuring Obligation: | 2019 | 2018 | ||||||
Balance of restructuring accrual at January 1, | $ | 879 | $ | 1,000 | ||||
Charges for employee severance, benefits and other | (169 | ) | 1,164 | |||||
Payments | (673 | ) | (1,261 | ) | ||||
Foreign currency impact | (9 | ) | (24 | ) | ||||
Balance of restructuring accrual at December 31, | $ | 28 | $ | 879 | ||||
The restructuring accrual at December 31, 2019 and 2018 is included the consolidated balance sheets in accrued expenses and other long-term liabilities.
NOTE 19: Equity Instruments and Share-Based Compensation
Capital Shares
We have 500,000 shares of authorized ordinary shares with a nominal value of $0.01 per ordinary share. As of December 31, 2019, we had 42,927 and 37,520 ordinary shares issued and outstanding, respectively. The Board of Directors is authorized to issue preferred shares in series, and with respect to each series, to fix its designation, relative rights (including voting, dividend, conversion, sinking fund, and redemption rights), preferences (including dividends and liquidation) and limitations. We have 50,000 shares of authorized preferred shares, $0.01 nominal value, none of which is currently issued and outstanding.
Share Repurchases
In March 2017, the Board of Directors approved an authorization to repurchase up to $25,000 of Avadel ordinary shares represented by ADSs. Under this authorization, which has an indefinite duration, share repurchases may be made in the open market, in block transactions on or off the exchange, in privately negotiated transactions, or through other means as determined by the Board of Directors and in accordance with the regulations of the Securities and Exchange Commission. The timing and amount of repurchases, if any, will depend on a variety of factors, including the price of our shares, cash resources, alternative investment opportunities, corporate and regulatory requirements and market conditions. This share repurchase program may be modified, suspended or discontinued at any time without prior notice. We may also from time to time establish a trading plan under Rule 10b5-1 of the Securities and Exchange Act of 1934 to facilitate purchases of our shares under this program. Additionally, on
-88-
February 12, 2018, the Board of Directors approved an authorization to repurchase up to $18,000 of Avadel ordinary shares represented by American Depository Shares in connection with our Convertible Notes Offering completed on February 16, 2018. See Note 11: Long-Term Debt. In March 2018, the Board of Directors approved an authorization to repurchase up to $7,000 of Avadel ordinary shares represented by American Depository Shares, bring the total authorization to $50,000. There were no share repurchases during the year ended December 31, 2019. As of December 31, 2019, the Company had repurchased 5,407 ordinary shares for $49,998.
Share-Based Compensation
Compensation expense included in the Company’s consolidated statements of (loss) income for all share-based compensation arrangements was as follows for the periods ended December 31, 2019, 2018 and 2017, respectively:
Share-based Compensation Expense: | 2019 | 2018 | 2017 | |||||||||
Research and development | $ | 429 | $ | 880 | $ | 672 | ||||||
Selling, general and administrative | 2,154 | 6,972 | 7,400 | |||||||||
Restructuring costs | (2,064 | ) | — | — | ||||||||
Total share-based compensation expense | $ | 519 | $ | 7,852 | $ | 8,072 | ||||||
As of December 31, 2019, the Company expects $2,695 of unrecognized expense related to granted, but non-vested share-based compensation arrangements to be incurred in future periods. This expense is expected to be recognized over a weighted average period of 2.4 years.
The excess tax benefit related to share-based compensation recorded by the Company was not material for the years ended December 31, 2019, 2018 and 2017.
Upon exercise of stock options or warrants, or upon the issuance of restricted share awards, the Company issues new shares.
At December 31, 2019, there were 565,716 shares authorized for stock option grants, warrant grants and restricted share award grants in subsequent periods.
Determining the Fair Value of Stock Options
The Company measures the total fair value of stock options on the grant date using the Black-Scholes option-pricing model and recognizes each grant’s fair value as compensation expense over the period that the option vests. Options are granted to employees of the Company and become exercisable ratably over four years following the grant date and expire ten years after the grant date. Prior to 2017, warrants were typically issued to the Company’s Board of Directors as compensation for services rendered and generally become exercisable within one year following the grant date, and expire four years after the grant date. Beginning in 2017, the Company issues stock options to our Board of Directors as compensation for services rendered and generally become exercisable within one year following the grant date, and expire four years after the grant date.
The weighted-average assumptions under the Black-Scholes option-pricing model for stock option grants as of December 31, 2019, 2018 and 2017, are as follows:
Stock Option Assumptions: | 2019 | 2018 | 2017 | ||||||
Stock option grants: | |||||||||
Expected term (years) | 6.25 | 6.25 | 6.25 | ||||||
Expected volatility | 56.48 | % | 56.59 | % | 58.82 | % | |||
Risk-free interest rate | 2.52 | % | 2.68 | % | 2.20 | % | |||
Expected dividend yield | — | — | — | ||||||
Expected term: The expected term of the options represents the period of time between the grant date and the time the options are either exercised or forfeited, including an estimate of future forfeitures for outstanding options. Given the limited historical data and the grant of stock options to a limited population, the simplified method has been used to calculate the expected life.
Expected volatility: The expected volatility is calculated based on an average of the historical volatility of the Company’s stock price for a period approximating the expected term.
-89-
Risk-free interest rate: The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant and a maturity that approximates the expected term.
Expected dividend yield: We have not distributed any dividends since our inception, and has no plan to distribute dividends in the foreseeable future.
Stock Options
A summary of the combined stock option activity and other data for the Company’s stock option plans for the year ended December 31, 2019 is as follows:
Stock Option Activity and Other Data: | Number of Stock Options | Weighted Average Exercise Price per Share | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||
Stock options outstanding, January 1, 2019 | 4,601 | $ | 11.39 | ||||||||||
Granted | 2,631 | 2.24 | |||||||||||
Exercised | — | — | |||||||||||
Forfeited | (1,333 | ) | 7.37 | ||||||||||
Expired | (778 | ) | 12.91 | ||||||||||
Stock options outstanding, December 31, 2019 | 5,121 | $ | 7.51 | 7.43 years | $ | 12,119 | |||||||
Stock options exercisable, December 31, 2019 | 2,636 | $ | 11.63 | 5.73 years | $ | 572 | |||||||
The aggregate intrinsic value of options exercisable at December 31, 2019, 2018 and 2017 was $572, $0, and $1,161, respectively.
The weighted average grant date fair value of options granted during the years ended December 31, 2019, 2018 and 2017 was $1.24, $3.60 and $5.20 per share, respectively.
Warrants
A summary of the combined warrant activity and other data for the year ended December 31, 2019 is as follows:
Warrant Activity and Other Data: | Number of Warrants | Weighted Average Exercise Price per Share | Weighted Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||
Warrants outstanding, January 1, 2019 | 596 | $ | 17.72 | ||||||||||
Granted | — | — | |||||||||||
Exercised | — | — | |||||||||||
Forfeited | — | — | |||||||||||
Expired | (305 | ) | 21.67 | ||||||||||
Warrants outstanding, December 31, 2019 | 291 | $ | 13.59 | 0.61 years | $ | — | |||||||
Warrants exercisable, December 31, 2019 | 291 | $ | 13.59 | 0.61 years | $ | — | |||||||
Each of the above warrants is convertible into one ordinary share. There was no aggregate intrinsic value of warrants exercised during the years ended December 31, 2019, 2018 and 2017.
There were no warrants granted during the years ended December 31, 2019, 2018 and 2017.
At January 1, 2018, an additional 3,300 warrants were outstanding and exercisable relative to consideration paid for the Company’s acquisition of Éclat Pharmaceuticals on March 13, 2012. These warrants are not considered share-based compensation and are therefore excluded from the above tables, and instead are addressed within Note 12: Long-Term Related Party Payable. On February 23, 2018, the related party exercised in full the warrant to purchase 2,200 ordinary shares. On March 12, 2018 the remaining warrants to purchase 1,100 ordinary shares expired.
Restricted Share Awards
Restricted share awards represent Company shares issued free of charge to employees of the Company as compensation for services rendered. The Company measures the total fair value of restricted share awards on the grant date using the Company’s stock price at the time of the grant. Restricted share awards granted during and after 2017 vest over a three-year period; two-thirds (2/3)
-90-
vesting on the second anniversary of the grant date and the remaining one-third (1/3) vesting on the third anniversary of the grant date. Beginning in 2018, the Company issues restricted share awards to our Board of Directors vesting over a three-year period; one-third (1/3) vesting on each of the three anniversaries of the grant date. Compensation expense for such awards granted during and after 2017 is recognized over the applicable vesting period.
A summary of the Company’s restricted share awards as of December 31, 2019, and changes during the year then ended, is reflected in the table below.
Restricted Share Activity and Other Data: | Number of Restricted Share Awards | Weighted Average Grant Date Fair Value | |||||
Non-vested restricted share awards outstanding, January 1, 2019 | 491 | $ | 7.20 | ||||
Granted | 251 | 2.47 | |||||
Vested | (153 | ) | 7.50 | ||||
Forfeited | (242 | ) | 5.65 | ||||
Non-vested restricted share awards outstanding, December 31, 2019 | 347 | $ | 4.73 | ||||
The weighted average grant date fair value of restricted share awards granted during the years ended December 31, 2019, 2018 and 2017 was $2.47, $5.87 and $8.95, respectively.
Employee Share Purchase Plan
In 2017, the Board of Directors approved of the Avadel Pharmaceuticals plc 2017 Avadel Employee Share Purchase Plan (“ESPP”). The total number of Company ordinary shares, nominal value $0.01 per share, or ADSs representing such ordinary shares (collectively, “Shares”) which may be issued under the ESPP is 1,000. The purchase price at which a Share will be issued or sold for a given offering period will be established by the Compensation Committee of the Board (“Committee”) (and may differ among participants, as determined by the Committee in its sole discretion) but will in no event be less than 85% of the lesser of: (a) the fair market value of a Share on the offering date; or (b) the fair market value of a Share on the purchase date. During the years ended December 31, 2019 and 2018, the Company issued 54 and 25 ordinary shares to employees, respectively. Expense related to the ESPP for the years ended December 31, 2019 and 2018 was immaterial.
NOTE 20: Net (Loss) Income Per Share
Basic net (loss) income per share is calculated by dividing net (loss) income by the weighted average number of shares outstanding during each period. Diluted net (loss) income per share is calculated by dividing net (loss) income by the diluted number of shares outstanding during each period. Except where the result would be anti-dilutive to net (loss) income, diluted net (loss) income per share would be calculated assuming the impact of the conversion of the 2023 Notes, the exercise of outstanding equity compensation awards, ordinary shares expected to be issued under our ESPP and the exercise of contingent consideration warrants, all of which have been exercised or have expired during the first quarter of 2018.
We have a choice to settle the conversion obligation under the 2023 Notes in cash, shares or any combination of the two. We utilize the if-converted method to reflect the impact of the conversion of the 2023 Notes, unless the result is anti-dilutive. This method assumes the conversion of the 2023 Notes into shares of our ordinary shares and reflects the elimination of the interest expense related to the 2023 Notes.
The dilutive effect of the warrants, stock options, RSU’s and ordinary shares expected to be issued under our ESPP has been calculated using the treasury stock method.
-91-
A reconciliation of basic and diluted net (loss) income per share, together with the related shares outstanding in thousands for the years ended December 31, 2019, 2018 and 2017, is as follows:
Net (Loss) Income Per Share: | 2019 | 2018 | 2017 | |||||||||
Net (loss) income | $ | (33,226 | ) | $ | (95,304 | ) | $ | 68,271 | ||||
Weighted average shares: | ||||||||||||
Basic shares | 37,403 | 37,325 | 40,465 | |||||||||
Effect of dilutive securities—employee and director equity awards outstanding and 2023 Notes | — | — | 1,300 | |||||||||
Diluted shares | 37,403 | 37,325 | 41,765 | |||||||||
Net (loss) income per share - basic | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.69 | ||||
Net (loss) income per share - diluted | $ | (0.89 | ) | $ | (2.55 | ) | $ | 1.63 | ||||
Potential ordinary shares of 16,740, 17,529, and 6,368 were excluded from the calculation of weighted average shares for the years ended December 31, 2019, 2018 and 2017, respectively, because their effect was considered to be anti-dilutive. For the years ended December 31, 2019 and 2018, the effects of dilutive securities were entirely excluded from the calculation of net (loss) income per share as a net loss was reported in these periods.
NOTE 21: Comprehensive (Loss) Income
The following table shows the components of accumulated other comprehensive (loss) income for the year ended December 31, net of immaterial tax effects:
Accumulated Other Comprehensive (Loss) Income: | 2019 | 2018 | 2017 | |||||||||
Foreign currency translation adjustment: | ||||||||||||
Beginning balance | $ | (23,621 | ) | $ | (23,202 | ) | $ | (23,336 | ) | |||
Net other comprehensive (loss) income | (117 | ) | (419 | ) | 134 | |||||||
Balance at December 31, | (23,738 | ) | (23,621 | ) | (23,202 | ) | ||||||
Unrealized gain (loss) on marketable securities, net | ||||||||||||
Beginning balance | 205 | (64 | ) | (229 | ) | |||||||
Net other comprehensive income, net of ($43), ($18), $28, tax, respectively | 727 | 269 | 165 | |||||||||
Balance at December 31, | 932 | 205 | (64 | ) | ||||||||
Accumulated other comprehensive loss at December 31, | $ | (22,806 | ) | $ | (23,416 | ) | $ | (23,266 | ) | |||
-92-
NOTE 22: Company Operations by Product, Customer and Geographic Area
We have determined that we operate in one segment, the development and commercialization of pharmaceutical products, including controlled-release therapeutic products based on our proprietary polymer based technology. The Company’s Chief Operating Decision Maker is the CEO. The CEO reviews profit and loss information on a consolidated basis to assess performance and make overall operating decisions as well as resource allocations. All products are included in one segment because the Company’s products have similar economic and other characteristics, including the nature of the products and production processes, type of customers, distribution methods and regulatory environment.
The following table presents a summary of total revenues by these products for the twelve months ended December 31, 2019, 2018, and 2017:
Revenue by Product: | 2019 | 2018 | 2017 | |||||||||
Bloxiverz | $ | 7,479 | $ | 20,850 | $ | 45,596 | ||||||
Vazculep | 33,152 | 42,916 | 38,187 | |||||||||
Akovaz | 18,642 | 33,759 | 80,617 | |||||||||
Other | (58 | ) | 3,898 | 8,441 | ||||||||
Product sales | 59,215 | 101,423 | 172,841 | |||||||||
License revenue | — | 1,846 | 404 | |||||||||
Total revenues | $ | 59,215 | $ | 103,269 | $ | 173,245 | ||||||
Concentration of credit risk with respect to accounts receivable is limited due to the high credit quality comprising a significant portion of the Company’s customers. Management periodically monitors the creditworthiness of our customers and believes that we have adequately provided for any exposure to potential credit loss.
The following table presents a summary of total revenues by significant customer for the twelve months ended December 31, 2019, 2018, and 2017:
Revenue by Significant Customer: | 2019 | 2018 | 2017 | |||||||||
Cardinal Health | $ | 15,088 | $ | 25,413 | $ | 37,965 | ||||||
McKesson Corporation | 14,900 | 26,794 | 44,762 | |||||||||
AmerisourceBergen | 12,059 | 18,620 | 25,691 | |||||||||
Others | 17,168 | 30,596 | 64,423 | |||||||||
Product sales | 59,215 | 101,423 | 172,841 | |||||||||
License revenue | — | 1,846 | 404 | |||||||||
Total revenues | $ | 59,215 | $ | 103,269 | $ | 173,245 | ||||||
In 2017, 31% of product sales was sold to PharMedium. In 2018 and 2019, product sales to PharMedium was less than 10%.
As of December 31, 2019, the Company had three customers, each of which are substantial wholesale distributors, and accounted for 10% or more of the accounts receivable balance. One customer accounted for 40%, or $3,346, a second customer accounted for 29% or $2,416, and a third customer accounted for 11% or $949. As of December 31, 2019, the Company had no significant past due account receivable balances.
The following table summarizes revenues by geographic region for the twelve months ended December 31, 2019, 2018, and 2017:
Revenue by Geographic Region: | 2019 | 2018 | 2017 | |||||||||
U.S. | $ | 59,215 | $ | 101,423 | $ | 172,841 | ||||||
Ireland | — | 1,846 | 404 | |||||||||
Total revenues | $ | 59,215 | $ | 103,269 | $ | 173,245 | ||||||
Currently, we depend on a single contract manufacturing organization for the manufacture of Bloxiverz and Vazculep and two contract manufacturing organizations for the manufacture of Akovaz, from which we derive a majority of our revenues. Additionally,
-93-
we purchase certain raw materials used in our products from a limited number of suppliers, including a single supplier for certain key ingredients.
Non-monetary long-lived assets primarily consist of property and equipment, goodwill and intangible assets. The following table summarizes non-monetary long-lived assets by geographic region as of December 31, 2019, 2018, and 2017:
Long-lived Assets by Geographic Region: | 2019 | 2018 | 2017 | |||||||||
U.S. | $ | 22,254 | $ | 27,761 | $ | 116,536 | ||||||
France | 196 | 1,365 | 2,257 | |||||||||
Ireland | 7,244 | 6,028 | 1,360 | |||||||||
Total | $ | 29,694 | $ | 35,154 | $ | 120,153 | ||||||
NOTE 23: Related Party Transactions
In March 2012, the Company acquired all of the membership interests of Éclat from Breaking Stick Holdings, L.L.C. (“Breaking Stick”, formerly Éclat Holdings), an affiliate of Deerfield Capital L.P (“Deerfield”), a significant shareholder of the Company. During the three months ended December 31, 2019, Deerfield sold all of their shares. At December 31, 2019, the remaining consideration obligation for this transaction consisted of commitments to make earnout payments to Breaking Stick of 20% of any gross profit generated by certain Éclat products (the “Products”). Breaking Stick is majority owned by Deerfield, with a minority interest owned by certain current and former employees. The Company entered into a Security Agreement dated March 13, 2012 with Breaking Stick, whereby Breaking Stick was granted a security interest in various tangible and intangible assets related to the Products to secure the obligations of Éclat and Avadel US Holdings, Inc., including the full and prompt payment of royalties to Breaking Stick under the Royalty Agreement.
As part of a February 2013 debt financing transaction conducted with Deerfield Management, Éclat entered into a Royalty Agreement with Horizon Santé FLML, Sarl and Deerfield Private Design Fund II, L.P., both affiliates of the Deerfield Entities (together, “Deerfield PDF/Horizon”). The Royalty Agreement provides for the Company to pay Deerfield PDF/Horizon 1.75% of the net sales of the Products sold by the Company and any of our affiliates until December 31, 2024, with royalty payments paid in arrears for each calendar quarter during the term of the Royalty Agreement. The Company has also entered into a Security Agreement dated February 4, 2013 with Deerfield PDF/Horizon, whereby Deerfield PDF/Horizon was granted a security interest in the various tangible and intangible assets related to the Products to secure the obligations of Éclat and Avadel US Holdings, Inc., including the full and prompt payment of royalties to Deerfield PDF/Horizon under the Royalty Agreement.
As part of a December 2013 debt financing transaction conducted with Broadfin Healthcare Master Fund (“Broadfin”), the Company also entered into a Royalty Agreement with Broadfin, a significant shareholder of the Company, dated as of December 3, 2013 (the “Broadfin Royalty Agreement”). During the three months ended December 31, 2019, Broadfin sold a majority of their shares and was not a significant shareholder at December 31, 2019. Pursuant to the Broadfin Royalty Agreement, the Company is required to pay a royalty of 0.834% on the net sales of certain products sold by the Company and any of our affiliates until December 31, 2024 with royalty payments paid in arrears for each calendar quarter during the term of the Royalty Agreement. We also entered into a Security Agreement dated December 3, 2013 with Broadfin, whereby Broadfin was granted a security interest in the various tangible and intangible assets related to the Products to secure the obligations of Éclat and Avadel US Holdings, Inc., including the full and prompt payment of royalties to Broadfin under the Royalty Agreement.
The Company entered into an agreement dated February 5, 2016 to acquire FSC Holdings, LLC (“FSC”), a specialty pharmaceutical company dedicated to providing innovative solutions to unmet medical needs for pediatric patients, from Deerfield CSF, LLC, a Deerfield Management company (“Deerfield”), a related party. Under the terms of the acquisition, which was completed on February 8, 2016, the Company was to pay $1,050 annually for five years with a final payment in January 2021 of $15,000 for a total of $20,250 to Deerfield for all of the equity interests in FSC. The Company will also pay Deerfield a 15% royalty per annum on net sales of the current FSC products, up to $12,500 for a period not exceeding ten years. These obligations were assumed by Cerecor in connection with the divestiture of the Company’s pediatric products on February 16, 2018. In connection with the divestiture, the Company provided its guarantee in favor of Deerfield and in return, Armistice Capital Master Fund, Inc., the majority shareholder of Cerecor, guaranteed to the Company the payment by Cerecor of the Assumed Obligations mentioned in Note 17: Divestiture of the Pediatric Assets. On October 10, 2019, Cerecor entered into a purchase and sale agreement with Aytu BioScience, Inc (“Buyer”) pursuant to which the Buyer will purchase certain assets from Cerecor and assume certain of Cerecor’s liabilities, including all of Cerecor’s liabilities assumed as part of the Purchase Agreement discussed in Note 17. As part of this transaction, on November 1, 2019, Armistice has agreed to deposit $15,000 in an escrow account governed by an escrow agreement between Armistice and Deerfield having the purpose of securing the $15,000 balloon payment due January 2021 as part of the Membership Interest Purchase Agreement. As part of the Cerecor transaction with the buyer, Deerfield contractually acknowledges
-94-
and agrees that it will seek payment from the escrow funds before requesting payment from the Company pursuant to the Deerfield Guarantee discussed above. Due to the change in circumstances, another valuation was performed based on an analysis of the possible timing of the updated possible cash flow which excludes the $15,000 that Armistice has deposited in an escrow account. See Note 17 for further discussion around the divestiture.
NOTE 24: Subsequent Events
Exela Litigation
On January 7, 2020, Exela filed a complaint against the Company and its subsidiary, Avadel Legacy in the United States District for the District of Delaware. The complaint alleges infringement of a certain Exela patent by Avadel’s NOURESS™ product, which has not yet been commercially launched in the U.S. Exela is most notably seeking i) a declaratory judgment that the NOURESS™ product infringes its patent, ii) an injunction (both preliminary and permanent) precluding the launch of NOURESS™, and iii) monetary damages (including enhanced damages, prejudgment interest and attorneys’ fees) in the event NOURESS™ is commercially launched and found to infringe Exela’s patent. The Company and Avadel Legacy have not yet been served with the complaint.
Shelf Registration Statement on Form S-3
In February 2020, we filed with the SEC a new shelf registration statement on Form S-3 (the 2020 Shelf Registration Statement) (File No. 333-236258) that allows issuance and sale by us, from time to time, of:
(a) | up to $250,000 in aggregate of ordinary shares, nominal value US$0.01 per share (the “Ordinary Shares”), each of which may be represented by ADSs, preferred shares, nominal value US$0.01 per share (the “Preferred Shares”), debt securities (the “Debt Securities”), warrants to purchase Ordinary Shares, ADSs, Preferred Shares and/or Debt Securities (the “Warrants”), and/or units consisting of Ordinary Shares, ADSs, Preferred Shares, one or more Debt Securities or Warrants in one or more series, in any combination, pursuant to the terms of the 2020 Shelf Registration Statement, the base prospectus contained in the 2020 Shelf Registration Statement (the “Base Prospectus”), and any amendments or supplements thereto (together, the “Securities”); including |
(b) | up to $50,000 of ADSs that may be issued and sold from time to time pursuant to the terms of an Open Market Sale AgreementSM (“the Sales Agreement”), entered into with Jefferies LLC on February 4, 2020 (the “Sales Agreement”), the 2020 Shelf Registration Statement, the Base Prospectus and the terms of the sales agreement prospectus contained in the 2020 Shelf Registration Statement. |
Securities Purchase Agreement (dollars and shares in thousands)
On February 21, 2020, we announced that we have entered into a definitive agreement for the sale of our ADSs and Series A Non-Voting Convertible Preferred Shares (“Series A Preferred”) in a private placement to a group of institutional accredited investors. The private placement has resulted in gross proceeds of approximately $65,000 before deducting placement agent and other offering expenses, and resulting in net proceeds of approximately $61,000.
Pursuant to the terms of the private placement, we issued 8,680 ADSs and 488 shares of Series A Preferred at a price of $7.09 per share, priced at-the-market under Nasdaq rules. Each share of non-voting Series A Preferred is convertible into one ADS, provided that conversion will be prohibited if, as a result, the holder and its affiliates would own more than 9.99% of the total number of Avadel ADSs outstanding. The closing of the private placement occurred on February 25, 2020. Proceeds from the private placement will be used to fund continued clinical and program development of FT218, including an open-label extension study for REST-ON, a switch study to evaluate patients switching from twice-nightly sodium oxybate to once-nightly FT218, as well as for general corporate purposes. If this transaction would have happened prior to December 31, 2019, ordinary shares issued and outstanding would have been 51,927 and 46,200, respectively.
-95-
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Avadel Pharmaceuticals plc
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Avadel Pharmaceuticals plc (the "Company") as of December 31, 2019 and 2018, the related consolidated statements of (loss) income, comprehensive (loss) income, shareholders’ (deficit) equity, and cash flows, for each of the three years in the period ended December 31, 2019, and the related notes and financial statement schedule listed in the Index at Item 15 (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 16, 2020, expressed an unqualified opinion on the Company's internal control over financial reporting.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Deloitte and Touche LLP
St. Louis, Missouri
March 16, 2020
We have served as the Company's auditor since 2016.
-96-
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Avadel Pharmaceuticals plc
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of Avadel Pharmaceuticals plc (the “Company”) as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated financial statements as of and for the year ended December 31, 2019, of the Company and our report dated March 16, 2020, expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Deloitte and Touche LLP
St. Louis, Missouri
March 16, 2020
-97-
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
Not applicable.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
As required by Rule 15d -15(b) of the Exchange Act, we have evaluated, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this Annual Report. Our disclosure controls and procedures are designed to provide reasonable assurance that the information required to be disclosed by us in reports that we file under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure and is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the U.S. Securities and Exchange Commission (the “SEC”). Based on that evaluation, our principal executive officer and principal financial officer concluded that as of the end of the period covered by this report our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended. The Company’s internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect all misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2019. In making this assessment, the Company’s management used the criteria set forth in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this assessment, management concluded that, as of December 31, 2019, the Company’s internal control over financial reporting is effective based on those criteria.
Changes in Internal Control Over Financial Reporting
During the year ended December 31, 2019 as part of our restructuring initiatives described in this Annual Report under Item 8 “Financial Statements and Supplementary Data - Note 18 (Restructuring Costs)” and Item 7 “Management Discussion and Analysis of Results of Operations and Financial Condition - Results of Operation,” we moved all of our Irish and a portion of our French accounting operations to St. Louis, Missouri. Further, as part of these restructuring initiatives, the Company completed the outsourcing of a majority of its Information Technology resources to a third party. These moves were made in order to consolidate our accounting systems, gain efficiencies of scale, reduce costs and make internal control over financial reporting more consistent across our various entities. These moves were not made in response to any identified deficiency or weakness in the Company’s internal control over financial reporting. Other than these changes, there has been no change in our internal control over financial reporting during the quarter or year ended December 31, 2019 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
Not applicable.
-98-
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K because we intend to file our definitive proxy statement for our 2019 annual general meeting of shareholders pursuant to Regulation 14A of the Securities Exchange Act of 1934 (our “Definitive 2019 Proxy Statement”), not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information to be included in our Definitive 2019 Proxy Statement is incorporated herein by reference.
Item 10. Directors, Executive Officers and Corporate Governance.
Information regarding Directors, Executive Officers and Corporate Governance is hereby incorporated by reference to our Definitive 2020 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2019.
Item 11. Executive Compensation.
Information regarding Executive Compensation is hereby incorporated by reference to our Definitive 2020 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2019.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Information regarding Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters is hereby incorporated by reference to our Definitive 2020 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2019.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Information regarding Certain Relationships and Related Transactions, and Director Independence is hereby incorporated by reference to our Definitive 2020 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2019.
Item 14. Principal Accountant Fees and Services.
Information regarding Principal Accountant Fees and Services is hereby incorporated by reference to our Definitive 2020 Proxy Statement, which we intend to file with the SEC within 120 days after December 31, 2019.
-99-
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) | Documents filed as part of this report: |
1. | Financial Statements |
See Item 8 - Financial Statements and Supplementary Data of Part II of this Report.
2. | Financial Statement Schedules |
See below for Schedule II: Valuation and Qualifying Accounts. All other schedules are omitted as they are not applicable, not required or the information is included in the consolidated financial statements or related notes to the consolidated financial statements.
Schedule II
Valuation and Qualifying Accounts
(In thousands)
Deferred Tax Asset Valuation Allowance: | Balance, Beginning of Period | Additions (a) | Deductions (b) | Other Changes (c) | Balance, End of Period | |||||||||||||||
2019 | $ | 21,199 | $ | 6,496 | $ | (4,762 | ) | $ | (5,896 | ) | $ | 17,037 | ||||||||
2018 | $ | 15,354 | $ | 6,089 | $ | (75 | ) | $ | (169 | ) | $ | 21,199 | ||||||||
2017 | $ | 7,599 | $ | 391 | $ | (664 | ) | $ | 8,028 | $ | 15,354 | |||||||||
a. | Additions to the deferred tax asset valuation allowance relate to movements on certain French, Irish and U.S. deferred tax assets where we continue to maintain a valuation allowance until sufficient positive evidence exists to support reversal. |
b. | Deductions to the deferred tax asset valuation allowance include movements relating to utilization of net operating losses and tax credit carryforwards, release in valuation allowance and other movements including adjustments following finalization of tax returns. |
c. | Other changes to the deferred tax asset valuation allowance including currency translation adjustments recorded directly in equity, account method changes and the impact of corporate restructuring. |
3. Exhibits required by Item 601 of Regulation S-K
The information required by this Section (a)(3) of Item 15 is set forth on the exhibit index that follows the Signatures page of this Form 10-K.
Index to Exhibits
Exhibit Number | Exhibit Description | |
3.1 | ||
3.2 | ||
4.1 | ||
-100-
4.2 | ||
4.3 | ||
4.4 | ||
4.5 | ||
4.6 | ||
10.1 | ||
10.2* | ||
10.3* | ||
10.4 | ||
10.5* | ||
10.6 | ||
10.7 | ||
10.8 | ||
10.9 | ||
-101-
10.10* | ||
10.11* | ||
10.12 | ||
10.13 | ||
10.14‡ | ||
10.15‡ | ||
10.16‡ | ||
10.17‡ | ||
10.18‡ | ||
10.19‡ | ||
10.20‡ | ||
10.21‡ | ||
10.22‡ | ||
10.23‡ | ||
10.24‡ | ||
-102-
10.25‡ | ||
10.26‡ | ||
10.28* | ||
10.29* | ||
10.30 | ||
10.31 | ||
10.32* | ||
10.33* | ||
10.34* | ||
10.35* | ||
10.36 | ||
10.37‡ | ||
-103-
10.38 | ||
10.39# | ||
10.40 | ||
14.1 | ||
14.2 | ||
21.1 | ||
23.1 | ||
31.1 | ||
31.2 | ||
32.1 | ||
32.2 | ||
101.INS | XBRL Instant Document | |
101.SCH | XBRL Taxonomy Extension Schema Document | |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |
* Confidential treatment has been requested for the redacted portions of this agreement. A complete copy of the agreement, including the redacted portions, has been filed separately with the Securities and Exchange Commission.
# The representations and warranties contained in this agreement were made only for purposes of the transactions contemplated by the agreement as of specific dates and may have been qualified by certain disclosures between the parties and a contractual standard of materiality different from those generally applicable under securities laws, among other limitations. The representations and warranties were made for purposes of allocating contractual risk between the parties to the agreement and should not be relied upon as a disclosure of factual information relating to the Company, the Investors or the transaction described in the Current Report on Form 8-K.
-104-
‡ Management contract or compensatory plan or arrangement filed pursuant to Item 15(b) of Form 10-K.
(1) This certification accompanies the Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the registrant under the Securities Act of 1933 or the Securities Exchange Act of 1934 (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing.
-105-
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
Avadel Pharmaceuticals plc | ||
Dated: March 16, 2020 | By: | /s/ Gregory J. Divis |
Name: Gregory J. Divis | ||
Title: Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each of each of Geoffrey M. Glass, Eric J. Ende, Mark A. McCamish, MD, Ph.D., Linda S. Palczuk, and Peter Thornton, by their respective signatures below, irrevocably constitutes and appoints Gregory J. Divis and Phillandas T. Thompson, and each of them individually acting alone without the other, his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either of them, or their or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
Signature | Title | Date | ||
/s/ Gregory J. Divis | Director, Chief Executive Officer and Principal Executive Officer | March 16, 2020 | ||
Gregory J. Divis | ||||
/s/ Thomas S. McHugh | Chief Financial Officer and Principal Financial and Accounting Officer | March 16, 2020 | ||
Thomas S. McHugh | ||||
/s/ Geoffrey M. Glass | Non-Executive Chairman of the Board and Director | March 16, 2020 | ||
Geoffrey M. Glass | ||||
/s/ Dr. Eric J. Ende | Director | March 16, 2020 | ||
Dr. Eric J. Ende | ||||
/s/ Mark A. McCamish, MD, Ph.D. | Director | March 16, 2020 | ||
Mark A. McCamish, MD, Ph.D. | ||||
/s/ Linda S. Palczuk | Director | March 16, 2020 | ||
Linda S. Palczuk | ||||
/s/ Peter Thornton | Director | March 16, 2020 | ||
Peter Thornton | ||||
-106-