Attached files
| file | filename |
|---|---|
| EX-31.2 - EX-31.2 - ENTELLUS MEDICAL INC | d882717dex312.htm |
| EX-32.2 - EX-32.2 - ENTELLUS MEDICAL INC | d882717dex322.htm |
| EX-31.1 - EX-31.1 - ENTELLUS MEDICAL INC | d882717dex311.htm |
| EX-23.1 - EX-23.1 - ENTELLUS MEDICAL INC | d882717dex231.htm |
| EX-32.1 - EX-32.1 - ENTELLUS MEDICAL INC | d882717dex321.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014
Or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 001-36814
Entellus Medical, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 20-4627978 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 3600 Holly Lane North, Suite 40 Plymouth, Minnesota |
55447 | |
| (Address of principal executive offices) | (Zip Code) | |
(763) 463-1595
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $0.001 par value per share | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ¨ No x
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (Section 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | x (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The registrant was not a public company as of the last business day of its most recently completed second fiscal quarter and, therefore, cannot calculate the aggregate market value of its voting and non-voting common equity held by non-affiliates as of such date.
As of March 6, 2015, there were 18,679,216 shares of the Registrant’s common stock, par value $0.001 per share, issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.
Table of Contents
Entellus Medical, Inc.
FORM 10-K
YEAR ENDED DECEMBER 31, 2014
As used in this report, the terms “we,” “us,” “our,” “Entellus Medical,” “Entellus” and the “Company” mean Entellus Medical, Inc., unless the context indicates another meaning.
Table of Contents
PART I
Cautionary Note Regarding Forward-Looking Statements
Except for historical information, the matters discussed in this Annual Report on Form 10-K are forward looking statements that involve risks, uncertainties and assumptions that, if they never materialize or if they prove incorrect, could cause our results to differ materially from those expressed or implied by such forward-looking statements. We make such forward-looking statements under the provision of the “Safe Harbor” section of the Private Securities Litigation Reform Act of 1995. Actual future results may vary materially from those projected, anticipated or indicated in any forward-looking statements as a result of various factors, including those set forth in Item 1A of this Annual Report on Form 10-K under the heading “Risk Factors.” Readers should also carefully review the risk factors described in the other documents that we file from time to time with the Securities and Exchange Commission, or the SEC. In this Annual Report on Form 10-K, the words “anticipates,” “believes,” “expects,” “intends,” “future,” “could,” “estimates,” “plans,” “would,” “should,” “potential,” “continues” and similar words or expressions (as well as other words or expressions referencing future events, conditions or circumstances) identify forward-looking statements. Forward-looking statements also include the assumptions underlying or relating to any of the foregoing statements. The forward-looking statements contained in this Annual Report include, but are not limited to, statements related to:
| • | estimates of our annual total addressable market, future revenue, expenses, capital requirements and our needs for additional financing; |
| • | our ability to obtain additional financing in the future; |
| • | the implementation of our business model and strategic plans for our products, technologies and businesses; |
| • | competitive companies and technologies and our industry; |
| • | our ability to manage and grow our business by expanding our sales to existing customers or introducing our products to new customers; |
| • | third-party payor reimbursement and coverage decisions; |
| • | our ability to establish and maintain intellectual property protection for our products or avoid claims of infringement; |
| • | extensive government regulation; |
| • | the timing or likelihood of regulatory filings and approvals; |
| • | our ability to hire and retain key personnel; |
| • | the volatility of the trading price of our common stock; |
| • | our expectations regarding the use of proceeds from our initial public offering; and |
| • | our expectations about market trends. |
Forward-looking statements are based on management’s current expectations, estimates, forecasts and projections about our business and the industry in which we operate, and management’s beliefs and assumptions are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. As a result, any or all of our forward-looking statements in this Annual Report on Form 10-K may turn out to be inaccurate. Furthermore, if the forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. Factors that may cause actual results to differ materially from current expectations include, among other things, those described in the section entitled “Risk Factors” and elsewhere in this Annual Report. Readers are urged to consider these factors carefully in evaluating these forward-looking statements. These forward-looking statements speak only as of the date of this Annual Report on Form 10-K. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
1
Table of Contents
| Item 1. | Business |
Overview
We are a medical technology company focused on the design, development and commercialization of products for the minimally invasive treatment of patients who are suffering from chronic sinusitis. Our XprESS family of products is used by ear, nose and throat, or ENT, physicians to open narrowed or obstructed sinus drainage pathways using balloon sinus dilation to treat patients with symptomatic inflammation of the nasal sinuses. When used as a stand-alone therapy in the doctor’s office, our balloon sinus dilation products are the only devices proven in a sufficiently powered prospective, multicenter, randomized, controlled trial to be as effective as functional endoscopic sinus surgery, or FESS. Patients treated with our products in this trial in the ENT physician office also experienced faster recovery, less bleeding at discharge, less use of prescription pain medication and fewer post-procedure debridements than patients receiving FESS. We estimate that physicians have treated over 75,000 patients with our XprESS products since the launch of the first XprESS product in February 2010.
Chronic sinusitis is a common medical condition that, according to estimates from the Centers for Disease Control and Prevention, or CDC, impacts approximately 12% of the adult population aged 18 and over in the United States, or approximately 29 million people based on 2013 U.S. Census Bureau data, making it more prevalent than asthma or heart disease. Our addressable patient population consists of a portion of patients who undergo FESS, as well as patients who fail medical management, but do not undergo sinus surgery. We currently estimate the annual total addressable market for our products in the United States to be approximately 630,000 patients, which, based on our estimate of the average revenue per procedure in the balloon sinus dilation market, represents an annual market opportunity of nearly $1.0 billion.
Minimally invasive balloon sinus dilation devices have enabled a shift towards office-based treatment of chronic sinusitis patients who are candidates for sinus surgery in the operating room. We believe this shift has been facilitated by our technology and clinical data, as well as procedure economics that are favorable to the healthcare system, patient and provider. Our XprESS family of products is used to treat patients with inflammation of the frontal, ethmoid, sphenoid and maxillary sinuses and is specifically designed for ease-of-use in the ENT physician office setting. Patients treated in the physician office setting with our products under local anesthesia report high levels of comfort during the procedure, fast recovery and durable symptom relief, all in a procedure that reduces costs to the patient and healthcare system compared to FESS. Our research and development efforts are focused on enhancing our XprESS family of products and broadening their indications for use.
We currently sell our products through a direct sales force in the United States. Our commercial organization consists of our sales, marketing and reimbursement personnel, and has grown from 47 people as of December 31, 2010, to 114 people as of December 31, 2014, which includes 69 full quota-carrying representatives as of December 31, 2014. We intend to continue to grow our sales force in order to further penetrate the sinusitis market. As of December 31, 2014, we estimate that over 1,400 accounts are purchasing our balloon sinus dilation products for use by the ENT surgeon. Health insurance coverage for stand-alone balloon sinus dilation performed in the physician office setting is in place with Medicare, Medicaid and other third-party payors cover approximately 218 million persons in the United States as of December 31, 2014. In addition, as of December 31, 2014, coverage for hybrid procedures involving balloon sinus dilation performed together with FESS in the operating room is available for approximately 245 million persons in the United States.
2
Table of Contents
Overview of Sinusitis and the Market
Sinuses are air-filled pockets within the bones of the face and skull. The four types of sinuses are frontal, ethmoid, sphenoid and maxillary. One of each type of sinus lies on either side of the face.
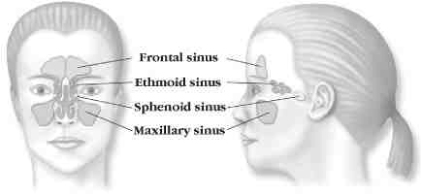
The sinuses are lined with soft, moist tissue, or mucosa, that is covered with a layer of mucus. Mucus moistens the nasal lining and protects the body from inhaled impurities such as dust, pollutants and bacteria. Each of the maxillary, sphenoid and frontal sinuses has a corresponding ostium, or opening, through which mucus drains. The ethmoid sinuses are a series of cells with multiple, often interconnected openings and drainage pathways. The surface tissues of the sinuses are covered with millions of cilia, which are small, hair-like structures that act in coordination to sweep the mucus through the ostium of each sinus cavity to the back of the throat. The drainage of mucus is a normal process that keeps the sinuses healthy.
Sinusitis is inflammation of the sinus cavities that may be caused by infections, allergies or environmental factors, as well as structural issues such as blockage of an ostium. If one or more sinus drainage pathways becomes blocked, normal mucus drainage is prevented and damage to ciliary function may occur. There are three categories of sinusitis: acute, recurrent acute and chronic. Acute sinusitis is transient in nature and lasts less than four weeks. Recurrent acute sinusitis is a type of chronic sinusitis that involves four or more episodes of acute sinusitis over a 12-month period. Chronic sinusitis is more severe and lasts 12 weeks or longer. We refer to recurrent acute sinusitis and chronic sinusitis together as chronic sinusitis. The symptoms of sinusitis include facial pain, pressure, nasal congestion, headaches, fatigue and loss of smell. When persistent, these symptoms can severely impact a patient’s day-to-day well-being, resulting in frequent doctor visits and can lead to reduced sleep function, chronic fatigue and depression. The condition significantly reduces work productivity, increases absenteeism and impairs daily activities.
According to estimates from the CDC, approximately 12% of the adult population aged 18 and over in the United States is affected by chronic sinusitis, or approximately 29 million people using 2013 U.S. Census Bureau data. Chronic sinusitis is associated with substantial healthcare utilization and expenditure and ranks as one of the ten costliest physical health conditions, contributing to an estimated $8.6 billion in direct healthcare costs in the United States. The latest CDC ambulatory medical surveys estimated that chronic sinusitis resulted in 12.3 million physician visits annually in the United States in each of 2009 and 2010, of which an estimated 8.1 million were to primary care physicians, 2.8 million were to ENT physicians, and the remainder were to hospital outpatient facilities and emergency rooms. We estimate that the 2.8 million annual patient visits to ENT physicians represent approximately 1.23 million distinct patients seen annually by ENT physicians. Primary care and ENT physicians rely on medical management for first-line therapy to treat chronic sinusitis. In cases where patients remain symptomatic despite multiple rounds of medical management, a physician may recommend FESS or balloon sinus dilation combined with FESS, or in cases of uncomplicated chronic sinusitis, may recommend stand-alone balloon sinus dilation. Uncomplicated chronic sinusitis is sinus disease that does not include fungus, extensive polyps, or inflammation and infection of the bones below the sinus mucosa. We
3
Table of Contents
estimate that, of the 1.23 million patients with chronic sinusitis who are expected to be seen by ENT physicians in 2015, 552,000 will undergo FESS, while 493,000 will have symptoms relieved by medical management and 188,000 will fail medical management but will elect not to undergo FESS.
We estimate the annual total addressable market for our products in the United States is approximately 630,000 patients, which, based on our estimate of the average revenue per procedure in the balloon sinus dilation market, represents an annual market opportunity of nearly $1.0 billion. Our addressable patient population consists of a portion of patients who undergo FESS, as well as patients who fail medical management but do not undergo sinus surgery. We believe that out of the total 552,000 estimated annual patients who are expected to undergo FESS procedures in 2015, approximately 55% are well-suited for office-based balloon treatment because they present with uncomplicated sinusitis and do not require a septoplasty procedure to correct a severely deviated septum, and approximately 30% are well-suited for treatment with a hybrid balloon sinus dilation procedure in the operating room along with a FESS procedure due to the location and nature of their sinusitis. In addition, we believe that approximately 85% of the 188,000 patients who fail medical management but do not undergo FESS represent a significant market opportunity, as these patients currently avoid sinus surgery, seek a non-surgical alternative procedure to FESS after fewer episodes of failed medical management, or are increasingly referred by primary care physicians to ENT physicians for their chronic sinusitis. We estimate this current market opportunity to be approximately 160,000 patients annually, and believe that patient population will increase and be amenable to balloon sinus dilation as the body of experience and clinical evidence supporting the benefits of office-based balloon sinus dilation continues to grow.
Current Treatments for Sinusitis and their Limitations
The treatment of chronic sinusitis is progressive in nature and involves therapies that attempt to achieve the most effective solution in the least invasive manner. Treatment typically begins with medical management, and if this is unsuccessful, an ENT physician may perform FESS or a balloon sinus dilation procedure to treat both symptoms and obstruction of the sinus drainage pathways.
Medical Management
Sinusitis patients often present with a sinus infection and nasal congestion. Therefore, first-line therapy typically involves the use of prescription medications, such as antibiotics and decongestants. If the condition persists, physicians may prescribe more aggressive medical management, including additional antibiotics, oral steroids or intra-nasal steroid sprays to reduce inflammation, or saline irrigations to loosen mucus. To facilitate increased delivery of steroids to inflamed sinus tissue, reduce symptoms and reduce recurrence of polyps, physicians may also prescribe off-label use of steroids added to nasal irrigation solutions.
While medical management can be effective, its effect is often temporary, and is unable to address the underlying anatomical issues such as chronically narrowed or obstructed sinus drainage pathways that may be contributing to the condition. Even where medical management is effective, prolonged medication use can give rise to undesirable side effects. For example, frequent use of antibiotics can lead to the development of antibiotic resistance and repeated use of decongestant sprays can cause nasal congestion rebound, or an increase in nasal congestion after stoppage of use. Similarly, prolonged use of oral steroids can produce potentially serious side effects such as aggressive behavior, sleeplessness, glaucoma, bone loss, weight gain, high blood pressure and psychosis. Medical management also represents an ongoing expense to patients and payors. Based upon published studies, we estimate that approximately 60% of chronic sinusitis patients who are seen by ENT physicians and receive medical management remain symptomatic.
Sinus Surgery
In cases where patients diagnosed with sinusitis are unresponsive to multiple rounds of medical management, an ENT physician may recommend surgical treatment. The primary surgical alternative is FESS, which attempts to open the sinus drainage pathways such as the ostia while attempting to preserve as much bone
4
Table of Contents
and sinus mucosa as possible. FESS is typically performed under general anesthesia in an operating room during a procedure that typically takes approximately two hours. During FESS, an ENT physician inserts an endoscope into the nasal cavity to view a patient’s anatomy. The physician typically uses rigid steel instruments and powered cutting tools to remove inflamed sinus tissue and underlying bone to create a larger passage through the nasal anatomy to the infected sinuses, where the physician then removes additional tissue and bone to open the sinus ostia. Surgeons may also perform additional surgery on a deviated nasal septum or enlarged turbinate, a bulbous structure in the nose that warms and humidifies air, in the nose to treat nasal obstruction or simply to gain surgical access to the sinuses. At the conclusion of the procedure, patients often have their nasal cavity packed with a material that acts as a spacer to prevent surgical adhesions and control bleeding. Patients typically require one or more follow-up debridement treatments in which the physician may remove more tissue, crusting, scabs or scar tissue at the area of surgery in order to keep the sinus drainage pathway open and promote proper healing.
While FESS is often effective at treating chronic sinusitis, FESS results in irreversible changes to the anatomy and significant postoperative pain, discomfort and recovery time. In addition, postoperative debridement is unpleasant for the patient and costly to the healthcare system. Although FESS is the standard of care, approximately 7% to 12% of FESS patients require revision surgery, often as a result of ongoing inflammation and scarring associated with the procedure. Within the first year following a FESS procedure, approximately 64% of patients experience recurrent symptoms. In addition, given the use of surgical tools in close proximity to the brain, eyes and other critical anatomy, the potential for significant complications is a concern of physicians and patients alike. The risks of FESS, particularly in the frontal sinuses, cause some ENT physicians to avoid performing surgery in the frontal sinus drainage pathway. Major complications, such as cerebral spinal fluid leaks, swelling of the eye or blindness, occur in approximately 1% of FESS procedures. We believe that patient concerns about undergoing sinus surgery, postoperative discomfort, multiple days of recovery time and potential complications can make patients hesitant to undergo FESS. As an example, in our REMODEL trial, eight of 53 patients randomized to the FESS arm of the trial refused surgical treatment and withdrew from the study, noting they did not want to undergo a FESS procedure, whereas only one of 52 patients randomized to balloon sinus dilation withdrew before treatment because of not wanting the balloon procedure.
Balloon Sinus Dilation
Balloon sinus dilation was developed to provide patients a minimally invasive treatment alternative that was more lasting and effective than medical management, while providing patients a safe and efficacious alternative to FESS that preserves sinus mucosa and future treatment options. By employing balloon sinus dilation as a non-surgical method to open blocked sinus pathways, balloon sinus dilation spares mucosa while increasing ventilation and mucus drainage, which preserves the natural function of the cilia and important natural anatomical structures, such as the uncinate process, which is a tissue bridge that prevents inhaled dust and bacteria from easily entering maxillary sinus ostia.
Balloon sinus dilation can be performed as a stand-alone procedure using local anesthesia in the ENT physician office, typically in approximately one hour. In a typical office-based balloon sinus dilation procedure, the physician uses topical anesthetics followed by an injection to numb the patient’s sinus lining. Once a patient’s nasal passages and ostia are numb, the physician inserts a balloon sinus dilation product into the nose using endoscopic visualization, and guides the balloon device into a narrowed sinus drainage pathway. Some balloon sinus dilation products use a guide catheter and guidewire to direct the balloon into a sinus drainage pathway. In conjunction with placing the balloon into the ostium, the physician confirms the balloon device has correctly accessed the sinus, often using methods such as direct endoscopic visualization or transcutaneous visualization of light emanating from the tip of a balloon device or guidewire. The balloon is then inflated to a high pressure, resulting in fracture and remodeling of the bones underlying the sinus mucosa. For patients with multiple obstructed sinus drainage pathways, the physician may repeat the process of placement, confirmation and inflation in each affected sinus. Balloon sinus dilation can also be used with FESS performed under general anesthesia in the operating room.
5
Table of Contents
ENT physicians are increasingly treating more complicated sinusitis patients in the office with a procedure known as mini-FESS. This procedure combines office-based balloon sinus dilation of the obstructed sinus drainage pathways with limited excision of adjacent tissue, including polyp removal, removal of enlarged ethmoid air cells, or reduction of enlarged turbinates. We expect the adoption of mini-FESS procedures will broaden the population of patients treatable with office-based balloon sinus dilation products. We believe this trend will be enhanced as ENT physicians prescribe on-label as well as off-label use of steroids in nasal irrigations to reduce polyp recurrence and promote healing following excision of sinus tissue.
Despite the advantages of a minimally invasive balloon sinus dilation procedure, we believe its adoption has been limited by a number of historical factors that include:
| • | initial perceptions among some ENT physicians that the primary indication for balloon sinus dilation was treatment of frontal sinuses in a hybrid procedure that added cost compared to FESS only; |
| • | reimbursement codes and payment for office-based stand-alone balloon sinus dilation did not come into effect until 2011, six years after sinus dilation balloons were initially launched; |
| • | until 2013, there was limited clinical evidence to support the use of balloon sinus dilation in a broader patient population and as a stand-alone treatment in the ENT physician office; and |
| • | a perception among some ENT physicians of patients’ inability to tolerate balloon sinus dilation procedures in the physician office setting. |
In addition, a limitation of balloon sinus dilation is that it does not remove irreversibly diseased sinus mucosa, which may be present in patients with sinusitis complicated by fungus, extensive polyps and infection of the bones below the sinus mucosa.
Our Competitive Strengths
We are focused exclusively on the treatment of chronic sinusitis, one of the most common medical conditions in the United States resulting in patient visits to physicians. Our XprESS Multi-Sinus Dilation devices and PathAssist tools represent a broad product line of minimally invasive technologies cleared by the U.S. Food and Drug Administration, or FDA, for treating chronic sinusitis patients after failed medical management. Our products are designed to further transition the treatment of uncomplicated chronic sinusitis from the operating room to the physician office setting. We believe our following competitive strengths will help drive further adoption of our products:
| • | Focus on physician office treatment of sinusitis—We believe the majority of sinusitis patients who fail medical management can be treated in the physician office with a stand-alone, office-based balloon sinus dilation procedure. To facilitate the adoption of these procedures, we have focused our product development on creating treatment solutions that are effective, easy to perform and well-tolerated by an awake patient. We educate physicians about our clinical data demonstrating efficacy in a broad spectrum of patients who are well-suited for treatment in the ENT physician office setting and the procedure economics that are favorable to the patient, provider and healthcare system. |
| • | Significant body of clinical data—We have developed a significant body of clinical data supporting the safety and effectiveness of our products. We have sponsored seven clinical studies in which a total of 604 patients were treated with our products and followed to assess safety, and of which 452 patients were followed for an extended period of six, 12 or 24 months to assess long-term efficacy. Our clinical data shows clinically meaningful results across a broad set of clearly defined patient groups. Our REMODEL randomized trial demonstrated that when our balloon sinus dilation devices were used as a stand-alone therapy in the physician office setting, patients experienced similar efficacy rates, faster recovery times, less bleeding at discharge, less use of prescription pain medication and fewer post-procedure debridements than patients receiving FESS. Our XprESS Multi-Sinus Study further demonstrated the efficacy of our products in a broader population of chronic sinusitis patients. |
6
Table of Contents
| Additionally, one of the challenges associated with trans-nasal balloon treatment of the maxillary sinus is the lack of visualization of the ostia. We have completed a cadaver study that demonstrated effective and accurate trans-nasal access to the maxillary ostia with our XprESS Pro device to demonstrate that our XprESS balloon sinus dilation device could consistently and effectively treat the maxillary ostia without puncturing tissue and creating a false channel into the sinus. We believe this data is unique to our product and responsible in part for driving adoption of office-based balloon sinus dilation in treating maxillary sinuses, the most commonly treated sinus during FESS. |
| • | Competitive advantages over other balloon sinus dilation products—Our products are intuitive and easy to use because they are placed into the diseased sinus like a sinus seeker, a tool commonly used by ENT physicians. Our XprESS family of devices provides depth markings and tactile feel with a controllable tip to allow a user to gently access the ostia and visualize the device insertion depth. Our XprESS products also have fewer components than other balloon sinus dilation products, and come with a shaping tool that can bend our XprESS device so that a single device can be used to treat all diseased sinuses in a single procedure. For accessing the approach to the maxillary sinus, our XprESS LoProfile provides a small profile, having a 1.75 millimeter diameter ball tip and an adjustable 135-degree angle, compared to a guide catheter used by our main competitor that is 77% larger in diameter and limited to a fixed 110-degree angle. Furthermore, in our XprESS devices, a slideable balloon is used enabling the balloon to be kept out of the way when the device is advanced into a sinus opening, which allows the user to better visualize and enter the sinuses. Our XprESS products enable unique confirmation methods such as controlled excursion of transilluminated light to confirm entry into the frontal sinus, and for accessing the maxillary ostia, observation of the ball tip as it deflects or transilluminates the uncinate process, thereby enhancing the accuracy of sinus access. Our XprESS LoProfile and XprESS Ultra Multi-Sinus Dilation Systems provide further convenience for office users by having a self-contained, battery-powered light source, eliminating the need for a separate light cord or xenon lamp light source associated with competing product offerings. Additionally, the use of our products does not require significant additional capital expenditures by ENT physicians associated with some competing products that must be used in conjunction with a CT image guidance system that can cost over $100,000. |
| • | Demonstrated to comfortably treat sinusitis patients—Our products are designed to provide patient comfort during office-based procedures. Four of our clinical studies demonstrate that awake patients treated in the physician office with our products are comfortable, with patient pain scores ranging from 1.8 to 3.2 on a zero to ten scale where zero is no pain. We also believe that our products facilitate comfortable patient treatment because, unlike our main competitor, they do not require the use of a guidewire that can contact the non-anesthetized interior of a treated sinus and cause patient discomfort. |
| • | Comprehensive and broad IP portfolio—As of December 31, 2014, we had 19 issued U.S. patents and 16 pending U.S. patent applications. Our intellectual property portfolio covers the current and future XprESS products with four issued U.S. patents, nine pending U.S. patent applications and multiple pending foreign applications. |
7
Table of Contents
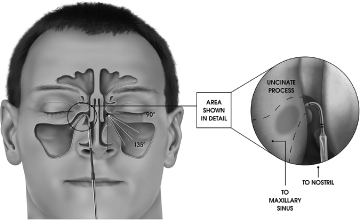
XprESS device accessing the maxillary sinus with 135° bend angle
and transillumination of the uncinate process
Our Products
Our XprESS Multi-Sinus Dilation family of devices and PathAssist tools represent a broad product line of FDA-cleared, minimally invasive products for treating chronic sinusitis patients. Our devices have been specifically designed to enable easy access to the sinus cavities under endoscopic visualization, and provide confirmation of sinus location and device placement using multiple methods. These products facilitate the access and dilation of blocked sinus ostia and pathways in order to reestablish proper mucus transport and drainage with retained ciliary function. Our XprESS devices are indicated for use to access and treat sinuses in adults, and the PathAssist Tools are indicated for use to locate and illuminate nasal and sinus structures in adults.
XprESS Multi-Sinus Dilation Product Family
Our XprESS Multi-Sinus Dilation family of products consists of our XprESS Pro device, our XprESS LoProfile device and our XprESS Ultra device. The XprESS LoProfile device and the XprESS Ultra device are always sold with our LED Light Fiber. These disposable devices open an obstructed or narrowed drainage pathway of a sinus cavity by means of trans-nasal balloon sinus dilation. These devices combine multiple functions in a hand-held, single, easy-to-use, cost-effective and minimally invasive device designed for efficient balloon sinus dilation procedures in the physician office or operating room. Our XprESS devices combine the features of balloon sinus dilation, suction, irrigation and light confirmation, all integrated into a single-use device shaped to feel like a sinus seeker, a tool used regularly by ENT physicians to probe for sinus openings and drainage pathways.
Our XprESS devices include an ergonomic handle attached to a hollow metal shaft with a malleable tip that can be quickly shaped with our disposable bending tool and easily steered into diseased frontal, sphenoid and maxillary sinuses without requiring a guidewire or purchase of another component. Along the shaft of the tip are marks at 1.0 and 2.0 centimeters to denote depth of insertion into the sinus cavities. A balloon is attached to a movable shaft that allows the balloon to be advanced and retracted along the metal shaft. This enables the balloon to be kept out of the way while the device is being placed into a sinus opening, allowing the ENT physician to better visualize and enter the diseased sinus drainage pathway. The handle contains a one-finger balloon slide mechanism that is used to position the balloon within the sinus ostia and an integrated finger suction vent that allows the ENT physician to control active suction in order to maintain good endoscopic visualization throughout the procedure. The proximal end of the handle contains two ports that connect to the balloon inflation lumen and the suction/irrigation lumen of the metal shaft. The port used for suction and irrigation tubing also can accommodate a connection of a light fiber for transillumination. The port for balloon inflation connects to a one-handed inflation syringe used to inflate the balloon to a controlled pressure of 12 atmospheres, or approximately 175 pounds per square inch.
8
Table of Contents
Our XprESS Ultra device is our fourth generation balloon sinus dilation device. In addition to the features of our XprESS Pro and LoProfile devices, this device offers the lowest profile re-shapeable atraumatic ball tip balloon device in the industry. The tip of this device measures 1.5 millimeters, compared to 2.0 millimeters for our XprESS Pro and 1.75 millimeters for our XprESS LoProfile devices, allowing for better access to tight spaces within the sinus cavities. Our XprESS Ultra device was launched in February 2015, our XprESS LoProfile device was launched in January 2013 and our XprESS Pro was launched in February 2010.
The XprESS family of products is available in three different balloon sizes and two different balloon lengths. The XprESS device used is dependent on physician preference. Our XprESS devices are sold in a kit that also contains an inflation syringe, two extension lines and a bending tool that contains three angle cut-outs that are specifically designed to shape the ball tip for access into a specific sinus cavity.
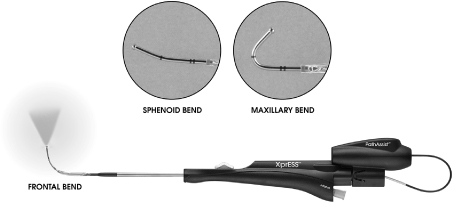
XprESS multi-sinus dilation device with LED Light Fiber
PathAssist Tools
Our PathAssist tools provide ENT physicians with an easy way to confirm sinus location and XprESS device placement. These tools enable users to screen symptomatic patients for in-office balloon sinus dilation and to more easily and efficiently perform balloon sinus dilation procedures in the physician office and operating room. In addition, ENT physicians may use other competing devices to confirm sinus location and placement of our device.
LED Light Fiber and Light Fiber
The LED Light Fiber is a single-use tool that provides real-time high intensity red transillumination of the sinus cavity with its own battery power, eliminating the need for extra light sources, cables and adapters. This sinus confirmation tool consists of a flexible illumination fiber that emits light from the distal tip. The instrument snaps on easily to the proximal end of the XprESS device. A reduced diameter LED light fiber was developed and is currently sold with our XprESS Ultra and LoProfile products.
Our Light Fiber is a single-use tool that provides real-time transillumination of the sinus cavity. The fiber is designed to be loaded into the XprESS device and can connect to various different cables and light sources. It allows for suction or irrigation to be performed through XprESS when it is connected to the XprESS device.
Other Tools
Our other PathAssist tools comprise our Light Seeker, Maxillary Seeker and Sphenoid Seeker/Freer. The Light Seeker is a tool with optical fibers embedded into the device to allow ENT physicians to access the frontal sinus and trans-illuminate the sinus cavity and forehead to confirm correct entry into the frontal sinus. The Light
9
Table of Contents
Seeker can be used to confirm non-surgical access to the frontal sinus prior to opening our XprESS balloon kit. The Maxillary Seeker is a tool that allows users to quickly find the correct angle to access the natural maxillary ostia. The Sphenoid Seeker/Freer is a two-in-one tool that enables ENT physicians to navigate easier access to the sphenoid ostium and to manipulate the middle turbinate. These stainless steel tools are reusable and can be used prior to balloon sinus dilation or as general sinus surgery tools.

PathAssist tools and XeroGel Nasal Packing Material
FinESS
We launched our initial product line, the FinESS sinus dilation system, or FinESS device, in 2009. The FinESS device was designed as a solution for treatment of the maxillary sinus drainage pathway using direct endoscopic visualization and dilation of these sinus ostia through a trans-antral approach.
In 2011, we introduced a technique for the trans-nasal balloon sinus dilation of the maxillary sinuses using our XprESS device and published these data in 2012. As a result, our XprESS device is now used in place of the FinESS device in over 99% of balloon sinus dilation procedures performed by our customers.
XeroGel Nasal Packing Material
We entered into a distribution agreement with CogENT Therapeutics in September 2013 and began exclusive distribution of XeroGel in October 2013 in the United States. XeroGel is indicated for use in patients undergoing nasal or sinus surgery as a space-occupying packing to separate tissue or structures compromised by surgical trauma, prevent adhesions between mucosal surfaces, help control bleeding following surgery or trauma, and act as an aid in the natural healing process. XeroGel is also indicated for use as a nasal packing to treat epistaxis, or nasal bleeding. XeroGel is a dissolvable co-polymer that contains chitosan, a naturally occurring element from chitin, or the exoskeleton of crustaceans. The co-polymer absorbs fluids, swells to conform to the treatment site and dissolves gradually, typically by the first postoperative visit. CogENT is responsible for all manufacturing, order fulfillment (for product shipped directly to customers), quality and regulatory requirements of XeroGel.
Future Products and Indications for Use
We currently have an approved Investigational Device Exemption from the FDA to conduct a pivotal study in pediatric patients using our XprESS device and PathAssist product in which we are targeting an indication for the treatment of the maxillary sinus in patients between the ages of 2 and 12, and for the treatment of the frontal, sphenoid and maxillary sinuses in patients aged 13 and older. If we receive FDA clearance, we believe we will be the only company with such a pediatric indication. We expect to receive this indication for use in 2016. Based on research published in Laryngoscope in 2006, we estimate that approximately six million Americans under the age of 18 suffer from chronic sinusitis.
10
Table of Contents
Other Products
In addition to our internal product development efforts, we may leverage our direct U.S. sales force by licensing or acquiring other products that can be used by ENT physicians for the treatment of sinusitis and related diseases.
Clinical Results and Studies
Overview
We have developed a significant body of clinical data supporting the safety and effectiveness of our products. We have sponsored seven clinical studies in which a total of 604 patients were treated with our products and followed to assess safety, and 452 patients were followed for an extended period of six to 24 months to assess long-term efficacy. Over 50 different clinical investigators participated in these clinical studies. Our clinical evidence includes the only published randomized trial with sufficient statistical power to demonstrate advantages of a nonsurgical treatment compared to traditional FESS for the treatment of chronic sinusitis. While we are aware of a limited number of studies comparing balloon dilation to FESS, these studies were small single-center studies that were under-powered, meaning their sample sizes were too small to prove their endpoints with sufficient statistical power. In contrast, the sample size for the REMODEL trial was calculated with sufficient statistical power (90%) at a one-sided alpha level of 0.025 to prove the two primary endpoints and hypotheses. The minimum required sample size was 72 patients (36 in each arm) and the trial exceed this requirement, with 89 patients completing 12 month follow-up. Our REMODEL prospective, multicenter, randomized, controlled trial demonstrated that when our balloon sinus dilation products were used as a stand-alone therapy performed in the ENT physician office, patients experienced better recovery outcomes and similar efficacy to FESS. Throughout our seven clinical studies, symptom reduction was shown in a broad set of clearly defined patient groups, including those presenting with chronic sinusitis, mild to moderate septal deviations, multi-sinus disease including disease in all eight sinuses or sinusitis present in any combination of the sinuses, and accessory ostia, which are multiple drainage pathways from the maxillary sinus. We also completed two additional studies in which our products were studied in cadaver specimens. The first cadaver study demonstrated effective and accurate trans-nasal entrance through the maxillary ostia with our XprESS device. The second cadaver study demonstrated that access to frontal sinuses using our XprESS device with transillumination is as effective as placement of a probe with a CT image guidance system.
Efficacy endpoints measured in our clinical studies included the success rate of balloon sinus dilation of the targeted sinus pathway, symptom improvement using the validated Sino-Nasal Outcomes Test, or SNOT-20, quality of life, or QOL, survey, ostial patency rate, change in the number of sinusitis episodes after treatment, and revision surgery rate. The SNOT-20 QOL survey is used with patients to rate the severity of 20 different sinusitis symptoms on a scale of 0, or no problem, to 5, or problem as bad as it can be. A decrease of at least 0.8 of the mean SNOT-20 score is considered a clinically meaningful improvement. Safety was assessed by measuring complication rates. Patient comfort and recovery outcomes were measured using pain scores for the office-based balloon sinus dilation procedure, time to return to normal activities, use of post-procedure pain medication, postoperative debridements, and short-term symptom improvement using SNOT-20 QOL survey scores. Debridements are a trans-nasal procedure performed within the nasal cavity to remove dead, contaminated, or adherent tissue or foreign material that may promote infection and impede healing. A debridement is performed in the physician office setting by the ENT physician during a follow-up visit after a sinus procedure. Changes in utilization of healthcare resources and patient activity impairment were assessed using the validated Rhinosinusitis Symptom Inventory, or RSI survey. The RSI survey measures patient-reported number of sinus infections, antibiotic prescriptions, physician visits for sinusitis, absenteeism, presenteeism and number of homebound days. The Work Productivity and Activity Impairment, or WPAI, survey measures percent impairment at work and in daily activity.
Our clinical study data shows clinically meaningful and persistent levels of symptom reduction at one, six, 12 and 24 months. A study published in 2010 demonstrated that symptom improvements measured with QOL surveys after treatment with FESS do not appear to change between six and 20 months and recommended that
11
Table of Contents
clinical trial designs incorporating QOL outcomes following FESS should consider the six-month time frame as an appropriate long-term primary endpoint. Therefore, we believe that six-month follow-up is sufficient to assess long-term outcomes of our products. All but one of our seven clinical studies followed patients for at least 12 months.
REMODEL Randomized Trial of Entellus Balloon Sinus Dilation versus FESS
We sponsored the REMODEL trial, which was a prospective, multicenter, randomized, controlled trial, in which 92 adult patients at 10 centers in the United States were treated between December 2011 and December 2012. The trial was designed to test the hypothesis that symptom improvement after balloon sinus dilation with our products in an ENT physician office was noninferior to FESS and that balloon sinus dilation with our products was superior to FESS for postoperative debridements. No study center contributed more than 20% of patient enrollments and results were consistent across sites. Patients were randomized one-to-one to either balloon sinus dilation or FESS.
The two primary endpoints in the REMODEL trial were long-term improvement in sinus symptoms as assessed by the mean change in overall SNOT-20 QOL survey score between treatment and six-month follow-up and the mean number of debridements following treatment. Secondary and additional endpoints included short-term symptom improvement scores, complication rates, recovery time, surgical revision rate, ostial patency, change in number of sinusitis episodes, and changes in activity impairment and work productivity. Post-procedure follow-up assessment of patients was performed at one week, and at months one, three, six and 12. A minimum of 36 patients were needed in each study arm to assess the primary endpoints with sufficient statistical power.
After randomization, 92 of 105 enrolled patients completed treatment. Eleven patients randomized to FESS withdrew their consent for treatment. Eight of these patients withdrew because they did not want to undergo surgical treatment while three withdrew due to other non-sinus medical conditions or a lack of time available for follow-up. Two patients in the balloon sinus dilation arm withdrew from the study with one of them doing so because they did not want to undergo the balloon procedure. Six-month follow-up was achieved in 91, or 99% of the patients. Twelve-month follow-up was achieved in 89, or 97% of the patients. Patients in this trial had suffered with chronic sinusitis symptoms for an average of over 12 years and had not undergone a previous sinus surgery or balloon dilation procedure before study enrollment. The mean SNOT-20 score prior to treatment in the REMODEL study was 2.54 in each arm of the study.
Both primary endpoints were achieved in this trial. Comparison of changes between each trial group confirmed the mean symptom improvement for patients undergoing balloon sinus dilation was noninferior to that of patients undergoing FESS. Both trial groups experienced clinically meaningful and statistically significant improvements in overall SNOT-20 QOL survey scores. The mean number of postoperative debridements per patient showed a significant difference between the trial groups, demonstrating the superiority of balloon sinus dilation where patients in this group received an average of 0.1 debridements compared to an average of 1.2 debridements in the FESS group.
12
Table of Contents
The trial demonstrated that when our balloon sinus dilation devices were used as a stand-alone therapy in the physician office setting, patients experienced similar efficacy rates in terms of symptom improvement, ostial patency, reduction of sinusitis episodes, and very low surgical revision rates, with faster recovery times, less bleeding at discharge, less use of prescription pain medication, and fewer post-procedure debridements than patients receiving FESS. The REMODEL trial six-month follow-up results were published in the American Journal of Rhinology and Allergy in August 2013. The one-year follow-up results of this trial were published in the same journal in May 2014. The following table summarizes the outcomes from the REMODEL trial:
| REMODEL Trial Outcome Measure |
Our Balloon Sinus Dilation Products (Mean or %) |
FESS (Mean or %) |
p-Value | Compared to FESS, Balloon Sinus Dilation is: | ||||||||||
| Primary Endpoints |
||||||||||||||
| Change in SNOT-20 symptom score at 6 months |
-1.67 | -1.60 | <0.001 | Noninferior1 | ||||||||||
| Change in SNOT-20 symptom score at 12 months |
-1.64 | -1.65 | <0.001 | Noninferior1 | ||||||||||
| Mean number of debridements per patient |
0.1 | 1.2 | <0.0001 | Superior2 | ||||||||||
| Secondary Outcomes (Recovery and Short Term) |
||||||||||||||
| Patients discharged with nasal bleeding |
28 | % | 55 | % | 0.011 | Significantly better3 | ||||||||
| Recovery time (days) |
1.6 | 4.8 | 0.002 | Significantly better3 | ||||||||||
| Duration of prescription pain medications (days) |
0.9 | 2.8 | <0.001 | Significantly better3 | ||||||||||
| Short-term change in SNOT-20 symptom score (one week/one month) |
-1.49/-1.70 | -0.96/-1.62 | 0.014 | Significantly better4 | ||||||||||
| Secondary Outcomes (1 Year) |
||||||||||||||
| Change in number of sinusitis episodes per patient |
-4.2 | -3.5 | NS | Not significantly different3 | ||||||||||
| Ostial patency |
97 | % | 99 | % | NS | Not significantly different4 | ||||||||
| Mean reduction of activity impairment due to chronic sinusitis |
78 | % | 79 | % | NS | Not significantly different3 | ||||||||
| Mean reduction in overall work impairment due to chronic sinusitis |
81 | % | 78 | % | NS | Not significantly different3 | ||||||||
| Complications |
0 | % | 0 | % | NS | Not significantly different3 | ||||||||
| Revision surgery rate |
2.1 | % | 2.4 | % | NS | Not significantly different3 | ||||||||
| 1 | Based on one-sided Student’s t-test for noninferiority. Values of p < 0.025 were considered statistically significant. |
| 2 | Based on one-sided two-sample Wilcoxon test for superiority. Value of p < 0.025 was considered statistically significant. |
| 3 | Based on two-sided two-sample t-tests. Values of p < 0.05 were considered statistically significant. |
| 4 | Based on a repeated measures regression model. Values of p < 0.05 were considered statistically significant. |
XprESS Multi-Sinus Study
We sponsored the XprESS Multi-Sinus Study, a prospective, multicenter, nonrandomized study, in which 81 adult patients at 10 different clinical centers in the United States were treated in 2012. The purpose of this study was to evaluate patient outcomes at one year following stand-alone balloon sinus dilation treatment in the physician office with our XprESS device for patients with maxillary sinusitis with or without sinusitis in the frontal, ethmoid, or sphenoid sinuses. A total of 307 of 313, or 98%, of sinus ostia were successfully dilated with our XprESS device under local anesthesia. Seventy-six of the 81 patients, or 94%, treated completed one-year follow-up. One-year patient follow-up showed a statistically significant mean reduction in the SNOT-20 QOL survey score of 1.57, nearly twice the level of 0.8 reduction needed for a clinically meaningful change in symptoms. Overall, 79% of all subjects experienced clinically meaningful sinus symptom improvement at one
13
Table of Contents
year. Clinically meaningful and statistically significant symptom improvement was also observed after balloon treatment of any combination of sinusitis including all sinuses, patients with ethmoid sinusitis, septal deviations, turbinate enlargement, and chronic or recurrent sinusitis. The in-office procedure was tolerated well among the patients with the mean pain score as rated by the patient equal to 2.8, on a scale of 0 being no pain and 10 being severe pain. Patients experienced a statistically significant reduction in all RSI survey measurements. Compared to the one year before balloon sinus dilation treatment, the number of sinus infections, antibiotic courses, and physician visits for sinusitis were reduced in the year following treatment by 77%, 71%, and 73%, respectively. Only one patient, or 1.3%, required a revision procedure. The one-year follow-up results of this study were published in the American Journal of Rhinology and Allergy in February 2014.
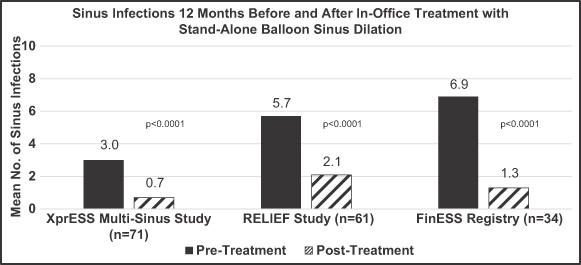
XprESS Registry Study
We sponsored the XprESS Registry Study, which was a prospective, multicenter, nonrandomized study, in which 175 adult patients at eight different clinical centers in the United States were treated between June 2010 and September 2011. The study was designed to evaluate the safety, sustained effectiveness, and one-year patency of the sinus drainage pathways dilated with our XprESS device. Over 96%, or 479 of 497, of the attempted sinus dilations were successfully completed, primarily in the frontal and sphenoid sinuses. A total of 448 dilations in 166 patients were completed during a hybrid procedure along with FESS while 31 dilations in nine patients underwent a stand-alone balloon sinus dilation procedure. The XprESS device demonstrated a high level of safety with no patients experiencing a serious adverse event. Efficacy was also demonstrated with clinically meaningful and statistically significant sinus symptom improvement as measured by SNOT-20 QOL survey scores and a 2.3% surgical revision rate among the 44 patients followed for one year post-procedure. Analysis of patient subgroups with and without nasal polyps also showed clinically meaningful and statistically significant improvement in sinus symptoms for each group. The one-year follow-up results of this study were published in the International Forum for Allergy and Rhinology in February 2013.
XprESS Maxillary Pilot Study
We sponsored the XprESS Maxillary Pilot Study, which was a prospective, dual-center, nonrandomized study, in which 21 adult patients at two clinical centers in the United States were treated between 2011 and 2012. The study was designed to evaluate symptom improvement after stand-alone trans-nasal balloon sinus dilation of the maxillary ostia with our XprESS device in the physician office setting. Patient comfort during the procedure was also assessed. Efficacy was demonstrated with clinically meaningful and statistically significant sinus symptom improvement as measured by SNOT-20 QOL survey scores. The in-office procedure was tolerated well
14
Table of Contents
among the patients with the overall mean pain score as rated by the patient of 1.8, on a scale of 0 being no pain and 10 being severe pain. The six-month results of this study were published in the ENT Journal in December 2012. As shown in the table below, in five separate studies observing patient tolerance of office-based balloon sinus dilation, four of which we sponsored and one of which was sponsored by a competitor, patients tolerated the procedure very well.
| Patient Tolerance of Office-Based Balloon Sinus Dilation |
||||||||
| Clinical Study |
N | Average Pain Score During Procedure (0=No Pain; 10=Severe Pain) |
||||||
| XprESS Multi-Sinus |
81 | 2.8 | ||||||
| XprESS Maxillary Pilot |
21 | 1.8 | ||||||
| RELIEF |
69 | 3.2 | ||||||
| BREATHE |
19 | 2.7 | ||||||
| ORIOS2 (Non-Entellus Study) |
198 | 4.5 | ||||||
XprESS Maxillary Cadaver Study
We sponsored the XprESS Maxillary Cadaver Study conducted in the United States in December 2012. The study was designed to evaluate the ability of our XprESS Pro device to successfully access the maxillary sinuses through a trans-nasal approach. One of the challenges associated with trans-nasal balloon sinus dilation of the maxillary sinuses is the lack of visualization of the ostia. As a result, inaccurate placement of the balloon device can lead to creation of false channels and ineffective patient treatment. Six ENT physicians, three who had significant prior experience with our XprESS Pro device and three newly trained physicians who had very little clinical experience with our device, attempted to access the natural maxillary ostia using our XprESS Pro device bent into a maxillary configuration. The study was done using trans-nasal endoscopic visualization and multiple other methods for confirming device tip position. These six ENT physicians were blinded to the trans-antral endoscopic views from inside the maxillary sinuses that were used to determine accuracy of entry. Thirty-nine of 40 ostium, or 97.5%, were successfully accessed and dilated when the recommended procedure was performed. There was one instance in which the device successfully accessed the natural maxillary ostium but was not considered an appropriate dilation because the device was inserted through a pre-existing hole in the uncinate process. We believe that the results of this study demonstrate that the XprESS Pro device has successfully addressed the unique challenges of maxillary sinus treatment with balloon sinus dilation. The results of this study were published in the American Journal of Rhinology and Allergy in December 2012.
Accuracy of Transillumination vs. CT Image Guidance System as a Positioning Method — Cadaver Study
We sponsored the Accuracy of Transillumination vs. CT Image Guidance System Positioning Method Cadaver study conducted in the United States in October 2013. This study was designed to compare transillumination using our XprESS LoProfile with LED Light Fiber device to a CT image guidance system probe attached to the FusionTM ENT Navigation System by Medtronic, Inc. for determining the accuracy of device tip placement in a frontal sinus. Two ENT physicians were asked to position our XprESS LoProfile device into a frontal sinus using standard techniques including transillumination and also to position a CT image guidance system probe into a frontal sinus. Each physician then reported their level of confidence that tip placement was in the frontal sinus for each attempt. Attempts to enter the frontal sinus were performed in 16 specimens and 32 frontal sinuses. The physicians were blinded to a separate camera view from inside the frontal sinus used to determine if the XprESS LoProfile device or CT image guidance system probe successfully accessed the sinus. The physicians reported their confidence at entering the frontal sinus, with 0 being no confidence and 4 being very confident. Access to the frontal sinus was successful in 30 of 32 attempts, or 94%, using transillumination and in 29 of 32 attempts, or 91%, using a CT image guidance system. When physician confidence at entering the frontal sinus was rated a 4, this correctly predicted frontal entry in 29 of 29, or 100%, of instances with transillumination and in 27 of 28, or 96%, of instances with a CT image guidance system. It was concluded from this study that confirmation of access to the frontal sinus is comparable whether using
15
Table of Contents
transillumination or a CT image guidance system. With both technologies, the positive predictive value of device placement in the frontal sinus is very high. The results of this study were presented as a poster at the Combined Otolaryngology Spring Meeting in April 2014.
The two leading ENT societies, the American Academy of Otolaryngology — Head and Neck Surgery and the American Rhinologic Society, do not recommend a CT image guidance system as a standard of care for sinus procedures, but suggest that a CT image guidance system should be reserved for complex anatomy. We believe that balloon sinus dilation has been proven to be safe and effective without the need for a CT image guidance system.
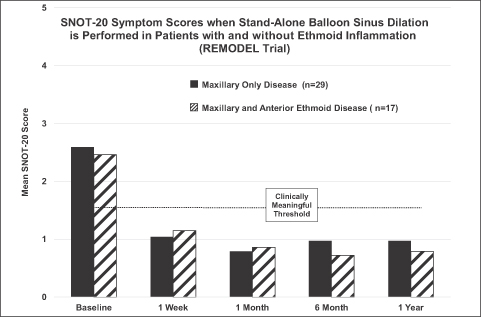
Evidence that Inflammation in the Ethmoid Sinuses Does Not Need Direct Treatment
Four of our clinical studies, BREATHE, RELIEF, XprESS Multi-Sinus, and REMODEL, demonstrated that stand-alone balloon dilation of sinus drainage pathways such as the maxillary sinuses or frontal sinuses significantly reduced sinusitis symptoms in patients with uncomplicated disease, irrespective of the presence or absence of inflammation in the adjacent ethmoid sinuses. A typical result is shown below and was replicated in three other studies that we sponsored with 12-month follow-up. The osteomeatal complex in the nose is a location where the frontal, maxillary, and anterior ethmoid sinuses typically drain. Studies have shown that balloon sinus dilation of the sinus drainage pathways of the maxillary and/or frontal sinuses reduces inflammation in those treated sinuses. We believe that lessening the disease burden in these major sinuses along with enhancing mucus transport in the osteomeatal complex may also improve mucus clearance from the adjacent anterior ethmoid air cells and reduce the inflammation in those lesser sinuses. Results of the BREATHE study were published in the International Forum of Allergy & Rhinology in May/June 2012. The results of the RELIEF study were published in the Annals of Otology, Rhinology & Laryngology in November 2013.
Trans-Antral Studies
We sponsored three clinical studies, BREATHE, FinESS Registry, and RELIEF, that examined the safety and effectiveness of our FinESS trans-antral product line that is used to dilate the maxillary outflow track after positioning an endoscope and balloon catheter into the maxillary sinus. Access to the maxillary sinus was achieved through a small puncture above the canine tooth in the direction of the maxillary sinus, in a dental-like
16
Table of Contents
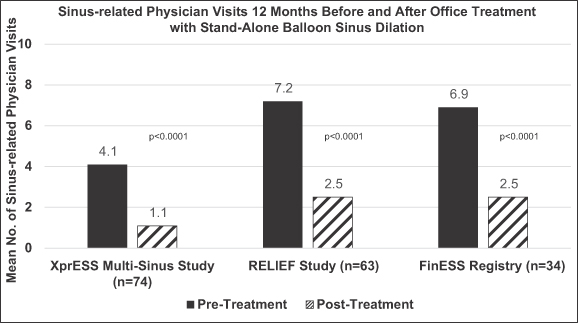
approach to the sinus. Since conducting these studies, we have optimized the ability of our XprESS Multi-Sinus device to access the maxillary sinus in a trans-nasal approach using the existing opening in the nostril. As a result, sales of our FinESS product line have largely been supplanted by sales of our XprESS family of products. However, the clinical outcomes from the trans-antral studies are relevant to all uses of balloon sinus dilation of the maxillary drainage pathway because the balloon treats the same anatomic region irrespective of the site of access. Our trans-antral studies show relief of sinus symptoms using the SNOT-20 symptom score through 12-month to 24-month follow-up periods with low surgical revision rates of 6.8%, 2.9%, and 5.8% in the 59, 137, and 66 patients followed in the BREATHE, FinESS Registry, and RELIEF studies, respectively. As a reference, the clinical literature shows revision surgery rates following FESS most commonly range from 7% to 12%. Additional outcomes data demonstrate that treatment with our trans-antral product results in patients needing fewer physician and nurse visits for nasal problems and requiring fewer courses of antibiotics compared to pretreatment. It has also been shown that dilation of the maxillary outflow path significantly relieves symptoms in patients with combination maxillary and anterior ethmoid disease. The results of the FinESS Registry were presented at the Combined Otolaryngology Spring Meeting in April 2011.
Sales and Marketing and Physician Training
We have focused our sales and marketing efforts on educating ENT physicians about the clinical value, ease-of-use, convenience and cost-effectiveness of office-based sinus dilation as both an alternative to FESS for uncomplicated patients, and for patients who repeatedly fail aggressive medical management and are progressing toward becoming a surgical candidate. We also sell our products to ENT physicians who perform balloon sinus dilation in a hospital outpatient operating room or surgery center.
We maintain a direct sales organization in the United States which as of December 31, 2014 consisted of 103 employees in direct sales and sales management, four employees in sales operations, six employees in marketing and three employees in customer service. We do not market our products outside the United States and Canada.
Our marketing group and clinically focused personnel support our sales personnel through the following initiatives:
| • | Education of ENT physicians is performed using a variety of methods and custom training tools including head model training, observation of video-taped procedures, online visualization of live |
17
Table of Contents
| procedures, cadaver lab training and visits to peers who perform office-based balloon sinus dilation. Our products are designed to be easy-to-use and because they are placed into a sinus drainage pathway in a manner similar to existing sinus seekers on the market, our products typically do not require extensive clinical training for most new physician customers. Approximately 15% of newly trained physicians opt to visit a peer for physician training whereas other new users are trained with our custom training tools by field trainers from our sales, clinical and R&D organizations. |
| • | Education of patients about the benefits of our sinus dilation procedure. We utilize online patient education information, and find-a-doctor resource to help patients who are exploring sinusitis treatment options to be able to discuss treatment options with their physician. |
| • | Promotion to physicians of the advantages of our products and the clinical outcomes they enable for patients. |
Reimbursement and Procedure Economics
When balloon sinus dilation is performed adjunctively with standard FESS as a hybrid procedure, existing FESS codes are used and insurance coverage is available for an estimated 86% of covered lives in the United States as of December 2014.
Effective January 1, 2011, when balloon sinus dilation is performed as a stand-alone procedure, providers use CPT codes 31295, -96, and -97 for dilation of the maxillary, frontal, and sphenoid sinus ostia, respectively. When a stand-alone balloon procedure is performed in the physician office, the reimbursement associated with these CPT codes includes a non-facility practice expense component of payment intended to cover the cost of equipment, supplies, and overhead associated with these procedures performed in the physician office, including the cost of our devices. In addition, physicians receive additional value from the convenience and efficiency of treating patients in their office setting compared to treatments in a hospital or surgery center. This, along with excellent clinical outcomes, motivates many ENT physicians and practices to adopt our procedure.
As an example of cost savings to the healthcare system from office balloon sinus dilation procedures, the 2015 national average Medicare payments to a physician performing bilateral maxillary sinus dilation in the office and performing a nasal endoscopy exam one week later is $3,351. By comparison, treatment of the same patient in an ASC, with FESS including ethmoidectomy and performing a debridement one week after FESS costs Medicare $4,139, or 24% more, including ENT physician professional fees of $969. Treatment of the same patient with FESS in a hospital out-patient department costs Medicare, $6,408, or 91% more than balloon sinus dilation office treatment. When more complex cases are performed involving dilation of four or six sinuses, the level of savings to the healthcare system for office sinus dilation increases. Publication in October 2014 by the Centers for Medicare & Medicaid Services, or CMS, the federal agency responsible for administering the Medicare program, of final 2015 payment rates for stand-alone balloon sinus dilation resulted in 2015 rates very similar to 2014 rates.
| Sinuses Dilated |
2015 Medicare National Average Payment Rates | |||||||||||||||
| Office Balloon Sinus Dilation |
FESS in Hospital | FESS in ASC | % FESS costs more than dilation: Hospital & ASC |
|||||||||||||
| Maxillary |
$ | 3,351 | $ | 6,408 | $ | 4,139 | 91% & 24 | % | ||||||||
| Maxillary+Frontal |
$ | 4,964 | $ | 10,790 | $ | 6,896 | 117% & 39 | % | ||||||||
| Maxillary+Frontal+Sphenoid |
$ | 6,527 | $ | 15,344 | $ | 9,583 | 135% & 47 | % | ||||||||
Note: National average payment rates include the total reimbursement made to physicians and facilities for the balloon sinus dilation or FESS procedure plus cost of post-FESS debridement or post-balloon sinus dilation nasal endoscopy. Ethmoidectomy is performed with all FESS procedures.
As of December 2014, insurance coverage for stand-alone balloon sinus dilation procedures is available for an estimated 218 million persons in the United States. Positive coverage decisions for stand-alone balloon sinus dilation performed in the physician office setting are in place with several third-party payors including Medicare,
18
Table of Contents
UnitedHealthcare, Aetna, Cigna, Humana, Kaiser Permanente, TRICARE, Health Net, Medicaid in 30 states, and Blue Cross Blue Shield plans in 17 states. Balloon sinus dilation has the support of the American Academy of Otolaryngology and the American Rhinological Society, both of which have written positive policy statements for balloon sinus treatment and participate in coverage obtainment efforts.
With the benefit of cost savings from office-based balloon sinus dilation procedures, we believe the combination of clinical and cost advantages will lead to additional coverage as payors become more knowledgeable about the full body of clinical evidence. However, there can be no assurance that payors will continue to cover stand-alone balloon sinus dilation, or that additional payors will issue positive coverage policies for the procedure.
Competition
Our industry is highly competitive, subject to change and significantly affected by activities of industry participants. We compete with companies and products used to treat chronic sinusitis, including traditional FESS, medical therapies and other balloon sinus dilation products. Our balloon sinus dilation products compete with medical management by providing a procedure and treatment option that may be chosen to be performed by ENT physicians after fewer rounds of medical management than historically employed due to the minimally disruptive nature of balloon sinus dilation as compared to FESS. Our balloon sinus dilation products compete with FESS by providing a comparably effective in-office alternative for patients with uncomplicated sinusitis. Accordingly, our balloon sinus dilation products are increasingly positioned in the middle of the treatment continuum between medical management and FESS, drawing patients from medical management who might not have chosen any treatment beyond medical management due to the invasive nature of FESS, as well as patients who would have chosen FESS if not for our less invasive alternative. Our balloon sinus dilation products also compete with other companies selling balloon sinus dilation products to ENT physicians. Our PathAssist products also compete with companies selling devices for use in balloon sinus dilation. Additionally, ENT physicians may use competing devices, instead of our PathAssist products, to confirm sinus location and placement of our XprESS device.
Many of the companies developing or marketing these competitive products are publicly traded or are divisions of publicly traded companies. These companies may enjoy several competitive advantages, including:
| • | greater financial and human capital resources; |
| • | significantly greater name recognition; |
| • | established relationships with ENT physicians, referring physicians, customers and third-party payors; |
| • | additional lines of products, and the ability to offer rebates or bundle products to offer greater discounts or incentives to gain a competitive advantage; and |
| • | more established sales, marketing and worldwide distribution networks. |
Companies that sell balloon sinus dilation products include the Acclarent division of Johnson and Johnson Inc., the market leader in balloon sinus dilation products, as well as the Xomed division of Medtronic, which launched its NuVent balloon sinus dilation system in August 2014, and the ArthroCare division of Smith & Nephew. In addition, a small private company, SinuSys Corporation, sells a non-balloon sinus dilation system that uses osmotic self-expansion technology. Companies that provide tools and devices for FESS include the Xomed division of Medtronic, the Gyrus ACMI division of Olympus and Stryker Corporation. Intersect ENT sells a steroid-eluting biodegradable stent that is used following FESS or in patients who have previously undergone an FESS procedure and is not currently a competitor. However, Intersect ENT is currently developing products for office-based treatment of sinusitis, which if approved may be competitive with us in the future.
We believe our continued ability to compete favorably depends on:
| • | successfully expanding our commercial operations; |
19
Table of Contents
| • | continuing to enjoy suitable reimbursement economics for our customers; |
| • | attracting and retaining skilled personnel; |
| • | continuing to innovate and demonstrate the advantages of our existing and new products in clinical studies; |
| • | our patent licensor successfully protecting their intellectual property in the U.S.; and |
| • | protecting our patents. |
Intellectual Property
We believe that the strength of our competitive position will depend substantially upon our ability to obtain and enforce intellectual property rights protecting our technology. We apply for patents for new patentable technologies relevant to our business and utilize other forms of intellectual property protection to strategically protect our intellectual property. Periodically, we may review and attempt to acquire rights in third-party patents and applications that are strategically valuable to us.
Patents
As of December 31, 2014, we owned 19 issued U.S. patents relating to the field of sinusitis treatment and have at least 16 pending patent applications for U.S. patents. Four of these issued patents includes claims directed towards the XprESS products and methods of using the XprESS products. As of December 31, 2014, at least nine of our pending patent applications currently include claims directed towards one or more aspects of our XprESS family of products.
Our issued patents that protect our commercial products and current product pipeline are derived from applications that were filed between 2006 and 2009. Since U.S. law provides for a 20-year patent term measured from the date of filing, many of our current patent holdings will begin to expire starting in 2026, though many of our patents enjoy patent term extensions granted by the U.S. Patent Office. While we have pursued and continue to pursue patent protection for our technologies, we may, from time to time, abandon certain patents and patent applications for business reasons.
Trademarks
Our XPRESS, PATHASSIST, FINESS, ENTELLUS MEDICAL, LIGHT FIBER, and LIGHT SEEKER marks are registered trademarks of our company in the United States.
Trade Secrets
We may rely, in some circumstances, on trade secret law to protect some of our technology. Trade secrets, however, can be difficult to protect.
We seek to protect our proprietary technology and manufacturing process, in part, by confidentiality and invention assignment agreements with employees, under which they are bound to assign to us inventions that are made during the term of their employment and relate to our business, unless otherwise excepted. These agreements further prohibit our employees from using, disclosing, or bringing onto the premises any proprietary information belonging to a third-party. In addition, most of our consultants, scientific advisors and contractors are required to sign agreements under which they must assign to us any inventions that relate to our business. These agreements also prohibit these third-parties from incorporating into any inventions the proprietary rights of third-parties without informing us. It is our policy to require all employees to document potential inventions and other intellectual property in laboratory notebooks and to disclose inventions to patent counsel. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
20
Table of Contents
While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants, contractors or collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
License Agreement with Acclarent, Inc.
In February 2011, we entered into a license agreement with Acclarent in connection with the settlement of a then-ongoing litigation in which Acclarent alleged that our FinESS and XprESS products infringed certain claims of five of their patents, which allegations we denied. In October 2012, we agreed with Acclarent to amend certain terms of our license agreement, or, as amended, the Acclarent License. Pursuant to the Acclarent License, Acclarent granted us a worldwide, non-exclusive, royalty-bearing license under certain patents held by Acclarent to manufacture, use and commercialize our currently marketed XprESS, PathAssist and FinESS products and certain future versions of those products, or, collectively, the Covered Products, in the field of the expansion of paranasal sinuses and sinus pathways.
Under the Acclarent License, we made an initial up-front, lump-sum payment to Acclarent as a retroactive royalty payment. We are also required to pay Acclarent an ongoing royalty in the low double-digit percentages, on a country-by-country basis, on net sales of Covered Products. The Acclarent License will expire upon the expiration or abandonment of the last to expire of the patents licensed to us under the agreement. Acclarent has the right to terminate the agreement upon 60 days’ notice in the event of our uncured material breach.
Manufacturing and Supply
We manufacture our XprESS Pro, XprESS LoProfile, XprESS Ultra and PathAssist products at our facility in Plymouth, Minnesota with components supplied by external suppliers. We perform inspections of these components before use in our manufacturing operations. Using these components, we assemble, inspect, test and package our products and send them to a third-party sterilization vendor. After sterilization, we perform additional inspections of the finished products, carton and label products internally and release the lot to inventory and then ship the finished product to customers.
We believe our manufacturing operations are in compliance with regulations mandated by the FDA. We are an FDA-registered medical device manufacturer. Our manufacturing facilities and processes are subject to periodic inspections and audits by various federal, state and foreign regulatory agencies. For example, our facilities were inspected by the FDA in August 2012, and no major nonconformance reports were issued as a result of that inspection. Our facility was last inspected in March 2015 by the British Standards Institute, or BSI Group, our Notified Body, and no major nonconformance reports were issued as a result of that inspection.
Certain components used in our products are supplied to us from single source suppliers. We rely on single-source suppliers for some of our balloon extrusions and molded components and some off-the-shelf components. We generally acquire our single-source components pursuant to purchase orders placed in the ordinary course of business. While our suppliers have generally met our demand for their products on a timely basis in the past, we are not certain that they will in the future be able to meet our demand for their products, either because of the nature of our agreements with those suppliers, our limited experience with those suppliers, or our relative importance as a customer to those suppliers.
Our suppliers manufacture the components they produce for us or test our components and devices to our specifications. We intend to maintain sufficient supplies of the components from these single-source suppliers in the event that one or more of these suppliers were to encounter delay in supply or end supply to enable us to continue to manufacture our products for a sufficient amount of time necessary to obtain another source of components.
21
Table of Contents
Government Regulation
The design, development, research, manufacture, testing, labeling, promotion, advertising, distribution, marketing, sale and export and import of our products are subject to regulation by numerous governmental authorities, principally the FDA and corresponding state and foreign regulatory agencies. Failure to obtain or maintain approval or clearance to market our products or to meet the ongoing requirements of these regulatory authorities could prevent us from marketing and continuing to market our products.
Regulation by the FDA
In the United States, the Federal Food, Drug, and Cosmetic Act, or FDCA, FDA regulations and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, device safety, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, record-keeping procedures, advertising and promotion, sales and distribution, export and import, recalls and field safety corrective actions, and post-market surveillance, including complaint handling and medical device reporting. The FDA regulates the design, manufacturing, servicing, sale and distribution of medical devices. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution. The FDA can also refuse to approve or clear pending applications.
Each medical device we wish to distribute commercially in the United States requires marketing authorization from the FDA prior to distribution either through the 510(k) premarket notification process or the more demanding premarket approval, or PMA, process. The type of marketing authorization necessary is generally linked to the classification of the device. The FDA classifies medical devices into one of three classes (Class I, II, or III) based on the degree of risk associated with a device and the level of regulatory control deemed necessary to ensure its safety and effectiveness. Low- and moderate-risk devices requiring fewer controls are placed in Class I or II. Class I devices that pose the least risk are subject only to the general controls applicable to all devices, such as requirements for device labeling, premarket notification and adherence to the FDA’s current good manufacturing practices, or cGMPs, as reflected in the Quality System Regulation, or QSR. Class II devices that pose a moderate risk are subject to general controls and may also be subject to special controls such as performance standards, product-specific guidance documents, special labeling requirements, patient registries, or post-market surveillance. Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through general or special controls, including devices that support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury.
Most Class I devices and some Class II devices are exempted by regulation from the 510(k) clearance requirement and can be marketed without prior authorization from the FDA. Some Class I devices that have not been so exempted and Class II devices are eligible for marketing through the 510(k) clearance pathway. By contrast, devices placed in Class III generally require PMA approval or 510(k) de novo clearance prior to commercial marketing. The PMA approval process is more stringent, time-consuming and expensive than the 510(k) clearance process; however, the 510(k) clearance process has also become increasingly stringent and expensive.
Medical Devices. Each of our current commercially available devices is classified as either a Class I device exempt for the requirement to submit a 510(k) notification, or as a Class I or Class II device requiring 510(k) clearance. None of our current commercially available devices is classified as a Class III device requiring PMA approval.
510(k) Clearance. To obtain 510(k) clearance for a medical device, an applicant must submit a premarket notification to the FDA demonstrating that the device is “substantially equivalent” to a legally marketed device, commonly known as the “predicate device.” A legally marketed predicate device may include a device that was
22
Table of Contents
legally marketed in the United States prior to May 28, 1976 for which a PMA is not required (commonly known as a “pre-amendment device” based on the date the Medical Device Amendments of 1976 were enacted), a device which the FDA has reclassified from Class III to Class II or I, or a device which has been found substantially equivalent through the 510(k) process. A device is substantially equivalent if, with respect to the predicate device, it has the same intended use and has either (i) the same technological characteristics, or (ii) different technological characteristics, but the information provided in the 510(k) submission demonstrates that the device does not raise new questions of safety and effectiveness and is at least as safe and effective as the predicate device. A showing of substantial equivalence sometimes, but not always, requires clinical data.
Before the FDA will accept a 510(k) submission for substantive review, the FDA will first assess whether the submission satisfies a minimum threshold of acceptability. If the FDA determines that the 510(k) submission is incomplete, the FDA will issue a “Refuse to Accept” letter which generally outlines the information the FDA believes is necessary to permit a substantive review and to reach a determination regarding substantial equivalence. An applicant must submit the requested information before the FDA will proceed with additional review of the submission. Once the 510(k) submission is accepted for review, by regulation, the FDA has 90 days to review and issue a determination. As a practical matter, clearance often takes longer. The FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence. Unless a specific exemption applies, 510(k) premarket notification submissions are subject to user fees.
Medical devices can be marketed only for the indications for which they are cleared or approved. After a device has received 510(k) clearance for a specific intended use, any change or modification that significantly affects its safety or effectiveness, such as a significant change in the design, materials, method of manufacture or intended use, may require a new 510(k) clearance or PMA approval and payment of an FDA user fee. The determination as to whether or not a modification could significantly affect the device’s safety or effectiveness is initially left to the manufacturer using available FDA guidance; however, the FDA may review this determination to evaluate the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and recall the modified device until 510(k) clearance or PMA approval is obtained. The manufacturer may also be subject to significant regulatory fines or penalties.
PMA Approval. A PMA must be submitted to the FDA if the device is classified in Class III or otherwise cannot be cleared through the 510(k) process (although the FDA has discretion to continue to allow certain pre-amendment Class III devices to use the 510(k) process). PMA applications must be supported by, among other things, valid scientific evidence demonstrating the safety and effectiveness of the device, which typically requires extensive data, including technical, preclinical, clinical and manufacturing data. A PMA application must also include, among other things, a complete description of the device and its components, a detailed description of the methods, facilities and controls used to manufacture the device, and proposed labeling. FDA regulations provide 180 days for the FDA to review a PMA application and make a determination, although the process typically takes significantly longer, and may require several years to complete. In addition, the FDA will conduct a pre-approval inspection of the applicant or its third-party manufacturers’ or suppliers’ manufacturing facilities to ensure compliance with the QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures.
None of our products are currently approved under a PMA approval, and we have no plans for any indication or system improvement or extension that we believe would require a PMA.
Clinical Trials
Clinical trials are generally required to support a PMA application and are sometimes required for 510(k) clearance. Such trials generally require submission of an investigational device exemption, or IDE, application to the FDA for a specified number of patients and study sites, unless the product is deemed a non-significant risk device eligible for more abbreviated IDE requirements. If an IDE is required, the FDA and the appropriate institutional review boards, or IRBs, at the clinical sites must approve the study before clinical trials may begin.
23
Table of Contents
Clinical trials are subject to extensive monitoring, record keeping and reporting requirements. Clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to good clinical practices. To conduct a clinical trial, the patient’s informed consent must be obtained in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. The clinical trial sponsor, the FDA or the IRB could suspend or terminate a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable health risk. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and effectiveness of the device or may otherwise not be sufficient to obtain FDA clearance or approval to market the product.
Continuing FDA Regulation
Any devices we manufacture or distribute pursuant to 510(k) clearance or PMA approval by the FDA are subject to pervasive and continuing regulation by the FDA and certain state agencies. These include product listing and establishment registration requirements, which help facilitate FDA inspections and other regulatory actions. We are required to adhere to applicable cGMP requirements, as set forth in the QSR, which require, manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all phases of the design and manufacturing process. Noncompliance with these regulations can result in, among other things, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, FDA refusal to grant 510(k) clearance or PMA approval to new devices, withdrawal of existing clearances or approvals, and criminal prosecution.
We must also comply with post-market surveillance regulations, including medical device reporting, or MDR, requirements which require that we review and report to the FDA any incident in which our products may have caused or contributed to a death or serious injury, and any incident in which our product has malfunctioned if that malfunction would likely cause or contribute to a death or serious injury if it were to recur. Labeling and promotional activities are subject to scrutiny by the FDA and, in certain circumstances, by the Federal Trade Commission. Medical devices approved or cleared by the FDA may not be promoted for unapproved or uncleared uses, otherwise known as “off-label” promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
Failure to comply with applicable regulatory requirements, including delays in or failures to report incidents to the FDA as required under the MDR regulations or off-label promotion, can result in enforcement action by the FDA which can include any of the following sanctions:
| • | warning letters, untitled letters, fines, injunctions, consent decrees and civil penalties; |
| • | customer notifications or repair, replacement, refunds, recall, administrative detention or seizure of our products; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | refusing or delaying requests for 510(k) clearance or PMA approval of new or modified products; |
| • | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| • | refusal to grant export approval for products; or |
| • | criminal prosecution. |
We cannot guarantee that we have adequately complied with all regulatory requirements. If any of these events were to occur, they could have a material adverse effect on our business.
24
Table of Contents
U.S. Anti-Kickback, False Claims and Other Healthcare Laws
We are also subject to healthcare regulation and enforcement by the federal government and the states and foreign governments and authorities in the locations in which we conduct our business. These other agencies include, without limitation, the Centers for Medicare and Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services, the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, as well as state and local governments. Such agencies enforce a variety of laws which include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, data privacy and security, and physician sunshine laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any good, facility, item or service reimbursable, in whole or part by Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value, including cash, improper discounts, and free or reduced price items and services. Among other things, the Anti-Kickback Statute has been interpreted to apply to arrangements between medical device manufacturers on the one hand and prescribers and purchasers on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the Anti-Kickback Statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate, in order to have committed a violation. Further, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute also constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to or approval by the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of medical device companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. In addition, the federal civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent
25
Table of Contents
statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the Anti-Kickback Statute a person or entity does not need to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation.
Also, many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s security standards directly applicable to business associates, defined as service providers of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, many state laws govern the privacy and security of health information in certain circumstances, many of which differ from HIPAA and each other in significant ways and may not have the same effect.
Additionally, there has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education and Reconciliation Act, or collectively, the Affordable Care Act, imposed, among other things, new annual reporting requirements for covered manufacturers for certain payments and “transfers of value” provided to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit timely, accurately and completely the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary penalties of up to an aggregate of $150,000 per year and up to an aggregate of $1 million per year for “knowing failures.” Covered manufacturers were required to report detailed payment data for the first reporting period (August 1, 2013 – December 31, 2013) under this law and submit legal attestation to the completeness and accuracy of such data by June 30, 2014. Thereafter, covered manufacturers must submit reports by the 90th day of each subsequent calendar year. In addition, certain states require implementation of commercial compliance programs and compliance with the device industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, impose restrictions on marketing practices, and/or tracking and reporting of gifts, compensation and other remuneration or items of value provided to physicians and other healthcare professionals and entities.
These laws impact the kinds of financial arrangements we may have with hospitals, physicians or other potential purchasers of our products. They particularly impact how we structure our sales offerings, including discount practices, customer support, education and training programs, physician consulting, research grants and other arrangements. If our operations are found to be in violation of any of the health regulatory laws described above or any other laws that apply to us, we may be subject to penalties, including potentially significant criminal, civil and/or administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of our operations.
Coverage and Reimbursement
Our currently cleared products are treated as supplies utilized in balloon sinus dilation procedures and if covered by third-party payors, are paid for as part of a stand-alone balloon sinus dilation procedure or as a part of FESS if used in conjunction with FESS. Failure by physicians, hospitals, ASCs and other users of our products to obtain sufficient coverage and reimbursement from healthcare payors for procedures in which our products are used, or adverse changes in government and private third-party payors’ policies would have a material adverse effect on our business.
26
Table of Contents
In the United States, third-party payors continue to implement initiatives that restrict the use of certain technologies to those that meet certain clinical evidentiary requirements. We are aware of a number of third-party payors in the United States, including Anthem/Wellpoint and certain Blue Cross Blue Shield plans that consider stand-alone balloon sinus dilation procedures to be experimental or investigational. Failure to obtain and maintain favorable coverage policies could have a material adverse effect on our business and operations.
In addition to uncertainties surrounding coverage policies, there are periodic changes to reimbursement. Third-party payors regularly update reimbursement amounts and also from time to time revise the methodologies used to determine reimbursement amounts. This includes annual updates to payments to physicians, hospitals and ASCs for procedures during which our products are used. Because the cost of our products generally is recovered by the healthcare provider as part of the payment for performing a procedure and not separately reimbursed, these updates could directly impact the demand for our products. An example of payment updates is the Medicare program’s updates to hospital and physician payments, which are done on an annual basis using a prescribed statutory formula. In the past, when the application of the formula resulted in lower physician payments, Congress has passed interim legislation to prevent the reductions. Most recently, the Protecting Access to Medicare Act of 2014, signed into law in April 2014, provided for a 0.5% update from 2013 payment rates under the Medicare Physician Fee Schedule through 2014 and a 0% update from January 1 until April 1, 2015. If Congress fails to intervene to prevent the negative update factor in future years, the resulting decrease in payment may adversely affect our business. Any changes in coverage and reimbursement that further restricts coverage of our products or lowers reimbursement for procedures using our devices could materially affect our business.
Healthcare Reform
Current and future legislative proposals to further reform healthcare or reduce healthcare costs may limit coverage for the procedures associated with the use of our products, or result in lower reimbursement for those procedures. The cost containment measures that payors and providers are instituting and the effect of any healthcare reform initiative implemented in the future could significantly reduce our revenues from the sale of our products.
By way of example, the Affordable Care Act substantially changed healthcare financing and delivery by both governmental and private insurers, and significantly impacted the pharmaceutical and medical device industries. The Affordable Care Act imposed, among other things, a new federal excise tax on the sale of certain medical devices, provided incentives to programs that increase the federal government’s comparative effectiveness research, and implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and will stay in effect through 2024 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012, was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
27
Table of Contents
Employees
As of December 31, 2014, we had 177 employees, of whom 20 were in manufacturing, 14 were in research and development and 114 were in sales, marketing and reimbursement.
Financial Information
We operate in one reportable segment, which we have presented as the aggregation of our chronic sinusitis product families. Within this reportable segment we are organized on the basis of the products and services we provide which are used for the treatment of chronic sinusitis. See Note B—Summary of Significant Accounting Policies—Segment, Geographical and Customer Concentration of the Notes to Financial Statements to our audited financial statements included elsewhere in this Annual Report on Form 10-K for financial information regarding segment reporting.
Other Information
We are incorporated in Delaware as Entellus Medical, Inc. Our offices are located at 3600 Holly Lane North, Suite 40, Plymouth, MN 55447. Our telephone number is (763) 463-1595. Our corporate website is www.entellusmedical.com. The information contained on or that can be accessed through our website is not incorporated by reference into this Annual Report on Form 10-K. We make available, free of charge through our website, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to these reports as soon as reasonably practicable after we electronically file these materials with, or otherwise furnish them to, the SEC.
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and, for so long as we are an emerging growth company, are eligible to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. These include, but are not limited to:
| • | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
| • | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| • | reduced disclosure obligations regarding executive compensation; and |
| • | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may remain an “emerging growth company” until as late as December 31, 2020, the fiscal year-end following the fifth anniversary of the completion of our initial public offering, or IPO, though we may cease to be an “emerging growth company” earlier under certain circumstances, including if (1) we have more than $1.0 billion in annual revenue in any fiscal year, (2) the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30 or (3) we issue more than $1.0 billion of non-convertible debt over a three-year period.
In addition, Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, or the Securities Act, for complying with new or revised accounting standards. In other words, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to take advantage of such extended transition period. Section 107 of the JOBS Act provides that we can elect to opt out of the extended transition period at any time, which election is irrevocable.
28
Table of Contents
| Item 1A. | Risk Factors |
Our business faces significant risks and uncertainties. Certain important factors may have a material adverse effect on our business prospects, financial condition and results of operations, and you should carefully consider them. Accordingly, in evaluating our business, we encourage you to consider the following discussion of risk factors, in its entirety, in addition to other information contained in or incorporated by reference into this Annual Report on Form 10-K and our other public filings with the SEC. Other events that we do not currently anticipate or that we currently deem immaterial may also affect our business, prospects, financial condition and results of operations.
Risks Related to Our Business
We have incurred significant operating losses since inception, we expect to incur operating losses in the future and we may not be able to achieve or sustain profitability.
We have incurred net losses since our inception in 2006. Our net loss for the year ended December 31, 2014 was $6.9 million. As of December 31, 2014, we had an accumulated deficit of $103.7 million. To date, we have financed our operations primarily through private placements of our convertible preferred securities, certain debt-related financing arrangements and from sales of our products and most recently with the completion of our IPO. We have devoted substantially all of our resources to research and development of our products, sales and marketing activities and clinical and regulatory initiatives to obtain clearances or approvals for our products. Our ability to generate sufficient revenue from our existing products or from any of our products in development, to transition to profitability and generate consistent positive cash flows, is uncertain. We also expect that our operating expenses will continue to increase as we continue to build our commercial infrastructure, develop, enhance and commercialize new products and incur additional operational costs associated with being a public company. As a result, we expect to continue to incur operating losses for the foreseeable future and may never achieve profitability. Furthermore, even if we do achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis. If we do not achieve profitability, it will be more difficult for us to finance our business and accomplish our strategic objectives.
Our revenue is primarily generated from our XprESS family of multi-sinus products and we are therefore highly dependent on a limited number of products.
We began selling our XprESS family of products in 2010, and these products accounted for 95% of our revenue for the year ended December 31, 2014. We expect that sales of these products will continue to account for the substantial majority of our revenue going forward. Therefore, our ability to execute our growth strategy and become profitable will depend not only upon a continued shift of more sinus procedures from traditional operating room settings to the ENT physician office setting, but also to the adoption of our XprESS family of products to perform those procedures. Some ENT physicians may have prior history with or preference for a competitor’s balloon sinus dilation products or be reluctant to alter practice patterns and undergo training required to use our products. If our XprESS products fail to achieve wide market acceptance for any reason, our business may be adversely affected.
If physicians or patients are not willing to change current practices and continue to adopt office-based balloon sinus dilation procedures, our products may fail to gain increased market acceptance, and our business will suffer.
Our primary strategy to grow revenues is to drive an increase in office-based balloon sinus dilation procedures and the adoption of our XprESS family of products to perform these procedures. While the number of ENT physicians performing sinus procedures using office-based balloon sinus dilation has increased in recent years, there is a significant group of ENT physicians who have not yet adopted these procedures, and additional ENT physicians may not adopt office-based balloon sinus dilation for a number of reasons, including:
| • | lack of significant experience with balloon sinus dilation as a treatment alternative; |
29
Table of Contents
| • | lack of availability of adequate insurance coverage or reimbursement for office-based balloon sinus dilation procedures; |
| • | perceived inadequacy of evidence supporting clinical benefits or cost-effectiveness of office-based balloon sinus dilation over existing alternatives; |
| • | a perception among some ENT physicians of patients’ inability to tolerate balloon sinus dilation procedures in the physician office setting; and |
| • | liability risks generally associated with the use of new products and procedures. |
If additional ENT physicians do not continue to adopt, or existing ENT physicians cease using office-based balloon sinus dilation procedures for any reason, including those listed above, our ability to execute our growth strategy would be impaired, and our business may be adversely impacted.
We believe recommendations and support of our products by notable ENT physicians can influence market acceptance and adoption. If we do not receive support from these influential ENT physicians, our ability to achieve broad market acceptance for our products may be impaired.
In addition, if patient receptivity toward treatment in an ENT physician office setting becomes less favorable in the future, this shift could negatively impact market acceptance of our products. Any negative change due to patient receptivity could also be compounded by patients reporting to physicians or other patients through word-of-mouth or social media.
Additionally, while it is currently more cost-effective to the healthcare system for providers to perform balloon sinus dilation in an ENT physician’s office than a FESS procedure in the operating room, healthcare economics are always subject to change. If the use of our balloon sinus dilation products were to cease being more cost-effective than FESS due to changes in reimbursement economics, our products may fail to gain market acceptance, our future growth would be limited and our business may be adversely affected. In addition, if payment rates for office-based balloon sinus dilation procedures were to be lowered, in particular by Medicare, which we believe would likely cause other third-party payors to implement comparable reductions to their reimbursement rates, ENT physicians may reduce the frequency of balloon sinus dilation procedures and our business may be adversely affected.
If we are unable to achieve and maintain adequate levels of coverage or reimbursement for the procedures using our products, or any future products we may seek to commercialize, their commercial success may be severely hindered.
Hospital, physician and other healthcare provider customers, including ambulatory surgery center, or ASC, customers that purchase our products typically bill various third-party payors to cover all or a portion of the costs and fees associated with the procedures in which our products are used and bill patients for any deductibles or co-payments. In the hospital and ASC settings, our products are often used in a hybrid procedure in conjunction with traditional FESS. Because there is often no separate reimbursement for supplies used in surgical procedures, and with respect to FESS, no separate reimbursement for balloon sinus dilation, the additional cost associated with the use of our products in these settings can impact the profit margin of the hospital or surgery center where the hybrid procedure is performed. Some of our target customers may be unwilling to adopt our products in light of the additional associated cost. Further, any decline in the amount payors are willing to reimburse our customers for balloon sinus dilation procedures in either the office or facility setting could make it difficult for existing customers to continue using or to adopt our products and could create additional pricing pressure for us. If we are forced to lower the price we charge for our products, our gross margins will decrease, which will materially adversely affect our business.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the United States, no uniform
30
Table of Contents
policy of coverage and reimbursement for procedures using our products exists among third-party payors. Therefore, coverage and reimbursement for procedures using our products can differ significantly from payor to payor. In addition, payors continually review new and existing technologies for possible coverage and can, without notice, deny or reverse coverage or alter pre-authorization requirements for new or existing products and procedures. Additionally, some third-party payors do not currently cover or reimburse balloon sinus dilation procedures because they have determined insufficient evidence of favorable clinical outcomes is available. We are actively working to avoid or to reverse these non-coverage decisions, as applicable, but cannot provide assurance that we will be successful in these efforts. If we are not successful in reversing existing noncoverage policies, or if third-party payors that currently cover or reimburse balloon sinus dilation procedures reverse or limit their coverage in the future, or if other third-party payors issue similar policies, this could have a material adverse impact on our business.
Further, we believe that future coverage and reimbursement may be subject to increased restrictions, such as additional preauthorization requirements, both in the United States and in international markets. Third-party coverage and reimbursement for procedures using our products or any of our products in development for which we may receive regulatory approval may not be available or adequate in either the United States or international markets, which could have an adverse impact on our business.
We compete and may compete in the future against other companies, some of which have longer operating histories, more established products or greater resources than we do, which may prevent us from achieving increased market penetration or improved operating results.
Our industry is highly competitive, subject to change and significantly affected by new product introductions and other activities of industry participants. We believe competitors have historically dedicated and will continue to dedicate significant resources to promote their products or develop new products or methods to treat sinusitis. We currently compete against companies providing alternative treatments to balloon sinus dilation, as well as companies that provide competing balloon sinus dilation products. For example, we are aware that the Xomed division of Medtronic has recently launched a balloon sinus dilation device that competes with our XprESS family of products. Additionally, Intersect ENT is currently developing products for office-based treatment of sinusitis, which if approved may be competitive with us in the future. Many of the companies developing or marketing these ENT products are publicly traded or are divisions of publicly traded companies, including the Xomed division of Medtronic, the Acclarent division of Johnson & Johnson and the ArthroCare division of Smith & Nephew, each of which produces competing balloon sinus dilation products, and the Gyrus ACMI division of Olympus, Stryker Corporation and Intersect ENT, each of which produces tools and devices for FESS. These companies may enjoy several competitive advantages, including:
| • | greater financial and human capital resources; |
| • | significantly greater name recognition; |
| • | control of key intellectual property, which could impact future products under development; |
| • | established relationships with ENT physicians, suppliers, referring physicians, customers and third-party payors; |
| • | additional lines of products, and the ability to offer rebates or bundle products to offer greater discounts or incentives to gain a competitive advantage; and |
| • | more established sales, marketing and worldwide distribution networks. |
To a lesser extent, we also have faced and may in the future face competition from pharmaceutical companies that develop medical therapies to treat sinusitis.
In addition, substantially all of our products are designed to be used exclusively during balloon sinus dilation or FESS procedures. If another company successfully develops an approach for the treatment of chronic
31
Table of Contents
sinusitis that would not benefit from the use of our products, or if another company develops a balloon sinus dilation device that is more efficacious, more cost-effective or easier to use than our products, sales of our products could be significantly and adversely affected, which could have a material adverse effect on our business.
Our business is substantially dependent on our license agreement with Acclarent, which exposes us to a variety of risks.
We are a party to a license agreement, or the Acclarent License, with Acclarent. Under the Acclarent License, Acclarent has granted us a non-exclusive license under patents held by Acclarent to manufacture, use and commercialize our currently marketed XprESS, PathAssist and FinESS products and certain future versions of those products, or, collectively, the Covered Products. We have certain obligations under the Acclarent License, including paying Acclarent a royalty on the sale of Covered Products. If we fail to comply with our obligations under the Acclarent License, Acclarent may terminate the Acclarent License, in which case our current products would no longer enjoy a license under Acclarent’s patents. Without the license, Acclarent might successfully enforce its patents against us based on the sale of our current products, possibly resulting in our being ordered to pay Acclarent monetary damages and/or in our being enjoined from selling our current products within any country where Acclarent has a valid and enforceable claim covering a given product. Furthermore, because the Acclarent License is non-exclusive, Acclarent could choose to also license the same intellectual property to other third-party competitors. Any of these outcomes could have a material adverse effect on our business.
In addition, we do not control the prosecution of the patent applications licensed to us under the Acclarent License. Therefore, these patents and applications may not be prosecuted or enforced in a manner consistent with the best interests of our business. If Acclarent fails to maintain the patents it has licensed to us under the Acclarent License, or loses its rights to those patents, the rights we have licensed may be reduced or eliminated, which could severely harm our ability to successfully commercialize our products.
Our long-term growth depends on our ability to develop and commercialize additional ENT products.
The medical device industry is highly competitive and subject to rapid change and technological advancements. Therefore, it is important to our business that we continue to enhance our product offerings and introduce new products. Developing products is expensive and time-consuming and could divert management’s attention away from our core balloon sinus dilation business. Even if we are successful in developing additional products, the success of any new product offering or enhancements to existing products will depend on several factors, including our ability to:
| • | properly identify and anticipate ENT physician and patient needs; |
| • | develop and introduce new products or product enhancements in a timely manner; |
| • | avoid infringing upon the intellectual property rights of third-parties; |
| • | demonstrate, if required, the safety and efficacy of new products with data from preclinical studies and clinical trials; |
| • | obtain the necessary regulatory clearances or approvals for new products or product enhancements; |
| • | be fully FDA-compliant with marketing of new devices or modified products; |
| • | provide adequate training to potential users of our products; |
| • | receive adequate coverage and reimbursement for procedures performed with our products; and |
| • | develop an effective and dedicated sales and marketing team. |
If we are unsuccessful in developing and commercializing new products, our ability to increase our revenue may be impaired.
32
Table of Contents
Our future growth depends on physician awareness and adoption of our balloon sinus dilation devices.
We focus our sales, marketing and training efforts on ENT physicians. However, the initial point of contact for many patients suffering from chronic sinusitis may be primary care physicians or other referring medical professionals, such as nurse practitioners or physician assistants, who commonly see patients with sinus infections and sinusitis and prescribe pharmaceuticals to address sinusitis symptoms. We believe that education of primary care physicians and other medical professionals about the clinical merits and patient benefits of office-based balloon sinus dilation procedures is an important element of the growth of office-based balloon sinus dilation procedures. If we fail to educate primary care physicians and other medical professionals, they may not refer sinusitis patients to an ENT physician to perform balloon sinus dilation procedures and, as a result, those patients may continue to receive only medical management. If this were to occur, our ability to increase our revenue may be impaired.
The efficacy of our products have been demonstrated in clinical studies sponsored by us, and insurance companies who do not cover stand-alone balloon sinus dilation may require additional or independently performed clinical studies prior to adopting or covering or maintaining coverage of procedures in which our products are used as a stand-alone therapy.
Our success depends on the medical and third-party payor community’s acceptance of our products as tools that are useful to ENT physicians treating patients with chronic sinusitis. We have sponsored seven clinical studies with over 600 patients to track outcomes of treatment with our products. While the results of these studies, collectively indicate that our products can be used to successfully treat uncomplicated chronic sinusitis equally effectively as FESS while also providing better recovery outcomes, if physicians or insurers do not find our data compelling or wish to wait for additional or independently performed studies, they may choose not to use or provide coverage and reimbursement for our products. Currently, we estimate there are insurance plans, including a number of large third-party payors, covering approximately 84 million lives that have determined balloon sinus dilation to be investigational and therefore do not cover it at this time.
In addition, the long-term effects of balloon sinus dilation with our products beyond 24 months are not known. Certain ENT physicians, hospitals, ASCs and insurers may prefer to see longer-term efficacy data than we have produced. We cannot make any assurances that any data that we or others generate will be consistent with that observed in our existing clinical studies.
It is difficult to forecast future performance and our financial results may vary from forecasts and may fluctuate from quarter to quarter.
Our limited operating history and commercial experience make it difficult for us to predict future performance. A number of factors over which we have limited control, such as seasonal variations in revenue, may contribute to fluctuations in our financial results. In the first quarter, our results can be impacted by adverse weather and by resetting of annual patient healthcare insurance plan deductibles, both of which may cause patients to delay elective procedures in which our products are used. In the second quarter, demand may be increased by the seasonal nature of allergies and the resultant onset of sinus-related symptoms. In the third quarter, we believe the number of elective surgeries and particularly FESS or balloon sinus dilation procedures nationwide is historically lower than other quarters as a result of summer vacations of ENT physicians and their patients. In the fourth quarter, demand typically increases with the onset of the cold and flu season and related symptoms, as well as the desire of patients to spend the remaining balances in their flexible-spending accounts or because of lower out-of-pocket costs to patients who have already met their annual deductibles under their health insurance plans.
Other factors that may cause fluctuation in our quarterly results include:
| • | ENT physician adoption of our products; |
| • | timing of new product offerings, acquisitions, licenses or other significant events by us or our competitors; |
33
Table of Contents
| • | unanticipated pricing pressure; |
| • | the hiring, retention and continued productivity of our sales representatives; |
| • | our ability to expand the geographic reach of our sales and marketing efforts; |
| • | our ability to obtain regulatory clearance or approval for our products in development or for our current products outside the United States; |
| • | results of clinical research and trials on our existing products and products in development; |
| • | delays in receipt of anticipated purchase orders; |
| • | delays in, or failure of, component and raw material deliveries by our suppliers; and |
| • | positive or negative coverage in the media or clinical publications of our products or products of our competitors or our industry. |
In the event our actual revenue and operating results do not meet our forecasts or the forecasts or estimates of the research analysts that cover us for a particular period, the market price of our common stock may decline substantially.
Consolidation in the healthcare industry or group purchasing organizations could lead to demands for price concessions, which may impact our ability to sell our products at prices necessary to support our current business strategies.
Healthcare costs have risen significantly over the past decade, which has resulted in or led to numerous cost reform initiatives by legislators, regulators and third-party payors. Cost reform has triggered a consolidation trend in the healthcare industry to aggregate purchasing power, which may create more requests for pricing concessions in the future. Additionally, group purchasing organizations, independent delivery networks and large single accounts may continue to use their market power to consolidate purchasing decisions for hospitals and ASCs. We expect that market demand, government regulation, third-party coverage and reimbursement policies and societal pressures will continue to change the healthcare industry worldwide, resulting in further business consolidations and alliances among our customers, which may exert further downward pressure on the prices of our products.
The size of the market for balloon sinus dilation products has not been established with precision, and may be smaller than we estimate.
Our estimate of the annual total addressable market for balloon sinus dilation products is based on a number of internal and third-party estimates, including, without limitation, the number of annual patient visits to ENT and primary care physicians, internal estimates regarding the number of certain types of FESS procedures performed annually, and our estimate of the number of people currently under medical management who would benefit from and be amenable to a balloon sinus procedure. In addition, our internal estimates are based in large part on current patterns of patient selection by ENT physicians for balloon sinus dilation. While we believe these factors have historically provided and may continue to provide us with effective tools in estimating the total market for balloon sinus dilation procedures and our products, these estimates may not be correct and the conditions supporting our estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. As a result, our estimates of the annual total addressable market for our products may prove to be incorrect. If the actual number of patients who would benefit from our products and the annual total addressable market for our products is smaller than we have estimated, it may impair our sales growth and have an adverse impact on our business.
34
Table of Contents
We have limited experience marketing and selling our products, and if we are unable to expand, manage and maintain our direct sales and marketing organizations we may not be able to generate anticipated revenue.
We began selling our first FDA-cleared product, FinESS, in April 2008. We subsequently began selling our XprESS devices and PathAssist tools in February 2010 and August 2011, respectively. As a result, we have limited experience marketing and selling our products. As of December 31, 2014, our direct sales organization, including marketing and reimbursement, consisted of 114 employees, having increased from 84 employees as of December 31, 2012. Our operating results are directly dependent upon the sales and marketing efforts of our employees. If our direct sales force fails to adequately promote, market and sell our products, our sales may suffer.
In order to generate future sales growth, we will need to expand the size and geographic scope of our direct sales organization. Accordingly, our future success will depend largely on our ability to continue to hire, train, retain and motivate skilled sales personnel with significant technical knowledge of sinus procedures and related products. Because the competition for their services is high, we cannot assure you we will be able to hire and retain additional personnel on favorable or commercially reasonable terms, if at all. Failure to hire or retain qualified sales personnel would prevent us from expanding our business and generating sales. If we are unable to expand our sales and marketing capabilities, we may not be able to effectively commercialize our products, which could have an adverse impact on our business.
We may be unable to manage our growth effectively.
Our past growth has provided, and our future growth may create, challenges to our organization. From December 31, 2012, to December 31, 2014, the number of our employees increased from 124 to 173. In the future, we expect to hire and train new personnel as we continue to grow and expand our operations. As a public company, we will need to support managerial, operational, financial and other resources. This growth may place significant strain on us. Successful growth is also dependent upon our ability to implement appropriate financial and management controls and systems and procedures. If we fail to manage these challenges effectively, there may be an adverse impact on our business.
Our ability to maintain our competitive position depends on our ability to attract and retain key executives and highly qualified personnel.
We believe that our continued success depends to a significant extent upon the efforts and abilities of our key executives. Although we have entered into employment letter agreements with certain of our executive officers, each of them may terminate their employment with us at any time. We expect that Brian Farley will transition from his role as Chief Executive Officer to Executive Chairman in the future. The replacement of any of our key personnel likely would involve significant time and costs and may significantly delay or prevent the achievement of our business objectives and could therefore have an adverse impact on our business. In addition, we do not carry any “key person” insurance policies that could offset potential loss of service under applicable circumstances.
Many of our employees have become or will soon become vested in a substantial amount of our common stock or a number of common stock options. Our employees may be more likely to leave us if the shares they own have significantly appreciated in value relative to the original purchase prices of the shares, or if the exercise prices of the options that they hold are significantly below the market price of our common stock, particularly after the expiration of the lock-up agreements described herein. Our future success also depends on our ability to continue to attract and retain additional executive officers and other key employees.
35
Table of Contents
We face the risk of product liability claims that could be expensive, divert management’s attention and harm our reputation and business. We may not be able to maintain adequate product liability insurance.
Our business exposes us to the risk of product liability claims that are inherent in the testing, manufacturing and marketing of medical devices. This risk exists even if a device is cleared or approved for commercial sale by the FDA and manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority. Our products are designed to affect, and any future products will be designed to affect, important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our products or our products in development could result in patient injury or death. The medical device industry has historically been subject to extensive litigation over product liability claims, and we cannot offer any assurance that we will not face product liability suits. We may be subject to product liability claims if our balloon sinus dilation or other products cause, or merely appear to have caused, patient injury or death. In addition, an injury that is caused by the activities of our suppliers, such as those who provide us with components and raw materials, may be the basis for a claim against us. Product liability claims may be brought against us by consumers, healthcare providers or others selling or otherwise coming into contact with our products, among others. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities and reputational harm. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| • | costs of litigation; |
| • | distraction of management’s attention from our primary business; |
| • | the inability to commercialize our existing or new products; |
| • | decreased demand for our products or, if cleared or approved, products in development; |
| • | damage to our business reputation; |
| • | product recalls or withdrawals from the market; |
| • | withdrawal of clinical trial participants; |
| • | substantial monetary awards to patients or other claimants; or |
| • | loss of revenue. |
While we may attempt to manage our product liability exposure by proactively recalling or withdrawing from the market any defective products, any recall or market withdrawal of our products may delay the supply of those products to our customers and may impact our reputation. We can provide no assurance that we will be successful in initiating appropriate market recall or market withdrawal efforts that may be required in the future or that these efforts will have the intended effect of preventing product malfunctions and the accompanying product liability that may result. Such recalls and withdrawals may also be used by our competitors to harm our reputation for safety or be perceived by patients as a safety risk when considering the use of our products, either of which could have an adverse impact on our business.
In addition, although we have product liability and clinical study liability insurance that we believe is appropriate, this insurance is subject to deductibles and coverage limitations. Our current product liability insurance may not continue to be available to us on acceptable terms, if at all, and, if available, coverage may not be adequate to protect us against any future product liability claims. If we are unable to obtain insurance at an acceptable cost or on acceptable terms or otherwise protect against potential product liability claims, we could be exposed to significant liabilities. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could have an adverse impact on our business.
We utilize third-party, single-source suppliers for some components and materials used in our products, and the loss of any of these suppliers could have an adverse impact on our business.
We rely on single-source suppliers for some components and materials used in our products. Our ability to supply our products commercially and to develop any future products depends, in part, on our ability to obtain these components in accordance with regulatory requirements and in sufficient quantities for commercialization
36
Table of Contents
and clinical testing. While our suppliers have generally met our demand for their products on a timely basis in the past, we cannot assure that they will in the future be able to meet our demand for their products, either because we do not have long-term agreements with those suppliers, our relative importance as a customer to those suppliers, or their ability to produce the components used in our products.
While we believe replacement suppliers exist for all components and materials we obtain from single sources, establishing additional or replacement suppliers for any of these components or materials, if required, may not be accomplished quickly. Even if we are able to find a replacement supplier, the replacement supplier would need to be qualified and may require additional regulatory authority approval, which could result in further delay. While we seek to maintain adequate inventory of the single-source components and materials used in our products in the event of disruption, those inventories may not be sufficient.
If our third-party suppliers fail to deliver the required commercial quantities of materials on a timely basis and at commercially reasonable prices, and we are unable to find one or more replacement suppliers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality on a timely basis, the continued commercialization of our products, the supply of our products to customers and the development of any future products would be delayed, limited or prevented, which could have an adverse impact on our business.
If the quality of our products does not meet the expectations of ENT physicians or patients, then our brand and reputation could suffer and our business could be adversely impacted.
In the course of conducting our business, we must adequately address quality issues that may arise with our products, as well as defects in third-party components included in our products. Although we have established internal procedures to minimize risks that may arise from quality issues, there can be no assurance that we will be able to eliminate or mitigate occurrences of these issues and associated liabilities. If the quality of our products does not meet the expectations of ENT physicians or patients, then our brand and reputation could suffer with those ENT physicians or patients and our business could be adversely impacted.
If we choose to acquire new and complementary businesses, products or technologies, we may be unable to complete these acquisitions or to successfully integrate them in a cost-effective and non-disruptive manner.
Our success depends on our ability to continually enhance and broaden our product offerings in response to changing customer demands, competitive pressures and advances in technologies. Accordingly, although we have no current commitments with respect to any acquisition or investment, we may in the future pursue the acquisition of, or joint ventures relating to, complementary businesses, products or technologies instead of developing them ourselves. We do not know if we will be able to successfully complete any future acquisitions or joint ventures, or whether we will be able to successfully integrate any acquired business, product or technology or retain any key employees related thereto. Integrating any business, product or technology we acquire could be expensive and time-consuming, disrupt our ongoing business and distract our management. If we are unable to integrate any acquired businesses, products or technologies effectively, our business will suffer. In addition, any amortization or charges resulting from the costs of acquisitions could increase our expenses.
The implementation of a new enterprise resource planning system could cause disruption to our business and operations.
We are in the process of implementing a new enterprise resource planning system, which we expect to be implemented during 2015. This system will integrate our operations, including supply-chain, order entry, manufacturing, inventory and financial reporting, among others. This project requires significant investment of capital and human resources, the re-engineering of many processes of our business, and the attention of many associates and managers who would otherwise be focused on other aspects of our business. If we fail to implement the system successfully or in the contemplated timeframe, or if the system does not perform in a satisfactory manner once implementation is complete, our business, including our ability to report financial results accurately and timely, could be adversely affected.
37
Table of Contents
We expect to incur significant additional costs as a result of being a public company, which may adversely affect our operating results and financial condition.
We expect to incur costs associated with corporate governance requirements that are newly applicable to us as a public company, including rules and regulations of the SEC, under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, or the Dodd-Frank Act, and the Securities Exchange Act of 1934, as amended, or the Exchange Act, as well as the rules of The Nasdaq Global Market. We expect that these rules and regulations will significantly increase our accounting, legal and financial compliance costs and make some activities more time-consuming. We also expect these rules and regulations to make it more expensive for us to maintain directors’ and officers’ liability insurance. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors or as executive officers. Accordingly, increases in costs incurred as a result of becoming a publicly traded company may adversely affect our operating results and financial condition.
Failure of a key information technology system, process or site could have an adverse impact on our business.
We rely extensively on information technology systems to conduct business. These systems impact, among other things, ordering and managing materials from suppliers, shipping products to customers, processing transactions, summarizing and reporting results of operations, complying with regulatory, legal or tax requirements, data security and other processes necessary to manage our business. If our systems are damaged or cease to function properly due to any number of causes, ranging from catastrophic events to power outages to security breaches, and our business continuity plans do not effectively compensate on a timely basis, we may suffer interruptions in our operations, which could have an adverse impact on our business.
In addition, we accept payments for many of our sales through credit and debit card transactions, which are handled through a third-party payment processor. As a result, we are subject to a number of risks related to credit and debit card payments. As a result of these transactions, we pay interchange and other fees, which may increase over time and could require us to either increase the prices we charge for our products or experience an increase in our costs and expenses. In addition, as part of the payment processing process, we transmit our customers’ credit and debit card information to our third-party payment processor. We may in the future become subject to lawsuits or other proceedings for purportedly fraudulent transactions arising out of the actual or alleged theft of our customers’ credit or debit card information if the security of our third-party credit card payment processor is breached. We and our third-party credit card payment processor are also subject to payment card association operating rules, certification requirements and rules governing electronic funds transfers, which could change or be reinterpreted to make it difficult or impossible for us to comply. If we or our third-party credit card payment processor fail to comply with these rules or requirements, we may be subject to fines and higher transaction fees and lose our ability to accept credit and debit card payments from our customers, and there may be an adverse impact on our business.
If our facilities are damaged or become inoperable, we will be unable to continue to research, develop and manufacture our products and, as a result, there will be an adverse impact on our business until we are able to secure a new facility.
We do not have redundant facilities. We perform substantially all of our research and development, manufacturing and back office activity and maintain all our raw material and finished goods inventory in a single location in Plymouth, Minnesota. Our facility and equipment would be costly to replace and could require substantial lead time to repair or replace. The facility may be harmed or rendered inoperable by natural or man-made disasters, including, but not limited to, tornadoes, flooding, fire and power outages, which may render it difficult or impossible for us to perform our research, development, manufacturing and commercialization activities for some period of time. The inability to perform those activities, combined with our limited inventory of reserve raw materials and finished product, may result in the inability to continue manufacturing our products during such periods and the loss of customers or harm to our reputation. Although we possess insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and this insurance may not continue to be available to us on acceptable terms, or at all.
38
Table of Contents
We have no prior experience selling our products outside of the United States and Canada and do not know if we will be successful in achieving adoption of our products and revenue growth outside of the United States in a timely manner. If we commercialize any products outside of the United States, a variety of risks associated with international operations could materially adversely affect our business.
We may establish a direct sales force outside of the United States or enter into agreements with third-parties to market our products outside the United States. We expect that we would be subject to additional risks related to entering into international business relationships, including:
| • | training of third-parties on our products and the procedures in which they are used; |
| • | reduced protection for intellectual property rights; |
| • | unexpected changes in tariffs, trade barriers and regulatory requirements; |
| • | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| • | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| • | foreign taxes, including withholding of payroll taxes; |
| • | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| • | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| • | international regulators and third-party payors may require additional clinical studies prior to approving or allowing reimbursement for our products; |
| • | disadvantages of competing against companies from countries that are not subject to U.S. laws and regulations, including the U.S. Foreign Corrupt Practices Act, regulations of the U.S. Office of Foreign Assets Controls, and U.S. anti-money laundering regulations, as well as exposure of our foreign operations to liability under these regulatory regimes; |
| • | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| • | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires. |
Risks Relating to Government Regulation
The FDA regulatory clearance process is expensive, time-consuming and uncertain, and the failure to obtain and maintain required regulatory clearances and approvals could prevent us from commercializing our products.
Before we can market or sell a new regulated product or a new use of or a claim for a cleared product in the United States, we must obtain clearance from the FDA through the 510(k) premarket notification process, unless an exemption applies. In addition, we may be required to seek FDA clearance for any changes or modifications to our products that could significantly affect their safety or effectiveness, or would constitute a change in intended use. The 510(k) clearance processes can be expensive, time-consuming and uncertain. In addition to the time required to conduct clinical trials, if necessary, it generally takes from three to six months from submission of an application to obtain 510(k) clearance; however, it may take longer and 510(k) clearance may never be obtained. Our ability to obtain additional regulatory clearances for new products and indications may be significantly delayed or may never be obtained. Moreover, any new product introduction or product modification could be subjected to a lengthier, more rigorous FDA review process.
39
Table of Contents
Delays in receipt of, or failure to obtain, regulatory clearances for any product enhancements or new products we develop would result in delayed or no realization of revenue from such product enhancements or new products and in substantial additional costs which could decrease our profitability.
In addition, we are required to continue to comply with applicable FDA and other regulatory requirements once we have obtained clearance for a product. We cannot assure you that we will successfully maintain the clearances we have received or may receive in the future. In addition, our existing clearances can be revoked if any issues arise that bring into question our products’ safety or effectiveness. Any failure to maintain compliance with FDA regulatory requirements could have an adverse impact on our business.
The misuse or off-label use of our products may harm our image in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.
The products we currently market have been cleared by the FDA for specific treatments. We train our marketing and direct sales force to not promote our products for uses outside of the FDA-cleared indications for use, known as “off-label uses.” We cannot, however, prevent a physician from using our products off-label, when in the physician’s independent professional medical judgment he or she deems it appropriate. There may be increased risk of injury to patients if physicians attempt to use our products off-label. Furthermore, the use of our products for indications other than those cleared by the FDA or approved by any foreign regulatory body may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients.
If the FDA or any foreign regulatory body determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, and the curtailment of our operations.
In addition, physicians may misuse our products or use improper techniques if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our products are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. Similarly, in an effort to decrease costs, physicians may also reuse those of our products that are intended for a single use or may purchase reprocessed products from third-party reprocessors in lieu of purchasing new products from us, which could result in product failure and liability. As described immediately above, product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance.
Our products may cause or contribute to adverse medical events that we are required to report to the FDA, and if we fail to do so, we would be subject to sanctions that would materially harm our business.
Our marketed products are subject to Medical Device Reporting, or MDR, obligations, which require that we report to the FDA any incident in which our products may have caused or contributed to a death or serious injury, or in which our products malfunctioned and, if the malfunction were to recur, it could likely cause or contribute to a death or serious injury. The timing of our obligation to report under the MDR regulations is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event
40
Table of Contents
or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA could take action including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearances, seizure of our products, or delay in clearance of future products.
We and any of our future contract manufacturers are subject to various governmental regulations related to the manufacturing of our products, and we may incur significant expenses to comply with, experience delays in our product commercialization as a result of, and be subject to material sanctions if we or our future contract manufacturers violate, these regulations.
The methods used in, and the facilities used for, the manufacture of our products must comply with the FDA’s Quality System Regulation, or QSR, which covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of medical devices. The FDA enforces the QSR through periodic announced or unannounced inspections of manufacturing facilities, and both we and any future contract manufacturers we may employ are subject to such inspections. We cannot guarantee that we or any future third-party manufacturers will take the necessary steps to comply with applicable regulations, which could cause delays in the delivery of our products.
Failure to comply with applicable FDA requirements, or later discovery of previously unknown problems with our products or our third-party manufacturers’ manufacturing processes, including any failure to take satisfactory corrective action in response to an adverse QSR inspection, can result in, among other things:
| • | administrative or judicially imposed sanctions; |
| • | injunctions or the imposition of civil penalties; |
| • | recall or seizure of our products; |
| • | total or partial suspension of production or distribution; |
| • | the FDA’s refusal to grant pending or future clearances or approvals for our products; |
| • | withdrawal or suspension of regulatory clearances or approvals; |
| • | clinical holds; |
| • | warning letters or untitled letters; |
| • | refusal to permit the import or export of our products; and |
| • | criminal prosecution of us or our employees. |
Any of these actions could prevent us from marketing, distributing or selling our products and would likely harm our business.
Our products may in the future be subject to product recalls. A recall of our products, either voluntarily or at the direction of the FDA or another governmental authority, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in their design or manufacture. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device would cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government-mandated or voluntary recall could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation and business, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be subject to liability claims, be required to bear other costs, or take other actions that may have a negative impact on our future sales and our ability to generate profits.
41
Table of Contents
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA. We may initiate voluntary recalls or corrections for our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls and we may be subject to enforcement action.
If clinical studies of our future products do not produce results necessary to support regulatory clearance or approval in the United States or, with respect to our current or future products, elsewhere, we will be unable to expand the indications for or commercialize these products.
We will likely need to conduct additional clinical studies in the future to support new indications for our products or for clearances of new product lines, or for the approval of the use of our products in some foreign countries. Clinical testing can take many years, can be expensive and carries uncertain outcomes. The initiation and completion of any of these studies may be prevented, delayed, or halted for numerous reasons.
Clinical failure can occur at any stage of testing. Our clinical studies may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and non-clinical testing in addition to those we have planned. Our failure to adequately demonstrate the safety and efficacy of any of our devices would prevent receipt of regulatory clearance or approval and, ultimately, the commercialization of that device or indication for use. Even if our future products are cleared in the United States, commercialization of our products in foreign countries would require approval by regulatory authorities in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials. Any of these occurrences could have an adverse impact on our business.
Healthcare regulatory reform may affect our ability to sell our products profitably.
In the United States and in certain foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the regulatory and healthcare systems in ways that could impact our ability to sell our products profitably. In the United States in recent years, new legislation has been proposed and adopted at the federal and state levels that is effecting major changes in the healthcare system. In addition, new regulations and interpretations of existing healthcare statutes and regulations are frequently adopted.
In March 2010, the Affordable Care Act was signed into law. While the goal of healthcare reform is to expand coverage to more individuals, it also involves increased government price controls, additional regulatory mandates and other measures designed to constrain medical costs. The Affordable Care Act substantially changes the way healthcare is financed by both governmental and private insurers, encourages improvements in the quality of healthcare items and services and significantly impacts the medical device industry. Among other things, the Affordable Care Act:
| • | imposes an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States, with limited exceptions (described in more detail below), although the effective rate paid may be lower; |
| • | establishes a new Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; |
| • | implements payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models; and |
| • | creates an independent payment advisory board that will submit recommendations to Congress to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
42
Table of Contents
In addition, third-party payors regularly update payments to physicians and hospitals where our products are used. For Medicare, annual updates to physician payments are made using a prescribed statutory formula. In the past, when the application of the formula resulted in lower payment, Congress has passed interim legislation to prevent the reductions. Most recently, the Protecting Access to Medicare Act of 2014, signed into law in April 2014, provided for a 0.5% increase from 2013 payment rates under the Medicare Physician Fee Schedule through 2014 and a 0% change from January 1 until April 1, 2015. If Congress fails to intervene to prevent the negative update factor in future years, the resulting decrease in payment could have an adverse impact on our business. In addition, the Budget Control Act of 2011 imposed reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013, and will stay in effect through 2024 unless additional Congressional action is taken. Because there is no separate reimbursement for our products, and with respect to FESS, no separate reimbursement for balloon sinus dilation, these payment updates could directly impact the demand for our products or any products we may develop in the future, if cleared or approved.
Furthermore, we believe that many individuals who have obtained insurance coverage through the health insurance exchanges which arose as a result of the Affordable Care Act have done so with policies that have significantly higher deductibles than policies they may have obtained prior to its enactment. Because the out-of-pocket costs of undergoing a procedure such as balloon sinus dilation for patients who have not met their deductible for a given year would be significantly higher than they historically would have been, these patients may be discouraged from undergoing such a procedure due to the cost. Any reluctance on the part of patients to undergo balloon sinus dilation procedures due to cost could impact our ability to expand sales of our products and could adversely impact our business.
We are subject to additional federal, state and foreign laws and regulations relating to our healthcare business; our failure to comply with those laws could have an adverse impact on our business.
Although we do not provide healthcare services, submit claims for third-party reimbursement, or receive payments directly from Medicare, Medicaid or other third-party payors for our products, we are subject to healthcare fraud and abuse regulation and enforcement by federal and state governments, which could adversely impact our business. Healthcare fraud and abuse and health information privacy and security laws potentially applicable to our operations include:
| • | the federal Anti-Kickback Statute, which applies to our marketing practices, educational programs, pricing policies and relationships with healthcare providers, by prohibiting, among other things, soliciting, receiving, offering or providing remuneration intended to induce the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare or Medicaid programs. A person or entity does not need to have actual knowledge of this statute or specific intent to violate it to have committed a violation; |
| • | federal civil and criminal false claims laws and civil monetary penalty laws, including civil whistleblower or qui tam actions that prohibit, among other things, knowingly presenting, or causing to be presented, claims for payment or approval to the federal government that are false or fraudulent, knowingly making a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay or transmit money or property to the federal government. The government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, and its implementing regulations, which created federal criminal laws that prohibit, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, also imposes certain regulatory and contractual requirements regarding the privacy, security and transmission of individually identifiable health information; |
43
Table of Contents
| • | federal “sunshine” requirements imposed by the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the Affordable Care Act, on device manufacturers regarding any “transfer of value” made or distributed to physicians and teaching hospitals. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately, and completely reported in an annual submission. The period between August 1, 2013 and December 31, 2013 was the first reporting period, and manufacturers were required to report aggregate payment data by March 31, 2014, and to report detailed payment data and submit legal attestation to the accuracy of such data by June 30, 2014. Thereafter, manufacturers must submit reports by the 90th day of each subsequent calendar year; |
| • | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require device companies to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state laws that require device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of certain health information, many of which differ from each other in significant ways and often are not preempted by HIPAA. |
The risk of our being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. We are unable to predict what additional federal or state legislation or regulatory initiatives may be enacted in the future regarding our business or the healthcare industry in general, or what effect such legislation or regulations may have on us. Federal or state governments may impose additional restrictions or adopt interpretations of existing laws that could have a material adverse effect on us.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under such laws, it is possible that some of our business activities, including certain sales and marketing practices and financial arrangements, including the provision of stock options as partial compensation for consulting services, with physicians, some of whom use or purchase our products, and other customers, could be subject to challenge under one or more of such laws. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from governmental healthcare programs, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely impact our business.
We may be unable to obtain or maintain international regulatory registrations or approvals for our current or future products and indications, which could adversely impact our business.
Sales of our devices outside the United States are subject to foreign regulatory requirements that vary widely from country to country. In addition, the FDA regulates exports of medical devices from the United States. Complying with international regulatory requirements can be an expensive and time-consuming process and approval is not certain. The time required to obtain registration or approvals, if required by other countries, may be longer than that required for FDA clearance, and requirements for such registrations or approvals may significantly differ from FDA requirements. In certain countries we intend to rely upon third-party
44
Table of Contents
distributors to obtain all required regulatory registrations and approvals, and these distributors may be unable to obtain or maintain such registrations or approvals. Our distributors in these countries may also incur significant costs in attempting to obtain and in maintaining foreign regulatory approvals or registrations, which could increase the difficulty of attracting and retaining qualified distributors. If these distributors experience delays in receiving necessary registrations or approvals to market our products outside the United States, or if they fail to receive those registrations or approvals, we may be unable to market our products or enhancements in certain international markets effectively, or at all.
Our operations involve the use of hazardous and toxic materials, and we must comply with environmental, health and safety laws and regulations, which can be expensive, and could have an adverse impact on our business.
Our operations use or generate small volumes of hazardous or toxic materials. We are therefore subject to a variety of federal, state and local regulations relating to the use, handling, storage, disposal and human exposure to hazardous and toxic materials. Liability under environmental laws can be joint and several and without regard to comparative fault, and environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations, which could have an adverse impact on our business. Although we believe that our activities conform in all material respects with environmental, health and safety laws, there can be no assurance that violations of environmental, health and safety laws will not occur in the future as a result of human error, accident, equipment failure or other causes. The failure to comply with past, present or future laws could result in the imposition of fines, third-party property damage and personal injury claims, investigation and remediation costs, the suspension of production or a cessation of operations. We also expect that our operations will be affected by other new environmental and health and safety laws and regulations on an ongoing basis. Although we cannot predict the ultimate impact of any such new laws and regulations, they will likely result in additional costs, and may require us to change how we manufacture our products, which could have an adverse impact on our business.
Risks Relating to Capital Requirements and Finances
We may need substantial additional funding and may be unable to raise capital when needed, which could force us to delay or reduce our commercialization efforts or product development programs.
We believe that the net proceeds from our recent IPO, together with our existing cash and cash equivalents and revenue will be sufficient to meet our capital requirements and fund our operations for at least 24 months. However, we have based these estimates on assumptions that may prove to be incorrect, and we could spend our available financial resources much faster than we currently expect. Any future funding requirements will depend on many factors, including:
| • | market acceptance of our products; |
| • | the scope, rate of progress and cost of our clinical studies; |
| • | the cost of our research and development activities; |
| • | the cost of filing and prosecuting patent applications and defending and enforcing our patent or other intellectual property rights; |
| • | the cost of defending, in litigation or otherwise, any claims that we infringe third-party patents or other intellectual property rights; |
| • | the cost and timing of additional regulatory clearances or approvals; |
| • | the cost and timing of establishing additional sales, marketing and distribution capabilities; |
| • | costs associated with any product recall that may occur; |
| • | the effect of competing technological and market developments; |
45
Table of Contents
| • | the extent to which we acquire or invest in products, technologies and businesses, although we currently have no commitments or agreements relating to any of these types of transactions; and |
| • | the costs of operating as a public company. |
If we raise additional funds by issuing additional equity securities, our stockholders may experience dilution. Any future debt financing into which we enter may impose upon us covenants that restrict our operations, including limitations on our ability to incur liens or additional debt, pay dividends, repurchase our common stock, make certain investments and engage in certain merger, consolidation or asset sale transactions. Any debt financing or additional equity that we raise may contain terms that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third-parties, it may be necessary to relinquish some rights to our technologies or our products, or grant licenses on terms that are not favorable to us.
Furthermore, we cannot be certain that additional funding will be available on acceptable terms, if at all. If we do not have, or are not able to obtain, sufficient funds, we may have to delay development or commercialization of our products or license to third-parties the rights to commercialize products or technologies that we would otherwise seek to commercialize. We also may have to reduce marketing, customer support or other resources devoted to our products or cease operations. Any of these factors could harm our operating results.
Our ability to use our net operating losses and research and development credit carryforwards to offset future taxable income may be subject to certain limitations.
In general, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, a corporation that undergoes an “ownership change,” generally defined as a greater than 50% change by value in its equity ownership over a three-year period, is subject to limitations on its ability to utilize its pre-change net operating losses, or NOLs, and its research and development credit carryforwards to offset future taxable income. Our existing NOLs and research and development credit carryforwards may be subject to limitations arising from previous ownership changes, and if we undergo an ownership change, our ability to utilize NOLs and research and development credit carryforwards could be further limited by Sections 382 and 383 of the Code. Future changes in our stock ownership, some of which might be beyond our control, could result in an ownership change under Section 382 of the Code. For these reasons, in the event we experience a change of control, we may not be able to utilize a material portion of the NOLs and research and development credit carryforwards, even if we attain profitability.
Risks Relating to Intellectual Property Matters
Intellectual property rights may not provide adequate protection and third-parties from which we license intellectual property may not enforce their intellectual property rights, which may permit third-parties to compete against us more effectively.
Our success depends significantly on our ability to protect our proprietary rights to the technologies and inventions used in, or embodied by, our products. To protect our proprietary technology, we rely on patent protection, as well as a combination of copyright, trade secret and trademark laws, as well as nondisclosure, confidentiality and other contractual restrictions in our consulting and employment agreements. These legal means afford only limited protection, however, and may not adequately protect our rights or permit us to gain or keep any competitive advantage. Furthermore, while we have no reason to believe that any third parties hold rights of ownership to the intellectual property we regard as our own, we cannot assure shareholders that third parties of which we are unaware do not possess such rights, or that we will not be subject to, and forced to defend against, third-party claims of ownership to our intellectual property in the future. In either case, our ability to compete effectively could be harmed.
46
Table of Contents
Patents
The process of applying for patent protection itself is time-consuming and expensive and we cannot assure you that all of our patent applications will issue as patents or that, if issued, they will issue in a form that will be advantageous to us. The rights granted to us under our patents, including prospective rights sought in our pending patent applications, may not be meaningful or provide us with any commercial advantage, and they could be opposed, contested, or circumvented by our competitors or declared invalid or unenforceable in judicial or administrative proceedings. We may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to patents licensed by third-parties. For example, we do not control the prosecution of the patent applications licensed to us under the Acclarent License described above. Therefore, these patents and applications may not be prosecuted or enforced in a manner consistent with the best interests of our business. If such licensors fail to maintain such patents, or lose rights to those patents, the rights we have licensed may be reduced or eliminated. In addition, because the Acclarent License is non-exclusive, Acclarent could choose to also license the same intellectual property to other third-party competitors, which would also have a material adverse effect on our business.
We own numerous issued patents and pending patent applications relating to our technology and products. The rights granted to us under these patents, including prospective rights sought in our pending patent applications, could be opposed, contested or circumvented by our competitors or declared invalid or unenforceable in judicial or administrative proceedings. If any of our patents are challenged, invalidated or legally circumvented by third-parties, and if we do not own other enforceable patents protecting our products, competitors could market products and use processes that are substantially similar to, or superior to, ours, and our business will suffer. In addition, the patents we own may not be of sufficient scope or strength to provide us with any meaningful protection or commercial advantage, and competitors may be able to design around our patents or develop products that provide outcomes comparable to ours without infringing on our intellectual property rights.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a “first-to-invent” system to a “first-to-file” system. Under a “first-to-file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The U.S. Patent and Trademark Office, or USPTO, recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition. In addition, patent reform legislation may pass in the future that could lead to additional uncertainties and increased costs surrounding the prosecution, enforcement, and defense of our patents and applications. We may be subject to a third-party preissuance submission of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or other patent office proceedings or litigation, in the United States or elsewhere, challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third-parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
47
Table of Contents
Moreover, the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance fees on issued patents often must be paid to the USPTO and foreign patent agencies over the lifetime of the patent. While an unintentional lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our products or procedures, we may not be able to stop a competitor from marketing products that are the same as or similar to our products, which would have a material adverse effect on our business.
Furthermore, we do not have patent rights in certain foreign countries in which a market may exist in the future, and the laws of many foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. Thus, we may not be able to stop a competitor from marketing and selling in foreign countries products that are the same as or similar to our products.
Trademarks
We rely on our trademarks as one means to distinguish our products from the products of our competitors, and have registered or applied to register many of these trademarks. Our trademark applications may not be approved, however. Third-parties may oppose our trademark applications, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. Our competitors may infringe our trademarks and we may not have adequate resources to enforce our trademarks.
Trade Secrets and Know-How
We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by consultants, vendors, former employees or current employees, despite the existence generally of confidentiality agreements and other contractual restrictions. Monitoring unauthorized uses and disclosures of our intellectual property is difficult, and we do not know whether the steps we have taken to protect our intellectual property will be effective.
Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Competitors could purchase our products and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If our intellectual property is not adequately protected so as to protect our market against competitors’ products and methods, our competitive position could be adversely affected, as could our business.
We may in the future be a party to patent and other intellectual property litigation and administrative proceedings that could be costly and could interfere with our ability to successfully market our products.
The medical device industry has been characterized by frequent and extensive intellectual property litigation and is highly competitive. Our competitors or other patent holders may assert that our products and/or the methods employed in our products are covered by their patents. If our products or methods are found to infringe, we could be prevented from manufacturing or marketing our products. In the event that we become involved in such a dispute, we may incur significant costs and expenses and may need to devote resources to resolving any claims, which would reduce the cash we have available for operations and may be distracting to management.
48
Table of Contents
We do not know whether our competitors or potential competitors have applied for, will apply for, or will obtain patents that will prevent, limit or interfere with our ability to make, use, sell, import or export our products. Competing products may also be sold in other countries in which our patent coverage might not exist or be as strong. If we lose a foreign patent lawsuit, alleging our infringement of a competitor’s patents, we could be prevented from marketing our products in one or more foreign countries. We may also initiate litigation against third-parties to protect our own intellectual property. Our intellectual property has not been tested in litigation. If we initiate litigation to protect our rights, we run the risk of having our patents invalidated, which would undermine our competitive position.
Litigation related to infringement and other intellectual property claims, with or without merit, is unpredictable, can be expensive and time-consuming, and can divert management’s attention from our core business. If we lose this kind of litigation, a court could require us to pay substantial damages, treble damages and attorneys’ fees, and could prohibit us from using technologies essential to our products, any of which would have a material adverse effect on our business. If relevant patents are upheld as valid and enforceable and we are found to infringe, we could be prevented from selling our products unless we can obtain licenses to use technology or ideas covered by such patents. We do not know whether any necessary licenses would be available to us on satisfactory terms, if at all. If we cannot obtain these licenses, we could be forced to design around those patents at additional cost or abandon the product altogether. As a result, our ability to grow our business and compete in the market may be harmed.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
We could in the future be subject to claims that we or our employees have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of former employers or competitors. In addition, we may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. If our defense to those claims fails, in addition to paying monetary damages, a court could prohibit us from using technologies or features that are essential to our products, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. An inability to incorporate technologies or features that are important or essential to our products could have an adverse impact on our business, and may prevent us from selling our products. In addition, we may lose valuable intellectual property rights or personnel. Any litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our products, which could have an adverse impact on our business.
Risks Related to Our Common Stock
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for our stockholders.
Our stock price is likely to be volatile. In recent years, the stock markets generally have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors may significantly affect the market price of our common stock, regardless of our actual operating performance. The market price of our common stock may fluctuate substantially due to many factors, including:
| • | the volume and timing of sales of our products; |
| • | the introduction of new products or product enhancements by us or others in our industry; |
| • | disputes or other developments with respect to our or others’ intellectual property rights; |
49
Table of Contents
| • | our ability to develop, obtain regulatory clearance or approval for, and market new and enhanced products on a timely basis; |
| • | product liability claims or other litigation; |
| • | quarterly variations in our results of operations or those of others in our industry; |
| • | media exposure of our products or of those of others in our industry; |
| • | changes in governmental regulations or in reimbursement; |
| • | changes in earnings estimates or recommendations by securities analysts; and |
| • | general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors. |
In addition, in the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Securities litigation brought against us following volatility in our stock price, regardless of the merit or ultimate results of such litigation, could result in substantial costs, which would hurt our financial condition and operating results and divert management’s attention and resources from our business.
Securities analysts may not publish favorable research or reports about our business or may publish no information at all, which could cause our stock price or trading volume to decline.
If a trading market for our common stock develops, the trading market will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We do not control these analysts. If any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding our stock price, our stock price could decline. If one or more of these analysts cease coverage of us or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price or trading volume to decline.
We are an “emerging growth company” and the reduced disclosure requirements applicable to “emerging growth companies” may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we may take, and intend to take, advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not “emerging growth companies.” In particular, while we are an “emerging growth company” (1) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (2) we will be exempt from any rules that could be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotations or a supplement to the auditor’s report on financial statements, (3) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and (4) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved.
In addition, while we are an “emerging growth company” we will not be required to comply with any new financial accounting standard until such standard is generally applicable to private companies. As a result, our financial statements may not be comparable to companies that are not “emerging growth companies” or elect not to avail themselves of this provision.
We may remain an “emerging growth company” until as late as December 31, 2020, the fiscal year-end following the fifth anniversary of the completion of our IPO, though we may cease to be an “emerging growth company” earlier under certain circumstances, including if (1) we have more than $1.0 billion in annual revenue in any fiscal year, (2) the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30 or (3) we issue more than $1.0 billion of non-convertible debt over a three-year period.
50
Table of Contents
The exact implications of the JOBS Act are still subject to interpretations and guidance by the SEC and other regulatory agencies, and we cannot assure you that we will be able to take advantage of all of the benefits of the JOBS Act. In addition, investors may find our common stock less attractive to the extent we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline or become more volatile.
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
As of March 6, 2015, our officers, directors and principal stockholders each holding more than 5% of our common stock, collectively control approximately 68.1% of our outstanding common stock. As a result, these stockholders, if they were to act together, would be able to control the management and affairs of our Company and most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change of control and might adversely affect the market price of our common stock. This concentration of ownership may not be in the best interests of our other stockholders.
In the event that our internal control over financial reporting is perceived as inadequate, or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results and the trading price of our common stock could decline.
We are subject to the periodic reporting requirements of the Exchange Act. As a result, our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with U.S. generally accepted accounting principles. As a result of becoming a public company, we are required, under Section 404(a) of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. We designed our disclosure controls and procedures to provide reasonable assurance that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. In preparing our financial statements for the nine months ended September 30, 2014, we determined that a material weakness in internal control over financial reporting existed that related to our having recorded revenue relating to sales orders that had been falsified by one of our sales representatives. Subsequent to identifying this material weakness, we implemented processes and controls designed to enhance our internal controls over the customer sales order process and to remediate the material weakness. As of December 31, 2014, we determined that we have remediated the material weakness. We believe that any disclosure controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur in the future and not be detected.
In addition, once we cease to be an “emerging growth company” under the federal securities laws, our auditors will be required to express an opinion on the effectiveness of our internal controls under Section 404(b) of the Sarbanes-Oxley Act. While we may remain an “emerging growth company” until as late as December 31, 2020, the fiscal year-end following the fifth anniversary of the completion of our IPO, we may cease to be an “emerging growth company” earlier under certain circumstances and that could accelerate our timeline for complying with Section 404(b).
51
Table of Contents
If we are unable to confirm that our internal control over financial reporting is effective, or if our auditors, when required, are unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, or be delayed in producing these financial reports, both of which could cause the price of our common stock to decline. We could also be subject to, among other things, regulatory or enforcement actions by the SEC and The Nasdaq Global Market and could be subject to securities litigation.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Because our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. Among others, these provisions include that:
| • | our board of directors has the exclusive right to expand the size of our board of directors and to elect directors to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; |
| • | our board of directors is divided into three classes, Class I, Class II and Class III, with each class serving staggered three-year terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors; |
| • | our stockholders may not act by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders; |
| • | a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; |
| • | our amended and restated certificate of incorporation prohibits cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
| • | our board of directors may alter our bylaws without obtaining stockholder approval; |
| • | the required approval of the holders of at least two-thirds of the shares entitled to vote at an election of directors to adopt, amend or repeal our bylaws or repeal the provisions of our amended and restated certificate of incorporation regarding the election and removal of directors; |
| • | stockholders must provide advance notice and additional disclosures in order to nominate individuals for election to the board of directors or to propose matters that can be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of our Company; and |
| • | our board of directors is authorized to issue shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer. |
52
Table of Contents
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. Furthermore, our amended and restated certificate of incorporation specifies that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for most legal actions involving actions brought against us by stockholders. We believe this provision benefits us by providing increased consistency in the application of Delaware law by chancellors particularly experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums and protection against the burdens of multi-forum litigation. However, the provision may have the effect of discouraging lawsuits against our directors and officers. The enforceability of similar choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is possible that, in connection with any applicable action brought against us, a court could find the choice of forum provisions contained in our amended and restated certificate of incorporation to be inapplicable or unenforceable in such action.
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, our current credit facility precludes, and any future debt agreements may preclude, us from paying dividends. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
53
Table of Contents
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We occupy approximately 32,500 square feet of leased office and warehouse space located in Plymouth, Minnesota. The terms of our lease expire in August 2018. We believe that our facilities are sufficient to meet our current needs and that suitable additional space will be available as and when needed on acceptable terms.
| Item 3. | Legal Proceedings |
We are not currently a party to any material legal proceedings. We may at times be involved in litigation and other legal claims in the ordinary course of business. When appropriate in our estimation, we may record reserves in our financial statements for pending litigation and other claims.
| Item 4. | Mine Safety Disclosures |
Not applicable.
54
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market Information
Our common stock has been listed on The Nasdaq Global Market under the symbol “ENTL” since January 29, 2015. As a result, we have not set forth quarterly information with respect to the high and low prices for our common stock for the two most recent fiscal years. In February 2015, the high and low prices for our common stock were $24.91 and $20.40, respectively. In March 2015 (through March 18), the high and low prices for our common stock were $22.05 and $19.56, respectively.
Holders
As of the close of business on March 6, 2015, there were approximately 146 holders of record of our common stock. Since many of the common shares are registered in “nominee” or “street” names, we believe that the total number of beneficial owners is considerably higher.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock, and we do not currently intend to pay any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to support operations and to finance the growth and development of our business. In addition, our existing credit facility restricts our ability to pay dividends on our capital stock. Any future determination to pay dividends will be made at the discretion of our board of directors subject to applicable laws and the restrictions set forth in our credit facility, and will depend upon, among other factors, our results of operations, financial condition, contractual restrictions and capital requirements. Our future ability to pay cash dividends on our capital stock may also be limited by the terms of any future debt or preferred securities or future credit facility.
Recent Sales of Unregistered Securities
The following list sets forth information as to all securities we sold during the year ended December 31, 2014 that were not registered under the Securities Act:
| 1. | We issued an aggregate of 465,258 shares of common stock to employees, directors and consultants for cash consideration in the aggregate amount of $575,356 upon the exercise of stock options. |
| 2. | We granted stock options to employees, directors and consultants under our 2006 Stock Incentive Plan, or the 2006 Plan, covering an aggregate of 1,396,173 shares of common stock, at an average exercise price of $10.55 per share. |
The historical common stock and per share information described above reflects the 1-for-4 reverse split of our common stock that was effected in January 2015.
The issuances of stock options and the shares of common stock issuable upon the exercise of the options described above were issued pursuant to written compensatory plans or arrangements with our employees, directors and consultants, in reliance on the exemption provided by Rule 701 promulgated under the Securities Act, or pursuant to Section 4(a)(2) under the Securities Act, relative to transactions by an issuer not involving any public offering, to the extent an exemption from such registration was required.
All of the foregoing securities are deemed restricted securities for purposes of the Securities Act. All certificates representing the issued shares of capital stock described in this Item 15 included appropriate legends setting forth that the securities have not been registered and the applicable restrictions on transfer.
55
Table of Contents
Purchases of Equity Securities by the Issuer and Affiliated Purchaser
None.
Use of Proceeds
On January 28, 2015, the SEC declared effective our Registration Statement on Form S-1 (File No. 333-201237), as amended, filed in connection with our IPO. Pursuant to the Registration Statement, as well as the Registration Statement on Form S-1 (File No. 333-201741) filed on January 28, 2015 to register additional securities, we issued and sold an aggregate of 5,294,117 shares of our common stock in the IPO at a price to the public of $17.00 per share (including 690,537 shares sold pursuant to an option granted to the underwriters). As a result of the IPO, we received gross proceeds of approximately $90.0 million, which resulted in net proceeds to us of approximately $81.2 million. None of the expenses associated with the IPO were paid to directors, officers, persons owning ten percent or more of any class of equity securities, or to their associates, or to our affiliates. Merrill Lynch, Pierce, Fenner & Smith Incorporated and Piper Jaffray & Co. acted as joint book-running managers and William Blair & Company, L.L.C. and Canaccord Genuity Inc. acted as co-managers for the offering. On February 3, 2015, following the sale of the 5,294,117 shares of common stock, the offering terminated.
We intend to use the net offering proceeds for product development and clinical research, sales, marketing, working capital, and general corporate purposes. Pending use of the net proceeds, we intend to invest the net proceeds in short-term, interest-bearing, investment grade securities, certificates of deposit or governmental securities. There has been no material change in the planned use of proceeds from our IPO from that described in the final prospectus, dated January 28, 2015, filed with the SEC pursuant to Rule 424(b).
56
Table of Contents
| Item 6. | Selected Financial Data |
The following tables set forth, for the periods and as of the dates indicated, our selected financial data. The statements of operations data for the years ended December 31, 2014, 2013 and 2012 and the balance sheet data as of December 31, 2014 and 2013 are derived from our audited financial statements included in this Annual Report on Form 10-K. The balance sheet data as of December 31, 2012 are derived from our audited financial statements that are not included in this Annual Report on Form 10-K. You should read the following information together with the more detailed information contained in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes included elsewhere in this Annual Report on Form 10-K. Our historical results are not indicative of the results to be expected in the future.
| Year Ended December 31, | ||||||||||||
| (in thousands, except per share data) | 2014 | 2013 | 2012 | |||||||||
| Statement of Operations Data: |
||||||||||||
| Revenue |
$ | 48,820 | $ | 32,545 | $ | 17,559 | ||||||
| Cost of goods sold |
10,754 | 7,808 | 4,784 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
38,066 | 24,737 | 12,775 | |||||||||
| Operating expense: |
||||||||||||
| Selling and marketing |
32,763 | 27,631 | 21,634 | |||||||||
| Research and development |
4,307 | 5,143 | 5,896 | |||||||||
| General and administrative |
6,097 | 4,311 | 4,080 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expense |
43,167 | 37,085 | 31,610 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(5,101 | ) | (12,348 | ) | (18,835 | ) | ||||||
| Other expense, net |
(1,828 | ) | (1,048 | ) | (161 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (6,929 | ) | $ | (13,396 | ) | $ | (18,996 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per share: |
||||||||||||
| Basic and diluted |
$ | (4.62 | ) | $ | (11.82 | ) | $ | (27.29 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average shares used in the calculation of loss per share: |
||||||||||||
| Basic and diluted |
1,499 | 1,133 | 696 | |||||||||
|
|
|
|
|
|
|
|||||||
| As of December 31, | ||||||||||||
| (in thousands) | 2014 | 2013 | 2012 | |||||||||
| Balance Sheet Data: |
||||||||||||
| Cash and cash equivalents |
$ | 3,484 | $ | 7,709 | $ | 6,681 | ||||||
| Working capital |
7,763 | 10,702 | 15,456 | |||||||||
| Total assets |
19,236 | 17,876 | 20,546 | |||||||||
| Convertible preferred stock warrant liability |
291 | 211 | 131 | |||||||||
| Total debt (credit facility) |
20,000 | 15,000 | 7,375 | |||||||||
| Total liabilities |
28,036 | 20,550 | 10,894 | |||||||||
| Convertible preferred stock |
91,554 | 91,554 | 91,554 | |||||||||
| Accumulated deficit |
(103,758 | ) | (96,829 | ) | (83,433 | ) | ||||||
| Total stockholders’ deficit |
(100,354 | ) | (94,228 | ) | (81,902 | ) | ||||||
57
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our financial statements and the related notes to those statements included elsewhere in this Annual Report on Form 10-K. In addition to historical financial information, the following discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. Some of the numbers included herein have been rounded for the convenience of presentation. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those discussed under “Risk Factors” and elsewhere in this Annual Report on Form 10-K.
Overview
We are a medical technology company focused on the design, development and commercialization of products for the minimally invasive treatment of patients who are suffering from chronic sinusitis. Our XprESS family of products is used by ear, nose and throat, or ENT, physicians to open narrowed or obstructed sinus drainage pathways using balloon sinus dilation to treat patients with symptomatic inflammation of the nasal sinuses. When used as a stand-alone therapy in the doctor’s office, our balloon sinus dilation products are the only devices clinically proven in a sufficiently powered prospective, multicenter, randomized, controlled trial to be as effective as functional endoscopic sinus surgery, or FESS. Patients treated with our products in this trial in the ENT physician office also experienced faster recovery, less bleeding at discharge, less use of prescription pain medication and fewer post-procedure debridements than patients receiving FESS.
Minimally invasive balloon sinus dilation devices have enabled a shift towards office-based treatment of chronic sinusitis patients who are candidates for sinus surgery in the operating room. We believe this shift has been facilitated by our technology and clinical data, as well as procedure economics that are favorable to the healthcare system, patient and provider. Our XprESS family of products is used to treat patients with inflammation of the frontal, ethmoid, sphenoid and maxillary sinuses and is specifically designed for ease-of-use in the ENT physician office setting. Our XprESS family of products includes our XprESS Pro device, our XprESS LoProfile device and our XprESS Ultra device. We derive substantially all of our revenue from our XprESS family of products. For the years ended December 31, 2014, 2013 and 2012, our XprESS family of products represented 95%, 95% and 85% of our revenues, respectively. Our research and development efforts are focused on enhancing our XprESS family of products and broadening their indications for use. We are also the exclusive distributor of XeroGel.
Our direct U.S. sales force engages in sales efforts and promotional activities focused on ENT physicians. As of December 31, 2014, we employed 103 full-time persons in our direct sales organization, an increase from 90 persons as of December 31, 2013. We have expanded our sales force to include 69 full quota-carrying representatives as of December 31, 2014, an expansion of 41% from our 49 full quota-carrying representatives as of December 31, 2013. For the fourth quarter of 2014 our full quota-carrying representatives sold at an annualized rate of approximately $750,000. We expect to continue to expand our sales force and staffing to further penetrate the sinusitis market. In addition, we invest substantial resources to educate ENT physicians and patients on the proven clinical advantages of stand-alone balloon sinus dilation. We have a diverse customer base of ENT physicians, hospitals and ASCs in the United States, with no single customer accounting for more than 5% of our revenue during the year ended December 31, 2014. Our customers are reimbursed by governmental and private health insurers for procedures using our products pursuant to reimbursement codes specific to the setting of the procedure. We manufacture all of our proprietary products at our 32,351 square foot facility in Plymouth, Minnesota with components supplied by external suppliers. As of December 31, 2014, our manufacturing organization included 20 people. We expect the capacity of our current facility to be able to meet expected demand through at least the end of 2017.
For the year ended December 31, 2014 we generated revenue of $48.8 million and had a net loss of $6.9 million compared to revenue of $32.5 million and a net loss of $13.4 million for the year ended December 31, 2013. We expect to continue to incur losses until at least 2016 as we expend resources to expand our organization to
58
Table of Contents
support planned sales growth and expansion while also continuing to invest in the development of next generation products and new products for the ENT market. As of December 31, 2014, we had an accumulated deficit of $103.8 million. Our primary sources of capital to date have been from sales of our products, private placements of our convertible preferred securities and amounts borrowed under our credit facility. We operate in one segment.
In February 2015, we completed our initial public offering, or IPO, by issuing 5,294,117 shares of common stock at an offering price of $17.00 per share, for net proceeds to us of approximately $81.2 million after deducting underwriting discounts and commissions and offering expenses.
Components of Our Results of Operation
Revenue
We derive substantially all of our revenue from the sale of our XprESS family of products to ENT physicians, hospitals and ASCs in the United States. We derive additional revenue from the sale of XeroGel products, of which we are the exclusive distributor. Recent revenue growth has been driven by, and we expect our revenue to continue to increase in the future as a result of, increased physician awareness of the clinical efficacy of stand-alone balloon sinus dilation and our products and increasing insurance coverage for balloon sinus dilation procedures. Any reversal in these recent trends, however, could have a negative impact on our future revenue. In addition, we have expanded our sales and marketing infrastructure to help us drive and support revenue growth and intend to continue this expansion. Our revenue has fluctuated, and we expect our revenue to continue to fluctuate, from quarter to quarter due to a variety of factors.
When used as a stand-alone balloon sinus dilation procedure in the physician office setting, our products are paid for as part of the practice expense component of the physician’s fee. When used in an operating room in an ASC or hospital, our products are typically utilized as tools used during FESS and are paid for as part of the FESS procedure.
Seasonality
Our business may be affected by seasonality. In the first quarter, our results can be impacted by adverse weather and by resetting of annual patient healthcare insurance plan deductibles, both of which may cause patients to delay elective procedures in which our products are used. In the second quarter, demand may be increased by the seasonal nature of allergies and the resultant onset of sinus-related symptoms. In the third quarter, the number of balloon sinus dilation and FESS procedures nationwide is historically lower than other quarters throughout the year, which we believe is attributable to the summer vacations of ENT physicians and their patients. In the fourth quarter, demand may be increased by the onset of the cold and flu season and related symptoms, as well as the desire of patients to spend the remaining balances in their flexible-spending accounts or because of lower out-of-pocket costs to patients who have met their annual deductibles under their health insurance plans.
Cost of Goods Sold and Gross Margin
Cost of goods sold consists primarily of manufacturing overhead costs, material costs and direct labor. A significant portion of our cost of goods sold consists of manufacturing overhead costs. These overhead costs include the cost of quality assurance, material procurement, inventory control, facilities, equipment and operations supervision and management. We expect overhead costs as a percentage of revenue to continue to decrease as our production volume increases. Cost of goods sold also includes depreciation expense for production equipment, amortization of leasehold improvements, shipping costs and royalty expense related to our licensing agreement with Acclarent, Inc. that is payable in connection with sales of substantially all of our products. We expect cost of goods sold to increase in absolute dollars primarily as, and to the extent, our revenue grows.
59
Table of Contents
We calculate gross margin as gross profit divided by revenue. Our gross margin has been and will continue to be affected by a variety of factors, primarily product sales mix and prices, production volumes, manufacturing costs and product yields, and to a lesser extent the implementation of cost-reduction strategies. Our gross margin may continue to increase over the long term as our production volume increases and as we spread the fixed portion of our manufacturing overhead costs over a greater number of units produced, thereby reducing our per unit manufacturing costs. However, our gross margin will likely fluctuate from quarter to quarter due to seasonality.
Selling and Marketing Expenses
Selling and marketing expenses consist primarily of compensation for personnel, including base salaries and variable compensation associated with sales results, spending related to marketing, reimbursement and customer service functions, and stock-based compensation. Other selling and marketing expenses include training, travel expenses, promotional activities, conferences, trade shows and consulting services. We expect selling and marketing expenses to continue to increase in absolute dollars as we expand our commercial infrastructure to both drive and support our planned growth in revenue. However, we expect selling and marketing expenses to decrease as a percentage of revenue.
Research and Development Expenses
Research and development, or R&D, expenses consist primarily of engineering, product development, clinical studies to develop and support our products, regulatory expenses, consulting services, materials, depreciation and other costs associated with products and technologies in development and next generation versions of current devices. These expenses include employee and non-employee compensation, including stock-based compensation, supplies, materials, quality assurance expenses allocated to R&D programs, consulting, related travel expenses and facilities expenses, in each case related to R&D programs. Clinical expenses include clinical trial management and monitoring, payments to clinical investigators, data management and travel expenses and the cost of manufacturing products for clinical trials. We expect R&D expenses to increase modestly, as increases in expenses related to developing, enhancing and commercializing new products and technologies will be only partially offset by the timing and extent of expenses relating to clinical studies. We expect R&D expenses as a percentage of revenue to vary over time depending on the level and timing of initiating new product development efforts as well as our clinical development activities.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for personnel, including base salaries and bonus compensation, spending related to finance, information technology, human resource functions and stock-based compensation. Other general and administrative expenses include travel expenses, credit card processing fees, professional services fees, audit fees, insurance costs and general corporate expenses, including allocated facilities-related expenses. We expect general and administrative expenses to continue to increase in absolute dollars as we expand our commercial infrastructure to both drive and support our planned growth in revenue. We also expect to incur additional legal, accounting, insurance and other professional service fees associated with being a public company, which may increase further when we are no longer able to rely on the “emerging growth company” exemption we are afforded under the Jump Start Our Business Startups Act of 2012, or JOBS Act. We expect general and administrative expenses to continue to decrease as a percentage of revenue.
Other Expense, Net
Other expense, net consists of fair value adjustments related to our convertible preferred stock warrants, which, prior to the IPO, were accounted for as a liability and marked-to-market at each reporting period, and interest expense payable under our credit facility. In connection with the IPO, the warrants converted to warrants to purchase shares of our common stock.
60
Table of Contents
Results of Operations
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2014 | 2013 | 2012 | |||||||||
| Revenue |
$ | 48,820 | $ | 32,545 | $ | 17,559 | ||||||
| Cost of goods sold |
10,754 | 7,808 | 4,784 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
38,066 | 24,737 | 12,775 | |||||||||
| Gross margin |
78% | 76% | 73% | |||||||||
| Operating expense: |
||||||||||||
| Selling and marketing |
32,763 | 27,631 | 21,634 | |||||||||
| Research and development |
4,307 | 5,143 | 5,896 | |||||||||
| General and administrative |
6,097 | 4,311 | 4,080 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expense |
43,167 | 37,085 | 31,610 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(5,101 | ) | (12,348 | ) | (18,835 | ) | ||||||
| Other expense, net |
(1,828 | ) | (1,048 | ) | (161 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (6,929 | ) | $ | (13,396 | ) | $ | (18,996 | ) | |||
|
|
|
|
|
|
|
|||||||
Comparison of Years Ended December 31, 2014 and 2013
Revenue
Revenue increased $16.3 million, or 50%, to $48.8 million during the year ended December 31, 2014, compared to $32.5 million during the year ended December 31, 2013. The growth in revenue was primarily attributable to an increase of approximately $15.7 million in sales of our XprESS family of products and $0.9 million in sales of our XeroGel products offset in part by a decrease of $0.3 million from all other products.
Cost of Goods Sold and Gross Margin
Cost of goods sold increased $2.9 million, or 38%, to $10.8 million during the year ended December 31, 2014, compared to $7.8 million during the year ended December 31, 2013. The increase in cost of goods sold was primarily attributable to the growth in sales of our XprESS family of products described above.
Gross margin for the year ended December 31, 2014 increased to 78%, compared to 76% for the year ended December 31, 2013. The increase in gross margin was primarily due to increased unit sales, which allowed us to spread the fixed portion of our manufacturing overhead costs over more production units. While average selling prices also increased, the effect of this increase was partially offset by increased cost of goods sold associated with the XprESS LoProfile.
Selling and Marketing Expenses
Selling and marketing expenses increased $5.1 million, or 19%, to $32.8 million during the year ended December 31, 2014, compared to $27.6 million during the year ended December 31, 2013. The increase was primarily attributable to a $2.9 million increase in compensation and other employee-related expenses and a $1.8 million increase in travel expenses as a result of increased headcount in our sales and marketing organizations. In addition, consulting and other selling and marketing expenses increased $0.7 million, offset in part by a decrease of $0.3 million in selling equipment expenses.
Research and Development Expenses
R&D expenses decreased $0.8 million, or 16%, to $4.3 million during the year ended December 31, 2014, compared to $5.1 million during the year ended December 31, 2013. The decrease in R&D expenses was
61
Table of Contents
primarily due to a decrease of $0.6 million in clinical trial costs for the REMODEL trial as enrollment had reached planned levels and data collection was completed during 2014. In addition, compensation and other employee-related expenses and R&D product development expenses each decreased by $0.1 million.
General and Administrative Expense
General and administrative expenses increased $1.8 million, or 41%, to $6.1 million during the year ended December 31, 2014, compared to $4.3 million during the year ended December 31, 2013. The increase was primarily due to an increase of $0.9 million in compensation and other employee-related expenses. In addition, legal and audit fees increased $0.3 million, credit card processing fees increased $0.2 million and federal excise tax on the sale of products increased $0.1 million. Other general and administrative expenses accounted for the remaining increase.
Other Expense, Net
Other expense, net, reflected an expense of $1.8 million during the year ended December 31, 2014 compared to an expense of $1.0 million during the year ended December 31, 2013. The increase in expense was primarily attributable to the fair value adjustment of our then-outstanding convertible preferred stock warrants, which are accounted for as a liability and marked-to-market at each reporting period, and to interest expense related to our credit facility.
Comparison of Years Ended December 31, 2013 and 2012
Revenue
Revenue increased $15.0 million, or 85%, to $32.5 million during the year ended December 31, 2013, compared to $17.6 million during the year ended December 31, 2012. The growth in revenue was primarily attributable to an increase of approximately $15.9 million in sales of our XprESS family of products offset in part by a decrease of $0.9 million from other non XprESS family products.
Cost of Goods Sold and Gross Margin
Cost of goods sold increased $3.0 million, or 63% to $7.8 million during the year ended December 31, 2013, compared to $4.8 million during the year ended December 31, 2012. The increase in cost of goods sold was primarily attributable to the growth in sales of our XprESS LoProfile products described above.
Gross margin for the year ended December 31, 2013 increased to 76%, compared to 73% for the year ended December 31, 2012. The increase in gross margin was primarily due to increased unit sales and changes in product sales mix described above, which allowed us to spread the fixed portion of our manufacturing overhead costs over more production units.
Selling and Marketing Expenses
Selling and marketing expenses increased $6.0 million, or 28%, to $27.6 million during the year ended December 31, 2013, compared to $21.6 million during the year ended December 31, 2012. The increase was primarily attributable to a $4.5 million increase in compensation and other employee-related expenses and a $1.2 million increase in travel expenses as a result of increased headcount in our sales and marketing organizations.
Research and Development Expenses
R&D expenses decreased $0.8 million, or 13%, to $5.1 million during the year ended December 31, 2013, compared to $5.9 million during the year ended December 31, 2012. The decrease in R&D expenses was primarily due to a decrease of $0.9 million in clinical trial costs for the REMODEL trial as enrollment had reached planned levels and data collection was nearing completion.
62
Table of Contents
General and Administrative Expenses
General and administrative expenses increased $0.2 million, or 6%, to $4.3 million during the year ended December 31, 2013, compared to $4.1 million during the year ended December 31, 2012. The increase was primarily due to an expense of $0.3 million due under a new federal excise tax on the sale of our products during the year ended December 31, 2013.
Other Expense, Net
Other expense, net, reflected an expense of $1.0 million during the year ended December 31, 2013, compared to an expense of $0.2 million during the year ended December 31, 2012. The increase in expense was primarily attributable to the fair value adjustment of our then-outstanding convertible preferred stock warrants, which are accounted for as a liability and marked-to-market at each reporting period, and to interest expense related to our credit facility.
Critical Accounting Policies and Estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenue, expenses and related disclosures. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
While our significant accounting policies are more fully described in Note B to our financial statements included elsewhere in this Annual Report on Form 10-K, we believe the following discussion addresses our most critical accounting policies, which are those that are most important to the portrayal of our financial condition and results of operations and require our most difficult, subjective and complex judgments.
Revenue Recognition
We derive revenue from the sale of our XprESS family of products to ENT physicians, hospitals and ASCs in the United States. We recognize revenue when persuasive evidence of a sales arrangement exists, delivery of goods has occurred through the transfer of title and risks and rewards of ownership, the selling price is fixed or determinable, and collection is reasonably assured. Revenue is reported net of the allowance for sales returns. The revenue derived on the sale of XeroGel products is recognized when the criteria above is met and is accounted for on primarily a net basis.
Stock-Based Compensation
We account for all stock-based compensation awards using a fair value method. We record stock options at their estimated grant date fair value using the Black-Scholes option pricing model. We recognize the fair value of each award as an expense on a straight-line basis over the requisite service period, generally the vesting period of the equity grant. We reflect excess tax benefits related to stock option exercises as financing cash inflows.
The valuation model for stock compensation expense requires us to make assumptions and judgments about the variables used in the calculation including the expected life (weighted-average period of time that the options granted are expected to be outstanding), the expected volatility of our common stock, an assumed risk-free interest rate, the dividend yield and the estimated forfeitures of unvested stock options. We have opted to use the “simplified method” for estimating the expected term of options, whereby the expected term equals the arithmetic average of the vesting term and the original contractual term of the option. Due to our limited
63
Table of Contents
operating history and a lack of company specific historical and implied volatility data, we have based our estimate of expected volatility on the historical volatility of a group of similar companies that are publicly traded. The historical volatility data was computed using the daily closing prices for the selected companies’ shares during the equivalent period of the calculated expected term of the share-based payments. We will continue to analyze the historical stock price volatility and expected term assumptions as more historical data for our common stock becomes available. The risk-free rate assumption is based on the U.S. Treasury instruments with maturities similar to the expected term of our stock options. The expected dividend assumption is based on our history of not paying dividends and our expectation that we will not declare dividends for the foreseeable future.
The assumptions used in the Black-Scholes option pricing model for the periods specified below are as follows:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Expected life in years |
5.76 | 5.72 | 5.97 | |||||||||
| Risk-free interest rate |
1.68% | 1.08% | 1.05% | |||||||||
| Expected dividend yield |
0% | 0% | 0% | |||||||||
| Expected volatility |
54% | 68% | 69% | |||||||||
Prior to our IPO, our board of directors, with the assistance of management determined the fair value of our common stock on the date of grant based on several factors, including:
| • | our financial condition and operating results, including our projected results; |
| • | our stage of development and business strategy; |
| • | the financial condition and operating results of publicly owned companies with similar lines of business and their historical volatility; |
| • | external market conditions that could affect companies in the life sciences and medical device sectors; |
| • | the prices of our convertible preferred stock sold to outside investors and the rights, preferences and privileges of our convertible preferred stock as compared to those of our common stock, including the liquidation preference of our convertible preferred stock; |
| • | the likelihood of a liquidity event such as an initial public offering, a merger or the sale of the Company; and |
| • | any recent valuations prepared in accordance with the American Institute of Certified Public Accountants Practice Aid entitled Valuation of Privately Held Company Equity Securities Issued as Compensation. |
For the options granted subsequent to our IPO, the exercise price of stock options is equal to the closing market price of the underlying common stock on the grant date.
Convertible Preferred Stock Warrant Liability
For a warrant classified as a derivative liability, we record the fair value of that warrant on the balance sheet at the inception of such classification and adjust to fair value at each financial reporting date. We record the changes in the fair value of the warrants in the statement of operations as a component of interest and other income or expense as appropriate. We will continue to adjust the carrying value of the convertible preferred stock warrant liability for changes in the fair value of the warrants until the earlier of: the exercise of the warrants, at which time we will reclassify the liability to temporary equity; the conversion of the underlying preferred stock into common stock, at which time we will reclassify the liability to stockholders’ deficit; or the expiration of the warrant, at which time we would reverse the entire amount and reflect it in the statement of operations. Our assumptions with regard to the warrant valuation are based on estimates of the valuation of the underlying preferred stock, volatility, interest rate and such estimates could vary significantly.
64
Table of Contents
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update No. 2014-09: Revenue from Contracts with Customers. The standard will eliminate the transaction and industry specific revenue recognition guidance under current U.S. generally accepted accounting principles and replace it with a principle-based approach for determining revenue recognition. The new guidance is effective for annual and interim periods beginning after December 15, 2017. We are still evaluating the impact of the adoption of this standard.
Jumpstart Our Business Startups Act of 2012 (JOBS Act)
In April 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We intend to take advantage of such extended transition period. Section 107 of the JOBS Act provides that we can elect to opt out of the extended transition period at any time, which election is irrevocable.
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements under the JOBS Act. Subject to certain conditions, as an emerging growth company, we may rely on certain of these exemptions, including without limitation, (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (ii) complying with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an emerging growth company until the earlier of (a) the last day of the fiscal year in which we have total annual gross revenues of $1.0 billion or more; (b) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous six years; or (c) the date on which we are deemed to be a large accelerated filer under the rules of the SEC; or (d) December 31, 2020, the last day of the fiscal year following the fifth anniversary of our IPO.
Liquidity and Capital Resources
Overview
As of December 31, 2014, we had cash and cash equivalents of $3.5 million and an accumulated deficit of $103.8 million, compared to cash and cash equivalents of $7.7 million and an accumulated deficit of $96.8 million as of December 31, 2013. Our primary sources of capital through the year ended December 31, 2014 have been from sales of our products, private placements of our convertible preferred securities and amounts borrowed under our credit facility. As of December 31, 2014, we have raised $92.1 million from private placements of our convertible preferred securities and had $20.0 million of borrowings outstanding and up to $5.0 million of additional availability under our credit facility, subject to achievement of certain revenue milestones.
In February 2015, we completed our IPO by issuing 5,294,117 shares of common stock at an offering price of $17.00 per share, for net proceeds to us of approximately $81.2 million after deducting underwriting discounts and commissions and offering expenses payable by us.
65
Table of Contents
Cash Flows
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2014 | 2013 | 2012 | |||||||||
| Net cash (used in) provided by: |
||||||||||||
| Operating activities |
$ | (8,777 | ) | $ | (13,211 | ) | $ | (20,432 | ) | |||
| Investing activities |
(998 | ) | 6,038 | 2,083 | ||||||||
| Financing activities |
5,550 | 8,201 | 17,361 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net (decrease) increase in cash and cash equivalents |
$ | (4,225 | ) | $ | 1,028 | $ | (988 | ) | ||||
|
|
|
|
|
|
|
|||||||
Net Cash Used in Operating Activities
Net cash used in operating activities for 2014 was $8.8 million, consisting primarily of a net loss of $6.9 million, offset by an increase in net operating assets of $3.1 million and by non-cash charges of $1.2 million. The increase in net operating assets was primarily due to increases in deferred offering costs, accounts receivable and inventory, offset by an increases in accounts payable due to offering costs and accrued compensation due to the growth in our sales organization and accrued royalties. Non-cash charges consisted primarily of depreciation, the accretion of the final payment fee on our credit facility, stock-based compensation and the change in fair value of convertible preferred stock warrants.
Net cash used in operating activities for 2013 was $13.2 million, consisting primarily of a net loss of $13.4 million, offset by an increase in net operating assets of $0.9 million and by non-cash charges of $1.1 million. The increase in net operating assets was primarily due to increases in accounts receivable and inventory, offset by an increases in accrued compensation due to the growth in our sales organization and accrued royalties. Non-cash charges consisted primarily of depreciation, stock-based compensation and the change in fair value of convertible preferred stock warrants.
Net cash used in operating activities for 2012 was $20.4 million, consisting primarily of a net loss of $19.0 million and an increase in net operating assets of $2.2 million, partially offset by non-cash charges of $0.8 million. The increase in net operating assets was primarily due to increases in accounts receivable and inventory as we accelerated commercialization of our XprESS family of products, partially offset by an increase in accrued royalties and a decrease in accrued compensation due to bonus payments paid in December 2012 rather than January 2013. Non-cash charges consisted primarily of depreciation and stock-based compensation.
Net Cash (Used in) Provided by Investing Activities
Net cash used in investing activities in 2014 was $1.0 million for purchases of property and equipment. Net cash provided by investing activities in 2013 and 2012 was $6.0 million and $2.1 million, consisting of proceeds from maturities of short-term investments of $6.6 million and $2.9 million, partially offset by purchases of property and equipment of $0.6 million and $0.8 million, respectively.
Net Cash Provided by Financing Activities
Net cash provided by financing activities in 2014 and 2013 was $5.6 million and $8.2 million, respectively, consisting of borrowing under our credit facility of $5.0 million and $7.5 million and proceeds from the exercise of stock options of $0.6 million and $0.7 million respectively.
Net cash provided by financing activities in 2012 was $17.4 million, consisting of net proceeds from the issuance of our Series E convertible preferred stock of $10.0 million, net borrowings under our credit facility of $7.1 million and proceeds from the exercise of stock options of $0.3 million.
Credit Facility
In October 2012, we entered into a credit facility with Oxford Finance LLC and borrowed $7.5 million. In December 2013, we amended and restated the credit facility, and in October 2014, we further amended the credit
66
Table of Contents
facility. We refer to the amended and restated credit facility, as amended, as the credit facility. Under the credit facility, we can borrow up to a total of $25.0 million in three tranches at a fixed rate of 9.40%. The first tranche of $15.0 million was borrowed in December 2013, a portion of which was used to refinance the $7.5 million previously outstanding under the credit facility. The second tranche of up to $5.0 million, or the 2014 tranche, was available through December 31, 2014, of which all was borrowed. The third tranche of up to $5.0 million is available at any time through December 31, 2015, subject to certain trailing six-month revenue milestones. The credit facility matures and all amounts borrowed thereunder are due on December 1, 2018. As a result of the $5.0 million borrowed during the three months ended December 31, 2014, all amounts borrowed under the credit facility are interest-only through January 2016 (24 months), after which we will make monthly payments of principal and interest. The interest-only period may be extended to 36 months based on funding and revenue milestones provided for in the credit facility.
In addition to the principal and interest payments under the credit facility, we are required to pay a final payment fee of 7.15% on all amounts outstanding, which is being accrued over the credit facility term and will be due at the earlier of maturity or prepayment. If we repay all of the amounts borrowed under the loan on or prior to maturity, we will also be required to pay a prepayment fee equal to 0.75%. We may make principal payments during any interest only periods to reduce, but not repay in full, the outstanding principal balance of the loan without incurring any prepayment penalties. In this case, however, we would still be required to pay the final payment fee.
Our obligations under the credit facility are secured by a first priority security interest in substantially all of our assets, other than our intellectual property. There are no financial covenants contained in the credit facility and we are in compliance with the affirmative and restrictive covenants as of December 31, 2014. We have agreed not to pledge or otherwise encumber our intellectual property assets, except for permitted liens, as such terms are defined in the credit facility and except as otherwise provided for in the credit facility.
As of December 31, 2014, we have borrowed and have outstanding $20.0 million of debt under the credit facility.
Off-Balance Sheet Arrangements
We do not maintain any off-balance sheet partnerships, arrangements or other relationships with unconsolidated entities or others, often referred to as structured finance or special-purpose entities that are established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Contractual Obligations
The following table sets out, as of December 31, 2014, our contractual obligations due by period.
| Payments Due by Period | ||||||||||||||||||||
| Less Than 1 Year |
1-3 Years |
3-5 Years |
More Than 5 Years |
Total | ||||||||||||||||
| (in thousands) |
||||||||||||||||||||
| Debt obligations (1) |
$ | — | $ | 5,708 | $ | 15,722 | $ | — | $ | 21,430 | ||||||||||
| Operating lease obligations (2) |
205 | 407 | 137 | — | 749 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| $ | 205 | $ | 6,115 | $ | 15,859 | $ | — | $ | 22,179 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (1) | The total amount outstanding under the credit facility was $20.0 million at December 31, 2014. Assumes a 36-month amortization period for repayment of the debt. The total debt amount is inclusive of the final payment fee of 7.15% due with the final payment. |
| (2) | We currently lease approximately 32,500 square feet for our headquarters and manufacturing facilities in Plymouth, Minnesota under leases that expire in August 2018. |
67
Table of Contents
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
Interest Rate Risk
Interest rate fluctuations have not had a material impact on our results of operations. Our cash and cash equivalents include cash in readily available checking and money market accounts. Because our cash equivalents are primarily short-term in duration, we believe that our exposure to interest rate risk is not significant and a 100 basis point movement in market interest rates would not have a significant impact on the total value of our cash and cash equivalents. Additionally, the interest rate on our credit facility is fixed and not subject to changes in market interest rates.
Inflation
Inflationary factors, such as increases in our cost of goods sold and selling and operating expenses, may adversely affect our operating results. Although we do not believe that inflation has had a material impact on our financial position or results of operations to date, a high rate of inflation in the future may have an adverse effect on our ability to maintain and increase our gross margin and selling and marketing and operating expenses as a percentage of our revenue if the selling prices of our products do not increase as much or more than these increased costs.
Credit Risk
As of December 31, 2014, our cash and cash equivalents were maintained with two financial institutions in the United States, and our current deposits are likely in excess of insured limits. We have reviewed the financial statements of these institutions and believe they have sufficient assets and liquidity to conduct their operations in the ordinary course of business with little or no credit risk to us.
Our accounts receivable primarily relate to revenue from the sale of our XprESS family of products to hospitals, ASCs and physician offices in the United States. For the year ended December 31, 2014, no single customer represented more than 5% of our revenue.
Foreign Currency Risk
Our business is conducted in U.S. dollars and foreign transactions have been minimal. Any transactions that may be conducted in foreign currencies are not expected to have a material effect on our results of operations, financial position or cash flows.
68
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
ENTELLUS MEDICAL, INC.
Index to Financial Statements
69
Table of Contents
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders of
Entellus Medical, Inc.
We have audited the accompanying balance sheets of Entellus Medical, Inc. (a Delaware corporation) (the “Company”) as of December 31, 2014 and 2013, and the related statements of operations and comprehensive loss, convertible preferred stock and stockholders’ deficit, and cash flows for each of the three years in the period ended December 31, 2014. Our audits of the basic financial statements included the financial statement schedule listed in the index appearing under Item 15(a)(2). These financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Entellus Medical, Inc. as of December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
/s/ Grant Thornton LLP
Minneapolis, Minnesota
March 19, 2015
70
Table of Contents
BALANCE SHEETS
(in thousands, except per share data)
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| ASSETS |
||||||||
| CURRENT ASSETS |
||||||||
| Cash and cash equivalents |
$ | 3,484 | $ | 7,709 | ||||
| Accounts receivable, net |
8,746 | 5,823 | ||||||
| Inventories |
2,439 | 1,869 | ||||||
| Prepaid expenses and other current assets |
883 | 816 | ||||||
|
|
|
|
|
|||||
| Total current assets |
15,552 | 16,217 | ||||||
| PROPERTY AND EQUIPMENT, NET |
1,730 | 1,349 | ||||||
| OTHER ASSETS |
||||||||
| Debt issuance costs, net |
237 | 291 | ||||||
| Other non-current assets |
1,717 | 19 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 19,236 | $ | 17,876 | ||||
|
|
|
|
|
|||||
| LIABILITIES, CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIT |
||||||||
| CURRENT LIABILITIES |
||||||||
| Accounts payable |
$ | 2,414 | $ | 1,202 | ||||
| Convertible preferred stock warrant liability |
291 | 211 | ||||||
| Accrued expenses |
5,084 | 4,102 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
7,789 | 5,515 | ||||||
| LONG-TERM LIABILITIES |
||||||||
| Long-term debt |
20,000 | 15,000 | ||||||
| Other noncurrent liabilities |
247 | 35 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
28,036 | 20,550 | ||||||
| COMMITMENTS AND CONTINGENCIES (see Note L) |
||||||||
| CONVERTIBLE PREFERRED STOCK (see Note G), issuable in series, $0.001 par value per share: actual: 44,567 shares authorized and 44,317 shares issued and outstanding at December 31, 2014 and 2013 |
91,554 | 91,554 | ||||||
| STOCKHOLDERS’ DEFICIT (see Note H) |
||||||||
| Common stock, $0.001 par value per share: |
2 | 2 | ||||||
| Additional paid-in capital |
3,402 | 2,599 | ||||||
| Accumulated deficit |
(103,758 | ) | (96,829 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ deficit |
(100,354 | ) | (94,228 | ) | ||||
|
|
|
|
|
|||||
| Total liabilities, convertible preferred stock and stockholders’ deficit |
$ | 19,236 | $ | 17,876 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
71
Table of Contents
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except per share data)
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Revenue |
$ | 48,820 | $ | 32,545 | $ | 17,559 | ||||||
| Cost of goods sold |
10,754 | 7,808 | 4,784 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
38,066 | 24,737 | 12,775 | |||||||||
| Operating expenses |
||||||||||||
| Selling and marketing |
32,763 | 27,631 | 21,634 | |||||||||
| Research and development |
4,307 | 5,143 | 5,896 | |||||||||
| General and administrative |
6,097 | 4,311 | 4,080 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
43,167 | 37,085 | 31,610 | |||||||||
| Loss from operations |
(5,101 | ) | (12,348 | ) | (18,835 | ) | ||||||
| Other income (expense) |
||||||||||||
| Interest income |
75 | 6 | 14 | |||||||||
| Interest expense |
(1,904 | ) | (1,082 | ) | (155 | ) | ||||||
| Other non-operating income (expense) |
1 | 28 | (20 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total other income (expense), net |
(1,828 | ) | (1,048 | ) | (161 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (6,929 | ) | $ | (13,396 | ) | $ | (18,996 | ) | |||
|
|
|
|
|
|
|
|||||||
| Other comprehensive loss |
||||||||||||
|
|
|
|
|
|
|
|||||||
| Unrealized loss on short term investments |
— | (1 | ) | (3 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive loss |
$ | (6,929 | ) | $ | (13,397 | ) | $ | (18,999 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per share, basic and diluted |
$ | (4.62 | ) | $ | (11.82 | ) | $ | (27.29 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average common shares used to compute net loss per share, basic and diluted |
1,499 | 1,133 | 696 | |||||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
72
Table of Contents
STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIT
(in thousands)
| Additional Paid-in Capital |
Accumulated Deficit |
Accumulated Other Comprehensive Income(Loss) |
Total Stockholders’ Deficit |
|||||||||||||||||||||||||||||||
| Convertible preferred stock |
Common stock | |||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||
| Balance at December 31, 2011 |
39,213 | $ | 81,564 | 635 | $ | 1 | $ | 971 | $ | (64,437 | ) | $ | 4 | $ | (63,461 | ) | ||||||||||||||||||
| Stock options exercised |
— | — | 231 | — | 254 | — | — | 254 | ||||||||||||||||||||||||||
| Issuance of Series E convertible preferred stock, net of issuance costs of $10 |
5,104 | 9,990 | — | — | — | — | — | — | ||||||||||||||||||||||||||
| Stock-based compensation expense |
— | — | — | — | 304 | — | — | 304 | ||||||||||||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (18,996 | ) | (3 | ) | (18,999 | ) | |||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2012 |
44,317 | 91,554 | 866 | 1 | 1,529 | (83,433 | ) | 1 | (81,902 | ) | ||||||||||||||||||||||||
| Stock options exercised |
— | — | 583 | 1 | 716 | — | — | 717 | ||||||||||||||||||||||||||
| Stock-based compensation expense |
— | — | — | — | 354 | — | — | 354 | ||||||||||||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (13,396 | ) | (1 | ) | (13,397 | ) | |||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2013 |
44,317 | 91,554 | 1,449 | 2 | 2,599 | (96,829 | ) | — | (94,228 | ) | ||||||||||||||||||||||||
| Stock options exercised |
— | — | 466 | — | 576 | — | — | 576 | ||||||||||||||||||||||||||
| Repurchase of common stock |
— | — | (28 | ) | — | (21 | ) | — | — | (21 | ) | |||||||||||||||||||||||
| Stock-based compensation expense |
— | — | — | — | 248 | — | — | 248 | ||||||||||||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (6,929 | ) | — | (6,929 | ) | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2014 |
44,317 | $ | 91,554 | 1,887 | $ | 2 | $ | 3,402 | $ | (103,758 | ) | $ | — | $ | (100,354 | ) | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
The accompanying notes are an integral part of these financial statements
73
Table of Contents
STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (6,929 | ) | $ | (13,396 | ) | $ | (18,996 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
618 | 460 | 432 | |||||||||
| Amortization of debt issuance costs |
59 | 82 | 21 | |||||||||
| Amortization of premium on investments |
— | 9 | 26 | |||||||||
| Accretion of final debt payment |
227 | — | — | |||||||||
| Deferred rent |
(13 | ) | 8 | 26 | ||||||||
| Accretion of debt discount |
— | 125 | 7 | |||||||||
| (Gain) loss on disposal of equipment |
(1 | ) | (1 | ) | 19 | |||||||
| Stock-based compensation |
248 | 354 | 304 | |||||||||
| Changes in fair value of convertible preferred stock warrants |
80 | 80 | (1 | ) | ||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accrued interest income |
— | 46 | (43 | ) | ||||||||
| Accounts receivables, net |
(2,923 | ) | (2,283 | ) | (1,417 | ) | ||||||
| Inventories |
(570 | ) | (297 | ) | (457 | ) | ||||||
| Prepaid expenses and other current assets |
(67 | ) | (340 | ) | (150 | ) | ||||||
| Other non-current assets |
(1,698 | ) | — | — | ||||||||
| Accounts payable |
1,212 | (54 | ) | (18 | ) | |||||||
| Accrued expenses |
980 | 1,996 | (185 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(8,777 | ) | (13,211 | ) | (20,432 | ) | ||||||
| Cash flows from investing activities: |
||||||||||||
| Purchase of property and equipment |
(999 | ) | (587 | ) | (876 | ) | ||||||
| Proceeds from sale of property and equipment |
1 | 1 | 13 | |||||||||
| Purchase of short-term investments |
— | — | (8,754 | ) | ||||||||
| Proceeds from maturities of short-term investments |
— | 6,624 | 11,700 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) provided by investing activities |
(998 | ) | 6,038 | 2,083 | ||||||||
| Cash Flows from Financing Activities |
||||||||||||
| Proceeds from long-term debt |
5,000 | 7,500 | 7,500 | |||||||||
| Debt issuance costs |
(5 | ) | (16 | ) | (377 | ) | ||||||
| Proceeds from stock options exercised |
576 | 717 | 254 | |||||||||
| Repurchase of common stock |
(21 | ) | — | — | ||||||||
| Proceeds from issuance of convertible preferred stock, net of issuance costs |
— | — | 9,990 | |||||||||
| Security deposit |
— | — | (6 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
5,550 | 8,201 | 17,361 | |||||||||
| Net increase (decrease) in cash and cash equivalents |
(4,225 | ) | 1,028 | (988 | ) | |||||||
| Cash and equivalents - beginning of period |
7,709 | 6,681 | 7,669 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents - end of period |
$ | 3,484 | $ | 7,709 | $ | 6,681 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental schedule of non-cash investing and financing activities: |
||||||||||||
| Debt issuance costs |
$ | 5 | $ | — | $ | 132 | ||||||
|
|
|
|
|
|
|
|||||||
| Cash paid for interest |
$ | 1,349 | $ | 626 | $ | 52 | ||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
74
Table of Contents
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
NOTE A – NATURE OF BUSINESS
Entellus Medical, Inc. (the “Company”) was incorporated in Minnesota in April 2006 and was reincorporated in Delaware in August 2006. The Company is focused on the design, development and commercialization of products for the minimally invasive treatment of patients suffering from chronic sinusitis. The Company’s XprESS family of products is used by ear, nose and throat (“ENT”) physicians to treat patients with chronic sinusitis by opening narrowed or obstructed sinus drainage pathways using balloon sinus dilation performed in the physician office or the operating room. The Company is located in Plymouth, Minnesota, and currently sells product throughout the United States.
NOTE B – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Initial Public Offering
In February 2015, the Company completed its initial public offering (“IPO”) by issuing 5,294 shares of common stock at an offering price of $17.00 per share, for net proceeds to the Company of approximately $81,200, after deducting underwriting discounts and commissions and offering expenses payable by the Company. In connection with the IPO, the Company’s outstanding shares of convertible preferred stock were automatically converted into an aggregate of 11,404 shares of common stock and warrants exercisable for convertible preferred stock were automatically converted into warrants to purchase up to an aggregate of 38 shares of common stock, resulting in the reclassification of the related redeemable convertible preferred stock warrant liability of $291 to additional paid-in-capital. See Note N—Subsequent Events.
1-for-4 Reverse Stock Split
In connection with the IPO, the Company’s board of directors and stockholders approved a reverse stock split of the Company’s common stock on a 1-for-4 basis. The reverse stock split became effective on January 12, 2015. The par value of the common stock was not adjusted as a result of the reverse stock split. Adjustments corresponding to the reverse stock split were made to the ratio at which the convertible preferred stock converted into common stock immediately prior to the closing of the IPO. Accordingly, all share and per share amounts for all periods presented in these financial statements and notes thereto have been adjusted retroactively, where applicable, to reflect the reverse stock split and adjustment of the preferred stock’s conversion ratios. See Note N—Subsequent Events.
Basis of Preparation
The financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”) and pursuant to the rules and regulations of the United States Securities and Exchange Commission (“SEC”).
JOBS Act Accounting Election
As an emerging growth company under the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”), the Company is eligible to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. The Company has elected to take advantage of the extended transition period for adopting new or revised accounting standards that have different effective dates for public and private companies until such time as those standards apply to private companies.
75
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers. This standard will eliminate the transaction and industry specific revenue recognition guidance under current U.S. GAAP and replace it with a principles based approach for determining revenue recognition. The new guidance for private companies is effective for annual and interim periods beginning after December 15, 2017. The Company is in the process of evaluating the impact of the adoption of this standard.
Cash and Equivalents
The Company considers all highly liquid investments purchased with an initial maturity of 90 days or less to be cash equivalents. At December 31, 2014 and 2013, cash equivalents consisted of money market funds, which are stated at cost and approximate fair value. The Company maintains cash in bank accounts which, at times, may exceed Federal Deposit Insurance Corporation (“FDIC”) limits. The Company has not experienced any losses from maintaining balances in excess of FDIC limits.
Fair Value of Financial Instruments
As of December 31, 2014 and 2013, the carrying amounts of cash and cash equivalents, accounts receivable, accounts payable and accrued expenses approximated their estimated fair value because of the short-term nature of the financial instruments. The Company determines the fair value of its assets and liabilities based on the exchange price that would be received for an asset or paid to transfer a liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value maximize the use of observable inputs and minimize the use of unobservable inputs. A fair value hierarchy with three levels of inputs, of which the first two are considered observable and the last unobservable, is used to measure fair value. The fair value hierarchy gives the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1). The next highest priority is based on quoted prices for similar assets or liabilities in active markets or quoted prices for identical or similar assets or liabilities in non-active markets or other observable inputs (Level 2). The lowest priority is given to unobservable inputs (Level 3).
Concentration of Credit Risk
Financial instruments, which potentially subject the Company to concentrations of credit risk, consist principally of cash equivalents and accounts receivable. The Company generally does not require collateral and losses on accounts receivable have historically been within management’s expectations.
The Company’s investment policy limits investments, when held, to certain types of debt securities issued by the U.S. government, its agencies, and institutions with investment-grade credit ratings, as well as corporate debt or commercial paper issued by the highest quality financial and non-financial companies, and places restrictions on maturities and concentration by type and issuer. The Company is exposed to credit risk in the event of a default by the financial institutions holding its cash and equivalents and issuers of debt securities to the extent recorded on the balance sheets.
Accounts Receivable
Credit is granted to customers in the normal course of business, generally without collateral or any other security to support amounts due. Accounts are considered past due generally after 30 days. The Company
76
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
maintains an allowance for doubtful accounts for estimated losses in the collection of accounts receivable. The Company makes estimates regarding the future ability of its customers to make required payments based on historical credit experience, delinquency and expected future trends. The Company will write off accounts receivable when it determines that the accounts receivable are uncollectible, typically upon customer bankruptcy or the customer’s non-response to continued collection efforts. The allowance for doubtful accounts was $150 and $135 as of December 31, 2014 and 2013, respectively. The Company also maintains an allowance for sales returns based on historical experience and evaluation of current sales and returns trends. The allowance for sales returns was $140 and $100 as of December 31, 2014 and 2013, respectively.
Inventories
Inventories are stated at the lower of cost or market with cost determined using the weighted-average cost method, which approximates the first-in, first-out method.
Property and Equipment
Property and equipment are recorded at cost. Depreciation of furniture, office, computer and laboratory equipment is calculated on a straight-line basis over the asset’s estimated useful life, which ranges from three to ten years. Tooling and molds are depreciated on a straight-line basis over three to five years. Leasehold improvements are amortized on a straight-line basis over the shorter of the remaining life of the lease or the useful life of the asset. Repair and maintenance expenditures are charged to earnings as incurred. Major repairs and maintenance expenditures that extend useful lives of property and equipment are capitalized.
Deferred Offering Costs
Deferred offering costs, primarily consisting of legal, accounting and other direct fees and costs relating to the initial public offering, are capitalized. The deferred offering costs were offset against the IPO proceeds upon the closing of the offering in February 2015. As of December 31, 2014, there was $1,698 in deferred offering costs capitalized in other non-current assets on the balance sheet. There were no deferred offering costs capitalized as of December 31, 2013.
Impairment of Long-Lived Assets
The Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceed the fair value of the assets. Assets to be disposed of are reported at the lower of the carrying amount or fair value less costs to sell. The Company has not recorded impairment charges on long-lived assets for the periods presented in these financial statements.
Deferred Rent
The Company accounts for rent expense related to operating leases by determining total minimum rent payments on the leases over their respective periods and recognizing the rent expense on a straight-line basis. The difference between the actual amount paid and the amount recorded as rent expense in each period presented is recorded as an adjustment to other non-current liabilities in the balance sheet.
77
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
Debt Issuance and Debt Discount
Debt issuance costs are stated at cost, net of accumulated amortization and included within other assets. Debt discount is recorded as a reduction of long-term debt. Amortization of debt issuance costs and the debt discount is calculated using the effective interest method over the term of the debt and is recorded in interest expense in the accompanying statements of operations and comprehensive loss.
Revenue Recognition
The Company recognizes revenue when persuasive evidence of a sales arrangement exists, delivery of goods has occurred through the transfer of title and risks and rewards of ownership, the selling price is fixed or determinable and collectability is reasonably assured. Revenue is reported net of the allowance for sales returns. Taxes assessed by a governmental authority that are directly imposed on revenue-producing transactions between a seller and a customer are not recorded as revenue.
The Company entered into a distribution agreement with CogENT Therapeutics in September 2013 and began exclusive distribution of XeroGel in October 2013 in the United States. For products shipped from the CogENT warehouse to customers, CogENT is responsible for all manufacturing, order fulfillment, quality and regulatory requirements of the product. For these direct shipment orders, the Company does not take title to CogENT products prior to products being ordered by customers or in the event CogENT products are returned by customers. The Company records transactions under this arrangement on a net basis based on the difference between the amounts billed to customers less the amounts paid to CogENT for the XeroGel product. The Company’s net revenue for the year ended December 31, 2014, 2013 and 2012 were approximately $839, $65 and $0, respectively. For products for which the Company takes title, ships the product and handles returns, the Company records transactions under these arrangements on a gross basis. The amounts of revenue recorded on a gross basis for the year ended December 31, 2014, 2013 and 2012 were approximately $150, $0 and $0, respectively.
Shipping and Handling
Shipping and handling costs charged to customers are included as a component of revenue and related costs are included in costs of goods sold.
Cost of Goods Sold
Cost of goods sold consists primarily of manufacturing overhead costs, material costs and direct labor. A significant portion of the Company’s cost of goods sold currently consists of manufacturing overhead costs. These overhead costs include the cost of quality assurance, material procurement, inventory control, facilities, equipment, operations supervision and management, depreciation expense for production equipment, amortization of leasehold improvements, shipping costs and royalty expense payable in connection with sales of certain products.
Research and Development Costs
Research and development expenses consist primarily of engineering, product development, clinical and regulatory affairs, consulting services, materials, depreciation and other costs associated with products and technologies in development. These expenses include employee and non-employee compensation, including stock-based compensation, supplies, materials, consulting, related travel expenses and facility costs. Clinical expenses include clinical trial design, clinical site reimbursement, data management and travel expenses, and the cost of manufacturing products for clinical trials. Expenditures for research and development activities are charged to operations as incurred.
78
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
Advertising Expense
Expenditures for advertising are charged to operations as incurred. Advertising expenses were $436, $481 and $388 during the years ended December 31, 2014, 2013 and 2012, respectively.
Common Stock Valuation and Stock-based Compensation
Prior to the IPO, the Company maintained its 2006 Stock Incentive Plan (the “2006 Plan”) to provide long-term incentives for employees, consultants and members of the board of directors. The 2006 Plan allowed for the issuance of non-statutory and incentive stock options to employees and non-statutory stock options to consultants and non-employee directors. In connection with the IPO, the 2006 Plan terminated, and the Company adopted a new equity incentive plan, the 2015 Incentive Award Plan (the “2015 Plan”). See Note N—Subsequent Events.
The Company is required to determine the fair value of equity incentive awards and recognize compensation expense for all equity incentive awards made to employees and directors, including employee stock options. The Company also determines the fair value of non-statutory stock options issued to consultants based on the fair value of the consultant’s commitment at the date of grant. Such expense is recognized over the requisite service period. In addition, stock-based compensation expense recognized in the statements of operations and comprehensive loss is based on awards expected to vest and therefore the amount of expense has been reduced for estimated forfeitures. The Company uses the straight-line method for expense attribution, except for awards issued with performance-based conditions which require an accelerated attribution method over the requisite performance and service periods.
Under the applicable accounting guidance for equity incentive awards issued to non-employees, the date at which the fair value of such awards is measured is equal to the earlier of: 1) the date at which a commitment for performance by the counterparty to earn the equity instrument is reached; or 2) the date at which the counterparty’s performance is complete. The Company recognizes stock-based compensation expense for the fair value of the vested portion of the non-employee awards in the statements of operations and comprehensive loss.
The valuation model used for calculating the fair value of awards for stock-based compensation expense is the Black-Scholes option-pricing model (the “Black-Scholes model”). The Black-Scholes model requires the Company to make assumptions and judgments about the variables used in the calculation, including the expected term (weighted average period of time that the options granted are expected to be outstanding), the volatility of common stock and an assumed risk-free interest rate. The Company uses the “simplified method” to determine the expected term of the stock option. Volatility is based on an average of the historical volatilities of the common stock of publicly-traded companies with characteristics similar to those of the Company. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected term of the option. The expected dividend assumption is based on the Company’s history of not paying dividends and its expectation that it will not declare dividends for the foreseeable future. Potential forfeitures of awards are estimated based on the Company’s historical forfeiture experience. The estimate of forfeitures will be adjusted over the service period to the extent that actual forfeitures differ, or are expected to differ, from prior estimates.
Convertible Preferred Stock Warrant Liability
For a warrant classified as a derivative liability, the fair value of that warrant is recorded on the balance sheet when granted and adjusted to fair value at each financial reporting date. The changes in the fair value of the warrants are recorded in the statement of operations and comprehensive loss as a component of interest income or expense as appropriate. In connection with the IPO, in February 2015, the warrants for convertible preferred stock were converted to warrants for common stock and the corresponding liability was reclassified to stockholders’ deficit.
79
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax base and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled.
The Company applies a recognition threshold for income tax positions taken or expected to be taken on a tax return. The impact of an uncertain tax position taken or expected to be taken on an income tax return are recognized in the financial statements at the largest amount that is more-likely-than not to be sustained upon audit by the relevant taxing authority. An uncertain tax position is not recognized in the financial statements unless it is more-likely-than not of being sustained. The Company has no reserve for unrecognized tax benefits at December 31, 2014 and 2013, and accordingly, there is no interest or penalties recorded on the balance sheet for such reserves. The Company is currently open to audit under the statute of limitations by the Internal Revenue Service and the appropriate state income taxing authorities for 2006 through 2014.
Net Loss per Share
Basic net loss per share is computed by dividing the net loss by the weighted average number of shares of common stock outstanding during the period. Diluted net loss per share is computed by dividing the net loss by the weighted average number of shares of common stock and dilutive potential shares of common stock outstanding during the period. Because the Company has reported a net loss for all periods presented, diluted net loss per share is the same as basic net loss per share for those periods as all potentially dilutive shares consisting of convertible preferred stock, stock options and warrants were antidilutive in those periods.
The Company allocates no loss to participating securities because they have no contractual obligation to share in the losses of the Company. The shares of the Company’s convertible preferred stock participate in any dividends declared by the Company and are therefore considered to be participating securities.
Comprehensive Loss
Comprehensive loss consists of net loss and changes in unrealized gains and losses on available-for-sale marketable securities. Accumulated other comprehensive income (loss) is presented in the accompanying balance sheets, when applicable.
Segment, Geographical and Customer Concentration
The Company operates in one segment. All of the Company’s assets and revenue are based in the United States and no single customer accounted for more than 10% of its revenue during the years ended December 31, 2014, 2013 and 2012.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
80
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
NOTE C – COMPOSITION OF CERTAIN FINANCIAL STATEMENT ITEMS
Inventories
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Finished goods |
$ | 859 | $ | 703 | ||||
| Work in process |
318 | 379 | ||||||
| Raw materials |
1,262 | 787 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 2,439 | $ | 1,869 | ||||
|
|
|
|
|
|||||
Property and Equipment
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Furniture and office equipment |
$ | 401 | 357 | |||||
| Computer hardware and software |
837 | 708 | ||||||
| Laboratory equipment |
1,427 | 709 | ||||||
| Tooling and molds |
1,237 | 1,185 | ||||||
| Leasehold improvements |
494 | $ | 458 | |||||
|
|
|
|
|
|||||
| $ | 4,396 | 3,417 | ||||||
| Less accumulated depreciation and amortization |
(2,768 | ) | (2,173 | ) | ||||
| Property and equipment in progress |
102 | 105 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 1,730 | $ | 1,349 | ||||
|
|
|
|
|
|||||
Depreciation and amortization expense was $618, $460 and $432 during the years ended December 31, 2014, 2013 and 2012, respectively.
Other Non-Current Assets
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Security deposit on operating lease |
$ | 19 | $ | 19 | ||||
| Deferred offering costs |
1,698 | — | ||||||
|
|
|
|
|
|||||
| $ | 1,717 | $ | 19 | |||||
|
|
|
|
|
|||||
Accrued Expenses
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Compensation and commissions payable |
$ | 3,001 | $ | 2,522 | ||||
| Royalty payable |
1,537 | 1,160 | ||||||
| Other accrued expenses |
546 | 420 | ||||||
|
|
|
|
|
|||||
| $ | 5,084 | $ | 4,102 | |||||
|
|
|
|
|
|||||
81
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
Interest Expense
| December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Interest expense |
$ | 1,750 | $ | 1,000 | $ | 155 | ||||||
| Increase in fair value of convertible preferred stock warrants |
154 | 82 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 1,904 | $ | 1,082 | $ | 155 | |||||||
|
|
|
|
|
|
|
|||||||
NOTE D – LIQUIDITY AND BUSINESS RISKS
As of December 31, 2014 and 2013, the Company had cash and equivalents of $3,484 and $7,709 and an accumulated deficit of $103,758 and $96,829, respectively. In February 2015, the Company completed its IPO by issuing 5,294 shares of common stock at an offering price of $17.00 per share, for net proceeds to the Company of approximately $81,200 after deducting underwriting discounts and commissions and offering expenses payable by the Company. See Note N—Subsequent Events. Prior to the IPO, the Company financed its operations with a combination of revenue, private placements of convertible preferred securities and amounts borrowed under its credit facility.
NOTE E – CONVERTIBLE PREFERRED STOCK WARRANT LIABILITY
As of December 31, 2014 and 2013, convertible preferred stock warrant liabilities, which represent financial instruments that were categorized as Level 3, according to the three-level fair value hierarchy.
The following table sets forth a summary of the changes in the estimated fair value of the Company’s convertible preferred stock warrant liability:
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Beginning of the period |
$ | 211 | $ | 131 | ||||
| Issued |
— | — | ||||||
| Exercised |
— | — | ||||||
| Expired |
— | — | ||||||
| Change in fair value |
80 | 80 | ||||||
|
|
|
|
|
|||||
| End of period |
$ | 291 | $ | 211 | ||||
|
|
|
|
|
|||||
The fair value of the convertible preferred stock warrant liability was determined using the option pricing method or the probability weighted expected return method using the following assumptions:
| Years Ended December 31, |
||||||||
| 2014 |
2013 | |||||||
| Expected life in years |
7.8 | 8.8 | ||||||
| Risk-free interest rate |
2.0 | % | 2.5 | % | ||||
| Expected dividend yield |
0.0 | % | 0.0 | % | ||||
| Expected volatility |
50 | % | 50 | % | ||||
82
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
NOTE F – DEBT
In October 2012, the Company entered into a loan and security agreement with Oxford Finance LLC (the “lender”) under which it could borrow up to a total of $17,000 in two tranches at a fixed rate floor of 8.35%. The first tranche of $7,500 was borrowed in October 2012.
In December 2013, the Company amended and restated the loan and security agreement and in October 2014, the Company further amended the loan and security agreement (as currently in effect, the “credit facility”). Under the credit facility, the Company can borrow up to a total of $25,000 in three tranches at a fixed rate of 9.40%. The first tranche of $15,000, including the refinance of $7,500 previously outstanding, was borrowed in December 2013. In connection with the funding of the first tranche, the Company paid a prorated final payment fee on the original credit facility of $143. The second tranche of up to $5,000 (the “2014 tranche”) was available through December 31, 2014, of which $5,000 was borrowed during the three months ended December 31, 2014. The third tranche of up to $5,000 is available at any time through December 31, 2015, subject to the achievement by the Company of certain revenue milestones. The credit facility matures and all amounts borrowed thereunder are due on December 1, 2018. As a result of the $5,000 borrowed in the three months ended December 31, 2014, all amounts borrowed under the credit facility are interest-only through January 2016 (24 months), after which the Company will make monthly payments of principal and interest. The interest-only period may be extended to 36 months based on funding and revenue milestones provided for in the agreement.
In addition to the principal and interest payments, under the credit facility the Company is required to pay a final payment fee of 7.15% on all amounts outstanding, which is being accrued over the credit facility term and shall be due at the earlier of maturity or prepayment. If the Company repays all of the amounts borrowed under the loan on or prior to maturity, the Company will also be required to pay a prepayment fee equal to 0.75%.
The credit facility includes affirmative and restrictive covenants and events of default, including the following events of default: payment defaults, breaches of covenants, judgment defaults, cross defaults to certain other contracts, certain events with respect to governmental approvals if such events could cause a material adverse change, a material impairment in the perfection or priority of the lender’s security interest or in the value of the collateral, a material adverse change in the business, operations or condition of the Company or any of its subsidiaries and a material impairment of the prospect of repayment of the loans. Upon the occurrence of an event of default, a default increase in the interest rate of an additional 5.00% could be applied to the outstanding loan balance and the lender could declare all outstanding obligations immediately due and payable and take such other actions as set forth in the credit facility.
The Company’s obligations under the credit facility are secured by a first priority security interest in substantially all of its assets, other than its intellectual property. There are no financial covenants contained in the credit facility and the Company is in compliance with the affirmative and restrictive covenants as of December 31, 2014 and 2013.
As of December 31, 2014, the Company has borrowed and had outstanding $20,000 of debt under the credit facility.
In association with the original credit facility and in conjunction with the close of the first tranche in October 2012, the Company issued the lender warrants to purchase 151 shares of Series E Preferred Stock at an exercise price of $1.9594 per share. In connection with the IPO in February 2015, the warrants became exercisable for up to 38 shares of the Company’s common stock with an exercise price of $7.84 per share. Each warrant may be exercised on a cashless basis in whole or in part. Using the Black-Scholes valuation model,
83
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
management estimated the fair value of these warrants to be approximately $132 at issuance of the warrant. The following assumptions were used to estimate the fair value: expected volatility of 45.00%, risk-free interest rate of 1.64%, and expected term of 10 years. These warrants were considered to be costs incurred as part of the credit facility and were recorded as a debt discount which was offset against the loan, and were to be amortized over the life of the original credit facility based on the effective interest method to interest expense. In connection with the amended credit facility, the Company amortized the remaining debt discount to interest expense. No stock warrants were issued in conjunction with the amended credit facility.
Assuming a 36-month amortization period as stated in the fourth paragraph above, the Company’s principal payments are due under the amended credit facility as follows for the years ending December 31, 2014:
| 2016 |
$ | 5,708 | ||
| 2017 |
6,812 | |||
| 2018 |
7,480 | |||
|
|
|
|||
| $ | 20,000 | |||
|
|
|
At December 31, 2014 and 2013, the carrying value of debt approximated fair market value. The fair value of the Company’s debt is considered a Level 3 measurement.
NOTE G – CONVERTIBLE PREFERRED STOCK
In October 2012, the Company issued 5,104 shares of Series E Preferred Stock for net proceeds of approximately $9,990.
During 2011, 2009, 2008, 2007 and 2006, the Company issued 12,759, 15,311, 3,717, 4,986 and 2,440 shares of Series E, D, C, B and A Preferred Stock, respectively. The Series A, B, C, D and E Preferred Stock are collectively referred to as the Convertible Preferred Stock.
As of December 31, 2014 and 2013, respectively, the Convertible Preferred Stock consists of the following (presented in actual amounts):
| Number of Shares authorized |
Number of shares issued and outstanding |
Carrying value | Liquidation preference per share |
8% dividend per share |
Conversion rate |
|||||||||||||||||||
| Series A |
2,440,000 | 2,440,000 | $ | 2,963,745 | $ | 1.2500 | $ | 0.100000 | 0.2500 | |||||||||||||||
| Series B |
4,986,188 | 4,986,188 | 8,988,134 | $ | 1.8100 | $ | 0.144800 | 0.2500 | ||||||||||||||||
| Series C |
3,717,329 | 3,717,329 | 14,937,389 | $ | 4.0350 | $ | 0.322800 | 0.3374 | ||||||||||||||||
| Series D |
15,310,943 | 15,310,943 | 29,822,047 | $ | 1.9594 | $ | 0.156752 | 0.2500 | ||||||||||||||||
| Series E |
18,112,611 | 17,862,611 | 34,842,241 | $ | 1.9594 | $ | 0.156752 | 0.2500 | ||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Total |
44,567,071 | 44,317,071 | $ | 91,553,556 | ||||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
The Series C Preferred Stock included an antidilution adjustment that resulted in the applicable conversion rate of the Series C Preferred Stock increasing from 0.2500 to 0.3374 as a result of the sale of the Series D Preferred Stock. The resulting incremental common stock to the Series C Preferred Stockholders did not result in any intrinsic value due, because the estimated fair market value of common stock when the Series C Preferred Stock was issued was greater than the estimated fair market value of common stock when the conversion rate
84
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
increased to 1.3498. If there had been intrinsic value, the Company would have recorded a deemed dividend, resulting in a reduction of retained earnings and increase to Series C Preferred Stock, equal to the fair value of the incremental shares of common stock.
The Company recorded its Convertible Preferred Stock at fair value on the dates of issuance, net of issuance costs. A redemption event would have occurred only upon the liquidation or winding up of the Company, a greater than 50% change in control, or sale of substantially all of the assets of the Company. As the redemption event was outside the control of the Company, all shares of Convertible Preferred Stock have been presented outside of permanent equity. Further, the Company also elected not to adjust the carrying values of the Convertible Preferred Stock to the redemption value of such shares, since it was uncertain whether or when a redemption event would occur. Subsequent adjustments to increase the carrying value to the redemption values will be made when it becomes probable that such redemption will occur. As of December 31, 2014 and 2013, respectively, it was not probable that such redemption would occur. In connection with the IPO, all series of the Company’s Convertible Preferred Stock converted to common stock. See Note N—Subsequent Events.
The terms of the Company’s Convertible Preferred Stock were as follows:
(a) Conversion and Redemption Rights
The Convertible Preferred Stock was convertible into common stock at the option of the holder immediately upon issuance. The conversion price per share was equal to a rate determined by dividing the initial purchase price by the then applicable conversion price. The initial purchase price was equal to $1.25, $1.81, $4.035, $1.9594 and $1.9594 for Series A, Series B, Series C, Series D and Series E Preferred Stock, respectively. The applicable conversion price was equal to $5.00, $7.24, $11.9576, $7.8376 and $7.8376 for Series A, Series B, Series C, Series D and Series E, respectively. The Convertible Preferred Stock would automatically convert into common stock upon a qualified public offering or by consent of the holders of at least a majority in interest of the then-outstanding Convertible Preferred Stock voting together, as a single class on an as-if-converted-to-common stock basis.
The Convertible Preferred Stock was not redeemable.
(b) Dividends
The holders of Series A, B, C, D and E Convertible Preferred Stock were entitled to receive dividends (as determined on a per annum basis and on an as-converted basis), from any assets legally available, prior and in preference to any declaration or payment of any dividend to the common stockholders. Such dividends were payable when and if declared by the Board of Directors and are not cumulative. No dividends were declared through December 31, 2014.
(c) Liquidation Preferences
In the event of any liquidation, dissolution, change in control or winding-up of the Company, the holders of Series E Preferred Stock would have been entitled to be paid out of the assets of the Company available for distribution to its stockholders on a pro rata basis, prior and in preference to the holders of Series A, B, C and D Preferred Stock and common stock, for the sum of $1.9594 per share of Series E held, plus unpaid dividends thereon, if declared and unpaid.
After the payment in full of the aggregate Series E Liquidation Preference, the holders of the Series D Preferred Stock would have been entitled to distribution on a pro rata basis, prior and in preference to the
85
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
holders of Series A, B and C Preferred Stock and common stock, for the sum of $1.9594 per share of Series D held, plus unpaid dividends thereon, if declared and unpaid.
Any remaining assets of the Company would then have been distributed on a pari passu basis among the holders of Series A, B and C on a pro rata basis, prior and in preference to the holders of common stock, for the sum of $1.25 per share of Series A held, $1.81 per share of Series B held and $4.035 per share of Series C held, plus unpaid dividends thereon, if declared and unpaid.
Any remaining assets of the Company would then have been distributed equally, on a per share basis, among the holders of the Series D Preferred Stock, Series E Preferred Stock and common stockholders on an as-if converted basis. The maximum aggregate proceeds that the holders of Series D Preferred Stock and Series E Preferred Stock may receive upon liquidation is equal to $3.223 per share of Series D and Series E held.
A liquidation of the Company includes a liquidation, dissolution, or winding up of the Company and, unless waived by the holders of a majority in interest of the Convertible Preferred Stock, the sale, transfer, exclusive licensing or other disposition of all or substantially all of the Company’s assets including intellectual property, or the merger or consolidation of the Company with another entity gaining greater than 50% ownership of the Company.
(d) Voting Agreement
For so long as at least 20% of the shares of Convertible Preferred Stock originally issued remained outstanding, the holders of Convertible Preferred Stock, voting separately as a class, were entitled to elect three members of the Board, the holders of common stock, voting separately as a class, were entitled to elect one member of the Board, and the holders of Preferred Stock and common stock, voting together, as a single class on an as-if-converted-to-common-stock basis, were entitled to elect one member of the Board.
For so long as at least any shares of Convertible Preferred Stock originally issued remained outstanding, the Company could not, without the affirmative vote of at least a majority in interest of the then outstanding Convertible Preferred Stock, (i) increase or decrease the number of authorized shares of Convertible Preferred Stock, (ii) create any new class or series of capital stock senior to the Convertible Preferred Stock, (iii) redeem any shares of Convertible Preferred Stock or common stock, (iv) declare, authorize or pay a dividend or distribution, (v) dispose of all or substantially all of the assets of the corporation, (vi) change the authorized number of directors, (vii) increase shares reserved for issuance under any stock-based compensation plan, (viii) fundamentally change the principal business of the corporation, (ix) acquire or invest in another corporation or entity, (x) alter or change the voting power, rights, preferences, privileges or restrictions of Convertible Preferred Stock, (xi) amend, repeal or waive any provision of the Company’s certificate of incorporation or bylaws in a manner adverse to the holders of Convertible Preferred Stock, (xii) create any debt security or incur any debt or lease obligation, other than equipment leases, bank lines of credit and venture debt transactions or (xiii) create or hold capital stock in any subsidiary that is not a wholly-owned subsidiary.
NOTE H – STOCKHOLDERS’ DEFICIT
Common Stock
In May 2014, the Company amended its Certificate of Incorporation to increase the authorized number of shares of common stock available for issuance from 57,000 to 57,565 shares of $0.001 par value common stock. In December 2014, the Company further amended its Certificate of Incorporation to increase the authorized number of common stock shares available for issuance to 62,276 shares.
86
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
The common stockholders are generally entitled to one vote for each share held on all matters submitted to a vote of the stockholders and do not have any cumulative voting rights. The Company’s common stockholders are entitled to receive proportionally any dividends declared by the Company’s Board of Directors, subject to any preferential dividend rights of outstanding preferred stock.
In the event of the Company’s liquidation or dissolution, the common stockholders are entitled to share ratably in all assets remaining after payment of all debts and other liabilities, subject to any preferential rights of any outstanding preferred stock. The common stockholders have no preemptive, subscription, redemption or conversion rights.
2006 Equity Incentive Plan
Under the 2006 Plan approved by the Company’s Board of Directors in August 2006, 3,541 shares of common stock have been reserved for the issuance of incentive stock options (“ISOs”), non-statutory stock options (“NSOs”), stock bonuses and rights to acquire restricted stock to employees, officers, directors and consultants of the Company as of December 31, 2014. ISOs and NSOs have been granted with exercise prices at no less than 100% of the fair value of the common stock on the date of grant. Options granted to a 10% stockholder shall be at no less than 110% of the fair value and ISO stock option grants to such 10% stockholders expire five years from the date of grant. ISOs granted under the 2006 Plan generally vest ratably over a 4 year vesting term and expire 10 years from the grant date, unless subject to provisions regarding 10% stockholders. NSOs vest per the specific agreement and expire 10 years from the date of grant. New shares are issued upon exercise of options under the 2006 Plan. At December 31, 2014, there were 23 shares available for issuance under the 2006 Plan. In connection with the IPO, the 2006 Plan terminated, and the Company adopted a new equity incentive plan, the 2015 Plan. See Note N—Subsequent Events.
A summary of the Company’s stock option activity and related information is as follows:
| Years Ended December 31, | ||||||||||||||||||||||||
| 2014 | 2013 | 2012 | ||||||||||||||||||||||
| Options | Weighted average exercise price |
Options | Weighted average exercise price |
Options | Weighted average exercise price |
|||||||||||||||||||
| Outstanding, beginning of period |
1,216 | $ | 1.18 | 1,505 | $ | 1.37 | 1,301 | $ | 1.53 | |||||||||||||||
| Granted |
1,396 | 10.55 | 437 | 1.33 | 486 | 0.76 | ||||||||||||||||||
| Exercised |
(466 | ) | 1.24 | (583 | ) | 1.23 | (231 | ) | 1.10 | |||||||||||||||
| Cancelled |
— | — | (112 | ) | 4.08 | — | — | |||||||||||||||||
| Forfeited |
(48 | ) | 2.43 | (31 | ) | 0.84 | (51 | ) | 1.05 | |||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Outstanding, end of period |
2,098 | 7.38 | 1,216 | 1.18 | 1,505 | 1.37 | ||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Exercisable |
485 | 1.35 | 683 | 1.24 | 879 | 1.64 | ||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
The weighted average remaining contractual life of options outstanding was 8.8 and 7.4 years as of December 31, 2014 and 2013, respectively. The weighted average remaining contractual life of options exercisable was 6.1and 6.5 years as of December 31, 2014 and 2013, respectively.
As of December 31, 2014 and 2013, the aggregate pre-tax intrinsic value of options outstanding and exercisable was approximately $4,860 and $599, respectively. As of December 31, 2014 and 2013, the aggregate
87
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
pre-tax intrinsic value of options outstanding was approximately $8,385 and $1,140, respectively. The aggregate pre-tax intrinsic value of options exercised was $4,197, $173 and $6 during the years ended December 31, 2014, 2013 and 2012, respectively. The aggregate pre-tax intrinsic value was calculated as the difference between the exercise prices of the underlying options and the estimated fair value of the common stock on the date of exercise or December 31, 2014, 2013 and 2012, as applicable. The total cash received upon the exercise of options was $576, $717 and $254 during the years ended December 31, 2014, 2013 and 2012, respectively.
Early Exercise of Stock Options
Stock options granted under the 2006 Plan provide option holders the right to elect to exercise unvested options in exchange for restricted common stock. During the year ended December 31, 2012, a director exercised options for 64 unvested shares. As of December 31, 2013, 41 shares remained unvested and subject to a repurchase right held by the Company at the original issuance price in the event the optionees’ service was voluntarily or involuntarily terminated. On October 31, 2014, a director resigned from the Board of Directors of the Company and the Company repurchased the remaining 28 unvested shares at a total purchase price of $21 in November 2014.
Reserved Shares
The Company is required to reserve and keep available out of its authorized but unissued shares of common stock a number of shares sufficient to effect the conversion of all outstanding shares of convertible preferred stock (and preferred stock warrants), plus options granted and available for grant under the incentive stock plan.
The number of such shares of common stock reserved for the conversion, exercise and issuance of the following options and shares outstanding as of December 31, 2014 and 2013, were as follows:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Convertible preferred stock: |
||||||||
| Series A outstanding |
610 | 610 | ||||||
| Series B outstanding |
1,246 | 1,246 | ||||||
| Series C outstanding |
929 | 929 | ||||||
| Series C anti-dilution shares |
325 | 325 | ||||||
| Series D outstanding |
3,828 | 3,828 | ||||||
| Series E outstanding |
4,466 | 4,466 | ||||||
| Warrants issued in connection with the credit facility |
38 | 38 | ||||||
| 2006 Stock Incentive Plan, options outstanding |
2,098 | 1,216 | ||||||
| Options available for future grant |
23 | 51 | ||||||
|
|
|
|
|
|||||
| 13,563 | 12,709 | |||||||
|
|
|
|
|
|||||
88
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
NOTE I – STOCK-BASED COMPENSATION EXPENSE
Stock-based compensation expense was reflected in the statement of operations and comprehensive loss for the years ended December 31, 2014, 2013 and 2012, as follows:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Cost of goods sold |
$ | 11 | $ | 15 | $ | 9 | ||||||
| Selling and marketing |
111 | 78 | 67 | |||||||||
| Research and development |
46 | 68 | 57 | |||||||||
| General and administrative |
80 | 193 | 171 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 248 | $ | 354 | $ | 304 | |||||||
|
|
|
|
|
|
|
|||||||
The amount of unearned stock-based compensation currently estimated to be expensed through the year 2018 related to unvested employee stock-based payment awards as of December 31, 2014, is approximately $6,012. The weighted average period over which the unearned stock-based compensation is expected to be recognized as of December 31, 2014, is approximately 3.2 years. If there are any modifications or cancellations of the underlying unvested securities, the Company may be required to accelerate, increase or cancel any remaining unearned stock-based compensation expense. Future stock-based compensation expense and unearned stock-based compensation will increase to the extent that the Company grants additional share-based payments.
The Company estimates the fair value of stock-based compensation on the date of grant using the Black-Scholes option-pricing model. The Black-Scholes model determines the fair value of stock-based payment awards based on the fair market value of the Company’s common stock on the date of grant and is affected by assumptions regarding a number of highly complex and subjective variables. These variables include, but are not limited to, the fair market value of the Company’s common stock, volatility over the expected term of the awards and actual and projected employee stock option exercise behaviors. The Company has opted to use the “simplified method” for estimating the expected term of options, whereby the expected term equals the arithmetic average of the vesting term and the original contractual term of the option. Due to the Company’s limited operating history and a lack of company-specific historical and implied volatility data, the Company has based its estimate of expected volatility on the historical volatility of a group of similar companies that are publicly traded. The historical volatility data was computed using the daily closing prices for the selected companies’ shares during the equivalent period of the calculated expected term of the share-based payments. The Company will continue to analyze the historical stock price volatility and expected term assumptions as more historical data for the Company’s common stock becomes available. The risk-free rate assumption is based on the U.S. Treasury instruments with maturities similar to the expected term of the Company’s stock options. The expected dividend assumption is based on the Company’s history of not paying dividends and its expectation that it will not declare dividends for the foreseeable future.
As stock-based compensation expense recognized in the financial statements is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from estimates. Forfeitures are estimated based on estimated future employee turnover and historical experience.
89
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
The fair value of the options granted to employees or directors during the years ended December 31, 2014, 2013 and 2012, was estimated as of the grant date using the Black-Scholes model assuming the weighted average assumptions listed in the following table:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Expected life in years |
5.76 | 5.72 | 5.97 | |||||||||
| Risk-free interest rate |
1.68% | 1.08% | 1.05% | |||||||||
| Expected dividend yield |
0% | 0% | 0% | |||||||||
| Expected volatility |
54% | 68% | 69% | |||||||||
| Weighted average fair value |
$ | 5.32 | $ | 3.04 | $ | 1.92 | ||||||
Performance-Based Options
Options issued during the year ended December 31, 2014, included grants to certain employees totaling 23 shares that contained vesting conditions contingent on the achievement of certain milestones. Assuming continued service by the employee, the option would vest immediately upon achievement of the performance criteria. At December 31, 2014, certain milestones were achieved and therefore 5 shares vested immediately. The Company recorded stock-based compensation expense totaling $11 related to these options. The remaining 18 shares were cancelled.
NOTE J – INCOME TAXES
During the years ended December 31, 2014 and 2013, the Company did not record a provision or benefit for current or deferred income taxes in the statement of operations and comprehensive loss due to its cumulative net losses.
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible.
Based on the level of historical taxable income and projections of future taxable income over the periods in which the deferred tax assets are deductible, management believes that it is more likely than not that the Company will not realize the benefits of these deductible differences. Accordingly, the Company has recorded a full valuation allowance against its net deferred tax assets as of December 31, 2014 and 2013. The Company’s valuation allowance increased by $2,429 in 2014 due to the increase in total deferred tax assets.
90
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
The tax effects of temporary differences that give rise to significant portions of the net deferred tax asset (liability) were as follows at December 31:
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Deferred tax asset: |
||||||||
| Accounts receivable |
$ | 103 | $ | 83 | ||||
| Inventories |
32 | 50 | ||||||
| Accrued expenses |
218 | 108 | ||||||
| Depreciation and amortization |
195 | 273 | ||||||
| Research and devleopment credits generated |
837 | 735 | ||||||
| Net operating loss carryforward |
35,231 | 32,969 | ||||||
| Organizational costs |
5 | 5 | ||||||
| Stock-based compensation expense (non-qualified options) |
211 | 220 | ||||||
| Other |
20 | 26 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
36,852 | 34,469 | ||||||
| Deferred tax liabilities: |
||||||||
| Prepaid expense |
(207 | ) | (253 | ) | ||||
|
|
|
|
|
|||||
| Total deferred tax liabilities |
(207 | ) | (253 | ) | ||||
|
|
|
|
|
|||||
| Total deferred tax assets, net |
36,645 | 34,216 | ||||||
| Less valuation allowance |
(36,645 | ) | (34,216 | ) | ||||
|
|
|
|
|
|||||
| Total deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
As of December 31, 2014 and 2013, the Company had federal tax net operating loss carryforwards of approximately $98,700 and $93,000, respectively, which will be available to offset earnings during the carryforward period. If not used, these carryforwards begin to expire in 2026. In addition, changes in ownership could put limitations on the availability of the net operating loss carryforwards.
NOTE K – EARNINGS PER SHARE
Basic net loss per share is computed by dividing the net loss by the weighted average number of shares of common stock outstanding during the period. Diluted net loss per share is computed by dividing the net loss by the weighted average number of shares of common stock and dilutive potential shares of common stock outstanding during the period. Because the Company has reported a net loss for all periods presented, diluted net loss per share is the same as basic net loss per share for those periods as all potentially dilutive shares consisting of convertible preferred stock, stock options and warrants were antidilutive in those periods.
The following table sets forth the computation of the Company’s net loss per share:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Net loss |
$ | (6,929 | ) | $ | (13,396 | ) | $ | (18,996 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average common stock outstanding |
1,499 | 1,133 | 696 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss per share, basic and diluted |
$ | (4.62 | ) | $ | (11.82 | ) | $ | (27.29 | ) | |||
|
|
|
|
|
|
|
|||||||
91
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
The following potentially dilutive securities outstanding have been excluded from the computations of diluted weighted average shares outstanding because such securities have an antidilutive impact due to losses reported:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Convertible preferred stock outstanding |
11,404 | 11,404 | 11,404 | |||||||||
| Convertible preferred stock warrants |
38 | 38 | 38 | |||||||||
| Common stock options |
2,098 | 1,216 | 1,505 | |||||||||
|
|
|
|
|
|
|
|||||||
| 13,540 | 12,658 | 12,947 | ||||||||||
|
|
|
|
|
|
|
|||||||
NOTE L – COMMITMENTS AND CONTINGENCIES
License Agreement with Acclarent, Inc.
In February 2011, the Company entered into a license agreement with Acclarent, Inc. (“Acclarent”) in connection with the settlement of a then-ongoing litigation in which Acclarent alleged that the Company’s FinESS and XprESS products infringed certain claims of five of their patents, which allegations the Company denied. In October 2012, the Company agreed with Acclarent to amend certain terms of the license agreement (as amended, the “Acclarent License”). Pursuant to the Acclarent License, Acclarent granted the Company a worldwide, non-exclusive, royalty-bearing license under certain patents held by Acclarent to manufacture, use and commercialize the Company’s currently marketed XprESS, PathAssist and FinESS products and certain future versions of those products (collectively, the “Covered Products”) in the field of the expansion of paranasal sinuses and sinus pathways.
Under the Acclarent License, the Company made an initial up-front, lump-sum payment to Acclarent as a retroactive royalty payment. The Company is also required to pay Acclarent an ongoing royalty in the low double-digit percentages, on a country-by-country basis, on net sales of Covered Products. Royalty expense is included in the costs of goods sold and is recognized in the period when revenue is recognized on Covered Products. The Acclarent License will expire upon the expiration or abandonment of the last to expire of the patents licensed to us under the agreement. Acclarent has the right to terminate the agreement upon 60 days’ notice in the event of the Company’s uncured material breach. Amounts due for royalties payable are recorded within accrued expenses on the balance sheets.
Operating Leases
As of December 31, 2014, the Company has two leased facilities under operating lease agreements. The Company entered into a 41-month lease in February 2012, effective April 1, 2012, for a facility with larger production and office space. The Company entered into a 50-month lease on June 30, 2014, effective July 1, 2014 for additional distribution space in a second facility. Rental payments on operating leases are charged to expense on a straight-line basis over the period of the lease. The lease agreement requires the Company to pay executory costs such as real estate taxes, insurance and repairs, and includes renewal and escalation clauses.
92
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
Approximate minimum future obligations which include monthly contractual lease payments under operating leases with initial terms in excess of one year are as follows:
| Period Ending December 31, |
||||
| 2015 |
$ | 151 | ||
| 2016 |
41 | |||
| 2017 |
36 | |||
| 2018 |
24 | |||
|
|
|
|||
| $ | 252 | |||
|
|
|
|||
Total lease expense was approximately $305, $234 and $233 for the years ended December 31, 2014, 2013 and 2012, respectively.
NOTE M – EMPLOYEE BENEFIT PLAN
The Company’s employees are eligible to participate in a defined contribution benefit plan. Employees may contribute a percentage of their wages, subject to limits established by the Internal Revenue Code. The Company may elect to make discretionary contributions to the plan. There were no discretionary contributions during the years ended December 31, 2014, 2013 and 2012.
NOTE N – SUBSEQUENT EVENTS
Initial Public Offering
In February 2015, the Company completed its IPO by issuing 5,294 shares of common stock at an offering price of $17.00 per share, for net proceeds to the Company of approximately $81,200, after deducting underwriting discounts and commissions of approximately $6,300 and offering expenses payable by the Company of approximately $2,500. In connection with the IPO, the Company’s outstanding shares of convertible preferred stock were automatically converted into an aggregate of 11,404 shares of common stock and warrants exercisable for convertible preferred stock were automatically converted into warrants to purchase up to an aggregate of 38 shares of common stock, resulting in the reclassification of the related redeemable convertible preferred stock warrant liability of $291 to additional paid-in-capital.
1-for-4 Reverse Stock Split
In connection with the IPO, the Company’s Board of Directors and stockholders approved a reverse stock split of the Company’s common stock on a 1-for-4 basis. The reverse stock split became effective on January 12, 2015. The par value of the common stock was not adjusted as a result of the reverse stock split. Adjustments corresponding to the reverse stock split were made to the ratio at which the Convertible Preferred Stock converted into common stock immediately prior to the closing of the IPO. Accordingly, all share and per share amounts for all periods presented in these financial statements and notes thereto have been adjusted retroactively, where applicable, to reflect the reverse stock split and adjustment of the preferred share conversion ratios.
2015 Incentive Award Plan
In December 2014, the Company’s Board of Directors adopted, and in January 2015 the Company’s stockholders approved, the 2015 Plan. The 2015 Plan became effective on the effective date of the IPO, at which
93
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
time the Company ceased making awards under the 2006 Plan. Under the 2015 Plan, the Company may grant stock options, stock appreciation rights, restricted stock, restricted stock units and certain other awards to individuals who are employees, officers, directors or consultants of the Company. A total of 1,346 shares of common stock were initially reserved for issuance under the 2015 Plan. In addition, the number of shares available for issuance under the 2015 Plan will be annually increased by an amount equal to the lesser of (A) 875 shares, (B) 4% of the outstanding shares of the Company’s common stock as of the last day of our immediately preceding fiscal year or, an amount determined by the Company’s Board of Directors.
2015 Employee Stock Purchase Plan
In December 2014, the Company’s board of directors adopted, and in January 2015 the Company’s stockholders approved, the 2015 Employee Stock Purchase Plan (“2015 ESPP”). The 2015 ESPP became effective on the effective date of the IPO. A total of 200 shares were initially reserved for issuance under the 2015 ESPP. In addition, the number of shares available for issuance under the ESPP will be annually increased on the first day of each fiscal year during the term of the ESPP, beginning with the 2016 fiscal year, by an amount equal to the lesser of (A) 1% of outstanding shares of the Company’s common stock or (B) an amount determined by the Company’s Board of Directors.
Lease Extension
On February 25, 2015, the Company exercised its right to extend the lease term of its manufacturing and corporate facility for an additional three years through August 2018. The lease extension includes renewal and escalation clauses. Minimum future contractual lease payments are as follows:
| Period Ending December 31, |
||||
| 2015 |
$ | 54 | ||
| 2016 |
163 | |||
| 2017 |
166 | |||
| 2018 |
112 | |||
|
|
|
|||
| $ | 495 | |||
|
|
|
|||
94
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
NOTE O – QUARTERLY RESULTS OF OPERATIONS (UNAUDITED)
The following is a summary of the Company’s unaudited quarterly results of operations for the years December 31, 2014 and 2013 (in thousands except per share amounts):
| Three Months Ended | ||||||||||||||||
| March 31, 2014 |
June 30, 2014 |
September 30, 2014 |
December 31, 2014 |
|||||||||||||
| Revenue |
$ | 10,175 | $ | 12,498 | $ | 11,691 | $ | 14,456 | ||||||||
| Cost of goods sold |
2,298 | 2,716 | 2,501 | 3,239 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Gross profit |
7,877 | 9,782 | 9,190 | 11,217 | ||||||||||||
| Operating expenses: |
||||||||||||||||
| Selling and marketing |
7,722 | 8,163 | 8,391 | 8,487 | ||||||||||||
| Research and development |
1,116 | 1,047 | 972 | 1,172 | ||||||||||||
| General and administrative |
1,158 | 1,222 | 1,387 | 2,330 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expense |
9,996 | 10,432 | 10,750 | 11,989 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(2,119 | ) | (650 | ) | (1,560 | ) | (772 | ) | ||||||||
| Other income (expense): |
||||||||||||||||
| Interest income |
4 | — | — | 71 | ||||||||||||
| Interest expense |
(421 | ) | (523 | ) | (473 | ) | (487 | ) | ||||||||
| Other non-operating income |
1 | — | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total other expense, net |
(416 | ) | (523 | ) | (473 | ) | (416 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss and comprehensive loss |
$ | (2,535 | ) | $ | (1,173 | ) | $ | (2,033 | ) | $ | (1,188 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per share, basic and diluted |
$ | (1.80 | ) | $ | (0.81 | ) | $ | (1.36 | ) | $ | (0.73 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted average common shares used to compute net loss per share, basic and diluted |
1,412 | 1,455 | 1,500 | 1,628 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
95
Table of Contents
ENTELLUS MEDICAL, INC.
NOTES TO FINANCIAL STATEMENTS
(in thousands, except per share data)
| Three Months Ended | ||||||||||||||||
| March 31, 2013 |
June 30, 2013 |
September 30, 2013 |
December 31, 2013 |
|||||||||||||
| Revenue |
$ | 6,244 | $ | 7,712 | $ | 7,889 | $ | 10,700 | ||||||||
| Cost of goods sold |
1,536 | 1,844 | 1,903 | 2,525 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Gross profit |
4,708 | 5,868 | 5,986 | 8,175 | ||||||||||||
| Operating expenses: |
||||||||||||||||
| Selling and marketing |
6,618 | 6,515 | 6,901 | 7,597 | ||||||||||||
| Research and development |
1,544 | 1,250 | 1,226 | 1,123 | ||||||||||||
| General and administrative |
1,049 | 1,070 | 970 | 1,222 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
9,211 | 8,835 | 9,097 | 9,942 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(4,503 | ) | (2,967 | ) | (3,111 | ) | (1,767 | ) | ||||||||
| Other income (expense): |
||||||||||||||||
| Interest income |
3 | 1 | 2 | — | ||||||||||||
| Interest expense |
(184 | ) | (236 | ) | (185 | ) | (477 | ) | ||||||||
| Other non-operating income |
— | — | — | 28 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total other expense, net |
(181 | ) | (235 | ) | (183 | ) | (449 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (4,684 | ) | $ | (3,202 | ) | $ | (3,294 | ) | $ | (2,216 | ) | ||||
| Other comprehensive loss |
||||||||||||||||
| Unrealized loss on short-term investments |
(1 | ) | — | — | — | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Comprehensive loss |
$ | (4,685 | ) | $ | (3,202 | ) | $ | (3,294 | ) | $ | (2,216 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per share, basic and diluted |
$ | (5.22 | ) | $ | (3.04 | ) | $ | (2.59 | ) | $ | (1.71 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted average common shares used to compute net loss per share, basic and diluted |
898 | 1,054 | 1,274 | 1,298 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
96
Table of Contents
SCHEDULE II
VALUATION AND QUALIFYING ACCOUNTS
| Beginning Balance |
Additions | Deductions | Ending Balance |
|||||||||||||
| Allowance for doubtful accounts receivable |
||||||||||||||||
| Year ended December 31, 2014 |
$ | 135 | $ | 49 | $ | 34 | $ | 150 | ||||||||
| Year ended December 31, 2013 |
125 | 69 | 59 | 135 | ||||||||||||
| Year ended December 31, 2012 |
78 | 47 | — | 125 | ||||||||||||
| Reserve for sales returns and allowances |
||||||||||||||||
| Year ended December 31, 2014 |
$ | 100 | $ | 120 | $ | 80 | $ | 140 | ||||||||
| Year ended December 31, 2013 |
62 | 135 | 97 | 100 | ||||||||||||
| Year ended December 31, 2012 |
24 | 65 | 27 | 62 | ||||||||||||
97
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Limitations on Effectiveness of Controls and Procedures
In designing and evaluating our disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated, as of the end of the period covered by this Annual Report on Form 10-K, the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act). Based on that evaluation, our chief executive officer and chief financial officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31, 2014.
Changes in Internal Control Over Financial Reporting
With the participation of our chief executive officer and our chief financial officer, management has evaluated whether any changes in our internal control over financial reporting that occurred during our last fiscal quarter have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. Based on the evaluation we conducted, management has implemented changes as described below.
Management previously identified and disclosed a material weakness in our internal control over financial reporting that existed as of September 30, 2014. The material weakness related to our having recorded revenue relating to sales orders that had been falsified by one of our sales representatives. Specifically, our controls did not require direct communication from our customers to indicate persuasive evidence of an arrangement for all sales transactions at the point of sale. While we were able to detect the falsified transactions, we determined that our internal controls were deficient because we did not detect these transactions within our financial statements on a timely basis. Management also identified a significant deficiency related to our field inventory reconciliation process and a significant deficiency related to our policies and procedures for evaluating the appropriate accounting treatment for our convertible preferred stock warrant liability. We implemented processes and controls designed to remediate this material weakness and the significant deficiency related to field inventory as of December 31, 2014, including (1) requiring customer printed name, signature, date, and contact information on all sales order forms for any transaction involving the sale of units from a sale representative’s field inventory, (2) requiring follow-up order confirmation emails or phone calls with customers who purchase field inventory from a sales representative, (3) requiring that all orders sent to our customer service department by one of our sales representatives require the use of a completed customer product ordering form and customer signature, and are dated prior to shipment of product to the customer; if any portion of this is not completed properly, customer service will contact the customer directly to verify an order prior to shipment and (4) periodic physical confirmation of field inventory in the possession of sales representatives. We also implemented additional policies and procedures designed to remediate the significant deficiency relating to the accounting treatment for our convertible preferred stock warrant liability as of December 31, 2014.
Management believes the measures that have been implemented to remediate the material weakness and significant deficiencies had a material impact on our internal control over financial reporting during the quarter ended December 31, 2014 and were sufficient to remediate the previously reported material weakness and significant deficiencies as of December 31, 2014.
98
Table of Contents
Management’s Annual Report on Internal Control Over Financial Reporting
This annual report does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of our independent registered public accounting firm due to a transition period established by rules of the SEC for newly public companies.
| Item 9B. | Other Information |
None.
99
Table of Contents
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
Executive Officers, Key Employees and Directors
The following table sets forth information regarding our executive officers, key employees and directors, as of December 31, 2014:
| Name |
Age | Position(s) | ||||
| Executive Officers and Key Employees |
||||||
| Brian E. Farley |
57 | Chief Executive Officer and Chairman of the Board | ||||
| Robert S. White |
53 | President and Chief Operating Officer | ||||
| Thomas E. Griffin |
51 | Chief Financial Officer | ||||
| Margaret A. Boiano |
60 | Vice President, Healthcare Policy and Reimbursement | ||||
| Kevin L. Mensink |
42 | Vice President, Marketing | ||||
| Stephen R. Paidosh |
52 | Vice President, Operations | ||||
| Karen E. Peterson |
56 | Vice President, Clinical, Regulatory and Quality | ||||
| Tim B. Petrick |
46 | Vice President, Research and Development | ||||
| James D. Surek |
47 | Vice President, Sales | ||||
| Non-Employee Directors |
||||||
| Joshua Baltzell(1)(3) |
45 | Director | ||||
| Shawn T McCormick(1)(3) |
50 | Director | ||||
| David B. Milne(2) |
52 | Director | ||||
| Guido Neels(1)(2) |
66 | Director | ||||
| (1) | Member of the audit committee. |
| (2) | Member of the compensation committee. |
| (3) | Member of the nominating and corporate governance committee. |
Executive Officers and Key Employees
Brian E. Farley has served as our Chief Executive Officer since March 2010 and as a member of our board of directors since November 2008, including as Chairman of the Board since November 2014. He also served as our President from March 2010 to November 2014. Prior to joining us, Mr. Farley was employed at VNUS Medical Technologies, Inc. (NASDAQ: VNUS) starting in 1995 and served as President and Chief Executive Officer of VNUS from January 1996 to June 2009 when VNUS was sold to Covidien Ltd. Prior to joining VNUS, Mr. Farley was employed from 1981 to 1995 at Guidant Corporation, in the Medical Device Division of Eli Lilly and Company, and in Lilly Research Labs in a variety of research and development, clinical research and business development leadership positions. Since November 2009, Mr. Farley has served as a director of Neuronetics, Inc., a private medical device company, including as Chairman of the Board from June 2011 to July 2014 and again since December 2014. Mr. Farley has also served on the board of directors of DFINE, Inc., a private medical device company, as well as its compensation committee, since March 2012. Mr. Farley holds both a B.S. in Engineering with an emphasis in Biomedical Engineering and an M.S. in Electrical Engineering from Purdue University.
We believe Mr. Farley’s experience in the industry, his role as our Chief Executive Officer and his knowledge of the Company enable him to make valuable contributions to our board of directors.
Robert S. White has served as our President and Chief Operating Officer since November 2014. Mr. White was the President and Chief Executive Officer of TYRX, Inc., or TYRX, which specialized in commercializing innovative, implantable combination drug and device products focused on infection control, from January 2010 to March 2014, when TRYX was sold to Medtronic. Prior to joining TYRX, Mr. White held several positions with Medtronic from February 2003 to November 2009, including serving as President of Medtronic Kyphon
100
Table of Contents
from April 2008 to August 2009 following the acquisition of Kyphon by Medtronic. Mr. White started his career with General Electric and joined Eli Lilly and Company in 1989. Mr. White serves on the Boards of Directors of several companies, including publicly traded companies Novadaq Technologies (September 2014 to present) and AtriCure, Inc. (March 2013 to present), as well as HyperBranch Medical Technology, Inc. (June 2010 to present). Mr. White holds a B.S. in Aerospace Engineering from the University of Missouri-Rolla and an M.B.A. from Cornell University’s Johnson Graduate School of Management.
Thomas E. Griffin has served as our Chief Financial Officer since December 2007. From July 2006 to December 2007, Mr. Griffin served as our acting Chief Financial Officer as a consultant. From September 2003 to December 2007, Mr. Griffin provided independent financial and strategic consulting to medical device companies. From December 1995 to April 2003, Mr. Griffin served as Chief Financial Officer and Secretary of Digital Gene Technologies, Inc., a privately held biotechnology company. Prior to that, Mr. Griffin was Controller for Centerpulse Spine-Tech, Inc. (now Zimmer Spine, Inc.) and CIMA Labs Inc. (now owned by Teva Pharmaceutical Industries Ltd.). Mr. Griffin holds a B.A. in Accounting from the University of Minnesota, Duluth, and an M.B.A. in Management from the University of St. Thomas. Mr. Griffin is a certified public accountant (inactive).
Margaret A. Boiano has served as our Vice President of Healthcare Policy and Reimbursement since August 2010. From November 2005 to June 2010, Ms. Boiano was the Director of Healthcare Policy and Reimbursement of VNUS. Prior to VNUS, Ms. Boiano held various reimbursement and payor relations positions with several medical companies, including Sleep Solutions Inc., CardioNet and CardGuard. Ms. Boiano holds a B.S. in Radiologic Technology, Administration and Teaching from Midwestern State University.
Kevin L. Mensink has served as our Vice President of Marketing since April 2013. From January 2007 to March 2013, Mr. Mensink served as Vice President of Marketing for GN ReSound Group, a division of GN Store Nord A/S, a Danish corporation, where he was responsible for North America marketing, product management and technical support and training. From October 2005 to January 2007, Mr. Mensink served as Director of Marketing Analytics for GN ReSound Corp. From May 2002 to October 2005, Mr. Mensink served as Marketing Manager at Thomson West in the Corporate Segment Division. Prior to that, Mr. Mensink served as a Senior Financial Analyst at Thomson West from October 1997 to May 2002. Mr. Mensink holds a B.S. in Business Administration, Accounting and Economics and an M.B.A. in Management from the University of St. Thomas.
Stephen R. Paidosh has served as our Vice President of Operations since December 2007. From July 1999 to December 2007, Mr. Paidosh served as Vice President of Operations at Myocor, Inc., a privately held medical device company. From December 1988 to July 1999, Mr. Paidosh held various leadership positions in engineering and operations at SCIMED Life Systems, Inc., or SCIMED, and Boston Scientific Corporation, or Boston Scientific, following the acquisition of SCIMED by Boston Scientific, ending as Director of International Vascular and Metallurgical Operations. Prior to that, Mr. Paidosh held positions in engineering with Pfizer Inc.’s subsidiary Schneider-Shiley (USA) Inc. and at St. Jude Medical, Inc. Mr. Paidosh holds a B.S. in Industrial Technology (Manufacturing Engineering) from the University of Wisconsin, Stout.
Karen E. Peterson has served as our Vice President of Clinical, Regulatory and Quality since July 2010 and our Compliance Officer since June 2011. Ms. Peterson also served as our Vice President of Regulatory and Quality from May 2010 to July 2010. From November 2006 to April 2010, Ms. Peterson served as Vice President of Clinical, Regulatory and Quality at Leptos Biomedical, Inc. Prior to that, Ms. Peterson held various clinical, regulatory and quality positions for Impres Medical, Inc., Carbon Medical Technologies, Inc., XRT Corporation and Guidant. Ms. Peterson holds a B.S. in Chemistry from the University of Wisconsin, Eau Claire including a Math minor and an M.S. in Biometry and Health Information Systems from the University of Minnesota.
Tim B. Petrick has served as our Vice President of Research and Development since June 2008 and served in the capacity of Manager or Director of Research and Development from February 2007 to May 2008. From October 2002 to December 2006 Mr. Petrick was a Project Engineer with Velocimed, Inc. which was acquired by
101
Table of Contents
St. Jude Medical, Inc. in 2005. From January 1998 to July 2002, Mr. Petrick held various positions in regulatory affairs and engineering with TERAMed Corporation, which was acquired by Cordis Corporation, a division of Johnson & Johnson, in 2001. Mr. Petrick holds a B.S. in Mechanical Engineering from North Dakota State University.
James D. Surek has served as our Vice President of Sales since January 2010. From February 2008 to January 2010, Mr. Surek was the Vice President of Sales in the Cochlear Implant Division at Advanced Bionics, LLC, which was sold to Sonova Holding AG in 2009. From July 2003 to February 2008, Mr. Surek was Vice President of Sales for Boston Scientific in the Neuromodulation/Pain Management Division. Mr. Surek served in various roles at Medtronic, Inc. in the Sofamor Danek Group from August 1994 to June 2003, ending as Vice President, Sales and Development. Mr. Surek holds a B.S. in Psychology from Loyola University in Chicago and a Masters of International Management from the American Graduate School of International Management, Thunderbird Campus.
Mr. White joined us on November 24, 2014 upon completion of a succession planning process initiated by our board of directors. Mr. White’s scope of responsibilities as President and Chief Operating Officer includes overseeing U.S. sales, marketing, international operations, research and development, manufacturing and customer service. Mr. Farley retains responsibility for the executives leading our finance, quality, clinical, regulatory, human resources and healthcare policy and reimbursement departments. In the future, we expect that Mr. Farley will transition to the position of Executive Chairman of our board of directors and that Mr. White will become our President and Chief Executive Officer.
Non-Employee Directors
Joshua Baltzell has served as a member of our Board of Directors since August 2006. Mr. Baltzell is a Venture Partner at Split Rock Partners, LLC and also serves as a Venture Partner at SightLine Partners LLC, or SightLine. He has been with Split Rock Partners, LLC, since May 2004 and with SightLine since July 2014. Mr. Baltzell has over 20 years of experience in the healthcare industry. Prior to his tenure in the venture capital industry, Mr. Baltzell held roles as an investment banker at Piper Jaffray Companies from 2000 to 2002, where he focused primarily on mergers and acquisitions in the medical device sector, as well as various marketing and business development positions with SCIMED and Boston Scientific. Mr. Baltzell currently serves on the boards of Colorescience, EBR Systems, Inc., DFINE, Inc. and RF Surgical Systems, Inc., all privately held companies, and of Histogenics Corporation (NASDAQ: HSGX). Mr. Baltzell holds a B.A. in Economics from St. Olaf College and an M.B.A. from the University of Minnesota’s Carlson School of Management.
We believe Mr. Baltzell’s experience in the industry and his knowledge of the Company enable him to make valuable contributions to our board of directors.
Shawn T McCormick has served as a member of our Board of Directors since November 2014. Mr. McCormick is the current Chief Financial Officer of Tornier N.V. and Tornier, Inc. and serves as Tornier’s Principal Accounting Officer. He has been with Tornier since September 2012. Mr. McCormick served as Chief Operating Officer of Lutonix Inc. from April 2011 to February 2012. He served as Chief Financial Officer and Senior Vice President of ev3, Inc. from January 19, 2009 to July 2010 and also served as its Principal Accounting Officer. He has more than 15 years of financial expertise and operational experience in the medical device industry. He served as Vice President of Corporate Development at Medtronic, Inc. from May 2008 to January 2009, where he was responsible to lead its worldwide business development activities and served in key corporate and divisional financial leadership roles within the Medtronic organization. He has been a Director of Nevro Corp. since September 2014. Mr. McCormick previously served on the board of LANX from August 2010 to November 2013, serving on the compensation committee and audit committee, before LANX was sold to Biomet in November 2013. Mr. McCormick is a Certified Public Accountant and holds a B.S. in Accounting/Finance from Arizona State University and an M.B.A. from the University of Minnesota’s Carlson School of Management.
102
Table of Contents
We believe that Mr. McCormick’s experience as a chief financial officer of a medical technology company and his background and sophistication in finance enable him to make valuable contributions to our board of directors.
David B. Milne has served as a member of our Board of Directors since August 2006. Mr. Milne is a Managing Partner at SV Life Sciences Advisers, LLC, or SVLS. He joined SVLS in 2005 and has 25 years of experience in the healthcare industry, having worked at several leading public and private medical technology companies. From 1999 until joining SVLS in 2005, he held the position of Vice President of Corporate Business Development at Boston Scientific. Mr. Milne currently sits on the board of AqueSys, Inc., Altura Medical, Inc., EBR Systems, Inc., ReShape Medical, Inc., and Spinal Kinetics, Inc., all privately held companies, and TransEnterix, Inc., a publicly traded medical device company, where he also serves on the corporate governance and nominating committee. In addition, Mr. Milne previously served on the board of Sadra Medical, Inc., MindFrame, Inc., NovaLign, Inc. and CardioMind, Inc., all privately held companies. Previously Mr. Milne worked at SCIMED, Becton, Dickinson and Company and Parker Laboratories, Inc. He holds an M.B.A. in Marketing/Finance from New York University and a B.S. in Biology from Rutgers University.
We believe that Mr. Milne’s experience in the industry and his knowledge of the Company enable him to make valuable contributions to our board of directors.
Guido Neels has served as a member of our Board of Directors since November 2009. Mr. Neels has been with Essex Woodlands Health Ventures, or Essex Woodlands, since 2006, where he is now a Venture Partner. Prior to joining Essex Woodlands, Mr. Neels served in a variety of management positions at Guidant, a developer of cardiovascular medical products. From July 2004 until retiring in November 2005, Mr. Neels served as Guidant’s Chief Operating Officer, where he was responsible for the global operations of Guidant’s four operating units: Cardiac Rhythm Management, Vascular Intervention, Cardiac Surgery and Endovascular Solutions. From December 2002 to July 2004, Mr. Neels served as Guidant’s Group Chairman, Office of the President, responsible for worldwide sales operations, corporate communications, corporate marketing, investor relations and government relations. In January 2000, Mr. Neels was named Guidant’s President, Europe, Middle East, Africa and Canada. In addition, Mr. Neels served as Guidant’s Vice President, Global Marketing, Vascular Intervention, from 1996 to 2000 and as Guidant’s General Manager, Germany and Central Europe, from 1994 to 1996. Mr. Neels has a business engineering degree from the University of Leuven in Belgium and an M.B.A. from the Stanford University Graduate School of Business. Mr. Neels served on the board of directors of Biopure Corporation, a publicly traded medical device company, from 2005 to 2009, Lemaitre Vascular, Inc., a publicly traded medical device company, from 2006 to 2008, and Nellix Inc., a privately held medical device company, from 2006 until its acquisition by Endologix in December 2010. Mr. Neels currently serves on the boards of directors of 480 Biomedical, Inc., Oraya Therapeutics, Inc., Arsenal Medical, Inc., EndGenitor Technologies, Inc., Bioventus LLC, and White Pine Medical LLC, all privately held medical device companies, and Christel House International, a not-for-profit organization. He also serves on the Board of Endologix, where he serves on the board’s compensation and nominating and governance committees.
We believe that Mr. Neels’ experience in the industry, familiarity with serving on the boards of public companies and his knowledge of the Company enable him to make valuable contributions to our board of directors.
Family Relationships
There are no family relationships among any of our directors or executive officers.
Board Committees
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee. The charter for each of these committees is available under the Corporate Governance section of our website at www.entellusmedical.com. Our board of directors may establish other
103
Table of Contents
committees to facilitate the management of our business. The composition and functions of the audit committee, the compensation committee and the nominating and corporate governance committee are described below. Members serve on these committees until their resignation or until otherwise determined by our board of directors.
Audit Committee
Our audit committee consists of Mr. McCormick, Mr. Baltzell and Mr. Neels. The chair of our audit committee is Mr. McCormick, whom our board of directors has determined is an “audit committee financial expert” as that term is defined under the SEC rules implementing Section 407 of the Sarbanes-Oxley Act, and possesses financial sophistication, as defined under the listing standards of The Nasdaq Global Market. Our board of directors has also determined that each member of our audit committee can read and understand fundamental financial statements in accordance with applicable requirements. In arriving at these determinations, the board of directors has examined each audit committee member’s scope of experience and the nature of their experience in the corporate finance sector.
The responsibilities of our audit committee include:
| • | appointing, approving the compensation of, and assessing the independence of our registered public accounting firm; |
| • | overseeing the work of our registered public accounting firm, including through the receipt and consideration of reports from such firm; |
| • | reviewing and discussing with management and the registered public accounting firm our annual and quarterly financial statements and related disclosures; |
| • | monitoring our internal control over financial reporting, disclosure controls and procedures and code of business conduct and ethics; |
| • | discussing our risk management policies; |
| • | reviewing and approving or ratifying any related person transactions; and |
| • | preparing the audit committee report required by SEC rules. |
Our board of directors has determined that Mr. McCormick and Mr. Baltzell, two of the three members of our audit committee, satisfy the independence standards for the audit committee established by applicable SEC rules and the listing standards of The Nasdaq Global Market and Rule 10A-3. Under applicable rules of The Nasdaq Global Market, we are permitted to phase in our compliance with the independent audit committee requirements on the same schedule as we are permitted to phase in our compliance with the independent audit committee requirements pursuant to Rule 10A-3, which require: (1) one independent member at the time of listing, (2) a majority of independent members within 90 days of listing and (3) all independent members within one year of listing. Within one year of our listing on The Nasdaq Global Select Market, we intend all members of our audit committee to be independent under the rules of The Nasdaq Global Market and Rule 10A-3. We do not believe that our reliance on the exemption provided in Rule 10A-3 materially adversely affects the ability of our audit committee to act independently and to satisfy the other requirements of Rule 10A-3.
Compensation Committee
Our compensation committee consists of Mr. Neels and Mr. Milne. The chair of our compensation committee is Mr. Neels.
The responsibilities of our compensation committee include:
| • | reviewing and approving, or recommending that our board of directors approve, the compensation of our chief executive officer and our other executive officers; |
104
Table of Contents
| • | reviewing and recommending to our board of directors the compensation of our directors; |
| • | selecting independent compensation consultants and advisers and assessing whether there are any conflicts of interest with any of the committees compensation advisers; and |
| • | reviewing and approving, or recommending that our board of directors approve, incentive compensation and equity plans. |
Our board of directors has determined that Mr. Neels and Mr. Milne are “outside directors” as that term is defined in Section 162(m) of the Code, or Section 162(m).
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Mr. Baltzell and Mr. McCormick. The chair of our nominating and corporate governance committee is Mr. Baltzell.
The responsibilities of our nominating and corporate governance committee include:
| • | identifying individuals qualified to become board members; |
| • | recommending to our board the persons to be nominated for election as directors and to each of the board’s committees; |
| • | reviewing and making recommendations to the board with respect to management succession planning; |
| • | developing and recommending to the board corporate governance principles; and |
| • | overseeing a periodic evaluation of the board. |
Each member of the nominating and corporate governance committee is independent within the meaning of the applicable NASDAQ listing standards, is a non-employee director and is free from any relationship that would interfere with the exercise of his independent judgment.
Role of the Board in Risk Oversight
The audit committee of the board of directors is primarily responsible for overseeing our risk management processes on behalf of the board of directors. Going forward, we expect that the audit committee will receive reports from management on at least a quarterly basis regarding our assessment of risks. In addition, the audit committee reports regularly to the board of directors, which also considers our risk profile. The audit committee and the board of directors focus on the most significant risks we face and our general risk management strategies. While the board of directors oversees our risk management, management is responsible for day-to-day risk management processes.
Code of Business Conduct and Ethics
Our board of directors has adopted a code of business conduct and ethics applicable to all officers, directors and employees, which is available under the Corporate Governance section of our website at www.entellusmedical.com. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding amendment to, or waiver from, a provision of our code of business conduct and ethics, as well as Nasdaq’s requirement to disclose waivers with respect to directors and executive officers, by posting such information on our website at the address and location specified above.
Section 16(a) Beneficial Ownership Reporting Compliance
We did not have any class of equity securities registered pursuant to Section 12 of the Exchange Act during our most recent fiscal year. As a result, none of our directors, executive officers or beneficial ownership of more than 10% of our equity securities were subject to Section 16 of the Exchange Act during such year.
105
Table of Contents
| Item 11. | Executive Compensation |
Executive Compensation
This section discusses the material components of the executive compensation program for our executive officers who are named in the “Summary Compensation Table” below. Our “named executive officers” and their positions for the year ended December 31, 2014 were as follows:
| • | Brian E. Farley, Chief Executive Officer; |
| • | Robert S. White, President and Chief Operating Officer; and |
| • | James D. Surek, Vice President, Sales. |
Summary Compensation Table
The following table sets forth certain information with respect to the compensation paid to our named executive officers during the years ended December 31, 2013 and December 31, 2014.
| Name and Principal Position |
Salary ($) | Bonus ($) | Options Awards ($)(1)(2) |
Non-Equity Incentive Plan Compensation ($)(2) |
All Other Compensation ($) |
Total ($) | ||||||||||||||||||||||
| Brian E. Farley |
2014 | 458,902 | 1,002,528 | 348,704 | (3) | 66 | (4) | 1,810,200 | ||||||||||||||||||||
| Chief Executive Officer |
2013 | 445,536 | 70,197 | 143,936 | 66 | 659,735 | ||||||||||||||||||||||
| Robert S. White |
2014 | 43,007 | 50,000 | (5) | 4,277,074 | 17,948 | (3) | 6 | (4) | 4,388,105 | ||||||||||||||||||
| President and Chief Operating Officer |
2013 | |||||||||||||||||||||||||||
| James D. Surek |
2014 | 290,000 | 235,575 | 96,636 | (6) | 66 | (4) | 622,277 | ||||||||||||||||||||
| Vice President, Sales |
2013 | 289,000 | 9,362 | 118,802 | 66 | 417,230 | ||||||||||||||||||||||
| (1) | Amounts reflect the full grant-date fair value of stock options granted during the applicable calendar year computed in accordance with ASC Topic 718, rather than the amounts paid to or realized by the named individual and, with respect to Mr. Farley, the incremental fair value ($20,569) associated with a stock option that was repriced in 2013. We provide information regarding the assumptions used to calculate the value of all stock awards and option awards made to executive officers in Note I to our financial statements included elsewhere in this Annual Report on Form 10-K. There can be no assurance that unvested awards will vest (and, absent vesting and exercise, no value will be realized by the executive for the award). |
| (2) | Amounts reported for 2014 in the “Option Awards” and “Non-Equity Incentive Plan Compensation” columns revise the amounts previously disclosed in our Summary Compensation Table contained in our final prospectus, dated January 28, 2015, filed with the SEC pursuant to Rule 424(b). |
| (3) | Amounts represent bonuses paid with respect to 2014 services under our 2014 corporate bonus plan. |
| (4) | Amounts represent life insurance premiums paid by our company. |
| (5) | Amount represents Mr. White’s one-time signing bonus of $50,000. |
| (6) | Amount represents a bonus paid with respect to 2014 services under our Vice President of Sales 2014 bonus plan. |
Narrative Disclosure to Summary Compensation Table
2014 Salaries
The named executive officers receive a base salary to compensate them for services rendered to our company. The base salary payable to each named executive officer is intended to provide a fixed component of compensation reflecting the executive’s skill set, experience, role and responsibilities.
In 2014, our compensation committee approved merit salary increases for Messrs. Farley and Surek of 3% and 0.3%, respectively, of the executive’s base salary (to $458,902 and $290,000, respectively). Mr. White’s base salary of $420,000 was determined as the result of an arms-length negotiation of his employment agreement.
106
Table of Contents
The 2014 base salaries paid to our named executive officers are disclosed in the Summary Compensation Table above. The following table sets forth 2015 base salaries for each of our named executive officers. For 2015, Mr. Farley received a 19.8% merit salary increase effective January 1, 2015.
| Named Executive Officer |
2015 Annual Base Salary | |||
| Brian E. Farley |
$ | 550,000 | ||
| Robert S. White |
$ | 420,000 | ||
| James D. Surek |
$ | 290,000 | ||
We expect that base salaries for the named executive officers will be reviewed periodically by the compensation committee, with adjustments expected to be made generally in accordance with the considerations described above and to maintain base salaries at competitive levels.
2014 Bonuses
In 2014, each of the named executive officers participated in an annual bonus compensation program under which cash bonuses were awarded.
Messrs. Farley and White participated in our corporate bonus plan, pursuant to which each was eligible to receive a bonus based on achievement of revenue, operating income and customer quality goals established by the compensation committee, which were weighted 80%, 15% and 5%, respectively. The 2014 target bonuses for Messrs. Farley and White were 75% and 50%, respectively, of the executive’s annual base salary paid in 2014.
Under the corporate bonus plan, Mr. Farley was eligible to receive (i) payout of a cash bonus of 30% to 65% of base salary based on attainment of 80% to 110% of the target revenue goal; (ii) payout of a cash bonus of 11.25% to 13.25% of base salary based on attainment of the target operating income goal to overachievement by $1 million of the target operating income goal, so long as the target revenue goal was also achieved; and (iii) 3.75% of base salary if customer quality goals were fully attained.
Under the corporate bonus plan, Mr. White was eligible to receive (i) payout of a cash bonus of 20% to 45% of base salary based on attainment of 80% to 110% of the target revenue goal; (ii) payout of a cash bonus of 7.5% to 9.5% of base salary based on attainment of the target operating income goal to overachievement by $1 million of the target operating income goal, so long as the target revenue goal was also achieved; and (iii) 2.5% of base salary if customer quality goals were fully attained.
As a member of our sales team, in 2014 Mr. Surek participated in our management business objectives bonus plan, which provided for quarterly and annual bonus opportunities upon the achievement of revenue targets established by the compensation committee for the particular year. Mr. Surek’s target bonus in 2014 was $90,000, which consisted of a $21,000 target bonus for each quarterly revenue goal and a $6,000 target bonus associated with an annual revenue goal. To the extent that actual revenue was less than targeted revenue for a relevant period, each 1% shortfall would reduce Mr. Surek’s actual bonus earned by 2.0% of the applicable target bonus. To the extent that actual revenue was greater than targeted revenue for a relevant period, each 1% excess would increase his actual bonus earned by 2.5% of the applicable target bonus.
The actual annual cash bonuses payable for 2014 are set forth in the Summary Compensation Table above in the column titled “Non-Equity Incentive Plan Compensation.” We currently expect that our 2015 corporate bonus plan for Messrs. Farley and White will have annual and quarterly revenue and corporate quality goals weighted at 95% and 5%, respectively, with the annual revenue goal and each of the quarterly revenue goals representing 20% of the revenue component of the corporate bonus plan. In 2015, Mr. Surek will participate in a bonus program with revenue and departmental objectives weighted at 85% and 15%, respectively. The 2015 target bonuses for Messrs. Farley, White and Surek are 75%, 50% and 50% respectively, of the executive’s annual base salary paid in 2015. Mr. Surek’s target bonus was subject to the completion of our initial public offering prior to March 1, 2015.
107
Table of Contents
In connection with entering into his employment agreement, Mr. White also received a one-time signing bonus of $50,000 that will be repayable on a pro rata basis if he resigns without “good reason” (as defined in the employment agreement) within 12 months following November 24, 2014.
Equity Compensation
In 2014, our Company maintained the 2006 Plan in order to provide additional incentives for our directors, employees and consultants and to enable the Company to obtain and retain the services of these individuals, which is essential to our long term success. The 2006 Plan provides for the grant of stock options, stock appreciation rights, restricted stock, performance units and stock bonuses.
Our board of directors has adopted the 2015 Incentive Award Plan, which we refer to as the 2015 Plan, in order to facilitate the grant of cash and equity incentives to directors, employees (including our named executive officers) and consultants of our company and certain of our affiliates and to enable our company and certain of our affiliates to obtain and retain the services of these individuals, which is essential to our long-term success. The 2015 Plan became effective in connection with our initial public offering, and upon its effectiveness no further grants will be made under the 2006 Plan.
Historically, we have granted stock options to our named executive officers. Stock options typically vest over a period of four years, or based on the achievement of specified performance goals. In 2014, we granted stock options to certain of our named executive officers. The following table sets forth the total number of stock options granted to our named executive officers in 2014.
| Named Executive Officer |
Number of 2014 Stock Options |
|||
| Brian E. Farley |
188,800 | |||
| Robert S. White |
733,600 | |||
| James D. Surek |
47,500 | |||
In connection with entering into his employment agreement in 2014, Mr. White was granted: (i) an option to purchase 463,325 shares of the Company’s common stock, vesting over four years with 25% of the shares vesting on the first anniversary of the grant date, and the balance vesting equally on a monthly basis over the next three years and (ii) an option to purchase 270,275 shares of the Company’s common stock, vesting in substantially equal monthly installments over the four-year period starting on the date Mr. White is appointed as the Chief Executive Officer of the Company.
In 2014, stock options covering 188,800 and 42,500 shares were granted to Messrs. Farley and Surek, respectively, and vest in substantially equal monthly installments over four years beginning December 1, 2014, subject to the completion of our initial public offering. In addition, a stock option granted to Mr. Surek in 2014 covering 10,000 shares were scheduled to vest upon the achievement of pre-determined 2014 revenue goals (of which 5,000 shares actually vested).
Other Elements of Compensation
We provide customary employee benefits to our full- and part-time employees, including our named executive officers, in the United States (in the case of part-time employees), including medical and dental benefits, short-term and long-term disability insurance, accidental death and dismemberment insurance and life insurance.
We also maintain a 401(k) retirement savings plan for our employees, including our named executive officers, who satisfy certain eligibility requirements. Under our 401(k) plan, eligible employees may defer a portion of their compensation, within prescribed tax code limits, on a pre-tax basis through contributions to the 401(k) plan.
108
Table of Contents
Outstanding Equity Awards as of December 31, 2014
The following table shows the number of shares of common stock underlying outstanding stock options for each of our named executive officers as of December 31, 2014. As of December 31, 2014, none of our named executive officers held any other outstanding equity incentive plan awards.
| Option Award | ||||||||||||||||||||
| Name |
Grant Date | Number of Securities Underlying Unexercised Options (#) Exercisable(11) |
Number of Securities Underlying Unexercised Options (#) Unexercisable(11) |
Option Exercise Price ($) |
Option Expiration Date |
|||||||||||||||
| Brian E. Farley |
3/24/2010 | (1) | 39,563 | — | 1.36 | 3/24/2020 | ||||||||||||||
| 2/8/2012 | (2) | 9 | 28,430 | 0.76 | 2/8/2022 | |||||||||||||||
| 3/19/2013 | (3) | — | 34,510 | 1.24 | 3/19/2023 | |||||||||||||||
| 12/16/2014 | (4) | — | 188,800 | 11.36 | 12/16/2024 | |||||||||||||||
| Robert S. White |
12/16/2014 | (5) | — | 463,325 | 11.36 | 12/16/2024 | ||||||||||||||
| 12/16/2014 | (6) | — | 270,275 | 11.36 | 12/16/2024 | |||||||||||||||
| James D. Surek |
2/1/2011 | (7) | 156 | 56 | 0.68 | 2/1/2021 | ||||||||||||||
| 2/8/2012 | (8) | 1,875 | 7,500 | 0.76 | 2/8/2022 | |||||||||||||||
| 3/19/2013 | (9) | 787 | 6,508 | 1.24 | 3/19/2023 | |||||||||||||||
| 1/17/2014 | (10) | 5,000 | — | 3.48 | 1/17/2024 | |||||||||||||||
| 12/16/2014 | (4) | — | 42,500 | 11.36 | 12/16/2024 | |||||||||||||||
| (1) | This stock option was fully vested as of January 1, 2014. |
| (2) | This stock option vested and continues to vest in 48 substantially equal monthly installments, commencing on February 8, 2012, and on the last day of each month thereafter (through and including December 31, 2015), subject to the executive’s continued service. |
| (3) | This stock option vested and continues to vest as to (i) 4,140 shares on April 1, 2013, (ii) 1,380 shares on May 1, 2013 and on the first day of each month thereafter (through and including December 1, 2016) and (iii) 1,390 shares on January 1, 2017, subject to the executive’s continued service. |
| (4) | These stock options vest in substantially equal monthly installments over the four-year period starting December 1, 2014, subject to the executive’s continued service and the completion of this offering. |
| (5) | This stock option vests as to (i) 115,831 shares on November 24, 2015 and (ii) in 36 substantially equal monthly installments thereafter, subject to the executive’s continued service. |
| (6) | This stock option vests in substantially equal monthly installments subject to Mr. White’s continued service to the Company on each applicable vesting date, over the four-year period beginning on the date Mr. White is appointed as the Chief Executive Officer of the Company. |
| (7) | This stock option vests in 48 substantially equal monthly installments, commencing on February 1, 2011 and on the first day of each month thereafter (through and including January 1, 2015), subject to the executive’s continued service. |
| (8) | This stock option vests in 48 substantially equal monthly installments, commencing on February 8, 2012 and on the last day of each month thereafter (through and including December 31, 2015), subject to the executive’s continued service. |
| (9) | This stock option vests in 48 substantially equal monthly installments, commencing on February 1, 2013 and on the first day of each month thereafter (through and including January 1, 2017), subject to the executive’s continued service. |
| (10) | This stock option vested in part in 2014 upon the achievement of pre-determined revenue goals. |
| (11) | Amounts reported in these columns revise the amounts previously disclosed in our final prospectus, dated January 28, 2015, filed with the SEC pursuant to Rule 424(b). |
109
Table of Contents
Employment Agreements and Severance and Change in Control Benefits
Severance Arrangements
Each of our named executive officers is party to a severance agreement (Farley, Surek) or a change in control severance agreement (White) that provides the executive with severance payments and benefits if the executive’s employment is terminated by the Company without “cause” or by the executive for “good reason” (each, as defined in the applicable agreement), in either case, within 12 months following a “change in control” of the Company (as defined in the applicable agreement) or prior to a change in control if the termination occurs in connection with the change in control. In any such event, the executive will receive, subject to the execution and non-revocation of a general release of claims in favor of the Company:
| • | a lump-sum payment in an amount equal to 12 months (or 18 months for Mr. Farley) of the executive’s annual base salary then in effect; |
| • | a lump-sum payment in an amount equal to the executive’s annual target bonus (or, with respect to Mr. Farley, 125% of his annual target bonus); and |
| • | Company-subsidized healthcare continuation coverage for the executive and his or her dependents for up to 12 months (or 18 months for Mr. Farley) after the termination date. |
In addition, each agreement also provides that if the executive’s employment is terminated without cause or for good reason following a change in control then (i) the executive’s then-outstanding equity awards (that had been assumed/replaced at the change in control) will accelerate and vest in full and (ii) each such stock option will remain exercisable until the one-year anniversary of the termination date (but not beyond the maximum term of the option). In addition, under the agreements, to the extent that any change in control payment or benefit would be subject to an excise tax imposed in connection with Section 4999 of the Code, such payments and/or benefits may be subject to a “best pay cap” reduction to the extent necessary so that the executive receives the greater of the (i) net amount of the change in control payments and benefits reduced such that such payments and benefits will not be subject to the excise tax and (ii) net amount of the change in control payments and benefits without such reduction.
Pursuant to Mr. Farley’s agreement in effect in 2014, if the Company had experienced a change in control on or before December 31, 2014 and Mr. Farley had remained employed until immediately prior to such change in control, then Mr. Farley would have been entitled to receive a bonus equal to (i) 0.25% of the Company’s “net proceeds” if the net proceeds are equal to a threshold amount or (ii) 0.5% of the Company’s net proceeds if the net proceeds are equal to a target amount.
In addition, the severance agreements with Messrs. Farley and Surek provide the executive with severance payments and benefits if the executive’s employment is terminated by the Company without cause or by the executive for good reason outside of the change in control context. In any such event, the executive will receive, subject to the execution and non-revocation of a general release of claims in favor of the Company:
| • | a lump-sum payment in an amount equal to six months (Surek) or 12 months (Farley) of the executive’s annual base salary; and |
| • | Company-subsidized healthcare continuation coverage for the executive and his or her dependents for up to six months (Surek) or 12 months (Farley) after the termination date. |
White Employment Agreement
On November 18, 2014, we entered into an employment agreement with Robert S. White, pursuant to which he serves as our President and Chief Operating Officer. Under the employment agreement, Mr. White is entitled to receive an annual base salary of $420,000 per year. The employment agreement also provides that Mr. White is eligible to receive an annual bonus of up to 50% of his annualized salary based on the achievement of certain objectives as determined by our board of directors in connection with the applicable bonus plan. In connection
110
Table of Contents
with entering into the employment agreement, Mr. White received a one-time signing bonus of $50,000 that will be repayable on a pro rata basis if he resigns without “good reason” (as defined in the employment agreement) within 12 months following November 24, 2014.
During his employment, Mr. White is eligible to participate in our health and welfare, retirement and other plans and programs that we make available to our senior executives from time to time. In addition, the employment agreement provides that Mr. White is entitled to reimbursement for all reasonable expenses incurred on behalf of the Company.
In connection with the execution of the employment agreement, Mr. White was granted: (i) an option to purchase 463,325 shares of the Company’s common stock, with 25% of the shares vesting on November 24, 2015, and the balance vesting equally on a monthly basis over the next 36 months subject to Mr. White’s continued service with the Company on each applicable vesting date, and (ii) an option to purchase 270,275 shares of the Company’s common stock, vesting in substantially equal installments over the four-year period starting on the date Mr. White is appointed as the Chief Executive Officer of the Company, subject to his continued service with the Company.
Mr. White’s employment agreement provides that if his employment is terminated by the Company without “cause” or by Mr. White for “good reason” (as “good reason” is defined in his employment agreement and “cause” is defined in his change of control agreement) outside of the change in control context, then Mr. White will receive, subject to the execution and non-revocation of a general release of claims in favor of the Company:
| • | 12 months’ continuation payments of his annual base salary then in effect over the 12-month period following the termination of employment; |
| • | a lump-sum payment in an amount equal to the annual bonus earned for the year of termination; and |
| • | Company-subsidized healthcare continuation coverage for Mr. White and his dependents for up to 12 months after the termination date. |
Mr. White’s employment agreement also provides that if his employment is terminated due to his death or disability, he will be entitled to receive an amount equal to three times his monthly salary then in effect, payable in a lump sum within 30 days after the date of his termination.
Mr. White’s employment agreement also contains customary confidentiality, non-competition and non-solicitation provisions.
2006 Plan Options
Each stock option held by Mr. Farley that is granted pursuant to our 2006 Plan will accelerate and vest in full as of a “change in control” (as defined in the 2006 Plan). In addition, the 2006 Plan provides that upon the executive’s termination of service due to the participant’s death, disability or retirement, then the executive’s outstanding stock options will remain exercisable for six months following the termination date.
Director Compensation
In 2014, each of our non-employee directors other than Mr. McDermott (who had previously resigned) was granted a stock option covering 15,000 shares that vests quarterly over the 36-month period beginning on December 1, 2014 (with the start of vesting contingent upon the completion of our initial public offering) or, with respect to Mr. McCormick, the date of his appointment to our board on November 9, 2014. In addition, Mr. McCormick received cash compensation following his appointment to our board. Our other non-employee directors did not receive any cash compensation for their services in 2014.
111
Table of Contents
The following table sets forth information concerning the compensation of our directors during the year ended December 31, 2014. Mr. Farley, our Chief Executive Officer, does not receive additional compensation for his service as a director, and therefore is not included in the table below:
| Name |
Fees Earned or Paid in Cash ($) |
Option Awards ($)(1)(2) |
Total ($) |
|||||||||
| Joshua Baltzell |
— | 79,650 | 79,650 | |||||||||
| Shawn McCormick |
6,875 | 89,988 | 96,863 | |||||||||
| John McDermott |
— | — | — | |||||||||
| David Milne |
— | 79,650 | 79,650 | |||||||||
| Guido Neels |
— | 79,650 | 79,650 | |||||||||
| (1) | Amounts reflect the full grant-date fair value of stock options granted during 2014 computed in accordance with ASC Topic 718, rather than the amounts paid to or realized by the named individual. We provide information regarding the assumptions used to calculate the value of all option awards made to directors in Note I to our financial statements included elsewhere in this Annual Report on Form 10-K. There can be no assurance that unvested awards will vest (and, absent vesting and exercise, no value will be realized by the executive for the award). As of December 31, 2014, each of our directors (other than Mr. McDermott) held an option covering 15,000 shares, all of which remained unvested. |
| (2) | Amounts reported for 2014 in the “Option Awards” revise the amounts previously disclosed in our final prospectus, dated January 28, 2015, filed with the SEC pursuant to Rule 424(b). |
In connection with our IPO, our board adopted a compensation program for our non-employee directors that consists of a combination of cash annual retainer fees and long-term equity-based compensation. The terms of the program are described below. The program became effective upon the completion of our IPO.
Cash Compensation
Under the program, each non-employee director will be entitled to receive annual cash retainers in the following amounts, pro-rated for any partial year of service:
| Director: |
$ | 35,000 | ||
| Chair of Audit Committee: |
$ | 20,000 | ||
| Chair of Compensation Committee: |
$ | 15,000 | ||
| Chair of Nominating and Corporate Governance Committee: |
$ | 10,000 | ||
| Audit Committee Member (Non-Chair): |
$ | 10,000 | ||
| Compensation Committee Member (Non-Chair): |
$ | 7,500 | ||
| Nominating and Corporate Governance Committee (Non-Chair): |
$ | 5,000 |
All annual retainers will be paid in cash quarterly in arrears promptly following the end of the applicable calendar quarter, but in no event more than thirty (30) days after the end of such quarter.
Equity Compensation
Under the program, each non-employee director who is initially elected or appointed to serve on our board automatically will receive an option to purchase 15,000 shares of common stock, which will vest quarterly over a three-year period, subject to continued service through the applicable vesting date.
Each non-employee director serving on our board as of the date of each annual shareholder meeting automatically will receive an option to purchase 7,500 shares of common stock, which will vest in full on the earlier to occur of the one-year anniversary of the grant date and the date of the annual meeting of our stockholders following the grant date, subject to continued service through the applicable vesting date.
112
Table of Contents
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee has ever been an officer or employee of the Company. None of our executive officers serves, or has served during the last fiscal year, as a member of the board of directors, compensation committee or other board committee performing equivalent functions of any entity that has one or more executive officers serving as one of our directors or on our compensation committee.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth information relating to the beneficial ownership of our common stock as of March 6, 2015, by:
| • | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our outstanding shares of common stock; |
| • | each of our directors; |
| • | each of our named executive officers; and |
| • | all directors and executive officers as a group. |
The number of shares beneficially owned by each entity, person, director or executive officer is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any shares over which the individual has sole or shared voting power or investment power as well as any shares that the individual has the right to acquire within 60 days of March 6, 2015 through the exercise of any stock option, warrants or other rights. Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all shares of common stock held by that person.
113
Table of Contents
The percentage of shares beneficially owned is computed on the basis of 18,679,216 shares of our common stock outstanding as of March 6, 2015. Shares of our common stock that a person has the right to acquire within 60 days of March 6, 2015 are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all directors and executive officers as a group. Unless otherwise indicated below, the address for each beneficial owner listed is c/o Entellus Medical, Inc., 3600 Holly Lane North, Suite 40, Plymouth, Minnesota 55447.
| Name and Address of Beneficial Owner |
Number of Shares Beneficially Owned |
Percentage of Shares Beneficially Owned |
||||||
| 5% Stockholders: |
||||||||
| Essex Woodlands Health Ventures(1) 21 WaterWay Avenue Suite 225 The Woodlands, Texas 77380 |
3,813,196 | 20.4 | % | |||||
| SV Life Sciences(2) One Boston Place 201 Washington Street Suite 3900 Boston, Massachusetts 02108 |
3,489,531 | 18.7 | % | |||||
| Split Rock Partners, LP(3) 10400 Viking Drive Suite 550 Eden Prairie, Minnesota 55344 |
3,156,731 | 16.9 | % | |||||
| Covidien Ventures S.a.r.l. 3b, bd Prince Henri Luxembourg L-1724 Luxembourg |
956,925 | 5.1 | % | |||||
| Directors and Named Executive Officers: |
||||||||
| Brian E. Farley(4) |
681,385 | 3.6 | % | |||||
| Joshua Baltzell(5) |
1,250 | * | ||||||
| David B. Milne(6) |
1,250 | * | ||||||
| Shawn McCormick(7) |
4,191 | * | ||||||
| Guido Neels(8) |
16,250 | * | ||||||
| Robert S. White |
5,882 | * | ||||||
| James D. Surek(9) |
141,135 | * | ||||||
| All directors and executive officer as a group (13 persons) (10) |
1,326,848 | 7.0 | % | |||||
| * | Indicates beneficial ownership of less than 1% of the total outstanding common stock. |
| (1) | Represents shares held by Essex Woodlands Health Ventures Fund VIII, L.P., Essex Woodlands Health Ventures Fund VIII-A, L.P. and Essex Woodlands Health Ventures Fund VIII-B, L.P., which we collectively refer to as the “Essex Stockholders.” Essex Woodlands Health Ventures VIII, L.P., a Delaware limited partnership, is the general partner of each of the Essex Stockholders and is referred to as the “Partnership,” and Essex Woodlands Health Ventures VIII, LLC, a Delaware limited liability company, is the general partner of the Partnership and is referred to as the “General Partner.” James L. Currie, Martin P. Sutter, Immanuel Thangaraj, Petri Vainio, Jeff Himawan, Ron Eastman, Guido Neels and Steve Wiggins are the managers of the General Partner, and each is referred to as a “Manager” and collectively as the “Managers.” The Partnership is deemed to have sole voting and dispositive power with respect to the shares held by each of the Essex Stockholders. The Managers are deemed to have shared voting and dispositive power with respect to the shares held by each of the Essex Stockholders by unanimous consent and through the Partnership. Each Manager disclaims beneficial ownership of such shares except to the extent of his pecuniary interest therein. The address of the Essex Stockholders is 21 WaterWay Avenue, Suite 225, The Woodlands, Texas 77380. |
114
Table of Contents
| (2) | Based solely on a Schedule 13D filed by SV Life Sciences, et al., on February 13, 2015. Represents shares held by: (i) SV Life Sciences Fund IV, L.P., or Fund IV; (ii) SV Life Sciences Fund IV Strategic Partners, L.P., or Fund IV Strategic; (iii) International Life Sciences Fund III (LP1), L.P., or ILSF LP1; (iv) International Life Sciences Fund III Co-Investment, L.P., or ILSF Co-Invest; and (v) International Life Sciences Fund III Strategic Partners, L.P., or ILSF Strategic. Fund IV and Fund IV Strategic are collectively referred to as the “Fund IV Entities.” ILSF LP1, ILSF Co-Invest and ILSF Strategic are collectively referred to as the “Fund III Entities.” |
International Life Sciences Fund III (GP), L.P. (“Fund III GP”) is the general partner of each of the Fund III Entities. ILSF III, LLC (the “ILSF General Partner”) is the general partner of Fund III GP and, through an investment committee comprised of James Garvey, Kate Bingham, Eugene D. Hill, III and Michael Ross, controls voting and investment decisions over the shares held by the Fund III Entities by majority vote. Each member of the investment committee of ILSF General Partner disclaims beneficial ownership over the shares held by the Fund III Entities, except to the extent of any pecuniary interest therein. Each of ILSF General Partner and Fund III GP disclaim beneficial ownership over the shares held by the Fund III Entities except to the extent of their respective pecuniary interest therein.
SV Life Sciences Fund IV (GP), L.P. (“Fund IV GP”) is the general partner of each of the Fund IV Entities. SVLSF IV, LLC (the “SVLS General Partner”) is the general partner of Fund IV GP and, through an investment committee comprised of David Milne (also a member of the Issuer’s board of directors), James Garvey, Kate Bingham, Eugene D. Hill, III and Michael Ross, controls voting and investment decisions over the shares held by the Fund IV Entities by a majority vote. Each member of the investment committee of SVLS General Partner disclaims beneficial ownership over the shares held by the Fund IV Entities, except to the extent of any pecuniary interest therein. Each of SVLS General Partner and Fund IV GP disclaim beneficial ownership over the shares held by the Fund IV Entities except to the extent of their respective pecuniary interest therein.
The address of each of the Fund III entities and each of the Fund IV entities is One Boston Place, Suite 3900, 201 Washington Street, Boston, Massachusetts 02108.
| (3) | Based solely on a Schedule 13D filed by Split Rock Partners, LP on February 11, 2015. Voting and investment power over the shares is delegated to Split Rock Partners Management, LLC, the general partner of Split Rock Partners, LP. Split Rock Partners Management, LLC has delegated voting and investment power to three individuals, Michael Gorman, James Simons and David Stassen, who require a two-thirds vote to act. Split Rock Partners Management, LLC disclaims beneficial ownership of the shares except to the extent of any pecuniary interest therein. |
| (4) | Includes 75,617 shares issuable upon the exercise of stock options granted to Mr. Farley that are exercisable within 60 days of March 6, 2015. |
| (5) | Includes 1,250 shares issuable upon the exercise of stock options granted to Mr. Baltzell that are exercisable within 60 days of March 6, 2015. |
| (6) | Includes 1,250 shares issuable upon the exercise of stock options granted to Mr. Milne that are exercisable within 60 days of March 6, 2015. |
| (7) | Includes 1,250 shares issuable upon the exercise of stock options granted to Mr. McCormick that are exercisable within 60 days of March 6, 2015. |
| (8) | Includes 1,250 shares issuable upon the exercise of stock options granted to Mr. Neels that are exercisable within 60 days of March 6, 2015. |
| (9) | Includes 16,104 shares issuable upon the exercise of stock options granted to Mr. Surek that are exercisable within 60 days of March 6, 2015. |
| (10) | Includes options to purchase 319,204 shares of common stock exercisable within 60 days of March 6, 2015 held by all of our executive officers and directors. |
115
Table of Contents
Equity Compensation Plan Information
The following table summarizes securities available under our equity compensation plans as of December 31, 2014(1).
| Plan Category | Shares Issuable Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Available for Future Issuance |
|||||||||
| Equity Compensation Plans Approved by Security Holders:(2) |
2,098,251 | $ | 7.38 | 22,541 | ||||||||
| Equity Compensation Plans Not Approved by Security Holders |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
2,098,251 | $ | 7.38 | 22,541 | ||||||||
|
|
|
|
|
|
|
|||||||
| (1) | In January 2015, we effected a 1-for-4 reverse split of our common stock. All historical common stock and per share information in this chart has been changed to reflect the stock split. |
| (2) | Consists of our 2006 Stock Incentive Plan, which was adopted in August 2006, and was subsequently amended and restated by the Company on November 12, 2009, and provides for awards of incentive stock options, nonqualified stock options, stock appreciation rights, restricted stock awards, performance units and stock bonuses to employees, directors and consultants of our Company. In connection with the closing of our IPO in February 2015, we adopted the 2015 Incentive Award Plan and the 2015 Employee Stock Purchase Plan. In connection with the effectiveness of the 2015 Incentive Award Plan, no new awards will be granted under the 2006 Stock Incentive Plan. |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
Certain Relationships and Related Transactions
Policies and Procedures for Related Person Transactions
Our board of directors has adopted a written related person transaction policy setting forth the policies and procedures for the review and approval or ratification of related person transactions. This policy covers, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we were or are to be a participant, where the amount involved exceeds $120,000 in any fiscal year and a related person had, has or will have a direct or indirect material interest, including without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction.
The following are certain transactions, arrangements and relationships with our directors, executive officers and stockholders owning 5% or more of our outstanding common stock. All of the transactions described in this section occurred prior to the adoption of the related person transaction policy.
Registration Rights Agreement
We are party to a registration rights agreement, or the Registration Rights Agreement, with certain of our stockholders that held our convertible preferred stock prior to the IPO, including certain holders of 5% of our capital stock and entities affiliated with certain of our directors, for the registration of shares of common stock that were issued upon conversion of such shares of convertible preferred stock at the closing of the IPO. Subject to certain limitations, these holders have the right to request that we prepare, file and maintain up to two
116
Table of Contents
registration statements on Form S-1 covering the sale of such shares of common stock and, once we are eligible to use a registration statement on Form S-3, up to two registration statements on Form S-3 in any 12-month period covering the sales of such shares of common stock. Additionally, these holders have unlimited “piggyback” registration rights to include these shares of common stock in future registration statements that we may initiate, subject to certain conditions and limitations (including customary cut-back rights). Under the Registration Rights Agreement, we will pay all expenses relating to such registrations, including the reasonable fees of one special counsel for the participating holders, and the holders will pay all underwriting discounts and commissions relating to the sale of their shares of common stock. The Registration Rights Agreement also contains other customary terms, including for indemnification.
Under the terms of the Registration Rights Agreement, we will not be required to file a registration statement pursuant to any demand by the holders in the 180-day period commencing with the closing of the IPO.
The Registration Rights Agreement will terminate upon the earlier of (1) the date that is five years after the closing of the IPO or (2) with respect to each stockholder following the closing of the IPO, at such time as such stockholder holds 1% or less of our common stock and can sell all of its shares pursuant to Rule 144 of the Securities Act during any three-month period.
Investor Rights Agreement
We are party to an investor rights agreement, or the Investor Rights Agreement, with certain of our stockholders that held our convertible preferred stock prior to the IPO, including certain holders of 5% of our capital stock and entities affiliated with certain of our directors. As a result of the IPO, most of the covenants and restrictions set forth in the Investor Rights Agreement that apply to us terminated, but we remain obligated to comply with reporting requirements under the Exchange Act.
Voting Agreement
We were party to a voting agreement, or the Voting Agreement, under which certain of our stockholders that held our convertible preferred stock prior to the IPO, including certain holders of 5% of our capital stock and entities affiliated with certain of our directors, agreed to vote in a certain way on certain matters, including with respect to the election of directors, and certain holders have the right to have a designated representative present at meetings of our board of directors (or any committee of the board). Pursuant to the Voting Agreement, each of SV Life Sciences, Essex Woodlands and Split Rock Partners had the right to designate one member of our board of directors. David B. Milne, Guido Neels and Joshua Baltzell were designated by SV Life Sciences, Essex Woodlands and Split Rock Partners, respectively, under the Voting Agreement. The Voting Agreement terminated by its terms in connection with the closing of the IPO. No stockholder has any continuing voting rights, including special rights regarding the election or designation of members of our board of directors, following the termination of the Voting Agreement.
Right of First Refusal and Co-Sale Agreement
We were party to a right of first refusal and co-sale agreement with our founders and certain of our stockholders that held our convertible preferred stock prior to the IPO, including certain holders of 5% of our capital stock and entities affiliated with certain of our directors, pursuant to which the holders of convertible preferred stock had a right of first refusal and co-sale in respect of certain sales of securities by our founders. The right of first refusal and co-sale agreement terminated in connection with the closing of the IPO.
Severance Arrangements
We have entered into employment agreements, severance agreements and/or change in control severance agreements with our executive officers that provide the respective executive with severance payments and benefits if the executive’s employment is terminated under certain circumstances as described in greater detail in the section of this Annual Report on Form 10-K titled “Item 11. Executive Compensation—Employment Agreements and Severance and Change in Control Benefits—Severance Agreements.”
117
Table of Contents
Indemnification Agreements
We have entered into indemnification agreements with our directors and executive officers, in addition to indemnification provided for in our amended and restated certificate of incorporation and amended and restated bylaws. These agreements, among other things, provide for indemnification of our directors and executive officers for expenses, judgments, fines and settlement amounts incurred by this person in any action or proceeding arising out of this person’s services as a director or executive officer or at our request.
Participation in the IPO
Certain of our directors and principal stockholders, including Essex Woodlands, SV Life Sciences, Split Rock and Guido Neels, purchased shares of our common stock in the IPO at the initial public offering price, on the same terms as the shares that were sold to the public generally. Split Rock and Essex Woodlands each purchased an aggregate of approximately $6.0 million in common stock. SV Life Sciences purchased approximately $5.4 million in common stock. Mr. Neels purchased approximately $0.3 million in common stock.
Director Independence
Our common stock has been listed on The Nasdaq Global Market since January 29, 2015 in connection with our IPO. Under the listing requirements and rules of The Nasdaq Global Market, independent directors must compose a majority of a listed company’s board of directors within one year of listing. In addition, the rules of The Nasdaq Global Market require that, subject to specified exceptions and phase in periods following its IPO, each member of a listed company’s audit and compensation, nominating and governance committee be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act, or Rule 10A-3. Under the rules of The Nasdaq Global Market, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
To be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of our audit committee, our board of directors or any other board committee: (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries; or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our board of directors has undertaken a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our board of directors has determined that none of Mr. McCormick, Mr. Baltzell, Mr. Milne and Mr. Neels has a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director, and that each of these directors is “independent” as that term is defined under the applicable rules and regulations of the listing requirements and rules of The Nasdaq Global Market. In making this determination, our board of directors considered the current and prior relationships that each non-employee director has with the Company and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director.
118
Table of Contents
| Item 14. | Principal Accountant Fees and Services |
Independent Registered Public Accountants’ Fees
The following table represents aggregate fees billed to us for services related to the fiscal years ended December 31, 2014 and 2013, respectively, by Grant Thornton LLP, our independent registered public accounting firm.
| Year Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Audit Fees(1) |
$ | 648,000 | $ | 39,000 | ||||
| Audit-Related Fees(2) |
8,000 | — | ||||||
| Tax Fees(3) |
24,000 | 53,000 | ||||||
| All Other Fees |
— | — | ||||||
|
|
|
|
|
|||||
| Total |
$ | 680,000 | $ | 92,000 | ||||
|
|
|
|
|
|||||
| (1) | Audit Fees consist of fees billed for professional services by Grant Thornton LLP for the audit of our annual financial statement and the review of our registration statement on Form S-1 and related services that are normally provided in connection with statutory and regulatory filings or engagements. |
| (2) | Audit-Related Fees consist of fees billed by Grant Thornton LLP for assurance and related services that are reasonably related to the performance of the audit or review of our financial statements. |
| (3) | Tax Fees consist of fees for professional services, including tax consulting and compliance performed by Grant Thornton LLP. |
Pre-Approval Policies and Procedures
Our audit committee has adopted a policy, or the Pre-Approval Policy, that sets forth the procedures and conditions pursuant to which audit and non-audit services proposed to be performed by the independent auditor may be pre-approved. The Pre-Approval Policy generally provides that we will not engage Grant Thornton LLP to render any audit, audit-related, tax or permissible non-audit service unless the service is either (i) explicitly approved by the audit committee (“specific pre-approval”) or (ii) entered into pursuant to the pre-approval policies and procedures described in the Pre-Approval Policy (“general pre-approval”). Unless a type of service to be provided by Grant Thornton LLP has received general pre-approval under the Pre-Approval Policy, it requires specific pre-approval by the audit committee. Any proposed services exceeding pre-approved cost levels or budgeted amounts will also require specific pre-approval. On an annual basis, the audit committee reviews and generally pre-approves the services (and related fee levels or budgeted amounts) that may be provided by Grant Thornton LLP without first obtaining specific pre-approval from our audit committee. Our audit committee may revise the list of general pre-approved services from time to time, based on subsequent determinations.
During 2014 and 2013, no services were provided to us by Grant Thornton LLP other than in accordance with the pre-approval policies and procedures described above.
119
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
| (a) | Documents filed as part of this report: |
| 1. | List of Financial Statements |
The financial statements required by this item are listed in Item 8, “Financial Statements and Supplementary Data” herein.
| 2. | List of Financial Statement Schedules |
The financial statement schedule required by this item is listed in Item 8, “Financial Statements and Supplementary Data” herein.
| 3. | List of Exhibits |
| Incorporated by Reference | ||||||||||||
| Exhibit |
Exhibit Description |
Filed/ Furnished Herewith |
Form | File No. | Exhibit | Filing Date | ||||||
| 3.1 |
Amended and Restated Certificate of Incorporation of Entellus Medical, Inc. | 8-K | 001-36814 | 3.1 | 02/03/15 | |||||||
| 3.2 |
Amended and Restated Bylaws of Entellus Medical, Inc. | 8-K | 001-36814 | 3.2 | 02/03/15 | |||||||
| 4.1 |
Form of Common Stock Certificate of Entellus Medical, Inc. | S-1/A | 333-201237 | 4.1 | 01/15/15 | |||||||
| 4.2 |
Fifth Amended and Restated Investor Rights Agreement, dated August 17, 2011, by and among Entellus Medical, Inc., the parties set forth therein and such other investors as may from time to time become a party thereto | S-1/A | 333-201237 | 4.3 | 01/28/15 | |||||||
| 4.3 |
Fifth Amended and Restated Registration Rights Agreement, dated August 17, 2011, by and among Entellus Medical, Inc., the parties set forth therein and such other parties as may from time to time become a party thereto | S-1/A | 333-201237 | 4.4 | 01/28/15 | |||||||
| 4.4 |
Warrant to Purchase Stock, dated October 18, 2012, issued by Entellus Medical, Inc. in favor of Oxford Finance LLC | S-1/A | 333-201237 | 4.6 | 01/28/15 | |||||||
| 4.5 |
Warrant to Purchase Stock, dated October 18, 2012, issued by Entellus Medical, Inc. in favor of Oxford Finance LLC | S-1/A | 333-201237 | 4.7 | 01/28/15 | |||||||
| 10.1# |
Entellus Medical, Inc. 2006 Stock Incentive Plan (as amended and restated November 12, 2009) | S-1/A | 333-201237 | 10.1 | 01/28/15 | |||||||
| 10.1(a)# |
Third Amendment to Entellus Medical, Inc. 2006 Stock Incentive Plan | S-1 | 333-201237 | 10.1(a) | 12/23/14 | |||||||
| 10.1(b)# |
Fourth Amendment to Entellus Medical, Inc. 2006 Stock Incentive Plan | S-1/A | 333-201237 | 10.1(b) | 01/15/15 | |||||||
120
Table of Contents
| Incorporated by Reference | ||||||||||||
| Exhibit |
Exhibit Description |
Filed/ Furnished Herewith |
Form | File No. | Exhibit | Filing Date | ||||||
| 10.2# |
Form of Incentive Stock Option Agreement pursuant to 2006 Stock Incentive Plan | S-1/A | 333-201237 | 10.2 | 01/28/15 | |||||||
| 10.3# |
Form of Non-Statutory Stock Option Agreement pursuant to 2006 Stock Incentive Plan | S-1/A | 333-201237 | 10.3 | 01/28/15 | |||||||
| 10.4# |
Entellus Medical, Inc. 2015 Incentive Award Plan | S-1/A | 333-201237 | 10.4 | 01/15/15 | |||||||
| 10.4(a)# |
Form of Entellus Medical, Inc. 2015 Incentive Award Plan Stock Option Agreement | S-1/A | 333-201237 | 10.4(a) | 01/15/15 | |||||||
| 10.4(b)# |
Form of Entellus Medical, Inc. 2015 Incentive Award Plan Restricted Stock Unit Award Grant Notice and Award Agreement | S-1/A | 333-201237 | 10.4(b) | 01/15/15 | |||||||
| 10.5# |
Entellus Medical, Inc. 2015 Employee Stock Purchase Plan | S-1/A | 333-201237 | 10.5 | 01/15/15 | |||||||
| 10.6 |
Form of Indemnification Agreement between Entellus Medical, Inc. and its directors and officers | S-1 | 333-201237 | 10.6 | 12/23/14 | |||||||
| 10.7† |
Confidential Settlement Agreement and Non-Exclusive Patent License Agreement, dated February 17, 2011, by and between Acclarent, Inc. and Entellus Medical, Inc. | S-1/A | 333-201237 | 10.7 | 01/15/15 | |||||||
| 10.8† |
Amendment No. 1 to the Confidential Settlement and Non-Exclusive Patent License Agreement, dated October 5, 2012, by and between Acclarent, Inc. and Entellus Medical, Inc. | S-1/A | 333-201237 | 10.8 | 01/28/15 | |||||||
| 10.9 |
Amended and Restated Loan and Security Agreement, dated December 20, 2013, among Oxford Finance LLC, the lenders listed therein and Entellus Medical, Inc. | S-1/A | 333-201237 | 10.9 | 01/28/15 | |||||||
| 10.9(a) |
First Amendment to Amended and Restated Loan and Security Agreement, dated October 31, 2014, by and among Oxford Finance LLC, the lenders listed therein and Entellus Medical, Inc. | S-1/A | 333-201237 | 10.9(a) | 01/28/15 | |||||||
| 10.10# |
Severance Agreement, effective January 1, 2015, between Entellus Medical, Inc. and Brian E. Farley | S-1/A | 333-201237 | 10.10 | 01/15/15 | |||||||
| 10.11# |
Change in Control Severance Agreement, dated November 24, 2014, between Entellus Medical, Inc. and Robert S. White | S-1/A | 333-201237 | 10.11 | 01/15/15 | |||||||
| 10.12# |
Severance Agreement, effective January 1, 2015, between Entellus Medical, Inc. and James Surek | S-1/A | 333-201237 | 10.12 | 01/15/15 | |||||||
| 10.13# |
Non-Employee Director Compensation Program | S-1/A | 333-201237 | 10.13 | 01/15/15 | |||||||
121
Table of Contents
| Incorporated by Reference | ||||||||||||||||||||||
| Exhibit |
Exhibit Description |
Filed/ Furnished Herewith |
Form | File No. | Exhibit | Filing Date |
||||||||||||||||
| 10.14# |
Letter Agreement, dated November 16, 2012, by and between Entellus Medical, Inc. and Thomas V. Ressemann | S-1 | 333-201237 | 10.15 | 12/23/14 | |||||||||||||||||
| 10.15# |
Assignment of Letter Agreement, dated November 26, 2012, by and among Entellus Medical, Inc., Thomas V. Ressemann and Ressemann and Associates, LLC | S-1 | 333-201237 | 10.16 | 12/23/14 | |||||||||||||||||
| 10.16# |
Employment Agreement, dated November 18, 2014, by and between Entellus Medical, Inc. and Robert S. White | S-1 | 333-201237 | 10.17 | 12/23/14 | |||||||||||||||||
| 23.1 |
Consent of Grant Thornton LLP | * | ||||||||||||||||||||
| 31.1 |
Rule 13a-14(a) / 15d-14(a) Certification of Chief Executive Officer | * | ||||||||||||||||||||
| 31.2 |
Rule 13a-14(a) / 15d-14(a) Certification of Chief Financial Officer | * | ||||||||||||||||||||
| 32.1 |
Section 1350 Certification of Chief Executive Officer | * | * | |||||||||||||||||||
| 32.2 |
Section 1350 Certification of Chief Financial Officer | * | * | |||||||||||||||||||
| * | Filed herewith. |
| ** | Furnished herewith. |
| # | Indicates management contract or compensatory plan. |
| † | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment pursuant to Rule 24b-2 under the Securities Exchange Act of 1934, as amended. |
122
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ENTELLUS MEDICAL, INC. | ||
| By: |
/s/ Brian E. Farley | |
| Name: | Brian E. Farley | |
| Title: | Chief Executive Officer | |
Date: March 19, 2015
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Brian E. Farley Brian E. Farley |
Chief Executive Officer and Chairman of the Board (Principal Executive Officer) |
March 19, 2015 | ||
| /s/ Thomas E. Griffin Thomas E. Griffin |
Chief Financial Officer (Principal Accounting Officer and Principal Financial Officer) |
March 19, 2015 | ||
| /s/ Joshua Baltzell Joshua Baltzell |
Director | March 19, 2015 | ||
| /s/ Shawn T McCormick Shawn T McCormick |
Director | March 19, 2015 | ||
| /s/ David B. Milne David B. Milne |
Director | March 19, 2015 | ||
| /s/ Guido Neels Guido Neels |
Director | March 19, 2015 | ||
123