Attached files
| file | filename |
|---|---|
| 8-K - 8-K - REGENERON PHARMACEUTICALS, INC. | a15-6672_28k.htm |
| EX-99.2 - EX-99.2 - REGENERON PHARMACEUTICALS, INC. | a15-6672_2ex99d2.htm |
Exhibit 99.1
Efficacy and safety of alirocumab 150 mg and 300 mg every 4 weeks in patients with poorly controlled hypercholesterolemia: the ODYSSEY CHOICE I and CHOICE II studies
Erik Stroes,(1) John R. Guyton,(2) Michel Farnier,(3) Daniel Rader,(4) Patrick M. Moriarty,(5) Jean Bergeron,(6) Gisle Langslet,(7) Norman Lepor,(8) Fernando Civeira,(9) Daniel Gaudet,(10) Gerald F. Watts,(11) Garen Manvelian,(12) Guillaume Lecorps,(13) Jian Zhao,(14) MarieT. Baccara-Dinet,(15) Eli M. Roth(16)
(1)Department of Vascular Medicine, Academic Medical Center, Amsterdam, The Netherlands; (2)Duke University Medical Center, Durham, NC, USA; (3)Point Médical, Dijon, France; (4)Perelman School of Medicine, University of Pennsylvania, Smilow Center for Translational Research, Philadelphia, PA, USA; (5)Department of Internal Medicine, Division of Clinical Pharmacology, University of Kansas Medical Center, Kansas City, KS, USA; (6)Clinique des Maladies Lipidiques de Québec Inc., Québec, Canada; (7)Oslo University Hospital, Lipid Clinic, Oslo, Norway; (8)Westside Medical Associates of Los Angeles, Beverly Hills, CA, USA; (9)Lipid Unit, Hospital Universitario Miguel Servet, Zaragoza, Spain; (10)ECOGENE-21 Clinical Trial Center and Department of Medicine, Université de Montréal, Chicoutimi, Quebec, Canada; (11)Lipid Disorders Clinic, Cardiovascular Medicine, Royal Perth Hospital, School of Medicine and Pharmacology, University of Western Australia, Australia; (12)Regeneron Pharmaceuticals Inc., Biostatistics & Data Management, Tarrytown, NY, USA; (13)Sanofi, Paris, France; (14)Regeneron Pharmaceuticals, Inc., Basking Ridge, NJ, USA; (15)Sanofi, Montpellier, France; (16)The Sterling Research Group, Cincinnati, OH, USA
Background
· Despite standard-of-care with statin therapy and addition of other lipid-lowering therapies (LLTs) such as ezetimibe, a large proportion of adult patients with hypercholesterolemia at risk of cardiovascular disease (CVD) do not reach their individual low-density lipoprotein cholesterol (LDL-C) goals.(1),(2) As such, these patients are characterized by a considerable residual CV risk.
· Many patients in need of LLTs are not on statin therapy or are on lower than optimal doses of statins, due to intolerance for (high-dose) statin therapy, mainly muscle-related statin intolerance (MRSI).(3)
· Alirocumab, a fully human monoclonal antibody (mAb) to proprotein convertase subtilisin/kexin type 9 (PCSK9), has been demonstrated to reduce LDL-C levels by 44—63% using doses of 75 or 150 mg every 2 weeks (Q2W) either on background of statins or as monotherapy.
· Every 4 weeks (Q4W) dosing may be a convenient, effective option for some patients, with or without concomitant statin therapy, as suggested by data from a Phase I study of alirocumab dosed Q4W as monotherapy and in combination with non-statin LLTs.(4)
· In the Phase 3 ODYSSEY CHOICE I (NCT01926782) and ODYSSEY CHOICE II (NCT02023879) studies, the efficacy and safety of different doses (150 mg and 300 mg) and dosing frequency of alirocumab (Q2W or Q4W) were investigated to evaluate the potential to offer tailored therapeutic options for patients with hypercholesterolemia, regardless of their current statin therapies and LLT.
Methods
· CHOICE I included patients with poorly controlled hypercholesterolemia and (1) moderate to very-high CV risk and receiving maximally tolerated statin, (2) moderate CV risk and not receiving statin, or (3) moderate to very-high CV risk and MRSI. The study was planned to include approximately two thirds of patients who were receiving statin therapy and one third who were not.
· CHOICE II included patients with hypercholesterolemia not receiving statin but receiving ezetimibe, fenofibrate or diet alone. Patients were only included in the study if they were (1) MRSI with moderate to very-high CV risk or (2) not MRSI with moderate CV risk.
· In both studies, MRSI was defined as the inability to tolerate >2 statins: one statin at the lowest daily starting dose and another statin at any dose, due to skeletal muscle-related symptoms.
· CV risk was defined as very high (documented coronary heart disease [CHD]/CVD), high (no CHD/CVD but with SCORE(5) 10-year risk of fatal CVD >5%, chronic kidney disease, diabetes or heterozygous familial hypercholesterolemia), or moderate (SCORE >1 and <5%).
· Patients were randomized in CHOICE I to alirocumab 300 mg Q4W, alirocumab 75 mg Q2W, or placebo, and in CHOICE II to alirocumab 150 mg Q4W, alirocumab 75 mg Q2W, or placebo. In both studies alirocumab 75 mg Q2W was included as calibrator arm, and in both studies and both alirocumab treatment groups dose/dosing frequency was adjusted at Week (W)12 to 150 mg Q2W if LDL-C goals were not reached at W8 (Figure 1).
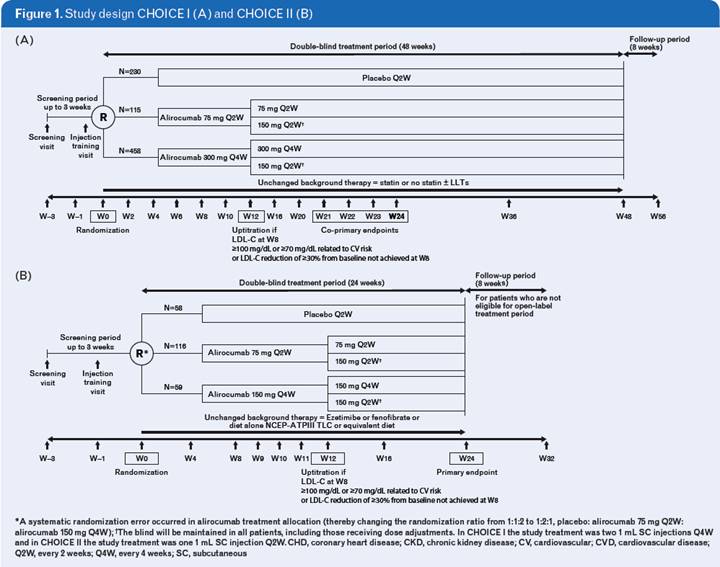
· At W12, the dose regimen changed (using a blinded process) in patients who either did not achieve their predetermined treatment goal (<70 or 100 mg/dL, depending on CV risk) or did not have >30% reduction in LDL-C from baseline at W8.
· The double-blind treatment periods of CHOICE I & II were 48 and 24 weeks, respectively. CHOICE I is ongoing, while CHOICE II has completed the double-blind treatment period, after which patients could opt to enter an open-label extension. Here, we present results from pre-planned analyses for each study (efficacy to W24; safety including all data collected up to cut-off of the last patient’s W24 visit).
· The primary efficacy endpoint for both CHOICE I & II was the % change in calculated LDL-C from baseline to W24.
· In CHOICE I, the co-primary efficacy endpoint was % change in calculated LDL-C from baseline to averaged LDL-C over W21–24.
· Safety parameters were assessed throughout the study.
Results
· Baseline patient characteristics and lipid parameters were generally similar and balanced in the treatment groups of each study (Table 1). Most patients did not have familial hypercholesterolemia; in CHOICE I, as planned, approximately two thirds of patients received statin therapy and one third did not (patients in CHOICE II did not receive statin therapy).
Table 1. Patient characteristics and lipid efficacy parameters at baseline
|
|
|
CHOICE I |
|
CHOICE II |
| ||||||||||||||
|
|
|
No statin group |
|
Statin group |
|
No statin received |
| ||||||||||||
|
|
|
|
|
Alirocumab |
|
|
|
Alirocumab |
|
|
|
Alirocumab |
| ||||||
|
|
|
Placebo |
|
75 mg Q2W |
|
300 mg Q4W |
|
Placebo |
|
75 mg Q2W |
|
300 mg Q4W |
|
Placebo |
|
75 mg Q2W |
|
150 mg Q4W |
|
|
Treatment group |
|
(N=73) |
|
(N=37) |
|
(N=146) |
|
(N=157) |
|
(N=78) |
|
(N=312) |
|
(N=58) |
|
(N=116) |
|
(N=59) |
|
|
Age, years, mean (SD) |
|
59.4 (10.2) |
|
59.3 (11.3) |
|
59.2 (10.8) |
|
61.6 (9.7) |
|
60.7 (9.1) |
|
61.6 (10.0) |
|
63.1 (10.7) |
|
62.5 (9.9) |
|
64.2 (10.0) |
|
|
Male, % |
|
54.8 |
|
37.8 |
|
45.2 |
|
64.3 |
|
65.4 |
|
60.9 |
|
53.4 |
|
59.5 |
|
50.8 |
|
|
Race, white, % |
|
84.9 |
|
86.5 |
|
84.2 |
|
87.3 |
|
87.2 |
|
89.4 |
|
96.6 |
|
93.1 |
|
93.2 |
|
|
BMI group >30 kg/m2, % |
|
63.0 |
|
43.2 |
|
50.7 |
|
47.8 |
|
48.7 |
|
51.6 |
|
35.1 |
|
39.7 |
|
28.8 |
|
|
HeFH, % |
|
1.4 |
|
0 |
|
1.4 |
|
7.6 |
|
7.7 |
|
8.3 |
|
8.6 |
|
12.9 |
|
15.3 |
|
|
Any LLT other than statins, % |
|
45.2 |
|
32.4 |
|
45.2 |
|
32.5 |
|
28.2 |
|
40. |
|
70.7 |
|
70.7 |
|
71.2 |
|
|
LDL-C (calculated), mg/dL, mean (SD) |
|
131.0 (30.4) |
|
148.4 (36.8) |
|
146.1 (33.5) |
|
112.1 (37.3) |
|
114.9 (36.0) |
|
112.4 (32.8) |
|
158.5 (47.3) |
|
154.5 (44.6) |
|
163.9 (69.1) |
|
BMI, body mass index; HeFH, heterozygous familial hypercholesterolemia; SD, standard deviation
· Both studies included a substantial number of patients defined as having MRSI. In CHOICE I, of the patients not receiving statin therapy 42.2% (n=108/256) were not receiving statin due to MRSI. In CHOICE II, none of the patients received a statin (n=233); 90.1% of patients fulfilled the MRSI criteria.
Efficacy results
· In CHOICE I, the % reduction in LDL-C from baseline to W24 was significantly greater with alirocumab 300 mg Q4W (with possible W12 adjustment to 150 mg Q2W) versus placebo both in patients not receiving concurrent statin (least-square (LS) mean difference [standard error, SE] –52.4 [3.3]%) or receiving statin (LS mean difference [SE] versus placebo, –58.7 [2.8]%) (both P<0.0001) (Figures 2A and 2B).
· Furthermore, the average % reduction in LDL-C from baseline to W21 to W24 was significantly greater for the alirocumab 300 mg Q4W arm compared to placebo (no statin group, –55.2%; statin group, –65.0%; both groups, P<0.0001) (Figures 2A and 2B).
· In patients receiving alirocumab 300 mg Q4W (with possible W12 adjustment to 150 mg Q2W), those on concurrent statin had a tendency for lower baseline LDL-C levels and therefore, as expected, achieved lower mean LDL-C levels at W24 compared with those not receiving statin (Figures 2A and 2B).
· In CHOICE II, alirocumab 150 mg Q4W (with possible W12 adjustment to 150 mg Q2W) demonstrated significantly greater LDL-C reductions from baseline to W24 compared with placebo (LS mean difference [SE] –56.4 [3.3]%; P<0.0001) (Figure 2C). The average % reduction in LDL-C from baseline to W9-12 was –55.5% versus placebo (P<0.0001).
· Patients requiring a dose adjustment at W12 (based on W8 LDL-C levels) to achieve guideline-based target levels for LDL-C generally demonstrated higher mean baseline LDL-C levels in both studies (Figure 3).
· Overall, in CHOICE I, only one in five to seven patients required an adjusted dosing regimen from alirocumab 300 mg Q4W to 150 mg Q2W (hence same monthly dose administered) to achieve target LDL-C levels.
· In CHOICE II, one in two patients required an adjusted dosing regimen from 150 mg Q4W to 150 mg Q2W (hence doubling of monthly dose) to achieve target LDL-C levels.
· In CHOICE I, adjusted dose regimen was associated with less variability in LDL-C response from week-to-week (as observed over W21–24), particularly for those patients receiving a statin (Figures 3A and 3B).
· In CHOICE II, for patients who required a dose adjustment from a starting dose of 150 mg Q4W to 150 mg Q2W at W12, an additional reduction in LDL-C of approximately 20% could be observed after dose modification, with patients achieving a mean LDL-C level of 79.0 mg/dL by W24 (from a mean baseline LDL-C level of 197.5 mg/dL) (Figure 3C).


Safety
· In CHOICE I, the rate of treatment emergent adverse events (TEAEs) ranged from 61.1% to 75.0% for the placebo group, and from 71.5% to 78.1% for the alirocumab 300 mg Q4W group. In CHOICE II, TEAEs occurred in 63.8% of placebo-treated patients and 77.6% of those receiving alirocumab 150 mg Q4W (Table 2).
· The frequency of muscle-related symptoms was low and similar between alirocumab- and placebo-treated patients within each study.
· No deaths were reported in either study.
· In both studies, the rate of injection site reactions (ISRs) was higher than previously experienced in other ODYSSEY trials; however, the intensity of most ISRs was mild and most patients experiencing these continued to receive study medication (Table 2).
Table 2. Safety summary
|
|
|
CHOICE I |
|
CHOICE II |
| ||||||||||||||
|
|
|
No statin group |
|
Statin group |
|
No statin received |
| ||||||||||||
|
|
|
|
|
Alirocumab* |
|
|
|
Alirocumab* |
|
|
|
Alirocumab* |
| ||||||
|
|
|
Placebo |
|
75 mg Q2W |
|
300 mg Q4W |
|
Placebo |
|
75 mg Q2W |
|
300 mg Q4W |
|
Placebo |
|
75 mg Q2W |
|
150 mg Q4W |
|
|
Treatment group |
|
(N=72) |
|
(N=37) |
|
(N=146) |
|
(N=157) |
|
(N=78) |
|
(N=312) |
|
(N=58) |
|
(N=116) |
|
(N=59) |
|
|
Subjects with any TEAEs, n (%) |
|
54 (75.0) |
|
30 (81.1) |
|
114 (78.1) |
|
96 (61.1) |
|
50 (64.1) |
|
223 (71.5) |
|
37 (63.8) |
|
84 (73.0) |
|
45 (77.6) |
|
|
Subjects with any treatment-emergent SAE, n (%) |
|
7 (9.7) |
|
3 (8.1) |
|
14 (9.6) |
|
16 (10.2) |
|
6 (7.7) |
|
25 (8.0) |
|
4 (6.9) |
|
6 (5.2) |
|
7 (12.1) |
|
|
Patients with any TEAE leading to discontinuation, n (%) |
|
4 (5.6) |
|
2 (5.4) |
|
10 (6.8) |
|
10 (6.4) |
|
3 (3.8) |
|
15 (4.8) |
|
2 (3.4) |
|
2 (1.7) |
|
4 (6.9) |
|
|
Most frequent TEAEs (recorded in >5% of patients in any group), n (%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Injection site reaction |
|
6 (8.3) |
|
2 (5.4) |
|
27 (18.5) |
|
9 (5.7) |
|
7 (9.0) |
|
48 (15.4) |
|
0 |
|
4 (3.5) |
|
8 (13.8) |
|
|
Headache |
|
4 (5.6) |
|
3 (8.1) |
|
16 (11) |
|
6 (3.8) |
|
3 (3.8) |
|
10 (3.2) |
|
3 (5.2) |
|
10 (8.7) |
|
5 (8.6) |
|
|
Upper respiratory tract infection |
|
4 (5.6) |
|
2 (5.4) |
|
13 (8.9) |
|
5 (3.2) |
|
5 (6.4) |
|
18 (5.8) |
|
4 (6.9) |
|
4 (3.5) |
|
3 (5.2) |
|
|
Sinusitis |
|
6 (8.3) |
|
3 (8.1) |
|
9 (6.2) |
|
2 (1.3) |
|
0 |
|
10 (3.2) |
|
1 (1.7) |
|
1 (0.9) |
|
0 |
|
|
Nasopharyngitis |
|
3 (4.2) |
|
2 (5.4) |
|
7 (4.8) |
|
10 (6.4) |
|
3 (3.8) |
|
23 (7.4) |
|
3 (5.2) |
|
10 (8.7) |
|
5 (8.6) |
|
|
Nausea |
|
3 (4.2) |
|
2 (5.4) |
|
9 (6.2) |
|
10 (6.4) |
|
5 (6.4) |
|
9 (2.9) |
|
2 (3.4) |
|
6 (5.2) |
|
3 (5.2) |
|
|
Arthralgia |
|
3 (4.2) |
|
1 (2.7) |
|
10 (6.8) |
|
9 (5.7) |
|
4 (5.1) |
|
14 (4.5) |
|
2 (3.4) |
|
7 (6.1) |
|
7 (12.1) |
|
|
Pain in extremity |
|
1 (1.4) |
|
1 (2.7) |
|
10 (6.8) |
|
2 (1.3) |
|
2 (2.6) |
|
8 (2.6) |
|
1 (1.7) |
|
4 (3.5) |
|
3 (5.2) |
|
|
Muscle spasms |
|
3 (4.2) |
|
1 (2.7) |
|
4 (2.7) |
|
10 (6.4) |
|
2 (2.6) |
|
4 (1.3) |
|
0 |
|
8 (7.0) |
|
3 (5.2) |
|
|
Fatigue |
|
2 (2.8) |
|
3 (8.1) |
|
7 (4.8) |
|
7 (4.5) |
|
0 |
|
6 (1.9) |
|
0 |
|
5 (4.3) |
|
4 (6.9) |
|
|
Hypertension |
|
6 (8.3) |
|
1 (2.7) |
|
5 (3.4) |
|
5 (3.2) |
|
2 (2.6) |
|
6 (1.9) |
|
2 (3.4) |
|
0 |
|
2 (3.4) |
|
|
Diarrhea |
|
5 (6.9) |
|
0 |
|
9 (6.2) |
|
9 (5.7) |
|
4 (5.1) |
|
12 (3.8) |
|
3 (5.2) |
|
5 (4.3) |
|
1 (1.7) |
|
|
Back pain |
|
5 (6.9) |
|
1 (2.7) |
|
3 (2.1) |
|
6 (3.8) |
|
3 (3.8) |
|
23 (7.4) |
|
0 |
|
6 (5.2) |
|
2 (3.4) |
|
|
Bronchitis |
|
4 (5.6) |
|
2 (5.4) |
|
3 (2.1) |
|
2 (1.3) |
|
2 (2.6) |
|
10 (3.2) |
|
0 |
|
1 (0.9) |
|
1 (1.7) |
|
|
Myalgia |
|
4 (5.6) |
|
1 (2.7) |
|
4 (2.7) |
|
3 (1.9) |
|
1 (1.3) |
|
9 (2.9) |
|
3 (5.2) |
|
7 (6.1) |
|
3 (5.2) |
|
|
Urinary tract infection |
|
2 (2.8) |
|
1 (2.7) |
|
7 (4.8) |
|
4 (2.5) |
|
5 (6.4) |
|
15 (4.8) |
|
1 (1.7) |
|
4 (3.5) |
|
4 (6.9) |
|
|
Dizziness |
|
3 (4.2) |
|
1 (2.7) |
|
5 (3.4) |
|
5 (3.2) |
|
2 (2.6) |
|
11 (3.5) |
|
4 (6.9) |
|
1 (0.9) |
|
4 (6.9) |
|
|
Rash |
|
2 (2.8) |
|
0 |
|
2 (1.4) |
|
0 |
|
1 (1.3) |
|
6 (1.9) |
|
0 |
|
1 (0.9) |
|
3 (5.2) |
|
|
Contusion |
|
2 (2.8) |
|
0 |
|
3 (2.1) |
|
8 (5.1) |
|
1 (1.3) |
|
6 (1.9) |
|
1 (1.7) |
|
0 |
|
1 (1.7) |
|
|
Fall |
|
1 (1.4) |
|
0 |
|
3 (2.1) |
|
2 (1.3) |
|
1 (1.3) |
|
7 (2.2) |
|
2 (3.4) |
|
6 (5.2) |
|
0 |
|
|
Arthropod bite |
|
0 |
|
2 (5.4) |
|
3 (2.1) |
|
1 (0.6) |
|
1 (1.3) |
|
2 (0.6) |
|
0 |
|
0 |
|
1 (1.7) |
|
*Dose/dosing frequency was adjusted at W12 to 150 mg Q2W if LDL-C goals were not reached at W8. SAE, serious adverse event.
Conclusions
· Alirocumab 150 mg and 300 mg Q4W, with possible dose regimen adjustment (to alirocumab 150 mg Q2W) at W12 if goals not reached at W8, demonstrated significant reductions in LDL-C levels versus placebo in patients with inadequately controlled baseline levels of LDL-C. Alirocumab was generally well-tolerated across the study groups.
· The higher ISR rate observed in CHOICE I and II versus earlier ODYSSEY studies could potentially be a factor of two 1 mL injections at each administration (CHOICE I only; previous ODYSSEY studies had one 1 mL injection at each administration) and/or more frequent study visits (both studies).
· Besides confirming good efficacy as well as safety for alirocumab, these data show that dosing of alirocumab aiming to achieve target LDL-C levels can be optimized according to the presence of background statin, baseline LDL-C level, and CV risk (affecting LDL-C target level).
· In aggregate, alirocumab Q4W and Q2W dosing regimens have the potential to allow physicians a choice in selecting an LDL-C-lowering regimen that is tailored to an individual patient’s requirements according to their background statin, LLT status, baseline LDL-C level, and CV risk.
References
1. Hess G, et al. Curr Med Res Opin. 2014;10:1—14
2. Huijgen R, et al. PLoS One. 2010;5:e9220
3. Rosenson RS, et al. J Clin Lipidol. 2014;8:S58—71.
4. Rey J, et al. 2014 ACC Abstract #1183/131 (http://content.onlinejacc.org/article. aspx?articleid=1855597)
5. Reiner Z, et al. Eur Heart J. 2011;32:1769—1818
Acknowledgments
The ODYSSEY CHOICE I and CHOICE II studies were funded by Sanofi and Regeneron. Medical writing support was provided by Susanne Ulm of Prime Medica Ltd., Knutsford, Cheshire, UK, supported by Sanofi and Regeneron.
Disclosures
E. Stroes received research/research grants from BMS, Amgen, Merck, and Sanofi.
J.R. Guyton received consulting/honoraria fees from Amgen Inc., ARMCO, Novella, and Regeneron, and research/research grants from Amarin, Amgen Inc., Regeneron, and Sanofi-Aventis.
M. Farnier has received research support from Amgen, Merck, and Sanofi, speaker’s bureau fees from Amgen, Sanofi, and Merck, honoraria from Abbott, Eli Lilly, and Pfizer, and consultant/advisory board fees from Astra Zenaca, Roche, Kowa, Recordati, SMB, Amgen, Sanofi, and Merck.
D. Rader received consultant/advisory board fees from Sanofi.
P. M. Moriarty received research grants from Pfizer, Catabasis, Espirion, B. Braun, Kaneka, Amgen, Kowa, Lilly, Novartis, Sanofi, Regeneron, and Genzyme, and received honoraria from Amarin and Kowa; is a consultant for Regeneron, Duke Clinical Research Institute, Lilly, Catabasis, B. Braun, Kaneka, and Genzyme.
J. Bergeron received consultant/advisory board fees from Amgen (Canada) and Sanofi (Canada), and gave educational lecture to GPs for Merck (Canada) and Valeant.
G. Langslet received consultant/advisory board fees from Amgen, Sanofi-Aventis, and Janssen Pharmaceuticals.
N. Lepor has received consultant fees/honoraria from Gilead, Quest Diagnostics, and Takeda, has a role in US Medical Innovations, received research/research grants from Amarin, Amgen, Gilead, Novartis, Regeneron, and Sanofi, and is a member of speaker’s bureau for Abbott, Arbor, Astellas Pharma US, Boerhinger-Ingelheim, Bristol Myers Squibb, Eli Lilly/Diachi Sankyo, Gilead, Pfizer, and Vivus.
F. Civeira received grants, consulting fees, and/or honoraria from Amgen, Merck, and Sanofi.
D. Gaudet received consultant/honoraria fees from Amgen, Catabasis, Chiesi, Novartis, Regeneron, and Sanofi-Aventis, and research/research grants from Aegerion Pharmaceuticals, Amgen, Astra Zeneca, Catabasis, Eli Lilly, Genzyme Corporation, ISIS Pharmaceuticals, Merck, Novartis, Pfizer, Regeneron, and Sanofi-Aventis.
G.F. Watts has nothing to disclose.
G. Manvelian is an employee and a stockholder of Regeneron.
G. Lecorps is an employee and stockholder of Sanofi.
J. Zhao is an employee of Regeneron (contractor).
M.T. Baccara-Dinet is an employee of Sanofi.
E.M. Roth is employed by a company that has received research funds and has received consulting fees from Regeneron, Sanofi, and Amgen.
Poster presented at the American College of Cardiology Congress, March 14—16, 2015, San Diego, CA, USA