Attached files
| file | filename |
|---|---|
| 8-K - 8-K - TETRALOGIC PHARMACEUTICALS Corp | a14-14878_28k.htm |
Exhibit 99.1
SUMMARY
Unless the context indicates otherwise, as used in this offering memorandum, the terms “TetraLogic,” “we,” “us,” “our,” “our company” and “our business” refer to TetraLogic Pharmaceuticals Corporation and its subsidiaries.
Overview
We are a clinical-stage biopharmaceutical company focused on discovering, acquiring and developing novel product candidates to treat cancer and infectious diseases. We currently have two clinical-stage product candidates in development: birinapant and SHAPE. Birinapant is designed to mimic the activity of an endogenous protein, the Second Mitochondrial Activator of Caspases, or SMAC, which is involved in the regulation of the apoptotic process within cells. Avoidance of apoptosis is a critical step in tumor development. SHAPE is a histone deacetylase inhibitor, or HDACi. Altered expression of histone deacetylases, or HDACs, has been linked to tumor development and several drugs approved for systemic use in the treatment of cancers take advantage of this mechanism.
Birinapant, our lead clinical-stage product candidate, was selected from our internally-discovered library of over 3,000 small-molecule compounds, including SMAC-mimetics, designed to inhibit Inhibitor of Apoptosis Proteins, or IAPs, and to cause or enable abnormal cells that are resistant to the body’s immune system to self-destruct. This mechanism of action is potentially applicable across a variety of cancers and viral and mycobacterial infections. Birinapant is currently being tested in Phase 1 and Phase 2 clinical trials for multiple solid tumors and hematological malignancies. Our clinical trials of birinapant as of March 31, 2014 have enrolled over 295 subjects. Our composition of matter patents on birinapant in the U.S. expire in 2030 without accounting for any potential patent term extensions.
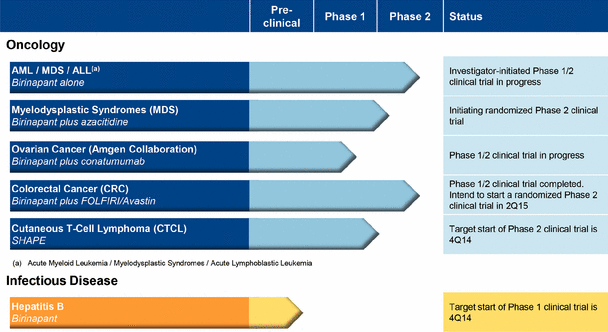
Our clinical and pre-clinical programs with birinapant are focused on:
· myelodysplastic syndromes, or MDS
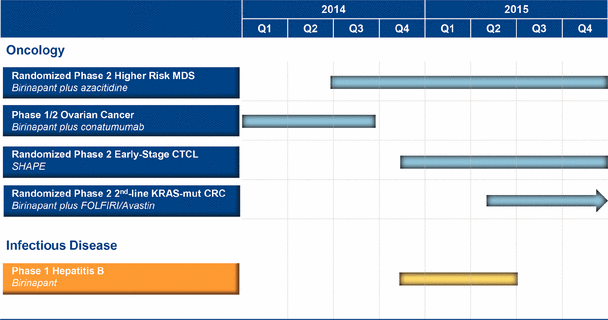
A Phase 1/2 clinical trial of birinapant as a single agent in various blood cancers is ongoing. We also have an ongoing Phase 1 clinical trial of birinapant administered with azacitidine in MDS and are initiating a randomized Phase 2 clinical trial of birinapant administered with azacitidine in MDS.
· colorectal cancer, or CRC
We completed a Phase 1/2 clinical trial of birinapant administered with irinotecan in CRC, and we intend to initiate in the first half of 2015 a randomized clinical trial of birinapant administered with a chemotherapy regimen containing irinotecan plus Avastin in second line KRAS-mutant CRC subjects.
· ovarian cancer
We are conducting a Phase 1/2 clinical trial of birinapant administered with conatumumab in ovarian cancer.
· hepatitis B virus, or HBV
We intend to initiate a Phase 1 clinical trial of birinapant in HBV in the fourth quarter of 2014.
In April 2014, we acquired, by merger, Shape Pharmaceuticals, a development-stage pharmaceutical company developing the product candidate SHAPE. We acquired this company to add a second clinical-stage oncology compound to the TetraLogic portfolio. We paid $13 million in cash at closing and entered into a contingent consideration arrangement that may require us to pay additional consideration, including milestone payments.
SHAPE, our second clinical-stage product candidate, is an HDAC inhibitor being developed for topical use for the treatment of cutaneous T-cell lymphoma, or CTCL. HDAC is a validated cancer target and HDAC inhibitors are a proven class of anti-cancer drugs. SHAPE is a novel therapeutic designed to maximize HDAC inhibition locally in the skin with limited systemic exposure. As a result, SHAPE has characteristics that could allow it to be used topically over large body surface areas with minimal systemic absorption. By potentially avoiding toxicities typical of orally-administered HDACi, SHAPE may provide a more favorable safety profile than an HDAC inhibitor delivered systemically. SHAPE has been evaluated in a randomized, dose escalation placebo-controlled Phase 1 clinical trial in early-stage CTCL subjects that met safety endpoints and demonstrated clinical activity. We plan to commence a randomized Phase 2 clinical trial of SHAPE in subjects with early-stage CTCL by the end of 2014. SHAPE’s composition of matter patent in the U.S. extends until at least 2028; in addition, SHAPE has been granted U.S. orphan drug designation for CTCL. We have acquired worldwide development and commercialization rights to SHAPE for all indications.
Overview of Clinical and Pre-Clinical Programs
The following table sets forth our highest priority clinical programs:
We incurred research and development expenses of $12.1 million and $9.5 million during the years ended December 31, 2012 and 2013, respectively, and $3.1 million during the three months ended March 31, 2014.
MDS
A Phase 1/2 investigator-initiated clinical trial in acute myelogenous leukemia, or AML, MDS and acute lymphoblastic leukemia, or ALL, is ongoing at the University of Pennsylvania and 20 study subjects have been treated with birinapant as the sole agent or administered with hydroxyurea (if deemed necessary by the treating physician). The majority of subjects enrolled are elderly (over 70 years) with AML secondary to MDS and have received multiple prior treatments. In preliminary data, the treatment-related adverse events included Grade 3 and Grade 4 increases in serum levels of the digestive enzymes amylase and lipase, as determined by laboratory testing, with no subject- reported symptoms of abdominal pain. The preliminary data also shows reductions in leukemic blasts (tumor bulk) in some subjects. There were increases in neutrophils with the first birinapant dose in some subjects. One subject continued on treatment with birinapant as sole agent for approximately ten months. Based on the synergy we observed in pre-clinical studies between birinapant and azacitidine, the current standard of care for MDS, and the action of birinapant in subjects with AML secondary to MDS, in August 2013, we initiated a Phase 1 clinical trial of birinapant administered with azacitidine in higher-risk MDS subjects who have relapsed or are refractory to azacitidine. We expanded this clinical trial to include subjects who have not been previously treated with, or are naïve to, azacitidine. The primary objective of this Phase 1 clinical trial is to characterize the safety and tolerability and determine the recommended phase 2 dose of birinapant when administered in combination with azacitidine. Additional objectives of the trial are to assess any preliminary indications of efficacy and pharmacodynamics of the combination.
Based on the currently available data, no dose limiting hematological toxicities of the combination have been reported. A number of subjects who received subcutaneous azacitidine experienced local injection site skin reactions or cellulitis. Although cellulitis is a known effect of azacitidine, several of these were considered to be of increased severity with the combination, possibly reflecting a localized synergistic pharmacodynamic effect in the skin. We believe this toxicity should be mitigated by the use of intravenous, or IV, azacitidine, as no subject whose route of administration has been changed to IV azacitidine experienced further injection site cellulitis. Also, based on data from this trial, the recommended dose of birinapant administered with IV azacitidine for further trials is 13mg/m2 twice weekly for three out of four weeks. Birinapant showed inhibition of NF-κB in circulating blast cells at this dose. Although too early to detect durable responses, preliminary data showed significant bone marrow blast count reductions in three of the first nine evaluable subjects. We are initiating a randomized Phase 2 clinical trial of birinapant administered with azacitidine versus azacitidine alone in first-line higher-risk MDS subjects.
Ovarian Cancer
In pre-clinical studies, we observed synergy between birinapant and TNF- related apoptosis-inducing ligand, or TRAIL, receptor agonist antibodies. In collaboration with Amgen, Inc., or Amgen, we are exploring the combination of birinapant administered with Amgen’s TRAIL receptor agonist antibody, conatumumab. We began dosing subjects in December 2013 in a Phase 1/2 ovarian cancer trial.
CRC
We have results of a Phase 1/2 clinical trial of birinapant administered with irinotecan in 71 CRC subjects who had previously failed standard chemotherapies. The clinical trial showed activity,
with six subjects (8%) showing partial responses, or PRs, defined as at least a 30% decrease in the sum of all measurable tumor lesions by Response Evaluation Criteria in Solid Tumors, or RECIST. RECIST is a set of published rules that define when cancer patients improve (or respond), stay the same (or stabilize), or worsen (or progress) during treatment. The median progression-free survival, or PFS, was 2.2 months. 34% of study subjects were alive without progression of their tumor at four months and 21% were alive without progression of their tumor at six months. The combination of birinapant administered with irinotecan was generally well tolerated. Compared to treatment with irinotecan alone, birinapant administered with irinotecan led to a modest increase in anemia (or a decrease in red blood cells) and a modest increase in thrombocytopenia (or a decrease in platelets). Irinotecan is one of the chemotherapies that induces Tumor Necrosis Factor, an extracellular signaling molecule that induces apoptosis. As the majority of subjects had disease progression on prior irinotecan treatment (65 of 71, or 92%), we believe that this data supports the view that the activity seen in this study is being driven by the synergistic effect of birinapant and irinotecan. In addition, of the 71 subjects in this Phase 1/2 clinical trial, 37 CRC subjects with KRAS-mutant tumors who had previously failed an irinotecan based therapy were treated with birinapant and irinotecan. Of these 37 subjects, three subjects (8%) demonstrated PRs, 38% of subjects were alive without progression of their disease at four months, and 24% of subjects were alive without progression of disease at six months. We believe that this data may suggest activity from the combination of birinapant administered with irinotecan in the KRAS-mutant population and provides a rationale for targeting this underserved group of patients who are not eligible for EGFR antibody therapy because of their KRAS-mutant status. Based on the clinical data that has emerged from the study of birinapant, we intend to commence in the first half of 2015 a randomized Phase 2 clinical trial of birinapant administered with the irinotecan-containing chemotherapy regimen FOLFIRI (5-fluorouracil, irinotecan, and leucovorin) plus Avastin in second-line KRAS-mutant CRC subjects, meaning those who have already failed a prior treatment regimen for advanced disease.
CTCL
SHAPE, our second clinical-stage product candidate, is an HDAC inhibitor being developed for topical use for the treatment of CTCL. SHAPE is a novel therapeutic intentionally designed to maximize HDAC inhibition locally in the skin with limited systemic exposure. As a result, SHAPE has characteristics that could allow it to be used topically over large body surface areas with minimal systemic absorption. By potentially avoiding toxicities typical of orally- administered HDACi, SHAPE may provide a more favorable safety profile than an HDAC inhibitor delivered systemically. SHAPE has been evaluated in a randomized, dose escalation placebo-controlled Phase 1 clinical trial in early-stage CTCL subjects that met safety endpoints and demonstrated clinical activity. Four of 15 patients receiving SHAPE attained an objective response as measured by a greater than 50% improvement in their Composite Assessment of Index Lesion Severity, or CAILS, score during and after 28 days of dosing. No placebo patients responded. We plan to commence a randomized Phase 2 clinical trial of SHAPE in subjects with early-stage CTCL by the end of 2014.
HBV
Hepatitis B is a liver disease that results from infection with HBV. In pre-clinical studies, birinapant significantly reduced HBV. The clearance was additive when given in combination with entecavir, the standard of care therapy for HBV. Birinapant caused a decline in HBV surface antigen whereas entecavir did not, implying a different mechanism of action. These pre-clinical studies are ongoing to understand the action of birinapant in greater detail in HBV, and to determine the spectrum of potential therapeutic activity of birinapant in other infectious diseases. Consistent with these results, birinapant demonstrated activity at clinically achievable study drug exposures in studies of human immunodeficiency virus, or HIV, in human blood cells in vitro. It was also active in a mouse model of tuberculosis and in a mouse model of Legionella. We intend to continue pre-clinical studies and
regulatory activities and intend to initiate a Phase 1 clinical trial in the fourth quarter of 2014. We have entered into a research collaboration with the Walter and Eliza Hall Institute of Medical Research, or WEHI, based in Melbourne, Australia, to examine SMAC-mimetics, including birinapant, in the treatment of infectious disease. We have entered into a license agreement with WEHI for worldwide exclusive rights to a patent application relating to a method of treating intracellular infections with administration of an IAP inhibitor.
The following table sets forth our expected timetable for our programs in oncology and infectious disease:
Our Strategy
Our goal is to maximize the potential value of both birinapant as a first- in-class and best-in-class SMAC-mimetic and SHAPE as a novel skin-targeted HDACi. The key elements of our strategy to achieve this goal include:
· pursuing regulatory approval for birinapant administered with other therapies for the treatment of first-line higher-risk MDS. The data from our randomized Phase 2 clinical trial will determine the size of the treatment effect of birinapant administered with azacitidine versus azacitidine alone and will form the basis of a Phase 3 clinical trial in first-line higher-risk MDS;
· pursuing regulatory approval for birinapant administered with FOLFIRI for treatment of second-line KRAS-mutant CRC. We plan to initiate a randomized clinical trial in the first half of 2015;
· continuing a Phase 1/2 clinical trial started in December 2013 with birinapant administered with conatumumab in ovarian cancer;
· continuing our pre-clinical studies of birinapant as a potential antiviral therapeutic agent, with the intent of starting an antiviral clinical trial in the fourth quarter of 2014;
· initiating a randomized Phase 2 clinical trial of SHAPE in subjects with early-stage CTCL, with a target start date in the fourth quarter of 2014;
· considering collaborations to accelerate development of our clinical programs outside of the U.S.; and
· in-licensing or acquisitions of assets and companies to expand our existing technologies and operations.
Other elements of our business strategy include exploiting our understanding of the role of SMAC-mimetics more broadly in infectious disease, leveraging our library of SMAC-mimetic compounds to develop novel molecules to expand the utility of this developing class and pursuing potential collaborations.
Our Corporate Information
We were originally incorporated in July 2001 as Apop Corp., a New Jersey corporation. Pursuant to a merger effective as of October 2003 with and into our wholly owned subsidiary, we became a Delaware corporation and commenced operations in 2003. Our name was changed to Apop Corporation in March 2004, to Gentara Corporation in June 2004 and to TetraLogic Pharmaceuticals Corporation in January 2006. Our website address is http://www.tlog.com. The inclusion of our website address here and elsewhere in this offering memorandum is, in each case, intended to be an inactive textual reference only and not an active hyperlink to our website. The information contained in, or that can be accessed through, our website is not part of this offering memorandum. Our main offices are located at 343 Phoenixville Pike, Malvern, Pennsylvania 19355, and our telephone number is (610) 889-9900.
SUMMARY FINANCIAL DATA
The summary of our financial data set forth below should be read together with our financial statements and accompanying notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report on Form 10-K for the year ended December 31, 2013 and in our Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2014, and our Current Report on Form 8-K including certain pro forma financial information related to our acquisition of Shape Pharmaceuticals and historical financial statements of Shape Pharmaceuticals filed with the SEC on April 14, 2014, which are incorporated by reference in this offering memorandum.
We have derived the below statement of operations data for the years ended December 31, 2012 and 2013 from our audited financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2013, incorporated by reference herein. We have derived the below statement of operations data for the three-month periods ended March 31, 2013 and 2014 and balance sheet data as of March 31, 2014 from our unaudited financial statements included in our Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2014, incorporated by reference herein.
The unaudited pro forma statement of operations data for the year ended December 31, 2013 and the three-month period ended March 31, 2014 and pro forma balance sheet data as of March 31, 2014 are based on our historical financial statements and Shape Pharmaceuticals’ historical consolidated financial statements to give effect to our acquisition of Shape Pharmaceuticals on April 14, 2014. The unaudited pro forma statement of operations data for the year ended December 31, 2013 and the three-month period ended March 31, 2014 give effect to the acquisition of Shape Pharmaceuticals as if it had occurred at the beginning of each such periods. The unaudited pro forma balance sheet data as of March 31, 2014 gives effect to the acquisition of Shape Pharmaceuticals as if it had occurred on March 31, 2014. The unaudited pro forma financial data reflect pro forma adjustments that are based upon available information and certain assumptions that management believes are reasonable. The unaudited pro forma financial data do not purport to represent our results of operations and financial position that would have resulted had the transactions to which pro forma effects are given been consummated as of the date or for the periods indicated.
Our historical results are not necessarily indicative of the results that may be expected in the future, and our interim period results are not necessarily indicative of results to be expected for a full year or any other interim period.
|
|
|
Year Ended December 31, |
|
Three Months Ended March 31, |
| ||||||||||||||
|
|
|
2012 |
|
2013 |
|
2013 |
|
2013 |
|
2014 |
|
2014 |
| ||||||
|
Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Revenue |
|
$ |
— |
|
$ |
— |
|
$ |
600,000 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
Expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
General and administrative |
|
4,075,649 |
|
8,467,467 |
|
8,871,611 |
|
977,918 |
|
2,508,285 |
|
2,654,612 |
| ||||||
|
Research and development |
|
12,096,278 |
|
9,523,427 |
|
11,431,580 |
|
2,243,464 |
|
3,122,749 |
|
3,465,491 |
| ||||||
|
Total expenses |
|
16,171,927 |
|
17,990,894 |
|
20,303,191 |
|
3,221,382 |
|
5,631,034 |
|
6,120,103 |
| ||||||
|
Loss from operations |
|
(16,171,927 |
) |
(17,990,894 |
) |
(19,703,191 |
) |
(3,221,382 |
) |
(5,631,034 |
) |
(6,120,103 |
) | ||||||
|
Change in fair value of derivative liabilities |
|
43,136 |
|
807,291 |
|
807,291 |
|
— |
|
285,021 |
|
285,021 |
| ||||||
|
Interest and other income |
|
2,694 |
|
629 |
|
3,288 |
|
5 |
|
11,946 |
|
11,946 |
| ||||||
|
Interest expense |
|
(73,353 |
) |
(2,767,422 |
) |
(2,767,422 |
) |
(207,295 |
) |
— |
|
(452 |
) | ||||||
|
Net loss |
|
(16,199,450 |
) |
(19,950,396 |
) |
(21,660,034 |
) |
(3,428,672 |
) |
(5,334,067 |
) |
(5,823,588 |
) | ||||||
|
Cumulative preferred stock dividends |
|
(3,453,412 |
) |
(3,249,948 |
) |
(3,249,948 |
) |
(851,526 |
) |
— |
|
— |
| ||||||
|
Net loss attributable to common stockholders |
|
$ |
(19,652,862 |
) |
$ |
(23,200,344 |
) |
$ |
(24,909,982 |
) |
$ |
(4,280,198 |
) |
$ |
(5,334,067 |
) |
$ |
(5,823,588 |
) |
|
Per share information: |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Net loss per share of common stock - basic |
|
$ |
(20.26 |
) |
$ |
(10.11 |
) |
$ |
(10.85 |
) |
$ |
(3.89 |
) |
$ |
(0.24 |
) |
$ |
(0.26 |
) |
|
Average weighted common shares outstanding - basic |
|
969,998 |
|
2,295,636 |
|
2,295,636 |
|
1,099,785 |
|
22,249,474 |
|
22,249,474 |
| ||||||
|
Net loss per share of common stock - diluted |
|
$ |
(20.26 |
) |
$ |
(10.11 |
) |
$ |
(10.85 |
) |
$ |
(3.89 |
) |
$ |
(0.25 |
) |
$ |
(0.27 |
) |
|
Average weighted common shares outstanding - diluted |
|
969,998 |
|
2,295,636 |
|
2,295,636 |
|
1,099,785 |
|
22,321,473 |
|
22,321,473 |
| ||||||
|
|
|
Three Months Ended March 31, 2014 |
| ||||
|
|
|
|
|
(Pro Forma) |
| ||
|
Balance Sheet Data: |
|
|
|
|
| ||
|
Cash, cash equivalents and investments |
|
$ |
48,838,530 |
|
$ |
35,990,612 |
|
|
Total assets |
|
50,648,100 |
|
78,320,863 |
| ||
|
Total liabilities |
|
4,524,007 |
|
32,196,770 |
| ||
|
Deficit accumulated during the development stage |
|
(95,206,305 |
) |
(95,206,305 |
) | ||
|
Total stockholders’ equity (deficit) |
|
46,124,093 |
|
46,124,093 |
| ||
RISK FACTORS
Risks Related to Our Business and Industry
Our future success is dependent on the successful clinical development, regulatory approval and commercialization of our product candidates, birinapant and SHAPE, which are currently undergoing clinical trials and will require significant capital resources and years of additional clinical development effort.
We do not have any products that have gained regulatory approval. Currently, our only clinical-stage product candidates are birinapant and SHAPE. As a result, our business is dependent on our ability to successfully complete clinical development of, obtain regulatory approval for, and, if approved, to successfully commercialize our product candidates in a timely manner. We cannot commercialize our product candidates in the U.S. without first obtaining regulatory approval from the FDA; similarly, we cannot commercialize our product candidates outside of the U.S. without obtaining regulatory approval from comparable foreign regulatory authorities. Before obtaining regulatory approvals for the commercial sale of our product candidates for any target indication, we must demonstrate with substantial evidence gathered in pre-clinical studies and well-controlled clinical trials, and, with respect to approval in the U.S., to the satisfaction of the FDA, that our product candidates are safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate. Even if our product candidates were to successfully obtain approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. If we are unable to obtain regulatory approval for our product candidates in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding or generate sufficient revenue to continue the development of any other product candidate that we may discover, in-license, develop or acquire in the future. Furthermore, even if we obtain regulatory approval for our product candidates, we will still need to develop a commercial organization, establish commercially viable pricing and obtain approval for adequate reimbursement from third-party and government payors. If we are unable to successfully commercialize our product candidates, we may not be able to earn sufficient revenues to continue our business.
We will need to grow the size and complexity of our organization as a result of our acquisition of Shape Pharmaceuticals, and we may experience difficulties in managing this growth or in maintaining our focus on both birinapant and SHAPE.
As a result of adding another product candidate to our portfolio, we will need to increase the size and complexity of our organization to assist with the expansion of our operations. This will involve adding resources to manage both our existing and our newly acquired product candidate. Our failure to manage this growth effectively could have an adverse effect on our ability to accomplish our development and commercialization objectives for our product candidates.
Because the results of pre-clinical studies or earlier clinical trials are not necessarily predictive of future results, our product candidates may not have favorable results in later clinical trials or receive regulatory approval.
Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will generate adequate data to demonstrate the efficacy and safety of our product candidates. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in clinical trials, even after seeing promising results in earlier clinical trials. Despite the results reported in earlier clinical trials for our product candidates, we do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market our product candidates in any particular jurisdiction. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for our product candidates may be adversely impacted.
The therapeutic efficacy of our product candidates is unproven in humans, and we may not be able to successfully develop and commercialize our product candidates pursuant to these programs.
Birinapant and SHAPE are novel compounds and their potential benefit as a therapeutic cancer or antiviral drug, as the case may be, is unproven. Our ability to generate revenues from our product candidates, which we do not expect will occur in the short term, if ever, will depend heavily on their successful development and commercialization after approval, if achieved, which is subject to many potential risks. For example, birinapant or SHAPE may not prove to be an effective inhibitor of the cancer or viral targets, as the case may be, that each is being designed to act against and may not demonstrate in study subjects any or all of the pharmacological data points that may have been demonstrated in pre-clinical studies or earlier clinical trials. Birinapant or SHAPE may interact with human biological systems in unforeseen, ineffective or harmful ways. If birinapant or SHAPE is associated with undesirable side effects or has characteristics that are unexpected, we may need to abandon its development or limit development to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Many compounds that initially showed promise in early stage testing for treating cancer or infectious diseases have later been found to cause side effects that prevented further development of such compounds. As a result of these and other risks described herein that are inherent in the development of novel therapeutic agents, we may never successfully develop, enter into or maintain third-party licensing or collaboration transactions with respect to, or successfully commercialize our product candidates, in which case we will not achieve profitability and the value of our stock may decline.
Clinical development of product candidates involves a lengthy and expensive process with an uncertain outcome.
Clinical testing is expensive, can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through pre-clinical studies and early clinical trials.
We may experience delays in our ongoing or future clinical trials and we do not know whether planned clinical trials will begin or enroll subjects on time, need to be redesigned or be completed on schedule, if at all. There can be no assurance that the FDA or other foreign regulator authority will not put clinical trials of birinapant, SHAPE or any other product candidates on clinical hold now or in the
future. Clinical trials may be delayed, suspended or prematurely terminated for a variety of reasons, such as:
· delay or failure in reaching agreement with the FDA or a comparable foreign regulatory authority on a trial design that we are able to execute;
· delay or failure in obtaining authorization to commence a trial or inability to comply with conditions imposed by a regulatory authority regarding the scope or design of a clinical trial;
· delay or failure in reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
· delay or failure in obtaining institutional review board, or IRB, approval or the approval of other reviewing entities, including comparable foreign regulatory authorities, to conduct a clinical trial at each site;
· withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials;
· delay or failure in recruiting and enrolling suitable study subjects to participate in a trial;
· delay or failure in study subjects completing a trial or returning for post-treatment follow-up;
· clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial;
· inability to identify and maintain a sufficient number of trial sites, many of which may already be engaged in other clinical trial programs, including some that may be for competing product candidates with the same indication;
· failure of our third-party clinical trial managers to satisfy their contractual duties or meet expected deadlines;
· delay or failure in adding new clinical trial sites;
· ambiguous or negative interim results or results that are inconsistent with earlier results;
· feedback from the FDA, the IRB, data safety monitoring boards, or a comparable foreign regulatory authority, or results from earlier stage or concurrent pre-clinical studies and clinical trials, that might require modification to the protocol for the trial;
· decision by the FDA, the IRB, a comparable foreign regulatory authority, or us, or recommendation by a data safety monitoring board or comparable foreign regulatory authority, to suspend or terminate clinical trials at any time for safety issues or for any other reason;
· unacceptable risk-benefit profile, unforeseen safety issues or adverse side effects or adverse events;
· failure of a product candidate to demonstrate any benefit;
· difficulties in manufacturing or obtaining from third parties sufficient quantities of a product candidate for use in clinical trials;
· lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional clinical studies or increased expenses associated with the services of our CROs and other third parties; or
· changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Study subject enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the subject population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, ability to obtain and maintain subject consents, risk that enrolled subjects will drop out before completion, competing clinical trials and clinicians’ and subjects’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved or product candidates that may be studied in competing clinical trials for the indications we are investigating. We rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials, and while we have agreements governing their committed activities, we have limited influence over their actual performance.
If we experience delays in the completion of any clinical trial of our product candidates, their commercial prospects might be harmed, and our ability to generate product revenues from our product candidates, if approved, will be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our development and approval process for our product candidates and jeopardize our ability to commence product sales and generate revenues. In addition, many of the factors that could cause a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
Our commercial success depends upon attaining significant market acceptance of our product candidates, if approved, among physicians, patients, healthcare payors and the major operators of cancer or infectious disease clinics.
Even if we obtain regulatory approval for our product candidates, they may not gain market acceptance among physicians, healthcare payors, patients or the medical community. Market acceptance of our product candidates, if we receive approval, depends on a number of factors, including the:
· efficacy and safety of such product candidate or such product candidate administered with other drugs, each as demonstrated in clinical trials and post-marketing experience;
· clinical indications for which such product candidate is approved;
· acceptance by physicians, major operators of cancer or infectious disease clinics and patients of the product candidate as a safe and effective treatment;
· potential and perceived advantages of such product candidate over alternative treatments;
· safety of such product candidate seen in a broader patient group, including its use outside the approved indications should physicians choose to prescribe for such uses;
· prevalence and severity of any side effects;
· product labeling or product insert requirements of the FDA or other regulatory authorities;
· timing of market introduction of such product candidate as well as competitive products;
· cost of treatment in relation to alternative treatments;
· availability of coverage and adequate reimbursement and pricing by third-party payors and government authorities;
· relative convenience and ease of administration; and
· effectiveness of our sales and marketing efforts.
Moreover, if either birinapant or SHAPE is approved but fails to achieve market acceptance among physicians, patients, or healthcare payors or the products or product candidates that are being administered with birinapant are restricted, withdrawn or recalled or fail to be approved, as the case may be, we may not be able to generate significant revenues, which would compromise our ability to become profitable.
Our commercial success could depend upon the continued marketing of an approved product, or the approval of a product candidate, to be administered with birinapant.
Some of our clinical trials involve products marketed, or product candidates being developed, by other pharmaceutical companies and some of the indications for which we are developing birinapant involve its use in combination with these other products and product candidates. These products or product candidates may be administered in a clinical trial in combination with birinapant. In the event that any of these pharmaceutical companies have unforeseen issues that negatively impact their clinical development or marketing approval for these products and product candidates or otherwise negatively affect their ability to continue to clinically develop or market these products and product candidates, our ability to complete our applicable clinical trials and/or evaluate clinical results and, ultimately, our ability to receive regulatory approval for birinapant for the indications we are pursuing may also be negatively impacted. As a result, this could adversely affect our ability to file for, gain or maintain regulatory approvals for birinapant on a timely basis, if at all.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any marketing approval.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authorities. For example, even though birinapant has generally been well tolerated by subjects in our earlier-stage clinical trials, in some cases there were side effects, some of which were severe. In clinical trials where birinapant was administered as monotherapy, treatment-related side effects that were Grade 3 or Grade 4, meaning they were more than mild or moderate in severity, include increase of serum levels of
amylase or lipase protein concentrations, fatigue, headache, hypophosphatemia, lymphopenia, nausea, rash, thrombocytopenia and vomiting. Treatment-related Grade 3 or Grade 4 side effects observed in trials where birinapant was administered with other cancer therapies included abdominal cellulitis, abdominal pain, alanine aminotransferase increase, amylase increase, anemia, aspartate aminotransferase increase, caecitis, dehydration, diarrhea, dyspnea, fatigue, febrile neutropenia, granulocytopenia, headache, hyponatraemia, hypotension, leukopenia, lipase increase, lymphocytopenia, mucositis, nausea, neutropenia, pancytopenia, sepsis, stomatitis, stress cardiomyopathy, thrombocytopenia, vomiting, and weight decrease. In dose escalation studies of birinapant combined with chemotherapies that deliberately sought to define the dose-limiting toxicities, and thus the maximum tolerated dose, the most common dose-limiting side effect was Grade 2 Bell’s Palsy, or weakness or inability to control facial muscles on one side of the face. Minor side effects have been reported in some cases in clinical trials of SHAPE, including blisters, erythema, itching and burning sensations.
As a result of these side effects or further safety or toxicity issues that we may experience in our clinical trials in the future, we may not receive approval to market our product candidates, which could prevent us from ever generating revenues or achieving profitability. Results of our trials could reveal an unacceptably high severity and prevalence of side effects. In such an event, our trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The study drug-related side effects could affect study subject recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims.
Additionally, if our product candidates receive marketing approval, and we or others later identify undesirable side effects caused by our product candidates, a number of potentially significant negative consequences could result, including:
· we may be forced to suspend marketing of the product candidate;
· regulatory authorities may withdraw their approvals of the product candidate;
· regulatory authorities may require additional warnings on the label that could diminish the usage or otherwise limit the commercial success of the product candidate;
· we may be required to conduct post-market studies;
· we could be sued and held liable for harm caused to subjects or patients; and
· our reputation may suffer.
Any of these events could prevent us from achieving or maintaining market acceptance of our product candidates, if approved.
Even if our product candidates receive regulatory approval, we may still face future development and regulatory difficulties.
Even if we obtain regulatory approval for our product candidates, they would be subject to ongoing requirements by the FDA and comparable foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-market information. The safety profile of our product candidates will continue to be closely
monitored by the FDA and comparable foreign regulatory authorities after approval. If new safety information becomes available after approval of our product candidates, the FDA or comparable foreign regulatory authorities may require labeling changes or establishment of a Risk Evaluation and Mitigation Strategy, or similar strategy, impose significant restrictions on indicated uses or marketing, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. For example, the label ultimately approved for our product candidates, if they achieve marketing approval, may include restrictions on use.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or cGMP, and other regulations. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, birinapant, SHAPE or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
· issue warning letters or untitled letters;
· mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners;
· require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance;
· seek an injunction or impose civil or criminal penalties or monetary fines;
· suspend or withdraw regulatory approval;
· suspend any ongoing clinical trials;
· refuse to approve pending applications or supplements to applications filed by us;
· suspend or impose restrictions on operations, including costly new manufacturing requirements; or
· seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall.
The occurrence of any event or penalty described above may inhibit or preclude our ability to commercialize our product candidates and generate revenue.
Advertising and promotion of any product candidate that obtains approval in the U.S. will be heavily scrutinized by, among others, the FDA, the Department of Justice, or DOJ, the Office of the Inspector General of the Department of Health and Human Services, or HHS, state attorneys general, members of Congress and the public. Violations, including promotion of our products for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil and criminal sanctions by the FDA or other government agencies. Additionally, advertising and promotion of any
product candidate that obtains approval outside of the U.S. will be heavily scrutinized by comparable foreign regulatory authorities.
In the U.S., engaging in impermissible promotion of our product candidates for off-label uses can also subject us to false claims litigation under federal and state statutes, and other litigation and/or investigation, which can lead to civil and criminal penalties and fines and agreements that materially restrict the manner in which we promote or distribute our drug products. These false claims statutes include the federal False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims, or causing to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government prevails in the lawsuit, the individual will share in any fines or settlement funds. Since 2004, these False Claims Act lawsuits against pharmaceutical companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements based on certain sales practices promoting off-label drug uses. This increasing focus and scrutiny has increased the risk that a pharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, agree to comply with burdensome reporting and compliance obligations, and be excluded from the Medicare, Medicaid and other federal and state healthcare programs. If we do not lawfully promote our approved products, we may become subject to such litigation and/or investigation and, if we are not successful in defending against such actions, those actions could compromise our ability to become profitable.
Failure to obtain regulatory approval in international jurisdictions would prevent our product candidates from being marketed abroad.
In order to market and sell our products in the European Union and many other jurisdictions, including Japan and South Korea, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the U.S. generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the U.S., it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the U.S. on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the U.S. does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market. If we are unable to obtain approval of our product candidates by regulatory authorities in the European Union, Japan, South Korea or another country or jurisdiction, the commercial prospects of our product candidates may be significantly diminished and our business prospects could decline.
Recently enacted and future legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
The regulations that govern, among other things, marketing approvals, coverage, pricing and reimbursement for new drug products vary widely from country to country. In the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to successfully sell our product candidates if we may obtain marketing approval.
In the U.S., the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or Medicare Modernization Act, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for physician administered drugs. In recent years, Congress has considered further reductions in Medicare reimbursement for drugs administered by physicians. The Centers for Medicare and Medicaid Services, the agency that runs the Medicare program, also has the authority to revise reimbursement rates and to implement coverage restrictions for some drugs. Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products, which in turn would affect the price we can receive for those products. While the Medicare Modernization Act and Medicare regulations apply only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010, or the Affordable Care Act, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers and impose additional health policy reforms. The Affordable Care Act expanded manufacturers’ rebate liability to include covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations, increased the minimum rebate due for innovator drugs from 15.1% of average manufacturer price, or AMP, to 23.1% of AMP and capped the total rebate amount for innovator drugs at 100.0% of AMP. The Affordable Care Act and subsequent legislation also changed the definition of AMP. Furthermore, the Affordable Care Act imposes a significant annual, nondeductible fee on companies that manufacture or import certain branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may affect our business practices with healthcare practitioners, and a significant number of provisions are not yet, or have only recently become, effective. Although it is too early to determine the effect of the Affordable Care Act, it appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. In August 2011, President Obama signed into law the Budget Control Act of 2011, which, among other things, creates the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of an amount greater than $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, starting in 2013. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several categories of healthcare providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. If we ever obtain regulatory approval and commercialization of our product candidates, these new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our financial operations. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates may be.
In the U.S., the European Union and other potentially significant markets for our product candidates, government authorities and third-party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Furthermore, the increased emphasis on managed healthcare in the U.S. and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general.
Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for one of our product candidates in a particular country, but then be subject to price regulations that delay our commercial launch of such product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of such product in that particular country. Adverse pricing limitations may hinder our ability to recoup our investment in our product candidates even if they obtain marketing approval.
Laws and regulations governing international operations may preclude us from developing, manufacturing and selling product candidates outside of the U.S. and require us to develop and implement costly compliance programs.
As we seek to expand our operations outside of the U.S., we must comply with numerous laws and regulations in each jurisdiction in which we plan to operate. The creation and implementation of international business practices compliance programs is costly and such programs are difficult to enforce, particularly where reliance on third parties is required.
The Foreign Corrupt Practices Act of 1977, or FCPA, prohibits any U.S. individual or business from paying, offering, authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the U.S. to comply with certain accounting provisions requiring such companies to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. The anti-bribery provisions of the FCPA are enforced primarily by the DOJ. The SEC is involved with enforcement of the books and records provisions of the FCPA.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the U.S., or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. Our
expanding presence outside of the U.S. will require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling our product candidates outside of the U.S., which could limit our growth potential and increase our development costs.
The failure to comply with laws governing international business practices may result in substantial penalties, including suspension or debarment from government contracting. Violation of the FCPA can result in significant civil and criminal penalties. Indictment alone under the FCPA can lead to suspension of the right to do business with the U.S. government until the pending claims are resolved. Conviction of a violation of the FCPA can result in long-term disqualification as a government contractor. The termination of a government contract or relationship as a result of our failure to satisfy any of our obligations under laws governing international business practices would have a negative impact on our operations and harm our reputation and ability to procure government contracts. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
Even if we are able to commercialize our product candidates, these products may not receive coverage and adequate reimbursement from third-party payors, which could harm our business.
Our ability to commercialize our product candidates successfully will depend, in part, on the extent to which coverage and adequate reimbursement for our product candidates and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical drugs. Third-party payors may also seek additional clinical evidence, beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations before covering product candidates for those patients. We cannot be sure that coverage and adequate reimbursement will be available for our product candidates and, if reimbursement is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, our product candidates, if we obtain marketing approval. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates, if we obtain marketing approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved
products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate any revenue.
We do not currently have an organization for the sale, marketing and distribution of pharmaceutical products and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market any products approved by the FDA or comparable foreign regulatory authorities, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable. We will be competing with many companies that currently have extensive and well-funded sales and marketing operations. Without an internal commercial organization or the support of a third party to perform sales and marketing functions, we may be unable to compete successfully against these more established companies.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may affect the business or financial arrangements and relationships through which we would market, sell and distribute our products. Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our operations (including our marketing, promotion, educational programs, pricing, and relationships with healthcare providers or other entities, among other things) and expose us to areas of risk include the following:
· the federal healthcare Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
· federal civil and criminal false claims laws and civil monetary penalty laws impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
· the Health Insurance Portability and Accountability Act of 1996, or HIPAA, and the rules and regulations promulgated thereunder, establish federal standards for maintaining the privacy and security of certain patient health information known as Protected Health Information. As amended by the Health Information Technology for Economic and
Clinical Health Act, HIPAA also establishes federal standards for administrative, technical and physical safeguards relevant to the electronic transmission of Protected Health Information and imposes certain notification obligations in the event of a breach of the privacy or security of Protected Health Information. In addition to adhering to the requirements of HIPAA, entities considered “covered entities” under HIPAA (such as health plans, health care clearinghouses, and certain health care providers) are also required to obtain assurances in the form of a written contract from certain business associates to which they transmit Protected Health Information to ensure that the privacy and security of such information is maintained in accordance with HIPAA requirements;
· HIPAA also criminalizes health care fraud and makes it a felony to knowingly and willfully execute or attempt to execute a scheme or artifice to defraud any health care benefit program or to obtain money or other property owned or controlled by a health care benefit program by means of false or fraudulent pretenses, representations, or promises;
· failure to comply with HIPAA can result in civil and criminal liability, including civil money penalties, fines and imprisonment;
· the federal physician sunshine requirements under the Affordable Care Act requires manufacturers of drugs, devices, biologics and medical supplies to report annually to HHS information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations; and
· analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our business arrangements with third parties are compliant with applicable healthcare laws and regulations will involve the expenditure of appropriate, and possibly significant, resources. Nonetheless, it is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct for our directors, officers and employees, or the Code of Conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our product candidates and will face competition with respect to any other product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of products for the treatment of the disease indications for which we are developing birinapant, as well as for the treatment of early-stage CTCL for which we are developing SHAPE. Some of these competitive products and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. For example, there are several companies developing product candidates that target the same cancer pathways that we are targeting or that are testing product candidates in the same cancer indications that we are testing. For example, Curis Inc., or Curis (Phase 1), Debiopharma SA, or Debiopharma (Phase 1), and Novartis AG, or Novartis (Phase 2), are all developing IAP inhibitors. Similarly, Merck (Zolinza) and Celgene (Istodax) have each received approval for HDAC inhibitors as therapeutic options for patients with advanced CTCL, and Eisai (Targretin) and Actelion (Valchlor) have been approved as topical treatments for early-stage CTCL. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Birinapant and SHAPE are presently being developed primarily as cancer therapeutics. There are a variety of available therapies and supportive care products marketed for cancer patients. Some of these other drugs are branded and subject to patent protection, some are in clinical development and not yet approved, and others are available on a generic basis. Many of the approved drugs are well established therapies or products and are widely accepted by physicians, patients and third-party payors. Insurers and other third-party payors may also encourage the use of generic products. In addition, birinapant is delivered intravenously, which will require a visit to an oncologist office or a hospital. Some of our competitors are seeking to develop drugs that can be administered by oral delivery, and
thus would not require a visit to a doctor for each administration. These factors may make it difficult for us to achieve market acceptance at desired levels and/or in a timely manner to ensure viability of our business.
More established companies may have a competitive advantage over us due to their greater size, cash flows and institutional experience. Compared to us, many of our competitors may have significantly greater financial, technical and human resources.
As a result of these factors, our competitors may obtain regulatory approval of their products before we are able to, which may limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are safer, more effective, more widely used and cheaper than ours, and may also be more successful than us in manufacturing and marketing their products. These appreciable advantages could render our product candidates obsolete or non-competitive before we can recover the expenses relating to the development and commercialization of our product candidates.
Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific, management and commercial personnel, establishing clinical trial sites and subject registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of the product candidates that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates by us or our investigators in human clinical trials and will face an even greater risk if we commercially sell our product candidates after obtaining regulatory approval. Product liability claims may be brought against us by study subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling such product candidate. If we cannot successfully defend ourselves against claims that such product candidate caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in, for example:
· decreased demand for such product candidate;
· termination of clinical trial sites or entire trial programs;
· injury to our reputation and significant negative media attention;
· withdrawal of clinical trial subjects;
· significant costs to defend the related litigation;
· substantial monetary awards to clinical trial subjects or patients;
· loss of revenue;
· diversion of management and scientific resources from our business operations;
· the inability to commercialize such product candidate; and
· increased scrutiny and potential investigation by, among others, the FDA, DOJ, the Office of the Inspector General of the HHS, state attorneys general, members of Congress and the public.
We currently have $10.0 million in product liability insurance coverage in the aggregate, which may not be adequate to cover all liabilities that we may incur. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. We intend to expand our product liability insurance coverage to include the sale of commercial products if we obtain marketing approval for our product candidates, but we may be unable to obtain commercially reasonable product liability insurance for either product candidate, if approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
We will need to grow the size of our organization, and we may experience difficulties in managing this growth.
Following our acquisition of Shape Pharmaceuticals, we had 22 full-time employees and one part-time employee, of whom 10 hold Ph.D. degrees and three hold M.D. (or international M.D.-equivalent) degrees as of April 30, 2014. As our development and commercialization plans and strategies develop, or as a result of any future acquisitions, we will need additional managerial, operational, sales, marketing, financial and other resources. Our management, personnel and systems currently in place may not be adequate to support this future growth. Future growth would impose significant added responsibilities on members of management, including:
· managing our clinical trials effectively;
· identifying, recruiting, maintaining, motivating and integrating additional employees;
· managing our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors and other third parties;
· improving our managerial, development, operational and finance systems; and
· expanding our facilities.
As our operations expand, we will need to manage additional relationships with various strategic partners, suppliers and other third parties. Our future financial performance and our ability to commercialize our product candidates, if either product candidate is approved, and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and sales and marketing personnel. Our failure to accomplish any of these tasks could prevent us from successfully growing our company.
Our future success depends on our ability to retain our executive officers and to attract, retain and motivate qualified personnel.
We are highly dependent upon J. Kevin Buchi, our President and Chief Executive Officer, Lesley Russell, M.B.Ch.B., M.R.C.P., our Chief Operating Officer and Chief Medical Officer, Pete A. Meyers, our Chief Financial Officer and Treasurer, and C. Glenn Begley, M.B.B.S., Ph.D., F.R.A.C.P.,
F.R.C.P.A., F.R.C.Path., our Chief Scientific Officer and Senior Vice President, Research & Development. The employment agreements we have with the persons named above do not prevent such persons from terminating their employment with us at any time. We do not maintain “key person” insurance for any of our executives or other employees. The loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives.
If we are unable to attract and retain highly qualified employees, we may not be able to grow effectively.
Our future growth and success depend on our ability to recruit, retain, manage and motivate our employees. The loss of any member of our senior management team or the inability to hire or retain experienced management personnel could compromise our ability to execute our business plan and harm our operating results.
Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the pharmaceutical field is intense and as a result, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business.
Our business and operations would suffer in the event of computer system failures.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, fire, terrorism, war and telecommunication and electrical failures. In addition, our systems safeguard important confidential personal data regarding our subjects. If a disruption event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological or hazardous materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Business disruptions could seriously harm our future revenues and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to produce our product candidates. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption. The ultimate impact on us, our significant suppliers and our general infrastructure of being in certain geographical areas is unknown, but our operations and financial condition could suffer in the event of a major earthquake, fire or other natural disaster.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct our pre-clinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We rely on third-party CROs to monitor and manage data for our ongoing pre-clinical and clinical programs. We rely on these parties for execution of our pre-clinical studies and clinical trials, and we control only some aspects of their activities. Nevertheless, we are responsible for ensuring that each of our pre-clinical studies and clinical trials are conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on the CROs does not relieve us of our regulatory responsibilities. We also rely on third parties to assist in conducting our pre-clinical studies in accordance with Good Laboratory Practices, and the Animal Welfare Act requirements. We and our CROs are required to comply with federal regulations and current Good Clinical Practices, or GCP, which are international standards meant to protect the rights and health of subjects that are enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for our product candidates and any future product candidates in clinical development. Regulatory authorities enforce GCP through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCP, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP requirements. In addition, our clinical trials must be conducted with product produced under cGMP requirements. Failure to comply with these regulations may require us to repeat pre-clinical studies and clinical trials, which would delay the regulatory approval process.
Our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot control whether or not they devote sufficient time and resources to our ongoing clinical, nonclinical and pre-clinical programs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our protocols, regulatory
requirements or for other reasons, our pre-clinical studies and clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
Because we have relied on third parties, our internal capacity to perform these functions is limited. Outsourcing these functions involves risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. We currently have a small number of employees, which limits the internal resources we have available to identify and monitor our third-party providers. To the extent we are unable to identify and successfully manage the performance of third-party service providers in the future, our business may be adversely affected. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
If we lose our relationships with CROs, our drug development efforts could be delayed.
We rely on third-party vendors and CROs for pre-clinical studies and clinical trials related to our drug development efforts. Switching or adding additional CROs would involve additional cost and requires management time and focus. Our CROs generally have the right to terminate their agreements with us in the event of an uncured material breach. In addition, some of our CROs have an ability to terminate their respective agreements with us and/or research projects pursuant to such agreements if the safety of the subjects participating in our clinical trials warrants such termination in accordance with the reasonable opinion of the relevant CRO, if we make a general assignment for the benefit of our creditors or if we are liquidated. Identifying, qualifying and managing performance of third-party service providers can be difficult, time consuming and cause delays in our development programs. In addition, there is a natural transition period when a new CRO commences work and the new CRO may not provide the same type or level of services as the original provider. If any of our relationships with our third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms.
Our experience manufacturing our product candidates is limited to the needs of our pre-clinical studies and clinical trials. We have no experience manufacturing on a commercial scale and have no manufacturing facility. We are dependent on third-party manufacturers for the manufacture of our product candidates as well as on third parties for our supply chain, and if we experience problems with any such third parties, the manufacturing of our product candidates could be delayed.
We do not own or operate facilities for the manufacture of our product candidates. We currently have no plans to build our own clinical or commercial scale manufacturing capabilities. We currently rely on contract manufacturing organizations, or CMOs, for the chemical manufacture of the active pharmaceutical ingredient for our product candidates and another CMO for the production of the birinapant intravenous formulation. To meet our projected needs for pre-clinical and clinical supplies to support our activities through regulatory approval and commercial manufacturing, the CMOs with whom we currently work will need to increase the scale of production. We may need to identify additional CMOs for continued production of supply for our product candidates. Although alternative third-party suppliers with the necessary manufacturing and regulatory expertise and facilities exist, it could be expensive and take a significant amount of time to arrange for alternative suppliers. If we are unable to arrange for alternative third-party manufacturing sources, or to do so on commercially reasonable terms or in a timely manner, we may not be able to complete development of our product candidates, or market or distribute our product candidates.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured our product candidates ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control, including a failure to synthesize and manufacture our product candidates or any products we may eventually commercialize in accordance with our specifications, and the possibility of termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or damaging to us. In addition, the FDA and other regulatory authorities would require that our product candidates and any products that we may eventually commercialize be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities of our product candidates in a timely manner, could lead to a delay in, or failure to obtain, regulatory approval of our product candidates. In addition, such failure could be the basis for the FDA to issue a warning letter, withdraw approvals for such product candidate previously granted to us, or take other regulatory or legal action, including recall or seizure of outside supplies of our product candidates, total or partial suspension of production, suspension of ongoing clinical trials, refusal to approve pending applications or supplemental applications, detention or product, refusal to permit the import or export of products, injunction, or imposing civil and criminal penalties.
Any significant disruption in our supplier relationships could harm our business. Any significant delay in the supply of our product candidates or their respective key materials for an ongoing pre-clinical study or clinical trial could considerably delay completion of such pre-clinical study or clinical trial, product testing and potential regulatory approval of our product candidates. If our manufacturers or we are unable to purchase these key materials after regulatory approval has been obtained for one of our product candidates, the commercial launch of such product candidate would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of that product candidate.
We may elect to enter into licensing or collaboration agreements with third parties to develop, obtain regulatory approvals for and commercialize our product candidates. Our dependence on such relationships may adversely affect our business.
Because we have limited resources, we may seek to enter into collaboration agreements with other pharmaceutical or biotechnology companies. Any failure by our partners to perform their obligations or any decision by our partners to terminate these agreements could negatively impact our ability to successfully develop, obtain regulatory approvals for and commercialize our product candidates. In the event we grant exclusive rights to such partners, we would be precluded from potential commercialization of our product candidates within the territories in which we have a partner. In addition, any termination of our collaboration agreements will terminate the funding we may receive under the relevant collaboration agreement and may impair our ability to fund further development efforts and our progress in our development programs.
Our commercialization strategy for our product candidates may depend on our ability to enter into agreements with collaborators to obtain assistance and funding for the development and potential commercialization of the relevant product candidate in the territories in which we seek to partner. Despite our efforts, we may be unable to secure additional collaborative licensing or other arrangements that are necessary for us to further develop and commercialize our product candidates. Supporting diligence activities conducted by potential collaborators and negotiating the financial and other terms of a collaboration agreement are long and complex processes with uncertain results. Even if we are successful in entering into one or more collaboration agreements, collaborations may involve greater uncertainty for us, as we have less control over certain aspects of our collaborative programs
than we do over our proprietary development and commercialization programs. We may determine that continuing a collaboration under the terms provided is not in our best interest, and we may terminate the collaboration. Our potential future collaborators could delay or terminate their agreements, and as a result our product candidates may never be successfully commercialized.
Further, our potential future collaborators may develop alternative products or pursue alternative technologies either on their own or in collaboration with others, including our competitors, and the priorities or focus of our collaborators may shift such that our product candidates receive less attention or resources than we would like, or they may be terminated altogether. We may also enter into agreements with collaborators to share in the burden of conducting clinical trials, manufacturing and marketing our product candidates. Any such actions by our potential future collaborators may adversely affect our business prospects and ability to earn revenues. In addition, we could have disputes with our potential future collaborators, such as the interpretation of terms in our agreements. Any such disagreements could lead to delays in the development or commercialization of our product candidates, or could result in time-consuming and expensive litigation or arbitration, which may not be resolved in our favor.
Risks Related to Our Intellectual Property
If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate for our technology and product candidates, our competitive position could be harmed.
Our commercial success will depend in large part on our ability to obtain and maintain patent and other intellectual property protection in the U.S. and other countries with respect to our proprietary technology and products. We rely on trade secret, patent, copyright and trademark laws, and confidentiality, licensing and other agreements with employees and third parties, all of which offer only limited protection. We seek to protect our proprietary position by filing and prosecuting patent applications in the U.S. and abroad related to our novel technologies and products that are important to our business.
The patent positions of biotechnology and pharmaceutical companies generally are highly uncertain, involve complex legal and factual questions and have in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patents, including those patent rights licensed to us by third parties, are highly uncertain. The steps we or our licensors have taken to protect our proprietary rights may not be adequate to preclude misappropriation of our proprietary information or infringement of our intellectual property rights, both inside and outside the U.S. Further, the examination process may require us or our licensors to narrow the claims for our pending patent applications, which may limit the scope of patent protection that may be obtained if these applications issue. The rights already granted under any of our currently issued patents or those licensed to us and those that may be granted under future issued patents may not provide us with the proprietary protection or competitive advantages we are seeking. If we or our licensors are unable to obtain and maintain patent protection for our technology and products, or if the scope of the patent protection obtained is not sufficient, our competitors could develop and commercialize technology and products similar or superior to ours, and our ability to successfully commercialize our technology and products may be adversely affected. It is also possible that we or our licensors will fail to identify patentable aspects of inventions made in the course of our development and commercialization activities before it is too late to obtain patent protection on them.
With respect to patent rights, we do not know whether any of the pending patent applications for any of our compounds will result in the issuance of patents that protect our technology or products, or if any of our or our licensors’ issued patents will effectively prevent others from commercializing
competitive technologies and products. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing or in some cases not at all, until they are issued as a patent. Therefore we cannot be certain that we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we or our licensors were the first to file for patent protection of such inventions.
Our pending applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. Because the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, issued patents that we own or have licensed from third parties may be challenged in the courts or patent offices in the U.S. and abroad. Such challenges may result in the loss of patent protection, the narrowing of claims in such patents or the invalidity or unenforceability of such patents, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection for our technology and products. Protecting against the unauthorized use of our or our licensors’ patented technology, trademarks and other intellectual property rights is expensive, difficult and may in some cases not be possible. In some cases, it may be difficult or impossible to detect third-party infringement or misappropriation of our intellectual property rights, even in relation to issued patent claims, and proving any such infringement may be even more difficult.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could harm our business.
Our commercial success depends upon our ability to develop, manufacture, market and sell our product candidates, and to use our related proprietary technologies. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our product candidates, including interference or derivation proceedings before the U.S. Patent and Trademark Office, or USPTO. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue commercializing our product candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Under certain circumstances, we could be forced, including by court order, to cease commercializing our product candidates. In addition, in any such proceeding or litigation, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business.
While our product candidates are in pre-clinical studies and clinical trials, we believe that the use of our product candidates in these pre-clinical studies and clinical trials falls within the scope of the exemptions provided by 35 U.S.C. Section 271(e) in the U.S., which exempts from patent infringement liability activities reasonably related to the development and submission of information to the FDA. As our product candidates progress toward commercialization, the possibility of a patent infringement claim against us increases. We attempt to ensure that birinapant, SHAPE and the methods we employ to manufacture them, as well as the methods for their use we intend to promote, do not infringe other parties’ patents and other proprietary rights. There can be no assurance they do not, however, and competitors or other parties may assert that we infringe their proprietary rights in any event.
In addition, we are testing birinapant administered with other product candidates and regulatory approved products that are covered by patents held by other companies or institutions. In the event that a labeling instruction is required in product packaging recommending that combination, we could be accused of, or held liable for, infringement of the third-party patents covering the product candidate or regulatory approved products recommended for administration with birinapant. In such a case, we could be required to obtain a license from the other company or institution to use the required or desired package labeling, which may not be available on commercially reasonable terms, or at all.
We are aware of certain U.S. and foreign patents owned by a certain third party with claims that are broadly directed to pro-apoptotic SMAC peptide mimetic monomer and dimer compounds, as well as to their use in treating cancer. These patents could be construed to cover birinapant. Generally, conducting clinical trials and other development activities in the U.S. is not considered an act of infringement. If and when birinapant is approved by the FDA, that certain third party may then seek to enforce its patents by filing a patent infringement lawsuit against us. In such lawsuit, we may incur substantial expenses defending our rights to commercialize birinapant, and in connection with such lawsuit and under certain circumstances, it is possible that we could be required to cease or delay the commercialization of birinapant and/or be required to pay monetary damages or other amounts, including royalties on the sales of birinapant. Moreover, such lawsuit may also consume substantial time and resources of our management team and board of directors. The threat or consequences of such a lawsuit may also result in royalty and other monetary obligations, which may adversely affect our results of operations and financial condition.
If we breach any of our license agreements with the respective exclusive licensors of our product candidates, or related licenses, this could have a material adverse effect on our commercialization efforts for birinapant, SHAPE or other related compounds which may be covered by the claims of our in- licensed patents.
In November 2003, we entered into an exclusive license agreement with Princeton University, subsequently amended in June 2004, August 2006 and October 2006, which grants us the rights to certain U.S. patents controlled by the university relating to SMAC-mimetic compounds, including birinapant, and a non-exclusive right to certain know-how and technology relating thereto. In October 2008, Shape Pharmaceuticals entered into an exclusive license agreement with Harvard University and Dana-Farber Cancer Institute, Inc., which grants us the worldwide rights to certain patent applications and patents issuing therefrom as defined in the license agreement, which include claims covering the composition of the SHAPE molecule. Additionally, in January 2014, we entered into a license agreement with the Walter and Eliza Hall Institute of Medical Research in Melbourne, Australia for worldwide exclusive rights to a patent application and any patents issuing therefrom relating to a method of treating intracellular infections involving the administration of an IAP antagonist. Each license agreement includes the right for us to sublicense.
If we materially breach or fail to perform any provision under any of our license agreements (including, generally, failure to make payments when due for royalties and other sub-license revenues, failure to use reasonable efforts to develop, test, obtain regulatory approval, manufacture, market and sell licensed products, failure to file annual progress reports, commencement of bankruptcy or insolvency proceedings against us or failure to prosecute and maintain the licensed patents), the licensor under each license may have the right to terminate the applicable license, and upon the effective date of such termination, our right to practice the applicable licensed patent rights (or our rights in the relevant patent application) would end. To the extent the licensed technology, methodology or patent rights relate to our product candidates or uses thereof, we would expect to exercise all rights and remedies available to us, including attempting to cure any breach by us, and otherwise seek to preserve our rights under the patent rights or other technology or methodologies
licensed to us, but we may not be able to do so in a timely manner, at an acceptable cost to us or at all. Any uncured, material breach under such licenses could result in our loss of rights to practice the patent rights, technology or methodology licensed to us under the applicable license agreement, and to the extent such patent rights or other technology or methodology relate to birinapant, SHAPE or other of our compounds, it could have a material adverse effect on our commercialization efforts for birinapant, SHAPE or such other compounds, as the case may be.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on birinapant, SHAPE and any future product candidates throughout the world would be prohibitively expensive, and our or our licensors’ intellectual property rights in some countries outside the U.S. can be less extensive than those in the U.S. In addition, the laws and practices of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the U.S. Consequently, we and our licensors may not be able to prevent third parties from practicing our and our licensors’ inventions in all countries outside the U.S., or from selling or importing products made using our and our licensors’ inventions in and into the U.S. or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products, and may export otherwise infringing products to territories where we or our licensors have patent protection, but where enforcement is not as strong as that in the U.S. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our or our licensor’s patents or marketing of competing products in violation of our proprietary rights generally in those countries. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business, could put our and our licensors’ patents at risk of being invalidated or interpreted narrowly and our and our licensors’ patent applications at risk of not issuing and could provoke third parties to assert claims against us or our licensors. We or our licensors may not prevail in any lawsuits that we or our licensors initiate and the damages or other remedies awarded, if any, may not be commercially meaningful.
The laws of certain foreign countries may not protect our rights to the same extent as the laws of the U.S., and these foreign laws may also be subject to change. For example, methods of treatment and manufacturing processes may not be patentable in certain jurisdictions, and the requirements for patentability may differ in certain countries, particularly developing countries. Furthermore, generic drug manufacturers or other competitors may challenge the scope, validity or enforceability of our or our licensors’ patents, requiring us or our licensors to engage in complex, lengthy and costly litigation or other proceedings. Generic drug manufacturers may develop, seek approval for, and launch generic versions of our products. Many countries, including European Union countries, India, Japan and China, have compulsory licensing laws under which a patent owner may be compelled under certain circumstances to grant licenses to third parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our and our licensors’ efforts to enforce intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license.
Patent term may be inadequate to protect our competitive position on our products for an adequate amount of time.
Given the amount of time required for the development, testing and regulatory review of new product candidates, such as birinapant and SHAPE, patents protecting such candidates might expire before or shortly after such candidates are commercialized. We expect to seek extensions of patent terms in the U.S. and, if available, in other countries where we are prosecuting patents. In the U.S., the Drug Price Competition and Patent Term Restoration Act of 1984 permits a patent term extension of up to five years beyond the normal expiration of the patent, which is limited to the approved indication (or any additional indications approved during the period of extension). However, the applicable authorities, including the FDA and the USPTO in the U.S., and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and pre-clinical data and launch their product earlier than might otherwise be the case.
Changes in patent law, including recent patent reform legislation, could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
As is the case with other pharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the pharmaceutical industry involve technological and legal complexity, and obtaining and enforcing pharmaceutical patents is costly, time-consuming, and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the U.S. and other countries may diminish the value of our patents or narrow the scope of our patent protection. For example, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our and our licensors’ ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our and our licensors’ ability to obtain new patents or to enforce existing patents and patents we and our licensors may obtain in the future. Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our and our licensors’ patent applications and the enforcement or defense of our or our licensors’ issued patents and those licensed to us.
In September 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. In particular, under the Leahy-Smith Act, the U.S. transitioned in March 2013 to a “first to file” system in which the first inventor to file a patent application will be entitled to the patent. Third parties are allowed to submit prior art before the issuance of a patent by the USPTO and may become involved in opposition, derivation, reexamination, inter-partes review or interference proceedings challenging our patent rights or the patent rights of our licensors. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our or our licensors’ patent rights, which could adversely affect our competitive position.
The USPTO is currently developing regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, did not become effective until March 16, 2013.
Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submissions, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non- compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our product candidates, our competitive position would be adversely affected.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful and have a material adverse effect on the success of our business.
Competitors may infringe our patents or misappropriate or otherwise violate our intellectual property rights. To counter infringement or unauthorized use, litigation may be necessary in the future to enforce or defend our intellectual property rights, to protect our trade secrets or to determine the validity and scope of our own intellectual property rights or the proprietary rights of others. Also, third parties may initiate legal proceedings against us or our licensors to challenge the validity or scope of intellectual property rights we own or control. These proceedings can be expensive and time consuming. Many of our current and potential competitors have the ability to dedicate substantially greater resources to defend their intellectual property rights than we can. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results. In addition, in an infringement proceeding, a court may decide that a patent owned by or licensed to us is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of shares of our common stock.
We may be subject to claims by third parties asserting that our licensors, employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our employees and our licensors’ employees, including our senior management, were previously employed at universities or at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these employees, including each member of our senior management, executed proprietary rights, non-disclosure and non-competition agreements, or similar agreements, in connection with such previous employment. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such third party. Litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel or sustain damages. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms or at all. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
· others may be able to make compounds that are the same as or similar to birinapant or SHAPE, as the case may be, but that are not covered by the claims of the patents that we own or have exclusively licensed;
· we or our licensors or any strategic partners might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own or have exclusively licensed;
· we or our licensors might not have been the first to file patent applications covering certain of our inventions;
· others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights;
· it is possible that our pending patent applications will not lead to issued patents;
· issued patents that we own or have exclusively licensed may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges;
· our competitors might conduct research and development activities in the U.S. and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
· we may not develop additional proprietary technologies that are patentable; and
· the patents of others may have an adverse effect on our business.
Risks Related to Our Financial Position and Capital Needs
We have incurred significant losses since our inception and anticipate that we will continue to incur losses in the future.
We are a clinical-stage biopharmaceutical company. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk of failure to gain regulatory approval or become commercially viable. We have two product candidates, birinapant and SHAPE, at the early stages of clinical development and all of our other compounds are pre-clinical. We do not have any products approved by regulatory authorities for marketing and have not generated any revenue from product sales, and we continue to incur significant research, development and other expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in every reporting period since our inception in 2003. For the year ended December 31, 2013 and the three months ended March 31, 2014, we reported a net loss of $20.0 million and $5.3 million, respectively, and we had an accumulated deficit of $89.9 million and $95.2 million at December 31, 2013 and March 31, 2014, respectively.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses to increase as we continue the research and development of, and seek regulatory approvals for, our product candidates, and potentially begin to commercialize our product candidates, if they receive regulatory approval. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. If our product candidates fail in clinical trials or do not gain regulatory approval, or if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
We currently have no source of product revenue and may never become profitable.
We have not generated any revenues from commercial product sales (and we have no commercial products). Our ability to generate revenue from product sales and achieve profitability will depend upon our ability to successfully gain regulatory approval and commercialize birinapant, SHAPE or other product candidates that we may develop, in-license or acquire in the future. Even if we are able to successfully achieve regulatory approval for our product candidates, we do not know when it will generate revenue from product sales for us, if at all. Our ability to generate revenue from product sales from birinapant, SHAPE or any other future product candidates also depends on a number of additional factors, including our ability to:
· successfully complete development activities, including enrollment of study participants and completion of the necessary clinical trials;
· complete and submit NDAs to the FDA and obtain regulatory approval for indications for which there is a commercial market;
· complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities;
· successfully commercialize any products, if approved;
· make or have made commercial quantities of our products at acceptable cost levels;
· develop a commercial organization capable of manufacturing, sales, marketing and distribution for any products we intend to sell ourselves in the markets in which we choose to commercialize on our own;
· find suitable partners to help us market, sell and distribute our approved products in other markets; and
· obtain adequate pricing, coverage and reimbursement from third parties, including government and private payors.
In addition, because of the numerous risks and uncertainties associated with product development, including that birinapant or SHAPE may not advance through development or achieve the endpoints of applicable clinical trials, we are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we are able to complete the development and regulatory process for our product candidates, we anticipate incurring significant costs associated with commercializing these products.
Even if we are able to generate revenues from the sale of birinapant, SHAPE or any future commercial products, we may not become profitable and will need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, and we are not successful in obtaining additional funding, then we may be unable to continue our operations at planned levels.
We intend to expend our limited resources to pursue birinapant and SHAPE, and may fail to capitalize on other technologies, product candidates or other indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we are focusing on research programs relating to birinapant and SHAPE, which concentrates the risk of product failure in the event birinapant or SHAPE proves to be unsafe or ineffective or the SMAC-mimetic class of product candidates is considered to be inadequate for clinical development or commercialization. As a result, we may forego or delay pursuit of opportunities with other technologies, product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on proprietary research and development programs relating to birinapant and SHAPE may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for birinapant or SHAPE, we may relinquish valuable rights to birinapant or SHAPE through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to birinapant or SHAPE.
We will require additional capital to fund our operations and if we fail to obtain necessary financing, we may be unable to complete the development and potential commercialization of birinapant or SHAPE.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to advance the clinical development of birinapant and SHAPE and launch and commercialize birinapant and SHAPE, if we receive regulatory approval. We will require additional capital for the further development and potential commercialization of birinapant and SHAPE, and may also need to raise additional funds sooner to pursue a more accelerated development of birinapant and SHAPE. If we are unable to raise capital when needed or on attractive
terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
As of the date of this offering memorandum, we anticipate that the net proceeds from this offering (assuming no exercise of the initial purchaser’s option to purchase additional notes) and cash on hand will enable us to fund our operating expenses and capital expenditures, including the payment of interest on the notes, through the end of 2015. We have based this estimate on assumptions that may prove to be wrong, and we could deploy our available capital resources sooner than we currently expect. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to the:
· initiation, progress, timing, costs and results of pre-clinical studies and clinical trials for birinapant, SHAPE or any other future product candidates;
· clinical development plans we establish for birinapant, SHAPE and any other future product candidates;
· our obligation to make royalty and non-royalty sublicense receipt payments to third-party licensors, if any, under our licensing agreements;
· number and characteristics of product candidates that we discover or in-license and develop;
· outcome, timing and cost of regulatory review by the FDA and comparable foreign regulatory authorities, including the potential for the FDA or comparable foreign regulatory authorities to require that we perform more studies than those that we currently expect;
· costs of filing, prosecuting, defending and enforcing any patent claims and maintaining and enforcing other intellectual property rights;
· effect of competing technological and market developments;
· costs and timing of the implementation of commercial-scale manufacturing activities; and
· costs and timing of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval.
If we are unable to obtain necessary financing, our ability to complete the development and potential commercialization of birinapant and SHAPE will be compromised.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies, birinapant or SHAPE.
Until we can generate substantial revenue from product sales, if ever, we expect to seek additional capital through a combination of private and public equity offerings, debt financings, strategic collaborations and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of existing stockholders will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of existing stockholders. Debt financing, if available, may involve agreements that include liens or other restrictive covenants limiting our ability to take important actions, such as
incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through strategic collaborations and alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies in particular countries, or grant licenses on terms that are not favorable to us. If we are unable to raise additional funds through equity or debt financing when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts or grant rights to develop and market our technologies that we would otherwise prefer to develop and market ourselves.
We may acquire other assets or businesses, or form collaborations or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of assets, including pre-clinical, clinical or commercial stage products or product candidates, or businesses, or strategic alliances and collaborations, to expand our existing technologies and operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any such transaction, any of which could have a detrimental effect on our financial condition, results of operations and cash flows. We have limited experience with acquiring other companies, products or product candidates, and limited experience with forming strategic alliances and collaborations. We may not be able to find suitable acquisition candidates, and if we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business and we may incur additional debt or assume unknown or contingent liabilities in connection therewith. Integration of an acquired company or assets may also disrupt ongoing operations, require the hiring of additional personnel and the implementation of additional internal systems and infrastructure, especially the acquisition of commercial assets, and require management resources that would otherwise focus on developing our existing business. We may not be able to find suitable strategic alliance or collaboration partners or identify other investment opportunities, and we may experience losses related to any such investments.
To finance any acquisitions or collaborations, we may choose to issue debt or shares of our common stock as consideration. Any such issuance of shares would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other assets or companies or fund a transaction using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
Risks Relating to Securities Markets and Investment in Our Common Stock
The market price of our common stock may be volatile, which may adversely impact your investment in the notes.
The trading price of our common stock is likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this offering memorandum or the documents we have incorporated by reference in this offering memorandum, these factors include:
· trading volatility of low-priced stock;
· the success of competitive products or technologies;
· regulatory actions with respect to our products or our competitors’ products;
· actual or anticipated changes in our growth rate relative to our competitors;
· announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments;
· results of clinical trials of birinapant, SHAPE or product candidates of our competitors;
· regulatory or legal developments in the U.S. and other countries;
· developments or disputes concerning patent applications, issued patents or other proprietary rights;
· the recruitment or departure of key personnel;
· the level of expenses related to our clinical development programs;
· the results of our efforts to in-license or acquire additional product candidates or products;
· actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts;
· variations in our financial results or those of companies that are perceived to be similar to us;
· fluctuations in the valuation of companies perceived by investors to be comparable to us;
· share price and volume fluctuations attributable to inconsistent trading volume levels of our shares;
· announcement or expectation of additional financing efforts;
· sales of our common stock by us, our insiders or our other stockholders;
· changes in the structure of healthcare payment systems;
· market conditions in the pharmaceutical and biotechnology sectors; and
· general economic, industry and market conditions.
In addition, the stock market in general, and pharmaceutical and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. Moreover, some institutional investors and mutual funds cannot invest in stocks priced below $5.00 per share. The realization of any of these risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our common stock.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell substantial amounts of common stock or securities convertible into or exchangeable for common stock. These future issuances of common stock or common stock-related securities, together with the exercise of stock options, warrants outstanding or granted in the future, any additional shares issued in connection with acquisitions and any shares issued upon the conversion of the notes offered hereby, if any, may result in material dilution to our investors. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to those of holders of our common stock.
In addition, persons who were our stockholders prior to the sale of shares in our initial public offering continue to hold a substantial number of shares of our common stock that they may be able to sell in the public market, subject to the limitations of Rule 144 of the Securities Act. Significant portions of these shares are held by a small number of stockholders. Sales by our current stockholders of a substantial number of shares, or the expectation that such sales may occur, could significantly reduce the market price of our common stock. Moreover, the holders of a substantial number of shares of common stock may have rights, subject to certain conditions, to require us to file registration statements to permit the resale of their shares in the public market or to include their shares in registration statements that we may file for ourselves or other stockholders.
Pursuant to our equity incentive plans, our compensation committee is authorized to grant equity-based incentive awards to our directors, executive officers and other employees and service providers. As of March 31, 2014, the maximum number of shares of our common stock that may be issued under our existing equity incentive plans is 2,745,541. Future equity incentive grants and issuances of common stock under our existing equity incentive plans may have an adverse effect on the market price of our common stock.
We have also registered all common stock that we may issue under our employee benefits plans. As a result, these shares can be freely sold in the public market upon issuance, subject to restrictions under the securities laws. In addition, our directors and executive officers may in the future establish programmed selling plans under Rule 10b5-1 of the Exchange Act for the purpose of effecting sales of our common stock. If any of these events cause a large number of our shares to be sold in the public market, the sales could reduce the trading price of our common stock and impede our ability to raise future capital.
We may be subject to securities litigation, which is expensive and could divert our management’s attention.
The market price of our common stock may be volatile, and in the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
Insiders have substantial influence over us and could delay or prevent a change in corporate control.
As of March 31, 2014, our executive officers, directors and beneficial owners of over 5% of our common stock held approximately 66.0% of our common stock. This concentration of ownership could harm the market price of our common stock by:
· delaying, deferring or preventing a change in control of our company;
· impeding a merger, consolidation, takeover or other business combination involving our company; or
· discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of our company.
The interests of this group of stockholders may not always coincide with your interests or the interests of other stockholders and they may act in a manner that advances their best interests and not necessarily those of other stockholders, including seeking a premium value for their common stock, and might negatively affect the prevailing market price for our common stock.
We have never paid dividends on our capital stock, and because we do not anticipate paying any cash dividends in the foreseeable future, capital appreciation, if any, of our common stock will be your sole source of gain on an investment in our stock if converted.
We have never paid any cash dividends on our common stock, and we currently intend to retain any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future after you have converted your notes.
We are an “emerging growth company” and we take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in our common stock being less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups, or JOBS, Act, and we take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we will rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company, which in certain circumstances could be for up to five years from the end of the fiscal year in which we completed our initial public offering, which was December 31, 2013.
Our status as an “emerging growth company” under the JOBS Act may make it more difficult to raise capital as and when we need it.
Because of the exemptions from various reporting requirements provided to us as an “emerging growth company” we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
We will remain an “emerging growth company” until the earliest of (a) the last day of the first fiscal year in which our annual gross revenues exceed $1.0 billion, (b) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our shares that are held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, (c) the date on which we have issued more than $1.0 billion in nonconvertible debt during the preceding three-year period and (d) the last day of our fiscal year containing the fifth anniversary of the date on which shares of our common stock become publicly traded in the U.S., which was December 31, 2013.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. Commencing with our Annual Report on Form 10-K for the year ending December 31, 2014, we will be required, under Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge, and compile the system and process documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or
significant deficiency in our internal control over financial reporting once that firm begin its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ Stock Market, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision- making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
As a result of our recent initial public offering, we now incur significant additional expenses as a result of being a public company, which negatively impact our financial performance and could cause our results of operations and financial condition to suffer.
In December 2013 we completed our initial public offering. As a result, we are now required to incur significant legal, accounting, insurance, compliance and other expenses as a result of being a public company. These expenses may increase even more after we are no longer an “emerging growth company.” We are obligated to file annual and quarterly information and other reports with the SEC. In addition, we also became subject to other reporting and corporate governance requirements which impose significant compliance obligations upon us. The Sarbanes-Oxley Act of 2002, together with related rules implemented by the SEC and by the NASDAQ Stock Market, have imposed increased regulation and disclosure and have required enhanced corporate governance practices of public companies. Our efforts to comply with these laws, rules and regulations, including compliance with Section 404 of the Sarbanes-Oxley Act as discussed in “—If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock” above, substantially increase our expenses, including our legal and accounting costs, and make some activities more time-consuming and costly. We estimate that we will incur approximately $2.0 to $3.0 million in incremental costs per year associated with being a publicly traded company, although it is possible that our actual incremental costs will be higher than we currently estimate.
We also expect these laws, rules and regulations to make it more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors or as officers. As a result of the foregoing, we have begun to incur substantial increases in legal, accounting and insurance compliance and we expect to incur certain other expenses in the future, which will negatively impact our financial performance and could cause our results of operations and financial condition to suffer.
If our provision of a preliminary draft of our Form S-1 registration statement to prospective preferred stock investors in an uncompleted private placement in August and September 2013 were held to be in violation of the Securities Act, we could be required to repurchase certain shares of our common stock sold in our initial public offering to those prospective preferred stock investors.
In August and September 2013, we attempted to raise capital through a private placement of a new series of preferred stock. In connection with these efforts, in order to permit prospective preferred stock investors to make an informed investment decision as to whether or not to invest in our preferred stock, we provided prospective preferred stock investors with certain business and financial information, including a preliminary draft of the Form S-1 registration statement from our initial public offering. We did not complete the private placement and we did not sell any preferred stock at that time, nor did we make any offer at that time to such prospective preferred stock investors to purchase shares of common stock in our initial public offering. Thereafter, following the filing of our Form S-1 registration statement from our initial public offering with the SEC, we provided our preliminary prospectuses dated November 6, 2013 and December 10, 2013 to certain of these prospective preferred stock investors offering to sell them common stock in our initial public offering. If our provision of the preliminary draft of the registration statement to prospective preferred stock investors in August and September 2013 were held by a court to be in violation of the Securities Act, we could be required to repurchase shares of our common stock, if any, sold to any such prospective preferred stock investor that purchases our common stock in our initial public offering at the original purchase price, plus statutory interest from the date of purchase, for a period of one year following the date of the violation. We would vigorously contest any such claim that a violation of the Securities Act occurred.
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our stockholders, or remove our current management. These include provisions that:
· permit our board of directors to issue up to 25 million shares of preferred stock, with any rights, preferences and privileges as it may designate;
· provide that all vacancies on our board of directors, including as a result of newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum;
· establish a classified board of directors such that only one of three classes of directors is elected each year;
· provide that directors can only be removed for cause;
· require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent;
· provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide advance notice in writing, and also specify requirements as to the form and content of a stockholder’s notice;
· require that the amendment of certain provisions of our certificate of incorporation and bylaws relating to anti-takeover measures may only be approved by a vote of 75% of our outstanding capital stock;
· not provide for cumulative voting rights, thereby allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election; and
· provide that special meetings of our stockholders may be called only by the board of directors or by such person or persons designated by a majority of the board of directors to call such meetings.
These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, who are responsible for appointing the members of our management.
Because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may discourage, delay or prevent someone from acquiring us or merging with us whether or not it is desired by or beneficial to our stockholders. Under Delaware law, a corporation may not, in general, engage in a business combination with any holder of 15.0% or more of its capital stock unless the holder has held the stock for three years or, among other things, the board of directors has approved the transaction. Any provision of our certificate of
incorporation or bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
If securities or industry analysts publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.