Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Brickell Biotech, Inc. | Financial_Report.xls |
| EX-32.1 - CERTIFICATON OF CHIEF EXECUTIVE OFFICER - Brickell Biotech, Inc. | d268604dex321.htm |
| EX-10.8 - THIRD AMENDMENT TO CONSULTING AGREEMENT - Brickell Biotech, Inc. | d268604dex108.htm |
| EX-32.2 - CERTIFICATON OF CHIEF FINANCIAL OFFICER - Brickell Biotech, Inc. | d268604dex322.htm |
| EX-31.2 - CERTIFICATON OF CHIEF FINANCIAL OFFICER - Brickell Biotech, Inc. | d268604dex312.htm |
| EX-31.1 - CERTIFICATON OF CHIEF EXECUTIVE OFFICER - Brickell Biotech, Inc. | d268604dex311.htm |
| EX-23.1 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - Brickell Biotech, Inc. | d268604dex231.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2011.
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission file number: 000-21088
VICAL INCORPORATED
(Exact name of registrant as specified in its charter)
| Delaware | 93-0948554 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
| 10390 Pacific Center Court, San Diego, California | 92121-4340 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (858) 646-1100
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $0.01 par value |
The Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ¨ Yes x No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. ¨ Yes x No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). x Yes ¨ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company.
Large accelerated filer ¨ Accelerated filer x Non-accelerated filer ¨ Smaller reporting company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ¨ Yes x No
The aggregate market value of the voting stock held by non-affiliates of the registrant, based upon the last sale price of the registrant’s common stock reported on the Nasdaq Global Market on June 30, 2011, was approximately $258,750,263.
The number of shares of common stock outstanding as of February 15, 2012, was 85,881,117.
Documents Incorporated by Reference:
| Document |
Part of Form 10-K | |
| Proxy Statement for the Annual Meeting of Stockholders to be held May 24, 2012 |
Part III |
Table of Contents
VICAL INCORPORATED
FORM 10-K
| PART I. |
||||||
| ITEM 1. |
3 | |||||
| ITEM 1A. |
27 | |||||
| ITEM 1B. |
38 | |||||
| ITEM 2. |
39 | |||||
| ITEM 3. |
39 | |||||
| ITEM 4. |
39 | |||||
| PART II. |
||||||
| ITEM 5. |
40 | |||||
| ITEM 6. |
42 | |||||
| ITEM 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
43 | ||||
| ITEM 7A. |
51 | |||||
| ITEM 8. |
53 | |||||
| ITEM 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
80 | ||||
| ITEM 9A. |
80 | |||||
| ITEM 9B. |
82 | |||||
| PART III. |
||||||
| ITEM 10. |
82 | |||||
| ITEM 11. |
82 | |||||
| ITEM 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
82 | ||||
| ITEM 13. |
Certain Relationships and Related Transactions, and Director Independence |
82 | ||||
| ITEM 14. |
82 | |||||
| PART IV. |
||||||
| ITEM 15. |
83 | |||||
| 88 | ||||||
FORWARD-LOOKING STATEMENTS
In addition to historical information, this Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, including statements regarding our business, our financial position, the research and development of biopharmaceutical products based on our patented DNA delivery technologies, and other statements describing our goals, expectations, intentions or beliefs. Such statements reflect our current views and assumptions and are subject to risks and uncertainties, particularly those inherent in the process of developing and commercializing biopharmaceutical products based on our patented DNA delivery technologies. Actual results could differ materially from those discussed in this Annual Report on Form 10-K. Factors that could cause or contribute to such differences include, but are not limited to, those identified in Item 1A entitled “Risk Factors” beginning on page 27 of this report, as well as those discussed in our other filings with the Securities and Exchange Commission, or SEC, including our Quarterly Reports on Form 10-Q. As a result, you are cautioned not to unduly rely on these forward-looking statements. We disclaim any duty to update any forward-looking statement to reflect events or circumstances that occur after the date on which such statement is made.
2
Table of Contents
PART I
Overview
We research and develop biopharmaceutical products based on our patented DNA delivery technologies for the prevention and treatment of serious or life-threatening diseases. We believe the following areas of research offer the greatest potential for near-term commercialization for us and our partners:
| • | Vaccines for use in high-risk populations for infectious disease targets for which there are significant needs; |
| • | Vaccines for general pediatric, adolescent and adult populations for infectious disease applications; |
| • | Cancer vaccines or immunotherapies that complement our existing programs and core expertise; and |
| • | Gene-based delivery of therapeutic proteins, such as angiogenic growth factors for treatment of cardiovascular diseases. |
We currently have three active independent clinical and preclinical development programs in the areas of infectious disease and cancer including:
| • | A fully enrolled ongoing Phase 3 clinical trial using our Allovectin® immunotherapeutic in patients with metastatic melanoma which has been funded, up to certain limits, by AnGes MG, Inc., or AnGes, through cash payments and equity investments under a research and development agreement; |
| • | A completed preclinical program, with an allowed investigational new drug application, or IND, using our CyMVectin™ prophylactic vaccine formulated with our proprietary Vaxfectin® adjuvant to prevent cytomegalovirus, or CMV, infection before and during pregnancy; and |
| • | A preclinical program with therapeutic and prophylactic vaccines for herpes simplex virus type 2 formulated with our proprietary Vaxfectin® adjuvant. |
We have leveraged our patented technologies through licensing and collaboration arrangements, such as our licensing arrangements with Astellas Pharma Inc., or Astellas, Merck & Co., Inc., or Merck, Sanofi, AnGes, Aqua Health Ltd. of Canada, or Aqua Health, an affiliate of Novartis Animal Health, and Merial Limited, or Merial, a subsidiary of Sanofi, among other biopharmaceutical companies.
In addition, we have licensed complementary technologies from leading research institutions and biopharmaceutical companies. We also have granted non-exclusive, academic licenses to our DNA delivery technology patent estate to 11 leading research institutions including Stanford, Harvard, Yale and the Massachusetts Institute of Technology, or MIT. The non-exclusive academic licenses allow university researchers to use our technology free of charge for educational and internal, non-commercial research purposes. In exchange, we have the option to exclusively license from the universities potential commercial applications arising from their use of our technology on terms to be negotiated.
Available Information
We were incorporated in Delaware in 1987. Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to these reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, are available free of charge on our website at www.vical.com as soon as reasonably practicable after such reports and amendments are electronically filed with or furnished to the SEC. We also make available copies of our news releases and other financial information about us on our website.
3
Table of Contents
Our Core Technology
The key discovery leading to our patented core DNA delivery technology was that muscle tissues can take up polynucleotide genetic material, such as DNA or RNA, directly, without the use of viral components or other delivery vehicles or technologies, and subsequently express the proteins encoded by the genetic material for periods ranging from weeks to more than a year. Our approach typically involves designing and constructing closed loops of DNA called plasmids, or pDNAs. These pDNAs contain a DNA segment encoding the protein of interest, as well as short segments of DNA that control protein expression. Plasmids can be manufactured using uniform methods of fermentation and processing. This could result in faster development and production times than technologies that require development of product-specific manufacturing processes.
Since the initial discovery of our DNA delivery technology, our researchers have improved the design of our plasmids to provide increases in efficiency of gene expression and immunogenicity. In addition, we continue to develop formulations, adjuvants, and delivery technologies, including the use of lipid molecules, synthetic polymers called poloxamers and other approaches to enhance DNA expression or increase the immune response to DNA vaccines. We own broad patent rights in the United States and in key foreign markets to certain non-viral polynucleotide delivery technologies. Our patents and patent applications cover, for example, DNA delivery for immunization and delivery of therapeutic proteins, specific DNA constructs and formulations of gene-based product candidates, methods for producing pharmaceutical-grade DNA, and several families of lipid molecules and their uses in DNA delivery. Benefits of our DNA delivery technologies may include the following, which may enable us to offer novel treatment alternatives for diseases that are currently poorly addressed:
| • | Broad Applicability. Our DNA delivery technologies may be useful in developing vaccines for infectious diseases, in which the expressed protein induces an immune response; novel therapies for cancer, in which the expressed protein is an immune system stimulant or tumor suppressor; and therapeutic protein delivery, in which the expressed protein is a therapeutic agent; |
| • | Convenience. Our DNA-based biopharmaceutical product candidates are intended to be administered on an outpatient basis; |
| • | Safety. Our product candidates contain no infectious components that may cause unwanted immune responses, infections, or malignant and permanent changes in the targeted cells’ genetic makeup; |
| • | Repeat Administration. Our product candidates contain no infectious components that may preclude multiple dosing with a single product or use in multiple products; |
| • | Ease of Manufacturing. Our product candidates are manufactured using uniform fermentation and purification procedures; and |
| • | Cost-Effectiveness. Our DNA delivery technologies may be more cost-effective than other approaches. They may also cause fewer potential side effects, which may reduce per patient treatment costs. |
Applications of DNA Technology
Our DNA delivery technology is currently being developed by us and our partners in four broad areas of application:
Infectious Diseases
DNA vaccines use portions of the genetic code of a pathogen to cause the host to produce proteins of the pathogen that may induce an immune response. Compared with conventional vaccines that use live, weakened, or dead pathogens to produce an immune response, this method potentially offers superior safety and ease of manufacturing, as well as convenient storage and handling characteristics. DNA vaccines have the potential to induce potent T-cell responses against target pathogens as well as trigger production of antibodies. Over the past decade, many scientific publications have documented the effectiveness of DNA vaccines in contributing to
4
Table of Contents
immune responses in dozens of species, including fish, nonhuman primates and humans. We believe important steps in the validation of DNA vaccines occurred in 2005 when our licensee Aqua Health received Canadian approval to market its proprietary product, Apex®-IHN, a DNA vaccine to protect farm-raised salmon against infectious hematopoietic necrosis virus, or IHNV, and again in late 2009, when our licensee, Merial, received approval from the USDA to sell a therapeutic DNA vaccine, ONCEPT™, designed to aid in extending the survival time of dogs with oral melanoma.
Vaccines are generally recognized as the most cost-effective approach for infectious disease healthcare. However, the technical limitations of conventional vaccine approaches have constrained the development of effective vaccines for many diseases. Development of vaccines based on conventional methods requires significant infrastructure in research and manufacturing. In addition, the safety risks associated with certain conventional vaccine approaches may offset their potential benefits. We believe our potential vaccine products may be simpler to manufacture than vaccines made using live viruses or protein subunit approaches, including those involving mammalian, avian or insect cell, or egg-based, culture procedures. In addition, our DNA delivery technologies may accelerate certain aspects of vaccine product development such as nonclinical evaluation and manufacturing.
In the broader vaccine marketplace, it is important to note a changing dynamic. Traditionally, vaccines have been predominantly focused on the pediatric market, intended to protect children from diseases that could cause them serious harm. Today, there is a growing interest in vaccines against diseases that may affect adolescents and adults, which include both sexually transmitted diseases and infections that strike opportunistically, such as during pregnancy or in immunocompromised individuals, including the geriatric population. We believe our technologies, because of their potential safety and development timeline advantages, could be ideally suited for this new generation of vaccines.
Cancer
Cancer is a disease of uncontrolled cell growth. When detected early and still confined to a single location, cancer may be cured by surgery or irradiation. However, neither surgery nor irradiation can cure cancer that has spread throughout the body. Although chemotherapy can sometimes effectively treat cancer that has spread throughout the body, a number of non-cancerous cells, such as bone marrow cells, are also highly susceptible to chemotherapy. As a result, chemotherapy often has fairly significant side effects. Finally, it is common to see cancer return after apparently successful treatment by each of these means.
Immunotherapy, a process which uses the patient’s own immune system to treat cancer, may have advantages over surgery, irradiation, and chemotherapy. Many cancers appear to have developed the ability to “hide” from the immune system. A treatment that can augment the immune response against tumor cells by making the cancer more “visible” to the immune system would likely represent a significant improvement in cancer therapy. Immune-enhancing proteins such as interleukin-2, or IL-2, and interferon-alpha, or IFN-µ, have shown encouraging results. However, these agents often require frequent doses that regularly result in severe side effects.
Two new drugs for metastatic melanoma were approved in 2011, both on the basis of increased survival. Yervoy®, a monoclonal antibody marketed by Bristol-Myers Squibb Co., increases the effectiveness of T-cells that can seek out and destroy melanoma cells. Zelboraf®, a B-Raf inhibitor marketed by Roche and Daiichi Sankyo, interrupts a key process in melanoma growth in patients with a particular melanoma mutation. Both drugs are associated with significant side effects, and neither is considered a cure for melanoma.
We have researched delivery enhancements that may complement our core DNA delivery technology and may help us develop cancer therapies. Our current clinical-stage approach consists of directly injecting solid tumors with plasmids which, upon uptake into cells, direct the production of the encoded immunostimulatory proteins to generate a local, regional and systemic effect. The plasmids are formulated with a cationic lipid-based
5
Table of Contents
delivery system. The ease of manufacture, convenience, and ability to repeat administration may offer advantages over current modalities of therapy. In addition, cancer therapies using non-viral DNA delivery may offer an added margin of safety compared with viral-based delivery, as no viral particles or other potentially infectious agents are contained in the formulation.
Human studies utilizing this approach demonstrated anti-tumor effects and were found to be safe with a very low incidence of treatment-related serious adverse events. In addition, studies in animals have demonstrated the potential efficacy of this approach. Further validation of DNA technology in cancer vaccine applications occurred in late 2009, when our licensee Merial received approval from the USDA to sell a therapeutic DNA vaccine, ONCEPT™, designed to aid in extending the survival time of dogs with oral melanoma.
Cardiovascular
Cardiovascular diseases represent the leading cause of death in the United States and in most Western countries. Cardiovascular disease refers to the class of diseases that involve the heart or blood vessels. Peripheral artery disease, or PAD, including critical limb ischemia, or CLI, and coronary artery disease, or CAD, also known as ischemic heart disease, or IHD, are the end result of arterial occlusive disease, which is most commonly known as atherosclerosis. Atherosclerosis affects only the inner lining of an artery and is characterized by fatty deposits that block the flow of blood.
PAD is caused by atherosclerosis in association with hypertension, hypercholesterolemia, cigarette smoking or diabetes. PAD is a common circulatory problem in which narrowed arteries reduce the blood flow to the limbs. Early symptoms of PAD include transient pain in the legs upon walking, a condition called intermittent claudication, which is caused by ischemia. Approximately 25% of ischemic patients will progress to develop CLI, which is associated with pain at rest and ulcers, and frequently requires amputation. Altogether approximately 30% of patients with PAD die within 5 years of developing PAD, rising to approximately 50% after 10 years, which represents a mortality rate exceeding most other vascular conditions including CAD. The number of therapeutic options for PAD remains very limited in comparison with other areas of cardiovascular medicine and the healthcare burden associated with amputations in the U.S. is estimated to be greater than $10 billion per year.
CAD occurs when the coronary arteries that supply blood to the heart muscle become hardened and narrowed. CAD is the most common type of heart disease and is the leading cause of death in the U.S. in both men and women. Over time, CAD can weaken the heart muscle and contribute to heart failure or arrhythmias. Current treatment regimens for CAD include drugs, catheter-based interventional therapies such as balloon angioplasty or stents, mechanical therapies such as atherectomy, and surgical procedures such as bypass surgery.
We believe PAD and CAD may be better treated by stimulating angiogenesis, which refers to the growth of new blood vessels from pre-existing vessels to replace those blocked by the disease. Our core DNA delivery technology may allow the targeted delivery of certain growth factors with potential therapeutic value in the emerging field of angiogenesis. Angiogenesis has been shown to occur by the exogenous administration of angiogenic growth factors. We believe that the localized and sustained expression of these growth factors from plasmids may be both safe and effective.
Veterinary
Prior to their development for human therapy, our DNA delivery technologies were extensively tested in animals. Research scientists have published numerous papers detailing positive results in many species and covering a broad range of disease indications. Animal health encompasses two distinct market segments: livestock, or animals bred and raised for food or other products, and companion animals, or pets. Through our collaborative partnerships there have been approvals within each of these market segments for vaccines utilizing our DNA delivery technology.
6
Table of Contents
Business Strategy
There are four basic elements to our business strategy:
Develop Products Independently
We currently focus our resources on the independent development of infectious disease vaccines and cancer immunotherapeutics. The selection of targets for our independent development programs is driven by three key criteria: the complexity of the product development program, competition, and commercial opportunities. We intend to retain significant participation in the commercialization of any independently developed proprietary DNA vaccines and therapeutics that receive regulatory approval, although we may choose to enlist the support of partners to accelerate product development and commercialization.
Infectious Disease Vaccines. Vaccines are perceived by government and medical communities as an efficient and cost-effective means of healthcare. According to the Centers for Disease Control and Prevention, or CDC, “Vaccines are among the very best protections we have against infectious diseases.” In the infectious disease area, we have primarily focused our resources on the development of DNA-based vaccines against CMV, pandemic influenza and herpes simplex type 2, or HSV-2. We believe our technologies may lead to the development of novel preventive or therapeutic vaccines for infectious disease targets. DNA vaccines may help combat diseases for which conventional vaccine methods have been unsuccessful.
Cancer Therapies. In the cancer area, we are focusing our resources on the development of Allovectin® initially as a potential treatment for metastatic melanoma, an aggressive form of skin cancer. Allovectin® is a plasmid/lipid formulation containing the DNA sequences encoding HLA-B7 and ß2 microglobulin, which together form a Major Histocompatibility Complex, or MHC, Class I. Injection of Allovectin® directly into tumors is designed to stimulate a systemic immune response against both local and distant metastatic tumors. In previous human clinical trials, Allovectin® has shown evidence of activity against other types of cancer, and could potentially be used to treat any injectable immunoreactive solid tumor.
Enhance and Expand Our Technologies
We are actively pursuing the refinement of our plasmids and formulations, the evaluation of potential enhancements to our core technologies and the exploration of additional DNA delivery technologies. We are developing future product candidates based on these technologies through nonclinical and clinical testing to determine their safety and effectiveness. We also seek to develop additional applications for our technologies by testing new approaches to disease control or prevention. These efforts could lead to further independent product development or additional licensing opportunities. In addition, we continually evaluate compatible technologies or products that may be of potential interest for in-licensing or acquisition. We license intellectual property from companies holding complementary technologies to leverage the potential of our own DNA delivery technologies and to further the discovery of innovative therapies for internal development.
Expand the Applications of Our Technologies through Strategic Collaborations
We collaborate with major pharmaceutical and biotechnology companies and government agencies, providing us access to complementary technologies or greater resources. These collaborations are intended to provide us with mutually beneficial opportunities to expand or advance our product pipeline and serve significant unmet medical needs. We license our intellectual property to other companies to leverage our technologies for applications that may not be appropriate for our independent product development.
Pursue Contract Manufacturing Opportunities
We selectively pursue contract manufacturing opportunities to leverage our infrastructure and expertise in pDNA manufacturing, to support advancement and application of our technologies by others, and to provide revenues that contribute to our independent research and development efforts.
7
Table of Contents
Product Development
We, together with our licensees and collaborators, are currently developing a number of DNA-based vaccines and therapeutics for the prevention or treatment of infectious diseases, cancer and cardiovascular diseases. The table below summarizes our independent programs and corporate and government collaborations.
| Product/Concept |
Intended Use |
Development Status1 |
Lead Developer | |||
| Independent Programs |
||||||
|
Allovectin® cancer immunotherapeutic |
First-line treatment for metastatic melanoma |
Phase 3 |
Vical | |||
|
|
|
|
| |||
| CyMVectin™ prophylactic vaccine for cytomegalovirus |
Prevent infection before pregnancy to preclude fetal transmission |
Preclinical complete |
Vical | |||
|
|
|
|
| |||
| Therapeutic and prophylactic vaccines for herpes simplex type 2 virus |
Prevent and protect against recurring flare-ups, reduce viral shedding and transmission |
Preclinical |
Vical | |||
| Corporate Collaborations |
||||||
| TransVax™ therapeutic vaccine for cytomegalovirus |
Protect against CMV infection after stem cell transplants |
Phase 3 preparation |
Astellas | |||
|
|
|
|
| |||
| TransVax™ therapeutic vaccine for cytomegalovirus |
Protect against CMV infection after solid organ transplants |
Phase 2 preparation |
Astellas | |||
|
|
|
|
| |||
| Collategene™ angiogenic therapy encoding Hepatocyte Growth Factor |
Induce local growth of blood vessels to restore blood flow to limbs affected by critical limb ischemia |
Phase 3 preparation |
AnGes | |||
|
|
|
|
| |||
|
Apex®-IHN prophylactic vaccine for infectious hematopoietic necrosis virus |
Prevent infection and disease in farm-raised salmon when exposed to infected wild salmon |
Marketed in Canada |
Aqua Health (Novartis) | |||
|
|
|
|
| |||
| ONCEPT™ therapeutic cancer vaccine encoding human tyrosinase |
Adjunct treatment to increase survival time of dogs with oral melanoma |
Marketed in the United States |
Merial | |||
| Government Collaboration |
||||||
| Prophylactic and/or therapeutic HIV vaccine |
Prevent and/or treat infection, disease, and/or viral shedding |
Phase 2b |
NIH | |||
|
|
|
|
| |||
| Tetravalent dengue vaccine |
Prevent dengue disease caused by all 4 dengue serotypes |
Phase 1 |
Naval Medical Research Center |
| 1 | “Research” indicates exploration and/or evaluation of a potential product candidate in a nonclinical laboratory setting. “Preclinical” indicates that a specific product candidate in a nonclinical setting has shown functional activity that is relevant to a targeted medical need, and is advancing toward initial human clinical testing. “Phase 1” clinical trials are typically conducted with a small number of patients or healthy subjects to evaluate safety, determine a safe dosage range, identify side effects, and, if possible, gain early evidence of effectiveness. “Phase 2” clinical trials are conducted with a larger group of patients to evaluate effectiveness of an investigational product for a defined patient population, and to determine common short-term side effects and risks associated with the product candidate. “Phase 3” clinical trials involve large scale, multi-center, comparative trials that are conducted with patients afflicted with a target disease to evaluate the overall benefit-risk relationship of the investigational product and to provide an adequate basis for product labeling. |
8
Table of Contents
Independent Programs Targeting Infectious Diseases
CMV Vaccines
CMV is a ubiquitous herpes virus that can cause serious complications in two distinct patient populations: immunocompromised transplant patients and children born to women initially infected during pregnancy. We and our collaborator are currently developing two CMV vaccines: TransVax™ and CyMVectin™. TransVax™ is designed to serve the first patient population by preventing CMV reactivation or infection in transplant recipients. In 2011 we licensed the right to develop and commercialize TransVax™ to Astellas. CyMVectin™ is designed to serve the second, much larger patient population by preventing congenital infection by vaccinating women before pregnancy, which we believe represents a major commercial opportunity and could potentially lead to widespread vaccination.
CyMVectin™
CMV is the most common congenital infection in the United States and the leading cause of infectious disease- related birth defects. If a woman becomes infected with CMV for the first time during pregnancy, there are no treatment alternatives. CyMVectin™ was designed to prevent CMV infection prior to pregnancy. We believe this may ultimately reduce birth defects caused by CMV.
CyMVectin™ consists of pDNA that encodes the human CMV gB antigen with pDNA that encodes the human CMV pp65 antigen. The product is formulated with our proprietary lipid-based adjuvant Vaxfectin®. Vaxfectin® has been shown in nonclinical studies by us and others to enhance immune responses, particularly antibody responses, to expressed immunogens. A gB protein-based vaccine, developed by others, has shown some protection against CMV infection in a Phase 2 clinical trial, so we believe a vaccine strategy for CMV prevention can be successful. In addition, prior maternal CMV infection is associated with protection against secondary infection in pregnant women and reduction of congenital infection. Nonclinical studies performed in rabbit, guinea pig and mouse animal models have demonstrated the ability to induce high titers of gB-specific antibodies in animals receiving the gB plasmid. Rabbit studies of gB pDNA administered intramuscularly demonstrated an approximate 10-fold enhancement of gB antibodies with Vaxfectin® as an adjuvant when compared to gB pDNA in phosphate-buffered saline. Similarly, mouse studies also demonstrated this added benefit of Vaxfectin® as an adjuvant. The results of these studies support both our past experience and other published studies which indicate that immune responses can be induced by pDNA vaccination and that formulation with Vaxfectin® enhances those immune responses. Phosphoprotein 65 (pp65) is a major CMV tegument protein that is recognized by both CD4+ and CD8+ T cells in CMV-seropositive subjects. Induction of pp65-specific T cells following vaccination may provide an additional antiviral mechanism that could act to reduce CMV replication and viral loads. Repeat dose safety studies in rabbits with Vaxfectin®-formulated gB and pp65 plasmids have enabled the allowance of an IND to initiate clinical evaluation of CyMVectin™.
About CMV
Cytomegalovirus infects between 50% and 80% of adults in the United States by 40 years of age. Although most healthy people who are infected by CMV after birth are asymptomatic, CMV can affect certain high-risk groups including immunocompromised individuals and prenatal or postnatal infants. Significant mortality and morbidity are observed in the immunocompromised populations, especially hematopoietic stem cell transplant, or HSCT, and solid organ transplant, or SOT, recipients. In CMV-seropositive HSCT recipients, the incidence of CMV reactivation in the first 6 months following transplantation is 50-70% in the absence of prophylaxis. The incidence is reduced to approximately 5% by the use of preemptive antiviral therapy, but currently available antiviral therapies are associated with drug toxicity, are costly, may lead to drug resistance and provide incomplete efficacy. Late-onset CMV reactivation may also occur after this initial period of heightened susceptibility.
Currently no vaccine is approved for the prevention of CMV infection. The only approved treatment for CMV in HSCT patients is ganciclovir, although other antivirals are used off label, such as valganciclovir,
9
Table of Contents
foscarnet, and cidofovir. We believe a vaccine that enables the patient’s immune system to control CMV infection, thereby reducing the need for antiviral therapy, would be a valuable therapeutic option for HSCT recipients. The control of CMV in immunocompromised persons is primarily associated with T-cell mediated immune responses.
CMV-seropositive HSCT and SOT recipients represent important populations for the prevention of CMV reactivation and reduction in antiviral therapy. Approximately 50,000 HCT transplants and 60,000 SOT transplants are performed annually in the United States and Europe. We believe these populations represent a significant market potential for our TransVax™ vaccine.
CMV is the most common intrauterine infection in the United States, occurring in about 1 in 150 live-born infants and resulting in permanent disabilities, such as mental retardation, hearing loss and vision loss, in approximately 8,000 children per year, and 400 deaths annually. Symptomatic congenital infections occur about one-third of the time following a primary maternal infection during pregnancy. Congenital infection can also occur in CMV-seropositive women, which results in even more cases. Prior maternal CMV infection is associated with partial protection against secondary infection in pregnant women and reduction of the frequency and severity of congenital infection. Contact with infected young children is the primary source of infection for pregnant women, especially those exposed to children in daycare environments.
DNA vaccine induction of CMV-specific antibodies and T-cell responses may prevent or limit CMV infection in women before and during pregnancy, which could impact perinatal transmission and newborn infections. We believe that there is a significant market potential for our CyMVectin™ vaccine, as there are more than 40 million women of childbearing age in the United States.
Herpes Simplex Virus-2
In April 2008 we were awarded a two-year, $2.0 million Phase II Small Business Technology Transfer grant from the National Institute of Allergy and Infectious Diseases, or NIAID. The grant funded the ongoing development of our prophylactic and therapeutic plasmid DNA vaccine against HSV-2. The HSV-2 vaccines were evaluated with our Vaxfectin® adjuvant. The grant period was extended to allow preclinical development to continue for a third year. We have completed preclinical studies funded by the HSV-2 grant and found that our prophylactic Vaxfectin®-formulated plasmid DNA vaccine protected mice against lethal challenge, provided sterilizing immunity, and inhibited viral counts at both the primary and latent infection sites. In guinea pigs, our prophylactic and therapeutic Vaxfectin®-formulated vaccines provided complete protection against both primary and recurrent HSV-2 disease, and significantly reduced genital lesion recurrence and viral shedding as well as latent infection in the central nervous system.
The initial results demonstrated that an appropriate dose of the prophylactic Vaxfectin®-formulated vaccine, which encoded the HSV-2 glycoprotein D (gD2) antigen elicited antibody responses in 100% of mice against the encoded antigen; protected 100% of mice against subsequent challenge with a lethal dose of live virus; reduced viral shedding in mice at both the primary and latent infection sites; and elicited sterilizing immunity in 80% of mice as evidenced by no detectable virus after challenge at either the primary (vagina) or latent (dorsal root ganglia) infection sites. The Vaxfectin® adjuvant substantially improved vaccine effectiveness in the preclinical studies. A therapeutic vaccine that encoded the gD2 antigen as well as the tegument proteins UL46/UL47 significantly reduced recurrence of HSV-2 lesions in a therapeutic model using guinea pigs with latent infection (p<0.05). Three doses of Vaxfectin®-formulated vaccine were administered after resolution of the primary infection. The studies were repeated and the results were confirmed.
Based on the success of our preclinical development, we are advancing toward clinical testing in humans with a Vaxfectin®-formulated therapeutic vaccine. After completing required preclinical safety/toxicology and biodistribution studies, we expect to file an IND application for a Phase 1/2 therapeutic vaccine trial with safety, immunogenicity and clinical endpoints designed to establish clear proof of concept and support potential future partnering discussions.
10
Table of Contents
About HSV -2
HSV-2 is a member of the herpes virus family and is the leading cause of genital herpes worldwide. Approximately one out of every six individuals in the United States and an estimated one out of every four individuals worldwide is infected by HSV-2 before age 50. HSV-2 infection also significantly increases the risk of acquiring HIV-1. In the United States, HSV-2 infects some 1.6 million new people per year, with approximately 320,000 of those suffering from disease symptoms. At least 40 million people in the United States are infected with HSV-2. Even higher infection rates are evident in developing countries, with further complications in people also infected with HIV. All HSV-2 infections are permanent and result in periodic virus shedding. There is no approved vaccine for HSV-2. Although antiviral regimens have become a standard of care, we believe their inconvenience, cumulative cost and potential for drug resistance underscore the need for safe, new approaches to reducing HSV-2 lesions, shedding, and transmission. Estimated annual costs of treating HSV-2 in the United States alone are close to $1 billion, primarily for drugs and outpatient medical care.
Other Infectious Disease Programs
In 2005, we applied our DNA delivery technology to the development of a pandemic influenza vaccine formulated with our proprietary adjuvant Vaxfectin®. Our approach is to include vaccine components which we believe will provide potential cross strain protection, particularly against severe disease and mortality, unlike conventional influenza vaccines which provide symptomatic relief through antibodies alone and are unlikely to protect against severe disease and mortality if the strain match is not correct.
In 2008, we completed two Phase 1 H5N1 pandemic influenza trials. The data demonstrated that a Vaxfectin®-formulated DNA vaccine can achieve significant immune responses against H5N1 pandemic influenza in humans. The data suggested that the vaccine was well-tolerated and achieved potentially protective levels of antibody responses (H5 hemagglutination inhibition, or HI, titers of at least 40 and at least a four-fold increase from baseline) in at least 47% and up to 67% of evaluable subjects in the higher H5 dose cohorts in the trials. The data also showed that in the highest H5 dose cohorts, responses peaked by Day 56 and were sustained in 80% to 100% of the responders through the end of the study at Day 182. The vaccines demonstrated cross-clade antibody responses against two different strains in the trials and also induced T-cell responses against a matching strain of influenza virus in 75% to 100% of the subjects for at least six months. No significant safety issues were observed at any of the doses tested.
In 2011, we completed a Phase 1 clinical trial of our Vaxfectin®-formulated DNA vaccine against A/H1N1 pandemic influenza. The double-blind, placebo-controlled H1N1 influenza vaccine Phase 1 trial enrolled approximately 30 healthy adult volunteers at a single U.S. clinical site. Subjects were randomized 2:1 into the vaccine or placebo arms of the trial. The data suggested that the vaccine was well-tolerated and that more than half of the subjects in the trial generated neutralizing antibodies of the type expected to provide protection against the disease.
At this time, we do not intend to pursue further development of our pandemic influenza vaccines without funding through research grants, collaborations or otherwise. We believe we can further optimize the vaccine dose and formulation, confirm safety and immunogenicity in a larger number of subjects, and leverage the proof of concept for our DNA vaccine platform and Vaxfectin® adjuvant into additional indications.
There are also two ongoing infectious disease programs being conducted by the U.S. government using our technology. The National Institutes of Health, or NIH, has clinical-stage vaccine programs based on our technology for HIV and is currently conducting a Phase 2b DNA-prime, adenovirus-boost study. The Naval Medical Research Center, or NMRC, has initiated a Phase 1 study of a tetravalent (serotype 1, 2, 3, and 4) dengue DNA vaccine formulated with Vaxfectin®.
In addition to these programs, we have developed several vaccines targeted at emerging diseases including malaria, anthrax, severe acute respiratory syndrome, or SARS, West Nile Virus, or WNV, and Ebola. We have
11
Table of Contents
performed preclinical work and completed a Phase 1 clinical trial targeting anthrax. The NIH, has completed Phase 1 clinical trials using our vaccines targeting SARS, WNV and Ebola. Due to the lack of commercial opportunities and government funding, we do not plan on further developing these vaccines at this time.
Independent Program Targeting Cancer
Allovectin®
Allovectin® is a plasmid/lipid formulation containing the DNA sequences encoding HLA-B7 and ß2 microglobulin, which together form a MHC Class I complex. We believe injection of Allovectin® directly into tumor lesions directs a local, regional and systemic immune response against metastatic tumors through several mechanisms. In HLA-B7 negative patients, a vigorous allogeneic immune response may be initiated against the foreign MHC class I antigen. In all patients,ß2 microglobulin may reconstitute normal class I antigen presentation and/or increase tumor antigen presentation to the immune system. In any patient, an innate pro-inflammatory response may occur that induces tumor responses following intralesional injection of the pDNA/lipid complex. The goal of all three of these mechanisms is to initially cause recognition of the tumor at the local site to allow a then-sensitized immune response to recognize un-injected tumors at distant metastatic sites.
In 2001, we began a high-dose, 2 mg, Phase 2 trial evaluating the Allovectin® immunotherapeutic alone for patients with stage III or stage IV melanoma, who have few other treatment options. The high-dose Phase 2 trial completed enrollment in 2003. The data showed that the trial had a total of 15 responders among the 127 patients receiving the high dose (11.8 %), with four of the patients having complete responses and 11 having partial responses. Data were recently updated after long-term follow-up. The Kaplan-Meier estimated median duration of response was 13.8 months, and all responses were durable with a range of 6 months to 77 months. The updated Kaplan-Meier median survival was 18.8 months. The safety profile was excellent with no reported Grade 3 or Grade 4 adverse events associated with Allovectin®.
Based on detailed guidance received from the U.S. Food and Drug Administration, or FDA, in an End-of-Phase 2 meeting, we subsequently completed a Special Protocol Assessment, or SPA, with the FDA for a Phase 3 trial of high-dose, 2 mg, Allovectin® for certain patients with recurrent Stage III or Stage IV melanoma. The SPA-agreed protocol specifies the trial objectives and design, clinical endpoints, and planned analyses expected to yield data that will support a license application for product approval.
Enrollment in the Phase 3 trial began in December 2006 and has involved more than 100 clinical sites. Patients could have been previously treated with surgery, adjuvant therapy, and/or biotherapy, but could not have been previously treated with chemotherapy. The patients were randomized on a 2:1 basis with 260 patients treated with Allovectin® and 130 treated with their physician’s choice of either of two chemotherapy agents, dacarbazine or temozolomide. The primary endpoint is overall response rate that compares the two trial arms for objective responses that are ongoing or commence at 24 weeks or more after randomization. The study will also evaluate safety and tolerability as well as survival as secondary endpoints. The trial is currently in the final stages of patient follow-up, data collection and analysis, which is expected to be completed in late 2012.
AnGes has provided funding for the clinical trial up to certain limits under a research and development agreement. The funding consisted of purchases by AnGes of $10.85 million of restricted shares of our common stock and additional non-refundable cash payments by AnGes of $11.75 million. All of the funding provided by AnGes, including those funds used to purchase our common stock, were required to be used for costs related to the Allovectin® Phase 3 trial. Under the agreement, we granted to AnGes exclusive marketing rights for Allovectin® in specified countries in Asia and AnGes has the opportunity to pursue regulatory approvals in those countries, subject to receipt by us of regulatory approval in the United States. We also granted AnGes certain royalty-bearing licenses to our technology and know-how. AnGes is obligated to pay royalties to us on sales of Allovectin® in specified countries in Asia. AnGes also obtained the right to receive royalties from us on all commercial sales of Allovectin® outside the specified Asian countries.
12
Table of Contents
In 1999, Allovectin® was granted orphan drug designation for the treatment of invasive and metastatic melanoma by the FDA’s Office of Orphan Products Development. Orphan drug designation provides certain tax benefits for qualifying expenses and can result in extended marketing exclusivity. In 2010, the FDA granted Fast Track designation of Allovectin® as a treatment for metastatic melanoma. Fast Track designation is intended to facilitate the development and expedite the review of drugs with demonstrated potential to address an unmet medical need for serious diseases. Fast Track designation allows for submission of a Biologics License Application, or BLA, on a “rolling” basis with ongoing FDA review during the submission process.
About Metastatic Melanoma
The American Cancer Society estimated that approximately 76,250 new diagnoses of, and approximately 9,180 deaths from, melanoma occur annually in the United States. The lifetime risk of getting melanoma has been growing and is now approximately 2% (or 1 in 50) for Caucasians in the United States. Currently, there are no consistently effective therapies for advanced cases of melanoma where the cancer has spread beyond its site of origin, or metastasized. Treatment for these patients normally includes a combination of chemotherapy, radiation therapy, and surgery. In patients with advanced metastatic melanoma, median survival typically ranges from six to ten months.
FDA-approved drugs for treatment of metastatic melanoma include: hydroxyurea, which is no longer commonly used as a single agent, dacarbazine, and IL-2. The toxicity associated with FDA-approved treatments such as dacarbazine or IL-2 is often significant, resulting in serious or life-threatening side effects in many of the patients treated. Patients with metastatic melanoma often are treated with drugs that are not approved for treatment of metastatic melanoma, such as IFN-µ, which is approved as adjuvant therapy to surgery, or temozolomide, which is approved for certain types of brain cancer.
Two new drugs for metastatic melanoma were approved in 2011, both on the basis of increased survival. Yervoy®, a monoclonal antibody marketed by Bristol-Myers Squibb, increases the effectiveness of T-cells that can seek out and destroy melanoma cells. Zelboraf®, a B-Raf inhibitor marketed by Roche and Daiichi Sankyo, interrupts a key process in melanoma growth in patients with a particular melanoma mutation. Both drugs are associated with significant side effects, and neither is considered a cure for melanoma.
Adjuvant Development
Vaxfectin®
Vaxfectin® is our proprietary, cationic lipid formulation optimized to increase the immune response to vaccines. Vaxfectin® formulations have demonstrated safety and adjuvant activity in pDNA vaccine applications in multiple animal models, including nonhuman primates, in addition to the animal and human influenza studies cited above. Studies of Vaxfectin®-formulated pDNA vaccines against CMV and measles have shown enhanced immunogenicity in rodents and nonhuman primates, respectively. Vaxfectin® has also demonstrated dose-sparing attributes as an adjuvant for protein-based influenza vaccines as well as increased T-cell responses and antitumor responses to formulated peptide-based cancer antigens. In addition to the studies outlined below, there have been a number of published non-clinical infectious disease studies utilizing Vaxfectin® as an adjuvant.
DNA Vaccines with Vaxfectin®
A study has been completed which demonstrated that a measles DNA vaccine formulated with Vaxfectin® adjuvant elicited sustained protective levels of neutralizing antibodies in infant (6—10 week old) nonhuman primates confirmed by complete protection following challenge one year after intradermal vaccination, with no clinical signs of disease and no culturable virus after challenge. Similar results were found in juvenile (1—2 year old) nonhuman primates. Both measles studies were conducted in collaboration with Diane E. Griffin, M.D., Ph.D., Alfred and Jill Sommer Professor and Chair of Molecular Microbiology and Immunology, Johns Hopkins Bloomberg School of Public Health, under a grant from the Bill and Melinda Gates Foundation.
13
Table of Contents
Protein Vaccines with Vaxfectin®
Data from a study in mice showed that a seasonal influenza vaccine, Sanofi Pasteur’s Fluzone® commercial vaccine, when formulated with Vaxfectin® generated up to 200-fold higher antibody responses than an unformulated vaccine at the same dose. Formulation of Fluzone® with Vaxfectin® also allowed a nearly 10-fold reduction in vaccine dose while generating equivalent or better antibody responses compared with unformulated vaccine, even at the lowest doses tested. In separate studies conducted by third parties, Sanofi Pasteur’s H5N1 pandemic influenza vaccine with no adjuvant achieved target antibody levels in less than half the subjects after two 90 mcg doses – which are six times the normal 15 mcg dose for each strain of seasonal influenza virus that provides 75% to 90% protection against seasonal influenza. In a separate study in mice, we evaluated the potential of Vaxfectin® to be used as a dose-sparing agent with a protein-based H5N1 pandemic influenza vaccine currently stockpiled by the U.S. government. We demonstrated that after a single injection, the Vaxfectin®-formulated vaccine yielded five-fold higher antibody responses at the same dose as an unformulated vaccine, and comparable or better antibody responses at one-third the dose of unformulated vaccine. After a second injection, the Vaxfectin®-formulated vaccine yielded nine-fold higher antibody responses at the same dose as the unformulated vaccine, and five-fold better antibody responses at one-third the dose of the unformulated vaccine. Dose-sparing ability could be critical in extending limited vaccine supplies to protect the greatest number of people in the event of a pandemic influenza outbreak.
We also announced data from mouse studies which demonstrated that a Vaxfectin®-formulated seasonal influenza vaccine generated broader, more balanced antibody responses than an unformulated vaccine, and also generated influenza-specific T-cell responses. Adjusting the ratio of Vaxfectin® to vaccine allowed substantial increases in either antibody or T-cell responses, without reducing the other type of response, compared with unformulated vaccine. The ability to favor primarily antibody or T-cell responses could provide important advantages in developing vaccines for specific applications.
Cancer Vaccines with Vaxfectin®
In a mouse study completed in 2008, a Vaxfectin®-formulated vaccine containing a peptide from tyrosinase-related protein 2, or TRP-2, an antigen commonly expressed by several types of tumors including glioma and melanoma, resulted in approximately a 100-fold increase in antigen-specific CD8+ T-cell responses compared with unformulated vaccine. CD8+ T cells are deployed by the immune system to identify and destroy infected or cancerous cells.
Collaboration and Licensing Agreements
We have entered into various arrangements with corporate, academic, and government collaborators, licensors, licensees, and others. In addition to the agreements summarized below, we conduct ongoing discussions with potential collaborators, licensors and licensees.
Out-licensing
Astellas. In July 2011, we entered into license agreements with Astellas, granting Astellas exclusive, worldwide, royalty-bearing licenses under certain of the Company’s know-how and intellectual property to develop and commercialize certain products containing plasmids encoding certain forms of glycoprotein B and/or phosphoprotein 65, including TransVax™ but excluding CyMVectin™. Under the agreements, Astellas is responsible for the worldwide development and commercialization of products in the licensed field, at its expense, and has agreed to use commercially reasonable efforts to develop, obtain regulatory approval for and commercialize at least one licensed product for use in certain immunocompromised patients in the licensed field in the United States and certain other major markets. Astellas is currently preparing to begin a TransVax™ Phase 3 trial in patients to protect against CMV infection after stem cell transplants and a Phase 2 trial to protect patients against CMV infection after SOTs. Both trials are expected to begin in 2012.
14
Table of Contents
Under the terms of the license agreements, Astellas paid a nonrefundable upfront license fee of $25.0 million. We are entitled to receive an additional $10.0 million upon finalization of the trial design for a Phase 3 registration trial of TransVax™ in hematopoietic stem cell transplant recipients. We are also entitled to receive additional cash payments potentially totaling $95.0 million for achievement of certain milestones through commercial launch and to receive double-digit royalties on net sales of products and we have an option to co-promote TransVax™ in the United States. Under the terms of a supply and services agreement entered into by us and Astellas on the same date, we agreed to perform certain development and regulatory activities, at Astellas’ expense, and to supply licensed products to Astellas, at Astellas’ expense, for use in development and initial commercialization activities in the licensed field. During the year ended December 31, 2011, we recognized $25.3 million in license revenue and $2.7 million of revenue related to contract services delivered.
Prior to licensing TransVax™ to Astellas we completed a multicenter Phase 2 trial in 94 CMV-seropositive HSCT recipients (14 donor-recipient pairs and 80 recipient-only subjects), randomized 1:1 for vaccine or placebo. Subjects enrolled were 18-65 years old, CMV-seropositive, and diagnosed with selected leukemias or lymphomas. Enrollment in the trial was completed in November 2008 and a 1-year follow up was completed in November 2009. In 2010, we released 12 month post-transplant data, and showed that significant reductions were achieved for key viral reactivation metrics. The final study results were published by Lancet Infectious Diseases in January 2012.
In 2005, the Office of Orphan Products Development of the FDA designated TransVax™ as an orphan drug for the prevention of clinically significant CMV viremia, CMV disease and associated complications in at-risk transplant populations. Orphan drug designation provides certain tax benefits for qualifying expenses and can result in extended marketing exclusivity.
We initially developed TransVax™ as a pathway to establish a CMV vaccine proof of concept in a relatively small patient population. We decided to specifically target patients who were susceptible to CMV reactivation in HSCT patients. Therefore we designed a vaccine that would primarily induce a cellular immune response. TransVax™ is a plasmid DNA vaccine that induces both T-cell and antibody responses by expressing two antigens, phosphoprotein 65, or pp65, and glycoprotein B, or gB. The tegument protein, pp65, is a major antigen recognized by T cells in CMV-infected individuals. The gB protein is a major surface antigen of CMV and a primary target of neutralizing antibodies. The gB protein is also a major CMV antigen recognized by both CD4+ and CD8+ T cells in CMV-seropositive subjects. Induction of gB-specific T cells following DNA vaccination may provide an additional antiviral mechanism that could act to reduce CMV replication and viral loads shortly after infection. The vaccine is also formulated with poloxamer CRL1005, which has been shown in nonclinical studies by us and others to enhance gene expression and immune responses.
AnGes. In 2005, we granted an exclusive worldwide license to AnGes for use of our core DNA delivery technology in the development and commercialization of DNA-based products encoding hepatocyte growth factor, or HGF, for cardiovascular applications. HGF is a human protein that causes angiogenesis in areas of ischemia.
AnGes is developing Collategene™, a DNA-based delivery of HGF for indications related to PAD, a severe condition caused by blockage of blood vessels feeding the foot and lower leg. AnGes completed a Phase 3 trial in Japan in 2007 with Collategene™ for CLI, an advanced form of PAD. AnGes completed two Phase 2 trials in the United States in 2006, with Collategene™ for PAD. AnGes has partnered with Daiichi Pharmaceutical Co., Ltd., a wholly owned subsidiary of Daiichi Sankyo Company Limited, for development and commercialization of DNA-based HGF for PAD in Japan. In addition, AnGes completed a Phase 1 trial in the United States for IHD in 2006.
In mid-2007, AnGes reported positive results following an interim analysis of data from the first 41 subjects to complete the Phase 3 CLI trial of Collategene™ in Japan. In the trial, 40 subjects with CLI were evaluated for efficacy. The primary endpoints, improvement of rest pain or ischemic ulcer size, at 12 weeks post dosing,
15
Table of Contents
showed 30.8% improvement in the placebo group and 70.4% improvement in the treatment group, a statistically significant difference. Based on the findings that the primary efficacy endpoint in the trial had been achieved with high statistical significance compared to a placebo and that there were no major safety concerns related to treatment in 41 patients evaluated, an Independent Data Monitoring Committee recommended stopping the trial early to prevent potential ethical issues involving the subjects in the placebo group. AnGes filed an application for Japanese marketing approval in March 2008. In September 2010, AnGes announced that after a series of extensive consultations with the Japanese Pharmaceuticals and Medical Devices Agency, it had decided to conduct an additional clinical trial of Collategene™ and that in the interim it would be withdrawing its new drug application, or NDA, in Japan.
In December 2010, AnGes released long-term follow-up data from its Phase 3 CLI trial in Japan. The follow-up analysis was performed 3 years after gene delivery. The rate of lower limb amputation after 1 year in patients with CLI is typically 30%. The results of the study showed lower rates of lower limb amputation after administration of Collategene: 5.4% after one year, 5.4% after two years, and 9.2% after three years. The mortality rates at 1, 2 and 3 years in patients treated for CLI is typically 25%, 25% and 52.9%. The results of the study showed lower mortality rates after administration of Collategene which was 5.1% after one year, 15.7% after two years, and 26.6% after three years. AnGes also reported that there were no clinically significant adverse events that were suspected of being related to gene therapy.
AnGes has obtained a SPA from the FDA for a Phase 3 clinical trial of Collategene™ for patients with CLI. The study will be multinational, randomized and placebo-controlled with a target population of 560 patients. The Phase 3 trial will enroll no option as well as poor option patients with chronic and severe ischemia of the lower limb. No option patients are those unable to receive an endovascular intervention or surgical bypass procedure due to inflow, conduit or outflow reasons or due to a severe and irreversible co-morbidity where surgery is contraindicated. Poor option patients are those unable to receive an endovascular intervention and at high risk for bypass surgery due to their vascular anatomy or severe co-morbid disease. AnGes believes inclusion of poor option patients will open the clinical trial to at least three to four times as many target patients compared to other trials that only include no option patients.
The agreement with AnGes expires upon the expiration of last to expire of the patent rights licensed to AnGes, or when the patent rights licensed to AnGes are held invalid or unenforceable, unless earlier terminated as set forth in the agreement. We may terminate the agreement early upon the bankruptcy or insolvency of, or the material breach of the agreement by, AnGes, upon prior written notice to AnGes. AnGes may terminate the agreement early upon prior written notice to us. Under the license agreement, we received an initial upfront payment of $1.0 million, and in 2008 we received an additional payment of $1.0 million. Further development may lead to additional milestone and royalty payments.
Sanofi. In 1999, Sanofi began testing the DNA delivery of a gene encoding fibroblast growth factor 1, or FGF-1, an angiogenic growth factor, in patients with CLI. In 2000, Sanofi licensed the rights to our core DNA delivery technology for cardiovascular applications using FGF-1.
Sanofi conducted a double-blind, placebo-controlled Phase 2 trial of its FGF-1 plasmid-based therapeutic, or NV1FGF, in the United States and Europe. In March 2006, Sanofi released encouraging data from the Phase 2 trial demonstrating improvement in amputation-free survival in patients with CLI. In 2007, Sanofi announced that it had begun a 500 patient global Phase 3 study of NV1FGF. The trial was conducted in patients with CLI, with combined trial endpoints of major amputation or death. In September 2010, Sanofi announced that NV1FGF did not meet the primary endpoint in the global Phase 3 trial and that it is evaluating all options with respect to NV1FGF development in light of the Phase 3 clinical trial results.
Our agreement with Sanofi specifies that we are eligible to receive milestone payments plus royalties if and when products advance through commercialization. The agreement with Sanofi expires on the expiration of Sanofi’s obligation to pay royalties under the agreement. The obligation to pay royalties is the longer of seven
16
Table of Contents
years after the first commercial sale or when the patent rights licensed to Sanofi are held invalid or unenforceable, unless earlier terminated as set forth in the agreement. We may terminate the agreement early for an uncured, material breach of the agreement by Sanofi, upon prior written notice to Sanofi. Sanofi may terminate the agreement early upon prior written notice us.
Merck. In 1991, we entered into an agreement with Merck, which was subsequently amended, providing Merck with certain exclusive rights to develop and commercialize vaccines using our core DNA delivery technology for specified human diseases. Under the agreement, as amended, Merck licensed our core DNA delivery technology for use in preventive and therapeutic human infectious disease vaccines.
In 2005, Merck exercised an option related to three cancer targets that were granted under an amendment to the agreement. In 2008, Merck initiated a Phase 1 clinical trial of a candidate vaccine based on our DNA gene delivery technology and encodes human telomerase reverse transcriptase, or hTERT. As a result of the ongoing development of this vaccine, we received milestone payments of $1.5 million in 2009.
Merck also has rights to use the licensed technology for HIV, hepatitis C virus, and hepatitis B virus. Merck is obligated to pay fees if certain research milestones are achieved, and royalties on net sales if any products covered by our agreement with Merck are commercialized. The agreement with Merck expires on the expiration of Merck’s obligation to pay royalties under the agreement. The obligation to pay royalties is the longer of five years after the first commercial sale or upon the expiration of the last to expire of the patent rights licensed to Merck, or when the patent rights licensed to Merck are held invalid or unenforceable, unless earlier terminated as set forth in the agreement. We may terminate the agreement early upon the bankruptcy or insolvency of, or a material breach of the agreement by, Merck, upon prior written notice to Merck. Merck may terminate the agreement early upon prior written notice to us.
As of December 31, 2011, the aggregate potential milestone payments that we were eligible to receive under each of our out-license agreements with Astellas, AnGes, Sanofi and Merck was equal to approximately $62.5 million, $2.0 million, $5.0 million and $36.0 million, respectively. These amounts assume that all remaining milestones associated with the milestone payments are met. Although we believe that some of the milestones contained in these out-license agreements may be achieved, it is highly unlikely that a significant number of them will be achieved. Because the milestones are highly contingent and we have limited control over whether the development and regulatory milestones will be achieved, we are not in a position to reasonably estimate how much, if any, of the potential milestone payments will ultimately be received, or when. Additionally, under these out-license agreements, many of the milestone events are related to progress in clinical trials which will take several years to achieve.
We are also eligible to receive royalty payments under these out-license agreements based on net sales of any products which incorporate the out-licensed technology. In each of our out-license agreements with Astellas, AnGes, Sanofi and Merck, these royalties are based on percentages of net sales in the single to low double digit range. Our receipt of any royalty payments under our out-license agreements is contingent upon the licensee successfully developing and commercializing products incorporating the licensed technology, and we have limited control over our licensees’ efforts in this regard. Consequently, we are not in a position to reasonably estimate when or to what extent we will receive any royalty payments under our out-license agreements.
Aqua Health. In 2003, we granted a non-exclusive license to Aqua Health for use in Canada of our core DNA delivery technology in a vaccine against a disease that affects both wild and farm-raised fish. In 2005, Aqua Health received notification of approval from the Canadian Food Inspection Agency to sell its proprietary product, Apex®-IHN, a DNA vaccine to protect farm-raised salmon against IHNV. We believe this approval is an important step in the validation of our DNA delivery technology. Aqua Health pays royalties to us on sales of the vaccine.
Merial. In 2004, we granted an exclusive license to Merial for use of our core DNA delivery technology in a therapeutic vaccine designed to aid in extending survival time of dogs with oral melanoma. Under the agreement, Merial is responsible for research and development activities. In March 2009, Merial received approval from the USDA to market the DNA vaccine, now called ONCEPT™. Merial pays royalties to us on sales of the vaccine.
17
Table of Contents
Academic Collaborators. We also have granted non-exclusive, academic licenses to our DNA delivery technology patent estate to 11 leading research institutions including Stanford, Harvard, Yale and MIT. The academic licenses are intended to encourage widespread commercial use of our innovative DNA delivery technologies in the development of new antibodies, vaccines, therapeutic proteins, and diagnostics. The academic licenses allow university researchers to use our technology free of charge for educational and internal, non-commercial research purposes. In exchange, we have the option to exclusively license from the universities potential commercial applications stemming from their use of the technology on terms to be negotiated.
In-licensing
CytRx. In 2001, we entered into an exclusive agreement with CytRx Corporation, or CytRx, which grants us rights to use or sublicense CytRx’s poloxamer technology to enhance viral or non-viral delivery of polynucleotides in all unexcluded preventive and therapeutic human and animal health applications, including CMV. In addition, the agreement permits our use of CytRx’s technology to enhance the delivery of proteins in prime-boost vaccine applications that involve the use of polynucleotides. As part of the agreement, we made a $3.8 million up-front payment and agreed to make potential future milestone and royalty payments.
The agreement with CytRx expires upon the expiration of our royalty obligations, unless earlier terminated as set forth in the agreement. Each party may terminate the agreement early upon the bankruptcy or insolvency of, or the material breach of the agreement by, the other party, upon prior written notice to the other party. Subject to certain conditions, we may terminate the agreement early upon prior written notice to CytRx.
City of Hope. In 2003, we licensed from the City of Hope on an exclusive basis various U.S. patents that provide protection for CMV-related polynucleotide based vaccines, including our TransVaxTM and CyMVectinTM vaccine candidates. As of December 31, 2011, we had paid the City of Hope $2.3 million under the agreement. The agreement expires upon the last to expire of the patent rights licensed by us under the agreement, unless earlier terminated as set forth in the agreement. The City of Hope may terminate the agreement early, in accordance with notice provisions set forth in the agreement, if we cease to operate, fail to make payments when due or materially breach the agreement. Subject to certain conditions, we may terminate the agreement early at any time upon prior written notice to the City of Hope. We are also obligated to pay a low double-digit percentage of any payments we receive from the sub-license of products that incorporate the licensed technology.
Wisconsin Alumni Research Foundation. Under a 1989 research agreement, scientists at the University of Wisconsin, Madison, and our scientists co-invented a core technology related to intramuscular DNA administration. In 1991, we licensed from the Wisconsin Alumni Research Foundation, or WARF, its interest in that technology. Pursuant to the license agreement with the WARF, we paid the WARF an initial license fee and agreed to pay the WARF up to 10% of certain initial upfront monetary payments and a small percentage of some royalty payments received from third parties under sublicense agreements. As of December 31, 2011, we had paid the WARF an aggregate of $2.9 million under this agreement. The agreement expires once we have fulfilled our royalty obligations thereunder, unless earlier terminated as set forth in the agreement. The WARF may terminate this agreement early, in accordance with notice provisions set forth in the agreement, if we fail to make payments when due, materially breach the agreement or commit any act of bankruptcy or become insolvent. Subject to certain conditions, we may terminate the agreement early at any time upon prior written notice to the WARF.
University of Michigan. In 1992, we licensed from the University of Michigan rights to various U.S. and international patents that provide additional protection for Allovectin® related to the injection of DNA-based therapeutics into tumors. In July 2005, we amended the agreement to exclude certain patents. In February 2006, we entered into an additional agreement with the University of Michigan which provides for rights to a composition of matter patent related to a polycistronic plasmid and the use of this plasmid for the treatment of solid tumors, which we believe provides additional protection for Allovectin®.
18
Table of Contents
As of December 31, 2011, we have paid the University of Michigan an aggregate of $0.5 million under our 1992 agreement with the University of Michigan. The agreement expires upon the last to expire of the patent rights licensed by us under the agreement, unless earlier terminated as set forth in the agreement. Either party may terminate the agreement early, in accordance with notice provisions set forth in the agreement, upon material breach of the agreement by the other party. Subject to certain conditions, we may terminate the agreement early at any time upon prior written notice to the University of Michigan.
As of December 31, 2011, we had paid the University of Michigan only de minimus amounts under our 2006 agreement with the University of Michigan. The agreement expires upon the last to expire of the patent rights licensed by us under this agreement, unless earlier terminated as set forth in the agreement. The University of Michigan may terminate the agreement early, in accordance with notice provisions set forth in the agreement, if we cease to operate, fail to make payments when due or materially breach the agreement. Subject to certain conditions, we may terminate the agreement early at any time upon prior written notice to the University of Michigan.
As of December 31, 2011, the aggregate potential milestone payments that we could be obligated to pay under our in-license agreements with City of Hope, CytRx and the WARF, our 1992 in-license agreement with the University of Michigan and our 2006 in-license agreement with the University of Michigan was equal to approximately $11.9 million, $2.2 million, $4.9 million, $30,000 and $25,000, respectively. These amounts assume that all remaining milestones associated with the milestone payments are met. Although we believe that some of the milestones contained in the in-license agreements may be achieved, it is highly unlikely that a significant number of them will be achieved. Because the milestones are highly contingent and we have limited control over whether the regulatory milestones will be achieved, we are not in a position to reasonably estimate how much, if any, of the potential milestone payments will ultimately be paid, or when. Additionally, under these in-license agreements, many of the milestone events are related to progress in clinical trials which will take several years to achieve.
Under these in-license agreements, we may also be obligated to pay royalties based on net sales of any products which incorporate the in-licensed technology. In each of our in-license agreements with the City of Hope, CytRx, the WARF and the University of Michigan, these royalties are based on percentages of net sales in the single digit range. We may also be obligated to make payments under our in-license agreements with the City of Hope, WARF and the University of Michigan based on amounts we receive from sub-licensees, if any. Our obligations to pay any royalty payments under our in-license agreements is contingent upon the successful development and commercialization of products incorporating the in-licensed technology. Before any products incorporating the in-licensed technology may be sold, such products must be approved by U.S. or foreign regulatory authorities, which will require a substantial amount of additional research and development. Even if such product candidates are advanced through clinical trials, the results of such trials may not support approval by the FDA or comparable foreign agencies. Consequently, we are not in a position to reasonably estimate when or to what extent we will be obligated to pay any royalties under our in-license agreements.
Contract Manufacturing
IPPOX Foundation
In 2010, we entered into an agreement to manufacture plasmid DNA vaccines against HIV under a $2.4 million contract with the IPPOX Foundation, a collaborating institution for the Poxvirus Vaccine Regimen Design led by the Centre Hospitalier Universitaire Vaudois under the auspices of the Collaboration for AIDS Vaccine Discovery. We delivered the vaccine and recognized $2.4 million in revenue related to this contract in 2010.
Naval Medical Research Center
In 2008, we entered into a contract with the NMRC to manufacture a dengue DNA vaccine formulated with our Vaxfectin® adjuvant. We manufactured the vaccine and the adjuvant under a $1.3 million contract, and
19
Table of Contents
provided regulatory and clinical expertise. The dengue vaccine was delivered to the NMRC in 2009. The NMRC is currently conducting a Phase 1 clinical trial using the vaccine. While we do not have any ongoing rights to the NMRC’s dengue DNA vaccine, the vaccine does incorporate our patented Vaxfectin® adjuvant. Therefore any commercialization of the vaccine would require a license to these patents.
Manufacturing Process Development
In 2005, we were awarded funding for a one-year, $0.5 million project for the Defense Advanced Research Projects Agency of the U.S. Department of Defense. The award funded feasibility studies of a new approach for rapidly manufacturing large quantities of DNA vaccines. In 2007, we were awarded funding for a three-year, $6.0 million grant from the NIAID for further development of a DNA vaccine manufacturing process with the potential to produce several million doses of vaccines in a matter of days. The grant period was extended to allow preclinical development to continue through May 2011. Our RapidResponse™ DNA vaccine manufacturing platform is intended to significantly reduce the time required to develop, manufacture and deploy vaccines against emerging diseases during the early stages of an infectious outbreak. The RapidResponse™ platform produces a small segment of DNA, called a linear expression cassette, which includes only those DNA sequences essential for the specific vaccine. The bacterial fermentation process typically used for DNA vaccines produces a closed loop of DNA, called a plasmid, which must include DNA sequences required in the manufacturing process.
Conventional vaccine development and manufacturing methods require prolonged effort after the emergence of a new pathogen for production of even a single dose for testing. Current DNA vaccine development and manufacturing processes allow initial production of vaccines in as little as three months after selection of a gene sequence associated with a pathogen, but quantities are limited by the batch-processing capacity of available manufacturing equipment. By using a cell-free manufacturing process, we believe that the RapidResponse™ DNA platform can overcome the time, capacity and cost challenges of manufacturing conventional vaccines for diseases such as influenza, which use killed or disabled viruses grown in chicken eggs or via cell culture, requiring months of production time in large, dedicated facilities.
NIH Vaccine Research Center
The NIH through its Dale and Betty Bumpers Vaccine Research Center, or VRC, has clinical stage vaccine programs based on our technology for HIV. The NIH has also completed Phase 1 studies based on our technology for Ebola, WNV and SARS. We were granted exclusive options to exclusively or nonexclusively license any inventions developed by the NIH under the respective Cooperative Research and Development Agreement, or CRADAS, for HIV, Ebola or WNV. The options and licenses that were exercised under the CRADAS have since expired or terminated due to the discontinuation of these programs by the NIH. The NIH has transferred the IND application for its SARS DNA vaccine to us and we continue to evaluate our options in continuing the development of that vaccine.
Based on encouraging results in prior studies, the NIH initiated a Phase 2a clinical trial in 2005 of the “prime-boost” vaccine approach against HIV in several hundred patients. In 2007, the NIH released results from its Phase 2a HIV vaccine trial using a DNA prime-adenoviral vector boost approach. The results showed the vaccine regimen was safe and well-tolerated, and was effective in inducing T-cell immune responses in up to 70% of the vaccine recipients. The NIH planned to further test the DNA prime-adenoviral vector boost approach in a trial known as the PAVE 100 study, which was designed to enroll 8,500 volunteers. We manufactured the DNA prime component of the vaccine to be used in the PAVE 100 study. The study was to begin recruitment in October 2007, but was postponed following the NIH’s review of interim data from an unrelated Phase 2b trial known as the STEP study which utilized an adenoviral vector vaccine alone. The NIH concluded that the adenoviral vector vaccine failed to prevent HIV infection or reduce viral load, and the vaccinated group in the STEP study exhibited a higher incidence of infection than the placebo group. In July 2008, after soliciting and considering broad input from the scientific and HIV communities, the NIH determined that it would not conduct the PAVE 100 study.
20
Table of Contents
In August 2009, the NIH opened a smaller version of the PAVE 100 study, HVTN 505. The HVTN 505 study is designed to enroll 2,200 HIV-seronegative men in a Phase 2b trial of a prime-boost vaccine regimen for HIV using three doses of DNA vaccine, previously manufactured by us for the PAVE 100 study, followed by a single dose of adenoviral vector vaccine. Enrollment in this study is ongoing.
Intellectual Property
Patents and other proprietary rights are essential to our business. We file patent applications to protect our technologies, inventions, and improvements to our inventions that we consider important to the development of our business. We believe we have a comprehensive patent portfolio in the United States and in key foreign markets. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position.
Our patents and patent applications cover, for example, DNA delivery for immunization and delivery of therapeutic proteins, specific DNA constructs and formulations of gene-based product candidates, methods for producing pharmaceutical-grade DNA, and several families of lipid molecules and their uses in DNA delivery, as described more fully below:
| • | Core DNA Delivery Technology. We and the WARF co-own rights to issued U.S. patents covering our core DNA delivery technology, including patents on methods of administering DNA sequences for the purposes of expressing therapeutic proteins or for inducing immune responses and the administration of DNA sequences into blood vessels and the heart. In 1991, the WARF exclusively licensed its rights in the core DNA delivery technology to us. All of our clinical programs are dependent upon this platform technology. The remaining patents in this family expire between December 3, 2013 and December 30, 2014, however, under the Hatch-Waxman Act, a patent term extension for up to five years may be available in the U.S. under certain conditions. |
| • | Lipid Technologies. We are the sole assignee of issued U.S. patents covering numerous examples of cationic lipid compounds that are used to facilitate delivery of gene therapies to some tissues. These patented compounds include the lipids contained in our Allovectin® product candidate, and the last to expire patent related to this family is scheduled to expire on October 17, 2012. Our Allovectin® product candidate, pandemic influenza vaccine candidates, CyMVectinTM prophylactic vaccine candidate for CMV as well as our Vaxfectin® adjuvant are protected in-part by lipid technology and/or lipid compound patents that expire between May 18, 2011 and March 24, 2020. Patent protection of these key lipids also has been obtained in Europe, Canada and Japan. Under the Hatch-Waxman Act, a patent term extension for up to 5 years may be available in the U.S. under certain conditions. |
| • | Specific DNA Therapeutics. We have supplemented the broad patent coverage described above with patents covering specific product applications of our technologies. To date, we have received patents in the United States and Europe, and have patents pending in Canada and Japan, relating to codon-optimized polynucleotide-based vaccines against human CMV infection. The issued patents expire between December 19, 2023 and May 12, 2025. These patents further protect both our TransVaxTM therapeutic vaccine candidate for CMV as well as our CyMVectinTM prophylactic vaccine candidate for CMV. We also have an issued U.S. patent relating specifically to our pandemic influenza vaccine programs and foreign counterparts pending in Australia, Canada, Europe and Japan all of which will expire on May 18, 2025. We and the University of Michigan are the co-assignees of patents directed towards compositions of matter related to a polycistronic plasmid and the use of this plasmid for the treatment of solid tumors, which we believe provide additional protection for Allovectin®. In 2006, the University of Michigan exclusively licensed its rights in these patents to us. These patents have issued in the U.S., Canada, Europe and Japan, and expire between May 27, 2014 and June 8, 2016. Our therapeutic vaccine candidate for herpes simplex type 2 virus is further augmented by an issued U.S. patent and foreign counterparts pending in Australia, Canada, Europe and Japan all of which will expire on July 20, 2027. These patents are co-owned with the University of Washington. Under the Hatch-Waxman Act, a patent term extension for up to 5 years may be available in the U.S. under certain conditions. |
21
Table of Contents
| • | DNA Process Technologies. As a result of our pioneering efforts to develop the use of DNA as a therapeutic agent, we have also developed manufacturing processes for producing pharmaceutical-grade DNA. To date, we are the exclusive assignee of patents issued in the U.S. and granted in Japan and Europe covering various steps involved in the process of economically producing pure plasmids for pharmaceutical use. This provides a further level of protection to each of our ongoing programs. Patents within this category expire between February 1, 2014 and November 24, 2023. Under the Hatch-Waxman Act, a patent term extension for up to 5 years may be available under certain conditions. |
| • | Licensed DNA Delivery Technologies. We and the University of Michigan are the co-assignees of patents directed towards compositions of matter related to a polycistronic plasmid and the use of this plasmid for the treatment of solid tumors, which we believe provides additional protection for Allovectin®. In 2006, the University of Michigan exclusively licensed its rights in these patents to us. These patents have issued in the U.S., Canada, Europe and Japan, and expire between May 27, 2014 and June 8, 2016. Under the Hatch-Waxman Act, a patent term extension for up to 5 years may be available in the U.S. under certain conditions. |
During 2011, we were issued four U.S. patents, three Japanese patents, two Canadian patents, and a European patent related to our core DNA delivery technology, enhancements of that technology, and applications of that technology:
| • | Canadian Patent No. 2,365,416 and Japanese Patent No. 4,800,485 covering our novel cationic lipid/co-lipid adjuvant, Vaxfectin® which expires March 24, 2020; |
| • | U.S. Patent No. 7,879,339 and No. 7,935,352, assigned to us and the University of Washington, covering DNA vaccines for herpes simplex virus type 2 which expires July 20, 2027; |
| • | European Patent No. 1337242 covering gene delivery formulations and methods for treatment of ischemic disease which expires October 19, 2021; |
| • | U.S. Patent No. 8,080,642 covering severe acute respiratory syndrome DNS vaccine composition and methods of use which expires May 12, 2024; |
| • | Canadian Patent No. 2,508,281covering methods for producing sterile polynucleotide based medicaments which expires December 2, 2023; |
| • | Japanese Patent No. 4723097 covering cytofectin dimers and methods of use which expires May 26, 2020; |
| • | Japanese Patent No. 4723254 covering a process for purification of plasmid DNA which expires November 24, 2023; and |
| • | U.S. Patent No. 7,888,112 covering DNA vaccines for CMV containing specific gene sequences and formulated with our Vaxfectin® adjuvant which expires December 19, 2023. |
As of December 31, 2011, we were the assignee or co-assignee of 72 issued U.S. and foreign patents. We maintain our issued patents by paying maintenance fees to the patent office in each country when due. Where appropriate, we participate in legal proceedings to vigorously defend against the revocation or withdrawal of our patents. The scope and nature of these proceedings generally differ depending on the country in which they are initiated.
Among these issued patents, a granted patent in Europe related to our core DNA delivery technology was opposed by eight parties and revoked under an initial ruling. We appealed this decision and in December 2010 our appeal was successful and the patent was reinstated by the Appeals Board and sent back to the Opposition Division for further processing. While this patent expired in March 2010, the reinstatement gives us the ability to prosecute any infringing activity that may have occurred prior to the patent expiring and within five years of
22
Table of Contents
initiation of the legal proceedings. We may use other issued patents and patent applications that are pending in Europe to protect our DNA technology. If we are not successful in defending our patents, we may lose all or part of our proprietary rights related to those patents in these geographic regions.
As of December 31, 2011, we were also prosecuting 61 pending patent applications in the United States and in foreign countries that cover various aspects of our proprietary technologies, not including patent applications for which we are a co-assignee and that are being prosecuted by our partners. One of the pending foreign patent applications is an international patent application under the Patent Cooperation Treaty, which preserves our right to pursue national-phase patent applications in a large number of foreign countries.
See “Item 3—Legal Proceedings,” for a discussion of patent-related disputes, oppositions, and prosecution status. See also “Risk Factors—Our patents and proprietary rights may not provide us with any benefit and the patents of others may prevent us from commercializing our products,” and “The legal proceedings to obtain and defend patents, and litigation of third-party claims of intellectual property infringement, could require us to spend money and could impair our operations.”
Commercialization and Manufacturing
Because of the broad potential applications of our technologies, we intend to develop and commercialize products both on our own and through our collaborators and licensees. We intend to develop and commercialize products in well-defined specialty markets, such as infectious diseases, oncology and other life-threatening diseases. Where appropriate, we intend to rely on strategic marketing and distribution alliances.
We believe our plasmids can be produced in commercial quantities through uniform methods of fermentation and processing that are applicable to all plasmids. In addition, our formulations consist of components that are synthesized chemically using traditional, readily scalable organic synthesis procedures.
We produce and supply our own plasmids for all of our research needs and clinical trials and intend to produce sufficient supplies for all foreseeable clinical investigations. In 2002, we signed a 15-year lease on our current facility, which we believe will be sufficient for our foreseeable commercial manufacturing requirements. The facility received a California Food and Drug Branch manufacturing facility license and began production in 2004. We also engage in contract manufacturing of plasmid investigational products for selected clients.
Competition
We are aware of several development-stage and established enterprises, including major pharmaceutical and biotechnology firms, which are actively engaged in infectious disease vaccine research and development. These include Sanofi, Novartis, GlaxoSmithKline plc, MedImmune, Inc., a wholly owned subsidiary of AstraZeneca, Merck and Pfizer Inc., among others. We may also experience competition from companies that have acquired or may acquire technologies from companies, universities and other research institutions. These companies may develop proprietary technologies which may materially and adversely affect our business.
In addition, a number of companies are developing products to address the same diseases that we are targeting. For example, Sanofi, MedImmune, Roche, GlaxoSmithKline, AlphaVax in conjunction with Novartis, and others have products or development programs for CMV treatment and prevention. Bristol-Myers Squibb, Roche in conjunction with Daiichi Sankyo through its acquisition of Plexxikon, GlaxoSmithKline, Amgen through its acquisition of BioVex, Celgene and others are developing treatments for melanoma. If these or any other companies develop products with efficacy or safety profiles significantly better than our products, we may not be able to commercialize our products, and sales of any of our commercialized products could be harmed.
Some of our competitors and potential competitors have substantially greater product development capabilities and financial, scientific, marketing and human resources than we do. Competitors may develop
23
Table of Contents
products earlier, obtain FDA approvals for products more rapidly, or develop products that are more effective than those under development by us. We will seek to expand our technological capabilities to remain competitive, however, research and development by others may render our technologies or products obsolete or noncompetitive, or result in treatments or cures superior to ours.
Our competitive position will be affected by the disease indications addressed by our product candidates and those of our competitors, the timing of market introduction for these products and the stage of development of other technologies to address these disease indications. For us and our competitors, proprietary technologies, the ability to complete clinical trials on a timely basis and with the desired results, and the ability to obtain timely regulatory approvals to market these product candidates are likely to be significant competitive factors. Other important competitive factors will include the efficacy, safety, ease of use, reliability, availability and price of products and the ability to fund operations during the period between technological conception and commercial sales.
The FDA and other regulatory agencies may expand current requirements for public disclosure of DNA-based product development data, which may harm our competitive position with foreign and U.S. companies developing DNA-based products for similar indications.
Government Regulation
Any products we develop will require regulatory clearances prior to clinical trials and additional regulatory approvals prior to commercialization. New gene-based products for vaccine or therapeutic applications are subject to extensive regulation by the FDA and comparable agencies in other countries. The precise regulatory requirements with which we will have to comply are uncertain at this time due to the novelty of the gene-based products and indications, or uses, that are currently under development. Our potential products will be regulated either as biological products or as drugs. In the United States, drugs are subject to regulation under the Federal Food, Drug and Cosmetic Act, or the FDC Act. Biological products, in addition to being subject to provisions of the FDC Act, are regulated in the United States under the Public Health Service Act. Both statutes and related regulations govern, among other things, testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising, and other promotional practices.
Obtaining FDA approval is a costly and time-consuming process. Generally, FDA approval requires that preclinical studies be conducted in the laboratory and in animal model systems to gain preliminary information on efficacy and to identify any major safety concerns. The results of these studies are submitted as a part of an IND application which the FDA must review and allow before human clinical trials can start. The IND application includes a detailed description of the proposed clinical investigations.
A company must submit an IND application for each proposed product and must conduct clinical studies to demonstrate the safety and efficacy of the product necessary to obtain FDA approval. The FDA receives reports on the progress of each phase of clinical testing and may require the modification, suspension, or termination of clinical trials if an unwarranted risk is presented to patients.
To obtain FDA approval prior to marketing a pharmaceutical product in the United States typically requires several phases of clinical trials to demonstrate the safety and efficacy of the product candidate. Clinical trials are the means by which experimental treatments are tested in humans, and are conducted following preclinical testing. Clinical trials may be conducted within the United States or in foreign countries. If clinical trials are conducted in foreign countries, the products under development as well as the trials are subject to regulations of the FDA and/or its counterparts in the other countries. Upon successful completion of clinical trials, approval to market the treatment for a particular patient population may be requested from the FDA in the United States and/or its counterparts in other countries.
Clinical trials for therapeutic products are normally done in three phases. Phase 1 clinical trials are typically conducted with a small number of patients or healthy subjects to evaluate safety, determine a safe dosage range,
24
Table of Contents
identify side effects, and, if possible, gain early evidence of effectiveness. Phase 2 clinical trials are conducted with a larger group of patients to evaluate effectiveness of an investigational product for a defined patient population, and to determine common short-term side effects and risks associated with the drug. Phase 3 clinical trials involve large scale, multi-center, comparative trials that are conducted to evaluate the overall benefit-risk relationship of the investigational product and to provide an adequate basis for product labeling. In some special cases where the efficacy testing of a product may present a special challenge to testing in humans, such as in the case of a vaccine to protect healthy humans from a life-threatening disease that is not a naturally occurring threat, effectiveness testing may be required in animals.
After completion of clinical trials of a new product, FDA marketing approval must be obtained. If the product is regulated as a biologic, a BLA is required. If the product is classified as a new drug, an NDA is required. The NDA or BLA must include results of product development activities, preclinical studies, and clinical trials in addition to detailed chemistry, manufacturing and control information.
Applications submitted to the FDA are subject to an unpredictable and potentially prolonged approval process. Despite good-faith communication and collaboration between the applicant and the FDA during the development process, the FDA may ultimately decide, upon final review of the data, that the application does not satisfy its criteria for approval or requires additional product development or further preclinical or clinical studies. Even if FDA regulatory clearances are obtained, a marketed product is subject to continual review, and later discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions.
Before marketing clearance for a product can be secured, the facility in which the product is manufactured must be inspected by the FDA and must comply with cGMP regulations. In addition, after marketing clearance is secured, the manufacturing facility must be inspected periodically for cGMP compliance by FDA inspectors, and, if the facility is located in California, by inspectors from the Food and Drug Branch of the California Department of Health Services.
In addition to the FDA requirements, the NIH has established guidelines for research involving human genetic materials, including recombinant DNA molecules. The FDA cooperates in the enforcement of these guidelines, which apply to all recombinant DNA research that is conducted at facilities supported by the NIH, including proposals to conduct clinical research involving gene therapies. The NIH review of clinical trial proposals and safety information is a public process and often involves review and approval by the Recombinant DNA Advisory Committee, or RAC, of the NIH.
Sponsors of clinical trials are required to register, and report results for, all controlled, clinical investigations, other than Phase 1 investigations, of a product subject to FDA regulation. Trial registration may require public disclosure of confidential commercial development data resulting in the loss of competitive secrets, which could be commercially detrimental.
We also are subject to various federal, state and local laws, regulations, and recommendations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals, and the use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research. The extent of government regulation that might result from any future legislation or administrative action cannot be accurately predicted.
Employees
As of December 31, 2011, we had 112 full-time employees, including 18 with doctorate degrees. Of these full-time employees, 85 were engaged in, or directly support, research and development and manufacturing activities, and 27 were in general and administrative positions. A significant number of our management and
25
Table of Contents
other employees have prior experience with pharmaceutical and/or biotechnology companies. None of our employees is covered by collective bargaining agreements, and our management considers relations with our employees to be good.
Executive Officers of the Registrant
Our executive officers and other executives are as follows:
| Name |
Age1 | Position | ||||
| Vijay B. Samant2 |
59 | President, Chief Executive Officer and Director | ||||
| Jill M. Broadfoot2 |
50 | Senior Vice President, Chief Financial Officer and Secretary | ||||
| Alain P. Rolland, Pharm.D., Ph.D. 2 |
52 | Executive Vice President, Product Development | ||||
| Igor P. Bilinsky, Ph.D. |
39 | Senior Vice President, Corporate Development | ||||
| Richard T. Kenney, M.D. |
53 | Vice President, Clinical Development | ||||
| Larry R. Smith, Ph.D. |
51 | Vice President, Vaccine Research | ||||
| 1 | As of December 31, 2011. |
| 2 | Executive officer. |
Vijay B. Samant joined us as President and Chief Executive Officer in November 2000. Previously, he held various positions at Merck & Co., Inc., from 1977 to 2000. From 1998 to 2000, he was Chief Operating Officer of the Merck Vaccine Division. From 1990 to 1998, he served in the Merck Manufacturing Division as Vice President of Vaccine Operations, Vice President of Business Affairs and Executive Director of Materials Management. From 1977 to 1990, Mr. Samant held a variety of positions of increasing responsibility in manufacturing, process engineering, production planning and control, business development and loss prevention in several Merck operating divisions. Mr. Samant holds a bachelor’s degree in chemical engineering from the University of Bombay, India, an M.S. degree in chemical engineering from Columbia University and an S.M. degree in management studies from the Sloan School of Management at MIT. Mr. Samant became a member of the board of directors of Raptor Pharmaceutical Corporation in 2011. Mr. Samant has been a member of the Board of Trustees for the National Foundation for Infectious Diseases and the International Vaccine Institute in Seoul, South Korea since 2008. Mr. Samant was also a Director of the Aeras Global TB Vaccine Foundation from 2001 to 2010.
Jill M. Broadfoot, joined us as Vice President, Chief Financial Officer and Secretary in October 2004 and was named Senior Vice President, Chief Financial Officer and Secretary in January 2009. From February 1999 until joining us, Ms. Broadfoot held various positions at DJO Global, Inc., a medical device company, most recently as Vice President of Finance and Controller with broad responsibilities in finance, accounting, treasury, risk management, and corporate governance. From September 1994 until joining DJO Global, Inc., Ms. Broadfoot served as an audit manager at Ernst & Young LLP, where her clients included life sciences, computer software and telecommunications companies as well as government contractors. From June 1990 until joining Ernst & Young, she was Division Controller at Medical Imaging Centers of America, Inc., a chain of freestanding imaging centers and mobile imaging centers, where she held divisional accounting and financial reporting responsibilities. Ms. Broadfoot received her bachelor’s degree in business administration and accounting from San Diego State University and is a Certified Public Accountant.
Alain P. Rolland, Pharm.D., Ph.D., joined us as Vice President, Product Development in August 2002. He was named Senior Vice President, Product Development in April 2004 and Executive Vice President, Product Development in January 2009. Dr. Rolland was Senior Vice President of Pre-Clinical Research and Development and Head of The Woodlands Center of Valentis, Inc. from 2000 to 2002. From 1993 to 1999, he served in several positions at a predecessor company to Valentis, Inc., where he progressed from Director of Gene Delivery to Vice President of Research. From 1989 to 1993, he was the Head of Formulation Research at the Research & Development Center of Galderma International in France. Prior to that, he was a scientist at the Advanced Drug
26
Table of Contents
Delivery Research Center of Ciba Geigy Pharmaceuticals in the United Kingdom. He received his Pharm.D., D.E.A., and Ph.D. degrees from Rennes University, France. Dr. Rolland holds several U.S. and European patents on advanced drug and gene delivery for medical applications. He has authored numerous publications and books in the area of nonviral gene delivery resulting from his active career in research and development. He also serves on the editorial board of several journals and was the founding Editor-in-Chief of “Current Pharmaceutical Biotechnology.”
Igor P. Bilinsky, Ph.D., joined us as Senior Vice President, Corporate Development in October 2010. Prior to joining Vical, Dr. Bilinsky was Vice President, Business Development and Special Operations at Halozyme Therapeutics, Inc. since January 2008, after joining the company in 2007 as Executive Director, Corporate Development and Special Operations. From 2005 to 2007, Dr. Bilinsky was Chief Executive Officer of Androclus Therapeutics, Inc., a privately-held biotechnology company developing novel therapeutics for autoimmune and inflammatory diseases. He joined Androclus in 2004 as Chief Operating Officer. From 1999 to 2004, Dr. Bilinsky served in positions of increasing responsibility as a management consultant, project leader and ultimately as principal in the healthcare practice of the Boston Consulting Group, where he advised companies in the biotechnology, pharmaceutical and life science industries on business strategy, operational performance and mergers and acquisitions. Prior to joining the Boston Consulting Group, Dr. Bilinsky also worked in research positions at Symyx Technologies, Inc. and the MIT Lincoln Laboratory. Dr. Bilinsky received his B.S. degree in physics from the Moscow Institute of Physics and Technology and his Ph.D. degree in physics from the MIT.
Richard T. Kenney, M.D., joined us as Vice President, Clinical Development, in December 2009. Prior to joining us, Dr. Kenney held key positions in influenza and biodefense vaccine development at GSK Biologicals (formerly ID Biomedical) from 2005 to 2009, most recently as Senior Director of Global Clinical R&D, Vaccines for Viral Diseases. His initial role at ID Biomedical was Vice President, Clinical & Regulatory Affairs. From 2001 to 2005, he served as Vice President, Clinical Development and then as Vice President, Medical & Regulatory Affairs at IOMAI Corporation. Dr. Kenney advanced through several positions from 1995 to 2001 as a researcher/reviewer at the U.S. Food and Drug Administration Center for Biologics Evaluation and Research, Office of Vaccine Research and Review. After completing his residency in internal medicine at Duke University Medical Center, Dr. Kenney pursued postdoctoral training at the NIAID, completing a fellowship in infectious diseases, and then in molecular parasitology and tropical medicine. He received board certifications in Internal Medicine and Infectious Diseases. He graduated with honors from George Washington University and earned his M.D. degree at Harvard Medical School in 1985.
Larry R. Smith, Ph.D., joined us as Executive Director, Vaccinology in September 2003, and was named Vice President, Vaccine Research in October 2006. Prior to joining Vical, Dr. Smith was Director of Viral Vaccines Research at Wyeth Vaccines, where he oversaw the immunogenicity testing of various viral vaccines including a number of recombinant viral vectors. Prior to joining Wyeth in 1996, Dr. Smith was a Scientific Investigator at Immune Response, where he identified autoreactive T-cell targets in psoriasis and multiple sclerosis which led to the clinical testing of several therapeutic vaccine candidates. Dr. Smith received a B.S. degree in Biology from Purdue University, a Ph.D. in Microbiology and Immunology from the University of Texas Medical Branch, and was a postdoctoral fellow in the Immunology Department at Scripps Clinic and Research Foundation.
You should consider carefully the risks described below, together with all of the other information included in this Annual Report on Form 10-K, and in our other filings with the SEC, before deciding whether to invest in or continue to hold our common stock. The risks described below are all material risks currently known, expected or reasonably foreseeable by us. If any of these risks actually occur, our business, financial condition, results of operations or cash flow could be seriously harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment.
27
Table of Contents
None of our independently developed product candidates has been approved for sale, and we have a limited number of independently developed product candidates in clinical trials. If we do not develop commercially successful products, we may be forced to curtail or cease operations.
All of our independently developed product candidates are either in research or development. We must conduct a substantial amount of additional research and development before any U.S. or foreign regulatory authority will approve any of our product candidates. Limited data exist regarding the efficacy of DNA vaccines or therapeutics compared with conventional vaccines or therapeutics. Results of our research and development activities may indicate that our product candidates are unsafe or ineffective. In this case, regulatory authorities will not approve them.
For example, our independently developed product candidates currently in clinical development include Allovectin®, for which we announced the completion of enrollment of a Phase 3 clinical trial in 2010. We have completed preclinical work on CyMVectin™ and have begun preclinical work on a HSV-2 vaccine. We may not meet the primary endpoint of the Allovectin® trial for which a Special Protocol Assessment agreement is in place with the FDA. We may not conduct Phase 1 CyMVectin™ or HSV-2 vaccine trials, and the future trials, if any, may not demonstrate sufficient efficacy to support further product development.
Additionally, we are in early stages of development with other product candidates. These product candidates will require significant costs to advance through the development stages. If such product candidates are advanced through clinical trials, the results of such trials may not support approval by the FDA or comparable foreign agencies. Even if approved, our products may not be commercially successful, particularly if they do not gain market acceptance among physicians, patients, healthcare payers and relevant medical communities. If we fail to develop and commercialize our products, we may be forced to curtail or cease operations.
We are dependent on our license agreements with Astellas to further develop and commercialize TransVax™. The failure to maintain these agreements, or the failure of Astellas to perform its obligations under these agreements, could negatively impact our business.
Pursuant to the terms of our license agreements with Astellas, we granted to Astellas exclusive worldwide rights to develop and commercialize certain products, including TransVax™ but excluding CyMVectin™, for the control and prevention of CMV infection in immunocompromised patients, including transplant recipients and transplant donors, and pursuant to the terms of our supply and services agreement with Astellas, we are obligated to perform certain development activities and supply Astellas with its product requirements for development and initial commercialization activities. Consequently, our ability to generate any revenues from TransVax™ depends on Astellas’ ability to develop, obtain regulatory approvals for and successfully commercialize TransVax™. We have limited control over the amount and timing of resources that Astellas will dedicate to these efforts.
We are subject to a number of other risks associated with our dependence on our license agreements with Astellas, including:
| • | Astellas may not comply with applicable regulatory guidelines with respect to developing or commercializing TransVax™, which could adversely impact sales or future development of TransVax™, |
| • | We and Astellas could disagree as to future development plans and Astellas may delay, fail to commence or stop future clinical trials or other development; |
| • | There may be disputes between us and Astellas, including disagreements regarding the license agreements, that may result in (1) the delay of or failure to achieve developmental, regulatory and commercial objectives that would result in milestone or royalty payments, (2) the delay or termination of any future development or commercialization of TransVax™, and/or (3) costly litigation or arbitration that diverts our management’s attention and resources; |
28
Table of Contents
| • | Astellas may not provide us with timely and accurate information regarding development, sales and marketing activities or supply forecasts, which could adversely impact our ability to comply with our service and supply obligations to Astellas and manage our own inventory of TransVax™, as well as our ability to generate accurate financial forecasts; |
| • | Business combinations or significant changes in Astellas’ business strategy may adversely affect Astellas’ ability or willingness to perform its obligations under our license agreements; |
| • | Astellas may not properly defend our intellectual property rights, or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property rights or expose us to potential litigation; |
| • | The royalties we are eligible to receive from Astellas may be reduced based upon Astellas’ and our ability to maintain or defend our intellectual property rights and the presence of generic competitors; |
| • | Limitations on our or an acquiror’s ability to maintain or pursue development or commercialization of products that are competitive with TransVax™ could deter a potential acquisition of us that our stockholders may otherwise view as beneficial; and |
| • | If Astellas is unsuccessful in developing, obtaining regulatory approvals for or commercializing TransVax™, we may not receive any additional milestone or royalty payments under the license agreements and our business prospects and financial results may be materially harmed. |
The license agreements and supply and services agreement are subject to early termination, including through Astellas’ right to terminate upon advance notice to us if Astellas reasonably determines that further development and/or commercialization will not be beneficial for Astellas. If the agreements are terminated early, we may not be able to find another collaborator for the commercialization and further development of TransVax™ on acceptable terms, or at all, and we may be unable to pursue continued development or commercialization of TransVax™ on our own.
Our revenues partially depend on the development and commercialization of products in collaboration with others to whom we have licensed our technologies. If our other collaborators or licensees do not successfully develop and commercialize products covered by these arrangements, or if we are unable to find collaborators or licensees in the future, we may not be able to derive revenues from these arrangements, we may lose opportunities to validate our DNA delivery technologies, or we may be forced to curtail our development and commercialization efforts in these areas.
In addition to our license agreements with Astellas, we have licensed, and may continue to license, our technologies to corporate collaborators and licensees for the research, development and commercialization of specified product candidates. Our revenues partially depend upon the ability of these collaborators and licensees to successfully develop and commercialize products covered by these arrangements. In addition, our licensees Astellas and AnGes have product candidates in advanced stage of clinical development, for which we believe regulatory approval would provide important further validation of our DNA delivery technologies. The development and commercialization efforts of our collaborators and licensees are subject to the same risks and uncertainties described above with respect to our independently developed product candidates.
Some collaborators or licensees may not succeed in their product development efforts. It is possible that AnGes or any of our other collaborators or licensees may be unable to obtain regulatory approval of product candidates using our technologies or successfully market and commercialize any such products for which regulatory approval is obtained. In September 2010, AnGes announced that after a series of extensive consultations with the Japanese Pharmaceuticals and Medical Devices Agency, it would be withdrawing its NDA in Japan. Also in September 2010, another one of our licensees, Sanofi announced that NV1FGF, an angiogenic growth factor therapeutic for which Sanofi had licensed our DNA delivery technology, did not meet the primary endpoint in a global Phase 3 trial. Other collaborators or licensees may not devote sufficient time or resources to the programs covered by these arrangements, and we may have limited or no control over the time or resources
29
Table of Contents
allocated by these collaborators or licensees to these programs. The occurrence of any of these events may cause us to derive little or no revenue from these arrangements, lose opportunities to validate our DNA delivery technologies, or force us to curtail or cease our development and commercialization efforts in these areas.
Our collaborators and licensees may breach or terminate their agreements with us, including some that may terminate their agreements without cause at any time subject to certain prior written notice requirements, and we may be unsuccessful in entering into and maintaining other collaborative arrangements for the development and commercialization of products using our technologies. If we are unable to maintain existing collaboration arrangements or enter into new ones, our ability to generate licensing, milestone or royalty revenues would be materially impaired.
Some of our independent product candidates and some of those under development by our sublicensees incorporate technologies we have licensed from others. If we are unable to retain rights to use these technologies, we or our sublicensees may not be able to market products incorporating these technologies on a commercially feasible basis, if at all.
We have licensed certain technologies from corporate collaborators and research institutions, and sublicensed certain of such technologies to others, for use in the research, development and commercialization of product candidates. Our product development efforts and those of our sublicensees partially depend upon continued access to these technologies. For example, we or our licensors may breach or terminate our agreements, or disagree on interpretations of those agreements, which could prevent continued access to these technologies. If we were unable to resolve such matters on satisfactory terms, or at all, we or our sublicensees may be unable to develop and commercialize our products, and we may be forced to curtail or cease operations.
We have a history of net losses. We expect to continue to incur net losses and we may not achieve or maintain profitability.
To date, we have not sold, or received approval to sell, any pharmaceutical products. We do not expect to sell any pharmaceutical products for at least the next several years. Our net losses were approximately $7.3 million, $30.4 million and $28.6 million for the years ended December 31, 2011, 2010 and 2009, respectively. As of December 31, 2011, we had incurred cumulative net losses totaling approximately $325.0 million. Moreover, we expect that our net losses will continue and may increase for the foreseeable future. We may not be able to achieve projected results if we generate lower revenues or receive lower investment income than expected, or we incur greater expenses than expected, or all of the above. We may never generate sufficient product revenue to become profitable. We also expect to have quarter-to-quarter fluctuations in revenues, expenses, and losses, some of which could be significant.
We may need additional capital in the future. If additional capital is not available, we may have to curtail or cease operations.
We may need to raise more money to continue the research and development necessary to bring our products to market and to establish marketing and additional manufacturing capabilities. We may seek additional funds through public and private stock offerings, government contracts and grants, arrangements with corporate collaborators, borrowings under lease lines of credit or other sources. We currently have on file a shelf registration statement that allows us to raise up to an additional $53.4 million from the sale of common stock, preferred stock, debt securities and/or warrants. However, we may not be able to raise additional funds on favorable terms, or at all. Conditions in the credit markets and the financial services industry may make equity and debt financing more difficult to obtain, and may negatively impact our ability to complete financing transactions. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants, such as limitations on our ability to incur additional indebtedness and other operating restrictions that could adversely impact our ability to conduct our business.
30
Table of Contents
If we are unable to obtain additional funds, we may have to scale back our development of new products, reduce our workforce or license to others products or technologies that we otherwise would seek to commercialize ourselves. The amount of money we may need would depend on many factors, including:
If we are unable to obtain additional funds, we may have to scale back our development of new products, reduce our workforce or license to others products or technologies that we otherwise would seek to commercialize ourselves. The amount of money we may need would depend on many factors, including:
| • | The progress of our research and development programs; |
| • | The scope and results of our preclinical studies and clinical trials; and |
| • | The time and costs involved in: obtaining necessary regulatory approvals; filing, prosecuting and enforcing patent claims; scaling up our manufacturing capabilities; and the commercial arrangements we may establish. |
The regulatory approval process is expensive, time consuming and uncertain, which may prevent us and our collaborators and licensees from obtaining required approvals for the commercialization of our products.
Our product candidates under development and those of our collaborators and licensees, including Astellas, are subject to extensive and rigorous regulations by numerous governmental authorities in the United States and other countries. The regulatory approval process takes many years and will require us to expend substantial resources. For example, the FDA has provided only limited guidelines concerning the size and scope of clinical trials required for gene-based therapeutic and vaccine products.
Therefore, U.S. or foreign regulations could prevent or delay regulatory approval of our products or limit our and our collaborators and licensees’ ability to develop and commercialize our products. Delays could:
| • | Impose costly procedures on our activities and those of our collaborators and licensees; |
| • | Delay or prevent our receipt of developmental or commercial milestones from our collaborators and licensees; |
| • | Diminish any competitive advantages that we or our products attain; or |
| • | Otherwise negatively affect our results of operations and cash flows. |
We have no experience in filing a Biologics License Application, or BLA, or NDA, with the FDA. Because a BLA or NDA must be submitted to and approved by the FDA before any of our product candidates may be commercialized, our lack of experience may impede our ability to obtain FDA approval in a timely manner, if at all, which in turn would delay or prevent us from commercializing those products. Similarly, our lack of experience with respect to obtaining regulatory approvals in countries other than the United States may impede our ability to commercialize our products in those countries.
We believe that the FDA and comparable foreign regulatory bodies will regulate separately each product containing a particular gene depending on its intended use. Presently, to commercialize any product we and our collaborators and licensees must file a regulatory application for each proposed use. We and our collaborators and licensees must conduct clinical studies to demonstrate the safety and efficacy of the product necessary to obtain FDA or foreign regulatory authority approval. The results obtained so far in our clinical trials and those of our collaborators and licensees may not be replicated in ongoing or future trials, or the results may be subject to varying interpretation on whether they are sufficient to support approval for commercialization. This may prevent any of our product candidates from receiving approval for commercial sale.
We use recombinant DNA molecules in our product candidates, and therefore we and our collaborators and licensees also must comply with guidelines instituted by the NIH and its Office of Biotechnology Activities. The NIH could restrict or delay the development of our product candidates.
31
Table of Contents
If any of our product candidates receive regulatory approval, the FDA or other foreign regulatory agencies may still impose significant restrictions on the indicated uses or marketing of our product candidates or impose ongoing requirements for potentially costly post-approval studies. In addition, regulatory agencies subject a product, its manufacturer and the manufacturer’s facilities to continual review and periodic inspections. If a regulatory agency discovers previously unknown problems with a product or a product class, including adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product or product class, our collaborators and licensees or us, including requiring withdrawal of a product from the market. Our product candidates will also be subject to ongoing FDA and other foreign regulatory agency requirements for the labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information on the product. If we or our collaborators and licensees fail to maintain regulatory compliance after receiving marketing approval, we or our collaborators and licensees may be unable to market our products and our business could suffer.
Adverse events or the perception of adverse events in the field of gene therapy, or with respect to our product candidates, may negatively impact regulatory approval or public perception of our products.
The commercial success of some of our product candidates will depend in part on public acceptance of the use of gene therapy for preventing or treating human diseases. Serious adverse events, including patient deaths, have occurred in clinical trials utilizing viral delivery systems to deliver therapeutic genes to the patient’s targeted cells. Although none of our current products or studies utilize viral delivery systems, these adverse events, as well as any other adverse events in the field of gene therapy that may occur in the future, may negatively influence public perception of gene therapy in general. If public perception is influenced by claims that gene therapy is unsafe, our product candidates may not be accepted by the general public or the medical community.
Future adverse events in gene therapy or the biotechnology industry could also result in greater governmental regulation, stricter labeling requirements and potential regulatory delays in the testing or approval of our potential products. Any increased scrutiny could delay or increase the costs of our product development efforts or clinical trials. In addition, any adverse events that may occur in our clinical trials and any resulting publicity may cause regulatory delays or otherwise affect our product development efforts or clinical trials.
Some of our potential products may be administered to patients who are suffering from, or are vulnerable to, serious diseases or other conditions which can themselves be life-threatening and often result in the death of the patient. For example, one patient in our Allovectin® Phase 2 trial conducted in 2000, died from progressive disease more than two months after receiving Allovectin® and other cancer therapies. The death was originally reported as unrelated to the treatment. Following an autopsy, the death was reclassified as “probably related” to the treatment because the possibility could not be ruled out. We do not believe Allovectin® was a significant factor in the patient’s death. Patient deaths in our clinical trials, even if caused by pre-existing diseases or conditions, could negatively affect the perception of our product candidates. In addition, in our TransVax™ Phase 2 trial, we administered TransVax™ to patients who were at risk of CMV reactivation. Although we do not believe our vaccine candidates could cause the diseases they are designed to protect against, a temporal relationship between vaccination and disease onset could be perceived as causal. Some of our products are designed to stimulate immune responses, and those responses, if particularly strong or uncontrolled, could result in local or systemic adverse events, including latent adverse events.
Our patents and proprietary rights may not provide us with any benefit and the patents of others may prevent us from commercializing our products.
As of December 31, 2011, we were the assignee or co-assignee of 72 issued U.S. and foreign patents. We maintain our issued patents by paying maintenance fees to the patent office in each country when due. Where appropriate, we participate in legal proceedings to vigorously defend against the revocation or withdrawal of our patents. The scope and nature of these proceedings generally differ depending on the country in which they are initiated. If we are not successful in defending our patents, we may lose all or part of our proprietary rights related to those patents in these geographic regions.
32
Table of Contents
As of December 31, 2011, we were also prosecuting 61 pending patent applications in the United States and in foreign countries that cover various aspects of our proprietary technologies, not including patent applications for which we are a co-assignee and that are being prosecuted by our partners.
We may not receive any patents from our current patent applications. Issued patents provide exclusivity for only a limited time period, after which they no longer serve to protect proprietary technologies or to provide any commercial advantage. Moreover, if patents are issued to us, governmental authorities may not allow claims sufficient to protect our technologies and products. Others may also challenge or seek to circumvent or invalidate our patents. In that event, the rights granted under our patents may be inadequate to protect our proprietary technologies or to provide any commercial advantage.
In addition, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was recently signed into law and includes a number of significant changes to United States patent law. These include changes in the way patent applications will be prosecuted and may also affect patent litigation. The U.S. Patent and Trademark Office is currently developing regulations and procedures to administer the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act will not become effective until one year or 18 months after its enactment. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the cost of prosecuting our patent applications, our ability to obtain patents based on our patent applications and our ability to enforce or defend our issued patents. An inability to obtain, enforce and defend patents covering our proprietary technologies would materially and adversely affect our business prospects and financial condition.
Some components of our gene-based product candidates are, or may become, patented by others. As a result, we may be required to obtain licenses to conduct research, to manufacture, or to market such products. Licenses may not be available on commercially reasonable terms, or at all, which may impede our ability to commercialize our products.
The NIH and the FDA jointly developed the Genetic Modification Clinical Research Information System, or GeMCRIS, an Internet-based database of human gene transfer trials. GeMCRIS enables individuals to easily view information on particular characteristics of clinical gene transfer trials. Although GeMCRIS includes special security features designed to protect patient privacy and confidential commercial information, these security features may be inadequately designed or enforced, potentially resulting in disclosure of confidential commercial information. In addition, the NIH, in collaboration with the FDA, has developed an Internet site, ClinicalTrials.gov, which provides public access to information on clinical trials and their results for a wide range of diseases and conditions. Future disclosures of such confidential commercial information may result in loss of advantage of competitive secrets.
The legal proceedings to obtain and defend patents, and litigation of third-party claims of intellectual property infringement, could require us to spend money and could impair our operations.
Our and our collaborators’, including Astellas’, success will depend in part on our, or our collaborators’, ability to obtain patent protection for our products and processes, both in the United States and in other countries. The patent positions of biotechnology and pharmaceutical companies, however, can be highly uncertain and involve complex legal and factual questions. Therefore, it is difficult to predict the breadth of claims allowed in the biotechnology and pharmaceutical fields.
We also rely on confidentiality agreements with our corporate collaborators, employees, consultants and certain contractors to protect our proprietary technologies. However, these agreements may be breached and we may not have adequate remedies for such breaches. In addition, our trade secrets may otherwise become known or independently discovered by our competitors.
Protecting intellectual property rights can be very expensive. Litigation may be necessary to enforce patents issued to us or to determine the scope and validity of third-party proprietary rights. If we or, as applicable, our commercialization partners, including Astellas pursuant to its first right to enforce patents licensed to it under our
33
Table of Contents
license agreements, choose to go to court to stop someone else from using our inventions, that individual or company has the right to ask the court to rule that the underlying patents are invalid and/or should not be enforced against that third party. Moreover, if a competitor were to file a patent application claiming technology also invented by us or our collaborators or licensees, we would have to participate in an interference proceeding before the U.S. Patent and Trademark Office to determine the priority of the invention. We or our collaborators or licensees may be drawn into interferences with third parties or may have to provoke interferences ourselves to unblock third-party patent rights to allow us or our collaborators or licensees to commercialize products based on our technologies. Litigation could result in substantial costs and the diversion of management’s efforts regardless of the results of the litigation. An unfavorable result in litigation could subject us to significant liabilities to third parties, require disputed rights to be licensed or require us to cease using some technologies.
Our products and processes may infringe, or be found to infringe, patents not owned or controlled by us. Patents held by others may require us to alter our products or processes, obtain licenses, or stop activities. If relevant claims of third-party patents are upheld as valid and enforceable, we or our collaborators or licensees could be prevented from practicing the subject matter claimed in the patents, or may be required to obtain licenses or redesign our products or processes to avoid infringement. In addition, we or our collaborators or licensees could be required to pay money damages. A number of genetic sequences or proteins encoded by genetic sequences that we are investigating are, or may become, patented by others. As a result, we or our collaborators or licensees may have to obtain licenses to test, use or market these products. Our business will suffer if we or our collaborators or licensees are not able to obtain licenses at all or on terms commercially reasonable to us or them and we or they are not able to redesign our products or processes to avoid infringement.
We have incurred costs in several legal proceedings involving our intellectual property rights in Europe, Japan and Canada. We may continue to incur costs to defend and prosecute patents and patent applications in these and other regions.
Competition and technological change may make our product candidates and technologies less attractive or obsolete.
We compete with companies, including major pharmaceutical and biotechnology firms that are pursuing other forms of treatment or prevention for diseases that we target. We also may experience competition from companies that have acquired or may acquire technologies from universities and other research institutions. As these companies develop their technologies, they may develop proprietary positions which may prevent us from successfully commercializing products.
Some of our competitors are established companies with greater financial and other resources than we have. Other companies may succeed in developing products and obtaining regulatory approval from the FDA or comparable foreign agencies faster than we do, or in developing products that are more effective than ours. Research and development by others may seek to render our technologies or products obsolete or noncompetitive or result in treatments or cures superior to any therapeutics developed by us.
If we lose our key personnel or are unable to attract and retain additional personnel, we may not be able to achieve our business objectives.
We are highly dependent on our principal scientific, manufacturing, clinical, regulatory and management personnel, including Vijay B. Samant, our President and Chief Executive Officer. The loss of the services of these individuals might significantly delay or prevent the achievement of our objectives. We do not maintain “key person” life insurance on any of our personnel. We depend on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We face competition for qualified individuals from other companies, academic institutions, government entities and other organizations in attracting and retaining personnel. To pursue our product development plans, we may need to hire additional management personnel and additional scientific personnel to perform research and development, as well as additional personnel with expertise in clinical trials, government regulation and manufacturing. However, due to the reasons noted above, we may not be successful in hiring or retaining qualified personnel and therefore we may not be able to achieve our business objectives.
34
Table of Contents
We have limited experience in manufacturing our product candidates in commercial quantities. We may not be able to comply with applicable manufacturing regulations or produce sufficient product for contract or commercial purposes.
The commercial manufacturing of vaccines and other biological products is a time-consuming and complex process, which must be performed in compliance with the FDA’s current Good Manufacturing Practices, or cGMP, regulations. We may not be able to comply with the cGMP regulations, and our manufacturing process may be subject to delays, disruptions or quality control problems. In addition, we may need to complete the installation and validation of additional large-scale fermentation and related purification equipment to produce the quantities of product expected to be required for commercial purposes. We have limited experience in manufacturing at this scale. Noncompliance with the cGMP regulations, the inability to complete the installation or validation of additional large-scale equipment, or other problems with our manufacturing process may limit or delay the development or commercialization of our product candidates, and cause us to breach our contract manufacturing service arrangements or our obligations under our agreements with collaborators, including our obligations under our supply and services agreement with Astellas.
We currently depend on third parties to conduct our clinical trials and may initially depend on third parties to manufacture our product candidates commercially.
We currently rely on third parties, including clinical research organizations, to perform critical services for us in connection with our clinical trials. Clinical research organizations are responsible for many aspects of the trials, including finding and enrolling subjects for testing and administering the trials. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its protocol and applicable regulations, including good clinical practices. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner, and we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. In addition, if such third parties fail to perform their obligations in compliance with our clinical trial protocols or applicable regulations, our clinical trials may not meet regulatory requirements or may need to be repeated. These risks also apply to the development activities of our collaborators and licensees, and we do not control our collaborators’ and licensees’ research and development, clinical trials or regulatory activities.
We may also initially depend on collaborators, licensees or other third parties to manufacture our product candidates in commercial quantities. There are a limited number of third parties that could manufacture our product candidates. We may be unable to enter into any arrangement for the commercial manufacture of our product candidates, and any arrangement we secure may not meet our requirements for manufacturing quality or quantity. Our dependence on third parties for the commercial manufacture of our product candidates may also reduce our profit margins and our ability to develop and deliver products in a timely manner.
We have no marketing or sales experience, and if we are unable to develop our own sales and marketing capability, we may not be successful in commercializing our products.
Our current strategy is to market our proprietary products directly in the United States, but we currently do not possess pharmaceutical marketing or sales capabilities. To market and sell our proprietary products, we will need to develop a sales force and a marketing group with relevant pharmaceutical industry experience, or make appropriate arrangements with strategic partners to market and sell these products. Developing a marketing and sales force is expensive and time-consuming and could delay any product launch. If we are unable to successfully employ qualified marketing and sales personnel or develop other sales and marketing capabilities, we may not be able to generate sufficient product revenue to become profitable.
35
Table of Contents
Healthcare reform and restrictions on reimbursement may limit our returns on potential products.
Our ability to earn sufficient returns on our products will depend in part on how much, if any, reimbursement for our products and related treatments will be available from:
| • | Government health administration authorities; |
| • | Government agencies procuring biodefense products for military or public use, including some for which we may become a sole-source vendor; |
| • | Private health coverage insurers; |
| • | Managed care organizations; and |
| • | Other organizations. |
If we fail to obtain appropriate reimbursement, we could be prevented from successfully commercializing our potential products. There are ongoing efforts by governmental and third-party payers to contain or reduce the costs of healthcare through various reform measures. In the United States, the Federal government passed comprehensive healthcare reform legislation in 2010. Many of the details regarding the implementation of this legislation are yet to be determined and we currently cannot predict whether or to what extent such implementation or adoption of reforms may impair our business.
Additionally, third-party payers are increasingly challenging the price of medical products and services. If purchasers or users of our products are not able to obtain adequate reimbursement for the cost of using our products, they may forego or reduce their use. Significant uncertainty exists as to the reimbursement status of newly approved healthcare products, and whether adequate third-party coverage will be available.
We use hazardous materials in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research and development processes involve the controlled storage, use and disposal of hazardous materials and biological materials. Our hazardous materials include certain compressed gases, flammable liquids, acids and bases, and other toxic compounds. We are subject to federal, state and local regulations governing the use, manufacture, storage, handling and disposal of materials and waste products. Although we believe that our safety procedures for handling and disposing of these hazardous materials comply with the standards prescribed by law and regulation, the risk of accidental contamination or injury from hazardous materials cannot be completely eliminated. In the event of an accident, we could be held liable for any damages that result. We have insurance that covers our use of hazardous materials with the following coverage limits: up to $50,000 per occurrence for losses related to contaminant clean-up or removal, $250,000 per occurrence for losses related to the contamination of our covered premises and $250,000 per occurrence for losses from radioactive contamination. Any liability could exceed the limits or fall outside the coverage of our insurance. We could incur significant costs to comply with current or future environmental laws and regulations.
We may have significant product liability exposure.
We face an inherent business risk of exposure to product liability and other claims in the event that our technologies or products are alleged to have caused harm. We also have potential liability for products manufactured by us on a contract basis for third parties. Although we currently maintain product liability insurance in the amount of $10 million in the aggregate plus additional coverage specific to the foreign countries where our clinical trials are being conducted, this insurance coverage may not be sufficient, and we may not be able to obtain sufficient coverage in the future at a reasonable cost. Our inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the commercialization of any products developed by us or our collaborators, or our ability to manufacture products for third parties. If we are sued for any injury caused by our technologies or products, or by third-party products that we manufacture, our liability could exceed our insurance coverage and total assets.
36
Table of Contents
Negative conditions in the global credit markets may impair the liquidity of a portion of our investment portfolio.
Our investment securities consist of high-grade auction rate securities, corporate debt securities and government agency securities. As of December 31, 2011, our long-term investments included (at par value) $6.5 million of auction rate securities secured by municipal bonds and student loans. At December 31 2011, the auction rate securities we held had Standard and Poor’s credit ratings of BBB or AAA. Our auction rate securities are debt instruments with a long-term maturity and with an interest rate that is reset in short intervals through auctions. Ongoing conditions in the global credit markets have prevented some investors from liquidating their holdings of auction rate securities because the amount of securities submitted for sale has exceeded the amount of purchase orders for such securities. If there is insufficient demand for the securities at the time of an auction, the auction may not be completed and the interest rates may be reset to predetermined rates. When auctions for these securities fail, the investments may not be readily convertible to cash until a future auction of these investments is successful or they are redeemed or mature.
Since February 2008, there has been insufficient demand at auction for all of our auction rate securities held at December 31, 2011. As a result, these affected securities are currently not liquid, and we could be required to hold them until they are redeemed by the issuer or to maturity. As of December 31, 2011, we had recognized $1.5 million of losses related to those auction rate securities by adjusting their carrying value. The market value of these securities has partially recovered from the lows that created the loss. As of December 31, 2011, we had recorded cumulative unrealized gains of $0.6 million. Any future decline in market value may result in additional losses being recognized.
In the event we need to access the funds that are in an illiquid state, we will not be able to do so without the possible loss of principal, until a future auction for these investments is successful or they are redeemed by the issuer or they mature. If we are unable to sell these securities in the market or they are not redeemed, then we may be required to hold them to maturity.
Our stock price could continue to be highly volatile and you may not be able to resell your shares at or above the price you pay for them.
The market price of our common stock, like that of many other life sciences companies, has been and is likely to continue to be highly volatile. From January 1, 2009, to December 31, 2011, our stock price has ranged from $1.20 to $5.51. The following factors, among others, could have a significant impact on the market price of our common stock:
| • | The results of our preclinical studies and clinical trials or announcements regarding our plans for future studies or trials, or those of our collaborators, licensees or competitors; |
| • | Evidence or lack of evidence of the safety or efficacy of our potential products or those of our collaborators, licensees or competitors; |
| • | The success of our collaborators and licensees, including Astellas, in the development or commercialization of our product candidates; |
| • | The announcement by us or our collaborators, licensees or competitors of technological innovations or new products; |
| • | Developments concerning our patent or other proprietary rights or those of our collaborators, licensees or competitors, including litigation and challenges to our proprietary rights; |
| • | Other developments with our collaborators or licensees, including our entry into new collaborative or licensing arrangements; |
| • | Geopolitical developments, natural or man-made disease threats, or other events beyond our control; |
| • | U.S. and foreign governmental regulatory actions; |
37
Table of Contents
| • | Changes or announcements in reimbursement policies; |
| • | Period-to-period fluctuations in our operating results; |
| • | Market conditions for life science stocks in general; |
| • | Changes in the collective short interest in our stock; |
| • | Changes in estimates of our performance by securities analysts; and |
| • | Our cash balances, need for additional capital, and access to capital. |
We are at risk of securities class action litigation due to our expected stock price volatility.
In the past, stockholders have brought securities class action litigation against a company following a decline in the market price of its securities. This risk is especially acute for us because life science companies have experienced greater than average stock price volatility in recent years and, as a result, have been subject to, on average, a greater number of securities class action claims than companies in other industries. To date, we have not been subject to class action litigation. However, we may in the future be the target of this litigation. Securities litigation could result in substantial costs and divert our management’s attention and resources, and could seriously harm our business.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Our certificate of incorporation and bylaws include anti-takeover provisions, such as a classified board of directors, a prohibition on stockholder actions by written consent, the authority of our board of directors to issue preferred stock without stockholder approval, and supermajority voting requirements for specified actions. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which generally prohibits stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years. These provisions may delay or prevent an acquisition of us, even if the acquisition may be considered beneficial by some stockholders. In addition, they may discourage or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
The issuance of preferred stock could adversely affect our common stockholders.
We currently have on file a shelf registration statement that allows us to raise up to an additional $53.4 million from the sale of common stock, preferred stock, debt securities and/or warrants and our restated certificate of incorporation authorizes us to issue up to 5,000,000 shares of preferred stock. The issuance of preferred stock could adversely affect the voting power of holders of our common stock, and reduce the likelihood that our common stockholders will receive dividend payments and payments upon liquidation. The issuance of preferred stock could also decrease the market price of our common stock, or have terms and conditions that could discourage a takeover or other transaction that might involve a premium price for our shares or that our stockholders might believe to be in their best interests.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not Applicable.
38
Table of Contents
We lease approximately 68,400 square feet of manufacturing, research laboratory and office space at a single site in San Diego, California, under a lease agreement that expires in August 2017.
European patent 1026253, or the ‘253 patent, covering a significant portion of our core DNA delivery technology, was granted in September 2004. In June 2005, the ‘253 patent was opposed by eight parties. This ‘253 patent was revoked on formal grounds in December 2008 under an initial ruling by the Opposition Division of the European Patent Office. We appealed this decision and in December 2010 our appeal was successful and the ‘253 patent was reinstated by the Appeals Board and sent back to the Opposition Division for further processing. The Opposition Division has scheduled hearings in March 2012 to determine if the patent should be revoked on other grounds. While the ‘253 patent expired in March 2010, the reinstatement gives us the ability to prosecute any infringing activity that may have occurred prior to the expiration of the ‘253 patent expiring and within five years of initiation of the legal proceedings. We may use other issued patents and patent applications that are pending in Europe to protect our DNA technology.
We prosecute our intellectual property estate vigorously to obtain the broadest valid scope for our patents. Due to uncertainty of the ultimate outcome of these matters, the impact on future operating results or our financial condition is not subject to reasonable estimates.
In the ordinary course of business, we may become a party to lawsuits involving various matters. We are unaware of any such lawsuits presently pending against us which, individually or in the aggregate, are deemed to be material to our financial condition or results of operations.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
39
Table of Contents
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is listed on the Nasdaq Global Market under the symbol VICL. The following table presents quarterly information on the range of high and low sales prices for our common stock during the periods presented.
| 2011 |
High | Low | ||||||
| First Quarter |
$ | 3.00 | $ | 1.70 | ||||
| Second Quarter |
4.99 | 2.85 | ||||||
| Third Quarter |
5.30 | 2.31 | ||||||
| Fourth Quarter |
4.82 | 2.22 | ||||||
| 2010 |
||||||||
| First Quarter |
$ | 4.12 | $ | 2.78 | ||||
| Second Quarter |
3.86 | 2.98 | ||||||
| Third Quarter |
4.05 | 2.20 | ||||||
| Fourth Quarter |
2.35 | 1.74 | ||||||
As of February 15, 2012, there were approximately 294 stockholders of record of our common stock and 85,881,117 shares of our common stock outstanding. We have never declared or paid any dividends and do not expect to pay any dividends in the foreseeable future. We did not repurchase any of our common stock in the fourth quarter of 2011.
40
Table of Contents
Performance Graph
The following performance graph and related information shall not be deemed “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act or Exchange Act, except to the extent that we specifically incorporate it by reference into such filing.
The following table compares total stockholder returns for Vical over the last five years to the Nasdaq U.S. and Foreign Index and the Nasdaq Pharmaceutical Stocks Index assuming a $100 investment made on December 31, 2006. Each of the two comparative measures of cumulative total return assumes reinvestment of dividends. The stock performance shown on the graph below is not necessarily indicative of future price performance.
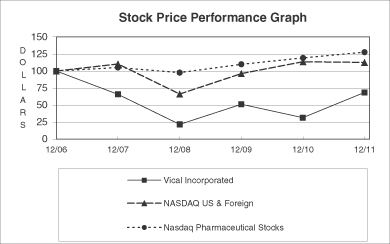
41
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA
The following table summarizes certain selected financial data derived from our audited financial statements. The information presented should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our audited financial statements and notes thereto appearing elsewhere in this Annual Report on Form 10-K.
| Years ended December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| (in thousands, except per share amounts) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Contract and grant revenue |
$ | 4,223 | $ | 6,249 | $ | 3,692 | $ | 2,146 | $ | 4,574 | ||||||||||
| License and royalty revenue |
25,795 | 2,462 | 8,994 | 5,810 | 938 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total revenues |
30,018 | 8,711 | 12,686 | 7,956 | 5,512 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
17,975 | 19,692 | 23,449 | 25,532 | 22,934 | |||||||||||||||
| Manufacturing and production |
10,267 | 11,436 | 10,354 | 11,046 | 13,762 | |||||||||||||||
| General and administrative |
9,598 | 8,798 | 7,469 | 8,721 | 9,078 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
37,840 | 39,926 | 41,272 | 45,299 | 45,774 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(7,822 | ) | (31,215 | ) | (28,586 | ) | (37,343 | ) | (40,262 | ) | ||||||||||
| Investment and other income, net |
539 | 830 | 30 | 468 | 4,464 | |||||||||||||||
| Interest expense |
— | — | (2 | ) | (21 | ) | (96 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
(7,283 | ) | $ | (30,385 | ) | $ | (28,558 | ) | $ | (36,896 | ) | $ | (35,894 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss per share (basic and diluted) |
$ | (0.10 | ) | $ | (0.51 | ) | $ | (0.61 | ) | $ | (0.93 | ) | $ | (0.92 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average shares used in per share calculation |
72,031 | 60,084 | 47,086 | 39,856 | 39,190 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance Sheet Data (at end of period): |
||||||||||||||||||||
| Cash, cash equivalents, marketable securities, long-term investments, including restricted |
$ | 56,355 | $ | 60,702 | $ | 52,562 | $ | 41,676 | $ | 71,489 | ||||||||||
| Working capital |
47,096 | 49,874 | 38,424 | 30,144 | 64,642 | |||||||||||||||
| Total assets |
68,773 | 72,907 | 67,372 | 59,057 | 90,585 | |||||||||||||||
| Long-term obligations , less current portion |
1,964 | 2,211 | 2,380 | 2,469 | 2,565 | |||||||||||||||
| Total stockholders’ equity |
60,348 | 64,362 | 54,982 | 48,614 | 79,912 | |||||||||||||||
42
Table of Contents
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We research and develop biopharmaceutical products based on our patented DNA delivery technologies for the prevention and treatment of serious or life-threatening diseases. We believe the following areas of research offer the greatest potential for near-term commercialization for us and our partners:
| • | Vaccines for use in high-risk populations for infectious disease targets for which there are significant needs; |
| • | Vaccines for general pediatric, adolescent and adult populations for infectious disease applications; |
| • | Cancer vaccines or immunotherapies which complement our existing programs and core expertise; and |
| • | Gene-based delivery of therapeutic proteins, such as angiogenic growth factors, for treatment of cardiovascular diseases. |
We currently have three active independent clinical and preclinical development programs in the areas of infectious disease and cancer including:
| • | A fully enrolled ongoing Phase 3 clinical trial using our Allovectin® immunotherapeutic in patients with metastatic melanoma which has been funded, up to certain limits, by AnGes MG, Inc., or AnGes, through cash payments and equity investments under a research and development agreement; |
| • | A completed preclinical program, with an allowed IND application, using our CyMVectin™ prophylactic vaccine formulated with our proprietary Vaxfectin® adjuvant to prevent CMV infection before and during pregnancy; and |
| • | A preclinical program with therapeutic and prophylactic vaccines for herpes simplex virus type 2 formulated with our proprietary Vaxfectin® adjuvant. |
We have leveraged our patented technologies through licensing and collaboration arrangements, such as our licensing arrangements with Astellas, Merck, Sanofi, AnGes, Aqua Health and Merial, among other biopharmaceutical companies.
In addition, we have licensed complementary technologies from leading research institutions and biopharmaceutical companies. We also have granted non-exclusive, academic licenses to our DNA delivery technology patent estate to 11 leading research institutions including Stanford, Harvard, Yale and MIT. The non-exclusive academic licenses allow university researchers to use our technology free of charge for educational and internal, non-commercial research purposes. In exchange, we have the option to exclusively license from the universities potential commercial applications arising from their use of our technology on terms to be negotiated.
43
Table of Contents
Research, Development and Manufacturing Programs
To date, we have not received revenues from the sale of our independently developed pharmaceutical products and have received minimal amounts of revenue from the sale of commercially marketed products by our licensees. We earn revenue by performing services under research and development contracts, grants, manufacturing contracts, and from licensing access to our proprietary technologies. Since our inception, we estimate that we have received approximately $198.2 million in revenue from these sources. Revenues by source for each of the three years ended December 31, 2011, were as follows (in millions):
| Source |
2011 | 2010 | 2009 | |||||||||
| Navy contracts |
$ | — | $ | 1.5 | $ | 2.0 | ||||||
| Astellas contract |
2.7 | — | — | |||||||||
| IPPOX contract |
— | 2.4 | — | |||||||||
| Manufacturing process development grant |
0.9 | 1.5 | 1.0 | |||||||||
| HSV grants |
0.2 | 0.7 | 0.6 | |||||||||
| Other contracts and grants |
0.4 | 0.1 | 0.1 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total contract and grant revenues |
4.2 | 6.2 | 3.7 | |||||||||
|
|
|
|
|
|
|
|||||||
| Astellas license |
25.3 | — | — | |||||||||
| Merck license |
— | — | 1.5 | |||||||||
| AnGes license |
— | 2.0 | 6.7 | |||||||||
| Life Technologies royalties |
0.3 | 0.3 | 0.4 | |||||||||
| Other royalties and licenses |
0.2 | 0.2 | 0.4 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total royalty and license revenues |
25.8 | 2.5 | 9.0 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
$ | 30.0 | $ | 8.7 | $ | 12.7 | ||||||
|
|
|
|
|
|
|
|||||||
Research, development, manufacturing and production costs by major program, as well as other expenses for each of the three years ended December 31, 2011, were as follows (in millions):
| Program |
2011 | 2010 | 2009 | |||||||||
|
Allovectin® |
$ | 17.3 | $ | 16.5 | $ | 21.0 | ||||||
| Pandemic influenza |
0.2 | 2.1 | 1.6 | |||||||||
| CMV |
7.5 | 6.7 | 5.0 | |||||||||
| Other research, development, manufacturing and production |
3.2 | 5.8 | 6.2 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research, development, manufacturing and production |
$ | 28.2 | $ | 31.1 | $ | 33.8 | ||||||
|
|
|
|
|
|
|
|||||||
From inception through December 31, 2011, we estimate that we have spent approximately $442 million on research, development, manufacturing and production. Our current independent development focus is on our cancer immunotherapeutic Allovectin®, a novel DNA vaccine for CMV, a vaccine to treat HSV-2, and other clinical and preclinical targets.
We are conducting a Phase 3 clinical trial using Allovectin® in patients with recurrent metastatic melanoma which has been funded, up to certain limits, by AnGes through cash payments and equity investments under a research and development agreement. We are also in the early stages of clinical development of vaccine candidates for CMV and HSV-2, and these programs will require significant additional funds to advance through development to commercialization. From inception through December 31, 2011, we have spent approximately $150 million on our Allovectin® program and $64 million on our CMV programs.
We have other product candidates in the research stage. It can take many years to develop product candidates from the initial decision to screen product candidates, perform preclinical and safety studies, and perform clinical trials leading up to possible approval of a product by the FDA or comparable foreign agencies.
44
Table of Contents
The outcome of the research is unknown until each stage of the testing is completed, up through and including the registration clinical trials. Accordingly, we are unable to predict which potential product candidates we may proceed with, the time and cost to complete development, and ultimately whether we will have a product approved by the FDA or comparable foreign agencies.
As a result, we expect to incur substantial operating losses for at least the next several years, due primarily to the advancement of our research and development programs, the cost of preclinical studies and clinical trials, spending for outside services, costs related to maintaining our intellectual property portfolio, costs due to manufacturing activities, costs related to our facilities, and possible advancement toward commercialization activities.
Critical Accounting Policies and Estimates
The preparation and presentation of financial statements in accordance with accounting principles generally accepted in the United States requires that management make a number of assumptions and estimates that affect the reported amounts of assets, liabilities, revenues and expenses in our financial statements and accompanying notes. Management bases its estimates on historical information and assumptions believed to be reasonable. Although these estimates are based on management’s best knowledge of current events and circumstances that may impact us in the future, they are inherently uncertain and actual results may differ materially from these estimates.
Our critical accounting policies are those that affect our financial statements materially and involve a significant level of judgment by management. Our critical accounting policies regarding revenue recognition are in the following areas: license and royalty agreements, manufacturing contracts, and grant revenues. Our critical accounting policies also include recognition of research and development expenses and the valuation of long-lived and intangible assets.
Revenue Recognition
Revenue is recognized when the four basic criteria of revenue recognition are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services rendered; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured. Certain of the our revenue is generated through manufacturing contracts and stand-alone license agreements.
We have entered into multiple-element arrangements. In order to account for the multiple-element arrangements, we identify the deliverables included within the agreement and evaluates which deliverables represent separate units of accounting. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation.
Multiple-element arrangements prior to January 1, 2011
Prior to adopting the revised multiple element guidance on January 1, 2011, we analyzed our multiple element arrangements to determine whether the identified deliverables could be accounted for individually as separate units of accounting. The delivered item(s) were considered a separate unit of accounting if all of the following criteria were met: (1) the delivered item(s) has value to the customer on a standalone basis; (2) there is objective and reliable evidence of the fair value of the undelivered item(s); and (3) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item(s) is considered probable and substantially in our control. If these criteria were not met, the deliverable was combined with other deliverables in the arrangement and accounted for as a combined unit of accounting.
45
Table of Contents
Multiple-element arrangements after January 1, 2011
Effective January 1, 2011, we followed the provisions of ASU No. 2009-13 for all multiple element agreements, including contract manufacturing, contract services and license agreements. Under the revised guidance, the delivered item(s) has value to the customer on a standalone basis and, if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
A delivered item is considered a separate unit of accounting when the delivered item has value to the partner on a standalone basis based on the consideration of the relevant facts and circumstances for each arrangement. Factors considered in this determination include the research capabilities of the partner and the availability of research expertise in this field in the general marketplace. Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence, or VSOE, of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, we use our best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement. If facts and circumstances dictate that the license has standalone value from the undelivered items, which generally include research and development services and the manufacture of drug products, the license is identified as a separate unit of accounting and the amounts allocated to the license are recognized upon the delivery of the license, assuming the other revenue recognition criteria have been met. However, if the amounts allocated to the license through the relative selling price allocation exceed the upfront license fee, the amount recognized upon the delivery of the license is limited to the upfront fee received. If facts and circumstances dictate that the license does not have standalone value, the transaction price, including any upfront license fee payments received, are allocated to the identified separate units of accounting and recognized as those items are delivered.
The terms of our partnership agreements provide for milestone payments upon achievement of certain regulatory and commercial events. Effective January 1, 2011, we adopted on a prospective basis the Milestone Method of accounting under ASU 2010-17. Under the Milestone Method, we recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following three criteria: 1) The consideration is commensurate with either the entity’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, 2) The consideration relates solely to past performance, and 3) The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to us.
Contract Services, Grant and Royalty Revenue
We recognize revenue from contract services and federal government research grants during the period in which the related expenditures are incurred and related payments for those services are received or collection is reasonably assured. Royalties to be received based on sales of licensed products by our partners incorporating our licensed technology are recognized when received.
46
Table of Contents
Accruals for Potential Disallowed Costs on Government Contracts
We have contracts with U.S. government agencies under which we bill for direct and indirect costs incurred. These billed costs are subject to audit by government agencies. We have established accruals of approximately $49,000 and $0.2 million at December 31, 2011 and 2010, respectively, to provide for potential disallowed costs. In the event that the final costs allowed are different from what we have estimated, we may need to make a change in our estimated accrual, which could also affect our results of operations and cash flow.
Research and Development Expenses
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, clinical trials, contract services and other outside expenses. Research and development expenses are charged to operations as they are incurred.
We assess our obligations to make milestone payments that may become due for licensed or acquired technology to determine whether the payments should be expensed or capitalized. We charge milestone payments to research and development expense when:
| • | The technology is in the early stage of development and has no alternative uses; |
| • | There is substantial uncertainty of the technology or product being successful; |
| • | There will be difficulty in completing the remaining development; and |
| • | There is substantial cost to complete the work. |
Capitalization and Valuation of Long-Lived and Intangible Assets
Intangible assets with finite useful lives consist of capitalized legal costs incurred in connection with patents, patent applications pending and technology license agreements. Payments to acquire a license to use a proprietary technology are capitalized if the technology is expected to have alternative future use in multiple research and development projects. We amortize costs of approved patents, patent applications pending and license agreements over their estimated useful lives, or terms of the agreements, whichever are shorter.
For patents pending, we amortize the costs over the shorter of a period of twenty years from the date of filing the application or, if licensed, the term of the license agreement. We re-assess the useful lives of patents when they are issued, or whenever events or changes in circumstances indicate the useful lives may have changed. For patents and patent applications pending that we abandon, we charge the remaining unamortized accumulated costs to research and development expense.
Intangible assets and long-lived assets are evaluated for impairment at least annually or whenever events or changes in circumstances indicate that their carrying value may not be recoverable. If the review indicates that intangible assets or long-lived assets are not recoverable, their carrying amount would be reduced to fair value. Factors we consider important that could trigger an impairment review include the following:
| • | A significant change in the manner of our use of the acquired asset or the strategy for our overall business; and/or |
| • | A significant negative industry or economic trend. |
In the event we determine that the carrying value of intangible assets or long-lived assets is not recoverable based upon the existence of one or more of the above indicators of impairment, we may be required to record impairment charges for these assets. As of December 31, 2011, our largest group of intangible assets with finite lives includes patents and patents pending for our DNA delivery technology, consisting of intangible assets with a net carrying value of approximately $2.8 million.
47
Table of Contents
Recent Accounting Pronouncements
For information on the recent accounting pronouncements which may impact our business, see Note 1 of the Notes to Financial Statements included in this Annual Report on Form 10-K.
Results of Operations
Year Ended December 31, 2011, Compared to Year Ended December 31, 2010
Total Revenues. Total revenues increased $21.3 million, or 244.6%, to $30.0 million in 2011 from $8.7 million in 2010. Our license and royalty revenue increased by $23.3 million which was primarily the result of the recognition of $25.3 million of revenue related to our TransVax™ license agreement with Astellas, which was partially offset by a decrease of $2.0 million in license revenue recognized related to our Allovectin® program. Our contract and grant revenue decreased by $2.0 million which was primarily the result of a $3.6 million decrease in revenue related to the manufacture of vaccines under contract as well as a $1.1 million decrease in grant revenue, which was partially offset by an increase of $2.7 million in contract service revenue recognized under our contracts with Astellas.
Research and Development Expenses. Research and development expenses decreased $1.7 million, or 8.7%, to $18.0 million for 2011 from $19.7 million for 2010. This decrease was primarily due to lower costs related to our clinical trials for TransVax™ and Allovectin® during the year ended December 31, 2011, which was partially offset by a sub-license payment we made to the City of Hope related to the license of our TransVax™ program in 2011.
Manufacturing and Production Expenses. Manufacturing and production expenses decreased $1.2 million, or 10.2%, to $10.3 million for 2011 from $11.4 million for 2010. This decrease was primarily the result of the capitalization of $1.2 million in manufacturing costs related to the production of TransVax™ under our service and supply agreement with Astellas.
General and Administrative Expenses. General and administrative expenses increased $0.8 million, or 9.1%, to $9.6 million for 2011 from $8.8 million for 2010. This increase was primarily the result of higher overall wages and higher consulting costs during the year ended December 31, 2011.
Investment and Other Income. Investment and other income decreased $0.3 million, or 35.1%, to $0.5 million for 2011 from $0.8 million for 2010. This decrease was primarily the result of our receipt of $0.5 million in income related to the Qualifying Therapeutic Discovery Project tax credit during the year ended December 31, 2010.
Year Ended December 31, 2010, Compared to Year Ended December 31, 2009
Total Revenues. Total revenues decreased $4.0 million, or 31.3%, to $8.7 million in 2010 from $12.7 million in 2009. Our license and royalty revenue decreased by $6.5 million which was primarily the result of a $4.7 million decrease in the recognition of revenue related to our Allovectin® license agreement with AnGes and a $1.5 million milestone payment received from Merck during the year ended December 31, 2009, related to its Phase 1 clinical-stage development of an investigational plasmid DNA cancer vaccine. Our contract and grant revenue increased by $2.6 million which was primarily the result of the recognition of revenue related to the delivery of an HIV vaccine to the IPPOX Foundation and the delivery of an H1N1 vaccine to the U.S. Navy.
Research and Development Expenses. Research and development expenses decreased $3.8 million, or 16.0%, to $19.7 million for 2010 from $23.5 million for 2009. This decrease was primarily the result of lower Allovectin® clinical trial related costs.
48
Table of Contents
Manufacturing and Production Expenses. Manufacturing and production expenses increased $1.1 million, or 10.5%, to $11.4 million for 2010 from $10.3 million for 2009. This increase was primarily the result of the recognition of costs related to the delivery of an H1N1 influenza vaccine in 2010 that we manufactured for the U.S. Navy.
General and Administrative Expenses. General and administrative expenses increased $1.3 million, or 17.8%, to $8.8 million for 2010 from $7.5 million for 2009. This increase was primarily the result of higher stock-based compensation expense during the year ended December 31, 2010, when compared to the prior year period.
Investment Income. Investment and other income increased $0.8 million, or 2,667%, to $0.8 million for 2010 from $30,000 for 2009. This increase was primarily the result of our receipt of $0.5 million in income related to the Qualifying Therapeutic Discovery Project tax credit during the year ended December 31, 2010.
Liquidity and Capital Resources
Since our inception, we have financed our operations primarily through private placements of preferred and common stock, public offerings of common stock, and revenues from our operations. From our inception through December 31, 2011, we have received approximately $198.2 million in revenues from performing services under research and development and manufacturing contracts, from grants and from licensing access to our proprietary technologies, and we have raised net proceeds of approximately $371.0 million from the sale of equity securities. Cash, cash equivalents, marketable securities, and long-term investments, including restricted securities, totaled $56.4 million at December 31, 2011, compared with $60.7 million at December 31, 2010. The decrease in our cash, cash equivalents and marketable securities for the year ended December 31, 2011, resulted primarily from the use of cash to fund our operations offset by the receipt of $25.0 million related to the license of our TransVax™ program to Astellas in 2011.
Net cash used in operating activities was $3.8 million and $28.2 million for the years ended December 31, 2011 and 2010, respectively. The decrease in net cash used in operating activities for the year ended December 31, 2011, compared with the prior year, was primarily the result of a decrease in our net loss.
Net cash (used in) provided by investing activities was $(4.8) million and $12.5 million for the years ended December 31, 2011 and 2010, respectively. The decrease in net cash provided by investing activities for the year ended December 31, 2011, compared with the prior year, was primarily the result of an increase in net purchases of investments.
Net cash provided by financing activities was $55,000 and $37.2 million for the years ended December 31, 2011 and 2010, respectively. The decrease in cash provided by financing activities for the year ended December 31, 2011, compared with the prior year, was primarily the result of net proceeds received from the sale of common stock and the exercise of warrants during the year ended December 31, 2010.
A discussion of our exposure to auction rate securities is included in Part II, Item 7A of this Annual Report on Form 10-K under the heading “Quantitative and Qualitative Disclosures About Market Risk.”
We expect to incur substantial additional research and development expenses, manufacturing and production expenses, and general and administrative expenses, including continued increases in costs related to personnel, preclinical and clinical testing, outside services, facilities, intellectual property and possible commercialization. Our future capital requirements will depend on many factors, including continued scientific progress in our research and development programs, the scope and results of preclinical testing and clinical trials, the time and costs involved in obtaining regulatory approvals, the costs involved in filing, prosecuting, enforcing and defending patent claims, the impact of competing technological and market developments, the cost of manufacturing scale-up and validation, and possible commercialization activities and arrangements. We may
49
Table of Contents
seek additional funding through research and development relationships with suitable potential corporate collaborators. We may also seek additional funding through public or private financings. We currently have on file a shelf registration statement that allows us to raise up to an additional $53.4 million from the sale of common stock, preferred stock, debt securities and/or warrants. However, additional financing may not be available on favorable terms or at all. If additional funding is not available, we anticipate that our current available cash and existing sources of funding will be adequate to satisfy our cash needs at least through December 31, 2013.
Contractual Obligations and Off-Balance Sheet Arrangements
The following table sets forth our contractual obligations, including all off-balance sheet arrangements, as of December 31, 2011 (in thousands):
| Payment Due by Period | ||||||||||||||||||||
| Total | Less than 1 Year |
2-3 Years |
4-5 Years |
After 5 Years |
||||||||||||||||
| Contractual Obligations1 |
||||||||||||||||||||
| Operating lease obligations |
$ | 19,742 | $ | 3,360 | $ | 6,849 | $ | 7,116 | $ | 2,417 | ||||||||||
| Unconditional purchase obligations2 |
425 | 425 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total contractual obligations |
$ | 20,167 | $ | 3,785 | $ | 6,849 | $ | 7,116 | $ | 2,417 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| 1 | Certain long-term liabilities reflected on our balance sheet are not presented in this table because they are already reflected in operating lease commitments or do not require cash settlement in the future. |
| 2 | Unconditional purchase obligations represent contractual commitments entered into for goods and services in the normal course of our business. The purchase obligations do not include potential severance payment obligations to our executive officers. For information regarding these severance arrangements, refer to the final paragraph in this Item 7. |
Under our Merck, Sanofi, AnGes, Merial and Aqua Health agreements, we are required to pay up to 10% of certain initial upfront monetary payments, and a small percentage of some royalty payments, to the WARF and/or the University of Michigan. Under our license agreements with Astellas, we are required to make certain payments to the City of Hope and CytRx in connection with the development and commercialization of our products licensed by Astellas. In addition, certain technology license agreements require us to make other payments if we or our sublicensees advance products through clinical development. For programs developed with the support of U.S. government funding, the U.S. government may have rights to resulting products without payment of royalties to us.
We may be required to make future payments to our licensors based on the achievement of milestones set forth in various in-licensing agreements. In most cases, these milestone payments are based on the achievement of development or regulatory milestones, including the exercise of options to obtain licenses related to specific disease targets, commencement of various phases of clinical trials, filing of product license applications, approval of product licenses from the FDA or a foreign regulatory agency, and the first commercial sale of a related product. Payment for the achievement of milestones under our in-license agreements is highly speculative and subject to a number of contingencies.
The aggregate amount of additional milestone payments that we could be required to pay under all of our in-license agreements in place at December 31, 2011, is approximately $23.4 million, of which approximately $15.5 million is related to our independent programs and corporate and government collaborations which are currently in clinical development. These amounts assume that all remaining milestones associated with the milestone payments are met. In the event that product license approval for any of the related products is obtained, we may be required to make royalty payments in addition to these milestone payments. Although we believe that some of the milestones contained in our in-license agreements may be achieved, it is highly unlikely that a significant number of them will be achieved. Because the milestones are highly contingent and we have limited
50
Table of Contents
control over whether the development and regulatory milestones will be achieved, we are not in a position to reasonably estimate how much, if any, of the potential milestone payments will ultimately be paid, or when. Additionally, under the in-license agreements, many of the milestone events are related to progress in clinical trials which will take several years to achieve.
In addition, we have undertaken certain commitments under license agreements with collaborators, and under indemnification agreements with our officers and directors. Under the license agreements with our collaborators, we have agreed to continue to maintain and defend the patent rights licensed to the collaborators and, in the case of our agreements with Astellas, have agreed to undertake certain development and manufacturing activites. Under the indemnification agreements with our officers and directors, we have agreed to indemnify those individuals for any expenses and liabilities in the event of a threatened, pending or actual investigation, lawsuit, or criminal or investigative proceeding.
We have employment agreements that contain severance arrangements with each of our three executive officers and three of our other executives. Under the agreements with the executive officers we are obligated to pay severance if we terminate the executive officer’s employment without “cause,” or if the executive officer resigns for “good reason,” as defined in the agreements, within the periods set forth therein. The severance for the executive officers consists of continued base salary payments at the then-current rate, including the payment of health insurance premiums, for the period specified in each agreement, which ranges from 12 to 18 months, plus a payment equal to between one and one and a half times the executive’s cash bonus in the previous year. In addition, the executive officers receive accelerated vesting on all their unvested stock awards as if they had remained employed by us for between 12 and 18 months from the date of termination. In the event that the termination occurs within 24 months of a “change in control,” as defined in the agreements, the severance for the executive officers consists of lump sum payments equal to between 18 and 24 months of base salary at the then-current rate, the payment of health insurance premiums for the period specified in each agreement, which ranges from 12 to 18 months, plus a payment equal to between one and one and a half times the executive’s cash bonus in the previous year. In addition, all outstanding unvested stock awards will vest immediately. The severance for the other executives consist of continued payments at the then-current base compensation rate for a period of six months. All of the agreements specify that any earnings from employment or consulting during this period will offset any salary continuation payments due from us. The maximum payments due under these employment agreements would have been $3.1 million if each such executive officer and other executives were terminated at December 31, 2011.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are subject to interest rate risk. Our investment portfolio is maintained in accordance with our investment policy which defines allowable investments, specifies credit quality standards and limits the credit exposure of any single issuer. Our investment portfolio consists of cash equivalents, both restricted and non-restricted, marketable securities and long-term investments. The average maturity of our investments, excluding our auction rate securities, is approximately four months. Our investments are classified as available-for-sale securities.
To assess our interest rate risk, we performed a sensitivity analysis projecting an ending fair value of our cash equivalents and current marketable securities using the following assumptions: a 12-month time horizon, a 9-month average maturity and a 150-basis-point increase in interest rates. This pro forma fair value would have been $143,000 lower than the reported fair value of our investments at December 31, 2011.
All of our investment securities are classified as available-for-sale and therefore reported on the balance sheet at market value. Our investment securities consist of high-grade auction rate securities, corporate debt securities and government agency securities. As of December 31, 2011, our long-term investments included (at par value) $6.5 million of auction rate securities secured by municipal bonds and student loans. At December 31, 2011, the auction rate securities we held had Standard and Poor’s credit ratings of BBB or AAA. Our auction rate
51
Table of Contents
securities are debt instruments with a long-term maturity and with an interest rate that is reset in short intervals through auctions. The recent conditions in the global credit markets have prevented some investors from liquidating their holdings of auction rate securities because the amount of securities submitted for sale has exceeded the amount of purchase orders for such securities. If there is insufficient demand for the securities at the time of an auction, the auction may not be completed and the interest rates may be reset to predetermined higher rates. When auctions for these securities fail, the investments may not be readily convertible to cash until a future auction of these investments is successful or they are redeemed or mature.
Since February 2008, there has been insufficient demand at auction for all of our auction rate securities held at December 31, 2011. As a result, these affected securities are currently not liquid, and we could be required to hold them until they are redeemed by the issuer or to maturity. As of December 31, 2011, we had recognized $1.5 million of losses related to those auction rate securities by adjusting their carrying value. The market value of these securities has partially recovered from the lows that created the losses. As of December 31, 2011, we had recorded cumulative unrealized gains of $0.6 million. Any future decline in market value may result in additional losses being recognized.
The valuation of our auction rate security investment portfolio is subject to uncertainties that are difficult to predict. The fair values of these securities are estimated utilizing a discounted cash flow analysis or other type of valuation model as of December 31, 2011. The key drivers of the valuation model include the expected term, collateralization underlying the security investments, the creditworthiness of the counterparty, the timing of expected future cash flows, rates of default of the underlying assets, discount rates, and the expectation of the next time the security is expected to have a successful auction. These securities were also compared, when possible, to other observable market data with similar characteristics to the securities held by us.
In the event we need to access the funds that are not currently liquid, we will not be able to do so without the possible loss of principal, until a future auction for these investments is successful or they are redeemed by the issuer or they mature. If we are unable to sell these securities in the market or they are not redeemed, then we may be required to hold them to maturity. We do not anticipate a need to access these funds for operational purposes for the foreseeable future. We will continue to monitor and evaluate these investments on an ongoing basis for impairment. Based on our ability to access our cash and other short-term investments, our expected operating cash flows, and our other sources of cash, we do not anticipate that the potential illiquidity of these investments will affect our ability to execute our current business plan.
52
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
INDEX TO FINANCIAL STATEMENTS
| Page | ||||
| 54 | ||||
| 55 | ||||
| 56 | ||||
| 57 | ||||
| 58 | ||||
| 59 | ||||
53
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Vical Incorporated
We have audited the accompanying balance sheets of Vical Incorporated as of December 31, 2011 and 2010, and the related statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2011. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Vical Incorporated at December 31, 2011 and 2010, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 1 to the financial statements, the Company changed its method of accounting for revenue recognition as a result of the adoption of the amendments to the FASB Accounting Standards Codification resulting from Accounting Standards Update No. 2009-13, Revenue Recognition (Topic 605): Multiple-Deliverable Revenue Arrangements, effective January 1, 2011.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Vical Incorporated’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 15, 2012 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 15, 2012
54
Table of Contents
VICAL INCORPORATED
(in thousands, except per share data)
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 38,696 | $ | 47,320 | ||||
| Marketable securities, available-for-sale |
8,733 | 5,037 | ||||||
| Restricted cash |
2,998 | 2,911 | ||||||
| Receivables and other assets |
3,130 | 940 | ||||||
|
|
|
|
|
|||||
| Total current assets |
53,557 | 56,208 | ||||||
| Long-term investments |
5,928 | 5,434 | ||||||
| Property and equipment, net |
6,226 | 7,560 | ||||||
| Intangible assets, net |
2,871 | 3,247 | ||||||
| Other assets |
191 | 458 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 68,773 | $ | 72,907 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable and accrued expenses |
$ | 6,362 | $ | 6,334 | ||||
| Deferred revenue |
99 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
6,461 | 6,334 | ||||||
| Long-term liabilities: |
||||||||
| Deferred rent |
1,964 | 2,211 | ||||||
| Commitments and contingencies (Notes 6 and 7) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.01 par value, 5,000 shares authorized, none issued and outstanding |
— | — | ||||||
| Common stock, $0.01 par value, 160,000 shares authorized, 71,913 and 71,640 shares issued and outstanding at December 31, 2011 and 2010, respectively |
719 | 716 | ||||||
| Additional paid-in capital |
384,087 | 380,929 | ||||||
| Accumulated deficit |
(325,038 | ) | (317,755 | ) | ||||
| Accumulated other comprehensive income |
580 | 472 | ||||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
60,348 | 64,362 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 68,773 | $ | 72,907 | ||||
|
|
|
|
|
|||||
See accompanying notes to financial statements
55
Table of Contents
VICAL INCORPORATED
(in thousands, except per share data)
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Revenues: |
||||||||||||
| Contract and grant revenue |
$ | 4,223 | $ | 6,249 | $ | 3,692 | ||||||
| License and royalty revenue |
25,795 | 2,462 | 8,994 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
30,018 | 8,711 | 12,686 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
17,975 | 19,692 | 23,449 | |||||||||
| Manufacturing and production |
10,267 | 11,436 | 10,354 | |||||||||
| General and administrative |
9,598 | 8,798 | 7,469 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
37,840 | 39,926 | 41,272 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(7,822 | ) | (31,215 | ) | (28,586 | ) | ||||||
| Other income (expense): |
||||||||||||
| Investment and other income, net |
539 | 830 | 30 | |||||||||
| Interest expense |
— | — | (2 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (7,283 | ) | $ | (30,385 | ) | $ | (28,558 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (0.10 | ) | $ | (0.51 | ) | $ | (0.61 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average shares used in computing basic and diluted net loss per share |
72,031 | 60,084 | 47,086 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to financial statements
56
Table of Contents
VICAL INCORPORATED
STATEMENTS OF STOCKHOLDERS’ EQUITY
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED DECEMBER 31, 2011
(in thousands)
| Common Stock | Additional Paid-in Capital |
Accumulated Deficit |
Accumulated Other Comprehensive Income/(loss) |
Total Stockholders’ Equity |
||||||||||||||||||||
| Number of Shares |
Amount | |||||||||||||||||||||||
| Balance at January 1, 2009 |
40,358 | $ | 403 | $ | 307,051 | $ | (258,812 | ) | $ | (28 | ) | $ | 48,614 | |||||||||||
| Net loss |
— | — | — | (28,558 | ) | — | (28,558 | ) | ||||||||||||||||
| Unrealized gain on marketable securities arising during holding period |
— | — | — | — | 544 | 544 | ||||||||||||||||||
| Reclassification of realized gain included in net loss |
— | — | — | — | (4 | ) | (4 | ) | ||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
(28,018 | ) | ||||||||||||||||||||||
| Issuance of common stock |
11,421 | 115 | 28,716 | — | — | 28,831 | ||||||||||||||||||
| Exercise of stock options, warrants and issuance of common stock underlying restricted stock units |
2,002 | 20 | 4,150 | — | — | 4,170 | ||||||||||||||||||
| Non-cash compensation expense related to grant of equity based compensation |
— | — | 1,385 | — | — | 1,385 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2009 |
53,781 | 538 | 341,302 | (287,370 | ) | 512 | 54,982 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss |
— | — | — | (30,385 | ) | — | (30,385 | ) | ||||||||||||||||
| Unrealized loss on marketable securities arising during holding period |
— | — | — | — | (35 | ) | (35 | ) | ||||||||||||||||
| Reclassification of realized gain included in net loss |
— | — | — | — | (5 | ) | (5 | ) | ||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
(30,425 | ) | ||||||||||||||||||||||
| Issuance of common stock |
15,368 | 153 | 32,026 | — | — | 32,179 | ||||||||||||||||||
| Exercise of stock options, warrants and issuance of common stock underlying restricted stock units |
2,491 | 25 | 4,965 | — | — | 4,990 | ||||||||||||||||||
| Non-cash compensation expense related to grant of equity based compensation |
— | — | 2,636 | — | — | 2,636 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2010 |
71,640 | 716 | 380,929 | (317,755 | ) | 472 | 64,362 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss |
— | — | — | (7,283 | ) | — | (7,283 | ) | ||||||||||||||||
| Unrealized gain on marketable securities arising during holding period |
— | — | — | — | 108 | 108 | ||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
(7,175 | ) | ||||||||||||||||||||||
| Exercise of stock options and issuance of common stock underlying restricted stock units |
273 | 3 | 52 | — | — | 55 | ||||||||||||||||||
| Non-cash compensation expense related to grant of equity based compensation |
— | — | 3,106 | — | — | 3,106 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2011 |
71,913 | $ | 719 | $ | 384,087 | $ | (325,038 | ) | $ | 580 | $ | 60,348 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
See accompanying notes to financial statements
57
Table of Contents
VICAL INCORPORATED
(in thousands)
| Year ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (7,283 | ) | $ | (30,385 | ) | $ | (28,558 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
2,298 | 2,808 | 2,629 | |||||||||
| Other than temporary loss on marketable securities and other assets |
— | — | 405 | |||||||||
| Write-off of abandoned patents |
75 | 148 | 86 | |||||||||
| Gain on sale of property and equipment |
— | — | (5 | ) | ||||||||
| Compensation expense related to stock options and awards |
3,106 | 2,636 | 1,385 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Receivables and other assets |
(2,190 | ) | 409 | 503 | ||||||||
| Other assets |
267 | — | — | |||||||||
| Accounts payable and accrued expenses |
(51 | ) | (1,771 | ) | 1,848 | |||||||
| Deferred revenue |
99 | (1,990 | ) | 281 | ||||||||
| Deferred rent |
(169 | ) | (83 | ) | (26 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(3,848 | ) | (28,228 | ) | (21,452 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Maturities of marketable securities—including restricted |
20,429 | 31,815 | 16,018 | |||||||||
| Purchases of marketable securities—including restricted |
(24,598 | ) | (18,549 | ) | (24,657 | ) | ||||||
| Purchases of property and equipment |
(256 | ) | (347 | ) | (337 | ) | ||||||
| Proceeds from the sale of property and equipment |
— | — | 10 | |||||||||
| Patent and licensed technology expenditures |
(406 | ) | (413 | ) | (315 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) provided by investing activities |
(4,831 | ) | 12,506 | (9,281 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from issuance of common stock |
214 | 37,261 | 33,038 | |||||||||
| Principal payments under equipment financing obligations |
— | — | (156 | ) | ||||||||
| Payment of withholding taxes for net settlement of restricted stock units |
(159 | ) | (92 | ) | (37 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
55 | 37,169 | 32,845 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net (decrease) increase in cash and cash equivalents |
(8,624 | ) | 21,447 | 2,112 | ||||||||
| Cash and cash equivalents at beginning of year |
47,320 | 25,873 | 23,761 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of year |
$ | 38,696 | $ | 47,320 | $ | 25,873 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental information: |
||||||||||||
| Interest paid |
$ | — | $ | — | $ | 2 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to financial statements
58
Table of Contents
VICAL INCORPORATED
1. Organization and Summary of Significant Accounting Policies
Organization and Business Activity
Vical Incorporated, or the Company, a Delaware corporation, was incorporated in April 1987 and has devoted substantially all of its resources since that time to its research and development programs. The Company researches and develops biopharmaceutical products based on its patented DNA delivery technologies for the prevention and treatment of serious or life-threatening diseases.
All of the Company’s potential products are in research and development phases. No revenues have been generated from the sale of any such products, nor are any such revenues expected for at least the next several years. The Company earns revenue from research and development agreements with pharmaceutical collaborators and grant and contract arrangements with government entities. Most of the Company’s product candidates will require significant additional research and development efforts, including extensive preclinical and clinical testing. All product candidates that advance to clinical testing will require regulatory approval prior to commercial use, and will require significant costs for commercialization. There can be no assurance that the Company’s research and development efforts, or those of its collaborators, will be successful. The Company expects to continue to incur substantial losses and not generate positive cash flows from operations for at least the next several years. No assurance can be given that the Company can generate sufficient product revenue to become profitable or generate positive cash flows from operations.
Basis of Presentation
These financial statements are prepared in conformity with accounting principles generally accepted in the United States of America. The Company has evaluated all subsequent events through the date and time its financial statements were issued.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the financial statements and disclosures made in the accompanying notes. Actual results could differ materially from those estimates.
Cash, Cash Equivalents and Marketable Securities
Cash and cash equivalents consist of cash and highly liquid securities with original maturities at the date of acquisition of ninety days or less. Investments with an original maturity of more than ninety days are considered marketable securities and have been classified by management as available-for-sale. Such investments are classified as current assets and carried at fair value, with unrealized gains and losses included as a separate component of stockholders’ equity. Realized gains and losses from the sale of available-for-sale securities or the amounts reclassified out of accumulated other comprehensive income, if any, are determined on a specific identification basis.
Restricted Cash and Marketable Securities
The Company is required to maintain a letter of credit securing an amount equal to twelve months of the current monthly installment of base rent for the term of the lease for its facilities, which ends in August 2017. Under certain circumstances the Company may be able to eliminate the need for the letter of credit. At December 31, 2011 and 2010, restricted cash of $3.0 million and $2.9 million, respectively, was pledged as collateral for the letter of credit.
59
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash equivalents, marketable securities and receivables. The Company invests its excess cash in debt instruments of financial institutions and of corporations with strong credit ratings, in U.S. government obligations, and in money market funds at financial institutions.
Property and Equipment
Property and equipment is recorded at cost and depreciation is computed using the straight-line method over the estimated useful lives of the assets. Assets acquired pursuant to capital lease arrangements and leasehold improvements are amortized using the straight-line method over the shorter of the life of the remaining lease term or the remaining useful life of the asset. Manufacturing equipment has estimated useful lives of ten years. All other property and equipment have estimated useful lives of 3 to 5 years. Maintenance and repairs of property and equipment is expensed as incurred.
Intangible Assets
Intangible assets include licensed technology rights and certain costs related to patent applications. The Company capitalizes license fees paid to acquire access to proprietary technology if the technology is expected to have alternative future use in multiple research and development projects. The cost of licensed technology rights is amortized using the straight-line method over the estimated useful life of the technology. Certain costs related to patent applications are amortized over the estimated economic lives of the patents, which is generally 20 years and typically commences at the time the patent application is filed. Amortization expense for licensed technology and capitalized patent cost is included in research and development expenses.
Impairment of Long-lived Assets
The Company reviews long-lived assets for impairment at least annually, quarterly for intangible assets, and whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Should an impairment exist, the impairment loss would be measured based on the excess of the carrying amount of the asset over the asset’s fair value and the loss recognized in current earnings. The Company recognized research and development expense of approximately $0.1 million for each of the years ended December 31, 2011, 2010 and 2009, related to patents for which the value was deemed to be impaired. The Company believes the future cash flows to be received from its remaining long-lived assets will exceed the assets’ carrying value, and accordingly has not recognized any additional impairment losses.
Revenue Recognition
Revenue is recognized when the four basic criteria of revenue recognition are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services rendered; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured. Certain of the Company’s revenue is generated through manufacturing contracts and stand-alone license agreements.
The Company has entered into multiple-element arrangements. In order to account for the multiple-element arrangements, the Company identifies the deliverables included within the agreement and evaluates which deliverables represent separate units of accounting. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation.
60
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
Multiple-element arrangements prior to January 1, 2011
Prior to adopting the revised multiple element guidance on January 1, 2011, the Company analyzed its multiple element arrangements to determine whether the identified deliverables could be accounted for individually as separate units of accounting. The delivered item(s) were considered a separate unit of accounting if all of the following criteria were met: (1) the delivered item(s) has value to the customer on a standalone basis; (2) there is objective and reliable evidence of the fair value of the undelivered item(s); and (3) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item(s) is considered probable and substantially in the Company’s control. If these criteria were not met, the deliverable was combined with other deliverables in the arrangement and accounted for as a combined unit of accounting.
Multiple-element arrangements after January 1, 2011
Effective January 1, 2011, the Company follows the provisions of ASU No. 2009-13 for all multiple element agreements, including contract manufacturing, contract services and license agreements. Under the revised guidance, the delivered item(s) has value to the customer on a standalone basis and, if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item(s) is considered probable and substantially in the Company’s control.
A delivered item is considered a separate unit of accounting when the delivered item has value to the partner on a standalone basis based on the consideration of the relevant facts and circumstances for each arrangement. Factors considered in this determination include the research capabilities of the partner and the availability of research expertise in this field in the general marketplace. Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence, or VSOE, of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, the Company uses its best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement. If facts and circumstances dictate that the license has standalone value from the undelivered items, which generally include research and development services and the manufacture of drug products, the license is identified as a separate unit of accounting and the amounts allocated to the license are recognized upon the delivery of the license, assuming the other revenue recognition criteria have been met. However, if the amounts allocated to the license through the relative selling price allocation exceed the upfront license fee, the amount recognized upon the delivery of the license is limited to the upfront fee received. If facts and circumstances dictate that the license does not have standalone value, the transaction price, including any upfront license fee payments received, are allocated to the identified separate units of accounting and recognized as those items are delivered.
The terms of the Company’s partnership agreements provide for milestone payments upon achievement of certain regulatory and commercial events. Effective January 1, 2011, the Company adopted on a prospective basis the Milestone Method of accounting under ASU 2010-17. Under the Milestone Method, the Company recognizes consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following three criteria: 1) The consideration is commensurate with either the entity’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, 2) The
61
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
consideration relates solely to past performance, and 3) The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to the Company.
Contract Services, Grant and Royalty Revenue
The Company recognizes revenues from contract services and federal government research grants during the period in which the related expenditures are incurred and related payments for those services are received or collection is reasonably assured. Royalties to be received based on sales of licensed products by the Company’s partners incorporating the Company’s licensed technology are recognized when received.
Accruals for Potential Disallowed Costs on Government Contracts
The Company has contracts with U.S. government agencies under which it bills for direct and indirect costs incurred. These billed costs are subject to audit by government agencies. The Company has established accruals of approximately $49,000 and $0.2 million at December 31, 2011 and 2010, respectively, to provide for potential disallowed costs. In the event that the final costs allowed are different from what the Company has estimated, the Company may need to make a change in its estimated accrual, which could also affect its results of operations and cash flow.
Accruals for Potential Disallowed Costs on Government Contracts
The Company has contracts with U.S. government agencies under which it bills for direct and indirect costs incurred. These billed costs are subject to audit by government agencies. The Company has established accruals of approximately $49,000 and $0.2 million at December 31, 2011 and 2010, respectively, to provide for potential disallowed costs. In the event that the final costs allowed are different from what the Company has estimated, the Company may need to make a change in its estimated accrual, which could also affect its results of operations and cash flow.
Research and Development Costs
Research and development costs are expensed as incurred. Research and development costs include salaries and personnel-related costs, supplies and materials, outside services, costs of conducting preclinical and clinical trials, facilities costs and amortization of intangible assets. The Company accounts for its clinical trial costs by estimating the total cost to treat a patient in each clinical trial, and accruing this total cost for the patient over the estimated treatment period, which corresponds with the period over which the services are performed, beginning when the patient enrolls in the clinical trial. This estimated cost includes payments to the site conducting the trial, and patient-related lab and other costs related to the conduct of the trial. Cost per patient varies based on the type of clinical trial, the site of the clinical trial, the method of administration of the treatment, and the number of treatments that a patient receives. Treatment periods vary depending on the clinical trial. The Company makes revisions to the clinical trial cost estimates in the current period, as clinical trials progress.
62
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
Manufacturing and Production Costs
Manufacturing and production costs include expenses related to manufacturing contracts and expenses related to the production of plasmid DNA for use in the Company’s research and development efforts. Manufacturing expenses related to manufacturing contracts are deferred and expensed when the related revenue is recognized. Production expenses related to the Company’s research and development efforts are expensed as incurred.
Net Loss Per Share
Basic and diluted net loss per share has been computed using the weighted-average number of shares of common stock outstanding during the period. The weighted-average number of shares used to compute diluted loss per share excludes any assumed exercise of stock options, and the assumed issuance of common stock under restricted stock units, or RSUs, as the effect would be antidilutive. Common stock equivalents of 1.4 million, 0.7 million and 1.1 million for the years ended December 31, 2011, 2010 and 2009, respectively, were excluded from the calculation because of their antidilutive effect.
Fair Value of Financial Instruments
The carrying amounts of cash, cash equivalents, restricted marketable securities, marketable securities, receivables, accounts payable and accrued expenses at December 31, 2011 and 2010, are considered to reasonably approximate fair value because of the short term nature of those items.
Income Taxes
Deferred income taxes result primarily from temporary differences between financial and tax reporting. Deferred tax assets and liabilities are determined based on the difference between the financial statement bases and the tax bases of assets and liabilities using enacted tax rates. A valuation allowance is established to reduce a deferred tax asset to the amount that is expected more likely than not to be realized.
Comprehensive Loss
Comprehensive loss consists of net loss and certain changes in equity that are excluded from net loss. Comprehensive loss for the years ended December 31, 2011, 2010 and 2009, has been reflected in the Statements of Stockholders’ Equity. Accumulated other comprehensive income (loss), which is included in stockholders’ equity, represents unrealized gains and losses on marketable securities.
Business Segments
The Company operates in one business segment, which is within the United States, and is dedicated to research and development of DNA delivery technology.
Recent Accounting Pronouncements
In June 2011, the Financial Accounting Standards Board, or FASB, issued authoritative guidance regarding comprehensive income. This newly issued accounting standard allows an entity to have the option to present the components of net income and comprehensive income in either one or two consecutive financial statements. The guidance eliminates the current option to report other comprehensive income and its components in the statement of changes in stockholders’ equity. While the new guidance changes the presentation of comprehensive income,
63
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
there are no changes to the components that are recognized in net income or other comprehensive income under current accounting guidance. The guidance is effective for fiscal years and interim periods beginning after December 15, 2011. While the new guidance will require us to change the manner in which we present other comprehensive income and its components on a retrospective basis, we do not believe our adoption of the guidance in the first quarter of 2012 will have a material impact on our financial position, results of operations or cash flows.
In May 2011, the FASB issued authoritative guidance regarding common fair value measurements and disclosure requirements in U.S. Generally Accepted Accounting Principles and International Financial Reporting Standards. This newly issued accounting standard clarifies the application of certain existing fair value measurement guidance and expands the disclosures for fair value measurements that are estimated using significant unobservable inputs. This guidance is effective on a prospective basis for annual and interim reporting periods beginning on or after December 15, 2011. The Company does not expect that the adoption of this standard will have a material impact on its financial position or results of operations.
In March 2010, the FASB ratified the milestone method of revenue recognition. Under this new standard, an entity can recognize contingent consideration earned from the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved and (iii) that would result in additional payments being due to the entity. This guidance is effective for annual periods beginning after June 15, 2010, but may be adopted earlier as of the beginning of an annual period. The Company adopted these provisions as of January 1, 2011. The adoption did not have a material impact on the Company’s financial position or results of operations.
In September 2009, the FASB issued authoritative guidance regarding multiple-deliverable revenue arrangements. This guidance requires an entity to allocate arrangement consideration at the inception of an arrangement to all of its deliverables based on their relative selling prices. The guidance eliminates the use of the residual method of allocation and requires the relative-selling-price method in all circumstances in which an entity recognizes revenue for an arrangement with multiple deliverables. The Company adopted these provisions as of January 1, 2011. The adoption allowed us to recognize the $25 million upfront license fee included in the Astellas license agreement as further discussed in footnote 6.
Share-Based Compensation
The Company records its compensation expense associated with stock options and other forms of equity compensation based on their fair value at the date of grant using the Black-Scholes-Merton option pricing model. Stock-based compensation includes amortization related to stock option awards based on the estimated grant date fair value. Stock-based compensation expense related to stock options includes an estimate for forfeitures and the portion that is ultimately expected to vest is recognized ratably over the vesting period of the option. In addition, the Company records expense related to RSUs granted based on the fair value of those awards on the grant date. The fair value related to the RSUs is amortized to expense over the vesting term of those awards. Stock-based compensation expense related to RSUs includes an estimate for forfeitures and is recognized over the expected term of the award using the straight-line method. The expected forfeiture rate of all equity based compensation is based on observed historical patterns of the Company’s employees and is estimated to be 11.2% annually for each of the years ended December 31, 2011, 2010 and 2009.
The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton valuation model using the assumptions noted in the following table. The expected life of options is based on the
64
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
Company’s observed historical exercise patterns. The expected volatility of stock options is based upon the historical volatility of the Company’s stock. The risk-free interest rate is based on the implied yield on a U.S. Treasury zero-coupon issue with a remaining term equal to the expected term of the option. The dividend yield reflects that the Company has not paid any cash dividends since inception and does not intend to pay any cash dividends in the foreseeable future.
| Years Ended December 31, | ||||||
| 2011 | 2010 | 2009 | ||||
| Assumptions: |
||||||
| Assumed risk-free interest rate |
1.90% | 2.44% | 1.62% | |||
| Assumed volatility |
68% | 66% | 60% | |||
| Average expected option life |
4.5 years | 4.5 years | 4.5 years | |||
| Expected dividend yield |
— | — | — | |||
2. Short-Term Marketable Securities
The following is a summary of short-term marketable securities classified as available-for-sale (in thousands):
| December 31, 2011 |
Amortized Cost |
Unrealized Gain |
Unrealized Loss |
Market Value |
||||||||||||
| U.S. treasuries |
$ | 1,008 | $ | 2 | $ | — | $ | 1,010 | ||||||||
| Government-sponsored enterprise securities |
7,200 | — | 2 | 7,198 | ||||||||||||
| Certificate of deposit |
525 | — | — | 525 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 8,733 | $ | 2 | $ | 2 | $ | 8,733 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| December 31, 2010 |
Amortized Cost |
Unrealized Gain |
Unrealized Loss |
Market Value |
||||||||||||
| U.S. treasuries |
$ | 1,002 | $ | — | $ | — | $ | 1,002 | ||||||||
| Government-sponsored enterprise securities |
2,999 | 1 | — | 3,000 | ||||||||||||
| Certificate of deposit |
1,035 | — | — | 1,035 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 5,036 | $ | 1 | $ | — | $ | 5,037 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
At December 31, 2011, $5.0 million of these securities are scheduled to mature outside of one year and are classified as current assets. There were no net realized gains (losses) on sales of available-for-sale securities for the years ended December 31, 2011, 2010 and 2009. None of these investments have been in a continuous unrealized loss position for more than 12 months as of December 31, 2011 and 2010.
3. Long-Term Investments
As of December 31, 2011, the Company held $6.5 million (at par value) of auction rate securities which were classified as long-term investments. With the liquidity issues experienced in global credit and capital markets, these auction rate securities have experienced multiple failed auctions since 2008, as the amount of securities submitted for sale has exceeded the amount of purchase orders, and as a result, these affected securities are currently not liquid. All of the Company’s auction rate securities are secured by either student loans or municipal bonds. The student loans are backed by the full faith and credit of the federal government (up to approximately 98% of the value of the student loan). At December 31, 2011, the auction rate securities held by the Company had Standard and Poor’s credit ratings of BBB or AAA. At December 31, 2010, the auction rate
65
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
securities held by the Company had Standard and Poor’s credit ratings of BBB or AAA. All of these securities continue to pay interest according to their stated terms. While it is not the Company’s intent to hold these securities until their stated ultimate maturity dates, these investments are scheduled to ultimately mature between 2038 and 2043.
The valuation of the Company’s auction rate security investment portfolio is subject to uncertainties that are difficult to predict. The fair values of these securities are estimated utilizing a discounted cash flow analysis. The key drivers of the valuation model include the expected term, collateralization underlying the security investments, the creditworthiness of the counterparty, the timing of expected future cash flows, rates of default of the underlying assets, discount rates, and the expectation of the next time the security is expected to have a successful auction. These securities were also compared, when possible, to other observable market data for securities with similar characteristics. Based on the valuation of the individual securities, the Company has recognized cumulative losses of $1.5 million as of December 31, 2011, none of which was realized during the year ended December 31, 2011. The losses when incurred are included in investment and other income. The market value of these securities has partially recovered. Included in other comprehensive income are net unrealized (losses)/gains of $0.5 million, $(23,000) and $0.5 million during the years ended December 31, 2011, 2010 and 2009, respectively. As of December 31, 2011, the Company had recorded cumulative unrealized gains of $0.6 million. The resulting carrying value of the auction rate securities at December 31, 2011 was $5.9 million, which is included in long-term investments. Any future decline in market value may result in additional losses being recognized.
At present, in the event the Company needed to liquidate its auction rate securities that are in an illiquid state, it may not be able to do so without the possible loss of principal until a future auction for these investments is successful, another secondary market evolves for these securities, they are redeemed by the issuer or they mature. If the Company is unable to sell these securities in the market or they are not redeemed, then the Company could be required to hold them to maturity. The Company does not have a need to access these funds for operational purposes in the foreseeable future. The Company will continue to monitor and evaluate these investments on an ongoing basis for impairment.
4. Fair Value Measurements
The Company measures fair value as an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability. Fair value measurements are based on a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value as follows:
| • | Level 1: Observable inputs such as quoted prices in active markets; |
| • | Level 2: Inputs, other than the quoted prices in active markets, that are observable either directly or indirectly; and |
| • | Level 3: Unobservable inputs for which there is little or no market data, which require the reporting entity to develop its own assumptions. |
66
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
Cash equivalents, marketable securities and long-term investments measured at fair value are classified in the table below in one of the three categories described above (in thousands):
| Fair Value Measurements | ||||||||||||||||
| December 31, 2011 |
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Certificates of deposit |
$ | 525 | $ | — | $ | — | $ | 525 | ||||||||
| Money market funds |
12,778 | — | — | 12,778 | ||||||||||||
| U.S. treasuries |
1,010 | — | — | 1,010 | ||||||||||||
| Government-sponsored enterprise securities |
— | 7,198 | — | 7,198 | ||||||||||||
| Auction rate securities |
— | — | 5,928 | 5,928 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 14,313 | $ | 7,198 | $ | 5,928 | $ | 27,439 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Fair Value Measurements | ||||||||||||||||
| December 31, 2010 |
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Certificates of deposit |
$ | 1,035 | $ | — | $ | — | $ | 1,035 | ||||||||
| Money market funds |
47,251 | — | — | 47,251 | ||||||||||||
| U.S. treasuries |
1,002 | — | — | 1,002 | ||||||||||||
| Government-sponsored enterprise securities |
— | 3,000 | — | 3,000 | ||||||||||||
| Auction rate securities |
— | — | 5,434 | 5,434 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 49,288 | $ | 3,000 | $ | 5,434 | $ | 57,722 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The Company’s investments in U.S. treasury securities, certificates of deposit and money market funds are valued based on publicly available quoted market prices for identical securities as of December 31, 2011. The Company determines the fair value of other government-sponsored enterprise related securities with the aid of valuations provided by third parties using proprietary valuation models and analytical tools. These valuation models and analytical tools use market pricing or similar instruments that are both objective and publicly available, including matrix pricing or reported trades, benchmark yields, broker/dealer quotes, issuer spreads, two-sided markets, benchmark securities, bids and/or offers. The Company validates the valuations received from its primary pricing vendors for its level 2 securities by examining the inputs used in that vendor’s pricing process and determines whether they are reasonable and observable. The Company also compares those valuations to recent reported trades for those securities The Company did not adjust any of the valuations received from these third parties with respect to any of its level 2 securities at December 31, 2011. The valuation of the Company’s investments in auction rate securities are more fully described in Note 3.
Activity for assets measured at fair value using significant unobservable inputs (Level 3) is presented in the table below (in thousands):
| Balance at December 31, 2010 |
$ | 5,434 | ||
| Total net unrealized gains included in other comprehensive income |
494 | |||
| Net transfers in and/out of Level 3 |
— | |||
|
|
|
|||
| Balance at December 31, 2011 |
$ | 5,928 | ||
|
|
|
|||
| Amount of total losses for the period included in net loss attributable to the change in unrealized gains or losses relating to assets still held at December 31, 2011 |
$ | — | ||
|
|
|
67
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
The Company transferred $6.5 million of its long-term investments into the Level 3 category during the year ended December 31, 2008. Total unrealized losses of $1.5 million relate to Level 3 assets still held as of December 31, 2011. Unrealized losses of zero, zero and $0.4 million related to Level 3 assets are included in earnings for the year ended December 31, 2011, 2010 and 2009, respectively, and are included in investment and other income.
5. Other Balance Sheet Accounts
Property and equipment consisted of the following at December 31 (in thousands):
| 2011 | 2010 | |||||||
| Equipment |
$ | 19,147 | $ | 18,959 | ||||
| Leasehold improvements |
7,992 | 7,992 | ||||||
|
|
|
|
|
|||||
| 27,139 | 26,951 | |||||||
| Less accumulated depreciation and amortization |
(20,913 | ) | (19,391 | ) | ||||
|
|
|
|
|
|||||
| $ | 6,226 | $ | 7,560 | |||||
|
|
|
|
|
|||||
Depreciation and amortization of equipment and leasehold improvements for the years ended December 31, 2011, 2010 and 2009, was $1.6 million, $2.0 million and $1.8 million, respectively. These amounts include depreciation related to equipment under equipment financing arrangements. See Note 7 for equipment financing arrangements.
Intangible assets consisted of the following at December 31 (in thousands):
| 2011 | 2010 | |||||||
| Licensed technology rights |
$ | 4,015 | $ | 4,015 | ||||
| Patent application costs |
6,093 | 5,787 | ||||||
|
|
|
|
|
|||||
| 10,108 | 9,802 | |||||||
| Accumulated amortization licensed technology rights |
(3,906 | ) | (3,538 | ) | ||||
| Accumulated amortization patent costs |
(3,331 | ) | (3,017 | ) | ||||
|
|
|
|
|
|||||
| $ | 2,871 | $ | 3,247 | |||||
|
|
|
|
|
|||||
Amortization of licensed technology rights and patent application costs for the years ended December 31, 2011, 2010 and 2009, was $0.7 million, $0.8 million and $0.8 million, respectively. Estimated annual amortization for these assets for each of the years in the period from 2012 to 2016 is $0.4 million, $0.4 million, $0.3 million, $0.3 million and $0.2 million, respectively.
Accounts payable and accrued expenses consisted of the following at December 31 (in thousands):
| 2011 | 2010 | |||||||
| Employee compensation |
$ | 3,653 | $ | 2,730 | ||||
| Clinical trial accruals |
1,681 | 2,338 | ||||||
| Accounts payable |
351 | 752 | ||||||
| Other accrued liabilities |
677 | 514 | ||||||
|
|
|
|
|
|||||
| $ | 6,362 | $ | 6,334 | |||||
|
|
|
|
|
|||||
68
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
6. Significant Contracts, Grants, License and Royalty Agreements
Contract and Grant Agreements
Astellas
In July 2011, the Company entered into license agreements with Astellas Pharma Inc., or Astellas, granting Astellas exclusive, worldwide, royalty-bearing licenses under certain of the Company’s know-how and intellectual property to develop and commercialize certain products containing plasmids encoding certain forms of glycoprotein B and/or phosphoprotein 65, including TransVax™ but excluding CyMVectin™. Under the agreements, Astellas is responsible for the worldwide development and commercialization of products in the licensed field, at its expense, and has agreed to use commercially reasonable efforts to develop, obtain regulatory approval for and commercialize at least one licensed product for use in certain immunocompromised patients in the licensed field in the United States and certain other major markets. Under the terms of the license agreements, Astellas paid a nonrefundable upfront license fee of $25.0 million.
The Company will be entitled to receive a $10.0 million milestone payment upon finalization of the trial design for a Phase 3 registration trial of TransVax™ in hematopoietic stem cell transplant recipients. Subsequent to the achievement of the trial design milestone, the Company is also entitled to receive additional cash payments potentially totaling $95.0 million for achievement of certain milestones through commercial launch and to receive double-digit royalties on net sales of products. In addition, the Company has an option to co-promote TransVax™ in the United States. Under the terms of a supply and services agreement entered into by the Company and Astellas on the same date, the Company agreed to perform certain development and regulatory activities, at Astellas’ expense, and to supply licensed products to Astellas, at Astellas’ expense, for use in development and initial commercialization activities in the licensed field.
The Company identified the deliverables at the inception of the agreements. The Company has determined that the license and related know-how, the development and regulatory services and the drug product supply individually represent separate units of accounting, because each deliverable has standalone value. The best estimated selling prices for these units of accounting was determined based on market conditions, the terms of comparable collaborative arrangements for similar technology in the pharmaceutical and biotechnology industry and entity-specific factors, such as the terms of the Company’s previous collaborative agreements, the Company’s pricing practices and pricing objectives and the nature of the research and development services to be performed for the partner. The arrangement consideration was allocated to the deliverables based on the relative selling price method.
However, the amount of allocable arrangement consideration is limited to amounts that are fixed or determinable; therefore, the amount allocated to the licenses at December 31, 2011 was limited to the extent of cash received. As a result, during the year ended December 31, 2011, the Company recognized $25.3 million related to the license fee and know-how. The Company will recognize the amounts allocated to research and development services as revenues under the agreements as the related services are delivered and as reimbursements are received. During the year ended December 31, 2011, the Company recognized $2.7 million of revenue related to contract services delivered. The Company will recognize as revenue the amounts allocated to the sales of drug product when the sale of that drug product has met all required specifications and the related title and risk of loss and damages have passed to Astellas. The Company is eligible to receive additional cash payments upon the achievement of specified regulatory and commercial milestones. The Company has determined that each of the regulatory and commercial milestones meets the definition of a milestone and that each milestone is substantive in accordance with the milestone method of revenue recognition. Accordingly, the Company expects to recognize such regulatory and commercial milestone payments as revenues under the agreements upon achievement of each milestone.
69
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
AnGes Research and Development Agreement
On May 25, 2006, the Company entered into a research and development agreement, or R&D Agreement, with AnGes MG, Inc., or AnGes, whereby AnGes agreed to fund the Company’s Allovectin® Phase 3 clinical trial. The funding consisted of purchases by AnGes of $10.85 million of restricted shares of the Company’s common stock and additional non-refundable cash payments by AnGes of up to $11.8 million. The Company and AnGes have agreed to share project costs, up to certain limits, that are in excess of $22.6 million. All of the funding provided by AnGes, including those funds used to purchase the Company’s common stock, must be used for actual and documented costs related to the conduct of the Allovectin® Phase 3 trial.
Under the R&D Agreement, the Company has granted to AnGes exclusive marketing rights for Allovectin® in specified countries in Asia and AnGes has agreed to pursue regulatory approvals in those countries, subject to receipt by the Company of regulatory approval in the United States. The Company has also granted AnGes certain royalty-bearing licenses to its technology and know-how. AnGes is obligated to pay royalties to the Company on sales of Allovectin® in specified countries in Asia. AnGes also obtained the right to receive royalties from the Company on any commercial sales of Allovectin® in the United States outside specified Asian countries. AnGes may also purchase supplies of Allovectin® from the Company for resale by AnGes in Asia.
The first equity installment of $6.9 million was received by the Company upon execution of the R&D Agreement and a related stock purchase agreement. In accordance with the terms of the stock purchase agreement AnGes was issued 1,061,538 shares of the Company’s restricted common stock at $6.50 per share in exchange for the first installment. The second equity installment of $3.95 million was received by the Company during 2008. In accordance with the terms of the stock purchase agreement AnGes was issued 1,109,550 shares of the Company’s restricted common stock at $3.56 per share in exchange for the second installment.
Under the stock purchase agreement, the Company has also granted AnGes limited rights to require the Company to register the shares of common stock under the Securities Act of 1933, as amended, upon the occurrence of certain events. AnGes has also agreed to certain transfer restrictions with respect to the shares of common stock sold under the stock purchase agreement and has further agreed to certain standstill provisions whereby AnGes will refrain from acquiring or taking certain other actions with respect to the Company’s common stock, subject to certain exceptions.
The Company has received total cash installments of $11.8 million from AnGes under the agreement. Revenue of $0.0 million, $2.0 million and $6.7 million has been recognized during the years ended December 31, 2011, 2010 and 2009, respectively, based on the ratio of actual costs incurred to total estimated costs expected to be incurred.
U.S. Navy
In 2008, the Company entered into a $1.3 million contract with the Naval Medical Research Center, or NMRC, to manufacture a dengue DNA vaccine formulated with the Company’s Vaxfectin® adjuvant. The Company manufactured the vaccine and the adjuvant and provided regulatory and clinical expertise. The NMRC utilized the vaccine in preclinical and Phase 1 clinical trials. The vaccine was delivered and $1.3 million of revenue was recognized in 2009.
In 2009, the Company entered into a Cooperative Research and Development Agreement, or CRADA, with the NMRC, to develop an H1N1 DNA vaccine formulated with the Company’s Vaxfectin® adjuvant. Under the
70
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
agreement, the Company was obligated to manufacture the vaccine and adjuvant, perform preclinical studies, file an IND with the FDA, and develop a clinical immunoassay. These activities were being funded under a $1.3 million contract with the U.S. government. NMRC plans to utilize the vaccine in a Phase 1 clinical trial which began in 2010. The Company recognized $0.7 million and $0.6 million in revenue related to this contract in 2010 and 2009, respectively.
In 2010, the Company entered into a $0.8 million contract with the U.S. Navy, through the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., to conduct a Phase 1 clinical trial of the company’s Vaxfectin®-formulated DNA vaccine against A/H1N1 pandemic influenza utilizing the vaccine the Company manufactured under the 2009 CRADA with the NMRC. The Company recognized $0.8 million in revenue related to this contract in 2010.
IPPOX Foundation
In 2010, the Company entered into an agreement to manufacture bulk plasmid DNA vaccines against HIV under a $2.4 million contract with the IPPOX Foundation, a collaborating institution for the Poxvirus Vaccine Regimen Design led by the Centre Hospitalier Universitaire Vaudois under the auspices of the Collaboration for AIDS Vaccine Discovery. The Company recognized $2.4 million in revenue related to this contract in 2010.
Government Grants
In 2008, the Company was awarded a two-year, $2.0 million Phase II Small Business Technology Transfer grant from National Institute of Allergy and Infectious Diseases, or NIAID, of the NIH. The grant period was extended to allow preclinical development to continue for a third year. The grant will fund the ongoing development of the Company’s immunotherapeutic plasmid DNA vaccine against herpes simplex virus type 2, or HSV-2. The Company recognized $0.2 million, $0.7 million and $0.6 million in revenue under this grant in 2011, 2010 and 2009, respectively.
In 2007, the Company was awarded funding for a three-year, $6.0 million grant from the NIAID for development of a DNA vaccine manufacturing process with the potential to produce several million doses of vaccines in a matter of days. The grant period was extended to allow preclinical development to continue for a fourth year. The Company recognized $0.9 million, $1.5 million and $1.0 million in revenue under this grant in 2011, 2010 and 2009, respectively.
License and Royalty Agreements
Merck
In 1991, the Company entered into an agreement with Merck, which was subsequently amended, providing Merck with certain exclusive rights to develop and commercialize vaccines using the Company’s core DNA delivery technology for specified human diseases. Under the agreement, as amended, Merck licensed the Company’s core DNA delivery technology for use in preventive and therapeutic human infectious disease vaccines.
In June 2005, Merck exercised an option related to three cancer targets that were granted under an amendment to the agreement. Merck initiated a Phase 1 clinical trial of a candidate vaccine based on our DNA gene delivery technology and encodes human telomerase reverse transcriptase, or hTERT.
Merck is obligated to pay fees if certain research milestones are achieved, and royalties on net sales if any products covered by the Company’s agreement with Merck are commercialized. Merck has the right to terminate
71
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
this agreement without cause upon 90 days prior written notice. Total revenue recognized under this agreement was $1.5 million in 2009. No revenues were recognized under this agreement in 2011 or 2010. Further development of these vaccines may lead to additional milestone and royalty payments.
In-licensing Agreements
City of Hope
In 2003, the Company licensed from the City of Hope on an exclusive basis various U.S. patents that provide protection for CMV related polynucleotide based vaccines, including the TransVaxTM and CyMVectinTM vaccine candidates. The agreement expires upon the last to expire of the patent rights licensed by the Company under the agreement, unless earlier terminated as set forth in the agreement. The City of Hope may terminate the agreement early, in accordance with notice provisions set forth in the agreement, if the Company ceases to operate, fails to make payments when due or materially breaches the agreement. Subject to certain conditions, the Company may terminate the agreement early at any time upon prior written notice to the City of Hope. The Company is also obligated to pay a low double-digit percentage of any payments it receives from the sub-license of products that incorporate the licensed technology. The Company paid the City of Hope $1.9 million, $0.1 million and $0.1 million under the agreement for the years ended December 31, 2011, 2010 and 2009, respectively.
CytRx
In 2001, the Company entered into an exclusive agreement with CytRx which grants to the Company the rights to use or sublicense CytRx’s poloxamer technology to enhance viral or non-viral delivery of polynucleotides in all preventive and therapeutic human and animal health applications, including CMV. The agreement excludes applications for four infectious disease vaccine targets that had been licensed to Merck and prostate-specific membrane antigen. In addition, the agreement permits the Company’s use of CytRx’s technology to enhance the delivery of proteins in prime-boost vaccine applications that involve the use of polynucleotides. As part of the agreement, the Company made a $3.8 million up-front payment and agreed to make potential future milestone and royalty payments. The license fee is fully amortized.
Wisconsin Alumni Research Foundation and University of Michigan License Agreements
The Company has research and exclusive license agreements with the Wisconsin Alumni Research Foundation, or WARF, and the University of Michigan for continuing research and license rights to technology related to DNA delivery. The agreements grant the Company the right to commercialize any product derived from specified technology. The fees paid by the Company under these agreements are expensed as incurred.
Under the Merck, Sanofi, AnGes, Merial and Aqua Health agreements, the Company is required to pay up to 10% of certain initial upfront monetary payments, and a small percentage of some royalty payments, to the WARF. The CytRx, University of Michigan, and other license agreements require the Company to make payments if the Company or its sublicensees advance products through clinical development. For programs developed with the support of U.S. government funding, the U.S. government may have rights to resulting products without payment of royalties.
Milestone Payments
The Company may be required to make future payments to its licensors based on the achievement of milestones set forth in various in-licensing agreements. In most cases, these milestone payments are based on the achievement of development or regulatory milestones, including the exercise of options to obtain licenses related
72
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
to specific disease targets, commencement of various phases of clinical trials, filing of product license applications, approval of product licenses from the FDA or a foreign regulatory agency, and the first commercial sale of a related product. Payment for the achievement of milestones under the Company’s in-license agreements is highly speculative and subject to a number of contingencies.
The aggregate amount of additional milestone payments that the Company could be required to pay under all of its in-license agreements in place at December 31, 2011, is approximately $23.4 million, of which approximately $15.5 million is related to the Company’s independent programs and corporate and government collaborations which are currently in clinical trials. These amounts assume that all remaining milestones associated with the milestone payments are met. In the event that product license approval for any of the related products is obtained, the Company may be required to make royalty payments in addition to these milestone payments. Although the Company believes that some of the milestones contained in its in-license agreements may be achieved, it is highly unlikely that a significant number of them will be achieved. Because the milestones are highly contingent and the Company has limited control over whether the development and regulatory milestones will be achieved, the Company is not in a position to reasonably estimate how much, if any, of the potential milestone payments will ultimately be paid. Additionally, under the in-license agreements, many of the milestone events are related to progress in clinical trials which will take several years to achieve.
7. Commitments and Contingencies
Facility Leases
The Company is currently leasing its facility which has approximately 68,400 square feet of manufacturing, research laboratory and office space. The lease expires in August 2017. The Company has the option to renew the lease for three additional five-year periods beyond its expiration.
The lease related to the facility is treated as an operating lease. The minimum annual rent on the facility is subject to increases specified in the lease. The Company is also required to pay taxes, insurance and operating costs under the facility lease. The Company recognizes level monthly rent for its facility lease over the entire lease period. The monthly rent is calculated by adding the total rent payments over the entire lease period and then dividing the result by the total term of the lease. The $2.2 million difference between the base rent paid and the rent expensed through December 31, 2011, is recorded as deferred rent in the balance sheet. Rent expense for the years ended December 31, 2011, 2010 and 2009, was $2.9 million, $2.9 million and $3.3 million, respectively.
At December 31, 2011, future minimum rental payments due under the Company’s facilities lease were as follows (in thousands):
| Year ending December 31, |
||||
| 2012 |
$ | 3,306 | ||
| 2013 |
3,372 | |||
| 2014 |
3,440 | |||
| 2015 |
3,509 | |||
| 2016 |
3,579 | |||
| Thereafter |
2,417 | |||
|
|
|
|||
| Total lease payments |
$ | 19,623 | ||
|
|
|
Other Contingencies
The Company prosecutes its intellectual property estate vigorously to obtain the broadest valid scope for its patents. Due to uncertainty of the ultimate outcome of these matters, the impact on future operating results or the Company’s financial condition is not subject to reasonable estimates.
73
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
In the ordinary course of business, the Company may become a party to lawsuits involving various matters. The Company is unaware of any such lawsuits presently pending against it which, individually or in the aggregate, are deemed to be material to the Company’s financial condition or results of operations.
8. Stockholders’ Equity
As of the date of this filing the Company has on file a shelf registration statement that allows it to raise up to an additional $53.4 million from the sale of common stock, preferred stock, debt securities and/or warrants. Specific terms of any offering under the shelf registration statements and the securities involved would be established at the time of sale.
In September 2010, the Company sold 15.0 million shares of its common stock in a public offering at a price to the public of $2.25 per share. Net proceeds from the offering, after deducting underwriting discounts and commissions and other offering expenses payable by the Company, totaled $31.5 million. In October 2010, the Company sold an additional 368,662 shares pursuant to the exercise of the underwriters’ overallotment option at a price to the public of $2.25 per share. Net proceeds from the overallotment option exercise, after deducting underwriting discounts and commissions and other offering expenses payable by the Company, totaled $0.8 million. All of the shares of common stock were offered pursuant to an effective shelf registration statement.
In the fourth quarter of 2009 and again in the first quarter of 2010, certain of the Company’s investors exercised warrants to purchase an aggregate of 1,967,689 and 2,365,644 shares of common stock, respectively. The Company received net proceeds of $4.2 million and $5.0 million as a result of these exercises in 2009 and 2010, respectively. The warrants were issued in connection with the Company’s May 2009 registered direct offering.
In July 2009, the Company completed a $10.0 million registered direct offering of its common stock to the Federated Kaufmann Funds. Under the terms of the Common Stock Purchase Agreement for the offering, the Company sold an aggregate of 2,754,821 shares of its common stock for a purchase price of $3.63 per share. Net proceeds from the offering, after deducting related expenses, totaled $9.9 million. All of the shares of common stock were offered pursuant to an effective shelf registration statement.
In May 2009, the Company completed a $20.0 million registered direct offering of its securities to three institutional investors. Under the terms of the Securities Purchase Agreements for the offering, the Company sold an aggregate of 8,666,667 shares of its common stock and warrants to purchase up to an aggregate of 4,333,333 additional shares of its common stock. Each unit, consisting of one share of common stock and a warrant to purchase approximately one-half of a share of common stock, was sold for a purchase price of $2.3125. Net proceeds from the offering, after deducting related expenses, totaled $18.9 million. The warrants to purchase additional shares became exercisable six months after issuance at $2.25 per share and have all been exercised. The estimated fair value of the warrants is included in additional paid in capital. All of the securities were offered pursuant to an effective shelf registration statement.
In June 2008, the Company received approximately $4.0 million in gross proceeds from the sale of approximately 1.1 million shares of its common stock at $3.56 per share in a private placement to AnGes. In June 2006, the Company received approximately $6.9 million in gross proceeds from the sale of approximately 1.1 million shares of its common stock at $6.50 per share in a private placement to AnGes. Both of the equity issuances to AnGes were pursuant to a research and development agreement and a stock purchase agreement as described in Note 6.
74
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
9. Stock Based Compensation
On December 31, 2011, the Company had two stock-based compensation plans, which are described below. Total stock-based compensation expense of $3.1 million, $2.6 million and $1.4 million was recognized for the years ended December 31, 2011, 2010 and 2009, respectively.
Total stock-based compensation expense was allocated to research and development, manufacturing and production and general and administrative expense as follows (in thousands):
| Years ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Research and development |
$ | 947 | $ | 727 | $ | 379 | ||||||
| Manufacturing and production |
165 | 281 | 154 | |||||||||
| General and administrative |
1,994 | 1,628 | 852 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation expense |
$ | 3,106 | $ | 2,636 | $ | 1,385 | ||||||
|
|
|
|
|
|
|
|||||||
| Cash received from options exercised |
$ | 212 | $ | 35 | $ | 6 | ||||||
|
|
|
|
|
|
|
|||||||
Stock Plan and Directors’ Stock Option Plan
The Company has a stock incentive plan, under which 12,700,000 shares of common stock, subject to adjustment as provided in the plan, are reserved for issuance to employees, non-employee directors and consultants of the Company. As of December 31, 2011 there were 11,016,619 shares reserved for future issuance under the plan. The plan provides for the grant of incentive and nonstatutory stock options and the direct award or sale of shares, including restricted stock. The exercise price of stock options must equal at least the fair market value of the underlying common stock on the date of grant. The maximum term of options granted under the plan is ten years. Except for annual grants to non-employee directors which vest at the next annual meeting, options generally vest 25% on the first anniversary of the date of grant, with the balance vesting quarterly over the remaining three years. The plan also limits the number of options that may be granted to any plan participant in a single calendar year to 1,300,000 shares.
The Company has granted RSUs to executive officers, other executives, and employees under the stock incentive plan. In 2011, 2010, and 2009 the Company granted RSUs covering an aggregate of 545,780, 438,329, and 330,000 shares of common stock, respectively. These RSUs vest 25% on the first anniversary date of the grant, with the remaining rights vesting quarterly over the remaining three years and, once vested, allow the participants to acquire the underlying shares of common stock at par value. The participants are not entitled to sell or transfer any unvested RSUs and are not entitled to vote or receive dividends on any shares of common stock covered by the RSUs prior to the acquisition of such shares. Granted but unvested RSUs are forfeited at termination of employment. Compensation expense related to the RSUs for the years ended December 31, 2011, 2010, and 2009 was approximately $1.1 million, $0.9 million and $0.5 million, respectively.
75
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
The following table summarizes stock option transactions under the Company’s stock incentive plans for the years ended December 31, 2011, 2010 and 2009:
| Shares | Weighted Average Exercise Price |
|||||||
| Outstanding December 31, 2008 |
3,757,720 | $ | 7.40 | |||||
| Granted |
1,276,051 | $ | 1.88 | |||||
| Exercised |
(1,677 | ) | $ | 3.72 | ||||
| Forfeited |
(731,742 | ) | $ | 7.97 | ||||
|
|
|
|||||||
| Outstanding December 31, 2009 |
4,300,352 | $ | 5.67 | |||||
| Granted |
1,722,204 | $ | 3.35 | |||||
| Exercised |
(20,123 | ) | $ | 1.75 | ||||
| Forfeited |
(816,328 | ) | $ | 11.10 | ||||
|
|
|
|||||||
| Outstanding December 31, 2010 |
5,186,105 | $ | 4.06 | |||||
| Granted |
2,052,000 | $ | 2.34 | |||||
| Exercised |
(74,386 | ) | $ | 2.85 | ||||
| Forfeited |
(208,619 | ) | $ | 5.77 | ||||
|
|
|
|||||||
| Outstanding December 31, 2011 |
6,955,100 | $ | 3.51 | |||||
|
|
|
|||||||
| Vested and unvested options expected to vest as of December 31, 2011 |
6,599,697 | $ | 3.57 | |||||
The number of underlying shares and weighted average exercise price of options exercisable at December 31, 2011, 2010 and 2009, were 3,895,520 shares at $4.26, 2,899,922 shares at $4.84, and 2,756,826 shares at $7.40, respectively. The weighted average remaining contractual term of options outstanding and options exercisable at December 31, 2011, was 6.6 years and 5.0 years, respectively. The weighted average remaining contractual term of vested and unvested options expected to vest at December 31, 2011, was 6.5 years. The aggregate intrinsic value of options outstanding and options exercisable at December 31, 2011, was $9.0 million and $3.3 million, respectively. As of December 31, 2011, the total unrecognized compensation cost related to unvested options was $1.8 million, which is expected to be recognized over a weighted-average period of 1.38 years.
The weighted average grant-date fair value of options granted during the years ended December 31, 2011, 2010 and 2009, was $1.21, $1.73 and $0.87 per share, respectively. The total intrinsic value of options exercised during the years ended December 31, 2011, 2010 and 2009, was $124,000, $9,000 and $2,000, respectively. At December 31, 2011, there were 3,020,650 shares available for grant under the Company’s stock incentive plans.
A summary of the outstanding RSUs as of December 31, 2011, and changes during the year then ended is presented below:
| Shares | Weighted Average Grant-Date Fair Value per Share |
|||||||
| Unvested at December 31, 2010 |
572,705 | $ | 3.06 | |||||
| Granted |
545,780 | $ | 2.41 | |||||
| Vested |
(307,594 | ) | $ | 3.12 | ||||
| Cancelled |
(6,667 | ) | $ | 2.60 | ||||
|
|
|
|||||||
| Unvested at December 31, 2011 |
804,224 | $ | 2.59 | |||||
|
|
|
|||||||
The aggregate grant-date fair value of RSUs granted during the years ended December 31, 2011, 2010 and 2009, was $1.3 million, $1.5 million and $0.6 million, respectively. As of December 31, 2011, the total unrecognized compensation cost related to unvested RSUs was $0.8 million, which is expected to be recognized
76
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
over a weighted average period of 1.37 years. The aggregate grant-date fair value of shares subject to RSUs vested during the years ended December 31, 2011, 2010 and 2009, was $1.0 million, $0.5 million and $0.3 million, respectively. As of December 31, 2011, there were 236,645 shares of common stock underlying RSUs that were fully vested but the issuance of such shares has been deferred.
10. Income Taxes
The impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. There were no unrecognized tax benefits recorded by the Company as of the date of adoption in 2007. There are no unrecognized tax benefits included in the balance sheets that would, if recognized, affect the effective tax rate.
At December 31, 2011, the Company had net deferred tax assets of $6.2 million. Due to uncertainties surrounding the Company’s ability to generate future taxable income to realize these assets, a full valuation allowance has been established to offset the net deferred tax asset. Additionally, the future utilization of the Company’s net operating loss and research and development credit carryforwards to offset future taxable income may be subject to an annual limitation, pursuant to Internal Revenue Code Sections 382 and 383, as a result of ownership changes that may have occurred previously or that could occur in the future. The Company has not completed its analysis to determine if an ownership change has occurred and what, if any, impact the ownership change would have on the Company’s ability to utilize its net operating losses. The Company has decided to wait until the ownership change analysis is complete and then update its Section 382 analysis regarding the limitation of the net operating loss and research and development credit carryforwards. Until this analysis has been completed the Company has removed the deferred tax assets for net operating losses of $108.7 million and tax credits of $29.0 million generated through 2011 from its deferred tax asset schedule and has recorded a corresponding decrease to its valuation allowance. When this analysis is finalized, the Company plans to update its unrecognized tax benefits. Due to the existence of the valuation allowance, future changes in the Company’s unrecognized tax benefits will not impact the Company’s effective tax rate.
The Company’s practice is to recognize interest and/or penalties related to income tax matters as income tax expense. The Company had no accrual for interest or penalties on its balance sheets at December 31, 2011 and 2010, and has not recognized interest and/or penalties in its statement of operations for any of the years ended December 31, 2011, 2010 or 2009.
The Company is subject to taxation in the United States and California. The Company’s tax years for 1992 and forward are subject to examination by the United States and California tax authorities due to the carryforward of unutilized net operating losses and R&D credits.
77
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
Significant components of the Company’s deferred tax assets as of December 31, 2011 and 2010 are listed below. A valuation allowance of $6.2 million and $6.6 million at December 31, 2011 and 2010, respectively, has been recognized to offset the net deferred tax assets as realization of such assets is uncertain. Amounts for the years ended December 31 were as follows (in thousands):
| Deferred Tax Assets |
2011 | 2010 | ||||||
| Capital loss carryover |
$ | 45 | $ | 45 | ||||
| Depreciation and amortization |
2,604 | 3,235 | ||||||
| Other |
3,182 | 2,916 | ||||||
| Accruals and reserves |
366 | 432 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
6,197 | 6,628 | ||||||
| Less valuation allowance |
(6,197 | ) | (6,628 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets (1) |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The reconciliation between the provision for income taxes and income taxes computed using the U.S. federal statutory corporate tax rate were as follows for the years ended December 31(in thousands):
| 2011 | 2010 | 2009 | ||||||||||
| Computed “expected” tax benefit |
$ | (2,606 | ) | $ | (10,331 | ) | $ | (9,709 | ) | |||
| State income taxes, net of federal benefit |
(545 | ) | (2,117 | ) | (1,687 | ) | ||||||
| Tax effect of: |
||||||||||||
| Change in valuation allowance (1) |
2,185 | 12,987 | 9,887 | |||||||||
| Expiration of prior year credits and net operating losses |
1,880 | 1,838 | 3,095 | |||||||||
| Research and development and other tax credits carryovers |
(1,468 | ) | (2,519 | ) | (1,791 | ) | ||||||
| Stock compensation |
297 | 294 | 188 | |||||||||
| Other |
257 | (152 | ) | 17 | ||||||||
|
|
|
|
|
|
|
|||||||
| Provision for income taxes |
$ | — | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| (1) | The removal of the valuation allowance related to the net operating loss carryforwards and tax credits are not included in the increase in the valuation allowance. See above explanation. |
As of December 31, 2011 and 2010, the Company had available federal net operating loss carryforwards of approximately $277.6 million and $279.4 million, respectively, which expire from 2012 through 2031. In addition, the Company had federal research and development credit and orphan drug credit carryforwards of $23.7 million and $21.8 million as of December 31, 2011 and 2010, respectively, to reduce future federal income taxes, if any. These carryforwards expire from 2012 through 2031 and are subject to review and possible adjustment by the Internal Revenue Service. The Company also has available California state net operating loss carryforwards of approximately $251.4 million and $230.1 million as of December 31, 2011 and 2010, respectively, which expire from 2012 to 2031. In addition, the Company had California research and development credits and manufacturers’ investment credits of approximately $8.1 million and $7.8 million as of December 31, 2011 and 2010, respectively, to reduce future California income tax, if any. The manufacturers’ investment credits expire from 2012 through 2014. The California research and development credits do not expire.
The Company generated windfall tax benefits from the settlement of certain stock awards. The tax benefit will be recorded as a credit to additional paid-in capital in the year the deduction reduces income taxes payable. The net operating loss carryforwards related to these windfall tax benefits of approximately $1.0 million are included in the net operating loss carryforwards disclosed above.
78
Table of Contents
VICAL INCORPORATED
NOTES TO FINANCIAL STATEMENTS—(Continued)
11. Employee Benefit Plan
The Company has a defined contribution savings plan under section 401(k) of the Internal Revenue Code. The plan covers substantially all employees. The Company matches employee contributions made to the plan according to a specified formula. The Company’s matching contributions totaled approximately $0.2 million for each the years ended 2011, 2010 and 2009.
12. Summary of Unaudited Quarterly Financial Information
The following is a summary of the Company’s unaudited quarterly results of operations for the years ended December 31 (in thousands, except per share amounts):
| 2011: |
March 31, | June 30, | Sept. 30, | Dec. 31, | ||||||||||||
| Total revenues |
$ | 649 | $ | 818 | $ | 26,619 | $ | 1,932 | ||||||||
| Total operating expenses |
9,366 | 9,269 | 10,227 | 8,978 | ||||||||||||
| Net income (loss) |
(8,694 | ) | (8,406 | ) | 16,431 | (6,614 | ) | |||||||||
| Basic net income (loss) per share (1) |
(0.12 | ) | (0.12 | ) | 0.23 | (0.09 | ) | |||||||||
| Diluted net income (loss) per share (1) |
(0.12 | ) | (0.12 | ) | 0.22 | (0.09 | ) | |||||||||
| 2010: |
March 31, | June 30, | Sept. 30, | Dec. 31, | ||||||||||||
| Total revenues |
$ | 1,463 | $ | 2,075 | $ | 2,257 | $ | 2,916 | ||||||||
| Total operating expenses |
10,092 | 10,580 | 9,067 | 10,187 | ||||||||||||
| Net loss |
(8,478 | ) | (8,380 | ) | (6,767 | ) | (6,760 | ) | ||||||||
| Basic and diluted net loss per share (1) |
(0.15 | ) | (0.15 | ) | (0.12 | ) | (0.09 | ) | ||||||||
| (1) | Net income (loss) per share is computed independently for each quarter and the full year based upon respective shares outstanding. Therefore, the sum of the quarterly loss per share amounts may not equal the annual amounts reported. |
13. Subsequent Event
In January 2012, the Company sold 13,333,334 shares of its common stock in a public offering at a price to the public of $3.75 per share. Net proceeds from the offering, after deducting underwriting discounts and commissions and other offering expenses payable by the Company, totaled $46.6 million. In February 2012, the Company sold an additional 576,358 shares pursuant to a partial exercise of the underwriters’ overallotment option at a price to the public of $3.75 per share. Net proceeds from the overallotment option exercise, after deducting underwriting discounts and commissions and other offering expenses payable by the Company, totaled approximately $2.0 million. All of the shares of common stock were offered pursuant to two effective shelf registration statements.
79
Table of Contents
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined in Rule 13a-15(e) promulgated under the Exchange Act, as of the end of the period covered by this Annual Report on Form 10-K. The evaluation of our disclosure controls and procedures included a review of the disclosure controls’ and procedures’ objectives, design, implementation and the effect of the controls and procedures on the information generated for use in this report. In the course of our evaluation, we sought to identify data errors, control problems or acts of fraud and to confirm the appropriate corrective actions, including process improvements, were being undertaken. Based on this evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this Annual Report on Form 10-K.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, as of December 31, 2011, the end of the period covered by this Annual Report on Form 10-K. Based on our evaluation under the framework in Internal Control – Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2011.
Ernst & Young LLP, the independent registered public accounting firm that audited the financial statements included in this Annual Report on Form 10-K, has issued an attestation report on the effectiveness of our internal control over financial reporting as of December 31, 2011. That report, which expresses an unqualified opinion on the effectiveness of our internal control over financial reporting as of December 31, 2011, is included herein.
Changes in Internal Controls
There has been no change in our internal control over financial reporting during the three months ended December 31, 2011, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
80
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Vical Incorporated
We have audited Vical Incorporated’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Vical Incorporated’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Vical Incorporated maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Vical Incorporated as of December 31, 2011 and 2010, and the related statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2011 of Vical Incorporated and our report dated March 15, 2012 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 15, 2012
81
Table of Contents
None.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item and not included in Part I, Item 1 of this Annual Report on Form 10-K is incorporated by reference from our Proxy Statement for our 2012 Annual Meeting of Stockholders, or our Proxy Statement.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated by reference from our Proxy Statement.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference from our Proxy Statement.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is incorporated by reference from our Proxy Statement.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is incorporated by reference from our Proxy Statement.
82
Table of Contents
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)(1) Financial Statements
The following independent auditors’ reports and financial statements are filed as part of this Annual Report:
Report of Independent Registered Public Accounting Firm
Balance Sheets as of December 31, 2011 and 2010
Statements of Operations for each of the three years in the period ended December 31, 2011
Statements of Stockholders’ Equity for each of the three years in the period ended December 31, 2011
Statements of Cash Flows for each of the three years in the period ended December 31, 2011
Notes to Financial Statements
(2) Financial Statement Schedules
The schedules required to be filed by this item have been omitted because of the absence of conditions under which they are required, or because the required information is included in the financial statements or the notes thereto.
(3) Exhibits
See the list in paragraph (b) below. Each management contract or compensatory plan or arrangement required to be identified by this item is so designated in such list.
(b) Exhibits
| Exhibit |
Description of Document | |
| 3.1(i)(7) | Restated Certificate of Incorporation. | |
| 3.2(ii)(2) | Amended and Restated Bylaws of the Company. | |
| 3.3(i)(2) | Certificate of Amendment to Restated Certificate of Incorporation. | |
| 3.4(i)(16) | Certificate of Amendment to Restated Certificate of Incorporation. | |
| 4.1(7) | Specimen Common Stock Certificate. | |
| 10.1(2)a | Amended and Restated Stock Incentive Plan of Vical Incorporated. | |
| 10.3(11)a | Form of Indemnity Agreement between the Company and its directors and officers. | |
| 10.4(24)a | Vical Incorporated Non-Employee Director Compensation Policy. | |
| 10.5(25)a | Consulting Agreement dated August 1, 2010, by and between the Company and Gary A. Lyons. | |
| 10.6(26)a | First amendment dated December 17, 2010 to Consulting Agreement dated August 1, 2010, by and between the Company and Gary A. Lyons. | |
| 10.7(27)a | Second amendment dated June 16, 2011 to Consulting Agreement dated August 1, 2010, by and between the Company and Gary A. Lyons. | |
| 10.8a | Third amendment effective December 31, 2011 to Consulting Agreement dated August 1, 2010, by and between the Company and Gary A. Lyons. | |
83
Table of Contents
| Exhibit |
Description of Document | |
| 10.9(4)b | Research Collaboration and License Agreement dated May 31, 1991, between the Company and Merck & Co., Inc. | |
| 10.12(1)b | License Agreement dated January 1, 1991, between the Company and Wisconsin Alumni Research Foundation. | |
| 10.14(1)b | License Agreement dated October 23, 1992, between the Company and the Regents of University of Michigan. | |
| 10.16(5) | Research, Option and License Agreement dated September 29, 1994, between the Company and Pasteur Mérieux Sérums & Vaccins (subsequently Sanofi Pasteur). | |
| 10.17(6) | Amendment dated April 27, 1994, to Research Collaboration and License Agreement dated May 31, 1991, between the Company and Merck & Co., Inc. | |
| 10.19(13)b | Amendment dated November 3, 1997, to Research Collaboration and License Agreement dated May 31, 1991, between the Company and Merck & Co., Inc. | |
| 10.23(9)a | Restated employment letter dated January 9, 2009, between the Company and Vijay B. Samant. | |
| 10.26(10)b | Amendment No. 4 dated December 7, 2001, to Research, Option and License Agreement between the Company and Sanofi Pasteur (formerly Pasteur Mérieux Sérums & Vaccins). | |
| 10.27(10) | Lease dated January 30, 2002, between the Company and Kilroy Realty, L.P. a Delaware Limited Partnership. | |
| 10.29(17)a | Restated employment letter dated January 9, 2009, between the Company and Alain Rolland. | |
| 10.30(11)b | Amendment No. 5 dated September 23, 2002, to Research, Option and License Agreement between the Company and Sanofi Pasteur (formerly Pasteur Mérieux Sérums & Vaccins). | |
| 10.32(12)b | Fourth Amendment dated August 20, 2003, to Research Collaboration and License Agreement dated May 31, 1991, between the Company and Merck & Co., Inc. | |
| 10.36(13)b | Amendment dated May 20, 2004, to License Agreement dated January 1, 1991, between the Company and the Wisconsin Alumni Research Foundation. | |
| 10.41(14)b | License Agreement dated May 24, 2005, between the Company and AnGes MG, Inc. | |
| 10.45(21)a | Restated employment letter dated January 9, 2009, by and between Vical Incorporated and Jill M. Broadfoot. | |
| 10.46(15)b | Fifth Amendment dated September 8, 2005, to Research Collaboration and License Agreement dated May 31, 1991, by and between Vical Incorporated and Merck & Co., Inc. | |
| 10.47(18)b | Amendment dated February 20, 2006, to License Agreement dated May 24, 2005, between the Company and AnGes MG, Inc. | |
| 10.50(19)b | Research and Development Agreement dated May 25, 2006, between the Company and AnGes MG, Inc. | |
| 10.54(8)b | First Amendment to Research and Development Agreement and Stock Purchase Agreement dated September 26, 2007, between the Company and AnGes MG, Inc. | |
| 10.56(20)b | License Agreement dated December 7, 2001, between the Company and CytRx Corporation. | |
| 10.57(22)b | License Agreement dated February 14, 2006, between the Company and the Regents of the University of Michigan. | |
84
Table of Contents
| Exhibit |
Description of Document | |
| 10.58(3)a | Form of Delayed Issuance Stock Purchase Election Agreement, as amended, under the Amended and Restated Stock Incentive Plan (with deferral election). | |
| 10.59(3)a | Form of Delayed Issuance Stock Purchase Election Agreement, as amended, under the Amended and Restated Stock Incentive Plan. | |
| 10.62(23) | Common Stock Purchase Agreement dated January 8, 2010 between Vical and Azimuth Opportunity Ltd. | |
| 10.63(28)b | U.S. License Agreement dated July 12, 2011, between the Company and Astellas Pharma Inc. | |
| 10.64(29)b | Ex-U.S. License Agreement dated July 12, 2011, between the Company and Astellas Pharma Inc. | |
| 10.65(30)b | Supply and Services Agreement dated July 12, 2011, between the Company and Astellas Pharma Inc. | |
| 10.66(31)b | Exclusive License Agreement dated February 3, 2003, between the Company and City of Hope. | |
| 10.67(32) | Underwriting Agreement, dated January 6, 2012. | |
| 10.68(33) | Letter agreement dated July 5, 2011, related to the License Agreement dated December 7, 2001, between the Company and CytRx Corporation. | |
| 10.69(34) | Letter agreement dated July 7, 2011, related to the Exclusive License Agreement dated February 3, 2003, between the Company and City of Hope. | |
| 10.70(35) | Letter agreement dated July 12, 2011, related to the U.S. License Agreement dated July 12, 2011, between the Company and Astellas Pharma Inc. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 31.1 | Certification of Vijay B. Samant, Chief Executive Officer, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of Jill M. Broadfoot, Chief Financial Officer, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification of Vijay B. Samant, Chief Executive Officer, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2 | Certification of Jill M. Broadfoot, Chief Financial Officer, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101.INS | XBRL Instance Document. | |
| 101.SCH | XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document. | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document. | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document. | |
| (1) | Incorporated by reference to the Company’s Registration Statement on Form S-1 (No. 33-56830) filed on January 7, 1993. |
| (2) | Incorporated by reference to the exhibit of the same number filed with the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2010. |
| (3) | Incorporated by reference to the exhibit of the same number to the Company’s Annual Report on Form 10-K for the year ended December 31, 2009. |
85
Table of Contents
| (4) | Incorporated by reference to Exhibit 10.9 of the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 1994 (No. 0-21088). |
| (5) | Incorporated by reference to Exhibit A of the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 1994. |
| (6) | Incorporated by reference to Exhibit A of the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 1994 (No. 0-21088). |
| (7) | Incorporated by reference to the exhibit of the same number filed with the Company’s Registration Statement on Form S-3 (No. 33-95812) filed on August 15, 1995. |
| (8) | Incorporated by reference to the exhibit of the same number to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2007. |
| (9) | Incorporated by reference to Exhibit 99.1 to the Company’s Current Report on Form 8-K filed on January 15, 2009. |
| (10) | Incorporated by reference to the exhibit of the same number to the Company’s Annual Report on Form 10-K for the year ended December 31, 2001. |
| (11) | Incorporated by reference to the exhibit of the same number to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2002. |
| (12) | Incorporated by reference to the exhibit of the same number to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2003. |
| (13) | Incorporated by reference to the exhibit of the same number to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2004. |
| (14) | Incorporated by reference to the exhibit of the same number to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2005. |
| (15) | Incorporated by reference to Exhibits 10.3 – 10.4 to the Company’s Current Report on Form 8-K filed on October 12, 2005. |
| (16) | Incorporated by reference to Exhibit 4.2 filed with the Company’s Registration Statement on Form S-8 (No. 333-135398) filed on June 23, 2006. |
| (17) | Incorporated by reference to Exhibit 99.2 to the Company’s Current Report on Form 8-K filed on January 15, 2009. |
| (18) | Incorporated by reference to the exhibit of the same number to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2006. |
| (19) | Incorporated by reference to the exhibit of the same number to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2006. |
| (20) | Incorporated by reference to Exhibit 99 to CytRx Corporation’s Current Report on Form 8-K filed on December 21, 2001. |
| (21) | Incorporated by reference to Exhibit 99.3 to the Company’s Current Report on Form 8-K filed on January 15, 2009. |
| (22) | Incorporated by reference to the exhibit of the same number to the Company’s Amendment No.1 on Form 10-K/A to the Company’s Annual Report on Form 10-K for the year ended December 31, 2007. |
| (23) | Incorporated by reference to Exhibit 10.1 filed with the Company’s Current Report on Form 8-K filed on January 8, 2010. |
| (24) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010. |
| (25) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2010. |
| (26) | Incorporated by reference to the exhibit of the same number to the Company’s Annual Report on Form 10-K for the year ended December 31, 2010. |
| (27) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2011. |
| (28) | Incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011. |
86
Table of Contents
| (29) | Incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011. |
| (30) | Incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011. |
| (31) | Incorporated by reference to Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011. |
| (32) | Incorporated by reference to Exhibit 1.1 to the Company’s Current Report on Form 8-K filed on January 9, 2012. |
| (33) | Incorporated by reference to Exhibit 99.2 to the Company’s Current Report on Form 8-K filed on January 5, 2012. |
| (34) | Incorporated by reference to Exhibit 99.3 to the Company’s Current Report on Form 8-K filed on January 5, 2012. |
| (35) | Incorporated by reference to Exhibit 99.4 to the Company’s Current Report on Form 8-K filed on January 5, 2012. |
| a | Indicates management contract or compensatory plan or arrangement. |
| b | Confidential treatment of certain portions of this agreement has been requested and/or received and such portions have been omitted and filed separately with the SEC pursuant to Rule 24b-2 under the Securities Exchange Act of 1934. |
87
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: March 15, 2012
| VICAL INCORPORATED | ||
| By: | /S/ VIJAY B. SAMANT | |
| Vijay B. Samant | ||
| President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| /S/ VIJAY B. SAMANT Vijay B. Samant |
President, Chief Executive Officer and Director (Principal Executive Officer) |
March 15, 2012 | ||
| /S/ JILL M. BROADFOOT Jill M. Broadfoot |
Senior Vice President, Chief Financial Officer, and Secretary (Principal Financial and Accounting Officer) |
March 15, 2012 | ||
| /S/ R. GORDON DOUGLAS, M.D. R. Gordon Douglas, M.D. |
Chairman of the Board of Directors |
March 15, 2012 | ||
| /S/ ROBERT H. CAMPBELL Robert H. Campbell |
Director |
March 15, 2012 | ||
| /S/ GARY A. LYONS Gary A. Lyons |
Director |
March 15, 2012 | ||
| /S/ ROBERT C. MERTON, PH.D. Robert C. Merton, Ph.D. |
Director |
March 15, 2012 | ||
88