UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
FORM 10-K
| |
|
|
|
(Mark One)
|
|
|
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal
year ended December 31, 2010
|
|
OR
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period from
to
|
Commission file number:
001-34637
ANTHERA PHARMACEUTICALS,
INC.
(Exact Name of Registrant as
Specified in Its Charter)
| |
|
|
|
Delaware
|
|
20-1852016
|
(State or Other Jurisdiction
of
Incorporation or Organization)
|
|
(I.R.S. Employer
Identification No.)
|
|
|
|
|
|
25801 Industrial Boulevard, Suite B
Hayward, California
(Address of Principal
Executive Offices)
|
|
94545
(Zip Code)
|
(510) 856-5600
(Registrant’s telephone
number, including area code)
Securities registered pursuant to Section 12(b) of the
Act:
| |
|
|
|
Title of Each Class
|
|
Name of Each Exchange on Which Registered
|
|
|
|
Common Stock, par value $0.001 per share
|
|
The NASDAQ Stock Market LLC
|
Securities registered pursuant to Section 12(g) of the
Act:
None
Indicate by a check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by a check mark if the registrant is not required to
file reports pursuant to Section 13 or 15(d) of the
Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
during the preceding 12 months (or for such shorter period
that the registrant was required to submit and post such
files). Yes o No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
FORM 10-K
or any amendment to this
FORM 10-K. þ
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in
Rule 12b-2
of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large accelerated
filer o
|
|
Accelerated
filer o
|
|
Non-accelerated
filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting
company þ
|
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). Yes o No þ
The aggregate market value of the registrant’s common stock
held by non-affiliates as of June 30, 2010 was
approximately $58,846,545, based upon the closing sales price of
the registrant’s common stock as reported on the NASDAQ
Global Market. Shares of common stock held by each executive
officer and director and by each person who owns 10 percent
or more of the outstanding common stock have been excluded in
that such persons may be deemed to be affiliates. This
determination of affiliate status is not necessarily a
conclusive determination for any other purpose.
As of February 28, 2011, the number of outstanding shares
of the registrant’s common stock, par value $0.001 per
share, was 32,906,412.
ANTHERA
PHARMACEUTICALS, INC.
FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2010
INDEX
2
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on
Form 10-K,
including the section entitled “Management’s
Discussion and Analysis of Financial Condition and Results of
Operations,” contains forward-looking statements regarding
future events and our future results that are subject to the
safe harbors created under the Securities Act of 1933, as
amended (the “Securities Act”), and the Securities
Exchange Act of 1934, as amended (the “Exchange Act”).
Forward-looking statements relate to future events or our future
financial performance. We generally identify forward-looking
statements by terminology such as “may,”
“will,” “would,” “should,”
“expects,” “plans,” “anticipates,”
“could,” “intends,” “target,”
“projects,” “contemplates,”
“believes,” “estimates,”
“predicts,” “assume,” “intend,”
“potential,” “continue” or other similar
words or the negative of these terms. These statements are only
predictions. We have based these forward-looking statements
largely on our current expectations and projections about future
events and financial trends that we believe may affect our
business, financial condition and results of operations. The
outcome of the events described in these forward-looking
statements is subject to risks, uncertainties and other factors
described in “Risk Factors” and elsewhere in this
report. Accordingly, you should not place undue reliance upon
these forward-looking statements. We cannot assure you that the
events and circumstances reflected in the forward-looking
statements will be achieved or occur, the timing of events and
circumstances and actual results could differ materially from
those projected in the forward looking statements.
Forward-looking statements contained in this report include, but
are not limited to, statements about:
|
|
|
| |
•
|
the progress of, timing of and amount of expenses associated
with our research, development and commercialization
activities;
|
| |
| |
•
|
the timing, conduct and success of our clinical studies for
our product candidates;
|
| |
| |
•
|
our ability to obtain U.S. and foreign regulatory
approval for our product candidates and the ability of our
product candidates to meet existing or future regulatory
standards;
|
| |
| |
•
|
our expectations regarding federal, state and foreign
regulatory requirements;
|
| |
| |
•
|
the therapeutic benefits and effectiveness of our product
candidates;
|
| |
| |
•
|
the accuracy of our estimates of the size and characteristics
of the markets that may be addressed by our product
candidates;
|
| |
| |
•
|
our ability to manufacture sufficient amounts of our product
candidates for clinical studies and products for
commercialization activities;
|
| |
| |
•
|
our intention to seek to establish strategic collaborations
or partnerships for the development or sale of our product
candidates;
|
| |
| |
•
|
our expectations as to future financial performance, expense
levels and liquidity sources;
|
| |
| |
•
|
the timing of commercializing our product candidates;
|
| |
| |
•
|
our ability to compete with other companies that are or may
be developing or selling products that are competitive with our
product candidates;
|
| |
| |
•
|
anticipated trends and challenges in our potential
markets;
|
| |
| |
•
|
our ability to attract and retain key personnel; and
|
| |
| |
•
|
other factors discussed elsewhere in this report.
|
The forward-looking statements made in this report relate
only to events as of the date on which the statements are made.
We have included important factors in the cautionary statements
included in this report, particularly in the section entitled
“Risk Factors” that we believe could cause actual
results or events to differ materially from the forward-looking
statements that we make. Our forward-looking statements do not
reflect the potential impact of any future acquisitions,
mergers, dispositions, joint ventures or investments we may
make. Except as required by law, we do not assume any intent to
update any forward-looking statements after the date on which
the statement is made, whether as a result of new information,
future events or circumstances or otherwise.
3
PART I
Unless the context otherwise requires, we use the terms
“Anthera Pharmaceuticals,” “Anthera,”
“we,” “us,” “the Company” and
“our” in this report to refer to Anthera
Pharmaceuticals, Inc. and its sole subsidiary. We use various
trademarks, service marks and trade names in our business,
including without limitation “Anthera Pharmaceuticals”
and “Anthera.” This report also contains trademarks,
services marks and trade names of other businesses that are the
property of their respective holders.
Overview
Anthera Pharmaceuticals, Inc. is a biopharmaceutical company
focused on developing and commercializing products to treat
serious diseases associated with inflammation, including
cardiovascular and autoimmune diseases. We currently have one
Phase 3 clinical program, varespladib, and two Phase 2 clinical
programs,
A-623 and
A-001. Two
of our product candidates, varespladib and
A-001, are
designed to inhibit a novel enzyme target known as secretory
phospholipase
A2,
or
sPLA2.
Elevated levels of
sPLA2
have been implicated in a variety of acute inflammatory
conditions, including acute coronary syndrome and acute chest
syndrome associated with sickle cell disease, as well as in
chronic diseases, including stable coronary artery disease, or
CAD. In addition,
A-623, a
phase 2 product candidate, targets elevated levels of
B-lymphocyte stimulator, or BLyS, also known as B-cell
Activating Factor, or BAFF, which has been associated with a
variety of B-cell mediated autoimmune diseases, including
systemic lupus erythematosus, or lupus, lupus nephritis, or LN,
rheumatoid arthritis, multiple sclerosis, Sjögren’s
Syndrome, Graves’ Disease and others.
We were incorporated in Delaware in 2004. Our corporate
headquarters are located at 25801 Industrial Boulevard,
Suite B, Hayward, California 94545 and our telephone number
is
(510) 856-5600.
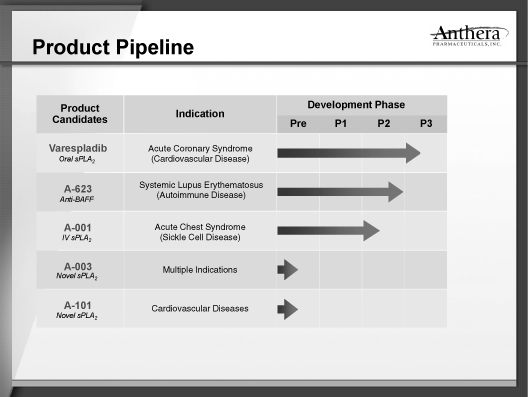
Summary
of Product Development Programs
4
We have worldwide rights to develop and commercialize our
products in all indications and markets, with the exception of
Japan where Shionogi & Co., Ltd. retains commercial
rights to our
sPLA2
product candidates. Our current development plans are focused on
acute treatment and orphan indications that may provide an
accelerated and cost-efficient path to regulatory approval and
commercialization. We believe that certain of these markets can
be commercialized through a limited specialty sales force. In
addition, we believe that our product candidates can also
address market opportunities in chronic indications and we may
seek development and commercialization partners to address
chronic, non-specialty and international markets.
Inflammation
and Diseases
The inflammatory process is a powerful and essential early line
of defense for protection against injury and to repair body
tissue. As a result, it is tightly regulated by the body to
ensure appropriate activation and prompt resolution. However,
under certain circumstances, the normal process can malfunction,
leading to acute or chronic inflammation or inappropriate
activation directed against the body’s own tissues. All of
these circumstances can cause significant damage to cells and
tissues, leading to a range of inflammatory disorders, such as
cardiovascular and autoimmune diseases.
Our
sPLA2
Inhibition Portfolio
Building upon our knowledge of the regulation of inflammatory
pathways and the growing body of evidence that links
inflammation to multiple disease states, we believe that we have
developed a leadership position in the field of
sPLA2
inhibition. Our
sPLA2
inhibitors have been studied in a number of inflammatory
disorders in multiple therapeutic areas. The effect of our
sPLA2
inhibitors on
sPLA2
concentration and activity have been implicated in acute
coronary syndrome and acute chest syndrome associated with
sickle cell disease. We currently have the two most advanced
sPLA2
inhibitors in clinical development.
Our lead product candidate, varespladib (an oral prodrug of
A-001), is a
broad-spectrum inhibitor of
sPLA2
enzymes and is being evaluated in a Phase 3 clinical study for
short-term (16-week) treatment of patients who have experienced
an acute coronary syndrome. The American Heart Association
defines acute coronary syndrome as any group of clinical
symptoms related to acute myocardial ischemia, including
unstable angina, or UA. Varespladib, when combined with Lipitor
(atorvastatin), is one of only a few therapeutics in development
with the potential to offer a unique and synergistic treatment
approach targeting inflammation, elevated lipid levels and
atherosclerosis as part of physician-directed standard of care.
Through its novel mechanism of action, varespladib may have
applications in a broad range of acute and chronic
cardiovascular diseases. Based on the successful results of our
completed Phase 2b clinical study, we initiated a Phase 3
clinical study, VISTA-16, in patients with acute coronary
syndrome in June 2010.
Our second product candidate, varespladib sodium,
A-001, is an
intravenously administered inhibitor of
sPLA2,which
we are planning to evaluate in a Phase 2 clinical study for the
prevention of acute chest syndrome associated with sickle cell
disease. Acute chest syndrome is a form of inflammation-induced
lung failure and is the most common cause of death in patients
with sickle cell disease. Given that there are currently no
approved drugs for the prevention of acute chest syndrome
associated with sickle cell disease, we have received orphan
drug designation and fast track status from the FDA for
A-001.
We also have a broad series of additional
sPLA2
inhibitors designed with distinct chemical scaffolds in
preclinical development. These product candidates are intended
to provide new
sPLA2
inhibitors for our existing target indications as well as new
candidates for other therapeutic areas. Our lead candidate
within the series,
A-003, is
chemically distinct from
A-001 and
varespladib and has shown increased potency against the target
enzymes and higher drug exposure after dosing in preclinical
studies. As a result,
A-003 may
confer beneficial pharmacodynamic effects in patients and can be
formulated for oral or intravenously administered use. We plan
to file an investigational new drug application, or IND, for
A-003 in the
future and we may continue to assess additional new compounds.
We have explored the use of our varespladib and
A-001
sPLA2
inhibitors as both topical and inhalation therapies in animal
models for the treatment of atopic dermatitis and asthma,
respectively. Results from a standard mouse model of edema
demonstrated that topically administered varespladib was
equivalent to the marketed
5
immunosuppressant Elidel in resolving inflammation. In a sheep
model of allergen-induced asthma, inhaled
A-001
demonstrated an improvement in lung function similar to inhaled
steroids.
sPLA2
Biology
sPLA2
is a family of enzymes directly involved in the acute and
chronic steps of an inflammatory response.
sPLA2
activity is highly elevated during the early stages of
inflammation, and its acute effects serve to substantially
amplify the inflammatory process. The
sPLA2
enzyme catalyzes the first step in the arachidonic acid pathway
of inflammation, one of the main metabolic processes for the
production of inflammatory mediators, which, when amplified, are
responsible for causing damage to cells and tissue.
Specifically,
sPLA2
breaks down phospholipids that result in the formation of fatty
acids such as arachidonic acid. Arachidonic acid is subsequently
metabolized to form several pro-inflammatory and thrombogenic
molecules.
In cardiovascular diseases such as acute coronary syndrome,
elevated levels of
sPLA2
mass and
sPLA2
activity have acute and chronic implications on disease
progression and patient outcomes. In published studies and our
own clinical studies, significant elevations in
sPLA2
activity and mass have been seen from 24 hours to two weeks
following an acute coronary syndrome and can persist for up to
an additional 12 weeks thereafter. Shortly after a heart
attack,
sPLA2
is dramatically elevated, amplifying inflammation that is
associated with more frequent and secondary cardiovascular
events. This resulting elevated level of inflammation is
problematic for acute coronary syndrome patients who are already
at higher risk of complications during the weeks following their
initial event. For example, increased inflammation can
destabilize vulnerable vascular lesions or atherosclerotic
plaque, destroy damaged but viable cardiac cells and adversely
modify lipids, any of which may lead to the recurrence of a
major adverse cardiovascular event, or MACE.
Historical and recent clinical results have demonstrated
circulating levels of
sPLA2
are significantly correlated with a well-established
inflammatory marker, C-reactive protein, or CRP. These and other
clinical studies have also demonstrated that
sPLA2
independently predicts coronary events in patients that have
recently experienced an acute coronary syndrome and patients
with stable CAD independent of other standard risk factors. In a
stable cardiovascular patient,
sPLA2
not only sustains chronic vascular inflammation as discussed
earlier, but it also adversely remodels lipoproteins such as
low-density lipoprotein cholesterol, or LDL-C.
sPLA2
interacts with LDL-C in a series of reactions that result in
smaller, more pro-atherogenic and pro-inflammatory LDL-C
particles. Moreover, these modified lipoproteins have a reduced
affinity for LDL-C receptors, which are responsible for removal
of cholesterol from the body. As a result, LDL-C remains in
circulation longer and has a greater tendency to deposit in the
artery wall. This increased LDL-C deposition and sustained
chronic vascular inflammation may contribute to the development
of atherosclerosis.
The family of
sPLA2
enzymes includes at least three forms that play a role in
inflammation and the development of cardiovascular disease or
lung injury. While
sPLA2enzymes
are a member of the phospholipase family that includes a
lipoprotein associated phospholipase
A2,
or
Lp-PLA2,
there are important distinctions. Although both are present in
blood,
Lp-PLA2
is mostly bound to LDL-C and high-density lipoprotein, or HDL,
while
sPLA2
enzymes are not. Based on our clinical studies, we believe that
our
sPLA2
inhibitor, varespladib, can be distinguished from other
PLA2
enzyme inhibitors such as those targeted at inhibiting
Lp-PLA2
because varespladib treatment:
|
|
|
| |
•
|
is synergistic with HMG-CoA reductase inhibitors, or statins,
including Lipitor (atorvastatin), in reducing LDL-C, total
cholesterol and non-HDL cholesterol in patients with CAD;
|
| |
| |
•
|
lowers circulating small, dense and pro-atherogenic, or
plaque-building LDL-C particles, while
Lp-PLA2
inhibition has not demonstrated similar effects;
|
| |
| |
•
|
has been shown to lower CRP, a well-established marker of
inflammation in a statistically significant manner; and
|
| |
| |
•
|
reduces plaque volume and aneurysms in standard rodent models of
atherosclerosis and has demonstrated synergistic reductions of
plaque volume in standard rodent models of atherosclerosis when
used in combination with statins.
|
6
In diseases such as acute chest syndrome, a very serious form of
lung injury associated with sickle cell disease,
sPLA2
acts acutely on a number of substrates that amplify the
inflammatory disease process. Sickle cell disease is a genetic
disorder which leads to the structural alteration, or
“sickling,” of otherwise healthy red blood cells.
Patients with sickle cell disease experience periods of intense
pain known as vaso-occlusive crisis, or VOC, as structurally
altered red blood cells bind together and occlude small blood
vessels that supply blood and nutrients to vital tissue and
bone.
sPLA2
levels are dramatically elevated in sickle cell patients during
an episode of VOC as well as within 24 to 48 hours of the
onset of acute chest syndrome. During VOC, microscopic fat
emboli, or droplets of fat from the bone marrow, are prevalent
and can break free and become lodged in the lung. These emboli
are substrates for
sPLA2
enzymes and provide fuel for an already established inflammatory
response, increasing lung injury. In addition,
sPLA2
has been demonstrated to degrade human lung surfactant, a
component necessary in maintaining appropriate lung function,
which further complicates lung injury.
We believe that early intervention with a drug designed to
inhibit
sPLA2
activity may offer a unique opportunity to reduce the
complications associated with certain inflammatory diseases such
as acute coronary syndrome in cardiovascular patients and acute
chest syndrome in patients with sickle cell disease.
Our
BAFF Antagonism Portfolio
BAFF has been associated with a wide range of B-cell mediated
autoimmune diseases including lupus, LN, rheumatoid arthritis,
multiple sclerosis, Sjögren’s Syndrome, Graves’
Disease and others. The role of BAFF in lupus and rheumatoid
arthritis has recently been validated in multiple clinical
studies with other BAFF antagonists. We are advancing the
development of our BAFF inhibitor molecule,
A-623, a
selective peptibody, to exploit its broad potential clinical
utility in autoimmune diseases. A peptibody is a novel fusion
protein that is distinct from an antibody. We have worldwide
rights to
A-623 in all
potential indications. We have initiated PEARL-SC, the Phase 2b
clinical study of
A-623, for
the treatment of Systemic Lupus Erythematosus (lupus). Lupus
patients suffer from a chronic autoimmune disease, which often
leads to severe skin rash, fatigue, joint pain, major organ
complications and cardiovascular disease.
A-623
demonstrates anti-BAFF activity and has shown statistically
significant reductions in B-cells in two Phase 1 clinical
studies in patients with lupus. We believe
A-623 may
offer a number of potential differentiations over other BAFF
antagonists, as well as other novel B-cell directed therapies
including:
|
|
|
| |
•
|
convenient, at-home, patient-administered subcutaneous dosing
with a range of dosing frequencies including monthly and weekly;
|
| |
| |
•
|
the ability to inhibit the activity of both membrane-bound and
soluble BAFF;
|
| |
| |
•
|
selective modulation and reduction of relevant B-cell
sub-types in
lupus patients;
|
| |
| |
•
|
a novel molecular structure, which may confer differentiating
pharmacokinetic and pharmacodynamic characteristics, potentially
providing efficacy and dosing benefits, as well as manufacturing
benefits and lower cost of goods based on a bacterial
fermentation manufacturing process; and
|
| |
| |
•
|
multiple binding domains achieve highest reported affinity for
inhibition of BAFF.
|
7
Product
Development Programs
We have focused our product development programs on
anti-inflammatory therapeutics for cardiovascular diseases,
lupus and other serious diseases for which we believe that
current treatments are either inadequate or non-existent. Our
current product development programs are listed in the table
below.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Worldwide
|
|
|
|
|
|
|
|
Development
|
|
Product
|
|
|
|
|
|
Product Candidate
|
|
Phase
|
|
Rights
|
|
Description
|
|
Next Milestone(s)
|
|
|
|
Lead Development Programs
|
|
|
|
|
|
|
|
|
|
Varespladib with Lipitor (atorvastatin)
|
|
Phase 3
|
|
Anthera(1)
|
|
• Orally administered
sPLA2
inhibitor
• Target indication for the prevention of
secondary MACE following an acute coronary syndrome
(16-week
treatment)
|
|
• Biomarker analysis on independent
markers of cardiovascular risk
• Data Safety Monitoring Board, or DSMB,
review of clinical data
• Interim efficacy review of MACE
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A-623
|
|
Phase 2b
|
|
Anthera
|
|
• Selective subcutaneous administered
“dual action” peptibody antagonist of soluble and
membrane bound BAFF cytokine
• Being developed for the treatment of
B-cell mediated autoimmune diseases
• Target indication for lupus
|
|
• Completion of technology transfer to
contract manufacturer
• Establish comparability plan with U.S.
FDA
• Initiation of Phase 3 manufacturing
campaign
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional Programs
|
|
|
|
|
|
|
|
|
|
A-001-varespladib
sodium
|
|
Phase 2
|
|
Anthera(1)
|
|
• Intravenous
sPLA2
inhibitor with orphan drug and fast track status
• Indicated for prevention of acute chest
syndrome in hospitalized patients with sickle cell disease
|
|
• Publish final impacts data
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A-623
|
|
Phase 2
|
|
Anthera
|
|
• Selective subcutaneous administered
antagonist of soluble and membrane bound BAFF for the treatment
of B-cell mediated disease e.g. Sjogren’s syndrome or small
vessel vasculitis or transplant rejection
|
|
• Submit orphan product protocol to U.S.
FDA
|
|
|
|
|
(1) |
|
Shionogi & Co., Ltd. retains product rights in Japan |
We have historically spent a significant portion of our capital
resources on research and development. Our research and
development expenses were $29.5, $8.4 and $10.9 million for
the years ended December 31, 2010, 2009 and 2008,
respectively.
Varespladib
Varespladib is an orally administered pro-drug of
A-001, which
is a broad-spectrum, once-daily inhibitor of the IIa, V and X
iso-forms of the
sPLA2
enzyme that has demonstrated potent anti-inflammatory,
lipid-lowering and lipid-modulating treatment effects in
multiple clinical studies. We have commenced the Phase 3
VISTA-16 study to
8
evaluate varespladib in combination with atorvastatin therapy,
specifically Lipitor, for the short-term (16-week) treatment of
acute coronary syndrome. We have an agreement with the FDA on a
Special Protocol Assessment, or SPA for the VISTA-16 study. An
SPA provides an opportunity for the clinical study sponsor to
receive feedback from the FDA regarding the potential adequacy
of a clinical study to meet certain regulatory and scientific
requirements if conducted in accordance with the SPA agreement.
An SPA is not a guarantee of an approval of a product candidate
or any permissible claims about the product candidate.
To date, over 1,000 patients and healthy volunteers in at
least 15 previous clinical studies have been exposed to
varespladib. Varespladib was generally well-tolerated in studies
where patients were exposed to a maximum of 48 weeks of
therapy. Varespladib has been studied in combination with
Lipitor (atorvastatin) in a Phase 2b clinical study in acute
coronary syndrome patients and two earlier Phase 2 clinical
studies in stable CAD patients, the majority of whom were on
various statin therapies.
We currently have all worldwide product rights to varespladib,
except in Japan where Shionogi & Co., Ltd. retains
rights. We originally licensed our
sPLA2
inhibitor portfolio, including varespladib and
A-001, from
Eli Lilly & Company, or Eli Lilly, and
Shionogi & Co., Ltd. in July 2006.
Market
Opportunity — Acute Coronary Syndrome
According to the American Heart Association, over
18 million people in the United States have experienced an
acute coronary syndrome and an estimated 1.5 million
Americans will have a new or recurrent heart attack. In
addition, the American Heart Association estimates that
worldwide, cardiovascular disease kills an estimated
17.5 million people each year. According to British Heart
Foundation statistics, CAD, which often leads to acute coronary
syndrome or heart attacks, accounts for 1.9 million deaths
in Europe annually. According to the World Health Organization,
or the WHO, cardiovascular disease is the most common cause of
death in the western world and a major cause of hospital
admissions. In addition, the American Heart Association provides
that for people over the age of 40, 20% of them will die within
one year following an initial heart attack, and over one-third
of them will die within the first five years of an initial heart
attack. These numbers are expected to increase given an aging
population, as well as the rising epidemics of diabetes and
obesity, two conditions known to increase the risk of acute
coronary syndrome.
The American Heart Association defines acute coronary syndrome
as any group of clinical signs and symptoms related to acute
myocardial ischemia. Acute myocardial ischemia can often present
as chest pain due to insufficient blood supply to the heart
muscle that results from CAD. Acute coronary syndrome covers a
spectrum of clinical conditions that include ST-elevated
myocardial infarction, or STEMI, non-ST-elevated myocardial
infarction, or NSTEMI, and UA. Both STEMI and NSTEMI are forms
of a heart attack, where damage to the heart muscle occurs due
to ischemia, which is lack of blood flow to tissues due to a
blockage of a vessel. Typically, UA results in chest pain from
ischemia, but does not cause permanent damage to the heart
muscle.
Furthermore, for any patient who experiences an acute coronary
syndrome, the risk of a secondary MACE is significantly
increased immediately following the initial event. Large
clinical outcome studies such as MIRACL and PROVE-IT have
previously reported, and data from our own FRANCIS Phase 2b
clinical study supports, the 16-week rate of secondary MACE in
acute coronary syndrome patients to be between 6.1% and 14.8%.
Current treatments for CAD other than interventional procedures
include a variety of medications such as aspirin, statins and
anti-platelet and anti-coagulant therapeutics. These medications
are used to offer both acute and chronic benefits to patients.
For patients presenting with acute coronary syndrome,
therapeutics are administered quickly to improve blood flow to
the heart and limit the risk associated with continued ischemia
and thrombosis, which is the formation of a blood clot inside a
vessel, which obstructs blood flow. In addition, interventional
procedures and other medications, such as statins that are
initiated early primarily for lipid benefits, are continued in
an attempt to provide chronic protection against secondary MACE
through improvement in lipid profiles such as lowering LDL-C.
Inflammation
in Cardiovascular Disease
In patients experiencing an acute coronary syndrome, the
relationship between higher levels of inflammation, as measured
by CRP,
sPLA2
and interleukin-6, or IL-6, and increased risk for MACE has been
demonstrated extensively. In numerous clinical studies with a
variety of therapeutic interventions, reductions in CRP have
been correlated with
9
reductions in subsequent MACE. We believe, if our Phase 3
pivotal study is successful, that varespladib, if approved by
the FDA, would represent the first anti-inflammatory therapeutic
approved for prevention of MACE.
CRP is one of the most commonly used marker of inflammation. It
has been correlated with adverse cardiovascular outcomes in
multiple clinical studies. Although a causative role for CRP has
not been established, inflammation is known to promote acute
coronary syndrome and CRP may play a direct role in both
vascular inflammation as well as plaque rupture.
Statins reduce the level of CRP and other markers of
inflammation in patients with stable CAD. In April 2001, the
Journal of the American Medical Association published results
from the MIRACL study describing the effect of statins in acute
coronary syndrome, where inflammation is greatly elevated. 3,086
were randomized within 96 hours of their index event to
treatment with high-dose Lipitor (atorvastatin) or placebo.
Lipitor (atorvastatin) significantly reduced secondary MACE
after 16 weeks. A second paper from the same study,
published in Circulation in 2003, described the rapid decline of
inflammatory markers in patients on statin treatment that was
associated with reduced MACE. After 16 weeks, Lipitor
(atorvastatin) reduced CRP levels by 34%.
More recently, in 2005, the New England Journal of Medicine
published data from the PROVE-IT study. A total of
3,745 patients were randomized to either intensive statin
therapy with 80 mg Lipitor (atorvastatin) or moderate
statin therapy with 40 mg pravastatin. Patients with low
CRP or LDL-C had fewer MACE than those with higher levels of
either CRP or LDL-C. Patients who had both LDL-C <
70 mg/dL and CRP < 1 mg/L had the fewest number
of secondary events overall.
LDL-C
in Cardiovascular Disease
The direct relationship between lower LDL-C levels and reduced
risk for major cardiovascular events has been consistently
demonstrated for over a decade in 18 outcome studies involving
over 119,000 patients. Results from large clinical outcome
studies demonstrate achieving incrementally lower LDL-C levels
reduces the risk of future cardiovascular events and provides
continued patient benefit. As a result, the lipid treatment
guidelines have been revised to establish more aggressive LDL-C
treatment goals over time. The most recent guidelines from the
National Cholesterol Education Program’s Adult Treatment
Panel III, or NCEP ATP III, updated in 2004 advocate treatment
goals for LDL-C below 100 mg/dL for high-risk patients and
70 mg/dL for very high-risk patients. Given the breadth of
more recent clinical data available, we believe that future
treatment guidelines from the NCEP will likely establish new
LDL-C treatment goals that apply the 70 mg/dL standard or
lower to a broader population of at-risk patients. Patients
enrolled in our FRANCIS Phase 2b clinical study and our planned
Phase 3 acute coronary syndrome study represent high-risk
patients as defined by the NCEP.
In order to achieve these more aggressive LDL-C targets, doctors
prescribe other approved lipid-lowering therapies such as
cholesterol absorption inhibitors, nicotinic acid and fish oils
in combination with statins to further reduce LDL-C. Still, many
acute coronary syndrome patients who represent the NCEP ATP III
guideline categories of high-risk and very high-risk do not
achieve these recommended lipid goals despite maximum
lipid-lowering therapies. Moreover, substantial residual risk
remains even among the group of patients who do achieve these
aggressive LDL-C goals suggesting additional biological
mechanisms, including inflammation, may be relevant.
This is exemplified in a November 2008 publication in the New
England Journal of Medicine that detailed the results from a
17,000 patient, multinational, primary prevention study
named JUPITER. The study randomized patients with relatively
normal levels of LDL-C, but elevated levels of inflammation
based on CRP, to statin or placebo therapy. The JUPITER study
was stopped early because those patients randomized to statin
therapy demonstrated a statistically significant reduction in
CRP, which also translated to a statistically significant
reduction in cardiovascular events versus those on placebo. The
reduction in events was well in excess of that which would be
predicted from historical data evaluating LDL-C reductions
alone. While these results were generated in a primary
prevention setting, we believe that the benefits of reducing
inflammation may prove to be even more meaningful in settings
where patients are in a hyper-inflammatory state, such as
following an acute coronary syndrome. As a result of these
studies, we believe that there is a substantial need for novel
therapies that provide meaningful reductions in inflammation
while also improving LDL-C levels in high-risk cardiovascular
patients beyond the benefits of statin therapy. Therefore, it is
our belief that targeting inflammation and elevated LDL-C with
sPLA2
inhibition during the early phase of an acute coronary syndrome
will further improve patient outcomes.
10
We believe that varespladib is one of only a few novel drugs in
development with the potential to offer a clinical benefit to
high-risk cardiovascular patients. Varespladib’s unique
mechanism provides potent anti-inflammatory activity, as
measured by reductions in
sPLA2,
CRP and IL-6; incremental lipid-lowering activity, as measured
by LDL-C; and lipid-modulating activity beyond that achievable
with statin therapy alone. Furthermore, because of their
complementary mechanisms, we believe that the combination of
statins and varespladib can provide synergistic
anti-inflammatory and lipid-lowering benefits. We also have
preliminary data to suggest that varespladib may be synergistic
with other cardiovascular therapeutic regimens, such as niacin.
Pivotal
VISTA-16 Study — Acute Coronary Syndrome
In 2008, based on the results from Phase 2 stable CAD studies,
as discussed below, we met with the FDA to discuss the next
steps of clinical development of varespladib during our end of
Phase 2 meeting. As a result of that meeting and the results
from our Phase 2b acute coronary syndrome study, we submitted an
SPA to the FDA for the Phase 3 VISTA-16 study of varespladib for
the short-term (16-week) treatment of patients who have recently
experienced an acute coronary syndrome. We reached agreement
with the FDA on all aspects of the VISTA-16 study protocol,
including patient inclusion/exclusion criteria, study size,
statistical considerations, efficacy endpoints, study duration,
randomization and lipid management strategies.
An independent DSMB will continually evaluate the performance of
the VISTA-16 study over time to ensure patient safety. In
addition, after a minimum of 1,000 patients have been
enrolled in the VISTA-16 study, an independent statistician not
involved with the conduct of the VISTA-16 study will conduct a
biomarker analysis to ensure patient levels of inflammation, as
measured by
sPLA2,
CRP and IL-6, and lipid profiles, as measured by LDL-C, have met
pre-specified reductions versus placebo at various time-points.
These markers of inflammation and lipid profiles are established
in the clinical community and pharmaceutical industry as
independent predictors of cardiovascular risk and, if positive,
will provide additional validation of our previous findings from
the FRANCIS Phase 2b clinical study. At the same time, the
independent DSMB will review all clinical data from the VISTA-16
study to ensure no emergent adverse safety signals.
In June 2010, we initiated enrollment in the VISTA-16 clinical
study. Pursuant to our SPA agreement with the FDA, our
multinational, randomized, double-blind, placebo-controlled
Phase 3 acute coronary syndrome VISTA-16 study will enroll up to
6,500 patients. We anticipate the study will be conducted
in up to 15 countries and up to 500 centers. However, enrollment
may be stopped anytime after a minimum of 385 adjudicated
endpoint events as described in the protocol have occurred. This
number of events could allow us to detect a treatment effect on
the composite endpoint as low as 18.1% with a p-value of less
than 0.05. We may increase the sample size if the adjudicated
endpoint events occur at a lower rate than we expect. Patients
will be randomized at entry to receive 16 weeks of either
500 mg once-daily of varespladib or placebo in addition to
a 20, 40 or 80 milligram dose of Lipitor (atorvastatin). The
dose of Lipitor (atorvastatin) may be adjusted after eight weeks
if the patient’s LDL-C level remains above 100 mg/dL.
Survival status will be obtained for patients six months after
the completion of dosing. The clinical study will attempt to
recruit a similar population of high-risk cardiovascular
patients with acute coronary syndrome to those enrolled in the
FRANCIS study. As in FRANCIS, randomization must occur within
96 hours of hospitalization for the acute coronary syndrome
event, or if already hospitalized, within 96 hours of event
diagnosis. Patient blood chemistry will be evaluated at
baseline, 24 hours and weeks one, two, four, eight and 16.
Randomization is being stratified by the presence or absence of
lipid-lowering therapy prior to the index event as well as the
type of acute coronary syndrome event, such as UA, NSTEMI or
STEMI. The number of subjects who undergo percutaneous coronary
intervention following the index event and prior to
randomization will be limited to no more than 55% of the total
patient population.
The primary endpoint of the VISTA-16 study is to determine
whether 16 weeks of once-daily treatment with varespladib
plus a dose of Lipitor (atorvastatin) is superior to placebo
plus Lipitor (atorvastatin) in the time to the first occurrence
of the combined endpoint of cardiovascular death, non-fatal
myocardial infarction, non-fatal stroke or documented UA with
objective evidence of ischemia requiring hospitalization as
defined by recent FDA draft guidance.
On July 22, 2009, the Center for Drug Education and
Research division of the FDA issued draft recommendations for
standardized definitions for cardiovascular outcomes trials. The
VISTA-16 clinical study endpoint definitions conform to these
guidelines.
11
Components
of VISTA-16 Primary (MACE) Endpoint
|
|
|
| |
•
|
Cardiovascular Death
|
| |
| |
•
|
Non-Fatal Myocardial Infarction
|
| |
| |
•
|
Non-Fatal Stroke
|
| |
| |
•
|
Documented UA with Objective Evidence of Ischemia Requiring
Hospitalization
|
A secondary endpoint for the VISTA-16 study is to determine
whether varespladib plus a dose of Lipitor (atorvastatin) is
superior to placebo plus Lipitor (atorvastatin) in the time to
the first occurrence of the combined endpoint of all cause
mortality, non-fatal myocardial infarction, non-fatal stroke or
documented UA with objective evidence of ischemia requiring
hospitalization. A comparison between treatment groups will also
be made for each component of the primary efficacy endpoint.
Additionally, the time to multiple occurrences of any non-fatal
component of the composite primary endpoint will also be
explored. The biomarkers CRP, IL-6, LDL-C and
sPLA2
will also be evaluated at each time point of the clinical study.
Historical
Clinical Studies
Phase 2b
Acute Coronary Syndrome Study — FRANCIS (Fewer
Recurrent Acute coronary events with Near-term Cardiovascular
Inflammation Suppression)
In July 2008, we initiated a randomized, double-blind,
placebo-controlled Phase 2b clinical study that enrolled 625
acute coronary syndrome patients across 35 centers in three
countries. Given the drug’s combined anti-inflammatory,
lipid-lowering and lipid-modulating effects, we evaluated the
effects of varespladib in acute coronary syndrome patients with
high levels of inflammation and dislipidemia. The clinical study
was designed to evaluate the safety and efficacy of varespladib
when co-administered with the highest dose (80 mg) of
Lipitor (atorvastatin). The clinical study randomized all
patients to a minimum of 24 weeks of treatment with either
500 mg once-daily of varespladib or placebo in combination
with 80 mg Lipitor (atorvastatin) and physician-directed
standard of care.
Patients were eligible for enrollment if they had a diagnosis of
UA, NSTEMI or STEMI. In addition, they must have had one of the
following risk factors: diabetes, body mass index (BMI)
> 25 kg/m2, CRP > 2 mg/L
(NSTEMI/STEMI) or CRP > 3 mg/L (UA) and
presence of three (pre-defined) characteristics of metabolic
syndrome. Subjects must have been randomized within
< 96 hours of hospital admission for the index
event, or, if already hospitalized, within <
96 hours of index event diagnosis. Any percutaneous
revascularization was required to occur prior to randomization.
In addition, because we wanted to assess the effects of
varespladib with the highest available dose of Lipitor
(atorvastatin), patients were not allowed to use any other
lipid-lowering therapies during the clinical study.
Follow-up
visits for evaluation occurred post-randomization at weeks two,
four, eight, 12, 16, 20, 24 and then monthly thereafter until
clinical study completion. All enrolled subjects remained on
treatment until all subjects had been treated for a minimum of
24 weeks or until the occurrence of MACE. Patients
randomized into the FRANCIS study had baseline characteristics
such as LDL-C indexed-event risk factors and demographics
similar to other studies of this type. All patients who
completed the clinical study received a final evaluation.
The primary efficacy endpoint evaluated the change in LDL-C
after 500 patients completed eight weeks of treatment.
LDL-C is the most widely recognized surrogate for predicting
cardiovascular risk where percentage reductions in LDL-C have
been highly correlated with reductions in future cardiovascular
risk. Secondary endpoints included:
|
|
|
| |
•
|
changes in established markers of inflammation such as
sPLA2,
CRP and IL-6; and
|
| |
| |
•
|
the occurrence of secondary MACE (for purposes of this clinical
study, all-cause mortality, non-fatal myocardial infarction,
documented UA requiring urgent hospitalization,
revascularization occurring > 60 days
post the index event or non-fatal stroke).
|
Results of the primary endpoint demonstrated a statistically
significant incremental LDL-C reduction of 5.7% (p = 0.0023) in
varespladib treated patients versus those treated with
80 mg Lipitor (atorvastatin) alone after eight weeks of
therapy. A p-value is a probability with a value ranging from 0
to 1, which indicates the likelihood that a clinical study is
different between treatment and control groups. P-values below
0.05 are typically referred to as
12
statistically significant. A statistically significant
difference was observed in LDL-C reduction from baseline as
early as two weeks after treatment. The treatment effect was
maintained throughout the observation period.
Secondary endpoints measured effects of varespladib on
sPLA2,
CRP and IL-6 levels, which are well-established markers of
inflammation. While the FRANCIS study was not designed to
demonstrate statistically significant changes in CRP and IL-6,
the results were consistent with previous studies, which
demonstrated improvement across these biomarkers and achieved
statistical significance at some time points.
sPLA2
concentration was statistically significantly reduced from the
earliest time point of two weeks through the 16-week time point
(p < 0.0001) as compared to high-dose statin (80 mg
Lipitor (atorvastatin)) therapy alone. While our first
sPLA2
measurement in this clinical study occurred at two weeks, data
from previous clinical studies utilizing varespladib or
A-001
demonstrated reductions in
sPLA2
as early as two days following treatment.
In addition, treatment-related reductions in CRP and IL-6 levels
were also greater in varespladib treated patients compared to
those treated with placebo at all time points in the clinical
study. The percent decrease in CRP at week two was nearly
two-fold greater among varespladib and 80 mg Lipitor
(atorvastatin) treated patients than those treated with placebo
and 80 mg Lipitor (atorvastatin) alone (-39% versus -20%, p
= 0.183), and at week 16, the difference between treatment
groups was statistically significant (-82% versus -73%, p =
0.0067). At weeks two, four, eight and 16, varespladib treated
patients had numerically reduced levels of CRP versus patients
treated with placebo.
The percent decrease in IL-6 in patients on varespladib at week
two was more than three times the reduction in IL-6 in patients
on placebo (-18% versus -5.1%, p = 0.18).
Treatment with varespladib resulted in more subjects with LDL-C
levels lower than 70 mg/dL and lower than 50 mg/dL
than those on placebo (80 mg Lipitor (atorvastatin) and
physician-directed standard of care) alone at eight, 16 and
24 weeks of treatment. As discussed above, the NCEP ATP III
guidelines have established an LDL-C of 70 mg/dL as an
optional target for very high-risk patients. As indicated in the
table below, the data suggests varespladib treatment helps
patients achieve their LDL-C target levels more quickly and
maintain them longer than with high-dose statin (80 mg
Lipitor (atorvastatin)) therapy alone.
Finally, given the importance of reducing inflammation as well
as LDL-C following an acute coronary syndrome event, we examined
the proportion of patients in the clinical study that were able
to achieve both LDL-C levels less than 70 mg/dL and CRP
levels below 1 mg/L. Significantly more patients at week
four and week 16 (p = 0.02 and p = 0.01) reached this combined
target when treated with varespladib and 80 mg Lipitor
(atorvastatin) than with placebo and 80 mg Lipitor
(atorvastatin) alone. (The actual proportion of subjects in the
varespladib group was 25% and 16% in the placebo group).
Additionally, in the PROVE-IT study a comparable proportion
(16%) of patients treated with 80 mg Lipitor (atorvastatin)
achieved these goals.
We also conducted an exploratory analysis of MACE in the
clinical study. At 16 weeks, there were 14 (4.2%) MACE in
the varespladib treated group as compared to 19 (6.1%) in the
placebo group. At the completion of the clinical study, all
patients had received at least six months of therapy and there
were 23 (7.4%) MACE in the varespladib treated group as compared
to 24 (7.7%) MACE in the placebo group. While the MACE analysis
was not designed to demonstrate any statistical differences
between the two treatment groups, we believe that the results
are encouraging and has helped us to design our VISTA-16 study.
Overall, varespladib was generally well-tolerated in this
clinical study and no imbalance was seen in dropouts due to drug
effects. After completing patient treatment, overall exposure to
varespladib was a mean of 30 weeks and median of
34 weeks. In total, 485 total patients completed six months
of treatment, with 167 subjects completing 40 weeks and 70
completing 44 weeks. There was no imbalance of overall
adverse events between the treatment arms. During the clinical
study, at week four and week eight, occasional mild and
transient elevations in liver enzymes, defined as elevations
three times the upper limit of normal, were seen among more
patients taking varespladib, but the frequency and magnitude of
the elevations were not meaningfully different between the
active and control groups at the end of the clinical study. The
frequency of the elevations was also similar to that reported
for Lipitor (atorvastatin) and other currently approved
lipid-lowering agents. Furthermore, there were no effects on
blood pressure or the QT interval, an electro-cardiographic
safety endpoint.
13
Summary data from FRANCIS was presented at the American College
of Cardiology meeting in 2010 and the detailed results from the
study were published in the Journal of the American College of
Cardiology in September 2010.
Phase 2
Stable Coronary Artery Disease Study — PLASMA
(Phospholipase Levels and Serological Markers of
Atherosclerosis): Varespladib Twice-Daily Versus
Placebo
Our Phase 2 PLASMA study was designed to confirm the safety and
effect of varespladib on
sPLA2
concentration, other inflammatory biomarkers and lipids in
patients with stable CAD. In October 2007, we completed a
randomized, double-blind, placebo-controlled study evaluating
four doses of varespladib administered twice-daily versus
placebo among 396 patients with stable CAD from 38 centers
in two countries. The clinical study enrolled patients more than
12 weeks after a myocardial infarction or six weeks after
an episode of UA. The varespladib doses tested were 50 mg,
100 mg, 250 mg and 500 mg administered twice per
day. Following randomization, patients were treated for eight
weeks and safety and efficacy evaluations were conducted at
weeks two, four and eight. Physician-directed standard of care
therapies were permitted during the clinical study, including
259 patients who were on background statin therapy.
The primary endpoint of the clinical study was the change in
sPLA2
concentration from baseline to week eight in varespladib, across
all doses, versus placebo patients. Secondary endpoints in the
clinical study included the change in lipids, including LDL-C,
lipoprotein subclasses and certain inflammatory biomarkers, from
baseline to each of weeks two, four and eight.
Our Phase 2 PLASMA results were selected for a late-breaking
presentation at the American Cardiology Conference and published
in the Lancet journal in February 2009. Results from the
clinical study demonstrated that treatment with varespladib led
to statistically significant reductions in
sPLA2,
LDL-C and various plaque-building and pro-inflammatory forms of
LDL-C. In patients receiving varespladib, there were incremental
reductions in CRP versus placebo (-55.6% versus -24.8%, p =
0.47) from baseline to eight weeks.
Among all patients treated with varespladib, median
sPLA2
concentration decreased by 86.7% from baseline to week eight, as
compared to 4.8% in the placebo group (p < 0.0001). Median
sPLA2
concentration decreased among the varespladib groups in a
dose-dependent manner.
At week eight, across all dosage groups, LDL-C was reduced by
9.7% versus placebo (p = 0.0035). In a subgroup of patients
taking statins with LDL-C > 70 mg/dL, LDL-C was
reduced by 12.0% (p = 0.0065) versus placebo at the eight week
time point. Notably, the reductions in LDL-C appear to be driven
primarily by a shift in the distribution of LDL-C particles with
fewer pro-atherogenic, pro-inflammatory small LDL-C particles
present in the circulation. In addition, statistically
significant reductions from baseline to week eight were seen in
total cholesterol and non-HDL cholesterol in the overall
clinical study population treated with varespladib.
Varespladib was generally well-tolerated among all patients
treated. In general, adverse effects were mild or moderate with
no imbalance of adverse events in the varespladib groups as
compared to placebo. The most common adverse effects seen in the
varespladib groups were headache (6.4%) and nausea (5.4%). There
were mild and transient elevations of liver function tests,
defined as elevations three times the upper limit of normal, in
patients taking varespladib.
Phase 2
Stable Coronary Artery Disease Study — PLASMA-2
(Phospholipase Levels and Serological Markers of Atherosclerosis
-2): Once-Daily of Varespladib Versus Placebo
Based on data from our first PLASMA study, we initiated a second
Phase 2 clinical study (PLASMA-2) to evaluate the effect of
once-daily varespladib treatment on inflammatory and lipid
biomarkers. In December 2007, we completed a randomized,
double-blind, placebo-controlled Phase 2 clinical study
evaluating two doses of varespladib versus placebo amongst
138 patients with stable CAD. The clinical study, conducted
in the United States, involved 13 clinical sites. Following
randomization to one of two doses of varespladib or placebo,
patients were treated for eight weeks with safety and efficacy
evaluations at weeks two, four and eight. Physician-directed
standard of care therapies were permitted during the clinical
study, including 123 patients (89.1%) who were on
background statin therapy.
14
The primary endpoint of the clinical study was a comparison
between once-daily doses of varespladib and placebo in changes
in
sPLA2
concentration at week eight. Secondary endpoints in the clinical
study included measurements of lipids including LDL-C and
certain other inflammatory biomarkers from baseline to each of
weeks two, four and eight.
Results of the primary endpoint,
sPLA2,
were statistically significant and consistent with those
generated from the first PLASMA study described above. Patients
on varespladib demonstrated a 77.8% reduction in
sPLA2
concentration as compared to an increase of 8.3% in placebo
treated patients (p < 0.0001). Pharmacokinetic data
indicated that once-daily dosing with varespladib would be
sufficient to achieve over 90% inhibition of
sPLA2
mass and activity over a
24-hour
period.
The anti-inflammatory, lipid-lowering and lipid-modulating
effects of varespladib treatment were consistent with those seen
in the first PLASMA study: LDL-C was decreased by 8.3% compared
to 0.7% in placebo (p = 0.014). Due to the small size
of this clinical study and the low baseline inflammation present
in these patients, no meaningful changes with CRP could be
detected between the active and control groups. As was observed
in the first clinical study, there were statistically
significant reductions from baseline to week eight in total
cholesterol and non-HDL cholesterol in the overall clinical
study population treated with varespladib.
The adverse effect profile for varespladib was consistent with
earlier studies and there was no imbalance of adverse events
among the varespladib groups and placebo. Varespladib was
generally well-tolerated. The most common effects seen in the
varespladib groups were diarrhea (6.7%), nausea (5.6%), any
increase in alanine aminotransferase (5.6%), which is an enzyme
that indicates liver cell injury, and any increase in aspartate
aminotransferase (5.6%), which is another enzyme that indicates
liver cell injury. However, mild and transient elevations of
these liver enzymes, defined as elevations three times the upper
limit of normal, were infrequent in patients taking varespladib.
Placebo-corrected Percent Decrease from Baseline to Week Eight
in Biomarkers
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LDL
|
|
|
Total
|
|
|
Non-HDL
|
|
|
Oxidized
|
|
|
|
|
|
|
|
sPLA2
|
|
|
Cholesterol
|
|
|
Cholesterol
|
|
|
Cholesterol
|
|
|
LDL-C
|
|
|
|
|
|
|
|
PLASMA
|
|
|
81.9
|
%
|
|
|
9.7
|
%
|
|
|
4.9
|
%
|
|
|
7.2
|
%
|
|
|
5.4
|
%
|
|
|
|
|
|
(All doses varespladib)
|
|
|
(p < 0.0001
|
)
|
|
|
(p = 0.0035
|
)
|
|
|
(p = 0.0069
|
)
|
|
|
(p = 0.0009
|
)
|
|
|
(p = 0.0065
|
)
|
|
|
|
|
|
PLASMA-2
|
|
|
86.1
|
%
|
|
|
13.9
|
%
|
|
|
9.2
|
%
|
|
|
14.2
|
%
|
|
|
7.3
|
%
|
|
|
|
|
|
(500 mg varespladib)*
|
|
|
(p < 0.0001
|
)
|
|
|
(p = 0.0007
|
)
|
|
|
(p = 0.0006
|
)
|
|
|
(p = 0.0001
|
)
|
|
|
(pNS
|
)(†)
|
|
|
|
|
|
|
|
|
* |
|
Dose selected for Phase 3 |
| |
|
† |
|
Probability not significant |
Investigator-Sponsored
Phase 2 Percutaneous Intervention Study — SPIDER-PCI
(sPLA2)
Inhibition to Decrease Enzyme Release After PCI): Varespladib
Once-Daily Versus Placebo for up to 10 Days.
In May 2007, Dr. Vladimir Dzavik at University Health
Network Hospital in Toronto, Ontario, Canada initiated an
investigator sponsored study with varespladib in patients
undergoing a percutaneous intervention, or PCI. The primary
endpoint of this study was to determine if inhibition of
sPLA2
with varespladib will result in a decrease in peri-PCI
myocardial necrosis, or heart muscle damage, as measured by
elevations of myocardial enzyme markers creatine kinase-MB, or
CK-MB, or troponin I. The study was to enroll a maximum of
164 patients who were scheduled to undergo PCI. Elevated
levels of troponin I following PCI are associated with an
increase in in-hospital complications and, in one study, were an
independent predictor of major cardiac events. After PCI,
circulating levels of
sPLA2
increase and patients with higher levels have an increased risk
of events after a two-year
follow-up.
This study explores the notion that
sPLA2
inhibition may reduce myocardial damage after PCI and improve
patient outcomes.
As of August 2009, enrollment and dosing in the SPIDER-PCI
investigator study were completed with 144 patients
evaluated for purposes of assessing the primary endpoint. On
December 11, 2009, we received a statistical analysis of
the patient evaluations, which showed that the primary endpoint
of the study, a reduction in the elevation of CK-MB or troponin
I above the upper limit of normal at six to eight hours or 18 to
24 hours, was not met (varespladib patients 57% versus
placebo patients 51%, p = 0.55). However, the results showed
statistically significant reductions of
sPLA2
as early as 18 hours post-PCI procedure, which persisted
throughout the five days of
15
dosing (-93.0%, p < 0.001). Consistent with results from
other clinical studies with varespladib, there were numerical
reductions in CRP from baseline versus placebo at three to five
days (-82.1%, p = 0.23).
Previous
Experience at Eli Lilly and Shionogi & Co.,
Ltd.
Eli Lilly and Shionogi & Co., Ltd. previously
conducted a series of clinical studies evaluating varespladib
and A-001 in
various inflammatory conditions. In total, at least 17 Phase 1
and Phase 2 clinical studies evaluated varespladib and
A-001 as a
treatment in sepsis, rheumatoid arthritis, asthma and ulcerative
colitis, an inflammatory bowel disease. Results from these
studies provide a large body of safety data for varespladib and
A-001 with
more than 1,000 healthy volunteers and subjects receiving
treatment.
Throughout these studies, varespladib was generally
well-tolerated.
Non-Clinical
Studies with Varespladib and
A-001
Approximately 150 preclinical pharmacology and toxicology
studies have been completed with varespladib and
A-001,
including two-year rat and mouse carcinogenicity studies,
one-year primate study and three-month rat study in combination
with Lipitor (atorvastatin).
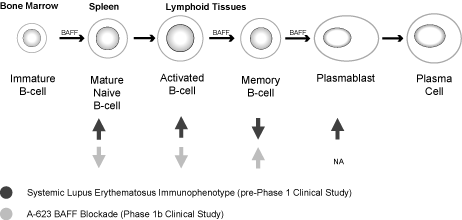
A-623
A-623 is a
peptibody antagonist of the BAFF cytokine that is initially
being developed as a treatment for lupus. BLyS, also known as
B-cell activating factor, or BAFF, is a tumor necrosis family
member and is critical to the development, maintenance and
survival of B-cells. It is primarily expressed by macrophages,
monocytes and dendritic cells and interacts with three different
receptors on B-cells including BAFF receptor, or BAFF-R, B-cell
maturation, or BCMA, and transmembrane activator and cyclophilin
ligand interactor, or TACI. The BAFF-R receptor is expressed
primarily on peripheral B-cells.
Two randomized, dose-ranging, placebo-controlled Phase 1
clinical studies for
A-623 in 104
lupus patients have already been completed. Results from these
studies demonstrated
A-623
generated anti-BAFF activity and showed statistically
significant reductions in B-cells of
50-70% (p
< 0.001) in lupus patients across multiple subcutaneous and
intravenous formulations.
After successfully reactivating our Investigational New Drug
Application, or IND, we initiated a Phase 2b clinical study with
A-623 for
the treatment of lupus in July 2010 called PEARL-SC. We may also
study A-623
in other B-cell mediated autoimmune diseases such as
Sjögren’s Syndrome or orphan indications such as
myasthenia gravis and pemphigus. We are actively pursuing a
partnership with major pharmaceutical companies to develop and
commercialize
A-623.
We intend to advance the development of our BAFF targeting
molecule,
A-623, a
selective peptibody, to exploit its broad clinical utility in
autoimmune diseases.
A-623, as a
peptibody directed against BAFF, was developed as an alternative
to antibodies and is produced in Escherichia coli
bacterial culture versus antibodies that are produced in
mammalian cell culture. In addition,
A-623 offers
a number of potential differentiations over other anti-BAFF
compounds, as well as other novel B-cell directed therapies,
including:
|
|
|
| |
•
|
convenient, at-home, patient-administered subcutaneous dosing
with a range of dosing frequencies including monthly and weekly;
|
| |
| |
•
|
ability to inhibit the activity of both membrane-bound and
soluble BAFF, which may confer differentiating pharmacodynamic
characteristics;
|
| |
| |
•
|
non-glycosylated protein that is produced in a bacterial
fermentation manufacturing process, which may reduce the
potential to be immunogenic and may provide manufacturing
benefits and lower cost of goods; and
|
| |
| |
•
|
multiple binding domains achieve highest reported affinity for
inhibition of BAFF.
|
16
Market
Opportunity
Lupus is an autoimmune disorder that involves inflammation that
causes swelling, pain and tissue damage throughout the body.
Lupus can affect any part of the body, but especially the skin,
heart, brain, lungs, joints and the kidneys. The course of the
disease is unpredictable, with periods of illness, called flares
alternating with remission. The Lupus Foundation estimates that
approximately 1.5 million people in the United States and
five million worldwide suffer from lupus. Although lupus may
affect people of either sex, women are 10 times more likely to
suffer from the disease than men, according to the Lupus
Foundation.
Patients with active lupus may have a broad range of symptoms
related to the inflammation. Inflammation of the brain may cause
seizures and other neurologic abnormalities. Inflammation of the
heart may cause heart failure or sudden death. Lung inflammation
causes shortness of breath. Lupus may also cause swollen joints
and severe rash. In addition, LN may lead to requiring kidney
dialysis or transplantation.
Although the cause of lupus is still not completely understood,
B-cell activation and autoantibody production are known to be
central to the process. Evidence has emerged that
over-expression of BAFF plays an important role in this disease
process. In preclinical studies, transgenic mice created to
over-express BAFF begin to exhibit symptoms similar to lupus. In
addition, treatment of these same mice with BAFF antagonists
appears to ameliorate the disease.
PEARL-SC
Phase 2b Clinical Study in Patients with Lupus
Based on positive results among 104 patients in our Phase
1a and 1b clinical studies, we initiated a Phase 2b clinical
study in lupus patients called PEARL-SC. PEARL-SC is a
randomized, placebo-controlled, phase 2b clinical study which
may enroll up to 600 patients in approximately 90 centers
worldwide. Subjects will be randomized into three active
subcutaneous treatment arms and one placebo treatment arm for a
minimum of 24 weeks. The primary endpoint of the PEARL-SC
study will be clinical improvement at 24 weeks in responder
rates of a composite systemic lupus erythematosus responder
index, or SRI, in the pooled treatment arms versus placebo. The
primary SRI endpoint is a composite score based upon changes in
SELENA and SLEDAI disease activity scale, Physician’s
Global Assessment scores and British Isles Lupus Assessment
Group scores, which are clinical standards for the measurement
of disease severity in lupus patients. Secondary endpoints will
include safety, improvement in other clinical assessment scores,
clinical response in patients with various baseline disease
severities, resolution of fatigue, steroid utilization and time
to flare. An interim biomarker analysis to establish the
appropriate drug effect on total B-cells is included early in
the study. Initially we intend to randomize only
540 patients. This total number of patients will
potentially allow us to detect a treatment effect of 14% with a
p-value of 0.05 between the pooled active arms and the placebo
arm.
In November 2010, we placed a voluntary hold on the PEARL-SC
study due to problems found with vials. Patient enrollment in
the study was temporarily suspended and dosing was discontinued
in patients who were enrolled in the study while we conducted an
analysis of the problem. We resolved the issues found with the
vials in December 2010. After analysis, simulation and
consultation with industry experts, we determined that shipping
on dry ice was the root cause of the issue. Shipping logistics
were modified and we reinitiated enrollment in
PEARL-SC and
dosing in January 2011. We have received no reports of
patient-related side effects or problems with drug
administration that could be attributed to the vial problem.
Future
Development of
A-623
A-623
Manufacturing Strategy
In May 2010, we successfully completed a manufacturing campaign
for a high concentration
A-623
injection formulation for subcutaneous administration.
Manufacturing was conducted per current good manufacturing
practices, or cGMP, and the product was released to clinical
sites in July 2010. In August 2010, we manufactured a second
batch of vials of the high-concentration
A-623
injection formulation from 34 liters of Amgen manufactured bulk
drug substance, which was subsequently released for clinical
use. We believe we now have sufficient clinical material, both
placebo and
A-623, to
dose up to 540 patients for a minimum of six months in the
PEARL-SC study.
In December 2010, we announced the selection of a CMO for our
larger scale GMP manufacturing, the Merck Biomanufacturing
Network (recently acquired by Fujifilm), and began the technical
transfer of the full
17
manufacturing of GMP bulk product for eventual pivotal clinical
studies and an optional expansion
and/or
extension of the PEARL-SC study.
The following chart outlines the basic manufacturing steps
required for the production of
A-623.
A-623
Regulatory Strategy
In July 2010, we received clearance from the FDA to begin
recruitment of lupus patients into the PEARL-SC clinical study.
The study protocol allows for enrollment of up to
600 patients treated for a maximum of 12 months.
Patients enrolled in this study will be randomized into three
active treatment arms and one placebo arm. Subsequent to this
clearance, the FDA requested additional information regarding
characterization and qualification of the manufactured vials of
A-623. In
addition, the FDA requested minor changes to aspects of the
PEARL-SC study including collection of ECG testing at the end of
dosing and a recommendation for a corticosteroid tapering
strategy. Neither of these changes are considered material to
the conduct of the PEARL-SC study. The FDA also recommended we
submit to the IND analytical and comparability data from our
recently completed manufacturing lot of
A-623 vials
and a comparability proposal for purposes of soliciting their
input prior to implementation. We submitted a response to the
FDA in October 2010.
It is our intent to discuss with the FDA comparability proposals
which will propose the method by which future batches of
material manufactured by our CMO would meet the FDA’s
standards for equivalence. We also intend to continuously submit
results of comparability testing from all of our manufacturing
campaigns to the FDA. If these batches meet specification and
the FDA agrees
A-623
product manufactured by our CMO meets comparability
requirements, we believe these batches could be used to further
extend and expand the PEARL-SC study
and/or
initiate identical Phase 3 clinical studies for purposes of
registration.
Historical
Clinical Studies
Prior to our in-licensing of
A-623, Amgen
completed two Phase 1 clinical studies of
A-623 in
lupus patients to evaluate the safety and pharmacokinetics of
single and multiple doses of the drug using intravenous and
subcutaneous formulations. Prior to conducting Phase 1 clinical
studies in lupus patients, Amgen conducted a pre-Phase 1
clinical study in lupus patients. In Amgen’s pre-Phase 1
clinical study, individual B-cell subsets, such as mature
naïve B-cells, activated B-cells and memory B-cells, all
therapeutic targets for
A-623, were
quantified in order to characterize the specific B-cell subset
abnormalities associated with lupus.
The randomized, placebo-controlled, dose-escalation Phase 1a
clinical study evaluated
A-623 as a
single intravenous or subcutaneous therapy among 56 lupus
patients. Intravenous doses included 1, 3 and 6 mg/kg, and
subcutaneous doses included 0.1, 0.3, 1 and 3 mg/kg. The
primary endpoint was to assess the safety and tolerability of
single dose administrations of
A-623.
Secondary endpoints were designed to assess the plasma
pharmacokinetic profile and immunogenicity of
A-623.
Results from this clinical study indicated the safety and
tolerability of
A-623
administered as single dose of intravenous or subcutaneous was
comparable to placebo. Single doses of
A-623
exhibited linear pharmacokinetics after both intravenous and
subcutaneous administration. There were comparable adverse
events between the
A-623 and
placebo groups with no deaths reported. In addition, no
neutralization antibodies were seen across all doses. The most
common adverse events were nausea (15%), headache (10%), upper
respiratory tract infection (10%) and diarrhea (8%).
A-623 was
evaluated in a randomized, placebo-controlled, multi-dose Phase
1b clinical study as an intravenous or subcutaneous therapy
among 63 lupus patients. The intravenous dose was 6 mg/kg,
and subcutaneous doses included 0.3, 1 and 3 mg/kg.
Patients received their doses of
A-623 or
placebo once-weekly for four weeks. The primary endpoint was to
assess the safety and tolerability of multiple dose
administrations of
A-623.
Secondary endpoints were designed to assess the plasma
pharmacokinetic profile and immunogenicity of
A-623 after
multiple doses. Results showed that multiple doses of
A-623
exhibited dose-proportional pharmacokinetics after both
intravenous and subcutaneous administration. Further, results
demonstrated a dose-dependent decrease in total B-cells as early
as 15 days of treatment, and total B-cell reduction (up to
approximately
60-70% of
baseline) reached its nadir after about 160 days of
therapy. By six months after treatment, the B-cell populations
had returned to baseline levels.
18
An experimental analysis was also conducted to assess B-cell
subsets in patients following multiple doses. Results
demonstrated that
A-623
selectively modulates certain B-cell subsets and induced trends
toward normalizing the B-cell abnormalities that were observed
in lupus patients in the pre-Phase 1 clinical study.
Results indicated that the tolerability of
A-623
administered as multiple doses of intravenous or subcutaneous
administration was generally comparable to placebo. There were
no deaths reported between the
A-623 and
placebo groups. Few neutralization antibodies were seen, and all
resolved in subsequent visits. Based on these results and
published data from competitor studies, we initiated a Phase 2b
clinical study evaluating
A-623 in
lupus patients during the second half of 2010.
A-001
A-001 is an
intravenously administered, potent, broad-spectrum inhibitor of
sPLA2,
including forms IIa, V and X.
A-001 will
be evaluated in a Phase 2 clinical study for the prevention of
acute chest syndrome associated with sickle cell disease in
at-risk patients. Substantial scientific evidence implicates
sPLA2
activity in the development of acute chest syndrome associated
with sickle cell disease, as well as other forms of acute lung
injury. The FDA granted orphan drug and fast-track designation
for A-001
for the prevention of acute chest syndrome in
at-risk
patients. We currently retain all worldwide product rights,
except in Japan where Shionogi & Co., Ltd. retains
rights. We also licensed
A-001 from
Eli Lilly and Shionogi & Co., Ltd. in July 2006.
sPLA2
levels increase in advance of acute chest syndrome episodes and
can be used alongside the presence of fever to strongly predict
an impending episode. There is a strong correlation between
levels of CRP and
sPLA2
in this patient population. Patients with acute chest syndrome
associated with sickle cell disease can exhibit levels of
sPLA2 that
can be 100 times greater than normal. We believe that early
intervention with
A-001 to
inhibit
sPLA2
activity may offer a novel preventative therapy to improve
outcome among sickle cell disease patients presenting with a
high risk of acute chest syndrome.
Market
Opportunity
Sickle cell disease is a lifelong genetic, blood disorder
typically diagnosed during early childhood. According to the
Sickle Cell Information Center, in the United States, over
70,000 people currently suffer from the disease and
approximately 1,000 children are born with the disease annually.
According to Medtech Insight, in Europe, there are over
200,000 people suffering from the disease, and the numbers
increase dramatically in Africa, where, according to the WHO,
200,000 children alone are born with sickle cell disease each
year. Life expectancy for these patients is significantly
shortened, with most expected to live only until their mid-40s.
The disease is characterized by structurally altered red blood
cells that assume an abnormal shape, similar to a sickle, and
produce an altered form of hemoglobin. These altered red blood
cells have a shortened life-cycle, become stiff and have
difficulty passing through the body’s small blood vessels.
At times, these abnormal cells may obstruct or block blood flow
through small blood vessels, leading to significant damage in
tissue and bone. This damage is more
19
commonly labeled as VOC. During VOC, blockage occurs within the
circulation of the long bones, causing microscopic bone damage.
Fragments of bone or bone fat may break free and embolize to the
lungs, causing lung injury.
VOC is a common trigger for the more serious complication of
acute chest syndrome associated with sickle cell disease. Acute
chest syndrome exhibits symptoms and characteristics similar to
acute lung injury. There are an estimated 10,000 episodes of
acute chest syndrome associated with sickle cell disease per
year in the United States. It represents the most common cause
of death in sickle cell patients and the second most common
cause of hospitalization among such patients. A majority of
sickle cell patients will experience at least one episode of
acute chest syndrome and repeated episodes can result in
progressive lung disease. The disorder is most common in the
two- to four- year age group and gradually declines in incidence
with age.
There are no marketed therapies targeting acute chest syndrome
associated with sickle cell disease. The most common treatment
regimen includes heavy doses of corticosteroids, opiates,
transfusion and antibiotics while the patient suffers through
the attack. In addition, hydroxyurea, a chemotherapy, was found
to reduce the frequency of VOC and the need for blood
transfusions in adult patients with sickle cell disease.
However, all of these therapeutics are associated with
significant adverse effects while only offering limited patient
benefit.
Phase 2B Clinical Study: Prevention of Acute Chest Syndrome in
Patients with Sickle Cell Disease
Our planned multinational, randomized, double-blind,
placebo-controlled Phase 3 clinical study will enroll up to
200 patients with sickle cell disease who are at an
elevated risk of developing acute chest syndrome as a result of
fever, VOC and CRP 5.0 mg/l
³
at the time of hospitalization. Patients will be randomized to
receive a continuous infusion of
A-001 or
placebo for 48 hours after randomization. The primary
endpoint of this study will be freedom from acute chest syndrome
as determined by physician assessment and independent review of
chest X-rays. This study represents a unique treatment approach
for a small, orphan designated indication. As a result the
appropriateness of the design and endpoints of this study for
purposes of registration will only be known at the conclusion of
the study and upon submission to the FDA.
Historical
Clinical Studies
Phase 2
Acute Chest Syndrome in Hospitalized Patients with Sickle Cell
Disease Study — Investigation of the Modulation of
Phospholipase in Acute Chest Syndrome, or IMPACTS.
In January 2007, we initiated a randomized, double-blind,
placebo-controlled Phase 2 clinical study to assess the safety
and tolerability of escalating doses of
A-001
therapy when administered as a
48-hour
continuous infusion. The clinical study was designed to enroll
up to 75 patients across approximately 30 sites in the
United States. This clinical study enrolls hospitalized sickle
cell disease patients at risk for acute chest syndrome on the
basis of VOC,
20
fever and serum
sPLA2
concentration level greater than 50 mg/mL. The primary
endpoint for the clinical study was designed to assess safety
and tolerability. Secondary endpoints included the absence of
acute chest syndrome, suppression of
sPLA2,
reduced need for blood transfusions and assessment of
pharmacokinetics.
The first group of patients was randomized 2:1 to receive low
dose A-001
or placebo as a
48-hour
continuous infusion. A pre-specified interim analysis was
conducted in February 2009 after the 30th patient completed
treatment to examine safety and adjust dosing schedules. The
interim data was balanced between two dosing arms of 30 55
µg/kg/hr (n = 11) and 55 µg/kg/hr (n = 6).
Interim results indicated serum levels of
A-001 when
dosed at 55 µig/kg/hr reduced
sPLA2
activity levels by more than 80% from baseline within
48 hours. Furthermore, the prevention of acute chest
syndrome associated with sickle cell disease appeared to be
related to the level of
sPLA2
activity. The DSMB recommended the clinical study continue based
on safety and tolerability. In addition, given the safety
profile, the DSMB approved the addition of a higher dose group
of 110 µg/kg/hr via continuous infusion during the second
half of the clinical study. We believe that the data suggests
A-001 can
suppress
sPLA2
at levels that may prevent the complication of acute chest
syndrome associated with sickle cell disease.
Reductions of
sPLA2
activity from baseline and incidence of acute chest syndrome
(including placebo patients and patients receiving
A-001).
Exploratory analysis to determine correlation between degree of
sPLA2
suppression and incidence of acute chest syndrome.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
48-Hour
sPLA2
Activity as
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a Percentage of Baseline
|
|
0.0% < 25.0%
|
|
|
³
25%
³
50%
|
|
|
³
50%
³
75%
|
|
|
³
75%
|
|
|
|
|
Number of Subjects
|
|
|
7
|
|
|
|
7
|
|
|
|
3
|
|
|
|
12
|
|
|
Number of Subjects Developing Acute Chest Syndrome (%)
|
|
|
0
|
(0)
|
|
|
2
|
(28)
|
|
|
1
|
(33)
|
|
|
4
|
(25)
|
Our
Strategy
Our objective is to develop and commercialize our product
candidates to treat serious diseases associated with
inflammation, including cardiovascular and autoimmune diseases.
To achieve these objectives, we intend to initially focus on:
Advancing
Varespladib Through Phase 3.
Inflammatory processes and lipid abnormalities are central to
the onset of acute coronary syndrome and the development of CAD.
Varespladib operates through a novel mechanism of action to
offer both targeted anti-inflammatory activity and incremental
lipid reductions, including LDL-C, when used in combination with
statins. Despite the benefits of statin therapy, many acute
coronary syndrome patients still remain at substantial risk of a
coronary event, suggesting additional biological mechanisms may
be relevant, including inflammation. We believe that combination
therapy with varespladib and statins will provide acute coronary
syndrome patients with a unique, short-term therapeutic option
unavailable with existing agents today. In addition, we believe
that an opportunity exists in the future to evaluate varespladib
in chronic indications such as CAD.
Advancing
Clinical Development of
A-623.
We are advancing the development of
A-623 to
exploit the broad potential clinical utility of BAFF antagonism.
We have initiated the Phase 2b clinical study known as PEARL-SC
in lupus patients. We may opportunistically enter into
collaborations with third parties for development of this
compound in lupus or in other B-cell mediated diseases, such as
multiple sclerosis, rheumatoid arthritis or Sjögren’s
Syndrome, that may benefit from BAFF antagonism, including
securing corporate partners whose capabilities complement ours.
Potential
Development of
A-00.1
In the future, if additional funds are available, we may develop
A-001, an
intravenous
sPLA2
inhibitor for prevention of acute chest syndrome associated with
sickle cell disease, because we identified that elevations in
sPLA2
activity are known to precede and predict disease progression.
Given that there are currently no approved drugs for the
prevention of acute chest syndrome associated with sickle cell
disease, we have received orphan drug designation and fast track
status from the FDA for
A-001.
21
Leveraging
Our
sPLA2
Expertise to Develop Products for Additional Disease
Indications.
We believe that we have developed a leadership position in the
field of
sPLA2
inhibition. Beyond our acute coronary syndrome and acute chest
syndrome program, we believe that
sPLA2
inhibition may have applications in other acute disease settings
where early intervention may have an impact and reduce
anti-inflammatory activity, such as acute lung injury.
Additionally, we believe that we can apply our
sPLA2
expertise to develop novel therapeutics for a number of chronic
diseases. For example,
sPLA2
has been shown to be involved in the development of such chronic
inflammatory diseases as atherosclerosis and dermatitis. We plan
to pursue these indications opportunistically and potentially in
collaboration with third parties.
We are also developing new and unique
sPLA2
inhibitor compounds for additional therapeutic areas.
A-003 is our
second generation lead candidate. We plan to continue
preclinical development of
A-003 for an
IND filing and we will continue to assess additional new
compounds.
Developing
Commercial Strategies Designed to Maximize Our Product
Candidates’ Market Potential.
Our primary product candidates are focused on either the acute
care setting in the hospital or highly-specialized physician
segments, such as rheumatologists. We believe that we can build
a small, focused sales force capable of marketing our products
effectively in acute care and orphan indications such as acute
coronary syndrome and acute chest syndrome associated with
sickle cell disease. In other chronic indications such as CAD,
we intend to seek commercial collaborations with companies that
have a large, dedicated sales force focused on general
practitioners and cardiologists and we plan to seek
commercialization partners for products in non-specialty and
international markets.
Competition
Our industry is highly competitive and subject to rapid and
significant technological change. Our potential competitors
include large pharmaceutical and biotechnology companies,
specialty pharmaceutical and generic drug companies, academic
institutions, government agencies and research institutions. We
believe that key competitive factors that will affect the
development and commercial success of our product candidates are
efficacy, safety and tolerability profile, reliability,
convenience of dosing, price and reimbursement.
Many of our potential competitors, including many of the
organizations named below, have substantially greater financial,
technical and human resources than we do and significantly
greater experience in the discovery and development of product
candidates, obtaining FDA and other regulatory approvals of
products and the commercialization of those products.
Accordingly, our competitors may be more successful than we may
be in obtaining FDA approval for drugs and achieving widespread
market acceptance. Our competitors’ drugs may be more
effective, or more effectively marketed and sold, than any drug
we may commercialize and may render our product candidates
obsolete or non-competitive before we can recover the expenses
of developing and commercializing any of our product candidates.
We anticipate that we will face intense and increasing
competition as new drugs enter the market and advanced
technologies become available. Finally, the development of new
treatment methods for the diseases we are targeting could render
our drugs non-competitive or obsolete.
The
sPLA2
product candidates we are currently developing, if approved,
will face intense competition, either as monotherapies or in
combination therapies. Although there are no
sPLA2
inhibitors currently approved by the FDA, we are aware of other
pharmaceutical companies, as described below that are developing
product candidates in this area for separate indications.
sPLA2
in Acute Coronary Syndrome
Our lead product candidate, varespladib, for the short-term
(16-week) treatment of acute coronary syndrome has a dual
mechanism of action that we believe confers anti-inflammatory
and lipid-lowering and lipid-modulating benefits. The market for
cardiovascular therapeutics and acute coronary syndrome,
specifically, is especially large and competitive. A wide range
of medications are typically administered to patients suffering
an acute coronary syndrome event in order to reduce ischemia and
thrombosis and improve blood flow. We expect that varespladib
for the treatment of acute coronary syndrome patients, if
approved, may compete with the following anti-inflammatory
therapeutics in development.
22
| |
|
|
|
|
|
|
|
|
|
Compound
|
|
Stage
|
|
Company
|
|
Indications
|
|
Notes
|
|
|
|
Darapladib
|
|
Phase 3
|
|
GlaxoSmithKline plc
|
|
Acute coronary syndrome
|
|
• Lp-PLA2
Inhibitor
• Collaboration with Human Genome
Sciences, Inc.
• Various back-up compounds
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
VIA-2291
|
|
Phase 2
|
|
Via Pharmaceuticals, Inc.
|
|
Acute coronary syndrome or atherosclerosis
|
|
• 5-lipoxygenase inhibitor
• Discussions on-going with FDA
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E-5555
|
|
Phase 2
|
|
Eisai Inc.
|
|
Acute coronary syndrome or atherosclerosis
|
|
• 600 patient study completed
October 2009
• Thrombin receptor antagonist
• Evaluating biomarkers and events
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
VT-111(a)
|
|
Phase 2b
|
|
Viron
|
|
Acute coronary syndrome
|
|
• SERPINS family glycoprotein
• Inhibits monocyte migration during
vascular inflammation
• Phase 2a completed
|
Other
Agents Under Development
Additionally, we are aware of other products in development that
are being tested for anti-inflammatory benefits in patients with
acute coronary syndrome such as Via Pharmaceuticals, Inc. and
its 5-lipoxygenase, or
5-LO,
inhibitor, which has been evaluated in Phase 2 clinical studies,
GlaxoSmithKline plc and its product candidate, darapladib, which
is an
Lp-PLA2
inhibitor currently being evaluated in Phase 3 clinical studies.
If approved, these products or others in development may compete
directly with varespladib.
Approved
Categories of Drugs
Statins — Treatment with varespladib is
designed to offer anti-inflammatory benefits for acute coronary
syndrome patients that are additive to treatment with statins.
However, statin therapy is thought to confer some element of
anti-inflammatory benefit as monotherapy. In certain
circumstances, it is possible the anti-inflammatory benefits of
statin monotherapy with products such as Lipitor (atorvastatin),
which is marketed by Pfizer Inc., Crestor (rosuvastatin), which
is marketed by AstraZeneca UK Limited and Zocor (simvastatin),
which is marketed by Merck & Co., Inc. may be viewed
as competitive to that offered by varespladib.
Other Lipid-Lowering
Therapies — Increasingly, additional
lipid-lowering agents are being administered either in
combination with statins or as monotherapy to help acute
coronary syndrome patients reduce levels of LDL-C. Varespladib
has demonstrated LDL-C lowering benefits when tested as
monotherapy and in combination with statin therapy. To the
extent acute coronary syndrome patients need additional LDL-C
lowering, varespladib may compete for use with other approved
agents such as Vytorin, which is a fixed dose combination
therapy combining ezetimibe and Zocor, Tricor (fenofibrate
tablets) and Niaspan (niacin), both of which are marketed by
Abbott Laboratories, Zetia (ezetimibe) and fish oils (omega-3).
Lupus
Human Genome Sciences, Inc.’s and partner GlaxoSmithKline
plc’s Benlysta is currently under review with the FDA and,
if approved, would be the first novel therapy in the last fifty
years. Current therapies such as non-steroidal anti-inflammatory
drugs, or NSAIDs, corticosteroids and immunosuppressants
generally act to hold back broadly the proliferation of many
types of cells, including white blood cells. However, use of
these agents is associated with significant adverse events and
broad immune suppression.
23
Several new biological agents under development have targeted
BAFF (or BLyS) and other B-cell related pathways for the
treatment of lupus. These product candidates include Benlysta
(belimumab) from Human Genome Sciences, Inc., LY2127399 from Eli
Lilly and Company, atacicept, or TACI-Ig, from ZymoGenetics Inc.
and epratuzumab from Immunomedics, Inc., as well as others
acting via non
B-cell
mechanisms, such as Lupuzor from Cephalon. We believe that
A-623 may
offer potential differentiation from these agents, including
demonstrated dosing flexibility with both subcutaneous and
intravenous delivery; selective modulation and reduction of
relevant B-cell types in lupus patients; the ability to inhibit
the activity of both membrane-bound and soluble BAFF; its
smaller size as compared to a full antibody, which may confer
differentiating pharmacokinetic and pharmacodynamic
characteristics; and distinct patent protection based on a novel
and proprietary technology developed and commercialized by
Amgen, which may also confer potential manufacturing advantages
with lower cost of goods based on a bacterial fermentation
manufacturing process.
| |
|
|
|
|
|
|
|
|
|
Compound
|
|
Stage
|
|
Company
|
|
Indications
|
|
Notes
|
|
|
Benlysta
(intravenous and
subcutaneous)
|
|
BLA Submission
(PDUFA in
March 2011)
|
|
Human Genome Sciences, Inc./
GlaxoSmithKline plc
|
|
Lupus
|
|
• Monoclonal antibody against BAFF, an
agent that demonstrated partial reduction in B-cells
• Inhibits soluble BAFF only
• Positive results reported in two Phase 3
clinical studies
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LY2127399
(subcutaneous)
|
|
Phase 3
|
|
Eli Lilly and Company
|
|
Lupus,
Rheumatoid
Arthritis,
Multiple
Myelomas
|
|
• Monoclonal antibody against BAFF
inhibits soluble and membrane-bound BAFF
• Recent positive results in RA study
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Atacicept
(intravenous)
|
|
Phase 3
|
|
ZymoGenetics Inc./Merck
Serono S.A.
|
|
Lupus
|
|
• Fusion protein against BAFF and APRIL;
Phase 3 clinical study in lupus on-going
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Epratuzumab
(intravenous)
|
|
Phase 3
|
|
Immunomedics, Inc./UCB S.A.
|
|
Lupus, Non-Hodgkin’s
Lymphoma
|
|
• Humanized antibody against
CD-22, an
agent that specifically targets
B-cells and
leads to partial depletion of peripheral B-cells
• Initiating Phase 3 clinical study
|
|
|
|
|
|
Lupuzor
(subcutaneous)
|
|
Phase 3
|
|
Cephalon, Inc./
ImmuPharma
PLC
|
|
Lupus
|
|
• Modulates CD 4 T cells
• Positive Phase 2b clinical study results reported
|
sPLA2
for Acute Chest Syndrome Associated with Sickle Cell
Disease
There are no currently approved agents for treatment or
prophylaxis of acute chest syndrome associated with sickle cell
disease. Droxia (hydroxyurea) is approved for prevention of VOC
in sickle cell disease and thus could reduce the pool of
patients with VOC at risk for acute chest syndrome. In addition,
there is evidence in the literature that blood transfusions may
prevent the occurrence of acute chest syndrome associated with
sickle cell disease, and a randomized clinical study is underway
by the National Heart, Lung and Blood Institute to explore this
possibility.
Intellectual
Property
Our policy is to pursue, maintain and defend patent rights,
developed internally and licensed from third parties, to protect
the technology, inventions and improvements that are
commercially important to the development of our business. We
also rely on trade secrets that may be important to the
development of our business.
Our success will depend significantly on our ability to:
|
|
|
| |
•
|
obtain and maintain patent and other proprietary protection for
the technology, inventions and improvements we consider
important to our business;
|
| |
| |
•
|
defend our patents;
|
| |
| |
•
|
preserve the confidentiality of our trade secrets; and
|
24
|
|
|
| |
•
|
operate our business without infringing the patents and
proprietary rights of third parties.
|
Varespladib
and
A-001
As of the date of this report, our licensed varespladib and
A-001 patent
portfolio includes:
|
|
|
| |
•
|
13 U.S. patents;
|
| |
| |
•
|
One pending U.S. non-provisional patent application;
|
| |
| |
•
|
Five European, or EP, patents, each validated in one or more of
Austria, Belgium, Denmark, France, Germany, Greece, Ireland,
Italy, Liechtenstein, Luxembourg, the Netherlands, Portugal,
Spain, Sweden, Switzerland and the United Kingdom;
|
| |
| |
•
|
One pending EP patent application;
|
| |
| |
•
|
20 non-EP foreign patents in Argentina, Australia, Brazil,
Canada, China, Finland, India, Malaysia, Mexico, the
Philippines, South Korea, Taiwan and Turkey; and
|
| |
| |
•
|
Three pending non-EP foreign patent applications in Brazil,
Japan and Thailand.
|
We hold exclusive worldwide licenses from Eli Lilly and
Shionogi & Co., Ltd. to all of these patents and
patent applications with the exception of licensing rights in
Japan, which Shionogi & Co., Ltd. retains. These
licenses are described below under
“— Licenses.” The patents and applications
described above contain claims directed to varespladib and
A-001
compositions of matter and to various methods of making and
using varespladib and
A-001,
including methods of treating various inflammatory conditions.
The U.S. patents are currently scheduled to expire between
2014 and 2021. The primary U.S. composition of matter
patent for varespladib and
A-00l
currently expires in August 2014. This patent is expected to be
eligible for a Hatch-Waxman term restoration of up to five
years, which could extend the expiration date to August 2019. We
intend to pursue pediatric exclusivity as well, which could add
an additional six months to the patent term. The primary
European composition of matter patent currently expires in March
2015. This patent is expected to be eligible for a Supplementary
Protection Certificate of up to five years, which could extend
the expiration date to March 2020.
As of the date of this report, our internally developed
varespladib and
A-001 patent
portfolio includes:
|
|
|
| |
•
|
Four pending U.S. non-provisional patent applications;
|
| |
| |
•
|
Two pending U.S. provisional patent applications;
|
| |
| |
•
|
Two pending Patent Cooperation Treaty, or PCT, patent
applications; and
|
| |
| |
•
|
National phase applications in the European Patent Office, the
Eurasian Patent Organization and 17 other countries (Australia,
Brazil, Canada, China, Hong Kong, India, Indonesia, Israel,
Japan, Malaysia, Mexico, New Zealand, The Philippines,
Singapore, South Africa, South Korea and Vietnam).
|
We own, and therefore hold all worldwide rights in and to, these
patent applications, which contain claims directed to
varespladib and
A-001
compositions of matter and methods of treating various
cardiovascular indications.
Several of the pending U.S. and
non-U.S.
patent applications include disclosure relating to the
combination of varespladib and
A-001 with
various cardiovascular drugs, including statins. Pending claims
in these applications are directed to both compositions of
matter and methods. Any patents issuing from these applications
would expire between 2028 and 2030.
A-003
As of the date of this report, our licensed
A-003 patent
portfolio includes:
|
|
|
| |
•
|
Two U.S. patents;
|
| |
| |
•
|
One licensed pending U.S. non-provisional patent
application (also listed above as covering varespladib and
A-001);
|
| |
| |
•
|
Five EP patents (two also listed above as covering varespladib
and A-001),
each validated in one or more of Albania, Austria, Belgium,
Denmark, Finland, France, Germany, Greece, Ireland, Italy,
Latvia, Liechtenstein, Lithuania, Luxembourg, the Netherlands,
Portugal, Romania, Slovenia, Spain, Sweden, Switzerland and the
United Kingdom;
|
25
|
|
|
| |
•
|
15 non-EP foreign patents (six also listed above as covering
varespladib and
A-001) in
Argentina, Australia, Canada, China, India, Mexico, South Korea
and Taiwan; and
|
| |
| |
•
|
One pending non-EP foreign patent application in Brazil (also
listed above as covering varespladib and
A-001).
|
We hold exclusive worldwide licenses from Eli Lilly and
Shionogi & Co., Ltd. to these patents and patent
applications with the exception of licensing rights in Japan,
which Shionogi & Co., Ltd. retains. These licenses are
described below under “— Licenses.” The
patents and applications listed above contain claims directed to
A-003
compositions of matter and to various methods of making and
using A-003,
including methods of treating various inflammatory indications.
The issued U.S. patents are currently scheduled to expire
between 2017 and 2018.
As of the date of this report, our internally developed
A-003 patent
portfolio includes:
|
|
|
| |
•
|
Three U.S. non-provisional patent applications (all also
listed above as covering varespladib and
A-001);
|
| |
| |
•
|
Two pending U.S. provisional patent applications (both also
listed above as covering varespladib and
A-001);
|
| |
| |
•
|
Two pending PCT patent applications (both also listed above as
covering varespladib and
A-001); and
|
| |
| |
•
|
National phase applications in the European Patent Office, the
Eurasian Patent Organization and 17 other countries (Australia,
Brazil, Canada, China, Hong Kong, India, Indonesia, Israel,
Japan, Malaysia, Mexico, New Zealand, The Philippines,
Singapore, South Africa, South Korea and Vietnam).
|
We own, and therefore hold all worldwide rights in and to, these
patent applications, which contain claims directed to
A-003
compositions of matter and methods of treating various
cardiovascular indications.
New
sPLA2
Compounds
As of the date of this report, our new
sPLA2
compound patent portfolio includes over 30 licensed
U.S. patents, one pending U.S. non-provisional patent
application, three EP patents, one pending EP patent
application, four non-EP foreign patents, and one pending non-EP
foreign patent application not listed above as covering
A-001,
varespladib or
A-003. We
hold exclusive worldwide licenses from Eli Lilly and
Shionogi & Co., Ltd. to these patents and patent
applications with the exception of licensing rights in Japan,
which Shionogi & Co., Ltd. retains. These licenses are
described below under “— Licenses.” The
patents and applications listed above contain claims directed to
various
sPLA2
second generation compounds, as well as methods of making and
using these new
sPLA2
compounds. The issued U.S. patents are currently scheduled
to expire between 2013 and 2024.
A-623
As of the date of this report, our
A-623 patent
portfolio includes:
|
|
|
| |
•
|
Two U.S. patents;
|
| |
| |
•
|
One pending U.S. non-provisional patent application;
|
| |
| |
•
|
One EP patent validated in Albania, Austria, Belgium, Cyprus,
Denmark, Finland, France, Germany, Greece, Ireland, Italy,
Latvia, Liechtenstein, Lithuania, Luxembourg, Monaco, the
Netherlands, Portugal, Romania, Slovenia, Spain, Sweden,
Switzerland, Turkey and the United Kingdom;
|
| |
| |
•
|
Two pending EP patent applications;
|
| |
| |
•
|
Eleven non-EP foreign patents in Australia, China, Estonia,
Eurasia (validated in all nine Eurasian countries), Japan, New
Zealand, the Philippines, Singapore, South Korea and South
Africa; and
|
| |
| |
•
|
13 pending non-EP foreign patent applications in Brazil,
Bulgaria, Canada, China, the Czech Republic, Hong Kong, Hungary,
Israel, Mexico, Norway, Poland, Serbia and Slovakia.
|
We hold exclusive worldwide licenses from Amgen to all of these
patents and patent applications. In addition, we hold a
non-exclusive worldwide license to one pending
U.S. non-provisional patent application, one EP patent, one
pending EP patent application, ten non-EP foreign patents, and
over 30 pending non-EP foreign patent applications relating to
general peptibody compositions and formulations.
The U.S. patents are currently scheduled to expire in May
2022. One of the U.S. patents is expected to be eligible
for a Hatch-Waxman term restoration of up to five years, which
could extend the expiration date to May
26
2027. We intend to pursue pediatric exclusivity as well, which
could add an additional six months to the patent term. The
European patent is currently scheduled to expire May 2022. This
patent is expected to be eligible for a Supplementary Protection
Certificate of up to five years, which could extend the
expiration date to May 2027.
The U.S. patent system permits the filing of provisional
and non-provisional patent applications. A non-provisional
patent application is examined by the U.S. Patent Office,
or USPTO, and can mature into a patent once the USPTO determines
that the claimed invention meets the standards for
patentability. A provisional patent application is not examined,
and automatically expires 12 months after its filing date.
As a result, a provisional patent application cannot mature into
a patent. The requirements for filing a provisional patent
application are not as strict as those for filing a
non-provisional patent application. Provisional applications are
often used, among other things, to establish an early filing
date for a subsequent non-provisional patent application.
The filing date of a non-provisional patent application is used
by the USPTO to determine what information is prior art when it
considers the patentability of a claimed invention. If certain
requirements are satisfied, a non-provisional patent application
can claim the benefit of the filing date of an earlier filed
provisional patent application. As a result, the filing date
accorded by the provisional patent application may remove
information that otherwise could preclude the patentability of
an invention.
Depending upon the timing, duration and specifics of FDA
approval of varespladib,
A-623,
A-001,
A-003 or one
or more new
sPLA2
compounds, one or more of the U.S. patents listed above may
be eligible for limited patent term restoration under the Drug
Price Competition and Patent Term Restoration Act of 1984,
commonly referred to as the Hatch-Waxman Act. See
“— Regulatory Matters — Patent Term
Restoration and Marketing Exclusivity.”
A-101
A-101 is
novel compound developed to treat cardiovascular disease.
As of the date of this report, our internally developed
A-101 patent
portfolio includes:
|
|
|
| |
•
|
One pending U.S. non-provisional patent application.
|
We own, and therefore hold all worldwide rights in and to, the
patent application, which contains claims directed to
A-101
compositions of matter and methods of treating various
cardiovascular indications.
Licenses
Eli Lilly
and Shionogi & Co., Ltd.
In July 2006, we entered into a license agreement with Eli Lilly
and Shionogi & Co., Ltd., pursuant to which we
obtained an exclusive license in all countries except for Japan
to certain technology and compounds relating to
sPLA2
inhibitors. The licensed technology was largely developed under
a research and development agreement between Eli Lilly and
Shionogi & Co., Ltd., which was entered into between
the parties in August 1992 and terminated in December 2004.
Under the agreement, we obtained exclusive rights to
(i) use licensed patent rights and know-how to identify and
develop
sPLA2
inhibitors, (ii) develop, make, have made, use, import,
offer for sale and sell licensed compounds and pharmaceutical
formulations thereof, including varespladib,
A-001,
A-003 and
other
sPLA2
inhibitors and (iii) grant sublicenses. The licensed patent
rights include a specific set of previously filed U.S. and
foreign patents and applications, as well as any applications
filed after the execution date by Eli Lilly or
Shionogi & Co., Ltd. that relate to licensed know-how.
Certain patents and applications within the licensed patent
rights are defined as “core patents.” Although the
agreement does not allow us to sell or offer for sale licensed
products in Japan, it does allow us to conduct preclinical and
clinical studies in Japan in support of applications for
marketing authorization outside of Japan, and to make and have
made licensed products in Japan for use or sale outside of
Japan. Eli Lilly and Shionogi & Co., Ltd. retain the
right to use licensed products for research purposes only. Eli
Lilly also retains the right to conduct studies of specific
compounds in animals for research purposes, but only with our
prior written approval. In addition, Shionogi & Co.,
Ltd. retains the non-exclusive right to make and have made
licensed products for supply to us, as well as its rights to
continue research, development and marketing of licensed
technology in Japan.
Upon entering into the license agreement, we assumed control of
all prosecution and maintenance of core patents prosecuted and
maintained by Eli Lilly prior to the agreement. All core patents
prosecuted and maintained
27
by Shionogi & Co., Ltd. prior to the agreement
remained under the control of Shionogi & Co., Ltd.
Licensed patent rights that were not classified as core remained
under the control of Eli Lilly and Shionogi & Co.,
Ltd. However, control of certain of these patents and
applications has since been transferred to us following the
decision by Eli Lilly or Shionogi & Co., Ltd. to
discontinue prosecution and maintenance.
Upon entering into the license agreement, we made one-time
payments of cash in the amount of $250,000 and issued shares of
convertible preferred stock with a total aggregate value of
$2.3 million to Eli Lilly and Shionogi & Co.,
Ltd. We are required to make various milestone payments and to
pay tiered royalty payments on net sales, which increase as a
percentage as net sales increase. Both the milestone and royalty
payment schedules vary depending on the specific formulation
(e.g., oral versus intravenously administered). For varespladib,
we are required to pay up to $32.0 million upon achievement
of certain approval and post-approval sales milestones. For
A-001, we
are required to pay up to $3.0 million upon achievement of
certain clinical development milestones and up to
$25.0 million upon achievement of certain approval and
post-approval sales milestones. For other product formulations
that we are not currently developing, we would be required to
pay up to $2.0 million upon achievement of certain clinical
development milestones and up to $35.5 million upon
achievement of certain approval and post-approval sales
milestones. Our royalty payments vary based upon type of
formulation and annual net sales, but generally range from the
mid-single digits to the low double digits. Our royalty payment
obligations for a particular licensed product in a particular
country begin on the date of the first commercial sale of the
licensed product in that country, and end upon the later of
10 years from the date of first commercial sale in that
country or the first date on which a generic version of the
licensed product reaches a 25% total market share in that
country.
The license agreement will remain in effect for the length of
our royalty obligation on a
product-by-product
and
country-by-country
basis, unless we elect to terminate earlier or until termination
by mutual agreement. Upon expiration of the agreement, our
license will remain in effect and will convert to an
irrevocable, perpetual royalty-free license. If we fail to meet
our obligations under the agreement, Eli Lilly or
Shionogi & Co., Ltd. can terminate the agreement,
resulting in a loss of our exclusive rights to the licensed
technology.
Amgen
In December 2007, we entered into a license agreement with
Amgen, which was amended in October 2009, pursuant to which we
obtained an exclusive worldwide license to certain technology
and compounds relating to
A-623, as
well as a non-exclusive worldwide license to technology relating
to certain peptibody compositions of matter and formulations.
Under the agreement, we obtained exclusive rights under the
licensed patents and know-how to research, develop, make, have
made, use, sell, offer for sale and import pharmaceutical
products containing
A-623, as
well as the right to grant sublicenses. The licensed patents
included a specific set of previously filed U.S. and
foreign patents and applications, as well as any applications
filed after the execution date by Amgen and covering licensed
know-how. During the period of the agreement, we are responsible
for the filing, prosecution, defense and maintenance of all
exclusively licensed
A-623
patents and applications. Amgen retains the right to review all
documents relating to said filing, prosecution, defense and
maintenance, and we are required to incorporate all reasonable
comments or suggestions that Amgen makes with regard to these
documents.
During the seven-year period after execution of the agreement,
Amgen is prohibited from clinically developing or
commercializing any BAFF peptibody. Similarly, we are prohibited
during the term of the agreement from clinically developing or
commercializing any molecule other than
A-623 that
modulates BAFF as the primary intended therapeutic mechanism of
action.
We paid a first installment fee of $3.0 million and a
second installment fee of $3.0 million. In addition, we are
required to make various milestone payments upon the achievement
of certain development, regulatory and commercial objectives,
including payment upon initiation of the first Phase 3 clinical
study for any
A-623
formulation. We are also required to pay up to
$10.0 million upon achievement of certain pre-approval
clinical development milestones and up to $23.0 million
upon achievement of certain post- approval milestones.
Furthermore, we are required to make tiered quarterly royalty
payments on net sales, which increase as a percentage from the
high single digits to the low double digits as net sales
increase. Our royalty payment obligations for a particular
product in a particular country begin on the date of the first
commercial sale of the licensed product in that country,
28
and end upon the later of 10 years from the date of first
commercial sale in that country or the expiration date of the
last valid claim of a licensed patent that covers the
manufacture, use or sale, offer to sell or import of the product.
The license agreement will remain in effect until we elect to
terminate, or until termination for material breach by either
party or insolvency on our part. Under these terms, Amgen can
terminate the agreement if we fail to meet our obligations,
resulting in a loss of our exclusive rights to the licensed
technology.
Manufacturing
and Supply
We currently rely on contract manufacturers to produce drug
substances and drug products required for our clinical studies
under cGMP with oversight by our internal managers. We plan to
continue to rely upon contract manufacturers and, potentially,
collaboration partners to manufacture commercial quantities of
our product candidates if and when approved for marketing by the
FDA. Should a supplier or a manufacturer on which we have relied
to produce a product candidate provide us with a faulty product
or such product is later recalled, we would likely experience
significant delays and material additional costs.
Sales and
Marketing
Given our stage of development, we have not developed a
commercial organization or distribution capabilities. We expect
that we would develop these capabilities once we receive Phase 3
data in contemplation of FDA approval and the commercial launch
of our product candidates. In order to commercialize any of our
product candidates, we must develop these capabilities
internally or through collaboration with third parties. In
selected therapeutic areas where we feel that any approved
products can be commercialized by a specialty sales force that
calls on a limited and focused group of physicians, acute care
and orphan indications such as acute coronary syndrome and acute
chest syndrome associated with sickle cell disease, we may seek
to commercialize these product candidates alone. In therapeutic
areas that require a large sales force selling to a large and
diverse prescribing population, such as chronic indications such
as CAD, we currently plan to partner with third parties to
commercialize our product candidates while retaining rights to
co-promote our products to a select audience of high prescribing
physicians in the United States, thereby supplementing or
enhancing the efforts of a commercial partner. We also plan to
seek commercialization partners for products in non-specialty
and international markets.
In North America and Western Europe, patients in the target
markets for our product candidates are largely managed by
medical specialists in the areas of cardiology and internal
medicine. Historically, companies have experienced substantial
commercial success through the deployment of specialized sales
forces that can address a majority of key prescribers,
particularly within the cardiovascular disease marketplace.
Therefore, we expect to utilize a specialized sales force in
North America for the sales and marketing of product candidates
that we may successfully develop. Based upon sales models, we
estimate that we could effectively promote (supplementing a
commercial partner’s sales efforts) the treatment of acute
coronary syndrome to 3,000 cardiologists with approximately 300
sales representatives in North America and Western Europe. If we
obtain additional label indications for varespladib or
A-001, we
may choose to increase our sales force size to promote these new
uses. Due to their concentrated and focused nature, specialty
target audiences may be reached with more focused and
cost-effective marketing campaigns. Outside of North America,
and in situations or markets where a more favorable return may
be realized through licensing commercial rights to a third
party, we may license a portion or all of our commercial rights
in a territory to a third party in exchange for one or more of
the following: up-front payments, research funding, development
funding, milestone payments and royalties on drug sales.
We intend to build the commercial infrastructure necessary to
bring varespladib,
A-623 and
A-001 to
market alone or in collaboration with a co-development or
co-promotion partner. In addition to a specialty sales force,
sales management, internal sales support and an internal
marketing group, we will need to establish capabilities to
manage key accounts, such as managed care organizations,
group-purchasing organizations, specialty pharmacies and
government accounts. We may also choose to employ medical sales
liaisons personnel to support the product.
Regulatory
Matters
Government
Regulation and Product Approval
Government authorities in the United States at the federal,
state and local level and other countries, extensively regulate,
among other things, the research, development, testing,
manufacture, quality control, approval, labeling, packaging,
storage, record-keeping, promotion, advertising, distribution,
marketing, export and import of products
29
such as those we are developing. Our product candidates must be
approved by the FDA through the new drug application, or NDA,
process, and our biological product candidate,
A-623, must
be approved by the FDA through the biologics license
application, or BLA, process before they may legally be marketed
in the United States.
United
States Drug Development Process
In the United States, the FDA regulates drugs under the Federal
Food, Drug, and Cosmetic Act, or FDCA, and implementing
regulations and biological products under both the FDCA and the
Public Health Service Act, or the PHSA, and implementing
regulations. The process of obtaining regulatory approvals and
compliance with appropriate federal, state, local and foreign
statutes and regulations require the expenditure of substantial
time and financial resources. Failure to comply with the
applicable U.S. requirements at any time during the product
development process, approval process, or after approval, may
subject an applicant to administrative or judicial sanctions.
These sanctions could include the FDA’s refusal to approve
pending applications, withdrawal of an approval, a clinical
hold, warning letters, product recalls, product seizures, total
or partial suspension of production or distribution,
injunctions, fines, refusals of government contracts,
restitution, disgorgement or civil or criminal penalties. The
process required by the FDA before a drug or biological product
may be marketed in the United States generally involves the
following:
|
|
|
| |
•
|
completion of preclinical laboratory tests, animal studies and
formulation studies according to Good Laboratory Practices
regulations;
|
| |
| |
•
|
submission to the FDA of an IND, which must become effective
before human clinical studies may begin;
|
| |
| |
•
|
performance of adequate and well-controlled human clinical
studies according to Good Clinical Practices, or GCP, to
establish the safety and efficacy of the proposed drug or
biological product for its intended use;
|
| |
| |
•
|
submission to the FDA of an NDA for a new drug or BLA for a
biological product;
|
| |
| |
•
|
satisfactory completion of an FDA inspection of the
manufacturing facility or facilities at which the drug or
biological product is produced to assess compliance with
cGMP; and
|
| |
| |
•
|
FDA review and approval of the NDA or BLA.
|
The testing and approval process requires substantial time,
effort and financial resources and we cannot be certain that any
approvals for our product candidates will be granted on a timely
basis, if at all.
Once a pharmaceutical or biological product candidate is
identified for development, it enters the preclinical testing
stage. Preclinical tests include laboratory evaluations of
product chemistry, toxicity, formulation and stability, as well
as animal studies to assess its potential safety and efficacy.
An IND sponsor must submit the results of the preclinical tests,
together with manufacturing information, analytical data and any
available clinical data or literature, to the FDA as part of the
IND. The sponsor will also include a protocol detailing, among
other things, the objectives of the initial clinical study, the
parameters to be used in monitoring safety and the effectiveness
criteria to be evaluated if the initial clinical study lends
itself to an efficacy evaluation. Some preclinical testing may
continue even after the IND is submitted. The IND automatically
becomes effective 30 days after receipt by the FDA, unless
the FDA places the clinical study on a clinical hold within that
30-day time
period. In such a case, the IND sponsor and the FDA must resolve
any outstanding concerns before the clinical study can begin.
Clinical holds may also be imposed by the FDA at any time before
or during clinical studies due to safety concerns or
non-compliance.
All clinical studies must be conducted under the supervision of
one or more qualified investigators in accordance with GCP
regulations. These regulations include the requirement that all
research subjects provide informed consent. Further, an
institutional review board, or IRB, must review and approve the
plan for any clinical study before it commences at any
institution. An IRB considers, among other things, whether the
risks to individuals participating in the studies are minimized
and are reasonable in relation to anticipated benefits. The IRB
also approves the information regarding the clinical study and
the consent form that must be provided to each clinical study
subject or to his or her legal representative and must monitor
the clinical study until completed.
Each new clinical protocol and any amendments to the protocol
must be submitted to the FDA for review, and to the IRBs for
approval. Protocols detail, among other things, the objectives
of the clinical study, dosing procedures, subject selection and
exclusion criteria, and the parameters to be used to monitor
subject safety.
30
Human clinical studies are typically conducted in three
sequential phases that may overlap or be combined:
|
|
|
| |
•
|
Phase 1. The product is initially introduced
into healthy human subjects and tested for safety, dosage
tolerance, absorption, metabolism, distribution and excretion.
In the case of some products for severe or life-threatening
diseases, especially when the product may be too inherently
toxic to ethically administer to healthy volunteers, the initial
human testing is often conducted in patients.
|
| |
| |
•
|
Phase 2. Involves studies in a limited patient
population to identify possible adverse effects and safety risks
to preliminarily evaluate the efficacy of the product for
specific targeted diseases and to determine dosage tolerance and
optimal dosage and schedule.
|
| |
| |
•
|
Phase 3. Clinical studies are undertaken to
further evaluate dosage, clinical efficacy and safety in an
expanded patient population at geographically dispersed clinical
study sites. These studies are intended to establish the overall
risk/benefit ratio of the product and provide an adequate basis
for product labeling.
|
Progress reports detailing the results of the clinical studies
must be submitted at least annually to the FDA and safety
reports must be submitted to the FDA and the investigators for
serious and unexpected adverse events. Phase 1, Phase 2 and
Phase 3 testing may not be completed successfully within any
specified period, if at all. The FDA or the sponsor may suspend
or terminate a clinical study at any time on various grounds,
including a finding that the research subjects or patients are
being exposed to an unacceptable health risk. Similarly, an IRB
can suspend or terminate approval of a clinical study at its
institution if the clinical study is not being conducted in
accordance with the IRB’s requirements or if the drug or
biological product has been associated with unexpected serious
harm to patients.
Concurrent with clinical studies, companies usually complete
additional animal studies and must also develop additional
information about the chemistry and physical characteristics of
the product and finalize a process for manufacturing the product
in commercial quantities in accordance with cGMP requirements.
The manufacturing process must be capable of consistently
producing quality batches of the product candidate and, among
other things, the manufacturer must develop methods for testing
the identity, strength, quality and purity of the final product.
Additionally, appropriate packaging must be selected and tested
and stability studies must be conducted to demonstrate that the
product candidate does not undergo unacceptable deterioration
over its shelf life.
U.S.
Review and Approval Processes
The results of product development, preclinical studies and
clinical studies, along with descriptions of the manufacturing
process, analytical tests conducted on the drug or biological
product, proposed labeling and other relevant information, are
submitted to the FDA as part of an NDA for a new drug or BLA for
a biological product, requesting approval to market the product.
The submission of an NDA or BLA is subject to the payment of a
substantial user fee; a waiver of such fee may be obtained under
certain limited circumstances.
In addition, under the Pediatric Research Equity Act of 2003, or
PREA, which was reauthorized under the Food and Drug
Administration Amendments Act of 2007, an NDA or BLA or
supplement to an NDA or BLA must contain data to assess the
safety and effectiveness of the drug or biological product for
the claimed indications in all relevant pediatric subpopulations
and to support dosing and administration for each pediatric
subpopulation for which the product is safe and effective. The
FDA may grant deferrals for submission of data or full or
partial waivers. Unless otherwise required by regulation, PREA
does not apply to any drug or biological product for an
indication for which orphan designation has been granted.
The FDA reviews all NDAs and BLAs submitted to ensure that they
are sufficiently complete for substantive review before it
accepts them for filing. The FDA may request additional
information rather than accept a NDA or BLA for filing. In this
event, the NDA or BLA must be re-submitted with the additional
information. The
re-submitted
application is also subject to review before the FDA accepts it
for filing. Once the submission is accepted for filing, the FDA
begins an in-depth substantive review. The FDA reviews an NDA to
determine, among other things, whether a product is safe and
effective for its intended use and whether its manufacturing is
cGMP-compliant
to assure and preserve the product’s identity, strength,
quality and purity. The FDA reviews a BLA to determine, among
other things, whether the product is safe, has an acceptable
purity profile and is adequately potent, and whether its
manufacturing meets standards designed to assure the
product’s continued identity, sterility, safety, purity and
potency. Before approving an NDA or BLA, the FDA will inspect
the facility or facilities where the product is manufactured.
The FDA will not approve an application unless it determines
that the manufacturing
31
processes and facilities are in compliance with cGMP
requirements and adequate to assure consistent production of the
product within required specifications. The FDA may refer the
NDA or BLA to an advisory committee for review, evaluation and
recommendation as to whether the application should be approved
and under what conditions. An advisory committee is a panel of
experts who provide advice and recommendations when requested by
the FDA on matters of importance that come before the agency.
The FDA is not bound by the recommendation of an advisory
committee but it generally follows such recommendations.
The approval process is lengthy and difficult and the FDA may
refuse to approve an NDA or BLA if the applicable regulatory
criteria are not satisfied or may require additional clinical
data or other data and information. Even if such data and
information is submitted, the FDA may ultimately decide that the
NDA or BLA does not satisfy the criteria for approval. Data
obtained from clinical studies are not always conclusive and the
FDA may interpret data differently than we interpret the same
data. The FDA will issue a complete response letter if the
agency decides not to approve the NDA or BLA in its present
form. The complete response letter usually describes all of the
specific deficiencies in the NDA or BLA identified by the FDA.
The deficiencies identified may be minor, for example, requiring
labeling changes, or major, for example, requiring additional
clinical studies. Additionally, the complete response letter may
include recommended actions that the applicant might take to
place the application in a condition for approval. If a complete
response letter is issued, the applicant may either resubmit the
NDA or BLA, addressing all of the deficiencies identified in the
letter, or withdraw the application
If a product receives regulatory approval, the approval may be
significantly limited to specific diseases and dosages or the
indications for use may otherwise be limited, which could
restrict the commercial value of the product. Further, the FDA
may require that certain contraindications, warnings or
precautions be included in the product labeling. In addition,
the FDA may require Phase 4 testing which involves clinical
studies designed to further assess a drug or biological
product’s safety and effectiveness after NDA or BLA
approval and may require testing and surveillance programs to
monitor the safety of approved products that have been
commercialized.
Patent
Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA
approval of the use of our product candidates, some of our
U.S. patents may be eligible for limited patent term
extension under the Drug Price Competition and Patent Term
Restoration Act of 1984, commonly referred to as the
Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a
patent restoration term of up to five years as compensation for
patent term lost during the FDA regulatory review process.
However, patent term restoration cannot extend the remaining
term of a patent beyond a total of 14 years from the
product’s approval date. The patent term restoration period
is generally one-half the time between the effective date of an
IND and the submission date of an NDA plus the time between the
submission date of an NDA and the approval of that application.
Only one patent applicable to an approved drug is eligible for
the extension and the application for the extension must be
submitted prior to the expiration of the patent. The USPTO, in
consultation with the FDA, reviews and approves the application
for any patent term extension or restoration. In the future, we
intend to apply for restorations of patent terms for some of our
currently owned or licensed patents to add patent life beyond
their current expiration dates, depending on the expected length
of the clinical studies and other factors involved in the filing
of the relevant NDA.
Market exclusivity provisions under the FDCA can also delay the
submission or the approval of certain competitor applications.
The FDCA provides a five-year period of non-patent marketing
exclusivity within the United States to the first applicant to
gain approval of an NDA for a new chemical entity. A drug is a
new chemical entity if the FDA has not previously approved any
other new drug containing the same active moiety, which is the
molecule or ion responsible for the action of the drug
substance. During the exclusivity period, the FDA may not accept
for review an abbreviated new drug application, or ANDA, or a
505(b)(2) NDA submitted by another company for another version
of such drug where the applicant does not own or have a legal
right of reference to all the data required for approval.
However, an application may be submitted after four years if it
contains a certification of patent invalidity or
non-infringement. The FDCA also provides three years of
marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to
an existing NDA if new clinical investigations, other than
bioavailability studies, that were conducted or sponsored by the
applicant are deemed by the FDA to be essential to the approval
of the application, for example new indications, dosages or
strengths of an existing drug. This three-year exclusivity
covers only the conditions associated with the new clinical
investigations and does not prohibit the FDA from approving
ANDAs for drugs containing the original active agent. Five-year
and three-year exclusivity
32
will not delay the submission or approval of a full NDA.
However, an applicant submitting a full NDA would be required to
conduct or obtain a right of reference to all of the preclinical
studies and adequate and well-controlled clinical studies
necessary to demonstrate safety and effectiveness. HR 3590
provides 12 years of data exclusivity for innovator
biologics. During this exclusivity period, competitors are
barred from relying on the innovator’s safety and efficacy
data to gain FDA approval. Therefore, a competitor seeking to
obtain marketing approval during this exclusivity period would
be required to conduct its own preclinical and clinical studies.
Pediatric exclusivity is another type of exclusivity in the
United States. Pediatric exclusivity, if granted, adds an
additional six months to an existing exclusivity or statutory
delay in approval resulting from a patent certification. This
six-month exclusivity, which runs from the end of other
exclusivity protection or patent delay, may be granted based on
the voluntary completion of a pediatric study in accordance with
an FDA-issued “Written Request” for such a study. The
current pediatric exclusivity provision was reauthorized in
September 2007.
Orphan
Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation
to a drug or biological product intended to treat a rare disease
or condition, which is generally a disease or condition that
affects fewer than 200,000 individuals in the United States, or
more than 200,000 individuals in the United States and for which
there is no reasonable expectation that the cost of developing
and making a drug or biological product available in the United
States for this type of disease or condition will be recovered
from sales of the product. Orphan product designation must be
requested before submitting an NDA or BLA. After the FDA grants
orphan product designation, the identity of the therapeutic
agent and its potential orphan use are disclosed publicly by the
FDA. Orphan product designation does not convey any advantage in
or shorten the duration of the regulatory review and approval
process.
If a product that has orphan designation subsequently receives
the first FDA approval for the disease or condition for which it
has such designation, the product is entitled to orphan product
exclusivity, which means that the FDA may not approve any other
applications to market the same drug or biological product for
the same indication, except in very limited circumstances, for
seven years. Competitors, however, may receive approval of
different products for the indication for which the orphan
product has exclusivity or obtain approval for the same product,
but for a different indication for which the orphan product has
exclusivity. Orphan product exclusivity could also block the
approval of one of our products for seven years if a competitor
obtains approval of the same drug or biological product as
defined by the FDA or if our product candidate is determined to
be contained within the competitor’s product for the same
indication or disease. If a drug or biological product
designated as an orphan product receives marketing approval for
an indication broader than what is designated, it may not be
entitled to orphan product exclusivity.
The FDA also administers a clinical research grants program,
whereby researchers may compete for funding to conduct clinical
studies to support the approval of drugs, biologics, medical
devices and medical foods for rare diseases and conditions. A
product does not have to be designated as an orphan product to
be eligible for the grant program. An application for an orphan
grant should propose one discrete clinical study to facilitate
FDA approval of the product for a rare disease or condition. The
clinical study may address an unapproved new product or an
unapproved new use for a product already on the market.
Expedited
Development and Review Programs
The FDA has a fast track program that is intended to expedite or
facilitate the process for reviewing new drugs and biological
products that meet certain criteria. Specifically, new drugs and
biological products are eligible for fast track designation if
they are intended to treat a serious or life-threatening
condition and demonstrate the potential to address unmet medical
needs for the condition. Fast track designation applies to the
combination of the product and the specific indication for which
it is being studied. For a fast track product, the FDA may
consider for review on a rolling basis sections of the NDA or
BLA before the complete application is submitted, if the sponsor
provides a schedule for the submission of the sections of the
NDA or BLA, the FDA agrees to accept sections of the NDA or BLA
and determines that the schedule is acceptable, and the sponsor
pays any required user fees upon submission of the first section
of the NDA or BLA.
A fast track product may also be eligible for other types of FDA
programs intended to expedite development and review, such as
priority review and accelerated approval. A fast track product
is eligible for priority review if it has the potential to
provide safe and effective therapy where no satisfactory
alternative therapy exists or a
33
significant improvement in the treatment, diagnosis or
prevention of a disease compared to marketed products. The FDA
will attempt to direct additional resources to the evaluation of
an application for a new drug or biological product designated
for priority review in an effort to facilitate the review.
Additionally, a fast track product may be eligible for
accelerated approval. Drug or biological products studied for
their safety and effectiveness in treating serious or
life-threatening illnesses and that provide meaningful
therapeutic benefit over existing treatments may receive
accelerated approval, which means that they may be approved on
the basis of adequate and well-controlled clinical studies
establishing that the product has an effect on a surrogate
endpoint that is reasonably likely to predict a clinical
benefit, or on the basis of an effect on a clinical endpoint
other than survival or irreversible morbidity. As a condition of
approval, the FDA may require that a sponsor of a drug or
biological product receiving accelerated approval perform
adequate and well-controlled post-marketing clinical studies.
Fast track designation, priority review and accelerated approval
do not change the standards for approval but may expedite the
development or approval process.
We have been granted fast track designation for our product
candidate,
A-001, for
the prevention of acute chest syndrome associated with sickle
cell disease in at-risk patients. Even though we have received
fast track designation for
A-001, the
FDA may later decide that
A-001 no
longer meets the conditions for qualification. In addition,
obtaining fast track designation may not provide us with a
material commercial advantage.
Post-Approval
Requirements
Any drug or biological products for which we receive FDA
approvals are subject to continuing regulation by the FDA,
including, among other things, record-keeping requirements,
reporting of adverse experiences with the product, providing the
FDA with updated safety and efficacy information, product
sampling and distribution requirements, complying with certain
electronic records and signature requirements and complying with
FDA promotion and advertising requirements. The FDA strictly
regulates labeling, advertising, promotion and other types of
information on products that are placed on the market. Drugs and
biological products may be promoted only for the approved
indications and in accordance with the provisions of the
approved label. Further, manufacturers of drugs and biological
products must continue to comply with cGMP requirements, which
are extensive and require considerable time, resources and
ongoing investment to ensure compliance. In addition, changes to
the manufacturing process generally require prior FDA approval
before being implemented and other types of changes to the
approved product, such as adding new indications and additional
labeling claims, are also subject to further FDA review and
approval.
Drug and biological product manufacturers and other entities
involved in the manufacturing and distribution of approved drugs
or biological products are required to register their
establishments with the FDA and certain state agencies, and are
subject to periodic unannounced inspections by the FDA and
certain state agencies for compliance with cGMP and other laws.
The cGMP requirements apply to all stages of the manufacturing
process, including the production, processing, sterilization,
packaging, labeling, storage and shipment of the drug or
biological product. Manufacturers must establish validated
systems to ensure that products meet specifications and
regulatory standards, and test each product batch or lot prior
to its release.
Manufacturers of biological products must also report to the FDA
any deviations from cGMP that may affect the safety, purity or
potency of a distributed product; or any unexpected or
unforeseeable event that may affect the safety, purity or
potency of a distributed product. The regulations also require
investigation and correction of any deviations from cGMP and
impose documentation requirements.
We rely, and expect to continue to rely, on third parties for
the production of clinical and commercial quantities of our
products. Future FDA and state inspections may identify
compliance issues at the facilities of our contract
manufacturers that may disrupt production or distribution or may
require substantial resources to correct.
The FDA may withdraw a product approval if compliance with
regulatory standards is not maintained or if problems occur
after the product reaches the market. Later discovery of
previously unknown problems with a product may result in
restrictions on the product or even complete withdrawal of the
product from the market. Further, the failure to maintain
compliance with regulatory requirements may result in
administrative or judicial actions, such as fines, warning
letters, holds on clinical studies, product recalls or seizures,
product detention or refusal to permit the import or export of
products, refusal to approve pending applications or
supplements, restrictions on marketing or manufacturing,
injunctions or civil or criminal penalties.
34
In addition, from time to time, legislation is drafted,
introduced and passed in Congress that could significantly
change the statutory provisions governing the approval,
manufacturing and marketing of products regulated by the FDA.
For example, in September 2007, the Food and Drug Administration
Amendments Act of 2007 was enacted, giving the FDA enhanced
post-market authority, including the authority to require
post-market studies and clinical studies, labeling changes based
on new safety information and compliance with a risk evaluation
and mitigation strategy, or REMS, approved by the FDA. In
determining whether a REMS is necessary, the FDA must consider
the size of the population likely to use the drug or biological
product, the seriousness of the disease or condition to be
treated, the expected benefit of the product, the duration of
treatment, the seriousness of known or potential adverse events
for varespladib and whether the product is a new molecular
entity. We have submitted a REMS as an appendix to the SPA. If
the FDA determines our REMS is necessary, we must submit a REMS
plan as part of an NDA or BLA. The FDA may require that a REMS
include various elements, such as a medication guide, patient
package insert, a communication plan to educate health care
providers, limitations on who may prescribe or dispense the
product, or other measures.
Failure to comply with any requirements under the new law may
result in significant penalties. The new law also authorizes
significant civil money penalties for the dissemination of false
or misleading
direct-to-consumer
advertisements and allows the FDA to require companies to submit
direct-to-consumer
television drug advertisements for FDA review prior to public
dissemination. Additionally, the new law expands the clinical
study registry so that sponsors of all clinical studies, except
for Phase 1 clinical studies, are required to submit certain
clinical study information for inclusion in the clinical study
registry data bank. In addition to new legislation, the FDA
regulations and policies are often revised or reinterpreted by
the agency in ways that may significantly affect our business
and our products. It is impossible to predict whether further
legislative or FDA regulation or policy changes will be enacted
or implemented and what the impact of such changes, if any, may
be.
Foreign
Regulation
In addition to regulations in the United States, we will be
subject to a variety of foreign regulations governing clinical
studies and commercial sales and distribution of our products to
the extent we choose to sell any products outside of the United
States. Whether or not we obtain FDA approval for a product, we
must obtain approval of a product by the comparable regulatory
authorities of foreign countries before we can commence clinical
studies or marketing of the product in those countries. The
approval process varies from country to country and the time may
be longer or shorter than that required for FDA approval. The
requirements governing the conduct of clinical studies, product
licensing, pricing and reimbursement vary greatly from country
to country.
In the European Union, our products are subject to extensive
regulatory requirements, which provide, among other things, that
no medicinal product may be placed on the market of a European
Union member state unless a marketing authorization has been
issued by the European Medicines Agency or a national competent
authority. European Union member states require both regulatory
clearance by the national competent authority and a favorable
ethics committee opinion prior to the commencement of a clinical
study.
Under the European Union regulatory systems, we may submit
marketing authorization applications either under a centralized
or decentralized procedure. The centralized procedure provides
for the grant of a single marketing authorization that is valid
for all European Union member states. The centralized procedure
is compulsory for medicines produced by certain biotechnological
processes, products with a new active substance indicated for
the treatment of certain diseases such as neurodegenerative
disorder or diabetes and products designated as orphan medicinal
products, and optional for those products which are highly
innovative or for which a centralized process is in the interest
of patients. The decentralized procedure of approval provides
for approval by one or more other, or concerned, member states
of an assessment of an application performed by one member
state, known as the reference member state. Under the
decentralized approval procedure, an applicant submits an
application, or dossier, and related materials (draft summary of
product characteristics, draft labeling and package leaflet) to
the reference member state and concerned member states. The
reference member state prepares a draft assessment and drafts of
the related materials within 120 days after receipt of a
valid application. Within 90 days of receiving the
reference member state’s assessment report, each concerned
member state must decide whether to approve the assessment
report and related materials. If a member state cannot approve
the assessment report and related materials on the grounds of
potential serious risk to public health, the disputed points may
eventually be referred to the European Commission, whose
decision is binding on all member states.
35
Reimbursement
Sales of pharmaceutical products depend significantly on the
availability of third-party reimbursement. Third-party payors
include government health administrative authorities, including
at the federal and state level, managed care providers, private
health insurers and other organizations. We anticipate
third-party payors will provide reimbursement for our products.
However, these third-party payors are increasingly challenging
the price and examining the cost-effectiveness of medical
products and services. In addition, significant uncertainty
exists as to the reimbursement status of newly approved health
care products. We may need to conduct expensive pharmacoeconomic
studies in order to demonstrate the cost-effectiveness of our
products. Our product candidates may not be considered
cost-effective. It is time consuming and expensive for us to
seek reimbursement from third-party payors. Reimbursement may
not be available or sufficient to allow us to sell our products
on a competitive and profitable basis.
In addition, the U.S. Congress and state legislatures from
time to time propose and adopt initiatives aimed at cost
containment, which could impact our ability to sell our products
profitably. For example, in March 2010, President Obama signed
into law the Patient Protection and Affordable Care Act, and the
associated reconciliation bill, which we refer to collectively
as the Health Care Reform Law, a sweeping law intended to
broaden access to health insurance, reduce or constrain the
growth of healthcare spending, enhance remedies against fraud
and abuse, add new transparency requirements for healthcare and
health insurance industries, impose new taxes and fees on the
health industry and impose additional health policy reforms.
Effective October 1, 2010, the Health Care Reform Law
revises the definition of “average manufacturer price”
for reporting purposes, which could increase the amount of
Medicaid drug rebates to states once the provision is effective.
Further, beginning in 2011, the new law imposes a significant
annual fee on companies that manufacture or import certain
branded prescription drug products and biologic agents.
Substantial new provisions affecting compliance also have been
enacted, which may require us to modify our business practices
with healthcare practitioners. We will not know the full effects
of the Health Care Reform Law until applicable federal and state
agencies issue regulations or guidance under the new law.
Although it is too early to determine the effect of the Health
Care Reform Law, the new law appears likely to continue the
pressure on pharmaceutical pricing, especially under the
Medicare program, and also may increase our regulatory burdens
and operating costs.
The Medicare Prescription Drug, Improvement, and Modernization
Act of 2003, or the MMA, imposed new requirements for the
distribution and pricing of prescription drugs for Medicare
beneficiaries, and included a major expansion of the
prescription drug benefit under a new Medicare Part D.
Medicare Part D went into effect on January 1, 2006.
Under Part D, Medicare beneficiaries may enroll in
prescription drug plans offered by private entities which will
provide coverage of outpatient prescription drugs. Part D
plans include both stand-alone prescription drug benefit plans
and prescription drug coverage as a supplement to Medicare
Advantage plans. Unlike Medicare Part A and B, Part D
coverage is not standardized. Part D prescription drug plan
sponsors are not required to pay for all covered Part D
drugs, and each drug plan can develop its own drug formulary
that identifies which drugs it will cover and at what tier or
level. However, Part D prescription drug formularies must
include drugs within each therapeutic category and class of
covered Part D drugs, though not necessarily all the drugs
in each category or class. Any formulary used by a Part D
prescription drug plan must be developed and reviewed by a
pharmacy and therapeutic committee.
It is not clear what effect the MMA will have on the prices paid
for currently approved drugs and the pricing options for new
drugs approved after January 1, 2006. Government payment
for some of the costs of prescription drugs may increase demand
for products for which we receive marketing approval. However,
any negotiated prices for our products covered by a Part D
prescription drug plan will likely be lower than the prices we
might otherwise obtain. Moreover, while the MMA applies only to
drug benefits for Medicare beneficiaries, private payors often
follow Medicare coverage policy and payment limitations in
setting their own payment rates. Any reduction in payment that
results from the MMA may result in a similar reduction in
payments from non-governmental payors.
There are also laws that govern a company’s eligibility to
participate in Medicare and Medicaid reimbursements. For
example, a company may be debarred from participation if it is
found to have violated federal anti-kickback laws, which could
have a significant effect on a company’s ability to operate
its business.
The cost of pharmaceuticals continues to generate substantial
governmental and third-party payor interest. We expect that the
pharmaceutical industry will experience pricing pressures due to
the trend toward managed
36
healthcare, the increasing influence of managed care
organizations, and additional legislative proposals. Indeed, we
expect that there will continue to be a number of federal and
state proposals to implement governmental pricing controls and
limit the growth of healthcare costs, including the cost of
prescription drugs. At the present time, Medicare is prohibited
from negotiating directly with pharmaceutical companies for
drugs. However, Congress is considering passing legislation that
would lift the ban on federal negotiations. While we cannot
predict whether such legislative or regulatory proposals will be
adopted, the adoption of such proposals could harm our business,
financial condition and results of operations.
Some third-party payors also require pre-approval of coverage
for new or innovative drug therapies before they will reimburse
healthcare providers that use such therapies. While we cannot
predict whether any proposed cost-containment measures will be
adopted or otherwise implemented in the future, the announcement
or adoption of these proposals could have a material adverse
effect on our ability to obtain adequate prices for our product
candidates and operate profitably.
In addition, in some foreign countries, the proposed pricing for
a drug must be approved before it may be lawfully marketed. The
requirements governing drug pricing vary widely from country to
country. For example, the European Union provides options for
its member states to restrict the range of medicinal products
for which their national health insurance systems provide
reimbursement and to control the prices of medicinal products
for human use. A member state may approve a specific price for
the medicinal product or it may instead adopt a system of direct
or indirect controls on the profitability of the company placing
the medicinal product on the market. In some countries, pricing
negotiations with governmental authorities can take six to
12 months or longer after the receipt of marketing
approval. To obtain reimbursement or pricing approval in some
countries, we may be required to conduct a clinical trial that
compares the cost-effectiveness of our product candidate to
other available therapies. There can be no assurance that any
country that has price controls or reimbursement limitations for
pharmaceutical products will allow favorable reimbursement and
pricing arrangements for any of our products.
Employees
As of December 31, 2010, we had 25 employees, eight of
whom hold an M.D., Ph.D. or Pharm. D. All of our employees
are engaged in administration, finance, clinical, regulatory and
business development functions. None of our employees are
represented by a labor union, and we believe that our relations
with our employees are good.
Other
Available Information
We are subject to the information requirements of the Exchange
Act. Therefore, we file periodic reports, proxy statements and
other information with the SEC, which may be obtained by
visiting the Public Reference Room of the SEC at
100 F Street, NE, Washington, D.C. 20549 or by
calling the SEC at
1-800-SEC-0330.
In addition, the SEC maintains a website (www.sec.gov) that
contains reports, proxy and information statements, and other
information regarding issuers that file electronically.
The mailing address of our headquarters is 25801 Industrial
Blvd, Hayward, CA 94545, and our telephone number at that
location is
510-856-5600.
Our website is www.anthera.com. Through a link on the
“Investors” section of our website (under “SEC
Filings” in the “Financial Information” section),
we make available, free of charge, the following filings as soon
as reasonably practicable after they are electronically filed
with or furnished to the SEC: our Annual Reports on
Form 10-K;
Quarterly Reports on
Form 10-Q;
Current Reports on
Form 8-K;
and any amendments to those reports filed or furnished pursuant
to Section 13(a) or 15(d) of the Exchange Act.
Before you decide to invest in our common stock, you should
carefully consider the risks described below, together with the
other information contained in this Annual Report on
Form 10-K,
including the financial statements and the related notes that
appear at the end of this report. We believe the risks described
below are the risks that are material to us as of the date of
this report. If any of the following risks occur, our business,
financial condition, results of operations and future growth
prospects would likely be materially and adversely affected. In
these circumstances, the market price of our common stock could
decline, and you may lose all or part of your investment.
37
Risks
Related to Our Financial Condition and Capital
Requirements
We
have incurred significant losses since our inception and
anticipate that we will incur continued significant losses for
the foreseeable future.
We are a development stage company with only six years of
operating history. We have focused primarily on developing our
three product candidates, varespladib,
A-623 and
varespladib sodium
(A-001). We
have financed our operations exclusively through equity
offerings and private placements of convertible debt and we have
incurred losses in each year since our inception in September
2004. Our net losses were approximately $8.7 million in
2006, $25.7 million in 2007, $18.1 million in 2008,
$12.2 million in 2009 and $40.4 million for the year
ended December 31, 2010. As of December 31, 2010, we
had an accumulated deficit of approximately $105.6 million.
Substantially all of our losses resulted from costs incurred in
connection with our product development programs and from
general and administrative costs associated with our operations.
We expect to incur additional losses over the next several
years, and these losses may increase if we cannot generate
revenues. Our historical losses, combined with expected future
losses, have had and will continue to have an adverse effect on
our stockholders’ equity and working capital. We expect our
development expenses, as well as our clinical product
manufacturing expenses, to increase in connection with our
pivotal Phase 3 clinical study named VISTA-16 for varespladib,
our Phase 2b clinical study named PEARL-SC for
A-623 and
other clinical studies related to the development of
A-623. In
addition, we will incur additional costs of operating as a
public company and, if we obtain regulatory approval for any of
our product candidates, we may incur significant sales,
marketing, in-licensing and outsourced manufacturing expenses as
well as continued product development expenses. As a result, we
expect to continue to incur significant and increasing losses
for the foreseeable future.
We
have never generated any revenue and may never be
profitable.
Our ability to generate revenue and achieve profitability
depends on our ability, alone or with collaborators, to
successfully complete the development of our product candidates,
conduct preclinical tests in animals and clinical studies in
human beings, obtain the necessary regulatory approvals for our
product candidates and commercialize any approved products. We
have not generated any revenue from our development-stage
product candidates, and we do not know when, or if, we will
generate any revenue. The commercial success of our
development-stage product candidates will depend on a number of
factors, including, but not limited to, our ability to:
|
|
|
| |
•
|
obtain favorable results for and advance the development of our
lead product candidate, varespladib, for the treatment of acute
coronary syndrome, including successfully launching and
completing the VISTA-16 study;
|
| |
| |
•
|
obtain favorable results for and advance the development of our
product candidate
A-623 for
the treatment of B-cell mediated autoimmune diseases, including
successfully launching and completing a Phase 2b clinical study
in patients with systemic lupus erythematosus, or lupus, or
other indications related to the development of
A-623;
|
| |
| |
•
|
obtain favorable results for and advance the development of our
product candidate
A-001 for
the prevention of acute chest syndrome associated with sickle
cell disease, including completing a multi-center Phase 2
clinical study;
|
| |
| |
•
|
successfully execute our planned preclinical studies in animals
and clinical studies in human beings for our other product
candidates;
|
| |
| |
•
|
obtain regulatory approval for varespladib,
A-623,
A-001 and
our other product candidates;
|
| |
| |
•
|
if regulatory approvals are obtained, begin the commercial
manufacturing of our product candidates with our third-party
manufacturers;
|
| |
| |
•
|
launch commercial sales and effectively market our product
candidates, either independently or in strategic collaborations
with third parties; and
|
| |
| |
•
|
achieve broad market acceptance of our product candidates in the
medical community and with third-party payors.
|
All of our product candidates are subject to the risks of
failure inherent in the development of therapeutics based on new
technologies. Currently, we have three product candidates in
clinical development: varespladib,
38
A-623 and
A-001. These
product candidates could fail in clinical studies if we are
unable to demonstrate that they are effective or if they cause
unacceptable adverse effects in the patients we treat. Failure
of our product candidates in clinical studies would have a
material adverse effect on our ability to generate revenue or
become profitable. If we are not successful in achieving
regulatory approval for our product candidates or are
significantly delayed in doing so, our business will be
materially harmed.
Additionally, all of our other product candidates are in
preclinical development. Our drug discovery efforts may not
produce any other viable or marketable product candidates. We do
not expect any of our potential product candidates to be
commercially available until at least 2013.
Even if our product candidates are approved for commercial sale,
the approved product candidate may not gain market acceptance or
achieve commercial success. Physicians, patients, payors or the
medical community in general may be unwilling to accept, utilize
or recommend any of our products. We would anticipate incurring
significant costs associated with commercializing any approved
product. Even if we are able to generate product sales, which we
cannot guarantee, we may not achieve profitability soon
thereafter, if ever. If we are unable to generate product
revenues, we will not become profitable and may be unable to
continue operations without additional funding.
We
will need substantial additional capital in the future to fund
our operations. If additional capital is not available, we will
have to delay, reduce or cease operations.
We will need to raise substantial additional capital to fund our
operations and to develop our product candidates. Our future
capital requirements could be substantial and will depend on
many factors including:
|
|
|
| |
•
|
the rate of progress of our Phase 3 clinical study named
VISTA-16 study for varespladib and our Phase 2b clinical study
named PEARL-SC for
A-623;
|
| |
| |
•
|
the scope, size, rate of progress, results and costs of our
preclinical studies, clinical studies and other development
activities for one or more of our other product candidates;
|
| |
| |
•
|
manufacturing campaign of
A-623
clinical matters, including formulation development and
enhancement;
|
| |
| |
•
|
non-clinical activities that we may pursue parallel to clinical
trials for each clinical compound;
|
| |
| |
•
|
the cost, timing and outcomes of regulatory proceedings;
|
| |
| |
•
|
payments received under any strategic collaborations;
|
| |
| |
•
|
the filing, prosecution and enforcement of patent claims;
|
| |
| |
•
|
the costs associated with commercializing our product candidates
if they receive regulatory approval, including the cost and
timing of developing sales and marketing capabilities, or
entering into strategic collaboration with others relating to
the commercialization of our product candidates; and
|
| |
| |
•
|
revenues received from approved products, if any, in the future.
|
As of the date of this report, we anticipate that our existing
cash, cash equivalents and short-term investments, will enable
us to maintain our currently planned operations through at least
the next 12 months. Changing circumstances may cause us to
consume capital significantly faster than we currently
anticipate. Additional financing may not be available when we
need it or may not be available on terms that are favorable to
us. If adequate funds are not available to us on a timely basis,
or at all, we may be required to:
|
|
|
| |
•
|
terminate, reduce or delay preclinical studies, clinical studies
or other development activities for one or more of our product
candidates; or
|
| |
| |
•
|
terminate, reduce or delay our (i) establishment of sales
and marketing capabilities, (ii) pursuit of strategic
collaborations with others relating to the sales, marketing and
commercialization of our product candidates or (iii) other
activities that may be necessary to commercialize our product
candidates, if approved for sale.
|
39
The
timing of the milestone and royalty payments we are required to
make to each of Eli Lilly and Company, Shionogi & Co.,
Ltd. and Amgen Inc. is uncertain and could adversely affect our
cash flows and results of operations.
In July 2006, we entered into a license agreement with Eli Lilly
and Company, or Eli Lilly, and Shionogi & Co., Ltd. to
develop and commercialize certain secretory phospholipase
A2,
or
sPLA2,
inhibitors for the treatment of cardiovascular disease and other
diseases. Pursuant to our license agreement with them, we have
an obligation to pay to each of Eli Lilly and
Shionogi & Co., Ltd. significant milestone and royalty
payments based upon how we develop and commercialize certain
sPLA2
inhibitors, including varespladib and
A-001, and
our achievement of certain significant corporate, clinical and
financial events. For varespladib, we are required to pay up to
$32.0 million upon achievement of certain approval and
post-approval sales milestones. For
A-001, we
are required to pay up to $3.0 million upon achievement of
certain clinical development milestones and up to
$25.0 million upon achievement of certain approval and
post-approval sales milestones. For other product formulations
that we are not currently developing, we would be required to
pay up to $2.0 million upon achievement of certain clinical
development milestones and up to $35.5 million upon
achievement of certain approval and post-approval sales
milestones. In addition, in December 2007, we entered into a
license agreement with Amgen Inc., or Amgen, pursuant to which
we obtained an exclusive worldwide license to certain technology
and compounds relating to
A-623.
Pursuant to our license agreement with Amgen, we are required to
make various milestone payments upon our achievement of certain
development, regulatory and commercial objectives for any
A-623
formulation. We are required to pay up to $10.0 million
upon achievement of certain pre-approval clinical development
milestones and up to $23.0 million upon achievement of
certain post-approval milestones. We are also required to make
tiered quarterly royalty payments on net sales, which increase
as a percentage from the high single digits to the low double
digits as net sales increase. The timing of our achievement of
these events and corresponding milestone payments becoming due
to Eli Lilly, Shionogi & Co., Ltd. and Amgen is
subject to factors relating to the clinical and regulatory
development and commercialization of certain
sPLA2
inhibitors or
A-623, as
applicable, many of which are beyond our control. We may become
obligated to make a milestone payment during a period in which
we do not have the cash on hand to make such payment, which
could require us to delay our clinical studies, curtail our
operations, scale back our commercialization and marketing
efforts, seek funds to meet these obligations at terms
unfavorable to us or default on our license agreements, which
could result in license termination.
Our
limited operating history makes it difficult to evaluate our
business and prospects.
We were incorporated in September 2004. Our operations to date
have been limited to organizing and staffing our company,
acquiring product and technology rights, conducting product
development activities for our primary product candidates,
varespladib,
A-623 and
A-001, and
performing research and development. We have not yet
demonstrated an ability to obtain regulatory approval for or
commercialize a product candidate. Consequently, any predictions
about our future performance may not be as accurate as they
could be if we had a history of successfully developing and
commercializing pharmaceutical products.
Risks
Associated with Development and Commercialization of Our Product
Candidates
We
depend substantially on the success of our three primary product
candidates, varespladib,
A-623 and
A-001, which
are still under clinical development. We cannot assure you that
these product candidates or any of our other product candidates
will receive regulatory approval or be successfully
commercialized.
To date, we have not obtained marketing approval for, or
marketed, distributed or sold any product candidates. The
success of our business depends primarily upon our ability to
develop and commercialize our three primary product candidates
successfully. Our lead product candidate is varespladib, which
has completed its Phase 2 clinical studies and for which we have
received (i) an agreement from the U.S. Food and Drug
Administration, or FDA, on a Special Protocol Assessment, or
SPA, for the VISTA-16 Phase 3 study protocol, and
(ii) scientific advice from the European Medicines Agency
on our European development strategy for varespladib. We
initiated the VISTA-16 study for varespladib in June 2010.
Our next product candidate is
A-623, which
has completed several Phase 1 clinical studies and recently
began enrollment for our Phase 2b clinical study. In July 2010,
we received clearance from the FDA to begin recruitment of lupus
patients into the PEARL-SC Phase 2b clinical study. In November
2010, we placed a voluntary hold on the PEARL-SC study due to
problems found with vials. Patient enrollment in the study was
temporarily suspended and dosing was discontinued in patients
who were enrolled in the study while we conducted an analysis of
the problem.
40
We resolved the issues found with the vials in December 2010.
After analysis, simulation and consultation with industry
experts, we determined that shipping on dry ice was the root
cause of the issue. Shipping logistics were modified and we
reinitiated enrollment in PEARL-SC and dosing in January 2011.
We have received no reports of patient-related side effects or
problems with drug administration that could be attributed to
the vial problem.
Our third product candidate, varespladib sodium
(A-001), is
an intravenously administered inhibitor of
sPLA2.
We have completed a Phase 2 clinical study for the prevention of
acute chest syndrome associated with sickle cell disease. A
pre-specified interim review of our Phase 2 clinical study
results by a Data Safety Monitoring Board, or DSMB, indicated
A-001, at a
certain dose, reduced
sPLA2
activity by more than 80% from baseline within 48 hours.
Furthermore, the incidence of acute chest syndrome appeared to
be related to the level of
sPLA2
activity.
Our product candidates are prone to the risks of failure
inherent in drug development. Before obtaining regulatory
approvals for the commercial sale of any product candidate for a
target indication, we must demonstrate with substantial evidence
gathered in preclinical and well-controlled clinical studies,
and, with respect to approval in the United States, to the
satisfaction of the FDA and, with respect to approval in other
countries, similar regulatory authorities in those countries,
that the product candidate is safe and effective for use for
that target indication and that the manufacturing facilities,
processes and controls are adequate. Despite our efforts, our
product candidates may not:
|
|
|
| |
•
|
offer therapeutic or other improvement over existing, comparable
therapeutics;
|
| |
| |
•
|
be proven safe and effective in clinical studies;
|
| |
| |
•
|
meet applicable regulatory standards;
|
| |
| |
•
|
be capable of being produced in sufficient quantities at
acceptable costs;
|
| |
| |
•
|
be successfully commercialized; or
|
| |
| |
•
|
obtain favorable reimbursement.
|
We are not permitted to market our varespladib and
A-001
product candidates in the United States until we receive
approval of a new drug application, or NDA, or with respect to
our A-623
product candidate, approval of a biologics license application,
or BLA, from the FDA, or in any foreign countries until we
receive the requisite approval from such countries. We have not
submitted an NDA or BLA or received marketing approval for any
of our product candidates.
Preclinical testing and clinical studies are long, expensive and
uncertain processes. We may spend several years completing our
testing for any particular product candidate, and failure can
occur at any stage. Negative or inconclusive results or adverse
medical events during a clinical study could also cause the FDA
or us to terminate a clinical study or require that we repeat it
or conduct additional clinical studies. Additionally, data
obtained from a clinical study are susceptible to varying
interpretations and the FDA or other regulatory authorities may
interpret the results of our clinical studies less favorably
than we do. The FDA and equivalent foreign regulatory agencies
have substantial discretion in the approval process and may
decide that our data are insufficient to support a marketing
application and require additional preclinical, clinical or
other studies.
Any
termination or suspension of, or delays in the commencement or
completion of, clinical testing of our product candidates could
result in increased costs to us, delay or limit our ability to
generate revenue and adversely affect our commercial
prospects.
Delays in the commencement or completion of clinical testing
could significantly affect our product development costs. We do
not know whether planned clinical studies will begin on time or
be completed on schedule, if at all. The commencement and
completion of clinical studies can be delayed for a number of
reasons, including delays related to:
|
|
|
| |
•
|
obtaining regulatory approval to commence a clinical study or
complying with conditions imposed by a regulatory authority
regarding the scope or design of a clinical study;
|
| |
| |
•
|
reaching agreement on acceptable terms with prospective clinical
research organizations, or CROs, and study sites, the terms of
which can be subject to extensive negotiation and may vary
significantly among different CROs and study sites;
|
41
|
|
|
| |
•
|
manufacturing, including manufacturing sufficient quantities of
a product candidate or other materials for use in clinical
studies;
|
| |
| |
•
|
obtaining institutional review board, or IRB, approval or the
approval of other reviewing entities to conduct a clinical study
at a prospective site;
|
| |
| |
•
|
recruiting and enrolling patients to participate in clinical
studies for a variety of reasons, including size of patient
population, nature of clinical study protocol, the availability
of approved effective treatments for the relevant disease and
competition from other clinical study programs for similar
indications;
|
| |
| |
•
|
severe or unexpected drug-related adverse effects experienced by
patients in a clinical study; and
|
| |
| |
•
|
retaining patients who have initiated a clinical study, but may
withdraw due to treatment protocol, adverse effects from the
therapy, lack of efficacy from the treatment, personal issues or
who are lost to further
follow-up.
|
Clinical studies may also be delayed, suspended or terminated as
a result of ambiguous or negative interim results, or results
that are inconsistent with earlier results. For example, the
independent statistician that is conducting the data review may
recommend that we stop our VISTA-16 study for varespladib if
certain biomarkers of inflammation and lipid profiles fail to
meet pre-specified reductions from a subset of the first 1,000
or more patients. In addition, a clinical study may be suspended
or terminated by us, the FDA, the IRB or other reviewing entity
overseeing the clinical study at issue, any of our clinical
study sites with respect to that site, or other regulatory
authorities due to a number of factors, including:
|
|
|
| |
•
|
failure to conduct the clinical study in accordance with
regulatory requirements or our clinical protocols;
|
| |
| |
•
|
inspection of the clinical study operations or study sites by
the FDA or other regulatory authorities resulting in the
imposition of a clinical hold;
|
| |
| |
•
|
unforeseen safety issues or any determination that a clinical
study presents unacceptable health risks; and
|
| |
| |
•
|
lack of adequate funding to continue the clinical study,
including the incurrence of unforeseen costs due to enrollment
delays, requirements to conduct additional clinical studies and
increased expenses associated with the services of our CROs and
other third parties.
|
Product development costs to us and our collaborators will
increase if we have delays in testing or approval of our product
candidates or if we need to perform more or larger clinical
studies than planned. For example, we may need to increase our
sample size for our VISTA-16 study for varespladib if the
overall major adverse cardiovascular event, or MACE, rate is
lower than expected. We typically rely on third-party clinical
investigators at medical institutions and health care facilities
to conduct our clinical studies and, as a result, we may face
additional delaying factors outside our control.
Additionally, changes in regulatory requirements and policies
may occur and we may need to amend clinical study protocols to
reflect these changes. Amendments may require us to resubmit our
clinical study protocols to IRBs for reexamination, which may
impact the costs, timing or successful completion of a clinical
study. If we experience delays in completion of, or if we, the
FDA or other regulatory authorities, the IRB or other reviewing
entities, or any of our clinical study sites suspend or
terminate any of our clinical studies, the commercial prospects
for our product candidates may be harmed and our ability to
generate product revenues will be delayed. In addition, many of
the factors that cause, or lead to, termination or suspension
of, or a delay in the commencement or completion of, clinical
studies may also ultimately lead to the denial of regulatory
approval of a product candidate. Also, if one or more clinical
studies are delayed, our competitors may be able to bring
products to market before we do, and the commercial viability of
our product candidates could be significantly reduced.
The
results of biomarker assays in earlier clinical studies in
varespladib are not necessarily predictive of future results,
and therefore the results of biomarker assays in the VISTA-16
study may not be similar to those observed
previously.
Success in our Phase 2 clinical studies in lowering low-density
lipoprotein cholesterol, or LDL-C, C-reactive protein, or CRP,
sPLA2
and interleukin-6, or IL-6, during treatment with varespladib
does not ensure that later clinical studies, such as our
VISTA-16 study, will demonstrate similar reductions in these
biomarkers. Each of these biomarkers has been associated with an
increased risk for secondary MACE following an acute coronary
syndrome.
42
Our inability to demonstrate similar biomarker effects in our
VISTA-16 study may reduce our ability to achieve our primary
endpoint to reduce MACE and to achieve regulatory approval of
varespladib. Even if we demonstrate similar biomarker effects in
our VISTA-16 study, those results do not necessarily equate with
reductions in MACE.
Because
the results of preclinical testing or earlier clinical studies
are not necessarily predictive of future results, varespladib,
A-623,
A-001 or any
other product candidate we advance into clinical studies may not
have favorable results in later clinical studies or receive
regulatory approval.
Success in preclinical testing and early clinical studies does
not ensure that later clinical studies will generate adequate
data to demonstrate the efficacy and safety of an
investigational drug or biologic. A number of companies in the
pharmaceutical and biotechnology industries, including those
with greater resources and experience, have suffered significant
setbacks in Phase 3 clinical studies, even after seeing
promising results in earlier clinical studies. Despite the
results reported in earlier clinical studies for our product
candidates, including varespladib,
A-623 and
A-001, we do
not know whether any Phase 3 or other clinical studies we may
conduct will demonstrate adequate efficacy and safety to result
in regulatory approval to market any of our product candidates.
If later stage clinical studies do not produce favorable
results, our ability to achieve regulatory approval for any of
our product candidates may be adversely impacted. Even if we
believe that our product candidates have performed
satisfactorily in preclinical testing and clinical studies, we
may nonetheless fail to obtain FDA approval for our product
candidates.
If we
breach the license agreements for our primary product
candidates, we could lose the ability to continue the
development and commercialization of our primary product
candidates.
We are party to an agreement with Eli Lilly and
Shionogi & Co., Ltd. containing exclusive, worldwide
licenses, except for Japan, of the composition of matter,
methods of making and methods of use for certain
sPLA2
inhibitors. We are also party to an agreement with Amgen
containing exclusive, worldwide licenses of the composition of
matter and methods of use for
A-623. These
agreements require us to make timely milestone and royalty
payments, provide regular information, maintain the
confidentiality of and indemnify Eli Lilly, Shionogi &
Co., Ltd. and Amgen under the terms of the agreements.
If we fail to meet these obligations, our licensors may
terminate our exclusive licenses and may be able to
re-obtain
licensed technology and aspects of any intellectual property
controlled by us that relate to the licensed technology that
originated from the licensors. Our licensors could effectively
take control of the development and commercialization of
varespladib,
A-623 and
A-001 after
an uncured, material breach of our license agreements by us or
if we voluntarily terminate the agreements. While we would
expect to exercise all rights and remedies available to us,
including seeking to cure any breach by us, and otherwise seek
to preserve our rights under the patents licensed to us, we may
not be able to do so in a timely manner, at an acceptable cost
or at all. Any uncured, material breach under the licenses could
result in our loss of exclusive rights and may lead to a
complete termination of our product development and any
commercialization efforts for varespladib,
A-623 or
A-001.
Our
industry is subject to intense competition. If we are unable to
compete effectively, our product candidates may be rendered
non-competitive or obsolete.
The pharmaceutical industry is highly competitive and subject to
rapid and significant technological change. Our potential
competitors include large pharmaceutical and more established
biotechnology companies, specialty pharmaceutical and generic
drug companies, academic institutions, government agencies and
other public and private research organizations that conduct
research, seek patent protection and establish collaborative
arrangements for research, development, manufacturing and
commercialization. All of these competitors currently engage in,
have engaged in or may engage in the future in the development,
manufacturing, marketing and commercialization of
pharmaceuticals and biotechnologies, some of which may compete
with our present or future product candidates. It is possible
that any of these competitors could develop technologies or
products that would render our product candidates obsolete or
non-competitive, which could adversely affect our revenue
potential. Key competitive factors affecting the commercial
success of our product candidates are likely to be efficacy,
safety profile, reliability, convenience of dosing, price and
reimbursement.
The market for inflammatory disease therapeutics is especially
large and competitive. All of the
sPLA2
inhibitor compounds we are currently developing, if approved,
will face intense competition, either as
43
monotherapies or in combination therapies. We are aware of other
companies with products in development that are being tested for
anti-inflammatory benefits in patients with acute coronary
syndrome, such as Via Pharmaceuticals, Inc. and its
5-lipoxygenase, or 5-LO, inhibitor, which has been evaluated in
Phase 2 clinical studies; and GlaxoSmithKline plc and its
product candidate, darapladib, which is a lipoprotein associated
phospholipase
A2,
or
Lp-PLA2,
inhibitor currently being evaluated in Phase 3 clinical studies.
Although there are no
sPLA2
inhibitor compounds currently approved by the FDA for the
treatment of acute chest syndrome associated with sickle cell
disease, Droxia, or hydroxyurea, is approved for the prevention
of vaso-occlusive crisis, or VOC, in sickle cell disease and
thus could reduce the pool of patients with VOC at risk for
acute chest syndrome. Further, we are aware of companies with
other products in development that are being tested for
potential treatment of lupus, including Human Genome Sciences,
Inc. and GlaxoSmithKline plc, who have a BAFF antagonist
monoclonal antibody product candidate, Benlysta, which recently
reported favorable results from two Phase 3 clinical studies in
lupus; ZymoGenetics, Inc. and Merck Serono S.A., whose dual
BAFF/APRIL antagonist fusion protein, Atacicept, is in a Phase 3
clinical study for lupus; and Immunomedics, Inc. and UCB S.A.,
who recently reported favorable results for their CD-22
antagonist humanized antibody, epratuzumab, which completed a
Phase 2b clinical study in lupus and has begun a Phase 3 study.
Many of our potential competitors have substantially greater
financial, technical and human resources than we do and
significantly greater experience in the discovery and
development of drug candidates, obtaining FDA and other
regulatory approvals of products and the commercialization of
those products. Accordingly, our competitors may be more
successful than we may be in obtaining FDA approval for drugs
and achieving widespread market acceptance. Our
competitors’ drugs may be more effective, have fewer
adverse effects, be less expensive to develop and manufacture or
be more effectively marketed and sold than any product candidate
we may commercialize and may render our product candidates
obsolete or non-competitive before we can recover the expenses
of developing and commercializing any of our product candidates.
We anticipate that we will face intense and increasing
competition as new drugs enter the market and advanced
technologies become available. These entities may also establish
collaborative or licensing relationships with our competitors.
Finally, the development of new treatment methods for the
diseases we are targeting could render our drugs non-competitive
or obsolete. All of these factors could adversely affect our
business.
Our
product candidates may cause undesirable adverse effects or have
other properties that could delay or prevent their regulatory
approval or limit the commercial profile of any approved
label.
Undesirable adverse effects caused by our product candidates
could cause us, IRBs or other reviewing entities, clinical study
sites, or regulatory authorities to interrupt, delay or halt
clinical studies and could result in the denial of regulatory
approval by the FDA or other regulatory authorities. Phase 2
clinical studies conducted by us with our product candidates
have generated differences in adverse effects and serious
adverse events. The most common adverse effects seen with any of
our product candidates versus placebo include diarrhea,
headache, nausea and increases in alanine aminotransferase,
which is an enzyme that indicates liver cell injury. The most
common serious adverse events seen with any of our product
candidates include death, VOC and congestive heart failure.
While none of these serious adverse events were considered
related to the administration of our product candidates by the
clinical investigators, if serious adverse events that are
considered related to our product candidates are observed in any
Phase 3 clinical studies, our ability to obtain regulatory
approval for our product candidates may be adversely impacted.
Further, if any of our product candidates receives marketing
approval and we or others later discover, after approval and use
in an increasing number of patients, that our products could
have adverse effect profiles that limit their usefulness or
require their withdrawal (whether or not the therapies showed
the adverse effect profile in Phase 1 through Phase 3 clinical
studies), a number of potentially significant negative
consequences could result, including:
|
|
|
| |
•
|
regulatory authorities may withdraw their approval of the
product;
|
| |
| |
•
|
regulatory authorities may require the addition of labeling
statements, such as warnings or contraindications;
|
| |
| |
•
|
we may be required to change the way the product is
administered, conduct additional clinical studies or change the
labeling of the product;
|
| |
| |
•
|
we could be sued and held liable for harm caused to
patients; and
|
| |
| |
•
|
our reputation may suffer.
|
44
Any of these events could prevent us from achieving or
maintaining market acceptance of the affected product candidate
and could substantially increase the costs of commercializing
our product candidates.
After
the completion of our clinical studies, we cannot predict
whether or when we will obtain regulatory approval to
commercialize our product candidates and we cannot, therefore,
predict the timing of any future revenue from these product
candidates.
Even if we project positive clinical results and file for
regulatory approval, we cannot commercialize any of our product
candidates until the appropriate regulatory authorities have
reviewed and approved the applications for such product
candidates. We cannot assure you that the regulatory agencies
will complete their review processes in a timely manner or that
we will obtain regulatory approval for any product candidate we
develop. Satisfaction of regulatory requirements typically takes
many years, is dependent upon the type, complexity and novelty
of the product and requires the expenditure of substantial
resources. In addition, we may experience delays or rejections
based upon additional government regulation from future
legislation or administrative action or changes in FDA policy
during the period of product development, clinical studies and
FDA regulatory review.
Our
agreement with the FDA on an SPA for our VISTA-16 study of
varespladib for the potential treatment of acute coronary
syndrome does not guarantee any particular outcome from
regulatory review of the study or the product
candidate.
The FDA’s SPA process creates a written agreement between
the sponsoring company and the FDA regarding clinical study
design and other clinical study issues that can be used to
support approval of a product candidate. The SPA is intended to
provide assurance that if the agreed upon clinical study
protocols are followed and the clinical study endpoints are
achieved, the data may serve as the primary basis for an
efficacy claim in support of an NDA. However, the SPA agreement
is not a guarantee of an approval of a product or any
permissible claims about the product. In particular, the SPA is
not binding on the FDA if public health concerns unrecognized at
the time of the SPA agreement is entered into become evident,
other new scientific concerns regarding product safety or
efficacy arise or if the sponsor company fails to comply with
the agreed upon clinical study protocols. Although we have an
agreement with the FDA on an SPA for our VISTA-16 clinical study
of varespladib for the potential short-term
(16-week)
treatment of acute coronary syndrome, we do not know how the FDA
will interpret the commitments under our agreed upon SPA, how it
will interpret the data and results or whether it will approve
our varespladib product candidate for the short-term (16-week)
treatment of acute coronary syndrome. Regardless of our SPA
agreement, we cannot guarantee any particular outcome from
regulatory review of our VISTA-16 study.
Even
if our product candidates receive regulatory approval, they may
still face future development and regulatory
difficulties.
Even if U.S. regulatory approval is obtained, the FDA may
still impose significant restrictions on a product’s
indicated uses or marketing or impose ongoing requirements for
potentially costly post-approval studies or post-market
surveillance. For example, the label ultimately approved for
varespladib, if any, may include restrictions on use. Further,
the FDA has indicated that long-term safety data on varespladib
may need to be obtained as a post-market requirement. Our
product candidates will also be subject to ongoing FDA
requirements governing the labeling, packaging, storage,
distribution, safety surveillance, advertising, promotion,
recordkeeping and reporting of safety and other post-market
information. In addition, manufacturers of drug products and
their facilities are subject to continual review and periodic
inspections by the FDA and other regulatory authorities for
compliance with current good manufacturing practices, or cGMP,
regulations. If we or a regulatory agency discovers previously
unknown problems with a product, such as adverse events of
unanticipated severity or frequency, or problems with the
facility where the product is manufactured, a regulatory agency
may impose restrictions on that product, the manufacturing
facility or us, including requiring recall or withdrawal of the
product from the market or suspension of manufacturing. If we,
our product candidates or the manufacturing facilities for our
product candidates fail to comply with applicable regulatory
requirements, a regulatory agency may:
|
|
|
| |
•
|
issue warning letters or untitled letters;
|
| |
| |
•
|
seek an injunction or impose civil or criminal penalties or
monetary fines;
|
| |
| |
•
|
suspend or withdraw regulatory approval;
|
| |
| |
•
|
suspend any ongoing clinical studies;
|
45
|
|
|
| |
•
|
refuse to approve pending applications or supplements to
applications filed by us;
|
| |
| |
•
|
suspend or impose restrictions on operations, including costly
new manufacturing requirements; or
|
| |
| |
•
|
seize or detain products, refuse to permit the import or export
of products, or require us to initiate a product recall.
|
The occurrence of any event or penalty described above may
inhibit our ability to commercialize our products and generate
revenue.
New
legal and regulatory requirements could make it more difficult
for us to obtain approvals for our product candidates and could
limit or make more burdensome our ability to commercialize any
approved products.
New federal legislation or regulatory requirements could affect
the requirements for obtaining regulatory approvals of our
product candidates or otherwise limit our ability to
commercialize any approved products or subject our products to
more rigorous post-approval requirements. For example, the FDA
Amendments Act of 2007, or FDAAA, granted the FDA new authority
to impose post-approval clinical study requirements, require
safety-related changes to product labeling and require the
adoption of risk management plans, referred to in the
legislation as risk evaluation and mitigation strategies, or
REMS. The REMS may include requirements for special labeling or
medication guides for patients, special communication plans to
health care professionals, and restrictions on distribution and
use. Pursuant to the FDAAA, if the FDA makes the requisite
findings, it might require that a new product be used only by
physicians with specified specialized training, only in
specified designated health care settings, or only in
conjunction with special patient testing and monitoring. The
legislation also included the following: requirements for
providing the public information on ongoing clinical studies
through a clinical study registry and for disclosing clinical
study results to the public through such registry; renewed
requirements for conducting clinical studies to generate
information on the use of products in pediatric patients; and
substantial new penalties, for example, for false or misleading
consumer advertisements. Other proposals have been made to
impose additional requirements on drug approvals, further expand
post-approval requirements, and restrict sales and promotional
activities. The new legislation, and the additional proposals if
enacted, may make it more difficult or burdensome for us to
obtain approval of our product candidates, any approvals we
receive may be more restrictive or be subject to onerous
post-approval requirements, our ability to successfully
commercialize approved products may be hindered and our business
may be harmed as a result.
If any
of our product candidates for which we receive regulatory
approval does not achieve broad market acceptance, the revenue
that we generate from its sales, if any, will be
limited.
The commercial success of our product candidates for which we
obtain marketing approval from the FDA or other regulatory
authorities will depend upon the acceptance of these products by
the medical community, including physicians, patients and health
care payors. The degree of market acceptance of any of our
approved products will depend on a number of factors, including:
|
|
|
| |
•
|
demonstration of clinical safety and efficacy compared to other
products;
|
| |
| |
•
|
the relative convenience, ease of administration and acceptance
by physicians and payors of varespladib in the treatment of
acute coronary syndrome,
A-623 in the
treatment of lupus and
A-001 in the
prevention of acute chest syndrome associated with sickle cell
disease;
|
| |
| |
•
|
the prevalence and severity of any adverse effects;
|
| |
| |
•
|
limitations or warnings contained in a product’s
FDA-approved labeling;
|
| |
| |
•
|
availability of alternative treatments, including, in the case
of varespladib, a number of competitive products being studied
for anti-inflammatory benefits in patients with acute coronary
syndrome or expected to be commercially launched in the near
future;
|
| |
| |
•
|
pricing and cost-effectiveness;
|
| |
| |
•
|
the effectiveness of our or any future collaborators’ sales
and marketing strategies;
|
| |
| |
•
|
our ability to obtain and maintain sufficient third-party
coverage or reimbursement from government health care programs,
including Medicare and Medicaid; and
|
| |
| |
•
|
the willingness of patients to pay
out-of-pocket
in the absence of third-party coverage.
|
46
If our product candidates are approved but do not achieve an
adequate level of acceptance by physicians, health care payors
and patients, we may not generate sufficient revenue from these
products, and we may not become or remain profitable. In
addition, our efforts to educate the medical community and
third-party payors on the benefits of our product candidates may
require significant resources and may never be successful.
Our
future success depends on our ability to retain our chief
executive officer and other key executives and to attract,
retain and motivate qualified personnel.
We are highly dependent on Mr. Paul F. Truex, our President
and Chief Executive Officer, Dr. Colin Hislop, our Senior
Vice President and Chief Medical Officer and the other principal
members of our executive team listed under “Executive
Officers” in Part III of this report. The loss of the
services of any of these persons might impede the achievement of
our research, development and commercialization objectives.
Recruiting and retaining qualified scientific personnel and
possibly sales and marketing personnel will also be critical to
our success. We may not be able to attract and retain these
personnel on acceptable terms given the competition among
numerous pharmaceutical and biotechnology companies for similar
personnel. We also experience competition for the hiring of
scientific personnel from universities and research
institutions. Failure to succeed in clinical studies may make it
more challenging to recruit and retain qualified scientific
personnel. In addition, we rely on consultants and advisors,
including scientific and clinical advisors, to assist us in
formulating our research and development and commercialization
strategy. Our consultants and advisors may be employed by
employers other than us and may have commitments under
consulting or advisory contracts with other entities that may
limit their availability to us.
Recently
enacted and future legislation or regulatory reform of the
health care system in the United States and foreign
jurisdictions may affect our ability to sell our products
profitably.
Our ability to commercialize our future products successfully,
alone or with collaborators, will depend in part on the extent
to which reimbursement for the products will be available from
government and health administration authorities, private health
insurers and other third-party payors. The continuing efforts of
the U.S. and foreign governments, insurance companies,
managed care organizations and other payors of health care
services to contain or reduce health care costs may adversely
affect our ability to set prices for our products which we
believe are fair, and our ability to generate revenues and
achieve and maintain profitability.
Specifically, in both the United States and some foreign
jurisdictions, there have been a number of legislative and
regulatory proposals to change the health care system in ways
that could affect our ability to sell our products profitably.
In March 2010, President Obama signed into law the Patient
Protection and Affordable Care Act, as amended by the Health
Care and Education Reconciliation Act, or collectively, the
Health Care Reform Law, a sweeping law intended to broaden
access to health insurance, reduce or constrain the growth of
healthcare spending, enhance remedies against fraud and abuse,
add new transparency requirements for healthcare and health
insurance industries, impose new taxes and fees on the health
industry and impose additional health policy reforms.
We will not know the full effects of the Health Care Reform Law
until applicable federal and state agencies issue regulations or
guidance under the new law. Although it is too early to
determine the effect of the Health Care Reform Law, the new law
appears likely to continue the pressure on pharmaceutical
pricing, especially under the Medicare program, and also may
increase our regulatory burdens and operating costs. We expect
further federal and state proposals and health care reforms to
continue to be proposed by legislators, which could limit the
prices that can be charged for the products we develop and may
limit our commercial opportunity.
Also in the United States, the Medicare Prescription Drug,
Improvement, and Modernization Act of 2003, also called the
Medicare Modernization Act, or MMA, changed the way Medicare
covers and pays for pharmaceutical products. The legislation
expanded Medicare coverage for drug purchases by the elderly and
introduced a new reimbursement methodology based on average
sales prices for drugs. In addition, this legislation authorized
Medicare Part D prescription drug plans to use formularies
where they can limit the number of drugs that will be covered in
any therapeutic class. As a result of this legislation and the
expansion of federal coverage of drug products, we expect that
there will be additional pressure to contain and reduce costs.
These cost reduction initiatives and other provisions of this
legislation could decrease the coverage and price that we
receive for any approved products and could seriously harm our
business. While the MMA applies only to drug benefits for
Medicare beneficiaries, private payors often follow Medicare
coverage policy and payment limitations in setting
47
their own reimbursement rates, and any reduction in
reimbursement that results from the MMA may result in a similar
reduction in payments from private payors.
The continuing efforts of government and other third-party
payors to contain or reduce the costs of health care through
various means may limit our commercial opportunity. It will be
time-consuming and expensive for us to go through the process of
seeking reimbursement from Medicare and private payors. Our
products may not be considered cost-effective, and government
and third-party private health insurance coverage and
reimbursement may not be available to patients for any of our
future products or sufficient to allow us to sell our products
on a competitive and profitable basis. Our results of operations
could be adversely affected by the MMA, the Health Care Reform
Law, and additional prescription drug coverage legislation, by
the possible effect of this legislation on amounts that private
insurers will pay and by other health care reforms that may be
enacted or adopted in the future. In addition, increasing
emphasis on managed care in the United States will continue to
put pressure on the pricing of pharmaceutical products. Cost
control initiatives could decrease the price that we or any
potential collaborators could receive for any of our future
products and could adversely affect our profitability.
In some foreign countries, including major markets in the
European Union and Japan, the pricing of prescription
pharmaceuticals is subject to governmental control. In these
countries, pricing negotiations with governmental authorities
can take six to 12 months or longer after the receipt of
regulatory marketing approval for a product. To obtain
reimbursement or pricing approval in some countries, we may be
required to conduct a clinical study that compares the
cost-effectiveness of our product candidates to other available
therapies. Such pharmacoeconomic studies can be costly and the
results uncertain. Our business could be harmed if reimbursement
of our products is unavailable or limited in scope or amount or
if pricing is set at unsatisfactory levels.
We
face potential product liability exposure, and, if successful
claims are brought against us, we may incur substantial
liability.
The use of our product candidates in clinical studies and the
sale of any products for which we obtain marketing approval
expose us to the risk of product liability claims. Product
liability claims might be brought against us by consumers,
health care providers, pharmaceutical companies or others
selling or otherwise coming into contact with our products. If
we cannot successfully defend ourselves against product
liability claims, we could incur substantial liabilities. In
addition, regardless of merit or eventual outcome, product
liability claims may result in:
|
|
|
| |
•
|
impairment of our business reputation;
|
| |
| |
•
|
withdrawal of clinical study participants;
|
| |
| |
•
|
costs of related litigation;
|
| |
| |
•
|
distraction of management’s attention from our primary
business;
|
| |
| |
•
|
substantial monetary awards to patients or other claimants;
|
| |
| |
•
|
the inability to commercialize our product candidates; and
|
| |
| |
•
|
decreased demand for our product candidates, if approved for
commercial sale.
|
Our product liability insurance coverage for our clinical
studies may not be sufficient to reimburse us for all expenses
or losses we may suffer. Moreover, insurance coverage is
becoming increasingly expensive, and, in the future, we may not
be able to maintain insurance coverage at a reasonable cost or
in sufficient amounts to protect us against losses due to
liability. If and when we obtain marketing approval for any of
our product candidates, we intend to expand our insurance
coverage to include the sale of commercial products; however, we
may be unable to obtain this product liability insurance on
commercially reasonable terms. On occasion, large judgments have
been awarded in class action lawsuits based on drugs that had
unanticipated adverse effects. A successful product liability
claim or series of claims brought against us could cause our
stock price to decline and, if judgments exceed our insurance
coverage, could decrease our cash and adversely affect our
business.
If we
use hazardous and biological materials in a manner that causes
injury or violates applicable law, we may be liable for
damages.
Our research and development activities involve the controlled
use of potentially hazardous substances, including toxic
chemical and biological materials. We could be held liable for
any contamination, injury or other damages resulting from these
hazardous substances. In addition, our operations produce
hazardous waste products.
48
While third parties are responsible for disposal of our
hazardous waste, we could be liable under environmental laws for
any required cleanup of sites at which our waste is disposed.
Federal, state, foreign and local laws and regulations govern
the use, manufacture, storage, handling and disposal of these
hazardous materials. If we fail to comply with these laws and
regulations at any time, or if they change, we may be subject to
criminal sanctions and substantial civil liabilities, which may
harm our business. Even if we continue to comply with all
applicable laws and regulations regarding hazardous materials,
we cannot eliminate the risk of accidental contamination or
discharge and our resultant liability for any injuries or other
damages caused by these accidents.
We
rely on third parties to conduct, supervise and monitor our
clinical studies, and those third parties may perform in an
unsatisfactory manner, such as by failing to meet established
deadlines for the completion of these clinical studies, or may
harm our business if they suffer a catastrophic
event.
We rely on third parties such as CROs, medical institutions and
clinical investigators to enroll qualified patients and conduct,
supervise and monitor our clinical studies. Our reliance on
these third parties for clinical development activities reduces
our control over these activities. Our reliance on these third
parties, however, does not relieve us of our regulatory
responsibilities, including ensuring that our clinical studies
are conducted in accordance with good clinical practices, or
GCP, and the investigational plan and protocols contained in the
relevant regulatory application, such as the investigational new
drug application, or IND. In addition, the CROs with whom we
contract may not complete activities on schedule, or may not
conduct our preclinical studies or clinical studies in
accordance with regulatory requirements or our clinical study
design. If these third parties do not successfully carry out
their contractual duties or meet expected deadlines, our efforts
to obtain regulatory approvals for, and to commercialize, our
product candidates may be delayed or prevented. In addition, if
a catastrophe such as an earthquake, fire, flood or power loss
should affect one of the third parties on which we rely, our
business prospects could be harmed. For example, if a central
laboratory holding all of our clinical study samples were to
suffer a catastrophic loss of their facility, we would lose all
of our samples and would have to repeat our studies.
Any
failure by our third-party manufacturers on which we rely to
produce our preclinical and clinical drug supplies and on which
we intend to rely to produce commercial supplies of any approved
product candidates may delay or impair our ability to
commercialize our product candidates.
We have relied upon a small number of third-party manufacturers
and active pharmaceutical ingredient formulators for the
manufacture of our material for preclinical and clinical testing
purposes and intend to continue to do so in the future. We also
expect to rely upon third parties to produce materials required
for the commercial production of our product candidates if we
succeed in obtaining necessary regulatory approvals. If we are
unable to arrange for third-party manufacturing sources, or to
do so on commercially reasonable terms, we may not be able to
complete development of our product candidates or market them.
Reliance on third-party manufacturers entails risks to which we
would not be subject if we manufactured product candidates
ourselves, including reliance on the third party for regulatory
compliance and quality assurance, the possibility of breach of
the manufacturing agreement by the third party because of
factors beyond our control (including a failure to synthesize
and manufacture our product candidates in accordance with our
product specifications) and the possibility of termination or
nonrenewal of the agreement by the third party, based on its own
business priorities, at a time that is costly or damaging to us.
In addition, the FDA and other regulatory authorities require
that our product candidates be manufactured according to cGMP
and similar foreign standards. Any failure by our third-party
manufacturers to comply with cGMP or failure to scale up
manufacturing processes, including any failure to deliver
sufficient quantities of product candidates in a timely manner,
could lead to a delay in, or failure to obtain, regulatory
approval of any of our product candidates. In addition, such
failure could be the basis for action by the FDA to withdraw
approvals for product candidates previously granted to us and
for other regulatory action, including recall or seizure, total
or partial suspension of production or injunction.
As part of our discussions to reactivate our US IND for
A-623, we
received a request from the FDA for additional information
regarding the characterization and qualification of the already
manufactured vials of
A-623 and
plans for any future manufactured vials of
A-623 that
we intend to use in clinical studies. In response to this
request, we provided the FDA additional analytical data
regarding all lots of previously manufactured
A-623 to be
utilized in the current PEARL-SC clinical study. In addition,
since new vials of
A-623 will
be manufactured at a new facility by our partner Merck
Biomanufacturing Network (recently acquired by Fujifilm ), we
are preparing and will
49
submit a comparability protocol to the FDA to establish
appropriate comparability and specifications requirements of
newly manufactured vials of
A-623 to be
included in any future clinical studies. We anticipate
submitting this protocol to the FDA in Q2 2011. Should the FDA
not agree with our comparability protocol proposal or if we are
unable to agree on the specifications for future
A-623
manufacturing, further clinical development of
A-623 beyond
the PEARL-SC clinical study would be substantially delayed and
we would incur substantial additional expense.
We rely on our manufacturers to purchase from third-party
suppliers the materials necessary to produce our product
candidates for our clinical studies. There are a small number of
suppliers for certain capital equipment and raw materials that
we use to manufacture our drugs. Such suppliers may not sell
these raw materials to our manufacturers at the times we need
them or on commercially reasonable terms. We do not have any
control over the process or timing of the acquisition of these
raw materials by our manufacturers. Moreover, we currently do
not have any agreements for the commercial production of these
raw materials. Although we generally do not begin a clinical
study unless we believe we have a sufficient supply of a product
candidate to complete the clinical study, any significant delay
in the supply of a product candidate or the raw material
components thereof for an ongoing clinical study due to the need
to replace a third-party manufacturer could considerably delay
completion of our clinical studies, product testing and
potential regulatory approval of our product candidates. If our
manufacturers or we are unable to purchase these raw materials
after regulatory approval has been obtained for our product
candidates, the commercial launch of our product candidates
would be delayed or there would be a shortage in supply of such
product candidates, which would impair our ability to generate
revenues from the sale of our product candidates.
Because of the complex nature of our compounds, our
manufacturers may not be able to manufacture our compounds at a
cost or in quantities or in a timely manner necessary to make
commercially successful products. If we successfully
commercialize any of our drugs, we may be required to establish
large-scale commercial manufacturing capabilities. In addition,
as our drug development pipeline increases and matures, we will
have a greater need for clinical study and commercial
manufacturing capacity. We have no experience manufacturing
pharmaceutical products on a commercial scale and some of these
suppliers will need to increase their scale of production to
meet our projected needs for commercial manufacturing, the
satisfaction of which on a timely basis may not be met.
If we
are unable to establish sales and marketing capabilities or
enter into agreements with third parties to market and sell our
product candidates, we may be unable to generate any
revenue.
We do not currently have an organization for the sales,
marketing and distribution of pharmaceutical products and the
cost of establishing and maintaining such an organization may
exceed the cost-effectiveness of doing so. In order to market
any products that may be approved by the FDA, we must build our
sales, marketing, managerial and other non-technical
capabilities or make arrangements with third parties to perform
these services. If we are unable to establish adequate sales,
marketing and distribution capabilities, whether independently
or with third parties, we may not be able to generate product
revenue and may not become profitable. We will be competing with
many companies that currently have extensive and well-funded
marketing and sales operations. Without an internal team or the
support of a third party to perform marketing and sales
functions, we may be unable to compete successfully against
these more established companies.
Guidelines
and recommendations published by various organizations may
adversely affect the use of any products for which we may
receive regulatory approval.
Government agencies issue regulations and guidelines directly
applicable to us and to our product candidates. In addition,
professional societies, practice management groups, private
health or science foundations and organizations involved in
various diseases from time to time publish guidelines or
recommendations to the medical and patient communities. These
various sorts of recommendations may relate to such matters as
product usage and use of related or competing therapies. For
example, organizations like the American Heart Association have
made recommendations about therapies in the cardiovascular
therapeutics market. Changes to these recommendations or other
guidelines advocating alternative therapies could result in
decreased use of any products for which we may receive
regulatory approval, which may adversely affect our results of
operations.
50
Risks
Related to Our Intellectual Property
If our
or our licensors’ patent positions do not adequately
protect our product candidates or any future products, others
could compete with us more directly, which would harm our
business.
As of the date of this report and as described in the section
entitled “Business — Intellectual Property”
on page 25, we hold a total of four pending
U.S. non-provisional patent applications, two pending
U.S. provisional patent applications and two pending Patent
Cooperation Treaty, or PCT, patent applications. Another PCT
application has entered the national phase in the European
Patent Office, the Eurasian Patent Organization and 17 other
countries. We have also entered into exclusive license
agreements for certain composition of matter, method of use and
method of making patents and patent applications for certain of
our development compounds. These license agreements encompass
(i) 13 U.S. patents, one pending
U.S. non-provisional patent application, five European, or
EP, patents, one pending EP patent application, 20 non-EP
foreign patents and three pending non-EP foreign patent
applications relating to varespladib and
A-001;
(ii) more than 30 U.S. patents, one pending
U.S. non-provisional patent application, six EP patents,
one pending EP patent application, 13 issued non-EP foreign
patents and one pending non-EP foreign patent applications
relating to other
sPLA2
inhibiting compounds including
A-003; and
(iii) two U.S. patents, one pending
U.S. non-provisional patent application, one EP patent, two
pending EP patent applications, eleven non-EP foreign patents
and 13 non-EP foreign patent applications relating to
A-623. Our
commercial success will depend in part on our and our
licensors’ ability to obtain additional patents and protect
our existing patent positions, particularly those patents for
which we have secured exclusive rights, as well as our ability
to maintain adequate protection of other intellectual property
for our technologies, product candidates and any future products
in the United States and other countries. If we or our licensors
do not adequately protect our intellectual property, competitors
may be able to use our technologies and erode or negate any
competitive advantage we may have, which could materially harm
our business, negatively affect our position in the marketplace,
limit our ability to commercialize our product candidates and
delay or render impossible our achievement of profitability. The
laws of some foreign countries do not protect our proprietary
rights to the same extent as the laws of the United States, and
we may encounter significant problems in protecting our
proprietary rights in these countries.
The patent positions of biotechnology and pharmaceutical
companies, including our patent position, involve complex legal
and factual questions, and, therefore, validity and
enforceability cannot be predicted with certainty. Patents may
be challenged, deemed unenforceable, invalidated or
circumvented. We and our licensors will be able to protect our
proprietary rights from unauthorized use by third parties only
to the extent that our proprietary technologies, product
candidates and any future products are covered by valid and
enforceable patents or are effectively maintained as trade
secrets.
The degree of future protection for our proprietary rights is
uncertain, and we cannot ensure that:
|
|
|
| |
•
|
we or our licensors were the first to make the inventions
covered by each of our pending patent applications;
|
| |
| |
•
|
we or our licensors were the first to file patent applications
for these inventions;
|
| |
| |
•
|
others will not independently develop similar or alternative
technologies or duplicate any of our technologies;
|
| |
| |
•
|
any of our or our licensors’ pending patent applications
will result in issued patents;
|
| |
| |
•
|
any of our or our licensors’ patents will be valid or
enforceable;
|
| |
| |
•
|
any patents issued to us or our licensors and collaborators will
provide a basis for commercially viable products, will provide
us with any competitive advantages or will not be challenged by
third parties;
|
| |
| |
•
|
we will develop additional proprietary technologies or product
candidates that are patentable; or
|
| |
| |
•
|
the patents of others will not have an adverse effect on our
business.
|
We may
be unable to adequately prevent disclosure of trade secrets and
other proprietary information.
We rely on trade secrets to protect our proprietary know-how and
technological advances, especially where we do not believe
patent protection is appropriate or obtainable. However, trade
secrets are difficult to protect. We rely in part on
confidentiality agreements with our employees, consultants,
outside scientific collaborators, sponsored researchers and
other advisors to protect our trade secrets and other
proprietary information. These agreements may
51
not effectively prevent disclosure of confidential information
and may not provide an adequate remedy in the event of
unauthorized disclosure of confidential information. In
addition, others may independently discover our trade secrets
and proprietary information. Costly and time-consuming
litigation could be necessary to enforce and determine the scope
of our proprietary rights. Failure to obtain or maintain trade
secret protection could enable competitors to use our
proprietary information to develop products that compete with
our products or cause additional, material adverse effects upon
our competitive business position.
We
license patent rights from third-party owners. If we, or such
owners, do not properly maintain or enforce the patents
underlying such licenses, our competitive position and business
prospects will be harmed.
We have obtained exclusive, worldwide licenses, except for
Japan, of the composition of matter, methods of making and
methods of use for certain
sPLA2
compounds from Eli Lilly and Shionogi & Co., Ltd. In
addition, we are party to a license agreement with Amgen that
provides exclusive and worldwide rights to develop and
commercialize
A-623, a
novel BAFF inhibitor, as well as non-exclusive rights to certain
technology relating to peptibody compositions and formulations.
We may enter into additional licenses to third-party
intellectual property in the future.
We depend in part on our licensors to protect the proprietary
rights covering our in-licensed
sPLA2
compounds and
A-623,
respectively. Our licensors are responsible for maintaining
certain issued patents and prosecuting certain patent
applications. We have limited, if any, control over the amount
or timing of resources that our licensors devote on our behalf
or the priority they place on maintaining these patent rights
and prosecuting these patent applications to our advantage. Our
licensors may also be notified of alleged infringement and be
sued for infringement of third-party patents or other
proprietary rights. We may have limited, if any, control or
involvement over the defense of these claims, and our licensors
could be subject to injunctions and temporary or permanent
exclusionary orders in the United States or other countries. Our
licensors are not obligated to defend or assist in our defense
against third-party claims of infringement. We have limited, if
any, control over the amount or timing of resources, if any,
that our licensors devote on our behalf or the priority they
place on defense of such third-party claims of infringement.
Our success will depend in part on the ability of us or our
licensors to obtain, maintain and enforce patent protection for
their intellectual property, in particular, those patents to
which we have secured exclusive rights. We or our licensors may
not successfully prosecute the patent applications which we have
licensed. Even if patents issue in respect of these patent
applications, we or our licensors may fail to maintain these
patents, may determine not to pursue litigation against other
companies that are infringing these patents or may pursue such
litigation less aggressively than we would. Without protection
for the intellectual property we license, other companies might
be able to offer substantially identical products for sale,
which could adversely affect our competitive business position
and harm our business prospects.
If we
do not obtain protection under the Hatch-Waxman Act and similar
foreign legislation to extend our licensed patent terms and to
obtain market exclusivity for our product candidates, our
business will be materially harmed.
The United States Drug Price Competition and Patent Term
Restoration Act of 1984, more commonly known as the
“Hatch-Waxman Act,” provides for an extension of
patent term for drug compounds for a period of up to five years
to compensate for time spent in the regulatory approval process.
Assuming we gain a five-year patent term extension for each of
our current product candidates in clinical development, and that
we continue to have rights under our license agreements with
respect to these product candidates, we would have exclusive
rights to varespladib’s U.S. “new chemical
entity” patent (the primary patent covering the compound as
a new composition of matter) until 2019 and to
A-623’s
U.S. new chemical entity patent until 2027. In Europe,
similar legislative enactments allow patent terms in the
European Union to be extended for up to five years through the
grant of a Supplementary Protection Certificate. Assuming we
gain such a five-year extension for each of our current product
candidates in clinical development, and that we continue to have
rights under our license agreements with respect to these
product candidates, we would have exclusive rights to
varespladib’s European new chemical entity patents until
2020 and to
A-623’s
European new chemical entity patents until 2027. In addition,
since varespladib has not been previously approved in the United
States, varespladib could be eligible for up to five years of
New Chemical
52
Entity, or NCE, exclusivity from the FDA. NCE exclusivity would
prevent the FDA from approving any generic competitor following
NDA approval independent of the patent status of varespladib.
Further, since
A-623 has
not been previously approved,
A-623 could
be eligible for 12 years of data exclusivity from the FDA.
During the data exclusivity period, competitors are barred from
relying on the innovator biologic’s safety and efficacy
data to gain approval. Similarly, the European Union provides
that companies who receive regulatory approval for a new small
molecule compound or biologic will have a
10-year
period of data exclusivity for that compound or biologic (with
the possibility of a further one-year extension) in most EU
countries, beginning on the date of such European regulatory
approval, regardless of when the European new chemical entity
patent covering such compound expires. A generic version of the
approved drug may not be marketed or sold during such market
exclusivity period. However, there is no assurance that we will
receive the extensions of our patents or other exclusive rights
available under the Hatch-Waxman Act or similar foreign
legislation. If we fail to receive such Hatch-Waxman extensions
or marketing exclusivity rights or if we receive extensions that
are materially shorter than expected, our ability to prevent
competitors from manufacturing, marketing and selling generic
versions of our products will be materially harmed.
Our
current patent positions and license portfolio may not include
all patent rights needed for the full development and
commercialization of our product candidates. We cannot be sure
that patent rights we may need in the future will be available
for license to us on commercially reasonable terms, or at
all.
We typically develop our product candidates using compounds for
which we have in-licensed and original composition of matter
patents and patents that claim the activities and methods for
such compounds’ production and use to the extent known at
that time. As we learn more about the mechanisms of action and
new methods of manufacture and use of these product candidates,
we may file additional patent applications for these new
inventions or we may need to ask our licensors to file them. We
may also need to license additional patent rights or other
rights on compounds, treatment methods or manufacturing
processes because we learn that we need such rights during the
continuing development of our product candidates.
Although our in-licensed and original patents may prevent others
from making, using or selling similar products, they do not
ensure that we will not infringe the patent rights of third
parties. We may not be aware of all patents or patent
applications that may impact our ability to make, use or sell
any of our product candidates or proposed product candidates.
For example, because we sometimes identify the mechanism of
action or molecular target of a given product candidate after
identifying its composition of matter and therapeutic use, we
may not be aware until the mechanism or target is further
elucidated that a third party has an issued or pending patent
claiming biological activities or targets that may cover our
product candidate. U.S. patent applications filed after
November 29, 2000 are confidential in the U.S. Patent
and Trademark Office for the first 18 months after such
applications’ earliest priority date, and patent offices in
non-U.S. countries
often publish patent applications for the first time six months
or more after filing. Furthermore, we may not be aware of
published or granted conflicting patent rights. Any conflicts
resulting from patent applications and patents of others could
significantly reduce the coverage of our patents and limit our
ability to obtain meaningful patent protection. If others obtain
patents with conflicting claims, we may need to obtain licenses
to these patents or to develop or obtain alternative technology.
We may not be able to obtain any licenses or other rights to
patents, technology or know-how from third parties necessary to
conduct our business as described in this report and such
licenses, if available at all, may not be available on
commercially reasonable terms. Any failure to obtain such
licenses could delay or prevent us from developing or
commercializing our drug candidates or proposed product
candidates, which would harm our business. Litigation or patent
interference proceedings may be necessarily brought against
third parties, as discussed below, to enforce any of our patents
or other proprietary rights or to determine the scope and
validity or enforceability of the proprietary rights of such
third parties.
Litigation
regarding patents, patent applications and other proprietary
rights may be expensive and time consuming. If we are involved
in such litigation, it could cause delays in bringing product
candidates to market and harm our ability to
operate.
Our commercial success will depend in part on our ability to
manufacture, use, sell and offer to sell our product candidates
and proposed product candidates without infringing patents or
other proprietary rights of third parties. Although we are not
currently aware of any litigation or other proceedings or
third-party claims of intellectual
53
property infringement related to our product candidates, the
pharmaceutical industry is characterized by extensive litigation
regarding patents and other intellectual property rights. Other
parties may obtain patents in the future and allege that the use
of our technologies infringes these patent claims or that we are
employing their proprietary technology without authorization.
Likewise, third parties may challenge or infringe upon our or
our licensors’ existing or future patents.
Proceedings involving our patents or patent applications or
those of others could result in adverse decisions regarding the
patentability of our inventions relating to our product
candidates or the enforceability, validity or scope of
protection offered by our patents relating to our product
candidates.
Even if we are successful in these proceedings, we may incur
substantial costs and divert management time and attention in
pursuing these proceedings. If we are unable to avoid infringing
the patent rights of others, we may be required to seek a
license, defend an infringement action or challenge the validity
of the patents in court. Patent litigation is costly and
time-consuming. We may not have sufficient resources to bring
these actions to a successful conclusion. In addition, if we do
not obtain a license, develop or obtain non-infringing
technology, fail to defend an infringement action successfully
or have our patents declared invalid, we may incur substantial
monetary damages; encounter significant delays in bringing our
product candidates to market; or be precluded from participating
in the manufacture, use or sale of our product candidates or
methods of treatment requiring licenses.
Risks
Related to the Securities Markets and Investment in Our Common
Stock
Market
volatility may affect our stock price and the value of your
investment.
The market price for our common stock has been, and is likely to
continue to be, volatile. In addition, the market price of our
common stock may fluctuate significantly in response to a number
of factors, most of which we cannot predict or control,
including:
|
|
|
| |
•
|
plans for, progress in and results from clinical studies for
varespladib,
A-623,
A-001 and
our other product candidates;
|
| |
| |
•
|
announcements of new products, services or technologies,
commercial relationships, acquisitions or other events by us or
our competitors;
|
| |
| |
•
|
developments concerning proprietary rights, including those
pertaining to patents held by Eli Lilly and Shionogi &
Co., Ltd. concerning our
sPLA2
inhibitors and Amgen concerning
A-623;
|
| |
| |
•
|
failure of any of our product candidates, if approved, to
achieve commercial success;
|
| |
| |
•
|
fluctuations in stock market prices and trading volumes of
securities of similar companies;
|
| |
| |
•
|
general market conditions and overall fluctuations in
U.S. equity markets;
|
| |
| |
•
|
variations in our operating results, or the operating results of
our competitors;
|
| |
| |
•
|
changes in our financial guidance or securities analysts’
estimates of our financial performance;
|
| |
| |
•
|
changes in accounting principles;
|
| |
| |
•
|
sales of large blocks of our common stock, including sales by
our executive officers, directors and significant stockholders;
|
| |
| |
•
|
additions or departures of any of our key personnel;
|
| |
| |
•
|
announcements related to litigation;
|
| |
| |
•
|
changing legal or regulatory developments in the United States
and other countries; and
|
| |
| |
•
|
discussion of us or our stock price by the financial press and
in online investor communities.
|
Although our common stock is listed for trading on the NASDAQ
Global Market, our securities have been relatively thinly
traded. Investor trading patterns could serve to exacerbate the
volatility of the price of the stock. Accordingly, it may be
difficult to sell shares of common stock quickly without
significantly depressing the value of the stock. Unless we are
successful in developing continued investor interest in our
stock, sales of our stock could result in major fluctuations in
the price of the stock. In addition, the stock market in
general, and The NASDAQ Global Market in particular, have
experienced substantial price and volume volatility that is
often seemingly unrelated to the operating performance of
particular companies. These broad market fluctuations may cause
the
54
trading price of our common stock to decline. In the past,
securities class action litigation has often been brought
against a company after a period of volatility in the market
price of its common stock. We may become involved in this type
of litigation in the future. Any securities litigation claims
brought against us could result in substantial expenses and the
diversion of our management’s attention from our business.
Because
a small number of our existing stockholders own a majority of
our voting stock, your ability to influence corporate matters
will be limited.
Our executive officers, directors and greater than 5%
stockholders, in the aggregate, own approximately 75% of our
outstanding common stock. As a result, such persons, acting
together, will have the ability to control our management and
affairs and substantially all matters submitted to our
stockholders for approval, including the election and removal of
directors and approval of any significant transaction. These
persons will also have the ability to control our management and
business affairs. This concentration of ownership may have the
effect of delaying, deferring or preventing a change in control,
impeding a merger, consolidation, takeover or other business
combination involving us, or discouraging a potential acquirer
from making a tender offer or otherwise attempting to obtain
control of our business, even if such a transaction would
benefit other stockholders.
Future
sales of our common stock may cause our stock price to
decline.
As of December 31, 2010, there were 32,880,353 shares
of our common stock outstanding. In addition, as of
December 31, 2010, we had outstanding options to purchase
shares of our common stock and restricted stock units of
1,578,491 that, if exercised or released, will result in these
additional shares becoming available for sale. A large portion
of these shares and outstanding equity awards are held by a
small number of persons and investment funds. Sales by these
stockholders or option holders of a substantial number of shares
could significantly reduce the market price of our common stock.
Moreover, certain holders of shares of common stock will have
rights, subject to some conditions, to require us to file
registration statements covering the shares they currently hold,
or to include these shares in registration statements that we
may file for ourselves or other stockholders.
We have registered all common stock that we may issue under our
Amended and Restated 2010 Stock Option and Incentive Plan (the
“2010 Plan”) and our Employee Stock Purchase Plan (the
“ESPP”). As of February 28, 2011, an aggregate of
1,778,261 shares of our common stock has been reserved for
future issuance under the 2010 Plan, plus any shares reserved
and unissued under our 2005 Equity Incentive Plan, and an
aggregate of 350,000 shares has been reserved for future
issuance under our ESPP. These shares can be freely sold in the
public market upon issuance, subject to the
lock-up
agreements referred to above. If a large number of these shares
are sold in the public market, the sales could reduce the
trading price of our common stock.
We may
need to raise additional capital to fund our operations, which
may cause dilution to our existing stockholders, restrict our
operations or require us to relinquish rights.
We may seek additional capital through a combination of private
and public equity offerings, debt financings and collaboration,
strategic and licensing arrangements. To the extent that we
raise additional capital through the sale of equity or
convertible debt securities, your ownership interest may be
diluted, and the terms may include liquidation or other
preferences that adversely affect your rights as a stockholder.
Debt financing, if available, may involve agreements that
include covenants limiting or restricting our ability to take
specific actions such as incurring debt, making capital
expenditures or declaring dividends. If we raise additional
funds through collaboration, strategic alliance and licensing
arrangements with third parties, we may have to relinquish
valuable rights to our technologies or product candidates or
grant licenses on terms that are not favorable to us.
Operating
as a public company increases our expenses and administrative
burden.
As a public company, we incur significant legal, accounting and
other expenses that we did not incur as a private company. In
addition, our administrative staff will be required to perform
additional tasks. For example, the Sarbanes-Oxley Act of 2002,
or the Sarbanes-Oxley Act, as well as rules subsequently
implemented by the Securities and Exchange Commission, or SEC,
and The NASDAQ Global Market, impose various requirements on
public companies, including establishment and maintenance of
effective disclosure and financial controls and changes in
corporate governance practices. We must also bear all of the
internal and external costs of preparing and distributing
periodic public reports in compliance with our obligations under
the securities laws.
55
In particular, the Sarbanes-Oxley Act requires, among other
things, that we maintain effective internal control over
financial reporting and disclosure controls and procedures.
Commencing in 2011, we must perform system and process
evaluation and testing of our internal control over financial
reporting to allow management and our independent registered
public accounting firm to report on the effectiveness of our
internal control over financial reporting, as required by
Section 404 of the Sarbanes-Oxley Act. Our compliance with
Section 404 will require that we incur substantial
accounting expense and expend significant management time on
compliance-related issues. Moreover, if we are not able to
comply with the requirements of Section 404 in a timely
manner, our stock price could decline, and we could face
sanctions, delisting or investigations by The NASDAQ Global
Market, or other material adverse effects on our business,
reputation, results of operations, financial condition or
liquidity.
We do
not intend to pay dividends on our common stock so any returns
will be limited to the value of our stock.
We have never declared or paid any cash dividend on our common
stock. We currently anticipate that we will retain future
earnings for the development, operation and expansion of our
business and do not anticipate declaring or paying any cash
dividends for the foreseeable future. Any return to stockholders
will therefore be limited to the value of their stock.
Anti-takeover
provisions in our charter documents and under Delaware law could
make an acquisition of us, which may be beneficial to our
stockholders, more difficult and may prevent attempts by our
stockholders to replace or remove our current
management.
Provisions in our amended and restated certificate of
incorporation and amended and restated bylaws may delay or
prevent an acquisition of us or a change in our management.
These provisions include:
|
|
|
| |
•
|
a classified and staggered board of directors whose members can
only be dismissed for cause;
|
| |
| |
•
|
the prohibition on actions by written consent of our
stockholders;
|
| |
| |
•
|
the limitation on who may call a special meeting of stockholders;
|
| |
| |
•
|
the establishment of advance notice requirements for nominations
for election to our board of directors or for proposing matters
that can be acted upon at stockholder meetings;
|
| |
| |
•
|
the ability of our board of directors to issue preferred stock
without stockholder approval, which would increase the number of
outstanding shares and could thwart a takeover attempt; and
|
| |
| |
•
|
the requirement of at least 75% of the outstanding common stock
to amend any of the foregoing provisions.
|
In addition, because we are incorporated in Delaware, we are
governed by the provisions of Section 203 of the Delaware
General Corporation Law, which limits the ability of
stockholders owning in excess of 15% of our outstanding voting
stock to merge or combine with us. Although we believe these
provisions collectively provide for an opportunity to obtain
greater value for stockholders by requiring potential acquirors
to negotiate with our board of directors, they would apply even
if an offer rejected by our board were considered beneficial by
some stockholders. In addition, these provisions may frustrate
or prevent any attempts by our stockholders to replace or remove
our current management by making it more difficult for
stockholders to replace members of our board of directors, which
is responsible for appointing the members of our management.
Our
ability to use our net operating loss carryforwards may be
subject to limitation and may result in increased future tax
liability to us.
Generally, a change of more than 50% in the ownership of a
corporation’s stock, by value, over a three-year period
constitutes an ownership change for U.S. federal income tax
purposes. An ownership change may limit a company’s ability
to use its net operating loss carryforwards attributable to the
period prior to such change. We have not performed a detailed
analysis to determine whether an ownership change under
Section 382 of the Internal Revenue Code has occurred after
each of our previous private placements of preferred stock and
convertible debt, or our previous issuances of common stock,
which if sufficient, taking into account prior or future shifts
in our ownership over a three-year period, could cause us to
undergo an ownership change. As a result, if we earn net taxable
income, our ability to use our pre-change net operating loss
carryforwards to offset U.S. federal taxable income may
become subject to limitations, which could potentially result in
increased future tax liability to us.
56
|
|
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS
|
Not applicable.
We are currently subleasing approximately 7,800 square feet
of office space in Hayward, California, which we occupy under a
sublease that commenced on October 1, 2008 and expired on
January 31, 2011. In January 2011, we renewed our sublease
through July 31, 2011. We believe our existing facilities
are adequate for our current needs and that any additional space
we need will be available in the future on commercially
reasonable terms.
|
|
|
ITEM 3.
|
LEGAL
PROCEEDINGS
|
From time to time, we may be involved in routine legal
proceedings, as well as demands, claims and threatened
litigation, which arise in the normal course of our business. We
believe there is no litigation pending that could, individually
or in the aggregate, have a material adverse effect on our
results of operations or financial condition.
|
|
|
ITEM 4.
|
REMOVED
AND RESERVED
|
57
PART II
|
|
|
ITEM 5.
|
MARKET
FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS
AND ISSUER PURCHASES OF EQUITY SECURITIES
|
Price
Range of Common Stock
Our common stock has been listed on The NASDAQ Global Market
under the symbol “ANTH” since our IPO. Prior to that
offering, there was no public market for our common stock. The
following table sets forth, for the periods indicated, the high
and low intraday sales prices of our common stock as reported by
The NASDAQ Global Market:
| |
|
|
|
|
|
|
|
|
|
|
|
High
|
|
|
Low
|
|
|
|
|
First Quarter 2010 (beginning March 1, 2010)
|
|
$
|
7.39
|
|
|
$
|
6.86
|
|
|
Second Quarter 2010
|
|
$
|
8.55
|
|
|
$
|
5.07
|
|
|
Third Quarter 2010
|
|
$
|
5.99
|
|
|
$
|
2.82
|
|
|
Fourth Quarter 2010
|
|
$
|
6.90
|
|
|
$
|
4.12
|
|
Holders
of our Common Stock
As of December 31, 2010, an aggregate of
32,880,353 shares of our common stock were issued and
outstanding and were held by approximately 850 holders of record
and beneficial holders, based on information provided by the
Company’s transfer agent.
58
Performance
Graph
The following graph shows a comparison of cumulative total
return of our common stock, the NASDAQ Composite Index and the
Nasdaq Biotech Index from March 1, 2010 (the date of our
IPO) through December 31, 2010. The graph and table assume
that $100 was invested on March 1, 2010 in each of our
common stock, the NASDAQ Composite Index and the NASDAQ Biotech
Index, and that all dividends were reinvested. The past
performance of our common stock is no indication of future
performance.
COMPARISON
OF TOTAL RETURN
Among Anthera Pharmaceuticals, Inc., The NASDAQ Composite
Index
And The NASDAQ Biotech Index
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3/1/2010
|
|
|
|
3/31/2010
|
|
|
|
6/30/2010
|
|
|
|
9/30/2010
|
|
|
|
12/31/2010
|
|
|
Anthera Pharmaceuticals, Inc.
|
|
|
$
|
100.00
|
|
|
|
$
|
99.71
|
|
|
|
$
|
76.46
|
|
|
|
$
|
59.77
|
|
|
|
$
|
69.61
|
|
|
Nasdaq Composite
|
|
|
$
|
100.00
|
|
|
|
$
|
105.47
|
|
|
|
$
|
92.77
|
|
|
|
$
|
104.18
|
|
|
|
$
|
116.68
|
|
|
Nasdaq Biotech
|
|
|
$
|
100.00
|
|
|
|
$
|
104.08
|
|
|
|
$
|
88.66
|
|
|
|
$
|
99.24
|
|
|
|
$
|
107.53
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dividend
Policy
We have never declared or paid any cash dividends on our common
stock. We currently intend to retain any future earnings to
finance the growth and development of our business. Therefore,
we do not anticipate declaring or paying any cash dividends in
the foreseeable future. Any future determination as to the
declaration and payment of dividends, if any, will be at the
discretion of our board of directors and will depend on then
existing conditions, including our financial condition,
operating results, contractual restrictions, capital
requirements, business prospects and other factors our board of
directors may deem relevant.
Securities
Authorized for Issuance under Equity Compensation
Plans
The information regarding securities authorized for issuance
under equity compensation plans is included in Part III of
this report.
Recent
Sales of Unregistered Securities
All information under this Item has been previously reported on
our Current Report on
Form 8-K,
filed with the Securities and Exchange Commission (the
“SEC”) on September 22, 2010.
Issuer
Purchases of Equity Securities
We did not repurchase any securities in the fourth quarter of
2010.
59
|
|
|
ITEM 6.
|
SELECTED
FINANCIAL DATA
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cumulative
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
2010
|
|
|
|
|
Statement of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
29,456,742
|
|
|
$
|
8,415,414
|
|
|
$
|
10,882,322
|
|
|
$
|
23,921,932
|
|
|
$
|
7,759,106
|
|
|
$
|
80,780,723
|
|
|
General and administrative
|
|
|
6,300,849
|
|
|
|
3,425,690
|
|
|
|
2,980,170
|
|
|
|
2,468,607
|
|
|
|
822,732
|
|
|
|
16,218,416
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
(35,757,591
|
)
|
|
|
(11,841,104
|
)
|
|
|
(13,862,492
|
)
|
|
|
(26,390,539
|
)
|
|
|
(8,581,838
|
)
|
|
|
(96,999,139
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other Income (Expense)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other expense and interest income, net
|
|
|
(859,733
|
)
|
|
|
(362,388
|
)
|
|
|
(118,174
|
)
|
|
|
696,962
|
|
|
|
92,592
|
|
|
|
(539,593
|
)
|
|
Mark-to-market
adjustment of warrant liability
|
|
|
(3,796,491
|
)
|
|
|
|
|
|
|
|
|
|
|
—
|
|
|
|
|
|
|
|
(3,796,491
|
)
|
|
Beneficial conversion feature
|
|
|
—
|
|
|
|
—
|
|
|
|
(4,118,544
|
)
|
|
|
—
|
|
|
|
(190,000
|
)
|
|
|
(4,308,544
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total other income (expense)
|
|
|
(4,656,224
|
)
|
|
|
(362,388
|
)
|
|
|
(4,236,718
|
)
|
|
|
696,962
|
|
|
|
(97,408
|
)
|
|
|
(8,644,628
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(40,413,815
|
)
|
|
$
|
(12,203,492
|
)
|
|
$
|
(18,099,210
|
)
|
|
$
|
(25,693,577
|
)
|
|
$
|
(8,679,246
|
)
|
|
$
|
(105,643,767
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share — basic and diluted(1)
|
|
$
|
(1.76
|
)
|
|
$
|
(8.06
|
)
|
|
$
|
(13.47
|
)
|
|
$
|
(28.15
|
)
|
|
$
|
(13.82
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average number of shares used in per share
calculation — basic and diluted(2)
|
|
|
22,909,802
|
|
|
|
1,513,598
|
|
|
|
1,343,420
|
|
|
|
912,668
|
|
|
|
627,904
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Diluted earnings per share, or EPS, is identical to basic EPS
since common equivalent shares are excluded from the
calculation, as their effect is anti-dilutive. |
| |
|
(2) |
|
For accounting purposes only, the number of issued and
outstanding shares for the years ended December 31, 2006,
2007, 2008, 2009 and 2010 do not include weighted-average shares
of unvested stock of 297,596, 261,649, 230,028, 110,079 and
47,654, respectively. These shares are subject to a risk of
repurchase by us until such shares are vested. See Note 2
of our financial statements for more information. |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
40,029,972
|
|
|
$
|
3,803,384
|
|
|
$
|
7,895,113
|
|
|
$
|
152,744
|
|
|
$
|
20,781,916
|
|
|
Short-term investments
|
|
|
23,350,922
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,825,000
|
|
|
|
—
|
|
|
Working capital
|
|
|
57,240,395
|
|
|
|
(14,344,436
|
)
|
|
|
(495,836
|
)
|
|
|
(2,907,995
|
)
|
|
|
19,629,639
|
|
|
Total assets
|
|
|
65,263,062
|
|
|
|
5,888,789
|
|
|
|
8,034,154
|
|
|
|
6,193,213
|
|
|
|
20,856,892
|
|
|
Total liabilities
|
|
|
8,005,382
|
|
|
|
18,167,645
|
|
|
|
8,494,417
|
|
|
|
12,058,184
|
|
|
|
1,174,621
|
|
|
Convertible preferred stock
|
|
|
—
|
|
|
|
52,123,859
|
|
|
|
52,123,859
|
|
|
|
28,892,004
|
|
|
|
28,892,004
|
|
|
Deficit accumulated during the development stage
|
|
|
(105,643,767
|
)
|
|
|
(65,229,952
|
)
|
|
|
(53,026,460
|
)
|
|
|
(34,927,250
|
)
|
|
|
(9,233,673
|
)
|
|
Total stockholders’ (deficit) equity
|
|
|
57,257,680
|
|
|
|
(12,278,856
|
)
|
|
|
(460,263
|
)
|
|
|
(5,864,971
|
)
|
|
|
19,682,271
|
|
60
|
|
|
ITEM 7.
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
The following discussion and analysis should be read together
with our financial statements and the related notes set forth
under “Item 8. Financial Statements and Supplementary
Data.” This discussion contains forward-looking statements
reflecting our current expectations that involve risks and
uncertainties. See “Special Note Regarding Forward-Looking
Statements” for a discussion of the uncertainties, risks
and assumptions associated with these statements. Actual results
and the timing of events could differ materially from those
discussed in our forward-looking statements as a result of many
factors, including those set forth under “Risk
Factors” and elsewhere in this report.
Overview
We are a biopharmaceutical company focused on developing and
commercializing products to treat serious diseases associated
with inflammation, including cardiovascular and autoimmune
diseases. We currently have one Phase 3 clinical program,
varespladib, and two Phase 2 clinical programs,
A-623 and
A-001. Two
of our product candidates, varespladib and
A-001, are
designed to inhibit a novel enzyme target known as secretory
phospholipase
A2,
or
sPLA2.
Elevated levels of
sPLA2
have been implicated in a variety of acute inflammatory
conditions, including acute coronary syndrome and acute chest
syndrome associated with sickle cell disease, as well as in
chronic diseases, including stable coronary artery disease. In
addition, our Phase 2 product candidate,
A-623,
targets elevated levels of B-cell activating factor, or BAFF,
which has been associated with a variety of B-cell mediated
autoimmune diseases, including systemic lupus erythematosus, or
lupus, lupus nephritis, rheumatoid arthritis, multiple
sclerosis, Sjögren’s Syndrome, Graves’ Disease
and others.
We were incorporated and commenced operations in September 2004.
Since our inception, we have generated significant losses. As of
December 31, 2010, we had an accumulated deficit of
approximately $105.6 million. As of the date of this
filing, we have never generated any revenue and have generated
only interest income from cash and cash equivalents and
short-term investments. We expect to incur substantial and
increasing losses for at least the next several years as we
pursue the development and commercialization of our product
candidates.
To date, we have funded our operations through equity offerings
and private placements of convertible debt, raising an aggregate
of approximately $145.7 million. We will need substantial
additional financing to continue to develop our product
candidates, obtain regulatory approvals and to fund operating
expenses, which we will seek to raise through public or private
equity or debt financings, collaborative or other arrangements
with third parties or through other sources of financing. We
cannot assure you that such funds will be available on terms
favorable to us, if at all. In addition to the normal risks
associated with development-stage companies, we may never
successfully complete development of any of our product
candidates, obtain adequate patent protection for our
technology, obtain necessary government regulatory approval for
our product candidates or achieve commercial viability for any
approved product candidates. In addition, we may not be
profitable even if we succeed in commercializing any of our
product candidates.
Revenue
To date, we have not generated any revenue. We do not expect to
generate revenue unless or until we obtain regulatory approval
of, and commercialize, our product candidates or in- license
additional products that generate revenue. We intend to seek to
generate revenue from a combination of product sales, up-front
fees and milestone payments in connection with collaborative or
strategic relationships and royalties resulting from the
licensing of the commercial rights to our intellectual property.
We expect that any revenue we generate will fluctuate from
quarter to quarter as a result of the nature, timing and amount
of milestone payments we may receive upon the sale of our
products, to the extent any are successfully commercialized, as
well as any revenue we may receive from our collaborative or
strategic relationships.
Research
and Development Expenses
Since our inception, we have focused our activities on our
product candidate development programs. We expense research and
development costs as they are incurred. Research and development
expenses consist of personnel costs, including salaries,
benefits and stock-based compensation, clinical studies
performed by contract research organizations, or CROs, materials
and supplies, licenses and fees and overhead allocations
consisting of
61
various administrative and facilities-related costs. Research
and development activities are also separated into three main
categories: licensing, clinical development and pharmaceutical
development. Licensing costs consist primarily of fees paid
pursuant to license agreements. Historically, our clinical
development costs have included costs for preclinical and
clinical studies. We expect to incur substantial clinical
development costs for our Phase 3 clinical study named VISTA- 16
for varespladib and for our Phase 2b clinical study named
PEARL-SC for
A-623, as
well as for the development of our other product candidates.
Pharmaceutical development costs consist of expenses incurred
relating to clinical studies and product formulation and
manufacturing.
We expense both internal and external research and development
costs as incurred. We are developing our product candidates in
parallel, and we typically use our employee and infrastructure
resources across several projects. Thus, some of our research
and development costs are not attributable to an individually
named project, but rather are allocated across our clinical
stage programs. These unallocated costs include salaries,
stock-based compensation charges and related “fringe
benefit” costs for our employees (such as workers
compensation and health insurance premiums), consulting fees and
travel.
The following table shows our total research and development
expenses for the years ended December 31, 2010, 2009 and
2008, and for the period from September 9, 2004 (Date of
Inception) through December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Period
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Allocated costs:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A-001
|
|
$
|
(11,688
|
)(1)
|
|
$
|
192,979
|
|
|
$
|
456,633
|
|
|
$
|
6,508,358
|
(1)
|
|
Varespladib
|
|
|
19,229,868
|
(1)(2)
|
|
|
5,535,529
|
|
|
|
7,370,850
|
|
|
|
47,090,513
|
(1)(2)(4)
|
|
A-623
|
|
|
5,826,475
|
(3)
|
|
|
34,179
|
|
|
|
100,851
|
|
|
|
11,969,892
|
(3)(5)
|
|
Unallocated costs
|
|
|
4,412,087
|
|
|
|
2,652,727
|
|
|
|
2,953,988
|
|
|
|
15,211,960
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total development
|
|
$
|
29,456,742
|
|
|
$
|
8,415,414
|
|
|
$
|
10,882,322
|
|
|
$
|
80,780,723
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes Qualifying Therapeutic Discovery Project Credit under
Section 48D of approximately $244,000. See Note 2 of
our financial statements for more information. |
| |
|
(2) |
|
Includes milestone payments of $3.5 million pursuant to
amendments to the license agreements with each of Eli Lilly and
Shionogi & Co. Ltd., which were paid in the form of
shares of common stock. |
| |
|
(3) |
|
Includes Qualifying Therapeutic Discovery Project Credit under
Section 48D of approximately $488,000. See Note 2 of
our financial statements for more information. |
| |
|
(4) |
|
Includes license fees of $4.0 million pursuant to a license
agreement with each of Eli Lilly and Shionogi & Co.
Ltd., which were paid in cash and shares of preferred stock in
2006. |
| |
|
(5) |
|
Includes a one-time license initiation fee of $6.0 million
pursuant to a license agreement with Amgen. |
We expect our research and development expenses to increase
significantly as we continue to develop our product candidates.
We began enrollment of patients in the VISTA-16 study of
varespladib for the treatment of patients experiencing acute
coronary syndrome in June 2010. We also initiated the PEARL-SC
study of
A-623 in
July 2010. We intend to fund our clinical studies with existing
cash and proceeds from potential future debt and equity
offerings.
We expect that a large percentage of our research and
development expenses in the future will be incurred in support
of our current and future clinical development programs. These
expenditures are subject to numerous uncertainties in timing and
cost to completion. As we obtain results from clinical studies,
we may elect to discontinue or delay clinical studies for
certain product candidates or programs in order to focus our
resources on more promising product candidates or programs.
Completion of clinical studies may take several years or more,
but the length of time generally varies according to the type,
complexity, novelty and intended use of a product candidate. The
cost of clinical studies may vary significantly over the life of
a program as a result of differences arising during clinical
development, including:
|
|
|
| |
•
|
the number of sites included in the studies;
|
62
|
|
|
| |
•
|
the length of time required to enroll suitable patient subjects;
|
| |
| |
•
|
the number of patients that participate in the studies;
|
| |
| |
•
|
the number of doses that patients receive;
|
| |
| |
•
|
the drop-out or discontinuation rates of patients; and
|
| |
| |
•
|
the duration of patient
follow-up.
|
Our expenses related to clinical studies are based on estimates
of the services received and efforts expended pursuant to
contracts with multiple research institutions and clinical
research organizations that conduct and manage clinical studies
on our behalf. The financial terms of these agreements are
subject to negotiation and vary from contract to contract and
may result in uneven payment flows. Generally, these agreements
set forth the scope of work to be performed at a fixed fee or
unit price. Payments under the contracts depend on factors such
as the successful enrollment of patients or the completion of
clinical study milestones. Expenses related to clinical studies
generally are accrued based on contracted amounts and the
achievement of milestones such as number of patients enrolled.
If timelines or contracts are modified based upon changes to the
clinical study design or scope of work to be performed, we
modify our estimates of accrued expenses accordingly on a
prospective basis.
None of our product candidates has received U.S. Food and
Drug Administration, or FDA, or foreign regulatory marketing
approval. In order to grant marketing approval, the FDA or
foreign regulatory agencies must conclude that clinical data
establishes the safety and efficacy of our product candidates
and that the manufacturing facilities, processes and controls
are adequate. Despite our efforts, our product candidates may
not offer therapeutic or other improvement over existing,
comparable drugs, be proven safe and effective in clinical
studies, or meet applicable regulatory standards.
As a result of the uncertainties discussed above, we are unable
to determine the duration and completion costs of our
development projects or when and to what extent we will receive
cash inflows from the commercialization and sale of an approved
product candidate, if ever.
General
and Administrative Expenses
General and administrative expenses consist primarily of
compensation for employees in executive and operational
functions, including clinical, chemical manufacturing,
regulatory, finance and business development. Other significant
costs include professional fees for legal services, including
legal services associated with obtaining and maintaining
patents. We will continue to incur significant general and
administrative expenses as a public company, including costs for
insurance, costs related to the hiring of additional personnel,
payment to outside consultants, lawyers and accountants and
complying with the corporate governance, internal controls and
similar requirements applicable to public companies.
Critical
Accounting Policies and Estimates
Our management’s discussion and analysis of our financial
condition and results of operations is based on our financial
statements, which have been prepared in accordance with
accounting principles generally accepted in the United States,
or GAAP. The preparation of these financial statements requires
us to make estimates and judgments that affect the reported
amounts of assets, liabilities and expenses. On an ongoing
basis, we evaluate these estimates and judgments, including
those described below. We base our estimates on our historical
experience and on various other assumptions that we believe to
be reasonable under the circumstances. These estimates and
assumptions form the basis for making judgments about the
carrying values of assets and liabilities that are not readily
apparent from other sources. Actual results and experiences may
differ materially from these estimates.
While our significant accounting policies are more fully
described in the accompanying notes to the financial statements,
we believe that the following accounting policies are the most
critical to aid you in fully understanding and evaluating our
reported financial results and affect the more significant
judgments and estimates that we use in the preparation of our
financial statements.
Accrued
Clinical Expenses
As part of the process of preparing our financial statements, we
are required to estimate our accrued expenses. This process
involves reviewing open contracts and purchase orders,
communicating with our applicable personnel
63
to identify services that have been performed on our behalf and
estimating the level of service performed and the associated
cost incurred for the service when we have not yet been invoiced
or otherwise notified of actual cost. The majority of our
service providers invoice us monthly in arrears for services
performed. We make estimates of our accrued expenses as of each
balance sheet date in our financial statements based on facts
and circumstances known to us at that time. We periodically
confirm the accuracy of our estimates with the service providers
and make adjustments if necessary. Examples of estimated accrued
clinical expenses include:
|
|
|
| |
•
|
fees paid to CROs in connection with clinical studies;
|
| |
| |
•
|
fees paid to investigative sites in connection with clinical
studies;
|
| |
| |
•
|
fees paid to contract manufacturers in connection with the
production of clinical study materials; and
|
| |
| |
•
|
fees paid to vendors in connection with the preclinical
development activities.
|
We base our expenses related to clinical studies on our
estimates of the services received and efforts expended pursuant
to contracts with multiple research institutions and CROs that
conduct and manage clinical studies on our behalf. The financial
terms of these agreements are subject to negotiation, vary from
contract to contract and may result in uneven payment flows.
Payments under some of these contracts depend on factors such as
the successful enrollment of patients and the completion of
clinical study milestones. In accruing service fees, we estimate
the time period over which services will be performed and the
level of effort to be expended in each period. If the actual
timing of the performance of services or the level of effort
varies from our estimate, we adjust the accrual accordingly. If
we do not identify costs that we have begun to incur or if we
underestimate or overestimate the level of services performed or
the costs of these services, our actual expenses could differ
from our estimates.
Stock-Based
Compensation
Effective January 1, 2006, we adopted the provisions of
FASB ASC 718, Compensation — Stock
Compensation, using the modified prospective method.
Compensation costs related to all equity instruments granted
after January 1, 2006 are recognized at the grant-date fair
value of the awards. Additionally, we are required to include an
estimate of the number of awards that will be forfeited in
calculating compensation costs, which are recognized over the
requisite service period of the awards on a straight-line basis.
We account for stock-based compensation using the Black-Scholes
option pricing model to estimate the fair value of each option
grant on the date of grant. Black-Scholes option pricing model
requires the input of highly subjective assumptions, including
the expected stock price volatility, expected term, and
forfeiture rate. Any changes in these highly subjective
assumptions significantly impact stock-based compensation
expense.
As of December 31, 2010, 1,578,491 shares of our
common stock were issuable upon exercise of stock options and
release of restricted stock units.
Fair
Value Measurements and Impairments
All of our
available-for-sale
investments are subject to periodic impairment review.
Investments are considered to be impaired when a decline in fair
value is judged to be
other-than-temporary.
This determination requires significant judgment. For publicly
traded investments, impairment is determined based upon the
specific facts and circumstances present at the time, including
factors such as current economic and market conditions, the
credit rating of the security’s issuer, the length of time
an investment’s fair value has been below our carrying
value, and our ability and intent to hold investments to
maturity or for a period of time sufficient to allow for any
anticipated recovery in fair value. If an investment’s
decline in fair value, caused by factors other than changes in
interest rates, is deemed to be
other-than-temporary,
we reduce its carrying value to its estimated fair value, as
determined based on quoted market prices, liquidation values or
other metrics.
Pursuant to the accounting guidance for fair value measurement
and its subsequent updates, fair value is defined as the price
that would be received to sell an asset or paid to transfer a
liability (i.e., the “exit price”) in an orderly
transaction between market participants at the measurement date.
The accounting guidance establishes a hierarchy for inputs used
in measuring fair value that minimizes the use of unobservable
inputs by requiring the use of observable market data when
available. Observable inputs are inputs that market participants
would use in pricing the asset or liability based on active
market data. Unobservable inputs are inputs that reflect the
assumptions market participants would use in pricing the asset
or liability based on the best information available in the
circumstances.
64
The fair value hierarchy is broken down into the three input
levels summarized below:
|
|
|
| |
•
|
Level 1 — Valuations are based on quoted
prices in active markets for identical assets or liabilities,
and readily accessible by us at the reporting date. Examples of
assets and liabilities utilizing Level 1 inputs are certain
money market funds, U.S. Treasuries and trading securities
with quoted prices on active markets.
|
| |
| |
•
|
Level 2 — Valuations based on inputs other
than the quoted prices in active markets that are observable
either directly or indirectly in active markets. Examples of
assets and liabilities utilizing Level 2 inputs are
U.S. government agency bonds, corporate bonds, commercial
paper, certificates of deposit and
over-the-counter
derivatives.
|
| |
| |
•
|
Level 3 — Valuations based on unobservable
inputs in which there is little or no market data, which require
us to develop our own assumptions. Examples of assets and
liabilities utilizing Level 3 inputs are cost method
investments, auction rate securities (ARS) and the Primary Fund.
|
We measure our
available-for-sale
securities at fair value on a recurring basis.
Available-for-sale
securities include U.S. Treasury securities,
U.S. government agency bonds, corporate bonds, commercial
paper, money market funds and certificates of deposit. Where
possible, we utilize quoted market prices to measure and such
items are classified as Level 1 in the hierarchy. When
quoted market prices for identical assets are unavailable,
varying valuation techniques are used. Such assets are
classified as Level 2 or Level 3 in the hierarchy. We
classify items in Level 2 if investments are valued using
observable inputs to quoted market prices, benchmark yields,
reported trades, broker/dealer quotes or alternative pricing
sources with reasonable levels of price transparency. We
classify items in Level 3 if investments are valued using a
pricing model, based on unobservable inputs in the market or
that require us to develop our own assumptions. Our assessment
of the significance of a particular input to the fair value
measurement in its entirety requires judgment and considers
factors specific to the investment.
We are also exposed to market risk relating to our
available-for-sale
investments due to uncertainties in the credit and capital
markets. The fair value of our investments may change
significantly due to events and conditions in the credit and
capital markets. These securities/issuers could be subject to
review for possible downgrade. Any downgrade in these credit
ratings may result in an additional decline in the estimated
fair value of our investments. We monitor and evaluate the
accounting for our investment portfolio on a quarterly basis for
additional
other-than-temporary
impairment charges.
We actively review current investment ratings, company specific
events, and general economic conditions in managing our
investments and determining whether there is a significant
decline in fair value that is
other-than-temporary.
As of December 31, 2010 our short-term in marketable
securities have been classified as
“available-for-sale”
and are carried at fair value.
Available-for-sale
investments with original maturities of greater than three
months at the date of purchases are classified as short-term
investments as these investments generally consist of marketable
securities that are intended to be available to meet current
cash requirements.
Recognized gains and losses on available for sale investments
during 2010, 2009 and 2008 were not material. Management
determines the appropriate classification of investments at the
time of purchase and reevaluates the classification at each
reporting date.
Comprehensive
Income (Loss)
Comprehensive income (loss) consists of other comprehensive
income and net loss. Other comprehensive income includes certain
changes in equity that are excluded from net income (loss).
Specifically, the Company includes unrealized gains (losses) on
available for sale securities in other comprehensive income
(loss). Comprehensive income (loss) for each period presented is
set forth in the Statement of Stockholders’ Equity
(Deficit) and Comprehensive Loss.
Results
of Operations
Comparison
of Years Ended December 31, 2010 and 2009
Research and development expenses. Research
and development expenses consist of personnel costs for
employees in clinical, chemical manufacturing and regulatory
functions, clinical studies performed by CROs, pharmaceutical
development costs including product formulation and
manufacturing, preclinical costs, license fees and overhead
allocations consisting of various administrative and
facilities-related costs.
65
Change in research and development expenses from the years ended
December 31, 2009 to 2010 (in millions):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
$ Change
|
|
|
% Change
|
|
|
|
|
Research and development expense
|
|
$
|
29.4
|
|
|
$
|
8.4
|
|
|
$
|
21.0
|
|
|
|
250
|
%
|
The increase in research and development expenses from 2009 to
2010 was primarily attributable to the recognition of a
$3.5 million non-cash charge related to milestone payments
recorded in connection with the initiation of our Phase 3
clinical study of varespladib, which was paid through the
issuance of 531,914 shares of common stock; and increased
CRO and manufacturing cost related to the launch of our Phase 3
clinical study of varespladib and Phase 2 clinical study of
A-623, as
well as increased headcount to support these clinical studies.
General and administrative expenses. General
and administrative expenses consist of personnel costs for
employees in executive, business development and operational
functions, professional service fees including corporate legal
fees, accountant fees and overhead allocations consisting of
various administrative and facilities-related costs.
Change in general and administrative expenses from the years
ended December 31, 2009 to 2010 (in millions):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
$ Change
|
|
|
% Change
|
|
|
|
|
General and administrative expense
|
|
$
|
6.3
|
|
|
$
|
3.4
|
|
|
$
|
2.9
|
|
|
|
85
|
%
|
The increase in general and administrative expenses from 2009 to
2010 was primarily attributable to increased headcount and
professional services incurred in connection with our financial
audit and other costs associated with operating as a public
company.
Other expense and interest income, net. Other
expense consists of primarily non-cash charge related to the
amortization of discounts,
mark-to-market
adjustment relating to warrants and embedded derivative
associated with our convertible promissory notes issued in July
and September of 2009, which were converted into shares of our
common stock upon the closing of our IPO in March 2010. Interest
income consists of interest earned on our cash, cash equivalents
and short-term investments.
Change in other expense and interest income, net, from the years
ended December 31, 2009 to 2010 (in millions):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
$ Change
|
|
|
% Change
|
|
|
|
|
Other expense and interest income, net
|
|
$
|
(0.9
|
)
|
|
$
|
(0.4
|
)
|
|
$
|
(0.6
|
)
|
|
|
138
|
%
|
|
Mark-to-market
adjustment of warrant liability
|
|
$
|
(3.8
|
)
|
|
|
—
|
|
|
$
|
(3.8
|
)
|
|
|
100
|
%
|
The increase in other expense and interest income, net, from
2009 to 2010 was primarily attributable to a non-cash charge of
$4.5 million recorded for the amortization of discounts on
our convertible promissory notes and the
mark-to-market
adjustment relating to warrants and embedded derivative
connected to these convertible promissory notes. The balance was
offset by interest income from 2009 to 2010 attributable to
higher cash and investment balances in 2010 resulting from
proceeds received from our IPO in March 2010, the exercise in
April 2010 of the overallotment option by our underwriters in
connection with our IPO, and the private placement offering in
September.
Comparison
of Years Ended December 31, 2009 and 2008
Research and development expenses. The
decrease in research and development expenses from 2008 to 2009
was primarily attributable to decreased activity in our Phase 2
clinical study designed to examine the impact of varespladib
when administered to patients within 96 hours of an acute
coronary syndrome event in the third quarter of 2009 as the
study progressed toward completion.
Change in research and development expenses from the years ended
December 31, 2008 to 2009 (in millions):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
$ Change
|
|
|
% Change
|
|
|
|
|
Research and development expense
|
|
$
|
8.4
|
|
|
$
|
10.9
|
|
|
$
|
(2.5
|
)
|
|
|
(23
|
)%
|
66
General and administrative expenses. The
increase in general and administrative expenses from 2008 to
2009 was primarily attributable to expenses relating to the
expansion of our intellectual property portfolio.
Change in general and administrative expenses from the years
ended December 31, 2008 to 2009 (in millions):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
$ Change
|
|
|
% Change
|
|
|
|
|
General and administrative expense
|
|
$
|
3.4
|
|
|
$
|
3.0
|
|
|
$
|
0.4
|
|
|
|
13
|
%
|
Other expense and interest income, net. The
increase in other expense and interest income, net, from 2008 to
2009 was primarily attributable to interest accrued for
convertible promissory notes and amortization of note discount
and debt issuance cost.
Change in other expense and interest income, net, from the years
ended December 31, 2008 to 2009 (in millions):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009
|
|
|
2008
|
|
|
$ Change
|
|
|
% Change
|
|
|
|
|
Other expense and interest income, net
|
|
$
|
(0.4
|
)
|
|
$
|
(0.1
|
)
|
|
$
|
0.3
|
|
|
|
200
|
%
|
Beneficial conversion feature. In connection
with the issuance of convertible promissory notes in 2008, we
recorded expense related to the beneficial conversion feature of
the notes in the amount of $4.1 million for the year ended
December 31, 2008. The expense was amortized from the
issuance date of the notes to the date of their conversion into
shares of
Series B-2
convertible preferred stock in August 2008. No such expense was
recognized in 2009 and 2010.
Liquidity
and Capital Resources
To date, we have funded our operations primarily through private
placements of preferred stock and common stock, convertible debt
and our IPO. As of December 31, 2010, we had received net
proceeds of approximately $119.5 million from the sale of
equity securities and approximately $26.2 million from the
issuance of convertible promissory notes. As of
December 31, 2010, we had cash, cash equivalents and
short-term investments of approximately $63.4 million.
Cash, cash equivalents and investments consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
40,029,972
|
|
|
$
|
3,803,384
|
|
|
Short-term investments
|
|
|
23,350,922
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
63,380,894
|
|
|
$
|
3,803,384
|
|
|
|
|
|
|
|
|
|
|
|
Our principal liquidity requirements are primarily to meet our
working capital needs, support ongoing business activities,
research and development, and our capital expenditure needs.
Cash
Flows
Year
ended December 31, 2010
For the year ended December 31, 2010, we incurred a net
loss of approximately $40.4 million.
Net cash used in operating activities was approximately
$27.8 million. The net loss is higher than cash used in
operating activities by $12.6 million. The primary drivers
for the difference are adjustments for non-cash charges such as
stock-based compensation of approximately $702,000, amortization
of note discount and debt issuance cost of approximately
$769,000, issuance of $3.5 million worth of common stock in
lieu of cash milestone payments due to Eli Lilly and
Shionogi & Co., Ltd., the conversion of approximately
$300,000 of accrued interest into shares of common stock upon
conversion of certain convertible promissory notes,
mark-to-market
adjustments relating to warrant and derivative liability of
$3.8 million, and increase in net operating assets and
liabilities of approximately $3.6 million.
Net cash used by investing activities was $23.4 million and
was primarily driven by the purchase of short-term investments
during the period.
67
Net cash provided by financing activities was approximately
$87.5 million and consisted of proceeds of
$61.2 million received from the issuance of common stock at
our IPO, the exercise of the overallotment option by our
underwriters in connection with our IPO, the release of funds
held in an escrow account concurrent with the closing of our
IPO, proceeds of $29.6 million received from the issuance
of common stock and warrants in connection with the September
private placement offering, and proceeds of approximately
$200,000 received from the exercise of stock options and
issuance of common stock through our ESPP, offset by
approximately $3.5 million of issuance cost paid during the
period.
Year
Ended December 31, 2009
For the year ended December 31, 2009, we incurred a net
loss of approximately $12.2 million.
Net cash used in operating activities was approximately
$17.2 million. The net loss is higher than cash used in
operating activities by $5.0 million. The primary drivers
for the difference are adjustments for non-cash charges such as
depreciation of $18,000, stock-based compensation of
approximately $342,000 and amortization of note discount and
debt issuance cost of approximately $216,000, a decrease in
current liabilities of approximately $598,000 primarily due to
payments made to CROs for the achievement of clinical milestones
and a $5.0 million license fee payment made to Amgen.
Net cash provided by financing activities was approximately
$13.0 million and consisted of net proceeds of
$13.3 million received from the issuance of convertible
promissory notes, partially offset by approximately $274,000 in
expense paid in connection with our IPO.
Contractual
Obligations and Commitments
The following table summarizes our long-term contractual
obligations and commitments as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Period
|
|
|
|
|
|
|
|
Less Than
|
|
|
|
|
|
|
|
|
After
|
|
|
|
|
Total
|
|
|
1 Year
|
|
|
1-3 Years
|
|
|
3-5 Years
|
|
|
5 Years
|
|
|
|
|
Operating lease obligations(1)
|
|
$
|
15,594
|
|
|
$
|
10,914
|
|
|
$
|
4,680
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
(1) |
|
Operating lease obligations reflect our obligation to make
payments in connection with a sublease that commenced in October
2008 and expired in January 2011, which sublease was
subsequently extended through July 2011, for approximately
7,800 square feet of office space, and office equipment
leases that commenced in October 2007 and will expire in June
2013. |
The above amounts exclude potential payments to be made under
our license agreements to our licensors that are based on the
progress of our product candidates in development, as these
payments are not determinable. Under our license agreement with
Eli Lilly and Shionogi & Co., Ltd. to develop and
commercialize certain
sPLA2
inhibitors, we are obligated to make additional milestone
payments upon the achievement of certain development,
regulatory, and commercial objectives. We are also obligated to
pay royalties on future net sales of products that are developed
and approved as defined by this collaboration. Our obligation to
pay royalties with respect to each licensed product in each
country will expire upon the later of (a) 10 years
following the date of the first commercial sale of such licensed
product in such country, and (b) the first date on which
generic version(s) of the applicable licensed product achieve a
total market share, in the aggregate, of 25% or more of the
total unit sales of wholesalers to pharmacies of licensed
product and all generic versions combined in the applicable
country.
Also excluded from the table above are potential milestone
payments on the development of
A-623. Under
our license agreement with Amgen to develop and commercialize
A-623, we
are obligated to make additional milestone payments upon the
achievement of certain development, regulatory, and commercial
objectives. We are also obligated to pay royalties on future net
sales of products that are developed and approved as defined by
this collaboration. Our royalty obligations as to a particular
licensed product will be payable, on a
country-by-country
and licensed
product-by-licensed
product basis, for the longer of (a) the date of expiration
of the last to expire valid claim within the licensed patents
that covers the manufacture, use or sale, offer to sell, or
import of such licensed product by us or a sublicensee in such
country, or (b) 10 years after the first commercial
sale of the applicable licensed product in the applicable
country.
68
Funding
Requirements
We expect to incur substantial expenses and generate significant
operating losses as we continue to advance our product
candidates into preclinical studies and clinical studies and as
we:
|
|
|
| |
•
|
continue clinical development of the Phase 3 VISTA-16 study for
varespladib;
|
| |
| |
•
|
continue clinical development of the Phase 2b PEARL-SC study for
A-623;
|
| |
| |
•
|
hire additional clinical, scientific and management
personnel; and
|
| |
| |
•
|
implement new operational, financial and management information
systems.
|
Our future capital uses and requirements depend on numerous
forward-looking factors. These factors include the following:
|
|
|
| |
•
|
the progress of preclinical development and clinical studies of
our product candidates;
|
| |
| |
•
|
the time and costs involved in obtaining regulatory approvals;
|
| |
| |
•
|
delays that may be caused by evolving requirements of regulatory
agencies;
|
| |
| |
•
|
the costs involved in filing and prosecuting patent applications
and enforcing or defending patent claims;
|
| |
| |
•
|
our ability to establish, enforce and maintain selected
strategic alliances; and
|
| |
| |
•
|
the acquisition of technologies, product candidates and other
business opportunities that require financial commitments.
|
To date, we have not generated any revenue. We do not expect to
generate revenue unless or until we obtain regulatory approval
of, and commercialize, our product candidates. We expect our
continuing operating losses to result in increases in cash used
in operations over the next several years. Our future capital
requirements will depend on a number of factors including the
progress and results of our clinical studies, the costs, timing
and outcome of regulatory review of our product candidates, our
revenue, if any, from successful development and
commercialization of our product candidates, the costs of
commercialization activities, the scope, progress, results and
costs of preclinical development, laboratory testing and
clinical studies for other product candidates, the emergence of
competing therapies and other market developments, the costs of
preparing, filing and prosecuting patent applications and
maintaining, enforcing and defending intellectual property
rights, the extent to which we acquire or invest in other
product candidates and technologies, and our ability to
establish collaborations and obtain milestone, royalty or other
payments from any collaborators.
We expect our existing resources as of the date of this report,
to be sufficient to fund our planned operations, including our
continued product candidate development, for at least the next
12 months. However, we may require significant additional
funds earlier than we currently expect to conduct additional or
extended clinical studies and seek regulatory approval of our
product candidates. Because of the numerous risks and
uncertainties associated with the development and
commercialization of our product candidates, we are unable to
estimate the amounts of increased capital outlays and operating
expenditures associated with our current and anticipated
clinical studies.
Additional funding may not be available to us on acceptable
terms or at all. In addition, the terms of any financing may
adversely affect the holdings or the rights of our stockholders.
For example, if we raise additional funds by issuing equity
securities or by selling debt securities, if convertible,
further dilution to our existing stockholders may result. To the
extent our capital resources are insufficient to meet our future
capital requirements, we will need to finance our future cash
needs through public or private equity offerings, collaboration
agreements, debt financings or licensing arrangements.
If adequate funds are not available, we may be required to
terminate, significantly modify or delay our development
programs, reduce our planned commercialization efforts, or
obtain funds through collaborators that may require us to
relinquish rights to our technologies or product candidates that
we might otherwise seek to develop or commercialize
independently. We may elect to raise additional funds even
before we need them if the conditions for raising capital are
favorable.
69
Off-Balance
Sheet Arrangements
We do not currently have, nor have we ever had, any
relationships with unconsolidated entities or financial
partnerships, such as entities often referred to as structured
finance or special purpose entities, established for the purpose
of facilitating off-balance sheet arrangements or other
contractually narrow or limited purposes. In addition, we do not
engage in trading activities involving non-exchange traded
contracts.
|
|
|
ITEM 7A.
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
Our primary exposure to market risk is interest income
sensitivity, which is affected by changes in the general level
of U.S. interest rates. We are exposed to market risk
related to fluctuations in interest rates, market prices, and
foreign currency exchange rates. However, since a majority of
our investments are in short-term certificates of deposit,
FDIC-insured corporate bonds and money market funds, we do not
believe we are subject to any material market risk exposure. As
of December 31, 2010, we did not have any material
derivative financial instruments. The fair value of our
marketable securities, including those included in cash
equivalents and short-term investments, was $47.8 million
as of December 31, 2010.
Our investment policy is to limit credit exposure through
diversification and investment in highly rated securities. We
actively review, along with our investment advisors, current
investment ratings, company specific events and general economic
conditions in managing our investments and in determining
whether there is a significant decline in fair value that is
other-than-temporary.
We will monitor and evaluate the accounting for our investment
portfolio on a quarterly basis for additional
other-than-temporary
impairment charges.
|
|
|
ITEM 8.
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
The financial statements required by this item are set forth
beginning in Item 15 of this report and are incorporated
herein by reference.
|
|
|
ITEM 9.
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND
FINANCIAL DISCLOSURE
|
Not applicable.
|
|
|
ITEM 9A.
|
CONTROLS
AND PROCEDURES
|
Evaluation
of Disclosure Controls and Procedures
Our management, with the participation of our chief executive
and financial officers, evaluated the effectiveness of our
disclosures controls and procedures, as defined in
Rules 13a-15(e)
and
15d-15(e)
under the Exchange Act, as of December 31, 2010. The term
“disclosure controls and procedures,” as defined in
Rules 13a-15(e)
and
15d-15(e)
under the Exchange Act, means controls and other procedures of a
company that are designed to ensure that information required to
be disclosed by a company in the reports that it files or
submits under the Exchange Act is recorded, processed,
summarized and reported within the time periods specified in the
SEC’s rules and forms. Disclosure controls and procedures
include, without limitation, controls and procedures designed to
ensure that information required to be disclosed by a company in
the reports that it files or submits under the Exchange Act is
accumulated and communicated to the company’s management,
including its principal executive and principal financial
officers, as appropriate, to allow timely decisions regarding
required disclosure. Management recognizes that any controls and
procedures, no matter how well designed and operated, can
provide only reasonable assurance of achieving their objectives
and management necessarily applies its judgment in evaluating
the cost-benefit relationship of possible controls and
procedures. Based on the evaluation of our disclosure controls
and procedures as of December 31, 2010, our principal
executive officer and principal financial officer concluded
that, as of such date, our disclosure controls and procedures
were effective at the reasonable assurance level.
70
Changes
in Internal Control over Financial Reporting
There was no change in our internal control over financial
reporting during the year ended December 31, 2010
identified in connection with the evaluation required by
Rule 13a-15(d)
and
15d-15(d) of
the Exchange Act that has materially affected, or is reasonably
likely to materially affect, our internal control over financial
reporting.
Management’s
Annual Report on Internal Control over Financial Reporting and
Attestation Report of the Registered Accounting Firm
This annual report does not include a report of
management’s assessment regarding internal control over
financial reporting or an attestation report of the
company’s registered public accounting firm due to a
transition period established by rules of the Securities and
Exchange Commission for newly public companies.
|
|
|
ITEM 9B.
|
OTHER
INFORMATION
|
Not applicable.
71
PART III
|
|
|
ITEM 10.
|
DIRECTORS,
EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
Members
of the Board of Directors
The biographies of our directors and their ages as of
March 1, 2011 are set forth below.
| |
|
|
|
|
|
|
|
Name
|
|
Age
|
|
Position
|
|
|
|
Paul F. Truex
|
|
|
42
|
|
|
Chief Executive Officer, President and Director
|
|
Christopher S. Henney, Ph.D.
|
|
|
70
|
|
|
Chairman of the Board of Directors
|
|
Annette Bianchi
|
|
|
52
|
|
|
Director
|
|
James I. Healy, M.D., Ph.D.
|
|
|
46
|
|
|
Director
|
|
Donald J. Santel
|
|
|
50
|
|
|
Director
|
|
Daniel K. Spiegelman
|
|
|
52
|
|
|
Director
|
|
David E. Thompson
|
|
|
63
|
|
|
Director
|
|
Peter A. Thompson, M.D.
|
|
|
51
|
|
|
Director
|
Our certificate of incorporation provides for a Board of
Directors that is divided into three classes, categorized as
Class I, Class II, and Class III. The term for
each class is three years, staggered over time. Class I
consists of Messrs. Santel and Thompson; Class II
consists of Ms. Bianchi and Drs. Healy and Thompson;
Class III consists of Dr. Henney and
Messrs. Spiegelman and Truex.
Paul F. Truex. Mr. Truex has served as
our President and Chief Executive Officer since our inception in
September 2004 and as a member of our Board of Directors since
November 2004. Prior to founding Anthera, Mr. Truex served
as a Director, President and Chief Executive Officer of
Peninsula Pharmaceuticals, Inc., a biopharmaceutical company,
from the commencement of its operations in October 2001. Prior
to Peninsula, Mr. Truex was Vice President of Commercial
Development for Vicuron, Inc. from April 2000 to September 2001.
From July 1997 to April 2000, Mr. Truex held various
positions at Eli Lilly and Company. Mr. Truex holds an
M.B.A. in marketing and finance from Indiana University and a
B.A. in economics from the University of Waterloo.
Mr. Truex is a director of Trius Therapeutics, Inc.
Christopher S.
Henney, Ph.D. Dr. Henney has served as
the Chairman of our Board of Directors since August 2008 and has
been a member of our Board of Directors since April 2005.
Dr. Henney served as Chairman and Chief Executive Officer
of Dendreon Corporation, a biotechnology company he co-founded,
from 1995 until his retirement in July 2004. Dr. Henney was
previously a founder of Immunex Corp. and Icos Corp.
Dr. Henney holds a B.Sc. with honors in medical
biochemistry, a Ph.D. in experimental pathology and a D.Sc. for
contributions to the field of immunology, all from the
University of Birmingham, England. Dr. Henney served as a
director of AVI BioPharma Inc. from March 2009 until June 2010
and is currently the Chairman and a director of Oncothyreon,
Inc. and is vice-chairman and a director of Cyclacel
Pharmaceuticals, Inc.
Annette Bianchi. Ms. Bianchi has served
as a member of our Board of Directors since August 2006.
Ms. Bianchi has served as a Managing Director at
VantagePoint Venture Partners, a venture capital firm, since
2004. From 1999 to 2004, Ms. Bianchi served as a Managing
Director at Pacific Venture Group, a dedicated health care fund.
From 1992 to 1999, Ms. Bianchi served as a General Partner
at Weiss, Peck & Greer Venture Partners, a venture
capital firm. From 1985 to 1992, Ms. Bianchi served as an
associate and a General Partner of Burr, Egan,
Deleage & Co., a venture capital firm. From 2005
through 2008, Ms. Bianchi served as a director of Conceptus
Inc. Ms. Bianchi holds a B.S.E. and an M.S.E. in Biomedical
Engineering from the University of Pennsylvania and an M.B.A.
from The Wharton School of the University of Pennsylvania.
James I.
Healy, M.D., Ph.D. Dr. Healy has
served as a member of our Board of Directors since August 2006.
Dr. Healy is a Managing Partner of Sofinnova Management VI,
LLC, the general partner of Sofinnova Venture Partners VI, L.P.,
a fund managed by Sofinnova Ventures, Inc., a venture capital
firm, a position he has held since June 2000. Prior to
Sofinnova, Dr. Healy began his private equity career at
Sanderling Ventures, and has been an early investor and board
member of numerous biopharmaceutical companies. Dr. Healy
holds a B.A. in molecular biology and a B.A. in Scandinavian
studies from the University of California at Berkeley, an M.D.
from Stanford
72
University School of Medicine and a Ph.D. in immunology from
Stanford University. Dr. Healy is a director of InterMune,
Inc. and Amarin Corporation plc, both biopharmaceutical
companies.
Donald J. Santel. Mr. Santel has served
as a member of our Board of Directors since October 2007. From
February 2000 until January 2007, Mr. Santel held various
positions in and was a member of the board of directors of
CoTherix, Inc., a pharmaceutical company he co-founded. From
October 2003 to August 2004, Mr. Santel served as President
and Chief Operating Officer of CoTherix and from August 2004
until January 2007, Mr. Santel served as Chief Executive
Officer. From June 2008 through June 2009, Mr. Santel
served as a consultant and from June 2009 until the present,
Mr. Santel has served as the Chief Executive Officer of
Hyperion Therapeutics, Inc., a pharmaceutical company.
Mr. Santel holds a B.S.E. in biomedical engineering from
Purdue University and an M.S. in electrical engineering from the
University of Minnesota.
Daniel K. Spiegelman. Mr. Spiegelman has
served as a member of our Board of Directors since February
2010. Currently, Mr. Spiegelman provides management and
financial consulting services to biotechnology companies. From
January 1998 to May 2009, Mr. Spiegelman served as Senior
Vice President and Chief Financial Officer of CV Therapeutics,
Inc., a biopharmaceutical company that was acquired by Gilead
Sciences, Inc. in April 2009. From July 1991 to January 1998,
Mr. Spiegelman served at Genentech, Inc., most recently as
Treasurer. Mr. Spiegelman also serves on the board of
directors of Affymax, Inc., Cyclacel Pharmaceuticals, Inc.,
Omeros Corporation and Oncothyreon, Inc., all of which are
publicly-traded biopharmaceutical companies. Mr. Spiegelman
also previously served on the board of directors of Xcyte
Therapies, Inc. from 2003 through 2006, a publicly-traded
company, until Cyclacel acquired Xcyte via reverse merger in
2006. Mr. Spiegelman holds a B.A. in economics from
Stanford University and an M.B.A. from the Stanford Graduate
School of Business.
David E. Thompson. Mr. Thompson has
served as a member of our Board of Directors since November
2005. Mr. Thompson served as Vice President of Corporate
Strategy Business Development for Eli Lilly and Company from
January 2001 until his retirement in July 2005. Thereafter, he
was a partner at VantagePoint Venture Partners from 2006 through
2008. Mr. Thompson holds a B.S. and an M.B.A. from Michigan
State University.
Peter A. Thompson, M.D. Dr. Thompson
has served as a member of our Board of Directors since February
2011. Dr. Thompson is currently a Venture Partner with
OrbiMed Advisors, LLC and has over 20 years of industry
experience. He co-founded Trubion Pharmaceuticals, and served as
CEO and Chairman from its inception through its IPO on NASDAQ
and as a public company until his retirement in 2009.
Dr. Thompson is the former Vice President and General
Manager of Chiron Informatics at Chiron Corporation and held
various executive positions at Becton Dickinson, including Vice
President, Research and Technology Department of BD Bioscience.
Dr. Thompson is a co-founder of iMetrikus, a clinical
decision support company, where he served as CEO and Chairman.
He is the founder and Managing Director of Strategicon Partners,
an investment and management services company. Dr. Thompson
is an Ernst & Young Entrepreneur of the Year awardee,
an inventor on numerous patents, a board-certified internist and
oncologist, and was on staff at the National Cancer Institute
following his internal medicine training at Yale University.
Dr. Thompson served on the Board of Directors of Trubion
Pharmaceuticals from 2006 through 2009 and currently serves on
the Board of Directors for Response Biomedical and CoDa
Therapeutics.
There are no family relationships between any of our directors
or executive officers.
Code of
Ethics
Our written Code of Ethics applies to all of our directors and
employees, including our principal executive officer, principal
financial officer, principal accounting officer or controller or
persons performing similar functions. The Code of Ethics is
available on our website at
http://www.anthera.com
in the Investors section under “Corporate Governance.”
Committees
of the Board of Directors
The Board of Directors has determined each of the following
current and former directors is an “independent
director” as such term is defined in NASDAQ Marketplace
Rule 5605(a)(2): Messrs. Santel, Spiegelman and
Thompson, Drs. Henney, Healy, Rachel A. Leheny, and
Thompson and Ms. Bianchi.
The Board of Directors has a standing Audit Committee,
Compensation Committee, and Nominating and Corporate Governance
Committee. Each of our Audit Committee, Compensation Committee
and Nominating and Corporate Governance Committee is composed
entirely of independent directors in accordance with current
Nasdaq
73
listing standards. Furthermore, our Audit Committee meets the
enhanced independence standards established by the
Sarbanes-Oxley Act of 2002 and related rulemaking of the SEC.
The Board of Directors has further determined that Daniel K.
Spiegelman, a member of the Audit Committee of the Board of
Directors, is an “Audit Committee Financial Expert,”
as such term is defined in Item 407(d)(5) of
Regulation S-K
promulgated by the SEC. Copies of our Audit Committee,
Nominating and Corporate Governance Committee and Compensation
Committee charters and our corporate governance guidelines are
available, free of charge, on our website at
http://www.anthera.com.
Audit Committee. The Audit Committee appoints,
approves the compensation of, and assesses the independence of
our independent registered public accounting firm and
pre-approves auditing and permissible non-audit services, and
the terms of such services, to be provided by our independent
registered public accounting firm. The Audit Committee is also
responsible for reviewing and discussing with management and the
independent registered public accounting firm our annual and
quarterly financial statements and related disclosures and
preparing the report required by the rules of the SEC to be
included in our annual proxy statement. The Audit Committee also
coordinates the oversight and reviews the adequacy of our
internal controls over financial reporting and establishes
policies and procedures for the receipt and retention of
accounting-related complaints and concerns. Currently, the Audit
Committee is comprised of Mr. Spiegelman (Chair),
Mr. Santel and Dr. Thompson.
Compensation Committee’s Role in Risk
Management. Our Compensation Committee
participates in the design of compensation structures that
create incentives that encourage a level of risk-taking behavior
consistent with the Company’s business strategy.
Executive
Officers
Set forth below are the names of each of our current executive
officers, their ages as of March 1, 2011, their positions
with the Company, and summaries of their backgrounds and
business experience. (For information on the business experience
of Mr. Truex, see the section above entitled “Members
of the Board of Directors.”)
| |
|
|
|
|
|
|
|
Name
|
|
Age
|
|
Position
|
|
|
|
Paul F. Truex
|
|
|
42
|
|
|
Chief Executive Officer, President and Director
|
|
Christopher P. Lowe
|
|
|
43
|
|
|
Chief Business Officer and Chief Financial Officer
|
|
Colin Hislop, M.D.
|
|
|
53
|
|
|
Senior Vice President and Chief Medical Officer
|
|
Debra Odink, Ph.D.
|
|
|
47
|
|
|
Senior Vice President, Pharmaceutical Research and Development
|
|
Georgina Kilfoil
|
|
|
42
|
|
|
Senior Vice President, Product Development and Clinical
Operations
|
Christopher P. Lowe. Mr. Lowe has served
as our Chief Business Officer since February 2011. Prior to that
time and since November 2007, he served as our Chief Financial
Officer and Vice President of Administration. Beginning in
September 2005 and up until he joined the company, Mr. Lowe
served as Vice President of Finance & Administration
and, beginning in January 2006, as Chief Financial Officer of
Asthmatx, Inc., a medical technology company. Previously,
Mr. Lowe was with Peninsula Pharmaceuticals, Inc., as
Corporate Controller from June 2004 to October 2004 and Chief
Accounting Officer from October 2004 until June 2005.
Mr. Lowe holds a B.S. in business administration from
California Polytechnic State University, San Luis Obispo
and an M.B.A. from Saint Mary’s University, Texas.
Mr. Lowe is a director of Hansen Medical Corporation, a
medical device company.
Colin Hislop, M.D. Dr. Hislop has
served as our Senior Vice President and Chief Medical Officer
since June 2010. Prior to that, he served as our Senior Vice
President of Cardiovascular Products since November 2005 and
also served as a consultant to the company from July 2005
through November 2005. From October 2004 until June 2005,
Dr. Hislop was Vice President, Clinical Development for
Peninsula Pharmaceuticals, Inc. where he oversaw three global
development programs for Peninsula’s anti-infective product
portfolio. From September 2001 until September 2004,
Dr. Hislop served as Vice President of Clinical Development
at CV Therapeutics, Inc., a biopharmaceutical company.
Dr. Hislop holds a B.Sc. in medical biochemistry from the
University of Surrey, and a degree in medicine from the
University of London.
Debra Odink, Ph.D. Dr. Odink was
promoted to Senior Vice President of Pharmaceutical Research and
Development in June 2010. Prior to that, she served as our Vice
President of Pharmaceutical Research and Development since
December 2005. From September 2002 until July 2005,
Dr. Odink served as Vice President of
74
Pharmaceutical Chemistry and Product Development at Peninsula
Pharmaceuticals, Inc., a biopharmaceutical company, where she
was responsible for manufacturing and product development
strategies for assets licensed to Peninsula. Dr. Odink
holds a B.S. in chemistry from California State University,
Stanislaus and a Ph.D. in inorganic chemistry from the
University of California at Davis.
Georgina Kilfoil. Ms. Kilfoil has served
as our Senior Vice President, Product Development and Clinical
Operations since March 2010. Prior to joining us,
Ms. Kilfoil was the Vice President, Alliances and Project
Management of Peninsula Pharmaceuticals, Inc. from 2004 to 2005.
From August 2000 to December 2003, Ms. Kilfoil was a
project management consultant with InClin, Inc., a consulting
company. Ms. Kilfoil holds a B.S. in pharmacology from the
University of Bristol, United Kingdom and an M.B.A. from the
Australian Graduate School of Management, Sydney, Australia.
Section 16(a)
Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our officers and
directors, and persons who own more than 10% of a registered
class of our equity securities, to file reports of ownership and
changes in ownership (Forms 3, 4 and 5) with the SEC.
Officers, directors and greater than 10% stockholders are
required to furnish us with copies of all such forms which they
file.
To our knowledge, based solely on our review of such reports or
written representations from certain reporting persons, we
believe that all of the filing requirements applicable to our
officers, directors, greater than 10% beneficial owners and
other persons subject to Section 16 of the Exchange Act
were complied with during the year ended December 31, 2010.
|
|
|
ITEM 11.
|
EXECUTIVE
COMPENSATION
|
Director
Compensation
Each of our non-employee directors receives a $40,000 annual
retainer fee instead of per-meeting fees. In consideration for
their services, the Chairman of our Board of Directors receives
an additional $40,000, the chairman of our Audit Committee
receives an additional $15,000 and the chairman of our
Compensation Committee receives an additional $10,000, each on
an annual basis.
In addition, since the completion of our initial public
offering, each new non-employee director receives a
non-qualified stock option to purchase 25,000 shares of our
common stock upon joining the Board, which vests over a
four-year period from the date of grant. In addition, each
non-employee director receives a non-qualified stock option to
purchase 12,000 shares of our common stock each year, which
vests over a one-year period from the date of grant. Any new
Chairman of our Board of Directors would receive a non-qualified
stock option to purchase 45,000 shares of our common stock
upon election to the Board, which would vest over a four-year
period from the date of grant. Our Chairman also receives a
non-qualified stock option to purchase 15,000 shares of our
common stock each year, which vests over a one-year period from
the date of grant.
All members of our Board of Directors are eligible to receive
full reimbursement for travel expenses arising from their
attendance of our board meetings.
75
Director
Compensation Table — 2010
The following table sets forth information with respect to the
compensation earned by our non-employee directors during the
fiscal year ended December 31, 2010.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fees Earned
|
|
|
Option
|
|
|
|
|
|
|
|
or Paid
|
|
|
Awards
|
|
|
|
|
|
Name
|
|
in Cash ($)
|
|
|
($)(1)
|
|
|
Total ($)
|
|
|
|
|
Christopher S. Henney, Ph.D. (Chairman)
|
|
$
|
80,000
|
|
|
$
|
37,089
|
(2)
|
|
$
|
117,089
|
|
|
Annette Bianchi
|
|
$
|
40,000
|
|
|
$
|
29,671
|
(3)
|
|
$
|
69,671
|
|
|
James I. Healy, M.D., Ph.D.
|
|
$
|
40,000
|
|
|
$
|
29,671
|
|
|
$
|
69,671
|
|
|
A. Rachel Leheny, Ph.D(4)
|
|
$
|
40,000
|
|
|
$
|
29,671
|
(5)
|
|
$
|
69,671
|
|
|
Donald J. Santel
|
|
$
|
41,250
|
|
|
$
|
29,671
|
(6)
|
|
$
|
70,921
|
|
|
Daniel K. Spiegelman(7)
|
|
$
|
50,417
|
|
|
$
|
29,671
|
(7)
|
|
$
|
80,088
|
|
|
David E. Thompson
|
|
$
|
50,000
|
|
|
$
|
29,671
|
(8)
|
|
$
|
79,671
|
|
|
Peter A. Thompson(9)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
(1) |
|
This column reflects the aggregate grant date fair value of
equity awards granted in 2010 and calculated in accordance with
FASB ASC 718, excluding the effect of estimated
forfeitures. See Note 11 to our financial statements for a
discussion of the assumptions made in determining the valuation
of option awards. |
| |
|
(2) |
|
Dr. Henney held 15,000 shares underlying stock options
as of December 31, 2010. |
| |
|
(3) |
|
Ms. Bianchi held 32,443 shares underlying stock
options as of December 31, 2010. |
| |
|
(4) |
|
Dr. Leheny resigned from our Board of Directors on
February 1, 2011. |
| |
|
(5) |
|
Dr. Leheny held 26,602 shares underlying stock options
as of December 31, 2010. |
| |
|
(6) |
|
Mr. Santel held 32,443 shares underlying stock options
as of December 31, 2010. |
| |
|
(7) |
|
Mr. Spiegelman joined our Board of Directors on
February 2, 2010. He held 37,000 shares underlying
stock options as of December 31, 2010. |
| |
|
(8) |
|
Mr. Thompson held 29,523 shares underlying stock
options as of December 31, 2010. |
| |
|
(9) |
|
Dr. Thompson joined our Board of Directors on
February 1, 2011. |
Compensation
Discussion and Analysis
This section discusses our executive compensation policies and
arrangements as they relate to our named executive officers who
are listed in the compensation tables set forth below. The
following discussion should be read together with the
compensation tables and related disclosures set forth below.
Background
and Objectives
We are a biopharmaceutical company focused on developing and
commercializing products to treat serious diseases associated
with inflammation, including cardiovascular and autoimmune
diseases. The success of development companies is significantly
influenced by the quality and motivation of their work forces.
As a result, we face significant competition for executives and
other talented employees from numerous pharmaceutical research
and development companies in the San Francisco Bay Area.
With this in mind, we strive to provide what we believe is a
competitive total compensation package to our executive officers
through a combination of base salary, short-term cash incentives
and long-term equity compensation, in addition to broad-based
employee benefits programs, in order to closely align the
interests of our executive officers with those of our
stockholders, to attract talented individuals to manage and
operate all aspects of our business, to reward these individuals
fairly and to retain those individuals who meet our high
expectations and support the achievement of our business
objectives.
Role of
Compensation Committee and Executive Officers
Our executive compensation program is administered by our board
of directors upon recommendation of our compensation committee.
Our compensation committee is responsible for overseeing our
executive compensation policies, plans and programs, reviewing
our achievements as a company and the achievements of our
individual officers, and recommending to our board of directors
the type and level of compensation for our named executive
officers and our directors. The primary goal of our compensation
committee is to closely align the interests of our
76
named executive officers with those of our stockholders. To
achieve this goal, our compensation committee relies on
compensation that is designed to attract and retain executives
whose abilities are critical to our long term success, that
motivates individuals to perform at their highest level and that
rewards achievement.
The annual responsibilities of our compensation committee
include the following:
|
|
|
| |
•
|
reviewing and recommending corporate goals and objectives
relevant to the compensation of our Chief Executive Officer and
named executive officers;
|
| |
| |
•
|
evaluating the performance of our Chief Executive Officer and
named executive officers in light of such corporate goals and
objectives and determining the compensation of our Chief
Executive Officer; and
|
| |
| |
•
|
reviewing and approving the level of equity awards, annual
salary and bonuses for our named executive officers and other
employees.
|
In reviewing and approving these matters, our compensation
committee considers such matters as it deems appropriate,
including our financial and operating performance, the alignment
of interests of our executive officers and our stockholders and
our ability to attract and retain qualified individuals. For
executive compensation decisions, including decisions relating
to the grant of equity awards to our named executive officers,
our compensation committee typically considers the
recommendations of Mr. Truex, our Chief Executive Officer.
Mr. Truex also generally participates in our compensation
committee’s deliberations about executive compensation
matters. However, Mr. Truex does not participate in the
deliberation or determination of his own compensation.
Our compensation committee has not established any formal
policies or guidelines for allocating compensation between
current and long-term equity compensation, or between cash and
non-cash compensation. In determining the amount and mix of
compensation elements and whether each element provides the
correct incentives and rewards for performance consistent with
our short-term and long-term goals and objectives, our
compensation committee relies on its judgment about each
individual’s performance in a rapidly changing business
environment rather than adopting a formulaic approach to
compensatory decisions that are too narrowly responsive to
short-term changes in business performance. In making
determinations about performance, our compensation committee
does not solely rely on formal goals or metrics, but rather
takes into account input from appropriate members of management
with respect to an individual’s performance, as well as its
own observations.
Role of
Compensation Consultant
Our compensation committee has the authority under its charter
to engage the services of any consulting firm or other outside
advisor to assist it. Prior to our IPO, in September 2009, our
compensation committee engaged J. Thelander Consulting, an
independent consulting firm selected by our compensation
committee, to review and provide comparative data on the base
salary, bonus and equity compensation of (i) chief
executive officers of private biotechnology companies with
funding levels between $50 to $70 million and
(ii) chief executive officers and other executive officers
of publicly traded biotechnology companies with a market
capitalization between $220 to $375 million. J. Thelander
Consulting also provided a review of the board compensation of
such publicly traded biotechnology companies. Our compensation
committee reviewed the report by J. Thelander Consulting, and in
April 2010, made certain changes to our executive compensation
as detailed below based, in part, on such report.
In December 2010, our compensation committee engaged Remedy
Compensation Consulting (which has since merged with Compensia),
an independent consulting firm specializing in the life sciences
and technology industries, to review and provide comparative
data on the base salary, bonus, equity compensation and total
direct compensation of our executive officers as compared
against 22 similar peer group public biotechnology companies as
well as Northern Californian companies with
50-149 employees
participating in the Radford Life Sciences survey. Our
compensation committee reviewed the report by Remedy
Compensation Consulting and made certain changes to our
executive compensation as detailed below, based in part, on such
report.
77
The companies in the peer group were as follows:
| |
|
|
|
|
|
|
|
•
|
|
Affymax, Inc.
|
|
•
|
|
Neurogesx Inc.
|
|
•
|
|
Alexza Pharmaceuticals, Inc.
|
|
•
|
|
Omeros Corp.
|
|
•
|
|
Amarin Corp.
|
|
•
|
|
Optimer Pharmaceuticals Inc.
|
|
•
|
|
Avanir Pharmaceuticals
|
|
•
|
|
Orexigen Therapeutics, Inc.
|
|
•
|
|
AVI BioPharma Inc.
|
|
•
|
|
Pain Therapeutics Inc.
|
|
•
|
|
Cell Therapeutics, Inc.
|
|
•
|
|
Pharmacyclics Inc.
|
|
•
|
|
Cytokinetics, Inc.
|
|
•
|
|
Sunesis Pharmaceuticals Inc.
|
|
•
|
|
Depomed Inc.
|
|
•
|
|
Supergen Inc.
|
|
•
|
|
DURECT Corporation
|
|
•
|
|
Transcept Pharmaceuticals Inc.
|
|
•
|
|
Ligand Pharmaceuticals Inc.
|
|
•
|
|
Vical, Inc.
|
|
•
|
|
Map Pharmaceuticals Inc.
|
|
•
|
|
Xenoport, Inc.
|
J. Thelander Consulting and Remedy Compensation Consulting
were retained by and reported directly to our compensation
committee and do not provide any other services to the Company.
Compensation
Elements
Base Salary. The base salaries of our named
executive officers are primarily established based on the scope
of their responsibilities and performance, taking into account
comparable company data from our compensation consultants and
based upon our compensation committee’s understanding of
compensation paid to similarly situated executives, and adjusted
as necessary to recruit or retain specific individuals. We
typically review the base salaries of our named executive
officers annually. We may increase the base salary of an
executive officer at other times if a change in the scope of the
executive’s responsibilities, such as promotion, justifies
such consideration. We believe that a competitive base salary
relative to the companies with which we compete for executives
is a necessary element of any compensation program that is
designed to attract and retain talented and experienced
executives. We also believe that attractive base salaries can
motivate and reward executives for their overall performance.
Base salaries are established in part based on experience,
skills and expected contributions of our executives and our
executives’ performance during the prior year. In making
determinations about the performance of our named executive
officers, our compensation committee takes into account the
achievement of corporate goals, which are set annually by our
compensation committee and generally include milestones related
to our preclinical and clinical studies and fundraising, as well
as informal individual goals, which are position-specific and
are communicated to the named executive officer over the course
of the year.
After our IPO and taking into consideration the comparative
compensation data from J. Thelander Consulting as mentioned
above, in April 2010, as part of its annual review of
compensation, the board of directors, upon the recommendation of
the compensation committee, approved annual base salary
adjustments for Company employees, including certain of the
Company’s named executive officers, which became effective
on May 1, 2010. The adjusted base salaries for such named
executive officers (and their titles at the time) are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
Annual Base Salary Prior
|
|
|
Annual Base Salary
|
|
|
|
|
to May 1,
|
|
|
Effective May 1,
|
|
|
Named Executive Officer
|
|
2010
|
|
|
2010
|
|
|
|
|
Paul F. Truex,
|
|
$
|
300,000
|
|
|
$
|
425,000
|
|
|
President and Chief Executive Officer
|
|
|
|
|
|
|
|
|
|
Christopher P. Lowe,
|
|
$
|
250,000
|
|
|
$
|
300,000
|
|
|
Chief Business Officer and Chief Financial Officer
|
|
|
|
|
|
|
|
|
|
Colin Hislop, M.D.,
|
|
$
|
270,000
|
|
|
$
|
320,000
|
|
|
Senior Vice President and Chief Medical Officer
|
|
|
|
|
|
|
|
|
|
Debra Odink, Ph.D.,
|
|
$
|
200,000
|
|
|
$
|
225,000
|
|
|
Senior Vice President, Pharmaceutical Research and Development
|
|
|
|
|
|
|
|
|
78
In connection with Dr. Odink’s promotion to Senior
Vice President, Pharmaceutical Research and Development in June
2010, her annual base salary was increased from $225,000 to
$250,000.
As part of its annual review of compensation and taking into
consideration the comparative compensation data from Remedy
Compensation Consulting as mentioned above, effective
February 1, 2011, our board of directors, upon the
recommendation of the compensation committee, approved annual
base salary adjustments for certain of our employees, including
certain of our named executive officers. Such adjustments were
targeted towards the 50th percentile of base compensation
in our peer group, based on the Remedy Compensation Consulting
data, taking into consideration adjustments for promotions
(including Mr. Lowe to Chief Business Officer). The
adjusted salaries for such named executive officers are as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
Currant Annual
|
|
|
Annual Base Salary
|
|
|
Named Executive Officer
|
|
Base Salary
|
|
|
Effective February 1, 2011
|
|
|
|
|
Paul F. Truex,
|
|
$
|
425,000
|
|
|
$
|
500,000
|
|
|
President and Chief Executive Officer
|
|
|
|
|
|
|
|
|
|
Christopher P. Lowe,
|
|
$
|
300,000
|
|
|
$
|
340,000
|
|
|
Chief Business Officer and Chief Financial Officer
|
|
|
|
|
|
|
|
|
|
Colin Hislop, M.D.,
|
|
$
|
320,000
|
|
|
$
|
340,000
|
|
|
Senior Vice President and Chief Medical Officer
|
|
|
|
|
|
|
|
|
|
Debra Odink, Ph.D.,
|
|
$
|
250,000
|
|
|
$
|
270,000
|
|
|
Senior Vice President, Pharmaceutical Research and Development
|
|
|
|
|
|
|
|
|
Cash Bonuses. Prior to our IPO, we did not
have a formal cash incentive program, although we have paid cash
bonuses based on the achievement of approved operational
milestones in the past. In March 2010, the board of directors
adopted the Company’s Executive Incentive Bonus Plan, or
the Bonus Plan, which applies to certain key executives, or the
Executives, that are recommended by the compensation committee
and selected by the board. The Bonus Plan provides for bonus
payments based upon the attainment of performance targets
established by the board and related to financial and
operational metrics with respect to the Company or any of its
subsidiaries, or the Performance Goals, which would include the
achievement of clinical study or operational milestones, results
of clinical studies and achievement of specified financial
metrics or objectives. Any bonuses paid under the Bonus Plan
shall be based upon objectively determinable bonus formulas that
tie such bonuses to one or more performance targets relating to
the Performance Goals. The bonus formulas shall be adopted in
each performance period by the Board and communicated to each
Executive. No bonuses shall be paid under the Bonus Plan unless
and until the Board makes a determination with respect to the
attainment of the performance objectives. Notwithstanding the
foregoing, the Company may adjust bonuses payable under the
Bonus Plan based on achievement of individual performance goals
or pay bonuses (including, without limitation, discretionary
bonuses) to Executives under the Bonus Plan based upon such
other terms and conditions as the Board may in its discretion
determine.
Each Executive is given a targeted bonus opportunity set for
each performance period. The maximum bonus payable to an
Executive under the Bonus Plan is 125% of the Executive’s
bonus opportunity. The Performance Goals will be measured at the
end of each fiscal year after the Company’s financial
reports have been published or such other appropriate time as
the board shall determine. If the Performance Goals are met,
payments will be made within 30 days thereafter, and if met
for the previous fiscal year, not later than March 31. An
Executive must be employed by the Company as of the payment date
in order to receive a bonus payment, provided that the board may
make exceptions to this requirement, in its sole discretion,
including, without limitation, in the case of an
Executive’s termination of employment, retirement, death or
disability.
In April 2010, our board of directors, upon recommendation of
the compensation committee, approved performance bonus awards
under the Bonus Plan based upon certain operational performance,
including successful negotiation and completion of the SPA with
the FDA, reactivation of the IND for
A-623
including submission and acceptance of the Phase II
protocol, completion of equity financings with proceeds to the
Company in excess of $50 million and completion and
submission of our FRANCIS study including a six-month
follow-up
evaluation. Such cash bonus awards were subject to a six-month
lapsing right of repayment in the event of termination or
employment. Our named executive officers received the following
performance cash bonus awards: Mr. Truex ($250,000),
Mr. Lowe ($100,000), Dr. Hislop ($65,000) and
Dr. Odink ($25,000).
79
In December 2010, our board of directors, upon recommendation of
the compensation committee, approved annual cash bonuses under
the Bonus Plan for performance in 2010. The annual target bonus
opportunities for 2010 (expressed as a percentage of base salary
and targeted towards the 50th percentile of our peer group)
for our named executive officers were as follows: Mr. Truex
(50%), Mr. Lowe (35%), Dr. Hislop (30%),
Dr. Odink (30%) and Ms. Kilfoil (30%). These bonus
payments were based upon the achievement of the following
corporate goals, which were equally weighted:
|
|
|
| |
•
|
continued development of our product candidates, including
initiating enrollment of our VISTA-16 clinical study for
varespladib and obtaining at least a certain level of enrollment
by the end of the year,
|
| |
| |
•
|
initiating Phase 2 enrollment for
A-623 and
completing enrollment by the end of the year,
|
| |
| |
•
|
considering strategic partnership opportunities for our product
candidates, and
|
| |
| |
•
|
opportunistically supporting our corporate development
objectives with appropriate financing and budgeting efforts.
|
In determining cash bonuses under the Bonus Plan, our board
determined that 70% of the 2010 corporate goals had been
achieved. Each individual’s target bonus was then adjusted
for personal performance. The following cash bonus amounts for
2010 performance were approved for our named executive officers:
Mr. Truex ($148,000), Mr. Lowe ($84,000),
Dr. Hislop ($67,000), Dr. Odink ($55,000) and
Ms. Kilfoil ($37,000).
Equity Incentive Compensation. We generally
grant stock options to our employees, including our named
executive officers, in connection with their initial employment
with us. We also typically grant stock options on an annual
basis as part of annual performance reviews of our employees.
Our compensation committee had previously established grant
guidelines for our employees, other than our Chief Executive
Officer (whose grants were made at the discretion of the board
of directors), based on an employee’s position, which
guidelines specify a range of equity grant amounts expressed as
a percentage of our common stock outstanding on a fully-diluted
basis, which range from 0.02% to 2.75%, depending on position.
We grant equity incentive compensation to our executive officers
because we believe doing so will motivate our executives by
aligning their interests more closely with the interests of our
stockholders. In the past, we granted restricted stock to
certain initial employees, including Mr. Truex, as we
believed that it was appropriate for our initial key employees
to have an immediate equity stake, and because we believed
owning restricted stock would more closely align the interests
of the recipient with those of our stockholders. Now that we are
a more mature company, we believe it is generally more
appropriate to grant stock options or restricted stock units to
employees, as is the general practice at other companies with
which we compete for talent, although we may continue to grant
restricted stock or grant other types of equity awards when we
deem it appropriate and in our stockholders’ best interests.
Prior to our initial public offering, equity incentive grants to
our named executive officers and other employees were made at
the discretion of our board of directors with the recommendation
of our compensation committee out of our 2005 Equity Incentive
Plan (the “2005 Plan”). In determining equity
incentive grants, the compensation committee considered the
grant guidelines it had established for each position, along
with the equity incentives already provided to an employee. Our
compensation committee also considered individual performance,
based on an informal evaluation of the individual’s
contribution to our corporate goals (which generally include
milestones related to our preclinical and clinical studies and
fundraising) and input received from management. Following our
initial public offering, all stock options continue to be
granted with an exercise price equal to the fair market value of
our common stock on the date of grant, which is defined as the
closing market price of a share of our common stock on the date
of grant. We do not currently have any program, plan or practice
of setting the exercise price based on a date or price other
than the fair value of our common stock on the grant date.
In 2010, our board of directors granted 333,000 restricted stock
units to our employees and options to purchase a total of
112,000 shares of common stock to our directors, including
213,500 restricted stock units to our named executive officers.
In exercising its discretion to determine the amount of each
grant for recommendation to our board of directors, the
compensation committee generally took into account each
individual’s contributions towards the achievement of our
annual corporate goals. In March 2010, our board of directors,
upon the compensation committee’s recommendation, approved
grants of restricted stock units to certain of our employees,
including Mr. Truex, Mr. Lowe, Dr. Hislop and
Dr. Odink, based upon the management team’s
contributions to our 2009 and
80
first quarter 2010 operational performance on a relative scale
dependent on such named executive officer’s job function
and responsibility. The amount of each grant was based upon
industry data as well as such named executive officer’s
current level of equity awards. All of these grants were made to
further motivate the recipients by aligning their interests more
closely with our stockholders over the next several years by
providing them with an equity interest in the company. In
December 2010, based upon the Remedy Compensation Consulting
data, our board of directors, upon recommendation of the
compensation committee, approved annual grants of stock options,
effective as of January 1, 2011, to certain of our
employees, including our named executive officers, which were
intended to target the 50th percentile of equity awards of
our peer group, after giving effect to the annual grants. Our
named executive officers received grants of stock options to
purchase the following shares: Mr. Truex
(260,000 shares), Mr. Lowe (160,000 shares),
Dr. Hislop (170,000 shares), Dr. Odink
(95,000 shares) and Ms. Kilfoil (100,000 shares).
Stock option awards provide our named executive officers and
other employees with the right to purchase shares of our common
stock at a fixed exercise price, subject to their continued
employment. Stock options are earned on the basis of continued
service and generally vest over four years, beginning with
vesting as to 25% of the award on the one-year anniversary of
the date of grant, and pro-rata vesting monthly thereafter.
Stock options granted under the 2010 Plan may not be exercised
prior to the award vesting in full. Stock Options previously
granted under the 2005 Plan may be exercised prior to the award
vesting in full, subject to our right of repurchase. In
addition, in the past, we have also granted options to purchase
smaller amounts of stock, typically fewer than
10,000 shares, which are immediately vested to recognize
employee contributions, including those of our named executive
officers. Furthermore, we generally grant incentive stock
options to employees up to the statutory limit, then
non-statutory options thereafter and non-statutory options to
non-employees. See the section entitled
“— Potential Payments Upon Termination or Change
in Control” for a discussion of the change in control
provisions related to stock options.
Restricted stock units provide our executive officers and other
employees to receive shares of stock upon the vesting of the
restricted stock units, subject to their continued employment.
Restricted stock units generally vest in equal annual
installments over four years. However, we also have granted
restricted stock units that vest in full after one year as a
short-term incentive after our successful initial public
offering. See the section below entitled
“— Potential Payments Upon Change in Control and
Termination” for a discussion of the change in control
provisions related to restricted stock.
After our IPO, we adopted an equity award grant policy that
formalized how we grant equity-based awards to officers and
employees. Under our equity award grant policy, all grants must
be approved by our board of directors or compensation committee.
All stock options will be awarded with an exercise price equal
to the fair value of our common stock and calculated based on
our closing market price on the last trading day of the quarter
in which the grant is approved.
Other Compensation. We currently maintain
broad-based benefits that are provided to all employees,
including health insurance, life and disability insurance,
dental insurance and a 401(k) plan.
As discussed below in “— Severance and Change in
Control Agreements” and in “— Potential
Payments Upon Change in Control and Termination,” we have,
for all named executive officers, agreements providing certain
benefits upon termination of their employment in relation to a
change in control, including the acceleration of vesting of
restricted stock and options. Our goal in providing severance
and change in control benefits is to offer sufficient cash
continuity protection such that our executives will focus their
full time and attention on the requirements of the business
rather than the potential implications for their respective
positions. We prefer to have certainty regarding the potential
severance amounts payable to the named executive officers under
certain circumstances, rather than negotiating severance at the
time that a named executive officer’s employment
terminates. We have also determined that accelerated vesting
provisions in connection with a termination following a change
of control are appropriate because they will encourage our
restricted stock and option holders, including our named
executive officers, to stay focused in such circumstances,
rather than the potential implications for them.
All of our named executive officers are party to severance
agreements that provide benefits upon termination of employment
in connection with a change of control.
Tax and Accounting Treatment of
Compensation. Section 162(m) of the Internal
Revenue Code places a limit of $1.0 million per person on
the amount of compensation that we may deduct in any one year
with respect to each of
81
our named executive officers other than the chief financial
officer. There is an exemption from the $1.0 million
limitation for performance-based compensation that meets certain
requirements. Grants of stock options and stock appreciation
rights under our 2010 Plan are intended to qualify for the
exemption. Restricted stock awards and restricted stock unit
awards under our 2010 Plan, as well as performance cash awards,
may qualify for the exemption if certain additional requirements
are satisfied. To maintain flexibility in compensating officers
in a manner designed to promote varying corporate goals, our
compensation committee has not adopted a policy requiring all
compensation to be deductible. Although tax deductions for some
amounts that we pay to our named executive officers as
compensation may be limited by section 162(m), that
limitation does not result in the current payment of increased
federal income taxes by us due to our significant net operating
loss carry-forwards. Our compensation committee may approve
compensation or changes to plans, programs or awards that may
cause the compensation or awards to exceed the limitation under
section 162(m) if it determines that such action is
appropriate and in our best interests.
We account for equity compensation paid to our employees under
the rules of FASB ASC 718, which requires us to estimate
and record an expense for each award of equity compensation over
the service period of the award. Accounting rules also require
us to record cash compensation as an expense at the time the
obligation is incurred.
Compensation
of Executive Officers
Summary
Compensation Table
The following table summarizes the compensation that we paid to
our Chief Executive Officer, Chief Financial Officer and each of
our three other most highly compensated executive officers
during the years ended December 31, 2010, 2009 and 2008. We
refer to these officers in this report as our named executive
officers.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-Equity
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock
|
|
|
Incentive Plan
|
|
|
|
|
Name and Principal Position as of
|
|
|
|
|
Salary
|
|
|
Bonus
|
|
|
Awards
|
|
|
Compensation
|
|
|
Total
|
|
|
December 31, 2010
|
|
Year
|
|
|
($)
|
|
|
($)
|
|
|
($)(1)
|
|
|
($)(2)
|
|
|
($)
|
|
|
|
|
Paul F. Truex
|
|
|
2010
|
|
|
$
|
383,333
|
|
|
|
|
|
|
$
|
460,960
|
|
|
$
|
398,000
|
|
|
$
|
1,242,293
|
|
|
President, Chief Executive Officer
|
|
|
2009
|
|
|
$
|
281,837
|
|
|
|
|
|
|
$
|
88,125
|
|
|
|
—
|
|
|
$
|
369,962
|
|
|
and Director
|
|
|
2008
|
|
|
$
|
300,000
|
|
|
|
|
|
|
|
—
|
|
|
|
—
|
|
|
$
|
300,000
|
|
|
Christopher P. Lowe
|
|
|
2010
|
|
|
$
|
283,333
|
|
|
|
|
|
|
$
|
214,400
|
|
|
$
|
184,000
|
|
|
$
|
681,733
|
|
|
Chief Business Officer and Chief
|
|
|
2009
|
|
|
$
|
241,174
|
|
|
|
|
|
|
$
|
23,500
|
|
|
|
—
|
|
|
$
|
264,674
|
|
|
Financial Officer
|
|
|
2008
|
|
|
$
|
250,000
|
|
|
|
|
|
|
$
|
117,411
|
|
|
|
—
|
|
|
$
|
367,411
|
|
|
Colin Hislop, M.D.
|
|
|
2010
|
|
|
$
|
303,333
|
|
|
|
|
|
|
$
|
214,400
|
|
|
$
|
132,000
|
|
|
$
|
649,733
|
|
|
Senior Vice President,
|
|
|
2009
|
|
|
$
|
259,621
|
|
|
$
|
1,247
|
|
|
$
|
28,795
|
|
|
|
—
|
|
|
$
|
289,663
|
|
|
Cardiovascular Products
|
|
|
2008
|
|
|
$
|
270,000
|
|
|
|
|
|
|
|
—
|
|
|
|
—
|
|
|
$
|
270,000
|
|
|
Debra Odink, Ph.D.
|
|
|
2010
|
|
|
$
|
232,179
|
|
|
|
|
|
|
$
|
134,000
|
|
|
$
|
80,000
|
|
|
$
|
446,179
|
|
|
Senior Vice President, Pharmaceutical
|
|
|
2009
|
|
|
$
|
158,580
|
|
|
$
|
3,996
|
|
|
$
|
29,375
|
|
|
|
—
|
|
|
$
|
191,951
|
|
|
Research and Development
|
|
|
2008
|
|
|
$
|
200,000
|
|
|
|
|
|
|
|
—
|
|
|
|
—
|
|
|
$
|
200,000
|
|
|
Georgina Kilfoil(3)
|
|
|
2010
|
|
|
$
|
197,917
|
|
|
|
|
|
|
$
|
120,600
|
|
|
$
|
37,000
|
|
|
$
|
355,517
|
|
|
Senior Vice President, Development
|
|
|
2009
|
|
|
|
—
|
|
|
|
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
And Clinical Operations
|
|
|
2008
|
|
|
|
—
|
|
|
|
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
(1) |
|
This column reflects the aggregate grant date fair value of
equity awards granted in 2010, 2009 and 2008. During 2010, we
granted restricted stock units to our executive officers and the
grant date fair value is calculated based on the closing sales
price of our common stock on the grant date and in accordance
with FASB ASC 718. During 2009 and 2008, we granted stock
options to our executive officers and the grant date fair value
is calculated in accordance with FASB ASC 718, excluding
the effect of estimated forfeitures. See Note 11 to our
financial statements for a discussion of the assumptions made in
determining the valuation of option awards. |
| |
|
(2) |
|
Includes performance cash bonus awards paid following our
initial public offering and annual cash performance bonus awards
paid in 2011 for 2010 performance. |
| |
|
(3) |
|
Ms. Kilfoil joined us on March 16, 2010. |
82
Grants of
Plan-Based Awards
The following table sets forth certain information with respect
to awards under our equity and non-equity incentive plans made
by us to our named executive officers and stock options awarded
to our named executive officers for the year ended
December 31, 2010.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Estimated Future Payouts
|
|
|
All Other Stock
|
|
|
|
|
|
|
|
|
|
|
Under Non-Equity Incentive
|
|
|
Awards: Number
|
|
|
Grant Date Fair
|
|
|
|
|
|
|
|
Plan Awards (4)
|
|
|
of Shares of Stock
|
|
|
Value of Stock
|
|
|
|
|
Grant
|
|
|
Target
|
|
|
Maximum
|
|
|
or Units
|
|
|
Awards
|
|
|
Name
|
|
Date
|
|
|
($)
|
|
|
($)(5)
|
|
|
(#)
|
|
|
($)(3)
|
|
|
|
|
Paul F. Truex
|
|
|
6/30/2010
|
(1)
|
|
|
|
|
|
|
|
|
|
|
50,000
|
|
|
$
|
268,000
|
|
|
|
|
|
6/30/2010
|
(2)
|
|
|
|
|
|
|
|
|
|
|
36,000
|
|
|
$
|
192,960
|
|
|
|
|
|
|
|
|
$
|
212,500
|
|
|
$
|
531,250
|
|
|
|
|
|
|
|
|
|
|
Christopher P. Lowe
|
|
|
6/30/2010
|
(1)
|
|
|
|
|
|
|
|
|
|
|
22,000
|
|
|
$
|
117,920
|
|
|
|
|
|
6/30/2010
|
(2)
|
|
|
|
|
|
|
|
|
|
|
18,000
|
|
|
$
|
96,480
|
|
|
|
|
|
|
|
|
$
|
105,000
|
|
|
$
|
375,000
|
|
|
|
|
|
|
|
|
|
|
Colin Hislop, M.D.
|
|
|
6/30/2010
|
(1)
|
|
|
|
|
|
|
|
|
|
|
30,000
|
|
|
$
|
160,800
|
|
|
|
|
|
6/30/2010
|
(2)
|
|
|
|
|
|
|
|
|
|
|
10,000
|
|
|
$
|
53,600
|
|
|
|
|
|
|
|
|
$
|
96,000
|
|
|
$
|
400,000
|
|
|
|
|
|
|
|
|
|
|
Debra Odink, Ph.D.
|
|
|
6/30/2010
|
(1)
|
|
|
|
|
|
|
|
|
|
|
20,000
|
|
|
$
|
107,200
|
|
|
|
|
|
6/30/2010
|
(2)
|
|
|
|
|
|
|
|
|
|
|
5,000
|
|
|
$
|
26,800
|
|
|
|
|
|
|
|
|
$
|
75,000
|
|
|
$
|
312,500
|
|
|
|
|
|
|
|
|
|
|
Georgina Kilfoil
|
|
|
6/30/2010
|
(1)
|
|
|
|
|
|
|
|
|
|
|
22,500
|
|
|
$
|
120,600
|
|
|
|
|
|
|
|
|
$
|
59,400
|
(6)
|
|
$
|
247,396
|
(6)
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
These restricted stock units vest in equal annual installments
over four years. The vesting commencement date of these grants
is June 30, 2010. |
| |
|
(2) |
|
These restricted stock units vest in one annual installment. The
vesting commencement date of these grants is June 30, 2010. |
| |
|
(3) |
|
The grant date fair value of each equity award is calculated
based on the closing sales price of our common stock on the date
of grant in accordance with FASB ASC 718, excluding the
effect of estimated forfeitures. |
| |
|
(4) |
|
The plan does not have a threshold. |
| |
|
(5) |
|
The plan allows for a bonus payment of up to 125% of the
executive officer’s salary. |
| |
|
(6) |
|
Ms. Kilfoil joined us on March 16, 2010. This number
is prorated based on her service for the year. |
83
Outstanding
Equity Awards at Fiscal Year-End
The following table sets forth certain information with respect
to outstanding equity awards as of December 31, 2010 with
respect to our named executive officers.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Option Awards
|
|
|
Stock Awards
|
|
|
|
|
Number of
|
|
|
Number of
|
|
|
|
|
|
|
|
|
Number of
|
|
|
Market Value
|
|
|
|
|
Securities
|
|
|
Securities
|
|
|
|
|
|
|
|
|
Shares or
|
|
|
of Shares
|
|
|
|
|
Underlying
|
|
|
Underlying
|
|
|
|
|
|
|
|
|
Units of
|
|
|
or Units
|
|
|
|
|
Unexercised
|
|
|
Unexercised
|
|
|
Option
|
|
|
|
|
|
Stock That
|
|
|
of Stock
|
|
|
|
|
Options
|
|
|
Options
|
|
|
Exercise
|
|
|
Option
|
|
|
Have Not
|
|
|
That Have
|
|
|
|
|
Exercisable
|
|
|
Unexercisable
|
|
|
Price
|
|
|
Expiration
|
|
|
Vested
|
|
|
Not Vested
|
|
|
Name
|
|
(#)
|
|
|
(#)*
|
|
|
($)
|
|
|
Date
|
|
|
(#)
|
|
|
($)(1)
|
|
|
|
|
Paul F. Truex
|
|
|
362,826
|
|
|
|
—
|
|
|
$
|
0.26
|
|
|
|
1/23/2017
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
52,548
|
|
|
|
13,828
|
(2)
|
|
$
|
1.51
|
|
|
|
2/18/2019
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
16,815
|
|
|
|
4,425
|
(3)
|
|
$
|
1.51
|
|
|
|
2/18/2019
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
50,000
|
(7)
|
|
$
|
244,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
36,000
|
(8)
|
|
$
|
175,680
|
|
|
Christopher P. Lowe
|
|
|
2,920
|
(4)
|
|
|
—
|
|
|
$
|
0.14
|
|
|
|
3/6/2016
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
57,725
|
|
|
|
17,161
|
(5)
|
|
$
|
1.34
|
|
|
|
2/21/2018
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
36,828
|
|
|
|
10,949
|
(6)
|
|
$
|
1.34
|
|
|
|
2/21/2018
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
18,497
|
|
|
|
4,867
|
(2)
|
|
$
|
1.51
|
|
|
|
2/18/2019
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
22,000
|
(7)
|
|
$
|
107,360
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18,000
|
(8)
|
|
$
|
87,840
|
|
|
Colin Hislop, M.D.
|
|
|
130,447
|
|
|
|
—
|
|
|
$
|
0.26
|
|
|
|
1/23/2017
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
18,497
|
|
|
|
4,867
|
(2)
|
|
$
|
1.51
|
|
|
|
2/18/2019
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
5,257
|
|
|
|
—
|
|
|
$
|
1.51
|
|
|
|
4/15/2019
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
30,000
|
(7)
|
|
$
|
146,400
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10,000
|
(8)
|
|
$
|
48,800
|
|
|
Debra Odink, Ph.D.
|
|
|
23,121
|
|
|
|
6,084
|
(2)
|
|
$
|
1.51
|
|
|
|
2/18/2019
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
20,000
|
(7)
|
|
$
|
97,600
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5,000
|
(8)
|
|
$
|
24,400
|
|
|
Georgina Kilfoil
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
22,500
|
(7)
|
|
$
|
109,800
|
|
|
|
|
|
* |
|
Unless otherwise noted in the footnotes, these options vest over
four years as follows: 25% of the shares vest one year following
the vesting commencement date, with the remaining 75% vesting in
equal monthly installments over the next three years. All
unvested options, pursuant to the 2005 Plan, contain an early
exercise feature subject to the Company’s right of
repurchase. |
| |
|
(1) |
|
Represents the closing market price of our common stock as of
December 31, 2010 ($4.88) multiplied by the number of
restricted stock units. |
| |
|
(2) |
|
This incentive stock option vests in equal monthly installments
over four years commencing on August 12, 2008. |
| |
|
(3) |
|
This non-statutory stock option vests in equal monthly
installments over four years commencing on August 12, 2008. |
| |
|
(4) |
|
These options were granted to Mr. Lowe on March 6,
2006 in his capacity as a consultant to the Company and vested
immediately on the grant date. |
| |
|
(5) |
|
The vesting commencement date of this incentive stock option is
November 26, 2007. |
| |
|
(6) |
|
The vesting commencement date of this non-statutory stock option
is November 26, 2007. |
| |
|
(7) |
|
These restricted stock units vest in equal annual installments
over four years. The vesting commencement date of these grants
is June 30, 2010. |
| |
|
(8) |
|
These restricted stock units vest in one annual installment. The
vesting commencement date of these grants is June 30, 2010. |
84
Option
Exercises and Stock Vested
Options
Exercised — 2010
The following table sets forth certain information with respect
to the options exercised during the year ended December 31,
2010 with respect to our named executive officers. There was no
vesting of stock awards during the year ended December 31,
2010 with respect to our named executive officers.
| |
|
|
|
|
|
|
|
|
|
|
|
Option Awards
|
|
|
|
|
Number of
|
|
|
|
|
|
|
|
Shares
|
|
|
|
|
|
|
|
Acquired
|
|
|
Value Realized
|
|
|
|
|
on Exercise
|
|
|
on Exercise
|
|
|
Name
|
|
(#)
|
|
|
($)(1)
|
|
|
|
|
Paul F. Truex
|
|
|
23,364
|
|
|
$
|
158,642
|
|
|
Christopher P. Lowe
|
|
|
—
|
|
|
|
—
|
|
|
Colin Hislop, M.D.
|
|
|
14,683
|
|
|
$
|
43,021
|
|
|
Debra Odink, Ph.D.
|
|
|
—
|
|
|
|
—
|
|
|
Georgina Kilfoil
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
(1) |
|
This column reflects the value realized for vested options
exercised in 2010, which represents the difference between the
market price of our common stock on the date of exercise and the
exercise price of the stock option. |
Stock and
Benefit Plans
2005
Equity Incentive Plan
Our 2005 Plan was adopted by our board of directors and approved
by our stockholders in April 2005. We have reserved
2,175,817 shares of our common stock for the issuance of
awards under the 2005 Plan.
Our 2005 Plan is administered by our board of directors, which
has the authority to delegate full power and authority to a
committee of the board. Our board of directors or any committee
delegated by our board of directors has the power to select the
individuals to whom awards will be granted, to make any
combination of awards to participants, to accelerate the
exercisability or vesting of any award, to provide substitute
awards and to determine the specific terms and conditions of
each award, subject to the provisions of the 2005 Plan.
The 2005 Plan permits us to make grants of incentive stock
options, non-qualified stock options, restricted stock awards
and stock appreciation rights to employees, directors and
consultants. Stock options granted under the 2005 Plan have a
maximum term of 10 years from the date of grant and
incentive stock options have an exercise price of no less than
the fair market value of our common stock on the date of grant.
Upon a sale event in which all awards are not assumed or
substituted by the successor entity, the vesting of awards under
the 2005 Plan shall be accelerated in full prior to the sale
event and all stock options issued thereunder will terminate.
All stock option awards that are granted to our named executive
officers are covered by a stock option agreement. Except as
noted above, under the stock option agreements, 25% of the
shares vest on the first anniversary of the grant date and the
remaining shares vest monthly over the following three years.
Our board of directors may accelerate the vesting schedule in
its discretion. We did not engage in any option repricing or
other modification to any of our outstanding equity awards
during the fiscal year ended December 31, 2010.
Our board of directors has determined not to grant any further
awards under the 2005 Plan after the completion of our initial
public offering. We have adopted the 2010 Plan effective upon
the consummation of our initial public offering in March 2010.
Amended
and Restated 2010 Stock Option and Incentive Plan
In February 2010, our board of directors, upon the
recommendation of our compensation committee, approved the 2010
Plan, which was also approved by our stockholders. Our board of
directors subsequently approved the amendment and restatement of
our 2010 Plan, which was approved by our stockholders at our
annual stockholders’ meeting held in July 2010. The 2010
Plan became effective upon the consummation of our initial
public offering and replaced the 2005 Plan, as our board of
directors determined not to make additional awards under that
plan once
85
the 2010 Plan became effective. The 2010 Plan provides
flexibility to our compensation committee to use various
equity-based incentive awards as compensation tools to motivate
our workforce.
We initially reserved 233,644 shares of our common stock
for the issuance of awards under the 2010 Plan plus an
additional 35,670 shares of common stock available for
grant under our 2005 Plan, which shares were added to the shares
reserved under our 2010 Plan, and an additional
200,000 shares that were added by the amendment and
restatement approved at our 2010 annual stockholders’
meeting. The 2010 Plan provides that the number of shares
reserved and available for issuance under the plan will
automatically increase each January 1, beginning in 2011,
by 4% of the outstanding number of shares of common stock on the
immediately preceding December 31. This number is subject
to adjustment in the event of a stock split, stock dividend or
other change in our capitalization.
The shares we issue under the 2010 Plan are authorized but
unissued shares or shares that we reacquire. The shares of
common stock underlying any awards that are forfeited, canceled,
held back upon exercise or settlement of an award to satisfy the
exercise price or tax withholding, reacquired by us prior to
vesting, satisfied without any issuance of stock, expire or are
otherwise terminated (other than by exercise) under the 2010
Plan are added back to the shares of common stock available for
issuance under the 2010 Plan.
The 2010 Plan is administered by our board of directors under
recommendation by our compensation committee. Our board of
directors has full power to select, from among the individuals
eligible for awards, the individuals to whom awards will be
granted, to make any combination of awards to participants and
to determine the specific terms and conditions of each award,
subject to the provisions of the 2010 Plan. The board of
directors may delegate to our compensation committee or our
Chief Executive Officer the authority to grant options to
certain individuals. Persons eligible to participate in the 2010
Plan will be those of our full or part-time officers, employees,
non-employee directors and other key persons (including
consultants and prospective employees) as selected from time to
time by our board of directors in its discretion.
The 2010 Plan permits the granting of (i) options to
purchase common stock intended to qualify as incentive stock
options under Section 422 of the Internal Revenue Code of
1986, as amended, and the regulations thereunder, or the Code,
and (ii) options that do not so qualify. The option
exercise price of each option will be determined by our
compensation committee but may not be less than 100% of the fair
market value of the common stock on the date of grant. The term
of each option will be fixed by our compensation committee and
may not exceed 10 years from the date of grant. Our
compensation committee will determine at what time or times each
option may be exercised.
Our board of directors may award stock appreciation rights
subject to such conditions and restrictions as our compensation
board of directors may determine. Stock appreciation rights
entitle the recipient to shares of common stock equal to the
value of the appreciation in the stock price over the exercise
price. The exercise price shall not be less than the fair market
value of the common stock on the date of grant.
Our board of directors may award restricted shares of common
stock to participants subject to such conditions and
restrictions as our compensation committee may determine. These
conditions and restrictions may include the achievement of
certain performance goals
and/or
continued employment with us through a specified restricted
period. Our compensation committee may award restricted stock
units to any participants. Restricted stock units are ultimately
payable in the form of shares of common stock and may be subject
to such conditions and restrictions as our compensation
committee may determine. These conditions and restrictions may
include the achievement of certain performance goals
and/or
continued employment through a specified vesting period. Our
board of directors may also grant shares of common stock which
are free from any restrictions under the 2010 Plan. Unrestricted
stock may be granted to any participant in recognition of past
services or other valid consideration and may be issued in lieu
of cash compensation due to such participant.
Our board of directors may grant performance share awards to any
participant which entitles the recipient to receive shares of
common stock upon the achievement of certain performance goals
and such other conditions as our compensation committee shall
determine.
Our board of directors may grant dividend equivalent rights to
participants which entitle the recipient to receive credits for
dividends that would be paid if the recipient had held specified
shares of common stock.
Our board of directors may grant cash bonuses under the 2010
Plan to participants. The cash bonuses may be subject to the
achievement of certain performance goals.
86
The 2010 Plan also provides that upon the effectiveness of a
“sale event” as defined in the 2010 Plan, except as
otherwise provided by our compensation committee in the award
agreement, all awards will automatically terminate, unless the
parties to the sale event agree that such awards will be assumed
or continued by the successor entity. Awards with conditions and
restrictions relating to the attainment of performance goals may
become vested and non-forfeitable in connection with a sale
event in the compensation committee’s discretion. In
addition, in the case of a sale event in which our stockholders
will receive cash consideration, we may make or provide for a
cash payment to participants holding options and stock
appreciation rights equal to the difference between the per
share cash consideration and the exercise price of the options
or stock appreciation rights.
No awards may be granted under the 2010 Plan after the date that
is 10 years from the date of stockholder approval.
2010
Employee Stock Purchase Plan
Our board of directors adopted the Anthera Pharmaceuticals, Inc.
2010 Employee Stock Purchase Plan (the “ESPP”), and
our stockholders approved the ESPP at our 2010 annual
stockholders’ meeting. Our Board of Directors subsequently
amended the ESPP on December 15, 2010. We have reserved
100,000 shares of common stock for issuance thereunder plus
on January 1, 2011 and each January 1 thereafter, the
number of shares of stock reserved and available for issuance
under the Plan shall be cumulatively increased by the lesser of
(i) one percent (1%) of the number of shares of common
stock issued and outstanding on the immediately preceding
December 31 or (ii) 250,000 shares of common stock.
Under the ESPP, eligible employees of the Company and certain
designated subsidiaries of the Company may authorize the Company
to deduct amounts from their compensation, which amounts are
used to enable the employees to purchase shares of the
Company’s common stock. The purpose of the ESPP is to
attract and retain key personnel, and encourage stock ownership
by the Company’s employees.
The ESPP is a broad-based employee stock purchase plan under
Section 423 of the Code.
The shares that are reserved under the ESPP have an aggregate
value of approximately $0.5 million based on the closing
price of the common stock as reported on The NASDAQ Global
Market on December 31, 2010.
The ESPP is administered by the person or persons appointed by
the Company’s board of directors. The ESPP provides that
all employees of the Company and any designated subsidiaries of
the Company who work at least 20 hours per week are
eligible to participate in the ESPP, except for persons who are
deemed under Section 423(b)(3) of the Code to own five
percent (5%) or more of the voting stock of the Company.
Participation by any eligible employee is voluntary. The number
of employees potentially eligible to participate in the ESPP is
approximately 20 persons.
The ESPP provides for two “offering periods” within
each year, and the first commenced on September 1, 2010 and
ended on December 31, 2010. Thereafter, the first offering
period in a year will commence on the first business day
occurring on or after each January 1 and ending on the last
business day occurring on or before the following June 30,
and the second will commence on the first business day occurring
on or after each July 1 and ending on the last business day
occurring on or before the following December 31. Eligible
employees may elect to become participants in the ESPP by
enrolling prior to each semi-annual date to purchase shares
under the ESPP. Shares are purchased through the accumulation of
payroll deductions of not less than one percent (1%) nor more
than ten percent (10%) of each participant’s compensation.
The maximum number of shares of common stock that can be
purchased under the ESPP during any one calendar year is that
number having a fair market value of $25,000 on the first day of
the purchase period pursuant to which the shares are purchased.
The number of shares to be purchased with respect to any
purchase period will be the lesser of (a) the number of
shares determined by dividing the participant’s balance in
the plan account on the last day of the purchase period by the
purchase price per share for the stock,
(b) 10,000 shares, and (c) such other lesser
maximum number of shares as shall have been established by the
administrator in advance of the offering. The purchase price per
share will be 85% of the fair market value of the common stock
as of the first date or the ending date of the applicable
semi-annual purchase period, whichever is less.
A participant’s right to purchase shares during a purchase
period under the ESPP is not transferable by the participant
except by will or by the laws of descent and distribution.
Employees may cease their participation in the offering at any
time during the offering period, and participation automatically
ceases on termination of employment with the Company.
87
The number of shares that are reserved for issuance under the
ESPP is subject to adjustment for stock splits and similar
events. The proceeds received by the Company from exercise under
the ESPP will be used for the general purposes of the Company.
Shares issued under the ESPP may be authorized but unissued
shares or shares reacquired by the Company and held in its
treasury.
The ESPP shall remain in full force and effect until suspended
or discontinued by our board of directors. Our board of
directors may, at any time, terminate the ESPP; provided, that
the ESPP shall automatically terminate in accordance with its
terms as of the tenth anniversary of its adoption by the board
of directors. Our board of directors may at any time, and from
time to time, amend the ESPP in any respect, except that without
the approval within 12 months of such board action by the
stockholders, no amendment may be made increasing the number of
shares approved for the ESPP or making any other change that
would require stockholder approval in order for the ESPP, as
amended, to qualify as an “employee stock purchase
plan” under Section 423(b) of the Code.
401(k)
Savings Plan
We have established a 401(k) plan to allow our employees to save
on a tax-favorable basis for their retirement. We do not match
any contributions made by any employees, including our named
executive officers, pursuant to the plan.
Pension
Benefits
None of our named executive officers participate in or have
account balances in pension benefit plans sponsored by us.
Nonqualified
Defined Contribution and Other Nonqualified Defined Compensation
Plans
None of our named executive officers participate in or have
account balances in non-qualified defined contribution plans or
other deferred compensation plans maintained by us.
Severance
and Change in Control Arrangements
We consider it essential to the best interests of our
stockholders to foster the continuous employment of our key
management personnel. In this regard, we recognize that the
possibility of a change in control may exist and that the
uncertainty and questions that it may raise among management
could result in the departure or distraction of management
personnel to the detriment of the Company and our stockholders.
In order to reinforce and encourage the continued attention and
dedication of certain key members of management, we have entered
into several change in control agreements and severance
agreements with certain of our executive officers.
In these agreements, the definition of “change in
control” generally means the occurrence, in a single
transaction or in a series of related transactions of any one or
more of the following events, subject to specified events:
(a) any Exchange Act Person (defined in the change in
control agreements generally as any natural person, entity, or
group not including the Company or any subsidiaries) becomes the
owner of securities representing more than 50% of the combined
voting power of our then outstanding securities; (b) a
merger, consolidation or similar transaction involving the
Company is consummated and immediately after the consummation of
such merger, consolidation, or similar transaction, our
stockholders immediately prior thereto do not own either
outstanding voting securities representing more than 50% of the
combined outstanding voting power of the surviving entity or
more than 50% of the combined outstanding voting power of the
parent of the surviving entity in such merger, consolidation, or
similar transaction; or (c) a sale, lease, license or other
disposition of all or substantially all of our consolidated
assets is consummated.
In these agreements, “cause” means: (a) gross
negligence or willful misconduct in the performance of duties
that is not cured within 30 days of written notice, where
such gross negligence or willful misconduct has resulted or is
likely to result in substantial and material damage to the
Company; (b) repeated unexplained or unjustified absence;
(c) a material and willful violation of any federal or
state law; (d) commission of any act of fraud with respect
to the Company; or (e) commission of an act of moral
turpitude or conviction of or entry of a plea of nolo contendere
to a felony.
“Constructive termination” means an
officer’s resignation within 180 days of the
occurrence of any of the following events without the
officer’s prior written consent, provided the officer
provides notice within 90 days of the first occurrence of
such event and such event remains uncured 30 days after
delivery of the written notice: (a) a
88
material diminution of such officer’s duties,
responsibilities or authority; (b) a material diminution of
base compensation; or (c) a material change in the
geographic location at which the officer provides services to us.
Paul
F. Truex
On October 15, 2009, we entered into an amended and
restated change in control agreement with Mr. Truex. Upon
the occurrence of a change in control or within 12 months
thereafter, if we terminate Mr. Truex’s employment for
any reason other than for cause or if there is a constructive
termination, in either case, Mr. Truex is entitled to
receive as severance compensation 100% of his then-current base
salary for a period of up to 12 months and payment of
continuation coverage premiums for health, dental, and vision
benefits for Mr. Truex and his covered dependents, if any,
for a period of 12 months pursuant to COBRA. In addition,
Mr. Truex is entitled to receive
(i) 12 months’ accelerated vesting of any
unvested options to purchase our common stock and (ii) the
immediate lapsing of any vesting restrictions on any restricted
stock awards as of the date of termination.
Christopher
P. Lowe
On October 12, 2009, we entered into an amended and
restated change in control agreement with Mr. Lowe. Upon
the occurrence of a change in control or within 12 months
thereafter, if we terminate Mr. Lowe’s employment for
any reason other than for cause or if there is a constructive
termination, in either case, Mr. Lowe is entitled to
receive as severance compensation 100% of his then-current base
salary for a period of up to 12 months and payment of
continuation coverage premiums for health, dental, and vision
benefits for Mr. Lowe and his covered dependents, if any,
for a period of 12 months pursuant to COBRA. In addition,
Mr. Lowe is entitled to receive
(i) 12 months’ accelerated vesting of any
unvested options to purchase our common stock and (ii) the
immediate lapsing of any vesting restrictions on any restricted
stock awards as of the date of termination.
Colin
Hislop, M.D.
On October 15, 2009, we entered into an amended and
restated change in control agreement with Dr. Hislop. Upon
the occurrence of a change in control or within 12 months
thereafter, if we terminate Dr. Hislop’s employment
for any reason other than for cause or if there is a
constructive termination, in either case, Dr. Hislop is
entitled to receive as severance compensation 100% of his
then-current base salary for a period of up to six months and
payment of continuation coverage premiums for health, dental,
and vision benefits for Dr. Hislop and his covered
dependents, if any, for a period of six months pursuant to
COBRA. In addition, Dr. Hislop is entitled to receive
(i) six months’ accelerated vesting of any unvested
options to purchase our common stock and (ii) the immediate
lapsing of any vesting restrictions on any restricted stock
awards as of the date of termination.
Debra
Odink, Ph.D.
On October 15, 2009, we entered into an amended and
restated change in control agreement with Dr. Odink. Upon
the occurrence of a change in control or within 12 months
thereafter, if we terminate Dr. Odink’s employment for
any reason other than for cause or if there is a constructive
termination, in either case, Dr. Odink is entitled to
receive as severance compensation 100% of her then-current base
salary for a period of up to six months and payment of
continuation coverage premiums for health, dental, and vision
benefits for Dr. Odink and her covered dependents, if any,
for a period of six months pursuant to COBRA. In addition,
Dr. Odink is entitled to receive (i) six months’
accelerated vesting of any unvested options to purchase our
common stock and (ii) the immediate lapsing of any vesting
restrictions on any restricted stock awards as of the date of
termination.
Georgina
Kilfoil
On July 7, 2010, we entered into a change in control
agreement with Ms. Kilfoil. Upon the occurrence of a change
in control or within 12 months thereafter, if we terminate
Ms. Kilfoil’s employment for any reason other than for
cause or if there is a constructive termination, in either case,
Ms. Kilfoil is entitled to receive as severance
compensation 100% of her then-current base salary for a period
of up to twelve months and payment of continuation coverage
premiums for health, dental, and vision benefits for
Ms. Kilfoil and her covered dependents, if any, for a
period of twelve months pursuant to COBRA. In addition,
Ms. Kilfoil is entitled to receive (i) twelve
months’ accelerated vesting of any unvested options to
purchase our common stock and (ii) the immediate lapsing of
any vesting restrictions on any restricted stock awards as of
the date of termination.
89
All payments and benefits are conditioned on the
executive’s execution and non-revocation of a general
release agreement at the time of termination. All payments due
upon termination (as discussed in this entire section) may be
delayed up to six months from the termination date if necessary
to avoid adverse tax treatment under Section 409A of the
Internal Revenue Code.
Potential
Payments Upon Change in Control and Termination
The tables below reflect potential payments and benefits
available for each of our named executive officers upon
termination in connection with a change in control or
termination, assuming the date of occurrence is
December 31, 2010. See section entitled
“— Severance and Change in Control
Agreements” above.
Named
Executive Officer Benefits and Payments Upon
Termination(1)
| |
|
|
|
|
|
|
|
Involuntary
|
|
|
|
Termination within
|
|
|
|
One Year of Change
|
|
Name
|
|
in Control(2)
|
|
|
|
Paul F. Truex
|
|
$
|
431,704
|
|
|
Christopher P. Lowe
|
|
$
|
305,847
|
|
|
Colin Hislop, M.D.
|
|
$
|
163,261
|
|
|
Debra Odink, Ph.D.
|
|
$
|
127,878
|
|
|
Georgina Kilfoil
|
|
$
|
256,265
|
|
|
|
|
|
(1) |
|
Assumes triggering event effective as of December 31, 2010.
Upon a voluntary termination or termination for cause, each
named executive officer would receive any earned but unpaid base
salary until December 31, 2010. These payments would be
available to all employees upon termination. |
| |
|
(2) |
|
Includes continuation of base salary determined as of
December 31, 2010 and health, dental and vision benefits
for 12 months for Mr. Truex, Mr. Lowe and
Ms. Kilfoil. All other named executive officers receive six
months’ continuation of base salary and benefits. |
Acceleration
of Vesting of Options and Stock upon
Termination(1)
| |
|
|
|
|
|
|
|
Number of Shares of
|
|
|
|
Accelerated Stock and Value
|
|
|
|
upon Involuntary
|
|
|
|
Termination and in
|
|
|
|
Connection with a
|
|
Name
|
|
Change in Control(2)
|
|
|
|
Paul F. Truex
|
|
$
|
456,588
|
(3)
|
|
Christopher P. Lowe
|
|
$
|
304,550
|
(4)
|
|
Colin Hislop, M.D.
|
|
$
|
200,120
|
(5)
|
|
Debra Odink, Ph.D.
|
|
$
|
128,150
|
(6)
|
|
Georgina Kilfoil
|
|
$
|
109,800
|
(7)
|
|
|
|
|
(1) |
|
Assumes triggering event effective as of December 31, 2010
and excludes vested stock held as of such date. The market value
of the accelerated option shares is based on the difference
between our closing price of $4.88 per share on
December 31, 2010 and the exercise price of such
accelerated options. The market value of the accelerated
restricted stock units is based on the closing price of $4.88
per share on December 31, 2010. |
| |
|
(2) |
|
Includes acceleration of options for 12 months for
Mr. Truex, Mr. Lowe and Ms. Kilfoil. All other
named executive officers have six months’ acceleration of
options. |
| |
|
(3) |
|
10,952 of Mr. Truex’s options and 86,000 shares
of Mr. Truex’s restricted stock units would accelerate
upon involuntary termination and in connection with a change of
control. |
| |
|
(4) |
|
31,030 of Mr. Lowe’s options and 40,000 shares of
Mr. Lowe’s restricted stock units would accelerate
upon involuntary termination and in connection with a change of
control. |
| |
|
(5) |
|
1,460 of Dr. Hislop’s options and 40,000 shares
of Dr. Hislop’s restricted stock units would
accelerate upon involuntary termination and in connection with a
change of control. |
90
|
|
|
|
(6) |
|
1,825 of Dr. Odink’s options and 25,000 shares of
Dr. Odink’s restricted stock units would accelerate
upon involuntary termination and in connection with a change of
control. |
| |
|
(7) |
|
22,500 shares of Ms. Kilfoil’s restricted stock
units would accelerate upon involuntary termination and in
connection with a change of control. |
Compensation
Committee Interlocks and Insider Participation
None of the members of the Compensation Committee is or has at
any time during the past fiscal year been an officer or employee
of the Company. None of the members of the Compensation
Committee has formerly been an officer of the Company. None of
our executive officers serve or in the past fiscal year has
served as a member of the board of directors or compensation
committee of any other entity that has one or more executive
officers serving as a member of our Board of Directors or
Compensation Committee.
The following Compensation Committee Report is not deemed
filed with the Securities and Exchange Commission.
Notwithstanding anything to the contrary set forth in any of the
Company’s filings made under the Securities Act of 1933 or
the Exchange Act that might incorporate filings made by the
Company under those statutes, the Compensation Committee Report
shall not be incorporated by reference into any prior filings or
into any future filings made by the Company under those
statutes.
Compensation
Committee Report
The Compensation Committee of the Board of Directors (the
“Compensation Committee”) has furnished this report on
executive compensation. None of the members of the Compensation
Committee is currently an officer or employee of the Company and
all are “non-employee directors” for purposes of
Rule 16b-3
under the Exchange Act and “outside directors” for
purposes of Section 162(m) of the Internal Revenue Code.
The Compensation Committee is responsible for designing,
recommending to the Board of Directors for approval and
evaluating the compensation plans, policies and programs of the
Company and reviewing and approving the compensation of the
Chief Executive Officer and other officers and directors.
This report, filed in accordance with Item 407(e)(5) of
Regulation S-K,
should be read in conjunction with the other information
relating to executive compensation which is contained elsewhere
in this report and is not repeated here.
In this context, the Compensation Committee hereby reports as
follows:
1. The Compensation Committee has reviewed and discussed
the Compensation Discussion and Analysis section contained
herein with management.
2. Based on the review and discussions referred to in
paragraph (1) above, the Compensation Committee recommended
to the Board of Directors, and the Board of Directors has
approved, that the Compensation Discussion and Analysis be
included in this report.
COMPENSATION COMMITTEE
David E. Thompson,
Chairman
Donald J. Santel
Peter A. Thompson, M.D.
91
|
|
|
ITEM 12.
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND
RELATED STOCKHOLDER MATTERS.
|
Securities
Authorized for Issuance Under Equity Compensation
Plans
The following table provides information regarding our equity
compensation plans in effect as of December 31, 2010.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity Compensation Plan Information
|
|
|
|
|
|
|
|
Number of
|
|
|
|
|
|
|
|
Securities
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
Weighted
|
|
Available for
|
|
|
|
Number of
|
|
Average
|
|
Future Issuance
|
|
|
|
Securities
|
|
Exercise
|
|
Under Equity
|
|
|
|
to be Issued
|
|
Price of
|
|
Compensation
|
|
|
|
Upon Exercise of
|
|
Outstanding
|
|
Plan (Excluding
|
|
|
|
Outstanding
|
|
Options,
|
|
Securities
|
|
|
|
Options, Warrants
|
|
Warrants
|
|
Referenced in
|
|
|
|
and Rights
|
|
and Rights
|
|
Column (a))
|
|
Plan Category
|
|
(a) (1)
|
|
(b) (2)
|
|
(c)
|
|
|
|
Equity compensation plans approved by security holders(3)
|
|
|
1,578,491
|
|
|
$
|
1.26
|
|
|
|
110,473
|
(4)
|
|
Equity compensation plans not approved by security holders
|
|
|
N/A
|
|
|
|
N/A
|
|
|
|
N/A
|
|
|
Total
|
|
|
1,578,491
|
|
|
$
|
1.26
|
|
|
|
110,473
|
(4)
|
|
|
|
|
(1) |
|
Includes 302,500 shares issuable upon settlement of outstanding
restricted stock units with a weighted average grant date fair
value of $5.13 per share. |
| |
|
(2) |
|
Does not take into account shares issuable upon settlement of
restricted stock units, which have no exercise price. |
| |
|
(3) |
|
Consists of our 2005 Plan, our Amended and Restated 2010 Plan
and our ESPP. |
| |
|
(4) |
|
This number includes 75,084 shares that were available for
future issuance under the ESPP, as of December 31, 2010.
The 2010 Plan provides that the number of shares reserved and
available for issuance under the plan will automatically
increases each January 1, beginning in 2011, by 4% of the
outstanding number of shares or common stock on the immediately
preceding December 31. This number is subject to adjustment
in the event of a stock split, stock dividend or other change in
our capitalization. Under the ESPP, we have reserved
100,000 shares of common stock for issuance thereunder plus
on January 1, 2011 and each January 1 thereafter, the
number of shares of stock reserved and available for issuance
under the plan is cumulatively increased by the lesser of
(i) one percent (1%) of the number of shares of common
stock issued and outstanding on the immediately preceding
December 31 or (ii) 250,000 shares of common stock. |
92
Security
Ownership of Certain Beneficial Owners and Management
The following table sets forth information with respect to the
beneficial ownership of shares of our common stock by
(i) each director, (ii) each named executive officer,
(iii) all directors and executive officers as a group, and
(iv) each person who we know beneficially owns more than 5%
of our common stock as of January 31, 2011, unless
otherwise indicated below.
Beneficial ownership is determined in accordance with the rules
of the SEC. These rules generally attribute beneficial ownership
of securities to persons who possess sole or shared voting power
or investment power with respect to those securities and include
shares of common stock issuable upon the exercise of stock
options that are immediately exercisable or exercisable within
60 days after January 31, 2011, but excludes unvested
stock options, which contain an early exercise feature. Except
as otherwise indicated, all of the shares reflected in the table
are shares of common stock and all persons listed below have
sole voting and investment power with respect to the shares
beneficially owned by them, subject to applicable community
property laws. The information is not necessarily indicative of
beneficial ownership for any other purpose.
In computing the number of shares of common stock beneficially
owned by a person and the percentage ownership of that person,
we deemed outstanding shares of common stock subject to options
or warrants held by that person that are currently exercisable
or exercisable within 60 days of January 31, 2011. We
did not deem these shares outstanding, however, for the purpose
of computing the percentage ownership of any other person.
Percentage ownership calculations for beneficial ownership for
each person or entity are based on 32,906,412 shares
outstanding as of January 31, 2011. Except as otherwise
indicated in the table below, addresses of named beneficial
owners are in care of Anthera Pharmaceuticals, Inc., 25801
Industrial Blvd., Suite B, Hayward, California 94545.
| |
|
|
|
|
|
|
|
|
|
|
|
Number of Shares
|
|
|
|
|
|
|
|
Beneficially
|
|
|
Percent of
|
|
|
Name of Beneficial Owner
|
|
Owned
|
|
|
Class
|
|
|
|
|
5% or Greater Stockholders:
|
|
|
|
|
|
|
|
|
|
VantagePoint Venture Partners IV, L.P. and affiliated entities,
or VantagePoint(1)
|
|
|
6,472,646
|
|
|
|
19.56
|
%
|
|
Caduceus Private Investments IV, LP(2)
|
|
|
4,783,068
|
|
|
|
13.97
|
%
|
|
Sofinnova Venture Partners VI, L.P. and affiliated entities, or
Sofinnova(3)
|
|
|
4,177,621
|
|
|
|
12.65
|
%
|
|
FRM LLC(4)
|
|
|
2,926,000
|
|
|
|
8.89
|
%
|
|
Visium Balanced Master Fund, Ltd.(5)
|
|
|
2,599,197
|
|
|
|
7.78
|
%
|
|
Columbia Wanger Asset Management, LLC(6)
|
|
|
1,823,600
|
|
|
|
5.54
|
%
|
|
A.M. Pappas Life Science Ventures III, L.P. and affiliated
entities(7)
|
|
|
1,813,140
|
|
|
|
5.47
|
%
|
|
HBM BioCapital, L.P. and affiliated entities(8)
|
|
|
1,702,471
|
|
|
|
5.15
|
%
|
|
All 5% or greater stockholders as a group
|
|
|
26,297,743
|
|
|
|
74.27
|
%
|
|
Named Executive Officers and Directors:
|
|
|
|
|
|
|
|
|
|
Paul F. Truex(9)
|
|
|
1,133,302
|
|
|
|
3.40
|
%
|
|
Christopher P. Lowe(10)
|
|
|
161,648
|
|
|
|
*
|
|
|
Colin Hislop, M.D.(11)
|
|
|
179,676
|
|
|
|
*
|
|
|
Debra Odink, Ph.D.(12)
|
|
|
120,961
|
|
|
|
*
|
|
|
Georgina Kilfoil(13)
|
|
|
87,616
|
|
|
|
*
|
|
|
Christopher S. Henney, Ph.D.(14)
|
|
|
122,408
|
|
|
|
*
|
|
|
Annette Bianchi(15)
|
|
|
23,332
|
|
|
|
*
|
|
|
James I. Healy, M.D., Ph.D.(16)
|
|
|
4,211,175
|
|
|
|
12.75
|
%
|
|
Donald J. Santel(17)
|
|
|
24,244
|
|
|
|
*
|
|
|
Daniel K. Spiegelman(18)
|
|
|
14,250
|
|
|
|
*
|
|
|
David E. Thompson(19)
|
|
|
37,690
|
|
|
|
*
|
|
|
Peter A. Thompson
|
|
|
—
|
|
|
|
—
|
|
|
All named executive officers and directors as a group
(12 persons)
|
|
|
6,116,302
|
|
|
|
17.64
|
%
|
93
|
|
|
|
* |
|
Represents beneficial ownership of less than 1% of the shares of
common stock. |
| |
|
(1) |
|
Includes (i) 5,695,228 shares of common stock and
147,861 shares of common stock issuable upon exercise of
warrants, all owned of record by VantagePoint Venture
Partners IV (Q), L.P., (ii) 570,147 shares of
common stock and 14,801 shares of common stock issuable
upon exercise of warrants, all owned of record by VantagePoint
Venture Partners IV, L.P., (iii) 20,739 shares of
common stock and 538 shares of common stock issuable upon
exercise of warrants, all owned of record by VantagePoint
Venture Partners IV Principals Fund, L.P., and
(iv) options to purchase an additional 23,332 shares
of common stock that are exercisable within 60 days of
January 31, 2011 that are owned of record by Annette
Bianchi, over which VantagePoint has sole voting and investment
power. Ms. Bianchi, a director of Anthera, is a Managing
Director at VantagePoint. Alan E. Salzman, through his authority
to cause the general partner of the limited partnerships that
directly hold such shares to act, may be deemed to have voting
and investment power with respect to such shares.
Mr. Salzman disclaims beneficial ownership with respect to
such shares except to the extent of his pecuniary interest
therein. The address for VantagePoint Venture Partners is 1001
Bayhill Drive, Suite 300, San Bruno, CA 94066. |
| |
|
(2) |
|
Includes 3,449,734 shares of common stock and
1,333,334 shares of common stock issuable upon exercise of
warrants, all owned of record by Caduceus Private Investments,
LP. OrbiMed Advisors LLC, a registered investment adviser under
the Investment Advisers Act of 1940, as amended, is the sole
managing member of OrbiMed Capital GP IV LLC, which is the sole
general partner of Caduceus Private Investments IV, LP. Samuel
D. Isaly is the owner of a controlling interesting in OrbiMed
Advisors LLC. As such, OrbiMed Advisors LLC, OrbiMed Capital GP
IV LLC and Mr. Isaly may be deemed to have voting and
investment power with respect to such shares. The address for
OrbiMed Advisors LLC is 767 Third Avenue, 30th Floor, New York,
New York 10017. |
| |
|
(3) |
|
Includes: (i) 3,360,574 shares of common stock and
86,996 shares of common stock issuable upon exercise of
warrants all owned of record by Sofinnova Venture Partners VI,
L.P.; (ii) 665,820 shares of common stock and
17,237 shares of common stock issuable upon exercise of
warrants all owned of record by Sofinnova Venture Partners VI
GmbH & Co. KG; and (iii) 45,809 shares of
common stock, all owned of record by Sofinnova Venture
Affiliates VI, L.P. Alain Azan, Eric Buatois, Michael Powell and
Dr. James I. Healy are the managing members of the general
partner of the limited partnership, that directly hold such
shares, and as such, may be deemed to share voting and
investment power with respect to such shares. Dr. Healy is
a director of Anthera. Messrs. Azan, Buatois and Powell and
Dr. Healy disclaim beneficial ownership, except to the
extent of their proportionate pecuniary interest in Sofinnova.
The address for Sofinnova Ventures is 850 Oak Grove Ave., Menlo
Park, CA 94025. |
| |
|
(4) |
|
Based on Schedule 13G filed on February 14, 2011 filed
on behalf of FMR LLC and Edward C. Johnson 3d, Chairman of FMR
LLC. Reported holdings consist of 2,926,000 shares of
common stock beneficially owned by Fidelity
Management & Research Company (“Fidelity”),
a wholly-owned subsidiary of FMR LLC. Edward C. Johnson 3d and
FMR LLC, through its control of Fidelity, and the funds each has
sole power to dispose of the 2,926,000 shares owned by the
funds. Members of the family of Edward C. Johnson 3d, Chairman
of FMR LLC, are the predominant owners, directly or through
trusts, of Series B voting common shares of FMR LLC,
representing 49% of the voting power of FMR LLC. The Johnson
family group and all other Series B shareholders have
entered into a shareholders’ voting agreement under which
all Series B voting common shares will be voted in
accordance with the majority vote of Series B voting common
shares. Accordingly, through their ownership of voting common
shares and the execution of the shareholders’ voting
agreement, members of the Johnson family may be deemed, under
the Investment Company Act of 1940, to form a controlling group
with respect to FMR LLC. Neither FMR LLC nor Edward C. Johnson
3d, Chairman of FMR LLC, has the sole power to vote or direct
the voting of the shares owned directly by the Fidelity Funds,
which power resides with the Funds’ Boards of Trustees.
Fidelity carries out the voting of the shares under written
guidelines established by the Funds’ Boards of Trustees.
The address for Fidelity is 82 Devonshire Street, Boston,
Massachusetts 02109. |
| |
|
(5) |
|
Based on Schedule 13G/A filed February 10, 2011 on
behalf of the following persons: Visium Balanced Master Fund,
LTD, a Cayman Islands corporation (“VBMF”),Visium
Asset Management, LP, a Delaware limited partnership
(“VAM”), JG Asset, LLC, a Delaware limited liability
company (“JG Asset”), and Jacob |
94
|
|
|
|
|
|
Gottlieb. Reported holdings consist of 2,099,197 shares of
common stock and 500,000 shares of commons stock issuable
upon exercise of warrants, all owned of record by VBMF. Jacob
Gottlieb, Managing Member of JG Asset, which is the General
Partner of VAM, which is the investment manager to pooled
investment funds, may be deemed to have voting and investment
power with respect to such shares. The address for Visium
Balanced Master Fund, Ltd. is 950 Third Avenue, New York, NY
10022. |
| |
|
(6) |
|
Based on Schedule 13G filed on February 10, 2011 on
behalf of Columbia Wanger Asset Management, LLC. |
| |
|
(7) |
|
Based on Schedule 13G/A filed February 10, 2011 on
behalf of the following persons: A.M. Pappas Life Science
Ventures III, LP, a Delaware limited partnership (“Pappas
Ventures III”), PV III CEO Fund, LP, a Delaware limited
partnership (the “CEO Fund”), AMP&A Management
III, LLC, a Delaware limited liability company (“AMP&A
Management”), and Arthur M. Pappas
(“Mr. Pappas”). Reported holdings consist of
(i) 1,505,394 shares of common stock and
201,638 shares of common stock issuable upon exercise of
warrants, all owned of record by Pappas Ventures III and
(ii) 93,572 shares of common stock and
12,536 shares of common stock issuable upon exercise of
warrants, all owned of record by the CEO Fund. Mr. Pappas,
in his role as chairman of the investment committee of
AMP&A Management, the general partner of Pappas
Ventures III and CEO Fund, has voting and investment
authority over these shares. Mr. Pappas disclaims
beneficial ownership of these shares except to the extent of his
pecuniary interest arising therein. The address for both Pappas
Ventures III and CEO Fund is 2520 Meridian Parkway,
Suite 400, Durham, NC 27713. |
| |
|
(8) |
|
Based on Schedule 13G/A filed February 14, 2011 on
behalf of the following persons: HBM BioCapital (EUR) L.P., a
Cayman Islands limited partnership (“BioCapital EUR”),
HBM BioCapital (USD) L.P., a Cayman Islands limited partnership
(“BioCapital USD”) and HBM BioCapital Ltd., a Cayman
Islands limited company (“BioCapital Ltd.”). Reporting
holdings consist of (i) 1,307,840 shares of common
stock and 139,263 shares of common stock issuable upon
exercise of warrants, all owned of record by BioCapital (EUR)
and (ii) 230,793 shares of common stock and
24,575 shares of common stock issuable upon exercise of
warrants, all owned of record by BioCapital (USD), together with
Biocapital (EUR), the HBM BioCapital Funds. The board of
directors of BioCapital Ltd., the general partner of the HBM
BioCapital Funds, has sole voting and dispositive power with
respect to such shares. The board of directors of BioCapital
Ltd. consists of John Arnold, Sophia Harris, Richard Coles,
Dr. Andreas Wicki and John Urquhart, none of whom has
individual voting or investment power with respect to the
shares. The address for the HBM BioCapital Funds is
c/o HBM
BioCapital Ltd., Centennial Towers, 3rd Floor, 2454 West
Bay Road, Grand Cayman, Cayman Islands. |
| |
|
(9) |
|
Includes 698,375 shares of common stock and options to
purchase an additional 434,927 shares of common stock that
are exercisable within 60 days of January 31, 2011,
owned of record by Paul F. Truex. |
| |
|
(10) |
|
Includes (i) 9,637 shares of common stock owned of
recorded by Dina Gonzalez, Mr. Lowe’s spouse,
(ii) 27,645 shares of common stock and
(iii) options to purchase 124,366 shares of common
stock that are exercisable within 60 days of
January 31, 2011, owned of record by Mr. Lowe. On
February 11, 2011, Mr. Lowe transferred his position
as manager of BioVest III for no consideration and
transferred 80,997 shares of common stock owned of record
by BioVest III. The address for BioVest III is 25801
Industrial Blvd., Suite B, Hayward, CA 94545. |
| |
|
(11) |
|
Includes (i) 20,851 shares of common stock,
(ii) 3,894 shares received by Dr. Hislop on
February 11, 2011 in a pro rata distribution to the
partners in Biovest III for no additional consideration,
and (iii) options to purchase an additional
154,931 shares of common stock that are exercisable within
60 days of January 31, 2011, owned of record by
Dr. Hislop. |
| |
|
(12) |
|
Includes 96,928 shares of common stock and options to
purchase an additional 24,033 shares of common stock that
are exercisable within 60 days of January 31, 2011,
all owned of record by the Debra A. Odink Living Trust, for
which Dr. Odink serves as trustee. |
| |
|
(13) |
|
Includes 87,616 shares of common stock held by certain
family members and for which Ms. Kilfoil is the beneficial
owner. |
| |
|
(14) |
|
Includes 112,408 shares of common stock, 9,979 shares
of which are subject to the Company’s right of repurchase
and options to purchase an additional 10,000 shares of
common stock that are exercisable within 60 days of
January 31, 2011, owned of record by Dr. Henney. |
95
|
|
|
|
(15) |
|
Includes options to purchase 23,332 shares of common stock
that are exercisable within 60 days of January 31,
2011, owned of record by Ms. Bianchi. VantagePoint has sole
voting and investment power with respect to these shares, and
Ms. Bianchi disclaims beneficial ownership thereof except
to the extent of her pecuniary interest in the shares of common
stock issuable upon exercise of the option. |
| |
|
(16) |
|
Includes 20,443 shares of common stock owned of record by
Dr. Healy, 5,111 shares of which are subject to the
Company’s right of repurchase and options to purchase an
additional 8,000 shares of common stock that are
exercisable within 60 days of January 31, 2011, owned
of record by Dr. Healy. |
| |
|
(17) |
|
Includes options to purchase 24,244 shares of common stock
that are exercisable within 60 days of January 31,
2011, owned of record by the Donald J. Santel and Kelly L.
McGinnis Revocable Living Trust. |
| |
|
(18) |
|
Includes options to purchase 14,250 shares of common stock
that are exercisable within 60 days of January 31,
2011, owned of record by Mr. Spiegelman. |
| |
|
(19) |
|
Includes 28,594 shares of common stock and options to
purchase an additional 9,096 shares of common stock that
are exercisable within 60 days of January 31, 2011,
owned of record by Mr. Thompson. |
|
|
|
ITEM 13.
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR
INDEPENDENCE
|
Transactions
with Related Persons, Promoters and Certain Control
Persons
Since January 1, 2010, we have engaged in the following
transactions with our directors, executive officers, holders of
more than 5% of our voting securities, each of whom we refer to
as a Beneficial Owner, or any member of the immediate family of
any of the foregoing persons.
Private
Placements of Securities
2009
Equity Financing and 2010 Amendments
On September 25, 2009, we entered into a stock purchase
agreement, as amended to add an additional purchaser on
November 3, 2009, with certain existing holders of our
preferred stock for the sale of shares of our common stock equal
to $20.5 million divided by the price per share at which
shares of our common stock were sold to the public in our IPO
minus any per-share underwriting discounts, commissions or fees.
We refer to this transaction as the 2009 equity financing.
Pursuant to the terms of the stock purchase agreement, the
investors deposited $20.5 million into an escrow account
for the purchase of the shares. On December 11, 2009, we
entered into a note purchase agreement and amended escrow
agreement with the investors to release $3.4 million of the
$20.5 million held in the escrow account and issued such
investors convertible promissory notes for the released amount,
which notes we refer to as the escrow notes and which are more
fully described below. The balance of the funds, or
$17.1 million, held in the escrow account would be released
simultaneously with the closing of an IPO in which the aggregate
net proceeds to us (after underwriting discounts, commissions
and fees) were at least $50.0 million. On February 24,
2010, we amended the stock purchase agreement and escrow
agreement with such holders to provide that the funds held in
the escrow account would be released simultaneously with the
closing of an IPO in which the aggregate net proceeds to us
(after underwriting discounts, commissions and fees) were at
least $20.0 million. The funds held in the escrow account
were released in connection with the closing of our IPO on
March 4, 2010.
96
The following table summarizes commitments made to participate
in the 2009 equity financing by any of our current directors,
executive officers, Beneficial Owners or any member of the
immediate family of any of the foregoing:
| |
|
|
|
|
|
|
|
|
|
|
|
Aggregate Consideration
|
|
|
Shares Issued
|
|
|
|
|
to be Paid upon Closing
|
|
|
upon Release of
|
|
|
Name
|
|
of the 2009 Equity Financing
|
|
|
Escrow Account(a)
|
|
|
|
|
VantagePoint Venture Partners IV, L.P. and affiliated entities,
or Vantage Point
|
|
$
|
7,586,035
|
(1)
|
|
|
1,152,891
|
|
|
Sofinnova Venture Partners VI, L.P. and affiliated entities, or
Soffinova
|
|
$
|
4,898,784
|
|
|
|
744,496
|
|
|
A.M. Pappas Life Sciences Ventures III, L.P. and affiliated
entities, or Pappas
|
|
$
|
1,279,265
|
(2)
|
|
|
194,416
|
|
|
HBM BioCapital, L.P. and affiliated entities, or HBM BioCapital
|
|
$
|
1,417,958
|
(3)
|
|
|
215,495
|
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL:
|
|
$
|
15,182,042
|
|
|
|
2,307,298
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(a) |
|
Numbers in this column calculated by dividing the
“Aggregate Consideration to be Paid upon Closing of the
2009 Equity Financing” by $6.58 (which equals the price per
share to the public in our initial public offering less the
underwriting discounts, commissions and fees). |
| |
|
(1) |
|
Includes approximately $6,872,948 to be paid by VantagePoint
Ventures IV (Q), L.P., approximately $688,053 to be paid by
VantagePoint Venture Partners IV, L.P. and approximately $25,034
to be paid by VantagePoint Venture Partners IV Principals
Fund, L.P. |
| |
|
(2) |
|
Includes approximately $1,204,428 to be paid by A.M. Pappas
Life Science Ventures III, L.P. and approximately $74,837 to be
paid by PV III CEO Fund, L.P. |
| |
|
(3) |
|
Includes approximately $1,205,264 to be paid by HBM BioCapital
(EUR) L.P. and approximately $212,694 to be paid by HBM
BioCapital (USD) L.P. |
One additional purchaser, Shionogi & Co., Ltd., who is
not a current director, executive officer, Beneficial Owner or a
member of the immediate family of any of the foregoing, had also
committed $0.5 million to our 2009 equity financing, and
thus received 75,987 shares upon release of the escrow
account.
2009
Escrow Notes and 2010 Amendments
On December 11, 2009, we sold convertible promissory notes,
or the escrow notes, that were secured by a first priority
security interest in all of our assets to purchase shares of our
equity securities to certain of our existing investors for an
aggregate purchase price of $3.4 million. The escrow notes
accrued interest at a rate of 8% per annum and had a maturity
date of the earlier of (i) July 17, 2010 or
(ii) an event of default pursuant to the terms of the
escrow notes. The escrow notes were automatically convertible
into common stock upon the consummation of an IPO in which the
aggregate net proceeds to us (after underwriting discounts,
commissions and fees) were at least $50.0 million, at the
price per share in which shares were sold to the public, minus
any per-share underwriting discounts, commissions or fees.
However, if an IPO was not consummated by January 31, 2010,
the escrow notes became exchangeable for exchange notes in the
same principal amount plus any accrued interest thereon, which
would be automatically convertible into the securities that were
sold in our next equity financing at a 25% discount to the price
in which such securities were sold to other investors, or they
were alternatively convertible into shares of our
Series B-2
convertible preferred stock in connection with a change of
control of the Company. In addition, each exchange note that
would be issued would be accompanied by a warrant, which would
be exercisable for the security into which the accompanying
exchange note, if any, was converted, at the price at which that
security was sold to other investors. Depending on when the
exchange notes are converted, each warrant may be exercisable
for a number of shares equal to the quotient obtained by
dividing (x) (i) 25% of the principal amount of the
accompanying exchange notes, in the event the conversion
occurred prior to April 1, 2010, or (ii) 50% of the
principal amount of the accompanying exchange notes, in the
event the conversion occurred on or after April 1, 2010, by
(y) the purchase price of the securities into which the
exchange note was ultimately converted. Furthermore, if a sale
of all or substantially all of our equity interests or assets
occurred prior to our next equity financing and any exchange
note had not converted, we were obligated to pay such exchange
note holder an amount equal to the accrued interest and
97
two times the outstanding principal amount on such note in
conjunction with the closing of such sale. On February 24,
2010, the note holders waived their right to exchange the escrow
notes for exchange notes and warrants unless our IPO was not
consummated by March 31, 2010. In addition, on
February 24, 2010, we amended the note purchase agreement
relating to the escrow notes to provide that the escrow notes
were automatically convertible into common stock upon the
consummation of an initial public offering in which the
aggregate net proceeds to us (after underwriting discounts,
commissions and fees) were at least $20.0 million. The
escrow notes automatically converted into common stock upon the
closing of our IPO on March 4, 2010, and thus no principal
or interest payments were ever made on the notes and no amounts
remain due under such notes. Moreover, because the escrow notes
were not exchanged, no warrants were ever issued in connection
with such notes.
The following table summarizes the participation in the 2009
escrow notes by any of our current directors, executive
officers, Beneficial Owners or any member of the immediate
family of any of the foregoing persons:
| |
|
|
|
|
|
|
|
|
|
|
|
Aggregate
|
|
|
Shares Issued upon
|
|
|
|
|
Consideration
|
|
|
Conversion
|
|
|
Name
|
|
Paid
|
|
|
of Escrow Notes(a)
|
|
|
|
|
VantagePoint
|
|
$
|
1,553,766
|
(1)
|
|
|
240,222
|
|
|
Sofinnova
|
|
$
|
1,003,366
|
|
|
|
155,127
|
|
|
Pappas
|
|
$
|
262,018
|
(2)
|
|
|
40,509
|
|
|
HBM BioCapital
|
|
$
|
290,425
|
(3)
|
|
|
44,901
|
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL:
|
|
$
|
3,109,575
|
|
|
|
480,759
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(a) |
|
Calculated by dividing (x) the sum of
(i) “Aggregate Consideration to be Paid” and
(ii) accrued interest by (y) $6.58 (which equals the
price per share to the public in our initial public offering
less the underwriting discounts, commissions and fees). |
| |
|
(1) |
|
Consists of (i) a convertible promissory note with a
principal amount of $1,407,712 purchased by VantagePoint Venture
Partners IV (Q), L.P., (ii) a convertible promissory
note with a principal amount of $140,927 purchased by
VantagePoint Venture Partners IV, L.P. and (iii) a
convertible promissory note with a principal amount of $5,127
purchased by VantagePoint Venture Partners IV Principals
Fund, L.P. |
| |
|
(2) |
|
Consists of (i) a convertible promissory note with a
principal amount of $246,690 purchased by A.M. Pappas Life
Science Ventures III, L.P. and (ii) a convertible
promissory note with a principal amount of $15,328 purchased by
PV III CEO Fund, L.P. |
| |
|
(3) |
|
Consists of (i) a convertible promissory note with a
principal amount of $246,861 purchased by HBM BioCapital (EUR)
L.P. and (ii) a convertible promissory note with a
principal amount of $43,564 purchased by HBM BioCapital (USD)
L.P. |
September
2010 Private Placement
On September 20, 2010, we entered into a securities
purchase agreement with certain accredited investors pursuant to
which we sold, on September 24, 2010, an aggregate of
10,500,000 units, with each unit consisting of one share of
our common stock and a Warrant to purchase 0.40 shares of
our common stock. The purchase price per unit was $3.00. Piper
Jaffray & Co. served as our lead placement agent and
Wedbush PacGrow Life Sciences served as co-placement agent in
the private placement. Each Warrant is exercisable in whole or
in party at any time until September 24, 2015 at a per
share exercise price of $3.30, subject to certain adjustments as
specified in the Warrant.
98
The following table summarizes the participation in the private
placement by any of our current directors, executive officers,
Beneficial Owners or any member of the immediate family of any
of the foregoing persons:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares of Common
|
|
|
|
|
Aggregate
|
|
|
Shares of Common
|
|
|
Stock Underlying
|
|
|
Name
|
|
Consideration Paid
|
|
|
Stock Issued
|
|
|
Outstanding Warrants
|
|
|
|
|
HBM BioCapital(1)
|
|
$
|
999,999
|
|
|
|
333,333
|
|
|
|
133,333
|
|
|
Pappas(2)
|
|
$
|
1,400,001
|
|
|
|
466,667
|
|
|
|
186,667
|
|
|
Caduceus Private Investments IV, LP
|
|
$
|
10,000,002
|
|
|
|
3,333,334
|
|
|
|
1,333,334
|
|
|
Visium Balanced Master Fund, Ltd.
|
|
$
|
3,750,000
|
|
|
|
1,250,000
|
|
|
|
500,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
16,150,002
|
|
|
|
5,383,334
|
|
|
|
2,153,334
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes 283,333 shares of common stock and a Warrant to
purchase 113,333 shares of common stock issued to HBM
BioCapital (EUR), L.P. and 50,000 shares of common stock
and a warrant to purchase 20,000 shares of common stock
issued to HBM BioCapital (USD), L.P. |
| |
|
(2) |
|
Includes 439,352 shares of common stock and a Warrant to
purchase 175,741 shares of common stock issued to
A.M. Pappas Life Science Ventures III, L.P. and
27,315 shares of common stock and a Warrant to purchase
10,926 shares of common stock issued to PV III CEO Fund,
L.P. |
Other
Related-Party Transaction
The spouse of Georgina Kilfoil, our Senior Vice President,
Product Development and Clinical Operations, is the Chief
Executive Officer of InClin, Inc., or InClin. Ms. Kilfoil
was a consultant for InClin until joining us in March 2010. We
use InClin’s clinical research organization services to
supplement the clinical research organization services we
receive from other providers. For the period from
September 9, 2004 (Date of Inception) to December 31,
2010, we paid Inclin $665,191 for the clinical research
organization services it provides to us.
Procedures
for Approval of Related Person Transactions
The Audit Committee shall conduct an appropriate review of all
related party transactions for potential conflict of interest
situations on an ongoing basis, and the approval of the Audit
Committee shall be required for all such transactions. The Audit
Committee may establish such policies and procedures as it deems
appropriate to facilitate such review.
Director
Independence
Please refer to Item 10 under the heading “Committees
of the Board of Directors.”
|
|
|
ITEM 14.
|
PRINCIPAL
ACCOUNTANT FEES AND SERVICES
|
Independent
Registered Public Accountants Fee Information
The following is a summary of fees billed by
Deloitte & Touche LLP for fiscal years ended December
2010 and 2009.
| |
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Audit Fees(1)
|
|
$
|
486,575
|
|
|
$
|
362,316
|
|
|
Audit Related Fees(2)
|
|
|
—
|
|
|
|
—
|
|
|
Tax Fees(3)
|
|
$
|
65,000
|
|
|
|
—
|
|
|
All Other Fees(4)
|
|
$
|
2,200
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
553,775
|
|
|
$
|
362,316
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes fees associated with the annual audit of our financial
statements, the reviews of our interim financial statements and
the issuance of consent and comfort letters in connection with
registration statements, including filing our registration
statement on
Form S-1
for our IPO. |
| |
|
(2) |
|
We did not incur any audit related fees. |
99
|
|
|
|
(3) |
|
Includes fees associated with tax advice and tax planning. |
| |
|
(4) |
|
Includes fees associated with our subscription to an online
library of accounting literatures. |
Audit
Committee Pre-Approval Policies
The Audit Committee is directly responsible for the appointment,
retention and termination, and for determining the compensation,
of the Company’s independent registered public accounting
firm. The Audit Committee shall pre-approve all auditing
services and the terms thereof and non-audit services (other
than non-audit services prohibited under Section 10A(g) of
the Exchange Act or the applicable rules of the SEC or the
Public Company Accounting Oversight Board), except that
pre-approval is not required for the provision of non-audit
services if the “de minimus” provisions of
Section 10A(i)(1)(B) of the Exchange Act are satisfied. The
Audit Committee may delegate to one or more designated members
of the Audit Committee the authority to grant pre-approvals for
non-audit services, provided such approvals are presented to the
Audit Committee at a subsequent meeting. All services provided
by Deloitte & Touche LLP during fiscal years 2010 and
2009 were pre-approved by the Audit Committee in accordance with
the pre-approval policy described above.
100
PART IV
| |
|
|
|
|
ITEM 15. EXHIBITS AND
FINANCIAL STATEMENT SCHEDULES
|
|
|
|
|
|
|
|
|
|
|
|
(a) The following documents are filed as part of this
report:
|
|
|
|
|
|
|
|
|
|
|
|
(1) Index list to Financial Statements:
|
|
|
|
|
|
|
|
|
Page
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
102
|
|
|
Audited Financial Statements:
|
|
|
|
|
|
|
|
|
103
|
|
|
|
|
|
104
|
|
|
|
|
|
105
|
|
|
|
|
|
107
|
|
|
|
|
|
108
|
|
|
|
|
| |
(2)
|
Financial Statement Schedules
|
All other schedules are omitted because they are not required or
the required information is included in the financial statements
or notes thereto.
The exhibits listed in the accompanying Exhibit Index are
filed or incorporated by reference as part of this report.
101
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Anthera Pharmaceuticals, Inc.
Hayward, California
We have audited the accompanying balance sheets of Anthera
Pharmaceuticals, Inc. (a development stage company) (the
“Company”) as of December 31, 2010 and 2009, and
the related statements of operations, stockholders’ equity
(deficit) and comprehensive loss, and cash flows for each of the
three years in the period ended December 31, 2010 and for
the period from September 9, 2004 (Date of Inception) to
December 31, 2010. These financial statements are the
responsibility of the Company’s management. Our
responsibility is to express an opinion on the financial
statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. The Company was not required to
have, nor were we engaged to perform, an audit of its internal
control over financial reporting. Our audits included
consideration of internal control over financial reporting as a
basis for designing audit procedures that are appropriate in the
circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such opinion. An
audit also includes examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating
the overall financial statement presentation. We believe that
our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all
material respects, the financial position of the Company as of
December 31, 2010 and 2009, and the results of its
operations and its cash flows for each of the three years in the
period ended December 31, 2010 and for the period from
September 9, 2004 (Date of Inception) to December 31,
2010, in conformity with accounting principles generally
accepted in the United States of America.
The Company is a development stage enterprise engaged in
developing therapeutics to treat diseases associated with
inflammation. As discussed in Note 1 to the financial
statements, successful completion of the Company’s
development programs and the attainment of profitable operations
are dependent upon future events, including obtaining adequate
financing to complete its development activities, obtaining
regulatory approvals, and achieving a level of sales adequate to
support the Company’s cost structure.
/s/ Deloitte & Touche LLP
San Francisco, California
March 7, 2011
102
ANTHERA
PHARMACEUTICALS, INC
(A Development Stage Company)
BALANCE SHEETS
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
ASSETS
|
|
CURRENT ASSETS:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
40,029,972
|
|
|
$
|
3,803,384
|
|
|
Short term investments
|
|
|
23,350,922
|
|
|
|
—
|
|
|
Prepaid expenses and other current assets
|
|
|
1,864,883
|
|
|
|
19,825
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
65,245,777
|
|
|
|
3,823,209
|
|
|
Property and equipment — net
|
|
|
17,285
|
|
|
|
12,994
|
|
|
Deferred financing cost
|
|
|
—
|
|
|
|
1,922,183
|
|
|
Other assets
|
|
|
—
|
|
|
|
130,403
|
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL
|
|
$
|
65,263,062
|
|
|
$
|
5,888,789
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ DEFICIT
|
|
CURRENT LIABILITIES:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
3,791,693
|
|
|
$
|
3,145,706
|
|
|
Accrued clinical study
|
|
|
3,136,786
|
|
|
|
565,034
|
|
|
Accrued liabilities
|
|
|
467,817
|
|
|
|
767,663
|
|
|
Accrued payroll and related costs
|
|
|
609,086
|
|
|
|
153,235
|
|
|
Warrant and derivative liabilities
|
|
|
—
|
|
|
|
406,130
|
|
|
Convertible promissory notes
|
|
|
—
|
|
|
|
13,129,877
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
8,005,382
|
|
|
|
18,167,645
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities
|
|
|
8,005,382
|
|
|
|
18,167,645
|
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and Contingencies (Note 5)
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity (deficit)
|
|
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value, 5,000,000 and
11,948,557 shares authorized, 0 and 8,146,308 shares
issued and outstanding as of December 31, 2010 and 2009,
respectively; (aggregate liquidation value of $0 and $52,597,692
as of December 31, 2010 and 2009, respectively)
|
|
|
—
|
|
|
|
8,146
|
|
|
Common stock, $0.001 par value, 95,000,000 and
18,443,341 shares authorized; 32,853,032 and
1,566,199 shares issued and outstanding as of
December 31, 2010 and 2009, respectively
|
|
|
32,853
|
|
|
|
1,566
|
|
|
Additional paid-in capital
|
|
|
162,919,216
|
|
|
|
52,941,384
|
|
|
Accumulated comprehensive loss
|
|
|
(50,622
|
)
|
|
|
—
|
|
|
Deficit accumulated the during the development stage
|
|
|
(105,643,767
|
)
|
|
|
(65,229,952
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity (deficit)
|
|
|
57,257,680
|
|
|
|
(12,278,856
|
)
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL
|
|
$
|
65,263,062
|
|
|
$
|
5,888,789
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
103
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cumulative
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
OPERATING EXPENSES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
29,456,742
|
|
|
$
|
8,415,414
|
|
|
$
|
10,882,322
|
|
|
$
|
80,780,723
|
|
|
General and administrative
|
|
|
6,300,849
|
|
|
|
3,425,690
|
|
|
|
2,980,170
|
|
|
|
16,218,416
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
35,757,591
|
|
|
|
11,841,104
|
|
|
|
13,862,492
|
|
|
|
96,999,139
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LOSS FROM OPERATIONS
|
|
|
(35,757,591
|
)
|
|
|
(11,841,104
|
)
|
|
|
(13,862,492
|
)
|
|
|
(96,999,139
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OTHER INCOME (EXPENSE):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other expense and interest income, net
|
|
|
(859,733
|
)
|
|
|
(362,388
|
)
|
|
|
(118,174
|
)
|
|
|
(539,593
|
)
|
|
Mark-to-market
adjustment of warrant liability
|
|
|
(3,796,491
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(3,796,491
|
)
|
|
Beneficial conversion features
|
|
|
—
|
|
|
|
—
|
|
|
|
(4,118,544
|
)
|
|
|
(4,308,544
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total other income (expense)
|
|
|
(4,656,224
|
)
|
|
|
(362,388
|
)
|
|
|
(4,236,718
|
)
|
|
|
(8,644,628
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS
|
|
$
|
(40,413,815
|
)
|
|
$
|
(12,203,492
|
)
|
|
$
|
(18,099,210
|
)
|
|
$
|
(105,643,767
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share — basic and diluted
|
|
$
|
(1.76
|
)
|
|
$
|
(8.06
|
)
|
|
$
|
(13.47
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average number of shares used in per share
calculation — basic and diluted
|
|
|
22,909,802
|
|
|
|
1,513,598
|
|
|
|
1,343,420
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
104
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
Accumulated
|
|
Total
|
|
|
|
Convertible
|
|
|
|
|
|
Additional
|
|
Other
|
|
During
|
|
Stockholders’
|
|
|
|
Preferred Stock
|
|
Common Stock
|
|
Paid-In
|
|
Comprehensive
|
|
Development
|
|
Equity
|
|
|
|
Shares
|
|
Amount
|
|
Shares
|
|
Amount
|
|
Capital
|
|
Loss
|
|
Stage
|
|
(Deficit)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DATE OF INCEPTION — September 9, 2004
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock to founders for cash
|
|
|
—
|
|
|
$
|
—
|
|
|
|
140,186
|
|
|
$
|
140
|
|
|
$
|
100
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
240
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock to founders for service
|
|
|
—
|
|
|
|
—
|
|
|
|
735,981
|
|
|
|
736
|
|
|
|
524
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,260
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Repurchase of common stock from founder
|
|
|
—
|
|
|
|
—
|
|
|
|
(73,014
|
)
|
|
|
(73
|
)
|
|
|
(52
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(125
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series A convertible preferred stock for cash
at $1.47 per share, net of issuance cost of $8,555
|
|
|
526,955
|
|
|
|
527
|
|
|
|
—
|
|
|
|
—
|
|
|
|
766,768
|
|
|
|
—
|
|
|
|
—
|
|
|
|
767,295
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series A convertible preferred stock in
exchange for service at $1.47 per share
|
|
|
25,575
|
|
|
|
25
|
|
|
|
—
|
|
|
|
—
|
|
|
|
37,631
|
|
|
|
—
|
|
|
|
—
|
|
|
|
37,656
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
33,292
|
|
|
|
33
|
|
|
|
4,527
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,560
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclass of early exercise of stock options to liability
|
|
|
—
|
|
|
|
—
|
|
|
|
(29,204
|
)
|
|
|
(29
|
)
|
|
|
(3,971
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(4,000
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense related to consultant options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
842
|
|
|
|
—
|
|
|
|
—
|
|
|
|
842
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(554,427
|
)
|
|
|
(554,427
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2005
|
|
|
552,530
|
|
|
|
552
|
|
|
|
807,241
|
|
|
|
807
|
|
|
|
806,369
|
|
|
|
—
|
|
|
|
(554,427
|
)
|
|
|
253,301
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of Series A convertible preferred stock to
Series A-1 convertible preferred stock at a ratio of 1:1
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series A-2 convertible preferred stock for cash
at $5.14 per share — net of issuance cost of $202,019
|
|
|
1,138,677
|
|
|
|
1,139
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,645,093
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,646,232
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series A-2 convertible preferred stock upon
conversion of convertible promissory notes at $3.85 and $5.14
per share
|
|
|
224,248
|
|
|
|
224
|
|
|
|
—
|
|
|
|
—
|
|
|
|
961,527
|
|
|
|
—
|
|
|
|
—
|
|
|
|
961,751
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series A-2 convertible preferred stock in
exchange for licensed technology at $5.14 per share
|
|
|
257,744
|
|
|
|
258
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,323,524
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,323,782
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Beneficial conversion feature related to conversion of
convertible promissory notes into Series A-1 convertible
preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
190,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
190,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series B convertible preferred stock for cash
at $7.28 per share — net of issuance cost of $20,930
|
|
|
2,619,568
|
|
|
|
2,620
|
|
|
|
—
|
|
|
|
—
|
|
|
|
19,036,450
|
|
|
|
—
|
|
|
|
—
|
|
|
|
19,039,070
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of Series B convertible preferred stock in
exchange for licensed technology at $7.28 per share
|
|
|
127,297
|
|
|
|
127
|
|
|
|
—
|
|
|
|
—
|
|
|
|
926,091
|
|
|
|
—
|
|
|
|
—
|
|
|
|
926,218
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
125,581
|
|
|
|
126
|
|
|
|
17,074
|
|
|
|
—
|
|
|
|
—
|
|
|
|
17,200
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclass of early exercise of stock options to liability
|
|
|
—
|
|
|
|
—
|
|
|
|
(36,810
|
)
|
|
|
(37
|
)
|
|
|
(5,006
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(5,043
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense related to consultant options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,358
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,358
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense related to employee options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,648
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,648
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(8,679,246
|
)
|
|
|
(8,679,246
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2006
|
|
|
4,920,064
|
|
|
$
|
4,920
|
|
|
|
896,012
|
|
|
$
|
896
|
|
|
$
|
28,910,128
|
|
|
$
|
—
|
|
|
$
|
(9,233,673
|
)
|
|
$
|
19,682,271
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
493,605
|
|
|
|
494
|
|
|
|
118,426
|
|
|
|
—
|
|
|
|
—
|
|
|
|
118,920
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclass of early exercise of stock options liability
|
|
|
—
|
|
|
|
—
|
|
|
|
(240,165
|
)
|
|
|
(240
|
)
|
|
|
(60,333
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(60,573
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock for service
|
|
|
—
|
|
|
|
—
|
|
|
|
16,355
|
|
|
|
16
|
|
|
|
2,434
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,450
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense related to consultant options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,489
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,489
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense related to employee options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
74,861
|
|
|
|
—
|
|
|
|
—
|
|
|
|
74,861
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Change in other comprehensive loss — unrealized loss
on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,812
|
)
|
|
|
—
|
|
|
|
(1,812
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(25,693,577
|
)
|
|
|
(25,693,577
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(25,695,389
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2007 (carried forward)
|
|
|
4,920,064
|
|
|
|
4,920
|
|
|
|
1,165,807
|
|
|
|
1,166
|
|
|
|
29,058,005
|
|
|
|
(1,812
|
)
|
|
|
(34,927,250
|
)
|
|
|
(5,864,971
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
105
Anthera Pharmaceuticals, Inc.
(A Development Stage Company)
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
AND COMPREHENSIVE LOSS — (Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
Accumulated
|
|
Total
|
|
|
|
Convertible
|
|
|
|
|
|
Additional
|
|
Other
|
|
During
|
|
Stockholders’
|
|
|
|
Preferred Stock
|
|
Common Stock
|
|
Paid-In
|
|
Comprehensive
|
|
Development
|
|
Equity
|
|
|
|
Shares
|
|
Amount
|
|
Shares
|
|
Amount
|
|
Capital
|
|
Loss
|
|
Stage
|
|
(Deficit)
|
|
|
|
BALANCE — December 31, 2007 (brought forward)
|
|
|
4,920,064
|
|
|
$
|
4,920
|
|
|
|
1,165,807
|
|
|
$
|
1,166
|
|
|
$
|
29,058,005
|
|
|
$
|
(1,812
|
)
|
|
$
|
(34,927,250
|
)
|
|
$
|
(5,864,971
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of Series B convertible preferred stock to
Series B-1
convertible preferred stock at a ratio of 1:1
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of
Series B-2
convertible preferred stock for cash at $7.28 per share
— net of issuance cost of $242,327 and warrants
issuance (below)
|
|
|
962,066
|
|
|
|
962
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,512,241
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,513,203
|
|
|
Issuance of
Series B-2
convertible preferred stock upon conversion of convertible
promissory notes at $5.46 per share
|
|
|
2,235,661
|
|
|
|
2,235
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,197,765
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,200,000
|
|
|
Issuance of
Series B-2
convertible preferred stock in lieu of interest payment at $5.46
per share
|
|
|
28,517
|
|
|
|
29
|
|
|
|
—
|
|
|
|
—
|
|
|
|
155,601
|
|
|
|
—
|
|
|
|
—
|
|
|
|
155,630
|
|
|
Issuance of warrants in connection with issuance of
Series B-2
convertible preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
244,478
|
|
|
|
—
|
|
|
|
—
|
|
|
|
244,478
|
|
|
Beneficial conversion feature related to conversion of
convertible promissory notes into
Series B-2
convertible preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,118,544
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,118,544
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
179,886
|
|
|
|
180
|
|
|
|
67,925
|
|
|
|
—
|
|
|
|
—
|
|
|
|
68,105
|
|
|
Release of early exercise of stock options liability
|
|
|
—
|
|
|
|
—
|
|
|
|
128,180
|
|
|
|
128
|
|
|
|
12,773
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,901
|
|
|
Repurchase of common stock upon employee termination
|
|
|
—
|
|
|
|
—
|
|
|
|
(18,983
|
)
|
|
|
(19
|
)
|
|
|
(4,856
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(4,875
|
)
|
|
Stock-based compensation expense related to consultant options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
51,874
|
|
|
|
—
|
|
|
|
—
|
|
|
|
51,874
|
|
|
Stock-based compensation expense related to employee options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
143,406
|
|
|
|
—
|
|
|
|
—
|
|
|
|
143,406
|
|
|
Change in other comprehensive loss — unrealized gain
on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
652
|
|
|
|
—
|
|
|
|
652
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(18,099,210
|
)
|
|
|
(18,099,210
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(18,098,558
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2008
|
|
|
8,146,308
|
|
|
|
8,146
|
|
|
|
1,454,890
|
|
|
|
1,455
|
|
|
|
52,557,756
|
|
|
|
(1,160
|
)
|
|
|
(53,026,460
|
)
|
|
|
(460,263
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
19,089
|
|
|
|
19
|
|
|
|
15,255
|
|
|
|
—
|
|
|
|
—
|
|
|
|
15,274
|
|
|
Release of early exercise of stock options liability
|
|
|
—
|
|
|
|
—
|
|
|
|
92,220
|
|
|
|
92
|
|
|
|
26,027
|
|
|
|
—
|
|
|
|
—
|
|
|
|
26,119
|
|
|
Stock-based compensation expense related to consultant options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
88,382
|
|
|
|
—
|
|
|
|
—
|
|
|
|
88,382
|
|
|
Stock-based compensation expense related to employee options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
253,964
|
|
|
|
—
|
|
|
|
—
|
|
|
|
253,964
|
|
|
Change in other comprehensive loss — unrealized gain
on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,160
|
|
|
|
—
|
|
|
|
1,160
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(12,203,492
|
)
|
|
|
(12,203,492
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(12,202,332
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance — December 31, 2009
|
|
|
8,146,308
|
|
|
$
|
8,146
|
|
|
|
1,566,199
|
|
|
$
|
1,566
|
|
|
$
|
52,941,384
|
|
|
$
|
—
|
|
|
$
|
(65,229,952
|
)
|
|
$
|
(12,278,856
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of convertible preferred stock to common stock at a
ratio of 1:1
|
|
|
(8,146,308
|
)
|
|
|
(8,146
|
)
|
|
|
8,146,308
|
|
|
|
8,146
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock for cash at $7.00 per share
— net of issuance cost of $3,039,257
|
|
|
—
|
|
|
|
—
|
|
|
|
6,000,000
|
|
|
|
6,000
|
|
|
|
37,075,034
|
|
|
|
—
|
|
|
|
—
|
|
|
|
37,081,034
|
|
|
Issuance of common stock upon conversion of convertible
promissory notes and accrued interest at $5.25 and $6.28 per
share
|
|
|
—
|
|
|
|
—
|
|
|
|
2,511,235
|
|
|
|
2,511
|
|
|
|
13,880,601
|
|
|
|
—
|
|
|
|
—
|
|
|
|
13,883,112
|
|
|
Issuance of common stock upon release of escrow funds
|
|
|
—
|
|
|
|
—
|
|
|
|
2,598,780
|
|
|
|
2,599
|
|
|
|
17,097,373
|
|
|
|
—
|
|
|
|
—
|
|
|
|
17,099,972
|
|
|
Issuance of common stock upon cashless exercise of warrants
|
|
|
—
|
|
|
|
—
|
|
|
|
194,474
|
|
|
|
194
|
|
|
|
218
|
|
|
|
—
|
|
|
|
—
|
|
|
|
412
|
|
|
Issuance of common stock to collaborator in lieu of milestone
payment
|
|
|
—
|
|
|
|
—
|
|
|
|
531,914
|
|
|
|
532
|
|
|
|
3,499,468
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,500,000
|
|
|
Issuance of common stock upon exercise of overallotment by
underwriters net of issuance cost of $17,291
|
|
|
—
|
|
|
|
—
|
|
|
|
604,492
|
|
|
|
605
|
|
|
|
3,959,661
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,960,266
|
|
|
Issuance of common stock upon exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
138,878
|
|
|
|
139
|
|
|
|
115,847
|
|
|
|
—
|
|
|
|
—
|
|
|
|
115,986
|
|
|
Issuance of common stock pursuant to employee stock purchase plan
|
|
|
—
|
|
|
|
—
|
|
|
|
24,916
|
|
|
|
25
|
|
|
|
81,201
|
|
|
|
|
|
|
|
|
|
|
|
81,226
|
|
|
Issuance of common stock upon private placement transaction, net
of issuance cost of $508,384
|
|
|
—
|
|
|
|
—
|
|
|
|
10,500,000
|
|
|
|
10,500
|
|
|
|
23,767,172
|
|
|
|
—
|
|
|
|
—
|
|
|
|
23,777,672
|
|
|
Issuance of warrants in conjunction with private placement
transaction
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,323,944
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,323,944
|
|
|
Net change of early exercise of stock options and liability
|
|
|
—
|
|
|
|
—
|
|
|
|
35,836
|
|
|
|
36
|
|
|
|
1,492
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,528
|
|
|
Reclass of warrant and derivative liability to equity in
conjunction with conversion of convertible promissory notes into
common stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,473,491
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,473,491
|
|
|
Stock-based compensation expense related to consultant options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8,399
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8,399
|
|
|
Stock-based compensation expense related to employee options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
693,931
|
|
|
|
—
|
|
|
|
—
|
|
|
|
693,931
|
|
|
Change in other comprehensive loss — unrealized loss
on investments and foreign currency translation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(50,622
|
)
|
|
|
—
|
|
|
|
(50,622
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(40,413,815
|
)
|
|
|
(40,413,815
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(40,464,437
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2010
|
|
|
—
|
|
|
|
—
|
|
|
|
32,853,032
|
|
|
$
|
32,853
|
|
|
$
|
162,919,216
|
|
|
$
|
(50,622
|
)
|
|
$
|
(105,643,767
|
)
|
|
$
|
57,257,680
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
106
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
CASH FLOW FROM OPERATING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(40,413,815
|
)
|
|
$
|
(12,203,492
|
)
|
|
$
|
(18,099,210
|
)
|
|
$
|
(105,643,767
|
)
|
|
Adjustments to reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation
|
|
|
17,281
|
|
|
|
18,451
|
|
|
|
21,997
|
|
|
|
89,608
|
|
|
Amortization of premium/(discount) on short-term investments
|
|
|
102,116
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(28,132
|
)
|
|
Realized loss on short-term investments
|
|
|
—
|
|
|
|
1,160
|
|
|
|
7,522
|
|
|
|
8,682
|
|
|
Realized gain from disposal of property and equipment
|
|
|
—
|
|
|
|
(214
|
)
|
|
|
—
|
|
|
|
(214
|
)
|
|
Stock-based compensation expense — employees
|
|
|
693,931
|
|
|
|
253,964
|
|
|
|
143,406
|
|
|
|
1,170,810
|
|
|
Stock-based compensation expense — consultants
|
|
|
8,399
|
|
|
|
88,382
|
|
|
|
51,874
|
|
|
|
166,344
|
|
|
Issuance of common stock for consulting service
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
41,366
|
|
|
Issuance of preferred stock for service and license fee
|
|
|
3,500,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,750,000
|
|
|
Issuance of preferred stock in lieu of interest payment
|
|
|
173,194
|
|
|
|
—
|
|
|
|
155,630
|
|
|
|
330,575
|
|
|
Beneficial conversion feature
|
|
|
—
|
|
|
|
—
|
|
|
|
4,118,544
|
|
|
|
4,308,544
|
|
|
Amortization of discount on convertible promissory notes
|
|
|
540,993
|
|
|
|
136,722
|
|
|
|
—
|
|
|
|
677,715
|
|
|
Amortization of debt issuance cost
|
|
|
227,955
|
|
|
|
79,644
|
|
|
|
—
|
|
|
|
307,599
|
|
|
Mark-to-market adjustment on warrant liability
|
|
|
3,796,491
|
|
|
|
(715
|
)
|
|
|
—
|
|
|
|
3,795,776
|
|
|
Changes in assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expenses and other assets
|
|
|
(1,845,059
|
)
|
|
|
51,437
|
|
|
|
31,182
|
|
|
|
(1,864,885
|
)
|
|
Accounts payable
|
|
|
2,664,378
|
|
|
|
(212,623
|
)
|
|
|
(2,176,982
|
)
|
|
|
4,049,054
|
|
|
Accrued clinical study
|
|
|
2,571,752
|
|
|
|
(896,145
|
)
|
|
|
65,013
|
|
|
|
3,136,786
|
|
|
Accrued liabilities
|
|
|
(271,223
|
)
|
|
|
473,889
|
|
|
|
135,137
|
|
|
|
460,969
|
|
|
Accrued payroll and related costs
|
|
|
455,851
|
|
|
|
37,190
|
|
|
|
(578,910
|
)
|
|
|
609,086
|
|
|
License fee payable
|
|
|
—
|
|
|
|
(5,000,000
|
)
|
|
|
(1,000,000
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(27,777,756
|
)
|
|
|
(17,172,350
|
)
|
|
|
(17,124,797
|
)
|
|
|
(82,634,084
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
INVESTING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment purchases
|
|
|
(21,572
|
)
|
|
|
(3,852
|
)
|
|
|
(6,752
|
)
|
|
|
(107,079
|
)
|
|
Proceeds from disposal of property and equipment
|
|
|
—
|
|
|
|
400
|
|
|
|
—
|
|
|
|
400
|
|
|
Purchase of short-term investments
|
|
|
(24,948,692
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(39,749,256
|
)
|
|
Proceeds from sale of short-term investments
|
|
|
1,610,000
|
|
|
|
—
|
|
|
|
5,818,132
|
|
|
|
16,532,132
|
|
|
Restricted cash
|
|
|
—
|
|
|
|
40,000
|
|
|
|
30,000
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
(23,360,264
|
)
|
|
|
36,548
|
|
|
|
5,841,380
|
|
|
|
(23,323,803
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FINANCING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of convertible notes
|
|
|
—
|
|
|
|
13,400,000
|
|
|
|
12,200,000
|
|
|
|
26,560,000
|
|
|
Payment of debt issuance cost
|
|
|
(210,282
|
)
|
|
|
(97,317
|
)
|
|
|
—
|
|
|
|
(307,599
|
)
|
|
Net proceeds from issuance of preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
6,757,681
|
|
|
|
32,210,278
|
|
|
Payment of financing cost for initial public and private offering
|
|
|
(3,246,255
|
)
|
|
|
(273,884
|
)
|
|
|
—
|
|
|
|
(3,520,139
|
)
|
|
Proceeds from issuance of common stock
|
|
|
90,788,900
|
|
|
|
—
|
|
|
|
—
|
|
|
|
90,789,015
|
|
|
Proceeds from issuance of common stock pursuant to employee
stock purchase plan
|
|
|
81,226
|
|
|
|
—
|
|
|
|
—
|
|
|
|
81,226
|
|
|
Proceeds from exercise of stock options
|
|
|
115,985
|
|
|
|
15,274
|
|
|
|
68,105
|
|
|
|
340,044
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
87,529,574
|
|
|
|
13,044,073
|
|
|
|
19,025,786
|
|
|
|
146,152,825
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effect of exchange rate changes on cash
|
|
|
(164,966
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(164,966
|
)
|
|
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS
|
|
|
36,226,588
|
|
|
|
(4,091,729
|
)
|
|
|
7,742,369
|
|
|
|
40,029,972
|
|
|
CASH AND CASH EQUIVALENTS — Beginning of period
|
|
|
3,803,384
|
|
|
|
7,895,113
|
|
|
|
152,744
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH AND CASH EQUIVALENTS — End of period
|
|
$
|
40,029,972
|
|
|
$
|
3,803,384
|
|
|
$
|
7,895,113
|
|
|
$
|
40,029,972
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SUPPLEMENTAL CASH DISCLOSURES OF CASH FLOW INFORMATION:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest paid
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
1,413
|
|
|
$
|
15,229
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Taxes paid
|
|
$
|
23,159
|
|
|
$
|
4,900
|
|
|
$
|
4,379
|
|
|
$
|
52,746
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NONCASH INVESTING AND FINANCING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of convertible promissory notes and accrued interest
into common stock,
Series A-2
convertible preferred stock and
Series B-2
convertible preferred stock
|
|
$
|
13,883,112
|
|
|
$
|
—
|
|
|
$
|
12,355,630
|
|
|
$
|
27,200,493
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Beneficial conversion feature
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
4,118,544
|
|
|
$
|
4,308,544
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unamortized debt discount charged to equity in conjunction with
conversion of promissory notes into common stock
|
|
$
|
185,883
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
185,883
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclassification of warrant and derivative liabilities to
additional paid-in capital
|
|
$
|
406,130
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
406,130
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance costs charged to equity
|
|
$
|
3,564,932
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
3,564,932
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accrued and deferred financing and debt issuance costs
|
|
$
|
27,503
|
|
|
$
|
1,761,029
|
|
|
$
|
—
|
|
|
$
|
27,503
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
107
|
|
|
1.
|
ORGANIZATION
AND DESCRIPTION OF BUSINESS
|
Anthera Pharmaceuticals, Inc. (the “Company” or
“Anthera”) was incorporated on September 9, 2004
in the state of Delaware. During 2006, the Company opened its
headquarters in San Mateo, California, and subsequently
moved to Hayward, California. Anthera is a biopharmaceutical
company focused on developing and commercializing therapeutics
to treat serious diseases associated with inflammation,
including cardiovascular and autoimmune diseases. Two of the
Company’s primary product candidates, varespladib and
A-001, are
inhibitors of the family of human enzymes known as secretory
phospholipase
A2,
or
sPLA2.
The Company’s other primary product candidate,
A-623,
targets elevated levels of B-cell activating factor, or BAFF.
The Company’s activities since inception have consisted
principally of acquiring product and technology rights, raising
capital, and performing research and development. Accordingly,
the Company is considered to be in the development stage as of
December 31, 2010, as defined by the Financial Accounting
Standard Board (“FASB”) Accounting Standard
Codification (“ASC”) 915. Successful completion of the
Company’s development programs and, ultimately, the
attainment of profitable operations are dependent on future
events, including, among other things, its ability to access
potential markets; secure financing; develop a customer base;
attract, retain and motivate qualified personnel; and develop
strategic alliances. Although management believes that the
Company will be able to successfully fund its operations, there
can be no assurance that the Company will be able to do so or
that the Company will ever operate profitably.
The accompanying financial statements have been prepared in
conformity with accounting principles generally accepted in the
United States, or GAAP. From September 9, 2004 (Date of
Inception) through December 31, 2010, the Company had an
accumulated a deficit of $105.6 million. During the year
ended December 31, 2010, the Company incurred a net loss of
$40.4 million and had negative cash flows from operations
of $27.8 million. The Company expects to continue to incur
substantial losses and negative cash flows over the next several
years during its clinical development phase. To fully execute
its business plan, the Company will need to complete certain
research and development activities and clinical studies.
Further, the Company’s product candidates will require
regulatory approval prior to commercialization. These activities
may span many years and require substantial expenditures to
complete and may ultimately be unsuccessful. Any delays in
completing these activities could adversely impact the Company.
The Company plans to meet its capital requirements primarily
through issuances of equity securities, debt financing, and in
the longer term, revenue from product sales. Failure to generate
revenue or raise additional capital would adversely affect the
Company’s ability to achieve its intended business
objectives.
On February 26, 2010, the Company’s Registration
Statement on
Form S-1
was declared effective for its initial public offering
(“IPO”), pursuant to which the Company sold
6,000,000 shares of its common stock at a public offering
price of $7.00 per share. The Company received net proceeds of
approximately $37.1 million from this transaction.
Concurrent with the closing of the IPO, the Company received an
aggregate of $17.1 million from the issuance of
2,598,780 shares of its common stock to certain of its
investors pursuant to a common stock purchase agreement.
On April 6, 2010, the Company sold 604,492 shares of
common stock pursuant to the exercise of the underwriters’
over-allotment option in connection with the Company’s IPO
and received net proceeds of approximately $4.0 million.
On September 24, 2010, the Company completed a private
placement transaction with certain accredited investors pursuant
to which the Company sold an aggregate of 10,500,000 units
at a purchase price of $3.00 per unit, with each unit consisting
of one share of common stock and a warrant to purchase an
additional 0.40 shares of common stock. Each warrant is
exercisable in whole or in part at any time until
September 24, 2015 at a per share exercise price of $3.30,
subject to certain adjustments as specified in the warrant. The
Company received net proceeds of approximately
$29.1 million.
108
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
|
|
|
2.
|
SUMMARY
OF SIGNIFICANT ACCOUNTING POLICIES
|
Use of
Estimates
The preparation of these financial statements in conformity with
U.S. GAAP requires us to make estimates and judgments that
affect the reported amounts of assets, liabilities, expenses,
and related disclosures. On an ongoing basis, management
evaluates its estimates, including critical accounting policies
or estimates related to clinical trial accruals, our tax
provision and stock-based compensation. The Company bases its
estimates on historical experience and on various other market
specific and other relevant assumptions that it believes to be
reasonable under the circumstances, the results of which form
the basis for making judgments about the carrying values of
assets and liabilities that are not readily apparent from other
sources. Actual results may differ significantly from these
estimates.
Cash
and Cash Equivalents
The Company considers all highly liquid instruments purchased
with an original maturity or remaining maturities of three
months or less at the date of purchase to be cash equivalents.
Cash equivalents consist primarily of money market funds, for
which the carrying amounts are reasonable estimates of fair
value. Cash equivalents are recognized at fair value.
Short-Term
Investments
The Company has designated its investments as available for sale
and the investment are carried at fair value. The Company
determines the appropriate classification of securities at the
time of purchase and reevaluates such classification as of each
balance sheet date. Securities with maturity exceeding three
months but less than one year are classified as short-term
investments. Realized gains and losses and declines in value
judged to be other-than-temporary are determined based on
specific identification method and are reported in the
statements of operations. The Company includes any unrealized
gains and losses on short-term investments in stockholders’
equity as a component of other comprehensive income (loss).
Concentration
of Credit Risk
Financial instruments that potentially subject the Company to
concentrations of credit risk consist primarily of cash, cash
equivalents and short-term investments. The Company’s cash
equivalents consist of certificates of deposit with maturities
less than three months and treasury money market funds. The
Company’s short-term investments consist of certificates of
deposit and corporate bonds with maturities exceeding three
months but less than one year. The Company has not experienced
any losses in such accounts. The Company believes it is not
exposed to significant credit risk related to cash, cash
equivalents and short-term investments.
Property
and Equipment — Net
Property and equipment are stated at cost, less accumulated
depreciation. Depreciation is computed over the estimated useful
lives of the respective assets, which range from three to five
years, using the straight-line method. Repairs and maintenance
costs are expensed as incurred. Leasehold improvements are
stated at cost and amortized using the straight-line method over
the term of the lease or the life of the related asset,
whichever is shorter.
Deferred
Financing Cost
Deferred financing costs included costs directly attributable to
the Company’s offering of its equity securities. In
accordance with FASB
ASC 340-10,
Other Assets and Deferred Costs, these costs are deferred
and capitalized as part of other assets. Costs attributable to
the equity offerings were charged against the proceeds of the
Company’s IPO.
109
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
Long-Lived
Assets
The Company’s long-lived assets and other assets are
reviewed for impairment in accordance with the guidance of the
FASB
ASC 360-10,
Property, Plant, and Equipment, whenever events or
changes in circumstances indicate that the carrying amount of
the asset may not be recoverable. Recoverability of an asset to
be held and used is measured by a comparison of the carrying
amount of an asset to the future undiscounted cash flows
expected to be generated by the asset. If such asset is
considered to be impaired, the impairment to be recognized is
measured by the amount by which the carrying amount of the asset
exceeds its fair value. Through December 31, 2010, the
Company had not experienced impairment losses on its long-lived
assets.
Fair
Value of Financial Instruments
Fair value is defined as the price that would be received to
sell an asset or paid to transfer a liability in an orderly
transaction between market participants at the measurement date.
Valuation techniques used to measure fair value, as required by
FASB
ASC 820-10,
Fair Value Measurements and Disclosures, must maximize
the use of observable inputs and minimize the use of
unobservable inputs.
The standard describes a fair value hierarchy based on three
levels of inputs, of which the first two are considered
observable and the last unobservable, that may be used to
measure fair value. The Company’s assessment of the
significance of a particular input to the fair value
measurements requires judgment, and may affect the valuation of
the assets and liabilities being measured and their placement
within the fair value hierarchy. The three levels of input are:
Level 1 — Quoted prices in active markets
for identical assets or liabilities.
Level 2 — Inputs other than Level 1
that are observable, either directly or indirectly, such as
quoted prices for similar assets or liabilities; quoted prices
in markets that are not active; or other inputs that are
observable or can be corroborated by observable market data for
substantially the full term of the assets or liabilities.
Level 3 — Unobservable inputs that are
supported by little or no market activity and that are
significant to the fair value of the assets or liabilities.
The following is a description of the Company’s valuation
methodologies for assets and liabilities measured at fair value.
Where quoted prices are available in an active market, fair
value is based upon quoted market prices, and are classified in
Level 1 of the valuation hierarchy. If quoted market prices
are not available, fair value is based upon observable inputs
such as quoted prices for similar assets or liabilities, quoted
prices in markets that are not active, or other inputs that are
observable or can be corroborated by observable market data, the
assets or liabilities are classified in Level 2 of the
valuation hierarchy. When quoted prices and observable inputs
are unavailable, fair values are based on internally developed
cash flow models and are classified in Level 3 of the
valuation hierarchy. The internally developed cash flow models
primarily use, as inputs, estimates for interest rates and
discount rates including yields of comparable traded instruments
adjusted for illiquidity and other risk factors, amount of cash
flows and expected holding periods of the assets. These inputs
reflect the Company’s own assumptions about the assumptions
market participants would use in pricing the assets including
assumptions about risk developed based on the best information
available in the circumstances.
Other financial instruments, including accounts payable and
accrued liabilities, are carried at cost, which the Company
believes approximates fair value because of the short-term
maturity of these instruments.
Research
and Development Costs
Research and development expenses consist of personnel costs,
including salaries, benefits and stock-based compensation,
clinical studies performed by contract research organizations,
or CROs, materials and supplies, licenses and fees, and overhead
allocations consisting of various administrative and facilities
related costs. Research and development activities are also
separated into three main categories: research, clinical
development, and
110
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
pharmaceutical development. Research costs typically consist of
preclinical and toxicology costs. Clinical development costs
include costs for Phase 1, 2 and 3 clinical studies.
Pharmaceutical development costs consist of expenses incurred in
connection with product formulation and chemical analysis.
The Company charges research and development costs, including
clinical study costs, to expense when incurred, consistent with
the guidance of FASB ASC 730, Research and
Development. Clinical study costs are a significant
component of research and development expenses. All of the
Company’s clinical studies are performed by third-party
CROs. The Company accrues costs for clinical studies performed
by CROs on a straight-line basis over the service periods
specified in the contracts and adjusts the estimates, if
required, based upon the Company’s ongoing review of the
level of effort and costs actually incurred by the CROs. The
Company monitors levels of performance under each significant
contract, including the extent of patient enrollment and other
activities through communications with the CROs, and adjusts the
estimates, if required, on a monthly basis so that clinical
expenses reflect the actual effort expended by each CRO.
All material CRO contracts are terminable by the Company upon
written notice and the Company is generally only liable for
actual effort expended by the CROs and certain noncancelable
expenses incurred at any point of termination.
Amounts paid in advance related to incomplete services will be
refunded if a contract is terminated. Some contracts include
additional termination payments that become due and payable if
the Company terminates the contract. Such additional termination
payments are only recorded if a contract is terminated.
In the fourth quarter of 2010, the Company was notified by the
Internal Revenue Service that its applications for projects
under the qualifying therapeutic discovery project tax credit
under Section 48D of the Internal Revenue Code (“48D
Program”) were approved. The 48D Program provided a total
of approximately $977,000 of grant monies for certain research
and development expenditures the Company incurred during 2009 or
2010. Under International Accounting Standard 20,
Accounting for Government Grants and Disclosure of Government
Assistance, there are two alternatives for accounting for
grants of this nature. They can be recorded as either
i) other income, in a separate line item under operating
income, or ii) a reduction in the related expense account
(research and development expense or other). The Company elected
to record the grants as an offset to the related research and
development expense for the year ended December 31, 2010.
Comprehensive
Income (Loss)
Comprehensive income (loss) consists of certain changes in
equity that are excluded from net income (loss). Specifically,
the Company includes unrealized gains (losses) on available for
sale securities and the effect of exchange rate changes on cash
equivalents and short-term investments in other comprehensive
income (loss). Comprehensive income (loss) for each period
presented is set forth in the Statement of Stockholders’
Equity (Deficit) and Comprehensive Loss.
Income
Taxes
The Company accounts for income taxes in accordance with FASB
ASC 740, Income Taxes. FASB ASC 740 prescribes
the use of the liability method whereby deferred tax asset and
liability account balances are determined based on differences
between the financial reporting and tax bases of assets and
liabilities and are measured using the enacted tax rates and
laws that will be in effect when the differences are expected to
reverse. The Company provides a valuation allowance, if
necessary, to reduce deferred tax assets to their estimated
realizable value.
Segments
The Company operates in only one segment. Management uses cash
flow as the primary measure to manage its business and does not
segment its business for internal reporting or decision-making.
111
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
Net
Loss Per Share
The Company computes net loss per share in accordance with FASB
ASC 260, Earnings Per Share, under which basic net
loss attributable to common stockholders per share is computed
by dividing income available to common stockholders (the
numerator) by the weighted-average number of common shares
outstanding (the denominator) during the period. Shares issued
during the period and shares reacquired during the period are
weighted for the portion of the period that they were
outstanding. The computation of diluted EPS is similar to the
computation of basic EPS except that the denominator is
increased to include the number of additional common shares that
would have been outstanding if the dilutive potential common
shares had been issued. In addition, in computing the dilutive
effect of convertible securities, the numerator is adjusted to
add back any convertible preferred dividends and the after-tax
amount of interest recognized in the period associated with any
convertible debt. The numerator also is adjusted for any other
changes in income or loss that would result from the assumed
conversion of those potential common shares, such as
profit-sharing expenses. Diluted EPS is identical to basic EPS
since common equivalent shares are excluded from the
calculation, as their effect is anti-dilutive.
The following table summarizes the Company’s calculation of
net loss per common share:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Net loss per share
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Numerator
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(40,413,815
|
)
|
|
$
|
(12,203,492
|
)
|
|
$
|
(18,099,210
|
)
|
|
Denominator
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average common shares outstanding
|
|
|
22,957,456
|
|
|
|
1,623,677
|
|
|
|
1,573,448
|
|
|
Less: Weighted-average shares subject to repurchase
|
|
|
(47,654
|
)
|
|
|
(110,079
|
)
|
|
|
(230,028
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Denominator for basic and diluted net loss per share
|
|
|
22,909,802
|
|
|
|
1,513,598
|
|
|
|
1,343,420
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share
|
|
$
|
(1.76
|
)
|
|
$
|
(8.06
|
)
|
|
$
|
(13.47
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following table shows weighted-average historical dilutive
common share equivalents outstanding, which are not included in
the above calculation, as the effect of their inclusion is
anti-dilutive during each period.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Options to purchase common stock
|
|
|
978,231
|
|
|
|
932,544
|
|
|
|
539,234
|
|
|
Common stock subject to repurchase
|
|
|
47,654
|
|
|
|
110,079
|
|
|
|
230,028
|
|
|
Warrants to purchase common stock
|
|
|
1,496,314
|
(2)
|
|
|
240,516
|
(1)
|
|
|
94,230
|
(1)
|
|
Convertible preferred stock (on an as-if-converted basis)
|
|
|
—
|
|
|
|
8,146,308
|
|
|
|
6,184,045
|
|
|
Restricted stock units
|
|
|
153,658
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,675,857
|
|
|
|
9,429,447
|
|
|
|
7,047,537
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
The warrants were exercised upon the closing of the
Company’s IPO in March 2010. |
| |
|
(2) |
|
Consists of 357,136 warrants which carry a contractual term of
five years and terminate upon the earlier of i) five years
from the date of issuance which will be July 17 or
September 9, 2014 and ii) upon certain corporate
transactions and 1,139,178 warrants which carry a contractual
term of five years expiring September 24, 2015. Each of the
warrants contains a customary net issuance feature, which allows
the warrant holder to pay the |
112
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
|
|
|
|
|
|
exercise price of the warrant by forfeiting a portion of the
executed warrant shares with a value equal to the aggregate
exercise price. |
Stock-Based
Compensation
Effective January 1, 2006, the Company adopted the
provisions of FASB ASC 718, Compensation —
Stock Compensation, using the modified prospective method.
Compensation costs related to all equity instruments granted
after January 1, 2006 are recognized at the grant-date fair
value of the awards. Additionally, the Company is required to
include an estimate of the number of awards that will be
forfeited in calculating compensation costs, which are
recognized over the requisite service period of the awards on a
straight-line basis.
The Company uses the Black-Scholes option pricing model as the
method for determining the estimated fair value for all
stock-based awards, including employee stock options, and rights
to purchase shares under our Employee Stock Purchase Plan based
on their estimated fair value, and recognize the costs in our
financial statements over the employees’ requisite service
period. The Black-Scholes option pricing model requires the use
of highly subjective and complex assumptions which determine the
fair value of share-based awards, including the option’s
expected term and the price volatility of the underlying stock.
Expected Term — The Company’s expected
term represents the period that the Company’s stock-based
awards are expected to be outstanding and is determined using
the simplified method.
Expected Volatility — Expected volatility is
estimated using comparable public company volatility for similar
terms.
Expected Dividend — The Black-Scholes option
pricing model calls for a single expected dividend yield as an
input and the Company has never paid dividends and has no plans
to pay dividends.
Risk-Free Interest Rate — The risk-free
interest rate used in the Black-Scholes option pricing method is
based on the U.S. Treasury zero-coupon issues in effect at
the time of grant for periods corresponding with the expected
term of option.
Estimated Forfeitures — The estimated
forfeiture rate is determined based on the Company’s
historical forfeiture rates to date. The Company monitors actual
expenses and periodically updates the estimate.
Equity instruments issued to nonemployees are recorded at their
fair value as determined in accordance with FASB
ASC 505-50,
Equity, and are periodically revalued as the equity
instruments vest and recognized as expense over the related
service period.
|
|
|
3.
|
DEFERRED
FINANCING COST
|
At December 31, 2009, the Company capitalized and deferred
$1,922,183 of financing cost attributable to the Company’s
IPO, which were charged against the proceeds upon the closing of
the Company’s IPO in March 2010.
|
|
|
4.
|
PREPAID
EXPENSES AND OTHER CURRENT ASSETS
|
Prepaid expenses and other current assets are comprised of the
following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Prepaid insurance
|
|
$
|
405,385
|
|
|
$
|
4,331
|
|
|
Grant receivable
|
|
|
977,917
|
|
|
|
—
|
|
|
Interest receivable
|
|
|
446,949
|
|
|
|
11
|
|
|
Other current assets
|
|
|
34,632
|
|
|
|
15,483
|
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expense and other current assets
|
|
$
|
1,864,883
|
|
|
$
|
19,825
|
|
|
|
|
|
|
|
|
|
|
|
113
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
|
|
|
5.
|
PROPERTY
AND EQUIPMENT
|
Property and equipment are comprised of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Computers and software
|
|
$
|
77,318
|
|
|
$
|
66,548
|
|
|
Office equipment and furniture
|
|
|
16,730
|
|
|
|
16,730
|
|
|
Leasehold improvements
|
|
|
10,802
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total property and equipment
|
|
|
104,850
|
|
|
|
83,278
|
|
|
Less accumulated depreciation
|
|
|
(87,565
|
)
|
|
|
(70,284
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment, net
|
|
$
|
17,285
|
|
|
$
|
12,994
|
|
|
|
|
|
|
|
|
|
|
|
The Company recorded the following depreciation expense in the
respective periods:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
Years Ended December 31,
|
|
December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
2010
|
|
|
|
Depreciation expense
|
|
$
|
17,281
|
|
|
$
|
18,451
|
|
|
$
|
21,997
|
|
|
$
|
89,608
|
|
|
|
|
6.
|
CASH
EQUIVALENTS AND INVESTMENTS
|
The Company’s cash equivalents and short-term investments
as of December 31, 2010 are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
|
Unrealized
|
|
|
|
|
|
|
|
Cost
|
|
|
Losses
|
|
|
Fair Value
|
|
|
|
|
Cash
|
|
$
|
15,499,182
|
|
|
$
|
—
|
|
|
$
|
15,499,182
|
|
|
Money market funds
|
|
|
19,467,096
|
|
|
|
—
|
|
|
|
19,467,096
|
|
|
Certificates of deposit
|
|
|
14,478,000
|
|
|
|
(6,765
|
)
|
|
|
14,471,235
|
|
|
Corporate bonds
|
|
|
4,010,563
|
|
|
|
(83
|
)
|
|
|
4,010,480
|
|
|
Investments in foreign sovereign debt
|
|
|
10,017,010
|
|
|
|
(84,109
|
)
|
|
|
9,932,901
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
63,471,851
|
|
|
|
(90,957
|
)
|
|
|
63,380,894
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Less amounts classified as cash and cash equivalents
|
|
|
(40,045,129
|
)
|
|
|
15,157
|
|
|
|
(40,029,972
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
23,426,722
|
|
|
$
|
(75,800
|
)
|
|
$
|
23,350,922
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Realized losses recorded for the years ended December 31,
2010 and 2009 were immaterial.
|
|
|
7.
|
FAIR
VALUE OF INSTRUMENTS
|
As of December 31, 2010, the Company held
$23.4 million short-term investments, which consisted of
certificates of deposit, FDIC insured corporate bonds and
investments in foreign sovereign debt. These securities were
classified as short-term based on their maturity terms being
less than one year. The Company included any unrealized gains
and losses on short-term investments in stockholders’
equity as a component of other comprehensive income (loss).
Individual securities with a fair value below the cost basis at
December 31, 2010 were evaluated to determine if they were
other-than-temporarily
impaired.
114
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
The following table presents the Company’s fair value
hierarchy for all its financial assets, including those in cash
and cash equivalents, measured at fair value on a recurring
basis as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Estimated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair Value
|
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
|
|
Money market funds
|
|
$
|
19,467,096
|
|
|
$
|
19,467,096
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Certificates of deposit
|
|
|
14,471,235
|
|
|
|
—
|
|
|
|
14,471,235
|
|
|
|
—
|
|
|
Corporate bonds
|
|
|
4,010,480
|
|
|
|
—
|
|
|
|
4,010,480
|
|
|
|
—
|
|
|
Investments in foreign sovereign debt
|
|
|
9,932,901
|
|
|
|
—
|
|
|
|
9,932,901
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
47,881,712
|
|
|
$
|
19,467,096
|
|
|
$
|
28,414,616
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company did not have any financial liabilities,
non-financial assets or non-financial liabilities that were
required to be measured at fair value as of December 31,
2010.
|
|
|
8.
|
COMMITMENTS
AND CONTINGENCIES
|
Leases
The Company leases its office facilities under an operating
lease that expired in January 2011, which was subsequently
renewed through July 2011. In addition to the facility lease,
the Company leases office equipment under operating lease
agreements, which began in 2007 and ends in 2013.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Office rental expense
|
|
$
|
134,231
|
|
|
$
|
165,016
|
|
|
$
|
111,506
|
|
|
$
|
532,256
|
|
|
Equipment rental expense
|
|
|
23,302
|
|
|
|
17,219
|
|
|
|
15,216
|
|
|
|
58,557
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
157,533
|
|
|
$
|
182,235
|
|
|
$
|
126,722
|
|
|
$
|
590,813
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Future minimum payments under the operating leases for the years
ending December 31, 2011, 2012 and 2013 are $10,914, $3,120
and $1,560, respectively.
Other
Commitments
In July 2006, the Company entered into a license agreement with
Shionogi & Co., Ltd. and Eli Lilly and Company, or Eli
Lilly, to develop and commercialize certain
sPLA2
inhibitors for the treatment of inflammatory diseases. The
agreement granted the Company commercialization rights to
Shionogi & Co., Ltd.’s and Eli Lilly’s
sPLA2
inhibitors, including varespladib and
A-001. Under
the terms of the agreement, the Company’s license is
worldwide, with the exception of Japan where
Shionogi & Co., Ltd. has retained rights. Pursuant to
this license agreement, the Company paid Shionogi &
Co., Ltd. and Eli Lilly a one-time license initiation fee of
$250,000 in the aggregate. Additionally, in consideration for
the licensed technology, the Company issued an aggregate of
257,744 shares of
Series A-2
convertible preferred stock at $5.14 per share and an aggregate
of 127,297 shares of
Series B-1
convertible preferred stock at $7.28 per share with a total
aggregate value of $2.3 million to Shionogi &
Co., Ltd. and Eli Lilly. As there is no future alternative use
for the technology and in accordance with the guidance of the
Research and Development topic of the FASB ASC, the Company
recorded the initiation and license fees in research and
development expenses during the year ended December 31,
2006. In March 2010, the Company paid $1.75 million each to
Eli Lilly and Shionogi & Co., Ltd. on the forms of the
Company’s common stock upon the commencement of the
Company’s Phase 3
VISTA-16
study of varespladib.
115
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
The Company is obligated to make additional milestone payments
of up to $97.5 million upon the achievement of certain
development and regulatory milestones. The Company is also
obligated to pay tiered royalties, which increase as a
percentage from the mid-single digits to the low double digits
as net sales increase, of up to $92.5 million on future net
sales of products that are developed and approved as defined by
this collaboration. The Company’s obligation to pay
royalties with respect to each licensed product in each country
will expire upon the later of (a) 10 years following
the date of the first commercial sale of such licensed product
in such country and (b) the first date on which generic
version(s) of the applicable licensed product achieve a total
market share, in the aggregate, of 25% or more of the total unit
sales of wholesalers to pharmacies of licensed product and all
generic versions combined in the applicable country.
On December 18, 2007, the Company entered into with Amgen,
Inc. (“Amgen”), a worldwide, exclusive license
agreement, or the Amgen Agreement, to develop and commercialize
A-623 for
the treatment of systemic lupus erythematosus
(“lupus”). Under the terms of the Amgen Agreement, the
Company was required to pay a nonrefundable, upfront license fee
of $6.0 million, payable in two installments with the first
installment due within 90 days from the effective date of
the Amgen Agreement and the second installment due on the
earlier of (i) termination of the Amgen Agreement by the
Company or (ii) February 1, 2009. As there is no
future alternative use for the technology, the Company expensed
the license fee in research and development expenses during the
year ended December 31, 2007.
Under the terms of the Amgen Agreement, the Company is obligated
to make additional milestone payments to Amgen of up to
$33.0 million upon the achievement of certain development
and regulatory milestones. The Company is also obligated to pay
tiered royalties on future net sales of products, ranging from
the high single digits to the low double digits, that are
developed and approved as defined by this collaboration. The
Company’s royalty obligations as to a particular licensed
product will be payable, on a
country-by-country
and licensed
product-by-licensed
product basis, for the longer of (a) the date of expiration
of the last to expire valid claim within the licensed patents
that covers the manufacture, use or sale, offer to sell, or
import of such licensed product by the Company or a sublicense
in such country or (b) 10 years after the first
commercial sale of the applicable licensed product in the
applicable country. There were no outstanding obligations due to
Amgen as of December 31, 2010 and December 31, 2009.
|
|
|
9.
|
CONVERTIBLE
PROMISSORY NOTES AND EQUITY FINANCING
|
Prior to our IPO, we used convertible debt as a method to
finance our clinical trials. In connection with the completion
of the Company’s IPO on March 4, 2010, all of the
Company’s outstanding convertible debt was converted to
shares of common stock. As of December 31, 2010 there is no
convertible debt outstanding.
In April 2006, the Company issued convertible promissory notes
to a group of individuals, or Holders, in exchange for an
aggregate principal amount of $570,000, or Bridge Loan. The
Bridge Loan was converted into
Series A-2
convertible preferred stock at a discount of 25% resulting in a
$3.85 per share price in August 2006. The interest on these
loans was 7% per annum and accrued interest of $13,816 was paid
out to the Holders upon closing of our
Series A-2
convertible preferred stock. In connection with the conversion
of the Bridge Loan, a beneficial conversion feature of $190,000
representing the difference between the conversion price and the
fair value of the preferred shares multiplied by the number of
shares converted was recorded as non-cash interest expense and
an increase in additional paid-in capital.
In June 2006, the Company issued two additional convertible
promissory notes to two new investors for an aggregate principal
amount of $390,000. The notes were converted into
Series A-2
convertible preferred stock at the issuance price of our
Series A-2
convertible preferred stock, or $5.14 per share, in August 2006.
The interest on these loans was 8% per annum. A portion of
accrued interest in the amount of $1,751 was converted into
Series A-2
convertible preferred stock and the remainder of accrued
interest was paid out to the investors.
During February and May 2008, the Company issued convertible
promissory notes to its existing investors in exchange for an
aggregate principal amount of $12.2 million. The interest
on these loans was 4.2% per annum. The notes and accrued
interest of $155,630 were converted into
Series B-2
convertible preferred stock at the issuance
116
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
price of our
Series B-2
convertible preferred stock, or $5.46 per share, in August 2008.
In connection with the terms of the convertible promissory
notes, a charge for the beneficial conversion feature of
$4.1 million representing the difference between the
conversion price and the fair value of the preferred shares
multiplied by the number of shares converted was recorded as
non-cash interest expense and an increase to additional paid-in
capital.
On August 12, 2008, the Company issued
2,267,178 shares of its
Series B-2
convertible preferred stock to certain of its existing investors
in exchange for conversion of $12.2 million of aggregate
principal amount of and $155,630 of aggregate interest accrued
upon convertible promissory notes and 962,066 shares of its
Series B-2
convertible preferred stock to two new investors in exchange for
$7.0 million of cash. In connection with the issuance of
our
Series B-2
convertible preferred stock, the Company issued warrants to
purchase 240,516 shares of the Company’s common stock
to those investors purchasing shares for cash.
In July and September 2009, the Company sold
(i) convertible promissory notes, or the 2009 notes, that
are secured by a first priority security interest in all of the
Company’s assets, and (ii) warrants, or the 2009
warrants, to purchase shares of the Company’s equity
securities to certain of its existing investors for an aggregate
purchase price of $10.0 million. These transactions are
collectively referred to as the 2009 bridge financing. The 2009
notes and accrued interest were converted into shares of the
Company’s common stock at a discount of 25% resulting in
$5.25 per share in March 2010 upon the closing of the
Company’s IPO.
In September 2009, the Company executed a stock purchase
agreement, which was amended to add an additional purchaser in
November 2009, with certain existing preferred stock holders for
the sale of shares of the Company’s common stock equal to
$20.5 million. In December 2009, the Company entered into a
note purchase agreement and amended the September 2009 stock
purchase and escrow agreements. The agreements provided for the
release of $3.4 million of the $20.5 million held in
the escrow account. The Company issued convertible promissory
notes, or the escrow notes, for the released amount to the
investors. The escrow funds, escrow notes and accrued interest
were converted into shares of the Company’s common stock at
$6.58 per share in March 2010 upon the closing of the
Company’s IPO. The conversion price reflected the
Company’s IPO price of $7.00 per share, minus per-share
underwriting discount and commission fees.
Common
Stock
At December 31, 2010, the Company is authorized to issue
100,000,000 shares of capital stock, of which
95,000,000 shares are designated as common stock, par value
$0.001 per share. Holders of common stock are entitled to one
vote per share on all matters to be voted upon by the
stockholders of the Company. Holders of common stock are
entitled to receive ratably dividends, if any, as may be
declared by the Board of Directors. No dividends have been
declared to date.
At December 31, 2010, the Company had reserved the
following shares for future issuance:
| |
|
|
|
|
|
Common stock warrants outstanding
|
|
|
4,557,136
|
|
|
Common stock options outstanding
|
|
|
1,275,991
|
|
|
Restricted stock units outstanding
|
|
|
302,500
|
|
|
Common stock options available for future grant under stock
option plan
|
|
|
35,389
|
|
|
|
|
|
|
|
|
Total
|
|
|
6,171,016
|
|
|
|
|
|
|
|
Convertible
Preferred Stock
In connection with the completion of the Company’s IPO on
March 4, 2010, all of the Company’s shares of
preferred stock outstanding at the time of the offering were
converted into an aggregate of 8,146,308 shares of common
stock. As of December 31, 2010, no liquidation preference
remained.
117
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
The Company’s Fifth Amended and Restated Certificate of
Incorporation designates 5,000,000 shares of the
Company’s capital stock as undesignated preferred stock.
Warrants
In August 2008, in connection with the issuance of
Series B-2
convertible preferred stock, the Company issued 240,516 warrants
for the purchase of common stock at $1.34 per share to two new
investors. The warrants expired upon the earliest of
(i) seven years from the issuance date, (ii) the
closing date of the Company’s IPO or (iii) upon
consummation by the Company of any consolidation or merger. The
Company valued the warrants using the Black-Scholes option
pricing model with the following assumptions: expected
volatility of 72%, risk-free interest rate of 3.46% and expected
term of seven years. The fair value of the warrants was
calculated to be $224,478 and recorded as issuance cost and an
increase to additional paid-in capital. As of December 31,
2009, 240,516, warrants remain outstanding. Each of the warrants
contained a net issuance feature, which allowed the warrant
holder to pay the exercise price of the warrant by forfeiting a
portion of the exercised warrant shares with a value equal to
the aggregate exercise. The warrants were exercised upon the
closing of the Company’s IPO on March 4, 2010.
In connection with the issuance of the 2009 notes for
$10.0 million, as discussed in Note 9, the Company
issued warrants to each note holder to purchase shares of its
equity securities. Each 2009 warrant is exercisable for the
security into which each 2009 note is converted, at the price at
which that security is sold to other investors. Depending on
when the 2009 notes are converted, each 2009 warrant may be
exercisable for a number of shares equal to the quotient
obtained by dividing (x) (i) 25% of the principal amount of
the accompanying 2009 notes, in the event the conversion occurs
prior to April 1, 2010, or (ii) 50% of the principal
amount of the accompanying 2009 notes, in the event the
conversion occurs on or after April 1, 2010, by
(y) the purchase price of the securities into which the
note is ultimately converted. The Company accounted for the 2009
warrants in accordance with FASB ASC 480, Distinguishing
Liabilities from Equity, which requires that a financial
instrument, other than outstanding shares, that, at inception,
is indexed to an obligation to repurchase the issuer’s
equity shares, regardless of the timing of the redemption
feature, and may require the issuer to settle the obligation by
transferring assets, be classified as liability through the
completion of the Company’s IPO. The Company measured the
fair value of the 2009 warrants using the Black-Scholes option
pricing model on issuance date and adjusted the fair value at
the end of each reporting period based on the following
assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
December 31,
|
|
|
September 30,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2009
|
|
|
|
|
Expected Volatility
|
|
|
94
|
%
|
|
|
78
|
%
|
|
|
78
|
%
|
|
Dividend Yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Risk-Free Interest Rate
|
|
|
2.28
|
%
|
|
|
2.34
|
%
|
|
|
2.38
|
%
|
|
Expected Term (years)
|
|
|
5.00
|
|
|
|
5.00
|
|
|
|
5.00
|
|
The Company then applied probability factors to the different
possible conversion scenarios and calculated the initial fair
value of the 2009 warrants to be $320,000, which amount was
recorded as a discount to the 2009 notes. The discount was
amortized as interest expense over the terms of the 2009 notes.
The Company re-measured the fair value of the 2009 warrants on
December 31, 2009 and recorded the change in fair value in
non-operating income. Upon conversion of the 2009 notes into
shares of common stock at the completion of the Company’s
IPO, the fair value of the 2009 warrants was re-measured again
by the Company and the change in fair value of $1.5 million
was recorded in non-operating expense during the year ended
December 31, 2010. Concurrent with the conversion of the
2009 notes, the Company calculated the number of warrant shares
to be 357,136 based on 25% of the principal amount of the
accompanying 2009 notes and the IPO price of the Company’s
common stock of $7.00 per share. The warrant liability and
unamortized discount were reclassified to additional
paid-in-capital
as a result of the conversion of the 2009 notes.
In connection with the issuance of the escrow notes for
$3.4 million, as discussed in Note 9, which were
exchangeable for exchange notes, each exchange note that was
issued would be accompanied by a warrant, which was exercisable
for the security into which the accompanying exchange note, if
any, was converted, at the price at
118
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
which that security was sold to other investors. Depending on
when the exchange notes are converted, each warrant may be
exercisable for a number of shares equal to the quotient
obtained by dividing (x) (i) 25% of the principal amount of
the accompanying exchange notes, in the event the conversion
occurs prior to April 1, 2010, or (ii) 50% of the
principal amount of the accompanying exchange notes, in the
event the conversion occurs on or after April 1, 2010, by
(y) the purchase price of the securities into which the
exchange note is ultimately converted. The Company accounts for
the potential issuance of the warrants in accordance with FASB
ASC 480. The Company measured the fair value of its
derivative using the Black-Scholes option pricing model with the
following assumptions: expected volatility of 78%, risk-free
interest rate of 2.34% and expected term of five years. The
Company then applied probability factors to the different
possible exchange and conversion scenarios and calculated the
fair value of the warrants to be $86,845, which amount was
recorded as a discount to the escrow notes. The discount was
amortized as interest expense over the terms of the escrow
notes. The escrow notes were converted into shares of the
Company’s common stock upon the closing of its IPO. As a
result of the conversion taking place prior to the exchange of
the escrow notes into exchange notes, the Company’s
obligation to issue the warrants was eliminated. Consequently,
the Company reclassified the unamortized discount into
additional paid-in capital and reduced the fair value of the
warrant liability to zero.
On September 24, 2010, the Company closed a private
placement transaction with certain accredited investors pursuant
to which the Company sold an aggregate of 10,500,000 units
at a purchase price of $3.00 per unit, with each unit consisting
of one share of common stock and a warrant to purchase an
additional 0.40 shares of common stock. Each warrant is
exercisable in whole or in part at any time until
September 24, 2015 at a per share exercise price of $3.30,
subject to certain adjustments as specified in the warrant. The
Company valued the warrant using the Black-Scholes option
pricing model with the following assumptions: expected
volatility of 64%, risk-free interest rate of 1.37% and expected
term of five years. The fair value of the warrants was
calculated to be $5.3 million and has been recorded in
additional paid-in capital. As of December 31, 2010,
4,200,000 warrants remain outstanding. Each of the warrants
contains a net issuance feature, which allows the warrant holder
to pay the exercise price of the warrant by forfeiting a portion
of the exercised warrant shares with a value equal to the
aggregate exercise.
Embedded
Derivative
The 2009 notes and the escrow notes discussed in Note 9
contained a contingent automatic redemption feature and a
contingent put option that meet the definition of an embedded
derivative as defined in FASB ASC 815, Derivatives and
Hedging, because these notes contain features with implicit
or explicit terms that affect some or all of the cash flows or
the value of other exchanges required by a contract in a manner
similar to a derivative instrument. As a result, the Company
evaluated these embedded derivative features under the guidance
of FASB ASC 815 and determined that the embedded derivative
features should be separated from the 2009 notes and escrow
notes and recognized as derivative instruments. Pursuant to the
guidance of FASB ASC 815, if a hybrid instrument contains
more than one embedded derivative feature that would
individually warrant separate accounting as a derivative
instrument, those embedded derivative features shall be bundled
together as a single, compound embedded derivative that shall
then be bifurcated and accounted for separately from the host
contract unless a fair value election is made. Since the Company
may not make a fair value election, the contingent automatic
redemption and the contingent put option should be bundled
together as a single, compound embedded derivative and separated
from the 2009 notes and escrow notes. The Company recognized the
bundled embedded derivative as a derivative liability with
initial and subsequent measurements at fair value and changes in
fair value recorded in earnings. Upon conversion of the 2009
notes and escrow notes into shares of common stock at the
completion of the Company’s IPO, the Company re-measured
the fair value of the embedded derivative and recorded a charge
of $2.5 million in non-operating expense during the year
ended December 31, 2010.
119
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
Option
Plan
The Company’s 2005 Equity Incentive Plan (the “2005
Plan”) was adopted by the board of directors in January
2005. The 2005 Plan permits the granting of incentive and
non-statutory stock options, restricted stock, stock
appreciation rights, performance units, performance shares and
other stock awards to eligible employees, directors and
consultants. The Company grants options to purchase shares of
common stock under the 2005 Plan at no less than the fair market
value of the underlying common stock as of the date of grant.
Options granted under the 2005 Plan have a maximum term of
10 years and generally vest over four years at the rate of
25% of total shares underlying the option. Selected grants vest
immediately or over a shorter vesting period.
The 2005 Plan allows the option holders to exercise their
options prior to vesting. Unvested shares are subject to
repurchase by the Company at the option of the Company. Unvested
shares subject to repurchase have been excluded from the number
of shares outstanding. Option activity in the table below
includes options exercised prior to vesting. At
December 31, 2008 and 2009 and December 31, 2010,
161,646, 69,424 and 27,321 shares were subject to
repurchase with a corresponding liability of $56,715, $31,131,
and $27,973, respectively.
On February 1, 2010, the Company’s board of directors
adopted the 2010 Stock Option and Incentive Plan (the “2010
Plan”) effective upon consummation of the IPO, which was
also approved by the Company’s stockholders. The Company
initially reserved 233,644 shares of common stock for
issuance under the 2010 Plan, plus 35,670 shares remaining
available for grant under the Company’s 2005 Plan, plus any
additional shares returned under the 2005 Plan as a result of
the cancellation of options or the repurchase of shares issued
pursuant to the 2005 Plan. On July 9, 2010, the
Company’s stockholders approved an increase to the
aggregate number of shares initially available for grant under
the 2010 Plan by 200,000 shares to 433,644 shares of
common stock, plus 35,670 shares remaining available for
grant under the 2005 Plan, plus any additional shares referred
under the 2005 Plan as a result of the cancellation of options
or repurchase of shares issued under the 2005 Plan. In addition,
the 2010 Plan provides for annual increases in the number of
shares available for issuance thereunder on the first day of
each fiscal year, beginning with the 2011 fiscal year, equal to
four percent (4%) of the outstanding shares of the
Company’s common stock on the last day of the immediately
preceding fiscal year. The maximum aggregate number of shares of
stock that may be issued in the form of incentive stock options
shall not exceed the lesser of (i) the number of shares
reserved and available for issuance under the Plan or
(ii) 1,460,280 shares of stock, subject in all cases
to adjustment including reorganization, recapitalization,
reclassification, stock dividend, stock split, reverse stock
split or other similar change in the Company’s capital
stock. The 2010 Plan permits the granting of incentive and
non-statutory stock options, restricted and unrestricted stock
awards, restricted stock units, stock appreciation rights,
performance share awards, cash-based awards and dividend
equivalent rights to eligible employees, directors and
consultants. The option exercise price of an option granted
under the 2010 Plan may not be less than 100% of the fair market
value of a share of the Company’s common stock on the date
the stock option is granted. Options granted under the 2010 Plan
have a maximum term of 10 years and generally vest over
four years. In addition, in the case of certain large
stockholders, the minimum exercise price of incentive options
must equal 110% of fair market value on the date of grant and
the maximum term is limited to five years.
The 2010 Plan does not allow the option holders to exercise
their options prior to vesting.
120
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
The following table summarizes stock option activity for the
Company from inception to December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
Weighted-
|
|
Average
|
|
|
|
|
|
|
|
|
Average
|
|
Remaining
|
|
Aggregate
|
|
|
|
Number of
|
|
|
Exercise
|
|
Contractual
|
|
Intrinsic
|
|
|
|
Options
|
|
|
Price
|
|
Life in Years
|
|
Value
|
|
|
|
Balance at December 31, 2007
|
|
|
847,735
|
|
|
|
$0.26
|
|
|
|
8.08
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options granted
|
|
|
327,973
|
|
|
|
$1.34
|
|
|
|
|
|
|
|
|
|
|
Options exercised
|
|
|
(179,886
|
)
|
|
|
$0.38
|
|
|
|
|
|
|
|
|
|
|
Options cancelled
|
|
|
(38,697
|
)
|
|
|
$0.42
|
|
|
|
|
|
|
|
|
|
|
Repurchase
|
|
|
—
|
|
|
|
$0.26
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
957,125
|
|
|
|
$0.60
|
|
|
|
8.28
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options granted
|
|
|
405,358
|
|
|
|
$1.69
|
|
|
|
|
|
|
|
|
|
|
Options exercised
|
|
|
(19,089
|
)
|
|
|
$0.80
|
|
|
|
|
|
|
|
|
|
|
Options cancelled
|
|
|
(19,618
|
)
|
|
|
$0.92
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
1,323,776
|
|
|
|
$0.92
|
|
|
|
7.94
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options granted
|
|
|
112,000
|
|
|
|
$4.82
|
|
|
|
|
|
|
|
|
|
|
Options exercised
|
|
|
(138,878
|
)
|
|
|
$0.84
|
|
|
|
|
|
|
|
|
|
|
Options cancelled
|
|
|
(20,907
|
)
|
|
|
$1.50
|
|
|
|
|
|
|
|
|
|
|
Repurchase
|
|
|
—
|
|
|
|
$0.26
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2010
|
|
|
1,275,991
|
|
|
|
$1.26
|
|
|
|
7.07
|
|
|
|
$4,700,543
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ending vested at December 31, 2010
|
|
|
1,049,865
|
|
|
|
$0.97
|
|
|
|
6.81
|
|
|
|
$4,138,497
|
|
|
Ending vested and expected to vest at December 31, 2010
|
|
|
1,275,991
|
|
|
|
$1.26
|
|
|
|
7.07
|
|
|
|
$4,700,543
|
|
The intrinsic value of stock options represents the difference
between the exercise price of stock options and the market price
of our stock on that day for all
in-the-money
options. Additional information related to our stock options is
summarized below (in millions except per share information):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Weighted-average fair value per share granted
|
|
$
|
3.10
|
|
|
$
|
1.01
|
|
|
$
|
0.96
|
|
|
$
|
0.57
|
|
|
Intrinsic value of options exercised
|
|
$
|
699,366
|
|
|
$
|
13,550
|
|
|
$
|
109,741
|
|
|
$
|
828,200
|
|
|
Proceeds received from the exercise of stock options
|
|
$
|
115,986
|
|
|
$
|
15,274
|
|
|
$
|
68,105
|
|
|
$
|
345,151
|
|
|
Grant date fair value of options vested
|
|
$
|
235,232
|
|
|
$
|
358,121
|
|
|
$
|
113,166
|
|
|
$
|
819,565
|
|
There was $500,483 of total unrecognized compensation expense as
of December 31, 2010 related to options. The unrecognized
compensation expense will be amortized on a straight-line basis
over a weighted-average remaining period of 1.31 years.
121
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
Information about stock options outstanding, vested and expected
to vest as of December 31, 2010, is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding, Vested and Expected to Vest
|
|
|
Options Vested
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
Weighted Average
|
|
|
|
|
|
|
|
Number of
|
|
|
Contractual Life
|
|
|
Exercise
|
|
|
Number of
|
|
|
Range of Exercise Price
|
|
Shares
|
|
|
(In Years)
|
|
|
Price
|
|
|
Shares
|
|
|
|
|
$0.14 — $0.14
|
|
|
4,672
|
|
|
|
3.33
|
|
|
$
|
0.14
|
|
|
|
4,672
|
|
|
$0.26 — $0.26
|
|
|
567,161
|
|
|
|
6.14
|
|
|
$
|
0.26
|
|
|
|
559,969
|
|
|
$1.34 — $1.34
|
|
|
246,352
|
|
|
|
7.16
|
|
|
$
|
1.34
|
|
|
|
180,053
|
|
|
$1.51 — $1.51
|
|
|
334,124
|
|
|
|
7.71
|
|
|
$
|
1.51
|
|
|
|
257,239
|
|
|
$4.19 — $7.70
|
|
|
123,682
|
|
|
|
9.56
|
|
|
$
|
5.09
|
|
|
|
47,932
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1,275,991
|
|
|
|
7.07
|
|
|
$
|
1.26
|
|
|
|
1,049,865
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, 2010, there are 35,389 shares
available for grant under the 2010 Plan.
Restricted
Stock Units
During 2010, the Company granted restricted stock unit awards
under its 2010 Plan representing an aggregate of
333,000 shares of common stock. The restricted stock units
granted represent a right to receive shares of common stock at a
future date determined in accordance with the participant’s
award agreement. An exercise price and monetary payment are not
required for receipt of restricted stock units or the shares
issued in settlement of the award. Instead, consideration is
furnished in the form of the participant’s services to the
Company. Substantially all of the restricted stock units vest
over four years. Compensation cost for these awards is based on
the closing price of the Company’s common stock on the date
of grant and recognized as compensation expense on a
straight-line basis over the requisite service period.
Compensation expense recognized was $371,283 for the year ended
December 31, 2010. At December 31, 2010, the
unrecognized compensation cost related to these awards was
$1.34 million, which is expected to be recognized on a
straight-line basis over 2.71 years.
The following table summarizes activity related to our
restricted stock units:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
Grant
|
|
|
|
|
|
|
|
Date Fair
|
|
|
|
|
Shares
|
|
|
Value
|
|
|
|
|
Outstanding at December 31, 2009
|
|
|
—
|
|
|
|
—
|
|
|
Restricted stock units granted
|
|
|
333,000
|
|
|
$
|
5.15
|
|
|
Restricted stock units vested
|
|
|
—
|
|
|
|
—
|
|
|
Restricted stock units forfeitures and cancellations
|
|
|
(30,500
|
)
|
|
$
|
5.36
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2010
|
|
|
302,500
|
|
|
$
|
5.13
|
|
|
|
|
|
|
|
|
|
|
|
Early
Exercise of Employee Options
Stock options granted under the Company’s 2005 Plan provide
employee option holders the right to elect to exercise unvested
options in exchange for restricted common stock. Unvested
shares, which amounted to 27,321 and 69,424 at December 31,
2010, and December 31, 2009, respectively, were subject to
a repurchase right held by the Company at the original issuance
price in the event the optionees’ employment is terminated
either voluntarily or involuntarily. For exercises of employee
options, this right lapses 25% on the first anniversary of the
vesting start date and in 36 equal monthly amounts thereafter.
These repurchase terms are considered to be a forfeiture
provision and do not result in variable accounting. The shares
purchased by the employees pursuant to the early exercise of
122
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
stock options are not deemed to be outstanding until those
shares vest. In addition, cash received from employees for
exercise of unvested options is treated as a refundable deposit
shown as a liability in the Company’s financial statements.
For the year ended December 31, 2010 and the year ended
December 31, 2009, cash received for early exercise of
options totaled $24,714 and $6,615, respectively. As the shares
vest, the shares and liability are released into common stock
and additional paid-in capital.
The activity of early exercise of options granted to employees
is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
Average
|
|
|
Unvested Shares
|
|
Shares
|
|
|
Grant Price
|
|
|
|
|
Balance as of December 31, 2007
|
|
|
289,824
|
|
|
$
|
0.24
|
|
|
Early exercise of options
|
|
|
59,191
|
|
|
$
|
0.62
|
|
|
Vested
|
|
|
(168,386
|
)
|
|
$
|
0.22
|
|
|
Repurchases
|
|
|
(18,983
|
)
|
|
$
|
0.26
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2008
|
|
|
161,646
|
|
|
$
|
0.34
|
|
|
Early exercise of options
|
|
|
4,381
|
|
|
$
|
1.51
|
|
|
Vested
|
|
|
(96,603
|
)
|
|
$
|
0.35
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2009
|
|
|
69,424
|
|
|
$
|
0.45
|
|
|
|
|
|
|
|
|
|
|
|
|
Early exercise of options
|
|
|
18,011
|
|
|
$
|
1.37
|
|
|
Vested
|
|
|
(53,847
|
)
|
|
$
|
0.34
|
|
|
Repurchases
|
|
|
(6,267
|
)
|
|
$
|
0.26
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2010
|
|
|
27,321
|
|
|
$
|
1.02
|
|
|
|
|
|
|
|
|
|
|
|
2010
Employee Stock Purchase Plan
In July 2010, the Company’s stockholders approved the ESPP.
The Company reserved 100,000 shares of common stock for
issuance thereunder plus on January 1, 2011 and each
January 1 thereafter, the number of shares of stock reserved and
available for issuance under the Plan shall be cumulatively
increased by the lesser of (i) one percent (1%) of the
number of shares of common stock issued and outstanding on the
immediately preceding December 31 or
(ii) 250,000 shares of common stock.
Under the ESPP, eligible employees of the Company and certain
designated subsidiaries of the Company may authorize the Company
to deduct amounts from their compensation, which amounts are
used to enable the employees to purchase shares of the
Company’s common stock. The purchase price per share is 85%
of the fair market value of the common stock as of the first
date or the ending date of the applicable semi-annual purchase
period, whichever is less (the “Look-Back Provision”).
The 15% discount and the Look-Back Provision make the ESPP
compensatory under
ASC 718-50-25-2,
Compensation — Stock Compensation —
Employee Share Purchase Plans — Recognition. The
Black-Scholes option pricing model was used to value the
employee stock purchase rights. For the year ended
December 31, 2010 and the period from September 9,
2004 (Inception Date) through
123
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
December 31, 2010, the following weighted-average
assumptions were used in the valuation of the stock purchase
rights:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
Year Ended
|
|
|
Inception) to
|
|
|
|
|
December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2010
|
|
|
|
|
Expected Volatility
|
|
|
67
|
%
|
|
|
67
|
%
|
|
Dividend Yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Risk-Free Interest Rate
|
|
|
0.16
|
%
|
|
|
0.16
|
%
|
|
Expected Term (years)
|
|
|
0.33
|
|
|
|
0.33
|
|
The Company received $81,226 in contribution from participants
during the year ended December 31, 2010. Compensation
expense recognized for the year ended December 31, 2010 was
$28,891. As of December 31, 2010, 24,916 shares have
been issued and 75,084 shares were available for future
purchase under the ESPP.
Stock-Based
Compensation Expense
Total employee stock-based compensation expense recognized under
FASB ASC 718 was as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Research and development
|
|
$
|
223,229
|
|
|
$
|
101,395
|
|
|
$
|
45,544
|
|
|
$
|
417,231
|
|
|
General and administrative
|
|
|
470,702
|
|
|
|
152,569
|
|
|
|
97,862
|
|
|
|
753,579
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total employee stock-based compensation
|
|
$
|
693,931
|
|
|
$
|
253,964
|
|
|
$
|
143,406
|
|
|
$
|
1,170,810
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The assumptions used in the Black-Scholes option-pricing model
are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Expected Volatility
|
|
|
69
|
%
|
|
|
74
|
%
|
|
|
81
|
%
|
|
|
79
|
%
|
|
Dividend Yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Risk-Free Interest Rate
|
|
|
1.91
|
%
|
|
|
2.10
|
%
|
|
|
3.08
|
%
|
|
|
3.86
|
%
|
|
Expected Term (years)
|
|
|
6.25
|
|
|
|
6.25
|
|
|
|
6.25
|
|
|
|
6.25
|
|
Nonemployee
Stock-Based Compensation
The Company accounts for stock options granted to nonemployees
as required by FASB ASC 718. In connection with stock options
granted to consultants, the Company recorded $51,874, $88,382,
$8,399 and $166,344 for nonemployee stock-based compensation
during the years ended December 31, 2008, 2009, 2010 and
for the period from September 9, 2004 (Date of Inception)
to December 31, 2010, respectively. These amounts were
based upon the fair value of the vested portion of the grants.
124
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
The assumptions used in the Black-Scholes option-pricing model
are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004 (Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Expected Volatility
|
|
|
98
|
%
|
|
|
98
|
%
|
|
|
98
|
%
|
|
|
98
|
%
|
|
Dividend Yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Risk-Free Interest Rate
|
|
|
3.16
|
%
|
|
|
3.57
|
%
|
|
|
3.67
|
%
|
|
|
3.67
|
%
|
|
Expected Term (years)
|
|
|
7.86
|
|
|
|
9.94
|
|
|
|
9.26
|
|
|
|
9.63
|
|
Amounts expensed during the remaining vesting period will be
determined based on the fair value at the time of vesting.
|
|
|
12.
|
EMPLOYEE
BENEFIT PLAN
|
The Company maintains a defined contribution 401(k) plan, or the
401(k) Plan. Employee contributions are voluntary and are
determined on an individual basis, limited by the maximum
amounts allowable under federal tax regulations. The Company has
made no contributions to the 401(k) Plan since its inception.
The Company has incurred net operating losses since inception.
The Company has not reflected any benefit of such net operating
loss carryforwards in the accompanying financial statements and
has established a full valuation allowance against its deferred
tax assets.
Deferred income taxes reflect the net tax effects of
(a) temporary differences between the carrying amounts of
assets and liabilities for financial reporting purposes and the
amounts used for income tax purposes, and (b) operating
losses and tax credit carryforwards.
The significant components of the Company’s deferred tax
assets for the years ended December 31, 2010 and 2009 are
as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Deferred tax assets:
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards
|
|
$
|
33,300,399
|
|
|
$
|
20,254,375
|
|
|
Tax credits
|
|
|
3,543,005
|
|
|
|
2,378,197
|
|
|
Intangible assets
|
|
|
3,014,136
|
|
|
|
3,279,699
|
|
|
Accrued bonus
|
|
|
230,189
|
|
|
|
61,040
|
|
|
Accrued liabilities
|
|
|
1,249,792
|
|
|
|
91,529
|
|
|
Stock-based compensation
|
|
|
269,661
|
|
|
|
68,439
|
|
|
Other
|
|
|
34,109
|
|
|
|
5,828
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax assets
|
|
|
41,641,291
|
|
|
|
26,139,107
|
|
|
Deferred tax liabilities
|
|
|
—
|
|
|
|
—
|
|
|
Valuation allowance
|
|
|
(41,641,291
|
)
|
|
|
(26,139,107
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax asset
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
125
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
A reconciliation of the statutory tax rates and the effective
tax rates for the years ended December 31, 2008, 2009 and
2010 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Statutory rate
|
|
|
34
|
%
|
|
|
34
|
%
|
|
|
34
|
%
|
|
State tax
|
|
|
7
|
%
|
|
|
6
|
%
|
|
|
5
|
%
|
|
Tax credit
|
|
|
2
|
%
|
|
|
1
|
%
|
|
|
2
|
%
|
|
Beneficial conversion feature
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
(8
|
)%
|
|
Other
|
|
|
(4
|
)%
|
|
|
(3
|
)%
|
|
|
0
|
%
|
|
Valuation allowance
|
|
|
(38
|
)%
|
|
|
(38
|
)%
|
|
|
(33
|
)%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effective tax rates
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Realization of the future tax benefits is dependent on the
Company’s ability to generate sufficient taxable income
within the carryforward period. Because of the Company’s
recent history of operating losses, management believes that the
deferred tax assets arising from the above-mentioned future tax
benefits are currently not likely to be realized and,
accordingly, has provided a full valuation allowance. The net
valuation allowance increased by $4,690,989 and $15,502,184 for
the years ended December 31, 2009 and 2010, and $41,641,291
for the period from September 9, 2004 (Date of Inception)
to December 31, 2010.
Net operating losses and tax return credit carryforwards as of
December 31, 2010, are as follows:
| |
|
|
|
|
|
|
|
|
|
Amount
|
|
|
Expiration Years
|
|
|
|
Net operating losses — federal
|
|
$
|
83,553,933
|
|
|
Beginning 2024
|
|
Net operating losses — state
|
|
$
|
83,848,589
|
|
|
Beginning 2014
|
|
Tax return credits — federal
|
|
$
|
2,491,822
|
|
|
Beginning 2024
|
|
Tax return credits — state
|
|
$
|
1,592,701
|
|
|
Not applicable
|
Utilization of the net operating loss carryforwards and credits
may be subject to a substantial annual limitation due to the
ownership change limitations provided by the Internal Revenue
Code of 1986, as amended, or the IRC, and similar state
provisions. The Company has not performed a detailed analysis to
determine whether an ownership change under Section 382 of
the IRC has occurred. The effect of an ownership change would be
the imposition of an annual limitation on the use of net
operating loss carryforwards attributable to periods before the
change.
As of December 31, 2010, the Company had unrecognized tax
benefits of $1,375,594, all of which would not currently affect
the Company’s effective tax rate if recognized due to the
Company’s deferred tax assets being fully offset by a
valuation allowance. The Company did not anticipate any
significant change to the unrecognized tax benefit balance as of
December 31, 2010. A reconciliation of the beginning and
ending amount of unrecognized tax benefits is as follows:
| |
|
|
|
|
|
|
|
Amount
|
|
|
|
|
Balance as December 31, 2007
|
|
$
|
646,181
|
|
|
Additions based on tax positions related to current year
|
|
|
162,381
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2008
|
|
|
808,562
|
|
|
Additions based on tax positions related to current year
|
|
|
83,848
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2009
|
|
|
892,410
|
|
|
Additions based on tax positions related to current year
|
|
|
469,098
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2010
|
|
$
|
1,361,508
|
|
|
|
|
|
|
|
126
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
The Company would classify interest and penalties related to
uncertain tax positions in income tax expense, if applicable.
There was no interest expense or penalties related to
unrecognized tax benefits recorded through December 31,
2010. The tax years 2004 through 2010 remain open to examination
by one or more major taxing jurisdictions to which the Company
is subject.
The Company does not anticipate that total unrecognized net tax
benefits will significantly change prior to the end of 2010.
|
|
|
14.
|
SELECTED
QUARTERLY FINANCIAL DATA (unaudited)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended
|
|
|
|
|
March 31,
|
|
|
June 30,
|
|
|
September 30,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OPERATING EXPENSES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
5,241,814
|
|
|
$
|
6,438,149
|
|
|
$
|
6,885,125
|
|
|
$
|
10,891,654
|
|
|
General and administrative
|
|
|
1,224,110
|
|
|
|
1,509,869
|
|
|
|
1,510,021
|
|
|
|
2,056,849
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LOSS FROM OPERATIONS
|
|
|
(6,465,924
|
)
|
|
|
(7,948,018
|
)
|
|
|
(8,395,146
|
)
|
|
|
(12,948,503
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other expense and interest income, net
|
|
|
(4,637,868
|
)
|
|
|
11,655
|
|
|
|
61,606
|
|
|
|
(91,617
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS
|
|
$
|
(11,103,792
|
)
|
|
$
|
(7,936,363
|
)
|
|
$
|
(8,333,540
|
)
|
|
$
|
(13,040,120
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share — basic and diluted
|
|
$
|
(0.83
|
)
|
|
$
|
(0.36
|
)
|
|
$
|
(0.36
|
)
|
|
$
|
(0.40
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used in computing basic and diluted net loss per share
|
|
|
13,344,231
|
|
|
|
22,223,941
|
|
|
|
22,964,279
|
|
|
|
32,828,697
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended
|
|
|
|
|
March 31,
|
|
|
June 30,
|
|
|
September 30,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OPERATING EXPENSES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
2,914,766
|
|
|
$
|
2,286,415
|
|
|
$
|
2,525,948
|
|
|
$
|
688,285
|
|
|
General and administrative
|
|
|
846,243
|
|
|
|
999,331
|
|
|
|
884,908
|
|
|
|
695,208
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LOSS FROM OPERATIONS
|
|
|
(3,761,009
|
)
|
|
|
(3,285,746
|
)
|
|
|
(3,410,856
|
)
|
|
|
(1,383,493
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other expense and interest income, net
|
|
|
(24,351
|
)
|
|
|
(50,310
|
)
|
|
|
(193,556
|
)
|
|
|
(94,171
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS
|
|
$
|
(3,785,360
|
)
|
|
$
|
(3,336,056
|
)
|
|
$
|
(3,604,412
|
)
|
|
$
|
(1,477,664
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share — basic and diluted
|
|
$
|
(2.57
|
)
|
|
$
|
(2.23
|
)
|
|
$
|
(2.37
|
)
|
|
$
|
(0.95
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used in computing basic and diluted net loss per share
|
|
|
1,470,722
|
|
|
|
1,496,011
|
|
|
|
1,520,875
|
|
|
|
1,557,708
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
127
ANTHERA
PHARMACEUTICALS, INC.
(A Development Stage Company)
NOTES TO FINANCIAL
STATEMENTS — (Continued)
|
|
|
15.
|
RELATED
PARTY TRANSACTIONS
|
The Company engaged an outside service provider where one of the
founders is employed for clinical management services. In
consideration for the services rendered, the Company paid the
following fees:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
September 9,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2004
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Date of
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Inception) to
|
|
|
|
|
Years Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2010
|
|
|
|
|
Project management fees
|
|
$
|
533,617
|
|
|
$
|
38,274
|
|
|
$
|
22,200
|
|
|
$
|
665,191
|
|
As of December 31, 2010, the Company had $519,386 payable
to this organization for services performed during the year.
The Company evaluated all events or transactions that occurred
after December 31, 2010. The Company did not have any
material subsequent events that require adjustment or disclosure
in these financial statements.
|
|
|
17.
|
RECENT
ACCOUNTING PRONOUNCEMENTS
|
There were no recent accounting pronouncements that have not yet
been adopted by the Company that are expected to have a material
impact on the financial statements.
128
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the registrant has duly caused
this report to be signed on its behalf by the undersigned
thereunto duly authorized.
ANTHERA PHARMACEUTICALS, INC.
Paul F. Truex
President and Chief Executive Officer
(Principal Executive Officer)
Dated: March 7, 2011
POWER OF
ATTORNEY
We, the undersigned officers and directors of Anthera
Pharmaceuticals, Inc., hereby severally constitute and appoint
Paul F. Truex and Christopher P. Lowe, and each of them singly
(with full power to each of them to act alone), our true and
lawful attorneys-in-fact and agents, with full power of
substitution and resubstitution in each of them for him and in
his name, place and stead, and in any and all capacities, to
sign for us and in our names in the capacities indicated below
any and all amendments to this report, and to file the same,
with all exhibits thereto and other documents in connection
therewith, with the Securities and Exchange Commission, granting
unto said attorneys-in-fact and agents, and each of them, full
power and authority to do and perform each and every act and
thing requisite or necessary to be done in and about the
premises, as full to all intents and purposes as he might or
could do in person, hereby ratifying and confirming all that
said attorneys-in-fact and agents or any of them, or their or
his substitute or substitutes, may lawfully do or cause to be
done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the registrant and in the capacities and on the
dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ Paul
F. Truex
Paul
F. Truex
|
|
President, Chief Executive
Officer and Director
(Principal Executive Officer)
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ Christopher
P. Lowe
Christopher
P. Lowe
|
|
Chief Financial Officer
(Principal Financial and Accounting Officer)
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ Christopher
S. Henney
Christopher
S. Henney
|
|
Chairman of the Board of Directors
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ Annette
Bianchi
Annette
Bianchi
|
|
Director
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ James
I. Healy
James
I. Healy
|
|
Director
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ Donald
J. Santel
Donald
J. Santel
|
|
Director
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ Daniel
K. Spiegelman
Daniel
K. Spiegelman
|
|
Director
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ David
E. Thompson
David
E. Thompson
|
|
Director
|
|
March 7, 2011
|
|
|
|
|
|
|
|
/s/ Peter
A. Thompson
Peter
A. Thompson
|
|
Director
|
|
March 7, 2011
|
129
Exhibit
Index
| |
|
|
|
|
|
Number
|
|
Description
|
|
|
|
|
3
|
.1
|
|
Fifth Amended and Restated Certificate of Incorporation(1)
|
|
|
3
|
.2
|
|
Amended and Restated Bylaws(2)
|
|
|
4
|
.1
|
|
Specimen certificate evidencing shares of common stock(3)
|
|
|
4
|
.2
|
|
Second Amended and Restated Investor Rights Agreement by and
among the Company and the other persons and entities party
thereto, dated as of July 17, 2009(3)
|
|
|
# 10
|
.1
|
|
2005 Equity Incentive Plan and form agreements thereunder(4)
|
|
|
# 10
|
.2
|
|
Amended and Restated 2010 Stock Option and Incentive Plan(5)
|
|
|
# 10
|
.3
|
|
Form of Amended and Restated Indemnification Agreement(4)
|
|
|
# 10
|
.4
|
|
Form of Amended and Restated Change in Control Agreement(6)
|
|
|
# 10
|
.5
|
|
Form of Amended and Restated Severance Benefits Agreement(6)
|
|
|
+ 10
|
.6
|
|
License Agreement among Eli Lilly and Company, Shionogi &
Co., Ltd. and the Company, dated as of July 31, 2006(4)
|
|
|
+ 10
|
.7
|
|
Agreement between Shionogi & Co., Ltd. and the Company,
dated as of September 7, 2009 (amending License Agreement among
Eli Lilly and Company, Shionogi & Co., Ltd. and the
Company, dated as of July 31, 2006)(4)
|
|
|
+ 10
|
.8
|
|
Agreement between Eli Lilly and Company and the Company, dated
as of September 15, 2009 (amending License Agreement among Eli
Lilly Company, Shionogi & Co., Ltd. and the Company, dated
as of July 31, 2006)(4)
|
|
|
+ 10
|
.9
|
|
Amended and Restated Technology Transfer Letter Agreement
between Eli Lilly and Company and the Company, dated as of July
12, 2006(4)
|
|
|
+ 10
|
.10
|
|
License Agreement between Amgen Inc. and the Company, dated as
of December 18, 2007(4)
|
|
|
10
|
.11
|
|
Consent to Sublease, by and among the Company, NewTower Trust
Company Multi-Employer Property Trust and Guava Technologies,
dated as of September 12, 2008(4)
|
|
|
10
|
.12
|
|
Sublease by and between the Company and Guava Technologies,
dated as of August 1, 2008(4)
|
|
|
10
|
.13
|
|
Note and Warrant Purchase Agreement by and among the Company and
the other persons and entities party thereto, dated as of July
17, 2009(4)
|
|
|
10
|
.14
|
|
Form of Senior Secured Promissory Note sold pursuant to that
Note and Warrant Purchase Agreement, dated as of July 17, 2009(4)
|
|
|
10
|
.15
|
|
Form of Stock Purchase Warrant sold pursuant to that Note and
Warrant Purchase Agreement, dated as of July 17, 2009(4)
|
|
|
10
|
.16
|
|
Stock Purchase Agreement by and among the Company and the other
persons and entities party thereto, dated as of September 25,
2009(6)
|
|
|
10
|
.17
|
|
Escrow Agreement by and among the Company, Fremont Bank and the
other persons and entities party thereto, dated as of September
25, 2009(6)
|
|
|
10
|
.18
|
|
Amendment No. 1 to License Agreement between Amgen Inc. and the
Company, dated as of October 16, 2009(6)
|
|
|
10
|
.19
|
|
Amendment No. 1 to Stock Purchase Agreement and Escrow Agreement
by and among the Company and the other persons and entities
party thereto, dated as of November 3, 2009(7)
|
|
|
10
|
.20
|
|
Amendment No. 2 to Stock Purchase Agreement and Escrow Agreement
by and among the Company and the other persons and entities
party thereto, dated as of December 11, 2009(3)
|
|
|
10
|
.21
|
|
Note Purchase Agreement by and among the Company and the other
persons and entities party thereto, dated as of December 11,
2009(3)
|
|
|
10
|
.22
|
|
Form of Senior Secured Promissory Note sold pursuant to that
certain Note Purchase Agreement, dated as of December 11, 2009(3)
|
|
|
10
|
.23
|
|
Form of Senior Secured Promissory Note to be exchanged pursuant
to that certain Note Purchase Agreement, dated as of December
11, 2009(3)
|
|
|
10
|
.24
|
|
Form of Stock Purchase Warrant issued pursuant to that certain
Note Purchase Agreement, dated as of December 11, 2009(3)
|
| |
|
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.25
|
|
Agreement between Eli Lilly and Company and the Company, dated
as of January 28, 2010 (amending Agreement between the parties,
dated as of September 15, 2009)(3)
|
|
|
10
|
.26
|
|
Agreement between Shionogi & Co., Ltd. and the Company,
dated as of February 24, 2010 (amending the Agreement between
the parties, dated as of September 7, 2009)(8)
|
|
|
10
|
.27
|
|
Amendment No. 3 to Stock Purchase Agreement and Escrow Agreement
by and among the Company and the other persons and entities
party thereto, dated as of February 24, 2010(8)
|
|
|
10
|
.28
|
|
Amendment No. 1 to Note Purchase Agreement by and between the
Company and the other persons and entities party thereto, dated
as of February 24, 2010(8)
|
|
|
# 10
|
.29
|
|
2010 Employee Stock Purchase Plan(9)
|
|
|
# 10
|
.30
|
|
Employment Agreement by and between the Company and James
Pennington, effective as of May 1, 2010(10)
|
|
|
# 10
|
.32
|
|
Form of Non-Qualified Stock Option Agreement for Company
Employees Under the Anthera Pharmaceuticals, Inc. 2010 Stock
Option and Incentive Plan(11)
|
|
|
# 10
|
.33
|
|
Form of Non-Qualified Stock Option Agreement for Non-Employee
Directors Under the Anthera Pharmaceuticals, Inc. 2010 Stock
Option and Incentive Plan(11)
|
|
|
# 10
|
.34
|
|
Form of Incentive Stock Option Agreement Under the Anthera
Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(11)
|
|
|
# 10
|
.35
|
|
Form of Restricted Stock Award Agreement Under the Anthera
Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(11)
|
|
|
# 10
|
.36
|
|
Restricted Stock Unit Award Agreement Under the Anthera
Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(12)
|
|
|
10
|
.37
|
|
Form of Securities Purchase Agreement, among the Company and the
purchasers thereto, dated September 20, 2010(13)
|
|
|
10
|
.38
|
|
Form of Registration Rights Agreement, between the Company and
the Holders thereto, dated September 20, 2010(14)
|
|
|
10
|
.39
|
|
Form of Warrant sold pursuant to that Securities Purchase
Agreement, among the Company and the purchasers thereto, dated
September 20, 2010(15)
|
|
|
10
|
.40
|
|
First Addendum to Sublease by and between the Company and
Millipore Corporation, as successor in interest to Guava
Technologies, dated as of September 24, 2010(16)
|
|
|
10
|
.41
|
|
Second Addendum to Sublease by and between the Company and
Millipore Corporation, as a successor in interest to Guava
Technologies, dated as of January 12, 2011.
|
|
|
10
|
.42
|
|
Amendment No. 1 to 2010 Employee Stock Purchase Plan
|
|
|
14
|
.1
|
|
Code of Ethics
|
|
|
21
|
.1
|
|
Subsidiary of Anthera Pharmaceuticals, Inc.(4)
|
|
|
23
|
.1
|
|
Consent of Deloitte & Touche LLP, independent registered
public accounting firm
|
|
|
24
|
.1
|
|
Power of Attorney (included on signature page hereto)
|
|
|
31
|
.1
|
|
Certification of Principal Executive Officer pursuant to Rule
13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of
1934, as amended.
|
|
|
31
|
.2
|
|
Certification of Principal Financial Officer pursuant to Rule
13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of
1934, as amended.
|
|
|
32
|
.1
|
|
Certification of Principal Executive Officer pursuant to Rule
13a-14(b) of the Securities Exchange Act of 1934, as amended,
and 18 U.S.C. Section 1350, as adopted pursuant to Section
906 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.2
|
|
Certification of Principal Financial Officer pursuant to Rule
13a-14(b) of the Securities Exchange Act of 1934, as amended,
and 18 U.S.C. Section 1350, as adopted pursuant Section 906
of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
+ |
|
Certain provisions of this Exhibit have been omitted pursuant to
a request for confidential treatment |
| |
|
# |
|
Indicates management contract or compensatory plan, contract or
agreement |
| |
|
(1) |
|
Filed as Exhibit 3.6 to the registrant’s Amendment
No. 4 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed February 3, 2010 and incorporated herein by reference. |
|
|
|
|
(2) |
|
Filed as Exhibit 3.7 to the registrant’s Amendment
No. 4 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed February 3, 2010 and incorporated herein by reference. |
| |
|
(3) |
|
Filed as the same numbered exhibit to the registrant’s
Amendment No. 3 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed January 29, 2010 and incorporated herein by reference. |
| |
|
(4) |
|
Filed as the same numbered exhibit to the registrant’s
Registration Statement on
Form S-1
(File
No. 333-161930),
filed September 15, 2009 and incorporated herein by
reference. |
| |
|
(5) |
|
Filed as Appendix A to the registrant’s Definitive
Proxy Statement on Schedule 14A filed June 8, 2010 and
incorporated herein by reference. |
| |
|
(6) |
|
Filed as the same numbered exhibit to the registrant’s
Amendment No. 1 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed October 19, 2009 and incorporated herein by reference. |
| |
|
(7) |
|
Filed as the same numbered exhibit to the registrant’s
Amendment No. 2 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed November 16, 2009 and incorporated herein by
reference. |
| |
|
(8) |
|
Filed as the same numbered exhibit to the registrant’s
Post-Effective Amendment No. 1 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed February 26, 2010 and incorporated herein by
reference. |
| |
|
(9) |
|
Filed as Appendix B to the registrant’s Definitive
Proxy Statement on Schedule 14A filed June 8, 2010 and
incorporated herein by reference. |
| |
|
(10) |
|
Filed as Exhibit 10.1 to the registrant’s Current
Report on
Form 8-K
filed June 4, 2010 and incorporated herein by reference |
| |
|
(11) |
|
Filed as Exhibit 10.2 to the registrant’s Amendment
No. 4 to Registration Statement on
Form S-1
(File
No. 333-161930),
filed February 3, 2010 and incorporated herein by reference. |
| |
|
(12) |
|
Filed as Exhibit 10.1 to the registrant’s Quarterly
Report on
Form 10-Q
filed May 14, 2010 and incorporated herein by reference. |
| |
|
(13) |
|
Filed as Exhibit 10.1 to the registrant’s Current
Report on
Form 8-K
filed September 22, 2010 and incorporated herein by
reference. |
| |
|
(14) |
|
Filed as Exhibit 10.2 to the registrant’s Current
Report on
Form 8-K
filed September 22, 2010 and incorporated herein by
reference. |
| |
|
(15) |
|
Filed as Exhibit 4.1 to the registrant’s Current
Report on
Form 8-K
filed September 22, 2010 and incorporated herein by
reference. |
| |
|
(16) |
|
Filed as the same numbered exhibit to the registrant’s
Registration Statement on
Form S-1
(File
No. 333-170099),
filed October 22, 2010 and incorporated herein by reference. |