Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the fiscal year ended December 31, 2015
OR
|
|
o
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from to
Commission file number: 001-34637
ANTHERA PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
|
Delaware
|
20-1852016
|
|
(State or Other Jurisdiction of
Incorporation or Organization)
|
(I.R.S. Employer
Identification No.)
|
|
25801 Industrial Boulevard, Suite B
Hayward, California
(Address of Principal Executive Offices)
|
94545 (Zip Code)
|
(510) 856-5600
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class
|
Name of Each Exchange on Which Registered
|
|
|
Common Stock, par value $0.001 per share
|
The NASDAQ Global Market
|
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by a check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by a check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this FORM 10-K or any amendment to this FORM 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer o
|
Accelerated filer x
|
Non-accelerated filer o
(Do not check if a smaller reporting company)
|
Smaller reporting company o
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).Yes o No x
The aggregate market value of the registrant’s common stock held by non-affiliates as of June 30, 2015 was approximately $276.0 million based upon the closing sales price of the registrant’s common stock as reported on the NASDAQ Global Market. Shares of common stock held by each executive officer and director and by each person who owns 10 percent or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for any other purpose.
As of February 29, 2016, the number of outstanding shares of the registrant’s common stock, par value $0.001 per share, was 40,004,037
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for the registrant’s 2015 Annual Meeting of Stockholders will be filed with the Securities and Exchange Commission within 120 days after the registrant’s fiscal year ended December 31, 2015 and are incorporated by reference in Part III of this report.
1
ANTHERA PHARMACEUTICALS, INC.
FORM 10-K FOR THE FISCAL YEAR ENDED
DECEMBER 31, 2015
|
Page
|
||
|
PART I
|
||
|
5
|
||
|
34
|
||
|
52
|
||
|
52
|
||
|
52
|
||
|
52
|
||
|
PART II
|
||
|
53
|
||
|
54
|
||
|
55
|
||
|
67
|
||
|
67
|
||
|
67
|
||
|
67
|
||
|
68
|
||
|
PART III
|
||
|
69
|
||
|
69
|
||
|
69
|
||
|
69
|
||
|
69
|
||
|
PART IV
|
||
|
70
|
||
|
92
|
||
Unless the context otherwise requires, we use the terms “Anthera Pharmaceuticals,” “Anthera,” “we,” “us,” “the Company” and “our” in this Annual Report on Form 10-K refer to Anthera Pharmaceuticals, Inc. and its subsidiaries. We use various trademarks, service marks and trade names in our business, including without limitation “Anthera Pharmaceuticals” and “Anthera.” This report also contains trademarks, services marks and trade names of other businesses that are the property of their respective holders.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, including the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements regarding future events and our future results that are subject to the safe harbors created under the Securities Act of 1933, as amended (the “Securities Act”), and the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements relate to future events or our future financial performance. We generally identify forward-looking statements by terminology such as “may,” “will,” “would,” “should,” “expects,” “plans,” “anticipates,” “could,” “intends,” “target,” “projects,” “contemplates,” “believes,” “estimates,” “predicts,” “assume,” “intend,” “potential,” “continue” or other similar words or the negative of these terms. These statements are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. The outcome of the events described in these forward-looking statements is subject to risks, uncertainties and other factors described in “Risk Factors” and elsewhere in this report. Accordingly, you should not place undue reliance upon these forward-looking statements. We cannot assure you that the events and circumstances reflected in the forward-looking statements will be achieved or occur, the timing of events and circumstances and actual results could differ materially from those projected in the forward looking statements. Forward-looking statements contained in this report include, but are not limited to, statements about:
|
|
•
|
the progress of, timing of and amount of expenses associated with our research, development and commercialization activities;
|
|
|
•
|
the timing, conduct and success of our clinical studies for our product candidates;
|
|
|
•
|
our ability to obtain U.S. and foreign regulatory approval for our product candidates and the ability of our product candidates to meet existing or future regulatory standards;
|
|
|
•
|
our expectations regarding federal, state and foreign regulatory requirements;
|
|
|
•
|
the therapeutic benefits and effectiveness of our product candidates;
|
|
|
•
|
the accuracy of our estimates of the size and characteristics of the markets that may be addressed by our product candidates;
|
|
|
•
|
our ability to manufacture sufficient amounts of our product candidates for clinical studies and products for commercialization activities;
|
|
|
•
|
our intention to seek to establish strategic collaborations or partnerships for the development or sale of our product candidates;
|
|
|
•
|
our expectations as to future financial performance, expense levels and liquidity sources;
|
|
|
•
|
the timing of commercializing our product candidates;
|
|
|
•
|
our ability to compete with other companies that are or may be developing or selling products that are competitive with our product candidates;
|
|
|
•
|
anticipated trends and challenges in our potential markets;
|
|
|
•
|
our ability to attract and retain key personnel; and
|
|
|
•
|
other factors discussed elsewhere in this report.
|
The forward-looking statements made in this report relate only to events as of the date on which the statements are made. We have included important factors in the cautionary statements included in this report, particularly in the section entitled “Risk Factors” that we believe could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments we may make. Except as required by law, we do not assume any intent to update any forward-looking statements after the date on which the statements are made, whether as a result of new information, future events or circumstances or otherwise.
PART I
Overview
Anthera Pharmaceuticals, Inc. is a biopharmaceutical company focused on advancing the development and commercialization of innovative medicines that benefit patients with unmet medical needs. We currently have two Phase 3 product candidates, liprotamase also known as SollpuraTM and blisibimod. We licensed liprotamase from Eli Lilly & Co (“Eli Lilly”) in July 2014. Sollpura is a novel non-porcine investigational Pancreatic Enzyme Replacement Therapy (“PERT”) intended for the treatment of patients with Exocrine Pancreatic Insufficiency (“EPI”), often seen in patients with cystic fibrosis and other conditions. We licensed blisibimod from Amgen, Inc. (“Amgen”) in December 2007. Blisibimod targets B-cell activating factor, or BAFF, which has been shown to be elevated in a variety of B-cell mediated autoimmune diseases, including systemic lupus erythematosus (“SLE”), or lupus, Immunoglobulin A nephropathy, or IgA nephropathy, lupus nephritis, and others.
Summary of Product Portfolio
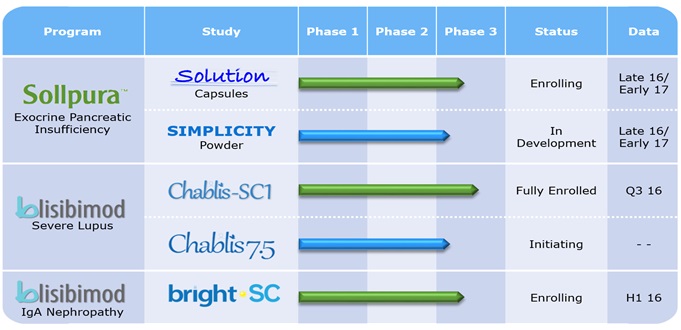
Summary of Product Development Programs
|
Product Candidate
|
Study Name
|
Development Stage
|
Indication
|
Next Milestone(s)
|
|
Sollpura
|
SOLUTION
|
Phase 3
|
EPI
|
Topline data towards end of 2016 or early 2017
|
|
Sollpura
|
SIMPLICITY
|
Phase 3
|
EPI
|
Initiate patient enrollment in first half of 2016
|
|
Blisibimod
|
CHABLIS-SC1
|
Phase 3
|
Lupus
|
Topline data in the second half of 2016
|
|
Blisibimod
|
CHABLIS-SC7.5
|
Phase 3
|
Lupus
|
Initiate patient enrollment first half of 2016
|
|
Blisibimod
|
BRIGHT-SC
|
Phase 2
|
IgA Nephropathy
|
Topline data first half of 2016
Follow-up data in second half of 2016
|
Our Pancreatic Enzyme Replacement Therapy Portfolio
Liprotamase is a novel non-porcine PERT that contains three biotechnology-derived digestive enzymes: a lipase, a protease and an amylase. Through enzyme cross-linking, the lipase enzyme in liprotamase is more stable than the porcine derived lipase in the low pH environment of the stomach and therefore liprotamase does not have an enteric polymer coating. Furthermore, since the three enzymes in liprotamase are biotechnology-derived, liprotamase does not contain porcine proteins or purines that may be associated with a risk of viral transmission or allergic reaction to proteins of porcine origin. The individual enzyme components of liprotamase are formulated at a fixed ratio of lipase, protease, and amylase. The liprotamase enzyme dose ratio was selected from preclinical efficacy studies conducted using a canine model of pancreatic insufficiency which demonstrated that the lipase enzyme in liprotamase was efficacious when administered at >500 units/kg per meal, and the protease doses >1000 units/kg per meal (Borowitz et al. 2006). This finding is further supported by the similar efficacy observed between liprotamase and Creon in pigs with surgically-induced pancreatic insufficiency.
Pancreatic enzyme replacement therapy is currently the mainstay of treatment for nutrient malabsorption in patients with digestive enzyme deficiencies known as EPI. EPI occurs when diseases such as cystic fibrosis, or CF, and chronic pancreatitis impede or destroy the exocrine function of the pancreas. Orally delivered porcine PERTs have been available for many years for the treatment of EPI. Unmet medical needs for the treatment of EPI remain. For example, as the porcine-derived proteins contained in the PERTs pass through the low pH environment of the stomach, enzyme activity rapidly diminishes and, as a consequence, large doses of porcine enzymes are often required. Since porcine lipase can become irreversibly deactivated in an acid environment, most products are enterically-coated. Patient-to-patient differences in the acidity of the intestine makes dissolution of enterically-coated products variable, and gives rise to alterations in the rate and extent to which enzymes are released from these products. Poor stability and variability in terms of potency and pharmaceutical properties have also been identified as important factors contributing to a poor response of some patients to PERTs.
Liprotamase is a novel PERT intended for the treatment of EPI. The exocrine pancreas is responsible for synthesis and secretion of digestive enzymes, including lipase, protease, and amylase. EPI occurs when diseases such as CF and chronic pancreatitis impede or destroy the exocrine function of the pancreas. A reduction in, or absence of the normally secreted pancreatic digestive enzymes, causes lipids, proteins, and carbohydrates to enter the distal gastrointestinal (“GI”) tract in unabsorbable forms, leading to GI pain and distention, maldigestion, and steatorrhea. Without appropriate therapy, patients with EPI may experience malnutrition, poor growth or weight loss, reduced quality of life, and, in severe cases, increased morbidity and early death.
We believe liprotamase is potentially the first soluble, stable and non-porcine derived enzyme product to offer a novel solution to patients who are unable to maintain appropriate nutritional health. Liprotamase’s chemical characteristics, unlike currently available PERTs, make it ideal for powder formulation as either a capsule, or sachet of powder for oral solution which can be conveniently administered in solution in a small volume of water.
Difference between Liprotamase and Current PERT

Our Phase 3 Development of Liprotamase in EPI
We initiated the SOLUTION study in the third quarter of 2015. SOLUTION is a Phase 3, randomized, open-label, assessor-blind, non-inferiority, active-comparator study evaluating the efficacy and safety of liprotamase in patients with cystic fibrosis-related exocrine pancreatic insufficiency. This pivotal study is intended to evaluate the non-inferiority of liprotamase compared with another commercially available PERT in a population enriched for PERT responders and will enroll up to 150 patients in the United States and Europe. Liprotamase will be supplied in capsule form for the SOLUTION study. A second, smaller Phase 3 study, SIMPLICITY, in younger pediatric subjects is planned for initiation in first half of 2016. The SIMPLICITY study will utilize sachets containing liprotamase powder for oral solution for ease of administration and will enroll approximately 60 patients. We believe the SOLUTION and SIMPLICITY studies may offer a number of potential opportunities for differentiation versus the currently marketed PERTs, including:
|
·
|
use of biotechnology-derived high-purity enzymes that are produced by fermentation processes rather than from mammalian organs which carry a label warning for viral transmission;
|
|
·
|
use of a novel chemically modified lipase drug substance that provides resistance to degradation;
|
|
·
|
a formulation containing an appropriate ratio of the three digestive enzymes (lipase, protease and amylase) to maximize relative efficacy while minimizing the potential for adverse events, such as fibrosing colonopathy;
|
|
·
|
a capsule formulation using known, safe, excipients that provides lower pill burden and good delivery performance. The pure, high-activity enzyme constituents, and absence of bulky enteric coating give rise to smaller, easy-to-swallow capsules with good disintegration once swallowed, and adequate storage stability compared with porcine PERTs of an equivalent lipase unit dose strength; and
|
|
·
|
a sachet formulation containing liprotamase power for oral solution which can be easily dissolved into water, and finally provides patients, especially young pediatric patients, with an easy-to-swallow dosing option.
|
Our BAFF Antagonism Portfolio
BAFF, or B-cell Activating Factor, (also known as B lymphocyte stimulator or BLyS), is a member of a tumor necrosis family of natural human proteins and is critical to the development, maintenance and survival of multiple B-cell lineages as well as plasma cells – all of which are critical to the human immune response. B-cells and plasma cells are a vital part of the human immune system, producing natural antibody responses to invading pathogens such as viruses, bacteria and other dangerous antigens. Abnormally high elevations of BAFF, B-cells and plasma cells have been associated with several autoimmune diseases, including lupus and IgA nephropathy. BAFF is primarily expressed by macrophages, monocytes and dendritic cells and interacts with three different receptors on B-cells and plasma cells including BAFF receptor, or BAFF-R, B-cell maturation antigen, or BCMA, and transmembrane activator and cyclophilin ligand interactor, or TACI. The potential role of BAFF inhibition and associated reductions in B-cell and plasma cell numbers in lupus and rheumatoid arthritis has been validated in multiple clinical studies with blisibimod and other BAFF antagonists.
Based on data from our Phase 2b clinical study, we have advanced the clinical development of our BAFF inhibitor, blisibimod, to exploit its potential clinical utility in a number of autoimmune diseases. Blisibimod, a peptibody directed against BAFF, was developed as an alternative to antibodies and is produced in Escherichia coli bacterial culture, as opposed to antibodies that are typically produced in mammalian cell culture. A peptibody is a novel fusion protein that is distinct from an antibody with several potential advantages, including ease of manufacture, potency and relatively small molecular weight. Blisibimod inhibits both soluble and membrane-bound BAFF.
In 2012, we completed the PEARL-SC Phase 2b clinical study, which evaluated the efficacy and safety of multiple doses of subcutaneous blisibimod versus placebo in patients with active and seropositive lupus. Lupus patients suffer from a chronic autoimmune disease, where an inappropriate or abnormal immune response often leads to severe skin rash, fatigue, joint pain, ulceration, major kidney complications, including proteinuria, and cardiovascular disease. Inhibition of BAFF is believed to reduce survival of B-cells and plasma cells and autoantibodies, leading to a reduction in severity of disease and resolution of lupus symptoms.
The development program for blisibimod is focused on evaluating the efficacy and safety of blisibimod in patients with lupus, and IgA nephropathy for which we believe current treatments are either inadequate or non-existent. Our current plan includes continuing the ongoing CHABLIS-SC1 registration clinical study in patients with active lupus and the BRIGHT-SC phase 2 clinical study in patients with IgA nephropathy, initiating CHABLIS-SC 7.5, and evaluating the potential of blisibimod in hematological diseases through clinical and nonclinical investigations. We have successfully manufactured blisibimod at launch-scale quantities. The blisibimod product is designed for at-home, self-administration and is presented as a pre-filled syringe for subcutaneous administration.
Our Phase 3 Development of Blisibimod in Systemic Lupus Erythematosus (Lupus)
In the third quarter of 2012 at an End of Phase 2 meeting with the United States Food and Drug Administration, or FDA, we presented the results of the PEARL-SC study and our plans for Phase 3 registration studies in patients with active lupus. As a result of this meeting we initiated patient enrollment in the initial Phase 3 CHABLIS-SC1 study in March 2013.
CHABLIS-SC1 is a multicenter, randomized, double-blind, placebo-controlled study designed to evaluate the efficacy, safety, tolerability and immunogenicity of blisibimod in patients with seropositive, clinically-active lupus (SELENA-SLEDAI ≥ 10) who require corticosteroid therapy in addition to standard-of-care for treatment of their disease. The study enrolled 442 SLE patients to receive either 200mg of blisibimod or placebo in addition to their standard-of-care medication for 52 weeks. Topline data is expected after the last enrolled patient completes 52 weeks of treatment around the third quarter of 2016. The primary endpoint of the CHABLIS-SC1 will be clinical improvement in the SLE Responder Index-6 (SRI-6) response at 52 weeks. Key secondary outcomes from the study, including SRI-8, reduction in the number of lupus flares and steroid use, are intended to provide more insight into blisibimod’s efficacy, and further differentiate blisibimod from currently available therapies. The demographics and disease characteristics of the patients enrolled in the CHABLIS-SC1 study are consistent with our goal to enroll patients with higher levels of lupus activity and positive biomarkers despite the stable use of corticosteroids. These characteristics were associated with improved outcomes in both our previous Phase 2 clinical study as well as in large Phase 3 studies conducted with other BAFF-inhibitors, belimumab (Benlysta) and tabalumab.
We believe the CHABLIS development program for blisibimod may offer a number of potential opportunities for differentiation versus the currently marketed BAFF antagonist and other novel B-cell directed therapies, including:
|
·
|
clinical differentiation:
|
|
Ø
|
potential for improved clinical response due to enriched patient selection;
|
|
Ø
|
a focus on requirement that patients in greater need of alternate therapy as defined by high disease activity and use of steroid therapy at time of randomization;
|
|
Ø
|
an improved clinical efficacy endpoint which requires a six-point reduction in the SELENA-SLEDAI;
|
|
Ø
|
earlier onset of clinical benefit by allowing earlier steroid taper and restriction of background medications;
|
|
Ø
|
potential to demonstrate reductions in lupus flares;
|
|
·
|
patient convenience: A convenient, at-home, patient-administered subcutaneous product;
|
|
·
|
mechanism of action: Blisibimod is able to inhibit the activity of both membrane-bound and soluble BAFF;
|
|
·
|
manufacturing: Blisibimod’s novel molecular structure, comprised of 2 identical peptide chains assembled into a covalent dimer, confers manufacturing benefits and lower cost of goods through a bacterial fermentation manufacturing process;
|
|
·
|
structure: Blisibimod’s 4 BAFF binding domains, compared to the typical 2 domains in a monoclonal antibody, domains, provides a 350-fold higher affinity for blisibimod (1 picomolar affinity) compared with the reported affinities for the anti-BAFF monoclonal antibody belimumab (Benlysta) ; and
|
|
·
|
fully human IgG1 Fc domain confers acceptable pharmacokinetic properties to support once-weekly dosing.
|
An independent Data Safety Monitoring Board (“DSMB”) meets regularly over the course of the study to assess patient safety. During these regular meetings, the DSMB reviews un-blinded safety data which include adverse events, suspected unexpected serious adverse reactions or SUSARs, deaths, laboratory data, and withdrawal data and compares trends between treatments. After the most recent scheduled meeting in November 2015, the DSMB recommended continuing the CHABLIS-SC1 and BRIGHT-SC clinical studies.
In February 2015, an interim analysis of CHABLIS-SC1 was conducted by an independent un-blinded statistician, who evaluated at a pre-specified time point, the proportion of responders to the systemic lupus erythematous SRI-6 responder index, and recommended the study to continue to completion as planned. This futility analysis was not intended to provide any rules for stopping for overwhelming efficacy, for a change in study sample size, or for an alteration of the study design. Rather, the analysis suggests that the observed data conforms with the assumptions upon which the trial was designed. The systemic lupus erythematosus response index is a recognized endpoint by the FDA for previously approved therapeutics. Prior to the interim analysis and on the advice of our Scientific Advisory Board who evaluated the published clinical data from other recent lupus studies with BAFF inhibitors, we modified the primary endpoint of CHABLIS-SC1 from a comparison of the proportion of responders to the SRI-8 responder index to a comparison of the proportion of responders to the SRI-6 responder index (previously a secondary endpoint of the study) as data, including emerging observations with tabalumab (Isenberg et al., Annual Meeting of the American College of Rheumatology, November 2014), suggest that SRI-6 responder rates are consistent and stable across multiple trials. Response rates to the SRI-8 responder index will remain a key secondary endpoint of the study. In addition to serving as a registration study for a potential lupus indication, observations in this study are intended to be included in marketing applications for blisibimod in IgA nephropathy and other indications.
We submitted a study protocol for our second lupus study, CHABLIS 7.5 (formerly named CHABLIS-SC2), to the FDA in the third quarter of 2015. The CHABLIS 7.5 study’s name emphasizes the intent to reduce background corticosteroid medication to ≤7.5mg prednisone in order to mitigate the risks associated with long-term steroid medication. This study will evaluate the effect of blisibimod on top of standard-of-care medication in patients with severe, seropositive SLE that is inadequately controlled with corticosteroids. Patient eligibility for this study is informed by responder traits identified in the Phase 2 trial with blisibimod as well as the large Phase 3 programs with other BAFF inhibitors, belimumab and tabalumab. We plan to initiate the CHABLIS 7.5 study in the first half of 2016. These two pivotal studies are anticipated to form the basis of submission for blisibimod as a treatment for active SLE that is not controlled by standard-of-care medication, including corticosteroids.
Our Phase 2 Development of Blisibimod for Immunoglobulin A Nephropathy
We partnered our development of IgA nephropathy with Zenyaku Kogyo Co., Ltd. (“Zenyaku”) through a collaborative and license agreement executed in December 2014, which agreement was terminated effective January 7, 2016. According to the National Organization of Rare Disease, IgA nephropathy, an orphan indication, is believed to affect approximately 130,000 people annually in the United States. In Asia, a higher reported prevalence is attributed to the routine urinalyses that are often performed for school children, and renal biopsies are performed for any patients with asymptomatic hematuria. According to the National Kidney and Urologic Diseases Information Clearinghouse, 25% of adults with IgA nephropathy eventually develop total kidney failure. IgA is a human antibody that helps the body fight infections. IgA nephropathy may occur when plasma B-cells express excessive amounts of abnormal IgA and subsequent immune complexes containing this immunogenic protein are deposited in the kidneys. These IgA deposits build up inside the small blood vessels of the kidney and as a result kidney glomeruli become inflamed and damaged, leading to leakage of blood and protein into urine. According to a recent publication in the New England Journal of Medicine (Wyatt & Julian, 2013), primary IgA nephropathy occurs at any age, most commonly with clinical onset in the second and third decades of life, and a large number of cases eventually progress to renal failure.
Similar to patients with other autoimmune diseases such as lupus, in IgA nephropathy, elevated levels of BAFF are associated with histological severity of disease in kidney tissue. In patients with IgA nephropathy, levels of BAFF are significantly higher than in healthy individuals. In IgA nephropathy, increased plasma B-cells express immunogenic IgA that forms immune complexes that deposit in renal tissue and lead to renal inflammation and damage that can progress to renal failure and end-stage renal disease. Significant reductions in plasma B-cells were observed in previous clinical studies of patients with lupus with another BAFF inhibitor antibody, belimumab. In our PEARL-SC Phase 2b study, significant reductions in total B-cells as well as significant improvements in proteinuria and increases in complement C3 were observed with blisibimod in lupus patients. We believe inhibition of BAFF may reduce proliferative B-cells and plasma B-cells, reduce serum levels of IgA and therefore reduce progressive renal damage in patients with IgA nephropathy.
The BRIGHT-SC study is a Phase 2 multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy, safety, tolerability and immunogenicity of blisibimod in IgA nephropathy. Enrollment criteria are biopsy-proven IgA nephropathy and proteinuria greater than one gram per 24 hours (1g/24hr). Patients must be receiving standard of care medication including angiotensin converting enzyme inhibitors and angiotensin receptor blockers. The BRIGHT-SC study was initiated in the second quarter of 2013. Patients enrolled in the BRIGHT-SC study receive 300mg weekly blisibimod or placebo subcutaneously during the first 8 weeks of therapy, the induction phase, followed by a minimum of 24 weeks of 200mg weekly blisibimod or placebo, the maintenance phase. The BRIGHT-SC clinical study has enrolled patients in Southeast Asia, the European Union (“EU”) and Eastern Europe.
In March 2015, an interim futility analysis of BRIGHT-SC study was conducted by an independent unblinded statistician, who evaluated several important biomarkers of renal disease in patients who had completed at least 8 weeks of treatment and recommended the study to continue to completion as planned. We plan to conduct an analysis of the clinical data in the first half of 2016 when more than 40 patients have completed 6 months of treatment, followed by a longer-term treatment data in the second half of 2016 when a majority of the patients have completed one year of treatment.
Market Opportunity
Sollpura (Liprotamase) for the treatment of Exocrine Pancreatic Insufficiency
According to IMS Health, EPI is a disease that affects an estimated 250,000 patients in the United States. The most common causes of EPI are chronic pancreatitis and cystic fibrosis, the former a longstanding inflammation of the pancreas altering the organ's normal structure and function that can result in malnutrition, heredity, or (in the western world especially), behavior (alcohol use and smoking), and the latter a recessive hereditary disease most common in Europeans and Ashkenazi Jews where the molecular culprit is an altered, CFTR-encoded chloride channel. In children, another common cause is Shwachman-Bodian-Diamond syndrome, a rare autosomal recessive genetic disorder resulting from mutation in the SBDS gene.
Blisibimod for the treatment of Systemic Lupus Erythematosus
Lupus is an autoimmune disorder that involves inflammation that causes swelling, pain and tissue damage throughout the body. Lupus can affect any part of the body, but especially the skin, heart, brain, lungs, joints and kidneys. The course of the disease is unpredictable, with periods of disease worsening, called flares. Although lupus may affect people of either sex, women are 10 times more likely to suffer from the disease than men, according to the Lupus Foundation of America. According to the Alliance for Lupus Research, it is estimated that up to 1.5 million people have lupus in the United States. Lupus Europe estimates that one in 750 women suffers from lupus in Europe and that lupus is a worldwide disease more common in some races than others. It is estimated that approximately 10% of people with lupus are treated in the United States. Based on the results from our PEARL-SC study, we believe patients in the moderate-to-severe lupus, approximately 30,000 to 40,000 people, are likely to benefit most from treatment with blisibimod. This is the population we are enrolling in our Phase 3 CHABLIS-SC studies.
Patients with active lupus may have a broad range of symptoms related to an abnormally active immune response in one or more organs. In the brain, lupus may cause seizures and other neurologic abnormalities. In the heart, lupus may cause heart failure or sudden death. Lung inflammation in the lung may cause shortness of breath, pleurisy and chest pain. Lupus may also cause swollen joints, arthritis, muscle aches, proteinuria, severe rash, oral ulcers and alopecia. In addition, patients with lupus nephritis may require kidney dialysis or eventual transplantation.
Although the cause of lupus is still not completely understood, B-cell activation and autoantibody production are known to be central to the process. Evidence has emerged that over-expression of BAFF correlates with disease severity in patients (Petri M., Stohl W., Chatham W., McCune W.J., Chevrier M., Ryel J., Recta V., Zhong J., Freimuth W., Association of Plasma B Lymphocyte Stimulator Levels and Disease Activity in Systemic Llupus Erythematosus. 2008, Arthritis Rheumatology, 58(8): 2453-9). In preclinical studies, transgenic mice created to over-express BAFF begin to exhibit symptoms similar to lupus. In addition, treatment of lupus-prone mice with blisibimod ameliorates the disease (Hsu H, Khare S.D., Lee F, Miner K, Hu YL, Stolina M., Hawkins N., Chen Q., Ho S.Y., Min H., Xiong F., Boone T., Zack D.J., A Novel Modality of BAFF-Specific Inhibitor AMG623 Peptibody Reduces B-cell Number and Improves Outcomes in Murine Models of Autoimmune Disease. 2012, Clinical and Experimental Rheumatology, 30(2): 197-201).
Blisibimod for the treatment of IgA Nephropathy
Immunoglobulin A (IgA) is a human antibody that plays a critical role in mucosal immunity, which is a portion of the immune system that provides protection to an organism’s various mucous membranes from invasion by infections. IgA nephropathy (also known as IgA nephritis or Berger's disease) is the most common form of primary glomerulonephritis (inflammation of the glomeruli of the kidney) throughout the world and a principal cause of end-stage renal disease. The prevalence of IgA nephropathy varies throughout the world, with the highest prevalence in Asia (Singapore, Japan and China), Australia, Finland and southern Europe (20 to 40% of all glomerulonephritis). IgA nephropathy occurs when too much of this protein, especially aberrant and immunogenic forms of IgA, is deposited in the kidneys. These immunogenic IgA immune complexes deposit inside the small blood vessels of the kidney and, as a result kidney glomeruli become inflamed and damaged, leading to leakage of blood and protein into urine. The classic presentation (in 40-50% of the cases) of signs and symptoms in patients with IgA nephropathy is episodic frank hematuria which usually starts within a day or two of a non-specific upper respiratory tract infection or (less commonly) gastrointestinal or urinary infection. All of these infections have in common the activation of mucosal defenses and hence IgA antibody production.
According to the National Organization for Rare Diseases, primary IgA nephropathy occurs at any age, most commonly with clinical onset in the second and third decades of life and a large number of cases eventually progress to renal failure. There is also a striking geographic variation in the prevalence of IgA nephropathy throughout the world. In the United States, IgA nephropathy is considered an orphan disease as it is believed to affect approximately 130,000 people annually. In Asia, routine urinalyses are performed for school children, and renal biopsies are performed for patients with asymptomatic hematuria and the reported prevalence of the disease is much higher. For example, in Japan, IgA nephropathy is estimated to affect over 350,000 people annually. According to the National Kidney and Urologic Diseases Information Clearinghouse, 25% of adults with IgA nephropathy eventually develop total kidney failure.
Manufacturing Strategy
Blisibimod
In December 2011, we completed the manufacturing site transfer from Amgen to our contract manufacturing organization (“CMO”), Fujifilm Diosynth Bioservices (“Fujifilm”). We also scaled up manufacturing from 300 liters up to 3,000 liters. Three batches of blisibimod produced under U.S. and EU good manufacturing procedures (“GMPs”), at the 3,000 liter scale passed all physical quality specifications and comparability assessments.
We have successfully manufactured blisibimod at launch scale volumes to support the CHABLIS-SC1, CHABLIS-7.5 and BRIGHT-SC studies. We are currently evaluating an auto injector strategy for use with the subcutaneous product presentation of blisibimod.
Liprotamase
Technology transfer to commercial scale active pharmaceutical ingredients, or API and drug product manufacturing CMOs was initiated in 2015 and is ongoing. The large scale fermentation and the associated down-stream processing for all three enzyme APIs are anticipated to be complete through process validation in 2016. In parallel, commercial scale capsule and sachet manufacturing are also being established at respective product CMOs.
Regulatory Strategy
Liprotamase
In 2013, our licensor, Eli Lilly, gained agreement from the FDA on the design of a Phase 3 trial that would provide adequate evaluation of efficacy and safety of liprotamase to respond to the FDA’s 2011 complete response letter. The Phase 3 study (SOLUTION) is a randomized, open-label, assessor-blind, non-inferiority, active-comparator study intended to evaluate the non-inferiority of liprotamase compared with another commercially available PERT. The SOUTION design also addresses the 2005 EMA protocol assistance comments, which were consistent with the FDA’s request for an active comparator trial.
Lupus
The Phase 3 program for blisibimod was presented to the European Medicines Agency (EMA, Scientific Advice) in the second quarter of 2012, to the FDA in the third quarter of 2012 (End-of-Phase 2 meeting) and to the FDA in a follow-up advice procedure in the third quarter of 2013 (Type C request to which the FDA provided written responses in lieu of a meeting). The Phase 3 CHABLIS-SC program incorporates feedback and advice obtained from both regulatory agencies. The Phase 3 studies (CHABLIS-SC1 and CHABLIS7.5) are planned to be multicenter, placebo-controlled, randomized, double-blind studies intended to evaluate the efficacy, safety, tolerability and immunogenicity of blisibimod in patients with clinically active lupus (SELENA-SLEDAI > 10) who require corticosteroid therapy in addition to standard of care for treatment of their disease. The design of the second Phase 3 study, CHABLIS-7.5, will be based on input advice from clinical experts as well as learnings from recently reported data from clinical trials in SLE. The CHABLIS-SC1 study has completed enrollment of 442 patients, and the CHABLIS 7.5 protocol was submitted to the FDA in the second half of 2015.
IgA Nephropathy
In September 2013, we met with the FDA who agreed to consider accepting proteinuria as an endpoint for Subpart E approval for blisibimod in IgA nephropathy. In April 2014, we met with the Japan Pharmaceuticals and Medical Devices Agency (“PMDA”) to discuss our registration program for blisibimod in IgA nephropathy. In this meeting we gained the PMDA’s agreement on the acceptability of proteinuria as the primary efficacy endpoint to support marketing approval in Japan and have amended the BRIGHT-SC study to include the specific data requirements of the PMDA. In December 2014 we met with the European Medicines Agency (“EMA”) as part of the scientific advice process for blisibimod. We reached an agreement with the EMA on the acceptability of proteinuria as the primary efficacy variable as well as the requirement for a single study to support a Conditional Marketing Authorization Application (“CMAA”) provided that confirmatory evidence from a second study, would be available post approval. The EMA also recommended the protocol to provide information on the required duration of treatment, duration of response and need for re-treatment. Data generated from the analysis of the BRIGHT-SC study in the first half of 2016, will be used to guide the final study design elements for the Phase 3 registration study protocol.
We continue to dose patients in our BRIGHT-SC clinical study for the treatment of patients with IgA nephropathy (“IgAN”), an orphan disease. Following results from previous studies in SLE, namely the PEARL-SC phase 2 clinical study, where patients, with baseline urinary protein excretion (“UPE”) of greater than one gram per 24 hours, demonstrated statistically significant reductions in urinary protein after 24 weeks, we initiated the BRIGHT-SC clinical study to evaluate the effects of blisibimod treatment in patients with IgAN. Following our receipt of a termination notice from Zenyaku and the subsequent expiration of the 120-day termination period on January 7, 2016, we elected to stop further enrollment in the BRIGHT-SC clinical study. We have enrolled 57 patients, 45 of which remained in the study. We plan to conduct a proof-of-concept efficacy analysis when substantially all patients have received a minimum of six months of therapy in Q2 2016. This analysis will examine the effects of blisibimod versus placebo in the proportion of qualifying patients who achieve a complete response (“CR”) and a partial response (“PR”) at six months. CR is defined as the proportion of patients with baseline UPE of greater than two grams per 24 hours, achieving UPE of less than one gram per 24 hours or a 50% reduction in UPE from baseline, and the proportion of patients with baseline UPE between one and two grams per 24 hours, achieving UPE of less than one gram per 24 hours and a 50% reduction from baseline. PR is defined as the proportion of patients achieving urinary protein excretion (“UPE”) of less than or equal to one gram per 24 hours. Based on our previous discussions with regulatory authorities, we believe an evaluation of the change in proteinuria could serve as a surrogate endpoint to support an accelerated or conditional approval in various key commercial geographies. Following the completion of the BRIGHT-SC study we intend to explore a Special Protocol Assessment from the US FDA to ensure final concurrence on the willingness to consider proteinuria as a surrogate endpoint, the timing for completion of enrollment of the post marketing confirmatory study and blisibimod’s eligibility to receive orphan designation.
Historical Clinical Studies by Licensor - Liprotamase
Liprotamase was studied from 2002 to 2009 in seven clinical trials, in which a total of 492 unique subjects received at least 1 dose of liprotamase. Three Phase 1 trials were conducted, 1 in healthy volunteers and 2 in subjects with EPI due to CF. Two short-term trials, the Phase 2 Study TC-2A, and the Phase 3 Study 726 evaluated the efficacy of liprotamase in subjects ≥7 years of age with EPI due to CF. Two long-term Phase 3 safety and tolerability trials were also conducted: Study 767 in subjects with EPI due to CF and Study 810 in subjects with EPI due to CP/pancreatectomy. Completed clinical trials demonstrated that dietary fat and nitrogen (protein) absorption are significantly increased in patients with cystic fibrosis and EPI who received liprotamase. In 2013, Eli Lilly gained agreement from the FDA on the design of a pivotal trial that would provide adequate evaluation of efficacy and safety.
The dose-ranging Phase 1 study TC-1B evaluated five dose levels across a 50-fold range, from 100-to-5000 units (U) per kg per meal in CF-EPI subjects. In this study, greater improvements in nutrients absorption, as measured using the percent change from baseline in the coefficient of fat absorption (CFA) and coefficient of nitrogen absorption (CNA), were observed at doses of 500 U per kg per meal and higher.
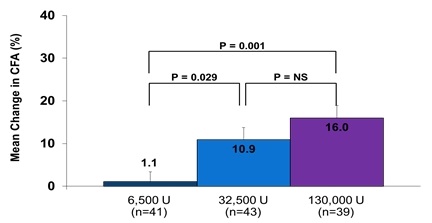
Study TC-2A, a Phase 2, randomized, double-blind, parallel group, dose-finding trial, was conducted in 125 pediatric and adult subjects with CF-related EPI who were treated with liprotamase in one of three dosing regimens containing 6,500 U, 32,500 U, and 130,000 U lipase administered per meal or snack. Observed mean CFAs at the end of study were 56.2%, 67.0%, and 69.7% in the 6,500, 32,500, and 130,000 U dose groups, respectively (one-way ANOVA p = 0.0032) with significant improvements in mean changes from baseline (one-way ANOVA p = 0.0005). Pairwise comparison of the CFA values showed that statistically greater improvements were observed at the higher doses of liprotamase compared with the lowest dose of 6,500 U (Figure 1). Similar improvements in the CNA, were observed with liprotamase at the two highest dose levels.
Figure 1: Mean Change from Baseline CFA (ITT) in the Phase II TC-2A Study
Study 726, a Phase 3, placebo-controlled, parallel design, multinational clinical, evaluated the effects of a single capsule of liprotamase (containing 32,500 U lipase, with protease and amylase in fixed ratios) or placebo administered with every meal or snack in subjects with cystic fibrosis-related EPI. Amongst the 138 subjects enrolled in this study, treatment with liprotamase resulted in a statistically significant improvement in the change from baseline in the CFA of 21.2% with liprotamase compared with 6.0% with placebo (p = 0.0011) (Figure 2). The median body weight of subjects in this trial was 50 kg, for which the corresponding liprotamase dose was 650 U lipase/kg/meal or snack.
Figure 2: Mean Change from Baseline CFA in Study 726

Two long-term studies were conducted to evaluate the safety and effects on nutritional status of liprotamase. Study 810 evaluated adult subjects with EPI due to CP or after pancreatectomy. Amongst the 214 subjects who were treated, an average dose of 5.5 capsules (containing 32,500 U lipase, with protease and amylase in fixed ratios) of liprotamase per day maintained nutritional status as assessed by serial measurement of height and weight, including age-appropriate growth and weight gain in children. Mean BMI z-scores for subjects in Study 767 were maintained over time on study (mean BMI z-score at baseline, Months 3, 6, and 12 were -0.503,-0.637, -0.688, and -0.655, respectively).
Blisibimod Development History
To date, four randomized, clinical studies have been conducted with blisibimod in patients with lupus: two Phase 1 dose-ranging studies in which a total of 104 patients were enrolled by our licensor, Amgen, a Phase 2b double-blind placebo-controlled dose-ranging clinical outcomes study (PEARL-SC) in which 547 subjects were enrolled, and a Phase 2 Open-Label Extension study (OLE) which evaluated the long-term safety of blisibimod in subjects previously enrolled in the Phase 2b PEARL-SC study, both conducted by us. As expected for a BAFF inhibitor, statistically significant reductions in total B-cells were observed in patients treated with blisibimod compared with placebo in all three of the placebo-controlled studies.
Phase 2b Study PEARL-SC Study in Patients with Lupus
Based on positive safety and pharmacodynamics results among 118 lupus patients in Amgen’s Phase 1a and 1b clinical studies, we conducted the PEARL-SC study, a Phase 2b randomized, double- blind, placebo-controlled study to evaluate the efficacy and safety of various subcutaneous doses of blisibimod in patients with seropositive lupus and active disease defined as SELENA-SLEDAI >6 at enrollment, which ran from mid-2010 to April 2012. The study enrolled 547 patients in 11 countries at 72 clinical sites. All patients completed the PEARL-SC study when the final enrolled patient completed six months of therapy.
In June and July of 2012 we announced results from the Phase 2b PEARL-SC study, which we believe support the initiation of a differentiated Phase 3 registration plan utilizing a 200mg weekly dose of blisibimod in patients with active lupus, despite the concomitant use of corticosteroids.
The primary endpoint of the PEARL-SC study was a clinical improvement at the lupus responder index, or SRI-5, at week 24 for the pooled blisibimod dose groups versus placebo groups. SRI-5 is defined as a five-point improvement in the SELENA-SLEDAI score, no new BILAG 1A or 2B scores, and no new increase in Physician’s Global Assessment of more than 0.3 points. The primary endpoint of this study was not met due to the lack of efficacy in the two lowest dose groups. However, the primary endpoint SRI-5 responder rates were numerically higher in subjects receiving blisibimod 200mg weekly (QW) compared with pooled placebo, from Week 16 (∆SRI 5 for blisibimod-placebo=8%, p= 0.14), through Week 24 (∆SRI 5=8.2%, p=0.15), reaching statistical significance at Week 20 (∆SRI 5=13.4%, p = 0.02). Treatment benefit was greater still when compared with the regimen-matched (QW) placebo. In pre-specified secondary analyses, benefit was observed at Week 24 with the 200mg QW group compared with matched placebo using modified SRI analyses in which responders attained SELENA-SLEDAI improvements of ≥7 or ≥8 (∆SRI 5 = 8.7% p=0.23; ∆SRI-7 =16.3% p=0.003; ∆SRI-8 =17.4% p=0.001). Blisibimod was also effective in a subgroup of patients with severe lupus with baseline SELENA- SLEDAI ≥10 and receiving corticosteroids at any dose (n=278) utilizing more stringent response thresholds (∆SRI-5=13.8%, p=0.18; ∆SRI-6=15.9%, p=0.12; ∆SRI-7=28.9%, p=0.002; ∆SRI- 8=31.1%, p<0.001, Figure 3 and Table 1). Based on SELENA SLEDAI definitions, 76 subjects (13.9%) had renal involvement at baseline and 54 subjects had proteinuria equivalent ≥1g/24hr. Compared with baseline, decreases in proteinuria were observed with blisibimod from Week 12 through Week 52 in the subgroup of subjects with proteinuria equivalent ≥1 g/24hr at enrollment (Figure 4). Similarly, decreases in proteinuria were observed in subjects with proteinuria equivalent ≥0.5 g/24hr through Week 44. Significantly greater reductions in proteinuria were also observed in a subgroup of subjects with active inflammation (i.e. low C3) and high anti-double-stranded DNA (anti-dsDNA).
Secondary endpoints included safety, improvements in variant forms of the SRI (e.g. defined by greater improvements in SELENA-SLEDAI), effects on clinical response in subgroup of patients with greater baseline disease severities, time to lupus disease flare, improvements in proteinuria, improvements in biomarkers of inflammation (e.g. anti-dsDNA, complement C3 and C4), and changes in B-cell counts. In June of 2012 we completed dosing in the PEARL-SC study.
Using a higher treatment threshold of an six-point reduction in the SELENA-SLEDAI, or an SRI-6 endpoint, in this enhanced subgroup population, the 200mg blisibimod treatment group demonstrated a 15.9% treatment difference compared to regimen-matched placebo (54.2% versus 46.4%, p=0.12). In this “severe” subgroup, separation of clinical response was evident as early as Week 8 and numerical improvements relative to placebo were maintained beyond Week 24. This “severe” subgroup is the population that we believe will benefit most from treatment with blisibimod, and is the population we are enrolling in our Phase 3 CHABLIS-SC studies. Significant and early decreases in proteinuria were also observed with blisibimod as early as Week 8 in subjects with baseline proteinuria equivalent ≥1g/24 hrs. Consistent pharmacodynamics responses were noted with blisibimod including reductions of B-cells, double-stranded-DNA antibodies and increases in complement C3 and C4. Blisibimod was safe and well-tolerated at all dose levels with no meaningful imbalances in serious adverse events. In addition to publications in earlier abstracts at key international rheumatology conferences, the efficacy and safety data from the PEARL-SC study were published by Dr. Richard Furie and colleagues (Ann Rheum Dis. 2015), and Dr. Scheinberg (International Journal of Clinical Rheumatology, 2014). Additional information and publications from the PEARL-SC study can be found at http://www.anthera.com/pipeline/clinical-studies/past-studies/pearl-sc.html. The information found on our website is not part of this or any other report we file or furnish with the SEC.
Figure 3: Systemic Lupus Erythematosus Responder Index-6 (SRI-6) in Subjects with Baseline SELENA-SLEDAI ≥10 and Receiving Steroids
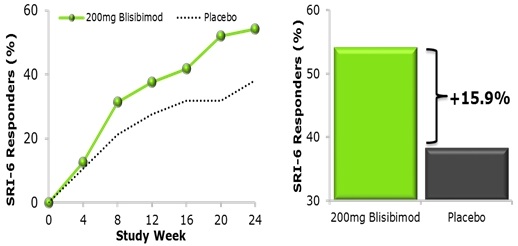
In the above figure, an SRI-6 responder achieved all of the following: ≥6 point improvement in SELENA-SLEDAI, and no new BILAG 1A or 2B organ domain scores, and no worsening (<0.3 increase) in Physician’s Global Assessment. In a subgroup analysis of patients with severe lupus disease (SELENA–SLEDAI≥10 and receiving steroid at baseline, n=278), the percent of subjects achieving the SRI-5 was higher in subjects receiving the highest dose of blisibimod (200mg QW) compared with placebo. The graph shows data for blisibimod (200mg QW) and regimen matched placebo administered subcutaneously for 24 weeks (Furie et al., Ann Rheum Dis. 2014).
Table 1: The Proportion of Subjects with Severe Lupus Who Attained the Criteria for the SLE Responder Index at Week 24
|
Pooled
Placebo
|
200mg QW
Placebo
|
200mg QW
blisibimod
|
Real Difference versus
Pooled Placebo
|
Real Difference
versus 200mg QW Placebo
|
|
|
Total Study N
|
N=269
|
N=92
|
N=92
|
||
|
Subgroup N
|
N=138
|
N=47
|
N=48
|
||
|
SRI-5
|
47.1%
N=65
|
40.4%
N=19
|
54.2%
N=26
|
+7.1%
p=0.48
|
+13.8%
p=0.18
|
|
SRI-5 + No Increase in Steroid Dose
|
43.5%
N=60
|
38.3%
N=18
|
52.1%
N=25
|
+8.6%
p=0.377
|
+13.8%
p=0.18
|
|
SRI-6 (Primary Endpoint of the CHABLIS –SC Clinical Study)
|
46.4%
N=64
|
38.3%
N=18
|
54.2%
N=26
|
+7.8%
p=0.43
|
+15.9%
p=0.12
|
|
SRI-7
|
28.3%
N=39
|
12.8%
N=6
|
41.7%
N=20
|
+13.4%
p=0.11
|
+28.9%
p = 0.002
|
|
SRI-8
|
26.1%
N=36
|
10.6%
N=5
|
41.7%
N=20
|
+15.6%
p=0.05
|
+31.1%
p < 0.001
|
Analyses were conducted in the subgroup of subjects with more severe lupus (SELENA-SLEDAI score ≥10) at enrollment. SLE Responder Index (SRI) is defined as patients who respond to treatment and achieve a reduction in SELENA-SLEDAI equal to or greater than the number indicated, no new BILAG 1A or 2B organ domain scores, and no increase in Physician's Global Assessment (PGA) of greater than 0.3 on a three point scale.
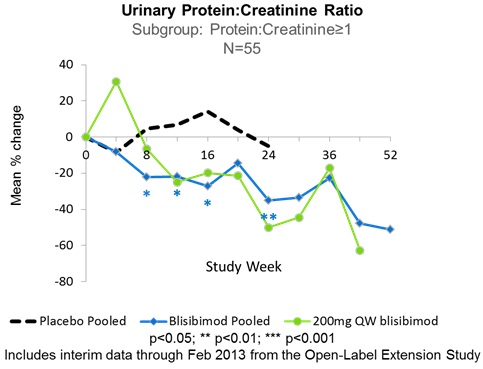
In subjects with baseline urinary protein excretion equivalent to 1-6g/24hrs (measured as urinary protein:creatinine ratio 1-6 mg/mg), treatment with blisibimod resulted in significantly greater reductions in proteinuria compared to placebo from Week 8 through Week 24. Furthermore, the observed treatment-related decreases in proteinuria resulted in near normalization of the proteinuria to ≤1g/24hrs in those subjects receiving blisibimod, and were durable through 52 weeks of continuous blisibimod dosing in the PEARL-SC and open-label extension studies. Figure 4 shows data from subgroups of patients defined by baseline proteinuria for blisibimod. Separately, data are shown for the subgroup of subjects with high inflammatory biomarker status at enrollment, defined by low C3 and high anti-dsDNA autoantibodies at baseline. Data are plotted for all pooled blisibimod dose levels as well as 200mg once-weekly and placebo administered subcutaneously for 24 weeks in the PEARL-SC trial. See data presented by Dr. Richard Furie at the European League Against Rheumatology Annual Conference, Madrid, Spain 2013 in Figure 4.
Figure 4: Effects of Blisibimod on Proteinuria in Subjects with SLE Enrolled in the PEARL-SC Study
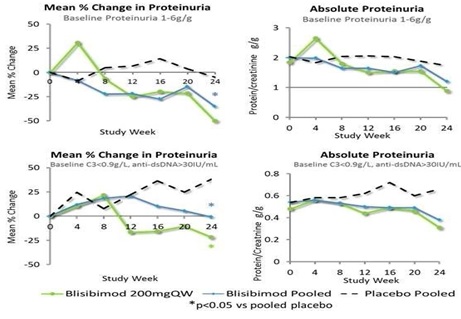
Effects of Blisibimod on Proteinuria in Subjects with SLE Enrolled in the PEARL-SC Study
In this study, statistically significant reductions in total B-cells were observed in patients treated with blisibimod compared with placebo due to its mechanism of BAFF inhibition. In addition, treatment with blisibimod was associated with significant improvements in lupus disease activity and lupus biomarkers, including anti-dsDNA antibodies, proteinuria, immunoglobulins IgG and IgM, and complement components C3 and C4. Lupus biomarker observations from the PEARL-SC trial are summarized in the next section, “Open-Label Extension Study in Patients Enrolled in PEARL-SC Study”, along with data from the open-label extension study.
Blisibimod was safe and well-tolerated at all dose levels evaluated in this study, with no meaningful imbalances in serious adverse events or infections compared with placebo. Amongst the adverse events reported, only injection site reactions were more frequent with blisibimod than placebo; these were never serious or severe.
The effects of blisibimod on patient-reported fatigue were evaluated in the PEARL-SC study using the Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue scale, and reported by Dr. Michelle Petri at the American College of Rheumatology Annual Conference, Boston MA, 2014. Improvements in patient self-reported fatigue were observed amongst subjects randomized to blisibimod based on the FACIT-Fatigue scale as shown in Figure 5, especially in the 200mg QW group (N=80) where favorable effects of blisibimod compared with placebo were observed as early as Week 8, and a mean 6.9-point improvement from baseline was reported at Week 24 compared to 4.4 with placebo (N=229). These effects meet the criteria for minimal clinically-important improvement difference of 5.9 defined by Goligher and colleagues (2008) for patients with SLE.
Figure 5: Effects of Blisibimod on Patient-Reported Fatigue in the PEARL-SC Study in Subjects with Lupus

Open-Label Extension Study in Patients Enrolled in PEARL-SC Study (“OLE”)
In order to evaluate longer-term safety of blisibimod, patients with lupus who completed the PEARL-SC study were able to participate in an OLE study in which they were treated with blisibimod. The OLE study was opened in the second quarter of 2011 and closed in the second quarter of 2013 after subjects had completed a minimum of one year of continuous therapy with blisibimod. Interim data from the combined PEARL-SC and OLE studies were presented at the 2013 Annual Scientific Meeting of the American College of Rheumatology and Association of Rheumatology Health Professionals (ACR/ARHP). In addition, a case report was published summarizing observations in a patient who was randomized to placebo in the PEARL-SC study then experienced a rapid improvement in serum cryoglobulins upon initiation of blisibimod therapy in the OLE study (Golima et al., Rheumatology, 2013).
The emerging data for the OLE study corroborate the effects of 24-week therapy with blisibimod observed in the PEARL-SC study. Specifically, the improvements in proteinuria observed in the subgroup of patients with abnormal proteinuria at enrollment were maintained over 52 weeks of continuous blisibimod therapy. In addition, the effects observed with blisibimod on peripheral B-cells, anti-dsDNA autoantibodies, and complement C3 and C4 and immunoglobulins IgG and IgM observed over 24 weeks of dosing in the PEARL-SC study were found to be durable over 52 weeks of therapy through the OLE study. Blisibimod was safe and well-tolerated at all dose levels through the PEARL-SC and OLE studies, with no associated increase in risk of severe infection as illustrated in the following table.
Table 2: Adverse Events and Serious Adverse Events Reported in the Phase 2 Placebo-controlled and OLE trials with Blisibimod in Patients with Lupus
|
PEARL-SC
|
Open-Label
|
||
|
Placebo
|
Blisibimod
|
Blisibimod
|
|
|
N=266
|
N=280
|
N=380
|
|
|
Overview (% incidence )
|
|||
|
AEs
|
85
|
82.5
|
81.3
|
|
Serious AEs
|
15.8
|
11.1
|
10.8
|
|
AEs Related to Study Drug
|
37.2
|
40
|
33.9
|
|
AEs Leading to Withdrawal
|
7.9
|
5.7
|
5.0
|
|
AEs Leading to Death
|
1.1
|
1.4
|
0
|
|
Severe Infection AEs
|
1.1
|
1.4
|
1.6
|
|
Severe Injection Site Reactions
|
0
|
0
|
0
|
|
Serious Adverse Events Occurring in >1 Subject, n(%)
|
|||
|
Herpes zoster
|
2 (0.8)
|
2 (0.7)
|
1(0.3)
|
|
Pneumonia
|
4 (1.5)
|
3 (1.1)
|
1 (0.3)
|
|
Urinary tract infections
|
2 (0.8)
|
2 (0.7)
|
0
|
|
SLE
|
3 (1.1)
|
2 (0.7)
|
0
|
|
Deep vein thrombosis
|
2 (0.8)
|
3 (1.1)
|
0
|
|
Cellulitis
|
1 (0.4)
|
0
|
4 (1.1)
|
|
Intervertebral disc
protrusion
|
0
|
0
|
2 (0.5)
|
|
Abortion spontaneous
|
0
|
1 (0.4)
|
4 (1.1)
|
|
Nephrolithiasis
|
1 (0.4)
|
0
|
2 (0.5)
|
The data from the Phase 2b (PEARL-SC) clinical program with blisibimod support the ongoing exploration of blisibimod’s efficacy and safety in the Phase 3 study in patients with lupus (CHABLIS-SC1) as well as the clinical study in patients with IgA nephropathy (BRIGHT-SC). Additional information for the OLE clinical studies can be found at http://www.anthera.com/pipeline/clinical-studies/past-studies/open-label-extension-study.html. The information found on our website is not part of this or any other report we file or furnish with the SEC.
The significant improvements in proteinuria in patients with lupus randomized to blisibimod compared with placebo observed in the PEARL-SC study were found to be durable through their continuing exposure to blisibimod in the OLE study.
Figure 6 shows data for blisibimod (all pooled dose levels as well as 200mg once-weekly) and placebo administered subcutaneously for 24 weeks in the PEARL-SC trial, and interim data for subjects who continued to receive blisibimod through the OLE trial are presented through Week 52 (presented by Dr. Furie at the American College of Rheumatology Annual Conference, San Diego, 2013).
Figure 6: Durable Effects of Blisibimod on Lupus
As expected for its mechanism of BAFF inhibition, blisibimod treatment was associated with significant reductions in the numbers of total B-cells, anti-dsDNA antibodies, as well as significant increases in complement components C3 and C4. In addition, significant reductions in immunoglobulins IgG and IgM were observed with blisibimod compared with placebo during the placebo-controlled PEARL-SC study. The effects of blisibimod on lupus biomarkers and serum immunoglobulins observed in the PEARL-SC study were found to be durable in subjects who continued to receive blisibimod through the OLE study. The effects on immunoglobulins were not associated with infection risk, nor were alterations in white blood cells, monocytes, or neutrophils associated with blisibimod. Figure 7 show data for blisibimod (all pooled dose levels as well as 200mg once-weekly) and placebo administered subcutaneously for 24 weeks in the PEARL-SC trial, and data for subjects who continued to receive blisibimod through the OLE trial are presented through Week 52 (interim data presented by Dr. Furie at the American College of Rheumatology Annual Conference, San Diego 2013).
Figure 7: Effects of Blisibimod on Lupus Biomarkers and Serum Immunoglobulins in the Phase 2 PEARL-SC and Open-Label Extension Trials

Historical Clinical Studies By Licensor - Blisibimod
Blisibimod, a peptibody directed against BAFF, was developed as an alternative to antibodies and is produced in Escherichia coli bacterial culture as opposed to antibodies that are typically produced in mammalian cell culture. Prior to our in-licensing of blisibimod, our licensor, Amgen, completed two Phase 1 clinical studies of blisibimod in lupus patients to evaluate the safety and pharmacokinetics of single and multiple doses of the drug using intravenous and subcutaneous formulations. The randomized, placebo-controlled, dose-escalation Phase 1a clinical study evaluated blisibimod or placebo as a single intravenous or subcutaneous therapy among 54 lupus patients. Intravenous doses included 1, 3 and 6mg/kg, and subcutaneous doses included 0.1, 0.3, 1 and 3mg/kg. The primary endpoint was to assess the safety and tolerability of single dose administrations of blisibimod. Secondary endpoints were designed to assess the plasma pharmacokinetic profile and immunogenicity of blisibimod. Results from this clinical study indicated the safety and tolerability of blisibimod administered as a single intravenous or subcutaneous dose were comparable to placebo. Single doses of blisibimod exhibited linear pharmacokinetics after both intravenous and subcutaneous administration. There were comparable adverse events between the blisibimod and placebo groups with no deaths reported. In addition, no neutralization antibodies were seen across all doses. The most common adverse events were nausea (15%), headache (10%), upper respiratory tract infection (10%) and diarrhea (8%). Observations from this trial were published in 2015 (Stohl and colleagues, Arthritis Research & Therapy).
Blisibimod was evaluated in a randomized, placebo-controlled, multi-dose Phase 1b clinical study as an intravenous or subcutaneous therapy among 64 lupus patients. The intravenous dose was 6mg/kg, and subcutaneous doses included 0.3, 1 and 3 mg/kg. Patients received their doses of blisibimod or placebo once-weekly for four weeks. The primary endpoint was to assess the safety and tolerability of multiple dose administrations of blisibimod. Secondary endpoints were designed to assess the plasma pharmacokinetic profile and immunogenicity of blisibimod after multiple doses. Blisibimod exhibited dose-proportional pharmacokinetics after repeat intravenous and subcutaneous administration. Further, a significant decrease in total B-cells as early as 15 days after beginning treatment, and total B-cell reduction (up to approximately 60-70% of baseline) reached its nadir after about 160 days of therapy. By six months after beginning treatment, the B-cell populations had returned to baseline levels. Further analyses of B-cell subsets found that naive B-cells and activated B-cells were significantly decreased while memory B-cells were transiently significantly increased following treatment with blisibimod. There were no deaths reported between the blisibimod and placebo groups. Few neutralization antibodies were seen, and all resolved in subsequent visits. Observations from this trial were published in 2015 (Stohl and colleagues, Arthritis Research & Therapy). Based on these results and published data from competitor studies, we conducted a Phase 2b clinical study evaluating blisibimod in lupus patients from the second half of 2010 to the third quarter of 2012.
Research and Development
Since our inception in 2004, we have focused primarily on developing our product candidates, which currently include liprotamase and blisibimod. We currently focus our clinical development and research efforts on liprotamase, as an enzyme replacement therapy for EPI, and blisibimod, which is being developed for autoimmune disease, including lupus and IgA nephropathy. In the years ended December 31, 2015, 2014 and 2013, we incurred $33.6 million, $21.8 million and $21.7 million, respectively, of research and development expense.
Our Strategy
Our objective is to develop and commercialize our product candidates to treat serious diseases associated with inflammation, including enzyme replacement therapies and autoimmune diseases. To achieve these objectives, we intend to initially focus on the following activities.
Advancing Clinical Development of Liprotamase and Blisibimod
We are advancing the development of liprotamase in a Phase 3 registration program in patients with cystic fibrosis-related EPI. We are also advancing the development of blisibimod to evaluate the broad potential clinical utility of BAFF antagonism. We have completed a Phase 2b clinical study with blisibimod in patients with lupus and plan to continue the advancement of blisibimod in our ongoing Phase 3 registration program in lupus and development in IgA nephropathy in 2016. We may opportunistically enter into collaborations with third parties for development of liprotamase and blisibimod, including securing corporate partners whose capabilities complement ours.
Developing Commercial Strategies Designed to Maximize Our Product Candidates’ Market Potential
Our product candidates are focused on highly-specialized physician segments, such as cystic fibrosis specialists, rheumatologists and nephrologists. We believe that we can build a small, focused sales force capable of marketing our products effectively in acute care and orphan indications.
Competition
Our industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions. Our primary competitors are described in further detail below, under “Approved Categories of Drugs”. We believe that key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement.
Many of our potential competitors have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than we may be in obtaining FDA approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any drug we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing our product candidates. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Finally, the development of new treatment methods for the diseases we are targeting could render our drugs non-competitive or obsolete.
Approved Categories of Drugs
Exocrine Pancreatic Insufficiency
There are currently several marketed products for EPI caused by cystic fibrosis, including Creon marketed by AbbVie, Inc., Pancreaze by Janssen Pharmaceuticals, Inc., Pertzye by Cornestone Therapeutics, Inc., and Ultresa and Zenpep by Aptalis Pharma US. Inc. Alcresta recently gained marketing approval for its product, Relizorb, which utilizes a digestive lipase enzyme, and is designed for use by adults on enteral tube feeding who have trouble breaking down and absorbing dietary fats. We are aware of one company with other products in development that are being tested for potential treatment of EPI caused by cystic fibrosis: Nordmark Arzneimittel GmbH & Co. KG’s compound, Burlulipase (also known as NM-BL), recently completed a Phase 1/Phase 2 study in patients with EPI.
Lupus
Human Genome Sciences, Inc. and partner GlaxoSmithKline plc (GSK) obtained FDA approval for Benlysta® (belimumab) in 2011 for the treatment of lupus. Benlysta®, the first novel therapy approved in the last 50 years, was acquired by GSK in July 2012. Other current therapies such as non-steroidal anti-inflammatory drugs, or NSAIDs, corticosteroids, anti-malarials and immunosuppressants are generally associated with limited efficacy or significant adverse events and broad immune suppression.
Emerging data from 2 large Phase 3 trials with the BAFF-targeted monoclonal antibody, tabalumab, were presented by Professor David Isenberg at the American College of Rheumatology Annual Conference, Boston, MA, November 2014. These data demonstrate that targeting BAFF remains an effective strategy for improving chronic disease activity in SLE. Several other agents under development target other B-cell related pathways or other inflammatory mechanisms for the treatment of lupus. These product candidates include atacicept, or TACI-Ig, from ZymoGenetics Inc. and anifrolumab from AstraZeneca, Other pathways targeting both B-cell and non-B-cell mechanisms are in earlier-phase clinical trials, such as Lupuzor from ImmuPharma plc; the anti-IFN gamma antibody AMG 811; toll-like receptor inhibitors; the anti-interleukin 6 antibodies sirukumab and PF-04236921; the phosphodiesterase 4 inhibitor CC-10004; the anti-CD74 monoclonal antibody milatuzumab; and inhibition of B7 related protein (B7RP-1) pathway with AMG 557. We believe that blisibimod may offer potential differentiation from these agents, including focused evaluation on the populations of lupus patients who were identified in prior trials with BAFF inhibitors to be responsive to this treatment modality; selective modulation and reduction of relevant B-cell types in lupus patients; the ability to inhibit the activity of both membrane-bound and soluble BAFF; the use of a bacterial expression platform which is expected to translate to lower manufacturing costs compared with therapeutic antibodies; and distinct patent protection based on a novel and proprietary technology developed and commercialized by Amgen.
PERT Drugs Approved or Under Late-Stage Clinical Development (other than liprotamase)
|
Compound
|
Stage
|
Company
|
Indications
|
Notes
|
|
Creon
|
Approved
|
Abbvie
|
EPI, CF and other
|
Ÿ Porcine, enteric coated
|
|
Zenpep
|
Approved
|
Actavis
|
EPI, CF and other
|
Ÿ Porcine, enteric coated
|
|
Ultrase
|
Approved
|
Actavis
|
EPI, CF and other
|
Ÿ Porcine, enteric coated
|
|
Pancreaze
|
Approved
|
Janssen/J&J
|
EPI, CF and other
|
Ÿ Porcine, enteric coated
|
|
Pertzye
|
Approved
|
Digestive care, Inc.
|
EPI, CF and other
|
Ÿ Porcine, enteric coated
|
|
Viokase
|
Approved
|
Actavis
|
EPI, chronic pancreatitis or pancreatectomy only
|
Ÿ Porcine, non-enteric coated
Ÿ In combination with proton pump inhibitor
|
|
Burlulipase
|
Phase 2
|
Nordmark Arzneimittel GmbH & Co. KG
|
EPI, CF
|
Ÿ Phase 2 completed Aug 2014
|
|
MS1819
|
Phase1/2a complete
|
Azzurx
|
EPI, CF and other
|
Ÿ Lipase only
|
Drugs Approved or Under Late-Stage Clinical Development in lupus (other than blisibimod)
|
Compound
|
Company
|
Stage
|
Indications
|
Notes
|
|
Benlysta®
(belimumab intravenous)
|
GlaxoSmithKline plc
|
Approved
|
Lupus (approved)
|
Ÿ 2 positive Phase 3 clinical trials
|
|
Phase 3
|
Other
|
Ÿ Additional registries or trials in other age
groups or populations ongoing
|
||
|
Belimumab (subcutaneous)
|
GlaxoSmithKline plc
|
Phase 3
|
Lupus
|
Ÿ Positive Phase 3 results reported
|
|
Atacicept (subcutaneous)
|
ZymoGenetics Inc./Merck Serono S.A.
|
Phase 2/3
|
Lupus
|
Ÿ Decreased incidence of flares in patients
with severe lupus
|
|
Phase 2b
|
Lupus
|
Ÿ Ongoing
|
||
|
Anifrolumab(subcutaneous)
|
AstraZeneca
|
Phase 2
|
Lupus
|
Ÿ Positive Phase 2 in lupus
|
|
Phase 3
|
Lupus
|
Ÿ 2 Phase 3 trials in lupus ongoing
|
||
|
Phase 3
|
Lupus nephritis
|
Ÿ 1 Phase 3 trial in lupus nephritis ongoing
|
Intellectual Property
Our policy is to pursue, maintain and defend patent rights, developed internally and licensed from third parties, to protect the technology, inventions and improvements that are commercially important to the development of our business. We also rely on trade secrets that may be important to the development of our business.
Our success will depend significantly on our ability to:
|
•
|
obtain and maintain patent and other proprietary protection for the technology, inventions and improvements we consider important to our business;
|
|
•
|
defend our patents;
|
|
•
|
preserve the confidentiality of our trade secrets; and
|
|
•
|
operate our business without infringing the patents and proprietary rights of third parties.
|
Liprotamase
As of the date of this report, our liprotomase portfolio is made up of exclusively licensed patents and patent applications from Eli Lilly, including:
|
•
|
Two issued U.S. patents;
|
|
•
|
Three issued European (EP) patents, each validated in one or more of Austria, Belgium, Bulgaria, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Great Britain, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Monaco, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, Switzerland and Turkey;
|
|
•
|
One pending EP patent application;
|
|
•
|
14 issued non-EP foreign patents in Australia, Canada, China, Hong Kong, India, Israel, Japan, Mexico, Russia, South Korea and Ukraine; and
|
|
•
|
Three pending non-EP foreign patent application.
|
We hold exclusive worldwide licenses from Eli Lilly to all of these patents and patent applications. The exclusively licensed U.S. patents are currently scheduled to expire in March 2025 and July 2028. Depending upon the timing, duration and specifics of FDA approval of liprotamase, one of these U.S. patents may be eligible for a patent term restoration of up to five years under Hatch-Waxman Act. See “—Regulatory Matters— Patent Term Restoration and Marketing Exclusivity.” This could extend the expiration date of the selected U.S. Patent to as late as March 2030 or July 2033, depending on which patent the term restoration is applied to. We intend to pursue pediatric exclusivity as well, which could add an additional six months to the patent term. The three exclusively licensed EP patents are currently scheduled to expire between February 2021 and October 2025. One of these patents may be eligible for a Supplementary Protection Certificate of up to five years, which could extend the expiration date to between February 2026 and October 2030.
Blisibimod
As of the date of this report, our blisibimod patent portfolio includes:
|
•
|
Four issued U.S. patents;
|
|
•
|
One pending U.S. non-provisional patent application;
|
|
•
|
Three issued European (EP) patents, each validated in one or more of Albania, Austria, Belgium, Cyprus, Denmark, Finland, France, Germany, Greece, Ireland, Italy, Latvia, Liechtenstein, Lithuania, Luxembourg, Monaco, the Netherlands, Portugal, Romania, Slovenia, Spain, Sweden, Switzerland, Turkey and the United Kingdom;
|
|
•
|
One pending EP patent application;
|
|
•
|
23 issued non-EP foreign patents in Australia, Bulgaria, Canada, China, the Czech Republic, Estonia, Eurasia (validated in all nine Eurasian countries), Hong Kong, Hungary, Israel, Japan, Mexico, New Zealand, Norway, the Philippines, Poland, Serbia, Singapore, Slovakia, South Korea and South Africa; and
|
|
•
|
Four pending non-EP foreign patent applications in Brazil, Hong Kong, Mexico, and Poland.
|
We hold exclusive worldwide licenses from Amgen to all of these patents and patent applications. In addition, we hold a non-exclusive worldwide license to one pending U.S. non-provisional patent application, one EP patent, one pending EP patent application, and over 50 non-EP foreign patents and pending patent applications relating to general peptibody compositions and formulations.
The four exclusively licensed U.S. patents are currently scheduled to expire in May 2022, March 2023 and November 2023. Depending upon the timing, duration and specifics of FDA approval of blisibimod, one of these U.S. patents (or another patent issuing from a related patent application) is expected to be eligible for a patent term restoration of up to five years under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. See “—Regulatory Matters— Patent Term Restoration and Marketing Exclusivity.” This could extend the expiration date of the U.S. Patent to as late as May 2027, March 2028 or November 2028, depending on which patent the term restoration is applied to. We intend to pursue pediatric exclusivity as well, which could add an additional six months to the patent term. The exclusively licensed EP patents are currently scheduled to expire in May 2022. One of these patents is expected to be eligible for a Supplementary Protection Certificate of up to five years, which could extend the expiration date to May 2027.
The U.S. patent system permits the filing of provisional and non-provisional patent applications. A non-provisional patent application is examined by the United States Patent and Trademark Office, or USPTO, and can mature into a patent once the USPTO determines that the claimed invention meets the standards for patentability. A provisional patent application is not examined, and automatically expires 12 months after its filing date. As a result, a provisional patent application cannot mature into a patent. The requirements for filing a provisional patent application are not as strict as those for filing a non-provisional patent application. Provisional applications are often used, among other things, to establish an early filing date for a subsequent non-provisional patent application.
The filing date of a non-provisional patent application is used by the USPTO to determine what information is prior art when it considers the patentability of a claimed invention. If certain requirements are satisfied, a non-provisional patent application can claim the benefit of the filing date of an earlier filed provisional patent application. As a result, the filing date accorded by the provisional patent application may remove information that otherwise could preclude the patentability of an invention.
We are aware of two families of third-party U.S. patents and pending foreign applications that contain broad claims related to BLyS or BAFF binding polypeptides. Based on our analyses, if these patents were asserted against us, we do not believe that blisibimod would be found to infringe any valid claim of these patents. If we were to challenge the validity of any issued U.S. patent in court, we would need to overcome the presumption of validity that attaches to every U.S. patent by presenting clear and convincing evidence as to the invalidity of the patent’s claims. There is no assurance that a court would find in our favor on questions of infringement or validity, and we could incur substantial costs in litigation if we are required to defend against patent suits brought by third parties or if we initiate these suits. If third-party patents are determined to be valid and construed to cover blisibimod, the development and commercialization of this program could be affected, subjecting us to potential liability for damages and possibly requiring us to obtain a license to continue marketing the affected product. Such a license may not be available on commercially acceptable terms, if at all.
Current License Agreements
Amgen
In December 2007, we entered into a license agreement with Amgen, which was amended in October 2009 and November 2014 (as amended, the “Amgen Agreement”), pursuant to which we obtained an exclusive worldwide license to certain technology and compounds relating to blisibimod, as well as a non-exclusive worldwide license to technology relating to certain peptibody compositions of matter and formulations.
Under the Amgen Agreement, we obtained exclusive rights under the licensed patents and know-how to research, develop, make, have made, use, sell, offer for sale and import pharmaceutical products containing blisibimod, as well as the right to grant sublicenses. The licensed patents included a specific set of previously filed U.S. and foreign patents and applications, as well as any applications filed after the execution date by Amgen and covering licensed know-how. During the period of the agreement, we are responsible for the filing, prosecution, defense and maintenance of all exclusively licensed blisibimod patents and applications. Amgen retains the right to review all documents relating to said filing, prosecution, defense and maintenance, and we are required to incorporate all reasonable comments or suggestions that Amgen makes with regard to these documents.
During the seven-year period after execution of the agreement, Amgen is prohibited from clinically developing or commercializing any BAFF peptibody. Similarly, we are prohibited during the term of the agreement from clinically developing or commercializing any molecule other than blisibimod that modulates BAFF as the primary intended therapeutic mechanism of action.
Pursuant to the terms of the Amgen Agreement, we have paid $6.0 million in license fees to Amgen for blisibimod. In addition, we are required to make various milestone payments upon the achievement of certain development, regulatory and commercial objectives, including payment upon commencement of the first Phase 3 clinical study for any blisibimod formulation in the United States, European Union or Japan. We are also required to pay up to $10.0 million upon achievement of certain pre-approval clinical development milestones and up to $23.0 million upon achievement of certain post-approval milestones. Furthermore, we are required to make tiered quarterly royalty payments on net sales, which increase as a percentage from the high single digits to the low double digits as net sales increase. Our royalty payment obligations for a particular product in a particular country begin on the date of the first commercial sale of the licensed product in that country, and end upon the later of 10 years from the date of first commercial sale in that country or the expiration date of the last valid claim of a licensed patent that covers the manufacture, use or sale, offer to sell or import of the product.
The Amgen Agreement will remain in effect until we elect to terminate, or until termination for material breach by either party or insolvency on our part. Under these terms, Amgen can terminate the agreement if we fail to meet our obligations, resulting in a loss of our exclusive rights to the licensed technology.
In connection with the collaborative arrangement with Zenyaku, we amended the Amgen Agreement in November 2014 to (i) adjust certain royalty and milestone payment obligations payable to Amgen in light of our collaboration with Zenyaku and (ii) provide that the sublicense granted by us to Zenyaku shall survive the termination of the Amgen Agreement. Under this amendment, we also agreed to grant Amgen that number of shares of our common stock equal $1.0 million divided by the volume weighted average price of our common stock for 20 trading days prior to issuance. We issued 420,751 shares of common stock to Amgen at $2.3767 per share on January 28, 2015 pursuant to a subscription agreement with Amgen, with the consideration paid by Amgen in the form of a waiver of a fee otherwise payable to Amgen under the Amgen Agreement.
Eli Lilly and Company
On July 11, 2014, we entered into a worldwide, exclusive license agreement with Eli Lilly (the “Lilly Agreement”), to develop and commercialize liprotamase, a Phase 3 novel investigational PERT for the treatment of patients with EPI, often seen in patients with cystic fibrosis and other conditions. Under the terms of the Lilly Agreement, we were not required to make any up-front payment but we are obligated to make milestone payments of up to $33.5 million for capsule products and $9.5 million for reformulated products upon the achievement of certain regulatory and commercial sales milestones, none of which have been achieved as of December 31, 2015. In addition, after sales of the licensed products exceed an aggregate of $100.0 million in the United States, we are obligated to pay tiered royalties on future net sales of products, ranging from the single digits to the mid-teens for products that are developed and approved as defined in the Lilly Agreement. Our royalty obligations as to a particular licensed product will be payable, on a licensed product-by-licensed product basis, for the longer of (a) the date of expiration of the last to expire valid claim within the licensed patents that covers the manufacture, use or sale, offer to sell, or import of such licensed product by us or a sublicense in such country, or (b) 12 years after the first commercial sale of the applicable licensed product in the applicable country.
Zenyaku Kogyo Co., Ltd.
On December 11, 2014, we entered into the Zenyaku Agreement with Zenyaku, pursuant to which we granted Zenyaku an exclusive license to certain patent rights, know-how and other intellectual property relating to blisibimod. In September 2015, Zenyaku provided us a notice of its intent to terminate the Zenyaku Agreement, effective January 7, 2016. The termination was “at will” and Zenyaku alleged no breach of the Zenyaku Agreement by us. We did not incur any termination penalties in connection with the early termination of the Zenyaku Agreement by Zenyaku. As of the date of termination, no patients had been enrolled in any blisibimod clinical studies in the Zenyaku territory and Zenyaku had not purchased any blisibimod product from us. As a result of the termination on January 7, 2016, we regained full worldwide rights for blisibimod.
Manufacturing and Supply
We currently rely on contract manufacturers to produce drug substances and drug products required for our clinical studies under current GMP with oversight by our internal managers. We plan to continue to rely upon contract manufacturers and, potentially, collaboration partners to manufacture commercial quantities of our product candidates if and when approved for marketing by the FDA. Should a supplier or a manufacturer on which we have relied to produce product candidates provide us with faulty products or such products are later recalled, we would likely experience significant delays and material additional costs.
Our contract manufacturers obtain the raw materials for the drug substances and drug products required for our clinical studies from a variety of sources. We believe that this will provide a sufficient supply of these raw materials and drug product to meet our needs for the foreseeable future. We do not have in place long term supply agreements with respect to all of the components of any of our pharmaceutical systems, however, and are subject to the risk that we may not be able to procure all required components in adequate quantities with acceptable quality, within acceptable time frames or at reasonable cost.
Our research and development activities involve the controlled use of potentially hazardous substances, including toxic chemical and biological materials. Accordingly, we are subject to federal, state and local laws governing the use, handling and disposal of these materials. We believe that our safety procedures for handling and disposing of these materials comply in all material respects with the standards prescribed by local, state and federal regulations.
Sales and Marketing
Given our stage of development, we have not developed a commercial organization or distribution capabilities. We expect that we would develop these capabilities once we receive Phase 3 data in contemplation of FDA approval and the commercial launch of our product candidates. In order to commercialize our product candidates, we must develop these capabilities internally or through collaboration with third parties. In selected therapeutic areas where we feel that any approved products can be commercialized by a specialty sales force that calls on a limited and focused group of physicians, we may seek to commercialize the product candidates alone. We also plan to seek commercialization partners for products in international markets.
We intend to build the commercial infrastructure necessary to bring our product candidates to market. In addition to a specialty sales force, sales management, internal sales support and an internal marketing group, we will need to establish capabilities to manage key accounts, such as managed care organizations, group-purchasing organizations, specialty pharmacies and government accounts. We may also choose to employ medical sales liaisons personnel to support the product.
Regulatory Matters
Government Regulation and Product Approval
Government authorities in the United States at the federal, state and local level and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, export and import of products such as those we are developing. Our product candidates must be approved by the FDA through the new drug application, or NDA, process, and our biological product candidate, blisibimod, must be approved by the FDA through the biologics license application, or BLA, process before they may legally be marketed in the United States.
United States Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations and biological products under both the FDCA and the Public Health Service Act, or the PHSA, and implementing regulations. The process of obtaining regulatory approvals and compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. The process required by the FDA before a drug or biological product may be marketed in the United States generally involves the following:
|
•
|
completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices regulations;
|
|
•
|
submission to the FDA of an IND, which must become effective before human clinical studies may begin;
|
|
•
|
performance of adequate and well-controlled human clinical studies according to Good Clinical Practices, or GCP, to establish the safety and efficacy of the proposed drug or biological product for its intended use;
|
|
•
|
submission to the FDA of an NDA for a new drug or BLA for a biological product;
|
|
•
|
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug or biological product is produced to assess compliance with cGMP; and
|
|
•
|
FDA review and approval of the NDA or BLA.
|
The testing and approval process requires substantial time, effort and financial resources and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. Once a pharmaceutical or biological product candidate is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies to assess its potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND. The sponsor will also include a protocol detailing, among other things, the objectives of the initial clinical study, dosing procedures, subject selection and exclusion criteria, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the initial clinical study lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical study on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin. Clinical holds may also be imposed by the FDA at any time before or during clinical studies due to safety concerns or non-compliance.
All clinical studies must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an institutional review board, or IRB, must review and approve the plan for any clinical study before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the studies are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical study and the consent form that must be provided to each clinical study subject or to his or her legal representative and must monitor the clinical study until completed. Each new clinical protocol and any amendments to the protocol must be submitted to the FDA for review, and to the IRBs for approval.
Human clinical studies are typically conducted in three sequential phases that may overlap or be combined:
|
•
|
Phase 1. The product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
|
|
•
|
Phase 2. Involves studies in a limited patient population to identify possible adverse effects and safety risks to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage and schedule.
|
|
•
|
Phase 3. Clinical studies are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling.
|
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA. Safety reports must be submitted to the FDA and the investigators within 15 days after the sponsor determines the information qualifies for reporting for serious and unexpected suspected adverse reactions, findings from other studies or animal or in vitro testing that suggest a significant risk in humans, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator’s brochure. A sponsor also must notify FDA of any unexpected fatal or life-threatening suspected adverse reaction within 7 days after the sponsor’s receipt of the information. Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA , a data safety monitoring board, or the sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical study at its institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the drug or biological product has been associated with unexpected serious harm to patients.
Concurrent with clinical studies, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidates and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidates do not undergo unacceptable deterioration over their shelf life.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical studies, along with descriptions of the manufacturing process, analytical tests conducted on the drug or biological product, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA for a new drug or BLA for a biological product, requesting approval to market the product. The submission of an NDA or BLA is subject to the payment of a substantial user fee; a waiver of such fee may be obtained under certain limited circumstances.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, which was reauthorized under the Food and Drug Administration Amendments Act of 2007, an NDA or BLA or supplement to an NDA or BLA must contain data to assess the safety and effectiveness of the drug or biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any drug or biological product for an indication for which orphan designation has been granted.
The FDA reviews all NDAs and BLAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept a NDA or BLA for filing. In this event, the NDA or BLA must be re-submitted with the additional information. The re-submitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP- compliant to assure and preserve the product’s identity, strength, quality and purity. The FDA reviews a BLA to determine, among other things, whether the product is safe, has an acceptable purity profile and is adequately potent, and whether its manufacturing meets standards designed to assure the product’s continued identity, sterility, safety, purity and potency. Before approving an NDA or BLA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may refer the NDA or BLA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. An advisory committee is a panel of experts who provide advice and recommendations when requested by the FDA on matters of importance that come before the agency. The FDA is not bound by the recommendation of an advisory committee but it generally follows such recommendations.
The approval process is lengthy and difficult and the FDA may refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical studies are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA or BLA in its present form. The complete response letter usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical studies. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical studies designed to further assess a drug or biological product’s safety and effectiveness after NDA or BLA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permit a patent restoration term of up to five years as compensation for patent term lost during the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent terms for some of our currently owned or licensed patents to add patent life beyond their current expiration dates, depending on the expected length of the clinical studies and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain competitor applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical studies necessary to demonstrate safety and effectiveness. HR 3590 provides 12 years of data exclusivity for innovator biologics. During this exclusivity period, competitors are barred from relying on the innovator’s safety and efficacy data to gain FDA approval. Therefore, a competitor seeking to obtain marketing approval during this exclusivity period would be required to conduct its own preclinical and clinical studies.
Pediatric exclusivity is another type of exclusivity in the United States. Pediatric exclusivity, if granted, adds an additional six months to an existing exclusivity or statutory delay in approval resulting from a patent certification. This six-month exclusivity, which runs from the end of other exclusivity protection or patent delay, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA or BLA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication, except in very limited circumstances, for seven years. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product, but for a different indication for which the orphan product has exclusivity. Orphan product exclusivity could also block the approval of one of our products for seven years if a competitor obtains approval of the same drug or biological product as defined by the FDA or if any of our product candidates is determined to be contained within the competitor’s product for the same indication or disease. If a drug or biological product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity.
The FDA also administers a clinical research grants program, whereby researchers may compete for funding to conduct clinical studies to support the approval of drugs, biologics, medical devices and medical foods for rare diseases and conditions. A product does not have to be designated as an orphan product to be eligible for the grant program. An application for an orphan grant should propose one discrete clinical study to facilitate FDA approval of the product for a rare disease or condition. The clinical study may address an unapproved new product or an unapproved new use for a product already on the market.
Expedited Development and Review Programs
The FDA has a fast track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. For a fast track product, the FDA may consider for review on a rolling basis sections of the NDA or BLA before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA or BLA, the FDA agrees to accept sections of the NDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA or BLA.
A fast track product may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A fast track product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a fast track product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well- controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies. Fast track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
The FDA Safety and Innovation Act, or FDASIA includes a provision that allows sponsors to request that their drug be designated as a Breakthrough Therapy. The goal of this program is to expedite the development and review of a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition if preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA actions to expedite the development of a Breakthrough Therapy include (a) holding meetings with the sponsor and the review team throughout the development of the drug, (b) providing timely advice to and interactive communication with the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable, (c) involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review, (d) assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor and (e) taking steps to ensure that the design of the clinical trials is as efficient as practicable, when scientifically appropriate, such as by minimizing the number of patients exposed to a potentially less efficacious treatment.
Post-Approval Requirements
Any drug or biological products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record- keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Drugs and biological products may be promoted only for the approved indications and in accordance with the provisions of the approved label. Further, manufacturers of drugs and biological products must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Drug and biological product manufacturers and other entities involved in the manufacturing and distribution of approved drugs or biological products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug or biological product. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release.
Manufacturers of biological products must also report to the FDA any deviations from cGMP that may affect the safety, purity or potency of a distributed product; or any unexpected or unforeseeable event that may affect the safety, purity or potency of a distributed product. The regulations also require investigation and correction of any deviations from cGMP and impose documentation requirements.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution or may require substantial resources to correct.
The FDA may withdraw a product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, warning letters, holds on clinical studies, product recalls or seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or civil or criminal penalties.
In addition, from time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA.
In addition to new legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical studies and commercial sales and distribution of our products to the extent we choose to sell any products outside of the United States. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical studies or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical studies, product licensing, pricing and reimbursement vary greatly from country to country.
In the European Union, our products are subject to extensive regulatory requirements, which provide, among other things, that no medicinal product may be placed on the market of a European Union member state unless a marketing authorization has been issued by the European Medicines Agency or a national competent authority. European Union member states require regulatory clearance by both the national competent authority and a favorable ethics committee opinion prior to the commencement of a clinical study.
Under the European Union regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The centralized procedure is compulsory for medicines produced by certain biotechnological processes, products with a new active substance indicated for the treatment of certain diseases such as neurodegenerative disorder or diabetes and products designated as orphan medicinal products, and optional for those products which are highly innovative or for which a centralized process is in the interest of patients. The decentralized procedure of approval provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state, known as the reference member state. Under the decentralized approval procedure, an applicant submits an application, or dossier, and related materials (draft summary of product characteristics, draft labeling and package leaflet) to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states.
Reimbursement
Sales of pharmaceutical products depend significantly on the availability of third-party reimbursement. Third-party payors include government health administrative authorities, including, at the federal and state level, managed care providers, private health insurers and other organizations. We anticipate third-party payors will provide reimbursement for our product. However, these third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved health care products. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. It is time consuming and expensive for us to seek reimbursement from third-party payors. Reimbursement may not be available or sufficient to allow us to sell our product on a competitive and profitable basis.
In addition, the U.S. Congress and state legislatures from time to time propose and adopt initiatives aimed at cost containment, which could impact our ability to sell our products profitably. For example, in March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, and the associated reconciliation bill, which we refer to collectively as the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Effective October 1, 2010, the Health Care Reform Law revises the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states once the provision is effective. Further, beginning in 2011, the new law imposes a significant annual fee on companies that manufacture or import certain branded prescription drug products and biologic agents. Substantial new provisions affecting compliance also have been enacted, which may require us to modify our business practices with healthcare practitioners. We will not know the full effects of the Health Care Reform Law until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effect of the Health Care Reform Law, the new law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and also may increase our regulatory burdens and operating costs.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries, and included a major expansion of the prescription drug benefit under a new Medicare Part D. Medicare Part D went into effect on January 1, 2006. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee.
Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
There are also laws that govern a company’s eligibility to participate in Medicare and Medicaid reimbursements. For example, a company may be debarred from participation if it is found to have violated federal anti-kickback laws, which could have a significant effect on a company’s ability to operate its business.
The cost of pharmaceuticals continues to generate substantial governmental and third-party payor interest. We expect that the pharmaceutical industry will experience pricing pressures due to the trend toward managed healthcare, the increasing influence of managed care organizations, and additional legislative proposals. Indeed, we expect that there will continue to be a number of federal and state proposals to implement governmental pricing controls and limit the growth of healthcare costs, including the cost of prescription drugs. At the present time, Medicare is prohibited from negotiating directly with pharmaceutical companies for drugs. However, Congress is considering passing legislation that would lift the ban on federal negotiations. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could harm our business, financial condition and results of operations.
Some third-party payors also require pre-approval of coverage for new or innovative drug therapies before they will reimburse healthcare providers that use such therapies. While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a material adverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. In some countries, pricing negotiations with governmental authorities can take six to 12 months or longer after the receipt of marketing approval. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidates to other available therapies. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for our product.
Employees
As of December 31, 2015, we had 25 employees. All of our employees are engaged in administration, finance, clinical, regulatory and business development functions. None of our employees are represented by a labor union, and we believe that our relations with our employees are good.
Other Available Information
We are subject to the information requirements of the Exchange Act. Therefore, we file periodic reports, proxy statements and other information with the Securities and Exchange Commission (SEC), which may be obtained by visiting the Public Reference Room of the SEC at 100 F Street, NE, Washington, D.C. 20549 or by calling the SEC at 1-800-SEC-0330. In addition, the SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically.
We were incorporated in Delaware in 2004. The mailing address of our headquarters is 25801 Industrial Blvd, Hayward, CA 94545, and our telephone number at that location is 510-856-5600. Our website is www.anthera.com. Through a link on the “Investors” section of our website (under “SEC Filings” in the “Financial Information” section), we make available, free of charge, the following filings as soon as reasonably practicable after they are electronically filed with or furnished to the SEC: our Annual Reports on Form 10-K; Quarterly Reports on Form 10-Q; Current Reports on Form 8-K; and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act. Information contained on, or that can be accessed through, our website does not constitute part of this Annual Report on Form 10-K.
You should carefully consider the risks described below, together with the other information contained in this Annual Report on Form 10-K, including the consolidated financial statements and the related notes that appear at the end of this report. If any of the following risks occur, our business, financial condition, results of operations and future growth prospects would likely be materially and adversely affected.
An investment in our securities involves a high degree of risk. We operate in a dynamic and rapidly changing industry that involves numerous risks and uncertainties. The risks and uncertainties described below are not the only ones we face. Other risks and uncertainties, including those that we do not currently consider material, may impair our business. If any of the risks discussed below actually occur, our business, financial condition, operating results or cash flows could be materially adversely affected. This could cause the value of our securities to decline, and you may lose all or part of your investment.
Risks Related to Our Financial Condition and Capital Requirements
We have incurred significant losses since our inception and anticipate that we will incur continued significant losses for the foreseeable future.
We are a clinical-stage biotechnology company with two clinical assets in the late stage of development. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect or an acceptable safety profile, gain regulatory approval and become commercially viable. We have no products approved for commercial sale and have not generated any revenue from product sales to date and we continue to incur significant research and development and other expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in each period since our inception in 2004. As of December 31, 2015, we had an accumulated deficit of $352.0 million. Substantially all of our losses resulted from costs incurred in connection with our product development programs and from general and administrative costs associated with our operations.
We expect to incur additional losses over the next several years, and these losses may increase if we cannot generate revenues. Our historical losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. In addition, if we obtain regulatory approval for our product candidates, we may incur significant sales, marketing, in-licensing and outsourced manufacturing expenses as well as continued product development expenses. As a result, we expect to continue to incur significant and increasing losses for the foreseeable future.
We have never generated any product revenue and may never be profitable.
Our ability to generate product revenue and achieve profitability depends on our ability, alone or with collaborators, to successfully complete the development of our product candidates, conduct clinical studies in patients, obtain the necessary regulatory approvals for our product candidates and commercialize any approved products. Although we recognized collaborative revenues from our license agreement with Zenyaku in 2015, we cannot guarantee we will recognize collaborative revenue in the future. For the year ended December 31, 2015, we recognized collaborative revenue related to the amortization of upfront license fee and reimbursement for our internal full time equivalent (“FTE”) employee and certain research and development expenses by Zenyaku. In September 2015, Zenyaku provided us a notice of its intent to terminate the Zenyaku Agreement, effective January 7, 2016. The termination was “at will” and Zenyaku alleged no breach of the Zenyaku Agreement by us. As a result of an early termination the Zenyaku Agreement, we do not expect to recognize revenues under the Zenyaku Agreement beyond January 2016. We have not generated any revenue from our development-stage product candidates, and we do not know when, or if, we will generate any revenue. The commercial success of our development-stage product candidates will depend on a number of factors, including, but not limited to, our ability to:
|
·
|
obtain favorable results for and advance the development of liprotamase, our product candidate for the treatment of patients with low digestive enzyme levels, or EPI, and potentially other diseases;
|
|
·
|
obtain favorable results for and advance the development of blisibimod, our product candidate for the treatment of B-cell mediated autoimmune diseases, including successfully launching and completing clinical studies in patients with systemic lupus erythematosus, or lupus, IgA nephropathy, or other indications related to the development of blisibimod;
|
|
·
|
obtain regulatory approval for liprotamase and blisibimod;
|
|
·
|
if regulatory approvals are obtained, begin the commercial manufacturing of our product candidates with third-party manufacturers;
|
|
·
|
launch commercial sales and effectively market our product candidates, either independently or in strategic collaborations with third parties; and
|
|
·
|
achieve broad market acceptance of our product candidates in the medical community and with third-party payors.
|
Our product candidates are subject to the risks of failure inherent in the development of therapeutics based on new technologies. Our product candidates could fail in clinical studies if we are unable to demonstrate that it is effective or if it causes unacceptable adverse effects in the patients we treat. Failure of our product candidates in clinical studies would have a material adverse effect on our ability to generate revenue or become profitable. If we are not successful in achieving regulatory approval for our product candidates or are significantly delayed in doing so, our business will be materially harmed.
We will need substantial additional capital in the future to fund our operations. If additional capital is not available, we will have to delay, reduce or cease operations.
We will need to raise substantial additional capital to fund our operations and to develop our product candidates. Our future capital requirements will depend on many factors including:
|
·
|
the scope, size, rate of progress, results and costs of our clinical studies and other development activities for our product candidates;
|
|
·
|
manufacturing campaign for liprotamase and blisibimod clinical materials, including formulation development and product enhancement;
|
|
·
|
non-clinical activities that we may pursue parallel to our clinical studies;
|
|
·
|
the cost, timing and outcomes of regulatory proceedings;
|
|
·
|
payments received under any strategic collaborations;
|
|
·
|
the filing, prosecution and enforcement of patent claims;
|
|
·
|
the costs associated with commercializing our product candidates if they receive regulatory approval, including the cost and timing of developing sales and marketing capabilities, or entering into strategic collaboration with others relating to the commercialization of our product candidates; and
|
|
·
|
revenues received from approved products, if any, in the future
|
As of the date of this report, we anticipate that our existing cash and access to additional capital through an equity purchase agreement and an at-the-market sales agreement are sufficient to fund our near term liquidity needs for at least the next 12 months. Changing circumstances may cause us to consume capital significantly faster than we currently anticipate. Additional financing may not be available when we need it or may not be available on terms that are favorable to us. If adequate funds are not available to us on a timely basis, or at all, we may be required to:
|
·
|
terminate, reduce or delay clinical studies or other development activities for our product candidates; or;
|
|
·
|
terminate, reduce or delay our (i) establishment of sales and marketing capabilities, (ii) pursuit of strategic collaborations with others relating to the sales, marketing and commercialization of our product candidates or (iii) other activities that may be necessary to commercialize our product candidates, if approved for sale.
|
The timing of the milestone and royalty payments we are required to make to our licensors is uncertain and could adversely affect our cash flows and results of operations.
In December 2007, we entered into the Amgen Agreement, pursuant to which we obtained an exclusive worldwide license to certain technology and compounds relating to blisibimod. Pursuant to the Amgen Agreement, we are required to make various milestone payments upon our achievement of certain development, regulatory and commercial objectives for any blisibimod formulation. We are required to pay up to $10.0 million upon achievement of certain pre-approval clinical development milestones and up to $23.0 million upon achievement of certain post-approval milestones. We are also required to make tiered quarterly royalty payments on net sales, which increase as a percentage from the high single digits to the low teens as net sales increase.
In July 2014, we entered into the Lilly Agreement, pursuant to which we obtained an exclusive worldwide license to certain technology and compounds relating to liprotamase. Pursuant to the Lilly Agreement, we are required to make various milestone payments upon our achievement of certain regulatory and commercial objectives for any liprotamase formulation. We are also required to make tiered royalty payments on net sales, which percentage increases from the high single digits to the mid-teens as net sales increase.
In March 2015, we received a research award of up to $3 million from Cystic Fibrosis Foundation Therapeutics Incorporated (“CFFT”) for the development of liprotamase. Under the research award agreement, we are obligated to pay royalties to CFFT as follows: i) a one-time royalty in an amount equal to the five times the award, payable in three installments between the first and second anniversaries of the first commercial sale of a product; ii) a one-time royalty in an amount equal to the actual award after net product sales reaches $100 million; and iii) in the event of a license, sale or other transfer of the product or a change of control transaction prior to the commercial sale of the product, a milestone payment equal to three times the actual award.
The timing of our achievement of these events and corresponding milestone payments becoming due to our licensors is subject to factors relating to the clinical and regulatory development and commercialization of our product candidates, as applicable, many of which are beyond our control. We may become obligated to make a milestone payment during a period in which we do not have the cash on hand to make such payment, which could require us to delay our clinical studies, curtail our operations, scale back our commercialization and marketing efforts, seek funds to meet these obligations at terms unfavorable to us or default on our license agreements, which could result in license termination.
Our future debt obligations could impair our liquidity and financial condition, and in the event we are unable to meet our debt obligations, the lenders could foreclose on our assets.
As a part of our financing strategy, we have in the past and we may in the future enter into credit agreements with lenders. In connection with these credit agreements, our debt obligations to the lenders could:
|
·
|
impair our liquidity and make it more difficult for us to satisfy our other obligations;
|
|
·
|
require us to dedicate cash flow to payments on our debt obligations, which would reduce the availability of our cash flow to fund working capital, capital expenditures and other corporate requirements;
|
|
·
|
impose restrictions on our ability to incur other indebtedness and grant liens on our assets, other than permitted indebtedness and permitted liens, and could impede us from obtaining additional financing in the future for working capital, capital expenditures, acquisitions and general corporate purposes;
|
|
·
|
impose restrictions on us with respect to the use of our available cash, including in connection with future acquisitions;
|
|
·
|
adversely affect our ability to enter into strategic transactions and similar agreements, or require us to obtain the consent of our lenders;
|
|
·
|
make us more vulnerable in the event of a downturn in our business prospects and could limit our flexibility to plan for, or react to, changes in our licensing markets; and
|
|
·
|
place us at a competitive disadvantage when compared to our competitors who are not similarly restricted.
|
We may be required to pledge substantially all of our assets to secure our obligations under credit agreements in the future. In the event that we were to fail to make any required payment under any credit agreement, or fail to comply with the covenants contained in the any credit agreement and other related agreements, we may be in default regarding that indebtedness. A debt default would enable the lender to foreclose on the assets securing such debt and could significantly diminish the market value and marketability of our common stock and could result in the acceleration of the payment obligations under all or a portion of our consolidated indebtedness.
Our limited operating history makes it difficult to evaluate our business and prospects.
We were incorporated in September 2004. Our operations to date have been limited to organizing and staffing our company, acquiring product and technology rights, conducting product development activities for our primary product candidates, liprotamase and blisibimod, and performing research and development. We have not yet demonstrated an ability to obtain regulatory approval for or commercialize a product candidate. Consequently, any predictions about our future performance may not be as accurate as they could be if we had a history of successfully developing and commercializing pharmaceutical products.
Risks Associated with Development and Commercialization of Our Product Candidates
We depend substantially on the success of our product candidates which are still under clinical development. We cannot assure you that our product candidates will receive regulatory approval or be successfully commercialized.
To date, we have not obtained marketing approval for, or marketed, distributed or sold any products. The success of our business depends primarily upon our ability to develop and commercialize our product candidates successfully.
Our product candidates are prone to the risks of failure inherent in drug development. Before obtaining regulatory approvals for the commercial sale of any product candidates for a target indication, we must demonstrate with substantial evidence gathered in preclinical and well- controlled clinical studies, and, with respect to approval in the United States, to the satisfaction of the U.S. FDA and, with respect to approval in other countries, similar regulatory authorities in those countries, that the product candidates are safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate. Despite our efforts, our product candidates may not:
|
·
|
offer therapeutic or other improvement over existing, comparable therapeutics;
|
|
·
|
be proven safe and effective in clinical studies;
|
|
·
|
meet applicable regulatory standards;
|
|
·
|
be capable of being produced in sufficient quantities at acceptable costs;
|
|
·
|
be successfully commercialized; or
|
|
·
|
obtain favorable reimbursement.
|
We are not permitted to market our product candidates in the United States until the U.S. FDA approves our biologics license applications, or BLAs, or in any foreign countries until we receive the requisite approval from such countries. We have not submitted any BLA or received marketing approval for our product candidates.
Preclinical testing and clinical studies are long, expensive and uncertain processes. We may spend several years completing our testing for any particular product candidate, and failure can occur at any stage. Negative or inconclusive results or adverse medical events during a clinical study could also cause the U.S. FDA or us to terminate a clinical study or require that we repeat it or conduct additional clinical studies. Additionally, data obtained from a clinical study are susceptible to varying interpretations and the U.S. FDA or other regulatory authorities may interpret the results of our clinical studies less favorably than we do. The U.S. FDA and equivalent foreign regulatory agencies have substantial discretion in the approval process and may decide that our data are insufficient to support a marketing application and require additional preclinical, clinical or other studies.
From time to time during the regulatory approval process of our product candidates, we engage in discussions with the U.S. FDA and other non-US regulatory authorities regarding the regulatory requirements for our development programs. We may receive informal verbal and or written guidance from these authority agencies which may help form the basis of our clinical trial designs. The U.S. FDA and other non-US regulatory agencies may change their position on such informal guidance prior to the approval of our product candidates. As a result, we are unable to determine whether the outcome of informal deliberations will become final. If we are unable to effectively and efficiently resolve and comply with inquires and requests from the U.S. FDA and other non-US regulatory authorities, the approval of our product candidates may be delayed and their value maybe be reduced.
Any termination or suspension of, or delays in the commencement or completion of, clinical testing of our product candidates could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospect.
Delays in the commencement or completion of clinical testing could significantly affect our product development costs. We do not know whether planned clinical studies will begin on time or be completed on schedule, if at all. The commencement and completion of clinical studies can be delayed for a number of reasons, including delays related to:
|
·
|
obtaining regulatory approval to commence a clinical study or complying with conditions imposed by a regulatory authority regarding the scope or design of a clinical study;
|
|
·
|
reaching agreement on acceptable terms with prospective clinical research organizations, or CROs, and study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and study sites;
|
|
·
|
manufacturing, including manufacturing sufficient quantities of product candidates or other materials for use in clinical studies;
|
|
·
|
obtaining IRB, approval or the approval of other reviewing entities to conduct a clinical study at prospective sites;
|
|
·
|
recruiting and enrolling patients to participate in clinical studies for a variety of reasons, including size of patient population, nature of clinical study protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical study programs for similar indications;
|
|
·
|
severe or unexpected drug-related adverse effects experienced by patients in a clinical study; and
|
|
·
|
retaining patients who have initiated a clinical study, but may withdraw due to treatment protocol, adverse effects from the therapy, lack of efficacy from the treatment, personal issues or who are lost to further follow-up.
|
Clinical studies may also be delayed, suspended or terminated as a result of ambiguous or negative interim results, or results that are inconsistent with earlier results. In addition, a clinical study may be suspended or terminated by us, the U.S. FDA, the IRB or other reviewing entity overseeing the clinical study at issue, any of our clinical study sites with respect to that site, or other regulatory authorities due to a number of factors, including:
|
·
|
failure to conduct the clinical study in accordance with regulatory requirements or our clinical protocols;
|
|
·
|
inspection of the clinical study operations or study sites by the U.S. FDA or other regulatory authorities resulting in the imposition of a clinical hold;
|
|
·
|
unforeseen safety issues or any determination that a clinical study presents unacceptable health risks; and
|
|
·
|
lack of adequate funding to continue the clinical study, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional clinical studies and increased expenses associated with the services of our CROs and other third parties.
|
Product development costs to us will increase if we have delays in testing or approval of our product candidates or if we need to perform more or larger clinical studies than planned. We typically rely on third-party clinical investigators at medical institutions and health care facilities to conduct our clinical studies and, as a result, we may face additional delays outside our control.
Additionally, changes in regulatory requirements and policies may occur and we may need to amend clinical development plans or clinical study protocols to reflect these changes. Amendments may require us to resubmit our clinical study protocols to IRBs for re- examination, which may impact the costs, timing or successful completion of a clinical study. If we experience delays in completion of, or if we, the U.S. FDA or other regulatory authorities, the IRB or other reviewing entities, or any of our clinical study sites suspend or terminate any of our clinical studies, the commercial prospects for our product candidates may be harmed and our ability to generate product revenues will be delayed. In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical studies may also ultimately lead to the denial of regulatory approval of our product candidates. Also, if one or more clinical studies are delayed, our competitors may be able to bring products to market before we do, and the commercial viability of our product candidates could be significantly reduced.
Because the results of preclinical testing or earlier clinical studies are not necessarily predictive of future results, any product candidate we advance into clinical studies may not have favorable results in later clinical studies or receive regulatory approval.
Success in preclinical testing and early clinical studies does not ensure that later clinical studies will generate adequate data to demonstrate the efficacy and safety of an investigational drug or biologic. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in Phase 3 clinical studies, even after seeing promising results in earlier clinical studies. Despite the results reported in earlier clinical studies for our product candidates, we do not know whether any Phase 3 or other clinical studies we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market our product candidates. If later stage clinical studies do not produce favorable results, our ability to achieve regulatory approval for any of our product candidates may be adversely impacted. Even if we believe that our product candidates have performed satisfactorily in preclinical testing and clinical studies, we may nonetheless fail to obtain U.S. FDA approval for our product candidates.
If we breach the license agreements for our product candidates, we could lose the ability to continue the development and commercialization of our product candidates.
We are party to the Amgen Agreement, which provides for the exclusive worldwide licenses of the compositions of matter and methods of use for blisibimod, as well as non-exclusive worldwide licenses of compositions of matter and methods of use relating to peptibodies generally. We are also party to the Lilly Agreement, which provides for an exclusive worldwide license of the compositions of matter, formulation, and methods of use patents for liprotamase. These agreements require us to make timely milestone and royalty payments, provide regular information, maintain the confidentiality of and indemnify our licensors under the terms of the agreements.
If we fail to meet these obligations, our licensors may terminate our licenses and may be able to re-obtain licensed technologies and aspects of any intellectual properties controlled by us that relate to the licensed technologies that originated from our licensors. Our licensors could effectively take control of the development and commercialization of the licensed product candidates after an uncured, material breach of our license agreements by us or if we voluntarily terminate the agreements. While we would expect to exercise all rights and remedies available to us, including seeking to cure any breach by us, and otherwise seek to preserve our rights under the patents and patent applications licensed to us, we may not be able to do so in a timely manner, at an acceptable cost or at all. Any uncured, material breach under the license agreements could result in our loss of exclusive rights and may lead to a complete termination of our product development and any commercialization efforts for our product candidates.
Our industry is subject to intense competition. If we are unable to compete effectively, our product candidates may be rendered non-competitive or obsolete.
The pharmaceutical industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large pharmaceutical and more established biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. All of these competitors currently engage in, have engaged in or may engage in the future in the development, manufacturing, marketing and commercialization of pharmaceuticals and biotechnologies, some of which may compete with our present or future product candidates. It is possible that any of these competitors could develop technologies or products that would render our product candidates obsolete or non-competitive, which could adversely affect our revenue potential. Key competitive factors affecting the commercial success of our product candidates are likely to be efficacy, safety profile, reliability, convenience of dosing, price and reimbursement.
The market for pancreatic enzyme replacement therapy is also highly competitive. There are currently several marketed products for EPI caused by cystic fibrosis, including Creon marketed by AbbVie, Inc., Pancreaze by Janssen Pharmaceuticals, Inc., Pertzye by Cornestone Therapeutics, Inc., and Ultresa and Zenpep by Aptalis Pharma US. Inc. We are also aware of companies with other products in development that are being tested for potential treatment of EPI caused by cystic fibrosis: Johnson and Johnson Research and Development LLC recently completed a Phase 3 study to assess the effectiveness and safety of oral pancrelipase MT in the treatment of adult and pediatric/adolescent cystic fibrosis patients with clinical symptoms of EPI; and Nordmark Arzneimittel GmbH & Co. KG’s compound, Burlulipase, is being tested in a Phase 3 study in patients with EPI.
The market for inflammatory disease therapeutics is especially large and competitive. For lupus, GlaxoSmithKline plc’s BAFF antagonist monoclonal antibody, Benlysta®, was approved by the U.S. FDA for treatment of lupus. In 2015, new efficacy data with a subcutaneous presentation of Benlysta were presented, along with safety and pharmacokinetics data for an autoinjector for the subcutaneous product. We assume that these presentations will become commercially-available in the future. Further, we are aware of companies with other products in development that are being tested for potential treatment of lupus: AstraZeneca is conducting 3 Phase 3 trials in lupus and lupus nephritis with their drug candidate, anifrolumab, Merck Serono S.A., whose dual BAFF/APRIL antagonist fusion protein, Atacicept, recently completed a Phase 2/3 clinical study for lupus. In addition, other companies including Janssen, Pfizer, have completed Phase 2 trials in lupus and may continue to advance their clinical development in lupus.
Many of our potential competitors have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of drug candidates, and in obtaining U.S. FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than we may be in obtaining U.S. FDA approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, have fewer adverse effects, be less expensive to develop and manufacture or be more effectively marketed and sold than any product candidates we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing our product candidates. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. These entities may also establish collaborative or licensing relationships with our competitors. Finally, the development of new treatment methods for the diseases we are targeting could render our drugs non-competitive or obsolete. All of these factors could adversely affect our business.
Our product candidates may cause undesirable adverse effects or have other properties that could delay or prevent their regulatory approval or limit the commercial profile of any approved label.
Undesirable adverse effects caused by our product candidates could cause us, IRBs or other reviewing entities, clinical study sites, or regulatory authorities to interrupt, delay or halt clinical studies and could result in the denial of regulatory approval by the U.S. FDA or other regulatory authorities. Phase 2 clinical studies conducted by us with blisibimod have generated differences in adverse effects and serious adverse events. The most common adverse effects seen with our product candidates versus placebo include injection site erythema and nasopharyngitis. The most common serious adverse events seen with blisibimod include Herpes zoster, pneumonia, urinary tract infections and deep vein thrombosis, cellulitis, intervertebral disc protrusion, spontaneous abortion, and kidney stones. During the placebo-controlled Phase 2 PEARL study, blisibimod was safe and well-tolerated at all dose levels with no meaningful imbalances in serious adverse events or infections between blisibimod and placebo. Discontinuation due to adverse event was lower amongst blisibimod-treated subjects (5.7%) compared to placebo (7.9%). Amongst the commonly-reported adverse events, imbalance was observed only with injection site reactions (200mg QW blisibimod = 15%, matched placebo = 7%), but these were not serious or severe and did not result in discontinuation of treatment. Studies conducted by our licensor on liprotamase have generated the following common adverse effects: general gastrointestinal disorder, such as abdominal pain, flatulence, loose stools and diarrhea.
Our second product candidate, liprotamase, which we licensed from Eli Lilly in July 2014, received a complete response letter (“CRL”) from the U.S. FDA while it was under development by Eli Lilly in April 2011. Eli Lilly has attempted to address the material items highlighted by the FDA in the CRL and worked directly with the U.S. FDA on a clinical development program for liprotamase which, if successful, could result in regulatory approval of liprotamase. There are still open items from the CRL that we will need to address with the U.S. FDA. While we plan to make reasonable efforts to accommodate and address the U.S. FDA’s inquires and request, we are unable to determine the final outcome of the CRL. Any delay in addressing the CRL to the satisfaction of the U.S. FDA may result in postponement of our Phase 3 clinical trial of liprotamase in patients with EPI.
If serious adverse events that are considered related to our product candidates are observed in any Phase 3 clinical studies, our ability to obtain regulatory approval for our product candidates may be adversely impacted. Further, if our product candidates receive marketing approval and we or others later discover, after approval and use in an increasing number of patients, that our products could have adverse effect profiles that limit their usefulness or require their withdrawal (whether or not the therapies showed the adverse effect profile in Phase 1 through Phase 3 clinical studies), a number of potentially significant negative consequences could result, including:
|
•
|
regulatory authorities may withdraw their approval of the products;
|
|
•
|
regulatory authorities may require the addition of labeling statements, such as warnings or contraindications;
|
|
•
|
we may be required to change the way the products are administered, conduct additional clinical studies or change the labeling of the products;
|
|
•
|
we could be sued and held liable for harm caused to patients; and
|
|
•
|
our reputation may suffer.
|
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidates and could substantially increase the costs of commercialization.
After the completion of our clinical studies, we cannot predict whether or when we will obtain regulatory approval to commercialize our product candidates and we cannot, therefore, predict the timing of any future revenue from the product candidates.
Even if we project positive clinical results and file for regulatory approval, we cannot commercialize any product candidate until the appropriate regulatory authorities have reviewed and approved the applications for such product candidate. We cannot assure you that the regulatory agencies will complete their review processes in a timely manner or that we will obtain regulatory approval for any product candidates we develop. Satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires the expenditure of substantial resources. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action or changes in U.S. FDA policy during the period of product development, clinical studies and U.S. FDA regulatory review.
Even if our product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if U.S. regulatory approval is obtained, the U.S. FDA may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. For example, the label ultimately approved for blisibimod or liprotamase, if any, may include restrictions on use. Further, the U.S. FDA has indicated that long-term safety data on blisibimod may need to be obtained as a post-market requirement. Our product candidates will also be subject to ongoing U.S. FDA requirements governing the labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of safety and other post-market information. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the U.S. FDA and other regulatory authorities for compliance with current good manufacturing procedures, or cGMP, regulations. If we or a regulatory agency discovers previously unknown problems with our product candidates, such as adverse events of unanticipated severity or frequency, or problems with the facility where the products are manufactured, a regulatory agency may impose restrictions on the products, the manufacturing facility or us, including requiring recall or withdrawal of the products from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
|
•
|
issue warning letters or untitled letters;
|
|
•
|
seek an injunction or impose civil or criminal penalties or monetary fines;
|
|
•
|
suspend or withdraw regulatory approval;
|
|
•
|
suspend any ongoing clinical studies;
|
|
•
|
refuse to approve pending applications or supplements to applications filed by us;
|
|
•
|
suspend or impose restrictions on operations, including costly new manufacturing requirements; or
|
|
•
|
seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall.
|
The occurrence of any event or penalty described above may inhibit our ability to commercialize our products and generate revenue.
New legal and regulatory requirements could make it more difficult for us to obtain approvals for our product candidates and could limit or make more burdensome our ability to commercialize any approved products.
New federal legislation or regulatory requirements could affect the requirements for obtaining regulatory approvals of our product candidates or otherwise limit our ability to commercialize any approved products or subject our products to more rigorous post-approval requirements. New legislation, and the additional proposals if enacted, may make it more difficult or burdensome for us to obtain approval of our product candidates, any approvals we receive may be more restrictive or be subject to onerous post-approval requirements, our ability to successfully commercialize approved products may be hindered and our business may be harmed as a result.
If our product candidates for which we receive regulatory approval do not achieve broad market acceptance, the revenue that we generate from their sales, if any, will be limited.
The commercial success of our product candidates for which we obtain marketing approval from the U.S. FDA or other regulatory authorities will depend upon the acceptance of these products by the medical community, including physicians, patients and health care payors. The degree of market acceptance of our approved products will depend on a number of factors, including:
|
•
|
demonstration of clinical safety and efficacy compared to other products;
|
|
•
|
the relative convenience, ease of administration and acceptance by physicians and payors of our product candidates;
|
|
•
|
the prevalence and severity of any adverse effects;
|
|
•
|
limitations or warnings contained in a product’s U.S. FDA-approved labeling;
|
|
•
|
availability of alternative treatments;
|
|
•
|
pricing and cost-effectiveness;
|
|
•
|
the effectiveness of our or any future collaborators’ sales and marketing strategies;
|
|
•
|
our ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare and Medicaid; and
|
|
•
|
the willingness of patients to pay out-of-pocket in the absence of third-party coverage.
|
If our product candidates are approved but do not achieve an adequate level of acceptance by physicians, health care payors and patients, we may not generate sufficient revenue from these products, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third- party payors on the benefits of our product candidates may require significant resources and may never be successful.
Our future success depends on our ability to retain our chief executive officer and other key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on Mr. Paul F. Truex, our Chief Executive Officer, Mr. Craig Thompson, our President and Chief Operating Officer, Dr. Colin Hislop, our Senior Vice President and Chief Medical Officer, Dr. Chuck Olson, our Chief Technology Officer, and Ms. Klara Dickinson-Eason, our Chief Regulatory Officer, and the other principal members of our executive team. The loss of the services of any of these persons might impede the achievement of our research, development and commercialization objectives. Recruiting and retaining qualified scientific personnel and possibly sales and marketing personnel will also be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific personnel from universities and research institutions. Failure to succeed in clinical studies may make it more challenging to recruit and retain qualified scientific personnel. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
Recently enacted and future legislation or regulatory reform of the health care system in the United States and foreign jurisdictions may affect our ability to sell our products profitably.
Our ability to commercialize our future products successfully, alone or with collaborators, will depend in part on the extent to which reimbursement for the products will be available from government and health administration authorities, private health insurers and other third-party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors of health care services to contain or reduce health care costs may adversely affect our ability to set prices for our products which we believe are fair, and our ability to generate revenues and achieve and maintain profitability.
Specifically, in both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably. In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
We will not know the full effects of the Health Care Reform Law until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effect of the Health Care Reform Law, the new law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and also may increase our regulatory burdens and operating costs. We expect further federal and state proposals and health care reforms to continue to be proposed by legislators, which could limit the prices that can be charged for any product we develop and may limit our commercial opportunity.
Also in the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, also called the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
The continuing efforts of government and other third-party payors to contain or reduce the costs of health care through various means may limit our commercial opportunity. It will be time-consuming and expensive for us to go through the process of seeking reimbursement from Medicare and private payors. Our products may not be considered cost-effective, and government and third- party private health insurance coverage and reimbursement may not be available to patients for any of our future products or sufficient to allow us to sell our products on a competitive and profitable basis. Our results of operations could be adversely affected by the MMA, the Health Care Reform Law and additional prescription drug coverage legislation, by the possible effect of this legislation on amounts that private insurers will pay and by other health care reforms that may be enacted or adopted in the future. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the pricing of pharmaceutical products. Cost control initiatives could decrease the price that we or any potential collaborators could receive for any of our future products and could adversely affect our profitability.
In some foreign countries, including major markets in the EU and Japan, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take six to 12 months or longer after the receipt of regulatory marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical study that compares the cost-effectiveness of our product candidates to other available therapies. Such pharmacoeconomic studies can be costly and the results uncertain. Our business could be harmed if reimbursement of our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels.
We face potential product liability exposure, and, if successful claims are brought against us, we may incur substantial liability.
The use of product candidates in clinical studies and the sale of any products for which we obtain marketing approval expose us to the risk of product liability claims. Product liability claims might be brought against us by consumers, health care providers, pharmaceutical companies or others selling or otherwise coming into contact with our products. If we cannot successfully defend ourselves against product liability claims, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in:
|
•
|
impairment of our business reputation;
|
|
•
|
withdrawal of clinical study participants;
|
|
•
|
costs of related litigation;
|
|
•
|
distraction of management’s attention from our primary business;
|
|
•
|
substantial monetary awards to patients or other claimants;
|
|
•
|
the inability to commercialize product candidates; and
|
|
•
|
decreased demand for product candidates, if approved for commercial sale.
|
Our product liability insurance coverage for our clinical studies may not be sufficient to reimburse us for all expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If and when we obtain marketing approval for any product candidate, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain this product liability insurance on commercially reasonable terms. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated adverse effects. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
If we use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including toxic chemical and biological materials. We could be held liable for any contamination, injury or other damages resulting from these hazardous substances. In addition, our operations produce hazardous waste products. While third parties are responsible for disposal of our hazardous waste, we could be liable under environmental laws for any required cleanup of sites at which our waste is disposed. Federal, state, foreign and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials. If we fail to comply with these laws and regulations at any time, or if they change, we may be subject to criminal sanctions and substantial civil liabilities, which may harm our business. Even if we continue to comply with all applicable laws and regulations regarding hazardous materials, we cannot eliminate the risk of accidental contamination or discharge and our resultant liability for any injuries or other damages caused by these accidents.
We rely on third parties to conduct, supervise and monitor our clinical studies, and those third parties may perform in an unsatisfactory manner, such as by failing to meet established deadlines for the completion of these clinical studies, or may harm our business if they suffer a catastrophic event.
We rely on third parties such as CROs, medical institutions and clinical investigators to enroll qualified patients and conduct, supervise and monitor our clinical studies. Our reliance on these third parties for clinical development activities reduces our control over these activities. Our reliance on these third parties, however, does not relieve us of our regulatory responsibilities, including ensuring that our clinical studies are conducted in accordance with good clinical practices, or GCP, and the investigational plan and protocols contained in the relevant regulatory application, such as the investigational new drug application, or IND. In addition, the CROs with whom we contract may not complete activities on schedule, or may not conduct our preclinical studies or clinical studies in accordance with regulatory requirements or our clinical study design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for, and to commercialize, our product candidates may be delayed or prevented. In addition, if a catastrophe such as an earthquake, fire, flood or power loss should affect one of the third parties on which we rely, our business prospects could be harmed. For example, if a central laboratory holding all of our clinical study samples were to suffer a catastrophic loss of their facility, we would lose all of our samples and would have to repeat our studies.
Any failure by our third-party manufacturers on which we rely to produce our preclinical and clinical drug supplies and on which we intend to rely to produce commercial supplies of any approved product candidates may delay or impair our ability to commercialize our product candidates.
We have relied upon a small number of third-party manufacturers and active pharmaceutical ingredient formulators for the manufacture of our material for preclinical and clinical testing purposes and intend to continue to do so in the future. We also expect to rely upon third parties to produce materials required for the commercial production of our product candidates if we succeed in obtaining necessary regulatory approvals. If we are unable to arrange for third-party manufacturing sources, or to do so on commercially reasonable terms, we may not be able to complete development of our product candidates or market them.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control (including a failure to synthesize and manufacture our product candidates in accordance with our product specifications) and the possibility of termination or nonrenewal of the agreement by the third party, based on its own business priorities, at a time that is costly or damaging to us. In addition, the U.S. FDA and other regulatory authorities require that our product candidates be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities in a timely manner, could lead to a delay in, or failure to obtain, regulatory approval. In addition, such failure could be the basis for action by the U.S. FDA to withdraw approvals previously granted to us and for other regulatory action, including recall or seizure, total or partial suspension of production or injunction.
We rely on our manufacturers to purchase from third-party suppliers the materials necessary to produce drug product for our clinical studies. There are a small number of suppliers, and in some instances, a single supplier for certain capital equipment and raw materials that we use to manufacture drug product. Such suppliers may not sell these raw materials and equipment to our manufacturers at the times we need them or on commercially reasonable terms. We do not have any control over the process or timing of the acquisition of these raw materials and equipment by our manufacturers. Moreover, we currently do not have any agreements for the commercial production of these raw materials. Although we generally do not begin a clinical study unless we believe we have a sufficient supply of product candidates to complete the clinical study, any significant delay in the supply of product candidates or the raw material components thereof for an ongoing clinical study due to the need to replace a third-party manufacturer could considerably delay completion of our clinical studies, product testing and potential regulatory approval. If our manufacturers or we are unable to purchase these raw materials after regulatory approval has been obtained, the commercial launch would be delayed or there would be a shortage in supply of such product candidates, which would impair our ability to generate revenues from the sale of such product candidates.
Because of the complex nature of our compounds, our manufacturers may not be able to manufacture our compounds at a cost or in quantities or in a timely manner necessary to make commercially successful products. If we successfully commercialize a product candidate, we may be required to establish large-scale commercial manufacturing capabilities. In addition, as our drug development pipeline increases and matures, we will have a greater need for clinical study and commercial manufacturing capacity. We have no experience manufacturing pharmaceutical products on a commercial scale and some of these suppliers will need to increase their scale of production to meet our projected needs for commercial manufacturing, which may not occur on a timely basis.
Some of our manufacturing suppliers are located overseas, and the transportation of drug supplies to or from these facilities to their intended destinations is subject to certain risks of loss and damage beyond our control. Additionally, the importation of drug supplies into and from foreign countries is subject to customs regulations that may require us to incur additional regulatory costs.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate any revenue.
We do not currently have an organization for the sales, marketing and distribution of pharmaceutical products and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market any products that may be approved by the U.S. FDA, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable. We will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
Guidelines and recommendations published by various organizations may adversely affect the use of any products for which we may receive regulatory approval.
Government agencies issue regulations and guidelines directly applicable to us and to our product candidates. In addition, professional societies, practice management groups, private health or science foundations and organizations involved in various diseases from time to time publish guidelines or recommendations to the medical and patient communities. These various sorts of recommendations may relate to such matters as product usage and use of related or competing therapies. Changes to these recommendations or other guidelines advocating alternative therapies could result in decreased use of any products for which we may receive regulatory approval, which may adversely affect our results of operations.
Risks Related to Our Intellectual Property
If our or our licensors’ patent positions do not adequately protect our product candidates or any future products, others could compete with us more directly or prevent us from commercializing our products, which would harm our business.
We hold license rights to numerous U.S. European Patents (“EP”), and non-EP foreign patents and patent applications relating to blisibimod and liprotamase. Our liprotomase portfolio is made up of exclusively licensed patents and patent applications from Eli Lilly. Our blisibimod portfolio is made up of exclusively and non-exclusively licensed patents and patent applications from Amgen, Inc.
Our commercial success will depend in part on our and our licensors’ ability to obtain additional patents and protect our existing patent positions, particularly those patents for which we have secured exclusive rights, as well as our ability to maintain adequate protection of other intellectual property for our technologies, product candidates and any future products in the United States and other countries. If we or our licensors do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could materially harm our business, negatively affect our position in the marketplace, limit our ability to commercialize our product candidates and delay or render impossible our achievement of profitability. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries.
The patent positions of biotechnology and pharmaceutical companies, including our patent position, involve complex legal and factual questions, and, therefore, validity and enforceability cannot be predicted with certainty. Patents may be challenged, deemed unenforceable, invalidated or circumvented. We and our licensors will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies, product candidates and any future products are covered by valid and enforceable patents or are effectively maintained as trade secrets.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
|
•
|
we or our licensors were the first to make the inventions covered by each of our pending patent applications;
|
|
•
|
we or our licensors were the first to file patent applications for these inventions;
|
|
•
|
others will not independently develop similar or alternative technologies or duplicate any of our technologies;
|
|
•
|
any of our or our licensors’ pending patent applications will result in issued patents;
|
|
•
|
any of our or our licensors’ patents will be valid or enforceable;
|
|
•
|
any patents issued to us or our licensors and collaborators will provide a basis for commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties;
|
|
•
|
we will develop additional proprietary technologies or product candidates that are patentable; or
|
|
•
|
the patents of others will not have an adverse effect on our business.
|
We are aware of two families of third party United States patents and pending foreign applications that contain broad claims related to BLyS or BAFF binding polypeptides. Based on our analyses, if these patents were asserted against us, we do not believe that blisibimod would be found to infringe any valid claim of these patents. If we were to challenge the validity of any issued United States patent in court, we would need to overcome the presumption of validity that attaches to every United States patent by presenting clear and convincing evidence as to the invalidity of the patent’s claims. There is no assurance that a court would find in our favor on questions of infringement or validity, and we could incur substantial costs in litigation if we are required to defend against patent suits brought by third parties or if we initiate these suits. If third party patents are determined to be valid and construed to cover blisibimod, the development and commercialization of this program could be affected, subjecting us to potential liability for damages and in addition may require us to obtain a license to continue marketing the affected product. Such a license may not be available on commercially acceptable terms, if at all.
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
We rely on trade secrets to protect our proprietary know-how and technological advances, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights. Failure to obtain or maintain trade secret protection could enable competitors to use our proprietary information to develop products that compete with our products or cause additional, material adverse effects upon our competitive business position.
We license patent rights from third-party owners. If we, or such owners, do not properly maintain or enforce the patents underlying such licenses, our competitive position and business prospects will be harmed.
We are party to a license agreement with Amgen that provides exclusive and worldwide rights to develop and commercialize the novel BAFF inhibitor blisibimod, as well as non-exclusive rights to certain technology relating to peptibody compositions and formulations. We are also a party to a license agreement with Eli Lilly and Company that provides exclusive and worldwide rights to develop and commercialize liprotamase, as well as non-exclusive rights to certain technology relating to liprotamase compositions and formulations.
We depend in part on our licensors to protect the proprietary rights covering blisibimod and liprotamase. Our licensors are responsible for maintaining certain issued patents and prosecuting certain patent applications. We have limited, if any, control over the amount or timing of resources that our licensors devote on our behalf or the priority they place on maintaining these patent rights and prosecuting these patent applications to our advantage. Our licensors may also be notified of alleged infringement and be sued for infringement of third-party patents or other proprietary rights. We may have limited, if any, control or involvement over the defense of these claims, and our licensors could be subject to injunctions and temporary or permanent exclusionary orders in the United States or other countries. Our licensors are not obligated to defend or assist in our defense against third-party claims of infringement. We have limited, if any, control over the amount or timing of resources, if any, that our licensors devote on our behalf or the priority they place on defense of such third-party claims of infringement.
Our success will depend in part on the ability of us or our licensors to obtain, maintain and enforce patent protection for their intellectual property, in particular, those patents to which we have secured exclusive rights. We or our licensors may not successfully prosecute the patent applications which we have licensed. Even if patents issue in respect of these patent applications, we or our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
If we do not obtain protection under the Hatch-Waxman Act and similar foreign legislation to extend our licensed patent terms and to obtain market exclusivity for our product candidates, our business will be materially harmed.
The Hatch-Waxman Act provides for an extension of patent term for drug compounds for a period of up to five years to compensate for time spent in the regulatory approval process. Assuming we gain a five-year patent term extension for blisibimod and that we continue to have rights under our license agreement with respect to blisibimod, we would have exclusive rights to blisibimod’s U.S. new chemical entity patent until 2027 or 2028. In Europe, similar legislative enactments allow patent terms in the European Union to be extended for up to five years through the grant of a Supplementary Protection Certificate. Assuming we gain such a five-year extension for blisibimod and that we continue to have rights under our license agreement with respect to blisibimod, we would have exclusive rights to blisibimod’s European new chemical entity patents until 2027. Further, since blisibimod has not been previously approved, blisibimod could be eligible for 12 years of data exclusivity from the U.S. FDA. During the data exclusivity period, competitors are barred from relying on the innovator biologic’s safety and efficacy data to gain approval.
Similarly, the European Union provides that companies who receive regulatory approval for a new small molecule compound or biologic will have a 10-year period of data exclusivity for that compound or biologic (with the possibility of a further one-year extension) in most EU countries, beginning on the date of such European regulatory approval, regardless of when the European new chemical entity patent covering such compound expires. A generic version of the approved drug may not be marketed or sold during such market exclusivity period. However, there is no assurance that we will receive the extensions of our patents or other exclusive rights available under the Hatch-Waxman Act or similar foreign legislation. If we fail to receive such Hatch-Waxman extensions or marketing exclusivity rights or if we receive extensions that are materially shorter than expected, our ability to prevent competitors from manufacturing, marketing and selling generic versions of our products will be materially harmed.
Our current patent positions and license portfolio may not include all patent rights needed for the full development and commercialization of our product candidates. We cannot be sure that patent rights we may need in the future will be available for license to us on commercially reasonable terms, or at all.
We typically develop product candidates using compounds for which we have in-licensed and original composition of matter patents and patents that claim the activities and methods for such compounds’ production and use to the extent known at that time. As we learn more about the mechanisms of action and new methods of manufacture and use of product candidates, we may file additional patent applications for these new inventions or we may need to ask our licensors to file them. We may also need to license additional patent rights or other rights on compounds, treatment methods or manufacturing processes because we learn that we need such rights during the continuing development of our product candidates.
Although our in-licensed and original patents may prevent others from making, using or selling similar products, they do not ensure that we will not infringe the patent rights of third parties. We may not be aware of all patents or patent applications that may impact our ability to make, use or sell our product candidates. For example, because we sometimes identify the mechanism of action or molecular target of a given product candidate after identifying its composition of matter and therapeutic use, we may not be aware until the mechanism or target is further elucidated that a third party has an issued or pending patent claiming biological activities or targets that may cover our product candidates. U.S. patent applications filed after November 29, 2000 are confidential in the U.S. Patent and Trademark Office for the first 18 months after such applications’ earliest priority date, and patent offices in non-U.S. countries often publish patent applications for the first time six months or more after filing. Furthermore, we may not be aware of published or granted conflicting patent rights. Any conflicts resulting from patent applications and patents of others could significantly reduce the coverage of our patents and limit our ability to obtain meaningful patent protection. If others obtain patents with conflicting claims, we may need to obtain licenses to these patents or to develop or obtain alternative technology.
We may not be able to obtain any licenses or other rights to patents, technology or know-how from third parties necessary to conduct our business as described in this report and such licenses, if available at all, may not be available on commercially reasonable terms. Any failure to obtain such licenses could delay or prevent us from developing or commercializing our product candidates or proposed product candidates, which would harm our business. Litigation or patent interference proceedings may be necessarily brought against third parties, as discussed below, to enforce any of our patents or other proprietary rights or to determine the scope and validity or enforceability of the proprietary rights of such third parties.
Litigation regarding patents, patent applications and other proprietary rights may be expensive and time consuming. If we are involved in such litigation, it could cause delays in bringing product candidates to market and harm our ability to operate.
Our commercial success will depend in part on our ability to manufacture, use, sell and offer to sell our product candidates and proposed product candidates without infringing patents or other proprietary rights of third parties. Although we are not currently aware of any litigation or other proceedings or third-party claims of intellectual property infringement related to our product candidates, the pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may obtain patents in the future and allege that the use of our technologies infringes these patent claims or that we are employing their proprietary technology without authorization. Likewise, third parties may challenge or infringe upon our or our licensors’ existing or future patents.
Proceedings involving our patents or patent applications or those of others could result in adverse decisions regarding the patentability of our inventions relating to our product candidates or the enforceability, validity or scope of protection offered by our patents relating to our product candidates.
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time-consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have our patents declared invalid, we may incur substantial monetary damages; encounter significant delays in bringing our product candidates to market; or be precluded from participating in the manufacture, use or sale of our product candidates or methods of treatment requiring licenses.
Risks Related to the Securities Markets and Investment in Our Common Stock
Market volatility may affect our stock price and the value of your investment.
The market price for our common stock has been and is likely to continue to be volatile. In addition, the market price of our common stock may fluctuate significantly in response to a number of factors, most of which we cannot predict or control, including:
|
·
|
plans for, progress in and results from clinical studies for our product candidates;
|
|
·
|
announcements of new products, services or technologies, commercial relationships, acquisitions or other events by us or our competitors;
|
|
·
|
developments concerning proprietary rights, including those pertaining to patents patent applications held by our licensors;
|
|
·
|
failure of any of our product candidates, if approved, to achieve commercial success;
|
|
·
|
fluctuations in stock market prices and trading volumes of securities of similar companies;
|
|
·
|
general market conditions and overall fluctuations in U.S. equity markets;
|
|
·
|
variations in our operating results, or the operating results of our competitors;
|
|
·
|
changes in our financial guidance or securities analysts’ estimates of our financial performance;
|
|
·
|
changes in accounting principles;
|
|
·
|
sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders;
|
|
·
|
additions or departures of any of our key personnel;
|
|
·
|
announcements related to litigation;
|
|
·
|
changing legal or regulatory developments in the United States and other countries; and
|
|
·
|
discussion of us or our stock price by the financial press and in online investor communities.
|
Although our common stock is listed for trading on The NASDAQ Global Market, our securities have been relatively thinly traded. Investor trading patterns could serve to exacerbate the volatility of the price of the stock. Accordingly, it may be difficult to sell shares of common stock quickly without significantly depressing the value of the stock. Unless we are successful in developing continued investor interest in our stock, sales of our stock could result in major fluctuations in the price of the stock. In addition, the stock market in general, and The NASDAQ Global Market in particular, have experienced substantial price and volume volatility that is often seemingly unrelated to the operating performance of particular companies. These broad market fluctuations may cause the trading price of our common stock to decline. In the past, securities class action litigation has often been brought against a company after a period of volatility in the market price of its common stock. We may become involved in this type of litigation in the future. Any securities litigation claims brought against us could result in substantial expenses and the diversion of our management’s attention from our business.
Because a small number of our existing stockholders own a material amount of our voting stock, your ability to influence corporate matters will be limited.
Our executive officers, directors and greater than 5% stockholders, in the aggregate, own approximately 27.63% of our outstanding common stock. As a result, such persons, acting together, will have the ability to influence our management and affairs and substantially all matters submitted to our stockholders for approval, including the election and removal of directors and approval of any significant transaction. These persons will also have the ability to influence our management and business affairs. This concentration of ownership may have the effect of delaying, deferring or preventing a change in control, impeding a merger, consolidation, takeover or other business combination involving us, or discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of our business, even if such a transaction would benefit other stockholders.
Future sales of our common stock may cause our stock price to decline.
As of December 31, 2015, there were 40,004,037 shares of our common stock outstanding. In addition, as of December 31, 2015, we had outstanding options and warrants to purchase 4,296,159 shares of our common stock that, if exercised, will result in these additional shares becoming available for sale. A large portion of these shares and outstanding equity awards are held by a small number of persons and investment funds. Sales by these stockholders or option holders of a substantial number of shares could significantly reduce the market price of our common stock. Moreover, certain holders of shares of common stock will have rights, subject to some conditions, to require us to file registration statements covering the shares they currently hold, or to include these shares in registration statements that we may file for ourselves or other stockholders.
We have registered or will register all common stock that we may issue under our 2013 Stock Option and Incentive Plan (the “2013 Plan”), our Amended and Restated 2010 Stock Option and Incentive Plan (the “2010 Plan”) and our Employee Stock Purchase Plan (the “ESPP”). As of December 31, 2015, an aggregate of 729,492 shares of our common stock have been reserved for future issuance under the 2013 Plan, plus any shares cancelled under our 2005 Equity Incentive Plan and 2010 Plan, and an aggregate of 55,929 shares of common stock have been reserved for future issuance under our ESPP. These shares can be freely sold in the public market upon issuance. If a large number of these shares are sold in the public market, the sales could reduce the trading price of our common stock.
In addition, we may sell shares of stock pursuant to an equity purchase agreement with LPC executed in March 2015, pursuant to which we have the right, but not the obligation, to sell to LPC up to an aggregate of $6.0 million of common stock until March 2017. We may also sell shares of stock pursuant to a Sales Agreement with Cowen and Company, LLC (“Cowen”) pursuant to an at-the-market equity program (“ATM”) under which we may from time to time offer and sell up to $25.0 million shares of our common stock, $3.2 million of which remained available to be sold as of the date of this report. Finally, we maintain an effective shelf registration statement on Form S-3 with the SEC for the issuance and sale from time to time of up to approximately $1.2 million of our equity and debt securities, which will expire in April 2016 and which we may replace at any time.
We will need to raise additional capital to fund our operations, which may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights.
We will need to seek additional capital through a combination of private and public equity offerings, debt financings and collaboration, strategic and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest may be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic alliance and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates or grant licenses on terms that are not favorable to us.
Operating as a public company increases our expenses and administrative burden.
As a public company, we incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, our administrative staff will be required to perform additional tasks. For example, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and The NASDAQ Global Market, impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. We must also bear all of the internal and external costs of preparing and distributing periodic public reports in compliance with our obligations under the securities laws.
In particular, the Sarbanes-Oxley Act requires, among other things, that we maintain effective internal control over financial reporting and disclosure controls and procedures. We must perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on the effectiveness of our internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management time on compliance-related issues. Moreover, if we are not able to comply with the requirements of Section 404 in a timely manner, our stock price could decline, and we could face sanctions, delisting or investigations by The NASDAQ Global Market, or other material adverse effects on our business, reputation, results of operations, financial condition or liquidity.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividend on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will therefore be limited to the value of their stock.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws may delay or prevent an acquisition of us or a change in our management. These provisions include:
|
·
|
a classified and staggered board of directors whose members can only be dismissed for cause;
|
|
·
|
the prohibition on actions by written consent of our stockholders;
|
|
·
|
the limitation on who may call a special meeting of stockholders;
|
|
·
|
the establishment of advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted upon at stockholder meetings;
|
|
·
|
the ability of our board of directors to issue preferred stock without stockholder approval, which would increase the number of outstanding shares and could thwart a takeover attempt; and
|
|
·
|
the requirement of at least 75% of the outstanding common stock to amend any of the foregoing provisions.
|
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which limits the ability of stockholders owning in excess of 15% of our outstanding voting stock to merge or combine with us. Although we believe these provisions collectively provide for an opportunity to obtain greater value for stockholders by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer rejected by our board were considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
Our ability to use our net operating loss carryforwards may be subject to limitation and may result in increased future tax liability to us.
Generally, a change of more than 50% in the ownership of a corporation’s stock, by value, over a three-year period constitutes an ownership change for U.S. federal income tax purposes. An ownership change may limit a company’s ability to use its net operating loss carryforwards attributable to the period prior to such change. We underwent an ownership change within the meaning of Section 382 ownership of the Internal Revenue Code during 2012 and as such, our net operating loss carryforwards are limited. In addition, the pre-change R&D tax credits have also been limited for federal tax purposes. If we earn net taxable income, our ability to use our pre-change net operating loss carryforwards to offset U.S. federal taxable income will be subject to limitations, which will result in increased future tax liability to us.
None.
We lease our main operating facility in Hayward, California. We occupy approximately 14,000 square feet under a facility lease agreement that expires in September 2017. We believe our existing facility is adequate for our current needs and that any additional space we need will be available in the future on commercially reasonable terms.
We are not subject to any material pending legal proceedings. From time to time, we may be involved in routine legal proceedings, as well as demands, claims and threatened litigation, which arise in the normal course of our business.
Not applicable.
PART II
Price Range of Common Stock
Our common stock has been listed on The NASDAQ Global Market under the symbol “ANTH” since our initial public offering (“IPO”). Prior to that offering, there was no public market for our common stock. The following table sets forth, for the periods indicated, the high and low intraday sales prices of our common stock as reported by The NASDAQ Global Market:
|
High
|
Low
|
|||||||
|
First Quarter 2014
|
$
|
3.79
|
$
|
2.70
|
||||
|
Second Quarter 2014
|
$
|
3.69
|
$
|
2.54
|
||||
|
Third Quarter 2014
|
$
|
3.50
|
$
|
1.53
|
||||
|
Fourth Quarter 2014
|
$
|
2.50
|
$
|
1.46
|
||||
|
First Quarter 2015
|
$
|
6.37
|
$
|
1.59
|
||||
|
Second Quarter 2015
|
$
|
9.89
|
$
|
4.05
|
||||
|
Third Quarter 2015
|
$
|
11.65
|
$
|
5.80
|
||||
|
Fourth Quarter 2015
|
$
|
7.15
|
$
|
4.25
|
||||
Holders of our Common Stock
As of December 31, 2015, an aggregate of 40,004,037 shares of our common stock were issued and outstanding and were held by 55 registered holders, based on information provided by the Company’s transfer agent. A significantly larger number of stockholders may be "street name" or beneficial holders, whose shares of record are held by banks, brokers and other financial institutions.
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently intend to retain any future earnings to finance the growth and development of our business. Therefore, we do not anticipate declaring or paying any cash dividends in the foreseeable future. Any future determination as to the declaration and payment of dividends, if any, will be at the discretion of our board of directors and will depend on then-existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant.
Securities Authorized for Issuance under Equity Compensation Plans
The information regarding securities authorized for issuance under equity compensation plans is included in Part III, Item 12 of this report.
Recent Sales of Unregistered Securities
All information under this Item has been previously reported on our Current Reports on Form 8-K, filed with the SEC.
Issuer Purchases of Equity Securities
We did not repurchase any securities during the quarter or year ended December 31, 2015.
The following tables reflect selected consolidated summary financial data for each of the last five fiscal years and are derived from our audited financial statements. This data should be read in conjunction with Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and Part II, Item 8, “Financial Statements and Supplementary Data”, appearing elsewhere in this Annual Report on Form 10-K.
|
Year Ended December 31,
|
||||||||||||||||||||
|
2015
|
2014
|
2013
|
2012
|
2011
|
||||||||||||||||
|
(in thousands, except share and per share data)
|
||||||||||||||||||||
|
Statement of Operations Data:
|
||||||||||||||||||||
|
Revenues
|
||||||||||||||||||||
|
License revenue
|
$ | 2,562 | $ | — | $ | — | $ | — | $ | — | ||||||||||
|
Collaborative revenue
|
623 | — | — | — | — | |||||||||||||||
|
Total revenues
|
3,185 | — | — | — | — | |||||||||||||||
|
Operating expenses
|
||||||||||||||||||||
|
Research and development
|
$ | 33,498 | $ | 21,839 | $ | 21,684 | $ | 49,219 | $ | 85,281 | ||||||||||
|
General and administrative
|
7,568 | 6,620 | 6,563 | 6,715 | 7,857 | |||||||||||||||
|
Research award (1)
|
(2,638 | ) | — | — | — | — | ||||||||||||||
|
Total operating expenses
|
38,428 | 28,459 | 28,247 | 55,934 | 93,138 | |||||||||||||||
|
Loss from operations
|
(35,243 | ) | (28,459 | ) | (28,247 | ) | (55,934 | ) | (93,138 | ) | ||||||||||
|
Other income (expense):
|
||||||||||||||||||||
|
Other income (expense)
|
23 | (96 | ) | (15 | ) | (111 | ) | 606 | ||||||||||||
|
Interest expense
|
— | (1,049 | ) | (2,599 | ) | (3,354 | ) | (2,803 | ) | |||||||||||
|
Mark-to-market adjustment of warrant liability
|
— | — | — | 14,070 | (3,738 | ) | ||||||||||||||
|
Total other income (expense)
|
23 | (1,145 | ) | (2,614 | ) | 10,605 | (5,935 | ) | ||||||||||||
|
Net loss
|
$ | (35,220 | ) | $ | (29,604 | ) | $ | (30,861 | ) | $ | (45,329 | ) | $ | (99,073 | ) | |||||
|
Net loss per share — basic and diluted (2)
|
$ | (0.99 | ) | $ | (1.36 | ) | $ | (1.69 | ) | $ | (6.27 | ) | $ | (21.18 | ) | |||||
|
Weighted average shares used in net loss per share -- basic and
diluted (3) |
35,631,237 | 21,776,269 | 18,267,413 | 7,225,406 | 4,677,210 | |||||||||||||||
|
December 31,
|
||||||||||||||||||||
|
2015
|
2014
|
2013
|
2012
|
2011
|
||||||||||||||||
|
(in thousands)
|
||||||||||||||||||||
|
Balance Sheet Data:
|
||||||||||||||||||||
|
Cash, cash equivalents and short-term investments
|
$
|
46,951
|
$
|
2,639
|
$
|
25,946
|
$
|
24,753
|
$
|
67,370
|
||||||||||
|
Restricted cash
|
—
|
—
|
10,000
|
—
|
—
|
|||||||||||||||
|
Working capital
|
39,394
|
(2,729
|
)
|
18,743
|
6,429
|
37,742
|
||||||||||||||
|
Total assets
|
48,125
|
3,490
|
37,417
|
26,445
|
69,493
|
|||||||||||||||
|
Total notes payable
|
—
|
—
|
17,875
|
20,550
|
24,331
|
|||||||||||||||
|
Total liabilities
|
8,468
|
5,751
|
22,659
|
29,971
|
66,747
|
|||||||||||||||
|
Common stock and additional paid-in capital
|
391,688
|
314,550
|
301,965
|
252,827
|
213,792
|
|||||||||||||||
|
Accumulated deficit
|
(352,031
|
)
|
(316,811
|
)
|
(287,207
|
)
|
(256,346
|
)
|
(211,017
|
)
|
||||||||||
|
Total stockholders’ equity (deficit)
|
39,657
|
(2,261
|
)
|
14,758
|
(3,526
|
)
|
2,746
|
|||||||||||||
|
(1)
|
In March 2015, we received a research award of up to $3 million from CFFT for our development of liprotamase. For the year ended December 31, 2015, we recognized $2.6 million in research award from CFFT in connection with achieving certain milestones specified in the award agreement. The amount has been recognized as a component of Operating Expenses.
|
|
(2)
|
Diluted earnings per share, or EPS, is identical to basic EPS since common equivalent shares are excluded from the calculation, as their effect is anti-dilutive.
|
|
(3)
|
Weighted average shares used in net loss per basic and diluted share in 2012 and 2011 have been adjusted to reflect a 1-for-8 reverse stock split effectuated by the Company on July 15, 2013.
|
The following discussion and analysis should be read together with our consolidated financial statements and the related notes set forth under “Item 8. Consolidated Financial Statements and Supplementary Data.” This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. See “Special Note Regarding Forward-Looking Statements” for a discussion of the uncertainties, risks and assumptions associated with these statements. Actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under “Risk Factors” and elsewhere in this report.
Overview
Anthera Pharmaceuticals, Inc. is a biopharmaceutical company focused on advancing the development and commercialization of innovative medicines that benefit patients with unmet medical needs. We currently have two compounds in development, liprotamase and blisibimod. We licensed liprotamase from Eli Lilly & Co (“Eli Lilly”) in July 2014. Liprotamase is a novel non-porcine investigational Pancreatic Enzyme Replacement Therapy (“PERT”) intended for the treatment of patients with Exocrine Pancreatic Insufficiency (“EPI”), often seen in patients with cystic fibrosis and other conditions. We licensed blisibimod from Amgen, Inc. (“Amgen”) in December 2007. Blisibimod targets B-cell activating factor, or BAFF, which has been shown to be elevated in a variety of B-cell mediated autoimmune diseases, including systemic lupus erythematosus, or lupus, Immunoglobulin A nephropathy, or IgA nephropathy, lupus nephritis, and others.
We were incorporated in September 2004. We have devoted substantially all of our resources to research and development of our product candidates. We have not generated any revenue from the commercial sales of our product candidates, and since inception we have funded our operations through equity offerings, private placements of convertible debt, debt financing, equity investment and cost reimbursement from a former collaborative partner, Zenyaku Kogyo Co., Ltd (“Zenyaku”), and a research award from Cystic Fibrosis Foundation Therapeutics Incorporated ("CFFT"). We will need substantial additional financing to continue to develop our product candidates, obtain regulatory approvals and to fund operating expenses, which we will seek to raise through public or private equity or debt financings, collaborative or other arrangements with third parties or through other sources of financing. We cannot assure you that such funds will be available on terms favorable to us, if at all. In addition to the normal risks associated with drug development companies, we may never successfully complete development of our product candidates, obtain adequate patent protection for our technology, obtain necessary government regulatory approval for our product candidates or achieve commercial viability for any approved product candidates. In addition, we may not be profitable even if we succeed in commercializing our product candidates.
In December 2014, we entered in an exclusive license agreement with Zenyaku for the development and commercialization of blisibimod in Japan and potentially other countries throughout Asia. In September 2015, Zenyaku provided us a notice of its intent to terminate the Zenyaku Agreement, effective January 7, 2016. The termination was “at will” and Zenyaku alleged no breach of the Zenyaku Agreement by us. There are no termination penalties incurred by us in connection with the early termination of the Zenyaku Agreement. As a result of the early termination of the Zenyaku Agreement, we do not expect to recognize revenues under the Zenyaku Agreement beyond January 2016. We also regained full worldwide rights for blisibimod and we are actively pursuing partnerships with pharmaceutical and biotech companies to further advance the development of blisibimod globally.
In March 2015, we received a research award of up to $3 million from CFFT for our development of liprotamase. We retain the right to develop and commercialize liprotamase and will owe royalties to CFFT on net sales of any drug candidate approved and commercialized under the collaboration. The funding is to be disbursed by CFFT to us upon our achievement of milestones specified in the agreement. At our discretion, we may choose to fund a particular stage of the liprotamase development plan without CFFT funds. Any CFFT funds not expended on the development program of liprotamase must be returned to CFFT and, upon such return, the amounts of such returned funds will not be included as part of the research award for the purpose of calculating royalties or other amounts owed by us to CFFT. To the extent CFFT provides or makes available any information, expertise, know-how or other intellectual property related to cystic fibrosis or the treatment, prevention or cure thereof (“CFFT Know-How”) to us, CFFT grants to us a non-exclusive, transferrable, sublicensable, worldwide rights and license under all of CFFT’s rights in such CFFT Know-How to assist us to research, develop, commercialize, make or have made, use, sell, have sold, offer for sale, import, export and otherwise exploit the product.
Our Phase 3 Development of Liprotamase in EPI
We initiated the SOLUTION study in the third quarter of 2015. SOLUTION is a Phase 3, randomized, open-label, assessor-blind, non-inferiority, active-comparator study evaluating the efficacy and safety of liprotamase in patients with cystic fibrosis-related exocrine pancreatic insufficiency. This pivotal study is intended to evaluate the non-inferiority of liprotamase compared with another commercially available PERT in a population enriched for PERT responders and will enroll approximately 130 patients in the United States and Europe. A second, smaller Phase 3 study, SIMPLICITY, in younger pediatric subjects is planned for initiation in first half of 2016. The SIMPLICITY study will use sachets containing a formulation of liprotamase powder for oral solution for ease of administration, and will enroll approximately 60 patients. We believe the SOLUTION and SIMPLICITY studies may offer a number of potential opportunities for differentiation versus the currently marketed PERTs, including:
|
•
|
Use of biotechnology-derived high-purity enzymes that are produced by fermentation processes rather than from mammalian organs which carry a label warning for viral transmission;
|
|
•
|
use of a novel chemically modified lipase drug substance that provides resistance to degradation;
|
|
•
|
a formulation containing an appropriate ratio of the three digestive enzymes (lipase, protease and amylase) to maximize relative efficacy while minimizing the potential for adverse events, such as fibrosing colonopathy;
|
|
•
|
a capsule formulation using known, safe, excipients that provides lower pill burden and good delivery performance. The pure, high-activity enzyme constituents, and absence of bulky enteric coating give rise to smaller, easy-to-swallow capsules with good disintegration once swallowed, and adequate storage stability compared with porcine PERTs of an equivalent lipase unit dose strength; and
|
|
•
|
a sachet formulation containing liprotamase power for oral solution which can be easily dissolved into water, and finally provides patients, especially young pediatric patients, with an easy-to-swallow dosing option.
|
Our Phase 3 Development of Blisibimod in Systemic Lupus Erythematosus (Lupus)
CHABLIS-SC1 is a multicenter, randomized, double-blind, placebo-controlled study designed to evaluate the efficacy, safety, tolerability and immunogenicity of blisibimod in patients with seropositive, clinically-active lupus (SELENA-SLEDAI ≥ 10) who require corticosteroid therapy in addition to standard-of-care for treatment of their disease. The study was designed to randomize up to 400 patients to receive either 200mg of blisibimod or placebo in addition to their standard-of-care medication for 52 weeks. We reached the enrollment target in June 2015. Topline data is expected after the last enrolled patient completes 52 weeks of treatment around the third quarter of 2016. The primary endpoint of the CHABLIS-SC1 will be clinical improvement in the SRI-6 response at 52 weeks. Key secondary outcomes from the study, including SRI-8, reduction in the number of lupus flares and steroid use, are intended to further differentiate blisibimod from currently available therapies. To date, enrolled patient demographics and disease characteristics for the CHABLIS-SC1 study are consistent with our goal to enroll patients with higher levels of lupus activity and positive biomarkers despite the stable use of corticosteroids. These characteristics were associated with improved outcomes in both our previous Phase 2 clinical study as well as in large Phase 3 studies conducted with other BAFF-inhibitors, belimumab (Benlysta) and tabalumab.
We believe the CHABLIS development program for blisibimod may offer a number of potential opportunities for differentiation versus the currently marketed BAFF antagonist and other novel B-cell directed therapies, including:
|
·
|
clinical differentiation:
|
|
Ø
|
potential for improved clinical response due to enriched patient selection;
|
|
Ø
|
a focus on requirement that patients in greater need of alternate therapy as defined by high disease activity and use of steroid therapy at time of randomization;
|
|
Ø
|
an improved clinical efficacy endpoint which requires a larger six-point reduction in the SELENA-SLEDAI;
|
|
Ø
|
earlier onset of clinical benefit by allowing earlier steroid taper and restriction of background medications;
|
|
Ø
|
potential to demonstrate reductions in lupus flares;
|
|
·
|
patient convenience: A convenient, at-home, patient-administered subcutaneous product;
|
|
·
|
mechanism of action: Blisibimod is able to inhibit the activity of both membrane-bound and soluble BAFF;
|
|
·
|
potential labeling differentiation: A second study (CHABLIS-7.5) will also aim to enroll patients with positive serology and low complements;
|
|
·
|
manufacturing: Blisibimod’s novel molecular structure, comprised of 2 identical peptide chains assembled into a covalent dimer, confers manufacturing benefits and lower cost of goods through a bacterial fermentation manufacturing process;
|
|
·
|
structure: Blisibimod’s 4 BAFF binding domains, compared to the typical 2 domains in a monoclonal antibody, domains, give rise a 350-fold higher affinity for blisibimod (1 picomolar affinity) compared with the reported affinity for the anti-BAFF monoclonal antibodies belimumab (Benlysta); and
|
|
·
|
fully human IgG1 Fc domain confers acceptable pharmacokinetic properties to support once-weekly dosing.
|
An independent Data Safety Monitoring Board (“DSMB”) meets regularly over the course of the study to assess patient safety. During these regular meetings, the DSMB reviews un-blinded safety data which include adverse events, suspected unexpected serious adverse reactions or SUSARs, deaths, laboratory data, and withdrawal data and compares trends between treatments. After the most recent scheduled meeting in November 2015, the DSMB recommended continuing the CHABLIS-SC1 and BRIGHT-SC clinical studies.
In February 2015, an interim analysis of CHABLIS-SC1 was conducted by an independent un-blinded statistician, who evaluated at a pre-specified time point, the proportion of responders to the systemic lupus erythematous SRI-6 responder index, and recommended the study to continue to completion as planned. This futility analysis was not intended to provide any rules for stopping for overwhelming efficacy, for a change in study sample size, or for an alteration of the study design. Rather, the analysis suggests that the observed data conforms with the assumptions upon which the trial was designed. The systemic lupus erythematosus response index is a recognized endpoint by the FDA for previously approved therapeutics. Prior to the interim analysis and on the advice of our Scientific Advisory Board who evaluated the published clinical data from other recent lupus studies with BAFF inhibitors, we modified the primary endpoint of CHABLIS-SC1 from a comparison of the proportion of responders to the SRI-8 responder index to a comparison of the proportion of responders to the SRI-6 responder index (previously a secondary endpoint of the study) as data, including emerging observations with tabalumab (Isenberg et al., Annual Meeting of the American College of Rheumatology, November 2014), suggest that SRI-6 responder rates are consistent and stable across multiple trials. Response rates to the SRI-8 responder index will remain a key secondary endpoint of the study. In addition to serving as a registration study for a potential lupus indication, observations in this study are intended to be included in marketing applications for blisibimod in IgA nephropathy and other indications.
We submitted a study protocol for our second lupus study, CHABLIS 7.5 (formerly named CHABLIS-SC2), to the FDA in the third quarter of 2015. The CHABLIS 7.5 study’s name emphasizes the intent to reduce background corticosteroid medication to ≤7.5mg prednisone in order to mitigate the risks associated with long-term steroid medication. This study will evaluate the effect of blisibimod on top of standard-of-care medication in patients with severe, seropositive SLE that is inadequately controlled with corticosteroids. Patient eligibility for this study is informed by responder traits identified in the Phase 2 trial with blisibimod as well as the large Phase 3 programs with other BAFF inhibitors, belimumab and tabalumab. We plan to initiate the CHABLIS 7.5 study in the first half of 2016. These two pivotal studies are anticipated to form the basis of submission for blisibimod as a treatment for active SLE that is not controlled by standard-of-care medication, including corticosteroids.
Our Phase 2 Development of Blisibimod for in IgA Nephropathy
The BRIGHT-SC study is a Phase 2 multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy, safety, tolerability and immunogenicity of blisibimod in IgA nephropathy. Enrollment criteria are biopsy-proven IgA nephropathy and proteinuria greater than one gram per 24 hours (1g/24hr). Patients must be receiving standard of care medication including angiotensin converting enzyme inhibitors and angiotensin receptor blockers. The BRIGHT-SC study was initiated in the second quarter of 2013. Patients enrolled in the BRIGHT-SC study receive 300mg weekly blisibimod or placebo subcutaneously during the first 8 weeks of therapy, the induction phase, followed by a minimum of 24 weeks of 200mg weekly blisibimod or placebo, the maintenance phase. The BRIGHT-SC clinical study has enrolled patients in Southeast Asia, the European Union (“EU”) and Eastern Europe.
In September 2013, we met with the U.S. FDA who agreed to consider accepting proteinuria as an endpoint for Subpart E approval for blisibimod in IgA nephropathy. In April 2014, we met with the Japan Pharmaceuticals and Medical Devices Agency (“PMDA”) to discuss our registration program for blisibimod in IgA nephropathy. In this meeting we gained the PMDA’s agreement on the acceptability of proteinuria as the primary efficacy endpoint to support marketing approval in Japan and have amended the BRIGHT-SC study to include the specific data requirements of the PMDA. In December 2014 we met with the European Medicines Agency (“EMA”) as part of the scientific advice process for blisibimod. We reached an agreement with the EMA on the acceptability of proteinuria as the primary efficacy variable as well as the requirement for a single study in support a Conditional Marketing Authorization Application (“CMAA”) provided that confirmatory evidence from a second study, would be available post approval. The EMA also recommended the protocol to provide information on the recommended duration of treatment, duration of response and need for re-treatment.
In March 2015, an interim futility analysis of BRIGHT-SC study was conducted by an independent unblinded statistician, who evaluated several important biomarkers of renal disease in patients who had completed at least 8 weeks of treatment and recommended the study to continue to completion as planned. We plan to conduct a six-month efficacy analysis in the first half of 2016 when approximately 40 patients have completed 6 months of treatment, followed by a topline analysis in the second half of 2016 when a majority of the patients have completed one year of treatment.
Revenue
We have not generated any revenue from the commercial sales of our product candidates since our inception and do not expect to generate any revenue from the commercial sales of our product candidates in the near term. However, as a result of the collaborative arrangement that we entered into with Zenyaku in December 2014 for the development of blisibimod, we began recognizing license fee revenue and collaborative revenue in 2015. The license fee from the collaborative arrangement with Zenyaku was initially amortized as revenue over the performance obligation period (product development period) while reimbursement for our FTEs was recorded as collaborative revenues as incurred. In September 2015, we received a termination notice from Zenyaku to terminate the collaborative arrangement, effective January 7, 2016. Consequently, we revised the amortization period of our deferred revenue to correspond with the shortened collaboration period and have fully amortized our deferred revenue as of January 7, 2016.
Research and Development Expenses
Since our inception, we have focused our activities on our product candidates development programs. We expense research and development costs as they are incurred. Research and development expenses consist of personnel costs, including salaries, benefits and stock-based compensation, clinical studies performed by contract research organizations, or CROs, materials and supplies, licenses and fees and overhead allocations consisting of various administrative and facilities-related costs. Research and development activities are also separated into three main categories: licensing, clinical development and pharmaceutical development. Licensing costs consist primarily of fees paid pursuant to license agreements. Historically, our clinical development costs have included costs for preclinical and clinical studies. We expect to incur substantial clinical development costs for the continued development of blisibimod. Pharmaceutical development costs consist of expenses incurred relating to clinical studies and product formulation and manufacturing.
We are developing our product candidates in parallel, and we typically use our employee and infrastructure resources across several projects. Thus, some of our research and development costs are not attributable to an individually named project. These unallocated costs include salaries, stock-based compensation charges and related “fringe benefit” costs for our employees (such as workers compensation and health insurance premiums), consulting fees and travel.
The following table shows our total research and development expenses for 2015, 2014, and 2013 (in thousands):
| Year Ended December 31, | ||||||||||||
| 2015 | 2014 | 2013 | ||||||||||
|
Allocated costs:
|
||||||||||||
|
Blisibimod (1) (2)
|
$
|
21,082
|
(1)
|
$
|
17,806
|
(2)
|
$
|
16,824
|
||||
|
Liprotamase
|
6,682
|
464
|
—
|
|||||||||
|
Varespladib and varespladib sodium (4)
|
—
|
(464
|
) (3)
|
323
|
||||||||
|
Unallocated costs
|
5,734
|
4,033
|
4,537
|
|||||||||
|
Total research and development expense
|
$
|
33,498
|
$
|
21,839
|
$
|
21,684
|
||||||
|
(1)
|
Includes reimbursed development costs totaling $1.5 million pursuant to the Zenyaku Agreement.
|
|
(2)
|
Included non-cash license fee $1.0 million in connection with an amendment to the Amgen Agreement.
|
|
(3)
|
Included refund of $0.5 million from a vendor for our VISTA-16 clinical study
|
|
(4)
|
All development efforts for Varespladib and Varespladib Sodium were terminated in 2012
|
We expect our research and development expenses to increase significantly as we continue to develop our product candidates. We intend to fund our clinical studies with existing cash and proceeds from potential future debt and equity offerings.
We expect that a large percentage of our research and development expenses in the future will be incurred in support of our current and future clinical development programs. These expenditures are subject to numerous uncertainties in timing and cost to completion. As we obtain results from clinical studies, we may elect to discontinue or delay clinical studies for certain clinical development programs in order to focus our resources on more promising clinical development programs. Completion of clinical studies may take several years or more, but the length of time generally varies according to the type, complexity, novelty and intended use of product candidates. The cost of clinical studies may vary significantly over the life of a program as a result of differences arising during clinical development, including:
|
•
|
the number of sites included in the studies;
|
|
•
|
the length of time required to enroll suitable patient subjects;
|
|
•
|
the number of patients that participate in the studies;
|
|
•
|
the number of doses that patients receive;
|
|
•
|
the drop-out or discontinuation rates of patients; and
|
|
•
|
the duration of patient follow-up.
|
Our expenses related to clinical studies are based on estimates of the services received and efforts expended pursuant to contracts with many research institutions, clinical research organizations and other service providers that conduct and manage clinical studies on our behalf. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price. Payments under the contracts are mainly driven by time and materials incurred by these service providers. Expenses related to clinical studies generally are accrued based on time and materials incurred by the service providers and in accordance with the contracts. If timelines or contracts are modified based upon changes to the clinical study design or scope of work to be performed, we modify our estimates of accrued expenses accordingly on a prospective basis.
None of our product candidates has received FDA or foreign regulatory marketing approval. In order to grant marketing approval, the FDA or foreign regulatory agencies must conclude that clinical data establishes the safety and efficacy of our product candidates and that the manufacturing facilities, processes and controls are adequate. Despite our efforts, our product candidates may not offer therapeutic or other improvement over existing, comparable drugs, be proven safe and effective in clinical studies, or meet applicable regulatory standards.
As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our development projects or when and to what extent we will receive cash inflows from the commercialization and sale of an approved product candidate, if ever.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational functions, including clinical, chemical manufacturing, regulatory, finance and business development. Other significant costs include professional fees for legal services, including legal services associated with obtaining and maintaining patents. We will continue to incur significant general and administrative expenses as a public company, including costs for insurance, costs related to the hiring of additional personnel, payment to outside consultants, lawyers and accountants and complying with the corporate governance, internal controls and similar requirements applicable to public companies.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. Generally Accepted Accounting Principles, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on our historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates.
While our significant accounting policies are more fully described in the accompanying notes to the consolidated financial statements included in this Annual Report on Form 10-K for the year ended December 31, 2015, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our reported financial results and affect the more significant judgments and estimates that we use in the preparation of our consolidated financial statements.
Revenue Recognition
During 2015, we had a collaborative arrangement with Zenyaku which provided for various types of payments to us, including development milestones, sales milestone, royalty, and reimbursement for a portion of the Company’s internal and external costs. All payments from Zenyaku were nonrefundable. The collaborative arrangement was on a best-efforts basis, did not require scientific achievement as a performance obligation and provided for payment to be made when costs were incurred or services were performed. The collaboration was terminated on January 7, 2016 pursuant to a termination notice from Zenyaku to us.
With respect to the collaborative arrangement with Zenyaku, we recognized revenue in accordance with FASB Accounting Standards Codification, or ASC 605 “ Revenue Recognition ”, subtopic ASC 605-25 “ Revenue with Multiple Element Arrangements ” and subtopic ASC 605-28 “ Revenue Recognition-Milestone Method ”, which provide accounting guidance for revenue recognition for arrangements with multiple deliverables and guidance on defining the milestone and determining when the use of the milestone method of revenue recognition for research and development transactions is appropriate, respectively. For multiple-element arrangements, each deliverable within a multiple deliverable revenue arrangement is accounted for as a separate unit of accounting if both of the following criteria are met: (1) the delivered item or items have value to the customer on a standalone basis and (2) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
The deliverables under the Zenyaku agreement had been determined to be a single unit of accounting and as such any license fees received were recorded as deferred revenue and recognized ratably over the term of the estimated performance period under the agreement, which was the product development period. As a result of the early termination of the Zenyaku Agreement, we revised the amortization period of our deferred revenue to correspond with the shortened collaboration period in the third quarter of 2015 and fully amortized our deferred revenue as of January 7, 2016.
For the collaborative research activities, we were entitled to reimbursement from Zenyaku for our internal personnel cost at a pre-determined full time equivalent (“FTE”) rate. Revenue related to FTE services was recognized as research services that were performed over the related performance periods. We were required to perform research and development activities as specified in the collaboration agreement. The payments received were not refundable and were based on a contractual reimbursement rate per FTE working on the project. Reimbursement for FTE cost was recorded as collaborative revenue as incurred.
Stock-Based Compensation
We account for stock options and stock purchase rights related to our equity incentive plans under the provisions of ASC 718 which requires the recognition of the fair value of stock-based compensation. The fair value of stock options is estimated using a Black-Scholes option valuation model. This model requires the input of subjective assumptions including expected stock price volatility, expected life and estimated forfeitures of each award. The fair value of equity-based awards is amortized ratably over the requisite service period of the award. Due to the limited amount of historical data available to us, particularly with respect to stock-price volatility, employee exercise patterns and forfeitures, actual results could differ from our assumptions.
We account for equity instruments issued to non-employees in accordance with the provisions of ASC 718 and ASC 505, “Equity.” As a result, the non-cash charge to operations for non-employee options with service or other performance criteria is affected each reporting period by changes in the estimated fair value of our common stock, as the underlying equity instruments vest. The two factors which most affect these changes are the price of the common stock underlying stock options for which stock-based compensation is recorded and the volatility of the stock price. If our estimates of the fair value of these equity instruments change, it may have the effect of significantly changing compensation expense
Accrued Clinical Expense
We make estimates of our accrued clinical expenses as of each balance sheet date in our consolidated financial statements based on facts and circumstances known to us at that time. This process involves reviewing open contracts and purchase orders, communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us at least monthly in arrears for services performed. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Examples of estimated accrued clinical expenses include:
|
•
|
fees paid to CROs in connection with clinical studies;
|
|
•
|
fees paid to investigative sites in connection with clinical studies;
|
|
•
|
fees paid to contract manufacturers in connection with the production of clinical study materials; and
|
|
•
|
fees paid to vendors in connection with preclinical development activities.
|
We base our accruals related to clinical studies on our estimates of the services received and efforts expended pursuant to contracts with many research institutions, clinical research organizations and other service providers that conduct and manage clinical studies on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price. Payments under the contracts are mainly driven by time and materials incurred by these service providers. In accruing for service fees, we estimate the time and materials incurred by these service providers in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual accordingly. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
Results of Operations
Comparison of Years Ended December 31, 2015 and 2014
Revenue
The following table summarizes our revenues for the years ended December 31, 2015 and 2014 (in thousands, except percentages):
|
2015
|
2014
|
$ Change
|
% Change
|
|||||||||||||
|
License revenue
|
$
|
2,562
|
$
|
—
|
$
|
2,562
|
100
|
%
|
||||||||
|
Collaborative revenue
|
623
|
—
|
623
|
100
|
%
|
|||||||||||
|
Total revenues
|
$
|
3,185
|
$
|
—
|
$
|
3,185
|
100
|
%
|
||||||||
We began to recognize revenues in 2015 as a result of the collaborative arrangement that we entered into with Zenyaku in December 2014 for the development of blisibimod. During the year ended December 31, 2015, we recorded $2.6 million for the amortization of the license fee and $0.6 million for the reimbursement of FTEs. License fee from the collaborative arrangement is amortized over the period of performance (product development period) while reimbursement for our FTEs is recorded as collaborative revenue. In September 2015, we received a termination notice from Zenyaku to terminate the collaborative arrangement, effective January 7, 2016. Consequently, we revised the amortization period of our deferred revenue to correspond with the shortened collaboration period and have fully amortized our deferred revenue as of January 7, 2016.
Research and Development Expense
The following table summarizes our research and development expenses for the years ended December 31, 2015 and 2014 (in thousands, except percentages):
|
2015
|
2014
|
$ Change
|
% Change
|
|||||||||||||
|
Research and development expense
|
$
|
33,498
|
$
|
21,839
|
$
|
11,659
|
53
|
%
|
||||||||
Research and development expenses increased in 2015 from 2014 primarily due to higher non-cash stock-based compensation recognized in 2015 as a result of options issued in 2015 and higher expense in manufacturing and CRO fees to support the ongoing CHABLIS-SC1 and BRIGHT-SC studies and enable the initiation of our Phase 3 SOLUTION study with liprotamase. The increase is partially offset by $1.5 million in expense reimbursements from Zenyaku for certain development costs associated with blisibimod.
General and Administrative Expense
The following table summarizes our general and administrative expenses for the years ended December 31, 2015 and 2014 (in thousands, except percentages):
|
2015
|
2014
|
$ Change
|
% Change
|
|||||||||||||
|
General and administrative expenses
|
$
|
7,568
|
$
|
6,620
|
$
|
948
|
14
|
%
|
||||||||
General and administrative expenses increased in 2015 from 2014 primarily due to higher non-cash stock-based compensation expense recognized in 2015 as a result of options issued in 2015 and higher expense related to professional services to support the growth of the Company.
Research Award
The following table summarizes our research award for the years ended December 31, 2015 and 2014 (in thousands, except percentages):
|
2015
|
2014
|
$ Change
|
% Change
|
|||||||||||||
|
Research Award
|
$
|
(2,638)
|
$
|
—
|
$
|
(2,638)
|
(100)
|
%
|
||||||||
In March 2015, we received a research award of up to $3 million from CFFT for our development of liprotamase. For the year ended December 31, 2015, we recognized $2.6 million in research award from CFFT in connection with achieving certain milestones specified in the award agreement. The amount has been recognized as a component of Operating Expenses. There was no research award recognized in 2014.
Other Income (Expense)
The following table summarizes our other income (expenses) for the years ended December 31, 2015 and 2014 (in thousands, except percentages):
|
2015
|
2014
|
$ Change
|
% Change
|
|||||||||||||
|
Other income (expense)
|
$
|
23
|
$
|
(96
|
)
|
$
|
119
|
124
|
%
|
|||||||
|
Interest expense
|
—
|
(1,049
|
)
|
1,049
|
100
|
%
|
||||||||||
|
Total other income (expense)
|
$
|
23
|
$
|
(1,145
|
)
|
$
|
1,168
|
102
|
%
|
|||||||
Income (expense) recorded in 2015 and 2014 was mainly a result of interest earned on our cash and cash equivalents and the net impact of realized gain or loss from foreign currency exchange fluctuations. There was no interest expense recorded in 2015 due to the full payment of our long-term debt in 2014.
Comparison of Years Ended December 31, 2014 and 2013
Research and Development Expense
The following table summarizes our research and development expenses for the years ended December 31, 2014 and 2013 (in thousands, except percentages):
|
2014
|
2013
|
$ Change
|
% Change
|
|||||||||||||
|
Research and development expense
|
$
|
21,839
|
$
|
21,684
|
$
|
155
|
1
|
%
|
||||||||
Research and development expense in the year ended December 31, 2014 included a non-cash charge of $1.0 million in license fee that we recognized in connection with an amendment to the Amgen Agreement. Excluding this non-cash license fee, research and development decreased by $0.9 million in 2014 from 2013. The decrease in research and development expense was mainly due to a decrease in clinical development expense as fewer subjects were treated in our Phase 3 CHABLIS SC1 and Phase 2 BRIGHT-SC studies as compared the PEARL-SC and Open Label Extension studies in 2013.
General and Administrative Expense
The following table summarizes our general and administrative expenses for the years ended December 31, 2014 and 2013 (in thousands, except percentages):
|
2014
|
2013
|
$ Change
|
% Change
|
|||||||||||||
|
General and administrative expenses
|
$
|
6,620
|
$
|
6,563
|
$
|
57
|
1
|
%
|
||||||||
The change in general and administrative expenses during the year ended December 31, 2014 was insignificant as compared to the year ended in 2013.
Other Income (Expense)
The following table summarizes our other income (expenses) for the years ended December 31, 2014 and 2013 (in thousands, except percentages):
|
2014
|
2013
|
$ Change
|
% Change
|
|||||||||||||
|
Other expense
|
$
|
(96
|
)
|
$
|
(15
|
)
|
$
|
(81
|
)
|
540
|
%
|
|||||
|
Interest expense
|
(1,049
|
)
|
(2,599
|
)
|
1,550
|
(60
|
)%
|
|||||||||
|
Total Other income (expense)
|
$
|
(1,145
|
)
|
$
|
(2,614
|
)
|
$
|
1,469
|
(56
|
)%
|
||||||
Other expense recorded in both 2014 and 2013 was a result of net loss realized from foreign currency exchange fluctuations. The decrease in interest expense in 2014 as compared to 2013 is primarily due to the refinancing of our debt from Hercules to MidCap and Square 1 Bank in April 2013, which resulted in a significantly reduced interest rate, and repayment of our debts to Midcap and Square 1 Bank in October and December 2014, respectively.
Liquidity and Capital Resources
To date, we have funded our operations primarily through private placements of preferred stock and common stock, convertible debt, debt financings and public offerings of common stock, equity investment and cost reimbursement from a collaborative partner, and a research award from CFFT. As of December 31, 2015, we had cash and cash equivalents of approximately $47.0 million.
Our principal liquidity requirements are primarily to meet our working capital needs, support ongoing business activities, research and development, and our capital expenditure needs.
On March 12, 2015, we executed an equity purchase agreement (as amended, the “Purchase Agreement”), with Lincoln Park Capital Fund, LLC (“LPC”) pursuant to which we have the right, but not the obligation, to sell to LPC up to an aggregate of $10.0 million of common stock over a period of two years in amounts as set forth in the Purchase Agreement. In July 2015, we amended the Purchase Agreement and reduced the amount LPC can purchase to an aggregate of $6.0 million of common stock over a period of two years. No sales of common stock have been made under the Purchase Agreement as of December 31, 2015. The Purchase Agreement will expire in March 2017.
On November 15, 2013, we entered into a Sales Agreement (the “Agreement”) with Cowen to create an at-the-market (“ATM’) offering program under which the Company from time to time may offer and sell shares of its common stock, par value $0.001 per share, having an aggregate offering price of up to $25.0 million through Cowen, as agent. As of December 31, 2015, we have sold $21.8 million of common stock through the ATM program, of which $12.4 million was sold during the year ended December 31, 2015, leaving a balance of $3.2 million available for future sale pursuant to the Agreement.
In April 2013, we filed a universal shelf registration statement with the SEC on Form S-3 (File No. 333-187780) for the proposed offering from time to time of up to $100.0 million of our securities, including common stock, preferred stock, debt securities and/or warrants. On November 15, 2013, we registered $25.0 million under the registration statement for the Cowen ATM. In 2015, we registered $10.0 million under the registration statement for the issuance of our common stock to Zenyaku and Amgen. In March 2015, we registered $10.3 million under the registration statement for the 2015 Purchase Agreement with LPC, which was subsequently reduced to $6.3 million in July 2015. In March and July 2015, we utilized $57.5 million of this registration statement through the issuance of 10,222,223 shares of common stock in two separate public offerings. As of December 31, 2015, there was $1.2 million available for future issuance under this shelf registration statement. The current shelf registration statement is scheduled to expire in April 2016 and we expect to file a replacement shelf registration statement to register up to $100.0 million of our securities prior to that date.
Based on the requirements of Form S-3, however, there are certain factors, such as volume of trading in our common stock and our stock price, which may limit the amount that can be raised in a short period of time through the Purchase Agreement and registration statements described above.
Cash Flows
Comparison of Years Ended December 31, 2015 and 2014
Cash flows during the years ended December 31, 2015 and 2014 consisted of the following (in thousands):
|
2015
|
2014
|
|||||||
|
Net cash used in operating activities
|
$
|
(30,907)
|
$
|
(25,601
|
)
|
|||
|
Net cash provided by (used in) investing activities
|
(80)
|
10,000
|
||||||
|
Net cash provided by (used in) financing activities
|
75,299
|
(7,706
|
)
|
|||||
|
Total
|
$
|
44,312
|
$
|
(23,307
|
)
|
|||
During 2015 and 2014, our operating activities used cash of $30.9 million and $25.6 million, respectively. The increase in cash used for operating activities in 2015 was mainly attributable to higher research and development expense due to the continued advancement of blisibimod in the clinics and the launch of a new registration enabling trial with liprotamase.
During the year ended December 31, 2015, investing activities used cash of $80,000 in capital expenditures. During the year ended December 31, 2014, investing activities provided cash of $10.0 million, which amount represented the release of $10.0 million in restricted cash as a result of repayment of our long-term debt to Square 1 Bank.
During the year ended December 31, 2015, financing activities provided cash of $75.3 million from the issuance of common stock through two rounds of public offerings, an ATM program, and equity investments by Zenyaku. During the year ended December 31, 2014, financing activities used cash of $7.7 million, which represented the net effect of $18.1 million used for the early retirement of our debt to MidCap, offset by $10.4 million in net proceeds received from the sale of stock to LPC and pursuant to an ATM program.
Comparison of Years Ended December 31, 2014 and 2013
Cash flow from operations during the years ended December 31, 2014 and 2013 consisted of the following (in thousands):
|
2014
|
2013
|
|||||||
|
Net cash used in operating activities
|
$
|
(25,601
|
)
|
$
|
(31,912
|
)
|
||
|
Net cash provided by (used in) investing activities
|
10,000
|
(4,687
|
)
|
|||||
|
Net cash provided by (used in) financing activities
|
(7,706
|
)
|
43,115
|
|||||
|
Effect of exchange rate changes on cash
|
—
|
(1
|
)
|
|||||
|
Total
|
$
|
(23,307
|
)
|
$
|
6,515
|
|||
During 2014 and 2013, our operating activities used cash of $25.6 million and $31.9 million, respectively. The decrease in cash used in 2014 compared to 2013 was mainly attributable to lower research and development expense due to the Company focusing exclusively on the development of blisibimod and liprotamase. In addition, interest expense also decreased significantly in 2014 as compared to 2013 as a result of our refinancing of our long-term debt from Hercules to Midcap and Square 1 Bank in early 2013 and repayment of our debts to Midcap and Square 1 Bank in October and December 2014, respectively.
During the year ended December 31, 2014, investing activities provided cash of $10.0 million, which amount represented the release of $10.0 million in restricted cash as a result of repayment of our long-term debt to Square 1 Bank. During the year ended December 31, 2013, investing activities used cash of $4.7 million, which amount represented the effect of the establishment of $10.0 million in restricted cash for the Square 1 Bank debt, offset by proceeds of $5.3 million from the maturities of short-term investments.
During the year ended December 31, 2014, financing activities used cash of $7.7 million, which represented the net effect of $18.1 million used for the early retirement of our debt to MidCap, offset by $10.4 million in net proceeds received from the sale of stock to LPC and pursuant to the ATM. During the year ended December 31, 2013, financing activities provided cash of $43.1 million, which represented the net effect of $45.7 million in net proceeds received from a public offering in January 2013 and $19.8 million in net proceeds received from the issuance of notes payable, offset by net principal payments of $22.4 million.
Contractual Obligations and Commitments
We have lease obligations consisting of an operating lease for our operating facility that expires in September 2017.
The following table summarizes our estimated scheduled future minimum contractual obligations and commitments as of December 31, 2015 (in thousands):
|
Payment Due by Period
|
||||||||||||||||||||
|
Contractual Obligations
|
Less than
1 year
|
1-3 years
|
3-5 years
|
More than
5 years |
Total
|
|||||||||||||||
|
Facility Lease
|
$
|
230
|
$
|
177
|
$
|
—
|
$
|
—
|
$
|
407
|
||||||||||
The above amounts exclude potential payments to be made under our license agreements to our licensors that are based on the progress of our product candidates in development, as these payments are not determinable.
Under the Amgen Agreement, we are obligated to make additional milestone payments upon the achievement of certain development, regulatory and commercial objectives. We are also obligated to pay royalties on future net sales of products that are developed and approved as defined by this collaboration. Our royalty obligations as to a particular licensed product will be payable on a country-by-country basis and licensed on a product-by-licensed-product basis, for the longer of (a) the date of expiration of the last-to-expire valid claim within the licensed patents that covers the manufacture, use or sale, offer to sell or import of such licensed product by us or a sublicensee in such country, or (b) 10 years after the first commercial sale of the applicable licensed product in the applicable country.
Under the Lilly Agreement, we are obligated to make milestone payments upon the achievement of certain regulatory and commercial sales milestones. In addition, after sales of the licensed products exceed an aggregate of $100.0 million in the United States, we are obligated to pay tiered royalties on future net sales, ranging from the single digits to the mid-teens, for products that are developed and approved as defined in the Lilly Agreement. Our royalty obligations as to a particular licensed product will be payable, on a licensed product-by-licensed product basis, for the longer of (a) the date of expiration of the last to expire valid claim within the licensed patents that covers the manufacture, use or sale, offer to sell, or import of such licensed product by the Company or a sublicense in such country, or (b) 12 years after the first commercial sale of the applicable licensed product in the applicable country.
Under the research award agreement with CFFT, we are obligated to pay royalties to CFFT as follows: i) a one-time royalty in an amount equal to five times the actual award, payable in three installments between the first and second anniversaries of the first commercial sale of a product; ii) a one-time royalty in an amount equal to the actual award after net product sales reaches $100 million; and iii) in the event of a license, sale or other transfer of the product or a change of control transaction prior to the commercial sale of the product, a milestone payment equal to three times the actual award.
Funding Requirements
To date, we have not generated any revenue. We expect to incur substantial expenses and generate significant operating losses over the next several years as we continue to advance our product candidates into clinical studies and as we:
|
•
|
continue clinical development of blisibimod;
|
|
•
|
hire additional clinical, scientific and management personnel; and
|
|
•
|
implement new operational, financial and management information systems.
|
Our future capital uses and requirements depend on numerous forward-looking factors. These factors include the following:
|
•
|
the progress of clinical studies of our product candidates;
|
|
•
|
the time and costs involved in obtaining regulatory approvals;
|
|
•
|
delays that may be caused by evolving requirements of regulatory agencies;
|
|
•
|
the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims;
|
|
•
|
our ability to establish, enforce and maintain selected strategic alliances; and
|
|
•
|
the acquisition of technologies, product candidates and other business opportunities that require financial commitments.
|
As of the filing of this report, we believe our existing cash, anticipated equity investment and access to an equity purchase agreement with LPC and our ATM program will enable us to meet our obligations and sustain our operations through at least the next 12 months. However, we may require significant additional funds earlier than we currently expect to conduct additional or extended clinical studies and seek regulatory approval of our product candidates. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical studies.
Additional funding may not be available to us on acceptable terms or at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities or by selling debt securities, if convertible, further dilution to our existing stockholders may result. To the extent our capital resources are insufficient to meet our future capital requirements, we will need to finance our future cash needs through public or private equity offerings, collaboration agreements, debt financings or licensing arrangements.
If adequate funds are not available, we may be required to terminate, significantly modify or delay our development programs, or obtain funds through collaborators that may require us to relinquish rights to our technologies or product candidates that we might otherwise seek to develop or commercialize independently. We may elect to raise additional funds even before we need them if the conditions for raising capital are favorable.
Off-Balance Sheet Arrangements
We do not currently have, nor have we ever had, any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts.
Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates. We are exposed to market risk related to fluctuations in interest rates, market prices, and foreign currency exchange rates. However, since a majority of our investments are in highly liquid money market funds, we do not believe we are subject to any material market risk exposure. As of December 31, 2015, we did not have any material derivative financial instruments. The fair value of our cash and cash equivalents was $47.0 million as of December 31, 2015.
Our investment policy is to limit credit exposure through diversification and investment in highly rated securities. We actively review, along with our investment advisors, current investment ratings, company specific events and general economic conditions in managing our investments and in determining whether there is a significant decline in fair value that is other-than-temporary.
The consolidated financial statements required by this item are set forth beginning in Item 15 of this report and are incorporated herein by reference.
Not applicable.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive officer and Principal Accounting Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2015. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities and Exchange Act of 1934, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Securities Exchange Act of 1934 is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Securities Exchange Act of 1934 is accumulated and communicated to that company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by SEC Rule 13a-15(e), we carried out an evaluation under the supervision and with the participation of our management, including our Chief Executive Officer and Principal Accounting Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the year covered by this report. Based on the foregoing, our Chief Executive Officer and Principal Accounting Officer concluded that, as of December 31, 2015, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act. Internal control over financial reporting is a process, effected by an entity’s board of directors, management and other personnel, designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with GAAP. Internal control over financial reporting includes those policies and procedures which pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of assets; provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with GAAP; provide reasonable assurance that receipts and expenditures are being made only in accordance with management’s and or the board of directors’ authorization; and provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our consolidated financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect material errors in our consolidated financial statements. Also, projection of any evaluation of the effectiveness of our internal control over financial reporting to future periods is subject to the risk that controls may become inadequate because of changes in conditions, because the degree of compliance with our policies and procedures may deteriorate.
Management, including our chief executive officer and principal accounting officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2015. In making its assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control-Integrated Framework (2013). Based on the results of our evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2015. We reviewed the results of management’s assessment with the Audit Committee of our board of directors. Based on this assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2015. BDO USA, LLP, our independent registered public accounting firm, has issued an attestation report on our internal control over financial reporting as of December 31, 2015, which is included herein.
Changes in Internal Control Over Financial Reporting
There have been no other changes in our internal control over financial reporting during the most recent quarter ended December 31, 2015 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
PART III
The information required by this Item 10 is incorporated by reference from our definitive Proxy Statement for our 2016 Annual Meeting of Stockholders (“Proxy Statement”), where it appears under the headings “Election of Directors”, “Executive Officers” and “Section 16(a) Beneficial Ownership Reporting Compliance.”
We have adopted a Code of Ethics that applies to all directors, officers and employees of the Company. We publicize the Code of Business Conduct and Ethics through posting the policy on our website, http://www.anthera.com.
The information required by this Item 11 is incorporated by reference from our Proxy Statement where it appears under the headings “Compensation Discussion and Analysis”, “Compensation of Executive Officers”, “Election of Directors” and “Compensation Committee Report.”
The information required by this Item 12 is incorporated by reference from our Proxy Statement where it appears under the headings “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information.”
The information required by this Item 13 is incorporated by reference from our Proxy Statement where it appears under the headings “Certain Relationships and Related Transactions” and “Election of Directors.”
The information required by this Item 14 is incorporated by reference from our Proxy Statement where it appears under the heading “Ratification of Auditors.”
PART IV
STATEMENT SCHEDULES
|
(a)
|
The following documents are filed as part of this report:
|
|
|
(1)
|
Index list to Consolidated Financial Statements:
|
|
Page
|
|
|
71
|
|
|
72
|
|
|
73
|
|
|
74
|
|
|
75
|
|
|
76
|
|
|
77
|
|
|
(2)
|
Consolidated financial statement Schedules
|
All other schedules are omitted because they are not required or the required information is included in the consolidated financial statements or notes thereto.
|
|
(3)
|
Exhibits
|
The exhibits listed in the accompanying Exhibit Index are filed or incorporated by reference as part of this report.
Board of Directors and Stockholders
Anthera Pharmaceuticals, Inc.
Hayward, California
We have audited the accompanying consolidated balance sheets of Anthera Pharmaceuticals, Inc. as of December 31, 2015 and 2014 and the related consolidated statements of operations and comprehensive loss, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Anthera Pharmaceuticals, Inc. at December 31, 2015 and 2014, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2015, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Anthera Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated March 14, 2016, expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
San Jose, California
March 14, 2016
Board of Directors and Stockholders
Anthera Pharmaceuticals, Inc.
Hayward, California
We have audited Anthera Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Anthera Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Item 9A, Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Anthera Pharmaceuticals, Inc.’s maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Anthera Pharmaceuticals, Inc. as of December 31, 2015 and 2014, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2015 and our report dated March 14, 2016, expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
San Jose, California
March 14, 2016
ANTHERA PHARMACEUTICALS, INC.
(in thousands, except per share data)
|
December 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Assets
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
46,951
|
$
|
2,639
|
||||
|
Accounts receivable
|
326
|
—
|
||||||
|
Prepaid expenses and other current assets
|
585
|
383
|
||||||
|
Total current assets
|
47,862
|
3,022
|
||||||
|
Property and equipment, net
|
263
|
468
|
||||||
|
Total assets
|
$
|
48,125
|
$
|
3,490
|
||||
|
Liabilities and Stockholders' Equity
|
||||||||
|
Liabilities
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$
|
5,259
|
$
|
2,232
|
||||
|
Accrued clinical studies
|
1,377
|
1,239
|
||||||
|
Accrued liabilities
|
98
|
211
|
||||||
|
Accrued payroll and related costs
|
1,596
|
1,069
|
||||||
|
Other current liabilities
|
—
|
1,000
|
||||||
|
Deferred revenue - current
|
138
|
—
|
||||||
|
Total current liabilities
|
8,468
|
5,751
|
||||||
|
Total liabilities
|
8,468
|
5,751
|
||||||
|
Commitments and contingencies (Note 7)
|
||||||||
|
Stockholders' equity (deficit):
|
||||||||
|
Common stock, $0.001 par value, 100,000,000 shares authorized; 40,004,037 and 23,005,209
shares issued and outstanding as of December 31, 2015 and 2014, respectively |
40
|
23
|
||||||
|
Additional paid-in capital
|
391,648
|
314,527
|
||||||
|
Accumulated deficit
|
(352,031)
|
(316,811
|
)
|
|||||
|
Total stockholders' equity (deficit)
|
39,657
|
(2,261)
|
||||||
|
Total liabilities and stockholders' equity
|
$
|
48,125
|
$
|
3,490
|
||||
See accompanying notes to consolidated financial statements.
ANTHERA PHARMACEUTICALS, INC.
(in thousands, except per share data)
|
Years ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
REVENUES:
|
||||||||||||
|
License revenue
|
$
|
2,562
|
$
|
—
|
$
|
—
|
||||||
|
Collaborative revenue
|
623
|
—
|
—
|
|||||||||
|
Total revenues
|
3,185
|
—
|
—
|
|||||||||
|
OPERATING EXPENSES:
|
||||||||||||
|
Research and development
|
$
|
33,498
|
$
|
21,839
|
$
|
21,684
|
||||||
|
General and administrative
|
7,568
|
6,620
|
6,563
|
|||||||||
|
Research award
|
(2,638)
|
—
|
—
|
|||||||||
|
Total operating expenses
|
38,428
|
28,459
|
28,247
|
|||||||||
|
LOSS FROM OPERATIONS
|
(35,243)
|
(28,459
|
)
|
(28,247
|
)
|
|||||||
|
OTHER INCOME (EXPENSE):
|
||||||||||||
|
Other income (expense)
|
23
|
(96
|
)
|
(15
|
)
|
|||||||
|
Interest expense
|
—
|
(1,049
|
)
|
(2,599
|
)
|
|||||||
|
Total other income (expense)
|
23
|
(1,145
|
)
|
(2,614
|
)
|
|||||||
|
NET LOSS
|
$
|
(35,220)
|
$
|
(29,604
|
)
|
$
|
(30,861
|
)
|
||||
|
Other comprehensive income (loss):
|
||||||||||||
|
Unrealized gain on short-term investments, net
|
—
|
—
|
17
|
|||||||||
|
COMPREHENSIVE LOSS
|
(35,220)
|
$
|
(29,604
|
)
|
(30,844
|
)
|
||||||
|
Net loss per share—basic and diluted
|
$
|
(0.99)
|
$
|
(1.36
|
)
|
$
|
(1.69
|
)
|
||||
|
Weighted-average number of shares used in
per share calculation—basic and diluted
|
35,631,237
|
21,776,269
|
18,267,413
|
|||||||||
See accompanying notes to consolidated financial statements.
ANTHERA PHARMACEUTICALS, INC.
(in thousands except share and per share amounts)
|
Common Stock
|
Additional | Accumulated Other |
Total | |||||||||||||||||||||
|
Shares
|
Amount
|
Paid-In
Capital |
Comprehensive
Loss |
Accumulated
Deficit |
Stockholders’
Equity (Deficit) |
|||||||||||||||||||
|
BALANCE—December 31, 2012
|
9,893,924 | $ | 10 | $ | 252,827 | $ | (17 | ) | $ | (256,346 | ) | $ | (3,526 | ) | ||||||||||
|
Issuance of common stock upon release
of restricted stock units
|
31,081 | — | 51 | — | — | 51 | ||||||||||||||||||
|
Issuance of common stock pursuant to
employee stock purchase plan
|
13,961 | — | 38 | — | — | 38 | ||||||||||||||||||
|
Issuance of common stock for cash at
$5.28 per share, net of issuance cost
of $474
|
8,712,119 | 9 | 42,737 | — | — | 42,746 | ||||||||||||||||||
|
Issuance of common stock pursuant to
an equity purchase agreement, net of
issuance cost of $398
|
764,816 | — | 2,959 | — | — | 2,959 | ||||||||||||||||||
|
Share-based compensation related to
equity awards
|
— | — | 3,054 | — | — | 3,054 | ||||||||||||||||||
|
Issuance of warrants in conjunction with
debt financing
|
— | — | 280 | — | — | 280 | ||||||||||||||||||
|
Unrealized loss on short-term
investments
|
— | — | — | 17 | — | 17 | ||||||||||||||||||
|
Net loss
|
— | — | — | — | (30,861 | ) | (30,861 | ) | ||||||||||||||||
|
BALANCE—December 31, 2013
|
19,415,901 | 19 | 301,946 | — | (287,207 | ) | 14,758 | |||||||||||||||||
|
Issuance of common stock upon release
of restricted stock units
|
66,704 | 1 | 195 | — | — | 196 | ||||||||||||||||||
|
Issuance of common stock pursuant to
exercise of stock options and
employee stock purchase plan
|
27,000 | — | 37 | — | — | 37 | ||||||||||||||||||
|
Issuance of common stock pursuant to
an equity purchase agreement, net of
issuance cost of $7
|
512,626 | — | 1,301 | — | — | 1,301 | ||||||||||||||||||
|
Issuance of common stock pursuant to
an at-the-market equity program, net
of issuance cost of $354
|
2,982,978 | 3 | 9,047 | — | — | 9,050 | ||||||||||||||||||
|
Share-based compensation related to
equity awards
|
— | — | 2,001 | — | — | 2,001 | ||||||||||||||||||
|
Net loss
|
— | — | — | (29,604 | ) | (29,604 | ) | |||||||||||||||||
|
BALANCE—December 31, 2014
|
23,005,209 | $ | 23 | $ | 314,527 | $ | — | $ | (316,811 | ) | $ | (2,261 | ) | |||||||||||
|
Issuance of common stock upon release
of restricted stock units
|
515 | — | 3 | — | — | 3 | ||||||||||||||||||
|
Issuance of common stock pursuant to
exercise of stock options and
employee stock purchase plan
|
140,662 | — | 410 | — | — | 410 | ||||||||||||||||||
|
Issuance of common stock pursuant to
an equity purchase agreement, net of
issuance cost of $60
|
59,338 | — | 75 | — | — | 75 | ||||||||||||||||||
|
Issuance of common stock pursuant to
an at-the-market equity program, net
of issuance cost of $379
|
3,208,529 | 3 | 12,055 | — | — | 12,058 | ||||||||||||||||||
|
Issuance of common stock for cash at
average $5.62 per share, net of
issuance cost of $3,744
|
10,222,223 | 10 | 53,746 | — | — | 53,756 | ||||||||||||||||||
|
Issuance of common stock to Zenyaku
Kogyo Co., Ltd. for cash
|
2,946,810 | 3 | 6,297 | — | — | 6,300 | ||||||||||||||||||
|
Issuance of common stock to Amgen to
settle a license fee obligation
|
420,751 | 1 | 999 | — | — | 1,000 | ||||||||||||||||||
|
Share-based compensation related to
equity awards
|
— | — | 3,536 | — | — | 3,536 | ||||||||||||||||||
|
Net loss
|
— | — | — | (35,220 | ) | (35,220 | ) | |||||||||||||||||
|
BALANCE—December 31, 2015
|
40,004,037 | $ | 40 | $ | 391,648 | $ | — | $ | (352,031 | ) | $ | 39,657 | ||||||||||||
See accompanying notes to consolidated financial statements.
ANTHERA PHARMACEUTICALS, INC.
(in thousands)
|
Year Ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net loss
|
$
|
(35,220
|
)
|
$
|
(29,604
|
)
|
$
|
(30,861
|
)
|
|||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Depreciation
|
285
|
344
|
353
|
|||||||||
|
Realized loss on short-term investments
|
—
|
—
|
12
|
|||||||||
|
Stock-based compensation expense
|
3,541
|
2,175
|
3,145
|
|||||||||
|
Common stock issued to settle a license fee obligation
|
—
|
1,000
|
—
|
|||||||||
|
Amortization of discount and deferred interest on notes payable
|
—
|
219
|
979
|
|||||||||
|
Amortization of debt issuance cost
|
—
|
226
|
190
|
|||||||||
|
Changes in operating assets and liabilities:
|
||||||||||||
|
Accounts receivable
|
(326
|
)
|
—
|
—
|
||||||||
|
Prepaid expenses and other current assets
|
(202
|
)
|
(26
|
)
|
66
|
|||||||
|
Accounts payable
|
3,027
|
(1,210
|
)
|
(1,833
|
)
|
|||||||
|
Accrued clinical studies
|
138
|
587
|
(2,724
|
)
|
||||||||
|
Accrued liabilities
|
(113
|
)
|
(46
|
)
|
(1,214
|
)
|
||||||
|
Accrued payroll and related costs
|
527
|
734
|
(25
|
)
|
||||||||
|
Deferred revenue
|
(2,564
|
)
|
—
|
—
|
||||||||
|
Net cash used in operating activities
|
(30,907)
|
(25,601
|
)
|
(31,912
|
)
|
|||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Property and equipment purchases
|
(80
|
)
|
—
|
(15
|
)
|
|||||||
|
Proceeds from maturities of short-term investments
|
—
|
—
|
5,328
|
|||||||||
|
Decrease (increase) in restricted cash
|
—
|
10,000
|
(10,000
|
)
|
||||||||
|
Net cash provided by (used in) investing activities
|
(80
|
)
|
10,000
|
(4,687
|
)
|
|||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Proceeds from issuance of notes payable, net of issuance costs
|
—
|
—
|
19,798
|
|||||||||
|
Principal payment against note payable
|
—
|
(18,094
|
)
|
(22,436
|
)
|
|||||||
|
Proceeds from issuance of common stock, net of offering costs
|
65,889
|
10,351
|
45,746
|
|||||||||
|
Proceeds from issuance of common stock to Zenyaku Kogyo Co., Ltd.
|
9,000
|
—
|
—
|
|||||||||
|
Proceeds from issuance of common stock pursuant to exercise of stock options
and employee stock purchase plan
|
410
|
37
|
39
|
|||||||||
|
Withholding taxes paid on vested restricted stock units
|
—
|
—
|
(32
|
)
|
||||||||
|
Net cash provided by (used in) financing activities
|
75,299
|
(7,706
|
)
|
43,115
|
||||||||
|
Effect of exchange rate changes on cash
|
—
|
—
|
(1
|
)
|
||||||||
|
Net increase (decrease) in cash and cash equivalents
|
44,312
|
(23,307
|
)
|
6,515
|
||||||||
|
Cash and cash equivalents, beginning of period
|
2,639
|
25,946
|
19,431
|
|||||||||
|
Cash and cash equivalents, end of period
|
$
|
46,951
|
$
|
2,639
|
$
|
25,946
|
||||||
|
SUPPLEMENTAL CASH DISCLOSURES OF CASH FLOW INFORMATION
|
||||||||||||
|
Cash paid for interest
|
$
|
—
|
$
|
655
|
$
|
2,477
|
||||||
|
Issuance of common stock as a commitment fee pursuant to an equity purchase agreement
|
$
|
60
|
$
|
7
|
$
|
398
|
||||||
|
Issuance of common stock to settle a license fee obligation
|
$
|
1,000
|
$
|
—
|
$
|
—
|
||||||
|
Issuance of warrants in conjunction with debt financing
|
$
|
—
|
$
|
—
|
$
|
280
|
||||||
See accompanying notes to consolidated financial statements.
ANTHERA PHARMACEUTICALS, INC.
|
1.
|
ORGANIZATION AND DESCRIPTION OF BUSINESS
|
Organization
Anthera Pharmaceuticals, Inc. (“the Company”) is a biopharmaceutical company focused on advancing the development and commercialization of innovative medicines that benefit patients with unmet medical needs. The Company currently has two compounds in development, liprotamase and blisibimod. The Company licensed liprotamase from Eli Lilly & Co (“Eli Lilly”) in July 2014. Liprotamase is a novel non-porcine investigational Pancreatic Enzyme Replacement Therapy (“PERT”) intended for the treatment of patients with Exocrine Pancreatic Insufficiency (“EPI”), often seen in patients with cystic fibrosis and other conditions. The Company licensed blisibimod from Amgen, Inc. (“Amgen”) in December 2007. Blisibimod targets B-cell activating factor or (“BAFF”) which has been shown to be elevated in a variety of B-cell mediated autoimmune diseases, including systemic lupus erythematosus (“SLE”), or lupus, Immunoglobulin A nephropathy, or IgA nephropathy, lupus nephritis, and others.
Liquidity and Need for Additional Capital
The Company’s planned principal operations are acquiring product and technology rights, raising capital and performing research and development activities. The Company is currently conducting research and development activities to treat autoimmune diseases and EPI. The Company’s activities are subject to significant risks and uncertainties. Successful completion of the Company’s development programs and, ultimately, the attainment of profitable operations are dependent on future events, including, among other things, its ability to access potential markets; secure financing; develop a customer base; attract, retain and motivate qualified personnel; and develop strategic alliances.
Since inception in 2004, the Company has funded its operations through equity offerings, private placements of convertible debt, debt financing, equity investment and cost reimbursement from a collaborative partner, and a research award from Cystic Fibrosis Foundation Therapeutics Incorporated ("CFFT"). As of the date of this report, the Company anticipates its existing cash, and access to additional cash through an equity purchase agreement with Lincoln Park Capital (“LPC”) and an at-the-market (“ATM”) Sales Agreement with Cowen & Company, LLC (“Cowen”) will be sufficient to fund its near term liquidity needs for at least the next 12 months.
To fully execute its business plan, the Company will need to complete certain research and development activities and clinical studies. Further, the Company’s product candidates will require regulatory approval prior to commercialization. These activities may span many years and require substantial expenditures to complete and may ultimately be unsuccessful. Any delays in completing these activities could adversely impact the Company. The Company will need substantial additional financing to conduct new trials in the development of its product candidates; such financing may not be available on terms favorable to the Company, if at all. The Company plans to meet its capital requirements primarily through issuances of equity securities, debt financing, potential partnerships and in the longer term, revenue from product sales. Failure to generate revenue or raise additional capital would adversely affect the Company’s ability to achieve its intended business objectives.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States, or GAAP.
|
2.
|
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
|
Revenue Recognition
During 2015, the Company had a collaboration with Zenyaku Kogyo Co., Ltd. (“Zenyaku”) which provided for various types of payments from Zenyaku, including development milestones, sales milestone, royalty, and reimbursement for a portion of the Company’s internal and external costs. All payments from Zenyaku are nonrefundable. The collaborative arrangement was on a best-efforts basis, did not require scientific achievement as a performance obligation and provided for payment to be made when costs were incurred or services were performed. The collaboration was terminated on January 7, 2016 pursuant to a termination notice from Zenyaku to the Company.
With respect to the collaborative arrangement with Zenyaku, the Company recognized revenue in accordance with FASB Accounting Standards Codification, or ASC 605 “ Revenue Recognition ”, subtopic ASC 605-25 “ Revenue with Multiple Element Arrangements ” and subtopic ASC 605-28 “ Revenue Recognition-Milestone Method ”, which provide accounting guidance for revenue recognition for arrangements with multiple deliverables and guidance on defining the milestone and determining when the use of the milestone method of revenue recognition for research and development transactions is appropriate, respectively. For multiple-element arrangements, each deliverable within a multiple deliverable revenue arrangement is accounted for as a separate unit of accounting if both of the following criteria are met: (1) the delivered item or items have value to the customer on a standalone basis and (2) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
The deliverables under the Zenyaku agreement had been determined to be a single unit of accounting and as such any license fees received were recorded as deferred revenue and recognized ratably over the term of the estimated performance period under the agreement, which was the product development period. As a result of an early termination of the Zenyaku agreement, the Company revised the amortization period of its deferred revenue to correspond with the shortened collaboration period in the third quarter of 2015 and had fully amortized its deferred revenue as of January 7, 2016.
For the collaborative research activities, the Company was entitled to reimbursement from Zenyaku for its internal personnel cost at a pre-determined full time equivalent (“FTE”) rate. Revenue related to FTE services was recognized as research services were performed over the related performance periods. The Company was required to perform research and development activities as specified in the collaboration agreement. The payments received were not refundable and were based on a contractual reimbursement rate per FTE working on the project. Reimbursement for FTE costs was recorded as collaborative revenue as incurred.
Use of Estimates
The preparation of these consolidated financial statements in conformity with GAAP requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, expenses, and related disclosures. On an ongoing basis, management evaluates its estimates, including critical accounting policies or estimates related to clinical trial accruals, tax provision, stock-based compensation, allocation of consideration to various elements under multiple-element arrangement and recognition of revenue. The Company bases its estimates on historical experience and on various other market specific and other relevant assumptions that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates.
Cash and Cash Equivalents
The Company considers all highly liquid instruments purchased with an original maturity or remaining maturities of three months or less at the date of purchase to be cash equivalents. Cash equivalents consist primarily of cash currencies and money market funds, for which the carrying amounts are reasonable estimates of fair value. Cash equivalents are recognized at fair value.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash, cash equivalents and short-term investments. The Company’s cash equivalents consist of certificates of deposit with maturities less than three months and treasury money market funds. The Company’s short-term investments consist of certificates of deposit with maturities exceeding three months but less than one year. The Company has not experienced any losses in such accounts. The Company believes it is not exposed to significant credit risk related to cash, cash equivalents and short-term investments.
Property and Equipment—Net
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is computed over the estimated useful lives of the respective assets, which range from three to five years, using the straight-line method. Repairs and maintenance costs are expensed as incurred. Leasehold improvements are stated at cost and amortized using the straight-line method over the term of the lease or the life of the related asset, whichever is shorter.
Long-Lived Assets
The Company’s long-lived assets and other assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. Recoverability of an asset to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted cash flows expected to be generated by the asset. If such asset is considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the asset exceeds its fair value. Through December 31, 2015, the Company had not experienced impairment losses on its long-lived assets.
Accrued Clinical Studies
Expenses related to clinical studies are based on estimates of the services received and efforts expended pursuant to contracts with many research institutions, clinical research organizations and other service providers that conduct and manage clinical studies on our behalf. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price. Payments under the contracts are mainly driven by time and materials incurred by these service providers. Expenses related to clinical studies are generally accrued based on time and materials incurred by the service providers and in accordance with the contracts. This process involves reviewing open contracts and purchase orders, communicating with applicable personnel to identify services that have been performed on the Company’s behalf and estimating the level of service performed and the associated cost incurred for the service when the Company has not yet been invoiced or otherwise notified of actual cost. The majority of service providers’ invoice at least monthly in arrears for services performed. The Company periodically confirms the accuracy of estimates with the service providers and makes adjustments if necessary. Examples of estimated accrued clinical expenses include:
|
|
•
|
fees paid to Contract Research Organizations, or CROs, in connection with clinical studies;
|
|
|
•
|
fees paid to investigative sites in connection with clinical studies;
|
|
|
•
|
fees paid to contract manufacturers in connection with the production of clinical study materials; and
|
|
|
•
|
fees paid to vendors in connection with preclinical development activities.
|
Research and Development Costs
Research and development expenses consist of personnel costs, including salaries, benefits and stock-based compensation, clinical studies performed by CROs, materials and supplies, licenses and fees, and overhead allocations consisting of various administrative and facilities related costs. Clinical study expenses are further separated into two main categories: clinical development and pharmaceutical development. Clinical development costs include costs for Phase 1, 2 and 3 clinical studies. Pharmaceutical development costs consist of expenses incurred in connection with manufacturing campaign, product formulation and chemical analysis.
The Company charges research and development costs, including clinical study costs, to expense when incurred. Clinical study costs are a significant component of research and development expenses. All of the Company’s clinical studies are performed by third-party CROs. The Company accrues costs for clinical studies performed by CROs based on patient enrollment activities and adjusts the estimates, if required, based upon the Company’s ongoing review of the level of effort and costs actually incurred by the CROs. The Company monitors levels of performance under each significant contract, including the extent of patient enrollment and other activities through communications with the CROs, and adjusts the estimates, if required, on a monthly basis so that clinical expenses reflect the actual effort expended by each CRO.
All material CRO contracts are terminable by the Company upon written notice and the Company is generally only liable for actual effort expended by the CROs and certain noncancelable expenses incurred at any point of termination.
Comprehensive Loss
Comprehensive loss consists of certain changes in equity that are excluded from net loss. Specifically, the Company includes unrealized gains and losses on available for sale securities in other comprehensive loss. Comprehensive loss for each period presented is set forth in the Statement of Operations and Comprehensive Loss.
Income Taxes
The Company accounts for income taxes in accordance with the liability method whereby deferred tax asset and liability account balances are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, if necessary, to reduce deferred tax assets to their estimated realizable value.
Segments
The Company operates in only one segment. Management uses cash flow as the primary measure to manage its business and does not segment its business for internal reporting or decision-making. The Company’s long-lived tangible assets consisted of mainly machinery purchased by the Company and installed by its contract manufacturing vendor, Fujifilm Diosynth Bioservices, in the United Kingdom. The machinery is used for all of the Company’s blisibimod product manufacturing campaigns.
Net Loss Per Share
Basic net loss attributable to common stockholders per share is computed by dividing income available to common stockholders (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period are weighted for the portion of the period that they were outstanding. The computation of diluted Earnings Per Share, or EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. In addition, in computing the dilutive effect of convertible securities, the numerator is adjusted to add back any convertible preferred dividends and the after-tax amount of interest recognized in the period associated with any convertible debt. The numerator also is adjusted for any other changes in income or loss that would result from the assumed conversion of those potential common shares, such as profit-sharing expenses. Diluted EPS is identical to basic EPS since common equivalent shares are excluded from the calculation, as their effect is anti-dilutive.
The following table summarizes the Company’s calculation of net loss per common share (in thousands except share and per share amounts):
|
Year Ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Numerator:
|
||||||||||||
|
Net loss
|
$ | (35,220 | ) | $ | (29,604 | ) | $ | (30,861 | ) | |||
|
Denominator:
|
||||||||||||
|
Weighted-average number of common shares outstanding
|
35,631,237 | 21,776,269 | 18,267,413 | |||||||||
|
Net loss per share:
|
||||||||||||
|
Basic
|
$ | (0.99 | ) | $ | (1.36 | ) | $ | (1.69 | ) | |||
As the Company incurred net losses for all of the periods presented, the following outstanding potentially dilutive securities were excluded from the computation of diluted net loss per share, as the effect of including them would have been antidilutive:
|
Year Ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Total options to purchase common stock
|
4,255,981
|
3,021,969
|
1,997,075
|
|||||||||
|
Total warrants to purchase common stock
|
40,178
|
556,838
|
675,006
|
|||||||||
|
Total restricted stock units
|
—
|
937
|
42,042
|
|||||||||
|
Total
|
4,296,159
|
3,579,744
|
2,714,123
|
|||||||||
Stock-Based Compensation
The Company uses the Black-Scholes option pricing model as the method for determining the estimated fair value for all stock-based awards, including employee stock options, and rights to purchase shares under the Company’s Employee Stock Purchase Plan, and recognizes the costs in its consolidated financial statements over the employees’ requisite service period. The Black-Scholes option pricing model requires the use of highly subjective and complex assumptions which determine the fair value of share-based awards, including the option’s expected term and the price volatility of the underlying stock. Additionally, the Company is required to include an estimate of the number of awards that will be forfeited in calculating compensation costs, which are recognized over the requisite service period of the awards on a straight-line basis.
Expected Term —The Company’s expected term represents the period that the Company’s stock-based awards are expected to be outstanding and is determined using the simplified method, which is computed as the arithmetic mean of weighted vested period and contractual life.
Expected Volatility —Expected volatility is estimated using comparable public company volatility for instruments with similar terms.
Expected Dividend —The Black-Scholes option pricing model calls for a single expected dividend yield as an input and the Company has never paid dividends and has no plans to pay dividends.
Risk-Free Interest Rate —The risk-free interest rate used in the Black-Scholes option pricing method is based on the U.S. Treasury zero-coupon issues in effect at the time of grant for periods corresponding with the expected term of option.
Estimated Forfeitures —The estimated forfeiture rate is determined based on the Company’s historical forfeiture rates to date. The Company monitors actual forfeitures and periodically updates the estimate.
Equity instruments issued to nonemployees are recorded at their fair value as determined in accordance with guidance provided by the Financial Accounting Standard Board (“FASB”) and are periodically revalued as the equity instruments vest and recognized as expense over the related service period.
Recent Accounting Pronouncements
In September 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-15, Presentation of Financial Statements - Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as Going Concern (“ASU 2014-15”). ASU 2014-15 defines when and how companies are required to disclose going concern uncertainties, which must be evaluated each interim and annual period. Specifically, the ASU requires management to determine whether substantial doubt exists regarding the entity’s going concern presumption. Substantial doubt about an entity’s ability to continue as a going concern exists when relevant conditions and events, considered in the aggregate, indication that it is probable that the entity will be unable to meet its obligations as they become within one year after the financial statements are issued (or available to be issued). If substantial doubt exists, certain disclosures are required; the extent of those disclosures depends on an evaluation of management’s plans (if any) to mitigate the going concern uncertainty. The guidance is effective for fiscal years beginning after December 15, 2016 and for interim periods within that fiscal year. The Company does not expect the adoption of this guidance to materially affect its consolidated financial statements.
In November 2015, the FASB issued guidance on the classification of deferred taxes, Accounting Standards Update No. 20154-17 (“ASU 2015-17”), Balance Sheet Classification of Deferred Taxes. ASU 2015-17 eliminates the guidance in Topic 740, Income Taxes, that required an entity to separate deferred tax liabilities and assets between current and noncurrent amounts in a classified balance sheet. The amendments require that all deferred tax liabilities and assets of the same tax jurisdiction or a tax filing group, as well as any related valuation allowance, be offset and presented as a single amount in a classified balance sheet. The amendments are effective for public business entities for fiscal years, and for interim periods within those fiscal years, beginning after December 15, 2016. Early adoption is permitted as of the beginning of any interim period or annual reporting period. The Company does not expect the adoption of this guidance to materially affect its consolidated financial statements.
In February 2016, the FASB issued ASU No. 2016-02 Leases (Topic 842). ASU 2016-02 impacts any entity that enters into a lease with some specified scope exceptions. The guidance updates and supersedes Topic 840, Leases. For public entities, ASU 2016-02 is effective for fiscal years, and interim periods with those years, beginning after December 15, 2018, and early adoption is permitted. We have not evaluated the impact of this guidance, but do not expect the adoption of this standard to have a material impact on our financial statements.
3. RESEARCH AWARD
In March 2015, the Company received a research award of up to $3 million from the CFFT for the Company's development of liprotamase. The Company retains the right to develop and commercialize liprotamase and will owe royalties to CFFT on net sales of any drug candidate approved and commercialized under the collaboration. The funding is to be disbursed by CFFT to the Company upon the Company’s achievement of milestones specified in the award agreement. At its discretion, the Company may choose to fund a particular stage of the liprotamase development plan without CFFT funds. Any CFFT funds not expended on the development program of liprotamase must be returned to CFFT and, upon such return, the amounts of such returned funds will not be included as part of the research award for the purpose of calculating royalties or other amounts owed by the Company to CFFT. To the extent CFFT provides or makes available any information, expertise, know-how or other intellectual property related to cystic fibrosis or the treatment, prevention or cure there-of (“CFFT Know-How”) to the Company, CFFT grants to the Company a non-exclusive, transferrable, sublicensable, worldwide rights and license under all of CFFT’s rights in such CFFT Know-How to assist the Company to research, develop, commercialize, make or have made, use, sell, have sold, offer for sale, import, export and otherwise exploit the product.
In consideration for CFFT’s research award and any licenses of intellectual property granted by CFFT, the Company agrees to pay royalties to CFFT as follows: i) a one-time royalty in an amount equal to five times the actual award, payable in three installments between the first and second anniversaries of the first commercial sale of a product; ii) a one-time royalty in an amount equal to the actual award after net product sales reaches $100 million; and iii) in the event of a license, sale or other transfer of the product or a change of control transaction prior to the commercial sale of the product, a milestone payment equal to three times the actual award.
For the year ended December 31, 2015, the Company recognized $2.6 million of research award from CFFT in connection with achieving certain milestones specified in the award agreement. As of December 31, 2015, there was $0.4 million available under the research award.
|
4.
|
FAIR VALUE OF INSTRUMENTS
|
Pursuant to the accounting guidance for fair value measurement and its subsequent updates, fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (i.e., the “exit price”) in an orderly transaction between market participants at the measurement date. The accounting guidance establishes a hierarchy for inputs used in measuring fair value that minimizes the use of unobservable inputs by requiring the use of observable market data when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on active market data. Unobservable inputs are inputs that reflect the assumptions market participants would use in pricing the asset or liability based on the best information available in the circumstances.
The fair value hierarchy is broken down into the three input levels summarized below:
|
|
•
|
Level 1 —Valuations are based on quoted prices in active markets for identical assets or liabilities and readily accessible by us at the reporting date. Examples of assets and liabilities utilizing Level 1 inputs are certain money market funds, U.S. Treasuries and trading securities with quoted prices on active markets.
|
|
|
•
|
Level 2 —Valuations based on inputs other than the quoted prices in active markets that are observable either directly or indirectly in active markets. Examples of assets and liabilities utilizing Level 2 inputs are U.S. government agency bonds, corporate bonds, commercial paper, certificates of deposit and over-the-counter derivatives.
|
|
|
•
|
Level 3 —Valuations based on unobservable inputs in which there are little or no market data, which requires the Company to develop its own assumptions.
|
The following tables present the Company’s fair value hierarchy for all its financial assets (including those in cash and cash equivalents), in thousands, by major security type measured at fair value on a recurring basis as of December 31, 2015 and 2014 (in thousands):
|
December 31, 2015
|
||||||||||||||||
|
Estimated
Fair Value
|
Level 1
|
Level 2
|
Level 3
|
|||||||||||||
|
Money market funds
|
$
|
45,156
|
$
|
45,156
|
$
|
—
|
$
|
—
|
||||||||
|
December 31, 2014
|
||||||||||||||||
|
Estimated
Fair Value
|
Level 1
|
Level 2
|
Level 3
|
|||||||||||||
|
Money market funds
|
$
|
2,354
|
$
|
2,354
|
$
|
—
|
$
|
—
|
||||||||
|
5.
|
PROPERTY AND EQUIPMENT
|
Property and equipment are comprised of the following (in thousands):
|
December 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Laboratory equipment
|
$
|
1,312
|
$
|
1,312
|
||||
|
Computer and software
|
50
|
50
|
||||||
|
Office furniture and fixture
|
140
|
60
|
||||||
|
Leasehold improvements
|
206
|
206
|
||||||
|
Total property and equipment
|
1,708
|
1,628
|
||||||
|
Less accumulated depreciation and amortization
|
(1,445
|
)
|
(1,160
|
)
|
||||
|
Property and equipment, net
|
$
|
263
|
$
|
468
|
||||
For the years ended December 31, 2015, 2014, and 2013, the Company recorded $285,000, $344,000, and $353,000 respectively, in depreciation and amortization expense.
|
6.
|
COLLABORATIVE ARRANGEMENT
|
Zenyaku Kogyo Co., Ltd.
In December 2014, the Company entered into an exclusive license agreement with Zenyaku (“Zenyaku Agreement”) for the development and commercialization of blisibimod in Japan and potentially other countries throughout Asia, while the Company retained full development and commercialization rights of blisibimod for all other global territories including North America and the European Union.
Under the terms of the Zenyaku Agreement, the Company had the right to receive an upfront and forgivable loan of $7.0 million, milestone payments of up to $22.0 million contingent upon the achievement of certain regulatory and commercial sales milestones, and up to $15.0 million in sales of the Company’s common stock at a purchase price equal to 1.3 times the volume weighted average price of the Company’s common stock for 20 trading days prior to the delivery of the closing notice (the “Premium Purchase Price”). Not all the conditions required to secure the forgivable loan were met and therefore, the Company had not exercised its right to receive the loan from Zenyaku. During 2015, the Company had exercised its right with respect to $11.0 million of the $15.0 million in equity puts to Zenyaku, of which $9.0 million was received through the issuance of 2,946,810 shares of common stock and the Company does not expect to receive the remaining $2.0 million.
Under the terms of the Zenyaku Agreement, Zenyaku was responsible for all development, marketing and commercialization costs in Japan and would reimburse Anthera for i) 100% of blisibimod development cost in Japan for IgA nephropathy; ii) 25% of global blisibimod development cost outside of Japan for IgA nephropathy; iii) a percentage of Anthera’s personnel costs at a pre-determined full-time equivalent (“FTE”) rate and iv) exclusive purchase of blisibimod clinical drug supplies at cost and blisibimod commercial drug products from the Company at a premium to the Company’s manufacturing cost.
In September 2015, Zenyaku provided the Company a notice of its intent to terminate the Zenyaku Agreement, effective January 7, 2016 (“Termination Notice”). The termination was “at will” and the Termination Notice alleged no breach of the Zenyaku Agreement by the Company. There were no termination penalties incurred by the Company in connection with the early termination of the Zenyaku Agreement by Zenyaku. No patients had been enrolled in any blisibimod clinical studies in the Zenyaku territory and Zenyaku. The Company regained global rights of blisibimod on January 7, 2016.
As a result of the Termination Notice received from Zenyaku in September 2015, the Company changed the amortization period of its deferred revenue to correspond with the shortened collaboration period beginning in September 2015 when the Company received the termination notice from Zenyaku. The effect of this change in estimate was a $2.0 million increase license revenue and $0.05 decrease in net loss per share as a result of accelerating the amortization of deferred revenue during the year ended December 31, 2015. During the year ended December 31, 2015, the Company recorded revenue of $3.2 million, which was comprised of $2.6 million for the amortization of the license fee revenue and $0.6 million for the reimbursement of FTEs, respectively. In addition, during the year ended December 31, 2015, the Company recorded $1.5 million as reduction to research and development expenses in connection with the reimbursement of qualifying costs under the collaboration agreement.
7. COMMITMENTS AND CONTINGENCIES
Leases
The Company leases its main operating facility in Hayward, California. The lease is for approximately 14,000 square feet and the lease agreement will expire in September 2017. For the years ended December 31, 2015, 2014 and 2013, the Company recognized $222,000, $244,000 and $215,000, respectively, in rental expense.
As of December 31, 2015, future minimum lease payments under noncancellable operating leases were as follows (in thousands):
|
2016
|
$
|
230
|
||
|
2017
|
177
|
|||
|
Total
|
$
|
407
|
Other Commitments
In December 2007, the Company and Amgen entered into a worldwide, exclusive license agreement (the “Amgen Agreement”) to develop and commercialize blisibimod in any indication, including for the treatment of systemic lupus erythematosus (“lupus”). Under the terms of the Amgen Agreement, the Company paid a nonrefundable, upfront license fee of $6.0 million. As there was no future alternative use for the technology, the Company expensed the license fee in research and development expenses during 2007. Under the terms of the Amgen Agreement, the Company is obligated to make additional milestone payments to Amgen of up to $33.0 million upon the achievement of certain development and regulatory milestones. The Company is also obligated to pay tiered royalties on future net sales of products, ranging from the high single digits to the low double digits, which are developed and approved as defined by this collaboration. The Company’s royalty obligations as to a particular licensed product will be payable, on a country-by-country and licensed product-by-licensed product basis, for the longer of (a) the date of expiration of the last to expire valid claim within the licensed patents that covers the manufacture, use or sale, offer to sell, or import of such licensed product by the Company or a sublicense in such country or (b) 10 years after the first commercial sale of the applicable licensed product in the applicable country.
In connection with the collaborative arrangement with Zenyaku pursuant to the Zenyaku Agreement, the Company amended the Amgen Agreement in November 2014 to (i) adjust certain royalty and milestone payment obligations payable to Amgen in light of the collaboration between Anthera and Zenyaku and (ii) provide that the sublicense granted by Anthera to Zenyaku shall survive the termination of the Amgen Agreement. Under this amendment, Anthera also agreed to grant Amgen that number of shares of its common stock equal $1.0 million divided by the volume weighted average price of the Company’s common stock for 20 trading days prior to issuance. The Company issued 420,751 shares of common stock to Amgen at $2.3767 per share on January 28, 2015, pursuant to a subscription agreement with Amgen, with the consideration paid by Amgen in the form of a waiver of a fee otherwise payable to Amgen under the Amgen Agreement. The Company accrued $1.0 million of license fees as research and development expense with a corresponding current liability in the year ended December 31, 2014.
On July 11, 2014, the Company and Eli Lilly and Company (“Eli Lilly”) entered into a worldwide, exclusive license agreement (the “Lilly Agreement”), to develop and commercialize liprotamase, a Phase 3 novel investigational Pancreatic Enzyme Replacement Therapy (“PERT”), for the treatment of patients with Exocrine Pancreatic Insufficiency, or EPI, often seen in patients with cystic fibrosis and other conditions. Under the terms of the Lilly Agreement, the Company was not required to make any up-front payment but is obligated to make milestone payments of up to up to $33.5 million for capsule products and $9.5 million for reformulated products upon the achievement of certain regulatory and commercial sales milestones, none of which have been achieved as of December 31, 2015. In addition, after sales of the licensed products exceed an aggregate of $100.0 million in the United States, the Company is obligated to pay tiered royalties on future net sales of products, ranging from the single digits to the mid-teens, that are developed and approved as defined in the Lilly Agreement. The Company’s royalty obligations as to a particular licensed product will be payable, on a licensed product-by-licensed product basis, for the longer of (a) the date of expiration of the last to expire valid claim within the licensed patents that covers the manufacture, use or sale, offer to sell, or import of such licensed product by the Company or a sublicense in such country, or (b) 12 years after the first commercial sale of the applicable licensed product in the applicable country.
See Note 3 – “Research Award” for discussion of commitments and contingencies associated with the research award received from the CFFT.
8. STOCKHOLDERS’ EQUITY (DEFICIT)
Preferred Stock
The Company’s Fifth Amended and Restated Certificate of Incorporation designates 5,000,000 shares of the Company’s capital stock as undesignated preferred stock. There were no preferred shares issued and outstanding as of December 31, 2015.
Common Stock
On April 5, 2013, the Company entered into an equity purchase agreement (the “2013 Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“LPC”), pursuant to which the Company has the right to sell to LPC up to an aggregate of $18.5 million in shares of the our common stock. The Company sold 40,000 shares of common stock pursuant to the 2013 Purchase Agreement and issued 150 shares of common stock to LPC as commitment fee in January 2015. On March 12, 2015, the Company terminated the 2013 Purchase Agreement with LPC, which had a remaining balance of $14.1 million and executed the 2015 Purchase Agreement with more favorable terms, pursuant to which the Company has the right, but not obligation, to sell to LPC up to an aggregate of $10.0 million in shares of common stock over a period of two years. Concurrent with the execution of the Purchase Agreement, the Company issued 19,188 shares of common stock to LPC as an upfront commitment fee. In July 2015, the Company amended the Purchase Agreement and reduced the amount to an aggregate of $6.0 million. No sales of common stock have been made under the Purchase Agreement as of December 31, 2015. The Purchase Agreement will expire in March 2017.
On November 15, 2013, the Company entered into a Sales Agreement (the “Agreement”) with Cowen to create an ATM program under which the Company from time to time may offer and sell shares of its common stock, par value $0.001 per share, having an aggregate offering price of up to $25.0 million through Cowen, as agent. As of December 31, 2015, the Company has sold $21.8 million of common stock through the ATM program of which $12.4 million was sold during the year ended December 31, 2015, leaving a balance of $3.2 million available for future sale pursuant to the Agreement.
In April 2013, the Company filed a universal shelf registration statement with the SEC on Form S-3 (File No. 333-187780) for the proposed offering from time to time of up to $100.0 million of its securities, including common stock, preferred stock, debt securities and/or warrants. On November 15, 2013, the Company registered $25.0 million under the registration statement for the Cowen ATM. In 2015, the Company registered $10.0 million under the registration statement for the issuance of its common stock to Zenyaku and Amgen. In March 2015, the Company registered $10.3 million under the registration statement for the Purchase Agreement with LPC, which was subsequently reduced to $6.3 million in July 2015. In March and July 2015, the Company registered an aggregate of $57.5 million in connection with the issuance of 10,222,223 shares of common stock in two separate public offerings. As of December 31, 2015, there was $1.2 million available for future issuance under this shelf registration statement.
Based on the requirements of Form S-3, however, there are certain factors, such as volume of trading in the Company’s common stock and stock price, which may limit the amount that can be raised in a short period of time through LPC Purchase Agreement and registration statements described above.
At December 31, 2015, the Company had reserved the following shares for future issuance:
|
Common stock options outstanding
|
4,255,981
|
|||
|
Common stock warrants outstanding
|
40,178
|
|||
|
Common stock available for future grant under ESPP plan
|
55,929
|
|||
|
Common stock options available for future grant under stock option plan
|
729,492
|
|||
|
Total
|
5,081,580
|
Warrants
In March 2011, the Company issued a seven-year warrant to purchase 40,178 shares of the Company’s common stock at an exercise price of $48.00 per share. The warrant was immediately exercisable and expires in March 2018. As of December 31, 2015, the warrant remained outstanding and exercisable.
On September 24, 2015, unexercised warrants to purchase 516,660 shares of common stock, which were issued in a prior financing transaction in September 2010, expired.
9. STOCK-BASED AWARDS
Option Plan
On March 25, 2013, the Company’s board of directors adopted the 2013 Stock Option and Incentive Plan (the “2013 Plan”), which was also approved by the Company’s stockholders at its annual general meeting on May 16, 2013. The Company initially reserved 1,750,000 shares of its common stock for the issuance of awards under the 2013 Plan, plus all shares remaining available for grant under the Company’s 2010 Stock Option and Incentive Plan (the “2010 Plan), plus any additional shares returned under the 2010 Plan or 2013 Plan as a result of the cancellation, forfeiture or other termination (other than by exercise) of awards issued pursuant to the 2010 Plan or 2013 Plan, subject in all cases to adjustment including reorganization, recapitalization, reclassification, stock dividend, stock split, reverse stock split or other similar change in the Company’s capital stock. In May 2015, the Company’s shareholders approved an increase of 1,790,818 shares to the 2013 Plan pool. Of the shares of common stock reserved for issuance under the 2013 Plan, no more than 750,000 shares will be issued to any individual participant as incentive options, non-qualified options or stock appreciation rights during any calendar year. The 2013 Plan permits the granting of incentive and non-statutory stock options, restricted and unrestricted stock awards, restricted stock units, stock appreciation rights, performance share awards, cash-based awards and dividend equivalent rights to eligible employees, directors and consultants. The option exercise price of an option granted under the 2013 Plan may not be less than 100% of the fair market value of a share of the Company’s common stock on the date the stock option is granted. Options granted under the 2013 Plan have a maximum term of 10 years and generally vest over four years. In addition, in the case of certain large stockholders, the minimum exercise price of incentive options must equal 110% of fair market value on the date of grant and the maximum term is limited to five years. Subject to overall Plan limitations, the maximum aggregate number of shares of common stock that may be issued in the form of incentive options shall not exceed 6,250,000 shares of common stock.
The 2013 Plan does not allow the option holders to exercise their options prior to vesting.
The following table summarizes stock option activity for 2013, 2014 and 2015:
|
Number of
Options
|
Weighted-
Average
Exercise
Price
|
Weighted-
Average
Remaining
Contractual
Life in
Years
|
Aggregate
Intrinsic
Value
(in thousands)
|
||||||||
|
Balance at December 31, 2012
|
300,468
|
$
|
24.15
|
2.06
|
$
|
126
|
|||||
|
Options granted
|
2,124,250
|
$
|
4.79
|
||||||||
|
Options exercised
|
—
|
$
|
—
|
||||||||
|
Options cancelled/forfeited
|
(427,643
|
)
|
$
|
16.41
|
|||||||
|
Balance at December 31, 2013
|
1,997,075
|
$
|
5.21
|
9.11
|
$
|
48
|
|||||
|
Options granted
|
1,389,743
|
$
|
2.00
|
||||||||
|
Options exercised
|
(2,000
|
)
|
$
|
2.08
|
|||||||
|
Options cancelled/forfeited
|
(362,849
|
)
|
$
|
5.78
|
|||||||
|
Balance at December 31, 2014
|
3,021,969
|
$
|
3.67
|
8.92
|
$
|
0
|
|||||
|
Options granted
|
1,392,499
|
$
|
7.10
|
||||||||
|
Options exercised
|
(80,218
|
)
|
$
|
3.73
|
|||||||
|
Options cancelled/forfeited
|
(78,269
|
)
|
$
|
4.01
|
|||||||
|
Balance at December 31, 2015
|
4,255,981
|
$
|
4.79
|
8.44
|
$
|
4,330
|
|||||
|
Ending vested at December 31, 2015
|
1,713,948
|
$
|
4.56
|
7.95
|
$
|
1,524
|
|||||
As of December 31, 2015, the vested and expected to vest stock options was 4,252,091.
The assumptions used in the Black-Scholes option-pricing model to value stock options are as follows:
| Years Ended December 31, | ||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Expected Volatility
|
94
|
%
|
91
|
%
|
94
|
%
|
||||||
|
Dividend Yield
|
0
|
%
|
0
|
%
|
0
|
%
|
||||||
|
Risk-Free Interest Rate
|
1.70
|
%
|
1.60
|
%
|
1.08
|
%
|
||||||
|
Expected Term (years)
|
5.92
|
5.22
|
5.79
|
|||||||||
|
Weighted-average fair value per option
|
$
|
5.40
|
$
|
1.35
|
$
|
3.55
|
||||||
The intrinsic value of stock options represents the difference between the exercise price of stock options and the market price of our stock on that day for all in-the-money options. Additional information related to our stock options is summarized below (in thousands except per share information):
|
Years Ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Intrinsic value of options exercised
|
$
|
140
|
$
|
3
|
$
|
—
|
||||||
|
Proceeds received from the exercise of stock options
|
$
|
299
|
$
|
4
|
$
|
—
|
||||||
|
Grant date fair value of options vested
|
$
|
3,414
|
$
|
1,932
|
$
|
550
|
||||||
There was $8.5 million of total unrecognized compensation expense as of December 31, 2015 related to stock options. The unrecognized compensation expense will be amortized on a straight-line basis over a weighted-average remaining period of 2.40 years.
Information about stock options outstanding, vested and exercisable as of December 31, 2015, was as follows:
|
Options Outstanding
|
Options Vested & Exercisable
|
|||||||||||||||
|
Range of Exercise Price
|
Number of
Shares
|
Weighted-
Average
Remaining
Contractual Life
(In Years)
|
Weighted
Average Exercise
Price
|
Number of
Shares
|
||||||||||||
|
$ 1.58 - $7.95
|
3,400,188 | 8.18 | $ | 3.78 | 1,543,224 | |||||||||||
|
$ 7.96 - $14.34
|
849,043 | 9.50 | $ | 9.98 | 163,974 | |||||||||||
|
$ 33.47 - $39.85
|
3,375 | 4.75 | $ | 33.52 | 3,375 | |||||||||||
|
$ 58.98 - $65.36
|
3,375 | 5.30 | $ | 65.36 | 3,375 | |||||||||||
|
Total
|
4,255,981 | 8.44 | $ | 4.56 | 1,713,948 | |||||||||||
As of December 31, 2015, there were 729,492 shares available for grant under the 2013 Plan.
2010 Employee Stock Purchase Plan
Effective July 2010, under the terms of the ESPP, eligible employees of the Company may authorize the Company to deduct amounts from their compensation, which amounts are used to enable the employees to purchase shares of the Company’s common stock. The Company initially reserved 12,500 shares of common stock for issuance thereunder plus on January 1, 2011 and each January 1 thereafter, the number of shares of stock reserved and available for issuance under the Plan shall be cumulatively increased by the lesser of (i) one percent (1%) of the number of shares of common stock issued and outstanding on the immediately preceding December 31 or (ii) 31,250 shares of common stock. During the year ended December 31, 2015 and 2014, 60,444 and 25,000 shares were issued pursuant to the purchase of common stock by our employees under the ESPP, respectively. As of December 31, 2015, there were 55,929 shares available for grant under the 2010 Employee Stock Purchase Plan.
Under the ESPP, eligible employees of the Company may authorize the Company to deduct amounts from their compensation, which amounts are used to enable the employees to purchase shares of the Company’s common stock. The purchase price per share is 85% of the fair market value of the common stock as of the first date or the ending date of the applicable semi-annual purchase period, whichever is less (the “Look-Back Provision”). The 15% discount and the Look-Back Provision make the ESPP compensatory. The Black-Scholes option pricing model was used to value the employee stock purchase rights. For the years ended December 31, 2015, 2014 and 2013, the following weighted-average assumptions were used in the valuation of the stock purchase rights:
|
Years Ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Expected Volatility
|
82
|
%
|
41
|
%
|
126
|
%
|
||||||
|
Dividend Yield
|
0
|
%
|
0
|
%
|
0
|
%
|
||||||
|
Risk-Free Interest Rate
|
0.08
|
%
|
0.05
|
%
|
0.11
|
%
|
||||||
|
Expected Term (years)
|
0.50
|
0.50
|
0.50
|
|||||||||
|
Weighted-average grant date fair value per right
|
$
|
1.04
|
$
|
1.04
|
$
|
2.46
|
||||||
Stock-Based Compensation Expense
Total stock-based compensation expense, including expense recorded for the ESPP, was as follows (in thousands):
|
Years Ended December 31,
|
||||||||||||
|
2015
|
2014
|
2013
|
||||||||||
|
Research and development
|
$
|
1,453
|
$
|
825
|
$
|
1,451
|
||||||
|
General and administrative
|
2,088
|
1,350
|
1,694
|
|||||||||
|
Total employee stock-based compensation
|
$
|
3,541
|
$
|
2,175
|
$
|
3,145
|
||||||
10. EMPLOYEE BENEFIT PLAN
The Company maintains a defined contribution 401(k) plan, or the 401(k) Plan. Employee contributions are voluntary and are determined on an individual basis, limited by the maximum amounts allowable under federal tax regulations. Prior to 2011, the Company had not made any contributions to the 401(k) Plan. In December 2012, the Company amended its 401(k) plan to provide for non-elective employer contribution at the Company’s discretion. During the years ended December 31, 2015 and 2014, the Company contributed approximately $284,000 and $185,000, respectively, in non-elective employer contribution into the employees’ 401(k) accounts.
|
11.
|
INCOME TAXES
|
The Company has incurred net operating losses since inception. The Company has not reflected any benefit of such net operating loss carryforwards in the accompanying consolidated financial statements and has established a full valuation allowance against its deferred tax assets.
Deferred income taxes reflect the net tax effects of (a) temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes, and (b) operating losses and tax credit carryforwards.
The significant components of the Company’s deferred tax assets for the years ended December 31, 2015 and 2014 are as follows (in thousands):
|
December 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Deferred tax assets:
|
||||||||
|
Net operating loss carryforwards
|
$
|
37,576
|
$
|
32,315
|
||||
|
Tax credits
|
3,023
|
2,307
|
||||||
|
Intangible assets
|
2,033
|
2,343
|
||||||
|
Capitalized R&D
|
35,514
|
28,105
|
||||||
|
Other
|
2,285
|
1,110
|
||||||
|
Total deferred tax assets
|
80,431
|
66,180
|
||||||
|
Deferred tax liabilities
|
—
|
—
|
||||||
|
Valuation allowance
|
(80,431
|
)
|
(66,180
|
)
|
||||
|
Net deferred tax asset
|
$
|
—
|
$
|
—
|
||||
A reconciliation of the statutory tax rates and the effective tax rate for the years ended December 2015, 2014, and 2013 is as follows:
|
2015
|
2014
|
2013
|
||||||||||
|
Statutory rate
|
34
|
%
|
34
|
%
|
34
|
%
|
||||||
|
State tax
|
6
|
%
|
6
|
%
|
6
|
%
|
||||||
|
Tax credit
|
2
|
%
|
1
|
%
|
2
|
%
|
||||||
|
Stock based compensation
|
(1
|
)%
|
(1
|
)%
|
(3
|
)%
|
||||||
|
Valuation allowance
|
(41
|
)%
|
(40
|
)%
|
(39
|
)%
|
||||||
|
Effective tax rates
|
0
|
%
|
0
|
%
|
0
|
%
|
||||||
Tax benefits of net operating losses, temporary differences and credit carryforwards are recorded as an asset to the extent that management assesses that realization is “more likely than not.” Realization of the future tax benefits is dependent on the Company’s ability to generate sufficient taxable income within the carryforward period. Because of the Company’s history of operating losses, management believes that the deferred tax assets arising from the above-mentioned future tax benefits are currently not likely to be realized and, accordingly, has provided a full valuation allowance. The net valuation allowance increased by $14.3 million in 2015, $11.7 million in 2014, and $11.9 million in 2013.
The amount of the valuation allowance for deferred tax assets associated with excess tax deductions from stock-based compensation arrangements that would be allocated to contributed capital if the future tax benefits are subsequently recognized is $1.5 million.
Net operating losses and tax return credit carryforwards as of December 31, 2015, are as follows (in thousands):
|
Amount
|
Expiration Years
|
||||
|
Net operating losses—federal
|
$
|
94,677
|
Beginning 2024
|
||
|
Net operating losses—state
|
$
|
93,913
|
Beginning 2016
|
||
|
Tax return credits—federal
|
$
|
1,510
|
Beginning 2032
|
||
|
Tax return credits—state
|
$
|
2,293
|
Do not expire
|
||
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an "ownership change," generally defined as a greater than 50% change (by value) in its equity ownership over a three year period, the corporation's ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes, such as research tax credits, to offset its post-change income may be limited.
Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, provide for annual limitations on the utilization of net operating loss and research and experimentation credit carryforwards if the Company were to undergo an ownership change, as defined in Section 382. In general, an ownership change occurs whenever the percentage of the shares of a corporation owned, directly or indirectly, by 5-percent shareholders, as defined in Section 382, increases by more than 50 percentage points over the lowest percentage of the shares of such corporation owned, directly or indirectly, by such 5-percent shareholders at any time over the preceding three years. The Company underwent an ownership change within the meaning of Section 382 and 383 during 2012 and as such, the Company’s net operating loss carryforwards are limited. In addition, the pre-change R&D tax credits have also been limited for federal tax purposes. As a result of the Section 382 limitation, $144.9 million of the Company’s net operating losses and $11.5 million of R&D credits have been written off. As of December 31, 2015, the Company had federal net operating losses of $70.8 million that are subject to an annual limitation of $4.8 million through 2016 and decreases to $1.1 million thereafter. Any unused annual limitation balance is available for carryforward to subsequent years for utilization. The federal and state R&D credits reported are not subject to limitation.
Since the Company’s last ownership change within the meaning of Section 382 and 383, which occurred in 2012, the Company has increased its outstanding shares of common stock from 19,415,901 as of December 31, 2012 to 40,004,034 as of December 31, 2015, which may have resulted in a further ownership change. Accordingly, the utilization of net operating loss and credit carryforwards which existed at that time could be limited. The Company has not completed a Section 382 analysis to assess whether an ownership change has occurred or whether there have been multiple ownership changes since the Company became a “loss corporation” under the Code. The Company has and will continue to evaluate alternative analyses permitted under Section 382 and IRS notices to determine whether or not any ownership changes have occurred and may occur (and if so, when they occurred) that would result in limitations on its net operating losses (“NOLs”) or certain other tax attributes.
As of December 31, 2015, the Company had unrecognized tax benefits of $1.3 million, all of which would not currently affect the Company’s effective tax rate if recognized due to the Company’s deferred tax assets being fully offset by a valuation allowance. The Company did not anticipate any significant change to the unrecognized tax benefit balance as of December 31, 2015. A reconciliation of unrecognized tax benefits is as follows (in thousands):
|
Amount
|
||||
|
Balance as of December 31, 2012
|
545
|
|||
|
Additions based on tax positions related to prior years
|
104
|
|||
|
Additions based on tax positions related to current year
|
179
|
|||
|
Balance as of December 31, 2013
|
828
|
|||
|
Additions based on tax positions related to prior year
|
—
|
|||
|
Additions based on tax positions related to current year
|
171
|
|||
|
Balance as of December 31, 2014
|
$
|
999
|
||
|
Additions based on tax positions related to prior year
|
—
|
|||
|
Additions based on tax positions related to current year
|
269
|
|||
|
Balance as of December 31, 2015
|
$
|
1,268
|
||
The Company would classify interest and penalties related to uncertain tax positions in income tax expense, if applicable. There was no interest expense or penalties related to unrecognized tax benefits recorded through December 31, 2015. The tax years 2004 through 2014 remain open to examination by one or more major taxing jurisdictions to which the Company is subject.
The Company does not anticipate that total unrecognized net tax benefits will significantly change prior to the end of 2016.
|
12.
|
SUMMARIZED QUARTERLY UNAUDITED FINANCIAL DATA
|
Quarterly results were as follows (in thousands, except per share data):
|
Quarter Ended
|
||||||||||||||||
|
March 31,
|
June 30,
|
September 30,
|
December 31,
|
|||||||||||||
|
2015
|
||||||||||||||||
|
REVENUES:
|
||||||||||||||||
|
License revenue
|
$ | 49 | $ | 146 | $ | 548 | $ | 1,819 | ||||||||
|
Collaborative revenue
|
196 | 143 | 185 | 99 | ||||||||||||
|
Total revenues
|
245 | 289 | 733 | 1,918 | ||||||||||||
|
OPERATING EXPENSES:
|
||||||||||||||||
|
Research and development
|
$ | 5,995 | $ | 8,539 | $ | 10,359 | $ | 8,605 | ||||||||
|
General and administrative
|
1,907 | 1,696 | 2,091 | 1,874 | ||||||||||||
|
Research award
|
— | (1,100 | ) | (367 | ) | (1,171 | ) | |||||||||
|
LOSS FROM OPERATIONS
|
(7,657 | ) | (8,846 | ) | (11,350 | ) | (7,390 | ) | ||||||||
|
Other income (expense)
|
(3 | ) | (49 | ) | 24 | 51 | ||||||||||
|
NET LOSS
|
$ | (7,660 | ) | $ | (8,895 | ) | $ | (11,326 | ) | $ | (7,339 | ) | ||||
|
Net loss per share—basic and diluted
|
$ | (0.28 | ) | $ | (0.25 | ) | $ | (0.29 | ) | $ | (0.18 | ) | ||||
|
Shares used in computing basic and diluted
net loss per share
|
27,595,081 | 35,817,794 | 39,241,738 | 39,947,036 | ||||||||||||
|
Quarter Ended
|
||||||||||||||||
|
March 31,
|
June 30,
|
September 30,
|
December 31,
|
|||||||||||||
|
2014
|
||||||||||||||||
|
OPERATING EXPENSES:
|
||||||||||||||||
|
Research and development
|
$ | 5,765 | $ | 5,279 | $ | 5,268 | $ | 5,527 | ||||||||
|
General and administrative
|
1,844 | 1,586 | 1,419 | 1,771 | ||||||||||||
|
LOSS FROM OPERATIONS
|
(7,609 | ) | (6,865 | ) | (6,687 | ) | (7,298 | ) | ||||||||
|
Other income (expense)
|
(48 | ) | (31 | ) | (14 | ) | (3 | ) | ||||||||
|
Interest income (expense)
|
(259 | ) | (360 | ) | (286 | ) | (144 | ) | ||||||||
|
NET LOSS
|
$ | (7,916 | ) | $ | (7,256 | ) | $ | (6,987 | ) | $ | (7,445 | ) | ||||
|
Net loss per share—basic and diluted
|
$ | (0.39 | ) | $ | (0.34 | ) | $ | (0.31 | ) | $ | (0.32 | ) | ||||
|
Shares used in computing basic and diluted
net loss per share
|
20,123,447 | 21,479,386 | 22,747,308 | 22,926,664 | ||||||||||||
|
13.
|
SUBSEQUENT EVENTS
|
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
ANTHERA PHARMACEUTICALS, INC.
|
|||
|
By:
|
/s/ Paul F. Truex
|
||
|
Paul F. Truex
|
|||
|
Chief Executive Officer
|
|||
|
(Principal Executive Officer)
|
|||
Dated: March 14, 2016
POWER OF ATTORNEY
We, the undersigned officers and directors of Anthera Pharmaceuticals, Inc., hereby severally constitute and appoint Paul F. Truex and May Liu, and each of them singly (with full power to each of them to act alone), our true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution in each of them for him or her and in his or her name, place and stead, and in any and all capacities, to sign for us and in our names in the capacities indicated below any and all amendments to this report, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as full to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
|
/s/ Paul F. Truex
|
Chief Executive Officer and Director (Principal Executive Officer)
|
March 14, 2016
|
|
Paul F. Truex
|
||
|
/s/ May Liu
|
Senior Vice President, Finance and Administration (Principal Accounting Officer)
|
March 14, 2016
|
|
May Liu
|
||
|
/s/ Christopher S. Henney
|
Chairman of the Board of Directors
|
March 14, 2016
|
|
Christopher S. Henney
|
||
|
/s/ Brian R. Mueller
|
Director
|
March 14, 2016
|
|
Brian R. Mueller
|
||
|
/s/ Philip T. Sager
|
Director
|
March 14, 2016
|
|
Philip T. Sager
|
||
|
/s/ Steven B. Engle
|
Director
|
March 14, 2016
|
|
Steven B. Engle
|
||
|
/s/ David E. Thompson
|
Director
|
March 14, 2016
|
|
David E. Thompson
|
||
|
/s/ Sanford S. Zweifach
|
Director
|
March 14, 2016
|
|
Sanford S. Zweifach
|
Exhibit Index
|
Number
|
Description
|
|
|
3.1
|
Fifth Amended and Restated Certificate of Incorporation(1)
|
|
|
3.2
|
Certificate of Amendment to the Fifth Amended and Restated Certificate of Incorporation filed October 12, 2012(2)
|
|
|
3.3
|
Certificate of Amendment to the Fifth Amended and Restated Certificate of Incorporation filed July 12, 2013 and effective July 15, 2013(3)
|
|
|
3.4
|
Amended and Restated Bylaws, as amended on May 21, 2015(4)
|
|
|
4.1
|
Specimen certificate evidencing shares of common stock(5)
|
|
|
4.2
|
Form of Warrant sold pursuant to that Securities Purchase Agreement, among the Company and the purchasers thereto, dated September 20, 2010(6)
|
|
|
4.3
|
Form of Warrant Agreement dated as of March 25, 2011(7)
|
|
|
#10.1
|
2005 Equity Incentive Plan and form agreements thereunder(8)
|
|
|
#10.2
|
Amended and Restated 2010 Stock Option and Incentive Plan(9)
|
|
|
#10.3
|
Certificate of Amendment to Amended and Restated 2010 Stock Option and Incentive Plan(10)
|
|
|
#10.4
|
Form of Non-Qualified Stock Option Agreement for Company Employees Under the Anthera Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(11)
|
|
|
#10.5
|
Form of Non-Qualified Stock Option Agreement for Non-Employee Directors Under the Anthera Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(11)
|
|
|
#10.6
|
Form of Incentive Stock Option Agreement Under the Anthera Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(11)
|
|
|
#10.7
|
Form of Restricted Stock Award Agreement Under the Anthera Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(11)
|
|
|
#10.8
|
Restricted Stock Unit Award Agreement Under the Anthera Pharmaceuticals, Inc. 2010 Stock Option and Incentive Plan(12)
|
|
|
#10.9
|
2010 Employee Stock Purchase Plan(13)
|
|
|
#10.10
|
Amendment No. 1 to 2010 Employee Stock Purchase Plan(14)
|
|
|
#10.11
|
Amendment No. 2 to 2010 Employee Stock Purchase Plan(15)
|
|
|
#10.12
|
2013 Stock Option and Incentive Plan(16)
|
|
|
#10.13
|
Form of Non-Qualified Stock Option Agreement for Company Employees Under the 2013 Stock Option and Incentive Plan(17)
|
|
|
#10.14
|
Form of Non-Qualified Stock Option Agreement for Non-Employees Directors Under the 2013 Stock Option and Incentive Plan(17)
|
|
|
#10.15
|
Form of Incentive Stock Option Agreement Under the 2013 Stock Option and Incentive Plan(17)
|
|
|
#10.16
|
Form of Restricted Stock Award Agreement Under the 2013 Stock Option and Incentive Plan(17)
|
|
|
#10.17
|
Form of Restricted Stock Unit Award Agreement Under the 2013 Stock Option and Incentive Plan(17)
|
|
|
#10.18
|
Form of Amended and Restated Indemnification Agreement(18)
|
|
#10.19
|
Form of Amended and Restated Change in Control Agreement(19)
|
|
|
#10.20
|
Form of Amended and Restated Severance Benefits Agreement(20)
|
|
|
+10.21
|
License Agreement between Amgen Inc. and the Company, dated as of December 18, 2007(21)
|
|
|
10.22
|
Amendment No. 1 to License Agreement between Amgen Inc. and the Company, dated as of October 16, 2009(22)
|
|
|
+10.23
|
Amendment No. 2 to License Agreement between Amgen Inc. and the Company, dated as of November 26, 2014(23)
|
|
|
10.24
|
Form of Securities Purchase Agreement, among the Company and the purchasers thereto, dated September 20, 2010(24)
|
|
|
10.25
|
Lease by and between the Company and MEPT Mount Eden LLC, dated as of May 4, 2011(25)
|
|
|
10.26
|
Lease Amendment by and between the Company and MEPT Mount Eden LLC, dated as of November 13, 2013(26)
|
|
|
+10.27
|
License Agreement between the Company and Eli Lilly, dated as of July 11, 2014(27)
|
|
|
+10.28
|
Collaboration and License Agreement between the Company and Zenyaku Kogyo Co., Ltd., dated as of December 11, 2014(28)
|
|
|
+10.29
|
Stock Purchase Agreement between the Company and Zenyaku Kogyo Co., Ltd., dated as of December 11, 2014(29)
|
|
|
+10.30
|
Subscription Agreement between the Company and Zenyaku Kogyo Co., Ltd., dated as of January 27, 2015(30)
|
|
|
+10.31
|
Subscription Agreement between the Company and Amgen Inc., dated as of January 27, 2015(31)
|
|
|
+10.32
|
Subscription Agreement between the Company and Zenyaku Kogyo Co., Ltd., dated as of August 14, 2015
|
|
|
#10.33
|
Deferred Compensation Election Form by and between the Company and Paul Truex, effective as of December 28, 2015
|
|
|
#10.34
|
Employment Offer Letter between the Company and Mr. Craig Thompson, dated as of December 4, 2015
|
|
|
#10.35
|
Employment Agreement, by and between the Company and Mr. Paul Truex, dated as of January 25, 2016
|
|
|
#10.36
|
Employment Agreement, by and between the Company and Mr. Craig Thompson, dated as of January 25, 2016
|
|
|
#10.37
|
Employment Agreement, by and between the Company and Dr. Colin Hislop, dated as of January 25, 2016
|
|
|
#10.38
|
Employment Agreement, by and between the Company and Ms. Klara Dickinson-Eason, dated as of January 25, 2016
|
|
|
#10.39
|
Employment Agreement, by and between the Company and Dr. Chuck Olson, dated as of January 25, 2016
|
|
|
#10.40
|
Employment Agreement, by and between the Company and Ms. May Liu, dated as of January 25, 2016
|
|
|
10.41
|
Stock Purchase Agreement between the Company and Lincoln Park Capital Fund, LLC, dated as of March 12, 2015 (32)
|
|
|
10.42
|
Amendment to the Stock Purchase Agreement between the Company and Lincoln Park Capital Fund LLC, dated as of July 8, 2015 (33)
|
|
|
14.1
|
Code of Ethics (34)
|
|
|
21.1
|
Subsidiaries of Anthera Pharmaceuticals, Inc.(35)
|
|
|
23.1
|
Consent of BDO USA LLP, independent registered public accounting firm
|
|
|
24.1
|
Power of Attorney (included on signature page hereto)
|
|
|
31.1
|
Certification of Principal Executive Officer pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended
|
|
31.2
|
Certification of Principal Financial Officer pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended
|
|
|
32.1
|
Certification of Principal Executive Officer pursuant to Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|
|
32.2
|
Certification of Principal Financial Officer pursuant to Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. Section 1350, as adopted pursuant Section 906 of the Sarbanes-Oxley Act of 2002
|
|
101.INS
|
XBRL Instance Document.
|
|
|
101.SCH
|
XBRL Taxonomy Extension Schema Document.
|
|
|
101.CAL
|
XBRL Taxonomy Extension Calculation Linkbase Document.
|
|
|
101.DEF
|
XBRL Taxonomy Extension DefinitionLinkbase Document.
|
|
|
101.LAB
|
XBRL Taxonomy Extension Label Linkbase Document.
|
|
|
101.PRE
|
XBRL Taxonomy Extension Presentation Linkbase Document.
|
|
+
|
Certain provisions of this Exhibit have been omitted pursuant to a request for confidential treatment.
|
|
#
|
Indicates management contract or compensatory plan, contract or agreement.
|
|
(1)
|
Filed as Exhibit 3.6 to the registrant’s Registration Statement on Form S-1/A (File No. 333-161930), filed with the SEC on February 3, 2010 and incorporated herein by reference.
|
|
(2)
|
Filed as the same numbered exhibit to the registrant’s Annual Report on Form 10-K, filed with the SEC on March 26, 2013 and incorporated herein by reference.
|
|
(3)
|
Filed as Exhibit 3.1 to the registrant Current Report on Form 8-K, filed with the SEC on July 16, 2013 and incorporated herein by reference.
|
|
(4)
|
Filed as Exhibit 3.4 to the registrant’s Quarterly Report on Form 10-Q filed with the SEC on August 10, 2015 and incorporated herein by reference.
|
|
(5)
|
Filed as the same numbered exhibit to the registrant’s Amendment No. 3 to Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on January 29, 2010 and incorporated herein by reference.
|
|
(6)
|
Filed as Exhibit 4.1 to the registrant’s Current Report on Form 8-K, filed with the SEC on September 22, 2010 and incorporated herein by reference.
|
|
(7)
|
Filed as Exhibit 10.2 to registrant’s Current Report on Form 8-K, filed with the SEC on March 29, 2011 and incorporated herein by reference.
|
|
(8)
|
Filed as the same numbered exhibit to the registrant’s Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on September 15, 2009 and incorporated herein by reference.
|
|
(9)
|
Filed as Appendix A to the registrant’s Definitive Proxy Statement on Schedule 14A, filed with the SEC on June 8, 2010 and incorporated herein by reference.
|
|
(10)
|
Filed as Exhibit 10.2 to registrant’s Quarterly Report on Form 10-Q, filed with the SEC on August 12, 2011 and incorporated herein by reference.
|
|
(11)
|
Filed as Exhibit 10.2 to the registrant’s Amendment No. 4 to Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on February 3, 2010 and incorporated herein by reference.
|
|
(12)
|
Filed as Exhibit 10.1 to the registrant’s Quarterly Report on Form 10-Q, filed with the SEC on May 14, 2010 and incorporated herein by reference.
|
|
(13)
|
Filed as Appendix B to the registrant’s Definitive Proxy Statement on Schedule 14A, filed with the SEC on June 8, 2010 and incorporated herein by reference.
|
|
(14)
|
Filed as Exhibit 10.42 to the registrant’s Annual Report on Form 10-K, filed with the SEC on March 7, 2011 and incorporated herein by reference.
|
|
(15)
|
Filed as Exhibit 10.34 to the registrant’s Annual Report on Form 10-K, filed with the SEC on March 26, 2013 and incorporated herein by reference.
|
|
(16)
|
Filed as Annex B to the registrants Definitive Proxy Statement on Schedule 14A, filed with the SEC on April 5, 2013 and incorporated herein by reference.
|
|
(17)
|
Filed as the same numbered exhibit to the registrant’s Annual Report on Form 10-K, filed with the SEC on March 28, 2014 and incorporated herein by reference.
|
|
(18)
|
Filed as Exhibit 10.3 to the registrant’s Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on September 15, 2009 and incorporated herein by reference.
|
|
(19)
|
Filed as the Exhibit 10.4 to the registrant’s Amendment No. 1 to Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on October 19, 2009 and incorporated herein by reference.
|
|
(20)
|
Filed as the Exhibit 10.5 to the registrant’s Amendment No. 1 to Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on October 19, 2009 and incorporated herein by reference.
|
|
(21)
|
Filed as Exhibit 10.10 to the registrant’s Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on September 15, 2009 and incorporated herein by reference.
|
|
(22)
|
Filed as Exhibit 10.18 to the registrant’s Amendment No. 1 to Registration Statement on Form S-1 (File No. 333-161930), filed with the SEC on October 19, 2009 and incorporated herein by reference.
|
|
(23)
|
Filed as Exhibit 10.23 to registrant’s Annual Report on Form 10-K, filed with the SEC on March 16, 2015, and incorporated herein by reference.
|
|
(24)
|
Filed as Exhibit 10.1 to registrant’s Current Report on Form 8-K, filed with the SEC on September 22, 2010 and incorporated herein by reference.
|
|
(25)
|
Filed as Exhibit 10.4 to registrant’s Quarterly Report on Form 10-Q, filed with the SEC on May 13, 2011, and incorporated herein by reference.
|
|
(26)
|
Filed as Exhibit 10.27 to registrant’s Annual Report on Form 10-K, filed with the SEC on March 28, 2014, and incorporated herein by reference.
|
|
(27)
|
Filed as Exhibit 10.1 to registrant’s Quarterly Report on Form 10-Q/A, filed with the SEC on December 12, 2014 and incorporated herein by reference
|
|
(28)
|
Filed as Exhibit 10.33 to registrant’s Annual Report on Form 10-K, filed with the SEC on March 16, 2015 and incorporated herein by reference
|
|
(29)
|
Filed as Exhibit 10.34 to registrant’s Annual Report on Form 10-K, filed with the SEC on March 16, 2015 and incorporated herein by reference
|
|
(30)
|
Filed as Exhibit 10.35 to registrant’s Annual Report on Form 10-K, filed with the SEC on March 16, 2015 and incorporated herein by reference
|
|
(31)
|
Filed as Exhibit 10.36 to registrant’s Annual Report on Form 10-K, filed with the SEC on March 16, 2015 and incorporated herein by reference
|
|
(32)
|
Filed as Exhibit 10.1 to the registrant’s Current Report on Form 8-K, filed with the SEC on March 16, 2015 (dated March 12, 2015) and incorporated herein by reference
|
|
(33)
|
Filed as Exhibit 10.1 to registrant’s Quarterly Report on Form 10-Q, filed with the SEC on August 10, 2015 and incorporated herein by reference
|
|
(34)
|
Filed as the same numbered exhibit to the registrant’s Annual Report on Form 10-K, filed with the SEC on March 7, 2011 and incorporated herein by reference
|
|
(35)
|
Filed as the same numbered exhibit to the registrant’s Annual Report on Form 10-K, filed with the SEC on March 14, 2012 and incorporated herein by reference
|
97