UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, DC 20549
Form 10-K
For
Annual and Transition Reports Pursuant to
Section 13 or 15(d) of the Securities Exchange Act of
1934
| |
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal
year ended December 31,
2009
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to
|
Commission File Number:
000-51820
Alexza Pharmaceuticals,
Inc.
(Exact name of Registrant as
specified in its charter)
| |
|
|
Delaware
(State or Other Jurisdiction
of Incorporation or Organization)
|
|
77-0567768
(I.R.S. Employer
Identification Number)
|
2091 Stierlin Court
Mountain View, California 94043
(Address of Principal Executive
Offices including Zip Code)
Registrant’s telephone number, including area code:
(650) 944-7000
Securities
registered pursuant to Section 12 (b) of the
Act:
| |
|
|
|
Title of Each Class
|
|
Name of Each Exchange on Which Registered
|
|
|
|
Common Stock, par value $0.0001 per share
|
|
Nasdaq Global Market
|
Securities registered pursuant to Section 12 (g) of
the Act:
None
Indicate by check mark if the Registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark if the Registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. Yes o No þ
Indicate by check mark whether the Registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the Registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
during the preceding 12 months (of for such shorter period
that the registrant was required to submit and post such
files). Yes o No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of
Form 10-K
or any amendments to this
Form 10-K. þ
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in
Rule 12b-2
of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large accelerated
filer o
|
|
Accelerated
filer þ
|
|
Non-accelerated
filer o
|
|
Smaller reporting
company o
|
|
|
|
(Do not check if a smaller
reporting company)
|
Indicate by check mark whether the Registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). Yes o No þ
The aggregate market value of the voting and non-voting stock
held by non-affiliates of the Registrant was $61,758,062 based
on the closing sale price of the Registrant’s common stock
on The NASDAQ Global Market on June 30, 2009. Shares of the
Registrant’s common stock beneficially owned by each
executive officer and director of the Registrant and by each
person known by the Registrant to beneficially own 10% or more
of its outstanding common stock have been excluded, in that such
persons may be deemed to be affiliates. This determination of
affiliate status is not necessarily a conclusive determination
for other purposes. The number of outstanding shares of the
Registrant’s common stock as of February 26, 2010 was
52,566,338.
DOCUMENTS
INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for
the 2010 Annual Meeting of Stockholders to be filed within
120 days after the end of the Registrant’s fiscal year
ended December 31, 2009 are incorporated by reference into
Part III of this Annual Report on
Form 10-K
to the extent stated therein.
ANNUAL
REPORT ON
FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2009
TABLE OF
CONTENTS
2
The names “Alexza” and “Staccato” are
trademarks of Alexza Pharmaceuticals, Inc. We have registered
the trademarks “Alexza Pharmaceuticals,”
“Alexza” and “Staccato” with the
U.S. Patent and Trademark Office. All other trademarks,
trade names and service marks appearing in this Annual Report on
Form 10-K
are the property of their respective owners.
PART I.
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS
Some of the statements under “Business,” “Risk
Factors,” “Management’s Discussion and Analysis
of Financial Condition and Results of Operations” and
elsewhere in this Annual Report constitute forward-looking
statements. In some cases, you can identify forward-looking
statements by the following words: “may,”
“will,” “could,” “would,”
“should,” “expect,” “intend,”
“plan,” “anticipate,” “believe,”
“estimate,” “predict,” “project,”
“potential,” “continue,” “ongoing”
or the negative of these terms or other comparable terminology,
although not all forward-looking statements contain these words.
Examples of these statements include, but are not limited to,
statements regarding the following: the prospects of us
receiving approval to market AZ-004, our anticipated timing for
receiving approval for our New Drug Application for AZ-004, the
implications of interim or final results of our clinical trials,
the progress and timing of our research programs, including
clinical testing, our anticipated timing for filing additional
Investigational New Drug Applications with the United States
Food and Drug Administration, the initiation or completion of
Phase 1, Phase 2 or Phase 3 clinical testing for our product
candidates, the extent to which our issued and pending patents
may protect our products and technology, the potential of such
product candidates to lead to the development of safe or
effective therapies, our ability to enter into collaborations,
our future operating expenses, our future losses, our future
expenditures, and the sufficiency of our cash resources. These
statements involve known and unknown risks, uncertainties and
other factors that may cause our actual results, levels of
activity, performance or achievements to be materially different
from the information expressed or implied by these
forward-looking statements. While we believe that we have a
reasonable basis for each forward-looking statement contained in
this Annual Report, we caution you that these statements are
based on a combination of facts and factors currently known by
us and our projections of the future, about which we cannot be
certain.
In addition, you should refer to the “Risk Factors”
section of this Annual Report for a discussion of other
important factors that may cause our actual results to differ
materially from those expressed or implied by our
forward-looking statements. As a result of these factors, we
cannot assure you that the forward-looking statements in this
Annual Report will prove to be accurate. Furthermore, if our
forward-looking statements prove to be inaccurate, the
inaccuracy may be material. In light of the significant
uncertainties in these forward-looking statements, you should
not regard these statements as a representation or warranty by
us or any other person that we will achieve our objectives and
plans in any specified time frame, or at all.
We undertake no obligation to publicly update any
forward-looking statements, whether as a result of new
information, future events or otherwise. You are advised,
however, to consult any further disclosures we make on related
subjects in our Quarterly Reports on
Form 10-Q,
Current Reports on
Form 8-K,
and our website.
We are a pharmaceutical company focused on the research,
development, and commercialization of novel proprietary products
for the acute treatment of central nervous system, or CNS,
conditions. All of our product candidates are based on our
proprietary technology, the Staccato system. The
Staccato system vaporizes an excipient-free drug to form
a condensation aerosol that, when inhaled, allows for rapid
systemic drug delivery. Because of the particle size of the
aerosol, the drug is quickly absorbed through the deep lung into
the bloodstream, providing speed of therapeutic onset that is
comparable to intravenous, or IV, administration but with
greater ease, patient comfort and convenience. In December 2009,
we submitted our first New Drug Application, or NDA, to the
U.S. Food and Drug Administration, or FDA, for our lead
product candidate, AZ-004 (Staccato loxapine). In
February 2010, we licensed the U.S. and Canadian
commercialization rights to AZ-004 to Biovail Laboratories
International SRL, or Biovail. We plan to seek additional
commercial partners for AZ-004 outside of the U.S. and
Canada.
3
We have five other product candidates in various stages of
clinical development, ranging from Phase 1 through late-stage
Phase 2. In January 2009 we reduced, and in some cases
suspended, the development of these product candidates in order
to concentrate our efforts on the clinical, regulatory,
manufacturing and commercial development of our lead product
candidate, AZ-004. During the first half of 2010, we expect to
conduct a review of our product candidate portfolio. In the
second half of 2010, we plan to advance the development of at
least one of these product candidates. We are seeking partners
to support the continued development of these product
candidates, but may develop one or more of these product
candidates without partner support.
Since our inception, we have screened more than 400 drug
compounds and we have identified approximately 200 drug
compounds that demonstrate initial vaporization feasibility for
delivery with our technology. We believe that a number of these
drug compounds, when delivered by the Staccato system,
would have a desirable therapeutic profile for the treatment of
various acute and intermittent conditions. We are initially
focusing on developing proprietary products by combining our
Staccato system with small molecule drugs that have been
in use for many years and are well-characterized to create
aerosolized forms of these drugs. We believe that we will be
able to reduce the development time and risks associated with
our product candidates, compared to the development of new
chemical entities.
Our clinical-stage product candidates are:
|
|
|
| |
•
|
AZ-004 (Staccato loxapine). We are developing
AZ-004 for the rapid treatment of agitation in patients with
schizophrenia or bipolar disorder. In December 2009, we
submitted our NDA to the FDA. In February 2010, the FDA accepted
our filing and provided us a Prescription Drug User Fee Act
(PDUFA) goal date of October 11, 2010. We believe that the
data generated from our clinical and non-clinical studies (and
is contained within our NDA submission) adequately demonstrate
the efficacy and safety of AZ-004 for the rapid treatment of
agitation in patients with schizophrenia or bipolar disorder.
|
In February 2010, we entered into a collaboration and license
agreement, or license agreement, and a manufacture and supply
agreement, collectively, the collaboration, with Biovail
Laboratories International SRL, or Biovail, for AZ-004
(Staccato®
loxapine) for the treatment of psychiatric
and/or
neurological indications and the symptoms associated with these
indications, including the initial indication of treating
agitation in schizophrenia and bipolar disorder patients. The
collaboration contemplates that we will be the exclusive
supplier of drug product for clinical and commercial uses and
have responsibility for the NDA for AZ-004 for the initial
indication of rapid treatment of agitation in patients with
schizophrenia or bipolar disorder, as well as responsibility for
any additional development and regulatory activities required
for use in these two patient populations in the outpatient
setting. Biovail will be responsible for commercialization for
the initial indication and, if it elects, development and
commercialization of additional indications for AZ-004 in the
U.S. and Canada.
Under the terms of the license agreement, Biovail paid us an
upfront fee of $40 million, and we may be eligible to
receive up to an additional $90 million in milestone
payments upon achievement of predetermined regulatory, clinical
and commercial manufacturing milestones. We may be subject to
certain payment obligations to Biovail, up to $5 million,
if we do not meet certain other milestones prior to a
termination of the license agreement. We are also eligible to
receive tiered royalty payments of 10% to 25% on any net sales
of AZ-004. We are responsible for conducting and funding all
development and regulatory activities associated with
AZ-004’s initial indication for the rapid treatment of
agitation in patients with schizophrenia or bipolar disorder as
well as for its possible use in the outpatient setting in these
two patient populations. Our obligation to fund the outpatient
development efforts is limited to a specified amount, none of
which is expected to be incurred in 2010. Biovail is responsible
for certain Phase 4 development commitments and related costs
and expenses. For additional indications, we have an obligation
regarding certain efforts and related costs and expenses, up to
a specified amount, and, if it elects, Biovail is responsible
for all other development commitments and related costs and
expenses.
Under the terms of the manufacture and supply agreement, we are
the exclusive supplier of AZ-004 and have responsibility for the
manufacture, packaging, labeling and supply for clinical and
commercial uses. Biovail will purchase AZ-004 from us at
predetermined transfer prices. The transfer prices depend on the
volume of AZ-004 purchases, subject to certain adjustments.
4
Either party may terminate the collaboration for the other
party’s uncured material breach or bankruptcy. In addition,
Biovail has the right to terminate the collaboration
(a) upon 90 days written notice for convenience;
(b) upon 90 days written notice if FDA does not
approve the AZ-004 NDA for the initial indication for the rapid
treatment of agitation in patients with schizophrenia or bipolar
disorder; (c) immediately upon written notice for safety
reasons or withdrawal of marketing approval; (d) upon
90 days written notice upon certain recalls of the product;
or (e) immediately upon written notice within 60 days
of termination of the supply agreement under certain
circumstances. The supply agreement automatically terminates
upon the termination of the license agreement.
|
|
|
| |
•
|
AZ-007 (Staccato zaleplon). We are
developing AZ-007 for the treatment of insomnia in patients who
have difficulty falling asleep, including patients who awake in
the middle of the night and have difficulty falling back asleep.
AZ-007 has completed Phase 1 testing. In the Phase 1 clinical
trial, AZ-007 delivered an IV-like pharmacokinetic profile with
a median time to peak drug concentration of 1.6 minutes.
Pharmacodynamics, measured as sedation assessed on a 100 mm
visual analog scale, showed onset of effect as early as 2
minutes after dosing with AZ-007.
|
| |
| |
•
|
AZ-001 (Staccato prochlorperazine). We are
developing AZ-001 to treat patients suffering from acute
migraine headaches. During the third quarter of 2008, we
conducted an
end-of-Phase
2 meeting with the FDA. We believe we have a clear understanding
of the development requirements for filing an NDA for this
product candidate.
|
| |
| |
•
|
AZ-104 (Staccato loxapine,
low-dose). We are developing AZ-104 to treat
patients suffering from acute migraine headaches. AZ-104 is a
lower-dose version of AZ-004. In September 2009, we announced
preliminary results from our 366 patient Phase 2b clinical
trial of AZ-104 in patients with migraine headache. The trial
was an outpatient, multi-center, randomized, double-blind,
single administration, placebo-controlled study. The study was
designed to evaluate the treatment of a single migraine attack
of moderate to severe intensity. Two doses of AZ-104,
1.25 mg and 2.5 mg, and placebo were evaluated in the
clinical trial. Both AZ-104 dose groups trended towards
statistical significance, but the study did not meet its primary
endpoint, which was defined as pain-relief at the
two-hour
time point, compared to placebo. There were no serious adverse
events in the clinical trial, and AZ-104 was generally safe and
well tolerated in this patient population.
|
| |
| |
•
|
AZ-002 (Staccato alprazolam). AZ-002 has
completed a Phase 1 clinical trial in healthy subjects and a
Phase 2a
proof-of-concept
clinical trial in panic disorder patients for the treatment of
panic attacks, an indication we are not planning to pursue.
However, given the safety profile, the successful and
reproducible delivery of alprazolam, and the IV-like
pharmacological effect demonstrated to date, we are assessing
AZ-002 for other possible indications and renewed clinical
development.
|
| |
| |
•
|
AZ-003 (Staccato fentanyl). We are
developing AZ-003 for the treatment of patients with acute pain,
including patients with breakthrough cancer pain and
postoperative patients with acute pain episodes. We have
completed and announced positive results from a Phase 1 clinical
trial of AZ-003 in opioid-naïve healthy subjects.
|
In August 2009, we completed the acquisition of Symphony
Allegro, Inc., an entity formed in 2006 by Symphony Capital LLC
and other investors, together the Allegro Investors, to fund
additional clinical and nonclinical development of AZ-002, and
AZ-004/104, through the exercise of our option to acquire all of
the outstanding equity of Symphony Allegro. In exchange for all
of the outstanding shares of Symphony Allegro, we:
(i) issued to the Allegro Investors 10 million shares
of common stock, (ii) issued to the Allegro Investors
five-year warrants to purchase 5 million shares of common
stock at an exercise price of $2.26 per share and canceled the
previously outstanding warrants to purchase 2 million
shares of common stock held by the Allegro Investors, and
(iii) agreed to pay certain percentages of cash payments
that may be generated from future partnering transactions for
AZ-004, AZ-104
and/or
AZ-002, the product candidates that were licensed to Symphony
Allegro. In February 2010, we paid Symphony $7.5 million of
the upfront fee that was received from Biovail pursuant to our
collaboration with them. Symphony will be entitled to receive a
portion of any future milestone and royalty payments we may
receive from Biovail pursuant to this agreement.
5
Other than those licensed to Biovail, we have retained all
rights to our product candidates and the Staccato system.
We eventually plan to build a United States-based specialty
sales force to commercialize our product candidates, other than
AZ-004, which are approved for marketing and which are intended
for specialty pharmaceutical markets. We plan to enter into
strategic partnerships with other companies to commercialize
products that are intended for certain markets in the United
States and for all of our product candidates in geographic
territories outside the United States.
Market
Opportunity for Acute and Intermittent Conditions
Acute and intermittent medical conditions are characterized by a
rapid onset of symptoms that are temporary and severe, and that
occur at irregular intervals, unlike the symptoms of chronic
medical conditions that continue at a relatively constant level
over time. Approved drugs for the treatment of many acute and
intermittent conditions, such as antipsychotics to treat
agitation, triptans to treat migraine headaches and
benzodiazepines to treat anxiety, are typically delivered either
in tablets or by injections. Traditional inhalation technologies
are also being developed to treat these conditions. These
delivery methods have the following advantages and disadvantages:
|
|
|
| |
•
|
Oral Tablets. Oral tablets or capsules are
convenient and cost effective, but they generally do not provide
rapid onset of action. Oral tablets may require at least one to
four hours to achieve peak plasma levels. Also, some drugs, if
administered as a tablet or capsule, do not achieve adequate or
consistent bioavailability due to the degradation of the drug by
the stomach or liver or inability to be absorbed into the
bloodstream.
|
| |
| |
•
|
Injections. Intravenous, or IV, or
intramuscular, or IM, injections provide a more rapid onset of
action than oral tablets and can sometimes be used to titrate
potent drugs with very rapid changes in effect. Titration refers
to the ability of a patient or care giver to administer an
initial dose of medication and then determine if the medication
is effective; if the medication is effective no further dosing
is required. However, if the medication is not yet effective,
another dose can be administered repeating this process until
the medication has had an adequate effect. However, with a few
exceptions, injections generally are administered by trained
medical personnel in a medical care setting. Other forms of
injections result in an onset of action that is generally
substantially slower than IV injection, although often
faster than oral administration. All forms of injections are
invasive, can be painful to some patients and are often
expensive. In addition, many drugs are not water soluble and can
be difficult to formulate in an injectable form.
|
| |
| |
•
|
Traditional Inhalation. Traditional dry powder
and aerosolized inhalation delivery systems have been designed
and used primarily for local delivery of drugs to the
respiratory airways, not to the deep lung for rapid systemic
drug delivery. Certain recent variants of these systems,
however, can provide systemic delivery of drugs, either for the
purpose of rapid onset of action or to enable noninvasive
delivery of drugs that are not orally bioavailable.
Nevertheless, many of these systems have difficulty in
generating appropriate drug particle sizes or consistent emitted
doses for deep lung delivery. To achieve appropriate drug
particle sizes and consistent emitted doses, most traditional
inhalation systems require the use of excipients and additives
such as detergents, stabilizers and solvents, which may
potentially cause toxicity or allergic reactions. Many
traditional inhalation devices require patient coordination to
deliver the correct drug dose, leading to potentially wide
variations in the drug delivered to a patient.
|
As a result of these limitations, we believe there is a
significant unmet medical and patient need for products for the
treatment of acute and intermittent conditions that can be
delivered in precise amounts, provide rapid therapeutic onset,
and are noninvasive and easy to use.
Our
Solution: Staccato System
Our Staccato system rapidly vaporizes an excipient-free
drug compound to form a proprietary condensation aerosol that is
inhaled and rapidly achieves systemic blood circulation via deep
lung absorption. The Staccato system consistently creates
aerosol particles averaging one to three and one-half microns in
size, which is the most appropriate size for deep lung
inhalation and absorption into the bloodstream.
6
We believe our Staccato system matches delivery
characteristics and product attributes to patient needs for
acute and intermittent conditions, with the following advantages:
|
|
|
| |
•
|
Rapid Onset. The aerosol produced with the
Staccato system is designed to be rapidly absorbed
through the deep lung with a speed of therapeutic onset
comparable to an IV injection, generally achieving peak
plasma levels of drug in two to five minutes.
|
| |
| |
•
|
Ease of Use. The Staccato system is
breath actuated, and a patient simply inhales to administer the
drug dose. Unlike injections, the Staccato system is
noninvasive and does not require caregiver assistance. The
aerosol produced with the Staccato system is relatively
insensitive to patient inhalation rates. Unlike many other
inhalation technologies, the patient does not need to learn a
special breathing pattern. In addition, the Staccato
device is small and easily portable.
|
| |
| |
•
|
Consistent Particle Size and Dose. The
Staccato system uses rapid heating of the drug film to
create consistent and appropriate particle sizes for deep lung
inhalation and absorption into the bloodstream. The Staccato
system also produces a consistent high emitted dose,
regardless of the patient’s breathing pattern.
|
| |
| |
•
|
Broad Applicability. We have screened over 400
drugs, and approximately 200 have exhibited initial vaporization
feasibility using our Staccato system. The Staccato
system can deliver both water soluble and water insoluble
drugs and eliminates the need for excipient and additives such
as detergents, stabilizers and solvents, avoiding the side
effects that may be associated with the excipient or additives.
|
| |
| |
•
|
Design Flexibility. The Staccato system
can incorporate multiple features, including lockout to
potentially enhance safety, the convenience of patient
titration, and a variety of dose administration regimens.
|
Drug
Candidates Based on the Staccato System
We combine small molecule drugs with our Staccato system
to create proprietary product candidates. We believe that the
drugs we are currently using are no longer eligible for patent
protection as chemical entities or have their patent protection
expiring in the next several years. These drugs have been widely
used, and we believe their biological activity and safety are
well understood and characterized. We have received composition
of matter patent protection on the Staccato aerosolized
forms of these drugs. We also intend to collaborate with
pharmaceutical companies to develop new chemical entities,
including compounds that might otherwise not be suitable for
development because of limitations of traditional delivery
methods.
Staccato
System
Our product candidates employing Staccato system consist
of three core components: (1) a heat source that includes
an inert metal substrate; (2) a thin film of an
excipient-free drug compound, also known as an active
pharmaceutical ingredient, or API, coated on the substrate; and
(3) an airway through which the patient inhales. The left
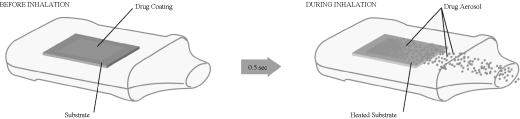
panel of the illustration below depicts these core components
prior to patient inhalation.
The right panel of the illustration below depicts the
Staccato system during patient inhalation: (1) the
heated substrate has reached peak temperature in less than one
half second after the start of patient inhalation; (2) the
thin drug film has been vaporized; and (3) the drug vapor
has subsequently cooled and condensed into excipient-free drug
aerosol particles that are being drawn into the patient’s
lungs. The entire Staccato system actuation occurs in
less than one second.
7
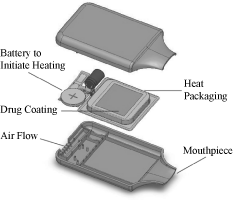
Five of our product candidates, AZ-004, AZ-007, AZ-001, AZ-104,
and AZ-002, use the same disposable, single-dose delivery
device. The single dose delivery device consists of a metal
substrate that is chemically heated through a battery-initiated
reaction of energetic materials. In the current design, the heat
package can be coated with up to 10 milligrams of API. The
device is portable and easy to carry, with dimensions of
approximately three inches in length, two inches in width, and
one inch in thickness. The device weighs approximately one
ounce. A diagram of the single dose delivery device is shown
below:
AZ-003 uses a multiple dose delivery device consisting of a
reusable controller and a disposable dose cartridge. We have
designed the multiple dose delivery device to meet the specific
needs of our AZ-003 product candidate. The dose cartridge
currently contains 25 separate metal substrates, each coated
with the API, which rapidly heat upon application of electric
current from the controller. In the current design, 25
micrograms of drug compound are coated on each metal substrate.
The device is portable and easy to carry, with dimensions of
approximately five inches in length, two and one-half inches in
width and one inch in thickness. The controller weighs
approximately four ounces, and the dose cartridge weighs
approximately one ounce.
We continue to undertake research and development efforts to
improve commercial manufacturability of our single dose device
and to develop future generations of the Staccato
technology.
8
Our
Pipeline
As indicated below, we have submitted an NDA for AZ-004, our
lead product candidate. We have five additional product
candidates; one product candidate has completed Phase 2 clinical
testing, two product candidates are in Phase 2 clinical testing,
and two product candidates have completed Phase 1 clinical
testing. In January 2009 we reduced, and in some cases
suspended, the development of our five additional product
candidates in order to concentrate our efforts on the clinical,
regulatory, manufacturing and commercial development of our lead
product candidate, AZ-004. During the first half of 2010, we
expect to conduct a review of our product candidate portfolio.
In the second half of 2010, we plan to advance the development
of at least one of these product candidates. We are seeking
partners to support continued development of these product
candidates, but may develop one or more of these product
candidates without partner support.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza
|
|
|
|
|
|
|
|
Development
|
|
Commercial
|
|
Product Candidate
|
|
API
|
|
Target Indication
|
|
Status
|
|
Rights
|
|
|
|
AZ-004
|
|
Loxapine
|
|
Agitation in schizophrenia or bipolar disorder patients
|
|
NDA submitted December 2009, PDUFA date of October 11, 2010.
|
|
Out-licensed U.S. and Canadian commercialization rights,
retained all other rights*
|
|
AZ-007
|
|
Zaleplon
|
|
Insomnia
|
|
Phase 1 completed
|
|
Worldwide
|
|
AZ-001
|
|
Prochlorperazine
|
|
Migraine headache
|
|
End of Phase 2 FDA meeting completed
|
|
Worldwide
|
|
AZ-104
|
|
Loxapine (low-dose)
|
|
Migraine headache
|
|
Phase 2
|
|
Worldwide
|
|
AZ-002
|
|
Alprazolam
|
|
Panic attacks and other CNS conditions
|
|
Phase 2
|
|
Worldwide
|
|
AZ-003
|
|
Fentanyl
|
|
Acute pain
|
|
Phase 1 completed
|
|
Worldwide
|
|
|
|
|
* |
|
Licensed to Biovail Laboratories International, SRL |
AGITATION
PROGRAM: AZ-004 (Staccato loxapine)
We are developing AZ-004 (Staccato loxapine) for the
rapid treatment of agitation in patients with schizophrenia or
bipolar disorder. Episodes of agitation afflict many people
suffering from major psychiatric disorders, including
schizophrenia, which affects approximately 2.4 million
adults in the United States, and bipolar disorder, which affects
approximately 5.7 million adults in the United States. More
than 90% of these patients will experience agitation in their
lifetimes.
Agitation generally escalates over time with patients initially
feeling uncomfortable, tense and restless. As the agitation
intensifies, their behavior appears more noticeable to others as
they become threatening and potentially violent, especially if
the agitation is not treated. While patients seek treatment at
different points along this agitation continuum, those with the
most severe symptoms generally require treatment with injectable
drugs in emergency medical settings, and currently are thought
to represent the agitation market. Alexza, however, believes the
therapeutic market for agitation is broader than only this
limited perspective of patients in severe crisis —
many more are in need of treatment for an agitation episode.
Market
Opportunity
Our primary market research indicates that approximately 50% of
treated acute agitation episodes are treated in emergency
settings. Another approximately 35% of the treated agitation
episodes suffered by schizophrenic and bipolar disorder patients
are treated in an inpatient setting (hospital and long-term
residential settings), and approximately 15% are treated in a
physician’s office. Our market research studies with
schizophrenia patient caregivers and bipolar disorder patients
indicate these patients currently experience an average of 11 to
12 episodes of agitation each year.
Agitation episodes are currently treated about 55% of the time
with oral antipsychotics and about 45% of the time with
intra-muscular, or IM, injections. Oral medications work
relatively slowly, but are easy to administer,
9
painless and are less threatening to patients. IM injections
have a faster onset of action and a higher predictability of
drug effect, but because they are invasive and can be
frightening to patients, IM injections are usually the treatment
option of last resort. Currently, no non-invasive therapies are
available that work faster than 30 minutes to help agitated
patients in need of treatment.
AZ-004 is an anti-agitation therapeutic that combines
Alexza’s proprietary Staccato system with loxapine,
a drug belonging to the class of compounds known generally as
antipsychotics. Loxapine is currently approved in oral and
injectable (intramuscular only) formulations in the United
States for the management of the manifestations of
schizophrenia. The Staccato system is a hand-held,
chemically-heated, single-dose inhaler that delivers a pure drug
aerosol to the highly vascularized tissues of the deep lung.
As an easy-to-use, patient-controlled, and highly reliable
therapeutic that provides rapid relief, onset of effect was 10
minutes in two Phase 3 trials, we believe AZ-004 meets the three
key treatment attributes for acute agitation specified in the
American Association of Emergency Psychiatrists’ Expert
Consensus Guidelines for the Treatment of Behavioral
Emergencies: speed of onset, reliability of medication delivery
and patient preference.
We believe that AZ-004, if approved, has the potential to change
the treatment practices for rapidly treating agitation, as the
only product available to meet both patient desires for comfort
and control, and the clinician goals of rapid and reliable
control of an agitation episode.
Development
Status
The AZ-004 NDA contains efficacy and safety data from more than
1,600 patients and subjects who have been studied in
thirteen different clinical trials, beginning with our first
Phase 1 study initiated in August 2005. During 2009, we
initiated and completed enrollment in five non-pivotal safety
and NDA-supporting studies for AZ-004, including a pulmonary
safety study in healthy subjects, a thorough QTc study in
healthy subjects, a smoker/non-smoker pharmacokinetic, or PK,
study in healthy subjects, a pulmonary safety study in subjects
with asthma and a pulmonary safety study in subjects with
chronic obstructive pulmonary disease, or COPD.
We completed a Phase 1 placebo-controlled study in 30 healthy
subjects to assess the pulmonary safety of AZ-004. We observed
that AZ-004, administered twice within a
24-hour
period, was safe and generally well tolerated in this study.
There were no systemic effects on pulmonary function versus
placebo, and no respiratory adverse events.
We completed the Phase 1 placebo-controlled thorough QTc study
in 48 healthy subjects. The purpose of a thorough QTc study is
to determine a drug’s potential effect on cardiac rhythms.
In this study, we found that the active control, moxifloxacin,
produced a positive QT/QTc signal that validated the sensitivity
of the clinical study. At all time points for the primary
analysis, the confidence intervals of the QTc for AZ-004 were
within the FDA standard 10 millisecond window, supporting the
cardiac safety of AZ-004.
We completed the Phase 1 smoker/non-smoker PK study in 35
healthy subjects. We observed comparable blood levels in smokers
and non-smokers for both AZ-004 and the metabolites of AZ-004.
Side-effect profiles were similar in smokers and non-smokers.
We completed Phase 1 placebo-controlled studies in 53 subjects
with predominantly
moderate-to-severe
COPD and in 52 subjects with
mild-to-moderate
persistent asthma to assess the pulmonary safety of AZ-004 in
these two populations. The studies employed double-blind,
parallel-group designs. In each study, subjects were given two
doses of Staccato placebo or two doses of 10 mg
AZ-004, ten hours apart. Spirometry testing and other safety
assessments were performed at several time points up to
24 hours after the second dose. The primary safety measure
was forced expiratory volume in one second, or
FEV1,
a standard test of lung function. Decreases in
FEV1
versus baseline, respiratory symptoms, and use of a quick-relief
bronchodilator occurred in both treatment groups, but were more
frequent in each study after treatment with AZ-004. There were
no serious or severe respiratory adverse events. All respiratory
symptoms developing after treatment were either self-limiting or
readily managed with the inhaled bronchodilator.
In December 2009, we submitted our NDA to the FDA. In
February 2010, the FDA accepted our filing and has established
our PDUFA goal date for the AZ-004 NDA as October 11, 2010.
In February 2010, we licensed the U.S. and Canadian
commercialization rights to AZ-004 to Biovail Laboratories
International SRL.
10
INSOMNIA
PROGRAM: AZ-007 (Staccato zaleplon)
We are developing AZ-007 for the treatment of insomnia in
patients who have difficulty falling asleep, including those
patients with middle of the night awakening who have difficulty
falling back asleep. Insomnia is the most prevalent sleep
disorder, and we believe that it affects at least 15% to 20% of
the United States population, with some estimates of up to 50%
of Americans reporting difficulty getting a good night’s
sleep at least a few nights a week. Insomnia can be due to any
variety of causes, including depression, grief or stress,
menopause, age, shift work, or environmental disruption.
Whatever the cause of insomnia, it can take its toll on both the
afflicted and the non-afflicted. Sleep disturbances have a major
negative impact on public health and economic productivity.
Costs for direct healthcare associated with insomnia are
estimated to be approximately $14 billion to
$15 billion each year.
Market
Opportunity
Insomnia is a prevalent disorder that drives almost
$5 billion in worldwide sales of prescription medications
each year. In a large survey conducted by the National Sleep
Foundation in 2009, results showed that 64% of the respondents
experienced a minimum of one symptom of insomnia at least a few
nights a week , with 41% reporting this occurring every night or
almost every night and 31% using some sort of sleep aid at least
a few nights per week, 18% of whom use a medication sleep aid.
Of those, respondents complained primarily of waking up feeling
unrefreshed (45%), being wake a lot during the night (46%),
having difficulty falling asleep (29%), and waking up too early
and not being able to get back to sleep (30%). Also, sleepy
Americans are creating a major public safety problem —
drowsy driving More than one-half of adults (54%) reported that
they have driven at least once while drowsy in the past year,
with almost a third (28%) reporting that they do so at least
once per month, and 28% have nodded off or fallen asleep while
driving. Of those who have driven drowsy, 38% use a sleep aid at
least a few night per week.
Although benzodiazepines have been the gold standard in
treatment for sleep disorders for decades, issues with drug
misuse and dependency are common and concerning. Other current
treatments for insomnia include non-benzodiazepine GABA-A
receptor agonists, which include Ambien, both immediate release
and controlled-release tablets, Sonata, and Lunesta, which have
less abuse potential and side effects than classical
benzodiazepines and can be used for longer term treatment.
Patients and physicians surveyed suggest that current oral forms
of these leading insomnia medications can take from
30-60
minutes to work, while promotions for insomnia medications cite
20-30
minutes. Compounds with a longer half-life that keep patients
asleep longer, or those that are dosed in the middle of the
night are also those that have residual side effects that can
cause a “hangover” feeling the next day.
We believe the opportunity in insomnia is achieving a balance in
treating patients so they can fall asleep quickly, whether at
bedtime or in the middle of the night, while enabling them to
function well the next day without a groggy feeling that can
impact driving, employment and leisure activities. We believe
there is a potentially significant clinical need for rapid and
predictable onset of sleep in patients with insomnia, coupled
with a predictable duration of sleep and rapid, clear awakening
that can be satisfied with AZ-007.
Development
Status
Clinical
Studies
In April 2008, we announced positive results from a Phase 1
clinical trial of AZ-007. The AZ-007 Phase 1 clinical trial
enrolled 40 healthy volunteers at a single U.S. clinical
center. The purpose of this trial was to assess the safety,
tolerability and pharmacokinetic parameters of a single dose of
AZ-007. Using a double blind, randomized, dose-escalation trial
design, 4 doses of AZ-007 (ranging from 0.5 to 4.0 mg) were
compared to placebo.
AZ-007 delivered an IV-like pharmacokinetic profile with a
median time to peak venous concentration, or Tmax, of 1.6
minutes. Zaleplon exposure was dose proportional across the 4
doses studied, as calculated by power analysis.
Pharmacodynamics, measured as sedation assessed on a 100 mm
visual-analog scale, showed onset of effect as early as 2
minutes after dosing with AZ-007.
The most common side effects, reported by at least 10% of the
patients in any treatment group, were dizziness and somnolence.
These side effects were generally mild to moderate in severity.
These data indicated a rapid onset
11
of effect, apparently directly related to the IV-like
pharmacokinetics, and showed that AZ-007 was generally safe and
well tolerated in this population of healthy volunteers
Preclinical
Studies
Zaleplon, the active pharmaceutical ingredient in AZ-007, has
been approved for marketing in oral form. There are publicly
available safety pharmacology, systemic toxicology,
carcinogenicity and reproductive toxicology data we will be able
to use for our regulatory filings. Therefore, our preclinical
development testing is primarily focused on assessing the local
tolerability of inhaled zaleplon. Our two preclinical inhalation
toxicology studies with zaleplon have indicated that it was
generally well tolerated.
MIGRAINE
HEADACHE PROGRAM: AZ-001 (Staccato prochlorperazine) and AZ-104
(Staccato loxapine, low-dose)
We are developing AZ-001 (Staccato prochlorperazine) and
AZ-104 (Staccato loxapine, low-dose) for the treatment of
acute migraine headaches. Although there are numerous products
available for the treatment of migraines, including simple
analgesics such as aspirin and acetaminophen, and nonsteroidal
anti-inflammatory drugs such as ibuprofen and naproxen, the
prescription market is dominated by a class of orally
administered medications commonly known as triptans.
Market
Opportunity
According to the National Headache Foundation, approximately
13 million people in the United States have been diagnosed
with migraine headaches that occur often, usually one to four
times per month and are treated with prescription medications
some of which can be addicting. According to a survey conducted
by the National Headache Foundation in 2007, 82% of migraine
respondents have taken more than one prescription medication for
their migraines, and the average number of medications a patient
has taken for migraines is four, which we believe speaks to the
need for more medication options that work for patients.
Of the estimated 29.5 million migraine sufferers, including
diagnosed and undiagnosed sufferers, there are at least two
groups of potential patients for whom we believe AZ-001 or
AZ-104 could be effective and safe in comparison to triptans.
Many migraine sufferers who do take triptans have an
insufficient therapeutic response to these medications. In
addition, according to the warning labels on triptans, patients
with hypertension or high cholesterol, or who smoke cigarettes,
are contraindicated for and should not take these medications
due to potential cardiovascular and cerebrovascular health risks.
AZ-001
(Staccato prochlorperazine)
The API of AZ-001 is prochlorperazine, a generic drug belonging
to the class of drugs known as phenothiazines. Prochlorperazine
is currently approved in oral, injectable and suppository
formulations in the United States for the treatment of several
indications, including nausea and vomiting. In several published
clinical studies, 10 mg of prochlorperazine administered
intravenously demonstrated effective relief of migraine pain.
Prochlorperazine is often administered intravenously to patients
with severe migraine headaches who come to emergency departments
or migraine treatment clinics. We believe the combination of
prochlorperazine with our Staccato system could
potentially result in a speed of therapeutic onset advantage
over oral tablets and a convenience and comfort advantage over
injections. In addition, AZ-001 may be appropriate for patients
who do not achieve effective relief with triptans or cannot take
triptans due to the cardiovascular risk sometimes associated
with the administration of triptans. For patients who do not
obtain adequate relief from current migraine therapies, AZ-001
may offer a new anti-migraine mechanism of action.
Development
Status
Regulatory
Status
During the third quarter of 2008, we conducted an
end-of-Phase
2 meeting with the FDA. We believe we have a clear understanding
of the development requirements for filing an NDA for this
product candidate.
12
Clinical
Studies
In December 2007, we completed enrollment of a thorough QT
clinical trial, in which two doses of AZ-001 (5 and 10 mg)
were compared to active control and to placebo. The purpose of a
thorough QT study is to determine a drug’s effect on
cardiac rhythms. With approximately 40 subjects per study arm,
we found that the active control, moxifloxacin, produced a
positive QT/QTc signal that verified the sensitivity of the
clinical study. Neither of the doses of AZ-001 produced a QT/QTc
prolongation that would suggest an increased risk of cardiac
arrhythmia.
We reported initial results of a Phase 2b clinical trial in
March 2007. The AZ-001 Phase 2b clinical trial was an
outpatient, multi-center, randomized, double blind,
placebo-controlled study. The study was designed to evaluate the
treatment of a single migraine attack in each of approximately
400 migraine patients, with and without aura. In the trial,
three doses of AZ-001 (5 mg, 7.5 mg and 10 mg
doses) and placebo were tested, with 100 patients assigned
to each treatment group. The primary efficacy endpoint for the
trial was headache pain relief at
2-hours
post-dose, as defined by the International Headache Society, or
IHS, 4-point headache pain rating scale. Secondary efficacy
endpoints for the trial included various additional measurements
of pain relief, as well as effects on nausea, vomiting,
phonophobia and photophobia. The clinical trial study period was
24 hours post dosing for each patient. All results were
considered statistically significant at the p < 0.05 level,
as compared to placebo, and all statistical analyses were made
on an
intent-to-treat
basis. Side effects were recorded throughout the clinical trial
study period, and a safety evaluation was made at each
patient’s closeout visit.
Primary Efficacy Endpoint. AZ-001 met the
primary efficacy endpoint of the clinical trial, which was pain
relief at
2-hours
post-dose using the IHS 4-point headache pain rating scale, for
all three doses of the drug compared to placebo. Statistically
significant improvements in pain response were observed in 66.0%
of patients at the 10 mg dose (p=0.0013), 63.7% of patients
at the 7.5 mg dose (p=0.0046) and 60.2% of patients at the
5 mg dose (p=0.0076), compared to 40.8% of patients
receiving placebo.
Additional Efficacy Endpoints. Another measure
of efficacy was the achievement of a pain-free response at
2 hours, where a patient has a pain score of 0, or
“no”, headache pain at the
2-hours
post-dose time point. In the trial, AZ-001 showed statistically
significant differences from placebo in this measure with 35.0%
of patients who received the 10 mg dose achieving pain-free
status (p=0.0019) and 29.7% of patients who received the
7.5 mg dose achieving pain-free status (p=0.0226). Patients
receiving the 5 mg dose (21.4%) did not achieve a
statistically significant pain-free response, compared to
placebo. The rate of pain-free response at 2 hours in
patients receiving placebo was 15.3%.
We believe duration of efficacy is an important consideration in
developing migraine therapeutics. A commonly used measure of
duration of efficacy is the sustained pain-free response,
whereby a patient reports a pain-free score at the
2-hour
post-dose time point and remains pain-free for the remainder of
the 24 hour study period. The 10 mg and 7.5 mg
doses of AZ-001 showed statistically-significant differences in
sustained pain-free response, compared to placebo. Sustained
pain-free outcomes through 24 hours were observed in 30.1%
and 23.1% of patients in the 10 mg and 7.5 mg dose
groups, respectively. The placebo group exhibited a sustained
pain-free response in 10.2% of patients.
AZ-001 exhibited rapid onset of pain relief. The 7.5 mg
dose showed statistically significant pain response, compared to
placebo, at 15 minutes (p=0.016). At 30 minutes, all three doses
of AZ-001 showed statistically significant pain response,
compared to placebo; 10 mg (p=0.0056), 7.5 mg
(p=0.0003) and 5 mg (p=0.0056).
Symptom management is an important consideration in the overall
efficacy of a migraine therapy. Important symptoms to be managed
in migraine patients are nausea, vomiting, photophobia
(sensitivity to light) and phonophobia (sensitivity to sound).
Survival analyses for nausea, photophobia and phonophobia over
the 2 hour time period post-dose showed a statistically
significant difference, compared to placebo. The total number of
patients with vomiting were too few to make conclusions about
drug effect.
Safety Evaluations. Side effects were recorded
throughout the clinical trial study period, and a safety
evaluation was made at each patient’s closeout visit. There
were no serious adverse events reported during the trial. The
most common side effects reported by at least 10% of the
patients in any treatment group were taste, throat irritation,
cough, and somnolence. These side effects appeared to be dose
related, with a lower incidence and
13
severity of the side effects generally seen at the lower doses
of AZ-001. These side effects were generally mild to moderate in
severity.
Preclinical
Studies
We have completed several preclinical studies of AZ-001
including inhalation toxicology studies in two animal species,
cardiovascular and respiratory safety studies in one species,
and in vitro and in vivo studies to assess
potential gene mutations. In animal toxicology studies of
prochlorperazine aerosols involving prolonged daily dosing, we
detected changes to, and increases in the number of, the cells
in the upper airway of the test animals. The terms for these
changes and increases are “squamous metaplasia” and
“hyperplasia,” respectively. We also observed lung
inflammation in some animals. Squamous metaplasia and
hyperplasia occurred at doses that were substantially greater
than those administered in our human clinical trials. In
subsequent toxicology studies of AZ-001 involving intermittent
dosing, we detected lower incidence and severity of squamous
metaplasia and hyperplasia in the upper airway of the test
animals compared to the daily dosing results. No lung
inflammation was observed with intermittent dosing. During the
second quarter of 2008, we completed a
28-day
repeat dose inhalation study in dogs. Consistent with previous
findings in shorter-term and higher dose studies, we observed
dose-related minimal to slight squamous metaplasia in the upper
respiratory tract, primarily in the lining of the nasal
passages, in all treated groups. These changes were partially
reversible by the end of a
28-day
post-treatment period. No lower respiratory tract or lung
findings were reported. We do not expect to observe these events
when AZ-001 is delivered intermittently and at proportionately
lower doses in future toxicology studies.
AZ-104
(Staccato loxapine, low-dose)
The API of AZ-104 is loxapine, a generic drug belonging to the
class of drugs known as antipsychotics. Loxapine is currently
approved in oral and injectable (intramuscular only)
formulations in the United States for the management of the
manifestations of schizophrenia.
Development
Status
Clinical
Trials
We reported initial results of the AZ-104 Phase 2b trial in
September 2009. This was an outpatient, multi-center,
randomized, double-blind, single administration,
placebo-controlled study. The study was designed to evaluate the
treatment of a single migraine attack of moderate to severe
intensity in each of approximately 360 migraine patients, with
or without aura. Two doses of AZ-104 (1.25 mg and
2.5 mg) and placebo were evaluated in the clinical trial.
The study enrolled a total of 366 patients:
125 patients in the placebo dose group, 121 patients
in the 1.25 mg dose group, and 120 patients in the
2.5 mg dose group. Both AZ-104 dose groups trended towards
statistical significance, but the study did not meet its primary
endpoint, which was defined as pain-relief at the
2-hour time
point, compared to placebo. There were no serious adverse events
in the clinical trial, and AZ-104 was generally safe and well
tolerated in this patient population.
Patients rated their headache pain using the IHS 4-point rating
scale. The primary efficacy endpoint was headache pain relief,
which is headache pain rated as mild or none, at 2 hours
post-dose. Secondary efficacy endpoints for the clinical trial
included various additional measurements of pain relief, as well
as effects on nausea, vomiting, phonophobia and photophobia. All
results were considered statistically significant at the p
< 0.05 level, as compared to placebo, and all analyses were
made on an
intent-to-treat
basis. Safety evaluations were also made throughout the clinical
trial period.
AZ-104 was numerically superior to placebo in pain-relief at
2-hours
post-dose, but these differences were not statistically
significant. Pain relief was observed in 56% of patients
receiving the 2.5 mg dose (p=0.11) and 54% of patients
receiving the 1.25 mg dose (p=0.12), as compared to 45% of
patients receiving placebo.
Another commonly used measure of efficacy in migraine studies is
the percentage of patients who are pain-free at 2 hours
post-dose. Again, AZ-104 was numerically superior to placebo in
this measure, but the differences were not statistically
significant. Pain-free responders were 31% of the patients
receiving the 2.5 mg dose and 27% of the patients receiving
the 1.25 mg dose, as compared to 23% of the patients
receiving placebo.
14
Preclinical
Studies
Loxapine has been approved for marketing in oral and injectable
forms. There are publicly available safety pharmacology,
systemic toxicology, carcinogenicity and reproductive toxicology
data we will be able to use for our regulatory filings.
Therefore, our preclinical development testing is primarily
focused on assessing the local tolerability of inhaled loxapine.
Our two preclinical inhalation toxicology studies with loxapine
have indicated that it was generally well tolerated.
ACUTE
PAIN PROGRAM: AZ-003 (Staccato fentanyl)
We are developing our product candidate AZ-003 (Staccato
fentanyl) for the treatment of patients with acute pain,
including patients with breakthrough cancer pain and
postoperative patients with acute pain episodes. Based on our
analysis of industry data and clinical literature, we believe
over 25 million postoperative patients experience
inadequate pain relief, despite receiving some form of pain
management and, according to a three month study on cancer pain
by Portenoy and Hagen (1990) and a cross-sectional study on
cancer pain by Caraceni (2004), approximately 65% of patients
diagnosed with cancer pain experience breakthrough cancer pain.
A patient controlled analgesia, or PCA, IV pump is often used
directly after surgery so the patient can achieve quick pain
relief as needed. The PCA pump approach generally works well,
but typically requires patients to remain in the hospital with
an IV line in place. Physicians generally treat cancer pain
using a combination of a chronic, long-acting drug and an acute
or rapid acting drug for breakthrough pain. Treating a
breakthrough pain episode with an oral medication is difficult
due to the slow onset of therapeutic effect. However, patients
usually also find more invasive, injectable treatments
undesirable. Based on preclinical testing and the results of our
Phase 1 clinical trial, we believe the PK of fentanyl delivered
using a Staccato system will be similar to the PK
of IV fentanyl administration. We believe many patients
would benefit from a noninvasive but fast acting therapy that
allows them to titrate the amount of pain medication to the
amount of pain relief required.
The API of AZ-003 is fentanyl, a generic drug belonging to the
class of drugs known as opioid analgesics. Fentanyl is currently
approved in three different formulations in the United States
for the management of various types of pain: injectable,
transmucosal, which deliver drugs through the mucous membranes
of the mouth or nose, and transdermal, which deliver drug
through the skin. Since the Staccato system can
incorporate lockout and multiple dose features, we believe that
AZ-003 will facilitate patient titration to the minimum
effective drug dose in a safe, convenient, easy to use and
simple delivery system. In addition, we believe the
incorporation of patient lockout features may be a significant
safety advantage and has the potential to prevent diversion, or
use by individuals who have not been prescribed the drug.
Development
Status
Clinical
Studies
We completed the initial analysis of the top-line results of our
Phase 1 clinical trial with AZ-003 in December 2006. The primary
aims of the Phase 1 clinical trial were to evaluate the arterial
PK and absolute bioavailability for AZ-003 by comparing the
AZ-003 profile to that of IV fentanyl, and to examine the
pharmacodynamics, tolerability and safety of AZ-003 in
opioid-naive healthy subjects. The trial enrolled 50 subjects
and was conducted at a single clinical center in two stages.
Stage 1 of the protocol was an open-label, crossover comparison
of a 25 g dose of AZ-003 by a single inhalation and the same
dose of fentanyl administered intravenously over five seconds.
Stage 2 of the protocol was a randomized double-blind,
placebo-controlled, dose escalation of AZ-003 evaluating
cumulative doses of 50 g, 100 g, 150 g and 300 g of fentanyl. A
25 g individual dose of fentanyl was inhaled once in Stage 1, or
2, 4 or 6 times at 4 minute intervals for the first four
different cohorts in Stage 2. A fifth cohort in Stage 2 received
a 150 g dosing sequence starting at time zero and then a second
150 g dosing sequence starting at 60 minutes after the first
dose, for a cumulative dose of 300 g. In addition to
comprehensive PK sample collection, pharmacodynamic data were
generated using pupillometry, a surrogate measure used to assess
the functional activity of opioids.
The AZ-003 PK was substantially equivalent to the IV
fentanyl PK, with similar peak plasma concentration, or Cmax,
time to maximum plasma concentration, or Tmax, and area under
the curve concentration, or AUC. These data suggest very high
absolute bioavailability of the inhaled dose. Mean peak arterial
plasma concentrations were observed within 30 seconds for both
administration routes. In Stage 2 of the clinical trial,
ascending doses of AZ-
15
003 controlled by the Staccato device, exhibited
dose-proportionality of fentanyl throughout the dosing range
from 50 mcg to 300 mcg, following an AUC analysis. There were no
serious adverse events attributable to AZ-003, and the results
from the clinical study showed that AZ-003 was generally safe
and well tolerated at all doses.
In October 2007, clinical data from the AZ-003 Phase 1 clinical
trial were presented in four different presentations at the
American Society of Anesthesiologists 2007 Annual Meeting, in
San Francisco, California. The four presentations were
entitled, “Pharmacokinetic Profiles of Fentanyl Delivered
by Intravenous and Inhaled Thermal Aerosol Routes”,
“Pharmacokinetic Profile of Multiple Doses of Fentanyl
Delivered by Inhaled Thermal Aerosol Route”,
“Pharmacodynamic Response to Fentanyl Delivered by
Intravenous and Inhaled Thermal Aerosol Routes” and
“Pharmacodynamic Response to Multiple Doses of Fentanyl
Delivered by Inhaled Thermal Aerosol Route”. This clinical
trial demonstrated that the pharmacokinetic profile of AZ-003 in
a single breath offers a speed of onset and consistency
equivalent to fentanyl administered intravenously over 5
seconds. This clinical trial also demonstrated that the
pharmacodynamic profile of AZ-003 in a single breath was
comparable to that of fentanyl administered by intravenous
administration.
Preclinical
Studies
Fentanyl is approved for marketing in injectable, transdermal
and transmucosal forms. We are able to use publicly available
safety pharmacology, systemic toxicology and reproductive
toxicology data for our regulatory filings. Therefore, our
preclinical development testing was primarily focused on
assessing the local tolerability of inhaled fentanyl. Our two
preclinical toxicology tests in two animal species with fentanyl
have indicated that it was generally well tolerated.
AZ-002
(Staccato alprazolam)
We were developing AZ-002 (Staccato alprazolam) for the
acute treatment of panic attacks associated with panic disorder,
a condition characterized by the frequent, unpredictable
occurrence of panic attacks. The API of AZ-002 is alprazolam, a
generic drug belonging to the class of drugs known as
benzodiazepines. Alprazolam is currently approved in oral
formulations in the United States for use in the management of
anxiety disorder, for the short term relief of symptoms of
anxiety, for anxiety associated with depression, and for the
treatment of panic disorder with or without agoraphobia, or
abnormal fear of being in public places. We will continue to
explore additional CNS indications for AZ-002 given its safety
profile, the successful and reproducible delivery of alprazolam,
and the IV-like pharmacological effect demonstrated,
Development
Status
Clinical
Trials
In June 2008, we released the preliminary results from our Phase
2a
proof-of-concept
clinical trial with
AZ-002 in
patients with panic disorder. The study did not meet its two
primary endpoints, which were the effect of
AZ-002 on
the incidence of a doxapram-induced panic attack and the effect
of AZ-002 on
the duration of a doxapram-induced panic attack, both as
compared with placebo. There were no serious adverse events in
the clinical trial, and
AZ-002 was
safe and well tolerated in the study patient population.
The AZ-002 Phase 2a clinical trial was an in-clinic, randomized,
double-blind, placebo-controlled
proof-of-concept
evaluation of patients with panic disorder. After an open-label
pilot phase, 40 patients were enrolled at 3
U.S. clinical centers, with 20 patients receiving
1 mg AZ-002 and 20 patients receiving Staccato
placebo. The primary aim of the clinical trial was to assess
the safety and efficacy of a single dose of AZ-002 in treating a
pharmacologically-induced panic attack. Two primary endpoints
were prospectively defined for the study, one to assess the
effect of treatment on the occurrence of a doxapram-induced
panic attack of sufficient intensity and a second to assess the
effect of treatment on the duration of the doxapram-induced
panic attack. Data for these two endpoints were based on the
Acute Panic Inventory, a commonly used 22-item self-report
questionnaire designed to measure panic-like response to
biological challenges or other stressful situations. After
receiving training and baseline assessments, all patients in the
double-blind phase of the study received a Staccato
device, randomized to either 1 mg AZ-002 or placebo,
and an intravenous administration of doxapram, a respiratory
stimulant used to induce a simulated panic attack.
16
Preclinical
Studies
Alprazolam has been approved for marketing in oral tablet form.
There are publicly available safety pharmacology, systemic
toxicology, carcinogenicity and reproductive toxicology data
that we will be able to use for our regulatory filings.
Therefore, our preclinical development plan is primarily focused
on assessing the local tolerability of inhaled alprazolam. To
date, our two preclinical inhalation toxicology studies with
inhaled alprazolam have indicated that it is generally well
tolerated.
Product
Candidate Selection
Since 2004, we have identified five drug compounds and have
successfully filed six INDs and one NDA for product candidates
using our Staccato system. At the end of 2009, in the
aggregate, we have dosed more than 2,400 patients and
subjects in 22 different clinical trials. In 2009, our primary
emphasis was on later stage clinical development of, and seeking
regulatory approval for, AZ-004, and not on new product
candidate identification. We believe our Staccato system
is broadly applicable to a large number of medically important
small molecule compounds that could be useful in the treatment
of acute and intermittent conditions.
Once we have established initial vaporization feasibility, we
conduct experiments and activities designed to identify viable
product candidates. These experiments and activities include
calculation of emitted doses, analysis of whether or not the
emitted dose would be therapeutic, particle size analyses, early
product stability studies and comprehensive medical and market
needs assessments. After completion of these experiments and
activities, a formal Product Selection Advisory Board, or PSAB,
composed of employees and outside experts, is convened to
evaluate these data.
After a positive PSAB decision, we initiate preclinical
pharmacology and toxicology studies, with the intent of filing
an IND upon successful completion of our preclinical studies.
During this preclinical period, we also manufacture toxicology
study supplies and initiate the manufacturing
scale-up to
move the product candidate through manufacturing design
verification testing and the production of clinical trial
materials. We believe that, with the current development status
of our single dose device, we can move a compound from initial
screening through filing of an IND in 12 to 18 months.
In January 2009, we consolidated our operations, with a primary
focus on the continued rapid development of AZ-004. As our
efforts will focus on the regulatory approval and commercial
launch of AZ-004, we do not anticipate moving any new product
candidates into the clinic in 2010.
Our
Strategy
We intend to develop an extensive portfolio of products. Key
elements of our strategy include:
|
|
|
| |
•
|
Focus on Acute and Intermittent Conditions. We
focus our development and commercialization efforts on product
candidates based on our Staccato system that are intended
to address important unmet medical and patient needs in the
treatment of acute and intermittent conditions in which rapid
onset, ease of use, noninvasive administration and, in some
cases, patient titration of dosage are required.
|
| |
| |
•
|
Establish Strategic Partnerships. We intend to
strategically partner with pharmaceutical and other companies to
provide development funding or to address markets that may
require a larger sales force, greater marketing resources or
specific expertise to maximize the value of some product
candidates. We also intend to seek international distribution
partners for our product candidates. We may also enter into
strategic partnerships with other pharmaceutical companies to
combine our Staccato system with their proprietary
compounds.
|
| |
| |
•
|
Retain and Control Product Manufacturing. We
own all manufacturing rights to our product candidates. We
intend to internally complete the final manufacture and assembly
of our product candidates and any future products, potentially
enabling greater intellectual property protection and economic
return from our future products. We also believe controlling the
final manufacture and assembly reduces the risk of supply
interruptions and allows more cost effective manufacturing.
|
17
Licensing
Collaboration
In February 2010, we entered into a collaboration and license
agreement, or license agreement, and a manufacture and supply
agreement, collectively, the collaboration, with Biovail
Laboratories International SRL, or Biovail, for AZ-004
(Staccato®
loxapine) for the treatment of psychiatric
and/or
neurological indications and the symptoms associated with these
indications, including the initial indication of treating
agitation in schizophrenia and bipolar disorder patients. The
collaboraton contemplates that we will be the exclusive supplier
of drug product for clinical and commercial uses and have
responsibility for the NDA for AZ-004 for the initial indication
of rapid treatment of agitation in patients with schizophrenia
or bipolar disorder, as well as responsibility for any
additional development and regulatory activities required for
use in these two patient populations in the outpatient setting.
Biovail will be responsible for commercialization for the
initial indication and, if it elects, development and
commercialization of additional indications for AZ-004 in the
U.S. and Canada.
Under the terms of the license agreement, Biovail paid us an
upfront fee of $40 million, and we may be eligible to
receive up to an additional $90 million in milestone
payments upon achievement of predetermined regulatory, clinical
and commercial manufacturing milestones. We may be subject to
certain payment obligation to Biovail, up to $5 million, if
we do not meet certain other milestones prior to a termination
of the license agreement. We are also eligible to receive tiered
royalty payments of 10% to 25% on any net sales of AZ-004. We
are responsible for conducting and funding all development and
regulatory activities associated with AZ-004’s initial
indication for the rapid treatment of agitation in patients with
schizophrenia or bipolar disorder as well as for its possible
use in the outpatient setting in these two patient populations.
Our obligation to fund the outpatient development efforts is
limited to a specified amount, none of which is expected to be
incurred in 2010. Biovail is responsible for certain Phase 4
development commitments and related costs and expenses. For
additional indications, we have an obligation regarding certain
efforts and related costs and expenses, up to a specified
amount, and, if it elects, Biovail is responsible for all other
development commitments and related costs and expenses.
Under the terms of the manufacture and supply agreement, we are
the exclusive supplier of AZ-004 and have responsibility for the
manufacture, packaging, labeling and supply for clinical and
commercial uses. Biovail will purchase AZ-004 from us at
predetermined transfer prices. The transfer prices depend on the
volume of AZ-004 purchases, subject to certain adjustments.
Either party may terminate the collaboration for the other
party’s uncured material breach or bankruptcy. In addition,
Biovail has the right to terminate the collaboration
(a) upon 90 days written notice for convenience;
(b) upon 90 days written notice if FDA does not
approve the AZ-004 NDA for the initial indication for the rapid
treatment of agitation in patients with schizophrenia or bipolar
disorder; (c) immediately upon written notice for safety
reasons or withdrawal of marketing approval; (d) upon
90 days written notice upon certain recalls of the product;
or (e) immediately upon written notice within 60 days
of termination of the supply agreement under certain
circumstances. The supply agreement automatically terminates
upon the termination of the license agreement.
Research
and Development
Research and development expenditures made to advance our
product candidates and other research efforts during the last
three years ended December 31, 2009, were as follows (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
Preclinical and Clinical Development:
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
AZ-004/104
|
|
$
|
30,084
|
|
|
$
|
26,789
|
|
|
$
|
15,524
|
|
|
AZ-003
|
|
|
1,631
|
|
|
|
17,070
|
|
|
|
1,474
|
|
|
AZ-001
|
|
|
—
|
|
|
|
1,151
|
|
|
|
8,163
|
|
|
AZ-002
|
|
|
181
|
|
|
|
1,898
|
|
|
|
3,795
|
|
|
AZ-007
|
|
|
—
|
|
|
|
1,773
|
|
|
|
8,214
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total preclinical and clinical development
|
|
|
31,896
|
|
|
|
48,681
|
|
|
|
37,170
|
|
|
Research
|
|
|
7,882
|
|
|
|
12,884
|
|
|
|
8,475
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total research and development
|
|
$
|
39,778
|
|
|
$
|
61,565
|
|
|
$
|
45,645
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18
Manufacturing
We manufacture our product candidates with components supplied
by vendors. We believe that manufacturing our product candidates
will potentially enable greater intellectual property protection
and economies of scale and decrease the risk of supply
interruptions.
After inspection and qualification, we assemble the components
of our product candidates and coat the exterior of the metal
substrate with a thin film of API. We then place the plastic
airway around the assembly and package a completed device in a
pharmaceutical-grade foil pouch. The controller for our multiple
dose delivery design includes the battery power source for
heating the individual metal substrates, a microprocessor that
directs the electric current to the appropriate metal substrate
at the appropriate time, and an icon-based liquid crystal
display that shows pertinent information to the user, for
example, the number of doses remaining in the dose cartridge and
the controller status. We may need to develop modified versions
of our devices for future product candidates.
We believe we have developed quality assurance and quality
control systems applicable to the design, manufacture,
packaging, labeling and storage of our product candidates in
compliance with applicable regulations. These systems include
extensive requirements with respect to quality management and
organization, product design, manufacturing facilities,
equipment, purchase and handling of components, production and
process controls, packaging and labeling controls, device
evaluation, distribution and record keeping.
We outsource the production of the components of our product
candidates, including the printed circuit boards, the molded
plastic airways and the heat packages used in the single dose
version of our Staccato system device. We currently use
single source suppliers for these components, as well as for the
API used in each of our product candidates. We do not carry a
significant inventory of these components, and establishing
additional or replacement suppliers for any of these components
may not be accomplished quickly, or at all, and could cause
significant additional expense. Any supply interruption from our
vendors would limit our ability to manufacture our product
candidates and could delay clinical trials for, and regulatory
approval of, our product candidates.
In 2007, we completed a current good manufacturing practices, or
cGMP, compliant pilot manufacturing facility located in Mountain
View, California. In November 2007, we received a pharmaceutical
manufacturing license from the California State Food and Drug
Branch for this facility. We believe this pilot manufacturing
facility will have sufficient capacity to manufacture materials
for toxicology studies and clinical trial materials for future
clinical trials. We also believe that this facility will be
sufficient to manufacture early commercial-scale batches of our
products. In January 2009, we renewed our pharmaceutical
manufacturing license from the California State Food and Drug
Branch for our Mountain View facility. This new license is valid
until January 31, 2011.
Under the terms of the manufacture and supply agreement, we are
the exclusive supplier of AZ-004 and have responsibility for the
manufacture, packaging, labeling and supply for clinical and
commercial uses. Biovail will purchase AZ-004 from us at
predetermined transfer prices. The transfer prices depend on the
volume of AZ-004 purchases, subject to certain adjustments.
Either party may terminate the collaboration for the other
party’s uncured material breach or bankruptcy. In addition,
Biovail has the right to terminate the collaboration
(a) upon 90 days written notice for convenience;
(b) upon 90 days written notice if FDA does not
approve the AZ-004 NDA for the initial indication for the rapid
treatment of agitation in patients with schizophrenia or bipolar
disorder; (c) immediately upon written notice for safety
reasons or withdrawal of marketing approval; (d) upon
90 days written notice upon certain recalls of the product;
or (e) immediately upon written notice within 60 days
of termination of the supply agreement under certain
circumstances. The supply agreement automatically terminates
upon the termination of the license agreement.
Autoliv
ASP, Inc.
On November 2, 2007, we entered into a manufacturing and
supply agreement, or the supply agreement, with Autoliv relating
to the commercial supply of chemical heat packages that can be
incorporated into our single dose Staccato device.
Autoliv had developed these chemical heat packages for us
pursuant to a development agreement executed in October 2005.
Under the terms of the supply agreement, Autoliv will develop a
manufacturing line capable of producing 10 million chemical
heat packages a year. We have an obligation to pay Autoliv
$12 million upon the earlier of December 31, 2011 or
60 days after the approval by the FDA of an NDA filed by
us. If either
19
party terminates the supply agreement, we will be required to
reimburse Autoliv for certain expenses related to the equipment
and tooling used in the production and testing of the chemical
heat packages up to $12 million. Upon either payment
Autoliv will be required to transfer possession and ownership of
such equipment and tooling to us.
Autoliv has agreed to manufacture, assemble and test the
chemical heat packages solely for us in conformance with our
specifications. We will pay Autoliv a specified purchase price,
which varies based on annual quantities ordered by us, per
chemical heat package delivered. The initial term of the supply
agreement expires on December 31, 2012 and may be extended
by written mutual consent. The supply agreement provides that
during the term of the supply agreement, Autoliv will be our
exclusive supplier of chemical heat packages. In addition, the
supply agreement grants Autoliv the right to negotiate for the
right to supply commercially any second generation chemical heat
package, or a second generation product, and provides that we
will pay Autoliv certain royalty payments if we manufacture
second generation products ourselves or if we obtain second
generation products from a third party manufacturer. Upon the
expiration or termination of the supply agreement we will also
be required, on an ongoing basis, to pay Autoliv certain royalty
payments related to the manufacture of the chemical heat
packages by us or third party manufacturers.
The supply agreement also contains certain provisions regarding
the rights and responsibilities of the parties with respect to
manufacturing specifications, forecasting and ordering, delivery
arrangements, payment terms, packaging requirements, change
orders, intellectual property rights confidentiality and
indemnification, as well as certain other customary matters.
Product
Commercialization
We have licensed all U.S. and Canadian commercialization
rights to Biovail for AZ-004, excluding the treatment of
migraine. Biovail paid us an upfront fee and will pay potential
additional milestone payments upon achievement of predetermined
regulatory and clinical milestones and will pay us royalties on
net sales of AZ-004. We have responsibility for the manufacture,
packaging, labeling and supply of AZ-004 to Biovail, and Biovail
will purchase AZ-004 from us at predetermined transfer prices.
Eventually, we may build a United States specialty sales force
to commercialize any of our other product candidates that are
approved and are intended for the specialty pharmaceutical type
markets. We plan to enter into strategic partnerships with other
companies to commercialize products that are intended for other
markets in the United States and for all of our product
candidates in geographic territories outside the United States.
Government
Regulation
The testing, manufacturing, labeling, advertising, promotion,
distribution, export and marketing of our product candidates are
subject to extensive regulation by governmental authorities in
the United States and other countries. Our product candidates
include drug compounds incorporated into our delivery device and
are considered “combination products” in the United
States. We have agreed with the FDA that our product candidates
will be reviewed by the FDA’s Center for Drug Evaluation
and Research. The FDA, under the Federal Food, Drug and Cosmetic
Act, or FDCA, regulates pharmaceutical products in the United
States. The steps required before a drug may be approved for
marketing in the United States generally include:
|
|
|
| |
•
|
preclinical laboratory studies and animal tests;
|
| |
| |
•
|
the submission to the FDA of an IND for human clinical testing,
which must become effective before human clinical trials
commence;
|
| |
| |
•
|
adequate and well controlled human clinical trials to establish
the safety and efficacy of the product;
|
| |
| |
•
|
the submission to the FDA of an NDA;
|
| |
| |
•
|
satisfactory completion of an FDA inspection of the
manufacturing facilities at which the product is made to assess
compliance with cGMP. In addition, the FDA may audit clinical
trial sites that generated the data in support of the
NDA; and
|
| |
| |
•
|
FDA review and approval of the NDA.
|
20
The testing and approval process requires substantial time,
effort and financial resources, and the receipt and timing of
any approval is uncertain. Preclinical studies include
laboratory evaluations of the product candidate, as well as
animal studies to assess the potential safety and efficacy of
the product candidate. The results of the preclinical studies,
together with manufacturing information and analytical data, are
submitted to the FDA as part of the IND, which must become
effective before clinical trials may be commenced. The IND will
become effective automatically 30 days after receipt by the
FDA, unless the FDA raises concerns or questions about the
conduct of the trials as outlined in the IND prior to that time.
In this case, the IND sponsor and the FDA must resolve any
outstanding concerns before clinical trials can proceed.
Clinical trials involve the administration of the product
candidates to healthy volunteers or patients under the
supervision of a qualified principal investigator. Further, each
clinical trial must be reviewed and approved by an independent
institutional review board, or IRB, at or servicing each
institution at which the clinical trial will be conducted. The
IRB will consider, among other things, ethical factors, the
safety of human subjects and the possible liability of the
institution.
Clinical trials typically are conducted in three sequential
phases prior to approval, but the phases may overlap. A fourth,
or post-approval, phase may include additional clinical studies.
These phases generally include the following:
|
|
|
| |
•
|
Phase 1. Phase 1 clinical trials involve the
initial introduction of the drug into human subjects, frequently
healthy volunteers. These studies are designed to determine the
metabolism and pharmacologic actions of the drug in humans, the
adverse effects associated with increasing doses and, if
possible, to gain early evidence of effectiveness. In Phase 1
clinical trials, the drug is usually tested for safety,
including adverse effects, dosage tolerance, absorption,
distribution, metabolism, excretion and pharmacodynamics.
|
| |
| |
•
|
Phase 2. Phase 2 clinical trials usually
involve studies in a limited patient population to
(1) evaluate the efficacy of the drug for specific,
targeted indications; (2) determine dosage tolerance and
optimal dosage; and (3) identify possible adverse effects
and safety risks. Although there are no statutory or regulatory
definitions for Phase 2a and Phase 2b, Phase 2a is commonly used
to describe a Phase 2 clinical trial evaluating efficacy,
adverse effects and safety risks and Phase 2b is commonly used
to describe a subsequent Phase 2 clinical trial that also
evaluates dosage tolerance and optimal dosage.
|
| |
| |
•
|
Phase 3. If a compound is found to be
potentially effective and to have an acceptable safety profile
in Phase 2 clinical trials, the clinical trial program will be
expanded to further demonstrate clinical efficacy, optimal
dosage and safety within an expanded patient population at
geographically dispersed clinical trial sites. Phase 3 clinical
trials usually include several hundred to several thousand
patients.
|
| |
| |
•
|
Phase 4. Phase 4 clinical trials are studies
required of, or agreed to by, a sponsor that are conducted after
the FDA has approved a product for marketing. These studies are
used to gain additional experience from the treatment of
patients in the intended therapeutic indication and to document
a clinical benefit in the case of drugs approved under
accelerated approval regulations. If the FDA approves a product
while a company has ongoing clinical trials that were not
necessary for approval, a company may be able to use the data
from these clinical trials to meet all or part of any Phase 4
clinical trial requirement. These clinical trials are often
referred to as Phase 3/4 post-approval clinical trials. Failure
to promptly conduct Phase 4 clinical trials could result in
withdrawal of approval for products approved under accelerated
approval regulations.
|
In the case of products for the treatment of severe or life
threatening diseases, the initial clinical trials are sometimes
conducted in patients rather than in healthy volunteers. Since
these patients are already afflicted with the target disease, it
is possible that such clinical trials may provide evidence of
efficacy traditionally obtained in Phase 2 clinical trials.
These trials are referred to frequently as Phase
1/2
clinical trials. The FDA may suspend clinical trials at any time
on various grounds, including a finding that the subjects or
patients are being exposed to an unacceptable health risk.
The results of preclinical studies and clinical trials, together
with detailed information on the manufacture and composition of
the product, are submitted to the FDA in the form of an NDA
requesting approval to market the product. Generally, regulatory
approval of a new drug by the FDA may follow one of three
routes. The most traditional of these routes is the submission
of a full NDA under Section 505(b)(1) of the FDCA. A second
route,
21
which is possible where an applicant chooses to rely in part on
the FDA’s conclusion about the safety and effectiveness of
previously approved drugs is to submit a more limited NDA
described in Section 505(b)(2) of the FDCA. The final route
is the submission of an Abbreviated New Drug Application for
products that are shown to be therapeutically equivalent to
previously approved drug products as permitted under
Section 505(j) of the FDCA. We do not expect any of our
product candidates to be submitted under Section 505(j).
Both Section 505(b)(1) and Section 505(b)(2)
applications are required by the FDA to contain full reports of
investigations of safety and effectiveness. However, in contrast
to a traditional NDA submitted pursuant to
Section 505(b)(1) in which the applicant submits all of the
data demonstrating safety and effectiveness, we believe an
application submitted pursuant to Section 505(b)(2) can
rely upon findings by the FDA that the parent drug is safe and
effective in that indication. As a consequence, the preclinical
and clinical development programs leading to the submission of
an NDA under Section 505(b)(2) may be less expensive to
carry out and can be concluded in a shorter period of time than
programs required for a Section 505(b)(1) application. In
its review of any NDA submissions, however, the FDA has broad
discretion to require an applicant to generate additional data
related to safety and efficacy, and it is impossible to predict
the number or nature of the studies that may be required before
the FDA will grant approval. Notwithstanding the approval of
many products by the FDA pursuant to Section 505(b)(2),
over the last few years certain brand-name pharmaceutical
companies and others have objected to the FDA’s
interpretation of Section 505(b)(2). If the FDA changes its
interpretation of Section 505(b)(2), this could delay or
even prevent the FDA from approving any Section 505(b)(2)
NDA that we submit.
To the extent that a Section 505(b)(2) applicant is relying
on the FDA’s findings for an already-approved product, the
applicant is required to certify to the FDA concerning any
patents listed for the approved product in the FDA’s Orange
Book publication. A certification that the new product will not
infringe the already approved products’ Orange Book-listed
patents or that such patents are invalid is called a
paragraph IV certification, and could be challenged in
court by the patent owner or holder of the application of the
already approved products. This could delay the approval of any
Section 505(b)(2) application we submit. In addition, any
period of marketing exclusivity applicable to the already
approved product might delay approval of any
Section 505(b)(2) application we submit. Any
Section 505(b)(1) or Section 505(b)(2) application we
submit for a drug product containing a previously approved API
might be eligible for three years of marketing exclusivity,
provided new clinical investigations that were conducted or
sponsored by the applicant are essential to the FDA’s
approval of the application. Five years of marketing exclusivity
is granted if FDA approves an NDA for a new chemical entity. In
addition, we can list in the FDA’s Orange Book publication
any of our patents claiming the drug product, drug substance or
that cover an approved
method-of-use.
In order for a generic applicant to rely on the FDA’s
approval of any NDA we submit, such generic applicant must
certify to any Orange Book listed patents and might be subject
to any marketing exclusivity covering our approved drug product.
In our AZ-004 NDA submission and future submissions for our
other product candidates, we intend to follow the development
pathway permitted under the FDCA that we believe will maximize
the commercial opportunities for these product candidates. We
are currently pursuing the Section 505(b)(2) application
route for our product candidates. As such, we have and intend to
continue to engage in discussions with the FDA to determine
which, if any, portions of our development program can be
modified, based on previous FDA findings of a drug’s safety
and effectiveness.
Before approving an NDA, the FDA will inspect the facilities at
which the product is manufactured, whether ours or our third
party manufacturers’, and will not approve the product
unless the manufacturing facility complies with cGMP. The FDA
reviews all NDA’s submitted before it accepts them for
filing and may request additional information rather than accept
an NDA for filing. Once the NDA submission has been accepted for
filing, the FDA begins an in-depth review of the NDA. Under the
goals and policies agreed to by the FDA under the Prescription
Drug User Fee Act, or PDUFA, the FDA has 10 months in which
to complete its initial review of a standard NDA and respond to
the applicant, and six months for a priority NDA. The FDA does
not always meet the PDUFA goal dates for standard and priority
NDA’s. The review process is often significantly extended
by FDA requests for additional information or clarification. The
FDA may delay approval of an NDA if applicable regulatory
criteria are not satisfied, require additional testing or
information
and/or
require post-marketing testing and surveillance to monitor
safety or efficacy of a product. FDA approval of any NDA
submitted by us will be at a time the FDA chooses. Also, if
regulatory approval of a product is granted, such approval may
entail limitations on the indicated
22
uses for which such product may be marketed. Once approved, the
FDA may withdraw the product approval if compliance with pre-and
post-marketing regulatory requirements and conditions of
approvals are not maintained or if problems occur after the
product reaches the marketplace. In addition, the FDA may
require post-marketing studies, referred to as Phase 4 clinical
trials, to monitor the effect of approved products and may limit
further marketing of the product based on the results of these
post-marketing studies.
If we obtain regulatory approval for a product, this approval
will be limited to those diseases and conditions for which the
product is effective, as demonstrated through clinical trials.
Even if this regulatory approval is obtained, a marketed
product, its manufacturer and its manufacturing facilities are
subject to continual review and periodic inspections by the FDA
and, in our case, the State of California. Discovery of
previously unknown problems with a medicine, device,
manufacturer or facility may result in restrictions on the
marketing or manufacturing of an approved product, including
costly recalls or withdrawal of the product from the market. The
FDA has broad post-market regulatory and enforcement powers,
including the ability to suspend or delay issuance of approvals,
seize or recall products, withdraw approvals, enjoin violations
and institute criminal prosecution.
In addition to regulation by the FDA and certain state
regulatory agencies, the United States Drug Enforcement
Administration, or DEA, imposes various registration,
recordkeeping and reporting requirements, procurement and
manufacturing quotas, labeling and packaging requirements,
security controls and a restriction on prescription refills on
certain pharmaceutical products under the Controlled Substances
Act. A principal factor in determining the particular
requirements, if any, applicable to a product is its actual or
potential abuse profile. The DEA regulates drug substances as
Schedule I, II, III, IV or V substances, with
Schedule I and II substances considered to present the
highest risk of substance abuse and Schedule V substances
the lowest risk. Alprazolam, the API in AZ-002, is regulated as
a Schedule IV substance, fentanyl, the API in AZ-003, is
regulated as a Schedule II substance, and zaleplon, the API
in AZ-007, is regulated as a Schedule IV substance. Each of
these product candidates are subject to DEA regulations relating
to manufacturing, storage, distribution and physician
prescription procedures, and the DEA regulates the amount of the
scheduled substance that would be available for clinical trials
and commercial distribution. As a Schedule II substance,
fentanyl is subject to additional controls, including quotas on
the amount of product that can be manufactured and limitations
on prescription refills. We have received necessary
registrations from the DEA for the manufacture of AZ-002, AZ-003
and AZ-007. The DEA periodically inspects facilities for
compliance with its rules and regulations. Failure to comply
with current and future regulations of the DEA could lead to a
variety of sanctions, including revocation, or denial of
renewal, of DEA registrations, injunctions, or civil or criminal
penalties, and could harm our business and financial condition.
The single dose design of our Staccato system uses what
we refer to as “energetic materials” to generate the
rapid heating necessary for vaporizing the drug while avoiding
degradation. Manufacture of products containing these types of
materials is controlled by the Bureau of Alcohol, Tobacco,
Firearms and Explosives, or ATF, under 18 United States Code
Chapter 40. Technically, the energetic materials used in
our Staccato system are classified as “low
explosives,” and we have been granted a license/permit by
the ATF for the manufacture of such low explosives.
Additionally, due to inclusion of the energetic materials in our
Staccato system, shipments of the single dose design of
our Staccato system are regulated by the Department of
Transportation, or DOT, under Section 173.56, Title 49
of the United States Code of Federal Regulations. The single
dose version of our Staccato device has been granted
“Not Regulated as an Explosive” status by the DOT.
We have received funding for one or more research projects from
a funding agency of the United States government, and inventions
conceived or first actually reduced to practice during the
performance of the research project are subject to the rights
and limitations of certain federal statutes and various
implementing regulations known generally and collectively as the
“Bayh-Dole Requirements.” As a funding recipient, we
are subject to certain invention reporting requirements, and
certain limitations are placed on assignment of the invention
rights. In addition, the federal government retains a
non-exclusive, irrevocable,
paid-up
license to practice the invention and, in exceptional cases, the
federal government may seek to take title to the invention.
We also will be subject to a variety of foreign regulations
governing clinical trials and the marketing of any future
products. Outside the United States, our ability to market a
product depends upon receiving a marketing authorization from
the appropriate regulatory authorities. The requirements
governing the conduct of clinical trials, marketing
authorization, pricing and reimbursement vary widely from
country to country. In any country, however,
23
we will only be permitted to commercialize our products if the
appropriate regulatory authority is satisfied that we have
presented adequate evidence of safety, quality and efficacy.
Whether or not FDA approval has been obtained, approval of a
product by the comparable regulatory authorities of foreign
countries must be obtained prior to the commencement of
marketing of the product in those countries. The time needed to
secure approval may be longer or shorter than that required for
FDA approval. The regulatory approval and oversight process in
other countries includes all of the risks associated with the
FDA process described above.
Pharmaceutical
Pricing and Reimbursement
In both domestic and foreign markets, our ability to
commercialize successfully and attract strategic partners for
our product candidates depends in significant part on the
availability of adequate coverage and reimbursement from
third-party payors, including, in the United States,
governmental payors such as the Medicare and Medicaid programs,
managed care organizations, and private health insurers.
Third-party payors are increasingly challenging prices charged
for medical products and services and examining their cost
effectiveness, in addition to their safety and efficacy. We may
need to conduct expensive pharmacoeconomic studies in order to
demonstrate the cost effectiveness of any future products. Even
with studies, our product candidates may be considered less
safe, less effective or less cost effective than existing
products, and third-party payors therefore may not provide
coverage and reimbursement for our product candidates, in whole
or in part.
Political, economic and regulatory influences are subjecting the
healthcare industry in the United States to fundamental changes.
There have been, and we expect there will continue to be, a
number of legislative and regulatory proposals to change the
healthcare system in ways that could significantly affect our
business. We anticipate that Congress, state legislatures and
the private sector will continue to consider and may adopt
healthcare policies intended to curb rising healthcare costs.
These cost containment measures include:
|
|
|
| |
•
|
controls on government funded reimbursement for medical products
and services;
|
| |
| |
•
|
controls on healthcare providers;
|
| |
| |
•
|
challenges to the pricing of medical products and services or
limits or prohibitions on reimbursement for specific products
and therapies through other means;
|
| |
| |
•
|
reform of drug importation laws; and
|
| |
| |
•
|
expansion of use of managed care systems in which healthcare
providers contract to provide comprehensive healthcare for a
fixed cost per person.
|
We are unable to predict what additional legislation,
regulations or policies, if any, relating to the healthcare
industry or third-party coverage and reimbursement may be
enacted in the future or what effect such legislation,
regulations or policies would have on our business. Any cost
containment measures, including those listed above, or other
healthcare system reforms that are adopted could have a material
adverse effect on our ability to operate profitably.
Patents
and Proprietary Rights
We actively seek to patent the technologies, inventions and
improvements we consider important to the development of our
business. In addition, we rely on trade secrets and contractual
arrangements to protect our proprietary information. Some areas
for which we seek patent protection include:
|
|
|
| |
•
|
the Staccato system and its components;
|
| |
| |
•
|
methods of using the Staccato system;
|
| |
| |
•
|
the aerosolized form of drug compounds produced by the
Staccato system; and
|
| |
| |
•
|
methods of making and using the drug containing aerosols,
including methods of administering the aerosols to a patient.
|
As of February 1, 2010, we held 192 issued and allowed
U.S. and international patents. Most of our patents are
directed to compositions for delivery of an aerosol comprising
drugs other than our lead product candidates
24
described below, and cover the process for producing these
aerosols using the Staccato system. As of that date, we
held 29 additional pending patent applications in the United
States. We also hold 59 pending corresponding foreign patent
applications or Patent Cooperation Treaty applications that will
permit us to pursue additional patents outside of the United
States. The claims in these various patents and patent
applications are directed to various aspects of our drug
delivery devices and their components, methods of using our
devices, drug containing aerosol compositions and methods of
making and using such compositions.
AZ-004/AZ-104
(Staccato loxapine)
One of our issued U.S. patents covers compositions for
delivery of a condensation aerosol comprising loxapine and
covers the process for producing such condensation aerosol using
the Staccato system technology. This patent will not
expire until 2022. Counterparts to this patent are pending in a
number of foreign jurisdictions, including Europe. We also have
three other U.S. patents directed to condensation aerosol
compositions for delivery of loxapine, kits containing devices
for forming such compositions and methods of administering such
compositions.
AZ-007
(Staccato zaleplon)
One of our issued U.S. patents covers compositions for
delivery of a condensation aerosol comprising zaleplon and
covers the process for producing such condensation aerosol using
the Staccato system technology. This patent will not
expire until 2022. Counterparts to this patent are pending in a
number of foreign jurisdictions, including Europe. We also have
three other U.S. patents directed to condensation aerosol
compositions for delivery of zaleplon, kits containing devices
for forming such compositions, and methods of administering such
compositions.
AZ-001
(Staccato prochlorperazine)
One of our issued U.S. patents covers compositions for
delivery of a condensation aerosol comprising prochlorperazine
and covers the process for producing such condensation aerosol
using the Staccato system technology. This patent will
not expire until 2022. Counterparts to this patent are pending
in a number of foreign jurisdictions, including Europe. We also
have three other U.S. patents directed to condensation
aerosol compositions for delivery of prochlorperazine, kits
containing devices for forming such compositions, and methods of
administering such compositions.
AZ-002
(Staccato alprazolam)
One of our issued U.S. patents covers compositions for
delivery of a condensation aerosol comprising alprazolam and
covers the process for producing such condensation aerosol using
the Staccato system technology. This patent will not
expire until 2022. Counterparts to this patent are pending in a
number of foreign jurisdictions, including Europe. We also have
three other U.S. patents directed to condensation aerosol
compositions for delivery of alprazolam, kits containing devices
for forming such compositions, and methods of administering such
compositions.
AZ-003
(Staccato fentanyl)
One of our issued U.S. patents covers compositions for
delivery of a condensation aerosol comprising fentanyl and
covers the process for producing such condensation aerosol using
the Staccato system technology. This patent will not
expire until 2022. Counterparts to this patent are pending in a
number of foreign jurisdictions, including Europe. We also have
three other U.S. patents directed to condensation aerosol
compositions for delivery of fentanyl, kits containing devices
for forming such compositions, and methods of administering such
compositions.
Competition
The pharmaceutical and biotechnology industries are intensely
competitive. Many pharmaceutical companies, biotechnology
companies, public and private universities, government agencies
and research organizations are actively engaged in research and
development of products targeting the same markets as our
product candidates. Many of these organizations have
substantially greater financial, research, drug development,
manufacturing and marketing resources than we have. Large
pharmaceutical companies in particular have extensive experience
in
25
clinical testing and obtaining regulatory approvals for drugs.
Our ability to compete successfully will depend largely on our
ability to:
|
|
|
| |
•
|
develop products that are superior to other products in the
market;
|
| |
| |
•
|
attract and retain qualified scientific, product development,
manufacturing, and commercial personnel;
|
| |
| |
•
|
obtain patent
and/or other
proprietary protection covering our future products and
technologies;
|
| |
| |
•
|
obtain required regulatory approvals; and
|
| |
| |
•
|
successfully collaborate with pharmaceutical and biotechnology
companies in the development and commercialization of new
products.
|
We expect any future products we develop to compete on the basis
of, among other things, product efficacy and safety, time to
market, price, extent of adverse side effects experienced and
convenience of treatment procedures. One or more of our
competitors may develop products based upon the principles
underlying our proprietary technologies earlier than we do,
obtain approvals for such products from the FDA more rapidly
than we do or develop alternative products or therapies that are
safer, more effective
and/or more
cost effective than any future products developed by us. In
addition, our ability to compete may be affected if insurers and
other third-party payors encourage the use of generic products
through other routes of administration.
Any future products developed by us would compete with a number
of alternative drugs and therapies, including the following:
|
|
|
| |
•
|
AZ-004 would compete with the injectable form of loxapine and
other antipsychotic drugs;
|
| |
| |
•
|
AZ-007 would compete with non-benzodiazepine GABA-A receptor
agonists;
|
| |
| |
•
|
AZ-001 and AZ-104 would compete with available triptan drugs
and IV prochlorperazine;
|
| |
| |
•
|
AZ-003 would compete with injectable and other forms of fentanyl
and various generic oxycodone, hydrocodone and morphine
products; and
|
| |
| |
•
|
AZ-002 would compete with the oral tablet form of alprazolam and
other benzodiazepines.
|
Many of these existing drugs have substantial current sales and
long histories of effective and safe use. As patent protection
expires for these drugs, we will also compete with their generic
versions. In addition to currently marketed drugs and their
generic versions, we believe there are a number of drug
candidates in clinical trials that, if approved in the future,
would compete with any future products we may develop.
Employees
As of February 8, 2010, we had 90 full time employees, 15
of whom held Ph.D. or M.D. degrees and 64 of whom were engaged
in full time research and development activities. None of our
employees are represented by a labor union, and we consider our
employee relations to be good.
Corporate
Information
We were incorporated in the state of Delaware on
December 19, 2000 as FaxMed, Inc. In June 2001, we changed
our name to Alexza Corporation and in December 2001 we became
Alexza Molecular Delivery Corporation. In July 2005, we changed
our name to Alexza Pharmaceuticals, Inc.
Available
Information
Our website address is www.alexza.com; however, information
found on, or that can be accessed through, our website is not
incorporated by reference into this Annual Report. We file
electronically with the SEC our Annual Report, quarterly reports
on
Form 10-Q,
current reports on
Form 8-K
and amendments to those reports filed or furnished pursuant to
Section 13(a) or 15(d) of the Securities Exchange Act of
1934. We make available free of charge on or through our website
copies of these reports as soon as reasonably practicable after
we electronically file such material with, or furnish it to, the
SEC. The SEC maintains an internet site that contains reports,
proxy and
26
information statements and other information regarding our
filings at www.sec.gov. You may also read and copy any of our
materials filed with the SEC at the SEC’s Public References
Room at 100 F Street, NW, Washington, DC 20549.
Information regarding the operation of the Public Reference Room
can be obtained by calling the SEC at
1-800-SEC-0330.
RISK
FACTORS
Investing in our common stock involves a high degree of risk.
You should carefully consider the risks described below,
together with all of the other information included in this
Annual Report, before deciding whether to invest in shares of
our common stock. Additional risks and uncertainties not
presently known to us or that we currently deem immaterial also
may impair our business operations. The occurrence of any of the
following risks could harm our business, financial condition or
results of operations. In such case, the trading price of our
common stock could decline, and you may lose all or part of your
investment.
Risks
Relating to Our Business
We
have a history of net losses. We expect to continue to incur
substantial and increasing net losses for the foreseeable
future, and we may never achieve or maintain
profitability.
We are not profitable and have incurred significant net losses
in each year since our inception, including net losses of
$56.1 million, $77.0 million, $55.9 million and
$309.7 million for the years ended December 31, 2009,
2008 and 2007, and the period from December 19, 2000
(inception) to December 31, 2009, respectively. As of
December 31, 2009, we had a deficit accumulated during
development stage of $264.6 million and a
stockholders’ deficit of $7.1 million. We expect our
expenses to decrease in 2010 compared to 2009 due to lower
expected clinical expenses with respect to our lead development
program. We expect to incur substantial net losses and negative
cash flow for the foreseeable future. These losses and negative
cash flows have had, and will continue to have, an adverse
effect on our stockholders’ equity and working capital.
Because of the numerous risks and uncertainties associated with
pharmaceutical product development and commercialization, we are
unable to accurately predict the timing or amount of future
expenses or when, or if, we will be able to achieve or maintain
profitability. Currently, we have no products approved for
commercial sale, and to date we have not generated any product
revenue. We have financed our operations primarily through the
sale of equity securities, capital lease and equipment
financing, collaboration and licensing agreements, and
government grants. The size of our future net losses will
depend, in part, on the rate of growth or contraction of our
expenses and the level and rate of growth, if any, of our
revenues. Revenues from strategic partnerships are uncertain
because we may not enter into any additional strategic
partnerships. We began to recognize revenues from our
partnership with Endo Pharmaceuticals, Inc. in the third quarter
of 2008, and we recognized approximately $9.5 million in
revenue in the three months ended March 31, 2009 as a
result of termination of the Endo license agreement in January
2009. If we are unable to develop and commercialize one or more
of our product candidates or if sales revenue from any product
candidate that receives marketing approval is insufficient, we
will not achieve profitability. Even if we do achieve
profitability, we may not be able to sustain or increase
profitability.
We are
a development stage company. Our success depends substantially
on our lead product candidates. If we do not develop
commercially successful products, we may be forced to cease
operations.
You must evaluate us in light of the uncertainties and
complexities affecting a development stage pharmaceutical
company. We have not completed clinical development for any of
our product candidates. We filed our NDA for AZ-004 in December
2009 and each of our other product candidates is at an earlier
stage of development. Each of our product candidates will be
unsuccessful if it:
|
|
|
| |
•
|
does not demonstrate acceptable safety and efficacy in
preclinical studies and clinical trials or otherwise does not
meet applicable regulatory standards for approval;
|
27
|
|
|
| |
•
|
does not offer therapeutic or other improvements over existing
or future drugs used to treat the same or similar conditions;
|
| |
| |
•
|
is not capable of being produced in commercial quantities at an
acceptable cost, or at all; or
|
| |
| |
•
|
is not accepted by patients, the medical community or third
party payors.
|
Our ability to generate product revenue in the future is
dependent on the successful development and commercialization of
our product candidates. We have not proven our ability to
develop and commercialize products. Problems frequently
encountered in connection with the development and utilization
of new and unproven technologies and the competitive environment
in which we operate might limit our ability to develop
commercially successful products. We do not expect any of our
current product candidates to be commercially available before
2011, if at all. If we are unable to make our product candidates
commercially available, we will not generate product revenues,
and we will not be successful.
We
will need substantial additional capital in the future. If
additional capital is not available, we will have to delay,
reduce or cease operations.
We will need to raise additional capital to fund our operations,
to develop our product candidates and to develop our
manufacturing capabilities. Our future capital requirements will
be substantial and will depend on many factors including:
|
|
|
| |
•
|
the scope, rate of progress, results and costs of our
preclinical studies, clinical trials and other research and
development activities, and our manufacturing development and
commercial manufacturing activities;
|
| |
| |
•
|
the cost, timing and outcomes of regulatory proceedings;
|
| |
| |
•
|
the cost and timing of developing manufacturing capacity;
|
| |
| |
•
|
the cost and timing of developing sales and marketing
capabilities prior to receipt of any regulatory approval of our
product candidates;
|
| |
| |
•
|
revenues received from any existing or future products;
|
| |
| |
•
|
payments received under any future strategic partnerships;
|
| |
| |
•
|
the filing, prosecution and enforcement of patent
claims; and
|
| |
| |
•
|
the costs associated with commercializing our product
candidates, if they receive regulatory approval.
|
We believe that with current cash, cash equivalents and
marketable securities along with interest earned thereon, the
proceeds from option exercises, purchases of common stock
pursuant to our Employee Stock Purchase Plan, and the proceeds
from our agreement with Biovail, we will be able to maintain our
currently planned operations through the first quarter of 2011
and will extend into 2012 if we achieve the eligible
milestones under the Biovail agreement during the coming
12 months. Changing circumstances may cause us to consume
capital significantly faster or slower than we currently
anticipate. We have based these estimates on assumptions that
may prove to be wrong, and we could utilize our available
financial resources sooner than we currently expect. The key
assumptions underlying these estimates include:
|
|
|
| |
•
|
expenditures related to continued preclinical and clinical
development of our product candidates during this period within
budgeted levels;
|
| |
| |
•
|
achievement of the milestone payments pursuant to our license
agreement with Biovail;
|
| |
| |
•
|
no unexpected costs related to the development of our
manufacturing capability; and
|
| |
| |
•
|
no growth in the number of our employees during this period.
|
We may never be able to generate a sufficient amount of product
revenue to cover our expenses. Until we do, we expect to finance
our future cash needs through public or private equity
offerings, debt financings, strategic partnerships or licensing
arrangements, as well as interest income earned on cash and
marketable securities balances and proceeds from stock option
exercises and purchases under our Employee Stock Purchase Plan.
Any
28
financing transaction may contain unfavorable terms. As a result
of the late filing of a current report on
Form 8-K
in the first quarter of 2009, we are currently ineligible to use
Form S-3
to register securities for sale by us or for resale by other
security holders until we have timely filed all required reports
under the Securities Exchange Act of 1934 until at least April
2010. In the meantime, for capital raising transactions, we must
use
Form S-1
to register securities with the SEC, or issue such securities in
a private placement, which could increase the costs and
adversely impact our ability to raise capital in a timely manner
during this period. If we raise additional funds by issuing
equity securities, our stockholders’ equity will be
diluted. If we raise additional funds through strategic
partnerships, we may be required to relinquish rights to our
product candidates or technologies, or to grant licenses on
terms that are not favorable to us.
The
process for obtaining approval of an NDA is time consuming,
subject to unanticipated delays and costs, and requires the
commitment of substantial resources. The FDA accepted our NDA in
February 2010 with a PDUFA goal date of October 11,
2010.
The FDA is conducting an in-depth review of the submission to
determine whether to approve AZ-004 for commercial marketing for
the indications we have proposed. If the FDA is not satisfied
with the information we provide, the agency may refuse to
approve our NDA or may require us to perform additional studies
or provide other information in order to secure approval. The
FDA may delay, limit or refuse to approve our NDA for many
reasons, including:
|
|
|
| |
•
|
the information we submit may be insufficient to demonstrate
that AZ-004 is safe and effective;
|
| |
| |
•
|
the FDA might not approve the processes or facilities that will
be used for the commercial manufacture of AZ-004; or
|
| |
| |
•
|
the FDA’s interpretation of the nonclinical, clinical or
manufacturing data we provided in our NDA may differ from our
own interpretation of such data.
|
If the FDA determines that the clinical trials of AZ-004 that
were submitted in support of our NDA were not conducted in full
compliance with the applicable protocols for these studies, as
well as with applicable regulations and standards, or if the
agency does not agree with our interpretation of the results of
such studies, the FDA may reject the data that resulted from
such studies. The rejection of data from clinical trials
required to support our NDA for AZ-004 could negatively impact
our ability to obtain marketing authorization for this product
candidate and would have a material adverse effect on our
business and financial condition.
In addition, our NDA may not be approved, or approval may be
delayed, as a result of changes in FDA policies for drug
approval during the review period. For example, although many
products have been approved by the FDA in recent years under
Section 505(b)(2) under the Federal Food, Drug and Cosmetic
Act, objections have been raised to the FDA’s
interpretation of Section 505(b)(2). If challenges to the
FDA’s interpretation of Section 505(b)(2) are
successful, the agency may be required to change its
interpretation, which could delay or prevent the approval of our
NDA for AZ-004.
Under goals set in accordance with the Prescription Drug User
Fee Act of 1992, as amended, or PDUFA, the FDA reviews most NDAs
within 10 months of submission. The review process may be
formally extended by three months or longer if the FDA requires
additional time to review any additional information that the
agency requests or that we elect to provide. If we are unable to
timely respond to the FDA’s requests for additional
information in the course of its review of the NDA for AZ-004,
the approval of the NDA would be delayed. In addition, other
companies have announced that the FDA has notified them that
their scheduled review dates were delayed due to the FDA’s
internal resource constraints. There can be no assurance that
the FDA will not impose such delays on the continuing review of
our NDA for AZ-004, and any failure or significant delay in
obtaining the required approval would have a material adverse
effect on our business and financial condition.
Unstable
market conditions may have serious adverse consequences on our
business.
The recent economic downturn and market instability has made the
business climate more volatile and more costly. Our general
business strategy may be adversely affected by unpredictable and
unstable market conditions. If the current equity and credit
markets deteriorate further, or do not improve, it may make any
necessary debt or
29
equity financing more difficult, more costly, and more dilutive.
While we believe that with current cash, cash equivalents and
marketable securities along with interest earned thereon, the
proceeds from option exercises, purchases of common stock
pursuant to our Employee Stock Purchase Plan, and the proceeds
from our agreement with Biovail, we will be able to maintain our
currently planned operations through the first quarter of 2011
and will extend into 2012 if we achieve the eligible
milestones under the Biovail agreement during the coming
12 months, we may obtain additional financing on less than
attractive rates or on terms that are excessively dilutive to
existing stockholders. Failure to secure any necessary financing
in a timely manner and on favorable terms could have a material
adverse effect on our growth strategy, financial performance and
stock price and could require us to delay or abandon clinical
development plans. There is a risk that one or more of our
current component manufacturers and partners may encounter
difficulties during challenging economic times, which would
directly affect our ability to attain our operating goals on
schedule and on budget.
Unless
our preclinical studies demonstrate the safety of our product
candidates, we will not be able to commercialize our product
candidates.
To obtain regulatory approval to market and sell any of our
product candidates, we must satisfy the FDA and other regulatory
authorities abroad, through extensive preclinical studies, that
our product candidates are safe. Our Staccato system
creates condensation aerosol from drug compounds, and there
currently are no approved products that use a similar method of
drug delivery. Companies developing other inhalation products
have not defined or successfully completed the types of
preclinical studies we believe will be required for submission
to regulatory authorities as we seek approval to conduct our
clinical trials. We may not have conducted or may not conduct in
the future the types of preclinical testing ultimately required
by regulatory authorities, or future preclinical tests may
indicate that our product candidates are not safe for use in
humans. Preclinical testing is expensive, can take many years
and have an uncertain outcome. In addition, success in initial
preclinical testing does not ensure that later preclinical
testing will be successful. We may experience numerous
unforeseen events during, or as a result of, the preclinical
testing process, which could delay or prevent our ability to
develop or commercialize our product candidates, including:
|
|
|
| |
•
|
our preclinical testing may produce inconclusive or negative
safety results, which may require us to conduct additional
preclinical testing or to abandon product candidates that we
believed to be promising;
|
| |
| |
•
|
our product candidates may have unfavorable pharmacology,
toxicology or carcinogenicity; and
|
| |
| |
•
|
our product candidates may cause undesirable side effects.
|
Any such events would increase our costs and could delay or
prevent our ability to commercialize our product candidates,
which could adversely impact our business, financial condition
and results of operations.
Preclinical
studies indicated possible adverse impact of pulmonary delivery
of AZ-001.
In our daily dosing animal toxicology studies of
prochlorperazine, the active pharmaceutical ingredient, or API,
in AZ-001, we detected changes to, and increases of, the cells
in the upper airway of the test animals. The terms for these
changes and increases are “squamous metaplasia” and
“hyperplasia,” respectively. We also observed lung
inflammation in some animals. These findings occurred in daily
dosing studies at doses that were proportionately substantially
greater than any dose we expect to continue to develop or
commercialize. In subsequent toxicology studies of AZ-001
involving intermittent dosing consistent with its intended use,
we detected lower incidence and severity of the changes to, and
increases of, the cells in the upper airway of the test animals
compared to the daily dosing results. We did not observe any
lung inflammation with intermittent dosing. In 2008, we
completed a
28-day
repeat dose inhalation study in dogs. Consistent with previous
findings in shorter-term and higher dose studies, we observed
dose-related minimal to slight squamous metaplasia in the upper
respiratory tract, primarily in the lining of the nasal
passages, in all treated groups. No lower respiratory tract or
lung findings were reported. These findings suggest that the
delivery of the pure drug compound of AZ-001 at the
proportionately higher doses used in daily dosing toxicology
studies may cause adverse consequences if we were to administer
prochlorperazine chronically for prolonged periods of time. If
we observe these findings in our clinical trials of AZ-001, it
could prevent further development or commercialization of AZ-001.
30
Failure
or delay in commencing or completing clinical trials for our
product candidates could harm our business.
We have not completed all the clinical trials necessary to
support an application with the FDA for approval to market any
of our product candidates other than what we believe to be
adequate clinical trials to support the marketing approval for
AZ-004 in the United States. Future clinical trials may be
delayed or terminated as a result of many factors, including:
|
|
|
| |
•
|
delays or failure in reaching agreement on acceptable clinical
trial contracts or clinical trial protocols with prospective
sites;
|
| |
| |
•
|
regulators or institutional review boards may not authorize us
to commence a clinical trial;
|
| |
| |
•
|
regulators or institutional review boards may suspend or
terminate clinical research for various reasons, including
noncompliance with regulatory requirements or concerns about
patient safety;
|
| |
| |
•
|
we may suspend or terminate our clinical trials if we believe
that they expose the participating patients to unacceptable
health risks;
|
| |
| |
•
|
we may experience slower than expected patient enrollment or
lack of a sufficient number of patients that meet the enrollment
criteria for our clinical trials;
|
| |
| |
•
|
patients may not complete clinical trials due to safety issues,
side effects, dissatisfaction with the product candidate, or
other reasons;
|
| |
| |
•
|
we may have difficulty in maintaining contact with patients
after treatment, preventing us from collecting the data required
by our study protocol;
|
| |
| |
•
|
product candidates may demonstrate a lack of efficacy during
clinical trials;
|
| |
| |
•
|
we may experience governmental or regulatory delays, failure to
obtain regulatory approval or changes in regulatory
requirements, policy and guidelines; and
|
| |
| |
•
|
we may experience delays in our ability to manufacture clinical
trial materials in a timely manner as a result of ongoing
process and design enhancements to our Staccato system.
|
Any delay in commencing or completing clinical trials for our
product candidates would delay commercialization of our product
candidates and harm our business, financial condition and
results of operations. It is possible that none of our product
candidates will successfully complete clinical trials or receive
regulatory approval, which would severely harm our business,
financial condition and results of operations.
Continuing
development of our single dose version device may delay
regulatory submissions and marketing approval for
AZ-004
A majority of our clinical studies to date for our product
candidates, other than AZ-003, have been completed using a
version of our single dose Staccato device we refer to as
the chemical single dose, or CSD, device. We are developing a
version of the CSD that is intended to cost less to manufacture
and is more scalable than the current version of CSD. We refer
to the newer version of this single dose device as the
commercial production device, or CPD, version. The CPD
incorporates the same basic chemical heat package and
electronics as the CSD. The four NDA-supporting studies
completed during 2009 were conducted with the CPD. Additionally,
we have conducted a device comparability/bioequivalence study in
normal volunteers using the CSD and the CPD versions of the
device to determine if the drug dose dispensed by the two
devices is comparable
and/or
bioequivalent. If the FDA determines that the results of this
study and the available analytical and other in vitro
data from these devices do not support the comparability
and/or
bioequivalency of the two devices, or if the FDA or foreign
regulatory authorities determine the CPD is unacceptable for any
other reason, we may be required to conduct additional clinical
research for AZ-004 with the CPD version of the device.
Conducting any additional clinical trials could delay any
potential marketing approval in the United States.
31
If our
product candidates do not meet safety and efficacy endpoints in
clinical trials, they will not receive regulatory approval, and
we will be unable to market them.
We have filed an NDA for AZ-004, however we have not yet
received regulatory approval from the FDA or any foreign
regulatory authority to market AZ-004, and our other product
candidates are in preclinical and clinical development. The
clinical development and regulatory approval process is
extremely expensive and takes many years. The timing of any
approval cannot be accurately predicted. If we fail to obtain
regulatory approval for our current or future product
candidates, we will be unable to market and sell them and
therefore we may never be profitable.
As part of the regulatory process, we must conduct clinical
trials for each product candidate to demonstrate safety and
efficacy to the satisfaction of the FDA and other regulatory
authorities abroad. The number and design of clinical trials
that will be required varies depending on the product candidate,
the condition being evaluated, the trial results and regulations
applicable to any particular product candidate. In June 2008, we
announced that our Phase 2a
proof-of-concept
clinical trial of AZ-002 (Staccato Alprazolam) did not
meet either of its two primary endpoints. In September 2009, we
announced that our Phase 2b clinical trial of AZ-104
(Staccato loxapine) for the treatment of migraine did not
meet its primary endpoint.
Prior clinical trial program designs and results are not
necessarily predictive of future clinical trial designs or
results. Initial results may not be confirmed upon full analysis
of the detailed results of a trial. Product candidates in later
stage clinical trials may fail to show the desired safety and
efficacy despite having progressed through initial clinical
trials with acceptable endpoints.
If our
product candidates fail to show a clinically significant benefit
compared to placebo, they will not be approved for
marketing.
The design of our clinical trials is based on many assumptions
about the expected effect of our product candidates, and if
those assumptions prove incorrect, the clinical trials may not
produce statistically significant results. Our Staccato
system is not similar to other approved drug delivery
methods, and there is no precedent for the application of
detailed regulatory requirements to our product candidates. We
cannot assure you that the design of, or data collected from,
the clinical trials of our product candidates will be sufficient
to support the FDA and foreign regulatory approvals.
Regulatory
authorities may not approve our product candidates even if they
meet safety and efficacy endpoints in clinical
trials.
The FDA and other foreign regulatory agencies can delay, limit
or deny marketing approval for many reasons, including:
|
|
|
| |
•
|
a product candidate may not be considered safe or effective;
|
| |
| |
•
|
the manufacturing processes or facilities we have selected may
not meet the applicable requirements; and
|
| |
| |
•
|
changes in their approval policies or adoption of new
regulations may require additional work on our part.
|
Any delay in, or failure to receive or maintain, approval for
any of our product candidates could prevent us from ever
generating meaningful revenues or achieving profitability.
Our product candidates may not be approved even if they achieve
their endpoints in clinical trials. Regulatory agencies,
including the FDA, or their advisors may disagree with our trial
design and our interpretations of data from preclinical studies
and clinical trials. Regulatory agencies may change requirements
for approval even after a clinical trial design has been
approved. Regulatory agencies also may approve a product
candidate for fewer or more limited indications than requested
or may grant approval subject to the performance of
post-marketing studies. In addition, regulatory agencies may not
approve the labeling claims that are necessary or desirable for
the successful commercialization of our product candidates.
32
Our
product candidates will remain subject to ongoing regulatory
review even if they receive marketing approval. If we fail to
comply with continuing regulations, we could lose these
approvals, and the sale of any future products could be
suspended.
Even if we receive regulatory approval to market a particular
product candidate, the FDA or a foreign regulatory authority
could condition approval on conducting additional costly
post-approval studies or could limit the scope of our approved
labeling. Moreover, the product may later cause adverse effects
that limit or prevent its widespread use, force us to withdraw
it from the market or impede or delay our ability to obtain
regulatory approvals in additional countries. In addition, we
will continue to be subject to FDA review and periodic
inspections to ensure adherence to applicable regulations. After
receiving marketing approval, the FDA imposes extensive
regulatory requirements on the manufacturing, labeling,
packaging, adverse event reporting, storage, advertising,
promotion and record keeping related to the product.
If we fail to comply with the regulatory requirements of the FDA
and other applicable U.S. and foreign regulatory
authorities or previously unknown problems with any future
products, suppliers or manufacturing processes are discovered,
we could be subject to administrative or judicially imposed
sanctions, including:
|
|
|
| |
•
|
restrictions on the products, suppliers or manufacturing
processes;
|
| |
| |
•
|
warning letters or untitled letters;
|
| |
| |
•
|
civil or criminal penalties or fines;
|
| |
| |
•
|
injunctions;
|
| |
| |
•
|
product seizures, detentions or import bans;
|
| |
| |
•
|
voluntary or mandatory product recalls and publicity
requirements;
|
| |
| |
•
|
suspension or withdrawal of regulatory approvals;
|
| |
| |
•
|
total or partial suspension of production; and
|
| |
| |
•
|
refusal to approve pending applications for marketing approval
of new drugs or supplements to approved applications.
|
If we
do not produce our devices cost effectively, we will never be
profitable.
Our Staccato system based product candidates contain
electronic and other components in addition to the active
pharmaceutical ingredients. As a result of the cost of
developing and producing these components, the cost to produce
our product candidates, and any approved products, will likely
be higher per dose than the cost to produce intravenous or oral
tablet products. This increased cost of goods may prevent us
from ever selling any products at a profit. In addition, we are
developing single dose and multiple dose versions of our
Staccato system. Developing multiple versions of our
Staccato system may reduce or eliminate our ability to
achieve manufacturing economies of scale. Developing multiple
versions of our Staccato system reduces our ability to
focus development resources on each version, potentially
reducing our ability to effectively develop any particular
version. We expect to continue to modify each of our product
candidates throughout their clinical development to improve
their performance, dependability, manufacturability and quality.
Some of these modifications may require additional regulatory
review and approval, which may delay or prevent us from
conducting clinical trials. The development and production of
our technology entail a number of technical challenges,
including achieving adequate dependability, that may be
expensive or time consuming to solve. Any delay in or failure to
develop and manufacture any future products in a cost effective
way could prevent us from generating any meaningful revenues and
prevent us from becoming profitable.
33
We
rely on third parties to conduct our preclinical studies and our
clinical trials. If these third parties do not perform as
contractually required or expected, we may not be able to obtain
regulatory approval for our product candidates, or we may be
delayed in doing so.
We do not have the ability to conduct preclinical studies or
clinical trials independently for our product candidates. We
must rely on third parties, such as contract research
organizations, medical institutions, academic institutions,
clinical investigators and contract laboratories, to conduct our
preclinical studies and clinical trials. We are responsible for
confirming that our preclinical studies are conducted in
accordance with applicable regulations and that each of our
clinical trials is conducted in accordance with its general
investigational plan and protocol. The FDA requires us to comply
with regulations and standards, commonly referred to as good
laboratory practices, or GLP, for conducting and recording the
results of our preclinical studies and good clinical practices
for conducting, monitoring, recording and reporting the results
of clinical trials, to assure that data and reported results are
accurate and that the clinical trial participants are adequately
protected. Our reliance on third parties does not relieve us of
these responsibilities. If the third parties conducting our
clinical trials do not perform their contractual duties or
obligations, do not meet expected deadlines, fail to comply with
the FDA’s good clinical practice regulations, do not adhere
to our clinical trial protocols or otherwise fail to generate
reliable clinical data, we may need to enter into new
arrangements with alternative third parties and our clinical
trials may be extended, delayed or terminated or may need to be
repeated, and we may not be able to obtain regulatory approval
for or commercialize the product candidate being tested in such
trials.
Problems
with the third parties that manufacture the active
pharmaceutical ingredients in our product candidates may delay
our clinical trials or subject us to liability.
We do not currently own or operate manufacturing facilities for
clinical or commercial production of the active pharmaceutical
ingredient, or API, used in any of our product candidates. We
have no experience in drug manufacturing, and we lack the
resources and the capability to manufacture any of the APIs used
in our product candidates, on either a clinical or commercial
scale. As a result, we rely on third parties to supply the API
used in each of our product candidates. We expect to continue to
depend on third parties to supply the API for our lead product
candidates and any additional product candidates we develop in
the foreseeable future.
An API manufacturer must meet high precision and quality
standards for that API to meet regulatory specifications and
comply with regulatory requirements. A contract manufacturer is
subject to ongoing periodic unannounced inspection by the FDA
and corresponding state and foreign authorities to ensure strict
compliance with current good manufacturing practice, or cGMP,
and other applicable government regulations and corresponding
foreign standards. Additionally, a contract manufacturer must
pass a pre-approval inspection by the FDA to ensure strict
compliance with cGMP prior to the FDA’s approval of any
product candidate for marketing. A contract manufacturer’s
failure to conform with cGMP could result in the FDA’s
refusal to approve or a delay in the FDA’s approval of a
product candidate for marketing. We are ultimately responsible
for confirming that the APIs used in our product candidates are
manufactured in accordance with applicable regulations.
Our third party suppliers may not carry out their contractual
obligations or meet our deadlines. In addition, the API they
supply to us may not meet our specifications and quality
policies and procedures. If we need to find alternative
suppliers of the API used in any of our product candidates, we
may not be able to contract for such supplies on acceptable
terms, if at all. Any such failure to supply or delay caused by
such contract manufacturers would have an adverse effect on our
ability to continue clinical development of our product
candidates or commercialize any future products.
If our third party drug suppliers fail to achieve and maintain
high manufacturing standards in compliance with cGMP
regulations, we could be subject to certain product liability
claims in the event such failure to comply resulted in defective
products that caused injury or harm.
If we
experience problems with the manufacturers of components of our
product candidates, our development programs may be delayed or
we may be subject to liability.
We outsource the manufacturing of the components of our
Staccato system, including the printed circuit boards,
the plastic airways, and the chemical heat packages to be used
in our commercial single dose device. We
34
have no experience in the manufacturing of components, other
than our current chemical heat packages, and we currently lack
the resources and the capability to manufacture them, on either
a clinical or commercial scale. As a result, we rely on third
parties to supply these components. We expect to continue to
depend on third parties to supply these components for our
current product candidates and any devices based on the Staccato
system we develop in the foreseeable future.
The third party suppliers of the components of our Staccato
system must meet high precision and quality standards for
those components to comply with regulatory requirements. A
contract manufacturer is subject to ongoing periodic unannounced
inspection by the FDA and corresponding state and foreign
authorities to ensure strict compliance with the FDA’s
Quality System Regulation, or QSR, which sets forth the
FDA’s current good manufacturing practice requirements for
medical devices and their components, and other applicable
government regulations and corresponding foreign standards. We
are ultimately responsible for confirming that the components
used in the Staccato system are manufactured in
accordance with the QSR or other applicable regulations.
Our third party suppliers may not comply with their contractual
obligations or meet our deadlines, or the components they supply
to us may not meet our specifications and quality policies and
procedures. If we need to find alternative suppliers of the
components used in the Staccato system, we may not be
able to contract for such components on acceptable terms, if at
all. Any such failure to supply or delay caused by such contract
manufacturers would have an adverse affect on our ability to
continue clinical development of our product candidates or
commercialize any future products.
In addition, the heat packages used in the single dose version
of our Staccato system are manufactured using certain
energetic, or highly combustible, materials that are used to
generate the rapid heating necessary for vaporizing the drug
compound while avoiding degradation. Manufacture of products
containing these types of materials is regulated by the
U.S. government. We have entered into a supply agreement
with Autoliv for the manufacture of the heat packages in the
commercial design of our single dose version of our Staccato
system. If Autoliv is unable to manufacture the heat
packages to our specifications, or does not carry out its
contractual obligations to supply our heat packages to us, our
clinical trials or commercialization efforts may be delayed,
suspended or terminated while we seek additional suitable
manufacturers of our heat packages, which may prevent us from
commercializing our product candidates that utilize the single
dose version of the Staccato system.
If we
do not establish additional strategic partnerships, we will have
to undertake development and commercialization efforts on our
own, which would be costly and delay our ability to
commercialize any future products.
A key element of our business strategy is our intent to
selectively partner with pharmaceutical, biotechnology and other
companies to obtain assistance for the development and potential
commercialization of our product candidates. In December 2006,
we entered into such a development relationship with Symphony
Allegro, and in December 2007 we entered into a strategic
relationship with Endo Pharmaceuticals, Inc. for the development
of AZ-003.
In January 2009, we mutually agreed with Endo to terminate our
agreement. In June 2009, we amended the terms of our option
agreement with Symphony Allegro, resulting in our acquisition of
Symphony Allegro and the termination of the agreement in August
2009. In February 2010, we entered into a collaboration with
Biovail for the commercialization of AZ-004 in the U.S. and
Canada. We intend to enter into additional strategic
partnerships with third parties to develop and commercialize our
product candidates. To date, other than Symphony Allegro, Endo
and Biovail, we have not entered into any strategic partnerships
for any of our product candidates. We face significant
competition in seeking appropriate strategic partners, and these
strategic partnerships can be intricate and time consuming to
negotiate and document. We may not be able to negotiate
additional strategic partnerships on acceptable terms, or at
all. We are unable to predict when, if ever, we will enter into
any additional strategic partnerships because of the numerous
risks and uncertainties associated with establishing strategic
partnerships. If we are unable to negotiate additional strategic
partnerships for our product candidates we may be forced to
curtail the development of a particular candidate, reduce or
delay its development program or one or more of our other
development programs, delay its potential commercialization,
reduce the scope of our sales or marketing activities or
undertake development or commercialization activities at our own
expense. In addition, we will bear all the risk related to the
development of that product candidate. If we elect to increase
our expenditures to fund development or commercialization
activities on our own, we may need to obtain additional capital,
which may not be available to us
35
on acceptable terms, or at all. If we do not have sufficient
funds, we will not be able to bring our product candidates to
market and generate product revenue.
If we
enter into additional strategic partnerships, we may be required
to relinquish important rights to and control over the
development of our product candidates or otherwise be subject to
terms unfavorable to us.
Due to our relationship with Biovail we are, and for any other
strategic partnerships or collaborations with pharmaceutical or
biotechnology companies we may establish, subject to a number of
risks including:
|
|
|
| |
•
|
we may not be able to control the amount and timing of resources
that our strategic partners devote to the development or
commercialization of product candidates;
|
| |
| |
•
|
strategic partners may delay clinical trials, provide
insufficient funding, terminate a clinical trial or abandon a
product candidate, repeat or conduct new clinical trials or
require a new version of a product candidate for clinical
testing;
|
| |
| |
•
|
strategic partners may not pursue further development and
commercialization of products resulting from the strategic
partnering arrangement or may elect to discontinue research and
development programs;
|
| |
| |
•
|
strategic partners may not commit adequate resources to the
marketing and distribution of any future products, limiting our
potential revenues from these products;
|
| |
| |
•
|
disputes may arise between us and our strategic partners that
result in the delay or termination of the research, development
or commercialization of our product candidates or that result in
costly litigation or arbitration that diverts management’s
attention and consumes resources;
|
| |
| |
•
|
strategic partners may experience financial difficulties;
|
| |
| |
•
|
strategic partners may not properly maintain or defend our
intellectual property rights or may use our proprietary
information in a manner that could jeopardize or invalidate our
proprietary information or expose us to potential litigation;
|
| |
| |
•
|
business combinations or significant changes in a strategic
partner’s business strategy may also adversely affect a
strategic partner’s willingness or ability to complete its
obligations under any arrangement;
|
| |
| |
•
|
strategic partners could independently move forward with a
competing product candidate developed either independently or in
collaboration with others, including our competitors; and
|
| |
| |
•
|
strategic partners could terminate the arrangement or allow it
to expire, which would delay the development and may increase
the cost of developing our product candidates.
|
If we
fail to gain market acceptance among physicians, patients,
third-party payors and the medical community, we will not become
profitable.
The Staccato system is a fundamentally new method of drug
delivery. Any future product based on our Staccato system
may not gain market acceptance among physicians, patients,
third-party payors and the medical community. If these products
do not achieve an adequate level of acceptance, we will not
generate sufficient product revenues to become profitable. The
degree of market acceptance of any of our product candidates, if
approved for commercial sale, will depend on a number of
factors, including:
|
|
|
| |
•
|
demonstration of efficacy and safety in clinical trials;
|
| |
| |
•
|
the existence, prevalence and severity of any side effects;
|
| |
| |
•
|
potential or perceived advantages or disadvantages compared to
alternative treatments;
|
| |
| |
•
|
perceptions about the relationship or similarity between our
product candidates and the parent drug compound upon which each
product candidate is based;
|
| |
| |
•
|
the timing of market entry relative to competitive treatments;
|
36
|
|
|
| |
•
|
the ability to offer any future products for sale at competitive
prices;
|
| |
| |
•
|
relative convenience, product dependability and ease of
administration;
|
| |
| |
•
|
the strength of marketing and distribution support;
|
| |
| |
•
|
the sufficiency of coverage and reimbursement of our product
candidates by governmental and other third-party payors; and
|
| |
| |
•
|
the product labeling or product insert required by the FDA or
regulatory authorities in other countries.
|
AZ-001
and other product candidates that we may develop may require
expensive carcinogenicity tests.
The API in AZ-001, prochlorperazine, was approved by the FDA in
1956 for the treatment of severe nausea and vomiting. At that
time, the FDA did not require the carcinogenicity testing that
is now generally required for marketing approval. It is unclear
whether we will be required to perform such testing prior to
filing our application for marketing approval of AZ-001 or
whether we will be allowed to perform such testing after we file
an application. Such carcinogenicity testing will be expensive
and require significant additional resources to complete and may
delay approval to market AZ-001. We may encounter similar
requirements with other product candidates incorporating drugs
that have not undergone carcinogenicity testing. Any
carcinogenicity testing we are required to complete will
increase the costs to develop a particular product candidate and
may delay or halt the development of such product candidate.
If
some or all of our patents expire, are invalidated or are
unenforceable, or if some or all of our patent applications do
not yield issued patents or yield patents with narrow claims,
competitors may develop competing products using our or similar
intellectual property and our business will
suffer.
Our success will depend in part on our ability to obtain and
maintain patent and trade secret protection for our technologies
and product candidates both in the United States and other
countries. We do not know whether any patents will issue from
any of our pending or future patent applications. In addition, a
third party may successfully circumvent our patents. Our rights
under any issued patents may not provide us with sufficient
protection against competitive products or otherwise cover
commercially valuable products or processes.
The degree of protection for our proprietary technologies and
product candidates is uncertain because legal means afford only
limited protection and may not adequately protect our rights or
permit us to gain or keep our competitive advantage. For example:
|
|
|
| |
•
|
we might not have been the first to make the inventions covered
by each of our pending patent applications and issued patents;
|
| |
| |
•
|
we might not have been the first to file patent applications for
these inventions;
|
| |
| |
•
|
others may independently develop similar or alternative
technologies or duplicate any of our technologies;
|
| |
| |
•
|
the claims of our issued patents may be narrower than as filed
and not sufficiently broad to prevent third parties from
circumventing them;
|
| |
| |
•
|
it is possible that none of our pending patent applications will
result in issued patents;
|
| |
| |
•
|
we may not develop additional proprietary technologies or drug
candidates that are patentable;
|
| |
| |
•
|
our patent applications or patents may be subject to
interference, opposition or similar administrative proceedings;
|
| |
| |
•
|
any patents issued to us or our potential strategic partners may
not provide a basis for commercially viable products or may be
challenged by third parties in the course of litigation or
administrative proceedings such as reexaminations or
interferences; and
|
| |
| |
•
|
the patents of others may have an adverse effect on our ability
to do business.
|
37
Even
if valid and enforceable patents cover our product candidates
and technologies, the patents will provide protection only for a
limited amount of time.
Our potential strategic partners’ ability to obtain patents
is uncertain because, to date, some legal principles remain
unresolved, there has not been a consistent policy regarding the
breadth or interpretation of claims allowed in patents in the
United States, and the specific content of patents and patent
applications that are necessary to support and interpret patent
claims is highly uncertain due to the complex nature of the
relevant legal, scientific and factual issues. Furthermore, the
policies governing pharmaceutical and medical device patents
outside the United States may be even more uncertain. Changes in
either patent laws or interpretations of patent laws in the
United States and other countries may diminish the value of our
intellectual property or narrow the scope of our patent
protection.
Even if patents are issued regarding our product candidates or
methods of using them, those patents can be challenged by our
competitors who can argue that our patents are invalid
and/or
unenforceable. Third parties may challenge our rights to, or the
scope or validity of, our patents. Patents also may not protect
our product candidates if competitors devise ways of making
these or similar product candidates without legally infringing
our patents. The Federal Food, Drug and Cosmetic Act and the FDA
regulations and policies provide incentives to manufacturers to
challenge patent validity or create modified, non-infringing
versions of a drug or device in order to facilitate the approval
of generic substitutes. These same types of incentives encourage
manufacturers to submit new drug applications that rely on
literature and clinical data not prepared for or by the drug
sponsor.
We also rely on trade secrets to protect our technology,
especially where we do not believe that patent protection is
appropriate or obtainable. However, trade secrets are difficult
to protect. The employees, consultants, contractors, outside
scientific collaborators and other advisors of our company and
our strategic partners may unintentionally or willfully disclose
our confidential information to competitors. Enforcing a claim
that a third party illegally obtained and is using our trade
secrets is expensive and time consuming and the outcome is
unpredictable. Failure to protect or maintain trade secret
protection could adversely affect our competitive business
position.
Our research and development collaborators may have rights to
publish data and other information in which we have rights. In
addition, we sometimes engage individuals or entities to conduct
research that may be relevant to our business. The ability of
these individuals or entities to publish or otherwise publicly
disclose data and other information generated during the course
of their research is subject to certain contractual limitations.
These contractual provisions may be insufficient or inadequate
to protect our trade secrets and may impair our patent rights.
If we do not apply for patent protection prior to such
publication or if we cannot otherwise maintain the
confidentiality of our technology and other confidential
information, then our ability to receive patent protection or
protect our proprietary information may be jeopardized.
Litigation
or other proceedings or third party claims of intellectual
property infringement could require us to spend time and money
and could shut down some of our operations.
Our commercial success depends in part on not infringing patents
and proprietary rights of third parties. Others have filed, and
in the future are likely to file, patent applications covering
products that are similar to our product candidates, as well as
methods of making or using similar or identical products. If
these patent applications result in issued patents and we wish
to use the claimed technology, we would need to obtain a license
from the third party. We may not be able to obtain these
licenses at a reasonable cost, if at all.
In addition, administrative proceedings, such as interferences
and reexaminations before the U.S. Patent and Trademark
Office, could limit the scope of our patent rights. We may incur
substantial costs and diversion of management and technical
personnel as a result of our involvement in such proceedings. In
particular, our patents and patent applications may be subject
to interferences in which the priority of invention may be
awarded to a third party. We do not know whether our patents and
patent applications would be entitled to priority over patents
or patent applications held by such a third party. Our issued
patents may also be subject to reexamination proceedings. We do
not know whether our patents would survive reexamination in
light of new questions of patentability that may be raised
following their issuance.
38
Third parties may assert that we are employing their proprietary
technology or their proprietary products without authorization.
In addition, third parties may already have or may obtain
patents in the future and claim that use of our technologies or
our products infringes these patents. We could incur substantial
costs and diversion of management and technical personnel in
defending our self against any of these claims. Furthermore,
parties making claims against us may be able to obtain
injunctive or other equitable relief, which could effectively
block our ability to further develop, commercialize and sell any
future products and could result in the award of substantial
damages against us. In the event of a successful claim of
infringement against us, we may be required to pay damages and
obtain one or more licenses from third parties. We may not be
able to obtain these licenses at a reasonable cost, if at all.
In that event, we could encounter delays in product
introductions while we attempt to develop alternative methods or
products. In the event we cannot develop alternative methods or
products, we may be effectively blocked from developing,
commercializing or selling any future products. Defense of any
lawsuit or failure to obtain any of these licenses would be
expensive and could prevent us from commercializing any future
products.
We review from time to time publicly available information
concerning the technological development efforts of other
companies in our industry. If we determine that these efforts
violate our intellectual property or other rights, we intend to
take appropriate action, which could include litigation. Any
action we take could result in substantial costs and diversion
of management and technical personnel in enforcing our patents
or other intellectual property rights against others.
Furthermore, the outcome of any action we take to protect our
rights may not be resolved in our favor.
Competition
in the pharmaceutical industry is intense. If our competitors
are able to develop and market products that are more effective,
safer or less costly than any future products that we may
develop, our commercial opportunity will be reduced or
eliminated.
We face competition from established as well as emerging
pharmaceutical and biotechnology companies, academic
institutions, government agencies and private and public
research institutions. Our commercial opportunity will be
reduced or eliminated if our competitors develop and
commercialize products that are safer, more effective, have
fewer side effects or are less expensive than any future
products that we may develop and commercialize. In addition,
significant delays in the development of our product candidates
could allow our competitors to bring products to market before
us and impair our ability to commercialize our product
candidates.
We anticipate that, if approved, AZ-004 would compete with the
available intramuscular, or IM, injectable form and oral forms
of loxapine and other forms, such as IM, oral tablets, or oral
solutions of available antipsychotic drugs for the treatment of
agitation.
We anticipate that, if approved, AZ-007 would compete with
non-benzodiazepine GABA-A receptor agonists. We are also aware
of more than 10 generic versions of zolpidem oral tablets and
one version of zaleplon that has received a Complete Response
letter from the FDA, as well as at least five insomnia products
that are under review by the FDA. Additionally, we are aware of
three products in Phase 3 development for the treatment of
insomnia.
We anticipate that, if approved, AZ-001 and AZ-104 would compete
with currently marketed triptan drugs and with other migraine
headache treatments, including intravenous, or IV, delivery of
prochlorperazine, the API in
AZ-001. In
addition, we are aware of at least 15 product candidates in
development for the treatment of migraines, including triptan
products.
We anticipate that, if approved, AZ-003 would compete with some
of the available forms of fentanyl, including injectable
fentanyl, oral transmucosal fentanyl formulations and
ionophoretic transdermal delivery of fentanyl. We are also aware
of three fentanyl products under review by regulatory agencies
either in the United States or abroad, and at least 19 products
in Phase 3 clinical trial development for acute pain, seven of
which are fentanyl products. There are two inhaled forms of
fentanyl products that are in Phase 2 development. In addition,
if approved, AZ-003 would compete with various generic opioid
drugs, such as oxycodone, hydrocodone and morphine, or
combination products including one or more of such drugs.
We anticipate that, if approved, AZ-002 would compete with the
oral tablet form of alprazolam and possibly intravenous and oral
forms of other benzodiazepines.
39
Many of our competitors have significantly greater financial
resources and expertise in research and development,
manufacturing, preclinical testing, conducting clinical trials,
obtaining regulatory approvals and marketing approved products
than we do. Established pharmaceutical companies may invest
heavily to discover quickly and develop novel compounds or drug
delivery technology that could make our product candidates
obsolete. Smaller or early stage companies may also prove to be
significant competitors, particularly through strategic
partnerships with large and established companies. In addition,
these third parties compete with us in recruiting and retaining
qualified scientific and management personnel, establishing
clinical trial sites and patient registration for clinical
trials, as well as in acquiring technologies and technology
licenses complementary to our programs or advantageous to our
business. Accordingly, our competitors may succeed in obtaining
patent protection, receiving FDA approval or discovering,
developing and commercializing products before we do. If we are
not able to compete effectively against our current and future
competitors, our business will not grow and our financial
condition will suffer.
If we
are unable to establish sales and marketing capabilities or
enter into additional agreements with third parties to market
and sell our product candidates, we may be unable to generate
significant product revenue.
We have entered into an agreement to grant Biovail the rights to
sell, market, and distribute AZ-004 in the U.S. and Canada.
We do not have a sales organization and have no experience in
the sales and distribution of pharmaceutical products. There are
risks involved with establishing our own sales capabilities and
increasing our marketing capabilities, as well as entering into
arrangements with third parties to perform these services.
Developing an internal sales force is expensive and time
consuming and could delay any product launch. On the other hand,
if we enter into arrangements with third parties to perform
sales, marketing and distribution services, our product revenues
or the profitability of these product revenues are likely to be
lower than if we market and sell any products that we develop
ourselves.
We may establish our own specialty sales force
and/or
engage additional pharmaceutical or other healthcare companies
with an existing sales and marketing organization and
distribution systems to sell, market and distribute any future
products. We may not be able to establish a specialty sales
force or establish sales and distribution relationships on
acceptable terms. Factors that may inhibit our efforts to
commercialize any future products without strategic partners or
licensees include:
|
|
|
| |
•
|
our inability to recruit and retain adequate numbers of
effective sales and marketing personnel;
|
| |
| |
•
|
the inability of sales personnel to obtain access to or persuade
adequate numbers of physicians to prescribe any future products;
|
| |
| |
•
|
the lack of complementary products to be offered by sales
personnel, which may put us at a competitive disadvantage
relative to companies with more extensive product lines; and
|
| |
| |
•
|
unforeseen costs and expenses associated with creating an
independent sales and marketing organization.
|
Because the establishment of sales and marketing capabilities
depends on the progress towards commercialization of our product
candidates and because of the numerous risks and uncertainties
involved with establishing our own sales and marketing
capabilities, we are unable to predict when, if ever, we will
establish our own sales and marketing capabilities. If we are
not able to partner with additional third parties and are
unsuccessful in recruiting sales and marketing personnel or in
building a sales and marketing infrastructure, we will have
difficulty commercializing our product candidates, which would
adversely affect our business and financial condition.
If we
lose our key personnel or are unable to attract and retain
additional personnel, we may be unable to develop or
commercialize our product candidates.
We are highly dependent on our President and Chief Executive
Officer, Thomas B. King, the loss of whose services might
adversely impact the achievement of our objectives. In addition,
recruiting and retaining qualified clinical, scientific and
engineering personnel to manage clinical trials of our product
candidates and to perform future research and development work
will be critical to our success. There is currently a shortage
of skilled
40
executives in our industry, which is likely to continue. As a
result, competition for skilled personnel is intense and the
turnover rate can be high. Although we believe we will be
successful in attracting and retaining qualified personnel,
competition for experienced management and clinical, scientific
and engineering personnel from numerous companies and academic
and other research institutions may limit our ability to do so
on acceptable terms. In addition, we do not have employment
agreements with any of our employees, and they could leave our
employment at will. We have change of control agreements with
our executive officers and vice presidents that provide for
certain benefits upon termination or a change in role or
responsibility in connection with a change of control of our
company. We do not maintain life insurance policies on any
employees. Failure to attract and retain personnel would prevent
us from developing and commercializing our product candidates.
If
plaintiffs bring product liability lawsuits against us, we may
incur substantial liabilities and may be required to limit
commercialization of the product candidates that we may
develop.
We face an inherent risk of product liability as a result of the
clinical testing of our product candidates in clinical trials
and will face an even greater risk if we commercialize any
products. We may be held liable if any product we develop causes
injury or is found otherwise unsuitable during product testing,
manufacturing, marketing or sale. Regardless of merit or
eventual outcome, liability claims may result in decreased
demand for any product candidates or products that we may
develop, injury to our reputation, withdrawal of clinical
trials, costs to defend litigation, substantial monetary awards
to clinical trial participants or patients, loss of revenue and
the inability to commercialize any products that we develop. We
have product liability insurance that covers our clinical trials
up to a $10 million aggregate annual limit. We intend to
expand product liability insurance coverage to include the sale
of commercial products if we obtain marketing approval for
AZ-004 or any other products that we may develop. However, this
insurance may be prohibitively expensive, or may not fully cover
our potential liabilities. Inability to obtain sufficient
insurance coverage at an acceptable cost or otherwise to protect
against potential product liability claims could prevent or
delay the commercialization of our product candidates. If we are
sued for any injury caused by any future products, our liability
could exceed our total assets.
Our
product candidates AZ-002, AZ-003 and AZ-007 contain drug
substances which are regulated by the U.S. Drug Enforcement
Administration. Failure to comply with applicable regulations
could harm our business.
The Controlled Substances Act imposes various registration,
recordkeeping and reporting requirements, procurement and
manufacturing quotas, labeling and packaging requirements,
security controls and a restriction on prescription refills on
certain pharmaceutical products. A principal factor in
determining the particular requirements, if any, applicable to a
product is its actual or potential abuse profile. The
U.S. Drug Enforcement Administration, or DEA, regulates
chemical compounds as Schedule I, II, III, IV or
V substances, with Schedule I substances considered to
present the highest risk of substance abuse and Schedule V
substances the lowest risk. Alprazolam, the API in AZ-002, is
regulated as a Schedule IV substance, fentanyl, the API in
AZ-003, is regulated as a Schedule II substance, and
zaleplon, the API in AZ-007, is regulated as a Schedule IV
substance. Each of these product candidates is subject to DEA
regulations relating to manufacture, storage, distribution and
physician prescription procedures, and the DEA regulates the
amount of the scheduled substance that would be available for
clinical trials and commercial distribution. As a
Schedule II substance, fentanyl is subject to more
stringent controls, including quotas on the amount of product
that can be manufactured as well as a prohibition on the
refilling of prescriptions without a new prescription from the
physician. The DEA periodically inspects facilities for
compliance with its rules and regulations. Failure to comply
with current and future regulations of the DEA could lead to a
variety of sanctions, including revocation, or denial of
renewal, or of DEA registrations, injunctions, or civil or
criminal penalties and could harm our business, financial
condition and results of operations.
The
single dose version of our Staccato system contains materials
that are regulated by the U.S. government, and failure to comply
with applicable regulations could harm our
business.
The single dose version of our Staccato system uses
energetic materials to generate the rapid heating necessary for
vaporizing the drug, while avoiding degradation. Manufacture of
products containing energetic materials is controlled by the
U.S. Bureau of Alcohol, Tobacco, Firearms and Explosives,
or ATF. Technically, the energetic
41
materials used in our Staccato system are classified as
“low explosives,” and the ATF has granted us a
license/permit for the manufacture of such low explosives.
Additionally, due to inclusion of the energetic materials in our
Staccato system, the Department of Transportation, or
DOT, regulates shipments of the single dose version of our
Staccato system. The DOT has granted the single dose
version of our Staccato system “Not Regulated as an
Explosive” status. Failure to comply with the current and
future regulations of the ATF or DOT could subject us to future
liabilities and could harm our business, financial condition and
results of operations. Furthermore, these regulations could
restrict our ability to expand our facilities or construct new
facilities or could require us to incur other significant
expenses in order to maintain compliance.
We use
hazardous chemicals and highly combustible materials in our
business. Any claims relating to improper handling, storage or
disposal of these materials could be time consuming and
costly.
Our research and development processes involve the controlled
use of hazardous materials, including chemicals. We also use
energetic materials in the manufacture of the chemical heat
packages that are used in our single dose devices. Our
operations produce hazardous waste products. We cannot eliminate
the risk of accidental contamination or discharge or injury from
these materials. Federal, state and local laws and regulations
govern the use, manufacture, storage, handling and disposal of
these materials. We could be subject to civil damages in the
event of an improper or unauthorized release of, or exposure of
individuals to, hazardous materials. In addition, claimants may
sue us for injury or contamination that results from our use or
the use by third parties of these materials and our liability
may exceed our total assets. We maintain insurance for the use
of hazardous materials in the aggregate amount of $1 million,
which may not be adequate to cover any claims. Compliance with
environmental and other laws and regulations may be expensive,
and current or future regulations may impair our research,
development or production efforts.
Certain of our suppliers are working with these types of
hazardous and energetic materials in connection with our
component manufacturing agreements. In the event of a lawsuit or
investigation, we could be held responsible for any injury
caused to persons or property by exposure to, or release of,
these hazardous and energetic materials. Further, under certain
circumstances, we have agreed to indemnify our suppliers against
damages and other liabilities arising out of development
activities or products produced in connection with these
agreements.
We
will need to implement additional finance and accounting
systems, procedures and controls in the future as we grow and to
satisfy new reporting requirements.
The laws and regulations affecting public companies, including
the current provisions of the Sarbanes-Oxley Act of 2002, or
Sarbanes-Oxley, and rules enacted and proposed by the SEC and by
the Nasdaq Global Market, will result in increased costs to us
as we continue to undertake efforts to comply with rules and
respond to the requirements applicable to public companies. The
rules make it more difficult and costly for us to obtain certain
types of insurance, including director and officer liability
insurance, and we may be forced to accept reduced policy limits
and coverage or incur substantially higher costs to obtain the
same or similar coverage as compared to the polices previously
available to public companies. The impact of these events could
also make it more difficult for us to attract and retain
qualified persons to serve on our board of directors or our
board committees or as executive officers.
As a public company, we need to comply with Sarbanes-Oxley and
the related rules and regulations of the SEC, including expanded
disclosure, accelerated reporting requirements and more complex
accounting rules. Compliance with Section 404 of
Sarbanes-Oxley and other requirements will continue to increase
our costs and require additional management resources. We have
been upgrading our finance and accounting systems, procedures
and controls and will need to continue to implement additional
finance and accounting systems, procedures and controls as we
grow to satisfy new reporting requirements. We currently do not
have an internal audit group. In addition, we may need to hire
additional legal and accounting staff with appropriate
experience and technical knowledge, and we cannot assure you
that if additional staffing is necessary that we will be able to
do so in a timely fashion.
42
Our
facilities are located near known earthquake fault zones, and
the occurrence of an earthquake or other catastrophic disaster
could damage our facilities and equipment, which could cause us
to curtail or cease operations.
Our facilities are located in the San Francisco Bay Area
near known earthquake fault zones and, therefore, are vulnerable
to damage from earthquakes. We are also vulnerable to damage
from other types of disasters, such as power loss, fire, floods
and similar events. If any disaster were to occur, our ability
to operate our business could be seriously impaired. We
currently may not have adequate insurance to cover our losses
resulting from disasters or other similar significant business
interruptions, and we do not plan to purchase additional
insurance to cover such losses due to the cost of obtaining such
coverage. Any significant losses that are not recoverable under
our insurance policies could seriously impair our business,
financial condition and results of operations.
Risks
Relating to Owning Our Common Stock
Our
stock price has been and may continue to be extremely
volatile.
Our common stock price has experienced large fluctuations. In
addition, the trading prices of life science and biotechnology
company stocks in general have experienced extreme price
fluctuations in recent years. The valuations of many life
science companies without consistent product revenues and
earnings are extraordinarily high based on conventional
valuation standards, such as price to revenue ratios. These
trading prices and valuations may not be sustained. Any negative
change in the public’s perception of the prospects of life
science or biotechnology companies could depress our stock price
regardless of our results of operations. Other broad market and
industry factors may decrease the trading price of our common
stock, regardless of our performance. Market fluctuations, as
well as general political and economic conditions such as
terrorism, military conflict, recession or interest rate or
currency rate fluctuations, also may decrease the trading price
of our common stock. In addition, our stock price could be
subject to wide fluctuations in response to various factors,
including:
|
|
|
| |
•
|
actual or anticipated regulatory approvals or disapprovals of
our product candidates or competing products;
|
| |
| |
•
|
actual or anticipated results and timing of our clinical trials;
|
| |
| |
•
|
changes in laws or regulations applicable to our product
candidates;
|
| |
| |
•
|
changes in the expected or actual timing of our development
programs, including delays or cancellations of clinical trials
for our product candidates;
|
| |
| |
•
|
period to period fluctuations in our operating results;
|
| |
| |
•
|
announcements of new technological innovations or new products
by us or our competitors;
|
| |
| |
•
|
changes in financial estimates or recommendations by securities
analysts;
|
| |
| |
•
|
sales results for AZ-004, if it is approved for marketing;
|
| |
| |
•
|
conditions or trends in the life science and biotechnology
industries;
|
| |
| |
•
|
changes in the market valuations of other life science or
biotechnology companies;
|
| |
| |
•
|
developments in domestic and international governmental policy
or regulations;
|
| |
| |
•
|
announcements by us or our competitors of significant
acquisitions, strategic partnerships, joint ventures or capital
commitments;
|
| |
| |
•
|
additions or departures of key personnel;
|
| |
| |
•
|
disputes or other developments relating to proprietary rights,
including patents, litigation matters and our ability to obtain
patent protection for our technologies;
|
| |
| |
•
|
sales of our common stock (or other securities) by us; and
|
| |
| |
•
|
sales and distributions of our common stock by our stockholders.
|
43
In the past, stockholders have often instituted securities class
action litigation after periods of volatility in the market
price of a company’s securities. If a stockholder files a
securities class action suit against us, we would incur
substantial legal fees, and our management’s attention and
resources would be diverted from operating our business in order
to respond to the litigation.
If we
sell shares of our common stock in future financings, existing
common stock holders will experience immediate dilution and, as
a result, our stock price may go down.
We will need to raise additional capital to fund our operations,
to develop our product candidates and to develop our
manufacturing capabilities. We may obtain such financing through
the sale of our equity securities from time to time. As a
result, our existing common stockholders will experience
immediate dilution upon any such issuance. For example, in
August 2009 we issued 10,000,000 shares of our common stock
and warrants to purchase an additional 5,000,000 shares of
our common stock in connection with the closing of our
acquisition of all of the equity of Symphony Allegro, and in
October 2009 we issued 8,107,012 shares of our common stock
and warrants to purchase an additional 7,296,312 shares of
our common stock in a private placement. If we enter into other
financing transactions in which we issue equity securities in
the future, our existing common stockholders will experience
immediate dilution upon any such issuance.
|
|
|
Item 1B.
|
Unresolved
Staff Comments
|
None.
We lease two buildings with an aggregate of 106,894 square
feet of manufacturing, office, and laboratory facilities in
Mountain View, California, which we began to occupy in the
fourth quarter of 2007. We currently occupy 87,560 square
feet of these facilities and sublease the remaining
19,334 square feet. On March 1, 2010, we initiated a
second sublease for an additional 20,956 square feet
reducing the space we occupy to 66,604 square feet. The
lease for both facilities expires on March 31, 2018, and we
have two options to extend the lease for five years each. Our
sublease agreements expire on April 30, 2010 with regards
to 19,334 square feet and on February 28, 2014 with
regards to 20,956 square feet. We believe that the Mountain
View facilities are sufficient for our office, manufacturing and
laboratory needs for at least the next three years.
|
|
|
Item 3.
|
Legal
Proceedings
|
None
PART II
|
|
|
Item 5.
|
Market
for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities
|
|
|
|
Item 5A.
|
Quarterly
Stock Price Information and Registered
Shareholders
|
Our common stock trades on the NASDAQ Global Market under the
symbol “ALXA.” The following table sets forth, for the
periods indicated, the high and low sales prices of our common
stock.
| |
|
|
|
|
|
|
|
|
|
2009
|
|
High
|
|
Low
|
|
|
|
First Quarter
|
|
$
|
3.40
|
|
|
$
|
1.40
|
|
|
Second Quarter
|
|
|
3.25
|
|
|
|
1.50
|
|
|
Third Quarter
|
|
|
3.01
|
|
|
|
1.90
|
|
|
Fourth Quarter
|
|
|
2.55
|
|
|
|
1.93
|
|
| |
|
|
|
|
|
|
|
|
|
2008
|
|
High
|
|
Low
|
|
|
|
First Quarter
|
|
$
|
8.16
|
|
|
$
|
5.76
|
|
|
Second Quarter
|
|
|
7.55
|
|
|
|
3.75
|
|
|
Third Quarter
|
|
|
6.20
|
|
|
|
3.85
|
|
|
Fourth Quarter
|
|
|
4.91
|
|
|
|
1.16
|
|
44
As of December 31, 2009, there were 201 holders of record
of our common stock. We have not paid cash dividends on our
common stock since our inception, and we do not anticipate
paying any in the foreseeable future.
|
|
|
Item 5B.
|
Use
of Proceeds from the Sale of Registered Securities
|
None
None
|
|
|
Item 6.
|
Selected
Financial Data
|
The data set forth below has been modified to reflect our
adoption of Accounting Standards Codification topic No. 810
regarding noncontrolling interests and should be read in
conjunction with Management’s Discussion and Analysis of
Financial Condition and Results of Operations and the
consolidated financial statements and related notes included
elsewhere herein.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 19,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Inception) to
|
|
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
2009
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
Consolidated Statement of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue
|
|
$
|
9,514
|
|
|
$
|
486
|
|
|
$
|
—
|
|
|
$
|
1,028
|
|
|
$
|
2,230
|
|
|
$
|
16,945
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development(1)
|
|
|
39,778
|
|
|
|
61,565
|
|
|
|
45,645
|
|
|
|
36,494
|
|
|
|
26,235
|
|
|
|
244,461
|
|
|
General and administrative(1)
|
|
|
15,406
|
|
|
|
17,641
|
|
|
|
14,888
|
|
|
|
9,969
|
|
|
|
9,654
|
|
|
|
78,110
|
|
|
Restructuring charges(1)
|
|
|
2,037
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,037
|
|
|
Acquired in-process research and development
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,916
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses(1)
|
|
|
57,221
|
|
|
|
79,206
|
|
|
|
60,533
|
|
|
|
46,463
|
|
|
|
35,889
|
|
|
|
328,524
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(47,707
|
)
|
|
|
(78,720
|
)
|
|
|
(60,533
|
)
|
|
|
(45,435
|
)
|
|
|
(33,659
|
)
|
|
|
(311,579
|
)
|
|
Loss on change in fair value of contingent consideration
liability
|
|
|
(7,983
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(7,983
|
)
|
|
Interest income/(expense) and other income/(expense), net
|
|
|
(375
|
)
|
|
|
1,679
|
|
|
|
4,623
|
|
|
|
1,909
|
|
|
|
1,257
|
|
|
|
9,850
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
(56,065
|
)
|
|
|
(77,041
|
)
|
|
|
(55,910
|
)
|
|
|
(43,526
|
)
|
|
|
(32,402
|
)
|
|
|
(309,712
|
)
|
|
Consideration paid in excess of carrying value of the
noncontrolling interest in Symphony Allegro, Inc.
|
|
|
(61,566
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(61,566
|
)
|
|
Loss attributed to noncontrolling interest in Symphony Allegro,
Inc.
|
|
|
13,987
|
|
|
|
18,591
|
|
|
|
10,791
|
|
|
|
1,720
|
|
|
|
—
|
|
|
|
45,089
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to Alexza common stockholders
|
|
$
|
(103,644
|
)
|
|
$
|
(58,450
|
)
|
|
$
|
(45,119
|
)
|
|
$
|
(41,806
|
)
|
|
$
|
(32,402
|
)
|
|
$
|
(326,189
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share attributable to Alexza
common stockholders
|
|
$
|
(2.68
|
)
|
|
$
|
(1.81
|
)
|
|
$
|
(1.58
|
)
|
|
$
|
(2.13
|
)
|
|
$
|
(18.98
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used to compute basic and diluted net loss per share
attributable to Alexza common stockholders
|
|
|
38,609
|
|
|
|
32,297
|
|
|
|
28,605
|
|
|
|
19,584
|
|
|
|
1,707
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes stock-based compensation as follows: |
45
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 19,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Inception) to
|
|
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Research and development
|
|
$
|
3,443
|
|
|
$
|
2,926
|
|
|
$
|
1,885
|
|
|
$
|
1,770
|
|
|
$
|
167
|
|
|
$
|
10,295
|
|
|
General and administrative
|
|
|
3,025
|
|
|
|
2,520
|
|
|
|
1,531
|
|
|
|
447
|
|
|
|
874
|
|
|
|
8,397
|
|
|
Restructuring expenses
|
|
|
56
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
56
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
6,524
|
|
|
$
|
5,446
|
|
|
$
|
3,416
|
|
|
$
|
2,217
|
|
|
$
|
1,041
|
|
|
$
|
18,748
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
2006
|
|
2005
|
|
|
|
(In thousands)
|
|
|
|
Consolidated Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and marketable securities
|
|
$
|
19,916
|
|
|
$
|
37,556
|
|
|
$
|
69,391
|
|
|
$
|
42,623
|
|
|
$
|
38,369
|
|
|
Investments held by Symphony Allegro, Inc.
|
|
|
—
|
|
|
|
21,318
|
|
|
|
39,449
|
|
|
|
49,956
|
|
|
|
—
|
|
|
Working capital
|
|
|
(3,830
|
)
|
|
|
42,771
|
|
|
|
106,092
|
|
|
|
79,649
|
|
|
|
30,760
|
|
|
Total assets
|
|
|
46,174
|
|
|
|
84,635
|
|
|
|
149,125
|
|
|
|
105,766
|
|
|
|
47,405
|
|
|
Noncurrent portion of equipment financing obligations
|
|
|
—
|
|
|
|
2,515
|
|
|
|
6,317
|
|
|
|
5,865
|
|
|
|
5,155
|
|
|
Convertible preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
107,194
|
|
|
Deficit accumulated during development stage
|
|
|
(264,623
|
)
|
|
|
(222,545
|
)
|
|
|
(164,095
|
)
|
|
|
(118,976
|
)
|
|
|
(77,170
|
)
|
|
Total stockholders’ (deficit) equity
|
|
|
(7,126
|
)
|
|
|
39,054
|
|
|
|
99,943
|
|
|
|
84,517
|
|
|
|
(74,385
|
)
|
|
|
|
Item 7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
The following Management’s Discussion and Analysis of
Financial Condition and Results of Operations contains
forward-looking statements that are based upon current
expectations. In some cases, you can identify forward-looking
statements by terminology such as “may,”
“will,” “should,” “expect,”
“plan,” “anticipate,” “believe,”
“estimate,” “predict,” “intend,”
“potential” or “continue” or the negative of
these terms or other comparable terminology. Forward-looking
statements involve risks and uncertainties. Our actual results
and the timing of events could differ materially from those
discussed in our forward-looking statements as a result of many
factors, including those set forth under “Risk
Factors” and elsewhere in this Annual Report on
Form 10-K.
Overview
We are a pharmaceutical company focused on the research,
development, and commercialization of novel proprietary products
for the acute treatment of central nervous system, or CNS,
conditions. All of our product candidates are based on our
proprietary technology, the Staccato system. The
Staccato system vaporizes an excipient-free drug to form
a condensation aerosol that, when inhaled, allows for rapid
systemic drug delivery. Because of the particle size of the
aerosol, the drug is quickly absorbed through the deep lung into
the bloodstream, providing speed of therapeutic onset that is
comparable to intravenous, or IV, administration but with
greater ease, patient comfort and convenience. In December 2009,
we submitted our first New Drug Application, or NDA, to the
U.S. Food and Drug Administration, or FDA, for our lead
product candidate, AZ-004. In February 2010, we licensed the
U.S. and Canadian commercialization rights to AZ-004 to
Biovail Laboratories International SRL. We plan to seek
additional commercial partners for AZ-004 outside of the
U.S. and Canada.
We have five other product candidates in various stages of
clinical development, ranging from Phase 1 through late-stage
Phase 2. In January 2009 we reduced, and in some cases
suspended, the development of these product
46
candidates in order to concentrate our efforts on the clinical,
regulatory, manufacturing and commercial development of our lead
product candidate. During the first half of 2010, we expect to
conduct a review of our product candidate portfolio. In the
second half of 2010, we plan to advance the development of at
least one of these product candidates. We are seeking partners
to support continued development of these product candidates,
but may develop one or more of these product candidates without
partner support.
Our clinical-stage product candidates are:
|
|
|
| |
•
|
AZ-004 (Staccato loxapine). We are developing
AZ-004 for the rapid treatment of agitation in patients with
schizophrenia or bipolar disorder. In December 2009, we
submitted our NDA to the FDA. In February 2010, the FDA accepted
our filing and provided us a Prescription Drug User Fee Act
(PDUFA) goal date of October 11, 2010. We believe that the
data generated from our clinical and non-clinical studies (and
contained within our NDA submission) adequately demonstrate the
efficacy and safety of AZ-004 for the rapid treatment of
agitation in patients with schizophrenia or bipolar disorder.
|
|
|
|
| |
|
In February 2010, we entered into a collaboration and license
agreement, or license agreement, and a manufacture and supply
agreement, collectively, the collaboration, with Biovail
Laboratories International SRL, or Biovail, for AZ-004
(Staccato®
loxapine) for the treatment of psychiatric
and/or
neurological indications and the symptoms associated with these
indications, including the initial indication of treating
agitation in schizophrenia and bipolar disorder patients. The
collaboration contemplates that we will be the exclusive
supplier of drug product for clinical and commercial uses and
have responsibility for the NDA for AZ-004 for the initial
indication of rapid treatment of agitation in patients with
schizophrenia or bipolar disorder, as well as responsibility for
any additional development and regulatory activities required
for use in these two patient populations in the outpatient
setting. Biovail will be responsible for commercialization for
the initial indication and, if it elects, development and
commercialization of additional indications for AZ-004 in the
U.S. and Canada.
|
| |
| |
|
Under the terms of the license agreement, Biovail paid us an
upfront fee of $40 million, and we may be eligible to
receive up to an additional $90 million in milestone
payments upon achievement of predetermined regulatory, clinical
and commercial manufacturing milestones. We may be subject to
certain payment obligation to Biovail, up to $5 million, if
we do not meet certain other milestones prior to a termination
of the license agreement. We are also eligible to receive tiered
royalty payments of 10% to 25% on any net sales of AZ-004. We
are responsible for conducting and funding all development and
regulatory activities associated with AZ-004’s initial
indication for the rapid treatment of agitation in patients with
schizophrenia or bipolar disorder as well as for its possible
use in the outpatient setting in these two patient populations.
Our obligation to fund the outpatient development efforts is
limited to a specified amount, none of which is expected to be
incurred in 2010. Biovail is responsible for certain Phase 4
development commitments and related costs and expenses. For
additional indications, we have an obligation regarding certain
efforts and related costs and expenses, up to a specified
amount, and, if it elects, Biovail is responsible for all other
development commitments and related costs and expenses.
|
| |
| |
|
Under the terms of the manufacture and supply agreement, we are
the exclusive supplier of AZ-004 and have responsibility for the
manufacture, packaging, labeling and supply for clinical and
commercial uses. Biovail will purchase AZ-004 from us at
predetermined transfer prices. The transfer prices depend on the
volume of AZ-004 purchases, subject to certain adjustments.
|
| |
| |
|
Either party may terminate the collaboration for the other
party’s uncured material breach or bankruptcy. In addition,
Biovail has the right to terminate the collaboration
(a) upon 90 days written notice for convenience;
(b) upon 90 days written notice if FDA does not
approve the AZ-004 NDA for the initial indication for the rapid
treatment of agitation in patients with schizophrenia or bipolar
disorder; (c) immediately upon written notice for safety
reasons or withdrawal of marketing approval; (d) upon
90 days written notice upon certain recalls of the product;
or (e) immediately upon written notice within 60 days
of termination of the supply agreement under certain
circumstances. The supply agreement automatically terminates
upon the termination of the license agreement.
|
47
|
|
|
| |
•
|
AZ-007 (Staccato zaleplon). We are
developing AZ-007 for the treatment of insomnia in patients who
have difficulty falling asleep, including patients who awake in
the middle of the night and have difficulty falling back asleep.
AZ-007 has completed Phase 1 testing. In the Phase 1 study,
AZ-007 delivered an IV-like pharmacokinetic profile with a
median time to peak drug concentration of 1.6 minutes.
Pharmacodynamics, measured as sedation assessed on a 100 mm
visual-analog scale, showed onset of effect as early as 2
minutes after dosing.
|
| |
| |
•
|
AZ-001 (Staccato prochlorperazine). We are
developing AZ-001 to treat patients suffering from acute
migraine headaches. During the third quarter of 2008, we
conducted an
end-of-Phase
2 meeting with the FDA. We believe we have a clear understanding
of the development requirements for filing an NDA for this
product candidate.
|
| |
| |
•
|
AZ-104 (Staccato loxapine,
low-dose). We are developing AZ-104 to treat
patients suffering from acute migraine headaches.
|
| |
| |
•
|
AZ-002 (Staccato alprazolam). AZ-002 has
completed a Phase 1 clinical trial in healthy subjects and a
Phase 2a
proof-of-concept
clinical trial in panic disorder patients for the treatment of
panic attacks, an indication we are not planning to pursue.
However, given the safety profile, the successful and
reproducible delivery of alprazolam, and the IV-like
pharmacological effect demonstrated to date, we are assessing
AZ-002 for other possible indications and renewed clinical
development.
|
| |
| |
•
|
AZ-003 (Staccato fentanyl). We are
developing AZ-003 for the treatment of patients with acute pain,
including patients with breakthrough cancer pain and
postoperative patients with acute pain episodes. We have
completed and announced positive results from a Phase 1 clinical
trial of AZ-003 in opioid-naïve healthy subjects.
|
In December 2006, we entered into a transaction involving a
series of related agreements providing for the financing of
additional clinical and nonclinical development of AZ-002,
Staccato alprazolam, and AZ-004/AZ-104, Staccato
loxapine. Pursuant to the agreements, Symphony Capital LLC
and other investors, which we refer to collectively as the
Allegro Investors, invested $50 million to
form Symphony Allegro, Inc., or Symphony Allegro, to fund
additional clinical and nonclinical development of Staccato
alprazolam and Staccato loxapine. We exclusively
licensed to Symphony Allegro certain intellectual property
rights related to Staccato alprazolam and Staccato
loxapine. We retained manufacturing rights to these product
candidates. In August 2009, we completed the acquisition of
Symphony Allegro through the exercise of an option to acquire
all of the outstanding equity of Symphony Allegro, as amended in
June 2009. In exchange for all of the outstanding shares of
Symphony Allegro, we: (i) issued to the Allegro investors
10 million shares of common stock, (ii) issued to the
Allegro investors five-year warrants to purchase 5 million
shares of common stock at an exercise price of $2.26 per share
and canceled the previously outstanding warrants to purchase
2 million shares of common stock held by the Allegro
investors, and (iii) agreed to pay certain percentages of
cash payments that may be generated from future partnering
transactions for AZ-004, AZ-104
and/or
AZ-002, the product candidates that were licensed to Symphony
Allegro. In February 2010, we paid Symphony $7.5 million of
the total proceeds that were received from Biovail pursuant to
the license and supply agreement. In addition, Symphony will be
entitled to receive a portion of future milestone and royalty
payments we may receive from Biovail pursuant to this agreement.
Other than those licensed to Biovail, we have retained all
rights to our product candidates and the Staccato system.
We eventually plan to build a United States-based specialty
sales force to commercialize our product candidates which are
approved for marketing and which are intended for specialty
pharmaceutical markets. We plan to enter into strategic
partnerships with other companies to commercialize products that
are intended for certain markets in the United States and for
all of our product candidates in geographic territories outside
the United States.
We were incorporated December 19, 2000. We have
funded our operations primarily through the sale of equity
securities, capital lease and equipment financings and
government grants. We have generated $6.9 million in
revenues from inception through December 31, 2009, through
United States Small Business Innovation Research grants and drug
compound feasibility studies and $10 million from the
license and development agreement with Endo. Prior to 2007, we
recognized governmental grant revenue and drug compound
feasibility revenue, however, we expect no grant revenue or drug
compound feasibility screening revenue in 2010. In January 2009,
we and Endo
48
mutually terminated the license agreement, at which time we
fulfilled our obligations under the agreement, and we recognized
the remaining $9.5 million of deferred revenues into
revenues in the first quarter of 2009. We do not expect any
material product revenue until at least 2011.
On October 5, 2009, we issued a total of
8,107,012 shares of our common stock and warrants to
purchase up to an additional 7,296,312 shares of our common
stock in a private placement. These securities were sold as
units with each unit consisting of one share of common stock and
a warrant to purchase 0.9 shares of common stock at a
purchase price of $2.4325 per unit. The net proceeds, after
deducting the payment of a placement agent fee, and other
offering expenses, were approximately $19.0 million. The
warrants issued are cash or net exercisable for a period of
seven years from October 5, 2009 and have an exercise price
of $2.77 per share.
We have incurred significant losses since our inception. As of
December 31, 2009, our deficit accumulated during
development stage was $264.6 million and total
stockholders’ deficit was $7.1 million. We recognized
net losses of $56.1 million, $77.0 million,
$55.9 million and $309.7 million in 2009, 2008 and
2007, and the period from December 19, 2000 (Inception) to
December 31, 2009, respectively. In January 2009, we
consolidated our operations to primarily focus our efforts on
the continued rapid development of AZ-004. We expect our net
losses to continue, however we expect a decreases in operating
expenses in 2010 as compared to 2009 due to our decreased
clinical activity.
The process of conducting preclinical studies and clinical
trials necessary to obtain FDA approval is costly and time
consuming. We consider the development of our product candidates
to be crucial to our long term success. If we do not complete
development of our product candidates and obtain regulatory
approval to market one or more of these product candidates, we
may be forced to cease operations. The probability of success
for each product candidate may be impacted by numerous factors,
including preclinical data, clinical data, competition, device
development, manufacturing capability, regulatory approval and
commercial viability. Our strategy is to focus our resources on
AZ-004. In February 2010, the FDA accepted, for filing, the NDA
that we submitted in December 2009 for this product candidate.
We have announced that we are seeking partnerships to continue
development of our other programs. If in the future we enter
into additional partnerships, third parties could have control
over preclinical development or clinical trials for some of our
product candidates. Accordingly, the progress of such product
candidate would not be under our control. We cannot forecast
with any degree of certainty which of our product candidates, if
any, will be subject to any future partnerships or how such
arrangements would affect our development plans or capital
requirements.
As a result of the uncertainties discussed above, the
uncertainty associated with clinical trial enrollments, and the
risks inherent in the development process, we are unable to
determine the duration and completion costs of the current or
future clinical stages of our product candidates or when, or to
what extent, we will generate revenues from the
commercialization and sale of any of our product candidates.
Development timelines, probability of success and development
costs vary widely. While we are currently focused on developing
our product candidates, we anticipate that we and our partners,
will make determinations as to which programs to pursue and how
much funding to direct to each program on an ongoing basis in
response to the scientific and clinical success of each product
candidate, as well as an ongoing assessment as to the product
candidate’s commercial potential. We do not expect any of
our current product candidates to be commercially available
before 2011, if at all.
Critical
Accounting Estimates and Judgments
Our management’s discussion and analysis of our financial
condition and results of operations is based on our financial
statements, which have been prepared in accordance with
U.S. generally accepted accounting principles. The
preparation of these financial statements requires us to make
estimates and judgments that affect the reported amounts of
assets and liabilities and the disclosure of contingent assets
and liabilities at the date of the financial statements, as well
as reported revenues and expenses during the reporting periods.
On an ongoing basis, we evaluate our estimates and judgments
related to development costs. We base our estimates on
historical experience and on various other factors that we
believe are reasonable under the circumstances, the results of
which form the basis for making assumptions about the carrying
value of assets and liabilities that are not readily apparent
from other sources. Actual results may differ from these
estimates under different assumptions or conditions.
49
While our significant accounting policies are more fully
described in Note 3 of the notes to consolidated financial
statements, we believe the following accounting policies are
critical to the process of making significant estimates and
judgments in preparation of our financial statements.
Preclinical
Study and Clinical Trial Accruals
We estimate our preclinical study and clinical trial expenses
based on our estimates of the services received pursuant to
contracts with multiple research institutions and clinical
research organizations that conduct and manage preclinical
studies and clinical trials on our behalf. The financial terms
of these agreements vary from contract to contract and may
result in uneven payment flows. Preclinical study and clinical
trial expenses include the following:
|
|
|
| |
•
|
fees paid to contract research organizations in connection with
preclinical studies;
|
| |
| |
•
|
fees paid to contract research organizations and other clinical
sites in connection with clinical trials; and
|
| |
| |
•
|
fees paid to contract manufacturers in connection with the
production of components and drug materials for preclinical
studies and clinical trials.
|
We record accruals for these preclinical study and clinical
trial costs based upon the estimated amount of work completed.
All such costs are charged to research and development expenses
based on these estimates. Costs related to patient enrollment in
clinical trials are accrued as patients are entered in the
trial. We monitor patient enrollment levels and related
activities to the extent possible through internal reviews,
correspondence and discussions with research institutions and
organizations. However, if we have incomplete or inaccurate
information, we may underestimate or overestimate activity
levels associated with various preclinical studies and clinical
trials at a given point in time. In this event, we could record
significant research and development expenses in future periods
when the actual activity level becomes known. To date, we have
not made any material adjustments to our estimates of
preclinical study and clinical trial costs. We make good faith
estimates which we believe to be accurate, but the actual costs
and timing of clinical trials are highly uncertain, subject to
risk and may change depending upon a number of factors,
including our clinical development plan.
Share-Based
Compensation
Our share-based compensation expense
includes: (a) compensation cost for share-based
payments granted prior to, but not yet vested as of
December 31, 2005 related to (i) employees, based on
the awards grant date intrinsic value, and
(ii) non-employees using the awards fair value, and
(b) compensation cost for all share-based payments granted
or modified subsequent to December 31, 2005, based on the
awards grant-date fair value.
We currently use the Black-Scholes option pricing model to
determine the fair value of stock options and purchase rights
issued under the employee stock purchase plan. The determination
of the fair value of share-based payment awards on the date of
grant using an option-pricing model is affected by our stock
price as well as assumptions regarding a number of complex and
subjective variables. These variables include our expected stock
price volatility over the term of the awards, actual and
projected employee stock option exercise behaviors, risk-free
interest rates and expected dividends.
The estimated fair value of restricted stock unit awards is
calculated based on the market price of our common stock on the
date of grant, reduced by the present value of dividends
expected to be paid on our common stock prior to vesting of the
restricted stock unit. Our current estimate assumes no dividends
will be paid prior to the vesting of the restricted stock unit.
We estimate the expected term of options based on the historical
term periods of options that have been granted but are no longer
outstanding and the estimated terms of outstanding options. We
estimate the volatility of our stock based on our actual
historical volatility since our initial public offering. We base
the risk-free interest rate that we use in the option pricing
model on U.S. Treasury zero-coupon issues with remaining
terms similar to the expected term on the options. We do not
anticipate paying any cash dividends in the foreseeable future
and therefore use an expected dividend yield of zero in the
option pricing model.
50
We are required to estimate forfeitures at the time of grant and
revise those estimates in subsequent periods if actual
forfeitures differ from those estimates. We use historical data
to estimate pre-vesting option forfeitures and record
share-based compensation expense only for those awards that are
expected to vest. All share-based payment awards are amortized
on a straight-line basis over the requisite service periods of
the awards, which are generally the vesting periods.
If factors change and we employ different assumptions for
estimating share-based compensation expense in future periods or
if we decide to use a different valuation model, the expenses in
future periods may differ significantly from what we have
recorded in the current period and could materially affect our
operating loss, net loss and net loss per share.
Symphony
Allegro, Inc.
On December 1, 2006 we entered into a transaction involving
a series of related agreements with Symphony Capital LLC, or
Symphony Capital, Symphony Allegro Holdings LLC, or Holdings,
and Holdings’ wholly owned subsidiary Symphony Allegro, to
fund the clinical development of AZ-002, Staccato
alprazolam, and AZ-004/104, Staccato loxapine, or the
programs. Symphony Capital and other investors, which we refer
to collectively as the Allegro Investors, invested
$50 million in Holdings, which then invested the
$50 million in Symphony Allegro. Pursuant to the
agreements, Symphony Allegro agreed to invest up to the full
$50 million to fund the clinical development of the
programs, and we licensed to Symphony Allegro certain
intellectual property rights related to these programs. We
retained manufacturing rights to these product candidates.
Pursuant to the agreements, we continued to be primarily
responsible for all preclinical, clinical and device development
efforts as well as maintenance of the intellectual property
portfolio for the programs. We and Symphony Allegro had
established a development committee to oversee the programs. We
participated in the development committee and had the right to
appoint one of the five board of director seats of Symphony
Allegro. Pursuant to the agreements, we had received an
exclusive purchase option, or the purchase option, that gave us
the right, but not the obligation, to acquire all, but not less
than all, of the outstanding equity of Symphony Allegro, and
reacquire the intellectual property rights that we licensed to
Symphony Allegro at certain fixed prices. In consideration for
the purchase option, we issued to Holdings a five-year warrant
to purchase 2,000,000 shares of our common stock at $9.91
per share and paid $2.85 million for structuring fees and
related expenses to Symphony Capital.
Prior to the acquisition of all of the outstanding equity of
Symphony Allegro pursuant to the amended purchase option on
August 26, 2009, as described below, we had concluded that
Symphony Allegro was by design a Variable Interest Entity, or
VIE,. because we had a purchase option to acquire its
outstanding voting stock at prices that were fixed based upon
the date the option is exercised. The fixed nature of the
purchase option price limited the returns of the Allegro
Investors, as the investors in Symphony Allegro. Parties to an
arrangement are deemed to be de facto agents if they cannot
sell, transfer, or encumber their interests without the prior
approval of an enterprise. Symphony Capital was considered to be
a de facto agent of ours pursuant to this provision, and because
we and the Allegro Investors, as a related party group, absorbed
a majority of Symphony Allegro’s variability, we evaluated
whether we are most closely associated with Symphony Allegro. We
concluded that we were most closely associated with Symphony
Allegro and should consolidate Symphony Allegro because
(i) we originally developed the technology that was
assigned to Symphony Allegro, (ii) we continued to oversee
and monitor the development program, (iii) our employees
continued to perform substantially all of the development work,
(iv) we significantly influenced the design of the
responsibilities and corporate structure of Symphony Allegro,
(v) Symphony Allegro’s operations were substantially
similar to our activities, and (vi) through the purchase
option, we had the ability to meaningfully participate in the
benefits of a successful development effort.
The Allegro Investors were required to absorb the development
risk for their equity investment in Symphony Allegro. The
Allegro Investors’ equity investment in Symphony Allegro
was classified as noncontrolling interest in our consolidated
balance sheets. The noncontrolling interest held by the Allegro
Investors was reduced by the $10.7 million fair value of
the warrants they received in consideration for the purchase
option and $2.85 million of fees we immediately paid to
Symphony Capital upon the transaction’s closing because the
total consideration provided by us to the Allegro Investors
effectively reduced the Allegro Investors’ at-risk equity
investment in Symphony Allegro. While we performed the research
and development on behalf of Symphony Allegro, our development
risk is limited to the consideration we provided to the Allegro
Investors (the warrants and fees).
51
Net losses incurred by Symphony Allegro and charged to the
noncontrolling interest were $14.0 million,
$18.6 million and $10.8 million for the years ended
December 31, 2009, 2008 and 2007, respectively. We ceased
to charge net losses incurred by Symphony Allegro against the
noncontrolling interest upon our acquisition of Symphony Allegro
on August 26, 2009.
In December 2007, the FASB issued new guidance that required:
(i) noncontrolling interests in subsidiaries be reported as
a component of stockholders’ equity in the consolidated
balance sheet, (ii) noncontrolling interests continue to be
attributed its share of losses even if that attribution results
in a deficit noncontrolling interest balance, (iii) that
earnings or losses attributed to the noncontrolling interests be
reported as part of consolidated earnings and not as a separate
component of income or expense, and (iv) disclosure of the
attribution of consolidated earnings to the controlling and
noncontrolling interests on the face of the consolidated
statement of operations. On January 1, 2009, we adopted
these provisions. Had the previous requirements been applied,
the net loss attributable to noncontrolling interests in
Symphony Allegro would have decreased by $8.6 million
during the year ended December 31, 2009.
In June 2009, we entered into an agreement with Holdings to
modify the provisions of and to exercise the purchase option. We
completed the acquisition of all of the outstanding equity of
Symphony Allegro pursuant to the amended purchase option on
August 26, 2009. In exchange for all of the outstanding
equity of Symphony Allegro, we: (i) issued to the Allegro
Investors 10 million shares of common stock,
(ii) issued to the Allegro Investors 5 year warrants
to purchase 5 million shares of common stock with an
exercise price of $2.26 per share, and (iii) will pay
Holdings certain percentages of cash payments that may be
generated from future partnering transactions for the programs.
The outstanding warrants to purchase 2 million shares of
common stock held by the Allegro Investors were cancelled.
We recorded the acquisition of all of the outstanding equity of
Symphony Allegro pursuant to the amended purchase option as a
return of equity to the noncontrolling interest. The acquisition
was accounted for as a capital transaction that did not affect
our net loss. However, because the acquisition was accounted for
as a capital transaction, the excess consideration transferred
over the carrying value of the noncontrolling interest in
Symphony Allegro was treated as a deemed dividend for purposes
of reporting net loss per share, increasing net loss per share
attributable to Alexza stockholders during the year ended
December 31, 2009.
The following table outlines the estimated fair value of
consideration transferred by us and the computation of the
excess consideration transferred over the carrying value of the
noncontrolling interest in Symphony Allegro (in thousands):
| |
|
|
|
|
|
Description
|
|
Fair Value
|
|
|
|
|
Fair value of consideration transferred:
|
|
|
|
|
|
10,000,000 shares of Alexza common stock
|
|
$
|
28,000
|
|
|
Warrant consideration, net
|
|
|
8,085
|
|
|
Contingent cash payments to Symphony Allegro stockholders
|
|
|
16,855
|
|
|
|
|
|
|
|
|
Total consideration transferred
|
|
|
52,940
|
|
|
Add: Deficit of noncontrolling interest in Symphony Allegro
|
|
|
8,626
|
|
|
|
|
|
|
|
|
Excess consideration transferred over the carrying value of the
noncontrolling interest in Allegro
|
|
$
|
61,566
|
|
|
|
|
|
|
|
The fair value of the Alexza common stock was based on the
closing sales price of our common stock on the NASDAQ Global
Market on August 26, 2009, the date the transaction was
completed. The estimated fair values of the warrant
consideration were calculated using the Black-Scholes valuation
model.
We estimated the fair value of the liability associated with the
contingent cash payments to the Symphony Allegro stockholders,
or contingent consideration liability, using a
probability-weighted discounted cash flow model. We derived
multiple cash flow scenarios for each of the product candidates
subject to the cash payments and applied a probability to each
of the scenarios. These cash flows were then discounted at an
18% rate.
52
Changes in the fair value of the contingent consideration
liability subsequent to the August 26, 2009 acquisition
date are recognized in earnings in the period of the change.
Certain events including, but not limited to, clinical trial
results, FDA approval or rejection of its submissions, such as
our NDA filed in December 2009, the timing and terms of a
strategic partnership, the commercial success of the programs,
and the discount rate used could have a material impact on the
fair value of the contingent consideration liability, and as a
result, our results of operations.
Revenue
Recognition
We recognize revenue in accordance with the SEC Staff Accounting
Bulletin (SAB) No. 101, Revenue Recognition in Financial
Statements, or SAB 101, as amended by Staff Accounting
Bulletin No. 104, Revision of Topic 13. or
SAB 104.
In determining the accounting for collaboration agreements, we
determine whether an arrangement involves multiple
revenue-generating deliverables that should be accounted for as
a single unit of accounting or divided into separate units of
accounting for revenue recognition purposes and, if this
division is required, how the arrangement consideration should
be allocated among the separate units of accounting. If the
arrangement represents a single unit of accounting, the revenue
recognition policy and the performance obligation period must be
determined, if not already contractually defined, for the entire
arrangement. If the arrangement represents separate units of
accounting, a revenue recognition policy must be determined for
each unit.
Revenues for non-refundable upfront license fee payments, where
we continue to have obligations, will be recognized as
performance occurs and obligations are completed.
Results
of Operations
Comparison
of Years Ended December 31, 2009 and 2008
Revenue. We had $9,514,000 and $486,000 of
revenues in 2009 and 2008, respectively. In the third quarter of
2008, we began to recognize revenues related to our Endo license
agreement. In January 2009, we mutually agreed with Endo to
terminate the license agreement, at which time we fulfilled our
obligations under the license agreement and recognized the
remaining $9.5 million of deferred revenues into revenues
in 2009.
Operating
Expenses
Our operating expenses were affected by our prospective method
of adoption fair value accounting for employee share-based
compensation. As a result, we believe reviewing our operating
expenses both inclusive and exclusive of share-based
compensation provides a better understanding of the growth of
our operations. The impact of share-based compensation on
operating expenses is outlined as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Non share-based compensation expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
36,335
|
|
|
$
|
58,639
|
|
|
$
|
43,760
|
|
|
General and administrative
|
|
|
12,381
|
|
|
|
15,121
|
|
|
|
13,357
|
|
|
Restructuring charges
|
|
|
1,981
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total non share-based compensation expenses
|
|
|
50,697
|
|
|
|
73,760
|
|
|
|
57,117
|
|
|
Share-based compensation expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
3,443
|
|
|
|
2,926
|
|
|
|
1,885
|
|
|
General and administrative
|
|
|
3,025
|
|
|
|
2,520
|
|
|
|
1,531
|
|
|
Restructuring charges
|
|
|
56
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total share-based compensation expenses
|
|
|
6,524
|
|
|
|
5,446
|
|
|
|
3,416
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
$
|
57,221
|
|
|
$
|
79,206
|
|
|
$
|
60,533
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
53
Research and Development Expenses. Research
and development expenses consist of costs associated with
research activities, as well as costs associated with our
product development efforts, conducting preclinical studies and
clinical trials and manufacturing development efforts. All
research and development costs, including those funded by third
parties, are expensed as incurred. Research and development
expenses include:
|
|
|
| |
•
|
external research and development expenses incurred under
agreements with third party contract research organizations and
investigational sites where a substantial portion of our
preclinical studies and all of our clinical trials are conducted;
|
| |
| |
•
|
third party supplier, consultant and employee related expenses,
which include salary and benefits; and
|
| |
| |
•
|
facilities, depreciation and other allocated expenses, which
include direct and allocated expenses for rent and maintenance
of facilities, depreciation of leasehold improvements and
equipment and laboratory and other supplies.
|
The table below sets forth our research and development expenses
for 2009, 2008 and 2007 and cumulative expenses for each of our
lead product candidates based on our internal records and
estimated allocations of employee time and related expenses:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
From
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 19,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Inception)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Through
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
Preclinical and Clinical Development:
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2009.
|
|
|
|
|
AZ-004/104
|
|
$
|
30,084
|
|
|
$
|
26,789
|
|
|
$
|
15,524
|
|
|
$
|
81,776
|
|
|
AZ-003
|
|
|
1,631
|
|
|
|
17,070
|
|
|
|
1,474
|
|
|
|
31,525
|
|
|
AZ-001
|
|
|
—
|
|
|
|
1,151
|
|
|
|
8,163
|
|
|
|
39,372
|
|
|
AZ-002
|
|
|
181
|
|
|
|
1,898
|
|
|
|
3,795
|
|
|
|
15,191
|
|
|
AZ-007
|
|
|
—
|
|
|
|
1,773
|
|
|
|
8,214
|
|
|
|
12,371
|
|
|
Other preclinical programs
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,243
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total preclinical and clinical development
|
|
|
31,896
|
|
|
|
48,681
|
|
|
|
37,170
|
|
|
|
183,478
|
|
|
Research
|
|
|
7,882
|
|
|
|
12,884
|
|
|
|
8,475
|
|
|
|
60,983
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total research and development
|
|
$
|
39,778
|
|
|
$
|
61,565
|
|
|
$
|
45,645
|
|
|
$
|
244,461
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and Development Expenses. Research
and development expenses decreased 35% to $39.8 million in
2009 from $61.6 million in 2008. The decreases were due
primarily to:
|
|
|
| |
•
|
decreased spending on our AZ-003 product candidate in connection
with the termination of the license agreement with Endo in
January 2009; and
|
| |
| |
•
|
the suspension of the development of our AZ-001, AZ-002 and
AZ-007 product candidates and decreased spending on basic
research in connection with our decision to focus our resources
on AZ-004.
|
These decreases were partially offset by:
|
|
|
| |
•
|
increased spending on our AZ-004/104 product candidates as we
continued our development of these product candidates under the
Symphony Allegro agreement, including our efforts to support an
NDA filing for AZ-004, which was filed in December 2009, and the
AZ-104 Phase 2b clinical trial which initiated in late 2008 and
completed in 2009.
|
We expect that research and development expenses will decrease
in 2010 as we expect lower clinical expenses for AZ-004/AZ-104,
a result of our completing our clinical studies to support the
NDA for AZ-004 and the completion of the Phase 2a clinical study
for AZ-104 in 2009. We also expect lower employee related costs
in 2010, a result of our headcount reduction in the first
quarter of 2009. We also expect our expenses for AZ-001, AZ-002,
AZ-003, and AZ-104 to be lower in 2010 as we do not intend to
continue development of these programs unless we can partner the
programs.
54
General and Administrative Expenses. General
and administrative expenses consist principally of salaries and
related costs for personnel in executive, finance, accounting,
business development, legal and human resources functions. Other
general and administrative expenses include facility and
information technology costs not otherwise included in research
and development expenses, patent related costs and professional
fees for legal, consulting and accounting services.
The decreases in general and administrative expenses were
primarily due to decreased headcount expenses as a result of our
restructuring in January 2009, reduced facility expenses as we
completed our move to our Mountain View facility in the first
half of 2008, and our efforts to reduce third party costs to
conserve cash balances. We expect our general and administrative
expenses in 2010 to remain relatively consistent with 2009
levels.
Restructuring Charges In January 2009, we restructured
our operations to focus our efforts on the continued rapid
development of our AZ-004 (Staccato loxapine) product
candidate. The restructuring included a workforce reduction of
50 employees, representing approximately 33% of our total
workforce and was completed in the second quarter of 2009. We
incurred restructuring expenses related to employee severance
and other termination benefits of $2.0 million, including a
non-cash charge related to modifications to share-based awards
of $56,000. As of December 31, 2009, we have made all of
our restructuring related payments.
Interest and Other Income, Net. Interest and
other income, net, primarily represents income earned on our
cash, cash equivalents, marketable securities balances, and
prior to August 26, 2009, marketable securities held by
Symphony Allegro. Interest and other income, net was $92,000 for
2009 and $2.6 million for 2008. The decrease was primarily
due to lower average cash, cash equivalent and marketable
securities balances and lower interest rates earned on such
balances. We expect to continue to earn low interest income
returns on our cash, cash equivalent and marketable securities
balances.
Interest Expense. Interest expense represents
interest on our equipment loans and was $467,000 in 2009 and
$935,000 in 2008. The decrease was due to decreases in the
outstanding balances of our equipment loan borrowings as we made
no additional borrowings in 2008 or 2009.
Change in the Fair Value of Contingent Consideration
Liability. In connection with our acquisition of
all of the outstanding equity of Symphony Allegro, we are
obligated to pay the Symphony Investors certain percentages of
cash payments that may be generated from future partnering
transactions for AZ-002, AZ-004
and/or
AZ-104. We measure the fair value of this contingent
consideration liability at each balance sheet date. Any changes
in the fair value of this contingent consideration liability
will be recognized in earnings in the period of the change.
Certain events including, but not limited to, clinical trial
results, FDA approval or disapproval of our submissions, such as
our NDA filed in December 2009, the timing and terms of
strategic partnerships, such as our agreement with Biovail
executed in February 2010, the commercial success of AZ-002,
AZ-004
and/or
AZ-104, and the discount rate assumption could have a material
impact on the fair value of the contingent liability, and as a
result, our results of operations.
In the third quarter of 2009, we announced preliminary results
from our Phase 2b clinical trial of AZ-104, where AZ-104 did not
meet the primary endpoint of the study. This change resulted in
a decrease in the expected cash flow resulting in a decrease in
the contingent consideration liability. In the fourth quarter of
2009, we modified our assumptions regarding the probability of
certain cash flow outcomes to reflect the negotiations with
Biovail to partner AZ-004 as well as the filing of our NDA. The
reduction in these uncertainties resulted in an increase in
probability of certain expected cash flow resulting in an
increase in the contingent consideration liability. These items
combined resulted in our incurring a loss on the change in fair
value of the contingent consideration liability of
$8.0 million during the year ended December 31, 2009.
Loss Attributed to Noncontrolling Interest in Symphony
Allegro. Prior to our purchase of Symphony
Allegro on August 26, 2009, pursuant to the agreements that
we entered into with Symphony Allegro in December 2006, we
consolidated Symphony Allegro’s financial condition and
results of operations. Accordingly, we deducted the losses
attributable to the noncontrolling interest from our net loss in
the consolidated statement of operations, and we reduced the
noncontrolling interest holders’ ownership interest in
Symphony Allegro in the consolidated balance sheet by the loss
attributed to the noncontrolling interests in Symphony Allegro.
The losses attributed to the noncontrolling interest holders was
$14.0 million in 2009 and $18.6 million in 2008. The
decrease was primarily
55
due to a full year of Symphony Allegro’s losses being
attributed to the noncontrolling interest in 2008 as compared to
approximately 8 months in 2009 as a result of our
acquisition of all of the outstanding equity of Symphony Allegro
in August 2009.
Comparison
of Years Ended December 31, 2008 and 2007
Revenue. We had $486,000 of revenues in 2008
and no revenues in 2007. In the third quarter of 2008, we began
to recognize revenues related to our Endo license agreement.
Research and Development Expenses. Research
and development expenses increased 35% to $61.6 million in
2008 from $45.6 million in 2007. The increases were due
primarily to:
|
|
|
| |
•
|
increased spending on our AZ-004/104 product candidates as we
continued development of these product candidates under the
Symphony Allegro agreement, including our first Phase 3 clinical
trial of AZ-004 which began enrollment in February 2008 and
completed enrollment in June 2008 and our second Phase 3
clinical trial of AZ-004 which began enrollment in July 2008 and
completed enrollment in October 2008,
|
| |
| |
•
|
increased spending on our AZ-003 product candidate as we
continued development of this product candidate under the Endo
agreement, and
|
| |
| |
•
|
increased research expenses as we increased our device
development and manufacturing process
scale-up
efforts.
|
These increases were partially offset by decreased spending on:
|
|
|
| |
•
|
our AZ-001 product candidate due to Phase 2b clinical trial
efforts and ongoing non clinical efforts occurring in 2007,
|
| |
| |
•
|
our AZ-002 product candidate due to higher development and
manufacturing efforts to modify the AZ-002 device and
manufacture clinical trial materials for the Phase 2a trial in
2007, and
|
| |
| |
•
|
our AZ-007 product candidate due to the preclinical and
regulatory efforts in 2007 to support and prepare the IND filing
that occurred in the fourth quarter of 2007.
|
General and Administrative Expenses General
and administrative expenses increased 18% to $17.6 million
in 2008 from $14.9 million in 2007. The increases were
primarily due to increased staffing to manage and support our
growth resulting in increased payroll and related expenses,
increased third party intellectual property expenses as we
continued to increase and maintain our intellectual property
portfolio, and higher facilities expenses to support our growth.
Interest and Other Income, Net. Interest and
other income, net, primarily represents income earned on our
cash, cash equivalents, marketable securities balances, and
marketable securities held by Symphony Allegro. Interest and
other income, net was $2.6 million for 2008 and
$5.6 million for 2007. The decrease was primarily due to
lower average cash, cash equivalent and marketable securities
balances and lower interest rates earned on such balances.
Interest Expense. Interest expense represents
interest on our equipment loans and was $0.9 million in
2008 and $1.0 million in 2007. The decrease was primarily
due to decreases in our equipment loan borrowings as we made no
additional borrowing under our equipment financing agreements in
2008.
Loss Attributed to Noncontrolling Interest in Symphony
Allegro. The losses attributed to the
noncontrolling interest holders was $18.6 million in 2008
and $10.8 million in 2007. The increase was primarily due
to increased spending on AZ-004, primarily the result of the two
Phase 3 clinical trials in 2008.
Liquidity
and Capital Resources
Since inception, we have financed our operations primarily
through private placements and public offerings of equity
securities receiving aggregate net proceeds from such sales
totaling $244.4 million, revenues primarily from a
licensing agreement and government grants totaling
$16.9 million, and payments from Symphony Allegro. We have
received additional funding from equipment financing
obligations, interest earned on investments, as
56
described below, and funds received upon exercises of stock
options and exercises of purchase rights under our Employee
Stock Purchase Plan. As of December 31, 2009, we had
$19.9 million in cash, cash equivalents and marketable
securities. Our cash and marketable security balances are held
in a variety of interest bearing instruments, including
obligations of United States government agencies, high credit
rating corporate borrowers and money market accounts. Cash in
excess of immediate requirements is invested with regard to
liquidity and capital preservation.
Net cash used in operating activities was $53.1 million,
$55.1 million, and $35.8 million in 2009, 2008 and
2007, respectively. The net cash used in each of these periods
primarily reflects net loss for these periods, offset in part by
depreciation, non-cash stock-based compensation, loss attributed
to noncontrolling interests, and non-cash changes in operating
assets and liabilities. In 2009, the decrease in deferred
revenue was related to the mutual termination of our license
agreement with Endo, at which time we recognized the remaining
$9.5 million of deferred revenue. The decreases in accounts
payables of $2.2 million and accrued clinical trial expense
and other accrued liabilities of $2.7 million was due to
the decrease in our operations. In 2008, the large decrease in
other receivables was due to the collection of a receivable of
$10.0 million from Endo in January 2008 related to the
license agreement signed in December 2007 and a
$2.1 million receivable related to the reimbursement of
leasehold improvements from the landlord of our Mountain View
facility in May 2008. In 2007, the large increase in other
receivables was affected by the above mentioned receivable from
Endo and the receivable relating to tenant improvements, which
were outstanding in 2007 and collected in 2008. In 2007, the
increase in other liabilities is primarily due to
$10.0 million of deferred revenues related to the Endo
license agreement, and $14.3 million of leasehold
improvement reimbursements from the Mountain View landlord
recorded as deferred rent in 2007.
Net cash provided by (used in) investing activities was
$20.1 million, $42.8 million, and $(20.0) million
in 2009, 2008 and 2007, respectively. Investing activities
consist primarily of purchases and maturities of marketable
securities and capital purchases. During 2009 and 2008 we had
maturities, net of purchases, of marketable securities of
$4.9 million and $27.2 million, respectively. During
2007 we purchased $11.4 million of marketable securities,
net of maturities. Maturities of marketable securities held by
Symphony Allegro, Inc. were $16.4 million,
18.1 million, and 10.5 million in 2009, 2008, and
2007, respectively. Purchases of property and equipment were
$1.2 million, $2.7 million, and $19.1 million in
2009, 2008 and 2007, respectively. In 2007, $16.5 million
of property and equipment purchases related to the leasehold
improvements made to our leased facility in Mountain View,
California.
Net cash provided by financing activities was
$20.4 million, $6.9 million, and $70.1 million in
2009, 2008 and 2007, respectively. Financing activities consist
primarily of proceeds from the sale of our common stock,
purchase of a noncontrolling interest, and equipment financing
arrangements. In 2009, 2008 and 2007, we received net proceeds
from the issuance of common stock of $19.7 million,
$11.2 million, and $67.8 million, respectively. In
2009 we had proceeds from the purchase of the noncontrolling
interest in Symphony Allegro, Inc. of $4.9 million. In 2009
and 2008, payments on equipment financing arrangements were
$4.1 million and $4.2 million, respectively. Proceeds
from equipment financing arrangements, net of payments, were
$2.3 million during 2007. There were no new borrowings
under any equipment financing arrangements in 2009 or 2008.
We believe that with current cash, cash equivalents and
marketable securities along with interest earned thereon, the
proceeds from option exercises, purchases of common stock
pursuant to our Employee Stock Purchase Plan, and the proceeds
received from our agreement with Biovail, we will be able to
maintain our currently planned operations through the first
quarter of 2011 and will extend into 2012 if we achieve the
eligible milestones under the Biovail agreement during the next
12 months. Changing circumstances may cause us to consume
capital significantly faster or slower than we currently
anticipate. We have based these estimates on assumptions that
may prove to be wrong, and we could utilize our available
financial resources sooner than we currently expect. The key
assumptions underlying these estimates include:
|
|
|
| |
•
|
achievement of the milestones in the Biovail agreement;
|
| |
| |
•
|
expenditures related to continued preclinical and clinical
development of our lead product candidates during this period
within budgeted levels;
|
57
|
|
|
| |
•
|
no unexpected costs related to the development of our
manufacturing capability; and
|
| |
| |
•
|
no growth in the number of our employees during this period.
|
Our forecast of the period of time that our financial resources
will be adequate to support operations is a forward-looking
statement and involves risks and uncertainties, and actual
results could vary as a result of a number of factors, including
the factors discussed in “Risk Factors.” In light of
the numerous risks and uncertainties associated with the
development and commercialization of our product candidates and
the extent to which we enter into strategic partnerships with
third parties to participate in their development and
commercialization, we are unable to estimate the amounts of
increased capital outlays and operating expenditures associated
with our current and anticipated clinical trials. Our future
funding requirements will depend on many factors, including:
|
|
|
| |
•
|
the scope, rate of progress, results and costs of our
preclinical studies, clinical trials and other research and
development activities;
|
| |
| |
•
|
the terms and timing of any distribution, strategic partnerships
or licensing agreements that we may establish;
|
| |
| |
•
|
the cost, timing and outcomes of regulatory approvals;
|
| |
| |
•
|
the number and characteristics of product candidates that we
pursue;
|
| |
| |
•
|
the cost and timing of establishing manufacturing, marketing and
sales capabilities;
|
| |
| |
•
|
the cost of establishing clinical and commercial supplies of our
product candidates;
|
| |
| |
•
|
the cost of preparing, filing, prosecuting, defending and
enforcing any patent claims and other intellectual property
rights; and
|
| |
| |
•
|
the extent to which we acquire or invest in businesses, products
or technologies, although we currently have no commitments or
agreements relating to any of these types of transactions.
|
We will need to raise additional funds to support our
operations, and such funding may not be available to us on
acceptable terms, or at all. If we are unable to raise
additional funds when needed, we may not be able to continue
development of our product candidates or we could be required to
delay, scale back or eliminate some or all of our development
programs, reduce our efforts to build our commercial
manufacturing capacity, and other operations. We may seek to
raise additional funds through public or private financing,
strategic partnerships or other arrangements. Any additional
equity financing may be dilutive to stockholders and debt
financing, if available, may involve restrictive covenants. If
we raise funds through collaborative or licensing arrangements,
we may be required to relinquish, on terms that are not
favorable to us, rights to some of our technologies or product
candidates that we would otherwise seek to develop or
commercialize ourselves. Our failure to raise capital when
needed may harm our business, financial condition, results of
operations, and prospects.
Contractual
Obligations
We lease two buildings with an aggregate of 106,894 square
feet of manufacturing, office and laboratory facilities in
Mountain View, California, which we began to occupy in the
fourth quarter of 2007. We currently occupy 87,560 square
feet of these facilities and sublease the remaining
19,334 square feet. Beginning March 1, 2010, we will
sublease an additional 20,956 square feet of these
facilities, reducing the space we occupy to 66,604 square
feet. The lease for both facilities expires on March 31,
2018, and we have two options to extend the lease for five years
each. Our sublease agreements expire on April 30, 2010 with
regards to 19,334 square feet and on February 28, 2014
with regards to 20,956 square feet. We believe that the
Mountain View facilities are sufficient for our office,
manufacturing and laboratory needs for at least the next three
years.
We have financed a portion of our equipment purchases through
various equipment financing agreements. Under the agreements,
equipment advances are to be repaid in 36 to 48 monthly
installments of principal and interest. The interest rate, which
is fixed for each draw, is based on the U.S. Treasuries of
comparable maturities and ranges from 9.2% to 10.6%. The
equipment purchased under the equipment financing agreement is
pledged as security.
58
On November 2, 2007, we entered into a manufacturing and
supply agreement, or the supply agreement, with Autoliv ASP,
Inc, or Autoliv, relating to the commercial supply of chemical
heat packages that can be incorporated into our Staccato
device. Autoliv had developed these chemical heat packages
for us pursuant to a development agreement executed in October
2005. Under the terms of the supply agreement, Autoliv will
develop a manufacturing line capable of producing
10 million chemical heat packages a year. We have an
obligation to pay Autoliv $12 million upon the earlier of
December 31, 2011 or 60 days after the approval by the
Food and Drug Administration of a new drug application filed by
us. If the agreement is terminated by either party, we will be
required to reimburse Autoliv up to $12 million for certain
expenses related to the equipment and tooling used in the
production and testing of the chemical heat packages. Upon
payment by us, Autoliv will be required to transfer possession
and ownership of such equipment and tooling to us. Each quarter,
with assistance from Autoliv, we estimate the amount of work
performed on the development of the manufacturing line and
recognize a portion of the total payment related to the
manufacturing line as a capital asset and a corresponding
non-current liability. Autoliv has also agreed to manufacture,
assemble and test the chemical heat packages solely for us in
conformance with our specifications. We will pay Autoliv a
specified purchase price, which varies based on annual
quantities ordered by us, per chemical heat package delivered.
The initial term of the supply agreement expires on
December 31, 2012 and may be extended by written mutual
consent. As of December 31, 2009, we recorded a fixed asset
and a current liability of $3,750,000, based on our PDUFA goal
date of October 11, 2010, related to our commitment to
Autoliv for the development of the manufacturing line.
Our future contractual payments, net of sublease income,
including interest at December 31, 2009 are as follows (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Period
|
|
|
|
|
|
|
|
Less Than
|
|
|
|
|
|
|
|
|
|
|
|
Contractual Obligations
|
|
Total
|
|
|
1 Year
|
|
|
1-3 Years
|
|
|
3-5 Years
|
|
|
Thereafter
|
|
|
|
|
(In thousands)
|
|
|
|
|
Equipment financing obligations
|
|
$
|
2,669
|
|
|
$
|
2,226
|
|
|
$
|
443
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Operating lease obligations
|
|
|
39,239
|
|
|
|
4,516
|
|
|
|
9,538
|
|
|
|
9,327
|
|
|
|
15,858
|
|
|
Autoliv payment
|
|
|
12,000
|
|
|
|
12,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
53,908
|
|
|
$
|
18,742
|
|
|
$
|
9,981
|
|
|
$
|
9,327
|
|
|
$
|
15,858
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Recently
Adopted Accounting Standards
Noncontrolling
interest
In December 2007, the Financial Accounting Standards Board
(FASB) issued new guidance which establishes accounting and
reporting standards for ownership interests in subsidiaries held
by parties other than the parent, the amount of consolidated net
income (loss) attributable to the parent and to the
noncontrolling interests, changes in a parent’s ownership
interest and the valuation of retained noncontrolling equity
investments when a subsidiary is deconsolidated. This guidance
requires that the noncontrolling interest continue to be
attributed its share of losses even if that attribution results
in a deficit noncontrolling interest balance. This guidance also
establishes additional reporting requirements that identify and
distinguish between the ownership interest of the parent and the
interest of the noncontrolling owners.
On January 1, 2009, we adopted this guidance and
reclassified the noncontrolling interest in Allegro from a
liability to stockholders’ equity on our Consolidated
Balance Sheets on a retrospective basis. Had the previous
requirements been applied, the net loss attributable to
noncontrolling interest would have decreased by $8,626,000
during the year ended December 31, 2009. In addition,
consolidated net loss has been adjusted to include the net loss
attributed to the noncontrolling interest in Allegro and
consolidated comprehensive income or loss has been adjusted to
include the comprehensive income or loss attributed to the
noncontrolling interest in Allegro.
59
Recently
Issued Accounting Standards
Revenue
Arrangements with Multiple Deliverables
In September 2009, the FASB ratified ASU
2010-13,
which eliminates the residual method of allocation and the
requirement to use the relative selling price method when
allocating revenue in a multiple deliverable arrangement. When
applying the relative selling price method, the selling price
for each deliverable shall be determined using vendor specific
objective evidence of selling price, if it exists, otherwise
third-party evidence of selling price. If neither vendor
specific objective evidence nor third-party evidence of selling
price exists for a deliverable, companies shall use its best
estimate of the selling price for that deliverable when applying
the relative selling price method. ASU
2010-13
shall be effective in fiscal years beginning on or after
June 15, 2010, with earlier application permitted.
Companies may elect to adopt this guidance prospectively for all
revenue arrangements entered into or materially modified after
the date of adoption, or retrospectively for all periods
presented. The Company is currently evaluating the potential
impact, if any, of the adoption of this guidance on its
financial position, results of operations and cash flows.
Off-Balance
Sheet Arrangements
None.
|
|
|
Item 7A.
|
Quantitative
and Qualitative Disclosures About Market Risk
|
Our exposure to market risk is confined to our cash, cash
equivalents, which have maturities of less than three months,
and marketable securities. The primary objective of our
investment activities is to preserve our capital to fund
operations. We also seek to maximize income from our investments
without assuming significant risk. To achieve our objectives, we
maintain a portfolio of cash equivalents and marketable
securities in a variety of securities of high credit quality. As
of December 31, 2009, we had cash, cash equivalents and
marketable securities of $19.9 million. The securities in
our investment portfolio are not leveraged, are classified as
available for sale and are, due to their very short-term nature,
subject to minimal interest rate risk. We currently do not hedge
interest rate exposure. Because of the short-term maturities of
our investments, we do not believe that an increase in market
rates would have a material negative impact on the realized
value of our investment portfolio. We actively monitor changes
in interest rates. We perform quarterly reviews of our
investment portfolio and believe we have no exposure related to
mortgage and other asset backed securities and no exposure to
auction rate securities.
60
|
|
|
Item 8.
|
Financial
Statements and Supplementary Data
|
ALEXZA
PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
61
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
Alexza Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of
Alexza Pharmaceuticals, Inc. (a development stage company) (the
“Company”) as of December 31, 2009 and 2008, and
the related consolidated statements of operations, convertible
preferred stock and stockholders’ (deficit) equity, and
cash flows for each of the three years in the period ended
December 31, 2009 and for the period from December 19,
2000 (inception) to December 31, 2009. These consolidated
financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the consolidated financial
statements are free of material misstatement. An audit includes
examining, on a test basis, evidence supporting the amounts and
disclosures in the financial statements. An audit also includes
assessing the accounting principles used and significant
estimates made by management, as well as evaluating the overall
financial statement presentation. We believe that our audits
provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above
present fairly, in all material respects, the consolidated
financial position of Alexza Pharmaceuticals, Inc. (a
development stage company) at December 31, 2009 and 2008,
and the consolidated results of its operations and its cash
flows for each of the three years in the period ended
December 31, 2009 and for the period from December 19,
2000 (inception) to December 31, 2009, in conformity with
U.S. generally accepted accounting principles.
As discussed in Note 3 to the consolidated financial
statements, the Company changed its method of accounting for and
presentation of noncontrolling interest in its consolidated
financial statements effective January 1, 2009.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States),
Alexza Pharmaceuticals, Inc.’s internal control over
financial reporting as of December 31, 2009, based on
criteria established in Internal Control — Integrated
Framework issued by the Committee of Sponsoring Organizations of
the Treadway Commission and our report dated March 9, 2010
expressed an unqualified opinion thereon.
Palo Alto, California
March 9, 2010
62
ALEXZA
PHARMACEUTICALS, INC
(a development stage company)
CONSOLIDATED BALANCE SHEETS
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
13,450
|
|
|
$
|
26,036
|
|
|
Marketable securities
|
|
|
6,466
|
|
|
|
11,520
|
|
|
Investments held by Symphony Allegro, Inc.
|
|
|
—
|
|
|
|
21,318
|
|
|
Other receivables
|
|
|
1,406
|
|
|
|
—
|
|
|
Prepaid expenses and other current assets
|
|
|
804
|
|
|
|
1,130
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
22,126
|
|
|
|
60,004
|
|
|
Property and equipment, net
|
|
|
23,598
|
|
|
|
24,152
|
|
|
Restricted cash
|
|
|
400
|
|
|
|
400
|
|
|
Other assets
|
|
|
50
|
|
|
|
79
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
46,174
|
|
|
$
|
84,635
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ (DEFICIT) EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
2,705
|
|
|
$
|
4,928
|
|
|
Accrued clinical trial liabilities
|
|
|
303
|
|
|
|
1,294
|
|
|
Other accrued liabilities
|
|
|
3,481
|
|
|
|
5,205
|
|
|
Current portion of contingent consideration liability
|
|
|
13,202
|
|
|
|
—
|
|
|
Other current liabilities
|
|
|
3,750
|
|
|
|
—
|
|
|
Current portion of equipment financing obligations
|
|
|
2,515
|
|
|
|
4,139
|
|
|
Deferred revenues
|
|
|
—
|
|
|
|
1,667
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
25,956
|
|
|
|
17,233
|
|
|
Deferred rent
|
|
|
15,708
|
|
|
|
17,386
|
|
|
Deferred revenue
|
|
|
—
|
|
|
|
7,847
|
|
|
Noncurrent portion of contingent consideration liability
|
|
|
11,636
|
|
|
|
10,439
|
|
|
Noncurrent portion of equipment financing obligations
|
|
|
—
|
|
|
|
2,515
|
|
|
Other noncurrent liabilities
|
|
|
—
|
|
|
|
600
|
|
|
Commitments (See Note 8)
|
|
|
|
|
|
|
|
|
|
Stockholders’ (deficit) equity:
|
|
|
|
|
|
|
|
|
|
Preferred stock, $0.0001 par value, 5,000,000 shares
authorized at December 31, 2009 and 2008; no shares issued
and outstanding at December 31, 2009 or 2008
|
|
|
—
|
|
|
|
—
|
|
|
Common stock, $0.0001 par value; 100,000,000 shares
authorized at December 31, 2009 and 2008; 52,411,356 and
32,820,874 shares issued and outstanding at
December 31, 2009 and 2008, respectively
|
|
|
5
|
|
|
|
3
|
|
|
Additional paid-in capital
|
|
|
257,493
|
|
|
|
256,426
|
|
|
Deferred stock compensation
|
|
|
—
|
|
|
|
(219
|
)
|
|
Other comprehensive income
|
|
|
(1
|
)
|
|
|
28
|
|
|
Deficit accumulated during development stage
|
|
|
(264,623
|
)
|
|
|
(222,545
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total Alexza Pharmaceuticals, Inc. stockholders’ (deficit)
equity
|
|
|
(7,126
|
)
|
|
|
33,693
|
|
|
Noncontrolling interest in Symphony Allegro, Inc.
|
|
|
—
|
|
|
|
5,361
|
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ (deficit) equity
|
|
|
(7,126
|
)
|
|
|
39,054
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ (deficit) equity
|
|
$
|
46,174
|
|
|
$
|
84,635
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes.
63
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 19,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2000 (Inception)
|
|
|
|
|
Year Ended December 31,
|
|
|
to December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2009
|
|
|
|
|
(In thousands, except per share amounts)
|
|
|
|
|
Revenue
|
|
$
|
9,514
|
|
|
$
|
486
|
|
|
$
|
—
|
|
|
$
|
16,945
|
|
|
Operating expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
39,778
|
|
|
|
61,565
|
|
|
|
45,645
|
|
|
|
244,461
|
|
|
General and administrative
|
|
|
15,406
|
|
|
|
17,641
|
|
|
|
14,888
|
|
|
|
78,110
|
|
|
Restructuring charges
|
|
|
2,037
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,037
|
|
|
Acquired in-process research and development
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,916
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
57,221
|
|
|
|
79,206
|
|
|
|
60,533
|
|
|
|
328,524
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(47,707
|
)
|
|
|
(78,720
|
)
|
|
|
(60,533
|
)
|
|
|
(311,579
|
)
|
|
Loss on change in fair value of contingent consideration
liability
|
|
|
(7,983
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(7,983
|
)
|
|
Interest and other income, net
|
|
|
92
|
|
|
|
2,614
|
|
|
|
5,626
|
|
|
|
13,898
|
|
|
Interest expense
|
|
|
(467
|
)
|
|
|
(935
|
)
|
|
|
(1,003
|
)
|
|
|
(4,048
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
(56,065
|
)
|
|
|
(77,041
|
)
|
|
|
(55,910
|
)
|
|
|
(309,712
|
)
|
|
Consideration paid in excess of carrying value of the
noncontrolling interest in Symphony Allegro, Inc.
|
|
|
(61,566
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(61,566
|
)
|
|
Loss attributed to noncontrolling interest in Symphony Allegro,
Inc.
|
|
|
13,987
|
|
|
|
18,591
|
|
|
|
10,791
|
|
|
|
45,089
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to Alexza common stockholders
|
|
$
|
(103,644
|
)
|
|
$
|
(58,450
|
)
|
|
$
|
(45,119
|
)
|
|
$
|
(326,189
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per share attributable to Alexza
common stockholders
|
|
$
|
(2.68
|
)
|
|
$
|
(1.81
|
)
|
|
$
|
(1.58
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used to compute basic and diluted net loss per share
attributable to Alexza common stockholders
|
|
|
38,609
|
|
|
|
32,297
|
|
|
|
28,605
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes.
64
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED
STOCK AND STOCKHOLDERS’ (DEFICIT) EQUITY
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
in
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Issuance of common stock to founders at $0.22 per share in
December 2000 in exchange for technology and cash of $8
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
454,536
|
|
|
$
|
—
|
|
|
$
|
100
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
100
|
|
|
Issuance of Series A preferred stock for cash at $0.40 per
share in July 2001, net of issuance costs of $9
|
|
|
2,500,000
|
|
|
|
991
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of Series A1 preferred stock at $1.55 per share in
December 2001, in connection with merger
|
|
|
1,610,250
|
|
|
|
2,496
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of Series B preferred stock for cash at $1.40 per
share in December 2001, net of issuance costs of $71
|
|
|
6,441,000
|
|
|
|
8,946
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock in connection with merger at $1.10 per
share in December 2001
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
868,922
|
|
|
|
—
|
|
|
|
956
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
956
|
|
|
Warrants assumed in merger transaction
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10
|
|
|
Issuance of common stock for cash at $0.22 per share upon
exercise of options in December 2001
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
9,090
|
|
|
|
—
|
|
|
|
2
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(5,652
|
)
|
|
|
—
|
|
|
|
(5,652
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2001 (carried forward)
|
|
|
10,551,250
|
|
|
$
|
12,433
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,332,548
|
|
|
$
|
—
|
|
|
$
|
1,071
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
(5,652
|
)
|
|
$
|
—
|
|
|
$
|
(4,581
|
)
|
See accompanying notes.
65
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
in
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2001 (brought forward)
|
|
|
10,551,250
|
|
|
$
|
12,433
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,332,548
|
|
|
$
|
—
|
|
|
$
|
1,071
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
(5,652
|
)
|
|
$
|
—
|
|
|
$
|
(4,581
|
)
|
|
Issuance of common stock for cash at $0.22 per share upon
exercise of options in February 2002
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10,606
|
|
|
|
—
|
|
|
|
3
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3
|
|
|
Issuance of warrants to purchase Series B preferred stock
in March 2002, in connection with equipment financing loan
|
|
|
—
|
|
|
|
27
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock for cash at $0.22 per share upon
exercise of options in July 2002
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,180
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock to stockholder at $0.99 per share in
exchange for promissory note in July 2002
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
53,156
|
|
|
|
—
|
|
|
|
53
|
|
|
|
(53
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of Series C preferred stock for cash at $1.56 per
share in September 2002, net of issuance costs of $108
|
|
|
28,870,005
|
|
|
|
44,892
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Repurchase of common stock for cash at $1.05 per share in
October 2002
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,634
|
)
|
|
|
—
|
|
|
|
(3
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3
|
)
|
|
Issuance of common stock for cash at $1.05 per share for
services upon exercise of warrants in December 2002
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
9,368
|
|
|
|
—
|
|
|
|
10
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10
|
|
|
Unrealized gain on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
51
|
|
|
|
—
|
|
|
|
—
|
|
|
|
51
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(8,163
|
)
|
|
|
—
|
|
|
|
(8,163
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2002 (carried forward)
|
|
|
39,421,255
|
|
|
$
|
57,352
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,405,224
|
|
|
$
|
—
|
|
|
$
|
1,144
|
|
|
$
|
(53
|
)
|
|
$
|
—
|
|
|
$
|
51
|
|
|
$
|
(13,815
|
)
|
|
$
|
—
|
|
|
$
|
(12,673
|
)
|
See accompanying notes.
66
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
in
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2002 (brought forward)
|
|
|
39,421,255
|
|
|
$
|
57,352
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,405,224
|
|
|
$
|
—
|
|
|
$
|
1,144
|
|
|
$
|
(53
|
)
|
|
$
|
—
|
|
|
$
|
51
|
|
|
$
|
(13,815
|
)
|
|
$
|
—
|
|
|
$
|
(12,673
|
)
|
|
Issuance of common stock for cash at $0.22, $0.99 and $1.10 per
share upon exercise of options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
74,903
|
|
|
|
—
|
|
|
|
47
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
47
|
|
|
Issuance of warrants to purchase Series C preferred stock
in connection with equipment financing loan in January 2003
|
|
|
—
|
|
|
|
35
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of warrants to purchase Series C preferred stock
in connection with equipment financing loan in September 2003
|
|
|
—
|
|
|
|
27
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Repurchase of common stock for cash at $1.05 per share in
January 2003
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,172
|
)
|
|
|
—
|
|
|
|
(1
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(1
|
)
|
|
Repurchase of common stock for cash at $0.22 per share in
November 2003
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(14,772
|
)
|
|
|
—
|
|
|
|
(3
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3
|
)
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
31
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
31
|
|
|
Deferred stock compensation expense related to modification of
consultant stock option
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1
|
|
|
|
—
|
|
|
|
(1
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(55
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(55
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(14,328
|
)
|
|
|
—
|
|
|
|
(14,328
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2003 (carried forward)
|
|
|
39,421,255
|
|
|
$
|
57,414
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,464,183
|
|
|
$
|
—
|
|
|
$
|
1,219
|
|
|
$
|
(53
|
)
|
|
$
|
(1
|
)
|
|
$
|
(4
|
)
|
|
$
|
(28,143
|
)
|
|
$
|
—
|
|
|
$
|
(26,982
|
)
|
See accompanying notes.
67
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
in
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2003 (brought forward)
|
|
|
39,421,255
|
|
|
$
|
57,414
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,464,183
|
|
|
$
|
—
|
|
|
$
|
1,219
|
|
|
$
|
(53
|
)
|
|
$
|
(1
|
)
|
|
$
|
(4
|
)
|
|
$
|
(28,143
|
)
|
|
$
|
—
|
|
|
$
|
(26,982
|
)
|
|
Cancellation of unvested common stock at $0.99 per share in
March 2004
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(24,365
|
)
|
|
|
—
|
|
|
|
(24
|
)
|
|
|
24
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Repayment of vested portion of stockholder note receivable for
cash
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
29
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
29
|
|
|
Issuance of warrants to purchase Series C preferred stock
in connection with equipment financing loan in April 2004
|
|
|
—
|
|
|
|
20
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock for cash at $0.22, $0.99 and $1.10 per
share upon exercise of options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
100,192
|
|
|
|
—
|
|
|
|
72
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
72
|
|
|
Repurchase of common stock for cash at $1.05 per share in
September 2004
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(404
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of Series D preferred stock at $1.29 per share in
November and December 2004, net of issuance costs of $2,239
|
|
|
40,435,448
|
|
|
|
49,760
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of warrants to purchase common stock in connection with
Series D financing in November 2004
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
91
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
91
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
40
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
40
|
|
|
Compensation expense related to employee stock option
modifications
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
19
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
19
|
|
|
Amortization of deferred stock compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1
|
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(41
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(41
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(16,625
|
)
|
|
|
—
|
|
|
|
(16,625
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2004 (carried forward)
|
|
|
79,856,703
|
|
|
$
|
107,194
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,539,606
|
|
|
$
|
—
|
|
|
$
|
1,417
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
(45
|
)
|
|
$
|
(44,768
|
)
|
|
$
|
—
|
|
|
$
|
(43,396
|
)
|
See accompanying notes.
68
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
in
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2004 (brought forward)
|
|
|
79,856,703
|
|
|
$
|
107,194
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,539,606
|
|
|
$
|
—
|
|
|
$
|
1,417
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
(45
|
)
|
|
$
|
(44,768
|
)
|
|
$
|
—
|
|
|
$
|
(43,396
|
)
|
|
Issuance of common stock upon exercise of options $0.22, $0.99,
$1.10, per share
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
380,508
|
|
|
|
—
|
|
|
|
357
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
357
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
195
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
195
|
|
|
Deferred stock compensation, net of $4 reversal in connection
with employee terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,329
|
|
|
|
—
|
|
|
|
(3,329
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Amortization of deferred stock compensation,
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
404
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
404
|
|
|
Variable compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
442
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
442
|
|
|
Unrealized gain on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
15
|
|
|
|
—
|
|
|
|
—
|
|
|
|
15
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(32,402
|
)
|
|
|
—
|
|
|
|
(32,402
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2005 (carried forward)
|
|
|
79,856,703
|
|
|
$
|
107,194
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,920,114
|
|
|
$
|
—
|
|
|
$
|
5,740
|
|
|
$
|
—
|
|
|
$
|
(2,925
|
)
|
|
$
|
(30
|
)
|
|
$
|
(77,170
|
)
|
|
$
|
—
|
|
|
$
|
(74,385
|
)
|
See accompanying notes.
69
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
in
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-in
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2005 (brought forward)
|
|
|
79,856,703
|
|
|
$
|
107,194
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
1,920,114
|
|
|
$
|
—
|
|
|
$
|
5,740
|
|
|
$
|
—
|
|
|
$
|
(2,925
|
)
|
|
$
|
(30
|
)
|
|
$
|
(77,170
|
)
|
|
$
|
—
|
|
|
$
|
(74,385
|
)
|
|
Issuance of common stock for cash and shares upon exercise of
options at a weighted average price of $1.28 per share
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
159,446
|
|
|
|
—
|
|
|
|
195
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
195
|
|
|
Issuance of common stock for cash under the Company’s
Employee Stock Purchase Plan
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
131,682
|
|
|
|
—
|
|
|
|
896
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
896
|
|
|
Issuance of common stock for shares upon exercise of warrant
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
85,359
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock for cash, net of offering costs of
$2,156
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,325,000
|
|
|
|
1
|
|
|
|
44,901
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
44,902
|
|
|
Conversion of convertible preferred stock into common stock
|
|
|
(79,856,703
|
)
|
|
|
(107,194
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
15,197,712
|
|
|
|
1
|
|
|
|
107,193
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
107,194
|
|
|
Purchase of noncontrolling interest by Symphony Allegro, Inc,
preferred shareholders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
36,463
|
|
|
|
36,463
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
145
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
145
|
|
|
Compensation expense related to fair value of employee share
based awards issued after January 1, 2006
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,601
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,601
|
|
|
Amortization of deferred stock compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
727
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
727
|
|
|
Reversal of deferred stock compensation in connection with
employee terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(495
|
)
|
|
|
—
|
|
|
|
495
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Variable compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(442
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(442
|
)
|
|
Issuance of warrant to Symphony Allegro Holdings LLC
|
|
|
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10,708
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10,708
|
|
|
Unrealized gain on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
39
|
|
|
|
—
|
|
|
|
—
|
|
|
|
39
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(41,806
|
)
|
|
|
(1,720
|
)
|
|
|
(43,526
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2006 (carried forward)
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
23,819,319
|
|
|
$
|
2
|
|
|
$
|
170,442
|
|
|
$
|
—
|
|
|
$
|
(1,703
|
)
|
|
$
|
9
|
|
|
$
|
(118,976
|
)
|
|
$
|
34,743
|
|
|
$
|
84,517
|
|
See accompanying notes.
70
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
In
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2006 (brought forward)
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
23,819,319
|
|
|
$
|
2
|
|
|
$
|
170,442
|
|
|
$
|
—
|
|
|
$
|
(1,703
|
)
|
|
$
|
9
|
|
|
$
|
(118,976
|
)
|
|
$
|
34,743
|
|
|
$
|
84,517
|
|
|
Issuance of common stock for cash and shares upon exercise of
options at a weighted average price of $1.28 per share
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
204,423
|
|
|
|
—
|
|
|
|
432
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
432
|
|
|
Issuance of common stock for cash under the Company’s
Employee Stock Purchase Plan
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
205,870
|
|
|
|
—
|
|
|
|
1,405
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,405
|
|
|
Issuance of common stock upon vesting of restricted stock units
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8,245
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock for cash, net of offering costs of
$4,743
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,900,000
|
|
|
|
1
|
|
|
|
65,981
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
65,982
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
75
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
75
|
|
|
Compensation expense related to fair value of employee share
based awards issued after January 1, 2006
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,733
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,733
|
|
|
Amortization of deferred stock compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
577
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
577
|
|
|
Reversal of deferred stock compensation in connection with
employee terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(387
|
)
|
|
|
—
|
|
|
|
387
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Unrealized gain on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
132
|
|
|
|
—
|
|
|
|
—
|
|
|
|
132
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(45,119
|
)
|
|
|
(10,791
|
)
|
|
|
(55,910
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007(carried forward)
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
31,137,857
|
|
|
$
|
3
|
|
|
$
|
240,681
|
|
|
$
|
—
|
|
|
$
|
(739
|
)
|
|
$
|
141
|
|
|
$
|
(164,095
|
)
|
|
$
|
23,952
|
|
|
$
|
99,943
|
|
See accompanying notes.
71
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
In
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2007 (brought forward)
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
31,137,857
|
|
|
$
|
3
|
|
|
$
|
240,681
|
|
|
$
|
—
|
|
|
$
|
(739
|
)
|
|
$
|
141
|
|
|
$
|
(164,095
|
)
|
|
$
|
23,952
|
|
|
$
|
99,943
|
|
|
Issuance of common stock and common stock warrant for cash
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,250,000
|
|
|
|
—
|
|
|
|
9,840
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
9,840
|
|
|
Issuance of common stock for cash upon exercise of options at a
weighted average price of $1.55 per share
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
104,428
|
|
|
|
—
|
|
|
|
161
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
161
|
|
|
Issuance of common stock for cash under the Company’s
Employee Stock Purchase Plan
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
305,146
|
|
|
|
—
|
|
|
|
1,172
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,172
|
|
|
Issuance of common stock upon vesting of restricted stock units
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
23,443
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
22
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
22
|
|
|
Compensation expense related to fair value of employee share
based awards issued after January 1, 2006
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,633
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,633
|
|
|
Amortization of deferred stock compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
437
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
437
|
|
|
Reversal of deferred stock compensation in connection with
employee terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(83
|
)
|
|
|
—
|
|
|
|
83
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(113
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(113
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(58,450
|
)
|
|
|
(18,591
|
)
|
|
|
(77,041
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
32,820,874
|
|
|
$
|
3
|
|
|
$
|
256,426
|
|
|
$
|
—
|
|
|
$
|
(219
|
)
|
|
$
|
28
|
|
|
$
|
(222,545
|
)
|
|
$
|
5,361
|
|
|
$
|
39,054
|
|
See accompanying notes.
72
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK AND
STOCKHOLDERS’ (DEFICIT) EQUITY —
(Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alexza Pharmaceuticals, Inc. Stockholders
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deficit
|
|
|
Noncontrolling
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
Interest
|
|
|
|
|
|
|
|
Convertible
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Stockholder
|
|
|
Deferred
|
|
|
Other
|
|
|
During the
|
|
|
In
|
|
|
Total
|
|
|
|
|
Preferred Stock
|
|
|
Preferred Stock
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Note
|
|
|
Stock
|
|
|
Comprehensive
|
|
|
Development
|
|
|
Symphony
|
|
|
Stockholders’
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Receivable
|
|
|
Compensation
|
|
|
(Loss) Income
|
|
|
Stage
|
|
|
Allegro, Inc.
|
|
|
(Deficit) Equity
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance at December 31, 2008 (brought forward)
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
32,820,874
|
|
|
$
|
3
|
|
|
$
|
256,426
|
|
|
$
|
—
|
|
|
$
|
(219
|
)
|
|
$
|
28
|
|
|
$
|
(222,545
|
)
|
|
$
|
5,361
|
|
|
$
|
39,054
|
|
|
Issuance of common stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
135,041
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of common stock and common stock warrants for cash
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8,107,012
|
|
|
|
1
|
|
|
|
18,989
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
18,990
|
|
|
Issuance of common stock and common stock warrants for the
purchase of noncontrolling interest in Symphony Allegro,
Inc.
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
10,000,000
|
|
|
|
1
|
|
|
|
36,084
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
36,085
|
|
|
Deemed dividend for purchase of noncontrolling interest in
Symphony Allegro, Inc.
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(61,566
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8,626
|
|
|
|
(52,940
|
)
|
|
Issuance of common stock for cash upon exercise of options at a
weighted average price of $1.20 per share
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
69,708
|
|
|
|
—
|
|
|
|
84
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
84
|
|
|
Issuance of common stock for cash under the Company’s
Employee Stock Purchase Plan
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
439,252
|
|
|
|
—
|
|
|
|
599
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
599
|
|
|
Issuance of common stock upon vesting of restricted stock units
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
839,469
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Compensation expense related to consultant stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
53
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
53
|
|
|
Compensation expense related to fair value of employee share
based awards issued after January 1, 2006
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,860
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,860
|
|
|
Amortization of deferred stock compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
183
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
183
|
|
|
Reversal of deferred stock compensation in connection with
employee terminations
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(36
|
)
|
|
|
—
|
|
|
|
36
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(29
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(29
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(42,078
|
)
|
|
|
(13,987
|
)
|
|
|
(56,065
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
|
—
|
|
|
$
|
—
|
|
|
|
—
|
|
|
$
|
—
|
|
|
|
52,411,356
|
|
|
$
|
5
|
|
|
$
|
257,493
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
(1
|
)
|
|
$
|
(264,623
|
)
|
|
$
|
—
|
|
|
$
|
(7,126
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
73
ALEXZA
PHARMACEUTICALS, INC
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 19,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Inception) to
|
|
|
|
|
Year Ended December 31,
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(56,065
|
)
|
|
$
|
(77,041
|
)
|
|
$
|
(55,910
|
)
|
|
$
|
(309,712
|
)
|
|
Adjustments to reconcile net loss to net cash used in operating
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share-based compensation expense
|
|
|
7,096
|
|
|
|
5,092
|
|
|
|
3,385
|
|
|
|
18,935
|
|
|
Change in fair value of contingent consideration liability
|
|
|
7,983
|
|
|
|
—
|
|
|
|
—
|
|
|
|
7,983
|
|
|
Extinguishment of officer note receivable
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,300
|
|
|
Issuance of common stock for intellectual property
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
92
|
|
|
Charge for acquired in-process research and development
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,916
|
|
|
Amortization of assembled workforce
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
222
|
|
|
Amortization of debt discount and deferred interest
|
|
|
29
|
|
|
|
38
|
|
|
|
49
|
|
|
|
391
|
|
|
Amortization of discount on
available-for-sale
securities
|
|
|
126
|
|
|
|
(797
|
)
|
|
|
(929
|
)
|
|
|
(601
|
)
|
|
Depreciation
|
|
|
4,850
|
|
|
|
5,294
|
|
|
|
4,016
|
|
|
|
21,609
|
|
|
Loss on disposal of property and equipment
|
|
|
43
|
|
|
|
17
|
|
|
|
23
|
|
|
|
126
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other receivables
|
|
|
(1,406
|
)
|
|
|
12,055
|
|
|
|
(12,055
|
)
|
|
|
(1,406
|
)
|
|
Prepaid expenses and other current assets
|
|
|
326
|
|
|
|
247
|
|
|
|
(114
|
)
|
|
|
(798
|
)
|
|
Other assets
|
|
|
—
|
|
|
|
(24
|
)
|
|
|
42
|
|
|
|
(2,625
|
)
|
|
Accounts payable
|
|
|
(2,223
|
)
|
|
|
(278
|
)
|
|
|
(727
|
)
|
|
|
2,576
|
|
|
Accrued clinical trial expense and other accrued liabilities
|
|
|
(2,715
|
)
|
|
|
111
|
|
|
|
898
|
|
|
|
84
|
|
|
Deferred revenues
|
|
|
(9,514
|
)
|
|
|
(486
|
)
|
|
|
10,000
|
|
|
|
—
|
|
|
Other liabilities
|
|
|
(1,678
|
)
|
|
|
701
|
|
|
|
15,494
|
|
|
|
19,098
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(53,148
|
)
|
|
|
(55,071
|
)
|
|
|
(35,828
|
)
|
|
|
(237,810
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of
available-for-sale
securities
|
|
|
(13,259
|
)
|
|
|
(47,111
|
)
|
|
|
(62,466
|
)
|
|
|
(338,172
|
)
|
|
Maturities of
available-for-sale
securities
|
|
|
18,158
|
|
|
|
74,329
|
|
|
|
51,064
|
|
|
|
332,307
|
|
|
Purchase of
available-for-sale
securities held by Symphony Allegro, Inc.
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(49,975
|
)
|
|
Maturities of
available-for-sale
securities held by Symphony Allegro, Inc.
|
|
|
16,436
|
|
|
|
18,131
|
|
|
|
10,507
|
|
|
|
45,093
|
|
|
Decrease (increase) in restricted cash
|
|
|
—
|
|
|
|
204
|
|
|
|
—
|
|
|
|
(400
|
)
|
|
Purchases of property and equipment
|
|
|
(1,189
|
)
|
|
|
(2,732
|
)
|
|
|
(19,059
|
)
|
|
|
(41,346
|
)
|
|
Proceeds from disposal of property and equipment
|
|
|
—
|
|
|
|
25
|
|
|
|
—
|
|
|
|
28
|
|
|
Cash paid for merger
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(250
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
20,146
|
|
|
|
42,846
|
|
|
|
(19,954
|
)
|
|
|
(52,715
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock and exercise of stock
options and stock purchase rights
|
|
|
19,673
|
|
|
|
11,173
|
|
|
|
67,819
|
|
|
|
145,158
|
|
|
Repurchase of common stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(8
|
)
|
|
Proceeds from issuance of convertible preferred stock
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
104,681
|
|
|
Proceeds from repayment of stockholder note receivable
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
29
|
|
|
Proceeds received from purchase of the noncontrolling interest
in Symphony Allegro, Inc.
|
|
|
4,882
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,882
|
|
|
Proceeds from purchase of non controlling interest by preferred
shareholders in Symphony Allegro, Inc., net of fees
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
47,171
|
|
|
Proceeds from equipment term loans
|
|
|
—
|
|
|
|
—
|
|
|
|
5,814
|
|
|
|
18,932
|
|
|
Payments of equipment term loans and leases
|
|
|
(4,139
|
)
|
|
|
(4,249
|
)
|
|
|
(3,546
|
)
|
|
|
(16,870
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
20,416
|
|
|
|
6,924
|
|
|
|
70,087
|
|
|
|
303,975
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
|
(12,586
|
)
|
|
|
(5,301
|
)
|
|
|
14,305
|
|
|
|
13,450
|
|
|
Cash and cash equivalents at beginning of period
|
|
|
26,036
|
|
|
|
31,337
|
|
|
|
17,032
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of period
|
|
$
|
13,450
|
|
|
$
|
26,036
|
|
|
$
|
31,337
|
|
|
$
|
13,450
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosures of cash flow information
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest
|
|
$
|
467
|
|
|
$
|
935
|
|
|
$
|
1,003
|
|
|
$
|
3,732
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non cash investing and financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of convertible preferred stock to common stock
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
107,194
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of shares and warrants, net of warrant cancellation in
conjunction with Symphony Allegro purchase
|
|
$
|
36,085
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
36,085
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of contingent consideration liability
|
|
$
|
16,855
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
16,855
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of warrants in conjunction with establishment of
Symphony Allegro
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
10,708
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes.
74
ALEXZA
PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
|
|
|
1.
|
The
Company and Basis of Presentation
|
Business
Alexza Pharmaceuticals, Inc. (“Alexza” or the
“Company”), was incorporated in the state of Delaware
on December 19, 2000 as FaxMed, Inc. In June 2001, the
Company changed its name to Alexza Corporation and in December
2001 became Alexza Molecular Delivery Corporation. In July 2005,
the Company changed its name to Alexza Pharmaceuticals, Inc.
The Company is a pharmaceutical development company focused on
the research, development, and commercialization of novel
proprietary products for the acute treatment of central nervous
system (“CNS”) conditions. The Company’s primary
activities since incorporation have been establishing its
offices, recruiting personnel, conducting research and
development, conducting preclinical studies and clinical trials,
performing business and financial planning, and raising capital.
Accordingly, the Company is considered to be in the development
stage and operates in one business segment.
Basis
of Consolidation
The consolidated financial statements include the accounts of
Alexza and its wholly-owned subsidiaries, Alexza Singapore Pte.
Ltd., Alexza Singapore Manufacturing Pte. Ltd., Alexza UK
Limited, and Symphony Allegro, Inc. (“Allegro”). On
August 26, 2009, Alexza acquired all of the outstanding
equity of Allegro (see Note 9). Prior to August 26,
2009, Alexza consolidated the financial results of Allegro, as
Allegro was deemed a variable interest entity and Alexza was
deemed the primary beneficiary. All significant intercompany
balances and transactions have been eliminated.
Registered
Direct Equity Issuance
In March 2008, the Company completed the sale of
1,250,000 shares of its registered common stock to
Biomedical Sciences Investment Fund Pte. Ltd.
(“Bio*One”) at a price of $8.00 per share. As outlined
in the agreement, if the average closing price of the
Company’s stock over a 45 consecutive day trading period
does not exceed $8.00 between the closing date and
December 31, 2008, Bio*One would receive 135,041 additional
shares, which would adjust the effective purchase price to $7.22
per share. The Company’s average stock price did not meet
this level during the specified period and the Company issued
the additional shares to Bio*One in January 2009.
In addition, the Company issued a warrant to Bio*One to purchase
up to 375,000 of additional shares of Alexza common stock at a
purchase price per share of $8.00. The warrant was subject to
the same price adjustment as the common stock sale, and
effective January 1, 2009 the warrant was automatically
adjusted to give Bio*One the right to purchase
415,522 shares at a purchase price of $7.22 per share. The
Company committed to initiate and maintain manufacturing
operations in Singapore, and the warrant was to become
exercisable only if the Company terminated operations in
Singapore or did not achieve certain performance milestones. In
December 2008, the Company did not meet its defined performance
milestone, and as a result the warrant became fully exercisable.
The warrant is cash or net exercisable for a period of
5 years. Net proceeds from the sale of the stock and
warrant were approximately $9.84 million after deducting
offering expenses and is classified as equity in the
consolidated balance sheets.
Unregistered
Direct Equity Issuance
On October 5, 2009, the Company issued an aggregate of
8,107,012 shares of its common stock and warrants to
purchase up to an additional 7,296,312 shares of its common
stock in a private placement. These securities were sold as
units with each unit consisting of one share of common stock and
a warrant to purchase 0.9 shares of common stock at a
purchase price of $2.4325 per unit. The net proceeds, after
deducting the payment of a placement agent fee and other
offering expenses, were approximately $19.0 million and is
classified as equity in the
75
consolidated balance sheets. The warrants are cash or net
exercisable for a period of seven years from October 5,
2009 and have an exercise price of $2.77 per share.
The Company granted to the investors certain registration rights
related to the shares of common stock sold in the private
placement and the shares of common stock underlying the
warrants. The Company filed with the SEC a registration
statement covering the resale of these shares, and the SEC
declared such registration statement effective on
October 27, 2009. The Company also agreed to other
customary obligations regarding registration, including
indemnification and maintenance of the registration statement.
If the Company does not maintain an effective registration
statement, it will be subject to liquidated damages of 2% for
each 30 day period the registration statement is not
effective. The Company believes the risk of payment of the
liquidated damages to be remote.
|
|
|
2.
|
Need to
Raise Additional Capital
|
The Company has incurred significant losses from operations
since its inception and expects losses to continue for the
foreseeable future. The Company will need to raise additional
capital to fund its operations, to develop its product
candidates and to develop its manufacturing capabilities.
Management plans to finance the Company’s operations
through the sale of equity securities, debt arrangements or
partnership or licensing collaborations. Such funding may not be
available or may be on terms which are not favorable to the
Company. The Company believes its cash, cash equivalents and
marketable securities are sufficient to fund its operations
through the first quarter of 2011.
|
|
|
3.
|
Summary
of Significant Accounting Policies
|
Use of
Estimates
The preparation of financial statements in conformity with
generally accepted accounting principles requires management to
make estimates and assumptions that affect the amounts reported
in the financial statements and accompanying notes. Actual
results could differ from those estimates.
Fair
Value of Financial Instruments
Fair value is defined as the exchange price that would be
received for an asset or paid to transfer a liability (an exit
price) in the principal or most advantageous market for the
asset or liability in an orderly transaction between market
participants on the measurement date. Valuation techniques used
to measure fair value must maximize the use of observable inputs
and minimize the use of unobservable inputs. Three levels of
inputs, of which the first two are considered observable and the
last unobservable, may be used to measure fair value which are
the following:
|
|
|
| |
•
|
Level 1 — Quoted prices in active markets for
identical assets or liabilities.
|
| |
| |
•
|
Level 2 — Inputs other than Level 1 that are
observable, either directly or indirectly, such as quoted prices
for similar assets or liabilities; quoted prices in markets that
are not active; or other inputs that are observable or can be
corroborated by observable market data for substantially the
full term of the assets or liabilities.
|
| |
| |
•
|
Level 3 — Unobservable inputs that are supported
by little or no market activity and that are significant to the
fair value of the assets or liabilities.
|
76
The following table represents the Company’s fair value
hierarchy for its financial assets (cash equivalents and
marketable securities) by major security type and contingent
consideration liability measured at fair value on a recurring
basis as of December 31, 2009 and 2008 (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
Assets
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds
|
|
$
|
10,421
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
10,421
|
|
|
Government-sponsored enterprises
|
|
|
—
|
|
|
|
5,217
|
|
|
|
—
|
|
|
|
5,217
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
3,500
|
|
|
|
—
|
|
|
|
3,500
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
10,421
|
|
|
$
|
8,717
|
|
|
$
|
—
|
|
|
$
|
19,138
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contingent consideration liability
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
24,838
|
|
|
$
|
24,838
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
24,838
|
|
|
$
|
24,838
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2008
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
Assets
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds
|
|
$
|
19,350
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
19,350
|
|
|
Money market funds held by Symphony Allegro, Inc.
|
|
|
21,318
|
|
|
|
—
|
|
|
|
—
|
|
|
|
21,318
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
9,649
|
|
|
|
—
|
|
|
|
9,649
|
|
|
Government securities
|
|
|
—
|
|
|
|
1,505
|
|
|
|
—
|
|
|
|
1,505
|
|
|
Government-sponsored enterprises
|
|
|
—
|
|
|
|
6,620
|
|
|
|
—
|
|
|
|
6,620
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
40,668
|
|
|
$
|
17,774
|
|
|
$
|
—
|
|
|
$
|
58,442
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contingent
consideration liability
In connection with the exercise of the Company’s option to
purchase all of the outstanding equity of Allegro, the Company
is obligated to make future contingent cash payments to the
former Allegro shareholders related to certain payments received
by the Company from future partnering agreements pertaining to
AZ-004/104 (Staccato loxapine) or AZ-002 (Staccato
alprazolam) (see Note 11). The Company estimated the
fair value of this contingent consideration liability using a
probability-weighted discounted cash flow model. The Company
derived multiple cash flow scenarios for each of the product
candidates and applied a probability to each of the scenarios.
These cash flows were then discounted at an 18% rate.
Subsequent to the August 26, 2009 acquisition date, changes
in the fair value of the contingent consideration liability will
be recognized in the statement of operations in the period of
the change. Certain events including, but not limited to,
clinical trial results, FDA approval or disapproval of its
submissions, such as the NDA for AZ-004 submitted in December
2009, the timing and terms of any strategic partnership
agreement, and the commercial success of AZ-004, AZ-104 or
AZ-002 could have a material impact on the fair value of the
contingent consideration liability, and as a result, the
Company’s results of operations and financial position.
Subsequent to the acquisition date, the Company modified its
assumptions regarding the probability of certain cash flow
outcomes to reflect the depth of negotiations with Biovail to
partner AZ-004 as well as the filing of our NDA. These changes
resulted in an increase in the expected cash flow resulting in
an increase in the contingent consideration liability.
Additionally, the Company announced preliminary results from its
Phase 2b clinical trial of AZ-104, where AZ-104 did not meet the
primary endpoint of the study. This change resulted in a
decrease in the expected cash flow resulting in a decrease in
the contingent consideration liability. These items combined
resulted in the Company incurring a loss on the change in fair
value of the contingent consideration liability of
$8.0 million during the year ended December 31, 2009.
77
The following table represents a reconciliation of the change in
the fair value measurement of the contingent consideration
liability for the year ended December 31, 2009 (in
thousands).
| |
|
|
|
|
|
|
|
Amount
|
|
|
|
|
Acquisition date fair value measurement —
August 26, 2009
|
|
$
|
16,855
|
|
|
Adjustments to fair value measurement
|
|
|
7,983
|
|
|
|
|
|
|
|
|
Ending balance — December 31, 2009
|
|
$
|
24,838
|
|
|
|
|
|
|
|
Concentration
of Credit Risk
Financial instruments that potentially subject the Company to
credit risk consist of cash, cash equivalents and marketable
securities and restricted cash to the extent of the amounts
recorded on the balance sheets. The Company’s cash, cash
equivalents, marketable securities and restricted cash are
placed with high credit-quality U.S. financial institutions
and issuers. All cash, cash equivalents, marketable securities
are maintained with financial institutions that the
Company’s management believes are high credit-quality. The
Company believes that its established guidelines for investment
of its excess cash maintain liquidity through its policies on
diversification and investment maturity.
Cash
Equivalents and Marketable Securities
Management determines the appropriate classification of its
investments at the time of purchase. These securities are
recorded as either cash equivalents or marketable securities.
The Company considers all highly liquid investments with
original maturities of three months or less from date of
purchase to be cash equivalents. Cash equivalents consist of
interest-bearing instruments including obligations of
U.S. government agencies, high credit rating corporate
borrowers and money market funds, which are carried at market
value.
All other investments are classified as
available-for-sale
marketable securities. The Company views its
available-for-sale
investments as available for use in current operations.
Accordingly, the Company has classified all investments as
short-term marketable securities, even though the stated
maturity date may be one year or more beyond the current balance
sheet date. Marketable securities are carried at estimated fair
value with unrealized gains or losses included in accumulated
other comprehensive income (loss) in stockholders’
(deficit) equity.
The amortized cost of debt securities is adjusted for
amortization of premiums and accretion of discounts to maturity.
Such amortization and accretion are included in interest and
other income (expense), net. Realized gains and losses, if any,
are also included in interest and other income (expense), net.
The cost of all securities sold is based on the
specific-identification method. Interest and dividends are
included in interest income.
The Company reviews its investments for other than temporary
decreases in market value on a quarterly basis. Through
December 31, 2009, the Company has not recorded an other
than temporary impairment.
Property
and Equipment
Property and equipment are stated at cost, less accumulated
depreciation. Property and equipment are depreciated using the
straight-line method over the estimated life of the asset,
generally three years for computer equipment and five years for
laboratory equipment and furniture. Leasehold improvements are
amortized over the estimated useful life or the remaining lease
term, whichever is shorter.
Restricted
Cash
The Company must maintain a letter of credit as security for
performance under its facility lease agreement. The letter of
credit is secured by a certificate of deposit for the same
amount, which is classified as restricted cash, a non-current
asset.
78
Impairment
of Long-Lived Assets
The Company reviews long-lived assets, including property and
equipment, for impairment whenever events or changes in business
circumstances indicate that the carrying amount of the assets
may not be fully recoverable. An impairment loss would be
recognized when estimated undiscounted future cash flows
expected to result from the use of the asset and its eventual
disposition are less than its carrying amount. The impairment
loss, if recognized, would be based on the excess of the
carrying value of the impaired asset over its respective fair
value. Impairment, if any, is assessed using discounted cash
flows. Through December 31, 2009, the Company has not
recorded an impairment of a long-lived asset.
Revenue
Recognition
The Company recognizes revenue in accordance with the Securities
and Exchange Commission (“SEC”) Staff Accounting
Bulletin (“SAB”) No. 101, Revenue Recognition
in Financial Statements (“SAB 101”), as
amended by Staff Accounting Bulletin No. 104,
Revision of Topic 13 (“SAB 104”).
Revenue has consisted primarily of amounts earned under research
grants with the National Institutes of Health and from the Endo
licensing agreement. The Company’s federal government
research grants provided for the reimbursement of qualified
expenses for research and development as defined under the terms
of each grant. Equipment purchased specifically for grant
programs was recorded at cost and depreciated over the grant
period. Revenue under grants was recognized when the related
qualified research and development expenses were incurred up to
the limit of the approval funding amounts.
In determining the accounting for collaboration agreements such
as the Endo licensing agreement, see Note 9, the Company
determines if the arrangement represents a single unit of
accounting or includes multiple units of accounting. If the
arrangement represents a single unit of accounting, the revenue
recognition policy and the performance obligation period must be
determined, if not already contractually defined, for the entire
arrangement. If the arrangement represents separate units of
accounting, a revenue recognition policy must be determined for
each unit. Revenues for non-refundable upfront license fee
payments, where the Company continues to have obligations, will
be recognized as performance occurs and obligations are
completed.
Research
and Development
Research and development expenses include personnel and
facility-related expenses, outside contracted services including
clinical trial costs, manufacturing and process development
costs, research costs and other consulting services. Research
and development costs are expensed as incurred.
Clinical development costs are a significant component of
research and development expenses. The Company has a history of
contracting with third parties that perform various clinical
trial activities on its behalf in the ongoing development of its
product candidates. The financial terms of these contracts are
subject to negotiations and may vary from contract to contract
and may result in uneven payment flow. The Company accrues and
expenses costs for clinical trial activities performed by third
parties based upon estimates of the percentage of work completed
over the life of the individual study in accordance with
agreements established with contract research organizations and
clinical trial sites.
Income
Taxes
The Company utilizes the asset and liability method of
accounting for income taxes. Under this method, deferred tax
assets and liabilities are determined based on differences
between financial reporting and tax basis of assets and
liabilities and are measured using enacted tax rates and laws
that will be in effect when the differences are expected to
reverse. A valuation allowance is provided when it is more
likely than not that some portion or all of a deferred tax asset
will not be realized.
The impact of an uncertain income tax position on the income tax
return must be recognized at the largest amount that is
more-likely-than-not to be sustained upon audit by the relevant
taxing authority. An uncertain income tax position will not be
recognized if it has less than a 50% likelihood of being
sustained.
79
Comprehensive
Loss Attributable to Alexza Common Stockholders
Comprehensive loss attributable to Alexza common stockholders is
comprised of net loss and unrealized gains (losses) on
marketable securities. Total comprehensive loss for the years
ended December 31, 2009, 2008 and 2007 is as follows (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Period from
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 19,
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Inception) to
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2009
|
|
|
|
|
Net loss
|
|
$
|
(56,065
|
)
|
|
$
|
(77,041
|
)
|
|
$
|
(55,910
|
)
|
|
$
|
(309,712
|
)
|
|
Change in unrealized (loss) on marketable securities, net of
taxes
|
|
|
(29
|
)
|
|
|
(113
|
)
|
|
|
132
|
|
|
|
(1
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
(56,094
|
)
|
|
|
(77,154
|
)
|
|
|
(55,778
|
)
|
|
|
(309,713
|
)
|
|
Comprehensive loss attributable to noncontrolling interest in
Symphony Allegro. Inc., net of taxes
|
|
|
13,987
|
|
|
|
18,591
|
|
|
|
10,791
|
|
|
|
45,089
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss attributable to Alexza common stockholders
|
|
$
|
(42,107
|
)
|
|
$
|
(58,563
|
)
|
|
$
|
(44,987
|
)
|
|
$
|
(264,624
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share-Based
Compensation
Employee share-based compensation cost recognized includes:
(a) compensation cost for all share-based payments granted
prior to, but not yet vested, as of December 31, 2005 for
(i) employees using the intrinsic value and
(ii) non-employees using the fair value in accordance with
the provisions of SFAS 123, and (b) compensation cost
for all share-based payments granted or modified subsequent to
December 31, 2005, based on the fair value estimated in
accordance with the provisions of SFAS 123R.
All share-based payment awards are amortized on a ratable basis
over the requisite service periods of the awards, which are
generally the vesting periods. There was no share-based
compensation capitalized as of December 31, 2009.
Employee
Share-Based Awards Granted Prior to January 1,
2006
Compensation cost for employee stock options granted prior to
January 1, 2006 are accounted for using the option’s
intrinsic value. The Company recorded the total valuation of
these options as a component of stockholders’ (deficit)
equity, which was amortized over the vesting period of the
applicable option on a straight line basis. During the years
ended December 31, 2009, 2008 and 2007, the Company
reversed $36,000, $83,000, and $387,000, respectively, of
deferred share-based compensation related to unvested options
cancelled as a result of employee terminations. As of
December 31, 2009, all deferred stock compensation had been
recognized.
Employee
Share-Based Awards Granted On or Subsequent to January 1,
2006
Compensation cost for employee share-based awards granted on or
after January 1, 2006 is based on the grant-date fair value
and will be recognized over the vesting period of the applicable
award on a straight-line basis. The Company issues employee
share-based awards in the form of stock options and restricted
stock units under the Company’s equity incentive plans and
stock purchase rights under the Company’s employee stock
purchase plan.
Stock
Options, Stock Purchase Rights and Restricted Stock
Units
During the years ended December 31, 2009, 2008 and 2007,
the weighted average fair value of the employee stock options
granted was $1.71, $3.04, and $6.22, respectively, the weighted
average fair value of stock purchase rights granted was $2.81,
$2.58, and $3.44, respectively, and the weighted average fair
value of restricted stock units granted was $2.19, $4.35, and
$8.89, respectively.
80
The estimated grant date fair values of the stock options and
stock purchase rights were calculated using the Black-Scholes
valuation model, and the following assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
Stock Option Plans
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average expected term
|
|
|
5.0 years
|
|
|
|
5.0 years
|
|
|
|
6.1 years
|
|
|
Expected volatility
|
|
|
86
|
%
|
|
|
67
|
%
|
|
|
73
|
%
|
|
Risk-free interest rate
|
|
|
1.76
|
%
|
|
|
3.14
|
%
|
|
|
4.72
|
%
|
|
Dividend yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
|
Employee Stock Purchase Plan
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average expected term
|
|
|
1.90 Years
|
|
|
|
1.65 years
|
|
|
|
1.42 years
|
|
|
Expected volatility
|
|
|
74
|
%
|
|
|
71
|
%
|
|
|
53
|
%
|
|
Risk-free interest rate
|
|
|
2.55
|
%
|
|
|
2.68
|
%
|
|
|
4.31
|
%
|
|
Dividend yield
|
|
|
0
|
%
|
|
|
0
|
%
|
|
|
0
|
%
|
Weighted-Average Expected Term Prior to
January 1, 2008, the expected term of options granted was
determined using the “shortcut” method, as illustrated
in the Securities and Exchange Commission’s Staff
Accounting Bulletin No. 107
(“SAB 107”). Under this approach, the expected
term was presumed to be the average of the vesting term and the
contractual term of the option. As detailed information about
the employees’ exercise behavior became available to the
Company, beginning on January 1, 2008, the Company no
longer used the above mentioned shortcut method and determines
the expected term of the options granted through a combination
of the Company’s own historical exercise experience and
expected future exercise activities and post-vesting termination
behavior. The change of approach in determining the estimated
weighted average expected life resulted in the assumption
decreasing from approximately 6.1 years to 5.0 years.
Under the Employee Stock Purchase Plan, the expected term of
employee stock purchase plan shares is the average of the
purchase periods under each offering period.
Volatility Prior to January 1, 2008, as
the Company considered itself a newly public entity with
insufficient historical data on volatility of its stock, the
expected volatility used was based on volatility of similar
entities (referred to as “guideline” companies). In
evaluating similarity, the Company considered factors such as
industry, stage of life cycle and size. Due to the availability
of historical volatility data of the Company’s own stock,
the Company began utilizing its historical volatility to
determine future volatility for the purpose of determining
share-based payments for all options granted on or after
January 1, 2008.
Risk-Free Interest Rate. The risk-free rate
that the Company uses in the Black-Scholes option valuation
model is based on U.S. Treasury zero-coupon issues with
remaining terms similar to the expected term of the options or
purchase rights on the respective grant dates.
Dividend Yield The Company has never declared
or paid any cash dividends and does not plan to pay cash
dividends in the foreseeable future, and, therefore, used an
expected dividend yield of zero in the valuation model.
Forfeiture Rate The Company uses historical
data to estimate pre-vesting option forfeitures and record
stock-based compensation expense only for those awards that are
expected to vest. All stock-based payment awards are amortized
on a straight-line basis over the requisite service periods of
the awards, which are generally the vesting periods. The Company
increased its estimated forfeiture rate during the three months
ended March 31, 2008 from approximately 5.9% at
December 31, 2007 to approximately 7.0%.
Restricted Stock Units The estimated fair
value of restricted stock units awards is calculated based on
the market price of Alexza’s common stock on the date of
grant, reduced by the present value of dividends expected to be
paid on Alexza common stock prior to vesting of the restricted
stock unit. The Company’s estimate assumes no dividends
will be paid prior to the vesting of the restricted stock unit.
As of December 31, 2009, there was $4,367,000, $486,000 and
$421,000 total unrecognized compensation costs related to
non-vested stock option awards, non-vested restricted stock
units and stock purchase rights,
81
respectively, which are expected to be recognized over a
weighted average period of 1.68 years, 1.92 years and
1.1 years, respectively.
Recently
Adopted Accounting Standards
Accounting
Standards Codification Topic No. 810 (“ASC
810”)
ASC 810 establishes accounting and reporting standards for
ownership interests in subsidiaries held by parties other than
the parent, the amount of consolidated net income (loss)
attributable to the parent and to the noncontrolling interests,
changes in a parent’s ownership interest and the valuation
of retained noncontrolling equity investments when a subsidiary
is deconsolidated. ASC 810 requires that the noncontrolling
interest continue to be attributed its share of losses even if
that attribution results in a deficit noncontrolling interest
balance. ASC 810 also establishes additional reporting
requirements that identify and distinguish between the ownership
interest of the parent and the interest of the noncontrolling
owners.
On January 1, 2009, the Company adopted the provisions of
ASC 810 and reclassified the noncontrolling interest in Allegro
from a liability to stockholders’ (deficit) equity on its
Consolidated Balance Sheets on a retrospective basis. Had the
previous requirements been applied, the net loss attributable to
noncontrolling interest would have decreased by $8,626,000
during the year ended December 31, 2009. In addition,
consolidated net loss has been adjusted to include the net loss
attributed to the noncontrolling interest in Allegro and
consolidated comprehensive income or loss has been adjusted to
include the comprehensive income or loss attributed to the
noncontrolling interest in Allegro.
Recently
Issued Accounting Standards
Accounting
Standards Update
No. 2010-13
(“ASU
2010-13”)
In September 2009, the FASB ratified ASU
2010-13,
which eliminates the residual method of allocation and the
requirement to use the relative selling price method when
allocating revenue in a multiple deliverable arrangement. When
applying the relative selling price method, the selling price
for each deliverable shall be determined using vendor specific
objective evidence of selling price, if it exists, otherwise
third-party evidence of selling price. If neither vendor
specific objective evidence nor third-party evidence of selling
price exists for a deliverable, companies shall use its best
estimate of the selling price for that deliverable when applying
the relative selling price method. ASU
2010-13
shall be effective in fiscal years beginning on or after
June 15, 2010, with earlier application permitted.
Companies may elect to adopt this guidance prospectively for all
revenue arrangements entered into or materially modified after
the date of adoption, or retrospectively for all periods
presented. The Company is currently evaluating the potential
impact, if any, of the adoption of this guidance on its
financial position, results of operations and cash flows.
|
|
|
4.
|
Net Loss
per Share Attributable to Alexza Common Stockholders
|
Basic and diluted net loss per share attributable to Alexza
common stockholders is calculated by dividing the net loss
attributable to Alexza common stockholders by the
weighted-average number of common shares outstanding for the
period less weighted average shares subject to repurchase, of
which there were none in 2009, 2008 or 2007. Outstanding stock
options, warrants, and unvested restricted stock units are not
included in the net loss per share attributable to Alexza common
stockholders calculation for the years ended December 31,
2009, 2008 and 2007 as the inclusion of such shares would have
had an anti-dilutive effect.
Potentially dilutive securities include the following (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2009
|
|
2008
|
|
2007
|
|
|
|
Outstanding stock options
|
|
|
4,570
|
|
|
|
3,710
|
|
|
|
2,927
|
|
|
Unvested restricted stock units
|
|
|
706
|
|
|
|
123
|
|
|
|
58
|
|
|
Warrants to purchase common stock
|
|
|
5,091
|
|
|
|
2,324
|
|
|
|
2,016
|
|
82
|
|
|
5.
|
Cash,
Cash Equivalents, and Marketable Securities
|
The following table outlines the amortized cost, fair value and
unrealized gain/(loss) for the Company’s financial assets
by major security type as of December 31, 2009 and 2008 (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unrealized
|
|
|
December 31, 2009
|
|
Amortized Cost
|
|
|
Fair Value
|
|
|
Gain/(Loss)
|
|
|
|
|
Cash
|
|
$
|
778
|
|
|
$
|
778
|
|
|
$
|
—
|
|
|
Money market funds
|
|
|
10,421
|
|
|
|
10,421
|
|
|
|
—
|
|
|
Government-sponsored enterprises
|
|
|
5,218
|
|
|
|
5,217
|
|
|
|
(1
|
)
|
|
Corporate debt securities
|
|
|
3,500
|
|
|
|
3,500
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
19,917
|
|
|
$
|
19,916
|
|
|
$
|
(1
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unrealized
|
|
|
December 31, 2008
|
|
Amortized Cost
|
|
|
Fair Value
|
|
|
Gain/(Loss)
|
|
|
|
|
Cash
|
|
$
|
432
|
|
|
$
|
432
|
|
|
$
|
—
|
|
|
Money market funds
|
|
|
19,350
|
|
|
|
19,350
|
|
|
|
—
|
|
|
Money market fund held by Symphony Allegro, Inc.
|
|
|
21,318
|
|
|
|
21,318
|
|
|
|
—
|
|
|
Corporate debt securities
|
|
|
9,633
|
|
|
|
9,649
|
|
|
|
16
|
|
|
Government securities
|
|
|
1,505
|
|
|
|
1,505
|
|
|
|
—
|
|
|
Government-sponsored enterprises
|
|
|
6,608
|
|
|
|
6,620
|
|
|
|
12
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
58,846
|
|
|
$
|
58,874
|
|
|
$
|
28
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, 2009 and 2008, the Company reported the
financial assets as:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
13,450
|
|
|
$
|
26,036
|
|
|
Investments held by Symphony Allegro, Inc.
|
|
|
—
|
|
|
|
21,318
|
|
|
Marketable securities
|
|
|
6,466
|
|
|
|
11,520
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
19,916
|
|
|
$
|
58,874
|
|
|
|
|
|
|
|
|
|
|
|
The Company had no sales of marketable securities during the
years ended December 31, 2009, 2008 or 2007. As of
December 31, 2009, all of the Company’s marketable
securities have a maturity of less than one year.
When determining if there are any
“other-than-temporary”
impairments on its investments, the Company evaluates:
(i) whether the investment has been in a continuous
realized loss position for over twelve months, (ii) the
duration to maturity of the Company’s investments,
(iii) the Company’s intention to hold the investments
to maturity and if it is not more likely than not that the
Company will be required to sell the investment before recovery
of the amortized cost bases, (iv) the credit rating of each
investment, and (v) the type of investments made. Through
December 31, 2009, the Company has not recognized any
“other-than-temporary”
losses on its investments. As of December 31, 2009, no
investments have been in a continuous realized loss position for
longer than twelve months.
83
|
|
|
6.
|
Property
and Equipment
|
Property and equipment consisted of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Lab equipment
|
|
$
|
11,702
|
|
|
$
|
11,019
|
|
|
Computer equipment and software
|
|
|
4,755
|
|
|
|
4,885
|
|
|
Furniture
|
|
|
1,060
|
|
|
|
1,123
|
|
|
Leasehold improvements
|
|
|
19,255
|
|
|
|
19,135
|
|
|
Construction in progress — manufacturing equipment
|
|
|
4,240
|
|
|
|
1,019
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
41,012
|
|
|
|
37,181
|
|
|
Less: accumulated depreciation
|
|
|
(17,414
|
)
|
|
|
(13,029
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
23,598
|
|
|
$
|
24,152
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment also includes equipment that secures the
Company’s equipment financing agreements of $7,813,000 and
$14,338,000 at December 31, 2009 and 2008, respectively.
Accumulated depreciation related to assets under the equipment
financing loans was $5,498,000 and $9,588,000 at
December 31, 2009 and 2008, respectively. Depreciation of
property and equipment under equipment financing agreements is
included in depreciation expense in the statement of cash flows.
|
|
|
7.
|
Other
Accrued Liabilities
|
Other accrued liabilities consisted of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Accrued compensation
|
|
$
|
2,174
|
|
|
$
|
4,012
|
|
|
Accrued professional fees
|
|
|
630
|
|
|
|
439
|
|
|
Other
|
|
|
677
|
|
|
|
754
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
3,481
|
|
|
$
|
5,205
|
|
|
|
|
|
|
|
|
|
|
|
Equipment
Financing Obligations
The Company finances a portion of its fixed asset acquisitions
through equipment financing agreements. Loans drawn from the
equipment financing agreement are secured by certain fixed
assets of the Company. Fixed asset purchases used to secure
draws on the equipment financing agreement are recorded on the
Company’s balance sheet at cost. A liability is recorded
upon the Company making a draw on the agreements.
The loans are repaid in 36 - 48 monthly installments,
from the date of each draw, of principal and interest. The
interest rate, which is fixed for each draw, is based on the
U.S. Treasuries of comparable maturities and ranges from
9.2% to 10.6%. The equipment purchased under the equipment
financing agreement is pledged as security. As of
December 31, 2009, no additional borrowings were available
under the agreements. The Company believes the carrying value of
the debt is equal it its fair value at December 31, 2009.
84
Future scheduled principal payments under the equipment
financing agreements as of December 31, 2009 are as follows
(in thousands):
| |
|
|
|
|
|
2010
|
|
$
|
2,088
|
|
|
2011
|
|
|
427
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
2,515
|
|
|
|
|
|
|
|
Due to a late payment, the Company may have been in default of
the terms of its equipment financing obligations as of
December 31, 2009. The Company does not believe it was in
default; however, if the Company was in default, the lender
would have the right to demand payment on all outstanding
obligations. As a result, the Company has classified all of the
outstanding equipment financing obligations as a current
liability as of December 31, 2009. Subsequent to the late
payment, the Company paid the installment that was at issue.
Operating
Leases
The Company leases two buildings in Mountain View, California,
which the Company began to occupy in the fourth quarter of 2007.
The Company recognizes rental expense on the facility on a
straight line basis over the initial term of the lease.
Differences between the straight line rent expense and rent
payments are classified as deferred rent liability on the
balance sheet. The lease for both facilities expires on
March 31, 2018, and the Company has two options to extend
the lease for five years each.
The Mountain View lease, as amended, included $15,964,000 of
tenant improvement reimbursements from the landlord. The Company
has recorded all tenant improvements as additions to property
and equipment and is amortizing the improvements over the
shorter of the estimated useful life of the improvement or the
remaining life of the lease. The reimbursements received from
the landlord are included in deferred rent liability and
amortized over the life of the lease as a contra-expense.
In May 2008, the Company entered into an agreement to sublease a
portion of its Mountain View facility. The sublease agreement,
as amended, expires on April 30, 2010, at which time it
will convert to a
month-to-month
lease.
In January 2010, the Company entered into an agreement to
sublease an additional portion of its Mountain View facility
from March 1, 2010 through February 28, 2014. The
sublessee has an option to extend the lease agreement for
12 months and a second option to extend the lease agreement
an additional 37 months. If the sublessee exercises these
options, the rent will be at fair market rates at the time the
option is exercised.
Future minimum lease payments under non-cancelable operating
leases, net of sublease income, at
December 31,
2009 were as follows (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Lease
|
|
|
Sublease
|
|
|
Net
|
|
|
|
|
Payments
|
|
|
Receipts
|
|
|
Payments
|
|
|
|
|
2010
|
|
$
|
5,016
|
|
|
$
|
(500
|
)
|
|
$
|
4,516
|
|
|
2011
|
|
|
5,138
|
|
|
|
(426
|
)
|
|
|
4,712
|
|
|
2012
|
|
|
5,263
|
|
|
|
(438
|
)
|
|
|
4,825
|
|
|
2013
|
|
|
4,919
|
|
|
|
(451
|
)
|
|
|
4,468
|
|
|
2014
|
|
|
4,934
|
|
|
|
(74
|
)
|
|
|
4,860
|
|
|
Thereafter
|
|
|
15,858
|
|
|
|
—
|
|
|
|
15,858
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total minimum payments
|
|
$
|
41,128
|
|
|
$
|
(1,889
|
)
|
|
$
|
39,239
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Rental expense, net of sublease income, was $3,050,000,
$4,778,000, $5,402,000, and $18,971,000, for the years ended
December 31, 2009, 2008 and 2007, and for the period from
December 19, 2000 (inception) to December 31, 2009,
respectively. Rental income from the sublease agreement was
$656,000, $430,000, and $1,211,000 for the years ended
December 31, 2009 and 2008 and for the period from
December 19, 2000 (inception) to December 31, 2009,
respectively. The Company received no rental income in the year
ended December 31, 2007.
85
Manufacturing
and Supply Agreement
On November 2, 2007, The Company entered into a
manufacturing and supply agreement, or the supply agreement,
with Autoliv ASP, Inc, or Autoliv, relating to the commercial
supply of chemical heat packages that can be incorporated into
the Company’s Staccato device. Autoliv had developed
these chemical heat packages for the Company pursuant to a
development agreement between Autoliv and the Company executed
in October 2005. Under the terms of the supply agreement,
Autoliv agreed to develop a manufacturing line capable of
producing 10 million chemical heat packages a year. The
Company agreed to pay Autoliv $12 million upon the earlier
of December 31, 2011 or 60 days after the approval by
the Food and Drug Administration of a new drug application filed
by the Company. If the agreement is terminated by either party,
the Company will be required to reimburse Autoliv up to
$12 million for certain expenses related to the equipment
and tooling used in the production and testing of the chemical
heat packages. Upon payment by the Company, Autoliv will be
required to transfer possession and ownership of such equipment
and tooling to the Company. Each quarter, the Company estimates
the amount of work performed on the development of the
manufacturing line and recognizes a portion of the total payment
related to the manufacturing line as a capital asset and a
corresponding non-current liability. As of December 31,
2009, the Company recorded a fixed asset and a current
liability, based on our a Prescription Drug User Fee Act goal
date of October 11, 2010, of $3,750,000 related to its
commitment to Autoliv for the development of the manufacturing
line. Autoliv has also agreed to manufacture, assemble and test
the chemical heat packages solely for the Company in conformance
with the Company’s specifications. The Company will pay
Autoliv a specified purchase price, which varies based on annual
quantities ordered by the Company, per chemical heat package
delivered. The initial term of the supply agreement expires on
December 31, 2012 and may be extended by written mutual
consent.
Autoliv has agreed to manufacture, assemble and test the
Chemical Heat Packages solely for the Company in conformance
with the Company’s specifications. The Company will pay
Autoliv a specified purchase price, which varies based on annual
quantities ordered by the Company, per Chemical Heat Package
delivered. The initial term of the Supply Agreement expires on
December 31, 2012 and may be extended by mutual written
consent. The Supply Agreement provides that during the term of
the Supply Agreement, Autoliv will be the Company’s
exclusive supplier of the Chemical Heat Packages. In addition,
the Supply Agreement grants Autoliv the right to negotiate for
the right to supply commercially any second generation chemical
heat package (a “Second Generation Product”) and
provides that the Company will pay Autoliv certain royalty
payments if the Company manufactures Second Generation Products
itself or if the Company obtains Second Generation Products from
a third party manufacturer. Upon the expiration or termination
of the Supply Agreement the Company will be required, on an
ongoing basis, to pay Autoliv certain royalty payments related
to the manufacture of the Chemical Heat Packages by the Company
or third party manufacturers. No Chemical Heat Packages have
been purchased under this agreement as of December 31, 2009.
Symphony
Allegro, Inc.
On December 1, 2006 (the “Closing Date”), the
Company entered into a series of related agreements with
Symphony Capital LLC (“Symphony Capital”), Symphony
Allegro Holdings LLC (“Holdings”) and Allegro,
providing for the financing of the clinical development of its
AZ-002, Staccato alprazolam, and the AZ-004/104,
Staccato loxapine, product candidates (the
“Programs”). Symphony Capital and other investors
(collectively, the “Allegro Investors”) invested
$50,000,000 in Holdings, which then invested the $50,000,000 in
Allegro. Pursuant to the agreements, Allegro agreed to invest up
to the full $50,000,000 to fund the clinical development of the
Programs, and the Company licensed to Allegro certain
intellectual property rights related to the Programs.
The Company continued to be primarily responsible for all
preclinical, clinical and device development efforts, as well as
for maintenance of the intellectual property portfolio for the
Programs. The Company had no further obligation beyond the items
described above and the Company had no obligation to the
creditors of Allegro as a result of the Company’s
involvement with Allegro. The investments held by Allegro were
to be used to fund the development of the Programs, and were not
available for general corporate expenses. The Company issued to
Holdings five-year warrants to purchase 2,000,000 shares of
the Company’s common stock at $9.91 per share. The
86
warrants, issued upon closing, were assigned a value of
$10.7 million using the Black-Scholes valuation model and
had been recorded in additional paid in capital.
In consideration for the warrants, the Company received an
exclusive purchase option (the “Purchase Option”) that
gave the Company the right, but not the obligation, to acquire
all, but not less than all, of the outstanding equity of
Allegro, thereby allowing the Company to reacquire all of the
Programs. Prior to the amendments of the terms of the Purchase
Option described below, this Purchase Option was exercisable at
any time from December 1, 2007 to December 1, 2010, at
predetermined prices that increased over time and ranged from
$67,500,000 starting December 31, 2007 to $122,500,000
through December 1, 2010.
In June 2009, the Company entered into an agreement with
Holdings to amend the provisions of and to exercise the Purchase
Option. The Company completed the acquisition of all of the
outstanding equity of Allegro pursuant to the amended Purchase
Option on August 26, 2009. In exchange for all of the
outstanding equity of Allegro, the Company, in lieu of the
consideration described above: (i) issued to the Allegro
Investors 10,000,000 shares of common stock
(ii) issued to the Allegro Investors warrants to purchase
5,000,000 shares of common stock at an exercise price of
$2.26 per share that are cash or net exercisable for a period of
5 years and canceled the warrants to purchase
2,000,000 shares of common stock held by the Allegro
Investors and (iii) will pay Holdings certain percentages
of cash payments that may be generated from future partnering
transactions for the Programs. Pursuant to a registration rights
agreement with Holdings, the Company filed with the SEC a
registration statement for these shares of common stock and the
shares of common stock underlying the warrants. The SEC declared
such registration statement effective on October 16, 2009
and, pursuant to the registration rights agreement with
Holdings, the Company has an obligation to take certain actions
as are necessary keep such registration statement effective.
Prior to the completion of the acquisition of all of the
outstanding equity of Allegro pursuant to the amended Purchase
Option, the Company had concluded that Allegro was by design a
variable interest entity as the Company had a purchase option to
acquire Allegro’s outstanding voting stock at prices that
were fixed based upon the date the option was exercised. The
fixed nature of the purchase option price limited the returns of
the Allegro Investors, as the investors in Allegro. Parties to
an arrangement are considered to be de facto agents if they
cannot sell, transfer, or encumber their interests without the
prior approval of an enterprise. Symphony Capital was considered
to be a de facto agent of the Company pursuant to this
provision, and because the Company and the Allegro Investors, as
a related party group, absorbed a majority of Allegro’s
variability, the Company evaluated whether the Company was most
closely associated with Allegro. The Company concluded that it
was most closely associated with Allegro and should consolidate
Allegro because (i) the Company originally developed the
technology that was assigned to Allegro, (ii) the Company
continued to oversee and monitor the development program,
(iii) the Company’s employees continued to perform
substantially all of the development work, (iv) the Company
significantly influenced the design of the responsibilities and
corporate structure of Allegro, (v) Allegro’s
operations were substantially similar to the Company’s
activities, and (vi) through the Purchase Option, the
Company had the ability to meaningfully participate in the
benefits of a successful development effort.
The noncontrolling interest in Symphony Allegro, Inc.,
represented an equity investment by the Allegro Investors in
Allegro of $50,000,000 reduced by $10,708,000 for the value of
the Purchase Option, and by $2,829,000 for a structuring fee and
related expenses that the Company paid to Symphony Capital in
connection with the closing of the Allegro transaction,
resulting in the recording of a net noncontrolling interest of
$36,463,000 on the effective date. The Company charged the
losses incurred by Allegro, prior to August 26, 2009, to
the noncontrolling interest in the determination of the net loss
attributable to the Alexza common stockholders in the
consolidated statements of operations, and the Company also
reduced the noncontrolling interest in the consolidated balance
sheets by Allegro’s losses. For the years ended
December 31, 2009, 2008, and 2007, and the period from
December 19, 2000 (inception) to December 31, 2009,
the net losses of Allegro charged to the noncontrolling interest
were $13,987,000 $18,591,000, $10,791,000, and $45,089,000
respectively.
Upon closing of the acquisition of all of the outstanding equity
of Allegro pursuant to the amended Purchase Option, the Company
recorded the acquisition as a capital transaction that did not
affect its net loss. However, because the acquisition was
accounted for as a capital transaction, the excess consideration
transferred over the carrying value of the noncontrolling
interest in Allegro was treated as a deemed dividend for
purposes of reporting
87
net loss per share, increasing net loss per share attributable
to Alexza stockholders by $61,566,000 during the year ended
December 31, 2009. In addition, upon the closing, the
Company ceased to charge net losses of Allegro against the
noncontrolling interest.
The following table outlines the estimated fair value of
consideration transferred by Alexza and the computation of the
excess consideration transferred over the carrying value of the
noncontrolling interest in Allegro at the acquisition date (in
thousands):
| |
|
|
|
|
|
Description
|
|
Fair Value
|
|
|
|
|
Fair value of consideration transferred:
|
|
|
|
|
|
10,000,000 shares of Alexza common stock
|
|
$
|
28,000
|
|
|
Warrant consideration, net
|
|
|
8,085
|
|
|
Fair value of contingent cash payments to Allegro stockholders
|
|
|
16,855
|
|
|
|
|
|
|
|
|
Total consideration transferred
|
|
|
52,940
|
|
|
Add: Deficit of noncontrolling interest in Allegro
|
|
|
8,626
|
|
|
|
|
|
|
|
|
Excess consideration transferred over the carrying value of the
noncontrolling interest in Allegro
|
|
$
|
61,566
|
|
|
|
|
|
|
|
The fair value of the Alexza common stock of $2.80 was based on
the closing sales price of the Company’s common stock on
the NASDAQ Global Market on August 26, 2009, which is the
date the transaction was completed.
The estimated fair values of the warrant consideration were
calculated using the Black-Scholes valuation model, and the
following assumptions:
| |
|
|
|
|
|
|
|
Warrant
|
|
Warrant
|
|
|
|
Issued
|
|
Cancelled
|
|
|
|
Number of Shares
|
|
5,000,000
|
|
2,000,000
|
|
Expected term
|
|
5.0 years
|
|
2.3 years
|
|
Expected volatility
|
|
89%
|
|
117%
|
|
Risk-free interest rate
|
|
2.46%
|
|
1.06%
|
|
Dividend yield
|
|
0%
|
|
0%
|
Endo
Pharmaceuticals, Inc.
On December 27, 2007, the Company entered into a license,
development and supply agreement (the “license
agreement”), with Endo Pharmaceuticals, Inc.
(“Endo”) for AZ-003 (Staccato fentanyl) and the
fentanyl class of molecules for North America. Under the terms
of the license agreement, Endo paid the Company a $10,000,000
non-refundable upfront fee and Endo was obligated to pay
potential additional milestone payments of up to $40,000,000
upon achievement of predetermined regulatory and clinical
milestones. Endo was also obligated to pay royalties to the
Company on net sales of the product, from which the Company
would be required to pay for the cost of goods for the
manufacture of the commercial version of the product. Under the
terms of the license agreement, the Company had primary
responsibility for the development and costs of the Staccato
Electronic Multiple Dose device and the exclusive right to
manufacture the product for clinical development and commercial
supply. Endo had the responsibility for future pre-clinical,
clinical and regulatory development, and, if AZ-003 was approved
for marketing, for commercializing the product in North America.
The Company recorded the $10,000,000 upfront fee it received
from Endo in January 2008 as deferred revenue. The Company was
unable to allocate a fair value to the each of the deliverables
outlined in the agreement and therefore accounted for the
deliverables as a single unit of accounting. The Company began
to recognize the $10,000,000 upfront payment as revenue in the
third quarter of 2008 over the estimated performance period of
six years, resulting in revenues of $486,000 in 2008.
In January 2009, the Company and Endo mutually agreed to
terminate the license agreement, with all rights to AZ-003
reverting back to the Company. The Company’s obligations
under the license agreement were fulfilled
88
upon the termination of the agreement, and the Company
recognized the remaining deferred revenue of $9,514,000 during
the three months ended March 31, 2009.
The Company had reserved shares of common stock for future
issuances as of December 31, 2009 as follows:
| |
|
|
|
|
|
Stock options outstanding
|
|
|
4,740,499
|
|
|
Unvested restricted stock units outstanding
|
|
|
196,270
|
|
|
2005 Equity Incentive Plan and 2005 Non Employee Director Stock
Option Plan — shares available for issuance
|
|
|
534,179
|
|
|
Employee Stock Purchase Plan — shares available for
issuance
|
|
|
156,243
|
|
|
Warrants outstanding
|
|
|
12,727,554
|
|
|
|
|
|
|
|
|
Total
|
|
|
18,354,745
|
|
|
|
|
|
|
|
In March 2002, in connection with an equipment financing
agreement, the Company issued immediately exercisable and fully
vested warrants to purchase 21,429 shares of Series B
preferred stock at a per share price of $1.40. The warrants
expire on April 8, 2013. The Company recorded a deferred
financing cost of $27,000 related to the issuance of these
warrants. The Company valued these warrants using the
Black-Scholes valuation model, assuming an exercise price and
fair value of $1.40, an expected volatility of 100%, an expected
life of 10 years, an expected dividend yield of 0%, and a
risk-free interest rate of 4.61%. The estimated fair value of
the warrants is recorded as debt discount. This amount was
amortized to interest expense over the commitment term of the
equipment financing agreement. In 2006, the warrant was
converted to purchase 4,116 shares of common stock at a
price of $7.29 per share. As of December 31, 2009, this
warrant remained outstanding and exercisable.
In January and September 2003, in connection with the
modifications of an equipment financing agreement, the Company
issued immediately exercisable and fully vested warrants to
purchase 24,058 and 19,247 shares of Series C
preferred stock, respectively, at a per share price of $1.56.
The warrants expire on April 8, 2013. The Company valued
these warrants using the Black-Scholes valuation model, assuming
an exercise price and fair value of $1.56, an expected
volatility of 100%, an expected life of 10 years, an
expected dividend yield of 0%, and risk-free interest rate of
4.05% and 4.45%, respectively. The estimated fair values of
$35,000 and $27,000, respectively, were recorded as debt
discount and was amortized to interest expense over the
remaining commitment term of the financing agreement. In 2006,
these warrants were converted into warrants to purchase
4,852 shares and 3,882 shares of common stock, both at
a price of $7.74 shares. As of December 31, 2009, both
of these warrants remained outstanding and exercisable.
In March 2004, in connection with the modifications of an
equipment financing agreement, the Company issued immediately
exercisable and fully vested warrants to purchase
14,232 shares of Series C preferred stock at a per
share price of $1.56. The warrants expire on April 8, 2013.
The Company valued these warrants using the Black-Scholes
valuation model, assuming an exercise price and fair value of
$1.56, an expected volatility of 100%, an expected life of
10 years, an expected dividend yield of 0%, and risk-free
interest rate of 4.35%. The estimated fair value of $20,000 was
recorded as debt discount and amortized to interest expense over
the remaining commitment term of the financing agreement. In
2006, the warrant was converted into a warrant to purchase
2,870 shares of common stock at a price of $7.74. As of
December 31, 2009, these warrants remained outstanding and
exercisable.
In December 2006, in connection with the Symphony Allegro
transaction (see Note 9), the Company issued to Holdings a
five-year warrant to purchase 2,000,000 shares of the
Company’s common stock at $9.91 per share. The warrants
issued upon closing were assigned a value of $10.7 million
in accordance with the Black-Scholes option valuation
methodology assuming an exercise price of $9.91, an expected
volatility of 80%, an expected life of 5 years, an expected
dividend yield of 0% and risk-free interest rate of 4.45%. This
fair value has been recorded as a reduction to the
noncontrolling interest in Symphony Allegro. In August 2009,
this warrant was cancelled in conjunction with the
Company’s purchase of Symphony Allegro.
89
In March 2008, in connection with the registered direct equity
issuance to Bio*One described in Note 1, the Company issued
a warrant to Bio*One to purchase up to 375,000 of additional
shares of Alexza common stock at a purchase price per share of
$8.00. As outlined in the agreement, the warrant was subject to
the same price adjustment as the common stock sale, and
effective January 1, 2009 the warrant was adjusted to
purchase 415,522 shares at a purchase price of $7.22 per
share. The Company committed to initiate and maintain
manufacturing operations in Singapore, and the warrant was to
become exercisable only if the Company terminates operations in
Singapore or does not achieve certain performance milestones.
The warrant has a maximum term of 5 years. Net proceeds
from the sale of the stock and warrant were approximately
$9.84 million after deducting offering expenses. In
December 2008, the Company did not meet its defined performance
milestone, and as a result the warrant became fully exercisable.
At December 31, 2009, this warrant remains outstanding and
exercisable.
In August 2009, in connection with the acquisition of Symphony
Allegro (See Note 9) the Company issued five year
warrants to the Allegro Investors to purchase
5,000,000 shares of Alexza common stock at a price per
share of $2.26. At December 31, 2009, the warrants remained
outstanding and exercisable.
In October 2009, in conjunction with a private equity issuance
(see Note 1), the Company issued seven year warrants to
purchase an aggregate of 7,296,312 shares of its common
stock with an exercise price per share of $2.77. The warrants
are cash or net exercisable for a period of seven years from
October 5, 2009 and have an exercise price of $2.77 per
share. The Company granted to the investors certain registration
rights related to the shares of common stock underlying the
warrants. The Company filed with the SEC a registration
statement covering the resale of these shares, and the SEC
declared such registration statement effective on
October 27, 2009. The Company also agreed to other
customary obligations regarding registration, including
indemnification and maintenance of the registration statement.
At December 31, 2009, the warrants remained outstanding and
exercisable.
|
|
|
12.
|
Equity
Incentive Plans
|
2005
Equity Incentive Plan
In December 2005, the Company’s Board of Directors adopted
the 2005 Equity Incentive Plan (the “2005 Plan”) and
authorized for issuance thereunder 1,088,785 shares of
common stock. The 2005 Plan became effective upon the closing of
the Company’s initial public offering on March 8,
2006. The 2005 Plan is an amendment and restatement of the
Company’s previous stock option plans. Stock options issued
under the 2005 Plan generally vest over 4 years, vesting is
generally based on service time, and have a maximum contractual
term of 10 years.
In the third quarter of 2006, the Company began issuing
restricted stock units to non-officer employees. Beginning in
2009, the Company began issuing restricted stock units to both
officers and to non-employee directors. Restricted stock unit
issuances to non-employee directors were made in lieu of paying
cash director fees. Restricted stock units granted to officer or
non-officer employees generally vest over a four-year period
from the grant date or upon completion of certain performance
milestones. Restricted stock units granted to non-employee
directors generally vest one year after the date of grant. Prior
to vesting, restricted stock units do not have dividend
equivalent rights, do not have voting rights and the shares
underlying the restricted units are not considered issued and
outstanding. Shares are issued on the date the restricted stock
units vest.
The 2005 Plan provides for annual reserve increases on the first
day of each fiscal year commencing on January 1, 2007 and
ending on January 1, 2015. The annual reserve increases
will be equal to the lesser of (i) 2% of the total number
of shares of the Company’s common stock outstanding on
December 31 of the preceding calendar year, or
(ii) 1,000,000 shares of common stock. The
Company’s Board of Directors has the authority to designate
a smaller number of shares by which the authorized number of
shares of common stock will be increased prior to the last day
of any calendar year. In May 2008, the Company’s
stockholders approved an amendment to the plan to increase the
number of shares of the Company’s stock reserved for
issuance under the 2005 Plan by an additional
1,500,000 shares.
90
2005
Non-Employee Directors’ Stock Option Plan
In December 2005, the Company’s Board of Directors adopted
the 2005 Non-Employee Directors’ Stock Option Plan (the
“Directors’ Plan”) and authorized for issuance
thereunder 250,000 shares of common stock. The
Directors’ Plan became effective immediately upon the
closing of the Company’s initial public offering on
March 8, 2006. The Directors’ Plan provides for the
automatic grant of nonstatutory stock options to purchase shares
of common stock to the Company’s non-employee directors,
which vest over four years and have a term of 10 years. The
Directors’ Plan provides for an annual reserve increase to
be added on the first day of each fiscal year, commencing on
January 1, 2007 and ending on January 1, 2015. The
annual reserve increases will be equal to the number of shares
subject to options granted during the preceding fiscal year less
the number of shares that revert back to the share reserve
during the preceding fiscal year. The Company’s Board of
Directors has the authority to designate a smaller number of
shares by which the authorized number of shares of common stock
will be increased prior to the last day of any calendar year.
The following table sets forth the summary of stock option
activity under the Equity Incentive Plans:
| |
|
|
|
|
|
|
|
|
|
|
|
Outstanding Options
|
|
|
|
Number of
|
|
Weighted Average
|
|
|
|
Shares
|
|
Exercise Price
|
|
|
|
Balance as of January 1, 2007
|
|
|
2,611,042
|
|
|
$
|
5.23
|
|
|
Options granted
|
|
|
1,054,656
|
|
|
$
|
9.10
|
|
|
Options exercised
|
|
|
(204,423
|
)
|
|
$
|
2.11
|
|
|
Options forfeited
|
|
|
(249,536
|
)
|
|
$
|
6.98
|
|
|
Options cancelled
|
|
|
(4,875
|
)
|
|
$
|
6.60
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2007
|
|
|
3,206,864
|
|
|
$
|
6.56
|
|
|
Options granted
|
|
|
1,472,171
|
|
|
$
|
5.26
|
|
|
Options exercised
|
|
|
(104,428
|
)
|
|
$
|
1.55
|
|
|
Options forfeited
|
|
|
(190,284
|
)
|
|
$
|
7.20
|
|
|
Options cancelled
|
|
|
(200,975
|
)
|
|
$
|
7.81
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2008
|
|
|
4,183,348
|
|
|
$
|
6.14
|
|
|
Options granted
|
|
|
1,394,632
|
|
|
$
|
2.48
|
|
|
Options exercised
|
|
|
(69,708
|
)
|
|
$
|
1.20
|
|
|
Options forfeited
|
|
|
(422,118
|
)
|
|
$
|
5.79
|
|
|
Options cancelled
|
|
|
(345,655
|
)
|
|
$
|
6.08
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31, 2009
|
|
|
4,740,499
|
|
|
$
|
5.17
|
|
|
|
|
|
|
|
|
|
|
|
|
Options exercisable at:
|
|
|
|
|
|
|
|
|
|
December 31, 2007
|
|
|
1,365,538
|
|
|
$
|
5.54
|
|
|
December 31, 2008
|
|
|
1,950,662
|
|
|
$
|
6.02
|
|
|
December 31, 2009
|
|
|
2,865,898
|
|
|
$
|
5.71
|
|
The total intrinsic value of options exercised during the years
ended December 31, 2009, 2008, and 2007 was $80,000,
$463,000, and $1,662,000, respectively. None of the
Company’s options have expired.
91
Information regarding the stock options outstanding at
December 31, 2009 is summarized below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding
|
|
|
Exercisable
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
|
|
|
|
|
Contractual
|
|
|
Aggregate
|
|
|
|
|
|
Contractual
|
|
|
Aggregate
|
|
|
|
|
Number
|
|
|
Life
|
|
|
Intrinsic
|
|
|
Number
|
|
|
Life
|
|
|
Intrinsic
|
|
|
Exercise Price
|
|
of Shares
|
|
|
(In Years)
|
|
|
Value
|
|
|
of Shares
|
|
|
(In Years)
|
|
|
Value
|
|
|
|
|
$1.10 – 1.38
|
|
|
520,882
|
|
|
|
4.94
|
|
|
$
|
630,000
|
|
|
|
515,458
|
|
|
|
4.93
|
|
|
$
|
624,000
|
|
|
1.69 – 2.37
|
|
|
775,724
|
|
|
|
9.66
|
|
|
|
139,000
|
|
|
|
127,523
|
|
|
|
9.17
|
|
|
|
40,000
|
|
|
2.64 – 3.13
|
|
|
495,965
|
|
|
|
9.05
|
|
|
|
—
|
|
|
|
226,130
|
|
|
|
9.06
|
|
|
|
—
|
|
|
3.30 – 4.35
|
|
|
674,735
|
|
|
|
8.38
|
|
|
|
—
|
|
|
|
268,622
|
|
|
|
8.15
|
|
|
|
—
|
|
|
4.41 – 7.00
|
|
|
505,829
|
|
|
|
7.77
|
|
|
|
—
|
|
|
|
320,099
|
|
|
|
7.54
|
|
|
|
—
|
|
|
7.20 – 7.90
|
|
|
340,560
|
|
|
|
7.05
|
|
|
|
—
|
|
|
|
279,521
|
|
|
|
7.03
|
|
|
|
—
|
|
|
8.00 – 8.00
|
|
|
575,457
|
|
|
|
4.56
|
|
|
|
—
|
|
|
|
562,775
|
|
|
|
4.53
|
|
|
|
—
|
|
|
8.01 – 8.89
|
|
|
547,908
|
|
|
|
7.48
|
|
|
|
—
|
|
|
|
343,299
|
|
|
|
7.45
|
|
|
|
—
|
|
|
8.91 – 11.70
|
|
|
303,439
|
|
|
|
7.13
|
|
|
|
—
|
|
|
|
222,471
|
|
|
|
7.09
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,740,499
|
|
|
|
7.47
|
|
|
$
|
769,000
|
|
|
|
2,865,898
|
|
|
|
6.63
|
|
|
$
|
664,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The intrinsic value is calculated as the difference between the
market value as of December 31, 2009 and the exercise price
of the shares. The market value as of December 31, 2009 was
$2.40 as reported by the NASDAQ Stock Market.
Information with respect to nonvested share units (restricted
stock units) as of December 31, 2009 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
Number
|
|
|
Average
|
|
|
|
|
of
|
|
|
Grant — Date
|
|
|
|
|
Shares
|
|
|
Fair Value
|
|
|
|
|
Outstanding at January 1, 2007
|
|
|
34,080
|
|
|
|
7.00
|
|
|
Granted
|
|
|
74,575
|
|
|
|
8.89
|
|
|
Released
|
|
|
(8,245
|
)
|
|
|
7.00
|
|
|
Forfeited
|
|
|
(7,285
|
)
|
|
|
7.71
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2007
|
|
|
93,125
|
|
|
|
8.42
|
|
|
Granted
|
|
|
112,423
|
|
|
|
4.35
|
|
|
Released
|
|
|
(23,443
|
)
|
|
|
8.33
|
|
|
Forfeited
|
|
|
(10,151
|
)
|
|
|
7.00
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2008
|
|
|
171,954
|
|
|
|
5.86
|
|
|
Granted
|
|
|
965,643
|
|
|
|
2.19
|
|
|
Released
|
|
|
(839,469
|
)
|
|
|
2.31
|
|
|
Forfeited
|
|
|
(101,858
|
)
|
|
|
4.03
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2009
|
|
|
196,270
|
|
|
|
3.90
|
|
|
|
|
|
|
|
|
|
|
|
The total intrinsic value of restricted stock units released
during the years ended December 31, 2009, 2008, and 2007
was $1,898,000, $131,000 and $68,000, respectively.
92
The Company authorized shares of common stock for issuance under
the Plans as follows.
| |
|
|
|
|
|
Year
|
|
Number of Shares
|
|
|
|
2001
|
|
|
363,636
|
|
|
2002
|
|
|
770,732
|
|
|
2003
|
|
|
454,545
|
|
|
2004
|
|
|
1,000,000
|
|
|
2005
|
|
|
25,544
|
|
|
2006
|
|
|
1,327,990
|
|
|
2007
|
|
|
676,386
|
|
|
2008
|
|
|
2,174,840
|
|
|
2009
|
|
|
656,417
|
|
As of December 31, 2009, 534,179 shares remained
available for issuance under the 2005 Plan and the
Directors’ Plan.
On January 1, 2010 an additional 1,037,500 shares were
authorized for issuance under the evergreen provisions of the
2005 Plan and the Directors’ Plan.
2005
Employee Stock Purchase Plan
In December 2005, the Company’s Board of Directors adopted
the 2005 Employee Stock Purchase Plan (“ESPP”) and
authorized for issuance thereunder 500,000 shares of common
stock. The ESPP allows eligible employee participants to
purchase shares of the Company’s common stock at a discount
through payroll deductions. The ESPP consists of a fixed
offering period, generally twenty-four months with four purchase
periods within each offering period. Purchases are generally
made on the last trading day of each October and April.
Employees purchase shares at each purchase date at 85% of the
market value of our common stock on their enrollment date or the
end of the purchase period, whichever price is lower. The
Company issued 439,252, 305,146, and 205,870, and shares at a
weighted average prices of $1.36, $3.84, and $6.83, 2009, 2008,
and 2007, respectively.
The ESPP provides for annual reserve increases on the first day
of each fiscal year commencing on January 1, 2007 and
ending on January 1, 2015. The annual reserve increases
will be equal to the lesser of (i) 1% of the total number
of shares of the Company’s common stock outstanding on
December 31 of the preceding calendar year, or
(ii) 250,000 shares of common stock. The
Company’s Board of Directors has the authority to designate
a smaller number of shares by which the authorized number of
shares of common stock will be increased prior to the last day
of any calendar year. On January 1, 2009, 2008 and 2007 an
additional 250,000, 250,000, and 238,193 shares,
respectively, were reserved for issuance under this provision.
At December 31, 2009, 156,243 shares are available for
issuance under the ESPP.
On January 1, 2010 an additional 250,000 shares were
reserved for issuance under the ESPP.
|
|
|
13.
|
Restructuring
Charges
|
In January 2009, the Company restructured its operations to
focus its efforts on the continued rapid development of its
AZ-004 (Staccato loxapine) product candidate. The
restructuring included a workforce reduction of
50 employees, representing approximately 33% of the
Company’s total workforce and was completed in the second
quarter of 2009. The Company incurred $2,037,000 of
restructuring expenses related to employee severance and other
termination benefits, including a non-cash charge of $56,000
related to modifications to share-based awards, and does not
expect to incur any additional expenses related to this
restructuring in future periods. As of December 31, 2009,
the Company had no outstanding amounts due related to the
restructuring.
The Company sponsors a 401(k) Plan that stipulates that eligible
employees can elect to contribute to the 401(k) Plan, subject to
certain limitations. Pursuant to the 401(k) Plan, the Company
does not match any employee contributions.
93
There is no provision for income taxes because the Company has
incurred operating losses since inception.
The reported amount of income tax expense attributable to
operations for the year differs from the amount that would
result from applying domestic federal statutory tax rates to
loss before income taxes from operations as summarized below (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
|
|
|
Federal tax benefit at statutory rate
|
|
$
|
(14,307
|
)
|
|
$
|
(19,873
|
)
|
|
$
|
(15,321
|
)
|
|
State tax benefit net of federal effect
|
|
|
(2,385
|
)
|
|
|
(3,402
|
)
|
|
|
(2,629
|
)
|
|
Research and development credits
|
|
|
(2,537
|
)
|
|
|
(2,693
|
)
|
|
|
(3,538
|
)
|
|
Other permanent differences
|
|
|
(31
|
)
|
|
|
19
|
|
|
|
20
|
|
|
Share-based compensation
|
|
|
1,112
|
|
|
|
1,450
|
|
|
|
274
|
|
|
Adjustment to basis in subsidiary
|
|
|
3,180
|
|
|
|
—
|
|
|
|
—
|
|
|
Change in valuation allowance
|
|
|
14,662
|
|
|
|
24,567
|
|
|
|
21,193
|
|
|
Other
|
|
|
306
|
|
|
|
(68
|
)
|
|
|
1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred income taxes reflect the net tax effects of the
temporary differences between the carrying amounts of assets and
liabilities for financial reporting purposes and the amount used
for income tax purposes. The deferred tax assets were calculated
using an effective tax rate of 40%. Significant components of
the Company’s deferred tax assets are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Federal and state net operating loss carryforwards
|
|
$
|
79,478
|
|
|
$
|
58,451
|
|
|
Federal and state research and development credit carryforwards
|
|
|
11,840
|
|
|
|
10,210
|
|
|
Accrued liabilities
|
|
|
9,410
|
|
|
|
2,736
|
|
|
Capitalized research and development costs
|
|
|
25,675
|
|
|
|
23,216
|
|
|
Other
|
|
|
49
|
|
|
|
42
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax assets
|
|
|
126,453
|
|
|
|
94,655
|
|
|
Valuation allowance
|
|
|
(126,453
|
)
|
|
|
(94,655
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax assets
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
The Company’s accounting for deferred taxes involves the
evaluation of a number of factors concerning the realizability
of the Company’s net deferred tax assets. The Company
primarily considered such factors as the Company’s history
of operating losses, the nature of the Company’s deferred
tax assets and the timing, likelihood and amount, if any, of
future taxable income during the periods in which those
temporary differences and carryforwards become deductible. At
present, the Company does not believe that it is more likely
than not that the deferred tax assets will be realized;
accordingly, a full valuation allowance has been established and
no deferred tax asset is shown in the accompanying balance
sheets. The valuation allowance increased by approximately
$31,798,000, $25,252,000, and $20,847,000 during the years ended
December 31, 2009, 2008, and 2007, respectively.
As of December 31, 2009 the Company had federal net
operating loss carryforwards of approximately $202,200,000. The
Company also had federal research and development tax credit
carryforwards of approximately $7,917,000. The net operating
loss and tax credit carryforwards will expire at various dates
beginning in 2020, if not utilized.
94
As of December 31, 2009, the Company had state net
operating loss carryforwards of approximately $193,800,000,
which will begin to expire in 2012. The Company also had state
research and development tax credit carryforwards of
approximately $3,845,000, which have no expiration, and a
Manufacturer’s Investment Credit of $78,000, which will
expire in 2010, if not utilized.
As of December 31, 2009, approximately $583,000 of deferred
tax assets is attributable to certain employee stock option
deductions and the federal and state net operating loss
carryforward has been adjusted accordingly. When realized, the
benefit of the tax deduction related to these options will be
accounted for as a credit to stockholders’ equity rather
than as a reduction of the income tax provision.
Utilization of the net operating loss carryforwards and credits
may be subject to an annual limitation with substantial effect,
due to the ownership change limitations provided by the Internal
Revenue Code that are applicable if the Company experiences an
“ownership change”. That may occur, for example, as a
result of the initial public offering aggregated with certain
other sales of the Company’s stock.
The Company recognized a decrease to the deferred tax assets in
2007, to increase its reserve for unrecognized tax benefits.
Because of the correlative reduction in the Company’s full
valuation allowance, this adjustment did not result in a credit
to deficit accumulated during the development stage. During
2008, the Company performed an analysis of its research and
development credits. As a result of this analysis, the Company
decreased its reserve for unrecognized tax benefits related to
research and development credits. Because of the correlative
reduction in the Company’s full valuation allowance, this
adjustment did not result in a credit to the statement of
operations.
A reconciliation of the beginning and ending amount of
unrecognized tax benefits is as follows:
| |
|
|
|
|
|
Balance at January 1, 2007
|
|
$
|
1,635
|
|
|
Additions based on tax positions taken during a prior period
|
|
|
—
|
|
|
Reductions based on tax positions taken during a prior period
|
|
|
—
|
|
|
Additions based on tax positions taken during the current period
|
|
|
611
|
|
|
Reductions based on tax positions taken during the current period
|
|
|
—
|
|
|
Reductions related to settlement of tax matters
|
|
|
—
|
|
|
Reductions related to a lapse of applicable statute of
limitations
|
|
|
—
|
|
|
|
|
|
|
|
|
Balance at December 31, 2007
|
|
$
|
2,246
|
|
|
Additions based on tax positions taken during a prior period
|
|
|
—
|
|
|
Reductions based on tax positions taken during a prior period
|
|
|
(1,067
|
)
|
|
Additions based on tax positions taken during the current period
|
|
|
401
|
|
|
Reductions based on tax positions taken during the current period
|
|
|
—
|
|
|
Reductions related to settlement of tax matters
|
|
|
—
|
|
|
Reductions related to a lapse of applicable statute of
limitations
|
|
|
—
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008
|
|
$
|
1,580
|
|
|
Additions based on tax positions taken during a prior period
|
|
|
645
|
|
|
Reductions based on tax positions taken during a prior period
|
|
|
—
|
|
|
Additions based on tax positions taken during the current period
|
|
|
385
|
|
|
Reductions based on tax positions taken during the current period
|
|
|
—
|
|
|
Reductions related to settlement of tax matters
|
|
|
—
|
|
|
Reductions related to a lapse of applicable statute of
limitations
|
|
|
—
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009
|
|
$
|
2,610
|
|
|
|
|
|
|
|
If the Company eventually is able to recognize these uncertain
tax positions, most of the unrecognized tax benefits would
reduce the effective tax rate.
The Company has not incurred any material interest or penalties
as of December 31, 2009. The Company does not anticipate
any significant change within 12 months of this reporting
date of its uncertain tax positions. The
95
Company is subject to taxation in the US and various states
jurisdictions. There are no ongoing examinations by taxing
authorities at this time. The Company’s various tax years
starting with 2000 to 2009 remain open in various taxing
jurisdictions.
In February 2010, the Company entered into a collaboration and
license agreement (“License Agreement) and a manufacture
and supply agreement, (“Supply Agreement” or
collectively, the “Collaboration”), with Biovail
Laboratories International SRL (“Biovail”), for AZ-004
for the treatment of psychiatric
and/or
neurological indications and the symptoms associated with these
indications, including the initial indication of treating
agitation in schizophrenia and bipolar disorder patients. The
Collaboration contemplates that the Company will be the
exclusive supplier of drug product for clinical and commercial
uses and have responsibility for the NDA for AZ-004 for the
initial indication for the of rapid treatment of agitation in
patients with schizophrenia or bipolar disorder, as well as
responsibility for any additional development and regulatory
activities required for use by these two patient populations in
the outpatient setting. Biovail will be responsible for
commercialization for the initial indication and, if it elects,
development and commercialization of additional indications for
AZ-004 in the U.S. and Canada.
Under the terms of the License Agreement, Biovail paid the
Company an upfront fee of $40 million, and the Company may
be eligible to receive up to an additional $90 million in
milestone payments upon achievement of predetermined regulatory,
clinical and commercial manufacturing milestones. The Company
may be subject to certain payment obligations to Biovail, up to
$5 million, if it does not meet certain other milestones
prior to a termination of the license agreement. The Company is
also eligible to receive tiered royalty payments of 10% to 25%
on any net sales of AZ-004. The Company is responsible for
conducting and funding all development and regulatory activities
associated with AZ-004’s initial indication for the rapid
treatment of agitation in patients with schizophrenia or bipolar
disorder as well as for its possible use in the outpatient
setting in these two patient populations. The Company’s
obligation to fund the outpatient development efforts is limited
to a specified amount. Biovail is responsible for certain Phase
4 development commitments and related costs and expenses. For
additional indications, the Company has an obligation regarding
certain efforts and related costs and expenses, up to a
specified amount, and if it elects, Biovail is responsible for
all other development commitments and related costs and expenses.
Under the terms of the Supply Agreement, the Company is the
exclusive supplier of AZ-004 and has responsibility for the
manufacture, packaging, labeling and supply for clinical and
commercial uses. Biovail will purchase AZ-004 from the Company
at predetermined transfer prices. The transfer prices depend on
the volume of AZ-004 purchases, subject to certain adjustments.
Either party may terminate the Collaboration for the other
party’s uncured material breach or bankruptcy. In addition,
Biovail has the right to terminate the Collaboration
(a) upon 90 days written notice for convenience;
(b) upon 90 days written notice if FDA does not
approve the AZ-004 NDA for the initial indication for the rapid
treatment of agitation in patients with schizophrenia or bipolar
disorder; (c) immediately upon written notice for safety
reasons or withdrawal of marketing approval; (d) upon
90 days written notice upon certain recalls of the product;
or (e) immediately upon written notice within 60 days
of termination of the Supply Agreement under certain
circumstances. The Supply Agreement automatically terminates
upon the termination of the license agreement.
96
|
|
|
17.
|
Quarterly
Results (Unaudited)
|
The following table is in thousands, except per share amounts:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended
|
|
|
|
March 31
|
|
June 30
|
|
September 30
|
|
December 31
|
|
|
|
Fiscal 2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
$
|
9,514
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Loss from operations
|
|
|
(6,843
|
)
|
|
|
(16,835
|
)
|
|
|
(13,007
|
)
|
|
|
(11,022
|
)
|
|
Net loss
|
|
|
(6,904
|
)
|
|
|
(16,969
|
)
|
|
|
(12,425
|
)
|
|
|
(19,767
|
)
|
|
Net loss attributable to Alexza common stockholders
|
|
|
(1,714
|
)
|
|
|
(9,740
|
)
|
|
|
(72,423
|
)
|
|
|
(19,767
|
)
|
|
Basic and diluted net loss per share attributable to Alexza
common stockholders
|
|
|
(0.05
|
)
|
|
|
(0.29
|
)
|
|
|
(1.95
|
)
|
|
|
(0.39
|
)
|
|
Shares used in computation of basic and diluted net loss per
share attributable to Alexza common stockholders
|
|
|
32,967
|
|
|
|
33,136
|
|
|
|
37,060
|
|
|
|
51,272
|
|
|
Fiscal 2008
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
69
|
|
|
$
|
417
|
|
|
Loss from operations
|
|
|
(19,155
|
)
|
|
|
(20,500
|
)
|
|
|
(21,067
|
)
|
|
|
(17,998
|
)
|
|
Net loss
|
|
|
(18,366
|
)
|
|
|
(20,013
|
)
|
|
|
(20,758
|
)
|
|
|
(17,904
|
)
|
|
Net loss attributable to Alexza common stockholders
|
|
|
(14,609
|
)
|
|
|
(14,122
|
)
|
|
|
(14,692
|
)
|
|
|
(15,027
|
)
|
|
Basic and diluted net loss per share attributable to Alexza
common stockholders
|
|
|
(0.47
|
)
|
|
|
(0.43
|
)
|
|
|
(0.45
|
)
|
|
|
(0.46
|
)
|
|
Shares used in computation of basic and diluted net loss per
share attributable to Alexza common stockholders
|
|
|
31,225
|
|
|
|
32,532
|
|
|
|
32,610
|
|
|
|
32,821
|
|
97
|
|
|
Item 9.
|
Changes
in and Disagreements With Accountants on Accounting and
Financial Disclosure
|
Not Applicable.
|
|
|
Item 9A.
|
Controls
and Procedures
|
Evaluation
of Disclosure Controls and Procedures:
We maintain disclosure controls and procedures that are designed
to ensure that information required to be disclosed in our
Exchange Act reports is recorded, processed, summarized and
reported within the time periods specified in the Securities and
Exchange Commission’s rules and forms and that such
information is accumulated and communicated to our management,
including our Chief Executive Officer and Chief Financial
Officer, as appropriate, to allow for timely decisions regarding
required disclosure. In designing and evaluating the disclosure
controls and procedures, management recognizes that any controls
and procedures, no matter how well designed and operated, can
provide only reasonable assurance of achieving the desired
control objectives, and in reaching a reasonable level of
assurance, management is required to apply its judgment in
evaluating the cost-benefit relationship of possible controls
and procedures.
As of December 31, 2009, the end of the period covered by
this report, we carried out an evaluation, under the supervision
and with the participation of our management, including our
Chief Executive Officer and our Chief Financial Officer, of the
effectiveness of the design and operation of our disclosure
controls and procedures. Based on the foregoing, our Chief
Executive Officer and Chief Financial Officer concluded that our
disclosure controls and procedures were effective at the
reasonable assurance level.
Management’s
Annual Report on Internal Control Over Financial
Reporting:
Our management is responsible for establishing and maintaining
adequate internal control over financial reporting, as such term
is defined in Exchange Act
Rules 13a-15(f)
and
15d-15(f).
Under the supervision and with the participation of our
management, including our Chief Executive Officer and Chief
Financial Officer, we conducted an evaluation of the
effectiveness of our internal control over financial reporting
as of December 31, 2009 based on the framework in
Internal Control — Integrated Framework issued
by the Committee of Sponsoring Organizations of the Treadway
Commission (COSO). Based on that evaluation, our management
concluded that our internal control over financial reporting was
effective as of December 31, 2009. Our independent
registered public accounting firm, Ernst &Young LLP,
audited the consolidated financial statements included in this
Annual Report on
Form 10-K
and have issued an audit report on the effectiveness of our
internal control over financial reporting. Their report on the
audit of internal control over financial reporting appears below.
98
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
Alexza Pharmaceuticals, Inc.
We have audited Alexza Pharmaceuticals, Inc.’s internal
control over financial reporting as of December 31, 2009,
based on criteria established in Internal Control —
Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission (the COSO criteria).
Alexza Pharmaceuticals, Inc.’s management is responsible
for maintaining effective internal control over financial
reporting, and for its assessment of the effectiveness of
internal control over financial reporting included in the
accompanying Management’s Annual Report on Internal Control
over Financial Reporting. Our responsibility is to express an
opinion on the company’s internal control over financial
reporting based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control
over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of
internal control over financial reporting, assessing the risk
that a material weakness exists, testing and evaluating the
design and operating effectiveness of internal control based on
the assessed risk, and performing such other procedures as we
considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s
internal control over financial reporting includes those
policies and procedures that (1) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the
company; (2) provide reasonable assurance that transactions
are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting
principles, and that receipts and expenditures of the company
are being made only in accordance with authorizations of
management and directors of the company; and (3) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
In our opinion, Alexza Pharmaceuticals, Inc. maintained, in all
material respects, effective internal control over financial
reporting as of December 31, 2009 based on the COSO
criteria.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), the
consolidated balance sheets of Alexza Pharmaceuticals, Inc. as
of December 31, 2009 and December 31, 2008 and the
related consolidated statements of operations, convertible
preferred stock and stockholders’ (deficit) equity, and
cash flows for each of the three years in the period ended
December 31, 2009 and for the period from December 19,
2000 (inception) to December 31, 2009 and our report dated
March 9, 2010 expressed an unqualified opinion thereon.
Palo Alto, California
March 9, 2010
99
Changes
in Internal Control Over Financial Reporting:
There has been no change in our internal control over financial
reporting during our most recent fiscal quarter that has
materially affected, or is reasonably likely to materially
affect, our internal control over financial reporting.
|
|
|
Item 9B.
|
Other
Information
|
None.
PART III
|
|
|
Item 10.
|
Directors
and Executive Officers of the Registrant
|
The information required by this Item concerning our directors
is incorporated by reference to the information to be set forth
in the sections entitled
“Proposal No. 1 — Election of
Directors” and “Section 16(a) Beneficial
Ownership Reporting Compliance” in our definitive Proxy
Statement for the 2010 Annual Meeting of Stockholders to be
filed within 120 days after the end of the
Registrant’s fiscal year ended December 31, 2009, or
the Proxy Statement. The information required by this Item
concerning our executive officers is incorporated by reference
to the information to be set forth in the section of the Proxy
Statement entitled “Executive Officers.” Information
regarding compliance with Section 16(a) of the Exchange
Act, our code of business conduct and ethics and certain
information related to our Audit Committee and Ethics Committee
is set forth under the heading “Information Regarding the
Board of Directors and Corporate Governance” in our Proxy
Statement, and is incorporated herein by reference thereto.
|
|
|
Item 11.
|
Executive
Compensation
|
The information required by this Item 11 is incorporated by
reference to the information under the caption “Executive
Compensation” in the Proxy Statement to be filed with the
Securities and Exchange Commission no later than 120 days
from the end of our last fiscal year.
|
|
|
Item 12.
|
Security
Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters
|
The information required by this Item 12 with respect to
stock ownership of certain beneficial owners and management and
securities authorized for issuance under equity compensation
plans are incorporated by reference to the information under the
caption “Security Ownership of Certain Beneficial Owners
and Management” in the Proxy Statement.
Securities
Authorized For Issuance Under Equity Compensation
Plans
We maintain the 2005 Equity Incentive Plan, 2005 Non-Employee
Directors’ Stock Option Plan and 2005 Employee Stock
Purchase Plan pursuant to which we may grant equity awards to
eligible persons.
The following table gives information about equity awards under
our 2005 Equity Incentive Plan, 2005 Non-Employee
Directors’ Stock Option Plan and 2005 Employee Stock
Purchase Plan as of December 31, 2009.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(a)
|
|
|
(b)
|
|
|
(c)
|
|
|
|
|
Number of Securities
|
|
|
Weighted-Average
|
|
|
Number of Securities Remaining
|
|
|
|
|
to be Issued upon
|
|
|
Exercise Price of
|
|
|
Available for Future Issuance
|
|
|
|
|
Exercise of
|
|
|
Outstanding
|
|
|
Under Equity Compensation
|
|
|
|
|
Outstanding Options,
|
|
|
Options, Warrants
|
|
|
Plans (Excluding Securities
|
|
|
Plan Category
|
|
Warrants and Rights
|
|
|
and Rights
|
|
|
Reflected in Column (a))
|
|
|
|
|
Equity compensation plans approved by security holders
|
|
|
4,936,769
|
|
|
$
|
3.66
|
|
|
|
690,422
|
(1)(2)
|
|
Equity compensation plans not approved by security holders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
4,936,769
|
|
|
$
|
3.66
|
|
|
|
690,422
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
100
|
|
|
|
(1) |
|
The 2005 Plan incorporates an evergreen formula pursuant to
which on each January 1, the aggregate number of shares
reserved for issuance under the 2005 Plan will increase by a
number equal to the lesser of (i) 1,000,000 shares,
(ii) 2% of the outstanding shares on December 31 of the
preceding calendar year, or (iii) an amount determined by
our Board. |
| |
|
|
|
The Directors’ Plan incorporates an evergreen formula
pursuant to which on each January 1, the aggregate number
of shares reserved for issuance under the Director’s Plan
will increase by the number of shares subject to options granted
during the preceding calendar year less the number of shares
that revert back to the share reserve during the preceding
calendar year. |
|
|
|
The ESPP incorporates an evergreen formula pursuant to which on
each January 1, the aggregate number of shares reserved for
issuance under the ESPP will increase by a number equal to the
lesser of (i) 250,000 shares, (ii) 1% of the
outstanding shares on December 31 of the preceding calendar
year, or (iii) an amount determined by our Board. |
|
(2) |
|
Of these shares, 156,243 shares remain available for
purchase under the ESPP. |
|
|
|
Item 13.
|
Certain
Relationships and Related Transactions and Director
Independence
|
The information required in this Item 13 is incorporated by
reference to the information under the caption “Certain
Relationships and Related Transactions and Director
Independence” in the Proxy Statement.
|
|
|
Item 14.
|
Principal
Accountant Fees and Services
|
The information required by this Item 14 under the caption
“Principal Accountant Fees and Services” is
incorporated by reference to the information in the Proxy
Statement.
PART IV
|
|
|
Item 15.
|
Exhibits
and Financial Statement Schedules
|
(a) 1. Financial Statements
See Index to Financial Statements under Item 8 on
page 61
(a) 2. Financial Statement Schedules
All schedules are omitted because they are not applicable or are
not required or the information required to be set forth therein
is included in the Financial Statements or notes thereto.
(a) 3. Exhibits
101
EXHIBIT INDEX
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Document
|
|
|
|
|
3
|
.5
|
|
Restated Certificate of Incorporation(1)
|
|
|
3
|
.7
|
|
Amended and Restated Bylaws(1)
|
|
|
3
|
.8
|
|
Amendment to Amended and Restated Bylaws(5)
|
|
|
4
|
.1
|
|
Specimen Common Stock Certificate(1)
|
|
|
4
|
.2
|
|
Second Amended and Restated Investors’ Rights Agreement
between Registrant and certain holders of Preferred Stock dated
November 5, 2004(1)
|
|
|
10
|
.2
|
|
Form of Director/Officer Indemnification Agreement entered into
between Registrant and each of its directors and officers(9)*
|
|
|
10
|
.3
|
|
Form of Change of Control Agreement*(18)
|
|
|
10
|
.4
|
|
2005 Equity Incentive Plan(8)*
|
|
|
10
|
.5
|
|
Form of Option Grant Notice, Form of Option Agreement and Form
of Notice of Exercise to 2005 Equity Incentive Plan(1)*
|
|
|
10
|
.6
|
|
2005 Non-Employee Directors’ Stock Option Plan(1)
|
|
|
10
|
.7
|
|
Form of Option Grant Notice, Form of Option Agreement and Form
of Notice of Exercise to 2005 Non-Employee Directors’ Stock
Option Plan(1)
|
|
|
10
|
.8
|
|
2005 Employee Stock Purchase Plan(1)*
|
|
|
10
|
.9
|
|
Form of Offering Document to 2005 Employee Stock Purchase
Plan(1)*
|
|
|
10
|
.13
|
|
Development Agreement between Registrant and Autoliv ASP, Inc.
dated October 3, 2005(1)
|
|
|
10
|
.14
|
|
Loan and Security Agreement between Registrant and Silicon
Valley Bank dated March 20, 2002, as amended on
January 7, 2003, September 3, 2003, March 18,
2004 and May 16, 2005(1)
|
|
|
10
|
.15
|
|
Master Security Agreement between Registrant and General
Electric Capital Corporation dated May 17, 2005, as amended
on May 18, 2005(1)
|
|
|
10
|
.16
|
|
Promissory Note between Registrant and General Electric Capital
Corporation dated June 15, 2005(1)
|
|
|
10
|
.17
|
|
Promissory Note between Registrant and General Electric Capital
Corporation dated August 24, 2005(1)
|
|
|
10
|
.20
|
|
Warrant to Purchase shares of Series B Preferred Stock
issued to Silicon Valley Bank dated March 20, 2002(1)
|
|
|
10
|
.21
|
|
Warrant to Purchase shares of Series C Preferred Stock
issued to Silicon Valley Bank dated January 7, 2003, as
amended on March 4, 2003(1)
|
|
|
10
|
.22
|
|
Warrant to Purchase shares of Series C Preferred Stock
issued to Silicon Valley Bank dated September 19, 2003(1)
|
|
|
10
|
.23
|
|
Warrant to Purchase shares of Series C Preferred Stock
issued to Silicon Valley Bank dated April 7, 2004(1)
|
|
|
10
|
.24
|
|
Lease Agreement between the Brittania, LLC and the Registrant
dated August 25, 2006(2)
|
|
|
10
|
.26†
|
|
Purchase Option Agreement by and among Symphony Allegro Holdings
LLC and Symphony Allegro, Inc. and Registrant dated
December 1, 2006(2)
|
|
|
10
|
.29†
|
|
Amended and Restated Research and Development Agreement by and
among Symphony Allegro Holdings LLC and Symphony Allegro, Inc.
and Registrant dated December 1, 2006(2)
|
|
|
10
|
.30
|
|
Registration Rights Agreement between Symphony Allegro Holdings
LLC and Registrant dated December 1, 2006(2)
|
|
|
10
|
.31†
|
|
Novated and Restated Technology License Agreement by and among
Symphony Allegro Holdings LLC and Symphony Allegro, Inc. and
Registrant dated December 1, 2006(2)
|
|
|
10
|
.32
|
|
Confidentiality Agreement by and among Symphony Allegro Holdings
LLC and Symphony Allegro, Inc. and Registrant dated
December 1, 2006(2)
|
|
|
10
|
.33
|
|
2007 Performance Bonus Program*(2)
|
|
|
10
|
.34
|
|
First Amendment to Lease between Britannia Hacienda VIII LLC and
the Registrant dated May 4, 2007(3)
|
102
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Document
|
|
|
|
|
10
|
.35†
|
|
Second Amendment to Lease between Britannia Hacienda VIII LLC
and the Registrant dated August 28, 2007(4)
|
|
|
10
|
.36†
|
|
Manufacturing and Supply Agreement between Registrant and
Autoliv ASP, Inc., dated November 2, 2007(5)
|
|
|
10
|
.38
|
|
Offer Letter between the Registrant and Michael Simms, dated
January 23, 2008(5)
|
|
|
10
|
.39
|
|
Stock and Warrant Purchase Agreement between Registrant and
Biomedical Investment Fund Pte Ltd., dated March 26,
2008(6)
|
|
|
10
|
.40
|
|
Warrant to Purchase shares of Common Stock issued to Biomedical
Investment Fund Pte Ltd. dated March 27, 2008(6)
|
|
|
10
|
.41
|
|
Common Stock Purchase Agreement between Registrant and Azimuth
Opportunity Ltd. dated March 31, 2008(7)
|
|
|
10
|
.42
|
|
Form of Notice of Grant of Award and Stock Unit Award Agreement
to 2005 Equity Incentive Plan(18)
|
|
|
10
|
.43
|
|
2006 Performance Bonus Program*(10)
|
|
|
10
|
.44
|
|
2008 Performance Bonus Program*(11)
|
|
|
10
|
.45
|
|
2009-2010
Performance Based Incentive Program*(12)
|
|
|
10
|
.46
|
|
Severance Agreement and Release Agreement between the Registrant
and Anthony Tebbutt, dated February 6, 2009 and
February 17, 2009, respectively(13)
|
|
|
10
|
.47
|
|
Amended and Restated Purchase Option Agreement by and among the
Company, Holdings and Symphony Allegro dated June 15,
2009*(14)
|
|
|
10
|
.48
|
|
Warrant Purchase Agreement between the Company and Holdings
dated June 15, 2009(14)
|
|
|
10
|
.49
|
|
Amended and Restated Registration Rights Agreement between the
Company and Holdings dated June 15, 2009(14)
|
|
|
10
|
.50
|
|
Form of Amendment to Change of Control Agreement*(15)
|
|
|
10
|
.51
|
|
Form of Warrants to Purchase Shares of Common Stock, dated
August 26, 2009(16)
|
|
|
10
|
.52
|
|
Letter Agreement among the Company, Symphony Allegro Holdings
LLC, Symphony Capital Partners, L.P. and Symphony Strategic
Partners, LLC, dated August 26, 2009(16)
|
|
|
10
|
.53
|
|
Securities Purchase Agreement by and among Alexza and the
purchasers identified therein, dated September 29, 2009(17)
|
|
|
10
|
.54
|
|
Form of Warrants to Purchase shares of Common Stock, dated
October 5, 2009(17)
|
|
|
10
|
.55††u
|
|
Biovail Collaboration and License Agreement, dated
February 9, 2010
|
|
|
10
|
.56††u
|
|
Biovail Manufacture and Supply Agreement dated February 9,
2010
|
|
|
14
|
.1
|
|
Alexza Pharmaceuticals, Inc. Code of Business Conduct for
Employees, Executive Officers and Directors(2)
|
|
|
21
|
.1u
|
|
Subsidiaries of Registrant
|
|
|
23
|
.1u
|
|
Consent of Independent Registered Public Accounting Firm
|
|
|
24
|
.1u
|
|
Power of Attorney included on the signature pages hereto
|
|
|
31
|
.1u
|
|
Section 302 Certification of CEO.
|
|
|
31
|
.2u
|
|
Section 302 Certification of CFO.
|
|
|
32
|
.1u
|
|
Section 906 Certifications of CEO and CFO.
|
|
|
|
|
* |
|
Management contract or compensation plan or arrangement. |
| |
|
u |
|
Filed herein |
| |
|
† |
|
Confidential treatment has been granted with respect to certain
portions of this exhibit. This exhibit omits the information
subject to this confidentiality request. Omitted portions have
been filed separately with the SEC. |
| |
|
†† |
|
Confidential treatment has been requested with respect to
certain portions of this exhibit. This exhibit omits the
information subject to this confidentiality request. Omitted
portions have been filed separately with the SEC. |
103
|
|
|
|
(1) |
|
Incorporated by reference to exhibits to our Registration
Statement on
Form S-1
filed on December 22, 2005, as amended (File
No. 333-130644). |
| |
|
(2) |
|
Incorporated by reference to our Annual Report on
Form 10-K
(File
No. 000-51820)
as filed with the SEC on March 29, 2007. |
| |
|
(3) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on August 13, 2007. |
| |
|
(4) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on November 1, 2007. |
| |
|
(5) |
|
Incorporated by reference to our Annual Report on
Form 10-K
(File
No. 000-51820)
as filed with the SEC on March 17, 2008. |
| |
|
(6) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on March 17, 2008. |
| |
|
(7) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on March 31, 2008. |
| |
|
(8) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on May 30, 2008. |
| |
|
(9) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on July 14, 2008. |
| |
|
(10) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on June 5, 2006. |
| |
|
(11) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on April 16, 2008. |
| |
|
(12) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on February 24, 2009. |
| |
|
(13) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on May 11, 2009. |
| |
|
(14) |
|
Incorporated by reference to our Current Report on
Form 8-K/A
(File
No. 000-51820)
as filed with the SEC on June 26, 2009. |
| |
|
(15) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on August 5, 2009. |
| |
|
(16) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on August 26, 2009. |
| |
|
(17) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on September 30, 2009. |
| |
|
(18) |
|
Incorporated by reference to our Current Report on Form
10-K (File
No.
000-51820)
as filed with the SEC on March 10, 2009. |
104
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, as amended, (the “Exchange
Act”) the registrant has duly caused this report to be
signed on its behalf by the undersigned, thereunto duly
authorized.
ALEXZA PHARMACEUTICALS, INC.
Thomas B. King
President and Chief Executive Officer
Dated:
March 9, 2010
POWER OF
ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose
signature appears below constitutes and appoints Thomas B. King
and August J. Moretti, and each of them, as his true and lawful
attorneys-in-fact and agents, with full power of substitution
and resubstitution, for him and in his name, place, and stead,
in any and all capacities, to sign any and all amendments to
this Report, and to file the same, with all exhibits thereto,
and other documents in connection therewith, with the Securities
and Exchange Commission, granting unto said attorneys-in-fact
and agents, and each of them, full power and authority to do and
perform each and every act and thing requisite and necessary to
be done in connection therewith, as fully to all intents and
purposes as he might or could do in person, hereby ratifying and
confirming that all said attorneys-in-fact and agents, or any of
them or their or his substitute or substitutes, may lawfully do
or cause to be done by virtue hereof.
Pursuant to the requirements of the Exchange Act, this report
has been signed below by the following persons on behalf of the
Registrant and in the capacities indicated on March 9, 2010.
| |
|
|
|
|
|
Signature
|
|
Title
|
|
|
|
|
|
|
|
/s/ THOMAS
B. KING
Thomas
B. King
|
|
President, Chief Executive Officer and Director (Principal
Executive Officer)
|
|
|
|
|
|
/s/ AUGUST
J. MORETTI
August
J. Moretti
|
|
Senior Vice President and Chief Financial Officer (Principal
Financial and Accounting Officer)
|
|
|
|
|
|
/s/ HAL
V. BARRON
Hal
V. Barron
|
|
Director
|
|
|
|
|
|
/s/ ANDREW
L. BUSSER
Andrew
L. Busser
|
|
Director
|
|
|
|
|
|
/s/ SAMUEL
D. COLELLA
Samuel
D. Colella
|
|
Director
|
|
|
|
|
|
/s/ ALAN
D. FRAZIER
Alan
D. Frazier
|
|
Director
|
105
| |
|
|
|
|
|
Signature
|
|
Title
|
|
|
|
|
|
|
|
/s/ DEEPIKA
R. PAKIANATHAN
Deepika
R. Pakianathan
|
|
Director
|
|
|
|
|
|
/s/ J.
LEIGHTON READ
J.
Leighton Read
|
|
Director
|
|
|
|
|
|
/s/ GORDON
RINGOLD
Gordon
Ringold
|
|
Director
|
|
|
|
|
|
/s/ ISAAC
STEIN
Isaac
Stein
|
|
Director
|
106
EXHIBIT INDEX
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Document
|
|
|
|
|
3
|
.5
|
|
Restated Certificate of Incorporation(1)
|
|
|
3
|
.7
|
|
Amended and Restated Bylaws(1)
|
|
|
3
|
.8
|
|
Amendment to Amended and Restated Bylaws(5)
|
|
|
4
|
.1
|
|
Specimen Common Stock Certificate(1)
|
|
|
4
|
.2
|
|
Second Amended and Restated Investors’ Rights Agreement
between Registrant and certain holders of Preferred Stock dated
November 5, 2004(1)
|
|
|
10
|
.2
|
|
Form of Director/Officer Indemnification Agreement entered into
between Registrant and each of its directors and officers(9)*
|
|
|
10
|
.3
|
|
Form of Change of Control Agreement*(18)
|
|
|
10
|
.4
|
|
2005 Equity Incentive Plan(8)*
|
|
|
10
|
.5
|
|
Form of Option Grant Notice, Form of Option Agreement and Form
of Notice of Exercise to 2005 Equity Incentive Plan(1)*
|
|
|
10
|
.6
|
|
2005 Non-Employee Directors’ Stock Option Plan(1)
|
|
|
10
|
.7
|
|
Form of Option Grant Notice, Form of Option Agreement and Form
of Notice of Exercise to 2005 Non-Employee Directors’ Stock
Option Plan(1)
|
|
|
10
|
.8
|
|
2005 Employee Stock Purchase Plan(1)*
|
|
|
10
|
.9
|
|
Form of Offering Document to 2005 Employee Stock Purchase
Plan(1)*
|
|
|
10
|
.13
|
|
Development Agreement between Registrant and Autoliv ASP, Inc.
dated October 3, 2005(1)
|
|
|
10
|
.14
|
|
Loan and Security Agreement between Registrant and Silicon
Valley Bank dated March 20, 2002, as amended on
January 7, 2003, September 3, 2003, March 18,
2004 and May 16, 2005(1)
|
|
|
10
|
.15
|
|
Master Security Agreement between Registrant and General
Electric Capital Corporation dated May 17, 2005, as amended
on May 18, 2005(1)
|
|
|
10
|
.16
|
|
Promissory Note between Registrant and General Electric Capital
Corporation dated June 15, 2005(1)
|
|
|
10
|
.17
|
|
Promissory Note between Registrant and General Electric Capital
Corporation dated August 24, 2005(1)
|
|
|
10
|
.20
|
|
Warrant to Purchase shares of Series B Preferred Stock
issued to Silicon Valley Bank dated March 20, 2002(1)
|
|
|
10
|
.21
|
|
Warrant to Purchase shares of Series C Preferred Stock
issued to Silicon Valley Bank dated January 7, 2003, as
amended on March 4, 2003(1)
|
|
|
10
|
.22
|
|
Warrant to Purchase shares of Series C Preferred Stock
issued to Silicon Valley Bank dated September 19, 2003(1)
|
|
|
10
|
.23
|
|
Warrant to Purchase shares of Series C Preferred Stock
issued to Silicon Valley Bank dated April 7, 2004(1)
|
|
|
10
|
.24
|
|
Lease Agreement between the Brittania, LLC and the Registrant
dated August 25, 2006(2)
|
|
|
10
|
.26†
|
|
Purchase Option Agreement by and among Symphony Allegro Holdings
LLC and Symphony Allegro, Inc. and Registrant dated
December 1, 2006(2)
|
|
|
10
|
.29†
|
|
Amended and Restated Research and Development Agreement by and
among Symphony Allegro Holdings LLC and Symphony Allegro, Inc.
and Registrant dated December 1, 2006(2)
|
|
|
10
|
.30
|
|
Registration Rights Agreement between Symphony Allegro Holdings
LLC and Registrant dated December 1, 2006(2)
|
|
|
10
|
.31†
|
|
Novated and Restated Technology License Agreement by and among
Symphony Allegro Holdings LLC and Symphony Allegro, Inc. and
Registrant dated December 1, 2006(2)
|
|
|
10
|
.32
|
|
Confidentiality Agreement by and among Symphony Allegro Holdings
LLC and Symphony Allegro, Inc. and Registrant dated
December 1, 2006(2)
|
|
|
10
|
.33
|
|
2007 Performance Bonus Program*(2)
|
|
|
10
|
.34
|
|
First Amendment to Lease between Britannia Hacienda VIII LLC and
the Registrant dated May 4, 2007(3)
|
107
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description of Document
|
|
|
|
|
10
|
.35†
|
|
Second Amendment to Lease between Britannia Hacienda VIII LLC
and the Registrant dated August 28, 2007(4)
|
|
|
10
|
.36†
|
|
Manufacturing and Supply Agreement between Registrant and
Autoliv ASP, Inc., dated November 2, 2007(5)
|
|
|
10
|
.38
|
|
Offer Letter between the Registrant and Michael Simms, dated
January 23, 2008(5)
|
|
|
10
|
.39
|
|
Stock and Warrant Purchase Agreement between Registrant and
Biomedical Investment Fund Pte Ltd., dated March 26,
2008(6)
|
|
|
10
|
.40
|
|
Warrant to Purchase shares of Common Stock issued to Biomedical
Investment Fund Pte Ltd. dated March 27, 2008(6)
|
|
|
10
|
.41
|
|
Common Stock Purchase Agreement between Registrant and Azimuth
Opportunity Ltd. dated March 31, 2008(7)
|
|
|
10
|
.42
|
|
Form of Notice of Grant of Award and Stock Unit Award Agreement
to 2005 Equity Incentive Plan(18)
|
|
|
10
|
.43
|
|
2006 Performance Bonus Program*(10)
|
|
|
10
|
.44
|
|
2008 Performance Bonus Program*(11)
|
|
|
10
|
.45
|
|
2009-2010
Performance Based Incentive Program*(12)
|
|
|
10
|
.46
|
|
Severance Agreement and Release Agreement between the Registrant
and Anthony Tebbutt, dated February 6, 2009 and
February 17, 2009, respectively(13)
|
|
|
10
|
.47
|
|
Amended and Restated Purchase Option Agreement by and among the
Company, Holdings and Symphony Allegro dated June 15,
2009*(14)
|
|
|
10
|
.48
|
|
Warrant Purchase Agreement between the Company and Holdings
dated June 15, 2009(14)
|
|
|
10
|
.49
|
|
Amended and Restated Registration Rights Agreement between the
Company and Holdings dated June 15, 2009(14)
|
|
|
10
|
.50
|
|
Form of Amendment to Change of Control Agreement*(15)
|
|
|
10
|
.51
|
|
Form of Warrants to Purchase Shares of Common Stock, dated
August 26, 2009(16)
|
|
|
10
|
.52
|
|
Letter Agreement among the Company, Symphony Allegro Holdings
LLC, Symphony Capital Partners, L.P. and Symphony Strategic
Partners, LLC, dated August 26, 2009(16)
|
|
|
10
|
.53
|
|
Securities Purchase Agreement by and among Alexza and the
purchasers identified therein, dated September 29, 2009(17)
|
|
|
10
|
.54
|
|
Form of Warrants to Purchase shares of Common Stock, dated
October 5, 2009(17)
|
|
|
10
|
.55††u
|
|
Biovail Collaboration and License Agreement, dated February 9,
2010
|
|
|
10
|
.56††u
|
|
Biovail Manufacture and Supply Agreement dated February 9,
2010
|
|
|
14
|
.1
|
|
Alexza Pharmaceuticals, Inc. Code of Business Conduct for
Employees, Executive Officers and Directors(2)
|
|
|
21
|
.1u
|
|
Subsidiaries of Registrant
|
|
|
23
|
.1u
|
|
Consent of Independent Registered Public Accounting Firm
|
|
|
24
|
.1u
|
|
Power of Attorney included on the signature pages hereto
|
|
|
31
|
.1u
|
|
Section 302 Certification of CEO.
|
|
|
31
|
.2u
|
|
Section 302 Certification of CFO.
|
|
|
32
|
.1u
|
|
Section 906 Certifications of CEO and CFO.
|
|
|
|
|
* |
|
Management contract or compensation plan or arrangement. |
| |
|
u |
|
Filed herein |
| |
|
† |
|
Confidential treatment has been granted with respect to certain
portions of this exhibit. This exhibit omits the information
subject to this confidentiality request. Omitted portions have
been filed separately with the SEC. |
| |
|
†† |
|
Confidential treatment has been requested with respect to
certain portions of this exhibit. This exhibit omits the
information subject to this confidentiality request. Omitted
portions have been filed separately with the SEC. |
108
|
|
|
|
(1) |
|
Incorporated by reference to exhibits to our Registration
Statement on
Form S-1
filed on December 22, 2005, as amended (File
No. 333-130644). |
| |
|
(2) |
|
Incorporated by reference to our Annual Report on
Form 10-K
(File
No. 000-51820)
as filed with the SEC on March 29, 2007. |
| |
|
(3) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on August 13, 2007. |
| |
|
(4) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on November 1, 2007. |
| |
|
(5) |
|
Incorporated by reference to our Annual Report on
Form 10-K
(File
No. 000-51820)
as filed with the SEC on March 17, 2008. |
| |
|
(6) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on March 17, 2008. |
| |
|
(7) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on March 31, 2008. |
| |
|
(8) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on May 30, 2008. |
| |
|
(9) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on July 14, 2008. |
| |
|
(10) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on June 5, 2006. |
| |
|
(11) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on April 16, 2008. |
| |
|
(12) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on February 24, 2009. |
| |
|
(13) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on May 11, 2009. |
| |
|
(14) |
|
Incorporated by reference to our Current Report on
Form 8-K/A
(File
No. 000-51820)
as filed with the SEC on June 26, 2009. |
| |
|
(15) |
|
Incorporated by reference to our Quarterly Report on
Form 10-Q
(File
No. 000-51820)
as filed with the SEC on August 5, 2009. |
| |
|
(16) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on August 26, 2009. |
| |
|
(17) |
|
Incorporated by reference to our Current Report on
Form 8-K
(File
No. 000-51820)
as filed with the SEC on September 30, 2009. |
| |
|
(18) |
|
Incorporated by reference to our Current Report on Form
10-K (File
No.
000-51820)
as filed with the SEC on March 10, 2009. |
109