Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended September 30, 2009
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 1-33428
Pharmasset, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 98-0406340 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 303-A College Road East Princeton, New Jersey |
08540 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code (609) 613-4100
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $0.001 Par Value Per Share | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer x | |
| Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) based on the last reported sale price of the common stock on March 31, 2009 was $250.0 million.
As of October 31, 2009, the registrant had 28,277,841 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive Proxy Statement for the 2010 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
Table of Contents
TABLE OF CONTENTS
| Page | ||||
| PART I | ||||
| Item 1. |
1 | |||
| Item 1A. |
26 | |||
| Item 1B. |
50 | |||
| Item 2. |
50 | |||
| Item 3. |
50 | |||
| Item 4. |
51 | |||
| PART II | ||||
| Item 5. |
52 | |||
| Item 6. |
54 | |||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
56 | ||
| Item 7A. |
70 | |||
| Item 8. |
70 | |||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
70 | ||
| Item 9A. |
70 | |||
| Item 9B. |
71 | |||
| PART III | ||||
| Item 10. |
72 | |||
| Item 11. |
72 | |||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
72 | ||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
72 | ||
| Item 14. |
72 | |||
| PART IV | ||||
| Item 15. |
73 | |||
The “Company,” “Pharmasset,” “we,” and “us” as used in this Annual Report on Form 10-K refer to Pharmasset, Inc., a Delaware corporation. Pharmasset and our logo are our trademarks, and Racivir is our registered trademark. Other trademarks mentioned in this Annual Report on Form 10-K are the property of their respective owners.
Table of Contents
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements. The forward-looking statements are principally contained in the sections entitled “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” These statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance, or achievements expressed or implied by the forward-looking statements. For this purpose, any statement that is not a statement of historical fact should be considered a forward-looking statement. We may, in some cases, use words such as “project,” “believe,” “anticipate,” “plan,” “expect,” “estimate,” “intend,” “potential,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. These forward-looking statements include statements about the following:
| • | our product development efforts, primarily with respect to the preclinical and clinical trial results and regulatory approval of RG7128, PSI-7851, PSI-938, and PSI-879 for the treatment of hepatitis C virus (“HCV”) and, secondarily, the development of RacivirTM for the treatment of human immunodeficiency virus (“HIV”); |
| • | the termination of our clevudine registration trials; |
| • | the initiation, termination, completion, or success of preclinical studies and clinical trials; |
| • | clinical trial initiation and completion dates, anticipated regulatory filing dates, and regulatory approval for our product candidates; |
| • | the commercialization of our product candidates; |
| • | our collaboration agreement with F. Hoffmann-LaRoche Ltd. and Hoffmann- La Roche Inc. (collectively, “Roche”), including potential milestone or royalty payments there under; |
| • | our intentions regarding the establishment of collaborations or the licensing of product candidates or intellectual property; |
| • | the scope and enforceability of our intellectual property rights, including claims that we or our collaborators may infringe third party intellectual property rights or be otherwise required to pay license fess under such third party rights; |
| • | our intentions to expand our capabilities and hire additional employees; |
| • | anticipated operating losses, future revenues, research and development expenses, and the need for additional financing; and |
| • | our financial performance. |
Forward-looking statements reflect our current views with respect to future events and are subject to risks and uncertainties. We discuss many of the risks and uncertainties associated with our business in greater detail under the heading “Risk Factors.” Given these risks and uncertainties, you should not place undue reliance on these forward-looking statements. All forward-looking statements represent our estimates and assumptions only as of the date of this Annual Report on Form 10-K.
You should read this Annual Report on Form 10-K and the documents that we reference in it completely and with the understanding that our actual future results may be materially different from what we expect. Our business, financial condition, results of operations, and prospects may change. We may not update these forward-looking statements, even though our situation may change in the future, unless we have obligations under the federal securities laws to update and disclose material developments related to previously disclosed information. The forward-looking statements contained in this Annual Report on Form 10-K are subject to the safe-harbor protection provided by the Private Securities Litigation Reform Act of 1995 and Section 21E of the Securities Exchange Act of 1934, as amended (“Exchange Act”).
Table of Contents
PART I
| ITEM 1. | BUSINESS |
Overview
We are a clinical-stage pharmaceutical company committed to discovering, developing, and commercializing novel drugs to treat viral infections. Our primary focus is on the development of nucleoside/tide analogs as oral therapeutics for the treatment of chronic hepatitis C virus (“HCV”) infection and secondarily, on the development of RacivirTM for the treatment of human immunodeficiency virus (“HIV”) infection. Nucleoside/tide analogs are a class of compounds which act as alternative substrates for the viral polymerase, thus inhibiting viral replication. We currently have three clinical-stage product candidates, two of which we are developing ourselves and one of which we are developing with a strategic partner. We are also advancing a series of preclinical candidates in preparation for clinical development. Our three clinical stage product candidates are:
| • | RG7128 (formerly R7128), is a first generation HCV nucleoside polymerase inhibitor we are developing through a strategic collaboration with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (collectively, “Roche”). RG7128 is in a Phase 2b clinical trial in combination with Pegasys® (pegylated interferon) plus Copegus® (ribavirin) and recently completed INFORM-1, a study designed to investigate the combination of two oral, direct acting antivirals (“DAAs”) in the absence of pegylated interferon and ribavirin. Both of these studies are being conducted by Roche.; |
| • | PSI-7851, a second generation HCV nucleotide polymerase inhibitor, which has completed two phase 1 studies providing adequate safety and efficacy data to support initiation of a Phase 2a clinical trial; and |
| • | Racivir, for the treatment of HIV in combination with other approved HIV drugs, has completed a Phase 2 clinical trial. |
In addition, we recently nominated PSI-352938 (“PSI-938”) and PSI-352879 (“PSI-879”) as third generation nucleotide development candidates for the treatment of HCV, each of which could potentially be used in combination with our current nucleoside/tides, RG7128 or PSI-7851. PSI-938 and PSI-879 are proprietary purine nucleotide analog inhibitors of HCV polymerase that are in preclinical studies required for submission of an Investigational New Drug (“IND”) application with the U.S. Food and Drug Administration (“FDA”) or equivalent foreign regulatory application.
We are continuing to research nucleoside/tide analogs (both pyrimidines and purines) with the intention of identifying additional product candidates that can potentially be used in combination with our current nucleoside/tides, RG7128 and PSI-7851, in combination with other classes of DAAs, or with pegylated interferon and/or ribavirin for the treatment of HCV. We have identified proprietary nucleotide prodrugs that are referred to as phosphate prodrugs because they have the ability to deliver the monophosphate forms of the compounds into infected cells, thus bypassing a rate-limiting step in the metabolic pathway to the active triphosphate form of the drug. The goal of these efforts is to identify compounds with improved potency, safety, convenience, oral bioavailability, and increased intrahepatic nucleotide triphosphate levels. Certain of these compounds have demonstrated exceptional in vitro anti-HCV activity, with up to 100 times greater potency than PSI-6130 (of which RG7128 is a prodrug). Early studies in animals indicate that several of these compounds can achieve concentrations of the active triphosphate form in the liver up to 1000 times higher than PSI-6130 at equivalent doses.
We are developing PSI-7851, PSI-938, PSI-879, and Racivir ourselves. We have a strategic collaboration with Roche for the development of PSI-6130 and its prodrugs, including RG7128. Under the collaboration, Roche pays all development costs and provides us with potential income from milestone payments that can be used to fund the advancement of our proprietary product candidates.
Despite significant advances, there remain significant unmet medical needs in HCV and HIV. In the treatment of HCV, pegylated interferon in combination with ribavirin is the standard of care and has
1
Table of Contents
demonstrated, for some patients, the ability to offer a sustained virologic response, or SVR, defined as HCV RNA levels that are below the limit of detection by a standard test utilizing polymerase chain reaction, or PCR, six months after discontinuation of therapy. However, pegylated interferon is available only as an injectable and is associated with side effects, some of them serious, which may include fatigue, bone marrow suppression, anemia, and neuropsychiatric effects. Many individuals with HCV infection are unable to be treated with interferon due to pre-existing co-morbidities such as advanced liver disease or psychiatric conditions. In the treatment of HCV, there is an unmet medical need for drugs that offer an improved SVR rate with an improved safety profile. For HIV patients, treatment also involves a chronic regimen of antiviral drugs. During such prolonged treatment, viral mutations occur that make the viruses resistant to the drugs being used. We believe there is an unmet medical need for new HIV drugs that are effective against resistant viruses and can replace existing therapies that have lost their effectiveness.
We believe nucleoside/tide analogs are well suited to treat viral diseases because they can be designed to be highly specific and potent inhibitors of HCV replication, are relatively simple to manufacture, and have the potential for oral administration. In HCV, nucleoside/tide analog drugs have demonstrated a higher barrier to viral resistance than non-nucleoside and protease inhibitors for HCV. In addition, this class of compounds has a well-established development and regulatory history. There are 14 nucleoside analogs for the treatment of HBV and HIV that have been approved by the FDA and as a class are standard of care. Additional nucleoside analogs have been approved by the FDA for the treatment of HCV, cytomegalovirus and herpes simplex virus. In addition to RG7128, PSI-7851, PSI-938, PSI-879, and Racivir, we also have discovery programs focused on other nucleoside/tide analogs for HCV. Our scientific team of virologists, biologists, and chemists has experience discovering and developing nucleoside/tide analog drugs for antiviral indications. Collectively, our management team’s product development experience includes approximately 50 therapeutic and diagnostic product approvals. Our discovery platform includes a library of nucleoside/tide analogs and a collection of viral and cellular assays that we use to evaluate new product candidates.
We were incorporated as Pharmasset, Inc. under the laws of Delaware on June 8, 2004.
Strategy
Our primary objective is to become a leader in discovering, developing, and commercializing novel antiviral therapeutics that provide a competitive advantage and address unmet medical needs. Our primary focus is on the discovery and development of nucleoside/tide analogs as oral therapeutics for the treatment of HCV and, secondarily, on the development of Racivir for the treatment of HIV. To achieve this goal, we are pursuing the following strategies:
| • | Focus on developing our current clinical-stage and preclinical-stage product candidates and advancing them toward marketing approval. We are increasing our internal clinical development capabilities to enhance our ability to advance our product candidates. Our development team is responsible for planning and managing our preclinical and clinical trials of PSI-7851, PSI-938, and PSI-879 and supporting our partner, Roche, in its current and future clinical trials of RG7128. |
| • | Maintain a broad pipeline of potential product candidates to diversify commercial opportunities and reduce our dependence on any one product candidate’s clinical or commercial success. Our development capabilities can not only advance our clinical-stage product candidates, but also can be used to evaluate product opportunities from sources outside our company. We intend to leverage our research and development capabilities to evaluate external opportunities and may in-license products or technologies that we believe will complement our antiviral therapeutic focus. By maintaining a broad pipeline, we hope to create a portfolio of products that reduces our dependence on any one product and creates synergy within our pipeline through potential combination products. |
| • | Leverage our core competency in nucleoside/tide chemistry for research innovation and the discovery of additional product candidates. Our core competency is the discovery and development of nucleoside/tide analogs for use as antiviral therapeutics. We believe our nucleoside/tide chemistry |
2
Table of Contents
| expertise and our nucleoside/tide library provide us with a strong foundation from which to identify additional product candidates. We intend to continue to invest in our nucleoside/tide research capabilities and expand our nucleoside/tide analog library. |
| • | Commercialize our products ourselves or through collaborations, where appropriate, to optimize economic returns while managing financial risk. We allocate our limited resources to efforts that we believe will provide the greatest returns. Accordingly, we enter into collaborations to leverage our development capabilities and capitalize on commercialization opportunities that we cannot accomplish by ourselves. We believe this strategy will enable us to obtain the greatest returns from our antiviral discovery and development efforts. |
Background on Viral Disease
A virus is a cellular parasite that cannot reproduce by itself and therefore must infect a susceptible host cell to replicate. A viral infection begins when the virus encounters a susceptible host cell and attaches to the cell membrane. The virus then enters the host cell and directs the host cell’s metabolic machinery to participate in copying the viral genetic information, which is either RNA or DNA, and to produce the proteins encoded by that genetic information. This viral genetic information is packaged within a shell of newly produced viral proteins, forming an immature virus. In the case of HCV or HIV, this immature virus then acquires a coating or envelope of specific viral proteins and cellular lipids, forming a mature virus particle that is capable of infecting other cells. There are a wide variety of viruses, some of which are associated with a low rate of morbidity and mortality, such as viruses causing the common cold, while others, including HCV and HIV, are associated with higher morbidity and mortality rates.
The challenge in developing antiviral therapies is to inhibit viral replication without injuring the host cell. For many years, it appeared that the development of safe and effective antiviral therapies would not be possible because the processes of viral replication were so intertwined with the cell’s metabolic processes that the inhibition of viral functions would result in cell death. A breakthrough occurred with the identification of viral enzymes, such as viral polymerases, which are required for viral replication. These enzymes differ enough from cellular enzymes to permit their selective inhibition and thus prevent viral replication without harming the cell. HCV, an RNA virus, has an RNA polymerase which makes new viral RNA strands from an RNA template. HIV, an RNA retrovirus, has a reverse transcriptase which makes viral DNA from an RNA template.
A major challenge of antiviral therapy is the emergence of viral mutations that result in forms of the virus that are resistant to current therapies. Viral mutations result from mistakes that occur during the natural viral replication process when the genetic information is copied. The mutated form of the virus infects other cells and replicates in its mutated form. Some mutations make the virus resistant to certain types of antiviral medications. When a drug-resistant form of virus first arises, it usually comprises a very small percentage of the virus circulating within the blood. As the original or wild-type virus continues to be suppressed by antiviral therapy and the drug-resistant virus continues to replicate, the mutated virus eventually becomes the dominant virus type. To reduce the likelihood of a dominant drug-resistant mutation, patients must comply with their treatment regimens; however, current studies show that at any given time only approximately 70% of patients strictly adhere to their therapy. Each of the FDA-approved oral antiviral therapies is susceptible to a mutation that confers drug resistance. New drug-resistant forms of virus continue to emerge, and as a result, new therapies to fight drug-resistant virus continue to be needed.
HCV and HIV patients are classified as treatment-naïve or treatment-experienced. Treatment-naïve patients have not previously been exposed to antiviral therapies. In HIV, once viral mutations begin to emerge and the virus demonstrates resistance to the therapy, physicians either switch treatment regimens or add new drugs to existing regimens for the now treatment-experienced patients. The role of resistance mutations in HCV is less well understood as there are no direct acting antivirals yet on the market.
3
Table of Contents
Our Product Candidates
Our research and development programs are primarily focused on discovering and developing drugs that treat HCV and, secondarily, on the development of Racivir for the treatment of HIV infections. Our product candidates are nucleoside/tide analogs that we believe have potential competitive advantages with respect to safety, efficacy, drug resistance, and/or convenience of dosing as compared to currently approved drugs and other known investigational agents. The following table summarizes the five product candidates on which we are focusing:
| Product Candidate |
Indication |
Status |
Next Expected Milestone |
Commercialization | ||||
| RG7128 |
HCV | In a Phase 2b clinical trial and recently completed the INFORM-1 study conducted by Roche | Complete enrollment of cohort 2 (remaining 300 patients) in Phase 2b clinical trial by the end of the first calendar quarter of 2010, and the INFORM-2 (Phase 2) study to be initiated by Roche during the first calendar quarter of 2010 | Roche | ||||
| PSI-7851 |
HCV | Recently completed Phase 1 study | Begin Phase 2a clinical trial during the first calendar quarter of 2010 | — | ||||
| PSI-938 |
HCV | In IND-enabling preclinical studies | Submit IND application during the first calendar quarter of 2010 | — | ||||
| PSI-879 |
HCV | In IND-enabling preclinical studies | Submit IND application during the fourth calendar quarter of 2010 | — | ||||
| Racivir |
HIV | Completed Phase 2 clinical trial | Engage a development partner for a combination drug study | — | ||||
Product Candidates for the Treatment of HCV
HCV Background
HCV is a leading cause of chronic liver disease and liver transplants. The World Health Organization, or WHO, estimates nearly 180 million people worldwide, or approximately 3% of the world’s population, are infected with HCV. About 130 million of these individuals are chronic HCV carriers who are at an increased risk of developing liver cirrhosis or liver cancer, approximately 15 million of whom are in the United States, Europe, and Japan. The Center for Disease Control and Prevention (“CDC”), has reported that 4.1 million people in the United States have been infected with HCV, of whom 3.2 million are chronically infected. Of those chronically infected, the majority are undiagnosed and unaware of their HCV infection. Separately, approximately 10% of diagnosed HCV patients in the United States are treated each year.
At least six major genotypes of HCV have been identified, each with multiple subtypes. Genotypes are designated with numbers (genotypes 1-6) and subtypes with letters. HCV genotypes 1, 2, 3, and 4 have a worldwide distribution, but their prevalence varies from one geographic area to another. Genotype 1 and its subtypes (1a and 1b) are the most common genotype globally, accounting for approximately 70% of infections. Patients with genotype 2 or 3 represent approximately 25% of the worldwide chronically infected HCV population and the remaining 5% is comprised of genotypes 4 through 6. Worldwide sales of HCV drugs in 2005 were approximately $2.2 billion and are forecasted to reach more than $8.0 billion in 2015. Historical sales of HCV drugs increased as new therapies were introduced that improved the sustained viral response, or SVR, defined as the inability to detect HCV RNA in a patient’s blood six months after discontinuation of therapy, with a standard PCR test.
Limitations of Current HCV Infection Therapy
Globally, the current standard of care is a combination of pegylated interferon plus ribavirin. Pegylated interferon is a modified version of alpha interferon, a protein that occurs naturally in the human body and boosts
4
Table of Contents
the immune system’s ability to fight viral infections. Roche, our collaboration partner in the development of RG7128, is the market leader in sales of pegylated interferon and branded ribavirin under the brand names Pegasys® and Copegus®, respectively.
Current pegylated interferons are given as a weekly injection, administered together with twice daily ribavirin tablets. The current standard of care, however, has limitations that result in less than optimal sustained virologic response rates. Substantial side effects can render treatment intolerable for many patients. For example, pegylated interferon and ribavirin treated patients can have difficulties with fatigue, bone marrow suppression, anemia, and neuropsychiatric effects. In addition, genotype 1 patients typically receive 48 weeks of pegylated interferon and ribavirin, but less than 50% of these genotype 1 patients achieve an SVR, which many physicians and patients consider a low rate of success. Between 60% and 80% of the genotype 2 and 3 patients treated with pegylated interferon and ribavirin for 24 weeks achieve an SVR. Pegylated interferon is given as a weekly injection, along with daily ribavirin tablets. The occurrence of side effects combined with the inconvenient treatment regimen can result in many patients not completing therapy. Furthermore, a majority of individuals with HCV infection are unable to be treated with interferon due to contraindications, such as advanced liver disease or psychiatric conditions. The less than optimal antiviral efficacy, potential for dose-limiting side effects (some of which can be serious), contraindications, and inconvenient dosing regimen illustrate the unmet medical need of the currently available standard of care. Current therapies may also not directly target the virus, suggesting additional patient benefit from agents which directly interfere with hepatitis C virus replication.
Nucleoside/tide Analogs and Other Direct Acting Antivirals for HCV
The hepatitis C virus has several viral specific enzymes that are essential for its replication, thus providing multiple opportunities for therapeutic intervention. Many drug developers have focused on two of these enzymes: the protease (“NS3”) and the polymerase (“NS5b”) of HCV. The goal of HCV drug development is to discover and develop molecules that have a high affinity for binding to these enzymes thereby inhibiting enzymatic activity. This, in turn, inhibits viral replication and prevents the virus from spreading within the infected individual. These compound classes are often referred to as protease inhibitors and polymerase inhibitors. There are two types of polymerase inhibitors which have different mechanisms of action. Nucleoside/tide polymerase inhibitors work by acting as alternative substrates that block the synthesis of HCV RNA, which are essential for the virus to replicate. The other type of polymerase inhibitor, non-nucleoside polymerase inhibitors, binds directly to the polymerase enzyme, causing a change in its shape. This conformational change inhibits its enzymatic activity.
Our research efforts focus on blocking HCV replication by discovering and developing nucleoside/tide analog polymerase inhibitors. A nucleoside is a basic building block of the nucleic acids, DNA and RNA, the genetic material of all living cells and viruses. Nucleosides consist of a molecule of sugar linked to a nitrogen-containing organic ring compound. In the most important naturally occurring nucleosides, the sugar is either ribose (used to construct RNA) or deoxyribose (used to construct DNA), and the nitrogen-containing organic ring compound, referred to as the base, is either a pyrimidine (cytosine, thymine, or uracil) or a purine (adenine or guanine). A nucleoside combined with a phosphate group becomes a nucleotide.
In biological systems, nucleotides are linked by enzymes, including the polymerase, in a specific order to make long, chainlike polynucleotides (DNA or RNA) of defined sequence to pass along genetic information for a specific protein, a gene, or an entire organism, a genome. A nucleoside analog is a synthetic molecule that resembles a naturally occurring nucleoside. Chemical modifications in either the sugar portion or the base portion allow these compounds, once phosphorylated, to inhibit or disrupt the activity of the polymerase. When a nucleotide analog is incorporated into viral DNA or RNA during replication, it acts to prevent production of new virus by blocking the complete synthesis of the new viral DNA or RNA genome.
Experiments in vitro conducted by us and others show that nucleoside/tide analogs have consistent antiviral activity across all HCV genotypes. This characteristic of the nucleoside/tide polymerase inhibitor class relates to its unique mechanism of action. Recent clinical studies of RG7128, as more fully described below, show
5
Table of Contents
comparable anti-HCV activity across genotypes 1, 2, and 3. Other classes of anti-HCV drugs (i.e., protease inhibitors and non-nucleoside polymerase inhibitors) tested clinically show diminished antiviral activity in genotypes other than genotype 1.
In monotherapy studies with three separate nucleoside/tide analogs (including RG7128) over 14 days, viral breakthrough while on therapy did not occur. In clinical studies of non-nucleoside polymerase and protease inhibitors, viral breakthrough was seen as early as three to four days into the 14-day treatment period. The relative rapidity of the breakthrough with these classes of drugs suggests that the HCV patients may have harbored virus that was not susceptible to the therapeutic regimen. With longer exposure to any DAA, drug resistant virus will likely be selected over time. The rapidity and frequency with which this occurs may have significant consequences for patients.
Summary of Nucleoside/tide Analogs and Their Potential Use as Future Therapy
Conventional combination therapy for HIV usually includes at least one nucleoside/tide analog. Therefore, nucleoside/tide analogs may be referred to as the “backbone” of HIV therapy. The frequency of emergence of resistance variants in HCV inhibitor monotherapy trials suggests that combinations of antivirals with different modes of action may be required in the treatment of HCV. The combinations may not include the current standard of care, pegylated interferon and ribavirin. In consultation with experts in the field and our advisors, we believe the combination of at least one nucleoside/tide analogs with, for example, protease inhibitors or non-nucleoside polymerase inhibitors, or one nucleoside/tide combined with a second complementary nucleoside/tide presents a potentially useful therapeutic regimen. These different classes of DAAs use different and complementary mechanisms of action, suggesting that they will not adversely affect or antagonize the antiviral activity of the other compound. In addition, nucleoside inhibitors have demonstrated in vitro the ability to suppress the resistant variants that emerge with partially-suppressive concentrations of protease inhibitors or non-nucleoside polymerase inhibitors. Clinical use of a combination of DAAs may provide improved antiviral activity across HCV genotypes, and may lead to interferon-sparing regimens which avoid the adverse side effects that are often associated with the current standard of care.
Our pyrimidine nucleoside/tide analog polymerase inhibitor candidates (RG7128 and PSI-7851) and our purine nucleotide analog polymerase inhibitor candidates (PSI-938 and PSI-879) are described below.
Pyrimidine Nucleoside/tide Product Candidates
RG7128
Phase 1 Studies. In October 2004, we entered into a collaboration with Roche for the development and commercialization of PSI-6130 (an oral cytidine nucleoside analog polymerase inhibitor which we discovered) and its prodrugs, including RG7128, for the treatment of HCV. A prodrug is a chemically modified form of a molecule designed to enhance the absorption, distribution, and metabolic properties of that molecule. Roche and we initiated an adaptive Phase 1 clinical trial with RG7128 in October 2006 under an IND filing. On October 12, 2007, we were informed by the FDA that RG7128 received fast track designation. During December 2008, we completed the Phase 1 clinical trial. Following is a review of the design and results of this trial.
This adaptive Phase 1 clinical trial of RG7128 was a multiple center, observer-blinded, randomized, and placebo-controlled study designed to investigate the pharmacokinetics, pharmacodynamics, safety, tolerability, and food effect of RG7128 in healthy subjects and in patients chronically infected with HCV genotypes 1, 2, or 3. This trial provided antiviral potency data over 14 and 28 days in patients chronically infected with HCV genotype 1, and following 28 days of treatment in patients chronically-infected with HCV genotypes 2 or 3 who had not responded to prior interferon-based therapy. This study included three parts:
| • | Part 1 was a single ascending dose study conducted in 46 healthy subjects. The primary objective of Part 1 was to assess the safety, tolerability, and pharmacokinetics of RG7128 following single |
6
Table of Contents
| ascending doses under fasting conditions. The secondary objective of Part 1 was to explore the effect of food on the pharmacokinetics of RG7128. Single oral doses of RG7128 were administered to 46 healthy subjects in five sequential dose groups (500 mg, 1500 mg, 4500 mg, 6000 mg, and 9000 mg) and one food effect group (1500 mg). Results from the single ascending dose portion of the study indicated: |
| • | All doses of RG7128 studied (500 mg to 9000 mg) were generally safe and well-tolerated. |
| • | All patients completed the study and none experienced gastrointestinal adverse events or serious adverse events during the study. |
| • | No hematological or other safety laboratory abnormalities of clinical significance were noted. |
| • | No maximum tolerated dose was identified. |
| • | Part 2 was a multiple ascending dose study conducted in 40 patients chronically infected with HCV genotype 1 who had previously failed interferon therapy. The primary objective of Part 2 was to assess the safety, tolerability, and pharmacokinetics of RG7128 after once-daily (“QD”) or twice-daily (“BID”) dosing for 14 days. The secondary objective was to assess antiviral efficacy by measuring the change from baseline in circulating HCV RNA. Results from the multiple ascending dose portion of the study indicated: |
| • | RG7128 demonstrated potent, dose-dependent antiviral activity in four patient cohorts (8 active and 2 placebo per cohort) receiving 750 mg or 1500 mg administered either QD or BID for 14 days as monotherapy. RG7128 demonstrated mean HCV RNA decreases from baseline of 0.9 log10 (87.4% reduction), 1.5 log10 (96.8% reduction), 2.1 log10 (99.2% reduction), and 2.7 log10 (99.8% reduction) in patients receiving 750 mg QD, 1500 mg QD, 750 mg BID, and 1500 mg BID, respectively. A maximum reduction in HCV RNA of 4.2 log10 (99.9% reduction) was demonstrated in a patient following 14 days of monotherapy with 1500 mg BID of RG7128, a value also below the limit of detection, which was less than 15 International Units per milliliter (IU/ml). |
| • | There was no evidence of drug resistance in any dose cohort during the 14 days of dosing. |
| • | RG7128 was generally safe and well tolerated over 14 days of treatment. |
| • | Part 3 was a 4-week study of RG7128 in combination with the current standard of care for chronic HCV infection, Pegasys® (pegylated interferon) plus Copegus® (ribavirin), in 81 treatment-naïve patients chronically infected with HCV genotype 1, and additionally, in 25 prior treatment non-responders, or patients who did not achieve an SVR with previous interferon-based therapy, who were chronically infected with HCV genotypes 2 or 3. The primary objective of this study was to assess the safety, tolerability, and pharmacokinetics of RG7128 in the clinically-relevant setting of combination therapy with the current standard of care for chronic HCV infection. The secondary objective of Part 3 was to evaluate the short-term change in HCV RNA. The study included three oral dose regimens of RG7128 (500 mg, 1000 mg, and 1500 mg BID—cohorts 1, 2, and 3, respectively) in patients chronically infected with HCV genotype 1 and one oral dose regimen of RG7128 (1500 mg BID—cohort 4) in patients chronically infected with HCV genotypes 2 or 3. |
The antiviral results for cohorts 1, 2, and 3 are summarized in the following table:
| RG7128 dose |
N | Mean HCV |
HCV RNA | |||
| 500mg BID + SoC | 20 | -3.8 | 6/20 (30%) | |||
| 1000mg BID + SoC | 25 | -5.1 | 22/25 (88%) | |||
| 1500mg BID + SoC | 20 | -5.1 | 17/20 (85%) | |||
| Placebo + SoC | 16 | -2.9 | 3/16 (19%) |
| (1)— | LLOD means lower limit of detection by Roche Taqman Assay |
7
Table of Contents
For cohorts 1, 2, and 3 in treatment-naïve genotype 1 patients, RG7128 was generally safe and well-tolerated when administered for 4 weeks in combination with Pegasus plus Copegus® in patients with HCV genotype 1.
The antiviral results for the 1500 mg BID dose cohort (cohort 4) in 25 prior treatment non-responders (patients who did not achieve an SVR with previous interferon-based therapy) who were chronically infected with HCV genotype 2 or 3 are summarized in the following table:
| RG7128 dose |
N | Mean HCV |
HCV RNA | |||
| 1500mg BID + SoC | 20 | -5.0 | 18/20 (90%) | |||
| Placebo + SoC | 5 | -3.7 | 3/5 (60%) |
| (1)— | LLOD means lower limit of detection by Roche Taqman Assay |
RG7128 was generally safe and well-tolerated when administered for 4 weeks in combination with Pegasys® plus Copegus® in patients with HCV genotype 2 or 3.
Phase 2b Study. On April 24, 2009, Roche began dosing in a Phase 2b study with RG7128. The Phase 2b trial is anticipated to enroll approximately 400 treatment-naive, genotype-1, or genotype-4 HCV-infected patients. The trial will evaluate the dose and duration of treatment of RG7128 in combination with Pegasys® plus Copegus®. The primary efficacy endpoint of the trial will be the proportion of patients that achieve an SVR. Subjects are being enrolled into one of 5 arms:
| • | 24 weeks of total treatment, with RG7128 500mg BID in combination with Pegasys® plus Copegus® for 12 weeks, followed by 12 weeks of Pegasys® plus Copegus®, |
| • | 24 weeks of total treatment, with RG7128 1000mg BID in combination with Pegasys® plus Copegus® for 12 weeks, followed by 12 weeks of Pegasys® plus Copegus® |
| • | 24 weeks of total treatment, with RG7128 1000mg BID in combination with Pegasys® plus Copegus® for 8 weeks, followed by a further 16 weeks of Pegasys® plus Copegus® |
| • | 48 weeks of total treatment, with RG7128 1000mg BID in combination with Pegasys® plus Copegus® for 12 weeks, followed by a further 36 weeks of Pegasys® plus Copegus®. |
| • | A control arm with only Pegasys® plus Copegus® for 48 weeks. |
Patients in the 24 week arms will discontinue treatment at week 24 if they achieved an RVR. Patients who do not achieve an RVR will continue on Pegasys® plus Copegus®, until week 48. Patients are expected to be enrolled as two cohorts, with randomization of the second cohort, of approximately 300 patients, being initiated based on an independent Data Monitoring Committee (“DMC”) review of 12 week safety data from the first approximately 100 patients.
During November 2009, the independent DMC recommended to Roche that it could proceed with enrolling the second cohort of approximately 300 patients in this Phase 2b study of RG7128. This recommendation was reached by the DMC after the scheduled review of all available safety data from the first cohort of approximately 100 patients that completed 8 or 12 weeks of RG7128 or matching placebo in combination with Pegasys® plus Copegus®. The DMC reviewed any potential drug discontinuations, incidence and details of adverse events, and selected laboratory assessments. No safety events in the DMC review were considered significantly different from those expected from HCV patients taking pegylated interferon and ribavirin treatment. The committee expressed no safety concerns that would preclude enrollment of the remaining 300 patients in the ongoing phase 2b study in the HCV positive genotype 1 and 4 population, and have not recommended modification of dose or duration of any RG7128 dosing regimens. Pre-screening of subjects for cohort 2 has been initiated and full enrollment of cohort 2 of this study is expected to be completed at the end of the first calendar quarter of 2010.
Roche plans to initiate additional, longer duration Phase 2b studies of RG7128 during the first half of 2010. Roche is planning to initiate an additional study of patients infected with HCV genotypes 2 and 3 later in 2010.
8
Table of Contents
INFORM Studies. During November 2008, Roche, InterMune, Inc., (“InterMune”) and we announced the initiation of a Phase 1 study to investigate the combination of two DAAs in the absence of interferon and ribavirin. This study, named INFORM-1, combined for the first time in patients naïve to therapy and in patients who previously failed therapy (TF) two oral DAAs, RG7128 and RG7227. RG7227 is an inhibitor of the HCV NS3/4 protease, which is being developed by InterMune in collaboration with Roche.
INFORM-1 was a randomized, double-blind, ascending dose Phase 1 trial that enrolled a total of 86 patients and was conducted by Roche. The principal objectives were to evaluate safety, tolerability, and antiviral activity of RG7128 and RG7227 administered in combination at increasing doses for up to 13 days. Results from this study demonstrated for the first time that the combination of an oral protease inhibitor and an oral nucleoside polymerase inhibitor resulted in significant circulating HCV RNA reduction in patients with HCV, as patients receiving the combination of RG7227 and RG7128 for 13 days (without pegylated interferon or ribavirin) experienced a median reduction in HCV RNA of -4.8 to -5.2 log10 IU/mL in the highest dose levels tested. In addition, no treatment-related serious adverse events, dose reductions, or discontinuations were reported during the study. Pharmacokinetic analysis also confirmed that there were no drug-drug interactions between the compounds. Following is a graph that presents by dose regimen the median log10 HCV RNA change from baseline over the 13 days of treatment.
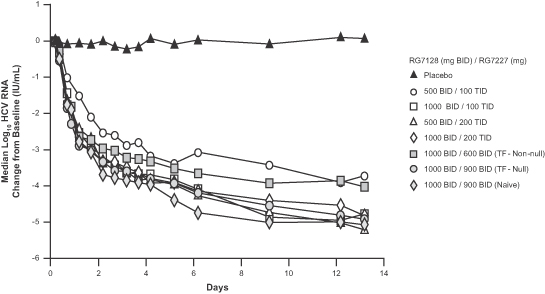
The antiviral results for each dose regimen are summarized in the following table.
| Regimen (RG7128 / RG7227 mg) |
N | Patient |
HCV RNA |
HCV RNA | ||||
| 500 BID/100 TID | 8 | Naïve | 1/8 (13%) | 1/8 (13%) | ||||
| 500 BID/200 TID | 8 | Naïve | 5/8 (63%) | 2/8 (25%) | ||||
| 1000 BID/100 TID | 7 | Naïve | 5/7 (71%) | 2/7 (29%) | ||||
| 1000 BID/200 TID | 8 | Naïve | 5/8 (63%) | 2/8 (25%) | ||||
| 1000 BID/600 BID | 8 | TF (non-null) | 4/8 (50%) | 1/8 (13%) | ||||
| 1000 BID/900 BID | 8 | TF (null) | 4/8 (50%) | 2/8 (25%) | ||||
| 1000 BID/900 BID | 8 | Naïve | 7/8 (88%) | 5/8 (63%) |
| (1)— | LLOQ means lower limit of quantification by Roche Taqman Assay |
| (2)— | LLOD means lower limit of detection by Roche Taqman Assay |
9
Table of Contents
The higher dose combination of RG7128 1000 mg and RG7227 900 mg administered twice daily without pegylated interferon or ribavirin for 13 days resulted in 88% of HCV-positive treatment-naïve patients achieving HCV RNA below the lower limit of quantification (“LLOQ”), and 63% of patients having HCV RNA below the lower limit of detection (“LLOD”). The same regimen in “null-responders” resulted in 50% of patients with HCV RNA below LLOQ and 25% of patients with HCV RNA below LLOD. “Null responders” were defined as patients with a documented failure to achieve a 1.0 log10 or greater drop in HCV RNA in 4 weeks or a 2.0 log10 or greater drop in HCV RNA in 12 weeks of prior treatment with pegylated interferon and ribavirin.
Roche expects to initiate a Phase 2 program of multiple INFORM studies during the first calendar quarter of 2010. Roche expects these INFORM-2 studies to investigate RVR achieved by twice-daily dosing of the DAAs RG7128 and RG7227 alone, in combination with Pegasys®, in combination with Copegus®, and in combination with both Pegasys® plus Copegus® (“QUAD”). Roche expects to initiate longer term studies evaluating SVR during the first half of 2010. Roche has announced that they expect no change to the above plans for the INFORM-2 resulting from Roche’s acceptance of an independent DMC’s recommendation to stop dosing and enrollment of the highest dose cohort of RG7227 in Roche’s clinical trial studying RG7227 in combination with Pegasys® and Copegus®, which is a trial that is separate from the INFORM studies and does not involve the use of RG7128.
PSI-7851
Phase 1 Study. PSI-7851 is a prodrug of a uridine nucleotide analog polymerase inhibitor and is in development for the treatment of chronic HCV infection. PSI-7851 has demonstrated in vitro anti-HCV activity with EC90 values of approximately 31 +/- 12 nM, approximately 15-fold more potent than the active metabolite of our first generation nucleoside polymerase inhibitor, PSI-6130. The in-vitro half-life of the triphosphate (the biologically active form of the molecule) in primary human hepatocytes is approximately 38 hours, which supports the exploration of once-daily dosing in early studies. Like RG7128, PSI-7851 has demonstrated in vitro activity against HCV genotypes 1, 2, 3, and 4.
During March 2009, we initiated a Phase 1 study of PSI-7851, which was a single ascending dose study that assessed the safety, tolerability, and pharmacokinetics of PSI-7851 in healthy subjects at doses ranging from 25mg to 800mg. Results from this study indicated there were:
| • | No serious adverse events or discontinuations, |
| • | No dose-related trends in adverse events, |
| • | No grade III/ IV lab abnormalities, and |
| • | No clinically significant changes in vital signs or ECGs. |
In June 2009, we initiated a Phase 1 multiple ascending dose study in HCV-infected patients. Subjects were enrolled at two U.S. centers and randomized to PSI-7851 (8 per cohort) or placebo (2 per cohort). The primary objective of this study was to assess the safety, tolerability, and pharmacokinetics of PSI-7851 after once-daily dosing for three days. The secondary objective of this study was to assess antiviral activity by measuring the change in circulating HCV RNA levels. Four dose cohorts of PSI-7851 (50mg QD, 100mg QD, 200mg QD, and 400mg QD) were evaluated. Results from this study indicated:
| • | PSI-7851 was generally safe and well tolerated across all cohorts with no discontinuations, no serious adverse events, and no dose-related trends in adverse events or laboratory abnormalities. |
| • | PSI-7851 demonstrated potent antiviral activity with a mean HCV RNA change from baseline of -0.49 log10 IU/mL or -0.61 log10 IU/mL in patients receiving 50 mg QD or 100 mg QD, respectively. |
| • | PSI-7851 200mg QD administered for 3 days resulted in a mean change from baseline HCV RNA of -1.01 log10 IU/mL, with 6 of 8 subjects achieving greater than a 1.0 log10 IU/mL decline from baseline. This antiviral effect met our threshold of approximately 1.0 log10 IU/mL decline over three days as established with the first-in-class nucleoside, RG7128. |
10
Table of Contents
| • | PSI-7851 400mg QD administered for 3 days resulted in a mean change from baseline HCV RNA of -1.95 log10 IU/mL, with 6 of 8 subjects achieving greater than a 1.5 log10 IU/mL decline from baseline. |
Purine Nucleotide Product Candidates
Two purine nucleotides, PSI-938 and PSI-879, were nominated as product candidates during 2009 as part of our strategy to identify new nucleoside/tides, which would be complementary to our pyrimidine analogs (RG7128 and PSI-7851) currently in clinical development. The new purine nucleotide analogs retain activity against HCV with the S282T mutation, a mutation which reduces the in vitro susceptibility of the virus to the pyrimidine analogs. The purines are phosphorylated by different enzymes than the pyrimidines, and thus do not compete with activity of the pyrimidines. In vitro, the combination of the purine analogs with the pyrimidine analogs provides additive to synergistic antiviral activity, presumably due to the fact that these classes of compounds both compete with naturally occurring nucleotides for incorporation into nascent HCV RNA. This combination increases the inhibitory potential at the level of the HCV NS5B enzyme. Such complementary activities offer the potential for a potent dual nucleoside/tide-based combination for the future treatment of HCV.
PSI-938
During June 2009, we nominated PSI-938 as a product candidate for the treatment of HCV. PSI-938 is a guanine nucleotide analog polymerase inhibitor for the treatment of chronic HCV infection. PSI-938 has demonstrated in vitro anti-HCV activity with EC50 values of approximately 0.2 ± 0.1 µM. This compound has demonstrated equivalent potency against the S282T mutant which has reduced sensitivity to several nucleoside inhibitors including RG7128, PSI-7851, NM283 and IDX184, and is metabolized to the active triphosphate form through a different phosphorylation pathway than our pyrimidine product candidates, RG7128 and PSI-7851, supporting a low likelihood of antagonism with the combination. The in vitro half-life of the triphosphate in primary human hepatocytes is approximately 12 hours, which supports the exploration of once-daily dosing in early development. PSI-938 is completing studies required for submission of an IND application with the FDA or equivalent foreign regulatory application. Our plan is to submit an IND application, or its foreign equivalent, during the first calendar quarter of 2010.
PSI-879
During September 2009, we nominated PSI-879 as a product candidate for the treatment of HCV. Our current plan is to submit an IND application, or its foreign equivalent, during the fourth calendar quarter of 2010. PSI-879 is a guanine nucleotide analog polymerase inhibitor for the treatment of chronic HCV infection. PSI-879 has demonstrated in vitro anti-HCV activity with EC50 values of approximately 0.04 ± 0.02 µM. PSI-879 uses a different prodrug strategy than PSI-938, but PSI-879 is metabolized to the same active triphosphate as PSI-938, it shares the same in vitro half-life, and retains activity against HCV with an S282T mutation. Given the similarities of PSI-938 and PSI-879, our plan is to select one of these product candidates for later-stage clinical development based upon a review of the early human clinical trial results of both PSI-938 and PSI-879.
Product Candidate for the Treatment of HIV
HIV Background
HIV destroys the body’s ability to fight infections by attacking cells of the immune system. In 1981, the first cases of Acquired Immunodeficiency Syndrome, or AIDS, were documented, and in 1983, HIV was identified as the cause of AIDS. According to a 2007 AIDS epidemic update by UNAIDS and the WHO, over 33 million people worldwide are living with HIV, and at least 25 million people worldwide have died from AIDS since the epidemic began. In the United States, the CDC has reported that the HIV mortality rate has steadily declined since the mid-to-late 1990s, while the incidence of infection continues to rise. This decrease in mortality can be
11
Table of Contents
attributed, in part, to the increased availability of HIV therapeutics used in the long-term treatment of HIV. According to a 2007 UNAIDS/WHO report, by the end of 2007, approximately 1.3 million people in North America and 760,000 people in Western and Central Europe were living with HIV and an additional 46,000 new patients in North America and 31,000 new patients in Western and Central Europe are diagnosed each year. In 2008, antiretroviral sales across the U.S. and five major European markets (France, Germany, Italy, Spain and the UK) totaled $10.5 billion, demonstrating a compound annual growth rate of 13.1% between 2005 and 2008.
The FDA has approved numerous single agents and fixed-dose combination therapies for the treatment of HIV. The single agents are classified as nucleoside reverse transcriptase inhibitors, or NRTIs, non-nucleoside reverse transcriptase inhibitors, or NNRTIs, protease inhibitors, or PIs, integrase inhibitors and entry inhibitors.
The standard treatment for HIV infection, as recommended by the U.S. Department of Health and Human Services, includes two NRTIs combined with a third drug from another class, either an NNRTI or a PI, to form a triple combination therapy known as Highly Active Anti-Retroviral Therapy, or HAART. The two NRTIs in HAART are usually analogs of different nucleosides. Typically, a cytidine analog is paired with a thymidine or an adenosine analog in an effort to ensure the broadest activity against viral mutations and delay the onset of drug resistance. Racivir is a cytidine analog. It has been estimated that lamivudine and emtricitabine currently are the most commonly prescribed cytidine analogs, alone or as components of fixed dose combination products.
Racivir
Racivir is an oral, once-daily deoxycytidine nucleoside analog that we are developing as an HIV therapy for use in combination with other approved HIV drugs. In a completed Phase 2 clinical trial, for the subset of patients carrying the M184V mutation and less than three thymidine analog mutations (“TAMs”), replacing lamivudine with Racivir in their existing combination therapies caused a mean decrease in plasma HIV RNA of 0.7 log10 (80% reduction) in the second week of treatment. Twenty-eight percent of these patients achieved an HIV RNA below the limit of detection (less than 400 copies per milliliter) and 64% of these patients achieved at least a 0.5 log10 decrease (68% reduction) in plasma HIV RNA.
Clinical Development. We have completed a Phase 2 clinical trial, Study 201, to assess the safety, tolerability, and antiviral effect of a 600 mg dose of Racivir head-to-head against lamivudine in HIV-infected, treatment-experienced patients with the M184V mutation who have been on lamivudine therapy. The study was a randomized, double-blind, placebo-controlled, multicenter study of 54 patients in the United States, Argentina, Mexico and Panama. Patients were randomized into two groups: one substituting Racivir in place of lamivudine in their existing therapies, and one continuing on their current lamivudine-containing therapy without any change. The study entry criteria included patients who were failing a HAART regimen. Specifically, participants were required to have received lamivudine as part of their antiviral therapy for the previous 60 days, to carry the M184V HIV mutation, and to have an HIV RNA viral load of greater than or equal to 2,000 viral copies per milliliter of blood plasma. The study had a blinded treatment period of up to 28 days, followed by an open label treatment period of up to 20 weeks. Patients were subsequently followed for an additional four weeks after the conclusion of the study treatment periods. The goal of this study was to evaluate the benefit of Racivir in patients carrying the M184V mutation by replacing lamivudine with Racivir in existing therapies.
In this study, 42 subjects were randomized to receive either Racivir (n=26) in place of lamivudine or to continue with lamivudine (n=16) in a double-blind manner for 28 days. HIV viral loads and genotypes were determined at baseline (mean viral load = 4.1 log) and throughout the study. After the blinded treatment period, subjects were allowed to continue Racivir in an open label manner with or without optimized background therapy for an additional 20 weeks, based on their primary care physicians’ advice. After 28 days of blinded treatment, the mean viral load rose by 0.13 log (a 34.9% increase) in the lamivudine group and dropped by 0.4 log (60.2% reduction) in the Racivir group (p=0.02). A subset analysis of the Racivir-treated group revealed that the change in viral load was largely due to a positive antiviral response in subjects who had an HIV mutation pattern that included M184V and less than 3 TAMs with or without NNRTI or PI mutations. In this subset of patients (n=14),
12
Table of Contents
replacing lamivudine with Racivir in their existing therapies caused a mean drop in viral load of 0.7 log (80% reduction, p=0.004) in the second week of treatment, with 28% of these patients achieving an undetectable level of virus (less than 400 copies per milliliter) and 64% of these patients achieving at least a 0.5 log (68%) drop in viral load. No serious adverse events attributed to therapy were noted in either group over the 28 days. Clonal genotypic analysis of virus from responders indicated that the M184V mutation was found in all clones in addition to multi-drug resistance-associated mutations observed with first line therapy failure.
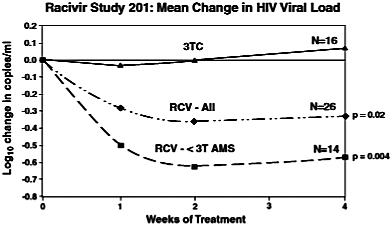
In summary, Racivir demonstrated antiviral activity in patients harboring HIV with the M184V mutation and less than three TAMs. These patients have genotypes consistent with first-line therapy failure and may be candidates for second line treatment regimens that contain Racivir. Any future studies would be designed to explore this potential use of Racivir in a combination therapy for second-line therapy.
Termination of Clevudine Registration Studies
Clevudine is an oral, once-daily pyrimidine nucleoside analog that we attempted to develop for the treatment of hepatitis B virus (“HBV”) pursuant to our license agreement with Bukwang Pharm. Co. Ltd., or Bukwang, a South Korean pharmaceutical company. On April 20, 2009, following consultations with our independent Data Safety Monitoring Board and the FDA, we voluntarily terminated our Phase 3 studies of clevudine after we became aware of a number of spontaneous Serious Adverse Event reports and Events of Special Interest in patients receiving clevudine as prescribed therapy for HBV in South Korea, where the drug is marketed by Bukwang under the trade name Levovir, and in Hong Kong, where clinical studies were being conducted under the sponsorship of Bukwang. Though only a small number of cases of mild to moderate myopathy, or muscle weakness associated with creatine kinase elevations, were reported in our clinical studies, many of the patients in South Korea and Hong Kong had longer exposures to clevudine than patients in our studies and have reported more serious myopathy than patients in our clinical trials. Given the number and severity of cases observed in South Korea and Hong Kong, we determined it was in the best interest of patients to voluntarily terminate the studies. Our obligations with regard to patients in this terminated study were complete on November 13, 2009. We expect to have this study termination process complete by December 31, 2009.
Our Research Programs
We have a library of cataloged nucleoside/tide analogs, as well as several other chemically diverse compounds. This library is the result of substantial collective effort, and we continue to enhance the compound library’s value through the addition of new compounds. We screen potential new targets against this library as a means of identifying promising chemical compounds to pursue for further development. We use preclinical discovery and development technologies and viral and cellular assays that we believe form a reasonable basis for anticipating clinical results. Developing additional compounds to treat HCV is the primary focus of our nucleoside/tide research and development activities.
13
Table of Contents
Collaborations and Licensing Agreements
Hoffmann-La Roche Inc.
Hoffmann-La Roche Inc. is the U.S. affiliate of F. Hoffmann-La Roche Ltd, a Swiss company (collectively “Roche”). In October 2004, we entered into a collaboration and license agreement with Roche to develop PSI-6130 and its prodrugs for the treatment of chronic HCV infections. Roche paid us an up-front payment of $8.0 million. Roche has also agreed to make milestone payments to us for PSI-6130 or a pro-drug of PSI-6130, including RG7128, of up to an aggregate of approximately $105 million, assuming successful development and regulatory approval in Roche’s territories. In addition, we will receive royalties paid as a percentage of total annual net product sales, if any, and we will be entitled to receive up to $30 million of one-time performance payments should net sales from the product exceed specified thresholds. Under this collaboration, Roche reimbursed us for all of the expenses associated with certain preclinical work, the IND filing, and the initial clinical trial, which we were responsible for and conducted. Roche has and will fund all of the expenses of, and be responsible for, other preclinical studies, future clinical development and commercialization of RG7128. In addition to the $8.0 million up-front payment, we have received milestone payments of $35.0 million and research reimbursement payments of $5.0 million from Roche under this agreement as of September 30, 2009.
We granted Roche worldwide rights, excluding Latin America and Korea, to which we refer as our retained territory, to PSI-6130 and its prodrugs. With respect to our retained territory, we may grant rights to a third party to distribute, promote, market, or sell a product covered by this collaboration agreement, as long as we first offer these rights to Roche, subject to certain exceptions. We retained certain co-promotion rights in the United States, including the right to market and promote products comprising these compounds to physicians who treat HIV patients. We will be required to pay to Roche royalties on our net product sales, if any, in the territories we have retained.
We have the right to prosecute, maintain, enforce, and defend patents that are owned by us and are subject to the Roche collaboration, while Roche has the same right with respect to certain designated territories if we choose not to exercise our rights. With respect to Roche’s patents that are the subject of this collaboration, Roche has the right to prosecute, maintain, enforce, and defend these patents, while we have the same right with respect to certain designated territories if Roche chooses not to exercise its rights. With respect to joint patents that are the subject of this collaboration, Roche and we are each responsible for prosecuting, maintaining, enforcing, and defending those joint patents in our respective territories. Subject to certain exceptions, we have agreed to share with Roche any damages, monetary awards, and other amounts recovered, after costs and expenses, in connection with patent litigation related to this collaboration.
This agreement will terminate once there are no longer any royalty or payment obligations. Additionally, Roche may terminate the agreement in whole or in part by providing six months’ written notice to us. In the event of termination, Roche must assign or transfer to us all regulatory filings, trademarks, patents, preclinical and clinical data related to this collaboration. Provided that Roche has not terminated the agreement, our royalty obligations under this agreement terminate on a product-by-product and country-by-country basis upon either the expiration of the last to expire patent that covers a licensed compound in such country, or 10 years from the launch of such licensed compound in such country, whichever occurs later. Otherwise, either party may terminate the agreement in whole or in part in connection with a material breach of this agreement by the other party that is not timely cured.
In conjunction with this agreement, Roche purchased 400,000 shares of our Series R redeemable convertible preferred stock and received warrants to purchase up to an additional 470,588 shares of our Series R-1 redeemable convertible preferred stock for $4.0 million. These shares and warrants were initially recorded at fair value for financial reporting purposes. The 400,000 shares of Series R redeemable convertible preferred stock were converted into 266,666 shares of our common stock on May 2, 2007 when we completed our initial public offering, or IPO, and the related warrants expired without exercise on October 26, 2006.
14
Table of Contents
University of Cincinnati
In October 2007, we entered in a three year research collaboration and license agreement with the University of Cincinnati (“UC”) on behalf of its Genome Research Institute (“GRI”) to identify active and selective compounds against antiviral targets for HBV, HCV and HIV. As part of the agreement, UC granted us access to the GRI Lead Generation Library, which includes over 250,000 compounds. We were also granted access to GRI’s drug discovery capabilities, including high-throughput screening, computational chemistry and in silico docking expertise. UC granted us commercial rights for any lead compounds that are identified for HBV, HIV and HCV. We are required to make an annual payment to UC in support of the research collaboration and are responsible for all development expenses of products that may result from the collaboration. If a lead compound progresses through clinical development activities and achieves regulatory approval, we will make certain milestone payments to UC and pay to UC a royalty on any net sales of the product.
During September 2009, we and UC agreed to terminate our research collaboration. The UC license agreement and related access to the GRI Lead Generation Library and GRI’s drug discovery capabilities as briefly described above remain unchanged. The annual maintenance fee for this license agreement is $75,000.
Apath, LLC
Apath, LLC (“Apath”) is a Missouri company that is engaged in the commercial application of molecular virology and viral genetics. On October 18, 2000, we entered into an agreement with Apath, as amended on January 30, 2004, pursuant to which Apath granted us a non-exclusive right to use its HCV Replicon technology for the design, discovery, development and commercialization of compounds inhibiting HCV in humans. This agreement required us to pay Apath royalties on sales of compounds discovered using this technology, and on any consideration received by us from a licensee of such compounds. We used this technology in the discovery of antivirals for the treatment of HCV.
We do not have the right to advise or to consult with Apath regarding the prosecution or maintenance of the licensed patent rights. We are one of several sublicensees of the licensed patents and have no rights to enforce such patents.
This agreement was terminated on August 26, 2005, on which date we entered into a new agreement with Apath. Under the terms of the new agreement, we paid Apath a one-time sublicense fee of $550,000 and an annual maintenance fee of $75,000, subject to annual renewals, retroactive to October 18, 2000. Our only obligation under the new agreement is to pay the annual maintenance fee for any year for which we choose to renew this agreement, and we will have no other financial obligations to Apath in connection with the design, discovery, development and commercialization of compounds inhibiting HCV in humans.
This agreement expires on the date of expiration of the last-to-expire U.S. patent in the licensed patent rights. The last expiration of these patents is scheduled to occur in March 2018. Apath retains no rights to the compounds we discover, and they will receive no payments or royalties for any of the compounds we discover. We are entitled to sublicense these compounds to a third party without Apath’s permission or consent. We may terminate the agreement for any reason or no reason by giving Apath 30 days prior written notice without any penalties. Apath is entitled to terminate the contract, but only should we breach the agreement, on 30 days notice in the event of any uncured breach.
Emory University
In December 1998, we entered into an exclusive, worldwide license agreement to license the active pharmaceutical ingredients in our product candidate, Racivir from Emory University (the “Racivir License Agreement”). Emory is a non-profit Georgia corporation located in Atlanta, Georgia. This Racivir License Agreement provided us with rights to make, have made, use, import, offer for sale and sell drug products based on a specified range of mixtures of (–) – FTC and (+) – FTC, or enriched FTC, which includes the mixture that
15
Table of Contents
we are developing as Racivir. As part of the consideration for this agreement, we issued to Emory 66,667 shares of our redeemable common stock valued at $1.50 per share, and agreed to pay Emory royalties as a percentage of net product sales. We subsequently issued to Emory an additional 13,307 shares of our redeemable common stock valued at $4.95 per share pursuant to an anti-dilution provision in our agreement. The 79,974 aggregate shares of our redeemable common stock were converted into 79,974 shares of our common stock on May 2, 2007 when we completed our IPO. We may also pay Emory up to an aggregate of $1.0 million in future marketing milestone payments. Beginning in the second year after NDA registration, these royalties are subject to specified minimums. The Racivir License Agreement permits us to sublicense these rights under certain circumstances, provided that we pay a percentage of milestone and royalty payments that we receive from a sublicensee.
Emory is primarily responsible for the patent prosecution and maintenance activities pertaining to the licensed patent applications and patents, while we are afforded, pursuant to a license agreement relating to emtricitabine that Emory entered into with Triangle Pharmaceuticals, now Gilead Sciences, Inc., in 1996, which we refer to as the Emory/Gilead License Agreement, reasonable opportunities to advise Emory on, and cooperate with Emory in, such activities. Pursuant to the Emory/Gilead License Agreement, in the event of any suspected infringement, (i) we and Emory may first agree to institute suit jointly, (ii) in the absence of such agreement, Emory may institute suit, and (iii) in the absence of agreement and if Emory does not institute suit, then we may institute suit.
The Racivir License Agreement will expire upon the expiration of all licensed patents. The last expiration of these patents is scheduled to occur in November 2020. Emory has the right to terminate the agreement if we fail to make required payments or reports when due, if we become insolvent or bankrupt, or if we materially breach the agreement. To exercise this right, Emory must give us 60 days’ written notice, after which time the agreement automatically terminates unless we have cured the breach. We have the right to terminate the Racivir License Agreement at our sole discretion on three months written notice.
In the Emory/Gilead License Agreement, Emory previously had granted a right of first refusal to Gilead that is applicable to any license or assignment relating to enriched FTC (which includes Racivir). The terms of this right of first refusal contains an exception permitting Emory to license or assign its rights in respect of enriched FTC to a permitted transferee, which includes any of the inventors (which included two of our founders) or to any corporate entity formed by or on behalf of the inventors for purposes of clinically developing enriched FTC so long as the licensee agrees in writing to be bound by the terms of Gilead’s right of first refusal to the same extent as Emory. Our license to Racivir was granted to us by Emory pursuant to this exception and therefore we are bound by the terms of Gilead’s right of first refusal to the same extent as Emory. The terms of this right of first refusal as set forth in the Emory/Gilead License Agreement require that, prior to the entry into any license or assignment agreement with a third party relating to any of Emory’s rights in respect of enriched FTC, Emory shall notify Gilead of the terms of the proposed agreement and provide a copy of the proposed agreement to Gilead together with all data and information in Emory’s possession relating to enriched FTC and its use as a therapeutic agent. Gilead has 30 days to accept or decline the offer. Although Emory considers us to be a permitted transferee under the Emory/Gilead License Agreement, Emory has subsequently taken the position that its grant of commercialization rights (i.e., the rights to offer for sale and sell Racivir) to us exceeded the rights that were permitted to be granted to a permitted transferee under its agreement with Gilead. While we believe that Gilead is aware of the Racivir License Agreement through both our and Emory’s communications with Gilead, Gilead has not contacted us regarding its interpretation of the terms of the Racivir License Agreement.
In March 2004, we entered into a supplemental agreement with Emory in which we and Emory agreed that, prior to any commercialization of enriched FTC by us, or by any licensee or assignee of our rights under the Racivir License Agreement, we and Emory would adhere strictly to the terms of the right of first refusal granted to Gilead in the Emory/Gilead License Agreement and offer to Gilead the same terms and conditions under which we, our licensee or our assignee, propose to commercialize enriched FTC. The supplemental agreement also outlines a procedure by which Emory and we would jointly offer the terms of a proposed license and commercialization agreement between us and a third party to Gilead after Emory has the opportunity to approve
16
Table of Contents
them. Therefore, before we could enter into a commercialization agreement for Racivir with a third party or commercialize Racivir on our own, we would be required to offer Gilead the opportunity to be our commercialization partner on the same terms on which we intend, or our prospective partner intends, to commercialize Racivir. It is uncertain whether a third party would be willing to negotiate the terms of a commercialization agreement with us, knowing that Gilead can take their place as licensee by accepting the negotiated terms and exercising its right of first refusal.
In 1998 and 2004, the Company entered into various license agreements, in addition to those described above, with UGARF, Emory University and/or the University of Alabama at Birmingham Research Foundation, Inc. (collectively, the “Universities”) to pursue the research, development, and commercialization of certain human antiviral, anticancer, and antibacterial applications and uses of certain specified technologies, including the agreement with Emory and UGARF that is the subject of an arbitration proceeding (See Item 3. Legal Proceedings). Under each of these agreements, the Universities have granted an exclusive right and license under the related patents to the Company. The Company and the Universities will share in any proceeds received by the Company related to internal development or sublicensing of the specified technologies, including milestone payments, fees, and royalties.
In April 2002, the license agreement between UGARF, Emory University, and the Company dated June 16, 1998 was selectively modified to terminate certain technologies and related rights and obligations.
These uncertainties related to our commercialization rights may result in our being prevented from obtaining the expected economic benefits from developing Racivir. In addition, we could become involved in litigation or arbitration related to our commercialization rights to Racivir in the future.
Bukwang Pharm. Co., Ltd.
Bukwang is a pharmaceutical, oral hygiene, and cosmetics company based in Seoul, Korea. On December 28, 1995, Bukwang entered into an exclusive, worldwide license agreement with the University of Georgia Research Foundation, or UGARF, and Yale University to develop and commercialize clevudine. On June 23, 2005, Bukwang granted us exclusive rights to develop, manufacture, and market clevudine in North America, Europe, Central and South America, the Caribbean, and Israel. Bukwang retained rights to the rest of the world, excluding those Asian territories that were licensed to Eisai in November 2004. The agreement permits us to sublicense these rights, without Bukwang’s consent, to any of the top one hundred pharmaceutical companies based on sales ranked by IMS Health Incorporated.
We paid Bukwang an up-front payment of $6.0 million and may pay up to an aggregate of $24.0 million in milestone payments related to development, regulatory and commercialization events, as well as future royalties on net sales, including a minimum royalty obligation in certain years. These future milestone payments are not expected to occur due to our termination of our Phase 3 registration studies of clevudine on April 20, 2009. We incurred our first milestone payment to Bukwang in the amount of $1.0 million upon initiation of our Phase 3 registration study during the quarter ended September 30, 2007. Other than the up-front payment and this $1.0 milestone payment, we have made no additional milestone payments through September 30, 2009 to Bukwang under this agreement. Subject to the rights of any third party, including Gilead, we granted Bukwang a right of first refusal for Racivir for the treatment of HBV in Korea, the royalties for which would be based on net sales, and without any license fee or milestone obligations.
University of Georgia Research Foundation, Inc. and Yale University
As part of the collaboration and license agreement with Bukwang, we sublicensed certain patents and technology related to clevudine. On June 23, 2005, we, along with UGARF and Yale University, signed a memorandum of understanding with regard to the patents and technology related to clevudine that had been exclusively licensed to Bukwang, and which we currently sublicense from Bukwang. The memorandum of understanding provides that UGARF and Yale will grant us a license to these patents and technology in the event
17
Table of Contents
that the primary license with Bukwang is terminated, under substantially the same terms as the primary license with Bukwang, including term, termination and financial provisions, provided that the reason for such termination does not relate to any breach of our sublicense by us or on our behalf. We made no up-front payments to UGARF or Yale University in connection with this agreement. The memorandum of understanding contains no stated termination date.
Boehringer Ingelheim Chemicals, Inc.
On August 8, 2008, we entered into a manufacturing services agreement with Boehringer Ingelheim Chemicals, Inc. (“BICI”) for the manufacture of clinical supplies of the active pharmaceutical ingredient (“API”) of clevudine. In addition to covering the clinical supply of the API, the Manufacturing Services Agreement also contains terms related to any eventual commercial manufacture and supply of the API by BICI. The Manufacturing Services Agreement has an initial term of five years and at our option may be extended for an additional three years, and under certain circumstances, may be terminated by either us or BICI in advance of the expiration of its term. We also entered into a license agreement with BICI pursuant to which BICI granted us a non-exclusive, worldwide, perpetual, royalty-free license, with the right to sublicense to contract manufacturing organizations solely for the purpose of manufacturing the API on behalf of us, to use and practice certain BICI technology related to the manufacture of the API (the “License”). We agreed that in exchange for the License, and in addition to certain cash payments we will pay to BICI, we may, if certain product development milestones are reached for products incorporating the API, source a certain minimum amount of its commercial requirements for the API exclusively from BICI.
Manufacturing and Supply
We do not have our own manufacturing capabilities and we rely on third-party manufacturers for supply of the APIs of our product candidates used in preclinical studies and clinical trials. We do not expect to establish our own manufacturing facilities and we will continue to rely on third-party manufacturers to produce commercial quantities of any product candidates we commercialize. We believe all of the materials required for the manufacture of our current product candidates could be obtained from more than one source. Roche has responsibility for establishing a single source of API for RG7128 for both the Roche territory and our territory. We also have the right to establish ourselves as the secondary source of API supply for RG7128, provided, however, that we may not supply in excess of 20% of the requirements for the global supply.
We may need to procure additional supplies of Racivir to complete our future preclinical studies and clinical trials. Our goal is to conduct future clinical trials with a collaborator that will study combination therapies that include Racivir for HIV patients receiving second-line therapy. If necessary, we plan to enter into a supply agreement after an evaluation of potential suppliers that could manufacture Racivir, including the company that manufactures our current supply of Racivir.
Government Regulation
Regulation by governmental authorities in the United States and other countries is a significant factor in the development, manufacture and marketing of pharmaceutical products and in research and development activities. Government authorities in the United States at the federal, state, and local levels and in foreign countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing, and import and export of pharmaceutical products, biologics, and medical devices. All of our products will require regulatory approval by governmental agencies prior to commercialization. Various federal, state, local, and foreign statutes and regulations also govern, among other things, testing, manufacturing, safety, labeling, marketing, storage, and record-keeping related to such products. The process of obtaining these approvals and subsequent process of maintaining substantial compliance with applicable federal, state, local, and foreign statutes and regulations require the expenditure of extensive time and financial resources. In addition, these statutes, rules, regulations, and policies may change and our products may be subject to new legislation or regulations.
18
Table of Contents
Pharmaceutical Regulation in the United States
In the United States, drugs are subject to rigorous regulation by the FDA. The Federal Food, Drug and Cosmetic Act and other federal and state statutes and regulations govern, among other things, the research, development, testing, safety, effectiveness, manufacture, quality control, storage, record keeping, labeling, promotion, marketing, and distribution of pharmaceutical products. The failure to comply with the applicable regulatory requirements may subject a company to a variety of sanctions such as the FDA’s refusal to approve pending applications, withdrawals of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of operations, injunctions, fines, civil penalties, or criminal prosecution. The FDA also administers certain controls over the export of drugs from the United States. Similar drug regulation exists in other regions such as the European Union (“EU”).
The steps ordinarily required before a new drug product may be marketed in the United States include preclinical laboratory tests, animal tests and formulation studies, the submission and activation of an IND, and adequate and well-controlled clinical trials to establish the safety and efficacy of the drug for each indication for which FDA approval is sought. Any advice granted by FDA to a company during drug development is the best available at that time. FDA may, at any time, change that advice and potentially cause substantial delay to the drug development or New Drug Approval (“NDA”) review timelines. The following is a general overview of the development and approval process for a new drug.
Preclinical Phase. Preclinical tests include laboratory evaluation of biological mechanisms and drug metabolism, product chemistry, formulation, and stability, as well as studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA.
An IND must be prepared and submitted to the FDA to request authorization to begin human testing of the drug. Upon receipt of an FDA IND Safe to Proceed Letter and Institutional Review Board (“IRB”) approval, the sponsor can begin that testing.
Preclinical testing continues through the clinical phase of development and is employed as a guide to help ensure the safe conduct of human subject clinical studies. Throughout the development program, the drug substance (active ingredient), drug product (formulation), and the attendant manufacturing processes are carefully controlled and regulated to ensure consistency.
Clinical Phase. The clinical phase of development involves the activities necessary to demonstrate safety and efficacy of the substance in humans. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study and the parameters to be used in assessing the safety and the efficacy of the drug. Each protocol must be submitted to the FDA, must be reviewed, approved, and conducted under the auspices of an IRB, and each trial includes each patient’s/subject’s informed consent. Sponsors, investigators, other clinical study staff, and IRBs also must satisfy compliance with Good Clinical Practice regulations. Some later trials may include an independent data safety monitoring committee to judge, in an ongoing fashion, the ability of the study to continue; decisions may be reached on the basis of predefined criteria or newly available information.
Clinical trials to support NDAs are typically conducted in three sequential phases, which might be compressed, might overlap, or might be omitted in some circumstances.
| • | Phase 1 Clinical Trials: After an IND becomes effective, Phase 1 human clinical trials can begin. These studies are controlled and closely monitored and initially conducted in a limited population to evaluate, among other things, a drug candidate’s safety, tolerability, and pharmacokinetics. |
| • | Phase 2 Clinical Trials: Phase 2 studies are generally conducted in a limited patient population to identify or further characterize possible adverse events and safety risks, to determine, among other things, the efficacy of the drug candidate for specific targeted indications and to determine the dosing regimen for Phase 3 studies. |
19
Table of Contents
| • | Phase 3 Clinical Trials: These are designed largely on the basis of Phase 2 data and conducted in the target patient population The goal of these studies is to obtain, statistical evidence of efficacy and a safety profile of the investigational new drug. |
In addition to ongoing IND communications with the FDA, a company may engage in meetings with the FDA at critical time points, such as the end of Phase 2, when the Phase 3 clinical program is discussed. Agreements and understandings reached at such meetings are important for ensuring the adequacy of an NDA.
New Drug Application. In order for a drug to be approved for marketing, an NDA is prepared and submitted to the FDA. An NDA is a modular compilation of data, reports, and analyses that includes, among other things, the results of all preclinical and clinical studies, chemistry, manufacturing, and quality information about the drug as well as draft labeling and a risk management plan, if it is required or appropriate. The cost of preparing and submitting an NDA is substantial. In most cases, the submission of an NDA is also subject to substantial application and establishment user fees. The manufacturer and/or sponsor under an approved NDA are also subject to annual product and establishment user fees.
The submission of the application is no guarantee that the FDA will find it complete and accept it for filing. It may refuse to file the application and request additional information rather than accept the application for filing, in which case, the application must be resubmitted with the supplemental information. Once an NDA is accepted, the FDA may refer the application to an advisory committee for review, evaluation, and an approval recommendation. The FDA is not bound by the opinion of the advisory committee. The current target NDA review time is 10 months. Drugs that successfully complete NDA review, and for which an approval letter is received, may be marketed in the United States, subject to all conditions imposed by the FDA and all applicable laws and regulations.
If the FDA determines that the data in the marketing application, the clinical study sites, or manufacturing facilities are not acceptable it will outline the deficiencies in a complete response letter and will often request additional testing or information. The length of time required to satisfy the deficiencies depends on the nature of the deficiencies.
Post-Approval Phase. As a condition of NDA approval, the FDA may require Phase 4 clinical trials to evaluate issues which may have arisen during the NDA review. Manufacturing facilities subject to the NDA are inspected on a routine basis and many changes to manufacturing processes must be submitted to FDA. Once a product is commercially launched, the requirements include FDA oversight of all promotional materials and timely reporting and analysis of safety information to FDA.
As part of the regulatory review process, an NDA may be granted patent term restoration for applicable patents, marketing exclusivity of three or five years duration, and, potentially, pediatric exclusivity. Separately, the product can also be designated an Orphan Drug, granting seven years exclusivity.
Foreign Regulatory Requirements
Outside the United States, our ability to market our products will also be contingent upon receiving marketing authorizations from the appropriate regulatory authorities and compliance with applicable post-approval regulatory requirements. The company currently conducts clinical trials in EU Member States (and other parts of the world) through the Clinical Trial Application (“CTA”) process.
As a general matter, foreign regulatory systems include risks similar to those associated with FDA regulation. In the EU, antiviral products are submitted as a centralized procedure in which approval of a single application to the European Medicines Evaluation Agency results in EU-wide marketing approval.
Even though global regulatory harmonization is a stated goal, regional (for example US vs. EU) regulatory requirements for approving an application may differ depending on issues such as different reviewers, local public health issues, and political situations. Additionally, as in the United States, post-approval regulatory
20
Table of Contents
requirements, such as those regarding product manufacturing, marketing, or distribution, would apply to any product that is approved in the EU.
Hazardous Materials
Our research and development processes involve the controlled use of numerous hazardous materials, chemicals and radioactive materials and produce waste products. We are subject to federal, state and local laws and regulations, and may be subject to foreign laws and regulations, governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products, including certain regulations promulgated by the U.S. Environmental Protection Agency, or EPA. The EPA regulations to which we are subject require that we register with the EPA as a generator of hazardous waste. Although we have safety procedures for handling and disposing of these materials, we cannot assure investors that accidental contamination or injury from these materials will not occur. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposures to blood-borne pathogens and the handling, transporting and disposing of biohazardous or radioactive materials. We do not expect the cost of complying with these laws and regulations to be material.
Competition
We face a broad range of current and potential competitors, from established global pharmaceutical companies with significant resources to development-stage companies. In addition, we face competition from academic and research institutions and government agencies for the discovery, development and commercialization of novel therapeutics to treat HCV and HIV. Many of our competitors, either alone or with their collaborative partners, have significantly greater financial, product development, technical, manufacturing, sales, and marketing resources than we do. In addition, many of our direct competitors are large pharmaceutical companies with internal research and development departments that have significantly greater experience in testing pharmaceutical products, obtaining FDA and other regulatory approvals of products, and achieving widespread market acceptance for those products.
We believe that a significant number of drugs are currently under development and will become available in the future for the treatment of HCV and HIV. We anticipate that we will face intense and increasing competition as new products enter the marketplace and advanced technologies become available. Our competitors’ products may be safer, more effective, or more effectively marketed and sold than any product we may commercialize. Competitive products may render one or more of our product candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our product candidates. It is also possible that the development of a cure, effective vaccine, or new treatment methods for HCV and HIV could render one or more of our product candidates non-competitive, obsolete, or reduce the demand for our product candidates.
We believe that our ability to compete depends, in part, upon our ability to develop products, complete the clinical trials and regulatory approval processes, and effectively market any products we develop. Further, we need to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary product candidates or processes, and secure sufficient capital resources for the substantial time period between the discovery of lead compounds and their commercial sales, if any.
HCV Therapeutics Competition
In the United States, the current standard of care for the treatment of HCV is a combination of interferon alfa and a nucleoside analog named ribavirin. Interferon alfa is approved in several chemically modified forms and is marketed by Roche, Merck (formerly Schering-Plough), and Three Rivers Pharmaceuticals. Roche, Merck, Three Rivers Pharmaceuticals and several generic manufacturers market ribavirin. We are aware that Roche and other companies are also developing new drugs for the treatment of HCV. For example, Novartis/Human Genome Sciences, Vertex, and Merck have advanced their drug candidates into Phase 3 clinical trials. The following table presents information about approved drugs and drug candidates for the treatment of HCV.
21
Table of Contents
FDA-Approved HCV Therapeutics or HCV Therapeutics in Development
| Brand Name |
Generic Name or |
Drug Class |
Phase of |
Company | ||||
| Pegasys® plus Copegus® | Peginterferon alfa-2a + ribavirin |
Interferon | Approved | Roche | ||||
| Peg-Intron plus Rebetol |
Peginterferon alfa-2b + ribavirin |
Interferon | Approved | Merck | ||||
| Infergen | Interferon alfacon-1 |
Interferon | Approved | Three Rivers Pharmaceuticals | ||||
| Zalbin | Albuferon/Albumin interferon alfa-2b | Interferon | Phase 3 /NDA Filed | Human Genome Sciences/ Novartis | ||||
| RG7128 | Nucleoside Polymerase Inhibitor | Phase 2b | Pharmasset/Roche | |||||
| PSI-7851 | Nucleotide Polymerase Inhibitor | Phase 1 | Pharmasset | |||||
| IDX184 | Nucleotide Polymerase Inhibitor | Phase 2a | Idenix | |||||
| Filibuvir | Non-nucleoside Polymerase Inhibitor | Phase 2 | Pfizer | |||||
| GS-9190 | Non-nucleoside Polymerase Inhibitor | Phase 2 | Gilead | |||||
| ABT-333 | Non-nucleoside Polymerase Inhibitor | Phase 2 | Abbott | |||||
| ANDS-598 | Non-nucleoside Polymerase Inhibitor | Phase 2 | Anadys | |||||
| BMS-790052 | NS5a Inhibitor | Phase 2 | Bristol-Myers Squibb | |||||
| Telaprevir | Protease Inhibitor | Phase 3 | Vertex/Johnson & Johnson | |||||
| Boceprevir | Protease Inhibitor | Phase 3 | Merck | |||||
| TMC435350 | Protease Inhibitor | Phase 2 | Medivir/Tibotec | |||||
| RG7227 | Protease Inhibitor | Phase 2 | InterMune/Roche | |||||
| MK-7009 | Protease Inhibitor | Phase 2b | Merck | |||||
| SCH-900518 | Protease Inhibitor | Phase 2 | Merck | |||||
| BMS-650032 | Protease Inhibitor | Phase 2a | Bristol-Myers Squibb | |||||
| BI 201335 | Protease Inhibitor | Phase 2b | Boehringer Ingelheim | |||||
| BI 207127 | Non-nucleoside Polymerase Inhibitor | Phase 2b | Boehringer Ingelheim | |||||
| ABT-450 | Protease Inhibitor | Phase 1 | Enanta/Abbott |
22
Table of Contents
HIV Therapeutics Competition
HAART, the standard of care for the treatment of HIV infection, generally includes two NRTIs combined with a third drug from another class (either an NNRTI or a protease inhibitor). Racivir is an NRTI and, therefore, our primary competitors are those companies that develop and market other NRTIs.
To provide additional convenience to HIV patients, companies have developed combination therapies that combine two or more NRTIs into a single tablet or capsule. These fixed-dose combination therapies have become leaders in the HIV marketplace.
Several of the FDA-approved individual NRTIs and combination products face patent expiration in the next several years. As a result, generic versions of these drugs may become available. We expect to face competition from these generic drugs, including price-based competition.
Intellectual Property
Our policy is to pursue patents and to otherwise endeavor to defend our technologies, inventions, and improvements to inventions that are commercially important to the development of our business. We seek U.S. and international patent protection on the novel compounds, product candidates, and therapeutic processes we discover or improve, as well as the chemical synthesis and manufacturing of such compounds and product candidates.
Clevudine. We have an exclusive sublicense in North, Central and South America, Europe, the Caribbean, and Israel from Bukwang to issued U.S. and corresponding foreign patents covering the composition of matter for Clevudine, methods of using clevudine to treat HBV and synthetic processes for clevudine, which expire between 2014 and 2022. Bukwang is the primary licensee from Yale University and the University of Georgia Research Foundation, Inc., who are the primary licensors that jointly own the intellectual property for this technology. Our sublicense with Bukwang also encompasses nonexclusive rights in the aforementioned territories to pending patents in the U.S. and certain foreign countries covering the use of clevudine in combination with other therapeutics for the treatment of HBV. Any patent issuing from these patent applications would expire no earlier than 2023. On April 20, 2009, we voluntarily terminated our Phase 3 registration studies of clevudine for the treatment of hepatitis B virus.
Racivir. On December 8, 1998, Emory University granted us an exclusive, worldwide license pursuant to the Racivir License Agreement to issued U.S. patents covering the composition of matter for Racivir, methods of synthesizing Racivir and methods of using Racivir to treat HIV and HBV, which expire between 2010 and 2020. This license also encompasses rights to corresponding patents and pending patent applications in Europe, Japan, South Africa, and other foreign countries.
In the Emory/Gilead License Agreement, Emory previously had granted a right of first refusal to Gilead that is applicable to any license or assignment relating to enriched FTC (which includes Racivir). The terms of this right of first refusal contains an exception permitting Emory to license or assign its rights in respect of enriched FTC to a permitted transferee, which includes any of the inventors (which included two of our founders) or to any corporate entity formed by or on behalf of the inventors for purposes of clinically developing enriched FTC so long as the licensee agrees in writing to be bound by the terms of Gilead’s right of first refusal to the same extent as Emory. Our license to Racivir was granted to us by Emory pursuant to this exception and therefore we are bound by the terms of Gilead’s right of first refusal to the same extent as Emory. The terms of this right of first refusal as set forth in the Emory/Gilead License Agreement require that, prior to the entry into any license or assignment agreement with a third party relating to any of Emory’s rights in respect of enriched FTC, Emory shall notify Gilead of the terms of the proposed agreement and provide a copy of the proposed agreement to Gilead together with all data and information in Emory’s possession relating to enriched FTC and its use as a therapeutic agent. Gilead has 30 days to accept or decline the offer. Although Emory considers us to be a permitted transferee under the Emory/Gilead License Agreement, Emory has subsequently taken the position that its grant of commercialization rights (i.e., the rights to offer for sale and sell Racivir) to us exceeded the rights
23
Table of Contents
that were permitted to be granted to a permitted transferee under its agreement with Gilead. While we believe that Gilead is aware of the Racivir license agreement through both our and Emory’s communications with Gilead, Gilead has not contacted us regarding its interpretation of the terms of the Racivir License Agreement.
In March 2004, we entered into a supplemental agreement with Emory in which we and Emory agreed that, prior to any commercialization of enriched FTC by us, or by any licensee or assignee of our rights under the Racivir License Agreement, we and Emory would adhere strictly to the terms of the right of first refusal granted to Gilead in the Emory/Gilead License Agreement and offer to Gilead the same terms and conditions under which we, our licensee or our assignee, propose to commercialize enriched FTC. The supplemental agreement also outlines a procedure by which Emory and we would jointly offer the terms of a proposed license and commercialization agreement between us and a third party to Gilead after Emory has the opportunity to approve them. Therefore, before we could enter into a commercialization agreement for Racivir with a third party or commercialize Racivir on our own, we would be required to offer Gilead the opportunity to be our commercialization partner on the same terms on which we intend, or our prospective partner intends, to commercialize Racivir. It is uncertain whether a third party would be willing to negotiate the terms of a commercialization agreement with us knowing that Gilead can take their place as licensee by accepting the negotiated terms and exercising its right of first refusal.
These uncertainties related to our commercialization rights may result in us being prevented from obtaining the expected economic benefits from developing Racivir. In addition, we could become involved in litigation or arbitration related to our commercialization rights to Racivir in the future.
RG7128. We own an issued U.S. patent (U.S. patent number 7,429,572) and pending U.S. and foreign patent applications directed to the PSI-6130 chemical compound, prodrugs (including RG7128), pharmaceutical formulations, therapeutic combinations and use to treat HCV infections. This patent was issued on September 30, 2008 and will expire on April 3, 2025. Any additional patents issuing from the pending applications would expire no earlier than 2024. We own an issued U.S. patent (U.S. patent number 7,601,820) and pending U.S. and foreign patent applications directed to the synthesis of PSI-6130 chemical compound, including synthetic intermediates thereof. This patent was issued on October 13, 2009 and will expire on November 13, 2025. Any additional patents issuing from the pending applications would expire no later than 2025.
Except where precise dates are given, the patent expiration dates stated above do not take into account any patent term adjustments that may accrue due to procedural delays by the United States Patent and Trademark Office. The patent expiration dates stated above do not take into account any patent term extensions that may accrue due to regulatory delays.
Attempts to obtain patent protection both in the United States and abroad can be expensive, take years to complete, and may not be successful. In addition, issued patents are subject to attack, may not be enforceable, and may otherwise fail to protect our business. Moreover, the trade secret laws and other sources of intellectual property protection may also be insufficient to protect our product candidates. For more information on these and other risks related to intellectual property rights, see “Risk Factors—Risks Related to Our Intellectual Property.”
In addition to protection offered by patent term and patent term restoration, regulatory authorities such as FDA and EMEA grant marketing exclusivity or data protection for new product approvals and certain supplemental applications. In addition, the US and EU may grant pediatric exclusivity which is dependent on the acceptance of a pediatric clinical program and conduct of trials in the pediatric population. Generic companies may litigate to modify or invalidate exclusivity and while the litigation may not be successful, should it occur, it would consume significant company resources.
Employees
As of September 30, 2009, we had 65 employees, 43 of whom performed research and development functions.
24
Table of Contents
Available Information
All periodic and current reports, registration statements, and other filings that we are required to file with the SEC, including our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) of the Securities Exchange Act of 1934 (Exchange Act), are available free of charge from the SEC website (www.sec.gov) or public reference room at 100 F Street N.E., Washington, DC 20549 (1-800-SEC-0330) or through our website at www.pharmasset.com. Such documents are available as soon as reasonably practicable after electronic filing of the material with the SEC. Copies of these reports (excluding exhibits) may also be obtained free of charge, upon written request to: Investor Relations, Pharmasset, Inc., 303-A College Road East, Princeton, NJ 08540. The website addresses included in this report are for identification purposes. The information contained therein or connected thereto are not intended to be incorporated into this report.
25
Table of Contents
| ITEM 1A. | RISK FACTORS |
Risks Related to Our Business
Risks Related to Drug Discovery, Development, and Commercialization
We are subject to significant regulatory requirements which could delay, prevent, or limit our ability to market our product candidates, including RG7128, PSI-7851, PSI-938, PSI-879, and Racivir.
Our research and development activities, preclinical studies, clinical trials, manufacturing, and anticipated marketing of our product candidates are subject to extensive regulation by a wide range of governmental authorities in the United States, including the FDA, and by comparable authorities in European and other countries. To date, none of our product candidates have been approved for sale by the FDA or any foreign regulatory authority. Neither we nor our collaborators, independently or collectively, will be able to commercialize any of our product candidates until we or they obtain FDA approval in the United States or approval by comparable regulatory agencies in Europe or other countries. To satisfy FDA or foreign regulatory approval standards for the commercial sale of our product candidates, we must, among other requirements, demonstrate in adequate and well-controlled clinical trials that our product candidates are safe and effective. The clinical trials of a drug candidate can be suspended at any time by us, a regulatory agency, institutional review board (“IRB”), an independent drug safety monitoring board, or others if there is a concern that subjects participating in the clinical trials are being exposed to unacceptable health risks or for other reasons. Adverse side effects of a product candidate on subjects in a clinical trial could result in the FDA or foreign regulatory authorities refusing to approve a particular drug candidate for any and all indications of use.
We or our collaborators have conducted initial preclinical studies and early-stage clinical trials of RG7128, PSI-7851, and Racivir and preclinical studies of PSI-938 and PSI-879. Many of these trials were not primarily designed to demonstrate the efficacy of these product candidates but, rather, to collect data on safety and assist in determining the appropriate dose. Even if our product candidates achieve positive results in preclinical and clinical trials, similar results may not be observed in subsequent trials and results may not prove to be statistically significant or demonstrate safety and efficacy to the satisfaction of the FDA or other regulatory agencies.
The FDA also regulates the manufacturing facilities of our collaborators and third-party manufacturers. Prior to approval, the FDA inspects manufacturing facilities to ensure compliance with current good manufacturing practice (“cGMP”), including quality control and record-keeping measures. Post-approval, the FDA and certain state agencies subject these facilities to unannounced inspections to ensure continued compliance with cGMP. Failure to satisfy the pre-approval inspection or subsequent discovery of problems with a product, or a manufacturing or laboratory facility used by us, our collaborators, or third-party manufacturers may result in an inability to receive approval, recall of products, delay in approval, or restrictions on the product or on the manufacturing post-approval, including a withdrawal of the drug from the market or suspension of manufacturing. Such inspections of third party manufacturers may adversely affect us whether or not our products are the cause of the inspection because other products or a general cGMP inspection was the initiating event for the inspection. Our collaborators and third-party manufacturers rely on a variety of suppliers of raw materials, equipment, and other supplies to comply with cGMP and other specifications and standards. The failure of a supplier to our collaborators and third-party manufacturers to meet such requirements could have a material adverse effect on our research, development, and future commercial activities. Foreign regulatory authorities have similar manufacturing compliance requirements which may result in similar outcomes to those noted above.
The FDA and foreign regulatory authorities also regulate the conduct of clinical trials, ensuring compliance with current good clinical practice regulations and guidance (“cGCP”). Clinical investigator sites contracted by us or any of our collaborators may be inspected, unannounced, by any regulatory authority at any time. Failure of the clinical site to successfully complete the regulatory inspection may adversely affect the company whether or not our trials are the cause of the inspection. This occurs because clinical investigators routinely conduct trials for other companies and inspection of those trials may uncover more systemic problems at the site which were not known to us.
26
Table of Contents
We also will be required to obtain foreign regulatory approval for the sale of our products outside of the United States. Foreign regulatory approval processes include all of the risks associated with the FDA approval processes described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by one regulatory agency, including the FDA, does not assure approval by other regulatory authorities. Many foreign regulatory authorities have different approval standards from each other and from those required by the FDA and may impose additional testing requirements for our product candidates. Furthermore, international ethical review boards may cause our clinical trials to be delayed pending their review of safety data, clinical procedures, and comments provided by foreign regulatory authorities. We have had limited interaction with foreign regulatory authorities. We may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to submit for foreign regulatory approvals, they may not accept the application and we may not receive necessary approvals to commercialize our existing and future product candidates in any market.
The regulatory approval process is expensive and, while the time required to complete clinical trials and for FDA and foreign regulatory approval processes is uncertain, it typically takes many years. Our analysis of data obtained from our preclinical studies and clinical trials is subject to confirmation and interpretation by different regulatory authorities who may have different views on the design, scope, or results of our clinical trials, which could delay, limit, or prevent regulatory approval. At any time, changes in regulatory policy during the development period of any of our product candidates, changes in, or the enactment of, additional regulations or statutes, or changes in regulatory review practices for a submitted product application may result in failure of the agency to accept our application for review, which could cause a delay in obtaining approval or result in the rejection of an application for regulatory approval. We could also encounter unanticipated delays or increased costs due to government regulation from future legislation or administrative action or changes in FDA or foreign regulatory policies during the period of product development, clinical trials, or regulatory review. The company seeks to ensure a productive dialogue with regulatory authorities throughout product development, application review and thereafter. We may reach the conclusion to not follow all of the regulatory authorities’ advice for the content of a marketing application and instead justify its position with supporting data and expert analyses contained in the original application. The regulatory authority(ies) may agree or disagree with this approach, which may affect acceptance of the application, the length of agency review, or other action on the application. The regulatory review process is subject to political, technical, economic, and other developments. This results in dynamic and unpredictable risks in drug development, regulatory compliance, and commercialization of pharmaceuticals.
As a result of these factors, our product candidates could require a significantly longer time to gain regulatory approval than expected or may never gain approval. We cannot assure you that, even after expending substantial time and resources, we will obtain regulatory approval for any of our product candidates. A delay or denial of regulatory approval could delay or prevent our ability to generate product revenues and to achieve profitability. If regulatory approval is obtained, our marketing of any product will be limited to its indicated uses, which will limit the size of the market for a product and affect our potential product revenues.
Our product candidates must undergo the conduct of rigorous clinical trials, the results of which are uncertain and could substantially delay or prevent us from bringing product candidates to market.
Before we can obtain regulatory approval for a product candidate, among other things, we must undertake extensive clinical trials in humans to demonstrate safety and efficacy to the satisfaction of the FDA or other regulatory agencies. Clinical trials of new product candidates sufficient to obtain regulatory marketing approval are complex and expensive and take years to complete. The results of earlier-stage testing may not be predictive of results in future trials. For example, estimates of detectable circulating virus reduction and activity against HCV (or HIV) obtained from preclinical studies and clinical trials are not necessarily indicative of results that could be achieved in subsequent clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier preclinical studies and clinical trials. We cannot assure you that we or our collaborators will successfully
27
Table of Contents
complete the planned clinical trials. Our collaborators or we may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our product candidates, including the following events:
| • | our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical studies, restrict investigation to limited patient populations, or to abandon development programs; |
| • | trial results may not meet the level of statistical significance required by the FDA or other regulatory agencies; |
| • | we, IRBs, independent safety monitors, or regulators may suspend or terminate clinical trials if the participating subjects are believed to be exposed to unacceptable health risks; and |
| • | the effects of our product candidates on subjects may not be the desired effects or may include undesirable side effects or other characteristics that may delay or preclude regulatory approval or limit their commercial use. |
We have limited experience in conducting clinical trials which could impair our timing or ability to obtain regulatory approval for our product candidates.
We have limited experience in conducting and managing the clinical trials necessary to obtain FDA approval or approval by other regulatory authorities. Our past clinical experience has been limited to a small number of product candidates in a limited number of therapeutic areas. By contrast, larger pharmaceutical companies often have substantial staffs experienced in conducting clinical trials with multiple product candidates across multiple indications. As a result, we may be at a competitive disadvantage that could, for example result in delays in obtaining regulatory approvals, if at all, for our product candidates for which we conduct or manage the clinical trial process.
Delays in conducting clinical trials could result in increased costs to us and delay our ability to obtain regulatory approval and commercialize our product candidates.
Significant delays in conducting clinical trials and related drug development programs could materially affect our product development costs and delay regulatory approval of our product candidates. We do not know whether planned clinical trials will begin on time, will need to be redesigned, or will be completed on schedule, if at all. A clinical trial can be delayed for a variety of reasons, including:
| • | delays or failures in obtaining regulatory authorization to commence a trial because of safety concerns of regulators relating to our product candidates or similar product candidates, competitive or comparator products or supportive care products or failure to follow regulatory guidelines; |
| • | delays or failures in obtaining clinical materials and manufacturing sufficient quantities of the product candidate for use in a trial; |
| • | delays or failures in reaching agreement on acceptable terms with prospective study sites or other contract research organizations; |
| • | delays or failures in obtaining approval of our clinical trial protocol from an IRB to conduct a clinical trial at a prospective study site; |
| • | delays in recruiting or enrolling subjects to participate in a clinical trial; |
| • | failure of a clinical trial or clinical investigators to be in compliance with Good Clinical Practices; |
| • | unforeseen safety issues; |
| • | inability to monitor subjects adequately during or after treatment; |
| • | difficulty monitoring multiple study sites; |
28
Table of Contents
| • | failure of our third-party clinical trial managers to satisfy their contractual duties, comply with regulations, or meet expected deadlines; and |
| • | determination by regulators that the clinical design of a trial is not adequate. |
Failure to recruit, enroll, and retain subjects for clinical trials may cause the development of our product candidates to be delayed or development costs to increase substantially.
We have experienced, and expect to experience in the future, delays in subject enrollment in our clinical trials for a variety of reasons. The timely completion of clinical trials in accordance with their protocol depends, among other things, on our ability to enroll a sufficient number of subjects who remain in the study until its conclusion. The enrollment of subjects depends on many factors, including:
| • | the subject eligibility criteria defined in the protocol; |
| • | the size of the subject population required for analysis of the trial’s primary endpoints; |
| • | the proximity of subjects to study sites; |
| • | the design of the trial; |
| • | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| • | our ability to obtain and maintain subject consents; |
| • | the risk that subjects enrolled in clinical trials will drop out of the trials before completion; and |
| • | competition for subjects by clinical trial programs for other treatments. |
Our clinical trials compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition reduces the number and types of subjects available to us, because some subjects who might have opted to enroll in our trials opt to enroll in a trial being conducted by one of our competitors. Since the number of qualified clinical investigators is limited, we conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which reduces the number of subjects who are available for our clinical trials in such clinical trial site. Delays in patient enrollment in the future as a result of these and other factors may result in increased costs or may affect the timing or outcome of our clinical trials, which could prevent us from completing these trials and adversely affect our ability to advance the development of our product candidates.
Our product candidates may demonstrate or be associated with undesirable side effects when used alone or in combination with other products that prevent their regulatory approval or limit their use if approved.
We must adequately define the safety profile of our product candidates to obtain regulatory approval. Although in clinical trials completed to date RG7128, PSI-7851, and Racivir were generally well tolerated, these trials involved a relatively small number of subjects and we may observe significant adverse events for these product candidates in the future. Roche is currently conducting larger clinical studies of RG7128 and we are planning to conduct additional clinical studies of PSI-7851. It is possible that any side effects associated with our product candidates may outweigh the benefits of our product candidates and prevent regulatory approval or demonstrate a risk/reward profile which would limit their market acceptance if they are approved. Recent developments in the pharmaceutical industry have prompted heightened government focus on safety reporting during both pre- and post-approval time periods and pharmacovigilance. Global health authorities may impose regulatory requirements to monitor safety that may burden our ability to commercialize our drug products.
Even if we receive regulatory approval to market our product candidates, the market may not be receptive to our product candidates, which would negatively affect our ability to achieve profitability.
29
Table of Contents
If approved for marketing, the commercial success of our product candidates will depend upon their acceptance by physicians and the medical community, patients, and private, government, and third-party payers as clinically safe and effective, cost-effective therapeutics. The degree of market acceptance of any of our approved products will depend upon a number of factors, including:
| • | the indication for which the product is approved and its approved labeling; |
| • | the acceptance in the medical community of the safety and efficacy of the product; |
| • | the incidence, prevalence, and severity of adverse side effects; |
| • | the presence of other competing approved therapies; |
| • | the potential advantages of the product over existing and future treatment methods; |
| • | the relative convenience and ease of administration of the product; |
| • | the strength of our sales, marketing, and distribution support; |
| • | the price and cost-effectiveness of the product; and |
| • | sufficient third-party reimbursement. |
We are aware that a significant number of product candidates are currently under development and may become available in the future for the treatment of HCV and HIV, and may be approved prior to any of our product candidates coming to market. Even if our products achieve market acceptance, we may not be able to maintain that market acceptance over time if new therapeutics are introduced that are more favorably received than our products or that render our products obsolete, or if unacceptable levels of drug resistance or significant adverse events occur. If our products do not achieve and maintain market acceptance, we will not be able to generate sufficient revenue from product sales to attain profitability.
Even if we obtain regulatory approvals, our marketed drugs will be subject to ongoing regulatory review. If we fail to comply with continuing U.S. and foreign regulations, we could lose our marketing approvals and our business would be seriously harmed.
Following initial regulatory approval of any drugs we or our collaborators may develop, we and our collaborators will be subject to continuing regulatory review by the FDA or other regulatory authorities, including the review of adverse drug events and clinical results that are reported after product candidates become commercially available. This would include results from any post-marketing follow-up studies or other reporting required as a condition to approval. The manufacture, distribution, sale, labeling, packaging, storage, advertising, promotion, reporting, and record-keeping related to the product will also be subject to extensive ongoing and regulatory requirements, which are subject to change. In addition, incidents of adverse drug reactions, unintended side effects, or misuse relating to our products could result in additional regulatory controls or restrictions or even lead to withdrawal of a product from the market.
Furthermore, our third-party manufacturers and the manufacturing facilities that they use to make our product candidates are regulated by the FDA and other governmental authorities (including foreign authorities). Quality control and manufacturing procedures must continue to conform to cGMP after approval. Pharmaceutical manufacturers and their subcontractors are required to register their facilities and/or products manufactured at time of submission of the marketing application and annually with the FDA and certain state and foreign agencies and are subject to periodic unannounced inspections by the FDA, state, and other foreign authorities. Any subsequent discovery of problems with a product, or a manufacturing or laboratory facility used by us or our collaborators, may result in restrictions on the product or on the manufacturing or laboratory facility, including marketed product recall, suspension of manufacturing, product seizure, or a voluntary withdrawal of the drug from the market. Any change to an approved product, including the way it is manufactured or promoted, often requires FDA or other regulatory authority notification or approval before the product, as modified, can be
30
Table of Contents
marketed. We and our third-party manufacturers will also be subject to ongoing FDA or other regulatory authority requirements for submission of safety and other post-market information. If we, our collaborators, or our third-party manufacturers fail to comply with applicable continuing regulatory requirements, our business could be seriously harmed because a regulatory agency may:
| • | issue warning letters which require corrective action; |
| • | suspend or withdraw our regulatory approval for approved products; |
| • | seize or detain products or recommend a product recall; |
| • | refuse to approve pending applications or supplements to approved applications submitted by us; |
| • | suspend any of our ongoing clinical trials; |
| • | impose restrictions on our operations, including costly new manufacturing requirements; |
| • | close the facilities of our contract manufacturers; or |
| • | impose civil or criminal penalties including fines, imprisonment, and disgorgement of profits. |
The FDA’s and foreign regulatory agencies’ policies are subject to change and additional federal, state, local, or foreign governmental regulations may be enacted that could affect our ability to maintain compliance. We cannot predict the likelihood, nature, or extent of adverse governmental regulation that may arise from future legislation or administrative action, either in the United States or abroad.
Our research and development efforts may not result in additional product candidates being discovered, which could limit our ability to generate revenues in the future.
Our research and development efforts may not lead to the discovery of any additional product candidates that would be suitable for further preclinical or clinical development. The discovery of additional product candidates requires significant research and preclinical studies as well as a substantial commitment of resources. Many lead compounds that appear promising in preclinical studies fail to progress to become product candidates in clinical trials. There is a great deal of uncertainty inherent in our research and development efforts and, as a consequence, in our ability to fill our drug development pipeline with promising additional product candidates.
We have no sales, marketing, or distribution experience. We will be required to invest significant amounts of financial and management resources in developing these resources.
If PSI-7851, PSI-938, and PSI-879 receive marketing approval in the United States, we intend to promote and commercialize certain of these products, in certain cases without a partner. To develop internal sales, distribution, and marketing capabilities, we will have to invest significant amounts of financial and management resources. As a result, we could face a number of risks, including:
| • | we may not be able to attract and build a significant marketing or sales force; |
| • | the cost of establishing, training, and providing compliance oversight for a marketing or sales force may not be justifiable in light of the revenues generated by any particular product; and |
| • | our direct sales and marketing efforts may not be successful. |
We and our collaborators will be subject to stringent federal, state, and foreign regulation of sales and marketing of any approved product candidate and a failure to comply with these regulations could result in substantial penalties.
The marketing and advertising of our drug products by our collaborators or us will be regulated by the FDA, certain state agencies, or foreign regulatory authorities. Violations of these laws and regulations, including promotion of our products for unapproved uses or failing to adequately disclose risk information, are punishable
31
Table of Contents
by criminal and civil sanctions and may result in the issuance of enforcement letters, corrective advertising or other enforcement action by the FDA, Department of Justice, state agencies, or foreign regulatory or other legal authorities that could jeopardize our ability to market the product.
In addition to FDA, state, and foreign regulations, the marketing of drug products by us or our collaborators will be regulated by federal, state, or foreign laws pertaining to health care “fraud and abuse,” such as the federal anti-kickback law prohibiting bribes, kickbacks, or other remuneration for the order or recommendation of items or services reimbursed by federal health care programs. Many states have laws applicable to items or services reimbursed by commercial insurers. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, imprisonment and exclusion from participation in federal and state health care programs, including Medicare, Medicaid, and Veterans Affairs healthcare programs. Because of the far-reaching nature of these laws, we will need regularly to evaluate and, as appropriate, potentially revise our practices to ensure compliance with these laws. Health care fraud and abuse regulations are complex, and even minor irregularities can potentially give rise to claims that a statute or prohibition has been violated. Any violations of these laws, or any action against us for violations of these laws, even if we successfully defend against it, could have a material adverse effect on our business, financial condition, and results of operations.
We could also become subject to false claims litigation under federal statutes, which can lead to civil money penalties, restitution, criminal fines, and imprisonment, and exclusion from participation in Medicare, Medicaid, and other federal and state health care programs. These false claims statutes include the False Claims Act, which allows any person to bring a suit on behalf of the federal government alleging submission of false or fraudulent claims, or causing to present such false or fraudulent claims, under federal programs or contracts claims or other violations of the statute and to share in any amounts paid by the entity to the government in fines or settlement. These suits against pharmaceutical companies have increased significantly in volume and breadth in recent years. Some of these suits against pharmaceutical companies have been brought on allegations that certain sales practices amount to the promotion of drug products for unapproved uses. This new growth in litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay fines or restitution, or be excluded from the Medicare, Medicaid, Veterans Affairs, and other federal and state healthcare programs as a result of an investigation arising out of such action. We may become subject to such litigation, the defense of which can be expensive, time consuming, and distracting. If we are not successful in defending against such actions, those actions may have a material adverse effect on our business, financial condition, and results of operations.
Risks Related to Our Financial Performance and Business Operations
We have incurred net losses since our inception and our future profitability is uncertain and we anticipate that we will incur significant continued net losses for the next several years.
We are a clinical-stage pharmaceutical company with a limited operating history upon which an investor can evaluate our operations and future prospects. We have incurred net losses in each year since our inception in 1998. For the years ended September 30, 2009, 2008, and 2007, we had net losses of $55.6 million, $54.7 million, and $5.1 million, respectively. As of September 30, 2009, we had an accumulated deficit of $167.4 million. We do not expect to generate significant sales revenue from our product candidates for at least the next several years and we expect to continue to incur significant operating losses in future periods. We expect to incur substantial costs to further our drug discovery and development programs and that our rate of spending will accelerate as a result of the increased costs and expenses associated with preclinical and clinical development of PSI-7851, PSI-938, PSI-879, Racivir, and other future product candidates. In addition, as we expand our operations, we will need to continue to improve our facilities and hire additional personnel. As a result, we expect that our annual operating losses will increase significantly over the next several years.
Our revenue and profit potential is unproven, and our limited operating history and the many risks inherent in drug development make our future operating results difficult to predict. To attain profitability, we and our collaborators will need to successfully develop products and effectively market and sell them. We have never
32
Table of Contents
generated revenue from the sale of products and there is no guarantee that we will be able to do so in the future. If our product candidates fail to show positive results in ongoing preclinical studies and clinical trials, if we or our collaborators do not receive regulatory approval, or if our product candidates do not achieve market acceptance even if approved, we may never become profitable. If we fail to become profitable, or if we are unable to continue to fund our continuing losses, we may be unable to continue our clinical development programs.
We will require substantial funds in the future and we may be unable to raise capital when needed, which could force us to delay, reduce, or eliminate some of our drug discovery, product development, and commercialization activities.
Developing product candidates, conducting clinical trials, and commercializing products is expensive and we will need to raise substantial additional funds to achieve our strategic objectives. Although we believe our existing cash resources as of September 30, 2009, together with anticipated payments under our existing collaboration agreement, will be sufficient to fund our projected cash requirements for the next 12 months, we will require significant additional financing in the future to fund our operations. Such financing may not be available on acceptable terms, if at all. Our future capital requirements will depend on many factors, including:
| • | the progress and costs of our preclinical studies, clinical trials, and other research and development activities; |
| • | the costs and timing of obtaining regulatory approval of our product candidates; |
| • | the scope, prioritization, and number of our clinical trials and other research and development programs; |
| • | the costs of the development and expansion of our operational infrastructure; |
| • | the ability of our collaborator(s) to achieve development milestones, marketing approval, and other events or developments under our collaboration agreement(s); |
| • | the amount of revenues we receive under our collaboration agreement(s); |
| • | the costs of filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; |
| • | the costs and timing of securing manufacturing arrangements for clinical or commercial production; |
| • | the costs of establishing sales and marketing capabilities or contracting with third parties to provide these capabilities for us; |
| • | the costs of acquiring or undertaking development and commercialization efforts for any future product candidates; |
| • | the magnitude of our general and administrative expenses; and |
| • | any costs that we may incur under current and future licensing arrangements relating to our product candidates. |
Our ability to raise additional funds will depend on financial, economic, and market conditions and other factors, many of which are beyond our control. Additional financing may not be available when we need it or, if available, may not be on terms that are favorable to us. If we are unable to obtain adequate funding on a timely basis, we may be required to delay, reduce the scope of, or eliminate one or more of our drug discovery or development programs.
33
Table of Contents
Raising additional capital may dilute our stockholders’ equity, limit our flexibility, or require us to relinquish rights.
We may need to raise additional capital to fund our operations through public or private equity offerings or debt financings. To the extent that we raise additional capital by issuing equity or equity-linked securities, our stockholders’ ownership will be diluted. Any debt financing we enter into may include covenants that limit our flexibility in conducting our business. We also could be required to seek funds through arrangements with collaborators or others, which might require us to relinquish valuable rights to our intellectual property or product candidates that we would have otherwise retained.
Our success depends in part on our ability to retain and recruit key personnel, and if we fail to do so, it may be more difficult for us to successfully develop our product candidates or achieve our business objectives.
Our success depends in part on our ability to attract, retain and motivate highly qualified management, clinical, and scientific personnel. We are highly dependent on our senior management and scientific staff, particularly P. Schaefer Price, our Chief Executive Officer, Kurt Leutzinger, our Chief Financial Officer, and M. Michelle Berrey, M.D., M.P.H., our Chief Medical Officer. We do not maintain key person insurance for our senior management or scientific staff. The loss of the services of any of our senior management or key members of our scientific staff may significantly delay or prevent the successful completion of our preclinical studies and clinical trials or the commercialization of our product candidates. To date, we are not aware that any member of our senior management or scientific staff plans to leave the company.
The employment of each of our employees with us is “at will” and each employee can terminate his or her employment with us at any time. We currently have an employment agreement in place with P. Schaefer Price.
Our success will also depend on our ability to hire and retain additional qualified scientific and management personnel. Competition for qualified individuals in the pharmaceutical field is intense, and we face competition from numerous pharmaceutical and biotechnology companies, universities and other research institutions. We may be unable to attract and retain qualified individuals on acceptable terms given the competition for such personnel. Furthermore, there is a possibility that a qualified candidate we are recruiting might opt to accept a position with one of our competitors instead of with us because our competitor may have products that are already on the market and generating revenue. If we are unsuccessful in our recruiting efforts, we may be unable to execute our strategy.
The requirements of being a public company may strain our administrative and operational infrastructure and increase our operating costs.
The obligations of being a public company require significant additional expenditures and place additional demands on our management, administrative, operational, internal audit, and accounting resources as we comply with the reporting requirements of a public company. If we are unable to continue to update our procedures and adapt our management, administrative, operational, and accounting functions in such a manner so as to meet such obligations in a timely and effective manner, our ability to comply with the rules that apply to public companies could be impaired. In meeting our obligations, we may need to upgrade our systems, implement additional financial and management controls, reporting systems and procedures, implement an internal audit function, and hire additional accounting, audit, and financial staff with appropriate public company experience and technical accounting knowledge, which will increase our general and administrative expenses and capital expenditures. The rules and any related regulations that may be proposed in the future that are applicable to public companies may make it more difficult and more costly for us to obtain certain types of insurance, including directors’ and officers’ liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher premiums to obtain the same or similar coverage. We cannot estimate the amount or timing of the additional costs we may incur as a result of the reporting requirements applicable to public companies, but we expect our operating results will be adversely affected by the costs of operating as a public company.
34
Table of Contents
We will need to increase the size of our organization, and we may encounter difficulties managing our growth.
As of September 30, 2009, we had 65 employees, 43 of whom perform research and development functions. We do not plan to hire a significant number of additional employees in the near future. As our product candidates continue to progress toward potential commercialization, we anticipate the need in the future to hire additional employees as required to add depth and specialized expertise to our team. This growth could place a strain on our administrative and operational infrastructure. If the product candidates that we are developing continue to advance in clinical trials, we will need to expand our development, regulatory, manufacturing, quality, marketing, and sales capabilities or contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to develop additional relationships with various collaborators, contract research organizations, suppliers, manufacturers, and other organizations. We may not be able to establish such relationships or may incur significant costs to do so. Our ability to manage our growth will also require us to continue to improve our operational, financial, and management controls, reporting systems and procedures, and other compliance programs and processes which will further increase our operating costs. If we are unable to successfully manage the expansion of our operations or operate on a larger scale, we will not achieve our strategic objectives.
Our debt obligations include covenants which may adversely affect us.
On September 30, 2007, we entered into a Venture Loan and Security Agreement (“Loan Agreement”) with a lender. As of September 30, 2009, we had a $20.1 million outstanding principal balance under the Loan Agreement. Under the Loan Agreement we agreed that in the event our market capitalization is below $90.0 million for 15 consecutive days in which the principal market for our common stock is open for trading to the public, we will repay 50% of the then outstanding principal balance of the loans. We further agreed under the Loan Agreement that in the event our market capitalization is below $40.0 million for 15 consecutive days in which the principal market for our common stock is open for trading to the public, we will be required to repay all of the then outstanding principal balance of the loans. In addition, all of our assets other than our intellectual property secure the loans. There is a risk that the lender could obtain rights to the secured assets in the event we default on our obligations under the Loan Agreement.
The Loan Agreement also contains covenants that, among other things, require us to obtain consent from the lender prior to paying dividends, making certain investments, changing the nature of our business, assuming or guaranteeing the indebtedness of another entity or individual, selling or otherwise disposing of a substantial portion of our assets, or merging or consolidating with another entity.
A breach of any of the covenants in the Loan Agreement could result in a default under that agreement. Upon the occurrence of an event of default, the lender could elect to declare all amounts outstanding under the Loan Agreement to be immediately due and payable.
Changes in foreign currency exchange rates could result in increased costs.
We are party to some agreements denominated, wholly or partly, in foreign currencies, and, in the future, we may enter into additional agreements denominated in foreign currencies. If the values of these currencies increase against the United States dollar, our costs would increase. To date, we have not entered into any contracts to reduce the risk of fluctuations in currency exchange rates. In the future, depending upon the amounts payable under any such agreements, we may enter into forward foreign exchange contracts to reduce the risk of unpredictable changes in these costs. However, due to the variability of timing and amount of payments under any such agreements, foreign exchange contracts may not mitigate the potential adverse impact on our financial results.
35
Table of Contents
Risks Related to Our Dependence on Third Parties
We have licensed PSI-6130 and its prodrugs, including RG7128, to Roche, and we will depend on Roche to continue its development and commercialization.
We are developing RG7128 under a collaborative licensing agreement that we entered into with Roche in October 2004. We are dependent on Roche to continue the development of RG7128 and successfully commercialize it. Roche may terminate its agreement with us without cause on six months notice. If Roche fails to aggressively pursue the development and marketing approval of RG7128, if a dispute arises with Roche over the terms or the interpretation of the collaboration agreement or an alleged breach of any provision of the agreement, or if Roche terminates its agreement, then the development and commercialization of RG7128, or our ability to receive the expected payments under this agreement, could be delayed or adversely affected.
Roche is subject to many of the same development and commercialization risks to which we are subject. If Roche decides to devote resources to alternative products, either on its own or in collaboration with other pharmaceutical companies, Roche may not devote sufficient resources to the development of RG7128. Further, if Roche decides to pursue additional therapies for HCV, future sales of RG7128 could be adversely affected. Any adverse development in Roche’s operations or financial condition could adversely affect the development and commercialization of RG7128 or other prodrugs of PSI-6130, and our receipt of future milestone payments and royalties on its sales.
We and our collaborators depend on third parties to conduct our clinical trials, which may result in costs and delays that prevent us from obtaining regulatory approval or successfully commercializing our product candidates.
We and our collaborators engage clinical investigators and medical institutions to enroll subjects in our clinical trials and contract research organizations to perform data collection and analysis and other aspects of our preclinical studies and clinical trials. As a result, we depend on these third parties to perform these activities on a timely basis in accordance with the clinical protocols, good laboratory practices, good clinical trial practices, and other regulatory requirements. Our reliance on these third parties for clinical development activities reduces our control over these activities. Accordingly, if these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed, terminated, or our data may be rejected by the FDA or other regulatory agencies. If it became necessary to replace a third party that was conducting one of our clinical trials, we believe that there are a number of other third-party contractors whom we could engage to continue these activities, although it may result in a delay of the applicable clinical trial. If there are delays in testing or obtaining regulatory approvals as a result of a third party’s failure to perform, our drug discovery and development costs will increase, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates.
Reliance on third parties to conduct and monitor clinical trials may result in increased financial and regulatory risks.
Most new drugs must undergo clinical trials on human subjects before they are approved for marketing in the United States or in other countries. In recent years, the number of clinical trials of new drugs has increased, due both to U.S. federal funding of biomedical research and to research investment by pharmaceutical and biotech companies and to increased global regulatory requirements such as long term safety testing prior to product approval. There has also been a trend toward increased monitoring and enforcement of laws and the issuance of industry guidelines applicable to clinical research, including those relating to fraud and abuse. Because we rely on third party contract research organizations (“CROs”), service providers, and other organizations and institutions to conduct and monitor our clinical studies we face risks related to fraud and abuse in the conduct of our clinical studies. Those risks include financial misconduct, misconduct associated with human subjects protection, and research misconduct.
36
Table of Contents
Several aspects of financial management in clinical trials particularly increase potential risks relating to fraud and abuse concerns, including third-party payor coverage for clinical trials and related costs and research sponsor payments for clinical trial services and costs, and federal grants oversight and management. For example, allegations of False Claims Act violations in connection with Medicare billing for clinical trial services have been asserted against a number of research sites and the Department of Health and Human Service (“HHS”) Office of Inspector General (“OIG”) has repeatedly expressed concern that some industry-sponsored clinical trials may be suspect under the anti-kickback statute if they are motivated by marketing objectives. A number of research institutions have entered settlement agreements with the federal government following False Claims Act charges relating to mismanagement of federal grant funds.
Human subjects protection in the United States is addressed in regulations and guidance issued by HHS and the FDA that focus on subjects’ informed consent for clinical trial participation, institutional review board review and monitoring the conduct of clinical trials, and identification and management of researchers’ conflicts of interest. Knowing failure to comply with these requirements has resulted in fraud claims against CROs, researchers and institutional review boards. Recent allegations of community clinical research center mismanagement, increased enforcement actions such as warning letters, and failure to comply with informed consent requirements may result in additional governmental monitoring, regulation, and enforcement applicable to us, our collaborators and the CROs, researchers, and institutional review boards we work with.
We maintain compliance programs through our clinical operations and development personnel working with finance and legal group support. Our clinical trial vendors are required to monitor and report to us the possible remedial action required for the conduct of clinical studies and we are obliged to take the appropriate action. We also monitor clinical trial vendors through our regulatory and quality assurance staff and service providers. It is our understanding that Roche has undertaken to monitor the clinical trials it sponsors; and that it devotes substantial resources to this effort. However, we cannot assure you that our or our collaborators’ programs and personnel will timely and fully discover any fraud or abuse that may occur in connection with our clinical trials. Such fraud or abuse, if it occurs, could have a material adverse affect on our research, development, or commercialization activities and results.
If parties on whom we rely to manufacture our product candidates do not manufacture the active pharmaceutical ingredients or finished products of satisfactory quality, in a timely manner, in sufficient quantities or at an acceptable cost, clinical development and commercialization of our product candidates could be delayed.
We do not own or operate manufacturing facilities. Consequently, we rely on third parties as sole suppliers of our product candidates. We do not expect to establish our own manufacturing facilities and we will continue to rely on third-party manufacturers to produce clinical supplies and commercial quantities of any drugs that we market or may supply to our collaborators. Our dependence on third parties for the manufacture of our product candidates may adversely affect our ability to develop and commercialize any product candidates on a timely and competitive basis. Our collaborators may decide to not accept the product that we supply them.
To date, our product candidates have only been manufactured in quantities sufficient for preclinical studies or initial clinical trials. We do not currently have any long-term supply agreements in place for our product candidates and will need to enter into supply agreements for additional supplies of each of our product candidates to complete clinical development and/or commercialize them. Additionally, in connection with our application for commercial approvals and if any product candidate is approved by the FDA or other regulatory agencies for commercial sale, we will need to procure commercial quantities from qualified third-party manufacturers. Neither we nor Roche may be able to contract for increased manufacturing capacity for any of our product candidates in a timely or economic manner or at all. A significant scale-up in manufacturing may require additional validation studies and commensurate financial investments by the contract manufacturers. If we or Roche are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage of supply, which could limit our sales and could initiate regulatory intervention to minimize the public health risk.
37
Table of Contents
Other risks associated with our reliance on contract manufacturers include the following:
| • | Contract manufacturers may encounter difficulties in achieving volume production, quality control, and quality assurance and also may experience shortages in qualified personnel and obtaining active ingredients for our product candidates. |
| • | If we or Roche need to change manufacturers, the FDA and foreign regulatory agencies must approve these manufacturers in advance. This requires prior approval of regulatory submissions as well as successful completion of pre-approval inspections to ensure compliance with FDA and foreign regulations and standards. |
| • | Contract manufacturers are subject to ongoing periodic, unannounced inspection by the FDA and state and foreign agencies or their designees to ensure strict compliance with cGMP and other governmental regulations and corresponding foreign standards. We do not have control over compliance by our contract manufacturers with these regulations and standards. Our contract manufacturers may not be able to comply with cGMP and other FDA requirements or other regulatory requirements outside the United States. Failure of contract manufacturers to comply with applicable regulations could result in delays, suspensions or withdrawal of approvals, seizures or recalls of product candidates, and operating restrictions, any of which could significantly and adversely affect our business. |
| • | Contract manufacturers may breach the manufacturing agreements that we or our development partners have with them because of factors beyond our control or may terminate or fail to renew a manufacturing agreement based on their own business priorities at a time that is costly or inconvenient to us. |
Changes to the manufacturing process during the conduct of clinical trials or after marketing approval also require regulatory submissions and the demonstration to the FDA or other regulatory authorities that the product manufactured under the new conditions complies with cGMP requirements. These requirements especially apply to moving manufacturing functions to another facility. In each phase of investigation, sufficient information about changes in the manufacturing process must be submitted to the regulatory authorities and may require prior approval before implementation with the potential of substantial delay or the inability to implement the requested changes.
We may experience difficulties in entering into contracts on favorable terms for supplies of our products for future preclinical studies and clinical trials, which could prevent us from completing these studies and delay the commercialization of our products.
Roche is supplying RG7128 for our joint clinical trials and development program. We are considering additional supply options of RG7128. [We have entered into an agreement with Roche for the purchase of clinical supplies of RG7128 and related clinical materials for our use in developing RG7128 outside of Roche’s licensed territory.] We will need to enter into supply agreements for additional supplies of each of our product candidates to complete clinical development and/or commercialize them. We cannot assure you that we will be able to do so on favorable terms, if at all.
If we conduct future preclinical or clinical studies of RG7128 in our retained territories, we will need to procure additional product and drug supplies. We are currently proceeding to identify and evaluate the qualifications of potential suppliers that could manufacture RG7128, but we have not yet entered into a definitive supply agreement with any company. We also anticipate the need to procure additional product and drug supplies, including qualifying potential additional suppliers, of PSI-7851, PSI-938, and PSI-879.
38
Table of Contents
If conflicts arise between our collaborators and us, our collaborators may act in their best interest and not in our best interest, which could adversely affect our business.
Conflicts may arise with our collaborators if they pursue alternative therapies for the diseases that we have targeted or develop alternative products either on their own or in collaboration with others. Competing products, either developed by our present collaborators or any future collaborators or to which our present collaborators or any future collaborators have rights, may result in development delays or the withdrawal of their support for one or more of our product candidates.
Additionally, conflicts may arise if there is a dispute about the progress of, or other activities related to, the clinical development of a product candidate, the achievement and payment of a milestone amount, or the ownership of intellectual property that is developed during the course of the collaborative arrangement. Similarly, the parties to a collaboration agreement may disagree as to which party owns newly developed products. If an agreement is terminated as a result of a dispute and before we have realized the benefits of the collaboration, our reputation could be harmed and we might not obtain revenues that we anticipated receiving.
We may rely on other collaborators in the future and if future collaborations are not successful, we may not be able to effectively develop and commercialize our product candidates.
We may decide to enter into future collaborations for the development and commercialization of PSI-7851 or other product candidates. We may not be successful in entering into any additional collaborations.
Relying on collaborative relationships poses a number of risks to us, including the following:
| • | we may be required to relinquish important rights, including intellectual property, marketing, and distribution rights; |
| • | we will not be able to control whether our collaborators will devote sufficient resources to the development or commercialization of the product candidates we license; |
| • | we will not have access to all information regarding the products being developed and commercialized by our collaborators, including information about clinical trial design and execution, safety reports from clinical trials or spontaneous safety reports if the product is marketed, regulatory affairs, process development, manufacturing, marketing, and other areas known by our collaborators. Thus, our ability to keep our stockholders informed about the status of our collaborated products will be limited by the degree to which our collaborators keep us informed; |
| • | business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness to actively pursue the development and commercialization of any products resulting from a collaboration; |
| • | a collaborator may separately move forward with a competing product candidate either developed independently or in collaboration with others, including our competitors; |
| • | collaborators with marketing rights may choose to devote fewer resources to the marketing of our products than they do to other products they are selling; |
| • | our collaborators may experience financial difficulties and may be unable to fund the clinical trials, fulfill their obligations under collaboration agreements with us or delay paying us agreed-upon milestone payments, reimbursements, royalties, or other committed amounts; and |
| • | disputes may arise between us and our collaborators delaying or terminating the research, development, or commercialization of our product candidates, resulting in litigation or arbitration that could be time-consuming and expensive. |
A collaborator may terminate its agreement with us or simultaneously pursue alternative products, therapeutic approaches, or technologies as a means of developing treatments for the diseases targeted by us or
39
Table of Contents
our collaborative effort. If a collaborator terminates its agreement with us, the development or commercialization of our product candidates could be delayed or terminated, or we could be required to undertake unforeseen additional responsibilities or devote unbudgeted additional resources to such development or commercialization.
If we fail to enter into additional in-licensing agreements or if these arrangements are unsuccessful, our ability to fill our clinical pipeline may be adversely affected.
In addition to entering into collaboration agreements with third parties for the development and commercialization of our product candidates, we intend to continue to explore opportunities to further enhance our discovery and development capabilities and develop our clinical pipeline by in-licensing product candidates that fit within our expertise and research and development capabilities. We face substantial competition for in-licensing opportunities from companies focused on antiviral therapies, many of which may have greater resources than we do. Additional in-licensing agreements for product candidates may not be available to us or, if available, the terms may not be favorable. We may also need to license additional technologies in order to continue to develop our clinical pipeline. If we are unable to enter into additional agreements to license product candidates or technologies, or if these arrangements are unsuccessful, our clinical pipeline may not contain a sufficient number of promising future product candidates and our research and development efforts could be delayed.
Risks Related to Our Intellectual Property
We licensed Racivir, one of our product candidates, from Emory University, and our rights to commercialize Racivir are subject to a right of first refusal held by Gilead, and uncertainties related to these rights may result in us being prevented from obtaining the expected economic benefits from developing Racivir.
We licensed Racivir from Emory University in 1998 pursuant to an exclusive, worldwide license agreement to make, have made, use, import, offer for sale, and sell Racivir, referred to as the Racivir License Agreement. In a license agreement relating to emtricitabine that Emory entered into with Triangle Pharmaceuticals, now Gilead Sciences, Inc. in 1996, which we refer to as the Emory/Gilead License Agreement, Emory previously had granted a right of first refusal to Gilead that is applicable to any license or assignment relating to a specified range of mixtures of (–) – FTC and (+) – FTC, referred to as enriched FTC (which includes Racivir). The terms of this right of first refusal contains an exception permitting Emory to license or assign its rights in respect of enriched FTC to a permitted transferee, which includes any of the inventors (which included two of our founders) or to any corporate entity formed by or on behalf of the inventors for purposes of clinically developing enriched FTC so long as the licensee agrees in writing to be bound by the terms of Gilead’s right of first refusal to the same extent as Emory. Our license to Racivir was granted to us by Emory pursuant to this exception. Therefore, we are bound by the terms of Gilead’s right of first refusal to the same extent as Emory. The terms of this right of first refusal as set forth in the Emory/Gilead License Agreement require that, before the entry into any license or assignment agreement with a third party relating to any of Emory’s rights in respect of enriched FTC, Emory shall notify Gilead of the terms of the proposed agreement and provide a copy of the proposed agreement to Gilead together with all data and information in Emory’s possession relating to enriched FTC and its use as a therapeutic agent. Gilead has 30 days to accept or decline the offer. Although Emory considers us to be a permitted transferee under the Emory/Gilead License Agreement, Emory has taken the position that its grant of commercialization rights (i.e., the rights to offer for sale and sell Racivir) to us exceeded the rights that were permitted to be granted to a permitted transferee under its agreement with Gilead. While we believe that Gilead is aware of the Racivir License Agreement through both our and Emory’s communications with Gilead, Gilead has not contacted us regarding its interpretation of the terms of the Racivir License Agreement.
In March 2004, we entered into a supplemental agreement with Emory in which we and Emory agreed that, before any commercialization of enriched FTC by us, or by any licensee or assignee of our rights under the Racivir License Agreement, we and Emory would adhere strictly to the terms of the right of first refusal granted to Gilead in the Emory/Gilead License Agreement and offer to Gilead the same terms and conditions under
40
Table of Contents
which we, our licensee or our assignee, propose to commercialize enriched FTC. The supplemental agreement also outlines a procedure by which Emory and we would jointly offer the terms of a proposed license and commercialization agreement between us and a third party to Gilead after Emory has the opportunity to approve them. Therefore, before we could enter into a commercialization agreement for Racivir with a third party or commercialize Racivir on our own, we would be required to offer Gilead the opportunity to be our commercialization partner on the same terms on which we intend, or our prospective partner intends, to commercialize Racivir. It is uncertain whether a third party would be willing to negotiate the terms of a commercialization agreement with us knowing that Gilead can take their place as licensee by accepting the negotiated terms and exercising its right of first refusal.
These uncertainties related to our commercialization rights may result in us being prevented from obtaining the expected economic benefits from developing Racivir. In addition, we could become involved in litigation or arbitration related to our commercialization rights to Racivir in the future.
If we are unable to obtain and maintain adequate patent protection for our product candidates, we may be unable to commercialize our product candidates or to prevent other companies from using our intellectual property in competitive products in certain countries.
Our commercial success will depend, in large part, on our ability and the ability of our licensors to obtain and maintain patents and proprietary intellectual property rights sufficient to prevent others from marketing our product candidates, as well as to successfully defend and enforce those patents against infringement and to avoid infringing the proprietary rights of others, both in the United States and in foreign countries. Roche and we have filed patent applications for RG7128, and we have filed and may in the future file our own patent applications for our other technology. We have also licensed certain patents, patent applications and other proprietary rights from third parties. We have licensed patents for Racivir. The patent covering the composition of matter for Racivir that we have licensed from Emory is scheduled to expire in September 2015. Except where precise dates are given, our current patent expiration dates do not take into account any patent term adjustments that may accrue due to procedural delays by the United States Patent and Trademark Office or patent term extensions that may accrue due to regulatory delays.
Our patent position, like that of many pharmaceutical and biotechnology companies, is uncertain and involves complex legal and factual questions for which important legal principles are unresolved. We may not develop or obtain rights to products or processes that are patentable. Even if we or our licensors do obtain patents, such patents may not adequately protect the products or technologies we own or have licensed. In addition, we generally do not control the patent prosecution of subject matter that we license from others. Generally, the patent holders are primarily responsible for the patent prosecution and maintenance activities pertaining to the licensed patent applications and patents, while we are afforded opportunities to advise the primary licensors on such activities with respect to our licensed territories. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we would over our own. Others may challenge, seek to invalidate, infringe, or circumvent any patents we own or license, and rights we receive under those patents may not provide competitive advantages to us. We cannot assure you as to the degree of protection that we will be afforded by any patents issued to, or licensed by, us. The laws of many countries may not protect intellectual property rights to the same extent as U.S. laws, and those countries may lack adequate rules and procedures for defending our intellectual property rights. For example, we may not be able to prevent a third party from infringing our patents in a country that does not recognize or enforce patent rights, or that imposes compulsory licenses on or restricts the prices of life-saving drugs. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property.
41
Table of Contents
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| • | we or our licensors might not have been the first to make the inventions covered by each of our or our licensors’ pending patent applications and issued patents, and we may have to participate in expensive and protracted interference proceedings to determine priority of invention; |
| • | we or our licensors might not have been the first to file patent applications for these inventions; |
| • | our or our licensors’ pending patent applications may not result in issued patents; |
| • | our or our licensors’ issued patents may not provide a basis for commercially viable products or may not provide us with any competitive advantages or may be challenged by third parties; |
| • | others may design around our or our licensors’ patent claims to produce competitive products which fall outside the scope of our or our licensors’ patents; or |
| • | the patents of others may prevent us from marketing one or more of our product candidates for one or more indications that may be valuable to our business strategy. |
An issued patent does not guarantee us the right to practice the patented technology or commercialize the patented product. Third parties may have or obtain rights to blocking patents that could be used to prevent us from commercializing our patented products and practicing our patented technology. Our issued patents and those that may be issued in the future may be challenged, invalidated, or circumvented, which could limit our ability to prevent competitors from marketing related product candidates or could limit the length of the term of patent protection of our product candidates. In addition, the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology and our competitors may independently develop similar technologies. Moreover, because of the extensive time required for development, testing, and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent or potential patent extension may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We may incur substantial costs or lose important rights as a result of litigation or other proceedings relating to patent and other intellectual property rights.
The defense and prosecution of intellectual property rights, U.S. Patent and Trademark Office interference proceedings and related legal and administrative proceedings in the United States and elsewhere are costly and time-consuming and their outcome is uncertain. In general, there is a substantial amount of litigation involving patent and other intellectual property rights in the biopharmaceutical industry. Litigation may be necessary to:
| • | assert or defend claims of infringement; |
| • | enforce patents we own or license; |
| • | protect trade secrets; or |
| • | determine the enforceability, scope, and validity of the proprietary rights of others. |
If we become involved in any litigation, interference, or other administrative proceeding, we will incur substantial expense and it will divert the efforts of our scientific and management personnel. Uncertainties resulting from the initiation and continuation of litigation, interference, or other administrative proceedings could have a material adverse effect on our ability to compete in the marketplace pending resolution of the disputed matters. An adverse determination may subject us to significant liabilities or require us to seek licenses that may not be available from third parties on commercially reasonable terms, if at all. We or our collaborators may be restricted or prevented from developing and commercializing our products in the event of an adverse determination in a judicial or administrative proceeding or if we fail to obtain necessary licenses. In such event, we may attempt to redesign our processes or technologies so that they do not infringe, which may not be commercially reasonable or technically possible.
42
Table of Contents
From time to time, we may become subject to third party intellectual property claims as part of our ordinary course of business. (See Item 3. Legal Proceedings.)
While our product candidates are in clinical trials, we believe that the use of our product candidates in these clinical trials falls within the scope of the exemptions provided by 35 U.S.C. Section 271(e) in the United States, which exempts from patent infringement liability activities reasonably related to the development and submission of information to the FDA. As our product candidates progress toward commercialization, the possibility of a patent infringement claim against us increases. We attempt to ensure that our product candidates and the methods we employ to manufacture them, as well as the methods for their use we intend to promote, do not infringe other parties’ patents and other proprietary rights. There can be no assurance they do not, however, and competitors or other parties may assert that we infringe their proprietary rights in any event.
If we find during clinical evaluation that our product candidates should be used in combination with a product that is covered by a patent held by another company or institution, and that a labeling instruction is required in product packaging recommending that combination, we could be accused of, or held liable for, infringement of the third-party patents covering the product recommended for co-administration with our product. In such a case, we could be required to obtain a license from the other company or institution to use the required or desired package labeling, which may not be available on commercially reasonable terms, or at all.
We may be subject to claims that our board members, employees, or consultants or we have used or disclosed alleged trade secrets or other proprietary information belonging to third parties and any such individuals that are currently affiliated with one of our competitors may disclose our proprietary technology or information.
As is commonplace in the biotechnology and pharmaceutical industries, some of our board members, employees, and consultants are or have been employed at, or associated with, other biotechnology or pharmaceutical companies that compete with us. While employed at or associated with these companies, these individuals may become exposed to or involved in research and technology similar to the areas of research and technology in which we are engaged. We may be subject to claims that we, or our employees, board members, or consultants have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of those companies. Litigation may be necessary to defend against such claims.
We have entered into confidentiality agreements with all of our employees. However, we do not have, and are not planning to enter into, any confidentiality agreements with our directors because they have a fiduciary duty of confidentiality as directors. There is the possibility that any of our former board members, employees, or consultants who are currently employed at, or associated with, one of our competitors may unintentionally or willfully disclose our proprietary technology or information.
The rights we rely upon to protect our unpatented trade secrets may be inadequate.
We rely on unpatented trade secrets, know-how, and technology. This intellectual property is difficult to protect, especially in the pharmaceutical industry, where much of the information about a product must be made public during the regulatory approval process. We seek to protect trade secrets, in part, by entering into confidentiality agreements with employees, consultants, and others. These parties may breach or terminate these agreements, and we may not have adequate remedies for such breaches. Furthermore, these agreements may not provide meaningful protection for our trade secrets or other proprietary information or result in the effective assignment to us of intellectual property, and may not provide an adequate remedy in the event of unauthorized use or disclosure of confidential information or other breaches of the agreements. Despite our efforts to protect our trade secrets, we or our collaboration partners, board members, employees, consultants, contractors, or scientific and other advisors may unintentionally or willfully disclose our proprietary information to competitors.
43
Table of Contents
There is a risk that our trade secrets could have been, or could, in the future, be shared by any of our former employees with and be used to the benefit of any company that competes with us. For example, a former director and founder of Pharmasset has, along with several of our former scientists, started a new pharmaceutical company to develop drugs to treat viral infections (including human retroviral and hepatitis infections), cancer, and dermatological conditions, which may compete with us in the future. These individuals left Pharmasset in 2005. We have a confidentiality agreement in place with our former director, and have both confidentiality agreements and covenant not to compete agreements in place with the former scientists. The term of the confidentiality agreements is indefinite with regard to any confidential information that is not subsequently made public. The covenant not to compete agreements expired on February 28, 2007.
If we fail to maintain trade secret protection, our competitive position may be adversely affected. Competitors may also independently discover our trade secrets. Enforcement of claims that a third party has illegally obtained and is using trade secrets is expensive, time consuming, and uncertain. If our competitors independently develop equivalent knowledge, methods, and know-how, we would not be able to assert our trade secrets against them and our business could be harmed.
Risks Related to Our Industry
Our industry is extremely competitive. If our competitors develop and market products that are equally or more effective, safer, or more affordable than ours, or obtain marketing approval before we do, our commercial opportunities may be limited.
Competition in the biotechnology and pharmaceutical industries is intense and continues to increase, particularly in the area of antiviral drugs. Many companies are pursuing the development of novel drugs that target the same diseases we are targeting. There are a significant number of drugs that are approved or currently under development that will become available in the future for the treatment of HCV and other viral infections. If any of the product candidates that our competitors are developing are successful, we will have difficulty gaining market share.
We face a broad range of current and potential competitors, from established global pharmaceutical companies with significant resources to development-stage companies. Listed below are some of the pharmaceutical and biotechnology companies developing compounds targeting HCV and other viral infections. Roche, Merck (formerly Schering-Plough), and Three Rivers Pharmaceuticals market alpha interferon, a component of the current standard of care, which is approved in several chemically modified forms. All three companies and several generic manufacturers market ribavirin, which is the other component of the current standard of care for HCV. Roche, Merck, and other companies, such as Abbott, Boehringer Ingelheim, Bristol Meyers Squibb, Gilead Sciences, Idenix, InterMune, Novartis/Human Genome Sciences, Pfizer and Vertex are also developing new drugs for the treatment of HCV.
In addition, we face competition from academic and research institutions and government agencies for the discovery, development and, commercialization of novel therapeutics to treat HCV. Some early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and biotechnology companies.
Many of our competitors have:
| • | significantly greater financial, technical, and human resources than we have and may be better equipped to develop, manufacture and market products; |
| • | more extensive experience in preclinical studies and clinical trials, obtaining regulatory approvals, and manufacturing and marketing products; and |
| • | products that have already been approved or are in the late stage of development and operate large, well-funded research and development programs. |
44
Table of Contents
Our competitors may succeed in developing or commercializing equally or more effective, safe, or affordable products, which would render our product candidates less competitive or noncompetitive. Our competitors may discover technologies and techniques, or enter into partnerships with collaborators, in order to develop competing products that are more effective or less costly than the products we develop. This may render our technology or products obsolete and noncompetitive. These competitors also compete with us to recruit and retain qualified scientific and management personnel, establish clinical trials sites and patient registration for clinical trials, as well as to acquire technologies and technology licenses complementary to our programs or advantageous to our business. Moreover, competitors that are able to achieve patent protection, obtain regulatory approvals, and commence commercial sales of their products before we do, and competitors who have already done so, will enjoy a significant competitive advantage. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
If we successfully develop and obtain approval for our product candidates, we will face competition for market share based on the safety and efficacy of our products, the timing and scope of regulatory approvals, the availability of supply, marketing and sales capability, reimbursement coverage, price, patent position, and other factors.
The development of direct acting antivirals to treat HCV may present additional risks beyond those inherent in drug development.
We are developing, alone and with collaborator(s), nucleoside/tides to treat HCV. The potential therapeutic regimens being tested or planned for testing include our nucleoside/tide in combination with:
| • | the current standard of care (pegylated interferon and ribavirin); |
| • | pegylated interferon; |
| • | ribavirin; |
| • | the current standard of care plus a direct acting antivirial (such as InterMune’s protease inhibitor, in studies conducted and planned by Roche); and; |
| • | with one or more direct acting antivirals without concomitant interferon or ribavirin therapy; |
These development programs and planned studies carry all the risks inherent in drug development activities, including the risk that they will fail to show efficacy or acceptable safety. However, these development programs are subject to regulatory, commercial, manufacturing, and other risks that may be additional to the risks described above. For example, regulatory guidelines for approval of direct acting antiviral drugs are evolving in the United States, Europe, and other countries. We anticipate that regulatory guidelines and regulatory agency responses to our, our collaborators’, and our competitors’ development programs will continue to change, resulting in the risk that our and our collaborators’ activities may not meet unanticipated new standards or requirements, which could lead to delay, additional expense, or potential failure of development activities. Our development programs, in addition, involve testing product candidates alone and in combination with approved drugs and with currently unapproved product candidates, the latter of which increases the risk of significant adverse effects or test failures. The timing, outcome, and cost of such testing are difficult to predict and dependent on a number of factors that are outside our reasonable control. To the extent that we, our collaborator, or our competitors successfully develop direct acting antivirals whose use improves the current standard of care or results in “interferon-sparing” treatment regimens, current HCV-treating physicians, HCV patients, healthcare payers, and others may not readily accept or pay for such improvements or new treatments. Because direct acting antivirals is an emerging field, the delay or failure of a competitor attempting to develop therapeutics that could have been combined with our product candidates or that are perceived to be similar to our product candidates could have a significant adverse effect on the commercial or regulatory environment for our product candidates or on the price of our stock. As we note elsewhere, other companies developing direct acting antivirals have more advanced development programs than we do. Their success or failure to successfully conclude clinical development and obtain marketing approval could have a material adverse affect on our development and commercialization plans and activities.
45
Table of Contents
If third-party payers do not adequately reimburse patients for any of our product candidates, that are approved for marketing, we may not be successful in selling them.
Our ability to commercialize any products successfully will depend in part on the extent to which reimbursement will be available from governmental and other third-party payers, both in the United States and in foreign markets. Even if we succeed in bringing one or more products to the market, the amount reimbursed for our products may be insufficient to allow our products to compete effectively with products that are reimbursed at a higher level. If the price we are able to charge for any products we develop is inadequate in light of our development costs, our profitability could be adversely affected.
Reimbursement by governmental and other third-party payers may depend upon a number of factors, including the governmental and other third-party payers’ determination that the use of a product is:
| • | a covered benefit under their health plan or part of their formulary; |
| • | safe, effective, and medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; and |
| • | neither experimental nor investigational. |
Obtaining reimbursement approval for a product from each third-party and government payer is a time-consuming and costly process that could require us to provide supporting scientific, clinical, and cost-effectiveness data for the use of our products to each payer. We may not be able to provide data sufficient to obtain reimbursement.
Eligibility for coverage does not imply that any drug product will be reimbursed in all cases or at a rate that allows us to make a profit. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not become permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on payments allowed for lower-cost drugs that are already reimbursed, may be incorporated into existing payments for other products or services, and may reflect budgetary constraints and/or Medicare or Medicaid data used to calculate these rates. Net prices for products also may be reduced by mandatory discounts or rebates required by government health care programs or by any future relaxation of laws that restrict imports of certain medical products from countries where they may be sold at lower prices than in the United States.
The health care industry is experiencing a trend toward containing or reducing costs through various means, including lowering reimbursement rates, limiting therapeutic class coverage, and negotiating reduced payment schedules with service providers for drug products. The Medicare Prescription Drug, Improvement and Modernization Act of 2003, or the MMA, created a broader prescription drug benefit for Medicare beneficiaries. The MMA also contains provisions intended to reduce or eliminate delays in the introduction of generic drug competition at the end of patent or nonpatent market exclusivity. The impact of the MMA on drug prices and new drug utilization in the future is unknown. The MMA also made adjustments to the physician fee schedule and the measure by which prescription drugs are presently paid. The effects of these changes are unknown but may include decreased utilization of new medicines in physician prescribing patterns, and further pressure on drug company sponsors to provide discount programs and reimbursement support programs. There have been, and we expect that there will continue to be, federal and state proposals to constrain expenditures for medical products and services, which may affect reimbursement levels for our future products. In addition, the Centers for Medicare and Medicaid Services frequently change product descriptors, coverage policies, product and service codes, payment methodologies, and reimbursement values. Third-party payers often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates and may have sufficient market power to demand significant price reductions. Future legislation may also limit the prices that can be charged for drugs we develop.
46
Table of Contents
Foreign governments tend to impose strict price controls which may adversely affect our future profitability.
In most foreign countries, particularly in the European Union, prescription drug pricing and/or reimbursement is subject to governmental control. In those countries that impose price controls, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, or if there is competition from lower priced cross-border sales, our profitability will be negatively affected.
Even if we achieve market acceptance for our products, we may experience downward pricing pressure on the price of our drugs because of generic and biosimilar competition and social pressure to lower the cost of drugs to treat HIV and HCV.
Several of the FDA-approved individual and combination products face patent expiration in the next several years. As a result, generic versions and biosimilars of these drugs and biologicals may become available. We expect to face competition from these products, including price-based competition.
Pressure from AIDS awareness and other social activist groups to reduce HIV drug prices may also put downward pressure on the prices of HIV drugs, including Racivir if it is commercialized. Similar trends of generic competition or social pressure may occur for HCV, which would result in downward pressure on the price for RG7128, PSI-7851, PSI-938, or PSI-879, if they are commercialized.
We face a risk of product liability claims and, if we are not able to obtain adequate liability insurance, a product liability claim could result in substantial liabilities.
Our business exposes us to the risk of significant potential product liability claims that are inherent in the manufacturing, testing, and marketing of human therapeutic products, and we will face a greater risk if our Collaborator(s) or we sell any products commercially. Regardless of their merit or eventual outcome, product liability claims may result in:
| • | delay or failure to complete our clinical trials; |
| • | withdrawal of clinical trial participants and difficulty in recruiting participants; |
| • | inability to commercialize our product candidates; |
| • | decreased demand for our product candidates; |
| • | injury to our reputation; |
| • | inability to establish new collaborations; |
| • | litigation costs; |
| • | substantial monetary awards against us; and |
| • | diversion of management or other resources from key aspects of our operations. |
Product liability claims could result in an FDA or other regulatory authority investigation of the safety or efficacy of our products, our manufacturing processes and facilities, our marketing programs, our internal safety reporting systems or our staff conduct. A regulatory authority investigation could also potentially lead to a recall of our products or more serious enforcement actions, limitations on the indications, for which they may be used, or suspension, or withdrawal of approval.
47
Table of Contents
We currently have product liability insurance that covers our clinical trials for up to $15.0 million for each occurrence and up to a $15.0 million annual aggregate limit, subject to deductibles of $50,000 per occurrence and $250,000 annual aggregate limit and coverage limitations. We intend to increase our insurance coverage and include the sale of commercial products if marketing approval is obtained. Because insurance coverage is becoming increasingly expensive, we may not be able to obtain or maintain adequate protection against potential product liabilities at a reasonable cost or at all, and the insurance coverage that we obtain may not be adequate to cover potential claims or losses.
We may incur significant costs to comply with laws regulating the protection of health and human safety and the environment, and failure to comply with these laws and regulations could expose us to significant liabilities.
Our research and development activities involve the controlled use of numerous hazardous materials, chemicals, and radioactive materials which produce waste products. We are subject to federal, state, and local laws and regulations, and may be subject to foreign laws and regulations, governing the use, manufacture, storage, handling, and disposal of hazardous materials and waste products, including certain regulations promulgated by the U.S. Environmental Protection Agency, or EPA. Our regulatory applications will include the request for an exemption to an environmental assessment for the impact of the product, materials, and processes subject to the application. The request could be denied, resulting in potentially substantial delays to our development time lines. The EPA regulations to which we are subject require that we register with the EPA as a generator of hazardous waste. The risk of accidental contamination or injury from the handling, transporting, and disposing of hazardous materials and waste products cannot be entirely eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health, and workplace safety laws and regulations, including those governing laboratory procedures, exposures to blood-borne pathogens, and the handling, transporting, and disposing of biohazardous or radioactive materials. Although we maintain workers’ compensation insurance to cover us for the costs and expenses we may incur if our employees are injured as a result of using these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain, nor do we plan to obtain, additional insurance coverage relating to damage claims arising from our use of hazardous materials. Further, we may be required to indemnify our collaborators or licensees against damages and other liabilities arising out of our development activities or products. Compliance with the applicable environmental and workplace laws and regulations is expensive. Future changes to environmental, health, workplace, and safety laws could cause us to incur additional expenses or may restrict our operations or impair our research, development, and production efforts.
Risks Related to our Common Stock
Our stock price is volatile.
The stock market in general and the market for clinical-stage pharmaceutical stocks in particular have experienced extreme volatility that has often been unrelated or disproportionate to the operating performance of particular companies. The volume and price per share of our common stock may be negatively affected by the results or activities of investors, which may be unrelated to our operating performance. In addition, broad market fluctuations may adversely affect the volume or trading price for our common stock.
In this market environment, the sale of a substantial number of shares of our common stock in the public market or the perception that such a sale might occur would likely have an adverse effect on the market price of our common stock. A number of investors hold relatively large positions in our securities. Particularly in light of the relatively low volume of trading in our common stock during many trading sessions, a decision by any of these investors to sell all or a block of its holdings of our common stock could cause our stock price to drop significantly.
48
Table of Contents
The market also continues to experience significant price and volume fluctuations, some of which are unrelated to the operating performance of particular companies. Since our IPO, the price of our common stock has fluctuated significantly and may continue to do so in the future. Many factors could have a significant effect on the market price for our common stock, including:
| • | adverse results or delays in our preclinical studies or clinical trials or the clinical trials of our collaborator(s) or others in the industry, including our competitors; |
| • | announcements of FDA or foreign regulatory non-approval of our product candidates, or delays in the FDA or other foreign regulatory agency review process; |
| • | adverse actions taken by regulatory agencies with respect to our product candidates, clinical trials, manufacturing processes, or sales and marketing activities; |
| • | introductions or announcements of new products or technological innovations or pricing by our competitors; |
| • | the loss of a significant collaborator; |
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters, and our ability to patent our product candidates and technologies; |
| • | changes in estimates of our financial performance by securities analysts or failure to meet or exceed securities analysts’ or investors’ expectations of our annual or quarterly financial results, clinical results, or our achievement of any milestones or changes in securities analysts’ recommendations regarding our common stock or other comparable companies or our industry generally; |
| • | fluctuations in stock market prices and trading volumes of similar companies or of the markets generally; |
| • | changes in accounting principles; |
| • | sales of large blocks of our common stock, or the expectation that such sales may occur, including sales by our executive officers, directors, and significant stockholders; |
| • | issuance of new shares of common stock in future offerings; |
| • | issuance of convertible debt, or warrants or stock options; |
| • | discussion of our business, products, financial performance, prospects, or our stock price by the financial and scientific press and online investor communities, such as chat rooms; |
| • | regulatory developments in the United States and abroad; |
| • | third-party healthcare reimbursement policies; |
| • | conditions or trends in the pharmaceutical and biotechnology industries; |
| • | departures of key personnel; |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, clinical trial results, joint ventures, or capital commitments; and |
| • | actual or anticipated variations in our annual or quarterly operating results. |
Any litigation brought against us as a result of this volatility could result in substantial costs and a diversion of our management’s attention and resources, which could negatively impact our financial condition, results of operations, and the price of our common stock.
If we raise additional capital by issuing equity securities in a fluctuating market, many or all of our existing stockholders may experience substantial dilution, and if we need to raise capital by issuing equity securities at a
49
Table of Contents
time when our stock price is lower, we may have difficulty raising sufficient capital to meet our requirements. If any of the risks described in these “RISK FACTORS” occurred, or if any unforeseen risk affected our performance, it could have a dramatic and adverse impact on the market price of our common stock.
Provisions of our amended and restated certificate of incorporation, bylaws, and Delaware law could delay or discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
Provisions of our amended and restated certificate of incorporation, bylaws, and Delaware law may discourage, delay, or prevent a merger or acquisition that stockholders may consider favorable. In addition, these provisions could make it more difficult for our stockholders to replace or remove our board of directors.
These provisions include:
| • | the application of a Delaware law prohibiting us from entering into a business combination with the beneficial owner of 15% or more of our outstanding voting stock for a period of three years after such 15% or greater owner first reached that level of stock ownership, unless we meet specified criteria; |
| • | authorizing the issuance of preferred stock with rights that may be senior to those of our common stock without any further vote or action by the holders of our common stock; |
| • | providing for a classified board of directors with staggered terms; |
| • | requiring that our stockholders provide advance notice when nominating our directors or proposing matters that can be acted on by stockholders at stockholders’ meetings; |
| • | eliminating the ability of our stockholders to convene a stockholders’ meeting; and |
| • | prohibiting our stockholders from acting by written consent. |
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | PROPERTIES |
On May 23, 2005, we entered into a lease for a 30,800 square foot building that has 12,000 square feet of laboratory space and approximately 18,000 square feet of administrative offices in Princeton, New Jersey. On June 9, 2009, we modified the lease and extended the term thereof for an additional five years, from May 22, 2010 through May 22, 2015. The annual occupancy expense under this lease is approximately $825,000. This facility is equipped to perform drug research activities. In April 2007, we also entered into a lease for office space in Durham, North Carolina. On February 2, 2009, we amended the lease term to extend through April 1, 2011. The annual occupancy expense under this lease is approximately $81,000.
| ITEM 3. | LEGAL PROCEEDINGS |
On July 28, 2009, Emory University and University of Georgia Research Foundation, Inc. (“Claimants”) filed a Demand for Arbitration and Relief (the “Demand”) with the American Arbitration Association in Atlanta, Georgia (the “Emory Arbitration”), claiming certain payments and seeking specific performance under the Company’s January 8, 2004 license agreement with Claimants (the “Emory License”).
The Demand alleges that payments Pharmasset has received under the Roche collaboration agreement are subject to the Emory License and that Pharmasset has not paid fees to Claimants based on such payments. In addition, the Demand alleges that Pharmasset has not complied with certain terms and conditions of the Emory
50
Table of Contents
License and that other Pharmasset product candidates are, or will be, covered by the Emory License. The Demand requests, among other things, specific performance of the Emory License, including the payment of license fees related to past payments received by Pharmasset. The Company’s response to the Demand was filed on August 14, 2009. The Company denies these allegations and intends to vigorously defend itself against the Demand.
| ITEM 4. | SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS |
We held a Special Meeting of Pharmasset, Inc. Stockholders on September 23, 2009 to ask our stockholders to approve an amendment to our 2007 Equity Incentive Plan to increase the number of shares issuable under such plan by 1,000,000. The proposed amendment was approved by our stockholders with 17,116,842 shares voting in favor of such proposal and 456,913 shares voting against such proposal.
51
Table of Contents
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock began trading on the Global Market of The NASDAQ Stock Market LLC (“NASDAQ”) on April 27, 2007 under the symbol “VRUS.” Prior to that time, there was no established public trading market for our common stock. The following table sets forth for the periods indicated the high and low sale prices per share of our common stock as reported by NASDAQ:
| Fiscal Year Ended September 30, 2009: |
High | Low | ||||
| Fourth fiscal quarter 2009 |
$ | 24.61 | $ | 10.86 | ||
| Third fiscal quarter 2009 |
$ | 11.72 | $ | 7.69 | ||
| Second fiscal quarter 2009 |
$ | 14.38 | $ | 7.62 | ||
| First fiscal quarter 2009 |
$ | 22.49 | $ | 11.72 | ||
| Fiscal Year Ended September 30, 2008: |
||||||
| Fourth fiscal quarter 2008 |
$ | 24.68 | $ | 17.62 | ||
| Third fiscal quarter 2008 |
$ | 20.47 | $ | 13.43 | ||
| Second fiscal quarter 2008 |
$ | 36.44 | $ | 13.50 | ||
| First fiscal quarter 2008 |
$ | 14.73 | $ | 11.96 | ||
Holders of Record
As of October 31, 2009, there were 32 holders of record of our common stock.
Comparative Stock Performance
The following graph and related information should not be deemed “soliciting material” or to be “filed” with the Securities and Exchange Commission, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
CUMULATIVE TOTAL RETURN
(Based on an initial investment of $100.00 on April 27, 2007 using
end of the quarter closing prices for each of the three investment options.)
52
Table of Contents
Dividends
We have never paid or declared any cash dividends on our common stock. We currently intend to retain any earnings for future growth and, therefore, do not expect to pay cash dividends in the foreseeable future. Moreover, under the terms of our Loan Agreement with our lender, we are not permitted to pay any dividends without its written consent.
53
Table of Contents
| ITEM 6. | SELECTED FINANCIAL DATA |
The following table presents our selected financial information. In 2005, we changed our fiscal year end from December 31 to September 30 for financial reporting purposes. The change was effective for the nine-month period ended September 30, 2005. The statement of operations data for the years ended September 30, 2009, 2008, and 2007 and the balance sheet data as of September 30, 2009 and 2008 have been derived from our audited financial statements included elsewhere in this Annual Report on Form 10-K. In the opinion of management, the unaudited selected financial data presented below reflect all adjustments necessary for a fair presentation of this data. The statement of operations data for the year ended September 30, 2006 and the nine months ended September 30, 2005, and the balance sheet data as of September 30, 2007, 2006, and 2005 have been derived from our audited financial statements that are not included in this Annual Report on Form 10-K. The results from the nine-month period ended September 30, 2005 are not indicative of results that would have been achieved for the twelve-month period ended September 30, 2005.
The selected financial data set forth below should be read together with our financial statements and the related notes to those financial statements, as well as “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” appearing elsewhere in this Annual Report on Form 10-K. The historical results are not necessarily indicative of results to be expected in any future period.
| Years Ended September 30, | Nine Months Ended September 30, 2005 |
|||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | |||||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Contract revenues |
$ | 13,293 | $ | 1,857 | $ | 22,009 | $ | 5,425 | $ | 3,719 | ||||||||||
| Government grant revenues |
— | — | — | — | — | |||||||||||||||
| Total revenues |
$ | 13,293 | $ | 1,857 | $ | 22,009 | $ | 5,425 | $ | 3,719 | ||||||||||
| Cost and expenses: |
||||||||||||||||||||
| Research and development |
52,552 | 42,996 | 20,319 | 10,498 | 10,468 | |||||||||||||||
| General and administrative |
13,365 | 13,289 | 9,211 | 7,912 | 8,096 | |||||||||||||||
| Total costs and expenses |
65,917 | 56,285 | 29,530 | 18,410 | 18,564 | |||||||||||||||
| Operating loss |
(52,624 | ) | (54,428 | ) | (7,521 | ) | (12,985 | ) | (14,845 | ) | ||||||||||
| Investment income |
221 | 1,986 | 2,471 | 1,659 | 1,136 | |||||||||||||||
| Interest expense |
(3,190 | ) | (2,216 | ) | (15 | ) | — | — | ||||||||||||
| Loss before income taxes |
(55,593 | ) | (54,658 | ) | (5,065 | ) | (11,326 | ) | (13,709 | ) | ||||||||||
| Provision for income taxes |
— | — | — | — | — | |||||||||||||||
| Net loss |
(55,593 | ) | (54,658 | ) | (5,065 | ) | (11,326 | ) | (13,709 | ) | ||||||||||
| Redeemable preferred stock accretion (1) |
— | — | 1,776 | 1,111 | 2,287 | |||||||||||||||
| Net loss attributable to common stockholders |
$ | (55,593 | ) | $ | (54,658 | ) | $ | (6,841 | ) | $ | (12,437 | ) | $ | (15,996 | ) | |||||
| Net loss per common share: |
||||||||||||||||||||
| Basic |
$ | (2.10 | ) | $ | (2.51 | ) | $ | (0.46 | ) | $ | (1.19 | ) | $ | (2.42 | ) | |||||
| Diluted |
$ | (2.10 | ) | $ | (2.51 | ) | $ | (0.46 | ) | $ | (1.19 | ) | $ | (2.42 | ) | |||||
| Weighted average number of shares used in per common share calculations: |
||||||||||||||||||||
| Basic (1) |
26,479,532 | 21,808,283 | 14,990,472 | 10,462,369 | 6,630,463 | |||||||||||||||
| Diluted (1) |
26,479,532 | 21,808,283 | 14,990,472 | 10,462,369 | 6,630,463 | |||||||||||||||
54
Table of Contents
| As of September 30, | ||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||
| (in thousands) | ||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||
| Cash and cash equivalents (1) |
$ | 58,408 | $ | 63,073 | $ | 68,746 | $ | 26,182 | $ | 33,442 | ||||||
| Short-term investments |
— | 497 | 1,252 | 1,250 | 12,007 | |||||||||||
| Working capital |
41,647 | 52,425 | 60,764 | 25,004 | 38,822 | |||||||||||
| Total assets (1) |
62,736 | 68,982 | 75,844 | 32,998 | 47,441 | |||||||||||
| Deferred revenue |
3,941 | 5,726 | 7,583 | 9,168 | 12,044 | |||||||||||
| Current portion of and long-term debt, net |
19,609 | 19,174 | — | — | — | |||||||||||
| Redeemable convertible preferred stock (1) |
— | — | — | 19,641 | 18,530 | |||||||||||
| Total stockholders’ equity (deficit) (1) |
$ | 28,898 | $ | 35,187 | $ | 58,936 | $ | (220 | ) | $ | 11,668 | |||||
| (1)— | On May 2, 2007, we completed our IPO of 5,050,000 shares of our common stock at a public offering price of $9.00 per share. Net cash proceeds from our IPO were $40.7 million after deducting offering costs paid during fiscal 2007 and $39.1 million after deducting additional offering costs paid in fiscal 2006. |
In connection with our IPO, the outstanding shares of our Series B, Series C, Series D, and Series R redeemable convertible preferred stock, our Series A convertible preferred stock, and our redeemable common stock were converted into 4,405,683 shares of our common stock as of May 2, 2007. In addition, holders of our Series D redeemable convertible preferred stock were entitled to receive quarterly dividends at a rate equal to 7.5% per annum of the purchase price per share. Such dividends accrued from February 4, 2006 through May 2, 2007 and were paid out in the form of 131,864 shares of our common stock. Our Series D-1 warrants were also exercised in full in connection with our IPO on a “net exercise” basis, which resulted in us issuing 822,689 shares of our common stock to the warrant holders.
55
Table of Contents
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our audited financial statements and related notes appearing elsewhere in this report. In addition to historical information, this discussion and analysis contains forward-looking statements based on current expectations that involve risks, uncertainties and assumptions, such as our plans, objectives, expectations, and intentions set forth in the “Cautionary Statement Regarding Forward-Looking Statements,” which can be found at the beginning of this report, and in Item 1A, Risk Factors. Our actual results and the timing of events may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth in the “Risk Factors” section and elsewhere in this report.
Overview
We are a clinical-stage pharmaceutical company committed to discovering, developing, and commercializing novel drugs to treat viral infections. Our primary focus is on the development of nucleoside/tide analogs as oral therapeutics for the treatment of chronic hepatitis C virus (“HCV”) infection and secondarily, on the development of RacivirTM for the treatment of human immunodeficiency virus (“HIV”) infection. Nucleoside/tide analogs are a class of compounds which act as alternative substrates for the viral polymerase, thus inhibiting viral replication. We currently have three clinical-stage product candidates, two of which we are developing ourselves and one of which we are developing with a strategic partner. We are also advancing a series of preclinical candidates in preparation for clinical development. Our three clinical stage product candidates are:
| • | RG7128 (formerly R7128), is a first generation HCV nucleoside polymerase inhibitor we are developing through a strategic collaboration with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (collectively, “Roche”). RG7128 is in a Phase 2b clinical trial in combination with Pegasys® (pegylated interferon) plus Copegus® (ribavirin) and recently completed INFORM-1, a study designed to investigate the combination of two oral, direct acting antivirals (“DAAs”) in the absence of pegylated interferon and ribavirin. Both of these studies are being conducted by Roche; |
| • | PSI-7851, a second generation HCV nucleotide polymerase inhibitor, which has completed two phase 1 studies providing adequate safety and efficacy data to support initiation of a Phase 2a clinical trial; and |
| • | Racivir, for the treatment of HIV in combination with other approved HIV drugs, has completed a Phase 2 clinical trial. |
In addition, we recently nominated PSI-352938 (“PSI-938”) and PSI-352879 (“PSI-879”) as third generation nucleotide development candidates for the treatment of HCV, each of which could potentially be used in combination with our current nucleoside/tides, RG7128 or PSI-7851. PSI-938 and PSI-879 are proprietary purine nucleotide analog inhibitors of HCV polymerase that are in preclinical studies required for submission of an Investigational New Drug (“IND”) application with the U.S. Food and Drug Administration (“FDA”) or equivalent foreign regulatory application.
We are continuing to research nucleoside/tide analogs (both pyrimidines and purines) with the intention of identifying additional product candidates that can potentially be used in combination with our current nucleoside/tides, RG7128 and PSI-7851, in combination with other classes of DAAs, or with pegylated interferon and/or ribavirin for the treatment of HCV. We have identified proprietary nucleotide prodrugs that are referred to as phosphate prodrugs because they have the ability to deliver the monophosphate forms of the compounds into infected cells, thus bypassing a rate-limiting step in the metabolic pathway to the active triphosphate form of the drug. The goal of these efforts is to identify compounds with improved potency, safety, convenience, oral bioavailability, and increased intrahepatic nucleotide triphosphate levels. Certain of these compounds have demonstrated exceptional in vitro anti-HCV activity, with up to 100 times greater potency than PSI-6130 (of which RG7128 is a pro-drug). Early studies in animals indicate that several of these compounds can achieve concentrations of the active triphosphate form in the liver up to 1000 times higher than PSI-6130 at equivalent doses.
56
Table of Contents
We are developing PSI-7851, PSI-938, PSI-879, and Racivir ourselves. We have a strategic collaboration with Roche for the development of PSI-6130 and its prodrugs, including RG7128. Under the collaboration, Roche pays all development costs and provides us with potential income from milestone payments that can be used to fund the advancement of our proprietary product candidates.
Despite significant advances, there remain significant unmet medical needs in HCV and HIV. In the treatment of HCV, pegylated interferon in combination with ribavirin is the standard of care and has demonstrated, for some patients, the ability to offer a sustained virologic response, or SVR, defined as HCV RNA levels that are below the limit of detection by a standard test utilizing polymerase chain reaction, or PCR, six months after discontinuation of therapy. However, pegylated interferon is available only as an injectable and is associated with side effects, some of them serious, which may include fatigue, bone marrow suppression, anemia, and neuropsychiatric effects. Many individuals with HCV infection are unable to be treated with interferon due to pre-existing co-morbidities such as advanced liver disease or psychiatric conditions. In the treatment of HCV, there is an unmet medical need for drugs that offer an improved SVR rate with an improved safety profile. For HIV patients, treatment also involves a chronic regimen of antiviral drugs. During such prolonged treatment, viral mutations occur that make the viruses resistant to the drugs being used. We believe there is an unmet medical need for new HIV drugs that are effective against resistant viruses and can replace existing therapies that have lost their effectiveness.
We believe nucleoside/tide analogs are well suited to treat viral diseases because they can be designed to be highly specific and potent inhibitors of HCV replication, are relatively simple to manufacture, and have the potential for oral administration. In HCV, nucleoside/tide analog drugs have demonstrated a higher barrier to viral resistance than non-nucleoside and protease inhibitors for HCV. In addition, this class of compounds has a well-established development and regulatory history. There are 14 nucleoside analogs for the treatment of HBV and HIV that have been approved by the FDA and as a class are standard of care. Additional nucleoside analogs have been approved by the FDA for the treatment of HCV, cytomegalovirus and herpes simplex virus. In addition to RG7128, PSI-7851, PSI-938, PSI-879, and Racivir, we also have discovery programs focused on other nucleoside/tide analogs for HCV. Our scientific team of virologists, biologists, and chemists has experience discovering and developing nucleoside/tide analog drugs for antiviral indications. Collectively, our management team’s product development experience includes approximately 50 therapeutic and diagnostic product approvals. Our discovery platform includes a library of nucleoside/tide analogs and a collection of viral and cellular assays that we use to evaluate new product candidates.
57
Table of Contents
Our Product Candidates
Our research and development programs are primarily focused on discovering and developing drugs that treat HCV and, secondarily, on the development of Racivir for the treatment of HIV infections. Our product candidates are nucleoside/tide analogs that we believe have potential competitive advantages with respect to safety, efficacy, drug resistance, and/or convenience of dosing as compared to currently approved drugs and other known investigational agents. The following table summarizes the five product candidates on which we are focusing:
| Product Candidate |
Indication |
Status |
Next Expected Milestone |
Commercialization | ||||
| RG7128 |
HCV | In a Phase 2b clinical trial and recently completed the INFORM-1 study conducted by Roche | Complete enrollment of cohort 2 (remaining 300 patients) in Phase 2b clinical trial by the end of the first calendar quarter of 2010, and the INFORM-2 (Phase 2) study to be initiated by Roche during the first calendar quarter of 2010 | Roche | ||||
| PSI-7851 |
HCV | Recently completed Phase 1 study | Begin Phase 2a clinical trial during the first calendar quarter of 2010 | — | ||||
| PSI-938 |
HCV | In IND-enabling preclinical studies | Submit IND application during the first calendar quarter of 2010 | — | ||||
| PSI-879 |
HCV | In IND-enabling preclinical studies | Submit IND application during the fourth calendar quarter of 2010 | — | ||||
| Racivir |
HIV | Completed Phase 2 clinical trial | Engage a development partner for a combination drug study | — | ||||
Financial History
We have incurred substantial operating losses since our inception because we have devoted substantially all of our resources to our research and development activities and have not generated any revenue from the sale of approved drugs. As of September 30, 2009, we had an accumulated deficit of $167.4 million. We expect our operating losses to increase for at least the next several years as we continue to pursue the clinical development of PSI-7851, PSI-938, and PSI-879 and as we expand our discovery and development pipeline.
We have funded our operations primarily through the sale of equity securities, payments received under collaboration agreements, borrowings under our Loan Agreement, and interest earned on investments. We expect to continue to fund our operations over the next several years through the net proceeds of our completed public offerings, our existing cash resources, borrowings under our Loan Agreement, potential future milestone payments that we expect to receive from Roche if certain conditions are satisfied, interest earned on our investments, and additional capital to be raised through public or private equity offerings or debt financings. We will require significant additional financing in the future to fund our operations. Additional financing may not be available on acceptable terms, if at all. As of September 30, 2009, we had $58.4 million of cash and cash equivalents.
Revenues
All of our product candidates are currently in development and, therefore, we do not expect to generate any direct revenues from drug product sales for at least the next several years, if at all. Our revenues to date have been generated primarily from milestone payments under our collaboration agreements, license fees, and research funding. We currently have one collaboration agreement with Roche for the development of RG7128. We entered into our collaboration agreement with Roche in October 2004. Roche subsequently paid us an up-front payment of $8.0 million. Pursuant to the terms of our collaboration agreement with Roche, we received
58
Table of Contents
a $10.0 million milestone payment during the quarter ended June 30, 2009. As of September 30, 2009, we had received an aggregate of $44.5 million in payments under the Roche collaboration agreement, including research funding and related fees as well as up-front and milestone payments.
Under the current terms of the Roche collaboration agreement, if we succeed in obtaining all of the regulatory approvals specified in the agreement for RG7128, as of September 30, 2009 the maximum future development and commercialization milestone payments payable to us are $105.0 million. Receipt of any additional milestone payments depends on many factors, some of which are beyond our control. We cannot assure you that we will receive any of these future payments.
We expect our revenues for the next several years to be derived primarily from payments under our current collaboration agreement with Roche and any additional collaborations that we may enter into in the future. In addition to the payments described above, we may receive future royalties on product sales, if any, under our collaboration agreement with Roche.
Research and Development Expenses
Our research and development expenses consist primarily of salaries and related personnel expenses, fees paid to external service providers, up-front and milestone payments under our license agreements, patent-related legal fees, costs of preclinical studies and clinical trials, drug and laboratory supplies, and costs for facilities and equipment. We use external service providers to manufacture our product candidates for clinical trials and for the majority of our preclinical and clinical development work. We charge all research and development expenses to operations as they are incurred. Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided.
Our research activities are primarily focused on discovering and developing novel drugs to treat HCV. Our development activities are primarily focused on the development of RG7128 (in collaboration with Roche), PSI-7851, PSI-938, and PSI-879 for the treatment of HCV and, secondarily, on the development of Racivir for the treatment of HIV. We are responsible for all costs incurred in the clinical development of PSI-7851, PSI-938, PSI-879, and Racivir, as well as the research costs associated with our other internal research programs. On April 20, 2009, we voluntarily terminated our Phase 3 registration studies of clevudine for the treatment of HBV.
Under our collaboration with Roche, Roche will fund the clinical development and commercialization of RG7128. Under this collaboration, Roche reimbursed us for all of the external expenses associated with, and we were responsible for, certain preclinical work, the IND filing, and the proof-of-concept clinical trial. During December 2008, we transferred the IND application for RG7128 to Roche. Roche will continue to fund all of the expenses of, and be responsible for, other preclinical studies and future clinical development of RG7128 in the territories licensed to Roche. We and Roche will continue to jointly oversee all development and marketing activities of RG7128 in the territories licensed to Roche. Roche received a license only to PSI-6130 and its pro-drugs, including RG7128.
We use our internal research and development resources, including our employees and discovery infrastructure, across various projects. Our related internal expenses are not attributable to a specific project, but are directed to broadly applicable research activities. Accordingly, we do not account for our internal research and development expenses on a project basis. We use external service providers to manufacture our product candidates for clinical trials and for the substantial majority of our preclinical and clinical development work. We have tracked some of these external research and development expenses on a project basis. To the extent that expenses are not attributable to a specific project, they are included in one of the “unattributed expenses” in the table below.
59
Table of Contents
The following table summarizes our research and development expenses for our current development projects, as well as clevudine, for each of the three years ended September 30, 2009, 2008, and 2007.
| Years Ended September 30, | Cumulative Project Costs | |||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Expenses attributed to projects: |
||||||||||||
| Clevudine Studies |
$ | 26,714 | $ | 26,452 | $ | 10,050 | $ | 71,610 | ||||
| Preclinical PSI-7851 Studies |
1,251 | 665 | — | 1,916 | ||||||||
| Phase 1 PSI-7851 Studies |
5,640 | 105 | — | 5,745 | ||||||||
| Preclinical PSI-938 Studies |
1,219 | — | — | 1,219 | ||||||||
| Phase 1 PSI-938 Studies |
94 | — | — | 94 | ||||||||
| Preclinical PSI-879 Studies |
3 | — | — | 3 | ||||||||
| Phase 2 Racivir Studies |
32 | 331 | 962 | 4,237 | ||||||||
| Total attributed expenses |
34,953 | 27,553 | 11,012 | |||||||||
| Unattributed expenses |
||||||||||||
| Salaries and related personnel expenses |
8,014 | 6,386 | 2,592 | |||||||||
| Non-cash stock compensation expense |
2,494 | 2,110 | 1,035 | |||||||||
| Legal expenses associated with patents |
1,657 | 1,549 | 1,238 | |||||||||
| Preclinical studies and new drug discovery services |
1,863 | 1,844 | 1,723 | |||||||||
| Drug and laboratory supplies |
930 | 1,234 | 621 | |||||||||
| Consulting expense |
117 | 443 | 203 | |||||||||
| Facility and other expenses |
2,524 | 1,877 | 1,895 | |||||||||
| Total unattributed expenses |
17,599 | 15,443 | 9,307 | |||||||||
| Total research and development expenses |
$ | 52,552 | $ | 42,996 | $ | 20,319 | ||||||
We will continue to make determinations as to which programs to pursue and how much funding to direct to each program on an ongoing basis. These determinations will be made in response to the scientific and clinical success of each product candidate, as well as an ongoing assessment as to the product candidate’s commercial potential. We do not believe that it is possible at this time to accurately project total program-specific expenses through commercialization for any of our product candidates, as there are numerous factors associated with the successful commercialization of any of our product candidates, including future trial design and various regulatory requirements such as competitive final product labeling and reasonable risk management programs, many of which cannot be determined with accuracy at this time based on our stage of development. Product candidates that may appear promising at early stages of development may not reach the market for a number of reasons. For example, product candidates may be found ineffective or may cause harmful side effects during clinical trials, may take longer to progress through clinical trials than anticipated, may fail to receive necessary regulatory approvals, or may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality. The lengthy process of seeking FDA and other regulatory agency approvals requires the expenditure of substantial resources. Any failure or delay in obtaining regulatory approvals could materially adversely affect our product development effort and financial condition. Because of these and other risks and uncertainties, we cannot predict when or whether we will successfully complete the development of any of our product candidates or the ultimate product development cost or whether we will obtain any approval required by the FDA or other regulatory agencies on a timely basis, if at all.
As we obtain results from clinical trials, we may elect to discontinue or delay preliminary studies or clinical trials for a product candidate or development program in order to focus our resources on more promising product candidates or programs. We are in the process of terminating our Phase 3 registration studies of clevudine and reducing the related expenses, which were $3.3 million in the quarter ended September 30, 2009, compared to $8.9 million, $7.4 million and $7.1 million in the first three quarters of fiscal 2009, respectively. We expect our level of expenses for these studies to be lower during the quarter ending December 31, 2009 than the level of
60
Table of Contents
expenses during the quarter ended September 30, 2009. We anticipate having this study termination process and all related expenses complete by December 31, 2009.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational functions, including accounting, finance, legal, business development, investor relations, information technology, and human resources. Other significant general and administration costs include facilities costs and professional fees for outside accounting and legal services, travel, insurance premiums, and depreciation.
Results of Operations
Year Ended September 30, 2009 Compared with Year Ended September 30, 2008
Revenues. Revenues were $13.3 million and $1.9 million during 2009 and 2008, respectively. Revenues during 2009 include a $10.0 million milestone payment from Roche for initiating a Phase 2b study of RG7128 and $1.4 million of research and development payments from Roche for activities related to holding the IND application for RG7128, for which we have no continuing performance obligations. Our performance obligations relating to the $10.0 million milestone payment consisted of successfully completing a Phase 1 study of RG7128, which led to the initiation of the Phase 2b study for RG7128 that triggered the milestone payment. Revenues during 2009 and 2008 also include $1.8 million and $1.9 million, respectively, of amortization of up-front and subsequent collaborative and license payments received from Roche previously recorded as deferred revenue.
The following is a reconciliation between cash payments received under contract revenue agreements and contract revenues reported:
| Years Ended September 30, | ||||||
| 2009 | 2008 | |||||
| (In thousands) | ||||||
| Cash received/receivable |
$ | 11,509 | $ | — | ||
| Deferred |
— | — | ||||
| Amortization |
1,784 | 1,857 | ||||
| Revenues |
$ | 13,293 | $ | 1,857 | ||
Research and Development Expenses. Research and development expenses increased to $52.6 million during 2009 from $43.0 million in 2008. This net increase of $9.6 million consists primarily of a $6.1 million increase for preclinical and clinical trial costs for our HCV product candidate, PSI-7851, an increase in compensation expenses of $2.0 million ($0.4 million of which was non-cash stock compensation expense) resulting from an increase in headcount, an increase of $1.3 million in preclinical study costs for our HCV product candidate, PSI-938, and a $0.3 million increase in Phase 3 registration clinical trial expenses for clevudine. Partially offsetting this increase was a $0.1 net reduction in our other research and development expenses.
On April 20, 2009, we voluntarily terminated our Phase 3 registration studies of clevudine for the treatment of hepatitis B virus. We expect our level of expenses for these studies to be lower during the quarter ending December 31, 2009 than the $3.3 million of expenses during the quarter ended September 30, 2009. We anticipate having this study termination process and the related expenses complete by December 31, 2009.
General and Administrative Expenses. General and administrative expenses were $13.4 million during 2009, an increase of $0.1 million from $13.3 million in 2008. The increase of $0.1 million was due primarily to increases in compensation expense of $1.7 million ($0.6 million of which was non-cash stock compensation expense), and marketing expense of $0.2 million. Partially offsetting this increase were reductions of $0.5 million in travel expenses, $0.3 million in audit and related fees (including consulting fees in support of our
61
Table of Contents
compliance with Section 404 of the Sarbanes-Oxley Act of 2002), $0.3 million in legal fees, $0.2 million in insurance expense, $0.2 million in recruiting expense, and $0.3 million in other administrative expenses.
Investment Income. Investment income decreased to $0.2 million in 2009 from $2.0 million in 2008. The decrease was primarily due to significantly lower rates of return on the average invested cash balances.
Interest Expense. Interest expense increased to $3.2 million in 2009 from $2.2 million in 2008. The increase in interest expense was due to interest on additional borrowings of long-term debt of $13.3 million ($10.0 million on March 28, 2008, and $3.3 million on December 12, 2008).
Income Taxes. As of September 30, 2009, we had United States federal net operating loss carryforwards of approximately $144.9 million available to offset future taxable income, if any. Of the federal net operating losses, $10.5 million was generated from windfall tax benefit stock option deductions. The tax benefit of this portion of the net operating loss will be accounted for directly to equity as additional paid in capital as the stock option related losses are utilized. As of September 30, 2009 we also had research and development tax credits of approximately $0.1 million available to offset future tax liabilities. As of September 30, 2009, we had a net deferred tax asset of $57.1 million, before consideration of a valuation allowance. We established a full valuation allowance on our net deferred tax asset as it is more likely than not that such tax benefits will not be realized. The loss carryovers and the research and development tax credits expire over a period of 2020 to 2029.
Under Section 382 of the Internal Revenue Code (the “Code”), utilization of the net operating loss and research and development tax credit carryforwards may be subject to a limitation if a change in ownership of the Company, as defined in the Code, occurred previously or could occur in the future. The Company completed a Section 382 analysis regarding limitation of its net operating loss and research and development credit carryforwards that covered the period three years prior to its IPO on May 2, 2007 through its most recent follow-on public offering of its common stock on February 5, 2009, and concluded that a change in control occurred at the Company during the quarter ended September 30, 2008. This change in control limits the future use of the Company’s net operating loss and research and development credit carryforwards from fiscal 2008 and prior years. However, based upon the Company’s financial projections, it does not believe that this limitation will result in the expiration of any of these net operating loss and research and development credit carryforwards before they are able to be utilized. Changes in ownership subsequent to this period could lead to a change in the limitation in future years, which could impact the use of the Company’s net operating loss and research and development credit carryforwards generated in the affected years. Any limitation may result in expiration of a portion of the NOL or research and development credit carryforwards before utilization, which would reduce the Company’s gross deferred tax assets. The Company will continue to monitor changes in its ownership and will update its Section 382 analysis when it believes a change in control may have occurred.
Year Ended September 30, 2008 Compared with Year Ended September 30, 2007
Revenues. Revenues were $1.9 million and $22.0 million during 2008 and 2007, respectively. Revenues during 2008 and 2007 reflect amortization of upfront and subsequent collaborative and license payments received from Roche previously recorded as deferred revenue of $1.9 million and $2.0 million, respectively, and the revenues from 2007 include milestone payments totaling $20.0 million received pursuant to our Roche collaboration.
62
Table of Contents
The following is a reconciliation between cash payments received under contract revenue agreements and contract revenues reported:
| Years Ended September 30, |
|||||||
| 2008 | 2007 | ||||||
| (in thousands) | |||||||
| Cash received/receivable |
$ | — | $ | 20,425 | |||
| Deferred |
— | (375 | ) | ||||
| Amortization |
1,857 | 1,959 | |||||
| Revenues |
$ | 1,857 | $ | 22,009 | |||
Research and Development Expenses. Research and development expenses increased to $43.0 million during 2008 from $20.3 million in 2007. This net increase of $22.7 million consists primarily of a $16.5 million increase in Phase 3 registration clinical trial expenses for clevudine, an increase in compensation expenses of $4.0 million ($1.1 million of which was non-cash stock compensation expense) resulting from an increase in headcount, and a $2.7 million increase in new drug discovery expenses, including PSI-7851. Partially offsetting this increase was a $0.5 million reduction in Phase 2 clinical trial expenses for Racivir during 2008, compared to the same period in 2007.
General and Administrative Expenses. General and administrative expenses were $13.3 million during 2008, an increase of $4.1 million from $9.2 million in 2007. The increase of $4.1 million was due primarily to increases in compensation expenses of $0.9 ($0.2 million of which was non-cash stock compensation expense), legal expenses of $0.7 million, insurance expenses of $0.6 million, audit and related fees of $0.6 million (including consulting fees in support of our compliance with Section 404 of the Sarbanes-Oxley Act of 2002), marketing expenses of $0.5 million, facility expenses of $0.2 million, and miscellaneous administrative expenses of $0.6 million.
Investment Income. Investment income decreased to $2.0 million in 2008 from $2.5 million in 2007. The decrease was primarily due to lower rates of return on the average invested cash balances.
Interest Expense. Interest expense increased to $2.2 million in 2008 from $0.0 million in 2007. The $2.2 million of interest expense in 2008 is from $20.0 million of long-term debt we incurred during 2008 ($10.0 million on October 5, 2007 and $10.0 million on March 28, 2008).
Income Taxes. As of September 30, 2008, we had United States federal net operating loss carryforwards of approximately $88.0 million available to offset future taxable income, if any. Of the federal net operating losses, $8.8 million was generated from windfall tax benefit stock option deductions. The tax benefit of this portion of the net operating loss will be accounted for directly to equity as additional paid in capital as the stock option related losses are utilized. As of September 30, 2008 we also had research and development tax credits of approximately $0.1 million available to offset future tax liabilities. As of September 30, 2008, we had a net deferred tax asset of $36.9 million, before consideration of a valuation allowance. We established a full valuation allowance on our net deferred tax asset as it is more likely than not that such tax benefits will not be realized. The loss carryovers and the research and development tax credits expire over a period of 2020 to 2029.
Preferred Stock Accretion. Preferred stock accretion was $1.8 million in 2007. There has been no redeemable preferred stock accretion subsequent to May 2, 2007 (when we completed our IPO) because all of the redeemable preferred stock outstanding was converted into common stock upon completion of our IPO.
Liquidity and Capital Resources
Since our inception, we have funded our operations primarily through public and private offerings of our equity securities, payments received under our collaboration agreements, and borrowings under our Loan Agreement. Since our inception, we have raised approximately $174.1 million in net proceeds from sales of our
63
Table of Contents
equity securities, including $43.4 million from our registered direct common stock offering completed on February 5, 2009. During the year ended September 30, 2009, we borrowed $3.3 million in addition to the $20.0 million we previously borrowed under our Loan Agreement entered into on September 30, 2007. At September 30, 2009, we held $58.4 million in cash and cash equivalents and have invested substantially all of our available cash and cash equivalents in a mutual fund, which invests in short-term U.S. Treasury and Agency Obligations, or in a money fund account.
Net cash provided by (used in) operating activities was ($49.5 million), ($52.1 million), and $0.9 million during the years ended September 30, 2009, 2008, and 2007, respectively. The $2.6 million decrease in net cash used in operating activities during 2009, as compared to 2008, was due primarily to favorable changes in our working capital. The $53.0 million increase in net cash used in operating activities during 2008, as compared to 2007, was due to (1) a decrease in revenues of $20.2 million, as the revenues from 2007 include milestone payments totaling $20.0 million from Roche, (2) an increase in cash outflows for operating expenses (including interest expense) of $27.6 million primarily resulting from our Phase 3 clinical trials for clevudine, and (3) an increase of $5.2 million of cash outflows associated with changes in operating assets and liabilities.
Net cash (used in) provided by investing activities was $0.1 million, ($0.2 million), and ($0.4 million) during the years ended September 30, 2009, 2008, and 2007, respectively. The net cash provided by investing activities during 2009 of $0.1 million resulted from the maturity of $0.5 million of short-term investments that was mostly offset by $0.3 million of purchases of equipment and leasehold improvements, and $0.1 million of restricted cash used as collateral for a letter of credit. The net cash used in investing activities during 2008 of $0.2 million consists of $0.9 million of purchases of equipment that were partially offset by cash proceeds received from the maturity of certain short-term investments of $0.7 million. The net cash used in investing activities in 2007 of $0.4 million was for the purchases of equipment.
Net cash provided by financing activities was $44.7 million, $46.6 million, and $42.1 million during the years ended September 30, 2009, 2008, and 2007, respectively. The net cash provided by financing activities during 2009 includes $43.4 million of net proceeds, after deducting placement agent fees and offering expenses, from the registered direct public offering we completed in February 2009, borrowings of long-term debt of $3.3 million, and proceeds from the exercise of stock options of $1.2 million, that were partially offset by principal payments on long-term debt and capital lease obligations of $3.3 million. The net cash provided by financing activities during the year ended September 30, 2008 includes $24.1 million of net proceeds, after deducting placement agent fees and offering expenses, from the registered direct public offering we completed in July 2008, borrowings of long-term debt of $20.0 million under the Loan Agreement we entered into during September 2007, and proceeds from the exercise of stock options of $2.6 million. The net cash provided by financing activities during 2007 includes net proceeds from our IPO of $40.7 million after deducting offering costs paid during fiscal 2007, along with proceeds from the exercise of stock options of $1.6 million.
On September 30, 2007, we entered into a Loan Agreement that allowed us to borrow up to $30.0 million in $10.0 million increments. We borrowed the first and second $10.0 million increments by signing two Secured Promissory Notes (“Notes A and B”) on October 5, 2007 and March 28, 2008. Notes A and B bear interest at 12%. On December 12, 2008, we amended the Loan Agreement and borrowed $3.3 million by signing a Secured Promissory Note (“Note C”). Note C bears interest at 12.5%. Notes A, B, and C are to be repaid over a 45-month period with the first 15 monthly payments representing interest only followed by 30 equal monthly payments of principal and interest. The principal monthly payments on each of the notes begin and end as follows:
| Note |
Begin | End | ||
| Note A |
March 1, 2009 | August 1, 2011 | ||
| Note B |
August 1, 2009 | January 1, 2012 | ||
| Note C |
May 1, 2010 | October 1, 2012 |
Prepayment of the loans made pursuant to the Loan Agreement is subject to penalty and substantially all of our tangible and intangible assets (except for intellectual property) are collateral for the Loan Agreement. Future
64
Table of Contents
total principal repayments of the three Notes amount to $7.5 million in fiscal 2010, $9.5 million in fiscal 2011, $3.0 million in fiscal 2012, and $0.1 million in fiscal 2013. There are no additional borrowings available under the Loan Agreement.
Under the Loan Agreement, we agreed that in the event our market capitalization is below $90.0 million for 15 consecutive days in which the principal market for our common stock is open for trading to the public, we will be required to repay 50% of the then outstanding principal balance of the loans. We further agreed that in the event our market capitalization is below $40.0 million for 15 consecutive days in which the principal market for our common stock is open for trading to the public, we will be required to repay all of the then outstanding principal balance of the loans.
The Loan Agreement also contains covenants that, among other things, require us to obtain consent from the lender prior to paying dividends, making certain investments, changing the nature of our business, assuming or guaranteeing the indebtedness of another entity or individual, selling or otherwise disposing of a substantial portion of our assets, or merging or consolidating with another entity.
Developing drugs, conducting clinical trials and commercializing products is expensive and we will need to raise additional funds to achieve our strategic objectives. Although we believe our existing cash resources, together with anticipated payments under our existing collaboration agreement will be sufficient to fund our projected cash requirements for the next 12 months, we will require significant additional financing in the future to complete our clinical trials for PSI-7851, PSI-938, and PSI-879, to fund our portion, if any, of the cost of clinical trials for RG7128 completed outside of the territories licensed by Roche, and to fund our other operations. Additional financing may not be available on acceptable terms, if at all. Our future capital requirements will depend on many factors, including:
| • | the progress and costs of our preclinical studies, clinical trials, and other research and development activities; |
| • | the scope, prioritization, and number of our clinical trials and other research and development programs; |
| • | the amount of revenues we receive under our existing collaboration agreement and any future collaboration agreements; |
| • | the costs of the development and expansion of our operational infrastructure; |
| • | the costs and timing of obtaining regulatory approval of our product candidates; |
| • | the ability of our collaborators to achieve development milestones, marketing approval, and other events or developments under our collaboration agreements; |
| • | the costs of filing, prosecuting, enforcing, and defending patent claims and other intellectual property rights; |
| • | the costs and timing of securing manufacturing arrangements for clinical or commercial production; |
| • | the costs of establishing sales and marketing capabilities or contracting with third parties to provide these capabilities for us; |
| • | the costs of acquiring or undertaking development and commercialization efforts for any future product candidates; |
| • | the magnitude of our general and administrative expenses; and |
| • | any costs that we may incur under current and future licensing arrangements relating to our product candidates. |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through payments received under our collaborations, debt or equity financings, or by out-licensing product candidates.
65
Table of Contents
We cannot be certain that additional funding will be available to us on acceptable terms, or at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts.
Contractual Obligations and Commitments
On May 23, 2005, we entered into an operating lease for office and laboratory space through May 22, 2010, in Princeton, New Jersey. On June 2, 2009, we entered into an extension and modification of the lease, extending the term of the lease for an additional five years, from May 22, 2010 through May 22, 2015. The annual occupancy expense under this lease is approximately $825,000. We also lease office space in Durham, North Carolina. Monthly lease payments began May 1, 2007 and, after amending the lease term on February 2, 2009, end on April 1, 2011. The annual occupancy expense under this lease is approximately $81,000. We executed three secured promissory notes totaling $23.3 million; $10.0 million in October 2007, $10.0 million in March 2008, and $3.3 million in December 2008. The secured promissory notes require payments of interest only for the first 15 months followed by 30 equal monthly payments of principal and interest. As of September 30, 2009, future payments under the three promissory notes and minimum future payments under non-cancellable operating leases (including the two lease extensions noted above) are as follows:
| Payments Due By Period | |||||||||||||||
| Total | Less than 1 year |
1-3 Years | 4-5 Years | After 5 Years | |||||||||||
| (In thousands) | |||||||||||||||
| Debt obligations |
|||||||||||||||
| Debt maturities |
$ | 20,080 | $ | 7,513 | $ | 12,439 | $ | 128 | $ | — | |||||
| Contractual interest |
3,013 | 1,870 | 1,142 | 1 | — | ||||||||||
| Capital lease obligations |
|||||||||||||||
| Debt maturities |
— | — | — | — | — | ||||||||||
| Contractual interest |
— | — | — | — | — | ||||||||||
| Operating leases |
4,640 | 714 | 2,554 | 1,372 | |||||||||||
| Purchase obligations |
— | — | — | — | — | ||||||||||
| Total contractual obligations |
$ | 27,733 | $ | 10,097 | $ | 16,135 | $ | 1,501 | $ | — | |||||
The above contractual obligations table does not include amounts for milestone payments related to development, regulatory, or commercialization events to licensors or collaboration partners, as the payments are contingent on the achievement of these milestones, which we have not achieved. Under our collaboration and license agreement with Bukwang for clevudine, up to an aggregate of $23.0 million in milestone payments are payable in the future if certain development, regulatory, and commercialization events occur, which are not expected to occur due to our termination of the registration studies of clevudine. Under our license agreement with Emory University for Racivir, we agreed to pay Emory University up to an aggregate of $1.0 million in future marketing milestone payments.
Off-Balance Sheet Transactions
To date, we have not had any relationships with unconsolidated entities or financial partnerships, such as entities referred to as structured finance or special purpose entities, which are established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the
66
Table of Contents
reported amounts of assets, liabilities, and expenses and related disclosures. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Our actual results may differ substantially from these estimates under different assumptions or conditions. Our significant accounting policies are described in more detail in Note 2 of the Notes to Financial Statements included elsewhere in this Annual Report on Form 10-K; however, we believe that the following accounting policies are critical to the judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
We recognize revenues when all of the following four criteria are present: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectibility is reasonably assured. For arrangements that involve the delivery or performance of multiple products, services, and/or rights to use assets, the elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the collaborator and whether there is objective and reliable evidence of the fair value of the undelivered obligation(s). The consideration received is allocated among the separate units either on the basis of each unit’s fair value or using the residual method, and the applicable revenue recognition criteria are applied to each of the separate units.
Our revenues are primarily related to our collaboration agreement with Roche. This agreement provides for various types of payments to us, including non-refundable upfront license fees, research and development payments, and milestone payments.
Where we have continuing performance obligations under the terms of a collaboration agreement, non-refundable upfront license payments received upon contract signing are recorded as deferred revenue and recognized as revenue as the related activities are performed. The period over which these activities are to be performed is based upon our estimate of the development period. Changes in our estimate could change the period over which revenues are recognized. Research and/or development payments are recognized as revenues as the related research and/or development activities are performed and when we have no continuing performance obligations related to the research and development payment received.
We recognize revenue from milestone payments when earned, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement and (ii) we do not have ongoing performance obligations related to the achievement of the milestone earned. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement and the related risk associated with the achievement of the milestone. Any amounts received under the agreements in advance of performance, if deemed substantive, are recorded as deferred revenue and recognized as revenue as we complete our performance obligations.
Where we have no continuing involvement under a collaborative arrangement, we record nonrefundable license fee revenues when we have the contractual right to receive the payment, in accordance with the terms of the license agreement, and records milestones upon appropriate notification to us of achievement of the milestones by the collaborative partner.
Deferred revenue associated with a non-refundable payment received under a collaborative agreement that is terminated prior to its completion results in an immediate recognition of the deferred revenues.
Accrued Expenses
We are required to estimate accrued expenses as part of our process of preparing financial statements. This process involves estimating the level of service performed on our behalf and the associated cost incurred in instances where we have not been invoiced or otherwise notified of actual costs. Examples of areas in which
67
Table of Contents
subjective judgments may be required include costs associated with services provided by contract organizations for preclinical development, clinical trials and manufacturing of clinical materials. We account for expenses associated with these external services by determining the total cost of a given study based on the terms of the related contract. We accrue for costs incurred as the services are being provided by monitoring the status of the trials and the invoices received from our external service providers. In the case of clinical trials, the estimated cost normally relates to the projected costs of having subjects enrolled in our trials, which we recognize over the estimated term of the trial according to the number of patients enrolled in the trial on an ongoing basis, beginning with patient enrollment. As actual costs become known to us, we adjust our accruals. To date, the number of clinical trials and related research service agreements has been relatively limited and our estimates have not differed significantly from the actual costs incurred. We expect, however, as clinical trials for PSI-7851, PSI-938, and PSI-879 advance, that our estimated accruals for clinical and research services will be more material to our operations in future periods.
Stock-based Compensation
We recognize stock compensation expense for awards of equity instruments to employees and directors based on the grant-date fair value of those awards (with limited exceptions). The grant-date fair value of the award is recognized as compensation expense over the life of the equity instruments issued. Equity instruments granted to consultants are periodically valued and recorded as stock compensation expense as the equity instrument vests.
Stock-based compensation expense is included in both research and development expenses and in general and administrative expenses in the statements of operations and comprehensive net income (loss). Since our stock was not publicly traded prior to April 27, 2007, the expected volatility was calculated for each equity award granted based on the “peer method.” We identified companies that traded publicly within the pharmaceutical industry that had similar SIC codes, employee count and revenues. Prior to October 1, 2006, we had chosen the weekly high price volatility for these companies for a period of five years. Subsequent to October 1, 2006, we have used the weekly high price for these companies for a period of six years to coincide with the expected term.
Recently Adopted Accounting Pronouncements
In May 2009, the Financial Accounting Standards Board (“FASB”) issued guidance that incorporates the accounting and disclosure requirements for subsequent events into U.S. generally accepted accounting principles. This guidance also introduces new terminology, defines a date through which management must evaluate subsequent events, and lists the circumstances under which an entity must recognize and disclose events or transactions occurring after the balance-sheet date. We adopted this guidance as of June 30, 2009, which was the required effective date, and its adoption did not affect our financial statements, other than the disclosures required by it, which can be found in Note 1 – Description of Business and Basis of Presentation.
In June 2009, the FASB issued guidance which divides nongovernmental U.S. GAAP into the authoritative Codification and guidance that is nonauthoritative. The Codification is not intended to change U.S. GAAP; however, it does significantly change the way in which accounting literature is organized and because it completely replaces existing standards, it will affect the way U.S. GAAP is referenced by most companies in their financial statements and accounting policies. The Codification is effective for financial statements issued for interim and annual periods ending after September 15, 2009. The adoption of the Codification did not have an impact on our consolidated financial statements.
Recently Issued Accounting Pronouncements
In September 2009, the FASB Emerging Issues Task Force, or EITF, issued guidance on accounting for multiple-deliverable revenue arrangements that provides principles and application guidance on whether multiple deliverables exist, how the arrangement should be separated and the consideration allocated. This guidance
68
Table of Contents
requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. The update eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. This guidance should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. We are currently evaluating the impact, if any, of adopting this guidance on our financial statements.
In June 2009, the FASB issued guidance to replace the quantitative-based risks and rewards calculation for determining which enterprise, if any, has a controlling financial interest in a variable interest entity and requires on-going reassessments of whether an enterprise is the primary beneficiary of a variable interest entity. It also requires the elimination of the quantitative approach for determining the primary beneficiary of a variable interest entity and amends certain guidance for determining whether an entity is a variable interest entity requiring enhanced disclosure that will provide users of financial statements with more transparent information about an enterprise’s involvement in a variable interest entity. Additionally, an enterprise is required to assess whether it has an implicit financial responsibility to ensure that a variable interest entity operates as designed when determining whether it has the power to direct the activities of the variable interest entity that most significantly impact the entity’s economic performance. This guidance is effective for financial statements issued for fiscal years after November 15, 2009. The adoption of this guidance is not expected to have a material impact on us.
In December 2007, the EITF issued guidance on accounting for collaborative arrangements. The EITF concluded on the definition of a collaborative arrangement and that revenues and costs incurred with third parties in connection with collaborative arrangements would be presented gross or net based on other accounting literature. Based on the nature of the arrangement, payments to or from collaborators would be evaluated and the terms, the nature of the entity’s business, and whether those payments are within the scope of other accounting literature would be presented. Companies are also required to disclose the nature and purpose of collaborative arrangements along with the accounting policies and the classification and amounts of significant financial statement amounts related to the arrangements. Activities in the arrangement conducted in a separate legal entity should be accounted for under other accounting literature; however the required disclosures above would apply to the entire collaborative agreement. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2008, and interim periods within those fiscal years, and is to be applied retrospectively to all periods presented for all collaborative arrangements existing as of the effective date. The adoption of this guidance is not expected to have a material impact on us.
In December 2007, the FASB issued guidance which changes the accounting for business acquisitions. This guidance requires the acquiring entity in a business combination to recognize all (and only) the assets acquired and liabilities assumed in the transaction and establishes the acquisition-date fair value as the measurement objective for all assets acquired and liabilities assumed in a business combination. Certain provisions of this standard will, among other things, impact the determination of acquisition-date fair value of consideration paid in a business combination (including contingent consideration); exclude transaction costs from acquisition accounting; and change accounting practices for acquired contingencies, acquisition-related restructuring costs, in-process research and development, indemnification assets, and tax benefits. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2008. The adoption of this guidance is not expected to have a material impact on us.
In December 2007, the FASB issued guidance which establishes new standards governing the accounting for and reporting of noncontrolling interests (“NCIs”) in partially owned consolidated subsidiaries and the loss of control of subsidiaries. Certain provisions of this standard indicate, among other things, that NCIs (previously referred to as minority interests) be treated as a separate component of equity, not as a liability; that increases and decrease in the parent’s ownership interest that leave control intact be treated as equity transactions, rather than as step acquisitions or dilution gains or losses; and that losses of a partially owned consolidated subsidiary be
69
Table of Contents
allocated to the NCI even when such allocation might result in a deficit balance. This standard also requires changes to certain presentation and disclosure requirements. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2008. The provisions of the standard are to be applied to all NCIs prospectively, except for the presentation and disclosure requirements, which are to be applied retrospectively to all periods presented. The adoption of this guidance is not expected to have a material impact on us.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Interest Rate Risk
We invest our excess cash in high quality, interest-bearing securities. The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we invest in highly liquid mutual and money market funds, and high quality marketable debt instruments of corporations, government agencies and financial institutions with maturities of less than two years. If a 10% change in interest rates were to have occurred on September 30, 2009, this change would not have had a material effect on the fair value of our investment portfolio as of that date. In addition, our secured promissory notes have fixed interest rates of 12% or 12.5%.
Foreign Currency Exchange Rate Risk
We have entered into some agreements denominated, wholly or partly, in foreign currencies, and, in the future, we may enter into additional, agreements denominated in foreign currencies. If the values of these currencies increase against the United States dollar, our costs would increase. To date, we have not entered into any contracts to reduce the risk of fluctuations in currency exchange rates. In the future, depending upon the amounts payable under any such agreements, we may enter into forward foreign exchange contracts to reduce the risk of unpredictable changes in these costs. However, due to the variability of timing and amount of payments under any such agreements, foreign exchange contracts may not mitigate the potential adverse impact on our financial results.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this Item is included in our Financial Statements and Supplementary Data listed in Item 15 of Part IV of this Annual Report on Form 10-K.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as such term is defined under Rule 13a-15(e) promulgated under the Exchange Act), as of September 30, 2009. Based on that evaluation, our principal executive officer and principal financial officer concluded that these controls and procedures are effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported as specified in SEC rules and forms. There were no changes in these controls or procedures identified in connection with the evaluation of such controls or procedures that occurred during our last fiscal quarter, or in other factors that have materially affected, or are reasonably likely to materially affect, these controls or procedures.
70
Table of Contents
Our disclosure controls and procedures are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in the rules and forms of the SEC. These disclosure controls and procedures include, among other things, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, our principal executive and principal financial and accounting officers and effected by our board of directors and management to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| • | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. generally accepted accounting principles, and that receipts and expenditures of our company are being made only in accordance with authorizations of our management and board of directors; and |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of September 30, 2009. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework. Based on our assessment, our management believes that, as of September 30, 2009, our internal control over financial reporting is effective. In addition, no changes in our internal control over financial reporting have occurred during the three months ended September 30, 2009 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Grant Thornton LLP, the independent registered public accounting firm that audited our financial statements included elsewhere in this Annual Report on Form 10-K, has issued an attestation report on our internal control over financial reporting. That report appears in Item 15 of Part IV of this Annual Report on Form 10-K and is incorporated by reference to this Item 9A.
| ITEM 9B. | OTHER INFORMATION |
Not applicable.
71
Table of Contents
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information regarding executive officers and directors required by this Item 10 will be included in the definitive Proxy Statement for our 2010 Annual Meeting, or 2010 Proxy Statement, under “Election of Directors”, “Executive Officers of the Company” and “Governance of the Company” and is incorporated herein by reference. Other information required by this Item 10 will be included in the 2010 Proxy Statement under “Section 16(a) Beneficial Ownership Reporting Compliance” and “Code of Ethics” and is incorporated herein by reference.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this item is incorporated herein by reference to the information contained under the sections captioned “Information About Executive and Director Compensation” and “Compensation Committee Interlocks and Insider Participation” of the 2010 Proxy Statement.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this item is incorporated herein by reference to the information contained under the sections captioned “Stock Ownership of Certain Beneficial Owners and Management” and “Securities Authorized for Issuance Under Our Equity Incentive Plans” of the 2010 Proxy Statement.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this item is incorporated herein by reference to the information contained under the sections captioned “Certain Relationships and Related Transactions” and “Board Determination of Independence” of the 2010 Proxy Statement.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this item is incorporated herein by reference to the information contained under the sections captioned “Fees of Independent Registered Public Accounting Firm” and “Pre-Approval Policies and Procedures” of the 2010 Proxy Statement.
72
Table of Contents
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a) (1) Financial Statements
The following documents are included on pages F-2 through F-28 attached hereto and are filed as part of this Annual Report on Form 10-K.
(a) (2) Financial Statement Schedules
Financial Statement Schedules have been omitted because they are either not applicable or the required information is included in the financial statements or notes thereto.
(a) (3) List of Exhibits
The following is a list of exhibits filed as part of this Annual Report on Form 10-K. We are incorporating by reference to our previous SEC filings each exhibit that contains a footnote. For exhibits incorporated by reference, the location of the exhibit in the previous filing is indicated in parentheses.
| Exhibit Number |
Description | |
| 3.1** | Third Amended and Restated Certificate of Incorporation of the Registrant (Exhibit 3.1) (5) | |
| 3.2** | Second Amended and Restated Bylaws, as amended, of Pharmasset, Inc. (Exhibit 3.1) (11) | |
| 4.1**# | Pharmasset, Ltd. 1998 Stock Plan, as amended (Exhibit 4.4) (3) | |
| 4.2**# | 2007 Equity Incentive Plan, as amended on September 23, 2009 (Exhibit 4) (14) | |
| 4.3**# | Form of agreement for awards under the 2007 Equity Incentive Plan (Exhibit 4.3) (5) | |
| 10.1**† | Collaboration Agreement, dated October 29, 2004, between F. Hoffmann-La Roche Ltd. and Hoffmann-La Roche Inc. and the Registrant (Exhibit 10.1) (4) | |
| 10.2**† | License Agreement, dated June 23, 2005, between Bukwang Pharm. Co., Ltd. and the Registrant (Exhibit 10.2) (4) | |
| 10.3** | Memorandum of Understanding, dated June 23, 2005, between The University of Georgia Research Foundation, Inc., Yale University and the Registrant (Exhibit 10.3) (1) | |
| 10.4**† | Non-Exclusive Sublicense Agreement, dated August 26, 2005, between Apath, L.L.C. and the Registrant (Exhibit 10.5) (4) | |
| 10.5**† | License Agreement, dated December 8, 1998, between Emory University and Pharmasset, Ltd. (Exhibit 10.9) (1) | |
73
Table of Contents
| Exhibit Number |
Description | |
| 10.6** | Termination and Reinstatement Agreement, dated as of June 9, 1999, between Emory University and Pharmasset, Ltd. (Exhibit 10.13) (1) | |
| 10.7** | Supplemental Agreement to the License Agreement, dated as of March 26, 2004, between Emory University and Pharmasset, Ltd. (Exhibit 10.14) (1) | |
| 10.8**# | Employment Agreement, dated as of June 15, 2004, between the Registrant and Peter Schaefer Price (Exhibit 10.15) (1) | |
| 10.9** | Lease, dated as of May 18, 2005, between 300 CRA LLC and the Registrant (Exhibit 10.18) (1) | |
| 10.10**# | Form of Indemnity Agreement for Directors and Officers (Exhibit 10.21) (3) | |
| 10.11**# | Consulting Agreement, dated June 28, 2005, between Michael K. Inouye and the Registrant (Exhibit 10.22) (1) | |
| 10.12**# | Form of Change of Control Severance Agreement (Exhibit 10.23) (2) | |
| 10.13**# | Severance Agreement, dated as of January 5, 2007, between the Registrant and Abel De La Rosa, Ph.D. (Exhibit 10.24) (2) | |
| 10.14** | Venture Loan and Security Agreement dated September 30, 2007 by and between the Registrant and Horizon Technology Funding V LLC (Exhibit 10.23) (8) | |
| 10.15** | Manufacturing Services Agreement dated August 8, 2008 between Pharmasset, Inc. and Boehringer Ingelheim Chemicals, Inc. (Exhibit 10.24) (9) | |
| 10.16** | License Agreement dated August 8, 2008 between Pharmasset, Inc. and Boehringer Ingelheim Chemicals, Inc. (Exhibit 10.25) (9) | |
| 10.17** | Secured Promissory Note, dated as of October 5, 2007, between the Registrant and Horizon Technology Funding Group V LLC (Exhibit 10.1) (6) | |
| 10.18** | Secured Promissory Note, dated as of March 28, 2008, between the Registrant and Horizon Technology Funding Group V LLC (Exhibit 10.1) (7) | |
| 10.19** | First Amendment of Venture Loan and Security Agreement and Warrant, dated as of December 12, 2008 between the Registrant and Horizon Technology Funding Group V LLC (Exhibit 10.1) (10) | |
| 10.20** | Secured Promissory Note, dated as of December 12, 2008, between the Registrant and Horizon Technology Funding Group V LLC (Exhibit 10.2) (11) | |
| 10.21** | Amendment No. 1 to License Agreement dated January 30, 2009, between Bukwang Pharm. Co. Ltd. and the Registrant (Exhibit 10.1) (15) | |
| 10.22** | First Extension and Modification of Lease dated June 2, 2009, by and between 300 CRA LLC and Pharmasset, Inc. (Exhibit 10.1) (16) | |
| 23.1 | Consent of Grant Thornton LLP | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended | |
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
74
Table of Contents
| **—Filed | Previously |
| †— | Portions of the Exhibit were omitted and filed separately with the Secretary of the SEC pursuant to an order of the SEC granting our application for confidential treatment filed pursuant to Rule 406 under the Securities Act. |
| #—Management | contract or compensatory plan or arrangement. |
| (1)— | Filed as an Exhibit to our Registration Statement on Form S-1 filed with the SEC on May 8, 2006. |
| (2)— | Filed as an Exhibit to our Registration Statement on Form S-1/A filed with the SEC on January 17, 2007. |
| (3)— | Filed as an Exhibit to our Registration Statement on Form S-1/A filed with the SEC on March 2, 2007. |
| (4)— | Filed as an Exhibit to our Registration Statement on Form S-1/A filed with the SEC on April 24, 2007. |
| (5)— | Filed as an Exhibit to our Annual Report on Form 10-K filed with the SEC on December 31, 2007. |
| (6)— | Filed as an Exhibit to our Current Report on Form 8-K filed with the SEC on October 11, 2007. |
| (7)— | Filed as an Exhibit to our Current Report on Form 8-K filed with the SEC on March 28, 2008. |
| (8)— | Filed as an Exhibit to our Annual Report on Form 10-K filed with the SEC on December 11, 2008. |
(9) and (10) Filed as an Exhibit to our Current Report on Form 8-K filed with the SEC on December 18, 2008.
| (11)—Filed | as an Exhibit to our Current Report on Form 8-K filed with the SEC on January 21, 2009. |
| (12)— | Filed as an Exhibit to our Current Report on Form 8-K filed with the SEC on February 5, 2009. |
| (13)— | Filed as an Exhibit to our Current Report on Form 8-K filed with the SEC on June 17, 2009. |
| (14)— | Filed as an Exhibit to our Registration Statement on Form S-8 filed with the SEC on October 6, 2009. |
75
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PHARMASSET, INC. | ||||||||
| November 25, 2009 | By: | /s/ P. SCHAEFER PRICE | ||||||
| P. Schaefer Price Chief Executive Officer | ||||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Each person, in so signing also makes, constitutes, and appoints Kurt Leutzinger and Paul Lubetkin as his true and lawful attorney-in-fact, with full power of substitution, in his name, place, and stead, to execute and cause to be filed with the Securities and Exchange Commission any or all amendments to this report.
| Name |
Title |
Date | ||
| /S/ P. SCHAEFER PRICE P. Schaefer Price |
Director, President and Chief Executive Officer (Principal Executive Officer) | November 25, 2009 | ||
| /S/ KURT LEUTZINGER Kurt Leutzinger |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | November 25, 2009 | ||
| /S/ G. STEVEN BURRILL G. Steven Burrill |
Chairman of the Board of Directors | November 25, 2009 | ||
| /S/ WILLIAM J. CARNEY William J. Carney, Esq. |
Director | November 25, 2009 | ||
| /S/ FREDRIC D. PRICE Fredric D. Price |
Director | November 25, 2009 | ||
| /S/ ELLIOT F. HAHN Elliot F. Hahn, Ph.D. |
Director | November 25, 2009 | ||
| /S/ MICHAEL K. INOUYE Michael K. Inouye |
Director | November 25, 2009 | ||
| /S/ ROBERT F. WILLIAMSON Robert F. Williamson III |
Director | November 25, 2009 | ||
| /S/ HERBERT J. CONRAD Herbert J. Conrad |
Director | November 25, 2009 | ||
76
Table of Contents
| Page | ||
| F-2, F-3 | ||
| Financial Statements as of September 30, 2009 and 2008, and for the Years Ended September 30, 2009, 2008, and 2007: |
||
| F-4 | ||
| F-5 | ||
| F-6 | ||
| F-7 | ||
| F-8 | ||
| F-9 |
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Pharmasset, Inc.
We have audited the accompanying balance sheets of Pharmasset, Inc. (the “Company”) as of September 30, 2009 and 2008, and the related statements of operations and comprehensive net loss, redeemable stock and warrants, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended September 30, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Pharmasset, Inc. as of September 30, 2009 and 2008, and the results of its operations and its cash flows for each of the three years in the period ended September 30, 2009 in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Pharmasset Inc.’s internal control over financial reporting as of September 30, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated November 25, 2009 expressed an unqualified opinion.
/s/ GRANT THORNTON LLP
Philadelphia, Pennsylvania
November 25, 2009
F-2
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Pharmasset, Inc.
We have audited Pharmasset, Inc.’s (the “Company”) internal control over financial reporting as of September 30, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Pharmasset, Inc. maintained, in all material respects, effective internal control over financial reporting as of September 30, 2009, based on criteria established in Internal Control—Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Pharmasset, Inc. as of September 30, 2009 and 2008, and the related statements of operations and comprehensive net loss, redeemable stock and warrants, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended September 30, 2009 and our report dated November 25, 2009 expressed an unqualified opinion.
/s/ GRANT THORNTON LLP
Philadelphia, Pennsylvania
November 25, 2009
F-3
Table of Contents
BALANCE SHEETS
(in thousands, except par value, share and per share amounts)
| As of September 30, | ||||||||
| 2009 | 2008 | |||||||
| ASSETS |
||||||||
| CURRENT ASSETS: |
||||||||
| Cash and cash equivalents |
$ | 58,408 | $ | 63,073 | ||||
| Short-term investments |
— | 497 | ||||||
| Amounts due from collaboration partner |
369 | 1,170 | ||||||
| Prepaid expenses and other assets |
1,656 | 1,008 | ||||||
| Total current assets |
60,433 | 65,748 | ||||||
| EQUIPMENT AND LEASEHOLD IMPROVEMENTS: |
||||||||
| Equipment |
3,613 | 3,362 | ||||||
| Leasehold improvements |
1,837 | 1,837 | ||||||
| 5,450 | 5,199 | |||||||
| Less accumulated depreciation and amortization |
(3,419 | ) | (2,432 | ) | ||||
| Total equipment and leasehold improvements, net |
2,031 | 2,767 | ||||||
| Restricted cash |
100 | — | ||||||
| Other assets |
172 | 467 | ||||||
| Total |
$ | 62,736 | $ | 68,982 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| CURRENT LIABILITIES: |
||||||||
| Current portion of long-term debt |
$ | 7,513 | $ | 2,652 | ||||
| Current portion of capital lease obligation |
— | 42 | ||||||
| Accounts payable |
2,533 | 2,466 | ||||||
| Accrued expenses |
7,675 | 6,182 | ||||||
| Deferred rent |
80 | 124 | ||||||
| Deferred revenue |
985 | 1,857 | ||||||
| Total current liabilities |
18,786 | 13,323 | ||||||
| Deferred rent |
— | 80 | ||||||
| Deferred revenue |
2,956 | 3,869 | ||||||
| Long-term debt, net |
12,096 | 16,523 | ||||||
| Total liabilities |
33,838 | 33,795 | ||||||
| Commitments and contingencies |
||||||||
| STOCKHOLDERS’ EQUITY: |
||||||||
| Common stock, $0.001 par value, 100,000,000 shares authorized, 28,268,004 and 23,340,498 shares issued and outstanding at September 30, 2009 and 2008, respectively |
28 | 23 | ||||||
| Warrants to purchase 127,248 and 116,183 shares of common stock for $12.05 per share as of September 30, 2009 and 2008, respectively |
1,230 | 1,140 | ||||||
| Additional paid-in capital |
195,025 | 145,819 | ||||||
| Accumulated other comprehensive (loss) income |
— | (3 | ) | |||||
| Accumulated deficit |
(167,385 | ) | (111,792 | ) | ||||
| Total stockholders’ equity |
28,898 | 35,187 | ||||||
| Total |
$ | 62,736 | $ | 68,982 | ||||
See notes to financial statements.
F-4
Table of Contents
STATEMENTS OF OPERATIONS AND COMPREHENSIVE NET LOSS
(in thousands except share and per share amounts)
| Years Ended September 30, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Revenues |
$ | 13,293 | $ | 1,857 | $ | 22,009 | ||||||
| COSTS AND EXPENSES: |
||||||||||||
| Research and development |
52,552 | 42,996 | 20,319 | |||||||||
| General and administrative |
13,365 | 13,289 | 9,211 | |||||||||
| Total costs and expenses |
65,917 | 56,285 | 29,530 | |||||||||
| Operating loss |
(52,624 | ) | (54,428 | ) | (7,521 | ) | ||||||
| Investment income |
221 | 1,986 | 2,471 | |||||||||
| Interest expense |
(3,190 | ) | (2,216 | ) | (15 | ) | ||||||
| Loss before income taxes |
(55,593 | ) | (54,658 | ) | (5,065 | ) | ||||||
| Provision for income taxes |
— | — | — | |||||||||
| Net loss |
(55,593 | ) | (54,658 | ) | (5,065 | ) | ||||||
| Redeemable preferred stock accretion |
— | — | 1,776 | |||||||||
| Net loss attributable to common stockholders |
$ | (55,593 | ) | $ | (54,658 | ) | $ | (6,841 | ) | |||
| COMPREHENSIVE NET LOSS: |
||||||||||||
| Net loss |
$ | (55,593 | ) | $ | (54,658 | ) | $ | (5,065 | ) | |||
| Unrealized gain (loss) on available-for-sale investments |
— | (7 | ) | 2 | ||||||||
| Comprehensive net loss |
$ | (55,593 | ) | $ | (54,665 | ) | $ | (5,063 | ) | |||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS PER SHARE: |
||||||||||||
| Basic |
$ | (2.10 | ) | $ | (2.51 | ) | $ | (0.46 | ) | |||
| Diluted |
$ | (2.10 | ) | $ | (2.51 | ) | $ | (0.46 | ) | |||
| WEIGHTED AVERAGE SHARES OUTSTANDING: |
||||||||||||
| Basic |
26,479,532 | 21,808,283 | 14,990,472 | |||||||||
| Diluted |
26,479,532 | 21,808,283 | 14,990,472 | |||||||||
See notes to financial statements.
F-5
Table of Contents
STATEMENTS OF REDEEMABLE STOCK AND WARRANTS
FOR THE YEARS ENDED SEPTEMBER 30, 2009, 2008, AND 2007
(in thousands)
| Series B Redeemable Convertible Preferred Stock |
Series C Redeemable Convertible Preferred Stock |
Series D Redeemable Convertible Preferred Stock |
Series R Redeemable Convertible Preferred Stock |
Series R-1 Warrants |
Redeemable Common Stock |
Total Redeemable Stock And Warrants |
|||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Number | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||
| BALANCE—September 30, 2006 |
368 | $ | 625 | 367 | $ | 1,996 | 2,506 | $ | 12,959 | 400 | $ | 3,797 | 471 | $ | 264 | 213 | $ | 1,190 | 20,831 | ||||||||||||||||||||||||||
| Accretion of redeemable common stock to redemption value |
729 | 729 | |||||||||||||||||||||||||||||||||||||||||||
| Expiration of Series R-1 warrants |
— | — | — | — | — | — | — | — | (471 | ) | (264 | ) | — | — | (264 | ) | |||||||||||||||||||||||||||||
| Accretion of redeemable convertible preferred stock to total redemption value |
— | 1 | — | 2 | — | 1,570 | — | 203 | — | — | — | — | 1,776 | ||||||||||||||||||||||||||||||||
| Payment of dividends in the form of common stock |
— | — | — | — | — | (1,187 | ) | — | — | — | — | — | — | (1,187 | ) | ||||||||||||||||||||||||||||||
| Return of excess dividend accretion to retained earnings |
— | — | — | — | — | (563 | ) | — | — | — | — | — | — | (563 | ) | ||||||||||||||||||||||||||||||
| Conversion of redeemable convertible preferred stock into common stock |
(368 | ) | (626 | ) | (367 | ) | (1,998 | ) | (2,506 | ) | (12,779 | ) | (400 | ) | (4,000 | ) | — | — | — | — | (19,403 | ) | |||||||||||||||||||||||
| Conversion of redeemable common stock into common stock |
— | — | — | — | — | — | — | — | — | — | (213 | ) | (1,919 | ) | (1,919 | ) | |||||||||||||||||||||||||||||
| BALANCE—September 30, 2007, 2008 and 2009 |
— | $ | — | — | $ | — | — | $ | — | — | $ | — | — | $ | — | — | $ | — | — | ||||||||||||||||||||||||||
See notes to financial statements.
F-6
Table of Contents
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
FOR THE YEARS ENDED SEPTEMBER 30, 2009, 2008, AND 2007
(in thousands)
| Series A Convertible Preferred Stock |
Accumulated Other Comprehensive Income (Loss) |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||||||||||||||||||||
| Series D-1 Warrants |
Additional Paid-in Capital |
|||||||||||||||||||||||||||||||||||||||
| Warrants | Common Stock | Accumulated Deficit |
||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Number | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| BALANCE—September 30, 2006 |
— | — | 2,640 | $ | 3 | $ | 1,255 | $ | 5,412 | 10,291 | $ | 10 | $ | 44,480 | $ | 2 | $ | (50,127 | ) | $ | (220 | ) | ||||||||||||||||||
| Expiration of Series R-1 warrants |
— | — | — | — | — | — | — | — | 264 | — | — | 264 | ||||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 532 | 1 | 1,500 | — | — | 1,501 | ||||||||||||||||||||||||||||
| Stock compensation |
— | — | — | — | — | — | — | — | 2,285 | — | — | 2,285 | ||||||||||||||||||||||||||||
| Accretion of redeemable preferred stock to redemption value |
— | — | — | — | — | — | — | — | — | — | (1,776 | ) | (1,776 | ) | ||||||||||||||||||||||||||
| Accretion of redeemable common stock to redemption value |
— | — | — | — | — | — | — | — | — | — | (729 | ) | (729 | ) | ||||||||||||||||||||||||||
| Payment of dividends in the form of common stock |
— | — | — | — | — | — | 132 | — | 1,187 | 1,187 | ||||||||||||||||||||||||||||||
| Conversion of convertible preferred stock into common stock |
— | — | (2,640 | ) | (3 | ) | — | — | 4,192 | 4 | 19,401 | — | — | 19,402 | ||||||||||||||||||||||||||
| Conversion of Series D-1 warrants into common stock |
— | — | — | — | (1,255 | ) | (5,412 | ) | 823 | 1 | 5,411 | — | — | — | ||||||||||||||||||||||||||
| Conversion of redeemable common stock into common stock |
— | — | — | — | — | — | 213 | — | 1,920 | — | — | 1,920 | ||||||||||||||||||||||||||||
| Return of excess dividend accretion to retained earnings |
— | — | — | — | — | — | — | — | — | — | 563 | 563 | ||||||||||||||||||||||||||||
| Net proceeds from initial public offering |
— | — | — | — | — | — | 5,050 | 5 | 39,070 | — | — | 39,075 | ||||||||||||||||||||||||||||
| Unrealized gain on available-for-sale investments |
— | — | — | — | — | — | — | — | — | 2 | — | 2 | ||||||||||||||||||||||||||||
| Sale of warrants in connection with debt financing |
66 | 527 | — | — | — | — | — | — | — | — | — | 527 | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (5,065 | ) | (5,065 | ) | ||||||||||||||||||||||||||
| BALANCE—September 30, 2007 |
66 | 527 | — | — | — | — | 21,233 | 21 | 115,518 | 4 | (57,134 | ) | 58,936 | |||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 617 | 1 | 2,642 | — | — | 2,643 | ||||||||||||||||||||||||||||
| Stock compensation and restricted shares issued |
— | — | — | — | — | — | 41 | — | 3,591 | — | — | 3,591 | ||||||||||||||||||||||||||||
| Net proceeds from registered direct offering |
— | — | — | — | — | — | 1,450 | 1 | 24,068 | — | — | 24,069 | ||||||||||||||||||||||||||||
| Unrealized loss on available-for-sale investments |
— | — | — | — | — | — | — | — | — | (7 | ) | — | (7 | ) | ||||||||||||||||||||||||||
| Grant of warrants in connection with debt financing |
50 | 613 | — | — | — | — | — | — | — | — | — | 613 | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (54,658 | ) | (54,658 | ) | ||||||||||||||||||||||||||
| BALANCE—September 30, 2008 |
116 | 1,140 | — | — | — | — | 23,340 | 23 | 145,819 | (3 | ) | (111,792 | ) | 35,187 | ||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 236 | — | 1,224 | — | — | 1,224 | ||||||||||||||||||||||||||||
| Stock compensation and restricted shares issued |
— | — | — | — | — | — | 14 | — | 4,562 | — | — | 4,562 | ||||||||||||||||||||||||||||
| Net proceeds from registered direct offering |
— | — | — | — | — | — | 4,678 | 5 | 43,420 | — | — | 43,425 | ||||||||||||||||||||||||||||
| Unrealized gain on available-for-sale investments |
— | — | — | — | — | — | — | — | — | 3 | — | 3 | ||||||||||||||||||||||||||||
| Grant of warrants in connection with debt financing |
11 | 90 | — | — | — | — | — | — | — | — | — | 90 | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (55,593 | ) | (55,593 | ) | ||||||||||||||||||||||||||
| BALANCE—September 30, 2009 |
127 | $ | 1,230 | — | $ | — | — | $ | — | 28,268 | $ | 28 | $ | 195,025 | — | $ | (167,385 | ) | $ | 28,898 | ||||||||||||||||||||
See notes to financial statements.
F-7
Table of Contents
STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended September 30, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||||||||
| Net loss |
$ | (55,593 | ) | $ | (54,658 | ) | $ | (5,065 | ) | |||
| Adjustments to reconcile net loss to net cash (used in) provided by operating activities: |
||||||||||||
| Depreciation |
498 | 506 | 403 | |||||||||
| Amortization |
489 | 489 | 489 | |||||||||
| Non-cash stock compensation |
4,562 | 3,591 | 2,285 | |||||||||
| Non-cash interest expense |
544 | 404 | — | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Amounts due from collaboration partner, prepaid expenses and other assets |
348 | (447 | ) | (1,338 | ) | |||||||
| Accounts payable |
67 | (816 | ) | 2,571 | ||||||||
| Accrued expenses |
1,493 | 837 | 3,291 | |||||||||
| Deferred rent |
(124 | ) | (124 | ) | (124 | ) | ||||||
| Deferred revenue |
(1,785 | ) | (1,857 | ) | (1,585 | ) | ||||||
| Net cash (used in) provided by operating activities |
(49,501 | ) | (52,075 | ) | 927 | |||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||||||||
| Proceeds from maturities or sale of investments |
500 | 750 | — | |||||||||
| Purchase of equipment and leasehold improvements |
(251 | ) | (900 | ) | (437 | ) | ||||||
| Restricted cash |
(100 | ) | — | — | ||||||||
| Net cash provided by (used in) investing activities |
149 | (150 | ) | (437 | ) | |||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||||||||
| Borrowings of long-term debt |
3,333 | 20,000 | — | |||||||||
| Proceeds from exercise of stock options |
1,224 | 2,642 | 1,621 | |||||||||
| Principal payments on long-term debt |
(3,253 | ) | — | — | ||||||||
| Principal payments on capital lease obligations |
(42 | ) | (159 | ) | (112 | ) | ||||||
| Proceeds from issuance of common stock, net of issuance costs, of $2,092, $1,813 and $1,548 paid during 2009, 2008 and 2007, respectively |
43,425 | 24,069 | 40,684 | |||||||||
| Payments related to purchase of common stock |
— | — | (120 | ) | ||||||||
| Net cash provided by financing activities |
44,687 | 46,552 | 42,073 | |||||||||
| Net (decrease) increase in cash and cash equivalents |
(4,665 | ) | (5,673 | ) | 42,563 | |||||||
| Cash and cash equivalents—Beginning of period |
63,073 | 68,746 | 26,183 | |||||||||
| Cash and cash equivalents—End of period |
$ | 58,408 | $ | 63,073 | $ | 68,746 | ||||||
| SUPPLEMENTAL DISCLOSURES: |
||||||||||||
| Cash paid during the period for: |
||||||||||||
| Interest |
$ | 2,646 | $ | 1,812 | $ | 15 | ||||||
| Noncash transactions: |
||||||||||||
| Accretion of redeemable convertible preferred stock to redemption value |
$ | — | $ | — | $ | 1,776 | ||||||
| Accretion of redeemable common stock to redemption value |
$ | — | $ | — | $ | 729 | ||||||
| Unrealized (loss) gain on available-for-sale investments |
$ | — | $ | (7 | ) | $ | 2 | |||||
| Conversion of redeemable convertible preferred stock, redeemable common stock, and convertible preferred stock into common stock |
$ | — | $ | — | $ | 21,325 | ||||||
| Exercise and conversion of Series D-1 warrants into common stock |
$ | — | $ | — | $ | 5,412 | ||||||
| Capital lease obligations incurred |
$ | — | $ | — | $ | 314 | ||||||
| Dividends paid on the Series D preferred stock in the form of common stock |
$ | — | $ | — | $ | 1,187 | ||||||
| Warrants granted in connection with debt financing |
$ | 90 | $ | 613 | $ | 527 | ||||||
See notes to financial statements.
F-8
Table of Contents
NOTES TO FINANCIAL STATEMENTS
1. DESCRIPTION OF BUSINESS AND BASIS OF PRESENTATION
We are a clinical-stage pharmaceutical company committed to discovering, developing, and commercializing novel drugs to treat viral infections. Our primary focus is on the development of nucleoside/tide analogs as oral therapeutics for the treatment of chronic hepatitis C virus (“HCV”) infection and secondarily, on the development of RacivirTM for the treatment of human immunodeficiency virus (“HIV”) infection. Nucleoside/tide analogs are a class of compounds which act as alternative substrates for the viral polymerase, thus inhibiting viral replication. We currently have three clinical-stage product candidates, two of which we are developing ourselves and one of which we are developing with a strategic partner. We are also advancing a series of preclinical candidates in preparation for clinical development. Our three clinical stage product candidates are:
| • | RG7128 (formerly R7128), is a first generation HCV nucleoside polymerase inhibitor we are developing through a strategic collaboration with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (collectively, “Roche”). RG7128 is in a Phase 2b clinical trial in combination with Pegasys® (pegylated interferon) plus Copegus® (ribavirin) and recently completed INFORM-1, a study designed to investigate the combination of two oral, direct acting antivirals (“DAAs”) in the absence of pegylated interferon and ribavirin. Both of these studies are being conducted by Roche; |
| • | PSI-7851, a second generation HCV nucleotide polymerase inhibitor, which has completed two phase 1 studies providing adequate safety and efficacy data to support initiation of a Phase 2a clinical trial; and |
| • | Racivir, for the treatment of HIV in combination with other approved HIV drugs, has completed a Phase 2 clinical trial. |
The Company is subject to risks common to companies in the pharmaceutical industry including, but not limited to, product development risks, protection of proprietary intellectual property, compliance with government regulations, dependence on key personnel, the need to obtain additional financing, uncertainty of market acceptance of products, and product liability. (See Part I, Item 1A.—Risk Factors for additional information.)
Basis of Presentation—We were incorporated as Pharmasset, Inc. under the laws of Delaware on June 8, 2004.
Management has evaluated subsequent events for disclosure or recognition in the accompanying financial statements up to the filing of this report, November 25, 2009.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates—The preparation of the Company’s financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents—Cash and cash equivalents represent cash and highly liquid investments purchased within three months of the maturity date and consist primarily of mutual and money market funds.
Investments—The Company invests available cash primarily in mutual and money market funds, bank certificates of deposit and investment-grade commercial paper, corporate notes, and government securities. All investments are classified as available-for-sale and are carried at fair market value with unrealized gains and losses recorded in accumulated other comprehensive (loss) income. For purposes of determining realized gains and losses, the cost of securities sold is based on specific identification.
F-9
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Deferred Offering Costs—Costs incurred in connection with an equity offering are deferred and, upon completion of the equity offering, are applied against the proceeds from the offering.
Deferred Financing Costs—Costs incurred in connection with debt offerings are deferred (and included in prepaid expenses and other current assets and other long-term assets on the balance sheet) and amortized as interest expense over the term of the related debt using the effective interest method. The amortization expense is included in interest expense in the statements of operations and comprehensive net (loss) income.
Equipment and Leasehold Improvements—Equipment and leasehold improvements are recorded at cost and are depreciated using the straight-line method over the following estimated useful lives of the assets: computer equipment—three years; laboratory and office equipment—seven years; and leasehold improvements—the lesser of the estimated life of the asset and the lease term. Expenditures for maintenance and repairs are expensed as incurred. Capital expenditures which improve and extend the life of the related assets are capitalized.
Intangible Assets—Intangible assets are recorded at cost and are amortized on a straight-line basis over the estimated useful life. The estimated useful life is determined based upon a review of several factors including the nature of the asset, its expected use, length of the agreement and the period over which benefits are expected to be received from the use of the asset.
Impairment of Long-Lived Assets—The Company continually evaluates whether events or circumstances have occurred that indicate the estimated remaining useful lives of long-lived assets may require revision or that the carrying value of these assets may be impaired. To determine whether assets have been impaired, the estimated undiscounted future cash flows for the estimated remaining useful life of the respective assets are compared to the carrying value. To the extent that the undiscounted future cash flows are less than the carrying value, a new fair value of the asset is required to be determined. If such fair value is less than the current carrying value, the asset is written down to its estimated fair value.
Fair Value of Financial Instruments—The Company categorizes its financial assets based on the priority of the inputs to the valuation technique into a three-level fair value hierarchy as set forth below. Except for the debt, the Company does not have any financial liabilities that are required to be measured at fair value on a recurring basis. If the inputs used to measure the financial instruments fall within different levels of the hierarchy, the categorization is based on the lowest level input that is significant to the fair value measurement of the instrument.
Financial assets recorded on the balance sheets are categorized as follows:
| • | Level 1—Financial assets whose values are based on unadjusted quoted prices for identical assets or liabilities in an active market which the company has the ability to access at the measurement date (examples include active exchange-traded equity securities and most U.S. Government and agency securities). |
| • | Level 2—Financial assets whose value are based on quoted market prices in markets where trading occurs infrequently or whose values are based on quoted prices of instruments with similar attributes in active markets. |
| • | Level 3—Financial assets whose values are based on prices or valuation techniques that require inputs that are both unobservable and significant to the overall fair value measurement. These inputs reflect management’s own assumptions about the assumptions a market participant would use in pricing the asset. |
F-10
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
As of September 30, 2009 and 2008, the Company did not have any Level 2 or 3 financial assets and the Company’s Level 1 financial assets were as follows:
| Level 1 | ||||||
| September 30, | ||||||
| 2009 | 2008 | |||||
| (in thousands) | ||||||
| Money Market Funds |
$ | 16,838 | $ | 2,165 | ||
| Mutual Funds (invested in short-term U.S. Treasury Obligations) |
41,570 | 60,908 | ||||
| Certificate of Deposit |
100 | — | ||||
| Corporate Bond |
— | 497 | ||||
| Total |
$ | 58,508 | $ | 63,570 | ||
The Certificate of Deposit included above as of September 30, 2009 is acting as cash collateral for a letter of credit that is in place to support a performance bond required to ensure payment of import duties on supplies used in the Company’s development programs, and is classified as Restricted Cash on the balance sheet as of September 30, 2009. The Corporate Bond listed in the above table as of September 30, 2008 represents an available-for-sale investment with an original cost of $499,914 that matured at its face value of $500,000 on January 26, 2009.
Concentrations of Credit Risk, Suppliers and Revenues—The Company’s financial instruments that potentially subject it to concentrations of credit risk are cash and cash equivalents, and investments. The Company invests cash that is not currently being used in operations in accordance with its investment policy. The policy allows for the purchase of low-risk, investment grade debt securities issued by the United States government and very highly-rated banks and corporations, subject to certain concentration limits. The policy allows for maturities that are not longer than two years for individual securities and an average of one year for the portfolio as a whole.
The Company relies on certain materials used in its development process, some of which are procured from a single source. The failure of a supplier, including a subcontractor, to deliver on schedule could delay or interrupt the development process and thereby adversely affect the Company’s operating results.
During each of the years ended September 30, 2009 and 2008, the Company derived all of its revenues from one customer, and during the year ended September 30, 2007, the Company derived substantially all of its revenue from one customer (See Note 4.)
Revenue Recognition—The Company recognizes revenues when all of the following four criteria are present: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. For arrangements that involve the delivery or performance of multiple products, services and/or rights to use assets, the elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the collaborator and whether there is objective and reliable evidence of the fair value of the undelivered obligation(s). The consideration received is allocated among the separate units either on the basis of each unit’s fair value or using the residual method, and the applicable revenue recognition criteria are applied to each of the separate units.
The Company’s revenues are primarily related to its collaboration agreement with Roche. This agreement provides for various types of payments to the Company, including non-refundable upfront license fees, research and/or development payments, and milestone payments.
F-11
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Where the Company has continuing performance obligations under the terms of a collaborative arrangement, non-refundable upfront license payments received upon contract signing are recorded as deferred revenue and recognized as revenue as the related activities are performed. The period over which these activities are to be performed is based upon management’s estimate of the development period. Changes in management’s estimate could change the period over which revenue is recognized. Research and/or development payments are recognized as revenues as the related research and/or development activities are performed and when the Company has no continuing performance obligations related to the research and development payment received.
The Company recognizes revenue from milestone payments when earned, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement and (ii) the Company does not have ongoing performance obligations related to the achievement of the milestone earned. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment is non-refundable; achievement of the milestone was not reasonably assured at the inception of the arrangement; substantive effort is involved to achieve the milestone; and the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement and the related risk associated with the achievement of the milestone. Any amounts received under the agreements in advance of performance, if deemed substantive, are recorded as deferred revenue and recognized as revenue as the Company completes its performance obligations.
Where the Company has no continuing involvement under a collaborative arrangement, the Company records nonrefundable license fee revenues when the Company has the contractual right to receive the payment, in accordance with the terms of the license agreement, and records milestones upon appropriate notification to the Company of achievement of the milestones by the collaborative partner.
Deferred revenue associated with a non-refundable payment received under a collaborative agreement that is terminated prior to its completion results in an immediate recognition of the deferred revenues.
Research and Development Expenses—Research and development expenses consist primarily of salaries and related personnel expenses, fees paid to external service providers, costs of preclinical studies and clinical trials, drug and laboratory supplies, costs for facilities and equipment and the costs of intangibles that are purchased from others for use in research and development activities, such as in-licensed product candidates, that have no alternative future uses. Research and development expenses are included in operating expenses when incurred. Reimbursements received from the Company’s collaborator(s) for third-party research and development expenses incurred by the Company on their behalf are recorded as a contra-expense. Amounts due from collaborators for reimbursement of research and development expenses are recorded on the balance sheets as “Amounts due from collaboration partner.”
Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided.
Stock-Based Compensation—The Company recognizes stock compensation expense for awards of equity instruments to employees and directors based on the grant-date fair value of those awards (with limited exceptions). The grant-date fair value of the award is recognized as compensation expense on a straight-line basis over the requisite service period. Equity instruments granted to consultants are periodically valued and recorded as stock compensation expense as the equity instrument vests.
F-12
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Stock-based compensation expense is included in both research and development expenses and in general and administrative expenses in the statements of operations and comprehensive net income (loss). Since the Company’s stock was not publicly traded prior to April 27, 2007, the expected volatility was calculated for each equity award granted based on the “peer method.” The Company identified companies that trade publicly within the pharmaceutical industry that have similar SIC codes, employee count and revenues. Prior to October 1, 2006, the Company had chosen the weekly high price volatility for these companies for a period of five years. Subsequent to October 1, 2006, the Company has used the weekly high price for these companies for a period of six years to coincide with the expected term.
Income Taxes—The Company accounts for income taxes under the asset and liability method. The Company provides deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the Company’s financial statement carrying amounts and the tax bases of assets and liabilities using enacted tax rates expected to be in effect in the years in which the differences are expected to reverse. A valuation allowance is provided to reduce the deferred tax assets to the amount that is expected to be realized.
On October 1, 2007, the Company adopted a new comprehensive model for how it recognizes, measure, presents and discloses in its financial statements uncertain tax positions that the company has taken or expects to take on a tax return (including a decision whether to file or not to file a return in a particular jurisdiction). Under this new comprehensive model, the financial statements reflect expected future tax consequences of such positions presuming the taxing authorities’ full knowledge of the position and all relevant facts.
As a result of adopting this new comprehensive model, there were no changes to the Company’s deferred tax assets as of October 1, 2007. The total amount of unrecognized tax benefits at October 1, 2007 was $126,000, all of which would favorably impact the Company’s effective tax rate if recognized. Since the unrecognized tax benefit has not been utilized on the Company’s tax returns, there is no liability recorded on the balance sheets. The Company does not have any interest or penalties accrued related to tax positions at adoption. In the event the Company determines that accrual of interest or penalties are necessary in the future, the amount will be presented as a component of income taxes.
Preferred Stock Accretion—Prior to the conversion of all of the Company’s redeemable convertible preferred stock into common stock in May 2007 when the Company completed its IPO, the Company used the interest method to increase the carrying amount of its redeemable convertible preferred stock on each balance sheet date, so that the carrying amount, initially the fair value of the security on the date of issue, would equal the redemption amount at the earliest redemption date. These periodic increases to the carrying amount also used the interest method to include amounts representing dividends not currently declared or paid (See Note 8.)
Comprehensive Net Income (Loss)—Components of comprehensive income (loss) include net income (loss) and unrealized gain (loss) on available-for-sale securities, net of tax. Comprehensive income (loss) is represented in the statements of operations and comprehensive net income (loss).
F-13
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Net Income (Loss) Per Common Share—Basic net income (loss) per common share is calculated by dividing net income (loss) attributable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net income (loss) per common share is calculated by dividing net income (loss) attributable to common stockholders by the weighted average number of common shares and other dilutive securities outstanding during the period. Dilutive potential common shares resulting from the assumed exercise of outstanding stock options and warrants are determined based on the treasury stock method.
| Years Ended September 30, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands, except per share amounts) |
||||||||||||
| Numerator: |
||||||||||||
| Net loss attributable to common stockholders |
$ | (55,593 | ) | $ | (54,658 | ) | $ | (6,841 | ) | |||
| Denominator: |
||||||||||||
| Weighted average common shares outstanding used in calculation of basic net loss per share |
26,480 | 21,808 | 14,990 | |||||||||
| Effect of dilutive securities: |
||||||||||||
| Common stock options |
— | — | — | |||||||||
| Common stock warrants |
— | — | — | |||||||||
| Weighted average common shares outstanding used in calculation of diluted net loss per share |
26,480 | 21,808 | 14,990 | |||||||||
| Net loss attributable to common stockholders per share: |
||||||||||||
| Basic |
$ | (2.10 | ) | $ | (2.51 | ) | $ | (0.46 | ) | |||
| Diluted |
$ | (2.10 | ) | $ | (2.51 | ) | $ | (0.46 | ) | |||
The following table summarizes the securities outstanding at the end of each period with the potential to become common stock that have been excluded from the computation of diluted net income (loss) attributable to common stockholders per share, as their effect would have been anti-dilutive.
| Years Ended September 30, | ||||||
| 2009 | 2008 | 2007 | ||||
| (In thousands) | ||||||
| Options to purchase common stock |
2,549 | 2,372 | 2,101 | |||
| Common stock warrants |
127 | 116 | 66 | |||
| Total |
2,676 | 2,488 | 2,167 | |||
Segment Reporting—Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker, or decision-making group, in making decisions regarding resource allocation and assessing performance. The Company, which uses financial information in determining how to allocate resources and assess performance, has determined that it operates in one segment, which focuses on developing nucleoside/tide analog drugs for the treatment of viral infections.
Recently Adopted Accounting Pronouncements—In May 2009, the FASB issued guidance that incorporates the accounting and disclosure requirements for subsequent events into U.S. generally accepted accounting principles. This guidance also introduces new terminology, defines a date through which management must evaluate subsequent events, and lists the circumstances under which an entity must recognize and disclose
F-14
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
events or transactions occurring after the balance-sheet date. The Company adopted this guidance as of June 30, 2009, which was the required effective date, and its adoption did not affect the Company’s financial statements, other than the disclosures required by it, which can be found in Note 1 – Description of Business and Basis of Presentation.
In June 2009, the FASB issued guidance which divides nongovernmental U.S. GAAP into the authoritative Codification and guidance that is nonauthoritative. The Codification is not intended to change U.S. GAAP; however, it does significantly change the way in which accounting literature is organized and because it completely replaces existing standards, it will affect the way U.S. GAAP is referenced by most companies in their financial statements and accounting policies. The Codification is effective for financial statements issued for interim and annual periods ending after September 15, 2009. The adoption of the Codification did not have an impact on the Company’s financial statements.
Recently Issued Accounting Pronouncements—In September 2009, the FASB Emerging Issues Task Force, or EITF, issued guidance on accounting for multiple-deliverable revenue arrangements that provides principles and application guidance on whether multiple deliverables exist, how the arrangement should be separated and the consideration allocated. This guidance requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. The update eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. This guidance should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. The adoption of this guidance may have a material impact on the Company’s financial position or results of operations for future collaborations arrangements.
In June 2009, the FASB issued guidance to replace the quantitative-based risks and rewards calculation for determining which enterprise, if any, has a controlling financial interest in a variable interest entity and requires on-going reassessments of whether an enterprise is the primary beneficiary of a variable interest entity. It also requires the elimination of the quantitative approach for determining the primary beneficiary of a variable interest entity and amends certain guidance for determining whether an entity is a variable interest entity requiring enhanced disclosure that will provide users of financial statements with more transparent information about an enterprise’s involvement in a variable interest entity. Additionally, an enterprise is required to assess whether it has an implicit financial responsibility to ensure that a variable interest entity operates as designed when determining whether it has the power to direct the activities of the variable interest entity that most significantly impact the entity’s economic performance. This guidance is effective for financial statements issued for fiscal years after November 15, 2009. The Company is currently evaluating the impact, if any, of adopting this guidance on its financial statements.
In December 2007, the EITF issued guidance on accounting for collaborative arrangements. The EITF concluded on the definition of a collaborative arrangement and that revenues and costs incurred with third parties in connection with collaborative arrangements would be presented gross or net based on other accounting literature. Based on the nature of the arrangement, payments to or from collaborators would be evaluated and the terms, the nature of the entity’s business, and whether those payments are within the scope of other accounting literature would be presented. Companies are also required to disclose the nature and purpose of collaborative arrangements along with the accounting policies and the classification and amounts of significant financial statement amounts related to the arrangements. Activities in the arrangement conducted in a separate legal entity should be accounted for under other accounting literature; however the required disclosures above would apply to the entire collaborative agreement. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2008, and interim periods within those fiscal years, and is to be applied
F-15
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
retrospectively to all periods presented for all collaborative arrangements existing as of the effective date. The adoption of this guidance is not expected to have a material impact on the Company.
In December 2007, the FASB issued guidance which changes the accounting for business acquisitions. This guidance requires the acquiring entity in a business combination to recognize all (and only) the assets acquired and liabilities assumed in the transaction and establishes the acquisition-date fair value as the measurement objective for all assets acquired and liabilities assumed in a business combination. Certain provisions of this standard will, among other things, impact the determination of acquisition-date fair value of consideration paid in a business combination (including contingent consideration); exclude transaction costs from acquisition accounting; and change accounting practices for acquired contingencies, acquisition-related restructuring costs, in-process research and development, indemnification assets, and tax benefits. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2008. The adoption of this guidance is not expected to have a material impact on the Company.
In December 2007, the FASB issued guidance which establishes new standards governing the accounting for and reporting of noncontrolling interests (“NCIs”) in partially owned consolidated subsidiaries and the loss of control of subsidiaries. Certain provisions of this standard indicate, among other things, that NCIs (previously referred to as minority interests) be treated as a separate component of equity, not as a liability; that increases and decrease in the parent’s ownership interest that leave control intact be treated as equity transactions, rather than as step acquisitions or dilution gains or losses; and that losses of a partially owned consolidated subsidiary be allocated to the NCI even when such allocation might result in a deficit balance. This standard also requires changes to certain presentation and disclosure requirements. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2008. The provisions of the standard are to be applied to all NCIs prospectively, except for the presentation and disclosure requirements, which are to be applied retrospectively to all periods presented. The adoption of this guidance is not expected to have a material impact on the Company.
3. ACCRUED EXPENSES
Accrued expenses consisted of the following:
| As of September 30, | ||||||
| 2009 | 2008 | |||||
| (In thousands) | ||||||
| Accrued clinical trial expenses |
$ | 4,274 | $ | 3,367 | ||
| Accrued compensation |
1,623 | 1,161 | ||||
| Accrued legal fees |
1,165 | 984 | ||||
| Other accrued expenses |
613 | 670 | ||||
| $ | 7,675 | $ | 6,182 | |||
F-16
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
4. CONTRACT REVENUE AGREEMENTS
The following is a reconciliation between cash payments received and receivable under contract revenue agreements and contract revenues reported:
| Years Ended September 30, | ||||||||||
| 2009 | 2008 | 2007 | ||||||||
| (In thousands) | ||||||||||
| Cash received/receivable |
$ | 11,509 | $ | — | $ | 20,425 | ||||
| Deferred |
— | — | (375 | ) | ||||||
| Amortization |
1,784 | 1,857 | 1,959 | |||||||
| Revenues |
$ | 13,293 | $ | 1,857 | $ | 22,009 | ||||
The Company recorded revenues from the collaboration agreement with Roche comprising 100%, 100%, and 99.8% of total revenues during the fiscal years ended September 30, 2009, 2008, and 2007. The $13.3 million of revenues during the year ended September 30, 2009 include a $10.0 million milestone payment received from Roche for initiating a Phase 2b study of RG7128 and $1.5 million of research and development payments from Roche for activities related to holding the IND application for RG7128, for which we have no continuing performance obligations. Also included is $1.8 million of amortization of up-front and subsequent collaborative and license payments received from Roche previously recorded as deferred revenue. The Company’s performance obligations relating to the $10.0 million milestone payment consisted of successfully completing a Phase 1 study of RG7128, which led to the initiation of the Phase 2b study for RG7128 that triggered the $10.0 million milestone payment.
Roche—In October 2004, the Company entered into a collaboration and license agreement with Roche to develop PSI-6130 and PSI-6130 pro-drugs (including RG7128) for treating chronic HCV infection, and to discover chemically related nucleoside polymerase inhibitors pursuant to a research collaboration. The research collaboration ended in December 2006. The Company granted Roche worldwide rights, excluding Latin America and Korea, to PSI-6130 and its pro-drugs. Roche paid the Company an up-front payment of $8.0 million and agreed to pay future research and development costs. The up-front payment has been recorded as deferred revenue and is being amortized over the estimated development period. The portion of the above payments recorded as deferred revenue on the Company’s balance sheets as of September 30, 2009 and 2008 was $3.9 million and $5.7 million, respectively. During the year ended September 30, 2009, Roche paid the Company a $10.0 million milestone payment for the initiation of a Phase 2b study for RG7128, as well as a $1.5 million payment for research and development activities related to holding the IND application for RG7128, all of which was recorded as revenue since there were no continuing performance obligations related to these payments. Roche is also required to make certain future payments to the Company for RG7128 upon the achievement of predefined development and marketing milestones in Roche’s territories. In addition, the Company will receive royalties paid as a percentage of total annual net product sales, if any, and the Company will be entitled to receive one time performance payments should net sales from the product exceed specified thresholds.
The Company retained certain co-promotion rights in the United States. The Company will be required to pay Roche royalties on net product sales, if any, in the territories the Company has retained. Prior to the transfer of the IND for RG7128 to Roche, which occurred during December 2008, Roche funded and the Company was responsible for preclinical work, the IND submission, and the initial clinical trial, while Roche managed other preclinical studies and clinical development. Roche reimbursed the Company $0.9 million, $5.3 million, and $4.2 million during the years ended September 2009, 2008, and 2007, respectively, under this agreement. Roche will continue to fund all of the expenses of, and be responsible for, other preclinical studies and future clinical
F-17
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
development of RG7128 in the territories licensed to Roche. Roche and the Company will continue to jointly oversee all development and marketing activities of RG7128 in the territories licensed to Roche.
The agreement will terminate once there are no longer any royalty or payment obligations. Additionally, Roche may terminate the agreement in whole or in part by providing six months’ written notice to the Company. Otherwise, either party may terminate the agreement in whole or in part in connection with a material breach of the agreement by the other party that is not timely cured. In the event of termination, Roche must assign or transfer to the Company all regulatory filings, trademarks, patents, and preclinical and clinical data related to this collaboration.
5. IN-LICENSE AGREEMENTS
University of Georgia Research Foundation, Inc., Emory University and UAB Research Foundation, Inc. —On December 8, 1998, Emory University granted the Company an exclusive, worldwide license pursuant to a license agreement (“Racivir License Agreement”) to make, have made, use, import, offer for sale, and sell drug products based on a specified range of mixtures of (–) – FTC and (+) – FTC (“enriched FTC”), which includes the mixture that the Company is developing as Racivir. As part of the consideration for this agreement, the Company issued to Emory University 66,667 shares of redeemable common stock, and agreed to pay Emory University royalties as a percentage of net product sales. The Company subsequently issued to Emory University an additional 13,307 shares of redeemable common stock pursuant to an anti-dilution provision in the agreement. The Company may also pay Emory University up to an aggregate of $1.0 million in future marketing milestone payments. The agreement will expire upon the expiration of all licensed patents.
In a license agreement relating to emtricitabine that Emory University entered into with Triangle Pharmaceuticals, now Gilead Sciences, Inc., or Gilead, in 1996 (“Emory/Gilead License Agreement”), Emory University previously had granted a right of first refusal to Gilead that is applicable to any license or assignment relating to enriched FTC (which includes Racivir). The terms of this right of first refusal contains an exception permitting Emory University to license or assign its rights in respect of enriched FTC to any of the inventors (which included two of the founders of the Company) or to any corporate entity formed by or on behalf of the inventors for purposes of clinically developing enriched FTC so long as the licensee agrees in writing to be bound by the terms of Gilead’s right of first refusal to the same extent as Emory University. The Company’s license to Racivir was granted to the Company by Emory University pursuant to this exception and therefore the Company is bound by the terms of Gilead’s right of first refusal to the same extent as Emory University.
Under this license agreement, the Company is obligated to make minimum royalty payments to Emory University if a new drug registration of a compound resulting from the licensed technology is obtained and the compound is subsequently commercialized. These minimum royalty payments would begin in the second year following a New Drug Application (“NDA”) Registration, and continue until the tenth year. The Company may also make up to $1.0 million in marketing milestone payments under this license agreement to Emory University if Racivir is commercialized and sold.
In March 2004, the Company entered into a supplemental agreement with Emory University in which it and Emory University agreed that, prior to any commercialization of enriched FTC by the Company, or by any licensee or assignee of the Company’s rights under the Racivir License Agreement, the Company and Emory University would adhere strictly to the terms of the right of first refusal granted to Gilead in the Emory/Gilead License Agreement and offer to Gilead the same terms and conditions under which the Company, its licensee or assignee, propose to commercialize enriched FTC. Therefore, before the Company could enter into a commercialization agreement for Racivir with a third party or commercialize Racivir on its own, it would be required to offer Gilead the opportunity to be its commercialization partner on the same terms in which the Company intends, or its prospective partner intends, to commercialize Racivir. It is uncertain whether a third
F-18
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
party would be willing to negotiate the terms of a commercialization agreement with the Company knowing that Gilead can take their place as licensee by accepting the negotiated terms and exercising its right of first refusal.
In 1998 and 2004, the Company entered into various license agreements in addition to those described above with UGARF, Emory University and the University of Alabama at Birmingham Research Foundation, Inc. (collectively, the “Universities”) to pursue the research, development, and commercialization of certain human antiviral, anticancer, and antibacterial applications and uses of certain specified technologies. Under each of these agreements, the Universities have granted an exclusive right and license under the related patents to the Company. The Company and the Universities will share in any proceeds received by the Company related to internal development or sublicensing of the specified technologies, including milestone payments, fees, and royalties.
In April 2002, the license agreement between UGARF, Emory University, and the Company dated June 16, 1998 was selectively modified to terminate certain technologies and related rights and obligations.
6. DEBT
On September 30, 2007, the Company entered into a Loan Agreement that allowed the Company to borrow up to $30.0 million in $10.0 million increments (“Loan Agreement”). The Company borrowed the first and second $10.0 million increments by signing two Secured Promissory Notes (“Notes A and B”) on October 5, 2007 and March 28, 2008, respectively. Notes A and B bear interest at 12%. On December 12, 2008, the Company amended the Loan Agreement and borrowed $3.3 million by signing a Secured Promissory Note (“Note C”). Note C bears interest at 12.5%. Notes A, B and C are to be repaid over a 45-month period with the first 15 monthly payments representing interest only followed by 30 equal monthly payments of principal and interest. The principal monthly repayments on each of the following notes begin and end as follows:
| Note |
Begin |
End | ||
| Note A |
March 1, 2009 | August 1, 2011 | ||
| Note B |
August 1, 2009 | January 1, 2012 | ||
| Note C |
May 1, 2010 | October 1, 2012 |
Prepayment of the loans made pursuant to the Loan Agreement is subject to penalty and substantially all of the Company’s tangible and intangible assets (except for intellectual property) are collateral for the Loan Agreement. Future total principal repayments of the three Notes amount to $7.5 million in fiscal 2010, $9.5 million in fiscal 2011, $3.0 million in fiscal 2012, and $0.1 million in fiscal 2013. There are no additional borrowings available under the Loan Agreement.
Under the Loan Agreement, the Company agreed that in the event its market capitalization is below $90.0 million for 15 consecutive days in which the principal market for its common stock is open for trading to the public, the Company will be required to repay 50% of the then outstanding principal balance of the loans. The Company further agreed that in the event its market capitalization is below $40.0 million for 15 consecutive days in which the principal market for its common stock is open for trading to the public, the Company will be required to repay all of the then outstanding principal balance of the loans.
In conjunction with entering into the Loan Agreement, the Company granted warrants to the lender to purchase shares of the Company’s common stock (See Note 8). Since these warrants were granted in conjunction with entering into the Loan Agreement and with the intention of executing promissory notes, the relative fair value of the warrant was recorded as equity and deferred interest as the warrants became exercisable and the deferred financing costs and debt discount are being amortized over the term of the promissory notes using the effective interest method.
F-19
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
7. STOCK COMPENSATION
The Company’s 1998 Stock Plan (“1998 Plan”), as amended, was originally adopted by its board of directors during 1998 and subsequently amended in 2000, 2004, and 2006. A maximum of 3,517,015 shares of the Company’s common stock are authorized for issuance under the 1998 Plan. The purpose of the 1998 Plan is to provide an incentive to officers, directors, employees, independent contractors, and to other persons who provide significant services to the Company. Upon the closing of the IPO, which occurred on May 2, 2007, the Company adopted the 2007 Equity Incentive Plan (“2007 Plan”). Upon the adoption of the 2007 Plan, no additional awards will be issued under the 1998 Plan and the shares remaining for future grant under the 1998 Plan were transferred to the 2007 Plan. On September 23, 2009, the Company’s stockholders approved amendments to the 2007 Plan to remove a provision that allowed for repricing stock options without stockholder approval, added certain minimum vesting periods for nonperformance based grants, and increased the number of shares authorized under the 2007 Plan by 1,000,000 shares (“the Revised 2007 Plan”). As of September 30, 2009, 1,224,450 shares of the Company’s common stock were reserved for future grants of stock options, stock appreciation rights, restricted stock, deferred stock, restricted stock units, performance shares, phantom stock, and similar types of stock awards as well as cash awards. Options granted under the Revised 2007 Plan may be either “incentive stock options,” as defined under Section 422 of the Code or nonstatutory stock options. Options granted under the Revised 2007 Plan shall be at per share exercise prices equal to the fair value of the shares on the dates of grant. The Revised 2007 Plan will terminate in fiscal 2017 unless it is extended or terminated earlier pursuant to its terms.
Stock Options—The assumptions used and weighted-average information for employee and director grants for the years ended September 30, 2009, 2008 and 2007 are as follows:
| Years Ended September 30, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Risk free interest rate |
3.19 | % | 4.01 | % | 4.55 | % | ||||||
| Expected dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Expected lives (years) |
5.98 | 6.03 | 5.94 | |||||||||
| Expected volatility |
54.39 | % | 56.76 | % | 54.33 | % | ||||||
| Weighted-average fair value of options granted |
$ | 9.97 | $ | 8.33 | $ | 2.79 | ||||||
Generally, stock options granted under these plans have a contractual life of ten years and vest pro rata over a four year term. A summary of the Company’s stock option activity during the year ended September 30, 2009 is as follows:
| Number of Shares |
Weighted Average Exercise Price | |||||
| Outstanding—September 30, 2008 |
2,371,861 | $ | 7.97 | |||
| Granted |
483,981 | $ | 18.43 | |||
| Exercised |
(235,507 | ) | $ | 5.20 | ||
| Forfeited |
(71,635 | ) | $ | 6.04 | ||
| Outstanding—September 30, 2009 |
2,548,700 | $ | 10.26 | |||
| Exercisable—September 30, 2009 |
1,481,788 | $ | 6.97 | |||
The range of exercise prices of stock options outstanding at September 30, 2009 was $3.00 to $32.00. The weighted average remaining contractual life of stock options outstanding at September 30, 2009 was 7.28 years. The total intrinsic value of options exercised during the year ended September 30, 2009 was $3,960,991. The
F-20
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Company recognized compensation expense of $3,590,663, $2,758,062, and $1,552,690, during the years ended September 30, 2009, 2008, and 2007, related to stock options issued to non-employees and employees. As of September 30, 2009 and 2008, $6,937,089 and $6,821,109, respectively, of deferred stock-based compensation expense related to employee stock options remained unamortized. The unamortized amount of $6,637,644 as of September 30, 2009 has a weighted-average period of approximately 1.32 years to be recognized.
| Outstanding as of September 30, 2009 | Exercisable as of September 30, 2009 | |||||||||||||
| Number of |
Exercise Price | Weighted Average Remaining Contractual Life (in Years) |
Weighted Average Exercise Price |
Number of Options |
Weighted Average Exercise Price | |||||||||
| 1,078,534 | 3.00–4.49 | 5.93 | $ | 3.46 | 951,171 | $ | 3.37 | |||||||
| 11,085 | 4.50–5.99 | 7.47 | $ | 5.52 | 3,137 | $ | 5.60 | |||||||
| 42,269 | 6.00–7.49 | 2.30 | $ | 6.44 | 42,268 | $ | 6.44 | |||||||
| 88,666 | 7.50–10.49 | 7.48 | $ | 8.87 | 79,916 | $ | 8.91 | |||||||
| 698,040 | 10.50–15.00 | 8.09 | $ | 13.68 | 298,233 | $ | 13.66 | |||||||
| 629,106 | 15.01–29.99 | 8.99 | $ | 18.65 | 106,688 | $ | 18.99 | |||||||
| 1,000 | 30.00–45.00 | 8.31 | $ | 32.00 | 375 | $ | 32.00 | |||||||
As of September 30, 2009, after considering estimated forfeitures there were 2,446,076 options outstanding that were either vested or expected to vest in the future, of which 1,481,788 options were currently exercisable, with weighted average exercise prices of $10.09 and $6.97 per share, aggregate intrinsic values of $27,055,626 and $21,006,067, and weighted average remaining contractual terms of 7.23 and 6.45 years, respectively.
Restricted Stock—During the fiscal year ended September 30, 2008, the Company issued a total of 40,666 shares of restricted stock to its non-employee directors and to a consultant. The restricted stock issued to each non-employee Director vested on July 16, 2009. The restricted stock issued to the consultant vests quarterly over a four year period. On March 24, 2009, the Company issued a total of 14,000 shares of restricted stock to its non-employee directors that vest one year from the date of grant, as long as the director continues to serve on the Company’s board of directors on that date, unless the failure to be so engaged is due solely to the fact that the director is nominated but not re-elected to serve as a director. As of September 30, 2009, holders were vested in 32,333 of the 54,666 restricted shares outstanding, leaving a total of 22,333 restricted shares unvested as of fiscal year end.
With regard to the restricted stock granted to the non-employee Directors, the fair value of the restricted stock issued was determined using the closing price of the Company’s common stock as reported on the Global Market of the NASDAQ Stock Market LLC (“NASDAQ”) on the date of grant and is recognized as stock-based compensation expense evenly over the vesting period. The weighted average fair value of the shares granted in 2008 and 2009 was $20.01 and $8.92, respectively, per share.
With regard to the restricted stock granted to the consultant, stock-based compensation expense equal to the fair value of the restricted shares that vest is recorded on a quarterly basis over the vesting period. The fair value of each of the restricted shares that vest is equal to the fair value of a share of the Company’s common stock as of each vesting date.
The Company recognized compensation expense of $431,971 during the year ended September 30, 2009 related to restricted stock issued to its non-employee directors and to the consultant. Unrecognized compensation expense for the restricted shares granted to the non-employee directors was $59,532 at September 30, 2009. This amount will be recognized over the remaining vesting period of the restricted shares.
F-21
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Valuation of Privately-Held Company Stock Options Issued as Compensation—During the years ended September 30, 2009, 2008, and 2007, the Company granted stock options to employees and directors at exercise and purchase prices deemed by the board of directors to be equal to the fair value of the common stock at the time of grant. Prior to January 1, 2006, the fair value of the common stock at the time of grant was determined by the board of directors at each stock option measurement date based on a variety of factors, including the Company’s financial position and historical financial performance, the status of developments within the Company, the composition and ability of the current research and development and management team, an evaluation and benchmark of the Company’s competitors, the current climate in the marketplace, the illiquid nature of the common stock, arm’s length sales of the Company’s capital stock (including redeemable convertible preferred stock), the effect of the rights and preferences of the preferred stockholders, and the prospects of a liquidity event, among others. In preparation for the Company’s planned initial public offering, a retrospective analysis of the fair value of the common stock at option grant dates during 2005 using the methodology favored by the guidelines of the American Institute of Certified Public Accountants (“AICPA”) titled “Valuation of Privately-Held Company Equity Securities Issued as Compensation” was performed by management. The methodology developed at that time was subsequently been applied by management to the valuation of all employee stock options granted since 2005 through April 11, 2007, the date on which the last options were granted prior to April 27, 2007 when the Company’s stock began trading on NASDAQ. The Company did not rely on an independent appraiser for stock option valuations because the Company used a methodology developed in accordance with AICPA guidelines and relied on the experience of management and members of its board of directors. Factors taken into consideration by this methodology included the judgment of management as to the probability of executing a successful initial public offering; the liquidation preferences of the Company’s preferred stock; the net balance of the Company’s cash, cash equivalents and short term investments; and the present value of the Company’s product candidates as estimated, when available, from the market value of publicly traded companies developing comparable product candidates, and when this was not available, by a discounted cash flow (“DCF”) analysis based on cost and revenue estimates provided by third party clinical research organizations, marketing consultants and management and using discount rates provided by an independent appraiser.
The application of the Company’s methodology for determining the fair value of the Company’s common stock at each issuance date from January 1, 2006 through April 11, 2007 is discussed below:
Between May 24, 2006 and July 10, 2006, the Company granted 447,400 options to employees and members of the Company’s board of directors. The Company’s technology value as a private company was based on clevudine alone due to the early stage of development of RG7128 and Racivir. A DCF analysis of clevudine was used due to the absence of a comparable program with an identifiable public market value. The value of the Company’s common stock to a private investor was then calculated by adding this private technology value to the Company’s net cash balance and subtracting the liquidation preference payments that would be made to the holders of the Company’s preferred stock out of the proceeds of a private sale of the Company prior to any participation in the proceeds by holders of the Company’s common stock. This process resulted in an estimate of the value of the Company’s common stock as a private company of $3.87 per share, which the Company’s board of directors deemed to be the appropriate fair value at which to set the exercise price of the options issued at that time. The theoretical value of the Company’s common stock had it been publicly traded at that time was calculated based on published academic research, the Company’s private technology value, and the Company’s net cash balance, then applying a premium to account for the value of the liquidity of a publicly traded stock. No subtraction was made for liquidity preferences, since all the preferred stock was convertible to common stock upon an IPO. This process resulted in an estimate of the theoretical public price of the Company’s common stock, based on clevudine alone, of $8.30 per share. The fair value of the Company’s common stock used for financial reporting purposes was a weighted average of the private and public values, with the weights equal to the probability of executing a successful initial public offering versus a private sale of the Company, as estimated
F-22
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
by management with the advice of investment bankers based on the recent experience of other biotechnology companies, market conditions and stockholder support for an initial public offering at that time. During this period, the probability of an IPO was considered to be 50%, resulting in a fair value of the Company’s common stock for financial reporting purposes of $6.09 per share.
On November 7, 2006, the Company granted 317,067 options to employees and members of the Company’s board of directors. The methodology used to determine the fair value of the Company’s common stock at that time was the same as that described above, so only variations in its application are discussed below. The estimated value of the Company’s common stock as a private company increased to $4.02 per share, based on the increase in the value of clevudine as it moved closer to market and the announcement of additional favorable clinical data that supported an increase in the revenue projection contained in the Company’s DCF analysis. This increase in clevudine’s value was partly offset by a reduction in the Company’s net cash balance. The theoretical public price of the Company’s common stock, based on clevudine alone, was estimated to be $8.75 at that time and the probability of an IPO was considered to be 50%, resulting in a weighted average fair value for financial reporting purposes of $6.38 per share.
From January 1, 2007 through April 11, 2007, the Company granted 147,500 options to employees and to a member of the Company’s board of directors. The methodology used to determine the fair values of the Company’s common stock during this time was the same as that described above, so only variations in its application are discussed below. The estimated value of the Company’s common stock as a private company increased to amounts ranging from $4.02 to $5.63 per share, based on the increase in the value of the RG7128 program as it advanced through Phase 1 clinical trials based on the valuation of a publicly traded company with an HCV program at a similar stage of development. The theoretical public price of the Company’s common stock, based on clevudine and RG7128, ranged from $8.84 to $11.28 during this time and the probability of an IPO was considered to be 90%, resulting in weighted average fair values for financial reporting purposes ranging from $6.53 to $10.71 per share.
During the year ended September 30, 2006, the Company granted 11,333 options to non-employees. The fair value of these awards on the initial grant date of $30,406 was estimated using the Black-Scholes option-pricing methodology.
8. STOCKHOLDERS’ EQUITY (DEFICIT), REDEEMABLE STOCK CONVERSIONS, AND WARRANTS
Common Stock—As of September 30, 2009, the Company had 100,000,000 shares of common stock authorized with a par value of $0.001 and the Company had reserved 2,548,700 shares of common stock for issuance upon the exercise of outstanding common stock options. Also, 1,224,450 shares of the Company’s common stock were reserved for future grants of stock options (or other similar equity instruments) under the Company’s Revised 2007 Plan as of September 30, 2009. In addition, 127,248 shares of the Company’s common stock were reserved for future exercise of outstanding warrants as of September 30, 2009.
During fiscal 2007, 13,333 shares of common stock were purchased at a fair value of $9.00 per share. Such shares were immediately retired.
Registered Direct Public Offerings—On February 5, 2009, the Company completed a registered direct public offering of 4,678,000 shares of its common stock to a select group of institutional investors at a price of $9.73 per share, resulting in $43.4 million in net proceeds after deducting the placement agent fee and estimated offering expenses. The Company intends to use the net proceeds from the sale of the shares for general corporate
F-23
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
purposes, which may include, but are not limited to, the acquisition of assets or businesses that are complementary to its existing business, the funding of clinical trials, and the funding of in-licensing agreements for product candidates, additional technologies, or other forms of intellectual property.
On July 21, 2008, the Company completed a registered direct public offering of 1,450,000 shares of its common stock to a select group of institutional investors at a price of $17.85 per share, resulting in $24.1 million in net proceeds after deducting placement agent fees and offering expenses. The Company intends to use the net proceeds from the sale of the shares for general corporate purposes, which may include, but are not limited to, the acquisition of assets or businesses that are complementary to its existing business, the funding of clinical trials and the funding of in-licensing agreements for product candidates, additional technologies or other forms of intellectual property.
Initial Public Offering—On May 2, 2007, the Company completed an IPO of 5,050,000 shares of its common stock (including the underwriters’ exercise of a portion of their over-allotment option) at a public offering price of $9.00 per share. Net cash proceeds from the IPO were $40.7 million after deducting offering costs paid during fiscal 2007 and $39.1 million after deducting additional offering costs paid in fiscal 2006.
Redeemable Stock Conversions—In conjunction with the completion of the IPO on May 2, 2007, and pursuant to the amended and restated by-laws of the Company, all outstanding shares of redeemable convertible preferred stock (Series B, C, D, and R) were converted into an aggregate of 2,432,569 shares of common stock as follows:
| Preferred Stock Series |
Amount of Common Shares | |
| Series B |
245,331 | |
| Series C |
250,120 | |
| Series D |
1,670,452 | |
| Series R |
266,666 | |
| 2,432,569 | ||
In addition, holders of the Series D redeemable convertible preferred stock were entitled to receive quarterly dividends at a rate equal to 7.5% per annum of the purchase price per share. Such dividends accrued from February 4, 2006 through May 2, 2007, the closing date of the IPO, and were paid out in the form of 131,864 shares of common stock. Also, all outstanding shares of redeemable common stock and Series A convertible preferred stock were converted into 213,306 and 1,759,808 shares of common stock, respectively on May 2, 2007.
Warrants—In conjunction with entering into a Loan Agreement and with executing three secured promissory notes (See Note 6), the Company granted warrants to the lender to purchase 127,248 shares of the Company’s Common Stock. The warrants expire seven years from the date of grant (or upon a change of control as defined in the Loan Agreement) as follows: 66,390 expire on September 30, 2014, 49,793 expire on March 28, 2015, and 11,065 expire on December 12, 2015.
During fiscal 2007, the Series D-1 warrants originally granted in August 2004 were exercised in full in connection with the IPO on a “net exercise” basis, which resulted in the Company issuing 822,689 shares of common stock to the warrant holders. Warrants to purchase up to 470,588 shares of Series R-1 redeemable convertible preferred stock granted in conjunction with Roche’s purchase of 400,000 shares of the Company’s Series R redeemable convertible preferred stock expired without exercise on October 26, 2006.
F-24
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Preferred Stock Accretion—Preferred stock accretion was $1,775,684 and $1,110,973 during the years ended September 30, 2007 and 2006, respectively. The accretion recorded during 2007 includes $1.0 million of accretion to bring the carrying amounts of the redeemable convertible preferred stock to their redemption values as of May 2, 2007, the date the Company completed the IPO and converted all of its redeemable convertible preferred stock outstanding into common stock.
9. INCOME TAXES
Income tax expense was $0 during the years ended September 30, 2009, 2008, and 2007.
The reconciliation between the federal statutory rate of 34.0% and the Company’s effective tax rate is as follows:
| Years Ended September 30, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Federal tax |
-34.0 | % | -34.0 | % | -34.0 | % | |||
| State tax |
-4.0 | % | -3.9 | % | -4.0 | % | |||
| Change in valuation allowance |
36.5 | % | 36.3 | % | 28.9 | % | |||
| Stock compensation |
1.5 | % | 1.6 | % | 8.8 | % | |||
| Other |
0.0 | % | 0.0 | % | 0.3 | % | |||
| Effective tax rate |
0.0 | % | 0.0 | % | 0.0 | % | |||
The Company was originally organized in 1998 as a Barbados limited company, Pharmasset, Ltd., under Section 10 of the International Business Companies Act of Barbados. The Company was subject to United States withholding tax of 5% under the United States-Barbados tax treaty for United States sourced royalties paid to a Barbados company.
Pharmasset Ltd. owned a Georgia subsidiary which conducted research and development in the United States under a contract research and development agreement with the Company. Prior to June 8, 2004, only the Georgia subsidiary was subject to United States income taxes. The Company became domesticated as a corporation under the laws of the State of Delaware on June 8, 2004 as Pharmasset, Inc., on a tax-free basis with a carryover of the tax basis of its assets, and Pharmasset, Ltd. was dissolved on June 21, 2004. A portion of the expenses incurred by Pharmasset, Ltd. prior to the domestication have been capitalized for tax purposes and are to be amortized to offset future taxable income, if any, in the United States and a portion of these losses cannot be utilized in the United States. On July 23, 2004, the Georgia subsidiary was merged into the Company.
F-25
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
Deferred tax assets and liabilities are determined based on the difference between financial statement and tax bases using enacted tax rates in effect for the year in which the differences are expected to reverse. The components of the deferred taxes consist of the following:
| As of September 30, | ||||||||
| 2009 | 2008 | |||||||
| Deferred tax assets: |
||||||||
| Capitalized research and development |
$ | 900 | $ | 1,219 | ||||
| Net operating loss carryforwards |
51,404 | 30,308 | ||||||
| Payments received in collaborations |
1,498 | 2,176 | ||||||
| Licensing agreements |
1,711 | 1,999 | ||||||
| Stock compensation |
1,033 | 684 | ||||||
| Accrued liabilities |
118 | 100 | ||||||
| Research and development tax credits |
138 | 138 | ||||||
| Deferred rent |
31 | 78 | ||||||
| Depreciation |
303 | 156 | ||||||
| Gross deferred tax assets |
$ | 57,136 | $ | 36,858 | ||||
| Valuation allowance |
(57,136 | ) | (36,858 | ) | ||||
| Net deferred tax asset |
$ | — | $ | — | ||||
As of September 30, 2009, the Company’s unrecognized tax benefits ($126,000 as of October 1, 2007) have not significantly changed. The Company does not expect any significant changes to the unrecognized tax benefits within 12 months of the reporting date.
The IRS could challenge tax positions taken by the Company for the periods for which there are open tax years. The Company is open to challenge for the periods of 2004-2008 from federal and state jurisdictions.
As of September 30, 2009, the Company has United States federal net operating loss carryforwards of approximately $144.9 million available to offset future taxable income, if any. Of the federal net operating losses, $10.5 million was generated from windfall tax benefit stock option deductions. The tax benefit of this portion of the net operating loss will be accounted for directly to equity as additional paid in capital as the stock option related losses are utilized. As of September 30, 2009, the Company also had research and development tax credits of approximately $138,159 available to offset future tax liabilities. The loss carryovers and the research and development tax credits expire over a period of 2020 to 2029. The Barbados net operating losses effectively do not carry over as the Company does not anticipate conducting future business in that country. The Company established a full valuation allowance on its net deferred tax assets as it is more likely than not that such tax benefits will not be realized.
Under Section 382 of the Internal Revenue Code (the “Code”), utilization of the net operating loss and research and development tax credit carryforwards may be subject to a limitation if a change in ownership of the Company, as defined in the Code, occurred previously or could occur in the future. The Company completed a Section 382 analysis regarding limitation of its net operating loss and research and development credit carryforwards that covered the period three years prior to its IPO on May 2, 2007 through its most recent follow-on public offering of its common stock on February 5, 2009, and concluded that a change in control occurred at the Company during the quarter ended September 30, 2008. This change in control limits the future use of the Company’s net operating loss and research and development credit carryforwards from fiscal 2008 and prior years. However, based upon the Company’s financial projections, it does not believe that this limitation will
F-26
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
result in the expiration of any of these net operating loss and research and development credit carryforwards before they are able to be utilized. Changes in ownership subsequent to this period could lead to a change in the limitation in future years, which could impact the use of the Company’s net operating loss and research and development credit carryforwards generated in the affected years. Any limitation may result in expiration of a portion of the NOL or research and development credit carryforwards before utilization, which would reduce the Company’s gross deferred tax assets. The Company will continue to monitor changes in its ownership and will update its Section 382 analysis when it believes a change in control may have occurred.
10. COMMITMENTS AND CONTINGENCIES
On May 23, 2005, the Company entered into an operating lease for office and laboratory space through May 22, 2010, in Princeton, New Jersey. Monthly lease payments began May 23, 2005. On June 2, 2009, the Company entered into an extension and modification of the lease, extending the term of the lease for an additional five years, from May 22, 2010 through May 22, 2015. The Company also leases office space in Durham, North Carolina. Monthly lease payments began May 1, 2007 and, after amending the lease term on February 2, 2009, end on April 1, 2011. In January 2007, the Company entered into a capital lease for lab equipment with principal and interest payments of $14,044 due monthly through December 2008.
As of September 30, 2009, minimum future payments under non-cancellable operating leases are as follows (in thousands):
| Fiscal 2010 |
$ | 714 | |
| Fiscal 2011 |
884 | ||
| Fiscal 2012 |
835 | ||
| Fiscal 2013 |
835 | ||
| Fiscal 2014 |
835 | ||
| Thereafter |
537 | ||
| Total minimum payments required |
$ | 4,640 | |
Rent expense under operating leases was $785,687, $825,024, and $777,939 during the years ended September 30, 2009, 2008, and 2007, respectively.
Under our collaboration and license agreement with Bukwang Pharmaceutical Company Ltd. for clevudine, up to an aggregate of $23.0 million in milestone payments are payable in the future if certain development, regulatory and commercialization events occur, which are not expected to occur due to the Company’s termination of the registration studies of clevudine. Under our license agreement with Emory University for Racivir, we agreed to pay Emory University up to an aggregate of $1.0 million in future marketing milestone payments. None of these potential future payments are included in our financial statements, as the payments are contingent on the achievement of milestones, which we have not yet achieved.
11. EMPLOYEE SAVINGS PLAN
The Company maintains a contributory employee savings plan (“401(k) Plan”) for its employees which provides for, among other things, a discretionary employer match of 50 cents on every dollar contributed by each employee under the plan up to a maximum annual amount of 6% of such employee’s salary up to a maximum annual match of $3,500 per employee, such discretionary match being made automatically unless action is taken by the compensation committee to cancel the match for a given year. Expense under the 401(k) Plan was $194,062, $141,888, and $122,263, during the years ended September 30, 2009, 2008, and 2007, respectively.
F-27
Table of Contents
PHARMASSET, INC.
NOTES TO FINANCIAL STATEMENTS—(Continued)
12. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
The following tables present unaudited quarterly financial data for the Company. The Company’s quarterly results of operations for these periods are not necessarily indicative of future results of operations.
| Three Months Ended | ||||||||||||||||
| Dec. 31, 2008 |
March 31, 2009 |
June 30, 2009 |
Sept. 30, 2009 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenues: |
$ | 464 | $ | 1,903 | $ | 10,501 | $ | 425 | ||||||||
| Net loss |
$ | (18,276 | ) | $ | (15,472 | ) | $ | (6,949 | ) | $ | (14,896 | ) | ||||
| Net loss attributable to common stockholders |
$ | (18,276 | ) | $ | (15,472 | ) | $ | (6,949 | ) | $ | (14,896 | ) | ||||
| Net loss per common share: |
||||||||||||||||
| Basic |
$ | (0.78 | ) | $ | (0.59 | ) | $ | (0.25 | ) | $ | (0.53 | ) | ||||
| Diluted |
$ | (0.78 | ) | $ | (0.59 | ) | $ | (0.25 | ) | $ | (0.53 | ) | ||||
| Three Months Ended | ||||||||||||||||
| Dec. 31, 2007 |
March 31, 2008 |
June 30, 2008 |
Sept. 30, 2008 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenues: |
$ | 464 | $ | 464 | $ | 464 | $ | 464 | ||||||||
| Net loss |
$ | (12,173 | ) | $ | (12,136 | ) | $ | (15,028 | ) | $ | (15,321 | ) | ||||
| Net loss attributable to common stockholders |
$ | (12,173 | ) | $ | (12,136 | ) | $ | (15,028 | ) | $ | (15,321 | ) | ||||
| Net loss per common share: |
||||||||||||||||
| Basic |
$ | (0.57 | ) | $ | (0.57 | ) | $ | (0.69 | ) | $ | (0.70 | ) | ||||
| Diluted |
$ | (0.57 | ) | $ | (0.57 | ) | $ | (0.69 | ) | $ | (0.70 | ) | ||||
Basic and diluted net loss per common share are identical since common equivalent shares are excluded from the calculation as their effect is antidilutive.
F-28
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 23.1 | Consent of Grant Thornton LLP | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended | |
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |