Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-Q
| x | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE QUARTERLY PERIOD ENDED DECEMBER 31, 2010
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM TO
Commission File Number: 1-33428
Pharmasset, Inc.
(Exact name of registrant as specified in its charter)
| DELAWARE | 98-0406340 | |
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) | |
| 303-A College Road East Princeton, New Jersey |
08540 | |
| (Address of registrant’s principal executive offices) | (Zip Code) | |
(609) 613-4100
(Telephone number, including area code)
N/A
(Former Name, Former Address and Former Fiscal Year, if Changed Since Last Report)
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ¨ Yes ¨ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer or a smaller reporting company. See definition of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ¨ Yes No x
The number of shares of the registrant’s common stock, $0.001 par value, outstanding as of January 31, 2011 was 37,022,855.
Table of Contents
FORM 10-Q
FOR THE QUARTER ENDED DECEMBER 31, 2010
INDEX
The “Company,” “Pharmasset,” “we,” and “us” as used in this Form 10-Q refer to Pharmasset, Inc., a Delaware corporation. Pharmasset, our logo and Racivir are our trademarks. Other trademarks mentioned in this Form 10-Q are the property of their respective owners.
Table of Contents
FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q contains forward-looking statements. The forward-looking statements are principally contained in the sections entitled “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” These statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to be materially different from any future results, performance, or achievements expressed or implied by the forward-looking statements. For this purpose, any statement that is not a statement of historical fact should be considered a forward-looking statement. We may, in some cases, use words such as “project,” “believe,” “anticipate,” “plan,” “expect,” “estimate,” “intend,” “potential,” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. These forward-looking statements include statements about the following:
| • | our product development efforts, primarily with respect to the preclinical studies, clinical trial results and regulatory approval of RG7128, PSI-7977, PSI-938, and PSI-661 for the treatment of hepatitis C virus (“HCV”); |
| • | the initiation, termination, completion, or success of preclinical studies and clinical trials for our product candidates; |
| • | clinical trial initiation and completion dates, anticipated regulatory filing dates, and regulatory approval for our product candidates; |
| • | the commercialization of our product candidates; |
| • | our collaboration agreement with F. Hoffmann-LaRoche Ltd. and Hoffmann-La Roche Inc. (collectively, “Roche”), including potential milestone or royalty payments thereunder, and our clinical collaboration agreement with Bristol Myers Squibb Company (“BMS”); |
| • | our intentions regarding the establishment of collaborations or the licensing of product candidates or intellectual property; |
| • | the scope and enforceability of our intellectual property rights, including claims that we or our collaborators may infringe third party intellectual property rights or be otherwise required to pay license fees under such third party rights; |
| • | our intentions to expand our capabilities and hire additional employees; |
| • | anticipated operating losses, future revenues, research and development expenses, and the need for additional financing; and |
| • | our financial performance. |
Forward-looking statements reflect our current views with respect to future events and are subject to risks and uncertainties. We discuss many of the risks and uncertainties associated with our business in greater detail in our Annual Report on Form 10-K for the fiscal year ended September 30, 2010 under the heading “Risk Factors.” Given these risks and uncertainties, you should not place undue reliance on these forward-looking statements. All forward-looking statements represent our estimates and assumptions only as of the date of this Quarterly Report on Form 10-Q.
You should read this Quarterly Report on Form 10-Q and the documents that we reference in it completely and with the understanding that our actual future results may be materially different from what we expect. You should assume that the information appearing in this Quarterly Report on Form 10-Q is accurate as of the date on the front cover of this Quarterly Report on Form 10-Q only. Our business, financial condition, results of operations, and prospects may change. We may not update these forward-looking statements, even though our situation may change in the future, unless we have obligations under the federal securities laws to update and disclose material developments related to previously disclosed information. The forward-looking statements contained in this Quarterly Report on Form 10-Q are subject to the safe-harbor protection provided by the Private Securities Litigation Reform Act of 1995 and Section 21E of the Securities Exchange Act of 1934, as amended (“Exchange Act”).
Table of Contents
| ITEM 1. | FINANCIAL STATEMENTS |
CONDENSED BALANCE SHEETS
(in thousands, except par value, share and per share amounts)
| As of December 31, 2010 |
As of September 30, 2010 |
|||||||
| (unaudited) | ||||||||
| ASSETS |
||||||||
| CURRENT ASSETS: |
||||||||
| Cash and cash equivalents |
$ | 102,651 | $ | 127,081 | ||||
| Amounts due from collaboration partner |
6 | 6 | ||||||
| Prepaid expenses and other current assets |
702 | 718 | ||||||
| Total current assets |
103,359 | 127,805 | ||||||
| EQUIPMENT AND LEASEHOLD IMPROVEMENTS: |
||||||||
| Equipment |
4,098 | 4,060 | ||||||
| Leasehold improvements |
1,837 | 1,837 | ||||||
| 5,935 | 5,897 | |||||||
| Less accumulated depreciation and amortization |
(4,327 | ) | (4,184 | ) | ||||
| Total equipment and leasehold improvements, net |
1,608 | 1,713 | ||||||
| Restricted cash |
100 | 100 | ||||||
| Other assets |
141 | 143 | ||||||
| Total |
$ | 105,208 | $ | 129,761 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| CURRENT LIABILITIES: |
||||||||
| Current portion of long-term debt |
$ | 7,796 | $ | 8,705 | ||||
| Accounts payable |
3,687 | 5,037 | ||||||
| Accrued expenses |
4,554 | 5,863 | ||||||
| Deferred rent |
25 | 25 | ||||||
| Deferred revenue |
985 | 985 | ||||||
| Total current liabilities |
17,047 | 20,615 | ||||||
| Deferred rent |
86 | 93 | ||||||
| Deferred revenue |
1,724 | 1,971 | ||||||
| Long-term debt, net of discount of $97 and $150 as of December 31, 2010 and September 30, 2010, respectively |
1,515 | 2,934 | ||||||
| Total liabilities |
20,372 | 25,613 | ||||||
| Commitments and contingencies |
||||||||
| STOCKHOLDERS’ EQUITY: |
||||||||
| Common stock, $0.001 par value, 100,000,000 shares authorized, 34,220,379 and 34,043,898 shares issued and outstanding at December 31, 2010 and September 30, 2010, respectively |
34 | 34 | ||||||
| Warrants to purchase 63,623 shares of common stock for $12.05 per share at December 31, 2010, and 127,248 shares of common stock for $12.05 per share at September 30, 2010 |
640 | 1,230 | ||||||
| Additional paid-in capital |
341,132 | 336,351 | ||||||
| Accumulated deficit |
(256,970 | ) | (233,467 | ) | ||||
| Total stockholders’ equity |
84,836 | 104,148 | ||||||
| Total |
$ | 105,208 | $ | 129,761 | ||||
See notes to financial statements.
4
Table of Contents
CONDENSED STATEMENTS OF OPERATIONS
(UNAUDITED)
(in thousands, except share and per share amounts)
| Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| Revenues |
$ | 247 | $ | 269 | ||||
| COSTS AND EXPENSES: |
||||||||
| Research and development |
18,768 | 9,197 | ||||||
| General and administrative |
5,076 | 4,239 | ||||||
| Total costs and expenses |
23,844 | 13,436 | ||||||
| Operating loss |
(23,597 | ) | (13,167 | ) | ||||
| Investment income |
3 | 4 | ||||||
| Other income |
489 | — | ||||||
| Interest expense |
(398 | ) | (706 | ) | ||||
| Loss before income taxes |
(23,503 | ) | (13,869 | ) | ||||
| Provision for income taxes |
— | — | ||||||
| Net loss |
$ | (23,503 | ) | $ | (13,869 | ) | ||
| Net loss per share: basic and diluted |
$ | (0.69 | ) | $ | (0.49 | ) | ||
| Weighed average shares outstanding: basic and diluted |
34,106,890 | 28,287,609 | ||||||
See notes to financial statements.
5
Table of Contents
CONDENSED STATEMENTS OF CASH FLOWS
(UNAUDITED)
(in thousands)
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||||
| Net loss |
$ | (23,503 | ) | $ | (13,869 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization |
143 | 250 | ||||||
| Non-cash stock compensation |
2,181 | 1,791 | ||||||
| Non-cash interest expense |
64 | 118 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Amounts due from collaboration partner, prepaid expenses and other assets |
6 | 1,398 | ||||||
| Accounts payable |
(1,351 | ) | (88 | ) | ||||
| Accrued expenses |
(1,309 | ) | (3,862 | ) | ||||
| Deferred rent |
(6 | ) | (31 | ) | ||||
| Deferred revenue |
(247 | ) | (246 | ) | ||||
| Net cash used in operating activities |
(24,022 | ) | (14,539 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||||
| Purchase of equipment and leasehold improvements |
(37 | ) | (25 | ) | ||||
| Net cash used in investing activities |
(37 | ) | (25 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||||
| Proceeds from exercise of stock options |
2,010 | 270 | ||||||
| Principal payments on long-term debt |
(2,381 | ) | (1,841 | ) | ||||
| Net cash used in financing activities |
(371 | ) | (1,571 | ) | ||||
| Net decrease in cash and cash equivalents |
(24,430 | ) | (16,135 | ) | ||||
| Cash and cash equivalents - Beginning of period |
127,081 | 58,408 | ||||||
| Cash and cash equivalents - End of period |
$ | 102,651 | $ | 42,273 | ||||
| SUPPLEMENTAL DISCLOSURES: |
||||||||
| Cash paid during the period for: |
||||||||
| Interest |
$ | 333 | $ | 588 | ||||
| Noncash transactions: |
||||||||
| Value of warrants exercised by converting warrants into shares of common stock (“net issuance method”) |
$ | 590 | $ | — | ||||
See notes to financial statements.
6
Table of Contents
Notes to Financial Statements (Unaudited)
1. DESCRIPTION OF BUSINESS AND BASIS OF PRESENTATION
Description of Business - Pharmasset, Inc. is a clinical-stage pharmaceutical company committed to discovering, developing, and commercializing novel drugs to treat viral infections. The Company’s primary focus is on the development of nucleoside/tide analogs as oral therapeutics for the treatment of chronic hepatitis C virus (“HCV”) infection. Nucleoside/tide analogs are a class of compounds which act as alternative substrates for the viral polymerase, thus inhibiting viral replication. The Company currently has three clinical-stage product candidates, two of which it is developing itself and one of which it is developing with a strategic partner. The Company is also advancing a series of preclinical candidates in preparation for clinical development. Pharmasset, Inc.’s three clinical stage product candidates are:
| • | RG7128, an HCV cytosine nucleoside polymerase inhibitor the Company is developing through a strategic collaboration with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (collectively, “Roche”). In October 2010, Roche presented data from a 12-week interim analysis from the Phase 2b “PROPEL” study of RG7128 in combination with Pegasys® (pegylated interferon) plus Copegus® (ribavirin), the standard of care for treating HCV (“SOC”) in patients with HCV genotypes 1 or 4. In addition, RG7128 is in a 24-week Phase 2b “JUMP-C” study in combination with SOC in patients with HCV genotypes 1 or 4. Roche is planning to conduct the next study of RG7128 in combination with ritonavir-boosted danoprevir. This “INFORM-SVR” study is part of a series of studies designed to investigate the combination of two oral, direct acting antivirals (“DAAs”) in the absence of pegylated interferon. Roche is also planning to conduct a Phase 2b study in patients with HCV genotypes 2 or 3. All of these studies are being, or are expected to be, conducted by Roche; |
| • | PSI-7977, an HCV uracil nucleotide analog polymerase inhibitor that is in a 12-week Phase 2b dose-finding study (“PROTON”) in combination with SOC in patients with HCV genotypes 1, 2 or 3. In addition, PSI-7977 recently began dosing in an exploratory Phase 2 study (“ELECTRON”) in combination with ribavirin, administered without and with varying durations of pegylated interferon, in patients with HCV genotypes 2 or 3; and |
| • | PSI-938, an HCV guanine nucleotide analog polymerase inhibitor that is in Part 2 of a Phase 1 study with PSI-7977 in patients with HCV genotype 1. |
The Company is subject to risks common to companies in the pharmaceutical industry including, but not limited to, risks relating to product development, protection of proprietary intellectual property, compliance with government regulations, collaboration partners, dependence on key personnel, the need to obtain additional financing, uncertainty of market acceptance of products, and product liability. (See Part II, Item 1A. - Risk Factors for additional information.)
Basis of Presentation - The Company was incorporated as Pharmasset, Inc. under the laws of Delaware on June 8, 2004. Management has evaluated subsequent events for disclosure or recognition in the accompanying financial statements up to the filing of this report.
The accompanying unaudited condensed financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America for interim financial information and with the instructions to Form 10-Q and Rule 10-01 of Regulation S-X. Accordingly, they do not contain all of the information and footnotes required for complete financial statements. In the opinion of management, the accompanying unaudited condensed financial statements reflect all adjustments, which include normal recurring adjustments, necessary to present fairly the Company’s interim financial information. The accompanying unaudited condensed financial statements and notes to the condensed financial statements should be read in conjunction with the audited financial
7
Table of Contents
statements for the fiscal year ended September 30, 2010 included in the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission (“SEC”) on November 23, 2010.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates - The preparation of the Company’s financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents - Cash and cash equivalents represent cash and highly liquid investments purchased within three months of the maturity date and consist primarily of mutual and/or money market funds.
Investments - The Company invests available cash primarily in mutual and money market funds, bank certificates of deposit and investment-grade commercial paper, corporate notes, and government securities. All investments are classified as available-for-sale and are carried at fair market value with unrealized gains and losses recorded in accumulated other comprehensive (loss) income. For purposes of determining realized gains and losses, the cost of securities sold is based on specific identification.
Deferred Offering Costs - Costs incurred in connection with an equity offering are deferred and, upon completion of the equity offering, are applied against the proceeds from the offering.
Deferred Financing Costs - Costs incurred in connection with debt offerings are deferred (and included in prepaid expenses and other current assets and other long-term assets on the balance sheet) and amortized as interest expense over the term of the related debt using the effective interest method. The amortization expense is included in interest expense in the statements of operations.
Equipment and Leasehold Improvements - Equipment and leasehold improvements are recorded at cost and are depreciated using the straight-line method over the following estimated useful lives of the assets: computer equipment—three years; laboratory and office equipment—seven years; and leasehold improvements—the lesser of the estimated life of the asset or the lease term. Expenditures for maintenance and repairs are expensed as incurred. Capital expenditures which improve and extend the life of the related assets are capitalized.
Intangible Assets - Intangible assets are recorded at cost and are amortized on a straight-line basis over the estimated useful life. The estimated useful life is determined based upon a review of several factors including the nature of the asset, its expected use, length of the agreement and the period over which benefits are expected to be received from the use of the asset.
Impairment of Long-Lived Assets - The Company continually evaluates whether events or circumstances have occurred that indicate the estimated remaining useful lives of long-lived assets may require revision or that the carrying value of these assets may be impaired. To determine whether assets have been impaired, the estimated undiscounted future cash flows for the estimated remaining useful life of the respective assets are compared to the carrying value. To the extent that the undiscounted future cash flows are less than the carrying value, a new fair value of the asset is required to be determined. If such fair value is less than the current carrying value, the asset is written down to its estimated fair value.
Fair Value of Financial Instruments - The Company categorizes its financial assets based on the priority of the inputs to the valuation technique into a three-level fair value hierarchy as set forth below. Except for its debt with its lender (See Note 6), the Company does not have any financial liabilities that are required to be measured at fair value on a recurring basis. If the inputs used to measure the financial instruments fall within different levels of the hierarchy, the categorization is based on the lowest level input that is significant to the fair value measurement of the instrument.
Financial assets recorded on the balance sheets are categorized as follows:
8
Table of Contents
| • | Level 1 – Financial assets whose values are based on unadjusted quoted prices for identical assets or liabilities in an active market which the company has the ability to access at the measurement date (examples include active exchange-traded equity securities and most U.S. Government and agency securities). |
| • | Level 2 – Financial assets whose values are based on quoted market prices in markets where trading occurs infrequently or whose values are based on quoted prices of instruments with similar attributes in active markets. |
| • | Level 3 – Financial assets whose values are based on prices or valuation techniques that require inputs that are both unobservable and significant to the overall fair value measurement. These inputs reflect management’s own assumptions about the assumptions a market participant would use in pricing the asset. |
As of December 31, 2010 and September 30, 2010, the Company did not have any Level 2 or 3 financial assets and the Company’s Level 1 financial assets were as follows:
| Level 1 | ||||||||
| December 31, 2010 |
September 30, 2010 |
|||||||
| (in thousands) | ||||||||
| Money Market Funds |
$ | 102,651 | $ | 127,081 | ||||
| Certificate of Deposit |
100 | 100 | ||||||
| Total |
$ | 102,751 | $ | 127,181 | ||||
The Certificate of Deposit included above as of December 31, 2010 and September 30, 2010 is for a letter of credit in place to support a performance bond required to ensure payment of import duties on supplies used in the Company’s development programs, and is classified as Restricted Cash on the balance sheet as of December 31, 2010 and September 30, 2010.
Concentrations of Credit Risk, Suppliers, and Revenues - The Company’s financial instruments that potentially subject it to concentrations of credit risk are cash and cash equivalents and investments. The Company invests cash that is not currently being used in operations in accordance with its investment policy. The policy allows for the purchase of low-risk, investment grade debt securities issued by the United States government and very highly-rated banks and corporations, subject to certain concentration limits. The policy allows for maturities that are not longer than two years for individual securities and an average of one year for the portfolio as a whole.
The Company relies on certain materials used in its development process, some of which are procured from a single source. The failure of a supplier, including a subcontractor, to deliver on schedule could delay or interrupt the development process and thereby adversely affect the Company’s operating results.
During the three months ended December 31, 2010 and 2009, the Company derived all of its revenues from one customer (See Note 4).
Revenue Recognition - The Company recognizes revenues when all of the following four criteria are present: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured.
The Company’s revenues are primarily related to its collaboration agreement with Roche. This agreement provides for various types of payments to the Company, including non-refundable upfront license fees, research and/or development payments, and milestone payments.
9
Table of Contents
Where the Company has continuing performance obligations under the terms of a collaborative arrangement, non-refundable upfront license payments received upon contract signing are recorded as deferred revenue and recognized as revenue as the related activities are performed. The period over which these activities are to be performed is based upon management’s estimate of the development period. Changes in management’s estimate could change the period over which revenue is recognized. Research and/or development payments are recognized as revenues as the related research and/or development activities are performed and when the Company has no continuing performance obligations related to the research and development payment received.
Where the Company has no continuing involvement under a collaborative arrangement, the Company records nonrefundable license fee revenues when the Company has the contractual right to receive the payment, in accordance with the terms of the collaboration agreement, and records milestones upon appropriate notification to the Company of achievement of the milestones by the collaborative partner.
Effective October, 1, 2010, the Company adopted the new accounting standards for determining whether the milestone method of revenue recognition is appropriate. The Company recognizes revenue from milestone payments when earned, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement and (ii) the Company does not have ongoing performance obligations related to the achievement of the milestone earned. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment (a) is commensurate with either the vendor’s performance to achieve the milestone or the enhancement of the value of the delivered item or items as a result of a specific outcome resulting from the vendor’s performance to achieve the milestone, (b) relates solely to past performance, and (c) is reasonable relative to all of the deliverables and payment terms (including other potential milestone consideration) within the arrangement. Any amounts received under the agreement in advance of performance, if deemed substantive, are recorded as deferred revenue and recognized as revenue as the Company completes its performance obligations.
Effective October 1, 2010, the Company also adopted the new accounting standards for revenue recognition for multiple deliverable revenue arrangements. Each deliverable within a multiple-deliverable revenue arrangement is accounted for as a separate unit of accounting under the guidance of the new authoritative guidance if both of the following criteria are met: (1) the delivered item or items have value to the customer on a standalone basis and (2) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
This new authoritative guidance amends previously issued guidance to eliminate the residual method of allocation for multiple-deliverable revenue arrangements, and requires that arrangement consideration be allocated at the inception of an arrangement to all deliverables using the relative selling price method. The new authoritative guidance also establishes a selling price hierarchy for determining the selling price of a deliverable, which includes (1) vendor-specific objective evidence, if available, (2) third-party evidence, if vendor-specific objective evidence is not available, and (3) estimated selling price if neither vendor-specific nor third-party evidence is available. Additionally, it expands the disclosure requirements related to a vendor’s multiple-deliverable revenue arrangements.
Deferred revenue associated with a non-refundable payment received under a collaborative agreement that is terminated prior to its completion results in an immediate recognition of the deferred revenue.
Research and Development Expenses - Research and development expenses consist primarily of salaries and related personnel expenses, fees paid to external service providers, costs of preclinical studies and clinical trials, drug and laboratory supplies, costs for facilities and equipment, and the costs of intangibles that are purchased from others for use in research and development activities, such as in-licensed product candidates, that have no alternative future uses. Research and development expenses are included in operating expenses when incurred. Reimbursements received from the Company’s collaborator(s) for third-party research and development expenses incurred by the Company on their behalf are recorded as a contra-expense. Amounts due from collaborators for reimbursement of research and development expenses are recorded on the balance sheets as “Amounts due from collaboration partner.”
Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided.
10
Table of Contents
Stock-Based Compensation - The Company recognizes stock compensation expense for awards of equity instruments to employees and directors based on the grant-date fair value of those awards (with limited exceptions). The grant-date fair value of the award is recognized as compensation expense on a straight-line basis over the requisite service period. Equity instruments granted to consultants are periodically valued and recorded as stock compensation expense as the equity instrument vests.
Stock-based compensation expense is included in both research and development expenses and in general and administrative expenses in the statements of operations. Since the Company’s stock was not publicly traded prior to April 27, 2007, the expected volatility was calculated for each equity award granted based on the “peer method.” The Company identified companies that trade publicly within the pharmaceutical industry that have similar SIC codes, employee count and revenues. Prior to October 1, 2006, the Company had chosen the weekly high price volatility for these companies for a period of five years. Subsequent to October 1, 2006, the Company has used the weekly high price for these companies for a period of six years to coincide with the expected term.
Income Taxes - The Company accounts for income taxes under the asset and liability method. The Company provides deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the Company’s financial statement carrying amounts and the tax bases of assets and liabilities using enacted tax rates expected to be in effect in the years in which the differences are expected to reverse. A valuation allowance is provided to reduce the deferred tax assets to the amount that is expected to be realized.
The Company uses a comprehensive model for how it recognizes, measures, presents, and discloses in its financial statements uncertain tax positions that the Company has taken or expects to take on a tax return (including a decision whether to file or not to file a return in a particular jurisdiction). Under this comprehensive model, the financial statements reflect expected future tax consequences of such positions presuming the taxing authorities’ full knowledge of the position and all relevant facts.
As a result of adopting this new comprehensive model, there were no changes to the Company’s deferred tax assets as of October 1, 2007. The total amount of unrecognized tax benefits at October 1, 2007 was $0.1 million, all of which would favorably impact the Company’s effective tax rate if recognized. Since the unrecognized tax benefit has not been utilized on the Company’s tax returns, there is no liability recorded on the balance sheets. The Company does not have any interest or penalties accrued related to tax positions at adoption. In the event the Company determines that accrual of interest or penalties are necessary in the future, the amount will be presented as a component of income taxes.
Net Income (Loss) Per Common Share - Basic net income (loss) per common share is calculated by dividing net income (loss) by the weighted average number of common shares outstanding during the period. Diluted net income (loss) per common share is calculated by dividing net income (loss) by the weighted average number of common shares and other dilutive securities outstanding during the period. Dilutive potential common shares resulting from the assumed exercise of outstanding stock options and warrants are determined based on the treasury stock method.
11
Table of Contents
| Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| (In thousands, except per share amounts) |
||||||||
| Numerator: |
||||||||
| Net loss |
$ | (23,503 | ) | $ | (13,869 | ) | ||
| Denominator: |
||||||||
| Weighted average common shares outstanding used in calculation of basic net loss per share |
34,107 | 28,288 | ||||||
| Effect of dilutive securities: |
||||||||
| Common stock options |
— | — | ||||||
| Common stock warrants |
— | — | ||||||
| Weighted average common shares outstanding used in calculation of diluted net loss per share |
34,107 | 28,288 | ||||||
| Net loss per share: basic and diluted |
$ | (0.69 | ) | $ | (0.49 | ) | ||
The following table summarizes the securities outstanding at the end of each period with the potential to become common stock that have been excluded from the computation of diluted net income (loss) per share, as their effect would have been anti-dilutive.
| Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| (In thousands) | ||||||||
| Options to purchase common stock |
3,167 | 3,058 | ||||||
| Common stock warrants |
64 | 127 | ||||||
| Total |
3,231 | 3,185 | ||||||
Segment Reporting - Operating segments are identified as components of an enterprise about which separate financial information is available for evaluation by the chief operating decision-maker, or decision-making group, in making decisions regarding resource allocation and assessing performance. The Company has determined that it operates in one segment, which focuses on developing nucleoside/tide analog drugs for the treatment of viral infections.
Recently Adopted Accounting Pronouncements - In October 2009, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2009-13, Multiple-Deliverable Revenue Arrangements. This ASU provides new accounting standards for determining whether multiple deliverables exist, how the arrangement should be separated, and how the consideration should be allocated. This guidance requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. The update eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. The Company adopted these new accounting standards on October 1, 2010 on a prospective basis. Adoption of these new accounting standards did not have any impact on the Company’s financial position or results of operations.
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition — Milestone Method. This ASU provides guidance on the criteria that should be met for determining whether the milestone method of revenue
12
Table of Contents
recognition is appropriate. Under the milestone method of revenue recognition, consideration that is contingent upon achievement of a milestone in its entirety can be recognized as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. This standard provides the criteria to be met for a milestone to be considered substantive which includes that: a) performance consideration earned by achieving the milestone be commensurate with either performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from performance to achieve the milestone; b) relate to past performance; and c) be reasonable relative to all deliverables and payment terms in the arrangement. The Company adopted these new accounting standards on October 1, 2010 on a prospective basis. Adoption of these new accounting standards did not have any impact on the Company’s financial position or results of operations.
3. ACCRUED EXPENSES
Accrued expenses consisted of the following:
| As of December 31, 2010 |
As of September 30, 2010 |
|||||||
| (In thousands) | ||||||||
| Accrued clinical trial expenses |
$ | 2,682 | $ | 1,771 | ||||
| Accrued compensation |
514 | 1,801 | ||||||
| Accrued legal fees |
824 | 1,770 | ||||||
| Other accrued expenses |
534 | 521 | ||||||
| $ | 4,554 | $ | 5,863 | |||||
4. CONTRACT REVENUE AGREEMENTS
The following is a reconciliation between cash payments received and receivable under contract revenue agreements and contract revenues reported:
| Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| (In thousands) | ||||||||
| Cash received/receivable |
$ | — | $ | 23 | ||||
| Deferred |
— | — | ||||||
| Amortization |
247 | 246 | ||||||
| Revenues |
$ | 247 | $ | 269 | ||||
The Company recorded revenues from the collaboration agreement with Roche, comprising 100% of total revenues during the three months ended December 31, 2010 and 2009. Revenues during each period primarily reflect amortization of up-front and subsequent collaborative and license payments received from Roche previously recorded as deferred revenue.
Roche - In October 2004, the Company entered into a collaboration and license agreement with Roche to develop PSI-6130 and PSI-6130 prodrugs (including RG7128) for treating chronic HCV infection, and to discover chemically related nucleoside polymerase inhibitors pursuant to a research collaboration. The research collaboration ended in December 2006. The Company granted Roche worldwide rights, excluding Latin America and Korea, to PSI-6130 and its prodrugs (including RG7128). Roche paid the Company an up-front payment of $8.0 million in 2004 and agreed to pay future research and development costs. The up-front payment has been recorded as deferred revenue and is being amortized over the estimated development period. The portion of the above payments recorded as deferred revenue on the Company’s balance sheets as of December 31, 2010 and September 30, 2010 was approximately $2.7 million and $3.0 million, respectively. Roche is also required to make certain future payments to
13
Table of Contents
the Company for RG7128 upon the achievement of predefined development and marketing milestones in Roche’s territories. In addition, the Company will receive royalties paid as a percentage of total annual net product sales, if any, in Roche’s territories, and the Company will be entitled to receive one time performance payments should net sales from the product exceed specified thresholds.
The Company retained certain co-promotion rights in the United States. The Company will be required to pay Roche royalties on net product sales, if any, in the territories the Company has retained. Prior to the transfer of the IND for RG7128 to Roche, which occurred during December 2008, Roche funded and the Company was responsible for preclinical work, the IND submission, and the initial clinical trial, while Roche managed other preclinical studies and clinical development. Roche reimbursed the Company approximately $14 thousand during the three months ended December 31, 2009 under this agreement. Roche will continue to fund all of the expenses of, and be responsible for, other preclinical studies and future clinical development of RG7128 in the territories licensed to Roche. Roche and the Company will continue to jointly oversee all development and marketing activities of RG7128 in the territories licensed to Roche.
The agreement will terminate once there are no longer any royalty or payment obligations. Additionally, Roche may terminate the agreement in whole or in part by providing six months’ written notice to the Company. Otherwise, either party may terminate the agreement in whole or in part in connection with a material breach of the agreement by the other party that is not timely cured. In the event of termination, Roche must assign or transfer to the Company all regulatory filings, trademarks, patents, and preclinical and clinical data related to this collaboration.
5. IN-LICENSE AGREEMENTS
In 1998 and 2004, the Company entered into various license agreements with the University of Georgia Research Foundation (“UGARF”), Emory University and the University of Alabama at Birmingham Research Foundation, Inc. (collectively, the “Universities”) to pursue the research, development, and commercialization of certain human antiviral, anticancer, and antibacterial applications and uses of certain specified technologies. Under each of these agreements, the Universities have granted an exclusive right and license under the related patents to the Company. The Company and the Universities will share in any proceeds received by the Company related to internal development or sublicensing of the specified technologies, including milestone payments, fees, and royalties.
In April 2002, the license agreement between UGARF, Emory University, and the Company dated June 16, 1998 was selectively modified to terminate certain technologies and related rights and obligations.
6. DEBT
On September 30, 2007, the Company entered into a Loan Agreement that allowed the Company to borrow up to $30.0 million in $10.0 million increments (“Loan Agreement”). The Company borrowed the first and second $10.0 million increments by signing two Secured Promissory Notes (“Notes A and B”) on October 5, 2007 and March 28, 2008, respectively. Notes A and B bear interest at 12%. On December 12, 2008, the Company amended the Loan Agreement and borrowed $3.3 million by signing a Secured Promissory Note (“Note C”). Note C bears interest at 12.5%. Notes A, B and C are to be repaid over a 45-month period with the first 15 monthly payments representing interest only followed by 30 equal monthly payments of principal and interest. The principal monthly repayments on each of the following notes begin and end as follows:
| Note |
Begin |
End | ||
| Note A |
March 1, 2009 | August 1, 2011 | ||
| Note B |
August 1, 2009 | January 1, 2012 | ||
| Note C |
May 1, 2010 | October 1, 2012 | ||
Prepayment of the loans made pursuant to the Loan Agreement is subject to penalty and substantially all of the Company’s tangible and intangible assets (except for intellectual property) are pledged as collateral for the Loan Agreement. Future total principal repayments of the three Notes amount to $6.3 million in fiscal 2011, $3.0 million in fiscal 2012, and $0.1 million in fiscal 2013. There are no additional borrowings available under the Loan Agreement.
14
Table of Contents
Under the Loan Agreement, the Company agreed that in the event its market capitalization is below $90.0 million for 15 consecutive days in which the principal market for its common stock is open for trading to the public, the Company will be required to repay 50% of the then outstanding principal balance of the loans. The Company further agreed that in the event its market capitalization is below $40.0 million for 15 consecutive days in which the principal market for its common stock is open for trading to the public, the Company will be required to repay all of the then outstanding principal balance of the loans.
In conjunction with entering into the Loan Agreement, the Company granted warrants to the lender to purchase shares of the Company’s common stock (See Note 8). Since these warrants were granted in conjunction with entering into the Loan Agreement and with the intention of executing promissory notes, the relative fair value of the warrant was recorded as equity and deferred interest as the warrants became exercisable and the deferred financing costs and debt discount are being amortized over the term of the promissory notes using the effective interest method.
7. STOCK COMPENSATION
The Company’s 1998 Stock Plan (“1998 Plan”), as amended, was originally adopted by its board of directors during 1998 and subsequently amended in 2000, 2004 and 2006. A maximum of 3,517,015 shares of the Company’s common stock are authorized for issuance under the 1998 Plan. Upon the closing of the IPO, which occurred on May 2, 2007, the Company adopted the 2007 Equity Incentive Plan (“2007 Plan”). Upon the adoption of the 2007 Plan, no additional awards will be issued under the 1998 Plan and the shares remaining for future grant under the 1998 Plan were transferred to the 2007 Plan. The purpose of the 2007 Plan is to provide an incentive to officers, directors, employees, independent contractors, and to other persons who provide significant services to the Company. On September 23, 2009, the Company’s stockholders approved amendments to the 2007 Plan to remove a provision that allowed for repricing stock options without stockholder approval, added certain minimum vesting periods for nonperformance based grants, and increased the number of shares authorized under the 2007 Plan by 1,000,000 shares (the “Revised 2007 Plan”). As of December 31, 2010, under the Revised 2007 Plan 209,826 shares of the Company’s common stock were reserved for future grants of stock options, stock appreciation rights, restricted stock, deferred stock, restricted stock units, performance shares, phantom stock, and similar types of stock awards as well as cash awards. The Company’s Board of Directors has approved an increase of 2,000,000 shares to the shares of the Company’s common stock reserved for future grants under the Revised 2007 Plan. This increase will be presented to the Company’s stockholders for their approval at the Company’s 2011 annual stockholders meeting to be held on March 23, 2011. Options granted under the Revised 2007 Plan may be either “incentive stock options,” as defined under Section 422 of the Internal Revenue Code or nonstatutory stock options. Options granted under the Revised 2007 Plan shall be at per share exercise prices equal to the fair value of the shares on the dates of grant. The Revised 2007 Plan will terminate in fiscal 2017 unless it is extended or terminated earlier pursuant to its terms.
Stock Options - The assumptions used and weighted-average information for employee and director grants for the three months ended December 31, 2010 and 2009 are as follows:
| Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| Risk free interest rate |
1.67 | % | 2.91 | % | ||||
| Expected dividend yield |
0.0 | % | 0.0 | % | ||||
| Expected lives (years) |
6.01 | 6.00 | ||||||
| Expected volatility |
62.21 | % | 64.25 | % | ||||
| Weighted-average fair value of options granted |
$ | 18.71 | $ | 13.11 | ||||
Generally, stock options granted under these plans have a contractual life of ten years and vest pro rata over three or four year terms. A summary of the Company’s stock option activity during the three months ended December 31, 2010 is as follows:
15
Table of Contents
| Number of Shares |
Weighted Average Exercise Price |
|||||||
| Outstanding - September 30, 2010 |
2,796,289 | $ | 12.66 | |||||
| Granted (unaudited) |
540,287 | $ | 32.46 | |||||
| Exercised (unaudited) |
(130,633 | ) | $ | 15.39 | ||||
| Forfeited (unaudited) |
(38,751 | ) | $ | 20.46 | ||||
| Outstanding - December 31, 2010 (unaudited) |
3,167,192 | $ | 15.83 | |||||
| Exercisable - September 30, 2010 |
1,788,794 | $ | 9.23 | |||||
| Exercisable - December 31, 2010 (unaudited) |
1,923,047 | $ | 10.25 | |||||
The range of exercise prices of stock options outstanding at December 31, 2010 was $3.00 to $40.84. The weighted average remaining contractual life of stock options outstanding at December 31, 2010 was 7.14 years. The total intrinsic value of options exercised during the three months ended December 31, 2010 was $3,598,184. The Company recognized compensation expense of $2,136,869 and $1,725,273 during the three months ended December 31, 2010 and 2009 respectively, related to stock options issued to non-employees and employees. As of December 31, 2010 and September 30, 2010, $14,743,030 and $8,048,915 respectively, of deferred stock-based compensation expense related to non-employee and employee stock options remained unamortized. The unamortized amount of $14,743,030 as of December 31, 2010 has a weighted-average period of approximately 1.60 years to be recognized.
| Outstanding as of December 31, 2010 | Exercisable as of December 31, 2010 | |||||||||||||||||||
| Number of |
Exercise Price | Weighted Average Remaining Contractual Life (in Years) |
Weighted Average Exercise Price |
Number of Options |
Weighted Average Exercise Price |
|||||||||||||||
| 926,136 | 3.00 - 4.49 | 4.66 | $ | 3.41 | 919,302 | $ | 3.41 | |||||||||||||
| 6,500 | 4.50 - 5.99 | 6.25 | $ | 5.58 | 5,479 | $ | 5.58 | |||||||||||||
| 6,668 | 6.00 - 7.49 | 1.79 | $ | 6.75 | 6,668 | $ | 6.75 | |||||||||||||
| 86,666 | 7.50 - 10.49 | 6.36 | $ | 8.88 | 84,166 | $ | 8.89 | |||||||||||||
| 563,229 | 10.50 - 15.00 | 6.82 | $ | 13.65 | 402,179 | $ | 13.64 | |||||||||||||
| 1,036,793 | 15.01 - 29.99 | 8.26 | $ | 20.13 | 475,252 | $ | 19.56 | |||||||||||||
| 541,200 | 30.00 - 45.00 | 9.78 | $ | 32.47 | 30,001 | $ | 32.33 | |||||||||||||
As of December 31, 2010, after considering estimated forfeitures, there were 3,048,010 options outstanding that were either vested or expected to vest in the future, of which 1,923,047 options were currently exercisable, with weighted average exercise prices of $15.50 and $10.25 per share, aggregate intrinsic values of $85,518,016 and $64,061,816 and weighted average remaining contractual terms of 7.08 and 6.09 years, respectively.
Restricted Stock - Restricted stock has been issued to the Company’s non-employee directors and to a consultant. Restricted stock issued to non-employee directors prior to fiscal 2010 vested no later than one year from the date of issuance, as long as the director remained in continuous service to the Company as of the vest date. Restricted stock issued to non-employee directors subsequent to fiscal 2009 vests 50% on the first anniversary of the date of grant, 25% on the second anniversary, and 25% on the third anniversary, provided that the director is and has remained in continuous service to the Company as a director as of such anniversary. Restricted stock issued to a consultant vests equally on a quarterly basis over four years.
With regard to restricted stock granted to non-employee directors, the fair value of the restricted stock issued was determined using the closing price of the Company’s common stock as reported on the Global Market of The NASDAQ Stock Market LLC (“NASDAQ”) on the date of grant and is recognized as stock-based compensation expense as the shares vest over the vesting period. With regard to the restricted stock granted to the consultant, stock-based compensation expense equal to the fair value of the restricted shares that vest is recorded on a quarterly
16
Table of Contents
basis over the vesting period of four years. The fair value of each of the restricted shares that vest is equal to the fair value of a share of the Company’s common stock as of each vesting date.
A summary of the Company’s restricted stock activity during the three months ended December 31, 2010 is as follows:
| Number of Shares |
||||
| Outstanding - September 30, 2010 |
66,666 | |||
| Granted |
— | |||
| Forfeited |
— | |||
| Outstanding - December 31, 2010 |
66,666 | |||
As of December 31, 2010, holders were vested in 54,666 of the 66,666 restricted shares outstanding, leaving a total of 12,000 restricted shares unvested as of quarter end. The weighted average fair value of the shares granted in fiscal 2010 was $29.01 per share.
The Company recognized compensation expense of $44 thousand and $66 thousand during the three months ended December 31, 2010 and 2009, respectively, related to restricted stock issued to its non-employee directors and to the consultant. Unrecognized compensation expense for the restricted shares granted to the non-employee directors was $211 thousand at December 31, 2010. This amount will be recognized over the remaining vesting period of the restricted shares.
8. STOCKHOLDERS’ EQUITY AND WARRANTS
Common Stock - As of December 31, 2010, the Company had 100,000,000 shares of common stock authorized with a par value of $0.001 per share and the Company had reserved 3,167,192 shares of common stock for issuance upon the exercise of outstanding common stock options. Also, 209,826 shares of the Company’s common stock were reserved for future grants of stock options (or other similar equity instruments) under the Company’s Revised 2007 Plan as of December 31, 2010. The Company’s Board of Directors has approved an increase of 2,000,000 shares to the shares of the Company’s common stock reserved for future grants under the Revised 2007 Plan. This increase will be presented to the Company’s stockholders for their approval at the Company’s 2011 annual stockholders meeting to be held on March 23, 2011. In addition, 63,623 shares of the Company’s common stock were reserved for future exercise of outstanding warrants as of December 31, 2010.
Warrants - In conjunction with entering into a Loan Agreement and with executing three secured promissory notes (See Note 6), the Company granted warrants to the lender to purchase 127,248 shares of the Company’s common stock at an exercise price of $12.05 per share. During the three months ended December 31, 2010, the lender elected to exercise 63,625 warrants using the net issuance method, which resulted in the issuance of 45,848 shares of common stock by the Company. The remaining warrants expire seven years from the date of grant (or upon a change of control as defined in the Loan Agreement) as follows: 22,130 expire on September 30, 2014, 30,428 expire on March 28, 2015, and 11,065 expire on December 12, 2015.
9. INCOME TAXES
There was no income tax expense during the three months ended December 31, 2010 and 2009. The Company’s effective tax rate for the three months ended December 31, 2010 and 2009 was 0% due to uncertainties related to the realizability of the deferred tax assets as a result of the Company’s history of operating losses. The net deferred tax asset as of December 31, 2010 remains fully offset by a valuation allowance since it is more likely than not that such tax benefits will not be realized.
17
Table of Contents
As of September 30, 2010, the Company had United States federal net operating loss (“NOL”) carryforwards of approximately $219.5 million available to offset future taxable income, if any. Of the federal NOLs, $14.1 million was generated from windfall tax benefit stock option deductions. The tax benefit of this portion of the NOL will be accounted for directly to equity as additional paid in capital as the stock option related losses are utilized. As of September 30, 2010, the Company also had research and development tax credits of $0.1 million available to offset future tax liabilities. The loss carryovers and the research and development tax credits expire over a period of 2020 to 2030.
As of September 30, 2010, the Company’s unrecognized tax benefits of $0.1 million have not significantly changed since October 1, 2007. The Company does not expect any significant changes to the unrecognized tax benefits within 12 months of the reporting date.
The Internal Revenue Service (“IRS”) could challenge tax positions taken by the Company for the periods for which there are open tax years. The Company is open to challenge for the periods of 2004-2009 from federal and state jurisdictions.
Under Section 382 of the Internal Revenue Code (the “Code”), utilization of the NOL and research and development tax credit carryforwards may be subject to a limitation if a change in ownership of the Company, as defined in the Code, occurred previously or could occur in the future. The Company completed a Section 382 analysis regarding limitation of its NOL and research and development tax credit carryforwards that covered the period three years prior to its IPO on May 2, 2007 through a public offering of its common stock on February 5, 2009, and concluded that a change in control occurred at the Company during the quarter ended September 30, 2008. This change in control limits the future use of the Company’s NOL and research and development tax credit carryforwards from fiscal 2008 and prior years. However, based upon the Company’s financial projections, it does not believe that this limitation will result in the expiration of any of these NOL and research and development tax credit carryforwards before they are able to be utilized. The Company is in the process of assessing whether another change in control occurred since the quarter ended September 30, 2008 and expects to disclose the results of this assessment when it is complete. Such a change and any future changes in ownership could impact the use of the Company’s NOL and research and development tax credit carryforwards generated in the affected years. Any limitation may result in expiration of a portion of the NOL or research and development tax credit carryforwards before utilization, which would reduce the Company’s gross deferred tax assets.
On October 29, 2010, the Company was awarded two grants ($244,479 each) totaling $489 thousand under the IRS Qualifying Therapeutic Discovery Project (QTDP) program, which was created by Congress as part of the Patient Protection and Affordable Care Act of 2010. The grants were received on November 12, 2010. One of the grants was awarded for the development of PSI-7977 and the other grant was awarded for the development of PSI-938 or PSI-661. All three of these product candidates are being developed for the treatment of HCV and all of the $489 thousand was recorded as Other income in the Statement of Operations during the three months ended December 31, 2010.
10. COMMITMENTS AND CONTINGENCIES
The Company has entered into an operating lease for office and laboratory space located in Princeton, New Jersey through May 22, 2015. The Company has also entered into an operating lease for office space located in Durham, North Carolina through December 31, 2015.
As of December 31, 2010, minimum future payments under non-cancellable operating leases are as follows:
18
Table of Contents
| December 31, 2010 | ||||
| (In thousands) | ||||
| Fiscal 2011 |
660 | |||
| Fiscal 2012 |
918 | |||
| Fiscal 2013 |
920 | |||
| Fiscal 2014 |
922 | |||
| Fiscal 2015 |
626 | |||
| Thereafter |
22 | |||
| Total minimum payments required |
$ | 4,068 | ||
Under a license agreement with Emory University for Racivir, the Company agreed to pay Emory University up to an aggregate of $1.0 million in future marketing milestone payments. None of these potential future payments are included in the Company’s financial statements, as the payments are contingent on the achievement of milestones, which it has not yet achieved.
On July 28, 2009, Emory University and University of Georgia Research Foundation, Inc. (“Claimants”) filed a Demand for Arbitration and Relief (the “Demand”) with the American Arbitration Association (“AAA”) in Atlanta, Georgia (the “Emory Arbitration”), claiming certain payments and seeking specific performance under the Company’s January 8, 2004 license agreement with Claimants (the “Emory License”).
The Demand alleged that payments Pharmasset had received under the Roche collaboration agreement were subject to the Emory License and that Pharmasset had not paid fees to Claimants based on such payments. In addition, the Demand alleged that Pharmasset had not complied with certain terms and conditions of the Emory License and that other Pharmasset product candidates were, or will be, covered by the Emory License. The Demand requested, among other things, specific performance of the Emory License, including the payment of license fees related to past payments received by Pharmasset. The Company’s response to the Demand was filed on August 14, 2009.
On December 6, 2010 a final arbitration award (the “Award”) was issued by a panel of AAA arbitrators. According to the Award, none of the payments the Company received under the Roche collaboration agreement were subject to the Emory License and, therefore, no license fees were owed to Emory based upon such payments. Furthermore, according to the Award, none of the other Company product candidates that were subject to the Demand are covered by the Emory License.
11. SUBSEQUENT EVENT
On January 26, 2011, the Company completed an underwritten public offering of 2,795,000 shares of the Company’s common stock, which includes the underwriter’s exercise in full of its over-allotment option of 495,000 shares and excludes 1,000,000 shares that were sold by selling stockholders, for a price to the public of $46.33 per share. The underwriter purchased the shares from the Company at a price of $44.25, pursuant to the underwriting agreement. The Company’s net proceeds from the sale of the shares, after deducting the underwriter’s discount and estimated offering expenses, was $123.4 million.
19
Table of Contents
| ITEM 2. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis should be read together with our condensed financial statements and the related notes to those condensed financial statements included elsewhere in this Quarterly Report on Form 10-Q.
Overview
We are a clinical-stage pharmaceutical company committed to discovering, developing, and commercializing novel drugs to treat viral infections. Our primary focus is on the development of nucleoside/tide analogs as oral therapeutics for the treatment of chronic hepatitis C virus (“HCV”) infection. Nucleoside/tide analogs are a class of compounds which act as alternative substrates for the viral polymerase, thus inhibiting viral replication. We currently have three clinical-stage product candidates, two of which we are developing ourselves and one of which we are developing with a strategic partner. We are also advancing a series of preclinical candidates in preparation for clinical development. Our three clinical stage product candidates are:
| • | RG7128, an HCV cytosine nucleoside polymerase inhibitor we are developing through a strategic collaboration with F. Hoffmann-La Roche Ltd and Hoffmann-La Roche Inc. (collectively, “Roche”). In October 2010, Roche presented data from a 12-week interim analysis from the Phase 2b “PROPEL” study of RG7128 in combination with Pegasys® (pegylated interferon) plus Copegus® (ribavirin), the standard of care for treating HCV (“SOC”) in patients with HCV genotypes 1or 4. In addition, RG7128 is in a 24-week Phase 2b “JUMP-C” study in combination with SOC in patients with HCV genotypes 1 or 4. Roche is planning to conduct the next study of RG7128 in combination with ritonavir-boosted danoprevir. This “INFORM-SVR” study is part of a series of studies designed to investigate the combination of two oral, direct acting antivirals (“DAAs”) in the absence of pegylated interferon. Roche is also planning to conduct a Phase 2b study in patients with HCV genotypes 2 or 3. All of these studies are being, or are expected to be, conducted by Roche; |
| • | PSI-7977, an HCV uracil nucleotide analog polymerase inhibitor that is in a 12-week Phase 2b dose-finding study (“PROTON”) in combination with SOC in patients with HCV genotypes 1, 2 or 3. In addition, PSI-7977 recently began dosing in an exploratory Phase 2 study (“ELECTRON”) in combination with ribavirin, administered without and with varying durations of pegylated interferon, in patients with HCV genotypes 2 or 3. We expect to initiate a 24-week Phase 2b study of PSI-7977 in combination with SOC during the second calendar quarter of 2011; and |
| • | PSI-938, an HCV guanine nucleotide analog polymerase inhibitor that is enrolling patients in Part 2 of a Phase 1 study with PSI-7977 in patients with HCV genotype 1. We plan to initiate a Phase 2 study of PSI-938 in combination with PSI-7977 during mid (calendar year) 2011 |
In addition, we are developing PSI-661, an HCV guanine nucleotide analog polymerase inhibitor we nominated as a development candidate in October 2009. PSI-661 is in preclinical studies required for submission of an Investigational New Drug (“IND”) application with the U.S. Food and Drug Administration (“FDA”) or equivalent foreign regulatory application. PSI-938 or PSI-661 could potentially be used in combination with our current nucleoside/tide analogs, RG7128 or PSI-7977, as well as other classes of DAAs. Given the similarities of PSI-938 and PSI-661, our plan is to select one of these product candidates for later-stage clinical development based upon a review of the early human clinical trial results of both PSI-938 and PSI-661.
We are continuing to research nucleoside/tide analogs (both pyrimidines and purines) with the intention of identifying additional product candidates that can potentially be used in combination with our nucleoside/tides, RG7128 and PSI-7977, in combination with other classes of DAAs, or with SOC for the treatment of HCV. We have identified proprietary nucleotide prodrugs that are referred to as phosphate prodrugs because they have the ability to
20
Table of Contents
deliver the biologically available monophosphate forms of the compounds into infected liver cells, thus bypassing a rate-limiting step in the metabolic pathway to the active triphosphate form of the drug. The goal of these efforts is to identify compounds with improved potency, safety, convenience, oral bioavailability, and increased intrahepatic nucleotide triphosphate levels. Certain of these compounds have demonstrated exceptional in vitro anti-HCV activity, with up to 100 times greater potency than PSI-6130 (of which RG7128 is a prodrug). Early studies in animals indicate that several of these compounds can achieve concentrations of the active triphosphate form in the liver up to 1000 times higher than PSI-6130 at equivalent doses.
We are developing PSI-7977, PSI-938, and PSI-661 ourselves. We have a strategic collaboration with Roche for the development of PSI-6130 and its prodrugs, including RG7128. Under the collaboration, Roche pays all development costs associated with RG7128 and provides us with potential income from milestone payments that can be used to fund the advancement of our proprietary product candidates.
21
Table of Contents
Our Product Candidates
Our research and development programs are primarily focused on discovering and developing drugs that treat HCV. Our product candidates are nucleoside/tide analogs that we believe have potential competitive advantages with respect to safety, efficacy, drug resistance, and/or convenience of dosing as compared to currently approved drugs and other known investigational agents. The following table summarizes the four product candidates on which we are focusing:
| Product Candidate |
Status |
Next Expected Milestone(s) |
Commercialization Partner | |||
| RG7128 | • Completing the Phase 2b “PROPEL” study and 24-week Phase 2b “JUMP C” study in patients with HCV genotypes 1 or 4. • Planning for an INFORM-SVR study and a Phase 2b study in patients with HCV genotypes 2 or 3. All of the above studies are being, or are expected to be, conducted by Roche. |
• Initiate INFORM-SVR study during the first calendar quarter of 2011. • Initiate a Phase 2b study in patients with HCV genotypes 2 or 3 during the first half of calendar year 2011. • Initiate a Phase 3 program during 2011. • All of the above studies are expected to be conducted by Roche. |
Roche | |||
| PSI-7977 | • In a 12-week Phase 2b dose-finding “PROTON” study in combination with SOC in patients with HCV genotypes 1, 2, or 3. • In a Phase 2 exploratory study “ELECTRON” in combination with ribavirin, administered without and with varying durations of pegylated interferon, in patients with HCV genotypes 2 or 3. |
• Report SVR121 results from the ongoing genotype 2/3 arm of the Phase 2b “PROTON” study during the second calendar quarter of 2011. • Report 12 week interim analysis from the genotype 1 arms of the Phase 2b study “PROTON” during the second calendar quarter of 2011. • Report interim data from the Phase 2 exploratory study “ELECTRON” in combination with ribavirin in the second half of calendar year 2011. • Initiate a 24-week Phase 2b study in combination with SOC during the second calendar quarter of 2011. • Initiate proof of concept study of PSI-7977 in combination with BMS-790052 during the first half of 20112. |
— | |||
| PSI-938 | • In Part 2 of a 14 day Phase 1 study with PSI-7977 in patients with HCV genotype 1. |
• Report preliminary results from Part 2 of the Phase 1 study during the quarter ending March 31, 2011. • Initiate a Phase 2 combination study with PSI-7977 in mid (calendar year) 2011. |
— | |||
| PSI-661 | • In IND-enabling preclinical studies. |
• Submit IND application during the first calendar quarter of 2011. • Initiate a Phase 1 study during the second calendar quarter of 2011. |
— | |||
| 1 | SVR12 – Sustained virologic response 12, or SVR12, is defined as a level of HCV RNA in a patient that is below the limit of detection (<15 IU/ml) 12 weeks after the discontinuation of therapy. |
| 2 | During January 2011, we entered into a clinical collaboration agreement with Bristol-Myers-Squibb Company (“BMS”) to evaluate the utility of PSI-7977 in combination with BMS-790052, BMS’s NS5a replication complex inhibitor, for the treatment of HCV. BMS is responsible for all costs of the study, except for the cost of PSI-7977, which will be supplied by Pharmasset. Neither party has licensed any commercial rights to the other party. |
22
Table of Contents
Product Candidates for the Treatment of HCV
HCV Background
HCV is a leading cause of chronic liver disease and liver transplants. The World Health Organization estimates nearly 180 million people worldwide, or approximately three percent of the world’s population, are infected with HCV. About 130 million of these individuals are chronic HCV carriers who are at an increased risk of developing liver cirrhosis or liver cancer, approximately 15 million of whom are in the United States, Europe, and Japan. The Centers for Disease Control and Prevention (“CDC”) has reported that 4.1 million people in the United States have been infected with HCV, of whom 3.2 million are chronically infected. Of those chronically infected, the majority are undiagnosed and unaware of their HCV infection. Separately, approximately ten percent of diagnosed HCV patients in the United States are treated each year.
At least six major genotypes of HCV have been identified, each with multiple subtypes. Genotypes are designated with numbers (genotypes 1-6) and subtypes with letters. HCV genotypes 1, 2, 3, and 4 have a worldwide distribution, but their prevalence varies from one geographic area to another. Genotype 1 and its subtypes (1a and 1b) are the most common genotype globally, accounting for approximately 70% of infections. In the United States, approximately 67% and 33% of all of the genotype 1 HCV infections are subtypes 1a and 1b, respectively. Patients with genotype 2 or 3 represent approximately 25% of the worldwide chronically infected HCV population and the remaining five percent is comprised of genotypes 4 through 6. Worldwide sales of HCV drugs in 2005 were approximately $2.2 billion and are forecasted to reach more than $8.0 billion in 2015. Historically, sales of HCV drugs increase as new therapies are introduced that improve the sustained virologic response (“SVR”), defined as the inability to detect HCV RNA in a patient’s blood six months after discontinuation of therapy, with a standard polymerase chain reaction (“PCR”) test, which measures the amount of HCV in the blood.
Limitations of Current HCV Infection Therapy
The current standard of care for treating HCV is a combination of pegylated interferon plus ribavirin. Pegylated interferon is a modified version of alpha interferon, a protein that occurs naturally in the human body and boosts the immune system’s ability to fight viral infections. Roche, our collaboration partner in the development of RG7128, is the market leader in sales of pegylated interferon and branded ribavirin under the brand names Pegasys® and Copegus®, respectively.
Patients currently being treated for HCV are given pegylated interferon as a weekly injection, administered together with twice daily ribavirin tablets. The current SOC, however, has limitations that result in less than optimal SVR rates. Substantial side effects can render treatment intolerable for many patients. For example, SOC-treated patients can have difficulties with fatigue, bone marrow suppression, anemia, and neuropsychiatric effects. In addition, genotype 1 patients typically receive 48 weeks of SOC, but less than 50% of these patients achieve an SVR, which many physicians and patients consider a low rate of success. Between 60% and 80% of the genotype 2 and 3 patients treated with SOC for 24 weeks achieve an SVR. The occurrence of side effects combined with the inconvenient treatment regimen can result in many patients not completing therapy. Furthermore, a majority of individuals with HCV are unable to be treated with interferon due to contraindications, such as advanced liver disease or psychiatric conditions. The less than optimal antiviral efficacy, potential for dose-limiting side effects (some of which can be serious), contraindications, and inconvenient dosing regimen illustrate the unmet medical need of the currently available SOC. Current therapies may also not directly target the virus, suggesting additional patient benefit from agents which directly interfere with HCV replication.
Nucleoside/tide Analogs and Other Direct Acting Antivirals for HCV
HCV has several viral specific enzymes that are essential for its replication, thus providing multiple opportunities for therapeutic intervention. Many drug developers have focused on three of the HCV enzymes: a protease (“NS3”), the polymerase (“NS5b”) and more recently, another protein, “NS5a”. The goal of HCV drug development is to discover and develop molecules that have a high affinity for binding to these enzymes thereby inhibiting enzymatic activity and, in turn, inhibiting viral replication. These compound classes are often referred to as protease inhibitors and polymerase inhibitors. There are two types of polymerase inhibitors, each with a different
23
Table of Contents
mechanism of action. Nucleoside/tide analog polymerase inhibitors work by acting as alternative substrates that block the synthesis of HCV RNA, which is essential for the virus to replicate. The other type of polymerase inhibitor, non-nucleoside polymerase inhibitors, binds directly to the polymerase enzyme, causing a change in its shape. This conformational change inhibits its enzymatic activity.
Our research efforts focus on blocking HCV replication by discovering and developing nucleoside/tide analog polymerase inhibitors. A nucleoside is a basic building block of the nucleic acids, DNA and RNA, the genetic material of all living cells and viruses. Nucleosides consist of a molecule of sugar linked to a nitrogen-containing organic ring compound. In the most important naturally occurring nucleosides, the sugar is either ribose (used to construct RNA) or deoxyribose (used to construct DNA), and the nitrogen-containing organic ring compound, referred to as the base, is either a pyrimidine (cytosine, thymine, or uracil) or a purine (adenine or guanine). A nucleoside combined with a phosphate group becomes a nucleotide.
In biological systems, nucleotides are linked by enzymes, including the polymerase, in a specific order to make long, chainlike polynucleotides (DNA or RNA) of defined sequence to pass along genetic information for a specific protein, a gene, or an entire organism, a genome. A nucleoside analog is a synthetic molecule that resembles a naturally occurring nucleoside. Chemical modifications in either the sugar portion or the base portion allow these compounds, once phosphorylated, to inhibit or disrupt the activity of the polymerase. When a nucleotide analog is incorporated into viral DNA or RNA during replication, it acts to prevent production of new virus by blocking the complete synthesis of the new viral DNA or RNA genome.
Experiments in vitro conducted by us and others show that nucleoside/tide analogs have conserved antiviral activity across all HCV genotypes. This characteristic of the nucleoside/tide analog class relates to its unique mechanism of action. Recent clinical studies of RG7128, as more fully described below, show comparable anti-HCV activity across HCV genotypes 1, 2, and 3. Other classes of anti-HCV drugs (i.e., protease inhibitors and non-nucleoside polymerase inhibitors) have not yet shown comparable activity across a broad spectrum of HCV genotypes.
In clinical monotherapy studies with three separate nucleoside/tide analogs (including RG7128) over 14 days, viral breakthrough while on therapy did not occur. In studies of non-nucleoside polymerase and protease inhibitors, viral breakthrough was seen as early as three to four days into the 14-day treatment period. The relative rapidity of the breakthrough with these classes of drugs suggests that the patients may have harbored HCV strains that were not susceptible to at least one component of the therapeutic regimen. With longer exposure to any DAA, drug resistant virus may be selected over time. The rapidity and frequency with which this occurs may have significant consequences for patients, including not obtaining an SVR.
Summary of Nucleoside/tide Analogs and Their Potential Use as Future Therapy
Current market research identifies the three most important attributes for improving HCV therapy, in order of importance, as: greater efficacy, improved tolerability, and shorter duration of treatment. Current guidance from regulatory authorities indicates that approval of DAAs for HCV must include their use in combination with SOC. Therefore, most efforts to improve SVR rates focus on adding a DAA to SOC. It is hoped that the addition of a DAA which directly inhibits viral growth will result in a more efficacious therapy without adding to the intolerability of interferon and ribavirin.
We and other developers of HCV DAAs are also currently investigating combination treatments with two or more DAAs in the absence of interferon. These DAA combinations may include a nucleoside/tide with a protease inhibitor, such as RG7128 with Roche’s ritonavir-boosted danoprevir, which are currently in the INFORM studies being conducted by Roche. Or they may include one nucleoside/tide combined with a second complementary nucleoside/tide, such as PSI-938 and PSI-7977. We believe the use of two DAAs would improve tolerability and may lead to a shorter duration of treatment. Due to the unique attributes of nucleoside/tides, including their ability to have complementary resistance profiles and to show comparable activity across a broad spectrum of HCV genotypes, we believe that dual nucleoside/tides combinations could possess advantages over other DAA combinations that do not contain a nucleoside/tide.
24
Table of Contents
Pyrimidine Nucleoside/tide Product Candidates
RG7128 Development
Phase 1 Studies. In October 2004, we entered into a collaboration with Roche for the development and commercialization of PSI-6130 (an oral cytosine nucleoside analog polymerase inhibitor which we discovered) and its prodrugs, including RG7128, for the treatment of HCV. A prodrug is a chemically modified form of a molecule designed to enhance the absorption, distribution, and metabolic properties of that molecule. Roche and we initiated an adaptive Phase 1 clinical trial with RG7128 in October 2006 under an IND filing. On October 12, 2007, we were informed by the FDA that RG7128 received fast track designation. During December 2008, we completed the Phase 1 clinical trial of RG7128. Following is a review of the design and results of this trial.
This adaptive Phase 1 clinical trial of RG7128 was a multiple center, observer-blinded, randomized, and placebo-controlled study designed to investigate the pharmacokinetics, pharmacodynamics, safety, tolerability, and food effect of RG7128 in healthy subjects and in patients chronically infected with HCV genotypes 1, 2, or 3. This trial provided antiviral potency data over 14 and 28 days in patients chronically infected with HCV genotype 1, and over 28 days of treatment in patients chronically-infected with HCV genotypes 2 or 3 who had not responded to prior interferon-based therapy. This study included three parts:
| • | Part 1 was a single ascending dose (“SAD”) study conducted in 46 healthy subjects. The primary objective of Part 1 was to assess the safety, tolerability, and pharmacokinetics of RG7128 following single ascending doses under fasting conditions. The secondary objective of Part 1 was to explore the effect of food on the pharmacokinetics of RG7128. Single oral doses of RG7128 were administered to 46 healthy subjects in five sequential dose groups (500mg, 1500mg, 4500mg, 6000mg, and 9000mg) and one food effect group (1500mg). Results from the single ascending dose portion of the study indicated: |
| • | All doses of RG7128 studied (500mg to 9000mg) were generally safe and well-tolerated. |
| • | All patients completed the study and none experienced gastrointestinal adverse events or serious adverse events during the study. |
| • | No hematological or other safety laboratory abnormalities of clinical significance were noted. |
| • | No maximum tolerated dose was identified. |
| • | Part 2 was a multiple ascending dose study (“MAD”) conducted in 40 patients chronically infected with HCV genotype 1 who had previously failed interferon therapy. The primary objective of Part 2 was to assess the safety, tolerability, and pharmacokinetics of RG7128 after once-daily (“QD”) or twice-daily (“BID”) dosing for 14 days. The secondary objective was to assess antiviral efficacy by measuring the change from baseline in circulating HCV RNA. Results from the multiple ascending dose portion of the study indicated: |
| • | RG7128 demonstrated potent, dose-dependent antiviral activity in four patient cohorts (8 active and 2 placebo per cohort) receiving 750mg or 1500mg administered either QD or BID for 14 days as monotherapy. RG7128 demonstrated mean HCV RNA decreases from baseline of 0.9 log10 (87.4% reduction), 1.5 log10 (96.8% reduction), 2.1 log10 (99.2% reduction), and 2.7 log10 (99.8% reduction) in patients receiving 750mg QD, 1500mg QD, 750mg BID, and 1500mg BID, respectively. A maximum reduction in HCV RNA of 4.2 log10 (99.9% reduction) was demonstrated in a patient following 14 days of monotherapy with RG7128 1500mg BID, a value also below the limit of detection, which was 15 International Units per milliliter (IU/ml). |
| • | There was no evidence of drug resistance in any dose cohort during the 14 days of dosing. |
| • | RG7128 was generally safe and well tolerated over 14 days of treatment. |
25
Table of Contents
| • | Part 3 was a 4-week study of RG7128 in combination with SOC in 81 treatment-naïve patients chronically infected with HCV genotype 1, and additionally, in 25 prior treatment non-responders, or patients who did not achieve an SVR with previous interferon-based therapy, who were chronically infected with HCV genotypes 2 or 3. The primary objective of this study was to assess the safety, tolerability, and pharmacokinetics of RG7128 in the clinically-relevant setting of combination therapy with SOC for chronic HCV infection. The secondary objective of Part 3 was to evaluate the short-term change in HCV RNA. The study included three oral dose regimens of RG7128 (500mg, 1000mg, and 1500mg BID – cohorts 1, 2, and 3, respectively) in patients chronically infected with HCV genotype 1 and one oral dose regimen of RG7128 (1500mg BID – cohort 4) in patients chronically infected with HCV genotypes 2 or 3. |
The antiviral results for cohorts 1, 2, and 3 are summarized in the following table:
| RG7128 dose |
N | Mean HCV RNA change from Baseline |
HCV
RNA <LLOD1 (<15IU/mL) N (%) |
|||||||||
| 500mg BID + SOC |
20 | -3.8 | 6/20 | (30%) | ||||||||
| 1000mg BID + SOC |
25 | -5.1 | 22/25 | (88%) | ||||||||
| 1500mg BID + SOC |
20 | -5.1 | 17/20 | (85%) | ||||||||
| Placebo + SOC |
16 | -2.9 | 3/16 | (19%) | ||||||||
1 – LLOD means lower limit of detection by Roche Taqman Assay
For cohorts 1, 2, and 3 in treatment-naïve genotype 1 patients, RG7128 was generally safe and well-tolerated when administered for 4 weeks in combination with SOC in patients with HCV genotype 1.
The antiviral results for the 1500mg BID dose cohort (cohort 4) in 25 prior treatment non-responders (patients who did not achieve an SVR with previous interferon-based therapy) who were chronically infected with HCV genotype 2 or 3 are summarized in the following table:
| RG7128 dose |
N | Mean HCV RNA change from Baseline |
HCV
RNA <LLOD1 (<15IU/mL) N (%) |
|||||||||
| 1500mg BID + SOC |
20 | -5.0 | 19/20 | (95%) | ||||||||
| Placebo + SOC |
5 | -3.7 | 3/5 | (60%) | ||||||||
1 – LLOD means lower limit of detection by Roche Taqman Assay
26
Table of Contents
RG7128 was generally safe and well-tolerated in cohort 4. After receiving 4 weeks of RG7128 in combination with SOC, the 20 patients infected with HCV genotype 2 or 3 continued to receive SOC alone for an additional 20 to 44 weeks as determined by their prior response and HCV genotype. Twenty-four weeks after the end of all treatment, 65% (13 of 20) of these patients demonstrated an SVR.
Phase 2b Study. In April 2009, Roche began dosing in a Phase 2b “PROPEL” study with RG7128. During May 2010, dosing of RG7128 triple combination therapy (RG7128 plus SOC) or placebo plus SOC in 408 treatment-naïve, genotype-1 or genotype-4 HCV-infected patients (cirrhotic and non-cirrhotic) was completed. The trial is evaluating the dose and duration of treatment of RG7128 in combination with SOC. The primary efficacy endpoint of the trial will be the proportion of patients that achieve an SVR. Subjects were equally randomized into one of 5 arms of the study:
| • | 24 weeks of total treatment, with RG7128 500mg BID in combination with SOC for 12 weeks, followed by 12 weeks of SOC (n = 80) |
| • | 24 weeks of total treatment, with RG7128 1000mg BID in combination with SOC for 12 weeks, followed by 12 weeks of SOC (n = 82) |
| • | 24 weeks of total treatment, with RG7128 1000mg BID in combination with SOC for 8 weeks, followed by 16 weeks of SOC (n = 81) |
| • | 48 weeks of total treatment, with RG7128 1000mg BID in combination with SOC for 12 weeks, followed by 36 weeks of SOC (n = 81) |
| • | A control arm with placebo in combination with SOC for 48 weeks (n = 84) |
Patients in the 24 week cohorts discontinued treatment at week 24 if they achieved a rapid virologic response (“RVR”), defined as HCV RNA below the limit of detection (<15 IU/mL as measured by Roche TaqMan assay) four weeks after the initiation of treatment, that is maintained until week 22, a strategy known as “response-guided” treatment. Patients who did not meet these virologic criteria will continue on SOC until week 48.
Results from an interim analysis of all 408 patients who had completed the first 12 weeks of the PROPEL study indicated the following:
| • | RG7128 1000mg BID with SOC for 12 weeks achieved a high rate of complete Early Virologic Response (“cEVR”, defined as HCV RNA below the level of detection 12 weeks after the initiation of treatment) of 83% with no on-treatment viral breakthrough, |
| • | The safety and tolerability of RG7128 1000mg BID with SOC were comparable to placebo/SOC with no renal or hematologic safety signals, and a discontinuation rate similar to placebo/SOC, and |
| • | No drug resistance was observed in patients treated with up to 12 weeks of RG7128. |
An amendment to the protocol for the PROPEL study has been implemented which allows patients who were initially randomized to the placebo/SOC arm and who are non-responders to receive open label RG7128 1000mg BID in combination with SOC for 24 weeks, followed by an additional 24 weeks of SOC. Non-response is defined as a patient who does not achieve at least a 2 log decline in HCV RNA by week 12 of therapy, or who has HCV RNA above the limit of detection (15 IU/mL) at week 24 of therapy. This amendment will provide longer-term treatment data on patients with prior non-response to SOC, including demonstrated null responders.
Roche is also conducting a 24-week Phase 2b “JUMP-C” study of RG7128 in combination with SOC in 168 treatment-naïve patients with HCV genotypes 1 and 4 to evaluate the safety and efficacy of RG7128 in combination with SOC. Patients with HCV RNA below the limit of detection at the end of week 4 through week 22 will stop all therapy (RG7128 and SOC) at week 24, while patients who do not meet this response guideline will receive a full 48 weeks of SOC. Supportive data from this study could provide the flexibility for longer dosing of RG7128 which may be required in some populations, as well as combinations of RG7128 with other direct acting antivirals currently in
27
Table of Contents
development. This study completed enrollment during the second calendar quarter of 2010 and is being conducted at sites in the U.S. and Canada. Patients were initially equally randomized into one of two arms of the study:
| • | 24 weeks of treatment, with RG7128 1000mg BID in combination with SOC |
| • | A control arm with placebo and SOC for 48 weeks |
An amendment to the protocol for this 24-week study has been implemented which allows patients who were initially randomized to the placebo/SOC arm and who are non-responders to receive open label RG7128 1000mg BID in combination with SOC for 24 weeks, followed by an additional 24 weeks of SOC. Non-response is defined as a patient who does not achieve at least a 2 log decline in HCV RNA by week 12 of therapy, or who has HCV RNA above the limit of detection (15 IU/mL) at week 24 of therapy. Patients from this study as well as the initial Phase 2b “PROPEL” study of 12 weeks’ RG7128 who are randomized to placebo/SOC (described above) will provide longer-term treatment data on patients with prior non-response to SOC, including demonstrated null responders.
In addition, Roche is planning to initiate a Phase 2b study of RG7128 in combination with SOC in patients with HCV genotypes 2 and 3 during the first half of calendar year 2011. RG7128 in combination with SOC has previously demonstrated antiviral activity in HCV genotypes 2 and 3 prior non-responders in a 28 day clinical trial, with an RVR of 95% and an SVR of 65%. Roche is also planning to initiate a Phase 3 program for RG7128 during 2011 and plans to submit a marketing application for RG7128 to one or more regulatory authorities in 2013.
PSI-7977 Development
PSI-7977 is an isomer of PSI-7851, a prodrug of a uracil nucleotide analog polymerase inhibitor we are developing for the treatment of chronic HCV infection. PSI-7851 demonstrated potent in vitro anti-HCV activity with EC90 values of 0.44 +/- 0.21 µM, between 14 and 17-fold more potent than the active metabolite of our cytosine nucleoside polymerase inhibitor, PSI-6130. The in vitro half-life of the triphosphate (the biologically active form of the molecule) in primary human hepatocytes is approximately 38 hours, which supports the exploration of once-daily dosing in early studies. Like RG7128, PSI-7851 has demonstrated in vitro activity against HCV genotypes 1, 2, 3, and 4.
Phase 1 Studies. In March 2009, we initiated a Phase 1 study of PSI-7851, which was a single ascending dose (“SAD”) study that assessed the safety, tolerability, and pharmacokinetics of PSI-7851 in 42 healthy subjects at doses ranging from 25mg to 800mg. Results from this study indicated there were:
| • | No dose-limiting toxicity, |
| • | No serious adverse events, and |
| • | No clinically significant changes in vital signs or electrocardiograph (“ECG”) readings. |
In June 2009, we initiated a Phase 1 multiple ascending dose (“MAD”) study in HCV-infected patients. Forty subjects were enrolled at two U.S. centers and randomized to PSI-7851 (8 per cohort) or placebo (2 per cohort). The primary objective of this study was to assess the safety, tolerability, and pharmacokinetics of PSI-7851 after once-daily dosing for three days. The secondary objective of this study was to assess antiviral activity by measuring the change in circulating HCV RNA levels. Four dose cohorts of PSI-7851 (50mg QD, 100mg QD, 200mg QD, and 400mg QD) were evaluated. Results from this study indicated:
| • | PSI-7851 was generally safe and well tolerated across all cohorts with no discontinuations, no serious adverse events, and no dose-related trends in adverse events or laboratory abnormalities. |
| • | PSI-7851 demonstrated potent antiviral activity with a mean HCV RNA change from baseline of -0.49 log10 IU/mL in patients receiving 50mg QD and -0.61 log10 IU/mL in patients receiving 100mg QD. |
28
Table of Contents
| • | PSI-7851 200mg QD administered for 3 days resulted in a mean change from baseline HCV RNA of -1.01 log10 IU/mL, with 6 of 8 subjects achieving greater than a 1.0 log10 IU/mL decline from baseline. This antiviral effect met our threshold of approximately 1.0 log10 IU/mL decline over three days as established with the first-in-class nucleoside, RG7128. |
| • | PSI-7851 400mg QD administered for 3 days resulted in a mean change from baseline HCV RNA of -1.95 log10 IU/mL, with 6 of 8 subjects achieving greater than a 1.5 log10 IU/mL decline from baseline. |
Selection of PSI-7977. PSI-7851 is a mixture of two molecules of identical chemical composition, PSI-7976 and PSI-7977, which only differ in the stereo-orientation of one of the atoms on the prodrug. Once inside a liver cell, both molecules are rapidly converted to the same active triphosphate. PSI-7977 demonstrated potent in vitro anti-HCV activity with EC90 values of 0.42 +/- 0.23 µM, between 14 and 17-fold more potent than the active metabolite of our cytidine nucleoside polymerase inhibitor, PSI-6130. The in vitro half-life of the triphosphate in primary human hepatocytes is approximately 38 hours, which supports the exploration of once-daily dosing in early studies. Like RG7128, PSI-7977 has demonstrated in vitro activity against HCV genotypes 1, 2, 3, and 4. Given the improvements in manufacturing and slightly better in vitro potency, we selected PSI-7977 for further clinical development. We also made improvements in the formulation, which we evaluated in a Phase 1 study in healthy volunteers. In August 2010, we were informed by the FDA that PSI-7977 received fast track designation. Following are results from the Phase 2a study of PSI-7977, along with the design of a Phase 2b study we initiated in August 2010.
Phase 2a Study. During January 2010, we initiated a 28-day Phase 2a study of PSI-7977 and enrolled 63 patients with genotype 1 chronic HCV infection who had not been treated previously. The primary goal of the study was to determine the safety and tolerability of PSI-7977 in combination with SOC. The primary efficacy endpoint of the trial was the proportion of patients who achieve an RVR. Patients will continue to be followed through an SVR endpoint. Patients were randomized to receive one of four treatments:
| • | PSI-7977 100mg QD in combination with SOC for 28 days, followed by 44 weeks of SOC alone (n=16) |
| • | PSI-7977 200mg QD in combination with SOC for 28 days, followed by 44 weeks of SOC alone (n=18) |
| • | PSI-7977 400mg QD in combination with SOC for 28 days, followed by 44 weeks of SOC alone (n=15) |
| • | A control arm with placebo in combination with SOC for 48 weeks (n=14) |
The baseline HCV RNA for patients enrolled in the study ranged from 6.3 to 6.6 log10 IU/mL, across the cohorts. Results from this study are summarized in the following table:
| Study Arm |
Mean decrease in HCV RNA |
Percentage of Patients with (<15 IU/mL) at Day 28 | ||
| 100mg PSI-7977 QD + SOC |
-5.32 | 88%(14/16) | ||
| 200mg PSI-7977 QD + SOC |
-5.06 | 94%(17/18) | ||
| 400mg PSI-7977 QD + SOC |
-5.33 | 93%(14/15) | ||
| Placebo + SOC |
-2.80 | 21%(3/14) |
1 – LLOD means lower limit of detection by Roche Taqman Assay
PSI-7977 treatment in combination with SOC for 28 days was generally safe and well tolerated with no dose discontinuations due to adverse events and no dose-related laboratory parameter changes.
Phase 2b Dose-Finding Study (“PROTON”). In August 2010, we began dosing of PSI-7977 in combination with SOC in a 12-week Phase 2b study “PROTON”. This study is evaluating PSI-7977 200mg QD and 400mg QD in
29
Table of Contents
combination with SOC in approximately 125 treatment-naïve patients with HCV genotype 1. The primary goal of the study is to assess the safety and tolerability of PSI-7977 in combination with SOC for 12 weeks. The primary efficacy endpoint of the study is the proportion of patients who achieve an SVR12 and SVR24, defined as HCV RNA below the limit of detection (<15 IU/ml) 12 and 24 weeks, respectively, after the discontinuation of therapy. Patients receiving PSI-7977 in combination with SOC for 12 weeks will discontinue treatment at week 24 if their HCV RNA is below the level of detection at week 4 through week 12; otherwise, patients are expected to continue on SOC through week 48. Patients are being randomized into one of three arms as follows:
| • | PSI-7977 200mg QD in combination with SOC for 12 weeks, followed by 12 or 36 weeks of SOC (n=50), |
| • | PSI-7977 400mg QD in combination with SOC for 12 weeks, followed by 12 or 36 weeks of SOC (n=50), and |
| • | A control arm with placebo in combination with SOC for 48 weeks (n=25). |
We expect to report interim results from the first 12 weeks of treatment for the above three arms of the study during the second quarter of calendar year 2011.
In a fourth, open label arm of the study, we enrolled 25 treatment-naïve patients with HCV genotypes 2 or 3. Twenty-four patients received 12 weeks of PSI-7977 400mg QD in combination with SOC with no SOC follow-up. One patient was lost to follow-up after the first visit. Preliminary results from this open label arm indicated:
| • | All 24 patients achieved an RVR and remained below the limit of detection through the 12 week treatment period; |
| • | There were no serious adverse events and no discontinuations due to adverse events; and |
| • | There were no clinically significant, treatment emergent trends in any clinical laboratory parameters. |
Patients in the open label arm are being monitored for an additional 24 weeks after discontinuation of therapy to assess whether they achieved an SVR at 12 weeks and 24 weeks after the discontinuation of therapy. We expect to report SVR12 results from the genotype 2 or 3 arm of this study and an interim analysis of the genotype 1 arms of this study during the second quarter of calendar year 2011. We also expect to initiate a 24-week Phase 2b study of PSI-7977 in combination with SOC during the second calendar quarter of 2011.
Phase 2 Exploratory Study (“ELECTRON”). In December 2010, we began dosing of PSI-7977 400mg QD in combination with ribavirin, administered with and without pegylated interferon, in patients with HCV genotypes 2 or 3. This study is expected to enroll approximately 40 treatment-naïve patients. The primary goal of the study is to assess the safety and tolerability of PSI-7977 in combination with ribavirin for 12 weeks, with and without pegylated interferon. The study is being conducted in New Zealand and patients are being randomized into one of four arms as follows:
| • | PSI-7977 400mg QD in combination with ribavirin for 12 weeks (no pegylated interferon); |
| • | PSI-7977 400mg QD in combination with ribavirin for 12 weeks, with only four weeks of pegylated interferon); |
| • | PSI-7977 400mg QD in combination with ribavirin for 12 weeks, with only eight weeks of pegylated interferon); |
| • | PSI-7977 400mg QD in combination with ribavirin and pegylated interferon for 12 weeks. |
We expect to report interim data from this exploratory study during the second half of calendar year 2011.
30
Table of Contents
Combination Treatment with Two or More Direct Acting Antivirals
The use of interferon in the current standard of care limits the number of patients who can or are willing to undergo therapy for HCV and may result in less compliance with treatment regimens. We believe that the combination of two or more DAAs may provide an SVR in the absence of interferon. We believe that the selection of the drugs in such combinations should focus on molecules which provide potent viral suppression, lack metabolic interaction and possess complementary resistance profiles.
The frequent emergence of resistant variants in HCV inhibitor monotherapy trials with some classes of DAAs suggests that combinations of DAAs with potent antiviral activity, complementary resistance profiles and differing metabolic pathways may be required to treat HCV. Based on consultations with experts in the field, we believe the combination of at least one nucleoside/tide analog with, for example, a protease inhibitor or NS5a enzyme inhibitor, or one nucleoside/tide combined with a second complementary nucleoside/tide, present potentially useful therapeutic regimens. These combinations of DAAs possess complementary resistance profiles and differing metabolic pathways, suggesting that they will not have clinically relevant antagonism. In addition, nucleoside/tide analogs have demonstrated in vitro the ability to suppress the resistant variants that emerge with partially-suppressive concentrations of protease inhibitors or non-nucleoside polymerase inhibitors. Clinical use of a combination of DAAs may provide improved antiviral activity across HCV genotypes and may lead to interferon-sparing regimens. Following are descriptions of DAA combination studies involving our product candidates, along with the results of these studies to date.
Nucleoside in Combination with a Protease Inhibitor
INFORM Studies – (our nucleoside analog polymerase inhibitor, RG7128, in combination with Roche’s protease inhibitor, danoprevir, or RG7227). During November 2008, Roche, InterMune, Inc. (“InterMune”), and we announced the initiation of a Phase 1 study to investigate the combination of two DAAs in the absence of interferon and ribavirin. This study, named INFORM-1, combined for the first time in patients naïve to therapy and in patients who previously failed therapy (“TF”) two oral DAAs, RG7128 and danoprevir (also known as RG7227 or ITMN 191). Danoprevir is an inhibitor of the HCV NS3/4 protease, which prior to October 2010, was being developed by InterMune in collaboration with Roche. During October 2010, Roche purchased the worldwide development and commercialization rights to danoprevir from InterMune and simultaneously terminated the exclusive license and collaboration agreement it had entered into with InterMune to develop danoprevir.
INFORM-1 was a randomized, double-blind, ascending dose Phase 1 trial that enrolled a total of 86 patients and was conducted by Roche. The principal objectives were to evaluate safety, tolerability, and antiviral activity of RG7128 and danoprevir administered in combination at increasing doses for up to 13 days. Results from this study demonstrated for the first time that the combination of an oral protease inhibitor and an oral nucleoside polymerase inhibitor resulted in significant circulating HCV RNA reduction in patients with HCV, as patients receiving the combination of danoprevir and RG7128 for 13 days (without pegylated interferon or ribavirin) experienced a median reduction in HCV RNA of -4.8 to -5.2 log10 IU/mL in the highest dose levels tested. In addition, no treatment-related serious adverse events, dose reductions, or discontinuations were reported during the study. Pharmacokinetic analyses also confirmed that there were no drug-drug interactions between the compounds. Following is a figure that presents by dose regimen the median log10 HCV RNA change from baseline over the 13 days of treatment.
31
Table of Contents
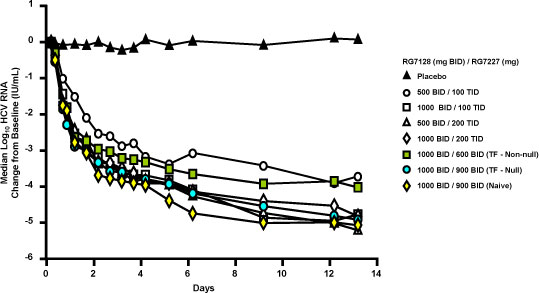
The antiviral results for each dose regimen are summarized in the following table.
| Regimen (RG7128 / RG7227 mg) |
N | Patient Population |
HCV
RNA <LLOQ1 (<43 IU/mL) N (%) |
HCV
RNA <LLOD2 (<15 IU/mL) N (%) |
||||||||||||
| 500 BID/100 TID |
8 | Naïve | 1/8(13 | %) | 1/8(13 | %) | ||||||||||
| 500 BID/200 TID |
8 | Naïve | 5/8(63 | %) | 2/8(25 | %) | ||||||||||
| 1000 BID/100 TID |
7 | Naïve | 5/7(71 | %) | 2/7(29 | %) | ||||||||||
| 1000 BID/200 TID |
8 | Naïve | 5/8(63 | %) | 2/8(25 | %) | ||||||||||
| 1000 BID/600 BID |
8 | TF (non-null | ) | 4/8(50 | %) | 1/8(13 | %) | |||||||||
| 1000 BID/900 BID |
8 | TF (null | ) | 4/8(50 | %) | 2/8(25 | %) | |||||||||
| 1000 BID/900 BID |
8 | Naïve | 7/8(88 | %) | 5/8(63 | %) | ||||||||||
1 – LLOQ means lower limit of quantification by Roche Taqman Assay
2 – LLOD means lower limit of detection by Roche Taqman Assay
The higher dose combination of RG7128 1000mg and danoprevir 900mg administered twice daily without pegylated interferon or ribavirin for 13 days resulted in 88% of HCV-positive treatment-naïve patients achieving HCV RNA below the lower limit of quantification (“LLOQ”), and 63% of patients having HCV RNA below the lower limit of detection (“LLOD”). The same regimen in “null-responders” resulted in 50% of patients with HCV RNA below LLOQ and 25% of patients with HCV RNA below LLOD. “Null responders” were defined as patients with a documented failure to achieve a 1.0 log10 or greater decline in HCV RNA in 4 weeks or a 2.0 log10 or greater decline in HCV RNA in 12 weeks of prior treatment with pegylated interferon and ribavirin.
As a result of Grade 4 ALT elevations experienced in a Phase 2b study of danoprevir, in February 2010 Roche announced that it would not conduct the previously planned 28 day INFORM-2 study, designed to evaluate the
32
Table of Contents
combination of RG7128 with danoprevir with and without pegylated interferon and ribavirin. During October 2010, Roche announced plans to conduct a longer duration study of RG7128 and ritonavir-boosted danoprevir. This “INFORM-SVR” study will have an SVR endpoint and is expected to begin during the first calendar quarter of 2011, after Roche identifies a generally safe and well-tolerated dose level for ritonavir-boosted danoprevir from ongoing studies.
Nucleotide in Combination with an NS5a Replication Complex Inhibitor
Clinical Collaboration Study – (our nucleotide analog polymerase inhibitor, PSI-7977, in combination with BMS’s NS5a replication complex inhibitor, BMS-790052). During January 2011, we entered into a clinical collaboration agreement with Bristol-Myers Squibb Company (“BMS”) to evaluate the utility of PSI-7977 in combination with BMS-790052, BMS’s NS5a replication complex inhibitor, for the treatment of chronic HCV. This collaboration represents the first cross-company collaboration combining two oral DAAs to address a significant unmet medical need in the treatment of HCV.
BMS is planning to initiate a proof of concept study of PSI-7977 in combination with BMS-790052 to evaluate the potential to achieve an SVR, defined as a level of HCV RNA in a patient that is below the limit of detection (<15 IU/ml) after the discontinuation of therapy, with an all oral, once-daily treatment regimen in patients across multiple HCV genotypes. The primary goal of the study is to assess the safety, pharmacokinetics and pharmacodynamics of PSI-7977 in combination with BMS-790052, with and without ribavirin, in treatment-naïve patients chronically infected with HCV genotypes 1, 2, and 3. The study is planned to start in the first half of calendar year 2011 and BMS will be conducting the study. BMS is responsible for all costs of the study, except for the cost of PSI-7977, which will be supplied by Pharmasset. Neither party has licensed any commercial rights to the other party.
Nucleotide in Combination with Another Nucleotide
Complementary Nucleotides in Combination Study – (our guanine nucleotide analog polymerase inhibitor, PSI-938, in combination with our uracil nucleotide analog polymerase inhibitor, PSI-7977). In late November 2010, we began dosing PSI-938 in Part 2 of a Phase 1 study that includes the first combinations of a purine (PSI-938) and a pyrimidine (PSI-7977) nucleotide analog for the treatment of HCV. The cohorts within Part 2 are evaluating PSI-938 QD, in the absence of interferon, as monotherapy and in combination with PSI-7977 QD. The primary objective of Part 2 of this study is to assess the safety, tolerability and pharmacokinetics of PSI-938 alone and in combination with PSI-7977 in the clinically-relevant setting of combination therapy for 14 days. The secondary objective of Part 2 of this study is to evaluate the short-term change in HCV RNA.
Forty patients with HCV genotype 1 are being enrolled into one of four cohorts (10 patients per cohort, n = 8 and placebo = 2) as follows:
| • | Cohort A – PSI-938 300mg QD administered alone for 14 days, |
| • | Cohort B1 – PSI-938 300mg QD for 7 days followed by the combination of PSI-938 300mg QD plus PSI-7977 400mg QD for 7 days, |
| • | Cohort B2 – PSI-7977 400mg QD for 7 days followed by the combination of PSI-7977 400mg QD plus PSI-938 300mg QD for 7 days, |
| • | Cohort C – patients will receive PSI-938 300mg QD plus PSI-7977 400mg QD for 14 days. |
Preliminary results from Cohort A indicated:
| • | PSI-938 was generally safe and well tolerated over 14 days; |
| • | There were no serious adverse events and no dose modifications or discontinuations; and |
| • | There were no clinically significant, treatment-emergent trends in any clinical laboratory parameters; |
33
Table of Contents
| • | PSI-938 demonstrated potent antiviral activity with a median HCV RNA change from baseline of 5.23 log10 IU/mL in the 8 patients receiving 300mg QD monotherapy for 14 days. HCV RNA declined rapidly and consistently throughout the 14 day dosing period, with no viral breakthrough noted. In the 8 subjects who received PSI-938 300mg QD monotherapy for 14 days, half (4 of 8) of the subjects achieved HCV RNA below the limit of detection (15 IU/mL), and 5 of 8 patients achieved HCV RNA below the limit of quantification (43 IU/mL). The median baseline HCV RNA in patients enrolled in Cohort A was approximately 1.0 log10 higher than in the Part 1 multiple ascending dose trial (6.95 log10 IU/mL versus 5.92 log10 IU/mL), allowing for a full assessment of the antiviral activity of the nucleotide analog. |
Patients are now being enrolled in Cohorts B1 and B2. Following the availability of data from these initial combination cohorts, the fourth and final cohort is expected to begin enrolling patients. We anticipate reporting further data from Part 2 of this study during the quarter ending March 31, 2011. We also expect to initiate a Phase 2 study of PSI-938 in combination with PSI-7977 during mid (calendar year) 2011. This Phase 2 study will explore dosing durations of PSI-938 and PSI-7977 with an SVR endpoint.
Pharmasset’s Proprietary DAA Combinations
Our drug discovery efforts are now primarily focused on the identification of purine nucleoside/tides that have resistance profiles that complement the resistance profiles of our pyrimidine analog product candidates, RG7128 and PSI-7977. Purines are phosphorylated by different enzymes than the pyrimidines, and thus should not antagonize the antiviral activity of the pyrimidines. In vitro, the combination of a purine analog with a pyrimidine analog provides additive to synergistic antiviral activity, potentially due to the fact that each of these classes of analogs compete with a different class of naturally occurring nucleotides for incorporation into nascent HCV RNA. Such complementary activities offer the potential for a potent dual nucleoside/tide analog-based combination for the future treatment of HCV.
Due to the unique attributes of nucleoside/tides, we believe a combination of two nucleoside/tides could possess a competitive advantage over SOC and other DAA combinations which do not contain a nucleoside/tide. We also believe that other combinations including a protease inhibitor or non-nucleoside polymerase inhibitor may be limited in their efficacy due to (1) their limited utility in genotypes other than Genotype 1, and (2) their lower barriers to resistance. Inclusion of a nucleoside/tide in such a combination may improve its barrier to resistance. A dual nucleoside/tide combination has the potential to provide therapeutic activity for all genotypes. In addition, since nucleoside/tides possess favorable resistance profiles, we believe that dual nucleoside/tide combinations could possess an advantage over other combinations that incorporate drug classes with less robust resistance profiles. This strategy of dual nucleoside/tide therapy underpins the current standard of care in HIV. Fixed-dose combinations of nucleoside/tides approved to treat HIV have long provided additional advantages, such as ease of compliance and reduced emergence of resistance.
Purine Nucleotide Product Candidates
Guanosine Program – PSI-938 and PSI-661
In 2007, we launched a purine nucleotide research and development program and in 2009, we nominated two guanine nucleotide analog polymerase inhibitors, PSI-938 and PSI-661, as development candidates. PSI-938 and PSI-661 have demonstrated potent in vitro anti-HCV activity with EC90 values of 1.43 +/- 0.67 µM and 0.01 +/- 0.005 µM, respectively. PSI-938 and PSI-661 have many of the benefits of our pyrimidine nucleoside/tide analogs, RG7128 and PSI-7977, because we believe that they:
| • | Have demonstrated similar in vitro activity across multiple HCV genotypes, |
| • | Have a higher barrier to resistance than other classes of HCV small molecules in development, and |
| • | Have a lower risk of drug interactions when combined with other direct acting antivirals targeting HCV. |
34
Table of Contents
PSI-938 and PSI-661 retain equivalent potency against wildtype HCV and virus with the S282T mutation associated with in vitro resistance to other nucleoside/tide analogs under development, such as RG7128, PSI-7977, IDX184 and INX189. Furthermore, the purines are metabolized to the same active triphosphate form through a different phosphorylation pathway than the pyrimidine analogs, RG7128 and PSI-7977, thus decreasing the risk of metabolic competition during phosphorylation. The in vitro half-life of the triphosphate in primary human hepatocytes is approximately 12 hours for PSI-938 and PSI-661, which supports the exploration of once-daily dosing in early development. The main difference between PSI-938 and PSI-661 is that PSI-661 uses a different prodrug strategy than PSI-938. Based on these characteristics, PSI-938 and PSI-661 have the potential to be combined with our pyrimidine analogs as part of a future regimen, which is expected to be the focus of upcoming trials. Our plan is to select one of these product candidates for later-stage clinical development based upon a review of the early human clinical trial results of both PSI-938 and PSI-661.
PSI-938 Development
Phase 1 Studies. In April 2010, we initiated a Phase 1 study of PSI-938, which was a SAD study to assess the safety, tolerability, and pharmacokinetics of PSI-938 following single oral administration in healthy subjects. Preliminary results from this study include:
| • | Single ascending doses of PSI-938 up to 1600mg were generally safe and well tolerated, |
| • | Based upon the long terminal half-life, once-daily dosing is likely. |
During July 2010, we initiated a Phase 1 MAD study of PSI-938 administered as monotherapy in treatment naïve patients with HCV genotype 1. Forty subjects were enrolled and randomized to PSI-938 (8 per cohort) or placebo (2 per cohort). The primary objective of the study was to assess the safety, tolerability, and pharmacokinetics of PSI-938 administered as monotherapy over seven days. The secondary objective of this study was to assess antiviral activity by measuring the change in circulating HCV RNA levels. Four dose cohorts of PSI-938 (100mg QD, 200mg QD, 300mg QD, and 100mg BID) were evaluated. Results from this study to date indicated PSI-938 was generally safe and well tolerated across all cohorts with no discontinuations, no serious adverse events, and no dose-related trends in adverse events or laboratory abnormalities. Antiviral results from this study are summarized in the following table:
| n | Median Change in HCV RNA at Day 8 (log10 IU/mL) |
Range (log10 IU/ |
Number of Subjects with HCV RNA |
|||||||||||||||
| Dose |
<LLOD 1 | <LLOQ 2 | ||||||||||||||||
| 100mg QD |
8 | -4.31 | -2.66 to -5.12 | 1 | 3 | |||||||||||||
| 200mg QD |
8 | -4.64 | -3.49 to -5.35 | 5 | 7 | |||||||||||||
| 300mg QD |
8 | -3.94 | -3.43 to -5.29 | 4 | 4 | |||||||||||||
| 100mg BID |
8 | -4.59 | -3.94 to -5.08 | 2 | 3 | |||||||||||||
| Placebo |
8 | -0.05 | +0.17 to -0.29 | 0 | 0 | |||||||||||||
1 – LLOD represents lower limit of detection by Roche Taqman Assay (<15 IU/mL)
2 – LLOQ represents lower limit of quantification by Roche Taqman Assay (<43 IU/mL)
PSI-938 demonstrated potent antiviral activity across all dose cohorts with a median HCV RNA change from baseline of 4.31 log10 IU/mL, 4.64 log10 IU/mL, 3.94 log10 IU/mL, and 4.59 log10 IU/mL in patients receiving 100mg
35
Table of Contents
QD, 200mg QD, 300mg QD and 100mg BID for 7 days, respectively. HCV RNA declined consistently throughout the 7-day dosing period, with no viral breakthrough. For the 16 subjects who received PSI-938 200 mg QD or 300 mg QD for 7 days, more than half (9 of 16) of the subjects on PSI-938 monotherapy achieved HCV RNA below the limit of detection (15 IU/mL) and 11 out of 16 patients achieved HCV RNA below the limit of quantification (43 IU/mL).
In late November 2010, we began dosing PSI-938 in Part 2 of a Phase 1 study that includes the first combinations of a purine (PSI-938) and a pyrimidine (PSI-7977) nucleotide analog for the treatment of HCV. A complete description of this study, along with the results of this study to date can be found under “Combination Treatment with Two or More Direct Acting Antivirals”, “Nucleotide in Combination with Another Nucleotide” located in the previous section.
PSI-661 Development
PSI-661 is a prodrug of a guanine nucleotide analog polymerase inhibitor which we are developing for the treatment of chronic HCV infection. It is in advanced preclinical development and our current plan is to submit an IND application, or its foreign equivalent, during the first quarter of calendar year 2011. We also plan to initiate a Phase 1 SAD study to assess the safety, tolerability, and pharmacokinetics of PSI-661 during the second quarter of calendar year 2011.
Clinical Collaboration
During January 2011, we entered into a clinical collaboration agreement with Bristol-Myers Squibb Company (“BMS”) to evaluate the utility of PSI-7977 in combination with BMS-790052, BMS's NS5a replication complex inhibitor, for the treatment of HCV. This collaboration represents the first cross-company collaboration combining two oral agents to address a significant unmet medical need in the treatment of HCV. We and BMS are planning to initiate a proof of concept study of PSI-7977 in combination with BMS-790052. The study, which is planned to start in the first half of calendar year 2011, will be conducted by BMS. BMS is responsible for all costs of the study, except for the cost of PSI-7977, which will be supplied by Pharmasset. Neither party has licensed any commercial rights to the other party.
Financial History
We have incurred substantial operating losses since our inception because we have devoted substantially all of our resources to our research and development activities and have not generated any revenue from the sale of approved drugs. As of December 31, 2010, we had an accumulated deficit of $257.0 million. We expect our operating losses to increase for at least the next few years as we continue to pursue the clinical development of PSI-7977, PSI-938, and PSI-661 and as we expand our discovery and development pipeline.
We have funded our operations primarily through the sale of equity securities, payments received under collaboration agreements, borrowings under our Loan Agreement, and interest earned on investments. We expect to continue to fund our operations over the next several years through our existing cash resources, potential future milestone payments that we expect to receive from Roche if certain conditions are satisfied, interest earned on our investments, and additional capital to be raised through public or private equity offerings or debt financings. We will require significant additional financing in the future to fund our operations. Additional financing may not be available on acceptable terms, if at all. As of December 31, 2010, we had $102.7 million of cash and cash equivalents.
Revenues
All of our product candidates are currently in development and, therefore, we do not expect to generate any direct revenues from product sales for at least the next few years, if at all. Our revenues to date have been generated primarily from milestone payments under our collaboration agreements, license fees, and research funding. We currently have a collaboration agreement with Roche for the development of RG7128. We entered into our collaboration agreement with Roche in October 2004. Roche subsequently paid us an up-front payment of $8.0 million.
36
Table of Contents
As of December 31, 2010, we had received an aggregate of $44.5 million in payments under the Roche collaboration agreement, including research funding and related fees as well as up-front and milestone payments.
Under the current terms of the Roche collaboration agreement, if we and Roche succeed in obtaining all of the regulatory approvals specified in the agreement for RG7128, as of December 31, 2010 the maximum future development and commercialization milestone payments payable to us is $105.0 million. Receipt of any additional milestone payments depends on many factors, some of which are beyond our control. We cannot assure you that we will receive any of these future payments.
We expect our revenues for at least the next few years to be derived primarily from payments under our current collaboration agreement with Roche and any additional collaborations that we may enter into in the future. In addition to the payments described above, we may receive future royalties on product sales, if any, under our collaboration agreement with Roche.
Research and Development Expenses
Our research and development expenses consist primarily of salaries and related personnel expenses, fees paid to external service providers, up-front and milestone payments under our license agreements, patent-related legal fees, costs of preclinical studies and clinical trials, drug and laboratory supplies, and costs for facilities and equipment. We use external service providers to manufacture our product candidates for clinical trials and for the majority of our preclinical and clinical development work. We charge all research and development expenses to operations as they are incurred. Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided.
Our research activities are primarily focused on discovering and developing novel drugs to treat HCV. Our development activities are primarily focused on the development of RG7128 (in collaboration with Roche), PSI-7977, PSI-938, and PSI-661 for the treatment of HCV. We are responsible for all costs incurred in the clinical development of PSI-7977, PSI-938, and PSI-661, as well as the research costs associated with our other internal research programs.
Under our collaboration with Roche, Roche will fund the clinical development and commercialization of RG7128. Under this collaboration, Roche reimbursed us for all of the external expenses associated with, and we were responsible for, certain preclinical work, the IND filing, and the proof-of-concept clinical trial. During December 2008, we transferred the IND application for RG7128 to Roche. Roche will continue to fund all of the expenses of, and be responsible for, other preclinical studies and future clinical development of RG7128 in the territories licensed to Roche. We and Roche will continue to jointly oversee all development and marketing activities of RG7128 in the territories licensed to Roche. Roche received a license only to PSI-6130 and its pro-drugs, including RG7128.
Under our clinical collaboration agreement with BMS, BMS will conduct and be responsible for all costs of the proof of concept study of PSI-7977 in combination with BMS-790052, except for the cost of PSI-7977 to be used in the study, which will be supplied by Pharmasset.
We use our internal research and development resources, including our employees and discovery infrastructure, across various projects. Our related internal expenses are not attributable to a specific project, but are directed to broadly applicable research activities. Accordingly, we do not account for our internal research and development expenses on a project basis. We use external service providers to manufacture our product candidates for clinical trials and for the substantial majority of our preclinical and clinical development work. We have tracked some of these external research and development expenses on a project basis. To the extent that expenses are not attributable to a specific project, they are included in one of the “unattributed expenses” in the table below.
The following table summarizes our research and development expenses for our current development programs for the three months ended December 31, 2010 and 2009:
37
Table of Contents
| Three months ended December 31, |
Cumulative Project Costs |
|||||||||||
| 2010 | 2009 | |||||||||||
| (In thousands) | ||||||||||||
| Expenses attributed to projects: |
||||||||||||
| RG7128 Studies (1) |
$ | — | $ | — | $ | — | ||||||
| PSI-7977 (including PSI-7851) Studies |
9,895 | 2,599 | 33,987 | |||||||||
| PSI-938 Studies |
3,138 | 375 | 12,401 | |||||||||
| PSI-661 (including PSI-879) Studies |
787 | 325 | 5,616 | |||||||||
| Clevudine Studies (2) |
— | 1,349 | ||||||||||
| Total attributed expenses |
13,820 | 4,648 | ||||||||||
| Unattributed expenses |
||||||||||||
| Salaries and related personnel expenses |
2,133 | 1,914 | ||||||||||
| Non-cash stock compensation expense |
1,286 | 1,042 | ||||||||||
| Legal expenses associated with patents |
361 | 394 | ||||||||||
| Preclinical studies and new drug discovery services |
482 | 438 | ||||||||||
| Drug and laboratory supplies |
262 | 243 | ||||||||||
| Consulting expense |
2 | 29 | ||||||||||
| Facility and other expenses |
422 | 489 | ||||||||||
| Total unattributed expenses |
4,948 | 4,549 | ||||||||||
| Total research and development expenses |
$ | 18,768 | $ | 9,197 | ||||||||
| (1) | Roche is responsible for all of the expenses associated with the research and development of RG7128. |
| (2) | In April 2009, we voluntarily terminated our Phase 3 registration studies of clevudine for the treatment of hepatitis B virus. We completed the termination process during the first quarter of fiscal 2010. |
We will continue to make determinations as to which programs to pursue and how much funding to direct to each program on an ongoing basis. These determinations will be made in response to the scientific and clinical success of each product candidate, as well as an ongoing assessment as to the product candidate’s commercial potential. We do not believe that it is possible at this time to accurately project total program-specific expenses through commercialization for any of our product candidates, as there are numerous factors associated with the successful commercialization of any of our product candidates, including future trial design and various regulatory requirements such as competitive final product labeling and reasonable risk management programs, many of which cannot be determined with accuracy at this time based on our stage of development. Product candidates that may appear promising at early stages of development may not reach the market for a number of reasons. For example, product candidates may be found ineffective or may cause harmful side effects during clinical trials, may take longer to progress through clinical trials than anticipated, may fail to receive necessary regulatory approvals, or may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality. The lengthy process of seeking FDA and other regulatory agency approvals requires the expenditure of substantial resources. Any failure or delay in obtaining regulatory approvals could materially adversely affect our product development effort and financial condition. Because of these and other risks and uncertainties, we cannot predict when or whether we will successfully complete the development of any of our product candidates or the ultimate product development cost or whether we will obtain any approval required by the FDA or other regulatory agencies on a timely basis, if at all.
As we obtain results from clinical trials, we may elect to discontinue or delay preclinical studies or clinical trials for a product candidate or development program in order to focus our resources on more promising product candidates or programs.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational functions, including accounting, finance, legal, business development, investor relations, information technology, and human resources. Other significant general and administration costs include facilities costs and professional fees for outside accounting and legal services, travel, insurance premiums, and depreciation.
38
Table of Contents
Results of Operations
Three Months Ended December 31, 2010 and 2009
Revenues. Revenues were $0.2 million during the quarter ended December 31, 2010, compared to $0.3 million during the quarter ended December 31, 2009. Revenues during each three month period primarily reflect amortization of up-front and subsequent collaborative and license payments received from Roche previously recorded as deferred revenue.
The following is a reconciliation between cash payments received under contract revenue agreements and contract revenues reported:
| Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| (In thousands) | ||||||||
| Cash received/receivable |
$ | — | $ | 23 | ||||
| Deferred |
— | — | ||||||
| Amortization |
247 | 246 | ||||||
| Revenues |
$ | 247 | $ | 269 | ||||
Research and Development Expenses. Research and development expenses increased to $18.8 million during the quarter ended December 31, 2010 from $9.2 million during the quarter ended December 31, 2009. This net increase of $9.6 million consists of a $7.3 million increase in preclinical and clinical trial costs for PSI-7977 (including an increase in active pharmaceutical ingredient (API) manufacturing (and related drug supply) costs and SOC costs of $4.4 million), a $2.8 million increase in preclinical and clinical trial costs for PSI-938 (including an increase in active pharmaceutical ingredient (API) manufacturing (and related drug supply) costs and SOC costs of $1.5 million), a $0.5 million increase in preclinical and clinical trial costs for PSI-661, and approximately a $0.3 million increase in other research and development expenses. Partially offsetting this $10.9 million increase was a $1.3 million decrease in clinical trial expenses for clevudine resulting from our voluntary termination of our Phase 3 registration studies of clevudine, which was completed as of December 31, 2009.
General and Administrative Expenses. General and administrative expenses were $5.1 million during the quarter ended December 31, 2010, an increase of $0.9 million from $4.2 million during the quarter ended December 31, 2009. The increase of $0.9 million was due to increases of $0.4 million in legal expenses incurred in connection with our defense against the Demand for Arbitration and Relief (see Part II., Item 1.—Legal Proceedings, for additional information), $0.4 million in compensation expenses ($0.1 million of which was non-cash stock compensation expense), and $0.1 million of other administrative expenses.
Other Income. On October 29, 2010, we were awarded two grants ($244,479 each) totaling $489 thousand under the IRS Qualifying Therapeutic Discovery Project (QTDP) program, which was created by Congress as part of the Patient Protection and Affordable Care Act of 2010. The grants were received on November 12, 2010. One of the grants was awarded for the development of PSI-7977 and the other grant was awarded for the development of PSI-938 or PSI-661. All three of these product candidates are being developed for the treatment of HCV and all of the $489 thousand was recorded as “Other income” in the Statement of Operations during the three months ended December 31, 2010.
Interest Expense. Interest expense decreased to $0.4 million during the quarter ended December 31, 2010 from $0.7 million during the quarter ended December 31, 2009. The decrease in interest expense was due to a lower amount of long-term debt outstanding during the quarter ended December 31, 2010 compared to the quarter ended December 31, 2009.
Liquidity and Capital Resources
Since our inception, we have funded our operations primarily through public and private offerings of our equity securities, payments received under our collaboration agreements, and borrowings under our Loan Agreement. We
39
Table of Contents
have raised approximately $435.3 million in net proceeds from sales of our equity securities, including $123.4 million from our common stock offering completed on January 26, 2011 (see Part 1. – Item 1. – Notes to Financial Statements – Note 11. Subsequent Event, for additional information), and borrowed a total of $23.3 million under our Loan Agreement entered into on September 30, 2007. At December 31, 2010, we held $102.7 million in cash and cash equivalents and have invested substantially all of our available cash and cash equivalents in a money market fund. Borrowings under our Loan Agreement were $9.4 million as of December 31, 2010.
Net cash used in operating activities was $24.0 million during the three months ended December 31, 2010 compared to $14.5 million during the three months ended December 31, 2009. The $9.5 million increase in net cash used in operating activities during 2010, as compared to 2009, was due primarily to higher operating expenses of $9.3 million, primarily resulting from the advancement of PSI-7977 into a series of Phase 2 studies during fiscal 2011.
Net cash used in investing activities of $37 thousand and $25 thousand during the three months ended December 31, 2010 and 2009, respectively, was for the purchase of equipment during each period.
Net cash used in financing activities was $0.4 million during the three months ended December 31, 2010, compared to $1.6 million during the three months ended December 31, 2009. The net cash used in financing activities during 2010 consisted of $2.4 million of principal payments on long-term debt that were mostly offset by $2.0 million in proceeds from the exercise of stock options. Net cash used in financing activities during 2009 consisted of $1.8 million of principal payments on long-term debt that were partially offset by $0.3 million in proceeds from the exercise of stock options.
On September 30, 2007, we entered into a Loan Agreement that allowed us to borrow up to $30.0 million in $10.0 million increments. We borrowed the first and second $10.0 million increments by signing two Secured Promissory Notes (“Notes A and B”) on October 5, 2007 and March 28, 2008. Notes A and B bear interest at 12%. On December 12, 2008, we amended the Loan Agreement and borrowed $3.3 million by signing a Secured Promissory Note (“Note C”). Note C bears interest at 12.5%. Notes A, B, and C are to be repaid over a 45-month period with the first 15 monthly payments representing interest only followed by 30 equal monthly payments of principal and interest. The principal monthly payments on each of the notes begin and end as follows:
| Note |
Begin | End | ||
| Note A | March 1, 2009 | August 1, 2011 | ||
| Note B | August 1, 2009 | January 1, 2012 | ||
| Note C | May 1, 2010 | October 1, 2012 |
Prepayment of the loans made pursuant to the Loan Agreement is subject to penalty and substantially all of our tangible and intangible assets (except for intellectual property) are pledged as collateral for the Loan Agreement. Future total principal repayments of the three Notes amount to $6.3 million in fiscal 2011, $3.0 million in fiscal 2012, and $0.1 million in fiscal 2013. There are no additional borrowings available under the Loan Agreement.
Under the Loan Agreement, we agreed that in the event our market capitalization is below $90.0 million for 15 consecutive days in which the principal market for our common stock is open for trading to the public, we will be required to repay 50% of the then outstanding principal balance of the loans. We further agreed that in the event our market capitalization is below $40.0 million for 15 consecutive days in which the principal market for our common stock is open for trading to the public, we will be required to repay all of the then outstanding principal balance of the loans.
The Loan Agreement also contains covenants that, among other things, require us to obtain consent from the lender prior to paying dividends, making certain investments, changing the nature of our business, assuming or guaranteeing the indebtedness of another entity or individual, selling or otherwise disposing of a substantial portion of our assets, or merging or consolidating with another entity.
Developing drugs, conducting clinical trials and commercializing products is expensive and we will need to raise additional funds to achieve our strategic objectives. Although we believe our existing cash resources, together with the $123.4 million in net proceeds from our common stock offering completed on January 26, 2011, will be sufficient to fund our projected cash requirements for at least the next 18 months, we will require significant additional financing in the future to complete our clinical trials for PSI-7977, PSI-938, and PSI-661, to fund our portion, if any,
40
Table of Contents
of the cost of clinical trials for RG7128 completed outside of the territories licensed by Roche, to supply PSI-7977 for the proof of concept study of PSI-7977 in combination with BMS-790052 under our clinical collaboration agreement with BMS, and to fund our other operations. Additional financing may not be available on acceptable terms, if at all. Our future capital requirements will depend on many factors, including:
| • | the progress and costs of our preclinical studies, clinical trials, and other research and development activities; |
| • | the scope, prioritization, and number of our clinical trials and other research and development programs; |
| • | the amount of cash we receive under our existing collaboration agreement with Roche and any future collaboration agreements; |
| • | the costs of the development and expansion of our operational infrastructure; |
| • | the costs and timing of obtaining regulatory approval of our product candidates; |
| • | the ability of our collaborators to achieve development milestones, marketing approval, and other events or developments under our collaboration agreements; |
| • | the costs of filing, prosecuting, enforcing, and defending patent claims and other intellectual property rights; |
| • | the costs and timing of securing manufacturing arrangements for clinical or commercial production; |
| • | the costs of establishing sales and marketing capabilities or contracting with third parties to provide these capabilities for us; |
| • | the costs of acquiring or undertaking development and commercialization efforts for any future product candidates; |
| • | the magnitude of our general and administrative expenses; and |
| • | any costs that we may incur under current and future licensing arrangements relating to our product candidates. |
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through payments received under our collaborations, debt or equity financings, or by out-licensing product candidates. We cannot be certain that additional funding will be available to us on acceptable terms, or at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts.
Contractual Obligations and Commitments
We entered into an operating lease for office and laboratory space located in Princeton, New Jersey through May 22, 2015. We also entered into an operating lease for office space located in Durham, North Carolina through April 2011, which as of January 1, 2011, was extended through December 31, 2015. We executed three secured promissory notes totaling $23.3 million; $10.0 million in October 2007, $10.0 million in March 2008, and $3.3 million in December 2008. The secured promissory notes require payments of interest only for the first 15 months followed by 30 equal monthly payments of principal and interest. As of December 31, 2010, future payments under the three promissory notes and minimum future payments under non-cancellable operating leases are as follows:
41
Table of Contents
| Total | Payments Due By Period | |||||||||||||||||||
| Less than 1 year |
1-3 Years | 4-5 Years | After 5 Years |
|||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Debt obligations |
||||||||||||||||||||
| Debt maturities |
$ | 9,408 | $ | 7,796 | $ | 1,612 | $ | — | $ | — | ||||||||||
| Contractual interest |
683 | 608 | 75 | — | — | |||||||||||||||
| Capital lease obligations |
||||||||||||||||||||
| Debt maturities |
— | — | — | — | — | |||||||||||||||
| Contractual interest |
— | — | — | — | — | |||||||||||||||
| Operating leases |
4,068 | 890 | 2,761 | 417 | ||||||||||||||||
| Purchase obligations |
— | — | — | — | — | |||||||||||||||
| Total contractual obligations |
$ | 14,159 | $ | 9,294 | $ | 4,448 | $ | 417 | $ | — | ||||||||||
The above contractual obligations table does not include amounts for milestone payments related to development, regulatory, or commercialization events to licensors or collaboration partners, as the payments are contingent on the achievement of these milestones, which we have not achieved. Under our license agreement with Emory University for Racivir, we agreed to pay Emory University up to an aggregate of $1.0 million in future marketing milestone payments.
Off-Balance Sheet Transactions
To date, we have not had any relationships with unconsolidated entities or financial partnerships, such as entities referred to as structured finance or special purpose entities, which are established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and related disclosures. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Our actual results may differ substantially from these estimates under different assumptions or conditions. Our significant accounting policies are described in more detail in Note 2 of the Notes to Financial Statements included elsewhere in this Quarterly Report on Form 10-Q; however, we believe that the following accounting policies are critical to the judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
We recognize revenues when all of the following four criteria are present: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured.
Our revenues are primarily related to our collaboration agreement with Roche. This agreement provides for various types of payments to us, including non-refundable upfront license fees, research and/or development payments, and milestone payments.
Where we have continuing performance obligations under the terms of a collaborative arrangement, non-refundable upfront license payments received upon contract signing are recorded as deferred revenue and recognized as revenue as the related activities are performed. The period over which these activities are to be performed is based upon management’s estimate of the development period. Changes in management’s estimate could change the period over which revenue is recognized. Research and/or development payments are recognized as
42
Table of Contents
revenues as the related research and/or development activities are performed and when we have no continuing performance obligations related to the research and development payment received.
Where we have no continuing involvement under a collaborative arrangement, we record nonrefundable license fee revenues when we have the contractual right to receive the payment, in accordance with the terms of the collaboration agreement, and record milestones upon appropriate notification to us of achievement of the milestones by the collaborative partner.
Effective October, 1, 2010, we adopted the new accounting standards for determining whether the milestone method of revenue recognition is appropriate. We recognize revenue from milestone payments when earned, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement and (ii) we do not have ongoing performance obligations related to the achievement of the milestone earned. Milestone payments are considered substantive if all of the following conditions are met: the milestone payment (a) is commensurate with either the vendor’s performance to achieve the milestone or the enhancement of the value of the delivered item or items as a result of a specific outcome resulting from the vendor’s performance to achieve the milestone, (b) relates solely to past performance, and (c) is reasonable relative to all of the deliverables and payment terms (including other potential milestone consideration) within the arrangement. Any amounts received under the agreement in advance of performance, if deemed substantive, are recorded as deferred revenue and recognized as revenue as we complete our performance obligations.
Effective October 1, 2010, the Company also adopted the new accounting standards for revenue recognition for multiple deliverable revenue arrangements. Each deliverable within a multiple-deliverable revenue arrangement is accounted for as a separate unit of accounting under the guidance of the new authoritative guidance if both of the following criteria are met: (1) the delivered item or items have value to the customer on a standalone basis and (2) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
This new authoritative guidance amends previously issued guidance to eliminate the residual method of allocation for multiple-deliverable revenue arrangements, and requires that arrangement consideration be allocated at the inception of an arrangement to all deliverables using the relative selling price method. The new authoritative guidance also establishes a selling price hierarchy for determining the selling price of a deliverable, which includes (1) vendor-specific objective evidence, if available, (2) third-party evidence, if vendor-specific objective evidence is not available, and (3) estimated selling price if neither vendor-specific nor third-party evidence is available. Additionally, it expands the disclosure requirements related to a vendor’s multiple-deliverable revenue arrangements.
Deferred revenue associated with a non-refundable payment received under a collaborative agreement that is terminated prior to its completion results in an immediate recognition of the deferred revenue.
Research and Development Expenses - Research and development expenses consist primarily of salaries and related personnel expenses, fees paid to external service providers, costs of preclinical studies and clinical trials, drug and laboratory supplies, costs for facilities and equipment, and the costs of intangibles that are purchased from others for use in research and development activities, such as in-licensed product candidates, that have no alternative future uses. Research and development expenses are included in operating expenses when incurred. Reimbursements received from the Company’s collaborator(s) for third-party research and development expenses incurred by the Company on their behalf are recorded as a contra-expense. Amounts due from collaborators for reimbursement of research and development expenses are recorded on the balance sheets as “Amounts due from collaboration partner.”
Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized. Such amounts are then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided.
Accrued Expenses
We are required to estimate accrued expenses as part of our process of preparing financial statements. This process involves estimating the level of service performed on our behalf and the associated cost incurred in instances
43
Table of Contents
where we have not been invoiced or otherwise notified of actual costs. Examples of areas in which subjective judgments may be required include costs associated with services provided by contract organizations for preclinical development, clinical trials and manufacturing of clinical materials. We account for expenses associated with these external services by determining the total cost of a given study based on the terms of the related contract. We accrue for costs incurred as the services are being provided by monitoring the status of the trials and the invoices received from our external service providers. In the case of clinical trials, the estimated cost normally relates to the projected costs of having subjects enrolled in our trials, which we recognize over the estimated term of the trial according to the number of patients enrolled in the trial on an ongoing basis, beginning with patient enrollment. As actual costs become known to us, we adjust our accruals. To date, the number of clinical trials and related research service agreements has been relatively limited and our estimates have not differed significantly from the actual costs incurred. We expect, however, as clinical trials for PSI-7977, PSI-938, and PSI-661 advance, that our estimated accruals for clinical and research services will be more material to our operations in future periods.
Stock-based Compensation
We recognize stock compensation expense for awards of equity instruments to employees and directors based on the grant-date fair value of those awards (with limited exceptions). The grant-date fair value of the award is recognized as compensation expense over the life of the equity instruments issued. Equity instruments granted to consultants are periodically valued and recorded as stock compensation expense as the equity instrument vests.
Stock-based compensation expense is included in both research and development expenses and in general and administrative expenses in the statements of operations and comprehensive net income (loss). Since our stock was not publicly traded prior to April 27, 2007, the expected volatility was calculated for each equity award granted based on the “peer method.” We identified companies that traded publicly within the pharmaceutical industry that had similar SIC codes, employee count and revenues. Prior to October 1, 2006, we had chosen the weekly high price volatility for these companies for a period of five years. Subsequent to October 1, 2006, we have used the weekly high price for these companies for a period of six years to coincide with the expected term.
Recently Adopted Accounting Pronouncements
In October 2009, the FASB issued ASU No. 2009-13, Multiple-Deliverable Revenue Arrangements. This ASU provides new accounting standards for determining whether multiple deliverables exist, how the arrangement should be separated, and how the consideration should be allocated. This guidance requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. The update eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. We adopted these new accounting standards on October 1, 2010 on a prospective basis. Adoption of these new accounting standards did not have any impact on our financial position or results of operations.
In April 2010, the FASB issued ASU No. 2010-17, Revenue Recognition — Milestone Method. This ASU provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. Under the milestone method of revenue recognition, consideration that is contingent upon achievement of a milestone in its entirety can be recognized as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. This standard provides the criteria to be met for a milestone to be considered substantive which includes that: a) performance consideration earned by achieving the milestone be commensurate with either performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from performance to achieve the milestone; b) relate to past performance; and (c) be reasonable relative to all deliverables and payment terms in the arrangement. We adopted these new accounting standards on October 1, 2010 on a prospective basis. Adoption of these new accounting standards did not have any impact on our financial position or results of operations.
44
Table of Contents
| ITEM 4. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
Our management, under the supervision and with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures pursuant to Rule 13a-15 promulgated under the Exchange Act as of December 31, 2010. Our disclosure controls and procedures are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in the rules and forms of the SEC. These disclosure controls and procedures include, among other things, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, management is required to apply its judgment in evaluating the benefits of possible disclosure controls and procedures relative to their costs to implement and maintain.
Based on management’s evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures are designed at a reasonable assurance level and are effective to provide reasonable assurance that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. There were no changes in our disclosure controls or procedures identified in connection with the evaluation of such controls or procedures that occurred during our last fiscal quarter, or in other factors that have materially affected, or are reasonably likely to materially affect, these disclosure controls or procedures.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the three months ended December 31, 2010 that have materially affected, or are reasonably likely to materially affect our internal control over financial reporting.
| ITEM 1. | LEGAL PROCEEDINGS |
On July 28, 2009, Emory University and University of Georgia Research Foundation, Inc. (“Claimants”) filed a Demand for Arbitration and Relief (the “Demand”) with the American Arbitration Association in Atlanta, Georgia (the “Emory Arbitration”), claiming certain payments and seeking specific performance under the Company’s January 8, 2004 license agreement with Claimants (the “Emory License”).
The Demand alleged that payments Pharmasset had received under the Roche collaboration agreement were subject to the Emory License and that Pharmasset had not paid fees to Claimants based on such payments. In addition, the Demand alleged that Pharmasset had not complied with certain terms and conditions of the Emory License and that other Pharmasset product candidates were, or will be, covered by the Emory License. The Demand requested, among other things, specific performance of the Emory License, including the payment of license fees related to past payments received by Pharmasset. The Company’s response to the Demand was filed on August 14, 2009.
On December 6, 2010 a final arbitration award (the “Award”) was issued by a panel of AAA arbitrators. According to the Award, none of the payments the Company received under the Roche collaboration agreement were subject to the Emory License and, therefore, no license fees were owed to Emory based upon such payments.
45
Table of Contents
Furthermore, according to the Award, none of the other Company product candidates that were subject to the Demand are covered by the Emory License.
| ITEM 1A. | RISK FACTORS |
There have been no material changes to the risk factors discussed in Part I, “Item 1A. Risk Factors,” in our Annual Report on Form 10-K for the year ended September 30, 2010 (“Form 10-K”). You should carefully consider the risks described in our Form 10-K, which could materially affect our business, financial condition or future results. The risks described in our Form 10-K are not the only risks we face. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition, and/or operating results. If any of the risks actually occur, our business, financial condition, and/or results of operations could be negatively affected.
| ITEM 2. | UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS |
On September 30, 2007, we entered into the Venture Loan and Security Agreement with Horizon Technology Funding Company V, LLC (the “Lender”) that allowed us to borrow up to $30.0 million in $10.0 million increments (“Loan Agreement”). In conjunction with entering into the Loan Agreement and with executing three secured promissory notes in connection therewith, we granted warrants to the Lender to purchase 127,248 shares of our common stock at an exercise price of $12.05 per share. During the three months ended December 31, 2010, the Lender elected to exercise 63,625 warrants using the “net issuance method,” which resulted in the issuance by us of 45,848 shares of our common stock. The remaining warrants expire seven years from the date of grant (or upon a change of control as defined in the Loan Agreement) as follows: 22,130 expire on September 30, 2014, 30,428 expire on March 28, 2015, and 11,065 expire on December 12, 2015. We did not receive any consideration from the issuance of shares of common stock to the Lender upon net exercise of the warrants.
The warrants and the common stock underlying the warrants were granted to the Lender in reliance upon the exemption from registration requirements under Regulation D and Rule 506 promulgated under the Securities Act of 1933, as amended (the “Securities Act”). In connection therewith, we relied, among other things, upon representations by the Lender regarding its intentions to acquire the securities for investment only and not with a view to distribute or sell in connection with any distribution. The warrants were affixed with appropriate restrictive legends at the time of grant. Certificates representing the 45,848 shares of common stock issued to the Lender upon net exercise of the warrants were issued without restrictive legends in accordance with the provisions of Rule 144 promulgated under the Securities Act.
| ITEM 6. | EXHIBITS |
| Exhibit Number |
Description | |
| 10.1(1) | Separation Agreement and General Release, dated November 12, 2010, between Paul Lubetkin and Pharmasset, Inc. | |
| 31.1* | Rule 13a-14(a)/15d-14(a) Certification | |
| 31.2* | Rule 13a-14(a)/15d-14(a) Certification | |
| 32.1* | Section 1350 Certification | |
| 32.2* | Section 1350 Certification | |
* - Filed herewith.
(1) - Filed as an Exhibit to our Current Report on Form 8-K filed with the SEC on November 18, 2010.
46
Table of Contents
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| PHARMASSET, INC. | ||||||
| Date: February 7, 2011 | By: | /s/ Kurt Leutzinger | ||||
| Kurt Leutzinger Chief Financial Officer (duly authorized officer and principal financial officer) | ||||||
47
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Description | |
| 31.1 | Rule 13a-14(a)/15d-14(a) Certification | |
| 31.2 | Rule 13a-14(a)/15d-14(a) Certification | |
| 32.1 | Section 1350 Certification | |
| 32.2 | Section 1350 Certification | |
48