Attached files
| file | filename |
|---|---|
| EX-32.2 - Provention Bio, Inc. | ex32-2.htm |
| EX-32.1 - Provention Bio, Inc. | ex32-1.htm |
| EX-31.2 - Provention Bio, Inc. | ex31-2.htm |
| EX-31.1 - Provention Bio, Inc. | ex31-1.htm |
| EX-23.1 - Provention Bio, Inc. | ex23-1.htm |
| EX-4.4 - Provention Bio, Inc. | ex4-4.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
[X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2019
OR
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE TRANSITION PERIOD FROM ___TO __.
Commission file number 001-38552
PROVENTION BIO, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 81-5245912 |
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
P.O. Box 666 Oldwick, New Jersey 08858
(Address of registrant’s principal executive offices)
(908) 336-0360
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Common Shares, $0.001 par value | PRVB | The Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X].
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes [ ] No [X].
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X]. No [ ].
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).Yes [X]. No [ ].
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer [ ] | Accelerated filer [X] | |
| Non-accelerated filer [ ] | Smaller reporting company [X] | |
| Emerging growth company [X] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]
Indicate by check mark whether the registrant is a shell company (as defined in 12b-2 of the Act). Yes [ ] No [X].
The aggregate market value of the common stock held by non-affiliates of the Registrant as of June 28, 2019, the last business day of the Registrant’s last completed second quarter, based upon the closing price of the common stock as reported by The Nasdaq Capital Market on such date was approximately $387.8 million.
On March 9, 2020, there were 47,709,636 shares of the registrant’s common stock, $0.0001 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of Provention Bio, Inc.’s definitive proxy statement to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year are incorporated by reference into Part III of this Form 10-K and certain documents are incorporated by reference into Part IV.
PROVENTION BIO, INC.
ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
| 1 |
This Annual Report on Form 10-K contains forward-looking statements that involve substantial risks and uncertainties. These forward-looking statements are made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements, other than statements of historical facts, contained in this Annual Report on Form 10-K, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management, are forward-looking statements. The words “believe,” “may,” “potentially,” “estimate,” “continue,” “anticipate,” “intend,” “could,” “would,” “project,” “plan,” “expect” and similar expressions that convey uncertainty of future events or outcomes are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These forward-looking statements include, but are not limited to, statements concerning the following:
| ● | our lack of operating history; | |
| ● | the expectation that we will incur operating losses for the foreseeable future; | |
| ● | our current and future capital requirements to support our development and commercialization efforts for our product candidates and our ability to satisfy our capital needs; | |
| ● | our dependence on our product candidates, which are still in preclinical or various stages of clinical development; | |
|
● | our ability to obtain, or delays in obtaining, regulatory approval from the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and other regulatory authorities for our product candidates, such as PRV-031 in the At-Risk indication, including due to insufficient clinical data, requirements to demonstrate the comparability of our current third party manufacturing process with that of previous manufacturing processes by other companies, and the complexity of the review process by the FDA; |
| ● | failure to maintain regulatory approval for our product candidates, if received;
| |
| ● | our, or that of our third-party manufacturers, ability to manufacture Good Manufacturing Practice, or GMP, batches of our product candidates as required for pre-clinical and clinical trials and, subsequently, our ability to manufacture commercial quantities of our product candidates;
| |
| ● | our ability to attract and retain key executives and medical, scientific and commercial personnel;
| |
| ● | our ability to complete required clinical trials for our product candidates and obtain approval from the FDA or other regulatory agencies in different jurisdictions;
| |
| ● | our ability to build a commercial organization, including sales and marketing, and successfully commercialize our product candidates if we obtain regulatory approval;
| |
| ● | our dependence on third-parties to manufacture our product candidates;
| |
| ● | our reliance on third-party contract research organizations (CROs) to conduct our clinical trials, including those in the U.S. and internationally;
| |
| ● | our ability to maintain or protect the validity of our licensed patents and other intellectual property;
| |
| ● | our ability to internally develop new inventions and intellectual property;
| |
| ● | our ability to compete within the market for our product candidates, if approved; | |
| ● | interpretations of current laws and the passages of future laws; | |
| ● | acceptance of our business model by investors; | |
| ● | the accuracy of our estimates regarding expenses and capital requirements; and | |
| ● | our ability to adequately support organizational and business growth. |
The foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or risk factors that we are faced with that may cause our actual results to differ from those anticipated in our forward-looking statements. Please see “Part I—Item 1A—Risk Factors” for additional risks which could adversely impact our business and financial performance.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference into this report. We have no obligation, and expressly disclaim any obligation, to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise. We have expressed our expectations, beliefs and projections in good faith and we believe they have a reasonable basis. However, we cannot assure you that our expectations, beliefs or projections will result or be achieved or accomplished.
| 2 |
Our Company
Overview
We are a clinical stage biopharmaceutical company, focused on the development and commercialization of novel therapeutics and solutions aimed at intercepting and preventing immune-mediated diseases. Since our inception, we have devoted substantially all of our efforts to research and development, business planning, recruiting management and technical staff, acquiring operating assets, partnering and raising capital. Our business is subject to significant risks and uncertainties and we will be dependent on raising substantial additional capital before it becomes profitable and it may never achieve profitability.
We have not generated any revenue to date and through December 31, 2019, we had an accumulated deficit of $79.1 million. We have financed our operations through a private offering of Series A Convertible Redeemable Preferred Stock in April 2017, our initial public offering, or IPO, in July 2018, and our underwritten public offering and concurrent private placement in September 2019.
In April 2017, we completed our private placement of Series A Convertible Redeemable Preferred Stock. We issued an aggregate 11,381,999 shares of Series A Convertible Redeemable Preferred Stock at $2.50 per share. We received net proceeds of $26.7 million.
In July 2018, we issued and sold an aggregate of 15,969,563 shares of common stock in our IPO at a public offering price of $4.00 per share. In connection with the IPO, we issued to MDB, the underwriter in the IPO, and its designees warrants to purchase 1,596,956 shares of Common Stock at an exercise price of $5.00 per share. We received net proceeds from the IPO of $59.3 million, after deducting underwriting discounts and commissions of approximately $3.7 million and other offering expenses of approximately $0.8 million. Upon the closing of the IPO, all of our shares of Series A Convertible Redeemable Preferred Stock outstanding at the time of the IPO were automatically converted into an aggregate of 11,381,999 shares of common stock. In addition, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock also converted to warrants for the purchase of an aggregate of 558,740 shares of our common stock.
In September 2019, we completed an underwritten public offering in which we sold 5,750,000 shares of common stock at a public offering price of $8.00 per share. The 5,750,000 shares sold included the full exercise of the underwriters’ option to purchase 750,000 shares at a price of $8.00 per share. Concurrent with the underwritten public offering, we sold 2,500,000 shares of common stock to Amgen, Inc. at the public offering price of $8.00 per share in a private placement, pursuant to the terms of our License and Collaboration Agreement with Amgen Inc, dated as of November 5, 2018. Aggregate net proceeds from the underwritten public offering and the concurrent private placement were $62.7 million, net of approximately $2.8 million in underwriting discounts and commissions and other offering expenses of $0.5 million.
We expect that over the next several years we will continue to incur losses from operations as we increase our expenditures in research and development in connection with our regulatory submissions, clinical trials and other development activities, as well as costs to support our commercialization efforts to launch teplizumab, if we receive regulatory approval in the U.S. If adequate funds are not available to us on a timely basis, or at all, we may be required to terminate or delay certain development activities.
Strategy
Provention preferentially sources, repositions, transforms and advances underdeveloped or deprioritized clinical-stage, or nearly clinical-stage, therapeutic candidates targeting the interception and prevention of immune-mediated disease. Our “predict” and “pre-empt” therapeutic approach focuses on identifying at-risk patients and intervening before the targeted disease begins, re-appears, exacerbates or progresses. We believe our experience and expertise in translational medicine, immunology, and the design and execution of rapid go/no-go clinical trials makes us unique in the field of immune-mediated disease.
| 3 |
We have access to relevant in-licensing opportunities from industry-leading pharmaceutical companies; innovative, development-stage biotechnology companies; and world-renowned academic centers. We have obtained exclusive worldwide rights to two product candidates from MacroGenics, Inc., the acquisition of a Phase 3 clinical-stage candidate for the interception, delay or prevention of T1D and the in-license of a Phase 1 candidate for the potential treatment of systemic lupus erythematosus (SLE). We also in-licensed an enterovirus vaccine platform, targeting the prevention of Coxsackie Virus B (CVB) infections and the onset of T1D and celiac disease, from Vactech Ltd., or Vactech, a Finnish biotechnology company. We in-licensed a Phase 2 clinical-stage candidate from Amgen, Inc. targeting celiac disease. Lastly, we in-licensed a Phase 2 clinical-stage candidate from an affiliated entity of Janssen Pharmaceuticals, Inc., or Janssen, which is a small molecule targeting an upstream pathological mechanism and believed to drive Crohn’s disease.
Our activities are subject to significant risks and uncertainties, including the need for additional capital, as described below. We have not yet commenced any revenue-generating operations, does not have any cash flows from operations, and will need to raise additional capital to finance our operations.
Recent Company Developments
Clinical Data & Safety
PRV-031 (teplizumab, anti-CD3 mAb)
In June 2019, we announced results from a Phase 2 clinical trial in patients with Stage 2 T1D (Teplizumab for Prevention of Type 1 Diabetes In Relatives “At-Risk”) conducted at TrialNet sites and sponsored by the NIDDK, part of the National Institutes of Health, or NIH, which evaluated PRV-031, a humanized, anti-CD3 mAb for the interception of T1D, in 76 patients with stage 2 T1D. The trial results showed that PRV-031 significantly delayed the median onset of clinical diabetes from 24.4 months (placebo) to 48.4 months (teplizumab) (p=0.006). PRV-031 is the first potential immune modulator therapy that has demonstrated a delay in the onset of clinical disease in T1D.
The results from At-Risk study were reported at the American Diabetes Association meeting in June 2019 and published in the New England Journal of Medicine. A total of 76 subjects were enrolled and randomized, 44 to teplizumab and 32 to placebo. The safety profile in At-Risk subjects who received a single course of teplizumab was consistent with those from subjects with newly-diagnosed clinical T1D who received two courses of the drug.
In August 2019, the U.S. Food and Drug Administration granted breakthrough therapy designation (“BTD”) to PRV-031 for the delay or prevention of clinical T1D in individuals at-risk of developing the disease. BTD is an FDA program designed to expedite the development and review of therapeutic candidates intended to treat serious or life-threatening diseases.
In October 2019, the European Medicines Agency (EMA) granted PRIority MEdicines (“PRIME”) eligibility to teplizumab (PRV-031) for the prevention or delay of clinical T1D in individuals at-risk of developing the disease. The PRIME initiative is designed to expedite the development and review of promising therapies that target an unmet need and show potential clinical benefit so the medicine can reach patients earlier. The designation offers the opportunity for enhanced interaction and dialogue with the EMA to optimize development, as well as the potential for accelerated assessment at the time of application for a marketing authorization.
In November 2019, we completed a Type B multidisciplinary meeting with the FDA to discuss the proposed contents of a BLA for PRV-031 (teplizumab) for the prevention or delay of T1D in individuals at-risk of developing T1D. Based on official FDA meeting minutes, Provention continues to expect that it will commence a rolling BLA submission for PRV-031 in the middle of 2020 and is targeting completion of the submission in the fourth quarter of 2020. We do not anticipate the need to conduct any additional clinical trials in the at-risk population prior to BLA submission.
PRV-3279 (humanized anti-CD32B and CD79B bispecific)
On March 12, 2020, we announced positive top-line results from the Phase 1b portion of the PREVAIL (PRV-3279 EVAluation In Lupus) study, which evaluated PRV-3279 in 16 healthy volunteers.
| 4 |
PRV-3279 was well-tolerated, with no serious adverse events, and as expected, did not deplete B cells and demonstrated profound and sustained binding to circulating B lymphocytes, with reduction of circulating immunoglobulin M levels in a dose-proportional manner. While anti-drug antibody production was observed at both dose levels tested, immunogenicity was found not to affect exposure, safety or pharmacodynamic parameters.
Based on the results, Provention plans to commence the Phase 2a portion of the PREVAIL study in lupus patients in the first half of 2021.
PRV-6527 (CSF-1R inhibitor)
In October 2019, we announced top-line results from our Phase 2a clinical trial (the PRINCE study), which evaluated PRV-6527, an oral CSF-1R small molecule inhibitor, in 93 patients with moderate-to-severe Crohn’s disease. The primary efficacy endpoint of the study was the change in the Crohn’s Disease Activity Index (CDAI) score at week 12. While PRV-6527 demonstrated a substantial improvement in this symptom driven score at week 12, it did not differentiate from placebo. This high placebo response is deemed to be possibly related to the standard of care and/or background medication used (~85%) in the study’s predominantly biologic-naïve population. PRV-6527 was associated with improvements in several key secondary objective endpoints in the steroid-free population (75% of study subjects), including mucosal endoscopy (as assessed by the Simple Endoscopic Score for Crohn’s Disease, SES-CD) and tissue histology (as measured by the Global Histological Activity Score, GHAS). Analysis of exploratory serum and tissue biomarkers showed that patients treated with PRV-6527 had significant reductions in circulatory inflammatory monocytes, as well as macrophages, dendritic cells and the CSF1 gene signature in colonic tissue, providing proof of mechanism in the interception of inflammatory myeloid cells. PRV-6527 was found to be generally safe and well tolerated, with no drug-related serious adverse events.
In December 2019, Janssen declined its option to buy back PRV-6527 from us for a one-time payment of $50.0 million and single-digit royalties on future net sales in inflammatory bowel disease indications. We retain the rights and are free to sublicense the program on a worldwide basis to another partner in the field of inflammatory bowel disease.
Our Focus and Pipeline
Inflammation is a natural consequence of most infections as it is the immune system’s first response to invading pathogens in the event of injury or acute illness. Most of the time, this response is beneficial and well-controlled; helping to repair tissue damage and clear pathogens from the body. In addition to directly damaging tissues and organs, an infection can sometimes result in the excessive release of toxic immune mediators leading to a potentially life-threatening acute pathological immune response. When patients have the requisite genetic predisposition, infections can also trigger chronic autoimmune responses that persist and progress long after the original insult has subsided. These sustained pathological responses have been linked to an increased susceptibility to chronic debilitating and potentially life-threatening diseases like inflammatory bowel disease, diabetes, cancer, and certain neurological disorders.
Our “predict” and “preempt” therapeutic approach is to intercept the underlying pathological immune and inflammatory responses in susceptible individuals. Our pipeline includes:
| ● | PRV-031: a humanized, anti-CD3 mAb for the interception of T1D in pediatric patients with newly-diagnosed T1D and for delaying and/or preventing disease progression in subjects at risk of developing clinical stage T1D. PRV-031 has been designated by the FDA as an orphan drug for the treatment of newly-diagnosed T1D. PRV-031 was also granted breakthrough therapy designation from the FDA in August 2019 and PRIME eligibility from the EMA in October 2019 for the delay or prevention of T1D;
| |
| ● | PRV-101: a CVB vaccine to prevent acute CVB infections and, in those patients at risk, preventing the CVB-triggered autoimmune damage to pancreatic beta cells that progress to T1D and damage to intestinal cells that lead to celiac disease; | |
| ● | PRV-3279: a humanized bispecific scaffold molecule targeting the B-cell surface proteins, CD32B and CD79B, for the treatment of systemic lupus erythematosus (SLE) and for the prevention of immunogenicity of biotherapeutics such as those used in gene therapy; | |
| ● | PRV-015: a human anti-interleukin 15, or IL-15, mAb for the treatment of gluten-free diet non-responsive celiac disease, or NRCD, intercepting the effects of contaminating gluten in the most common autoimmune disorder without any approved medication; and
| |
| ● | PRV-6527: an oral small molecule CSF-1R inhibitor targeting the differentiation and activation of antigen-presenting cells, or APCs, to prevent chronic inflammatory responses and progression or relapse in Crohn’s disease. |
| 5 |
The table below summarizes the current status and anticipated milestones for our principal product candidates:
| Product Candidate / Indication | Status | Next Expected Milestone | ||
| PRV-031 (teplizumab, anti-CD3 mAb) for the interception of T1D. | Top-line results from a National Institute of Diabetes and Digestive and Kidney Diseases-sponsored study conducted at TrialNet sites for preventing disease progression in patients at-risk of developing T1D were publicly reported in June 2019. |
We expect to file a BLA for the At-Risk indication in Q4 2020.
We expect to complete enrollment of the PROTECT study by the end of 2020. | ||
| In August 2019, the FDA granted BTD to PRV-031 for the delay or prevention of clinical T1D in individuals at-risk of developing the disease. | We expect to report top-line results from the Phase 3 PROTECT study in 2022. | |||
| In October 2019, the EMA granted PRIME eligibility to teplizumab to PRV-031 for the delay or prevention of clinical T1D in individuals at-risk of developing the disease. | ||||
| We commenced a Phase 3 clinical trial (the PROTECT study) in approximately 300 pediatric and adolescent patients with early onset type one diabetes. The first patient was dosed in the second quarter of 2019. | ||||
| PRV-101 (polyvalent CVB vaccine) for the prevention of acute CVB and the prevention of CVB triggered T1D and celiac disease. | We are developing a polyvalent vaccine at Intravacc, our strategic partner in vaccine manufacturing process development. | We expect to commence a first-in-human safety study in the second half of 2020. We expect to report top-line first-in-human data in 2021. | ||
| PRV-3279 (humanized anti-CD32B and CD79B bispecific) for the treatment of SLE and for the prevention of immunogenicity biotherapeutics such as gene therapy. | We announced positive top-line results from our Phase 1b clinical trial (the PREVAIL study), which evaluated PRV-3279 in 16 healthy volunteers. |
We expect to commence the phase 2a portion of the PREVAIL study in 2021. | ||
| PRV-015 (anti-IL-15 mAb) for the treatment of gluten-free diet non-responding celiac disease. | We designed a Phase 2b clinical trial (the PROACTIVE trial) in celiac patients with gluten-free diet non-responsive celiac disease and completed additional chronic toxicology studies to support this trial as needed. | We expect to commence the Phase 2b PROACTIVE study in the second quarter of 2020. | ||
| PRV-6527 (oral CSF-1R inhibitor) for the treatment of Crohn’s disease. | We announced top-line results from our Phase 2a clinical trial (the PRINCE study), which evaluated PRV-6527 in 93 patients with moderate-to-severe Crohn’s disease. In December 2019, Janssen declined its option to buy back PRV-6527. |
We expect to engage with third parties for the potential sublicense of PRV-6527. |
We intend to leverage our distinctive competences and drug development strategy; advance our carefully selected portfolio of product candidates; in-license or acquire additional targeted development assets and apply our disease interception and prevention approach to multiple autoimmune and immune-mediated inflammatory diseases.
| 6 |
OUR PRODUCT CANDIDATES
PRV-031 (teplizumab; anti-CD3 antibody) for T1D
Type 1 Diabetes Background Information
T1D is the end result of immune-mediated destruction of the insulin-producing beta cells of the pancreas and is one of the most common and serious chronic conditions occurring in childhood. T1D patients require life-long dependence on insulin products delivered through multiple daily injections or continuous infusion pumps. While the disease presents in children and adults, the vast majority of T1D is diagnosed in children, with more than half of T1D patients diagnosed before the age of 14 years. The life-expectancy of individuals with younger-onset disease is on average 16 years shorter than non-diabetic individuals. Individuals diagnosed before the age of 10 years have a 30-times greater risk of serious cardiovascular outcomes than the general population resulting in decreased life expectancy, compared to healthy individuals. It is believed the loss of beta cells, which is more severe and rapid in younger individuals leading to increased glycemic load, is the cause of increased cardiovascular-related deaths. The disease is believed to occur in genetically susceptible individuals upon exposure to environmental triggers. In addition, because of a similar genetic predisposition, patients with T1D are at high risk of developing celiac disease. Celiac disease is characterized by autoimmunity in the gut and other organs triggered by consumption of gluten and can lead to malnutrition and other complications including a form of cancer called lymphoma. There is no approved therapy for celiac disease.
Lack of insulin secretory capacity has serious consequences, even when patients receive insulin replacement therapy. The complications of T1D include eye disease, nerve damage, kidney disease and heart disease. Diabetic retinopathy has a prevalence of approximately 80% among patients with T1D and is the leading cause of vision impairment and blindness among adults. Moreover, about 60% to 70% of people with diabetes present some form of neuropathy that can induce numbness, weakness and blood pressure dysregulation. In addition, diabetic nephropathy is the leading cause of chronic kidney disease and affects about 30% of T1D patients. Diabetes can also cause severe heart complications and adults with diabetes are two to four times more likely to die from heart disease than adults without diabetes.
In summary, people with T1D experience substantial morbidity and mortality owing to chronic complications.
Current Treatment Options and Their Limitations
So far, no curative treatment exists for T1D. Patients with T1D still need to use daily insulin injections to manage blood sugar to within a normal range. However, it is estimated that fewer than one-third of people with T1D in the U.S. achieve target blood glucose levels and insulin injections often cause hypoglycemia (low blood sugar). While insulin injections or infusion allow a person with T1D to stay alive, they do not cure the disease, nor do they necessarily reduce the risk of serious effects and long-term complications of T1D.
While pancreatic and islet cell transplantation offer the ability to normalize glucose levels and remove the dependence on insulin products, there are significant risks. There is risk associated with mandatory immunosuppression, which commonly results in the development of infections that may be life-threatening. Furthermore, pancreas transplantation may be associated with technical complications (vascular thrombosis, pancreatitis, infection, fistulas) as well as acute and chronic organ rejection. Islet cell transplantation can provide better glycemic control and protect patients from hypoglycemic episodes, but only approximately 50% of patients are insulin-free after three years of follow-up.
New approaches are still required and could significantly enhance patient care. In particular, there is a strong need for new preventive or curative treatments. Among the different possible strategies, primary prevention through vaccination seems to be the best candidate considering the potential efficacy and safety balance that needs to be achieved.
| 7 |
T1D Market
According to the International Diabetes Federation, 8.8% of the adult population worldwide has diabetes, among whom 10-15% have T1D. According to the Juvenile Diabetes Research Foundation (JDRF), it is estimated that 1.6 million people in the U.S. have T1D and approximately five million are expected to have T1D by 2050, including nearly 600,000 under 15 years of age. The annual economic burden of T1D has been estimated by JDRF to be greater than $30.0 billion in the U.S. and greater than $90.0 billion globally.
Moreover, the incidence of T1D is increasing worldwide and it is estimated that more than 90,000 children are diagnosed each year in the U.S. and the largest five European countries. In the period between 2005 and 2020, epidemiologists predict a 70% increase in the incidence of T1D in children in Europe, with the age of onset decreasing and the number of cases in children younger than five years old doubling.
Overview of T1D Biology and PRV-031 Mechanism of Action
T1D is an autoimmune disease. Specialized white blood cells of our immune system, known as self-reactive T cells (also called auto-reactive), are triggered, presumably by CVB viral infection in at least 50% of cases, to attack and destroy beta cells of the pancreas, thus causing a decline in the natural production of insulin. Simultaneously, another type of T cell, regulatory T cells, or Tregs, which normally suppress the activity of self-reactive T cells, fail to do so effectively.
The clinical progression of T1D is relatively well understood and predictable, as it is a continuum marked by clinically-relevant biomarkers which identify stages of the disease. In an individual with normoglycemia with genetic risk (primarily driven by human leukocyte antigen (HLA) haplotypes), the natural evolution of T1D has been described in stages (see figure below).
| ● | Stage 1: emergence of T1D-related autoantibodies which reflect the initiation of the autoimmune process; this stage is associated with normoglycemia. |
| ● | Stage 2: persistence T1D-related autoantibodies, but with further loss of beta cell function and development of dysglycemia. |
| ● | Stage 3: symptomatic or clinical T1D, when remaining beta cell capacity is insufficient to maintain glucose metabolism. |
Stages of Type 1 Diabetes
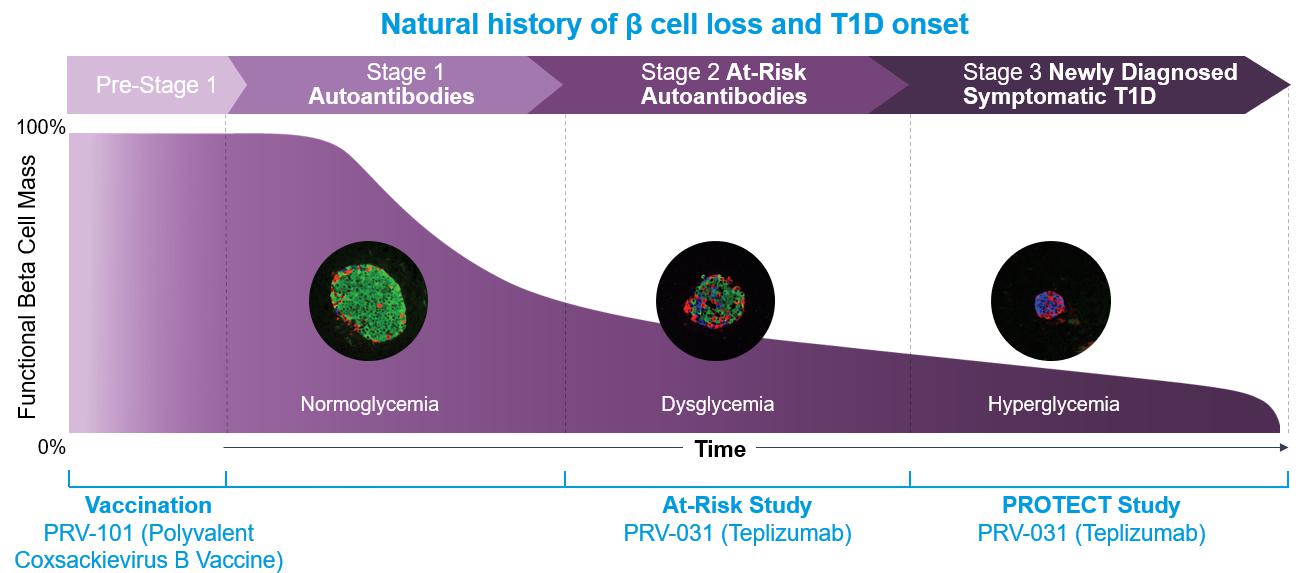
It is important to note that once subjects develop two or more T1D-related autoantibodies (Stage 1), the progression to clinical T1D (Stage 3) is not a matter of “if” but “when” as greater than 95 percent of the Stage 1 subjects and virtually all of the Stage 2 subjects will progress to Stage 3 necessitating insulin dependence. The progression of Stage 1 to Stage 3 is 44% in 5 years, and of Stage 2 to Stage 3 is 75% in 5 years.
| 8 |
PRV-031 (teplizumab) is a humanized mAb that binds with high specificity to a cell surface protein called CD3. The CD3 protein is a co-receptor that helps activate T cells and direct different kinds of immune responses. Preclinical data suggests that binding of PRV-031 to CD3 triggers events that differentially inhibit the activation of self-reactive T cells without affecting Tregs. This restores the important state of immune tolerance and may prevent self-reactive T cells from attacking beta cells in the pancreas. When administered to Stage 2 subjects, teplizumab delayed progression and onset of clinical T1D, and when administered to Stage 3 subjects, teplizumab slowed down the loss of beta cell function in all studies conducted to date. Therefore, we believe PRV-031 has the potential to intercept the T1D disease process along the entire continuum, slowing or preventing the destruction of insulin-producing pancreatic beta cells.
Clinical Development Program
Results from a Phase 2 clinical trial in patients with Stage 2 T1D (Teplizumab for Prevention of Type 1 Diabetes In Relatives “At-Risk”) conducted at TrialNet sites and sponsored by the NIDDK, part of the National Institutes of Health, or NIH were reported at the American Diabetes Association meeting on June 9, 2019 and published in the New England Journal of Medicine. A total of 76 subjects were enrolled and randomized, 44 to teplizumab and 32 to placebo. The data show that teplizumab significantly delayed the median onset of clinical diabetes from 24.4 months (placebo) to 48.4 months (teplizumab) (p=0.006). The safety profile in At-Risk subjects who received a single course of teplizumab was consistent with those from subjects with recent-onset clinical T1D who received two courses of the drug. Teplizumab is the first potential immune modulator therapy that has demonstrated a delay in the onset of clinical disease in T1D.
In August 2019, the FDA granted breakthrough therapy designation to PRV-031 for the delay or prevention of clinical T1D in individuals at-risk of developing the disease. BTD is an FDA program designed to expedite the development and review of therapeutic candidates intended to treat serious or life-threatening diseases.
In October 2019, the European Medicines Agency (EMA) granted PRIority MEdicines (“PRIME”) eligibility to teplizumab (PRV-031) for the prevention or delay of clinical T1D in individuals at-risk of developing the disease. The PRIME initiative is designed to expedite the development and review of promising therapies that target an unmet need and show potential clinical benefit so the medicine can reach patients earlier. The designation offers the opportunity for enhanced interaction and dialogue with the EMA to optimize development, as well as the potential for accelerated assessment at the time of application for a marketing authorization.
In addition, we commenced a Phase 3 clinical trial in pediatric and adolescent patients with newly-diagnosed T1D (Stage 3) and a minimum level of pancreatic beta-cell function based on C-peptide levels at study entry. The study is a randomized, controlled, multi-center study being conducted in North America and Europe. The primary endpoint will evaluate the effect of PRV-031, as compared with placebo, in preserving beta cell function, measured by C-peptide secretion. Secondary endpoints will measure insulin use, HbA1c levels, time-in-range of blood glucose levels and hypoglycemic episodes. We expect to enroll approximately 300 patients. The first patient was dosed in April 2019 and we expect to report top line results in 2022.
We believe that combination therapy may enhance the therapeutic benefit of PRV-031 by increasing efficacy, enhancing the durability of response, or restoring insulin production by beta cells. Combination therapies may include beta-cell autoantigens, tolerogenic cytokines, other modalities which could enhance better depletion of self-reactive lymphocytes or increasing the function of Tregs, or agents that could restore beta cell function or mass. We are collaborating with Precigen (formerly Intrexon) and its subsidiary, ActoBio Therapeutics, to explore the combination of PRV-031 and the oral administration of a Lactococcus lactis strain genetically engineered to secrete human proinsulin and human interleukin-10, an anti-inflammatory cytokine. We plan to explore other combination therapies as the Phase 3 program progresses.
Clinical Evaluation of PRV-031
To date, clinical development of PRV-031 has included both academic and biopharmaceutical sponsors. More than 1,000 subjects have been enrolled in PRV-031 clinical trials, with over 800 subjects receiving PRV-031. These studies represent various doses, formulations, and indications and includes earlier smaller investigator-sponsored studies. The enrollment of patients by therapeutic indication includes approximately 1,050 T1D patients, eight renal or renal-pancreatic allograft rejection patients, 20 induction immunotherapy in pancreatic islet transplant recipients, 11 psoriatic arthritis patients, one plaque psoriasis patient and 64 high-risk patients for the prevention or delay of onset of developing T1D.
In T1D patients, ten studies have been conducted, of which nine involved intravenous dosing (two Phase 1, three Phase 2, two Phase 3 and a Phase 3 extension study) and one subcutaneous dosing (Phase 1).
| 9 |
Among the T1D studies of PRV-031:
| ● | In Stage 2, the At-Risk study enrolled Stage 2 individuals who were characterized as having at least two T1D autoantibodies and evidence of hyperglycemia | |
| ● | In Stage 3, five studies (Study 1, Study 2, Study 3, Study 4 “AbATE”, and Study 5 “Delay”) were completed under the direction of Dr. Kevan Herold (currently at Yale University) and collaborators. Studies 2, 3 and 4 were sponsored by the Immune Tolerance Network. Four additional studies were conducted by MacroGenics: three with intravenous administration (“Protégé”, “Protégé Extension”, and “Protégé Encore”) and one with subcutaneous administration (SUBCUE) of PRV-031. Among these studies, “Protégé” and “Protégé Encore” were Phase 3 studies. Protégé was the largest completed study for treatment of T1D, which enrolled 516 patients (aged 8 to 35 years and T1D diagnosis within 12 weeks of study entry) and randomized into three PRV-031 dosing regimens compared to placebo. PRV-031 showed promising immunological and clinical activities in these studies and was well tolerated. In particular, PRV-031 treatment showed promising data on the preservation of C-peptide levels and the reduction of exogenous insulin use. |
Stage 2 Program
Stage 2: At-Risk Study
The At-Risk study was developed and conducted by Type 1 Diabetes TrialNet, funded by the National Institutes of Health, the American Diabetes Association, or the ADA, and the Juvenile Diabetes Research Foundation. The objective of the study was to determine whether treatment of at-risk subjects with teplizumab results in a delay or prevention of T1D. The primary endpoint was completed in 2018. The study was conducted in 18 sites in the United States, Canada and Germany.
Participants over eight years of age with Stage 2 T1D (presence of at least two T1D autoantibodies and dysglycemia, who were non-diabetic relatives of T1D individuals) were randomized 1:1 to receive teplizumab or placebo. Dysglycemia was defined on oral glucose tolerance test (OGTT) as: (a) Fasting plasma glucose ≥ 110mg/dL, and <126mgdL, or (b) 2-hour plasma glucose ≥140mg/dL, and <200mg/dL, or (c) 30, 60, or 90-minute value on OGTT ≥200mg/dL.
The primary endpoint was the time from randomization to the clinical diagnosis of diabetes, using ADA criteria. Criteria for T1D onset are, based on glucose testing, or the presence of unequivocal hyperglycemia with acute metabolic decompensation (diabetic ketoacidosis). One of the following criteria must be met on two occasions as soon as possible but no less than one day apart for diabetes to be defined:
| ● | Symptoms of diabetes plus casual plasma glucose concentration > 200 mg/dL (11.1 mmol/l). Casual is defined as any time of day without regard to time since last meal. The classic symptoms of diabetes include polyuria, polydipsia, and unexplained weight loss. | |
| ● | Fasting plasma glucose ≥ 126 mg/dL (7 mmol/l). Fasting is defined as no caloric intake for at least eight hours. | |
| ● | 2-hour plasma glucose ≥ 200 mg/dL (11.1 mmol/l). The test should be performed using a glucose load containing the equivalent of 1.75g/kg body weight to a maximum of 75 g anhydrous glucose dissolved in water. |
Teplizumab was administered over a 14-day course: 51 μg/m2, 103 μg/m2, 207 μg/m2, and 413 μg/m2 on study days 0–3, respectively, and 826 μg/m2 on each of study days four through 13. A total of 112 participants were screened and 76 were randomized, 44 to teplizumab and 32 to placebo. The baseline characteristics were balanced for age (median ~13-14 years of age), relationship to the relative with T1D, type of T1D autoantibodies and HbA1c.
Treatment with a single course of teplizumab delayed the time to T1D (see figure below): 19 of the 44 (43%) teplizumab-treated and 23 of the 32 (72%) placebo-treated participants were diagnosed with T1D. The annualized rates of T1D development were 14.9% and 35.9% per year, for the teplizumab and placebo groups, respectively. The median time to T1D was 24.4 months in the placebo and 48.4 months in the teplizumab groups (hazard ratio = 0.412 (95% CI: 0.216, 0.783) p=0.006 (2-sided)).
| 10 |
Time to T1D

In pre-specified analyses, the effects of teplizumab on the primary outcome based on baseline characteristics were evaluated. Although subgroup analyses had small sample sizes and need to be taken with caution, participants without anti-ZnT8 antibodies showed a greater response to teplizumab compared to those with those who did not have the antibody. The presence of HLA-DR4 and absence of HLA-DR3 were associated with more robust responses to teplizumab, and the response to teplizumab was greater in participants whose C-peptide responses to the OGTT at baseline were below the median (1.75 nmol/L).
Teplizumab treatment was associated with few adverse events, described in the table below. Similar to previous studies with teplizumab in newly-diagnosed T1D patients, the lymphocyte count declined to a nadir on day five by 72.3% (IQR 82.1, 68.4%) (p<0.0001). Fifteen (34.1%) of the grade 3 events in the teplizumab group involved lymphopenia during the first 30 days after study drug administration. The lymphocyte counts recovered quickly: Lymphopenia resolved in all participants by day 45 except in one, whose counts returned on day 105. A spontaneously resolving rash, as previously noted, occurred in 36% of drug treated participants. The rates of infection were similar in the two treatment arms. The adverse events below were determined to be possibly, probably or definitely related to the study drug.
Adverse Events
| Adverse Effect Category | Teplizumab | Placebo | ||||||||||
| No. of Events | No. of Subjects (%) | No. of Events | No. of Subjects (%) | |||||||||
| Blood/Bone Marrow*** | 45 | 33 (75) | 2 | 2 (6.2) | ||||||||
| Dermatology/Skin*** | 17 | 16 (36.4) | 1 | 1 (3.1) | ||||||||
| Pain | 11 | 5 (11.4) | 5 | 3 (9.4) | ||||||||
| Infection | 8 | 5 (11.4) | 5 | 3 (9.4) | ||||||||
| Gastrointestinal | 5 | 4 (9.1) | 3 | 3 (9.4) | ||||||||
| Metabolic/Laboratory | 7 | 4 (9.1) | 2 | 2 (6.2) | ||||||||
| Pulmonary/Upper Respiratory | 6 | 4 (9.1) | 0 | 0 (0) | ||||||||
| Constitutional Symptoms | 3 | 2 (4.5) | 0 | 0 (0) | ||||||||
| Allergy/Immunology | 2 | 2 (4.5) | 0 | 0 (0) | ||||||||
| Cardiac General | 1 | 1 (2.3) | 1 | 1 (3.1) | ||||||||
| Endocrine | 0 | 0 (0) | 2 | 2 (6.2) | ||||||||
| Vascular | 1 | 1 (2.3) | 1 | 1 (3.1) | ||||||||
| Neurology | 1 | 1 (2.3) | 0 | 0 (0) | ||||||||
| Ocular/Visual | 1 | 1 (2.3) | 0 | 0 (0) | ||||||||
| Musculoskeletal/Soft Tissue | 2 | 1 (2.3) | 0 | 0 (0) | ||||||||
| Hepatobiliary/Pancreas | 0 | 0 (0) | 1 | 1 (3.1) | ||||||||
| Syndromes | 1 | 1 (2.3) | 0 | 0 (0) | ||||||||
| Hemorrhage/Bleeding | 1 | 1 (2.3) | 0 | 0 (0) | ||||||||
| Total Events and Subjects | 112 | 44 (100) | 23 | 32 (100) | ||||||||
*** p < 0.001 Teplizumab vs placebo
| 11 |
Anti-CD3 mAb treatment has been associated with Epstein Barr virus, or EBV, reactivation. At entry, 30 participants (39%) (16 teplizumab and 14 placebo) had antibodies against EBV. At weeks 3-6 after study drug treatment, there was quantifiable EBV DNA in whole blood in eight of the seropositive participants – all in the teplizumab group, one of whom had symptoms of pharyngitis, rhinorrhea, and cough on day 38. In these participants, the EBV DNA levels were below the level of quantification between day 43 and 134 (average 77 days). At entry, 17 participants (ten teplizumab and seven placebo) had antibodies against cytomegalovirus, or CMV. One teplizumab participant, who was CMV seropositive, had detectable levels of CMV DNA at day 20 that was undetectable by day 42.
These results demonstrate that a single course of teplizumab significantly delayed the progression to clinical T1D in high risk Stage 2 non-diabetic relatives who had at least two autoantibodies and dysglycemia. The median delay in the diagnosis of diabetes was approximately two years, and at the conclusion of the trial, the frequency of diabetes-free subjects was double in the drug (57%) vs placebo-treated subjects (28%). The relatively rapid rate of progression to clinical diabetes in the placebo group, consistent with the previously reported natural history, reflects the very high risk of these individuals and reflects the inevitability of progression from Stage 2 to Stage 3 disease, consistent with observations of high rates of beta cell killing in these subjects. The rapid development of clinical T1D may also reflect the enrichment of pediatric participants (72.4%) in whom the rate of progression is rapid.
Stage 3 Program
Protégé Study
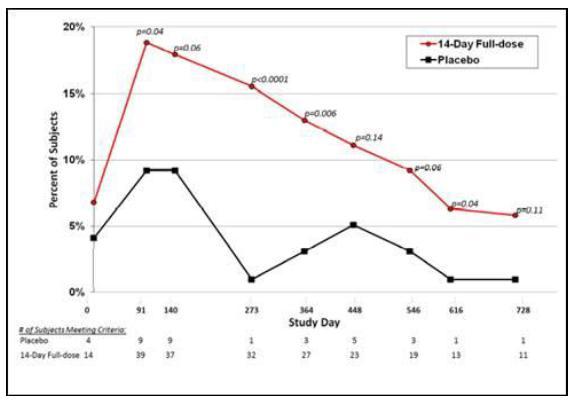
Protégé was a randomized, controlled Phase 3 clinical trial conducted in 83 centers in North America (U.S., Canada, Mexico), India, Israel, and Europe (Czech Republic, Estonia, Germany, Latvia, Poland, Romania, Spain, Sweden, Ukraine). Patients aged eight to 35 years with recently diagnosed T1D (≤12 weeks) were followed for 12 months (Protégé) and continued to 24 months (Protégé Extension). Three dose regimens of PRV-031 were administered to 417 patients as intravenous infusions for six to 14 days; 99 patients received placebo. At 12 months, the primary efficacy endpoint, the proportion of patients with insulin use <0.5 U/kg per day and HbA1c <6.5%, ranged from 13.7% to 20.8% patients in the PRV-031 groups, depending on dosing regimen, and 20.4% in the placebo group. The difference between PRV-031-treated patients and placebo-treated patients was not significant. The change in HbA1c from baseline also did not show a significant difference between PRV-031 and placebo. However, subgroup analyses indicated the following findings:
| 12 |
| ● | The primary endpoint could have been achieved if cut-offs were changed to insulin use of <0.25 U/kg per day and HbA1c <7.0%, not only at 12 months but also at 24 months (figure below). |
| 13 |
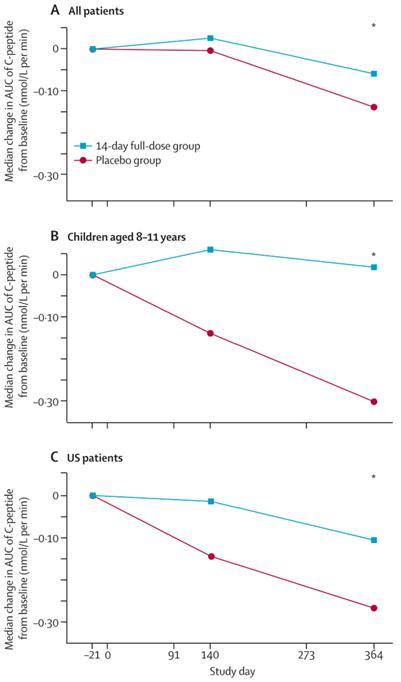
| ● | C-peptide levels significantly improved in the PRV-031 group compared with placebo group in all patients, and further analyses indicated that this difference was more pronounced in younger patients (aged eight to 11 years) and patients enrolled in U.S. sites. These findings are consistent with other clinical trials, showing a stronger effect in T1D patients who are younger (<17 years), more recently diagnosed (<10 weeks), and with higher C-peptide levels at baseline. |
Protégé Encore Study
Protégé Encore was a randomized, controlled Phase 3 clinical trial conducted in 125 centers in 16 countries. Patients aged eight to 35 years with recently diagnosed T1D were to be followed for 24 months. Three dose regimens of PRV-031, given as intravenous infusions for six to 14 days, were compared with placebo. The primary endpoint, the proportion of patients with insulin use <0.5 U/kg per day and HbA1c <6.5% at 12 months, was not met. Study enrollment was stopped at 254 patients (400 planned) when the Protégé study showed that the primary endpoint was not met. Efficacy analyses were not conducted in this study.
| 14 |
A summary of the C-peptide data in the completed Phase 2 clinical trials and Phase 3 Protégé study are shown in the table below. All these studies have shown consistent and significant C-peptide benefit. Furthermore, subgroup analysis of the Protégé data indicated that younger patients (aged eight to 17 years) with minimum baseline beta cell function (C-peptide >0.2 pmol/mL) along with even more robust data in newly-diagnosed T1D (diagnosis under six weeks, Study 1), informed the inclusion criteria that will be applied in our planned Phase 3 study, PROTECT.
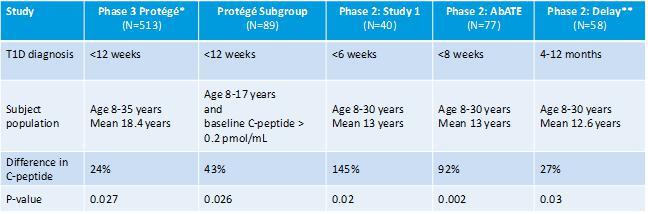
| * | Full 9.0 mg/m2/course 14-Day regimen was explored in 205 treated patients and 98 placebos; |
| ** | Delay study based on 12-month time-point. All other studies based on 24-month time-points |
SUBCUE Study
SUBCUE was a randomized, controlled Phase 1 clinical trial to evaluate the safety and tolerability, pharmacokinetic, or PK, and pharmacodynamics, or PD of subcutaneously injected PRV-031. Patients aged 18 to 35 years who were diagnosed with T1D within 12 months were to be given three dosing regimens of PRV-031 or placebo. Patients were to be followed for 91 days. However, the study was stopped after one subject was enrolled, upon the Protégé study results.
Safety Data
The majority of safety data for PRV-031 comes from two completed Phase 3 studies: Protégé and Protégé Encore. In PRV-031 and placebo-treated subjects, there were no major differences in the overall adverse events, or AEs (99.7% and 100%), and serious adverse events, or SAEs (13.2% (85 out of 645 subjects), and 9.4% (15 out of 160 subjects)), although there were more severe adverse events in PRV-031 subjects (63% and 30%). In the Protégé study, 261 of 415 (62.9%) subjects had severe adverse events compared with placebo, 28 out of 98 subjects (28.6%). In Protégé Encore, 121 of 192 (63%) subjects had severe adverse events compared with placebo, 16 out of 62 subjects (25.8%).
The most common AEs were decreased white blood cells including lymphopenia, leukopenia and neutropenia. Leukopenia/lymphopenia and rash were experienced most frequently by PRV-031-treated subjects. Lymphopenia was expected based on the mechanism of action of PRV-031 and was observed in approximately 70% of type-1 diabetes patients who received PRV-031; lymphopenia was reported in approximately 14% of placebo subjects. It was commonly mild to moderate and resolved within 14 days. In the Protégé study, approximately 50% and 20% of PRV-031- and placebo-treated patients, respectively, reported rash or pruritus. In PRV-031-treated patients, the rash was predominantly mild to moderate and usually resolved within one to two weeks. Laboratory abnormalities were also reported as AEs. The main differences in PRV-031 and placebo subjects were changes in lymphocyte counts (30.1% and 9.4%) and liver function test (alanine aminotransferase, 30.9% and 14.1%). These abnormalities usually resolved within 14 days of dose completion and did not cause significant or lasting clinical concern. Cytokine release syndrome, which may include symptoms of rash, headache, nausea, vomiting, and chills/fever, occurred in fewer than 6% of PRV-031-treated patients and was mild to moderate in severity.
| 15 |
The most common SAEs reported in the Protégé and Protégé Encore studies were related to diabetes control including diabetic ketoacidosis, hypoglycemic seizures/unconsciousness, hyperglycemia, hypoglycemia (consistent with the underlying disorder) and were reported in 6.2% and 2.5% of PRV-031 and placebo subjects, respectively. Three deaths were observed and categorized by the principal investigator (in accordance with International Conference on Harmonisation/Good Clinical Practice guidelines) and included in the Investigator Brochure for PRV-031 filed with the FDA. The relationship between each death and PRV-031 is listed in the Investigator Brochure as follows: one death, “none”; one death “not related”; and one death “unlikely.” The specific causes of deaths were (1) unknown for which the relationship was listed as “none” in the Investigator Brochure, (2) anterior myocardial infarction with ventricular tachycardia and cardio-respiratory arrest for which the relationship was listed as “not related” in the Investigator Brochure and (3) diabetic ketoacidosis for which the relationship was listed as “unlikely” in the Investigator Brochure. AEs of infections (most commonly gastroenteritis) were reported in 3.6% and 2.5% of PRV-031 and placebo subjects, respectively. Fifteen of 85 SAEs and five of 15 SAEs were deemed related to PRV-031 and placebo treatment, respectively.
The following table reflects all the SAEs reported in the Investigator Brochure for the Protégé and Protégé Encore studies:
| SAE by Organ System |
Placebo N=160 n (%) |
Any PRV-031 N=645 n (%) |
| At least one event | 15 (9.4%) | 85 (13.2%)* |
|
Blood and Lymphatic disorders ● Neutropenia ● Lymphopenia |
1 (0.6%) ● 1 (0.6%) |
4 (0.6%) ● 2 (0.3%) ● 2 (0.3%) |
|
Cardiac disorders ● Acute myocardial infarction ● Angina pectoris ● Cardio-respiratory arrest ● Coronary artery disease ● Ventricular tachycardia |
0 |
2 (0.3%)** ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) |
|
Ear and Labyrinth disorders ● Deafness neurosensory |
0 |
1 (0.2%) ● 1 (0.2%) |
|
Eye disorders ● Cataract subcapsular ● Corneal erosion ● Iritis |
0 |
3 (0.5%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) |
|
Gastrointestinal disorders ● Gastritis ● Abdominal pain ● Abdominal pain upper ● Intestinal obstruction ● Nausea ● Peritonitis ● Vomiting |
0 |
10 (1.6%) ● 4 (0.6%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) |
|
General disorders and administration site disorders ● Pyrexia ● Death*** ● Non-cardiac chest pain ● Pain |
2 (1.3%)
● 2 (1.3%) |
4 (0.6%) ● 2 (0.3%) ● 1 (0.2%)
● 1 (0.2%) |
|
Hepatobiliary disorders ● Biliary dyskinesia ● Biloma ● Chlolecystitis acute ● Hepatosplenomegaly |
0 |
3 (0.5%)** ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) |
|
Immune system disorders ● Cytokine release syndrome ● Hypersensitivity |
0 |
4 (0.6%) ● 3 (0.5%) ● 1 (0.2%) |
| 16 |
|
Infections and Infestations ● Gastroenteritis ● Gastroenteritis viral ● Anal abscess ● Appendicitis ● Appendicitis perforated ● Bronchitis ● Dengue fever ● Gastritis viral ● Hepatic amoebiasis ● Hepatitis A ● Infection ● Infectious mononucleosis ● Pharyngotonsillitis ● Pilonidal cyst ● Pneumonia ● Pulmonary tuberculosis ● Pyelonephritis ● Renal abscess ● Sepsis ● Staphylococcal sepsis ● Urinary tract infection ● Varicella ● Cellulitis ● Paronychia ● Tuberculosis |
4 (2.5%) ● 1 (0.6%)
● 1 (0.6%) ● 1 (0.6%) ● 1 (0.6%) |
23 (3.6%)** ● 3 (0.5%) ● 2 (0.3%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%)
|
|
Injury poisoning and procedural complications ● Caustic injury ● Compression fracture ● Fall ● Fibula fracture ● Foot fracture ● Splenic rupture ● Upper limb fracture ● Facial bones fracture |
1 (0.6%)
● 1 (0.6%) |
4 (0.6%)** ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%) ● 1 (0.2%)
|
|
Investigations ● Alanine aminotransferase increased ● Aspartate aminotransferase increased ● Nuclear magnetic resonance imaging brain abnormal |
0 |
3 (0.5%)** ● 2 (0.3%) ● 2 (0.3%) ● 1 (0.2%)
|
|
Metabolism and nutrition disorders ● Diabetic ketoacidosis ● Hypoglycemic seizures ● Hyperglycemia ● Diabetes mellitus out of control ● Hypoglycemic unconsciousness ● Hypoglycemia ● Dehydration ● Ketoacidosis ● Ketosis |
4 (2.5%)
● 3 (1.9%)
● 1 (0.6%)
● 1 (0.6%) |
40 (6.2%)** ● 21 (3.3%) ● 7 (1.1%) ● 5 (0.8%) ● 4 (0.6%) ● 2 (0.3%) ● 2 (0.3%) ● 1 (0.2%) ● 1 (0.2%)
|
|
Neoplasms benign, malignant and unspecified (including cysts and polyps) ● Metastatic malignant melanoma |
0 |
1 (0.2%)
● 1 (0.2%) |
|
Nervous system disorders ● Hypoglycemic coma |
1 (0.6%) ● 1 (0.6%) |
1 (0.2%) ● 1 (0.2%) |
|
Pregnancy, puerperium and perinatal conditions ● Abortion spontaneous ● Complications of pregnancy |
1 (0.6%)
● 1 (0.6%) |
1 (0.2%) ● 1 (0.2%) |
|
Psychiatric disorders ● Mental disorder ● Suicide attempt |
0 |
2 (0.3%) ● 1 (0.2%) ● 1 (0.2%) |
|
Renal and urinary disorders ● Intercapillary glomerulosclerosis ● Ketonuria ● Microalbuminuria |
1 (0.6%)
● 1 (0.6%) |
2 (0.3%) ● 1 (0.2%) ● 1 (0.2%) |
|
Reproductive system and breast disorders ● Epididymitis |
0 |
1 (0.2%) ● 1 (0.2%) |
|
Skin and subcutaneous tissue disorders ● Rash |
0 |
2 (0.3%) ● 2 (0.3%) |
|
Vascular disorders ● Subclavian vein thrombosis |
0 |
1 (0.2%) ● 1 (0.2%) |
| * | Note: there are 112 events observed in 85 subjects |
| ** | Note: subject may have more than one adverse event |
| *** | Note: because the cause of death was unknown, “death” is reported as an adverse event |
| 17 |
The most common SAE occurring in at least 10% of subjects in both treatment groups in the Protégé study was decreased white blood cell counts (lymphopenia/neutropenia) observed in 47% (196 out of 415 subjects) and 10% (10 out of 98 subjects) of PRV-031 and placebo subjects, respectively. In Protégé Encore, lymphopenia/neutropenia was also the most frequently observed severe adverse event, occurring in 24% (46 out of 192 subjects) and 6% of PRV-031 (four out of 62 subjects and placebo subjects, respectively. This severe adverse event is consistent with the mechanism of action of PRV-031.
Overall, infections were not increased following PRV-031 treatment. However, in Protégé there were ten cases of herpes zoster infections (a virus that usually causes chicken pox or shingles) in PRV-031-treated patients that were possibly dose-related, and none in the placebo group. All of these cases were resolved. In the Protégé Encore study, only one patient, who was randomized to placebo had herpes zoster. A link between PRV-031 and herpes infections remains unclear. Other herpes virus infections (e.g., cytomegalovirus and Epstein-Barr virus) were not increased with PRV-031 treatment.
Phase 3 Clinical Trial of PRV-031 in Pediatric Patients Newly-Diagnosed T1D (PROTECT Study)
The PROTECT study (PROvention T1D trial Evaluating C-peptide with Teplizumab) is a randomized, double-blind, placebo-controlled, multicenter Phase 3 clinical trial in pediatric and adolescent patients (aged eight to17 years) with recent-onset T1D. Patients with minimum beta-cell cell function (C-peptide >0.2 pmol/mL) and within six weeks of T1D diagnosis will receive two courses of teplizumab, six months apart. Each course will consist of 12 days of teplizumab administered intravenously. The primary endpoint is the change in C-peptide at 18 months. Secondary endpoints including insulin use, HbA1C levels, hypoglycemic events and safety will also be evaluated. The study is expected to enroll approximately 300 patients with 2:1 randomization (200 active: 100 placebo) and enrollment commenced in the second quarter of 2019. We expect to complete enrollment by the end of 2020 and report top line results for the Phase 3 PROTECT study in 2022.
Phase 2 Clinical Trial of PRV-031 in combination with AG019 in newly diagnosed T1D patients
This is a Phase 1b/2a clinical trial being conducted in collaboration with ActoBio which will explore the combination of teplizumab with ActoBio’s AG019 in participants with recent-onset T1D. AG019 is a capsule consisting of engineered Lactococcus lactis specifically modified to deliver human proinsulin and the tolerance-enhancing cytokine human interleukin-10 to the mucosal lining of the gastro-intestinal tissues. The primary objective of the study is to assess the safety and tolerability of different doses of AG019 alone as well as AG019 in association with teplizumab. The secondary objectives of this study are: to obtain PD data of AG019 alone as well as AG019 in association with teplizumab; and PK data to determine the potential presence of AG019 in systemic circulation (safety - systemic exposure) and the presence of L. lactis bacteria in fecal excretion (local exposure). The study will enroll 48 participants and will be conducted in two phases:
| ● | Phase 1b: open-label part of the study which will investigate the safety and tolerability of two different doses of AG019 in two age groups (18 to 40 years of age and 12 to 17 years of age). | |
| ● | Phase 2a: randomized, double-blind part of the study which will investigate the safety and tolerability of AG019, in association with teplizumab, in two age groups (18 to 40 years of age and 12 to 17 years of age). |
The Phase 1b part of the study commenced in October 2018.
| 18 |
Pre-Clinical Evaluation of PRV-031
PRV-031 binds specifically to human T cells with CD3 on the surface. It also binds to CD3+ T cells in chimpanzees, an endangered species that is inappropriate for extensive experimentation, but does not bind to CD3+ T cells of any other animal species. Due to this lack of feasible animal models, nonclinical pharmacology, pharmacokinetic, and toxicology studies are limited. Nonetheless, consistent with its mechanism of action and binding to CD3, PRV-031-treated chimpanzees showed reversible reductions in circulating T cells and a dose-dependent increase in various immune signaling molecules (TNF-α, IL-6, IL-10 and IFN-γ). At very high PRV-031 doses, approximately 450-fold higher than the highest daily dose administered in humans (826 μg/m2), chimpanzees developed B cell lymphoproliferative disease (similar to lymphoma) and Epstein-Barr virus-like infection. In human tissues, PRV-031 binds to T cells in multiple human tissues without unanticipated binding to other cell types. These results indicate that PRV-031 has a low probability of producing unexpected and unintended toxicities in human clinical trials.
Regulatory Strategy for PRV-031
In November 2019, we completed a Type B multidisciplinary meeting with the U.S. Food and Drug Administration (FDA) to discuss the proposed contents of a Biologics License Application (BLA) for PRV-031 (teplizumab) for the prevention or delay of T1D in individuals at-risk of developing T1D. Based on official FDA meeting minutes, Provention continues to expect that it will commence a rolling BLA submission for PRV-031 in the middle of 2020 and is targeting completion of the submission in the fourth quarter of 2020. We do not anticipate the need to conduct any additional clinical trials in the at-risk population prior to BLA submission.
For the Chemistry, Manufacturing and Controls (CMC) module, the FDA confirmed that it would require the demonstration of comparability between the study drug previously manufactured by MacroGenics and Eli Lilly and the to-be-commercialized drug substance and drug product scheduled for production by Provention and its contract manufacturing partners.
The Type B meeting discussion with the FDA supports Provention’s belief that results from the “At-Risk” study, together with adequate confirmatory evidence from prior teplizumab studies in early onset T1D, will be sufficient for a BLA submission. The FDA provided guidance on specific analyses of data from our clinical database of over 800 patients for inclusion in the BLA submission, specifically the impact of PRV-031 on C-peptide levels in T1D patients. C-peptide is a byproduct of endogenous insulin production and a universally accepted measure of the amount of insulin naturally produced by functional beta cells in the pancreas.
The FDA also confirmed that the safety database from the “At Risk” study and prior teplizumab clinical studies in patients with early onset T1D appears adequate to support the submission and review of a BLA.
Market Opportunity for PRV-031
Estimated Patient Prevalence of At-Risk Indications (all ages)
| Market | Stage 1 / Stage 2 >2 Autoantibodies | Stage 2 >2 Autoantibodies and Dysglycemia | Stage 2 Familial Direct Relatives of T1D Patients who exhibit >2 Autoantibodies and Dysglycemia | |||||||||
| United States | 300,000 | 200,000 | 30,000 | |||||||||
| European Union | 180,000-300,000 | 120,000-200,000 | 18,000-30,000 | |||||||||
Our potential launch of PRV-031 would initially focus on familial direct relatives of T1D patients, with two or more autoantibodies and dysglycemia, and then expand to universal screening.
| 19 |
Our commercial priorities are as follows:
Near-Term Focus
| ● | Commercial Planning in the United States for the At-Risk indication | |
| ● | Type 1 Diabetes disease awareness and risk factors | |
| ● | Health Care Professional (HCP) and Key Opinion Leader (KOL) Engagement | |
| ● | Patient Advocacy Group Partnerships | |
| ● | Screening of familial direct relatives of known T1D patients |
Mid-Term Focus
| ● | Commercial Planning / Partnership in European Union | |
| ● | Newly Diagnosed indication launch |
Long-Term Focus
| ● | Market expansion with broader population screening | |
| ● | Addition of age groups and multiple course of treatment for both At-Risk and Newly Diagnosed indications. |
T1D Lifecycle Management and other potential indications
We are currently exploring and supporting research and lifecycle management programs for T1D beyond the current treatment regimen and patient population including, repeat dosing and age expansion for at-risk patients and newly diagnosed patients, as well as subcutaneous formulations and combination therapies potentially with antigens, metabolic drugs, immune modulators and beta cell transplants. Other potential indications for PRV-031 may include GI immunology disorders such as Crohn’s disease, Celiac disease and autoimmune hepatitis or rheumatology disorders such as psoriatic arthritis and rheumatoid arthritis. These initiatives may include new clinical studies sponsored by us or investigator-initiated studies, which are clinical studies initiated and sponsored by physicians or research institutions with funding from us.
PRV-101 (CVB Vaccine) for Acute Infection and T1D
Overview of CVB Infection of the Pancreas, T1D and PRV-101’s Mechanism of Action
Longitudinal studies of more than 200,000 children studied for up to two decades in Finland by our technology licensor, Vactech, and its collaborators (DIPP Study), identified CVB infection as a likely environmental trigger in the onset of T1D autoimmunity and T1D-associated celiac disease (CD) autoimmunity. Subsequent full-virome analysis of the TEDDY Study (400,000 children international study) confirmed that CVB is the only virus whose persistent infection is associated with the development of T1D and celiac disease autoimmunity (T1D-associated and also independent of T1D).
CVB infection is very common and is responsible for various symptoms and complications ranging from mild respiratory disease, gastrointestinal disturbances and hand-foot-mouth disease to life-threatening cardiomyopathy and meningitis. However, in patients with a certain genetic background, CVB appears responsible for the development of autoimmunity. The T1D association with CVB infection has been observed in independent cohorts in 15 countries, including in North America, Europe and Australasia. These epidemiological observations have been substantiated by biological experimentation. Insulin-producing beta cells in the pancreas express specialized receptors associated with the transport, storage and release of insulin. These receptors appear to be used by CVB to preferentially infect these cells and polymorphism in these receptors are associated with development of T1D autoimmunity. Infection by enteroviruses can be detected in the pancreatic beta cells of approximately 60% of type-1 diabetes patients and in the gut of most patients with T1D-associated CD. Importantly, if mothers have anti-CVB immunity at the time of the pregnancy, a 50% reduction in the onset of T1D autoimmunity (T1D-associated auto-antibodies) has been observed in their offspring, presumably due to protection by maternal antibodies passed on to the fetus. This observation strongly suggests the potential efficacy of CVB vaccination for children and/or mothers, resulting in the development of protective antibodies potentially capable of preventing or delaying the onset of T1D.
An analysis of stool samples collected from these individuals identified enterovirus infections prior to the first detection of T1D auto-antibodies. Enterovirus RNA was also detected in stool samples. Examination of antibodies present in DIPP children who developed at least two islet cell auto-antibodies (sign of incipient T1D) and/or progressed to T1D confirmed that among all enteroviruses, only CVB was significantly associated with initiation of beta cell autoimmunity.
| 20 |
Enterovirus RNA in Blood is Linked to the Development of T1D
OR: odd ratio; CI: confidence interval; EV: enterovirus

Proposed First in Human Phase 1 Clinical Trial of PRV-101
PRV-101 is expected to be a polyvalent (more than one strain) prophylactic CVB vaccine intended to prevent acute CVB infection and the development of CVB-induced T1D and celiac autoimmunity. We believe that, if successful, PRV-101 may prevent up to 50% of T1D cases and up to 20% of celiac cases. The vaccine is currently in a Clinical Trial Application (CTA)-enabling stage, requiring manufacturing and nonclinical studies prior to initiation of the FIH study. Animal safety and efficacy modeling studies completed to date by Vactech demonstrate that CVB triggers diabetes in two animal models of T1D and that vaccination against CVB protects mice from acute infection as well as prevents the onset of diabetes triggered by CVB infection.
We plan to commence a FIH Phase 1 clinical trial in the second half 2020 in healthy adult volunteers, with top-line data available in 2021. The primary objective of this FIH clinical trial is to evaluate the safety and tolerability of multiple doses of PRV-101 administered at two different dose levels in adult healthy volunteers. A secondary objective is to evaluate the immunogenicity (ability to elicit antibodies to CVB) of PRV-101.
Preclinical Data for PRV-101
The mechanism of action and efficacy of PRV-101 is supported by the results of several in vivo studies. Inactivated CVB-based viral vaccines efficiently protect mice from CVB infections and from viral spread to the pancreas, as seen for CVB1 and CVB3 vaccines. Similar experiments conducted with a vaccine covering all six CVB serotypes demonstrated that it can induce a strong neutralizing anti-CVB response in mice and protect the animals against multiple CVB infections from the corresponding live viruses. Independent experiments confirm that CVB infection can accelerate T1D onset in T1D susceptible NOD (Non-obese diabetic) or SOCS-1-Tg (suppressor of cytokine signaling 1 transgenic) mice, suggesting that protection from CVB infection would therefore protect against T1D development. This hypothesis has been recently confirmed in experiments conducted by the Karolinska Institute (Sweden) and the University of Tampere (Finland), demonstrating that a CVB1 vaccine indeed protected SOCS-1-Tg mice against T1D induced by CVB1. These mice develop T1D after CVB1 infection as a consequence of a direct infection of insulin-producing beta cells in the pancreas and the subsequent immune response against the beta cells, mimicking human T1D. A three-injection vaccination course induced robust neutralizing antibody responses against CVB1 and protected mice from both CVB1 infection and CVB1-driven T1D. CVB1 infection led to a loss of insulin-producing cells in unvaccinated mice, which also was prevented by the vaccine. These data strongly support the development of PRV-101 for the prevention of T1D.
| 21 |
Formalin-Inactivated CVB1 Vaccine is effective against CVB1-Induced T1D in a Mouse Model.
As seen in the left panel below, CVB1 infection led to loss of insulin-producing cells, and this pathology was completely prevented by the CVB1 vaccine (right panel). In this experiment, while 50% of unvaccinated mice develop T1D as a consequence of CVB1 infection, all vaccinated mice were protected (not shown).
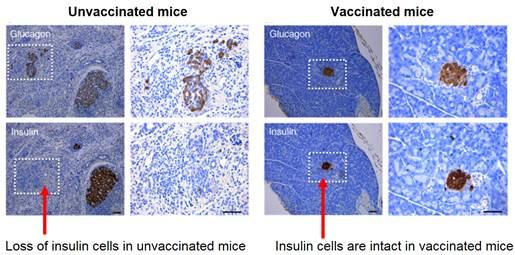
Important from a safety point of view, the formalin-inactivated CVB1 vaccines did not cause any undesirable effects in the pancreas. There was no vaccine-induced pancreatic pathological change, islet autoimmunity or diabetes in the vaccinated mice.
Finally, maternal CVB infection during gestation in mice protects the offspring from CVB infection and subsequent T1D development, presumably through transfer of specific antibodies from the mother to the fetus, corroborating previous findings in humans in the DIPP study and further supporting the use of a prophylactic vaccine to protect against CVB-associated-T1D.
CTA-Enabling Program to Support FIH Study
The planned CVB vaccine toxicology program will consist of Good Laboratory Practices, or GLP, and non-GLP safety and immunogenicity studies conducted in mice. These studies are designed to identify and characterize potential toxicities associated with PRV-101 treatment, including those arising from the immune responses induced by the product. They will mirror the administration regimen that will be used in the proposed FIH study by same route of administration.
Pharmacology studies will be conducted to determine the exact composition of the vaccine. It is currently considered that such CVB vaccine should ideally be a polyvalent vaccine (encompassing several CVB serotypes). After completion of these studies, we plan to undertake GMP manufacturing of the final vaccine for clinical trials.
CVB Infection Market
Enteroviruses are responsible for an estimated ten to 15 million symptomatic infections in the U.S. annually. CVB contributes to a major part of the healthcare costs of enteroviruses as they cause the most serious complications and are among the most frequently reported enteroviral infections according to the CDC. Acute CVB infection is usually asymptomatic or causes common cold-type symptoms. It often leads also to a febrile illness associated by rash, hand-foot-mouth disease and/or mild GI distress. However, CVB infections cause also more severe manifestations including pericarditis, myocarditis, meningitis and pancreatitis.
| - | Myocarditis: CVB is the most common etiologic agents for myocarditis in the Western world, responsible for up to 33% of cases of myocarditis. Myocarditis is an important cause of sudden unexpected death: the prevalence of myocarditis in children and adolescents leading to sudden unexpected death has been reported to be as high as 8% to 42%. In certain individuals, acute myocarditis progresses to chronic myocarditis and dilated cardiomyopathy, which is a severe life-threatening condition. |
| 22 |
| - | Otitis media: otitis media (middle ear inflammation) may develop in patients with upper respiratory disease caused by enterovirus. Otitis media constitutes 18% of physician visits in the U.S. (largest single reason in children). The costs of otitis media treatment in the U.S. were estimated to be approximately $3 billion in 2014. | |
| - | Meningitis: CVB is a common cause of enteroviral meningitis. Meningitis beyond the neonatal period is characterized by the sudden onset of fever of 38-40°C. Headache and photophobia are almost universally reported in these patients. Reports on the incidence of viral meningitis vary from approximately 50,000 hospitalized cases to over 2 million cases of aseptic meningitis per year. Based on 300,000 annual cases of aseptic meningitis in the United States (of which enteroviruses, and coxsackie viruses in particular, are the most common cause), the economic impact is estimated to be $1.5 billion in direct costs alone. |
PRV-3279 (Humanized CD32B x CD79B Dual Affinity Biologic for SLE and Other Autoimmune Diseases)
Overview
SLE is a chronic autoimmune disorder that can affect nearly every major organ system, causing inflammation, tissue injury, organ damage, and in some patients, organ failure. The prognosis of SLE is highly variable in individual patients, often waxing and waning throughout their lifetime. The natural history of SLE ranges from relatively benign disease to rapidly progressive and even fatal disease. Comorbidities, such as infections, malignancies, hypertension, lipid disorders and diabetes increase the risk of disability and death in patients with SLE. Organ systems commonly affected by SLE include the central nervous system, kidneys, gastrointestinal system, mucous membranes, heart, skin, hematologic system, musculoskeletal system and lungs, with specific organ involvement defining subsets of the disease (e.g., lupus nephritis). According to the Lupus Foundation of America, at least 1.5 million Americans are afflicted by SLE and more than 16,000 new cases of lupus are reported annually. It is estimated that five million people throughout the world suffer from some form of lupus. Lupus affects primarily women of childbearing age (15 to 44 years). However, men, children, and teenagers can also develop lupus.
The pathogenesis of SLE is characterized by an abnormal overactivation of B cells and subsequent pathologic production of auto-antibodies (antibodies that attack one’s own cells and tissues). Uncontrolled activation of B cells is normally terminated when the activating stimulus is exhausted and when a negative feedback loop is triggered by the engagement of an inhibitory Fc receptor, or FcR, known as FcgammaRIIb, or CD32B. Mutations in the CD32B gene in humans are associated with an increased likelihood of SLE, and reduced expression of CD32B is apparent in B cells from SLE patients. It is thought that activation of this inhibitory pathway could ameliorate the overactive B cell-driven pathology of SLE and other autoimmune diseases. In addition, the excess auto-antibodies produced bind to target antigens and form immune complexes.
When the B cell receptor, or BCR, (which is the “Y” shaped molecule, resembling an antibody in the figure below) is bound and activated by an antigen, it initiates a cascade of biochemical changes necessary for the activation of the CD32B inhibitory pathway, thus triggering the negative feedback loop. CD79B is a subunit of the BCR that plays a key role in this process when it is close to CD32B. Therefore, if a pharmacologic treatment is to activate the CD32B inhibitory pathway, it also has to simultaneously bind to CD79B. PRV-3279 (formerly MGD010), is a humanized CD32B x CD79B DART protein developed originally by MacroGenics as a bi-specific therapy with these properties, and thus a potential treatment for SLE and other similar diseases. It is designed to simultaneously bind to CD32B and CD79B on B cells.
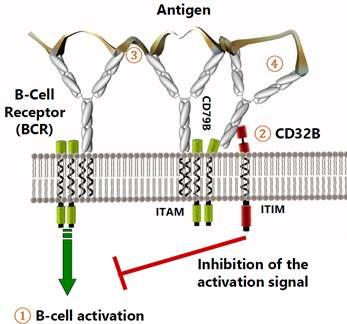
| 23 |
PRV-3279 and related molecules have shown inhibitory effects on BCR-induced B cell proliferation and antibody secretion (including B cells obtained from SLE patients) as well as beneficial effects in mouse models of autoimmunity. PRV-3279 is expected to boost the negative feedback loop on B cells by robustly engaging the available CD32B and CD79B.
PRV-3279 has been studied in humans and was shown to be well tolerated. Proof of mechanism and PRV-3279’s inhibitory effect on antibody immune responses were demonstrated in a Phase 1a single ascending dose study in healthy volunteers, including a cohort demonstrating inhibition of the immunogenicity of the hepatitis A vaccine. Immunogenicity of PRV-3279 was also observed, but had no impact on mechanistic effects, safety or pharmacokinetics, and decreased with increasing doses of PRV-3279, possibly a reflection of its mechanism of action.
We are developing PRV-3279 for the treatment of SLE in the PREVAIL (PRV-3279 EVAluation In Lupus) study. PREVAIL is a multiple ascending dose Phase 1b/2a clinical trial in two parts: Part 1 was a recently completed Phase 1b clinical trial in 16 healthy volunteers, and Part 2 will be a Phase 2a clinical trial in SLE patients. Part 2 will have a treatment duration of at least 12 weeks and will evaluate clinical and biomarker endpoints.
Our ultimate goal is to determine if PRV-3279 can intercept the pathophysiology of SLE by preventing the production of auto-antibodies by abnormally active B cells.
Current Treatment Options for SLE and Their Limitations
The treatment and management of SLE depends on disease severity and disease manifestations. Hydroxychloroquine plays a central role in the long-term treatment of SLE and is the cornerstone of SLE therapy. Corticosteroids, nonsteroidal anti-inflammatory drugs (NSAIDs), and immunosuppressive agents (e.g., azathioprine, cyclophosphamide, cyclosporine, methotrexate, and mycophenolate mofetil) have also been used in the treatment and management of SLE. These treatments are only modestly effective and present safety and/or immune suppression concerns with prolonged use. The B cell-depleting antibody rituximab (Rituxan), while not approved for treatment of SLE, appears to be beneficial in certain subsets of patients.
In 2011, the FDA approved belimumab (Benlysta), an antibody that targets B lymphocyte stimulator, for the treatment of mild to moderate SLE in combination with standard therapy, providing additional clinical validation of the therapeutic benefit of B cell-targeted therapy for autoimmune diseases. However, the modest therapeutic benefit of belimumab and delayed onset of disease intervention indicate the need for additional therapeutic strategies to inhibit overactive B cells. We believe PRV-3279 can fulfill that requirement and is uniquely differentiated to allow for rapid inhibition of activated B cells (potentially more effective than belimumab), while sparing non-activated B cells from depletion or inactivation (potentially safer than rituximab).
| 24 |
Overview of CD32B Biology
CD32B is expressed widely on the surface of human B cells. In addition to its expression on B cells, CD32B is also expressed on other immune cells such as dendritic cells, macrophages, neutrophils, and mast cells. It is a single-chain protein with a portion that sits outside of the cell membrane, which can be bound by chemical signals.
CD32B is the only known inhibitory FcR in the immune system. It plays an important role not only for innate and adaptive immune responses, but also in the maintenance of immune tolerance and controlling autoimmunity. Mice deficient in CD32B have increased antibody responses due in part to chronic B cell activation, and as a result, develop autoimmune disease similar to human SLE. In contrast, B cell-specific overexpression of CD32B reduces the incidence and severity of lupus in a mouse lupus model. In humans, mutations and decreased expression of the CD32B gene are associated with an increased likelihood of SLE. These results underscore the important role of CD32B in regulating the antibody immune response and suggest that drug-mediated engagement of CD32B could provide therapeutic benefit in autoimmune diseases by dampening the effects of chronically activated B cells and reducing the production of auto-antibodies. In particular, preventing the production of auto-antibodies could intercept the disease course in lupus nephritis, a subtype of lupus driven by accumulation of auto-antibodies and immune complexes (a mass of antibodies and other molecules) in the kidneys.
Mechanism of Action of PRV-3279
PRV-3279 is in a new class of bispecific scaffold antibody-like molecules called DARTs. It is designed to simultaneously bind to CD32B and CD79B on B cells. The simultaneous binding of both CD32B and CD79B triggers CD32B-coupled immunoreceptor tyrosine-based inhibitory motif signaling, which leads to the suppression of B cells activated to produce auto-antibodies, while not causing broad B cell depletion.
To prolong its half-life in the body, PRV-3279 contains a human IgG1 Fc region (a specific antibody fragment) that is manipulated to eliminate its effector function. As a molecule designed to inhibit immune responses, PRV-3279 does not activate any part of the immune system either in the body or in laboratory tests. PRV-3279 also does not bind to platelets, a unique feature compared to competing molecules targeting CD32B that are associated with toxicity due to binding to platelets.
Multiple Ascending Dose Phase 1b/2a clinical trial of PRV-3279 in Healthy Volunteers and Patients with Lupus
We are conducting a two-part study in SLE, the PREVAIL (PRV-3279 EVAluation In Lupus) study. PREVAIL is a randomized, double-blind, placebo-controlled Phase 1b/2a clinical trial to evaluate the safety, tolerability, PK, PD, and immunogenicity of multiple ascending doses of PRV-3279 in 16 healthy adult volunteers (Part 1) and the efficacy of PRV-3279 in patients with lupus (Part 2).
On March 12, 2020, we announced positive top-line results from the Phase 1b portion of the PREVAIL study. PRV-3279 was well-tolerated, with no serious adverse events, and as expected, did not deplete B cells and demonstrated profound and sustained binding to circulating B lymphocytes, with reduction of circulating immunoglobulin M levels in a dose-proportional manner. While anti-drug antibody production was observed at both dose levels tested, immunogenicity was found not to affect exposure, safety or pharmacodynamic parameters.
Based on these results, we plan to commence the Phase 2a portion of the PREVAIL study in lupus patients in the first half of 2021.
Our ultimate goal is to determine if PRV-3279 can intercept the pathophysiology of SLE by preventing the production of auto-antibodies by abnormally active B cells. Part 2 will have a treatment duration of at least 12 weeks and endpoints will include lupus clinical assessments and biomarker measurements. Clinical endpoints will include the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), the British Isles Lupus Assessment Group score, urine protein to creatinine ratio, and daily glucocorticoid use. Additional biomarkers will include urinary/renal markers (e.g., serum creatinine, estimated glomerular filtration rate) and blood/circulating markers (e.g., auto-antibodies, complement (C3 and C4), B cell function/phenotype, including CD32B expression/response relationship).
| 25 |
Preclinical Evaluation of PRV-3279
The only nonhuman species that PRV-3279 binds to is chimpanzees. An initial non-GLP study with PRV-3279 in chimpanzees demonstrated it to be well tolerated at all doses, with an assigned no observed-adverse-effect level, or NOAEL, of 10 mg/kg.
Due to the lack of target binding, chronic four-week and three-month repeat-dose GLP toxicology studies were performed using a surrogate DART molecule similar to PRV-3279 that was designed to target human CD32B and mouse CD79B in a transgenic mouse line that expresses human CD32B. A NOAEL at the highest dose of 50 mg/kg was assigned in the three-month study. These studies support the advancement of PRV-3279 in long-term efficacy studies in humans (up to three months).
Clinical Evaluation and Proof of Mechanism for PRV-3279
To date, there have been two clinical trials completed with PRV-3279. The first study was conducted at a single site in the U.S., from February 2015 to February 2017 and was a first in human, or FIH, double-blind, placebo-controlled Phase 1a clinical trial to evaluate the safety, tolerability, PK, PD, and immunogenicity of PRV-3279 in healthy adult volunteers.
A total of 49 subjects were randomized; 12 received placebo and 37 received PRV-3279 intravenously at escalating doses from 0.1 mg/kg to 10 mg/kg in six cohorts. PRV-3279 was well tolerated over the range of doses, with only mild adverse events that resolved quickly, including headache, somnolence (sleepiness), upper respiratory tract infection, folliculitis and night sweats. Target binding and proof of mechanism were demonstrated by measuring functional B cell inhibition at doses of 1 mg/kg or higher, without broader B cell activation or depletion observed.
Subsequently, proof of mechanism was further confirmed in a dose escalation extension of the study in which single doses of PRV-3279 at 3 mg/kg and 10 mg/kg (16 subjects) were compared with placebo (eight subjects) for the ability to affect B cell responses to a hepatitis A vaccine, which was administered to participants who had no previous hepatitis A immunity, on day two of the study. At both doses, PRV-3279 reduced the proportion of volunteers who generated an immune response against the vaccine, as well as the amount of antibody they produced, in both cases as compared to placebo.
The second study was the Phase 1b portion of the PREVAIL study, which was a double-blind, placebo-controlled, multiple ascending dose study in 16 health volunteers.
On March 12, 2020, we announced positive top-line results from the Phase 1b portion of the PREVAIL study. PRV-3279 was well-tolerated, with no serious adverse events, and as expected, did not deplete B cells and demonstrated profound and sustained binding to circulating B lymphocytes, with reduction of circulating immunoglobulin M levels in a dose-proportional manner. While anti-drug antibody production was observed at both dose levels tested, immunogenicity was found not to affect exposure, safety or pharmacodynamic parameters.
Pharmacokinetics and Immunogenicity of PRV-3279
PRV-3279 exhibited an approximate half-life of seven days after a single dose. A majority (~86%) of study participants developed antibodies against PRV-3279 (i.e., immunogenicity) after receiving the 3 mg/kg dose, but no detrimental effect was observed on the pharmacokinetics of PRV-3279. The proportion of participants developing antibodies against PRV-3279 decreased with increasing dose (29% in the 10 mg/kg dose) and such antibodies did not occur in the multiple dose chimpanzee study, suggesting that PRV-3279 may limit its own immunogenicity at therapeutic doses, which is consistent with its mechanism of action.
SLE Market and Other Opportunities for PRV-3279
Sales of therapies to treat SLE are expected to climb to nearly $2 billion in 2019, approximately 17% annual growth from 2009. This growth is driven primarily by the entry and uptake of novel treatments that target B cells such as belimumab, the possible approval of new mechanisms (e.g. Type I interferon inhibitors) and off-label use of rituximab. The uptake of belimumab has been driven largely by safety rather than substantial efficacy, supporting the unmet need and potential for novel and safe non-depleting B cell therapies with greater efficacy.
| 26 |
In addition to SLE, PRV-3279 has the potential to treat other B cell- and auto-antibody-driven autoimmune diseases. Such diseases include multiple sclerosis and RA, where B cell therapies rituximab and recently approved ocrelizumab (Ocrevus) have sales in excess of $1 billion. Several niche/orphan indications may also be explored, including T1D (potentially in combination with PRV-031), Sjogren’s syndrome, vasculitis (e.g., polymyalgia rheumatica, giant cell arteritis, Behçets disease), myasthenia gravis, pemphigus, neuromyelitis optica, anti-NMDA receptor encephalitis, Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, Grave’s ophthalmopathy, IgG4-related disease, and idiopathic thrombocytopenic purpura.
Finally, PRV-3279 also has the potential to prevent or reduce the immunogenicity of biotherapeutics, including but not limited to gene therapy vectors and transgenes (new proteins expressed as a result of the gene therapy). Provention Bio intends to explore collaborations with leaders in gene therapy and other biotherapeutic modalities.
PRV-015 (human anti-interleukin 15 mAb) for NRCD
Overview
Celiac disease is a systemic autoimmune disease triggered by gluten consumption in genetically susceptible individuals. Approximately 1% of the European and North American population is affected by celiac disease. Gluten is ubiquitous in food and elicits autoimmune responses in celiac patients, with damage to the mucosal lining of the small intestine. Celiac disease causes debilitating symptoms and serious medical complications, as the small bowel damage can lead to nutrient malabsorption and results in a range of subsequent intestinal and extra-intestinal clinical manifestations. The stimulation of intestinal lymphocytes for decades can lead to the development of lymphoma, with increased mortality.
The pro-inflammatory cytokine interleukin 15 (IL-15) has been identified as a major mediator in the pathophysiology of celiac disease. PRV-015, a fully human mAb, binds to and inhibits IL-15 and has emerged as a leading candidate for the treatment of nonresponsive celiac disease, in which patients continue to have disease activity despite ongoing gluten free diet, or GFD. PRV-015 was initially developed by Genmab A/S as HuMax-IL15, and by Amgen and Celimmune LLC as AMG 714. PRV-015 has undergone clinical testing in approximately 250 subjects who have received PRV-015 across two Phase 1 (healthy volunteers and psoriasis, rheumatoid arthritis, or RA) and three Phase 2 clinical trials (celiac disease, RCD-II, RA). No serious adverse events deemed related to PRV-015 were observed that would preclude further clinical development. Proof of Mechanism and/or proof of concept was demonstrated in RA, celiac disease and refractory celiac disease Type II. The effect of PRV-015 in celiac disease was evidenced by reduction in inflammation and symptoms after a controlled gluten challenge in a Phase 2a clinical trials with 63 celiac patients. We plan to initiate a 220-patient Phase 2b clinical trial in NCRD, in 2020, after completion of chronic toxicology studies.
Celiac Disease Background Information
Celiac disease is a systemic autoimmune disease triggered by gluten consumption in genetically susceptible individuals. Approximately 1% of the western population is affected by celiac disease. This prevalence has been reported to be doubling every 20 years. Gluten, the antigen responsible for celiac disease, is the main protein present in some of the most common cereals (wheat, barley, rye). Modern diets are increasingly enriched with gluten and it is also used as an additive in processed foods, cosmetics and oral medications. Gluten is also present in trace amounts in foods labeled as “gluten-free”, as a tableting excipient, and in products such as toothpaste and lipstick. As little as 50mg/day of gluten triggers the disease. A normal diet contains >10 g/day, 200 times the amount that causes damage and intestinal histological abnormalities. As such, celiac patients face enormous challenges to follow a strict GFD.
The pathophysiology of celiac disease is characterized by an abnormal immune response to gluten. Humans lack enzymes to fully digest gluten, which against the right genetic background triggers inflammation and autoimmunity in the intestine and in other organs. An adaptive immune response is triggered when gluten peptides are deamidated in the extracellular space, by the enzyme tissue transglutaminase, normally an intracellular enzyme that is released by damaged cells. This deamidation renders gluten peptides high-avidity binders to HLA-DQ2 and HLA-DQ8, which present these peptides to intestinal CD4+ T cells, thereby activating these T cells and initiating the inflammatory cascade. The innate immune system’s intraepithelial lymphocytes, or IELs, primarily CD8+, are able to directly lyse and destroy intestinal epithelial cells, damaging the mucosal lining of the small intestine, in response to IL-15 release stimulated by gluten peptides. In healthy individuals, the activated T cells are controlled by Tregs, but this does not happen in celiac disease as IL-15 confers the effector CD4+ T cells resistance to suppression by Tregs.
| 27 |
Celiac disease causes debilitating symptoms and serious medical complications. In many patients, gastrointestinal symptoms derived from intestinal mucosal damage dominate the patient reported symptoms at diagnosis. The normal villi (absorptive finger-like prolongations) present in the gut of healthy individuals are lost in active celiac disease as a result of mucosal atrophy and crypt enlargement. Small bowel damage often leads to nutrient malabsorption that can result in a range of further clinical manifestations (anemia, osteopenia, failure to thrive in children). In addition, extra-intestinal symptoms and systemic manifestations are often present, such as dermatitis, infertility, or neurological and skeletal disorders. Mortality is increased in subjects with persistent intestinal mucosal damage.
The most serious complication of celiac disease is the development of an in situ small bowel T cell lymphoma after many years of exposure, voluntary or inadvertent, to gluten. This malignant complication of celiac disease, which appears to be independent of gluten and unresponsive to a strict GFD, is termed RCD-II when the percentage of aberrant IELs is >20% and Type I refractory celiac disease when the percentage is <20%. In RCD-II, aberrant IELs proliferate in what represents a slow-growing non-Hodgkin lymphoma localized (in situ) in the small bowel, primarily in the epithelial compartment. RCD-II affects approximately 0.5% of celiac patients and can lead to overt and systemic enteropathy-associated T cell lymphoma, with very poor prognosis and >80% mortality in five years.
Current Treatment Options and Their Limitations
Celiac disease is the only common autoimmune disorder with no approved medication. The only current available strategy for the management of celiac disease is a lifelong total avoidance of gluten. While simple in theory, the ubiquity of gluten in foodstuffs, medications, household substances, cosmetics, and gluten-free items makes total avoidance of gluten difficult, if not impossible.
The main challenge to the successful maintenance of a GFD is that cereal flours are widely used in the food industry and are present in numerous food products either naturally or as additives. Although gluten-free products can be purchased, commercially manufactured gluten-free products may be difficult to find, tend to be less flavorful and are more expensive than regular gluten containing foods. In addition, labeling of food products is deficient in many countries. Even in countries with superior labeling guidelines foods labeled “gluten-free” may nevertheless contain gluten. For example, in northern European countries amounts of up to 100 parts per million are permitted in gluten-free products designated apt for celiac sufferers.
For these reasons, celiac sufferers are regularly exposed to gluten contamination in the food and beverages they consume. This exposure to gluten contamination and the associated physiological and psychological consequences results in a self-limitation of social activities and/or a reduction in the variety of foods consumed. Thus, the only currently available management option of a GFD presents both a considerable challenge and substantial burden for patients. A study by Shah and collaborators (2014) found the burden of celiac disease and GFD on patient quality of life to be very high, second only to end-stage renal disease – a condition that requires multiple, weekly dialysis treatments.
As a result of the difficulty in maintaining total avoidance of gluten while on a GFD, gluten contamination causes 50% or more of all diagnosed celiac patients on a GFD to continue to experience disease activity. Patients who continue to have symptoms despite attempting to maintain a GFD are deemed to have NRCD. NRCD has been defined as “persistent symptoms, signs or laboratory abnormalities typical of celiac disease despite 6–12 months of dietary gluten avoidance”. As requested by patient support groups and experts, alternative treatment options that can be administered independently or in combination with a GFD, as well as treatments for refractory celiac disease, are required in order to improve the quality of life for celiac patients.
Overview of IL-15 Biology and PRV-015 Mechanism of Action
IL-15 is a pro-inflammatory cytokine that serves as a potent growth, survival, and activation factor for T cells, particularly IELs, and for natural killer, or NK, cells. Increased expression of IL-15 has been demonstrated in a variety of inflammatory conditions, including celiac disease, RA, and psoriasis. IL-15 is considered a central regulator of celiac disease immunopathology and a non-redundant driver of lymphomagenesis in RCD-II.
| 28 |
Substantial evidence suggests a pathophysiological role for IL-15 in celiac disease:
Innate immunity:
| ● | IL-15 is an essential, non-redundant growth and activation factor for the IELs which destroy the intestinal mucosa; | |
| ● | The expression of IL-15 in the intestinal epithelium is necessary for villous atrophy; and | |
| ● | In some patients, IL-15 drives progression towards lymphomagenesis and potentially fatal RCD-II. |
Adaptive immunity:
| ● | IL-15 enhances the presentation of deamidated gluten peptides by APCs; | |
| ● | IL-15 renders the activated CD4+ T cells resistant to inhibition by Tregs; and | |
| ● | IL-15 has been proven to be a key factor in the loss of tolerance to food antigens. |
By activating the IELs, IL-15 is believed to be the main mediator in the mucosal damage that ensues in response to gluten exposure in celiac disease. The expression of IL-15 in the intestinal epithelium is necessary for villous atrophy in animal models of celiac disease and circumstantial evidence suggests this to be the case in humans, as well. In addition, IL-15 renders effector T cells resistant to inhibition by Tregs, promoting loss of tolerance to food antigens.
One of the studied mouse models of celiac disease is an IL-15-transgenic mouse, in which IL-15 overexpression by gut epithelial cells leads to celiac-like disease, including T and B cell-mediated pathology. IEL apoptosis has been observed in this animal model after treatment with anti-IL-15 or anti-IL-15-receptor monoclonal antibodies.
Figure 1. Multiple actions of IL-15 in the pathophysiology of celiac and refractory celiac disease
PRV-015 (formerly AMG 714 and HuMax-IL15), is a fully human immunoglobulin (IgG1κ) mAb which binds to and inhibits the function of IL-15 in all its forms (cis, trans, soluble IL-15 bound to IL-15Rα). PRV-015 inhibits IL-15-induced T cell proliferation and shows a dose-dependent inhibition of IL-15-induced TNF-α production. PRV-015 underwent preclinical testing and was subsequently evaluated in a Phase 1 and Phase 2 study in subjects with RA, in a Phase 1 study in healthy volunteers and in patients with psoriasis, and in two Phase 2a studies in celiac disease and refractory celiac disease Type-II.
| 29 |
Phase 2b Clinical Trial of PRV-015 in Celiac Disease (PROACTIVE Study)
We and our partner Amgen conducted chronic toxicology studies in 2019 which will continue into the first half of 2020 and intend to conduct a Phase 2b clinical trial (the PROACTIVE study) in approximately 220 patients with gluten-free diet NRCD. We expect to commence the Phase 2b PROACTIVE study in the second quarter of 2020. Based on feedback from regulators, we do not expect that we will need to conduct any pediatric PK clinical studies.
The PROACTIVE study (PROvention Amgen Celiac ProtecTIVE Study) will be a randomized, double-blind, placebo-controlled, parallel-group, multicenter Phase 2 clinical trial in adult patients with NRCD. PRV-015 will be administered every two weeks via subcutaneous route. The hypothesis of this study is that PRV-015 will be superior to the GFD at intercepting the effects of contaminating gluten exposure in celiac patients following a GFD, as measured by symptoms and objective signs of intestinal inflammation after 24 weeks of treatment. Approximately 220 subjects are planned to be enrolled. We expect to report top line results for the Phase 2b PROACTIVE study in 2022.
Pre-clinical Evaluation of PRV-015
The nonclinical development of PRV-015 consisted of a series of in vitro studies demonstrating the binding properties of PRV-015 against human IL-15; in vitro and in vivo studies providing proof-of-concept for the benefit of blocking the IL-15 pathway in celiac disease; and a series of GLP studies evaluating the nonclinical safety profile of Hu714MuXHu, the PRV-015 surrogate molecule which is active in macaques.
Pharmacology
PRV-015 was found to be efficacious in a mouse model of celiac disease triggered by the transgenic expression of human IL-15 in the gut epithelium. In this model, PRV-015 prevented IEL activation and proliferation, as well as histological abnormalities. In addition, PRV-015 was able to induce apoptosis of human IELs in ex vivo culture of small intestinal explants from active celiac disease and RCD-II patients. In this culture experiment, PRV-015 resulted in a suppression of IL-15-driven anti-apoptotic signaling via JAK3 and STAT5.
Toxicology
In vitro studies demonstrated that PRV-015 had high binding affinity for human IL-15, but lower affinity for macaque IL-15. Additionally, PRV-015 neutralized human IL-15 but did not efficiently neutralize macaque IL-15. To enable preclinical and toxicology studies in macaques, a surrogate antibody, Hu714MuXHu, was developed by Amgen by fusing the F(ab) portion of a mouse anti-human IL-15 mAb known to neutralize macaque IL-15, M111, with human IgG1 Fc. Hu714MuXHu was shown to neutralize macaque IL-15 with approximately the same potency as PRV-015 neutralizes human IL-15.
There was a decrease in NK cell counts and NK cell activity following administration of Hu714MuXHu to monkeys, reflecting a PD response to IL-15 blockade in this species, given the known role of IL-15 in NK cell biology in animal models (rodents and non-human primates). Of note, no changes in absolute or relative numbers of NK cells were observed in any of the human studies. This difference between observations in preclinical studies and clinical trials appears related to a differential sensitivity of human versus cynomolgus monkey NK cells to IL-15 deprivation. Human NK cells are not dependent on IL-15 for their survival, possibly due to the redundant role of IL-2 on human NK cells.
Pharmacokinetics of PRV-015
The PK of PRV-015 was consistent with a typical human immunoglobulin G1 antibody with no apparent target-mediated disposition within the investigated dosing range. The mean half-life in human studies has been 20 to 22 days, potentially enabling monthly dosing.
There was no development of anti-drug antibodies to PRV-015 in healthy volunteers, patients with psoriasis or patients with refractory celiac disease. Only one RA patient in the phase 2b study was positive for ADA. Approximately 14% of patients with celiac disease developed ADA in the Phase 2a clinical trial, with an additional 10% presenting pre-existing ADA, a reflection of the abnormal antibody responses which characterize celiac disease. The ADA were not associated with injection reactions or adverse events, and they were non-neutralizing, with no impact on PK.
| 30 |
Clinical Proof of Mechanism for PRV-015
Although PRV-015 missed the primary endpoint, approximately 60% of patients in both Phase 1 and Phase 2 studies versus a response of approximately 30% of patients in the placebo groups demonstrated a response to treatment. PRV-015 also led to decreases in inflammatory biomarkers such as C-reactive protein and erythrocyte sedimentation rate. PRV-015 was not effective in psoriasis, suggesting PRV-015’s action is selective, unlike that of broad systemic immune suppressants.
Upon gluten challenge in a Phase 2 clinical trial in celiac disease, PRV-015 did not prevent gluten-induced architectural mucosal injury, and thus missed the primary endpoint, yet the high dose of PRV-015, 300 mg, showed statistically significant attenuation of gluten’s effects on the change from baseline in intestinal inflammation, in patient-reported symptom questionnaires (the Celiac Disease Patient Reported Endpoint, CeD PRO, a registrational endpoint in NRCD) and in diarrhea, compared with placebo. The totality of the results from the patients who had gluten challenge indicate that 300 mg AMG 714 given every two weeks can ameliorate the inflammation and symptoms caused by substantial gluten exposure, the first demonstration of such dual benefit in intestinal inflammation and symptoms for any experimental medication for celiac disease. The results suggest that PRV-015 can be a potential adjunctive treatment for NRCD to the GFD to ameliorate or resolve persistent inflammation seen in the majority of celiac patients already on GFD.
In the Phase 2a clinical trial in RCD-II, the primary endpoint (reduction in intestinal IELs) was not achieved, yet PRV-015 showed statistically significant benefit over placebo in reducing T cell receptor clonality (no increase in clonality with PRV-015) and symptoms (diarrhea). Other endpoints, such as histology, did not reach statistically significant differences between groups, but the results consistently favored PRV-015 numerically. PRV-015 was well tolerated, with no observed immunogenicity.
Summary data of the Phase 2a clinical trial as presented at Digestive Disease Week in June 2018 is shown below:
| 31 |
The CELIM-NRCD-001 study included 62 randomized celiac patients on a gluten-free diet, of which 49 patients underwent a substantial gluten challenge of 2.5 grams per day for 10 weeks in order to assess the ability of PRV-015 to ameliorate the effects of gluten. Upon gluten challenge, PRV-015 did not prevent gluten-induced architectural mucosal changes, the primary endpoint in the study. However, in secondary efficacy assessments, the PRV-015 300 mg dose consistently attenuated the effects of gluten in intestinal inflammation (intraepithelial lymphocyte density, p=0.03), and in gastrointestinal symptoms as measured by three independent endpoints: the Celiac Disease Patient Reported Endpoint (CeD-PRO, p=0.02), the Celiac Disease Gastrointestinal Symptom Rating Scale (CeD-GSRS, p=0.07) and the Bristol Stool Form Scale (BSFS/diarrhea, p=0.0002). The CeD-PRO is a validated endpoint acceptable for registrational trials. In addition, patients in the PRV-015 300 mg arm had a significantly improved Physician Global Assessment of disease (PGA, p=0.03). The totality of the results demonstrated proof-of-concept for PRV-015 300 mg given subcutaneously every two weeks in the amelioration of inflammation and symptoms caused by the consumption of gluten by celiac patients. Importantly, PRV-015 was well tolerated, and only 14% of patients developed anti-drug antibodies, which were non-neutralizing and not correlated with impact of efficacy or safety. The PK profile was consistent with a monoclonal antibody, and potentially enables future monthly dosing.
Summary of clinical trials
| Study Number (Phase; Sponsor) | Key Design Features | Dose Route, Duration | Study Population | |||
Hx-IL15-001 (Phase 1; Genmab) |
Double-blind, placebo-controlled, single SC infusion, dose escalation, study with open-label, repeat-dose (4 weekly doses) follow-up | Initial single dose: 0 or 0.15 to 8 mg/kg SC infusion Repeated dose: 0.5 to 4 mg/kg SC infusion once weekly for four weeks. five doses over eight weeks |
30 subjects with RA | |||
20030210 (Phase 2; Genmab/ Amgen) |
Double-blind, placebo-controlled, multiple SC infusion, parallel-group, multicenter study | 0 or 40 to 280 mg SC infusion dose every two weeks for 12 weeks with initial 200% loading dose | 180 subjects with RA | |||
20050193 (Phase 1; Amgen) |
Double-blind, placebo-controlled, single SC or IV doses, dose-escalation study | SC doses: 0, 30, 100, 300 or 700 mg (cohorts 1 to 4) IV dose: 0 or 100 mg (cohort 5) | 40 healthy subjects | |||
20060349 (Phase 1b/2a; Amgen) |
Double-blind, placebo-controlled, multiple SC doses, dose-escalating study | 0 or 150 mg SC (cohort 1) 0 or 300 mg SC (cohort 2) Every two weeks for 12 weeks |
22 subjects with moderate to severe psoriasis | |||
| CELIM-NRCD-001 (Phase 2a; Celimmune) | Double-blind, placebo- controlled, SC, parallel group, multicenter study | 0, 150 mg or 300 mg PRV-015 once every two weeks for six consecutive doses over ten weeks | 63 subjects with NRCD | |||
| CELIM-RCD-002 (Phase 2a; Celimmune) | Double-blind, placebo controlled IV infusion, parallel group, multicenter study | 0 or 8 mg/kg IV, a total of 7 times over ten weeks | 24 subjects with Type II refractory celiac disease |
Safety of PRV-015
Approximately 250 subjects have been exposed to PRV-015 to date, including approximately 200 subjects for 12 weeks of biweekly dosing. In all studies to date, PRV-015 was generally well tolerated by healthy volunteers, patients with active RA, and patients with celiac disease or RCD-II. While PRV-015 could, theoretically, increase susceptibility to infections as is the case with immune modulators, PRV-015 has not demonstrated this effect in the six clinical trials completed to date. No deaths or clinically significant changes in laboratory parameters were observed, including no NK cell depletion.
| 32 |
The only adverse events clearly increased in PRV-015-treated patients have been injection site reactions, which were more commonly reported in subjects exposed to PRV-015, in a dose-dependent fashion (up to ~52% on PRV-015 vs ~26% in placebo in the celiac Phase 2a clinical trial), and nasopharyngitis (most cases with suspected allergic origin at a single site in RCD-II). In the population which will be studied in Phase 2b, celiac disease, there were no SAEs in the Phase 2a clinical trial, while there were six SAEs (five on PRV-015 and one on placebo) in the RCD-II, a much sicker patient population with immune suppression at baseline. Of the five SAE on PRV-015 in RCD-II patients, two were infections (both resolved while on PRV-015). There was one mild balance disorder (considered unlikely to be related to PRV-015 and resolved while on the drug.) Another patient had mild cerebellar syndrome which lead to discontinuation from the study.
Celiac Disease Market
There is no approved drug for CD. The annual healthcare utilization by NRCD patients in 2013 was $18,206, for $4,796 in matched controls due to the extra costs of uncontrolled CD investigations and treatment of complications. Given the large prevalence (15 to 20 million patients world-wide, 1% of the population in the Western world and 0.5% in Asia), and the unmet need (50% of patients on GFD continue to suffer from disease activity due to contaminating gluten in the diet), NRCD is considered a substantial opportunity for pharmaceutical development of an effective and well-tolerated adjunctive treatment to the GFD.
PRV-6527 (Small Molecule CSF-1R Inhibitor) for CD
Overview
CD, a type of chronic IBD characterized by inflammation of the gastrointestinal, or GI tract, can affect any part of the GI tract from the mouth to the anus but is more commonly found towards the end of the small intestine. It can also affect the eyes, skin, and joints. Myeloid cells, which originate in the bone marrow, are specialized immune cells, also called APCs believed to play a central role in CD. CSF-1 binds to its receptor (CSF-1R) on myeloid cells and drives the differentiation and maturation of these cells into inflammatory dendritic cells and macrophages, which then populate the gut and other tissues. In the gut, these differentiated myeloid cells present antigens from intestinal bacteria (the microbiome) to white blood cells and trigger inflammatory processes. Based on pre-clinical and human non-clinical data with an inhibitor of CSF-1R in an animal model of IBD, it is reasonable to hypothesize that an inhibitor of CSF-1R will “intercept” the differentiation of these inflammatory cells, preventing their migration from the bone marrow to the intestinal mucosa (i.e., the gut lining) in CD. It is anticipated that significant clinical benefits, such as preventing the relapse or progression of CD, as well as durable benefit (extended pharmacodynamic effect) may result from targeting the upstream pathologic mechanism, since APCs are the necessary initiators of the abnormal immune response.
PRV-6527 (previously known as JNJ-40346527) is a highly potent and selective small-molecule oral inhibitor of CSF-1R. It was developed by Janssen and has undergone clinical testing in 178 subjects to date, across Phase 1 (healthy volunteers; 94 received single dose up to 600 mg or two doses of 450 mg) and two Phase 2 clinical trials (RA 63 patients received 200 mg/day for 12 weeks; and 21 Hodgkin’s lymphoma, or HL), patients received 150 mg/day to 650 mg/day for at least three weeks). No serious adverse events deemed related to PRV-6527 were observed that would preclude further clinical development and proof of mechanism was demonstrated based on inhibition of CSF-1R signaling and myeloid cell counts in blood. While clinical data in the RA study was inconclusive and did not demonstrate efficacy in this disease, PRV-6527 ameliorates Crohn’s-like disease in mouse models, and CSF-1R and its pathway are upregulated in CD.
In the first quarter of 2018, we initiated a Phase 2a proof of concept clinical trial, referred to as the PRINCE study, in approximately 80 patients with CD to assess a clinical and histologic/tissue (gut mucosa) anti-inflammatory effects after 12 weeks of treatment with PRV-6527. This study evaluated doses and dosing duration that were previously tested by Janssen. We completed enrollment of the Phase 2a PRINCE study in April 2019.
In October 2019, we announced top-line results from our Phase 2a clinical trial the PRINCE study. The primary efficacy endpoint of the study was the change in the Crohn’s Disease Activity Index (CDAI) score at week 12. While PRV-6527 demonstrated a substantial improvement in this symptom driven score at week 12, it did not differentiate from placebo. This high placebo response was deemed to be possibly related to the standard of care and/or background medication used (~85%) in the study’s predominantly biologic-naïve population. PRV-6527 was associated with improvements in several key secondary objective endpoints in the steroid-free population (75% of study subjects), including mucosal endoscopy (as assessed by the Simple Endoscopic Score for Crohn’s Disease, SES-CD) and tissue histology (as measured by the Global Histological Activity Score, GHAS). Analysis of exploratory serum and tissue biomarkers showed that patients treated with PRV-6527 had significant reductions in circulatory inflammatory monocytes, as well as macrophages, dendritic cells and the CSF1 gene signature in colonic tissue, providing proof of mechanism in the interception of inflammatory myeloid cells. PRV-6527 was found to be generally safe and well tolerated, with no drug-related serious adverse events.
| 33 |
In December 2019, Janssen declined to exercise its right to buy back PRV-6527 from us for a one-time payment of $50.0 million and single-digit royalties on future net sales in inflammatory bowel disease indications. We retain the rights and are free to sublicense the program on a worldwide basis to another partner in the field of inflammatory bowel disease.
Significant Contracts and Agreements Related to Research and Development Activities
License and Acquisition Agreements
MacroGenics Asset Purchase Agreement
In May 2018, we entered into an Asset Purchase Agreement with MacroGenics pursuant to which we acquired MacroGenics’ interest in teplizumab (renamed PRV-031), a humanized mAb for the treatment of T1D. As partial consideration for the MacroGenics Asset Purchase Agreement, we granted MacroGenics a warrant to purchase 2,162,389 shares of our common stock at an exercise price of $2.50 per share. In July 2019, these warrants were exercised by MacroGenics on a cashless basis. We are obligated to pay MacroGenics contingent milestone payments totaling $170.0 million upon the achievement of certain regulatory approval milestones, including $60.0 million payable upon approval of a BLA in the United States. In addition, we are obligated to make contingent milestone payments to MacroGenics totaling $225.0 million upon the achievement of certain sales milestones. We have also agreed to pay MacroGenics a single-digit royalty on net sales of the product. We have also agreed to pay third-party obligations, including low single-digit royalties, a portion of which is creditable against royalties payable to MacroGenics, aggregate milestone payments of up to approximately $1.3 million and other consideration, for certain third-party intellectual property under agreements we are assuming pursuant to the Asset Purchase Agreement. Further, we are required to pay MacroGenics a low double-digit percentage of certain consideration to the extent it is received in connection with a future grant of rights to PRV-031 by us to a third party. We are obligated to use reasonable commercial efforts to develop and seek regulatory approval for PRV-031.
Vactech License
In April 2017, we entered into a License Agreement with Vactech, pursuant to which Vactech granted us exclusive global rights for the purpose of developing and commercializing the CVB vaccine platform technology. In consideration of the licenses and other rights granted by Vactech, we issued two million shares of our common stock to Vactech. We recorded the issuance of the shares at their estimated fair value of approximately $1.70 per share for a total of $3.4 million as a license fee expense included as part of research and development expenses for the year ended December 31, 2017. We paid Vactech a total of approximately $0.5 million for transition and advisory services during the first 18 months of the term of the agreement. Vactech is obligated to transition its intellectual property, provide reference samples, assist with the technology transfer to a third-party contract manufacturer, and participate on our scientific advisory board. In addition, we may be obligated to make a series of contingent milestone payments to Vactech totaling up to an additional $24.5 million upon the achievement of certain clinical development and regulatory filing milestones. In addition, we have agreed to pay Vactech tiered single-digit royalties on net sales of any approved product based on the CVB platform technology and three additional payments totaling $19.0 million upon the achievement of certain annual net sales levels. The Vactech Agreement may be terminated by us on a country by country basis without cause (in which case the exclusive global rights to the technology will transfer back to Vactech) and by either party upon a material breach or insolvency of the other party. If we terminate the agreement with respect to two or more specified European countries, the agreement will be deemed terminated with respect to all of the EU, and if we terminate the agreement with respect to the United States, the agreement will be deemed terminated with respect to all of North America and expires upon the expiration of our last obligation to make royalty payments to Vactech.
| 34 |
MacroGenics License Agreement
In May 2018, we entered into a License Agreement with MacroGenics, pursuant to which MacroGenics granted us exclusive global rights for the purpose of developing and commercializing MGD010 (renamed PRV-3279), a humanized protein and a potential treatment for SLE and other similar diseases. As partial consideration for the MacroGenics License Agreement, we granted MacroGenics a warrant to purchase 270,299 shares of our common stock at an exercise price of $2.50 per share. In July 2019, these warrants were exercised by MacroGenics on a cashless basis. We are obligated to make contingent milestone payments to MacroGenics totaling $42.5 million upon the achievement of certain developmental and approval milestones for the first indication, and an additional $22.5 million upon the achievement of certain regulatory approvals for a second indication. In addition, we are obligated to make contingent milestone payments to MacroGenics totaling $225.0 million upon the achievement of certain sales milestones. We have also agreed to pay MacroGenics a single-digit royalty on net sales of the product. Further, we are required to pay MacroGenics a low double-digit percentage of certain consideration to the extent received in connection with a future grant of rights to PRV-3279 by us to a third party. We are obligated to use commercially reasonable efforts to develop and seek regulatory approval for PRV-3279. The license agreement may be terminated by either party upon a material breach or bankruptcy of the other party, by us without cause upon prior notice to MacroGenics, and by MacroGenics in the event that we challenge the validity of any licensed patent under the agreement, but only with respect to the challenged patent.
Amgen License and Collaboration Agreement
In November 2018, we entered into a License and Collaboration Agreement (the “Amgen Agreement”) with Amgen, Inc. (“Amgen”) for PRV-015 (formerly AMG 714), a novel anti-IL-15 monoclonal antibody being developed for the treatment of gluten-free diet non-responsive celiac disease (NRCD). Under the terms of the agreement, we will conduct and fund a Phase 2b trial in NRCD and lead the development and regulatory activities for the program. Amgen agreed to make an equity investment of up to $20.0 million in us, subject to certain terms and conditions set forth in the agreement. Amgen is also responsible for the manufacturing of PRV-015. Upon completion of the Phase 2b trial, a $150.0 million milestone payment is due from Amgen to us, plus an additional regulatory milestone payment, and single digit royalties on future sales. If Amgen elects not to pay the $150.0 million milestone, AMG 714 rights will be transferred to us pursuant to a termination license agreement between Amgen and us. We will be obligated to make certain contingent milestone payments to Amgen and other third parties totaling up to $70.0 million upon the achievement of certain clinical and regulatory milestones and a low double-digit royalty on net sales of any approved product based on the IL-15 technology. We may terminate the agreement without cause (in which case the exclusive global rights to the technology will transfer back to Amgen) and by either party upon a material breach. The agreement expires upon the expiration of Amgen’s last obligation to make royalty payments to Provention (or, our last obligation to make royalty payments to Amgen, if the program rights are transferred to us). In September 2019, in a private placement completed concurrently with our underwritten public offering, Amgen purchased 2,500,000 shares of our common stock at the underwritten public offering price of $8.00 per share, for a total investment of $20.0 million.
Janssen License CSF-1R
In April 2017, we entered into a License, Development and Commercialization Agreement, pursuant to which Janssen Pharmaceutica NV granted us exclusive global rights for the purpose of developing and commercializing JNJ-40346527 (renamed PRV-6527), a colony stimulating factor 1 receptor (CSF-1R) inhibitor for IBDs including CD. We evaluated PRV-6527 in a recently completed Phase 2a proof-of-mechanism and proof-of-concept clinical trial for the CD indication. See Item 1. Business – Recent Company Developments for disclosure of top-line results of the PRINCE study. In December 2019, Janssen declined its option to buy back the rights for future development of PRV-6527 and all rights remain with us. We will be obligated to make contingent milestone payments to Janssen totaling $35.0 million upon the achievement of certain clinical and regulatory milestones for the first indication and an additional $20.0 million upon the achievement of certain clinical and regulatory milestones for a second indication. In addition, we have agreed to pay Janssen tiered single-digit royalties on net sales of any approved product based on the CSF-1R technology and three additional payments totaling $100.0 million upon the achievement of certain annual net sales levels. The CSF-1R License Agreement may be terminated by us without cause (in which case the exclusive global rights to the technology will transfer back to Janssen) and by either party upon a material breach and expires upon the expiration of our last obligation to make royalty payments to Janssen.
| 35 |
Intravacc Development Services Agreement
In March 2018, we entered into a Development Services Agreement with The Institute of Translational Vaccinology, or Intravacc, pursuant to which Intravacc will provide services related to process development, non-GMP and GMP manufacturing of our polyvalent CVB vaccine, including providing proprietary technology for manufacturing purposes. We will pay Intravacc approximately €10.0 million for their services over the development and manufacturing period which we expect will last for approximately 24 to 30 months. Each party retains its existing intellectual property and will share newly developed intellectual property via a fully-paid non-exclusive license between the parties for all development work through phase 1 clinical trials. Any future use, including commercial use, of Intravacc’s technology will be subject to a separate nonexclusive license agreement. The Intravacc Development Services Agreement may be terminated by us with ninety days’ notice without cause and by either party upon a material breach or insolvency of the other party.
AGC Biologics Agreement
In February 2019, we entered into services agreement with AGC Biologics, or AGC, to manufacture and supply teplizumab, PRV-031, for our anticipated clinical supply needs. We may terminate the agreement or any statement of work thereunder with approximately 12 months’ prior written notice to AGC. If we provide less than 12 months’ notice of termination, we may incur a cancellation fee depending on the timing of such notice. AGC may terminate the agreement if we do not, over a specified period, purchase and take delivery from AGC of a specified minimum quantity of API for teplizumab. Each party also has the right to terminate the agreement for other customary reasons such as material breach and bankruptcy. The agreement contains provisions relating to compliance by AGC with current Good Manufacturing Practices, cooperation by AGC in connection with potential marketing applications for PRV-031, indemnification, confidentiality, dispute resolution and other customary matters for an agreement of this kind.
Parexel Services Agreement
In February 2019, we entered into a services agreement with Parexel, or the Parexel Services Agreement, pursuant to which we retained Parexel to perform implementation and management services in connection with the PROTECT study of PRV-031. We may terminate the services agreement or any work order for any reason and without cause with 90 days’ written notice. Either party may terminate the agreement in the event of a material breach or, bankruptcy petition by the other party or, if any approval from a regulatory authority is revoked, suspended or expires without renewal. We currently anticipate that aggregate costs relating to all work orders under the Parexel Services Agreement for the PROTECT study will be approximately $42.0 million over the period of the study.
Intellectual Property
We believe that our current patent applications and any future patents and other proprietary rights that we own, or control through licensing, are and will be essential to our business. We believe that these intellectual property rights will affect our ability to compete effectively with others. We also rely and will rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to develop, maintain and strengthen our competitive position. We seek to protect these, in part, through confidentiality agreements with certain employees, consultants, advisors and other parties. Our success will depend in part on our ability, and the ability of our licensor, to obtain, maintain (including making periodic filings and payments) and enforce patent protection for our/their intellectual property, including those patent applications to which we have secured exclusive rights.
We plan to spend considerable resources and focus in the future on obtaining U.S. and foreign patents. We have and will continue to actively protect our intellectual property. No assurances can be given that any of our patent applications will result in the issuance of a patent or that the examination process will not require us to narrow our claims. In addition, any issued patents may be contested, circumvented, found unenforceable or invalid, and we may not be able to successfully enforce our patent rights against third parties. No assurance can be given that others will not independently develop a similar or competing technology or design around any patents that may be issued to us. We intend to expand our international operations in the future and our patent portfolio, copyright, trademark and trade secret protections may not be available or may be limited in foreign countries.
| 36 |
PRV-031 (teplizumab anti-CD3 antibody)
Through our agreement with MacroGenics, we have acquired a patent portfolio that includes seven issued patents, including two U.S. patents and five ex-U.S. patents in Australia, Israel, Mexico and Singapore. The issued patents are set to expire no earlier than dates ranging from 2026 and 2028, subject to any disclaimers, patent term adjustments or extensions available under the law. These issued patents cover use of certain humanized antibodies that bind to CD3 in the treatment of autoimmune disorders, including T1D and RA.
We have additionally filed one U.S. provisional patent application directed to new use of anti-CD3 antibodies, including teplizumab. If issued, patents claiming priority to this provisional application will expire no earlier than 2040, subject to any disclaimers, patent term adjustments or extensions available under the law.
PRV-101 (CVB/T1D)
Through our agreement with Vactech, we have a licensed patent portfolio that includes one issued U.S. patent, two pending U.S. patent applications, and 38 patents in various European countries (i.e., one granted European patent validated in 38 European Patent Convention member states). The pending U.S. patent applications and the European country patents disclose use of a CVB vaccine composition in the prevention or treatment of T1D.
The patents issued in the U.S. and various European countries generally have terms of 20 years from their respective priority filing dates, subject to available extensions, and are thus set to expire no earlier than 2032, subject to any disclaimers, patent term adjustments or extensions available under the law.
PRV-3279 (CD32B/CD79B diabody)
Through our agreement with MacroGenics, we have licensed a patent portfolio that includes: i) 219 issued patents, including 11 U.S. patents, 142 patents in European countries, and 66 patents in other ex-U.S. jurisdictions; and ii) 48 pending patent applications, including eight pending U.S. patent applications, four pending European patent applications, and 36 pending patent applications in other ex-U.S. jurisdictions.
The patents and patent applications disclose a platform technology for making diabodies, specific anti-CD32B antibodies, specific anti-CD79B antibodies, specific diabodies that co-ligate both CD32B and CD79B, as well as use of these antibodies and diabodies in treating various disorders, including cancer, autoimmune disorder, inflammatory disorder, and IgE-mediated allergic disorder.
The issued patents in the U.S. and various ex-U.S. countries generally have terms of 20 years from their respective priority filing dates, subject to available extensions, and are thus set to expire no earlier than dates ranging from 2023 and 2034, subject to any disclaimers, patent term adjustments or extensions available under the law. In the event that the pending patent applications issue as patents, although there can be no assurance that the patent applications will issue, the patents would be set to expire no earlier than dates ranging from 2023 and 2037, subject to any disclaimers, patent term adjustments or extensions available under the law.
We have additionally filed one U.S. provisional patent application directed to new use of B cell inhibitors, including PRV-3279. If issued, patents claiming priority to this provisional application will expire no earlier than 2040, subject to any disclaimers, patent term adjustments or extensions available under the law.
PRV-015 (IL-15)
Through our agreement with Amgen, we have licensed a patent portfolio that includes: i) 78 issued patents, including seven U.S. patents, 42 patents in European countries, and 29 patents in other ex-U.S. jurisdictions; and ii) 21 pending patent applications, including three pending U.S. patent applications, one pending European patent application, and 17 pending patent applications in other ex-U.S. jurisdictions.
The patents and patent applications disclose anti-IL-15 antibodies, methods of using the same, manufacturing conditions and dosages of the same.
The issued patents are set to expire no earlier than dates ranging from 2022 and 2027, subject to any disclaimers or extensions under the law. In the event that the pending patent applications issue as patents, although there can be no assurance that the patent applications will issue, the patents would be set to expire no earlier than dates ranging from 2026 and 2037, subject to any disclaimers, patent term adjustments of extensions available under the law.
| 37 |
PRV-6527 (CSF-1R)
Through our agreement with Janssen Pharmaceutica NV, we have licensed a patent portfolio that includes: i) 73 issued patents, including one U.S. patent, one patent in European countries, and 71 patents in other ex-U.S. jurisdictions; and ii) three pending patent applications, one pending U.S. patent application, one pending European patent application, and one pending patent applications in other ex-U.S. jurisdictions. The issued patents are set to expire no earlier than dates ranging from 2027 and 2030, subject to any disclaimers, patent term adjustments or extensions under the law. In the event that the pending patent applications issue as patents, although there can be no assurance that the patent applications will issue, the patents would be set to expire no earlier than dates ranging from 2027 and 2030, subject to any disclaimers, patent term adjustments or extensions under the law.
Sales and Marketing
We are a clinical stage company without a history of revenue or marketing experience. Because commercialization is expensive and time consuming, we intend to explore multiple commercialization strategies, including:
| ● | commercialize teplizumab ourselves in the U.S. while exploring strategic partners for markets outside the U.S.; | |
| ● | developing drug candidates through the earlier stages of clinical development with the objectives of rapid, cost effective risk reduction and value creation and then establishing strategic partnership for late stage clinical development and subsequent commercialization; | |
| ● | developing a robust pipeline of promising drug candidates at various stages of the development process to establish optionality and regular value inflection opportunities and revenue(s); | |
| ● | strategically entering into co-development partnership(s) to retain potential for commercialization rights on selected drug candidate(s) and market opportunities; and | |
| ● | partnering with industry participants to incorporate our technology into new and existing drugs. |
We expect that partnering with pharmaceutical or biotherapeutic companies may accelerate product acceptance into target market areas outside the U.S. and gain the sales and marketing advantages of the partner’s distribution infrastructure. We intend to continue to strengthen our market position and solidify our leadership position in immunotherapy by continuing to improve our technology, broadening our clinical and therapeutic applications, identifying new clinical and therapeutic applications and forming strategic relationships with our licensors.
Manufacturing
We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of any of our product candidates. Although we rely and intend to continue to rely upon third–party contract manufacturers to produce our products and product candidates, we have recruited personnel and consultants with experience to manage these third–party contract manufacturers. In certain cases, our collaboration partners for each respective program are responsible for providing clinical drug supply or drug product for those program’s clinical trials. In other cases, we have engaged third-party manufacturers to provide services related to process development, non-GMP and GMP manufacturing and other related services.
The table below lists the third-party responsible for manufacturing drug supply for each of our programs:
| Product Candidate | Supplier | Party Responsible for Costs | ||
| PRV-031 | Existing drug supply
– MacroGenics Future drug supply – AGC Biologics | MacroGenics Provention | ||
| PRV-101 | Intravacc | Provention | ||
| PRV-3279 | Existing drug supply
– MacroGenics Future drug supply – vendors being evaluated | MacroGenics Provention | ||
| PRV-015 | Amgen | Amgen |
We have historically relied upon an existing supply of PRV-031 produced by MacroGenics for use in our clinical trials of PRV-031. This existing supply is insufficient to fully supply the ongoing PROTECT study to completion. In February 2019, we entered into an agreement with AGC Biologics to manufacture PRV-031 for our anticipated clinical trial needs. In order to obtain regulatory approval, the PROTECT study will need to include data from a sufficient number of subjects dosed with the supply of PRV-031 produced by AGC Biologics to demonstrate safety and efficacy. In addition, we will require AGC Biologics to satisfy process validation requirements with regard to PRV-031 and we will be required to demonstrate to the FDA and other regulatory authorities the comparability of AGC Biologics product to MacroGenics product, in order for PRV-031 to obtain regulatory approval.
| 38 |
Competition
We face substantial competition from well-established large pharmaceutical companies, as well as innovative new entrants. Nevertheless, we believe our strategic intent is sufficiently differentiated in that we are focusing on intercepting or potentially preventing the onset and progression of immune-mediated and inflammatory diseases by selecting and developing product candidates that are aimed at relevant and predominantly upstream pathophysiological targets.
The symptomatic treatment of T1D is a highly competitive market with large incumbents such as Sanofi, Novo Nordisk and Eli Lilly providing insulin and blood glucose monitoring products and working on new ways to manage the disease. Our goal is to delay or prevent the onset of T1D and spare patients the need to live with blood glucose monitoring and daily insulin injections and this therapy’s many complications and clinically relevant shortfalls. We believe our enteroviral vaccine approach is unique in that it aims to prevent the onset of T1D prior to the rise of immune cells and auto-antibodies programmed to attack insulin producing beta cells. We are aware of competitive vaccine technologies in development that are attempting to alter the autoimmune cycle once these auto-antibodies have been detected. However, we believe our vaccine approach may intercept the process prior to this cycle being initiated (primary prevention). Other immunomodulation therapies have shown preservation of beta cell function in early phase studies of new-onset T1D including anti-thymocyte globulin (ATG), CTLA4-Ig (costimulatory blocker), anti-CD20 (Rituxan) and LFA3-Ig (alefacept). All of these Phase 2 studies were conducted by the academic community or by T1D networks and do not appear to be in active development by industry sponsors. The most recent data were reported with Thymoglobulin which is an approved anti-thymocyte globulin obtained by immunization of rabbits with human thymocytes and is indicated for the treatment of renal transplant acute rejection in conjunction with concomitant immunosuppression and for induction in adult renal transplant recipients. Low dose ATG was administered intravenously for 2 days in early onset T1D (within 100 days of diagnosis). While C-peptide preservation was observed, due to the risk of serum sickness, ATG was administered during a 2-3 day hospitalization and required pre-medication, including intravenous corticosteroids.
PRV-015 has the potential to be the first drug ever approved for CD since it is the only medication which has shown simultaneous improvement in symptoms and inflammation to date, and since most clinical-stage products are early in development and have not established proof-of-concept. Competition includes experimental medications in development by Innovate (INN-202/larazotide acetate, Phase 2b study completed in 2014; Phase 3 started in 2019), ImmunogenX (IMGX-003/latiglutenase, Phase 2b study completed in 2016 missed primary endpoint, new phase 2a started in 2019), Zedira/Dr. Falk (ZED1227, phase 2a on-going), Cour (NP-GLI, Phase 1/2 completed in 2019) and PvP Biologics (KumaMax, Phase 1 completed in 2019).
The market for lupus is currently led by large pharmaceutical companies commercializing older, off-patent products such as steroids, immunosuppressive agents including azathioprine, cyclosphosphamide, cyclosporine and mycophenolate. In addition, Glaxo SmithKline (GSK) and Roche offer recently approved B cell-targeted agents. GSK received approval for belimumab (Benlysta) in 2011, the first drug approval in lupus in 50 years. Despite modest efficacy and slow onset of effect, belimumab’s annual sales are currently approximately $800 million. Roche’s rituximab (Rituxan), a blockbuster drug, is used off-label in lupus despite not having been approved in SLE. The lupus field is competitive and new experimental drugs are being tested in late stage trials by large pharmaceutical companies and early to mid-stage biotech companies and include: the anti-interferon alpha receptor anifrolumab (Astra Zeneca), which posted a positive Phase 3 trial in late 2019; anti-CD19/CD32B XmAb5871 (Xencor), which has not moved forward after mixed Phase 2a results in 2018; and calcineurin inhibitor voclosporin (Aurinia Pharmaceuticals), also with a positive Phase 3 study in lupus nephritis in late 2019. We expect that PRV-3279 will be differentiated from the competition because of greater and faster-onset efficacy, better safety (PRV-3279 does not deplete B cells and is not expected to be immune-suppressive), and less side effects (since PRV-3279 is a highly specific mAb with likely minimal off-target side effects).
| 39 |
Government Regulation
Our business activities, including the manufacturing, research, development and marketing of our product candidates, are subject to extensive regulation by numerous governmental authorities in the United States and other countries. Before marketing in the United States, any new drug developed by us or our collaborators must undergo rigorous preclinical testing, clinical trials and an extensive regulatory clearance process implemented by the FDA under the Federal Food, Drug, and Cosmetic Act, as amended. The FDA regulates, among other things, the development, testing, manufacture, safety, efficacy, record keeping, labeling, storage, approval, advertising, promotion, import, export, sale and distribution of biopharmaceutical products. The regulatory review and approval process, which includes preclinical testing and clinical trials of each product candidate, is lengthy, expensive and uncertain. Moreover, government coverage and reimbursement policies will both directly and indirectly impact our ability to successfully commercialize any future approved products, and such coverage and reimbursement policies will be impacted by enacted and any applicable future healthcare reform and drug pricing measures. In addition, we are subject to state and federal laws, including, among others, anti-kickback laws, false claims laws, data privacy and security laws, and transparency laws that restrict certain business practices in the pharmaceutical industry.
In the United States, drug product candidates intended for human use undergo laboratory and animal testing until adequate proof of safety is established. Clinical trials for new product candidates are then typically conducted in humans in three sequential phases that may overlap. Phase 1 trials involve the initial introduction of the product candidate into healthy human volunteers. The emphasis of Phase 1 trials is on testing for safety or adverse events, dosage, tolerance, metabolism, distribution, excretion and clinical pharmacology. Phase 2 involves studies in a limited patient population to determine the initial efficacy of the compound for specific targeted indications, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks. Once a compound shows evidence of effectiveness and is found to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to more fully evaluate clinical outcomes. Before commencing clinical investigations in humans, we or our collaborators must submit an IND to the FDA.
Regulatory authorities, Institutional Review Boards and Data Monitoring Committees may require additional data before allowing clinical trials to commence, continue or proceed from one phase to another, and could demand that studies be discontinued or suspended at any time if there are significant safety issues. We have in the past and may in the future rely on assistance from our third-party collaborators and contract service providers to file our INDs and generally support our development and regulatory activities approval process for our potential products. Clinical testing must also meet requirements for clinical trial registration, institutional review board oversight, informed consent, health information privacy, and GCPs. Additionally, the manufacture of our drug product, must be done in accordance with current GMPs.
To establish a new product candidate’s safety and efficacy, the FDA requires companies seeking approval to market a drug product to submit extensive preclinical and clinical data, along with other information, for each indication for which the product will be labeled. The data and information are submitted to the FDA in the form of a New Drug Application, or NDA, or Biologics License Application, or BLA, which must be accompanied by payment of a significant user fee unless a waiver or exemption applies. Generating the required data and information for regulatory approval takes many years and requires the expenditure of substantial resources. Information generated in this process is susceptible to varying interpretations that could delay, limit or prevent regulatory approval at any stage of the process. The failure to demonstrate adequately the quality, safety and efficacy of a product candidate under development would delay or prevent regulatory approval of the product candidate. Under applicable laws and FDA regulations, each NDA or BLA submitted for FDA approval is given an internal administrative review within 60 days following submission of the NDA or BLA. If deemed sufficiently complete to permit a substantive review, the FDA will “file” the NDA or BLA. The FDA can refuse to file any NDA and BLA that it deems incomplete or not properly reviewable. The FDA has established internal goals of eight months from submission for priority review of NDAs or BLAs that cover product candidates that offer major advances in treatment or provide a treatment where no adequate therapy exists, and 12 months from submission for the standard review of NDAs and BLAs. However, the FDA is not legally required to complete its review within these periods, these performance goals may change over time and the review is often extended by FDA requests for additional information or clarification. Moreover, the outcome of the review, even if generally favorable, may not be an actual approval but a “complete response letter” that describes additional work that must be done before the NDA or BLA can be approved. Before approving an NDA or BLA, the FDA can choose to inspect the facilities at which the product is manufactured and will not approve the product unless the manufacturing facility complies with GMPs. The FDA may also audit sites at which clinical trials have been conducted to determine compliance with GCPs and data integrity. The FDA’s review of an NDA or BLA may also involve review and recommendations by an independent FDA advisory committee, particularly for novel indications. The FDA is not bound by the recommendation of an advisory committee.
| 40 |
In addition, delays or rejections may be encountered based upon changes in regulatory policy, regulations or statutes governing product approval during the period of product development and regulatory agency review.
Before receiving FDA approval to market a potential product, we or our collaborators must demonstrate through adequate and well-controlled clinical trials that the potential product is safe and effective in the patient population that will be treated. In addition, under the Pediatric Research Equity Act, or PREA, an NDA or BLA or supplement thereto must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective, unless a waiver applies. If regulatory approval of a potential product is granted, this approval will be limited to those disease states and conditions for which the product is approved. Marketing or promoting a drug for an unapproved indication is generally prohibited. Furthermore, FDA approval may entail ongoing requirements for risk management, including post-marketing, or Phase 4, studies. Even if approval is obtained, a marketed product, its manufacturer and its manufacturing facilities are subject to payment of significant annual fees and continuing review and periodic inspections by the FDA. Discovery of previously unknown problems with a product, manufacturer or facility may result in restrictions on the product or manufacturer, including labeling changes, warning letters, costly recalls or withdrawal of the product from the market.
Any drug is likely to produce some toxicities or undesirable side effects in animals and in humans when administered at sufficiently high doses and/or for sufficiently long periods of time. Unacceptable toxicities or side effects may occur at any dose level at any time in the course of studies in animals designed to identify unacceptable effects of a product candidate, known as toxicological studies, or during clinical trials of our potential products. The appearance of any unacceptable toxicity or side effect could cause us or regulatory authorities to interrupt, limit, delay or abort the development of any of our product candidates. Further, such unacceptable toxicity or side effects could ultimately prevent a potential product’s approval by the FDA or foreign regulatory authorities for any or all targeted indications or limit any labeling claims and market acceptance, even if the product is approved.
In addition, as a condition of approval, the FDA may require an applicant to develop a Risk Evaluation and Mitigation Strategy, or REMS. A REMS uses risk minimization strategies beyond the professional labeling to ensure that the benefits of the product outweigh the potential risks. To determine whether a REMS is needed, the FDA will consider the size of the population likely to use the product, seriousness of the disease, expected benefit of the product, expected duration of treatment, seriousness of known or potential adverse events, and whether the product is a new molecular entity. REMS can include medication guides, physician communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU may include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The FDA may require a REMS before approval or post-approval if it becomes aware of a serious risk associated with use of the product. The requirement for a REMS can materially affect the potential market and profitability of a product.
Any trade name that we intend to use for a potential product must be approved by the FDA irrespective of whether we have secured a formal trademark registration from the U.S. Patent and Trademark Office. The FDA conducts a rigorous review of proposed product names and may reject a product name if it believes that the name inappropriately implies medical claims or if it poses the potential for confusion with other product names. The FDA will not approve a trade name until the NDA or BLA for a product is approved. If the FDA determines that the trade names of other products that are approved prior to the approval of our potential products may present a risk of confusion with our proposed trade name, the FDA may elect to not approve our proposed trade name. If our trade name is rejected, we will lose the benefit of any brand equity that may already have been developed for this trade name, as well as the benefit of our existing trademark applications for this trade name.
We and our collaborators and contract manufacturers also are required to comply with the applicable FDA GMP regulations. GMP regulations include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Manufacturing facilities are subject to inspection by the FDA. These facilities must be approved before we can use them in commercial manufacturing of our potential products and must maintain ongoing compliance for commercial product manufacture. The FDA may conclude that we or our collaborators or contract manufacturers are not in compliance with applicable GMP requirements and other FDA regulatory requirements, which may result in delay or failure to approve applications, warning letters, product recalls and/or imposition of fines or penalties.
| 41 |
If a product is approved, we must also comply with post-marketing requirements, including, but not limited to, compliance with advertising and promotion laws enforced by various government agencies, including the FDA’s Office of Prescription Drug Promotion, through such laws as the Prescription Drug Marketing Act, federal and state anti-fraud and abuse laws, including anti-kickback and false claims laws, healthcare information privacy and security laws, post-marketing safety surveillance, and disclosure of payments or other transfers of value to healthcare professionals and entities. In addition, we are subject to other federal and state regulation including, for example, the implementation of corporate compliance programs.
If we elect to distribute our products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain.
Outside of the United States, our ability to market a product is contingent upon receiving a marketing authorization from the appropriate regulatory authorities, including the EMA. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the European Community, centralized registration procedures are available to companies wishing to market a product in more than one European Community member state. If the regulatory authority is satisfied that adequate evidence of safety, quality and efficacy has been presented, marketing authorization will be granted. This foreign regulatory development and approval process involves all of the risks associated with achieving FDA marketing approval in the U.S. as discussed above. In addition, foreign regulations may include applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or other transfers of value to healthcare professionals and entities.
Reimbursement
Potential sales of any of our product candidates, if approved, will depend, at least in part, on the extent to which such products will be covered by third-party payors, such as government health care programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly managing access and using restrictive measures to contain costs. If a third-party payor decides to provide coverage for an approved drug product, patient access and reimbursement is not certain. Further, one payor’s determination to provide coverage for a drug product does not assure that other payors will also provide coverage for the drug product. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our future revenues and results of operations. Decreases in third-party reimbursement or a decision by a third-party payor to not cover a product candidate, if approved, or any future approved products could reduce physician usage of our products, and have a material adverse effect on our sales, results of operations and financial condition.
In the United States, Medicare is a program that is administered by the federal government that covers individuals age 65 and over as well as those with certain disabilities. Medicare Part B generally covers drugs that must be administered by physicians and pays for such drugs under a payment methodology based on the average sales price (ASP) of the drugs. Reimbursement levels and reimbursement methodologies have come under scrutiny and may be subject to change. We do not know whether our product candidates, if approved, will be eligible for coverage under Medicare Part B, but individual Medicare Part B plans offer coverage subject to various factors such as those described above. Furthermore, private payors often follow Medicare coverage and reimbursement policies in setting their own coverage policies.
| 42 |
Healthcare Laws and Regulations
Sales of our product candidates, if approved, or any other future product candidate will be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we might conduct our business. The healthcare laws and regulations that may affect our ability to operate include the following:
| ● | The federal Anti-Kickback Statute makes it illegal for any person or entity to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration that is in exchange for or to induce the referral of business, including the purchase, order, lease of any good, facility, item or service for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. The term “remuneration” has been broadly interpreted to include anything of value. |
| ● | Federal false claims and false statement laws, including the federal civil False Claims Act, prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, for payment to, or approval by, federal programs, including Medicare and Medicaid, claims for items or services, including drugs, that are false or fraudulent. |
| ● | The U.S. federal Health Insurance Portability and Accountability Act of 1996 (HIPAA) created additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors or making any false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (HITECH) and their implementing regulations, impose obligations on certain types of individuals and entities regarding the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information. |
| ● | The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Center for Medicare & Medicaid Services information related to payments or other transfers of value made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. |
Also, many states have similar laws and regulations, such as anti-kickback and false claims laws that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, we may be subject to state laws that require pharmaceutical companies to comply with the federal government’s and/or pharmaceutical industry’s voluntary compliance guidelines, state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, as well as state and foreign laws governing the privacy and security of health information, many of which differ from each other in significant ways and often are not preempted by HIPAA.
Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
Segments and Geographic Information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. We view our operations and manage our business in one operating and reporting segment.
Employees
As of March 1, 2020, we had 19 full-time employees. We are a virtual company and all of our employees work remotely. None of our employees are represented by a labor union or covered by a collective bargaining agreement, and we believe our relationship with our employees is good. Additionally, we utilize independent contractors and other third parties to assist with various aspects of our drug and product development.
Our Corporate Information
We were incorporated under the laws of the State of Delaware in 2016. We are a virtual company and do not own or lease any office space. We maintain a mailing address at P.O. Box 666, Oldwick, NJ 08858 and our telephone number is (908) 336-0360. Our Internet website is http://www.proventionbio.com.
| 43 |
Available Information
We make available free of charge through our website our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act. We make these reports available through our website as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the SEC. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the SEC’s public reference room. You can review our electronically filed reports and other information that we file with the SEC on the SEC’s web site at http://www.sec.gov. We also make available, free of charge on our website, the reports filed with the SEC by our executive officers, directors and 10% stockholders pursuant to Section 16 under the Exchange Act as soon as reasonably practicable after copies of those filings are provided to us by those persons. The information contained on, or that can be accessed through, our website is not a part of or incorporated by reference in this Annual Report on Form 10-K.
Certain factors may have a material adverse effect on our business, financial condition and results of operations, and you should carefully consider them. Accordingly, in evaluating our business, we encourage you to consider the following discussion of risk factors in its entirety, in addition to other information contained in this Annual Report on Form 10-K, as well as our other public filings with the SEC. The risks and uncertainties described below are those we currently believe to be material, but they are not the only ones we face. If any of the following risks, or any other risks and uncertainties that we have not yet identified or that we currently consider not to be material, actually occur or become material risks, our business and financial condition could be materially and adversely affected.
Risks Related to Our Business
We are a clinical stage biopharmaceutical company with a limited operating history.
We are a clinical-stage biopharmaceutical company formed in October 2016 and have a limited operating history. We do not lease or own any corporate office or laboratory space and our employees all work remotely. We outsource our manufacturing, clinical trial, information technology, payroll, legal and certain other functions.
We have acquired or in-licensed five clinical stage assets and a late stage preclinical enteroviral vaccine platform as described herein, which will be our product candidates unless we in-license or acquire additional development assets. Marketing approval of our product candidates will require extensive clinical testing data to support safety and efficacy requirements, as well as pharmaceutical development, manufacturing and preclinical data, all of which are needed for regulatory approval. The likelihood of success of our business plan must be considered in light of the challenges, substantial expenses, difficulties, complications and delays frequently encountered in connection with developing and expanding early-stage businesses and the regulatory and competitive environment in which we operate. Biopharmaceutical product development is a highly speculative undertaking, involves a substantial degree of risk, and is a capital-intensive business.
Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially clinical-stage biopharmaceutical companies such as ours. Potential investors should carefully consider the risks and uncertainties that a company with a limited operating history will face. In particular, you should consider that we cannot assure you that we will be able to:
| ● | successfully implement or execute our current business plan, or that our business plan is sound; | |
| ● | successfully start and complete clinical trials and obtain regulatory approval for the marketing of our product candidates; | |
| ● | successfully contract for the manufacture of our clinical drug products and establish a commercial drug supply; | |
| ● | secure market exclusivity and/or adequate intellectual property protection for our product candidates; | |
| ● | attract and retain an experienced management and advisory team; | |
| ● | raise sufficient funds in the capital markets to effectuate our business plan, including clinical development, regulatory approval and commercialization for our product candidates; | |
| ● | successfully recruit and retain a sales and marketing organization; | |
| ● | successfully launch teplizumab in the U.S. ourselves; and | |
| ● | successfully establish a strategic partnership to launch teplizumab outside the U.S. |
| 44 |
If we cannot successfully execute any one of the foregoing, our business may not succeed, and your investment will be adversely affected.
We expect to incur substantial expenses and may never become profitable or be able to sustain profitability.
We expect to incur substantial expenses without corresponding revenues unless and until we are able to obtain regulatory approval and successfully commercialize our product candidates. We expect to incur significant expense to complete our clinical programs for our product candidates in the United States and elsewhere. We may never be able to obtain regulatory approval for the marketing of our product candidates in any indication in the United States or internationally. Even if we are able to commercialize our product candidates, there can be no assurance that we will generate significant revenues or ever achieve profitability.
We expect to have significant research and development expenses as we advance clinical trials for our product candidates. As a result, we expect to incur substantial losses for the foreseeable future, and these losses will be increasing. We are uncertain when or if we will be able to achieve or sustain profitability. If we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Failure to become and remain profitable may impair our ability to sustain operations and adversely affect our business and our ability to raise capital.
We need to raise additional funding. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate certain or all of our product development programs or commercialization efforts.
We expect our operating costs to be substantial as we incur costs related to our expected BLA submission for PRV-031 for the At-Risk indication, costs to support our commercialization efforts for PRV-031, including costs related to the buildout of an internal commercial infrastructure, and our ongoing and planned clinical trials for PRV-031 and our other product candidates. We will operate at a loss for the foreseeable future or until such time as we obtain regulatory approval for and execute a successful commercial launch of teplizumab. For the years ended December 31, 2019 and 2018, we had a net loss of $43.3 million and $26.8 million, respectively, and as of December 31, 2019, we had an accumulated deficit of $79.1 million and $85.4 million in cash, cash equivalents and marketable securities. We believe our current cash, cash equivalents and marketable securities will be sufficient to fund projected operating requirements for at least 12 months from the issuance of these financial statements. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect. We will need substantial additional capital to fund the clinical development program for all of our product candidates. In particular, we will need additional capital to fund the completion of the PROTECT study, a pivotal Phase 3 clinical trial of PRV-031 in type 1 diabetes, or T1D, commenced in the second quarter of 2019 with top-line data, based on current estimates, not available until 2022, including to produce additional drug supply in order to complete the PROTECT study.
We do not have any prospective financing arrangements or credit facilities as a source of future funds, and there can be no assurance that we will be able to raise sufficient additional capital on acceptable terms, or at all. We may seek additional capital through a combination of private equity offerings, public equity offerings, debt financings and strategic collaborations. If we raise additional funds through the issuance of equity or convertible debt securities, the percentage ownership of our stockholders could be significantly diluted, and these newly issued securities may have rights, preferences or privileges senior to those of existing stockholders. Debt financing, if obtained, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, could increase our expenses and could require that our assets secure such debt. Moreover, any debt we incur must be repaid regardless of our operating results. If we choose to pursue additional indications and/or geographies for our product candidates, in-license or acquire additional development assets, or otherwise expand more rapidly than we presently anticipate, we may also need to raise additional capital sooner than expected.
If we do not raise additional capital when required or on acceptable terms, we may need to significantly delay, scale back or discontinue the development or commercialization of one or more of our product candidates or cease operations altogether, or relinquish or license on unfavorable terms, our rights to technologies or any future product candidates that we otherwise would seek to develop or commercialize.
Our forecast of the period of time through which our financial resources will adequately support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed elsewhere in this Risk Factors section. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
| 45 |
We may not be able to correctly estimate or control our future operating expenses, which could lead to cash shortfalls.
Our operating expenses may fluctuate significantly in the future as a result of a variety of factors, many of which are outside of our control. These factors include:
| ● | the success of our development strategy; | |
| ● | the time, resources, and expense required to develop and conduct clinical trials and seek regulatory approvals for our product candidates; | |
| ● | the cost of preparing, filing, prosecuting, defending, and enforcing patent claims and other patent related costs, including litigation costs and the results of such litigation; | |
| ● | the cost of manufacturing and maintaining sufficient inventories of our products to meet anticipated demand; | |
| ● | any product liability or other lawsuits related to our product candidates and the costs associated with defending them or the results of such lawsuits; | |
| ● | the cost of growing our ongoing development operations and establishing commercialization operations; | |
| ● | the cost to attract and retain personnel with the skills required for effective operations; | |
| ● | the costs associated with being a public company; and | |
| ● | the costs associated with commercialization. |
Any material increases in our operating expenses will have a material impact on our financial condition and business operations. In addition, if we are unable to correctly estimate or control our future operating expenses, we may need to raise additional capital or delay or cease development of one or more of our product candidates, which could have a material adverse effect on our business, operating results and prospects.
Risks Related to Product Development, Regulatory Approval, Manufacturing and Commercialization
The results of the PRV-031 CMC comparability plan may be unacceptable to the regulatory authorities. This may require further CMC development activities and could affect the timing of BLA submission.
In November 2019, we completed a Type B multidisciplinary meeting with the FDA to discuss the proposed contents of a BLA for PRV-031 (teplizumab) for the prevention or delay of type 1 diabetes (T1D) in individuals at-risk of developing T1D. Based on official FDA meeting minutes, we do not anticipate the need to conduct any additional clinical trials in the at-risk population prior to BLA submission. However, for the CMC module, the FDA confirmed that it would require the demonstration of comparability between the study drug previously manufactured by MacroGenics and Eli Lilly and the to-be-commercialized drug substance and drug product scheduled for production by Provention and its contract manufacturing partners.
If we are unable to demonstrate the ability to manufacture drug substance and drug product that is comparable to the study drug previously manufactured by MacroGenics and Eli Lilly, the timing of the PRV-031 BLA submission could be delayed, which could have a material impact on our business.
We depend entirely on the success of our product candidates, one of which did not meet its primary endpoint in a prior Phase 3 clinical trial and two of which have not yet demonstrated efficacy for their target indication or any other indications in Phase 2 clinical trials. If we are unable to generate revenues from our product candidates, our ability to create stockholder value will be limited.
Our product candidates are either in various stages of clinical development or late stages of preclinical development. We do not generate revenues from any approved drug products. We are or will be conducting human clinical trials of our product candidates.
Our clinical trial of PRV-031 in T1D is currently being conducted in the United States. We plan to add sites in Canada and European countries. PRV-031 has an active Investigational New Drug Application, or IND, with the U.S. Food and Drug Administration, or FDA, and Clinical Trial Application, or CTA, the foreign equivalent of an IND. We are in the process of submitting CTAs in all intended countries.
| 46 |
Our Phase 2b clinical trial of PRV-015 in non-responsive celiac disease will be conducted in the United States, Canada and European Union countries. PRV-015 has an active IND with the U.S. FDA, and we will file CTAs in all intended countries upon completion of on-going toxicology studies.
We have submitted amendments to the IND for PRV-3279 for clinical development in B-cell directed immune diseases, and plan to file the appropriate documentation to the IND as well as CTAs for the planned Phase 2a study in lupus, now that the Phase 1b study has been completed.
We plan on submitting a CTA to the Finnish Medicines Agency (Fimea), seeking approval to initiate a First-in-Human Phase 1 clinical trial in normal healthy adult volunteers for our coxsackie B virus, or CVB, vaccine (PRV-101) for the prevention of acute CVB infection, which has been linked to the development of T1D and T1D-associated celiac disease.
We will be required to submit our clinical trial protocols and receive approvals from the regulatory authorities before we can commence any clinical trials with PRV-101 and PRV-015, and any additional studies with PRV-3279. Nonclinical study results for our product candidates, including toxicology studies, may not support the filing of an IND or foreign equivalent for the product candidate.
Moreover, we may not be successful in obtaining acceptance from the regulatory authorities to start our clinical trials. If we do not obtain such acceptance, the time in which we expect to commence clinical programs for any product candidate will be extended and such extension will increase our expenses and increase our need for additional capital.
There is no guarantee that our clinical trials will be successful or that we will continue clinical development in support of an approval from the regulatory authorities for any indication. We note that most drug candidates never reach the clinical development stage and even those that do commence clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval.
Our business currently depends entirely on the successful development, regulatory approval and commercialization of our product candidates. We cannot assure you that our product candidates will be successfully developed or commercialized. If we are unable to develop, or obtain regulatory approval for, or, if approved, to successfully commercialize our product candidates, it could have a material adverse effect on our business, operating results and prospects.
We may encounter substantial delays in completing our clinical trials which in turn will require additional costs, or we may fail to demonstrate adequate safety and efficacy to the satisfaction of applicable regulatory authorities.
It is impossible to predict if or when any of our product candidates will prove safe or effective in humans or will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Clinical testing is expensive, time-consuming and uncertain as to outcome. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
| ● | delays in reaching, or failing to reach, a consensus with regulatory agencies on study design; | |
| ● | delays in reaching, or failing to reach, agreement on acceptable terms with a sufficient number of prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | delays in obtaining required Institutional Review Board, or IRB or Ethics Committee (EC) approval at each clinical trial site; | |
| ● | delays in recruiting a sufficient number of suitable patients to participate in our clinical trials; | |
| ● | imposition of a clinical hold by regulatory agencies; | |
| ● | failure by our CROs, other third parties or us to adhere to clinical trial, regulatory or legal requirements; | |
| ● | failure to perform in accordance with the FDA’s good clinical practices, or GCP, or applicable regulatory guidelines in other countries; | |
| ● | delays in the testing, validation, manufacturing and delivery of sufficient quantities of our product candidates to the clinical sites; | |
| ● | delays in having patients’ complete participation in a study or return for post-treatment follow-up; | |
| ● | clinical study sites or patients dropping out of a study; | |
| ● | delay or failure to address any patient safety concerns that arise during the course of a trial; | |
| ● | unanticipated costs or increases in costs of clinical trials of our product candidates; | |
| ● | occurrence of serious adverse events associated with the product candidate that are viewed to outweigh its potential benefits; or | |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols. |
| 47 |
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs or ECs of the institutions in which such trials are being conducted, by an independent Safety Review Board, or SRB, for such trial or by the FDA, EMA, or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, EMA, or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, if we make manufacturing or formulation changes to our product candidates, we may need to conduct additional studies to bridge our modified product candidates to earlier versions.
Clinical trial delays could also shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
The outcome of preclinical studies and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Further, preclinical and clinical data are often susceptible to various interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval. If the results of our clinical trials are inconclusive or if there are safety concerns or adverse events associated with our other product candidates, we may:
| ● | be delayed in obtaining marketing approval for our product candidates, if approved at all; | |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; | |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; | |
| ● | be required to change the way the product is administered; | |
| ● | be required to perform additional preclinical or clinical trials to support approval or be subject to additional post-marketing testing requirements; | |
| ● | have regulatory authorities withdraw their approval of a product or impose restrictions on its distribution in the form of a modified risk evaluation and mitigation strategy; | |
| ● | be sued; or | |
| ● | experience damage to our reputation. |
Additionally, our product candidates could potentially cause other adverse events that have not yet been predicted. The inclusion of ill patients in our clinical trials may result in deaths or other adverse medical events due to other therapies or medications that such patients may be using. As described above, any of these events could prevent us from obtaining marketing approval or achieving or maintaining market acceptance of our product candidates and impair our ability to commercialize our products.
| 48 |
We have conducted and are conducting clinical trials outside the United States and anticipate conducting additional clinical trials outside the United States, and the FDA may not accept data from such trials.
We are currently conducting clinical trials for our product candidates in countries outside of the United States and we anticipate that we will conduct additional clinical trials in countries outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such study data by the FDA is subject to certain conditions. For example, the clinical trial must be conducted in accordance with GCP requirements and the FDA must be able to validate the data from the clinical trial through an onsite inspection if it deems such inspection necessary. Where data from foreign clinical trials are intended to serve as the sole basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless those data are considered applicable to the U.S. patient population and U.S. medical practice, the clinical trials were performed by clinical investigators of recognized competence, and the data is considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. In addition, such clinical trials would be subject to the applicable local laws of the foreign jurisdictions where the clinical trials are conducted. A description of any studies related to overdosage is also required, including information on dialysis, antidotes, or other treatments, if known. There can be no assurance the FDA will accept data from clinical trials conducted outside of the United States. If the FDA does not accept any such data, it would likely result in the need for additional clinical trials, which would be costly and time-consuming and delay aspects of our development plan.
Risks inherent in conducting international clinical trials include, but are not limited to:
| ● | foreign regulatory requirements that could burden or limit our ability to conduct our clinical trials; | |
| ● | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; | |
| ● | foreign currency fluctuations which could negatively impact our financial condition since certain payments are paid in local currencies; | |
| ● | manufacturing, customs, shipment and storage requirements; | |
| ● | cultural differences in medical practice and clinical research; and | |
| ● | diminished protection of intellectual property in some countries. |
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing of drug product candidates is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of pre-clinical trials and early clinical trials may not be predictive of the results of later-stage clinical trials. We cannot assure you that the FDA, EMA or comparable foreign regulatory authorities will view the results as we do or that any future trials of any of our product candidates will achieve positive results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through pre-clinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Any future clinical trial results for our product candidates may not be successful.
In addition, a number of factors could contribute to a lack of favorable safety and efficacy results for any of our product candidates. For example, such trials could result in increased variability due to varying site characteristics, such as local standards of care, differences in evaluation period and surgical technique, and due to varying patient characteristics including demographic factors and health status.
If we are not able to obtain any required regulatory approvals for our product candidates, we will not be able to commercialize our product candidates and our ability to generate revenue will be limited.
We must successfully complete clinical trials for our product candidates before we can apply for marketing approval. Even if we complete our clinical trials, it does not assure marketing approval. Our clinical trials may be unsuccessful, which would materially harm our business. Even if our initial clinical trials are successful, we are required to conduct additional clinical trials to establish our product candidates’ safety and efficacy, before a marketing application (New Drug Application, or NDA or Biologics License Application, or BLA, or their foreign equivalents) can be filed with the FDA, EMA, or comparable foreign regulatory authorities for marketing approval of our product candidates.
Clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in early phases of pre-clinical and clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates. Our product candidates may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude us from obtaining regulatory approval or prevent or limit commercial use with respect to one or all intended indications.
| 49 |
The research, testing, manufacturing, labeling, packaging, storage, approval, sale, marketing, advertising and promotion, pricing, export, import and distribution of drug products are subject to extensive regulation by the FDA, EMA, and other regulatory authorities in the United States, European Union, and other countries, where regulations differ from country to country. We are not permitted to market our product candidates as prescription pharmaceutical products in the United States until we receive approval of an NDA or a BLA from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the United States, the FDA generally requires the completion of clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA or a BLA is approved. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA or a BLA to the FDA or other regulatory authorities and even fewer are eventually approved for commercialization. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for a submitted product application may cause delays in the approval or rejection of an application.
We have not submitted an NDA or a BLA to the FDA or comparable applications to other regulatory authorities. We have only limited experience in filing the applications necessary to gain regulatory approvals and expect to rely on consultants and third party CROs with expertise in this area to assist us in this process. If our development efforts for our product candidates, including regulatory approval, are not successful for their planned indications, our business will be materially adversely affected.
Our success depends on the receipt of regulatory approval and the issuance of such regulatory approvals is uncertain and subject to a number of risks, including the following:
| ● | the results of nonclinical or toxicology studies may not support the filing of an IND or foreign equivalent for our CVB vaccine product candidate; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities or IRBs or ECs may disagree with the design or implementation of our clinical trials; | |
| ● | we may not be able to provide acceptable evidence of our product candidates’ safety and efficacy; | |
| ● | the results of our clinical trials may not be satisfactory or may not meet the level of statistical or clinical significance required by the FDA, EMA, or other regulatory agencies for marketing approval; | |
| ● | the dosing of our product candidates in a particular clinical trial may not be at an optimal level; | |
| ● | patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to our product candidates; | |
| ● | the data collected from clinical trials may not be sufficient to support the submission of an NDA, BLA or other marketing application or to obtain regulatory approval in the United States or elsewhere; | |
| ● | the potential for the FDA, EMA or other regulatory authorities to require additional studies, including a second phase 3 study for the PRV-031 program in T1D, which may require us to raise additional capital and may delay the approval of our product candidates; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities may disagree on the design or implementation of our clinical trials, including the methodology used in our studies, our chosen endpoints, our statistical analysis, or our proposed product indication; | |
| ● | our failure to demonstrate to the satisfaction of the FDA, EMA, or comparable regulatory authorities that a product candidate is safe and effective for its proposed indication; | |
| ● | we may fail to demonstrate that a product candidate’s clinical benefits outweigh its safety risks; | |
| ● | immunogenicity might affect a product candidate efficacy and/or safety; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities may disagree with our interpretation of data from nonclinical studies or clinical trials; | |
| ● | data collected from clinical trials of our product candidates may be insufficient to support the submission and filing of a marketing application or to obtain marketing approval. For example, the FDA may require additional studies to show that our product candidates are safe or effective; | |
| ● | the FDA, EMA, or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies; | |
| ● | we may fail to obtain approval of the manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies; | |
| ● | the third-party manufacturers with whom we contract for clinical and commercial supplies may be unable to produce sufficient quantities of our product candidates in a timely manner; | |
| ● | there may be changes in the approval policies or regulations that render our nonclinical and clinical data insufficient for approval; or | |
| ● | the FDA, EMA or comparable foreign regulatory authority may require more information, including additional nonclinical or clinical data to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. |
| 50 |
Failure or delay in obtaining regulatory approval for our product candidates for the foregoing, or any other reasons, will prevent or delay us from commercializing our product candidates, and our ability to generate revenue will be materially impaired. We cannot guarantee that regulators will agree with our assessment of the results of the clinical trials we have conducted or intend to conduct in the future or that such future trials will be successful. The FDA, EMA and other regulators have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional clinical trials, or pre-clinical or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of our product candidates.
Even if we obtain marketing approval for any of our product candidates, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates could be subject to labeling and other restrictions and withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates.
Even if we obtain regulatory approval for any of our product candidates for an indication, the FDA, EMA, or foreign equivalent may still impose significant restrictions on their indicated uses or marketing or the conditions of approval or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials, post-market surveillance to monitor safety and efficacy and a Risk Evaluation and Mitigation Strategy, or REMS. For example, we intend to initially seek regulatory approval for our CVB vaccine product candidate for the prevention of acute CVB infection. The results of longitudinal studies demonstrating the connection between CVB and T1D and T1D-associated celiac disease will be necessary to expand the indicated use of this vaccine to T1D. These studies must be completed and submitted to the FDA or EMA prior to receiving approval in the United States or European Union to market the CVB vaccine to prevent T1D. Such studies will be costly and time consuming and may not demonstrate to the FDA’s satisfaction the connection between the CVB virus and the onset of T1D.
Our product candidates will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market information. These requirements include registration with the FDA, as well as continued compliance with current Good Clinical Practices regulations, or cGCPs, for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current Good Manufacturing Practices, or cGMP, requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
With respect to sales and marketing activities by us or any future licensor, advertising and promotional materials must comply with FDA rules in addition to other applicable federal, state and local laws in the United States and similar legal requirements in other countries. In the United States, the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug Marketing Act. Application holders must obtain FDA approval for product and manufacturing changes, depending on the nature of the change. We may also be subject, directly or indirectly through our customers and licensors, to various fraud and abuse laws, including, without limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act, and similar state laws, which impact, among other things, our proposed sales, marketing, and scientific/educational grant programs. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of the U.S. Department of Veterans Affairs, or other government drug programs, we will be subject to complex laws and regulations regarding reporting and payment obligations. All of these activities are also potentially subject to U.S. federal and state consumer protection and unfair competition laws. Similar requirements exist in many of these areas in other countries.
| 51 |
In addition, if any of our product candidates are approved for a particular indication, our product labeling, advertising and promotion would be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for our product candidates, physicians may nevertheless legally prescribe our products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees of permanent injunctions under which specified promotional conduct is changed or curtailed.
If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, problems with the facility where the product is manufactured, or we or our manufacturers fail to comply with applicable regulatory requirements, we may be subject to the following administrative or judicial sanctions:
| ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; | |
| ● | issuance of warning letters or untitled letters; | |
| ● | clinical holds; | |
| ● | injunctions or the imposition of civil or criminal penalties or monetary fines; | |
| ● | suspension or withdrawal of regulatory approval; | |
| ● | suspension of any ongoing clinical trials; | |
| ● | refusal to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals; | |
| ● | suspension or imposition of restrictions on operations, including costly new manufacturing requirements; or | |
| ● | product seizure or detention or refusal to permit the import or export of product. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenue. Adverse regulatory action, whether pre- or post-approval, can also potentially lead to product liability claims and increase our product liability exposure.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials, as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. If we fail to comply with the regulatory requirements in international markets and/ or to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed.
| 52 |
Current and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell our product candidates. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In the United States, the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for our product candidates and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 or, collectively, the Health Care Reform Law, is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. Further, the law imposed a significant annual fee on companies that manufacture or import branded prescription drug products.
The Health Care Reform Law remains subject to legislative efforts to repeal, modify or delay the implementation of the law. However, if the Health Care Reform Law is repealed or modified, or if implementation of certain aspects of the Health Care Reform Law are delayed, such repeal, modification or delay may materially adversely impact our business, strategies, prospects, operating results or financial condition. We are unable to predict the full impact of any repeal, modification or delay in the implementation of the Health Care Reform Law on us at this time. Due to the substantial regulatory changes that will need to be implemented by CMS and others, and the numerous processes required to implement these reforms, we cannot predict which healthcare initiatives will be implemented at the federal or state level, the timing of any such reforms, or the effect such reforms or any other future legislation or regulation will have on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the Health Care Reform Law was enacted. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce or eliminate our profitability.
If we fail to successfully commercialize any of our product candidates, we may need to acquire additional product candidates and our business will be adversely affected.
We have never developed and obtained approval for any product candidates or commercialized any product candidates. We have limited product candidates and do not have any other compounds in pre-clinical testing, lead optimization or lead identification stages beyond our product candidates. We cannot be certain that any of our product candidates will prove to be sufficiently effective and safe to meet applicable regulatory standards for any indication. If we fail to successfully obtain regulatory approval or commercialize any of our product candidates for their targeted indications, whether as stand-alone therapies or in combination with other therapeutic agents, and if we are unable to acquire additional product candidates in the future, our business will be adversely affected.
| 53 |
Even if we receive regulatory approval for any of our product candidates, we may not be able to successfully commercialize the product and the revenue that we generate from its sales, if any, may be limited.
If approved for marketing, the commercial success of our product candidates will depend upon each product’s acceptance by the medical community, including physicians, patients and health care payors. The degree of market acceptance for any of our product candidates will depend on a number of factors, including:
| ● | demonstration of clinical safety and efficacy; | |
| ● | relative convenience, dosing burden and ease of administration; | |
| ● | the prevalence and severity of any adverse events; | |
| ● | the willingness of physicians to prescribe our product candidates, and the target patient population to try new therapies; | |
| ● | efficacy of our product candidates compared to competing products; | |
| ● | the introduction of any new products that may in the future become available targeting indications for which our product candidates may be approved; | |
| ● | new procedures or therapies that may reduce the incidences of any of the indications in which our product candidates may show utility; | |
| ● | pricing and cost-effectiveness; | |
| ● | the inclusion or omission of our product candidates in applicable therapeutic and vaccine guidelines; | |
| ● | the effectiveness of our own or any future collaborators’ sales and marketing strategies; | |
| ● | limitations or warnings contained in approved labeling from regulatory authorities; | |
| ● | our ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare and Medicaid, private health insurers and other third-party payors or to receive the necessary pricing approvals from government bodies regulating the pricing and usage of therapeutics; and | |
| ● | the willingness of patients to pay out-of-pocket in the absence of third-party coverage or reimbursement or government pricing approvals. |
If any of our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, health care payors, and patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
In addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize our product candidates successfully. For example, if the approval process takes too long, we may miss market opportunities and give other companies the ability to develop competing products or establish market dominance. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render our product candidates not commercially viable. For example, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve any of our product candidates with a label that does not include the labeling claims necessary or desirable for the successful commercialization for that indication. Further, the FDA or comparable foreign regulatory authorities may place conditions on approvals or require risk management plans or a REMS, to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS; the FDA will not approve the NDA or BLA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA may also require a REMS for an approved product when new safety information emerges. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of our product candidates. Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of our product candidates.
We currently have a limited commercial organization. If we are unable to establish satisfactory sales and marketing capabilities or secure a sales and marketing partner, we may not successfully commercialize any of our product candidates.
We have just recently begun to build out our commercial infrastructure and at present, we have only limited sales or marketing personnel. In order to commercialize products that are approved for commercial sales, we must either develop our own sales and marketing infrastructure or collaborate with third parties that have such commercial infrastructure. If we are not successful entering into appropriate collaboration arrangements or recruiting sales and marketing personnel or in building a sales and marketing infrastructure, we will have difficulty successfully commercializing our product candidates, which would adversely affect our business, operating results and financial condition.
| 54 |
We may not be able to enter into collaboration agreements on terms acceptable to us or at all. In addition, even if we enter into such relationships, we may have limited or no control over the sales, marketing and distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties. If we elect to establish a sales and marketing infrastructure, we may not realize a positive return on this investment. In addition, we will have to compete with established and well-funded pharmaceutical and biotechnology companies to recruit, hire, train and retain sales and marketing personnel. Factors that may inhibit our efforts to commercialize our product candidates without strategic partners or licensees include:
| ● | our inability to recruit and retain adequate numbers of effective sales and marketing personnel; | |
| ● | the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe any of our product candidates; | |
| ● | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and | |
| ● | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
Amgen has the right to assume control over the activities of our anti-IL-15 mAb product candidate.
Pursuant to our License and Collaboration Agreement with Amgen, or the Amgen Agreement, Amgen reserves the right, at any time until 120 days after the delivery of the final data package relating to the Phase 2b PROACTIVE study that we expect to commence in the second quarter of 2020, to assume control over all activities with respect to our anti-IL-15 monoclonal antibody, or mAb, product candidate, including pricing and marketing decisions, after the payment of a $150.0 million milestone. There can be no assurance that Amgen’s strategic direction will be in line with ours should it assume control of activities, or that their decisions will have a positive impact on our results of operations. Moreover, we may not realize the full economic benefit of this agreement.
We face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have existing competitors and will have potential new competitors in a number of jurisdictions, many of which have or will have substantially greater name recognition, commercial infrastructures and financial, technical and personnel resources than we have. Established competitors may invest heavily to quickly discover and develop novel compounds that could make any of our product candidates obsolete or uneconomical. Any new product that competes with an approved product may need to demonstrate compelling advantages in efficacy, cost, convenience, tolerability and safety to be commercially successful. Other competitive factors, including generic competition, could force us to lower prices or could result in reduced sales. In addition, new products developed by others could emerge as competitors to our product candidates. If we are not able to compete effectively against our current and future competitors, our business will not grow, and our financial condition and operations will suffer.
Our potential competitors both in the United States and throughout the world include companies developing and/or marketing drugs and therapeutic solutions for immune-mediated diseases, including oncological, autoimmune and inflammatory diseases, as well as companies working in our specific fields, including T1D, enteroviral and emerging viral diseases, lupus, and inflammatory bowel diseases, such as CD.
There can be no assurance that our product candidates will be more effective or achieve greater market acceptance than competitive products, or that our competitors will not succeed in developing products and technologies that are more effective than those being developed by us or that would render our products and technologies less competitive or obsolete. Additionally, there can be no assurance that the development by others of new or improved products will not make our product candidates superfluous or obsolete.
| 55 |
Our product candidates may face competition sooner than expected.
We intend to seek data exclusivity or market exclusivity for our monoclonal antibodies PRV-031 and PRV-015, our DART molecule PRV-3279 and our PRV-101 CVB vaccine product candidates provided under the Federal Food, Drug and Cosmetic Act, or FDCA, and similar laws in other countries. We believe that these product candidates will qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which was enacted as part of the Health Care Reform Law. Under the BPCIA, an application for a biosimilar product or BLA cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. The law is complex and the processes the FDA establishes to implement the law could have a material adverse effect on the future commercial prospects for our biological product candidates. There is also a risk that Congress could repeal or amend the BPCIA to shorten this exclusivity period, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection. Moreover, the extent to which a biosimilar, once approved, will be substituted for any reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Our product candidates that are not, or are not considered, biologics that would qualify for exclusivity under the BPCIA may be eligible for market exclusivity as drugs under the FDCA. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA, submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent.
Even if, as we expect, our product candidates are considered to be reference products eligible for 12 years of exclusivity under the BPCIA or five years of exclusivity under the FDCA, another company could market competing products if the FDA approves a full BLA or full NDA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products. Moreover, an amendment or repeal of the BPCIA could result in a shorter exclusivity period for our product candidates, which would have a material adverse effect on our business.
Our future growth depends, in part, on our ability to penetrate international markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability will depend, in part, on our ability to commercialize our product candidates in international markets for which we intend to rely on collaborations with third parties. If we commercialize any of our product candidates in international markets, we would be subject to additional risks and uncertainties, including:
| ● | our customers’ ability to obtain reimbursement for our product candidates in international markets; | |
| ● | our inability to directly control commercial activities because we are relying on third parties; | |
| ● | the burden of complying with complex and changing international regulatory, tax, accounting and legal requirements; | |
| ● | different medical practices and customs in foreign countries affecting acceptance in the marketplace; | |
| ● | import or export licensing requirements; | |
| ● | longer accounts receivable collection times; | |
| ● | longer lead times for shipping; | |
| ● | language barriers for technical training; | |
| ● | reduced protection of intellectual property rights in some foreign countries; | |
| ● | foreign currency exchange rate fluctuations; and | |
| ● | the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute. |
International sales of our product candidates could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs, any of which may adversely affect our results of operations.
| 56 |
If we market any of our product candidates in a manner that violates healthcare fraud and abuse laws, or if we violate government price reporting laws, we may be subject to civil or criminal penalties.
The FDA enforces laws and regulations which require that the promotion of pharmaceutical products be consistent with the approved prescribing information. While physicians may prescribe an approved product for a so-called “off label” use, it is unlawful for a pharmaceutical company to promote its products in a manner that is inconsistent with its approved label and any company which engages in such conduct can subject that company to significant liability. The federal government has levied large civil and criminal fines and/or other penalties against companies for alleged improper promotion and has investigated and/or prosecuted several companies in relation to off-label promotion. The FDA has also requested that certain companies enter consent decrees or permanent injunctions under which specified promotional conduct is changed, curtailed or prohibited. Similarly, industry codes in the EU and other foreign jurisdictions prohibit companies from engaging in off-label promotion and regulatory agencies in various countries enforce violations of the code with civil penalties. While we intend to ensure that our promotional materials are consistent with our label, regulatory agencies may disagree with our assessment and may issue untitled letters, warning letters or may institute other civil or criminal enforcement proceedings. In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal healthcare fraud and abuse laws have been applied in recent years to restrict certain marketing practices in the pharmaceutical industry. These laws include the U.S. Anti-Kickback Statute, U.S. False Claims Act and similar state laws. Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of these laws.
The U.S. Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, in cash or in kind, to induce, or in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under federal and state healthcare programs such as Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted broadly to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are several statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not, in all cases, meet all of the criteria for safe harbor protection from anti-kickback liability. Moreover, recent health care reform legislation has strengthened these laws. For example, the Health Care Reform Law, among other things, amends the intent requirement of the U.S. Anti-Kickback Statute and criminal health care fraud statutes; a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the Health Care Reform Law provides that the government may assert that a claim including items or services resulting from a violation of the U.S. Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the U.S. False Claims Act. Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid. Several other countries, including the United Kingdom, have enacted similar anti-kickback, fraud and abuse, and healthcare laws and regulations.
Over the past few years, several pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of alleged promotional and marketing activities, such as: allegedly providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion that caused claims to be submitted to Medicare or Medicaid for non-covered, off-label uses; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates. Most states also have statutes or regulations similar to the U.S. Anti-Kickback Statute and the U.S. False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include substantial civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, substantial criminal fines and imprisonment.
| 57 |
We are completely dependent on third parties to manufacture our product candidates, and our commercialization of our product candidates could be halted, delayed or made less profitable if those third parties fail to obtain manufacturing approval from the FDA or comparable foreign regulatory authorities, fail to provide us with sufficient quantities of our product candidates or fail to do so at acceptable quality levels or prices.
We do not currently have, nor do we plan to acquire, the capability or infrastructure to manufacture the bulk drug substance or the active pharmaceutical ingredient, or API, in our product candidates for use in our clinical trials or for commercial products, if any. As a result, we are obligated to rely on contract manufacturers for clinical supplies of our product candidates and will be obligated, if and when any of our product candidates are approved for commercialization, to rely on contract manufacturers for commercial supply. We have not entered into an agreement with any contract manufacturers for commercial supply and may not be able to engage a contract manufacturer for commercial supply of any of our product candidates on acceptable terms to us, or at all.
The facilities used by our contract manufacturers to manufacture our product candidates must be approved by the FDA, EMA or comparable foreign regulatory authorities pursuant to inspections that will be conducted after we submit an NDA or BLA to the FDA or equivalent applications to other relevant regulatory authorities. We will not control the manufacturing process of, and will be completely dependent on, our contract manufacturers for compliance with cGMPs for manufacture of both active drug substances and finished drug products. These cGMP regulations cover all aspects of the manufacturing, testing, quality control and record keeping relating to our product candidates. If our contract manufacturers do not successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, EMA or other regulatory authorities, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. If the FDA, EMA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
Our contract manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. We do not have control over our contract manufacturers’ compliance with these regulations and standards. Failure by any of our contract manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure to grant approval to market any of our product candidates, delays, suspensions or withdrawals of approvals, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business. In addition, we do not have control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. Failure by our contract manufacturers to comply with or maintain any of these standards could adversely affect our ability to develop, obtain regulatory approval for or market any of our product candidates.
If, for any reason, these third parties are unable or unwilling to perform, we may not be able to terminate our agreements with them, to the extent applicable, and we may not be able to locate alternative manufacturers or formulators or enter into favorable agreements with them and we cannot be certain that any such third parties will have the manufacturing capacity to meet future requirements. If these manufacturers or any alternate manufacturer of finished drug product experiences any significant difficulties in its respective manufacturing processes for our API or finished products or should cease doing business with us, we could experience significant interruptions in the supply of any of our product candidates or may not be able to create a supply of our product candidates at all. Were we to encounter manufacturing issues, our ability to produce a sufficient supply of any of our product candidates might be negatively affected. Our inability to coordinate the efforts of our third-party manufacturers, or the lack of capacity available at our third-party manufacturers, could impair our ability to supply any of our product candidates at required levels. Because of the significant regulatory requirements that we would need to satisfy in order to qualify a new bulk or finished product manufacturer, if we face these or other difficulties with our current manufacturers, we could experience significant interruptions in the supply of any of our product candidates if we decided to transfer the manufacture of any of our product candidates to one or more alternative manufacturers in an effort to deal with the difficulties.
| 58 |
Any manufacturing problem or the loss of a contract manufacturer could be disruptive to our operations and result in lost sales. Additionally, we rely on third parties to supply the raw materials needed to manufacture our potential products. Any reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to a future contract manufacturer caused by problems at suppliers could delay shipment of any of our product candidates, increase our cost of goods sold and result in lost sales.
We cannot guarantee that our future manufacturers and suppliers will be able to reduce the costs of commercial scale manufacturing of any of our product candidates over time. If the commercial-scale manufacturing costs of any of our product candidates are higher than expected, these costs may significantly impact our operating results. In order to reduce costs, we may need to develop and implement process improvements. However, in order to do so, we will need, from time to time, to notify or make submissions with regulatory authorities, and the improvements may be subject to approval by such regulatory authorities. We cannot be sure that we will receive these necessary approvals or that these approvals will be granted in a timely fashion. We also cannot guarantee that we will be able to enhance and optimize output in our commercial manufacturing process. If we cannot enhance and optimize output, we may not be able to reduce our costs over time.
We have no experience manufacturing PRV-031. As a result, if we encounter manufacturing issues, delays or increased costs in the PROTECT study may occur.
We have no experience manufacturing PRV-031. We currently rely on a single third-party manufacturer to supply us with PRV-031 drug substance on a purchase order basis. In order to obtain regulatory approval for PRV-031, this third-party manufacturer will be required to consistently produce the active pharmaceutical ingredient used in PRV-031 in commercial quantities and of specified quality on a repeated basis and document its ability to do so. This is referred to as process validation. In addition, in November 2019 the FDA confirmed that it would require the demonstration of comparability between the study drug previously manufactured by MacroGenics and Eli Lilly and the to-be-commercialized drug substance and drug product scheduled for production by Provention and its contract manufacturing partners. If this third-party manufacturer is unable to satisfy this requirement, our business will be materially and adversely affected.
Our current PRV-031 drug supply will not fully supply the PROTECT study to completion and we will need to produce new drug supply to complete the PROTECT study and to demonstrate that the new drug supply is comparable to the old supply. Further, the PROTECT study will need to include data from a sufficient number of subjects dosed with this new drug supply in this clinical trial to demonstrate safety and efficacy in order to obtain regulatory approval. If our third-party manufacturer is unable to produce a sufficient and comparable new drug supply to complete the PROTECT study in a timely manner, we may be required to suspend the PROTECT study until sufficient amounts of the new drug supply are produced. This may result in increased clinical trial costs and delays in obtaining regulatory approval for PRV-031. Such delays would in turn delay the marketing and commercialization of PRV-031, which would materially and adversely affect our business.
Our third-party manufacturer has not made any lots of PRV-031 to date. The manufacturing processes for PRV-031 have never been tested at commercial scale, and the process validation requirement has not yet been satisfied. These manufacturing processes and our third-party manufacturer’s facility will be subject to inspection and approval by the FDA before we can commence the manufacture and sale of PRV-031. If our third-party manufacturer is unable to pass such inspection and otherwise satisfactorily complete the FDA approval regimen, our business will be materially and adversely affected.
Also, as we or any manufacturer we engage scales up manufacturing of any approved product, we may encounter unexpected issues relating to the manufacturing process or the quality, purity and stability of the product, and we may be required to refine or alter our manufacturing processes to address these issues. Resolving these issues could result in significant delays and may result in significantly increased costs. If we experience significant delays or other obstacles in producing any approved product for commercial scale, our ability to market and sell any approved products may be adversely affected and our business could suffer.
| 59 |
Changes in product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates are developed through preclinical studies to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and results. During the course of a development program, sponsors may also change the contract manufacturers used to produce the product candidates. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of clinical trials. Such changes may also require additional testing, notification or approval by the FDA, EMA or other regulatory authorities. This could delay completion of clinical trials; require the conduct of bridging clinical trials or studies, or the repetition of one or more clinical trials; increase clinical trial costs; delay approval of our product candidates and jeopardize our ability to commence product sales and generate revenue.
We expect to rely on third parties to conduct clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize any of our product candidates and our business would be substantially harmed.
We rely on third-party CROs and vendors to conduct and manage our clinical programs including contracting with clinical sites to perform our clinical trials. We plan to rely heavily on these parties for execution of clinical trials for our product candidates and will control only certain aspects of their activities. Nevertheless, we will be responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on CROs and clinical sites will not relieve us of our regulatory responsibilities. We and our CROs are required to comply with cGCPs, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for any products in clinical development. The FDA and its foreign equivalents enforce these cGCP regulations through periodic inspections of trial sponsors, principal investigators and trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA or other regulatory authorities will determine that any of our clinical trials comply with cGCPs. In addition, our clinical trials must be conducted with products produced under cGMP regulations and will require a large number of test subjects. Our failure or the failure of our CROs or clinical sites to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
Although we design the clinical trials for our product candidates in consultation with CROs, we expect that the CROs will manage all of the clinical trials conducted at contracted clinical sites. As a result, many important aspects of our drug development programs would be outside of our direct control. In addition, the CROs and clinical sites may not perform all of their obligations under arrangements with us or in compliance with regulatory requirements. If the CROs or clinical sites do not perform clinical trials in a satisfactory manner, breach their obligations to us or fail to comply with regulatory requirements, the development and commercialization of any of our product candidates for the subject indication may be delayed or our development program materially and irreversibly harmed. We cannot control the amount and timing of resources these CROs and clinical sites will devote to our program or any of our product candidates. If we are unable to rely on clinical data collected by our CROs, we could be required to repeat, extend the duration of, or increase the size of our clinical trials, which could significantly delay commercialization and require significantly greater expenditures.
If any of our relationships with these third-party CROs or clinical sites terminate, we may not be able to enter into arrangements with alternative CROs or clinical sites. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any such clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for any of our product candidates would be harmed, our costs could increase and our ability to generate revenue could be delayed.
| 60 |
Any termination or suspension of, or delays in the commencement or completion of, any necessary studies of any of our product candidates for any indications could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects.
The commencement and completion of clinical trials can be delayed for a number of reasons, including delays related to:
| ● | the FDA, EMA or a comparable foreign regulatory authority failing to grant permission to proceed and placing the clinical trial on hold; | |
| ● | subjects failing to enroll or remain in our trials at the rate we expect; | |
| ● | a facility manufacturing any of our product candidates being ordered by the FDA, EMA or other government or regulatory authorities to temporarily or permanently shut down due to violations of cGMP requirements or other applicable requirements, or cross-contaminations of product candidates in the manufacturing process; | |
| ● | any changes to our manufacturing process that may be necessary or desired; | |
| ● | subjects choosing an alternative treatment for the indications for which we are developing our product candidates, or participating in competing clinical studies; | |
| ● | subjects experiencing severe or unexpected drug-related adverse effects; | |
| ● | reports from clinical testing on similar technologies and products raising safety and/or efficacy concerns; | |
| ● | third-party clinical investigators losing their license or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or employing methods consistent with the clinical trial protocol, cGMP requirements, or other third parties not performing data collection and analysis in a timely or accurate manner; | |
| ● | inspections of clinical study sites by the FDA, comparable foreign regulatory authorities, or IRBs finding regulatory violations that require us to undertake corrective action, result in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study, or that prohibit us from using some or all of the data in support of our marketing applications; | |
| ● | third-party contractors becoming debarred or suspended or otherwise penalized by the FDA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or any of the data produced by such contractors in support of our marketing applications; | |
| ● | one or more IRBs refusing to approve, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | deviations of the clinical sites from trial protocols or dropping out of a trial; | |
| ● | adding new clinical trial sites; | |
| ● | the inability of our CRO to execute any clinical trials for any reason; and | |
| ● | government or regulatory delays or “clinical holds” requiring suspension or termination of a trial. |
Product development costs for any of our product candidates will increase if we have delays in testing or approval or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to the FDA, comparable foreign regulatory authorities, and IRBs for reexamination, which may impact the costs, timing or successful completion of that study. If we experience delays in completion of, or if we, the FDA or other regulatory authorities, the IRB, or other reviewing entities, or any of our clinical trial sites suspend or terminate any of our clinical trials of any of our product candidates, its commercial prospects may be materially harmed and our ability to generate product revenues will be delayed. Any delays in completing our clinical trials will increase our costs, slow down our development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. In addition, if one or more clinical trials are delayed, our competitors may be able to bring products to market before we do, and the commercial viability of any of our product candidates could be significantly reduced.
Even though we may obtain or apply for orphan drug designation for a product candidate, we may not be able to obtain orphan drug marketing exclusivity.
The treatment of recent-onset T1D is an orphan indication, and PRV-031 has been designated as an orphan drug for this indication by the FDA. Some of the subsets of lupus erythematosus are orphan indications (e.g., lupus nephritis).
| 61 |
However, there is no guarantee that the FDA, EMA or its foreign equivalents will grant any future application for orphan drug designation for any of our other product candidates, including PRV-031 in the At-Risk indication, which would make us ineligible for the additional exclusivity and other benefits of orphan drug designation.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States or for which there is no reasonable expectation that the cost of developing and making a drug available in the Unites States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA or a BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of regulatory review and approval process. In addition to the potential period of exclusivity, orphan designation makes a company eligible for grant funding of up to $400,000 per year for four years to defray costs of clinical trial expenses, tax credits for clinical research expenses and potential exemption from the FDA application user fee.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. There can be no assurance that we will receive orphan drug designation for any of our product candidates in the indications for which we think they might qualify, if we elect to seek such applications.
As a result, even though we have received orphan drug exclusivity for PRV-031 for the treatment of recent-onset T1D in the United States, the FDA can still approve other drugs that have a different active ingredient for use in treating the same indication. Furthermore, the FDA can waive orphan drug exclusivity if we are unable to manufacture sufficient supply of PRV-031 or if the FDA finds that a subsequent applicant for recent-onset T1D demonstrates clinical superiority to PRV-031.
Although we may pursue expedited regulatory approval pathways for a product candidate, it may not qualify for expedited development or, if it does qualify for expedited development, it may not actually lead to a faster development or regulatory review or approval process.
Although we believe there may be an opportunity to accelerate the development of certain of our product candidates through one or more of the FDA’s expedited programs, such as fast track, breakthrough therapy, accelerated approval or priority review, we cannot be assured that any of our product candidates will qualify for such programs.
For example, a drug may be eligible for designation as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Although breakthrough designation or access to any other expedited program may expedite the development or approval process, it does not change the standards for approval. If we apply for breakthrough therapy designation or any other expedited program for our product candidates, the FDA may determine that our proposed target indication or other aspects of our clinical development plans do not qualify for such expedited program. Even if we are successful in obtaining a breakthrough therapy designation or access to any other expedited program, we may not experience faster development timelines or achieve faster review or approval compared to conventional FDA procedures. For example, the time required to identify and resolve issues relating to chemistry, manufacturing and controls, the acquisition of a sufficient supply of our product for clinical trial purposes or the need to conduct additional preclinical or clinical studies may delay approval by the FDA, even if the product candidate qualifies for a breakthrough therapy designation or access to any other expedited program. Access to an expedited program may also be withdrawn by the FDA if it believes that the designation is no longer supported by data from our clinical development program. Additionally, qualification for any expedited review procedure does not ensure that we will ultimately obtain regulatory approval for such product candidate.
| 62 |
Third-party coverage and reimbursement and health care cost containment initiatives and treatment guidelines may constrain our future revenues.
Our ability to successfully market our product candidates will depend in part on the level of reimbursement that government health administration authorities, private health coverage insurers and other organizations provide for the cost of our products and related treatments. Countries in which any of our product candidates are sold through reimbursement schemes under national health insurance programs frequently require that manufacturers and sellers of pharmaceutical products obtain governmental approval of initial prices and any subsequent price increases. In certain countries, including the United States, government-funded and private medical care plans can exert significant indirect pressure on prices. We may not be able to sell our product candidates profitably if adequate prices are not approved or coverage and reimbursement is unavailable or limited in scope. Increasingly, third-party payors attempt to contain health care costs in ways that are likely to impact our development of products including:
| ● | failing to approve or challenging the prices charged for health care products; | |
| ● | introducing reimportation schemes from lower priced jurisdictions; | |
| ● | limiting both coverage and the amount of reimbursement for new therapeutic products; | |
| ● | denying or limiting coverage for products that are approved by the regulatory agencies but are considered to be experimental or investigational by third-party payors; and | |
| ● | refusing to provide coverage when an approved product is used in a way that has not received regulatory marketing approval. |
Moreover, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their commercial products. There have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the cost of drugs under Medicare, and reform government program reimbursement methodologies for drugs. While any proposed measures will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Risks Relating to Our Intellectual Property Rights
We depend on rights to certain pharmaceutical compounds that are licensed to us. We do not control these pharmaceutical compounds and any loss of our rights to them could prevent us from selling our products.
We are dependent on licenses from third parties for all but one of our pharmaceutical compounds. We do not own the patents that underlie these licenses. Our rights to use the pharmaceutical compounds we license are subject to the continuation of and compliance with the terms of those licenses. Thus, the patents and patent applications applicable to our product candidates were not written by us or our attorneys, and we did not have control over the drafting and prosecution. The former patent owners and our licensors might not have given the same attention to the drafting and prosecution of these patents and applications as we would have if we had been the owners of the patents and applications and had control over the drafting. Moreover, under certain of our licenses, patent prosecution activities remain under the control of the licensor. We cannot be certain that drafting of the licensed patents and patent applications, or patent prosecution, by the licensors have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents and other intellectual property rights.
Our rights to develop and commercialize the product candidates we license are subject to the validity of the owner’s intellectual property rights. Enforcement of our licensed patents or defense or any claims asserting the invalidity of these patents is often subject to the control or cooperation of our licensors. Legal action could be initiated against the owners of the intellectual property that we license and an adverse outcome in such legal action could harm our business because it might prevent such companies or institutions from continuing to license intellectual property that we may need to operate our business. In addition, such licensors may resolve such litigation in a way that benefits them but adversely affects our ability to develop and commercialize our product candidates.
| 63 |
In addition, our rights to practice the inventions claimed in the licensed patents and patent applications are subject to our licensors abiding by the terms of those licenses and not terminating them. Our licenses may be terminated by the licensor if we are in material breach of certain terms or conditions of the license agreement or in certain other circumstances. Certain of our licenses contained in our agreements with Janssen and Vactech contain provisions that allow the licensor to terminate the license if (i) we breach any payment obligation or other material provision under the agreement and fail to cure the breach within a fixed time following written notice of termination, (ii) we or any of our affiliates, licensees or sublicensees directly or indirectly challenge the validity, enforceability, or extension of any of the licensed patents, (iii) we declare bankruptcy or dissolve, (iv) we fail to maintain a licensed product in active development or fail to use commercially reasonable efforts to develop or commercialize a licensed product. Our rights under the licenses are subject to our continued compliance with the terms of the license, including the payment of royalties due under the license. Termination of these licenses could prevent us from marketing some or all of our products. Because of the complexity of our products and the patents we have licensed, determining the scope of the license and related royalty obligations can be difficult and can lead to disputes between us and the licensor. An unfavorable resolution of such a dispute could lead to an increase in the royalties payable pursuant to the license. If a licensor believed we were not paying the royalties due under the license or were otherwise not in compliance with the terms of the license, the licensor might attempt to revoke the license. If such an attempt were successful, we might be barred from producing and selling some or all of our products.
It is difficult and costly to protect our intellectual property rights, and we cannot ensure the protection of these rights.
Our commercial success will depend, in part, on obtaining and maintaining patent protection for our technologies, products and processes, successfully defending these patents against third-party challenges and successfully enforcing these patents against third party competitors. The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal, scientific and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowable or enforceable in our patents. We currently own 8 issued patents, license 389 issued patents, and license 149 pending patents for our product candidates, in which the pending applications may never be approved by United States or foreign patent offices. The existing patents and patent applications relating to our product candidates and related technologies may be challenged, invalidated or circumvented by third parties and might not protect us against competitors with similar products or technologies.
The degree of future protection for our proprietary rights is uncertain, because legal means afford only limited protection and may not adequately protect our rights, permit us to gain or keep our competitive advantage, or provide us with any competitive advantage at all. For example, others have filed, and in the future are likely to file, patent applications covering products and technologies that are similar, identical or competitive to any of our product candidates, or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed by us, or that we will not be involved in interference, opposition or invalidity proceedings before United States or foreign patent offices.
In the future we may rely on know-how and trade secrets to protect technology, especially in cases when we believe patent protection is not appropriate or obtainable. However, know-how and trade secrets are difficult to protect. While we intend to require employees, academic collaborators, consultants and other contractors to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary or licensed information. Typically, research collaborators and scientific advisors have rights to publish data and information in which we may have rights. If we cannot maintain the confidentiality of our proprietary technology and other confidential information, our ability to receive patent protection and our ability to protect valuable information owned by us may be imperiled. Enforcing a claim that a third-party entity illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts are sometimes less willing to protect trade secrets than patents. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
If we fail to obtain or maintain patent protection or trade secret protection for our product candidates or our technologies, third parties could use our proprietary information, which could impair our ability to compete in the market and adversely affect our ability to generate revenues and attain profitability.
| 64 |
We may also rely on the trademarks we may develop to distinguish our products from the products of our competitors. We cannot guarantee that any trademark applications filed by us or our licensors will be approved. Third parties may also oppose such trademark applications, or otherwise challenge our use of the trademarks. In the event that the trademarks we use are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. Further, we cannot provide assurance that competitors will not infringe the trademarks we use, or that we will have adequate resources to enforce these trademarks.
Our product candidates may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our development and commercialization efforts.
Our success depends in part on avoiding infringement of the proprietary technologies of others. The pharmaceutical industry has been characterized by frequent litigation regarding patent and other intellectual property rights. Identification of third-party patent rights that may be relevant to our proprietary technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. Additionally, because patent applications are maintained in secrecy until the application is published, we may be unaware of third-party patents that may be infringed by commercialization of any of our product candidates or any future product candidate. There may be certain issued patents and patent applications claiming subject matter that we may be required to license in order to research, develop or commercialize any of our product candidates, and we do not know if such patents and patent applications would be available to license on commercially reasonable terms, or at all. Any claims of patent infringement asserted by third parties would be time-consuming and may:
| ● | result in costly litigation; | |
| ● | divert the time and attention of our technical personnel and management; | |
| ● | prevent us from commercializing a product until the asserted patent expires or is held finally invalid or not infringed in a court of law; | |
| ● | require us to cease or modify our use of the technology and/or develop non-infringing technology; or | |
| ● | require us to enter into royalty or licensing agreements. |
Third parties may hold proprietary rights that could prevent any of our product candidates from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin commercial activities relating to any of our product candidates or our processes could subject us to potential liability for damages and require us to obtain a license to continue to manufacture or market any of our product candidates or any future product candidates. We cannot predict whether we would prevail in any such actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all. In addition, we cannot be sure that we could redesign our product candidates or any future product candidates or processes to avoid infringement, if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from developing and commercializing any of our product candidates or a future product candidate, which could harm our business, financial condition and operating results.
A number of companies, including several major pharmaceutical companies, have conducted, or are conducting, research in immune-mediated diseases within the therapeutic fields in which we intend to operate, which has resulted, or may result, in the filing of many patent applications related to this research. If we were to challenge the validity of these or any issued United States patent in court, we would need to overcome a statutory presumption of validity that attaches to every issued United States patent. This means that, in order to prevail, we would have to present clear and convincing evidence as to the invalidity of the patent’s claims. If we were to challenge the validity of these or any issued United States patent in an administrative trial before the Patent Trial and Appeal Board in the United States Patent and Trademark Office, we would have to prove that the claims are unpatentable by a preponderance of the evidence. There is no assurance that a jury and/or court would find in our favor on questions of infringement, validity or enforceability.
We may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used or disclosed alleged confidential information or trade secrets of their former employers.
As is commonplace in our industry, we will employ individuals who were previously employed at other pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject in the future to claims that our employees or prospective employees are subject to a continuing obligation to their former employers (such as non-competition or non-solicitation obligations) or claims that our employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
| 65 |
General Company-Related Risks
We will need to grow the size of our organization, and we may experience difficulties in managing this growth.
As our development and commercialization plans and strategies continue to develop, we intend to expand the size of our employee and consultant/contractor base. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate and integrate additional employees. In addition, our management may have to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Our future financial performance and our ability to develop and commercialize our product candidates and any other future product candidates and our ability to compete effectively will depend, in part, on our ability to effectively manage our future growth.
Future capital raises may dilute our existing stockholders’ ownership and/or have other adverse effects on our operations.
If we raise additional capital by issuing equity securities, our existing stockholders’ percentage ownership will be reduced, and these stockholders may experience substantial dilution. We may also issue equity securities that provide for rights, preferences and privileges senior to those of our common stock. If we raise additional funds by issuing debt securities, these debt securities would have rights senior to those of our common stock and the terms of the debt securities issued could impose significant restrictions on our operations, including liens on our assets. If we raise additional funds through collaborations and licensing arrangements, we may be required to relinquish some rights to our technologies or candidate products, or to grant licenses on terms that are not favorable to us.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy. In addition, the loss of the services of our co-founders would adversely impact our business prospects.
Our management team has expertise in many different aspects of drug development and commercialization. However, our ability to compete in the highly competitive pharmaceuticals industry depends in large part upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We will need to hire additional personnel as we further develop our product candidates. Competition for skilled personnel in our market is intense and competition for experienced scientists may limit our ability to hire and retain highly qualified personnel on acceptable terms. Despite our efforts to retain valuable employees, members of our management, scientific and medical teams may terminate their employment with us on short notice. We have entered into employment agreements with certain of our executive officers. However, these employment arrangements will provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. Moreover, there can be no assurance that anyone we expect to employ in a key management position will be available to join our team when we expect them to, if at all. The loss of the services of any of our executive officers or other key employees, or our inability to hire targeted executives, could potentially harm our business, operating results or financial condition. In particular, we believe that the loss of the services of our co-founders would have a material adverse effect on our business. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level, and senior managers as well as junior, mid-level, and senior scientific and medical personnel.
Other pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles, and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success at which we can develop and commercialize product candidates would be limited.
| 66 |
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face a potential risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any of our product candidates or any other future product. For example, we may be sued if any product we develop, including any of our product candidates, or any materials that we use in our products allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. In the US, claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for any of our product candidates or any future products that we may develop; | |
| ● | injury to our reputation; | |
| ● | withdrawal of clinical trial participants; | |
| ● | costs to defend the related litigation; | |
| ● | a diversion of management’s time and our resources; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
| ● | the inability to commercialize some or all of our product candidates; and | |
| ● | a decline in the value of our stock. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. We maintain product liability insurance covering our clinical trials. Although we will maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction.
We are a virtual company and may be unable to adequately protect our information technology systems from cyber-attacks, which could result in the disclosure of confidential information, damage our reputation, and subject us to significant financial and legal exposure.
We are a virtual company and may be unable to adequately protect our information technology systems from cyber-attacks, which could result in the disclosure of confidential information, damage our reputation, and subject us to significant financial and legal exposure.
Cyber-attacks are increasing in their frequency, sophistication and intensity, and have become increasingly difficult to detect. Cyber-attacks could include wrongful conduct by hostile foreign governments, industrial espionage, deployment of harmful malware, denial-of-service, and other means to threaten data confidentiality, integrity and availability. A successful cyber-attack could cause serious negative consequences for our company, including the disruption of operations, the misappropriation of confidential business information and trade secrets, and the disclosure of corporate strategic plans. To date, we have not experienced threats to our data and information technology systems. However, although we devote resources to protect our information technology systems, we realize that cyber-attacks are a threat, and there can be no assurance that our efforts will prevent information security breaches that would result in business, legal or reputational harm to us, or would have a material adverse effect on our operating results and financial condition.
| 67 |
We rely on the proper function, availability and security of our information technology systems to operate our business and a cyber-attack or other breach or disruption of these systems could have a material adverse effect on our business and results of operations.
We rely on information technology systems to process, transmit and store electronic information in our day-to-day operations. The form and function of such systems may change over time as our business needs change. The nature of our business involves the receipt and storage of personal and financial information regarding our customers. We use our information technology systems to manage or support a variety of business processes and activities, including sales, procurement and supply chain, manufacturing and accounts payable. In addition, we use enterprise information technology systems to record, process, and summarize transactions and other financial information and results of operations for internal reporting purposes and to comply with regulatory financial reporting, legal, and tax requirements. Our information technology systems may be susceptible to damage, disruptions or shutdowns due to computer viruses, attacks by computer hackers, failures during the process of upgrading or replacing software, databases or components thereof, power outages, hardware failures, telecommunication failures, user errors or catastrophic events. Any failure by us to maintain or protect our information technology systems and data integrity, including from cyber-attacks, intrusions, disruptions or shutdowns, could result in the unauthorized access to personally identifiable information, theft of intellectual property or other misappropriation of assets or the loss of key data and information, or otherwise compromise our confidential or proprietary information and disrupt our operations. If our information technology systems are breached or suffer severe damage, disruption or shutdown and we are unable to effectively resolve the issues in a timely manner, our business and operating results may be materially and adversely affected.
If our efforts to maintain the privacy and security of our patient, employee, supplier or Company information are not successful, we could incur substantial additional costs and become subject to litigation, enforcement actions and reputational damage.
Our business, like that of most biopharmaceutical companies, involves the receipt, storage and transmission of patient information, as well as confidential information about our employees, our suppliers and our Company. Our information systems are vulnerable to an increasing threat of continually evolving cybersecurity risks. Unauthorized parties may attempt to gain access to our systems or information through fraud or other means of deceiving our employees or third-party service providers. Hardware, software or applications we develop or obtain from third parties may contain defects in design or manufacture or other problems that could unexpectedly compromise information and device security. The methods used to obtain unauthorized access, disable or degrade service or sabotage systems are also constantly changing and evolving, and may be difficult to anticipate or detect for long periods of time. The ever-evolving threats mean we must continually evaluate and adapt our systems and processes, and our efforts may not be adequate to safeguard against all data security breaches, misuse of data or sabotage of our systems. Any future significant compromise or breach of our data security, whether external or internal, or misuse of patient, employee, supplier or Company data, could result in additional significant costs, lost sales, fines, lawsuits and damage to our reputation. In addition, as the regulatory environment related to information security, data collection and use, and privacy becomes increasingly rigorous, with new and constantly changing requirements applicable to our business, compliance with those requirements could also result in additional costs.
We face business disruption and related risks resulting from the recent outbreak of the novel coronavirus 2019 (COVID-19), which could have a material adverse effect on our business plan.
The development of our product candidates could be disrupted and materially adversely affected by the recent outbreak of COVID-19. As a result of measures imposed by the governments in affected regions, businesses and schools have been suspended due to quarantines intended to contain this outbreak. The spread of COVID-19 from China to other countries has resulted in the Director General of the World Health Organization declaring the outbreak of COVID-19 as a Public Health Emergency of International Concern (PHEIC), based on the advice of the Emergency Committee under the International Health Regulations (2005), and the Centers for Disease Control and Prevention in the U.S. issued a warning on February 25, 2020 regarding the likely spread of COVID-19 to the U.S. While the COVID-19 outbreak is still in very early stages, international stock markets have begun to reflect the uncertainty associated with the slow-down in the Chinese economy and the reduced levels of international travel experienced since the beginning of January and the significant declines in the Dow Industrial Average at the end of February and beginning of March 2020 was largely attributed to the effects of COVID-19. We are still assessing our business plans and the impact COVID-19 may have on our ability to recruit candidates for clinical trials or to raise financing to support the development of our product candidates, but there can be no assurance that this analysis will enable us to avoid part or all of any impact from the spread of COVID-019 or its consequences, including downturns in business sentiment generally or in our sector in particular.
Risks Related to Ownership of our Common Stock
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for stockholders.
The market price of our common stock has been volatile and can be subject to wide fluctuations in response to various factors, some of which are beyond our control, including, the reporting of results of our clinical trials or partner-sponsored clinical trials involving our programs. These factors include those discussed in this “Risk Factors” section of this Annual Report on Form 10-K and others such as:
| ● | our commercialization, marketing and manufacturing prospects; | |
| ● | our intentions and our ability to establish collaborations and/or partnerships; | |
| ● | the timing or likelihood of regulatory filings and approvals; | |
| ● | our development, commercialization, marketing and manufacturing capabilities; |
| 68 |
| ● | our expectations regarding the potential market size and the size of the patient populations for our product candidates; | |
| ● | the implementation of our business model and strategic plans for our business and technology; | |
| ● | the scope of protection we are able to establish and maintain for intellectual property rights covering our product candidates, along with any product modifications and improvements; | |
| ● | estimates of our expenses, future revenue, capital requirements, our needs for additional financing and our ability to obtain additional capital; | |
| ● | our financial performance; and | |
| ● | developments and projections relating to our competitors and our industry, including competing therapies and procedures. |
In addition, the stock markets in general, and the markets for biopharmaceutical and biotechnology stocks in particular, have experienced extreme volatility that may have been unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the market price or liquidity of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
An active, liquid and orderly market for our common stock may not develop, which could result in substantial losses for stockholders.
Prior to our IPO, there was no public market for shares of our common stock. Although our common stock is listed on The Nasdaq Global Select Market, or Nasdaq, the market for our shares has demonstrated varying levels of trading activity and an active public market for our shares may not be sustained. The lack of an active market may impair the ability to sell shares at the time a shareholder wish to sell them or at a price that a shareholder may consider reasonable. An inactive market may also impair our ability to raise capital by selling shares and may impair our ability to acquire other businesses, applications, or technologies using our shares as consideration.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our stock, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. We only recently obtained research coverage by securities and industry analysts. If there are insufficient securities or industry analysts covering us, the market price for our stock would be negatively impacted. If any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our clinical trials and operating results fail to meet the expectations of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
Our failure to meet the continued listing requirements of Nasdaq could result in a delisting of our common stock.
If we fail to satisfy the continued listing requirements of Nasdaq, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock. Such a delisting would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with Nasdaq’s listing requirements.
| 69 |
We are an “emerging growth company” and as a result of the reduced disclosure and governance requirements applicable to emerging growth companies, our common stock may be less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. Investors may be unable to compare our business with other companies in our industry if they believe that our reporting is not as transparent as other companies in our industry. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of our IPO, (b) in which we have total annual gross revenue of at least $1.07 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
If we sell shares of our common stock in future financings, stockholders may experience immediate dilution and, as a result, our stock price may decline.
We may from time to time issue additional shares of common stock at a discount from the current market price of our common stock. As a result, our stockholders would experience immediate dilution upon the purchase of any shares of our common stock sold at such discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred stock or common stock. If we issue common stock or securities convertible into common stock, our common stockholders would experience additional dilution and, as a result, our stock price may decline.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates beneficially owned approximately 28% of our voting stock as of December 31, 2019. Therefore, these stockholders will have the ability to influence us through this ownership position. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to decline.
Sales of a substantial number of shares of our common stock in the public market could cause the market price of our common stock to decline. If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the market price of our common stock could decline.
In addition, as of December 31, 2019, approximately 8,955,000 shares of common stock were subject to outstanding options, reserved for future issuance under our equity incentive plans or subject to outstanding warrants. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the market price of our common stock could decline.
We are incurring significant increased costs as a result of becoming a public company that reports to the Securities and Exchange Commission (SEC) and our management is required to devote substantial time to meet compliance obligations.
As a newly public company, we are incurring significant legal, accounting and other expenses that we did not incur as a private company. We are subject to reporting requirements of the Exchange Act and the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and Nasdaq that impose significant requirements on public companies, including requiring the establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. In addition, on July 21, 2010, the Dodd-Frank Wall Street Reform and Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Act that are expected to increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and may also place undue strain on our personnel, systems and resources. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. In addition, these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified people to serve on our board of directors, our board committees or as executive officers.
| 70 |
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our amended and restated certificate of incorporation and bylaws contains provisions that could delay or prevent changes in control or changes in our management without the consent of our board of directors. These provisions include the following:
| ● | no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; | |
| ● | the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; | |
| ● | the ability of our board of directors to authorize the issuance of shares of preferred stock and to determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer; | |
| ● | the ability of our board of directors to amend our bylaws without obtaining stockholder approval; | |
| ● | the required approval of at least 66 2/3% of the shares entitled to vote at an election of directors to adopt, amend or repeal our bylaws or repeal the provisions of our amended and restated certificate of incorporation regarding the election and removal of directors; | |
| ● | the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and | |
| ● | advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us. |
In addition, these provisions would apply even if we were to receive an offer that some stockholders may consider beneficial.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation may not, in general, engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful third-party claims against us and may reduce the amount of money available to us.
Our amended and restated certificate of incorporation and bylaws provide that we will indemnify our directors and officers, in each case to the fullest extent permitted by Delaware law.
In addition, as permitted by Section 145 of the Delaware General Corporation Law, our bylaws became effective immediately prior to the completion of the IPO and our indemnification agreements that we have entered into with our directors and officers provide that:
| ● | we will indemnify our directors and officers for serving us in those capacities or for serving other business enterprises at our request, to the fullest extent permitted by Delaware law. Delaware law provides that a corporation may indemnify such person if such person acted in good faith and in a manner such person reasonably believed to be in or not opposed to the best interests of the registrant and, with respect to any criminal proceeding, had no reasonable cause to believe such person’s conduct was unlawful; | |
| ● | we may, in our discretion, indemnify employees and agents in those circumstances where indemnification is permitted by applicable law; |
| 71 |
| ● | we are required to advance expenses, as incurred, to our directors and officers in connection with defending a proceeding, except that such directors or officers shall undertake to repay such advances if it is ultimately determined that such person is not entitled to indemnification; | |
| ● | we will not be obligated pursuant to our bylaws to indemnify a person with respect to proceedings initiated by that person against us or our other indemnitees, except with respect to proceedings authorized by our board of directors or brought to enforce a right to indemnification; | |
| ● | the rights conferred in our bylaws are not exclusive, and we are authorized to enter into indemnification agreements with our directors, officers, employees and agents and to obtain insurance to indemnify such persons; and | |
| ● | we may not retroactively amend our bylaw provisions to reduce our indemnification obligations to directors, officers, employees and agents. |
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on our behalf, any action asserting a breach of fiduciary duty, any action asserting a claim against us arising pursuant to the Delaware General Corporation Law, our certificate of incorporation or our bylaws, any action to interpret, apply, enforce, or determine the validity of our certificate of incorporation or bylaws, or any action asserting a claim against us that is governed by the internal affairs doctrine. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. As a result, the exclusive forum provision will not apply to suits brought to enforce any duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. In addition, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. As a result, the exclusive forum provision will not apply to suits brought to enforce any duty or liability created by the Securities Act or any other claim for which the federal and state courts have concurrent jurisdiction.
The choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find the choice of forum provision contained in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
We do not intend to pay dividends on our common stock, and, consequently, the ability to achieve a return on investment will depend on appreciation in the price of our common stock.
We do not intend to pay any cash dividends on our common stock for the foreseeable future. We intend to invest our future earnings, if any, to fund our growth. Therefore, shareholders are not likely to receive any dividends on their common stock for the foreseeable future. Since we do not intend to pay dividends, the ability to receive a return on investment will depend on any future appreciation in the market value of our common stock. There is no guarantee that our common stock will appreciate or even maintain the price at which our holders have purchased it.
If we are unable to implement and maintain effective internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our securities may decrease.
Beginning with our annual report for the year ending December 31, 2019, Section 404 of the Sarbanes-Oxley Act will require that we evaluate and determine the effectiveness of our internal control over financial reporting and provide a management report on our internal control over financial reporting which will need to be attested to by our independent registered public accounting firm once our status as an emerging growth company ceases.
If we are unable to comply with the requirements of Section 404 in a timely manner, if we are unable to assert that our internal control over financial reporting is effective or, once required, provide an attestation report from our independent registered public accounting firm, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could decrease. We could also become subject to stockholder or other third-party litigation as well as investigations by the stock exchange on which our securities are listed, the SEC or other regulatory authorities, which could require additional financial and management resources and could result in fines, trading suspensions or other remedies.
ITEM 1B. Unresolved Staff Comments
None.
We are a virtual company and do not own or lease any office space. We maintain a mailing address at P.O. Box 666, Oldwick, NJ 08858.
We are not a party to any pending legal proceedings.
ITEM 4. Mine Safety Disclosures
Not applicable.
| 72 |
ITEM 5. Market for the Registrant’s Common Equity, Related Shareholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock was publicly traded under the symbol “PRVB” on The Nasdaq Capital Market from July 24, 2018 until December 22, 2019, at which time our common stock was transitioned to and began trading on The Nasdaq Global Select Market. Prior to July 24, 2018, there was no public market for our common stock.
Holders of Record
As of March 1, 2020, there were approximately 21 holders of record of our common stock. This number does not include beneficial owners whose shares are held by nominees in street name. The actual number of holders of our Common Stock is greater than this number of record holders and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers or held by other nominees.
Dividends
We have never declared or paid cash dividends on our common stock, and we do not expect to pay any cash dividends on our common stock in the foreseeable future. Any future determination to pay dividends will be made at the discretion of our board of directors and will depend on various factors, including applicable laws, our results of operations, financial condition, future prospects, then applicable contractual restrictions and any other factors deemed relevant by our board of directors.
Equity Compensation Plans
The information required by this item will be set forth in our Proxy Statement for the 2020 Annual Meeting of Stockholders and is incorporated in this Annual Report on Form 10-K by reference.
Recent Sales of Unregistered Securities
During the year ended December 31, 2019, there were no sales of our securities that were not reported in a Current Report on Form 8-K or our Quarterly Report on Form 10-Q.
Use of Proceeds from Initial Public Offering of Common Stock
The offer and sale of the shares in the IPO was registered under the Securities Act pursuant to registration statements on Form S-1 (File No. 333-224801), which was filed with the Securities and Exchange Commission, or the SEC, on May 9, 2018 and amended subsequently and declared effective on July 3, 2018, and Form S-1MEF (File No. 333-226198), which was filed with the SEC on July 16, 2018 and was effective on filing with the SEC. These registration statements, collectively, registered a maximum offering amount of $65.5 million of our common stock in connection with our IPO, pursuant to which we sold 15,969,563 shares of common stock to the public at a price of $4.00 per share. The IPO closed on July 19, 2018, generating net proceeds of approximately $59.3 million after deducting underwriting commissions of approximately $3.7 million and other offering expenses of $0.8 million payable by us. The offering terminated before all of the securities registered on the registration statements had been sold.
| 73 |
MDB Capital Group, LLC acted as sole book-running manager for the offering. Dougherty & Company LLC acted as qualified independent underwriter for the offering. Anthony DiGiandomenico, a member of our board of directors, is a co-founder of MDB.
As of December 31, 2019, we have used approximately $46.8 million of the net offering proceeds primarily to fund ongoing development activities for PRV-6527, PRV-300, PRV-031, PRV-101, PRV-3279 and for working capital and other general corporate purposes. We are holding a significant portion of the balance of the net proceeds from the offering in a variety of capital preservation investments, including short-term, investment grade, interest bearing investments in U.S. government securities. There has been no material change in our planned use of the net proceeds from the offering as described in our final prospectus filed with the SEC on July 17, 2018 pursuant to Rule 424(b) under the Securities Act.
We have not used any of the net proceeds from the offering to make payments by us, directly or indirectly, to any director or officer of ours (other than payment in the ordinary course of business of salaries and/or director fees) or to any of their associates, or to any persons owning ten percent or more of our common stock or to their associates, or to our affiliates, other than the underwriting commission paid to MDB Capital Group and underwriter warrants issued to MDB.
Issuer Purchases of Equity Securities
There were no repurchases of equity securities during the fourth quarter of 2019.
ITEM 6. Selected Financial Data
The following selected financial data should be read together with our financial statements and the related notes appearing elsewhere in this Annual Report on Form 10-K and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section of this Annual Report on Form 10-K. We have derived the statements of operations data for the years ended December 31, 2019, 2018, 2017 and for the period from October 4, 2016 (inception) through December 31, 2016 and the balance sheet data as of December 31, 2019, 2018, 2017 and 2016 from our audited financial statements included elsewhere in this Annual Report on Form 10-K, which have been audited by EisnerAmper LLP, an independent registered accounting firm. Our historical results for any prior period are not necessarily indicative of results to be expected in any future period, and our results for any interim period are not necessarily indicative of results to be expected for a full fiscal year.
| For the Years Ended December 31, | For
Period October 4, 2016 (inception) through December 31, | |||||||||||||||
| 2019 | 2018 | 2017 | 2016 | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 36,359 | $ | 22,649 | $ | 7,684 | $ | — | ||||||||
| General and administrative | 8,013 | 4,165 | 1,456 | 165 | ||||||||||||
| Total operating expenses | 44,372 | 26,814 | 9,140 | 165 | ||||||||||||
| Loss from operations | (44,372 | ) | (26,814 | ) | (9,140 | ) | (165 | ) | ||||||||
| Interest income | 1,087 | 669 | 132 | — | ||||||||||||
| Change in fair value of warrant liability | — | (520 | ) | (125 | ) | — | ||||||||||
| Loss before income tax benefit | (43,285 | ) | (26,665 | ) | (9,133 | ) | (165 | ) | ||||||||
| Income tax benefit | — | 187 | — | — | ||||||||||||
| Net loss | (43,285 | ) | (26,478 | ) | (9,133 | ) | (165 | ) | ||||||||
| Accretion on Series A Convertible Redeemable Preferred Stock | — | (276 | ) | (343 | ) | — | ||||||||||
| Net loss attributable to common stockholders | $ | (43,285 | ) | $ | (26,754 | ) | $ | (9,476 | ) | $ | (165 | ) | ||||
| Net loss per common share, basic and diluted | $ | (1.06 | ) | $ | (1.19 | ) | $ | (1.01 | ) | $ | (0.02 | ) | ||||
| Weighted average common shares outstanding, basic and diluted | 40,747 | 22,441 | 9,370 | 7,836 | ||||||||||||
| 74 |
| December 31, | ||||||||||||||||
| 2019 | 2018 | 2017 | 2016 | |||||||||||||
| (in thousands) | ||||||||||||||||
| Balance Sheet Data: | ||||||||||||||||
| Cash, cash equivalents and marketable securities | $ | 85,373 | $ | 58,539 | $ | 21,834 | $ | — | ||||||||
| Total assets | 85,996 | 61,529 | 22,428 | 125 | ||||||||||||
| Total liabilities | 3,840 | 1,871 | 2,276 | 290 | ||||||||||||
| Series A Convertible Redeemable Preferred Stock | — | — | 26,185 | — | ||||||||||||
| Accumulated deficit | (79,061 | ) | (35,776 | ) | (9,298 | ) | (165 | ) | ||||||||
| Total stockholders’ equity (deficit) | $ | 82,156 | $ | 59,658 | $ | (6,033 | ) | $ | (165 | ) | ||||||
ITEM 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Overview
We are a clinical stage biopharmaceutical company, focused on the development and commercialization of novel therapeutics and innovative approaches aimed at intercepting and preventing immune-mediated diseases. Since our inception, we have devoted substantially all of our efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets partnering and raising capital. We have not yet commenced any revenue-generating operations, do not have any positive cash flows from operations and we will need to raise additional capital to finance our operations. Our business is subject to significant risks and uncertainties and we will be dependent on raising substantial additional capital before we become profitable and we may never achieve profitability.
We have not generated any revenue to date and through December 31, 2019, we had an accumulated deficit of $79.1 million. We have financed our operations through a private offering of Series A Convertible Redeemable Preferred Stock in April 2017, our initial public offering, or IPO in July 2018 and our underwritten public offering and concurrent private placement in September 2019.
In April 2017, we completed our private placement of Series A Convertible Redeemable Preferred Stock. We issued an aggregate 11,381,999 shares of Series A Convertible Redeemable Preferred Stock at $2.50 per share. We received net proceeds of $26.7 million.
In July 2018, we issued and sold an aggregate of 15,969,563 shares of common stock in our IPO at a public offering price of $4.00 per share. In connection with the IPO, we issued to MDB, the underwriter in the IPO, and its designees warrants to purchase 1,596,956 shares of Common Stock at an exercise price of $5.00 per share. We received net proceeds from the IPO of $59.3 million, after deducting underwriting discounts and commissions of approximately $3.7 million and other offering expenses of approximately $0.8 million. Upon the closing of the IPO, all of our shares of Series A Convertible Redeemable Preferred Stock outstanding at the time of the IPO were automatically converted into 11,381,999 shares of common stock. In addition, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock also converted to warrants for the purchase of 558,740 shares of our common stock.
In September 2019, we completed an underwritten public offering in which we sold 5,750,000 shares of common stock at a public offering price of $8.00 per share. The 5,750,000 shares sold included the full exercise of the underwriters’ option to purchase 750,000 shares at a price of $8.00 per share. Concurrent with the underwritten public offering, we sold 2,500,000 shares of common stock to Amgen, Inc. at the public offering price of $8.00 per share in a private placement, pursuant to the terms of our License and Collaboration Agreement with Amgen Inc, dated as of November 5, 2018. Aggregate net proceeds from the underwritten public offering and the concurrent private placement were $62.7 million, net of approximately $2.8 million in underwriting discounts and commissions and other offering expenses of $0.5 million.
We expect that over the next several years we will continue to incur losses from operations as we increase our expenditures in research and development in connection with our planned BLA submission and our commercialization efforts for PRV-031 in the At-Risk indication, as well as our ongoing and planned clinical trials and other development activities. If adequate funds are not available to us on a timely basis, or at all, we may be required to terminate or delay certain development activities.
| 75 |
Our Focus and Pipeline
Inflammation is a natural consequence of most infections as it is the immune system’s first response to invading pathogens in the event of injury or acute illness. Most of the time, this response is beneficial and well-controlled; helping to repair tissue damage and clear pathogens from the body. In addition to directly damaging tissues and organs, an infection can sometimes result in the excessive release of toxic immune mediators leading to a potentially life-threatening acute pathological immune response. When patients have the requisite genetic predisposition, infections can also trigger chronic autoimmune responses that persist and progress long after the original insult has subsided. These sustained responses have been linked to an increased susceptibility to chronic debilitating and potentially life-threatening diseases like inflammatory bowel disease, diabetes, cancer, and certain neurological disorders.
Our “predict” and “preempt” therapeutic approach is to intercept the underlying pathological immune and inflammatory responses in susceptible individuals. Our pipeline includes:
| ● | PRV-031: a humanized, anti-CD3 mAb for the interception of T1D in pediatric patients with newly-diagnosed T1D and for delaying and/or preventing disease progression in subjects at risk of developing clinical stage T1D. PRV-031 has been designated by the FDA as orphan drug for the treatment of newly-diagnosed T1D. PRV-031 was granted breakthrough therapy designation from the FDA in August 2019 and PRIME eligibility from the EMA in October 2019 for the delay or prevention of T1D; | |
| ● | PRV-101: a CVB vaccine to prevent acute CVB infections and, in those patients at risk, preventing the CVB-triggered autoimmune damage to pancreatic beta cells that progress to T1D and damage to intestinal cells that lead to celiac disease; | |
| ● | PRV-3279: a humanized bispecific scaffold molecule targeting the B-cell surface proteins, CD32B and CD79B, for the treatment of systemic lupus erythematosus (SLE) and for the prevention of immunogenicity of biotherapeutics such as those used in gene therapy; | |
| ● | PRV-015: a human anti-interleukin 15, or IL-15, mAb for the treatment of gluten-free diet non-responsive celiac disease, or NRCD, intercepting the effects of contaminating gluten in the most common autoimmune disorder without any approved medication; and
| |
| ● | PRV-6527: an oral small molecule CSF-1R inhibitor targeting the differentiation and activation of antigen-presenting cells, or APCs, to prevent chronic inflammatory responses and progression or relapse in Crohn's disease. |
| 76 |
The table below summarizes the current status and anticipated milestones for our principal product candidates:
| Product Candidate / Indication | Status | Next Expected Milestone | ||
| PRV-031 (teplizumab, anti-CD3 mAb) for the interception of T1D. |
Top-line results from a National Institute of Diabetes and Digestive and Kidney Diseases-sponsored study conducted at TrialNet sites for preventing disease progression in patients at-risk of developing T1D were publicly reported in June 2019.
In August 2019, the FDA granted BTD to PRV-031 for the delay or prevention of clinical T1D in individuals at-risk of developing the disease.
In October 2019, the EMA granted PRIME eligibility to teplizumab to PRV-031 for the delay or prevention of clinical T1D in individuals at-risk of developing the disease.
We commenced a Phase 3 clinical trial (the PROTECT study) in approximately 300 pediatric and adolescent patients with early onset type one diabetes. The first patient was dosed in the second quarter of 2019.
|
We expect to file a BLA for the At-Risk indication in Q4 2020.
We expect to complete enrollment of the PROTECT study by the end of 2020.
We expect to report top-line results from the Phase 3 PROTECT study in 2022.
| ||
PRV-101 (polyvalent CVB vaccine) for the prevention of acute CVB and the prevention of CVB triggered T1D and celiac disease.
|
We are developing a polyvalent vaccine at Intravacc, our strategic partner in vaccine manufacturing process development. | We expect to commence a first-in-human safety study in the second half of 2020. We expect to report top-line first-in-human data in 2021. | ||
| PRV-3279 (humanized anti-CD32B and CD79B bispecific) for the treatment of SLE and for the prevention of immunogenicity biotherapeutics such as gene therapy. | We announced positive top-line results from our Phase 1b clinical trial (the PREVAIL study), which evaluated PRV-3279 in 16 healthy volunteers.
|
We expect to commence the Phase 2a portion of the PREVAIL study in 2021. | ||
| PRV-015 (anti-IL-15 mAb) for the treatment of gluten-free diet non-responding celiac disease. | We designed a Phase 2b clinical trial (the PROACTIVE trial) in celiac patients with gluten-free diet non-responsive celiac disease and completed additional chronic toxicology studies to support this trial as needed.
|
We expect to commence the Phase 2b PROACTIVE study in the second quarter of 2020. | ||
| PRV-6527 (oral CSF-1R inhibitor) for the treatment of Crohn’s disease. | We announced top-line results from our Phase 2a clinical trial (the PRINCE study), which evaluated PRV-6527 in 93 patients with moderate-to-severe Crohn’s disease. In December 2019, Janssen declined its option to buy back PRV-6527. |
We expect to engage with third parties for the potential sublicense of PRV-6527. |
| 77 |
We intend to leverage our distinctive competences and drug development strategy; advance our carefully selected portfolio of product candidates; in-license or acquire additional targeted development assets, and apply our disease interception and prevention approach to multiple autoimmune and immune-mediated inflammatory diseases.
Financial operations overview
Research and Development Expenses
Research and development expenses consist primarily of clinical studies, the cost of manufacturing our drug candidates for clinical study, regulatory costs, other internal operating expenses, and the cost of conducting preclinical activities. Expenses also include the cost of salaries, benefits and other related costs, including stock-based compensation, for personnel serving in our research and development functions. In addition, our research and development expenses include payments to third parties, as well as the fair value of equity issuances to third parties for the license rights to products in development (prior to marketing approval). Our expenses related to clinical trials are primarily related to activities at contract research organizations, or CROs, that design, obtain regulatory approval, and conduct clinical trials on our behalf. Our expenses related to the production of drug substance or drug product for our clinical trials and development programs are primarily related to activities performed by licensors, strategic partners or contract manufacturing organizations, or CMOs, on our behalf. Our development efforts from inception through December 31, 2019, were principally related to the acquisition and development of our programs detailed in the Pipeline description immediately above.
All research and development expenses are charged to operations as incurred in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic, or ASC, 730, Research and Development. We account for non-refundable advance payments for goods and services that will be used in future research and development activities as expenses when the service has been performed or when the goods have been received, rather than when the payment is made.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries, benefits and other related costs, including stock-based compensation, for our personnel serving in our executive, business development and finance and accounting functions. General and administrative expenses also include professional fees for legal, including patent-related expenses, consulting, insurance, board of director fees, tax and accounting services. We anticipate that we will incur increased general and administrative expenses related to audit, legal, regulatory, and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, director and officer insurance premiums, and investor relations costs associated with being a public company. In addition, we expect that our general and administrative expenses will increase in the future as a result of the build out of our commercial organization.
Interest Income
Interest income consists of interest income earned on our cash, cash equivalents and marketable securities.
Change in Fair Value of Warrant Liability
Change in fair value of warrant liability represents the re-measurement of liability classified warrants using the Black-Scholes option-pricing model at each financial reporting period. The fair value was affected by changes in inputs to the model including the fair value of our Series A Convertible Redeemable Preferred Stock, expected stock price volatility, the estimated term until exercise, and the risk-free interest rate.
Upon the completion of the IPO in July 2018, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock converted to warrants for the purchase of 558,740 shares of our common stock. The liability associated with these warrants was revalued just prior to the completion of the IPO, with the change in the fair value of the warrant liability charged to earnings. The warrant liability was then reclassified to additional paid-in capital upon the completion of the IPO in July 2018, as the warrants no longer contain redemption provisions outside our control.
| 78 |
RESULTS OF OPERATIONS
Comparison of years ended December 31, 2019 and 2018
| For the years ended December 31, | ||||||||||||
| 2019 | 2018 | Increase (Decrease) | ||||||||||
| (in thousands, except per share data) | ||||||||||||
| Statement of Operations Data: | ||||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 36,359 | $ | 22,649 | $ | 13,710 | ||||||
| General and administrative | 8,013 | 4,165 | 3,848 | |||||||||
| Total operating expenses | 44,372 | 26,814 | 17,558 | |||||||||
| Loss from operations | (44,372 | ) | (26,814 | ) | 17,558 | |||||||
| Interest income | 1,087 | 669 | 418 | |||||||||
| Change in fair value of warrant liability | — | (520 | ) | (520 | ) | |||||||
| Loss before income tax benefit | (43,285 | ) | (26,665 | ) | 16,620 | |||||||
| Income tax benefit | — | 187 | (187 | ) | ||||||||
| Net loss | (43,285 | ) | (26,478 | ) | 16,807 | |||||||
| Accretion on Series A Convertible Redeemable Preferred Stock | — | (276 | ) | (276 | ) | |||||||
| Net loss attributable to common stockholders | $ | (43,285 | ) | $ | (26,754 | ) | $ | 16,531 | ||||
| Net loss per common share, basic and diluted | $ | (1.06 | ) | $ | (1.19 | ) | ||||||
| Weighted average common shares outstanding, basic and diluted | 40,747 | 22,441 | ||||||||||
Research and Development Expenses
Research and development expenses were $36.4 million for the year ended December 31, 2019, an increase of $13.8 million, compared to $22.6 million for the year ended December 31, 2018. Research and development expenses for the year ended December 31, 2019 primarily included external clinical development expenses of $22.6 million for PRV-031, PRV-6527, PRV-300, and PRV-3279, development costs of $6.4 million for PRV-101 and internal personnel costs, including stock-based compensation, of $5.1 million.
Research and development expenses for the year ended December 31, 2018 included external clinical development expenses of $10.7 million for PRV-6527, PRV-300 and PRV-031, development costs of $4.1 million for PRV-101, non-cash product acquisition costs of $4.0 million, which represented the fair value of warrants issued in connection with the acquisition of PRV-031 and PRV-3279 in May 2018, and internal personnel costs, including stock-based compensation, of $2.9 million.
General and Administrative Expenses
General and administrative expenses were $8.0 million for the year ended December 31, 2019, an increase of $3.8 million, compared to $4.2 million for the year ended December 31, 2018. General and administrative expenses for the year ended December 31, 2019 primarily included $2.8 million in personnel costs, including stock-based compensation, $2.7 million in professional fees and legal expenses and $2.0 million in insurance and other costs associated with being a public company. General and administrative expenses were $4.2 million for the year ended December 31, 2018 and were primarily comprised of $1.6 million in personnel costs, including stock-based compensation, $1.5 million in professional fees and legal expenses and $0.7 million in insurance and other costs associated with being a public company.
Change in fair value of warrant liability
Change in fair value of warrant liability was a loss of approximately $0.5 million during the year ended December 31, 2018. This loss represents the change in fair value of our warrant liability using a Black-Scholes option-pricing model with updated assumptions as of July 18, 2018, the date just prior to the completion of our IPO, and was primarily impacted by a change in the fair value of our Series A Convertible Redeemable Preferred Stock.
Upon the completion of the IPO in July 2018, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock converted to warrants for the purchase of 558,740 shares of our common stock. The warrant liability was then reclassified to additional paid-in capital upon the completion of the IPO in July 2018, as the warrants no longer contain redemption provisions outside our control. As such, we did not record any gain or loss related to the warrant liability during the year ended December 31, 2019.
| 79 |
Interest Income
Interest income was $1.1 million during the year ended December 31, 2019 compared to $0.7 million during the year ended December 31, 2018. The increase in interest income in 2019 related primarily to an increase in average cash, cash equivalents and marketable securities balances that resulted from the net proceeds from our IPO in July 2018 and our underwritten public offering and concurrent private placement in September 2019.
Income Tax Benefit
We did not record any income tax benefit or provision during the year ended December 31, 2019. We recorded an income tax benefit of $0.2 million during the year ended December 31, 2018, which related to the proceeds from the sale of certain of our prior year New Jersey net operating losses.
Comparison of years ended December 31, 2018 and 2017
| For the years ended December 31, | ||||||||||||
| 2018 | 2017 | Increase (Decrease) | ||||||||||
| (in thousands, except per share data) | ||||||||||||
| Statement of Operations Data: | ||||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 22,649 | $ | 7,684 | $ | 14,965 | ||||||
| General and administrative | 4,165 | 1,456 | 2,709 | |||||||||
| Total operating expenses | 26,814 | 9,140 | 17,674 | |||||||||
| Loss from operations | (26,814 | ) | (9,140 | ) | 17,674 | |||||||
| Interest income | 669 | 132 | 537 | |||||||||
| Change in fair value of warrant liability | (520 | ) | (125 | ) | 395 | |||||||
| Loss before income tax benefit | (26,665 | ) | (9,133 | ) | 17,532 | |||||||
| Income tax benefit | 187 | — | 187 | |||||||||
| Net loss | (26,478 | ) | (9,133 | ) | 17,345 | |||||||
| Accretion on Series A Convertible Redeemable Preferred Stock | (276 | ) | (343 | ) | (67 | ) | ||||||
| Net loss attributable to common stockholders | $ | (26,754 | ) | $ | (9,476 | ) | $ | 17,278 | ||||
| Net loss per common share, basic and diluted | $ | (1.19 | ) | $ | (1.01 | ) | ||||||
| Weighted average common shares outstanding, basic and diluted | 22,441 | 9,370 | ||||||||||
Research and Development Expenses
Research and development expenses were $22.6 million for the year ended December 31, 2018, an increase of $14.9 million, compared to $7.7 million for the year ended December 31, 2017. Research and development expenses for the year ended December 31, 2018 included external clinical development expenses of $10.7 million for PRV-6527, PRV-300 and PRV-031, development costs of $4.1 million for PRV-101, non-cash product acquisition costs of $4.0 million, which represented the fair value of warrants issued in connection with the acquisition of PRV-031 and PRV-3279 in May 2018, and internal personnel costs, including stock-based compensation, of $2.9 million.
Research and development expenses during the year ended December 31, 2017 included non-cash product acquisition costs of $3.4 million, which represented the fair value of shares of common stock issued in connection with the Vactech License Agreement for PRV-101 in April 2017, $3.1 million in external clinical development expenses primarily related to PRV-6527 and PRV-300, and $1.1 million of internal clinical development costs consisting mostly of compensation and related expenses, including stock-based compensation.
General and Administrative Expenses
General and administrative expenses were $4.2 million for the year ended December 31, 2018, an increase of $2.7 million, compared to $1.5 million for the year ended December 31, 2017. General and administrative expenses for the year ended December 31, 2018 primarily included $1.6 million in personnel costs, including stock-based compensation, $1.5 million in professional fees and legal expenses and $0.7 million in insurance and other costs associated with being a public company. General and administrative expenses were $1.5 million for the year ended December 31, 2017 and were primarily comprised of $0.7 million in personnel costs, including stock-based compensation, and $0.6 million in professional fees and legal expenses.
| 80 |
Change in fair value of warrant liability
Change in fair value of warrant liability was a loss of approximately $0.5 million during the year ended December 31, 2018, compared to a loss of $0.1 million during the year ended December 31, 2017. This loss represents the change in fair value of our warrant liability using a Black-Scholes option-pricing model with updated assumptions as of July 18, 2018, the date just prior to the completion of our IPO, and was primarily impacted by a change in the fair value of our Series A Convertible Redeemable Preferred Stock.
Upon the completion of the IPO in July 2018, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock converted to warrants for the purchase of 558,740 shares of our common stock. The warrant liability was then reclassified to additional paid-in capital upon the completion of the IPO in July 2018, as the warrants no longer contain redemption provisions outside our control.
Interest Income
Interest income was $0.7 million during the year ended December 31, 2018 compared to $0.1 million during the year ended December 31, 2017. The increase in interest income in 2018 related primarily to an increase in average cash and cash equivalents balances that resulted from the net proceeds from our IPO in July 2018.
Income Tax Benefit
We recorded an income tax benefit of $0.2 million during the year ended December 31, 2018, which related to the proceeds from the sale of certain of our prior year New Jersey net operating losses We did not record any income tax benefit or provision during the year ended December 31, 2017.
LIQUIDITY AND CAPITAL RESOURCES
Overview
There is considerable time and cost associated with developing a potential drug or pharmaceutical product to the point of regulatory approval and commercialization. We have funded our operations to date through offerings of equity securities. We expect to continue to incur losses, as we plan to continue to fund development activities.
As of December 31, 2019, we had cash, cash equivalents and marketable securities of $85.4 million. In September 2019, we completed an underwritten public offering of 5,750,000 shares and a concurrent private placement with Amgen of 2,500,000 of our common stock, at a price of $8.00 per share, resulting in aggregate net cash proceeds from the sale of the shares, after deducting the underwriting discounts offering expenses, of $62.7 million. We believe that our current cash, cash equivalents and marketable securities will be sufficient to fund our projected operating requirements for at least 12 months from the issuance of these financial statements.
We will need to raise additional capital to fund our operations, to develop and commercialize PRV-031, PRV-015, PRV-3279 and PRV-101 and to develop, acquire, or in-license other products. We plan to raise additional capital through equity offerings. Such additional funding will be necessary to continue to develop our potential product candidates, to pursue the license or purchase of other technologies, to commercialize our product candidates or to purchase other products. We may seek to sell common or preferred equity or convertible debt securities, enter into a credit facility or another form of third-party funding, or seek other debt financing. In addition, we may consider raising additional capital to fund operating activities, to expand our business, to pursue strategic investments, to take advantage of financing opportunities, or for other reasons. The sale of equity and convertible debt securities may result in dilution to our stockholders and those securities may have rights senior to those of our common shares. If we raise additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict our operations. Any other third-party funding arrangement could require us to relinquish valuable rights. We may require additional capital beyond our currently anticipated amounts. Additional capital may not be available on reasonable terms, or at all. If we are unable to obtain sufficient additional funds when required, we may be forced to delay, restrict or eliminate all or a portion of our development programs, dispose of assets or technology or cease operations.
| 81 |
Our cash requirements in 2020 and beyond will be impacted by a number of factors, the most significant of which are expenses related to PRV-031, including the PROTECT clinical trial, which commenced in April 2019, CMC activities, and costs related to our expected BLA submission in the At-Risk indication. Other factors include costs related to our planned Phase 2b clinical study of PRV-015 and our continued development efforts for PRV-101.
In April 2017, we completed a private offering of 11,381,999 shares of our Series A Convertible Redeemable Preferred Stock, at a price of $2.50 per share, resulting in net cash proceeds from the sale of the shares, after deducting the underwriter’s discount and offering expenses, of $26.7 million.
In July 2018, we closed our initial public offering of 15,969,563 shares of common stock at a price of $4.00 per share. Our net proceeds were $59.3 million, after deducting underwriters’ commissions of approximately $3.7 million and other offering expenses of approximately $0.8 million. In connection with the closing of the IPO, all of our shares of redeemable convertible preferred stock outstanding at the time of the IPO were automatically converted into 11,381,999 shares of common stock. In addition, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock also converted to warrants for the purchase of 558,740 shares of our common stock.
In August 2019, we entered into the Sales Agreement with SVB Leerink LLC (“SVBLeerink”) and Cantor Fitzgerald & Co. (“Cantor”) pursuant to which SVBLeerink and Cantor will serve as our sales agent to sell up to $50.0 million of shares of our common stock through an “at the market offering.” To date, there have been no sales under the Sales Agreement.
In September 2019, we completed an underwritten public offering in which we sold 5,750,000 shares of common stock at a public offering price of $8.00 per share. The 5,750,000 shares sold included the full exercise of the underwriters’ option to purchase 750,000 shares at a price of $8.00 per share. Concurrent with the underwritten public offering, we sold 2,500,000 shares of common stock to Amgen, Inc. at the public offering price of $8.00 per share in a private placement, pursuant to the terms of our License and Collaboration Agreement with Amgen Inc, dated as of November 5, 2018. Aggregate net proceeds from the underwritten public offering and the concurrent private placement were $62.7 million, net of approximately $2.8 million in underwriting discounts and commissions and other offering expenses of $0.5 million.
Cash Flows
As of December 31, 2019, we had cash, cash equivalents and marketable securities totaling $85.4 million. We currently have invested our cash and cash equivalents in money market funds.
The following table shows a summary of our cash flows for the years ended December 31, 2019, 2018 and 2017:
| For the years ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| (in thousands) | ||||||||||||
| Net cash (used in) provided by: | ||||||||||||
| Operating activities | $ | (36,065 | ) | $ | (22,642 | ) | $ | (4,882 | ) | |||
| Investing activities | (46,250 | ) | — | — | ||||||||
| Financing activities | 62,941 | 59,347 | 26,716 | |||||||||
| Net change in cash and cash equivalents | $ | (19,374 | ) | $ | 36,705 | $ | 21,834 | |||||
Cash Flows from Operating Activities
Net cash used in operating activities was $36.1 million for the year ended December 31, 2019 and primarily related to cash used to fund clinical development activities for PRV-031, PRV-6527, PRV-3279 and PRV-300, development activities for PRV-101, and increased personnel costs to support our clinical programs and our public company infrastructure. Our working capital was $82.2 million as of December 31, 2019.
Net cash used in operating activities was $22.6 million for the year ended December 31, 2018 and primarily related to cash used to fund clinical development activities for PRV-6527, PRV-300 and PRV-031, development activities for PRV-101, and increased personnel costs to support our recently acquired programs and our new public company infrastructure. Our working capital was $59.7 million as of December 31, 2018.
| 82 |
Net cash used in operating activities was $4.9 million for the year ended December 31, 2017 and primarily related to start-up activities for our operations, including clinical development programs for PRV-6527 and PRV-300 and the development program for PRV-101, each of which were acquired in the second quarter of 2017.
Cash Flows from Investing Activities
Net cash used in investing activities for the year ended December 31, 2019 was $46.2 million and relates to the purchase of marketable securities during the period.
There was no cash used in or provided by investing activities for the year ended December 31, 2018 and 2017, respectively.
Cash Flows from Financing Activities
Net cash provided by financing activities was $62.9 million for the year ended December 31, 2019 and primarily related to the net proceeds we received from the completion of our underwritten public offering and concurrent private placement in September 2019.
Net cash provided by financing activities was $59.3 million for the year ended December 31, 2018 and primarily related to the net proceeds we received from the completion of our IPO in July 2018.
Net cash provided by financing activities for the year ended December 31, 2017 related to the completion of a private placement of our Series A Convertible Redeemable Preferred Stock, which yielded net proceeds of approximately $26.7 million.
Future Funding Requirements
To date, we have not generated revenue, and we do not know when, or if, we will generate revenue. We do not expect to generate revenue unless or until we obtain marketing approval of, secure reimbursement for, and commercialize, our product candidates. We will need to raise additional capital to fund our operations, to develop and commercialize our product candidates, and to develop, acquire, in-license or co-promote other products. Our future capital requirements may be substantial and will depend on many factors.
Commitments and Contractual Obligations
In May 2018, we entered into the MacroGenics Asset Purchase Agreement with MacroGenics pursuant to which we acquired MacroGenics’ interest in teplizumab (renamed PRV-031), a humanized mAb for the treatment of T1D. We are obligated to pay MacroGenics contingent milestone payments totaling $170.0 million upon the achievement of certain regulatory approval milestones, including $60.0 million payable upon approval of a BLA in the United States. In addition, we are obligated to make contingent milestone payments to MacroGenics totaling $225.0 million upon the achievement of certain sales milestones. We have also agreed to pay MacroGenics a single-digit royalty on net sales of the product. We have also agreed to pay third-party obligations, including low single-digit royalties, a portion of which is creditable against royalties payable to MacroGenics, aggregate milestone payments of up to approximately $1.3 million and other consideration, for certain third-party intellectual property under agreements we are assuming pursuant to the Asset Purchase Agreement. Further, we are required to pay MacroGenics a low double-digit percentage of certain consideration to the extent it is received in connection with a future grant of rights to PRV-031 by us to a third party. We are obligated to use reasonable commercial efforts to develop and seek regulatory approval for PRV-031. As of December 31, 2019, we have not achieved any milestones that would trigger payments to MacroGenics under the MacroGenics Asset Purchase Agreement.
| 83 |
In April 2017, we entered into the Vactech License Agreement, pursuant to which Vactech Ltd. (Vactech) granted us exclusive global rights for the purpose of developing and commercializing the group B coxsackie virus vaccine (CVB) platform technology. In consideration of the licenses and other rights granted by Vactech, we issued two million common shares to Vactech. We paid Vactech a total of approximately $0.5 million for transition and advisory services during the first 18 months of the term of the agreement. In addition, we are obligated to make a series of contingent milestone payments to Vactech totaling up to an additional $24.5 million upon the achievement of certain clinical development and regulatory filing milestones. In addition, we have agreed to pay Vactech tiered single-digit royalties on net sales of any approved product based on the CVB platform technology and three additional payments totaling $19.0 million upon the achievement of certain annual net sales levels. As of December 31, 2019, we have not achieved any milestones that would trigger payments to Vactech.
In March 2018, we entered into the Intravacc Development Services Agreement with The Institute of Translational Vaccinology, pursuant to which Intravacc will provide services related to process development, non-GMP and GMP manufacturing of our polyvalent coxsackie virus B vaccine (CVB), including providing proprietary technology for manufacturing purposes. We will pay Intravacc approximately 10 million euros for their services over the development and manufacturing period which we currently expect will last for approximately 24 to 30 months. Each party retains its existing intellectual property and will share newly developed intellectual property via a fully-paid non-exclusive license between the parties for all development work through phase 1 clinical trials. Any future use, including commercial use, of Intravacc’s technology will be subject to a separate nonexclusive license agreement. The Intravacc Development Services Agreement may be terminated by us with ninety-days’ notice without cause and by either party upon a material breach or insolvency of the other party. As of December 31, 2019, we have paid Intravacc a total of approximately 8.0 million euros, or approximately $9.4 million, for services provided by Intravacc under the Intravacc Development Services Agreement.
In May 2018, we entered into the MacroGenics License Agreement with MacroGenics, Inc., pursuant to which MacroGenics granted us exclusive global rights for the purpose of developing and commercializing MGD010 (renamed PRV-3279), a humanized protein and a potential treatment for SLE and other similar diseases. We are obligated to make contingent milestone payments to MacroGenics totaling $42.5 million upon the achievement of certain developmental and approval milestones for the first indication, and an additional $22.5 million upon the achievement of certain regulatory approvals for a second indication. In addition, we are obligated to make contingent milestone payments to MacroGenics totaling $225.0 million upon the achievement of certain sales milestones. We have also agreed to pay MacroGenics a single-digit royalty on net sales of the product. Further, we are required to pay MacroGenics a low double-digit percentage of certain consideration to the extent received in connection with a future grant of rights to PRV-3279 by us to a third party. We are obligated to use commercially reasonable efforts to develop and seek regulatory approval for PRV-3279. The license agreement may be terminated by either party upon a material breach or bankruptcy of the other party, by Provention without cause upon prior notice to MacroGenics, and by MacroGenics in the event that we challenge the validity of any licensed patent under the agreement, but only with respect to the challenged patent. As of December 31, 2019, we have not achieved any milestones that would trigger payments to MacroGenics under the MacroGenics License Agreement.
In November 2018, we entered into the Amgen Agreement with Amgen for PRV-015 (formerly AMG 714), a novel anti-IL-15 monoclonal antibody being developed for the treatment of gluten-free diet NRCD. Under the terms of the agreement, we will conduct and fund a Phase 2b trial in NRCD and lead the development and regulatory activities for the program. Amgen has agreed to make an equity investment of up to $20.0 million in us, which was completed in September 2019. See Note 5 – License and Other Agreements for further details. Amgen is also responsible for the manufacturing of PRV-015. Upon completion of the Phase 2b trial, a $150.0 million milestone payment is due from Amgen to us, plus an additional regulatory milestone payment, and single digit royalties on future sales. If Amgen elects not to pay the $150.0 million milestone, AMG 714 rights will be transferred to us pursuant to a termination license agreement from Amgen and us. We will be obligated to make certain contingent milestone payments to Amgen and other third parties totaling up to $70.0 million upon the achievement of certain clinical and regulatory milestones and a low double-digit royalty on net sales of any approved product based on the IL-15 technology. We may terminate the agreement without cause (in which case the exclusive global rights to the technology will transfer back to Amgen) and by either party upon a material breach. The agreement expires upon the expiration of Amgen’s last obligation to make royalty payments to Provention (or, our last obligation to make royalty payments to Amgen, if the program rights are transferred to us). As of December 31, 2019, we have not achieved any milestones that would trigger payments to Amgen under the Amgen Agreement.
| 84 |
In April 2017, we entered into the Janssen CSF-1R License Agreement, pursuant to which Janssen Pharmaceutica NV granted us exclusive global rights for the purpose of developing and commercializing a colony stimulating factor 1 receptor (CSF-1R) inhibitor named JNJ-40346527 (renamed PRV-6527) for inflammatory bowel diseases including Crohn’s disease and UC. We evaluated PRV-6527 for Crohn’s disease in a recently completed Phase 2a clinical trial (the PRINCE study). See Item 1 – Business, Recent Developments for the announcement of top-line results of the PRINCE study. In December 2019, Janssen declined its option to buy back the rights to PRV-6527. and as such, all rights will remain with us. We will be obligated to make contingent milestone payments to Janssen totaling $35.0 million upon the achievement of certain clinical and regulatory milestones for the first indication and an additional $20.0 million upon the achievement of certain clinical and regulatory milestones for a second indication. In addition, we have agreed to pay Janssen tiered single-digit royalties on net sales of any approved product based on the CSF-1R technology and three additional payments totaling $100.0 million upon the achievement of certain annual net sales levels. As of December 31, 2019, no milestones have been achieved that would trigger payments to Janssen under the CSF-1R License Agreement.
In February 2019, we entered into a services agreement with AGC Biologics (“AGC”) to manufacture and supply teplizumab, PRV-031, for our anticipated clinical supply needs. We may terminate the agreement or any statement of work thereunder with approximately12 months’ prior written notice to AGC. If we provide less than 12 months’ notice of termination, we may incur a cancellation fee depending on the timing of such notice. AGC may terminate the agreement if we do not, over a specified period, purchase and take delivery from AGC of a specified minimum quantity of API for teplizumab. Each party also has the right to terminate the agreement for other customary reasons such as material breach and bankruptcy. The agreement contains provisions relating to compliance by AGC with current Good Manufacturing Practices, cooperation by AGC in connection with potential marketing applications for PRV-031, indemnification, confidentiality, dispute resolution and other customary matters for an agreement of this kind.
The table below presents a summary of our contractual obligations as of December 31, 2019:
| Payments Due By Period | |||||||||||||||||||
| Within | More than | ||||||||||||||||||
| Total | 1 year | 1-3 Years | 3-5 Years | 5 years | |||||||||||||||
| (In thousands) | |||||||||||||||||||
| Purchase Obligations (1) | $ | 4,140 | $ | 4,140 | $ | — | $ | — | $ | — | |||||||||
| Total (2) | $ | 4,140 | $ | 4,140 | $ | — | $ | — | $ | — | |||||||||
| (1) | Purchase obligations represent our commitments under binding forecasts, and purchase orders (inclusive of cancellation fees), including those provided under our agreement(s) with AGC. The actual amounts incurred will be determined based on the amount of goods purchased and the pricing then in effect under the applicable arrangement. |
| (2) | This table does not include (a) any milestone payments which may become payable to third parties under license agreements as the timing and likelihood of such payments are not known with certainty, (b) any royalty payments to third parties as the amounts, timing and likelihood of such payments are not known, and (c) contracts that are entered into in the ordinary course of business that are not material in the aggregate in any period presented above. |
In addition, in the course of normal business operations, we have agreements with contract service providers to assist in the performance of our research and development and manufacturing activities. Expenditures to CROs, CMOs and other clinical development related vendors represent significant costs in clinical development. Subject to required notice periods and our obligations under binding purchase orders, we can elect to discontinue the work under these agreements at any time. We could also enter into additional collaborative research, contract research, manufacturing, and supplier agreements in the future, which may require upfront payments and even long-term commitments of cash.
We also have employment agreements with certain employees which require the funding of a specific level of payments, if certain events, such as a change in control or termination without cause, occur.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future material effect on our financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources. We do not have any interest in special purpose entities, structured finance entities or other variable interest entities.
Critical Accounting Policies and Estimates
Preparation of financial statements in accordance with generally accepted accounting principles in the US requires us to make estimates and assumptions affecting the reported amounts of assets, liabilities, and expenses and the disclosures of contingent assets and liabilities. We use our historical experience and other relevant factors when developing our estimates and assumptions. We continually evaluate these estimates and assumptions. The amounts of assets and liabilities reported in our balance sheets and the amounts of expenses reported in our statements of comprehensive loss are affected by estimates and assumptions, which are used for, but not limited to, the accounting for research and development, stock-based compensation, accrued expenses and both liability-classified and equity-classified warrants. The accounting policies discussed below are considered critical to an understanding of our financial statements because their application places the most significant demands on our judgment. Actual results could differ from our estimates. For additional accounting policies, see Note 3 to our Financial Statements— Summary of Significant Accounting Policies.
| 85 |
Research and Development
Research and development expenses consist primarily of salaries, benefits and other related costs, including stock-based compensation, for personnel serving our development functions, and other internal operating expenses, the cost of clinical studies, and the cost of our drug candidates for clinical study. In addition, research and development expenses include payments to third parties for the development of our product candidates and the estimated fair value for the issuance of equity for the license rights to products in development (prior to marketing approval). Our expenses related to clinical trials are primarily related to activities at contract research organizations that design, gain approval for and conduct clinical trials on our behalf. Such amounts are then recognized as an expense as the related goods are delivered or the services are performed.
Stock-Based Compensation
We recognize stock-based compensation expense for awards of equity instruments based on the grant-date fair value of those awards. The grant-date fair value of the award is recognized as compensation expense ratably over the requisite service period, which generally equals the vesting period of the award. We also grant performance-based stock options. The grant-date fair value of the performance-based stock options is recognized as compensation expense once it is probable that the performance condition will be achieved. We record actual forfeitures in the period the forfeiture occurs.
In June 2018, the FASB issued ASU No. 2018-07, Improvements to Nonemployee Share-Based Payment Accounting, which supersedes ASC 505-50 and expands the scope of ASC 718 to include all share-based payments arrangements related to the acquisition of goods and services from both employees and nonemployees. During the third quarter of 2018, we early adopted ASU 2018-07. After the adoption of ASU 2018-07, the measurement date for non-employee awards is the date of grant. Compensation expense for non-employees is recognized, without changes to the fair value of the award, over the requisite service period, which is the vesting period of the respective award. The adoption of ASU 2018-07 did not have a material impact our financial statements or footnote disclosures.
Prior to the third quarter of 2018, we accounted for awards of equity instruments issued to non-employees in accordance with ASC Topic 505-50 “Equity-Based Payment to Non-Employees” and accordingly the value of the stock compensation to non-employees was based upon the measurement date as determined at either a) the date at which a performance commitment was reached, or b) at the date at which the necessary performance to earn the equity instruments was complete, which was normally the end of the vesting period. At the end of each financial reporting period prior to completion of the service, the fair value of these awards was remeasured using updated assumption inputs in the Black-Scholes option-pricing model.
We used the Black-Scholes option-pricing model to estimate the fair value of option awards with the following weighted-average assumptions for the period indicated:
| Years ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Exercise Price | $ | 10.34 | $ | 3.93 | $ | 1.81 | ||||||
| Expected volatility | 72 | % | 61 | % | 64 | % | ||||||
| Expected dividends | — | — | — | |||||||||
| Expected term (in years) | 6.3 | 6.2 | 6.5 | |||||||||
| Risk-free interest rate | 1.90 | % | 2.78 | % | 1.96 | % | ||||||
| 86 |
The weighted-average valuation assumptions were determined as follows:
| ● | Risk-free interest rate: we base the risk-free interest rate on the interest rate payable on U.S. Treasury securities in effect at the time of grant for a period that is commensurate with the assumed expected option term. | |
| ● | Expected annual dividends: the estimate for annual dividends is 0%, because we have not historically paid, and do not expect for the foreseeable future to pay, a dividend. | |
| ● | Expected stock price volatility: the expected volatility used is based on historical volatilities of similar entities within our industry which were commensurate with our expected term assumption. | |
| ● | Expected term of options: the expected term of options represents the period of time options are expected to be outstanding. The expected term of the options granted to employees is derived from the “simplified” method as described in Staff Accounting Bulletin 107 relating to stock-based compensation, whereby the expected term is an average between the vesting period and contractual period due to our limited operating history. The expected term for options granted to non-employees is equal to the contractual term of the awards. |
Stock-based compensation expense is included in both research and development expenses and general and administrative expenses in the Statement of Operations.
Determination of Fair Value of Common Stock
Prior to our IPO, there was no public market for our common stock. The estimated fair value of our common stock had been determined by our board of directors as of the date of each option grant and quarter end, with input from management, considering our most recently available third-party valuations of common stock. These factors included, but were not limited to: our most recently available valuations of our common stock by an unrelated third party; the price at which we sold shares of our convertible redeemable preferred stock to outside investors in arms-length transactions; the rights, preferences and privileges of our convertible preferred stock relative to those of our common stock; our results of operations, financial position and capital resources; current business conditions and projections; the lack of marketability of our common stock; the hiring of key personnel and the experience of management; the risk inherent in the development of our products; our stage of development and material risks related to our business; the fact that the option grants involved illiquid securities in a private company; and the likelihood of achieving a liquidity event, such as our IPO or sale, in light of prevailing market conditions.
Previously, we determined the estimated fair value of our common stock at various dates using contemporaneous valuations performed in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation, or the Practice Aid. The Practice Aid identifies various available methods for allocating enterprise value across classes and series of capital stock to determine the estimated fair value of common stock at each valuation date. In accordance with the Practice Aid, our board of directors considered the following methods:
| ● | Current Value Method. Under the Current Value Method, or CVM, value is determined based on our balance sheet. This value is first allocated based on the liquidation preference associated with preferred stock issued as of the valuation date, and then any residual value is assigned to the common stock. | |
| ● | Option-Pricing Method. Under the option-pricing method, or OPM, shares valued by creating a series of call options with exercise prices based on the liquidation preferences and conversion terms of each equity class. The estimated fair values of the preferred and common stock are inferred by analyzing these options. | |
| ● | Probability-Weighted Expected Return Method. The probability-weighted expected return method, or PWERM, is a scenario-based analysis that estimates value per share based on the probability-weighted present value of expected future investment returns, considering each of the possible outcomes available to us, as well as the economic and control rights of each share class. |
Prior to our IPO, we determined that a PWERM was the most appropriate method for allocating our enterprise value to determine the estimated fair value of our common stock. Our common stock valuations as of April 25, 2017, June 30, 2017, September 11, 2017, September 30, 2017, December 1, 2017, December 31, 2017, March 1, 2018, March 31, 2018, May 7, 2018, June 30, 2018 and July 18, 2018 were prepared using the PWERM.
| 87 |
Our board of directors and management developed best estimates based on application of these approaches and the assumptions underlying these valuations, giving careful consideration to the advice from our third-party valuation expert. Such estimates involved inherent uncertainties and the application of significant judgment. As a result, if factors or expected outcomes change and we use significantly different assumptions or estimates, our equity-based compensation could be materially different.
Subsequent to the completion of our IPO, our board of directors determines the fair market value of our common stock based on its closing price as reported on the date of grant on the primary stock exchange on which our common stock is traded.
Accrued Research and Development Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued expenses. This process involves reviewing quotations and contracts, identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. The significant estimates in our accrued research and development expenses are related to expenses incurred with respect to CROs, CMOs and other vendors in connection with research and development and manufacturing activities.
We base our expenses related to CROs and CMOs on our estimates of the services received and efforts expended pursuant to quotations and contracts with such vendors that conduct research and development and manufacturing activities on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the applicable research and development or manufacturing expense. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual or prepaid expense accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and could result in us reporting amounts that are too high or too low in any particular period. There have been no material changes in estimates for the periods presented.
Warrant Liability
In April 2017, we issued warrants to purchase shares of Series A Convertible Redeemable Preferred Stock in connection with the issuance of the Series A Preferred Stock financing. We accounted for these warrants as a liability in the financial statements because the underlying instrument into which the warrants were exercisable contained redemption provisions that were outside our control.
Upon the completion of the IPO in July 2018, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock converted to warrants for the purchase of 558,740 shares of our common stock and no longer contain these redemption provisions outside our control. The liability associated with these warrants was revalued just prior to the completion of the IPO, with the change in the fair value of the warrant liability charged to earnings. The warrant liability was then reclassified to additional paid-in capital upon the completion of the IPO in July 2018, as the warrants no longer contain redemption provisions outside our control.
The fair value of the warrants was determined using the Black-Scholes option-pricing model and were re-measured to fair value at each financial reporting period date with any changes in fair value recognized in the statements of operations. See Note 8 to our financial statements in Part II-Item 8 of this Annual Report on Form 10-K for further information.
| 88 |
Recent Accounting Pronouncements
See Note 3, “Significant Accounting Policies”, in the accompanying notes to financial statements, which is incorporated herein by reference.
ITEM 7A. Quantitative and Qualitative Disclosures about Market Risk
Not applicable.
ITEM 8. Financial Statements and Supplementary Data
The financial information required by Item 8 is contained in Part IV, Item 15 of this Annual Report on Form 10-K.
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
ITEM 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
As of the end of the period covered by this report, management performed, with the participation of our principal executive and principal financial officers, an evaluation of the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act. Our disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, to allow timely decisions regarding required disclosures. Based on the evaluation, our principal executive and principal financial officers concluded that, as of December 31, 2019, our disclosure controls and procedures were effective.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed by, or under the supervision of our Chief Executive Officer and our Chief Financial Officer, and effected by our board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2019. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in the original Internal Control—Integrated Framework updated in 2013. Based on that assessment, our management concluded that, as of December 31, 2019, our internal control over financial reporting was effective.
This Annual Report on Form 10-K does not include an attestation report of our independent registered public accounting firm because we are an “emerging growth company,” and may take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act.
Changes in Internal Control over Financial Reporting
There were no significant changes in our internal control over financial reporting for the quarterly period or year ended December 31, 2019 identified in connection with the evaluation required by Rules 13a-15(e) and 15d-15(e) of the Exchange Act that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
| 89 |
ITEM 10. Directors, Executive Officers and Corporate Governance
Information required under this item relating to our Board of Directors, executive officers and corporate governance will be included in our definitive proxy statement for the 2020 Annual Meeting of Stockholders, to be filed with the SEC within 120 days after the end of the year ended December 31, 2019 (the “2020 Proxy Statement”), and such required information is incorporated herein by reference.
ITEM 11. Executive Compensation
Information required under this item relating to executive compensation is incorporated herein by reference from information included in the 2020 Proxy Statement.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters
Information required under this item relating to securities authorized for issuance under equity compensation plans and to security ownership of certain beneficial owners and management is incorporated herein by reference from information included in the 2020 Proxy Statement.
ITEM 13. Certain Relationships and Related Transactions and Director Independence
Information required under this item relating to certain relationships and transactions with related parties and about director independence is incorporated herein by reference from information included in the 2020 Proxy Statement.
ITEM 14. Principal Accounting Fees and Services
Information required under this item relating to the fees for professional services rendered by our independent accountants in 2019 and 2018 is incorporated herein by reference from information included in the 2020 Proxy Statement.
| 90 |
ITEM 15. Exhibits, Financial Statements Schedules
(a) Financial Statements
See accompanying index to Financial Statements.
(b) Financial Statement Schedules
All schedules have been omitted because the required information is included in the financial statements or the notes thereto, or is not applicable.
(c) Index to Exhibits
The following exhibits are filed or incorporated by reference as part of this Annual Report on Form 10-K:
| 91 |
+ Compensation Related Contract.
† Confidential treatment received for certain portions of this exhibit.
Not applicable.
| 92 |
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| Registrant: | ||
| Provention Bio, Inc. | ||
| Date: March 12, 2020 | By: | /s/ Andrew Drechsler |
| Andrew Drechsler | ||
Chief Financial Officer (Authorized
Officer and Principal Financial | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Ashleigh Palmer | Chief Executive Officer (Principal Executive Officer), Director | March 12, 2020 | ||
| Ashleigh Palmer | ||||
| /s/ Andrew Drechsler | Chief Financial Officer (Principal Financial Officer, Principal Accounting Officer) | March 12, 2020 | ||
| Andrew Drechsler | ||||
| /s/ Jeffrey Bluestone | Director | March 12, 2020 | ||
Jeffrey Bluestone |
||||
| /s/ Avery Catlin | Director | March 12, 2020 | ||
| Avery Catlin | ||||
| /s/ Anthony DiGiandomenico | Director | March 12, 2020 | ||
| Anthony DiGiandomenico | ||||
| /s/ Sean Doherty | Director | March 12, 2020 | ||
| Sean Doherty | ||||
| /s/ Wayne Pisano | Director | March 12, 2020 | ||
| Wayne Pisano |
| 93 |
PROVENTION BIO, INC.
INDEX TO FINANCIAL STATEMENTS
| F-1 |
Report of Independent Registered Public Accounting Firm
To The Board of Directors and Stockholders of Provention Bio, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Provention Bio, Inc. (the “Company”) as of December 31, 2019 and 2018, and the related statements of operations, stockholders’ equity (deficit), and cash flows for each of the years in the three-year period ended December 31, 2019, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2019, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since 2016.
EISNERAMPER LLP
Iselin, New Jersey
March 12, 2020
| F-2 |
PROVENTION BIO, INC.
(in thousands of U.S. dollars, except share and per share data)
| December 31, 2019 | December 31, 2018 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 39,165 | $ | 58,539 | ||||
| Marketable securities, current | 46,208 | — | ||||||
| Prepaid expenses and other current assets | 623 | 2,990 | ||||||
| Total assets | $ | 85,996 | $ | 61,529 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 1,775 | $ | 568 | ||||
| Accrued expenses | 2,065 | 1,303 | ||||||
| Total current liabilities | 3,840 | 1,871 | ||||||
| Commitments and Contingencies (Note 5) | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.0001 par value; 25,000,000 shares authorized; no shares issued or outstanding at December 31, 2019 and December 31, 2018 | — | — | ||||||
| Common stock, $0.0001 par value; 100,000,000 shares authorized; 47,658,361 shares issued and outstanding at December 31, 2019; 37,361,562 shares issued and outstanding at December 31, 2018 | 5 | 4 | ||||||
| Additional paid-in capital | 161,212 | 95,430 | ||||||
| Accumulated deficit | (79,061 | ) | (35,776 | ) | ||||
| Total stockholders’ equity | 82,156 | 59,658 | ||||||
| Total liabilities, preferred stock and stockholders’ equity | $ | 85,996 | $ | 61,529 | ||||
The accompanying notes are an integral part of the financial statements.
| F-3 |
PROVENTION BIO, INC.
(in thousands of U.S. dollars, except per share data)
| For the Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 36,359 | $ | 22,649 | $ | 7,684 | ||||||
| General and administrative | 8,013 | 4,165 | 1,456 | |||||||||
| Total operating expenses | 44,372 | 26,814 | 9,140 | |||||||||
| Loss from operations | (44,372 | ) | (26,814 | ) | (9,140 | ) | ||||||
| Interest income | 1,087 | 669 | 132 | |||||||||
| Change in fair value of warrant liability | — | (520 | ) | (125 | ) | |||||||
| Loss before income tax benefit | (43,285 | ) | (26,665 | ) | (9,133 | ) | ||||||
| Income tax benefit | — | 187 | — | |||||||||
| Net loss | (43,285 | ) | (26,478 | ) | (9,133 | ) | ||||||
| Accretion on Series A Convertible Redeemable Preferred Stock | — | (276 | ) | (343 | ) | |||||||
| Net loss attributable to common stockholders | $ | (43,285 | ) | $ | (26,754 | ) | $ | (9,476 | ) | |||
| Net loss per common share, basic and diluted | $ | (1.06 | ) | $ | (1.19 | ) | $ | (1.01 | ) | |||
| Weighted average common shares outstanding, basic and diluted | 40,747 | 22,441 | 9,370 | |||||||||
The accompanying notes are an integral part of the financial statements.
| F-4 |
PROVENTION BIO, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
(in thousands of U.S. dollars)
| Convertible
Redeemable Preferred Stock | Common Stock | Additional Paid-In | Accumulated | Total
Stockholders’ Equity | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||||||||
| Balance at December 31, 2016 | — | $ | — | 8,000 | $ | — | $ | — | $ | (165 | ) | $ | (165 | ) | ||||||||||||||
| Issuance of Series A Convertible Redeemable Preferred Stock, net of issuance costs of $1,740 | 11,382 | 26,715 | — | — | — | — | — | |||||||||||||||||||||
| Fair value of warrants issued in connection with Series A Convertible Redeemable Preferred Stock, reclassifed to warrant liability | — | (873 | ) | — | — | — | — | — | ||||||||||||||||||||
| Accretion of Series A Convertible Redeemable Preferred Stock to redemption value | — | 343 | — | — | (343 | ) | — | (343 | ) | |||||||||||||||||||
| Issuance of common stock | — | 2,000 | — | 3,400 | — | 3,400 | ||||||||||||||||||||||
| Payment of subscription | — | — | — | 1 | — | — | 1 | |||||||||||||||||||||
| Stock-based compensation | — | — | — | — | 207 | — | 207 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (9,133 | ) | (9,133 | ) | |||||||||||||||||||
| Balance at December 31, 2017 | 11,382 | $ | 26,185 | 10,000 | $ | 1 | $ | 3,264 | $ | (9,298 | ) | $ | (6,033 | ) | ||||||||||||||
| Issuance of common stock upon conversion of Series A Convertible Redeemable Preferred Stock | (11,382 | ) | (26,461 | ) | 11,382 | 1 | 26,460 | — | 26,461 | |||||||||||||||||||
| Accretion of Series A Convertible Redeemable Preferred Stock to redemption value | — | 276 | — | — | (276 | ) | — | (276 | ) | |||||||||||||||||||
| Reclassification of Series A warrants liability | — | — | — | — | 1,518 | — | 1,518 | |||||||||||||||||||||
| Issuance of common stock in initial public offering, net of issuance costs | — | — | 15,970 | 2 | 56,281 | — | 56,283 | |||||||||||||||||||||
| Issuance of common stock for stock options exercises | — | — | 10 | — | 25 | — | 25 | |||||||||||||||||||||
| Fair value of warrants issued in connection with initial public offering of common stock | — | — | — | — | 3,039 | — | 3,039 | |||||||||||||||||||||
| Fair value of warrants issued in connection with the acquisition of product rights | — | — | — | — | 3,978 | — | 3,978 | |||||||||||||||||||||
| Stock-based compensation | — | — | — | — | 1,141 | — | 1,141 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (26,478 | ) | (26,478 | ) | |||||||||||||||||||
| Balance at December 31, 2018 | — | $ | — | 37,362 | $ | 4 | $ | 95,430 | $ | (35,776 | ) | $ | 59,658 | |||||||||||||||
| Issuance of common stock in connection with public follow on offering, net of issuance costs | — | — | 5,750 | 1 | 42,650 | — | 42,651 | |||||||||||||||||||||
| Issuance of common stock in connection with private placement with Amgen | — | — | 2,500 | — | 20,000 | — | 20,000 | |||||||||||||||||||||
| Issuance of common stock in connection with stock option exercises | — | — | 79 | — | 290 | — | 290 | |||||||||||||||||||||
| Issuance of common stock in connection with warrant exercises | — | — | 1,967 | — | — | — | — | |||||||||||||||||||||
| Stock-based compensation | — | — | — | — | 2,842 | — | 2,842 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (43,285 | ) | (43,285 | ) | |||||||||||||||||||
| Balance at December 31, 2019 | — | $ | — | 47,658 | $ | 5 | $ | 161,212 | $ | (79,061 | ) | $ | 82,156 | |||||||||||||||
The accompanying notes are an integral part of the financial statements.
| F-5 |
PROVENTION BIO, INC.
(in thousands of U.S. dollars)
| For the Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Operating Activities | ||||||||||||
| Net loss | $ | (43,285 | ) | $ | (26,478 | ) | $ | (9,133 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Stock-based compensation expense | 2,842 | 1,141 | 207 | |||||||||
| Stock-based consideration in connection with acquisition of product rights (Note 5) | — | 3,978 | 3,400 | |||||||||
| Change in fair value of warrant liability | — | 520 | 125 | |||||||||
| Amortization of premium and discounts on marketable securities | (16 | ) | — | — | ||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Prepaid expenses and other current assets | 2,367 | (2,396 | ) | (469 | ) | |||||||
| Accounts payable | 1,207 | 109 | 459 | |||||||||
| Accrued interest receivable | 58 | — | — | |||||||||
| Accrued expenses | 762 | 484 | 529 | |||||||||
| Net cash used in operating activities | (36,065 | ) | (22,642 | ) | (4,882 | ) | ||||||
| Investing activities | ||||||||||||
| Purchases of marketable securities | (46,250 | ) | — | — | ||||||||
| Net cash used in investing activities | (46,250 | ) | — | — | ||||||||
| Financing activities | ||||||||||||
| Proceeds from underwritten public offering, net | 42,651 | — | — | |||||||||
| Proceeds from private placement with Amgen | 20,000 | — | — | |||||||||
| Proceeds from initial public offering, net | — | 59,322 | — | |||||||||
| Proceeds from issuance of Series A Convertible Redeemable Preferred Stock, net | — | — | 26,715 | |||||||||
| Proceeds from issuance of common stock | 290 | 25 | 1 | |||||||||
| Net cash provided by financing activities | 62,941 | 59,347 | 26,716 | |||||||||
| Net (decrease) increase in cash and cash equivalents | (19,374 | ) | 36,705 | 21,834 | ||||||||
| Cash and cash equivalents at beginning of period | 58,539 | 21,834 | — | |||||||||
| Cash and cash equivalents at end of period | $ | 39,165 | $ | 58,539 | $ | 21,834 | ||||||
| Supplemental disclosure of non-cash financing transactions: | ||||||||||||
| Conversion of Series A Preferred Stock to common stock upon completion of IPO | $ | — | $ | 26,461 | $ | — | ||||||
| Reclassification of Series A Preferred Stock warrant liability to stockholders’ equity | $ | — | $ | 1,518 | $ | — | ||||||
| Fair value of warrant issued in connection with Series A Convertible Redeemable Preferred Stock | $ | — | $ | — | $ | 873 | ||||||
| Accretion of Series A Convertible Redeemable Preferred Stock | $ | — | $ | 276 | $ | 343 | ||||||
The accompanying notes are an integral part of the financial statements.
| F-6 |
PROVENTION BIO, INC.
(tabular dollars and shares in thousands, except per share data)
1. DESCRIPTON OF BUSINESS AND BASIS OF PRESENTATION
Business
Provention Bio, Inc. (the “Company”) was incorporated on October 4, 2016 under the laws of the State of Delaware. The Company is a clinical stage biopharmaceutical company, focused on the development and commercialization of novel therapeutics and innovative approaches to intercept and prevent immune-mediated diseases. Since its inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital. The Company’s business is subject to significant risks and uncertainties and will be dependent on raising substantial additional capital before it becomes profitable and it may never achieve profitability.
Basis of Presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) and include all adjustments necessary for the fair presentation of the Company’s financial position for the periods presented.
2. LIQUIDITY
The accompanying financial statements have been prepared assuming the Company will continue as a going concern, which contemplates continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred recurring losses since inception and as of December 31, 2019, the Company had an accumulated deficit of $79.1 million. To date, the Company has not generated any revenues and has financed its operations primarily through a private offering of Series A Convertible Redeemable Preferred Stock in April 2017, its initial public offering (“IPO”) in July 2018, and an underwritten public offering and concurrent private placement in September 2019.
In April 2017, the Company completed its private placement of Series A Convertible Redeemable Preferred Stock (the “Series A Offering”). The Company issued an aggregate 11,381,999 shares of Series A Convertible Redeemable Preferred Stock at $2.50 per share. The Company received net proceeds of $26.7 million.
In July 2018, the Company issued and sold an aggregate of 15,969,563 shares of common stock in its IPO at a public offering price of $4.00 per share. In connection with the IPO, the Company issued to MDB Capital Group, LLC (“MDB”), the underwriter in the IPO, and its designees warrants to purchase 1,596,956 shares of Common Stock at an exercise price of $5.00 per share. The Company received net proceeds from the IPO of $59.3 million, after deducting underwriting discounts and commissions of approximately $3.7 million and other offering expenses of approximately $0.8 million. Upon the closing of the IPO, all of the Company’s shares of redeemable convertible preferred stock outstanding at the time of the offering were automatically converted into 11,381,999 shares of common stock. In addition, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock also converted to warrants for the purchase of 558,740 shares of the Company’s common stock.
In September 2019, the Company completed an underwritten public offering in which it sold 5,750,000 shares of common stock at a public offering price of $8.00 per share. The 5,750,000 shares sold included the full exercise of the underwriters’ option to purchase 750,000 shares at a price of $8.00 per share. Concurrent with the underwritten public offering, the Company sold 2,500,000 shares of common stock to Amgen, Inc. at the public offering price of $8.00 per share in a private placement, pursuant to the terms of the Company’s License and Collaboration Agreement with Amgen Inc, dated as of November 5, 2018. Aggregate net proceeds from the underwritten public offering and the concurrent private placement were $62.7 million, net of approximately $2.8 million in underwriting discounts and commissions and other offering expenses of $0.5 million.
| F-7 |
The Company has devoted substantially all of its financial resources and efforts to research and development and expects to continue to incur significant expenses and increasing operating losses over the next several years due to, among other things, costs related to research funding, development of its product candidates and its preclinical programs, strategic alliances and the development of its administrative and commercial organization. The Company’s net losses may fluctuate significantly from quarter to quarter and year to year.
The Company will require substantial additional financing to fund its operations and to continue to execute its strategy. The Company intends to raise capital through public or private equity financings. The sale of equity and other securities may result in dilution to the Company’s stockholders and certain of those securities may have rights senior to those of the Company’s existing shares. If the Company raises additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict the Company’s operations. Any other third-party funding arrangement could require the Company to relinquish valuable rights. The source, timing and availability of any future financing will depend principally upon market conditions, and, more specifically, on the progress of the Company’s clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to the Company. Lack of necessary funds may require the Company, among other things, to delay, scale back or eliminate some or all of the Company’s planned operations.
Based on the Company’s business plans, management believes that its cash, cash equivalents and marketable securities on hand at December 31, 2019 are sufficient to meet the Company’s obligations for at least 12 months from the issuance of these financial statements.
3. SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies followed by the Company in the preparation of the financial statements is as follows:
Use of estimates
The process of preparing financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of assets and liabilities at the date of financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates and changes in estimates may occur.
Segment and geographic information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating and reporting segment.
Cash, cash equivalents and concentration of credit risk
The Company considers only those investments which are highly liquid, readily convertible to cash, or that mature within three months from date of purchase to be cash equivalents. Marketable securities are those investments with original maturities in excess of three months. The carrying amounts reported in the balance sheets for cash and cash equivalents are valued at cost, which approximates their fair value
The Company has no significant off-balance-sheet concentration of credit risk such as foreign exchange contracts, option contracts or other hedging arrangements. The Company holds cash and cash equivalents in banks in excess of FDIC insurance limits. However, the Company believes risk of loss is minimal as the cash and cash equivalents are held by large, highly-rated financial institutions.
Marketable Securities
The Company considers securities with original maturities of greater than three months to be available for sale securities. Available for sale securities with original maturities of greater than one year are recorded as non-current assets. Available for sale securities are recorded at fair value and unrealized gains and losses are recorded within accumulated other comprehensive income. The estimated fair value of the available for sale securities is determined based on quoted market prices or rates for similar instruments. In addition, the cost of debt securities in this category is adjusted for amortization of premium and accretion of discount to maturity.
| F-8 |
The Company reviews the status of each security quarterly to determine whether an other-than-temporary impairment has occurred. In making its determination, the Company considers a number of factors, including: (1) the significance of the decline; (2) whether the security was rated below investment grade; (3) how long the security has been in an unrealized loss position; and (4) the Company’s ability and intent to retain the investment for a sufficient period of time for it to recover.
The Company’s available for sale securities are solely invested in U.S. Treasury securities.
Financial instruments
Cash, cash equivalents and marketable securities are reflected in the accompanying financial statements at fair value. The carrying amount of accounts payable and accrued expenses, including accrued research and development expenses, approximates fair value due to the short-term nature of those instruments.
Net loss per common share
Net loss per share information is determined using the two-class method, which includes the weighted-average number of shares of common stock outstanding during the period and other securities that participate in dividends (a participating security). The Company considered the Series A Preferred Stock, all of which were automatically converted to common stock upon the Company’s IPO, to be participating securities because they included rights to participate in dividends, if any, with the common stock.
Under the two-class method, basic net loss per share attributable to common stockholders is computed by dividing the net income (loss) attributable to common stockholders by the weighted-average number of shares of common stock outstanding during the period. The net loss attributable to common stockholders is calculated by adjusting the net loss of the Company for the accretion on the Preferred Stock. Net losses are not allocated to preferred stockholders as they do not have an obligation to share in the Company’s net losses. In periods with net income attributable to common stockholders, the Company would allocate net income first to preferred stockholders based on dividend rights under the Company’s certificate of incorporation and then to preferred and common stockholders based on ownership interests. Diluted net loss per share attributable to common stockholders is computed using the more dilutive of (1) the two-class method or (2) the if-converted method.
Diluted net loss per share attributable to common stockholders is the same as basic net loss per share attributable to common stockholders for all periods presented since the effect of potentially dilutive securities are anti-dilutive given the net loss of the Company.
Foreign Currency Translation
The Company considers the U.S. dollar to be its functional currency. Expenses denominated in foreign currencies are translated at the exchange rate on the date the expense is incurred. The effect of exchange rate fluctuations on translating foreign currency assets and liabilities into U.S. dollars is included in the Statements of Operations. Foreign exchange transaction gains and losses are included in the results of operations and are not material in the Company’s financial statements.
Research and development expenses
Research and development expenses primarily consist of costs associated with the preclinical and clinical development of our product candidate portfolio, including the following:
| ● | external research and development expenses incurred under arrangements with third parties, such as contract research organizations (CROs) and other vendors and contract manufacturing organizations (CMOs) for the production of drug substance and drug product; and | |
| ● | employee-related expenses, including salaries, benefits and share-based compensation expense. |
Research and development expenses also include costs of acquired product licenses and related technology rights where there is no alternative future use, costs of prototypes used in research and development, consultant fees and amounts paid to certain of our collaborative partners.
| F-9 |
All research and development expenses are charged to operations as incurred in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic, or ASC, 730, Research and Development. The Company accounts for non-refundable advance payments for goods and services that will be used in future research and development activities as expenses when the service has been performed or when the goods have been received, rather than when the payment is made.
Accrued Research and Development Expenses
As part of the process of preparing our financial statements, the Company is required to estimate its accrued expenses. This process involves reviewing quotations and contracts, identifying services that have been performed on the Company’s behalf and estimating the level of service performed and the associated cost incurred for the service when the Company has not yet been invoiced or otherwise notified of the actual cost. The majority of the Company’s service providers invoice us monthly in arrears for services performed or when contractual milestones are met. The Company makes estimates of its accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to the Company at that time. The Company periodically confirms the accuracy of its estimates with the service providers and make adjustments if necessary. The significant estimates in the Company’s accrued research and development expenses are related to expenses incurred with respect to CROs, CMOs and other vendors in connection with research and development and manufacturing activities.
The Company bases its expense related to CROs and CMOs on its estimates of the services received and efforts expended pursuant to quotations and contracts with such vendors that conduct research and development and manufacturing activities on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to the Company’s vendors will exceed the level of services provided and result in a prepayment of the applicable research and development or manufacturing expense. In accruing service fees, the Company estimates the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from its estimate, the Company adjusts the accrual or prepaid expense accordingly. Although the Company does not expect its estimates to be materially different from amounts actually incurred, the Company’s understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and could result in us reporting amounts that are too high or too low in any particular period. There have been no material changes in estimates for the periods presented.
Stock-based compensation expense
The Company follows the provisions of ASC 718, Compensation—Stock Compensation, which requires the measurement and recognition of compensation expense for all share-based payment awards made to employees and non-employees, including stock options. Stock-based compensation expense is based on the grant date fair value estimated in accordance with the provisions of ASC 718 and is generally recognized as an expense over the requisite service period. For grants containing performance-based vesting provisions, the grant-date fair value of the performance-based stock options is recognized as compensation expense once it is probable that the performance condition will be achieved. The Company accounts for actual forfeitures in the period the forfeiture occurs.
Stock Options
The Company estimates the fair value of stock options on the date of grant using the Black-Scholes option-pricing model. Due to the lack of trading history, the Company’s computation of stock-price volatility is based on the volatility rates of comparable publicly held companies over a period equal to the expected term of the options granted by the Company. The Company’s computation of expected term is determined using the “simplified” method, which is the midpoint between the vesting date and the end of the contractual term. The Company believes that it does not have sufficient reliable exercise data in order to justify the use of a method other than the “simplified” method of estimating the expected exercise term of employee stock option grants. The Company utilizes a dividend yield of zero based on the fact that the Company has never paid cash dividends to stockholders and has no current intentions to pay cash dividends. The risk-free interest rate is based on the zero-coupon U.S. Treasury yield at the date of grant for a term equivalent to the expected term of the option.
| F-10 |
In June 2018, the FASB issued ASU No. 2018-07, Improvements to Nonemployee Share-Based Payment Accounting, (“ASU 2018-07”) which supersedes ASC 505-50 and expands the scope of ASC 718 to include all share-based payments arrangements related to the acquisition of goods and services from both employees and nonemployees. During the third quarter of 2018, the Company early adopted ASU 2018-07. After the adoption of ASU 2018-07, the measurement date for non-employee awards is the date of grant. Compensation expense for non-employees is recognized, without changes to the fair value of the award, over the requisite service period, which is the vesting period of the respective award. The adoption of ASU 2018-07 did not have a material impact the Company’s financial statements or footnote disclosures.
Prior to the third quarter of 2018, the Company accounted for awards of equity instruments issued to non-employees in accordance with ASC Topic 505-50, Equity-Based Payment to Non-Employees, (“ASC 505-50”) and accordingly the value of the stock compensation to non-employees was based upon the measurement date as determined at either a) the date at which a performance commitment was reached, or b) at the date at which the necessary performance to earn the equity instruments was completed, which was normally the end of the vesting period. At the end of each financial reporting period prior to completion of the service, the fair value of these awards was remeasured using updated assumption inputs in the Black-Scholes option-pricing model.
Stock-based compensation expense is included in both research and development expenses and general and administrative expenses in the Statements of Operations.
Warrant liability
In April 2017, the Company issued warrants to purchase shares of Series A Convertible Redeemable Preferred Stock in connection with the issuance of the Series A Preferred Stock. The Company accounted for these warrants as a liability in the financial statements because the underlying instrument into which the warrants were exercisable contained redemption provisions that were outside the Company’s control.
Upon the completion of the IPO in July 2018, the warrants issued in connection with the Series A Convertible Redeemable Preferred Stock converted to warrants for the purchase of 558,740 shares of our common stock and the warrants no longer contain redemption provisions that are outside the Company’s control. The liability associated with these warrants was revalued just prior to the completion of the IPO, with the change in the fair value of the warrant liability charged to earnings. The warrant liability was then reclassified to additional paid-in capital upon the completion of the IPO in July 2018.
The fair value of the warrants was determined using the Black-Scholes option-pricing model and were re-measured to fair value at each financial reporting period date with any changes in fair value recognized in the statements of operations. See Note 8 to our financial statements for further information.
Income taxes
The Company utilizes the liability method of accounting for deferred income taxes, as set forth in ASC 740, Income Taxes. Under this method, deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the carrying amounts and the tax basis of assets and liabilities. A valuation allowance is established against deferred tax assets when, based on the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. The Company’s policy is to record interest and penalties on uncertain tax positions as income tax expense.
Recent Accounting Pronouncements
The Company considers the applicability and impact of all Accounting Standards Updates (“ASUs”), ASUs not discussed below were assessed and determined to be either not applicable or are expected to have minimal impact on our consolidated balance sheets or statements of operations.
| F-11 |
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”), which requires lessees to recognize assets and liabilities for the rights and obligations created by most leases on their balance sheet. Effective January 1, 2019, the Company adopted ASU 2016-02. The adoption had no impact of the Company’s financial statements and related disclosures as the Company does not have any lease agreements.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820), which modifies the disclosure requirements on fair value measurements in Topic 820, Fair Value Measurement, including, among other changes, the consideration of costs and benefits when evaluating disclosure requirements. For public companies, the amendments are effective for annual reporting periods beginning after December 15, 2019, including interim periods within those annual periods. Early adoption is permitted. The Company is currently assessing the impact that adopting this new accounting guidance will have on its financial statements and footnote disclosures.
In August 2018, the FASB issued ASU No. 2018-15, Intangibles - Goodwill and Other - Internal-Use Software (Subtopic 350-40): Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract (a consensus of the FASB Emerging Issues Task Force). ASU 2018-15 aligns the requirements for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software (and hosting arrangements that include an internal-use software license). This guidance is effective for fiscal years beginning after December 15, 2019, and for interim periods within those fiscal years, with early adoption permitted. The Company is currently assessing the impact that adopting this new accounting guidance will have on its financial statements and footnote disclosures.
In November 2018, the FASB issued ASU No. 2018-18, Collaborative Arrangements (Topic 808), which clarifies the interaction between the guidance for collaborative arrangements (Topic 808) and the new revenue recognition standard (Topic 606). For public companies, the amendments are effective for annual reporting periods beginning after December 15, 2019, including interim periods within those annual periods. Early adoption is permitted. The Company is currently assessing the impact that adopting this new accounting guidance will have on its financial statements and footnote disclosures.
4. CAPITALIZATION
As of December 31, 2019, the Company had authorized 100,000,000 shares of Common Stock, $0.0001 par value per share, of which 47,658,361 shares were issued and outstanding. In addition, as of December 31, 2019, the Company had authorized 25,000,000 shares of Preferred Stock, $0.0001 par value per share, of which none were issued and outstanding.
As of December 31, 2018, the Company had authorized 100,000,000 shares of Common Stock, $0.0001 par value per share, of which 37,361,562 shares were issued and outstanding. In addition, as of December 31, 2018, the Company had authorized 25,000,000 shares of Preferred Stock, $0.0001 par value per share, of which none were issued and outstanding.
5. LICENSE AND OTHER AGREEMENTS
In May 2018, the Company entered into an Asset Purchase Agreement with MacroGenics (the “MacroGenics Asset Purchase Agreement”) pursuant to which the Company acquired MacroGenics’ interest in teplizumab (renamed PRV-031), a humanized mAb for the treatment of Type 1 Diabetes (T1D). As partial consideration for the MacroGenics Asset Purchase Agreement, the Company granted MacroGenics a warrant to purchase 2,162,389 shares of the Company’s common stock at an exercise price of $2.50 per share (See Note 11). The Company is obligated to pay MacroGenics contingent milestone payments totaling $170.0 million upon the achievement of certain regulatory approval milestones, including $60.0 million payable upon approval of a Biologics License Application (“BLA”) in the United States. In addition, the Company is obligated to make contingent milestone payments to MacroGenics totaling $225.0 million upon the achievement of certain sales milestones. The Company has also agreed to pay MacroGenics a single-digit royalty on net sales of the product. We have also agreed pay third-party obligations, including low single-digit royalties, a portion of which is creditable against royalties payable to MacroGenics, aggregate milestone payments of up to approximately $1.3 million and other consideration, for certain third-party intellectual property under agreements the Company is assuming pursuant to the MacroGenics Asset Purchase Agreement. Further, the Company is required to pay MacroGenics a low double-digit percentage of certain consideration to the extent it is received in connection with a future grant of rights to PRV-031 by the Company to a third party. The Company is obligated to use reasonable commercial efforts to develop and seek regulatory approval for PRV-031. As of December 31, 2019, the Company has not achieved any milestones that would trigger payments to MacroGenics.
| F-12 |
In May 2018, the Company entered into a License Agreement with MacroGenics, Inc. (the “MacroGenics License Agreement”), pursuant to which MacroGenics, Inc. (“MacroGenics”) granted the Company exclusive global rights for the purpose of developing and commercializing MGD010 (renamed PRV-3279), a humanized protein and a potential treatment for systemic lupus erythematosus (SLE) and other similar diseases. As partial consideration for the MacroGenics License Agreement, the Company granted MacroGenics a warrant to purchase 270,299 shares of the Company’s common stock at an exercise price of $2.50 per share (See Note 11). The Company is obligated to make contingent milestone payments to MacroGenics totaling $42.5 million upon the achievement of certain developmental and approval milestones for the first indication, and an additional $22.5 million upon the achievement of certain regulatory approvals for a second indication. In addition, the Company is obligated to make contingent milestone payments to MacroGenics totaling $225.0 million upon the achievement of certain sales milestones. The Company has also agreed to pay MacroGenics a single-digit royalty on net sales of the product. Further, the Company is required to pay MacroGenics a low double-digit percentage of certain consideration to the extent received in connection with a future grant of rights to PRV-3279 by the Company to a third party. The Company is obligated to use commercially reasonable efforts to develop and seek regulatory approval for PRV-3279. The license agreement may be terminated by either party upon a material breach or bankruptcy of the other party, by Provention without cause upon prior notice to MacroGenics, and by MacroGenics in the event that the Company challenges the validity of any licensed patent under the agreement, but only with respect to the challenged patent.
The Company recorded the warrants issued under the MacroGenics Asset Purchase Agreement and the MacroGenics License Agreement at an estimated fair value of $1.64 per share, approximately $4.0 million in the aggregate, as license fee expense included as part of Research & Development Expense during the second quarter of 2018.
In July 2019, MacroGenics elected to exercise its warrants for an aggregate of 2,432,688 shares on a cashless basis, resulting in the Company’s net issuance of 1,948,474 shares. Following the MacroGenics’ July 2019 warrant exercises, the there are no additional warrants outstanding in connection with the MacroGenics License Agreement and the MacroGenics Asset Purchase Agreement. See Note 11 of these financial statements for further details of the warrants issued to MacroGenics.
In November 2018, the Company entered into a License and Collaboration Agreement (the “Amgen Agreement”) with Amgen, Inc. (“Amgen”) for PRV-015 (formerly AMG 714), a novel anti-IL-15 monoclonal antibody being developed for the treatment of gluten-free diet non-responsive celiac disease (NRCD). Under the terms of the agreement, the Company will conduct and fund a Phase 2b trial in NRCD and lead the development and regulatory activities for the program. Amgen agreed to make an equity investment of up to $20.0 million in the Company, subject to certain terms and conditions set forth in the agreement. Amgen is also responsible for the manufacturing of PRV-015. Upon completion of the Phase 2b trial, a $150.0 million milestone payment is due from Amgen to the Company, plus an additional regulatory milestone payment, and single digit royalties on future sales. If Amgen elects not to pay the $150.0 million milestone, AMG 714 rights will be transferred to Company pursuant to a termination license agreement from Amgen and the Company. The Company will be obligated to make certain contingent milestone payments to Amgen and other third parties totaling up to $70.0 million upon the achievement of certain clinical and regulatory milestones and a low double-digit royalty on net sales of any approved product based on the IL-15 technology. The agreement may be terminated by the Company without cause (in which case the exclusive global rights to the technology will transfer back to Amgen) and by either party upon a material breach. The agreement expires upon the expiration of Amgen’s last obligation to make royalty payments to Provention (or, the Company’s last obligation to make royalty payments to Amgen, if the program rights are transferred to the Company). In September 2019, in a private placement completed concurrently with the Company’s underwritten public offering, Amgen purchased 2,500,000 shares of the Company’s common stock at the underwritten public offering price of $8.00 per share, for a total investment of $20.0 million.
| F-13 |
In April 2017, the Company entered into a License Agreement with Vactech Ltd. (the “Vactech License Agreement”), pursuant to which Vactech Ltd. (“Vactech”) granted the Company exclusive global rights for the purpose of developing and commercializing the group B coxsackie virus vaccine (CVB) platform technology. In consideration of the licenses and other rights granted by Vactech, the Company issued two million shares of its common stock to Vactech. The Company recorded the issuance of the shares at their estimated fair value of approximately $1.70 per share for a total of $3.4 million as a license fee expense included as part of Research & Development Expense for the year ended December 31, 2017. Provention paid Vactech a total of approximately $0.5 million for transition and advisory services during the first 18 months of the term of the agreement. In addition, Provention may be obligated to make a series of contingent milestone payments to Vactech totaling up to an additional $24.5 million upon the achievement of certain clinical development and regulatory filing milestones. In addition, the Company has agreed to pay Vactech tiered single-digit royalties on net sales of any approved product based on the CVB platform technology and three additional payments totaling $19.0 million upon the achievement of certain annual net sales levels. The Vactech Agreement may be terminated by the Company on a country by country basis without cause (in which case the exclusive global rights to the technology will transfer back to Vactech) and by either party upon a material breach or insolvency of the other party. If the Company terminates the agreement with respect to two or more specified European countries, the agreement will be deemed terminated with respect to all of the EU, and if the Company terminates the agreement with respect to the United States, the agreement will be deemed terminated with respect to all of North America. The agreement expires upon the expiration of the Company’s last obligation to make royalty payments to Vactech. As of December 31, 2019, the Company has not achieved any milestones that would trigger payments to Vactech.
In March 2018, the Company entered into a Development Services Agreement with The Institute of Translational Vaccinology (the “Intravacc Development Services Agreement”), pursuant to which The Institute of Translational Vaccinology (“Intravacc”) will provide services related to process development, non-GMP and GMP manufacturing of the Company’s polyvalent coxsackie virus B vaccine (CVB), including providing proprietary technology for manufacturing purposes. The Company will pay Intravacc approximately 10 million euros for their services over the development and manufacturing period which we the Company currently expects will last for approximately 24 to 30 months. Each party retains its existing intellectual property and will share newly developed intellectual property via a fully-paid non-exclusive license between the parties for all development work through phase 1 clinical trials. Any future use, including commercial use, of Intravacc’s technology will be subject to a separate nonexclusive license agreement. The Intravacc Development Services Agreement may be terminated by us with ninety days’ notice without cause and by either party upon a material breach or insolvency of the other party. As of December 31, 2019, the Company had paid Intravacc a total of approximately 8.0 million euros, or approximately $9.4 million, for services provided by Intravacc under the Intravacc Development Services Agreement.
In April 2017, the Company entered into the Janssen CSF-1R License Agreement, pursuant to which Janssen Pharmaceutica NV granted the Company exclusive global rights for the purpose of developing and commercializing a colony stimulating factor 1 receptor (CSF-1R) inhibitor named JNJ-40346527 (renamed PRV-6527) for inflammatory bowel diseases including Crohn’s disease and UC. The Company evaluated PRV-6527 for Crohn’s disease in a recently completed Phase 2a clinical trial (the PRINCE study). See Item 1 – Business, Recent Developments of this Annual Report on Form 10-k for the announcement of top-line results of the PRINCE study. In December 2019, Janssen declined its option to buy back the rights to PRV-6527. and as such, all rights will remain with the Company. The Company will be obligated to make contingent milestone payments to Janssen totaling $35.0 million upon the achievement of certain clinical and regulatory milestones for the first indication and an additional $20.0 million upon the achievement of certain clinical and regulatory milestones for a second indication. In addition, the Company has agreed to pay Janssen tiered single-digit royalties on net sales of any approved product based on the CSF-1R technology and three additional payments totaling $100.0 million upon the achievement of certain annual net sales levels. As of December 31, 2019, no milestones have been achieved that would trigger payments to Janssen under the CSF-1R License Agreement.
In April 2017, the Company entered into a License, Development and Commercialization Agreement with Janssen Sciences Ireland UC (the “TLR3 License Agreement”), pursuant to which Janssen Sciences Ireland UC granted the Company exclusive global rights for the purpose of developing and commercializing JNJ-42915925 (renamed PRV-300), an anti-Toll-Like Receptor 3 (TLR3) antibody. The Company evaluated PRV-300 for UC in a recently completed Phase 1b trial (the PULSE study). The Company returned the rights to PRV-300 to Janssen and exercised its termination rights under the TLR3 License Agreement in the third quarter of 2019.
| F-14 |
6. NET LOSS PER SHARE OF COMMON STOCK
The following table sets forth the computation of basic and diluted loss per share for the years ended December 31, 2019, 2018, and 2017:
| For the Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Net loss, basic | $ | (43,285 | ) | $ | (26,478 | ) | $ | (9,133 | ) | |||
| Accretion of Series A Convertible Redeemable Preferred Stock | — | (276 | ) | (343 | ) | |||||||
| Net loss attributable to common stockholders | $ | (43,285 | ) | $ | (26,754 | ) | $ | (9,476 | ) | |||
| Weighted average common shares outstanding, basic and diluted | 40,747 | 22,441 | 9,370 | |||||||||
| Net loss per common share, basic and diluted | $ | (1.06 | ) | $ | (1.19 | ) | $ | (1.01 | ) | |||
The following potentially dilutive securities have been excluded from the computation of diluted weighted average shares outstanding as they would be antidilutive:
| For the Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Series A Convertible Redeemable Preferred Stock | — | — | 11,382 | |||||||||
| Warrants | 2,125 | 4,588 | 559 | |||||||||
| Stock options | 5,894 | 3,476 | 2,708 | |||||||||
7. ACCRUED EXPENSES
Accrued expenses consisted of the following:
| December 31, 2019 | December 31, 2018 | |||||||
| Accrued research and development costs | $ | 1,700 | $ | 1,023 | ||||
| Accrued professional fees | 221 | 126 | ||||||
| Other accrued liabilities | 90 | 154 | ||||||
| Accrued compensation | 54 | — | ||||||
| Total accrued expenses | $ | 2,065 | $ | 1,303 | ||||
8. FAIR VALUE OF ASSETS AND LIABILITIES
The carrying amounts reported in the balance sheet for cash and cash equivalents, accounts payable and accrued expenses approximate fair value based on the short-term nature of these items.
In accordance with accounting principles generally accepted in the United States, fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. A three-level hierarchy prioritizes the inputs used to measure fair value as follows:
Level 1 – Valuation is based on quoted prices in active markets for identical assets or liabilities. Level 1 assets and liabilities generally include debt and equity securities that are traded in an active exchange market. Valuations are obtained from readily available pricing sources for market transactions involving identical assets or liabilities.
Level 2 – Valuation is based on observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3 – Valuation is based on unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires significant management judgment or estimation.
| F-15 |
The Company had no assets or liabilities classified as Level 1 or Level 2 as of December 31, 2018. Liability classified warrants issued in April 2017 to the placement agent in conjunction with the Series A Preferred Stock offering (see Note 9) were previously classified as Level 3. During the year ended December 31, 2018, the Company recorded $0.5 million of expense related to the change in the fair value (an increase) of the Series A Preferred Stock warrant liability. Upon the completion of the IPO in July 2018, these warrants converted to warrants for the purchase of 558,740 shares of our common stock and no longer contain redemption provisions outside the Company’s control. The liability associated with these warrants was revalued just prior to the completion of the IPO, with the change in the fair value of the warrant liability charged to earnings. The warrant liability was then reclassified to additional paid-in capital upon the completion of the IPO in July 2018.
The following table sets forth a summary of the changes in the fair value of the Company’s level 3 financial instruments during the year ended December 31, 2018:
| Balance at December 31, 2017 | $ | 998 | ||
| Change in fair value during the period | 520 | |||
| Reclassification to equity upon completion of IPO | (1,518 | ) | ||
| Balance at December 31, 2018 | $ | — |
The Company had no level 3 financial instruments during the year ended December 31, 2019.
The following is a summary of assets and their related classifications under the fair value hierarchy:
| December 31, 2019 | ||||||||||||||||
| Financial Instruments Carried at Fair Value | ||||||||||||||||
| Quoted prices in | ||||||||||||||||
| active markets for | Significant other | Significant | ||||||||||||||
| identical items | observable inputs | unobservable inputs | ||||||||||||||
| (Level 1) | (Level 2) | (Level 3) | Total | |||||||||||||
| Assets: | ||||||||||||||||
| Cash and cash equivalents1 | $ | 39,165 | $ | — | $ | — | $ | 39,165 | ||||||||
| Investments in U.S. Treasury securities2 | 46,208 | — | — | 46,208 | ||||||||||||
| December 31, 2018 | ||||||||||||||||
| Financial Instruments Carried at Fair Value | ||||||||||||||||
| Quoted prices in | ||||||||||||||||
| active markets for | Significant other | Significant | ||||||||||||||
| identical items | observable inputs | unobservable inputs | ||||||||||||||
| (Level 1) | (Level 2) | (Level 3) | Total | |||||||||||||
| Assets: | ||||||||||||||||
| Cash and cash equivalents 1 | $ | 58,539 | $ | — | $ | — | $ | 58,539 | ||||||||
(1) Cash and cash equivalents primarily include investments in money market funds and U.S. Treasury securities with maturity dates less than three months from the purchase date
(2) Investments in U.S. Treasury securities are classified as available for sale securities
The Company recognizes transfers between levels within the fair value hierarchy, if any, at the end of each quarter. There were no transfers in or out of Level 1, Level 2 or Level 3 during the years ended December 31, 2019 and 2018.
The Company’s unrealized gain (loss) on its available for sale securities was de minimis during the year ended December 31, 2019.
| F-16 |
9. SERIES A CONVERTIBLE REDEEMABLE PREFERRED STOCK
In April 2017, the Company issued 11,381,999 shares of Series A Convertible Redeemable Preferred Stock for $2.50 per share which raised gross proceeds of $28.5 million and net cash proceeds of $26.7 million after deducting certain issuance costs including warrants. The Company classified the Series A Convertible Redeemable Preferred Stock outside of permanent equity based upon the terms of the instrument. See Note 11 for a description of the warrants issued to the placement agent in connection with this transaction.
In connection with the closing of the Company’s IPO in July 2018, all of the Company’s shares of redeemable convertible preferred stock outstanding at the time of the offering were automatically converted into 11,381,999 shares of common stock.
10. STOCK OPTIONS
In 2017, the Company adopted the Provention Bio, Inc. 2017 Equity Incentive Plan (the “2017 Plan”). Pursuant to the 2017 Plan, the Company’s Board of Directors may grant incentive stock options, nonqualified stock options, and restricted stock to employees, officers, directors, consultants and advisors. As of December 31, 2019, there were options to purchase an aggregate of 5,893,751 shares of Common Stock outstanding under the 2017 Plan. Options issued under the 2017 Plan are exercisable for up to 10 years from the date of issuance. There were 937,154 shares available for future grants as of December 31, 2019.
In connection with the evergreen provisions of the 2017 Plan, the number of shares available for issuance under the 2017 Plan was increased by 3,000,000 shares, as determined by the board of directors under the provisions described above, effective as of January 1, 2020. As of January 1, 2020, there were approximately 3,937,154 shares available for future grants.
In connection with the completion of its IPO, the Company amended and restated its 2017 Plan to, among other things, include an evergreen provision, which would automatically increase the number of shares available for issuance under the 2017 Plan in an amount equal to (1) the difference between (x) 18% of the total shares of the Company’s common stock outstanding, on a fully diluted basis, on December 31st of the preceding calendar year, and (y) the total number of shares of the Company’s common stock reserved under the 2017 Plan on December 31st of such preceding calendar year or (2) an amount less than this calculated increase as determined by the board of directors.
Stock-based compensation
Total stock-based compensation expense recognized for both employees and non-employees was as follows:
| Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Research and development | $ | 1,296 | $ | 629 | $ | 112 | ||||||
| General and administrative | 1,546 | 512 | 95 | |||||||||
| Total share-based compensation expense | $ | 2,842 | $ | 1,141 | $ | 207 | ||||||
Option activity
The Company grants options with service-based vesting requirements as well as options with performance-based vesting requirements. Generally, the service-based requirements vest over a four-year period in multiple tranches. Each tranche of the performance-based component vests upon the achievement of a specific milestone. These milestones are related to the Company’s clinical trials, manufacturing activities, regulatory activities, and certain other performance metrics.
| F-17 |
A summary of option activity for the years ended December 31, 2019 and 2018 are presented below:
| Weighted- | ||||||||||||||||
| Weighted- | Average | |||||||||||||||
| Average | Remaining | |||||||||||||||
| Underlying | Exercise | Contractual | Intrinsic | |||||||||||||
| Stock Option Awards | Shares | Price | Term | Value | ||||||||||||
| Outstanding at January 1, 2018 | 2,708 | $ | 2.50 | |||||||||||||
| Granted | 855 | $ | 3.94 | |||||||||||||
| Exercised | (10 | ) | $ | 2.50 | ||||||||||||
| Forfeited or expired | (77 | ) | $ | 2.50 | ||||||||||||
| Outstanding at December 31, 2018 | 3,476 | $ | 2.85 | 8.8 years | $ | — | ||||||||||
| Granted | 2,797 | $ | 10.34 | |||||||||||||
| Exercised | (79 | ) | $ | 3.66 | ||||||||||||
| Forfeited or expired | (300 | ) | $ | 4.19 | ||||||||||||
| Outstanding at December 31, 2019 | 5,894 | $ | 6.33 | 8.5 years | $ | 50,530 | ||||||||||
| Exercisable at December 31, 2019 | 1,901 | $ | 2.67 | 7.7 years | $ | 23,260 | ||||||||||
The weighted average grant-date fair value of options granted during the year ended December 31, 2019 was $6.81 per share. As of December 31, 2019, there were approximately 1,579,000 unvested options subject to performance-based vesting criteria with approximately $9.3 million of unrecognized compensation expense. This expense will be recognized when each milestone becomes probable of occurring. In addition, as of December 31, 2019, there were approximately 2,414,000 options outstanding subject to time-based vesting with approximately $9.7 million of unrecognized compensation expense which will be recognized over a period of 3.2 years.
Cash proceeds from, and the aggregate intrinsic value of, stock options exercised during the periods presented below were as follows:
| Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Cash proceeds from options exercised | $ | 290 | $ | 25 | $ | — | ||||||
| Aggregate intrinsic value of options exercised | $ | 551 | $ | 5 | $ | — | ||||||
The Company uses the Black-Scholes option-pricing model to estimate the fair value of option awards with the following weighted-average assumptions for the period indicated:
| Years ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Exercise Price | $ | 10.34 | $ | 3.93 | $ | 1.81 | ||||||
| Expected volatility | 72 | % | 61 | % | 64 | % | ||||||
| Expected dividends | — | — | — | |||||||||
| Expected term (in years) | 6.3 | 6.2 | 6.5 | |||||||||
| Risk-free interest rate | 1.90 | % | 2.78 | % | 1.96 | % | ||||||
The weighted-average valuation assumptions were determined as follows:
| ● | Risk-free interest rate: The Company bases the risk-free interest rate on the interest rate payable on U.S. Treasury securities in effect at the time of grant for a period that is commensurate with the assumed expected option term. | |
| ● | Expected annual dividends: The estimate for annual dividends is 0%, because the Company has not historically paid, and does not expect for the foreseeable future to pay, a dividend. |
| F-18 |
| ● | Expected stock price volatility: The expected volatility used is based on historical volatilities of similar entities within the Company’s industry which were commensurate with the Company’s expected term assumption. | |
| ● | Expected term of options: The expected term of options represents the period of time options are expected to be outstanding. The expected term of the options granted to employees is derived from the “simplified” method as described in Staff Accounting Bulletin 107 relating to stock-based compensation, whereby the expected term is an average between the vesting period and contractual period due to the limited operating history. The expected term for options granted to non-employees is equal to the contractual term of the awards. |
11. WARRANTS
In connection with the April 2017 sale of Series A Convertible Redeemable Preferred Stock, the Company issued warrants to MDB, the Placement Agent, and its designees to purchase 558,740 shares of Series A Convertible Redeemable Preferred Stock with an exercise price of $2.50 per share with a seven-year term. The underlying instrument into which the warrants are exercisable contained redemption provisions that were outside the Company’s control. Accordingly, these warrants were considered liabilities and at issuance, the fair value of $0.9 million was recorded as a warrant liability against a reduction to the proceeds from the issuance of the Series A Convertible Redeemable Preferred Stock. Upon completion of the IPO in July 2018, the warrants automatically became warrants for the purchase of 558,740 shares of the Company’s common stock and because the warrants no longer contain redemption provisions outside the Company’s control, the Company determined that equity classification was appropriate. See Note 8 for further information. In July 2019, certain warrant holders from the Series A Offering elected to exercise warrants for an aggregate of 4,065 shares on a cashless basis, resulting in the Company’s net issuance of 3,167 shares. As of December 31, 2019, there were 554,675 warrants outstanding related to the Series A Offering.
In May 2018, in connection with the MacroGenics License Agreement and the MacroGenics Asset Purchase Agreement, the Company issued warrants to MacroGenics to purchase 2,432,688 shares of the Company’s common stock at an exercise price of $2.50 per share. These warrants had a seven-year term. The Company recorded the warrants issued under MacroGenics License Agreement and the MacroGenics Asset Purchase Agreement at an estimated fair value of $1.64 per share, approximately $4.0 million in the aggregate, as license fee expense included as part of Research & Development Expense during the year ended December 31, 2018.
The Company used valuation methods and assumptions that consider, among other factors, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants issued to MacroGenics. The fair values of these instruments were determined using models based on inputs that require management judgment and estimates.
The fair value of the warrants issued to MacroGenics was measured at issuance on May 7, 2018 using the Black-Scholes option pricing model based on the following assumptions:
| Equity value upon issuance on May 7, 2018 | $ | 2.58 | ||
| Exercise Price | $ | 2.50 | ||
| Expected volatility | 62.0 | % | ||
| Expected dividends | — | |||
| Contractual term (in years) | 7.0 | |||
| Risk-free interest rate | 2.86 | % |
The MacroGenics warrants were evaluated under ASC 480, Distinguishing Liabilities from Equity, (“ASC 480”) and ASC 815, Derivatives and Hedging, (“ASC 815) and the Company determined that equity classification was appropriate.
In July 2019, MacroGenics elected to exercise its warrants for an aggregate of 2,432,688 shares on a cashless basis, resulting in the Company’s net issuance of 1,948,474 shares. Following the MacroGenics’ July 2019 warrant exercises, there were no additional warrants outstanding in connection with the MacroGenics License Agreement and the MacroGenics Asset Purchase Agreement.
| F-19 |
In connection with the Company’s completion of its IPO in July 2018, the Company issued to MDB, the underwriter in the IPO, and its designees warrants to purchase 1,596,956 shares of the Company’s common stock at an exercise price of $5.00 per share. These warrants have a five-year term.
The Company used valuation methods and assumptions that consider, among other factors, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants. The fair values of these instruments were determined using models based on inputs that require management judgment and estimates.
The fair value of the warrants issued to MDB were measured at issuance on July 19, 2018 using the Black-Scholes option-pricing model based on the following assumptions:
| Equity value upon issuance on July 19, 2018 | $ | 4.00 | ||
| Exercise Price | $ | 5.00 | ||
| Expected volatility | 60.0 | % | ||
| Expected dividends | — | |||
| Contractual term (in years) | 5.0 | |||
| Risk-free interest rate | 2.74 | % |
The Company estimated the fair value of the warrants issued to MDB to be $1.90 per share, approximately $3.0 million in the aggregate, which was recorded as a cost of the IPO. The MDB warrants were evaluated under ASC 480 and ASC 815 and the Company determined that equity classification was appropriate.
In July 2019, certain warrant holders from the IPO elected to exercise warrants for an aggregate of 27,063 shares on a cashless basis, resulting in the Company’s net issuance of 15,746 shares. As of December 31, 2019, there were 1,569,893 warrants outstanding related to the IPO.
12. INCOME TAXES
The Company recorded no provision or benefit for federal income taxes, as the Company has incurred a net loss for all periods presented, and the Company has provided a full valuation allowance against its deferred tax assets. For the year ended December 31, 2019, the Company also recorded no provision or benefit for state income taxes. The Company did record a benefit from state income taxes of approximately $0.2 million for the year ended December 31, 2018. The benefit from state income taxes solely reflects the reversal of valuation allowances previously recorded against the Company’s New Jersey State net operating losses (‘NOLs”) that resulted from the Company’s sale of approximately $2.3 million of its New Jersey state NOLs under the State of New Jersey’s Technology Business Tax Certificate Transfer Program (the “Program”) for cash proceeds of $0.2 million. The Program allows qualified technology and biotechnology businesses in New Jersey to sell unused amounts of NOLs and defined research and development tax credits.
At December 31, 2019, the Company had Federal net operating loss carryforwards of approximately $63.4 million, of which approximately $5.4 million will expire in varying amounts beginning in 2036. The balance of the Federal net operating loss carryforwards generated in 2018 and prospectively can be carried forward indefinitely. The Company has state NOLs in various jurisdictions of approximately $60.9 million, certain of which begin to expire beginning in 2036. Utilization of net operating losses may be subject to substantial annual limitations due to the “change in ownership” provisions of the Internal Revenue Code, and similar state provisions. The Company had research and development tax credit carryforwards, including orphan drug tax credits, of approximately $5.3 million at December 31, 2019.
| F-20 |
Income tax benefits computed using the federal statutory income tax rate differs from the Company’s effective tax rate primarily due to the following:
| For the Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Tax Provision at statutory tax rate | 21.0 | % | 21.0 | % | 34.0 | % | ||||||
| Change in federal tax rate | - | - | (29.0 | ) | ||||||||
| Change in valuation allowance | (36.5 | ) | (26.9 | ) | (12.0 | ) | ||||||
| Orphan drug credits | 10.4 | - | - | |||||||||
| State income taxes, net of federal benefit | 6.5 | 6.2 | 6.0 | |||||||||
| Sale of NJ net operating losses | - | 0.7 | - | |||||||||
| Permanent items | (2.1 | ) | (0.5 | ) | - | |||||||
| Other | 0.7 | 0.2 | 1.0 | |||||||||
| Effective Tax Rate | 0.0 | % | 0.7 | % | 0.0 | % | ||||||
The components of income tax benefit are as follows:
| For the Years Ended December 31, | ||||||||||||
| 2019 | 2018 | 2017 | ||||||||||
| Current: | ||||||||||||
| Federal | $ | — | $ | — | $ | — | ||||||
| State | — | (187 | ) | — | ||||||||
| Deferred: | ||||||||||||
| Federal | — | — | — | |||||||||
| State | — | — | — | |||||||||
| Income tax benefit | $ | — | $ | (187 | ) | $ | — | |||||
The income tax benefit for the year ended December 31, 2018 represents the sale by the Company of its 2017 New Jersey state net operating losses, from which the Company realized $0.2 million.
Significant components of the Company’s net deferred tax assets are as follows:
| December 31, | ||||||||
| 2019 | 2018 | |||||||
| Deferred tax assets: | ||||||||
| NOL Carryforward | $ | 17,520 | $ | 7,287 | ||||
| Federal research and development credits | 5,045 | 118 | ||||||
| Product License | 1,768 | 1,904 | ||||||
| Equity compensation | 1,018 | 376 | ||||||
| Other | 213 | 70 | ||||||
| Total deferred tax assets | 25,564 | 9,755 | ||||||
| Less valuation allowance | (25,564 | ) | (9,755 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
The Company’s gross deferred tax assets of $25.6 million and $9.8 million at December 31, 2019 and 2018, respectively, primarily consist of net operating loss carryforwards for income tax purposes. A valuation allowance is required to be recorded when it is not more likely than not that some portion or all of the net deferred tax assets will be realized. Since the Company cannot be assured of generating taxable income and thereby realizing the net deferred tax assets, a full valuation allowance has been recorded. The Company has no uncertain tax positions at December 31, 2019. Since the Company is in a loss carryforward position, the Company is generally subject to US federal and state income tax examinations by tax authorities for all years for which a loss carryforward is available. The Company has never been, nor is currently under audit. The Company’s policy is to recognize interest accrued related to unrecognized tax benefits and penalties in income tax expense, however the Company has recorded no such expense to date.
| F-21 |
13. RELATED PARTY TRANSACTIONS
In connection with the Company’s Asset Purchase Agreement with MacroGenics, the Company made payments of approximately $0.3 million to Tolerance Therapeutics, Inc. during the year ended December 31, 2018. The Company did not make any payments to Tolerance Therapeutics Inc. during the year ended December 31, 2019. Dr. Jeffrey Bluestone, who was appointed to our board of directors in March 2019, is a majority stockholder in Tolerance Therapeutics, Inc.
In July 2018, MDB served as the underwriter for the Company’s IPO and the Company paid MDB cash underwriters’ fees and expenses of $3.6 million. Further, the Company issued to MDB underwriter warrants to purchase 1,596,956 shares of the Company’s common stock. The warrants are exercisable, beginning July 3, 2019 for five years at $5.00 per share. At the time of the IPO, MDB beneficially owned greater than 5% of the Company’s common stock and one employee of which is a member of the Board of Directors of the Company. See Notes 2, 4 and 11 for further information.
The Company paid transition service fees of approximately $0.3 million and $0.2 million to Vactech for the years ended December 31, 2018 and 2017 respectively. The Company made only de minimis payments to Vactech during the year ended December 31, 2019. Until the completion of the Company’s financing in September 2019, Vactech beneficially owned greater than 5% of the Company’s common stock.
14. RETIREMENT SAVINGS PLAN
The Company has an employee savings and retirement plan which is qualified under Section 401(k) of the Internal Revenue Code. The Company made matching contributions for the years ended December 31, 2019 and 2018, of approximately $0.1 million and $0.1 million, respectively. The Company’s matching contributions for the year ended December 31, 2017 were de minimis.
15. SUBSEQUENT EVENTS
On March 12, 2020, the Company announced positive top-line results from the Phase 1b portion of the PREVAIL (PRV-3279 EVAluation In Lupus) study, which evaluated PRV-3279 in healthy volunteers.
PRV-3279 is a humanized diabody targeting the B-cell surface proteins, CD32B and CD79B, which has the potential to intercept the pathophysiology of systemic lupus erythematosus (SLE) and other B cell-mediated autoimmune diseases and prevent or reduce the immunogenicity of biotherapeutics, including gene therapy products.
PRV-3279 was well-tolerated, with no serious adverse events, and as expected, did not deplete B cells and demonstrated profound and sustained binding to circulating B lymphocytes, with reduction of circulating immunoglobulin M levels in a dose-proportional manner. While anti-drug antibody production was observed at both dose levels tested, immunogenicity was found not to affect exposure, safety or pharmacodynamic parameters.
Based on the results, Provention plans to commence the Phase 2a portion of the PREVAIL study in lupus patients in the first half of 2021.
| F-22 |