Attached files
| file | filename |
|---|---|
| EX-99.2 - EXHIBIT 99.2 - Zomedica Corp. | exh_992.htm |
| 8-K - FORM 8-K - Zomedica Corp. | f8k_122018.htm |
Exhibit 99.1
BUSINESS
Overview
We are a development stage veterinary diagnostic and pharmaceutical company creating products for companion animals (canine, feline, and equine) by focusing on the unmet needs of clinical veterinarians. We believe that we have identified and are developing diagnostics and therapeutics that have the potential to significantly improve the diagnosis and treatment of various diseases affecting companion animals. We believe that there are significant unmet medical needs for pets, and that the pet diagnostic and therapeutic segments of the animal health industry are likely to grow substantially as new diagnostic tools and treatments are identified, developed, and marketed specifically for companion animals.
Together with our strategic partners, we are developing three diagnostic platforms, a Bulk Acoustic Wave sensor-based veterinary point-of-care diagnostic platform for performing immunodiagnostic testing a Raman spectroscopy-based point-of-care diagnostic platform for the detection of pathogens, and liquid biopsy assays for the detection of cancer, along with related consumables. We believe that the regulatory pathway to approval of companion animal diagnostics is significantly shorter than for similar diagnostic products intended for human use. In certain cases, pre-market clearance may be unnecessary, depending on the intended use of the diagnostic.
We also have identified a number of drugs which have proven safe and effective in humans that we are developing for use in canines and felines. We believe this development approach enables us to reduce the risks associated with obtaining regulatory approval for unproven product candidates and shortens the development timeline necessary to bring our product candidates to market. We have four drug product candidates in early development and have identified several other potential product candidates for further investigation.
In addition, we are investigating the development of alternative drug delivery technologies for our drug product candidates. Many of the human-approved therapeutics used in companion animals are only available in pill or injectable form. However, it can be difficult to give a companion animal an injection or to assure that the animal has swallowed a pill. As a result, we believe that compliance with treatment regimens is a significant problem for veterinarians and pet owners. The challenges associated with medicating pets are unique, and we believe that developing product candidates that can be easily taken by the pet or easily administered by pet owners will help increase compliance.
Market Opportunity
U.S. consumers will spend an estimated $72 billion on their pets in 2018, according to the American Pet Products Association, or APPA, an increase of approximately 4% from 2017. The veterinary care segment is expected to account for an estimated $17 billion in revenue in 2018, an increase of approximately 2.4%% from 2017. According to dvm360 Magazine’s State of the Profession survey for 2015, diagnostics comprise 18%, and vaccinations, pharmaceuticals and biologicals comprise 25% of gross revenue at the veterinary practice level.
The dvm360 Magazine survey also states that 61% of respondents indicated that they were providing more diagnostic services than the prior year. Similarly, a 2016 Credit Suisse survey of veterinarians found that 73% of respondents expected their diagnostic testing to increase over the next 12 months. According to MarketsandMarkets™, the veterinary diagnostics market is expected to grow at a CAGR of 9.3% between 2017 and 2022, reaching $3.62 billion in sales by 2022, with North America accounting for the largest market share in 2016. The companion animal segment is expected to register the highest growth during the forecast period.
Packaged Facts’ Pet Medications, in its U.S. report for 2017, estimated the size of the U.S. pet medication market, the largest companion animal market worldwide, at $8.6 billion in 2017, up from $7 billion in 2015. Future Market Insights estimates that the global companion animal drug market is expected to grow at a compounded annual growth rate of 4.9% from 2015 - 2025.
We believe that several factors have contributed and will continue to contribute to an increase in spending on pet therapeutics. Companion animals are generally living longer, with the average lifespan for dogs increasing by half a year to 11 years between 2002 and 2012, according to a study by Banfield Pet Hospital. In 2015, the American Animal Hospital Association estimated that the average dog will account for approximately $3,600 in veterinary bills over its lifespan. According to Pet Supplies Plus, baby boomers are adopting pets in record numbers. In its December 2015 issue, Pet Business magazine predicted that the millennial generation would continue the trend of the baby boomers in their enthusiasm for and interest in their pets and pet products and services. This, we believe, along with the increasing awareness of, as the U.S. Public Health Service states, “the mental and emotional benefits of companion animals” and our use of companion animals to address or assist in a range of health and wellness issues including post-traumatic stress disorder and autism, will bolster the growth and development of the pet therapeutics and diagnostics market.
Pet owners in the United States generally pay for diagnostics and therapeutics for their companion animals out-of-pocket. According to statistics from the North American Pet Health Insurance Association, only about 2.0 million dogs and cats in the United States and Canada were covered by an insurance plan in 2018. This represents less than 1% of the nearly 184 million dogs and cats that the American Pet Products Association estimates are owned in the United States alone. We believe that this results in less pricing pressure than in human health care, although the limited adoption of insurance may also reduce the ability of pet owners to pay for diagnostics and therapeutics recommended by their veterinarians.
Development of Companion Animal Diagnostics
The development of companion animal diagnostics continues to evolve with the addition of new technologies to diagnostic portfolios. We believe that these new technologies may allow for the following:
| · |
Enhanced capability to detect the frequency of occurrence and severity of diseases and conditions that impact companion animals; |
| · | Increased accuracy and faster means to obtain test results; |
| · | Wider availability of new diagnostic tools; and |
| · | Enhanced economic benefits for veterinarians. |
Compared to human diagnostic development, the development of companion animal diagnostics is generally faster and less expensive since it typically requires smaller clinical studies, with fewer subjects. We believe that the lower cost of developing companion animal diagnostics enables us to pursue multiple diagnostic candidates simultaneously and to spread the risk of failure across a number of candidates, rather than concentrating all of our resources on one diagnostic candidate that may ultimately fail to achieve regulatory approval or market acceptance.
Development of Companion Animal Therapeutics
Compared to human drug development, the development of companion animal therapeutics is generally faster and less expensive since it requires fewer clinical studies involving fewer subjects and can be conducted directly in the target species. Based on our progress since commencing business in May 2015, we believe that we will be able to develop product candidates, from the initial opening of an INAD with the FDA-CVM through to marketing approval, in approximately five years at a cost of approximately $6 million per product candidate. According to the Tufts Center for the Study of Drug Development, the successful development of a new drug for use in humans can take more than 10 years and requires an average out-of-pocket expenditure of approximately $1.4 billion. The lower cost associated with the development of companion animal therapeutics permits us to pursue multiple product candidates simultaneously and to spread the risk of failure across a number of product candidates, rather than concentrating all of our resources on one novel candidate that may ultimately fail to achieve regulatory approval or market acceptance.
Because we are developing product candidates based on drugs that have been successfully developed and approved for human use—as opposed to drugs based on new active pharmaceutical ingredients (APIs)—we believe that we will be able to avoid or minimize the expenses associated with the human drug development process and more rapidly advance our development programs, while continuing to comply with current good manufacturing practices, or cGMP, for our product candidates. Since we are not pursuing entirely new chemical entities with our drug product candidates, we believe the risk of failure of a specific drug product candidate is significantly lower compared to developing a novel compound.
The respective businesses of developing and commercializing therapeutics for companion animals and humans share a number of characteristics, including the need to:
| · | Demonstrate safety and efficacy in clinical trials; |
| · | Obtain FDA-CVM or other regulatory approval for marketing; |
| · | Manufacture the therapeutics in facilities compliant with cGMP requirements; and |
| · | Market the therapeutics only for their intended indication based on claims permitted in the product label, and not for other uses, which is referred to as “off-label” use. |
However, despite these similarities, there are a number of important differences between the companion animal therapeutics and human therapeutics businesses, including:
| · | Faster, less expensive and more predictable development. The development of therapeutics for companion animals requires fewer clinical studies in fewer subject animals than the development of human therapeutics and, unlike human therapeutics, studies are conducted directly in the target species. We believe that our strategy of selecting APIs with demonstrated efficacy and safety in humans and that are currently being used by veterinarians in their human compounded form enhances the predictability of results and probability of success of our pivotal trials relative to novel compounds that have not been previously validated. |
| · | Role and incentives for veterinary practices. In the United States, veterinarians generally serve the dual role of doctor and pharmacist, and pet owners typically purchase medications directly from their veterinarians. However, veterinarians often are required to have human drugs specially compounded by third-party compounding pharmacies for use in smaller companion animals, resulting in the loss of much of the associated prescription revenue and an increase in the uncertainty around precise dosing and administration. We believe that therapeutics specifically developed for companion animals will enable veterinarians to provide potentially superior treatment options, while also increasing revenue streams from the sale of these therapeutics. |
| · | Less generic competition and strong brand loyalty. There is less generic competition in the companion animal therapeutics industry than in the human health care industry. According to the Generic Animal Drug Alliance, 86% of FDA-approved animal drugs do not have a generic version. We believe that stronger brand loyalty and a lack of the mandatory generic drug substitution that exists in the human pharmaceutical market, partially explains the low penetration of generics in veterinary medicine. |
Unmet Medical Needs
Diagnostics
We believe that there is a significant unmet medical need for cost-effective and accurate disease/condition detection solutions for veterinarians. We believe that we have identified potential diagnostic assays that have the potential to satisfy unmet needs or improve upon existing diagnostic processes frequently used by companion animal veterinarians.
For example, cancer is a prevalent disease in canines that can be difficult and costly to diagnose using existing diagnostic testing. According to the Veterinary Cancer Society, 50% of all dogs over the age of 10 will develop cancer and one in four dogs will develop cancer at some stage in their life. Diagnosing certain cancers in canines is difficult because the location of the tumor may make it difficult or risky to obtain cell material through a biopsy. In addition, the overall health of a canine may increase the risk of performing a biopsy. Other diagnostic technologies, such as advanced imaging, are expensive while others, such as histopathologic examination, may take several days or more to provide a definitive diagnosis. Many more canine cancer cases may go undetected due to cost constraints and other factors. To address these shortcomings, we are developing a circulating tumor cell detection assay for use in the detection of certain cancers in companion animals.
Therapeutics
Despite the growing market for pet therapeutics, there are relatively few treatment options approved for use in companion animals, as compared to those approved for humans. As a result, veterinarians often must resort to prescribing products approved for use in humans, but not approved or formulated for use in companion animals. Based on our own research, we estimate that more than half of the therapeutics used in animals are unapproved for such use. As a result, veterinarians must rely upon trial and error or untested rules of thumb to assess the proper dosage needed to be effective in the particular species without undue risk of side effects. The veterinarian must also find a way to administer the human product to animals and determine the actual dosage amount, tasks which are important and potentially overlooked as practical considerations in the treatment of companion animals. To do this, veterinarians often rely on compounding pharmacies to formulate human drugs into species’ appropriate doses and formulations. As a result, veterinarians are forced to rely on therapeutics not proven safe and effective for their patients and on formulations for which no regulatory approval has been obtained. At the same time, the use of compounding pharmacies results in the veterinary clinic’s loss of much of the associated prescription revenue.
We believe that therapeutics specifically developed for companion animals can extend and improve the quality of the lives of such animals, help veterinarians achieve improved medical outcomes, and make the process of administering therapeutics to companion animals much safer and more convenient. Advances in human medicines have created new therapeutics for managing many chronic diseases. Pets often suffer from many of these same diseases. In many cases, the biology of these diseases in companion animals is very similar to that in humans, which explains why animal efficacy models are used for human drug development. Because of the similarity of the diseases and their symptoms and effects, many human drugs, when formulated properly and administered in proper doses, are effective in companion animals. However, most human drugs are not specially formulated or approved for use in animals.
Many of the human therapeutics used in companion animals are only available in pill or injectable form. However, it can be difficult to give a companion animal a shot or to assure that it has swallowed a pill. It can also be difficult to divide human pills into small enough portions to achieve an appropriate dosage for companion animals. Consequently, we believe that compliance with treatment regimens is a significant problem for veterinarians and pet owners. The challenges associated with medicating pets are unique, and we believe that developing product candidates that can be easily taken by the pet or that can be easily administered by pet owners will help increase compliance.
Product Pipeline

Diagnostics
We are developing with our strategic partner a veterinary diagnostic assay, ZM-024, which is a Bulk Acoustic Wave sensor-based veterinary point-of-care diagnostic platform for performing immunodiagnostic testing. The diagnostic platform uses our partner’s differentiated Bulk Acoustic Wave (BAW) sensor, derived from the fundamental BAW filter technology that is deployed in millions of mobile devices worldwide, to enable a non-optical and fluorescence-free detection system. The final product is expected to be comprised of a table-top instrument that uses disposable assay cartridges to test a range of samples including whole blood, serum, plasma, and urine. Our partner has conducted preliminary analytical and functional sensitivity testing on its investigational BAW platform as well as feasibility testing for certain initial immunoassay candidates in its other development work. We believe ZM-024 may have potential utility in other veterinary diagnostic areas such as molecular diagnostics and multiplexing capabilities. The joint development work initially targets five assay cartridge candidates to detect the following thyroid and adrenal disorders in dogs and cats, which currently require reference lab immunoassay testing for reliable diagnostic results: hypothyroidism in dogs, one of the most common endocrine diseases, hyperthyroidism in cats, a significant cause of morbidity in older cats, and Cushing's disease in dogs, another common endocrine disorder.
We expect to complete assay verifications for ZM-024 in the fourth quarter of 2019, followed by validations in first quarter of 2020. Assuming the development work is successful, we expect to commence the marketing of this platform in the first half of 2020 for the initial five assay candidates, which we believe do not require pre-market regulatory approval by U.S. regulators.
Together with our strategic partner, we are developing a novel pathogen detection system in the form of an innovative point-of-care diagnostic instrument, ZM-020. We believe ZM-020 may deliver multiple benefits, including speed of results and an enhanced workflow with minimal sample preparation time. We believe that ZM-020 does not require pre-market regulatory approval for use with companion animals in the United States. We expect that ZM-020 will use Raman spectral measurements to provide real-time, reagentless and automated identification of pathogens and disease indicators. We expect that ZM-020 will use recent advances in the field of Raman spectroscopy, a laser-based spectroscopy technique, to enable the identification of biological and biochemical signatures in complex biological samples, beginning with the examination of urine and fecal samples. We intend to develop additional applications for the ZM-020 platform including further development of the pathogen detection library for urine and fecal analysis as well as for respiratory and dermatological analysis. ZM-020 is comprised of a bench-top instrument and consumables intended to analyze unprocessed biological samples.
In our early development work the ZM-020 platform has successfully detected 13 unique urine pathogen signatures in water including seven different gram positive and gram negative bacteria species and three types of crystals with greater than 93.93 percent sensitivity and 99.32 percent specificity in over 6,000 samples. Our next development phase will seek to further optimize these results by moving beyond “spiked” water samples to automated detection of these signatures in urine samples. If development work progresses as anticipated, we expect to commence validation for the UTI assay and verification for the fecal assay in the first quarter of 2019. Assuming our development work is successfully completed we expect to commence marketing ZM-020 in the first half of 2020.
Together with our strategic partner, we are developing a circulating tumor cell, or CTC, assay, ZM-017, also known as a “liquid biopsy,” for use by veterinarians as a cancer diagnostic. The liquid biopsy is a blood test that we believe has the potential to detect the presence of CTCs, which are cells that have shed from a primary tumor into neighboring blood vessels and are transported throughout the body’s circulatory system. Diagnosing certain cancers in canines is difficult because the location of the tumor may make it difficult or risky to obtain cell material through a biopsy. In addition, the overall health of a canine may increase the risk of performing a biopsy. We are focusing our initial development work on testing for difficult to biopsy cancers such as hemangiosarcoma and osteosarcoma in canines. Other diagnostic technologies, such as advanced imaging, are expensive while others, such as histopathologic examination, may take several days or more to provide a definitive diagnosis. We believe that the detection of CTCs in the blood could provide strong clinical support for a cancer diagnosis without the need for an invasive tissue biopsy or other expensive or time-consuming diagnostic test. If we successfully develop ZM-017, we expect that ZM-017 will provide veterinarians with a faster, more affordable, and less invasive test for certain cancers in canines compared to existing detection methods. We expect to initiate verification and validation efforts for a lymphoma assay in 2019 as well. According to The Merck Veterinary Manual canine lymphoma is reported to be the most common blood-borne cancer in dogs with an estimated incidence rate approaching 0.1%.
Zomedica extended validation of its initial cancer assay after continued verification efforts, performed in parallel with early clinical validation steps during 2018, revealed opportunities to further optimize the assay to achieve broader commercial potential. Assuming successful completion of the clinical validation, we expect to commence the marketing of ZM-017 during the second half of 2020.
Therapeutics
We have four drug product candidates. Our lead drug product candidate is ZM-007, an oral suspension formulation of metronidazole targeting the treatment of acute diarrhea in small dog breeds and puppies under nine pounds or four kilograms. Metronidazole suspension is only available as a compounded drug and is not approved by the FDA-CVM. An Investigational New Animal Drug, or INAD, was opened for ZM-007 with the Food and Drug Administration’s Center for Veterinary Medicine, or FDA-CVM, in October 2016. The API in ZM-007 is metronidazole, which has been the subject of multiple studies in humans and has been approved for use in humans for decades. We do not believe that the API in ZM-007 is protected by any patents or other proprietary rights of third parties in the U.S. We had a pre-submission meeting on December 13, 2017 with the FDA-CVM specific to the product development strategy for ZM-007 and ZM-012, a bioequivalent to ZM-007. Based on the feedback received from the FDA-CVM at that meeting and in light of additional market research demonstrating approved alternatives to compounded drugs, we have decided to prioritize development of ZM-007 over ZM-012. We expect to commence a pivotal safety study of ZM-007 in the first half of 2019.
Our second drug product candidate is ZM-012, a novel tablet formulation of metronidazole and a complementary formulation to ZM-007, targeting the treatment of acute diarrhea in dogs. Metronidazole tablets are currently only available as human generics. An INAD was opened for ZM-012 with the FDA-CVM in April 2016. We have finalized the formulation and completed pilot testing of ZM-012 as a beef-flavored oral tablet intended for dogs greater than nine pounds or four kilograms and we completed pilot testing of ZM-012 in the fourth quarter of 2017. We intend to pursue regulatory approval of ZM-012 as a bioequivalent to ZM-007 following approval of ZM-007 by FDA-CVM. Drugs that are considered to be bioequivalent are, for regulatory purposes, essentially the same, meaning the absence of significant difference between the extent and rate of absorption over the course of a specific period of time at the same dose and under the same conditions. The implementation of this bioequivalent strategy is contingent on FDA-CVM approval of the new animal drug application (NADA) for ZM-007. If the FDA-CVM permits us to rely on the bioequivalence of ZM-012 to ZM-007, we anticipate that this regulatory pathway will conserve significant development costs because a bioequivalence study could replace the need for pivotal safety and efficacy studies for ZM-012.
Our third drug product candidate is ZM-006, a transdermal gel formulation of methimazole targeting the chronic treatment of hyperthyroidism in cats. Hyperthyroidism is one of the most commonly diagnosed endocrine disorders in middle-aged to older cats according to the American Association of Feline Practitioners. We are investigating ZM-006 pursuant to an INAD opened with the FDA-CVM in June 2016. The API in ZM-006, methimazole, has been the subject of multiple studies in humans and has been approved for oral use in humans for decades. Our transdermal gel formulation is intended to provide an alternative to an oral tablet formulation already approved by the FDA-CVM for cats. We do not believe that the API in ZM-006 is protected by any patents or other proprietary rights of third parties. ZM-006 is intended for application to the inside of the cat’s ear . The formulation of ZM-006 has been completed. We completed pilot testing of ZM-006 to support our pivotal safety study in the fourth quarter of 2018 and are analyzing the results. We expect to present and confirm the regulatory strategy and development plan for ZM-006 with the FDA-CVM in the first quarter of 2019. Assuming pilot testing is successful, we intend to commence a pivotal safety study of ZM-006 in the first half of 2019. We also intend to initiate a pilot efficacy study in the first half of 2019.
Our fourth drug product candidate is ZM-011, a transdermal gel formulation of fluoxetine, most commonly known as Prozac®, its human pharmaceutical brand name. We believe that Fluoxetine in pill or compounded form is frequently prescribed by veterinarians to treat feline behavioral disorders such as inappropriate urination. We are investigating ZM-011 pursuant to an INAD opened with the FDA-CVM in January 2017. The API, fluoxetine, has been the subject of multiple studies in humans and has been approved for use in humans for decades. We do not believe that the API in ZM-011 is protected by any patents or other proprietary rights of third parties. ZM-011 is a transdermal gel formulation intended for application to the inside of the cat’s ear. The formulation of ZM-011 has been completed. We completed pilot testing of ZM-011 to support our pivotal safety study in the fourth quarter of 2018 and are analyzing the results. Assuming such pilot testing is successful, we intend to commence our pivotal safety study of ZM-011 in the second half of 2019.
License Agreements
In November 2018, we entered into a development and supply agreement with Qorvo Biotechnologies, LLC, or Qorvo, a wholly-owned subsidiary of Qorvo, Inc. focused on bringing Qorvo’s piezo-electric BAW sensor to the veterinary health sector. Under the terms of this agreement, we have exclusive, global rights to develop and market Qorvo's investigational point-of-care diagnostic platform for veterinary use. Under the agreement, Qorvo and we will collaborate on the development of veterinary diagnostic assays. The joint development work initially targets five assay cartridge candidates to detect the thyroid and adrenal disorders in dogs and cats. Qorvo is responsible for the development of the assay cartridges and the instrument. We have agreed to pay for the associated non-recurring engineering costs of up to $500,000 per assay cartridge and the instrument, and are responsible for the validation of the assay cartridges and the instrument. Qorvo will supply us, on an exclusive basis, with the instruments and the related assay cartridges to be developed under the agreement pursuant to a rolling forecast, subject to specified minimum purchase requirements, at prices specified in the agreement. We will be responsible for the marketing and sale of the disposable assay cartridges and instruments.
The agreement, which is exclusive worldwide in the practice of veterinary medicine for the health and wellbeing of any non-human animal, has an initial term of ten years (subject to early termination and extension in certain circumstances).
We paid Qorvo $1.0 million and issued to Qorvo unregistered common shares having a value of $3.9 million, consisting of an aggregate of 2,565,789 common shares with an ascribed price of $1.52 per share. We have agreed to pay Qorvo additional milestone payments in cash or, if elected by Qorvo, additional unregistered common shares having a value calculated as specified in the agreement. The total amount of additional milestone payments (if all milestones are met) will be $10 million (if paid entirely with cash) or up to $10.9 million (consisting of cash in the amount of $7 million and unregistered common shares having a value of $3.9 million, if Qorvo elects to receive compensation partially in equity). In connection with the agreement, we entered into a registration rights agreement providing Qorvo with certain registration rights with respect to the common shares to be issued by us under the agreement.
In May 2018, we entered into a development, commercialization and exclusive distribution agreement with Seraph Biosciences, Inc., or Seraph, a human biomedical device company. Under the terms of this agreement, we have exclusive global veterinary industry rights to develop and market a novel pathogen detection system in the form of a point-of-care diagnostic instrument. The agreement covers development and validation of ZM-020. We are responsible for development and validation, and their associated costs. Seraph will supply us, on an exclusive basis, with the hardware platform, associated software and the consumables to be developed under the agreement, pursuant to a rolling forecast, at prices specified in the agreement. We will be responsible for the marketing and sale of the hardware platform, associated software and the consumables. The agreement, which is exclusive to the field of global veterinary diagnostic applications, has a term of seven years (subject to adjustment in certain circumstances) and automatically renews for additional one-year terms thereafter.
We paid Seraph up-front fees of $500,000 and issued to Seraph unregistered common shares having a value of $1,250,000, consisting of an aggregate of 641,717 common shares at an ascribed price of $1.9479 per share. Seraph is entitled to additional payments for development costs. Seraph will be entitled to receive up to an additional $7,000,000, payable 50 percent in cash and 50 percent in additional unregistered common shares, upon the achievement of a series of staged, specified milestones, including completion of laboratory studies and field studies, production and commercial shipment of products. Future issuances of shares will be subject to TSX-V approval and will be priced relative to market at the time of issuance. Seraph is entitled to certain registration rights with respect to the common shares to be issued by us under the agreement. In addition, we have agreed to pay Seraph license fees based on a percentage of gross profit from commercial sales of ZM-020.
In January 2017, we entered into a collaborative research agreement with Celsee, Inc., or Celsee, a developer of diagnostics for the detection and quantification of cells and other markers. Subsequent to this agreement, in December 2017, we entered into a license and supply agreement with Celsee for exclusive global rights to develop and market Celsee’s liquid biopsy platform. The agreement with Celsee covers the development and commercialization of liquid biopsy assays and related consumables for the detection of cancer in companion animals. We are responsible for the clinical development and commercialization of the assays. Celsee will supply us on an exclusive basis with the assays and the consumables for the products to be developed under the agreement pursuant to a rolling forecast to be provided by us at prices specified in the agreement. We will be responsible for the marketing and sale of the assays and the related consumables. The agreement, which is exclusive in the field of veterinary cancer diagnostic applications, has a term of seven years (subject to termination in certain circumstances) and automatically renews for additional one-year terms thereafter.
We paid Celsee up-front fees of $500,000 and issued to Celsee unregistered common shares having a value of $250,000, consisting of an aggregate of 112,314 common shares at an ascribed price of $2.2259 per share. Celsee is entitled to additional payments totaling up to an additional $1 million, payable 50 percent in cash and 50 percent in additional unregistered common shares, upon the achievement of specified milestones—namely, completion of product development (in respect of 50 percent of the foregoing cash and share payments) and upon successful completion of manufacturing milestones (as to the remaining 50 percent of the foregoing cash and share payments). Future issuances of shares will be subject to TSX-V approval and will be priced relative to market at the time of issuance. Celsee is entitled to certain registration rights with respect to the common shares issued by us under the agreement.
In April 2016, we entered into a collaboration agreement with CTX Technology, Inc., or CTX, which has developed a peptide-based skin penetration platform technology for the topical delivery of a range of APIs. Under this agreement, we have an option to obtain an exclusive worldwide license to use CTX’s technology platform in animals. In the event that we exercise the option, we would be required to pay CTX a one-time license fee of $20,000 and to pay CTX a royalty in the low single digits on any products that we sell that incorporates their technology. Unless we exercise our option prior thereto, this agreement will terminate on March 1, 2019.
Research and Development
Together with our strategic partners, we are performing development work on our diagnostic platforms. Our drug product candidate development programs focus on the development of product candidates for target indications that have already demonstrated safety and efficacy in humans and the development of therapeutics based on these drugs for appropriate target indications in companion animals. In addition, we are investigating the development of alternative drug delivery systems for our drug product candidates. We use various contract research organizations, or CROs, to assist in performing our research and development activities.
In connection with these activities, we have incurred and will continue to incur significant research and development expenses. Our research and development expenses were $3,765,332 and $1,586,179 for the nine months ended September 30, 2018 and September 30, 2017, respectively, and $2,751,326 and $1,518,589 for the years ended December 31, 2017and December 31, 2016, respectively.
Sales and Marketing
We intend to commercialize any product candidate for which we receive regulatory approval in the United States with a direct sales force. We intend to sell products directly to veterinarians whom we believe are self-motivated to utilize advanced diagnostics and prescribe innovative therapeutics that are safe, effective, and supported by reliable clinical data and regulatory approval in order to improve the health of companion animals, while also generating additional revenue.
We also intend to market certain of our products to reference labs. Our commercialization strategy is to sell ZM-024 and ZM-020 to veterinarians, and to sell ZM-017 to reference lab(s), while driving utilization of the tests by veterinarians. We believe this strategy is consistent with the current practice of veterinarians who perform some of their own diagnostic tests and send other diagnostic samples to reference labs for analysis.
We also intend to selectively utilize distributors, which we believe will enable us to expand our commercial reach to a majority of all veterinarians in our chosen markets. We believe that we can compete effectively with a combination of our own direct sales force and complementary distributors.
To support our marketing efforts, we introduced a unique “Voice of the Vet™” program in the fourth quarter of 2016 to gather insights and better understand the needs of veterinarians and their practices, and to gauge interest for potential future product offerings, while building brand awareness as a valued veterinary partner. Our Voice of the Vet™ program allows veterinarians, practice managers and veterinary technicians to participate in conversations where they can share ideas and experiences with each other, as well as with us through an interactive platform.
During 2018, we have increased our investment to build brand and product awareness as a valued veterinary partner with clinical practitioners. In the fourth quarter of 2018, we initiated a strategic customer development initiative, which includes expansion activities for its Voice of the Vet™ programming for veterinarians, veterinary technicians and nurses, practice managers and hospital administrators, as well as veterinary students.
Additionally, we are continuing to conduct comprehensive market research across the United States with private, corporate and institutional clinics along with key opinion leaders and academia to obtain feedback on our product development efforts and to build relationships with key market influencers. We are also finalizing science-based educational white papers for our ZM-017 canine cancer diagnostic platform and our ZM-20 point-of-care pathogen detection platform.
During 2019, we expect to increase our product marketing efforts. Our goal is to be in a position toward the end of 2019 to begin soliciting and accepting commercial orders and deposits for our point-of-care diagnostic products for delivery in 2020 as described elsewhere herein.
Manufacturing
We have no internal manufacturing capabilities for our diagnostic and therapeutic product candidates.
Under our license and supply agreements, Qorvo, Seraph and Celsee are responsible for the manufacture and supply of the equipment and consumables to us. Our strategic partners have primary responsibility for assuring that all products will be manufactured in accordance with applicable laws and meet all agreed upon specifications.
To ensure a dependable and high quality supply of the APIs for our pilot studies and pivotal trials, we rely on cGMP-compliant contract manufacturers. Because the APIs in our drug product candidates are used in human drugs that are no longer subject to patent protection, we believe that there are multiple contract manufacturers for our drug product candidates that have demonstrated the ability to provide high-quality formulated products more cost effectively than we could on our own. We believe that the contract manufacturers of our trial supplies will be able to provide commercial supplies of any of our drug product candidates that are approved for marketing.
While we and our contract manufacturers have historically been able to obtain supplies of the APIs for development of our drug product candidates, neither we nor our contract manufacturers have long-term supply agreements with the API manufacturers. We also have no agreements for commercial-scale supply of the API or manufacture of any of our drug product candidates.
Intellectual Property
We intend to rely primarily upon a combination of in-licensing exclusive rights, regulatory exclusivity, proprietary know-how, and confidentiality agreements to protect our diagnostic assays, product formulations, processes, methods and other technologies and to preserve any trade secrets and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. We currently do not own any issued patents.
Our diagnostic technologies are dependent on intellectual property developed by our strategic partners and licensed to us. We do not own the intellectual property rights that underlie these licenses. Our rights to use the technology we license are subject to the negotiation of, continuation of and compliance with the terms of our licenses. However, we have filed three provisional patents to date, two of which cover methods of using antibody based cancer detection and another compositions and method patent for identifying lymphoma all of which relate to our ZM-017 platform.
Because our drug product candidates are based on approved human drugs that no longer are subject to patent protection, there is little, if any, composition-of-matter patent protection available for the API in these product candidates. Where feasible, however, we intend to pursue the broadest intellectual property protection possible for our compounds and any proprietary technology through enhanced formulations of our drug product candidates. However, even intellectual property protection, if available, may not afford us with complete protection against competitors.
We depend upon the skills, knowledge and experience of our management personnel, as well as that of our other employees, advisors, consultants and contractors, none of which are patentable. To help protect our know-how, and any inventions for which patents may be difficult to obtain or enforce, we require all of our employees, consultants, advisors and other contractors to enter into customary confidentiality and inventions agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
Competition
Diagnostics
Our potential competitors include large human pharmaceutical and medical diagnostics companies, small businesses focused on animal health, and reference laboratory services provided by academic institutions and in-clinic product providers. These competitors include Idexx Laboratories, Inc., Antech Diagnostics, a unit of VCA Inc., Abaxis, Inc., a wholly-owned subsidiary of Zoetis Inc., Heska Corporation and Zoetis Inc.
Therapeutics
If our drug product candidate is the first one approved by the FDA-CVM for use in animals, it may be eligible for between three and seven years of regulatory exclusivity in the United States, depending on the type of product and its intended use. However, while there are fewer competitors in the pet therapeutics industry than in the human pharmaceutical industry, the development and commercialization of new animal health medicines is highly competitive, and we expect competition from major pharmaceutical, biotechnology and specialty animal health medicine companies.
Our potential competitors include large animal health companies, which currently derive a significant portion of their revenue from livestock medications. Large animal health companies include Merck Animal Health, the animal health division of Merck & Co., Inc.; Elanco; Bayer Animal Health, the animal health division of Bayer AG; Novartis Animal Health, the animal health division of Novartis AG; Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH; and Zoetis, Inc., as well as European companies such as Virbac S.A., Vetoquinol S.A., and Dechra Pharmaceuticals PLC. We are also aware of several smaller early stage companies that are developing products for use in the pet therapeutics market, including Kindred Biosciences, Inc., Aratana Therapeutics, Inc., Parnell Pharmaceuticals Holdings Ltd., and Jaguar Animal Health, Inc. Our drug product candidates will also face competition from medicines and products approved for use in humans that are used off-label for pets. Private organizations, academic institutions and government agencies conducting animal health product research are also considered potential competitors.
General
Many of our competitors and potential competitors have substantially more financial, technical, and human resources than we do. Many also have far more experience in the development, manufacture, regulation and worldwide commercialization of animal diagnostics and animal health medicines, including pet therapeutics. We also expect to compete with academic institutions, governmental agencies and private organizations that are conducting research in the fields of animal diagnostics and animal health medicines. If such competing products achieve regulatory approval and commercialization prior to our product candidates, or if our intellectual property protection and efforts to obtain regulatory exclusivity fail to provide us with exclusive marketing rights for some of our products, we may be unable to effectively compete in the markets in which we participate.
Government Regulation
Diagnostic Product Candidates
Our diagnostic product candidates may be subject to regulatory review by the USDA-CVB and/or post-marketing oversight by the USDA-CVB or FDA-CVM. Generally speaking, full diagnostic kits aimed at the detection or diagnosis of an infectious disease in animals, including the materials required for testing along with instructions for use and interpretation of results, used at the point-of-care, including in-office diagnostic tests, may be subject to pre-market regulatory review and approval by the USDA-CVB. The USDA-CVB’s review process for diagnostics is subject to some variability based on the type of diagnostic kit being reviewed, however, the USDA-CVB will generally review the results of specific tests that are required to be conducted in accordance with the USDA-CVB’s testing criteria. These include diagnostic sensitivity/specificity studies, conducted using a large number of samples of U.S. origin, reproducibility/repeatability/suitability studies used to evaluate test kits under field conditions in participating laboratories and ruggedness studies in which manufacturers measure the ruggedness or robustness of the diagnostic test kits based on the capacity of the assay to remain unaffected by small variations in or deviations from the instructions for use (for example, not allowing the samples to reach the designated temperature). Diagnostic products and testing kits that do not claim to detect or diagnose an infectious disease and that are not designed for use at the point-of-care are generally subject only to post-marketing oversight by the FDA-CVM or the USDA-CVB. While the sale of these products does not require premarket approval by the FDA-CVM and does not subject us to the FDA-CVM’s cGMP requirements, these products must not be adulterated, mislabeled or misbranded under the Federal Food, Drug and Cosmetic Act, or the FDC Act, and are subject to post-marketing review.
Drug Product Candidates
The FDA-CVM regulates animal pharmaceuticals under the FDC Act. In order to obtain regulatory approval to market a drug product candidate in the U.S., an applicant must demonstrate that the product candidate is safe, effective and produced by a consistent method of manufacture. Post-approval monitoring of products is required by law, with reports being provided to the FDA-CVM's Surveillance and Compliance group. Reports of product quality defects, adverse events or unexpected results are required in accordance with the law.
Prior to commencing testing of a drug product candidate, an applicant is required to open an INAD with the FDA-CVM. Formulation work and pilot testing occurs once the INAD is opened. This is followed by a pre-submission conference with the FDA-CVM to discuss and agree on a proposed development plan, including the design of pivotal safety and clinical trials that would support approval of a new animal drug application, or NADA.
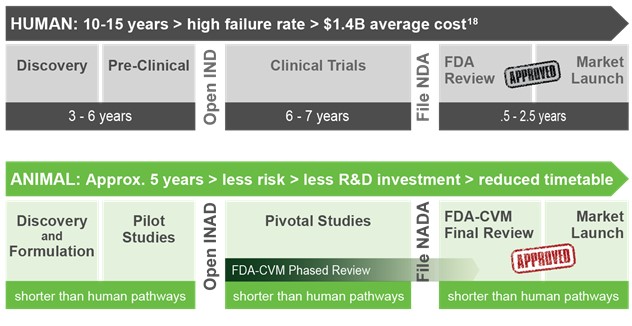
Early pilot studies may be conducted in laboratory animals to establish clinical endpoints and the dose range for a new drug product candidate. Data on how well the drug is absorbed when dosed by different routes of administration and the relationship of the dose to the effectiveness are studied.
During development, the applicant will usually submit a proposed pivotal trial protocol to the FDA-CVM for review and concurrence prior to conducting the trial. The applicant must gather and submit data on manufacturing, safety and effectiveness to the FDA-CVM for review, which will be conducted according to timelines specified in the Animal Drug User Fee Act, or ADUFA. ADUFA also imposes certain fees including a sponsor fee of $125,990 per year, an application fee of $449,348 per product candidate submission, and certain administrative application and manufacturing fees imposed per product candidate per year based on sales.
The pivotal clinical trial must be conducted with the formulation of the drug product candidate that is intended to be commercialized, and is a multi-site, randomized, controlled study, generally with a placebo control. To reduce bias in the study, individuals doing the assessment are not told whether the subject is in the group receiving the treatment being tested or the placebo group.
Once all data have been submitted and reviewed for each technical section - safety, effectiveness and chemistry, manufacturing and controls, or CMC - the FDA-CVM issues a “technical section complete letter” as each section review is completed, and when all three letters have been issued, the applicant prepares a draft of the Freedom of Information Summary, the proposed labeling, and all other relevant information, and submits these for FDA review. An administrative NADA is a NADA that is submitted after all of the technical sections that fulfill the requirements for the approval of the new drug product candidate have been reviewed by FDA-CVM and FDA-CVM has issued a technical section complete letter for each of those technical sections. Although this process is not required and submission of a non-administrative NADA is also acceptable, we plan to take advantage of the administrative NADA process to obtain a timelier phased review. Because FDA-CVM has already reviewed the individual technical sections before the administrative NADA is filed, FDA-CVM is committed under ADUFA to reviewing and acting on 90% of administrative NADAs within 60 days after submission. The FDA-CVM user fee goal is to review and act on 90% of non-administrative NADAs within 180 days after submission. After approval, we will be required to collect reports of adverse events and submit them on a regular basis to the FDA.
Other Regulatory Considerations
Regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to our product candidates because our product candidates are not intended for use in food animals or food production animals.
Advertising and promotion of animal health products is controlled by regulations in the United States. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and authorized by the FDA-CVM.
Any drug product candidate, if approved, may eventually face generic competition in the United States. In the United States, a generic animal drug may be approved pursuant to an Abbreviated New Animal Drug Application, or ANADA. Instead of demonstrating the drug’s safety and effectiveness in the target species as required in a NADA, a generic applicant must only show that the proposed generic product is the same as, and bioequivalent to, the approved brand name product. However, if any of our drug product candidates is the first one approved by the FDA-CVM for use in animals, it will be eligible for between three and seven years of regulatory exclusivity in the United States, depending on the type of product and its intended use.
We will be required to conduct post-approval monitoring of any approved product and to submit reports of product quality defects, adverse events or unexpected results, and be subject to regulatory inspection from time to time. Safety, quality, or efficacy concerns can lead to product recalls, withdrawals or suspended or declining sales, as well as product liability and other claims.
Employees
As of December 15, 2018, we had 27 employees including one employee who is a doctor of veterinary medicine. Of our employees, ten are engaged in research and development activities, seven are engaged in business development and marketing activities, and ten are engaged in corporate and administrative activities. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Properties
Our corporate headquarters and research and development laboratory is located in Ann Arbor, Michigan, where we lease approximately 26,500 square feet pursuant to a lease that expires February 2022. We have the option to extend that lease for two, five year renewal periods. We believe that our facilities are sufficient for our existing and expected future needs.
Legal Proceedings
We are not currently a party to any material legal proceedings.
Corporate Information
Zomedica Pharmaceuticals Corp. (formerly, Wise Oakwood Ventures Inc.) was originally incorporated as Wise Oakwood Ventures Inc. on January 7, 2013 under the Business Corporations Act (Alberta). On October 28, 2013, we completed our initial public offering in Canada and became classified as a Capital Pool Company, as defined under the rules of the TSX Venture Exchange, or TSX-V. On April 21, 2016, we changed our name to Zomedica Pharmaceuticals Corp. and consolidated our common shares on a one-for-two and one-half (2½) basis. ZoMedica Pharmaceuticals Inc., or ZoMedica Inc., was incorporated on May 14, 2015 under the Canada Business Corporations Act. On April 21, 2016, we completed a qualifying transaction, or the Qualifying Transaction, under TSX-V Policy 2.4 – Capital Pool Companies, consisting of a three-cornered amalgamation among our company, ZoMedica Inc. and our wholly-owned subsidiary. Under the Qualifying Transaction, ZoMedica Inc. and our subsidiary were amalgamated to form Zomedica Pharmaceuticals Ltd., or Zomedica Ltd. As consideration for the amalgamation, shareholders of ZoMedica Inc. became the owners of 97.6% (non-diluted) of our common shares, and ZoMedica Ltd. became our wholly-owned subsidiary. Subsequent to the Qualifying Transaction, Zomedica Ltd. was vertically amalgamated into our company. We have one wholly-owned subsidiary, Zomedica Pharmaceuticals, Inc., a Delaware company. ZoMedica Inc. entered into the Qualifying Transaction in order to accomplish the following:
| · | Enable its shareholders to own shares in a company that was publicly traded on the TSX-V; |
| · | Expand its shareholder base to include the public shareholders of Wise Oakwood; and |
| · | Obtain access to the cash resources raised by Wise Oakwood in its initial public offering. |
On November 10, 2017, our shares were approved for listing on the NYSE American under the symbol “ZOM”. On November 20, 2017 the U.S. Securities and Exchange Commission declared our registration statement on Form S-1 effective. Our common shares commenced trading on the NYSE American on November 21, 2017.
Our principal executive offices are located at 100 Phoenix Drive, Suite 190, Ann Arbor, MI 48108, and our telephone number is (734) 369-2555. Our website address is www.zomedica.com. The information contained in, or accessible through, our website is not part of the registration statement of which this prospectus forms a part.