Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Zomedica Corp. | exh_321.htm |
| EX-31.2 - EXHIBIT 31.2 - Zomedica Corp. | exh_312.htm |
| EX-31.1 - EXHIBIT 31.1 - Zomedica Corp. | exh_311.htm |
| EX-23.1 - EXHIBIT 23.1 - Zomedica Corp. | exh_231.htm |
| EX-10.23 - EXHIBIT 10.23 - Zomedica Corp. | exh_1023.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
| (Mark One) | FORM 10-K |
| [X] | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ______________________________ to ______________________________
Commission file number:
001-38298
ZOMEDICA PHARMACEUTICALS CORP.
(Exact name of registrant as specified in its charter)
| Alberta, Canada | N/A | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| Incorporation or organization) | Identification No.) |
| 100 Phoenix Drive, Suite 190, Ann Arbor, Michigan | 48108 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (734) 369-2555
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Shares, no par value | NYSE American | |
| _____________________________________ | ___________________________________________ |
Securities registered pursuant to section 12(g) of the Act: None
______________________________________________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
☐ Yes ☒ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
☐ Yes ☒ No
Indicate by checkmark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ☒ Yes ☐ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
Large accelerated filer ¨
Accelerated filer ¨
Non-accelerated filer ¨ (Do not check if a smaller reporting company)
Smaller reporting company ☒
Emerging growth company ☒
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☒
Indicate by a check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☒ No
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter.
Note.—If a determination as to whether a particular person or entity is an affiliate cannot be made without involving unreasonable effort and expense, the aggregate market value of the common stock held by non-affiliates may be calculated on the basis of assumptions reasonable under the circumstances, provided that the assumptions are set forth in this Form.
As of June 30, 2017, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $67.2 million based on the closing sale price as reported on the TSX Venture Exchange based on the exchange rate of CDN$1.2977 to U.S.$1.00 as published by the Bank of Canada as at June 30, 2017. Our common shares began trading on the NYSE American on November 21, 2017.
Indicate the number of shares outstanding of each of the registrant’s classes of common stock, as of the latest practicable date.
The number of the registrant’s common shares outstanding as of February 27, 2018, was 90,449,869.
| -2- |
TABLE OF CONTENTS
| -3- |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are based on management’s beliefs and assumptions and on information currently available to management. Some of the statements under “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business” and elsewhere in this Annual Report on Form 10-K contain forward-looking statements. In some cases, you can identify forward-looking statements by the following words: “may,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue,” “ongoing” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words.
These statements involve risks, uncertainties and other factors that may cause actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this prospectus, we caution you that these statements are based on a combination of facts and factors currently known by us and our projections of the future, about which we cannot be certain. Forward-looking statements in this prospectus include, but are not limited to, statements about:
| • | the success, cost and timing of our research and development activities, validation studies and pivotal trials, including with respect to our lead product candidates, ZM-017, ZM-012, ZM-006, ZM-007 and ZM-011; |
| • | our ability to obtain regulatory approval from the FDA-CVM and/or the USDA-CVB for our pharmaceutical and diagnostic product candidates, as applicable; |
| • | our ability to obtain funding for our operations; |
| • | the ability of our CROs to appropriately conduct our safety studies and certain development activities; |
| • | the ability of our CMOs to manufacture and supply our product candidates in accordance with cGMP and our clinical needs; |
| • | our plans to develop and commercialize any product candidates for which we receive regulatory approval; |
| • | our ability to develop and commercialize product candidates that can compete effectively against the product candidates developed and commercialized by our competitors; |
| • | the size and growth of the veterinary diagnostics and therapeutics markets; |
| • | our ability to obtain and maintain intellectual property protection for our current and future product candidates; |
| • | regulatory developments in the United States; |
| • | the loss of key scientific or management personnel; |
| • | our expectations regarding the period during which we will be an “emerging growth company” under the JOBS Act; |
| • | the accuracy of our estimates regarding expenses, future revenues, capital requirements and needs for additional financing; and |
| • | our status as a PFIC for U.S. federal income tax purposes. |
In addition, you should refer to the “Risk Factors” section of this Annual Report on Form 10-K for a discussion of other important factors that may cause actual results to differ materially from those expressed or implied by these forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 10-K will prove to be accurate. Furthermore, if the forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
| -4- |
| Item 1. | Business. |
BUSINESS
Overview
We are a development stage veterinary diagnostic and pharmaceutical company creating products for companion animals (canine, feline, and equine) by focusing on the unmet needs of clinical veterinarians. We believe that we have identified and are developing diagnostics and therapeutics that have the potential to significantly improve the diagnosis and treatment of various diseases affecting companion animals. We believe that there are significant unmet medical needs for pets, and that the pet diagnostic and therapeutic segments of the animal health industry are likely to grow substantially as new diagnostic tools and treatments are identified, developed, and marketed specifically for companion animals.
Together with our strategic partner, we are developing liquid biopsy assays and related consumables for the detection of cancer in companion animals. The regulatory pathway to obtain pre-market regulatory approval of companion animal diagnostics is significantly shorter than for similar diagnostic products intended for human use. In certain cases, pre-market regulatory approval may be unnecessary, depending on the intended use of the diagnostic.
We also have identified a number of drugs that have proven safe and effective in humans that we are developing for use in companion animals. We believe this development approach enables us to reduce the risks associated with obtaining regulatory approval for unproven product candidates and shortens the development timeline necessary to bring our product candidates to market. We have four drug product candidates in early development and have identified several other potential product candidates for further investigation.
In addition, we are investigating the development of alternative drug delivery technologies for our drug product candidates. Many of the human-approved therapeutics used in companion animals are only available in pill or injectable form. However, it can be difficult to give a companion animal an injection or to assure that the animal has swallowed a pill. As a result, we believe that compliance with treatment regimens is a significant problem for veterinarians and pet owners. The challenges associated with medicating pets are unique, and we believe that developing product candidates that can be easily taken by the pet or easily administered by pet owners will help increase compliance.
Market Opportunity
U.S. consumers spent an estimated $69.4 billion on their pets in 2017, according to the American Pet Products Association, or APPA, an increase of 4% from 2016. The veterinary care segment has been among the fastest growing segments of the overall U.S. pet market. This segment accounted for an estimated $16.6 billion in revenue in 2017, an increase of 4% from 2016. According to dvm360 Magazine’s State of the Profession survey for 2015, diagnostics comprise 18%, and vaccinations, pharmaceuticals and biologicals comprise 25% of gross revenue at the veterinary practice level.
The dvm360 Magazine survey also states that 61% of respondents indicated that they were providing more diagnostic services than the prior year. Similarly, a 2016 Credit Suisse survey of veterinarians found that 73% of respondents expected their diagnostic testing to increase over the next 12 months. According to MarketsandMarkets™, the veterinary diagnostics market is expected to grow at a CAGR of 9.3% between 2017 and 2022, reaching $3.62 billion in sales by 2022, with North America accounting for the largest market share in 2016. The companion animal segment is expected to register the highest growth during the forecast period.
Packaged Facts’ Pet Medications, in its U.S. report for 2017, estimated the size of the U.S. pet medication market, the largest companion animal market worldwide, at $8.6 billion in 2017, up from $7 billion in 2015. Future Market Insights estimates that the global companion animal drug market is expected to grow at a compounded annual growth rate of 4.9% from 2015 - 2025.
We believe that several factors have contributed and will continue to contribute to an increase in spending on pet therapeutics. Companion animals are generally living longer, with the average lifespan for dogs increasing by half a year to 11 years between 2002 and 2012, according to a study by Banfield Pet Hospital. As a result, companion animals increasingly require medical treatment. In 2015, the American Animal Hospital Association estimated that the average dog will account for approximately $3,600 in veterinary bills over its lifespan. According to Pet Supplies Plus, baby boomers are adopting pets in record numbers. In its December 2015 issue, Pet Business magazine predicted that the millennial generation would continue the trend of the baby boomers in their enthusiasm for and interest in their pets and pet products and services. This, we believe, along with the increasing awareness of, as the U.S. Public Health Service states, “the mental and emotional benefits of companion animals” and our use of companion animals to address or assist in a range of health and wellness issues including post-traumatic stress disorder and autism, will bolster the growth and development of the pet therapeutics market.
| -5- |
Pet owners in the United States generally pay for diagnostics and therapeutics for their companion animals out-of-pocket. According to statistics from the North American Pet Health Insurance Association, only about 1.8 million dogs and cats were covered by an insurance plan in 2016. This represents less than 1% of the nearly 184 million dogs and cats that the American Pet Products Association estimates are owned in the United States. We believe that this results in less pricing pressure than in human health care, although the limited adoption of insurance may also reduce pet owners ability to pay for diagnostics and therapeutics recommended by their veterinarians.
Development of Companion Animal Diagnostics
The development of companion animal diagnostics continues to evolve with the addition of new technologies to diagnostic portfolios. We believe that these new technologies may allow for the following:
| · | Enhanced capability to detect the frequency of occurrence and severity of diseases and conditions that impact companion animals; |
| · | Increased accuracy, lower cost and faster means to obtain test results; |
| · | Wider availability of new diagnostic tools; and |
| · | Enhanced economic benefits for veterinarians. |
Compared to human diagnostic development, the development of companion animal diagnostics is generally faster and less expensive since it typically requires smaller clinical studies, with fewer subjects. We believe that the lower cost of developing companion animal diagnostics enables us to pursue multiple diagnostic candidates simultaneously and to spread the risk of failure across a number of candidates, rather than concentrating all of our resources on one diagnostic candidate that may ultimately fail to achieve regulatory approval or market acceptance.
Development of Companion Animal Therapeutics
Compared to human drug development, the development of companion animal therapeutics is generally faster, more predictable, and less expensive, since it requires fewer clinical studies involving fewer subjects and can be conducted directly in the target species. Based on our progress since commencing business in May 2015, we believe that we will be able to develop a product candidate, from the initial opening of an INAD with the FDA-CVM through to marketing approval, in approximately three to five years at a cost of approximately $3 million to $5 million per product candidate. According to the Tufts Center for the Study of Drug Development, the successful development of a new drug for use in humans can take more than 10 years and requires an average out-of-pocket expenditure of approximately $1.4 billion. The lower cost associated with the development of companion animal therapeutics permits us to pursue multiple product candidates simultaneously and to spread the risk of failure across a number of product candidates, rather than concentrating all of our resources on one novel candidate that may ultimately fail to achieve regulatory approval or market acceptance.
Because we are developing product candidates based on drugs that have been successfully developed and approved for human use—as opposed to drugs based on new active pharmaceutical ingredients (APIs)—we believe that we will be able to avoid or minimize the expenses associated with the human drug development process and more rapidly advance our development programs, while continuing to comply with current good manufacturing practices, or cGMP, for our product candidates. Since we are not pursuing entirely new chemical entities with our drug product candidates, we believe the risk of failure of a specific drug product candidate is significantly lower compared to developing a novel compound.
| -6- |
The respective businesses of developing and commercializing therapeutics for companion animals and humans share a number of characteristics, including the need to:
| · | Demonstrate safety and efficacy in clinical trials; |
| · | Obtain FDA-CVM or other regulatory approval for marketing; |
| · | Manufacture the therapeutics in facilities compliant with cGMP requirements; and |
| · | Market the therapeutics only for their intended indication based on claims permitted in the product label, and not for other uses, which is referred to as “off-label” use. |
However, despite these similarities, there are a number of important differences between the companion animal therapeutics and human therapeutics businesses, including:
| · | Faster, less expensive and more predictable development. The development of therapeutics for companion animals requires fewer clinical studies in fewer subject animals than the development of human therapeutics and, unlike human therapeutics, studies are conducted directly in the target species. We believe that our strategy of selecting APIs with demonstrated efficacy and safety in humans and that are currently being used by veterinarians in their human compounded form enhances the predictability of results and probability of success of our pivotal trials relative to novel compounds that have not been previously validated. |
| · | Role and incentives for veterinary practices. In the United States, veterinarians generally serve the dual role of doctor and pharmacist, and pet owners typically purchase medications directly from their veterinarians. However, veterinarians often are required to have human drugs specially compounded by third-party compounding pharmacies for use in smaller companion animals, resulting in the loss of much of the associated prescription revenue and an increase in the uncertainty around precise dosing and administration. We believe that therapeutics specifically developed for companion animals will enable veterinarians to provide potentially superior treatment options, while also increasing revenue streams from the sale of these therapeutics. |
| · | Less generic competition and strong brand loyalty. There is less generic competition in the companion animal therapeutics industry than in the human health care industry. According to the Generic Animal Drug Alliance, 86% of FDA-approved animal drugs do not have a generic version. We believe that stronger brand loyalty and a lack of the mandatory generic drug substitution that exists in the human pharmaceutical market, partially explains the low penetration of generics in veterinary medicine. |
Unmet Medical Needs
Diagnostics
We believe that there is a significant unmet medical need for cost-effective and accurate disease/condition detection solutions for veterinarians. We believe that we have identified potential diagnostic assays that have the potential to satisfy unmet needs or improve upon existing diagnostic processes frequently used by companion animal veterinarians.
For example, cancer is a prevalent disease in canines that can be difficult and costly to diagnose using existing diagnostic testing. According to the Veterinary Cancer Society, 50% of all dogs over the age of 10 will develop cancer and one in four dogs will develop cancer at some stage in their life. Diagnosing certain cancers in canines is difficult because the location of the tumor may make it difficult or risky to obtain cell material through a biopsy. In addition, the overall health of a canine may increase the risk of performing a biopsy. Other diagnostic technologies, such as advanced imaging, are expensive while others, such as histopathologic examination, may take several days or more to provide a definitive diagnosis. Many more canine cancer cases may go undetected due to cost constraints and other factors. To address these shortcomings, we are developing a circulating tumor cell detection assay for use in the detection of certain cancers in companion animals.
| -7- |
Therapeutics
Despite the growing market for pet therapeutics, there are relatively few treatment options approved for use in companion animals, as compared to those approved for humans. As a result, veterinarians often must resort to prescribing products approved for use in humans, but not approved or formulated for use in companion animals. According to the FDA’s Electronic Animal Drug Product Listing Directory, approximately 54% of the therapeutics used in animals are unapproved for such use. As a result, veterinarians must rely upon trial and error or untested rules of thumb to assess the proper dosage needed to be effective in the particular species without undue risk of side effects. The veterinarian must also find a way to administer the human product to animals and determine the actual dosage amount, tasks which are important and potentially overlooked as practical considerations in the treatment of companion animals. To do this, veterinarians often rely on compounding pharmacies to formulate human drugs into species’ appropriate doses and formulations. As a result, veterinarians are forced to rely on therapeutics not proven safe and effective for their patients and on formulations for which no regulatory approval has been obtained. At the same time, the use of compounding pharmacies results in the veterinary clinic’s loss of much of the associated prescription revenue.
We believe that therapeutics specifically developed for companion animals can extend and improve the quality of the lives of such animals, help veterinarians achieve improved medical outcomes, and make the process of administering therapeutics to companion animals much safer and more convenient. Advances in human medicines have created new therapeutics for managing many chronic diseases. Pets often suffer from many of these same diseases. In many cases, the biology of these diseases in companion animals is very similar to that in humans, which explains why animal efficacy models are used for human drug development. Because of the similarity of the diseases and their symptoms and effects, many human drugs, when formulated properly and administered in proper doses, are effective in companion animals. However, most human drugs are not specially formulated or approved for use in animals.
Many of the human therapeutics used in companion animals are only available in pill or injectable form. However, it can be difficult to give a companion animal a shot or to assure that it has swallowed a pill. It can also be difficult to divide human pills into small enough portions to achieve an appropriate dosage for companion animals. Consequently, we believe that compliance with treatment regimens is a significant problem for veterinarians and pet owners. The challenges associated with medicating pets are unique, and we believe that developing product candidates that can be easily taken by the pet or that can be easily administered by pet owners will help increase compliance.
| -8- |
Product Pipeline
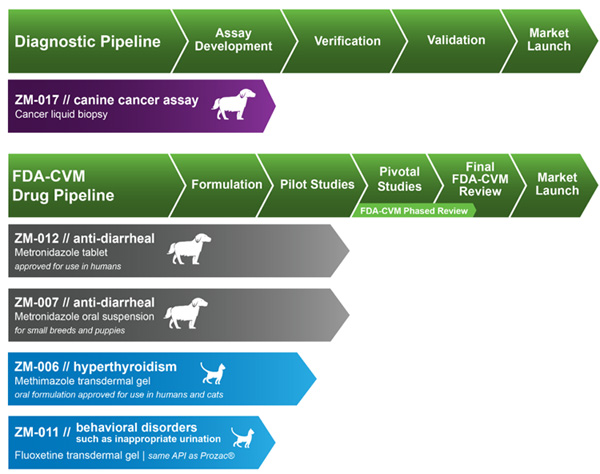
Diagnostics
Together with our strategic partner, we are developing a circulating tumor cell, or CTC, assay, also known as a “liquid biopsy,” for use by veterinarians as a cancer diagnostic. The liquid biopsy is a blood test that we believe has the potential to detect the presence of CTCs, which are cells that have shed from a primary tumor into neighboring blood vessels and are transported throughout the body’s circulatory system. Diagnosing certain cancers in canines is difficult because the location of the tumor may make it difficult or risky to obtain cell material through a biopsy. In addition, the overall health of a canine may increase the risk of performing a biopsy. Other diagnostic technologies, such as advanced imaging, are expensive while others, such as histopathologic examination, may take several days or more to provide a definitive diagnosis. We believe that the detection of CTCs in the blood could provide strong clinical support for a cancer diagnosis without the need for an invasive tissue biopsy or other expensive or time-consuming diagnostic test. We intend to develop and market ZM-017, a liquid biopsy for the detection of certain cancers in canines. If we successfully develop ZM-017, we expect that ZM-017 will provide veterinarians with a faster, more affordable, and less invasive test for certain cancers in canines compared to existing detection methods.
We expect to commence clinical validation of ZM-017 in the first half of 2018. Assuming that we successfully complete that clinical validation, we expect to commence the marketing of ZM-017 during the second half of 2018.
Therapeutics
We have four drug product candidates. Our lead drug product candidate is ZM-012, a novel tablet formulation of metronidazole targeting the treatment of acute diarrhea in dogs. An Investigational New Animal Drug, or INAD, was opened for ZM-012 with the Food and Drug Administration’s Center for Veterinary Medicine, or FDA-CVM, in April 2016. The API in ZM-012 is metronidazole, which has been the subject of multiple studies in humans and has been approved for use in humans for decades. We do not believe that the API in ZM-012 is protected by any patents or other proprietary rights of third parties in the U.S. We have finalized the formulation of ZM-012 as a beef-flavored oral tablet intended for dogs greater than nine pounds or four kilograms and we completed pilot testing of ZM-012 in the fourth quarter of 2017. In December 2017, we had a pre-submission meeting with the FDA-CVM to present the regulatory strategy and development plan for ZM-012. Based on the feedback we received from the FDA-CVM, we are making certain changes to the development plan for ZM-012 which we believe will not delay its development. We expect to commence a pivotal safety study of ZM-012 in the first half of 2018, which we expect to complete in the second half of 2018.
| -9- |
Our second drug product candidate is ZM-007, an oral suspension formulation of metronidazole and a complementary formulation to ZM-012, targeting the treatment of acute diarrhea in small dog breeds and puppies under nine pounds or four kilograms. An INAD was opened for ZM-007 with the FDA-CVM in October 2016. We have finalized the formulation and completed pilot testing of ZM-012. We expect to hold a pre-submission meeting in the first half of 2018 with the FDA-CVM specific to the product development strategy for ZM-007 as a bioequivalent to ZM-012. Drugs that are considered to be bioequivalent are, for regulatory purposes, essentially the same, meaning the absence of significant difference between the extent and rate of absorption over the course of a specific period of time at the same dose and under the same conditions. If deemed acceptable by the FDA-CVM, the implementation of this bioequivalent strategy is contingent on FDA-CVM approval of the new animal drug application (NADA) for ZM-012. If the FDA-CVM permits us to rely on the bioequivalence of ZM-007 to ZM-012, we anticipate that this regulatory pathway will conserve significant development costs because a bioequivalence study could replace the need for pivotal safety and efficacy studies for ZM-007.
Our third drug product candidate is ZM-006, a transdermal gel formulation of methimazole targeting hyperthyroidism in cats. Hyperthyroidism is one of the most commonly diagnosed endocrine disorders in middle-aged to older cats according to the American Association of Feline Practitioners. We are investigating ZM-006 pursuant to an INAD opened with the FDA-CVM in June 2016. The API in ZM-006, methimazole, has been the subject of multiple studies in humans and has been approved for oral use in humans for decades. Our transdermal gel formulation is intended to provide an alternative to an oral tablet formulation already approved by the FDA-CVM for cats. We do not believe that the API in ZM-006 is protected by any patents or other proprietary rights of third parties. ZM-006 is intended for application to the cat’s ear using an applicator pen. The formulation of ZM-006 has been completed. We expect to complete pilot testing of ZM-006 in the first half of 2018, and assuming that such pilot testing is successful, we intend to commence and complete a pivotal safety study of ZM-006 in the second half of 2018.
Our fourth drug product candidate is ZM-011, a transdermal gel formulation of fluoxetine, most commonly known as Prozac®, its human pharmaceutical brand name. Fluoxetine in pill or compounded form is frequently prescribed by veterinarians to treat feline behavioral disorders such as inappropriate urination. We are investigating ZM-011 pursuant to an INAD opened with the FDA-CVM in January 2017. The API, fluoxetine, has been the subject of multiple studies in humans and has been approved for use in humans for decades. We do not believe that the API in ZM-011 is protected by any patents or other proprietary rights of third parties. ZM-011 is a transdermal gel formulation intended for application to the cat’s ear using an applicator pen. The formulation of ZM-011 has been completed. We expect to complete pilot testing of ZM-011 in the second half of 2018.
License Agreements
In January 2017, we entered into a collaborative research agreement with Celsee, Inc., or Celsee, a developer of diagnostics for the detection and quantification of cells and other markers. Subsequent to this agreement, in December 2017, we entered into a license and supply agreement with Celsee for exclusive global rights to develop and market Celsee’s liquid biopsy platform. The agreement with Celsee covers the development and commercialization of liquid biopsy assays and related consumables for the detection of cancer in companion animals. We will be responsible for the clinical development and commercialization of the assays. Celsee will supply us on an exclusive basis with the assays and the consumables for the products to be developed under the agreement pursuant to a rolling forecast to be provided by us at prices specified in the agreement. We will be responsible for the marketing and sale of the assays and the related consumables. The agreement, which is exclusive in the field of veterinary cancer diagnostic applications, has a term of seven years (subject to termination in certain circumstances) and automatically renews for additional one-year terms thereafter.
| -10- |
We have agreed to pay Celsee up-front fees of $500,000 and to issue to Celsee unregistered common shares having a value of $250,000, consisting of an aggregate of 112,314 common shares to be issued at an ascribed price of $2.2259. Celsee is entitled to additional payments totaling up to an additional $1 million, payable 50 percent in cash and 50 percent in additional unregistered common shares, upon the achievement of specified milestones—namely, completion of product development (in respect of 50 percent of the foregoing cash and share payments) and upon successful completion of manufacturing milestones (as to the remaining 50 percent of the foregoing cash and share payments). Future issuances of shares will be subject to TSX-V approval and will be priced relative to market at the time of issuance. Celsee is entitled to certain registration rights with respect to the common shares issued by us under the agreement.
In April 2016, we entered into a collaboration agreement with CTX Technology, Inc., or CTX, which has developed a peptide-based skin penetration platform technology for the topical delivery of a range of APIs. Under this agreement, we have an option to obtain an exclusive worldwide license to use CTX’s technology platform in animals. In the event that we exercise the option, we would be required to pay CTX a one-time license fee of $20,000 and to pay CTX a royalty in the low single digits on any products that we sell that incorporates their technology. Unless we exercise our option prior thereto, this agreement will terminate on March 1, 2019.
Research and Development
Together with our strategic partner, we are performing development work on a liquid biopsy diagnostic platform for potential use in companion animals. Our drug product candidate development programs focus on the development of product candidates for target indications that have already demonstrated safety and efficacy in humans and the development of therapeutics based on these drugs for appropriate target indications in companion animals. In addition, we are investigating the development of alternative drug delivery systems for our drug product candidates. We use various contract research organizations, or CROs, to assist in performing our research and development activities.
In connection with these activities, we have incurred and will continue to incur significant research and development expenses. Our research and development expenses were $2,751,326 for the year ended December 31, 2017 and $1,518,589 for the year ended December 31, 2016.
Sales and Marketing
We intend to commercialize any product candidate for which we receive regulatory approval in the United States with a direct sales force. We intend to sell products directly to veterinarians, who typically mark up the diagnostics and therapeutics that they prescribe for pet owners. We believe that veterinarians are self-motivated to prescribe innovative therapeutics that are safe, effective, and supported by reliable clinical data and regulatory approval in order to improve the health of companion animals, while also generating additional revenue.
We will focus on marketing directly to both reference labs and the end user, veterinarians. It is common utility for veterinarians to send diagnostic samples to reference labs for analysis. Our strategic plan will be to sell the ZM-017 diagnostic equipment to a reference lab(s), while driving utilization of the test with the end user, veterinary community/market, back to the lab.
We also intend to selectively utilize distributors, which we believe will enable us to expand our commercial reach to a majority of all veterinarians in our chosen markets. We believe that we can compete effectively with a combination of our own direct sales force and complementary distributors.
To support our marketing efforts, we introduced a unique “Voice of the Vet™” program in the fourth quarter of 2016 to gather insights and better understand the needs of veterinarians and their practices, and to gauge interest for potential future product offerings, while building brand awareness as a valued veterinary partner. Our Voice of the Vet™ program allows veterinarians, practice managers and veterinary technicians to participate in conversations where they can share ideas and experiences with each other, as well as with us through an interactive platform. As part of our commercialization strategy, we also plan to participate in large veterinary meetings and to establish partnerships with leading veterinary colleges.
| -11- |
Manufacturing
We have no internal manufacturing capabilities for our diagnostic and therapeutic product candidates.
Under our license and supply agreement, Celsee is responsible for the manufacture and supply of the CTC platform technology and consumables to us. Celsee has primary responsibility for assuring that all products will be manufactured in accordance with applicable laws and meet all agreed upon specifications.
To ensure a dependable and high quality supply of the APIs for our pilot studies and pivotal trials, we rely on cGMP-compliant contract manufacturers. Because the APIs in our drug product candidates are used in human drugs that are no longer subject to patent protection, we believe that there are multiple contract manufacturers for our drug product candidates that have demonstrated the ability to provide high-quality formulated products more cost effectively than we could on our own. We believe that the contract manufacturers of our trial supplies will be able to provide commercial supplies of any of our drug product candidates that are approved for marketing.
While we and our contract manufacturers have historically been able to obtain supplies of the APIs for development of our drug product candidates, neither we nor our contract manufacturers have long-term supply agreements with the API manufacturers. We also have no agreements for commercial-scale supply of the API or manufacture of any of our drug product candidates.
Intellectual Property
We intend to rely primarily upon a combination of in-licensing exclusive rights, regulatory exclusivity, proprietary know-how, and confidentiality agreements to protect our diagnostic assays, product formulations, processes, methods and other technologies and to preserve any trade secrets and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. We currently have no issued patents.
Because our drug product candidates are based on approved human drugs that no longer are subject to patent protection, there is little, if any, composition-of-matter patent protection available for the API in these product candidates. Where feasible, however, we intend to pursue the broadest intellectual property protection possible for our compounds and any proprietary technology through enhanced formulations of our drug product candidates. However, even intellectual property protection, if available, may not afford us with complete protection against competitors.
We depend upon the skills, knowledge and experience of our management personnel, as well as that of our other employees, advisors, consultants and contractors, none of which are patentable. To help protect our know-how, and any inventions for which patents may be difficult to obtain or enforce, we require all of our employees, consultants, advisors and other contractors to enter into customary confidentiality and inventions agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
Competition
Diagnostics
Our potential competitors include large human pharmaceutical and medical diagnostics companies, small businesses focused on animal health, and reference laboratory services provided by academic institutions and in-clinic product providers. These competitors include Idexx Laboratories, Inc., Antech Diagnostics, a unit of VCA Inc., Abaxis, Inc., Heska Corporation and Zoetis Inc.
| -12- |
Therapeutics
If our drug product candidate is the first one approved by the FDA-CVM for use in animals, it may be eligible for between three and seven years of regulatory exclusivity in the United States, depending on the type of product and its intended use. However, while there are fewer competitors in the pet therapeutics industry than in the human pharmaceutical industry, the development and commercialization of new animal health medicines is highly competitive, and we expect competition from major pharmaceutical, biotechnology and specialty animal health medicine companies.
Our potential competitors include large animal health companies, which currently derive a significant portion of their revenue from livestock medications. Large animal health companies include Merck Animal Health, the animal health division of Merck & Co., Inc.; Elanco, the animal health division of Eli Lilly and Company; Bayer Animal Health, the animal health division of Bayer AG; Novartis Animal Health, the animal health division of Novartis AG; Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH; and Zoetis, Inc., as well as European companies such as Virbac S.A., Vetoquinol S.A., and Dechra Pharmaceuticals PLC. We are also aware of several smaller early stage companies that are developing products for use in the pet therapeutics market, including Kindred Biosciences, Inc., Aratana Therapeutics, Inc., Parnell Pharmaceuticals Holdings Ltd., and Jaguar Animal Health, Inc. Our drug product candidates will also face competition from medicines and products approved for use in humans that are used off-label for pets. Private organizations, academic institutions and government agencies conducting animal health product research are also considered potential competitors.
General
Many of our competitors and potential competitors have substantially more financial, technical, and human resources than we do. Many also have far more experience in the development, manufacture, regulation and worldwide commercialization of animal diagnostics and animal health medicines, including pet therapeutics. We also expect to compete with academic institutions, governmental agencies and private organizations that are conducting research in the fields of animal diagnostics and animal health medicines. If such competing products achieve regulatory approval and commercialization prior to our product candidates, or if our intellectual property protection and efforts to obtain regulatory exclusivity fail to provide us with exclusive marketing rights for some of our products, we may be unable to effectively compete in the markets in which we participate.
Government Regulation
Diagnostic Product Candidates
Our diagnostic product candidates may be subject to regulatory review by the USDA-CVB and/or post-marketing oversight by the USDA-CVB or FDA-CVM. Generally speaking, full diagnostic kits aimed at the detection or diagnosis of an infectious disease in animals, including the materials required for testing along with instructions for use and interpretation of results, used at the point-of-care, including in-office diagnostic tests, may be subject to pre-market regulatory review and approval by the USDA-CVB. The USDA-CVB’s review process for diagnostics is subject to some variability based on the type of diagnostic kit being reviewed, however, the USDA-CVB will generally review the results of specific tests that are required to be conducted in accordance with the USDA-CVB’s testing criteria. These include diagnostic sensitivity/specificity studies, conducted using a large number of samples of U.S. origin, reproducibility/repeatability/suitability studies used to evaluate test kits under field conditions in participating laboratories and ruggedness studies in which manufacturers measure the ruggedness or robustness of the diagnostic test kits based on the capacity of the assay to remain unaffected by small variations in or deviations from the instructions for use (for example, not allowing the samples to reach the designated temperature). Diagnostic products and testing kits that do not claim to detect or diagnose an infectious disease and that are not designed for use at the point-of-care are generally subject only to post-marketing oversight by the FDA-CVM or the USDA-CVB. While the sale of these products does not require premarket approval by the FDA-CVM and does not subject us to the FDA-CVM’s cGMP requirements, these products must not be adulterated, mislabeled or misbranded under the Federal Food, Drug and Cosmetic Act, or the FDC Act, and are subject to post-marketing review.
| -13- |
Drug Product Candidates
The FDA-CVM regulates animal pharmaceuticals under the FDC Act. In order to obtain regulatory approval to market a drug product candidate in the U.S., an applicant must demonstrate that the product candidate is safe, effective and produced by a consistent method of manufacture. Post-approval monitoring of products is required by law, with reports being provided to the FDA-CVM's Surveillance and Compliance group. Reports of product quality defects, adverse events or unexpected results are required in accordance with the law.
Prior to commencing testing of a drug product candidate, an applicant is required to open an INAD with the FDA-CVM. Formulation work and pilot testing occurs once the INAD is opened. This is followed by a pre-submission conference with the FDA-CVM to discuss and agree on a proposed development plan, including the design of pivotal safety and clinical trials that would support approval of a new animal drug application, or NADA.

Early pilot studies may be conducted in laboratory animals to establish clinical endpoints and the dose range for a new drug product candidate. Data on how well the drug is absorbed when dosed by different routes of administration and the relationship of the dose to the effectiveness are studied.
During development, the applicant will usually submit a proposed pivotal trial protocol to the FDA-CVM for review and concurrence prior to conducting the trial. The applicant must gather and submit data on manufacturing, safety and effectiveness to the FDA-CVM for review, which will be conducted according to timelines specified in the Animal Drug User Fee Act, or ADUFA. ADUFA also imposes certain fees including a sponsor fee of $75,150 per year, an application fee of $238,100 per product candidate submission, and certain administrative application and manufacturing fees imposed per product candidate per year based on sales.
The pivotal clinical trial must be conducted with the formulation of the drug product candidate that is intended to be commercialized, and is a multi-site, randomized, controlled study, generally with a placebo control. To reduce bias in the study, individuals doing the assessment are not told whether the subject is in the group receiving the treatment being tested or the placebo group.
Once all data have been submitted and reviewed for each technical section - safety, effectiveness and chemistry, manufacturing and controls, or CMC - the FDA-CVM issues a “technical section complete letter” as each section review is completed, and when all three letters have been issued, the applicant prepares a draft of the Freedom of Information Summary, the proposed labeling, and all other relevant information, and submits these for FDA review. An administrative NADA is a NADA that is submitted after all of the technical sections that fulfill the requirements for the approval of the new drug product candidate have been reviewed by FDA-CVM and FDA-CVM has issued a technical section complete letter for each of those technical sections. Although this process is not required and submission of a non-administrative NADA is also acceptable, we plan to take advantage of the administrative NADA process to obtain a timelier phased review. Because FDA-CVM has already reviewed the individual technical sections before the administrative NADA is filed, FDA-CVM is committed under ADUFA to reviewing and acting on 90% of administrative NADAs within 60 days after submission. The FDA-CVM user fee goal is to review and act on 90% of non-administrative NADAs within 180 days after submission. After approval, we will be required to collect reports of adverse events and submit them on a regular basis to the FDA.
Other Regulatory Considerations
Regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to our product candidates because our product candidates are not intended for use in food animals or food production animals.
| -14- |
Advertising and promotion of animal health products is controlled by regulations in the United States. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and authorized by the FDA-CVM.
Any drug product candidate, if approved, may eventually face generic competition in the United States. In the United States, a generic animal drug may be approved pursuant to an Abbreviated New Animal Drug Application, or ANADA. Instead of demonstrating the drug’s safety and effectiveness in the target species as required in a NADA, a generic applicant must only show that the proposed generic product is the same as, and bioequivalent to, the approved brand name product. However, if any of our drug product candidates is the first one approved by the FDA-CVM for use in animals, it will be eligible for between three and seven years of regulatory exclusivity in the United States, depending on the type of product and its intended use.
We will be required to conduct post-approval monitoring of any approved product and to submit reports of product quality defects, adverse events or unexpected results, and be subject to regulatory inspection from time to time. Safety, quality, or efficacy concerns can lead to product recalls, withdrawals or suspended or declining sales, as well as product liability and other claims.
Employees
As of December 31, 2017, we had 20 employees, including one employee who is a doctor of veterinary medicine. Of our employees, four are engaged in research and development activities, five are engaged in business development and marketing activities, and eleven are engaged in corporate and administrative activities. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Properties
Our corporate headquarters is located in Ann Arbor, Michigan, where we lease approximately 7,900 square feet pursuant to a lease that expires February 2022. Our research and development laboratory is also located in Ann Arbor, Michigan, where we lease approximately 4,800 square feet pursuant to a lease that expires in August 2018. We have the option to extend that lease for three additional years. We believe that our facilities are sufficient for our existing and expected future needs.
Legal Proceedings
We are not currently a party to any material legal proceedings.
Corporate Information
Zomedica Pharmaceuticals Corp. (formerly, Wise Oakwood Ventures Inc.) was originally incorporated as Wise Oakwood Ventures Inc. on January 7, 2013 under the Business Corporations Act (Alberta). On October 28, 2013, we completed our initial public offering in Canada and became classified as a Capital Pool Company, as defined under the rules of the TSX Venture Exchange, or TSX-V. On April 21, 2016, we changed our name to Zomedica Pharmaceuticals Corp. and consolidated our common shares on a one-for-two and one-half (2½) basis. ZoMedica Pharmaceuticals Inc., or ZoMedica Inc., was incorporated on May 14, 2015 under the Canada Business Corporations Act. On April 21, 2016, we completed a qualifying transaction, or the Qualifying Transaction, under TSX-V Policy 2.4 – Capital Pool Companies, consisting of a three-cornered amalgamation among our company, ZoMedica Inc. and our wholly-owned subsidiary. Under the Qualifying Transaction, ZoMedica Inc. and our subsidiary were amalgamated to form Zomedica Pharmaceuticals Ltd., or Zomedica Ltd. As consideration for the amalgamation, shareholders of ZoMedica Inc. became the owners of 97.6% (non-diluted) of our common shares, and ZoMedica Ltd. became our wholly-owned subsidiary. Subsequent to the Qualifying Transaction, Zomedica Ltd. was vertically amalgamated into our company. We have one wholly-owned subsidiary, Zomedica Pharmaceuticals, Inc., a Delaware company. ZoMedica Inc. entered into the Qualifying Transaction in order to accomplish the following:
| · | Enable its shareholders to own shares in a company that was publicly traded on the TSX-V; |
| · | Expand its shareholder base to include the public shareholders of Wise Oakwood; and |
| · | Obtain access to the cash resources raised by Wise Oakwood in its initial public offering. |
| -15- |
On November 10, 2017, our shares were approved for listing on the NYSE American under the symbol “ZOM”. On November 20, 2017 the U.S. Securities and Exchange Commission declared our registration statement on Form S-1 effective. Our common shares commenced trading on the NYSE American on November 21, 2017.
Our principal executive offices are located at 100 Phoenix Drive, Suite 190, Ann Arbor, MI 48108, and our telephone number is (734) 369-2555. Our website address is www.zomedica.com. The information contained in, or accessible through, our website is not part of the registration statement of which this prospectus forms a part.
| Item 1A. | Risk Factors. |
RISK FACTORS
Risks Related to Our Business
We have a limited operating history, are not profitable and may never become profitable.
We are a development stage veterinary diagnostic and pharmaceutical company creating products for companion animals (canine, feline, and equine) by focusing on the unmet needs of clinical veterinarians. Since the commencement of our business in May 2015, our operations have been primarily limited to the identification of product candidates and research and development of our diagnostic and drug product candidates, ZM-017, a non-invasive diagnostic assay or blood test for the detection of certain cancers in canines, ZM-012 and ZM-007, an anti-diarrheal in pill form and oral suspension respectively that is intended for use in dogs, ZM-006, a transdermal gel treatment for hyperthyroidism, a metabolic disorder, which is intended for use in cats and ZM-011, a transdermal gel treatment for behavioral disorders intended for use in cats. As a result, we have limited historical operations upon which to evaluate our business and prospects and we have not yet demonstrated an ability to obtain approval for any of our product candidates or successfully overcome the risks and uncertainties frequently encountered by companies in emerging fields such as the companion animal pharmaceuticals and health care solutions industry.
We also have not generated any revenue to date, and we expect to continue to incur significant research and development costs and other expenses. Our net loss and comprehensive loss for the years ended December 31, 2017 and December 31, 2016 was $8,065,072 and $5,740,492, respectively. Our accumulated deficit as of December 31, 2017 was $15,626,100. As of December 31, 2017, we had total shareholders' equity of $4,387,085. We expect to continue to incur losses for the foreseeable future, which will increase significantly from historical levels as we expand our product development activities (including conducting required clinical studies and trials), seek necessary approvals for our product candidates, and begin commercialization activities. Even if we succeed in developing and broadly commercializing one or more of our product candidates, we expect to continue to incur losses for the foreseeable future, and we may never become profitable. If we fail to achieve or maintain profitability, then we may be unable to continue our operations at planned levels and be forced to reduce or cease operations.
We will need to raise additional capital to achieve our goals.
We do not have any products approved for sale. Although we believe we do not require pre-market approval from the U.S. Food and Drug Administration’s Center for Veterinary Medicine, or the FDA-CVM, to market and sell ZM-017, the CTC diagnostic assay we are developing, we do not expect to commence marketing of ZM-017 until the second half of 2018.
Until, and unless, we receive approval from the FDA-CVM for our drug product candidates, we cannot market or sell our drug products in the United States and will have no material drug product revenue. Our lead drug product candidates, ZM-012, ZM-007, ZM-006 and ZM-011 are in the formulation, optimization and/or pilot study stage, and we have not yet begun pivotal trials. We anticipate that each of our drug product candidates will require from three to five years of development at a cost of approximately $3 million to $5 million per drug product candidate before we expect to be able to apply for marketing approval in the United States.
We are also seeking to identify potential complementary opportunities in the veterinary diagnostics and therapeutics sectors. We will continue to expend substantial resources for the foreseeable future to develop our existing product candidates and any other product candidates we may develop or acquire. These expenditures will include: costs of identifying additional potential product candidates; costs associated with drug formulation; costs associated with conducting pilot and pivotal trials and clinical studies; costs associated with completing other research and development activities; costs associated with payments to technology licensors and maintaining other intellectual property; costs of obtaining regulatory approvals; costs associated with securing contract manufacturers to meet our commercial manufacturing and supply capabilities; and costs associated with marketing and selling any of our products approved for sale. We also may incur unanticipated costs. Because the outcome of our development activities and commercialization efforts is inherently uncertain, the actual amounts necessary to successfully complete the development and commercialization of our existing or future product candidates may be greater or less than we anticipate.
| -16- |
As a result, we will need to obtain additional capital to fund the development of our business. Except for our $5,000,000 unsecured working capital loan facility, we have no existing agreements or arrangements with respect to any financings, and any such financings may result in dilution to our shareholders, the imposition of debt covenants and repayment obligations or other restrictions that may adversely affect our business or the value of our common shares.
Our future capital requirements depend on many factors, including, but not limited to:
| • | the scope, progress, results and costs of researching and developing our existing or future diagnostics and product candidates; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for any of our existing or future diagnostics or product candidates; |
| • | the number and characteristics of the diagnostics and/or product candidates we pursue; |
| • | the cost of contract manufacturers to manufacture our existing and future diagnostics and product candidates and any products we successfully commercialize; |
| • | the cost of commercialization activities if any of our existing or future diagnostics and product candidates are approved for sale, including marketing, sales and distribution costs; |
| • | the expenses needed to attract and retain skilled personnel; |
| • | the costs associated with being a public company; |
| • | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; and |
| • | the costs involved in preparing and filing patent applications, maintaining any successfully obtained patents and protecting and enforcing any such patents. |
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate one or more of our product development programs or any future commercialization efforts.
We are substantially dependent on the success of our lead product candidates, and cannot be certain that any of them will be approved for marketing, to the extent applicable, or successfully commercialized.
We have no products approved for sale in any jurisdiction and are focused primarily on the development of our lead diagnostic and drug product candidates, ZM-017, ZM-012, ZM-007, ZM-006 and ZM-011. Accordingly, our near-term prospects, including our ability to generate material product revenue, or enter into potential strategic transactions, will depend heavily on the successful development and commercialization of one or more of our lead candidates, which in turn will depend on a number of factors, including the following:
| • |
the successful completion of clinical validation of our diagnostic product candidate which may take significantly longer than we anticipate and will depend, in part, upon the satisfactory performance of third-party contractors; | |
| • | the successful completion of pilot testing and pivotal efficacy and safety trials of one or more of our drug product candidates, which may take significantly longer than we anticipate and will depend, in part, upon the satisfactory performance of third-party contractors; |
| • | our ability to demonstrate to the satisfaction of the FDA-CVM or the USDA Center for Veterinary Biologics, or USDA-CVB, as applicable, the safety and efficacy of our drug product candidates and to obtain regulatory approvals; |
| • | the ability of our third-party contract manufacturers to manufacture supplies of any of our product candidates and to develop, validate and maintain viable commercial manufacturing processes that are compliant with Good Manufacturing Practices or GMP; |
| • | our ability to successfully market any product candidate for which marketing approval is received, whether alone or in collaboration with others; |
| • | the availability, perceived advantages, relative cost, relative safety and relative efficacy of our product candidates compared to alternative and competing treatments; |
| • | the acceptance of our product candidates as safe and effective by veterinarians, pet owners and the animal health community; |
| -17- |
| • | our ability to achieve and maintain compliance with all regulatory requirements applicable to our business; and |
| • | our ability to obtain and enforce our intellectual property rights and obtain marketing exclusivity for our product candidates, and avoid or prevail in any third-party patent interference, patent infringement claims or administrative patent proceedings initiated by third parties or the United States Patent and Trademark Office (“USPTO”). |
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will be successful in developing or commercializing any of our product candidates. If we are unsuccessful or are significantly delayed in developing and commercializing ZM-017, ZM-012, ZM-007, ZM-006 or ZM-011 or any of our other product candidates, our business and prospects will be materially adversely affected and you may lose all or a portion of your investment.
We face unproven markets for our products candidates.
The companion animal therapeutic and diagnostic markets are less developed than the human therapeutic and diagnostic markets and as a result no assurance can be given that our product candidates will be successful. Veterinarians, pet owners or other veterinary health providers in general may not accept or utilize any products that we may develop.
The companion animal care industry is subject to rapidly changing technology, which could make our product candidates obsolete.
The companion animal care industry is characterized by rapid technological changes, frequent new product introductions and enhancements, and evolving industry standards, all of which could make our product candidates obsolete. Our future success will depend on our ability to keep pace with the evolving needs of our customers on a timely and cost-effective basis and to pursue new market opportunities that develop as a result of technological and scientific advances. We must continuously enhance our product offerings to keep pace with evolving standards of care. If we do not update our product offerings to reflect new scientific knowledge or new standards of care, our product candidates could become obsolete, which would have a material adverse effect on our business, financial condition, and results of operations.
Our ability to successfully develop and commercialize our existing and any future product candidates will depend on several factors, including:
| • | our ability to convince the veterinary community of the clinical utility of our products and their potential advantages over existing tests and therapies; |
| • | the willingness or ability by pet owners to pay for our products and the willingness of veterinarians to recommend our products; |
| • | the willingness of veterinarians to utilize our diagnostic tests; and |
| • | where applicable, the willingness of testing labs to buy our assay equipment. |
Our dependence on suppliers could limit our ability to develop and commercialize certain products
We rely on third-party suppliers to provide components in our product candidates, manufacture products that we do not manufacture ourselves and perform services that we do not provide ourselves. Because these suppliers are independent third parties with their own financial objectives, actions taken by them could have a materially negative effect on our results of operations. The risks of relying on suppliers include our inability to enter into contracts with third-party suppliers on reasonable terms, inconsistent or inadequate quality control, relocation of supplier facilities, supplier work stoppages and suppliers’ failure to comply with applicable regulations or their contractual obligations. Problems with suppliers could materially negatively impact our ability to complete development, supply the market, lead to higher costs or damage our reputation with our customers.
| -18- |
In addition, we currently purchase many products and materials from sole or single sources. Some of the products that we purchase from these sources are proprietary and, therefore, cannot be readily or easily replaced by alternative sources. To mitigate risks associated with sole and single source suppliers, we will seek when possible to enter into long-term contracts that provide for an uninterrupted supply of products at predictable prices. However, some suppliers may decline to enter into long-term contracts and we are required to purchase products with short term contracts or on a purchase order basis. There can be no assurance that suppliers with which we do not have contracts will continue to supply our requirements for products, that suppliers with which we do have contracts will always fulfill their obligations under these contracts, or that any of our suppliers will not experience disruptions in their ability to supply our requirements for products. In cases where we purchase sole and single source products or components under purchase orders, we are more susceptible to unanticipated cost increases or changes in other terms of supply. In addition, under some contracts with suppliers we have minimum purchase obligations, and our failure to satisfy those obligations may result in loss of some or all of our rights under these contracts or require us to compensate the supplier. If we are unable to obtain adequate quantities of products in the future from sole and single source suppliers, we may be unable to supply the market, which could have a material adverse effect on our results of operations.
The commercial potential of our product candidates is difficult to predict. The market for any product candidate, or for companion animal diagnostics and therapeutics overall, is uncertain and may be smaller than we anticipate, which could significantly and negatively impact our revenue, results of operations and financial condition.
We believe that the emerging nature of our industry and our unproven business plan make it difficult to estimate the commercial potential of any of our product candidates. The market for any product that we seek to commercialize will depend on important factors such as the cost, utility and ease of use of our diagnostic assays, the safety and efficacy of our drug candidates compared to other available treatments, including potentially less expensive human pharmaceutical alternatives with similar efficacy profiles, changing standards of care, preferences of veterinarians, the willingness of pet owners to pay for such products, and the availability of competitive alternatives that may emerge either during the product development process or after commercial introduction. If the market potential for our product candidates is less than we anticipate due to one or more of these factors, it could negatively impact our business, financial condition and results of operations. Further, the willingness of pet owners to pay for our product candidates, if approved, may be less than we anticipate, and may be negatively affected by overall economic conditions. Because relatively few pet owners purchase insurance for their companion animals, pet owners are more likely to have to pay for our products directly and may be unwilling or unable to pay for any such products.
All of our drug product candidates are based on APIs already demonstrated safe and effective in humans, and other companies may develop substantially similar products that may compete with our products.
Our lead drug product candidates, ZM-012, ZM-007, ZM-006 and ZM-011 include APIs already demonstrated safe and effective in humans and we expect that our future drug product candidates will be similarly based on such APIs. We do not engage in research or discovery of novel therapeutics, but focus on drug product candidates with APIs that have been successfully commercialized or demonstrated to be safe and effective in humans, which we sometimes refer to as validated. We expect that there will be little, if any, third-party patent protection of the APIs in our drug product candidates. As a result, our drug product candidates may face competition from their human equivalents in situations where such equivalents are available and used in unapproved animal indications, which is known as off-label use. There is no assurance that the eventual prices of our drug products will be lower than or competitive with the prices of the human equivalents used off-label, or that a palatable, easy-to-administer formulation will be sufficient to differentiate them from their human equivalents.
| -19- |
Our product candidates will face significant competition and may be unable to compete effectively.
The development and commercialization of veterinary diagnostics and pharmaceuticals is highly competitive and our success depends on our ability to compete effectively with other products in the market.
There are a number of competitors in the diagnostic market that have substantially greater financial and operational resources and established marketing, sales and service organizations. We expect to compete primarily with commercial clinical laboratories and hospitals’ clinical laboratories. Our principal competitors in the veterinary diagnostic market are Idexx Laboratories, Inc., Antech Diagnostics, a unit of VCA Inc., Abaxis, Inc., Heska Corporation and Zoetis Inc. We must develop our distribution channels and build our direct sales force in order to compete effectively in these markets. If we are unable to effectively manage our distribution channels in our highly competitive industry, we may fail to retain customers or obtain new customers and our business will suffer.
If our drug product candidates are approved, we expect to compete with large animal health companies including Merck Animal Health, the animal health division of Merck & Co., Inc.; Elanco, the animal health division of Eli Lilly and Company; Bayer Animal Health, the animal health division of Bayer AG; Novartis Animal Health, the animal health division of Novartis AG; Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH; and Zoetis, Inc., as well as European companies such as Virbac S.A., Vetoquinol S.A., and Dechra Pharmaceuticals PLC We are also aware of several smaller early stage companies that are developing products for use in the pet therapeutics market, including Kindred Biosciences, Inc., Aratana Therapeutics, Inc., Parnell Pharmaceuticals Holdings Ltd., and Jaguar Animal Health, Inc. We also expect to compete with academic institutions, governmental agencies and private organizations that are conducting research in the field of animal health medicines.
We target drug product candidates for which the API, while having been approved for use in human drugs, has not been previously approved for use in animals. If we are the first to gain approval for the use of such API in animals, our drug products will enjoy between three and seven years of marketing exclusivity in the United States for the approved indication. We also plan to differentiate our products where possible with alternative drug delivery systems that are more conducive to dosing for the target companion animal species, but we cannot assure you that we will be able to prevent our competitors from developing substantially similar products and bringing those products to market earlier than we are able to.
Our drug product candidates will face competition from various products approved for use in humans that are used off-label in animals, and all of our products will face potential competition from new products in development. These and other potential competing products may benefit from greater brand recognition and brand loyalty than our drug product candidates may achieve.
Many of our competitors and potential competitors have substantially more financial, technical and human resources than we do. Many also have far more experience than we have in the development, manufacture, regulation and worldwide commercialization of animal health medicines, including pet therapeutics. We also expect to compete with academic institutions, governmental agencies and private organizations that are conducting research in the fields of animal diagnostics and animal health. If such competing products are commercialized prior to our product candidates, or if our intellectual property protection and efforts to obtain regulatory exclusivity fail to provide us with exclusive marketing rights for some of our therapeutic products, we may be unable to compete effectively in the markets in which we participate.
Our ability to develop, manufacture and commercialize our drug product candidates is dependent on our establishing and maintaining relationships with GMP-compliant third party manufacturers.
We have no internal manufacturing capabilities and we do not plan to develop such capabilities. As a result, our ability to manufacture and commercialize our product candidates is substantially dependent on our ability to ensure a dependable and high quality supply of the APIs required for our pilot studies and pivotal trials and for future commercial manufacturing. We currently believe that, because the APIs used in our drug product candidates have been used in human drugs, there are multiple GMP-compliant manufacturers available that will be able to supply these APIs and that the contract manufacturers we currently use for our trial supplies will be able to provide commercial supplies of any of our drug product candidates. While we have historically been able to obtain the necessary supplies of our APIs for our development work, we cannot be certain that either we or our contract manufacturers will continue to be able to provide the necessary API supply. Neither we nor our contract manufacturers have long-term supply contracts with API manufacturers and we have no agreements in place for the commercial-scale supply of any API or the manufacture of any of our drug product candidates. If we are unable to procure the requisite apply of an API or to contract with a GMP-complaint third-party manufacturer, we may be unable to continue to develop, manufacture or commercialize any of our product candidates and our business may fail to grow or develop.
| -20- |
The results of earlier studies may not be predictive of the results of our pivotal trials, and we may be unable to obtain regulatory approval for our existing or future drug product candidates under applicable regulatory requirements or maintain any regulatory approval obtained. The denial, delay or loss of any regulatory approval would prevent or delay our commercialization efforts and adversely affect our financial condition and results of operations.
The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of our product candidates are subject to extensive regulation. We will not be permitted to market our drug product candidates in the United States until we receive approval of a New Animal Drug Application, or NADA, from the FDA-CVM and may not be able to market and sell any point-of-care diagnostic products without pre-marketing approval from the USDA-CVB. To gain approval to market a pet pharmaceutical or point-of-care diagnostic product kit for a particular species, we must provide the FDA-CVM or the USDA-CVB, as applicable, with efficacy data from pivotal trials that adequately demonstrate that our product candidates are safe and effective in the target species for the intended indications. In addition, we must provide manufacturing data. For the FDA-CVM, we must provide data from safety testing and clinical data, also called target animal safety studies. Similarly, for the USDA-CVB, we must provide the results of specific tests required to be conducted in accordance with the USDA-CVB’s guidelines demonstrating the sensitivity/specificity, reproducibility/repeatability/suitability and the ruggedness or robustness of the relevant diagnostic kit. Either of the FDA-CVM or the USDA-CVB may also require us to conduct costly post-approval testing and/or collect post-approval safety data to maintain our approval for any product candidate or diagnostic. The results of our pivotal studies and other initial development activities, and the results of any previous studies in humans or animals conducted by us or third parties, may not be predictive of future results of pivotal trials or other future studies, and failure can occur at any time during or after pivotal studies and other development activities by us or our contract research organizations or CROs. Our pivotal studies may fail to show the desired safety or efficacy of our product candidates despite promising initial data or the results in previous human or animal studies conducted by others, and the success of a product candidate in prior animal studies, or in the treatment of human beings, does not ensure success in subsequent studies. Clinical trials in humans and pivotal trials in animals sometimes fail to show a benefit even for drugs that are effective, because of statistical limitations in the design of the trials or other statistical anomalies. Therefore, even if our studies and other development activities are completed as planned, the results may not be sufficient to obtain regulatory approval for our product candidates.
The FDA-CVM or the USDA-CVB can delay, limit, deny or revoke approval of any of our product candidates for many reasons, including:
| • | if the FDA-CVM or USDA-CVB disagrees with our interpretation of data from our pivotal studies or other development efforts; |
| • | if we are unable to demonstrate to the satisfaction of the FDA-CVM or the USDA-CVB that the product candidate is safe and effective for the target indication; |
| • | if the FDA-CVM or USDA-CVB requires additional studies or changes its approval policies or regulations; |
| • | if the FDA-CVM or USDA-CVB does not approve of the formulation, labeling or the specifications of our existing and future product candidates; |
| • | if the FDA-CVM or USDA-CVB fails to approve the manufacturing processes of our third-party contract manufacturers; and |
| • | if any approved product candidate subsequently fails post-approval testing required by the FDA-CVM or the USDA-CVB. |
Further, even if we receive approval of our product candidates, such approval may be for a more limited indication than we originally requested, the FDA-CVM or USDA-CVB may not approve the labeling that we believe is necessary or desirable for the successful commercialization of our product candidates and we may be required to conduct costly post-approval testing. Any delay or failure in obtaining applicable regulatory approval for the intended indications of our product candidates would delay or prevent commercialization of such product candidates and would materially adversely impact our business and prospects.
| -21- |
Development of product candidates for use in companion animal health is an inherently expensive, time-consuming and uncertain, and any delay or discontinuance of validation or pivotal studies for our current or future product candidates would significantly harm our business and prospects.
Development of product candidates for use in companion animals is an inherently lengthy, expensive and uncertain process, and there is no assurance that our development activities will be successful. We do not know whether the validation study of ZM-017 or the pivotal studies of ZM-012, ZM-007, ZM-006 or ZM-011, or of any of our other product candidates, will begin or conclude on time, and they may be delayed or discontinued for a variety of reasons, including if we are unable to:
| • | address any safety concerns that arise during the course of the studies; |
| • | complete the studies due to deviations from the study protocols, the occurrence of adverse events or, in the case of our validation studies, sensitivity and selectivity results that vary from our expectations; |
| • | add new study sites; |
| • | address any conflicts with new or existing laws or regulations; or |
| • | reach agreement on acceptable terms with study sites, which can be subject to extensive negotiation and may vary significantly among different sites. |
Any delays in completing our development efforts will increase our costs, delay our product candidate development and any regulatory approval process and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, factors that may cause a delay in the commencement or completion of our development efforts may also ultimately lead to the denial of regulatory approval of our product candidates which, as described above, would materially, adversely impact our business and prospects.
We will rely on third parties to conduct certain of our development activities. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize our product candidates.
We have used CROs to conduct our research and development activities and expect to continue to do so, including with respect to our clinical validation, pilot studies and pivotal trials of ZM-017, ZM-012, ZM-007, ZM-006 and ZM-011. These CROs are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs or manage the risks associated with their activities on our behalf. We are responsible to regulatory authorities for ensuring that each of our studies is conducted in accordance with the development plans and trial protocols, and any failure by our CROs to do so may adversely affect our ability to obtain regulatory approvals, subject us to penalties, or harm our credibility with regulators. The FDA-CVM also requires us and our CROs to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, and good laboratory practices, or GLPs, for conducting, monitoring, recording and reporting the results of our studies to ensure that the data and results are scientifically credible and accurate.
Our agreements with our CROs may allow termination by the CROs in certain circumstances with little or no advance notice to us. These agreements generally will require our CROs to reasonably cooperate with us at our expense for an orderly winding down of the CROs’ services under the agreements. If the CROs conducting our studies do not comply with their contractual duties or obligations to us, or if they experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our development protocols or GCPs or for any other reason, we may need to secure new arrangements with alternative CROs, which could be difficult and costly. In such event, our studies also may need to be extended, delayed or terminated as a result, or may need to be repeated. If any of the foregoing were to occur, regulatory approval and commercialization of our product candidates may be delayed and we may be required to expend substantial additional resources.
| -22- |
The failure of any CRO to perform adequately or the termination of any arrangements with any of them may adversely affect our business.
We rely on third-party manufacturers to produce our product candidates. If we experience problems with any of these suppliers, the manufacturing of our product candidates or products could be delayed.
We do not have the capability to manufacture our product candidates and do not intend to develop that capability. As a result, we rely on CMOs to produce our product candidates. We expect to enter into contracts with CMOs for the commercial scale production of the products we intend to commercialize. Reliance on CMOs involves risks, including:
| • | the inability to meet our product specifications and quality requirements consistently; |
| • | inability to access production facilities on a timely basis; |
| • | inability or delay in increasing manufacturing capacity; |
| • | manufacturing and product quality issues related to the scale-up of manufacturing; |
| • | costs and validation of new equipment and facilities required for commercial level activity; |
| • | a failure to satisfy any applicable FDA-CVM cGMP requirements on a consistent basis; |
| • | the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms; |
| • | termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; |
| • | the reliance on a single sources of supply which, if unavailable, would delay our ability to complete the development and testing and commercialization of our products; |
| • | the lack of qualified backup suppliers for supplies that are currently purchased from a single source supplier; |
| • | operations of our CMOs or suppliers could be disrupted by conditions unrelated to our business or operations, including the bankruptcy of the CMO or supplier; |
| • | carrier disruptions or increased costs that are beyond our control; and |
| • | the failure to deliver products under specified storage conditions and in a timely manner. |
Any of these risks could cause the delay of validation studies, clinical trials, regulatory submissions, the receipt of any required approvals or the commercialization of our products, cause us to incur higher costs and prevent us from commercializing our product candidates successfully. Manufacturing of our product candidates and any approved products subject to cGMP could be disrupted or halted if our CMOs do not comply with cGMP, even if the compliance failure does not relate to our product candidates or approved products. Furthermore, if our CMOs fail to deliver the required commercial quantities of finished product on a timely basis and at commercially reasonable prices and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the FDA-CVM in the event that such approval is required.
Even if our product candidates obtain regulatory approval, they may never achieve market acceptance or commercial success.
Even if we obtain FDA-CVM, USDA-CVB or other regulatory approvals, our product candidates may not achieve market acceptance among veterinarians and pet owners, and may not be commercially successful. Market acceptance of any of our product candidates for which we receive approval depends on a number of factors, including:
| • | the safety of our products as demonstrated in our target animal studies; | |
| • | the indications for which our products are approved; | |
| • | the acceptance by veterinarians and pet owners of the product as a safe and effective treatment; |
| -23- |
| • | the proper training and administration or use of our products by veterinarians; | |
| • | the potential and perceived advantages of our product candidates over alternative treatments or diagnostics, including products approved for use by humans that are used off label; | |
| • | the cost of treatment in relation to alternative treatments and willingness to pay for our products, if approved, on the part of veterinarians and pet owners; | |
| • | the willingness of pet owners to pay for our treatments, relative to other discretionary items, especially during economically challenging times; | |
| • | the relative convenience and ease of administration; | |
| • | the prevalence and severity of adverse side effects; and | |
| • | the effectiveness of our sales and marketing efforts. |
If our approved products fail to achieve market acceptance or commercial success, our business could fail and you could lose your entire investment.
Pharmaceuticals for companion animals, like human pharmaceuticals, are subject to unanticipated post-approval safety or efficacy concerns, which may harm our business and reputation.
The success of our commercialization efforts will depend upon the perceived safety and effectiveness of pharmaceuticals for companion animals, in general, and of our products, in particular. Unanticipated safety or efficacy concerns can arise with respect to approved therapeutics after they enter into commerce, which may result in product recalls or withdrawals or suspension of sales, as well as product liability and other claims. It is also possible that the occurrence of significant adverse side effects in approved human compounds upon which our drug product candidates are based could impact our products. Any safety or efficacy concerns, or recalls, withdrawals or suspensions of sales of our products or other pet therapeutics, or of their human equivalents, could harm our reputation, in particular, or pet therapeutics, generally, and materially, adversely affect our business and prospects or the potential growth of the pet therapeutics industry, regardless of whether such concerns or actions are justified.
Changes in the distribution channels for companion animal products could negatively impact our market share, margins and distribution of our products.
In most markets, pet owners typically purchase their animal health products directly from veterinarians. In recent years, pet owners have increasingly been afforded the option to purchase animal health products from sources other than veterinarians, such as Internet-based retailers, “big-box” retail stores or other over-the-counter distribution channels. Pet owners also could decrease their reliance on, and visits to, veterinarians as they rely more on Internet-based animal health information. Since we intend to market our products through the veterinarian distribution channel, any decrease in visits to veterinarians by pet owners could reduce our market share for such products and materially adversely affect our operating results and financial condition. In addition, pet owners may substitute human health products for animal health products if human health products are deemed to be lower-cost alternatives.
We do not currently carry liability insurance; however, as we continue our development and commercialization activities, future federal and state legislation may result in increased exposure to product liability claims, which could result in substantial losses to us.
We do not currently carry any product liability insurance. Under existing federal and state laws, companion animals are generally considered to be the personal property of their owners and, as such, pet owners’ recovery for product liability claims involving their companion animals may be limited to the replacement value of the animals. Pet owners and their advocates, however, have filed lawsuits from time to time seeking non-economic damages such as pain and suffering and emotional distress for harm to their companion animals based on theories applicable to personal injuries to humans. If new legislation is passed to allow recovery for such non-economic damages, or if precedents are set allowing for such recovery, we could be exposed to increased product liability claims that could result in substantial losses to us if successful. We do not currently have product liability insurance and we may not be able to obtain or maintain this type of insurance in the future.
| -24- |
If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our existing or future product candidates, if approved, or generate product revenue.
We do not currently have a sales organization. We intend to commercialize any product candidate for which we received regulatory approval in the United States with a direct sales force and through third-party distributors. To achieve this, we will be required to build a direct sales organization and to establish relationships with distributors of veterinary products. We also will have to build our marketing, sales, managerial and other non-technical capabilities and make arrangements with third parties for distribution and to perform certain of these other services, and we may not be successful in doing so. Building an internal sales organization is time consuming and expensive and will significantly increase our compensation expense. We may be unable to secure third-party distribution contracts with distributors on favorable terms or at all. We have no prior experience in the marketing, sale and distribution of pharmaceuticals or diagnostic products for companion animals and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain and motivate qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively oversee a geographically dispersed sales and marketing team. If we are unable to build an effective sales organization and/or if we are unable to secure relationships with third-party distributors for our product candidates, we will not be able to successfully commercialize any product for which we receive marketing approval, our future product revenue will suffer and we would incur significant additional losses.
In jurisdictions outside of the United States we intend to utilize companies with an established commercial presence to market our products in those jurisdictions, but we may be unable to enter into such arrangements on acceptable terms, it at all.
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop any of our existing or future product candidates, conduct our in-licensing and development efforts and commercialize any of our existing or future drug candidates.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Gerald Solensky, Jr., our President and Chief Executive Officer, Bruk Herbst, our Chief Commercial Officer, Stephanie Morley, DVM, our Chief Operations Officer and Vice President of Product Development, and Shameze Rampertab, CPA, CA, our Chief Financial Officer. The loss of services of any of these individuals could delay or prevent the successful development of our existing or future product pipeline, completion of our planned development efforts or the commercialization of our product candidates. Although we have entered into employment agreements with Dr. Morley and Mr. Herbst for one year terms (automatically extending for one year terms thereafter) there can be no assurance that either of Dr. Morley or Mr. Herbst will extend their terms of service. We have also entered into employment agreements with Mr. Solensky and Mr. Rampertab, each without a fixed term of service.
Consolidation of our customers could negatively affect the pricing of our products.
Veterinarians will be our primary customers for any approved products. In recent years, there has been a trend towards the consolidation of veterinary clinics and animal hospitals. If this trend continues, these large clinics and hospitals could attempt to leverage their buying power to obtain favorable pricing from us and other companion animal pharmaceutical and diagnostic products companies. Any resulting downward pressure on the prices of any of our approved products could have a material adverse effect on our results of operations and financial condition.
We will need to increase the size of our organization and may not successfully manage our growth.
We will need to significantly expand our organization and systems to support our future expected growth. If we fail to manage our growth effectively, we will not be successful and our business could fail.
Our research and development relies on testing in animals, which is controversial and may become subject to bans or additional regulations.
We must test our product candidates in target animals to obtain marketing approval. Although our animal testing will be subject to GLP and GCP requirements, as applicable, animal testing in the human pharmaceutical industry and in other industries has been the subject of controversy and adverse publicity. Some organizations and individuals have sought to ban animal testing or encourage the adoption of additional regulations applicable to animal testing. To the extent that such bans or regulations are imposed, our research and development activities, and by extension our operating results and financial condition, could be materially adversely affected. In addition, negative publicity about animal practices by us or in our industry could harm our reputation among potential customers for our products.
| -25- |
Because our directors may serve as directors or officers of other companies, they may have a conflict of interest in making decisions for our business.
Our directors may serve as directors or officers of other companies or have significant shareholdings in other veterinary pharmaceutical or diagnostic products companies and, to the extent that such other companies may participate in ventures in which we may participate, our directors may have a conflict of interest in negotiating and concluding terms respecting the extent of such participation. In the event that such a conflict of interest arises at a meeting of our directors, we expect that the director who has such a conflict will declare his conflict, abstain from voting for or against the approval of such participation or such terms and, if deemed necessary or advisable, recuse himself from any discussion concerning the matters in question. In some circumstances, a director may be unable to manage such conflicts and may therefore need to resign. Our directors are required to act honestly, in good faith and in our best interests. In determining whether or not we will participate in a particular business opportunity or enter into a particular business arrangement, we expect that the directors and officers will be guided by their fiduciary duties and take into account such matters as they deem relevant, including considering the degree of risk to which we may be exposed and our financial position at that time.
We may seek to raise additional funds in the future through debt financing which may impose operational restrictions on our business and may result in dilution to existing or future holders of our common shares.
We expect that we will need to raise additional capital in the future to help fund our business operations. Debt financing, if available, may require restrictive covenants, which may limit our operating flexibility and may restrict or prohibit us from:
| • | paying dividends and/or making certain distributions, investments and other restricted payments; |
| • | incurring additional indebtedness or issuing certain preferred shares; |
| • | selling some or all of our assets; |
| • | entering into transactions with affiliates; |
| • | creating certain liens or encumbrances; |
| • | merging, consolidating, selling or otherwise disposing of all or substantially all of our assets; and |
| • | designating our subsidiaries as unrestricted subsidiaries. |
Debt financing may also involve debt instruments that are convertible into or exercisable for our common shares. The conversion of the debt to equity financing may dilute the equity position of our existing shareholders.
We may not be able to obtain or maintain sufficient insurance on commercially reasonable terms or with adequate coverage against potential liabilities in order to protect ourselves against product liability claims.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing and marketing of veterinary therapeutic and diagnostic products. We may become subject to product liability claims resulting from the use of our product candidates. We do not currently have product liability insurance and we may not be able to obtain or maintain this type of insurance for any future trials or product candidates. In addition, product liability insurance is becoming increasingly expensive. Being unable to obtain or maintain product liability insurance in the future on acceptable terms or with adequate coverage against potential liabilities could have a material adverse effect on our business.
| -26- |
We may acquire other businesses or form joint ventures that may be unsuccessful and could adversely dilute your ownership of our company.
As part of our business strategy, we may pursue in-licenses or acquisitions of other complementary assets and businesses and may also pursue strategic alliances. We have no experience in acquiring other assets or businesses and have limited experience in forming such alliances. We may not be able to successfully integrate any acquisitions into our existing business, and we could assume unknown or contingent liabilities or become subject to possible stockholder claims in connection with any related-party or third-party acquisitions or other transactions. We also could experience adverse effects on our reported results of operations from acquisition-related charges, amortization of acquired technology and other intangibles and impairment charges relating to write-offs of goodwill and other intangible assets from time to time following an acquisition. Integration of an acquired company requires management resources that otherwise would be available for ongoing development of our existing business. We may not realize the anticipated benefits of any acquisition, technology license or strategic alliance.
To finance future acquisitions, we may choose to issue shares of our common stock as consideration, which would dilute your ownership interest in us. Alternatively, it may be necessary for us to raise additional funds through public or private financings. Additional funds may not be available on terms that are favorable to us and, in the case of equity financings, may result in dilution to our stockholders.
Risks Related to Government Regulation
Various government regulations could limit or delay our ability to develop and commercialize our products or otherwise negatively impact our business
In the U.S., the manufacture and sale of certain of our diagnostic product candidates are regulated by agencies such as the USDA, the FDA or the EPA. While our diagnostic test for canine cancer does not require approval by the USDA prior to sale in the U.S., our diagnostic test will be subject to post-marketing oversight by the FDA-CVM. In addition, delays in obtaining regulatory approvals for new products or product upgrades could have a negative impact on our growth and profitability.
The manufacture and sale of our products, as well as our research and development processes, are subject to similar and potentially more stringent laws in foreign countries.
We are also subject to a variety of federal, state, local and international laws and regulations that govern, among other things, the importation and exportation of products; our business practices in the U.S. and abroad, such as anti-corruption and anti-competition laws; and immigration and travel restrictions. These legal and regulatory requirements differ among jurisdictions around the world and are rapidly changing and increasingly complex. The costs associated with compliance with these legal and regulatory requirements are significant and likely to increase in the future.
Any failure to comply with applicable legal and regulatory requirements could result in fines, penalties and sanctions; product recalls; suspensions or discontinuations of, or limitations or restrictions on, our ability to design, manufacture, market, import, export or sell our products; and damage to our reputation.
Even if we receive regulatory approval for a product candidate, we will be subject to ongoing FDA-CVM or USDA-CVB obligations and continued regulatory oversight, which may result in significant additional expense. Additionally, any product candidates, if approved, will be subject to labeling and manufacturing requirements and could be subject to other restrictions. Failure to comply with these regulatory requirements or the occurrence of unanticipated problems with our products could result in significant penalties.
If the FDA-CVM or USDA-CVB approves any of our existing or future therapeutic product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, establishment registration, and product listing, as well as continued compliance with GMP, GLP and GCP for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| • | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary product recalls; |
| • | fines, warning letters or holds on target animal studies; |
| • | refusal by the FDA-CVM or USDA-CVB to approve pending applications or supplements to approved applications filed by us or our strategic collaborators, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; and |
| • | injunctions or the imposition of civil or criminal penalties. |
| -27- |
The FDA-CVM’s or USDA-CVB’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business.
Our ability to market our drug candidates in the United States, if approved, will be limited to use for the treatment of the indications for which they are approved, and if we want to expand the indications for which we may market our product candidates, we will need to obtain additional FDA-CVM approvals, which may not be granted.
We expect to seek FDA-CVM approval in the United States for our lead product candidates, ZM-012, an anti-infective in pill form that is intended for use in canines, ZM-007, an anti-infective oral suspension for use as an anti-diarrheal in smaller dogs, ZM-006, a transdermal gel treatment for the metabolic disorder hyperthyroidism intended for use in cats, and ZM-011, a transdermal gel formulation treatment for behavioral disorders intended to use in cats.. If these drug product candidates are approved, the FDA-CVM will restrict our ability to market or advertise them for the treatment of indications other than the indications for which they are approved, which could limit their adoption by veterinarian and pet owners. We may attempt to develop, promote and commercialize new treatment indications and protocols for our drug product candidates in the future, but we cannot predict when or if we will receive the approvals required to do so. In addition, we would be required to conduct additional target animal studies to support our applications, which would utilize additional resources and may produce results that do not result in FDA-CVM approvals. If we do not obtain additional FDA-CVM approvals, our ability to expand our business in the United States will be limited.
If approved, any of our existing or future therapeutic products may cause or contribute to adverse medical events that we are required to report to regulatory authorities and, if we fail to do so, we could be subject to sanctions that would materially harm our business.
If we are successful in commercializing any of our existing or future therapeutic product candidates, we will be required to report adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the regulatory authorities could take action including criminal prosecution, seizure of our products or delay in approval or clearance of future products.
| -28- |
Legislative or regulatory reforms with respect to veterinary pharmaceuticals or health care solutions may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our existing or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, FDA-CVM and USDA-CVB regulations and guidance are often revised or reinterpreted by the FDA-CVM and USDA-CVB in ways that may significantly affect our business and our products. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States may impose additional costs or lengthen review times of any of our existing or future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
| • | changes to manufacturing methods; |
| • | recall, replacement or discontinuance of certain products; and |
| • | additional record-keeping. |
Each of these would likely entail substantial time and cost and could materially harm our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
Risks Related to Intellectual Property
Our ability to obtain intellectual property protection for our product candidates is uncertain.
Insofar as our business strategy is to develop APIs already approved for use in humans for veterinary use, our ability to obtain a proprietary intellectual property position for our product candidates is uncertain. We do not have any issued patents for our lead product candidates. We have not filed patent applications for any of our other product candidates to date. Our current and future patent applications may never result in the issuance of patents, and/or patents issued to us may be dominated by the patents of third parties, including for example, patents issued to analogous human drugs or biological compositions and their usages. Furthermore, even if any future patents are unchallenged by third parties, our patents, if issued, may not adequately protect our intellectual property or prevent others from designing around them. It is possible that we will not receive patents to cover any future approved products, and/ or that we will have little to no commercial protection against competing products. In such cases, we would then have to rely solely on other forms of exclusivity, such as regulatory exclusivity provided by the FDA-CVM approval, which may provide less protection to our competitive position.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of any future patent applications and the enforcement or defense of any patents that issue. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a “first-to-invent” system to a “first-to-file” system. Under a “first-to-file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that issue, all of which could have a material adverse effect on our business and financial condition.
Some of our products may or may not be covered by a patent. Further if an application is filed, it is not certain that a patent will be granted or if granted whether it will be held to be valid. All of which may impact our market share and ability to prevent others (competitor third parties) from making, selling, or using our products.
We intend to rely upon a combination of regulatory exclusivity periods, patents, trade secret protection, confidentiality agreements, and license agreements to protect the intellectual property related to our current product candidates and our development programs. We may not be successful in protecting our intellectual property rights, including our unpatented proprietary know-how and trade secrets, or in avoiding claims that we infringed on the intellectual property rights of others. In addition to relying on patent and trademark rights, we rely on unpatented proprietary know-how and trade secrets, and employ various methods, including confidentiality agreements with employees and consultants, customers and suppliers to protect our know-how and trade secrets. However, these methods and our patents and trademarks may not afford complete protection and there can be no assurance that others will not independently develop the know-how and trade secrets or develop better production methods than us. Further, we may not be able to deter current and former employees, contractors and other parties from breaching confidentiality agreements and misappropriating proprietary information and it is possible that third parties may copy or otherwise obtain and use our information and proprietary technology without authorization or otherwise infringe on our intellectual property rights. In the future, we may also rely on litigation to enforce our intellectual property rights and contractual rights, and, if not successful, we may not be able to protect the value of our intellectual property. Any litigation could be protracted and costly and could have a material adverse effect on our business and results of operations regardless of its outcome.
| -29- |
We have pending trademark applications in Canada and the European Union however trademark registration is not yet complete, and failure to finally secure these registrations could adversely affect our business.
We have pending trademark applications for our company name and design marks and our “Voice of the Vet” program in Canada and the European Union. We cannot make assurances that these trademarks will become registered in any pending jurisdiction. We may face rejections to one or more of our pending trademark applications. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. Additionally, we may need to enforce our trademark rights against third parties and expend significant additional resources to enforce such rights against infringements. Moreover, any name we propose to use with our product candidates in the United States must be approved by the FDA-CVM or the USDA-CVB regardless of whether we have registered it, or applied to register it, as a trademark. The FDA-CVM typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA-CVM or the USDA-CVB object to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA-CVM and the USDA-CVB.
Third parties may have intellectual property rights, which may require us to obtain a license or other applicable rights to make, sell or use our products. If such rights are not granted or obtained, I could have a material adverse effect on our business, financial condition and results of operations.
Our success depends in part on our ability to obtain, or license from third parties, patents, trademarks, trade secrets and similar proprietary rights without infringing on the proprietary rights of third parties. Although we believe our intellectual property rights are sufficient to allow us to conduct our business without incurring liability to third parties, our products may infringe on the intellectual property rights of such persons. Furthermore, no assurance can be given that we will not be subject to claims asserting the infringement of the intellectual property rights of third parties seeking damages, the payment of royalties or licensing fees and/or injunctions against the sale of our products. Any such litigation could be protracted and costly and could have a material adverse effect on our business, financial condition and results of operations.
Our diagnostic assay technology depends on certain technology that is licensed to us. We do not control this technology and any loss of our rights to them could prevent us from marketing our diagnostic assay product candidates.
Our diagnostic assay technology is dependent on a license from Celsee. We do not own the intellectual property rights that underlie this license. Our rights to use the technology we license are subject to the negotiation of, continuation of and compliance with the terms of our license. We do not control the prosecution, maintenance, or filing of the patents and other intellectual property licensed to us, or the enforcement of these intellectual property rights against third parties. The patents and patent applications underlying our license were not written by us or our attorneys, and we do not have control over the drafting and prosecution of such rights. Celsee might not have given the same attention to the drafting and prosecution of patents and patent applications as we would have if we had been the owners of the intellectual property rights and had control over such drafting and prosecution. We cannot be certain that drafting and/or prosecution of the licensed patents and patent applications has been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents and other intellectual property rights.
| -30- |
Our intellectual property agreements with third parties may be subject to disagreements over contract interpretation, which could narrow the scope of our rights to the relevant intellectual property or technology or increase our financial or other obligations to our licensors.
Certain provisions in our intellectual property agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could affect the scope of our rights to the relevant intellectual property or technology, or affect financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact conceives or develops intellectual property that we regard as our own. Our assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other pharmaceutical or animal health companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise improperly used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against any such claims. Even if we are successful in defending against any such claims, such litigation could result in substantial cost and be a distraction to our management and employees.
Risks Related to Our Common Shares
We believe that we will be a “passive foreign investment company,” or PFIC for the current taxable year, which could subject certain U.S. shareholders to materially adverse U.S. federal income tax consequences.
We believe we were classified as a PFIC during our taxable year ended 2015, and based on current business plans and financial expectations, we believe we may be a PFIC for the current and future taxable years. If we are a PFIC for any year in which you hold shares and you are a U.S. Holder (as defined below, in “Material United States Federal Income Tax Considerations”), unless you make a timely and effective Qualified Electing Fund election, or QEF Election or a mark-to-market election, or Mark-to-Market Election with respect to our common shares, you will not be eligible for the reduced tax rates associated with “qualified dividend income” with respect to distributions made to you or long-term capital gain upon a disposition of your common shares. Instead, all such distributions and gain will be taxable to you at the higher rates for ordinary income. In addition, a portion of any gain and distribution may be allocated to prior years during which you have owned our common shares and subjected to tax at the highest tax rate applicable to ordinary income in each such year. You would also be required to pay an interest charge on that portion of such gain or distribution.
If you are a U.S. Holder and make a timely and effective QEF Election, you generally must report on a current basis your share of our net capital gain and ordinary earnings for any year in which we are a PFIC, whether or not we distribute any amount to you, thus giving rise to so-called “phantom income” and to a potential tax liability.
| -31- |
If you are a U.S. Holder and make a timely and effective Mark-to-Market Election, you generally must include as ordinary income each year the excess of the fair market value of your common shares over your tax basis therein, thus also possibly giving rise to phantom income and a potential tax liability. Ordinary loss generally is recognized only to the extent of net mark-to-market gains previously included in income.
Each U.S. shareholder should consult its own tax advisors regarding the PFIC rules and the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares.
If the Internal Revenue Service determines that we are not a PFIC and you previously paid taxes pursuant to a QEF Election or a Mark-to-Market Election, you may pay more taxes than you legally owe.
If the Internal Revenue Service, or the IRS, makes a determination that we are not a PFIC and you previously paid taxes pursuant to a QEF Election or Mark-to-Market Election, then you may have paid more taxes than you legally owed due to such election. If you do not, or are unable to, file a refund claim before the expiration of the applicable statute of limitations, you will not be able to claim a refund for those taxes.
If securities or industry analysts do not publish research or reports about our company, or if they issue adverse or misleading opinions regarding us or our stock, our stock price and trading volume could decline.
Although we have research coverage by securities and industry analysts, if coverage is not maintained, the market price for our stock may be adversely affected. Our stock price also may decline if any analyst who covers us issues an adverse or erroneous opinion regarding us, our business model, our intellectual property or our stock performance, or if our target animal studies and operating results fail to meet analysts’ expectations. If one or more analysts cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline and possibly adversely affect our ability to engage in future financings.
We expect that the price of our common shares will fluctuate substantially.
You should consider an investment in our common shares risky and invest only if you can withstand a significant loss and wide fluctuations in the market value of your investment. The price of our common shares that will prevail in the market after the sale of our common shares by a selling shareholder may be higher or lower than the price you have paid. Numerous factors, including many over which we have no control, may have a significant impact on the market price of our common shares. These risks include those described or referred to in this “Risk Factors” section and elsewhere in this report as well as, among other things:
| • | any delays in, or suspension or failure of, our existing and future studies; |
| • | announcements of regulatory approval or disapproval of any of our existing or future product candidates or of regulatory actions affecting us or our industry; |
| • | delays in the commercialization of our existing or future product candidates; |
| • | manufacturing and supply issues related to our development programs and commercialization of our existing or future product candidates; |
| • | quarterly variations in our results of operations or those of our competitors; |
| • | changes in our earnings estimates or recommendations by securities analysts or adverse publicity about us or our product candidates; |
| • | announcements by us or our competitors of new product candidates, significant contracts, commercial relationships, acquisitions or capital commitments; |
| • | announcements relating to future development or license agreements including termination of such agreements; |
| • | adverse developments with respect to our intellectual property rights or those of our principal collaborators; |
| -32- |
| • | commencement of litigation involving us or our competitors; |
| • | any major changes in our board of directors or management; |
| • | new legislation in the United States relating to the prescription, sale, distribution or pricing of pet pharmaceuticals or diagnostic products; |
| • | product liability claims, other litigation or public concern about the safety of our product candidates or future products; |
| • | market conditions in the animal health industry, in general, or in the pet therapeutics sector, in particular, including performance of our competitors; and |
| • | general economic conditions in the United States and abroad. |
In addition, the stock market, in general, or the market for stocks in our industry, in particular, may experience broad market fluctuations, which may adversely affect the market price or liquidity of our common shares. Any sudden decline in the market price of our common shares could trigger securities class-action lawsuits against us. If any of our shareholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the time and attention of our management would be diverted from our business and operations. We also could be subject to damages claims if we are found to be at fault in connection with a decline in our stock price.
Our management owns a significant percentage of our common shares and will be able to exert significant control over matters subject to shareholder approval.
Based on shares outstanding as of February 27, 2018, our executive officers and directors and their respective affiliates beneficially own 56,733,040 or 59.2% of our voting shares. These shareholders will have the ability to influence us through this ownership position and may be able to determine all matters requiring shareholder approval. For example, these shareholders may be able to control elections of directors, amendments of our organizational documents, or approvals of any merger, sale of assets or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common shares that you may feel are in your best interest as one of our shareholders.
We are an “emerging growth company,” as defined under the JOBS Act and if we take advantage of reduced disclosure requirements applicable to “emerging growth companies,” our common shares could be less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, as amended, or the JOBS Act, and, for as long as we continue to be an “emerging growth company,” we may choose to take advantage of certain exemptions from various reporting requirements applicable to other public companies but not to “emerging growth companies,” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002, as amended, or SOX, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an “emerging growth company” for up to five years, or until the earliest of (i) the last day of the first fiscal year in which our annual gross revenues exceed $1.07 billion, (ii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common shares that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, or (iii) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three year period. We cannot predict if investors will find our common shares less attractive if we choose to continue to rely on these exemptions. If some investors find our common shares less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our common shares and our stock price may be more volatile.
In addition, Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. An “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have chosen to “opt out” of such extended transition period, however, and, as a result, we are required to comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
| -33- |
Our Articles of Incorporation (as amended and restated) authorize us to issue an unlimited number of common shares and preferred shares without shareholder approval and we may issue additional equity securities, or engage in other transactions that could dilute your ownership interest, which may adversely affect the market price of our common shares
Our Articles of Incorporation (as amended or restated) authorize our Board of Directors, subject to the provisions of the ABCA, to issue an unlimited number of common shares and preferred shares without shareholder approval. Our Board of Directors may determine from time to time to raise additional capital by issuing common shares, preferred shares or other equity securities. We are not restricted from issuing additional securities, including securities that are convertible into or exchangeable for, or that represent the right to receive, common shares or preferred shares. Because our decision to issue securities in any future offering will depend on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing, or nature of any future offerings, or the prices at which such offerings may be affected. Additional equity offerings may dilute the holdings of our existing shareholders or reduce the market price of our common shares, or both. Holders of our common shares are not entitled to pre-emptive rights or other protections against dilution. New investors also may have rights, preferences and privileges that are senior to, and that adversely affect, the then-current holders of our common shares. Additionally, if we raise additional capital by making offerings of debt or preference shares, upon our liquidation, holders of our debt securities and preferred shares, and lenders with respect to other borrowings, may receive distributions of our available assets before the holders of our common shares.
We will incur significant costs as a result of operating as a U.S. public company, and our management will devote substantial time to new compliance initiatives.
As a Canadian public company, we were not required to comply with certain U.S. corporate governance and financial reporting practices and policies required of a U.S. publicly-traded company. As a U.S. publicly-traded company, we will incur significant legal, accounting and other expenses that we were not required to incur in the recent past, particularly after we are no longer an “emerging growth company” as defined under the JOBS Act. In addition, new and changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act and the rules and regulations promulgated and to be promulgated thereunder, as well as under the Sarbanes-Oxley Act, the JOBS Act, and the rules and regulations of the U.S. Securities and Exchange Commission, or SEC, have created uncertainty for U.S. public companies and increased our costs and time that our board of directors and management must devote to complying with these rules and regulations. We expect these rules and regulations to increase our legal and financial compliance costs and lead to a diversion of management time and attention from revenue generating activities.
Furthermore, the need to establish the corporate infrastructure demanded of a U.S. public company may divert management’s attention from implementing our growth strategy, which could prevent us from improving our business, results of operations and financial condition. We have made, and will continue to make, changes to our internal controls and procedures for financial reporting and accounting systems to meet our reporting obligations as a U.S. public company. However, the measures we take may not be sufficient to satisfy our obligations as a U.S. public company.
For as long as we remain an “emerging growth company” as defined in the JOBS Act, we may choose to take advantage of certain exemptions from various reporting requirements that are applicable to other U.S. public companies that are not “emerging growth companies.” These exceptions provide for, but are not limited to, relief from the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, less extensive disclosure obligations regarding executive compensation in our periodic reports and proxy statements, exemptions from the requirements to hold a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved and an extended transition period for complying with new or revised accounting standards. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We may remain an “emerging growth company” for up to five years. See “JOBS Act” in this report. To the extent we are no longer eligible to use exemptions from various reporting requirements under the JOBS Act, we may be unable to realize our anticipated cost savings from those exemptions.
| -34- |
Our internal control over financial reporting does not meet the standards required by Section 404 of the Sarbanes-Oxley Act, and failure to achieve and maintain effective internal control over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act could have a material adverse effect on our business and share price.
As a Canadian public company, we were not required to evaluate our internal control over financial reporting in a manner that meets the standards of U.S. public companies required by Section 404 of the Sarbanes-Oxley Act, or Section 404. We were required to meet these standards in the course of preparing our financial statements as of and for the year ended December 31, 2017, and our management has reported on the effectiveness of our internal control over financial reporting for such year. Additionally, under the recently enacted JOBS Act, our independent registered public accounting firm is not required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act until we are no longer an “emerging growth company.” The rules governing the standards that must be met for our management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation.
In connection with the implementation of the necessary procedures and practices related to internal control over financial reporting, we may identify deficiencies that we may not be able to remediate in time to meet the deadline imposed by the Sarbanes-Oxley Act for compliance with the requirements of Section 404. In addition, we may encounter problems or delays in completing the implementation of any requested improvements and receiving a favorable attestation in connection with the attestation provided by our independent registered public accounting firm. We will be unable to issue securities in the public markets through the use of a shelf registration statement if we are not in compliance with Section 404. Furthermore, failure to achieve and maintain an effective internal control environment could have a material adverse effect on our business and share price and could limit our ability to report our financial results accurately and timely.
If we sell common shares in future financings, shareholders may experience immediate dilution and, as a result, our share price may decline.
We may from time to time issue additional common shares at a discount from the existing trading price of our common shares. As a result, our shareholders would experience immediate dilution upon the sale of any shares of our common shares at such discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred shares or common shares. If we issue common shares or securities convertible into common shares, our common shareholders would experience additional dilution and, as a result, our share price may decline.
We have never and do not, in the future, intend to pay dividends on our common shares, and your ability to achieve a return on your investment will depend on appreciation in the market price of our common shares.
We have never paid and do not expect to pay dividends on our common shares in the future. We intend to invest our future earnings, if any, to fund our growth and not to pay any cash dividends on our common shares. Since we do not intend to pay dividends, your ability to receive a return on your investment will depend on any future appreciation in the market price of our common shares. There is no assurance that our common shares will appreciate in price.
An active, liquid and orderly market for our common shares may not develop or be sustained, and you may not be able to sell your common shares.
Our common shares trade on the TSX-V and NYSE American exchanges. We cannot assure you that an active trading market for our common shares will develop or be sustained. The lack of an active market may impair your ability to sell the common shares at the time you wish to sell them or at a price that you consider reasonable. An inactive market may also impair our ability to raise capital by selling common shares and may impair our ability to acquire other businesses, applications or technologies using our common shares as consideration, which, in turn, could materially adversely affect our business.
| -35- |
We can provide no assurance that our common shares will continue to meet NYSE American listing requirements. If we fail to comply with the continuing listing standards of the NYSE American, our common shares could be delisted.
If we fail to satisfy the continued listing requirements of the NYSE American, such as the corporate governance requirements or the minimum closing bid price requirement, the NYSE American may take steps to delist our common shares. Such a delisting would likely have a negative effect on the price of our common shares and would impair your ability to sell or purchase common shares when you wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow our common shares to become listed again, stabilize the market price or improve the liquidity of our common shares, prevent our common shares from dropping below the NYSE American minimum bid price requirement or prevent future non-compliance with NYSE American’s listing requirements.
| Item 1B. | Unresolved Staff Comments. |
Not applicable.
| Item 2. | Properties. |
Our corporate headquarters are located in Ann Arbor, Michigan where we lease and occupy approximately 7,900 square feet pursuant to a lease that expires February 2022. We lease approximately 4,800 square feet of research and development and office space in another facility in Ann Arbor, Michigan, pursuant to a lease that expires in August 2018. We have the option to extend that lease three additional years.
| Item 3. | Legal Proceedings. |
We are not currently a party to any material proceedings.
| Item 4. | Mine Safety Disclosures. |
Not applicable.
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market Information
Our common shares commenced trading on the NYSE American on November 21, 2017 under the symbol “ZOM”. The following table sets forth the high and low sale prices for our common shares for the periods indicated as reported on the NYSE American:
| Fiscal Year 2017: | High | Low | ||||||
| Fourth Quarter (from November 21, 2017) | $ | 2.47 | $ | 1.88 | ||||
Common Stock Information
As of February 27, 2018, there were 90,449,869 common shares outstanding held of record by approximately 200 holders.
| -36- |
Dividends
We have never declared or paid any cash dividends on our common shares. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future. Any future determination related to dividend policy will be made at the discretion of our board of directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the board of directors deems relevant, and subject to the restrictions contained in any future credit facilities or other financing arrangements.
Equity Compensation Plan Information
| Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights |
Weighted-average exercise price of outstanding options, warrants and rights |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
| (a) | (b) | (c) | |
| Equity compensation plans approved by security holders |
8,080,000 |
$0.96 |
942,586 |
| Equity compensation plans not approved by security holders |
Nil |
N/A |
Nil |
| Total | 8,080,000 | $0.96 | 942,586 |
| Item 6. | Selected Financial Data. |
Not applicable.
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATION
Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to help the reader understand the results of operations and financial condition of the Company. The Management’s Discussion and Analysis of Financial Condition and Results of Operations should be read in conjunction with our audited consolidated financial statements and notes thereto for the year ended December 31, 2017. In addition to historical information, this Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which are intended to be covered by the safe harbors created thereby. See “Cautionary Note Regarding Forward-Looking Statements” in this Annual Report on Form 10-K. Our actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under the “Part I – Item 1A Risk Factors” section and elsewhere in this Annual Report on Form 10-K, as well as, in other reports and documents we file with the Securities and Exchange Commission from time to time. Except as required by law, we undertake no obligation to update any forward-looking statements to reflect events or circumstances occurring after the date of this Annual Report on Form 10-K.
Overview
We are a development stage veterinary diagnostic and pharmaceutical company creating products for companion animals (canine, feline, and equine) by focusing on the unmet needs of clinical veterinarians. We believe that we have identified and are developing diagnostics and therapeutics that have the potential to significantly improve the diagnosis and treatment of various diseases affecting companion animals. We believe that there are significant unmet medical needs for pets, and that the pet diagnostic and therapeutic segments of the animal health industry are likely to grow substantially as new diagnostic tools and treatments are identified, developed, and marketed specifically for companion animals.
| -37- |
Together with our strategic partner, we are developing liquid biopsy assays and related consumables for the detection of cancer in companion animals. The regulatory pathway to obtain pre-market regulatory approval of companion animal diagnostics is significantly shorter than for similar diagnostic products intended for human use. In certain cases, pre-market regulatory approval may be unnecessary, depending on the intended use of the diagnostic.
We also have identified a number of drugs that have proven safe and effective in humans that we are developing for use in companion animals. We believe this development approach enables us to reduce the risks associated with obtaining regulatory approval for unproven product candidates and shortens the development timeline necessary to bring our product candidates to market. We have four drug product candidates in early development and have identified several other potential product candidates for further investigation.
In addition, we are investigating the development of alternative drug delivery technologies for our drug product candidates. Many of the human-approved therapeutics used in companion animals are only available in pill or injectable form. However, it can be difficult to give a companion animal an injection or to assure that the animal has swallowed a pill. As a result, we believe that compliance with treatment regimens is a significant problem for veterinarians and pet owners. The challenges associated with medicating pets are unique, and we believe that developing product candidates that can be easily taken by the pet or easily administered by pet owners will help increase compliance.
We are a development-stage company with no products approved for marketing and sale, and we have not generated any revenue. We have incurred significant net losses since our inception. We incurred net losses of $8,065,072 and $5,740,492 for the year ended December 31, 2017 and December 31, 2016, respectively. These losses have resulted principally from costs incurred in connection with investigating and developing our product candidates, research and development activities and general and administrative costs associated with our operations. As of December 31, 2017, we had an accumulated deficit of $15,626,100 and cash and cash equivalents of $3,448,147.
| -38- |
For the foreseeable future, we expect to continue to incur losses, which will increase significantly from historical levels as we expand our product development activities, commercialize them if they do not require U.S. Food and Drug Administration’s Center for Veterinary Medicine, or FDA-CVM, pre-market approval, seek regulatory approvals for our product candidates where required from the FDA-CVM or the United States Department of Agriculture Center for Veterinary Biologics, or the USDA-CVB.
For further information on the regulatory, business and product pipeline, please see the “Business” section of this Annual Report on Form 10-K. For further information on the risk factors, please see the “Risk Factors” section of this Annual Report on Form 10-K.
Revenue
We do not have any products approved for sale, have not generated any revenue from product sales since our inception and do not expect to generate any revenue from the sale of products in the near future. If our development efforts result in clinical success and regulatory approval or collaboration agreements with third parties for any of our product candidates, we may generate revenue from those product candidates.
Operating Expenses
The majority of our operating expenses to date have been for the general and administrative activities related to general business activities, capital market activities and stock-based compensation, and research and development activities related to our lead product candidates.
Research and Development Expense
All costs of research and development are expensed in the period in which they are incurred. Research and development costs primarily consist of salaries and related expenses for personnel, stock-based compensation expense, fees paid to consultants, outside service providers, professional services, travel costs and materials used in clinical trials and research and development.
We have a non-invasive diagnostic assay or blood test, ZM-017, that we are developing as an aid for veterinarians in diagnosing cancer in canines.
We have four drug product candidates in development. Our lead drug product candidate is ZM-012, a novel tablet formulation of metronidazole targeting the treatment of acute diarrhea in dogs. Our second drug product candidate is ZM-007, an oral suspension formulation of metronidazole and a complementary formulation to ZM-012, targeting the treatment of acute diarrhea in small breeds and puppies under nine pounds or four kilograms. Our third drug product candidate is ZM-006, a transdermal gel formulation of methimazole targeting hyperthyroidism in cats. Our fourth drug product candidate is ZM-011, a transdermal gel formulation of fluoxetine, most commonly known as Prozac®, its human pharmaceutical brand name.
We are also investigating the development of alternative drug delivery systems for our drug product candidates. We typically use our employee and infrastructure resources across multiple development programs. We track outsourced development costs by product candidate, but do not allocate personnel or other internal costs related to development to specific programs or product candidates.
General and Administrative Expense
General and administrative expense consists primarily of personnel costs, including salaries, related benefits and stock-based compensation for employees, consultants and directors. General and administrative expenses also include rent and other facilities costs and professional and consulting fees for legal, accounting, tax services and other general business services.
| -39- |
Professional Fees
Professional fees include attorney’s fees, accounting fees and consulting fees incurred in connection with product investigation and analysis, regulatory analysis, government relations, audit, securities offerings, investor relations, and general corporate and intellectual property advice.
Income Taxes
As of December 31, 2017, we had net operating loss carryforwards for federal and state income tax purposes of $5,008,180 and non-capital loss carryforwards for Canada of approximately $6,526,850 respectively, which will begin to expire in fiscal year 2035. We have evaluated the factors bearing upon the realizability of our deferred tax assets, which are comprised principally of net operating loss carryforwards and non-capital loss carryforwards. We concluded that, due to the uncertainty of realizing any tax benefits as of December 31, 2017, a valuation allowance was necessary to fully offset our deferred tax assets.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or U.S. GAAP. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, and revenue, costs and expenses and related disclosures during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those described below. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 3 of the notes to our financial statements appearing elsewhere in this document, we believe that the estimates and assumptions involved in the following accounting policies may have the greatest potential impact on our financial statements.
JOBS Act
The Jumpstart Our Business Startups Act, or the JOBS Act, contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company.” We have irrevocably elected not to avail ourselves of the JOBS Act provision that an emerging growth company may delay adopting new or revised accounting standards until such times as those standards apply to private companies.
In addition, we are in the process of evaluating the benefits of relying on the other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, if as an “emerging growth company” we choose to rely on such exemptions, we may not be required to, among other things, (i) provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404, and (ii) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis). These exemptions will apply until December 31, 2022 or until we no longer meet the requirements of being an “emerging growth company,” whichever is earlier.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the year. Actual results could differ from those estimates.
| -40- |
Areas where significant judgment is involved in making estimates are: the determination of the functional currency; the fair values of financial assets and liabilities; the determination of fair value of stock-based compensation; and forecasting future cash flows for assessing the going concern assumption.
Research and Development Costs
Research and development expenses comprise costs incurred in performing research and development activities, including salaries and benefits, safety and efficacy studies and contract manufacturing costs, contract research costs, patent procurement costs, materials and supplies and occupancy costs. Research and development activities include internal and external activities associated with research and development studies of current product candidates and advancing product candidates towards a goal of obtaining regulatory approval to manufacture and market the product candidate.
Research and development costs related to continued research and development programs are expensed as incurred in accordance with ASC topic 730.
Translation of Foreign Currencies
The functional currency, as determined by management, is U.S. dollars, which is also our reporting currency. Transactions denominated in currencies other than U.S. dollars and the monetary value of assets and liabilities are translated at the period end exchange rates. Revenue and expenses are translated at rates of exchange prevailing on the transaction dates. All of the exchange gains or losses resulting from these other transactions are recognized in the consolidated statements of operations and comprehensive loss.
Stock-Based Compensation
We measure the cost of equity-settled transactions by reference to the fair value of the equity instruments at the date at which they are granted if the fair value of the goods or services received by us cannot be reliably estimated.
We calculate stock-based compensation using the fair value method, under which the fair value of the options at the grant date is calculated using the Black-Scholes Option Pricing Model, and subsequently expensed over the vesting period of the option. The provisions of our stock-based compensation plans do not require us to settle any options by transferring cash or other assets, and therefore we classify the awards as equity. Stock-based compensation expense recognized during the period is based on the value of stock-based payment awards that are ultimately expected to vest. We estimate forfeitures at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Volatility is determined based on volatilities of comparable companies when the Company does not have its own trading history. The expected term, which represents the period of time that options granted are expected to be outstanding, is estimated based on an average of the term of the options. The risk-free rate assumed in valuing the options is based on the Canadian treasury yield curve in effect at the time of grant for the expected term of the option. The expected dividend yield percentage at the date of grant is Nil as we are not expected to pay dividends in the foreseeable future.
Loss Per Share
Basic loss per share, or EPS, is computed by dividing the loss attributable to common shareholders by the weighted average number of common shares outstanding. Diluted EPS reflects the potential dilution that could occur from common shares issuable through the exercise or conversion of stock options, restricted stock awards, warrants and convertible securities. In certain circumstances, the conversion of options, warrants and convertible securities are excluded from diluted EPS if the effect of such inclusion would be anti-dilutive.
The dilutive effect of stock options is determined using the treasury stock method. Stock options and warrants to purchase our common shares issued during the period were not included in the computation of diluted EPS, as the effect would be anti-dilutive.
Comprehensive Loss
We follow ASC topic 220. This statement establishes standards for reporting and display of comprehensive (loss) income and its components. Comprehensive loss is net loss plus certain items that are recorded directly to shareholders' equity. We currently have no other comprehensive loss items.
| -41- |
Results of Operations
Year ended December 31, 2017 compared to year ended December 31, 2016
Our results of operations for the year ended December 31, 2017 and December 31, 2016 are as follows:
| Year ended | Year Ended | |||||||||||||||
| December 31, 2017 | December 31, 2016 | Change | ||||||||||||||
| $ | $ | $ | % | |||||||||||||
| Expenses | ||||||||||||||||
| Research and development | 2,751,326 | 1,518,589 | 1,232,737 | 81 | % | |||||||||||
| General and administrative | 3,946,270 | 2,916,604 | 1,029,666 | 35 | % | |||||||||||
| Professional fees | 1,294,044 | 1,245,182 | 48,862 | 4 | % | |||||||||||
| Amortization | 2,797 | 2,690 | 107 | 4 | % | |||||||||||
| Depreciation | 89,613 | 43,131 | 46,482 | 108 | % | |||||||||||
| Loss from operations | 8,084,050 | 5,726,196 | 2,357,854 | 41 | % | |||||||||||
| Gain on settlement of liabilities | (5,000 | ) | - | (5,000 | ) | N/A | ||||||||||
| Foreign exchange loss (gain) | (13,978 | ) | 14,296 | (28,274 | ) | -198 | % | |||||||||
| Loss before income taxes | 8,065,072 | 5,740,492 | 2,324,580 | 40 | % | |||||||||||
| Income tax expense | - | - | - | N/A | ||||||||||||
| Net loss and comprehensive loss | 8,065,072 | 5,740,492 | 2,324,580 | 40 | % | |||||||||||
Revenue
We did not generate any revenue during the years ended December 31, 2017 and December 31, 2016.
Research and Development
Research and development expense for the year ended December 31, 2017 was $2,751,326 compared to $1,518,589 for the year ended December 31, 2016, an increase of $1,232,737 or 81%. The increase was primarily due to the licensing fees paid upon entering into a license and supply agreement with Celsee Inc. as part of our development of ZM-017, ramping up of R&D activities related to the establishment of labs, the hiring of additional full-time employees, new product candidate development and contracted outsourcing activities. More specifically, contracted outsourced activities of $821,927, salaries of $620,694, licensing fees of $480,131, consultant fees of $325,388, and supplies of $239,292 relating to an increased level of contracted outsourced activities, lab activities, including in vitro and in vivo work, to support the further development of our product candidates ZM-017, ZM-012, ZM-006, ZM-007 and ZM-011. We expect that our R&D expenditures in 2018 will be significantly higher than in 2017, due to the initiation of pilot and pivotal studies related to our four INADs, work related to verification and validation of ZM-017, and additional veterinary pharmaceutical candidates, diagnostic developments and technologies.
General and Administrative
General and administrative expense for the year ended December 31, 2017 was $3,946,270, compared to $2,916,604 for the year ended December 31, 2016, an increase of $1,029,666 or 35%. The increase was primarily due to expenses related to the addition of personnel, accounting for salaries of $2,703,865, which included share-based compensation expense of $849,679, primarily as a result of the granting of options to purchase an aggregate of 535,000 common shares in February 2017, all of which vested immediately upon the date of grant, and the granting of options to purchase an aggregate of 1,280,000 common shares in August 2017, of which 1,242,500 have vested. Other expenses included travel and accommodation of $338,738, office expenses of $199,843, insurance costs of $182,753, marketing and investor relations costs of $168,623, rent of $164,250, and regulatory expense of $138,289. We expect that general and administrative expense will increase in 2018 and future periods as we increase our level of activity.
| -42- |
Professional Fees
Professional fees for the year ended December 31, 2017 were $1,294,044 compared to $1,245,182 for the year ended December 31, 2016, an increase of $48,862 or 4%. The increase was primarily due to expenses in connection with the preparation of our initial U.S. registration statement and work on our application to list our common shares on the NYSE American. Professional fees for the 2016 period consisted primarily of consulting fees incurred in connection with establishing our initial operations and preparing to execute our business plan, as well as legal fees incurred in connection with the Qualifying Transaction and our initial fundraising efforts.
Net Loss
Our net loss for the year ended December 31, 2017 was $8,065,072, or $0.09 per share, compared with a net loss of $5,740,492, or $0.07 per share, for the year ended December 31, 2016, an increase of $2,324,580 or 40%. The net loss in each period was attributed to the matters described above. We expect to continue to record net losses in future periods until such time as have sufficient revenue from our product candidates to offset our operating expenses.
Cash Flows
Year ended December 31, 2017 compared to year ended December 31, 2016
The following table shows a summary of our cash flows for the periods set forth below:
| Year ended | Year ended | |||||||||||||||
| December 31, 2017 | December 31, 2016 | Change | ||||||||||||||
| $ | $ | $ | % | |||||||||||||
| Cash flows used in operating activities | (7,093,017 | ) | (4,562,168 | ) | (2,530,849 | ) | 55 | % | ||||||||
| Cash flows provided by financing activities | 7,486,220 | 4,786,353 | 2,699,867 | 56 | % | |||||||||||
| Cash flows used in investing activities | (171,736 | ) | (241,215 | ) | 69,479 | -29 | % | |||||||||
| Increase (decrease) in cash | 221,467 | (17,030 | ) | 238,497 | -1400 | % | ||||||||||
| Cash and cash equivalents, beginning of year | 3,226,680 | 3,243,710 | (17,030 | ) | -1 | % | ||||||||||
| Cash and cash equivalents, end of year | 3,448,147 | 3,226,680 | 221,467 | 7 | % | |||||||||||
Operating Activities
Net cash used in operating activities for the year ended December 31, 2017 was $7,093,017, compared to $4,562,168 for the year ended December 31, 2016, an increase of $2,530,849, or 55%. The increase resulted primarily from our net loss of $8,065,072 for the year ended December 31, 2017, compared to our net loss of $5,740,492 for the year ended December 31, 2016. The largest uses of cash stemmed from an increase in salaries, bonus and benefits as we had 20 employees at December 31, 2017 compared to 11 employees at December 31, 2016. Other significant increases in uses of cash include a licensing fee payment of $500,000 to Celsee, Inc., professional fees and consulting expenses related to the preparation of our initial U.S. registration statement, work on our application to list our common shares on the NYSE American, and an increase in the current portion of the prepaid expenses and deposits. The increase in prepaid expenses and deposits was due to increased deposits with our contract manufacturing organization and increased prepaid health insurance premiums from an overall increase in the number of employees.
Net cash used in operating activities for the year ended December 31, 2016 was $4,562,168, which resulted primarily from our net loss of $5,740,492. The largest uses of cash were for employee salaries, bonus and benefits, fees paid to various consultants related to the Qualifying Transaction, and an increase in prepaid expenses and deposits. In 2016, we entered into lease agreement for our new headquarters in Ann Arbor, Michigan and prepaid $801,973 in rent for the entire five-year term. The increase in prepaid expenses and deposits was also due to increased deposits with our contract manufacturing organization and increased prepaid health insurance premiums from an overall increase in the number of employees.
| -43- |
Financing Activities
Net cash provided by financing activities for the year ended December 31, 2017 was $7,486,220, compared to net cash provided by financing activities of $4,786,353 for the year ended December 31, 2016, an increase of $2,699,867, or 56%. The increase resulted primarily from the private placement of $6,570,000 of our common shares in 2017, and proceeds from the exercise of stock options for $979,522, partially offset by stock issuance costs of $56,576 and repayment on a shareholder loan of $6,726.
Net cash provided by financing activities for the year ended December 31, 2016 was $4,786,353, which relates to cash acquired in the Qualifying Transaction, partially offset by cash paid for the stock issuance in the Qualifying Transaction.
Investing Activities
Net cash used in investing activities for the year ended December 31, 2017 was $171,736, compared to $241,215 for the year ended December 31, 2016, a decrease of $69,479, or 29%. The decrease resulted primarily from reduced leasehold improvements and reduced purchases of furniture and equipment for our additional office space in Ann Arbor.
Net cash used in investing activities for the year ended December 31, 2016 was $241,215, which primarily resulted from the investment in research equipment in support of the expanding R&D activities.
Liquidity and Capital Resources
We have incurred losses and negative cash flows from operations and have not generated any revenue since our inception in May 2015. As of December 31, 2017, we had an accumulated deficit of $15,626,100. We have funded our working capital requirements primarily through the sale of our common shares and the exercise of stock options. At December 31, 2017, we had cash and cash equivalents of $3,448,147.
Working capital (defined as current assets minus current liabilities) was $3,433,955 as at December 31, 2017. This was primarily due to cash and cash equivalents of $3,448,147 and prepaid expenses and deposits of $786,273, partially offset by accounts payables and accrued liabilities of $828,737.
On October 17, 2017 we entered into a five-year $5,000,000 unsecured working capital facility with Equidebt LLC, one of our shareholders (the “Equidebt Facility”). Amounts borrowed under the Equidebt Facility bear interest at a rate of 14% per annum payable at maturity. All amounts borrowed under the Equidebt Facility become due and payable on October 17, 2022. We can make two borrowing per month under the Equidebt Facility, each of which must be for a minimum of $250,000. The Equidebt Facility is unsecured; however Gerald A. Solensky Jr., our Chairman of the Board, President and Chief Executive Officer, has personally guaranteed our obligations under the Equidebt Facility.
We believe that our existing cash and available borrowings under the Equidebt Facility will be sufficient to fund our operations through the next twelve months. Our ability to continue as a going concern is ultimately dependent upon our ability to achieve sustainable positive cash flow from operations. However, we do not expect to generate revenue from the sale of our product candidates for the foreseeable future. To the extent that we do not generate sufficient cash flow from our operations, we intend to finance our working capital requirements through equity and/or debt financings, development agreements or marketing license agreements, the collection of revenues resulting from future commercialization activities and/or new strategic partnership agreements. There can be no assurance that we will be able to obtain any such capital on terms or in amounts sufficient to meet our needs or at all. The availability of equity or debt financing will be affected by, among other things, the results of our research and development activities, our ability to obtain regulatory approvals, market acceptance of any products for which we receive marketing approval, conditions in the capital markets generally and in the veterinary products industry, strategic alliance agreements and other relevant commercial considerations.
| -44- |
If we raise additional funds by issuing equity securities, our existing security holders will likely experience dilution, and the incurring of indebtedness would result in increased debt service obligations and could require us to agree to operating and financial covenants that could restrict operations. In the event that we are unable to obtain sufficient capital to meet our working capital requirements, we may be required to change or curtail current or planned operations in order to conserve cash until such time, if ever, that sufficient proceeds from operations are generated. In such an event, we may not be able to take advantage of business opportunities, and may have to terminate or delay safety and efficacy studies, curtail our product development programs, or sell or assign rights to our product candidates, products and technologies.
Based on the closing price of our common shares on December 31, 2017, the market price of our common shares exceeded the exercise price of our outstanding stock options. To the extent that some or all of such stock options are exercised, we would receive the proceeds of such exercises which would provide additional capital for our company. However no assurance can be given that any of such stock options will be exercised or as to the proceeds and timing of any exercises that do occur. The willingness of option holders to exercise their options depends on a number of factors, including, without limitation: the future market price of our common shares; the availability of capital to fund the payment of the exercise price of such options, the tax consequences of any such exercises and the ability of such option holders to resell some or all of the common shares received upon such exercises.
Our future capital requirements depend on many factors, including, but not limited to:
| • | the scope, progress, results and costs of researching and developing our current or future product candidates; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for any of our current or future product candidates; |
| • | the number and characteristics of the product candidates we pursue; |
| • | the cost of manufacturing our current and future product candidates and any products we successfully commercialize; |
| • | the cost of commercialization activities if any of our current or future product candidates are approved for sale, including marketing, sales, service, customer support and distribution costs; |
| • | the expenses needed to attract and retain skilled personnel; |
| • | the costs associated with being a public company; |
| • | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; and |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation. |
Off Balance Sheet Arrangements
Since inception, we have not engaged in the use of any off-balance sheet arrangements, such as structured finance entities, special purpose entities or variable interest entities.
| -45- |
Quantitative and Qualitative Disclosures about Liquidity and Market Risk
Liquidity risk is the risk that we will encounter difficulty raising liquid funds to meet our commitments as they fall due. In meeting our liquidity requirements, we closely monitor our forecasted cash requirements with expected cash drawdown.
We are exposed to interest rate risk, which is affected by changes in the general level of interest rates. Due to the fact that our cash is deposited with major financial institutions in an interest-bearing savings account, we do not believe that the results of operations or cash flows would be affected to any significant degree by a sudden change in market interest rates given their relative short-term nature.
We are also exposed to credit risk at period end from the carrying value of our cash. We manage this risk by maintaining bank accounts with a Canadian Chartered Bank and a U.S. bank that is a member of the Federal Deposit Insurance Corporation. Our cash is not subject to any external restrictions.
We are exposed to changes in foreign exchange rates between the Canadian and United States dollar which could affect the value of our cash. We had no foreign currency hedges or other derivative financial instruments as of December 31, 2017. We do not enter into financial instruments for trading or speculative purposes and do not currently utilize derivative financial instruments.
We have balances denominated in Canadian dollars that give rise to exposure to foreign exchange (“FX”) risk relating to the impact of translating certain non-U.S. dollar balance sheet accounts as these statements are presented in U.S. dollars. A strengthening U.S. dollar will lead to a FX loss, while a weakening U.S. dollar will lead to a FX gain. For each Canadian dollar balance of $1.0 million, a +/- 10% movement in the Canadian currency held by us versus the U.S. dollar would affect our loss and other comprehensive loss by $100,000.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers, requiring an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The updated standard will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective. In March 2016 the FASB issued ASU No. 2016-08 to clarify the implementation guidance on considerations of whether an entity is a principal or an agent, impacting whether an entity reports revenue on a gross or net basis. In April 2016, the FASB issued ASU No. 2016-10 to clarify guidance on identifying performance obligations and the implementation guidance on licensing. In May 2016, the FASB issued amendments ASU No. 2016-11 and 2016-12 to amend certain aspects of the new revenue guidance (including transition, collectability, noncash consideration and the presentation of sales and other similar taxes) and provided certain practical expedients. The guidance is effective for annual reporting periods beginning after December 15, 2017 (including interim reporting periods). Early adoption is permitted but not before the annual reporting period (and interim reporting period) beginning January 1, 2017. Entities have the option of using either a full retrospective or a modified approach to adopt the guidance. We are in the process of evaluating the amendments to determine if they have a material impact on our financial position, results of operations, cash flows or disclosures.
In January 2016, the FASB issued ASU No. 2016-01, which makes limited amendments to the guidance in U.S. GAAP on the classification and measurement of financial instruments. The new standard significantly revises an entity’s accounting related to (1) the classification and measurement of investments in equity securities and (2) the presentation of certain fair value changes for financial liabilities measured at fair value. It also amends certain disclosure requirements associated with the fair value of financial instruments. ASU No. 2016-01 is effective for fiscal years beginning after December 15, 2017, and interim periods within those annual periods. We are in the process of evaluating the amendments to determine if they have a material impact on our financial position, results of operations, cash flows or disclosures.
In February 2016, the FASB issued new guidance, ASU No. 2016-02, Leases (Topic 842). The main difference between current U.S. GAAP and the new guidance is the recognition of lease liabilities based on the present value of remaining lease payments and corresponding lease assets for operating leases under current U.S. GAAP with limited exception. Additional qualitative and quantitative disclosures are also required by the new guidance. Topic 842 is effective for annual reporting periods (including interim reporting periods) beginning after December 15, 2018. Early adoption is permitted. We are in the process of evaluating the amendments to determine if they have a material impact on our financial position, results of operations, cash flows or disclosures.
| -46- |
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230) Classification of Certain Cash Receipts and Cash Payments, which will make eight targeted changes to how cash receipts and cash payments are presented and classified in the Statement of Cash Flows. ASU 2016-15 will be effective on May 1, 2018, and will require adoption on a retrospective basis unless it is impracticable to apply, in which case we would be required to apply the amendments prospectively as of the earliest date practicable. We are in the process of evaluating the amendments to determine if they have a material impact on our financial position, results of operations, cash flows or disclosures.
In August 2016, the FASB issued ASU 2017-01 that changes the definition of a business to assist entities with evaluating when a set of transferred assets and activities is a business. The guidance requires an entity to evaluate if substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or a group of similar identifiable assets; if so, the set of transferred assets and activities is not a business. ASU 2017-01 also requires a business to include at least one substantive process and narrows the definition of outputs by more closely aligning it with how outputs are described in ASC 606.1. ASU 2017-01 is effective for public business entities for fiscal years beginning after December 15, 2017, and interim periods within those years. Early adoption is permitted. We are in the process of evaluating the amendments to determine if they have a material impact on our financial position, results of operations, cash flows or disclosures.
In May 2017, FASB issued ASU 2017-09 in relation to Compensation —Stock Compensation (Topic 718), Modification Accounting. The amendments provide guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. The amendments are effective for all entities for annual periods, and interim periods within those annual periods, beginning after December 15, 2017. Early adoption is permitted, including adoption in any interim period, for (1) public business entities for reporting periods for which financial statements have not yet been issued and (2) all other entities for reporting periods for which financial statements have not yet been made available for issuance. The amendments should be applied prospectively to an award modified on or after the adoption date. We are in the process of evaluating the amendments to determine if they have a material impact on our financial position, results of operations, cash flows or disclosures.
| Item 8. | Financial Statements and Supplementary Data. |
Our financial statements, together with the independent registered public accounting firm report thereon, are incorporated by reference from the applicable information set forth in Part IV Item 15, “Exhibits, Financial Statement Schedules” of this Annual Report on Form 10-K.
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. |
None.
| Item 9A. | Controls and Procedures. |
Evaluation of Our Disclosure Controls
We maintain disclosure controls and procedures that are designed to provide reasonable assurance that material information required to be disclosed in our periodic reports filed under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and to provide reasonable assurance that such information is accumulated and communicated to our management, our chief executive officer and our chief financial officer, to allow timely decisions regarding required disclosure. We carried out an evaluation, under the supervision and with the participation of our management, including our principal executive and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rule 13(a)-15(e) under the Exchange Act. Based on this evaluation, our principal executive officer and principal financial officer concluded that, as of December 31, 2017, our disclosure controls and procedures were effective.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in the framework in “Internal Control — Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2017.
| -47- |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
This Annual Report on Form 10-K does not include an attestation report of our independent registered public accounting firm because we are an “emerging growth company,” and may take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act.
Changes in Internal Controls over Financial Reporting
During the year ended December 31, 2017, there have been no changes in our internal control over financial reporting that have materially affected or are reasonably likely to materially affect our internal controls over financial reporting. From time to time, we make changes to our internal control over financial reporting that are intended to enhance its effectiveness and which do not have a material effect on our overall internal control over financial reporting.
| Item 9B. | Other Information. |
None.
| Item 10. | Directors, Executive Officers and Corporate Governance. |
MANAGEMENT
Executive Officers and Directors
The following table sets forth the name, age, position and tenure of each of our directors and executive officers as of December 31, 2017:
| Name | Age | Position |
| Gerald Solensky Jr. | 44 | Chairman of the Board, President and Chief Executive Officer |
| Shameze Rampertab | 51 | Chief Financial Officer, Corporate Secretary and Director |
| Stephanie Morley | 42 | Chief Operations Officer and Vice President of Product Development |
| Robert DiMarzo | 61 | Executive Vice President of Global Strategy |
| Bruk Herbst | 48 | Chief Commercial Officer |
| James LeBar (1)(2)(3) | 65 | Director |
| Rodney Williams (1)(2)(3) | 56 | Director |
| Jeffrey Rowe (1)(3) | 62 | Director |
| Thomas Robitaille (1)(2) | 55 | Director |
| Jane Eagleson (3) | 66 | Director |
_________________________________________________________________
| (1) | Member of the Audit Committee | |
| (2) | Member of the Compensation Committee | |
| (3) | Member of the Nominating and Corporate Governance Committee |
| -48- |
Management
Gerald Solensky Jr. is the founder of our business. He has been our President and Chief Executive Officer since May 2015. He has been the Chairman of our board of directors since May 2016. From 2013 to 2015, Mr. Solensky worked on developing our business model, authored a consumer financial education program and completed over 800 hours of observation time with a board-certified veterinary surgeon to garner a more complete understanding of our veterinary customers and their associated needs. From 2010 to 2013, he was a consultant for business turnarounds and capital raising. We selected Mr. Solensky to serve on and lead our board of directors due to his track-record building successful operations within start-up, turnaround and rapid-change environments.
Shameze Rampertab, CPA, CA has been our Chief Financial Officer since March 2016. In April 2016, he took on the roles of Corporate Secretary and Director. Mr. Rampertab acted as an independent consultant for a number of companies, including us, in respect of which he provided general financial advisory and accounting services prior to his appointment as Chief Financial Officer, from November 2015 to March 2016. He was the Chief Financial Officer of multiple publicly-traded health care companies including Profound Medical Corp. from October 2014 to November 2015 and Intellipharmaceutics International Inc. from October 2010 to October 2014. Mr. Rampertab is a chartered professional accountant and chartered accountant with twenty years of experience in capital markets, strategic planning and analysis. He holds an MBA from McMaster University and a Bachelor’s degree in molecular genetics and molecular biology from the University of Toronto. We selected Mr. Rampertab to serve on our board of directors due to his strong experience in the financial, medical and scientific arenas.
Stephanie Morley, DVM has been our Chief Operations Officer and Vice President of Product Development since July 2017. From October 2015 until July 2017, she served as our Chief Operating Officer. Prior thereto, from July 2015 until October 2015, Dr. Morley was a consultant for us providing strategic and tactical support. From December 2013 to August 2015 Dr. Morley served as Associate Director of Business Development with the University of Michigan Medical School. From April 2006 to August 2013 Dr. Morley held several positions of increasing responsibility with MPI Research, a contract research organization, including Vice President of Operations. Dr. Morley is a trained veterinarian, having earned her DVM degree from Michigan State University. After earning her DVM degree, Dr. Morley was a practicing veterinarian with Oakwood Animal Hospital in Kalamazoo, MI and Adobe Animal Medical Center in Albuquerque, NM where she assumed dual roles of both clinical practitioner and operations management.
Robert DiMarzo has been our Executive Vice President of Global Strategy since February 2017. Mr. DiMarzo was intermittently a Principal Consultant with DiMarzo Business Consulting from November 2007 to February 2017, including advising our company. From August 2015 through January 2016, Mr. DiMarzo was Vice President of Commercial Development and Product Category Management with the global animal health group at Henry Schein, Inc. Prior to that, he was Executive Chairman of the U.S. animal health distributor Ivesco Holdings, LLC from April 2010 to October 2013. Before that, Mr. DiMarzo was Executive Vice President of Sales and Marketing for the veterinary diagnostic startup Scandinavian Micro Biodevices from July 2008 to April 2010. From 1992 to 2007 Mr. DiMarzo held several director-level and executive leadership position with Pfizer Animal Health, including President of U.S. Operations.
Bruk Herbst has been our Chief Commercial Officer since July 2017. From October 2015 to December 2016 Mr. Herbst was the Executive Senior Vice President of Sales and Marketing at i4C Innovations Inc. d/b/a Voyce, an animal health and wellness company. From October 2007 to September 2015, he served as Executive Senior Director and Head of U.S. Sales at IDEXX Laboratories, Inc, a developer, manufacturer and distributor of products and services for the companion animal veterinary and other markets, where he was responsible for in-clinic and reference lab diagnostics, point of care, information technologies and digital radiology. From January 1999 to October 2007 Mr. Herbst also held commercial leadership roles in patient monitoring, pharmacy and diagnostics with Omnicare Specialty Care Group and Life Systems. He holds a Bachelor of Science degree in business from the University of Arizona.
Non-Management Directors
James LeBar has been a Director and the Chairman of our Compensation Committee since April 2016. Mr. LeBar also served as a director on the board of Zomedica Pharmaceuticals, Inc. from May 2015 until the completion of our Qualifying Transaction in April 2016. From March 2011 until his retirement in January 2016, Mr. LeBar served as a turnaround consultant for Nationwide Placement Inc., a specialized health training company. We selected Mr. LeBar to serve on our board of directors due to his experience as an entrepreneur and executive leader, an expert in building and operating start-up companies and establishing corporate structures for profitability and success.
| -49- |
Rodney Williams, MBA has served as a Director and the Chair of our Corporate Governance Committee (now called the Nominating and Corporate Governance Committee) since April 2016. He is currently employed as Corporate Global Vice President Portfolio and Services for publicly-traded Align Technologies (ALGN) as of February 1, 2017. Previously, Mr. Williams was an entrepreneur-in-residence with PTV Healthcare Capital, a private equity investment firm and he has been with PTV since October 2015. Prior to PTV, he was President and CEO of Heart Rhythm Society Consulting Services from January 2013 through August 2015. From January 2008 through January 2013, Mr. Williams served as Senior Vice President of Global Product Planning and Marketing at St. Jude Medical Inc. Mr. Williams also served in commercial leadership roles in sales and marketing at GE Healthcare, Johnson and Johnson, and Bausch & Lomb. Mr. Williams earned both his MBA and Bachelor of Science degrees from the University of Southern California and attended the General Management Executive Leadership Program at The Wharton School of Business. We selected Mr. Williams to serve on our board of directors due to his experience with both large and small-cap medical technology and related health care companies and his global commercialization expertise.
Jeffrey Rowe has served as a Director and the Chairman of our Audit Committee since April 2016. Until his retirement in October 2015, Mr. Rowe served as Executive Vice President and a Director of Diplomat Pharmacy, Inc., the largest independent specialty pharmacy company in the U.S. During his tenure with Diplomat, the company grew from a single location with less than $5 million in revenue, to sixteen locations and $3 billion in sales, and became publicly traded on the New York Stock Exchange. Prior to his career with Diplomat, Mr. Rowe owned two successful community pharmacies in Genesee County, Michigan. He holds a Bachelor of Pharmacy degree from Ferris State University. We selected Mr. Rowe to serve on our board of directors due to his financial expertise and his extensive experience in pharmaceutical operations, the specialty pharmacy industry and fundamental business strategies involving accreditation, contracting, cybersecurity and regulation, combined with an expertise in compounding and integrative medicine.
Thomas Robitaille has been a Director since October 2016. Mr. Robitaille is an independent management consultant specialized in animal health. From February 2016 to April 2017, he was the Vice President of Veterinary Channel Development at Blue Buffalo Company, a premium, all-natural pet food company. From October 2006 to October 2015, Mr. Robitaille was the Director of the Americas for the animal health pharmaceutical company Vetoquinol SA Inc. As the Director of the Americas he managed affiliated companies and regional distributors in Canada, the United States, Mexico and Brazil. He was responsible for veterinary pharmaceutical operations in the United Kingdom, Ireland, Belgium, and the Netherlands as Managing Director and also served as Director of International Development, where he contributed to an increase in sales and profit for in Eastern Europe, Asia Pacific, Africa, and Latin America. He has a Master of Business Administration degree from the University of Warwick and Bachelor of Science degree from Concordia University. We selected Mr. Robitaille to serve on our board of directors due to his lengthy experience in the animal health industry and his skills in the areas of product development, sales and marketing and mergers and acquisitions.
Jane Eagleson, has been a Director since October 2016. Dr. Eagleson is a veterinarian with more than 30 years of experience in animal health pharmaceutical development. She has owned Bleecker Street Consulting, a consulting firm specializing in global animal health pharmaceutical product development strategy since January 2013. From November 2014 until the company’s acquisition by Zoetis Inc. in July 2017, she served as Vice President of Clinical and Regulatory Affairs at Nexvet US, Inc., a veterinary biotherapeutics company, where she was responsible for the clinical and regulatory phases of global biopharmaceutical product development. From September 2007 through December 2012, Dr. Eagleson was the General Manager of Research & Development and subsequently the Head of Growth Strategies for Argenta Limited, a specialist animal health contract manufacturing organization based on Auckland, New Zealand, through December 2010 and New Jersey thereafter. At Argenta, Dr. Eagleson was responsible for the management of a subsidiary of the company, Alcherabio, an animal health clinical contract research organization in New Jersey, the development staff in New Zealand and the overall management of Argenta’s strategic plans. She has a Master of Veterinary Science in immunology from Massey University and Bachelor of Veterinary Science (U.S. DVM equivalent) from the University of Sydney. She has also authored a number of publications in peer reviewed journals. We selected Dr. Eagleson to serve on our board of directors due to her in depth knowledge of the animal health industry and regulatory agencies in developed markets including the United States, European Union and Oceania.
Board Composition
Our board of directors currently consists of seven members. Our bylaws provide that our directors will hold office until the close of the first annual meeting of shareholders following his or her election unless the director is elected for a stated term. Our board of directors is responsible for the business and affairs of our company and considers various matters that require its approval.
| -50- |
Our board of directors is comprised of a majority of directors who are “independent” (as discussed below), and the Board has established an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. We have adopted charters for our each of these committees and a code of ethics and business conduct, or Code of Ethics. Our Code of Ethics is available on our website at www.zomedica.com. The committee charters are also available for review on our website.
Director Independence
Our board of directors has determined that all of our directors, other than Messrs. Solensky and Rampertab, are “independent,” as defined under the NYSE American. For purposes of the NYSE American rules, an independent director means a person other than an executive officer or employee of our company or any other individual having a relationship which, in the opinion of our board of directors, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director, subject to certain additional limitations. Such directors are also deemed to be “independent” under applicable Canadian securities laws.
Code of Ethics
Our board of directors has adopted the Code of Ethics, which applies to all officers, directors and employees. Our Code of Ethics is available on our website at www.zomedica.com. Information contained in, or accessible through, our website does not constitute part of this Form 10-K. We intend to disclose any amendments to our Code of Ethics, or waivers of its requirements, on our website or in our filings under the Exchange Act.
Board Committees
Our board of directors has three standing committees: the Audit Committee, the Compensation Committee and the Nominating and Corporate Governance Committee. All of our committee members are “independent,” as defined under the NYSE American rules and for purposes of Canadian securities laws.
Each of our committee charters is available on our website at www.zomedica.com.
Audit Committee
Our audit committee is currently comprised of four members, Mr. Rowe (Chairman), Mr. Williams, Mr. LeBar and Mr. Robitaille. Each member of our audit committee is a non-employee member of our board of directors. We have designated Mr. Rowe as our “audit committee financial expert,” as defined under Item 407 of Regulation S-K. All of the members of our audit committee are “independent” members of our board of directors, as required by the NYSE American rules and Canadian securities laws.
The purpose of our audit committee of our board of directors is to oversee (i) the integrity of our company’s financial statements, our company’s accounting and financial reporting processes and financial statement audits; (ii) our company’s compliance with applicable legal and regulatory requirements; (iii) our company’s systems of internal control over financial reporting and disclosure controls and procedures; (iv) the independent auditor’s engagement, qualifications, performance, compensation and independence; (v) review of related party transactions; and (vi) compliance with the company’s corporate policies. The audit committee’s function is one of oversight, whereas the planning and conduct of the audit is the responsibility of the independent auditor, and the financial statements are the responsibility of the company’s management.
Each member of the audit committee has experience reviewing financial statements and dealing with related accounting and auditing issues and is “financially literate” within the meaning of Canadian securities laws.
The audit committee has the sole authority to pre-approve all audit and permitted non-audit services provided by the independent auditor.
| -51- |
Compensation Committee
Our compensation committee is currently comprised of three members, Mr. LeBar (Chairman), Mr. Williams and Mr. Robitaille. All of the members of our compensation committee are “independent” directors, as defined under the NYSE American rules and for purposes of Canadian securities laws.
The purpose of our compensation committee is to (i) make recommendations to our board of directors relating to evaluation and compensation of our executives, (ii) oversee incentive, equity-based and other compensatory plans in which executive officers and key employees of our company participate, (iii) review and participate in determining director compensation and (iv) prepare any report on executive compensation required by the rules and regulations of the Commission and the listing standards of NYSE American.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee is currently comprised of four members, Mr. Williams (Chairman), Mr. LeBar, Mr. Rowe and Dr. Eagleson. All of the members of our corporate governance committee are “independent” directors, as defined under the NYSE American rules and for the purposes of Canadian securities laws.
The purpose of our nominating and corporate governance committee of our board of directors is to carry out the responsibilities delegated by the board of directors relating to the our director nominations process, developing and maintaining our company’s corporate governance policies, and any related matters required by the federal securities laws or by the applicable listing rules of the NYSE American.
Board Leadership Structure and Role in Risk Oversight
Although we have not adopted a formal policy on whether the Chairman and Chief Executive Officer positions should be separate or combined, we have determined that it is in our best interests and the best interests of our shareholders to combine these roles. Mr. Solensky currently serves as our Chief Executive Officer and Chairman of our board of directors. Due to our small size and our early development stage, we believe it is currently most effective to have the Chairman and Chief Executive Officer positions combined.
Our board of directors is primarily responsible for overseeing our risk management processes. The board of directors receives and reviews periodic reports from management, auditors, legal counsel, and others, as considered appropriate regarding our assessment of risks. The board of directors focuses on the most significant risks facing our general risk management strategy, and us and also ensures that risks undertaken by us are consistent with the board’s appetite for risk. While the board oversees our risk management, management is responsible for day-to-day risk management processes. We believe that this division of responsibilities is the most effective approach for addressing the risks facing us and that our board leadership structure supports this approach.
| -52- |
| Item 11. | Executive Compensation. |
EXECUTIVE AND DIRECTOR COMPENSATION
The following table shows the compensation for each of the years ended December 31, 2017 and December 31, 2016 awarded to or earned by our principal executive officer and our two other most highly compensated executive officers who were serving as executive officers as of December 31, 2017. The persons listed in the following table are referred to herein as the “named executive officers.”
| Salary | Bonus | Option | All Other | Total | ||||||||
| Name and Principal Position | ($) | ($) | Awards(4) | Compensation(5) | ($) | |||||||
| ($) | ($) | |||||||||||
| Gerald Solensky Jr.(1) | 2017 | 285,000 | 30,000 | - | 24,000 | 339,000 | ||||||
| Chairman of the Board, President and Chief Executive Officer | 2016 | 252,918 | 40,000 | 323,501 | 900 | 617,319 | ||||||
| Stephanie Morley(2) | 2017 | 187,500 | 30,000 | 272,089 | 12,000 | 501,589 | ||||||
| Chief Operations Officer and Vice President of Product Development | 2016 | 175,001 | 40,000 | 250,306 | - | 465,307 | ||||||
| Robert DiMarzo(3) | 2017 | 188,125 | 18,000 | 287,064 | 67,958 | 561,147 | ||||||
| Executive Vice President of Global Strategy | 2016 | - | - | 34,053 | 25,000 | 59,053 |
__________________
(1) Mr. Solensky entered into an employment agreement in December 2016 which was amended in August 2017 pursuant to which he receives an annual salary of $285,000 and a monthly car allowance of $2,000. Mr. Solensky received a grant of options to purchase 950,000 common shares at an exercise price of $1.19 in 2016.
(2) Dr. Morley entered into an employment agreement with us in October 2015 pursuant to which she receives an annual salary of $150,000 per annum, which was increased to $200,000 effective July 2017. Effective in July 2017, Dr. Morley also receives a monthly allowance of $2,000 for vehicle and tax preparation. Dr. Morley received a grant of options to purchase 1,100,000 common shares at an exercise price of $0.20 in 2016, a grant of options to purchase 600,000 common shares at an exercise price of $1.19 in 2016, and a grant of options to purchase 500,000 common shares at an exercise price of $2.18.
(3) Mr. DiMarzo began serving as a consultant in October 2016 and received consulting fees of $50,958 in cash and options to purchase 100,000 common shares at an exercise price of $1.19. He was appointed Executive Vice President of Global Strategy of Zomedica Pharmaceuticals, Inc. in February 2017. Mr. DiMarzo entered into an employment agreement with us in February 2017 pursuant to which he receives an annual salary of $215,000 and a monthly allowance of $4,000 for vehicle, insurance and tax preparation. Mr. DiMarzo received a grant of options to purchase 500,000 common shares at an exercise price of $1.19 in 2017 and a grant of options to purchase 250,000 common shares at an exercise price of $2.18 in 2017.
(4) Represents the aggregate grant date fair value for grants made in 2017 and 2016, respectively, computed in accordance with FASB ASC Topic 718. The assumptions we used in valuing options are described in Note 10 to our financial statements included in this Annual Report on Form 10-K.
(5) All Other Compensation represents consulting fees and monthly allowances.
Employment and Consulting Agreements
Gerald Solensky Jr.
In December 2016, we entered into an employment agreement with Mr. Solensky, which was amended in August 2017 pursuant to which Mr. Solensky serves as our President and Chief Executive Officer. Mr. Solensky’s amended employment agreement has an unspecified term and provides him with an annual base salary of $285,000 plus quarterly bonuses and participation in our employee benefit plan. In addition, we agreed to pay Mr. Solensky a $2,000 monthly car allowance and four weeks of paid vacation. Pursuant to Mr. Solensky’s amended employment agreement, any options granted to him will be subject to accelerated vesting upon a change of control, a resolution of our board in anticipation of a change of control, our termination of his employment without cause or his resignation for good reason. Mr. Solensky’s employment agreement also includes customary non-solicitation, confidentiality and assignment of inventions provisions. If we terminate Mr. Solensky’s employment without cause or he resigns for good reason, we are required to pay him twelve months base salary and any quarterly bonus allocable or payable prior to termination.
| -53- |
Stephanie Morley
In connection with her appointment as Chief Operations Officer and Vice President of Product Development, effective July 1, 2017, we entered into an employment agreement with Dr. Morley that superseded and replaced her earlier employment agreement with us. The agreement is effective for a period of one year and automatically extends for one year terms unless either party elects to terminate it. Dr. Morley’s employment agreement provides her with an annual base salary of $200,000 and quarterly bonuses upon the achievement of certain specified objectives. In addition, we agreed to pay Dr. Morley a $2,000 monthly allowance in respect of the following items: (i) vehicle and (ii) tax preparation. Dr. Morley is entitled to three weeks paid vacation time. We granted Dr. Morley options to purchase 500,000 common shares at an exercise price of $2.20 per share and, under her employment agreement, Dr. Morley is eligible to receive additional options to purchase 500,000 common shares in the fourth quarter of 2017. All such grants will have an exercise price of not less than fair market value on the date of grant. Pursuant to Dr. Morley’s employment agreement, any options granted to her will be subject to accelerated vesting upon our termination of Dr. Morley’s employment without cause. Dr. Morley’s employment agreement also includes customary non-solicitation, confidentiality and assignment of inventions provisions. In the event that Dr. Morley has a “separation from service” within the meaning of Section 409A of the Internal Revenue Code of 1986, as amended, or the Code, Dr. Morley would have the right to exercise all of her options, and we would be required to pay her a lump sum equal to 12 months of her base salary and any quarterly bonus allocable or payable prior to the date of termination.
Robert DiMarzo
Effective February 1, 2017, we entered into an employment agreement with Robert DiMarzo. Mr. DiMarzo serves as our Executive Vice President of Global Strategy. The agreement is effective for a period of one year and automatically extends for one year terms unless either party elects to terminate it. Mr. DiMarzo’s employment agreement provides for an annual base salary of $215,000. In addition, Mr. DiMarzo is eligible to receive up to four quarterly bonuses totaling $36,000 upon the achievement of certain specified objectives. In addition, we agreed to pay Mr. DiMarzo a $4,000 monthly allowance in respect of the following items: (i) vehicle; (ii) insurance (medical, dental, vision) premiums; and (iii) tax preparation. Mr. DiMarzo is entitled to three weeks paid vacation time, and five business days’ vacation during the period between December 25 and December 31 of each year. We granted Mr. DiMarzo options to purchase 500,000 common shares at an exercise price of $1.20 per common share and, under his employment agreement, he is eligible to receive additional options to purchase 250,000 common shares upon the six month anniversary upon achievement of six month performance objectives, and to receive further options to purchase 250,000 common shares upon the twelve month anniversary upon achievement of twelve month performance objectives. All such grants will have an exercise price of not less than fair market value on the date of grant. Pursuant to Mr. DiMarzo’s employment agreement, any options granted to him will be subject to accelerated vesting upon our termination of Mr. DiMarzo’s employment without cause or resignation by Mr. DiMarzo for good reason. Mr. DiMarzo’s employment agreement also includes customary non-solicitation, confidentiality and assignment of inventions provisions. If we terminate Mr. DiMarzo’s employment other than for cause or Mr. DiMarzo resigns for good reason, we are required to pay Mr. DiMarzo twelve months base salary and any quarterly bonus amounts payable.
| -54- |
Outstanding Equity Awards at Fiscal Year End
The following table sets forth certain information, on an award-by-award basis, concerning outstanding equity awards for each named executive officer as of December 31, 2017.
| Option awards | Stock awards | ||||||||||||||||||||||||||||||||||
| Name | Number of securities underlying unexercised options (#) exercisable | Number of securities underlying options (#) unexercisable | Equity incentive plan awards: Number of securities underlying unexercised unearned options (#) | Option exercise price ($) | Option expiration date | Number of shares or units of stock that have not vested (#) | Market Value of shares of units of stock that have not vested ($) | Equity incentive plan awards: Number of unearned shares, units or other rights that have not vested (#) | Equity incentive plan awards: Market or payout value of unearned shares, units or other rights that have not vested ($) | ||||||||||||||||||||||||||
| Jerry Solensky Jr. (2) | 200,000 | - | - | 1.19 | 12/21/2018 | - | - | - | - | ||||||||||||||||||||||||||
| Dr. Stephanie Morley (1) | 1,100,000 | - | - | 0.20 | 4/21/2018 | - | - | - | - | ||||||||||||||||||||||||||
| Dr. Stephanie Morley (2) | 600,000 | - | - | 1.19 | 12/21/2018 | - | - | - | - | ||||||||||||||||||||||||||
| Dr. Stephanie Morley (4) | 500,000 | - | - | 2.18 | 8/14/2019 | - | - | - | - | ||||||||||||||||||||||||||
| Robert DiMarzo(2) | 100,000 | - | - | 1.19 | 12/21/2018 | - | - | - | - | ||||||||||||||||||||||||||
| Robert DiMarzo(3) | 500,000 | - | - | 1.19 | 2/24/2019 | - | - | - | - | ||||||||||||||||||||||||||
| Robert DiMarzo(4) | 250,000 | - | - | 2.18 | 8/14/2019 | - | - | - | - | ||||||||||||||||||||||||||
__________________
| (1) Stock options vest immediately upon issue, with an issue date of March 28, 2016, and expire on April 21, 2018. |
| (2) Stock options vest immediately upon issue, with an issue date of December 21, 2016, and expire on December 21, 2018. |
| (3) Stock options vest immediately upon issue, with an issue date of February 24, 2017, and expire on February 24, 2019. |
| (4) Stock options vest immediately upon issue, with an issue date of August 14, 2017, and expire on August 14, 2019. |
Equity Compensation Plan Information
The following table provides information, as of December 31, 2017, with respect to all compensation arrangements maintained by us, including individual compensation arrangements, under which shares are authorized for issuance.
| Plan Category | Number of Securities to be issued upon outstanding options rights (a) | Weighted-average exercise price outstanding options and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in columns (a)) (c) | |||||||||
| Equity compensation plans approved by shareholders | 8,080,000 | $ | 0.96 | 942,586 | ||||||||
| Equity compensation plans not approved by shareholders | Nil | N/A | Nil | |||||||||
| Total | 8,080,000 | $ | 0.96 | 942,586 | ||||||||
| -55- |
Stock Option Plans
As of December 31, 2015, Zomedica Pharmaceuticals Corp (formerly, Wise Oakwood Ventures Inc.), had a shareholder-approved option plan, or the WOW Plan, pursuant to which options to purchase 200,000 common shares were outstanding. The terms of the WOW Plan were substantially similar to those of our current Stock Option Plan. In connection with the Qualifying Transaction, these options were consolidated into options to purchase 80,000 common shares of Zomedica Pharmaceuticals Corp. and fully exercised and the WOW Plan was terminated.
In April 2016, concurrent with the completion of the Qualifying Transaction, we adopted a new equity stock option plan, the Stock Option Plan. The Stock Option Plan was approved by our shareholders. The purpose of the Stock Option Plan is to attract and retain employees, consultants, officers and directors to our company and to motivate them to advance the interests of our company by affording them with the opportunity, through share options, to acquire an equity interest in our company and benefit from its growth.
Administration. The Stock Option Plan is administered by our board of directors. Our board of directors may grant options to purchase shares of our common shares or such other shares as may substitute therefore in the capital of Zomedica Pharmaceuticals Corp. Our board of directors also has authority to determine the terms and conditions of each award, prescribe, amend and rescind rules and regulations relating to the Stock Option Plan, and amend the terms of awards (provided that no amendment may materially prejudice the rights of a participant without consent such participant’s consent). Our board of directors may delegate authority to a committee of our directors or to an officer. Our board or directors may terminate the Stock Option Plan.
Eligibility. Persons eligible to receive awards under the Stock Option Plan include any person who is an employee, officer, director or consultant provided that any consultant has performed and/or continues to perform services for our company under a written agreement and on an ongoing basis or is expected to provide a service to our company.
Shares Subject to the Stock Option Plan. The aggregate number of shares of common shares available for issuance in connection with options and awards granted under the Stock Option Plan is ten percent of the total number of issued and outstanding common shares calculated on a non-diluted basis. If any award of options granted under the Stock Option Plan expires or terminates without having been fully exercised, that number of common shares shall become available for the purpose of future grants under the Stock Option Plan.
Terms and Conditions of Options. Our board of directors will determine the exercise price of options granted under the Stock Option Plan. The exercise price of stock options may not be less than that from time to time permitted under the rules of any stock exchange on which the common shares are then listed. In addition, the exercise price of an option must be paid in cash.
The number of common shares subject to each option shall be determined by our board of directors with the following limitations. The number of common shares reserved for issuance to any one individual, consultant, person conducting investor relations or insider (as defined in the Securities Act (Alberta)) in a 12 month period may not exceed 5%, 2%, 2% and 10%, respectively, of the issued and outstanding common shares at the time of the grant.
No option may be exercisable for more than ten years from the date of grant. Options granted under the Stock Option Plan will be exercisable at such time or times as our board of directors prescribes at the time of grant. Options shall only be exercised by the participant as long as the optionee remains or was within the last ninety days an employee, officer, director or consultant, if the optionee dies, within one year of the optionee's death or if an optionee is engaged in investor relations activities, within 30 days of being so engaged by our company.
All benefits, rights and options accruing under the Stock Option Plan are non-transferrable and non-assignable unless specifically provided in the grant. During the lifetime of a participant, any options granted under the Stock Option Plan may only be exercised by the participant and in the event of the death of a participant, by the person or persons to whom the participant's rights under the option pass by the participant's will or applicable law.
Effect of Certain Corporate Transactions. In the event of a sale by our company of all or substantially all of its assets or in the event of a change of control (as defined in the Stock Option Plan) of our company, each participant shall be entitled to exercise, in whole or in part, the options granted to such participant under the Stock Option Plan, either during the term of the option or within ninety days after the date of the sale or change of control, whichever first occurs.
Director Compensation
We have not established a formal compensation policy for our outside directors. We did not compensate our outside directors for their service in 2017.
| -56- |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The table below sets forth certain information with respect to beneficial ownership of our securities as of February 27, 2018 by:
| · | each person known by us to be the beneficial owner of more than 5% of our issued and outstanding common shares; |
| · | each of our executive officers and directors; and |
| · | all executive officers and directors as a group. |
The number of shares beneficially owned by each shareholder is determined in accordance with SEC rules. Under these rules, beneficial ownership includes any shares as to which a person has sole or shared voting power or investment power. Percentage ownership is based on 90,449,869 common shares outstanding on February 27, 2018. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, common shares subject to stock options, warrants or other rights held by such person that are currently convertible or exercisable or will become convertible or exercisable within 60 days of February 27, 2018 are considered outstanding, although these shares are not considered outstanding for purposes of computing the percentage ownership of any other person.
Unless otherwise stated, the address of each 5% or greater beneficial holder is c/o Zomedica Pharmaceuticals Corp., 100 Phoenix Drive, Suite 190, Ann Arbor, Michigan 48108. We believe, based on information provided to us, that each of the shareholders listed below has sole voting and investment power with respect to the shares beneficially owned by the shareholder unless noted otherwise, subject to community property laws where applicable.
| Beneficial Ownership | ||||||||
| Number of Shares Beneficially Owned | Percent of Total Outstanding Common Shares | |||||||
| Name and Address of Beneficial Owner: | ||||||||
| Gerald Solensky Jr. (1) | 38,151,100 | 42.1 | % | |||||
| Jeffrey Rowe(2) | 12,240,480 | 13.5 | % | |||||
| Stephanie Morley(3) | 3,060,580 | 3.3 | % | |||||
| Shameze Rampertab(4) | 1,093,000 | 1.2 | % | |||||
| Robert DiMarzo(5) | 984,880 | 1.1 | % | |||||
| Bruk Herbst(6) | 203,000 | * | ||||||
| James LeBar(7) | 420,000 | * | ||||||
| Rodney Williams(8) | 380,000 | * | ||||||
| Jane Eagleson(9) | 100,000 | * | ||||||
| Thomas Robitaille(10) | 100,000 | * | ||||||
| All executive officers and directors as a group (ten persons)(11) | 56,733,040 | 59.2 | % | |||||
| * | Less than one percent. |
| (1) | Includes options to purchase 200,000 common shares. |
| (2) | Includes 11,120,000 shares are held in the Rowe Family GST Trust, 664,480 shares held by the Jeffrey M. Rowe U/T/A dated November 5, 2004 (the “Jeffrey M. Rowe Living Trust”) and 181,000 shares held by Mr. Rowe through his IRA. Mr. Rowe’s sister, Michele Ramo, serves as trustee to the Rowe Family GST Trust, with Mr. Rowe’s oversight and Mr. Rowe serves as trustee to the Jeffrey M. Rowe Living Trust. Mr. Rowe exclusively makes all investment decisions on behalf of this trust. Mr. Rowe also has options to purchase 275,000 common shares. |
| -57- |
| (3) | Includes options to purchase 2,200,000 common shares. Includes 5,000 common shares held by Dr. Morley’s children. |
| (4) | Includes options to purchase 926,000 common shares. Includes 3,000 common shares held by Mr. Rampertab’s children. |
| (5) | Includes options to purchase 850,000 common shares. |
| (6) | Includes options to purchase 200,000 common shares. Includes 3,000 common shares held by Mr. Herbst’s children. |
| (7) | Includes options to purchase 200,000 common shares. |
| (8) | Includes 40,000 shares held by Entrust Group Inc. FBO Rodney James Williams IRA and options to purchase 340,000 common shares. |
| (9) | Includes options to purchase 85,000 common shares. |
| (10) | Includes options to purchase 100,000 common shares. |
| (11) | In the aggregate, this includes options to purchase 5,376,000 common shares. |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
Other than compensation arrangements for our named executive officers and directors, we describe below each transaction or series of transactions, since January 1, 2017, to which we were a party or will be a party, in which:
| · | the amounts involved exceeded or will exceed $120,000; and |
| · | any of our directors, executive officers or holders of more than 5% of our common shares, or any member of the immediate family of the foregoing persons, had or will have a direct or indirect material interest. |
Compensation arrangements for our named executive officers and directors are described in Item 11 of this Annual Report on Form 10-K.
Equidebt Working Capital Facility
On September 1, 2017, Equidebt LLC, or Equidebt, one of our shareholders, entered into a Loan Agreement, or the Loan Agreement, with Mr. Solensky pursuant to which Equidebt agreed to provide Mr. Solensky with an unsecured line of credit in the amount of $5,000,000 for the purpose of enabling Mr. Solensky to exercise options to purchase up to 950,000 common shares expiring on December 21, 2018 and to purchase additional common shares from us from time to time, or the line of credit. Amounts borrowed under the line of credit were to bear interest at a rate of 14% per annum payable at maturity. In addition, Mr. Solensky was required to pay Equidebt a monthly maintenance fee of $6,250 per month payable at maturity. All amounts borrowed under the line of credit were to become due and payable on September 1, 2022. Upon the occurrence of an Event of Default (defined in the Loan Agreement to include Mr. Solensky’s failure to make payments under the line of credit or his other indebtedness when due, the occurrence of certain insolvency events relating to Mr. Solensky or the occurrence of a substantial change in the existing or prospective financial condition or net worth of Mr. Solensky which Equidebt determines to be materially adverse), Equidebt had the right to declare all amounts outstanding under the line of credit immediately due and payable. We were not a party to the line of credit, which was full recourse against Mr. Solensky.
| -58- |
As a result of discussions with the NYSE American in connection with our application to list our common shares, we restructured and replaced the line of credit. Accordingly, on October 17, 2017, we entered into a Loan Agreement, or the Working Capital Loan Agreement, with Equidebt pursuant to which Equidebt agreed to provide us with a five-year $5,000,000 unsecured working capital line of credit, or the working capital line of credit. Amounts borrowed under the working capital line of credit bear interest at a rate of 14% per annum payable at maturity. All amounts borrowed under the line of credit become due and payable on October 17, 2022. Upon the occurrence of an Event of Default (defined in the Working Capital Loan Agreement to include our failure to make payments under the working capital line of credit or our other indebtedness when due, the occurrence of certain insolvency events relating to us, Equidebt has the right to declare all amounts outstanding under the working capital line of credit immediately due and payable. The working capital line of credit is unsecured; however Mr. Solensky has personally guaranteed our obligations under the working capital line of credit. In connection with the establishment of the working capital line of credit, the line of credit provided by Equidebt to Mr. Solensky was cancelled without further liability or obligation of either party.
| Item 14. | Principal Accounting Fees and Services. |
The following table represents aggregate fees billed to the Company for the years ended December 31, 2017 and 2016 by MNP LLP, the Company’s independent registered public accounting firm.
| Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| (in thousands) | ||||||||
| Audit Fees | $ | 59,519 | $ | 60,020 | ||||
| Audit Related Fees | 72,831 | 70,661 | ||||||
| Tax Fees | 11,754 | 34,416 | ||||||
| All Other Fees | 21,279 | 20,809 | ||||||
| Total Fees | $ | 165,383 | $ | 185,906 | ||||
Audit Fees consist of fees for professional services and expenses relating to the audit of our annual financial statements, the audit of our internal control over financial reporting and the review of our quarterly financial information.
Audit Related Fees consist of fees for professional services and expenses reasonably relating to the audit of our annual financial statements and are not reported as Audit Fees.
Tax Fees are for tax-related services related primarily to tax compliance, tax advice and tax planning.
All Other Fees consist of fees for products and services which are not included in the previous three categories.
The Audit Committee pre-approves all auditing services and any non-audit services that the independent registered public accounting firm is permitted to render under Section 10A(h) of the Exchange Act.
The Audit Committee has considered whether the provision of non-audit services is compatible with maintaining the independence of MNP LLP, and has concluded that the provision of such services is compatible with maintain the indepence of our auditors. All such services were approved by the Audit Committee pursuant to Rule 2-01 of Regulation S-X under the Exchange Act to the extent that rule was applicable.
| Item 15. | Exhibits, Financial Statement Schedules. |
(a) The following documents are included in this Annual Report on Form 10-K
(1)-(2) Financial Statements
Index to Consolidated Financial Statements
| -59- |
| -60- |
| 101.INS | XBRL Instance Document | |
| 101.SCH | XBRL Taxonomy Extension Schema Document | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document |
| # | The registrant has received confidential treatment for certain portions of this exhibit. |
| ## | The registrant has sought confidential treatment with respect to certain portions of this exhibit. |
| + | Indicates management contract or compensatory plan. |
| * | Filed herewith. |
| ** | Furnished herewith. |
| Item 16. | Form 10-K Summary. |
None.
| -61- |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ZOMEDICA PHARMACEUTICALS CORP. | ||||
| By: | /s/ Gerald Solensky Jr. | |||
| Name: | Gerald Solensky Jr. | |||
| Title: | Chairman of the Board, President and Chief Executive Officer | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Gerald Solensky Jr. | ||||
| Gerald Solensky Jr. | Chairman of the Board, President, Chief Executive Officer | February 28, 2018 | ||
| (principal executive officer) | ||||
| /s/ Shameze Rampertab | ||||
| Shameze Rampertab | Chief Financial Officer, Corporate Secretary and Director | February 28, 2018 | ||
| (principal financial and accounting officer) | ||||
| /s/ James LeBar | ||||
| James LeBar | Director | February 28, 2018 | ||
| /s/ Rodney Williams | ||||
| Rodney Williams | Director | February 28, 2018 | ||
| /s/ Jeffrey Rowe | ||||
| Jeffrey Rowe | Director | February 28, 2018 | ||
| /s/ Thomas Robitaille | ||||
| Thomas Robitaille | Director | February 28, 2018 | ||
| /s/ Jane Eagleson | ||||
| Jane Eagleson | Director | February 28, 2018 | ||
| -62- |
Zomedica Pharmaceuticals Corp.
Consolidated financial statements
For the years ended December 31, 2017 and 2016
(Expressed in United States Dollars, except as otherwise noted)
Report of the Independent Registered Public Accounting Firm
To the Shareholders of Zomedica Pharmaceuticals Corp.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated financial statements of Zomedica Pharmaceuticals Corp. (the “Company”), which comprise the consolidated balance sheets as at December 31, 2017 and 2016, and the consolidated statements of operations and comprehensive loss, changes in equity and cash flows for the years then ended, and the related notes, comprising a summary of significant accounting policies and other explanatory information (collectively referred to as the consolidated financial statements).
In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as at December 31, 2017 and 2016, and its consolidated financial performance and its consolidated cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
Management’s Responsibility for the Consolidated Financial Statements
Management is responsible for the preparation and fair presentation of these consolidated financial statements in accordance with accounting principles generally accepted in the United States of America, and for such internal control as management determines is necessary to enable the preparation of consolidated financial statements that are free from material misstatement, whether due to error or fraud.
Auditor’s Responsibility
Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We conducted our audits in accordance with Canadian generally accepted auditing standards and the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free from material misstatement, whether due to error or fraud. Those standards also require that we comply with ethical requirements, including independence. We are required to be independent with respect to the Company in accordance with the ethical requirements that are relevant to our audits of the consolidated financial statements in Canada, the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. We are a public accounting firm registered with the PCAOB.
An audit includes performing procedures to assess the risks of material misstatements of the consolidated financial statements, whether due to error or fraud, and performing procedures to respond to those risks. Such procedures include obtaining and examining, on a test basis, audit evidence regarding the amounts and disclosures in the consolidated financial statements. The procedures selected depend on our judgment, including the assessment of the risks of material misstatement of the consolidated financial statements, whether due to error or fraud. In making those risk assessments, we consider internal control relevant to the entity’s preparation and fair preparation of the consolidated financial statements in order to design audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the entity’s internal control. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Accordingly, we express no such opinion.
An audit also includes evaluating the appropriateness of accounting policies and principles used and the reasonableness of accounting estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements.
We believe that the audit evidence we have obtained in our audits is sufficient and appropriate to provide a reasonable basis for our audit opinion.
| F-2 |
| /s/ MNP LLP |
| We have served as the Company's auditor since 2015. |
| February 28, 2018 | Chartered Professional Accountants | |
| Toronto, Ontario | Licensed Public Accountants |
| F-3 |
Zomedica Pharmaceuticals Corp.
Consolidated Balance Sheets
As at December 31, 2017 and 2016
(Stated in United States dollars)
| Note | December 31, 2017 | December 31, 2016 | ||||||||
| Assets | ||||||||||
| Current assets: | ||||||||||
| Cash and cash equivalents | $ | 3,448,147 | $ | 3,226,680 | ||||||
| Prepaid expenses and deposits | 5 | 786,273 | 332,611 | |||||||
| Trade and other receivable | 28,272 | 18,921 | ||||||||
| 4,262,692 | 3,578,212 | |||||||||
| Prepaid expenses and deposits | 5 | 566,832 | 690,374 | |||||||
| Property and equipment | 6 | 371,157 | 289,034 | |||||||
| Intangible assets | 7 | 15,141 | 17,938 | |||||||
| $ | 5,215,822 | $ | 4,575,558 | |||||||
| Liabilities and shareholders' equity | ||||||||||
| Current liabilities: | ||||||||||
| Accounts payable and accrued liabilities | $ | 828,737 | $ | 734,431 | ||||||
| Shareholder loans payable | 18 | - | 6,726 | |||||||
| 828,737 | 741,157 | |||||||||
| Shareholders' equity: | ||||||||||
| Capital stock | ||||||||||
| Authorized | ||||||||||
| Unlimited common shares without par value | ||||||||||
| Issued and outstanding | ||||||||||
| 90,225,869 common shares (2016 - 83,964,569) | 9 | 18,244,659 | 10,189,973 | |||||||
| Additional paid-in capital | 10 | 1,768,526 | 1,205,456 | |||||||
| Accumulated deficit | (15,626,100 | ) | (7,561,028 | ) | ||||||
| 4,387,085 | 3,834,401 | |||||||||
| $ | 5,215,822 | $ | 4,575,558 | |||||||
Signed on behalf of the Board:
| “Gerald Solensky” | “Jeff Rowe” | |
| Chairman of the Board | Director |
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
Zomedica Pharmaceuticals Corp.
Consolidated statements of operations and comprehensive loss
For the years ended December 31, 2017 and 2016
(Stated in United States dollars)
| Note | December 31, 2017 | December 31, 2016 | ||||||||
| Expenses: | ||||||||||
| Research and development | 15 | $ | 2,751,326 | $ | 1,518,589 | |||||
| General and administrative | 15 | 3,946,270 | 2,916,604 | |||||||
| Professional fees | 15 | 1,294,044 | 1,245,182 | |||||||
| Amortization | 7 | 2,797 | 2,690 | |||||||
| Depreciation | 6 | 89,613 | 43,131 | |||||||
| Loss from operations | 8,084,050 | 5,726,196 | ||||||||
| Gain on settlement of liabilities | (5,000 | ) | - | |||||||
| Foreign exchange (gain) loss | (13,978 | ) | 14,296 | |||||||
| Loss before income taxes | 8,065,072 | 5,740,492 | ||||||||
| Income tax expense | 11 | - | - | |||||||
| Net loss and comprehensive loss | $ | 8,065,072 | $ | 5,740,492 | ||||||
| Weighted average number of common shares - basic and diluted | 87,400,255 | 80,158,312 | ||||||||
| Loss per share - basic and diluted | $ | (0.09 | ) | $ | (0.07 | ) | ||||
Nature of operations and going concern (Note 1)
Commitments and contingencies (Note 12)
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
Zomedica Pharmaceuticals Corp.
Consolidated statements of shareholders’ equity
For the years ended December 31, 2017 and 2016
(Stated in United States dollars)
| Note | Number of common stock | Capital stock | Additional paid-in capital | Accumulated deficit | Total | |||||||||||||||||
| Balance at December 31, 2015 | 77,370,716 | $ | 5,214,691 | $ | 19,890 | $ | (1,820,536 | ) | $ | 3,414,045 | ||||||||||||
| Stock issuance due to recapitalization, net of cost | 19 | 1,900,000 | 196,534 | - | - | 196,534 | ||||||||||||||||
| Stock issuance for financing, net of cost | 9 | 4,133,853 | 4,717,570 | - | - | 4,717,570 | ||||||||||||||||
| Stock issuance for services | 9 | 80,000 | 15,741 | - | - | 15,741 | ||||||||||||||||
| Excess of purchase price over net asset value | 19 | - | - | (272,354 | ) | - | (272,354 | ) | ||||||||||||||
| Stock-based compensation | 10 | - | - | 1,467,934 | - | 1,467,934 | ||||||||||||||||
| Stock issued due to exercise of options | 10 | 480,000 | 45,437 | (10,014 | ) | - | 35,423 | |||||||||||||||
| Net loss | - | - | - | (5,740,492 | ) | (5,740,492 | ) | |||||||||||||||
| Balance at December 31, 2016 | 83,964,569 | $ | 10,189,973 | $ | 1,205,456 | $ | (7,561,028 | ) | $ | 3,834,401 | ||||||||||||
| Stock issuance for services | 155,927 | 275,131 | - | - | 275,131 | |||||||||||||||||
| Stock-based compensation | 10 | - | - | 849,679 | - | 849,679 | ||||||||||||||||
| Stock issuance for financing, net of cost | 9 | 4,405,373 | 6,513,424 | - | - | 6,513,424 | ||||||||||||||||
| Stock issued due to exercise of options | 10 | 1,700,000 | 1,266,131 | (286,609 | ) | - | 979,522 | |||||||||||||||
| Net loss | - | - | - | (8,065,072 | ) | (8,065,072 | ) | |||||||||||||||
| Balance at December 31, 2017 | 90,225,869 | $ | 18,244,659 | $ | 1,768,526 | $ | (15,626,100 | ) | $ | 4,387,085 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
Zomedica Pharmaceuticals Corp.
Consolidated statements of cash flows
For the years ended December 31, 2017 and 2016
(Stated in United States dollars)
| Note | 2017 | 2016 | ||||||||
| Cash flows used in operating activities: | ||||||||||
| Net loss | $ | (8,065,072 | ) | $ | (5,740,492 | ) | ||||
| Adjustments for | ||||||||||
| Depreciation | 6 | 89,613 | 43,131 | |||||||
| Amortization | 7 | 2,797 | 2,690 | |||||||
| Stock issued for services | 9 | 275,131 | 15,741 | |||||||
| Stock-based compensation | 10 | 849,679 | 1,467,934 | |||||||
| Change in non-cash operating working capital | ||||||||||
| Trade and other receivable | (9,351 | ) | (21,031 | ) | ||||||
| Prepaid expenses | (205,143 | ) | 7,393 | |||||||
| Deposits | (124,977 | ) | (890,142 | ) | ||||||
| Accounts payable and accrued liabilities | 94,306 | 552,608 | ||||||||
| (7,093,017 | ) | (4,562,168 | ) | |||||||
| Cash flows from financing activities: | ||||||||||
| Cash proceeds from financing | 9 | 6,570,000 | 4,755,586 | |||||||
| Cash paid on stock issuance costs | (56,576 | ) | (115,635 | ) | ||||||
| Cash received from stock option exercises | 979,522 | 35,423 | ||||||||
| Cash received on amalgamation | 19 | - | 108,966 | |||||||
| Repayments (advances) of shareholder loan | (6,726 | ) | 2,013 | |||||||
| 7,486,220 | 4,786,353 | |||||||||
| Cash flows used in investing activities: | ||||||||||
| Investment in intangibles | 7 | - | (9,611 | ) | ||||||
| Investment in property and equipment | 6 | (171,736 | ) | (231,604 | ) | |||||
| (171,736 | ) | (241,215 | ) | |||||||
| Increase (decrease) in cash and cash equivalents during the year | 221,467 | (17,030 | ) | |||||||
| Cash and cash equivalents, beginning of year | 3,226,680 | 3,243,710 | ||||||||
| Cash and cash equivalents, end of year | $ | 3,448,147 | $ | 3,226,680 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-7 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
1. Nature of operations and going concern
Zomedica Pharmaceuticals Corp. ("Zomedica" or the “Company”) was incorporated on January 7, 2013 under the Business Corporations Act (Alberta) as Wise Oakwood Ventures Inc. (“WOW”) and was classified as a capital pool company, as defined in Policy 2.4 of the TSX Venture Exchange. ZoMedica Pharmaceuticals Inc. was incorporated on May 14, 2015 under the Canada Business Corporations Act.
On April 21, 2016, the Company closed its qualifying transaction (“Transaction”), consisting of the acquisition of ZoMedica Pharmaceuticals Inc. (“ZoMedica”) pursuant to a three-cornered amalgamation, whereby ZoMedica was amalgamated with 9674128 Canada Inc. (which was wholly-owned by WOW) and common shares and options of the Company were issued to former holders of ZoMedica securities as consideration. The amalgamated company changed its name to Zomedica Pharmaceuticals Ltd. and WOW subsequently changed its name to Zomedica Pharmaceuticals Corp. Prior to completion of the Transaction, WOW consolidated its common shares on the basis of the one post-consolidation common share for every 2.5 pre-consolidation common shares. The Transaction constituted WOW’s qualifying transaction under TSX Venture Exchange Policy 2.4 – Capital Pool Companies. The shares of Zomedica Pharmaceuticals Corp. began trading on the TSX Venture Exchange under the new symbol “ZOM” on Monday, May 2, 2016. On June 21, 2016, the Company filed Articles of Amalgamation and vertically amalgamated with its wholly-owned subsidiary, Zomedica Pharmaceuticals Ltd.
Zomedica has one corporate subsidiary, Zomedica Pharmaceuticals, Inc., a Delaware company whose results and operations are included in these consolidated financial statements. The Company is a biopharmaceutical company targeting health and wellness solutions for the companion pet through a ground-breaking approach that focuses on the needs of the veterinarians themselves. Zomedica's head office is located at 100 Phoenix Drive, Suite 190, Ann Arbor, MI 48108 and its registered office is located at Suite 1250, 639 – 5th Avenue S.W., Calgary, Alberta T2P 0M9.
On November 20, 2017, Zomedica announced that its registration statement on Form S-1 was declared effective by the U.S. Securities and Exchange Commission (SEC) and on November 21, 2017, the Company’s common shares began trading on the NYSE under the symbol “ZOM”.
Going concern
The consolidated financial statements are prepared on a going concern basis, which assumes that the Company will be able to meet its obligations and continue its operations for the next twelve months.
| 2. | Basis of preparation |
The accounting policies set out below have been applied consistently in the consolidated financial statements.
Basis of consolidation
These consolidated financial statements include the accounts of the Company and its wholly owned operating subsidiary, ZoMedica Pharmaceuticals Inc.
All inter-company accounts and transactions have been eliminated on consolidation.
| F-8 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 3. | Significant accounting policies |
Use of estimates
The preparation of the consolidated financial statements in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the period. Actual results could differ from those estimates.
Areas where significant judgment is involved in making estimates are: the fair values of financial assets and liabilities; the determination of fair value of stock-based compensation; the useful lives of property and equipment; and forecasting future cash flows for assessing the going concern assumption.
Basis of measurement
The consolidated financial statements have been prepared on the historical cost basis except as otherwise noted.
Functional and reporting currencies
The Company’s and subsidiary’s functional currency, as determined by management, is US dollars, which is also the Company’s reporting currency.
The accounting policies set out below have been applied consistently to all periods and companies presented in the consolidated financial statements.
Cash and cash equivalents
The Company considers all highly liquid securities with an original maturity of three months or less to be cash equivalents. Cash and cash equivalents comprises cash on hand and cash held in trust related to share issuances. The cash held in trust is readily available to the Company and is classified as current.
The financial risks associated with these instruments are minimal and the Company has not experienced any losses from investments in these securities. The carrying amount of cash and cash equivalents approximates its fair value due to its short-term nature.
Property and equipment
Property and equipment are carried at historical cost less accumulated depreciation and any accumulated impairment losses. Each component of an item of property and equipment with a cost that is significant in relation to the total cost of the item is depreciated separately. Maintenance and repair expenditures that do not improve or extend the life are expensed in the period incurred.
Depreciation is recognized so as to write off the cost or valuation of assets (other than land) less their residual values over their useful lives, using the straight-line method. The estimated useful lives, residual values and depreciation methods are reviewed at the end of each year, with the effect of any changes in estimate accounted for on a prospective basis.
| F-9 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 3. | Significant accounting policies (continued) |
Property and equipment (continued)
An item of property and equipment is derecognized upon disposal or when no future economic benefits are expected to arise from the continued use of the asset. Any gain or loss arising on the disposal or retirement of an item of property, plant and equipment is determined as the difference between the sales proceeds and the carrying amount of the asset and is recognized in profit or loss.
Estimated useful lives for the principal asset categories are as follows:
| Computer equipment (years) | 3 | ||
| Furniture and equipment (years) | 5 | - | 7 |
| Laboratory equipment (years) | 5 | - | 7 |
| Leasehold improvements | Over shorter of estimated useful life and lease term |
Impairment of long-lived assets
Long-lived assets are reviewed for impairment when events or circumstances indicate that the carrying value of an asset may not be recoverable. For assets that are to be held and used, impairment is recognized when the sum of estimated undiscounted cash flows associated with the asset or group of assets is less than its carrying value. If impairment exists, an adjustment is made to write the asset down to its fair value, and a loss is recorded as the difference between the carrying value and fair value.
Research and development
Research and development costs related to continued research and development programs are expensed as incurred in accordance with ASC topic 730.
Share issue costs
Share issue costs are recorded as a reduction of the proceeds from the issuance of capital stock.
Translation of foreign currencies
In respect of other transactions denominated in currencies other than the Company and its wholly owned operating subsidiaries’ functional currencies, the monetary assets and liabilities are translated at the period end rates. Revenue and expenses are translated at rates of exchange prevailing on the transaction dates. All of the exchange gains or losses resulting from these other transactions are recognized in the consolidated statements of operations and comprehensive loss.
Stock-based compensation
The Company measures the cost of equity-settled transactions by reference to the fair value of the equity instruments at the date at which they are granted if the fair value of the goods or services received by the Company cannot be reliably estimated.
| F-10 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 3. | Significant accounting policies (continued) |
Stock-based compensation (continued)
The Company calculates stock-based compensation using the fair value method, under which the fair value of the options at the grant date is calculated using the Black-Scholes Option Pricing Model, and subsequently expensed over the vesting period of the option. The provisions of the Company's stock-based compensation plans do not require the Company to settle any options by transferring cash or other assets, and therefore the Company classifies the awards as equity. Stock-based compensation expense recognized during the period is based on the value of stock-based payment awards that are ultimately expected to vest.
The Company estimates forfeitures at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
Loss per share
Basic loss per share (“EPS”) is computed by dividing the loss attributable to common shareholders by the weighted average number of common shares outstanding. Diluted EPS reflects the potential dilution that could occur from common shares issuable through the exercise or conversion of stock options, restricted stock awards, warrants and convertible securities. In certain circumstances, the conversion of options are excluded from diluted EPS if the effect of such inclusion would be anti-dilutive.
The dilutive effect of stock options is determined using the treasury stock method. Stock options to purchase common shares of the Company during fiscal 2017 and 2016 were not included in the computation of diluted EPS because the Company has incurred a loss for the year ended December 31, 2017 and 2016 as the effect would be anti-dilutive.
Comprehensive loss
The Company follows ASC topic 220. This statement establishes standards for reporting and display of comprehensive (loss) income and its components. Comprehensive loss is net loss plus certain items that are recorded directly to shareholders' equity. The Company has no other comprehensive loss items.
Intangible assets
Intangible assets with finite useful lives that are acquired separately are carried at cost less accumulated amortization and accumulated impairment losses. Amortization is recognized on a straight-line basis over their estimated useful lives. The estimated useful lives and amortization methods are reviewed at the end of each year, with the effect of any changes in estimate being accounted for on a prospective basis. Intangible assets with indefinite useful lives that are acquired separately are carried at cost less accumulated impairment losses.
| Computer software (years) | 3 |
| Trademarks (years) | 15 |
| F-11 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 3. | Significant accounting policies (continued) |
Fair value measurement
Under ASC topic 820, fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date (i.e., an exit price). ASC topic 820 establishes a hierarchy for inputs to valuation techniques used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs that reflect assumptions market participants would use in pricing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company's own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. There are three levels to the hierarchy based on the reliability of inputs, as follows:
| l | Level 1 - Observable inputs that reflect quoted prices (unadjusted) for identical assets or liabilities in active markets. |
| l | Level 2 - Inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 inputs include quoted prices for similar assets or liabilities in active markets, or quoted prices for identical or similar assets and liabilities in markets that are not active. |
| l | Level 3 - Unobservable inputs for the asset or liability. |
The degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3.
Income taxes
The Company accounts for income taxes in accordance with Accounting Standard Codification 740, Income Taxes ("ASC 740"), on a tax jurisdictional basis. The Company files income tax returns in the United States and its subsidiary files income tax returns in Canada and the Province of Ontario.
Deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the tax bases of assets and liabilities and their financial statement reported amounts using enacted tax rates and laws in effect in the year in which the differences are expected to reverse. A valuation allowance is provided against deferred tax assets when it is determined to be more likely than not that the deferred tax asset will not be realized.
The Company assesses the likelihood of the financial statement effect of a tax position that should be recognized when it is more likely than not that the position will be sustained upon examination by a taxing authority based on the technical merits of the tax position, circumstances, and information available as of the reporting date. The Company is subject to examination by taxing authorities in jurisdictions such as the United States and Canada. Management does not believe that there are any uncertain tax positions that would result in an asset or liability for taxes being recognized in the accompanying consolidated financial statements. The Company recognizes tax-related interest and penalties, if any, as a component of income tax expense.
ASC 740 prescribes recognition threshold and measurement attributes for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. ASC 740 also provides guidance on de-recognition, classification, interest and penalties, accounting in periods, disclosure and transition. At December 31, 2017 and 2016, the Company has not taken any tax positions that would require disclosure under ASC 740.
| F-12 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 3. | Significant accounting policies (continued) |
Segmented reporting
The Company currently operates as a single segment. Its principal business relates to the discovery, development and commercialization of innovative pharmaceuticals for the companion pet.
Future accounting pronouncements
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers, requiring an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The updated standard will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective. In March 2016, the FASB issued ASU No. 2016-08 to clarify the implementation guidance on considerations of whether an entity is a principal or an agent, impacting whether an entity reports revenue on a gross or net basis. In April 2016, the FASB issued ASU No. 2016-10 to clarify guidance on identifying performance obligations and the implementation guidance on licensing. In May 2016, the FASB issued amendments ASU No. 2016-11 and 2016-12 to amend certain aspects of the new revenue guidance (including transition, collectability, noncash consideration and the presentation of sales and other similar taxes) and provided certain practical expedients. The guidance is effective for annual reporting periods beginning after December 15, 2017 (including interim reporting periods). Early adoption is permitted but not before the annual reporting period (and interim reporting period) beginning January 1, 2017. Entities have the option of using either a full retrospective or a modified approach to adopt the guidance. The Company is in the process of evaluating the amendments to determine if they have a material impact on the Company’s financial position, results of operations, cash flows or disclosures.
In January 2016, the FASB issued ASU No. 2016-01, which makes limited amendments to the guidance in U.S. GAAP on the classification and measurement of financial instruments. The new standard significantly revises an entity’s accounting related to (1) the classification and measurement of investments in equity securities and (2) the presentation of certain fair value changes for financial liabilities measured at fair value. It also amends certain disclosure requirements associated with the fair value of financial instruments. ASU No. 2016-01 is effective for fiscal years beginning after December 15, 2017, and interim periods within those annual periods. The Company is in the process of evaluating the amendments to determine if they have a material impact on the Company’s financial position, results of operations, cash flows or disclosures.
In February 2016, the FASB issued new guidance, ASU No. 2016-02, Leases (Topic 842). The main difference between current U.S. GAAP and the new guidance is the recognition of lease liabilities based on the present value of remaining lease payments and corresponding lease assets for operating leases under current U.S. GAAP with limited exception. Additional qualitative and quantitative disclosures are also required by the new guidance. Topic 842 is effective for annual reporting periods (including interim reporting periods) beginning after December 15, 2018. Early adoption is permitted. The Company is in the process of evaluating the amendments to determine if they have a material impact on the Company’s financial position, results of operations, cash flows or disclosures.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230) Classification of Certain Cash Receipts and Cash Payments, which will make eight targeted changes to how cash receipts and cash payments are presented and classified in the Statement of Cash Flows. ASU 2016-15 will be effective on May 1, 2018 and will require adoption on a retrospective basis unless it is impracticable to apply, in which case the Company would be required to apply the amendments prospectively as of the earliest date practicable. The Company is in the process of evaluating the amendments to determine if they have a material impact on the Company’s financial position, results of operations, cash flows or disclosures.
| F-13 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 3. | Significant accounting policies (continued) |
Future accounting pronouncements (continued)
In August 2016, the FASB issued ASU 2017-01 that changes the definition of a business to assist entities with evaluating when a set of transferred assets and activities is a business. The guidance requires an entity to evaluate if substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or a group of similar identifiable assets; if so, the set of transferred assets and activities is not a business. ASU 2017-01 also requires a business to include at least one substantive process and narrows the definition of outputs by more closely aligning it with how outputs are described in ASC 606.1. ASU 2017-01 is effective for public business entities for fiscal years beginning after December 15, 2017, and interim periods within those years. Early adoption is permitted. The Company is in the process of evaluating the amendments to determine if they have a material impact on the Company’s financial position, results of operations, cash flows or disclosures.
In May 2017, FASB issued ASU 2017-09 in relation to Compensation —Stock Compensation (Topic 718), Modification Accounting. The amendments provide guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. The amendments are effective for all entities for annual periods, and interim periods within those annual periods, beginning after December 15, 2017. Early adoption is permitted, including adoption in any interim period, for (1) public business entities for reporting periods for which financial statements have not yet been issued and (2) all other entities for reporting periods for which financial statements have not yet been made available for issuance. The amendments should be applied prospectively to an award modified on or after the adoption date. The Company is in the process of evaluating the amendments to determine if they have a material impact on the Company’s financial position, results of operations, cash flows or disclosures.
| 4. | Critical accounting judgments and key sources of estimation uncertainty |
The preparation of financial statements requires management to make judgments, estimates and assumptions that affect the application of policies and reported amounts of assets and liabilities, and revenue and expenses. The estimates and associated assumptions are based on historical experience and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis of making the judgments about carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
The estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period or in the period of the revision and further periods if the review affects both current and future periods.
Critical areas of estimation and judgements in applying accounting policies include the following:
Going concern
These consolidated financial statements have been prepared in accordance with U.S GAAP on a going concern basis, which assumes the realization of assets and discharge of liabilities in the normal course of business within the foreseeable future. Management uses judgment in determining assumptions for cash flow projections, such as anticipated financing, anticipated sales and future commitments to assess the Company’s ability to continue as a going concern. A critical judgment is that the Company continues to raise funds going forward and satisfy their obligations as they become due.
| F-14 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 4. | Critical accounting judgments and key sources of estimation uncertainty (continued) |
Useful lives of property and equipment
As described in Note 3 above, the Company reviews the estimated useful lives of property and equipment with definite useful lives at the end of each year and assesses whether the useful lives of certain items should be shortened or extended, due to various factors including technology, competition and revised service offerings. During the year ended December 31, 2017 and 2016, the Company was not required to adjust the useful lives of any assets based on the factors described above.
Deferred income taxes
The calculation of deferred income taxes is based on assumptions which are subject to uncertainty as to timing and which tax rates are expected to apply when temporary differences reverse. Deferred tax recorded is also subject to uncertainty regarding the magnitude of non-capital losses available for carry forward and of the balances in various tax pools. By their nature, these estimates are subject to measurement uncertainty, and the effect on the financial statements from changes in such estimates in future period could be material. Deferred tax assets are recognized to the extent that it is probable that they will be able to be utilized against future taxable income. Deferred tax assets are reviewed at each balance sheet date and adjusted to the extent that it is no longer probable that the related tax benefit will be realized.
Stock-based payments
The Company estimates the fair value of convertible securities such as options using the Black-Scholes option-pricing model which requires significant estimation around assumptions and inputs such as expected term to maturity, expected volatility and expected dividends.
| 5. | Prepaid expenses and deposits |
The Company entered into a lease agreement with Wickfield Phoenix LLC effective on August 23, 2016. The Company prepaid the full outstanding balance of $801,973 on August 26, 2016 and recorded the prepaid rent due within a year as current. As at December 31, 2017, the Company has classified $155,220 as a current asset in the consolidated balance sheet (December 31, 2016 - $160,395).
| F-15 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 6. | Property and equipment |
| Computer equipment | Furniture and equipment | Laboratory equipment | Leasehold improvements | Total | ||||||||||||||||
| Cost | ||||||||||||||||||||
| Balance at December 31, 2015 | $ | 51,795 | $ | 7,364 | $ | 32,665 | $ | 14,735 | $ | 106,559 | ||||||||||
| Additions | 9,803 | - | 210,864 | 10,937 | 231,604 | |||||||||||||||
| Balance at December 31, 2016 | 61,598 | 7,364 | 243,529 | 25,672 | 338,163 | |||||||||||||||
| Additions | 89,557 | 68,694 | 2,200 | 11,285 | 171,736 | |||||||||||||||
| Balance at December 31, 2017 | 151,155 | 76,058 | 245,729 | 36,957 | 509,899 | |||||||||||||||
| Accumulated depreciation | ||||||||||||||||||||
| Balance at December 31, 2015 | 3,163 | 438 | 1,578 | 819 | 5,998 | |||||||||||||||
| Depreciation | 10,695 | 1,052 | 28,205 | 3,179 | 43,131 | |||||||||||||||
| Balance at December 31, 2016 | 13,858 | 1,490 | 29,783 | 3,998 | 49,129 | |||||||||||||||
| Depreciation | 28,944 | 10,355 | 45,092 | 5,222 | 89,613 | |||||||||||||||
| Balance at December 31, 2017 | 42,802 | 11,845 | 74,875 | 9,220 | 138,742 | |||||||||||||||
| Net book value as at: | ||||||||||||||||||||
| December 31, 2016 | $ | 47,740 | $ | 5,874 | $ | 213,746 | $ | 21,674 | $ | 289,034 | ||||||||||
| December 31, 2017 | $ | 108,353 | $ | 64,213 | $ | 170,854 | $ | 27,737 | $ | 371,157 | ||||||||||
| 7. | Intangible assets |
| Computer software | Trademarks | Total | ||||||||||
| Cost | ||||||||||||
| Balance at December 31, 2015 | $ | 5,143 | $ | 6,625 | $ | 11,768 | ||||||
| Additions | - | 9,611 | 9,611 | |||||||||
| Balance at December 31, 2016 | 5,143 | 16,236 | 21,379 | |||||||||
| Additions | - | - | - | |||||||||
| Balance at December 31, 2017 | 5,143 | 16,236 | 21,379 | |||||||||
| Accumulated amortization | ||||||||||||
| Balance at December 31, 2015 | 714 | 37 | 751 | |||||||||
| Amortization | 1,714 | 976 | 2,690 | |||||||||
| Balance at December 31, 2016 | 2,428 | 1,013 | 3,441 | |||||||||
| Amortization | 1,715 | 1,082 | 2,797 | |||||||||
| Balance at December 31, 2017 | 4,143 | 2,095 | 6,238 | |||||||||
| Net book value as at: | ||||||||||||
| December 31, 2016 | $ | 2,715 | $ | 15,223 | $ | 17,938 | ||||||
| December 31, 2017 | $ | 1,000 | $ | 14,141 | $ | 15,141 | ||||||
| F-16 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 8. | Loan Arrangements |
On October 18, 2017, the Company entered into a loan arrangement with a shareholder of the Company, pursuant to which such shareholder has agreed to provide a loan facility to the Company, whereby the Company may borrow up to $5,000,000, with the proceeds to be used for working capital and general corporate purposes. The term of the loan facility is five (5) years, with principal and interest payments being due only at the time of maturity. Under the loan agreement, the Company may borrow in one or more advances, provided however that a minimum amount of $250,000 must be borrowed at any one time and not more than two advances may occur per month. Interest shall accrue at a rate of fourteen percent (14%) per annum, payable upon maturity. As of December 31, 2017, no amounts have been borrowed.
| 9. | Capital stock |
The Company is authorized to issue an unlimited number of common stock, all without par value.
Issued and outstanding common stock:
| Number of common stock | Capital stock | |||||||
| Balance at December 31, 2015 | 77,370,716 | $ | 5,214,691 | |||||
| Stock issued to effect the recapitalization (Note 19) | 1,900,000 | 196,534 | ||||||
| Stock issued due to option exercises related to amalgamation (Note 10) | 80,000 | 22,058 | ||||||
| Stock issued to Everfront Capital Corp | 80,000 | 15,741 | ||||||
| Stock issued for financing (i and ii) | 4,133,853 | 4,717,570 | ||||||
| Stock issued due to exercise of options (Note 10) | 400,000 | 23,379 | ||||||
| Balance at December 31, 2016 | 83,964,569 | 10,189,973 | ||||||
| Stock issuance for services (iii and iv) | 155,927 | 275,131 | ||||||
| Stock issued from financing (v and vi) | 4,405,373 | 6,513,424 | ||||||
| Stock issued due to exercise of options (Note 10) | 1,700,000 | 1,266,131 | ||||||
| Balance at December 31, 2017 | 90,225,869 | $ | 18,244,659 | |||||
| i) | On August 25, 2016, the Company issued 3,342,480 common shares for gross proceeds of $3,875,500. The Company recorded $29,310 of share issuance costs as an offset to capital stock. |
| ii) | On December 29, 2016, the Company issued 791,373 common shares for gross proceeds of $880,086. The Company recorded $8,706 of share issuance costs as an offset to capital stock. |
| iii) | On March 14, 2017, the Company settled $50,000 of amounts due to a vendor by issuing 43,613 common shares valued at $45,000 at the date of issuance. The Company recorded a $5,000 gain on the settlement of liabilities. |
| iv) | On December 22, 2017, the Company issued 112,314 common shares in accordance with a License and Supply Agreement with Celsee, Inc. and recognized $230,131 as a research and development expense in the consolidated statements of operations and comprehensive loss. |
| v) | On April 7, 2017, the Company issued 2,902,682 common shares for gross proceeds of $3,250,000. The Company recorded $32,754 of share issuance costs as an offset to capital stock. |
| vi) | On July 28, 2017, the Company issued 1,502,691 common shares for gross proceeds of $3,320,000. The Company recorded $23,822 of share issuance costs as an offset to capital stock. |
| F-17 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 10. | Stock-based compensation |
During the year ended December 31, 2017, the Company issued 1,815,000 stock options, each option entitling the holder to purchase one common share of the Company. During the year ended December 31, 2017, an aggregate of 1,700,000 options were exercised.
During the year ended December 31, 2016, the Company issued 7,375,000 stock options, each option entitling the holder to purchase one common share of the Company. The Company also had 80,000 options issued as part of the qualifying transaction as disclosed in Note 19. These options were exercised immediately after the close of the qualifying transaction on April 21, 2016. During the year ended December 31, 2016, 400,000 of additional options were exercised.
The continuity of stock options are as follows:
| Number of Options | Weighted Avg Exercise Price (CDN$) | |||||||
| Balance at December 31, 2015 | 1,000,000 | $ | 0.05 | |||||
| Options issued | 3,500,000 | $ | 0.25 | |||||
| Options issued through amalgamation | 80,000 | $ | 0.25 | |||||
| Options exercised on April 21, 2016 | (80,000 | ) | $ | 0.25 | ||||
| Options exercised on August 15, 2016 | (400,000 | ) | $ | 0.05 | ||||
| Options issued on December 21, 2016 | 3,875,000 | $ | 1.50 | |||||
| Balance at December 31, 2016 | 7,975,000 | $ | 0.84 | |||||
| Stock options exercised on February 21, 2017 | (10,000 | ) | $ | 0.25 | ||||
| Stock options exercised on February 21, 2017 | (400,000 | ) | $ | 0.05 | ||||
| Options issued on February 24, 2017 | 535,000 | $ | 1.50 | |||||
| Stock options exercised on May 8, 2017 | (7,060 | ) | $ | 1.50 | ||||
| Stock options cancelled on May 17, 2017 | (10,000 | ) | $ | 1.50 | ||||
| Stock options exercised on May 23, 2017 | (80,000 | ) | $ | 0.25 | ||||
| Stock options exercised on July 6, 2017 | (200,000 | ) | $ | 0.05 | ||||
| Stock options exercised on July 17, 2017 | (220,000 | ) | $ | 0.25 | ||||
| Options issued on August 14, 2017 | 1,280,000 | $ | 2.75 | |||||
| Stock options exercised on August 29, 2017 | (7,940 | ) | $ | 1.50 | ||||
| Stock options exercised on December 19, 2017 | (25,000 | ) | $ | 0.25 | ||||
| Stock options exercised on December 19, 2017 | (750,000 | ) | $ | 1.50 | ||||
| Balance at December 31, 2017 | 8,080,000 | $ | 1.21 | |||||
As at December 31, 2017, details of the issued and outstanding stock options are as follows:
| Grant date | Exercise price (CDN$) | Number of options issued and outstanding | Number of vested options outstanding | Weighted Avg Remaining Life (years) | ||||||||||||
| March 28, 2016 | $ | 0.25 | 3,165,000 | 3,165,000 | 0.24 | |||||||||||
| December 21, 2016 | $ | 1.50 | 3,100,000 | 3,100,000 | 0.97 | |||||||||||
| February 24, 2017 | $ | 1.50 | 535,000 | 535,000 | 1.15 | |||||||||||
| August 14, 2017 | $ | 2.75 | 1,205,000 | 1,205,000 | 1.62 | |||||||||||
| August 14, 2017 | $ | 2.75 | 75,000 | 37,500 | 0.62 | |||||||||||
| F-18 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 10. | Stock-based compensation (continued) |
The fair value of options granted during the year ended December 31, 2017 was estimated using the Black-Scholes option pricing model to determine the fair value of options granted using the following assumptions:
| March 28, 2016 | April 21, 2016 | December 21, 2016 | ||||||||||
| Volatility | 63 | % | 63 | % | 58 | % | ||||||
| Risk-free interest rate | 0.56 | % | 1.12 | % | 0.81 | % | ||||||
| Expected life (years) | 2.06 | 1 | 2 | |||||||||
| Dividend yield | 0 | % | 0 | % | 0 | % | ||||||
| Common share price | CDN $0.20 | CDN $0.20 | CDN $1.45 | |||||||||
| Strike price | CDN $0.25 | CDN $0.25 | CDN $1.50 | |||||||||
| Forfeiture rate | nil | nil | nil | |||||||||
| February 24, 2017 | August 14, 2017 | August 14, 2017 | ||||||||||
| Volatility | 59 | % | 59 | % | 83 | % | ||||||
| Risk-free interest rate | 0.81 | % | 1.22 | % | 1.22 | % | ||||||
| Expected life (years) | 2 | 2 | 1 | |||||||||
| Dividend yield | 0 | % | 0 | % | 0 | % | ||||||
| Common share price | CDN $1.35 | CDN $2.40 | CDN $2.40 | |||||||||
| Strike price | CDN $1.50 | CDN $2.75 | CDN $2.75 | |||||||||
| Forfeiture rate | nil | nil | nil | |||||||||
The Company recorded $849,679 of stock-based compensation for the year ended December 31, 2017 and $1,467,934 of stock-based compensation for the year ended December 31, 2016. During the year ended December 31, 2017, the Company recorded the cash receipt of $979,522 as capital stock and reclassified $286,609 of stock-based compensation to capital stock due to the exercise of 1,700,000 options disclosed above. During the year ended December 31, 2016, the Company recorded the cash receipt of $35,423 as capital stock and reclassified $10,014 of stock-based compensation to capital stock due to the exercise of 480,000 options disclosed above.
Volatility is determined based on volatilities of comparable companies when the Company does not have its own sufficient trading history. The expected term, which represents the period of time that options granted are expected to be outstanding, is estimated based on an average of the term of the options.
The risk-free rate assumed in valuing the options is based on the Canadian treasury yield curve in effect at the time of grant for the expected term of the option. The expected dividend yield percentage at the date of grant is Nil as the Company is not expected to pay dividends in the foreseeable future. The Company has estimated its stock option forfeitures to be Nil for the year ended December 31, 2017 and 2016.
| F-19 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 11. | Income taxes |
The reconciliation of the combined Canadian federal and provincial statutory income tax rate of 27% (2016- 27%) to the effective tax rate is as follows:
| For the year ended December 31, 2017 | For the year ended December 31, 2016 | |||||||
| Loss before income taxes | $ | (8,065,072 | ) | $ | (5,740,492 | ) | ||
| Expected income tax expense (recovery) | (2,177,570 | ) | (1,549,930 | ) | ||||
| Difference in foreign tax rates | (297,460 | ) | (162,210 | ) | ||||
| Tax rate changes and other adjustments | 808,260 | (43,960 | ) | |||||
| Stock based compensation and non-deductible expenses | 236,350 | 398,930 | ||||||
| Change in valuation allowance | 1,430,420 | 1,357,170 | ||||||
| Total income tax expense | $ | - | $ | - | ||||
The following table summarizes the components of deferred tax:
| Deferred Tax Assets | 2017 | 2016 | ||||||
| Property, plant and equipment | $ | 157,920 | $ | - | ||||
| Intangible assets | 90 | 104,340 | ||||||
| Intangible assets transferred on amalgamation | - | 14,980 | ||||||
| Share issuance costs | 89,970 | 23,120 | ||||||
| Reserves | 14,030 | 7,760 | ||||||
| Non-capital losses carried forward - Canada | 1,762,250 | 973,670 | ||||||
| Net operating losses carried forward - US | 1,289,100 | 887,890 | ||||||
| Investment Tax Credits | 87,200 | 42,200 | ||||||
| Total deferred tax assets | $ | 3,400,560 | $ | 2,053,960 | ||||
| Deferred Tax Liabilities | ||||||||
| Property and equipment | (57,370 | ) | (59,680 | ) | ||||
| Intangible assets | - | (520 | ) | |||||
| Total deferred tax liabilities | $ | (57,370 | ) | $ | (60,200 | ) | ||
| Valuation allowance | $ | 3,343,190 | $ | 1,993,760 | ||||
| Net deferred tax asset | $ | - | $ | - | ||||
No deferred tax asset has been recognized, as it is not more likely than not to be realized. Consequently, a valuation allowance has been applied against the net deferred tax asset. The Canadian non-capital loss carry forwards expire as noted in the table below.
| 2035 | 465,790 | |||
| 2036 | 1,945,020 | |||
| 2037 | 4,116,040 | |||
| $ | 6,526,850 |
| F-20 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 11. | Income taxes (continued) |
The Company’s US non-operating income tax losses expire as follows:
| 2035 | 856,300 | |||
| 2036 | 1,484,640 | |||
| 2037 | 2,667,240 | |||
| $ | 5,008,180 |
| 12. | Commitments and Contingencies |
Total future annual lease payments for the premises are as follows:
| 2018 | 34,784 | |||||
| Total | $ | 34,784 |
| 13. | Financial instruments |
| (a) | Fair values |
The Company follows ASC topic 820, “Fair Value Measurements” which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements. The provisions of ASC topic 820 apply to other accounting pronouncements that require or permit fair value measurements. ASC topic 820 defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants at the measurement date; and establishes a three level hierarchy for fair value measurements based upon the transparency of inputs to the valuation of an asset or liability as of the measurement date. Inputs refers broadly to the assumptions that market participants would use in pricing the asset or liability, including assumptions about risk. To increase consistency and comparability in fair value measurements and related disclosures, the fair value hierarchy prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The three levels of the hierarchy are defined as follows:
Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities.
Level 2 inputs are inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly for substantially the full term of the financial instrument.
Level 3 inputs are unobservable inputs for asset or liabilities.
The categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
| (i) | The Company calculates expected volatility based on historical volatility of the Company’s peer group that is publicly traded for options. |
An increase/decrease in the volatility would have resulted in an increase/decrease in the fair value of the options.
The carrying values of cash, trade and other receivable, accounts payable and accrued liabilities and shareholder loans payable approximates their fair values because of the short-term nature of these instruments.
| F-21 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 13. | Financial instruments (continued) |
| (b) | Interest rate and credit risk |
Interest rate risk is the risk that the value of a financial instrument might be adversely affected by a change in interest rates. The Company does not believe that the results of operations or cash flows would be affected to any significant degree by a sudden change in market interest rates, relative to interest rates on cash and cash equivalents, due to related parties due to the short-term nature of these balances.
The Company is also exposed to credit risk at period end from the carrying value of its cash. The Company manages this risk by maintaining bank accounts with a Canadian Chartered Bank. The Company’s cash is not subject to any external restrictions.
| (c) | Foreign exchange risk |
The Company has balances in Canadian dollars that give rise to exposure to foreign exchange (“FX”) risk relating to the impact of translating certain non-U.S. dollar balance sheet accounts as these statements are presented in U.S. dollars. A strengthening U.S. dollar will lead to a FX loss while a weakening U.S. dollar will lead to a FX gain. For each Canadian dollar balance of $1.0 million, a +/- 10% movement in the Canadian currency held by the Company versus the U.S. dollar would affect the Company’s loss and other comprehensive loss by $0.1 million.
| (d) | Liquidity risk |
Liquidity risk is the risk that the Company will encounter difficulty raising liquid funds to meet commitments as they fall due. In meeting its liquidity requirements, the Company closely monitors its forecasted cash requirements with expected cash drawdown.
The following are the contractual maturities of the undiscounted cash flows of financial liabilities as at December 31, 2017 and 2016:
| December 31, 2017 | ||||||||||||||||||||||||
| Less than 3 months | 3 to 6 months | 6 to 9 months | 9 months 1 year | Greater than 1 year | Total | |||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Third parties | ||||||||||||||||||||||||
| Accounts payable and accrued liabilities | 828,737 | - | - | - | - | 828,737 | ||||||||||||||||||
| Related parties | ||||||||||||||||||||||||
| Shareholder's loan payable | - | - | - | - | - | - | ||||||||||||||||||
| 828,737 | - | - | - | - | 828,737 | |||||||||||||||||||
| December 31, 2016 | ||||||||||||||||||||||||
| Less than 3 months | 3 to 6 months | 6 to 9 months | 9 months 1 year | Greater than 1 year | Total | |||||||||||||||||||
| $ | $ | $ | $ | $ | $ | |||||||||||||||||||
| Third parties | ||||||||||||||||||||||||
| Accounts payable and accrued liabilities | 734,431 | - | - | - | - | 734,431 | ||||||||||||||||||
| Related parties | ||||||||||||||||||||||||
| Shareholder's loan payable | 6,726 | - | - | - | - | 6,726 | ||||||||||||||||||
| 741,157 | - | - | - | - | 741,157 | |||||||||||||||||||
| F-22 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 14. | Segmented information |
The Company's operations comprise a single reportable segment engaged in the research, development targeting health and wellness solutions for the companion pet. As the operations comprise a single reportable segment, amounts disclosed in the financial statements for loss for the period, depreciation and total assets also represent segmented amounts. In addition, all of the Company's long-lived assets are in the United States of America (“US”).
| December 31, 2017 | December 31, 2016 | |||||||
| $ | $ | |||||||
| Total assets | ||||||||
| Canada | 3,519,918 | 114,912 | ||||||
| US | 1,695,904 | 4,460,646 | ||||||
| Total property and equipment | ||||||||
| US | 371,157 | 289,034 | ||||||
| 15. | Schedule of expenses |
| For the year ended December 31, 2017 | ||||||||||||
| Research and Development | Professional Fees | General and Administrative | ||||||||||
| Salaries, bonus and benefits | $ | 620,694 | $ | - | $ | 2,703,865 | ||||||
| Contracted expenditures | 821,927 | - | 5,610 | |||||||||
| Marketing and investor relations | - | - | 168,623 | |||||||||
| Travel and accommodation | 11,815 | - | 338,738 | |||||||||
| Insurance | 76,628 | - | 182,753 | |||||||||
| License fees | 480,131 | - | - | |||||||||
| Office | 33,222 | - | 199,844 | |||||||||
| Consultants | 325,388 | 1,294,044 | - | |||||||||
| Regulatory | 103,100 | - | 138,289 | |||||||||
| Rent | 39,129 | - | 164,250 | |||||||||
| Supplies | 239,292 | - | 44,298 | |||||||||
| Total | $ | 2,751,326 | $ | 1,294,044 | $ | 3,946,270 | ||||||
For the year ended December 31, 2016 | ||||||||||||
| Research and Development | Professional Fees | General and Administrative | ||||||||||
| Salaries, bonus and benefits | $ | 549,556 | $ | - | $ | 2,298,476 | ||||||
| Contracted expenditures | 322,165 | - | - | |||||||||
| Marketing and investor relations | - | - | 194,187 | |||||||||
| Travel and accommodation | - | - | 87,265 | |||||||||
| Insurance | 47,207 | - | 133,827 | |||||||||
| Office | 12,455 | - | 124,693 | |||||||||
| Consultants | 308,582 | 1,245,182 | 23,904 | |||||||||
| Regulatory | 101,100 | - | - | |||||||||
| Transfer agent and filing fees | - | - | 25,357 | |||||||||
| Rent | 19,264 | - | 28,895 | |||||||||
| Supplies | 158,260 | - | - | |||||||||
| Total | $ | 1,518,589 | $ | 1,245,182 | $ | 2,916,604 | ||||||
| F-23 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 16. | Capital risk management |
The capital of the Company includes equity, which is comprised of issued common capital stock, additional paid-in capital, and accumulated deficit. The Company's objective when managing its capital is to safeguard the ability to continue as a going concern in order to provide returns for its shareholders, and other stakeholders and to maintain a strong capital base to support the Company's core activities.
| 17. | Loss per share |
| For the year ended December 31, 2017 | For the year ended December 31, 2016 | |||||||
| Numerator | ||||||||
| Net loss for the year | $ | 8,065,072 | $ | 5,740,492 | ||||
| Denominator | ||||||||
| Weighted average shares - basic | 87,400,255 | 80,158,312 | ||||||
| Stock options | - | - | ||||||
| Denominator for diluted loss per share | 87,400,255 | 80,158,312 | ||||||
| Loss per share - basic and diluted | $ | (0.09 | ) | $ | (0.07 | ) | ||
For the above-mentioned periods, the Company had securities outstanding which could potentially dilute basic earnings per share in the future, but were excluded from the computation of diluted loss per share in the periods presented, as their effect would have been anti-dilutive.
| 18. | Related party transactions and key management compensation |
During the years ended December 31, 2017 and 2016, the Company incurred the following related party transactions:
| · | As at December 31, 2016, the Company owed $6,726 to a director and executive officer, which is recorded as shareholder loans payable. The loan is unsecured and has no specific repayment terms. As at December 31, 2017, this loan has been repaid. |
Key management personnel are comprised of the Company’s directors and executive officers. In addition to their salaries, key management personnel also receive share-based compensation. Key management personnel compensation is as follows:
| For the year ended December 31, 2017 | For the year ended December 31, 2016 | |||||||
| Salaries and benefits, including bonuses | $ | 1,357,264 | $ | 912,640 | ||||
| Stock-based compensation | 749,615 | 964,506 | ||||||
| Total | $ | 2,106,879 | $ | 1,877,146 | ||||
| F-24 |
Zomedica Pharmaceuticals Corp.
Notes to the consolidated financial statements
For the years ended December 31, 2017 and 2016
| (Stated in United States dollars) |
| 19. | Recapitalization involving a public shell |
On April 21, 2016, Wise Oakwood Ventures Inc. (“WOW”), a corporation existing under the laws of the Province of Alberta, closed its qualifying transaction with ZoMedica Pharmaceuticals Inc. The transaction proceeded by way of a three-cornered amalgamation, pursuant to which Zomedica Pharmaceuticals Inc. amalgamated with 9674128 Canada Inc., a wholly-owned subsidiary of WOW formed solely for the purposes of facilitating the transaction. The amalgamated company changed its name to Zomedica Pharmaceuticals Ltd. The transaction constituted WOW’s qualifying transaction under TSX Venture Exchange Policy 2.4 – Capital Pool Companies.
In accordance with the approvals of the Company’s shareholders at its annual and special meeting on April 21, 2016, WOW changed its name to Zomedica Pharmaceuticals Corp. and completed the consolidation of its outstanding common shares on a two and one-half (2½) pre-consolidated share for each one (1) post-consolidated share basis. As a result of the transaction, Zomedica Pharmaceuticals Ltd. became a wholly-owned subsidiary of Zomedica Pharmaceuticals Corp. The shares of Zomedica Pharmaceuticals Corp. began trading under the new symbol “ZOM” on Monday May 2, 2016 on the TSX Venture Exchange.
WOW's share capital of CDN $309,589, contributed surplus of CDN $32,467 and deficit of CDN $232,984 were all eliminated. The Company has accounted for the transaction as a recapitalization involving a nonoperating public shell with ZoMedica Pharmaceuticals Inc. being the accounting acquirer and WOW being the accounting acquiree. The transaction was not considered a business combination because the accounting acquiree, WOW did not meet the definition of a business under ASC standards. Under U.S GAAP, any excess of the fair value of the shares issued by the private entity over the value of the non-monetary assets of the public shell corporation is recognized as a reduction in equity.
As part of the transaction, WOW’s previously issued 200,000 stock options were converted to 80,000 post consolidation options. These options were exercised immediately after the close of the qualifying transaction as disclosed in Note 10.
| CDN | USD | |||||||
| Issuance of 1,900,000 Zomedica Pharmaceuticals Corp. shares | $ | 475,000 | $ | 373,207 | ||||
| Issuance of 80,000 options | 2,737 | 2,058 | ||||||
| Total issuance | $ | 477,737 | $ | 375,265 | ||||
| Cash | $ | 138,687 | $ | 108,966 | ||||
| Prepaid fees | 94,778 | 74,467 | ||||||
| Accounts payable and accrued liabilities | (102,485 | ) | (80,522 | ) | ||||
| Excess of purchase price over net asset value | 346,757 | 272,354 | ||||||
| $ | 477,737 | $ | 375,265 | |||||
| 20. | Subsequent events |
Subsequent to December 31, 2017, 224,000 stock options were exercised for proceeds of $45,289.
F-25