UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of
the Securities Exchange Act of 1934
Date of report (Date of earliest event reported): December 10, 2018
RA PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
001- 37926 |
|
26-2908274 |
|
(State or other jurisdiction of |
|
(Commission |
|
(I.R.S. Employer |
|
87 Cambridge Park Drive |
|
02140 |
|
(Address of principal executive offices) |
|
(Zip Code) |
(617) 401-4060
(Registrant’s telephone number, include area code)
Not Applicable
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions
o Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
o Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
o Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
o Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company x
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. x
Item 8.01. Other Events.
On December 10, 2018, Ra Pharmaceuticals, Inc. (the “Company”) announced positive topline results from its Phase 2 clinical trial evaluating zilucoplan for the treatment of generalized myasthenia gravis, or gMG. The Phase 2, multi-center, randomized, double-blind, placebo-controlled trial was designed to evaluate the safety, tolerability, and preliminary efficacy of zilucoplan in patients with gMG, regardless of prior therapies, who had a Myasthenia Gravis Foundation of America, or MGFA, Disease Class of II-IVa at screening and a Quantitative Myasthenia Gravis, or QMG, score of at least 12 at screening and randomization.
The Company enrolled a total of 44 patients in the Phase 2 clinical program in the United States and Canada. At the outset of the 12-week treatment period, patients were randomized in a 1:1:1 ratio to receive daily, SC doses of 0.1 mg/kg of zilucoplan, 0.3 mg/kg of zilucoplan, or matching placebo. All 44 patients completed the 12-week study and, of these, 43, or 98%, elected to enter a long-term extension to receive active study drug, with one patient in the placebo group not entering the extension. The diagram below summarizes the Phase 2 trial design:

Figure 1. Summary of Phase 2 Clinical Program
The pre-specified primary efficacy endpoint was the change in QMG score, a physician-administered assessment of MG-related muscle weakness, from baseline to week 12. The key secondary efficacy endpoint was the change in MG Activities of Daily Living, orMG-ADL, score, a patient-reported outcome measure, from baseline to week 12. Significance testing was pre-specified at a 1-sided alpha of 0.1.
Zilucoplan achieved clinically meaningful and statistically significant reductions in both the primary and key secondary endpoints for both zilucoplan dose groups tested versus placebo at 12 weeks. Zilucoplan dosed at 0.3 mg/kg SC daily achieved a mean reduction from baseline of 6.0 points in the QMG score (placebo-corrected change = -2.8; p=0.05), and a mean reduction from baseline of 3.4 points in the MG-ADL score (placebo-corrected change = -2.3; p=0.04), in each case resulting in a clinically meaningful and statistically significant improvement over placebo, as shown in the figures below.

Figure 2. Primary and Key Secondary Endpoints with High Dose Zilucoplan (0.3 mg/kg)
QMG and MG-ADL outcomes for the 0.1 mg/kg SC daily dose were similar to, but less pronounced than, the 0.3 mg/kg SC daily dose, also achieving pre-specified statistical significance on both endpoints, as shown in the figures below.
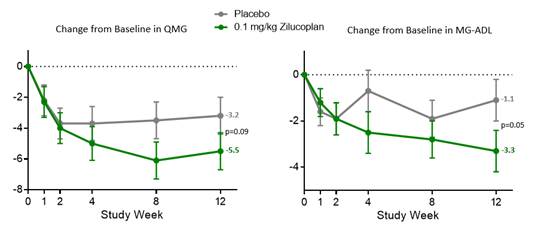
Figure 3. Primary and Key Secondary Endpoints with Low Dose Zilucoplan (0.1 mg/kg)
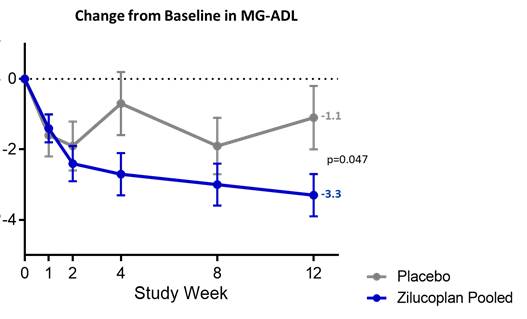
A prespecified analysis of the pooled active arms versus placebo showed a statistically significant difference in MG-ADL (2-sided p=0.047), as shown in the figure below, achieving the statistical significance threshold (2-sided p<0.05) used in pivotal Phase 3 clinical trials.
![]()
Figure 4. Pre-specified Pooled Analysis of Approval Endpoint (MG-ADL) Satisfies 2-Sided p<0.05
In addition, as shown in the figure below, patients dosed at 0.3 mg/kg SC daily exhibited the largest point improvement in each of the QMG score and the MG-ADL score, as compared to placebo.
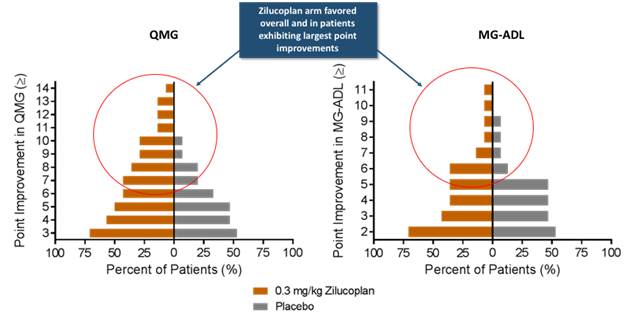
Figure 5. Point Improvements in the QMG Score and MG-ADL Score for Patients Dosed with 0.3mg/kg SC daily over Placebo
Furthermore, as depicted below, with respect to minimal symptom expression, or MSE, defined as achieving an MG-ADL score of zero or one, 23% of patients in the zilucoplan 0.3 mg/kg arm achieved this endpoint by week 12 versus 19.7% on eculizumab in the REAGIN Study by week 26.
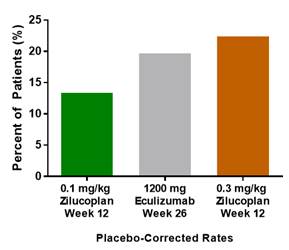
Figure 6. Minimal Symptom Expression (MG-ADL of 0 or 1) by Week 12
Finally, rescue therapy with intravenous immunoglobulin or plasma exchange was required by three of the 15 patients, or 20%, in the placebo arm, one of 15 patients, or 7%, in the 0.1 mg/kg zilucoplan arm, and zero patients in the 0.3 mg/kg zilucoplan arm. Treatment with zilucoplan was observed to have a favorable safety and tolerability profile that was consistent with previously-completed Phase 1 and Phase 2 clinical trials. The majority of adverse events, or AEs, reported were mild and were not considered by the investigators to be related to study drug. There were no serious AEs observed related to treatment with zilucoplan.
Based on these data, the Company plans to engage with regulatory agencies, including the U.S. Food and Drug Administration, or FDA, in the first half of 2019 regarding the design of a potential Phase 3 clinical trial evaluating zilucoplan versus placebo in
patients with gMG. The Company plans to release an expanded Phase 2 data set in the first half of 2019 and expects to initiate the Phase 3 clinical trial in the second half of 2019.
In December 2018, the Company received a development milestone payment under its collaboration agreement with Merck & Co., Inc. The milestone payment is associated with its collaboration for a non-complement cardiovascular target with a large market opportunity.
Forward-Looking Statements Disclaimer
This Current Report on Form 8-K (the “Current Report”) contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. All statements contained in this Current Report that do not relate to matters of historical fact should be considered forward-looking statements, including without limitation statements regarding our potential to transform treatment paradigms across multiple complement-mediated disorders, the potential safety, efficacy and regulatory and clinical progress of our product candidates, including without limitation zilucoplan, plans to engage with regulatory agencies, beliefs regarding clinical trial data, and plans for the presentation of clinical data. These forward-looking statements are based on management’s current expectations. These statements are neither promises nor guarantees, but involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements, including, but not limited to, the risk that our product candidates, including zilucoplan, will not successfully be developed or commercialized, in the timeframe we expect or at all, and the risk that top-line results as of November 29, 2018 from the Company’s global Phase 2 clinical program evaluating zilucoplan for the treatment of gMG may not be indicative of final study results. These and other important factors discussed under the caption “Risk Factors” in our Annual Report on Form 10-K filed with the Securities and Exchange Commission, or SEC, on March 14, 2018 and our other reports filed with the SEC could cause actual results to differ materially from those indicated by the forward-looking statements made in this Current Report. Any such forward-looking statements represent management’s estimates as of the date of this Current Report. While we may elect to update such forward-looking statements at some point in the future, we disclaim any obligation to do so, even if subsequent events cause our views to change. These forward-looking statements should not be relied upon as representing our views as of any date subsequent to the date of this Current Report.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
|
|
RA PHARMACEUTICALS, INC. | |
|
|
| |
|
Date: December 10, 2018 |
By: |
/s/ David C. Lubner |
|
|
|
David C. Lubner |
|
|
|
Executive Vice President and Chief Financial Officer |