Attached files
| file | filename |
|---|---|
| EX-31.2 - EXHIBIT 31.2 - MEDICINES CO /DE | mdcoex31212312017-q42017.htm |
| EX-32.2 - EXHIBIT 32.2 - MEDICINES CO /DE | mdcoex32212312017-q42017.htm |
| EX-32.1 - EXHIBIT 32.1 - MEDICINES CO /DE | mdcoex32112312017-q42017.htm |
| EX-31.1 - EXHIBIT 31.1 - MEDICINES CO /DE | mdcoex31112312017-q42017.htm |
| EX-23.1 - EXHIBIT 23.1 - MEDICINES CO /DE | mdcoex2312312017-q42017.htm |
| EX-21.1 - EXHIBIT 21.1 - MEDICINES CO /DE | mdcoex2112312017-q42017.htm |
| EX-10.29 - EXHIBIT 10.29 - MEDICINES CO /DE | mdcoex1029123117-q42017.htm |
| EX-10.9 - EXHIBIT 10.9 - MEDICINES CO /DE | mdcoex10912312017.htm |
| EX-3.2 - EXHIBIT 3.2 - MEDICINES CO /DE | mdcoex3212312017-q42017.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_____________________________________________
Form 10-K
(Mark One) | ||
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the fiscal year ended: December 31, 2017 | ||
Or | ||
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to | ||
_____________________________________________
THE MEDICINES COMPANY
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | 04-3324394 (I.R.S. Employer Identification No.) | |
8 Sylvan Way Parsippany, New Jersey (Address of principal executive offices) | 07054 (Zip Code) | |
Registrant’s telephone number, including area code: (973) 290-6000
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Name of Each Exchange on Which Registered | |
Common Stock, $.001 Par Value Per Share | NASDAQ Global Select Market | |
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes þ No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or Section 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer þ | Accelerated filer o | Non-accelerated filer o | Smaller reporting company o |
Emerging growth company o | (Do not check if a smaller reporting company) | ||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No þ
The aggregate market value of voting Common Stock held by non-affiliates of the registrant on June 30, 2017 was approximately $2,417,163,772 based on the last reported sale price of the Common Stock on The NASDAQ Global Select Market on June 30, 2017 of $38.01 per share.
Number of shares of the registrant’s class of Common Stock outstanding as of February 26, 2018: 73,283,509
DOCUMENTS INCORPORATED BY REFERENCE
The registrant intends to file a proxy statement pursuant to Regulation 14A within 120 days of the end of the fiscal year ended December 31, 2017. Portions of the proxy statement are incorporated herein by reference into the following parts of this Annual Report on Form 10-K:
Part III, Item 10. Directors, Executive Officers and Corporate Governance;
Part III, Item 11. Executive Compensation;
Part III, Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters;
Part III, Item 13. Certain Relationships and Related Transactions, and Director Independence; and
Part III, Item 14. Principal Accounting Fees and Services.
THE MEDICINES COMPANY
ANNUAL REPORT ON FORM 10-K
For the Fiscal Year Ended December 31, 2017
TABLE OF CONTENTS
Page | ||
EX-3.2 | ||
EX-10.9 | ||
EX-10.29 | ||
EX-21 | ||
EX-23 | ||
EX-31.1 | ||
EX-31.2 | ||
EX-32.1 | ||
EX-32.2 | ||
EX-101 INSTANCE DOCUMENT | ||
EX-101 SCHEMA DOCUMENT | ||
EX-101 CALCULATION LINKBASE DOCUMENT | ||
EX-101 LABELS LINKBASE DOCUMENT | ||
EX-101 PRESENTATION LINKBASE DOCUMENT | ||
EX-101 DEFINITION LINKBASE DOCUMENT | ||
The Medicines Company® name and logo, Angiomax®, Angiox® and Ionsys® are either registered trademarks or trademarks of The Medicines Company in the United States and/or other countries. All other trademarks, service marks or other tradenames appearing in this Annual Report on Form 10-K are the property of their respective owners. Except where otherwise indicated, or where the context may otherwise require, references to “Angiomax” in this Annual Report on Form 10-K mean Angiomax and Angiox, collectively. References to the Company, “we,” “us” or “our” mean The Medicines Company, a Delaware corporation, and its subsidiaries.
This Annual Report on Form 10-K includes forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and Section 27A of the Securities Act of 1933, as amended, or the Securities Act. For this purpose, any statements contained herein regarding our strategy, future operations, financial position, future revenues, potential transactions, projected costs, products in development, future clinical trials, prospects, plans and objectives of management, other than statements of historical facts, are forward-looking statements. The words “anticipates,” “believes,” “estimates,” “expects,” “intends,” “may,” “plans,” “projects,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We cannot guarantee that we actually will achieve the plans, intentions or expectations expressed or implied in our forward-looking statements. There are a number of important factors that could cause actual results, levels of activity, performance or events to differ materially from those expressed or implied in the forward-looking statements we make. These important factors include our “critical accounting estimates” described in Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Result of Operations of this Annual Report on Form 10-K and the factors set forth under the caption “Risk Factors” in Part I, Item 1A. of this Annual Report on Form 10-K. Although we may elect to update forward-looking statements in the future, we specifically disclaim any obligation to do so, even if our estimates change, and readers should not rely on our forward-looking statements as representing our views as of any date subsequent to the date of this Annual Report on Form 10-K.
1
PART I
Item 1. Business.
Our Company
Overview
We are a biopharmaceutical company driven by an overriding purpose - to save lives, alleviate suffering and contribute to the economics of healthcare. Our goal is to create transformational solutions to address the most pressing healthcare needs facing patients, physicians and providers in cardiovascular care. We are focused on inclisiran, an investigational agent which is potentially a first-in-class lipid-lowering drug, to reduce LDL-cholesterol, or LDL-C, which is commonly referred to as “bad” cholesterol, in patients with atherosclerotic cardiovascular disease, or ASCVD, or cardiovascular risk-equivalents. We believe that inclisiran possesses favorable attributes that competitive products do not possess, would satisfy unmet medical needs and has the potential to improve the economics of healthcare. We have the right to develop, manufacture and commercialize inclisiran under our collaboration agreement with Alnylam Pharmaceuticals, Inc., or Alnylam. In addition, we market Angiomax® (bivalirudin) in the United States primarily through a supply and distribution agreement with Sandoz Inc., or Sandoz, under which we granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax.
Business Strategy
On November 3, 2015, we announced that we were in the process of evaluating our operations with a goal of unlocking and maximizing stockholder value. In particular, we stated our intention was to explore strategies for optimizing our capital structure and liquidity position and to narrow our operational focus by strategically separating non-core businesses and products in order to generate non-dilutive cash and reduce associated cash burn and capital requirements.
As a result of our decision to narrow our operational focus, we have completed the following transactions:
• | On February 1, 2016, we completed the sale of our hemostasis portfolio, consisting of PreveLeak, Raplixa and Recothrom, to wholly owned subsidiaries of Mallinckrodt plc, or Mallinckrodt. At the completion of the sale, we received approximately $174.1 million in cash and may receive up to an additional $235.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of PreveLeak and Raplixa. |
• | On June 21, 2016, we completed the sale of Cleviprex, Kengreal and rights to Argatroban for Injection, which we refer to collectively as Non-Core ACC Assets, to Chiesi USA, Inc., or Chiesi USA, and its parent company Chiesi Farmaceutici S.p.A., or Chiesi. At the completion of the sale, we received approximately $263.8 million in cash, which included the value of product inventory, and may receive up to an additional $480.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of each of Cleviprex and Kengreal. |
• | On January 5, 2018, we completed the sale of our infectious disease portfolio, consisting of the products Vabomere, Orbactiv and Minocin IV and line extensions thereof, and substantially all of the assets related thereto, other than certain pre-clinical assets, to Melinta Therapeutics, Inc., or Melinta. At the completion of the sale, we received approximately $166.4 million and 3,313,702 shares of Melinta common stock having a market value, based on Melinta's closing share price on January 5, 2018, of approximately $54.5 million. In addition, we are entitled to receive (i) a cash payment payable 12 months following the closing of the transaction equal to $25 million; (ii) a cash payment payable 18 months following the closing of the transaction equal to $25 million; and (iii) tiered royalty payments of 5% to 25% on worldwide net sales of (a) Vabomere and (b) Orbactiv and Minocin IV, collectively. |
The transactions above are described in more detail in Part II, Item 7, Management’s Discussion and Analysis of Financial Condition and Results of Operations - Business Development Activity of this Annual Report on Form 10-K.
Consistent with our intentions announced in November 2015, in January 2017 we announced that we were seeking opportunities to partner or divest Ionsys (fentanyl iontophoretic transdermal system). Although we continue to seek a partnership or divestiture transaction for Ionsys, in June 2017 we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open. In addition, in August 2017, we announced that we are discontinuing the clinical development program for MDCO-700, an investigational anesthetic agent.
As a result of these transactions, we are now focused on the development of inclisiran as a transformative treatment for ASCVD.
2
Further, as a result of the rapid and significant evolution our business and the narrowing of our operational focus onto inclisiran, our board of directors determined to restructure the board, adding directors with relevant expertise and experience and reducing the board’s overall size from twelve to seven members, providing a more agile and efficient structure. Accordingly, the board of directors has determined that the slate of directors to be nominated for election as directors at the 2018 annual meeting of shareholders will consist of: Alexander J. Denner, Fredric N. Eshelman, Geno J. Germano, John C. Kelly, Clive A. Meanwell, Paris Panayiotopoulos, and Sarah J. Schlesinger.
Inclisiran
Overview
Inclisiran is a subcutaneously administered proprotein convertase subtilisin/kexin type 9, or PCSK9, synthesis inhibitor which works through RNA interference, or RNAi, and is being developed for the potential treatment of hypercholesterolemia. We obtained rights to this product candidate under a license and collaboration agreement that we entered into with Alnylam in February 2013 to develop, manufacture and commercialize RNAi therapeutics targeting the PCSK9 gene for the treatment of hypercholesterolemia and other human diseases. RNAi is a naturally occurring biological pathway within cells for selectively silencing and regulating the translation of specific mRNAs. PCSK9 is a protein involved in the regulation of low-density lipoprotein, or LDL, receptor levels on hepatocytes and the metabolism of LDL-C. Inclisiran is designed to inhibit the synthesis of PCSK9 and results in lower LDL-C levels.
PCSK9 and PCSK9 inhibition
PCSK9, a member of the serine protease family, plays a key role in controlling the levels of LDL receptors on the surface of hepatocytes. PCSK9 is expressed and secreted into the bloodstream predominantly by the liver, binds LDL receptors both intracellularly and extracellularly and promotes the lysosomal degradation of these receptors in hepatocytes, thereby increasing the circulating LDL-C levels. Loss of function mutations in PCSK9 have been found to lead to increased LDL receptors expression on hepatocytes, reduced serum LDL-C levels, and a lower risk for coronary heart disease, with no apparent negative health consequences.
Recently-developed and approved PCSK9-blocking monoclonal antibodies, Amgen’s Repatha® (evolocumab) and Sanofi’s Praluent® (alirocumab), reduce circulating PCSK9 levels and lower LDL-C levels. Preliminary reports and analyses of Amgen’s FOURIER trial with Repatha, the first large cardiovascular outcomes trial, indicated that treatment with such antibodies can lead to the reduction of cardiovascular events compared with placebo. Results from the FOURIER study were reported by Amgen in March 2017 in which Repatha significantly reduced the risk of cardiovascular events. The FOURIER study, which had approximately 27,000 subjects with clinically evident ASCVD, met its primary composite endpoint (cardiovascular death, nonfatal myocardial infarction, or MI, nonfatal stroke, hospitalization for unstable angina or coronary revascularization) and the key secondary composite endpoint (cardiovascular death, nonfatal MI or nonfatal stroke).
RNA interference
RNA interference, or RNAi, is a natural cellular process of gene silencing that represents one of the most promising and rapidly advancing frontiers in biology and drug development today. Its discovery was recognized with the award of the 2006 Nobel Prize for Physiology or Medicine. By harnessing the natural biological process of RNAi occurring in our cells, a major new class of medicines, known as RNAi therapeutics, is being developed. Small interfering RNA, or siRNA, the molecules that mediate RNAi and comprise Alnylam's RNAi therapeutic platform, and which yielded inclisiran, function upstream of today’s medicines by potently silencing messenger RNA, or mRNA - the genetic precursors - that encode for disease-causing proteins, thus preventing them from being made. This is a revolutionary approach with the potential to transform the care of patients with genetic and other diseases.
Clinical Development
Overview
Under our license and collaboration agreement with Alnylam, we and Alnylam initially collaborated on the development of inclisiran and ALN-PCS02, an intravenously administered earlier RNAi therapeutic. Alnylam was responsible for the development of these product candidates until Phase 1 was completed. We have assumed all other responsibility for the development and commercialization of all product candidates under our agreement with Alnylam. In October 2013, we and Alnylam selected a lead subcutaneously administered development candidate, now referred to as inclisiran, for development for the potential treatment of hypercholesterolemia. In December 2014, under the terms of our agreement with Alnylam, Alnylam initiated a Phase 1 clinical trial of inclisiran in the United Kingdom. Data from the Phase 1 trial was presented at the European Society of Cardiology meeting in August 2015 and at the American Heart Association meeting in November 2015, and was published in the New England Journal of Medicine.
3
In January 2016, we began enrolling patients in the ORION-1 Phase 2 dose finding trial. ORION-1 was conducted as a placebo-controlled, double-blind, randomized trial of single or multiple subcutaneous injections of inclisiran in a total of 501 patients with ASCVD or ASCVD-risk equivalents (e.g., diabetes and familial hypercholesterolemia), and elevated LDL-C despite maximum tolerated doses of LDL-C lowering therapies. The study compared the effect of different doses of inclisiran and evaluated the potential for an infrequent dosing regimen. The primary endpoint of the study was the percentage change in LDL-C from baseline at Day 180.
In March 2017, we reported positive final results from the ORION-1 Phase 2 study of inclisiran. Efficacy data presented reaffirmed inclisiran’s significant LDL-C lowering effects following a starting dose of 300 mg given on Day-1 and Day-90, after which the mean LDL-C showed reduction by an average of 52.6% and up to 81% at Day-180. For the subsequent six-month period - from Day-90 to Day-270 - the time-averaged LDL-C reduction was 51%. These robust data underscore the selection of a six-monthly maintenance dose of 300 mg in the inclisiran Phase 3 clinical program. With completion of one-year follow-up, safety data for inclisiran from the Phase 2 ORION-1 study includes 370 subject-years of observation, including at least 300 subject-years of inclisiran effects. No material safety issues were observed on inclisiran in ORION-1, which demonstrated an adverse event profile similar to placebo.
We developed a dose-pharmacodynamic, or dose-PD, model based on the ORION-1 data to perform modeling and simulation experiments to support the selection of the Phase 3 dose and dose regimen. The dose-PD modeling and simulation supported the clinical observations from ORION-1 that a 300 mg dose given subcutaneously on Day-1, Day-90 and then every six months thereafter is the optimal dose and dose regimen for further testing in Phase 3. This dose and dose regimen maintains a time-averaged LDL-C reduction of >50%. Our initial Phase 3 ORION program, described below, will test this dose and dose regimen in high risk patients with ASCVD, ASCVD risk equivalents and familial hypercholesterolemia, or FH. Further dose-PD modeling and simulation demonstrated that a 300 mg dose given once a year would result in a time-averaged LDL-C reduction of approximately 43-45%. We believe that this once a year dose regimen of 300 mg of inclisiran could be tested in patient populations at lower risk for cardiovascular events such as primary prevention of cardiovascular disease.
In January 2017, we initiated the ORION-2 and ORION-3 studies. ORION-2 examines the efficacy, safety and tolerability of inclisiran in patients with homozygous familial hypercholesterolemia, or HoFH. The ORION-3 study is an open label extension study with the objective to evaluate the efficacy, safety and tolerability of long-term dosing of inclisiran and will also measure the effects of treatment, including a comparison of the effects of inclisiran and evolocumab (trade named Repatha) on certain clinical and patient-reported endpoints, as well as the effects of switching from evolocumab to inclisiran.
Phase 3 Clinical Program - ORION 5, 9, 10 and 11 clinical trials.
In the fourth quarter of 2017, we initiated the Phase 3 LDL-C lowering program for inclisiran. The Phase 3 program is comprised of four pivotal clinical trials in patients with ASCVD, ASCVD risk equivalents and FH. We anticipate that data from the four trials will support the submission of an NDA in the United States and a marketing authorization application, or MAA, in the European Union at or around the end of 2019. In the ORION-9, ORION-10 and ORION-11 trials, patients will be studied for 18 months and the dose of inclisiran will be 300 mg given subcutaneously on Day-1, Day-90 and then every six months thereafter for a total of four doses of inclisiran during the 18-month study period. We expect patients in the ORION-5 trial to have a shorter comparative treatment window than the patients in the other ORION Phase 3 trials. The four Phase 3 clinical trials are further described below:
Study | Sites | Main inclusion criteria | Patients | |
ORION-5 | US, EU, South Africa (SA) | Homozygous familial hypercholesterolemia, or HoFH | 45 | |
ORION-9 | US, EU, SA | Heterozygous familial hypercholesterolemia, or HeFH | 400 | |
ORION-10 | US | ASCVD | 1,500 | |
ORION-11 | EU, SA | ASCVD and risk equivalent patients | 1,500 | |
3,445 | ||||
ORION-5 is planned to be a two-part (double-blind, placebo-controlled/open label) multicenter study to evaluate safety, tolerability, and efficacy of inclisiran in approximately 45 subjects with HoFH. We expect to commence the ORION-5 trial in 2018 following FDA review of the protocol. On January 23, 2018, the FDA granted orphan drug designation for inclisiran for the treatment of HoFH.
4
ORION-9 is a placebo-controlled, double-blind, randomized study of inclisiran versus placebo (1:1) in approximately 400 patients with HeFH. The primary endpoint of ORION-9 study is LDL-C reduction from baseline at Day-510. The ORION-9 trial commenced in November 2017. In February 2018, we announced that this trial had exceeded its target enrollment of 400 patients.
ORION-10 is a placebo-controlled, double-blind, randomized study of inclisiran versus placebo (1:1) in approximately 1,500 patients with ASCVD and elevated LDL-C levels above 70 mg/dL despite maximum tolerated doses of LDL-C lowering therapies including statins. The primary endpoint of ORION-10 study is LDL-C reduction from baseline at Day-510. The ORION-10 trial commenced in November 2017 and we expect to complete enrollment during the first half of 2018.
ORION-11 is a placebo-controlled, double-blind, randomized study of inclisiran versus placebo (1:1) in approximately 1,500 patients with ASCVD or ASCVD-risk equivalents and elevated LDL-C levels above 70 mg/dL or 100 mg/dL, respectively, despite maximum tolerated doses of LDL-C lowering therapies including statins. The primary endpoint of the study is LDL-C reduction from baseline at Day-510. The ORION-11 trial commenced in November 2017. In January 2018, we announced that this trial had exceeded its target enrollment of 1,500 patients.
Cardiovascular Outcomes Trial - ORION-4
We also expect to perform a cardiovascular outcomes trial in approximately 15,000 patients with ASCVD on a background of standard of care lipid-lowering therapy (usually high intensity statins), to determine the effects inclisiran on cardiovascular outcomes. We expect to initiate enrollment in the trial in 2018. The overall design of the ORION-4 outcomes trial has been agreed to with the FDA and EMA. The ORION-4 study will be conducted in close collaboration with the academic groups, Clinical Trial Service Unit and Epidemiological Studies Unit of the University of Oxford and Thrombolysis In Myocardial Infarction (TIMI) Study Group of the Brigham and Women’s Hospital, Boston, Massachusetts, as well as other scientific experts. The protocol is currently under review by the FDA. The primary efficacy endpoint of the trial will be a composite endpoint of coronary heart disease death, non-fatal myocardial infarction, fatal or non-fatal ischemic stroke and urgent coronary revascularization. These endpoints have been demonstrated to be modifiable in previous, similar outcomes trials with lipid modifying therapies. The duration of the outcomes trial will be long enough, with a median of four to five years follow-up, to accumulate a sufficient number of events to provide overwhelming statistical power to ascertain treatment group differences and maximize the clinical effect size associated with LDL-C lowering. We anticipate that, if inclisiran is approved for sale and the outcomes trial is successful, we will submit the results of the outcomes trial to the FDA as a supplemental New Drug Application, or sNDA, and as a variation to the MAA with the European Medicines Agency, or EMA.
Medical Need
Despite advances in treatment, cardiovascular disease is the leading cause of death worldwide, resulting in over 17 million deaths annually. Eighty percent of all cardiovascular disease deaths are due to coronary heart disease or strokes. Elevated LDL-C is a major risk factor for the development of cardiovascular disease and has recently been described as causative. Lowering LDL-C has been shown to reduce the risk of cardiovascular death or heart attack, and within the range of effects achieved so far, the clinical risk reduction is linearly-proportional to absolute LDL-C reduction.
Approximately 100 million people worldwide are treated with lipid lowering therapies, predominantly statins, to reduce LDL-C and the associated risk of death, nonfatal MI and nonfatal stroke or associated events. There is an unmet need for additional treatment options beyond currently-available treatments for lowering of the LDL-C level to reduce cardiovascular risk, as residual risk for cardiovascular events remains and statins are associated with well-known limitations. First, not all subjects reach LDL-C levels associated with optimal protection against clinical events. This is particularly true in patients with pre-existing coronary heart disease and/or diabetes or a history of FH, who are at the highest risk and require the most intensive management Second, not all subjects tolerate statins or are able to take statins at sufficiently-intensive doses. Third, observational studies have demonstrated that >50% of patients do not adhere to statin therapy for more than six months.
We believe that new effective treatments to significantly lower LDL-C are needed. Clinical studies performed with inclisiran, as well as clinical studies performed with monoclonal antibodies to PCSK9, with or without statins, have demonstrated that therapies that act on PCSK9 lower LDL-C by as much as 50%, and therefore have the potential to meet this unmet need for additional significant LDL-C reduction. In addition, we believe that the ease of dosing of inclisiran - small volume, subcutaneous injections twice a year, most likely given by healthcare professionals - may improve patient adherence to lipid therapy, which has been a significant problem with all other approaches.
Approved Products
Angiomax
Angiomax is an intravenous direct thrombin inhibitor that is a peptide compound. We licensed Angiomax from Biogen Idec, Inc., or Biogen Idec, in 1997 and have exclusive license rights to develop, market and sell Angiomax worldwide. Angiomax is
5
approved in the United States for use as an anticoagulant in combination with aspirin in patients with unstable angina undergoing PTCA and for patients undergoing percutaneous coronary intervention, or PCI, with provisional use of glycoprotein IIb/IIIa receptor inhibitors, or GP IIb/IIIa inhibitors, including patients with or at risk of heparin-induced thromboticytopenia, or HIT, or heparain-induced thrombotic thrombocytopenia syndrome, or HITTS.
We sell Angiomax in the United States under our name as a branded Angiomax product, and, on July 2, 2015, entered into a supply and distribution agreement with Sandoz under which we granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax (bivalirudin). We entered into the supply and distribution agreement as a result of the July 2, 2015 U.S. Court of Appeals for the Federal Circuit, or Federal Circuit Court, ruling against us in our patent infringement litigation with Hospira, Inc., or Hospira, with respect to U.S. Patent No. 7,582,727, or the ‘727 patent, and U.S. Patent No. 7,598,343, or the ‘343 patent, covering a more consistent and improved Angiomax drug product and the processes by which it is made. In addition to Hospira, other generic firms have entered the market. Mylan Pharmaceuticals, Inc., or Mylan, commenced marketing its generic bivalirudin product following a decision by the Federal Circuit Court in Mylan’s appeal that reversed an earlier district court decision that found that Mylan’s abbreviated new drug application, or ANDA, product infringed all of the asserted claims of the ‘727 patent. APP Pharmaceuticals LLC, or APP, through its affiliated company, Fresenius Kabi, commenced selling its generic version of Angiomax under provisions of a settlement agreement triggered by the Federal Circuit Court’s July 2, 2015 decision in the Hospira matter. Apotex Inc. and Dr. Reddy’s Laboratories have each also commenced commercialization of generic bivalirudin products upon receiving final approval if their respective ANDA filings by the FDA even though we remain in active litigation against Apotex and only recently settled with Dr. Reddy’s Laboratories. In addition, in January 2018 Baxter International Inc., or Baxter, announced that the FDA approved Baxter’s ready-to-use formulation of bivalirudin for use as an anticoagulant in patients undergoing PCI.
A number of companies in addition to Hospira, Mylan, APP, Apotex Inc. and Dr. Reddy’s Laboratories have filed ANDAs for their generic versions of Angiomax. In addition to the generic versions and the ready-to-use version of bivalirudin currently being sold, Angiomax could be subject to further generic competition in the United States from Teva Pharmaceuticals USA, Inc. and its affiliates, or Teva, and other generic ANDA filers that we have settled with, under the circumstances set forth in our respective settlement agreements with such parties and upon a final approval of each company's ANDA filings by the FDA. Pliva Hrvatska DOO, an affiliate of Teva, currently has tentative approval for its ANDA filing for its generic version of Angiomax. Other ANDA filers may commercialize their products ‘at risk’ if they receive final approval of their respective ANDA filings and are not subject to a Hatch-Waxman 30-month stay. Further, we remain in infringement litigation involving the ‘727 patent and ‘343 patent with the other ANDA filers. See Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K for descriptions of our litigation with ANDA filers and related settlements. There can be no assurance as to the outcome of our infringement litigation. We may continue to incur further legal expenses related to these matters.
Angiomax is approved in the European Union for use as an anticoagulant in adult patients undergoing PCI, including patients with ST elevation myocardial infarction, or STEMI, undergoing primary PCI. The approval for acute coronary syndrome in Europe also includes treatment of adult patients with unstable angina or non-STEMI planned for urgent or early intervention, when used with aspirin and clopidogrel. In Europe, the principal patent covering Angiomax expired in August 2015 and as a result, we do not have market exclusivity for Angiomax in Europe. In addition, Angiomax is also approved for use in Australia, Canada, New Zealand, Russia, India and a number of countries in Central America, South America and the Middle East for PCI indications similar to those approved by the FDA or EMA. In addition, Angiomax is approved in Canada for the treatment of patients with HIT/HITTS undergoing cardiac surgery. We are in the process of voluntarily discontinuing and withdrawing Angiomax from the market outside of North America and have ceased related commercialization activities.
In 2017, our total net revenues related to branded Angiomax and authorized generic Angiomax (bivalirudin) were approximately $44.6 million. Net product revenues related to sales of Angiomax in 2017 totaled approximately $18.8 million, including approximately $11.2 million of net product sales in the United States. Royalty revenues in 2017 related to the authorized generic sale of Angiomax (bivalirudin) by Sandoz totaled approximately $25.8 million.
Ionsys
Ionsys (fentanyl iontophoretic transdermal system) is a compact, needle-free patient-controlled system for the short term management of acute postoperative pain for adults requiring opioid analgesia in the hospital. In April 2015, we received FDA approval of our sNDA for Ionsys, and in November 2015, the European Commission granted marketing authorization for Ionsys in the European Union. In June 2017, we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open as we continue to explore opportunities to partner or divest Ionsys.
In 2017, our net product sales of Ionsys totaled approximately $0.1 million.
6
Research and Development Infectious Disease Portfolio
We maintain a small infectious disease research unit in San Diego focused on the discovery of novel antibiotics. Our research programs include next-generation beta-lactamase inhibitors, or BLIs, as well as other innovative classes to treat resistant gram-negative infections. Our BLI research program focuses on identifying compounds with improved attributes such as broadened coverage of resistance mechanisms and oral bioavailability. Our programs are at the pre-clinical stage of development and are supported in part through our government contracts and partnerships including the Biomedical Advanced Research and Development Authority, or BARDA, of the U.S. Department of Health and Human Services, or HHS. This agreement, which we refer to as the BARDA OTA agreement, was established under HHS’s Other Transaction Authority, known as OTA.
Sales and Distribution
On July 2, 2015, we entered into a supply and distribution agreement with Sandoz, which we amended in July 2017, under which we granted Sandoz the exclusive right to sell bivalirudin (250 mg/ml) in the United States under our approved NDA for Angiomax but labeled and sold under the Sandoz name. We refer to this product herein as authorized generic Angiomax (bivalirudin). Under the agreement, we agreed to supply Sandoz and Sandoz agreed to purchase exclusively from us authorized generic Angiomax (bivalirudin). Sandoz has agreed to use commercially reasonable efforts to market, distribute and sell authorized generic Angiomax (bivalirudin) in the United States during the term of the agreement. Sandoz has agreed to pay us a set price per vial and Sandoz will pay us on a quarterly basis a high double digit percentage of its net profits (net sales less our cost of goods and certain agreed expenses of Sandoz) on sales of authorized generic Angiomax (bivalirudin). The term of the agreement will continue until July 2, 2020 and will automatically renew for successive one-year periods thereafter unless either party provides notice of non-renewal at least six months prior to the end of the applicable term. Either party may terminate the agreement at any time if the other party is in material breach of the agreement and does not cure such breach within 60 days, the other party undergoes bankruptcy events, the other party is unable to perform its obligations under the agreement for more than 120 consecutive days due to a force majeure event, compliance with the agreement would violate law or net profits related to sales of the authorized generic Angiomax in any month fall below a low double digit percentage of net sales of the authorized generic Angiomax in such month. We may also terminate the agreement at any time that no other pharmaceutical product containing bivalirudin in a lyophilized form as its sole active ingredient is being sold in the United States.
We distribute our branded Angiomax product through a sole source distribution model with Integrated Commercialization Solutions, or ICS. Under this model, we currently sell our branded Angiomax product to our sole source distributor, ICS. ICS then sells branded Angiomax to a limited number of national medical and pharmaceutical wholesalers with distribution centers located throughout the United States and, in certain cases, directly to hospitals.
Our agreement with ICS, which we initially entered into in February 2007 and have subsequently amended from time to time, provides that ICS will be our exclusive distributor of our branded Angiomax product. Under the terms of this fee-for-service agreement, ICS places orders with us for sufficient quantities of branded Angiomax to maintain an appropriate level of inventory based on our wholesalers’ historical purchase volumes. ICS assumes all credit and inventory risks, is subject to our standard return policy and has sole responsibility for determining the prices at which it sells branded Angiomax, subject to specified limitations in the agreement. The agreement terminates on February 28, 2019 and will automatically renew for additional one-year periods unless either party gives notice at least 90 days prior to the automatic extension. Either party may terminate the agreement at any time and for any reason upon 180 days prior written notice to the other party. In addition, either party may terminate the agreement upon an uncured default of a material obligation by the other party and other specified conditions. ICS’s payment terms under our distribution agreement with them are 45 days, which can be further extended to 49 days if ICS pays by wire transfer.
We also market and sell Angiomax outside the United States, principally through distributor relationships. These distributors include Sandoz Canada Inc., which distributes Angiomax in Canada, and affiliates of Grupo Ferrer Internacional who distribute Angiox in Cyprus, Greece, Portugal and Spain and in a number of countries in Central America and South America. We also have agreements with other third parties for other countries outside of the United States. We are in the process of voluntarily discontinuing and withdrawing Angiomax from the market outside of North America and ceasing related commercialization activities, including terminating our agreements with our distributors. We have also entered into a strategic collaboration with SciClone Pharmaceuticals, or SciClone, under which we granted SciClone a license and the exclusive rights to promote, market and sell Angiomax in China.
Manufacturing
We do not have a manufacturing infrastructure and we do not intend to develop one. We are a party to agreements with contract manufacturers for the supply of bulk drug substance for our products and with other third parties for the formulation, packaging and distribution of our products. Our product manufacturing operation is comprised of professionals with expertise in pharmaceutical
7
manufacturing, product development, logistics and supply chain management and quality management and supply chain compliance. These professionals oversee the manufacturing and distribution of our products by third-party companies.
Inclisiran
Under our agreement with Alnylam, Alnylam supplied the quantity of finished product required for the conduct of the first Phase 1 clinical trial and the first Phase 2 clinical trial of inclisiran. Alnylam bore the costs of these activities, subject to certain agreed-upon caps. We have the sole right and responsibility to manufacture and supply licensed product for further development and commercialization under our development plan. We and Alnylam entered into a development supply agreement under which Alnylam agreed to transfer the manufacturing technology for the product to us or our third-party manufacturers. We have entered into agreements with two contract manufacturing organizations for the manufacture of clinical supplies of drug substance, and another manufacturing organization for the supply of drug product for use in clinical and non-clinical studies. Subsequent to the completion of Phase 2 all clinical and non-clinical materials have been directly sourced from suppliers by us.
Bulk Drug Substance. On October 27, 2016, we entered into a services and supply agreement with Agilent Technologies, or Agilent, to supply inclisiran sodium manufactured by a chemical solid phase oligonucleotide based process. Agilent has supplied a number of batches using this process that have been used in drug product manufacture for clinical studies. Further on December 9, 2015, we entered into a services and supply agreement, as amended on July 27, 2016, with Nitto Denko Avecia for the technical transfer and manufacture of inclisiran sodium. We have an agreement with Alnylam for the supply of GalNAc-resin, a key starting material through process validation. Additionally, we and Alnylam are transferring technology and relationships for the manufacturer of GalNAc-resin and associated components to third parties jointly selected by us and Alnylam for the manufacture of commercial supplies of GalNAc-succinate and GalNAc-resin.
Drug Product. On June 3, 2016, we entered into a master service agreement with Alcami Corporation to develop processes and methods for the manufacture of inclisiran drug product. Under the agreement, Alcami has manufactured all of the inclisiran sodium vials and placebo vials used to date in clinical and non-clinical studies.
Additionally, on September 25, 2017, we entered into a technology transfer and manufacturing service agreement with Corden Pharma for the development and manufacture of pre-filled syringes of inclisiran sodium and placebo for use in clinical studies. To date pre-filled syringes have not been used in clinical studies.
Angiomax
Bulk Drug Substance. In December 1999, we entered into a commercial development and supply agreement with Lonza Braine, S.A., or Lonza Braine, which was formerly known as UCB Bioproducts S.A., for the development and supply of Angiomax bulk drug substance. Together with Lonza Braine, we developed a second generation chemical synthesis process to improve the economics of manufacturing Angiomax bulk drug substance. This process, which was approved by the FDA in May 2003 and is used in the manufacture of Angiomax bulk drug substance today, is known as the Chemilog process. We have agreed that, during the term of the agreement, we will purchase a substantial portion of our Angiomax bulk drug substance manufactured using the Chemilog process from Lonza Braine at agreed upon prices. Following the expiration of the agreement or if we terminate the agreement prior to its expiration, Lonza Braine has agreed to transfer the development technology to us. If we engage a third party to manufacture Angiomax for us using the Chemilog process prior to bivalirudin becoming a generic drug in the United States, we will be obligated to pay Lonza Braine a royalty based on the amount paid by us to the third-party manufacturer. In June 2015, we amended the agreement with Lonza providing for the transition of the manufacture of Angiomax bulk drug substance from the Chemilog process to a solid phase peptide synthesis process. The amendment extends the expiration date of the agreement to December 31, 2019, subject to automatic renewals of consecutive three-year periods unless either party provides notice of non-renewal within eighteen months prior to the expiration of the initial term or any renewal term. We may only terminate the agreement prior to its expiration in the event of a material breach by Lonza Braine, if such breach is not cured within 30 days. In December 2016, Lonza Braine agreed to sell its peptide business, including the production and supply of Angiomax bulk drug substance, to Polypeptide Laboratories Holding AB.
Drug Product. In March 2011, we entered into a master agreement with Patheon International A.G., or Patheon International, for the manufacture of Angiomax drug product. Pursuant to the agreement, as amended, Patheon International conducts the fill-finish of Angiomax drug product for our commercial sale supply in accordance with binding commitments in a forecast provided by us. Our agreement with Patheon International expires in December 2018, subject to automatic renewals for successive terms of two years each unless either party gives written notice to the other party of its intention to terminate the agreement at least 18 months prior to the end of the then current term. Either party may terminate the agreement for material breach by the other party, if the material breach is not cured within 60 days after written notice, unless the breach by its nature is not curable. In such case, the non-breaching party has the right to terminate the agreement immediately upon providing written notice as long as the written notice is provided within 30 days of the terminating party receiving notice of the breach. We have the right to terminate the agreement upon
8
30 days’ prior written notice in the event that any governmental agency takes any action, or raises any objection, that prevents us from importing, exporting, purchasing or selling Angiomax.
In January 2012, we entered into a contract manufacturing agreement with APP. Under the contract manufacturing agreement, as amended, we agreed to purchase from APP a specified minimum percentage of our requirements for Angiomax finished product for the sale of the Angiomax product in the United States. We agreed to pay APP a fixed price per vial supplied and to reimburse APP for specified development costs and capital expenditures made by APP. The term of the contract manufacturing agreement ends on April 30, 2021, but may be extended, at our sole option, for an additional term of two years. If a generic form of bivalirudin for injection is marketed by APP or another third party during the term of the contract manufacturing agreement, we have the right to renegotiate the price and minimum quantity terms of the contract manufacturing agreement and, if such terms cannot be agreed to by the parties, we will have the right to terminate the contract manufacturing agreement upon 90 days prior written notice. Either party may terminate the contract manufacturing agreement in the event of a material breach by the other party, effective immediately in the case of a non-curable breach and effective upon 60 days prior written notice in the case of a curable breach if such breach is not cured within such 60-day period. Either party may also terminate the contract manufacturing agreement if the other party undergoes bankruptcy events. We may terminate the contract manufacturing agreement upon at least 12 months’ prior written notice if we decide to discontinue marketing the Angiomax product in the United States or upon 30 days’ prior written notice in the event that any government or regulatory authority prevents us from purchasing or selling the Angiomax product in the United States.
Competition
The development and commercialization of new drugs is highly competitive. We face competition from pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Many of our competitors are substantially larger than we are and have substantially greater capital resources, research and development capabilities and experience, and financial, technical, manufacturing, marketing and human resources than we have. Additional mergers and acquisitions in the pharmaceutical industry may result in even more resources being concentrated in our competitors.
In addition, our competitors may develop, market or license products or other novel technologies that are more effective, safer or less costly than any that have been or are being developed by us, or may obtain marketing approval for their products from the FDA or equivalent foreign regulatory bodies more rapidly than we may obtain approval for ours. We compete, in the case of our Angiomax, and expect to compete, in the cases of inclisiran, on the basis of product efficacy, safety, ease of administration, price and economic value compared to drugs used in current practice or currently being developed.
Inclisiran
The market targeting hypercholesterolemia is highly competitive. First line therapy consisting of HMG-CoA reductase inhibitors, commonly known as statins, which block production of cholesterol by the liver and increase clearance of LDL-C from the bloodstream, are widely prescribed and subject to generic competition. If approved, we expect inclisiran to compete with the two currently approved and marketed anti-PCSK9 antibodies, Amgen’s Repatha and Sanofi’s Praluent, which are indicated for the treatment of hypercholesterolemia in the United States and Europe. In addition, we believe other PCSK9-targeted approaches are in development at a number of companies. Oral products that lower LDL-C, if approved, including Bempedoic Acid (ETC-1002), which is being developed by Esperion Therapeutics Inc., and gemcabene, which is being developed by Gemphire Therapeutics Inc. and antisense oligonucleotide-based therapies in development may also be competitive with inclisiran, if approved.
Angiomax
Angiomax has been subject to generic competition since 2015. Hospira, Mylan, APP, Apotex Inc. and Dr. Reddy’s Laboratories are currently selling generic versions of Angiomax. In addition, a number of companies in addition to Hospira, Mylan, APP, Apotex Inc. and Dr. Reddy’s Laboratories have filed ANDAs for their generic versions of Angiomax. In addition to the generic versions and the ready-to-use version of bivalirudin currently being sold, Angiomax could be subject to further generic competition in the United States from Teva and other generic ANDA filers that we have settled with under the circumstances set forth in our respective settlement agreements with such parties and upon a final approval of each company's ANDA filings by the FDA. Pliva Hrvatska DOO, an affiliate of Teva, currently has tentative approval for its ANDA filing for its generic version of Angiomax. Other ANDA filers may commercialize their products ‘at risk’ if they receive final approval of their respective ANDA filings and are not subject to a Hatch-Waxman 30-month stay. Further, we remain in infringement litigation involving the ‘727 patent and ‘343 patent with the other ANDA filers. See Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K for descriptions of our litigation with ANDA filers and related settlements. There can be no assurance as to the outcome of our infringement litigation. We may continue to incur further legal expenses related to these matters. In addition, in January 2018 Baxter announced that the FDA approved Baxter’s ready-to-use formulation of bivalirudin.
9
In addition to generic versions and the ready-to-use version of Angiomax, Angiomax competes with heparin and treatment regimens combining heparin and GP IIb/IIIa inhibitors. Heparin is widely used in patients with ischemic heart disease, including PCI procedures. Heparin is manufactured and distributed by a number of companies as a generic product and is sold at a price that is significantly less than the price for Angiomax. Although their use may have decreased in recent years, GP IIb/IIIa inhibitors are widely used and some physicians believe they offer superior efficacy in high risk patients as compared to Angiomax.
In some circumstances, Angiomax competes with other anticoagulant drugs for the use of hospital financial resources. For example, many U.S. hospitals receive a fixed reimbursement amount per procedure for the angioplasties and other treatment procedures they perform. As this amount is not based on the actual expenses the hospital incurs, hospitals may choose to use either Angiomax or heparin or a combination of heparin and a GP IIb/IIIa inhibitor but not necessarily more than one of these drugs.
Patents, Proprietary Rights and Licenses
Our success will depend in part on our ability to protect the products we acquire or license by obtaining and maintaining patent protection both in the United States and in other countries. We rely upon trade secrets, know-how, continuing technological innovations, contractual restrictions and licensing opportunities to develop and maintain our competitive position. We plan to prosecute and defend patents or patent applications we file, acquire or license.
Inclisiran. We have exclusively licensed from Alnylam patents covering RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases for purposes of developing and commercializing such RNAi therapeutics. Some of these patents are directed to general RNAi technology and expire between 2020 and 2028 in the United States. Other patents are directed to compositions of the inclisiran product being developed under our license from Alnylam and to methods of treatment using such inclisiran product and the patents expire in 2027 and 2028 in the United States. In addition, Alnylam has filed and is prosecuting a number of patent applications in the United States and in certain foreign countries. One of these applications, which, if issued, expires in December 2033, contains claims directed to specific compositions of the inclisiran product we are developing and methods of administrating such compositions.
Angiomax. We have exclusively licensed from Biogen Idec and Health Research Inc., or HRI, patents and patent applications covering Angiomax and Angiomax analogs and other novel anticoagulants as compositions of matter, and processes for using Angiomax and Angiomax analogs and other novel anticoagulants. We also own two U.S. patents covering a more consistent and improved Angiomax drug product and the processes by which it is made.
The principal U.S. patents covering Angiomax include the ‘727 patent and the ‘343 patent and previously included the ‘404 patent. The ‘404 patent covered the composition of matter of Angiomax. The ‘404 patent was set to expire in March 2010, but the term was extended to December 15, 2014 by the U.S. Patent and Trademark Office, or PTO, under the Hatch-Waxman Act. As a result of our study of Angiomax in the pediatric setting, we had an additional six-month period of pediatric exclusivity following expiration of the ‘404 patent. This period of exclusivity expired in June 2015.
In the second half of 2009, the PTO issued to us the ‘727 patent and the ‘343 patent, covering a more consistent and improved Angiomax drug product and the processes by which it is made. The ‘727 patent and the ‘343 patent are set to expire in July 2029, which includes pediatric exclusivity. In response to Paragraph IV Certification Notice letters we received with respect to ANDAs filed by a number of parties with the FDA seeking approval to market generic versions of Angiomax, we have filed lawsuits against the ANDA filers alleging patent infringement of the ‘727 patent and ‘343 patent. We have settled our patent infringement litigations with Teva, APP, Sun, Dr. Reddy’s, Aurobindo Pharma Limited, Sagent Pharmaceuticals Inc. and Accord Healthcare Inc., but remain in infringement litigation involving the ‘727 patent and ‘343 patent with the other ANDA filers. Our patent infringement litigation involving the ‘727 patent and ‘343 patent and related settlements are described in more detail in Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K.
In Europe, the principal patent covering Angiomax expired in August 2015. This patent covered the composition of matter of Angiomax. As a result, we do not have market exclusivity for Angiomax in Europe. We are in the process of voluntarily discontinuing and withdrawing Angiomax from the market outside of North America and ceasing related commercial activities.
Ionsys. As a result of our acquisition of Incline Therapeutics, Inc., or Incline, in 2013, we acquired a portfolio of patents and patent applications covering the Ionsys device and its uses. Some of these patents and patent applications were exclusively licensed from ALZA. The expiration dates of patents covering the Ionsys device and its use range from January 2019 to February 2033 in the United States. In Europe, the expiration dates of patents covering the Ionsys device and its use range from October 2019 to March 2033. We are also currently prosecuting patent applications relating to Ionsys in the United States and in certain foreign countries. In June 2017, we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open as we continue to explore opportunities to partner or divest Ionsys.
10
The patent positions of pharmaceutical and biotechnology firms like us can be uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued. Consequently, we do not know whether any of the patent applications we acquire, license or file will result in the issuance of patents or, if any patents are issued, whether they will provide significant proprietary protection or will be challenged, circumvented or invalidated. Because unissued U.S. patent applications filed prior to November 29, 2000 and patent applications filed within the last 18 months are maintained in secrecy until patents issue, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain of the priority of inventions covered by pending patent applications. Moreover, we may have to participate in interference proceedings declared by the PTO to determine priority of invention, or in opposition proceedings in a foreign patent office. Participation in these proceedings could result in substantial cost to us, even if the eventual outcome is favorable to us. Even issued patents may not be held valid by a court of competent jurisdiction. An adverse outcome could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties or require us to cease using such technology.
The development of hypercholesterolemia products and RNAi therapeutics are intensely competitive. A number of pharmaceutical companies, biotechnology companies, universities and research institutions have filed patent applications or received patents in these fields. Some of these patent applications could be competitive with applications we have acquired or licensed, or could conflict in certain respects with claims made under our applications. Such conflict could result in a significant reduction of the coverage of the patents we have acquired or licensed, which would have a material adverse effect on our business, financial condition and results of operations. In addition, if patents are issued to other companies that contain competitive or conflicting claims with claims of our patents and such claims are ultimately determined to be valid, we may not be able to obtain licenses to these patents at a reasonable cost, or develop or obtain alternative technology.
We also rely on trade secret protection for our confidential and proprietary information. However, others may independently develop substantially equivalent proprietary information and techniques. Others may also otherwise gain access to our trade secrets or disclose such technology. We may not be able to meaningfully protect our trade secrets.
It is our policy to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements generally provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees and consultants, the agreements provide that all inventions conceived by the individual shall be our exclusive property. These agreements may not provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
We have a number of trademarks that we consider important to our business. The Medicines Company® name and logo, and Angiomax®, Angiox® and Ionsys® names and logos are either our registered trademarks or our trademarks in the United States and other countries. We have also registered some of these marks in a number of foreign countries. Although we have a foreign trademark registration program for selected marks, we may not be able to register or use such marks in each foreign country in which we seek registration. We believe that our products are identified by our trademarks and, thus, our trademarks are of significant value. Each registered trademark has a duration of 10 to 15 years, depending on the date it was registered and the country in which it is registered, and is subject to an infinite number of renewals for a like period upon continued use and appropriate application. We intend to continue the use of our trademarks and to renew our registered trademarks based upon each trademark’s continued value to us.
License Agreements
A summary of our licenses for our products and products in development is set forth below.
Alnylam License Agreement. In February 2013, we entered into a license and collaboration agreement with Alnylam to develop, manufacture and commercialize therapeutic products targeting the human PCSK9 gene based on certain of Alnylam’s RNAi technology. Under the terms of the agreement, we obtained the exclusive, worldwide right under Alnylam’s technology to develop, manufacture and commercialize PCSK9 products for the treatment, palliation and/or prevention of all human diseases. We paid Alnylam $25.0 million in an initial license payment and agreed to pay up to $180.0 million in success-based development and commercialization milestones. In December 2014, we paid a development milestone payment of $10.0 million based upon the initiation of a Phase 1 clinical trial for inclisiran and in January 2018 we paid a development milestone payment of $20 million in connection with the first dosing of a subject in a pivotal study for inclisiran. In addition, Alnylam will be eligible to receive scaled double-digit royalties based on annual worldwide net sales of PCSK9 products by us or our affiliates and sublicensees. Royalties to Alnylam are payable on a product-by-product and country-by-country basis until the last to occur of the expiration of patent rights in the applicable country that cover the applicable product, the expiration of non-patent regulatory exclusivities for such product in such country, or the twelfth anniversary of the first commercial sale of the product in such country. The royalties are subject to reduction in specified circumstances. We are also responsible for paying royalties, and in some cases milestone payments, owed by Alnylam to its licensors with respect to intellectual property covering these products.
11
The agreement expires when the last royalty term expires under the agreement, unless earlier terminated. We may terminate the agreement at any time with four months prior written notice to Alnylam. Either party may terminate the agreement on 60 days (10 days in the event of a payment breach) prior written notice if the other party materially breaches the agreement and fails to cure such breach within the applicable notice period. Such cure period may be extended in certain circumstances. If the agreement is terminated by us for convenience or by Alnylam for our uncured material breach or challenge of the patents licensed from Alnylam, we have agreed to grant a license to Alnylam under certain of its technology developed in the course of our activities under the agreement, subject to a royalty to be negotiated between the parties, and we will provide certain other assistance to Alnylam to continue the development and commercialization of the products. The exclusivity restrictions imposed on us will survive termination of the agreement for specified periods of time if we terminate the agreement for convenience or if Alnylam terminates the agreement for cause or for a patent challenge by us.
Angiomax. In March 1997, we entered into an agreement with Biogen, Inc., a predecessor of Biogen Idec, for the license of the anticoagulant pharmaceutical bivalirudin, which we have developed and market as Angiomax. Under the terms of the agreement, we acquired exclusive worldwide rights to the technology, patents, trademarks, inventories and know-how related to Angiomax. In exchange for the license, we paid $2.0 million on the closing date and are obligated to pay up to an additional $8.0 million upon the first commercial sales of Angiomax for the treatment of AMI in the United States and Europe. In addition, we are obligated to pay royalties on sales of Angiomax and on any sublicense royalties on a country-by-country basis earned until the later of the date 12 years after the date of the first commercial sales of the product in a country and the date on which the product or its manufacture, use or sale is no longer covered by a valid claim of the licensed patent rights in such country. The royalty rate due to Biogen Idec on sales increases as annual sales of Angiomax increase. Under the agreement, we are obligated to use commercially reasonable efforts to develop and commercialize Angiomax in specified European markets, including for PTCA and AMI indications. The license and rights under the agreement remain in force until our obligation to pay royalties ceases. As of December 15, 2014, we no longer owe royalties to Biogen Idec or HRI relating to sales of Angiomax in the United States. Either party may terminate the agreement for material breach by the other party, if the material breach is not cured within 90 days’ after written notice. In addition, we may terminate the agreement for any reason upon 90 days’ prior written notice. We are currently in a dispute with Biogen regarding Angiomax royalties under our license agreement; see Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K for additional details. During 2017, we incurred approximately $0.6 million in royalties related to Angiomax sales outside the United States under our agreement with Biogen Idec.
In March 1997, in connection with entering into the Biogen Idec license, Biogen Idec assigned to us a license agreement with HRI under which Biogen Idec had licensed HRI’s right to a specified patent application held jointly with Biogen Idec which resulted in a series of U.S. patents including the ‘404 patent. Under the terms of the agreement, we have exclusive worldwide rights to HRI’s rights to the licensed patent application and patents arising from the licensed patent application, other than rights for noncommercial research and educational purposes, which HRI retained. We are obligated to pay royalties on sales of Angiomax and on any sublicense income we earn. The royalty rate due to HRI on sales increases as annual sales of Angiomax increase. Under the agreement, we are obligated to use commercially reasonable efforts to research and develop, obtain regulatory approval and commercialize Angiomax. The license and rights under the agreement remain in force until the expiration of the last remaining patent granted under the licensed patent application. HRI may terminate the agreement for a material breach by us, if the material breach is not cured within 90 days after written notice or, in the event of bankruptcy, liquidation or insolvency, immediately on written notice. In addition, we may terminate the agreement for any reason upon 90 days’ prior written notice upon payment of a termination fee equal to the minimum royalty fee payable under the license agreement. As of December 15, 2014, we no longer owe royalties to HRI relating to sales of Angiomax in the United States. During 2017, we incurred approximately $0.2 million in royalties related to Angiomax sales outside the United States under the agreement with HRI.
Ionsys. As a result of our acquisition of Incline, we are a party to a license agreement with ALZA through our Incline subsidiary. Under the terms of the agreement, Incline acquired from ALZA certain rights to the Ionsys product and ALZA transferred to Incline specified trademarks, know-how, domain names and tangible assets relating to the Ionsys product. ALZA also granted Incline worldwide licenses under specified patent rights and know-how to develop, manufacture and commercialize iontophoretic transdermal systems providing delivery under the influence of an electric current which is from a source external to the human body of specified fentanyl analogs. The licenses granted by ALZA under the agreement are exclusive with respect to specified patent rights and know-how and nonexclusive under other specified patent rights.
We, through our subsidiary, Incline have the sole responsibility for the development and commercialization of licensed products under the agreement, and are required to use commercially reasonable efforts to develop and commercialize at least one licensed product in the United States, United Kingdom, Germany, France, Italy and Spain.
In addition to the other rights and licenses granted to Incline under the ALZA Agreement, if, at any time during the 10-year period following the date of the agreement, ALZA wishes to grant a license under specified licensed patents to a third party, other than in connection with the settlement of litigation, to develop, manufacture and/or commercialize specified systems that deliver opioid compounds or combinations of opioid compounds with fentanyl analogs or generic compounds, in each case that do not
12
contain any active compound that is proprietary to, licensed by or otherwise controlled by the third party or, except for specified fentanyl analogs, by ALZA, then we will have a right of first negotiation to obtain the proposed license.
If, at any time during the 10-year period following the date of the agreement, we wish to obtain from ALZA a license under specified licensed patents to develop, manufacture and/or commercialize specified systems that deliver generic compounds, combinations of generic compounds with fentanyl analogs or compounds exclusively owned, licensed or otherwise controlled by Incline, alone or in combination with generic compounds or specified fentanyl analogs, in each case that do not contain any active compound, other than specified fentanyl analogs, that is proprietary to, licensed by or otherwise controlled by ALZA or that is a generic drug owned, licensed or controlled by ALZA, then upon notice to ALZA of our desire to obtain the license, ALZA will be obligated to negotiate in good faith with Incline to grant the proposed license.
Under the ALZA Agreement, Incline paid ALZA an upfront payment and we will be obligated to pay ALZA up to an aggregate of $32.5 million in regulatory and commercial launch milestone payments and up to an aggregate of $83.0 million in sales milestone payments if certain specified milestones are met. ALZA is also entitled to specified royalties based on net sales of licensed products, on a licensed product-by-licensed product and country-by-country basis, during the period commencing on the first commercial sale of the licensed product in the applicable country and ending on the latest of the expiration of the licensed patents covering the licensed product, the expiration of applicable regulatory exclusivity or the 20th anniversary of the first commercial sale of the licensed product in the applicable country. We will also be required to pay amounts that become payable, if any, under specified ALZA third party licenses as a result of our development and commercialization of licensed products.
Either ALZA or we may terminate the agreement due to the other party’s material breach of the agreement if such breach is not cured within 60 days of notice of the breach except that if the breach relates solely to the United States, any country in Europe or any other country in the world, the termination right shall apply to the United States, applicable countries in Europe or the rest of the world (other than the US and Europe), as the case may be. ALZA may also terminate the agreement due to our bankruptcy. Neither party has any discretionary right to terminate the agreement. If not terminated earlier pursuant to its terms, the agreement terminates upon the expiration and satisfaction of all payment obligations under the agreement.
Although we continue to seek a partnership or divestiture transaction for Ionsys, in June 2017, we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open.
Customers
In the United States, we currently sell branded Angiomax, and we sold Minocin IV and Orbactiv until our divesture of the products to Melinta in January 2018, to our sole source distributor, ICS. ICS accounted for 5%, 48% and 87% of our net product revenue for 2017, 2016 and 2015, respectively. At December 31, 2017 and 2016, amounts due from ICS represented approximately $2.9 million and $2.2 million, or 27% and 11%, of gross accounts receivable, respectively. We also have a supply and distribution arrangement with Sandoz under which Sandoz sells authorized generic Angiomax (bivalirudin) in the United States. We generate total net revenue under the sales and distribution arrangement with Sandoz by making products sales to Sandoz (which is recorded in net product revenues) and receiving royalty payments from Sandoz in respect of Sandoz’s sales of authorized generic Angiomax (bivalirudin) (which is recorded in royalty revenues). Product sales to Sandoz accounted for 55% and 23% of our net product revenues for 2017 and 2016, respectively. At December 31, 2017 and 2016, amounts due from Sandoz related to product sales were approximately $0.9 million or 8% and $5.6 million or 27%, respectively, of gross accounts receivable. At December 31, 2017 and 2016, amounts due from Sandoz related to royalty revenues represented approximately $4.2 million or 40% and $9.1 million or 43%, respectively, of gross accounts receivable.
Government Regulation
Government authorities in the United States and other countries extensively regulate the research, testing, manufacturing, labeling, safety, advertising, promotion, storage, sales, distribution, import, export and marketing, among other things, of our products and product candidates. In the United States, the FDA regulates drugs and biologics, under the Federal Food, Drug, and Cosmetic Act and the Public Health Service Act respectively and their implementing regulations. We cannot market or commercially distribute a drug until we have submitted an application for marketing authorization to the FDA, and the FDA has approved it. Both before and after approval is obtained, violations of regulatory requirements may result in various adverse consequences, including, among other things, clinical holds, untitled letters, warning letters, fines and other monetary penalties, the FDA’s delay in approving or refusal to approve a product, product recall or seizure, suspension or withdrawal of an approved product from the market, interruption of production, operating restrictions, injunctions and the imposition of civil or criminal penalties. The steps required before a drug may be approved by the FDA and marketed in the United States generally include:
• | pre-clinical laboratory tests, animal studies and formulation studies; |
13
• | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin; |
• | adequate and well-controlled clinical trials to establish the safety and efficacy of the drug for each indication; |
• | submission to the FDA of an NDA or BLA; |
• | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practices, or cGMP; and |
• | FDA review and approval of the NDA or BLA. |
Pre-Clinical Tests
Pre-clinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. The results of the pre-clinical tests, together with manufacturing information, analytical data, clinical study protocol(s), and other information, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA puts the trial on clinical hold because of concerns or questions about issues such as the design of the clinical trial(s) or the safety of the drug for administration to humans. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. Submission of an IND does not necessarily result in the FDA allowing clinical trials to commence. In addition, the FDA may impose a clinical hold at any time which includes during an ongoing clinical trial if, for example, safety concerns arise, in which case the trial cannot recommence without the FDA’s authorization. A clinical hold can result in a substantial delay and expense.
Clinical Trials
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring subject safety, and the effectiveness criteria, or endpoints, to be evaluated. Each protocol intended to study investigational new drugs in the United States must be submitted to the FDA as part of the IND, and the FDA may or may not allow that trial to proceed. Each trial also must be reviewed and approved by an independent Institutional Review Board, or IRB, at each proposed study site before it can begin.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined.
• | Phase 1 usually involves the initial introduction of the investigational drug into people to evaluate its safety, dosage tolerance, pharmacokinetics, and, if possible, to gain an early indication of its effectiveness. |
• | Phase 2 usually involves trials in a limited patient population to: evaluate dosage tolerance and appropriate dosage; identify possible adverse effects and safety risks; and evaluate preliminarily the efficacy of the drug for specific indications. |
• | Phase 3 trials usually involve administration of the drug to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to evaluate safety, and statistically evaluate the efficacy of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
We cannot guarantee that Phase 3 testing of inclisiran will be completed successfully within any specified period of time, if at all. Furthermore, we, the IRB, or the FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Sponsors are required to publicly disseminate information about ongoing and completed clinical trials on a government website administered by the National Institutes of Health, or NIH, and are subject to civil money penalties and other civil and criminal sanctions for failing to meet these obligations.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the pre-clinical studies and of the clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of an NDA or BLA requesting approval to market the product for one or more indications. The submission of an NDA or BLA typically requires the payment of a significant user fee to FDA. The FDA conducts a preliminary review of all NDAs or BLAs within the first 60 days after submission before accepting them for filing to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA or BLA for filing. If the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Before approving an application, the FDA
14
usually will inspect the facility or the facilities at which the drug is manufactured, and will not approve the product unless cGMP compliance is satisfactory. The FDA also often inspects one or more sites at which the pivotal clinical trial or trials were conducted to ensure the integrity of the data and compliance with Good Clinical Practice, or GCP, requirements. If the FDA determines the application, data or manufacturing facilities are not acceptable, the FDA may outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If the FDA evaluation of the NDA and the various inspections are favorable, the FDA may issue an approval letter, which authorizes commercial marketing of the drug with specific prescribing information for a specific indication. As a condition of approval of an application, the FDA may request or require post-market testing and surveillance to monitor the drug’s safety or efficacy. The FDA also may impose requirements designed to ensure the safety of the drug up to and including distribution and use restrictions under a Risk Evaluation and Mitigation Strategy, or REMS. After approval, certain changes to the approved product, such as adding new indications, manufacturing changes, or additional labeling claims, are subject to further FDA review and approval before the changes can be implemented. The testing and approval process requires substantial time, effort and financial resources, and we cannot be sure that any approval will be granted on a timely basis, if at all. Product approvals may be further limited or withdrawn if compliance with regulatory standards is not maintained or safety or other problems are identified following initial marketing.
The FDA regulates combinations of products that cross FDA centers, such as drug, biologic or medical device components that are physically, chemically or otherwise combined into a single entity, as a combination product. The FDA center with primary jurisdiction for the combination product will take the lead in the premarket review of the product, with the other center consulting or collaborating with the lead center, and often will require approval of only a single application, such as an NDA or BLA. The FDA’s Office of Combination Products, or OCP, determines which center will have primary jurisdiction for the combination product based on the combination product’s “primary mode of action.” A mode of action is the means by which a product achieves an intended therapeutic effect or action. The primary mode of action is the mode of action that provides the most important therapeutic action of the combination product, or the mode of action expected to make the greatest contribution to the overall intended therapeutic effects of the combination product.
Manufacturing Requirements
After the FDA approves a product, we, our suppliers, and our contract manufacturers must comply with a number of post-approval requirements. For example, holders of an approved NDA or BLA are required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with certain requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, we and our contract manufacturers must continue to expend time, money, and effort to maintain compliance with cGMP and other aspects of regulatory compliance. In addition, discovery of problems such as safety problems may result in changes in labeling, imposition or modification of a REMS, or other restrictions on a product manufacturer, or NDA or BLA holder, including removal of the product from the market.
We use and will continue to use third-party manufacturers to produce our products in clinical and commercial quantities, and we cannot be sure that future FDA inspections will not identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product may result in restrictions on a product, manufacturer, or holder of an approved NDA or BLA, including withdrawal of the product from the market. Also, new government requirements may be established that could delay or prevent regulatory approval of our products under development.
Abbreviated New Drug Applications and Section 505(b)(2) New Drug Applications
Once an NDA is approved, the product covered thereby becomes a listed drug that can, in turn, be relied upon by potential competitors in support of approval of an ANDA or 505(b)(2) application. The FDA may approve an ANDA if the product is the same in important respects as the listed drug or if the FDA has declared it suitable for an ANDA submission. In these situations, applicants must submit studies showing that the product is bioequivalent to the listed drug, meaning that the rate and extent of absorption of the drug does not show a significant difference from the rate and extent of absorption of the listed drug. ANDA applicants are not required to conduct or submit results of preclinical or clinical tests to prove the safety or effectiveness of their drug product, other than the requirement for bioequivalence testing. Conducting bioequivalence studies is generally less time-consuming and costly than conducting pre-clinical and clinical trials necessary to support an NDA or BLA. Drugs approved via ANDAs on the basis that they are the “same” as a listed drug are commonly referred to as “generic equivalents” to the listed drug, and can often be and are substituted by pharmacists under prescriptions written for the original listed drug. A number of ANDAs have been filed and approved with respect to Angiomax. The regulations governing marketing exclusivity and patent protection are complex, and until the outcomes of our effort to extend the patent term and our patent infringement litigation, we may not know the disposition of such ANDA submissions.
15
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s product or a method of using the product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. An ANDA applicant relying upon a listed drug is required to certify to the FDA concerning any patents listed for the listed drug product in the FDA’s Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval. Specifically, the applicant must certify with respect to each patent that:
• | the required patent information has not been filed; |
• | the listed patent has expired; |
• | the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
• | the listed patent is invalid, unenforceable, or will not be infringed by the new product. |
A certification that the proposed generic product will not infringe the already approved product’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the ANDA applicant does not challenge the listed patents or indicate that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of a Paragraph IV certification notice automatically prevents the FDA from granting final approval to the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA also will not be approved until any applicable non-patent exclusivity period, such as exclusivity for obtaining approval of a new chemical entity, for the referenced product has expired, unless the exclusivity period protects an indication or other aspect of labeling that can be “carved out” of the labeling for the proposed generic product. Federal law provides a period of five years following approval of a drug containing no previously approved active moiety during which ANDAs for generic versions of those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original product approval. Federal law provides for a period of three years of exclusivity during which the FDA cannot grant effective approval of an ANDA if a listed drug contains a previously approved active moiety but FDA requires as a condition of approval new clinical trials conducted by or for the sponsor. This three-year exclusivity period often protects changes to a previously approved product, such as a new dosage form, route of administration, combination, or indication. Under the Best Pharmaceuticals for Children Act, federal law also provides that periods of patent and non-patent marketing exclusivity listed in the Orange Book for a drug may be extended by six months if the NDA sponsor conducts pediatric studies identified by the FDA in a written request. For written requests issued by the FDA after September 27, 2007, the date of enactment of the Food and Drug Administrative Amendment Act (FDAAA), the FDA must grant pediatric exclusivity no later than nine months prior to the date of expiration of patent or non-patent exclusivity in order for the six-month pediatric extension to apply to that exclusivity period.
Most drug products obtain FDA marketing approval pursuant to an NDA or an ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the FDA’s previous approval of a similar product, or published literature, in support of its application. 505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. If the 505(b)(2) applicant can establish that reliance on the FDA’s previous approval is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all or some of the labeled indications for which the referenced product has been approved, as well as for any new indication(s) sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would be required to do so. As a result, approval of a 505(b)(2) NDA can be prevented until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
16
Biologics Price Competition and Innovation Act
Under the Biologics Price Competition and Innovation Act, or BPCIA, enacted in the United States in 2010, the FDA now has the authority to approve biosimilar and interchangeable versions of previously-approved biological products through an abbreviated pathway following periods of data and marketing exclusivity. A competitor seeking approval of a biosimilar must file an application to show its molecule is highly similar to an approved innovator biologic, also known as a reference product, address the challenges of biologics manufacturing, and include a certain amount of safety and efficacy data which the FDA will evaluate on a case-by-case basis. A competitor seeking approval of an interchangeable biological product must demonstrate not only biosimilarity but also that the products can be expected to produce the same clinical effects in any given patient. Under the data protection provisions of this law, the FDA cannot accept a biosimilar application until four years, or approve a biosimilar application until 12 years, after initial marketing approval of the reference product. Although the FDA has issued draft guidance documents, to date it has not issued any regulations or final guidance explaining how it will implement the abbreviated BLA or biosimilar provisions enacted in 2010 under the BPCIA, including the exclusivity provisions for reference products. Regulators in the European Union and other countries also have been given the authority to approve biosimilars. The extent to which a biosimilar, once approved, will be approved as interchangeable with or substituted for the innovator biologic in a way that is similar to traditional generic substitution for non-biologic products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. A number of states have recently considered and, in some cases, adopted legislation governing the substitution of interchangeable biosimilars for the reference product.
U.S. Healthcare Reform
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, or PPACA, which was amended by the Health Care and Education Reconciliation Act of 2010. The PPACA, as amended, contains numerous provisions that impact the pharmaceutical and healthcare industries and it empowers the HHS to implement a number of related healthcare reform measures that are likely to have a broad impact on the pharmaceutical and healthcare industry. We are continually evaluating the impact of the PPACA and other healthcare reform-related programs and regulations on our business, including potential PPACA repeal and replacement. As of the date of this Annual Report on Form 10-K, we have not identified any provisions that currently materially impact our business and results of operations. However, the potential impact of the PPACA and other healthcare reform measures on our business and results of operations is inherently difficult to predict because many of the details regarding the implementation of this legislation have not been determined. In addition, the impact on our business and results of operations may change as and if our business evolves. On December 22, 2017, Congress passed and President Trump signed bill entitled “To provide for reconciliation pursuant to titles II and V of the concurrent resolution on the budget for fiscal year 2018,” which, among other things, repealed the PPACA individual mandate. President Trump and HHS Secretary Azar have announced support for regulatory provisions that would limit PPACA and number of healthcare reform programs initiated under the Obama administration. It remains unclear whether replacement programs will include similar limitations affecting reimbursement, although scrutiny over drug pricing and government costs is expected to continue. Similarly, efforts in Congress to reform Medicare and Medicaid may impact the pharmaceutical and healthcare industries.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we obtain regulatory approval. Sales of inclisiran, if approved, will depend, in part, on the extent to which the costs of the product will be covered by third-party payers, including government health programs such as Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a payer will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payer will pay for the drug product once coverage is approved. Third-party payers may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the approved drugs for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive health economic studies in order to demonstrate the economics of the product, in addition to incurring the costs required to obtain FDA or other comparable regulatory approvals. Even if a drug product is covered, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. Third-party payers are increasingly challenging the prices charged for medical products and services and examining the medical necessity and economic benefit of medical products and services, in addition to their safety and efficacy. If these third-party payers do not consider inclisiran to be economically beneficial compared to other available therapies, they may not cover it after approval as a benefit under their plans. Third-party payers may provide coverage, but place stringent limitations on such coverage, such as requiring alternative treatments to be tried first. The U.S. government, state legislatures and foreign
17
governments have shown significant interest in implementing cost-containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
Pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of inclisiran to currently available therapies. For example, the European Union provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a drug product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. There can be no assurance that any country that has price controls or reimbursement limitations for drug products will allow favorable reimbursement and pricing arrangements for inclisiran.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on drug pricing. Coverage policies, third party reimbursement rates and drug pricing regulation may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Foreign Regulations
In addition to regulations in the United States, we are subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In Europe, for example, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee for each clinical trial, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, the clinical trial may proceed in that country.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with Good Clinical Practices, or GCPs, and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational drug or biological product under European Union regulatory systems, we must submit a marketing authorization application. The application used to file the NDA or BLA in the United States is similar to that required in Europe, with the exception of, among other things, country-specific document requirements.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Drugs can be authorized in the European Union by using either the centralised authorisation procedure or national authorization procedures.
Centralised EMA Procedure. The EMA, formerly the EMEA, implemented the centralised procedure for the approval of human medicines to facilitate marketing authorisations that are valid throughout the European Union. This procedure results in a single marketing authorisation issued by the EMA that is valid across the European Union, as well as Iceland, Liechtenstein and Norway. The centralised procedure is compulsory for human medicines that are derived from biotechnology processes, contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions, and officially designated orphan medicines.
18
For drugs that do not fall within these categories, an applicant has the option of submitting an application for a centralised marketing authorisation to the EMA, as long as the drug concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health.
National Procedures. There are also three other possible routes to authorize medicinal products outside the scope of the centralised procedure and the EMA:
• | National procedures. A medicine is authorised in one European Union member state in accordance with the national procedures of that country. If a marketing authorisation holder wishes to apply subsequently for additional marketing authorisations in other member states for that product, the mutual recognition procedure must be used. |
• | Decentralised procedure. Using the decentralised procedure, an applicant may apply for simultaneous authorization in more than one European Union country of medicinal products that have not yet been authorized in any European Union country and that do not fall within the mandatory scope of the centralised procedure. |
• | Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one European Union member state, in accordance with the national procedures of that country, as described above. Following this, further marketing authorizations can be sought from other European Union countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization. |
Research and Development
Our research and development expenses, excluding discontinued operations, totaled $138.4 million in 2017, $92.1 million in 2016 and $90.4 million in 2015.
Employees
As of February 28, 2018 we employed approximately 170 persons worldwide. We believe that our success depends greatly on our ability to identify, attract and retain capable employees. We have assembled a management team with significant experience in drug development and commercialization. Our employees are not represented by any collective bargaining unit, and we believe our relations with our employees are good.
Workforce Restructuring
In 2017 and 2018, we conducted a series of workforce reductions, as described below. Our intention is to reduce our personnel to less than 60 employees as we announced in October 2017. Upon signing release agreements, affected employees have received, or are eligible to receive, a severance package, including reduction payments and fully paid health care coverage and outplacement services for six months to a year.
In June 2017, in connection with our voluntary discontinuation and withdrawal of Ionsys from the market in the United States, we commenced a workforce reduction, which resulted in the reduction of 57 employees, representing approximately 15% of our workforce.
Commencing in December 2017 and continuing through 2018, we are implementing a series of workforce reductions to focus on inclisiran, improve efficiencies and better align costs and structure. All employees who will be impacted by these reductions have been informed as to their respective timing of their departures. Through February 27, 2018, 27 employees have been terminated and 136 employees were transferred as part of the sale of the infectious disease business unit to Melinta. An additional 115 employees will be terminated through the remainder of the year, with the vast majority by midyear. Included in the 115 employees are 23 employees based in San Diego who are working on early stage infectious disease projects. We expect to sell or spin out those assets and employees by midyear. These workforce reductions are expected to reduce headcount costs included in operating expenses by approximately $74.0 million on an annualized basis.
Segments and Geographic Information
We have one reportable segment. For information regarding revenue and other information regarding our results of operations, including geographic segment information, for each of our last three fiscal years, please refer to our consolidated financial statements and Note 18 to our consolidated financial statements, which are included in Part II, Item 8. Financial Statements and Supplementary Data of this Annual Report on Form 10-K, and Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations included in this Annual Report on Form 10-K.
19
Our Corporate Information
We were incorporated in Delaware on July 31, 1996. Our principal executive offices are located at 8 Sylvan Way, Parsippany, New Jersey 07054, and our telephone number is (973) 290-6000.
Available Information
Our Internet address is http://www.themedicinescompany.com. The contents of our website are not part of this Annual Report on Form 10-K, and our Internet address is included in this document as an inactive textual reference only. We make our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports available free of charge on our website as soon as reasonably practicable after we file such reports with, or furnish such reports to, the Securities and Exchange Commission, or SEC.
Item 1A. | Risk Factors. |
Investing in our common stock involves a high degree of risk. You should consider carefully the risks and uncertainties described below in addition to the other information included or incorporated by reference in this Annual Report on Form 10-K. If any of the following risks actually occur, our business, financial condition or results of operations would likely suffer. In that case, the trading price of our common stock could decline. In addition to the risk factors identified under the captions below, the operation and results of our business are subject to risks and uncertainties identified elsewhere in this Annual Report on Form 10-K as well as general risks and uncertainties such as those relating to general economic conditions and demand in the market for our products.
Risks Related to Development, Approval and Commercialization of Inclisiran
We are almost entirely dependent on the success of inclisiran, our only significant drug candidate, which is currently in Phase 3 of clinical development, and we cannot be certain that inclisiran will receive regulatory approval or be successfully commercialized even if we receive regulatory approval.
We currently only commercialize Angiomax, which is subject to generic competition, and we may never be able to develop inclisiran as a marketable product. We expect that a substantial majority of our efforts and expenditures over the next few years will be devoted to inclisiran.
Accordingly, our future business, including the ability to generate revenue, finance our operations and repay our indebtedness, depends almost entirely on the successful development, regulatory approval and commercialization of inclisiran. We cannot be certain that inclisiran will receive regulatory approval or be successfully commercialized even if we receive regulatory approval. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drug products are and will remain subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries that each have differing regulations. We are not permitted to market inclisiran or any of our drug candidates in the United States until they receive approval of an NDA or BLA from the FDA, or in any foreign countries until they receive the requisite approval from such countries. We have not submitted an NDA or BLA to the FDA or comparable applications to other regulatory authorities. Obtaining approval of an NDA or BLA is an extensive, lengthy, expensive and inherently uncertain process, and the FDA may delay, limit or deny approval of a drug candidate for many reasons, including:
• | we may not be able to demonstrate that inclisiran or any other drug candidate is safe and effective as a treatment for our targeted indications to the satisfaction of the FDA; |
• | the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA for marketing approval; |
• | a clinical research organization, or CRO, that we retain to conduct clinical trials or any other third parties involved in the conduct of trials may take actions outside of our control that materially adversely impact our clinical trials; |
• | the FDA may not find the data from pre-clinical studies and clinical trials sufficient to demonstrate that the clinical and other benefits of inclisiran or any other drug candidate outweigh the safety risks; |
• | the FDA may disagree with our interpretation of data from our pre-clinical studies and clinical trials or may require that we conduct additional studies or trials; |
20
• | the FDA may not accept data generated at our clinical trial sites; |
• | if our NDA is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
• | the advisory committee may recommend that the FDA require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
• | the FDA may require development of a Risk Evaluation and Mitigation Strategy as a condition to approval; |
• | the FDA may identify deficiencies in the manufacturing processes or facilities of our third-party manufacturers; or |
• | the FDA may change its approval policies or adopt new regulations. |
If inclisiran gains regulatory approval, the commercial launch will require significant efforts from us. Our ability to successfully commercially launch inclisiran will depend on our ability to:
• | train, deploy and support a qualified sales force to market and sell our newly launched products; |
• | have third parties manufacture and release the products in sufficient quantities; |
• | implement and maintain agreements with wholesalers and distributors; |
• | receive adequate levels of coverage and reimbursement for these products from governments and third-party payors; |
• | develop and execute marketing and sales strategies and programs for the products; and |
• | enter into suitable partnerships with third parties, as needed, to provide a viable platform to commercialize inclisiran. |
We expect that the revenues from inclisiran, if approved, will represent a very significant portion of our revenues in the future, particularly given that Angiomax is subject to generic competition. As a result, if we are unable to successfully commercialize inclisiran, our business, results of operations and financial condition would be materially harmed.
If we are unable to successfully develop our business infrastructure and operations, our ability to generate future product revenue will be adversely affected and our business, results of operations and financial condition may be adversely affected.
Our ability to support the sales and marketing of our products in the United States and globally will depend on our ability to properly scale our internal organization and infrastructure to accommodate the development and, upon approval, commercialization of inclisiran. To manage our existing and future growth and the breadth and complexity of our activities, we need to properly invest in personnel, infrastructure, information management systems and other operational resources. If we are unable to scale global operations successfully and in a timely manner, the growth of our business may be limited. Developing our business infrastructure and operations may be more difficult, more expensive or take longer than we anticipate.
Future development of our business infrastructure and operations could strain our operational, human and financial resources. In order to manage the development of our business infrastructure and global operations, we must:
• | continue to improve operating, administrative, and information systems; |
• | accurately predict future personnel and resource needs to meet contract commitments; |
• | track the progress of ongoing projects; and |
• | attract and retain qualified management, sales, professional, scientific and technical operating personnel. |
21
If we do not take these actions and are not able to manage our business, then our operations may be less successful than anticipated.
Risks Related to Our Financial Results
We will need additional funds to support our operations, and such funding may not be available to us on acceptable terms, or at all, which raises substantial doubt about our ability to continue as a going concern.
We are focused on the advancement of our product candidate, inclisiran. The completion of the development and the potential commercialization of inclisiran, should it receive regulatory approval, will require substantial funds. We will need to obtain substantial additional sources of funding to develop and, if approved, commercialize inclisiran.
Due to the introduction of generic competition against Angiomax and the divestiture of certain of our non-core products, our revenues generated from product sales have declined significantly since 2014. Revenues are expected to continue to decline as generic competition for Angiomax increases. We have incurred net losses and negative cash flows from operations since 2014 and had an accumulated deficit of $1.3 billion as of December 31, 2017. We expect to incur significant expenses and operating losses for the foreseeable future as we continue to develop, seek regulatory approval for and commercially launch inclisiran. We believe our existing cash and cash equivalents and available for sale securities of approximately $151.4 million as of December 31, 2017, together with the upfront and guaranteed cash proceeds from the Melinta transaction and cash flows we expect to generate from product sales, will be sufficient to satisfy our anticipated operating and other funding requirements for the next twelve months from March 1, 2018 (the date of filing this Form 10-K).
Because we expect to continue to incur negative cash flows from operations, we may need to raise additional funds through other sources of liquidity from the Melinta transaction and from asset sales, including asset sales of products or businesses that generate a material portion of our revenues, engage in other strategic transactions, sell additional equity or debt securities, or seek additional financing through other arrangements in order to meet our anticipated operating and funding requirements prior to the filing of an NDA for inclisiran. There can be no assurances that asset sales or public or private financings may be available in amounts or on terms acceptable to us, if at all. Our ability to obtain additional debt financing may be limited by market conditions. If we are unable to consummate asset sales, obtain additional financing or otherwise increase our cash resources, we may be required to delay, reduce the scope of, or eliminate one or more of our planned research, development or commercialization activities.
We no longer have market exclusivity for Angiomax and face generic and other competition that will cause our net revenue to decline significantly.
A substantial majority of our historic revenue has come from sales of Angiomax (bivalirudin) in the United States. Angiomax is now subject to generic competition. In the United States, we sell Angiomax under our name as a branded Angiomax product, and, on July 2, 2015, entered into a supply and distribution agreement with Sandoz, under which we granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax (bivalirudin). We entered into the supply and distribution agreement as a result of the July 2, 2015 Federal Circuit Court ruling against us in our patent infringement litigation with Hospira with respect to the ‘727 patent and the ‘343 patent covering a more consistent and improved Angiomax drug product and the processes by which it is made. In addition to Hospira, other generic firms have entered the market. Mylan commenced marketing its generic bivalirudin product following a decision by the Federal Circuit Court in Mylan’s appeal that reversed an earlier district court decision that found that Mylan’s abbreviated ANDA product infringed all of the asserted claims of the ‘727 patent. APP Pharmaceuticals LLC, or APP, through its affiliated company, Fresenius Kabi, commenced selling its generic version of Angiomax under provisions of a settlement agreement triggered by the Federal Circuit Court’s July 2, 2015 decision in the Hospira matter. Apotex Inc. and Dr. Reddy’s Laboratories have each also commenced commercialization of generic bivalirudin products upon receiving final approval if their respective ANDA filings by the FDA even though we remain in active litigation against Apotex and only recently settled with Dr. Reddy’s Laboratories. In addition, in January 2018 Baxter announced that the FDA approved Baxter’s ready-to-use formulation of bivalirudin for use as an anticoagulant in patients undergoing PCI.
A number of companies in addition to Hospira, Mylan, APP, Apotex Inc. and Dr. Reddy’s Laboratories have filed ANDAs for their generic versions of Angiomax. In addition to the generic versions and the ready-to-use version of bivalirudin currently being sold, Angiomax could be subject to further generic competition in the United States from Teva and other generic ANDA filers that we have settled with under the circumstances set forth in our respective settlement agreements with such parties and upon a final approval of each company's ANDA filings by the FDA. Pliva Hrvatska DOO, an affiliate of Teva, currently has tentative approval for its ANDA filing for its generic version of Angiomax. Other ANDA filers may commercialize their products ‘at risk’ if they receive final approval of their respective ANDA filings and are not subject to a Hatch-Waxman 30-month stay. Further, we remain
22
in infringement litigation involving the ‘727 patent and ‘343 patent with the other ANDA filers. See Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K for descriptions of our litigation with ANDA filers and related settlements. There can be no assurance as to the outcome of our infringement litigation. We may continue to incur further legal expenses related to these matters.
The principal patent covering Angiomax in Europe expired in August 2015. As a result, we face generic competition in Europe. We are in the process of voluntarily discontinuing and withdrawing Angiomax from the market outside of North America and have ceased related commercialization activities.
Net product revenues from sales of Angiomax, which includes our sales of branded Angiomax and the sales of authorized generic Angiomax (bivalirudin) by Sandoz, decreased from $50.6 million for the year ended December 31, 2016 to $18.8 million for the year ended December 31, 2017. We expect that net product revenues from sales of Angiomax will continue to decline in 2018 and in future years due to generic and other competition. Although we have entered into a supply and distribution agreement with Sandoz to sell an authorized generic version of Angiomax, the royalty income from the sale of the authorized generic, which for the year ended December 31, 2017 was approximately $25.8 million, is expected to only partially offset the expected further decline in Angiomax net product revenues.
We have a history of net losses and may not achieve profitability in future periods.
We have incurred net losses in many years and on a cumulative basis since our inception, and we expect to continue to incur net losses. As of December 31, 2017, we had an accumulated deficit of approximately $1.3 billion. In those periods in which we were able to achieve profitability, our profitability was based on revenue from sales of Angiomax, and a substantial majority of our historic revenue has been generated from sales of Angiomax in the United States. However, generic competition for Angiomax commenced in the United States in July 2015 and we lost market exclusivity for Angiomax in Europe in August 2015. We expect that net revenue from sales of Angiomax will continue to decline in future years due to competition from generic versions of Angiomax, including our authorized generic being marketed by Sandoz and other generic versions of Angiomax which have been and may be approved by the FDA.
We expect to make substantial expenditures to further develop and commercialize inclisiran, including costs and expenses associated with research and development, clinical trials, nonclinical and preclinical studies, regulatory approvals and commercialization. We will need to generate significant revenue in future periods from inclisiran in order to achieve and maintain profitability. If we are unable to generate significant revenue, we may not achieve profitability in future periods. Our ability to generate future revenue will be substantially dependent on our ability to successfully commercialize inclisiran. If we fail to achieve profitability within the time frame expected by investors or securities analysts, the market price of our common stock may decline.
We review our inventory, including inventory purchase commitments, and provide reserves, as appropriate, against the carrying amount of inventory. For example, for the year ended December 31, 2015, we recorded a $29.5 million inventory obsolescence charge and a charge of $12.1 million for potential losses on future inventory purchases primarily due to the loss of exclusivity of Angiomax. We also recorded an $8.5 million reserve for potential inventory obsolescence during the year ended December 31, 2016. As of December 31, 2017, our inventory of Angiomax was $5.6 million and there are no further inventory-related purchase commitments for Angiomax bulk drug substance. We recorded a $16.7 million reserve for potential inventory obsolescence during the year ended December 31, 2017. If sales of Angiomax decline more than our current expectations, we could be required to make an additional allowance for excess or obsolete inventory, increase our accrual for product returns or increase our deferred tax valuation allowance, or we could incur other costs related to operating our business, each of which could negatively impact our results of operations and our financial condition.
We may need to raise additional capital. If we are unable to obtain such capital on favorable terms or at all, we may not be able to execute on our business plans and our business, financial condition and results of operations may be adversely affected.
On November 3, 2015, we announced that our current intention was to explore strategies for optimizing our capital structure and liquidity position. At December 31, 2017, we had approximately $151.4 million in cash and cash equivalents. We expect to devote substantial financial resources to our research and development efforts, clinical trials, nonclinical and preclinical studies and regulatory approvals and to our commercialization and manufacturing programs associated with our products and our products in development. We also will require cash to pay interest on the $400.0 million aggregate principal amount of the 2022 notes and the $402.5 million aggregate principal amount of the 2023 notes, and to make principal payments on the 2022 notes and 2023 notes at maturity or upon conversion (other than the 2023 notes upon conversion, in which case we will have the option to settle entirely in shares of our common stock). In addition, as part of our business development strategy, we generally structure our license agreements and acquisition agreements so that a significant portion of the total license or acquisition cost is contingent upon the successful achievement of specified development, regulatory or commercial milestones. As a result, we will require cash
23
to make payments upon achievement of these milestones under the license agreements and acquisition agreements to which we are a party.
As of February 27, 2018, we may have to make contingent cash payments upon the achievement of specified development, regulatory or commercial milestones of up to:
• | $150.0 million for the license and collaboration agreement with Alnylam; |
• | $69.3 million relating to our research and development infectious disease portfolio acquired in our Rempex acquisition (and which was not divested in the Melinta transactions); and |
• | $2.2 million for other transaction milestones. |
As of February 27, 2018, our total potential milestone payment obligations related to development, regulatory and commercial milestones for our products and products in development under our license agreements and acquisition agreements, assuming all milestones are achieved in accordance with the terms of these agreements, would be approximately $221.3 million. Of this amount, approximately $49.4 million relates to development milestones, $71.9 million relates to regulatory approval milestones and $100.0 million relates to commercial milestones. These amounts do not include milestone payments of up to $175.8 million related to Ionsys, which was discontinued and withdrawn from the market in the United States in June 2017 and has also been discontinued in Europe, and the MDCO-700 development program, which we discontinued in August 2017.
Based on our anticipated timeline for the achievement of development, regulatory and commercial milestones, we expect that we would make $5.0 million of milestone payments under our license agreements and acquisition agreements during the remainder of 2018. We may pay additional milestone payments under our license agreements and acquisition agreements during 2018 if we achieve additional development, regulatory and commercial milestones during the year.
Total net revenues from sales of Angiomax were significantly lower in the year ended December 31, 2017 than in previous comparable periods, and we expect these revenues will decline further. These reduced revenues are likely to significantly impact our cash and cash equivalents and how we fund our future capital requirements.
We continually evaluate our liquidity requirements, capital needs and availability of resources in view of, among other things, alternative sources and uses of capital, debt service requirements, the cost of debt and equity capital and estimated future operating cash flow. We may raise additional capital; enter into licenses or collaborations with third parties to develop and commercialize inclisiran; sell assets, including asset sales of products or businesses that generate a material portion of our current revenue; restructure or refinance outstanding debt; repurchase material amounts of outstanding debt or equity; or take a combination of such steps or other steps to increase or manage our liquidity and capital resources. Any such actions or steps could have a material effect on us.
Our future capital requirements will depend on many factors, including:
• | the progress, level, timing and cost of our research and development activities related to our clinical trials and non-clinical studies with respect to inclisiran; |
• | whether we develop and commercialize inclisiran on our own or through licenses and collaborations with third parties and the terms and timing of such arrangements, if any; |
• | the extent to which our submissions and planned submissions for regulatory approval of inclisiran are approved on a timely basis, if at all; |
• | the decline in Angiomax sales and the extent to which royalties on sales of the authorized generic of Angiomax are generated; |
• | if inclisiran receives regulatory approval, the extent to which it is commercially successful; |
• | the continuation or termination of third-party manufacturing, distribution and sales and marketing arrangements; |
• | the size, cost and effectiveness of our sales and marketing programs, including scaling our operations in anticipation of a potential launch of inclisiran; |
• | the amounts of our payment obligations to third parties with respect to inclisiran or other assets; and |
24
• | our ability to defend and enforce our intellectual property rights. |
With respect to both our short-term and long-term cash requirements, if our existing cash resources, together with cash that we generate from sales of our products and other sources, are insufficient to satisfy our research and development, clinical trial, product commercialization and other funding requirements, including obligations under our convertible notes, we will need to sell additional equity or debt securities, engage in asset sales, including asset sales of products or businesses that generate a material portion of our revenue, engage in other strategic transactions, or seek additional financing through other arrangements, any of which could be material. Any sale of additional equity or convertible debt securities may result in dilution to our stockholders. Public or private financing may not be available in amounts or on terms acceptable to us, if at all. If we seek to raise funds through collaboration or licensing arrangements with third parties, we may be required to relinquish rights to products, products in development or technologies that we would not otherwise relinquish or grant licenses on terms that may not be favorable to us. Moreover, our ability to obtain additional debt financing may be limited by the 2022 notes and the 2023 notes, market conditions or otherwise. If we are unable to obtain additional financing or otherwise increase our cash resources, we may be required to delay, reduce the scope of, or eliminate one or more of our planned research, development and commercialization activities, which could adversely affect our business, financial condition and operating results.
If we seek to raise additional capital by selling equity or debt securities or through other arrangements in the future, our stockholders could be subject to dilution and we may become subject to financial restrictions and covenants, which may limit our activities.
If we determine that raising capital would be in the interest of the company and our stockholders, we may seek to sell equity or debt securities or seek financing through other arrangements. Any sale of equity or debt securities may result in dilution to our stockholders and increased liquidity requirements. Debt financing may involve covenants limiting or restricting our ability to take specific actions, such as incurring additional debt or making capital expenditures. Our ability to comply with these financial restrictions and covenants could be dependent on our future performance, which is subject to prevailing economic conditions and other factors, including factors that are beyond our control such as foreign exchange rates, interest rates and changes in the level of competition. Failure to comply with the financial restrictions and covenants would adversely affect our business, financial condition and operating results.
We may not realize the anticipated benefits of past or future acquisitions or product licenses and integration of these acquisitions and any products and product candidates acquired or licensed may disrupt our business and management.
We have in the past and may in the future acquire or license additional development-stage compounds, clinical-stage product candidates, approved products, technologies or businesses. For example, we entered into a license and collaboration agreement with Alnylam with respect to inclisiran. We may not realize the anticipated benefits of an acquisition, license, or collaboration, each of which involves numerous risks. These risks include:
• | difficulty in integrating the operations, products or product candidates and personnel of an acquired company; |
• | entry into markets in which we have no or limited direct prior experience and where competitors in such markets have stronger market positions; |
• | failure to successfully further develop the acquired or licensed business, product, compounds, programs or technology or to achieve strategic objectives, including commercializing and marketing successfully the development stage compounds and clinical stage candidates that we acquire or license; |
• | disruption of our ongoing business and distraction of our management and employees from other opportunities and challenges; |
• | inadequate or unfavorable clinical trial results from acquired or contracted for products in development; |
• | inability to retain personnel, key customers, distributors, vendors and other business partners of the acquired company, or acquired or licensed product or technology; |
• | potential failure of the due diligence processes to identify significant problems, liabilities or other shortcomings or challenges of an acquired company, or acquired or licensed product or technology, including but not limited to, problems, liabilities or other shortcomings or challenges with respect to intellectual property, product quality, revenue recognition or other accounting practices, employee, customer or partner disputes or issues and other legal and financial contingencies and known and unknown liabilities; |
25
• | liability for activities of the acquired company or licensor before the acquisition or license, including intellectual property infringement claims, violations of laws, commercial disputes, tax liabilities, and other known and unknown liabilities; |
• | exposure to litigation or other claims in connection with, or inheritance of claims or litigation risk as a result of, an acquisition or license, including but not limited to, claims from terminated employees, customers, former stockholders or other third-parties; and |
• | difficulties in the integration of the acquired company’s departments, systems, including accounting, human resource and other administrative systems, technologies, books and records, and procedures, as well as in maintaining uniform standards, controls, including internal control over financial reporting required by the Sarbanes-Oxley Act of 2002 and related procedures and policies. |
Acquisitions and licensing arrangements are inherently risky, and ultimately, if we do not complete an announced acquisition or license transaction or integrate an acquired business, or an acquired or licensed product or technology successfully and in a timely manner, we may not realize the benefits of the acquisition or license to the extent anticipated and the perception of the effectiveness of our management team and our company may suffer in the marketplace. In addition, even if we are able to achieve the long-term benefits associated with our strategic transactions, our expenses and short-term costs may increase materially and adversely affect our liquidity and short-term profitability. Further, if we cannot successfully integrate an acquired business, or acquired or licensed products or technologies, we may experience material negative consequences to our business, financial condition or results of operations. Further, if we sell products that have been acquired through acquisitions or licensing arrangements, we may incur losses depending on the consideration received and structure of the transaction. For example, in connection with our sale of our hemostasis business, which we completed on February 1, 2016, we incurred impairment charges of $133.3 million, including $24.5 million related to goodwill. Future acquisitions or licenses could also result in dilutive issuances of our equity securities, the incurrence of debt, contingent liabilities, or amortization expenses, or impairment of goodwill and intangible assets, and restructuring charges, any of which could harm our business, financial condition or results of operations.
Risks Related to Our Notes
We have incurred substantial indebtedness, and our leverage and maintenance of high levels of indebtedness may adversely affect our business, financial condition and results of operations. Servicing this debt, including the 2022 notes and the 2023 notes, will require a significant amount of cash, and we may not have sufficient cash flow from our business to pay the interest on or principal of the 2022 notes, the 2023 notes or other debt we may incur.
We have incurred a significant amount of indebtedness. Our maintenance of this level of indebtedness could have adverse consequences, including:
• | requiring us to dedicate a substantial portion of cash flow from operations to the payment of interest on, and principal of, our debt, which will reduce the amounts available to fund working capital, capital expenditures, product development efforts and other general corporate purposes; |
• | increasing our vulnerability to general adverse economic, industry and market conditions; |
• | limiting our ability to obtain additional financing in the future or engage in certain strategic transactions without securing bondholder consent; |
• | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we compete; and |
• | placing us at a possible competitive disadvantage to less leveraged competitors and competitors that have less debt, better debt servicing options or better access to capital resources. |
In addition, our ability to make scheduled payments of the principal of, to pay interest on or to refinance the remaining amount outstanding under the 2022 notes or the 2023 notes depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. Our business may not generate cash flow from operations in the future sufficient to service our debt, including the notes. If we are unable to generate cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be unfavorable to us or highly dilutive, any of which may be material to the holders of our common stock. Our ability to refinance our indebtedness will depend
26
on the capital markets and our financial condition at the time we seek to refinance such indebtedness. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
We may not have the ability to raise the funds necessary to settle conversions of the 2022 notes or to repurchase the 2022 notes or the 2023 notes upon a fundamental change, and our future debt may contain limitations on our ability to pay cash upon conversion of the 2022 notes or repurchase of the 2022 notes or 2023 notes.
Holders of the 2022 notes and the 2023 notes will have the right to require us to repurchase their notes upon the occurrence of a fundamental change, as defined in the applicable indenture, at a repurchase price equal to 100% of their principal amount, plus accrued and unpaid interest, if any, as described in the applicable indenture. In addition, upon conversion of the 2022 notes, we will be required to make with respect to each $1,000 in principal amount of notes converted cash payments of at least the lesser of $1,000 and the sum of the daily conversion values as described in the applicable indenture. Upon conversion of the 2023 notes, we will have the option to settle such conversions in cash, shares of our common stock or a combination thereof. However, we may not have enough available cash or be able to obtain financing at the time we are required to repurchase notes, to pay the notes at maturity or to pay cash upon conversions of such notes. In addition, our ability to repurchase notes or to pay cash upon conversions of such notes may be limited by law, by regulatory authority or by agreements governing our existing indebtedness (including, in the case of the 2022 notes or the 2023 notes, the indenture governing any other series of notes) and future indebtedness. Our failure to repurchase notes at a time when the repurchase is required by the applicable indenture or to pay any cash payable on future conversions of the notes as required by the applicable indenture would constitute a default under the applicable indenture. A default under the applicable indenture governing the 2022 notes or the 2023 notes, respectively, or the fundamental change itself could also lead to a default under agreements governing our existing indebtedness (including, in the case of the 2022 notes or the 2023 notes, the indenture governing any other series of notes) and future indebtedness. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and repurchase the notes or make cash payments upon conversions thereof.
The conditional conversion feature of the 2022 notes or the 2023 notes, if triggered, may adversely affect our financial condition and operating results.
In the event the conditional conversion feature of the 2022 notes or the 2023 notes is triggered, holders of such notes will be entitled to convert the notes at any time during specified periods at their option, which are set forth in the applicable indenture. If one or more holders elect to convert their 2022 notes, we would be required, with respect to each $1,000 principal amount of 2022 notes, to make cash payments equal to the lesser of $1,000 and the sum of the daily conversion values, which could adversely affect our liquidity. If the holders of all of the 2022 notes were able to exercise their conversion option, we would not have sufficient cash to satisfy our payment obligations with respect to all of the 2022 notes and meet our anticipated funding requirements for a year from March 1, 2018. With respect to the 2023 notes, we have the option to settle conversions entirely in cash, in common stock or a combination thereof. In addition, even if holders do not elect to convert their notes, we are required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the notes as a current rather than long‑term liability, which results in a material reduction of our net working capital.
The accounting method for convertible debt securities that may be settled in cash, such as the 2022 notes and the 2023 notes, could have a material effect on our reported financial results.
Under Accounting Standards Codification 470-20, “Debt with Conversion and Other Options”, which we refer to as ASC 470-20, an entity must separately account for the liability and equity components of the convertible debt instruments that may be settled entirely or partially in cash upon conversion (such as the 2022 notes and the 2023 notes) in a manner that reflects the issuer’s economic interest cost. The effect of ASC 470-20 on the accounting for the 2022 notes and the 2023 notes is that the equity component is required to be included in the additional paid in capital section of stockholders’ equity on our consolidated balance sheet, and the value of the equity component would be treated as original issue discount for purposes of accounting for the debt component of the 2022 notes and the 2023 notes. As a result, we will be required to record a greater amount of non-cash interest expense in current periods presented as a result of the amortization of the discounted carrying value of the notes to their face amount over the term of the 2022 notes and the 2023 notes. We will report lower net income in our financial results because ASC 470-20 will require interest to include both the current period’s amortization of the debt discount and the instrument’s coupon interest, which could adversely affect our reported or future financial results, the market price of our common stock and the trading price of the 2022 notes and the 2023 notes.
In addition, under certain circumstances, convertible debt instruments that may be settled entirely or partly in cash (such as the 2022 notes or the 2023 notes) are currently accounted for utilizing the treasury stock method, the effect of which is that the shares issuable upon conversion of the notes are not included in the calculation of diluted earnings per share except to the extent that the conversion value of the notes exceeds their principal amount. Under the treasury stock method, for diluted earnings per
27
share purposes, the transaction is accounted for as if the number of shares of common stock that would be necessary to settle such excess are issued. We cannot be sure that the accounting standards in the future will continue to permit the use of the treasury stock method. If we are unable to use the treasury stock method in accounting for the shares issuable upon conversion of the 2022 notes or the 2023 notes, then our diluted earnings per share would be adversely affected.
We may incur substantially more debt or take other actions which would intensify the risks discussed above.
We and our subsidiaries may be able to incur substantial additional debt in the future, some of which may be secured debt. We and our subsidiaries are not restricted under the terms of the applicable indenture governing the 2022 notes or the 2023 notes from incurring additional debt, securing existing or future debt, recapitalizing our debt or taking a number of other actions that are not limited by the terms of the applicable indenture governing the 2022 notes or the 2023 notes that could have the effect of diminishing our ability to make payments on the notes when due.
Additional Risks Related to Commercialization
We face substantial competition, which may result in others discovering, developing or commercializing competing products before or more successfully than we do.
Our industry is highly competitive. Competitors in the United States and other countries include major pharmaceutical companies, specialty pharmaceutical companies and biotechnology firms, universities and other research institutions. Many of our competitors are substantially larger than we are and have substantially greater research and development capabilities and experience, and greater manufacturing, marketing and financial resources, than we do.
Our competitors may develop, market or license products or novel technologies that are more effective, safer, more convenient or less costly than any that have been or are being developed or sold by us, or may obtain marketing approval for their products from the FDA or equivalent foreign regulatory bodies more rapidly than we may obtain approval for ours.
There are well established products, including in many cases generic products, that are approved and marketed for the indications for which Angiomax is approved and the markets and indications for which we are developing inclisiran. In addition, competitors are developing products for such markets and indications. A description of the competition for inclisiran and Angiomax is included in “Part I, Item 1. Business-Competition” of this Annual Report on Form 10‑K for the year ended December 31, 2017.
We expect to compete, in the case of inclisiran, and we compete, in the case of Angiomax, on the basis of product efficacy, safety, ease of administration, price and economic value compared to drugs used in current practice or currently being developed. If we are not successful in demonstrating these attributes, physicians and other key healthcare decision makers may choose other products over our products, switch from our products to new products or choose to use our products only in limited circumstances, which could adversely affect our business, financial condition and results of operations.
If reimbursement by government payers or other third-party payers is not available or limited for our products, pricing is delayed or set at unfavorable levels or access to our products is reduced or terminated by governmental and other third-party payers, our ability to generate revenue would be adversely affected.
Acceptable levels of coverage and reimbursement of drug treatments by government payers, such as Medicare and Medicaid programs, private health insurers and other organizations, have a significant effect on our ability to successfully commercialize our products. Reimbursement in the United States, Europe or elsewhere may not be available for any products we may develop or, if already available, may be decreased in the future. We may not get reimbursement or reimbursement may be limited if government payers, private health insurers and other organizations are influenced by the prices of existing drugs in determining whether our products will be reimbursed and at what levels. If reimbursement is not available or is available only at limited levels, we may not be able to commercialize our products, or may not be able to obtain a satisfactory financial return on our products.
In certain countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals and the level of reimbursement are subject to governmental control. In some countries, pricing and reimbursement are set with limited, if any, participation in the process by the marketing authorization holder. In addition, it can take an extended period of time after the receipt of initial approval of a product to establish and obtain reimbursement or pricing approval. Reimbursement approval also may be required at the individual patient level, which can lead to further delays. In addition, in some countries, it may take an extended period of time to collect payment even after reimbursement has been established. If prices are set at unsatisfactory levels, such prices may negatively impact our revenues from sales in those countries. An increasing number of countries are taking initiatives to attempt to reduce large budget deficits by focusing cost-cutting efforts on pharmaceuticals for their state-run health care systems. These international price control efforts have impacted all regions of the world, but have been most drastic in the
28
European Union. Further, a number of European Union countries use drug prices from other countries of the European Union as “reference prices” to help determine pricing in their own countries. Consequently, a downward trend in drug prices for some countries could contribute to similar occurrences elsewhere. If reimbursement of our future products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
Third-party payers, including Medicare and Medicaid, increasingly are challenging prices charged for and the cost-effectiveness of medical products and services and they increasingly are limiting both coverage and the level of reimbursement for drugs. If these third-party payers do not consider our products to be economically beneficial compared to other available therapies, they may not cover our products after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit. Third-party payers may provide coverage, but place stringent limitations on such coverage, such as requiring alternative treatments to be tried first. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. There exists a broader trend in health care in which the government and other payors are seeking to move from individualized “fee for service” payments toward a system focused on “bundled” payments for more comprehensive packages of services and episodes of care. Also, the trend toward managed health care in the United States and the changes in health insurance programs may result in lower prices for pharmaceutical products and health care reform.
Health care reform measures such as those outlined above, and others consistent with these trends, could, among other things, increase pressure on pricing. Additionally, health care reform efforts undertaken during the Trump administration may result in additional reductions in Medicare, Medicaid and other healthcare funding. In addition to federal legislation, state legislatures and foreign governments have also shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. The establishment of limitations on patient access to our drugs, adoption of price controls and cost-containment measures in new jurisdictions or programs, and adoption of more restrictive policies in jurisdictions with existing controls and measures could adversely impact our business and future results. If governmental organizations and third-party payers do not consider our products to be cost-effective compared to other available therapies, they may not reimburse providers or consumers of our products or, if they do, the level of reimbursement may not be sufficient to allow us to sell our products on a profitable basis.
Use or misuse of our products may result in serious injuries or even death to patients and may subject us to significant claims for product liability. If we are unable to obtain insurance at acceptable costs and adequate levels or otherwise protect ourselves against potential product liability claims, we could be exposed to significant liability.
Our business exposes us to potential significant product liability risks which are inherent in the testing, manufacturing, marketing and sale of human healthcare products. Product liability claims might be made by patients in clinical trials, consumers, health care providers or pharmaceutical companies or others that sell our products. These claims may be made even with respect to those products that are manufactured in licensed and regulated facilities or otherwise possess regulatory approval for commercial sale or study.
These claims could expose us to significant liabilities that could prevent or interfere with the development or commercialization of our products. Product liability claims could require us to spend significant time and money in litigation or pay significant damages. With respect to our commercial sales and our clinical trials, we are covered by product liability insurance in the amount of $10.0 million per occurrence and $10.0 million annually in the aggregate on a claims-made basis. This coverage may not be adequate to cover all or any product liability claims that we face.
As we continue to commercialize Angiomax and develop inclisiran, we may wish to increase our product liability insurance. Product liability coverage is expensive. In the future, we may not be able to maintain or obtain such product liability insurance on reasonable terms, at a reasonable cost or in sufficient amounts to protect us against losses due to product liability claims.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants.
Our industry has experienced a high rate of turnover of management personnel in recent years. We are highly dependent on our ability to attract and retain qualified personnel for the acquisition, development and commercialization activities we conduct or sponsor. If we lose one or more of the members of our senior management, including our Chief Executive Officer, Clive A. Meanwell, or other key employees or consultants, our ability to implement successfully our business strategy could be seriously harmed. Our ability to replace these key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to acquire, develop and commercialize
29
products successfully. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate such additional personnel.
Risks Related to our Dependence on Third Parties for Manufacturing, Research and Development, and Distribution Activities
We do not have manufacturing or supply capabilities and are completely dependent on third parties for the manufacture and supply of our products. We depend on a limited number of suppliers for the production of bulk drug substance for inclisiran and Angiomax and to carry out fill-finish activities. If any of these suppliers does not or cannot fulfill its manufacturing or supply obligations to us, our ability to conduct clinical trials of inclisiran and to meet commercial demands for Angiomax could be impaired and our business could be harmed.
We do not manufacture inclisiran or Angiomax, and do not plan to develop any capacity to manufacture them. We currently rely on a limited number of manufacturers and other third parties for bulk substance and to carry out fill-finish activities for inclisiran and Angiomax. We expect to continue this manufacturing strategy for the foreseeable future.
In the event that any third-party is unable or unwilling to carry out its respective manufacturing or supply obligations or terminates or refuses to renew its arrangements with us, we may be unable to obtain alternative manufacturing or supply on commercially reasonable terms on a timely basis or at all. In such cases, the third-party manufacturers have made no commitment to supply the drug product to us on a long-term basis and could reject our purchase orders. Only a limited number of manufacturers are capable of manufacturing inclisiran and Angiomax. Consolidation within the pharmaceutical manufacturing industry could further reduce the number of manufacturers capable of producing our products, or otherwise affect our existing contractual relationships.
If we were required to transfer manufacturing processes to other third-party manufacturers and we were able to identify an alternative manufacturer, we would still need to satisfy various regulatory requirements. Satisfaction of these requirements could cause us to experience significant delays in receiving an adequate supply of inclisiran and Angiomax and could be costly. Moreover, we may not be able to transfer processes that are proprietary to the manufacturer. Any delays in the manufacturing process may adversely impact our ability to supply product for clinical trials of inclisiran, which could affect our ability to complete clinical trials of inclisiran on a timely basis and our ability to meet commercial demands for our products on a timely basis, which could reduce our revenue.
If third parties on whom we rely to manufacture and support the development and commercialization of inclisiran and Angiomax do not fulfill their obligations or we are unable to establish or maintain such arrangements, the development and commercialization of our products may be terminated or delayed, and the costs of development and commercialization may increase.
Our development and commercialization strategy involves entering into arrangements with corporate and academic collaborators, contract research organizations, distributors, third-party manufacturers, licensors, licensees and others to conduct development work, manage or conduct our clinical trials, manufacture our products and market and sell our products outside of the United States. We do not have the expertise or the resources to conduct many of these activities on our own and, as a result, are particularly dependent on third parties in many areas.
We may not be able to maintain our existing arrangements with respect to the commercialization or manufacture of Angiomax or establish and maintain arrangements to develop, manufacture and, if approved, commercialize inclisiran or any additional product candidates or products we may acquire on terms that are acceptable to us. Any current or future arrangements for development and commercialization may not be successful. If we are not able to establish or maintain agreements relating to Angiomax, inclisiran or any additional products or product candidates we may acquire, our results of operations would be materially adversely affected.
Third parties may not perform their obligations as expected. The amount and timing of resources that third parties devote to developing, manufacturing and commercializing our products are not within our control. Our collaborators may develop, manufacture or commercialize, either alone or with others, products and services that are similar to or competitive with the products that are the subject of the collaboration with us. Furthermore, our interests may differ from those of third parties that manufacture or commercialize our products. Our collaborators may reevaluate their priorities from time to time, including following mergers and consolidations, and change the focus of their development, manufacturing or commercialization efforts. Disagreements that may arise with these third parties could delay or lead to the termination of the development or commercialization of our product candidates, or result in litigation or arbitration, which would be time consuming and expensive.
30
If any third party that manufactures or supports the development or commercialization of our products breaches or terminates its agreement with us, fails to commit sufficient resources to our collaboration or conduct its activities in a timely manner, or fails to comply with regulatory requirements, such breach, termination or failure could:
• | delay or otherwise adversely impact the manufacturing, development or commercialization of Angiomax, inclisiran or any additional products or product candidates that we may acquire or develop; |
• | require us to seek a new collaborator or undertake unforeseen additional responsibilities or devote unforeseen additional resources to the manufacturing, development or commercialization of our products; or |
• | result in the termination of the development or commercialization of our products. |
Our reliance on third-party manufacturers and suppliers to supply Angiomax and inclisiran may increase the risk that we will not have appropriate supplies of each product or that sanctions may be imposed on us or the manufacturer due to a manufacturer’s failure to comply with regulation requirements, either of which could adversely affect our business, results of operations and financial condition.
Reliance on third-party manufacturers and suppliers entails risks to which we would not be subject if we manufactured Angiomax or inclisiran ourselves, including:
• | reliance on the third party for regulatory compliance and quality assurance; |
• | the possible breach of the manufacturing or supply agreement by the third party; and |
• | the possible termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us. |
Angiomax and inclisiran may compete with products of third parties for access to manufacturing facilities. If we are not able to obtain adequate supplies of our products, it will be more difficult for us to compete effectively, market and sell Angiomax and develop inclisiran.
Our manufacturers are subject to ongoing, periodic, unannounced inspection by the FDA and corresponding state and foreign agencies or their designees to evaluate compliance with the FDA’s current good manufacturing practices, or cGMP, regulations and other governmental regulations and corresponding foreign standards. We cannot be certain that our present or future manufacturers will be able to comply with cGMP regulations and other FDA regulatory requirements or similar regulatory requirements outside the United States. We do not control compliance by our manufacturers with these regulations and standards. Failure of our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on the manufacturer or us, including fines and other monetary penalties, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products in development, delays, suspension or withdrawal of approvals, suspension of clinical trials, license revocation, seizures or recalls of products in development or products, interruption of production, warning letters, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our Angiomax and inclisiran.
We may depend on collaborations with third parties for the development and commercialization of inclisiran. If those collaborations, if entered into, are not successful, we may not be able to capitalize on the market potential of inclisiran.
We may seek to develop and commercialize inclisiran through a variety of types of collaboration arrangements. Our likely collaborators for any marketing, distribution, development, licensing or broader collaboration arrangements include large and mid‑size pharmaceutical companies, regional and national pharmaceutical companies and biotechnology companies. We may not be able to enter into these types of arrangements on a timely basis, on favorable terms or at all. Our ability to enter into such arrangements with respect to inclisiran that are subject to licenses may be limited by the terms of those licenses. If we do enter into any such arrangements with any third parties in the future, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of inclisiran. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
31
Collaborations involving inclisiran could pose a number of risks to us, including:
• | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
• | collaborators may not pursue development and commercialization of inclisiran or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities; |
• | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon inclisiran, repeat or conduct new clinical trials or require a new formulation of inclisiran for clinical testing; |
• | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products in development if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
• | a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; |
• | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or otherwise expose us to potential litigation; |
• | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
• | disputes may arise with respect to the ownership of intellectual property developed pursuant to our collaborations; |
• | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our products or products in development or that result in costly litigation or arbitration that diverts management attention and resources; and |
• | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable products and products in development. |
Collaboration agreements may not lead to development or commercialization of products in development in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
If we use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages or subject to fines and penalties.
We conduct research and development activities that involve the controlled use of potentially hazardous substances, including chemical, biological and radioactive materials and viruses. In addition, our operations produce hazardous waste products. Federal, state and local laws and regulations in the United States and Canada govern the use, manufacture, storage, handling and disposal of hazardous materials. We may incur significant additional costs to comply with applicable laws in the future. Also, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials and we may incur liability as a result of any such contamination or injury. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. We have only limited insurance for liabilities arising from hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may restrict our research, development and production efforts, which could harm our business, operating results and financial condition.
32
Additional Risks Related to Regulatory Matters
Clinical trials of product candidates are expensive and time-consuming, and the results of these trials are uncertain. If we are unable to conduct clinical trials that continue to demonstrate the safety and efficacy of inclisiran on a timely basis, then our costs of developing inclisiran may increase and we may not be able to obtain regulatory approval for inclisiran on a timely basis or at all.
Before we can obtain regulatory approvals to market inclisiran, we will be required to complete extensive clinical trials in humans to demonstrate the safety and efficacy of such product for such indication.
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in pre-clinical testing or early clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. An unexpected result in one or more of our clinical trials can occur at any stage of testing. For example, in November 2016, we voluntarily discontinued our clinical development program for MDCO-216, an investigational cholesterol efflux promoter, and in August 2017 we voluntarily discontinued our clinical development program for MDCO-700, an investigational anesthetic agent.
We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our inclisiran, including:
• | our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials which even if undertaken cannot ensure we will gain approval; |
• | data obtained from pre-clinical testing and clinical trials may be subject to varying interpretations, which could result in the FDA or other regulatory authorities deciding not to approve a product in a timely fashion, or at all; |
• | the cost of clinical trials may be greater than we currently anticipate; |
• | regulators, ethics committees or institutional review boards may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
• | we, or the FDA or other regulatory authorities, might suspend or terminate a clinical trial at any time on various grounds, including a finding that participating patients are being exposed to unacceptable health risks. For example, we have in the past voluntarily suspended enrollment in one of our clinical trials to review an interim analysis of safety data from the trial; and |
• | the effects of inclisiran may not be the desired effects or may include undesirable side effects or inclisiran may have other unexpected characteristics. |
The rate of completion of clinical trials depends in part upon the rate of enrollment of patients. Patient enrollment is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the existence of competing clinical trials and the availability of alternative or new treatments. Delays in patient enrollment in any of our current or future clinical trials may result in increased costs and program delays.
If we or the contract manufacturers manufacturing inclisiran and Angiomax fail to comply with the extensive regulatory requirements to which we, our contract manufacturers and inclisiran and Angiomax are subject, the development of inclisiran could be jeopardized and Angiomax could be subject to restrictions or withdrawal from the market, and we could be subject to penalties.
The research, testing, manufacturing, labeling, safety, advertising, promotion, storage, sales, distribution, import, export and marketing, among other things, of our products, both before and after approval, are subject to extensive regulation by governmental authorities in the United States, Europe and elsewhere throughout the world. Both before and after approval of a product, quality control and manufacturing procedures must conform to cGMP. Regulatory authorities, including the FDA, periodically inspect manufacturing facilities to assess compliance with cGMP. Our failure or the failure of contract manufacturers to comply with the laws administered by the FDA, the EMA or other governmental authorities could result in, among other things, any of the following:
• | delay in approving or refusal to approve a product; |
• | product recall or seizure; |
33
• | suspension or withdrawal of an approved product from the market; |
• | delays in, suspension of or prohibition of commencing, clinical trials of inclisiran; |
• | interruption of production; |
• | operating restrictions; |
• | untitled or warning letters; |
• | injunctions; |
• | fines and other monetary penalties; |
• | the imposition of civil or criminal penalties; |
• | disruption of importing and exporting activities; and |
• | unanticipated expenditures. |
We may incur significant liability if it is determined that we are promoting the “off-label” use of Angiomax, or, if approved, inclisiran.
Physicians may prescribe drug products for uses that are not described in the product’s labeling and that differ from those approved by the FDA or other applicable regulatory agencies. Off-label uses are common across medical specialties. Although the FDA and other regulatory agencies do not regulate a physician’s choice of treatments, the FDA and other regulatory agencies do restrict communications on the subject of off-label use. Companies may not promote drugs for off-label uses. The FDA and other regulatory and enforcement authorities actively enforce laws and regulations prohibiting promotion of off-label uses and the promotion of products for which marketing approval has not been obtained. A company that is found to have promoted off-label uses may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions.
Notwithstanding the regulatory restrictions on off-label promotion, the FDA and other regulatory authorities allow companies to engage in truthful, non-misleading, and non-promotional scientific exchange concerning their products. We engage in medical education activities and communicate with investigators and potential investigators regarding our clinical trials. If the FDA or another regulatory or enforcement authority determines that our communications regarding our marketed products are not in compliance with the relevant regulatory requirements and that we have improperly promoted off-label uses, we may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions.
If we do not comply with federal, state and foreign laws and regulations relating to the health care business, we could face substantial penalties.
We and our customers are subject to extensive regulation by the federal government, and the governments of the states and foreign countries in which we may conduct our business. In the United States, the laws that directly or indirectly affect our ability to operate our business include the following:
• | the Federal Anti-Kickback Law, which prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce either the referral of an individual or furnishing or arranging for a good or service for which payment may be made under federal health care programs such as Medicare and Medicaid; |
• | other Medicare laws and regulations that prescribe the requirements for coverage and payment for services performed by our customers, including the amount of such payment; |
• | the Federal False Claims Act, which imposes civil and criminal liability on individuals and entities who submit, or cause to be submitted, false or fraudulent claims for payment to the government; |
34
• | the Federal False Statements Act, which prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with delivery of or payment for health care benefits, items or services; and |
• | various state laws that impose similar requirements and liability with respect to state healthcare reimbursement and other programs. |
If our operations are found to be in violation of any of the laws and regulations described above or any other law or governmental regulation to which we or our customers are or will be subject, we may be subject to civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs and the curtailment or restructuring of our operations. Similarly, if our customers are found to be non-compliant with applicable laws, they may be subject to sanctions, which could also have a negative impact on us. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business and our financial results. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
Failure to comply with the U.S. Foreign Corrupt Practices Act, or FCPA, as well as the anti-bribery laws of the nations in which we conduct business, could subject us to penalties and other adverse consequences.
We are subject to the FCPA, which generally prohibits U.S. companies from engaging in bribery or other prohibited payments to foreign officials for the purpose of obtaining or retaining business and requires companies to maintain accurate books and records and internal controls, including at foreign-controlled subsidiaries. In addition, we are subject to other anti-bribery laws of the nations in which we conduct business that apply similar prohibitions as the FCPA. Our employees or other agents may engage in prohibited conduct without our knowledge under our policies and procedures and the FCPA and other anti-bribery laws that we may be subject to for which we may be held responsible. If our employees or other agents are found to have engaged in such practices, we could suffer severe penalties and other consequences that may have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Intellectual Property
If we breach any of the agreements under which we license rights to products or technology from others, we could lose license rights that are material to our business or be subject to claims by our licensors.
We license rights to products and technology that are important to our business, and we expect to enter into additional licenses in the future. For instance, we have exclusively licensed patents and patent applications from Alnylam covering RNAi therapeutics. Under our agreement with Alnylam, we are subject to a range of commercialization and development, sublicensing, royalty, patent prosecution and maintenance, insurance and other obligations.
Any failure by us to comply with any of these obligations or any other breach by us of our license agreements could give the licensor the right to terminate the license in whole, terminate the exclusive nature of the license or bring a claim against us for damages. Any such termination or claim could have a material adverse effect on our financial condition, results of operations, liquidity or business. Even if we contest any such termination or claim and are ultimately successful, such dispute could lead to delays in the development or commercialization of potential products and result in time-consuming and expensive litigation or arbitration. In addition, on termination we may be required to license to the licensor any related intellectual property that we developed.
If we are unable to obtain or maintain protection for the intellectual property relating to our products, the value of our products will be adversely affected.
The patent positions of pharmaceutical companies like us are generally uncertain and involve complex legal, scientific and factual issues. We cannot be certain that our patents and patent applications, including our own and those that we have rights to through licenses from third parties, will adequately protect our intellectual property and value of our products. Our success protecting our intellectual property depends significantly on our ability to:
• | obtain and maintain U.S. and foreign patents, including defending those patents against adverse claims; |
• | secure patent term extension for the patents covering our approved products; |
35
• | protect trade secrets; |
• | operate without infringing the proprietary rights of others; and |
• | prevent others from infringing our proprietary rights. |
We may not have any additional patents issued from any patent applications that we own or license. If additional patents are granted, the claims allowed may not be sufficiently broad to protect our technology. In addition, issued patents that we own or license may be challenged in contested proceedings such as opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings and may be narrowed, invalidated or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of term of patent protection we may have for our products, and we may not be able to obtain patent term extension to prolong the terms of the principal patents covering our approved products. Changes in patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection.
In addition, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. This combination of events has created uncertainty with respect to the value of patents, once obtained, and with regard to our ability to obtain patents in the future. Depending on decisions by the U.S. Congress, the federal courts, and the PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Our patents also may not afford us protection against competitors with similar technology. Because patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind actual discoveries, neither we nor our licensors can be certain that others have not filed or maintained patent applications for technology used by us or covered by our pending patent applications without our being aware of these applications.
We exclusively license patents and patent applications for inclisiran and we own patents and patent applications for Angiomax. The patents covering inclisiran and Angiomax are currently set to expire at various dates.
Inclisiran. We have exclusively licensed from Alnylam patents and patent applications covering RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases for purposes of developing and commercializing such RNAi therapeutics. Some of these patents are directed to general RNAi technology and expire between 2020 and 2028 in the United States. Other patents are directed to compositions of the inclisiran product being developed under our license from Alnylam and to methods of treatment using such inclisiran product and the patents expire in 2027 and 2028 in the United States. In addition, Alnylam has filed and is prosecuting a number of patent applications in the United States and in certain foreign countries. One of these applications, which, if issued, expires in December 2033, contains claims directed to specific compositions of the inclisiran product we are developing and methods of administrating such compositions.
With respect to the portfolio of patents licensed from Alnylam, it is possible that one or more companies hold patent rights that could be asserted against us or patent rights to which we may need a license. If a court rules that we need such patent rights that have been asserted against us and/or we are not able to obtain a license on reasonable terms, we may be forced to pay excessive license fees or may be unable to market inclisiran, which in either case could have a material adverse effect on our business. For example, in October 2017 Silence Therapeutics plc and Silence Therapeutics GmbH, which we refer to together as Silence, served a claim in the High Court of Justice, Chancery Division, Patents Court in the United Kingdom, naming The Medicines Company UK Ltd., our wholly owned subsidiary, Alnylam and Alnylam UK Limited, as co-defendants. In Silence’s claim, it seeks a determination that it is entitled to supplementary protection certificates, or SPCs, based on its European Patent No. 2,258,847, or the ‘847 patent, and the prospective European regulatory approvals for inclisiran and for certain of Alnylam’s product candidates. This is based on Silence’s assertion that inclisiran and the cited Alnylam product candidates fall within the scope of the ‘847 patent. An SPC is an intellectual property right that could extend the life of the Silence patent in relation to a specified product for a period of up to five additional years bringing the expiration date up to 2028. In addition, Silence is seeking costs, interest and other unspecified relief. On October 31, 2017, we acknowledged service of the claim served by Silence and on November 30, 2017, submitted substantive defenses to the claim. On October 27, 2017, we and Alnylam filed and served a claim against Silence Therapeutics GmbH and Silence in the High Court seeking revocation of the ‘847 patent, as well as a declaration of non-infringement by inclisiran and certain of Alnylam’s product candidates of the ‘847 patent, and costs and interest among other potential remedies. On November 14, 2017, Silence filed a defense to our claim along with counterclaims alleging infringement of the ‘847 patent by inclisiran and certain of Alnylam’s product candidates. On December 11, 2017, we filed an answer and defense to the counter claims. The High Court has set a trial date of December 3, 2018 for all claims between Silence and the Defendants. Although we
36
believe the ‘847 patent is invalid and not infringed by inclisiran and we will vigorously defend any claim brought against us by Silence, litigation is subject to inherent uncertainty.
Angiomax. The principal U.S. patents covering Angiomax currently include the ‘727 patent and the ‘343 patent and previously included U.S. Patent No. 5,196,404, or the ‘404 patent. The ‘404 patent covered the composition of matter of Angiomax. The ‘404 patent was set to expire in March 2010, but the term was extended to December 15, 2014 by the PTO under the Hatch-Waxman Act. As a result of our study of Angiomax in the pediatric setting, we had an additional six-month period of pediatric exclusivity following expiration of the ‘404 patent. This period of exclusivity expired in June 2015.
In the second half of 2009, the PTO issued to us the ‘727 patent and the ‘343 patent, covering a more consistent and improved Angiomax drug product and the processes by which it is made. The ‘727 patent and the ‘343 patent are set to expire in January 2029, which includes pediatric exclusivity. In response to Paragraph IV Certification Notice letters we received with respect to ANDAs filed by a number of parties with the FDA seeking approval to market generic versions of Angiomax, we filed lawsuits against the ANDA filers alleging patent infringement of the ‘727 patent and ‘343 patent and have since entered into settlement agreements with respect to our suits against three ANDA filers, Teva, APP and Sun.
In our lawsuit against Hospira, on July 2, 2015, the Federal Circuit Court ruled against us, finding the ‘727 patent and ‘343 patent invalid under the Section 102(b) “on sale” bar. In November 2015, our petition for en banc review of the Federal Circuit Court's July 2, 2015 decision was granted and the Federal Circuit Court vacated its July 2, 2015 decision. In July 2015, as a result of the Federal Circuit Court's now vacated July 2, 2015 decision, we entered into a supply and distribution agreement with Sandoz under which we granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax (bivalirudin). On July 15, 2015, Hospira's ANDAs for its generic versions of Angiomax were approved by the FDA and Hospira began selling its generic versions of Angiomax. On July 11, 2016, in an unanimous decision, the en banc Federal Circuit Court ruled in our favor by finding that the ‘727 patent and the ‘343 patent were not invalid under the “on sale” bar. The remaining issues on appeal that were not decided by the original panel were remanded back to the same panel for consideration. On February 6, 2018, the Federal Circuit Court issued a decision affirming the District Court’s finding of noninfringement of the ‘727 patent and ‘343 patent and remanding the case back to the District Court to determine whether there was an offer for sale of the invention under Section 102(b). Our patent infringement litigation with Mylan was ordered to be a companion appeal to the Hospira appeal and was heard by the same judges as the Hospira appeal. On April 6, 2017, the Federal Circuit Court found that Mylan’s ANDA for a generic bivalirudin product does not infringe either the ‘727 patent and ‘343 patent. On June 28, 2017, the district court entered an amended final judgment in favor of Mylan.
In addition to Hospira, other generic firms have entered the market. Mylan commenced marketing its generic bivalirudin product following a decision by the Federal Circuit Court in Mylan’s appeal that reversed an earlier district court decision that found that Mylan’s ANDA product infringed all of the asserted claims of the ‘727 patent. APP, through its affiliated company, Fresenius Kabi, commenced selling its generic version of Angiomax under provisions of a settlement agreement triggered by the Federal Circuit Court’s July 2, 2015 decision in the Hospira matter. Apotex Inc. and Dr. Reddy’s Laboratories have each also commenced commercialization of generic bivalirudin products upon receiving final approval if their respective ANDA filings by the FDA even though we remain in active litigation against Apotex and only recently settled with Dr. Reddy’s Laboratories.
A number of companies in addition to Hospira, Mylan, APP, Apotex Inc. and Dr. Reddy’s Laboratories have filed ANDAs for their generic versions of Angiomax. In addition to the generic versions and Baxter’s ready-to-use version of bivalirudin currently being sold, Angiomax could be subject to further generic competition in the United States from Teva and other generic ANDA filers that we have settled with, under the circumstances set forth in our respective settlement agreements with such parties and upon a final approval of each company's ANDA filings by the FDA. Pliva Hrvatska DOO, an affiliate of Teva, currently has tentative approval for its ANDA filing for its generic version of Angiomax. Other ANDA filers may commercialize their products ‘at risk’ if they receive final approval of their respective ANDA filings and are not subject to a Hatch-Waxman 30-month stay. Further, we remain in infringement litigation involving the ‘727 patent and ‘343 patent with the other ANDA filers. See Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K for descriptions of our litigation with ANDA filers and related settlements. There can be no assurance as to the outcome of our infringement litigation. We may continue to incur further legal expenses related to these matters. Following our settlements with Teva, APP, Sun, Dr. Reddy’s, Aurobindo and Accord, we submitted the settlement documents for each settlement to the U.S. Federal Trade Commission, or the FTC, and the U.S. Department of Justice, or the DOJ. The FTC, the DOJ and state attorney general offices could seek to challenge our settlements with Teva, APP, Sun, Dr. Reddy’s, Aurobindo and Accord or a third party could initiate a private action under antitrust or other laws challenging our settlements with Teva, APP, Sun, Dr. Reddy’s, Aurobindo and Accord. While we believe our settlements are lawful, we may not prevail in any such challenges or litigation, in which case the other party might obtain injunctive relief, remedial relief, or such other relief as a court may order. In any event, we may incur significant costs in the event of an investigation or in defending any such action and our business and results of operations could be materially impacted if we fail to prevail against any such challenges.
37
In Europe, the principal patent covering Angiomax expired in August 2015. This patent covered the composition of matter of Angiomax. As a result, we face generic competition in Europe. We are in the process of voluntarily discontinuing and withdrawing Angiomax from the market outside of North America and have ceased related commercialization activities.
We plan to file applications for patent term extension for inclisiran upon its approval. If we do not receive patent term extensions for the periods requested by us or at all, our patent protection for inclisiran could be limited.
Among other proceedings, we are a party to a number of lawsuits that we brought against pharmaceutical companies that have notified us that they have filed ANDAs seeking approval to market generic versions of Angiomax. We cannot predict the outcome of these lawsuits and proceedings. Involvement in litigation and other proceedings, regardless of its outcome, is time-consuming and expensive and may divert our management’s time and attention. During the period in which these matters are pending, the uncertainty of their outcome may cause our stock price to decline. An adverse result in these matters, whether appealable or not, may cause our stock price to decline.
In addition to seeking to enforce our patent rights, we have in the past and may in the future seek to enforce our other intellectual property rights, including, for example, our trademark rights in order to prevent third parties from using the same or confusingly similar trademarks. We may not be successful in enforcing such rights and preventing such use. Further, certain of our trademark rights are licensed to us by third parties and, in certain circumstances, on a non-exclusive basis, which does not afford us the right to prevent third parties from using such trademarks. Failure to adequately pursue and enforce our intellectual property rights could damage our brands, enable others to compete with our products and impair our competitive position.
If we are not able to keep our trade secrets confidential, our technology and information may be used by others to compete against us.
We rely significantly upon unpatented proprietary technology, information, processes and know-how. We seek to protect this information by confidentiality agreements and invention assignment agreements with our employees, consultants and other third-party contractors, as well as through other security measures. We may not have adequate remedies for any breach by a party to these confidentiality agreements or invention assignment agreements. In addition, our competitors may learn or independently develop our trade secrets. If our confidential information or trade secrets become publicly known, they may lose their value to us.
If we infringe or are alleged to infringe intellectual property rights of third parties, our business may be adversely affected.
Our research, development and commercialization activities, as well as any product candidates or products resulting from these activities, may infringe or be claimed to infringe patents or patent applications under which we do not hold licenses or other rights. Third parties may own or control these patents and patent applications in the United States and abroad. These third parties could bring claims against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
As a result of patent infringement claims, or in order to avoid potential claims, we or our collaborators may choose or be required to seek a license from the third party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. This could harm our business significantly.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including reexamination, inter partes review, post-grant review, and interference proceedings declared by the PTO and opposition proceedings in the European Patent Office, regarding intellectual property rights with respect to our products and technology. Patent litigation and other proceedings may also absorb significant management time. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time.
38
Risks Related to Our Common Stock
Fluctuations in our operating results could affect the price of our common stock.
Our operating results may vary from period to period based on factors, including the timing, expenses and results of clinical trials, announcements regarding clinical trial results and product introductions by us or our competitors, the availability and timing of third-party reimbursement, including in Europe, sales and marketing expenses and the timing of regulatory approvals. If our operating results do not meet the expectations of investors and securities analysts as a result of these or other factors, the trading price of our common stock will likely decrease.
The capped call transactions may affect the value of the 2023 notes and our common stock.
In connection with the issuance of the 2023 notes, we entered into capped call transactions with respect to the 2023 notes with certain hedge counterparties. The capped call transactions will cover, subject to customary anti-dilution adjustments, the aggregate number of shares of common stock underlying the 2023 notes and are expected generally to reduce potential dilution to the common stock upon conversion of the 2023 notes in excess of the principal amount of such converted 2023 notes. In connection with establishing their initial hedges of the capped call transactions, the hedge counterparties (or their affiliates) entered into various derivative transactions with respect to the common stock concurrently with, and/or purchased the common stock shortly after, the pricing of the 2023 notes. The hedge counterparties (or their affiliates) are likely to modify their hedge positions by entering into or unwinding various derivative transactions with respect to the common stock and/or by purchasing or selling the common stock or other securities of ours in secondary market transactions prior to the maturity of the 2023 notes (and are likely to do so during the settlement averaging period under the capped call transactions, which precedes the maturity date of the 2023 notes, and on or around any earlier conversion date related to a conversion of the 2023 notes). The effect, if any, of any of these transactions and activities on the market price of our common stock or the 2023 notes will depend in part on market conditions and cannot be ascertained at this time, but any of these activities could adversely affect the value of our common stock, which could affect the value of the 2023 notes and the value of our common stock, if any, that the 2023 note holders receive upon any conversion of the 2023 notes.
Our stock price has been and may in the future be volatile. This volatility may make it difficult for you to sell common stock when you want or at attractive prices.
Our common stock has been and in the future may be subject to substantial price volatility. From January 1, 2015 to March 1, 2018, the last reported sale price of our common stock ranged from a high of $55.95 per share to a low of $24.32 per share. The value of your investment could decline due to the effect upon the market price of our common stock of any of the following factors, many of which are beyond our control:
• | announcements of results of clinical trials or nonclinical studies by us or third parties relating to inclisiran or Angiomax or products of our competitors or of regulatory proceedings by us or our competitors; |
• | approval or rejection of submissions for marketing approval for inclisiran; |
• | regulatory actions by the FDA limiting or revoking the use of Angiomax; |
• | changes in securities analysts’ estimates of our financial performance; |
• | changes in valuations of similar companies; |
• | variations in our operating results; |
• | acquisitions and strategic partnerships; |
• | announcements of technological innovations or new commercial products by us or our competitors or the filing of ANDAs, NDAs or BLAs for products competitive with ours; |
• | the timing, amount and receipt of revenue from and margins on sales of Angiomax; |
• | changes in governmental regulations; |
39
• | developments in patent rights or other proprietary rights; |
• | the extent to which our products are commercially successful globally; |
• | developments in our ongoing litigation and significant new litigation; |
• | developments or issues with our contract manufacturers; |
• | changes in our management; and |
• | general market conditions. |
We believe that period-to-period comparisons of our financial results will not necessarily be indicative of our future performance. If our revenues in any particular period do not meet expectations, we may not be able to adjust our expenditures in that period, which could cause our operating results to suffer. If our operating results in any future period fall below the expectations of securities analysts or investors, our stock price may fall by a significant amount.
The stock markets in general, and The NASDAQ Global Select Market and the market for biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations recently. These fluctuations often have been unrelated or disproportionate to the operating performance of these companies. These broad market and industry factors may adversely affect the market price of our common stock, regardless of our actual operating performance.
We have been subject to securities class action litigation and may be subject to similar or other litigation in the future, which may divert management’s attention and have a material adverse effect on our business, financial condition and results of operations.
In February 2014, a class action lawsuit was filed against us and certain of our current and former officers alleging, among other things, that we and certain of our current and former officers violated federal securities laws because we and certain current and former officers allegedly made misrepresentations or did not make proper disclosures regarding the results of clinical trials which tested the efficacy and safety of one of our recently divested products. On February 12, 2016, the parties executed a stipulation for a proposed class settlement, subject to court approval, and on June 7, 2016, the Court granted final approval of the settlement.
There may be additional suits or proceedings brought in the future. Monitoring and defending against legal actions, whether or not meritorious, is time-consuming for our management and detracts from our ability to fully focus our internal resources on our business activities, and we cannot predict how long it may take to resolve these matters. In addition, we may incur substantial legal fees and costs in connection with litigation. Although we have insurance, coverage could be denied or prove to be insufficient.
Our corporate governance structure, including provisions in our certificate of incorporation and by-laws and Delaware law, may prevent a change in control or management that security holders may consider desirable.
The General Corporation Law of the State of Delaware and our certificate of incorporation and by-laws contain provisions that might enable our management to resist a takeover of our company or discourage a third party from attempting to take over our company. These provisions include:
• | Section 203 of the Delaware General Corporation Law, which provides that we may not enter into a business combination with an interested stockholder for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in the manner prescribed in Section 203; |
• | our board of directors has the authority to issue, without a vote or action of stockholders, up to 5,000,000 shares of a new series of preferred stock and to fix the price, rights, preferences and privileges of those shares, each of which could be superior to the rights of holders of our common stock; |
• | currently and until such time after our 2018 annual meeting of stockholders that our board of directors ceases to be classified, our directors may be removed only for cause and then only by the affirmative vote of the holders of at least 75% of the votes which all stockholders would be entitled to cast in any annual election of directors, and at all times after our board ceases to be classified, our directors may be removed with or without cause (but subject to the same 75% voting requirement as currently in effect); |
40
• | the size of our board of directors is determined by resolution of the board of directors; |
• | any vacancy on our board of directors, however occurring, including a vacancy resulting from an enlargement of our board, may only be filled by vote of a majority of our directors then in office, even if less than a quorum; |
• | only our board of directors may call special meetings of stockholders; |
• | our by-laws may be amended, altered or repealed by (i) the affirmative vote of a majority of our directors, subject to any limitations set forth in the by-laws, or (ii) the affirmative vote of the holders of at least 75% of the votes which all the stockholders would be entitled to cast in any annual election of directors; |
• | stockholders must provide us with advance notice, and certain information specified in our by-laws, in connection with nominations or proposals by such stockholder for consideration at an annual meeting; |
• | stockholders may not take any action by written consent in lieu of a meeting; and |
• | our certificate of incorporation may only be amended or repealed by the affirmative vote of a majority of our directors and the affirmative vote of the holders of at least 75% of the votes which all the stockholders would be entitled to cast in any annual election of directors (and plus any separate class vote that might in the future be required pursuant to the terms of any series of preferred stock that might be outstanding at the time any of these amendments are submitted to stockholders). |
These provisions could have the effect of delaying, deferring, or preventing a change in control of us or a change in our management that stockholders may consider favorable or beneficial. These provisions could also discourage proxy contests and make it more difficult for stockholders to elect directors and take other corporate actions. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock or our other securities.
Our business could be negatively affected as a result of the actions of activist shareholders.
Proxy contests have been waged against many companies in the biopharmaceutical industry over the last few years. If faced with a proxy contest, we may not be able to successfully defend against the contest, which would be disruptive to our business. Even if we are successful, our business could be adversely affected by a proxy contest because:
• | responding to proxy contests and other actions by activist shareholders may be costly and time-consuming and may disrupt our operations and divert the attention of management and our employees; |
• | perceived uncertainties as to our future direction may result in our inability to consummate potential acquisitions, collaborations or in-licensing opportunities and may make it more difficult to attract and retain qualified personnel and business partners; and |
• | if individuals are elected to our board of directors with a specific agenda different from ours, it may adversely affect our ability to effectively and timely implement our strategic plan and create additional value for our stockholders. |
Item 1B. | Unresolved Staff Comments. |
None.
Item 2. | Properties. |
We lease our principal office in Parsippany, New Jersey, U.S. The lease for Parsippany office covers 173,146 square feet and expires January 2024. We also lease 63,000 square feet of office and laboratory space in San Diego, California. This lease expires in September 2028. On January 11, 2018, we entered into an agreement to sublease 32,039 square feet of the office and laboratory space in San Diego. The sublease agreement has a term of 84 months.
We believe that all of our facilities are in good condition and are well maintained and that our current arrangements will be sufficient to meet our needs for the foreseeable future.
41
Item 3. Legal Proceedings.
From time to time we are party to legal proceedings in the course of our business in addition to those described below. We do not, however, expect such other legal proceedings to have a material adverse effect on our business, financial condition or results of operations.
‘727 Patent and ‘343 Patent Litigations
Hospira, Inc.
In July 2010, we were notified that Hospira, Inc., or Hospira, had submitted two ANDAs seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 patent and ‘343 patent. On August 19, 2010, we filed suit against Hospira in the U.S. District Court for the District of Delaware for infringement of the ‘727 patent and ‘343 patent. On August 25, 2010, the case was reassigned in lieu of a vacant judgeship to the U.S. District Court for the Eastern District of Pennsylvania. Hospira’s answer denied infringement of the ‘727 patent and ‘343 patent and raised counterclaims of non-infringement and invalidity of the ‘727 patent and ‘343 patent. On September 24, 2010, we filed a reply denying the counterclaims raised by Hospira. The Hospira action was consolidated for discovery purposes with the then pending and now settled cases against Teva and APP. The case was reassigned back to the U.S. District Court for the District of Delaware. A Markman hearing was held on December 5, 2012. On July 12, 2013, the district court issued its Markman decision as to the claim construction of the ‘727 patent and the ‘343 patent. The district court’s decision varied from the other Markman decisions that we have received in our other patent infringement litigations. On July 22, 2013, we filed a motion for reconsideration of the district court’s claim construction ruling on the grounds that the district court (i) impermissibly imported process limitations disclosed in a preferred embodiment into the claims, (ii) improperly transformed product claims into product-by-process claims, (iii) improperly rendered claim language superfluous and violated the doctrine of claim differentiation, and (iv) improperly construed limitations based on validity arguments that have not yet been presented. On August 22, 2013, the district court denied the motion for reconsideration. A three day bench trial was held in September 2013 and a post-trial briefing was completed in December 2013. On March 31, 2014, the district court issued its trial opinion. With respect to patent validity, the district court held that the ‘727 and ‘343 patents were valid on all grounds. Specifically, the district court found that Hospira had failed to prove that the patents were either anticipated and/or obvious. The district court further held that the patents satisfied the written description requirement, were enabled and were not indefinite. With respect to infringement, based on its July 2013 Markman decision, the district court found that Hospira’s ANDAs did not meet the “efficient mixing” claim limitation and thus did not infringe the asserted claims of the ‘727 and ‘343 patents. The district court found that the other claim limitations in dispute were present in Hospira’s ANDA products. The district court entered a final judgment on April 15, 2014. On May 9, 2014, we filed a notice of appeal to the Federal Circuit Court. On May 23, 2014, Hospira filed a notice of cross-appeal. We filed our opening appeal brief on August 13, 2014. Hospira filed its opening appeal brief on September 26, 2014 asserting that the claim constructions and non-infringement findings were correct. Hospira also sought to overturn the finding of patent validity. Briefing was completed in December 2014. An oral argument before the Federal Circuit Court was held on March 6, 2015. On July 2, 2015, the Federal Circuit Court issued an opinion finding the asserted claims of the ‘727 patent and ‘343 patent invalid under the Section 102(b) “on sale” bar. The decision was based on a finding that third-party manufacturer, Ben Venue Laboratories, “sold” manufacturing services for three validation batches to us before a critical date. On July 15, 2015, Hospira received final approval for its ANDAs. On July 31, 2015, we filed with the Federal Circuit Court a combined petition for panel rehearing and rehearing en banc. On August 24, 2015, the Federal Circuit Court invited Hospira to respond to the petition. On September 8, 2015, Hospira filed a response. On November 13, 2015, the Federal Circuit Court granted our petition for rehearing en banc and vacated its earlier July 2, 2015 decision. The Federal Circuit Court set a briefing schedule, specified specific questions to be answered, invited the DOJ to file a brief expressing the views of the United States and also invited any other amicii curiae to file briefs on the en banc issues raised. Hospira filed its opening brief on January 11, 2016. We filed our response on February 24, 2016 and Hospira filed its reply brief on March 10, 2016. Nine amicus briefs were filed: Department of Justice, American Intellectual Property Law Association, Intellectual Property Owners Association, a Texas law firm, Miller Patti Pershern PLLC, Pharmaceutical Research and Manufacturers of America, Biotechnology Innovation Organization, Gilead Sciences, Inc., an individual, Roberta J. Morris, Esq., and Houston Intellectual Property Law Association. The Federal Circuit Court sitting en banc heard oral argument from the parties and the government on May 5, 2016. On July 11, 2016, in an unanimous decision, the en banc Federal Circuit Court affirmed the district court holding that our transaction with Ben Venue Laboratories did not constitute an invalidating sale under the “on sale” bar. The remaining issues on appeal that were not decided by the original panel were remanded back to the same panel for consideration. In a subsequent order of July 18, 2016, the parties were directed to file new appeal briefs taking into account the en banc decision. The parties submitted revised briefs and this briefing was completed in October 2016. The Federal Circuit Court heard oral argument on December 6, 2016. On February 6, 2018, the Federal Circuit Court issued a decision affirming the district court’s finding of noninfringement of the ‘727 patent and ‘343 patent and remanding the case back to the district court to determine whether there was an offer for sale of the invention under Section 102(b). We are evaluating next steps in the matter.
42
Mylan Pharmaceuticals, Inc.
In January 2011, we were notified that Mylan Pharmaceuticals, Inc. had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 patent and ‘343 patent. On February 23, 2011, we filed suit against Mylan Inc., Mylan Pharmaceuticals Inc. and Bioniche Pharma USA, LLC, which we refer to collectively as Mylan, in the U.S. District Court for the Northern District of Illinois for infringement of the ‘727 patent and ‘343 patent. Mylan’s answer denied infringement of the ‘727 patent and ‘343 patent and raised counterclaims of non-infringement and invalidity of the ‘727 patent and ‘343 patent. On April 13, 2011, we filed a reply denying the counterclaims raised by Mylan. On May 4, 2011, the district court set a pretrial schedule. Following a joint request, the district court issued an amended scheduling order on September 22, 2011. On November 29, 2011, Mylan moved to amend its answer to add counterclaims and affirmative defenses of inequitable conduct and unclean hands. Following motion practice, the district court granted Mylan’s request to add counterclaims and affirmative defenses of inequitable conduct and to add affirmative defenses of unclean hands. On March 7, 2012, we filed a reply denying these counterclaims. A Markman hearing was held on July 30, 2012. The district court issued a Markman Order on August 6, 2012. The parties have completed fact and expert discovery. On June 21, 2013, Mylan filed a summary judgment motion of non-infringement of the ‘727 and ‘343 patents and alternatively that the ‘727 patent was invalid. The district court’s decision granted non-infringement of the ‘343 patent and denied the motion with respect to non-infringement and invalidity of the ‘727 patent. A six day trial directed to the ‘727 patent was completed on June 18, 2014. Post-trial briefs were filed on July 1, 2014 and July 11, 2014. On October 27, 2014, the district court issued an opinion and order finding that Mylan’s ANDA product infringes all of the asserted claims of the ‘727 patent. The district court further found that Mylan failed to prove that the same asserted claims of the ‘727 patent are invalid or unenforceable. Specifically, the district court found that Mylan failed to prove its allegations of anticipation, obviousness, non-enablement and unenforceability due to inequitable conduct. On October 28, 2014 and November 13, 2014, Mylan filed Notices of Appeal to the Federal Circuit Court. On November 25, 2014, we filed a Notice of Cross Appeal of the district court’s summary judgment of noninfringement of the asserted claims of the ‘343 patent that it had issued on December 16, 2013 and the district court’s Markman Order on August 6, 2012. Appellate briefing was completed in April 2015. An oral argument before the Federal Circuit Court was scheduled for September 11, 2015. On July 29, 2015, following a Mylan motion for disposition of its appeal in view of the July 2, 2015 Hospira decision, the Federal Circuit Court granted the motion (1) reversing the district court’s judgment as to the ‘727 patent (2) dismissing as moot our cross-appeal (3) vacating the district court’s entry of an injunction, and (4) holding that each party shall bear its own costs. On August 27, 2015, we filed a petition for panel rehearing. Following the November 13, 2015 decision granting our en banc hearing request in the Hospira appeal and vacating the July 2, 2015 decision, we moved to vacate the Federal Circuit Court’s July 29, 2015 Order terminating the Mylan appeal. Following briefing, the Federal Circuit Court granted our motion and reopened the appeal, vacated its July 29, 2015 Order and then stayed the Mylan appeal pending resolution of the Hospira appeal. Following the en banc decision in the Hospira appeal described above, the Federal Circuit Court lifted the stay. The Mylan appeal was ordered to be a companion appeal to the Hospira appeal and was decided by the same judges as the Hospira appeal. The parties were ordered to file new briefs incorporating the en banc decision. The parties submitted revised briefs and this briefing was completed in October 2016. The Federal Circuit Court heard oral argument on December 6, 2016. Mylan’s ANDA received tentative approval from the FDA in February 2017. On April 6, 2017, the Federal Circuit issued a decision reversing the district court’s finding of infringement of the ‘727 patent and affirming the lower court’s summary judgment of non-infringement of the ‘343 patent. On April 7, 2017, Mylan filed an emergency motion to accelerate the time for any petition for rehearing and issuance of a mandate. On April 11, 2017, we opposed this motion and on April 12, 2017 the Federal Circuit Court denied Mylan’s request. On May 5, 2017, we filed with the Federal Circuit Court a petition for rehearing or en banc review. On May 12, 2017, the Federal Circuit Court invited Mylan to respond which they did on May 19, 2017. On May 23, 2017, we filed a motion to file a reply brief. On May 30, 2017, the Federal Circuit Court denied the motion for a reply and on June 6 denied our petition for panel rehearing. The Federal Circuit Court then issued its mandate on June 13, 2017. On June 9, 2017, Mylan filed in the district court a motion to amend the court’s October 27, 2014 judgment. On June 22, 2017, we filed our opposition to amend the final judgment and also moved for a new trial on the doctrine of equivalents of the ‘727 patent. On June 25, 2017, Mylan opposed the motion for a new trial and we filed our reply on June 26th. On June 28, 2017, the district court issued an order granting Mylan’s motion to amend the final judgment and denied our motion for a new trial. The district court entered an amended final judgment on June 28, 2017. Following further briefing by the parties, on October 30, 2017 the district court determined that Mylan was the prevailing party and awarded certain court costs.
Dr. Reddy’s Laboratories, Inc.
In March 2011, we were notified that Dr. Reddy’s Laboratories, Ltd. and Dr. Reddy’s Laboratories, Inc. had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On April 28, 2011, we filed suit against Dr. Reddy’s Laboratories, Ltd., Dr. Reddy’s Laboratories, Inc. and Gland Pharma, Inc., which
43
we refer to collectively as Dr. Reddy’s, in the U.S. District Court for the District of New Jersey for infringement of the ‘727 patent and ‘343 patent. Dr. Reddy’s answer denied infringement of the ‘727 patent and ‘343 patent and raised counterclaims of non-infringement and invalidity of the ‘727 patent and ‘343 patent. On May 11, 2012, Dr. Reddy’s filed a motion for summary judgment. On October 2, 2012, the district court held oral argument on Dr. Reddy’s summary judgment motion and conducted a Markman hearing. On October 15, 2012, the district court denied Dr. Reddy’s summary judgment motion. A Markman decision was issued by the district court on January 2, 2013. On January 25, 2013, Dr. Reddy’s filed a second summary judgment motion this time for non-infringement. At the direction of the district court, on May 13, 2013, the motion was withdrawn by Dr. Reddy’s. We have pending motions seeking further fact discovery of Dr. Reddy’s. The parties have yet to enter the expert phase of the case. On May 12, 2015 the district court issued a Stipulation and Order staying the case as Dr. Reddy's had yet to respond to an FDA Complete Response Letter dated December 7, 2012. In June 2016, Dr. Reddy’s responded to the FDA’s Complete Response Letter. As a result, following a joint submission by the parties, the district court on July 22, 2016 ordered the stay vacated and reopened discovery of Dr. Reddy’s ANDA. Following the decision by the Federal Circuit in the above Mylan appeal, the district court set a schedule for the exchange of expert reports and additional fact discovery. Following settlement discussions, the case was settled and a final judgment finding the ‘727 and ‘343 patents valid, enforceable and infringed by Dr. Reddy’s ANDA product was entered by the district court in December 2017. In connection with the Dr. Reddy’s settlement, we entered into a license agreement with Dr. Reddy’s under which Dr. Reddy’s agreed to pay us a one-time license fee and we granted Dr. Reddy’s a non-exclusive license under the ‘727 patent and ‘343 patent to sell a generic bivalirudin for injection product under Dr. Reddy’s ANDA in the United States. The settlement documents were submitted to the FTC and DOJ in December 2017.
Sun Pharmaceutical Industries LTD
In October 2011, we were notified that Sun Pharmaceutical Industries LTD had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On November 21, 2011, we filed suit against Sun Pharma Global FZE, Sun Pharmaceutical Industries LTD., Sun Pharmaceutical Industries Inc., and Caraco Pharmaceutical Laboratories, LTD., which we refer to collectively as Sun, in the U.S. District Court for the District of New Jersey for infringement of the ‘727 patent and ‘343 patent. The case has been assigned to the same judge and magistrate judge as the Dr. Reddy’s action. Sun’s answer denied infringement of the ‘727 patent and ‘343 patent. On June 7, 2012, the district court held an initial case scheduling conference. The parties proceeded with fact discovery. Following a December 20, 2013 status conference, the parties began discussing a stay in the case. Following further conferences with the district court a stipulation to stay the case was submitted and subsequently entered by the district court on April 1, 2014. Following settlement discussions, the case was settled and a final judgment finding the ‘727 and ‘343 patents valid, enforceable and infringed by Sun’s ANDA product was entered by the district court on March 27, 2015. In connection with the Sun settlement, we entered into a license agreement with Sun under which we granted Sun a non-exclusive license under the ‘727 patent and ‘343 patent to sell a generic bivalirudin for injection product under Sun’s ANDA in the United States beginning on June 30, 2019 or earlier in certain circumstances. The settlement documents were submitted to the FTC and DOJ in March 2015.
Apotex Inc.
In March 2013, we were notified that Apotex Inc. had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On May 1, 2013, we filed suit against Apotex Inc. and Apotex Corp., which we refer to collectively as Apotex, in the U.S. District Court for the District of New Jersey for infringement of the ‘727 and ‘343 patents. The case has been assigned to the same judge and magistrate judge as the Dr. Reddy’s and Sun actions. Apotex filed its answer on July 19, 2013 and raised counterclaims of non-infringement and invalidity. A scheduling conference before the magistrate judge was held on December 16, 2013. Following a subsequent conference on April 15, 2014 and further directions from the district court to resubmit a discovery schedule, the district court entered a revised discovery schedule on July 17, 2014. A Markman hearing commenced on January 22, 2015 and was completed on March 3, 2015. Following the July 2, 2015 Hospira decision, the parties requested and the district court entered an order staying the case until the Federal Circuit Court issues a mandate in the Hospira appeal. Following the Hospira en banc decision in July 2016, we moved the district court to lift the stay to resume fact discovery of Apotex’s ANDA, which Apotex opposed. The magistrate judge granted our request and issued an order on September 13, 2016 reinstating the case and ordered certain discovery to proceed. On September 23, 2016, Apotex filed a motion to vacate the September 13th order. Oral argument on the motion was held on October 17, 2016 and the district court entered an order that ANDA discovery could proceed. In addition, in October 2016, the district court ordered Apotex to give us 10-days’ notice before any at risk launch. The parties requested and the district court agreed to stay this case pending the above discussed Hospira and Mylan appeals. The district court conducted a status conference on February 6, 2018 and ordered the parties to exchange certain documents. A settlement conference has been set for March 21, 2018. The district court has not set a schedule for expert phase or a trial date.
44
Exela Pharma Sciences, LLC
In March 2014, we were notified that Exela Pharma Sciences, LLC, had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On April 25, 2014, we filed suit against Exela Pharma Sciences, LLC, Exela PharmSci, Inc. and Exela Holdings, Inc., which we collectively refer to as Exela, in the U.S. District Court for the Western District of North Carolina for infringement of the ‘727 and ‘343 patents. Exela filed its answer on June 3, 2014 and raised counterclaims of non-infringement, invalidity and unenforceability due to inequitable conduct. We filed a reply on July 11, 2014. The parties have conducted a Rule 26 conference. The district court has set a pretrial schedule through a June 2015 Markman hearing. On November 4, 2014, Exela filed a motion for judgment on the pleadings based on noninfringement. The motion was fully briefed on December 23, 2014. Claim construction discovery was under way. Following the July 2, 2015 Hospira decision, the parties requested and the court entered an order staying the case until the Federal Circuit Court issues a mandate in the Hospira appeal. On January 29, 2016, even though no mandate from the Hospira appeal has issued, Exela filed a motion to lift the stay and resume claim construction proceedings and other pretrial matters. On February 29, 2016, the district court denied Exela’s motion to lift the stay on the case. Following the Hospira en banc decision in July 2016, we moved to lift the stay. Exela opposed the motion but indicated it would agree to lift the stay under certain conditions. In a September 29, 2016 order, the magistrate judge ruled the case should remain stayed. On September 1, 2017, the case was reassigned to another judge, also of the Western District of North Carolina.
Accord Healthcare Inc., USA
In June 2014, we were notified that Accord Healthcare Inc., or Accord, had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On July 24, 2014, we filed suit against Accord and its parent, Intas Pharmaceuticals Ltd., or Intas, in the U.S. District Court for the Middle District of North Carolina for infringement of the ‘727 patent and ‘343 patent. On September 26, 2014, Accord and Intas filed an answer denying infringement and asserting that the ‘727 and ‘343 patents are invalid. The parties have conducted a Rule 26 conference. The district court has set February 17, 2016 for the close of all discovery and October 3, 2016 as a trial date. Following the July 2, 2015 Hospira decision, the parties requested and the district court entered an order staying the case until the Federal Circuit Court issues a mandate in the Hospira appeal. Following discussions, the parties agreed to a settlement and an order for judgment and permanent injunction was filed in February 2018, pursuant to which the parties seek a finding that the ‘727 and ‘343 patents are valid, enforceable and infringed by Accord’s ANDA product. In connection with the Accord settlement, we entered into a license agreement with Accord under which Accord agreed to pay us a royalty on net sales Accord’s bivalirudin product and we granted Accord a non-exclusive license under the ‘727 patent and ‘343 patent to sell a generic bivalirudin for injection product under its ANDA in the United States. The settlement documents were submitted to the FTC and DOJ in February 2018. Accord’s ANDA received tentative approval from the FDA in April 2016.
Aurobindo Pharma Limited
In March 2014, we were notified that Aurobindo Pharma Limited had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On April 11, 2014, we filed suit against Aurobindo Pharma Limited and Aurobindo Pharma USA, Inc., which we refer to collectively as Aurobindo, in the U.S. District Court for the District of New Jersey for infringement of the ‘727 and ‘343 patents. The case has been assigned to the same judge and magistrate judge as the Dr. Reddy’s, Sun and Apotex actions. Aurobindo filed its answer on July 3, 2014 and raised counterclaims of non-infringement and invalidity. A scheduling conference before the magistrate judge was held on November 20, 2014. The parties engaged in fact discovery and claim construction exchanges. On April 6, 2015, the district court entered a revised fact and expert discovery schedule. Thereafter, the parties proposed a stay of the case pending a decision in the above-referenced Hospira appeal to the district court, which the district court entered on April 15, 2015. Following the July 2, 2015 Hospira decision, the district court was informed of the decision and the parties requested the present stay to remain in effect until Federal Circuit Court issues a mandate in the Hospira appeal. The district court entered this request on July 20, 2015. On April 27, 2017, Aurobindo filed a motion to lift the stay. We filed an opposition on May 22, 2017 and in the alternative proposed a schedule to complete fact and expert discovery. On May 30, 2017, Aurobindo filed a reply and on August 16, 2017, the district court lifted the stay. On September 8, 2017, Aurobindo filed an amended answer adding additional counterclaims and defenses. On October 6, 2017, we filed our response to these new claims. On October 9, 2017, Aurobindo filed a motion for judgment on the pleadings pursuant to Rule 12(c). Following settlement discussions, the case was settled and a final judgment finding the ‘727 and ‘343 patents valid, enforceable and infringed by Aurobindo’s ANDA product was entered by the district court on November 22, 2017. In connection with the Aurobindo settlement, we entered into a license agreement with Aurobindo under which Aurobindo agreed to pay us a royalty on net sales Aurobindo’s bivalirudin product and we granted Aurobindo a non-exclusive license under the ‘727 patent and ‘343 patent to sell a generic bivalirudin for injection product under its ANDA in the United States. The settlement documents
45
were submitted to the FTC and DOJ in November 2017. Aurobindo’s ANDA received tentative approval from the FDA in December 2015.
Sagent Pharmaceuticals Inc.
In July 2015, we were notified that Sagent Pharmaceuticals Inc., or Sagent, had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 patent and ‘343 patent. On August 26, 2015, we filed suit against Sagent in the U.S. District Court for the Northern District of Illinois for infringement of the ‘727 patent and ‘343 patent. Sagent filed its answer on November 30, 2015 and raised counterclaims of non-infringement and invalidity. We filed a reply on December 22, 2015. A scheduling conference was held on January 21, 2016. The case has been stayed pending resolution of the Hospira en banc appeal. At a September 13, 2016 status conference, the parties jointly requested the stay be lifted and discovery proceed on our claim that Sagent’s ANDA infringes the ‘727 and ‘343 patents. In addition to a proposed case schedule, the parties submitted a joint partial judgment wherein Sagent acknowledged that the claims at issue in the Hospira and Mylan appeals, if found valid, will be valid in this case and Sagent’s invalidity claims are dismissed with prejudice. To the extent the Federal Circuit Court in the Hospira and Mylan matters finds any claim invalid, the parties agreed that the partial judgment will be vacated. Sagent’s ANDA received tentative approval in March 2015, but is subject to a Hatch-Waxman 30-month stay until 2018. Following settlement discussions, the case was settled. On December 20, 2017, the district court entered a stipulation of dismissal and judgment dismissing both the complaint and Sagent’s declaratory judgment counterclaims with prejudice.
Akorn, Inc.
In October 2016, we were notified that Akorn, Inc. had submitted an ANDA seeking permission to market its generic version of Angiomax prior to the expiration of the ‘727 and ‘343 patents. On November 15, 2016, we filed suit against Akorn in the U.S. District Court for the District of New Jersey for infringement of the ‘727 and ‘343 patents. The case has been assigned to the same judge and magistrate judge as the Dr. Reddy’s, Sun, Apotex and Aurobindo actions. Akorn filed its answer on December 27, 2016 and raised counterclaims of non-infringement and invalidity. A scheduling conference before the magistrate judge was scheduled for February 14, 2017. The parties jointly requested the case be stayed pending the Federal Circuit Court appeals involving the ‘727 and ‘343 patents. On January 10, 2017, the district court ordered the case stayed.
Biogen Idec Litigation
On September 15, 2015, Biogen Idec, notified us that after completing an audit of our books and records for the fourth quarter of 2014, Biogen Idec believes it is owed additional royalties relating to Angiomax under our license agreement with Biogen Idec. On September 23, 2015, we filed suit against Biogen Idec in the United States District Court for the District of New Jersey seeking, inter alia, declaratory judgments that we have satisfied our obligations under the license agreement. On November 12, 2015, Biogen Idec answered the complaint denying our claims and asserting counterclaims for breach of contract. In February 2017, Biogen’s claim for audit costs was voluntarily dismissed. The parties have completed fact and expert discovery. A trial date has not been set by the district court. We believe we will prevail in this suit, however, there can be no assurance that we will be successful. An adverse resolution could have a material adverse effect on our business, financial condition or results of operations.
Eagle Litigation
On February 2, 2016, we filed suit against Eagle Pharmaceuticals, Inc., or Eagle, SciDose LLC and TherDose Pharma Pvt. Ltd. for infringement of U.S. Patent Nos. 7,713,928, or the ’928 patent, and 7,803,762, or the ’762 patent, by Eagle’s New Drug Application No. 208298 for ready-to-use bivalirudin. In the lawsuit, we assert that the ‘928 and ‘762 patents are co-owned by us and Eagle and are exclusively licensed to us. The complaint also seeks a declaration that we are an owner and exclusive licensee of U.S. Patent Application No. 14/711,359 pursuant to the parties’ License and Development Agreement, which Eagle represents covers the product described in its NDA No. 208298. On March 25, 2016 defendants filed a motion to dismiss. On April 18, 2016 we filed an amended complaint reasserting the original claims and raising additional claims of, inter alia, trademark infringement, unfair competition and tortious interference. The trademark infringement claim asserts that Eagle’s mark for its ready-to-use bivalirudin, Kangio, infringes our Angiomax® mark and the Kengreal® mark. On May 23, 2016 defendants filed a second motion to dismiss, which we opposed. On July 8, 2016, the Court entered a stipulation of dismissal of the trademark related claims in which defendants represented that they have abandoned their U.S. trademark applications for Kangio, they will not use the Kangio trademark in U.S. commerce for goods and services related to bivalirudin and/or anticoagulants, and that they have and/or will remove any reference to Kangio from any and all promotional and marketing material and any applicable labeling and
46
packaging. On July 21, 2016, defendants filed a motion to bifurcate and stay our patent infringement claims. On August 18, 2016 the Court denied defendants’ second motion to dismiss on all counts and on September 9, 2016 the Court denied defendants’ motion to bifurcate and stay the patent infringement claims. On October 10, 2016, defendants filed a motion for summary judgment on the same grounds advanced in the motion to dismiss, which we have opposed. On March 15, 2017, the Court denied defendants’ motion for summary judgment. Defendants informed us that they are prepared and will deliver to us any actual physical materials and assign any intellectual property or sNDA related to the ready-to-use bivalirudin and, on October 4, 2017, based on the argument that this offer would resolve all federal claims in dispute, defendants filed a motion to dismiss the remaining claims for lack of subject matter jurisdiction. On October 16, 2017, defendants filed a motion to stay discovery pending a resolution on their motion to dismiss. On November 6, 2017, we filed an opposition to the defendants’ motion to dismiss and an opposition to defendants’ motion to stay discovery. Following settlement discussions, the parties agreed to settle the case and entered into a joint stipulation and order of dismissal with prejudice pursuant to which the case was dismissed with prejudice. As part of the settlement, Eagle agreed to make a one-time payment to us and assign to us all of Eagle’s respective rights, title, and interest, including intellectual property, to Eagle’s sNDA No. 208298.
SymBio Arbitration
On October 11, 2017, SymBio filed a Request for Arbitration with the International Chamber of Commerce’s International Court of Arbitration against us and our wholly owned subsidiary, Incline. In the Request for Arbitration, SymBio claims that we failed to provide adequate assurances of performance of, or, alternatively, have rendered ourselves unable to perform, our obligations under the license agreement between us, Incline and SymBio relating to the development and commercialization of IONSYS in Japan. As a result, SymBio seeks compensatory damages in an amount of $82 million. On December 15, 2017, we filed an Answer and Counterclaim denying SymBio’s allegations, asserting defenses to SymBio’s claims, and bringing a counterclaim for breach of contract. We are seeking compensatory damages in an amount of $10 million. We intend to defend ourselves vigorously in this matter and pursue all relief to which we are entitled.
Silence Therapeutics Litigation
In July 2017, Silence issued, and in October 2017 served, a claim in the High Court of Justice, Chancery Division, Patents Court in the United Kingdom, naming The Medicines Company UK Ltd., our wholly owned subsidiary, Alnylam and Alnylam UK Limited, as co-defendants. In Silence’s claim, it seeks a determination that it is entitled to supplementary protection certificates, or SPCs, based on Silence’s European Patent No. 2,258,847, or the ‘847 patent, and the prospective European regulatory approvals for inclisiran and for certain of Alnylam’s product candidates. This is based on Silence’s assertion that inclisiran and the cited Alnylam product candidates fall within the scope of the ‘847 patent. An SPC is an intellectual property right that could extend the life of the ‘847 patent in relation to a specified product for a period of up to five additional years bringing the expiration date up to 2028. In addition, Silence is seeking costs, interest and other unspecified relief. On October 31, 2017, we acknowledged service of the claim served by Silence and on November 30, 2017, submitted substantive defenses to the claim.
On October 27, 2017, we and Alnylam filed and served a claim against Silence Therapeutics GmbH and Silence in the High Court seeking revocation of the ‘847 patent, as well as a declaration of non-infringement by inclisiran and certain of Alnylam’s product candidates of the ‘847 patent, and costs and interest among other potential remedies. On November 14, 2017, Silence filed a defense to our claim along with counterclaims alleging infringement of the ‘847 patent by inclisiran and certain of Alnylam’s product candidates. On December 11, 2017, we filed an answer and defense to the counter claims.
The High Court has set a trial date of December 3, 2018 for all claims between Silence and the Defendants.
Item 4. | Mine Safety Disclosures. |
Not applicable.
47
PART II
Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Our common stock trades on The NASDAQ Global Select Market under the symbol “MDCO”. The following table reflects the range of the high and low sale price per share of our common stock, as reported on The NASDAQ Global Select Market for the periods indicated. These prices reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not necessarily represent actual transactions.
Common Stock Price | |||||||
High | Low | ||||||
Year Ended December 31, 2017 | |||||||
First Quarter | $ | 55.28 | $ | 32.61 | |||
Second Quarter | $ | 55.95 | $ | 36.91 | |||
Third Quarter | $ | 43.79 | $ | 32.39 | |||
Fourth Quarter | $ | 39.44 | $ | 25.40 | |||
Year Ended December 31, 2016 | |||||||
First Quarter | $ | 37.48 | $ | 27.50 | |||
Second Quarter | $ | 39.08 | $ | 31.15 | |||
Third Quarter | $ | 41.79 | $ | 33.29 | |||
Fourth Quarter | $ | 41.07 | $ | 30.80 | |||
American Stock Transfer & Trust Company is the transfer agent and registrar for our common stock. As of the close of business on February 26, 2018, we had 154 holders of record of our common stock.
Dividends
We have never declared or paid cash dividends on our common stock. We anticipate that we will retain all of our future earnings, if any, for use in the expansion and operation of our business and do not anticipate paying cash dividends in the foreseeable future. Payment of future dividends, if any, will be at the discretion of our board of directors.
48
Performance Graph
The graph below matches our cumulative five-year total return on common equity with the cumulative total returns of The NASDAQ Composite Index and The NASDAQ Biotechnology Index. The graph tracks the performance of a $100 investment in our common stock and in each of the indexes (with the reinvestment of all dividends) from December 31, 2012 to December 31, 2017. The stock price performance included in this graph is not necessarily indicative of future stock price performance.
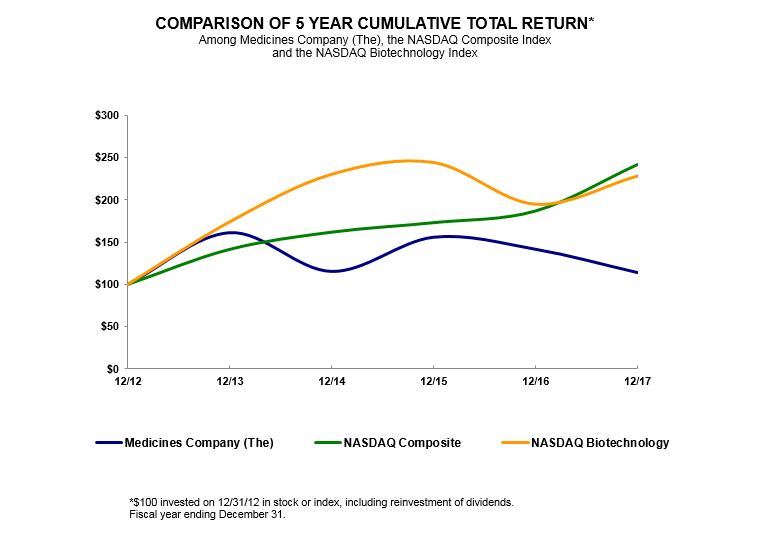
12/12* | 12/13* | 12/14* | 12/15* | 12/16* | 12/17* | |||||||
The Medicines Company | 100.00 | 161.12 | 115.44 | 155.78 | 141.59 | 114.06 | ||||||
NASDAQ Composite | 100.00 | 141.63 | 162.09 | 173.33 | 187.19 | 242.29 | ||||||
NASDAQ Biotechnology | 100.00 | 174.05 | 230.33 | 244.29 | 194.95 | 228.29 | ||||||
* | Fiscal year ended December 31. | |
This performance graph shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or incorporated by reference into any of our filings under the Securities Act or the Exchange Act, except as shall be expressly set forth by specific reference in such filing.
49
Item 6. | Selected Financial Data. |
In the table below, we provide you with our selected consolidated financial data for the periods presented. We have prepared this information using our audited consolidated financial statements for the years ended December 31, 2017, 2016, 2015, 2014, and 2013. We have made certain reclassifications to the selected financial data associated with our presentation of the infectious disease business and hemostasis business as discontinued operations. Refer to Note 23 “Discontinued Operations,” in Appendix A to this Annual Report on Form 10-K.
You should read the following selected consolidated financial data in conjunction with our consolidated financial statements and related notes included in this Annual Report on Form 10-K and “Part II. Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this Annual Report on Form 10-K.
Year Ended December 31, | |||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
(In thousands, except per share data) | |||||||||||||||||||
Statements of Operations Data | |||||||||||||||||||
Net product revenues | $ | 18,980 | $ | 71,956 | $ | 240,688 | $ | 657,533 | $ | 624,519 | |||||||||
Royalty revenues | 25,809 | 71,205 | 53,859 | — | — | ||||||||||||||
Total net revenues | 44,789 | 143,161 | 294,547 | 657,533 | 624,519 | ||||||||||||||
Operating expenses: | |||||||||||||||||||
Cost of product revenues | 47,193 | 60,653 | 103,986 | 228,514 | 216,607 | ||||||||||||||
Asset impairment charges | 392,097 | — | — | — | — | ||||||||||||||
Research and development | 138,370 | 92,107 | 90,388 | 96,257 | 104,354 | ||||||||||||||
Selling, general and administrative | 132,225 | 212,482 | 285,300 | 281,818 | 252,233 | ||||||||||||||
Total operating expenses | 709,885 | 365,242 | 479,674 | 606,589 | 573,194 | ||||||||||||||
(Loss) income from operations | (665,096 | ) | (222,081 | ) | (185,127 | ) | 50,944 | 51,325 | |||||||||||
Co-promotion and license income | 7,549 | 3,854 | 10,132 | 24,236 | 17,383 | ||||||||||||||
Gain on remeasurement of equity investment | — | — | 22,597 | — | — | ||||||||||||||
Gain on sale of investment | — | — | 19,773 | — | — | ||||||||||||||
Gain on sale of assets | — | 288,301 | — | — | — | ||||||||||||||
Loss on extinguishment of debt | — | (5,380 | ) | — | — | — | |||||||||||||
Legal settlement | — | — | 5,000 | 25,736 | — | ||||||||||||||
Loss in equity investment | — | — | — | (1,711 | ) | — | |||||||||||||
Investment impairment | — | — | — | (7,500 | ) | — | |||||||||||||
Interest expense | (48,564 | ) | (44,463 | ) | (37,092 | ) | (15,701 | ) | (15,531 | ) | |||||||||
Other income | 1,840 | 346 | 188 | 918 | 1,420 | ||||||||||||||
(Loss) income from continuing operations before income taxes | (704,271 | ) | 20,577 | (164,529 | ) | 76,922 | 54,597 | ||||||||||||
Benefit from (provision for) income taxes | 96,576 | (67 | ) | 29,733 | (18,808 | ) | (17,394 | ) | |||||||||||
(Loss) income from continuing operations | (607,695 | ) | 20,510 | (134,796 | ) | 58,114 | 37,203 | ||||||||||||
Loss from discontinued operations, net of tax | (100,678 | ) | (139,682 | ) | (217,950 | ) | (90,462 | ) | (21,943 | ) | |||||||||
Net (loss) income | (708,373 | ) | (119,172 | ) | (352,746 | ) | (32,348 | ) | 15,260 | ||||||||||
Net loss (income) attributable to non-controlling interest | — | 54 | (10 | ) | 138 | 252 | |||||||||||||
Net (loss) income attributable to The Medicines Company | $ | (708,373 | ) | $ | (119,118 | ) | $ | (352,756 | ) | $ | (32,210 | ) | $ | 15,512 | |||||
Basic (loss) earnings per common share attributable to The Medicines Company: | |||||||||||||||||||
(Loss) earnings from continuing operations | $ | (8.40 | ) | $ | 0.29 | $ | (2.02 | ) | $ | 0.90 | $ | 0.64 | |||||||
Loss from discontinued operations | (1.39 | ) | (2.00 | ) | (3.26 | ) | (1.40 | ) | (0.38 | ) | |||||||||
Basic (loss) income per share | $ | (9.79 | ) | $ | (1.71 | ) | $ | (5.28 | ) | $ | (0.50 | ) | $ | 0.26 | |||||
Diluted (loss) earnings per common share attributable to The Medicines Company: | |||||||||||||||||||
(Loss) earnings from continuing operations | $ | (8.40 | ) | $ | 0.28 | $ | (2.02 | ) | $ | 0.87 | $ | 0.60 | |||||||
Loss from discontinued operations | $ | (1.39 | ) | (1.91 | ) | (3.26 | ) | (1.36 | ) | (0.35 | ) | ||||||||
Diluted (loss) income per share | $ | (9.79 | ) | $ | (1.63 | ) | $ | (5.28 | ) | $ | (0.49 | ) | $ | 0.25 | |||||
Shares used in computing basic (loss) earnings per common share | 72,356 | 69,909 | 66,809 | 64,473 | 58,096 | ||||||||||||||
Shares used in computing diluted (loss) earnings per common share | 72,356 | 73,022 | 66,809 | 66,668 | 62,652 | ||||||||||||||
50
As of December 31, | |||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||
(In thousands) | |||||||||||||||||
Balance Sheet Data | |||||||||||||||||
Cash and cash equivalents, available for sale securities and accrued interest receivable | $ | 151,359 | 541,835 | 373,173 | $ | 370,741 | $ | 376,727 | |||||||||
Working capital | 387,812 | 409,328 | 298,670 | 220,071 | 417,188 | ||||||||||||
Total assets* | 872,983 | 1,705,211 | 1,795,516 | 1,881,769 | 1,736,014 | ||||||||||||
Long-term liabilities* | 672,577 | 807,570 | 512,406 | 557,855 | 669,600 | ||||||||||||
Accumulated deficit | (1,257,356 | ) | (548,983 | ) | (429,865 | ) | (77,109 | ) | (44,899 | ) | |||||||
Total stockholders’ equity | 24,914 | 651,983 | 731,774 | 920,091 | 892,161 | ||||||||||||
* Reclassified debt issuance costs of $2.4 million and $9.0 million related to the 2017 Notes and 2022 Notes, respectively, as of December 31, 2016 and $3.9 million, $5.3 million and $6.4 million as of December 31, 2015, 2014 and 2013, respectively, related to the 2017 Notes from Total assets and Long-term liabilities in connection with the adoption of ASU 2015-03.
51
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
You should read the following discussion and analysis of our financial condition and results of operations together with “Selected Consolidated Financial Data” and our financial statements and accompanying notes included elsewhere in this Annual Report on Form 10-K. In addition to the historical information, the discussion in this Annual Report on Form 10-K contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated by the forward-looking statements due to our critical accounting estimates discussed below and important factors set forth in this Annual Report on Form 10-K, including under “Risk Factors” in Item 1A of this Annual Report on Form 10-K.
Overview
Our Business
We are a biopharmaceutical company driven by an overriding purpose - to save lives, alleviate suffering and contribute to the economics of healthcare. Our goal is to create transformational solutions to address the most pressing healthcare needs facing patients, physicians and providers in cardiovascular care. We are focused on inclisiran, an investigational agent which is potentially a first-in-class lipid-lowering drug, to reduce LDL-cholesterol, or LDL-C, which is commonly referred to as “bad” cholesterol, in patients with atherosclerotic cardiovascular disease, or ASCVD, or cardiovascular risk-equivalents. We believe that inclisiran possesses favorable attributes that competitive products do not possess, would satisfy unmet medical needs and has the potential to improve the economics of healthcare. We have the right to develop, manufacture and commercialize inclisiran under our collaboration agreement with Alnylam Pharmaceuticals, Inc., or Alnylam. In addition, we market Angiomax® (bivalirudin) in the United States primarily through a supply and distribution agreement with Sandoz Inc., or Sandoz, under which we granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax.
On November 3, 2015, we announced that we were in the process of evaluating our operations with a goal of unlocking and maximizing stockholder value. In particular, we stated our intention was to explore strategies for optimizing our capital structure and liquidity position and to narrow our operational focus by strategically separating non-core businesses and products in order to generate non-dilutive cash and reduce associated cash burn and capital requirements.
As a result of our decision to narrow our operational focus, we have completed the following transactions:
• | On February 1, 2016, we completed the sale of our hemostasis portfolio, consisting of PreveLeak, Raplixa and Recothrom, to wholly owned subsidiaries of Mallinckrodt plc, or Mallinckrodt. At the completion of the sale, we received approximately $174.1 million in cash, and may receive up to an additional $235.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of PreveLeak and Raplixa. |
• | On June 21, 2016, we completed the sale of Cleviprex, Kengreal and rights to Argatroban for Injection, which we refer to collectively as Non-Core ACC Assets, to Chiesi USA, Inc., or Chiesi USA, and its parent company Chiesi Farmaceutici S.p.A., or Chiesi. At the completion of the sale, we received approximately $263.8 million in cash, which included the value of product inventory, and may receive up to an additional $480.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of each of Cleviprex and Kengreal. |
• | On January 5, 2018, we completed the sale of our infectious disease portfolio, consisting of the products Vabomere, Orbactiv and Minocin IV and line extensions thereof, and substantially all of the assets related thereto, other than certain pre-clinical assets, to Melinta Therapeutics, Inc., or Melinta. At the completion of the sale, we received approximately $166.4 million and 3,313,702 shares of Melinta common stock having a market value, based on Melinta's closing share price on January 5, 2018, of approximately $54.5 million. In addition, we are entitled to receive (i) a cash payment payable 12 months following the closing of the transaction equal to $25 million; (ii) a cash payment payable 18 months following the closing of the transaction equal to $25 million; and (iii) tiered royalty payments of 5% to 25% on worldwide net sales of (a) Vabomere and (b) Orbactiv and Minocin IV, collectively. |
Consistent with our intentions announced in November 2015, in January 2017 we announced that we were seeking opportunities to partner or divest Ionsys (fentanyl iontophoretic transdermal system). Although we continue to seek a partnership or divestiture transaction for Ionsys, in June 2017 we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open. Concurrent with this market withdrawal, we commenced implementation of a workforce reduction, which resulted in the reduction of 57 employees, representing approximately 15% of our workforce. Our workforce reductions are described in more detail below. In addition, in
52
August 2017, we announced that we are discontinuing the clinical development program for MDCO-700, an investigational anesthetic agent.
Our revenues to date have been generated primarily from sales of Angiomax in the United States. In 2017, we had net product revenues from sales of Angiomax of approximately $18.8 million. During this period, net product revenues from sales of Angiomax decreased by $31.8 million from 2016. As a result of our July 2015 supply and distribution agreement with Sandoz, we recognized $25.8 million of royalty revenues related to the authorized generic sales of Angiomax (bivalirudin) in 2017. We expect that net product revenues from sales of Angiomax will continue to decline in 2017 and in future years due to competition from generic versions of Angiomax following the loss of market exclusivity in the United States in July 2015 and in Europe in August 2015. Based on our current business, we expect to incur net losses for the foreseeable future.
Cost of product revenues represents expenses in connection with contract manufacture of our products sold and logistics, product costs, royalty expenses and amortization of the costs of license agreements, amortization and impairments of product rights and other identifiable intangible assets from product and business acquisitions and expenses related to excess inventory. Research and development expenses represent costs incurred for licenses of rights to products, clinical trials, nonclinical and preclinical studies, regulatory filings and manufacturing development efforts. We outsource much of our clinical trials, nonclinical and preclinical studies and all of our manufacturing development activities to third parties to maximize efficiency and minimize our internal overhead. We expense our research and development costs as they are incurred. Selling, general and administrative expenses consist primarily of salaries and related expenses, costs associated with general corporate activities, changes in fair value of contingent purchase price obligations related to our acquisitions, and costs associated with marketing and promotional activities. Research and development expense, selling, general and administrative expense and cost of revenue also include share-based compensation expense, which we allocate based on the responsibilities of the recipients of the share-based compensation.
Business Development Activity
Sale of Infectious Disease Products. On January 5, 2018, we completed the sale of our infectious disease portfolio, consisting of the products Vabomere, Orbactiv and Minocin IV and line extensions thereof, and substantially all of the assets related thereto, other than certain pre-clinical assets, to Melinta. At the completion of the sale, we received approximately $166.4 million and 3,313,702 shares of Melinta common stock having a market value, based on Melinta's closing share price on January 5, 2018, of approximately $54.5 million. In addition, we are entitled to receive (i) a cash payment payable 12 months following the closing of the transaction equal to $25 million; (ii) a cash payment payable 18 months following the closing of the transaction equal to $25 million; and (iii) tiered royalty payments of 5% to 25% on worldwide net sales of (a) Vabomere and (b) Orbactiv and Minocin IV, collectively.
Sale of Non-Core Cardiovascular Products. On June 21, 2016, we completed the sale of our Non-Core ACC Assets to Chiesi USA and Chiesi. Under the terms of the purchase and sale agreement, Chiesi and Chiesi USA acquired Cleviprex, Kengreal and rights to Argatroban for Injection and related assets, and assumed substantially all of the liabilities arising out of the operation of the businesses and the acquired assets after closing, including any obligations with respect to future milestones relating to Cleviprex, Kengreal and rights to Argatroban for Injection. At the completion of the sale, we received approximately $263.8 million in cash, which included the value of product inventory, and may receive up to an additional $480.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of each of Cleviprex and Kengreal. As part of the transaction to sell Non-Core ACC Assets, we sublicensed to Chiesi all of our rights to Cleviprex and Kengreal under our license from AstraZeneca. Subsequent to the completion of the sale, these sublicenses from us to Chiesi were terminated, Chiesi purchased from AstraZeneca all or substantially all of AstraZeneca’s assets relating to Cleviprex and Kengreal, the parties released certain claims against one another, and we paid Chiesi $7.5 million.
Sale of Hemostasis Business. On February 1, 2016, we completed the sale of our hemostasis business, consisting of PreveLeak, Raplixa and Recothrom products to wholly-owned subsidiaries of Mallinckrodt plc, or Mallinckrodt. Under the terms of the purchase and sale agreement, Mallinckrodt acquired all of the outstanding equity of Tenaxis Medical, Inc. and ProFibrix B.V. and assets exclusively related to the Recothrom product. Mallinckrodt assumed all liabilities arising out of Mallinckrodt's operation of the businesses and the acquired assets after closing, including all obligations with respect to milestones relating to the PreveLeak and Raplixa products. At the completion of the sale, we received approximately $174.1 million in cash from Mallinckrodt, and may receive up to an additional $235.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of PreveLeak and Raplixa. The amount paid at closing was subject to a post-closing purchase price adjustment process with respect to the Recothrom inventory and the net working capital of the hemostasis business as of the date of the closing.
Alnylam License Agreement. In February 2013, we entered into a license and collaboration agreement with Alnylam to develop, manufacture and commercialize therapeutic products targeting the human proprotein convertase subtilisin/kexin type 9, or PCSK9,
53
gene based on certain of Alnylam’s RNA interference technology. Under the terms of the agreement, we obtained the exclusive, worldwide right under Alnylam’s technology to develop, manufacture and commercialize PCSK9 products for the treatment, palliation and/or prevention of all human diseases. We paid Alnylam $25.0 million in an initial license payment and agreed to pay up to $180.0 million in success-based development, regulatory and commercialization milestones. In December 2014, we paid a development milestone payment of $10.0 million based upon the initiation of a Phase 1 clinical trial for inclisiran and in January 2018 we paid a development milestone payment of $20.0 million based upon the initiation of our phase 3 study for inclisiran. In addition, Alnylam will be eligible to receive scaled double-digit royalties based on annual worldwide net sales of PCSK-9 products by us or our affiliates and sublicensees. Royalties to Alnylam are payable on a product-by-product and country-by-country basis until the last to occur of the expiration of patent rights in the applicable country that cover the applicable product, the expiration of non-patent regulatory exclusivities for such product in such country, and the twelfth anniversary of the first commercial sale of the product in such country. The royalties are subject to reduction in specified circumstances. We are also responsible for paying royalties, and in some cases milestone payments, owed by Alnylam to its licensors with respect to intellectual property covering these products. Alnylam was responsible for developing the lead product through the end of the first Phase 1 clinical trial and to supply the lead product for the first Phase 1 clinical trial and the first phase 2 clinical trial. Alnylam will bear the costs for these activities, subject to certain caps on its costs. If Alnylam's development and supply costs exceed the applicable cap, Alnylam need not bear any additional development and supply costs except for costs directly caused by Alnylam's gross negligence and we shall have the option to assume such excess costs. We will direct and pay for all other development, manufacturing and commercialization activities under the agreement.
Workforce Restructuring
In 2017 and 2018, we conducted a series of workforce reductions, as described below. Our intention is to reduce our personnel to less than 60 employees as we announced in October 2017. Upon signing release agreements, affected employees have received, or are eligible to receive, a severance package, including reduction payments and fully paid health care coverage and outplacement services for six months to a year.
In June 2017, in connection with our voluntary discontinuation and withdrawal of Ionsys from the market in the United States, we commenced a workforce reduction, which resulted in the reduction of 57 employees, representing approximately 15% of our workforce.
Commencing in December 2017 and continuing through 2018, we are implementing a series of workforce reductions to focus on inclisiran, improve efficiencies and better align costs and structure. All employees who will be impacted by these reductions have been informed as to their respective timing of departure. Through February 27, 2018, 27 employees have been terminated and 136 employees were transferred as part of the sale of the infectious disease business unit to Melinta. An additional 115 employees will be terminated through the remainder of the year, with the vast majority by midyear. Included in the 115 employees are 23 employees based in San Diego who are working on early stage infectious disease projects. We expect to sell or spin out those assets and employees by midyear. These workforce reductions are expected to reduce headcount costs included in operating expenses by approximately $74.0 million on an annualized basis.
Angiomax Developments
We sell Angiomax in the United States under our name as a branded Angiomax product, and, on July 2, 2015, entered into a supply and distribution agreement with Sandoz under which we granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax (bivalirudin). We entered into the supply and distribution agreement as a result of the July 2, 2015 U.S. Court of Appeals for the Federal Circuit, or Federal Circuit Court, ruling against us in our patent infringement litigation with Hospira, Inc., or Hospira, with respect to U.S. Patent No. 7,582,727, or the ‘727 patent, and U.S. Patent No. 7,598,343, or the ‘343 patent, covering a more consistent and improved Angiomax drug product and the processes by which it is made. In addition to Hospira, other generic firms have entered the market. APP Pharmaceuticals LLC, or APP, through its affiliated company, Fresenius Kabi, commenced selling its generic version of Angiomax under provisions of a settlement agreement with us triggered by the Federal Circuit Court’s July 2, 2015 decision in the Hospira matter. Apotex Inc. and Dr. Reddy’s Laboratories have each also commenced commercialization of generic bivalirudin products upon receiving final approval if their respective ANDA filings by the FDA even though we remain in active litigation against Apotex and only recently settled with Dr. Reddy’s Laboratories. In addition, Mylan Pharmaceuticals, Inc., or Mylan, commenced marketing its generic bivalirudin product following a decision by the Federal Circuit Court in Mylan’s appeal that reversed an earlier district court decision that found that Mylan’s ANDA product infringed all of the asserted claims of the ‘727 patent. In addition, in January 2018 Baxter International Inc., or Baxter, announced that the FDA approved Baxter’s ready-to-use formulation of bivalirudin for use as an anticoagulant in patients undergoing percutaneous coronary intervention, or PCI.
54
A number of companies in addition to Hospira, Mylan, APP, Apotex Inc. and Dr. Reddy’s Laboratories have filed ANDAs for their generic versions of Angiomax. In addition to the generic versions and the ready-to-use version of bivalirudin currently being sold, Angiomax could be subject to further generic competition in the United States from Teva Pharmaceuticals USA, Inc. and its affiliates, or Teva, and other generic ANDA filers that we have settled with, under the circumstances set forth in our respective settlement agreements with such parties and upon a final approval of each company's ANDA filings by the FDA. Pliva Hrvatska DOO, an affiliate of Teva, currently has tentative approval for its ANDA filing for its generic version of Angiomax. Other ANDA filers may commercialize their products ‘at risk’ if they receive final approval of their respective ANDA filings and are not subject to a Hatch-Waxman 30-month stay. Further, we remain in infringement litigation involving the ‘727 patent and ‘343 patent with the other ANDA filers. See Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K for descriptions of our litigation with ANDA filers and related settlements. . There can be no assurance as to the outcome of our infringement litigation. We may continue to incur further legal expenses related to these matters.
The principal patent covering Angiomax in Europe expired in August 2015. As a result, we face generic competition in Europe. We are in the process of voluntarily discontinuing and withdrawing Angiomax from the market outside of North America and ceasing related commercial activities.
Agreements with Biomedical Advanced Research and Development Authority (BARDA)
2016 BARDA OTA Agreement. In September 2016, we entered into an agreement with the Biomedical Advanced Research and Development Authority, or BARDA, of the U.S. Department of Health and Human Services, or HHS. This agreement, which we refer to as the BARDA OTA agreement, was established under HHS’s Other Transaction Authority, known as OTA. Under the BARDA OTA agreement, we have the potential to receive up to $132.0 million in funding to support the development of early and late stage antibacterial candidates. The BARDA OTA agreement is a cost-sharing arrangement that consists of an initial base period and four option periods that BARDA may exercise in its sole discretion pursuant to the agreement. The BARDA OTA agreement provides for an initial commitment by BARDA of $32.0 million for the base period, and up to an additional $100.0 million if the remaining four options are exercised by BARDA. As of December 31, 2017, BARDA has committed $32.0 million for the base period and no additional options have been exercised. As of December 31, 2017, approximately $29.4 million of funds obligated during the exercised option periods remain available for reimbursement under the BARDA OTA agreement. Under this cost-sharing arrangement, we will be responsible for a portion of the costs associated with each period of work. If all option periods are exercised by BARDA, the estimated period of performance is expected to end in 2021, unless extended by the parties. Either party is entitled to terminate the agreement for convenience, in whole or in part upon 90 days written notice, and BARDA’s future period obligations are subject to Congressionally approved annual appropriations. We expect to use the total award under the BARDA OTA agreement to support non-clinical development activities, non-clinical toxicology, clinical studies, manufacturing, program management, and associated regulatory activities designed to advance a portfolio of potential new antibiotic drug candidates targeting drug resistant bacteria. Under our purchase and sale agreement with Melinta, we are reasonably cooperating with Melinta so that BARDA can enter into a new agreement with Melinta with respect to the provisions of the BARDA OTA that are related to Vabomere.
2014 BARDA Agreement. In February 2014, our former subsidiary Rempex entered into a cost-sharing agreement with BARDA, which we refer to as the 2014 BARDA agreement. As part of the divestiture of our infectious disease portfolio to Melinta, Melinta acquired Rempex and its assets, including the 2014 BARDA agreement and any obligations under the agreement. We are in the process of working with BARDA and Melinta to transfer the 2014 BARDA agreement to Melinta.
Convertible Senior Note Offerings
2023 Notes
On June 10, 2016, we completed our private offering of $402.5 million aggregate principal amount of our 2.75% convertible senior notes due 2023, or the 2023 notes, and entered into an indenture with Wells Fargo Bank, National Association, a national banking association, as trustee, governing the 2023 notes. The net proceeds from the offering were $390.8 million, after deducting the initial purchasers’ discounts and commissions and our offering expenses.
The 2023 notes bear cash interest at a rate of 2.75% per year, payable semi-annually on January 15 and July 15 of each year, beginning on January 15, 2017. The 2023 notes will mature on July 15, 2023. The 2023 notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, incurrence of other indebtedness, or issuance or repurchase of securities by us.
Holders may convert their 2023 notes at their option at any time prior to the close of business on the business day immediately preceding April 15, 2023 only under the following circumstances: (1) during any calendar quarter commencing on or after September
55
30, 2016 (and only during such calendar quarter), if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; (2) during the five business day period after any five consecutive trading day period, or measurement period, in which the trading price, as defined in the indenture governing the 2023 notes, per $1,000 principal amount of 2023 notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate on each such trading day; (3) during any period after we have issued notice of redemption until the close of business on the scheduled trading day immediately preceding the relevant redemption date; or (4) upon the occurrence of specified corporate events. On or after April 15, 2023, until the close of business on the second scheduled trading day immediately preceding the maturity date, holders may convert their 2023 notes at any time, regardless of the foregoing circumstances. Upon conversion, we will pay or deliver, as the case may be, cash, shares of our common stock or a combination of cash and shares of our common stock, at our election based upon a daily conversion value calculated on a proportionate basis for each trading day in a 50 trading day observation period (as more fully described in the 2023 notes indenture).
The conversion rate for the 2023 notes was initially, and remains, 20.4198 shares of our common stock per $1,000 principal amount of the 2023 notes, which is equivalent to an initial conversion price of approximately $48.97 per share of our common stock. The conversion rate and the conversion price are subject to customary adjustments for certain events, including, but not limited to, the issuance of certain stock dividends on our common stock, the issuance of certain rights or warrants, subdivisions, combinations, distributions of capital stock, indebtedness, or assets, cash dividends and certain issuer tender or exchange offers, as described in the indenture governing the 2023 notes.
We may not redeem the 2023 notes prior to July 15, 2020. We may redeem for cash all or any portion of the 2023 notes, at our option, on or after July 15, 2020 if the last reported sale price of our common stock has been at least 130% of the conversion price then in effect on the last trading day of, and for at least 19 other trading days (whether or not consecutive) during, any 30 consecutive trading day period ending on, and including, the trading day immediately preceding the date on which we provides notice of redemption, at a redemption price equal to 100% of the principal amount of the 2023 notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. However, no redemption date may be designated that falls on or after the 52nd scheduled trading date prior to maturity. No sinking fund is provided for the 2023 notes, which means that we are not required to redeem or retire the 2023 notes periodically.
If we undergo a fundamental change, as defined in the indenture governing the 2023 notes, subject to certain conditions, holders of the 2023 notes may require us to repurchase for cash all or part of their 2023 notes at a repurchase price equal to 100% of the principal amount of the 2023 notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. Following certain corporate transactions that constitute a change of control, we would increase the conversion rate for a holder who elects to convert the 2023 notes in connection with such change of control in certain circumstances.
The 2023 notes are our senior unsecured obligations and will rank senior in right of payment to our future indebtedness that is expressly subordinated in right of payment to the 2023 notes; equal in right of payment to our existing and future unsecured indebtedness that is not so subordinated (including the 2022 notes); effectively junior in right of payment to any of our secured indebtedness to the extent of the value of the assets securing such indebtedness; and structurally junior to all existing and future indebtedness and other liabilities (including trade payables) incurred by our subsidiaries.
The indenture governing the 2023 notes contains customary events of default with respect to the 2023 notes, including that upon certain events of default (including our failure to make any payment of principal on the 2023 notes when due and payable or our failure to make any interest payment on the 2023 notes when due and payable and such failure continues for a period of thirty days) occurring and continuing, the trustee for the 2023 notes by notice to us, or the holders of at least 25% in principal amount of the outstanding 2023 notes by notice to us and the trustee for the 2023 notes, may, and the trustee at the request of such holders (subject to the provisions of the indenture governing the 2023 notes) shall, declare 100% of the principal of and accrued and unpaid interest, if any, on all the 2023 notes to be due and payable. In case of certain events of bankruptcy, insolvency or reorganization, involving us or a significant subsidiary, 100% of the principal of and accrued and unpaid interest on the 2023 notes will automatically become due and payable. Upon such a declaration of acceleration, such principal and accrued and unpaid interest, if any, will be due and payable immediately.
Capped Call Transactions
To minimize the impact of potential dilution upon conversion of the 2023 Notes, we entered into capped call transactions separate from the issuance of the 2023 Notes with certain counterparties. The capped calls have a strike price of $48.97 per share and a cap price of $64.68 per share and are exercisable when and if the 2023 Notes are converted. If upon conversion of the 2023 Notes, the price of our common stock is above the strike price of the capped calls, the counterparties will deliver shares of our common stock and/or cash with an aggregate value equal to the difference between the price of our common stock at the conversion date and the strike price, multiplied by the number of shares of our common stock related to the capped calls being exercised. We paid $33.9 million for these capped call transactions.
56
For any conversions of the 2023 Notes prior to the close of business on the 52nd scheduled trading day immediately preceding the stated maturity date of the 2023 Notes, including without limitation upon an acquisition of the Company or similar business combination, a corresponding portion of the capped calls will be terminated. Upon such termination, the portion of the capped calls being terminated will be settled at fair value (subject to certain limitations), as determined by the counterparties to the capped calls and no payments will be due from us to such counterparties. The capped calls expire on the earlier of (i) the last day on which any Convertible Securities remain outstanding and (ii) the second “Scheduled Trading Day” (as defined in the indenture) immediately preceding the “Maturity Date” (as defined in the indenture).
2022 Notes
On January 13, 2015, we completed our private offering of $400.0 million aggregate principal amount of our 2.50% convertible senior notes due 2022, or the 2022 notes, and entered into an indenture with Wells Fargo Bank, National Association, a national banking association, as trustee, governing the 2022 notes. The aggregate principal amount of 2022 notes sold reflects the exercise in full by the initial purchasers of the 2022 notes of their option to purchase up to an additional $50.0 million in aggregate principal amount of the 2022 notes. The net proceeds from the offering were $387.2 million, after deducting the initial purchasers’ discounts and commissions and our offering expenses.
The 2022 notes bear cash interest at a rate of 2.50% per year, payable semi-annually on January 15 and July 15 of each year, beginning on July 15, 2015. The 2022 notes will mature on January 15, 2022. The 2022 notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, incurrence of other indebtedness, or issuance or repurchase of securities by us.
Holders may convert their 2022 notes at their option at any time prior to the close of business on the business day immediately preceding October 15, 2021 only under the following circumstances: (1) during any calendar quarter commencing on or after March 31, 2015 (and only during such calendar quarter), if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; (2) during the five business day period after any five consecutive trading day period, or measurement period, in which the trading price, as defined in the indenture governing the 2022 notes, per $1,000 principal amount of 2022 notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate on each such trading day; (3) during any period after we have issued notice of redemption until the close of business on the scheduled trading day immediately preceding the relevant redemption date; or (4) upon the occurrence of specified corporate events.
On or after October 15, 2021, until the close of business on the second scheduled trading day immediately preceding the maturity date, holders may convert their 2022 notes at any time, regardless of the foregoing circumstances. Upon conversion, we will pay cash up to the aggregate principal amount of the 2022 notes to be converted and deliver shares of our common stock in respect of the remainder, if any, of its conversion obligation in excess of the aggregate principal amount of 2022 notes being converted, subject to a daily share cap, as described in the indenture governing the 2022 notes. Holders of 2022 notes will not receive any additional cash payment or additional shares representing accrued and unpaid interest, if any, upon conversion of a note, except in limited circumstances. Instead, accrued but unpaid interest will be deemed to be paid by the cash and shares, if any, of our common stock, together with any cash payment for any fractional share, paid or delivered, as the case may be, upon conversion of a 2022 note.
The conversion rate for the 2022 notes was initially, and remains, 29.8806 shares of our common stock per $1,000 principal amount of the 2022 notes, which is equivalent to an initial conversion price of approximately $33.47 per share of our common stock. The conversion rate and the conversion price are subject to customary adjustments for certain events, including, but not limited to, the issuance of certain stock dividends on our common stock, the issuance of certain rights or warrants, subdivisions, combinations, distributions of capital stock, indebtedness, or assets, cash dividends and certain issuer tender or exchange offers, as described in the indenture governing the 2022 notes.
We may not redeem the 2022 notes prior to January 15, 2019. We may redeem for cash all or any portion of the 2022 notes, at our option, on or after January 15, 2019 if the last reported sale price of our common stock has been at least 130% of the conversion price then in effect on the last trading day of, and for at least 19 other trading days (whether or not consecutive) during, any 30 consecutive trading day period ending on, and including, the trading day immediately preceding the date on which we provides notice of redemption, at a redemption price equal to 100% of the principal amount of the 2022 notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. No sinking fund is provided for the 2022 notes, which means that we are not required to redeem or retire the 2022 notes periodically.
If we undergo a fundamental change, as defined in the indenture governing the 2022 notes, subject to certain conditions, holders of the 2022 notes may require us to repurchase for cash all or part of their 2022 notes at a repurchase price equal to 100% of the principal amount of the 2022 notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental
57
change repurchase date. Following certain corporate transactions that constitute a change of control, we would increase the conversion rate for a holder who elects to convert the 2022 notes in connection with such change of control in certain circumstances.
The 2022 notes are our senior unsecured obligations and will rank senior in right of payment to our future indebtedness that is expressly subordinated in right of payment to the 2022 notes; equal in right of payment to our existing and future unsecured indebtedness that is not so subordinated (including the 2023 notes); effectively junior in right of payment to any of our secured indebtedness to the extent of the value of the assets securing such indebtedness; and structurally junior to all existing and future indebtedness and other liabilities (including trade payables) incurred by our subsidiaries.
The indenture governing the 2022 notes contains customary events of default with respect to the 2022 notes, including that upon certain events of default (including our failure to make any payment of principal or interest on the 2022 notes when due and payable) occurring and continuing, the trustee for the 2022 notes by notice to us, or the holders of at least 25% in principal amount of the outstanding 2022 notes by notice to us and the trustee for the 2022 notes, may, and the trustee at the request of such holders (subject to the provisions of the indenture governing the 2022 notes) shall, declare 100% of the principal of and accrued and unpaid interest, if any, on all the 2022 notes to be due and payable. In case of certain events of bankruptcy, insolvency or reorganization, involving us or a significant subsidiary, 100% of the principal of and accrued and unpaid interest on the 2022 notes will automatically become due and payable. Upon such a declaration of acceleration, such principal and accrued and unpaid interest, if any, will be due and payable immediately.
2017 Notes
On June 11, 2012, we completed our private offering of $275.0 million aggregate principal amount of our 1.375% convertible senior notes due 2017, or the 2017 notes. The net proceeds from the offering were $266.2 million, after deducting the initial purchasers’ discounts and commissions and our offering expenses. The 2017 notes were our senior unsecured obligations and paid cash interest at a rate of 1.375% per year, payable semi-annually on June 1 and December 1 of each year. The conversion rate for the 2017 notes was 35.8038 shares of our common stock per $1,000 principal amount of 2017 notes, which is equivalent to an initial conversion price of $27.93 per share of our common stock.
In June 2016, we used approximately $323.2 million of the net proceeds of the 2023 notes to repurchase $220.0 million in aggregate principal amount of the 2017 notes in privately negotiated transactions effected through the initial purchasers of the 2017 notes. As part of the June 2016 repurchase of the 2017 notes, we settled a proportionate amount of outstanding bond hedge and warrants related to the bonds that were repurchased for a net cash receipt of $12.6 million.
The remaining 2017 notes matured on June 1, 2017. In connection with the maturity of 2017 Notes, the holders converted substantially all of the outstanding principal amount of the 2017 notes (other than $14,000 of principal amount of 2017 notes which was not converted and which amount was paid in full to the holders thereof), we paid cash to the converting 2017 note holders equal to $55.0 million in respect of principal, interest and fractional shares on the 2017 notes converted and delivered 819,901 shares of our common stock in respect of the remainder of our conversion obligation in excess of the aggregate principal amount of the 2017 notes converted.
Convertible Note Hedge and Warrant Transactions
In connection with the offering of the 2017 notes, on June 5, 2012, we entered into convertible note hedge transactions and warrant transactions with several of the initial purchasers of the 2017 notes, their respective affiliates and other financial institutions, which we refer to as the hedge counterparties. We used approximately $19.8 million of the net proceeds from the offering of the 2017 notes to pay the cost of the convertible note hedge transactions, after such cost was partially offset by the proceeds to us from the sale of warrants in the warrant transactions.
As part of the June 2016 repurchase of $220.0 million in aggregate principal amount of the 2017 Notes, we settled the related hedges and warrants for a net cash receipt of $12.6 million. On June 1, 2017, in connection with the maturity of the 2017 notes, we settled the note hedges and received from the note hedge counterparties approximately 820,000 shares of our common stock at an average price of $48.79 per share. The redemption offset the dilution with respect to the 819,901 shares of our common stock that were issued upon the conversion of the 2017 notes. The shares delivered to us in connection with the redemption of the 2017 note hedges are held by us as treasury shares. The remaining warrants, which were settled in December 2017, and the concurrent redemption of the note hedges, provide the holders the right to purchase up to approximately two million shares of our common stock, subject to customary antidilution adjustments, at a strike price of $34.20 per share. The warrants had a dilutive effect with respect to our common stock to the extent that the market price per share of our common stock, as measured under the terms of the warrants, exceeds the applicable strike price. The warrants were net-settled issuing common stock. The holders of the 2017 Warrants exercised 787,680 warrants on a net basis and as a result we issued 44,283 shares of common stock.
58
U.S. Healthcare Reform
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, or PPACA, which was amended by the Health Care and Education Reconciliation Act of 2010. The PPACA, as amended, contains numerous provisions that impact the pharmaceutical and healthcare industries and it empowers the Department of Health and Human Services, or HHS, to implement a number of related healthcare reform measures that are likely to have a broad impact on the pharmaceutical and healthcare industry. We are continually evaluating the impact of the PPACA and other healthcare reform-related programs and regulations on our business, including potential PPACA repeal and replacement. As of the date of this Annual Report on Form 10-K, we have not identified any provisions that currently materially impact our business and results of operations. However, the potential impact of the PPACA and other healthcare reform measures on our business and results of operations is inherently difficult to predict because many of the details regarding the implementation of this legislation have not been determined. In addition, the impact on our business and results of operations may change as and if our business evolves. On December 22, 2017, Congress passed and President Trump signed bill entitled “To provide for reconciliation pursuant to titles II and V of the concurrent resolution on the budget for fiscal year 2018,” which, among other things, repealed the PPACA individual mandate. President Trump and HHS Secretary Azar have announced support for regulatory provisions that would limit PPACA and number of healthcare reform programs initiated under the Obama administration. It remains unclear whether replacement programs will include similar limitations affecting reimbursement, although scrutiny over drug pricing and government costs is expected to continue. Similarly, efforts in Congress to reform Medicare and Medicaid may impact the pharmaceutical and healthcare industries.
59
Results of Operations
Years Ended December 31, 2017 and 2016
Total Net Revenues:
Total net revenues decreased 68.7% to $44.8 million in 2017 as compared to $143.2 million in 2016.
Year Ended December 31, | ||||||||||||||
2017 | 2016 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Net product revenues | $ | 18,980 | $ | 71,956 | $ | (52,976 | ) | (73.6 | )% | |||||
Royalty revenues | 25,809 | 71,205 | (45,396 | ) | (63.8 | )% | ||||||||
Total net revenues | $ | 44,789 | $ | 143,161 | $ | (98,372 | ) | (68.7 | )% | |||||
Net Product Revenues:
The following table reflects the components of net product revenues for 2017 and 2016:
Year Ended December 31, | ||||||||||||||
2017 | 2016 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Angiomax | $ | 18,842 | $ | 50,596 | $ | (31,754 | ) | (62.8 | )% | |||||
Other products | 138 | 21,360 | (21,222 | ) | (99.4 | )% | ||||||||
Net product revenues | $ | 18,980 | $ | 71,956 | $ | (52,976 | ) | (73.6 | )% | |||||
Net product revenues decreased by $53.0 million, or 73.6%, to $19.0 million in 2017 compared to $72.0 million in 2016, reflecting decreases of $49.0 million in the United States and of $3.9 million in international markets.
Angiomax. Net product revenues from sales of Angiomax decreased by $31.8 million, or 62.8%, to $18.8 million in 2017 compared to $50.6 million in 2016. The decrease in 2017 was due to further declines in price and volume as a result of the launch of generic versions of Angiomax in the United States in July 2015 by Hospira following a July 2, 2015 Federal Circuit Court decision in Hospira’s favor. Due to the Federal Circuit Court’s July 2, 2015 decision and our resulting entry into a supply and distribution agreement with Sandoz, Angiomax is now subject to generic competition with the authorized generic and five generic bivalirudin products. In addition, in January 2018 Baxter announced that the FDA approved Baxter’s ready-to-use formulation of bivalirudin for use as an anticoagulant in patients undergoing PCI. Of the $18.8 million and $50.6 million of net product revenues from sales of Angiomax in 2017 and 2016, respectively, $10.4 million and $16.9 million, respectively, related to shipments of generic Angiomax to Sandoz.
Net product revenues in the United States in 2017 and 2016 reflect chargebacks related to the 340B Drug Pricing Program and rebates related to the PPACA. Under the 340B Drug Pricing Program, we offer qualifying entities a discount off the commercial price of Angiomax for patients undergoing PCI on an outpatient basis. Chargebacks related to the 340B Drug Pricing Program decreased to $2.9 million in 2017 compared to $7.4 million in 2016 primarily due to the reduction in wholesaler purchases. Rebates related to the PPACA increased to $1.4 million in 2017 compared to $1.3 million in 2016.
Other Products. Net product revenues from sales of Cleviprex, ready-to-use Argatroban, Kengreal and Ionsys decreased by $21.2 million, or 99.4%, to $0.1 million in 2017 from $21.4 million in 2016, primarily due to the sale of the Non-Core ACC Products in June 2016.
Royalty Revenues:
In 2017 and 2016, we recognized $25.8 million and $71.2 million, respectively, in royalty revenues related to the authorized generic sale of Angiomax to hospitals by Sandoz. Royalty revenues may decline in 2018 and in future years due to increased competition from other generic versions of Angiomax.
60
Cost of Product Revenues:
Cost of product revenues in 2017 were $47.2 million, or 105.4% of net product revenues, compared to $60.7 million, or 42.4% of net product revenues in 2016.
Cost of product revenues during these periods consisted of:
• | expenses in connection with the manufacture of our products sold, including expenses related to excess inventory offset by the positive impact of sales of previously reserved units; |
• | royalty expenses under our agreement with Biogen and HRI related to Angiomax, our agreement with AstraZeneca related to Cleviprex and our agreement with Eagle Pharmaceuticals, Inc., or Eagle, related to ready-to-use Argatroban; |
• | amortization of the costs of selling rights agreements, product licenses, developed product rights and other identifiable intangible assets, which result from product and business acquisitions; |
• | logistics costs related to Angiomax, Cleviprex, ready-to-use Argatroban, Kengreal and Ionsys, including distribution, storage, and handling costs; and |
• | expenses associated with the discontinuance and market withdrawal of Ionsys in the United States market, including a write-off of inventory, severance and other exit costs. |
Year Ended December 31, | |||||||||||||
2017 | % of Total | 2016 | % of Total | ||||||||||
(in thousands) | (in thousands) | ||||||||||||
Manufacturing/Logistics | $ | 25,232 | 53.5 | % | $ | 38,302 | 63.1 | % | |||||
Royalties | 810 | 1.7 | % | 3,960 | 6.5 | % | |||||||
Impairment of inventory and amortization of acquired product rights and intangible assets | 21,151 | 44.8 | % | 18,391 | 30.4 | % | |||||||
Total cost of product revenues | $ | 47,193 | 100.0 | % | $ | 60,653 | 100.0 | % | |||||
Cost of product revenues decreased by $13.5 million in 2017 compared to 2016. This decrease was mainly due to manufacturing and logistics costs, and royalty costs incurred in 2016 associated with Non-Core ACC products prior to those products being sold. For further details, see Note 22, “Dispositions,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K. These decreases were partially offset by increases in impairment of inventory of $8.2 million to $16.7 million in 2017 compared to $8.5 million in 2016, mainly attributed to Angiomax. These reserves were taken since we project that inventory will expire prior to the expected future sales. Manufacturing/logistics expenses also decreased in 2017 due to the reduction in Angiomax product sales.
Asset Impairment Charges:
In 2017 we recognized impairment charges of $226.5 million, $26.2 million and $11.4 million to reduce the carrying amounts of the product licenses, developed product rights, and fixed assets, respectively, associated with Ionsys to their estimated fair values of zero as a result of the discontinuation and market withdrawal of Ionsys which became effective on June 19, 2017. In the second quarter of 2017, we recognized impairment charges of $65.0 million to reduce the carrying amount of the in-process research and development associated with MDCO-700 to an estimated fair value of zero as a result of management’s decision to discontinue the MDCO-700 trials. In the fourth quarter of 2017, we recognized impairment charges of $63.0 million associated with changes in fair value of the contingent purchase price for Raplixa. See Note 14 “Fair Value Measurements,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K. These impairment charges were recorded in asset impairment charges in the accompanying consolidated statements of operations. For further details, see Note 1, “Nature of Business,” and Note 2, “Significant Accounting Policies,” in the accompanying notes to the consolidated financial statements included in this Annual Report on Form 10-K, for details regarding the Ionsys, MDCO-700 and contingent purchase price impairments.
61
Research and Development Expenses:
Year Ended December 31, | |||||||||||||
2017 | % of Total | 2016 | % of Total | ||||||||||
(in thousands) | (in thousands) | ||||||||||||
Marketed products | |||||||||||||
Ionsys | $ | 3,951 | 2.9 | % | $ | 6,159 | 6.7 | % | |||||
Angiomax | (11 | ) | — | % | 1,646 | 1.8 | % | ||||||
Other | (250 | ) | (0.1 | )% | 3,377 | 3.7 | % | ||||||
Total marketed products | 3,690 | 2.7 | % | 11,182 | 12.1 | % | |||||||
Research and development product candidates | |||||||||||||
Inclisiran | 118,721 | 85.8 | % | 26,707 | 29.0 | % | |||||||
MDCO-216 | 479 | 0.3 | % | 33,856 | 36.8 | % | |||||||
Other | 15,480 | 11.2 | % | 20,362 | 22.1 | % | |||||||
Total research and development product candidates | 134,680 | 97.3 | % | 80,925 | 87.9 | % | |||||||
Total research and development expenses | $ | 138,370 | 100.0 | % | $ | 92,107 | 100.0 | % | |||||
Research and development expenses increased $46.3 million in 2017 compared to 2016. The increase in research and development expenses during 2017 compared to 2016 was primarily due to increases in expenses associated with inclisiran. Research and development expenses related to inclisiran increased $92.0 million due to the acceleration of clinical trials and related manufacturing development costs. These increases were partially offset by decreases in research and development costs of $45.7 million associated products that were terminated or sold.
We expect research and development expenses in 2018 to increase primarily due to increased costs related to clinical trials for inclisiran.
Selling, General and Administrative Expenses:
Year Ended December 31, | ||||||||||||||
2017 | 2016 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Selling, marketing and promotional | $ | 40,763 | $ | 103,560 | $ | (62,797 | ) | (60.6 | )% | |||||
General corporate and administrative | 91,462 | 108,922 | (17,460 | ) | (16.0 | )% | ||||||||
Total selling, general and administrative expenses | $ | 132,225 | $ | 212,482 | $ | (80,257 | ) | (37.8 | )% | |||||
Selling, general and administrative expenses decreased by $80.3 million in 2017 compared to 2016. This decrease is due to a decrease of $62.8 million in selling, marketing and promotional expenses and $17.5 million in general corporate and administrative expenses in 2017.
Selling, marketing and promotional expenses decreased by $62.8 million in 2017 primarily due to the sale of the Non-Core ACC Products, the discontinuation and market withdrawal of Ionsys and overall shift in corporate strategy and increased focus on research and development with respect to inclisiran.
General corporate and administrative expenses decreased by $17.5 million in 2017 primarily due to reorganization costs, reductions due to the implementation of workforce reduction initiatives from prior periods and the sale of the Non-Core ACC Products in 2016.
We expect our selling, general and administrative expenses will continue to decrease in 2018 due to a further decrease in headcount from the workforce reduction and the decrease in marketed products as a result of our sale of the infectious disease business to Melinta in early 2018.
62
Co-promotion and License Income:
Year Ended December 31, | Change | Change | ||||||||||||
2017 | 2016 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Co-promotion and license income | $ | 7,549 | $ | 3,854 | $ | 3,695 | 95.9 | % | ||||||
During 2017 and 2016, we recognized $6.9 million and $2.5 million, respectively, in license income under our collaboration agreement with SymBio Pharmaceuticals Ltd., or SymBio. The increase in license income was due to the write-off of deferred income as a result of the termination of the agreement during the fourth quarter of 2017. The agreement terminated in connection with a legal dispute with SymBio, as described in Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K.
During 2017 and 2016, we recorded license income of $0.6 million and $0.6 million, respectively, under our collaboration agreement with SciClone Pharmaceuticals, or SciClone. During 2016, we recorded license income of $0.8 million in co-promotion income under our license agreement with Eagle related to ready-to-use Argatroban. The decrease in Eagle revenue was due to the sale of the Non-Core ACC Products in June 2016.
Gain on Sale of Assets:
Year Ended December 31, | ||||||||||||||
2017 | 2016 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Gain on sale of assets | $ | — | $ | 288,301 | $ | (288,301 | ) | (100.0 | )% | |||||
During 2016, we recorded a gain of $288.3 million from the sale of the Non-Core ACC Products. For further details, see Note 22, “Dispositions,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Loss on Extinguishment of Debt:
Year Ended December 31, | Change | Change | ||||||||||||
2017 | 2016 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Loss on extinguishment of debt | $ | — | $ | (5,380 | ) | $ | 5,380 | (100.0 | )% | |||||
During 2016, we recorded a loss of $5.4 million on the extinguishment of debt for the repurchase of $220.0 million principal amount of the 2017 notes. For further details, see Note 9, “Convertible Senior Notes,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Interest Expense:
Year Ended December 31, | Change | Change | ||||||||||||
2017 | 2016 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Interest expense | $ | (48,564 | ) | $ | (44,463 | ) | $ | (4,101 | ) | (9.2 | )% | |||
During 2017, we recorded approximately $48.6 million in interest expense related to the 2017 Notes, 2022 Notes, and 2023 Notes as compared to $44.5 million during 2016. The increase in interest expense in 2017 was due to a higher effective interest rates on the 2023 Notes. We expect a decrease in interest expense in 2018 as compared to 2017 as a result of lower debt as a result of the maturity of the 2017 Notes.
63
Other Income:
Year Ended December 31, | Change | Change | ||||||||||||
2017 | 2016 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Other income | $ | 1,840 | $ | 346 | $ | 1,494 | 431.8 | % | ||||||
Other income, which is comprised of interest income and foreign currency transactions, increased by $1.5 million to $1.8 million in 2017, from $0.3 million in 2016. This increase was primarily due to interest income in 2017.
Benefit from (provision for) Income Taxes:
Year Ended December 31, | Change | Change | |||||||||||
2017 | 2016 | $ | % | ||||||||||
(In thousands) | |||||||||||||
Benefit from (provision for) income taxes | $ | 96,576 | $ | (67 | ) | $ | 96,643 | * | |||||
* Represents an increase in excess of 100%
Our income tax benefit, deferred tax assets and liabilities, and reserves for unrecognized tax benefits reflect management’s best assessment of estimated future taxes to be paid. We are subject to income taxes in both the United States and numerous foreign jurisdictions. We recorded a benefit from income taxes of $96.6 million and a provision for income taxes of $0.1 million in 2017 and 2016, respectively, based on a loss from continuing operations before income taxes of $704.3 million and income from continuing operations before taxes of $20.6 million in 2017 and 2016, respectively. The 2017 income tax benefit is primarily a result of the commercialization of Vabomere and impairment of in-process research and development, or IPR&D, associated with MDCO-700, which created a discrete benefit of $89.7 million and the recognition of refundable corporate alternative minimum tax, or AMT, credits of $4.9 million as a result of the Tax Cuts and Jobs Act, or TCJA, and the reduction of accruals related to the settlement of foreign tax audits of $1.4 million. For further details regarding the impairment of IPR&D, see Note 7, “Intangible Assets and Goodwill,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K. Our effective income tax rates in 2017 and 2016 were approximately 13.7% and 0.3%, respectively. This change in the effective tax rate was primarily driven by the discrete benefits from the commercialization of Vabomere, impairment of IPR&D associated with MDCO-700, the recognition of refundable AMT credits and the reversal of foreign uncertain tax positions. The effective tax rates in 2017 and 2016 included the non-cash tax impact arising from changes in contingent consideration related to the acquisitions of Incline Therapeutics, Inc., or Incline, and Annovation BioPharma, Inc., or Annovation.
At December 31, 2017, we had a $239.5 million valuation allowance which fully offsets our net deferred tax assets. Deferred income taxes arise from temporary differences between the tax and financial statement recognition of revenue and expense. In evaluating our ability to realize our deferred tax assets within the jurisdiction from which they arise, we consider all available positive and negative evidence on a periodic basis in light of changing facts and circumstances. These include, without limitation, the status of litigation with respect to the Angiomax patents and the potential impact to projections of future taxable income, scheduled reversal of deferred tax liabilities, tax planning strategies, tax legislation, rulings by relevant tax authorities, the progress of ongoing tax audits, the regulatory approval of products currently under development and the ability to achieve future anticipated revenues. These assumptions require significant judgment about the forecasts of future taxable income and are consistent with the plans and estimates we are using to manage the underlying businesses.
On December 22, 2017, the TCJA was enacted which significantly reforms the Internal Revenue Code of 1986, as amended. The TCJA, among other things, reduces the U.S. federal corporate tax rate from 35% to 21%, repeals the corporate alternative minimum tax (AMT), imposes additional limitations on the deductibility of interest, allows for the expensing of capital expenditures, and puts into effect the migration from a “worldwide” system of taxation to a territorial system. As a result of this legislation, we remeasured our deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21%. However, we are still analyzing certain aspects of the TCJA and refining its calculations, which could potentially affect the measurement of these balances or potentially give rise to new deferred tax amounts. The provisional amount recorded related to the remeasurement of our deferred tax balances was $126.5 million which was offset fully by the provisional amount recorded related to the reversal of previously established valuation allowances against these deferred tax balances. The TCJA also permits any remaining AMT tax attribute carryforwards to be used to offset future taxable income and/or be refundable over the next several years. As a result, we recognized a provisional benefit of $4.9 million during the year ended December 31, 2017
64
related to the reversal of a previously established valuation allowance against our AMT tax attribute carryforwards and the related refundable amount has been classified in other assets in the accompanying consolidated balance sheet. In addition, based on our preliminary analysis, we do not believe that we have offshore earnings that would be subject to the mandatory transition tax.
While we have completed our provisional analysis of the income tax effects of the TCJA and recorded a reasonable estimate of such effects, the amounts recorded related to the TCJA may differ, possibly materially, due to, among other things, further refinement of our calculations, changes in interpretations and assumptions that we have made, additional guidance that may be issued by the U.S. Government, and actions and related accounting policy decisions we may take as a result of the TCJA. We will complete our analysis over a one-year measurement period ending no later than December 22, 2018, and any adjustments during this measurement period will be included in loss from continuing operations as an adjustment to income tax expense/benefit in the reporting period when such adjustments are determined.
The calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws and regulations in a multitude of jurisdictions across our global operations.
Loss from Discontinued Operations, net of tax:
Year Ended December 31, | Change | Change | |||||||||||
2017 | 2016 | $ | % | ||||||||||
(In thousands) | |||||||||||||
Loss from discontinued operations, net of tax | $ | (100,678 | ) | $ | (139,682 | ) | $ | 39,004 | * | ||||
* Represents a decrease in excess of 100%
For details on discontinued operations see Note 23 “Discontinued Operations,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Years Ended December 31, 2016 and 2015
Net Revenues:
Total net revenues decreased 51.4% to $143.2 million in 2016 as compared to $294.5 million in 2015.
Year Ended December 31, | ||||||||||||||
2016 | 2015 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Net product revenues | $ | 71,956 | $ | 240,688 | $ | (168,732 | ) | (70.1 | )% | |||||
Royalty revenues | 71,205 | 53,859 | 17,346 | 32.2 | % | |||||||||
Total net revenues | $ | 143,161 | $ | 294,547 | $ | (151,386 | ) | (51.4 | )% | |||||
Net Product Revenues:
The following table reflects the components of net product revenues for 2016 and 2015:
Year Ended December 31, | ||||||||||||||
2016 | 2015 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Angiomax | $ | 50,596 | $ | 211,970 | $ | (161,374 | ) | (76.1 | )% | |||||
Other products | 21,360 | 28,718 | (7,358 | ) | (25.6 | )% | ||||||||
Net product revenues | $ | 71,956 | $ | 240,688 | $ | (168,732 | ) | (70.1 | )% | |||||
Net product revenues decreased by $168.7 million, or 70.1%, to $72.0 million in 2016 compared to $240.7 million in 2015, reflecting decreases of $160.9 million in the United States and of $7.8 million in international markets.
Angiomax. Net product revenues from sales of Angiomax decreased by $161.4 million, or 76.1%, to $50.6 million in 2016 compared to $212.0 million in 2015. The decrease in 2016 was due to further declines in price and volume as a result of the launch of generic versions of Angiomax in the United States in July 2015 by Hospira following a July 2, 2015 Federal Circuit Court decision in Hospira’s favor. Due to the Federal Circuit Court’s July 2, 2015 decision and our resulting entry into a supply and distribution agreement with Sandoz, Angiomax is now subject to generic competition with the authorized generic and five generic bivalirudin products. In addition, in January 2018 Baxter announced that the FDA approved Baxter’s ready-to-use formulation of bivalirudin for use as an anticoagulant in patients undergoing PCI. Of the $50.6 million and $212.0 million of net product revenues from sales of Angiomax in 2016 and 2015, respectively, $16.9 million and $10.9 million, respectively, related to shipments of generic Angiomax to Sandoz.
Net product revenues in the United States in 2016 and 2015 reflect chargebacks related to the 340B Drug Pricing Program and rebates related to the PPACA. Under the 340B Drug Pricing Program, we offer qualifying entities a discount off the commercial price of Angiomax for patients undergoing PCI on an outpatient basis. Chargebacks related to the 340B Drug Pricing Program decreased to $7.4 million in 2016 compared to $49.6 million in 2015 primarily due to the reduction in wholesaler purchases. Rebates related to the PPACA decreased to $1.3 million in 2016 compared to $1.6 million in 2015.
Other Products. Net product revenues from sales of Cleviprex, ready-to-use Argatroban, Kengreal and Ionsys decreased by $7.4 million, or 25.6%, to $21.4 million in 2016 from $28.7 million in 2015, primarily due to decreases in ready-to-use Argatroban and Cleviprex net product revenues due to the sale of the Non-Core ACC Products in June 2016.
Royalty Revenues:
In 2016 and 2015, we recognized $71.2 million and $53.9 million, respectively, in royalty revenues related to the authorized generic sale of Angiomax to hospitals by Sandoz. Royalty revenues may decline in 2017 and in future years due to competition from other generic versions of Angiomax.
Cost of Product Revenues:
Cost of product revenues in 2016 were $60.7 million, or 84.3% of net product revenues, compared to $104.0 million, or 43.2% of net product revenues in 2015.
Cost of product revenues during these periods consisted of:
• | expenses in connection with the manufacture of our products sold, including expenses related to excess inventory offset by the positive impact of sales of previously reserved units; |
• | royalty expenses under our agreement with Biogen and HRI related to Angiomax, our agreement with AstraZeneca related to Cleviprex and our agreement with Eagle related to ready-to-use Argatroban; |
• | amortization of the costs of selling rights agreements, product licenses, developed product rights and other identifiable intangible assets, which result from product and business acquisitions; and |
• | logistics costs related to Angiomax, Cleviprex, ready-to-use Argatroban, Kengreal and Ionsys, including distribution, storage, and handling costs. |
Year Ended December 31, | |||||||||||||
2016 | % of Total | 2015 | % of Total | ||||||||||
(in thousands) | (in thousands) | ||||||||||||
Manufacturing/Logistics | $ | 38,302 | 63.1 | % | $ | 43,002 | 41.4 | % | |||||
Royalties | 3,960 | 6.5 | % | 9,283 | 8.9 | % | |||||||
Impairment of inventory and amortization of acquired product rights and intangible assets | 18,391 | 30.4 | % | 51,701 | 49.7 | % | |||||||
Total cost of product revenues | $ | 60,653 | 100.0 | % | $ | 103,986 | 100.0 | % | |||||
Cost of product revenues decreased by $43.3 million in 2016 compared to 2015. This decrease was mainly due to reserves of $37.2 million recorded in 2015 for potential Angiomax inventory obsolescence costs and potential losses on future inventory
66
purchase commitments, respectively. In 2016, we recognized a reduction in cost of product revenues of $6.4 million related to Angiomax units sold through to hospitals that were previously reserved. The cost of product revenues decrease in 2016 was partially offset by an $8.5 million reserve for potential inventory obsolescence relating to Ionsys taken in 2016. This reserve was taken for Ionsys since we project that certain components of Ionsys will expire prior to the expected future sales. Manufacturing/logistics expenses also decreased in 2016 due to the reduction in Angiomax product sales as well as the sale of the Non-Core ACC Products.
Research and Development Expenses:
Year Ended December 31, | |||||||||||||
2016 | % of Total | 2015 | % of Total | ||||||||||
(in thousands) | (in thousands) | ||||||||||||
Marketed products | |||||||||||||
Ionsys | $ | 6,159 | 6.7 | % | $ | 5,355 | 5.9 | % | |||||
Angiomax | 1,646 | 1.8 | % | 11,314 | 12.5 | % | |||||||
Other | 3,377 | 3.7 | % | 3,666 | 4.1 | % | |||||||
Total marketed products | 11,182 | 12.1 | % | 20,335 | 22.5 | % | |||||||
Registration stage product candidates | |||||||||||||
Ionsys | — | — | % | 3,225 | 3.6 | % | |||||||
Other | — | — | % | 2,232 | 2.5 | % | |||||||
Total registration stage product candidates | — | — | % | 5,457 | 6.0 | % | |||||||
Research and development product candidates | |||||||||||||
Inclisiran | 26,707 | 29.0 | % | 8,379 | 9.3 | % | |||||||
MDCO-216 | 33,856 | 36.8 | % | 37,052 | 41.0 | % | |||||||
Other | 20,362 | 22.1 | % | 19,165 | 21.2 | % | |||||||
Total research and development product candidates | 80,925 | 87.9 | % | 64,596 | 71.5 | % | |||||||
Total research and development expenses | $ | 92,107 | 100.0 | % | $ | 90,388 | 100.0 | % | |||||
Research and development expenses increased $15.7 million in 2016 compared to 2015. The increase in research and development expenses during 2016 compared to 2015 was primarily due to increases in expenses associated with inclisiran. Research and development expenses related to inclisiran increased by $18.3 million due to increased costs in support of the ongoing Phase 2 clinical trial. The increase was partially offset by decreases in research and development costs for Angiomax of $9.7 million due to the suspension of our research and development efforts and expenditures starting in the third quarter of 2015. Research and development expenses associated with MDCO-216 decreased by $3.2 million as a result of the termination of the clinical trials in the fourth quarter of 2016.
Selling, General and Administrative Expenses:
Year Ended December 31, | ||||||||||||||
2016 | 2015 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Selling, marketing and promotional | $ | 103,560 | $ | 129,936 | $ | (26,376 | ) | (20.3 | )% | |||||
General corporate and administrative | 108,922 | 155,364 | (46,442 | ) | (29.9 | )% | ||||||||
Total selling, general and administrative expenses | $ | 212,482 | $ | 285,300 | $ | (72,818 | ) | (25.5 | )% | |||||
Selling, general and administrative expenses decreased by $72.8 million in 2016 compared to 2015. This decrease is primarily due to a decrease of $46.4 million in general corporate and administrative expenses, and $26.4 million in selling, marketing and promotional expenses in 2016.
Selling, marketing and promotional expenses decreased by $26.4 million in 2016 primarily due to the sale of the Non-Core ACC Products and overall shift in corporate strategy and increased focus on research and development.
67
General corporate and administrative expenses decreased by $46.4 million in 2016 primarily due to adjustments to the fair value of the contingent consideration of $58.7 million partially offset by increases in reorganization costs of $12.4 million due to a reduction in workforce initiated in June 2016 related to the sale of the Non-Core ACC Products. See Note 15, “Restructuring,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K for further details on the reduction in workforce. General corporate and administrative expenses in 2016 included a $7.5 million payment paid to Chiesi. See Note 1, “Nature of Business,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K for further details on this payment to Chiesi.
Legal Settlement:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Legal settlement | $ | — | $ | 5,000 | $ | (5,000 | ) | (100.0 | )% | |||||
In 2015, we recorded $5.0 million of income relating to the extension of the ‘404 patent for Angiomax.
Co-promotion and License Income:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Co-promotion and license income | $ | 3,854 | $ | 10,132 | $ | (6,278 | ) | (62.0 | )% | |||||
During 2016 and 2015, we recorded license income of $0.6 million and $8.2 million, respectively, under our collaboration agreement with SciClone and $0.8 million and $1.3 million, respectively, in co-promotion income under our license agreement with Eagle related to ready-to-use Argatroban. The decrease in SciClone revenue was due to the one-time revenue recognized in the 2015 related to commercialization rights.
During 2016 and 2015, we recognized $2.5 million and $0.6 million, respectively, in license income under our collaboration agreement with SymBio.
Gain on Remeasurement of Equity Investment:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Gain on remeasurement of equity investment | $ | — | $ | 22,597 | $ | (22,597 | ) | (100.0 | )% | |||||
We completed the acquisition of Annovation in February 2015 and Annovation became our wholly owned subsidiary. We accounted for our acquisition of Annovation as a step acquisition which required that we remeasure the fair value of our existing 35.8% ownership interest (previously accounted for as an equity method investment). The fair value of our interest in Annovation was $25.9 million upon the closing of the acquisition, resulting in a non-cash pre-tax gain of $22.6 million in 2015.
Gain on Sale of Investment:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Gain on sale of investment | $ | — | $ | 19,773 | $ | (19,773 | ) | (100.0 | )% | |||||
In the second quarter of 2015, we sold an investment in a specialty pharmaceutical company that had a zero cost basis as the carrying amount was deemed impaired in 2009 and realized a net gain on sale of approximately $19.8 million.
68
Gain on Sale of Assets:
Year Ended December 31, | ||||||||||||||
2016 | 2015 | Change $ | Change % | |||||||||||
(in thousands) | ||||||||||||||
Gain on sale of assets | $ | 288,301 | $ | — | $ | 288,301 | 100.0 | % | ||||||
During 2016, we recorded a gain of $288.3 million from the sale of the Non-Core ACC Products. For further details, see Note 22, “Dispositions,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Loss on Extinguishment of Debt:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Loss on extinguishment of debt | $ | (5,380 | ) | $ | — | $ | (5,380 | ) | (100.0 | )% | ||||
During 2016, we recorded a loss of $5.4 million on the extinguishment of debt for the repurchase of $220.0 million principal amount of the 2017 notes. For further details, see Note 9, “Convertible Senior Notes,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Interest Expense:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Interest expense | $ | (44,463 | ) | $ | (37,092 | ) | $ | (7,371 | ) | (19.9 | )% | |||
During 2016, we recorded approximately $44.5 million in interest expense related to the 2017 Notes, 2022 Notes, and 2023 Notes as compared to $37.1 million in interest expense related to the 2017 Notes and 2022 Notes during 2015. The increase in interest expense in 2016 was due to an increase in the debt as well as higher effective interest rates. We expect an increase in interest expense in 2017 as compared to 2016 as a result of a higher effective interest rate on the 2023 notes.
Other Income:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Other income | $ | 346 | $ | 188 | $ | 158 | 84.0 | % | ||||||
Other income, which is comprised of interest income and gains and losses on sales of fixed assets and foreign currency transactions, increased by $0.1 million to $0.3 million in 2016, from $0.2 million in 2015. This increase was primarily due to a changes in foreign currency transactions.
69
(Provision for) Benefit from Income Taxes:
Year Ended December 31, | Change | Change | |||||||||||
2016 | 2015 | $ | % | ||||||||||
(In thousands) | |||||||||||||
(Provision for) benefit from income taxes | $ | (67 | ) | $ | 29,733 | $ | (29,800 | ) | * | ||||
* Represents a decrease in excess of 100%
Our income tax benefit, deferred tax assets and liabilities, and reserves for unrecognized tax benefits reflect management’s best assessment of estimated future taxes to be paid. We are subject to income taxes in both the United States and numerous foreign jurisdictions. We recorded a provision for income taxes of $0.1 million and a benefit for income taxes of $29.7 million in 2016 and 2015, respectively, based on income (loss) from continuing operations before income taxes of $20.6 million and $(164.5) million in 2016 and 2015, respectively. Our effective income tax rates in 2016 and 2015 were approximately (0.1)% and 18.1%, respectively. This change in the effective tax rate was primarily driven by our loss for the year 2016 and our inability to realize any benefit from this loss due to the establishment of a valuation allowance against substantial portions of our deferred tax assets during the fourth quarter of 2015. The effective tax rates in 2016 and 2015 included the non-cash tax impact arising from changes in contingent consideration related to the Targanta, Incline, Rempex and Annovation acquisitions. In 2016, our provision for income taxes was the result of state tax minimums and taxes due by profitable foreign subsidiaries. Our benefit from income taxes for 2015 is attributed to a discrete benefit recognized for a partial release in a valuation allowance related to Dutch net operating losses associated with ProFibrix B.V. as a result of the regulatory approvals of Raplixa in both the United States and European Union.
At December 31, 2015, we recorded a $67.9 million valuation allowance against $161.7 million of deferred tax assets. Deferred income taxes arise from temporary differences between the tax and financial statement recognition of revenue and expense. In evaluating our ability to realize our deferred tax assets within the jurisdiction from which they arise, we consider all available positive and negative evidence on a periodic basis in light of changing facts and circumstances. These include, without limitation, the status of litigation with respect to the Angiomax patents and the potential impact to projections of future taxable income, scheduled reversal of deferred tax liabilities, tax planning strategies, tax legislation, rulings by relevant tax authorities, the progress of ongoing tax audits, the regulatory approval of products currently under development and the ability to achieve future anticipated revenues. These assumptions require significant judgment about the forecasts of future taxable income and are consistent with the plans and estimates we are using to manage the underlying businesses.
Changes in tax laws and rates could also affect recorded deferred tax assets and liabilities in the future. Management is not aware of any such changes that would have a material effect on our results of operations, cash flows, or financial position.
The calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws and regulations in a multitude of jurisdictions across our global operations.
Loss from Discontinued Operations, net of tax:
Year Ended December 31, | Change | Change | ||||||||||||
2016 | 2015 | $ | % | |||||||||||
(In thousands) | ||||||||||||||
Loss from discontinued operations, net of tax | $ | (139,682 | ) | $ | (217,950 | ) | $ | 78,268 | (35.9 | )% | ||||
For details on discontinued operations see Note 23 “Discontinued Operations,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Liquidity and Capital Resources
Sources of Liquidity
Since our inception, we have financed our operations principally through revenues from sales of Angiomax and our other products and the sale of common stock, convertible promissory notes and warrants. Revenue from sales of Angiomax has decreased significantly in recent years due to generic competition and we expect revenues from sales of Angiomax to decrease further. This
70
reduced revenue has and will likely continue to significantly impact our cash and cash equivalents and how we finance our operations. We had $151.4 million in cash and cash equivalents as of December 31, 2017.
On January 5, 2018, we completed the sale of its infectious disease business, consisting of the products Vabomere, Orbactiv and Minocin IV and line extensions thereof, and substantially all of the assets related thereto, other than certain pre-clinical assets, to Melinta. At the completion of the sale, we received approximately $166.4 million and 3,313,702 shares of Melinta common stock having a market value, based on Melinta's closing share price on January 5, 2018, of approximately $54.5 million. In addition, we are entitled to receive (i) a cash payment payable 12 months following the closing of the transaction equal to $25 million; (ii) a cash payment payable 18 months following the closing of the transaction equal to $25 million; and (iii) tiered royalty payments of 5% to 25% on worldwide net sales of (a) Vabomere and (b) Orbactiv and Minocin IV, collectively.
Cash Flows
As of December 31, 2017, we had $151.4 million in cash and cash equivalents, as compared to $541.8 million as of December 31, 2016. The decrease in cash and cash equivalents was primarily due to $368.3 million, $5.0 million and $17.1 million in net cash used in operating activities, investing activities and financing activities, respectively. For further details on cash flows related to discontinued operations, see Note 23 “Discontinued Operations,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Net cash used in operating activities was $368.3 million in 2017. The cash used in operating activities in 2017 primarily relates to a net loss of $708.4 million, non-cash items of $383.5 million and working capital items of $43.4 million. Non-cash items consist of asset impairment charges, stock compensation expenses, amortization of debt discounts, depreciation and amortization and reserve for excess or obsolete inventory, offset by deferred tax benefits.
Net cash used in operating activities was $323.3 million in 2016. The cash used in operating activities in 2016 primarily relates to a net loss of $119.2 million, non-cash items of $163.6 million and working capital items of $40.5 million. Non-cash items consist of gain on the sale of the Non-Core ACC products, offset by depreciation and amortization, amortization of debt discount, stock compensation expense, extinguishment of debt, changes in contingent consideration obligations and reserve for excess or obsolete inventory.
Net cash used in operating activities was $198.0 million in 2015. The decrease in cash was primarily due to our loss of patent protection on Angiomax and the introduction of generic bivalirudin competition as well as several large inventory purchases during 2015. The cash used in operating activities in 2015 primarily relates to non-cash items of $219.4 million offset by a net loss of $352.7 million and $64.6 million decrease from changes in working capital adjustments. Non-cash items consist of depreciation and amortization, asset impairment charges, share-based compensation expense and adjustments in contingent consideration. The changes in working capital items reflect a decrease in contingent purchase price of $78.9 million, primarily due to milestones paid to the former shareholders of Incline offset by an increase from accounts receivable of $103.1 million due to a decrease in receivables outstanding related to Angiomax.
Net cash used in investing activities was $5.0 million in 2017, which was primarily due to the purchase of available for sale securities of $131.6 million and the purchase of fixed assets of $4.5 million partially offset by proceeds from maturities and sales of available for sale securities of $131.5 million.
Net cash provided by investing activities was $422.0 million in 2016, which was primarily due to the sale of the hemostasis business completed in February 2016 and the sale of the Non-Core ACC Products completed in June 2016.
During 2015, $123.5 million in net cash was used in investing activities, primarily due to the payment of $88.1 million in connection with our acquisition of the remaining Recothrom assets in February 2015, $28.4 million in connection with our acquisition of Annovation in February 2015 and $24.5 million in milestone payments to third parties upon FDA approval and the commercial launch of Ionsys and Kengreal, partially offset by $19.8 million from the sale of an investment. Fixed asset purchases during 2015 were approximately $2.6 million.
Net cash used in financing activities was $17.1 million in 2017, which is primarily due to $55.0 million for the repayment of the 2017 Notes and $10.5 million in payments on contingent purchase price partially offset by $48.6 million of proceeds from issuance of common stock and purchases of stock under our employee stock purchase plan.
Net cash provided by financing activities was $70.6 million in 2016, which reflected the net proceeds from the issuance of the 2023 Notes of $390.8 million, offset by the repurchase of $220.0 million of the 2017 Notes for approximately $323.2 million and the purchase of the capped call in connection with the 2023 Notes for approximately $33.9 million. As part of the repurchase of the 2017 Notes, we settled the outstanding bond hedge and warrants related to the bonds repurchased for a net
71
cash receipt of $12.6 million. Net cash provided by financing activities also included $33.8 million of proceeds from issuance of common stock and purchases of stock under our employee stock purchase plan, offset by $9.4 million in payments on contingent purchase price.
Net cash provided by financing activities was $324.8 million in 2015, which reflected $387.2 million in net proceeds from the issuance of convertible notes in January 2015 and $95.2 million of proceeds from issuance of common stock and purchases of stock under our employee stock purchase plan, offset by $157.6 million in payments on contingent purchase price.
Funding Requirements
We expect to devote substantial financial resources to our research and development efforts, clinical trials, nonclinical and preclinical studies and regulatory approvals and to our commercialization and manufacturing programs associated with inclisiran. We also will require cash to pay interest on the $400.0 million aggregate principal amount of the 2022 notes, and the $402.5 million aggregate principal amount of the 2023 notes, and to make principal payments on the 2022 notes and 2023 notes at maturity or upon conversion (other than the 2023 notes upon conversion, in which case we will have the option to settle entirely in shares of our common stock). In addition, as part of our business development strategy, we generally structure our license agreements and acquisition agreements so that a significant portion of the total license or acquisition cost is contingent upon the successful achievement of specified development, regulatory or commercial milestones. As a result, we will require cash to make payments upon achievement of these milestones under the license agreements and acquisition agreements to which we are a party. As of February 26, 2018, we may have to make contingent cash payments upon the achievement of specified development, regulatory or commercial milestones of up to:
• | $150.0 million for the license and collaboration agreement with Alnylam; |
• |
• | $69.3 million relating to our research and development infectious disease portfolio acquired in our Rempex acquisition (and which was not divested in the Melinta transactions); and |
• | $2.2 million for other transaction milestones. |
As of February 26, 2018, our total potential milestone payment obligations related to development, regulatory and commercial milestones for our products and products in development under our license agreements and acquisition agreements, assuming all milestones are achieved in accordance with the terms of these agreements, would be approximately $221.3 million. Of this amount, approximately $49.4 million relates to development milestones, $71.9 million relates to regulatory approval milestones and $100.0 million relates to commercial milestones. These amounts do not include milestone payments of up to $175.8 million related to the Ionsys product, which was discontinued and withdrawn from the United States in June 2017 and which has also been discontinued in Europe, and the MDCO-700 development program, which we discontinued in August 2017.
Based on our anticipated timeline for the achievement of development, regulatory and commercial milestones, we expect that we would make milestone payments of $5.0 million under our license agreements and acquisition agreements during the remainder of 2018. We may pay additional milestone payments under our license agreements and acquisition agreements during 2018 if we achieve additional development, regulatory and commercial milestones during the year.
Total net revenues from sales of Angiomax were significantly lower in the year ended December 31, 2017 than in previous comparable periods, and we expect these revenues will decline further. These reduced revenues are likely to significantly impact our cash and cash equivalents and how we fund our future capital requirements.
We continually evaluate our liquidity requirements, capital needs and availability of resources in view of, among other things, alternative sources and uses of capital, debt service requirements, the cost of debt and equity capital and estimated future operating cash flow. We may raise additional capital; enter into licenses or collaborations with third parties to develop and commercialize inclisiran; sell assets, including asset sales of products or businesses that generate a material portion of our revenue; restructure or refinance outstanding debt; repurchase material amounts of outstanding debt or equity; or take a combination of such steps or other steps to increase or manage our liquidity and capital resources. Any such actions or steps could have a material effect on us.
Our future capital requirements will depend on many factors, including:
• | the progress, level, timing and cost of our research and development activities related to our clinical trials and non-clinical studies with respect to inclisiran; |
• | whether we develop and commercialize inclisiran on our own or through licenses and collaborations with third parties and the terms and timing of such arrangements, if any; |
72
• | the extent to which our submissions and planned submissions for regulatory approval of inclisiran are approved on a timely basis, if at all; |
• | the decline in Angiomax sales and the extent to which royalties on sales of the authorized generic of Angiomax are generated; |
• | if inclisiran receives regulatory approval, the extent to which it is commercially successful; |
• | the extent to which we are able to realize additional funds through other sources of liquidity from the Melinta transaction; |
• | the continuation or termination of third-party manufacturing, distribution and sales and marketing arrangements; |
• | the size, cost and effectiveness of our sales and marketing programs, including scaling our operations in anticipation of a potential launch of inclisiran; |
• | the amounts of our payment obligations to third parties with respect to inclisiran or other assets; and |
• | our ability to defend and enforce our intellectual property rights. |
We believe that our existing cash and cash equivalents on hand together with the cash flows we expect to generate from product sales and royalties and the proceeds received from Melinta at the closing of the sale of the infectious disease business, will be sufficient to meet our anticipated funding requirements for the next twelve months.
With respect to both our short-term and long-term cash requirements, if our existing cash resources, together with cash that we generate from sales of our products and other sources, are insufficient to satisfy our research and development, clinical trial, product commercialization and other funding requirements, including obligations under our convertible notes, we will need to sell additional equity or debt securities, engage in asset sales, including asset sales of products or businesses that generate a material portion of our revenue, engage in other strategic transactions, or seek additional financing through other arrangements, any of which could be material. Any sale of additional equity or convertible debt securities may result in dilution to our stockholders. Public or private financing may not be available in amounts or on terms acceptable to us, if at all. If we seek to raise funds through collaboration or licensing arrangements with third parties, we may be required to relinquish rights to products, products in development or technologies that we would not otherwise relinquish or grant licenses on terms that may not be favorable to us. Moreover, our ability to obtain additional debt financing may be limited by the 2022 notes and the 2023 notes, market conditions or otherwise. If we are unable to obtain additional financing or otherwise increase our cash resources, we may be required to delay, reduce the scope of, or eliminate one or more of our planned research, development and commercialization activities, which could adversely affect our business, financial condition and operating results.
Certain Contingencies
We may be, from time to time, a party to various disputes and claims arising from normal business activities. We accrue for loss contingencies when available information indicates that it is probable that a liability has been incurred and the amount of such loss can be reasonably estimated. In the cases where we believe that a reasonably possible loss exists, we disclose the facts and circumstances of the litigation, including an estimable range, if possible.
Currently, we are party to the legal proceedings as described in Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K, which include patent litigation matters, and litigation related to a license agreement. We have assessed such legal proceedings and do not believe that it is probable that a liability has been incurred and the amount of such liability can be reasonably estimated. As a result, we have not recorded a loss contingency related to these legal proceedings. Particularly with respect to the litigation related to a company license agreement, we are presently unable to predict the outcome of such lawsuit or to reasonably estimate the possible loss, or range of potential losses, if any, related to such lawsuit. While it is not possible to determine the outcome of the matters described in Part I, Item 3, Legal Proceedings, of this Annual Report on Form 10-K, we believe it is possible that the resolution of all such matters could have a material adverse effect on our business, financial condition or results of operations.
73
Contractual Obligations
Our long-term contractual obligations include commitments and estimated purchase obligations entered into in the normal course of business. These obligations include commitments related to purchases of inventory of our products, research and development service agreements, income tax contingencies, operating leases, selling, general and administrative obligations, leased office space for our principal office in Parsippany, New Jersey and our leased office space in San Diego, California, royalties, and milestone payments and other contingent payments due under our license and acquisition agreements. These obligations also include our obligations under the 2022 Notes and 2023 Notes.
Future estimated contractual obligations as of December 31, 2017 are:
Less Than | More Than | |||||||||||||||||||
Contractual Obligations (in thousands) (1) (2) | 1 Year | 1 - 3 Years | 4 - 5 Years | 5 Years | Total | |||||||||||||||
Inventory related commitments | $ | 52,111 | $ | 16,167 | $ | — | $ | — | $ | 68,278 | ||||||||||
Long-term debt obligations | 21,069 | 42,138 | 437,135 | 413,569 | 913,911 | |||||||||||||||
Research and development | 71,115 | 68,387 | 34,508 | 30,409 | 204,419 | |||||||||||||||
Operating leases | 7,185 | 15,029 | 15,428 | 27,928 | 65,570 | |||||||||||||||
Selling, general and administrative | 3,924 | 1,379 | — | — | 5,303 | |||||||||||||||
Total contractual obligations | 155,404 | $ | 143,100 | $ | 487,071 | $ | 471,906 | $ | 1,257,481 | |||||||||||
(1) | This table does not include any milestone and royalty payments which may become payable to third parties for which the timing and likelihood of such payments are not known, as discussed below. |
(2) | This table includes commitments related to our infectious disease business which was sold on January 5, 2018 and the related commitments were assumed by Melinta. These commitments include $68.3 million of inventory related commitments, $7.3 million for research and development service agreements and $1.8 million for selling general and administrative obligations. See Note 23 “Discontinued Operations,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K for further details. |
All of the inventory related commitments included above are non-cancellable. Of the total estimated contractual obligations for research and development and selling, general and administrative activities, $5.3 million are non-cancellable.
Our long-term debt obligations reflect our obligations under the 2022 Notes and 2023 Notes to pay interest on the $400.0 million and $402.5 million, respectively, aggregate principal amount of the 2022 Notes and 2023 Notes and to make principal payments on the 2022 Notes and 2023 Notes at maturity or upon conversion (other than the 2023 Notes upon conversion, in which case we will have the option to settle entirely in shares of our common stock).
We lease our principal office in Parsippany, New Jersey. The lease covers 173,146 square feet and expires January 2024. On October 1, 2014, we entered into an agreement to lease 63,000 square feet of office space with ARE-SD Region No. 35, LLC for new office and laboratory space in San Diego, California. This lease has a term of 144 months. The commencement date was in February 2017. The lease qualifies for operating lease treatment with recorded annual rent expense from commencement date to expiration. Our remaining obligation for this space is $36.6 million. On January 11, 2018, we entered into an agreement to sublease 32,039 square feet of the office and laboratory space in San Diego, California to Gossamer Bio, Inc. The sublease agreement has a term of 84 months, and will offset our remaining obligation for this space by $10.0 million.
Approximately 99.7% of the total operating lease commitments above relate to our principal office building in Parsippany, New Jersey and our office space in San Diego, California. Also included in total property lease commitments are automobile leases, computer leases and other property leases that we entered into while expanding our global infrastructure.
Aggregate rent expense under our property leases was approximately $9.6 million in 2017, $7.6 million in 2016 and $7.3 million in 2015.
In addition to the amounts shown in the above table, we are contractually obligated to make potential future success-based development, regulatory and commercial milestone payments and royalty payments in conjunction with collaborative agreements or acquisitions we have entered into with third-parties. These contingent payments include royalty payments with respect to Angiomax (other than relating to sales of Angiomax in the United States) under our license agreements with Biogen and HRI, royalty and/or milestone payments with respect to inclisiran, Ionsys and MDCO-700. Each of these payments is contingent upon the occurrence of certain future events and, given the nature of those events, it is unclear when, if ever, we may be required to make such payments and with respect to royalty payments, what the total amount of such payments will be. Further, the timing of any of the foregoing future payments is not reasonably estimable. For those reasons, these contingent payments have not been
74
included in the table above. We may have to make these significant contingent cash payments in connection with our acquisition and licensing activities upon the achievement of specified regulatory, sales and other milestones as follows:
• | Under the license agreement with Alnylam, we may have to make contingent cash payments of up to $170.0 million upon achievement of specified milestones for the PCSK9 products. We have also agreed to pay to Alnylam specified royalties on net sales of the PCSK9 products. In addition to these obligations to Alnylam, in connection with the license, we also agreed to make payments to third parties on sales of the PCSK9 products. |
• | In connection with our acquisition of Incline, we may have to make contingent cash payments of up to $60.0 million, less certain expenses, upon achievement of specified milestones with respect to Ionsys. We also agreed to make payments to third parties of up to $83.0 million upon achievement of specified milestones and are obligated to pay specified royalties based on net sales of Ionsys. While we continue to seek a partnership or divestiture transaction for Ionsys, in June 2017 we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open. |
• | In connection with our acquisition of Rempex, we may have to make contingent cash payments of up to $224.3 million, less certain expenses and employer taxes owing because of such payments, upon achievement of specified milestones. This includes up to approximately $155 million of contingent cash payments that were assumed by Melinta as a result of the sale of our infectious disease business on January 5, 2018. See Note 23 “Discontinued Operations,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K for further details. |
• | In connection with our acquisition of Targanta, we may have to make contingent cash payments up to $49.4 million to the former shareholders of Targanta and up to $25.0 million in additional payments to Eli Lilly and InterMune upon achievement of specified milestones. As a result of the Targanta acquisition, we are a party to a license agreement with Eli Lilly through our Targanta subsidiary. We are required to make payments to Eli Lilly upon reaching specified regulatory and sales milestones. In addition, we are obligated to pay royalties to Eli Lilly based on net sales of products containing Orbactiv or the other compounds in any jurisdiction in which we hold license rights to a valid patent. These commitments were assumed by Melinta as a result of the sale of our infectious disease business on January 5, 2018. See Note 23 “Discontinued Operations,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-Kfor further details. |
• | In connection with our acquisition of Annovation and its lead product candidate, MDCO-700, we may have to make contingent cash payments of up to $26.3 million upon achievement of specified milestones and up to $6.5 million in additional payments to other third parties. In August 2017, we announced that we are discontinuing the clinical development program for MDCO-700. |
Recent Accounting Pronouncements
For detailed information regarding recently issued accounting pronouncements and the expected impact on our financial statements, see Note 2 “Significant Accounting Policies,” in the accompanying notes to consolidated financial statements included in this Annual Report on Form 10-K.
Application of Critical Accounting Estimates
The discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect our reported assets and liabilities, revenues and expenses, and other financial information. Actual results may differ significantly from these estimates under different assumptions and conditions. In addition, our reported financial condition and results of operations could vary due to a change in the application of a particular accounting standard.
We regard an accounting estimate or assumption underlying our financial statements as a “critical accounting estimate” where:
• | the nature of the estimate or assumption is material due to the level of subjectivity and judgment necessary to account for highly uncertain matters or the susceptibility of such matters to change; and |
• | the impact of the estimates and assumptions on financial condition or operating performance is material. |
Our significant accounting policies are more fully described in Note 2 to our consolidated financial statements included in this Annual Report on Form 10-K. Not all of these significant accounting policies, however, require that we make estimates and assumptions that we believe are “critical accounting estimates.” We have discussed our accounting policies with the audit committee
75
of our board of directors, and we believe that our estimates relating to revenue recognition, inventory, share-based compensation, income taxes, in-process research and development, contingent purchase price from business combinations and impairment of long-lived assets described below are “critical accounting estimates.”
Revenue Recognition
Product Sales. We distribute our branded Angiomax in the United States through a sole source distribution model with ICS. We sold Cleviprex, Kengreal and ready-to-use Argatroban and Minocin, Orbactiv and Vabomere under this model up until the sale of these products to Chiesi and Melinta, respectively. See Note 22, “Dispositions,” in this Annual Report on Form 10-K for further details regarding the products sold to Chiesi and Note 23, “Discontinued Operations,” in this Annual Report on Form 10-K for further details regarding the products sold to Melinta. ICS then primarily sells branded Angiomax, and previously sold the other products, to a limited number of national medical and pharmaceutical wholesalers with distribution centers located throughout the United States. We also distributed Ionsys through a sole source distribution model with Cardinal Health, Inc.
Prior to July 1, 2015, sales of Angiomax in the United States were recognized upon shipment to ICS. As a result of the entrance of generic products in the marketplace beginning in the third quarter of 2015, we could not reasonably estimate our chargebacks with respect to branded Angiomax between July 1, 2015 and August 30, 2017, and sales of branded Angiomax in the United States were recognized under a deferred revenue model during that period. Under the deferred revenue model, we did not recognize revenue upon product shipment of branded Angiomax to ICS. Instead, upon product shipment, we invoiced ICS, recorded deferred revenue at gross invoice sales price, classified the cost basis of the product held by ICS as finished goods inventory held by others and included such cost basis amount within prepaid expenses and other current assets on the consolidated balance sheets. We recognized revenue when hospitals purchased the products and the transaction consideration became fixed or determinable. Beginning September 1, 2017, we had sufficient market information to reasonably estimate our chargebacks, returns and other adjustments to gross revenues associated with branded Angiomax and began recognizing sales upon shipment to ICS. This change in estimate did not materially impact net product revenues or cost of product revenues for the year ended December 31, 2017, and is not expected to materially impact net product revenues or costs of product revenues in future periods.
Effective July 2, 2015, we entered into a supply and distribution agreement with Sandoz under which we have granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax (bivalirudin). We recognize sales of generic Angiomax to Sandoz under a deferred revenue model. In accordance with the Sandoz agreement, we receive a royalty based on Sandoz’s gross margin, as defined in the agreement, of the authorized generic product sold by Sandoz to hospitals. We recognize royalty revenue on an accrual basis in the period it is reported by Sandoz. During 2017 and 2016, we recognized royalty revenue of $25.8 million and $71.2 million, respectively.
Our agreement with ICS provides that ICS will be our exclusive distributor of branded Angiomax in the United States. Under the terms of this fee-for-service agreement, ICS places orders with us for sufficient quantities to maintain an appropriate level of inventory based on our customers’ historical purchase volumes. ICS assumes all credit and inventory risks, is subject to our standard return policy and has sole responsibility for determining the prices at which it sells these products, subject to specified limitations in the agreement. The agreement terminates on February 28, 2019 and will automatically renew for additional one-year periods unless either party gives notice at least 90 days prior to the automatic extension. Either party may terminate the agreement at any time and for any reason upon 180 days’ prior written notice to the other party.
In Europe, we market and sell Angiomax, which we market under the trade name Angiox. We recognize revenue from such sales when hospitals purchase the product. We had deferred revenue of $0.2 million and $1.7 million as of December 31, 2017 and 2016, respectively, associated with sales of Angiomax to wholesalers outside of the United States.
We do not recognize revenue from product sales until there is persuasive evidence of an arrangement, delivery has occurred, the price is fixed or determinable, the buyer is obligated to pay us, the obligation to pay is not contingent on resale of the product, the buyer has economic substance apart from us, we have no obligation to bring about the sale of the product, the amount of returns can be reasonably estimated and collectability is reasonably assured.
We record allowances for chargebacks and other discounts or accruals for product returns, rebates and fee-for-service charges at the time of sale, and report revenue net of such amounts. In determining the amounts of certain allowances and accruals, we must make significant judgments and estimates. For example, in determining these amounts, we estimate hospital demand, buying patterns by hospitals and group purchasing organizations from wholesalers and the levels of inventory held by wholesalers and by ICS. Making these determinations involves estimating whether trends in past wholesaler and hospital buying patterns will predict future product sales. We receive data periodically from ICS and wholesalers on inventory levels and levels of hospital purchases and we consider this data in determining the amounts of these allowances and accruals.
76
• | Product returns. Our customers have the right to return any unopened product during the 18-month period beginning six months prior to the labeled expiration date and ending 12 months after the labeled expiration date. As a result, in calculating the accrual for product returns, we must estimate the likelihood that product sold might not be used within six months of expiration and analyze the likelihood that such product will be returned within 12 months after expiration. We consider all of these factors and adjust the accrual periodically throughout each quarter to reflect actual experience. When customers return product, they are generally given credit against amounts owed. The amount credited is charged to our product returns accrual. |
In estimating the likelihood of product being returned, we rely on information from ICS and wholesalers regarding inventory levels, measured hospital demand as reported by third-party sources and internal sales data. We also consider the past buying patterns of ICS and wholesalers, the estimated remaining shelf life of product previously shipped, the expiration dates of product currently being shipped, price changes of competitive products and introductions of generic products.
At December 31, 2017 and 2016, our accrual for product returns was $4.3 million and $1.6 million, respectively. A 10% change in our accrual for product returns would have had an approximately $0.4 million effect on our reported net revenue for the year ended December 31, 2017.
• | Chargebacks and rebates. Although we primarily sell Angiomax to ICS in the United States, we typically enter into agreements with hospitals, either directly or through group purchasing organizations acting on behalf of their hospital members, in connection with the hospitals’ purchases of Angiomax. |
Based on these agreements, most of our hospital customers have the right to receive a discounted price for Angiomax and volume-based rebates on Angiomax purchases. In the case of discounted pricing, we typically provide a credit to ICS, or a chargeback, representing the difference between ICS’s acquisition list price and the discounted price. In the case of the volume-based rebates, we typically pay the rebate directly to the hospitals.
We also participate in the 340B Drug Pricing Program under the Public Health Services Act. Under the 340B Drug Pricing Program, we offer qualifying entities a discount off the commercial price of Angiomax for patients undergoing PCI on an outpatient basis.
As a result of these agreements, at the time of product shipment, we estimate the likelihood that product sold to ICS might be ultimately sold to a contracting hospital or group purchasing organization. We also estimate the contracting hospital’s or group purchasing organization’s volume of purchases.
We base our estimates on industry data, hospital purchases and the historic chargeback data we receive from ICS, most of which ICS receives from wholesalers, which detail historic buying patterns and sales mix for particular hospitals and group purchasing organizations, and the applicable customer chargeback rates and rebate thresholds.
Our allowance for chargebacks was $5.9 million and $1.9 million at December 31, 2017 and 2016, respectively. A 10% change in our allowance for chargebacks would have had an approximate $0.0 million effect on our reported net revenue for the year ended December 31, 2017. We did not have any significant allowance for rebates at December 31, 2017 and 2016.
• | Fees-for-service. We offer discounts to certain wholesalers and ICS based on contractually determined rates for certain services. We estimate our fee-for-service accruals and allowances based on historical sales, wholesaler and distributor inventory levels and the applicable discount rate. Our discounts are accrued at the time of the sale and are typically settled with the wholesalers or ICS within 60 days after the end of each respective quarter. Our fee-for-service accruals and allowances were $0.9 million and $0.8 million at December 31, 2017 and 2016, respectively. A 10% change in our fee-for-service accruals and allowances would have had an approximately $0.0 million effect on our net revenue for the year ended December 31, 2017. |
We have adjusted our allowances for chargebacks and accruals for product returns, rebates and fees-for-service in the past based on actual sales experience, and we will likely be required to make adjustments to these allowances and accruals in the future. We continually monitor our allowances and accruals and make adjustments when we believe actual experience may differ from our estimates.
77
The following table provides a summary of activity with respect to our sales allowances and accruals during 2017, 2016 and 2015 (amounts in thousands):
Cash Discounts | Returns | Chargebacks | Rebates | Fees-for- Service | |||||||||||||||
Balance at January 1, 2015 | $ | 4,142 | $ | 3,349 | $ | 44,399 | $ | — | $ | 924 | |||||||||
Allowances for sales during 2015 | 9,212 | 12,143 | 107,564 | 833 | 14,249 | ||||||||||||||
Actual credits issued for prior year’s sales | (3,927 | ) | (3,528 | ) | (40,419 | ) | — | (1,179 | ) | ||||||||||
Actual credits issued for sales during 2015 | (8,540 | ) | (3,221 | ) | (95,828 | ) | (733 | ) | (11,314 | ) | |||||||||
Balance at December 31, 2015 | 887 | 8,743 | 15,716 | 100 | 2,680 | ||||||||||||||
Allowances for sales during 2016 | 1,854 | (1,424 | ) | 36,197 | (6 | ) | 3,166 | ||||||||||||
Actual credits issued for prior year’s sales | (887 | ) | (5,233 | ) | (15,610 | ) | (50 | ) | (2,655 | ) | |||||||||
Actual credits issued for sales during 2016 | (1,573 | ) | (502 | ) | (34,408 | ) | (29 | ) | (2,365 | ) | |||||||||
Balance at December 31, 2016 | 281 | 1,584 | 1,895 | 15 | 826 | ||||||||||||||
Allowances for sales during 2017 | 1,746 | 4,439 | 17,395 | 271 | 3,085 | ||||||||||||||
Actual credits issued for prior year’s sales | (281 | ) | (1,464 | ) | (1,246 | ) | (15 | ) | (865 | ) | |||||||||
Actual credits issued for sales during 2017 | (775 | ) | (220 | ) | (12,172 | ) | (126 | ) | (2,152 | ) | |||||||||
Balance at December 31, 2017 | $ | 971 | $ | 4,339 | $ | 5,872 | $ | 145 | $ | 894 | |||||||||
International Distributors. Under our agreements with our primary international distributors, we sell Angiomax to these distributors at a fixed price. The established price is typically determined once per year, prior to the first shipment of Angiomax to the distributor each year. The minimum selling price used in determining the price is 50% of the average net unit selling price.
Revenue associated with sales to our international distributors during 2017, 2016 and 2015 was $0.2 million, $1.1 million and $1.1 million, respectively.
Inventory
We record inventory upon the transfer of title from our vendors. Inventory is stated at the lower of cost or net realizable value and valued using first-in, first-out methodology. Angiomax bulk substances are classified as raw materials and their costs are determined using acquisition costs from our contract manufacturers. We record work-in-progress costs of filling, finishing and packaging against specific product batches.
We review inventory, including inventory purchase commitments, for slow moving or obsolete amounts based on expected revenues. We review our projected market share as well as current buying patterns from our customers. We analyze our ability to sell the inventory on hand and committed to customers prior to the expiration period of the respective inventory. Significant judgment is employed in determining the appropriateness of our ability to sell inventory on hand and commitments based on our sales projections. If annual and expected volumes are less than expected, we may be required to make additional allowances for excess or obsolete inventory in the future.
For the year ended December 31, 2016, upon review of expected future product sales volumes and the projected expiration of certain components of Ionsys, we recorded an $8.5 million reserve for potential inventory obsolescence. We have experienced difficulties in getting the product included on hospitals formulary lists and therefore we have experienced lower sales volumes than expected. We project that these components will reach their expiration date prior to the projected sales of the product.
For the year ended December 31, 2017, upon review of expected future product sales volumes and the projected expiration of inventory, we recorded a $16.7 million reserve for potential inventory obsolescence, mainly related to Angiomax. We have experienced further declines in price and volume as a result of the launch of generic versions of Angiomax in the United States in July 2015. We project that a portion of our inventory will reach their expiration date prior to the projected sales of the product.
Share-Based Compensation
We have established equity compensation plans for our employees, directors and certain other individuals. All grants and terms are authorized by our Board of Directors or the Compensation Committee of our Board of Directors, as appropriate. We may grant non-qualified stock options, restricted stock awards, stock appreciation rights and other share-based awards under our 2013 Stock Incentive Plan.
78
We account for share-based compensation in accordance with Financial Accounting Standards Board, or FASB, Accounting Standards Codification (ASC) 718-10, or ASC 718-10, and recognize expense using the accelerated expense attribution method. ASC 718-10 requires companies to recognize compensation expense in an amount equal to the fair value of all share-based awards granted to employees.
We estimate the fair value of each option on the date of grant using the Black-Scholes closed-form option-pricing model based on assumptions for the expected term of the stock options, expected volatility of our common stock, and prevailing interest rates. ASC 718-10 also requires us to estimate forfeitures in calculating the expense relating to share-based compensation as opposed to only recognizing forfeitures and the corresponding reduction in expense as they occur.
We have based our assumptions on the following:
Assumption | Method of Estimating | |
• Estimated expected term of options | • Employees’ historical exercise experience | |
• Expected volatility | • Historical price of our common stock | |
• Risk-free interest rate | • Yields of U.S. Treasury securities corresponding with the expected life of option grants | |
• Forfeiture rates | • Historical forfeiture data | |
Of these assumptions, the expected term of the option and expected volatility of our common stock are the most difficult to estimate since they are based on the exercise behavior of the employees and expected performance of our common stock. Increases in the term and the volatility of our common stock will generally cause an increase in compensation expense.
Income Taxes
We account for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.
We record net deferred tax assets to the extent we believe these assets will more likely than not be realized. On a periodic basis, we evaluate the realizability of our deferred tax assets net of deferred tax liabilities and adjust such amounts in light of changing facts and circumstances, including but not limited to our level of past and future taxable income, the current and future expected utilization of tax benefit carryforwards, any regulatory or legislative actions by relevant authorities with respect to the Angiomax patents, and the status of litigation with respect to those patents. We consider all available evidence, both positive and negative, to determine whether, based on the weight of that evidence, a valuation allowance is required to reduce the net deferred tax assets to the amount that is more likely than not to be realized in future periods.
Our annual effective tax rate is based on pre-tax earnings (loss) adjusted for differences between GAAP and income tax accounting, existing statutory tax rates, limitations on the use of net operating loss and tax credit carryforwards and tax planning opportunities available in the jurisdictions in which we operate.
We record uncertain tax positions on the basis of a two-step process whereby (1) we determine whether it is more likely than not that a tax position will be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position; and (2) for tax positions that meets the more-likely-than-not recognition threshold, we recognize the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement with the relevant tax authority. Significant judgment is required in evaluating our tax position. Settlement of filing positions that may be challenged by tax authorities could impact the income tax position in the year of resolution. Our liability for uncertain tax positions is reflected as a reduction to our deferred tax assets in our consolidated balance sheet.
Contingent Purchase Price From Sale of Business
We have contingent assets for certain specified calendar year net sales milestones as part of the sales of the hemostasis business and the Non-Core ACC Products. In determining the fair value of these sales milestones, considerable judgment is required to interpret the market data used to develop the assumptions and estimates. We utilize either the “income method” or a risk adjusted revenue simulation model. The income method applies a probability weighting that considers the estimated future net sales of each of the respective products to determine the probability that each sale milestone will be met. These projections were based on factors such as relevant market size, patent protection, historical pricing of similar products and expected industry trends. In
79
a risk adjusted revenue simulation model, the chances of achieving many different revenue levels are estimated and then adjusted to reflect the results of similar products and companies in the market to calculate the fair value of each milestone payment. The breadth of all possible revenue scenarios is captured in an estimate of revenue volatility - a measure that can be estimated from performance of similar companies in the market. We estimated revenue volatility as the delivered asset volatility observed in comparable companies’ historical performance, where the delivering asset was based on operational leverage of us. Under each of these possible scenarios, different amounts of the sales-based milestone payments are calculated, and the average of the payments across a range of possible scenarios is deemed to be the expected value of the earn-out payments. We will recognize any increases in the carrying amount or impairments of the contingent purchase price if and when the milestones are achieved or determined to have no value. These increases in carrying amount or impairments would be recorded in operating expenses in the consolidated statements of operations.
In-Process Research and Development
The cost of IPR&D acquired directly in a transaction other than a business combination is capitalized if the projects have an alternative future use; otherwise they are expensed. The fair values of IPR&D projects acquired in business combinations are capitalized. Several methods may be used to determine the estimated fair value of the IPR&D acquired in a business combination. The Company utilizes the “income method,” which applies a probability weighting that considers the risk of development and commercialization to the estimated future net cash flows that are derived from projected sales revenues and estimated costs. These projections are based on factors such as relevant market size, patent protection, historical pricing of similar products and expected industry trends. The estimated future net cash flows are then discounted to the present value using an appropriate discount rate. This analysis is performed for each project independently. These assets are treated as indefinite-lived intangible assets until completion or abandonment of the projects, at which time the assets are amortized over the remaining useful life or written off, as appropriate. These are tested at least annually or when a triggering event occurs that could indicate a potential impairment.
Contingent Purchase Price from Business Combinations
We record contingent consideration resulting from a business combination at its fair value on the acquisition date. We estimate the fair value of the contingent consideration liabilities related to the achievement of future development, regulatory and commercial milestones using either a probability-weighted discounted cash flow approach or a risk adjusted revenue simulation model. Changes to contingent consideration obligations can result from adjustments to discount rates and periods, updates in the assumed achievement or timing of any development or commercial milestone or changes in the probability of certain clinical events, the passage of time and changes in the assumed probability associated with regulatory approval. Each reporting period thereafter, we revalue these obligations and record increases or decreases in their fair value as an adjustment to net change in operating expenses within the accompanying consolidated statement of operations. Significant judgment is employed in determining the appropriateness of these assumptions as of the acquisition date and for each subsequent period. Accordingly, any change in the assumptions described above, could have a material impact on the amount of the net change in contingent consideration obligation that we record in any given period.
Impairment of Long-Lived Assets and Goodwill
Long-lived assets, such as property, plant and equipment and certain other long-term assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset or asset group to the estimated undiscounted future cash flows expected to be generated by the asset or asset group. If the carrying amount of the assets exceed their estimated future undiscounted net cash flows, an impairment charge is recognized for the amount by which the carrying amount of the assets exceed the fair value of the assets.
Goodwill represents the excess consideration in a business combination over the fair value of identifiable net assets acquired. Goodwill is not amortized, but subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We determine whether goodwill may be impaired by comparing the carrying value of its reporting unit to the fair value of its reporting unit.
In 2015, as a result of the sale of the hemostasis business, we are accounting for the assets and liabilities of the hemostasis business to be sold as held for sale. As a result of the classification as held for sale, we recorded impairment charges in 2015 of $133.3 million, including $24.5 million related to goodwill, to reduce the hemostasis business disposal group’s carrying value to its estimated fair value, less costs to sell.
80
Item 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
Market risk is the risk of change in fair value of a financial instrument due to changes in interest rates, equity prices, creditworthiness, financing, exchange rates or other factors. Our primary market risk exposure relates to changes in interest rates in our cash, cash equivalents and available for sale securities. We place our investments in high-quality financial instruments, primarily money market funds, corporate debt securities, asset backed securities and U.S. government agency notes with maturities of less than two years, which we believe are subject to limited interest rate and credit risk. We currently do not hedge interest rate exposure. At December 31, 2017, we held $151.4 million in cash and cash equivalents, which had an average interest rate of approximately 0.76%. A 10% change in such average interest rate would have had an approximate $0.1 million impact on our annual interest income. At December 31, 2017, all cash and cash equivalents were due on demand or within one year and 89.7% is held in the United States.
Most of our transactions are conducted in U.S. dollars. We do have certain agreements with parties located outside the United States. Transactions under certain of these agreements are conducted in U.S. dollars, subject to adjustment based on significant fluctuations in currency exchange rates. Transactions under certain other of these agreements are conducted in the local foreign currency. As of December 31, 2017, we had receivables denominated in currencies other than the U.S. dollar. A 10% change in foreign exchange rates would have had an approximate $0.4 million impact on our other income and cash.
Item 8. | Financial Statements and Supplementary Data. |
All financial statements and schedules required to be filed hereunder are filed as Appendix A to this Annual Report on Form 10-K and incorporated herein by this reference.
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. |
None.
Item 9A. | Controls and Procedures. |
Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2017. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2017, our chief executive officer and chief financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Annual Report on Internal Control Over Financial Reporting
The report required to be filed hereunder is included in Appendix A to this Annual Report on Form 10-K and incorporated herein by this reference.
Attestation Report of Independent Registered Public Accounting Firm
The report required to be filed hereunder is included in Appendix A to this Annual Report on Form 10-K and incorporated herein by this reference.
Changes in Internal Control Over Financial Reporting
No change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) occurred during the fiscal quarter ended December 31, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. | Other Information. |
None.
81
PART III
Pursuant to Paragraph G(3) of the General Instructions to Form 10-K, the information required by Part III (Items 10, 11, 12, 13 and 14) is being incorporated by reference herein from our proxy statement to be filed with the Securities and Exchange Commission within 120 days of the end of the fiscal year ended December 31, 2017 in connection with our 2018 annual meeting of stockholders. We refer to such proxy statement herein as our 2018 Proxy Statement.
Item 10. | Directors, Executive Officers and Corporate Governance. |
The information required by this item will be contained in our 2018 Proxy Statement under the captions “Discussion of Proposals,” “Information About Corporate Governance,” “Information About Our Executive Officers” and “Section 16(a) Beneficial Ownership Reporting Compliance” and is incorporated herein by this reference.
We have adopted a code of business conduct and ethics applicable to all of our directors and employees, including our principal executive officer, principal financial officer and our controller. The global code of conduct and ethics, as amended, is available on the corporate governance section of “About” of our website, www.themedicinescompany.com.
Any waiver of the code of business conduct and ethics for directors or executive officers, or any amendment to the code that applies to directors or executive officers, may only be made by the board of directors. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding an amendment to, or waiver from, a provision of this code of ethics by filing a Form 8-K disclosing such waiver, or, to the extent permitted by applicable NASDAQ regulations, by posting such information on our website, at the address and location specified above. To date, no such waivers have been requested or granted.
Item 11. | Executive Compensation. |
The information required by this item will be contained in our 2018 Proxy Statement under the captions “Information About Corporate Governance” and “Information About Our Executive Officers” and is incorporated herein by this reference.
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The information required by this item will be contained in our 2018 Proxy Statement under the captions “Principal Stockholders, “Information About Our Executive Officers” and “Equity Compensation Plan Information” and is incorporated herein by this reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item will be contained in our 2018 Proxy Statement under the captions “Information About Corporate Governance” and “Information About Our Executive Officers” and is incorporated herein by this reference.
Item 14. | Principal Accounting Fees and Services. |
The information required by this item will be contained in our 2018 Proxy Statement under the captions “Independent Registered Public Accounting Firm Fees and Other Matters” and “Discussion of Proposals” and is incorporated herein by this reference.
82
PART IV
Item 15. | Exhibits and Financial Statement Schedules. |
(a) Documents filed as part of this Annual Report on Form 10-K:
(1) Financial Statements. The Consolidated Financial Statements are included as Appendix A hereto and are filed as part of this Annual Report on Form 10-K. The Consolidated Financial Statements include:
Page | |
(2) Exhibits. The exhibits set forth on the Exhibit Index following the signature page to this annual report are filed as part of this Annual Report on Form 10-K. This list of exhibits identifies each management contract or compensatory plan or arrangement required to be filed as an exhibit to this Annual Report on Form 10-K.
83
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 1, 2018.
THE MEDICINES COMPANY
By: | /s/ Clive A. Meanwell |
Clive A. Meanwell | |
Chief Executive Officer | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
84
Signature | Title(s) | Date | ||
/s/ Clive A. Meanwell | Chief Executive Officer and Director | March 1, 2018 | ||
Clive A. Meanwell | (Principal Executive Officer) | |||
/s/ William B. O’Connor | Chief Financial Officer | March 1, 2018 | ||
William B. O’Connor | (Principal Financial and Accounting Officer) | |||
/s/ William W. Crouse | Director | March 1, 2018 | ||
William W. Crouse | ||||
/s/ Alexander J. Denner | Director | March 1, 2018 | ||
Alexander J. Denner | ||||
/s/ Fredric N. Eshelman | Executive Chairman and Director | March 1, 2018 | ||
Fredric N. Eshelman | ||||
/s/ Geno J. Germano | Director | March 1, 2018 | ||
Geno J. Germano | ||||
/s/ John C. Kelly | Director | March 1, 2018 | ||
John C. Kelly | ||||
/s/ Armin M. Kessler | Director | March 1, 2018 | ||
Armin M. Kessler | ||||
/s/ Paris Panayiotopoulos | Director | March 1, 2018 | ||
Paris Panayiotopoulos | ||||
/s/ Sarah J. Schlesinger | Director | March 1, 2018 | ||
Sarah J. Schlesinger | ||||
/s/ Hiroaki Shigeta | Director | March 1, 2018 | ||
Hiroaki Shigeta | ||||
/s/ Melvin K. Spigelman | Director | March 1, 2018 | ||
Melvin K. Spigelman | ||||
/s/ Elizabeth H.S. Wyatt | Director | March 1, 2018 | ||
Elizabeth H.S. Wyatt | ||||
85
APPENDIX A
INDEX TO THE CONSOLIDATED FINANCIAL STATEMENTS OF
THE MEDICINES COMPANY
Page | |
F - 1
Management’s Report on Consolidated Financial Statements and
Internal Control over Financial Reporting
The management of The Medicines Company has prepared, and is responsible for, The Medicines Company’s consolidated financial statements and related footnotes. These consolidated financial statements have been prepared in conformity with U.S. generally accepted accounting principles.
The Medicines Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of the Company’s principal executive and principal financial officers and effected by the Company’s board of directors, management, and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
• | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of The Medicines Company; |
• | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of The Medicines Company are being made only in accordance with authorizations of management and directors of The Medicines Company; and |
• | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of The Medicines Company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Medicines Company’s management assessed the Company’s internal control over financial reporting as of December 31, 2017. Management’s assessment was based upon the criteria established in “Internal Control — Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework). Based on its assessment, management concluded that, as of December 31, 2017, The Medicines Company’s internal control over financial reporting is effective based on those criteria.
The Company’s independent auditors, Ernst & Young LLP, a registered public accounting firm, are appointed by the Audit Committee, subject to ratification by the Company’s stockholders. Ernst & Young LLP have audited and reported on the consolidated financial statements of the Company and the effectiveness of the Company’s internal control over financial reporting. The reports of the independent auditors are contained in this Annual Report on Form 10-K.
/s/ Clive A. Meanwell | /s/ William B. O’Connor | |
Chief Executive Officer | Chief Financial Officer | |
Dated March 1, 2018
F - 2
Report of Independent Registered Public Accounting Firm
The Stockholders and Board of Directors of The Medicines Company
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of The Medicines Company (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 1, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 1996.
Iselin, New Jersey
March 1, 2018
F - 3
Report of Independent Registered Public Accounting Firm
The Stockholders and Board of Directors of The Medicines Company
Opinion on Internal Control over Financial Reporting
We have audited The Medicines Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, The Medicines Company (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2017 and 2016, the related consolidated statements of operations, comprehensive loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and our report dated March 1, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Consolidated Financial Statements and Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Iselin, New Jersey
March 1, 2018
F - 4
THE MEDICINES COMPANY
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
December 31, | |||||||
2017 | 2016 | ||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 151,359 | $ | 541,835 | |||
Accounts receivable, net of allowances of approximately $7.1 million and $2.9 million at December 31, 2017 and December 31, 2016, respectively | 3,496 | 18,171 | |||||
Inventory, net | 5,559 | 28,486 | |||||
Prepaid expenses and other current assets | 11,688 | 14,875 | |||||
Current assets held for sale | 391,202 | 50,586 | |||||
Total current assets | 563,304 | 653,953 | |||||
Fixed assets, net | 17,254 | 30,961 | |||||
In-process research & development | — | 65,000 | |||||
Product licenses, net | — | 26,987 | |||||
Developed product rights, net | — | 230,198 | |||||
Goodwill | 200,571 | 200,571 | |||||
Restricted cash | 5,541 | 5,032 | |||||
Contingent purchase price from sale of businesses | 80,700 | 143,700 | |||||
Other assets | 5,613 | 715 | |||||
Noncurrent assets held for sale | — | 348,094 | |||||
Total assets | $ | 872,983 | $ | 1,705,211 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 10,244 | $ | 20,578 | |||
Accrued expenses | 95,197 | 66,838 | |||||
Current portion of contingent purchase price | 4,995 | — | |||||
Convertible senior notes | — | 53,749 | |||||
Deferred revenue | 4,476 | 16,435 | |||||
Current liabilities held for sale | 60,580 | 87,025 | |||||
Total current liabilities | 175,492 | 244,625 | |||||
Contingent purchase price | 14,655 | 31,832 | |||||
Convertible senior notes | 649,198 | 623,584 | |||||
Deferred tax liabilities | — | 89,992 | |||||
Other liabilities | 8,724 | 11,705 | |||||
Noncurrent liabilities held for sale | — | 50,457 | |||||
Total liabilities | 848,069 | 1,052,195 | |||||
Equity component of currently redeemable convertible senior notes (Note 9) | — | 1,033 | |||||
Stockholders’ equity: | |||||||
Preferred stock, $1.00 par value per share, 5,000,000 shares authorized; no shares issued and outstanding | — | — | |||||
Common stock, $0.001 par value per share, 187,500,000 authorized; 76,191,958 issued and 73,178,815 outstanding at December 31, 2017 and 73,212,545 issued and 71,019,563 outstanding at December 31, 2016 | 76 | 73 | |||||
Additional paid-in capital | 1,377,393 | 1,256,890 | |||||
Treasury stock, at cost; 3,013,143 and 2,192,982 shares at December 31, 2017 and December 31, 2016, respectively | (90,016 | ) | (50,000 | ) | |||
Accumulated deficit | (1,257,356 | ) | (548,983 | ) | |||
Accumulated other comprehensive loss | (5,183 | ) | (5,479 | ) | |||
Total The Medicines Company stockholders’ equity | 24,914 | 652,501 | |||||
Non-controlling interest in joint venture | — | (518 | ) | ||||
Total stockholders’ equity | 24,914 | 651,983 | |||||
Total liabilities and stockholders’ equity | $ | 872,983 | $ | 1,705,211 | |||
See accompanying notes to consolidated financial statements.
F - 5
THE MEDICINES COMPANY
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Net product revenues | $ | 18,980 | $ | 71,956 | $ | 240,688 | |||||
Royalty revenues | 25,809 | 71,205 | 53,859 | ||||||||
Total net revenues | 44,789 | 143,161 | 294,547 | ||||||||
Operating expenses: | |||||||||||
Cost of product revenues | 47,193 | 60,653 | 103,986 | ||||||||
Asset impairment charges | 392,097 | — | — | ||||||||
Research and development | 138,370 | 92,107 | 90,388 | ||||||||
Selling, general and administrative | 132,225 | 212,482 | 285,300 | ||||||||
Total operating expenses | 709,885 | 365,242 | 479,674 | ||||||||
Loss from operations | (665,096 | ) | (222,081 | ) | (185,127 | ) | |||||
Legal settlement | — | — | 5,000 | ||||||||
Co-promotion and license income | 7,549 | 3,854 | 10,132 | ||||||||
Gain on remeasurement of equity investment | — | — | 22,597 | ||||||||
Gain on sale of investment | — | — | 19,773 | ||||||||
Gain on sale of assets | — | 288,301 | — | ||||||||
Loss on extinguishment of debt | — | (5,380 | ) | — | |||||||
Interest expense | (48,564 | ) | (44,463 | ) | (37,092 | ) | |||||
Other income | 1,840 | 346 | 188 | ||||||||
(Loss) income from continuing operations before income taxes | (704,271 | ) | 20,577 | (164,529 | ) | ||||||
Benefit from (provision for) income taxes | 96,576 | (67 | ) | 29,733 | |||||||
(Loss) income from continuing operations | (607,695 | ) | 20,510 | (134,796 | ) | ||||||
Loss from discontinued operations, net of tax | (100,678 | ) | (139,682 | ) | (217,950 | ) | |||||
Net loss | (708,373 | ) | (119,172 | ) | (352,746 | ) | |||||
Net loss (income) attributable to non-controlling interest | — | 54 | (10 | ) | |||||||
Net loss attributable to The Medicines Company | $ | (708,373 | ) | $ | (119,118 | ) | $ | (352,756 | ) | ||
Amounts attributable to The Medicines Company: | |||||||||||
(Loss) income from continuing operations | $ | (607,695 | ) | $ | 20,564 | $ | (134,806 | ) | |||
Loss from discontinued operations, net of tax | (100,678 | ) | (139,682 | ) | (217,950 | ) | |||||
Net loss attributable to The Medicines Company | $ | (708,373 | ) | $ | (119,118 | ) | $ | (352,756 | ) | ||
Basic (loss) earnings per common share attributable to The Medicines Company: | |||||||||||
(Loss) earnings from continuing operations | $ | (8.40 | ) | $ | 0.29 | $ | (2.02 | ) | |||
Loss from discontinued operations | (1.39 | ) | (2.00 | ) | (3.26 | ) | |||||
Basic loss per share | $ | (9.79 | ) | $ | (1.71 | ) | $ | (5.28 | ) | ||
Diluted (loss) earnings per common share attributable to The Medicines Company: | |||||||||||
(Loss) earnings from continuing operations | $ | (8.40 | ) | $ | 0.28 | $ | (2.02 | ) | |||
Loss from discontinued operations | (1.39 | ) | (1.91 | ) | (3.26 | ) | |||||
Diluted loss per share | $ | (9.79 | ) | $ | (1.63 | ) | $ | (5.28 | ) | ||
Weighted average number of common shares outstanding: | |||||||||||
Basic | 72,356 | 69,909 | 66,809 | ||||||||
Diluted | 72,356 | 73,022 | 66,809 | ||||||||
See accompanying notes to consolidated financial statements.
F - 6
THE MEDICINES COMPANY
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Net loss | $ | (708,373 | ) | $ | (119,172 | ) | $ | (352,746 | ) | ||
Other comprehensive income (loss): | |||||||||||
Foreign currency translation adjustment | 296 | 213 | 1,445 | ||||||||
Amounts reclassified from accumulated other comprehensive income | — | (9,665 | ) | — | |||||||
Other comprehensive income (loss) | 296 | (9,452 | ) | 1,445 | |||||||
Comprehensive loss | (708,077 | ) | (128,624 | ) | (351,301 | ) | |||||
Less: comprehensive loss (income) attributable to non-controlling interest | — | 54 | (10 | ) | |||||||
Comprehensive loss attributable to The Medicines Company | $ | (708,077 | ) | $ | (128,570 | ) | $ | (351,311 | ) | ||
See accompanying notes to consolidated financial statements.
F - 7
THE MEDICINES COMPANY
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands)
Common Stock | Treasury Stock | Additional Paid-in | Accumulated | Accumulated Comprehensive Income | Non- controlling Interest | Total Stockholders’ | |||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Capital | Deficit | (Loss) | in JV | Equity | |||||||||||||||||||||||||
Balance at January 1, 2015 | 67,667 | $ | 68 | (2,193 | ) | $ | (50,000 | ) | $ | 1,045,078 | $ | (77,109 | ) | $ | 2,528 | $ | (474 | ) | $ | 920,091 | |||||||||||||
Employee stock purchases | 2,989 | 3 | 65,235 | 65,238 | |||||||||||||||||||||||||||||
Issuance of restricted stock awards | 166 | — | — | — | |||||||||||||||||||||||||||||
Issuance of common stock | 945 | 1 | 29,963 | 29,964 | |||||||||||||||||||||||||||||
Non-cash stock compensation | 30,605 | 30,605 | |||||||||||||||||||||||||||||||
Equity component of the convertible notes issuance, net | 37,177 | 37,177 | |||||||||||||||||||||||||||||||
Net loss | (352,756 | ) | 10 | (352,746 | ) | ||||||||||||||||||||||||||||
Currency translation adjustment | 1,445 | 1,445 | |||||||||||||||||||||||||||||||
Balance at December 31, 2015 | 71,767 | $ | 72 | (2,193 | ) | $ | (50,000 | ) | $ | 1,208,058 | $ | (429,865 | ) | $ | 3,973 | $ | (464 | ) | $ | 731,774 | |||||||||||||
Employee stock purchases | 1,313 | 1 | 33,775 | 33,776 | |||||||||||||||||||||||||||||
Issuance of restricted stock awards | 132 | — | — | ||||||||||||||||||||||||||||||
Non-cash stock compensation | 30,987 | 30,987 | |||||||||||||||||||||||||||||||
Reclassification from mezzanine equity | 16,056 | 16,056 | |||||||||||||||||||||||||||||||
Equity component of 2017 Notes repurchased | (108,725 | ) | (108,725 | ) | |||||||||||||||||||||||||||||
Purchase of capped call transactions | (33,931 | ) | (33,931 | ) | |||||||||||||||||||||||||||||
Equity component of 2023 Notes issuance, net | 98,085 | 98,085 | |||||||||||||||||||||||||||||||
Settlement of hedges | (87,874 | ) | (87,874 | ) | |||||||||||||||||||||||||||||
Settlement of warrants | 100,459 | 100,459 | |||||||||||||||||||||||||||||||
Net (loss) income | (119,118 | ) | (54 | ) | (119,172 | ) | |||||||||||||||||||||||||||
Currency translation adjustment | 213 | 213 | |||||||||||||||||||||||||||||||
Amounts reclassified from accumulated other comprehensive income | (9,665 | ) | (9,665 | ) | |||||||||||||||||||||||||||||
Balance at December 31, 2016 | 73,212 | $ | 73 | (2,193 | ) | $ | (50,000 | ) | $ | 1,256,890 | $ | (548,983 | ) | $ | (5,479 | ) | $ | (518 | ) | $ | 651,983 | ||||||||||||
Employee stock purchases | 1,949 | 2 | 48,619 | 48,621 | |||||||||||||||||||||||||||||
Purchase of shares from non-controlling interest | (685 | ) | 518 | (167 | ) | ||||||||||||||||||||||||||||
Issuance of restricted stock awards | 166 | — | — | ||||||||||||||||||||||||||||||
Non-cash stock compensation | 31,520 | 31,520 | |||||||||||||||||||||||||||||||
Equity component of 2022 Notes repurchased | 1,031 | 1,031 | |||||||||||||||||||||||||||||||
Equity component of 2017 Notes repurchased | 3 | 3 | |||||||||||||||||||||||||||||||
Settlement of 2017 Notes | 820 | 1 | 1 | ||||||||||||||||||||||||||||||
Settlement of hedges | (820 | ) | (40,016 | ) | 40,015 | (1 | ) | ||||||||||||||||||||||||||
Settlement of warrants | 44 | — | — | ||||||||||||||||||||||||||||||
Net loss | (708,373 | ) | — | (708,373 | ) | ||||||||||||||||||||||||||||
Currency translation adjustment | 296 | 296 | |||||||||||||||||||||||||||||||
Balance at December 31, 2017 | 76,191 | $ | 76 | (3,013 | ) | $ | (90,016 | ) | $ | 1,377,393 | $ | (1,257,356 | ) | $ | (5,183 | ) | $ | — | $ | 24,914 | |||||||||||||
See accompanying notes to consolidated financial statements.
F - 8
THE MEDICINES COMPANY
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Cash flows from operating activities: | |||||||||||
Net loss | $ | (708,373 | ) | $ | (119,172 | ) | $ | (352,746 | ) | ||
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||||||
Depreciation and amortization | 21,974 | 31,042 | 34,837 | ||||||||
Asset impairment charges | 392,097 | — | 29,413 | ||||||||
Impairment on divestiture | — | — | 133,273 | ||||||||
Amortization of debt discount | 26,868 | 26,182 | 23,676 | ||||||||
Unrealized foreign currency transaction losses (gains), net | 2,180 | (941 | ) | (173 | ) | ||||||
Stock compensation expense | 31,520 | 30,987 | 30,605 | ||||||||
Loss on disposal of fixed assets | 105 | 521 | 543 | ||||||||
Deferred tax benefit | (89,895 | ) | (23 | ) | (53,292 | ) | |||||
Extinguishment of debt | — | 5,380 | — | ||||||||
Gain on sale of businesses | — | (289,305 | ) | — | |||||||
Gain on sale of investment | — | — | (19,773 | ) | |||||||
Gain on remeasurement of equity investment | — | — | (22,597 | ) | |||||||
Reserve for excess or obsolete inventory | 17,453 | 8,533 | 42,599 | ||||||||
Changes in contingent purchase price | (18,787 | ) | 23,981 | 20,278 | |||||||
Changes in operating assets and liabilities: | |||||||||||
Accounts receivable | 9,180 | 30,144 | 103,100 | ||||||||
Inventory, net | 6,511 | (15,653 | ) | (69,318 | ) | ||||||
Prepaid expenses and other assets | 534 | 569 | (5,286 | ) | |||||||
Accounts payable | (17,222 | ) | (7,398 | ) | 16,362 | ||||||
Accrued expenses | 31,526 | (37,233 | ) | (39,501 | ) | ||||||
Deferred revenue | (13,757 | ) | 1,568 | 8,386 | |||||||
Payments on contingent purchase price | (52,543 | ) | (1,045 | ) | (78,900 | ) | |||||
Other liabilities | (7,659 | ) | (11,446 | ) | 549 | ||||||
Net cash used in operating activities | (368,288 | ) | (323,309 | ) | (197,965 | ) | |||||
Cash flows from investing activities: | |||||||||||
Purchases of available for sale securities | (131,560 | ) | — | — | |||||||
Proceeds from sale of fixed assets | — | — | 250 | ||||||||
Proceeds from sale of investment | — | — | 19,773 | ||||||||
Proceeds from maturities and sales of available for sale securities | 131,535 | — | — | ||||||||
Purchases of fixed assets | (4,525 | ) | (2,176 | ) | (2,555 | ) | |||||
Payments for intangible assets | — | (10,000 | ) | (112,617 | ) | ||||||
Acquisition of business, net of cash acquired | — | — | (28,397 | ) | |||||||
Proceeds from sale of businesses | — | 437,875 | — | ||||||||
Change in restricted cash | (478 | ) | (3,656 | ) | 35 | ||||||
Net cash (used in) provided by investing activities | (5,028 | ) | 422,043 | (123,511 | ) | ||||||
Cash flows from financing activities: | |||||||||||
Proceeds from issuances of common stock, net | 48,621 | 33,776 | 95,198 | ||||||||
Payments on contingent purchase price | (10,523 | ) | (9,404 | ) | (157,601 | ) | |||||
Proceeds from the issuance of convertible senior notes | — | 402,500 | 400,000 | ||||||||
Repayments of convertible senior notes | (55,000 | ) | (323,225 | ) | — | ||||||
Purchase of capped call transactions related to convertible senior notes | — | (33,931 | ) | — | |||||||
Proceeds from settlement of bond hedges related to convertible senior notes | — | 100,459 | — | ||||||||
Settlement of warrants | (2 | ) | (87,874 | ) | — | ||||||
Debt and equity issuance costs | — | (11,725 | ) | (12,769 | ) | ||||||
Purchase of shares of non-controlling interest | (167 | ) | — | — | |||||||
Net cash (used in) provided by financing activities | (17,071 | ) | 70,576 | 324,828 | |||||||
Effect of exchange rate changes on cash | (89 | ) | (648 | ) | (920 | ) | |||||
(Decrease) increase in cash and cash equivalents | (390,476 | ) | 168,662 | 2,432 | |||||||
Cash and cash equivalents at beginning of period | 541,835 | 373,173 | 370,741 | ||||||||
Cash and cash equivalents at end of period | $ | 151,359 | $ | 541,835 | $ | 373,173 | |||||
Supplemental disclosure of cash flow information: | |||||||||||
Interest paid | $ | 22,561 | $ | 12,269 | $ | 8,837 | |||||
Taxes paid | $ | 575 | $ | 36 | $ | 114 | |||||
Non-cash investing and financing activities | |||||||||||
Issuance of common stock upon conversion of convertible notes | $ | 32,018 | $ | — | $ | — | |||||
Receipt of common stock upon settlement of 2017 Note hedge | $ | 40,015 | $ | — | $ | — | |||||
Issuance of common stock upon the exercise of the 2017 Warrants | $ | 1,638 | $ | — | $ | — | |||||
See accompanying notes to consolidated financial statements.
F - 9
THE MEDICINES COMPANY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of Business
The Medicines Company (the Company) is a biopharmaceutical company driven by an overriding purpose - to save lives, alleviate suffering and contribute to the economics of healthcare. The Company’s goal is to create transformational solutions to address the most pressing healthcare needs facing patients, physicians and providers in cardiovascular care. The Company is focused on inclisiran, an investigational agent which is potentially a first-in-class lipid-lowering drug, to reduce LDL-cholesterol (LDL-C), which is commonly referred to as “bad” cholesterol, in patients with atherosclerotic cardiovascular disease (ASCVD) or cardiovascular risk-equivalents. The Company believes that inclisiran possesses favorable attributes that competitive products do not possess, would satisfy unmet medical needs and has the potential to improve the economics of healthcare. The Company has the right to develop, manufacture and commercialize inclisiran under its collaboration agreement with Alnylam Pharmaceuticals, Inc. (Alnylam). In addition, the Company markets Angiomax® (bivalirudin) in the United States primarily through a supply and distribution agreement with Sandoz Inc. (Sandoz), under which the Company granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax.
On November 3, 2015, the Company announced that it was in the process of evaluating its operations with a goal of unlocking stockholder value. In particular, the Company stated its current intention was to explore strategies for optimizing its capital structure and liquidity position and to narrow the Company’s operational focus by strategically separating non-core businesses and products in order to generate non-dilutive cash and reduce associated cash burn and capital requirements.
On February 1, 2016, the Company completed the sale of its hemostasis portfolio, consisting of PreveLeak (surgical sealant), Raplixa (fibrin sealant) and Recothrom Thrombin topical (Recombinant) (the Hemostasis Business), to wholly owned subsidiaries of Mallinckrodt plc (collectively, Mallinckrodt) pursuant to the purchase and sale agreement dated December 18, 2015 between the Company and Mallinckrodt. At completion of the sale, the Company received approximately $174.1 million in cash from Mallinckrodt, and may receive up to an additional $235.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of PreveLeak and Raplixa. As a result of the transaction, the Company accounted for the assets and liabilities of the Hemostasis Business as held for sale at December 31, 2015. As a result of the classification as held for sale, the Company recorded impairment charges of $133.3 million, including $24.5 million related to goodwill, to reduce the Hemostasis Business disposal group’s carrying value to its estimated fair value, less costs to sell for the year ended December 31, 2015. Further, the financial results of the Hemostasis Business held for sale were reclassified to discontinued operations for all periods presented in our consolidated financial statements. See Note 23 “Discontinued Operations” for further details.
On June 21, 2016, the Company completed the sale of three non-core cardiovascular products, Cleviprex (clevidipine) injectable emulsion, Kengreal (cangrelor) and rights to Argatroban for Injection (collectively the Non-Core ACC Products) and related assets, to Chiesi USA, Inc. (Chiesi USA) and its parent company Chiesi Farmaceutici S.p.A. (Chiesi) pursuant to the purchase and sale agreement dated May 9, 2016 by and among the Company, Chiesi and Chiesi USA. At the completion of the sale, the Company received approximately $263.8 million in cash, which included the value of product inventory, and may receive up to an additional $480.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of each of Cleviprex and Kengreal. As part of the transaction, we sublicensed to Chiesi all of our rights to Cleviprex and Kengreal under our license from AstraZeneca. Subsequent to the completion of the sale, these sublicenses from us to Chiesi were terminated, Chiesi purchased from AstraZeneca all or substantially all of AstraZeneca’s assets relating to Cleviprex and Kengreal, we and Chiesi released certain claims against one another, and we paid Chiesi $7.5 million. See Note 22, “Dispositions,” for further details. Consistent with the Company’s intentions announced in November 2015, in January 2017 the Company announced that it was seeking opportunities to partner or divest Ionsys and is exploring alternatives for monetizing, in whole or in part, the Company’s infectious disease business.
Although the Company continues to seek a partnership or divestiture transaction for Ionsys, in June 2017 the Company commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open. Concurrent with this market withdrawal, the Company commenced implementation of a workforce reduction, which resulted in the reduction of 57 employees, representing approximately 15% of the Company’s workforce at that time. In the second quarter of 2017, the Company recorded a pre-tax charge of approximately $276.9 million associated with the discontinuation and market withdrawal of Ionsys in the United States market, of which $268.1 million was a non-cash impairment charge (including a write-off of inventory of $5.3 million) and $8.8 million relates to cash severance and other exit costs. The non-cash impairment charge includes $11.4 million to reduce the carrying amount of the fixed assets associated with Ionsys to an estimated fair value of zero. The Company has also discontinued and withdrawn Ionsys in the
F - 10
European market. Until October 2017, the Company had an exclusive license with SymBio Pharmaceuticals Ltd. (SymBio) to develop and commercialize Ionsys in Japan. That agreement terminated in connection with a legal dispute with SymBio, as described in Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K.
In August 2017, the Company announced that it discontinued the clinical development program for MDCO-700, an investigational anesthetic agent. In connection with this decision, the Company’s consolidated statement of operations for the year ended December 31, 2017 includes the following non-cash adjustments that were recorded during the second quarter of 2017: $65.0 million of asset impairment charges to in-process research and development (IPR&D) acquired from Annovation BioPharma, Inc. (Annovation), a $14.7 million decrease in the carrying value of the contingent purchase price to an estimated fair value of zero, and a $23.0 million benefit for income taxes due to a reduction in the Company’s recorded valuation allowance against its deferred tax assets as a result of the impairment charge.
On January 5, 2018, the Company completed the sale of its infectious disease business, consisting of the products Vabomere, Orbactiv and Minocin IV and line extensions thereof, and substantially all of the assets related thereto, other than certain pre-clinical assets, to Melinta Therapeutics, Inc. (Melinta) pursuant to a purchase and sale agreement dated November 28, 2017 between the Company and Melinta. At the completion of the sale, the Company received approximately $166.4 million and 3,313,702 shares of Melinta common stock having a market value, based on Melinta's closing share price on January 5, 2018, of approximately $54.5 million. In addition, the Company is entitled to receive (i) a cash payment payable 12 months following the closing of the Transaction equal to $25.0 million; (ii) a cash payment payable 18 months following the closing of the Transaction equal to $25.0 million; and (iii) tiered royalty payments of 5% to 25% on worldwide net sales of (a) Vabomere and (b) Orbactiv and Minocin IV, collectively. As a result of the transaction, the Company accounted for the assets and liabilities of the infectious disease business that were sold as held for sale at December 31, 2016. Further, the financial results of the infectious disease business held for sale were reclassified to discontinued operations for all periods presented in the consolidated financial statements. See Note 23 “Discontinued Operations” for further details.
2. Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP). Any reference in these notes to applicable guidance is meant to refer to the authoritative U.S. GAAP as found in the Accounting Standards Codification (ASC) and Accounting Standards Updates (ASU) of the Financial Accounting Standards Board (FASB). The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All significant intercompany balances and transactions have been eliminated in consolidation. The Company records net income (loss) attributable to non-controlling interest in the Company’s consolidated financial statements equal to percentage of ownership interest retained in the respective operations by the non-controlling parties. The Company has no unconsolidated subsidiaries.
Going Concern
Due to the introduction of generic competition against Angiomax and the divestiture of certain of the Company’s non-core products, the Company’s revenues generated from product sales have declined significantly since 2014. Revenues are expected to continue to decline as generic competition for Angiomax increases. The Company has incurred net losses and negative cash flows from operations since 2014 and had an accumulated deficit of $1.3 billion as of December 31, 2017. The Company expects to incur significant expenses and operating losses for the foreseeable future as it continues to develop, seek regulatory approval for and commercially launch inclisiran. As a result of the completion of the sale of the Company’s infectious disease business on January 5, 2018, the Company believes that its existing cash and cash equivalents of approximately $151.4 million as of December 31, 2017, together with the cash flows it expects to generate from product sales and royalties and the proceeds received from Melinta at the closing of the sale of the infectious disease business, will be sufficient to satisfy its anticipated operating and other funding requirements for the next twelve months from March 1, 2018 (the date of filing this Form 10-K).
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, costs, expenses and accumulated other comprehensive income/(loss) that are reported in the consolidated financial statements and accompanying disclosures. Actual results may be different.
F - 11
Investments
The Company accounts for its common stock investment in a minority interest of a company that does not have a readily determinable fair value over which it does not exercise significant influence on the cost method. Under the cost method, an investment is carried at cost until it is sold or there is evidence that changes in the business environment or other facts and circumstances suggest it may be other than temporarily impaired. Investments in which the Company has at least a 20%, but not more than a 50%, interest are generally accounted for under the equity method. These non-marketable securities have been classified as investments and included in other assets on the accompanying consolidated balance sheets. The Company’s proportionate share of the operating results is recorded as loss in equity investment in the Company’s consolidated statement of operations. On February 2, 2015, the Company completed the acquisition of Annovation, and Annovation became the Company’s wholly owned subsidiary. See Note 6 “Acquisition” for further details.
Inventory
The Company records inventory upon the transfer of title from the Company’s vendors. Inventory is stated at the lower of cost or net realizable value and valued using first-in, first-out methodology. Angiomax and Ionsys bulk substances are classified as raw materials and their costs are determined using acquisition costs from the Company’s contract manufacturers. The Company records work-in-progress costs of filling, finishing and packaging against specific product batches.
Fixed Assets
Fixed assets are stated at cost. Depreciation is provided using the straight-line method based on estimated useful lives or, in the case of leasehold improvements, over the lesser of the useful lives or the lease terms. Repairs and maintenance costs are expensed as incurred.
Treasury Stock
Treasury stock is recognized at the cost to reacquire the shares. Shares issued from treasury are recognized utilizing the first-in first-out method.
Intangible Assets with Definite Useful Lives
Intangible assets with definite useful lives are amortized over their estimated useful lives and reviewed for impairment if certain events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable.
In-Process Research and Development
The cost of in-process research and development (IPR&D) acquired directly in a transaction other than a business combination is capitalized if the projects have an alternative future use; otherwise it is expensed. The fair values of IPR&D projects acquired in business combinations are capitalized. Several methods may be used to determine the estimated fair value of the IPR&D acquired in a business combination. The Company utilizes the “income method,” which applies a probability weighting that considers the risk of development and commercialization to the estimated future net cash flows that are derived from projected sales revenues and estimated costs. These projections are based on factors such as relevant market size, patent protection, historical pricing of similar products and expected industry trends. The estimated future net cash flows are then discounted to the present value using an appropriate discount rate. This analysis is performed for each project independently. The Company also considers qualitative factors such as development of competing drugs, status in the development cycle of the product, regulatory developments and other qualitative factors.
These assets are treated as indefinite-lived intangible assets until completion or abandonment of the projects, at which time the assets are amortized over the remaining useful life or written off, as appropriate. These are tested at least annually or when a triggering event occurs that could indicate a potential impairment.
In August 2017, the Company announced that it discontinued the clinical development program for MDCO-700. In connection with this decision, the Company recorded impairment charges of $65.0 million related to IPR&D acquired from Annovation.
Goodwill
Goodwill represents the excess consideration in a business combination over the fair value of identifiable net assets acquired. Goodwill is not amortized, but subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. The Company determines whether goodwill may be impaired by comparing the carrying value of its
F - 12
reporting unit to the fair value of its reporting unit. A reporting unit is defined as an operating segment or one level below an operating segment. Based on the Company’s evaluation, goodwill was not impaired as of December 31, 2017.
Contingent Purchase Price From Sale of Business
The Company has contingent assets for certain specified calendar year net sales milestones as part of the sales of the Hemostasis Business and the Non-Core ACC Products. In determining the fair value of these sales milestones, considerable judgment is required to interpret the market data used to develop the assumptions and estimates. The Company utilizes either the “income method” or a risk adjusted revenue simulation model. The income method applies a probability weighting that considers the estimated future net sales of each of the respective products to determine the probability that each sale milestone will be met. These projections were based on factors such as relevant market size, patent protection, historical pricing of similar products and expected industry trends. In a risk adjusted revenue simulation model, the chances of achieving many different revenue levels are estimated and then adjusted to reflect the results of similar products and companies in the market to calculate the fair value of each milestone payment. The breadth of all possible revenue scenarios is captured in an estimate of revenue volatility - a measure that can be estimated from performance of similar companies in the market. The Company estimated revenue volatility as the delivered asset volatility observed in comparable companies’ historical performance, where the delivering asset was based on operational leverage of the Company. Under each of these possible scenarios, different amounts of the sales-based milestone payments are calculated, and the average of the payments across a range of possible scenarios is deemed to be the expected value of the earn-out payments. The Company will recognize any increases in the carrying amount or impairments of the contingent purchase price if and when the milestones are achieved or determined to have no value.
In the fourth quarter of 2017, the Company decreased the carrying value of the contingent purchase price from the sale of the Hemostasis Business by $63.0 million to an estimated fair value of zero, which is a Level 3 fair value measurement, as a result of the discontinuation of Raplixa by Mallinckrodt. The Company noted no indicators of impairment on the carrying amounts of the remaining contingent assets. In addition, the Company determined that the fair values of these contingent payments to be received from the buyers are not readily determinable at December 31, 2017, as the estimated future net sales of each of the respective products are determined by the future actions of the buyers.
Long-Lived Assets
Long-lived assets, such as property, plant and equipment and certain other long-term assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset or asset group to the estimated undiscounted future cash flows expected to be generated by the asset or asset group. If the carrying amount of the assets exceed their estimated future undiscounted net cash flows, an impairment charge is recognized for the amount by which the carrying amount of the assets exceed the fair value of the assets.
Contingent Purchase Price from Business Combinations
Subsequent to the acquisition date, the Company measures the fair value of the acquisition-related contingent consideration at each reporting period, with changes in fair value recorded in selling, general and administrative in the accompanying consolidated statements of operations. Changes to contingent consideration obligations can result from adjustments to discount rates and periods, updates in the assumed achievement or timing of any development or commercial milestone or changes in the probability of certain clinical events, the passage of time and changes in the assumed probability associated with regulatory approval. The fair value measurement is based on significant inputs not observable in the market and thus represents a Level 3 measurement as defined in fair value measurement accounting.
Risks and Uncertainties
The Company is subject to risks common to companies in the pharmaceutical industry including, but not limited to, uncertainties related to commercialization of products, regulatory approvals, dependence on key products, dependence on key customers and suppliers, and protection of intellectual property rights.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk include cash, cash equivalents and accounts receivable. The Company believes it minimizes its exposure to potential concentrations of credit risk by placing investments with high quality institutions. At December 31, 2017 and 2016, approximately $12.1 million and $56.1 million,
F - 13
respectively, of the Company’s cash and cash equivalents was invested in a single fund, the Dreyfus Cash Management Money Market Fund, a no-load money market fund with Capital Advisors Group.
The Company currently sells branded Angiomax in the United States to a sole source distributor, Integrated Commercialization Solutions, Inc. (ICS). ICS accounted for 5%, 48% and 87% of the Company’s net product revenues for 2017, 2016 and 2015, respectively. At December 31, 2017 and 2016, amounts due from ICS represented approximately $2.9 million and $2.2 million, or 27% and 11%, of gross accounts receivable, respectively. Product sales to Sandoz accounted for 55% and 23% of the Company’s net product revenues for 2017 and 2016, respectively. At December 31, 2017 and 2016, amounts due from Sandoz related to product sales were approximately $0.9 million or 8% and $5.6 million or 27%, respectively, of gross accounts receivable. At December 31, 2017 and 2016, amounts due from Sandoz related to royalty revenues were approximately $4.2 million or 40% and $9.1 million or 43%, respectively, of gross accounts receivable.
Contingencies
The Company may be, from time to time, a party to various disputes and claims arising from normal business activities. The Company continually assesses litigation to determine if an unfavorable outcome would lead to a probable loss or reasonably possible loss which could be estimated. In accordance with the guidance of the FASB on accounting for contingencies, the Company accrues for all contingencies at the earliest date at which the Company deems it probable that a liability has been incurred and the amount of such liability can be reasonably estimated. If the estimate of a probable loss is a range and no amount within the range is more likely than another, the Company accrues the minimum of the range. In the cases where the Company believes that a reasonably possible loss exists, the Company discloses the facts and circumstances of the litigation, including an estimable range, if possible.
Revenue Recognition
Product Sales. The Company distributes branded Angiomax in the United States through a sole source distribution model with Integrated Commercialization Solutions (ICS). The Company sold Cleviprex, Kengreal and ready-to-use Argatroban and Minocin, Orbactiv and Vabomere under this model up until the sale of these products to Chiesi and Melinta, respectively. See Note 22, “Dispositions,” for further details regarding the products sold to Chiesi and Note 23, “Discontinued Operations,” for further details regarding the products sold to Melinta. ICS then primarily sells branded Angiomax, and previously sold the other product, to a limited number of national medical and pharmaceutical wholesalers with distribution centers located throughout the United States.
Prior to July 1, 2015, sales of Angiomax in the United States were recognized upon shipment to ICS. As a result of the entrance of generic products in the marketplace beginning in the third quarter of 2015, the Company could not reasonably estimate its chargebacks with respect to branded Angiomax between July 1, 2015 and August 30, 2017, and sales of branded Angiomax in the United States were recognized under a deferred revenue model during that period. Under the deferred revenue model, the Company did not recognize revenue upon product shipment of branded Angiomax to ICS. Instead, upon product shipment, the Company invoiced ICS, recorded deferred revenue at gross invoice sales price, classified the cost basis of the product held by ICS as finished goods inventory held by others and included such cost basis amount within prepaid expenses and other current assets on the consolidated balance sheets. The Company recognized revenue when hospitals purchased the products and the transaction consideration became fixed or determinable. Beginning September 1, 2017, the Company had sufficient market information to reasonably estimate its chargebacks, returns and other adjustments to gross revenues associated with branded Angiomax and recognizes sales upon shipment to ICS. This change in estimate did not materially impact net product revenues or cost of product revenues for the year ended December 31, 2017, and is not expected to materially impact net product revenues or costs of product revenues in future periods.
Effective July 2, 2015, the Company entered into a supply and distribution agreement with Sandoz under which it has granted Sandoz the exclusive right to sell in the United States an authorized generic of Angiomax (bivalirudin). The Company recognizes sales of generic Angiomax to Sandoz under a deferred revenue model. In accordance with the Sandoz agreement, the Company receives a royalty based on Sandoz’s gross margin, as defined in the agreement, of the authorized generic product sold by Sandoz to hospitals. The Company recognizes royalty revenue on an accrual basis in the period it is reported by Sandoz. During 2017 and 2016, the Company recognized royalty revenue of $25.8 million and $71.2 million, respectively.
The Company’s agreement with ICS provides that ICS will be the Company’s exclusive distributor of branded Angiomax and acute care generic products in the United States. Under the terms of this fee-for-service agreement, ICS places orders with the Company for sufficient quantities to maintain an appropriate level of inventory based on the Company’s customers’ historical purchase volumes. ICS assumes all credit and inventory risks, is subject to the Company’s standard return policy and has sole
F - 14
responsibility for determining the prices at which it sells these products, subject to specified limitations in the agreement. The agreement terminates on February 28, 2019 and will automatically renew for additional one-year periods unless either party gives notice at least 90 days prior to the automatic extension. Either party may terminate the agreement at any time and for any reason upon 180 days’ prior written notice to the other party.
In Europe, the Company markets and sells Angiomax, which the Company markets under the trade name Angiox. The Company recognizes revenue from such sales when hospitals purchase the product. The Company had deferred revenue of $0.2 million and $1.7 million as of December 31, 2017 and 2016, respectively, associated with sales of Angiomax to wholesalers outside of the United States.
The Company does not recognize revenue from product sales until there is persuasive evidence of an arrangement, delivery has occurred, the price is fixed or determinable, the buyer is obligated to pay the Company, the obligation to pay is not contingent on resale of the product, the buyer has economic substance apart from the Company, the Company has no obligation to bring about the sale of the product, the amount of returns can be reasonably estimated and collectability is reasonably assured.
The Company records allowances for chargebacks and other discounts or accruals for product returns, rebates and fee-for-service charges at the time of sale, and reports revenue net of such amounts. In determining the amounts of certain allowances and accruals, the Company must make significant judgments and estimates. For example, in determining these amounts, the Company estimates hospital demand, buying patterns by hospitals and group purchasing organizations from wholesalers and the levels of inventory held by wholesalers and by ICS. Making these determinations involves estimating whether trends in past wholesaler and hospital buying patterns will predict future product sales. The Company receives data periodically from ICS and wholesalers on inventory levels and levels of hospital purchases and the Company considers this data in determining the amounts of these allowances and accruals.
The specific considerations the Company uses in estimating these amounts are as follows:
• | Product returns. The Company’s customers have the right to return any unopened product during the 18-month period beginning six months prior to the labeled expiration date and ending 12 months after the labeled expiration date. As a result, in calculating the accrual for product returns, the Company must estimate the likelihood that product sold might not be used within six months of expiration and analyze the likelihood that such product will be returned within 12 months after expiration. The Company considers all of these factors and adjusts the accrual periodically throughout each quarter to reflect actual experience. When customers return product, they are generally given credit against amounts owed. The amount credited is charged to the Company’s product returns accrual. |
In estimating the likelihood of product being returned, the Company relies on information from ICS and wholesalers regarding inventory levels, measured hospital demand as reported by third-party sources and internal sales data. The Company also considers the past buying patterns of ICS and wholesalers, the estimated remaining shelf life of product previously shipped, the expiration dates of product currently being shipped, price changes of competitive products and introductions of generic products.
At December 31, 2017 and 2016, the Company’s accrual for product returns was $4.3 million and $1.6 million, respectively.
• | Chargebacks and rebates. Although the Company primarily sells Angiomax to ICS in the United States, the Company typically enters into agreements with hospitals, either directly or through group purchasing organizations acting on behalf of their hospital members, in connection with the hospitals’ purchases of Angiomax. |
Based on these agreements, most of the Company’s hospital customers have the right to receive a discounted price for Angiomax and volume-based rebates on Angiomax purchases. In the case of discounted pricing, the Company typically provides a credit to ICS, or a chargeback, representing the difference between ICS’ acquisition list price and the discounted price. In the case of the volume-based rebates, the Company typically pays the rebate directly to the hospitals.
The Company also participates in the 340B Drug Pricing Program under the Public Health Services Act. Under the 340B Drug Pricing Program, the Company offers qualifying entities a discount off the commercial price of Angiomax for patients undergoing percutaneous coronary intervention on an outpatient basis.
As a result of these agreements, at the time of product shipment, the Company estimates the likelihood that product sold to ICS might be ultimately sold to a contracting hospital or group purchasing organization. The Company also estimates the contracting hospital’s or group purchasing organization’s volume of purchases.
F - 15
The Company bases its estimates on industry data, hospital purchases and the historic chargeback data it receives from ICS, most of which ICS receives from wholesalers, which details historic buying patterns and sales mix for particular hospitals and group purchasing organizations, and the applicable customer chargeback rates and rebate thresholds. With the entrance of generic products and their impact on pricing in the marketplace, the Company is no longer able to reasonably estimate these chargebacks with respect to Angiomax.
The Company’s allowance for chargebacks was $5.9 million and $1.9 million at December 31, 2017 and 2016, respectively. The Company’s allowance for rebates was not material at December 31, 2017 and 2016.
• | Fees-for-service. The Company offers discounts to certain wholesalers, Cardinal Health Inc. and ICS based on contractually determined rates for certain services. The Company estimates its fee-for-service accruals and allowances based on historical sales, wholesaler and distributor inventory levels and the applicable discount rate. The Company’s discounts are accrued at the time of the sale and are typically settled within 60 days after the end of each respective quarter. The Company’s fee-for-service accruals and allowances were $0.9 million and $0.8 million at December 31, 2017 and 2016, respectively. |
The Company has adjusted its allowances for chargebacks and accruals for product returns, rebates and fees-for-service in the past based on actual sales experience, and the Company will likely be required to make adjustments to these allowances and accruals in the future. The Company continually monitors its allowances and accruals and makes adjustments when it believes actual experience may differ from its estimates.
The following table provides a summary of activity with respect to the Company’s sales allowances and accruals during 2017, 2016 and 2015 (amounts in thousands):
Cash Discounts | Returns | Chargebacks | Rebates | Fees-for- Service | |||||||||||||||
Balance at January 1, 2015 | $ | 4,142 | $ | 3,349 | $ | 44,399 | $ | — | $ | 924 | |||||||||
Allowances for sales during 2015 | 9,212 | 12,143 | 107,564 | 833 | 14,249 | ||||||||||||||
Actual credits issued for prior year’s sales | (3,927 | ) | (3,528 | ) | (40,419 | ) | — | (1,179 | ) | ||||||||||
Actual credits issued for sales during 2015 | (8,540 | ) | (3,221 | ) | (95,828 | ) | (733 | ) | (11,314 | ) | |||||||||
Balance at December 31, 2015 | 887 | 8,743 | 15,716 | 100 | 2,680 | ||||||||||||||
Allowances for sales during 2016 | 1,854 | (1,424 | ) | 36,197 | (6 | ) | 3,166 | ||||||||||||
Actual credits issued for prior year’s sales | (887 | ) | (5,233 | ) | (15,610 | ) | (50 | ) | (2,655 | ) | |||||||||
Actual credits issued for sales during 2016 | (1,573 | ) | (502 | ) | (34,408 | ) | (29 | ) | (2,365 | ) | |||||||||
Balance at December 31, 2016 | 281 | 1,584 | 1,895 | 15 | 826 | ||||||||||||||
Allowances for sales during 2017 | 1,746 | 4,439 | 17,395 | 271 | 3,085 | ||||||||||||||
Actual credits issued for prior year’s sales | (281 | ) | (1,464 | ) | (1,246 | ) | (15 | ) | (865 | ) | |||||||||
Actual credits issued for sales during 2017 | (775 | ) | (220 | ) | (12,172 | ) | (126 | ) | (2,152 | ) | |||||||||
Balance at December 31, 2017 | $ | 971 | $ | 4,339 | $ | 5,872 | $ | 145 | $ | 894 | |||||||||
International Distributors. Under the Company’s agreements with its primary international distributors, the Company sells Angiomax to these distributors at a fixed price. The established price is typically determined once per year, prior to the first shipment of Angiomax to the distributor each year. The minimum selling price used in determining the price is 50% of the average net unit selling price.
Revenue associated with sales to the Company’s international distributors during 2017, 2016 and 2015 was $0.2 million, $1.1 million and $1.1 million, respectively.
Cost of Product Revenues
Cost of product revenues consists of expenses in connection with the manufacture of Angiomax, Cleviprex, ready-to-use Argatroban, Kengreal and Ionsys, royalty expenses under the Company’s agreements with Biogen Idec (Biogen) and Health Research Inc. (HRI) related to Angiomax, with AstraZeneca AB (AstraZeneca) related to Cleviprex and with Eagle Pharmaceuticals, Inc. (Eagle) related to ready-to-use Argatroban and the logistics costs related to Angiomax, Cleviprex, ready-to-use Argatroban,
F - 16
Kengreal and Ionsys including distribution, storage and handling costs. Amounts billed for shipping and handling are recorded as revenue. Shipping and handling expenses are recorded as a component of cost of product revenue.
Advertising Costs
The Company expenses advertising costs as incurred. Advertising costs were approximately $1.2 million for the year ended December 31, 2015. Advertising costs for the years ended December 31, 2017 and 2016 were de minimis.
Research and Development
Research and development costs are expensed as incurred. Clinical study costs are accrued over the service periods specified in the contracts and adjusted as necessary based upon an ongoing review of the level of effort and costs actually incurred. Payments for a product license prior to regulatory approval of the product and payments for milestones achieved prior to regulatory approval of the product are expensed in the period incurred as research and development. Milestone payments made in connection with regulatory approvals are capitalized and amortized to cost of revenue over the remaining useful life of the asset.
The Company performs research and development for US government agencies under a cost-reimbursable contract in which the Company is reimbursed for direct costs incurred plus allowable indirect costs. The Company recognizes the reimbursements under research contracts when a contract has been executed, the contract price is fixed and determinable, delivery of services or products has occurred and collection of the contract price is reasonably assured. The reimbursements are classified as an offset to research and development expenses. Payments received in advance of work performed are deferred. The Company recorded approximately $9.0 million, $15.8 million and $22.5 million of reimbursements by the government as a reduction of research and development expenses for the years ended December 31, 2017, 2016 and 2015, respectively.
Share-Based Compensation
The Company recognizes expense using the accelerated expense attribution method in an amount equal to the fair value of all share-based awards granted to employees. The Company estimates the fair value of its options on the date of grant using the Black-Scholes closed-form option-pricing model.
Expected volatilities are based principally on historic volatility of the Company’s common stock. The Company uses historical data to estimate forfeiture rate. The expected term of options represents the period of time that options granted are expected to be outstanding. The Company has made a determination of expected term by analyzing employees’ historical exercise experience. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant corresponding with the expected life of the options.
Foreign Currencies
The functional currencies of the Company’s foreign subsidiaries primarily are the local currencies: Euro, Swiss franc, and British pound sterling. The Company’s assets and liabilities are translated using the current exchange rate as of the balance sheet date. Stockholders’ equity is translated using historical rates at the balance sheet date. Revenues and expenses and other items of income are translated using a weighted average exchange rate over the period ended on the balance sheet date. Adjustments resulting from the translation of the financial statements of the Company’s foreign subsidiaries into U.S. dollars are excluded from the determination of net earnings (loss) and are accumulated in a separate component of stockholders’ equity. Foreign exchange transaction gains and losses are included in other income (loss) in the Company’s results of operations.
Income Taxes
The Company accounts for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.
The Company records net deferred tax assets to the extent it believes these assets will more likely than not be realized. On a periodic basis, the Company evaluates the realizability of its deferred tax assets net of deferred tax liabilities and adjusts such amounts in light of changing facts and circumstances, including but not limited to its level of past and future taxable income, the current and future expected utilization of tax benefit carryforwards, any regulatory or legislative actions by relevant authorities with respect to the Angiomax patents, and the status of litigation with respect to those patents. The Company considers all available
F - 17
evidence, both positive and negative, to determine whether, based on the weight of that evidence, a valuation allowance is required to reduce the net deferred tax assets to the amount that is more likely than not to be realized in future periods.
The Company’s annual effective tax rate is based on pre-tax earnings adjusted for differences between GAAP and income tax accounting, existing statutory tax rates, limitations on the use of net operating loss and tax credit carryforwards and tax planning opportunities available in the jurisdictions in which it operates.
The Company records uncertain tax positions on the basis of a two-step process whereby (1) it determines whether it is more likely than not that a tax position will be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position; and (2) for tax positions that meets the more-likely-than-not recognition threshold, the Company recognizes the largest amount of benefit that is greater than 50% likely of being realized upon ultimate settlement with the relevant tax authority. Significant judgment is required in evaluating the Company’s tax position. Settlement of filing positions that may be challenged by tax authorities could impact the income tax position in the year of resolution. The Company’s liability for uncertain tax positions is reflected as a reduction to its deferred tax assets on its consolidated balance sheet.
Comprehensive Income (Loss)
The Company’s accumulated comprehensive income (loss) is comprised of unrealized gains and losses on available for sale securities (if any), which are recorded and presented net of income tax, and foreign currency translation.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued a comprehensive new revenue recognition Accounting Standards Update (ASU), “Revenue from Contracts with Customers (Topic 606)” (ASU No. 2014-09). ASU No. 2014-09 provides guidance to clarify the principles for recognizing revenue. This guidance includes the required steps to achieve the core principle that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. In August 2015, the FASB deferred the effective date of the revenue recognition guidance to reporting periods beginning after December 15, 2017. Early adoption of the standard is permitted but not before the original effective date, which was for reporting periods beginning after December 15, 2016. The FASB has further amended guidance related to recording revenue on a gross versus a net basis and on identifying performance obligations and licensing. The FASB has also rescinded certain SEC guidance primarily related to ASC Topic 815, “Derivatives and Hedging,” and has issued additional improvements and practical expedients to the standard.
The Company, has analyzed the impacts of ASU No. 2014-09 on its revenue streams, specifically focusing on its product revenues, including its arrangement with Sandoz to sell in the United States an authorized generic version of Angiomax (bivalirudin). The Company’s assessment included a review of current accounting policies and practices to identify potential differences that would result from applying the guidance. Currently, the Company uses a deferred revenue model for sales of its authorized generic version of Angiomax (bivalirudin) as the price is not fixed or determinable and recognizes royalty revenue on an accrual basis in the period in which Sandoz reports sales to its customers. The Company currently records revenue recognized from sales of bivalirudin to Sandoz in both net product revenues and royalty revenues in its consolidated statements of operations. Under the new standard, the promise to provide bivalirudin to Sandoz and the promise to provide exclusivity to Sandoz to distribute bivalirudin in the United States will constitute one performance obligation; as a result, under the new standard, revenue recognized from sales of bivalirudin to Sandoz will be recorded solely in net product revenues in the Company’s consolidated statements of operations upon transfer of control of product to Sandoz. The transaction price will reflect the amount the Company expects to be entitled to in connection with the sale of bivalirudin to Sandoz, which will include an estimate of the variable amount of the consideration subject to the constraint that it must be probable that a significant reversal of revenue will not occur. This may result in revenue being recognized earlier provided the Company can reliably estimate the ultimate price expected to be realized from Sandoz’s customer. The Company has concluded that the adoption of this guidance will not have a material impact on its consolidated financial statements. However, the Company will be required to provide additional disclosures in the notes to the consolidated financial statements upon adoption. The Company will adopt the standard using the modified retrospective method.
In January 2016, the FASB issued ASU No. 2016-01, “Financial Instruments - Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities” (ASU No. 2016-01). ASU No. 2016-01 amends certain aspects of accounting and disclosure requirements of financial instruments, including the requirement that equity investments with readily determinable fair values be measured at fair value with changes in fair value recognized in a company’s results of operations. The new standard does not apply to investments accounted for under the equity method of accounting or those that result in consolidation of the investee. Equity investments that do not have readily determinable fair values may be measured at fair value or at cost minus
F - 18
impairment adjusted for changes in observable prices. A financial liability that is measured at fair value in accordance with the fair value option is required to be presented separately in other comprehensive income for the portion of the total change in the fair value resulting from change in the instrument-specific credit risk. In addition, a valuation allowance should be evaluated on deferred tax assets related to available-for-sale debt securities in combination with other deferred tax assets. ASU No. 2016-01 will be effective for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2017. Based on current investment holdings, the Company does not believe the adoption of this standard is expected to have a material impact on the consolidated balance sheets and statements of operations.
In February 2016, the FASB issued ASU No. 2016-02, “Leases (Topic 842)” (ASU No. 2016-02). ASU No. 2016-02 will require organizations that lease assets with lease terms of more than 12 months to recognize assets and liabilities for the rights and obligations created by those leases on their balance sheets. The ASU will also require new qualitative and quantitative disclosures to help investors and other financial statement users better understand the amount, timing, and uncertainty of cash flows arising from leases. ASU No. 2016-02 will be effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018, with early adoption permitted. The Company expects to adopt this guidance when effective and is currently evaluating the effect that the updated standard will have on its consolidated financial statements and related disclosures.
In March 2016, the FASB issued ASU No. 2016-09, “Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting” (ASU No. 2016-09). This ASU makes several modifications to Topic 718 related to the accounting for forfeitures, employer tax withholding on share-based compensation, and the financial statement presentation of excess tax benefits or deficiencies. ASU No. 2016-09 also clarifies the statement of cash flows presentation for certain components of share-based awards. On January 1, 2017, the Company adopted ASU No. 2016-09 and has elected to continue its determination of compensation costs recognized in each period based upon an estimate of expected future forfeitures. Upon the settlement of awards during the year ended December 31, 2017, the Company recorded excess tax benefits of $3.0 million but was unable to recognize any benefit due to the establishment of a valuation allowance on its net operating loss carry forward deferred tax assets. There was no net impact on the Company’s opening accumulated deficit upon application of this guidance using the modified retrospective transition method as the total cumulative-effect adjustment for previously deferred excess tax benefits was offset by a related change in the valuation allowance. The other amended requirements of ASU No. 2016-09 did not have a material impact on the Company’s consolidated financial statements and related disclosures.
In August 2016, the FASB issued ASU No. 2016-15, “Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments” (ASU No. 2016-15). This guidance clarifies how certain cash receipts and payments should be presented in the statement of cash flows and is effective for interim and annual reporting periods beginning after December 15, 2017, with early adoption permitted. The Company does not believe that this guidance will have an impact on the consolidated financial statements and related disclosures.
In November 2016, the FASB issued ASU 2016-18, “Statement of Cash Flows (Topic 230): Restricted Cash” (ASU No. 2016-18). This amends the guidance in ASC 230, including providing additional guidance related to transfers between cash and restricted cash and how entities present, in their statement of cash flows, the cash receipts and cash payments that directly affect the restricted cash accounts. ASU 2016-18 is effective for annual reporting periods beginning after December 15, 2017, and interim periods within those years, with early adoption permitted. The Company does not believe that this guidance will have an impact on the consolidated financial statements and related disclosures.
In January 2017, the FASB issued ASU 2017-01, Business Combinations (Topic 805): Clarifying the Definition of a Business, which clarifies the definition of a business with the objective of adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions (or disposals) of assets or businesses. ASU 2017-01 is effective for annual reporting periods beginning after December 15, 2017, and interim periods within those years, with early adoption permitted. The Company will apply the guidance to applicable transactions after the adoption date. The impact on the Company’s consolidated financial statements will depend on the facts and circumstances of any specific future transactions.
In January 2017, the FASB issued ASU 2017-04, Intangibles-Goodwill and Other, Simplifying the Test for Goodwill Impairment, which eliminates Step 2 from the goodwill impairment test. Under the revised test, an entity should perform its annual, or interim, goodwill impairment test by comparing the fair value of a reporting unit with its carrying amount. An entity should recognize an impairment charge for the amount by which the carrying amount exceeds the reporting unit’s fair value; however, the loss recognized should not exceed the total amount of goodwill allocated to that reporting unit. This ASU is effective for any interim or annual impairment tests for fiscal years beginning after December 15, 2019, with early adoption permitted. The Company does not believe that this guidance will have an impact on the consolidated financial statements and related disclosures.
F - 19
3. Inventory
The major classes of inventory were as follows:
2017 | 2016 | |||||||
(In thousands) | ||||||||
Raw materials | $ | 1,389 | $ | 18,714 | ||||
Work-in-progress | 3,608 | 8,397 | ||||||
Finished goods | 562 | 1,375 | ||||||
Total | $ | 5,559 | $ | 28,486 | ||||
The Company reviews inventory, including inventory purchase commitments, for slow moving or obsolete amounts based on expected product sales volume and provides reserves against the carrying amount of inventory as appropriate. For the year ended December 31, 2017, upon review of expected future product sales volumes and the projected expiration of inventory, the Company recorded a $16.7 million reserve for potential inventory obsolescence, mainly related to Angiomax.
For the year ended December 31, 2016, upon review of expected future product sales volumes and the projected expiration of certain components of Ionsys, the Company recorded an $8.5 million reserve for potential inventory obsolescence.
4. Fixed Assets
Fixed assets consist of the following:
Estimated | December 31, | ||||||||
Life (Years) | 2017 | 2016 | |||||||
(In thousands) | |||||||||
Furniture, fixtures and equipment | 2-15 | $ | 20,603 | $ | 25,132 | ||||
Computer software | 2-5 | 3,524 | 3,722 | ||||||
Computer hardware | 2-5 | 3,054 | 3,795 | ||||||
Leasehold improvements | 2-15 | 33,064 | 30,702 | ||||||
60,245 | 63,351 | ||||||||
Less: Accumulated depreciation | (42,991 | ) | (32,390 | ) | |||||
$ | 17,254 | $ | 30,961 | ||||||
Depreciation expense was approximately $6.8 million, $4.5 million and $5.1 million for the years ended December 31, 2017, 2016 and 2015, respectively.
5. | Cash, Cash Equivalents and Restricted Cash |
The Company considers all highly liquid investments purchased with original maturities at the date of purchase of three months or less to be cash equivalents. At December 31, 2017 and 2016, the Company had cash and cash equivalents of $151.4 million and $541.8 million, respectively, which consisted of cash of $139.3 million and $485.7 million and money market funds with maturities less than three months of $12.1 million and $56.1 million at December 31, 2017 and 2016, respectively.
Restricted Cash
The Company had restricted cash of $5.5 million and $5.0 million at December 31, 2017 and 2016, respectively, which included $4.1 million and $3.7 million reserved for an outstanding letter of credit associated with foreign taxes at December 31, 2017 and 2016, respectively, $1.0 million at both December 31, 2017 and 2016 for an outstanding letter of credit associated with the lease for the office space in Parsippany, New Jersey. The funds are invested in certificates of deposit. The letter of credit permits draws by the landlord to cure defaults by the Company. In addition, as a result of the acquisition of Targanta Therapeutics Corporation (Targanta) in 2009, the Company had restricted cash of $0.2 million and $0.1 million at December 31, 2017 and 2016, respectively, in the form of a guaranteed investment certificate collateralizing an available credit facility. The Company also had restricted cash of $0.3 million and $0.2 million at December 31, 2017 and 2016, respectively, related to certain foreign tender requirements.
F - 20
6. Acquisition
Annovation
On February 2, 2015, the Company completed the acquisition of Annovation, and Annovation became the Company’s wholly owned subsidiary. As a result of the acquisition of Annovation, the Company acquired MDCO-700, a novel intravenous anesthetic.
Under the terms of the acquisition agreement, the Company paid to the holders of Annovation’s capital stock and the holders of options to purchase shares of Annovation’s capital stock, which the Company refers to collectively as the Annovation equityholders, an aggregate of approximately $28.4 million in cash. In addition, the Company may be obligated to pay Annovation’s equityholders up to an additional $26.3 million in milestone payments subsequent to the closing if the Company achieves certain development and regulatory approval milestones at the times and on the conditions set forth in the acquisition agreement. The Company has also agreed to pay Annovation equityholders a low single digit percentage of worldwide net sales, if any, of certain Annovation products, including ABP-700, during a specified earnout period.
The Company accounted for this transaction as a step acquisition which required that the Company remeasure its then existing 35.8% ownership interest (previously accounted for as an equity method investment) to fair value at the acquisition date based upon the total enterprise value, adjusting for a control premium. The fair value of the Company’s interest in Annovation was $25.9 million at closing, resulting in a non-cash pre-tax gain of $22.7 million, recorded as gain on remeasurement of equity investment in the Company’s accompanying consolidated statements of operations. The Company’s previously recorded equity method investment in Annovation was derecognized from the Company’s consolidated balance sheets. Since the date of the step acquisition, the financial results of Annovation were included within the Company’s consolidated financial statements. In accordance with the acquisition method of accounting, the Company allocated the acquisition cost for the Annovation transaction to the underlying assets acquired and liabilities assumed by the Company, based upon estimated fair values of those assets and liabilities at the date of acquisition and classified the fair value of acquired IPR&D as indefinite-lived assets until the successful completion or abandonment of the associated research and development efforts.
The goodwill recorded as part of the acquisition is primarily related to establishing a deferred tax liability for the IPR&D intangible asset which has no tax basis and, therefore, will not result in a future tax deduction. The Company does not expect any portion of this goodwill to be deductible for tax purposes. The Company did not incur any significant acquisition related costs in connection with the Annovation acquisition during 2015.
In addition, as a result of the Company’s acquisition of Annovation, it, through its subsidiary Annovation, is a party to a license agreement with The General Hospital Corporation. Under the agreement, the Company will be obligated to pay General Hospital Corporation up to an aggregate of $6.5 million upon achievement of specified development, regulatory and sales milestones. The Company will also be obligated to pay General Hospital Corporation low single-digit percentage royalties on a product-by-product and country-by-country basis based on net sales of ABP-700 products until the later of the duration of the licensed patent rights which are necessary to manufacture, use or sell ABP-700 products in a country and the date ten years from the Company’s first commercial sale of ABP-700 products in such country.
Total purchase price, in thousands, is summarized as follows:
Upfront cash consideration | $ | 28,397 | ||
Fair value of existing equity interest in Annovation | 25,886 | |||
Total cash consideration and fair value of existing equity interest | 54,283 | |||
Fair value of contingent cash payment | 18,000 | |||
Total purchase price | $ | 72,283 | ||
F - 21
Below is a summary which details, in thousands, the allocation of assets acquired and liabilities assumed as a result of this acquisition:
Assets acquired: | ||||
Cash and cash equivalents | $ | 1,482 | ||
Other current assets | 692 | |||
IPR&D | 65,000 | |||
Goodwill | 24,530 | |||
Total assets | $ | 91,704 | ||
Liabilities assumed: | ||||
Accrued expenses | $ | 398 | ||
Contingent purchase price | 18,000 | |||
Deferred tax liability, net | 19,023 | |||
Total liabilities | $ | 37,421 | ||
Total cash price paid upon acquisition and fair value of existing equity interest | $ | 54,283 | ||
Pro forma results of operations for the acquisition of Annovation have not been presented because this acquisition is not material to the Company’s consolidated results of operations.
In August 2017, the Company announced that it discontinued the clinical development program for MDCO-700. In connection with this decision, the Company’s consolidated statement of operations for the year ended December 31, 2017 includes the following non-cash adjustments that were recorded during the second quarter of 2017: $65.0 million of asset impairment charges related to IPR&D acquired from Annovation, a $14.7 million decrease in the carrying value of the contingent purchase price to an estimated fair value of zero, and a $23.0 million benefit for income taxes due to a reduction in the Company’s recorded valuation allowance against its deferred tax assets as a result of the impairment charge.
7. | Intangible Assets and Goodwill |
The following table sets forth the carrying amounts and accumulated amortization of the Company’s intangible assets:
As of December 31, 2017 | As of December 31, 2016 | |||||||||||||||||||||||
Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | |||||||||||||||||||
(In thousands) | ||||||||||||||||||||||||
Amortizable intangible assets | ||||||||||||||||||||||||
Product licenses(1) | $ | — | $ | — | $ | — | $ | 30,000 | $ | (3,013 | ) | $ | 26,987 | |||||||||||
Developed product rights(2) | — | — | — | 250,000 | (19,802 | ) | 230,198 | |||||||||||||||||
Total | $ | — | $ | — | $ | — | $ | 280,000 | $ | (22,815 | ) | $ | 257,185 | |||||||||||
Intangible assets not subject to amortization: | ||||||||||||||||||||||||
In-process research & development | — | — | — | 65,000 | — | 65,000 | ||||||||||||||||||
Total intangible assets not subject to amortization: | — | — | — | 65,000 | — | 65,000 | ||||||||||||||||||
Total intangible assets | $ | — | $ | — | $ | — | $ | 345,000 | $ | (22,815 | ) | $ | 322,185 | |||||||||||
_______________________________________
(1) | The Company amortizes intangible assets related to the product licenses over their expected useful lives. |
(2) | The Company amortizes intangible assets related to developed product rights over the remaining life of the patents. |
F - 22
In the second quarter of 2017, the Company recorded impairment charges of $226.5 million and $26.2 million to reduce the unamortized carrying amounts of the developed product rights and product licenses, respectively, associated with Ionsys to their estimated fair values of zero which is a Level 3 fair value measurement, as a result of the discontinuation and market withdrawal of Ionsys which became effective on June 19, 2017. In the second quarter of 2017, the Company also recorded impairment charges of $65.0 million to reduce the carrying amount of the IPR&D associated with MDCO-700 to an estimated fair value of zero, which is a Level 3 fair value measurement, in connection with management’s decision to discontinue the MDCO-700 trials. These impairment charges were recorded in asset impairment charges in the accompanying consolidated statements of operations. See Note 1, “Nature of Business,” for further details and Note 14, “Fair Value Measurements,” for definitions of hierarchy levels.
Amortization expense was $4.5 million, $17.5 million and $10.0 million for the years ended December 31, 2017, 2016 and 2015, respectively. The Company records amortization expense in cost of revenue in the accompanying consolidated statements of operations.
The changes in the carrying amount of goodwill for the years ended December 31, 2017 and 2016 are as follows:
December 31, 2017 | December 31, 2016 | ||||||
(In thousands) | |||||||
Balance at beginning of period | $ | 200,571 | $ | 234,383 | |||
Allocation of goodwill to the Non-Core ACC Products | — | (33,812 | ) | ||||
Balance at end of period | $ | 200,571 | $ | 200,571 | |||
In the second quarter of 2016, the Company allocated approximately $33.8 million of its goodwill to the sale of the Non-Core ACC Products. See Note 22, “Dispositions,” for further details on the sale of the Non-Core ACC Products.
8. | Accrued Expenses |
Accrued expenses consisted of the following at December 31, 2017 and 2016:
2017 | 2016 | ||||||
(In thousands) | |||||||
Royalties | $ | 1,039 | $ | 739 | |||
Research and development services | 43,496 | 11,477 | |||||
Compensation related | 25,621 | 28,802 | |||||
Product returns, rebates and other fees | 5,363 | 2,336 | |||||
Legal, accounting and other | 6,162 | 4,821 | |||||
Manufacturing, logistics and related fees | 1,984 | 6,200 | |||||
Sales and marketing | 1,875 | 1,575 | |||||
Interest | 9,657 | 10,888 | |||||
Total | $ | 95,197 | $ | 66,838 | |||
9. Convertible Senior Notes
Convertible Senior Notes Due 2023
In June 2016, the Company issued, at par value, $402.5 million aggregate principal amount of 2.75% convertible senior notes due 2023 (the 2023 Notes). The 2023 Notes bear cash interest at a rate of 2.75% per year, payable semi-annually on January 15 and July 15 of each year, beginning on January 15, 2017. The 2023 Notes will mature on July 15, 2023. The net proceeds to the Company from the offering were $390.8 million after deducting the initial purchasers’ discounts and commissions and the offering expenses payable by the Company.
The 2023 Notes are governed by an indenture (the 2023 Notes Indenture) with Wells Fargo Bank, National Association, a national banking association, as trustee (the 2023 Notes Trustee).
F - 23
The 2023 Notes are senior unsecured obligations of the Company and will rank senior in right of payment to the Company’s future indebtedness that is expressly subordinated in right of payment to the 2023 Notes; equal in right of payment to the Company’s existing and future unsecured indebtedness that is not so subordinated; effectively junior in right of payment to any of the Company’s secured indebtedness to the extent of the value of the assets securing such indebtedness; and structurally junior to all existing and future indebtedness and other liabilities (including trade payables) incurred by the Company’s subsidiaries.
Holders may convert their 2023 Notes at their option at any time prior to the close of business on the business day immediately preceding April 15, 2023 only under the following circumstances:
• | during any calendar quarter commencing on or after September 30, 2016 (and only during such calendar quarter), if the last reported sale price of the Company’s common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; |
• | during the five business day period after any five consecutive trading day period (the ‘‘measurement period’’) in which the trading price (as defined in the 2023 Notes Indenture) per $1,000 principal amount of notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of the Company’s common stock and the conversion rate on each such trading day; |
• | during any period after the Company has issued notice of redemption until the close of business on the scheduled trading day immediately preceding the relevant redemption date; or |
• | upon the occurrence of specified corporate events. |
On or after April 15, 2023, until the close of business on the second scheduled trading day immediately preceding the maturity date, holders may convert their 2023 Notes at any time, regardless of the foregoing circumstances. Upon conversion, the Company will pay or deliver, as the case may be, cash, shares of the Company’s common stock or a combination thereof, at the Company’s option, based upon a daily conversion value calculated on a proportionate basis for each trading day in a 50 trading day observation period (as more fully described in the 2023 Notes Indenture). The conversion rate for the 2023 Notes was initially, and remains, 20.4198 shares of the Company’s common stock per $1,000 principal amount of the 2023 Notes, which is equivalent to an initial conversion price of approximately $48.97 per share of the Company’s common stock.
The Company may not redeem the 2023 Notes prior to July 15, 2020. The Company may redeem for cash all or any portion of the 2023 Notes, at its option, on or after July 15, 2020 if the last reported sale price of its common stock has been at least 130% of the conversion price then in effect on the last trading day of, and for at least 19 other trading days (whether or not consecutive) during, any 30 consecutive trading day period ending on, and including, the trading day immediately preceding the date on which the Company provides notice of redemption, at a redemption price equal to 100% of the principal amount of the 2023 Notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. No redemption date may be designated that falls on or after the 52nd scheduled trading date prior to maturity. No sinking fund is provided for the 2023 Notes, which means that the Company is not required to redeem or retire the 2023 Notes periodically.
If the Company undergoes a fundamental change (as defined in the 2023 Notes Indenture), subject to certain conditions, holders of the 2023 Notes may require the Company to repurchase for cash all or part of their 2023 Notes at a repurchase price equal to 100% of the principal amount of the 2023 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date.
The 2023 Notes Indenture governing the 2023 Notes contains customary events of default with respect to the 2023 Notes, including that upon certain events of default (including the Company’s failure to make any payment of principal or interest on the 2023 Notes when due and payable) occurring and continuing, the 2023 Notes Trustee by notice to the Company, or the holders of at least 25% in principal amount of the outstanding 2023 Notes by notice to the Company and the 2023 Notes Trustee, may, and the 2023 Notes Trustee at the request of such holders (subject to the provisions of the 2023 Notes Indenture) shall, declare 100% of the principal of and accrued and unpaid interest, if any, on all the 2023 Notes to be due and payable. In case of certain events of bankruptcy, insolvency or reorganization, involving the Company or a significant subsidiary, 100% of the principal of and accrued and unpaid interest on the 2023 Notes will automatically become due and payable. Upon such a declaration of acceleration, such principal and accrued and unpaid interest, if any, will be due and payable immediately.
F - 24
In accounting for the issuance of the 2023 Notes, the Company separated the 2023 Notes into liability and equity components. The carrying amount of the liability component was calculated by measuring the fair value of a similar liability that does not have an associated convertible feature. The carrying amount of the equity component representing the conversion option was determined by deducting the fair value of the liability component from the par value of the 2023 Notes as a whole. The excess of the principal amount of the liability component over its carrying amount, referred to as the debt discount, is amortized to interest expense over the seven-year term of the 2023 Notes. The equity component is not re-measured as long as it continues to meet the conditions for equity classification. The equity component related to the 2023 Notes is $101.0 million and is recorded in additional paid-in capital on the accompanying consolidated balance sheet.
In accounting for the transaction costs related to the issuance of the 2023 Notes, the Company allocated the total costs incurred to the liability and equity components of the 2023 Notes based on their relative values. Transaction costs attributable to the liability component are amortized to interest expense over the seven-year term of the 2023 Notes, and transaction costs attributable to the equity component are netted with the equity components in stockholders’ equity. Additionally, the Company initially recorded a net deferred tax liability of $33.5 million in connection with the 2023 Notes.
The 2023 Notes consist of the following:
Liability component | December 31, 2017 | December 31, 2016 | ||||||
(in thousands) | ||||||||
Principal | $ | 402,500 | $ | 402,500 | ||||
Less: Debt discount, net(1) | (90,552 | ) | (103,162 | ) | ||||
Net carrying amount | $ | 311,948 | $ | 299,338 | ||||
_______________________________________
(1) | Included in the accompanying consolidated balance sheets within convertible senior notes (due 2023) and amortized to interest expense over the remaining life of the 2023 Notes using the effective interest rate method. |
The fair value of the 2023 Notes was approximately $377.1 million as of December 31, 2017. The Company estimates the fair value of its 2023 Notes utilizing market quotations for debt that have quoted prices in active markets. Since the 2023 Notes do not trade on a daily basis in an active market, the fair value estimates are based on market observable inputs based on borrowing rates currently available for debt with similar terms and average maturities, which are classified as Level 2 measurements within the fair value hierarchy. See Note 14, “Fair Value Measurements,” for definitions of hierarchy levels. As of December 31, 2017, the remaining contractual life of the 2023 Notes is approximately 5.5 years.
The following table sets forth total interest expense recognized related to the 2023 Notes:
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(in thousands) | |||||||||||
Contractual interest expense | $ | 11,060 | $ | 6,158 | $ | — | |||||
Amortization of debt discount | 12,610 | 6,648 | — | ||||||||
Total | $ | 23,670 | $ | 12,806 | $ | — | |||||
Effective interest rate of the liability component | 7.5 | % | 7.5 | % | — | % | |||||
Capped Call Transactions
In June 2016, the Company entered into capped call transactions with certain counterparties of the 2023 Notes or their respective affiliates or other financial institutions. The Company used approximately $33.9 million of the net proceeds from the offering to pay the cost of the capped call transactions, which is included as a net reduction to additional paid-in capital on the accompanying consolidated balance sheet.
The capped call transactions are expected to reduce the potential dilution with respect to shares of the Company’s common stock upon any conversion of the 2023 Notes and/or offset any cash payments the Company is required to make in excess of the principal amount of converted 2023 Notes, as the case may be, if the market price of the Company’s common stock is then greater than the strike price of the capped call transactions. Such reduction of potential dilution or offset of cash payments is subject to a cap based on the cap price of the capped call transactions. The cap price of the capped calls is currently $64.68.
F - 25
For any conversions of the 2023 Notes prior to the close of business on the 52nd scheduled trading day immediately preceding the stated maturity date of the 2023 Notes, including without limitation upon an acquisition of the Company or similar business combination, a corresponding portion of the capped calls will be terminated. Upon such termination, the portion of the capped calls being terminated will be settled at fair value (subject to certain limitations), as determined by the counterparties to the capped calls and no payments will be due from the Company to such counterparties. The capped calls expire on the earlier of (i) the last day on which any Convertible Securities remain outstanding and (ii) the second “Scheduled Trading Day” (as defined in the 2023 Notes Indenture) immediately preceding the “Maturity Date” (as defined in the 2023 Notes Indenture).
Convertible Senior Notes Due 2022
In January 2015, the Company issued, at par value, $400.0 million aggregate principal amount of 2.5% convertible senior notes due 2022 (2022 Notes). The 2022 Notes bear cash interest at a rate of 2.5% per year, payable semi-annually on January 15 and July 15 of each year, beginning on July 15, 2015. The 2022 Notes will mature on January 15, 2022. The net proceeds to the Company from the offering were $387.2 million after deducting the initial purchasers’ discounts and commissions and the offering expenses payable by the Company.
The 2022 Notes are governed by an indenture (the 2022 Notes Indenture) with Wells Fargo Bank, National Association, a national banking association, as trustee (the 2022 Notes Trustee).
The 2022 Notes are senior unsecured obligations of the Company and will rank senior in right of payment to the Company’s future indebtedness that is expressly subordinated in right of payment to the 2022 Notes; equal in right of payment to the Company’s existing and future unsecured indebtedness that is not so subordinated; effectively junior in right of payment to any of the Company’s secured indebtedness to the extent of the value of the assets securing such indebtedness; and structurally junior to all existing and future indebtedness and other liabilities (including trade payables) incurred by the Company’s subsidiaries.
Holders may convert their 2022 Notes at their option at any time prior to the close of business on the business day immediately preceding October 15, 2021 only under the following circumstances:
• | during any calendar quarter commencing on or after March 31, 2015 (and only during such calendar quarter), if the last reported sale price of the Company’s common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; |
• | during the five business day period after any five consecutive trading day period (the measurement period) in which the trading price (as defined in the 2022 Notes Indenture) per $1,000 principal amount of 2022 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of the Company’s common stock and the conversion rate on each such trading day; |
• | during any period after the Company has issued notice of redemption until the close of business on the scheduled trading day immediately preceding the relevant redemption date; or |
• | upon the occurrence of specified corporate events. |
On or after October 15, 2021, until the close of business on the second scheduled trading day immediately preceding the maturity date, holders may convert their 2022 Notes at any time, regardless of the foregoing circumstances. Upon conversion, the Company will pay cash up to the aggregate principal amount of the 2022 Notes to be converted and deliver shares of its common stock in respect of the remainder, if any, of its conversion obligation in excess of the aggregate principal amount of 2022 Notes being converted, subject to a daily share cap.
The conversion rate for the 2022 Notes was initially, and remains, 29.8806 shares of the Company’s common stock per $1,000 principal amount of the 2022 Notes, which is equivalent to an initial conversion price of approximately $33.47 per share of the Company’s common stock.
The Company may not redeem the 2022 Notes prior to January 15, 2019. The Company may redeem for cash all or any portion of the 2022 Notes, at its option, on or after January 15, 2019 if the last reported sale price of its common stock has been at least 130% of the conversion price then in effect on the last trading day of, and for at least 19 other trading days (whether or not consecutive) during, any 30 consecutive trading day period ending on, and including, the trading day immediately preceding the date on which the Company provides notice of redemption, at a redemption price equal to 100% of the principal amount of the
F - 26
2022 Notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. No sinking fund is provided for the 2022 Notes, which means that the Company is not required to redeem or retire the 2022 Notes periodically.
If the Company undergoes a “fundamental change” (as defined in the Indenture governing the 2022 Notes Indenture), subject to certain conditions, holders of the 2022 Notes may require the Company to repurchase for cash all or part of their 2022 Notes at a repurchase price equal to 100% of the principal amount of the 2022 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date.
The 2022 Notes Indenture contains customary events of default with respect to the 2022 Notes, including that upon certain events of default (including the Company’s failure to make any payment of principal or interest on the 2022 Notes when due and payable) occurring and continuing, the 2022 Notes Trustee by notice to the Company, or the holders of at least 25% in principal amount of the outstanding 2022 Notes by notice to the Company and the 2022 Notes Trustee, may, and the 2022 Notes Trustee at the request of such holders (subject to the provisions of the 2022 Notes Indenture) shall, declare 100% of the principal of and accrued and unpaid interest, if any, on all the 2022 Notes to be due and payable. In case of certain events of bankruptcy, insolvency or reorganization, involving the Company or a significant subsidiary, 100% of the principal of and accrued and unpaid interest on the 2022 Notes will automatically become due and payable. Upon such a declaration of acceleration, such principal and accrued and unpaid interest, if any, will be due and payable immediately.
In accounting for the issuance of the 2022 Notes, the Company separated the 2022 Notes into liability and equity components. The carrying amount of the liability component was calculated by measuring the fair value of a similar liability that does not have an associated convertible feature. The carrying amount of the equity component representing the conversion option was determined by deducting the fair value of the liability component from the par value of the 2022 Notes as a whole. The excess of the principal amount of the liability component over its carrying amount, referred to as the debt discount, is amortized to interest expense over the seven-year term of the 2022 Notes. The equity component is not re-measured as long as it continues to meet the conditions for equity classification. The equity component related to the 2022 Notes is $88.9 million and is recorded in additional paid-in capital on the accompanying consolidated balance sheets.
In accounting for the transaction costs related to the issuance of the 2022 Notes, the Company allocated the total costs incurred to the liability and equity components of the 2022 Notes based on their relative values. Transaction costs attributable to the liability component are amortized to interest expense over the seven-year term of the 2022 Notes, and transaction costs attributable to the equity component are netted with the equity components in stockholders’ equity. Additionally, the Company initially recorded a net deferred tax liability of $31.8 million in connection with the 2022 Notes.
The 2022 Notes consist of the following:
Liability component | December 31, 2017 | December 31, 2016 | ||||||
(In thousands) | ||||||||
Principal | $ | 399,997 | $ | 400,000 | ||||
Less: Debt discount, net(1) | (62,747 | ) | (75,754 | ) | ||||
Net carrying amount | $ | 337,250 | $ | 324,246 | ||||
_______________________________________
(1) | Included on the accompanying consolidated balance sheets within convertible senior notes (due 2022) and amortized to interest expense over the remaining life of the 2022 Notes using the effective interest rate method. |
The fair value of the 2022 Notes was approximately $417.6 million as of December 31, 2017. The Company estimates the fair value of its 2022 Notes utilizing market quotations for debt that have quoted prices in active markets. Since the 2022 Notes do not trade on a daily basis in an active market, the fair value estimates are based on market observable inputs based on borrowing rates currently available for debt with similar terms and average maturities, which are classified as Level 2 measurements within the fair value hierarchy. See Note 14, “Fair Value Measurements,” for definitions of hierarchy levels. As of December 31, 2017, the remaining contractual life of the 2022 Notes is approximately 4.0 years.
F - 27
The following table sets forth total interest expense recognized related to the 2022 Notes:
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Contractual interest expense | $ | 10,000 | $ | 10,000 | $ | 9,639 | |||||
Amortization of debt discount | 13,007 | 12,139 | 10,942 | ||||||||
Total | $ | 23,007 | $ | 22,139 | $ | 20,581 | |||||
Effective interest rate of the liability component | 6.50 | % | 6.50 | % | — | % | |||||
Convertible Senior Notes Due 2017
In June 2012, the Company issued, at par value, $275.0 million aggregate principal amount of 1.375% convertible senior notes due June 1, 2017 (2017 Notes). The 2017 Notes bore cash interest at a rate of 1.375% per year, payable semi-annually on June 1 and December 1 of each year, beginning on December 1, 2012. The 2017 Notes matured on June 1, 2017. The net proceeds to the Company from the offering were $266.2 million after deducting the initial purchasers’ discounts and commissions and the offering expenses payable by the Company.
In June 2016, the Company used approximately $323.2 million of the net proceeds of the 2023 Notes to repurchase $220.0 million in aggregate principal amount of the 2017 Notes in privately negotiated transactions effected through the initial purchasers of the 2017 Notes. As part of the repurchase of the 2017 Notes, the Company settled a proportionate amount of outstanding bond hedges and warrants related to the 2017 Notes for a net cash receipt of $12.6 million. The Company recorded a loss of $5.4 million on the extinguishment of debt in the accompanying consolidated statements of operations during the year ended December 31, 2016 and accounted for the difference of $108.7 million between the consideration transferred to the holder and the fair value of the liability component of the 2017 Notes as a reduction of additional paid-in capital on the accompany consolidated balance sheet.
The 2017 Notes that remained outstanding after the 2016 repurchase matured on June 1, 2017. In connection with the maturity of the 2017 Notes, the holders converted substantially all of the outstanding principal amount of the 2017 Notes, the Company paid cash to the converting 2017 Note holders equal to $55.4 million in respect of principal, interest and fractional shares on the 2017 Notes to be converted and delivered 819,901 shares of the Company’s common stock.
The 2017 Notes consisted of the following:
Liability component | December 31, 2017 | December 31, 2016 | ||||||
(In thousands) | ||||||||
Principal | $ | — | $ | 55,000 | ||||
Less: Debt discount, net(1) | — | (1,251 | ) | |||||
Net carrying amount | $ | — | $ | 53,749 | ||||
_______________________________________
(1) | Included on the accompanying consolidated balance sheets within convertible senior notes (due 2017) and amortized to interest expense over the remaining life of the 2017 Notes using the effective interest rate method. |
The following table sets forth total interest expense recognized related to the 2017 Notes:
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Contractual interest expense | $ | 315 | $ | 2,101 | $ | 3,781 | |||||
Amortization of debt discount | 1,251 | 7,395 | 12,734 | ||||||||
Total | $ | 1,566 | $ | 9,496 | $ | 16,515 | |||||
Effective interest rate of the liability component | 6.02 | % | 6.02 | % | 6.02 | % | |||||
F - 28
Note Hedges
In June 2012, the Company paid an aggregate amount of $58.2 million for the 2017 Note Hedges, which was recorded as a reduction of additional paid-in-capital in stockholders’ equity. As part of the repurchase of $220.0 million in aggregate principal amount of the 2017 Notes, the Company settled the related hedges and received cash of approximately $100.5 million. The remaining 2017 Note Hedges covered approximately two million shares of the Company’s common stock, subject to anti-dilution adjustments substantially similar to those applicable to the 2017 Notes, had a strike price that corresponds to the initial conversion price of the 2017 Notes, and were exercisable upon conversion of the 2017 Notes. The 2017 Note Hedges were separate transactions entered into by the Company with the 2017 Hedge Counterparties and were not part of the terms of the 2017 Notes or the 2017 Warrants. Holders of the 2017 Notes and 2017 Warrants did not have any rights with respect to the 2017 Note Hedges. On June 1, 2017, in connection with the maturity of the 2017 Notes, the Company redeemed the 2017 Note Hedges and received from the Note Hedge counterparties 820,161 shares at a weighted average price of $48.79 per share. The redemption offset the dilution with respect to shares of the Company’s common stock issued upon the conversion of the 2017 Notes. The shares delivered to the Company in connection with the redemption of the 2017 Notes Hedges are held by the Company as treasury shares.
Warrants
In June 2012, the Company received aggregate proceeds of $38.4 million from the sale of warrants to the 2017 Hedge Counterparties, which the Company recorded as additional paid-in-capital in stockholders’ equity. The 2017 Warrants were separate transactions entered into by the Company with the 2017 Hedge Counterparties and are not part of the terms of the 2017 Notes or 2017 Note Hedges. Holders of the 2017 Notes and 2017 Note Hedges did not have any rights with respect to the 2017 Warrants. The 2017 Warrants also meet the definition of a derivative. Because the 2017 Warrants were indexed to the Company’s common stock and recorded in equity in the Company’s consolidated balance sheets, the 2017 Warrants were exempt from the scope and fair value provisions related to accounting for derivative instruments.
As part of the June 2016 repurchase of $220 million in aggregate principal amount of the 2017 Notes, the Company paid $87.9 million to settle the related warrants. The remaining 2017 Warrants, which continued to remain outstanding after the maturity of the 2017 Notes, were to purchase up to approximately two million shares of the Company’s common stock, subject to customary anti-dilution adjustments, at a strike price of $34.20 per share. The 2017 Warrants had a dilutive effect with respect to the Company’s common stock. The 2017 Warrants expired beginning in August 2017 through a series of expiration dates ending in December 2017. The holders of the 2017 Warrants exercised 787,680 warrants on a net basis and as a result the Company issued 44,283 shares of common stock.
10. | Stockholders’ Equity |
Preferred Stock
The Company has 5,000,000 shares of preferred stock (Preferred Stock) authorized, none of which are issued.
Common Stock
Common stockholders are entitled to one vote per share and dividends when declared by the Company’s Board of Directors, subject to the preferential rights of any outstanding shares of Preferred Stock.
Employees and directors of the Company purchased 1,949,117, 1,312,812 and 2,989,324 shares of common stock during the years ended December 31, 2017, 2016 and 2015, respectively, pursuant to option exercises and the Company’s employee stock purchase plan. The aggregate net proceeds to the Company resulting from these purchases were approximately $48.6 million, $33.8 million, and $65.2 million during the years ended December 31, 2017, 2016 and 2015, respectively, and are included within the financing activities section of the accompanying consolidated statements of cash flows. The Company issued 166,103, 132,344 and 166,042 shares under restricted stock awards during the years ended December 31, 2017, 2016 and 2015, respectively.
On May 29, 2015, the Company filed a certificate of amendment to its Third Amended and Restated Certificate of Incorporation with the Secretary of State of the state of Delaware that increased the number of authorized shares of common stock from 125,000,000 shares to 187,500,000 shares.
In August 2015, the Company issued 944,537 shares of its common stock in a private placement. Cash received from the August 2015 private placement totaled $30.0 million and is included within the financing activities section of the accompanying
F - 29
consolidated statements of cash flows. These shares are included in the Company’s weighted average number of common stock outstanding.
Treasury Stock
On June 5, 2012, the Company’s Board of Directors authorized the Company to use a portion of the net proceeds of the 2017 Notes offering to repurchase up to an aggregate of $50.0 million of its common stock. The Company repurchased 2,192,982 shares of its common stock in the second quarter of 2013 for an aggregate cost of $50.0 million.
On June 1, 2017, in connection with the maturity of the 2017 Notes, the Company redeemed the 2017 Note Hedges and received from the Note Hedge counterparties 820,161 shares at a weighted average price of $48.79 per share. The redemption offset the dilution with respect to shares of the Company’s common stock issued upon the conversion of the 2017 Notes. The shared delivered to the Company in connection with the redemption of the 2017 Notes Hedges are held by the Company as treasury shares.
As of December 31, 2017, there were 3,013,143 shares of the Company’s common stock held in treasury.
11. | Share-Based Compensation |
Stock Plans
The Company has adopted the following stock incentive plans under which awards remain outstanding:
• | the 2013 Stock Incentive Plan (the 2013 Plan); |
• | the 2009 Equity Inducement Plan; |
• | the 2007 Equity Inducement Plan; and |
• | the 2004 Stock Incentive Plan. |
These plans provide for the grant of stock options, other stock-based awards (including restricted stock awards, restricted stock units and stock appreciation rights) and cash-based awards to employees, officers, directors, consultants and advisors of the Company and its subsidiaries, including any individuals who have accepted an offer of employment. Stock option grants have an exercise price equal to the fair market value of the Company’s common stock on the date of grant and generally, for employee grants, have a 10-year term and vest 25% one year after grant and thereafter in equal monthly installments over a three-year period. The fair value of stock option grants is recognized, net of an estimated forfeiture rate, using an accelerated method over the vesting period of the options, which is generally four years for employee grants and one year for director grants.
As of December 31, 2017, the Company had granted an aggregate of 28,493,250 shares as restricted stock or subject to issuance upon exercise of stock options under all of the plans, of which 7,043,490 shares remained subject to outstanding options. The Company currently only grants stock options and restricted stock awards from the 2013 Plan. In accordance with ASC 718-10, the Company recorded approximately $31.5 million, $31.0 million and $30.6 million of share-based compensation expense related to the options, restricted stock and ESPP for the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, there was approximately $27.8 million of total unrecognized compensation costs related to non-vested share-based employee compensation arrangements granted under the Company’s equity compensation plans. This cost is expected to be recognized over a weighted average period of 1.22 years.
F - 30
Stock Option and Restricted Stock Award Activity
The following table presents a summary of option activity and data under the Company’s stock incentive plans as of December 31, 2017:
Number of Shares | Weighted-Average Exercise Price Per Share | Weighted- Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | |||||||||
Balance at January 1, 2017 | 8,023,696 | $ | 28.27 | |||||||||
Granted | 1,693,309 | $ | 49.89 | |||||||||
Exercised | (1,854,644 | ) | $ | 24.57 | ||||||||
Forfeited and expired | (818,871 | ) | $ | 37.24 | ||||||||
Outstanding, December 31, 2017 | 7,043,490 | $ | 33.40 | 5.85 | $ | 11,705,399 | ||||||
Vested and expected to vest, December 31, 2017 | 6,842,097 | $ | 33.13 | 5.81 | $ | 11,699,950 | ||||||
Exercisable, December 31, 2017 | 4,190,582 | $ | 28.97 | 5.05 | $ | 11,487,732 | ||||||
Available for future grant at December 31, 2017 | 3,202,401 | |||||||||||
Aggregate intrinsic value is the sum of the amounts by which the quoted market price of the Company’s common stock exceeded the exercise price of the options at December 31, 2017, for those options for which the quoted market price was in excess of the exercise price. The weighted-average grant date fair value of options granted during the years ended December 31, 2017, 2016 and 2015 were $18.46, $11.72, and $11.18, respectively. The total intrinsic value of options exercised during the years ended December 31, 2017, 2016 and 2015 were $34.1 million, $12.7 million, and $40.0 million, respectively.
The Company recorded approximately $22.6 million, $23.2 million, and $23.0 million in compensation expense related to options in the years ended December 31, 2017, 2016 and 2015. The remaining expense of approximately $21.4 million will be recognized over a period of 1.25 years.
For purposes of performing the valuation, employees were separated into two groups according to patterns of historical exercise behavior; the weighted average assumptions below include assumptions from the two groups of employees exhibiting different behavior.
The Company estimated the fair value of each option on the date of grant using the Black-Scholes closed-form option-pricing model applying the weighted average assumptions in the following table.
Years Ended December 31, | ||||||||
2017 | 2016 | 2015 | ||||||
Expected dividend yield | — | % | — | % | — | % | ||
Expected stock price volatility | 39.14 | % | 37.90 | % | 41.49 | % | ||
Risk-free interest rate | 1.867 | % | 1.249 | % | 1.436 | % | ||
Expected option term (years) | 5.00 | 4.93 | 5.01 | |||||
The following table presents a summary of the Company’s outstanding shares of restricted stock awards granted as of December 31, 2017:
Number of Shares | Weighted Average Grant-Date Fair Value | |||||
Balance at January 1, 2017 | 375,301 | $ | 31.88 | |||
Awarded | 196,462 | 50.51 | ||||
Vested | (172,076 | ) | 32.80 | |||
Forfeited | (30,359 | ) | 44.03 | |||
Outstanding, December 31, 2017 | 369,328 | $ | 40.37 | |||
F - 31
The restricted stock granted to employees generally vests in equal increments of 25% per year on an annual basis commencing twelve months after grant date. The restricted stock granted to non-employee directors generally vests on the first anniversary date after the grant date. Expense of approximately $6.5 million, $6.6 million and $6.1 million was recognized related to restricted stock awards in the years ended December 31, 2017, 2016 and 2015, respectively. The remaining expense of approximately $6.3 million will be recognized over a period of 1.13 years. The weighted average grant date fair value of restricted stock awarded during the years ended December 31, 2017, 2016 and 2015 were $50.51, $33.63, and $28.37, respectively. The total fair value of the restricted stock that vested during the years ended December 31, 2017, 2016 and 2015 were $8.4 million, $8.7 million and $7.1 million, respectively.
2010 ESPP
The Company has adopted the 2010 Employee Stock Purchase Plan (the 2010 ESPP), which, as amended, provides for the issuance of up to 2,000,000 shares of common stock. The 2010 ESPP permits eligible employees to purchase shares of common stock at the lower of 85% of the fair market value of the common stock at the beginning or at the end of each offering period. Employees who own 5% or more of the common stock are not eligible to participate in the 2010 ESPP. Participation in the 2010 ESPP is voluntary.
The Company issued 94,473, 136,378, and 184,432 shares under the 2010 ESPP during the years ended December 31, 2017, 2016 and 2015, respectively. The Company recorded approximately $1.0 million, $1.2 million and $1.5 million in compensation expense related to the 2010 ESPP in the years ended December 31, 2017, 2016 and 2015, respectively.
The fair value of each option element of the 2010 ESPP is estimated on the date of grant using the Black-Scholes closed-form option-pricing model applying the weighted average assumptions in the following table. Expected volatilities are based on historical volatility of the Company’s common stock. Expected term represents the six-month offering period for the 2010 ESPP. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant.
Years Ended December 31, | ||||||||
2017 | 2016 | 2015 | ||||||
Expected dividend yield | — | % | — | % | — | % | ||
Expected stock price volatility | 43.03 | % | 48.80 | % | 44.91 | % | ||
Risk-free interest rate | 0.89 | % | 0.34 | % | 0.15 | % | ||
Expected option term (years) | 0.5 | 0.5 | 0.5 | |||||
Common Stock Reserved for Future Issuance
At December 31, 2017, there were 972,959 shares of common stock available for grant under the 2010 ESPP and 3,202,401 shares of common stock available for grant under the 2013 Plan.
F - 32
12. | Earnings per Share |
The following table sets forth the computation of basic and diluted earnings per share for the years ended December 31, 2017, 2016 and 2015.
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands, except per share amounts) | |||||||||||
Amounts attributable to The Medicines Company: | |||||||||||
(Loss) income from continuing operations | $ | (607,695 | ) | $ | 20,564 | $ | (134,806 | ) | |||
Loss from discontinued operations, net of tax | (100,678 | ) | (139,682 | ) | (217,950 | ) | |||||
Net loss attributable to The Medicines Company | $ | (708,373 | ) | $ | (119,118 | ) | $ | (352,756 | ) | ||
. | |||||||||||
Weighted average common shares outstanding, basic | 72,356 | 69,909 | 66,809 | ||||||||
Plus: net effect of dilutive stock options, warrants, restricted common shares and shares issuable upon conversion of Notes | — | 3,113 | — | ||||||||
Weighted average common shares outstanding, diluted | 72,356 | 73,022 | 66,809 | ||||||||
Basic (loss) earnings per common share attributable to The Medicines Company: | |||||||||||
(Loss) earnings from continuing operations | $ | (8.40 | ) | $ | 0.29 | $ | (2.02 | ) | |||
Loss from discontinued operations | (1.39 | ) | (2.00 | ) | (3.26 | ) | |||||
Basic loss per share | $ | (9.79 | ) | $ | (1.71 | ) | $ | (5.28 | ) | ||
Diluted (loss) earnings per common share attributable to The Medicines Company: | |||||||||||
(Loss) earnings from continuing operations | $ | (8.40 | ) | $ | 0.28 | $ | (2.02 | ) | |||
Loss from discontinued operations | (1.39 | ) | (1.91 | ) | (3.26 | ) | |||||
Diluted loss per share | $ | (9.79 | ) | $ | (1.63 | ) | $ | (5.28 | ) | ||
Basic (loss) income per share is computed by dividing consolidated net (loss) income attributable to The Medicines Company by the weighted average number of shares of common stock outstanding during the period, excluding unvested restricted common shares. The potentially dilutive effect of the Company’s stock options, unvested restricted common stock, stock purchase warrants, the 2017 Notes (which matured on June 1, 2017) and 2022 Notes on earnings per share is computed under the treasury stock method. In 2016, the Company analyzed the potential dilutive effect of the 2023 Notes on its earnings per share under the treasury stock method. Beginning in 2017, the Company analyzes the potential dilutive effect of the 2023 Notes on earnings per share under the “if converted” method, in which it is assumed that the outstanding security converts into common stock at the beginning of the period.
For periods of income from continuing operations when the effects are not anti-dilutive, diluted earnings per share is computed by dividing the net income attributable to The Medicines Company by the weighted average number of shares outstanding and the impact of all potential dilutive common shares, consisting primarily of stock options, unvested restricted common stock, shares issuable upon conversion of the 2017 Notes, 2022 Notes and 2023 Notes and stock purchase warrants.
For periods of loss from continuing operations, diluted loss per share is calculated similar to basic loss per share as the effect of including all potentially dilutive common share equivalents is anti-dilutive. Due to the period of loss from continuing operations attributable to The Medicines Company, the calculation of diluted loss per share for the year ended December 31, 2017 and 2016 excluded 12,803,033 and 3,724,272, respectively, of potentially dilutive stock options, warrants, restricted common shares, and shares issuable upon conversion of the 2017 Notes, 2022 Notes and 2023 Notes, as their inclusion would have an anti-dilutive effect.
For periods of income from continuing operations when the effects are not anti-dilutive, diluted earnings per share is computed by dividing the Company’s net income by the weighted average number of shares outstanding and the impact of all potential
F - 33
dilutive common shares, consisting primarily of stock options, unvested restricted common stock, shares issuable upon conversion of the 2017 Notes, 2022 Notes, 2023 Notes and stock purchase warrants.
To minimize the impact of potential dilution upon conversion of the 2023 Notes, the Company entered into capped call transactions separate from the issuance of the 2023 Notes with certain counterparties. The capped calls have a strike price of $48.97 and a cap price of $64.68 and are exercisable when and if the 2023 Notes are converted. If upon conversion of the 2023 Notes, the price of the Company’s common stock is above the strike price of the capped calls, the counterparties will deliver shares of the Company’s common stock and/or cash with an aggregate value equal to the difference between the price of the Company’s common stock at the conversion date and the strike price, multiplied by the number of shares of the Company’s common stock related to the capped calls being exercised. The capped call transactions that are part of the 2023 Notes are not considered for purposes of calculating the total shares outstanding under the basic and diluted net income per share, as their effect would be anti-dilutive.
In June 2012, the Company issued the 2017 Notes (see Note 9, “Convertible Senior Notes”). In connection with the issuance of the 2017 Notes, the Company entered into convertible note hedge transactions with respect to its common stock (2017 Note Hedges) with several of the initial purchasers of the 2017 Notes, their affiliates and other financial institutions (2017 Hedge Counterparties). The options that were part of the 2017 Note Hedges were not considered for purposes of calculating the total shares outstanding under the basic and diluted net income per share, as their effect would be anti-dilutive. In June 2016, as part of the repurchase of $220.0 million in aggregate principal amount of the 2017 Notes, the Company settled the hedges related to the repurchased bonds. On June 1, 2017, in connection with the maturity of the 2017 Notes, the Company redeemed the 2017 Note Hedges and received from the Note Hedge counterparties 819,901 shares at a weighted average price of $48.79 per share. The redemption offset the dilution with respect to shares of the Company’s common stock issued upon the conversion of the 2017 Notes. The shares delivered to the Company in connection with the redemption of the 2017 Notes Hedges are held by the Company as treasury shares. For the year ended December 31, 2015, the number of shares of common stock issuable upon conversion of the 2017 Notes were excluded from the calculation of diluted loss per share as their inclusion would have been anti-dilutive.
In addition, in connection with the 2017 Note Hedges, the Company entered into warrant transactions with the 2017 Hedge Counterparties, pursuant to which the Company sold warrants (2017 Warrants) to the Hedge Counterparties to purchase, subject to customary anti-dilution adjustments, up to two million shares of the Company’s common stock at a strike price of $34.20 per share. The 2017 Warrants had a dilutive effect with respect to the Company’s common stock to the extent that the market price per share of the Company’s common stock, as measured under the terms of the 2017 Warrants, exceeded the applicable strike price of the 2017 Warrants. The Company elected to settle all of the 2017 Warrants in common stock. In June 2016, as part of the repurchase of $220.0 million in aggregate principal amount of the 2017 Notes, the Company settled the warrants related to the repurchased bonds.
13. | Income Taxes |
The benefit from (provision for) income taxes for continuing operations in 2017, 2016 and 2015 consists of current and deferred federal, state and foreign taxes based on income as follows:
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Current: | |||||||||||
Federal | $ | 4,859 | $ | — | $ | (5 | ) | ||||
State | (31 | ) | (33 | ) | (182 | ) | |||||
Foreign | 1,757 | (34 | ) | (229 | ) | ||||||
6,585 | (67 | ) | (416 | ) | |||||||
Deferred: | |||||||||||
Federal | $ | 88,556 | $ | — | $ | 28,011 | |||||
State | 1,435 | — | 2,138 | ||||||||
Foreign | — | — | — | ||||||||
89,991 | — | 30,149 | |||||||||
Total benefit from (provision for) taxes | $ | 96,576 | $ | (67 | ) | $ | 29,733 | ||||
F - 34
The Company’s deferred benefit from income taxes of $89.9 million was primarily attributable to a reduction in the Company’s recorded valuation allowance against its deferred tax assets as a result of the commencement of amortization of IPR&D associated with Vabomere upon approval by the FDA and the impairment of IPR&D associated with MDCO-700.
The components of (loss) income from continuing operations attributable to The Medicines Company before income taxes consisted of:
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Domestic | $ | (704,814 | ) | $ | 22,289 | $ | (163,772 | ) | |||
International | 543 | (1,712 | ) | (757 | ) | ||||||
Total | $ | (704,271 | ) | $ | 20,577 | $ | (164,529 | ) | |||
The difference between tax expense and the amount computed by applying the statutory federal income tax rate of 35% in 2017, 2016, and 2015 to income before income taxes is as follows:
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Statutory rate applied to pre-tax (loss) income from continuing operations | $ | (246,495 | ) | $ | 7,202 | $ | (57,589 | ) | |||
(Deduct) add: | |||||||||||
State income taxes, net of federal benefit | (913 | ) | 21 | (1,273 | ) | ||||||
Foreign | 53 | 442 | 287 | ||||||||
Revaluation of contingent purchase price | (5,366 | ) | (10,244 | ) | 10,272 | ||||||
Tax credits | (3,539 | ) | (967 | ) | (305 | ) | |||||
Lobbying costs | — | — | 35 | ||||||||
Meals and entertainment | 372 | 605 | 810 | ||||||||
Uncertain tax positions | (1,635 | ) | (2,064 | ) | 61 | ||||||
Bargain purchase | — | — | (7,310 | ) | |||||||
Loss on extinguishment of debt | — | 1,403 | — | ||||||||
Loss on ACC goodwill | — | 11,834 | — | ||||||||
Excess stock option benefit | (4,589 | ) | — | — | |||||||
Change in federal tax rate due to the Tax Cuts and Jobs Act | 126,502 | — | — | ||||||||
Other | 785 | (485 | ) | 1,239 | |||||||
Tax benefit of operating loss carryforwards | 11,509 | (105,045 | ) | — | |||||||
Valuation allowances | 26,740 | 97,365 | 24,040 | ||||||||
Income tax benefit | $ | (96,576 | ) | $ | 67 | $ | (29,733 | ) | |||
F - 35
The significant components of the Company’s deferred tax assets are as follows:
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Deferred tax assets: | |||||||
Net operating loss carryforwards | $ | 235,852 | $ | 230,775 | |||
Tax credits | 20,096 | 20,973 | |||||
Stock based compensation | 19,611 | 28,268 | |||||
Other | 26,113 | 26,889 | |||||
Total deferred tax assets | 301,672 | 306,905 | |||||
Valuation allowance | (239,536 | ) | (162,892 | ) | |||
Total deferred tax assets net of valuation allowance | 62,136 | 144,013 | |||||
Deferred tax liabilities: | |||||||
Fixed assets | $ | (568 | ) | $ | (4,997 | ) | |
Intangible assets | (30,664 | ) | (81,877 | ) | |||
Convertible debt | (30,904 | ) | (57,430 | ) | |||
Indefinite lived intangible assets | — | (89,701 | ) | ||||
Total deferred tax liabilities | (62,136 | ) | (234,005 | ) | |||
Net deferred tax liabilities | $ | — | $ | (89,992 | ) | ||
During 2017 and 2016, the Company recorded a net increase to its valuation allowance of $76.6 million and $95.0 million, respectively. At December 31, 2017 and 2016, the Company recorded a valuation allowance of $239.5 million and $162.9 million respectively, principally against net operating loss carryforwards in domestic and foreign jurisdictions. The Company considered positive and negative evidence including its level of past and future operating income, the utilization of carryforwards, the status of litigation with respect to the Angiomax patents and other factors in arriving at its decision to recognize its deferred tax assets. The Company continues to evaluate the realizability of its deferred tax assets and liabilities on a periodic basis and will adjust such amounts in light of changing facts and circumstances including, but not limited to, future projections of taxable income, tax legislation, rulings by relevant tax authorities, the progress of ongoing tax audits, the regulatory approval of products currently under development and the extension of patent rights relating to Angiomax. Any changes to the valuation allowance or deferred tax assets in the future would impact the Company’s effective tax rate.
On December 22, 2017, the “Tax Cuts and Jobs Act” (TCJA) was enacted which significantly reforms the Internal Revenue Code of 1986, as amended. The TCJA, among other things, reduces the U.S. federal corporate tax rate from 35% to 21%, repeals the corporate alternative minimum tax (AMT), imposes additional limitations on the deductibility of interest, allows for the expensing of capital expenditures, and puts into effect the migration from a “worldwide” system of taxation to a territorial system. As a result of this legislation, the Company remeasured its deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21%. However, the Company is still analyzing certain aspects of the TCJA and refining its calculations, which could potentially affect the measurement of these balances or potentially give rise to new deferred tax amounts. The provisional amount recorded related to the remeasurement of the Company’s deferred tax balances was $126.5 million which was offset fully by the provisional amount recorded related to the reversal of previously established valuation allowances against these deferred tax balances. The TCJA also permits any remaining AMT tax attribute carryforwards to be used to offset future taxable income and/or be refundable over the next several years. As a result, the Company recognized a provisional benefit of $4.9 million during the year ended December 31, 2017 related to the reversal of a previously established valuation allowance against its AMT tax attribute carryforwards and the related refundable amount has been classified in other assets in the accompanying consolidated balance sheet. In addition, based on its preliminary analysis, the Company does not believe that it has offshore earnings that would be subject to the mandatory transition tax.
While the Company has completed its provisional analysis of the income tax effects of the TCJA and recorded a reasonable estimate of such effects, the amounts recorded related to the TCJA may differ, possibly materially, due to, among other things, further refinement of the Company’s calculations, changes in interpretations and assumptions that the Company has made, additional guidance that may be issued by the U.S. Government, and actions and related accounting policy decisions the Company may take as a result of the TCJA. The Company will complete its analysis over a one-year measurement period ending no later
F - 36
than December 22, 2018, and any adjustments during this measurement period will be included in loss from continuing operations as an adjustment to income tax expense/benefit in the reporting period when such adjustments are determined.
In 1998 and 2002, the Company experienced a change in ownership as defined in Section 382 of the Internal Revenue Code. However, based on the market value of the Company at such dates, the Company believes that these ownership changes will not significantly impact its ability to use net operating losses or tax credits in the future to offset taxable income. During 2013 the Company acquired the stock of Incline and became the successor of certain net operating losses and tax credit carryforwards. These tax attributes are also subject to a limitation under Internal Revenue Code Section 382 and these amounts, combined with those of the Company in the table below, have been reduced appropriately for such utilization limitations. In addition, utilization of these net operating loss and tax credit carryforwards is dependent upon the Company achieving profitable results. To the extent the Company’s use of net operating loss and tax credit carryforwards is further limited by Section 382 as a result of any future ownership changes, the Company’s income would be subject to cash payments of income tax earlier than it would if the Company was able to fully use its net operating loss and tax credit carryforwards in the U.S.
At December 31, 2017, the Company has federal net operating loss carryforwards available to reduce taxable income and federal research and development tax credit carryforwards available to reduce future tax liabilities. They expire approximately as follows:
Year of Expiration | Federal Net Operating Loss Carryforwards | Federal Research and Development Tax Credit Carryforwards | ||||||
(In thousands) | ||||||||
2027 | $ | 6,256 | $ | 840 | ||||
2028 | 38,954 | 2,108 | ||||||
2029 | 4,755 | 1,149 | ||||||
2030 | 1,030 | 1,162 | ||||||
2031 | 605 | 3,097 | ||||||
2032 | 1,533 | 3,666 | ||||||
2033 | 37,209 | 3,178 | ||||||
2034 | 4,353 | 1,861 | ||||||
2035 | 195,416 | 752 | ||||||
2036 | 293,661 | 1,739 | ||||||
2037 | 423,531 | 3,515 | ||||||
$ | 1,007,303 | $ | 23,067 | |||||
At December 31, 2017 the Company has the following additional carryforwards: Refundable Alternative Minimum Tax Credits of $4.9 million with no expiration date and foreign net operating losses of approximately $49.5 million. The foreign net operating losses expire in varying amounts beginning in 2018.
The Company does not anticipate a significant change in its unrecognized tax benefits in the next twelve months. The Company is no longer subject to federal, state or foreign income tax audits for tax years prior to 2013. However applicable taxing authorities can review and adjust net operating loss or tax credit carryforwards originating in a closed tax year if utilized in an open tax year. The Company concluded an audit of its 2010 Italy tax filing resulting in a tax assessment of approximately $0.5 million. During 2017, the Company reduced its liability for unrecognized tax benefits by approximately $1.4 million for the difference between the amount previously accrued and the final assessment resulting from that audit. The Company is not under examination by any taxing authorities. However, while tax examinations are often complex, as tax authorities may disagree with the treatment of items reported requiring several years to resolve, the Company believes that it has adequately provided for all uncertain tax provisions for open tax years by tax jurisdiction. The Company classifies interest and penalties related to unrecognized tax benefits in income tax expense. The Company has not accrued any interest or penalties as of December 31, 2017. The total amount of unrecognized tax benefits that, if recognized, would affect the Company’s effective tax rate was $0.0 million, $1.9 million and $1.9 million as of December 31, 2017, 2016 and 2015.
F - 37
A reconciliation of the beginning and ending amount of gross unrecognized tax benefits is as follows:
Gross Unrecognized Tax Benefits | |||
(In thousands) | |||
Balance at January 1, 2015 | $ | 8,022 | |
Additions related to current year tax positions | 61 | ||
Balance at December 31, 2015 | 8,083 | ||
Additions related to current year tax positions | 193 | ||
Balance at December 31, 2016 | 6,018 | ||
Additions related to current year tax positions | 708 | ||
Reductions for prior year tax positions | (2,843 | ) | |
Balance at December 31, 2017 | $ | 3,883 | |
The Company provides income taxes on the earnings of foreign subsidiaries to the extent those earnings are taxable or are expected to be remitted. As of December 31, 2017, the Company’s accumulated foreign unremitted earnings have been immaterial. The Company’s policy is to invest indefinitely its unremitted foreign earnings outside the United States.
14. | Fair Value Measurements |
The Company applies a fair value framework in order to measure and disclose its financial assets and liabilities. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The fair value hierarchy requires an entity to maximize the use of observable inputs, where available, and minimize the use of unobservable inputs when measuring fair value. There are three levels of inputs that may be used to measure fair value:
Level 1 | Quoted prices in active markets for identical assets or liabilities. The Company’s Level 1 asset consists of money market investments. |
Level 2 | Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. Fair values are determined by utilizing quoted prices for similar assets and liabilities in active markets or other market observable inputs such as interest rates and yield curves. |
Level 3 | Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. The Company’s Level 3 assets and liabilities consist of the contingent purchase prices associated with the Company’s dispositions and business combinations, respectively. The fair value of certain development or regulatory milestone based contingent purchase prices was determined in a discounted cash flow framework by probability weighting the future contractual payment with management's assessment of the likelihood of achieving these milestones and present valuing them using a risk-adjusted discount rate. Certain sales milestone based payments were determined in a discounted cash flow framework where risk-adjusted revenue scenarios were estimated using Monte Carlo simulation models to compute contractual payments which were present valued using a risk-adjusted discount rate. |
Financial assets measured at fair value on a nonrecurring basis
As part of the purchase and sale agreement with Mallinckrodt, the Company may receive up to an additional $235.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of PreveLeak and Raplixa. In evaluating this information, considerable judgment is required to interpret the market data used to develop the assumptions and estimates. The Company utilized the “income method,” which applies a probability weighting that considers the estimated future net sales of each of the respective products to determine the probability that each sale milestone will be met. These projections were based on factors such as relevant market size, patent protection, historical pricing of similar products and expected industry trends. The Company anticipated payment from Mallinckrodt on these sales milestones between
F - 38
2018 and 2022 with probabilities of achievement ranging from 15% to 85%. The Company also considers qualitative factors such as development of competing drugs, regulatory developments and other qualitative factors. The Company determined the year in which it believes each of the sales milestones will be achieved. The respective milestones were then discounted to the present value using a discount rate of 10%. Any changes to fair value will be recorded if and when the sales milestones are achieved. The Company initially calculated the fair values of these contingent payments to be received from Mallinckrodt as $78.0 million, which are reflected as a contingent purchase price from sale of business on the accompanying consolidated balance sheet. The Company classified these contingent payments as Level 3 assets. Any increases in the carrying amount or impairments of sales milestones would be recognized in operating expenses if and when the milestones are achieved or determined to have no value. On December 31, 2017, the Company decreased the carrying value of the contingent purchase price from the sale of the Hemostasis Business by $63.0 million as a result of the discontinuation of Raplixa by Mallinckrodt.
As part of the purchase and sale agreement with Chiesi USA and Chiesi, the Company may receive up to an additional $480.0 million in the aggregate from Chiesi following the achievement of certain specified calendar year net sales milestones with respect to net sales of each of Cleviprex and Kengreal. In evaluating this information, considerable judgment is required to interpret the market data used to develop the assumptions and estimates. The Company utilized a risk adjusted revenue simulation model. In this simulation, the chances of achieving many different revenue levels are estimated and then adjusted to reflect the results of similar products and companies in the market to calculate the fair value of each milestone payment. The breadth of all possible revenue scenarios is captured in an estimate of revenue volatility - a measure that can be estimated from performance of similar companies in the market. The Company estimated revenue volatility as the delivered asset volatility observed in comparable companies’ historical performance, where the delivering asset was based on operational leverage of the Company. Under each of these possible scenarios, different amounts of the sales-based milestone payments are calculated, and the average of the payments across a range of possible scenarios is deemed to be the expected value of the earn-out payments. The Company compared the estimated revenue volatility to the delivered asset volatility to arrive at adjusted revenue volatilities between 30% and 41%. The Company then discounted the expected future value of the earn-out payments using a range of discount rates between 3.1% and 6.9%. The Company calculated the fair values of these contingent payments to be received from Chiesi as $65.7 million, which continue to be reflected as a contingent purchase price from sale of business on the accompanying consolidated balance sheet at December 31, 2017. The Company classified these contingent payments as Level 3 assets. Any increases in the carrying amount or impairments of sales milestones would be recognized in selling, general and administrative expenses if and when the milestones are achieved or determined to have no value. The Company noted no indicators of impairment on the contingent payments to be received from Chiesi.
Financial assets and liabilities measured at fair value on a recurring basis
Financial assets and liabilities measured at fair value are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment and considers factors specific to the asset or liability.
F - 39
Except for the Company’s Level 2 liabilities which are discussed in Note 9, “Convertible Senior Notes,” the following table sets forth the Company’s assets and liabilities that are measured at fair value on a recurring basis at December 31, 2017 and 2016, by level, within the fair value hierarchy:
As of December 31, 2017 | As of December 31, 2016 | |||||||||||||||||||||||||||||||
Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | Balance at December 31, | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | Balance at December 31, | |||||||||||||||||||||||||
Assets and Liabilities | (Level 1) | (Level 2) | (Level 3) | 2017 | (Level 1) | (Level 2) | (Level 3) | 2016 | ||||||||||||||||||||||||
(In thousands) | ||||||||||||||||||||||||||||||||
Assets: | ||||||||||||||||||||||||||||||||
Cash and cash equivalents | $ | 12,100 | $ | — | $ | 12,100 | $ | 56,097 | $ | — | $ | — | $ | 56,097 | ||||||||||||||||||
Total assets at fair value | $ | 12,100 | $ | — | $ | — | $ | 12,100 | $ | 56,097 | $ | — | $ | — | $ | 56,097 | ||||||||||||||||
Liabilities: | ||||||||||||||||||||||||||||||||
Contingent purchase price | $ | — | $ | — | $ | 19,650 | $ | 19,650 | $ | — | $ | — | $ | 31,832 | $ | 31,832 | ||||||||||||||||
Total liabilities at fair value | $ | — | $ | — | $ | 19,650 | $ | 19,650 | $ | — | $ | — | $ | 31,832 | $ | 31,832 | ||||||||||||||||
There were no transfers of assets between Level 1 and Level 2 of the fair value measurement hierarchy that occurred during 2017.
Level 3 disclosures
The Company measures contingent purchase price at fair value based on significant inputs not observable in the market, which causes it to be classified as a Level 3 measurement within the fair value hierarchy. The valuation of contingent purchase price uses assumptions and estimates the Company believes would be made by a market participant in making the same valuation. The Company assesses these assumptions and estimates on an on-going basis as additional data impacting the assumptions and estimates are obtained. Changes in the fair value of contingent purchase price related to updated assumptions and estimates are recognized within selling, general and administrative expenses in the accompanying consolidated statements of operations.
The contingent purchase price may change significantly as additional data is obtained, impacting the Company’s assumptions regarding probabilities of successful achievement of related milestones used to estimate the fair value of the liability. In evaluating this information, considerable judgment is required to interpret the market data used to develop the assumptions and estimates. The estimates of fair value may not be indicative of the amounts that could be realized in a current market exchange. Accordingly, the use of different market assumptions and/or different valuation techniques may have a material effect on the estimated fair value amounts, and such changes could materially impact the Company’s results of operations in future periods.
The following table provides quantitative information associated with the fair value measurements of the Company’s Level 3 liabilities:
Fair Value as of December 31, 2017 | Valuation Technique | Unobservable Input | Range (Weighted Average) | |||||||
(In thousands) | ||||||||||
Rempex: | ||||||||||
Contingent purchase price: Event-based milestones | $ | 19,650 | Probability-adjusted discounted cash flow | Probabilities of successes | 18% - 90% (71%) | |||||
Period in which milestones are expected to be achieved | 2018 - 2024 | |||||||||
Discount rate | 4.8% - 7.5% | |||||||||
F - 40
Fair Value as of December 31, 2016 | Valuation Technique | Unobservable Input | Range (Weighted Average) | |||||||
(In thousands) | ||||||||||
Incline: | ||||||||||
Contingent purchase price | $ | 1,269 | Probability-adjusted discounted cash flow | Probabilities of successes | 5% | |||||
Period in which milestones are expected to be achieved | 2019 | |||||||||
Discount rate | 18% | |||||||||
Rempex: | ||||||||||
Contingent purchase price: Event-based milestones | $ | 16,500 | Probability-adjusted discounted cash flow | Probabilities of successes | 18% - 95% (78%) | |||||
Discount rate | 6.6% - 8.2% | |||||||||
Annovation: | ||||||||||
Contingent purchase price | $ | 14,063 | Probability-adjusted discounted cash flow | Probabilities of successes | 9% - 50% (34%) | |||||
Period in which milestones are expected to be achieved | 2018 - 2031 | |||||||||
Discount rate | 6.0% - 10.0% | |||||||||
The fair value of the contingent purchase price represents the fair value of the Company’s liability for potential payments under the Company’s acquisition agreements for Incline Therapeutics, Inc. (Incline), Rempex Pharmaceuticals, Inc. (Rempex) and Annovation. There were no changes to the potential future payments under the Company’s acquisition agreements. As of December 31, 2017, the remaining potential future payments to the former equity holders of Rempex were up to $224.3 million. The remaining potential future payments under the Company’s acquisition agreements do not include payments of $175.8 million related to the Ionsys product, which was discontinued and withdrawn in the U.S in June 2017 and which has also been discontinued in Europe, and the MDCO-700 development program, which was discontinued in August 2017. The significant unobservable inputs used in the fair value measurement of the Company’s contingent purchase prices are the probabilities of successful achievement of development, regulatory, and sales milestones that would trigger payments under the Incline, Rempex and Annovation agreements, probabilities as to the periods in which the milestones are expected to be achieved and discount rates. Significant changes in any of the probabilities of success or periods in which milestones will be achieved would result in a significantly higher or lower fair value measurement.
The changes in fair value of the Company’s Level 3 contingent purchase price during the year ended December 31, 2017 and 2016 were as follows:
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Balance at beginning of period | $ | 31,832 | $ | 18,300 | |||
Payments | — | (10,449 | ) | ||||
Fair value adjustments to contingent purchase prices included in net loss | (12,182 | ) | 23,981 | ||||
Balance at end of period | $ | 19,650 | $ | 31,832 | |||
For the year ended December 31, 2017, changes in the carrying value of the contingent purchase price obligations resulted from changes in the fair value of the contingent consideration due to either the passage of time, changes in discount rates, changes in probabilities of success, or milestone payments.
No other changes in valuation techniques or inputs occurred during the year ended December 31, 2017 and 2016.
F - 41
15. | Restructuring |
2017 Workforce Reduction
In June 2017, the Company commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open. Concurrent with this market withdrawal, the Company commenced implementation of a workforce reduction, which resulted in the reduction of 57 employees, representing approximately 15% of the Company’s workforce at that time. The Company recorded a pre-tax charge of approximately $276.9 million associated with the discontinuation and market withdrawal of Ionsys in the United States market, of which $268.1 million was a non-cash impairment charge (including a write-off of inventory), $5.8 million relates to cash severance and $3.0 million relates to other exit costs. The Company has also discontinued Ionsys in the European market.
The impacted employees are eligible to receive severance payments in specified amounts, health benefits and outplacement services. The Company has and will record these charges in cost of goods sold, research and development and selling, general and administrative expenses based on responsibilities of the impacted employees.
2016 Workforce Reduction
On June 21, 2016, in connection with the sale of the Non-Core ACC Products, the Company commenced implementation of a reorganization intended to improve efficiency and better align the Company’s costs and employment structure with its strategic plans. The reorganization includes a workforce reduction. As a result, the Company reduced its personnel by 162 employees. Upon signing appropriate release agreements, impacted employees were eligible to receive severance payments in specified amounts, health benefits, outplacement services, and an extension of the exercise period for all vested options up to one year from their respective termination date. The Company incurred charges of approximately $17.2 million related to this reorganization in the aggregate. The Company has and will record these charges in cost of goods sold, research and development and selling, general and administrative expenses based on responsibilities of the impacted employees.
The following tables set forth details regarding the activities described above during the years ended December 31, 2017 and 2016:
Balance as of January 1, 2017 | Expenses, Net | Cash | Noncash | Balance as of December 31, 2017 | |||||||||||||||
(in thousands) | |||||||||||||||||||
Employee severance and other personnel benefits: | |||||||||||||||||||
2017 Workforce reduction | $ | — | $ | 5,897 | $ | (5,768 | ) | $ | (129 | ) | $ | — | |||||||
2016 Workforce reduction | 1,854 | — | (1,038 | ) | (816 | ) | — | ||||||||||||
Total | $ | 1,854 | $ | 5,897 | $ | (6,806 | ) | $ | (945 | ) | $ | — | |||||||
Balance as of January 1, 2016 | Expenses, Net | Cash | Noncash | Balance as of December 31, 2016 | |||||||||||||||
(in thousands) | |||||||||||||||||||
Employee severance and other personnel benefits: | |||||||||||||||||||
2016 Workforce reduction | $ | — | $ | 17,162 | $ | (14,697 | ) | $ | (611 | ) | $ | 1,854 | |||||||
Total | $ | — | $ | 17,162 | $ | (14,697 | ) | $ | (611 | ) | $ | 1,854 | |||||||
16. | Commitments and Contingencies |
The Company’s long-term contractual obligations include commitments and estimated purchase obligations entered into in the normal course of business. These obligations include commitments related to purchases of inventory of the Company’s products, research and development service agreements, operating leases, selling, general and administrative obligations, leased office space for its principal office in Parsippany, New Jersey and additional leased office space in San Diego, California, royalties, milestone payments and other contingent payments due under the Company’s license and acquisition agreements.
F - 42
Future estimated contractual obligations as of December 31, 2017 are:
Contractual Obligations (1) (2) | Less Than 1 Year | 1-3 Years | 3-5 Years | More Than 5 Years | Total | |||||||||||||||
(In thousands) | ||||||||||||||||||||
Inventory related commitments | $ | 52,111 | $ | 16,167 | $ | — | $ | — | $ | 68,278 | ||||||||||
Research and development | 71,115 | 68,387 | 34,508 | 30,409 | 204,419 | |||||||||||||||
Operating leases | 7,185 | 15,029 | 15,428 | 27,928 | 65,570 | |||||||||||||||
Selling, general and administrative | 3,924 | 1,379 | — | — | 5,303 | |||||||||||||||
Total contractual obligations | $ | 134,335 | $ | 100,962 | $ | 49,936 | $ | 58,337 | $ | 343,570 | ||||||||||
_______________________________________
(1) | This table does not include any milestone and royalty payments which may become payable to third parties for which the timing and likelihood of such payments are not known, as discussed below. It also does not include the long-term debt obligations. See Note 9 “Convertible Senior Notes” for further details. |
(2) | This table includes commitments related to the Company’s infectious disease business which was sold on January 5, 2018 and the related commitments were assumed by Melinta. These commitments include $68.3 million of inventory related commitments, $7.3 million for research and development service agreements and $1.8 million for selling, general and administrative obligations. See Note 23 “Discontinued Operations” for further details. |
All of the inventory related commitments included above are non-cancellable. Included within the inventory related commitments above are purchase commitments for 2018 totaling $66.9 million, $1.2 million and $0.2 million for Vabomere, Minocin and Orbactiv bulk drug substances, respectively. Of the total estimated contractual obligations for research and development and selling, general and administrative activities, $5.3 million are non-cancellable.
The Company leases its principal offices in Parsippany, New Jersey. The lease covers 173,146 square feet and expires January 2024. On October 1, 2014, the Company entered into an agreement to lease 63,000 square feet of office space with ARE-SD Region No. 35, LLC for new office and laboratory space in San Diego. This lease has a term of 144 months. The commencement date was February 2017. The lease qualifies for operating lease treatment with recorded annual rent expense from commencement date to expiration. The Company’s remaining obligation for this space is $36.6 million.
Approximately 99.7% of the total operating lease commitments above relate to the Company’s principal office building in Parsippany, New Jersey and the Company’s office in San Diego, California. Also included in total operating lease commitments are automobile leases, computer leases and other property leases that the Company entered into while expanding its global infrastructure.
Aggregate rent expense under the Company’s property leases in 2017, 2016 and 2015 was approximately $9.6 million, $7.6 million and $7.3 million, respectively.
In addition to the amounts shown in the above table, the Company is contractually obligated to make potential future success-based development, regulatory and commercial milestone payments and royalty payments in conjunction with collaborative agreements or acquisitions it has entered into with third-parties. These contingent payments include royalty payments with respect to Angiomax under the Company’s license agreements with Biogen and HRI, royalty and/or milestone payments with respect to Vabomere, inclisiran, Ionsys, MDCO-700 and Orbactiv. In 2017, 2016 and 2015, the Company incurred aggregate royalties to Biogen and HRI of $0.8 million, $0.8 million and $1.8 million, respectively, and royalties to AstraZeneca with respect to Cleviprex of $0.0 million, $0.6 million and $1.3 million. As a result of the sale of its Non-Core ACC Products, the Company no longer owes royalties to AstraZeneca relating to sales of Cleviprex. See Note 22, “Dispositions,” for further details.
The Company may have to make these significant contingent cash payments in connection with its acquisition and licensing activities upon the achievement of specified regulatory, sales and other milestones as follows:
• | $49.4 million due to the former equityholders of Targanta and up to $25.0 million in additional payments to other third parties related to the Targanta transaction; |
• | up to $224.3 million for the Rempex transaction; |
• | up to $170.0 million for the Alnylam license and collaboration agreement with Alnylam; and |
F - 43
• | $2.2 million for other transaction milestones. |
Given the nature of these events, it is unclear when, if ever, the Company may be required to pay such amounts. Accordingly, these contingent payments have not been included in the table above as the timing of any future payment is not reasonable estimable.
These amounts do not include milestone payments of up to $175.8 million related to the Ionsys product, which was discontinued and withdrawn in the United States in June 2017 and which has also been discontinued in Europe, and the MDCO-700 development program, which was discontinued in August 2017. The milestone payments above include $229.4 million related to the Targanta and Rempex transactions that were included in the sale of the infectious disease business and assumed by Melinta. See Note 23 “Discontinued Operations” for further details.
Contingencies
The Company may be, from time to time, a party to various disputes and claims arising from normal business activities. The Company accrues for loss contingencies when information available indicates that it is probable that a liability has been incurred and the amount of such loss can be reasonably estimated.
The Company is currently party to the other legal proceedings described in Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K, which are principally patent litigation matters. The Company has assessed such legal proceedings and does not believe that it is probable that a liability has been incurred or that the amount of any potential liability can be reasonably estimated. As a result, the Company did not record any loss contingencies for any of these matters. While it is not possible to determine the outcome of the matters described in Part I, Item 3. Legal Proceedings of this Annual Report on Form 10-K, the Company believes that the resolution of all such matters will not have a material adverse effect on its consolidated financial position or liquidity, but could possibly be material to its consolidated results of operations in any one accounting period.
17. | Employee Benefit Plan |
The Company has an employee savings and retirement plan which is qualified under Section 401(k) of the Internal Revenue Code. The Company made matching contributions in 2017, 2016 and 2015 of $1.5 million, $1.7 million and $2.5 million, respectively.
18. | Segment and Geographic Information |
The Company manages its business and operations as one segment and is focused on advancing the treatment of acute and intensive care patients through the delivery of innovative, cost-effective medicines to the worldwide hospital marketplace. The Company allocates resources and assesses financial performance on a consolidated basis. Revenues reported in 2017, 2016 and 2015 are derived primarily from sales of Angiomax in the United States, including royalty revenue from Sandoz.
The geographic segment information provided below is classified based on the major geographic regions in which the Company operates. Long-lived assets are comprised of the Company’s noncurrent assets.
Years Ended December 31, | ||||||||||||||||||||
2017 | 2016 | 2015 | ||||||||||||||||||
(In thousands) | ||||||||||||||||||||
Net revenue: | ||||||||||||||||||||
United States | $ | 37,131 | 82.9 | % | $ | 131,572 | 91.9 | % | $ | 275,118 | 93.4 | % | ||||||||
Europe | 7,239 | 16.2 | % | 9,331 | 6.5 | % | 16,745 | 5.7 | % | |||||||||||
Other | 419 | 0.9 | % | 2,258 | 1.6 | % | 2,684 | 0.9 | % | |||||||||||
Total net revenue | $ | 44,789 | $ | 143,161 | $ | 294,547 | ||||||||||||||
F - 44
Years Ended December 31, | |||||||||||||
2017 | 2016 | ||||||||||||
(In thousands) | |||||||||||||
Long-lived assets: | |||||||||||||
United States | $ | 308,843 | 99.7 | % | $ | 699,004 | 99.4 | % | |||||
Europe | 836 | 0.3 | % | 4,160 | 0.6 | % | |||||||
Total long-lived assets | $ | 309,679 | $ | 703,164 | |||||||||
19. Collaboration Agreements
Alnylam Pharmaceuticals, Inc.
In February 2013, the Company entered into a license and collaboration agreement with Alnylam Pharmaceuticals, Inc. (Alnylam) to develop, manufacture and commercialize therapeutic products targeting the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene, based on certain of Alnylam’s RNA interference (RNAi) technology. Under the terms of the agreement, the Company obtained the exclusive, worldwide right under Alnylam’s technology to develop, manufacture and commercialize PCSK-9 products for the treatment, palliation and/or prevention of all human diseases. Alnylam is responsible for the development costs of the products, subject to an agreed upon limit, until the completion of Phase 1 clinical studies. The Company is responsible for completing and funding the development costs of the products through commercialization, if successful. The Company paid Alnylam $25.0 million in an initial license payment and an additional $10.0 million upon the achievement of a milestone, which payments the Company recorded as research and development expenses in the accompanying consolidated statements of operations. The Company has also agreed to pay up to an aggregate of $180.0 million in success-based development and commercialization milestones. In addition, the Company has agreed to pay specified royalties on net sales of these products. Royalties to Alnylam are payable by the Company on a product-by-product and country-by-country basis until the last to occur of the expiration of patent rights in the applicable country that cover the applicable product, the expiration of non-patent regulatory exclusivities for such product in such country, or the twelfth anniversary of the first commercial sale of the product in such country, subject to reduction in specified circumstances. The Company is also responsible for paying royalties, and in some cases, milestone payments, owed by Alnylam to its licensors with respect to intellectual property covering these products. In December 2014, under the terms of the license and collaboration agreement with Alnylam, Alnylam initiated a Phase 1 clinical trial of ALN-PCSsc in the UK. Upon initiation of the Phase I clinical trial, the Company incurred a $10.0 million milestone. In November 2017, in connection with the first dosing of a subject in a pivotal study, the Company incurred a $20 million milestone.
SciClone Pharmaceuticals
On December 16, 2014, the Company entered into strategic collaboration agreements with SciClone Pharmaceuticals (SciClone) under which the Company granted SciClone licenses and the exclusive rights to promote, market and sell Angiomax and Cleviprex in China. As a result of the Company’s divestiture of Cleviprex to Chiesi, the Company is no longer a party to the strategic collaboration agreement with SciClone covering Cleviprex. Under the terms of the collaboration regarding Angiomax, SciClone will be responsible for all aspects of commercialization, including pre- and post-launch activities, in the China market (excluding Hong Kong and Macau) and will assist the Company in the registration process in China. The Company has filed in China for marketing approval of Angiomax. SciClone has paid the Company an upfront payment of $10.0 million and agreed to pay a product support services fee and regulatory/commercial success milestone payments of up to an aggregate of $50.5 million and royalties based on net sales of Angiomax in China.
Activities under the SciClone agreement were evaluated to determine if they represented a multiple element revenue arrangement. The SciClone agreement includes the following deliverables: (1) an exclusive license to commercialize Angiomax in China, excluding Hong Kong and Macau; (2) the Company’s obligation to conduct research and development activities related to the approvals of Angiomax; and (3) the Company’s obligation to participate on the joint operating committee established under the terms of the SciClone agreement and related subcommittees. All of these deliverables were deemed to have stand-alone value and to meet the criteria to be accounted for as separate units of accounting. Factors considered in this determination included, among other things, the subject of the licenses and the research and development and commercial capabilities of SciClone. Accordingly, each unit will be accounted for separately. For the years ended December 31, 2017 and 2016, the Company recorded $0.6 million and $0.6 million, respectively, of revenue associated with the SciClone agreement as co-promotion and license income.
The Company believes the regulatory approval milestones that may be achieved under the SciClone agreement are consistent with the definition of a milestone. Accordingly, the Company will recognize payments related to the achievement of such milestone, if any, when the applicable milestone is achieved. Factors considered in this determination included scientific and regulatory risks
F - 45
that must be overcome to achieve each milestone, the level of effort and investment required to achieve each milestone, and the monetary value attributed to each milestone.
SymBio Pharmaceuticals Limited
On October 2, 2015, the Company entered into strategic collaboration with SymBio Pharmaceuticals Limited (SymBio) under which the Company granted SymBio a license and the exclusive rights to promote, market and sell Ionsys in Japan. Under the terms of the collaboration, SymBio will be responsible for all aspects of commercialization, including pre- and post-launch activities, for both products in the Japan market and will assist the Company in the registration process for Ionsys. SymBio has paid the Company an upfront payment of $10.0 million and agreed to pay regulatory/commercial success milestone payments of up to an aggregate of $20.9 million, and royalties based on net sales of Ionsys in Japan. The agreement was terminated in connection with a legal dispute with SymBio effective in the fourth quarter of 2017.
Factors considered in the determination of deliverables included, among other things, the subject of the licenses and the research and development and commercial capabilities of Symbio. For the year ended December 31, 2017 and 2016, the Company recorded $6.9 million and $2.5 million, respectively, of revenue associated with the SymBio agreement as co-promotion and license income.
20. Accumulated Other Comprehensive Loss
The following table provides a reconciliation of the components of accumulated other comprehensive loss, net of tax, attributable to The Medicines Company:
Foreign currency translation adjustment | Unrealized (gain) loss on available for sale securities | Total | ||||||||||
(In thousands) | ||||||||||||
Balance at January 1, 2015 | $ | 2,479 | $ | 49 | $ | 2,528 | ||||||
Other comprehensive income before reclassifications | 1,445 | — | 1,445 | |||||||||
Total other comprehensive income | 1,445 | — | 1,445 | |||||||||
Balance at December 31, 2015 | $ | 3,924 | $ | 49 | $ | 3,973 | ||||||
Other comprehensive income before reclassifications | 213 | — | 213 | |||||||||
Amounts reclassified from accumulated other comprehensive income(1) (2) | (9,616 | ) | (49 | ) | (9,665 | ) | ||||||
Total other comprehensive loss | (9,403 | ) | (49 | ) | (9,452 | ) | ||||||
Balance at December 31, 2016 | $ | (5,479 | ) | $ | — | $ | (5,479 | ) | ||||
Other comprehensive income before reclassifications | 296 | 296 | ||||||||||
Total other comprehensive income | 296 | — | 296 | |||||||||
Balance at December 31, 2017 | $ | (5,183 | ) | $ | — | $ | (5,183 | ) | ||||
(1) | Amounts were reclassified to other income in the accompanying consolidated statements of operations. There is generally no tax impact related to foreign currency translation adjustments, as earnings are considered permanently reinvested. In addition, there were no material tax impacts related to unrealized gains or losses on available for sale securities in the periods presented. |
(2) | See Note 23, “Discontinued Operations,” for a discussion of this reclassification of foreign currency translation adjustment. |
F - 46
21. Selected Quarterly Financial Data (Unaudited)
The following table presents selected quarterly financial data for the years ended December 31, 2017 and 2016.
Three Months Ended | |||||||||||||||||||||||||||||||
March 31, 2017 | June 30, 2017 | Sept. 30, 2017 | Dec. 31, 2017 | March 31, 2016 | June 30, 2016 | Sept. 30, 2016 | Dec. 31, 2016 | ||||||||||||||||||||||||
(1) | (2) | (3) | |||||||||||||||||||||||||||||
(In thousands, except per share data) | |||||||||||||||||||||||||||||||
Total net revenues | $ | 17,465 | $ | 10,861 | $ | 7,868 | $ | 8,595 | $ | 46,075 | $ | 48,573 | $ | 31,084 | $ | 17,429 | |||||||||||||||
Cost of product revenues | 9,978 | 12,490 | 4,287 | 20,438 | 16,042 | 12,061 | 18,213 | 14,337 | |||||||||||||||||||||||
Total operating expenses | 76,879 | 393,195 | 71,129 | 168,682 | 104,248 | 120,817 | 51,881 | 88,296 | |||||||||||||||||||||||
Loss from operations | (59,414 | ) | (382,334 | ) | (63,261 | ) | (160,087 | ) | (58,173 | ) | (72,244 | ) | (20,797 | ) | (70,867 | ) | |||||||||||||||
(Loss) income from continuing operations attributable to The Medicines Company | $ | (70,996 | ) | $ | (370,065 | ) | $ | (7,218 | ) | $ | (159,416 | ) | $ | (67,125 | ) | $ | 201,912 | $ | (31,444 | ) | $ | (82,779 | ) | ||||||||
(Loss) income from discontinued operations, net of tax attributable to The Medicines Company | (31,674 | ) | (27,203 | ) | (22,957 | ) | (18,844 | ) | (25,324 | ) | (19,470 | ) | (54,815 | ) | (40,073 | ) | |||||||||||||||
Net (loss) income attributable to The Medicines Company | $ | (102,670 | ) | $ | (397,268 | ) | $ | (30,175 | ) | $ | (178,260 | ) | $ | (92,449 | ) | $ | 182,442 | $ | (86,259 | ) | $ | (122,852 | ) | ||||||||
Diluted (loss) earnings per common share attributable to The Medicines Company: | |||||||||||||||||||||||||||||||
(Loss) earnings from continuing operations | $ | (1.00 | ) | $ | (5.15 | ) | $ | (0.10 | ) | $ | (2.19 | ) | $ | (0.97 | ) | $ | 2.78 | $ | (0.45 | ) | $ | (1.17 | ) | ||||||||
(Loss) income from discontinued operations | (0.45 | ) | (0.38 | ) | (0.31 | ) | (0.26 | ) | (0.37 | ) | (0.27 | ) | (0.78 | ) | (0.57 | ) | |||||||||||||||
Diluted loss per share | $ | (1.45 | ) | $ | (5.53 | ) | $ | (0.41 | ) | $ | (2.45 | ) | $ | (1.34 | ) | $ | 2.51 | $ | (1.23 | ) | $ | (1.74 | ) | ||||||||
______________________________________
(1) | In June 2017, we commenced a voluntary discontinuation and withdrawal of Ionsys from the market and ceased related commercialization activities, with the regulatory authorizations for Ionsys remaining open. Concurrent with this market withdrawal, the Company commenced a workforce reduction, which resulted in the reduction of 57 employees, representing approximately 15% of the Company’s workforce at that time. The Company recorded a pre-tax charge of approximately $276.9 million associated with the discontinuation and market withdrawal of Ionsys in the United States market. |
In August 2017, the Company announced that it discontinued the clinical development program for MDCO-700 and recorded the following non-cash adjustments during the second quarter of 2017: $65.0 million of asset impairment charges to in-process research and development (IPR&D) acquired from Annovation, a $14.7 million decrease in the carrying value of the contingent purchase price to an estimated fair value of zero, and a $23.0 million benefit for income taxes due to a reduction in the Company’s recorded valuation allowance against its deferred tax assets as a result of the impairment charge.
(2) | In the fourth quarter of 2017, the Company decreased the carrying value of the contingent purchase price from the sale of the Hemostasis Business by $63.0 million as a result of the discontinuation of Raplixa by Mallinckrodt. |
(3) | On June 21, 2016, the Company completed the sale of its Non-Core ACC Products pursuant to the purchase and sale agreement dated May 9, 2016 by and among the Company, Chiesi USA and Chiesi. As a result of this sale, the Company realized a gain on sale of business of $288.3 million. |
22. Dispositions
On June 21, 2016, the Company completed the sale of its Non-Core ACC Products pursuant to the purchase and sale agreement dated May 9, 2016 by and among the Company, Chiesi USA and Chiesi. At the completion of the sale, the Company received approximately $263.8 million in cash, which included the value of product inventory, and may receive up to an additional $480.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of each of Cleviprex and Kengreal.
F - 47
The following table presents the consideration received, major classes of assets sold and the gain recognized on the sale of the Non-Core ACC Products:
(in thousands) | |||
Sale price: | |||
Cash | $ | 263,807 | |
Contingent purchase price from sale of business | 65,700 | ||
Total sale price | 329,507 | ||
Assets: | |||
Inventory | 2,184 | ||
Intangibles | 5,210 | ||
Goodwill | 33,812 | ||
Total assets sold | 41,206 | ||
Gain on sale of business | $ | 288,301 | |
The Company recognized a gain on sale of business of approximately $288.3 million in 2016 in continuing operations in the accompanying consolidated statements of operations. Disposition related costs during 2016 of approximately $7.9 million for advisory, legal and regulatory fees incurred in connection with the sale of the Non-Core ACC Products were recorded in selling, general and administrative expenses in the accompanying consolidated statements of operations. See Note 14, “Fair Value Measurements,” for further details on the contingent purchase price from sale of businesses.
23. Discontinued Operations
Sale of Infectious Disease Business
On January 5, 2018, the Company completed the sale of its infectious disease business, consisting of the products Vabomere, Orbactiv and Minocin IV and line extensions thereof, and substantially all of the assets related thereto, other than certain pre-clinical assets, to Melinta. At the completion of the sale, the Company received approximately $166.4 million and 3,313,702 shares of Melinta common stock having a market value, based on Melinta's closing share price on January 5, 2018, of approximately $54.5 million. In addition, the Company is entitled to receive (i) a cash payment payable 12 months following the closing of the Transaction equal to $25.0 million; (ii) a cash payment payable 18 months following the closing of the Transaction equal to $25.0 million; and (iii) tiered royalty payments of 5% to 25% on worldwide net sales of (a) Vabomere and (b) Orbactiv and Minocin IV, collectively. The fair value of the total consideration received was estimated to be approximately $500.0 million.
As a result of the transaction, the Company accounted for the assets and liabilities of the infectious disease business that were sold as held for sale at December 31, 2017.
Financial results of the infectious disease business are presented as “Loss from discontinued operations, net of tax” on the accompanying consolidated statements of operations for years ended 2017, 2016 and 2015. Assets and liabilities of the infectious disease business to be disposed of are presented as “Current assets held for sale,” “Noncurrent assets held for sale,” “Current liabilities held for sale,” and “Noncurrent liabilities held for sale” on the accompanying consolidated balance sheet as of December 31, 2017 and 2016.
The following table presents key financial results of the infectious disease business included in “Loss from discontinued operations, net of tax” for years ended 2017, 2016 and 2015.
F - 48
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Net product revenues | $ | 34,622 | $ | 24,673 | $ | 14,460 | |||||
Operating expenses: | |||||||||||
Cost of product revenue | 20,060 | 10,693 | 15,945 | ||||||||
Research and development | 39,984 | 47,155 | 33,218 | ||||||||
Selling, general and administrative | 74,346 | 106,670 | 52,643 | ||||||||
Total operating expenses | 134,390 | 164,518 | 101,806 | ||||||||
Loss from operations | (99,768 | ) | (139,845 | ) | (87,346 | ) | |||||
Other expense, net | (906 | ) | (19 | ) | 212 | ||||||
Loss from discontinued operations before income taxes | (100,674 | ) | (139,864 | ) | (87,134 | ) | |||||
Provision (benefit) for income taxes | 4 | 2 | (10 | ) | |||||||
Loss from discontinued operations, net of tax | $ | (100,678 | ) | $ | (139,866 | ) | $ | (87,124 | ) | ||
The following table presents the major classes of assets and liabilities at December 31, 2017 and 2016 related to the infectious disease business which were reclassified as held for sale:
December 31, | December 31, | |||||
2017 | 2016 | |||||
(In thousands) | ||||||
Assets: | ||||||
Accounts receivable, net | $ | 9,595 | $ | 3,916 | ||
Inventory | 41,412 | 42,411 | ||||
Other receivables | 2,740 | 4,259 | ||||
Intangibles, net | 282,398 | — | ||||
Goodwill | 55,057 | — | ||||
Current assets held for sale | 391,202 | 50,586 | ||||
Intangibles, net | — | 293,036 | ||||
Goodwill | — | 55,057 | ||||
Total assets held for sale | $ | 391,202 | $ | 398,679 | ||
Liabilities: | ||||||
Accounts payable | $ | 1,127 | $ | 7,872 | ||
Accrued expenses | 22,945 | 21,686 | ||||
Contingent purchase price – current | 24,650 | 55,000 | ||||
Deferred Revenue | 723 | 2,467 | ||||
Contingent purchase price – noncurrent | 11,135 | — | ||||
Current liabilities held for sale | 60,580 | 87,025 | ||||
Contingent purchase price – noncurrent | — | 50,457 | ||||
Total liabilities held for sale | $ | 60,580 | $ | 137,482 | ||
F - 49
Depreciation and amortization was ceased upon determination that the held for sale criteria were met in the fourth quarter of 2017. The significant cash flow items from discontinued operations for years ended 2017, 2016 and 2015 were as follows:
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Amortization from discontinued operations | $ | 10,638 | $ | 17,858 | $ | 6,798 | |||||
Changes in contingent purchase price | (3,456 | ) | 53,249 | 1 | |||||||
Reserve for excess or obsolete inventory | (435 | ) | (2,066 | ) | 4,228 | ||||||
Payments on contingent purchase price | (63,066 | ) | (10,449 | ) | (18 | ) | |||||
In connection with the divestiture of the infectious disease business unit to Melinta, the Company commenced a workforce reduction, which resulted in the reduction of 36 employees, representing approximately10% of its workforce.
Acquisitions prior to Sale of Hemostasis Business
Recothrom
In February 2013, pursuant to a master transaction agreement with Bristol-Myers Squibb Company (BMS), the Company acquired the right to sell, distribute and market Recothrom on a global basis for the collaboration term and BMS transferred to the Company certain limited assets exclusively related to Recothrom, primarily the biologics license application for Recothrom and certain related regulatory assets. BMS also granted to the Company, under the master transaction agreement, an option to purchase from BMS and its affiliates, following the expiration or earlier termination of the collaboration term, certain other assets, including certain patent and trademark rights, contracts, inventory, equipment and related books and records, held by BMS which are exclusively related to Recothrom. On February 6, 2015, the Company completed the acquisition of the remaining assets held by BMS which were exclusively related to Recothrom. Upon closing the exercise of the option in February 2015, the Company paid BMS approximately $132.4 million in the aggregate, including approximately $44.0 million for inventory and reclassified the value of the purchase option and additional amounts paid to BMS to Developed Product Rights and commenced amortizing.
Sale of Hemostasis Business
On February 1, 2016, the Company completed the sale of its Hemostasis Business to Mallinckrodt pursuant to the purchase and sale agreement dated December 18, 2015 between the Company and Mallinckrodt. At the completion of the sale, the Company received approximately $174.1 million in cash from Mallinckrodt, and may receive up to an additional $235.0 million in the aggregate following the achievement of certain specified calendar year net sales milestones with respect to net sales of PreveLeak and Raplixa. As a result of the transaction, the Company accounted for the assets and liabilities of the Hemostasis Business that were sold as held for sale at December 31, 2015. As a result of the classification as held for sale, the Company recorded impairment charges of $133.3 million, including $24.5 million related to goodwill, to reduce the Hemostasis Business disposal group’s carrying value to its estimated fair value, less costs to sell for the year ended December 31, 2015. The determination of fair value for these assets was based on the best information available that resided within Level 3 of the fair value hierarchy, including internal cash flow estimates discounted at an appropriate interest rate.
Financial results of the Hemostasis Business are presented as “Loss from discontinued operations, net of tax” on the accompanying consolidated statements of operations for years ended 2017, 2016 and 2015.
F - 50
The following table presents key financial results of the Hemostasis business included in “Loss from discontinued operations, net of tax” for years ended December 31, 2016 and 2015.
Year Ended December 31, | |||||||
2016 | 2015 | ||||||
(In thousands) | |||||||
Net product revenues | $ | 1,275 | $ | 65,754 | |||
Operating expenses: | |||||||
Cost of product revenue | 1,424 | 75,889 | |||||
Research and development | 90 | 7,568 | |||||
Selling, general and administrative | 542 | 560 | |||||
Impairment | — | 133,266 | |||||
Total operating expenses | 2,056 | 217,283 | |||||
Income (loss) from operations | (781 | ) | (151,529 | ) | |||
Gain from sale of business | 1,004 | — | |||||
Other expense, net | (39 | ) | (745 | ) | |||
Income (loss) from discontinued operations before income taxes | 184 | (152,274 | ) | ||||
Benefit for income taxes | — | (21,448 | ) | ||||
Loss from discontinued operations, net of tax | $ | 184 | $ | (130,826 | ) | ||
Cumulative translation adjustment (CTA) gains or losses of foreign subsidiaries related to divested businesses are reclassified into income once the liquidation of the respective foreign subsidiaries is substantially complete. At the completion of the sale of the Hemostasis Business, the Company reclassified $9.6 million, net of tax, of CTA gains from accumulated comprehensive loss to the Company’s results of discontinued operations. Of this amount, $8.4 million was included in the impairment loss recorded to reduce the Hemostasis Business disposal group’s carrying value to its estimated fair value, less costs to sell as of December 31, 2015 and $1.2 million was included in “Gain from sale of business” for the year ended December 31, 2016.
Cost of product revenue for the three months ended September 30, 2015 included a charge of $25.8 million to reduce the carrying value of the product rights associated with PreveLeak to their estimated fair value as a result of a reduction in expected future cash flows.
Depreciation and amortization was ceased upon determination that the held for sale criteria were met in the fourth quarter of 2015. The significant cash flow items from discontinued operations for years ended December 31, 2016 and 2015 were as follows:
Year Ended December 31, | |||||||
2016 | 2015 | ||||||
(In thousands) | |||||||
Depreciation from discontinued operations | $ | — | $ | 371 | |||
Amortization from discontinued operations | — | 42,278 | |||||
Gain on sale of business | (1,004 | ) | — | ||||
Asset impairment charges | — | 25,800 | |||||
Reserve for excess or obsolete inventory | — | 876 | |||||
Change in contingent consideration obligation | — | 8,743 | |||||
Proceeds from sale of businesses | 174,068 | — | |||||
Capital expenditures | — | 738 | |||||
24. Subsequent Events
As previously announced, the Company is implementing a series of workforce reductions to focus on inclisiran, improve efficiencies and better align costs and structure. Through February 27, 2018 our workforce restructuring plan has been finalized and communicated to substantially all employees who will be impacted. The Company estimates it will incur approximately $20 million to $26 million related to cash severance and other employee costs associated with these workforce reductions.
F - 51
INDEX TO EXHIBITS
Number | Description | ||
2.1# | |||
2.2#† | |||
2.3† | |||
2.4#† | |||
2.5#† | |||
3.1 | |||
3.2 | |||
4.2 | |||
4.3 | |||
10.1† | |||
10.2† | |||
10.3† | |||
10.4† | |||
10.5† | |||
10.6† | |||
10.7† | |||
10.8† | |||
10.9† | |||
10.10† | |||
10.11† | |||
10.12† | |||
10.13† | |||
10.14† | |||
10.15† | |||
10.16† | |||
10.17† | |||
10.18† | |||
10.19† | |||
10.20 | |||
10.21† | |||
10.22† | |||
10.23† | |||
10.24 | |||
10.25 | |||
10.26† | |||
10.27† | |||
10.28* | |||
10.29* | |||
10.30* | |||
10.31* | |||
10.32* | |||
10.33* | |||
10.34* | |||
10.35* | |||
10.36* | |||
10.37* | |||
10.38* | |||
10.39* | |||
10.40* | |||
10.41* | |||
10.42* | |||
10.43* | |||
10.44* | |||
10.45* | |||
10.46* | |||
10.47* | |||
10.48* | |||
10.49* | |||
10.50* | |||
10.51* | |||
10.52* | |||
10.53* | |||
10.54* | |||
10.55* | |||
10.56 | |||
10.57 | |||
10.58 | |||
10.59 | |||
10.60 | |||
10.61 | |||
10.62 | |||
10.63 | |||
10.64 | |||
21 | |||
23.1 | |||
31.1 | |||
31.2 | |||
32.1 | |||
32.2 | |||
101 | The following materials from The Medicines Company Annual Report on Form 10-K for the year ended December 31, 2017, formatted in XBRL (Extensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Comprehensive (Loss) Income, (iv) the Consolidated Statements of Stockholders’ Equity, (v) the Consolidated Statements of Cash Flows, and (vi) Notes to Consolidated Financial Statements. | ||
# | Schedules (and similar attachments) have been omitted pursuant to Item 601(b)(2) of Regulation S-K. The Company agrees to furnish supplementally copies of any of the omitted schedules (or similar attachments) to the Securities and Exchange Commission upon request. | |
* | Management contract or compensatory plan or arrangement filed as an exhibit to this form pursuant to Items 15(a) and 15(c) of Form 10-K | |
† | Confidential treatment requested as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission unless otherwise indicated, the exhibits incorporated herein by reference were filed under Commission file number 000-31191. | |