Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - ORASURE TECHNOLOGIES INC | d519538dex322.htm |
| EX-32.1 - EX-32.1 - ORASURE TECHNOLOGIES INC | d519538dex321.htm |
| EX-31.2 - EX-31.2 - ORASURE TECHNOLOGIES INC | d519538dex312.htm |
| EX-31.1 - EX-31.1 - ORASURE TECHNOLOGIES INC | d519538dex311.htm |
| EX-24 - EX-24 - ORASURE TECHNOLOGIES INC | d519538dex24.htm |
| EX-23 - EX-23 - ORASURE TECHNOLOGIES INC | d519538dex23.htm |
| EX-3.2 - EX-3.2 - ORASURE TECHNOLOGIES INC | d519538dex32.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 001-16537
ORASURE TECHNOLOGIES, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 36-4370966 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) | |
| 220 East First Street Bethlehem, Pennsylvania |
18015 | |
| (Address of Principal Executive Offices) | (Zip Code) | |
| (610) 882-1820 | ||
| (Registrant’s Telephone Number, Including Area Code): | ||
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
| Common Stock, $0.000001 par value per share | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the Registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405) during the preceding 12 months (or for such shorter period that the Registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☒ | Accelerated filer | ☐ | |||
| Non-accelerated filer | ☐ | Smaller reporting company | ☐ | |||
| Emerging Growth Company | ☐ |
If an emerging growth company, indicate by check mark if the Registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
State the aggregate market value of the voting and non-voting common equity held by nonaffiliates, computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the Registrant’s most recently completed second fiscal quarter (June 30, 2017): $1,010,050,592
Indicate the number of shares outstanding of each of the Registrant’s classes of common stock, as of February 23, 2018: 60,966,866 shares.
Documents Incorporated by Reference:
Portions of the Registrant’s Definitive Proxy Statement for the 2018 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report.
Table of Contents
Table of Contents
This Report contains certain “forward-looking statements,” within the meaning of the Federal securities laws. These may include statements about our expected revenues, earnings, losses, expenses or other financial performance, future product performance or development, expected regulatory filings and approvals, planned business transactions, expected manufacturing performance, views of future industry, competitive or market conditions, and other factors that could affect our future operations, results of operations or financial position. These statements often include words, such as “believes,” “expects,” “anticipates,” “intends,” “plans,” “estimates,” “may,” “will,” “should,” “could,” or similar expressions.
Forward-looking statements are not guarantees of future performance or results. Known and unknown factors that could cause actual performance or results to be materially different from those expressed or implied in these statements include, but are not limited to: ability to market and sell products, whether through our internal, direct sales force or third parties; ability to manufacture products in accordance with applicable specifications, performance standards and quality requirements; ability to obtain, and timing and cost of obtaining, necessary regulatory approvals for new products or new indications or applications for existing products; ability to comply with applicable regulatory requirements; ability to effectively resolve warning letters, audit observations and other findings or comments from the U.S. Food and Drug Administration (“FDA”) or other regulators; changes in relationships, including disputes or disagreements, with strategic partners or other parties and reliance on strategic partners for the performance of critical activities under collaborative arrangements; ability to meet increased demand for the Company’s products; impact of significant customer concentration in the genomics business; impact of increased reliance on U.S. government contracts; failure of distributors or other customers to meet purchase forecasts, historic purchase levels or minimum purchase requirements for our products; impact of replacing distributors; inventory levels at distributors and other customers; ability of the Company to achieve its financial and strategic objectives and continue to increase its revenues, including the ability to expand international sales; ability to identify, complete, integrate and realize the full benefits of future acquisitions; impact of competitors, competing products and technology changes; impact of negative economic conditions; reduction or deferral of public funding available to customers; competition from new or better technology or lower cost products; ability to develop, commercialize and market new products; market acceptance of oral fluid testing, collection or other products; changes in market acceptance of products based on product performance or other factors, including changes in testing guidelines, algorithms or other recommendations by the Centers for Disease Control and Prevention (“CDC”) or other agencies; ability to fund research and development and other products and operations; ability to obtain and maintain new or existing product distribution channels; reliance on sole supply sources for critical products and components; availability of related products produced by third parties or products required for use of our products; ability to maintain sustained profitability; ability to utilize net operating loss carry forwards or other deferred tax assets; volatility of the Company’s stock price; uncertainty relating to patent protection and potential patent infringement claims; uncertainty and costs of litigation relating to patents and other intellectual property; availability of licenses to patents or other technology; ability to enter into international manufacturing agreements; obstacles to international marketing and manufacturing of products; ability to sell products internationally, including the impact of changes in international funding sources and testing algorithms; adverse movements in foreign currency exchange rates; loss or impairment of sources of capital; ability to meet financial covenants in credit agreements; ability to attract and retain qualified personnel; exposure to product liability and other types of litigation; changes in international, federal or state laws and regulations; customer consolidations and inventory practices; equipment failures and ability to obtain needed raw materials and components; the impact of terrorist attacks and civil unrest; and general political, business and economic conditions. These and other factors that could affect our results are discussed more fully under Item 1A, entitled “Risk Factors,” and elsewhere in this Annual Report. Although forward-looking statements help to provide information about future prospects, readers should keep in mind that forward-looking statements may not be reliable. Readers are cautioned not to place undue reliance on the forward-looking statements. The forward-looking statements are made as of the date of this Annual Report and we undertake no duty to update these statements.
Investors should also be aware that while we do, from time to time, communicate with securities analysts, it is against our policy to disclose any material non-public information or other confidential commercial information.
1
Table of Contents
Accordingly, stockholders should not assume that we agree with any statement or report issued by any analyst irrespective of the content of the statement or report. Furthermore, we have a policy against issuing or confirming financial forecasts or projections issued by others. Thus, to the extent that reports issued by securities analysts contain any projections, forecasts or opinions, such reports are not the responsibility of OraSure.
References in this Annual Report to “OraSure” mean OraSure Technologies, Inc. References in this Annual Report to “we,” “us,” “our,” or the “Company” mean OraSure and its consolidated subsidiaries, unless otherwise indicated.
2
Table of Contents
PART I
| ITEM 1. | Business. |
Our business is made up of two principal segments. The first is our “OSUR” business, which consists of the development, manufacture, marketing and sale of oral fluid diagnostic products and specimen collection devices using our proprietary technologies, as well as other diagnostic products including immunoassays and other in vitro diagnostic tests that are used on other specimen types. Our diagnostic products include tests that are performed on a rapid basis at the point of care and tests that are processed in a laboratory. These products are sold in the United States and internationally to various clinical laboratories, hospitals, clinics, community-based organizations and other public health organizations, distributors, government agencies, physicians’ offices, and commercial and industrial entities. We also manufacture and sell medical devices used for the removal of benign skin lesions by cryosurgery or freezing. These cryosurgical products are sold in both professional and over-the-counter (“OTC”) markets in North America, Europe, Central and South America, and Australia.
The second segment is our “DNAG” or molecular collection systems business and is operated primarily through our subsidiary, DNA Genotek Inc. (“DNAG”), a company based in Ottawa, Canada. In the DNAG business, we manufacture and sell kits that are used to collect, stabilize, transport and store samples of genetic material for molecular testing in the consumer genetic, clinical genetic, academic research, pharmacogenomics, personalized medicine, microbiome and animal genetics markets. Our Oragene® DNA sample collection kit provides an all-in-one system for the collection, stabilization, transportation and storage of DNA from human saliva. We also sell research use only products into the microbiome and tuberculosis markets and we offer our customers a suite of genomics and microbiome services called “GenoFINDTM”, which range from package customization and study design optimization to extraction, analysis and reporting services. We serve customers in many countries worldwide, including many leading research universities and hospitals.
OraSure was formed in May 2000 under Delaware law solely for the purposes of combining two companies, STC Technologies, Inc. (“STC Technologies”) and Epitope, Inc. (“Epitope”), and changing the state of incorporation of Epitope from Oregon to Delaware. STC Technologies and Epitope were merged into OraSure on September 29, 2000. Our principal offices are located at 220 East First Street, Bethlehem, Pennsylvania 18015, and our telephone number is (610) 882-1820.
Additional information about us can be found on our website, www.orasure.com. We make available free of charge through a link provided at such website our Annual Reports on Form 10-K, our Quarterly Reports on Form 10-Q, our Current Reports on Form 8-K and our other filings with the Securities and Exchange Commission (“SEC”), as well as any amendments to those Reports and filings. These Reports and filings are made available as soon as reasonably practicable after they are filed or furnished to the SEC. Our Internet website and the information contained in or connected to that website are not intended to be incorporated by reference into this Annual Report.
3
Table of Contents
Products
The following is a summary of our principal products and services and their regulatory and commercial status:
| Product/Service |
Description |
Regulatory Status |
Commercial | |||
| OraQuick ADVANCE® HIV-1/2 |
A rapid, point-of-care qualitative test for antibodies to the Human Immunodeficiency Virus Type 1 (“HIV-1”) and Type 2 (“HIV-2” and together with HIV-1, “HIV-1/2”) that can be visually read in approximately 20 minutes.
|
Premarket approval (“PMA”) by the FDA for use with oral fluid, finger-stick and venous whole blood, and plasma.
CLIA (Clinical Laboratory Improvement Amendments of 1988) waived for use with oral fluid, finger-stick and venous whole blood.
|
Marketed
Marketed | |||
| CE mark (European Union) approved for use with oral fluid, finger-stick and venous whole blood, serum and plasma.
|
Marketed | |||||
| Registered in various other countries.
|
Marketed | |||||
| OraQuick® HIV – 1/2 (Export Only) |
World Health Organization (“WHO”) pre-qualification.
|
Marketed | ||||
| Registered in various foreign countries.
|
Marketed
| |||||
| OraQuick® In-Home HIV Test | A rapid, point-of-care qualitative oral fluid HIV-1/2 test for OTC use that can be visually read in approximately 20 minutes.
|
PMA approved for OTC use.
CE Mark (European Union) approved for OTC use.
|
Marketed
Not Marketed | |||
| OraQuick® HIV Self-Test | Rapid point-of-care qualitative and oral fluid HIV-1/2 Self-Test that can be visually read in approximately 20 minutes.
|
Registered in various countries.
WHO pre-qualification. |
Marketed | |||
| OraQuick® HCV | A rapid, point-of-care qualitative test for antibodies to the hepatitis C virus (“HCV”) that can be visually read in approximately 20 minutes.
|
PMA approved and CLIA waived for use with venous whole blood and finger-stick whole blood specimens. | Marketed | |||
| CE mark (European Union) approved for use with oral fluid, finger-stick and venous whole blood, serum and plasma. Also registered in various other countries.
|
Marketed | |||||
| Registered in various other countries.
|
Marketed
| |||||
| WHO pre-qualification.
|
Marketed
|
4
Table of Contents
| Product/Service |
Description |
Regulatory Status |
Commercial | |||
| OraQuick® Ebola | A rapid point-of-care qualitative test for Ebola antigen that can be visually read in approximately 30 minutes. | Emergency Use Authorization (“EUA”) for use with finger stick and venous whole blood specimens from live patients.
|
Marketed | |||
| EUA for use with oral fluid specimens from cadavers.
|
Marketed | |||||
| Emergency Use Assessment and Listing by WHO for whole blood samples and oral fluid samples from cadavers.
|
Marketed | |||||
| OraQuick® Zika | A rapid point-of-care qualitative test for Zika antibodies that can be read in approximately 20 minutes.
|
Under development. | Not Marketed | |||
| OraSure QuickFlu® Rapid Flu A&B Test |
A rapid, point-of-care qualitative test for antibodies to influenza (flu) Types A and B, including H1N1 infections, with results available in 10 minutes. | FDA 510(k) cleared for use with nasal swab, nasopharyngeal swab and nasal aspirate/wash.
CLIA waived for use with nasal and nasopharyngeal swabs.
|
Marketed
Marketed | |||
| OraSure® | Oral fluid collection device for detection of HIV-1 antibodies, cocaine and cotinine in a laboratory setting. | PMA approved for detection of HIV-1 antibodies with approved laboratory enzyme immunoassay test and registered as a Class I Medical device in the U.S. for detection of cocaine and cotinine.
|
Marketed | |||
| Oragene® • DX | Non-invasive all-in-one system for the collection, stabilization, transportation and storage of human DNA from saliva.
|
FDA 510(k) cleared for use with FDA-cleared or exempt molecular tests, including OTC use. | Marketed | |||
| Oragene® • DNA | Non-invasive all-in-one system for the collection, stabilization, transportation, and storage of human DNA from saliva.
|
CE marked and registered as Class 1 Medical Device in Canada.
Registered in various other countries. |
Marketed | |||
| Oragene® • RNA | Non-invasive all-in-one system for the collection, stabilization and transportation of RNA from human saliva.
|
Research use only product. | Marketed | |||
| ORAcollect® • DX | All-in-one system for the collection, stabilization, transportation, and storage of human DNA from saliva.
|
FDA 510(k) cleared. | Marketed | |||
| ORAcollect® • DNA | All-in-one system for the collection, stabilization, transportation, and storage of human DNA from saliva.
|
CE marked and registered as Class 1 Medical Device.
|
Marketed | |||
| Registered in various foreign countries.
|
Marketed
| |||||
| OMNIgene® • DISCOVER |
Non-invasive all-in-one system for the collection, stabilization, transportation, and storage of microbial DNA from saliva.
|
Research use only product. | Marketed |
5
Table of Contents
| Product/Service |
Description |
Regulatory Status |
Commercial | |||
| Performagene |
All-in-one non-invasive swab kit for the collection, stablization and transportation of animal DNA samples.
|
Animal research use only. | Marketed | |||
| OMNIgene® • GUT | All-in-one system for the collection, stabilization, transportation and storage of microbial DNA in stool samples. | Research use only product.
CE marked and registered in certain countries.
|
Marketed
Marketed | |||
| OMNIgene® • SPUTUM | Reagent for liquefying, decontaminating, transporting and preserving tuberculosis bacteria in sputum samples.
|
Research use only product. CE marked and registered in certain countries. |
Marketed | |||
| OMNIgene® •VAGINAL | Device for self-collection and stabilization of DNA and RNA from the vagina.
|
Research use only. | Marketed | |||
| OMNIgene® • ORAL | Device for self-collection and stabilization of DNA and RNA from the mouth.
|
Research use only. | Marketed | |||
| Hemagene® | Reagent to stabilize high molecular weight DNA in buffy coat samples at ambient temperature
|
Research use only. | Marketed | |||
| PrepIT® | Reagents for extraction and preparation of DNA from saliva.
|
CE marked and registered in the U.S., Canada and various other countries.
|
Marketed | |||
| GenoFINDTM | End-to-end services for genomics and microbiome analysis | Provided by CLIA and College of American Pathologists (“CAP”) certified lab.
|
Marketed | |||
| Intercept® | Oral fluid collection device for oral fluid drugs-of-abuse (“DOA”) testing in a laboratory setting.
|
FDA 510(k) cleared for use with nine MICRO-PLATE DOA assays. | Marketed | |||
| CE marked and registered in certain countries.
|
Marketed | |||||
| MICRO-PLATE DOA Assays | Used to detect the following drugs in an oral fluid sample collected with Intercept® device: tetrahydrocannabinol (“THC” or marijuana), cocaine, opiates, amphetamines, methamphetamines, phencyclidine (“PCP”), benzodiazepines, barbiturates and methadone.
|
Nine drug assays – FDA 510(k) cleared.
Assays CE marked and registered in certain countries.
|
Marketed
Marketed | |||
| Intercept i2® | Oral fluid collection device for oral fluid DOA testing in a laboratory setting using fully-automated, high-throughput oral fluid DOA assays.
|
Forensic use only product.
Generic device CE marked and registered as Class I Medical Device in the U.S.
|
Marketed
Marketed | |||
| Homogeneous DOA Assays | Fully-automated high-throughput oral fluid DOA assays jointly developed with Thermo Fisher for use on oral fluid samples collected | Forensic use only. | Marketed |
6
Table of Contents
| Product/Service |
Description |
Regulatory Status |
Commercial | |||
| with an Intercept i2® device to detect PCP, opiates, cocaine, methamphetamines amphetamines, and THC.
|
||||||
| Cryosurgical Systems - Professional | Cryosurgical (freezing) system for the removal of warts and other benign skin lesions, marketed under the Histofreezer® tradename primarily to the physicians’ office market.
|
FDA 510(k) cleared for nine types
CE marked and registered in certain countries.
|
Marketed
Marketed | |||
| Cryosurgical Systems – OTC | Cryosurgical system for the removal of common and plantar warts and skin tags, sold in various OTC markets under certain brand names and on a private label basis.
|
FDA 510(k) cleared for common and plantar warts.
Registered in Canada for warts and skin tags.
|
Marketed
Marketed | |||
| CE marked and registered for warts in certain countries under Scholl Freeze Spray® and POINTTS® names and for skin tags under CryoTag name.
|
Marketed | |||||
| CE marked for skin tags.
|
Marketed |
In addition to the above products, we sell certain immunoassay tests and reagents for insurance risk assessment, substance abuse testing and forensic toxicology applications; an oral fluid Western blot HIV-1 confirmatory test for confirming positive HIV-1 test results obtained from the use of our OraSure® collection device; and the FDA 510(k) cleared Q.E.D.® rapid point-of-care saliva alcohol test.
OraQuick® Rapid HIV Test
OraQuick® is our rapid point-of-care test platform designed to test oral fluid, whole blood (i.e., both finger-stick and venous), plasma and serum samples for the presence of various antibodies or analytes. The device uses a porous flat pad to collect an oral fluid specimen. After collection, the pad is inserted into a vial containing a pre-measured amount of developer solution and allowed to develop. When blood, plasma or serum is to be tested, a loop collection device is used to collect a drop of the specimen and mix it in the developer solution, after which the collection pad is inserted into the solution and allowed to develop. In all cases, the specimen and developer solution then flow through the testing device where test results are observable in approximately 20 minutes. The OraQuick® device is a screening test and generally requires a confirmation test where an initial positive result is obtained.
This product is sold under the OraQuick ADVANCE® name in North America, Europe and certain other countries and under the OraQuick® name in other developing countries. The test has received PMA approval from the FDA for the detection of antibodies to both HIV-1 and HIV-2 in oral fluid, finger-stick whole blood, venous whole blood and plasma. This test is available for use by laboratories located in the United States certified under the Clinical Laboratory Improvements Amendment of 1988, or CLIA, to perform moderately complex tests. We have also received a CLIA waiver for use of the test with oral fluid and finger-stick and venous whole blood. As a result, the test can be used by numerous additional sites in the United States not certified under CLIA to perform moderately complex tests, such as outreach clinics, community-based organizations and physicians’ offices.
On the international front, we have obtained a CE mark for our OraQuick ADVANCE® test so that we can sell this product in Europe and other countries accepting the CE mark for commercialization and this product is
7
Table of Contents
registered in other countries. We have distributors in place for several countries and are seeking to increase awareness and expand our distribution network for this product throughout the world. We have also received WHO pre-qualification for our export only version of this product.
OraQuick® In-Home HIV Test
The OraQuick® In-Home HIV test is an over-the-counter oral-fluid only version of our OraQuick ADVANCE® HIV 1/2 Antibody Test. We received PMA approval to sell this test in the U.S. OTC market. We have also received a CE mark for the OraQuick® In-Home HIV test, although this product is not currently sold in the European Union. The In-Home test is performed in the same manner as the OraQuick ADVANCE® test, except that it has product labeling and instructions designed for consumers. In addition, we have established a toll free, 24/7, 365-day per year customer call center to provide additional information and referral support for consumers.
OraQuick® HIV Self-Test
The OraQuick® HIV Self-Test has the same diagnostic capabilities as our U.S. approved OraQuick® In-Home HIV test but is sold into certain foreign countries at a lower cost to meet the needs of those markets. We are working with Population Services International (“PSI”), a leading global health organization, along with UNITAID, the WHO and health officials from several African countries to deploy our self-test through the UNITAID-PSI HIV Self-Testing in Africa (“STAR”) project. The OraQuick® Self-Test has labeling and instructions specifically tailored for the African marketplace. The purpose of the STAR project is to generate crucial information about how best to deliver HIV self-testing, how to generate demand for HIV testing in this manner and what the potential public health impact of self-testing will be. Our OraQuick® Self-Test was chosen by PSI because of its quality, ease-of-use and oral fluid option. This product has received WHO pre-qualification and is eligible for procurement by purchasing entities entitled to access funding and other resources from the Global Fund, UNITAID and other agencies.
OraQuick® HCV Rapid Antibody Test
Another test available on the OraQuick® platform is the OraQuick® HCV rapid antibody test. Like the OraQuick® HIV test, this product is a qualitative test that can detect antibodies to the hepatitis C virus, or HCV, in a variety of sample types. The OraQuick® HCV test operates in substantially the same manner as the OraQuick® HIV test.
We have received FDA approval and CLIA waiver for use of the test in detecting HCV antibodies in venous whole blood and finger-stick whole blood specimens, making it the first and only rapid HCV test approved by the FDA for use in the United States. The OraQuick® HCV test has received a CE mark for use with oral fluid, venous whole blood, finger-stick whole blood, plasma and serum and is sold in Europe. This product is also registered and sold in other foreign countries and has received WHO pre-qualification.
OraQuick® Ebola Rapid Antigen Test
In 2015, we completed development of our new rapid Ebola test. This product utilizes the OraQuick® technology platform for the detection of Ebola antigen. This test has received EUAs from the FDA for emergency use by laboratories and facilities adequately equipped, trained and capable of testing for Ebola infection (including treatment centers and public health clinics) on finger-stick and venous whole blood samples collected from live patients, as well as oral fluid specimens collected from cadavers. The WHO has also issued an Emergency Use Assessment and Listing for this product with respect to the same specimen types.
OraSure QuickFlu® Rapid Flu A&B Test
The OraSure QuickFlu® Rapid Flu A&B test is an FDA 510(k) cleared rapid qualitative test for the detection of influenza (flu) Types A and B, including H1N1 viral infections. The test utilizes specimen collected with a nasal
8
Table of Contents
swab, nasopharyngeal swab or nasal aspirate/wash. A reagent is first inserted into a test cartridge, the specimen is added and the test is allowed to flow. Results are available in as little as ten minutes. This product is manufactured for us under an agreement with Princeton BioMeditech Corporation (“PBM”) and is currently sold in certain U.S. markets. PBM has also obtained a CLIA waiver for this test for use with nasal and nasopharyngeal swabs.
OraSure® Collection Device
Our OraSure® oral fluid collection device is used in conjunction with screening and confirmatory tests for HIV-1 antibodies. The generic version of this product can be used for other analytes. This device consists of a small, treated cotton-fiber pad on a handle that is placed in a person’s mouth for two to five minutes. The device collects oral mucosal transudate (“OMT”), a serum-derived fluid that contains higher concentrations of certain antibodies and analytes than saliva. As a result, OMT testing is a highly accurate method for detecting HIV-1 infection and other analytes.
The OraSure® collection device is FDA approved for use in the detection of HIV-1 antibodies. The generic version is a Class I medical device for the detection of cocaine and cotinine in oral fluid specimens. HIV-1 antibody detection using the OraSure® collection device involves three steps:
| • | Collection of an oral fluid specimen using the OraSure® device; |
| • | Screening of the specimen for HIV-1 antibodies at a laboratory with an enzyme immunoassay (“EIA”) screening test approved by the FDA for use with the OraSure® device; and |
| • | Laboratory confirmation of any positive screening test results with either our oral fluid Western blot HIV-1 confirmatory test (described below) or a blood based nucleic acid test. |
A trained health care professional then conveys test results and provides appropriate counseling to the individual who was tested.
Tuberculosis (“TB”) Products
OMNIgene® • SPUTUM is a non-toxic and highly stable reagent that liquefies and decontaminates sputum samples at the point-of-collection or in the lab while preserving the viability of TB bacteria for at least eight (8) days at ambient temperatures. Optimized samples are compatible with all routine TB tests, enable cost-effective sample transport and simplify laboratory workflows while eliminating the need for lab reagents that require daily mixing and quality control. We believe OMNIGENE® • SPUTUM can improve laboratory and operational workflows compared to current approaches, and improve overall test results. This product is being offered to TB laboratories for evaluation.
The OMNIgene® • SPUTUM product is CE marked and is gaining greater interest for tuberculosis testing. Healthcare providers from more than 60 countries have expressed interest in evaluating this product and business entities, ranging from Ministries of Health, non-government organizations, donor agencies and diagnostic test developers, have begun their product evaluations. Our tuberculosis products are well positioned to support the U.S. government’s National Action Plan for combatting multi-drug resistant tuberculosis, by providing much needed solutions to developing countries that are at the highest risk for multi-drug resistant tuberculosis.
Molecular Collection Systems
We sell a number of genomic products that provide all-in-one systems for the collection, stabilization, transportation, and storage of DNA and/or RNA from human and animal biologic samples. Our lead product is sold under the Oragene® brand and is used to collect DNA from human saliva. These products are currently sold to thousands of academic, research and commercial customers in many countries worldwide.
9
Table of Contents
Our molecular collection products are available in several different configurations and contain proprietary chemical solutions that are optimized for the specific application for which each product is designed. Product physical design is focused on ease-of-use and reliability for self or assisted collection of samples. For example, several of the Oragene® products require users to simply hold the product close to their mouth and spit into the collection device. When the container is closed, the reagents stored in the lid of the container are mixed with the captured saliva and immediately protect the nucleic acids in the sample. This non-invasive collection method yields nucleic acid that remains stable at ambient temperature for extended periods. The stabilizing technology results in high quality and high quantity nucleic acids that are required for most genetic testing and analysis methods.
We also market several microbiome collection products designed to collect, stabilize and transport the microbial profile from multiple sample types. Unlike genomic DNA, the microbiome of a sample can change over time, especially when exposed to temperature and environment fluctuations. In order to optimize and standardize sample results, a reliable method that captures and preserves (“snapshots”) the microbiome after collection, until analysis is required. We believe our products provide such a reliable method.
Our OMNIgene® • GUT product is an all-in-one system designed to enable an individual to easily self-collect high quality microbial DNA from feces or stool samples for gut microbiome profiling for use in clinical laboratory and research use settings. The product ensures that the fecal sample is fully stabilized immediately upon collection and maintains an accurate and reliable bacterial profile for weeks at room temperature. Current methodologies for gut microbiome profiling have distinct shortcomings due to the introduction of bias, leading to a lack of reproducibility in the field. We have also begun marketing other microbiome collection kits for DNA or RNA collected from the vagina and oral cavity.
We believe these products provide significant advantages over competing DNA and RNA collection methods such as blood collection or buccal swabs, particularly in human genetic applications. Benefits include the reliable collection of high quality and stable genetic samples, use of simple non-invasive collection methods, the ability to store and transport collected samples for extended periods at ambient temperatures and compatibility with fully-automated laboratory testing systems.
Our molecular business also offers customers our GenoFINDTM services, a suite of genomic and microbiome services that range from package customization and study design optimization to extraction, analysis and reporting services. We believe our GenoFINDTM offering will become an increasingly valuable tool for meeting the needs of our molecular customers.
Our molecular collection products historically have been sold primarily as Class I medical devices for use by research and academic institutions. We have received FDA 510(k) clearance of the Oragene®•DX product for use with the eSensor® Warfarin sensitivity saliva test. A separate 510(k) clearance permits self-collection by consumers when the sample is to be tested with either an exempt or 510(k) cleared molecular test. Our ORAcollect® product similarly received 510(k) clearance from the FDA. We have also received CE mark approval for the Oragene®•DNA, ORAcollect® and OMNIgene®•GUT collection kits.
Intercept® Drug Testing System
A collection device that is substantially similar to the OraSure® device is sold under the name Intercept®, and is used to collect oral mucosal transudate or OMT for oral fluid drug testing. We have received FDA 510(k) clearance to use the Intercept® collection device with laboratory-based EIAs to test for drugs-of-abuse commonly identified by the National Institute for Drug Abuse (“NIDA”) as the NIDA-5 (i.e., tetrahydrocannabinol (“THC” or marijuana), cocaine, opiates, amphetamines/methamphetamines and phencyclidine (“PCP”), and for barbiturates, methadone and benzodiazepines. Each of these EIAs is also FDA 510(k) cleared for use with the Intercept® device. Our Intercept® device and oral fluid assays are sold in the U.S. primarily through laboratory distributors.
10
Table of Contents
We believe that the Intercept® device has several advantages over competing urine and other drugs-of-abuse testing products, including its lower total testing cost, its non-invasive nature, mobility and accuracy, the ease of maintaining a chain-of-custody, the treatment of test subjects with greater dignity, no requirement for specially-prepared collection facilities and difficulty of sample adulteration. The availability of an oral fluid test is intended to allow our customers to test for drug impairment and eliminate scheduling costs and inconvenience, thereby streamlining the testing process.
During 2014, we completed development of a next generation collection device, which we are marketing under the tradename “Intercept i2® he”. This device offers several important advantages over our original Intercept® device, including a sample adequacy indicator that provides a visual prompt when the appropriate volume of oral fluid has been collected, the ability to collect a larger sample required by current laboratory testing protocols and a more optimized chemistry that results in improved recovery of the targeted drug analytes. The Intercept i2®he device is currently being sold as a forensic use only device within the criminal justice and drug treatment markets along with a NIDA-5 panel of fully-automated high-throughput oral fluid drug assays that we distribute under an agreement with Thermo Fisher Scientific (“Thermo Fisher”).
Cryosurgical Systems (Skin Lesion Removal Products)
The Histofreezer® cryosurgical removal system is a low-cost alternative to liquid nitrogen and other methods for removal of warts and other benign skin lesions by physicians. The Histofreezer® product mixes three cryogenic gases in a small aerosol canister. When released, these gases are delivered to a specially designed foam bud, cooling the bud to a maximum of –50°C to –55°C. The frozen bud is then applied to the wart or lesion for 15 to 40 seconds (depending on the type of lesion) creating localized destruction of the target area by freezing. We have received 510(k) clearance for use of the Histofreezer® product to remove common warts and eight other types of benign skin lesions, and this product has been CE marked and registered for distribution in Canada, throughout Europe and in certain other foreign countries. We also supply this product on a private label basis for resale by one of our physician office distributors.
Internationally, we sell an OTC cryosurgical product through our distributor Genomma Labs (“Genomma”), under the POINTTS tradename, in Mexico and a number of South and Central American countries. We sell a CE marked cryosurgical wart removal product into the OTC foot care market in Europe, Australia and New Zealand through our distributor, Reckitt Benckiser (“Reckitt”), under the Scholl and Dr. Scholl trademarks. Reckitt is the owner of the Scholl and Dr. Scholl trademarks in countries outside North and South America. We also sell OTC cryosurgical products to retailers on a private label basis for the treatment of warts in the U.S., for the treatment of both warts and skin tags in Canada and for the treatment of skin tags in the U.K.
Immunoassay Tests and Reagents
We develop and sell immunoassay tests in formats, known as MICRO-PLATE and AUTO-LYTE®, to meet the specific needs of our customers. We also sell fully-automated high-throughput oral fluid drug assays developed under our agreement with Thermo Fisher.
In a MICRO-PLATE kit, the sample to be tested is placed into a small plastic receptacle, called a microwell, along with the reagents. The result of the test is determined by the color of the microwell upon completion of the reaction. Controlling the reaction involves the use of reagents by laboratory personnel. Test results are analyzed by any of a variety of commercially available laboratory instruments, which we may also provide to our laboratory customers. MICRO-PLATE tests can be performed on commonly used instruments and can detect drugs in urine, serum and sweat specimens. MICRO-PLATE tests are also used as part of the Intercept® product line to detect drugs-of-abuse in oral fluid specimens.
AUTO-LYTE® tests are sold in the form of bottles of liquid reagents. These reagents are run on commercially available laboratory-based automated analytical instruments, which are manufactured by a variety of third
11
Table of Contents
parties. AUTO-LYTE® is typically used in high volume, automated, commercial reference insurance laboratories to detect certain drugs or chemicals in urine. Test results are produced quickly, allowing for high-throughput. Our AUTO-LYTE® tests continue to face strong competition from cheaper “home-brew” tests developed internally by our laboratory customers. As a result, we may eventually stop selling our AUTO-LYTE® tests.
We previously entered into an agreement with Thermo Fisher under which Thermo Fisher would develop and supply up to 12 fully-automated high-throughput oral fluid drug assays for use with our Intercept i2® device. Under this agreement, we are currently selling a NIDA-5 panel of assays supplied by Thermo Fisher in the U.S. forensic market. The parties are discussing certain changes to this agreement and assessing their ability to complete development of several additional assays and pursue FDA 510(k) clearance, CE marking and other regulatory approvals of the Intercept i2® device for use with a 12-assay panel. Subject to these further discussions with Thermo Fisher, we believe the offering of an Intercept i2® device with a full menu of fully-automated high-throughput oral fluid assays will better meet the needs of our laboratory drug testing customers and allow us to compete more effectively against fully automated urine drug assays that dominate the drug testing market.
Western blot HIV-1 Confirmatory Test
We sell an oral fluid Western blot HIV-1 confirmatory test that received premarket approval from the FDA in 1996. This test uses the original specimen collected with the OraSure® oral fluid collection device to confirm positive results of initial oral fluid HIV-1 EIA screening tests. After re-evaluating the market demand for this product, we decided to stop manufacturing our western blot HIV-1 confirmatory test in the summer of 2017. We believe existing inventory levels of this product will be exhausted in the first quarter of 2018, after which we will no longer sell this product. We are providing support to assist customers in seeking appropriate confirmatory testing services for positive results from the use of our OraQuick® collection device.
Q.E.D.® Saliva Alcohol Test
Our Q.E.D.® saliva alcohol test is a point-of-care test device that is a cost-effective alternative to breath or blood alcohol testing. The test is a quantitative, saliva-based method for the detection of ethanol, has been cleared for sale by the FDA and has received a CLIA waiver. The U.S. Department of Transportation (“DOT”) has also approved the test.
Each Q.E.D.® test kit contains a collection stick that is used to collect a sample of saliva and a disposable detection device that displays results in a format similar to a thermometer. The Q.E.D.® device is easy to operate and instrumentation is not required to read the result. The product has a testing range of 0 to 0.145% blood alcohol and produces results in approximately two minutes.
Products Under Development
Infectious Disease
One recent area of focus for product development has been our OraQuick® Ebola rapid antigen test. In July 2015, we received an Emergency Use Authorization or EUA for our Ebola test from the FDA. This authorization allows the use of the product for the duration of the U.S. Secretary of the Department of Health and Human Services (“HHS”) August 5, 2014 declaration regarding the emergency use of in vitro diagnostic tests for the detection of the Ebola virus. Under this authorization, the test can be used on finger-stick and venous whole blood samples collected from live patients.
In June 2015, we entered into a contract with the Biomedical Advanced Research Development Authority (“BARDA”) within the HHS for up to $10.4 million of funding for our OraQuick® Ebola test. The three-year, multi-phased contract included an initial commitment of $1.8 million and options for up to an additional $8.6 million to fund certain clinical and regulatory activities. In September 2015 and July 2017, BARDA exercised an option to provide $7.2 million and $1.3 million, respectively, in additional funding for our OraQuick® Ebola test.
12
Table of Contents
In March 2016, we received an EUA for use of the Ebola test on oral fluid samples collected from cadavers. The WHO has issued an Emergency Use Assessment and Listing, or EUAL, for use of the product with whole blood samples collected from live patients and with oral fluid samples collected from cadavers. These approvals will allow expanded use of the product, particularly in Africa. We also intend to seek 510(k) clearance of our Ebola test from the FDA in the future.
Our recent product development efforts have also been focused on addressing global concerns regarding the Zika virus. As a result, we have been developing a rapid Zika antibody test on our OraQuick® platform. This virus is believed to be spread primarily through infected mosquitoes and has been linked as a possible cause of microcephaly in newborn babies whose mothers are infected. There has also been some potential correlation reported with Guillain-Barre syndrome in certain other infected patients.
In August 2016, we entered into a contract with BARDA for up to $16.6 million of funding for our OraQuick® Zika test. The six-year, multi-phased contract included an initial commitment of $7.0 million and options for up to an additional $9.6 million to fund the evaluation of additional product enhancements, and certain clinical and regulatory activities. In May 2017, BARDA exercised an option to provide $2.6 million in additional funding for our rapid Zika test.
Molecular Collections
In order to intersect evolving customer needs within the academic and commercial markets, our molecular business product development pipeline is focused on extending offerings across different sample types and analytes within both the genomics and microbiome areas. Genomic customers are demonstrating an increasing demand for RNA collection and stabilization. On the microbiome front, we continue to focus research and development work on collecting and stabilizing microbial DNA and RNA from multiple sample types including gut, skin, and saliva. We are also evolving the physical design and features of our products to further enable high throughput processing through improved interoperability with automated platforms.
Research and Development
In 2017, our research and development activities focused primarily on development of our new rapid Ebola and Zika tests, an improvement to our rapid HIV test, expanded molecular collection product offerings for the microbiome and genomics markets, our next generation Intercept i2® collection device, and clinical and technical support for our existing products, with significant focus in tuberculosis market support. From time to time, we have contracted with third parties to conduct research and development activities and we may do so in the future.
Research and development expenses were $13.4 million in 2017, $9.8 million in 2016, and $11.7 million in 2015. These expenses include our costs associated with research and development, regulatory affairs, clinical trials and product support.
Sales and Marketing
We attempt to reach our major target markets through a combination of direct sales, strategic arrangements and independent distributors. Our marketing strategy is to create or raise awareness through a full array of marketing activities, which include trade shows, print advertising, special programs, distributor promotions, telemarketing and the use of digital and social media in order to stimulate sales in each target market.
We market our products in the United States and internationally. Consolidated net revenues attributable to customers in the United States were $121.5 million, $99.8 million, and $96.5 million in 2017, 2016 and 2015 respectively. Consolidated net revenues attributable to international customers amounted to $45.6 million, $28.4 million, and $23.2 million, or 27%, 22% and 19% of our total revenues, in 2017, 2016 and 2015, respectively. For more information about our revenues and long-lived assets attributable to U.S. and international customers, please see Note 10 to our consolidated financial statements included elsewhere in this Annual Report.
13
Table of Contents
Infectious Disease Testing - Professional
We market the OraQuick ADVANCE® rapid HIV-1/2 antibody test directly to customers in the public health market for HIV testing. This market consists of a broad range of clinics and laboratories and includes states, counties, and other governmental agencies, family planning clinics, colleges and universities, correctional facilities and the military. There are also a number of organizations in the public health market, such as AIDS service organizations and various community-based organizations that are set up primarily for the purpose of encouraging and enabling HIV testing. We also sell our OraQuick ADVANCE® test directly to hospitals in the U.S. and through distributors into the U.S. physician office market and to retail clinics operated by pharmacies. In addition, we distribute our OraQuick® HIV test in certain other foreign countries.
Our OraQuick® HCV test is sold primarily to the same markets where our OraQuick® ADVANCE HIV test is sold, including public health organizations, hospitals, physicians and retail clinics. We also sell this test in other countries through distributors.
A major focus for our HCV business has been, and will continue to be, to market our test to foreign countries that intend to implement broad scale testing or country-wide HCV elimination programs.
We currently sell our OraQuick® Ebola test under an EUA and our only customer to date has been the CDC, which has purchased the product for field testing in Africa. There were minimal Ebola sales in 2017. Our ability to expand sales of this test to other customers will likely depend on the timing and extent of future outbreaks of the disease, the availability of government or other funding and whether we are able to obtain FDA 510(k) clearance and pre-qualification with the WHO for our product.
We market the OraSure® oral fluid collection device for HIV-1 testing, on its own and as a kit in combination with laboratory testing services. To better serve our public health customers, we have contracted a commercial laboratory to provide prepackaged OraSure® test kits, with prepaid laboratory testing and specimen shipping costs included. We also sell the OraSure® device in the international public health market.
We have distribution rights to an FDA 510(k) cleared and CLIA waived rapid flu A&B test, which we market under our proprietary OraSure QuickFlu® tradename. Under our agreement with the supplier of this product, we are permitted to sell this product into the U.S. hospital and public health markets.
We have also been working to commercialize the OMNIgene® • SPUTUM and PrepIT® • MAX products. TB is a major global health issue, with long established relationships among public health agencies, NGO’s, and suppliers. We have a focused sales and market development team working with these key players to have our products evaluated and adopted where possible.
Infectious Disease Testing - OTC
We sell our OraQuick® In-Home test in the U.S. retail or consumer market. Retailers carrying the product include CVS, Walgreens, Rite Aid and Wal-Mart. The product is also available for purchase on-line through certain retailers and our website, www.oraquick.com. The primary target population for our HIV-OTC test is comprised of young, sexually active adults, with greater purchase intent found in high-risk sub-groups, such as men who have sex with men, African Americans and Latino Americans.
To support individuals that purchase and use our test, we offer toll-free customer support on a 24/7, 365-day per year basis. This service provides consumers with access to trained representatives who can answer questions about HIV/AIDS and the use of our test, and refer consumers to appropriate resources for follow-up confirmatory testing, counseling and medical treatment.
In 2016, we began selling our OraQuick® HIV Self-Test to PSI (Population Services International) for use in a self-testing pilot program called the HIV Self-Testing in Africa, or STAR, project. Under Phase I of the project,
14
Table of Contents
750,000 OraQuick® HIV self-tests were sold to PSI for use in three African countries, Malawi, Zambia and Zimbabwe. More recently, we were selected to participate in Phase II of the project under which approximately 4 million HIV self-tests are expected to be deployed over a two-year period. We believe that we will supply the vast majority of tests under this phase of the project.
In June 2017, we entered into a charitable support agreement with the Bill & Melinda Gates Foundation (“Gates Foundation”) that will enable us to offer our OraQuick® HIV Self-Test at an affordable price in 50 developing countries in Africa and Asia with funding from the Gates Foundation. The funding will consist of support payments tied to the volume of product we sell and reimbursement of certain related costs. The agreement has a four-year term and will enable non-governmental organizations in the eligible countries that receive funding from government or public sector agencies and donors to access our HIV self-test at reduced pricing. The funding from the Gates Foundation will be in an aggregate amount not to exceed $20.0 million over the four-year term or $6.0 million each year of the agreement.
In July 2017, our OraQuick® HIV Self-Test was prequalified by the WHO. WHO prequalification aims to ensure that diagnostic tests for high burden diseases meet global standards of quality, safety, and efficacy in order to optimize use of health resources and improve health outcomes. WHO prequalification enables governmental organizations implementing HIV self-test pilots and programs to access international funding to purchase our test.
Molecular Collection Systems
DNAG sells its products directly to its customers, primarily through its own internal sales force. In some countries distributors are used. Over half of DNAG’s employees work in the areas of sales, marketing, business development or product management. The significant majority of employees who deal directly with customers have molecular science backgrounds, which we believe is useful in selling and marketing molecular collection products, and more importantly, in identifying and evaluating new market and business opportunities.
Most of DNAG’s revenues are derived from product sales to commercial customers and into the academic and research markets. Sales to commercial customers providing consumer genetics and clinical diagnostic services have been increasing and account for a majority of DNAG’s revenues. A significant portion of DNAG’s sales are derived from repeat customers, in both markets. DNAG also has a number of established global customers in the livestock market, including breed associations and research institutions.
DNAG has expanded its market focus by developing new collection devices for the emerging microbiome market, which is focused on the study of microbes and their effect on human health. DNAG’s primary product offering, OMNIgene® • GUT, is focused on the human gut microbiome (microbes living in human stool). DNAG is leveraging its existing sales force and global research connections to engage microbiome customers around the world to establish itself as the leader in ease-of-collection, stabilization and transport of this challenging sample type.
Substance Abuse Testing
Our substance abuse testing products are marketed to laboratories serving the workplace testing, forensic toxicology, criminal justice and drug rehabilitation markets in the U.S. and in certain international markets.
We have entered into agreements for the distribution of our Intercept® collection device and associated MICRO-PLATE assays for drugs-of-abuse testing in the workplace testing market in the United States and Canada through several laboratory distributors and internationally for workplace, criminal justice and forensic toxicology testing through other distributors. We also market the Intercept® collection device on its own and as a kit in combination with laboratory testing services. To better serve our workplace customers, we have contracted with commercial laboratories to provide prepackaged Intercept® test kits, with prepaid laboratory testing and specimen shipping costs included.
15
Table of Contents
The criminal justice market in the United States for our substance abuse testing products consists of a wide variety of entities in the criminal justice system that require drug screening, such as pre-trial services, parole and probation offices, police forces, drug courts, prisons, drug treatment programs and community/family service programs. The forensic toxicology market consists of several hundred laboratories including federal, state and county crime laboratories, medical examiner laboratories and reference laboratories.
As discussed above, we have also launched our next generation Intercept i2® collection device with a NIDA-5 panel of fully-automated high-throughput oral fluid assays developed with Thermo Fisher for the detection of PCP, THC, opiates, cocaine, methamphetamines and amphetamines. These products are currently sold into the criminal justice and drug treatment markets. Subject to our further discussions with Thermo Fisher, we plan to obtain FDA 510(k) clearance of our Intercept i2® device for use with the NIDA-5 assay panel, along with an additional six fully-automated high-throughput assays in order to expand sales of this product line into the workplace testing market and other markets that require 510(k) cleared drug tests. We expect that the 510(k) cleared Intercept i2® device and related fully-automated high-throughput assays will eventually replace our original Intercept® collector and MICRO-PLATE assays in the drug testing market.
We distribute our Q.E.D.® saliva alcohol test primarily through various distributors in the United States and internationally. The markets for alcohol testing are relatively small and fragmented with a broad range of legal and procedural barriers to entry. Markets range from law enforcement testing to workplace testing of employees in safety sensitive occupations. Typical usage situations include pre-employment, random, post-accident, reasonable-cause and return-to-duty testing.
Cryosurgical Systems
Most of our Histofreezer® sales occur in the United States to distributors that, in turn, resell the product to primary care physicians and podiatrists in the United States. Our major U.S. distributors include Cardinal Healthcare, McKesson Medical-Surgical, AmerisourceBergen Corporation, and Henry Schein. We have engaged a manufacturers’ representative organization to help our U.S. distributors promote and sell Histofreezer®. We also provide a private label version of our professional Histofreezer® product to one of our U.S. distributors. Internationally, we sell the Histofreezer® product through a network of distributors in more than 20 countries worldwide.
We distribute cryosurgical wart removal products in the OTC foot care market in Europe, Australia and New Zealand through our distributor, Reckitt Benckiser, under its Scholl and Dr. Scholl tradenames, and in the OTC markets in Mexico and several Central and South American countries under the POINTTS tradename through our distributor, Genomma. We sell our product in the U.K. through another distributor, Appia, under the CryoTag tradename, for the removal of skin tags. We also sell OTC private label cryosurgical products for the removal of warts and skin tags in Canada and for the removal of warts in the U.S.
Insurance Risk Assessment
We currently market the OraSure® oral fluid collection device for use in screening life insurance applicants in the United States and internationally to test for three of the most important underwriting risk factors: HIV-1, cocaine and cotinine (a metabolite of nicotine). Devices are sold to insurance testing laboratories, which in turn sell the devices to insurance companies, usually in combination with testing services.
We also promote use of the OraSure® device directly to insurance companies for life insurance risk assessment. Insurance companies then make their own decision regarding which laboratory to use to supply their collection devices and testing services. We sell our OraSure® Western blot confirmatory test directly to insurance testing laboratories for use in confirming oral fluid specimens collected with our OraSure® device that initially test positive for HIV-1.
16
Table of Contents
There exists a wide range of policy limits where our OraSure® product is being used. In general, many (but not all) of our insurance company customers use the OraSure® device in connection with life insurance policies having face amounts of up to $250,000, with some customers using the device for policies of up to $500,000 in amount. Some insurance companies have chosen to extend their testing to lower policy limits where they did not test at all before, while others have used OraSure® to replace some of their blood and urine-based testing. In recent years, some insurance customers have adopted a “Simplified Issues” policy, where lab testing is no longer required and instead the applicant completes a questionnaire about personal behaviors.
We also sell our AUTO-LYTE® assays and reagents in the insurance testing market directly to certain laboratories.
Significant Products and Customers
Several different products have contributed significantly to our financial performance, accounting for 10% or more of our total revenues during the past three years. The table below shows a breakdown of those product revenues (dollars in thousands).
| For the years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Oragene® collection systems |
$ | 71,610 | $ | 31,078 | $ | 29,385 | ||||||
| OraQuick® HIV |
35,149 | 33,068 | 34,358 | |||||||||
| OraQuick® HCV |
25,409 | 14,066 | 11,386 | |||||||||
| Cryosurgical systems |
12,279 | 13,234 | 11,920 | |||||||||
One of our customers accounted for approximately 25% of our net consolidated revenues in 2017. Another customer accounted for approximately 15% and 12% of our net consolidated revenues in 2016 and 2015, respectively.
Financial Information by Segment
We operate our business within two reportable segments. The first is our “OSUR” business, which consists of the development, manufacture and sale of diagnostic products, specimen collection devices, and medical devices. The second is our “DNAG” or molecular collection systems business, which consists primarily of the development, manufacture and sale of oral fluid collection devices that are used to collect, stabilize, and store samples of genetic material for molecular testing.
OSUR revenues consist primarily of product sold into the United States and internationally to various clinical laboratories, hospitals, clinics, community-based organizations, public health organizations, distributors, government agencies, physicians’ offices, and commercial and industrial entities. OSUR also derives revenues from the sale of OTC products to retail pharmacies and mass merchandisers, and to consumers over the internet and from licensing and product development activities. DNAG revenues consist of product sold into the academic research, consumer genetics, clinical genetic testing, pharmacogenomics, personalized medicine, microbiome and animal genetics markets. For more information about our revenues from external customers, income and total assets, please see the sections entitled “Selected Consolidated Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and Note 10 to the consolidated financial statements, included elsewhere in this Annual Report on Form 10-K.
Supply and Manufacturing
Our OraQuick ADVANCE® HIV test, OraQuick® In-Home HIV test, OraQuick® HCV test, OraQuick® Ebola test, OraSure®, Intercept® and Intercept i2® collection devices, AUTOLYTE and MICRO-PLATE assays and QED® saliva alcohol test are all manufactured in our Bethlehem, Pennsylvania facilities. We expect to continue to manufacture these products at this location for the foreseeable future.
17
Table of Contents
We have contracted with a third party in Thailand for the assembly of the OraQuick® HIV device and the OraQuick® HIV Self-Test in order to supply certain international markets. This supply agreement had an initial term of one year, and automatically renews for additional annual periods unless either party provides a timely notice of termination prior to the end of an annual period. We believe that other firms would be able to manufacture these OraQuick® tests on terms no less favorable than those set forth in the agreement if the Thailand contractor would be unable or unwilling to continue manufacturing this product.
We can purchase the HIV antigens, the nitrocellulose and certain other critical components used in the OraQuick® HIV product lines and the HCV antigens used in the OraQuick® HCV test only from a limited number of sources. If for any reason these suppliers are unwilling or no longer able to supply our antigen or nitrocellulose needs, we believe that alternative supplies could be obtained at a competitive cost. However, a change in any of the antigens, the nitrocellulose or other critical components used in our products would require FDA approval and some additional development work. This in turn could require significant time to complete, increase our costs and disrupt our ability to manufacture and sell the affected products.
Our MICROPLATE and AUTO-LYTE assays require the production of highly specific and sensitive antibodies corresponding to the antigen of interest. Substantially all our antibody requirements are provided by contract suppliers. We believe that we have adequate reserves of antibody supplies and that we have access to sufficient raw materials for these products.
Our OraSure QuickFlu® test is manufactured and supplied by a third party, Princeton BioMeditech. There is no other supply source for this product.
The fully-automated high-throughput oral fluid drug assays sold with our new Intercept i2® collection device are manufactured and supplied under a long-term agreement with Thermo Fisher. There is no other supply source for these products.
The Histofreezer® product and our OTC cryosurgical products are assembled by a U.S. vendor with certain components sourced from international suppliers. We believe that additional suppliers for our cryosurgical products are available on terms no less favorable than the terms of our existing supply agreements in the event that our current suppliers would be unable or unwilling to continue supplying components and manufacturing finished products.
DNAG has two long-term contract manufacturing relationships to supply virtually all of its products, including the Oragene® product line. Many of the raw materials and components used in these products are also purchased from third parties, including one critical component that is purchased from a sole source supplier. We believe there are other suppliers that can manufacture and supply the raw materials and components for the DNAG products.
Employees
As of December 31, 2017, we had 377 full-time employees (including 133 employees at our subsidiary, DNAG). Of this total, there were 131 in sales, marketing and client services; 43 in research and development; 140 in operations, manufacturing, quality control, information systems, purchasing and shipping; 33 in quality assurance and regulatory affairs; and 30 in administration and finance. This compares to 325 employees as of December 31, 2016. Our employees are not currently represented by a collective bargaining agreement.
Competition
The diagnostic industry is a multi-billion dollar international industry and is intensely competitive. Many of our competitors are substantially larger than we are, and have greater financial, research, manufacturing and marketing resources than we do.
18
Table of Contents
Important competitive factors for our products include price, quality, performance, ease of use, customer service and reputation. Industry competition is based on these and the following additional factors:
| • | Scientific and technological capability; |
| • | Proprietary know-how; |
| • | The ability to develop and market products and processes; |
| • | The ability to obtain FDA or other regulatory approvals; |
| • | The ability to manufacture products that meet applicable FDA or other applicable regulatory requirements; |
| • | Commercial execution and strength of distribution; |
| • | Access to adequate capital; |
| • | The ability to attract and retain qualified personnel; and |
| • | The availability of patent protection. |
A few large corporations produce a wide variety of diagnostic tests and other medical devices and equipment. A larger number of mid-size companies generally compete only in the diagnostic industry and a significant number of small companies produce only a few diagnostic products. As a result, the diagnostic test industry is highly fragmented and segmented.
The future market for diagnostic products is expected to be characterized by greater cost consciousness, the development of new technologies, tighter reimbursement policies and consolidation. The purchasers of diagnostic products are expected to place increased emphasis on lowering costs, reducing inventory levels, obtaining better performing products, automation, service and volume discounts. The increased complexity of the market is expected to force many competitors to enter into joint ventures or license certain products or technologies.
We expect competition to intensify as technological advances are made and become more widely known, and as new products reach the market. Furthermore, new testing methodologies could be developed in the future that render our products impractical, uneconomical or obsolete. There can be no assurance that our competitors will not succeed in developing or marketing technologies and products that are more effective than those we develop or that would render our technologies and products obsolete or otherwise commercially unattractive. In addition, there can be no assurance that our competitors will not succeed in obtaining regulatory approval for these products, or introduce or commercialize them, before we can do so. These developments could have a material adverse effect on our business, financial condition and results of operations.
Several companies market or have announced plans to market oral specimen collection devices and tests both within and outside the United States. We expect the number of devices competing with our OraQuick®, OraSure®, Intercept® and Intercept i2® devices to increase as the benefits of oral fluid-based testing become more widely accepted.
Competition in the U.S. market for infectious disease testing in medical settings is intense and is expected to increase. Our principal competition for HIV testing in the professional market comes from existing and new point-of-care rapid blood tests, automated laboratory-based blood tests, or other oral fluid-based tests. One of our competitors has received FDA approval and a CLIA waiver for a rapid oral fluid HIV test and another sells a rapid HIV antigen/antibody test that is both FDA approved and CLIA waived. Our OraQuick® rapid HCV test competes against laboratory-based blood tests in the U.S., as there currently are no other rapid HCV testing products approved by the FDA.
19
Table of Contents
Our competitors in the domestic infectious disease testing market include medical diagnostic companies and specialized biotechnology firms, as well as pharmaceutical companies with biotechnology divisions. Competing tests are often sold at a lower price than we charge for our products. This competition can result in lost sales and degradation of the price (and therefore the applicable profit margins) we can charge for our HIV and HCV tests.
Outside the U.S., our rapid HIV and HCV tests compete against other rapid and laboratory-based tests. Significant sales of these products in Europe have not materialized principally because of differences in European healthcare systems compared to U.S. systems. Unlike the U.S., adoption of rapid point-of-care diagnostics is not widespread in Europe because laboratory testing is entrenched and healthcare systems are structured around centralized testing models. In addition, many competing tests in international markets are sold at very low prices. We intend to continue to build awareness and develop strategies to expand sales of our OraQuick® HIV and HCV tests in European and other international markets.
Our OraQuick® In-Home HIV oral fluid test is the only rapid HIV test approved by the FDA for sale in the U.S. OTC market. We compete against one other non-rapid HIV blood test available in the OTC market, which requires consumers to self-collect a blood sample and then send it to a laboratory for testing. The OraQuick® HIV Self-Test that we sell in certain international markets is expected to face competition from other blood-based rapid HIV self-tests.
Competition for our OraQuick® Ebola test includes government and commercially-developed laboratory and point-of-care molecular tests, along with a small number of rapid antigen tests sold under FDA Emergency Use Authorization (EUA). Our Ebola test is the only product with regulatory authorization for use on both whole blood samples from living patients and oral fluid samples from cadavers.
The OraSure QuickFlu® test competes primarily against other rapid flu tests sold by various third parties in the U.S. hospital and public health markets.
Our Oragene® collection system competes against other types of collection devices used for molecular testing, such as blood collection devices and buccal swabs, which often are sold for prices lower than the prices charged for the Oragene® products. Although we believe the Oragene® device offers a number of advantages over these other products, the availability of lower price competitive devices can result in lost sales and degradation in pricing and profit margin. Our Oragene® product is also facing increasing competition from similarly designed collection systems which are beginning to enter the market.
OMNIgene® • GUT is being sold in the emerging microbiome market and competes with a variety of non-standard in-house solutions developed by various researchers, including simply freezing the sample after collection. The microbiome market is expected to require standardization in the methods used for collection and stabilization in order to derive more accurate and repeatable results. To date, DNAG is one of the few vendors to offer a solution that fully meets these requirements.
The OMNIgene® • SPUTUM product is unique and has no direct competition in terms of a comparable product. The primary competition for this product is the incumbent methodologies that are widely adopted for collecting sputum samples and have been used in labs globally for many years.
In the substance abuse testing market, our Intercept® drug testing systems competes with laboratory-based drug testing products using sample matrices such as urine, hair, sweat and oral fluid. We expect competition for our products to intensify, particularly from other domestic and international companies that have developed, or may develop, competing oral fluid drug testing products.
There are at least two competitors that sell fully-automated high-throughput oral fluid drug testing products in unregulated settings in the United States. These competitors sell these assays for use with either their own oral fluid collector or a collector manufactured by another party. These offerings compete against our Intercept® and Intercept i2® collection devices and related oral fluid assays.
20
Table of Contents
Our MICRO-PLATE oral fluid drug assays, which are sold for use with the original Intercept® collector and our OraSure® collection device, also continue to come under increasing competitive pressure from “home-brew” assays developed internally by our laboratory customers. Our oral fluid MICRO-PLATE assays also compete with urine-based homogeneous assays that are run on fully-automated, random access analyzers. These tests provide strong competitive pressure because they provide the benefits of automation, including lower costs and short turn-around times.
Our MICRO-PLATE drugs-of-abuse reagents sold in the forensic toxicology market are targeted to forensic testing laboratories where sensitivity, automation and “system solutions” are important. In the past, these laboratories have typically had to rely on radioimmunoassay test methods to provide an adequate level of sensitivity. Radioimmunoassays require radioactive materials, which have a short shelf-life and disposal problems. Our MICRO-PLATE tests meet the laboratories’ sensitivity needs, run on automated equipment, are not radioimmunoassays, and are offered to the laboratory as a complete system solution of reagents, instrumentation and software to meet the specific needs of each customer. We compete with both homogeneous and heterogeneous tests manufactured by many companies.
Sales of our AUTO-LYTE® urine assays have declined substantially over a number of years, primarily due to competition from “home-brew” assays developed internally by our laboratory customers, which can be produced at a cost lower than the price typically paid for our products. Many of our customers no longer purchase our AUTO-LYTE® assays, and we may eventually stop selling this product line.
Q.E.D.® competes against other semi-quantitative saliva-based alcohol tests that have received U.S. Department of Transportation approval as well as breath alcohol tests. Although there are lower priced tests on the market that use oral fluid or breath as a test medium, these tests are qualitative tests that are believed to be substantially lower in quality and provide fewer benefits than our Q.E.D.® test.
Our professional cryosurgical product is sold primarily to physicians, including family practitioners, pediatricians and podiatrists. This product primarily competes against other portable cryosurgical systems used for the removal of benign skin lesions in both the U.S. and Europe. In addition, certain of our distributors sell private label cryosurgical products that compete with our Histofreezer® product. Our OTC cryosurgical products compete against other cryosurgical products offered in the U.S. OTC market and certain international OTC markets.
Patents and Proprietary Information
We seek patents and other intellectual property rights to protect and preserve our proprietary technology and our right to capitalize on the results of our research and development activities. We also rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to provide competitive advantages for our products in our markets and to accelerate new product introductions. We regularly search for third-party patents in fields related to our business to shape our own patent and product commercialization strategies as effectively as possible and to identify licensing opportunities. United States patents generally have a maximum term of 20 years from the date an application is filed.
We have five United States patents and numerous foreign patents for the OraSure® and Intercept® collection devices and technology relating to oral fluid collection, containers for oral fluids, methods to test oral fluid, formulations for the manufacture of synthetic oral fluid, and methods to control the volume of oral fluid collected and dispersed. The U.S. patents expire from March 2018 to December 2026. We have also applied for additional patents, in both the United States and certain foreign countries, on such products and technology.
We have five United States patents for our OraQuick® platform, as well as corresponding related international patents. We also have patent applications pending internationally. Four of the U.S. patents expire from March to July 2019 and the fifth in July 2028.
21
Table of Contents
We hold, through our subsidiary, DNAG, eighteen United States patents and numerous foreign patents issued for compositions, methods and apparatuses for the collection, stabilization, transportation and storage of nucleic acids (DNA and RNA) from oral fluid and other bodily fluids and tissues. These patents expire from July 2019 through March 2034.
We have three United States patents and numerous foreign patents issued for apparatuses and methods for the topical removal of skin lesions relating to our cryosurgical wart removal products, and we have pending patent applications related to these products in the United States and in certain foreign countries. These patents expire from September 2025 to February 2032.
We require our employees, consultants, outside collaborators and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed by or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees and certain consultants, the agreements also provide that all inventions conceived by the individual during his or her tenure with us or the performance by the consultant of services for us will be our exclusive property.
We own rights to trademarks and service marks that we believe are necessary to conduct our business as currently operated. In the United States, we own a number of trademarks, including the OraSure®, Intercept®, Intercept i2®, OraQuick®, OraQuick ADVANCE?, Histofreezer®, OraSure QuickFlu®, Q.E.D.®, Oragene®, ORAcollect®, OMNIgene® and AUTO-LYTE® trademarks. We also own many of these marks and others in several foreign countries. With respect to our international OTC cryosurgical products, the Scholl and Dr. Scholl tradenames are owned by Reckitt Benckiser in Europe, Australia, New Zealand and other countries outside North and South America, and the POINTTS tradename is owned by Genomma.
Although important, the issuance of a patent or existence of trademark or trade secret protection does not in itself ensure the success of our business. Competitors may be able to produce products competing with our patented products without infringing our patent rights. Issuance of a patent in one country generally does not prevent manufacture or sale of the patented product in other countries. The issuance of a patent is not conclusive as to validity or as to the enforceable scope of the patent. The validity or enforceability of a patent or trademark can be challenged by litigation after its issuance or registration. If the outcome of such litigation is adverse to the owner of the patent, the owner’s rights could be diminished or withdrawn. Trade secret protection does not prevent independent discovery and exploitation of the secret product or technique.
Government Regulation
General
Most of our products are regulated by the U.S. Food and Drug Administration, or the FDA, along with other federal, state and local agencies and comparable regulatory bodies in other countries. This regulated environment governs almost all aspects of development, production and marketing, including product design and testing, authorizations to market, labeling, advertising and promotion, manufacturing, distribution, post-market surveillance and reporting, and recordkeeping. We believe that our products and procedures are in material compliance with all applicable FDA regulations, but the regulations regarding the manufacture and sale of our products may be unclear and are subject to change. We cannot predict the effect, if any, that these changes might have on our business, financial condition or results of operations.
Many of our FDA-regulated products require some form of review and action by the FDA before they can be marketed in the United States. After approval or clearance by the FDA, we must continue to comply with other FDA requirements applicable to marketed products and are subject to periodic inspections by the FDA and other regulatory bodies. Both before and after approval or clearance, failure to comply with the FDA’s requirements
22
Table of Contents
can lead to significant penalties or could disrupt our ability to manufacture and sell these products. In addition, the FDA could refuse permission to obtain certificates needed to export our products if the agency determines that we are not in compliance.
Domestic Regulation
Most of our products are regulated in the United States as in vitro diagnostic and medical devices.
There are several mechanisms by which regulated devices can be placed on the market in the United States. Some products may qualify for clearance under Section 510(k) of the Federal Food, Drug and Cosmetic Act. To obtain this clearance from the FDA, the manufacturer must submit to the FDA a premarket notification that it intends to begin marketing the product, and show that the product is substantially equivalent to another legally marketed predicate device (i.e., a device that has been cleared through the 510(k) process; a device that was legally marketed prior to May 28, 1976; a device that has been downclassified by the FDA; or a device that FDA has previously determined to be exempt from the 510(k) process). To be substantially equivalent, an applicant must show that when compared to a predicate, the new device has the same intended use and same technology, or if different technology, that the new device is as safe and effective as the predicate and does not raise different questions of safety and effectiveness. In all cases, data from some form of performance testing is required and in some cases, the submission must include data from human clinical studies. Marketing may only commence when the FDA issues a clearance letter finding that the new device is substantially equivalent to the predicate device. An applicant must submit a 510(k) application at least 90 days before commercial distribution of the affected product commences. The standards and data requirements necessary for the clearance of a new device may be unclear or may be subject to change. Although FDA clearance usually takes from four to twelve months, in some cases more than a year may be required before clearance is obtained, if at all.
If the device does not qualify for the 510(k) procedure, either because it is not substantially equivalent to a legally marketed predicate device or because it is classified by the FDA as a high risk device, the FDA must approve either a request for de novo classification or a premarket approval application, or PMA, before marketing can begin. A de novo classification is an alternate pathway to classify novel devices of low to moderate risk for which no substantially equivalent predicate device exists. Clearance of a de novo request generally takes six months to one year from the time of submission of the de novo request, although it can take longer. PMAs are generally required for devices that are determined to be high risk and must demonstrate, among other matters, that the medical device provides a reasonable assurance of safety and effectiveness for the intended use(s) of the device. A PMA is typically a complex submission, supported by valid scientific evidence, including the results of preclinical and clinical studies, detailed information about the manufacturing process for the device, and other data and information. Preparing a PMA is a resource-intensive and time-consuming process. Once a PMA has been submitted, the FDA is required to review the submission within 180 days. However, the FDA’s review may be, and often is, much longer, in many cases requiring one to three years or more, and may include requests for additional data, review by an independent panel of experts, and facility inspections before approval is granted, if at all.
If the FDA approves the PMA, it may place restrictions on the device. If the FDA’s evaluation of the PMA or the manufacturing facility is not favorable, the FDA may deny approval of the PMA application or issue a “not approvable” letter. The FDA may also require additional clinical trials, which can delay the PMA approval process by several years or prevent a PMA approval from being obtained.
If the FDA discovers that an applicant has submitted false or misleading information in any application or notification, the FDA may refuse to review submissions until certain requirements are met pursuant to its Application Integrity Policy (AIP). Delays in receipt of or failure to receive such clearances or approvals, the loss of previously received clearances or approvals, or the failure to comply with existing or future regulatory requirements could have a material adverse effect on our business, financial condition and results of operations.
23
Table of Contents
If there are any modifications made to our marketed devices, a new premarket notification or PMA supplement may be required to be submitted to, and cleared or approved by, the FDA, before the modified device may be marketed. A new PMA or a PMA supplement is required for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device’s intended use(s), manufacturing process, manufacturing facility, critical components, labeling and design. Likewise, a new 510(k) clearance is required for any modification that could significantly affect the safety or effectiveness of the device, e.g. a significant change or modification in design, material, chemical composition, energy source, or manufacturing process or a major change or modification in the intended use(s) of the device.
A clinical trial may be required in support of a 510(k) submission and generally is required for a de novo or PMA application. These trials generally require an Investigational Device Exemption, or IDE, application approved in advance by the FDA for a specified number of patients, unless the proposed study is deemed to be exempt from the IDE requirements. In addition, if a study meets the requirements for a non-significant risk study, it may be eligible for compliance with “abbreviated” IDE requirements, which include a subset of the requirements applicable to significant risk medical device studies. If a full IDE is required, an IDE application must be supported by appropriate data, such as laboratory testing results, protocols for the proposed investigation, and other information demonstrating that the device is appropriate for use with humans in a clinical study. Clinical trials may begin if the IDE application is approved by the FDA and the appropriate institutional review boards, or IRBs, at the clinical trial sites. Submission of an IDE application does not give assurance that the FDA will issue the IDE. If the IDE application is approved, there can be no assurance the FDA will determine that the data derived from the trial(s) support the ultimate approval or clearance of the device or warrant the continuation of clinical trials. An IDE supplement must be submitted to and approved by the FDA before a sponsor or investigator may make a change to the investigational plan in such a way that may affect its scientific soundness, study indication or the rights, safety or welfare of human subjects. The trial must also comply with the FDA’s regulations, including the requirement that informed consent be obtained from each subject, and with clinical trial reporting regulations that require submission of information on certain clinical trials to a database maintained by the National Institutes of Health. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA clearance to market the product in the United States.
Some of our products are used for research only or other nonclinical or non-diagnostic purposes. Our molecular collection products are sold to many academic and research institutions for research purposes and our drugs-of-abuse products are sold to laboratories and clinics for forensic or other non-medical uses. The FDA does not currently regulate products used for these purposes, although other state and federal regulatory requirements may apply.
Another option for marketing a product in the U.S. is through Emergency Use Authorization, or EUA, that is granted by the FDA as a result of the Secretary of Health and Human Services declaring an emergency justifying the authorization of emergency use of certain in vitro diagnostic devices to aid in addressing the emergency. Typically, analytical and clinical studies are completed as required by the FDA. Products are exempt from design controls and other quality requirements in order to expedite development of diagnostic tools to aid in the diagnosis of viral pathogens that have the potential to affect public health.
Most devices distributed in the United States must comply with the FDA’s Quality System Regulations (“QSRs”), including current good manufacturing practices. These regulations govern the entire lifecycle of a medical device, including design, manufacture, testing, release, packaging, distribution, documentation and purchasing as well as complaint handling, corrective and preventative actions, and internal auditing. In complying with the QSRs, manufacturers must continue to expend time, money and effort in the area of production, quality, and postmarket surveillance to ensure full compliance.
Companies are also subject to other post-market and general requirements, including product listing and establishment regulations, which help facilitate FDA inspections and other regulatory action, post-market
24
Table of Contents
surveillance requests, restrictions imposed on marketed products, promotional standards and requirements for recordkeeping and reporting of certain adverse reactions and malfunctions. Device reporting regulations require that manufacturers report if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur.
The FDA regularly inspects companies to determine compliance with the QSRs and other post-market requirements. Failure to comply with statutory requirements and the FDA’s regulations can result in an FDA Form 483 (which is issued by the FDA at the conclusion of an inspection when an investigator has observed any conditions that may constitute violations), public warning letters, monetary penalties against a company or its officers and employees, suspension or withdrawal of regulatory approvals, operating restrictions, total or partial suspension of production, injunctions, product recalls, product detentions, refusal to provide export certificates, seizure of products and criminal prosecution. We believe that our facilities and procedures are in material compliance with the FDA’s OSR regulations and other postmarket requirements, but the regulations are subject to change or may be unclear, and we cannot be sure that FDA investigators will agree with our compliance with the FDA’s postmarket requirements.
On December 23, 2013, our molecular collection systems subsidiary, DNAG, received a warning letter from the FDA. The warning letter primarily focused on DNAG’s response to two Form 483 observations issued by the FDA as a result of an inspection of DNAG’s Ottawa, Canada facilities in September 2013. Specifically, the warning letter indicated the need for additional documentation regarding design and development activities for DNAG’s products and focused in particular on the design planning and design history file for DNAG’s 510(k)-cleared Oragene®• DX collection device. In addition, the warning letter requested additional documentation related to finished product acceptance testing activities for DNAG’s ORAcollect® collection device. The letter further noted that DNAG does not currently have in place an approved PMA or 510(k) clearance for its ORAcollect® device.
DNAG submitted a formal response to the FDA to address the issues referenced in the warning letter and the warning letter was cleared in August 2017. In addition, DNAG received 510(k) clearance of its ORAcollect® device on April 18, 2016.
The Clinical Laboratory Improvement Amendments of 1988, or CLIA, prohibit any facility that conducts laboratory testing on specimens derived from humans from providing information for the diagnosis, prevention or treatment of any disease or impairment of, or the assessment of, the health of human beings, unless there is in effect for such facility a certificate issued by the U.S. Department of Health and Human Services applicable to the category of examination or procedure performed. Tests may be waived from this regulatory oversight if they meet certain requirements established under CLIA. We consider the applicability of CLIA requirements in the design and development of our products. We have obtained a waiver of the CLIA requirements for our OraQuick ADVANCE® rapid HIV-1/2 antibody test, our OraQuick® HCV rapid antibody test and our Q.E.D.® alcohol saliva test and may seek similar waivers for certain other products. In addition, the supplier of the OraSure Quick-Flu® test has obtained a CLIA waiver for that product. A CLIA waiver allows certain customers to use the waived products that may not have been able to use them without complying with applicable quality control and other requirements.
Certain of our products may also be affected by state regulations in the United States, which can restrict the use and sale of certain diagnostic products. We are presently working with legislators or regulators in certain of these states in an effort to modify or remove any restrictions affecting our ability to sell products.
Advertising and Promotion
Advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the Federal Trade Commission (“FTC”) and by other federal and state regulatory and enforcement authorities,
25
Table of Contents
including the Department of Justice (“DOJ”), the Office of Inspector General of the Department of Health and Human Services, and various state attorneys general. Although physicians are permitted to exercise medical judgment to use medical devices for indications other than those cleared or approved by the FDA, we may not promote our products for such “off-label” uses and can only market our products for cleared or approved uses. Promotional activities for FDA-regulated products of other companies have also been the subject of enforcement actions brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims. If the FDA determines that our promotional materials or training constitute promotion of an uncleared or unapproved use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a notice of violation, a warning letter, injunction, seizure, civil fine or criminal penalties. FTC enforcement actions often result in consent decrees that constrain future actions. DOJ prosecutions can result in significant criminal and civil penalties, including exclusion from the Medicare and Medicaid programs. If an enforcement action is brought by the FDA or FTC, our reputation could be damaged and sales of our products could be impaired.
Import and Export Requirements
Products for export from the United States are subject to foreign countries’ import requirements and the exporting requirements of the FDA, as applicable. In particular, international sales of medical devices manufactured in the United States that are not approved or cleared by the FDA for use in the United States, or are banned or deviate from lawful performance standards, are subject to FDA export requirements.
Foreign countries often require, among other things, an FDA certificate for products for export, also called a Certificate for Foreign Government (“CFG”). To obtain this certificate from the FDA, the device manufacturer must apply to the FDA. The FDA certifies that the product has been granted clearance or approval in the United States and that the manufacturing facilities were in compliance with QSR regulations at the time of the last FDA inspection. If the FDA determines that our facilities or procedures do not comply with the QSR regulations, it may refuse to provide such certificates until we resolve the issues to the FDA’s satisfaction. Failure to obtain a CFG could inhibit our ability to export our products to countries that require such certificates.
International
We are also subject to regulations in foreign countries governing products, human clinical trials and marketing, and may need to obtain approval (or pre-qualification or endorsement) from international public health agencies, such as the World Health Organization, in order to sell products in certain countries. Approval processes vary from country to country, and the length of time required for approval or to obtain other clearances may in some cases be longer than that required for U.S. governmental approvals. We generally pursue approval only in those countries that we believe have a significant market opportunity.
The International Organization for Standardization (“ISO”) is a worldwide federation of national standards bodies from some 130 countries, established in 1947. The mission of the ISO is to promote the development of standardization and related activities in the world with a view to facilitating the international exchange of goods and services. ISO certification indicates that our quality system complies with standards applicable to activities ranging from initial product design and development through production and distribution.
In the European Union (“EU”), products that fall under the scope of the Medical Devices Directive (“MDD”) and the In Vitro Diagnostic Directive (“IVDD”) are not subject to the prior approval of a regulatory authority, but, depending on the class of product, may require prior review by a notified body. Notified bodies are accredited and supervised by national regulatory authorities to conduct conformity assessment procedures of medical devices or other products. Such products must comply with certain essential requirements listed in those directives. ISO certification creates a rebuttable presumption that the product satisfies the applicable
26
Table of Contents
requirements. Compliance with these requirements allows us to complete the applicable conformity assessment procedure, involving a notified body where necessary, and to affix the CE mark to our products, without which they may not be placed on the market in the EU.
We have completed the applicable conformity assessment procedures and obtained the right to use the CE mark for the OraQuick ADVANCE® HIV-1/2 test, the OraQuick® HCV test, our Histofreezer® product line, our OTC cryosurgical removal product and certain of the Oragene® collection kits and OMNIgene® products sold by DNAG.
We must also comply with certain registration and licensing requirements as dictated by Health Canada, prior to commencing sales in Canada. We have completed this process for several of our current products and may do so with respect to other products in the future. In addition, Canadian law requires manufacturers of medical devices to have a quality management system that meets various ISO requirements in order to obtain a license to sell their devices in Canada.
We have obtained WHO pre-qualification for our OraQuick® HIV Self Test and OraQuick® HCV test and we will likely seek WHO pre-qualification or endorsement for certain other products sold into international markets.
Anti-Kickback and Other Fraud and Abuse Laws
The Federal Anti-Kickback Statute prohibits the knowing and willful offer, payment, solicitation, or receipt of any form of remuneration in return for, or to induce:
| • | The referral of an individual to a person for the furnishing or arranging for the furnishing of items or services reimbursable under Medicare, Medicaid or other governmental healthcare programs; or |
| • | The purchase, lease, or order of, or the arrangement or recommendation of the purchasing, leasing, or ordering of any item or service reimbursable under Medicare, Medicaid, or other governmental healthcare programs. |
Our products are or may be purchased by customers that will seek or receive reimbursement under Medicare, Medicaid or other governmental healthcare programs. Noncompliance with the federal anti-kickback statute can result in exclusion from Medicare, Medicaid or other governmental healthcare programs, and/or restrictions on our ability to operate in certain jurisdictions, as well as civil and criminal penalties, any of which could have an adverse effect on our business and results of operations.
The Federal Civil Monetary Penalties Law prohibits the offering or transferring of remuneration to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary’s selection of a particular provider, practitioner or supplier of Medicare or Medicaid payable items or services. Noncompliance can result in civil monetary penalties for each wrongful act, assessment of three times the amount claimed for each item or service and exclusion from the Federal healthcare programs.
Many states have also adopted some form of anti-kickback laws. A determination of liability under such laws could result in fines and penalties, restrictions on our ability to operate in these jurisdictions and significant damage to our reputation.
We are also subject to other federal and state laws targeting fraud and abuse in the healthcare industry, including false claims laws, marketing conduct laws and laws constraining the sales, marketing and other promotional activities of manufacturers of medical devices by limiting the kinds of financial arrangements, including sales programs, such manufacturers can enter into with physicians, hospitals, laboratories and other potential purchasers of medical devices. Violations of these laws may be punishable by criminal or civil sanctions, including substantial fines, imprisonment and exclusion from participation in government healthcare programs such as Medicare and Medicaid. These laws and regulations are wide ranging and subject to changing interpretation and application. In recent years, there has been greater scrutiny of marketing practices in the medical device industry which has resulted in several government investigations by various government
27
Table of Contents
authorities and the introduction and/or passage of federal and state legislation regulating interactions between medical device manufacturers and healthcare professionals and providers and requiring the disclosure by medical device manufacturers of payments to certain healthcare professionals and providers. For example, under the Sunshine Act provisions of the Affordable Care Act, device manufacturers are subject to new federal reporting and disclosure requirements with regard to payments or other transfers of value made to physicians and teaching hospitals. Reports submitted under the Sunshine Act are placed in a public database. Device manufacturers are required to submit annual reports by March 31 which cover the prior calendar year. To be in compliance with such disclosure laws, we have implemented necessary systems to accurately track gifts and other payments.
We have implemented a written Policy on Interactions with Health Care Professionals, which is based on the Code of Conduct for Interactions with Health Care Professionals promulgated by the Advanced Medical Technology Association, or AdvaMed, a leading trade association representing medical device manufacturers. The Policy applies to all employees and is intended to comply with applicable state and federal laws, regulations and government guidance. The Policy addresses interactions related to sales and marketing practices, research and development, product training and education, grants and charitable contributions, support of third-party educational conferences, and consulting arrangements. While we believe that our practices are in compliance with the Anti-Kickback and other fraud and abuse laws, the standards for compliance with such statutes can be unclear and subject to change.
Foreign Corrupt Practices Act and Other Anti-Corruption Laws
The U.S. Foreign Corrupt Practices Act (“FCPA”) prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to use any means of interstate commerce corruptly in the furtherance of any offer, payment, promise to pay or authorization of payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. Our present and future business has and will continue to be subject to the FCPA and various other laws, rules and/or regulations applicable to us as a result of our international sales. Those laws include the U.K. Bribery Act (the “Bribery Act”), which proscribes giving and receiving bribes in the public and private sectors, bribing a foreign public official, and failing to have adequate procedures to prevent employees and other agents from giving bribes. U.S. companies that conduct business in the United Kingdom generally will be subject to the Bribery Act. Penalties under the Bribery Act include potentially unlimited fines for companies and criminal sanctions for corporate officers under certain circumstances.
Environmental Regulation
Because of the nature of our current and proposed research, development, and manufacturing processes, we are subject to stringent federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge and handling and disposal of solid wastes, hazardous materials and hazardous wastes. Products that we sell in Europe are subject to regulation in European Union, or EU, markets under the Restriction of the Use of Hazardous Substances Directive, or RoHS. RoHS prohibits companies from selling products which contain certain hazardous materials, including lead, mercury, cadmium, chromium, polybrominated biphenyls and polybrominated diphenyl ethers, in EU member states. In addition, the EU’s Registration, Evaluation, Authorization, and Restriction of Chemicals Directive also restricts substances of very high concern in products.
Future environmental laws may require us to alter our manufacturing processes, thereby increasing our manufacturing costs. We believe that our products and manufacturing processes at our facilities comply in all material respects with applicable environmental laws and worker health and safety laws; however, the risk of environmental liabilities cannot be completely eliminated.
The foregoing discussion of our business should be read in conjunction with the consolidated financial statements and accompanying notes included in Item 15 of this Annual Report.
28
Table of Contents
| ITEM 1A. | Risk Factors |
You should carefully consider the risks and uncertainties described below, together with all of the other information included in this Annual Report and our other SEC filings, in considering our business and prospects. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not disclosed or not presently known to us or that we currently deem immaterial also may impair our business operations. The occurrence of any of the following risks could harm our business, financial condition or results of operations.
Regulatory Risks
The Need to Obtain Regulatory Approvals Could Increase Our Costs and Adversely Affect Our Financial Performance.
Many of our proposed and existing products are subject to regulation by the FDA and other governmental or public health agencies. In particular, we are subject to strict governmental controls on the development, manufacture, labeling, distribution and marketing of our products.
The process of obtaining required approvals, clearances or premarket authorizations can involve lengthy and detailed laboratory testing, human clinical trials, sampling activities and other costly, time-consuming procedures. These approvals, clearances and other premarket authorizations can require the submission of a large amount of clinical data which can be expensive and may require significant time to obtain. It is also possible that a product will not perform at a level needed to generate the clinical data required to obtain such premarket authorizations. The submission of an application to the FDA or other regulatory authority does not guarantee that an authorization to market or import the product will be received. A regulatory authority may impose requirements as a condition to granting an approval, clearance, or other premarket authorization that may include significant restrictions or limitations. The regulatory authority may delay or refuse to grant premarket authorization, even though a product has been approved or registered without restrictions or limitations in another country or by another agency. Delays in receipt or failure to receive such approvals, clearances or other premarket authorization could have a material adverse effect on our business, financial condition and results of operations.
Regulated devices must also meet ongoing FDA requirements after an initial pre-market authorization is received. These requirements include manufacturing quality control and quality assurance registration and listing, adverse event and other reporting and labeling, promotion and advertising requirements.
All in vitro diagnostic products that are to be sold in the EU must bear the CE mark indicating conformance with the essential requirements of the IVDD. We have obtained the CE mark for several of our existing products. We also intend to apply for CE marks for certain of our future products and are not aware of any material reason why we would be unable to obtain those marks. However, there can be no assurance that compliance with all provisions of the IVDD will be demonstrated and the CE mark will be obtained or maintained for all products that we desire to sell in the EU. The failure to obtain or maintain the CE mark for one or more of our products could lead to the termination of strategic alliances and agreements for sales of those products in the EU.
In addition, we or our distributors are often required to obtain premarket authorization or product registration with foreign governments or regulatory bodies before we can import and sell our products in foreign countries. We may also be required to obtain WHO pre-qualification or endorsement in order to sell certain products in international markets or enable our customers to access interested funding sources for our products. We may have difficulty obtaining such authorizations, registrations, pre-qualifications or endorsements and, if obtained, such authorizations, registrations, pre-qualifications or endorsements may contain restrictions that limit our ability to market and sell our products in the relevant country. In addition, any change in our arrangement with a foreign distributor could result in the loss of or delay in transfer of any applicable product registrations, thereby interrupting our ability to sell those products in the affected markets.
29
Table of Contents
Failure to Comply With FDA or Other Regulatory Requirements May Require Us to Suspend Production or Sale of Our Products or Institute a Recall Which Could Result in Higher Costs and a Loss of Revenues.
Regulation by the FDA and other federal, state and foreign regulatory agencies impacts many aspects of our operations, and the operations of our suppliers and distributors, including manufacturing, labeling, packaging, adverse event reporting, recalls, distribution, storage, advertising, promotion and record keeping. We are subject to routine inspection by the FDA and other agencies to determine compliance with QSR and FDA regulatory requirements in the United States and other applicable regulations worldwide, including but not limited to ISO standards. We believe that our facilities and procedures are in material compliance with the FDA requirements and ISO standards, but the regulations may be unclear and are subject to change, and we cannot be sure that the FDA or other regulators will agree with our compliance with these requirements. The FDA and foreign regulatory agencies may require post-marketing testing and surveillance to monitor the performance of approved or cleared products or impose conditions on any product clearances or approvals that could restrict the distribution or commercial applications of those products. Regulatory agencies may impose restrictions on our or our distributors’ advertising and promotional activities or preclude these activities altogether if a noncompliance is believed to exist. In addition, the subsequent discovery of previously unknown problems with a product may result in restrictions on the product or additional regulatory actions, including withdrawal of the product from the market.
Failure to comply with the applicable requirements of the FDA can result in, among other things, 483 notices, warning letters, administrative or judicially imposed sanctions such as injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal to grant premarket clearance or PMA approval for devices, withdrawal of product registrations, marketing clearances or approvals, or criminal prosecution. The ability of our suppliers to supply critical components or materials and of our distributors to sell our products could also be adversely affected if their operations are determined to be out of compliance. Such actions by the FDA and other regulatory bodies could adversely affect our revenues, costs and results of operations.
Some of our products, particularly those sold by DNAG, are sold for research purposes in the U.S. We do not promote these products for clinical diagnostic use and they are labeled “For Research Use Only”, or RUO. If the FDA were to disagree with our RUO designation of a product, we could be forced to stop selling the product until appropriate regulatory clearance or approval has been obtained.
In the ordinary course of business, we must frequently make subjective judgments with respect to compliance with applicable laws and regulations. If regulators subsequently disagree with the manner in which we have sought to comply with these regulations, we could be subjected to substantial civil and criminal penalties, as well as product recall, seizure or injunction with respect to the sale of our products. The assessment of any civil and criminal penalties against us could severely impair our reputation within the industry and any limitation on our ability to manufacture and market our products could have a material adverse effect on our business.
Our Inability to Respond to Changes in Regulatory Requirements Could Adversely Affect Our Business.
We believe that our products and procedures are in material compliance with all applicable FDA regulations, ISO requirements, and other applicable regulatory requirements, but the regulations regarding the manufacture and sale of our products, the Quality System Regulation (“QSR”) and ISO requirements, and other requirements may be unclear and are subject to change. Newly promulgated regulations could require changes to our products, necessitate additional clinical trials or procedures, or make it impractical or impossible for us to market our products for certain uses, in certain markets, or at all. The FDA and other regulatory authorities also have the ability to change the requirements for obtaining product approval and/or impose new or additional requirements as part of the approval process. These changes or new or additional requirements may occur after the completion of substantial clinical work and other costly development activities. The implementation of such changes or new or additional requirements may result in additional clinical trials and substantial additional costs and could delay
30
Table of Contents
or make it more difficult or complicated to obtain approvals and sell our products. In addition, the FDA may revoke an Emergency Use Authorization under which our products are sold, where it is determined that the underlying health emergency no longer exists or warrants such authorization. Such revocation would preclude the sale of our affected products unless and until a further regulatory approval or authorization is obtained. We cannot predict the effect, if any, that these changes might have on our business, financial condition or results of operations.
Our Inability to Manufacture Products in Accordance With Applicable Specifications, Performance Standards or Quality Requirements Could Adversely Affect Our Business.
The materials and processes used to manufacture our products must meet detailed specifications, performance standards and quality requirements to ensure our products will perform in accordance with their label claims, our customers’ expectations and applicable regulatory requirements. As a result, our products and the materials used in their manufacture or assembly undergo regular inspections and quality testing. Factors such as defective materials or processes, mechanical failures, human errors, environmental conditions, changes in materials or production methods, and other events or conditions could cause our products or the materials used to produce or assemble our products to fail inspections and quality testing or otherwise not perform in accordance with our label claims or the expectations of our customers.
Any failure or delay in our ability to meet the applicable specifications, performance standards, quality requirements or customer expectations could adversely affect our ability to manufacture and sell our products or comply with regulatory requirements. These events could, in turn, adversely affect our revenues and results of operations.
We Are Subject to Numerous Government Regulations in Addition to FDA Requirements, Which Could Increase Our Costs and Affect Our Operations.
In addition to the FDA and other regulations described previously, laws and regulations in some states may restrict our ability to sell products in those states. While we intend to work with state legislators and regulators to remove or modify any applicable restrictions, there is no guarantee we will be successful in these efforts.
We must also comply with numerous laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, disposal of hazardous substances, labor or employment practices and the configuration and operation of the websites through which we advertise our products. Compliance with these laws or any new or changed laws regulating our business could result in substantial costs. Because of the number and extent of the laws and regulations affecting our industry, and the number of governmental agencies whose actions could affect our operations, it is impossible to reliably predict the full nature and impact of these requirements. To the extent the costs and procedures associated with complying with these laws and requirements are substantial or it is determined that we do not comply, our business and results of operations could be adversely affected.
Failure To Comply With Privacy, Security and Breach Notification Regulations May Increase Our Costs.
The Company believes it is neither a covered entity nor a business associate of a covered entity and is not responsible for complying with the Health Insurance Portability and Accountability Act of 1996, as amended (“HIPAA”). However, the Company has in place certain administrative, technical and physical safeguards to protect the privacy and security of consumers’ personal information and endeavors to comply with all applicable state and federal laws with respect to the protection of consumers’ personal information. The Company is required to comply with varying state privacy, security and breach reporting laws. If we do not comply with existing or new laws and regulations related to properly transferring data containing consumers’ personal information, we could be subject to monetary fines, civil penalties or criminal sanctions. In addition to other federal and state laws that protect the privacy and security of consumers’ personal information, we may be
31
Table of Contents
subject to enforcement and interpretations by various governmental authorities and courts resulting in complex compliance issues. For example, we could incur damages under state laws pursuant to an action brought by a private party for the wrongful use or disclosure of consumers’ personal information.
Compliance With Regulations Governing Public Company Corporate Governance and Reporting is Complex and Expensive.
Many laws and regulations impose obligations on public companies, and these obligations have increased the scope, complexity and cost of corporate governance, reporting and disclosure practices. Examples include the Sarbanes-Oxley Act of 2002, the requirements of The NASDAQ Global Market, The Dodd-Frank Wall Street Reform and Consumer Protection Act, the SEC’s requirements for public companies to provide financial statements in interactive data format using the eXtensible Business Reporting Language (“XBRL”), and the International Financial Reporting Standards conversion requirements. Our implementation of certain aspects of these laws and regulations has required and will continue to require substantial management time and oversight and may require us to incur significant additional accounting and legal costs. We continually evaluate and monitor developments with respect to new and proposed rules and cannot predict or estimate the ultimate amount of additional costs we may incur or the timing of such costs. These laws and regulations are also subject to varying interpretations, in many cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. Although we are committed to maintaining high standards of corporate governance and public disclosure, if we fail to comply with any of these requirements, legal proceedings may be initiated against us, which may adversely affect our business.
FDA Regulation of Laboratory-Developed Tests and Genetic Testing Could Affect Demand For Our Products.
The FDA has regulatory responsibility over instruments, test kits, reagents and other devices used to perform diagnostic testing by clinical laboratories. In the past, the FDA has taken the position that it has regulatory authority over laboratory-developed tests, or LDTs, but has exercised enforcement discretion in not regulating most LDTs performed by high complexity CLIA-certified laboratories. LDTs are tests designed, developed, and performed in-house by a laboratory. Such laboratories are subject to regulation under CLIA but have not been subject to regulation by FDA under the agency’s medical device requirements. A significant portion of the total volume of genetic or molecular testing is performed with LDTs.
In mid-2010, the FDA announced that it would begin regulating LDTs, including laboratory developed molecular tests, and in October 2014 issued proposed guidance on the regulation of LDTs for public comment. On January 13, 2017, the FDA released a discussion paper synthesizing public comments on the 2014 draft guidance documents and outlining a possible approach to regulation of LDTs. The discussion paper has no legal status and does not represent a final version of the LDT draft guidance documents. We cannot predict what policies will be adopted with respect to regulating LDTs.
Our subsidiary, DNAG, sells its DNA collection systems to certain laboratories and other customers for use with LDTs. The FDA’s increased regulation of LDTs could make it more difficult for laboratories and other customers to continue offering LDTs that involve genetic or molecular testing. This, in turn, could increase costs, delay the introduction of new LDT’s and reduce demand for DNAG’s products and adversely impact our revenues.
Evolving Legislative, Judicial and Ethical Standards on the Use of Technology and Biotechnology Could Affect Our Molecular Collection Systems Business.
The adoption of genetic testing is occurring within the broader context of a myriad of decisions related to genetic patenting and genotyping. Issues associated with regulatory requirements, health insurance, data access and
32
Table of Contents
privacy, intellectual property protection, national and international legislative initiatives and other variables impact the widespread adoption of genetic testing or specific segments or tests within the genetic testing market. These developments could impact sales of our molecular collection systems products.
Federal and State Laws Pertaining to Healthcare Fraud and Abuse Could Adversely Affect Our Business, Financial Condition and Results of Operations.
We are subject to various federal and state laws targeting fraud and abuse in the healthcare industry, including anti-kickback laws, false claims laws, laws constraining the sales, marketing and promotion of medical devices by limiting the kinds of financial arrangements that manufacturers of these products may enter into with physicians, hospitals, laboratories and other potential purchasers of medical devices, and laws requiring the reporting of certain transactions between manufacturers and healthcare professionals. Violations of these laws are punishable by criminal or civil sanctions, including substantial fines, imprisonment and exclusion from participation in government healthcare programs such as Medicare and Medicaid. Many of the existing requirements have not been definitively interpreted by state authorities or courts, and available guidance is limited. Unless and until we are in full compliance with these laws, we could face enforcement action and fines and other penalties, and could receive adverse publicity, all of which could materially harm our business. In addition, changes in or evolving interpretations of these laws, regulations, or administrative or judicial interpretations, may require us to change our business practices or subject our business practices to legal challenges, which could have a material adverse effect on our business, financial condition and results of operations.
Our International Sales Create Potential Exposure Under Anti-Corruption Laws.
In 2017, approximately $45.6 million of our consolidated net revenues were generated from sales in a variety of foreign countries. These international activities subject us to the U.S. Foreign Corrupt Practices Act (“FCPA”), the U.K. Bribery Act and other laws that prohibit improper payments or offers of payments to foreign governments and their officials and political parties by business entities for the purpose of obtaining or retaining business. We have operations, agreements with third parties and make sales in countries known to experience corruption. Further international expansion may create increased exposure to such practices. Our activities in these countries create the risk of unauthorized payments or offers of payments by one of our employees, consultants, sales agents or distributors that could be in violation of various laws, including the FCPA, even though these parties are not always subject to our control. It is our policy to implement safeguards to discourage these practices by our employees and distributors, including employee training, contracts requiring compliance with the FCPA and similar rules, and standard reviews of our distributors. However, our existing safeguards and any future improvements may not prove to be effective, and our employees, consultants, sales agents or distributors may engage in conduct for which we might be held responsible. Violations of the FCPA and other laws may result in criminal or civil sanctions, which could be severe and we may be subject to other liabilities, which could negatively affect our reputation, business, results of operations and financial condition.
Risks Relating to Our Industry, Business and Strategy
Our Ability to Sell Products Could be Adversely Affected by Competition From New and Existing Products.
The markets we serve are highly competitive and rapidly changing and we expect competition to intensify as technological advances are made and become more widely known, and as new products reach the market. Many of our principal competitors have considerably greater financial, technical and marketing resources than we do. As new products enter the market, our products may become obsolete or a competitor’s products may be more effective or more effectively marketed and sold than ours. In addition, there can be no assurance that our competitors will not succeed in obtaining regulatory approval for new products that would render our
33
Table of Contents
technologies and products obsolete or otherwise commercially unattractive, or introduce or commercialize such products, before we can do so. If we fail to convince our customers of the advantages and economic value of our products or otherwise maintain and enhance our competitive position, our customers may decide to use products developed by competitors which could result in a loss of revenues. These developments could have a material adverse effect on our business, financial condition and results of operations.
We also face competition from products that are sold at a lower price. Where this occurs, customers may choose to buy lower cost products from third parties or we may be forced to sell our products at a lower price, both of which could result in a loss of revenues or a lower gross margin contribution from the sale of our products. We may also be required to increase our marketing efforts in order to compete effectively, which would increase our costs.
Consolidation in the Healthcare Industry Could Adversely Affect Our Future Revenues and Operating Results.
The healthcare industry has experienced a significant amount of consolidation. As a result of this consolidation, competition to provide goods and services to customers has increased. In addition, group purchasing organizations and integrated health delivery networks have served to concentrate purchasing decisions for some customers, which has also placed pricing pressure on medical device suppliers. We may not be able to compete successfully in such a consolidated industry. We believe industry consolidation may continue as companies attempt to strengthen or hold their market positions and as more companies are acquired or cease operating. Further consolidation in the industry could exert additional pressure on the prices of our products.
Our Research, Development and Commercialization Efforts May Not Succeed and Our Competitors May Develop and Commercialize More Effective or Successful Products.
In order to remain competitive, we must regularly commit substantial resources to research and development and the commercialization of new or enhanced products. The research and development process generally takes a significant amount of time from product inception to commercial launch. This process is conducted in various stages. During each stage there is a substantial risk that we will not achieve our goals on a timely basis, or at all, and we may have to abandon a new or enhanced product in which we have invested substantial time and money.
During 2017, 2016 and 2015 we incurred $13.4 million, $9.8 million and $11.7 million, respectively, in research and development expenses. We expect to continue to incur significant costs related to our research and development activities.
Successful products require significant development and investment, including testing to demonstrate their performance capabilities, cost-effectiveness or other benefits prior to commercialization. Regulatory approval must be obtained before most products may be sold and additional development efforts on these products may be required before any regulatory authority will review them. As noted above, regulatory authorities may not approve these products for commercial sale or may substantially delay or condition approval. Even if a product is developed and all applicable regulatory approvals are obtained, there may be little or no market for the product and entry into or development of new markets for our products may require an investment of substantial resources, such as new employees, offices and manufacturing facilities. Moreover, we may spend a significant amount of money on manufacturing facilities, advertising or other activities and fail to develop a market for the product. Other factors that could affect the success of our efforts include our ability to manufacture products in a cost-effective manner, whether we can obtain necessary intellectual property rights and protection and our ability to obtain reimbursement authorizations in the markets where the product will be sold.
Accordingly, if we fail to develop and gain commercial acceptance for our products, or if competitors develop more effective products or a greater number of successful new products, customers may decide not to purchase our products or may purchase and use products developed by our competitors. This would result in a loss of revenues and adversely affect our results of operations, cash flow and business.
34
Table of Contents
Failure to Successfully Commercialize Rapid Point–of-Care Ebola and Zika Tests Could Adversely Affect Our Results of Operations and Business Prospects.
We have completed development of a rapid Ebola antigen test using our OraQuick® technology platform. In 2015 and 2016, under an EUA (Emergency Use Authorization) received from the FDA, we sold $2.5 million of product to the CDC for field testing. There were minimal sales of this product during 2017. It is uncertain whether, and to what extent, we will be successful in obtaining additional or sustainable purchase commitments for our Ebola test. Significant efforts are under way to develop a vaccine for the Ebola virus. If a vaccine is developed and widely deployed in the countries at risk for Ebola, the need for testing could be reduced as could the demand for our product.
There is also no assurance that our Ebola test will perform at a level necessary to receive all of the regulatory approvals required for its use. In addition, it is possible that the FDA may revoke our EUA if it is determined that Ebola is no longer an emergency warranting that authorization. Failure to successfully obtain the required regulatory approvals or receive sustainable and significant purchase commitments for our Ebola test could adversely affect our revenues and results of operations.
During 2016 and 2017, we have worked to complete development of a rapid Zika antibody test, also using our OraQuick® technology platform. Once development is complete and a prototype test is finalized, we intend to seek EUA for this product. There is no assurance that we will be able to successfully complete development of our Zika test or that we will be able to obtain EUA or other necessary regulatory approvals required to commercialize this product. In addition, even if our development efforts are successful and such approvals are received, there is no assurance that we will be able to obtain significant or sustainable purchase commitments for our Zika test. Our failure to successfully complete development, obtain the required regulatory approvals or receive substantial purchase commitments for our Zika test could adversely affect our revenues and results of operations.
Failure to Achieve Our Financial and Strategic Objectives Could Have a Material Adverse Impact on Our Business Prospects.
As a result of any number of risk factors identified in this Annual Report, no assurance can be given that we will be successful in implementing our financial and strategic objectives, including our efforts to increase sales of our products or continue growing our business. In addition, the funds for research, clinical development and other projects have in the past come primarily from our business operations. If our business slows and we have less money available to fund research and development and clinical programs, we will have to decide at that time which programs to cut, and by how much. Similarly, if adequate financial, personnel, equipment or other resources are not available, we may be required to delay or scale back our business. Our operations will be adversely affected if our total revenue and gross profits do not correspondingly increase or if our technology, product, clinical and market development efforts are unsuccessful or delayed. Furthermore, our failure to successfully introduce new or enhanced products and develop new markets could have a material adverse effect on our business and prospects.
If We Lose Our Key Personnel or Are Unable to Attract and Retain Qualified Personnel as Necessary, Our Business Could be Harmed.
Our success depends to a large extent upon the contributions of our executive officers, management and sales, marketing, operations and scientific staff. Our business may be harmed by the loss of a significant number of our executive officers or senior managers. We may not be able to attract or retain a sufficient number of qualified employees in the future due to the intense competition for qualified personnel among medical products and other life science businesses. Our ability to recruit such employees will depend on a number of factors, including compensation, benefits, work location, the prospects of our Company, and the possibility for advancement within our organization. We generally do not enter into employment agreements requiring our employees to work for us for any specified period.
35
Table of Contents
If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will adversely affect our ability to effectively manufacture, sell and market our products, to meet the demands of our strategic partners in a timely fashion, or to support research, development and clinical programs. Although we believe we will be successful in attracting and retaining qualified personnel, competition for experienced scientists and other qualified personnel from numerous companies and academic and other research institutions may limit our ability to do so on acceptable terms.
If We Do Not Successfully Manage the Transition Associated with the Appointment of a New Chief Executive Officer and Chief Financial Officer, Our Business May Be Harmed
In January 2018, we announced the retirement of our President and Chief Executive Officer, which will become effective as of April 1, 2018, and the retirement of our Chief Financial Officer and Chief Operating Officer, which will become effective on or before June 30, 2018, with the exact date to be determined based on the timing for our appointment of a new Chief Financial Officer. Any changes that may result from hiring a new Chief Executive Officer or Chief Financial Officer can create uncertainty and may negatively impact our ability to execute our business strategy quickly and effectively. Management transition periods can be difficult as the new management gains detailed knowledge of our operations, and friction or further management changes or disruptions could result from changes in strategy and management style. Until we fully integrate our new Chief Executive Officer and Chief Financial Officer, we may be unable to successfully manage and grow our business, and our results of operations and financial condition could suffer as a result. Any changes in our organization as a result of this management transition may have a disruptive impact and could have a material adverse effect on our business, financial condition and results of operations.
Acquisitions or Investments May Not Generate the Expected Benefits and Could Disrupt Our Ongoing Business, Distract Our Management, Increase Our Expenses and Adversely Affect Our Business.
We may pursue strategic acquisitions or investments as a way to expand our business. These activities, and their impact on our business, are subject to many risks, including the following:
| • | Suitable acquisitions or investments may not be found or consummated on terms or schedules that are satisfactory to us or consistent with our objectives; |
| • | We may be unsuccessful in competing for acquisitions with other entities, some of which have greater financial resources or may be better able to realize synergies with a potential target; |
| • | The benefits expected to be derived from an acquisition or investment may not materialize and could be affected by numerous factors, such as regulatory developments, insurance reimbursement, our inexperience with new businesses or markets, general economic conditions and increased competition; |
| • | We may be unable to successfully integrate an acquired company’s personnel, assets, management, information technology systems, accounting policies and practices, products and/or technology into our business; |
| • | Worse than expected performance of an acquired business may result in the impairment of intangible assets; |
| • | Acquisitions may require substantial expense and management time and could disrupt our business; |
| • | We may not be able to accurately forecast the performance or ultimate impact of an acquired business; |
| • | We may have difficulties in coordinating geographically separate organizations; |
| • | We may fail to successfully manage relationships with customers, distributors and suppliers of an acquired business; |
| • | An acquisition may result in a diversion of resources from our existing products, business and technologies; |
36
Table of Contents
| • | An acquisition and subsequent integration activities may require greater capital and other resources than originally anticipated at the time of acquisition; |
| • | An acquisition may result in the incurrence of unexpected expenses, stockholder lawsuits, the dilution of our earnings or our existing stockholders’ percentage ownership, or potential losses from undiscovered liabilities not covered by an indemnification from the seller(s) of the acquired business; |
| • | An acquisition may result in the loss of our or the acquired company’s key personnel, customers, distributors or suppliers; and |
| • | An acquisition of a foreign business may involve additional risks, including, but not limited to, foreign currency exposure, liability or restrictions under foreign laws or regulations, and our inability to successfully assimilate differences in foreign business practices or overcome language or cultural barriers and other inherent risks of operating in unfamiliar legal and regulatory environments. |
The occurrence of one or more of the above or other factors may prevent us from achieving all or a significant part of the benefits expected from an acquisition or investment. This may adversely affect our financial condition, results of operations and ability to grow our business or otherwise achieve our financial and strategic objectives.
Our Revenues Could be Affected by Third-Party Reimbursement Policies and Potential Cost Constraints.
The end-users of our products include hospitals, physicians and other healthcare providers. Use of our products could be adversely impacted if these end-users do not receive adequate reimbursement for the cost of our products from their patients’ healthcare insurers or payors. Our net sales could also be adversely affected by changes in reimbursement policies of governmental or private healthcare payors, including in particular the level of reimbursement for our products.
In the United States, hospitals, physicians and other healthcare providers who purchase diagnostic products generally rely on third-party payors, such as private health insurance plans, Medicare and Medicaid, to reimburse all or part of the cost of the product and procedure. The overall escalating cost of medical products and services has led to, and will continue to lead to, increased pressures on the healthcare industry, both foreign and domestic, to reduce the cost of products and services. Given the efforts to control and reduce healthcare costs in the United States in recent years, currently available levels of reimbursement may not continue to be available in the future for our existing products or products under development. Third-party reimbursement and coverage may not be available or adequate in either the United States or international markets, current reimbursement amounts may be decreased in the future and future legislation, and regulation or reimbursement policies of third-party payors, may reduce the demand for our products or our ability to sell our products on a profitable basis.
Changes in Healthcare Regulation Could Affect Our Revenues, Costs and Financial Condition.
In recent years, there have been numerous initiatives at the federal and state level for comprehensive reforms affecting the payment for, the availability of and reimbursement for healthcare services in the United States. These initiatives have ranged from proposals to fundamentally change federal and state healthcare reimbursement programs, including providing comprehensive healthcare coverage to the public under government-funded programs, to minor modifications to existing programs. One example is the Patient Protection and Affordable Care Act, the Federal healthcare reform law enacted in 2010 (the “Affordable Care Act”). Similar reforms may occur internationally.
Legislative and regulatory bodies are likely to continue to pursue healthcare reform initiatives in many forms and may continue to reduce funding in an effort to lower overall federal healthcare spending. The U.S. government recently enacted legislation that eliminated what is known as the “individual mandate” under the Affordable Care Act and may enact other changes in the future. The ultimate content and timing of any of these types of changes
37
Table of Contents
in other healthcare reform legislation and the resulting impact on us are impossible to predict. If significant reforms are made to the healthcare system in the United States, or in other jurisdictions, those reforms may increase our costs or otherwise have an adverse effect on our financial condition and results of operations.
The Affordable Care Act imposes a 2.3% excise tax on certain transactions, including U.S. sales of many medical devices, which includes domestic sales of certain of our products. This new tax became effective in January 2013. However, the Consolidated Appropriations Act of 2016, which was enacted late 2015, suspended the tax beginning January 1, 2016, and this suspension was recently extended through December 31, 2019. It is unclear whether and to what extent this tax will impact our business, if and when the suspension is lifted.
New or Changed Testing Guidelines Could Affect Sales of Our Diagnostic Products.
From time to time, governmental agencies such as the Centers for Disease Control and Prevention, or CDC, issue diagnostic testing guidelines or recommendations, which can affect the usage of our HIV and HCV testing products. For example, domestic professional OraQuick® HIV sales have decreased in recent years in part due to customer migration to automated fourth generation HIV immunoassays performed in a laboratory, as recommended under testing guidelines issued by the CDC. In addition, some states have promulgated, or may in the future promulgate, laws and regulations that affect HIV or HCV testing. The issuance of new laws or guidelines, or changes in existing laws or guidelines, and the manner in which these new or changed laws and guidelines are interpreted and applied by healthcare practitioners, could impact the degree to which our OraQuick® rapid HIV and HCV testing products are used. New or changed laws or guidelines could affect the number of people tested, the frequency of testing and whether testing products such as our OraQuick® HIV and HCV tests are used broadly for screening large populations or in a more limited capacity as a confirmatory test or otherwise. These factors could in turn affect the level of sales of our products and our results of operations.
Reductions in Government Funding and Research Budgets Could Adversely Affect Our Business and Financial Results.
We sell our OraQuick ADVANCE® HIV-1/2 and OraQuick® HCV tests into the public health market which consists of state, county and other governmental public health agencies, community based organizations, service organizations and similar entities. We also sell these products into the hospital market. Many of these customers depend to a significant degree on grants or funding provided by governmental agencies to run their operations including programs that use our products. In international markets, we often sell our products to or through foreign governmental agencies or parties funded by such agencies.
Many of our molecular collection products are sold to researchers at academic institutions, pharmaceutical and biotechnology companies, government laboratories and private foundations. Many research customers are dependent for their funding on grants from U.S. governmental agencies such as the U.S. National Institutes of Health and agencies in other countries.
The level of available government grants or funding in the U.S. and elsewhere is unpredictable and may be affected by various factors including economic conditions, legislative and regulatory developments, political changes, civil unrest and changing priorities for research and development activities. Any reduction or delay in government funding as a result of legislative or regulatory changes or other factors, could cause our customers to delay, reduce or forego purchases of our products.
Certain Changes by the Current Administration Could Affect our Business.
The current Administration has promised to repeal and replace the Affordable Care Act, has expressed concerns with respect to existing trade agreements such as the North American Free Trade Agreement, or NAFTA, and has indicated a desire to make other regulatory changes. Changes in regulatory or economic conditions or in the laws and policies governing foreign trade, taxes, manufacturing, and development in the United States could impact
38
Table of Contents
our business. For example, changes in the level of reimbursement or the availability of funding for health care could create uncertainty for our business and negatively impact our revenues and results of operations. Similarly a substantial part of DNAG’s revenues represent sales of product from Canada into the United States. The imposition of tariffs or other changes in the trade relationship between Canada and the United States could negatively impact DNAG’s performance. Economic and regulatory changes could also affect foreign currency exchange rates which, in turn, could affect our reported financial results and our competitiveness on a worldwide basis.
Developments Related To The U.K.’s Referendum On Membership in The E.U. Could Adversely Affect Us.
On June 23, 2016, the United Kingdom (“U.K.”) held a referendum and voted in favor of exiting the European Union, or E.U. This “Brexit” referendum has created political and economic uncertainty, particularly in the U.K. and E.U. and this uncertainty may last for years. Until the precise terms and timing of the U.K.’s exit from the E.U. are determined, it will be difficult to predict the impact of the Brexit referendum. Our business in the U.K., the E.U. and world-wide could be affected during this period of uncertainty, and perhaps longer, by the impact of the Brexit referendum. The U.K.’s decision to exit the E.U. could cause volatility in global financial markets, including in global currency exchange rates, resulting in a slow-down in economic activity in the U.K., Europe or globally, negatively impact new trade agreements between the U.K and other countries, including the United States, and result in significant regulatory changes and uncertainty. One or more of these events could make it more difficult or costly to sell our products, particularly in the U.K. and Europe, and negatively affect our revenues and results of operations. The Brexit referendum may also influence other countries and result in additional countries deciding to leave the E.U. This in turn could result in additional changes and uncertainty, any or all of which could negatively impact our business.
Increases in Demand for Our Products Could Require Us to Expend Considerable Resources or Harm Our Customer Relationships if We are Unable to Meet That Demand.
If we experience significant or unexpected increases in the demand for our products, we and our suppliers may not be able to meet that demand without expending additional capital resources. These capital resources could involve the cost of new machinery or new manufacturing facilities. This would increase our capital costs, which could adversely affect our earnings. Our suppliers may be unable or unwilling to expend the necessary capital resources or otherwise expand their capacity. In addition, new manufacturing equipment or facilities may require FDA approval before they can be used to manufacture our products. To the extent we are unable to obtain or are delayed in obtaining such approvals, our ability to meet the demand for our products could be adversely affected.
If we or our suppliers are unable to develop necessary manufacturing capabilities in a timely manner, our sales could be adversely affected. If we fail to increase production volumes in a cost effective manner or if we experience lower than anticipated yields or production problems as a result of changes that we or our suppliers make in our manufacturing processes to meet increased demand, we could experience shipment delays or interruptions and increased manufacturing costs, which could also have a material adverse effect on our revenues and profitability.
Unexpected increases in demand for our products may require us to obtain additional raw materials in order to manufacture products to meet the demand. Some raw materials require significant ordering lead time and some are currently obtained from a sole supplier or a limited group of suppliers. We have long-term supply agreements with certain of these suppliers, but these long-term agreements involve risks for us, such as our potential inability to obtain an adequate supply of raw materials and components and our reduced control over pricing, quality and timely delivery. It is also possible that one or more of these suppliers may become unwilling or unable to deliver materials to us. Any shortfall in our supply of raw materials and components, or our inability to quickly and cost-effectively obtain alternative sources for this supply, could have a material adverse effect on our ability to meet increased demand for our products. This could negatively affect our total revenues or cost of sales and related profits.
39
Table of Contents
Our inability to meet customer demand for our products could also harm our customer relationships and impair our reputation within the industry. This, in turn, could have a material adverse effect on our business and prospects.
We Rely on Information Technology in Our Operations and Any Material Failure, Inadequacy, Interruption or Security Breach of that Technology Could Harm Our Ability to Efficiently Operate Our Business.
We rely heavily on enterprise resource planning and other complex information technology systems across our operations and on the internet, including for management of inventory, purchase orders, invoices, shipping, interactions with our third-party logistics provider, revenue and expense accounting, online business, consumer call support, and various other processes and transactions. Our ability to effectively manage our business, coordinate the production, distribution and sale of our products, respond to customer inquiries, and ensure the timely and accurate recording and disclosure of financial information depends significantly on the reliability and capacity of these systems and the internet.
The failure of any of the foregoing systems to operate effectively, problems with transitioning to upgraded or replacement systems, or disruptions in the operation of the internet, could cause delays in product sales and reduced efficiency of our operations. Significant expenditures could be required to remediate any such problem.
Security Breaches and Other Disruptions Could Compromise Our Information, Expose Us To Liability and Harm Our Reputation and Business.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, personal information, our proprietary business information and that of our customers, suppliers and business partners, and personally identifiable information of our employees in our data centers and on our networks. Secure maintenance and transmission of this information is critical to our operations business strategy. We generally rely on commercially available systems, software, tools and domestically available monitoring to provide security for processing, transmitting and storing this sensitive date.
Cyber-attacks could result in unauthorized access to our computer systems or our third party IT service provider’s systems and, if successful, misappropriate personal or confidential information. In addition, a contractor or other third party with whom we do business may attempt to circumvent our security measures or obtain such information, and may purposefully or inadvertently cause a breach involving sensitive information. While we will continue to evaluate and implement additional protective measures to reduce the risk and detect cyber incidents, cyberattacks are becoming more sophisticated and frequent and the techniques used in such attacks change rapidly. Despite our cybersecurity measures (including employee and third party training, monitoring of networks and systems and maintenance of back up of protective systems) which are continuously reviewed and upgraded, our information technology networks and infrastructure may still be vulnerable to damage, disruptions or shut downs due to attack by hackers or breaches, voyeur or malfeasance. Even the most well protected IT networks, systems and facilities remain potentially vulnerable because the techniques used in attempted security breaches are continually evolving and generally are not recognized until launched against a target or, in some cases, are designed not to be detected and, in fact, may not be detected. Any such compromise of our or our third party’s IT service providers’ data security and access, public disclosure, or loss of personal or confidential business information, could result in legal claims and proceedings, liability under laws to protect privacy of personal information, and regulatory penalties, and could disrupt our operations, require significant management attention and resources to remedy any damages that result, and damage our reputation and customers willingness to transact business with us, any of which could adversely affect our business.
40
Table of Contents
Risks Relating to Collaborators
The Use of Sole Supply Sources or Third-Party Suppliers For Critical Components of Our Products Could Adversely Affect Our Business.
We currently purchase certain critical components of our products from sole supply sources or other third-party suppliers. For example, the biological antigens and antibodies, nitrocellulose and certain other components required to make our OraQuick HIV, HCV and Ebola products are currently purchased from sole source suppliers. Our OraSure QuickFlu® test and the fully automated high-throughput drug assays sold with our Intercept i2® device are manufactured and supplied by sole source suppliers and the conjugates used in our MICROPLATE oral fluid drugs-of-abuse assays are obtained from third party suppliers.
We have contracted with a third party in Thailand for the assembly of OraQuick® HIV device and the OraQuick® HIV Self-Test in order to supply certain international markets. In addition, our subsidiary, DNAG, uses two third-party manufacturers to supply virtually all of its products, including its Oragene® line of collection kits. Many of the raw materials and components used in its products are also purchased from third parties, a critical one of which is obtained from a sole source supplier.
If our third-party suppliers are unable or unwilling to supply or manufacture a required component or product or if they make changes to a component, product or manufacturing process or do not supply materials meeting our specifications, we may need to find another source and/or manufacturer. This could require that we perform additional development work and it may be difficult to find such an alternate supply source in a reasonable time period or on commercially reasonable terms, if at all. We may also need to obtain FDA or other regulatory approvals for the use of an alternative component or for changes to our products or manufacturing process. Completing that development and obtaining such approvals could require significant time and expense and such approvals may not occur at all. The availability of critical components and products from sole supply sources or other third parties could also reduce our control over pricing, quality and timely delivery. These events could either disrupt our ability to manufacture and sell certain of our products into one or more markets or completely prevent us from doing so, and could increase our costs. Any such event could have a material adverse effect on our results of operations, cash flow and business.
Our Failure to Maintain Existing Distribution Channels, or Develop New Distribution Channels, May Result in Lower Revenues.
We have marketed many of our products by collaborating with laboratories, diagnostic companies and distributors. Our sales depend to a substantial degree on our ability to sell products to these customers and on the marketing and distribution abilities of the companies with which we collaborate.
Relying on distributors or others to market and sell our products could harm our business for various reasons, including:
| • | We may not be able to find suitable distributors to distribute our products on satisfactory terms, or at all; |
| • | Our distributors or other customers may not fulfill their contractual obligations to us or otherwise market and distribute our products in the manner or at the levels we expect; |
| • | We do not control the incentives provided by our distributors to their sales personnel and the effectiveness of these incentives could affect sales of our products; |
| • | Agreements with distributors may terminate prematurely due to disagreements or may result in litigation between the parties; |
| • | We may not be able to renew existing distribution agreements on acceptable terms, or at all; |
| • | Our distributors may not devote sufficient resources or priority to the sale of our products; |
41
Table of Contents
| • | Our distributors may prioritize their own private label products that compete with our products; |
| • | Our existing distributor relationships or contracts may preclude or limit us from entering into arrangements with other distributors; and |
| • | We may not be able to negotiate future distribution agreements on acceptable terms, or at all. |
Although we will try to maintain and expand our business with distributors and customers and require that they fulfill their contractual obligations, there can be no assurance that such companies will do so or that new distribution channels will be available on satisfactory terms. As a result, our revenues and business could be adversely affected.
We May Need Strategic Partners to Assist in Developing and Commercializing Some of Our Products.
Although we may elect to pursue some product opportunities independently, opportunities that require a technology controlled by a third party, a significant level of investment for development and commercialization or a distribution network beyond our existing sales force may necessitate involving one or more strategic partners. Our strategy for development and commercialization of products may entail entering into arrangements with distributors or other corporate parties, universities, research laboratories, government agencies, licensees and others. Relying on collaborative relationships could be risky to our business for a number of reasons, including:
| • | We may be required to transfer material rights to such strategic collaborators, government agencies, licensees and others; |
| • | Our collaborators may not devote sufficient resources or attach a sufficiently high priority to the success of our collaboration; |
| • | Our collaborators may not obtain regulatory approvals necessary to continue the collaborations in a timely manner; |
| • | Our collaborators may be acquired by another company, decide to terminate our collaborative arrangement or become insolvent; |
| • | Our collaborators may develop technologies or components competitive with our products; |
| • | Our collaborators may fail to deliver technologies or components that satisfy market requirements or such products may fail to perform properly; |
| • | Disagreements with collaborators could result in the termination of the relationship or litigation; |
| • | Collaborators may not have sufficient capital resources; and |
| • | We may not be able to negotiate future collaborative arrangements, or renewals of existing collaborative agreements, on acceptable terms or at all. |
While we generally expect that our collaborative partners will have an economic motivation to succeed in performing their contractual responsibilities, there is no assurance that they will do so, either at the level required or at all, and the amount and timing of resources to be devoted to these activities will be controlled by others. Reliance on strategic agreements can also make it difficult to accurately forecast our future revenues operating results. There can be no assurance that the expected revenues or profits will be fully derived from such arrangements.
Actions of Third-Party Inventory Management and Logistics Providers Could Adversely Affect Our Ability to Supply Products to Our Customers.
We use third-party logistics providers to store and manage certain of our finished goods inventory and ship finished product to our customers. We have selected highly reputable providers with extensive experience in the
42
Table of Contents
logistics field for these services. However, in the event any of our providers lose or damage our products, experience a casualty or catastrophic event at a warehouse or otherwise fail to provide safe storage and timely handling and delivery of our products, we could incur additional costs, experience difficulty in supplying our products to our customers or suffer damage to our reputation in the industry. These events could, in turn, reduce our revenues and adversely affect our results of operations.
Risks Relating to Intellectual Property
Our Success Depends on Our Ability to Protect Our Proprietary Technology.
Our industry places considerable importance on obtaining patent, trademark and trade secret protection, as well as other intellectual property rights, for new technologies, products and processes. Our success depends, in part, on our ability to develop and maintain a strong intellectual property portfolio or obtain licenses to patents and technologies, both in the United States and in other countries. If we cannot continue to develop, obtain and protect intellectual property rights, our revenues and gross profits could be adversely affected. Moreover, our current and future licenses or other rights to patents and other technologies may not be adequate for the operation of our business.
As appropriate, we intend to file patent applications and obtain patent protection for our proprietary technology. These patent applications and patents will cover, as applicable, compositions of matter for our products, methods of making those products, methods of using those products and apparatuses relating to the use or manufacture of those products. However, there have been changes to the patent laws and proposed changes to the rules of the U.S. Patent and Trademark Office, which may impact our ability to protect our technology and enforce our intellectual property rights. For example, in 2011, the U.S. enacted sweeping changes to the U.S. patent system under the Leahy-Smith America Invents Act (the “AIA”), including changes that would transition the U.S. from a “first-to-invent” system to a “first-to-file” system and alter the processes for challenging issued patents. These changes could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
We also rely on trade secrets, know-how and continuing technological advancements to protect our proprietary technology. We have entered, and will continue to enter, into confidentiality agreements with our employees, consultants, advisors and collaborators. Our employees and third-party consultants also sign agreements requiring that they assign to us interests in inventions and original expressions and any patents or copyrights arising from their work. However, these parties may not honor these agreements.
We cannot guarantee that the process of filing patents, the laws governing trade secrets and proprietary information, or any agreements we enter into with employees, consultants, advisors or collaborators will provide adequate protection of our intellectual property rights. For example, employees, consultants and others who participate in the development of our products may breach their agreements with us regarding our intellectual property, and we may not have adequate remedies for the breach. We also may not be able to effectively protect our intellectual property rights in some foreign countries, as many countries do not offer the same level of legal protection for intellectual property as the United States. Furthermore, for a variety of reasons, we may decide not to file for patent, copyright or trademark protection outside of the U.S. Our trade secrets could become known through other unforeseen means. Notwithstanding our efforts to protect our intellectual property, our competitors may independently develop similar or alternative technologies or products that are equal or superior to our technology. Our competitors may also develop similar products without infringing on any of our intellectual property rights or design around our proprietary technologies.
Moreover, issued patents remain in effect for a fixed period and after expiration will not provide protection of the inventions they cover. Once our patents expire, we may be faced with increased competition, which could reduce our revenues. We may also not be able to successfully protect our rights to unpatented trade secrets and know-how.
43
Table of Contents
Some of our employees, including scientific and management personnel, were previously employed by competing companies. Although we encourage and expect all of our employees to abide by any confidentiality agreement with a prior employer, competing companies may allege trade secret violations and similar claims against us. In addition, some of these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants or advisers have prior employment or consulting relationships.
We may collaborate with universities and governmental research organizations which, as a result, may acquire part of the rights to any inventions or technical information derived from our collaboration with them.
To facilitate development and commercialization of a proprietary technology base, we may need to obtain licenses to patents or other proprietary rights from other parties. Obtaining and maintaining such licenses may require the payment of substantial amounts. In addition, if we are unable to obtain these types of licenses, our product development and commercialization efforts may be delayed or precluded.
We May Become Involved in Intellectual Property Disputes, Which Could Increase our Costs and Limit or Eliminate Our Ability to Sell Products or Use Certain Technologies.
From time to time, we may seek to enforce our patents or other intellectual property rights through litigation. In addition, there are a large number of patents and patent applications in our product areas, and additional patents may be issued to third parties relating to our product areas. We, our customers or our suppliers may be sued for infringement of patents or misappropriation of other intellectual property rights with respect to one or more of our products. Litigation in our industry regarding patent and other intellectual property rights is prevalent and is expected to continue. We may also have disputes with parties that license patents to us if we believe the license is no longer needed for our products or the licensed patents are no longer valid or enforceable.
Our industry is characterized by a large number of patents, and the claims of these patents appear to overlap in many cases. As a result, there is a significant amount of uncertainty regarding the extent of patent protection and infringement. Companies may have pending patent applications, which are typically confidential for the first eighteen months following filing, that cover technologies we incorporate in our products. Accordingly, we may be subjected to substantial damages for past infringement or be required to modify our products or stop selling them if it is ultimately determined that our products infringe a third party’s proprietary rights. In addition, governmental agencies could commence investigations or criminal proceedings against our employees or us relating to claims of misuse or misappropriation of another party’s proprietary rights.
Intellectual property litigation is costly. As such, our involvement in litigation or other legal proceedings with respect to patents or other intellectual property and proprietary technology, either as a plaintiff or defendant, could adversely affect our revenues, market share, results of operations and business because:
| • | As is common with major litigation, it could consume a substantial portion of managerial and financial resources; |
| • | Its outcome would be uncertain and a court may find that our patents are invalid or unenforceable in response to claims by another party or that the third-party patent claims are valid and infringed by our products; |
| • | An adverse outcome could subject us to the loss of the protection of our patents or to liability in the form of past royalty payments, penalties, reimbursement of litigation costs and legal fees, special and punitive damages, or future royalty payments, any of which could significantly affect our future earnings; |
| • | Governmental agencies may commence investigations or criminal proceedings against our employees, former employees and us relating to claims of misappropriation or misuse of another party’s proprietary rights; |
44
Table of Contents
| • | Failure to obtain a necessary license upon an adverse outcome could prevent us from selling our current products or other products we may develop or acquire; |
| • | We may be required to alter our product, given the proprietary rights of others; |
| • | The pendency of any litigation may in and of itself cause our distributors and customers to reduce or terminate purchases of our products; and |
| • | A court could award a preliminary and/or permanent injunction, which would prevent us from selling our current or future products. |
We may indemnify some customers and strategic partners under our agreements with such parties if our products or activities have actually or allegedly infringed upon, misappropriated or misused another party’s proprietary rights. Further, our products may contain technology provided to us by other parties, such as contractors, suppliers or customers, and we may have little or no ability to determine in advance whether such technology infringes the intellectual property rights of a third party. These other parties may also not be required or financially able to indemnify us in the event that an infringement or misappropriation claim is asserted against us.
We may also become involved in other types of disputes regarding intellectual property rights, including state, federal or foreign court litigation, and patent interference, patent reexamination, patent reissue, or trademark opposition proceedings in the United States Patent and Trademark Office. Opposition or revocation proceedings could be instituted in a foreign patent office as well. Under the AIA, various forms of post issuance patent review proceedings have been authorized, including an inter-parties review process. These proceedings permit certain persons to challenge the validity of a patent on the grounds that it was known from the prior art. The filing of such proceedings, or the issuance of an adverse decision in such proceedings, could result in the loss of valuable patent rights that could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Risks Relating to Products, Marketing and Sales
Our Future Success Depends Upon Market Acceptance of Our Existing and Future Products.
We believe successful new product introductions provide a significant competitive advantage because customers make an investment of time in selecting and learning to use a new product and are reluctant to switch thereafter. Our future success will depend, in part, on the market acceptance, and the timing of such acceptance, of new products such as our OraQuick® HIV Self-Test , OraQuick® Ebola test, OraQuick® Zika test, OMNIgene® • GUT and OMNIgene® • SPUTUM product offerings, and other new products or technologies that may be developed or acquired. To achieve market acceptance, we and/or our distributors will likely be required to undertake substantial marketing efforts and spend significant funds to inform potential customers and the public of the existence and perceived benefits of these products. In addition, governmental funding for the purchase of our products may be needed to help create market acceptance and expand the use of our products.
There may be limited evidence on which to evaluate the market reaction to products that may be developed and our marketing efforts for new products may not be successful. It is also possible that governmental funding may be limited for new products, such as our OraQuick® Ebola and Zika tests or the new sample collection and stabilization products being commercialized by DNAG. As such, there can be no assurance that any products will obtain significant market acceptance and fill the market need that is perceived to exist on a timely basis, or at all.
If Acceptance and Adoption of Oral Fluid Testing and Collection Products Does Not Continue, Our Future Results May Suffer.
We have made significant progress in gaining acceptance of oral fluid testing products, particularly for (i) HIV testing in the public health, hospital, insurance and other markets, and (ii) drugs-of-abuse testing in the
45
Table of Contents
workplace and criminal justice markets. Our subsidiary, DNAG, has also made significant progress in gaining acceptance of oral fluid collection products that are used with molecular testing applications. However, the degree of acceptance for these products is uncertain, and one or more markets may resist the adoption of oral fluid products as a replacement for other testing or collection methods in use today. As a result, there can be no assurance that we will be able to expand the use of our oral fluid testing products in these or other markets.
Our Customers May Resist Adoption of Rapid Point-of-Care Diagnostic Testing.
Sales of our rapid point-of-care diagnostic products, such as our OraQuick ADVANCE® HIV-1/2, OraQuick® HCV and OraQuick® In-Home HIV tests, are an important part of our business. Rapid point-of-care tests are beneficial because, among other things, they can be administered by healthcare providers in their own facilities or used by consumers at home without sending samples to central laboratories and can help ensure that test results are delivered to the individuals being tested.
However, clinical reference laboratories and hospital-based laboratories currently provide the majority of diagnostic tests used by physicians and other healthcare providers in the U.S. In certain international markets such as Europe, diagnostic testing is performed primarily by centralized laboratories. Our future sales will depend, in part, on our ability to expand market acceptance of rapid point-of-care testing by physicians, other healthcare providers and consumers and successfully compete against laboratory testing methods and products. We expect that clinical reference and other hospital-based laboratories will continue to compete vigorously against our rapid point-of-care products. Even if we can demonstrate that our products are more cost effective, save time, or have better performance or other benefits, physicians, other healthcare providers and consumers may resist changing to rapid point-of-care tests and instead may choose to obtain diagnostic results through laboratory tests. Our failure to achieve and expand market acceptance of our rapid point-of-care diagnostic tests with customers would have a negative effect on our future sales growth.
We Expect to Face Intense Competition From Other Providers of Diagnostic Tests and Sample Collection Products.
Our rapid point-of-care tests compete with similar point-of-care products made by our competitors. This competition is particularly evident with respect to our OraQuick ADVANCE® HIV-1/2 test. In addition, the Oragene® product line sold by our subsidiary, DNAG, competes against other molecular collection products, such as blood collection kits and buccal swabs. There are a number of competitors making investments in competing technologies and products, and a number of our competitors may have a competitive advantage because of their greater financial, technical, research and other resources. Some competitors offer broader product lines, aggressively discount prices for their products and may have greater name recognition than we have. We also face competition from certain of our distributors or former customers that have created or may decide to create, their own products to compete with ours. If our competitors’ products take market share from our products through more effective marketing or competitive pricing, our revenues, margins and operating results could be adversely affected. In addition, our revenues and operating results could be negatively impacted if some of our customers internally develop or acquire their own sample collection devices and use those devices in place of our products in order to reduce costs.
Customer Concentration and Purchase Seasonality Creates Risk for Our Business.
We have experienced strong revenue growth during the past several years, primarily due to increasing sales of our OraQuick® rapid HCV test and our Oragene® saliva DNA collection kit. The higher sales of these products have been driven by increased demand by an increasing number of large customers.
Our molecular collections sales grew from $32.2 million in 2016 to $75.1 million in 2017, largely as a result of increased orders of our Oragene® collection systems by our largest customer serving the consumer genomics market. In November 2017, we announced the execution of a multi-year purchase agreement for the sale of $144 million of our Oragene® product to a large consumer genomics customer.
46
Table of Contents
In our infectious disease business, sales of our HCV test rose from $14.1 million in 2016 to $25.4 million in 2017. This growth was primarily attributable to sales under a previously announced $18.0 million contract with a foreign government in support of a country-wide HCV elimination program. This agreement was largely completed in 2017 and this customer unexpectedly decided not to renew the agreement for an additional 12 months.
We expect that sales to the large consumer genetics customer will continue to be a significant growth driver for our molecular business. It is also possible that other countries implementing broad based HCV testing or elimination programs could have a significant impact on our business. As a result, certain parts of our business will likely continue to have a high customer concentration and depend disproportionately on a few large customers. In addition, demand from a large consumer genomics customer increased significantly in 2016 and 2017 due to special promotions towards the end of these years. To the extent that such a large customer fails to meet its purchase commitments, does not continue such promotional programs or otherwise does not continue to purchase our products, or if we experience difficulty in meeting the high demand by these customers for our products, our revenues and results of operations could be adversely affected.
Our Inability to Carry Out Certain of Our Marketing and Sales Plans May Make it Difficult for Us to Grow or Maintain Our Business.
We have implemented in the past, and we intend to implement in the future, an aggressive sales and marketing plan to expand sales of our products. If we are unable to successfully implement these programs or modify these programs in response to evolving market and economic conditions, we may be unable to grow and our business could suffer.
Our Sales Cycles Can be Lengthy, and May Depend on Public Funding, Which Can Cause Variability and Unpredictability in Our Operating Results.
The sales cycles for certain of our products can be lengthy and unpredictable, which makes it more difficult to accurately forecast revenues in a given period and may cause revenues and operating results to vary from period to period. Sales of our products often involve purchasing decisions by large public and private institutions, may require many levels of approval and may be dependent on economic or political conditions and the availability of grants or funding from governmental or public health agencies which can vary from period to period in both amount and timing. For example, in past years our OraQuick ADVANCE® HIV-1/2 test has been purchased through bulk procurement or other funding provided by governmental agencies. Our OraQuick® HCV test has been purchased by customers who receive government funding, and we believe increased funding from the CDC and other agencies will be required to substantially increase the volume of HCV testing, especially in the public health market. More recently, we have sold large quantities of our OraQuick® HCV test to foreign governments and agencies for use in broad-based or country-wide HCV elimination programs. There can be no assurance that purchases or funding from these agencies will occur or continue. As a result, we may expend considerable resources on unsuccessful sales efforts or we may not be able to complete transactions at all or on a schedule and in an amount consistent with our objectives.
We May Face Product Liability Claims for Injuries Resulting From the Use of Our Products.
We may be held liable if any of our products, or any product which is made with the use or incorporation of any of our technologies, causes injury of any type or is found otherwise unsuitable during product testing, manufacturing, marketing, sale or usage. There is no assurance that we would be successful in defending any product liability lawsuits brought against us. Regardless of merit or eventual outcome, product liability claims could result in:
| • | Decreased demand for our products; |
| • | Lost revenues; |
47
Table of Contents
| • | Damage to our image or reputation; |
| • | Costs related to litigation; |
| • | Increased product liability insurance costs; |
| • | Diversion of management time and attention; and |
| • | Incurrence of damages payable to plaintiffs. |
We are selling cryosurgical products in the consumer or OTC market in the United States and certain countries and we may expand OTC sales of these products into other countries. We also sell the OraQuick® In-Home HIV test in the United States OTC market, and we offer HIV self-tests to consumers internationally. We believe the sale of products in the OTC market increases our potential exposure to product liability and other claims.
The Insurance We Purchase to Cover Our Potential Business Risks May be Inadequate.
Although we believe that our present product liability and other insurance coverage is sufficient to cover our current estimated exposures, we cannot be sure that we will not incur liabilities in excess of our policy limits. In addition, although we believe that we will be able to continue to obtain adequate coverage in the future, there is no assurance that we will be able to do so at acceptable costs.
We Could Suffer Monetary Damages, Incur Substantial Costs or be Prevented From Using Technologies Important to Our Products as a Result of Legal Proceedings.
We have been, and in the future may become, involved in various legal proceedings arising out of our businesses. These may include commercial disputes, negligence claims or various other lawsuits arising in the ordinary course of our business, including employment matters. Such lawsuits can seek damages, sometimes in substantial amounts, for commercial or personal injuries allegedly suffered and can include claims for punitive or other special damages. An adverse ruling or rulings in one or more such lawsuits could, individually or in the aggregate, result in the termination or modification of a material contract or otherwise have a material adverse effect on our sales, operations or financial performance.
Performance of Our Products May Affect Our Revenues, Stock Price and Reputation.
Our products are generally sold with labeling that contains performance claims approved or cleared by the FDA or other regulators. However, our products may not perform as expected. For example, a defect in one of our diagnostic products or a failure by a customer to follow proper testing procedures, may cause the product to report inaccurate information such as a false positive result or a false negative result. A false positive or negative result can also occur even when there is no apparent product defect and the customer has apparently used our product properly. Identifying the root cause of a product performance or quality issue can be difficult and time consuming.
If our products fail to perform in accordance with the applicable label claims or otherwise in accordance with the expectations or needs of our customers, customers may switch to a competing product or otherwise stop using our products, and our revenues could be adversely affected. Under such circumstances, we may be required to implement shipment holds or product recalls and incur warranty obligations, which would increase our costs. In addition, poor performance by one or more of our products and publicity surrounding such performance could have an adverse effect on our reputation, our continuing ability to sell products and the prevailing market price of our Common Stock.
Our Ability To Expand International Sales Could Adversely Affect Our Business and Results of Operations.
One of our strategic priorities is to substantially expand our product sales internationally. An opportunity to accomplish this objective is with the sale of our OraQuick® HIV Self-Test in support of large self-testing
48
Table of Contents
programs in certain African countries and elsewhere. While we believe international sales of our HIV self-test and other products represent attractive long-term opportunities with significant growth potential, there is no guarantee that these opportunities will continue or increase. Among other factors, competition from other cheaper products and the uncertainties of available funding could negatively impact the success of these opportunities. If international sales of these products do not continue or increase or if we are otherwise unable to expand international sales of our other products, our revenues and results of operations could be negatively impacted.
In addition, market conditions in many countries often require that we sell our products at a price below our typical U.S. or European pricing in order to participate in these markets. As a result, sales in certain countries may contribute lower profit margins to our business. To the extent these international sales comprise a large or increasing part of our business, our gross margins will be negatively affected. In addition, we may have difficulty selling our products at a sufficiently low price to maintain or increase this business over the long term without funding support from public health entities, government agencies or other sources. If we are unable to obtain or continue this funding support at sufficient levels, or at all, our revenues and results of operations could be negatively affected.
Our International Presence May Increase Our Risks and Expose Our Business to Regulatory, Cultural or Other Restraints.
We seek to increase revenue derived from international sales of our products. Our international sales accounted for $45.6 million or 27% of consolidated net revenues in 2017, $28.4 million or 22% of consolidated net revenues in 2016 and $23.2 million or 19% of consolidated net revenues in 2015. In addition, our molecular collection systems business, which accounted for $75.1 million or 45% of consolidated net revenues in 2017, is operated in Canada.
A number of factors could adversely affect the performance of our business and/or cause us to incur substantially increased costs because of our international presence and sales, including those set forth below:
| • | Uncertainty in the application of foreign laws and the interpretation of contracts with foreign parties; |
| • | The potential for inconsistent imposition of legal and regulatory requirements; |
| • | Cultural and political differences that favor local competitors or make it difficult to effectively market, sell and gain acceptance of our products; |
| • | Inexperience in international markets and territories and difficulties in staffing and managing foreign operations; |
| • | Exchange rates, currency fluctuations, tariffs and other barriers, extended payment terms and dependence on international distributors or representatives; |
| • | Regulatory requirements (including compliance with applicable customs regulations) and the need for reimbursement approvals; |
| • | Trade protection measures, trade sanctions and import/export licensing requirements; |
| • | The inability to obtain or maintain ISO certification for our or our suppliers’ manufacturing facilities; |
| • | Our inability to obtain or maintain regulatory approvals or registrations for our products; |
| • | Our inability to identify international distributors and negotiate acceptable terms for distribution agreements; |
| • | Diversion to the U.S. of our products that are sold at lower prices into international markets; |
| • | The loss of one or more distributors and difficulties or delays in obtaining new or transferred product registrations or approvals for use by a replacement distributor; |
| • | Multiple jurisdictions and differing tax laws, as well as changes in those laws; |
49
Table of Contents
| • | An increase of withholding and other taxes on remittances and other payments by a foreign subsidiary; |
| • | The creditworthiness of foreign distributors and customers and difficulty in collecting foreign accounts receivable; |
| • | Difficulty of enforcing contractual obligations or recovering damages under foreign legal systems; |
| • | Difficulty collecting amounts owed by foreign governments or other customers; |
| • | Economic conditions, political instability, the absence of available funding sources, terrorism, civil unrest, war and natural disasters in foreign countries; |
| • | Long sales cycles in international markets, especially for sales to foreign governments, quasi-governmental agencies and international public health agencies; |
| • | The sale of competing products by foreign competitors at prices at or below the prices we offer for our products; |
| • | Restrictions on our ability to repatriate investments and earnings from foreign operations; |
| • | Changes in shipping costs; |
| • | The unavailability of licenses to certain patents in force in a foreign country which cover our products; and |
| • | Reduced protection for, or enforcement of, our patents and other intellectual property rights in foreign countries. |
In addition, we have contracted with a third party in Thailand for the manufacture of a portion of our OraQuick® HIV-1/2 tests, and all of DNAG’s products are produced in Canada. We may enter into agreements to manufacture these or other products in additional foreign countries as well. However, economic, cultural and political conditions and foreign regulatory requirements may slow or prevent the manufacture of our products in countries other than the United States. Interruption of the supply of our products could reduce revenues or cause us to incur significant additional expenses in finding an alternative source of supply. Foreign currency fluctuations and economic conditions in foreign countries could also increase the costs of manufacturing our products in foreign countries.
Our U.S. Government Contracts Require Compliance With Numerous Laws and Increases Our Risk and Liability.
We are currently receiving funding from the U.S. government related to our OraQuick® Ebola rapid antigen test and our OraQuick® Zika rapid antibody test and we sell some of our products to the federal government. As a result of our U.S. government funding and product sales to the U.S. government, we must comply with laws and regulations relating to the award, administration and performance of U.S. government contracts. U.S. government contracts typically contain a number of extraordinary provisions that would not typically be found in commercial contracts and which may create a disadvantage and additional risks to us as compared to competitors that do not rely on government contracts. As a U.S. government contractor, we are subject to increased risks of investigation, criminal prosecution and other legal actions and liabilities to which purely private sector companies are not. The results of any such actions could adversely impact our business and have an adverse effect on our consolidated financial performance.
A violation of specific laws and regulations could result in the imposition of fines and penalties or the termination of our contracts, as well as suspension or debarment. The suspension or debarment in any particular case may be limited to the facility, contract or subsidiary involved in the violation or could be applied to our entire enterprise in certain severe circumstances. Even a narrow scope suspension or debarment could result in negative publicity that could adversely affect our ability to renew contracts and to secure new contracts, both with the U.S. government and private customers, which could materially and adversely affect our business and
50
Table of Contents
results of operations. Fines and penalties could be imposed for failing to follow procurement integrity and bidding rules, employing improper billing practices or otherwise failing to follow rules relating to billing on cost-plus contracts, receiving or paying kickbacks, or filing false claims, among other potential violations. In addition, we could suffer serious reputational harm and the value of our common stock could be negatively affected if allegations of impropriety related to such contracts are made against us.
Our U.S. Government Contracts May Affect our Intellectual Property Rights.
Provisions in our U.S. government contracts may affect our intellectual property rights. Certain of our activities have been funded, and may in the future be funded, by the U.S. government, including our contracts with BARDA. When new technologies are developed with U.S. government funding, the government obtains certain rights in any resulting patents, including the right to a nonexclusive license authorizing the government to use the invention. These rights may permit the government to disclose our confidential information to third parties and to exercise “march-in” rights to use and allow third parties to use our patented technology. The government can exercise its march-in rights if it determines that action is necessary because we fail to achieve practical application of the U.S. government-funded technology, because action is necessary to alleviate health or safety needs, to meet requirements of federal regulations, or to give preference to U.S. industry. In addition, government-funded inventions must be reported to the government, government funding must be disclosed in any resulting patent applications, and our rights in such inventions may be subject to certain requirements to manufacture products in the United States.
Our U.S. Government Contracts Are Subject to Future Funding and the Government’s Choice to Exercise Options, and May be Terminated for the Government’s Convenience.
Our contracts with the U.S. government are subject to future funding and are subject to the right of the government to terminate the contracts in whole or in part for its convenience. There is pressure for the U.S. government to reduce spending. The non-appropriation of funds or the termination for the government’s convenience of our contracts could negatively affect our financial results.
For U.S. government contracts that include options, such as our BARDA contracts, the U.S. government generally has the unilateral right not to exercise such options and may not exercise an option if the agency is not satisfied with our performance under the contract or does not receive funding to continue the program, among other reasons. If levels of U.S. government expenditures and authorizations for emerging diseases decrease or shift to programs in areas where we do not offer products or are not developing product candidates, or if the U.S. government otherwise declines to exercise its options under its contracts with us, our business, revenue and operating results would suffer.
Our U.S. Government Contracts Are Subject To Numerous Requirements in Order to be Reimbursed by the Federal Government.
Our current contracts with BARDA are cost plus fixed fee contracts and potential future contracts with the U.S. government may also be structured this way. Under our cost plus fixed fee contracts, we are allowed to recover our approved costs plus a fixed fee. The total price on a cost plus fixed fee contract is based primarily on allowable costs incurred, but generally is subject to contract funding limitations. U.S. government regulations require us to notify our customer of any cost overruns or underruns on a cost plus contract. If we incur costs in excess of the funding limitation specified in the contract we may not be able to recover those cost overruns.
Our U.S. Government Contracts and Related Administrative Processes Are Subject to Audits and Cost Adjustments by the Federal Government.
Federal government agencies can audit and investigate government contracts and the administrative processes and systems of government contractors. These agencies can review our performance on government contracts,
51
Table of Contents
pricing practices, cost structure, and compliance with applicable laws, regulations and standards. They can also review our compliance with government regulations and policies and the adequacy of our internal control systems and policies, including our purchasing, accounting, estimating, compensation and management information processes and systems. Any costs found to be improperly allocated to a specific government contract, unallowable or unreasonable will not be reimbursed, and any such costs already reimbursed may be required to be refunded and certain penalties may be imposed. Adjustments arising from government audits and reviews could have a material adverse effect on our business, financial condition, results of operations and prospects.
Moreover, if any administrative process or system related to such contracts is found not to comply with governmental requirements, we may be subjected to government scrutiny that could delay or otherwise adversely affect our ability to compete for or perform government contracts or collect our revenue in a timely manner. An unfavorable outcome of an audit of our government contracts could adversely affect our results of operations.
Risks Relating to the Economy, Our Financial Results, Investments, Credit Facilities and Need for Financing
Economic Volatility and Disruption Could Adversely Affect Our Results of Operations, Cash Flow and Financial Condition or Those of Our Customers and Suppliers.
Volatile economic conditions may occur again or continue in the future. These conditions could adversely affect our financial performance and condition or those of our customers and suppliers. These circumstances could also adversely affect our access to liquidity needed to conduct or expand our business or conduct future acquisitions or make other discretionary investments. Many of our customers rely on public funding provided by federal, state and local governments, and this funding has been and may continue to be reduced or deferred as a result of economic conditions or other factors. These circumstances may adversely impact our customers and suppliers, which, in turn, could adversely affect their ability to purchase our products or supply us with necessary equipment, raw materials or components. Even with the improvement of economic conditions, it may take time for our customers and suppliers to establish new budgets and return to normal purchasing and shipping patterns. We cannot predict the reoccurrence of any economic slowdown or the strength or sustainability of an economic recovery.
We Have Experienced Losses in the Recent Past and May Not Be Able to Maintain Profitable Operations.
We experienced annual net losses during the five years prior to 2015. In addition, as of December 31, 2017, the Company had an accumulated deficit of $119.5 million. Even though we achieved profitability since 2015, there can be no assurance that we will be able to sustain this profitability in the future.
Our ability to continue profitable operations in the future will be dependent upon a number of factors including, without limitation, the following:
| • | Our ability to continue growing our molecular collection systems business; |
| • | Our ability to mitigate declining sales of our OraQuick ADVANCE® HIV-1/2 test in the United States and expand sales of our OraQuick® HIV Self-Test internationally; |
| • | The level of expenditures we are required to make in order to develop, obtain regulatory approvals for and successfully commercialize our new products; |
| • | Our ability to successfully commercialize our OraQuick® Ebola and Zika tests and other new products. |
| • | Our ability to improve manufacturing efficiencies; |
| • | Our ability to successfully launch new products after receipt of required regulatory approvals or the acquisition of rights to those products; |
52
Table of Contents
| • | The degree to which our major distributors and customers comply with their contractual obligations, including minimum purchase commitments; |
| • | Whether we are successful in obtaining and maintaining required regulatory approvals and registrations for our new products; |
| • | The level of competition, including the degree to which competitors sell lower priced products or more attractive offerings to compete with our products; |
| • | Changes in economic conditions in domestic or international markets, such as economic downturns, reduced demand, inflation and currency fluctuations; |
| • | Failure to achieve our revenue growth targets; |
| • | Changes in distributor buying patterns or a buildup of significant quantities in our distributors’ inventories or distribution channels; and |
| • | The costs and results of patent infringement, product liability and other litigation or claims asserted by or against us. |
We May Experience Fluctuations in Our Financial Results or Fail to Meet Our Financial Projections.
Our operating results can fluctuate from quarter to quarter and year to year, which could cause our growth or financial performance to fall below the expectations of investors and securities analysts. Our financial projections for future periods are based on a number of assumptions, including estimated demand for our products. However, sales to our distributors and other customers may fall short of expectations because of lower than estimated demand or other factors, including continued volatility and disruption in economic conditions, increasing competition, reduced governmental funding and other circumstances described elsewhere in this Annual Report. Infrequent, unusual or unexpected changes in revenues or costs could also contribute to the variability of our financial results.
Customers in certain of the markets we serve often submit a high percentage of purchase orders in the third month of a calendar quarter. Although this can vary from quarter to quarter, many customers make purchase decisions late in a quarter due to budgetary or financial requirements. In addition, certain governmental customers must fully spend budgeted funds by the end of their fiscal year or risk losing these funds, which can contribute to fluctuations in our sales from year-to-year. This can make it difficult to accurately forecast whether we will achieve our quarterly sales forecasts and can cause variability in our operating results.
In addition, our products provide different contributions to our gross margin. Accordingly, our operating results could also fluctuate and be affected by the mix of products sold and the relative prices and gross margin contribution of those products. Failure to achieve operating results consistent with the expectations of investors and securities analysts could adversely affect our reputation and the price of our Common Stock.
Changes in Foreign Currency Exchange Rates Could Negatively Affect Our Operating Results.
Our financial statements are stated in U.S. Dollars and, historically, most of our international sales have also been denominated in U.S. Dollars. As a result, in the past our exposure to foreign currency exchange rate risk has not been material. Nonetheless, these sales are subject to currency risks, since changes in the values of foreign currencies relative to the value of the U.S. dollar can render our products comparatively more expensive. These exchange rate fluctuations could negatively impact international sales of our products, as could changes in the general economic conditions in those markets.
In addition, the revenues and expenses of our subsidiary, DNAG, are recorded in Canadian Dollars and certain of its international sales are denominated in local currencies, including the Euro, British Pound and Australian Dollar. Revenues and expenses denominated in foreign currencies are translated into U.S. dollars for purposes of
53
Table of Contents
reporting our consolidated financial results. Our expectation is that the DNAG business will continue to grow and our exposure to foreign currency exchange rates may be more significant than in past years.
Exchange rate fluctuations may affect DNAG’s revenues and expenses and the translation of DNAG’s financial results into U.S. dollars. Favorable movement in exchange rates have benefited us in prior periods. However, where there are unfavorable currency exchange rate fluctuations, our consolidated financial statements including our balance sheet, revenues and results of operations, could be negatively affected. In addition, fluctuations in exchange rates could affect year-to-year comparability of operating results. In the past, we have not generally entered into hedging instruments to manage our currency exchange rate risk, but we may need to do so in the future. However, our attempts to hedge against these risks may not be successful. If we are unable to successfully hedge against unfavorable foreign currency exchange rate movements, our consolidated financial results may be adversely impacted.
Tax Matters, Including the Changes in Corporate Tax Rates, Disagreements with Taxing Authorities and Imposition of New Taxes Could Impact our Results of Operations and Financial Condition.
We are subject to income and other taxes in the U.S. and our operations, plans and results are affected by tax and other initiatives. On December 22, 2017, the U.S. Government enacted comprehensive tax legislation known as the Tax Cuts and Jobs Act (the “Tax Act”). This legislation makes broad and complex changes to the U.S. tax code, including but not limited to reducing the corporate tax rate from 35% to 21%, requiring a one-time transition tax on certain unrepatriated earnings of foreign subsidiaries and accelerating first year expensing of certain capital expenditures. The Tax Act also introduces new tax laws that may impact our taxable income in 2018, which will include, but not be limited to, a new provision designed to tax global intangible low taxed income (GILTI), limitations on the deductibility of certain executive compensation, creating a base erosion anti-abuse tax (BEAT) and modifying or repealing many deductions and credits. The impact of many provisions of the Tax Act lack clarity and is subject to interpretation until additional Treasury Department guidance is issued. The ultimate impacts of the Tax Act may differ from the Company’s estimates due to changes in the interpretations and assumptions made, as well as any forthcoming regulatory guidance. The Tax Act could also impact changes to our valuation of deferred tax assets and liabilities. Any such change in valuation could have a material impact on our income tax expense and deferred tax balances.
We are also subject to regular reviews, examinations, and audits by the Internal Revenue Service and other taxing authorities with respect to our taxes. Although we believe our tax estimates are reasonable, if a taxing authority disagrees with the positions we have taken, we could face additional tax liability, including interest and penalties. There can be no assurance that payment of such additional amounts upon final adjudication of any disputes will not have a material impact on our results of operations and financial position.
We also need to comply with new, evolving or revised tax laws and regulations. The enactment of new or revised foreign or domestic tax regulations or increases in tariffs or changes in the application or interpretation of the Tax Act, could have an adverse effect on our business or on our results of operations.
Our Revolving Credit Facility Contains Certain Financial Covenants Which, if Not Satisfied, Could Limit Our Ability to Borrow in the Future or Result in the Acceleration of the Amounts Borrowed Under This Facility.
Our revolving credit facility with a commercial bank contains various financial and other covenants with which we must comply on an ongoing or periodic basis. If we enter into new or additional credit facilities or loan agreements, we would expect those arrangements would contain similar types of covenants. Although we do not expect to violate these covenants and obligations, if such a violation were to occur, our ability to borrow funds in the future may be adversely affected. To the extent we borrow funds and a subsequent violation occurs, the outstanding debt under our credit facility or other arrangement could become immediately due and payable and our lender(s) could proceed against any collateral securing such indebtedness.
54
Table of Contents
We May Require Future Additional Capital.
Our future liquidity and ability to meet our future capital requirements will depend on numerous factors, including, but not limited to, the following:
| • | The costs, scope and timing of strategic acquisitions; |
| • | The costs and timing of expansion of sales and marketing activities; |
| • | The timing and success of the commercial launch of new products; |
| • | The extent to which we gain or expand market acceptance for existing, new or enhanced products; |
| • | The costs and timing of the expansion of our manufacturing capacity; |
| • | The success of our research and product development efforts; |
| • | The time, cost and degree of success of conducting clinical trials and obtaining regulatory approvals; |
| • | The magnitude of capital expenditures; |
| • | Changes in existing and potential relationships with distributors and other business partners; |
| • | The costs involved in obtaining and enforcing patents, proprietary rights and necessary licenses; |
| • | The costs and liability associated with patent infringement or other types of litigation; and |
| • | Competing technological and market developments; |
If additional financing is needed, we may seek to raise funds through the sale of equity or other securities or through bank borrowings. There can be no assurance that financing through the sale of securities, bank borrowings or otherwise will be available to us on satisfactory terms, or at all.
Terrorist Attacks or Natural Disasters May Adversely Affect Our Business.
Terrorist attacks or natural disasters, and subsequent governmental responses to these events, could cause economic instability. These actions could adversely affect economic conditions both within and outside the United States and reduce demand for our products. These events could disrupt the operations of our customers and suppliers and eliminate, reduce or delay our customers’ ability to purchase and use our products and our suppliers’ ability to provide raw materials and finished products.
Although we have business interruption insurance, our facilities, including some pieces of manufacturing equipment and our computer systems, may be difficult to replace and could require substantial replacement lead-time. Various types of disasters, including earthquakes, fires, floods and acts of terrorism, may affect our manufacturing facilities and computer systems. In the event our existing manufacturing facilities or computer systems are affected by man-made or natural disasters, we may have difficulty operating our business and may be unable to manufacture products for sale or meet customer demands or sales projections. If our manufacturing operations were curtailed or shut down entirely, it would seriously harm our business. Moreover, we may incur incremental costs following an unforeseen event which could adversely affect our results of operation.
Risks Relating to Our Common Stock
Our Stock Price Could Continue to be Volatile.
Our stock price has been volatile, has fluctuated substantially in the past, may be volatile in the future and could experience substantial declines. The following factors, among others, could have a significant impact on the market for our Common Stock:
| • | The performance of our business, including our efforts to increase sales of our OraQuick® HIV, HCV and molecular collection systems products and our OraQuick® In-Home HIV test and HIV self-test; |
55
Table of Contents
| • | Future announcements concerning us and our products, including with respect to significant acquisitions, strategic collaborations and joint ventures; |
| • | Clinical results with respect to our products or those of our competitors; |
| • | The status of clinical studies and pending submissions for required regulatory approvals; |
| • | The announcement of regulatory or enforcement actions by the FDA or other agencies against us, our products or one or more of our customers; |
| • | The gain or loss of significant contracts and availability of funding for the purchase of our products; |
| • | Delays in the development, regulatory approval or commercialization of new or enhanced products; |
| • | Legislative developments and industry or competitive trends; |
| • | Biological or medical discoveries; |
| • | Disputes or developments with key customers, distributors or suppliers; |
| • | Developments in patent or other proprietary rights; |
| • | Litigation or threatened litigation; |
| • | Complaints or concerns about the performance or safety of our products and publicity about those issues, including publicity expressed through social media or otherwise over the internet; |
| • | Failure to achieve, or changes in, financial estimates by securities analysts and comments or opinions about us by securities analysts or major stockholders; |
| • | Governmental regulation; |
| • | Changes in the level of competition; |
| • | Loss of or declines in sales to major distributors or customers or changes in the mix of products sold; |
| • | Period-to-period fluctuations in our operating results; |
| • | Additions or departures of key personnel; |
| • | General market and economic conditions; and |
| • | Terrorist attacks, civil unrest, war and national disasters. |
In addition, the stock market in general has experienced extreme price and volume fluctuations that have affected the market price of our Common Stock, as well as the stock of many companies in the diagnostics and life sciences industries. Often, price fluctuations are unrelated to the operating performance of the specific companies whose stock is affected.
In the past, following periods of volatility in the market price of a company’s stock, securities class action litigation has occurred against the issuing company. If we were subject to this type of litigation in the future, we could incur substantial costs and experience a subsequent diversion of our management’s attention and resources, each of which could have a material adverse effect on our revenue and earnings. Any adverse determination in this type of litigation could also subject us to significant liabilities.
Future Sales of Our Common Stock by Existing Stockholders, Executive Officers or Directors Could Depress the Market Price of Our Common Stock and Make It More Difficult For Us to Sell Stock in the Future.
Sales of our Common Stock in the public market, or the perception that such sales may occur, could negatively impact the market price of our Common Stock. We are unable to estimate the number of shares of our Common Stock that may actually be resold in the public market since this will depend on the market price for our Common Stock, the individual circumstances of the sellers and other factors.
56
Table of Contents
We have a number of institutional stockholders that own significant blocks of our Common Stock. If one or more of these stockholders sell large portions of their holdings in a relatively short time, for liquidity or other reasons, the prevailing market price of our Common Stock could be negatively affected. In addition, it is possible that one or more of our executive officers or non-employee members of our Board of Directors could sell shares of our Common Stock during an open trading window or pursuant to a 10b5-1 sales plan under our Insider Trading Policy. These transactions and the perceived reasons for these transactions could have a negative effect on the prevailing market price of our Common Stock.
Investor Confidence and Share Value May be Adversely Impacted if We and/or Our Independent Registered Public Accounting Firm Conclude That Our Internal Control Over Financial Reporting is Not Effective.
As directed by Section 404 of the Sarbanes-Oxley Act of 2002, the SEC adopted rules requiring us, as a public company, to include a report in our Annual Reports on Form 10-K that contains an assessment by management of the effectiveness of our internal control over financial reporting. In addition, our independent registered public accounting firm must report on the effectiveness of these internal controls.
We expect that our internal controls will continue to evolve as our business activities change. Although we seek to diligently and vigorously review our internal control over financial reporting in an effort to ensure compliance with the Section 404 requirements, any control system, regardless of how well designed, operated and evaluated, can provide only reasonable, not absolute, assurance that its objectives will be met. In addition, the overall quality of our internal controls may be affected by the internal control over financial reporting implemented by any business we acquire and our ability to assess and successfully integrate the internal controls of any such business.
If, during any year, our independent registered public accounting firm is not satisfied with our internal control over financial reporting or the level at which our controls are documented, designed, operated, tested or assessed, or if the independent registered public accounting firm interprets the requirements, rules or regulations differently than we do, then it may issue a report that is qualified. We also could conclude that our internal control over financial reporting is not effective. These events could result in an adverse reaction in the financial marketplace due to a loss of investor confidence in the reliability of our financial statements and effectiveness of our internal controls, which ultimately could negatively impact the market price of our Common Stock.
Because We Do Not Intend to Pay Cash Dividends on Our Common Stock, an Investor in Our Common Stock Will Benefit Only if Our Stock Appreciates in Value.
We currently intend to retain our current earnings and future earnings, if any, to finance the expansion of our business and do not expect to pay any cash dividends on our Common Stock in the foreseeable future. As a result, the success of an investment in our Common Stock will depend entirely upon any future appreciation. There is no guarantee that our Common Stock will appreciate in value or even maintain the price at which investors purchased their shares.
Certain Provisions in Our Certificate of Incorporation and Bylaws and Under Delaware Law Could Make a Third-Party Acquisition of Us Difficult.
Our Certificate of Incorporation and Bylaws contain provisions that could make it more difficult for a third party to acquire us, even if doing so would be beneficial to our stockholders. We are also subject to certain provisions of Delaware law that could delay, deter or prevent a change in control of us. These provisions could limit the price investors might be willing to pay in the future for shares of our Common Stock.
57
Table of Contents
Future Sales of Shares of Our Common Stock Could Adversely Affect the Trading Price of Our Common Stock and Our Ability to Raise Funds in New Equity Offerings.
Future sales of a substantial number of our shares of Common Stock or equity-related securities in the public market or privately, or the perception that such sales may occur, could adversely affect prevailing trading prices of our Common Stock, and could impair our ability to raise capital through future offerings of equity or equity-related securities. No prediction can be made as to the effect, if any, that future sales of shares of Common Stock or the availability of shares of Common Stock for future sale will have on the trading price of our Common Stock.
| ITEM 1B. | Unresolved Staff Comments. |
Not Applicable.
| ITEM 2. | Properties. |
We own a 48,000 square foot facility which is OraSure’s primary corporate office and manufacturing facility, a 31,700 square foot facility that houses our sales and marketing, research and development, human resources, and regulatory and quality offices, and a 33,500 square foot facility which is used for manufacturing activities. Each of these facilities is located in Bethlehem, Pennsylvania. We also rent additional warehouse space on an as-needed basis, including a 70,000 square foot warehouse in Bethlehem Township, Northampton County, Pennsylvania. In addition, our subsidiary, DNAG, leases a 35,883 square foot facility in Ottawa, Canada, which is used as its primary corporate office and houses sales and marketing, research and development, and regulatory and quality operations.
We believe that the facilities described above are adequate for our current requirements.
| ITEM 3. | Legal Proceedings. |
From time to time, we are involved in certain legal actions arising in the ordinary course of business. In management’s opinion, the outcomes of such actions, either individually or in the aggregate, are not expected to have a material adverse effect on our future financial position or results of operations.
| ITEM 4. | Mine Safety Disclosures. |
Not Applicable.
58
Table of Contents
PART II
| ITEM 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market Information
Our Common Stock is listed for trading on the Global Select Market tier of The Nasdaq Stock Market LLC (“NASDAQ”) under the symbol OSUR. High and low sales prices reported by NASDAQ during the periods indicated are shown below.
| Year ended December 31, | Year ended December 31, | |||||||||||||||
| 2017 | 2016 | |||||||||||||||
| High | Low | High | Low | |||||||||||||
| First Quarter |
$ | 12.97 | $ | 8.39 | $ | 7.39 | $ | 5.09 | ||||||||
| Second Quarter |
$ | 17.96 | $ | 12.03 | $ | 7.99 | $ | 5.81 | ||||||||
| Third Quarter |
$ | 22.79 | $ | 17.06 | $ | 9.05 | $ | 5.57 | ||||||||
| Fourth Quarter |
$ | 23.01 | $ | 12.86 | $ | 9.53 | $ | 7.09 | ||||||||
On February 20, 2018, there were 369 holders of record and approximately 16,426 holders in street name of our Common Stock, and the closing price of our Common Stock was $17.86 per share.
Dividends
We have never paid any cash dividends and our Board of Directors does not anticipate paying cash dividends in the foreseeable future. We intend to retain any future earnings to provide funds for the operation and expansion of our business.
Share Repurchases and Retirements
| Period |
Total number of shares purchased |
Average price paid per Share |
Total number of shares purchased as part of publicly announced plans or programs |
Maximum number (or approximate dollar value) of shares that may yet be repurchased under the plans or programs (1, 2) |
||||||||||||
| October 1, 2017 - October 31, 2017 |
254 | (3) | $ | 19.92 | N/A | 11,984,720 | ||||||||||
| November 1, 2017 - November 30, 2017 |
— | — | — | 11,984,720 | ||||||||||||
| December 1, 2017 - December 31, 2017 |
— | — | — | 11,984,720 | ||||||||||||
|
|
|
|
|
|||||||||||||
| 254 | — | |||||||||||||||
|
|
|
|
|
|||||||||||||
| (1) | On August 5, 2008, our Board of Directors approved a share repurchase program pursuant to which we are permitted to acquire up to $25.0 million of outstanding shares. This share repurchase program may be discontinued at any time. |
| (2) | This column represents the amount that remains available under the $25.0 million repurchase plan, as of the period indicated. We have made no commitment to purchase any shares under this plan. |
| (3) | Pursuant to the OraSure Technologies, Inc. Stock Award Plan, and in connection with the vesting of restricted shares, these shares were retired to satisfy minimum tax withholdings. |
59
Table of Contents
Performance Graph
The performance graph set forth below shall not be deemed “soliciting material” or “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to liability under that Section. This graph will not be deemed “incorporated by reference” into any filing under the Securities Act of 1933, as amended, or the Exchange Act, whether such filing occurs before or after the date hereof, regardless of any general incorporation language in such filing.
The following graph compares the cumulative total returns to investors in the Company’s Common Stock, the NASDAQ Composite Index and the NASDAQ Biotechnology Index for the period from December 31, 2012 through December 31, 2017. The graph assumes that $100 was invested on December 31, 2012 in the Company’s Common Stock and in each of the above-mentioned indices, and that all dividends, if any, were reinvested.
The NASDAQ Composite Index was chosen because it is a broad index of companies whose equity securities are traded on NASDAQ. The NASDAQ Biotechnology Index was chosen because it includes a number of our competitors. Stockholders are cautioned that the graph shows the returns to investors only as of the dates noted and may not be representative of the returns for any other past or future period.
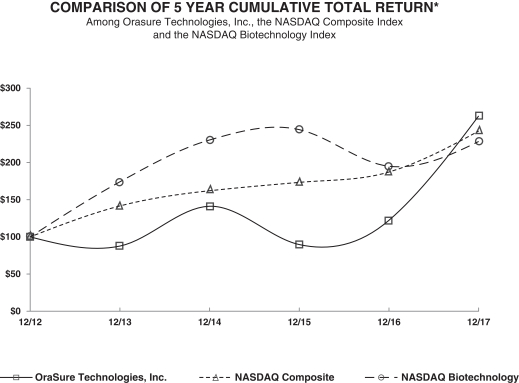
| * | $100 invested on 12/31/12 in stock or index, including reinvestment of dividends. Fiscal year ending December 31. |
| 12/12 | 12/13 | 12/14 | 12/15 | 12/16 | 12/17 | |||||||||||||||||||
| OraSure Technologies, Inc. |
100.00 | 87.60 | 141.23 | 89.69 | 122.28 | 262.67 | ||||||||||||||||||
| NASDAQ Composite |
100.00 | 141.63 | 162.09 | 173.33 | 187.19 | 242.29 | ||||||||||||||||||
| NASDAQ Biotechnology |
100.00 | 174.05 | 230.33 | 244.29 | 194.95 | 228.29 | ||||||||||||||||||
60
Table of Contents
Securities Authorized for Issuance Under Equity Compensation Plans
For certain information concerning securities authorized for issuance under our equity compensation plan, see Item 12, “Securities Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.”
| ITEM 6. | Selected Consolidated Financial Data. |
The following table sets forth selected consolidated financial data of the Company. This information should be read in conjunction with the consolidated financial statements and notes thereto included in Item 15 and the information set forth in Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Selected Consolidated Financial Data
(In thousands, except per share data)
| Years ended December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Operating Results: |
||||||||||||||||||||
| Net revenues |
$ | 167,064 | $ | 128,198 | (2) | $ | 119,719 | $ | 106,464 | $ | 98,940 | (4) | ||||||||
| Costs and expenses |
126,826 | (1) | 107,933 | 111,661 | 111,266 | (3) | 111,102 | (5) | ||||||||||||
| Operating income (loss) |
40,238 | 20,265 | 8,058 | (4,802 | ) | (12,162 | ) | |||||||||||||
| Other income, net |
794 | 58 | 774 | 531 | 200 | |||||||||||||||
| Income tax expense (benefit) |
10,084 | 603 | 665 | 343 | (772 | ) | ||||||||||||||
| Net income (loss) |
30,948 | 19,720 | 8,167 | (4,614 | ) | (11,190 | ) | |||||||||||||
| Earnings (loss) per share |
||||||||||||||||||||
| Basic |
$ | 0.52 | $ | 0.35 | $ | 0.14 | $ | (0.08 | ) | $ | (0.20 | ) | ||||||||
| Diluted |
$ | 0.51 | $ | 0.35 | $ | 0.14 | $ | (0.08 | ) | $ | (0.20 | ) | ||||||||
| Shares used in computing earnings (loss) per share |
||||||||||||||||||||
| Basic |
59,050 | 55,615 | 56,397 | 55,949 | 55,555 | |||||||||||||||
| Diluted |
61,024 | 56,513 | 56,846 | 55,949 | 55,555 | |||||||||||||||
| Cash Flow: |
||||||||||||||||||||
| Cash flows provided by operating activities |
$ | 28,156 | (1) | $ | 24,598 | $ | 15,773 | $ | 7,526 | (3) | $ | 8,385 | (5) | |||||||
| December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Financial Position: |
||||||||||||||||||||
| Cash, cash equivalents, and investments |
$ | 176,587 | $ | 120,950 | $ | 101,319 | $ | 97,867 | $ | 93,191 | ||||||||||
| Working capital |
189,668 | 139,106 | 111,480 | 104,752 | 100,590 | |||||||||||||||
| Total assets |
296,201 | 207,935 | 189,321 | 189,633 | 184,245 | |||||||||||||||
| Accumulated deficit |
(119,510 | ) | (150,458 | ) | (170,178 | ) | (178,345 | ) | (173,731 | ) | ||||||||||
| Stockholders’ equity |
258,081 | 185,850 | 159,436 | 158,701 | 161,146 | |||||||||||||||
| (1) | Includes a $12.5 million gain associated with the settlement of our litigation with Ancestry.com DNA LLC and its contract manufacturer, which was recorded as a reduction of operating expenses in the indicated period. |
| (2) | Includes an additional $5.4 million of exclusivity payments recognized in other revenues as a result of the early termination of our HCV co-promotion agreement with AbbVie, bringing total exclusivity payments recognized in 2016 to $18.9 million. |
| (3) | Includes a $5.5 million gain from the termination of the Company’s oral fluid assay agreement with Roche Diagnostics, which was recorded as a reduction of operating expenses in the indicated period. |
| (4) | Includes a non-recurring net favorable $2.5 million adjustment to account for a change in the Company’s revenue recognition policy related to its OraQuick® In-Home HIV test. |
| (5) | Includes an $8.3 million gain from the termination of the Company’s oral fluid assay agreement with Roche Diagnostics, which was recorded as a reduction of operating expenses in the indicated period. |
61
Table of Contents
| ITEM 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
Statements below regarding future events or performance are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Our actual results could be quite different from those expressed or implied by the forward-looking statements. Factors that could affect results are discussed more fully under the Item 1A, entitled “Risk Factors,” and elsewhere in this Annual Report. Although forward-looking statements help to provide complete information about us, readers should keep in mind that forward-looking statements may not be reliable. Readers are cautioned not to place undue reliance on the forward-looking statements. We undertake no duty to update any forward-looking statements made herein after the date of this Annual Report.
The following discussion should be read in conjunction with the consolidated financial statements contained herein and the notes thereto, along with the Section entitled “Critical Accounting Policies and Estimates,” set forth below.
Overview and Business Segments
Our business is comprised of two segments: Our “OSUR” business consists of the development, manufacture, marketing and sale of oral fluid diagnostic products and specimen collection devices using our proprietary technologies, as well as other diagnostic products including immunoassays and other in vitro diagnostic tests that are used on other specimen types. Our molecular collections systems or “DNAG” business consists of the manufacture and sale of specimen collection kits that are used to collect, stabilize, transport and store samples of genetic material for molecular testing in the consumer genetic, clinical genetic, academic research, pharmacogenomics, personalized medicine, microbiome and animal genetics markets.
Our OSUR diagnostic products include tests that are performed on a rapid basis at the point of care and tests that are processed in a laboratory. These products are sold in the United States and internationally to various clinical laboratories, hospitals, clinics, community-based organizations and other public health organizations, distributors, government agencies, physicians’ offices, and commercial and industrial entities. We also manufacture and sell medical devices used for the removal of benign skin lesions by cryosurgery or freezing. These cryosurgical products are sold in both professional and over-the-counter (“OTC”) markets in North America, Europe, Central and South America, and Australia.
Our DNAG or molecular collection systems business is operated by our subsidiary, DNA Genotek Inc. (“DNAG”), a company based in Ottawa, Canada. DNAG’s Oragene® DNA sample collection kit provides an all-in-one system for the collection, stabilization, transportation and storage of DNA from human saliva. We also sell research use only sample collection products into the microbiome and tuberculosis markets and we offer our customers a suite of genomics and microbiome services called “GenoFINDTM”, which range from package customization and study design optimization to extraction, analysis and reporting services. We serve customers worldwide, including many leading research universities and hospitals.
Recent Developments
Litigation Settlement
Effective February 6, 2017, we settled our patent infringement and breach of contract litigation against Ancestry.com DNA LLC (“Ancestry”) and its contract manufacturer. Under a Settlement and License Agreement executed by the parties, Ancestry agreed to pay a settlement fee of $12.5 million which we received in the first quarter of 2017 and recorded as a gain on litigation settlement in our consolidated statement of income. In addition, we granted Ancestry a royalty-bearing, non-exclusive, worldwide license to certain patents and patent applications related to the collection of DNA in human saliva. The license granted to Ancestry is limited to saliva DNA collection kits sold or used as part of Ancestry’s genetic testing service offerings and does not cover the sale or use of collection kits outside of Ancestry’s business. The Settlement and License Agreement also provides us with a royalty-free, non-exclusive license to patents related to Ancestry’s existing saliva DNA collection kit.
62
Table of Contents
Gates Foundation
In June 2017, we entered into a charitable support agreement with the Bill & Melinda Gates Foundation (“Gates Foundation”) that enables us to offer our OraQuick® HIV self-test at an affordable price in 50 developing countries in Africa and Asia with funding from the Gates Foundation. The funding will consist of support payments tied to the volume of product we sell and reimbursement of certain related costs. The agreement has a four-year term and will enable non-governmental organizations in the eligible countries that receive funding from government or public sector agencies and donors to access our HIV self-test at reduced pricing. The funding from the Gates Foundation will be in an aggregate amount not to exceed $20.0 million over the four-year term or $6.0 million each year of the agreement.
WHO Prequalification
In July 2017, our OraQuick® HIV self-test was prequalified by the World Health Organization (“WHO”). WHO prequalification indicates that diagnostic tests for high burden diseases meet global standards of quality, safety, and efficacy, in order to optimize use of health resources and improve health outcomes. WHO prequalification enables governmental organizations implementing HIV self-test pilots and programs to access international funding to purchase our test.
Self-Testing Project in Africa
In July 2017, our OraQuick® HIV self-test was selected by UNITAID and Population Services International (“PSI”) for use in Phase II of the HIV Self-Testing in Africa (“STAR”) project. Phase II of the project began late in 2017 and will continue over a two-year period. Approximately 4.0 million HIV self-tests are expected to be deployed in Phase II. We believe that we will supply the vast majority of these tests. The OraQuick® HIV self-test has been used in Phase I of the STAR Project, which represented the largest evaluation of HIV self-testing to date. Upon completion of Phase I of the project, 750,000 OraQuick® HIV self-tests will have been distributed.
New Genomics Contract
In November 2017, we entered into a new $144.0 million agreement for the supply of our Oragene®• Dx devices to a leading consumer genomics customer. The agreement provides for the supply of product over several years with minimum annual purchases by the customer.
HCV Foreign Supply Contract
In 2016, we entered into a contract to supply a foreign government with $18.0 million of product, primarily to support a nationwide HCV testing and treatment program. Through December 31, 2017, we have supplied $14.1 million of product to this customer under the contract. The contract includes a renewal option under which the government may purchase up to 100% of the original quantities of product on the same terms and conditions contained in the contract. During the second half of 2017, we engaged in renewal discussions with this customer, including with respect to specific quantities of product to be supplied. Subsequent to these discussions in late 2017, we were informed that the government decided to move to an all laboratory testing solution and that another party had been selected to provide that solution based on cost. In light of these developments, it is unclear whether this government will fulfill the remaining purchase obligations under our existing contract.
Management Succession Plan
In January 2018, we announced the retirement of our President and Chief Executive Officer (“CEO”) and our Chief Financial Officer (“CFO”) and Chief Operating Officer. Stephen S. Tang, Ph.D., who currently serves as Chairman of the Board of Directors (the “Board”), was appointed as the Company’s new President and CEO, effective as of April 1, 2018. Dr. Tang will replace Douglas A. Michels, who will retire as President and CEO,
63
Table of Contents
and as a member of the Board, on March 31, 2018. In addition, Ronald H. Spair, our CFO and Chief Operating Officer, will be retiring on or before June 30, 2018, with his exact retirement date to be determined based on the timing for the Company’s appointment of a new CFO.
Current Consolidated Financial Results
During the year ended December 31, 2017, our consolidated net revenues were $167.1 million, compared to $128.2 million for the year ended December 31, 2016. Net product revenues during the year ended December 31, 2017 increased 51% when compared to 2016, primarily due to higher sales of our molecular and OraQuick® HCV products and increased international sales of our OraQuick® HIV self-test. Partially offsetting these increases were lower domestic sales of our professional OraQuick® HIV product and lower domestic and OTC sales of our cryosurgical products. Other revenues for 2017 were $5.1 million compared to $21.3 million in 2016. Other revenues in 2017 included $4.4 million recognized in connection with funding received from the U.S. Department of Health and Human Services Office of the Assistant Secretary for Preparedness and Response’s Biomedical Advanced Research and Development Authority (“BARDA”) related to our Ebola and Zika products and $689,000 of cost reimbursement under our charitable support agreement with the Gates Foundation. Other revenues in 2016 included $2.3 million of BARDA funding and $18.9 million of exclusivity revenues recognized under our HCV co-promotion agreement with AbbVie, which terminated on December 31, 2016.
Our consolidated net income for the year ended December 31, 2017 was $31.0 million, or $0.51 per share on a fully-diluted basis, compared to consolidated net income of $19.7 million, or $0.35 per share on a fully-diluted basis for the year ended December 31, 2016. Results for the current year include a pre-tax gain of $12.5 million associated with the settlement of our litigation against Ancestry and its contract manufacturer in the first quarter of 2017.
Cash provided by operating activities for the year ended December 31, 2017 was $28.2 million and included the $12.5 million litigation settlement noted above. Cash provided by operating activities during the year ended December 31, 2016 was $24.6 million. As of December 31, 2017, we had $176.6 million in cash (including restricted cash), cash equivalents, and available-for-sale securities, compared to $120.9 million at December 31, 2016.
Results of Operations
YEAR ENDED DECEMBER 31, 2017 COMPARED TO DECEMBER 31, 2016
CONSOLIDATED NET REVENUES
The table below shows a breakdown of total net revenues (dollars in thousands) generated by each of our business segments.
| Year Ended December 31, | ||||||||||||||||||||
| Dollars | Percentage of Total Net Revenues |
|||||||||||||||||||
| 2017 | 2016 | % Change |
2017 | 2016 | ||||||||||||||||
| OSUR |
$ | 86,889 | $ | 74,710 | 16 | % | 52 | % | 58 | % | ||||||||||
| DNAG |
75,099 | 32,214 | 133 | 45 | 25 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net product revenues |
161,988 | 106,924 | 51 | 97 | 83 | |||||||||||||||
| Other |
5,076 | 21,274 | (76 | ) | 3 | 17 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net revenues |
$ | 167,064 | $ | 128,198 | 30 | % | 100 | % | 100 | % | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
Consolidated net product revenues increased 51% to $162.0 million in 2017 from $106.9 million in 2016. Higher sales of our molecular and OraQuick® HCV products and higher international sales of our OraQuick® HIV self-
64
Table of Contents
test were partially offset by lower domestic sales of our professional OraQuick® HIV product and lower domestic and OTC sales of our cryosurgical products. In 2017, we recognized $4.4 million as other revenues in connection with funding from BARDA related to our Ebola and Zika products and $689,000 of cost reimbursement under the Gates Foundation agreement. Other revenues in 2016 included $18.9 million in exclusivity payments received under our AbbVie HCV co-promotion agreement and $2.3 million in BARDA funding. Our co-promotion agreement with AbbVie was terminated on December 31, 2016 and no further revenues were recognized under this agreement after that date.
Consolidated net revenues from products sold to customers outside of the United States were $45.6 million and $28.4 million, or 27% and 22% of total net revenues, during the years ended December 31, 2017 and 2016, respectively. Because the majority of our international sales are denominated in U.S. dollars, the impact of fluctuating foreign currency exchange rates was not material to our total consolidated net revenues.
Net Revenues by Segment
OSUR Segment
The table below shows the amount of total net revenues (dollars in thousands) generated by our OSUR segment.
| Year Ended December 31, | ||||||||||||||||||||
| Dollars | Percentage of Total Net Revenues |
|||||||||||||||||||
| Market |
2017 | 2016 | % Change |
2017 | 2016 | |||||||||||||||
| Infectious disease testing |
$ | 61,951 | $ | 48,408 | 28 | % | 67 | % | 50 | % | ||||||||||
| Risk assessment testing |
12,659 | 13,068 | (3 | ) | 14 | 14 | ||||||||||||||
| Cryosurgical systems |
12,279 | 13,234 | (7 | ) | 13 | 14 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net product revenues |
86,889 | 74,710 | 16 | 94 | 78 | |||||||||||||||
| Other |
5,076 | 21,274 | (76 | ) | 6 | 22 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net revenues |
$ | 91,965 | $ | 95,984 | (4 | ) % | 100 | % | 100 | % | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
Infectious Disease Testing Market
Sales to the infectious disease testing market increased 28% to $61.9 million in 2017 from $48.4 million in 2016. This increase resulted from higher sales of our OraQuick® HCV product and higher international sales of our OraQuick® HIV self-test, partially offset by a decline in domestic sales of our professional OraQuick® HIV product.
The table below shows a breakdown of our total net OraQuick® HIV and HCV product revenues (dollars in thousands) during 2017 and 2016.
| Year Ended December 31, | ||||||||||||
| Market |
2017 | 2016 | % Change |
|||||||||
| Domestic HIV |
$ | 17,015 | $ | 21,499 | (21 | )% | ||||||
| International HIV |
11,301 | 5,248 | 115 | |||||||||
| Domestic OTC HIV |
6,832 | 6,320 | 8 | |||||||||
|
|
|
|
|
|||||||||
| Net HIV revenues |
35,148 | 33,067 | 6 | |||||||||
|
|
|
|
|
|||||||||
| Domestic HCV |
8,448 | 7,436 | 14 | |||||||||
| International HCV |
16,961 | 6,630 | 156 | |||||||||
|
|
|
|
|
|||||||||
| Net HCV revenues |
25,409 | 14,066 | 81 | |||||||||
|
|
|
|
|
|||||||||
| Net OraQuick® revenues |
$ | 60,557 | $ | 47,133 | 28 | % | ||||||
|
|
|
|
|
|||||||||
65
Table of Contents
Domestic OraQuick® HIV sales decreased 21% to $17.0 million for the year ended December 31, 2017 from $21.5 million for the year ended December 31, 2016. This reduction was primarily the result of competitive losses tied to pricing, the loss of sales to point-of-care HIV tests perceived to be more sensitive, and the CDC’s testing guidelines recommending the use of competing fourth generation automated HIV immunoassays performed in a laboratory. We anticipate that future domestic sales of our professional HIV product will continue to be negatively affected by the CDC testing guidelines, changes in government funding, and continued product and price competition.
International sales of our OraQuick® HIV products during 2017 rose 115% to $11.3 million from $5.2 million in 2016. This increase was largely due to the continued shipment of product in support of an HIV self-testing program in Africa, and higher sales of our professional product into certain areas of Africa, Asia, and Latin America, partially offset by a decline in sales in the Middle East and Europe. Funding under the charitable support agreement with the Gates Foundation began in the third quarter of 2017 and product revenues in 2017 included approximately $1.0 million of support payments under that agreement.
Sales of our OraQuick® In-Home HIV test in 2017 of $6.8 million increased 8% compared to $6.3 million in 2016, largely due to expansion of public health programs that use our In-Home test and additional shelf placement of the product in the home diagnostic section of certain retail pharmacies.
Domestic OraQuick® HCV sales increased 14% to $8.4 million in 2017 from $7.4 million in 2016 primarily due to higher sales to our U.S. public health customers in support of HCV testing program expansion, higher sales to non-acute healthcare offices, and increased sales to hospitals. International OraQuick® HCV sales increased 156% to $17.0 million in 2017 from $6.6 million in 2016, largely due to continued product shipments to a foreign government to support a nationwide HCV testing and treatment program and increased sales in Asia and Africa, partially offset by the loss of a multi-national humanitarian organization customer who switched to a competitive product due to pricing. As discussed above, we were recently notified that our supply contract with the foreign government will not be renewed and it is unclear whether this government will fulfill the remaining purchase obligations under the existing contract. During 2017, we recorded $11.8 million in revenue from this contract. It is uncertain if we will record revenues of this magnitude in 2018, if at all.
Risk Assessment Market
Sales to the risk assessment market decreased 3% to $12.6 million for the year ended December 31, 2017 from $13.1 million for the year ended December 31, 2016, primarily as a result of a reduction of inventory maintained by one of our largest customers and lab closings due to consolidations occurring in the laboratory service industry, partially offset by higher sales resulting from increased drug testing in the pain management and drug treatment markets.
Cryosurgical Systems Market
Sales of our cryosurgical systems products (which includes both the physicians’ office and OTC markets) decreased 7% to $12.3 million in 2017 from $13.2 million in 2016.
66
Table of Contents
The table below shows a breakdown of our total net cryosurgical systems revenues (dollars in thousands) generated in each market during 2017 and 2016.
| Year Ended December 31, | ||||||||||||
| Market |
2017 | 2016 | % Change |
|||||||||
| Domestic professional |
$ | 5,400 | $ | 5,545 | (3 | )% | ||||||
| International professional |
797 | 771 | 3 | |||||||||
| Domestic OTC |
1,230 | 1,350 | (9 | ) | ||||||||
| International OTC |
4,852 | 5,568 | (13 | ) | ||||||||
|
|
|
|
|
|||||||||
| Net cryosurgical systems revenues |
$ | 12,279 | $ | 13,234 | (7 | )% | ||||||
|
|
|
|
|
|||||||||
Sales of our Histofreezer® product to physicians’ offices in the United States decreased 3% to $5.4 million in the 2017 from $5.5 million 2016, primarily due to timing of orders placed by our distributors and loss of sales due to competing private label products. These declines in sales were partially offset by increased sales into new markets and additional business generated by our new mid-tier distributors. International sales of our Histofreezer® product increased 3% to $797,000 in 2017 from $771,000 in 2016.
Sales of our private-label wart removal product in the U.S. retail market decreased to $1.2 million in 2017 from $1.4 million in 2016. Sales volume in 2016 was higher as a result of initial stocking orders for a new large pharmacy customer during that period.
Sales of our international OTC cryosurgical products during 2017 decreased 13% to $4.9 million compared to $5.6 million in 2016, largely due to lower sales into Europe as a result of competitive pressures and lower sales in Latin America due to the economic instability of certain countries in that region.
Other revenues
Other revenues in 2017 decreased 76% to $5.1 million from $21.3 million in 2016. Other revenues in 2016 included AbbVie exclusivity revenues of $18.9 million. There are no similar revenues in 2017 due to the termination of our AbbVie HCV co-promotion agreement on December 31, 2016. Revenues related to funding from BARDA increased to $4.4 million in 2017 compared to $2.3 million in 2016. Revenues in 2017 also include $689,000 in reimbursement of certain costs under our charitable support agreement with the Gates Foundation. No such reimbursement occurred in 2016.
DNAG Segment
Molecular Collection Systems
Sales of our molecular collection systems products increased 133% to $75.1 million in 2017 from $32.2 million in 2016.
The table below shows a breakdown of our total net molecular collection systems revenues (dollars in thousands) generated in each market during 2017 and 2016.
| Year Ended December 31, | ||||||||||||
| Market |
2017 | 2016 | % Change |
|||||||||
| Commercial genomics |
$ | 61,594 | $ | 20,767 | 197 | % | ||||||
| Academic genomics |
10,017 | 10,312 | (3 | ) | ||||||||
| Microbiome |
3,488 | 1,135 | 207 | |||||||||
|
|
|
|
|
|||||||||
| Net molecular collection systems revenues |
$ | 75,099 | $ | 32,214 | 133 | % | ||||||
|
|
|
|
|
|||||||||
67
Table of Contents
Sales of our Oragene® product in the commercial market rose 197% to $61.6 million in 2017 compared to $20.8 million in 2016, largely as a result of higher customer demand primarily from a large customer in the consumer genetics market. Sales of our Oragene® product in the academic market decreased 3% to $10.0 million in 2017 compared to $10.3 million in 2016 largely due to the non-recurrence of a large order which occurred in 2016. Microbiome sales increased 207% to $3.5 million in 2017 compared to $1.1 million in 2016 as interest in our microbiome product offering continues to grow with both new and existing customers.
CONSOLIDATED OPERATING RESULTS
Consolidated gross margin was 59% for the year ended December 31, 2017 compared to 69% for the comparable period of 2016. Gross margin for 2017 decreased primarily due to the absence of AbbVie exclusivity revenues, an increase in lower margin international product sales, and higher scrap and spoilage costs.
Consolidated operating income in 2017 was $40.2 million, a $19.9 million improvement from $20.3 million of operating income reported in 2016. Operating income in 2017 benefited from increased product revenues, the Ancestry litigation settlement gain, and lower sales and marketing costs, partially offset by the loss of AbbVie exclusivity revenues recorded in the prior year and higher research and development and general and administrative expenses in the current year.
OPERATING INCOME (LOSS) BY SEGMENT
OSUR Segment
OSUR’s gross margin was 58% in 2017 compared to 69% in 2016. OSUR’s margin was negatively impacted in 2017 primarily by the absence of exclusivity revenues under our AbbVie agreement ($18.9 million was recorded in 2016 versus none in 2017), an increase in lower margin international product sales, and an increase in scrap and spoilage costs.
Research and development expenses increased 50% to $10.5 million in 2017 from $7.0 million in 2016, largely as a result of the inclusion in 2016 of a $1.4 million payment received to settle a claim against one of our raw material suppliers. This settlement was recorded as a reduction in research and development expense in 2016. During 2017, research and development expenses also increased due to higher supply costs associated with the development of our Ebola and Zika products and increased staffing expenses.
Sales and marketing expenses decreased 11% to $18.9 million in 2017 from $21.3 million in 2016, primarily as a result of the termination of our AbbVie HCV co-promotion agreement on December 31, 2016. During 2016, we incurred $1.1 million of detailing and co-promotion expenses associated with the AbbVie agreement. No such costs were incurred for 2017. For the year ended December 31, 2017, we also experienced lower staffing costs, partially offset by higher external commissions payable to certain international distributors.
General and administrative expenses increased 16% to $26.1 million in 2016 from $22.6 million in 2016 due to higher staffing costs and an increase in accrued bonuses as a result of Company’s strong performance.
All of the above contributed to OSUR’s operating loss of $2.7 million for 2017, which included non-cash charges of $3.2 million for depreciation and amortization and $6.4 million for stock-based compensation.
DNAG Segment
DNAG’s gross margin was 61% in 2017 compared to 67% in 2016. The decline in gross margin was primarily due to an increase in lower margin sales in 2017 as compared to 2016.
68
Table of Contents
Research and development expenses increased 4% to $2.8 million in 2017 from $2.7 million in 2016 due to higher staffing costs.
Sales and marketing expenses increased 15% to $9.6 million in 2017 compared to $8.3 million in 2016 due to higher staffing and commission expenses.
General and administrative expenses decreased 45% to $3.2 million in 2017 compared to $5.7 million in 2016, largely due to lower legal costs as our lawsuit with Ancestry and its contract manufacturer was settled in the first quarter of 2017.
Operating expenses in 2017 were offset by the $12.5 million pre-tax gain associated with the settlement of the Ancestry litigation.
All of the above contributed to DNAG’s operating income of $43.0 million for 2017, which included non-cash charges of $3.2 million for depreciation and amortization and $543,000 for stock-based compensation.
CONSOLIDATED INCOME TAXES
We continue to believe the full valuation allowance established in 2008 against OSUR’s total U.S. deferred tax asset is appropriate as the facts and circumstances necessitating the allowance have not changed. For the year ended December 31, 2017, we recorded state income tax expense of $0 compared to $250,000 in the year ended December 31, 2016. Canadian income tax expense of $10.1 million and $353,000 was recorded in 2017 and 2016, respectively. Canadian taxes in 2017 included the additional taxes due as a result of the $12.5 million Ancestry litigation settlement gain and DNAG’s increased pre-tax income. On December 22, 2017, the U.S. enacted the Tax Cuts and Jobs Act (“Tax Act”) that instituted fundamental changes to the taxation on multinational corporations. The impact of the Tax Act on our current and future financial results is discussed in more detail in Note 8 of the Notes to the consolidated financial statements included herein.
YEAR ENDED DECEMBER 31, 2016 COMPARED TO DECEMBER 31, 2015
CONSOLIDATED NET REVENUES
The table below shows a breakdown of total net revenues (dollars in thousands) generated by each of our business segments.
| Year Ended December 31, | ||||||||||||||||||||
| Dollars | Percentage of Total Net Revenues |
|||||||||||||||||||
| 2016 | 2015 | % Change |
2016 | 2015 | ||||||||||||||||
| OSUR |
$ | 74,710 | $ | 74,534 | 0 | % | 58 | % | 62 | % | ||||||||||
| DNAG |
32,214 | 29,924 | 8 | 25 | 25 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net product revenues |
106,924 | 104,458 | 2 | 83 | 87 | |||||||||||||||
| Other |
21,274 | 15,261 | 39 | 17 | 13 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net revenues |
$ | 128,198 | $ | 119,719 | 7 | % | 100 | % | 100 | % | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
Consolidated net product revenues increased 2% to $106.9 million in 2016 from $104.5 million in 2015, primarily as a result of higher international sales of our OraQuick® HIV and HCV tests and higher sales of our molecular collection systems and cryosurgical system products. These increases were partially offset by lower domestic sales of our OraQuick® HIV test and lower sales of our Ebola product. Other revenues in 2016
69
Table of Contents
increased 39% to $21.3 million compared to $15.3 million during 2015, largely due to higher exclusivity revenue recognized under our HCV co-promotion agreement with AbbVie. AbbVie revenues in 2016 included an additional $5.4 million recognized due to the early termination of the contract at the end of that year.
Consolidated net revenues derived from products sold to customers outside the U.S. were $28.4 million and $23.2 million, or 22% and 19% of total consolidated net revenues during the years ended December 31, 2016 and 2015, respectively. Because the majority of our international sales are denominated in U.S. dollars, the impact of fluctuating foreign currency exchange rates was not material to our total consolidated net revenues.
Net Revenues by Segment
OSUR Segment
The table below shows the amount of total net revenues (dollars in thousands) generated by our OSUR segment.
| Year Ended December 31, | ||||||||||||||||||||
| Dollars | Percentage of Total Net Revenues |
|||||||||||||||||||
| Market |
2016 | 2015 | % Change |
2016 | 2015 | |||||||||||||||
| Infectious disease testing |
$ | 48,408 | $ | 49,129 | (1 | )% | 50 | % | 55 | % | ||||||||||
| Risk assessment testing |
13,068 | 13,485 | (3 | ) | 14 | 15 | ||||||||||||||
| Cryosurgical systems |
13,234 | 11,920 | 11 | 14 | 13 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net product revenues |
74,710 | 74,534 | 0 | 78 | 83 | |||||||||||||||
| Other |
21,274 | 15,261 | 39 | 22 | 17 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net revenues |
$ | 95,984 | $ | 89,795 | 7 | % | 100 | % | 100 | % | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
Infectious Disease Testing Market
Sales to the infectious disease testing market decreased 1% to $48.4 million in 2016 from $49.1 million in 2015, primarily due to lower domestic sales of our OraQuick® HIV product and lower sales of our OraQuick® Ebola and In-Home HIV tests, partially offset by higher international sales of our OraQuick® HIV and HCV tests.
The table below shows a breakdown of our total net OraQuick® HIV and HCV product revenues (dollars in thousands) during 2016 and 2015.
| Year Ended December 31, | ||||||||||||
| Market |
2016 | 2015 | % Change |
|||||||||
| Domestic HIV |
$ | 21,499 | $ | 24,956 | (14 | )% | ||||||
| International HIV |
5,248 | 2,410 | 118 | |||||||||
| Domestic OTC HIV |
6,320 | 6,992 | (10 | ) | ||||||||
|
|
|
|
|
|||||||||
| Net HIV revenues |
33,067 | 34,358 | (4 | ) | ||||||||
|
|
|
|
|
|||||||||
| Domestic HCV |
7,436 | 7,502 | (1 | ) | ||||||||
| International HCV |
6,630 | 3,884 | 71 | |||||||||
|
|
|
|
|
|||||||||
| Net HCV revenues |
14,066 | 11,386 | 24 | |||||||||
|
|
|
|
|
|||||||||
| Net OraQuick® revenues |
$ | 47,133 | $ | 45,744 | 3 | % | ||||||
|
|
|
|
|
|||||||||
Domestic OraQuick® HIV sales decreased 14% to $21.5 million for the year ended December 31, 2016 from $24.9 million for the year ended December 31, 2015. This decrease was primarily the result of the continued loss
70
Table of Contents
of sales to competing fourth generation automated HIV immunoassays performed in a laboratory, as recommended under testing guidelines issued by the Centers for Disease Control and Prevention (“CDC”), the loss of sales to point-of-care HIV tests perceived to be more sensitive, reduced program funding, and the timing of orders placed by our customers. International sales of our OraQuick® HIV test in 2016 increased 118% to $5.2 million from $2.4 million in 2015. This increase was largely due to the shipment of product in support of a new HIV self-testing program in Africa, new shipments into the Middle East, sales to a new distributor in Russia, and increased sales in Asia due to a new military testing project.
Sales of our OraQuick® In-Home HIV test decreased 10% to $6.3 million in 2016 from $7.0 million in 2015. This decline was primarily the result of a reduction in promotions run in 2016 as compared to 2015 and a fourth quarter 2015 sales increase immediately following a celebrity’s announcement that he had tested positive for the HIV virus. A comparable event did not occur in 2016. The decline in sales was partially offset by the positive impact of a price increase implemented in the third quarter of 2015.
Domestic OraQuick® HCV sales decreased 1% to $7.4 million in 2016 from $7.5 million in 2015, largely due to the inclusion in 2015 of a $1.3 million order for a U.S. government HCV testing program that did not recur in 2016, partially offset by the expansion of existing HCV testing programs and the addition of new programs in the public health market. International OraQuick® HCV sales increased 71% to $6.6 million in 2016 from $3.9 million in 2015, largely due the first shipments of $1.4 million of product to a foreign government to support a nationwide HCV testing and treatment program, the expansion of our business in Asia, and increased sales in Africa.
Risk Assessment Market
Sales to the risk assessment market decreased 3% to $13.1 million for the year ended December 31, 2016 from $13.5 million for the year ended December 31, 2015, primarily as a result of lower sales of our OraSure® oral fluid collection device into the domestic life insurance market.
Cryosurgical Systems Market
Sales of our cryosurgical systems products (which includes both the physicians’ office and OTC markets) increased 11% to $13.2 million in 2016 from $11.9 million in 2015.
The table below shows a breakdown of our total net cryosurgical systems revenues (dollars in thousands) generated in each market during 2016 and 2015.
| Year Ended December 31, | ||||||||||||
| Market |
2016 | 2015 | % Change |
|||||||||
| Domestic professional |
$ | 5,545 | $ | 4,311 | 29 | % | ||||||
| International professional |
771 | 916 | (16 | ) | ||||||||
| Domestic OTC |
1,350 | 390 | 246 | |||||||||
| International OTC |
5,568 | 6,303 | (12 | ) | ||||||||
|
|
|
|
|
|||||||||
| Net cryosurgical systems revenues |
$ | 13,234 | $ | 11,920 | 11 | % | ||||||
|
|
|
|
|
|||||||||
Sales of our Histofreezer® product to physicians’ offices in the United States increased 29% to $5.5 million in 2016 from $4.3 million in 2015, primarily due to the recovery of business previously lost to competition from private label brands, the initiation of a distributor expansion strategy, and the timing of orders by certain customers. International sales of Histofreezer® decreased 16% to $771,000 in 2016 from $916,000 in the same period of the prior year, primarily due to lower sales in Asia.
71
Table of Contents
Sales of our domestic private-label OTC wart removal products in the U.S. retail market increased to $1.4 million in 2016 from $390,000 in 2015, due to the launch of private-label products in two additional large pharmacy chains early in 2016.
Sales of our international OTC cryosurgical products during 2016 decreased 12% to $5.6 million compared to $6.3 million in 2015, largely due to lower sales into Europe, partially offset by higher sales in Latin America.
Other revenues
Other revenues in 2016 increased 39% to $21.3 million from $15.3 million in 2015. AbbVie exclusivity revenues increased to $18.9 million in 2016 from $13.5 million in 2015 due to the early termination of our co-promotion agreement with AbbVie which we agreed to as of June 30, 2016. The agreement terminated on December 31, 2016, and as a result of the shortened term, an additional $5.4 million of associated deferred revenue which existed as of June 30, 2016 was recognized into revenue over the remaining six months of 2016. Following the termination of our HCV co-promotion agreement with AbbVie on December 31, 2016, AbbVie has no further financial obligations to us. Also included in other revenues for 2016 was $2.3 million in BARDA funding, a 30% increase from the $1.8 million in BARDA funding in 2015. This increase was due to new funding for our Zika product.
DNAG Segment
Molecular Collection Systems
Net molecular collection systems revenues increased 8% to $32.2 million in 2016 from $29.9 million in 2015.
The table below shows a breakdown of our total net molecular collection systems revenues (dollars in thousands) generated in each market during 2016 and 2015.
| Year Ended December 31, | ||||||||||||
| Market |
2016 | 2015 | % Change |
|||||||||
| Commercial genomics |
$ | 20,767 | $ | 19,513 | 6 | % | ||||||
| Academic genomics |
10,312 | 9,872 | 4 | |||||||||
| Microbiome |
1,135 | 539 | 111 | |||||||||
|
|
|
|
|
|||||||||
| Net molecular collection systems revenues |
$ | 32,214 | $ | 29,924 | 8 | % | ||||||
|
|
|
|
|
|||||||||
Sales of our Oragene® product in the commercial market increased 6% to $20.8 million in 2016 compared to $19.5 million in 2015, primarily as a result of sales to new customers and ordering patterns of existing customers, partially offset by the loss of two U.S. customers that previously filed for bankruptcy protection in 2015. These customers contributed approximately $2.9 million in sales during 2015. The Company has no unreserved collection exposure related to these two customers. Sales of our Oragene® product in the academic market rose 4% to $10.3 million in 2016 compared to $9.8 million in 2015, due to the shipment of product to support a study on autism. Sales in 2016 also included $1.1 million in sales of our gut microbiome product compared to $539,000 in the same period of 2015 as interest in our microbiome product offering continued to grow with both new and existing customers.
CONSOLIDATED OPERATING RESULTS
Consolidated gross margin was 69% for the year ended December 31, 2016 compared to 67% for the comparable period of 2015. Gross margin for 2016 increased primarily due to the higher AbbVie exclusivity revenues.
72
Table of Contents
Consolidated operating income in 2016 was $20.3 million, a $12.2 million increase from the $8.1 million of operating income reported in 2015. Operating income in 2016 benefited as compared to the prior year from higher revenues, improved gross margins and lower sales and marketing and research and development costs, partially offset by higher general and administrative expenses.
OPERATING INCOME BY SEGMENT
OSUR Segment
OSUR’s gross margin was 69% in 2016 compared to 66% in 2015. OSUR’s margin in 2016 was positively impacted by the increase in other revenues and lower scrap and spoilage costs.
Research and development expenses decreased 21% to $7.0 million in 2016 from $8.9 million in 2015, largely as a result of a $1.4 million payment received to settle a claim against one of our raw material suppliers. This settlement was recorded as a reduction in research and development expense.
Sales and marketing expenses decreased 20% to $21.3 million in 2016 from $26.6 million in 2015, primarily as a result of lower detailing and other expenses associated with our OraQuick® HCV co-promotion agreement with AbbVie, as well as lower staffing costs.
General and administrative expenses increased 12% to $22.6 million in 2015 from $20.1 million in 2015 due increased consulting, severance, and other staffing-related costs.
All of the above contributed to OSUR’s operating income of $15.6 million for 2016, which included non-cash charges of $2.7 million for depreciation and amortization and $5.6 million for stock-based compensation.
DNAG Segment
DNAG’s gross margin was 67% in 2016 compared to 70% in 2015. This decrease was attributable to a higher volume of lower margin sales experienced in 2016 when compared to 2015 coupled with the inclusion of severance costs in DNAG’s costs of goods sold in 2016.
Research and development expenses were $2.7 million in both 2016 and 2015.
Sales and marketing expenses decreased 1% to $8.3 million in 2016 compared to $8.4 million in 2015 due to lower bad debt expense.
General and administrative expenses increased 7% to $5.7 million in 2016 compared to $5.3 million in 2015, largely due to higher legal costs.
All of the above contributed to DNAG’s operating income of $4.6 million for 2016, which included non-cash charges of $3.0 million for depreciation and amortization and $467,000 for stock-based compensation.
CONSOLIDATED INCOME TAXES
We continue to believe the full valuation allowance established in 2008 against our total U.S. deferred tax asset is appropriate as the facts and circumstances necessitating the allowance have not changed. For the year ended December 31, 2016, state income tax expense of $250,000 was recorded compared to $0 in 2015. Canadian income tax expense of $353,000 and $665,000 was recorded in 2016 and 2015, respectively.
73
Table of Contents
Liquidity and Capital Resources
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| (In thousands) | ||||||||
| Cash, cash equivalents and restricted cash |
$ | 72,869 | $ | 109,790 | ||||
| Available for sale securities |
103,718 | 11,160 | ||||||
| Working capital |
189,668 | 139,106 | ||||||
Our cash, cash equivalents, restricted cash and available-for-sale securities increased to $176.6 million at December 31, 2017 from $120.9 million at December 31, 2016. Our working capital increased to $189.7 million at December 31, 2017 from $139.1 million at December 31, 2016.
During 2017, we generated $28.2 million in cash from operating activities. Our net income of $31.0 million included non-cash charges for stock-based compensation expense of $6.9 million and depreciation and amortization expense of $6.4 million, as well as non-cash benefits of $518,000. Additional sources of cash include increases in accounts payable of $5.2 million largely due to inventory purchases that were invoiced at the end of the year, accrued expenses and other liabilities of $9.2 million largely due to an increase in Canadian income taxes payable and a higher year-end accrual for management incentive bonuses. Uses of cash in operating activities during the year included an increase in accounts receivable of $22.1 million largely resulting from an increase in product orders placed during the fourth quarter of 2017, an increase in inventory balances of $7.4 million related to higher raw material purchases for our HIV self-test and HCV products, and an increase in prepaid expenses and other assets of $384,000.
We used $96.3 million in investing activities in 2017 to purchase $161.3 million of investments and $4.3 million to acquire property and equipment, partially offset by proceeds from the maturities and redemptions of investments of $69.3 million.
Net cash provided by financing activities was $30.4 million in 2017, which resulted from $31.7 million in proceeds received from the exercise of stock options partially offset by $1.2 million used for the repurchase of common stock to satisfy withholding taxes related to the vesting of restricted shares.
In September 30, 2016, we entered into a credit agreement (the “Credit Agreement”) with a commercial bank, which was amended in December 2017. The Credit Agreement provides for revolving extensions of credit in an initial aggregate amount of up to $10.0 million (inclusive of a letter of credit subfacility of $2.5 million), with an option to request, prior to the second anniversary of the closing date, that lenders, at their election, provide up to $5.0 million of additional revolving commitments. Obligations under the Credit Agreement are secured by a first priority security interest in certain eligible accounts receivable, 65% of the equity of our subsidiary, DNAG, and certain related assets. There were no borrowings outstanding under the Credit Agreement at December 31, 2017 or 2016.
Borrowings under the Credit Agreement are subject to compliance with borrowing base limitations tied to eligibility of accounts receivable. Interest under the Credit Agreement is payable at the London Interbank Offered Rate for one, two, three or six-month loans, as selected by the Company, plus 2.50% per year. The Credit Agreement will be subject to an unused line fee of 0.375% per annum on the unused portion of the commitment under the Credit Agreement during the revolving period. The maturity date of the Credit Agreement is September 30, 2019.
In connection with the Credit Agreement, under certain circumstances, we must comply with a minimum fixed charge coverage ratio of 1.10 to 1.00, measured as of the last day of each fiscal month and for the twelve-fiscal month period ending on such date. As of December 31, 2017 and 2016, we were in compliance with all applicable covenants under the Credit Agreement.
Our current balances of cash and cash equivalents and available-for-sale securities and our available borrowing capacity are expected to be sufficient to fund our current operating and capital needs for the foreseeable future.
74
Table of Contents
Our cash requirements, however, may vary materially from those now planned due to many factors, including, but not limited to, the scope and timing of future strategic acquisitions, the progress of our research and development programs, the scope and results of clinical testing, the cost of any future litigation, the magnitude of capital expenditures, changes in existing and potential relationships with business partners, the timing and cost of obtaining regulatory approvals, the timing and cost of future stock purchases, the costs involved in obtaining and enforcing patents, proprietary rights and any necessary licenses, the cost and timing of expansion of sales and marketing activities, market acceptance of new products, competing technological and market developments, the impact of the current economic environment and other factors. In addition, $43.7 million, or 25% of our $176.6 million in cash, cash equivalents, restricted cash and available-for-sale securities, belongs to our Canadian subsidiary. Repatriation of such cash into the United States exceeding certain levels could have adverse tax consequences.
Contractual Obligations and Commercial Commitments
The following sets forth our approximate aggregate obligations as of December 31, 2017 (in thousands) for future payments under contracts and other contingent commitments, for the year 2018 and beyond:
| Payments due by December 31, | ||||||||||||||||||||||||||||
| Contractual Obligations |
Total | 2018 | 2019 | 2020 | 2021 | 2022 | Thereafter | |||||||||||||||||||||
| Operating leases1 |
$ | 5,157 | $ | 617 | $ | 791 | $ | 828 | $ | 834 | $ | 845 | $ | 1,242 | ||||||||||||||
| Employment contracts2 |
2,059 | 1,729 | 330 | — | — | — | — | |||||||||||||||||||||
| Purchase obligations3 |
15,028 | 14,262 | 383 | 383 | — | — | — | |||||||||||||||||||||
| Minimum commitments under contracts4 |
292 | 292 | — | — | — | — | — | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total contractual obligations |
$ | 22,536 | $ | 16,900 | $ | 1,504 | $ | 1,211 | $ | 834 | $ | 845 | $ | 1,242 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| 1 | Represents payments required under our operating leases. See Note 11 of the Notes to the consolidated financial statements included herein. |
| 2 | Represents salary payments payable under the terms of employment agreements executed by us with certain executives. See Note 11 of the Notes to the consolidated financial statements included herein. |
| 3 | Represents payments required by non-cancellable purchase orders or supply agreements related to inventory, supplies, capital expenditures and other goods or services. The supply agreements are cancellable within a specified number of days of written notice to the supplier. See Note 11 of the Notes to the consolidated financial statements included herein. |
| 4 | Represents payments required pursuant to certain licensing agreements executed by the Company. These agreements are cancellable within a specified number of days after communication by the Company of its intent to terminate. See Note 11 of the Notes to the consolidated financial statements included herein. |
| 5 | Does not include any obligations under our revolving credit facility, which was undrawn as of December 31, 2017. See Note 7 of the Notes to the consolidated financial statements included herein. |
Off-Balance Sheet Arrangements. We do not have any off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of Regulation S-K under the Securities Exchange Act of 1934, as amended.
Critical Accounting Policies and Estimates
This Management’s Discussion and Analysis of Financial Condition and Results of Operations discusses our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires that we make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. On an on-going basis, we evaluate our judgments and estimates, including those related to bad debts, customer sales returns, inventories, intangible assets, income taxes, revenue recognition,
75
Table of Contents
performance-based compensation, contingencies and litigation. We base our judgments and estimates on historical experience and on various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in Note 2 of the Notes to the consolidated financial statements included in Item 15 of this Annual Report. We consider the following accounting policies, which have been discussed with our Audit Committee, to be most critical in understanding the more complex judgments that are involved in preparing our financial statements and the uncertainties that could impact our results of operations, financial condition and cash flows.
Revenue Recognition. We recognize product revenues when there is persuasive evidence that an arrangement exists, the price is fixed or determinable, title has passed and collection is reasonably assured. Product revenues are recorded net of allowances for any discounts or rebates. We do not grant price protection or product return rights to our customers except for sales of our OraQuick® In-Home HIV test in the retail market and for warranty returns. Historically, returns arising from warranty issues have been infrequent and immaterial. Accordingly, we expense warranty returns as incurred.
Our net revenues recorded on sales of the OraQuick® In-Home HIV test represent total gross revenues, less an allowance for expected returns, and customer allowances for cooperative advertising, discounts, rebates, and chargebacks. The allowance for expected returns is an estimate established by management, based upon currently available information, and is adjusted to reflect known changes in the factors that impact this estimate. Other customer allowances are at contractual rates and are recorded as a reduction of gross revenue when recognized in our consolidated statements of income.
We record shipping and handling charges billed to our customers as product revenue and the related expense as cost of products sold. Taxes assessed by governmental authorities, such as sales or value-added taxes, are excluded from product revenues.
On June 10, 2014, we entered into a Master Program Services and Co-Promotion Agreement with AbbVie to co-promote our OraQuick® HCV test in the United States. This agreement with AbbVie was terminated effective December 31, 2016. Accordingly, we did not record any revenue from this co-promotion agreement during 2017. During 2016 and 2015, $18.9 million and $13.5 million, respectively, in exclusivity revenues were recognized and recorded as other revenue in our consolidated statements of income.
On June 12, 2015, we were awarded a grant for up to $10.4 million in total funding from the U.S. Department of Health and Human Services (“HHS”) Office of the Assistant Secretary for Preparedness and Response’s Biomedical Advanced Research and Development Authority (“BARDA”) related to our OraQuick® Ebola rapid antigen test. The three-year, multi-phased grant included an initial commitment of $1.8 million and options for up to an additional $8.6 million to fund certain clinical and regulatory activities. In September 2015 and July 2017, BARDA exercised options to provide $7.2 million and $1.3 million, respectively, in additional funding for our OraQuick® Ebola test. Amounts related to this grant are recorded as other revenue in our consolidated statements of income as the activities are being performed and the related costs are incurred. During 2017, 2016 and 2015, $2.0 million, $1.7 million and $1.8 million, respectively, were recognized in connection with this grant.
In August 2016, we were awarded a contract for up to $16.6 million in total funding from BARDA related to our rapid Zika test. The six-year, multi-phased contract includes an initial commitment of $7.0 million and options for up to an additional $9.6 million to fund the evaluation of additional product enhancements and clinical and regulatory activities. In May 2017, BARDA exercised an option to provide $2.6 million in additional funding for
76
Table of Contents
our rapid Zika test. Funding received under this contract is recorded as other revenue in our consolidated statements of income as the activities are being performed and the related costs are incurred. During 2017 and 2016, $2.4 million and $616,000, respectively, were recognized in connection with this grant.
In June 2017, we entered into a four-year Charitable Support Agreement with the Bill & Melinda Gates Foundation (“Gates Foundation”) that will enable us to offer our OraQuick® HIV self-test at an affordable price in 50 developing countries with funding from the Gates Foundation. The funding will consist of support payments tied to volume of product sold by us and reimbursement of certain related costs. The funding from the Gates Foundation will be in an aggregate amount not to exceed $20.0 million over the four-year term or $6.0 million each year of the agreement. Funding received under this agreement in the form of support payments for product purchases is recorded as a component of product revenue. During 2017, $1.0 million of support payments were recognized in product revenue in connection with this agreement. Funding received in the form of reimbursement of certain related costs in the amount of $689,000 was also recorded as other revenue in our consolidated statements of income.
Inventories. Our inventories are valued at the lower of cost or net realizable value, with cost determined on a first-in first-out basis, and include the cost of raw materials, labor and overhead. The majority of our inventories are subject to expiration dating. We continually evaluate quantities on hand and the carrying value of our inventories to determine the need for reserves for excess and obsolete inventories based primarily on the estimated forecast of product sales. When, in the opinion of management, factors indicate that impairment has occurred, either a reserve is established against the inventories’ carrying value or the inventories are completely written off, as in the case of lapsing expiration dates. In addition to reserving for these items identified through specific identification procedures, we also reserve for unidentified scrap or spoilage under a fixed-formula methodology. During 2017, 2016, and 2015 we wrote-off inventory which had a cost of $3.3 million, $739,000 and $2.6 million, respectively. These write-offs were a result of quality, scrap and product expiration issues. Although we make every effort to ensure the accuracy of our forecasts of future product demand, any significant unanticipated changes in demand could have a significant impact on the carrying value of our inventories and reported operating results.
Deferred Tax Assets and Liabilities. We re-measured our Federal net deferred tax asset based on the federal rate at which they were expected to reverse in the future which is 21% pursuant to the recently enacted Tax Act. We also re-measured our state net deferred asset since the future Federal benefit was reduced to 21%.
At December 31, 2017, we had federal Net Operating Loss (“NOL”) carryforwards of $66.2 million. The net deferred tax assets, before the valuation allowance, associated with these NOLs and other temporary differences were $27.3 million at December 31, 2017. Net operating losses will begin to expire in 2020. In assessing the realizability of deferred tax assets, we consider whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the period in which those temporary differences become deductible or the NOLs and credit carryforwards can be utilized. We consider the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies in making this assessment.
We currently have a full valuation allowance recorded against our total U.S. deferred tax asset as we had determined in 2008 that it was more likely than not that we would not realize the benefits associated with our deferred tax asset in the immediate future. Each year, we continue to reevaluate our valuation allowance position and believe that it is more likely than not that our U.S. deferred income tax asset will not be realized in the immediate future. As such, we maintain a full valuation allowance as of December 31, 2017 and 2016 against our deferred tax assets associated with the operations subject to income tax in the U.S.
The Tax Reform Act of 1986 contains provisions under Internal Revenue Code (“IRC”) Section 382 that limit the annual amount of federal and state NOLs available to be used in any given year in the event of a significant change in ownership. Our ability to use our federal and state NOL carryforwards to offset future federal income tax obligations could be limited by changes in the ownership of our stock. The Company does not believe,
77
Table of Contents
however, that there is a Section 382 limitation that will impair our future ability to utilize NOLs to offset our future taxable income and the Company continues to review ownership changes on an annual basis.
| ITEM 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
We do not hold any amounts of derivative financial instruments or derivative commodity instruments and, accordingly, we have no material derivative risk to report under this Item.
As of December 31, 2017, we did not have any foreign currency exchange contracts or purchase currency options to hedge local currency cash flows. Sales denominated in foreign currencies comprised 3.5% of our total revenues for the year ended December 31, 2017. We do have foreign currency exchange risk related to our operating subsidiary in Canada. While the majority of their revenues are recorded in U.S. dollars, almost all of their operating expenses are denominated in Canadian dollars. Fluctuations in the exchange rate between the U.S. dollar and the Canadian dollar could affect year-to-year comparability of operating results and cash flows. Our Canadian subsidiary had net assets, subject to translation, of $102.6 million Canadian dollars ($81.6 million USD), which are included in the Company’s consolidated balance sheet as of December 31, 2017. A 10% unfavorable change in the Canadian-to-U.S. dollar exchange rate would have decreased our comprehensive income by $8.2 million in 2017.
| ITEM 8. | Consolidated Financial Statements and Supplementary Data. |
Information with respect to this Item is contained in our Consolidated Financial Statements included in Item 15 of this Annual Report on Form 10-K.
78
Table of Contents
| ITEM 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. |
Not applicable.
| ITEM 9A. | Controls and Procedures. |
(a) Evaluation of Disclosure Controls and Procedures.
The Company’s management, with the participation of the Company’s Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of the Company’s disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934) as of December 31, 2017. Based on that evaluation, the Company’s management, including such officers, concluded that as of December 31, 2017 the Company’s disclosure controls and procedures were effective to provide reasonable assurance that information required to be disclosed by the Company in the reports that we file or submit under the Securities Exchange Act of 1934 is accumulated and communicated to the Company’s management, including the Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosure and is recorded, processed, summarized, and reported within the time periods specified in the rules and forms of the Securities and Exchange Commission.
(b) Management’s Report on Internal Control Over Financial Reporting.
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934. Under the supervision and with the participation of the Company’s management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework, our management concluded that our internal control over financial reporting was effective to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles as of December 31, 2017.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by KPMG LLP, an independent registered public accounting firm, as stated in their report, which is included below.
(c) Changes in Internal Control Over Financial Reporting.
There were no changes in our internal control over financial reporting during the fiscal quarter ended December 31, 2017 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
(d) Report of Independent Registered Public Accounting Firm.
The Board of Directors and Stockholders
OraSure Technologies, Inc.:
We have audited OraSure Technologies, Inc.’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of
79
Table of Contents
Sponsoring Organizations of the Treadway Commission (COSO). OraSure Technologies, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, OraSure Technologies, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of OraSure Technologies, Inc. and subsidiaries as of December 31, 2017 and 2016, and the related consolidated statements of income, comprehensive income, stockholders’ equity and cash flows for each of the years in the three-year period ended December 31, 2017, and our report dated February 28, 2018 expressed an unqualified opinion on those consolidated financial statements.
/s/ KPMG LLP
Philadelphia, Pennsylvania
February 28, 2018
| ITEM 9B. | Other Information. |
Not applicable.
80
Table of Contents
PART III
We have omitted from Part III the information that will appear in our Definitive Proxy Statement for our 2018 Annual Meeting of Stockholders (the “Proxy Statement”), which will be filed within 120 days after the end of our fiscal year pursuant to Regulation 14A.
| ITEM 10. | Directors, Executive Officers and Corporate Governance. |
Certain information required by this Item is incorporated by reference to the information under the captions “Proposal No. 1. Election of Directors,” “Corporate Governance - Governance Guidelines and Code of Conduct,” “Corporate Governance – Committees of the Board,” “Executive Officers,” and “Section 16(a) Beneficial Ownership Reporting Compliance” in the Proxy Statement.
Our Board of Directors has adopted a Code of Business Conduct and Ethics that applies to our principal executive officer, principal financial officer and principal accounting officer, as well as to the members of our Board of Directors and our other officers and employees. This Code of Business Conduct and Ethics is available on our website at www.orasure.com. We intend to satisfy the amendment and waiver disclosure requirements under applicable securities regulations by posting any amendments of, or waivers to, the Code of Business Conduct and Ethics on our website.
| ITEM 11. | Executive Compensation. |
The information required by this Item is incorporated by reference to the information under the captions “Compensation Committee Matters,” (including the “Compensation Committee Report”), “Compensation Discussion and Analysis,” “Compensation Tables,” “Employment Agreements and Potential Payments Upon Termination or Change in Control,” and “Director Compensation” in the Proxy Statement.
| ITEM 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The information required by this Item with respect to the securities ownership of certain beneficial owners and management, and equity compensation plan information, is incorporated by reference to the information under the captions “Stock Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in the Proxy Statement.
| ITEM 13. | Certain Relationships and Related Transactions, and Director Independence. |
The information required by this Item is incorporated by reference to the information under the captions “Transactions with Related Persons” and “Corporate Governance - Director Independence” in the Proxy Statement.
| ITEM 14. | Principal Accountant Fees and Services. |
The information required by this Item is incorporated by reference to the information under the caption “Audit Committee Matters” in the Proxy Statement.
81
Table of Contents
PART IV
| ITEM 15. | Exhibits and Consolidated Financial Statement Schedules. |
(a)(1) and (a)(2). Consolidated Financial Statements and Schedules. For a list of the consolidated financial statements filed herewith, see the Index to Consolidated Financial Statements following the signature page to this Annual Report. No schedules are included with the consolidated financial statements because the required information is inapplicable or is presented in the consolidated financial statements or related notes thereto.
(a)(3). Exhibits. See Index to Exhibits following the consolidated financial statements in this Annual Report.
| ITEM 16. | Form 10-K Summary. |
None
82
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on February 28, 2018.
| ORASURE TECHNOLOGIES, INC. | ||
| By: | /s/ Douglas A. Michels | |
| Douglas A. Michels President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed on February 28, 2018, by the following persons on behalf of the Registrant and in the capacities indicated.
| SIGNATURE | TITLE | |
| /s/ Douglas A. Michels Douglas A. Michels |
President, Chief Executive Officer and Director (Principal Executive Officer) | |
| /s/ Ronald H. Spair Ronald H. Spair |
Chief Operating Officer, Chief Financial Officer and Director (Principal Financial Officer) | |
| /s/ Mark L. Kuna Mark L. Kuna |
Senior Vice President, Finance and Controller (Principal Accounting Officer) | |
| *MARA ASPINALL Mara Aspinall |
Director | |
| *MICHAEL CELANO Michael Celano |
Director | |
| *EAMONN P. HOBBS Eamonn P. Hobbs |
Director | |
| *RONNY B. LANCASTER Ronny B. Lancaster |
Director | |
| *CHARLES W. PATRICK Charles W. Patrick |
Director | |
| *ARADHANA SARIN Aradhana Sarin |
Director | |
| *STEPHEN S. TANG Stephen S. Tang |
Director | |
| *By: | /s/ Jack E. Jerrett | |
| Jack E. Jerrett | ||
| (Attorney-in-Fact) |
83
Table of Contents
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors
OraSure Technologies, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of OraSure Technologies, Inc. and subsidiaries (the Company) as of December 31, 2017 and 2016, the related consolidated statements of income, comprehensive income, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2017, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated February 28, 2018 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ KPMG LLP
We have served as the Company’s auditor since 2002.
Philadelphia, Pennsylvania
February 28, 2018
F-2
Table of Contents
ORASURE TECHNOLOGIES, INC. AND SUBSIDIARIES
(in thousands, except per share amounts)
| December 31, 2017 | December 31, 2016 | |||||||
| ASSETS |
||||||||
| CURRENT ASSETS: |
||||||||
| Cash and cash equivalents |
$ | 71,029 | $ | 107,959 | ||||
| Restricted cash |
1,840 | 1,831 | ||||||
| Short-term investments |
83,028 | 11,160 | ||||||
| Accounts receivable, net of allowance for doubtful accounts of $471 and $484 |
42,521 | 19,827 | ||||||
| Inventories |
19,343 | 11,799 | ||||||
| Prepaid expenses |
1,658 | 1,722 | ||||||
| Other current assets |
2,486 | 2,143 | ||||||
|
|
|
|
|
|||||
| Total current assets |
221,905 | 156,441 | ||||||
| PROPERTY AND EQUIPMENT, net |
21,372 | 20,033 | ||||||
| INTANGIBLE ASSETS, net |
8,223 | 10,337 | ||||||
| GOODWILL |
20,083 | 18,793 | ||||||
| LONG TERM INVESTMENTS |
20,690 | — | ||||||
| OTHER ASSETS |
3,928 | 2,331 | ||||||
|
|
|
|
|
|||||
| $ | 296,201 | $ | 207,935 | |||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| CURRENT LIABILITIES: |
||||||||
| Accounts payable |
$ | 10,228 | $ | 4,633 | ||||
| Deferred revenue |
1,314 | 1,388 | ||||||
| Accrued expenses |
20,695 | 11,314 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
32,237 | 17,335 | ||||||
|
|
|
|
|
|||||
| OTHER LIABILITIES |
3,932 | 2,304 | ||||||
|
|
|
|
|
|||||
| DEFERRED INCOME TAXES |
1,951 | 2,446 | ||||||
|
|
|
|
|
|||||
| COMMITMENTS AND CONTINGENCIES (Note 11) |
||||||||
| STOCKHOLDERS’ EQUITY |
||||||||
| Preferred stock, par value $.000001, 25,000 shares authorized, none issued |
— | — | ||||||
| Common stock, par value $.000001, 120,000 shares authorized, 60,662 and 56,001 shares issued and outstanding |
— | — | ||||||
| Additional paid-in capital |
387,931 | 350,528 | ||||||
| Accumulated other comprehensive loss |
(10,340 | ) | (14,220 | ) | ||||
| Accumulated deficit |
(119,510 | ) | (150,458 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
258,081 | 185,850 | ||||||
|
|
|
|
|
|||||
| $ | 296,201 | $ | 207,935 | |||||
|
|
|
|
|
|||||
See accompanying notes to the consolidated financial statements.
F-3
Table of Contents
ORASURE TECHNOLOGIES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF INCOME
(in thousands, except per share amounts)
| For the years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| NET REVENUES: |
||||||||||||
| Product |
$ | 161,988 | $ | 106,924 | $ | 104,458 | ||||||
| Other |
5,076 | 21,274 | 15,261 | |||||||||
|
|
|
|
|
|
|
|||||||
| 167,064 | 128,198 | 119,719 | ||||||||||
| COST OF PRODUCTS SOLD |
68,108 | 40,171 | 39,426 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
98,956 | 88,027 | 80,293 | |||||||||
|
|
|
|
|
|
|
|||||||
| OPERATING EXPENSES: |
||||||||||||
| Research and development |
13,365 | 9,754 | 11,654 | |||||||||
| Sales and marketing |
28,532 | 29,652 | 35,088 | |||||||||
| General and administrative |
29,321 | 28,356 | 25,493 | |||||||||
| Gain on litigation settlement |
(12,500 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| 58,718 | 67,762 | 72,235 | ||||||||||
|
|
|
|
|
|
|
|||||||
| Operating income |
40,238 | 20,265 | 8,058 | |||||||||
| OTHER INCOME |
794 | 58 | 774 | |||||||||
|
|
|
|
|
|
|
|||||||
| Income before income taxes |
41,032 | 20,323 | 8,832 | |||||||||
| INCOME TAX EXPENSE |
10,084 | 603 | 665 | |||||||||
|
|
|
|
|
|
|
|||||||
| NET INCOME |
$ | 30,948 | $ | 19,720 | $ | 8,167 | ||||||
|
|
|
|
|
|
|
|||||||
| EARNINGS PER SHARE: |
||||||||||||
| BASIC |
$ | 0.52 | $ | 0.35 | $ | 0.14 | ||||||
|
|
|
|
|
|
|
|||||||
| DILUTED |
$ | 0.51 | $ | 0.35 | $ | 0.14 | ||||||
|
|
|
|
|
|
|
|||||||
| SHARES USED IN COMPUTING EARNINGS PER SHARE: |
||||||||||||
| BASIC |
59,050 | 55,615 | 56,397 | |||||||||
|
|
|
|
|
|
|
|||||||
| DILUTED |
61,024 | 56,513 | 56,846 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to the consolidated financial statements.
F-4
Table of Contents
ORASURE TECHNOLOGIES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
(in thousands)
| For the years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| NET INCOME |
$ | 30,948 | $ | 19,720 | $ | 8,167 | ||||||
| OTHER COMPEHENSIVE INCOME (LOSS) |
||||||||||||
| Currency translation adjustments |
4,433 | 1,419 | (7,791 | ) | ||||||||
| Unrealized loss on marketable securities |
(553 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| COMPREHENSIVE INCOME |
$ | 34,828 | $ | 21,139 | $ | 376 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to the consolidated financial statements.
F-5
Table of Contents
ORASURE TECHNOLOGIES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
For the years ended December 31, 2017, 2016 and 2015
(in thousands)
|
Common Stock |
Additional Paid-in Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
|||||||||||||||||||||
| Shares | Amount | Total | ||||||||||||||||||||||
| Balance at January 1, 2015 |
56,187 | $ | — | $ | 344,894 | $ | (7,848 | ) | $ | (178,345 | ) | $ | 158,701 | |||||||||||
| Common stock issued upon exercise of options |
74 | — | 404 | — | — | 404 | ||||||||||||||||||
| Vesting of restricted stock |
354 | — | — | — | — | — | ||||||||||||||||||
| Purchase and retirement of common shares |
(910 | ) | — | (6,090 | ) | — | — | (6,090 | ) | |||||||||||||||
| Compensation cost for restricted stock |
— | — | 2,642 | — | — | 2,642 | ||||||||||||||||||
| Compensation cost for stock option grants |
— | — | 3,403 | — | — | 3,403 | ||||||||||||||||||
| Net income |
— | — | — | — | 8,167 | 8,167 | ||||||||||||||||||
| Currency translation adjustments |
— | — | — | (7,791 | ) | — | (7,791 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2015 |
55,705 | — | 345,253 | (15,639 | ) | (170,178 | ) | 159,436 | ||||||||||||||||
| Common stock issued upon exercise of options |
405 | — | 2,656 | — | — | 2,656 | ||||||||||||||||||
| Vesting of restricted stock |
446 | — | — | — | — | — | ||||||||||||||||||
| Purchase and retirement of common shares |
(555 | ) | — | (3,444 | ) | — | — | (3,444 | ) | |||||||||||||||
| Compensation cost for restricted stock |
— | — | 2,861 | — | — | 2,861 | ||||||||||||||||||
| Compensation cost for stock option grants |
— | — | 2,691 | — | — | 2,691 | ||||||||||||||||||
| Compensation cost for performance stock units |
— | — | 511 | — | — | 511 | ||||||||||||||||||
| Net income |
— | — | — | — | 19,720 | 19,720 | ||||||||||||||||||
| Currency translation adjustments |
— | — | — | 1,419 | — | 1,419 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2016 |
56,001 | — | 350,528 | (14,220 | ) | (150,458 | ) | 185,850 | ||||||||||||||||
| Common stock issued upon exercise of options |
4,413 | — | 31,670 | — | — | 31,670 | ||||||||||||||||||
| Vesting of restricted stock |
378 | — | — | — | — | — | ||||||||||||||||||
| Purchase and retirement of common shares |
(130 | ) | — | (1,240 | ) | — | — | (1,240 | ) | |||||||||||||||
| Compensation cost for restricted stock |
— | — | 2,705 | — | — | 2,705 | ||||||||||||||||||
| Compensation cost for stock option grants |
— | — | 2,045 | — | — | 2,045 | ||||||||||||||||||
| Compensation cost for performance stock units |
— | — | 2,223 | — | — | 2,223 | ||||||||||||||||||
| Net income |
— | — | — | — | 30,948 | 30,948 | ||||||||||||||||||
| Currency translation adjustments |
4,433 | 4,433 | ||||||||||||||||||||||
| Unrealized loss on marketable securities |
— | — | — | (553 | ) | — | (553 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2017 |
60,662 | $ | — | $ | 387,931 | $ | (10,340 | ) | $ | (119,510 | ) | $ | 258,081 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
See accompanying notes to the consolidated financial statements.
F-6
Table of Contents
ORASURE TECHNOLOGIES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| For the years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| OPERATING ACTIVITIES: |
||||||||||||
| Net income |
$ | 30,948 | $ | 19,720 | $ | 8,167 | ||||||
| Adjustments to reconcile net income to net cash provided by operating activities: |
||||||||||||
| Stock-based compensation |
6,973 | 6,063 | 6,045 | |||||||||
| Depreciation and amortization |
6,402 | 5,640 | 5,806 | |||||||||
| Unrealized foreign currency loss |
156 | 122 | 367 | |||||||||
| Loss (gain) on sale of fixed assets |
(25 | ) | (28 | ) | 80 | |||||||
| Deferred income taxes |
(649 | ) | (529 | ) | 48 | |||||||
| Changes in assets and liabilities |
||||||||||||
| Accounts receivable |
(22,116 | ) | (576 | ) | (3,739 | ) | ||||||
| Inventories |
(7,391 | ) | 1,476 | 2,348 | ||||||||
| Prepaid expenses and other assets |
(384 | ) | (652 | ) | (1,415 | ) | ||||||
| Accounts payable |
5,157 | 46 | (2,648 | ) | ||||||||
| Deferred revenue |
(93 | ) | (8,350 | ) | 1,706 | |||||||
| Accrued expenses and other liabilities |
9,178 | 1,666 | (992 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by operating activities |
28,156 | 24,598 | 15,773 | |||||||||
|
|
|
|
|
|
|
|||||||
| INVESTING ACTIVITIES: |
||||||||||||
| Purchases of investments |
(161,269 | ) | (34,133 | ) | (26,895 | ) | ||||||
| Proceeds from maturities and redemptions of investments |
69,253 | 30,421 | 23,934 | |||||||||
| Purchases of property and equipment |
(4,337 | ) | (4,353 | ) | (3,744 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(96,353 | ) | (8,065 | ) | (6,705 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| FINANCING ACTIVITIES: |
||||||||||||
| Payments for debt issue costs |
— | (367 | ) | — | ||||||||
| Proceeds from exercise of stock options |
31,675 | 2,656 | 404 | |||||||||
| Repurchase of common stock |
(1,240 | ) | (3,444 | ) | (6,090 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) financing activities |
30,435 | (1,155 | ) | (5,686 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| EFFECT OF FOREIGN EXCHANGE RATE CHANGES ON CASH |
841 | 318 | (2,155 | ) | ||||||||
| NET (DECREASE) INCREASE IN CASH, CASH EQUIVALENTS, AND RESTRICTED CASH |
(36,921 | ) | 15,696 | 1,227 | ||||||||
| CASH, CASH EQUIVALENTS, AND RESTRICTED CASH, BEGINNING OF PERIOD |
109,790 | 94,094 | 92,867 | |||||||||
|
|
|
|
|
|
|
|||||||
| CASH, CASH EQUIVALENTS, AND RESTRICTED CASH, END OF PERIOD |
$ | 72,869 | $ | 109,790 | $ | 94,094 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to the consolidated financial statements.
F-7
Table of Contents
ORASURE TECHNOLOGIES, INC. AND SUBSIDIARIES
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
(in thousands, except per share amounts, unless otherwise indicated)
| 1. | THE COMPANY: |
Our business is comprised of two segments: Our “OSUR” business consists of the development, manufacture, marketing and sale of oral fluid diagnostic products and specimen collection devices using our proprietary technologies, as well as other diagnostic products including immunoassays and other in vitro diagnostic tests that are used on other specimen types. Our molecular collections systems or “DNAG” business consists of the manufacture and sale of specimen collection kits that are used to collect, stabilize, transport and store samples of genetic material for molecular testing in the consumer genetic, clinical genetic, academic research, pharmacogenomics, personalized medicine, microbiome and animal genetics markets.
Our OSUR diagnostic products include tests that are performed on a rapid basis at the point of care and tests that are processed in a laboratory. These products are sold in the United States and internationally to various clinical laboratories, hospitals, clinics, community-based organizations and other public health organizations, distributors, government agencies, physicians’ offices, and commercial and industrial entities. We also manufacture and sell medical devices used for the removal of benign skin lesions by cryosurgery or freezing. These cryosurgical products are sold in both professional and over-the-counter (“OTC”) markets in North America, Europe, Central and South America, and Australia.
Our “DNAG” or molecular collection systems business is operated by our subsidiary, DNA Genotek Inc., a company based in Ottawa, Canada. DNAG’s Oragene® DNA sample collection kit provides an all-in-one system for the collection, stabilization, transportation and storage of DNA from human saliva. We also sell research use only sample collection products into the microbiome and tuberculosis markets and we offer our customers a suite of genomics and microbiome services called “GenoFINDTM”, which range from package customization and study design optimization to extraction, analysis and reporting services. We serve customers worldwide, including many leading research universities and hospitals.
| 2. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES: |
Principles of Consolidation and Basis of Presentation
The consolidated financial statements include the accounts of OraSure Technologies, Inc. (“OraSure”) and its wholly-owned subsidiary, DNA Genotek Inc. (“DNAG”). All intercompany transactions and balances have been eliminated. References herein to “we,” “us,” “our,” or the “Company” mean OraSure and its consolidated subsidiary, unless otherwise indicated.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions about future events. These estimates and underlying assumptions affect the amounts of assets and liabilities reported, disclosures about contingent assets and liabilities, and reported amounts of revenues and expenses. Such estimates include the valuation of accounts receivable and inventories and assumptions utilized in impairment testing for intangible assets and goodwill, as well as calculations related to accruals, taxes, and performance-based compensation expense, among others. These estimates and assumptions are based on management’s best estimates and judgment. Management evaluates its estimates and assumptions on an ongoing basis, using historical experience and other factors, which management believes to be reasonable under the circumstances, including the current economic environment. As future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Changes in those estimates resulting from continuing changes in the economic environment and other factors will be reflected in the financial statements in those future periods.
F-8
Table of Contents
Supplemental Cash Flow Information
In 2017, 2016 and 2015, we paid income taxes of $4,309, $1,123 and $719, respectively.
In 2017, 2016 and 2015, we recorded through the consolidated statements of income an increase in our allowance for doubtful accounts of $172, $40 and $341, respectively. We had $200 and $369 in write-offs against the allowance for doubtful accounts in 2017 and 2016, respectively. We had no material write-offs against the allowance for doubtful accounts in 2015.
As of December 31, 2017, 2016 and 2015, we had accruals for purchases of property and equipment of $449, $302 and $1,256, respectively.
Investments
We consider all investments to be available-for-sale securities. These securities are comprised of guaranteed investment certificates and corporate bonds with purchased maturities greater than ninety days. Available-for-sale securities are carried at fair value, based upon quoted market prices, with unrealized gains and losses, if any, reported in stockholders’ equity as a component of accumulated other comprehensive loss.
The following is a summary of our available-for-sale securities as of December 31, 2017 and 2016:
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value | |||||||||||||
| December 31, 2017 |
||||||||||||||||
| Guaranteed investment certificates |
$ | 22,261 | $ | — | $ | — | $ | 22,261 | ||||||||
| Corporate bonds |
82,010 | — | (553 | ) | 81,457 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total available-for-sale securities |
$ | 104,271 | $ | — | $ | (553 | ) | $ | 103,718 | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
| December 31, 2016 |
||||||||||||||||
| Guaranteed investment certificates |
$ | 11,160 | $ | — | $ | — | $ | 11,160 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total available-for-sale securities |
$ | 11,160 | $ | — | $ | — | $ | 11,160 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| At December 31, 2017 maturities of our available-for-sale securities were as follows: |
||||||||||||||||
| Less than one year |
$ | 83,403 | $ | — | $ | (375 | ) | $ | 83,028 | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Greater than one year |
$ | 20,868 | $ | — | $ | (178 | ) | $ | 20,690 | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
Accounts Receivable
Accounts receivable have been reduced by an estimated allowance for amounts that may become uncollectible in the future. This estimated allowance is based primarily on management’s evaluation of specific balances as they become past due, the financial condition of our customers and our historical experience related to write-offs.
Inventories
Inventories are stated at the lower of cost or net realizable value, with cost determined on a first-in, first-out basis, and include the cost of raw materials, labor and overhead. The majority of our inventories are subject to expiration dating. We continually evaluate quantities on hand and the carrying value of our inventories to determine the need for reserves for excess and obsolete inventories, based primarily on the estimated forecast of product sales. When factors indicate that impairment has occurred, either a reserve is established against the inventories’ carrying value or the inventories are completely written off, as in the case of lapsing expiration dates. In addition to reserving for these items identified through specific identification procedures, we also reserve for unidentified scrap or spoilage under a fixed-formula methodology.
F-9
Table of Contents
Property and Equipment
Property and equipment are stated at cost. Additions or improvements are capitalized, while repairs and maintenance are charged to expense. Depreciation and amortization are provided using the straight-line method over the estimated useful lives of the related assets. Buildings are depreciated over twenty to forty years, while computer equipment, machinery and equipment, and furniture and fixtures are depreciated over two to ten years. Building improvements are amortized over their estimated useful lives. When assets are sold, retired, or discarded, the related property amounts are relieved from the accounts, and any gain or loss is recorded in the consolidated statements of income.
Intangible Assets
Intangible assets consist of a customer list, patents and product rights, acquired technology and tradenames. Patents and product rights consist of costs associated with the acquisition of patents, licenses and product distribution rights. Intangible assets are amortized using the straight-line method over their estimated useful lives of seven to fifteen years.
Impairment of Long-Lived Assets
We assess the recoverability of our long-lived assets, which include property and equipment and intangible assets, by determining whether the carrying value of such assets can be recovered through the sum of the undiscounted future cash flows generated from the use and eventual disposition of the asset. If indicators of impairment exist, we measure the amount of such impairment by comparing the carrying value of the assets to the fair value of these assets, which is generally determined based on the present value of the expected future cash flows associated with the use of the assets.
Goodwill
Goodwill represents the excess of the purchase price we paid over the fair value of the net tangible and identifiable intangible assets acquired and liabilities assumed in our acquisition of DNAG in August 2011. Goodwill is not amortized but rather is tested annually for impairment or more frequently if we believe that indicators of impairment exist. Current U.S. generally accepted accounting principles permit us to make a qualitative evaluation about the likelihood of goodwill impairment. If we conclude that it is more likely than not that the carrying value of a reporting unit is greater than its fair value, then we would be required to recognize an impairment charge for the amount by which the carrying amount exceeds the reporting unit’s fair value, provided the impairment charge does not exceed the total amount of goodwill allocated to the reporting unit.
We performed our annual impairment assessment as of July 31, 2017 utilizing a qualitative evaluation and concluded that it was more likely than not that the fair value of our DNAG reporting unit is greater than its carrying value. We believe we have made reasonable estimates and assumptions to calculate the fair value of our reporting unit. If actual future results are not consistent with management’s estimates and assumptions, we may have to take an impairment charge in the future related to our goodwill. Future impairment tests will continue to be performed annually in the fiscal third quarter, or sooner if a triggering event occurs. As of December 31, 2017, we believe no indicators of impairment exist.
Revenue Recognition
We recognize product revenues when there is persuasive evidence that an arrangement exists, the price is fixed or determinable, title has passed and collection is reasonably assured. Product revenues are recorded net of allowances for any discounts or rebates. Other than for sales of our OraQuick® In-Home HIV test to the retail trade, we do not grant price protection or product return rights to our customers except for warranty returns. Historically, returns arising from warranty issues have been infrequent and immaterial. Accordingly, we expense warranty returns as incurred.
F-10
Table of Contents
Our net revenues recorded on sales of the OraQuick® In-Home HIV test represent total gross revenues, less an allowance for expected returns, and customer allowances for cooperative advertising, discounts, rebates, and chargebacks. The allowance for expected returns is an estimate established by management, based upon currently available information, and is adjusted to reflect known changes in the factors that impact this estimate. Other customer allowances are at contractual rates and are recorded as a reduction of gross revenue when recognized in our consolidated statements of income.
We record shipping and handling charges billed to our customers as product revenue and the related expense as cost of products sold. Taxes assessed by governmental authorities, such as sales or value-added taxes, are excluded from product revenues.
On June 10, 2014, we entered into a Master Program Services and Co-Promotion Agreement with AbbVie to co-promote our OraQuick® HCV test in the United States. On June 30, 2016, we mutually agreed to terminate our agreement effective December 31, 2016. Accordingly, we did not record any revenue from this co-promotion agreement during 2017. During 2016 and 2015, $18,951 and $13,479, respectively, in exclusivity revenues were recognized and recorded as other revenue in our consolidated statements of income.
On June 12, 2015, we were awarded a grant for up to $10,400 in total funding from the U.S. Department of Health and Human Services (“HHS”) Office of the Assistant Secretary for Preparedness and Response’s Biomedical Advanced Research and Development Authority (“BARDA”) related to our OraQuick® Ebola rapid antigen test. The three-year, multi-phased grant included an initial commitment of $1,800 and options for up to an additional $8,600 to fund certain clinical and regulatory activities. In September 2015 and July 2017, BARDA exercised options to provide $7,200 and $1,330, respectively, in additional funding for our OraQuick® Ebola test. Amounts related to this grant are recorded as other revenue in our consolidated statements of income as the activities are being performed and the related costs are incurred. During 2017, 2016 and 2015, $1,999, $1,707 and $1,783, respectively, were recognized in connection with this grant.
In August 2016, we were awarded a contract for up to $16,600 in total funding from BARDA related to our rapid Zika test. The six-year, multi-phased contract includes an initial commitment of $7,000 and options for up to an additional $9,600 to fund the evaluation of additional product enhancements, and clinical and regulatory activities. In May 2017, BARDA exercised an option to provide $2,600 in additional funding for our rapid Zika test. Funding received under this contract is recorded as other revenue in our consolidated statements of income as the activities are being performed and the related costs are incurred. During 2017 and 2016, $2,388 and $616, respectively, were recognized in connection with this grant.
In June 2017, we entered into a four-year Charitable Support Agreement with the Bill & Melinda Gates Foundation (“Gates Foundation”) that will enable us to offer our OraQuick® HIV self-test at an affordable price in 50 developing countries with funding from the Gates Foundation. The funding will consist of support payments tied to volume of product sold by us and reimbursement of certain related costs. The funding from the Gates Foundation will be in an aggregate amount not to exceed $20,000 over the four-year term or $6,000 each year of the agreement. Funding received under this agreement in the form of support payments for product purchases is recorded as a component of product revenue. During 2017, $1,047 of support payments were recognized in product revenue in connection with this agreement. Funding received in the form of reimbursement of certain related costs is recorded as other revenue in our consolidated statements of income. During 2017, $689 was recognized in other revenue in connection with this agreement.
Customer Sales Returns and Allowances
We do not grant return rights to our customers for any product, except for our OraQuick® In-Home HIV test. Accordingly, we have recorded an estimate of expected returns as a reduction of gross OraQuick® In-Home HIV product revenues in our consolidated statements of income. This estimate reflects our historical sales experience to retailers and consumers, as well as other retail factors, and is reviewed regularly to ensure that it reflects
F-11
Table of Contents
potential product returns. As of December 31, 2017 and 2016, the reserve for sales returns and allowances was $217. If actual product returns differ materially from our reserve amount, or if a determination is made that this product’s distribution would be discontinued in whole or in part by certain retailers, then we would need to adjust our reserve. Should the actual level of product returns vary significantly from our estimates, our operating and financial results could be materially affected.
Deferred Revenue
We record deferred revenue when funds are received prior to the recognition of the associated revenue. Deferred revenue as of December 31, 2017 and 2016 includes customer prepayments of $1,314 and $1,388, respectively.
Customer and Vendor Concentrations
One of our customers accounted for 37% and 15% of our accounts receivable as of December 31, 2017 and 2016, respectively. The same customer accounted for approximately 25% of our net consolidated revenues for the year ended December 31, 2017. Another customer accounted for approximately 15% and 12% of our net consolidated revenues for the years ended December 31, 2016 and 2015, respectively.
We currently purchase certain products and critical components of our products from sole-supply vendors. If these vendors are unable or unwilling to supply the required components and products, we could be subject to increased costs and substantial delays in the delivery of our products to our customers. Also, our subsidiary, DNAG, uses two third-party suppliers to manufacture its products. Our inability to have a timely supply of any of these components and products could have a material adverse effect on our business, as well as our financial condition and results of operations.
Research and Development
Research and development expenses consist of costs incurred in performing research and development activities, including salaries and benefits, facilities expenses, overhead expenses, clinical trial and related clinical manufacturing expenses, contract services and other outside expenses. Research and development costs are charged to expense as incurred.
Advertising Expenses
Advertising costs are charged to expense as incurred. During 2017, 2016, and 2015, we incurred $717, $626, and $623, respectively, in advertising expenses.
Stock-Based Compensation
We account for stock-based compensation to employees and directors using the fair value method. We recognize compensation expense for stock option and restricted stock awards issued to employees and directors on a straight-line basis over the requisite service period of the award. We recognize compensation expense related to performance-based restricted stock units based on assumptions as to what percentage of each performance target will be achieved. We evaluate these target assumptions on a quarterly basis and adjust compensation expense related to these awards, as appropriate. To satisfy the exercise of options, issuance of restricted stock, or redemption of performance-based restricted stock units, we issue new shares rather than purchase shares in the open market.
Income Taxes
We follow the asset and liability method for accounting for income taxes. Under this method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial
F-12
Table of Contents
statement carrying amounts of existing assets and liabilities and the respective tax basis of assets and liabilities, as well as operating loss and credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates for the respective taxing jurisdiction that are expected to apply to taxable income in the years in which those temporary differences and operating loss and credit carryforwards are expected to be recovered, settled or utilized. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
We assess the realizability of our net deferred tax assets on a quarterly basis. If, after considering all relevant positive and negative evidence, it is more likely than not that some portion or all of the net deferred tax assets will not be realized, we reduce our net deferred tax assets by a valuation allowance. The realization of the net deferred tax assets is dependent on several factors, including the generation of sufficient taxable income prior to the expiration of our net operating loss carryforwards.
Foreign Currency Translation
The assets and liabilities of our foreign operations are translated into U.S. dollars at current exchange rates as of the balance sheet date, and revenues and expenses are translated at average exchange rates for the period. Resulting translation adjustments are reflected in accumulated other comprehensive loss, which is a separate component of stockholders’ equity.
Transaction gains and losses resulting from exchange rate changes on transactions denominated in currencies other than functional currency are included in our consolidated statements of income in the period in which the change occurs. Net foreign exchange (losses) gains resulting from foreign currency transactions that are included in other income (expense) in our consolidated statements of income were $(1,442), $(607), and $1,005 for the years ended December 31, 2017, 2016, and 2015, respectively.
Earnings Per Share
Basic earnings per share is computed by dividing net income by the weighted-average number of shares of common stock outstanding during the period. Diluted earnings per share is computed in a manner similar to basic earnings per share except that the weighted-average number of shares outstanding is increased to include incremental shares from the assumed vesting or exercise of dilutive securities, such as common stock options, unvested restricted stock or performance stock units, unless the impact is antidilutive. The number of incremental shares is calculated by assuming that outstanding stock options were exercised and unvested restricted shares and performance stock units were vested, and the proceeds from such exercises or vesting were used to acquire shares of common stock at the average market price during the reporting period.
The computations of basic and diluted earnings per share are as follows:
| Year ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Net income |
$ | 30,948 | $ | 19,720 | $ | 8,167 | ||||||
|
|
|
|
|
|
|
|||||||
| Weighted average shares of common stock outstanding: |
||||||||||||
| Basic |
59,050 | 55,615 | 56,397 | |||||||||
| Dilutive effect of stock options, restricted stock, and performance stock units |
1,974 | 898 | 449 | |||||||||
|
|
|
|
|
|
|
|||||||
| Diluted |
61,024 | 56,513 | 56,846 | |||||||||
|
|
|
|
|
|
|
|||||||
| Earnings per share: |
||||||||||||
| Basic |
$ | 0.52 | $ | 0.35 | $ | 0.14 | ||||||
|
|
|
|
|
|
|
|||||||
| Diluted |
$ | 0.51 | $ | 0.35 | $ | 0.14 | ||||||
|
|
|
|
|
|
|
|||||||
F-13
Table of Contents
For the years ended December 31, 2017, 2016, and 2015, outstanding common stock options, unvested restricted stock, and unvested performance stock units representing 180, 2,546, and 4,314 shares, respectively, were excluded from the computation of diluted earnings per share as their inclusion would have been anti-dilutive.
Accumulated Other Comprehensive Loss
We classify items of other comprehensive income (loss) by their nature and disclose the accumulated balance of other comprehensive loss separately from accumulated deficit and additional paid-in capital in the stockholders’ equity section of our consolidated balance sheets.
We have defined the Canadian dollar as the functional currency of our Canadian subsidiary, DNAG, and as such, the results of its operations are translated into U.S. dollars, which is the reporting currency of the Company. Accumulated other comprehensive loss at December 31, 2017 consists of $9,787 of currency translation adjustments and $553 of net unrealized losses on marketable securities, which represents the fair market value adjustment for our investments portfolio. Accumulated other comprehensive loss at December 31, 2016 consists of $14,220 of currency translation adjustments.
Fair Value of Financial Instruments
As of December 31, 2017 and 2016, the carrying values of cash and cash equivalents, restricted cash, accounts receivable, and accounts payable approximate their respective fair values based on their short-term nature.
Fair value measurements of all financial assets and liabilities that are being measured and reported on a fair value basis are required to be classified and disclosed in one of the following three categories:
| Level 1: | Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities; | |
| Level 2: | Quoted prices in markets that are not active, or inputs which are observable, either directly or indirectly, for substantially the full term of the asset or liability; and | |
| Level 3: | Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable (i.e., supported by little or no market activity). | |
All of our available-for-sale securities are measured as Level 1 instruments as of December 31, 2017 and 2016.
Included in cash and cash equivalents at December 31, 2017 and 2016, was $40,760 and $83,704 invested in government money market funds. These funds have investments in government securities and are measured as Level 1 instruments.
We offer a nonqualified deferred compensation plan for certain eligible employees and members of our Board of Directors. The assets of the plan are held in the name of the Company at a third-party financial institution. Separate accounts are maintained for each participant to reflect the amounts deferred by the participant and all earnings and losses on those deferred amounts. The assets of the plan are held in mutual funds and Company stock. The fair value of the plan assets as of December 31, 2017 and 2016 was $3,514 and $1,980, respectively, and was calculated using the quoted market prices of the assets as of those dates. All investments in the plan are classified as trading securities and measured as Level 1 instruments. The fair value of plan assets is included in other assets with the same amount included in other liabilities in the accompanying consolidated balance sheets.
In 2017, we purchased certificates of deposit (“CDs”) from a commercial bank. The CDs bear interest at rates ranging from 0.89% to 1.03% and mature periodically through January 22, 2018. The carrying values of the CDs approximate their fair value. These CDs serve as collateral for certain standby letters of credit and are reported as restricted cash on the accompanying consolidated balance sheets. Also see Note 11 – Commitments and Contingencies.
F-14
Table of Contents
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued converged guidance on recognizing revenue in contracts with customers, ASU 2014-09, Revenue from Contracts with Customers. The intent of the new standard is to improve financial reporting and comparability of revenue globally. The core principle of the standard is for a company to recognize revenue in a manner that depicts the transfer of goods or services to customers in an amount that reflects the consideration which the company expects to receive in exchange for those goods or services. The guidance provides a five-step analysis of transactions to determine when and how revenue is recognized. Other major provisions include capitalization of certain contract costs, consideration of the time value of money in the transaction price, and in certain circumstances, allowing estimates of variable consideration to be recognized before contingencies are resolved. The guidance also requires enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue and cash flows arising from an entity’s contracts with customers. The standard will be effective for the first interim period within annual reporting periods beginning after December 15, 2017.
We have completed our evaluation of the new standard and have assessed the impacts of adoption on our consolidated financial statements and disclosures. Based on our evaluation of our current contracts and revenue streams, revenue recognition is mostly consistent under both the previous and new standard, with the exception of one revenue stream within our drug testing kits segment, which is described below.
We believe the key changes in the standard that impact our revenue recognition relate to the allocation of the transaction price to performance obligations and the estimate of unexercised rights (“breakage”) associated with the contracts. Prior to the adoption of ASU 2014-09, we used the residual value method to allocate the transaction prices related to our drug testing kits. With the issuance of ASU 2014-09, we have determined the more appropriate method to be based on the stand-alone selling price. This change in methodology also impacts our estimated breakage amount.
The FASB allows two adoption methods under ASU 2014-09. The guidance may be adopted through either retrospective application to all periods presented in the consolidated financial statements (full retrospective) or through a cumulative effect adjustment to retained earnings at the effective date (modified retrospective). We adopted the new standard effective January 1, 2018 using the modified retrospective transition method. Under that method, we applied the rules to all contracts existing as of January 1, 2018. We estimate the cumulative effect recorded to the opening balance of retained earnings of the above impacted revenue stream is approximately $163.
The disclosures in our notes to the consolidated financial statements related to revenue recognition will be significantly expanded under the new standard, specifically around the quantitative and qualitative information about performance obligations, changes in contract assets and liabilities, and disaggregation of revenue.
In July 2015, the FASB issued ASU 2015-11, Simplifying the Measurement of Inventory, which requires an entity that uses the first-in, first-out method for inventory measurement to report inventory cost at the lower of cost and net realizable value versus the current measurement principle of lower of cost or market. The ASU requires prospective adoption for inventory measurements for fiscal years beginning after December 15, 2016. We adopted ASU 2015-11 on January 1, 2017. The adoption of this standard did not have a material impact on our consolidated financial statements and related disclosures.
In February 2016, the FASB issued ASU 2016-02, Leases, which requires entities to begin recording assets and liabilities from leases on the balance sheet. The new guidance will also require significant additional disclosures about the amount, timing and uncertainty of cash flows from leases. The standard will be effective for the first interim period within annual reporting periods beginning after December 15, 2018, using a modified retrospective approach. Early adoption is permitted. We are evaluating the effect that ASU 2016-02 may have on our consolidated financial statements and related disclosures.
F-15
Table of Contents
In March 2016, the FASB issued authoritative guidance under ASU 2016-09, Compensation-Stock Compensation (Topic 718) Improvements to Employee Share-Based Payment Accounting. ASU 2016-09 provides for simplification of several aspects of the accounting for share-based payment transactions, including income tax consequences, classification of awards as either equity or liabilities and classification on the statement of cash flows. We adopted ASU 2016-09 on January 1, 2017. Since we have a full valuation allowance against our U.S. net deferred tax assets, the adoption of this standard for recognition of the tax effect of deductions for employee share awards in excess of compensation costs (“windfall”) did not have a material impact on our consolidated financial statements and related disclosures. See Note 8 – Income Taxes, for additional information. Should our full valuation allowance be reversed in future periods, the adoption of this new guidance will introduce more volatility in the calculation of our effective tax rate, depending on the Company’s share price at exercise or vesting of share-based awards as compared to grant date. The other provisions of ASU 2016-09 did not have a material impact on our consolidated financial statements and related disclosures.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments, which provides guidance related to cash flows presentation and is effective for annual reporting periods beginning after December 15, 2017, subject to early adoption. The majority of the guidance in ASU 2016-15 is consistent with our current cash flow classifications and we do not expect the adoption of this standard will have a material impact on our consolidated financial statements.
In January 2017, the FASB issued ASU 2017-04, Intangibles-Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment, which requires an entity to no longer perform a hypothetical purchase price allocation to measure goodwill impairment. Instead, impairment will be measured using the difference between the carrying amount and the fair value of the reporting unit. This update will be effective for annual and interim periods in fiscal years beginning after December 15, 2019. Early adoption is permitted. We adopted ASU 2017-04 in the second quarter of 2017. The adoption of this standard did not have a material impact on our consolidated financial statements and related disclosures.
In March 2017, the FASB issued ASU 2017-08, Receivables-Nonrefundable Fees and Other Costs (Subtopic 310-20): Premium Amortization on Purchased Callable Debt Securities, which shortens the premium amortization period for purchased non-contingently callable debt securities. Shortening the amortization period is generally expected to more closely align the interest income recognition with the expectations incorporated in the market pricing on the underlying securities. This ASU is effective for annual and interim periods in fiscal years beginning after December 15, 2018. Early adoption is permitted. We are evaluating the effect that ASU 2017-08 may have on our consolidated financial statements and related disclosures, but do not anticipate the adoption will have a material impact on our financial results.
In May 2017, the FASB issued ASU No. 2017-09, Compensation-Stock Compensation (Topic 718): Scope of Modification Accounting, to provide clarity to which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. This update will be effective for annual periods and interim periods in fiscal years beginning after December 15, 2017 with early adoption permitted. We do not expect the adoption will have a material effect on our consolidated financial statements.
| 3. | INVENTORIES: |
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Raw materials |
$ | 10,299 | $ | 5,399 | ||||
| Work in process |
199 | 1,034 | ||||||
| Finished goods |
8,845 | 5,366 | ||||||
|
|
|
|
|
|||||
| $ | 19,343 | $ | 11,799 | |||||
|
|
|
|
|
|||||
F-16
Table of Contents
| 4. | PROPERTY AND EQUIPMENT: |
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Land |
$ | 1,118 | $ | 1,118 | ||||
| Buildings and improvements |
20,726 | 20,179 | ||||||
| Machinery and equipment |
26,685 | 22,764 | ||||||
| Computer equipment and software |
9,131 | 8,357 | ||||||
| Furniture and fixtures |
2,194 | 2,113 | ||||||
| Construction in progress |
897 | 1,569 | ||||||
|
|
|
|
|
|||||
| 60,751 | 56,100 | |||||||
| Less accumulated depreciation |
(39,379 | ) | (36,067 | ) | ||||
|
|
|
|
|
|||||
| $ | 21,372 | $ | 20,033 | |||||
|
|
|
|
|
|||||
Depreciation expense was $3,434, $3,149, and $2,992 for 2017, 2016, and 2015, respectively.
| 5. | GOODWILL AND OTHER INTANGIBLE ASSETS: |
The changes in goodwill are as follows:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Balance as of January 1 |
$ | 18,793 | $ | 18,250 | ||||
| Change related to foreign currency translation |
1,290 | 543 | ||||||
|
|
|
|
|
|||||
| Balance as of December 31 |
$ | 20,083 | $ | 18,793 | ||||
|
|
|
|
|
|||||
Intangible assets consist of the following:
| December 31, 2017 | ||||||||||||||||
| Amortization Period (Years) |
Gross | Accumulated Amortization |
Net | |||||||||||||
| Customer list |
10 | $ | 9,960 | $ | (6,122 | ) | $ | 3,838 | ||||||||
| Patents and product rights |
10 | 5,400 | (4,258 | ) | 1,142 | |||||||||||
| Acquired technology |
7 | 7,737 | (6,690 | ) | 1,047 | |||||||||||
| Tradename |
15 | 3,818 | (1,622 | ) | 2,196 | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 26,915 | $ | (18,692 | ) | $ | 8,223 | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| December 31, 2016 | ||||||||||||||||
| Amortization Period (Years) |
Gross | Accumulated Amortization |
Net | |||||||||||||
| Customer list |
10 | $ | 9,321 | $ | (4,830 | ) | $ | 4,491 | ||||||||
| Patents and product rights |
10 | 5,400 | (3,808 | ) | 1,592 | |||||||||||
| Acquired technology |
7 | 7,240 | (5,279 | ) | 1,961 | |||||||||||
| Tradename |
15 | 3,573 | (1,280 | ) | 2,293 | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 25,534 | $ | (15,197 | ) | $ | 10,337 | ||||||||||
|
|
|
|
|
|
|
|||||||||||
F-17
Table of Contents
Patents and products rights are made up of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| HIV-related |
$ | 900 | $ | 900 | ||||
| HCV-related |
4,500 | 4,500 | ||||||
|
|
|
|
|
|||||
| 5,400 | 5,400 | |||||||
| Less accumulated amortization |
(4,258 | ) | (3,808 | ) | ||||
|
|
|
|
|
|||||
| $ | 1,142 | $ | 1,592 | |||||
|
|
|
|
|
|||||
Amortization expense for 2017, 2016, and 2015 was $2,641, $2,538, and $2,704, respectively.
Amortization expense for each of the five succeeding fiscal years and beyond is estimated as follows:
| 2018 |
$ | 2,639 | ||
| 2019 |
1,626 | |||
| 2020 |
1,417 | |||
| 2021 |
1,173 | |||
| 2022 |
246 | |||
| Beyond |
1,122 | |||
|
|
|
|||
| $ | 8,223 | |||
|
|
|
| 6. | ACCRUED EXPENSES: |
| December 31, 2017 | December 31, 2016 | |||||||
| Payroll and related benefits |
$ | 9,265 | $ | 7,685 | ||||
| Income taxes payable (receivable) |
6,469 | (39 | ) | |||||
| Professional fees |
1,064 | 982 | ||||||
| Royalties |
845 | 715 | ||||||
| Other |
3,052 | 1,971 | ||||||
|
|
|
|
|
|||||
| $ | 20,695 | $ | 11,314 | |||||
|
|
|
|
|
|||||
| 7. | CREDIT FACILITY: |
On September 30, 2016, we entered into a credit agreement (the “Credit Agreement”) with a commercial bank. The Credit Agreement, as amended on December 20, 2017, provides for revolving extensions of credit in an initial aggregate amount of up to $10,000 (inclusive of a letter of credit sub-facility of $2,500), with an option to request, prior to the second anniversary of the closing date, that the lender, at its election, provide up to $5,000 of additional revolving commitments. Obligations under the Credit Agreement are secured by a first priority security interest in certain eligible accounts receivable, 65% of the equity of our subsidiary, DNAG, and certain related assets. There were no borrowings under the facility during 2017 and 2016.
Borrowings under the Credit Agreement are subject to compliance with borrowing base limitations tied to eligibility of accounts receivable. Interest under the Credit Agreement is payable at the London Interbank Offered Rate for one, two, three or six-month loans, as selected by the Company, plus 2.50% per year. The Credit Agreement is subject to an unused line fee of 0.375% per year on the unused portion of the commitment under the Credit Agreement during the revolving period. The maturity date of the Credit Agreement is September 30, 2019.
F-18
Table of Contents
In connection with the Credit Agreement, under certain circumstances, we must comply with a minimum fixed charge coverage ratio of 1.10 to 1.00, measured as of the last day of each fiscal month and for the twelve-fiscal month period ending on such date. As of December 31, 2017 and 2016, we were in compliance with all applicable covenants in the Credit Agreement.
| 8. | INCOME TAXES: |
Income before income tax expense consists of the following:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| United States |
$ | 2,270 | $ | 18,492 | $ | 5,302 | ||||||
| Canada |
38,762 | 1,831 | 3,530 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 41,032 | $ | 20,323 | $ | 8,832 | |||||||
|
|
|
|
|
|
|
|||||||
The components of income tax expense are as follows:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Current |
||||||||||||
| Federal |
$ | — | $ | — | $ | — | ||||||
| State |
— | 250 | — | |||||||||
| Canada |
10,733 | 882 | 617 | |||||||||
|
|
|
|
|
|
|
|||||||
| 10,733 | 1,132 | 617 | ||||||||||
|
|
|
|
|
|
|
|||||||
| Deferred |
||||||||||||
| Federal |
10,297 | 6,508 | 1,511 | |||||||||
| State |
(827 | ) | 569 | 311 | ||||||||
| Canada |
(649 | ) | (529 | ) | 48 | |||||||
|
|
|
|
|
|
|
|||||||
| 8,821 | 6,548 | 1,870 | ||||||||||
| Decrease in valuation allowance |
(9,470 | ) | (7,077 | ) | (1,822 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| (649 | ) | (529 | ) | 48 | ||||||||
|
|
|
|
|
|
|
|||||||
| Total income tax expense |
$ | 10,084 | $ | 603 | $ | 665 | ||||||
|
|
|
|
|
|
|
|||||||
For the years ended December 31, 2017, 2016, and 2015 we recorded foreign income tax expense of $10,084, $353, and $665, respectively.
The Tax Cuts and Jobs Act
On December 22, 2017, the U.S. enacted the Tax Cuts and Jobs Act (“Tax Act”) that instituted fundamental changes to the taxation on multinational corporations. Although the Tax Act is generally effective January 1, 2018, U.S. generally accepted accounting principles requires recognition of the tax effects of new legislation during the reporting period that includes the enactment date, which was December 22, 2017. Among the numerous provisions, the Tax Act includes two provisions in particular which require consideration for 2017 financial reporting: a permanent reduction in the corporate tax rate to 21%; and a one-time transition tax imposed on a U.S. shareholder’s historical undistributed earnings of foreign affiliates.
The reduction in the U.S. corporate tax rate to 21% is effective in 2018. However, this change results in a re-measurement of our net deferred tax balance at the end of 2017 with no net financial statement impact due to a comparable decrease in our deferred tax valuation allowance.
F-19
Table of Contents
The one-time tax on undistributed and previously untaxed post-1986 foreign earnings and profits (E&P) of foreign affiliates owned by U.S. shareholders as of December 31, 2017 was analyzed for our Canadian subsidiary. As of December 31, 2017, our U.S. federal taxable loss exceeded and offset the provisional mandatory repatriation net inclusion and as such, no additional taxes will be due as a result of the deemed repatriation of these foreign earnings.
In addition, the Tax Act also includes changes to the taxation of foreign earnings by implementing a dividend exemption system, expansion of the current anti-deferral rules, a minimum tax on low-taxed foreign earnings and new measures to deter base erosion and promote U.S. production. The Tax Act also includes repeal of the corporate alternative minimum tax, expensing of capital investment, changes to the Federal net operating loss (“NOL”) utilization and carryforward rules, and limitations of the deduction for interest expense and certain employee compensation.
As a result of the complex impact of the Tax Act, the SEC provided guidance under Staff Accounting Bulletin No. 118 (“SAB 118”) that allows the Company to record provisional amounts as of December 31, 2017 for the impact of the Tax Act, provided that the provisional amounts can be reasonably determined and with the requirement that the final accounting be completed in a period not to exceed one year from the date of enactment. As of December 31, 2017, the Company has not completed the accounting for the tax effects of the Tax Act, and therefore, has recorded provisional amounts which include:
| (a) | An estimate of the impact of the one-time transition tax on existing net operating loss and foreign tax credit carryforwards generated, as well as the related offsetting valuation allowances. These results are subject to change as there is limited information for federal and state taxing authorities regarding the application and interpretation of the recently enacted legislation. In addition, the results are based on estimates and are affected by the interaction of various provision of the Tax Act on future taxable income. The Company will disclose the impact to the provisional amount in the reporting period in which the accounting is completed, which will not exceed one year from the date of the enactment. |
| (b) | The valuation allowance for the Company’s deferred tax assets as of December 31, 2017, is primarily dependent on forecasted future taxable income in the U.S. and Canada and may be impacted by the provisions of the Tax Act. Any increase or reduction in estimated forecasted future taxable income may require the Company to adjust the valuation allowance against the remaining deferred tax asset. The Company will continue to assess the impact of the Tax Act on the valuation allowance during the provisional period. |
The Tax Act imposes a U.S. tax on global intangible low taxed income (“GILTI”) that is earned by certain foreign affiliates owned by a U.S. shareholder effective in 2018. GILTI is generally intended to impose tax on the earnings of a foreign corporation that are deemed to exceed a certain threshold return relative to the underlying tangible property. The computation of GILTI is still subject to interpretation and additional clarifying guidance is expected. The Company has not yet made a policy election related to its treatment of GILTI as either a current period expense in the reporting period in which the tax is incurred or recognizing deferred taxes when a basis differences exists that is expected to affect the amount of the GILTI inclusion upon reversal. The Company is still in the process of analyzing the expected impact of GILTI in future periods.
F-20
Table of Contents
A reconciliation of the statutory United States federal income tax rate to our effective tax rate for each of the years ended December 31, 2017, 2016, and 2015 is as follows:
| 2017 | 2016 | 2015 | ||||||||||
| Statutory U.S. federal income tax rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| Deemed repatriation tax |
7.8 | — | — | |||||||||
| Statutory rate change, deferred tax impact |
29.4 | — | — | |||||||||
| Excess tax benefits for share-based payment awards |
(16.0 | ) | — | — | ||||||||
| Tax effect of Canadian items |
(9.8 | ) | (4.9 | ) | (13.1 | ) | ||||||
| State income taxes, net of federal benefit |
(1.1 | ) | 2.0 | 1.5 | ||||||||
| Nondeductible expenses and other |
3.4 | 6.7 | 5.7 | |||||||||
| Change in valuation allowance, federal and state |
(23.1 | ) | (34.8 | ) | (20.6 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Effective tax rate |
24.6 | % | 3.0 | % | 7.5 | % | ||||||
|
|
|
|
|
|
|
|||||||
Deferred income taxes reflect the tax effects of temporary differences between the basis of assets and liabilities recognized for financial reporting purposes and tax purposes, and net operating loss and tax credit carryforwards. Significant components of our deferred tax assets (liabilities) as of December 31, 2017 and 2016 are as follows:
| 2017 | 2016 | |||||||
| Deferred tax assets (liabilities): |
||||||||
| Net operating loss carryforwards |
$ | 15,338 | $ | 17,673 | ||||
| Inventories |
1,179 | 1,680 | ||||||
| Capitalized research and development costs |
2,402 | 4,711 | ||||||
| Accruals and reserves currently not deductible |
3,103 | 3,282 | ||||||
| Acquired intangible assets |
(1,825 | ) | (2,268 | ) | ||||
| Depreciation and amortization |
(621 | ) | (212 | ) | ||||
| Stock-based compensation |
976 | 6,110 | ||||||
| Tax credit carryforwards |
6,722 | 1,751 | ||||||
|
|
|
|
|
|||||
| Net deferred tax asset |
27,274 | 32,727 | ||||||
| Valuation allowance |
(29,225 | ) | (35,173 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax liability |
$ | (1,951 | ) | $ | (2,446 | ) | ||
|
|
|
|
|
|||||
In assessing the realizability of our deferred tax asset, we consider all relevant positive and negative evidence in determining whether it is more likely than not that some portion or all of the deferred income tax assets will not be realized. The realization of the gross deferred tax assets is dependent upon several factors, including the generation of sufficient taxable income prior to the expiration of the NOL carryforwards. In 2008, we established a full valuation allowance against our U.S. deferred tax asset, and management believes the full valuation allowance is still appropriate as of December 31, 2017 and 2016 since the facts and circumstances necessitating the allowance have not changed. As a result, no U.S. federal income tax benefit was recorded for the years ended December 31, 2017, 2016, or 2015. The valuation allowance primarily decreased by $9,470 in 2017 due to its re-measurement to the new corporate tax rate of 21% effective in 2018.
Our Federal NOL carryforwards expire as follows:
| Year of Expiration |
NOLs | |||
| 2020 - 2025 |
$ | 16,268 | ||
| 2026 - 2030 |
11,956 | |||
| 2031 - 2037 |
37,937 | |||
|
|
|
|||
| $ | 66,161 | |||
|
|
|
|||
F-21
Table of Contents
The new accounting guidance under ASU 2016-09 allows for the recognition of excess tax benefits associated with stock based compensation awards regardless of whether the deduction reduces taxes payable. On January 1, 2017, we recorded a cumulative adjustment to retained earnings of $3,391 to recognize the increase in our net operating loss carryforwards from the cumulative excess tax benefits not recognized in periods prior to January 1, 2017. A corresponding $3,391 increase to our valuation allowance associated with this tax benefit was also recorded to retained earnings thereby resulting in no impact to retained earnings.
The Tax Reform Act of 1986 contains provisions under Internal Revenue Code (“IRC”) Section 382 that limit the annual amount of federal and state NOL carryforwards that can be used in any given year in the event a significant change in ownership. The Company does not believe that there is a Section 382 limitation that will impair our future ability to utilize NOLs to offset our future taxable income. The Company continues to review ownership changes on an annual basis and we do not believe we have had a subsequent ownership change that would impact the NOLs.
Effective January 1, 2018, there is a transition to a participation exemption system whereby distributions from foreign subsidiaries to U.S. shareholders are generally exempt from taxation; therefore, the deferred tax liability on undistributed earnings is limited to any foreign withholding or other local taxes. The Company’s intention is to continue to permanently reinvest the historical undistributed earnings of our Canadian subsidiary to the extent that we will not incur any additional tax expense associated with foreign withholding or other local tax expense on the future cash transfers.
As of December 31, 2017, our gross unrecognized tax benefits totaled $1,663, and based upon the valuation allowance for our U.S. operations, the recognition of any tax benefit would not impact our effective tax rate. We record interest and penalties related to unrecognized tax benefits as a component of income tax expense. Interest and penalties were immaterial in 2017, 2016 and 2015. As a result of our net operating loss carryforward position, we are subject to audit by the Internal Revenue Service since our inception, as well as by several state jurisdictions for the years ended September 30, 1998 through December 31, 2017.
A reconciliation of our unrecognized tax benefits is as follows:
| 2017 | 2016 | 2015 | ||||||||||
| Balance as of January 1 |
$ | 2,084 | $ | 2,088 | $ | 2,073 | ||||||
| Additions for tax positions of prior periods |
— | — | 22 | |||||||||
| Reductions for tax positions of prior periods |
(421 | ) | (4 | ) | (7 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Balance as of December 31 |
$ | 1,663 | $ | 2,084 | $ | 2,088 | ||||||
|
|
|
|
|
|
|
|||||||
| 9. | STOCKHOLDERS’ EQUITY: |
Stock-Based Awards
We grant stock-based awards under the OraSure Technologies, Inc. Stock Award Plan, as amended (the “Stock Plan”). The Stock Plan permits stock-based awards to employees, outside directors and consultants or other third-party advisors. Awards which may be granted under the Stock Plan include qualified incentive stock options, nonqualified stock options, stock appreciation rights, restricted awards, performance awards and other stock-based awards.
As of December 31, 2017, 4,355 shares were available for future grants under the Stock Plan.
Under the terms of the Stock Plan, nonqualified stock options may be granted to eligible employees, including our officers at a price not less than 75 percent of the fair market value of a share of common stock on the date of grant. The option term and vesting schedule of such awards may be either unlimited or have a specified period in which to vest and be exercised. To date, options generally have been granted with ten-year exercise periods
F-22
Table of Contents
and an exercise price not less than the fair market value on the date of grant. Options generally vest over four years, with one quarter of the options vesting one year after grant and the remainder vesting on a monthly basis over the next three years.
The fair value of each stock option was estimated on the date of the grant using the Black-Scholes option-pricing model using the following weighted-average assumptions:
| Years Ended December 31, | ||||||||||||
| Black-Scholes Option Valuation Assumptions | 2017 | 2016 | 2015 | |||||||||
| Risk-free interest rate(1) |
2.22 | % | 1.76 | % | 1.49 | % | ||||||
| Expected dividend yield |
— | — | — | |||||||||
| Expected stock price volatility(2) |
43 | % | 46 | % | 51 | % | ||||||
| Expected life of stock options (in years)(2) |
7 | 7 | 7 | |||||||||
| (1) | Based on the constant maturity interest rate of U.S. Treasury securities whose term is consistent with the expected life of our stock options. |
| (2) | Based upon historical experience. |
The weighted-average grant date fair value of stock options granted during the years ended December 31, 2017, 2016 and 2015 was $4.71, $2.91 and $4.86, respectively.
Compensation expense recognized in the financial statements related to stock options was as follows:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Total compensation cost during the year |
$ | 2,045 | $ | 2,691 | $ | 3,403 | ||||||
| Amounts capitalized into inventory during the year |
(371 | ) | (270 | ) | (153 | ) | ||||||
| Amounts recognized in cost of products sold for amounts previously capitalized |
276 | 212 | 153 | |||||||||
|
|
|
|
|
|
|
|||||||
| Amounts charged against income |
$ | 1,950 | $ | 2,633 | $ | 3,403 | ||||||
|
|
|
|
|
|
|
|||||||
The aggregate intrinsic value of options exercised during the years ended December 31, 2017, 2016, and 2015 (the amount by which the market price of the stock on the date of exercise exceeded the exercise price) was $35,631, $763, and $329, respectively.
F-23
Table of Contents
The following table summarizes the stock option activity under the Stock Plan:
| Options | Weighted-Average Exercise Price Per Share |
Weighted-Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding on January 1, 2015 |
5,740 | $ | 7.22 | |||||||||||||
| Granted |
773 | 9.28 | ||||||||||||||
| Exercised |
(74 | ) | 5.46 | |||||||||||||
| Expired |
(163 | ) | 8.55 | |||||||||||||
| Forfeited |
(60 | ) | 8.07 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding on December 31, 2015 |
6,216 | 7.46 | ||||||||||||||
| Granted |
347 | 6.00 | ||||||||||||||
| Exercised |
(405 | ) | 6.57 | |||||||||||||
| Expired |
(479 | ) | 9.17 | |||||||||||||
| Forfeited |
(223 | ) | 7.31 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding on December 31, 2016 |
5,456 | 7.28 | ||||||||||||||
| Granted |
231 | 9.92 | ||||||||||||||
| Exercised |
(4,413 | ) | 7.20 | |||||||||||||
| Expired |
(27 | ) | 8.36 | |||||||||||||
| Forfeited |
(46 | ) | 7.79 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding on December 31, 2017 |
1,201 | $ | 8.04 | 6.5 | $ | 12,986 | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Vested or expected to vest as of December 31, 2017 |
1,201 | $ | 8.04 | 6.5 | $ | 12,986 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Exercisable on December 31, 2017 |
659 | $ | 7.81 | 5.2 | $ | 7,280 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
As of December 31, 2017, there was $1,950 of unrecognized compensation expense related to unvested option awards that is expected to be recognized over a weighted-average period of 1.9 years.
Net cash proceeds from the exercise of stock options were $31,675, $2,656 and $404 for the years ended December 31, 2017, 2016, and 2015, respectively. As a result of our net operating loss carryforward position, no actual income tax benefit was realized from stock option exercises for these periods.
F-24
Table of Contents
The following table summarizes information about stock options outstanding as of December 31, 2017:
| Options outstanding |
Options exercisable | |||||||||||||||||||
| Range of exercise prices |
Number Outstanding |
Weighted- Average Remaining Contractual Term (in years) |
Weighted- Average Exercise Price Per Share |
Number Exercisable |
Weighted- Average Exercise Price Per Share |
|||||||||||||||
| $2.79 -$5.19 |
19 | 0.3 | $ | 3.46 | 19 | $ | 3.46 | |||||||||||||
| $5.37 |
158 | 8.0 | 5.37 | 45 | 5.37 | |||||||||||||||
| $5.47 |
3 | 7.7 | 5.47 | 1 | 5.47 | |||||||||||||||
| $5.71 |
149 | 5.9 | 5.71 | 116 | 5.71 | |||||||||||||||
| $5.79 - $7.05 |
138 | 4.5 | 6.73 | 130 | 6.74 | |||||||||||||||
| $7.28 - $8.33 |
122 | 6.4 | 7.81 | 95 | 7.74 | |||||||||||||||
| $8.33 - $8.63 |
41 | 1.9 | 8.62 | 41 | 8.62 | |||||||||||||||
| $8.87 |
167 | 8.9 | 8.87 | — | 0.00 | |||||||||||||||
| $8.93 |
6 | 9.0 | 8.93 | — | 0.00 | |||||||||||||||
| $9.31 - $14.95 |
398 | 6.5 | 10.32 | 212 | 10.40 | |||||||||||||||
|
|
|
|
|
|||||||||||||||||
| 1,201 | 6.5 | $ | 8.04 | 659 | $ | 7.81 | ||||||||||||||
|
|
|
|
|
|||||||||||||||||
The Stock Plan also permits us to grant restricted shares of our common stock to eligible employees, including officers, and our outside directors. Generally, these shares are nontransferable until vested and are subject to vesting requirements and/or forfeiture, as determined by our Compensation Committee or Board of Directors. The market value of these shares at the date of grant is recognized on a straight-line basis over the period during which the restrictions lapse. Compensation cost of $2,705, $2,861 and $2,642 related to restricted shares was recognized during the years ended December 31, 2017, 2016, and 2015, respectively.
The following table summarizes restricted stock award activity under the Stock Plan:
| Units | Weighted-Average Grant Date Fair Value |
|||||||
| Issued and unvested, January 1, 2015 |
707 | $ | 6.50 | |||||
| Granted |
362 | 8.25 | ||||||
| Vested |
(354 | ) | 6.94 | |||||
| Forfeited |
(18 | ) | 8.01 | |||||
|
|
|
|||||||
| Issued and unvested, December 31, 2015 |
697 | 7.14 | ||||||
| Granted |
632 | 5.63 | ||||||
| Vested |
(446 | ) | 6.65 | |||||
| Forfeited |
(133 | ) | 6.43 | |||||
|
|
|
|||||||
| Issued and unvested, December 31, 2016 |
750 | 6.28 | ||||||
| Granted |
329 | 9.77 | ||||||
| Vested |
(378 | ) | 6.47 | |||||
| Forfeited |
(30 | ) | 7.57 | |||||
|
|
|
|||||||
| Issued and unvested, December 31, 2017 |
671 | $ | 7.83 | |||||
|
|
|
|
|
|||||
| Issued and expected to vest, December 31, 2017 |
671 | $ | 7.83 | |||||
|
|
|
|
|
|||||
As of December 31, 2017, there was $2,910 of unrecognized compensation expense related to unvested restricted stock awards that is expected to be recognized over a weighted average period of 1.7 years.
F-25
Table of Contents
In connection with the vesting of restricted shares and exercise of stock options during the years ended December 31, 2017, 2016 and 2015, we purchased and immediately retired 130, 132 and 133 shares with aggregate values of $1,240, $784 and $1,164, respectively, in satisfaction of minimum tax withholding and exercise obligations.
Commencing in 2016, we granted performance-based restricted stock units (“PSUs”) to certain executives. Vesting of these PSUs is dependent upon achievement of performance-based metrics during a one-year or three-year period, from the date of grant. Assuming achievement of each performance-based metric, the executive must also remain in our service for three years from the grant date. Performance during the one-year period will be based on a one-year earnings per share target. If the one-year target is achieved, the PSUs will then vest three years from grant date. Performance during the three-year period will be based on achievement of a three-year compound annual growth rate for consolidated product revenues. If the three-year target is achieved, the corresponding PSUs will then vest three years from grant date. PSUs are converted into shares of our common stock once vested. Upon grant of the PSUs, we recognize compensation expense related to these awards based on assumptions as to what percentage of each target will be achieved. The Company evaluates these target assumptions on a quarterly basis and adjusts compensation expense related to these awards, as appropriate.
Compensation cost of $2,223 and $511 related to the PSUs was recognized during the years ended December 31, 2017 and 2016, respectively.
The following table summarizes PSU activity under the Stock Plan:
| Units | Weighted- Average Grant Date Fair Value |
|||||||
| Issued and unvested, January 1, 2017 |
456 | 5.46 | ||||||
| Granted |
424 | 8.19 | ||||||
| Vested |
0 | 0.00 | ||||||
| Forfeited |
(28 | ) | 6.71 | |||||
|
|
|
|||||||
| Issued and unvested, December 31, 2017 |
852 | $ | 6.78 | |||||
|
|
|
|
|
|||||
| Issued and expected to vest, December 31, 2017 |
852 | $ | 6.78 | |||||
|
|
|
|
|
|||||
Share Repurchase Program
On August 5, 2008, our Board of Directors approved a share repurchase program pursuant to which we are permitted to acquire up to $25,000 of our outstanding common shares. No shares were purchased and retired in 2017. During the years ended December 31, 2016 and 2015, we purchased and retired 423 and 777 shares of common stock at an average price of $6.29 and $6.34 per share for a total cost of $2,660 and $4,926, respectively.
| 10. | BUSINESS SEGMENT INFORMATION: |
Our business is comprised of two segments: Our “OSUR” business consists of the development, manufacture, marketing and sale of oral fluid diagnostic products and specimen collection devices using our proprietary technologies, as well as other diagnostic products including immunoassays and other in vitro diagnostic tests that are used on other specimen types. Our molecular collections systems or “DNAG” business consists of the manufacture and sale of specimen collection kits that are used to collect, stabilize, transport and store samples of genetic material for molecular testing in the consumer genetic, clinical genetic, academic research, pharmacogenomics, personalized medicine, microbiome and animal genetics markets.
We organized our operating segments according to the nature of the products included in those segments. The accounting policies of the segments are the same as those described in the summary of significant accounting
F-26
Table of Contents
policies (see Note 2). We evaluate performance of our operating segments based on revenue and operating income. We do not allocate interest income, interest expense, other income, other expenses or income taxes to our operating segments. Reportable segments have no inter-segment revenues and inter-segment expenses have been eliminated.
The following table summarizes operating segment information for the years ended December 31, 2017, 2016, and 2015, and asset information as of December 31, 2017 and 2016:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Net revenues: |
||||||||||||
| OSUR |
$ | 91,965 | $ | 95,984 | $ | 89,795 | ||||||
| DNAG |
75,099 | 32,214 | 29,924 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 167,064 | $ | 128,198 | $ | 119,719 | ||||||
|
|
|
|
|
|
|
|||||||
| Operating income (loss): |
||||||||||||
| OSUR |
$ | (2,706 | ) | $ | 15,606 | $ | 3,558 | |||||
| DNAG |
42,944 | 4,659 | 4,500 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 40,238 | $ | 20,265 | $ | 8,058 | ||||||
|
|
|
|
|
|
|
|||||||
| Depreciation and amortization: |
|
|||||||||||
| OSUR |
$ | 3,170 | $ | 2,751 | $ | 3,015 | ||||||
| DNAG |
3,232 | 2,889 | 2,791 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 6,402 | $ | 5,640 | $ | 5,806 | ||||||
|
|
|
|
|
|
|
|||||||
| Capital expenditures: |
||||||||||||
| OSUR |
$ | 2,752 | $ | 2,145 | $ | 1,645 | ||||||
| DNAG |
1,585 | 2,208 | 2,099 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 4,337 | $ | 4,353 | $ | 3,744 | ||||||
|
|
|
|
|
|
|
|||||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Total assets: |
||||||||
| OSUR |
$ | 192,352 | $ | 151,719 | ||||
| DNAG |
103,849 | 56,216 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 296,201 | $ | 207,935 | ||||
|
|
|
|
|
|||||
The following table represents total net revenues by geographic area, based on the location of the customer:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| United States |
$ | 121,458 | $ | 99,758 | $ | 96,522 | ||||||
| Europe |
11,827 | 11,646 | 13,535 | |||||||||
| Other regions |
33,779 | 16,794 | 9,662 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 167,064 | $ | 128,198 | $ | 119,719 | |||||||
|
|
|
|
|
|
|
|||||||
F-27
Table of Contents
The following table represents total long-lived assets by geographic area:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| United States |
$ | 16,160 | $ | 15,737 | ||||
| Canada |
5,083 | 4,286 | ||||||
| Other regions |
129 | 10 | ||||||
|
|
|
|
|
|||||
| $ | 21,372 | $ | 20,033 | |||||
|
|
|
|
|
|||||
| 11. | COMMITMENTS AND CONTINGENCIES: |
Sublicense Agreement
In June 2004, we entered into a sublicense agreement with a third party pursuant to which we have been granted a limited, worldwide, non-exclusive sublicense to certain HIV-2 patents held by such party. Under the terms of this sublicense agreement, we are obligated to pay royalties based on a percentage of our net sales of certain products, which incorporate the technology covered by the licensed patents. Royalties are expensed as an element of cost of goods sold. Future minimum payments under this agreement are $292 for 2018. Royalties from our commercial sale of products covered by the sublicense can be credited against these minimum royalty obligations.
Leases
We lease office space for our Canadian subsidiary and domestic warehouse facilities under operating lease agreements. Future payments required under these non-cancelable leases are as follows:
| 2018 |
$ | 617 | ||
| 2019 |
791 | |||
| 2020 |
828 | |||
| 2021 |
834 | |||
| 2022 |
845 | |||
| Thereafter |
1,242 | |||
|
|
|
|||
| $ | 5,157 | |||
|
|
|
Rent expense for 2017, 2016 and 2015 was $852, $715, and $481, respectively.
Purchase Commitments
As of December 31, 2017, we had outstanding non-cancelable purchase commitments related to inventory, supplies, capital expenditures, and other goods or services as follows:
| 2018 |
14,262 | |||
| 2019 |
383 | |||
| 2020 |
383 | |||
|
|
|
|||
| $ | 15,028 | |||
|
|
|
Employment Agreements
Under terms of employment agreements with certain employees, which extend through 2019, we are required to pay each individual a base salary for continuing employment with us. The agreements require payments totaling $1,729 and $330 in 2018 and 2019, respectively.
F-28
Table of Contents
Standby Letters of Credit
We established standby letters of credit in the amount of $1,840, naming international customers as the beneficiaries. These letters of credit were required as a performance guarantee of our obligations under our product supply contracts with those customers and are collateralized by certificates of deposit maintained at a commercial bank. The standby letters of credit are recorded in restricted cash in the accompanying consolidated balance sheet.
Litigation
From time to time, we are involved in certain legal actions arising in the ordinary course of business. In management’s opinion, the outcomes of such actions, either individually or in the aggregate, are not expected to have a material adverse effect on our future financial position or results of operations.
| 12. | RETIREMENT PLANS: |
Substantially all of our U.S. employees are eligible to participate in the OraSure Technologies, Inc. 401(k) Plan (the “401(k) Plan”). The 401(k) Plan permits voluntary employee contributions to be excluded from an employee’s current taxable income under provisions of Internal Revenue Code Section 401(k) and the regulations thereunder. The 401(k) Plan also provides for us to match employee contributions up to $4 per year. We contributed $617, $619 and $587 to the 401(k) Plan, net of forfeitures, in 2017, 2016, and 2015, respectively.
In addition to our 401(k) plan, we offer a nonqualified deferred compensation plan to permit eligible directors and highly compensated employees of the Company to defer receipt and taxation of their compensation each year. We also may make discretionary contributions to the accounts of the participating employees in any amount either in cash or stock. Participants in the plan may not purchase OraSure stock as an investment vehicle. As of December 31, 2017 and 2016, the value of the assets associated with this plan was $3,514 and $1,980, respectively, and is included in other assets in our consolidated balance sheets. Our obligation related to the deferred compensation plan is included in other liabilities in our consolidated balance sheets. As of December 31, 2017 and 2016, our total obligation under this plan was $3,514 and $1,980, respectively.
Substantially all regular full-time Canadian employees are eligible to participate in the DNA Genotek Registered Retirement Savings Plan (the “RRSP”). The RRSP permits voluntary employee contributions to be excluded from an employee’s current taxable income and receive tax preferred treatment with Revenue Canada. The RRSP also provides for DNAG to match employee contributions up to $2 per year. We contributed $145, $134 and $134 to the RRSP in 2017, 2016, and 2015, respectively.
F-29
Table of Contents
| 13. | QUARTERLY DATA (Unaudited): |
The following tables summarize the quarterly results of operations for each of the quarters in 2017 and 2016. These quarterly results are unaudited, but in the opinion of management, have been prepared on the same basis as our audited financial information and include all adjustments (consisting only of normal recurring adjustments) necessary for a fair presentation of the information set forth herein.
| 2017 Results | ||||||||||||||||
| Three months ended | ||||||||||||||||
| March 31, 2017 |
June 30, 2017 |
September 30, 2017 |
December 31, 2017 |
|||||||||||||
| Net revenues |
$ | 32,546 | $ | 40,176 | $ | 42,314 | $ | 52,028 | ||||||||
| Costs and expenses |
16,675 | (1) | 33,289 | 34,995 | 41,867 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating income |
15,871 | (1) | 6,887 | 7,319 | 10,161 | |||||||||||
| Other income, net |
467 | 96 | 113 | 118 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Income before income taxes |
16,338 | 6,983 | 7,432 | 10,279 | ||||||||||||
| Income tax expense |
3,897 | 1,555 | 1,669 | 2,963 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income |
$ | 12,441 | $ | 5,428 | $ | 5,763 | $ | 7,316 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Earnings per share |
||||||||||||||||
| Basic |
$ | 0.22 | $ | 0.09 | $ | 0.10 | $ | 0.12 | (2) | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted |
$ | 0.21 | $ | 0.09 | $ | 0.09 | $ | 0.12 | (2) | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
| 2016 Results | ||||||||||||||||
| Three months ended | ||||||||||||||||
| March 31, 2016 |
June 30, 2016 |
September 30, 2016 |
December 31, 2016 |
|||||||||||||
| Net revenues |
$ | 29,089 | $ | 31,359 | $ | 32,251 | (3) | $ | 35,499 | (3) | ||||||
| Costs and expenses |
26,390 | 27,010 | 26,107 | 28,426 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating income |
2,699 | 4,349 | 6,144 | 7,073 | ||||||||||||
| Other income (expense), net |
(192 | ) | (340 | ) | 498 | 92 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Income before income taxes |
2,507 | 4,009 | 6,642 | 7,165 | ||||||||||||
| Income tax expense (benefit) |
61 | 173 | 400 | (31 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net income |
$ | 2,446 | $ | 3,836 | $ | 6,242 | $ | 7,196 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Earnings per share |
||||||||||||||||
| Basic |
$ | 0.04 | $ | 0.07 | $ | 0.11 | $ | 0.13 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted |
$ | 0.04 | $ | 0.07 | $ | 0.11 | $ | 0.13 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| (1) | Includes a $12,500 gain associated with the settlement of our litigation with Ancestry.com DNA LLC and its contract manufacturer, which was recorded as a reduction of operating expenses in the indicated period. |
| (2) | The summation of the quarterly amounts may not equal the year-end amounts as included in the consolidated financial statements due to the use of weighted-average shares for each period. |
| (3) | Revenues in the last six months of 2016 include an additional $5,436 of exclusivity payments recognized in other revenues as a result of the early termination of our HCV co-promotion agreement with AbbVie. |
| 14. | SUBSEQUENT EVENTS: |
In January 2018, we announced the retirement of our President and Chief Executive Officer (“CEO”) and our Chief Financial Officer (“CFO”) and Chief Operating Officer. Stephen S. Tang, Ph.D., who currently serves as
F-30
Table of Contents
Chairman of the Board of Directors (the “Board”), was appointed as the Company’s new President and CEO, effective as of April 1, 2018. Dr. Tang will replace Douglas A. Michels, who will retire as President and CEO, and as a member of the Board, on March 31, 2018. In addition, Ronald H. Spair, our CFO and Chief Operating Officer, will be retiring on or before June 30, 2018, with his exact retirement date to be determined based on the timing for the Company’s appointment of a new CFO. Charges associated with these transitions are expected to total $8,590 in 2018. These charges primarily reflect non-cash charges associated with modifications to existing stock grants held by the retiring executives and expenses associated with the onboarding of the Company’s new President and CEO.
F-31
Table of Contents
INDEX TO EXHIBITS
Table of Contents
Table of Contents
Table of Contents
| 101.INS | XBRL Instance Document |
| 101.SCH | XBRL Taxonomy Extension Schema Document |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase document |
| * | Management contract or compensatory plan or arrangement. |