Attached files
| file | filename |
|---|---|
| EX-99.1 - EXHIBIT 99.1 - 9 METERS BIOPHARMA, INC. | tv484364_ex99-1.htm |
| EX-10.3 - EXHIBIT 10.3 - 9 METERS BIOPHARMA, INC. | tv484364_ex10-3.htm |
| EX-10.2 - EXHIBIT 10.2 - 9 METERS BIOPHARMA, INC. | tv484364_ex10-2.htm |
| EX-10.1 - EXHIBIT 10.1 - 9 METERS BIOPHARMA, INC. | tv484364_ex10-1.htm |
| EX-4.2 - EXHIBIT 4.2 - 9 METERS BIOPHARMA, INC. | tv484364_ex4-2.htm |
| EX-4.1 - EXHIBIT 4.1 - 9 METERS BIOPHARMA, INC. | tv484364_ex4-1.htm |
| EX-3.2 - EXHIBIT 3.2 - 9 METERS BIOPHARMA, INC. | tv484364_ex3-2.htm |
| EX-3.1 - EXHIBIT 3.1 - 9 METERS BIOPHARMA, INC. | tv484364_ex3-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of
The Securities Exchange Act of 1934
February 2, 2018
Date of Report (Date of earliest event reported)
| INNOVATE BIOPHARMACEUTICALS, INC. |
| (Exact name of registrant as specified in its charter) |
| Delaware | 001-37797 | 27-3948465 | ||||||
| (State or other jurisdiction of incorporation) |
(Commission File Number) |
(I.R.S. Employer Identification No.) |
8480 Honeycutt Road, Suite 120
Raleigh, North Carolina 27615
(Address of principal executive offices)
(919) 275-1933
(Registrant's telephone number, including area code)
Monster Digital, Inc.
2655 First Street, Suite 250
Simi Valley, California 93065
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company x
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Item 2.01. Completion of Acquisition or Disposition of Assets.
On January 29, 2018, Monster Digital, Inc. (the “Company”), completed its reverse recapitalization with Innovate Biopharmaceuticals, Inc., which changed its name in connection with the transaction to “IB Pharmaceuticals Inc.” (“Innovate”), in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated as of July 3, 2017, by and among the Company, Monster Merger Sub., Inc. (“Merger Sub”), and Innovate (the “Merger Agreement”), pursuant to which Merger Sub merged with and into Innovate, with Innovate surviving as a wholly owned subsidiary of the Company (the “Merger”).
Also, on January 29, 2018, in connection with and immediately prior to the effective time of the Merger (the “Effective Time”), the Company (i) effected a reverse stock split at a ratio of one new share for every ten shares of its common stock outstanding (the “Reverse Stock Split”), (ii) increased the number of authorized shares of the Company’s common stock from 100,000,000 to 350,000,000 and (iii) changed its name to “Innovate Biopharmaceuticals, Inc.” Following the completion of the Merger, the business conducted by the Company became primarily the business conducted by Innovate, which is a clinical stage biotechnology company focused on developing novel autoimmune/inflammation therapeutic drugs.
Under the terms of the Merger Agreement, the Company issued shares of its common stock to Innovate’s stockholders, at an exchange ratio of 0.37813802 of a share of common stock (post Reverse Stock Split), in exchange for each share of Innovate common stock outstanding as of the Effective Time. The Company also assumed all of the stock options issued and outstanding under the 2015 Stock Incentive Plan (the “Innovate Plan”), with such stock options henceforth representing the right to purchase a number of shares of the Company’s common stock equal to 0.37813802 multiplied by the number of shares of Innovate’s common stock previously represented by such stock options.
Immediately prior to the Merger, Innovate issued and sold an aggregate of approximately $18.1 million of shares of Innovate common stock, including the conversion of $9,229,819 in outstanding convertible debt, to certain current stockholders of Innovate and certain new investors. Additionally, Innovate issued five-year warrants to each cash purchaser of common stock with a price per exercise price of $3.1764 after giving effect the exchange ratio.
Immediately following the Effective Time, there were approximately 25.8 million shares of the Company’s common stock outstanding. Immediately following the Effective Time, the former Innovate security holders owned approximately 94% of the fully-diluted common stock of the Company, with the Company’s security holders immediately prior to the Merger owning approximately 6% of the fully-diluted common stock of the Company.
The Company’s shares of common stock, which were previously listed on The Nasdaq Capital Market and traded through the close of business on January 31, 2018 under the ticker symbol “MSDI,” commenced trading on The Nasdaq Capital Market, under the symbol “INNT” on February 1, 2018. The Company’s common stock has a new CUSIP number, 45782F105.
The descriptions of the Merger and Merger Agreement included herein are not complete and are subject to and qualified in their entirety by reference to the Merger Agreement, a copy of which was attached as Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the SEC on July 6, 2017, and incorporated herein by reference.
On January 30, 2018, the Company issued a press release announcing the completion of the Merger. A copy of the press release is attached hereto as Exhibit 99.1.
Item 2.03. Creation of a Direct Financial Obligation or an Obligation under an Off-Balance Sheet Arrangement of a Registrant.
On January 29, 2018, the Company entered into a Note Purchase Agreement (the “Note Purchase Agreement”) between the Company and Gustavia Capital Partners LLC (the “Lender”). The Note Purchase Agreement included customary representations and warranties of the Company and Lender and customary closing conditions.
The Company issued a senior promissory note (the “Note”) pursuant to the Note Purchase Agreement in the aggregate principal amount of $4,800,000, against receipt of a cash payment by the Lender to the Company equal to $3,000,000 (less certain expense deductions).
On September 30, 2018 (the “Maturity Date”), the Company shall pay the Lender an amount in cash equal to 105% of the outstanding principal amount of the Note plus accrued and unpaid interest and Later Charges (as defined in the Note). The Company may not prepay the Note, however, the Company may, at any time prior to the Maturity Date, redeem all of the Outstanding Amount (as defined in the Note) at a price equal to 105% of the Outstanding Amount.
The Note bears interest at per annum rate of interest equal to 12.5%, compounded quarterly, and is due and payable in arrears on each Interest Date (as defined in the Note), with the first Interest Date on March 30, 2018.
The Note contains customary affirmative and negative covenants, including among others, covenants limiting the ability of the Company and its subsidiaries to transfer or dispose of any assets, incur indebtedness, change the nature of its business, grant liens, make investments, make certain restricted payments, issue securities, and enter into transactions with affiliates, in each case subject to certain exceptions.
Upon an event of default, the Lender may require the Company to redeem all or a portion of the Note. The Company shall redeem at a cash price equal to 105% of the Outstanding Amount. The events of default under the Note Purchase Agreement include, among others, payment defaults, covenant defaults, a material adverse effect default, bankruptcy and insolvency defaults, a failure to consummate the Merger (as defined in the Note), cross-defaults to other material indebtedness, judgment defaults, and defaults related to inaccuracy of representations and warranties. A default interest rate will apply on all obligations during the existence of an event of default under the Note at a per annum rate of interest equal to 18.0%.
The foregoing description of the Note Purchase Agreement and Note is qualified in its entirety by reference to the full text of the Note Purchase Agreement and Note which the Company is filing with this Form 8-K.
| 2 |
Item 3.02. Unregistered Sales of Equity Securities.
Pursuant to the terms of the Merger Agreement and in connection with the Merger, the Company issued shares of its common stock to Innovate’s stockholders. The number of shares issued, the nature of the transaction and the nature and amount of consideration received by the Company are described in Item 2.01 of this Form 8-K, which is incorporated by reference into this Item 3.02. The shares of Company Common Stock issued in connection with the Merger were not registered under the Securities Act of 1933, as amended (the “Securities Act”), in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act and/or Regulation D and Rule 506 promulgated thereunder.
Item 3.03. Material Modification to Rights of Security Holders.
To the extent required by Item 3.03 of Form 8-K, the information contained in Item 2.01 of this Current Report on Form 8-K is incorporated by reference herein.
On January 29, 2018, immediately prior to the Effective Time, the Company amended and restated its certificate of incorporation to (i) effect the Reverse Stock Split, (ii) increase the number of authorized shares of the Company’s common stock from 100,000,000 to 350,000,000 and (ii) change the Company’s name to “Innovate Biopharmaceuticals, Inc.” The amendment of the Company’s certificate of incorporation was approved by the Company’s stockholders at a special meeting of its stockholders on November 9, 2017.
The foregoing descriptions of the Company’s amended and restated certificate of incorporation are not complete and are subject to and qualified in their entirety by reference Company’s amended and restated certificate of incorporation, a copy of which is attached as Exhibit 3.1 hereto and is incorporated herein by reference.
| 3 |
Item 5.01. Changes in Control of Registrant.
The information set forth in Item 2.01 of this Current Report on Form 8-K is incorporated by reference into this Item 5.01.
In accordance with the Merger Agreement, on January 29, 2018, effective as of the Effective Time, David H. Clarke, Jonathan Clark, Robert B. Machinist, Christopher M. Miner and Steven Barre resigned from the Board and any respective committees of the Board to which they belonged. Also on January 29, 2018, the Board appointed, effective as of the Effective Time, Sandeep Laumas, M.D., Christopher Prior, Ph.D., Jay Madan, M.S., Roy Proujanksy, M.D., Lorin Johnson, Ph.D., Anthony Maida, Ph.D., M.A., M.B.A, and Anna Kazanchyan, M.D. as directors of the Company whose terms expire at the Registrant’s next annual meeting of stockholders.
Item 5.02. Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers.
(b) Pursuant to the Merger Agreement, on January 29, 2018, effective as of the Effective Time, David H. Clarke, Jonathan Clark, Robert B. Machinist, Christopher M. Miner and Steven Barre resigned from the Board and any respective committees of the Board on which they served, which resignations were not the result of any disagreements with the Company relating to the Company’s operations, policies or practices.
Also, pursuant to the Merger Agreement, on January 29, 2018, effective as of the Effective Time, David H. Clarke, the Company’s Chief Executive Officer, Jonathan Clark, the Company’s Interim President, David Olert, the Company’s Vice President of Finance and Chief Financial Officer, and Stephen R. Brownsell, the Company’s Vice President, resigned as officers of the Company.
(c) Effective as of the Effective Time, the Board appointed Sandeep Laumas as the Company’s Executive Chairman, Christopher Prior as the Company’s Chief Executive Officer, and Jay Madan as the Company’s President. There are no family relationships among any of the Company’s directors and executive officers. The information set forth in Item 8.01 of this Current Report on Form 8-K regarding the biographical information, compensation arrangements and related party transaction information for the newly appointed executive officers of the Company is incorporated by reference to this Item 5.02(c). Each of the newly appointed executive officers of the Company entered into the Company’s standard form of indemnification agreement with the Company on January 29, 2018, the form of which is attached hereto as Exhibit 10.3 and incorporated herein by reference.
(d) The information set forth in Item 5.01 of this Current Report on Form 8-K with respect to the appointment of directors to the Company’s board of directors pursuant to and in accordance with the Merger Agreement is incorporated by reference into this Item 5.02(d). The information set forth in Item 8.01 of this Current Report on Form 8-K regarding the related party transaction information for the newly appointed directors of the Company is incorporated by reference to this Item 5.02(d). Each of Roy Proujansky, Lorin Johnson, Anthony Maida, and Anna Kazanchyan entered into the Company’s standard form of indemnification agreement with the Company on January 29, 2018, the form of which is attached hereto as Exhibit 10.3 and incorporated herein by reference.
Audit Committee
On January 29, 2018, Anthony Maida, Lorin Johnson and Anna Kazanchyan were appointed to the Audit Committee. Anthony Maida will serve as the chair of the Audit Committee.
| 4 |
Compensation Committee
On January 29, 2018, Anna Kazanchyan, Lorin Johnson and Anthony Maida were appointed to the compensation committee of the Board. Anna Kazanchyan will serve as chair of the compensation committee.
Nominating and Corporate Governance Committee
On January 29, 2018, Lorin Johnson, Anna Kazanchyan and Anthony Maida were appointed to the nominating and corporate governance committee of the Board. Lorin Johnson will serve as chair of the nominating and corporate governance committee.
Item 5.03 Amendments to Articles of Incorporation or Bylaws; Change in Fiscal Year.
(a) To the extent required by Item 5.03 of Form 8-K, the information contained in Item 2.01 and Item 3.03 of this Current Report on Form 8-K is incorporated by reference herein.
Effective as of the Effective Time, the Board approved the amendment of the Company’s Bylaws to (i) amend Section 10 of Article I to eliminate the right of stockholders of the Company to act by written consent and (ii) conform to the name of the Company to “Innovate Biopharmaceuticals, Inc.” The foregoing description is qualified in its entirety by reference to the Bylaws, as amended, attached hereto as Exhibit 3.2 and incorporated herein by reference.
Item 8.01 Other Events.
In connection with the Merger and related transactions described in this Current Report on Form 8-K, the Company provides the following information related to Innovate set forth in this Item 8.01.
TABLE OF CONTENTS
Cautionary Statement Concerning Forward-Looking Statements
The information in Item 8.01 of this Current Report on Form 8-K, particularly in the sections entitled “Innovate Business,” and “Innovate Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the information incorporated herein by reference, include forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. These forward-looking statements are based on current expectations and beliefs and involve numerous risks and uncertainties that could cause actual results to differ materially from expectations. These forward-looking statements should not be relied upon as predictions of future events as we cannot assure you that the events or circumstances reflected in these statements will be achieved or will occur. When used in this report, the words “believe,” “may,” “could,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect,” “indicate,” “seek,” “should,” “would” and similar expressions are intended to identify forward-looking statements, though not all forward-looking statements contain these identifying words. All statements, other than statements of historical fact, are statements that could be deemed forward-looking statements.
If any of these risks or uncertainties materializes or any of these assumptions proves incorrect, our results could differ materially from the forward-looking statements in this report. All forward-looking statements in this report are current only as of the date of this report. We do not undertake any obligation to publicly update any forward-looking statement to reflect events or circumstances after the date on which any statement is made or to reflect the occurrence of unanticipated events.
The Company was incorporated in Delaware in November 2010 under the name “Monster Digital, Inc.” In January 2018, the Company merged its wholly-owned subsidiary, Monster Merger Sub, Inc., with and into IB Pharmaceuticals Inc. and changed the name of the Company to “Innovate Biopharmaceuticals, Inc.”
| 5 |
Overview
Innovate is a clinical-stage biopharmaceutical company developing novel therapies for autoimmune and inflammatory disorders. Innovate’s lead program, larazotide acetate (larazotide or INN-202), is entering Phase 3 registration trials and has the potential to be the first-to-market therapeutic for celiac disease, an unmet medical need, which effects an estimated 1% of the North American population or approximately 3 million individuals. Celiac patients have no treatment alternative other than a strict lifelong adherence to a gluten-free diet (GFD), which is difficult to maintain and can result in a lack in nutrients. Additionally, current FDA labeling standards allow up to 20 parts per million of gluten in GFD-labelled foods, which can be sufficient in many patients to cause celiac symptoms, including abdominal pain, cramping, bloating, gas, headaches, ataxia, ‘‘brain fog’’ and fatigue. Long-term sequelae of celiac disease include non-Hodgkin lymphoma, osteoporosis and anemia. Innovate’s second clinical program, INN-108, is in development for the treatment of mild-to-moderate ulcerative colitis (UC), an inflammatory bowel disease (IBD) with more than 1.25 million people affected in the major markets.
Innovate is led by an executive management team and board of directors that have held senior positions at leading pharmaceutical and biotechnology companies and that possess substantial experience across the spectrum of drug discovery, development and commercialization. Innovate’s CEO co-founded two successful biotechnology companies which were acquired by biotech and large pharmaceutical companies. Innovate’s medical and regulatory team conducted the Phase 2b trial for larazotide and have managed multiple other large scale clinical trials with successful New Drug Application (NDA) submissions and approvals. Members of Innovate’s board of directors have played key roles at Johnson & Johnson, Bristol-Myers Squibb, and AstraZeneca. One of Innovate’s directors was a co-founder of Salix Pharmaceuticals, the largest gastroenterology-focused company which was acquired for $16 billion in 2015.
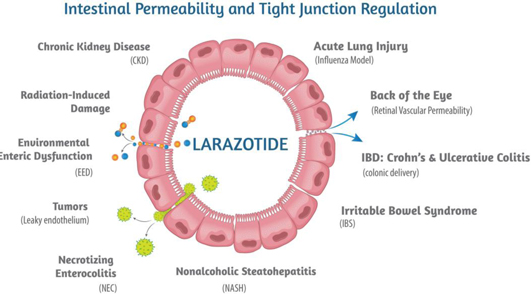
Figure 1: Larazotide’s mechanism of action is applicable to several other diseases.
| 6 |
Larazotide is an 8-amino acid synthetic peptide orally administered as a capsule which has been tested in more than 500 celiac patients with proven safety and efficacy in clinical trials. The FDA has granted larazotide Fast Track Designation for celiac disease. Larazotide’s proven safety profile is due to its lack of systemic absorption into the blood circulation which allows it to act locally in the small bowel. Additionally, larazotide has a first-in-class mechanism of action (MoA) as a tight junction regulator. Pre-clinical studies showed larazotide causes a reduction in permeability across the epithelial barrier, making it the only drug known to Innovate which is in clinical trials with this MoA. Increased intestinal permeability is a MoA which underlies several other diseases, including Crohn’s disease, irritable bowel syndrome (IBS) and non-alcoholic steatohepatitis (NASH), among others (Figure 1). Innovate is engaging in academic collaborations to expand larazotide’s clinical indications with a shorter time to clinical proof-of-concept due to larazotide’s safety profile and exposure in more than 800 subjects.
With the release of the Phase 2b trial data in 342 patients as a late-breaker presentation at the 2014 Digestive Disease Week (DDW), larazotide became the first and the only drug for the treatment of celiac disease (published data) yet to meet its primary endpoint with statistical significance. The Phase 2b data showed clinically meaningful and statistically significant (p=0.022) reduction in abdominal and non-GI (headache) symptoms. After a successful End-of-Phase 2 meeting with the FDA, which confirmed the regulatory path forward to approval, Innovate is preparing to launch the Phase 3 registration program later this year with topline pivotal data expected by 2019.
For celiac patients, larazotide is being investigated as an adjunct to a GFD for patients who continue to experience symptoms despite following a GFD. As a result of the difficulty of maintaining a gluten-free lifestyle due to access to and cost of gluten-free foods, contamination from gluten and social pressures, more than half the celiac population experiences multiple, potentially debilitating symptoms per month. A recent study from the UK indicates that more than 70% of patients diagnosed with celiac disease consume gluten either intentionally or inadvertently (Hall et al. 2013). In academic studies and proprietary market research, there is a clear need for a therapeutic for this large and growing population.
Innovate’s second drug candidate, INN-108, is in development for the treatment of mild-to-moderate UC. It is an oral tablet that uses an azo-bonded pro-drug approach linking mesalamine to 4-APAA (approved as Actarit in Japan in 1994 for the treatment of rheumatoid arthritis) and is entering a proof-of-concept Phase 2 trial in 2018, after having completed a successful Phase 1 trial demonstrating safety at currently approved doses of mesalamine. The azo-bond protects INN-108 (Figure 2) from the low pH in the stomach, allowing it to transit to the colon where the UC lesions are primarily located. In the colon, the azo bond is broken enzymatically, leading to mesalamine and 4-APAA releasing and having a synergistic anti-inflammatory effect. Although the majority of patients present with mild-to-moderate UC, which can progress to severe UC, the focus of drug development has been in moderate-to-severe UC with little innovation or drug development for mild-to-moderate UC. The mainstay of treatment for mild-to-moderate UC remains various oral reformulations of mesalamine or 5-ASA (5-amino salicylic acid) such as Shire’s Lialda (approved 2007) and Pentasa (approved 1993), Allergan’s Asacol HD (approved 2008) and Valeant/Salix’s Apriso (approved 2008).
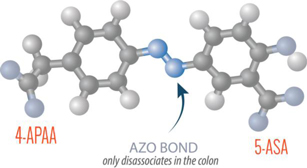
Figure 2: 4-APAA is covalently bonded to 5-ASA via a high energy azo-bond which is only cleaved enzymatically in the colon.
| 7 |
Innovate also owns the global rights to INN-329, a proprietary formulation of secretin, a peptide hormone which is used to improve visualization in magnetic resonance cholangiopancreatography (MRCP) procedures. Secretin is a 27-amino acid long hormone which rapidly stimulates release of pancreatic secretions, thus improving visualization of the pancreatic ducts during imaging procedures. Secretin has also been tested in a variety of central nervous system conditions such as autism.
Innovate is based in Raleigh, North Carolina, was incorporated under the laws of North Carolina under the name ‘‘GI Therapeutics, Inc.’’ in 2012, and changed its name when it converted to a Delaware corporation in 2014.
Innovate’s Strategy
Innovate’s goal is to become a leading biopharmaceutical company by developing novel therapeutics that have the potential to transform current treatment paradigms for patients and to address unmet medical needs. Innovate is currently pursuing the development of oral drugs for autoimmune and inflammatory diseases that target established biological pathways. The critical components of Innovate’s strategy are as follows:
| • | Advance larazotide for celiac disease into Phase 3 clinical trials. Innovate’s immediate priority is to initiate the Phase 3 trials for larazotide for the treatment of celiac disease. Innovate had a successful End-of-Phase 2 meeting with the FDA in 2017. With the guidance and agreement reached with the FDA, Innovate plans to initiate its Phase 3 trials by mid-2018. |
| • | Accelerate Development of larazotide for NASH. The mechanism of action of larazotide to decrease intestinal permeability is one of the key recognized pathogenic factors in NASH. Innovate is initiating development of larazotide in combination with select NASH therapies in clinical trials with the potential for synergistic therapeutic benefit. |
| • | Further the Development of larazotide for Crohn’s Disease. The mechanism of action of larazotide to decrease intestinal permeability can have a beneficial therapeutic effect in IBD. In an IL-10 knockout animal model, larazotide showed promising data which can position it for a proof-of-concept study alone and in combinations with select immunological therapies. |
| • | Advance INN-108 for ulcerative colitis into a proof-of-concept Phase 2 trial. Innovate is currently developing the plans to initiate the proof of concept Phase 2 trials for INN-108 for the treatment of ulcerative colitis. INN-108 will be initially developed for mild-to-moderate ulcerative colitis in adults. |
| • | Seek Partnerships to Commercialize Late Stage Pipeline Drugs. With large addressable markets, such as celiac disease, Innovate plans to seek out partners with an established presence and history of successful commercialization. |
| • | Leverage and protect Innovate’s existing intellectual property portfolio and secure patents for additional indications. Innovate intends to continue to expand its intellectual property protection strategy, grounded in securing composition of matter patents and method of use patents for newer indications. Innovate plans to develop newer formulations for the product candidates for other indications and improved performance of existing indications. |
| • | In-license additional intellectual property and pipeline drugs to expand Innovate’s presence in the treatment of autoimmune and inflammatory diseases. In addition to broadening its current pipeline through indication expansion, Innovate plans to explore expansion of its product pipeline through strategic partnerships and product acquisitions, as it did in 2016 through its inlicensing of larazotide, Alba Therapeutics, Inc.’s celiac program. Future pipeline expansion decisions will be based on the unmet medical needs within the gastrointestinal disease area including, but not limited to, celiac disease and ulcerative colitis, the commercial opportunity, and the ability to rapidly develop and commercialize a product candidate. |
| • | Leverage the expertise of Innovate’s management team and network of scientific advisors and key opinion leaders. Innovate is led by a strong management team with deep experience in drug development, collaborations, operations, and corporate finance. Innovate’s team has been involved in a broad spectrum of R&D activities leading to successful outcomes, including FDA approvals and drug launches. Innovate will continue to leverage the collective experience and talent of its management team, network of leading scientific experts, and key opinion leaders (KOLs) to strategize and implement its development and eventually its commercialization strategy. |
| 8 |
| • | Out-license Innovate’s non-core assets/indications and establish research collaborations. Innovate continually reviews its internal research priorities and therapeutic focus areas and may decide to out-license non-core assets/indications that arise from current and future available data. Innovate may seek research collaborations that leverage the capabilities of its core assets in order to monetize and expand upon the breadth of opportunities that may be uniquely accessible through its drug candidates. |
| • | Outsource capital intensive operations. Innovate plans to continue to outsource capital intensive operations, including most clinical development and all manufacturing operations of its product candidates in order to facilitate the rapid development of its pipeline by using high quality specialist vendors and consultants in a capital efficient manner. |
| 9 |
Innovate’s Drug Product Pipeline
Innovate’s current pipeline is focused on two clinical stage assets, one for celiac disease and one for ulcerative colitis. Innovate continues to leverage additional proof-of-concept work for larazotide to expand into additional indications, including irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD). The following table summarizes key information about Innovate’s pipeline of drug product candidates to date (Table 1):
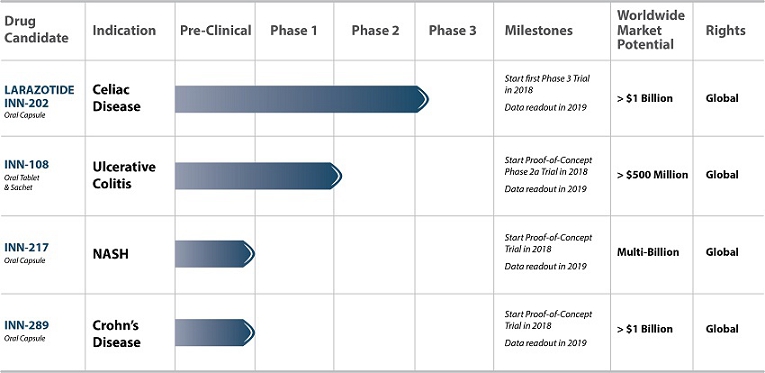
Table 1: Innovate’s key pipeline products are clinical stage with an established safety profile, large markets for chronically dosed therapies, and key milestones during the next 24 months.
Larazotide (INN-202) for Celiac Disease
Larazotide has been developed for the treatment of celiac disease and has successfully completed a Phase 2b trial showing clinically meaningful and statistically significant reduction in abdominal and non-GI (headache) symptoms. Innovate is planning to launch the Phase 3 trials in mid-2018.
Larazotide is an orally administered, locally acting, non-systemic, synthetic 8-amino acid (Figure 3), first-in-class tight junction regulator being investigated as an adjunct to a gluten-free diet in celiac disease patients who still experience persistent GI symptoms despite being on a gluten-free diet. Larazotide’s established safety profile and the lack of absorption into the blood stream are advantages for a chronically dosed lifetime medication.
The larazotide drug product is an enteric coated (EC) drug product formulated as enteric-coated multiparticulate beads filled into hard gelatin capsules for oral delivery. The enteric coating is designed to allow the bead particles to bypass the stomach and release larazotide upon entry into the small intestine (duodenum). A mixed bead formulation is used to allow partial release of larazotide upon entry into the duodenum and to release the remaining larazotide approximately 30 minutes later. In clinical trials, larazotide has been dosed 15 minutes before meals allowing time for its effect in the small bowel before exposure to gluten.
| 10 |
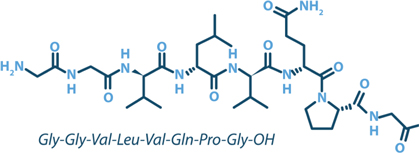
Figure 3: Larazotide acetate is an 8-amino acid peptide formulated into a proprietary oral capsule
Larazotide’s Mechanism of Action
In research studies supportive of the mechanism of action, larazotide has been shown to stimulate recovery of mucosal barrier function via the regulation of tight junctions both in vitro and in vivo, including in the celiac disease mouse model (Gopalakrishnan, 2012; Gopalakrishnan, 2012). In doing so, it is proposed that larazotide reduces the signs and symptoms associated with celiac disease.
In several autoimmune diseases, this increased intestinal permeability or paracellular leakage allows increased exposure to a triggering antigen and a consequent inflammatory response, the characteristics of which are determined by the particular disease and the genetic makeup of the individual. A new paradigm for autoimmune disease is that there are three contributing factors to the development of disease:
| 1. | A genetically susceptible immune system that allows the host to react abnormally to an environmental antigen; |
| 2. | An environmental antigen that triggers the disease process; and |
| 3. | The ability of the environmental antigen to interact with the immune system. |
Larazotide inhibits tight junction opening triggered by both gluten and inflammatory cytokines, thus reducing uptake of gluten. Larazotide disrupts the intestinal permeability-inflammation loop, and reduces symptoms associated with celiac disease.
Larazotide’s Unique Dose Response
Previously published in vitro work has shown a wide linear dose response using Caco-2 cells, larazotide has been shown in numerous clinical trials to exhibit significant benefit at reducing symptoms but only at the lower doses while inhibition of this activity occurs at the higher doses. To explain this observation, Dr. Anthony Bliksager from North Carolina State University, evaluated the pharmacology of larazotide at the luminal surface of the small intestine in an ex vivo pig model. A section of the gut was ligated, placed in an Ussing chamber and changes in permeability measured by electrical resistance. The data confirmed full length larazotide is capable of fully restoring intestinal wall integrity to that of the non-ischemic control following an ischemic insult. Subsequently, it was discovered a specific aminopeptidase only located within the brush borders of the intestinal epithelium which cleaved larazotide into two fragments missing the either one or both N-terminus glycine (G) residues (GGVLVQPG). Both cleaved fragments, GVLVQPG and VLVQPG, are inactive in this ex vivo pig model. Moreover, when these two fragments are combined with the active full length larazotide, activity is abolished. These data confirm that local buildup of these inactive fragments derived from higher doses of larazotide compete and block function of larazotide after threshold concentration. The in vitro experiments using Caco-2 monolayers did not show the same pharmacology as they are missing the brush border and thus lack the aminopeptidase to degrade larazotide. These data also provide an explanation for the clinical observations of an optimal low dose of larazotide which avoids the reservoir of competing inactive fragments generated at high doses of larazotide.
| 11 |

Figure 4: An aminopeptidase in the brush border cleaves larazotide into two fragments, #1 and #2, which then act as inhibitors of larazotide
| 12 |
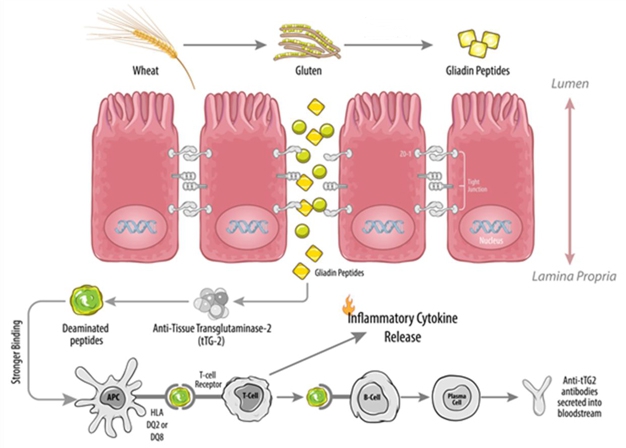
Figure 5: Illustrative effect of gluten ingestion, breakdown to gliadin which can cross a defective epithelial barrier in the small bowel thus activating the intestinal-inflammatory loop and causing symptoms and villous atrophy.
The Intestinal Barrier, Tight Junctions, and Intestinal Permeability
The intestine is the largest interface between a person and his or her environment, and an intact intestinal barrier is essential in maintaining overall health. An important function of the intestinal barrier is to regulate the trafficking of macromolecules between the environment and the host. Together with gut-associated lymphoid tissue (GALT) and the neuroendocrine network, the intestinal epithelial barrier controls the equilibrium between tolerance and immunity to non self-antigens. When the finely tuned trafficking of macromolecules is dysregulated, both intestinal and extra-intestinal autoimmune disorders can occur in genetically susceptible individuals (Figure 5).
Transcellular fluxes (through the cell membrane) allow nutrients and small molecules to enter the cell from the luminal side of the intestine and exit on the serosal side (internal milieu). Paracellular fluxes (between cells) in contrast are limited by size and charge constraints imposed by the tight junctions between epithelial cells. The paracellular pathway is the key regulator of intestinal permeability to larger more complex macromolecules that may be immunogenically significant.
Intestinal epithelial cells adhere to each other through junction complexes. The tight junction, also referred to as zonula occludens, represents the major barrier to diffusion within the paracellular space between intestinal cells. Multiple proteins that make up the tight junction have been identified including occludin, claudin family members, and junctional adhesion protein (JAM). These interact with cytosolic proteins (ZO-1, ZO-2, and ZO-3) that function as adaptors between the tight junction proteins and actin and myosin contractile elements within the cell. Acting together, they open and close the paracellular junctions between cells. It is now apparent that tight junctions are dynamic structures that are involved in developmental, physio logical, and pathological processes.
The role of tight junction dysfunction in the pathogenesis of autoimmune diseases is under active investigation. Many autoimmune populations have increased intestinal permeability and it is believed that this may play a fundamental role in the development of autoimmunity. In susceptible populations, the opening of tight junctions between intestinal epithelial cells may lead to exposure to oral antigens via paracellular transport and a consequent autoimmune response. A wide range of gastrointestinal and systemic inflammatory diseases are associated with abnormal intestinal permeability including celiac disease, type 1 diabetes, inflammatory bowel diseases (Crohn’s disease and ulcerative colitis), and ankylosing spondylitis.
| 13 |
Summary of Key Clinical Trials using Larazotide in Celiac Disease
Larazotide has been administered to humans in seven (7) clinical trials. These include three Phase 1 trials: (two trials in healthy subjects and a Phase 1b proof of concept (PoC) trial in subjects with celiac disease), two (2) Phase 2 gluten challenge studies in subjects with controlled celiac disease, and additionally two (2) Phase 2 trials in subjects with active celiac disease (Table 2). After demonstrating safety in the Phase 1 studies, larazotide was tested to explore which endpoint would be suitable for celiac disease. After looking at permeability changes in the gut, which turned out to be highly variable in a large trial setting, and then mucosal healing, which likely requires a longer-term study, symptom reduction showed the most consistent and reliable reduction both in a gluten challenge and a ‘‘real-life’’ trial. Importantly, after exposure in more than 800 subjects, the safety profile of larazotide remained similar to placebo due to its lack of absorption into the bloodstream — a critical advantage for a chronically dosed drug.
| Trial | Clinical Trial | No. of Subjects | ||
| -001 | Phase 1: Single Escalating Doses in Healthy Volunteers | 24 | ||
| -002 | Phase 1b: Multiple Dose POC in Celiac Patients – Gluten Challenge | 21 | ||
| -003 | Phase 1: Multiple Escalating Dose in Volunteers | 24 | ||
| -004 | Phase 2a: Multiple Dose POC in Celiac Patients Gluten Challenge 2 weeks | 86 | ||
| -006 | Phase 2b: Dose Ranging, in Celiac Patients Gluten Challenge, 6 weeks | 184 | ||
| -011 | Phase 2b: POC and Dose Ranging in Active Celiac Patients | 105 | ||
| -06B | Phase 2b: Similar to -006, in Celiac Patients | 42 | ||
| -012 | Phase 2b: Multiple dose in Celiac patients with Symptoms on a GFD | 342 |
Table 2: Significant drug exposure in more than 800 subjects in multiple clinical trials consistently showed a solid safety profile similar to placebo, which is a critical advantage for chronic lifetime administration.
Clinical Trial (‘006) Results Revealed Key Insight into Symptom Reduction as a Primary Endpoint
A Phase 2b study with a gluten challenge (CLIN1001-006) was conducted in 184 subjects with well-controlled celiac disease on a GFD. Subjects were randomized to one of four treatment groups, (placebo, 1 mg, 4 mg, or 8 mg larazotide) and asked to take treatment 15 minutes prior to each meal (TID). Nine hundred (900) mg of gluten was taken with each meal. Subjects remained on their GFD throughout the duration of the trial. The trial results revealed key insight into how to move the program forward by focusing on reduction of symptoms. The 1-mg dose prevented the development of gluten-induced symptoms as measured by GSRS (a patient-reported outcome (PRO) devised and validated by AstraZeneca), and all drug treatment groups had lower anti-transglutaminase antibody levels than the placebo group. Results of pre-specified secondary endpoints suggest that larazotide reduced antigen exposure as manifested by reduced production of anti-tTG levels and immune reactivity towards gluten and gluten-related gastrointestinal symptoms in subjects with celiac disease undergoing a gluten challenge.
| 14 |
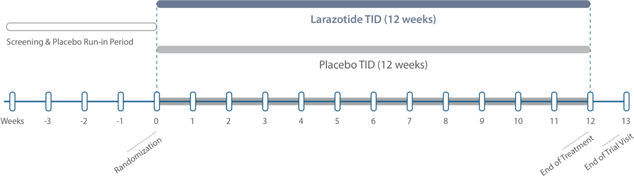
Figure 6: Trial design for Phase 2b and Phase 3 is the same with a screening period followed by 12 weeks of randomization, larazotide acetate vs. placebo.
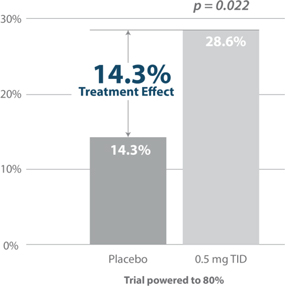
Figure 7: Responder Rate Analysis: Larazotide is the only celiac drug to meet its primary endpoint with statistical significance and a clinically meaningful improvement in the copyrighted CeD PRO (celiac disease patient reported outcome), an FDA-agreed upon primary endpoint for Phase 3 (shown above).
Source: Gastroenterology 2015; 148:1311–1319; p. 1315
Clinical Trial (‘012) Met the Primary Endpoint with Statistical Significance (CeD-GSRS/CeD PRO)
The purpose of the ‘012 study was to assess the efficacy (reduction and relief of signs and symptoms of celiac disease) of 3 different doses of larazotide (0.5 mg, 1 mg, and 2 mg TID) versus placebo for the treatment of celiac disease in adults as an adjunct to a GFD. Larazotide or placebo which was administered TID, 15 minutes prior to each meal. After a screening period, subjects were asked to continue following their current GFD diets into a placebo-run in phase for 4 weeks after which they were randomized to drug versus placebo. Subjects maintained an electronic diary capturing: daily symptoms celiac disease patient reported outcome (CeD-PRO), weekly symptoms (CeD-GSRS), bowel moments (BSFS), and a self-reported daily general well-being assessment (Figure 6).
The primary endpoint of average on-treatment CeD GSRS score throughout the treatment period was met at the 0.5 mg TID dose. In addition, a number of pre-specified secondary and exploratory endpoints, such as symptomatic days and symptom-free days, collectively demonstrated that a dose of 0.5 mg TID was superior to placebo and higher doses of larazotide. No difference was observed between the two higher dose levels (1 and 2 mg TID) or placebo, suggesting a narrow dose range around the 0.5mg dose which seems to correlate with pre-clinical data.
| 15 |
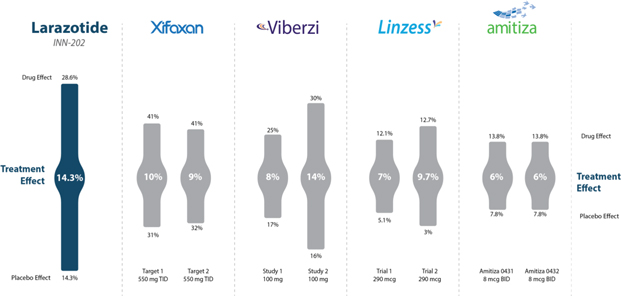
Figure 8: Treatment effect of larazotide acetate from the Phase 2b trial (‘012) compared to approved IBS/CIC drugs with varying treatment effects mostly in the mid to high single digit range.
Source: Gastroenterology 2015; 148:1311–1319; p. 1315 and FDA Drug Labels
The CeD PRO, a copyrighted PRO created specifically for celiac disease and wholly owned by Innovate, showed a statically significant (p=0.022) result with a treatment effect of 14.3% (drug responder rate minus placebo responder rate). Although, there are no celiac drugs approved as a comparator, the treatment effect was greater than several other GI dugs approved for irritable bowel syndrome (IBS) and chronic idiopathic constipation (CIC) which use a similar clinical trial design and have GI/abdominal symptoms similar to celiac disease (Figure 8).
Clinical Path Forward to Phase 3 Trials
After a successful End-of-Phase 2 meeting with the FDA, agreements were reached on the key aspects of the Phase 3 trials. The FDA agreed on using the previously validated CeD PRO as the primary endpoint with two doses of larazotide which bracket the range of efficacy in previous trials. Two Phase 3 trials with a size of about 450 patients each would allow for more than a 90% power to replicate the Phase 2b trial results. Most other criteria such as inclusion, exclusion, site selection/coordination will remain the same as the ‘012 Phase 2b trial. One of the leading causes of Phase 3 trial failure is toxicity which appears when drugs are tested in larger populations and in the case of larazotide, we believe this risk is diminished due to larazotide’s lack of systemic absorption into the blood circulation.
| 16 |
About Celiac Disease
Celiac disease is a genetic autoimmune disease triggered by the ingestion of gluten-containing foods such as wheat, barley, and rye. Individuals with celiac disease have increased intestinal permeability, commonly referred to as a ‘‘leaky’’ gut. This allows macromolecules that normally remain on the luminal side of the intestine to pass through to the serosal side through tight junctions via paracellular diffusion (Figure 9). In the case of celiac disease, this permeability may allow gluten break-down products, the triggering antigens of celiac disease, to reach gut-associated lymphoid tissue (GALT), initiating an inflammatory response. Celiac disease is characterized by chronic inflammation of the small intestinal mucosa that may result in diverse symptoms, malabsorption, atrophy of intestinal villi, and a variety of clinical manifestations.
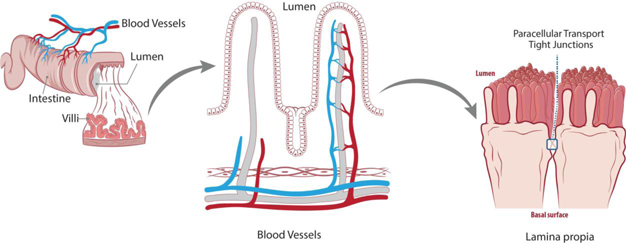
Figure 9: The epithelial barrier separates the intestinal content from the immune system (lamina propria) and the vasculature.
| 17 |
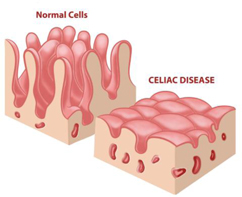
Figure 10: Intestinal villi atrophy in celiac patients, a characteristic finding upon biopsy of the duodenum.
Large Population — Unmet Need (no drug approved); Serious Long-Term Sequelae
Celiac disease affects an estimated 1% of the Western population (Dubé, 2005). Currently, there are no therapeutics available to treat celiac disease, and the current management of celiac disease is a life-long adherence to a gluten-free diet. Changes in dietary habits are difficult to maintain, and foods labeled as gluten-free may still contain small amounts of gluten (up to 20 ppm per FDA labeling standards). Dietary compliance is imperfect in a large fraction of patients (Rostom, 2006) and difficult to adhere to on an ongoing basis (Green, 2007). In a recent survey conducted in the United Kingdom non-adherence to the gluten-free diet was found to be as high as 70% (Hall, 2013).
There are serious long-term consequences to exposure to gluten in patients with celiac disease, including the risk of developing osteoporosis, stomach, esophageal, or colon cancer, and T-cell lymphoma (Green 2003, Green 2007). The continuous GI symptoms often result in significant morbidity with a substantial reduction in quality of life. In addition, not all patients respond to a gluten-free diet. Patients with known celiac disease may continue to have or re-develop symptoms despite being on a gluten-free diet (Rostom 2006). This suggests a need for a therapeutic agent for the treatment of celiac disease (Green, 2007; Hall, 2013).
Celiac disease represents a unique model of an autoimmune disorder in that the following elements are known:
| 1. | The triggering environmental factor is glutenin or gliadin, the proline, glutamine and glycine rich glycoprotein fractions of gluten; |
| 2. | There is a close genetic association with HLA haplotypes DQ2 and/or DQ8; and |
| 3. | A highly specific humoral autoimmune response occurs. |
| 18 |
Genetics of Celiac Disease
The high incidence of celiac disease in first degree relatives of celiac patients (10 − 15%) and high concordance rate in monozygotic twins (80%) suggest a strong genetic component. Gliadin deamidation by tissue transglutaminase (tTG) enhances the recognition of gliadin peptides by human leukocyte antigen (HLA) DQ2 and DQ8 T cells in genetically predisposed subjects, which in turn may initiate the cascade of autoimmune reactions responsible for mucosal destruction. This interaction implies that gliadin and/or its breakdown peptides in some way cross the intestinal epithelial barrier and reach the lamina propria of the intestinal mucosa where they are recognized by antigen-presenting cells. The enhanced paracellular permeability of individuals with celiac disease would allow passage of macromolecules through the paracellular spaces with resulting autoimmune inflammation. There is a strong genetic predisposition to celiac disease, with major risk associated with HLA DQ2 (approximately 95% of celiac disease patients) and HLA-DQ8 (approximately 5% of celiac disease patients). The prevalence of celiac disease in the U.S. is estimated to be approximately 1%; however approximately 30% of the general U.S. population is HLA DQ2 positive (Figure 11), indicating that additional factors are involved in the development of celiac disease.
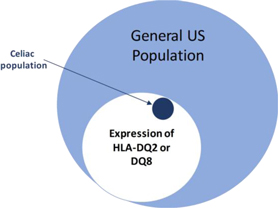
Figure 11: Distribution of HLA-DQ2/DQ8 in the general US population and in celiac disease.
Source: J. Clin. Invest. 2007 Jan 2;117(1):41.
In celiac disease, an inflammatory reaction occurs in the intestine that is characterized by infiltration of immune cells in the lamina propria and epithelial compartments with chronic inflammatory cells and progressive architectural changes to the mucosa. Both adaptive and innate branches of the immune system are involved. The adaptive response is mediated by gluten-reactive CD4+ T cells in the lamina propria that recognize gluten-derived peptides when presented by the HLA class II molecules DQ2 or DQ8. The CD4+ T cells then produce pro-inflammatory cytokines such as interferon gamma. This results in an inflammatory cascade with the release of cytokines, anti-tTG antibodies, T cells, and other tissue-damaging mediators leading to villous injury and crypt hyperplasia in the intestine. Anti-human tissue transglutaminase (anti-tTG) antibodies are also produced, which form the basis of serological diagnosis of celiac disease.
Anti-tTG Antibodies: Highly Sensitive and Specific Blood-based ELISA Diagnostic Test
The current approach for diagnosis of celiac disease, is to use anti-tissue transglutaminase-2 (tTG-2) antibody tests as an initial screen with definitive diagnosis from biopsy of the small intestine mucosa. The diagnosis of celiac disease is confirmed by demonstration of characteristic histologic changes in the small intestinal mucosa, which are scored based on criteria initially put forth by Marsh and later modified. In 2012, the European Society of Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) Guidelines allowed symptomatic children with serum anti-tTG antibody levels ≥10 times upper limit of normal (ULN) to avoid duodenal biopsies after positive HLA test and serum anti-endomysial antibodies (EMAs). It’s likely with further improvement in diagnostic testing, the guidelines will expand to include adults in the EU and eventually in the US as well, making it easier to quickly and cost effectively screen the broader population.
| 19 |
The need for multiple clinical and laboratory findings to diagnose celiac disease makes monitoring disease progression difficult. International guidelines give standardized definitions and criteria for the diagnosis of celiac disease, however there are not clear standards for follow-up and monitoring of treatment. This is particularly true for celiac patients diagnosed as adults, who respond differently and less completely to a GFD than do celiac patients diagnosed as children. It is not clear who should perform follow-up of patients with celiac disease and at what frequency but the American College of Gastroenterology suggests that an annual follow-up seems reasonable. Recommendations for monitoring disease progression include assessing symptoms and dietary compliance, and repeating serology tests. Markers of celiac disease progression and improvement that are both validated and provide a timely assessment of disease activity are lacking.
Role of Tissue Transglutaminase in Celiac Disease
Anti-tTG-2 antibodies are produced in the small-intestinal mucosa (Picarelli et al. 1996), where they can bind tTG-2 present in the basement membrane and around blood vessels and form deposits characteristic of the disease. tTG-2 has been implicated in a variety of human disorders including several neurodegenerative conditions and cancer. Transglutaminases (TGs) were first discovered in the 1950s and are a family of enzymes which catalyze Ca2+-dependent post-translational modification of proteins. Of the seven isoforms discovered so far all share the same basic four-domain tertiary structure, with minor variations, although their catalytic mechanism is conserved, resembling that of the cysteine proteases. tTGs cause transamidation, esterification, and hydrolysis; all of which lead to post-translational modifications in the target proteins. Characteristically, tTG’s mediate selective protein cross-linking by forming covalent isopeptide linkages between two target proteins. The resulting cross-linked products in many cases have high molecular masses and are unusually resistant to proteolytic degradation and mechanical strain. As in the case of the gliadin fragments in celiac disease, they are able to pass thru the leaky paracellular pathway from the lumen to the lamina propria, where the immune cells reside and are then activated.
Gliadin fragments, in addition to being rich in proline, also have high glutamine content, which makes them suitable substrates for tTG-2, which targets glutamine residues. For augmented DQ2/8 binding, the conversion of glutamine residues to glutamic acid is catalyzed by tTG-2 as a deamidation reaction. After deamidation, the gliadin peptides become highly negatively charged in key anchor positions, thereby increasing their affinity to the HLA molecules. CD4+ T cells recognize the deamidated gliadin peptides bound to the HLA DQ2 or DQ8 molecules by their T cell receptors, thus activating intestinal inflammation leading to villous atrophy.
Gluten and Food Labeling
Gluten is a complex molecule contained in several grains such as wheat, rye and barley. Gluten can be subdivided into two major protein subgroups according to their solubility in alcohol and aqueous solutions. These subclasses consist of gliadins, soluble in 40 − 70% ethanol and glutenins which are large, polymeric molecules insoluble in both alcohol and aqueous solutions. The gliadins and glutenins can be further subdivided into groups according to their molecular weight. Glutenins can be subdivided into low and high molecular weight proteins, while the gliadin protein family contains α-, β-, γ- and ω- types. Both glutenins and gliadins are characterized by a high amount of prolines (20%) and glutamines (40%) that protect them from complete degradation in the gastrointestinal tract and make them difficult to digest. Currently 31 nine-amino acid peptide sequences in the prolamins of wheat and related species have been defined as being celiac toxic or celiac ‘‘epitopes.’’ These epitopes are located in the repetitive domains of the prolamins, which are proline and glutamine-rich, and the high levels of proline make the peptide resistant to proteolysis. In addition, the prolamin-reactive T cells also recognize these epitopes to a greater extent when specific glutamine residues in their sequences have been deamidated to glutamic acid by tTG-2. The immunodominant sequence after wheat challenge corresponds to a well-characterized 33 residue peptide from α-gliadin, ‘‘33-mer,’’ that is resistant to gastrointestinal digestion (with pepsin and trypsin) and was initially identified as the major celiac toxic peptide in the gliadins.
| 20 |
The FDA finalized a standard definition of ‘‘gluten-free’’ in August 2013. As of August 5, 2014, all manufacturers of FDA-regulated packaged food making a gluten-free claim must comply with the guidelines outlined by the FDA (www.fda.gov/gluten-freelabeling). A ‘‘gluten free’’ claim still allows up to 20 ppm of gluten which leads to more than 100mg/day up to 500 mg/day of gluten exposure. Due to presence of gluten in foods, beer/liquor, cosmetics and household products, exposure is virtually impossible to completely avoid, and with cross-contamination, celiac patients cannot avoid exposure to gluten therefore, making symptoms more frequent than expected.
|
CNS |
Endocrine | Oncology/Heme | Skin | Other | ||||
| Headaches | Type 1 Diabetes |
Enteropathy associated T-cell lymphoma (EATL) |
Dermatitis herpetiformis | Rheumatoid arthritis (RA) | ||||
| Gluten ataxia |
Autoimmune thyroid |
Anemia | Alopecia areata |
Reduced bone density | ||||
| Peripheral neuropathies | Addison’s disease | Vitiligo | Sjogren’s syndrome |
Non-GI Manifestations of Celiac Disease and Co-Morbidities
Table 3: Diseases associated with celiac disease
Headache, Gluten Ataxia: Nervous System Manifestation of Celiac Disease
The association between celiac disease and neurologic disorders has been supported by numerous studies over the past 40 years. While peripheral neuropathy and ataxia have been the most frequently reported neurologic extra-intestinal manifestations of celiac disease a growing body of literature has established headache as a common presentation of celiac disease as well. The exact prevalence of headache among patients ranges from about 30% to 6% (Lebwohl, 2016).
Dermatitis herpetiformis: Skin Manifestation of Celiac Disease
Dermatitis herpetiformis (DH) is an inflammatory cutaneous disease characterized by intensely pruritic polymorphic lesions with a chronic-relapsing course, first described by Duhring in 1884. DH’s only treatment is a strict lifelong GFD, for achieving and maintaining a permanent control. It appears in around 25% patients with CD, at any age of life, mainly in adults and is a very characteristic clinical presenting symptom.
| 21 |
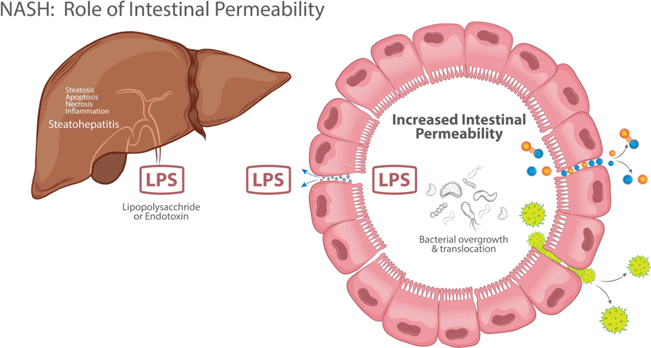
Figure 12: LPS (Lipopolysaccharide) or Endotoxin produced by bacteria in the “leaky gut” has been implicated in the pathogenesis of NASH. Larazotide can prevent LPS translocation to the liver via the portal circulation.
Non-alcoholic steatohepatitis (NASH)
NASH is a growing disorder (up to 25%) in the general population though its incidence is elevated 6-fold in celiac patients. It has been suggested several times that NASH is associated with increased gut permeability caused by disruption of intercellular tight junctions in the intestine allowing lipopolysaccharide (LPS) from bacteria to pass into the portal circulation to the liver. Explosive growth in the market for NASH therapeutics is expected according to Global Data across the seven major markets of the U.S., France, Germany, Italy, Spain, the UK, and Japan, with such market set to grow to around $25.3 billion by 2026. Larazotide can be used in combination with the multitude of NASH drugs in clinical trials as a safe drug with a different and potentially synergistic therapeutic effect.
Enteropathy-associated T-cell lymphoma (EATL): High Mortality Rate and Unmet Need
Cancer associated with celiac disease occurs in about 2% − 3% of the celiac population, with the most common representing approximately 2⁄3 of the cases, being Enteropathy-associated T-cell lymphoma (EATL). EATL effects approximately 1% of the celiac population. EATL is an intestinal tumor of intraepithelial T lymphocytes found throughout the small intestines and increased in number in celiac disease. Intestinal intraepithelial α-β T-cells, in various stages of transformation, are thought to be the normal-cell counterpart for EATL. Currently there are no standardized treatment regimens and surgery and/or radiation maybe indicated depending on tumor bulk and spread followed by anthracycline-containing chemotherapy such as CHOP is often used. Relapses after CHOP or CHOP-like chemotherapy occur 1 − 60 months from diagnosis in ∼80% of responsive patients, with a mortality of 85% due to progressive disease or complications.
Refractory Celiac Disease
Refractory celiac disease is clinically defined as the persistence of pathologic changes in the intestine consistent with celiac disease despite a strict gluten-free diet for more than 12 months. These pathologic changes include increased intraepithelial lymphocytes, villous atrophy, and crypt hyperplasia. Cases of refractory celiac disease are commonly divided into two types:
| 22 |
| • | Type I refractory celiac disease — These lesions typically show no atypia in the intraepithelial lymphocytes, normal surface T cell receptor, CD3 and CD8 expression by intraepithelial T lymphocytes and a polyclonal pattern on T cell receptor gene rearrangement studies. |
| • | Type II refractory celiac disease — These lesions also have no atypia in the intraepithelial lymphocytes, but demonstrate a loss of surface T cell receptor, CD3, or CD8 expression and may have a monoclonal T cell receptor gene rearrangement. |
The clinical significance of refractory celiac disease type is incompletely understood. While type I refractory celiac disease is unlikely to progress to EATL, type II may be a precursor lesion to EATL. Imaging with computed tomography, positron emission tomography, and video capsule endoscopy may help to identify cases of refractory celiac disease that have progressed to EATL.
Type I Diabetes Mellitus
Celiac disease is overrepresented in type 1 diabetes (T1DM), which shares a mutual genetic predisposition with celiac disease; both diseases are associated with the HLA class II genes (HLA-DQB1) on chromosome 6p21 (Smyth et al. 2008). The incidence of celiac disease is increased by 8 to 10-fold in the T1DM population versus the overall population incidence. In fact, T1DM patients with the HLADQ2/8 haplotype are also positive for anti-tTG antibodies, an established highly sensitive test for celiac disease.
The association of Human Leukocyte Antigens (HLA) with type 1 diabetes was first reported in the 1970s. Classical HLA molecules are cell-surface proteins that bind and present peptide antigens for recognition by the T cell receptor (TCR). The shape of the peptide binding groove and charges within it determine the type of peptides which bind to a given HLA and the combination then activates the TCR and in celiac disease leads to the intestinal-inflammatory loop causing villous atrophy and in T1DM causing destruction of the insulin secreting pancreatic β - cells.
In addition to the genetic link, several studies over the past two decades have implicated a leaky or damaged intestinal barrier leading to passage of toxins and/or viruses as a potential causative factor in T1DM. (Vaarala, 2008). Increased intestinal permeability, also common to celiac disease, and reports of gliadin activated lymphocytes trafficking to the pancreas lead to a link amongst autoimmunity, intestinal permeability, intestinal inflammation and potential role of gliadin. A growing body of evidence also suggests the beneficial effects of a GFD (for patients with concomitant celiac disease) may actually protect against the long-term complications of T1DM. such as retinopathy, nephropathy and others. Heretofore, until larazotide, no drug, to Innovate’s knowledge, with an MoA to decrease intestinal permeability has advanced into clinical trials and with its established safety profile as an oral capsule, a targeted exploratory clinical study could help further elucidate the role of intestinal permeability as a therapeutic modality for T1DM as well.
Non-Celiac Gluten Sensitivity: Large Growing Patient population with Celiac Symptoms
Non-celiac gluten sensitivity (NCGS) is a syndrome diagnosed in patients with symptoms that respond to removal of gluten from the diet, after celiac disease and wheat allergy have been excluded. NCGS patients lack the villous atrophy of the small intestine, yet biopsies show reduced numbers of T-regulatory cells, which may indicate that the innate immune system is involved. Anti-tTG antibodies are not elevated in NCGS as in celiac disease which is another key difference between the two diseases. Anti-gliadin antibodies (AGA) may be an indicator of NCGS as up to 50% of such patients presenting to gastroenterologists have detectable circulating levels, primarily of IgG AGA.
The ‘‘classical’’ presentation of NCGS is a combination of GI symptoms including abdominal pain, bloating, bowel habit abnormalities (either diarrhea or constipation), and extra-intestinal symptoms such as ‘‘brain fog,’’ depression, headache, fatigue, and leg or arm numbness. NCGS has been described in the literature since the 1980s and is distinct from celiac disease, however, both share symptomatic relief from a GFD, implying benefit from preventing gluten from passing through the epithelial barrier is beneficial.
| 23 |
Other Indications using Larazotide’s Mechanism of Action
Larazotide for Crohn’s Disease: IL-10 Knockout Mouse Model
The effect of larazotide on disease attenuation in an IL-10 knockout mouse study was studied (Arrieta, 2009). Larazotide was placed in the drinking water of the mice at a low dose (0.1 mg/ml) or high dose (1.0 mg/ml) during the period from 4 to 17 weeks of age. Results were compared to wild type mice, IL-10 knockout mice with no treatment, and IL-10 knockout mice treated with probiotics. Intestinal and colonic permeability was significantly reduced in the high dose larazotide treatment group, but not in the untreated IL-10 knockout group. Larazotide treatment caused a reduction in all tissue markers of colonic inflammation (IFNγ and TNFα) and in histological inflammation.
Larazotide for Environmental Enteric Dysfunction (EED): Positive in vitro Data; Potential for PriorityReview Voucher
Environmental enteric dysfunction (EED) is a rare pediatric tropical disease in the US and Europe, however, more than 165 million children in developing countries in Africa and Asia suffer from it. As per section 524 of the Federal Food, Drug, and Cosmetic Act (FD&C) Act, EED would likely fall under ‘‘Current List of Tropical Disease’’ number ‘S,’ thus making a drug approved for EED in the US eligible for a Priority Review Voucher (PRV). PRVs save time to approval for a drug and can be sold to a large pharma company. The most recent PRV sold by BioMarin Pharmaceuticals Inc. yielded them $125 million.
The histological presentation of EED is very similar to celiac disease with villous atrophy and chronic inflammation of the small bowel and the pathogenesis of EED is linked to increased intestinal permeability. We have tested Larazotide against some of the pathogens commonly found in EED (unpublished) and found positive in vitro results which will need to be confirmed in animal models before starting a clinical trial in EED.
INN-108: Mild-to-Moderate Ulcerative Colitis
INN-108 is in development for mild-to-moderate ulcerative colitis (UC) and is expected to enter a proof-of-concept Phase 2 trial in the second half of 2018 after a successful Phase 1 trial demonstrating safety at currently approved doses of mesalamine. UC is an inflammatory bowel disorder (IBD) that affects more than 1.25 million people in the major markets and is characterized by inflammation and ulcers in the colon and rectum. UC is a chronic disease that can be debilitating and sometimes lead to life-threatening complications. While poorly understood, a multitude of environmental factors and genetic vulnerabilities are thought to lead to the dysregulation of the immune response via a defective epithelial barrier. Although the majority of patients present with mild-to-moderate UC which can progress to severe UC, the focus of drug development has been in moderate-to-severe UC with little innovation or drug development for mild-to-moderate UC. The mainstay of treatment for mild-to-moderate UC remain various oral reformulations of mesalamine or 5-ASA (5-amino salicylic acid) such as Shire’s Lialda (approved 2007) and Pentasa (approved 1993), Allergan’s Asacol HD (approved 2008) and Valeant/Salix’s Apriso (approved 2008).
| 24 |
INN-108 uses an azo-bonded pro-drug approach linking mesalamine to 4-APAA. Mitsubishi Pharma developed 4-APAA as Actarit in Japan which was approved in 1994 for rheumatoid arthritis. IBD drugs were all originally approved for rheumatoid arthritis (RA), from the oldest 5-ASA, sulfasalazine, to the latest biologics, Humira and Enbrel. 4-APAA has more than two decades of safety data as a standalone drug and has an MoA which is differentiated from mesalamine though the ultimate effect for both is anti-inflammatory (Figure 13). Taken orally as a tablet, the azo-bond protects INN-108 from the low pH in the stomach, thus allowing it to transit to the colon where the UC lesions are located. In the colon, the azo bond is broken enzymatically leading to the release of mesalamine and 4-APAA which have a synergistic anti-inflammatory effect. With the addition of 4-APAA, which is not approved in the U.S. or EU, to the already approved mesalamine, the synergistic effect could lead to superior clinical efficacy over the currently approved oral mesalamines.
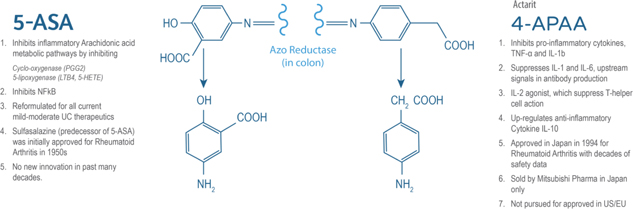
Figure 13: 4-APAA is covalently bonded to 5-ASA via a high energy azo-bond which is only cleaved enzymatically in the colon. The anti-inflammatory effect of each of 5-ASA and 4-APAA via different pathways which could lead to a potential synergistic anti-inflammatory effect as seen in animal studies.
| 25 |
INN-108: UC Animal Model Data Shows Synergy between 4-APAA and Mesalamine
The effects of chronic treatment with INN-108 on Clostridium diffıcile toxin A — induced colitis of the colon is shown in Figure 14. Orally administered INN-108 was significantly more potent than sulfasalazine or 4-APAA alone (McVey, 2005).
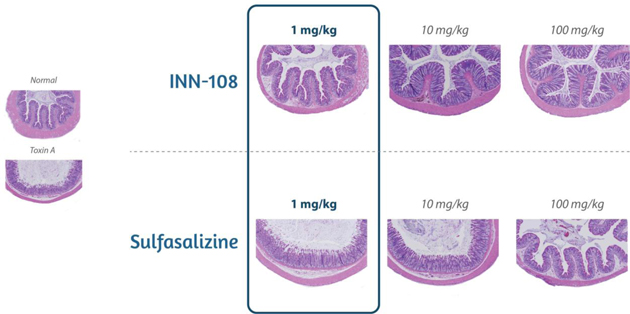
Figure 14: A rat UC model using toxin A induced-colitis as the insult leads to sloughing of the colonic epithelium with increasing doses. Using sulfasalazine vs. INN-108 to protect against the toxin A injury showed INN-108 was significantly more potent that sulfasalazine.
Source: McVey DC et al. Digestive Diseases and Sciences. 2005 Mar 1;50(3):565-73.
INN-108 Clinical Development Pathway
After completing a Phase 1 study with 24 subjects, safety was established with dosing of mesalamine and 4-APAA at 2 grams each for a total of 4 grams TID. The typical dose of the various approved mesalamine formulations range from 1.5g to 2.4g per day, thus INN-108’s mesalamine content is within the established approved dose range. The addition of 4-APAA is thought to improve the efficacy above mesalamine, which would allow INN-108 to be used either after or instead of current mesalamines. In a Phase 2 trial, Innovate plans to compare INN-108 to mesalamine seeking to demonstrate a greater clinical effect than mesalamine alone.
Source: McVey DC et al. Digestive Diseases and Sciences. 2005 Mar 1;50(3):565-73.
Ulcerative Colitis: Lack of Innovation in New Drug Development for Past Several Decades
Conventional therapies broadly inhibit mechanisms involved in the inflammatory process and are commonly used to effectively treat patients experiencing a mild-to-moderate form of the disease. For mild-to-moderate UC, oral mesalamine has an established efficacy and safety profile. However, gastroenterologists cite the need for new therapies for mild-to-moderate UC.
Patients who do not respond to mesalamine are eventually transitioned to biologics. The primary targets for biologics have been to control the immune response and inflammatory cascade, by inhibiting or downregulating molecules such as TNF-α, NF-κB, IL-1β and IFN1-γ. We believe INN-108 bridges the gap between mesalamine and biologics by its mechanism of action of both inhibiting the inflammatory process as well as down-regulating the cytokines.
| 26 |
Branded mesalamine formulations didn’t face any generics and have been a profit generator until July 2017, when Zydus Cadila launched the first generic of Shire’s Lialda. Within weeks generic Lialda grabbed about 40% unit market share as per IMS. This rapid generic penetration would be expected over time for the remaining mesalamines as well. Thus, if INN-108 shows improved efficacy over mesalamine, it could convert branded mesalamines, some generics as well as steroids/AZA/6-MP and early use of biologics. INN-108 could offer a more cost effective and more efficacious profile than mesalamines and avoid or delay the need for high-cost biologics (Figure 15).

Figure 15: INN-108’s market opportunity improved over mesalamine without the side effects from Steroids/AZA/6-MP
About Ulcerative Colitis
UC is a chronic intermittent relapsing inflammatory disorder of the large intestine and rectum. While poorly understood, a multitude of environmental factors and genetic vulnerabilities are thought to lead to the dysregulation of the immune response via a defective epithelial barrier. As a result, chronic inflammation and ulceration of the colon occurs. UC is specific to the colon and affects only the mucosal lining of the colon. Common symptoms of UC include diarrhea, bloody stools, and abdominal pain. The majority of patients are intermittent in their disease course, in that they experience a relapse among periods of remission. However, some patients experience only a single episode of the disease prior to maintaining remission whereas other patients are chronically symptomatic and may require a proctocolectomy to treat their condition.
History of Drug Development in Mild-to-Moderate Ulcerative Colitis
The original compound used in UC was sulfasalazine (Azulfidine), a conjugate of 5-ASA linked to sulfapyridine by an azo bond, which is split into the two molecules by bacterial azoreductases in the colon. The 5-ASA component or mesalamine is the active therapeutic moiety of sulfasalazine, with sulfapyridine thought to have little if any therapeutic effect. Sulfapyridine, however, is the cause of most of the significant adverse side effects of sulfasalazine.
| 27 |
This led to the development of other 5-ASA preparations utilizing azo chemistry to deliver high concentrations of mesalamine or 5-ASA to the colon by preventing early absorption of the drug in the small intestine. Such preparations include olsalazine (Dipentum), consisting of two molecules of 5-ASA bonded together by an azo bond, and balsalazide (Colazal), consisting of 5-ASA azo bonded to an inert carrier (4-aminobenzoyl-β-alanine). The efficacy of these newer oral forms of 5-ASA is comparable to that of sulfasalazine, but they are better tolerated. However, some side effects persist which prevent wider use. In each of these preparations, the only active moiety is mesalamine or 5-ASA, an anti-inflammatory agent.
INN-329
INN-329 is a proprietary formulation of secretin, a peptide hormone which is used to improve visualization in a magnetic resonance MRCP procedures. Secretin is a 27-amino acid long hormone which rapidly stimulates release of pancreatic secretions, thus improving visualization of the pancreatic ducts during imaging procedures. Secretin has also been tested in a variety of central nervous system conditions such as autism, though currently approved only for pancreatic function testing and imaging with endoscopic retrograde cholangiopancreatography (ERCP). The currently marketed synthetic secretin, approved by the FDA in 2004, is not approved by the FDA or the EMEA for Secretin-MRCP (S-MRCP) procedures. Innovate acquired the assets of secretin from Repligen Corporation in December 2014.
MRCP has been used for more than 20 years as a non-invasive tool for imaging pancreatic ducts. With the addition of secretin, pancreatic secretions are increased leading to significantly improved visualization of the pancreatic ducts for detection of abnormalities, including pancreatic cancer. The gold standard for pancreatic duct imaging had been ERCP, an expensive and invasive procedure with complications such as pancreatitis (3 − 5%), bleeding (1 − 2%), perforation (1%), infection (1 − 2%) and death (1/250). More than a half-million ERCP procedures are performed annually in the US and as the role of ERCP diminishes for screening, it will further the need for approval of secretin for S-MRCP. Innovate expects to repeat a Phase 3 trial with a partner, if and when secured, as per previous discussion with the FDA to look at improvement in visualization of the pancreatic duct via MRCP with and without secretin.
Innovate’s Intellectual Property
Innovate strives to protect the proprietary technology that it believes is important to its business, including its product candidates and its processes. Innovate seeks patent protection in the United States and internationally for its products, their methods of use, and processes of manufacture and any other technology to which Innovate has rights, as appropriate. Additionally, Innovate has licensed the rights to intellectual property related to certain of its product candidates, including patents and patent applications that cover the products or their methods of use or processes of manufacture. The terms of the licenses are described below under the heading ‘‘Licensing Agreements.’’ The patent families related to the intellectual property covered by the licenses include 29 U.S. patents and 107 foreign patents with expirations dates ranging from 2018 to 2035. Innovate also relies on trade secrets that may be important to the development of its business.
Innovate’s success will in part depend on the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to its business, the validity and enforceability of its patents, the continued confidentiality of its trade secrets as well as its ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. Innovate also relies on continuing technological innovation and in-licensing opportunities to develop and maintain its proprietary position.
| 28 |
Innovate cannot be sure that patents will be granted with respect to any of its pending patent applications or with respect to any patent applications it may own or license in the future, nor can Innovate be sure that any of its existing patents or any patents it may own or license in the future will be useful in protecting its technology and products. For this and more comprehensive risks related to Innovate’s intellectual property, please see ‘‘Risk Factors — Risks Related to Innovate’s Intellectual Property.’’
CeD PRO: Copyrighted Primary Endpoint for Celiac Disease Tested in a Successful Clinical Trial
The patient reported outcome (PRO) primary end point for celiac disease (CeD PRO). was developed based on FDA guidance and is copyrighted. Innovate believes that if larazotide is the first approved drug for celiac disease, the CeD PRO will become part of the drug label creating another barrier to entry for potential competitors. Subsequently, any company seeking to develop a drug for celiac disease would either need to develop their own PRO or would be required to license the CeD PRO from Innovate.
Strategic Collaborations and License Agreements
Innovate has entered into collaboration agreements with several academic institutions and other contract research organizations to investigate pre-clinical studies for the use of its drug candidates in potential other indications or to further broaden its understanding of its current indications.
Licensing Agreements
License with Alba Therapeutics Corporation
In February 2016, Innovate entered into a license agreement (the ‘‘Alba License’’) with Alba Therapeutics Corporation (‘‘Alba’’) to obtain an exclusive worldwide license to certain intellectual property relating to larazotide and related compounds.
Innovate’s initial area of focus for this asset relates to the treatment of celiac disease. This program is now referred to as INN-202 by Innovate. The license agreement gives Innovate the rights to (i) the patent families owned by University of Maryland, Baltimore (UMB) and licensed to Alba, (ii) the patent families (ALB-015, ALB-062, and ALB-065) are owned by Alba Therapeutics Corporation, and (iii) one patent family (ALB-030) that is jointly owned. In connection with the Alba License, Innovate also entered into a sublicense agreement with Alba under which Alba sublicensed the UMB patents to Innovate (the ‘‘Alba Sublicense’’).
As consideration for the Alba License, Innovate agreed to pay a one-time, non-refundable fee at the time of execution and set payments upon the achievement of certain milestones in connection with the development of the product, including the dosing of the first patient in the Phase 3 clinical trial, acceptance and approval of the New Drug Application, the first commercial sale, and the achievement of certain net sales targets. The last milestone payment is due upon the achievement of annual net sales of INN-202 in excess of $1.5 billion. Upon the first commercial sale of INN-202, the license becomes perpetual and irrevocable. The term of the Alba Sublicense extends until the earlier of (i) the termination of the Alba License, (ii) the termination of the underlying license agreement, or (iii) an assignment of the underlying license agreement to Innovate. After Innovate makes the first milestone payment after the dosing of the first patient in the Phase 3 clinical trial and is able to demonstrate sufficient financial resources to complete the trial, Innovate has the exclusive option to purchase the assets covered by the license. During the term of the Alba License, Alba has the right to sell the covered assets to Innovate upon delivering a notice to Innovate of such intent.
License with Seachaid Pharmaceuticals, Inc.
In April 2013, Innovate entered into a license agreement (the ‘‘Seachaid License’’) with Seachaid Pharmaceuticals, Inc. (‘‘Seachaid’’) to further develop and commercialize the licensed product, the compound known as APAZA. This program is now referred to as INN-108 by Innovate.
| 29 |
The license agreement gives Innovate the exclusive rights to (i) commercialize products covered by the patents owned or controlled by Seachaid related to the composition, formulation or use of any APAZA compound in the territory that includes the U.S., Canada, Japan, and most countries in Europe, and (ii) use, research, develop, export and make products worldwide for the purposes of such commercialization.
As consideration for the Seachaid License, Innovate agreed to pay a one-time, non-refundable fee at the earlier of the time Innovate met certain financing levels or 18 months following the execution of the agreement and set payments upon the achievement of certain milestones in connection with the development of the product, filing of the New Drug Application, the first commercial sale, and the achievement of certain net sales targets. There are future royalty payments based on achieving sales targets, and Innovate is required to pay Seachaid a portion of any sublicense revenue. The royalty payments continue for each licensed product and in each applicable country until the earlier of (i) the date of expiration of the last valid claim for such products to expire or (ii) the date that one or more generic equivalents if such product makes up 50 percent or more of sales in the applicable country. The term of the Seachaid License extends on a product-by-product and country-by-country basis until the expiration of the royalty period for the applicable product in the applicable country.
Asset Purchase Agreement
In December 2014, Innovate entered into an Asset Purchase Agreement (the ‘‘Asset Purchase Agreement’’) with Repligen Corporation (‘‘Repligen’’) to acquire Repligen’s RG-1068 program for the development of secretin for the pancreatic imaging market and MRCP) procedures. This program is now referred to as INN-329 by the Innovate. As consideration for the Asset Purchase Agreement, Innovate agreed to make a non-refundable cash payment on the date of the agreement and future royalty payments consisting of a percentage of annual net sales, with the royalty payment percentage increasing as annual net sales increase. The royalty payments are made on a product-by-product and country-by-country basis and the obligation to make the payments expires with respect to each country upon the later of (i) the expiration of regulatory exclusivity for the product in that country or (ii) ten years after the first commercial sale in that country. The royalty amount is subject to reduction in certain situations, such as the entry of generic competition in the market.
Manufacturing and Supply
Innovate contracts with third parties for the manufacturing of all of its product candidates, including INN-108, INN-202 and INN-329 for pre-clinical and clinical studies, and intends to continue to do so in the future. Innovate does not own or operate any manufacturing facilities and Innovate has no plans to build any owned clinical or commercial scale manufacturing capabilities. Innovate believes that the use of contract manufacturing organization (CMOs) eliminates the need to directly invest in manufacturing facilities, equipment and additional staff. Although Innovate relies on contract manufacturers, its personnel and consultants have extensive manufacturing experience overseeing CMOs.
As Innovate furthers its molecules, Innovate will consider secondary or back-up manufacturers for both active pharmaceutical ingredient and drug product manufacturing. To date, Innovate’s third-party manufacturers have met the manufacturing requirements for the product candidates in a timely manner. Innovate expects third-party manufacturers to be capable of providing sufficient quantities of Innovate’s product candidates to meet anticipated full-scale commercial demands but Innovate has not assessed these capabilities beyond the supply of clinical materials to date. Innovate currently engages CMOs on a ‘‘fee for services’’ basis based on Innovate’s current development plans. Innovate plans to identify CMOs and enter into longer term contracts or commitments as Innovate moves its product candidates into Phase 3 clinical trials.
Innovate believes there are alternate sources of manufacturing that have been and could be engaged and enabled to satisfy its clinical and commercial requirements, however Innovate cannot guarantee that identifying and establishing alternative relationships with such sources will be successful, cost effective, or completed on a timely basis without significant delay in the development or commercialization of Innovate’s product candidates. All of the vendors used by Innovate conduct their operations under current Good Manufacturing Practices, or cGMP, a regulatory standard for the manufacture of pharmaceuticals.
| 30 |
Commercialization
Innovate owns exclusive rights to all three of its product candidates in the United States and all other major markets, including Japan and the European Union. Innovate plans to pursue regulatory approvals for its products in the United States and the European Union, and may independently commercialize these products in the United States. In doing so, Innovate may engage with strategic partners to help implement optimal sales and promotion activities.
Innovate’s commercialization strategy will target key prescribing physicians including specialists such as gastroenterologists, as well as provide patients with support programs to ensure product access. Outside of the United States, Innovate plans to seek partners to commercialize its products via out-licensing agreements or other similar commercial arrangements.
Competition
The pharmaceutical industry is highly competitive and characterized by intense and rapidly changing competition to develop new technologies and proprietary products. Innovate’s potential competitors include both major and specialty pharmaceutical companies worldwide.
The competitive landscape in celiac disease is currently limited, which we believe is due to lack of significant past R&D investments and lack of recognition and education around the disease. To Innovate’s knowledge, there are no late stage competitors entering Phase 3 clinical trials or any who have successfully completed Phase 2 studies to date. However, in recent years large pharmaceutical companies have begun to expand their focus areas to autoimmune diseases such as celiac disease, and given the unmet medical needs in these areas, Innovate anticipates increasing competition. A few early stage programs are active, with time to enter Phase 1 clinical trials still several years away, including Roche/Genetech’s RG7625 (cathepsin S inhibitor), Takeda/PvP’s KumaMax (gluten degrading enzyme), Celimmune/Amgen’s AMG-714 (an IL-15 MAb) and Dr. Falk Pharma/Zeria’s ZED-1227 (a tTG-2 inhibitor). ImmunogenX’s IMGX003 (two gluten degrading enzymes) failed to meet its primary endpoint in a Phase 2b trial in 2015.
| Product | Status | Mechanism | Company | Route | Product Type | |||||
| AMG 714 | Phase 2 |
Anti-IL-15 MAb |
Celimmune/ Amgen |
Subcutaneous; 2x/month |
MAb (humanized) | |||||
| ZED-1227 | Phase 1b | TGase-2 inhibitor |
Zedira GmbH/ Dr Falk Pharma |
Oral |
Small molecule (peptidomimetic) | |||||
| Nexvax2 | Phase 1 | Tolerizing vaccine | ImmusanT | Intradermal | 3 gliadin epitopes (peptides) | |||||
| KumaMax | Pre-clinical | Enzymatic degradation of gluten |
Takeda/PvP Biologics |
Oral | Recombinant enzyme |
Table 4: Current celiac drugs in development are still in pre-clinical to early Phase 2 proof-of-concept stage. No drugs have completed a successful Phase 2b other than larazotide.
| 31 |
Ulcerative colitis drug development has historically been primarily focused on the moderate-to-severe UC population with little investment and R&D in mild-to-moderate UC, which is the majority of the patient populations. Current treatments for mild-to-moderate UC include the mesalamine reformulations that are pictured in Figure 16 and described above under the heading ‘‘History of Drug Development in Mild to Moderate Ulcerative Colitis,’’ as well as Lialda, Pentasa, Asacol HD and Apriso, Valeant/Salix’s Uceris (oral MMX-formulated budesonide; a corticosteroid) and 5-mercaptopurine (severe side effects). Eventually, half of the mild-to-moderate UC patients progress from mesalamine to the more expensive biologics, which creates a significant potential market opportunity for any drug that is more effective than mesalamine and less expensive than the biologics.
Figure 16: Other than various reformulations of mesalamine which have been used for the past several decades, no new drugs have been approved for mild-to-moderate UC
| 32 |
Government Regulations
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of drugs, such as those Innovate is developing. Innovate, along with third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which it wishes to conduct studies or seek approval or licensure of its product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
Government Regulation of Drugs
The process required by the FDA before drug product candidates may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices, or GLP, regulation; |
| • | submission to the FDA of an Investigational New Drug application, or IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| • | approval by an independent Institutional Review Board, or IRB, or ethics committee for each clinical site before a clinical trial can begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed product candidate for its intended purpose; |
| • | reparation of and submission to the FDA of a New Drug Application, or NDA, after completion of all required clinical trials; |
| • | a determination by the FDA within 60 days of its receipt of a NDA to file the application for review; |
| 33 |
| • | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with current Good Manufacturing Practices, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the product’s continued safety, purity and potency, and of selected clinical investigational sites to assess compliance with current Good Clinical Practices, or cGCPs; and |
| • | FDA review and approval of the NDA to permit commercial marketing of the product for particular indications for use in the United States, which must be updated annually and when significant changes are made. |
The testing and approval processes require substantial time, effort and financial resources, and Innovate cannot be certain that any approvals for its product candidates will be granted on a timely basis, if at all. Prior to beginning the first clinical trial with a product candidate, Innovate must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent Institutional Review Board, or IRB, for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site, and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
For purposes of NDA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| • | Phase 1. The drug product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, the initial human testing is often conducted in patients. |
| • | Phase 2. The drug product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| 34 |
| • | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling. |
| • | Phase 4. In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be required as a condition to approval of the NDA. |
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the drug characteristics of the product candidate, and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
NDA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a NDA requesting approval to market the product for one or more indications. The NDA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a NDA requires payment of a substantial User Fee to FDA, and the sponsor of an approved NDA is also subject to annual product and establishment user fees. These fees are typically increased annually. A waiver of user fees may be obtained under certain limited circumstances.
Within 60 days following submission of the application, the FDA reviews an NDA to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any NDA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the NDA must be resubmitted with the additional information. Once a NDA has been filed, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA reviews a NDA to determine, among other things, whether a product is safe and effective for the indication being pursued, and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety and effectiveness. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a NDA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a NDA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
| 35 |
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and Innovate may encounter difficulties or unanticipated costs in its efforts to secure necessary governmental approvals, which could delay or preclude us from marketing its products. After the FDA evaluates a NDA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may request additional information or clarification. The FDA may delay or refuse approval of a NDA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the NDA with a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of its products under development.
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs that meet certain criteria. Specifically, new drug products are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a fast track product, the FDA may consider sections of the NDA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A fast track designated product candidate may also qualify for priority review, under which the FDA sets the target date for FDA action on the NDA at six months after the FDA accepts the application for filing. Priority review is granted when there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the accelerated approval program, the FDA may approve an NDA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the biologic’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit. In addition, the Food and Drug Administration Safety and Innovation Act, or FDASIA, which was enacted and signed into law in 2012, established breakthrough therapy designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Sponsors may request the FDA to designate a breakthrough therapy at the time of or any time after the submission of an IND, but ideally before an end-of-phase 2 meeting with FDA. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the therapy; providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and considering alternative clinical trial designs when scientifically appropriate, which may result in smaller or more efficient clinical trials that require less time to complete and may minimize the number of patients exposed to a potentially less efficacious treatment. Breakthrough designation also allows the sponsor to file sections of the NDA for review on a rolling basis. Innovate may seek designation as a breakthrough therapy for some or all of its product candidates.
| 36 |
Fast Track designation, priority review and breakthrough therapy designation do not change the standards for approval but may expedite the development or approval process.
Orphan Drug Status
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drug candidates intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that costs of research and development of the drug for the indication can be recovered by sales of the drug in the United States. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Although there may be some increased communication opportunities, orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a drug candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same drug for the same indication for seven years, except in very limited circumstances, such as if the second applicant demonstrates the clinical superiority of its product or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
Orphan drug exclusivity could block the approval of Innovate’s drug candidates for seven years if a competitor obtains approval of the same product as defined by the FDA or if Innovate’s drug candidate is determined to be contained within the competitor’s product for the same indication or disease.
As in the United States, designation as an orphan drug for the treatment of a specific indication in the European Union, must be made before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including up to 10 years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product.
The FDA and foreign regulators expect holders of exclusivity for orphan drugs to assure the availability of sufficient quantities of their orphan drugs to meet the needs of patients. Failure to do so could result in the withdrawal of marketing exclusivity for the orphan drug.
Post-Approval Requirements
Any products manufactured or distributed by Innovate pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMP, which impose certain procedural and documentation requirements upon Innovate and its third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third-party manufacturers that Innovate may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. Innovate cannot be certain that it or its present or future suppliers will be able to comply with the cGMP regulations and other FDA regulatory requirements. If Innovate’s present or future suppliers are not able to comply with these requirements, the FDA may, among other things, halt its clinical trials, require them to recall a product from distribution, or withdraw approval of the NDA.
| 37 |
Future FDA and state inspections may identify compliance issues at Innovate’s facilities or at the facilities of its contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing.
The FDA may withdraw approval of an NDA if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market, or product recalls; |
| • | fines, warning letters, or holds on post-approval clinical studies; |
| • | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; or |
| • | injunctions or the imposition of civil or criminal penalties. |
The FDA closely regulates the marketing, labeling, advertising and promotion of drugs and biologics. A company can make only those claims relating to safety and efficacy that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by Innovate and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.
Other Healthcare Laws and Compliance Requirements
Innovate’s sales, promotion, medical education, clinical research and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services and state and local governments. Innovate’s promotional and scientific/educational programs and interactions with healthcare professionals must comply with the federal Anti-Kickback Statute, the civil False Claims Act, physician payment transparency laws, privacy laws, security laws, and additional federal and state laws similar to the foregoing.
| 38 |
The federal Anti-Kickback Statute prohibits, among other things, the knowing and willing, direct or indirect offer, receipt, solicitation or payment of remuneration in exchange for or to induce the referral of patients, including the purchase, order or lease of any good, facility, item or service that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, formulary managers, and beneficiaries on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to increased scrutiny and review if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal Anti-Kickback Statute has been violated. The government has enforced the federal Anti-Kickback Statute to reach large settlements with healthcare companies based on sham research or consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. Many states have similar laws that apply to their state health care programs as well as private payers.
Federal false claims and false statement laws, including the federal civil False Claims Act, or FCA, impose liability on persons and/or entities that, among other things, knowingly present or cause to be presented claims that are false or fraudulent or not provided as claimed for payment or approval by a federal health care program. The FCA has been used to prosecute persons or entities that “cause” the submission of claims for payment that are inaccurate or fraudulent, by, for example, providing inaccurate billing or coding information to customers, promoting a product off-label, submitting claims for services not provided as claimed, or submitting claims for services that were provided but not medically necessary. Actions under the FCA may be brought by the Attorney General or as a qui tam action by a private individual, or whistleblower, in the name of the government. Violations of the FCA can result in significant monetary penalties and treble damages. The federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other illegal sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, certain companies that were found to be in violation of the FCA have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or corporate integrity agreements, restricting the manner in which they conduct their business.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers; knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services; and willfully obstructing a criminal investigation of a healthcare offense. Like the federal Anti-Kickback Statute, the Affordable Care Act amended the intent standard for certain healthcare fraud statutes under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
| 39 |
Given the significant size of actual and potential settlements, it is expected that the federal government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws. Many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payer, in addition to items and services reimbursed under Medicaid and other state programs. To the extent that Innovate’s products, once commercialized, are sold in a foreign country, Innovate may be subject to similar foreign laws.
There has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposed new reporting requirements on certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, for payments or other transfers of value made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Covered manufacturers are required to collect and report detailed payment data and submit legal attestation to the accuracy of such data to the government each year. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Additionally, entities that do not comply with mandatory reporting requirements may be subject to a corporate integrity agreement. Certain states also mandate implementation of commercial compliance programs, impose restrictions on covered manufacturers’ marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare professionals.
Innovate may be subject to data privacy and security regulation by both the federal government and the states in which it conducts its business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations impose specified requirements on certain health care providers, plans and clearinghouses (collectively, “covered entities”) and their “business associates,” relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain states have their own laws that govern the privacy and security of health information in certain circumstances, many of which differ from each other and/or HIPAA in significant ways and may not have the same effect, thus complicating compliance efforts.
If Innovate’s operations are found to be in violation of any of such laws or any other governmental regulations that apply to them, Innovate may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, disgorgement, the curtailment or restructuring of its operations, exclusion from participation in federal and state healthcare programs, imprisonment, contractual damages, reputational harm, and diminished profits and future earnings, any of which could adversely affect its ability to operate its business and its financial results.
In addition to the foregoing health care laws, Innovate is also subject to the U.S. Foreign Corrupt Practices Act, or FCPA, and similar worldwide anti-bribery laws, which generally prohibit companies and their intermediaries from making improper payments to government officials or private-sector recipients for the purpose of obtaining or retaining business. Innovate has plans to adopt an anti-corruption policy, which will become effective upon the completion of this transaction, and expects to prepare and implement procedures to ensure compliance with such policy. The anti-corruption policy mandates compliance with the FCPA and similar anti-bribery laws applicable to its business throughout the world. However, Innovate cannot assure you that such a policy or procedures implemented to enforce such a policy will protect them from intentional, reckless or negligent acts committed by its employees, distributors, partners, collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on its business, results of operations and reputation.
| 40 |
Coverage and Reimbursement
Sales of pharmaceutical products depend significantly on the extent to which coverage and adequate reimbursement are provided by third-party payers. Third-party payers include state and federal government health care programs, managed care providers, private health insurers and other organizations. Although Innovate currently believes that third-party payers will provide coverage and reimbursement for its product candidates, if approved, Innovate cannot be certain of this. Third-party payers are increasingly challenging the price, examining the cost-effectiveness, and reducing reimbursement for medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. The U.S. government, state legislatures and foreign governments have continued implementing cost containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit Innovate’s net revenue and results. Innovate may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of its products. The product candidates that Innovate develops may not be considered cost-effective and thus may not be covered or sufficiently reimbursed. It is time consuming and expensive for them to seek coverage and reimbursement from third-party payers, as each payer will make its own determination as to whether to cover a product and at what level of reimbursement. Thus, one payer’s decision to provide coverage and adequate reimbursement for a product does not assure that another payer will provide coverage or that the reimbursement levels will be adequate. Moreover, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Reimbursement may not be available or sufficient to allow them to sell its products on a competitive and profitable basis.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could materially affect Innovate’s ability to sell its products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
By way of example, in 2010 the Affordable Care Act was signed into law, intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the Affordable Care Act of importance to Innovate’s potential drug candidates are:
| • | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively; |
| • | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| 41 |
| • | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include, among others, the Budget Control Act of 2011, which mandates aggregate reductions to Medicare payments to providers of up to 2% per fiscal year effective in 2013, and, due to subsequent legislative amendments, will remain in effect through 2024, unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for Innovate’s product candidates, if approved, and, accordingly, its financial operations.
Innovate expects that healthcare reform measures that may be adopted in the future, including the possible repeal and replacement of the Affordable Care Act which the Trump administration has stated is a priority, are unpredictable, and the potential impact on Innovate’s operations and financial position are uncertain, but may result in more rigorous coverage criteria and lower reimbursement, and place additional downward pressure on the price that it receives for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent Innovate from being able to generate revenue, attain profitability or commercialize their drugs.
Foreign Regulation
In addition to regulations in the United States, Innovate will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of its products to the extent Innovate chooses to develop or sell any products outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Innovate’s Employees
As of December 31, 2017, Innovate had 4 full-time employees. Innovate also engages consultants to provide services to Innovate, including clinical development, manufacturing support, regulatory support, business development, and general business operational support.
Facilities
Innovate’s main office is based in Raleigh, North Carolina, where the company leases approximately 2480 square feet of office space. The lease expires on September 30, 2020.
Innovate believes that its existing facilities are adequate for its near-term needs. Innovate believes that suitable alternative space would be available if required in the future on commercially reasonable terms.
Legal Matters
Innovate is not currently a party to any legal or governmental regulatory proceedings, nor is Innovate’s management currently aware of any pending or threatened legal or governmental regulatory proceedings proposed to be initiated against Innovate that would have a material adverse effect on its business, financial condition or operating results. Innovate’s industry is characterized by frequent claims and litigation including securities litigation, claims regarding patent and other intellectual property rights and claims for product liability. As a result, in the future, Innovate may be involved in various legal proceedings from time to time.
| 42 |
Risks Related to Innovate’s Capital Requirements and Financial Condition
Innovate has a limited operating history and has incurred significant losses since inception, and expects that it will continue to incur losses for the foreseeable future, which makes it difficult to assess Innovate’s future viability.
Innovate is a clinical development-stage biopharmaceutical company with a limited operating history upon which to evaluate its business and prospects. Innovate has not been profitable since it commenced operations in 2012, and may never achieve or sustain profitability. In addition, Innovate has limited history as an organization and has not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical industry. Drug development is a highly speculative undertaking and involves a substantial degree of risk. Innovate has not yet obtained any regulatory approvals for any of its product candidates, commercialized any of its product candidates, or generated any revenue from sales of products. Innovate has devoted significant resources to research and development and other expenses related to its ongoing clinical trials and operations, in addition to acquiring product candidates.
Since inception, most of Innovate’s resources have been dedicated to the acquisition and development of its product candidates, INN-202 (larazotide acetate), INN-108 and INN-329 (secretin). Innovate will require significant additional capital to continue operations and to execute on its current business strategy to develop INN-202 through to regulatory approval and further develop INN-108 and INN-329 for eventually seeking regulatory approval. Innovate cannot estimate with reasonable certainty the actual amounts necessary to successfully complete the development and commercialization of its product candidates and there is no certainty that Innovate will be able to raise the necessary capital on reasonable terms or at all.
Innovate’s auditor has expressed substantial doubt about its ability to continue as a going concern.
The audit report on Innovate’s financial statements for the years ended December 31, 2016 and 2015, includes an explanatory paragraph related to Innovate’s recurring losses from operations and dependence on additional financing to continue as a going concern. Innovate has incurred net losses for the years ended December 31, 2016 and 2015, and had an accumulated deficit of $7.7 million as of December 31, 2016. In view of these matters, Innovate’s ability to continue as a going concern is dependent upon its ability to raise additional debt or equity financing or enter into strategic partnerships. Since its inception, Innovate has financed its operations through convertible debt financings. Innovate intends to continue to finance its operations through debt or equity financing and/or strategic partnerships. The failure to obtain sufficient financing or strategic partnerships could adversely affect Innovate’s ability to achieve its business objectives and continue as a going concern.
Innovate will require substantial additional financing to obtain regulatory approval for INN-202 for celiac disease, and for further development of INN-108 (for ulcerative colitis) and INN-329 (for magnetic resonance cholangiopancreatography or MRCP), and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force Innovate to delay, limit, reduce or terminate Innovate’s product development efforts or other operations.
For the year ended December 31, 2016, and the nine months ended September 30, 2017, Innovate incurred losses from operations of $5.4 million and $8.9 million, respectively, and net cash used in operating activities was $2.2 million and $3.2 million, respectively. At September 30, 2017, Innovate had an accumulated deficit of $17 million, its cash, cash equivalents and investment securities were $1.5 million, and its working capital deficit was $11.1 million. Innovate expects to continue to incur substantial operating losses for the next several years as it advances its product candidates through clinical development, US and other regional regulatory approvals, and commercialization. No revenue from operations will likely be available until, and unless, one of its product candidates is approved by the FDA or another regulatory agency and successfully marketed, or Innovate enters into an arrangement that provides for licensing revenue or other partnering-related funding, outcomes which Innovate may not achieve on a timely basis, or at all.
| 43 |
Innovate’s capital requirements for the foreseeable future will depend in large part on, and could increase significantly as a result of, its expenditures on its development programs. Future expenditures on its development programs are subject to many uncertainties, and will depend on, and could increase significantly as a result of, many factors, including:
| · | the number, size, complexity, results and timing of its drug development programs; |
| · | the number of clinical and nonclinical studies necessary to demonstrate acceptable evidence of the safety and efficacy of its product candidates; |
| · | the terms of any collaborative or other strategic arrangement that Innovate may establish; |
| · | changes in standards of care which could increase the size and complexity of clinical studies; |
| · | the ability to locate patients to participate in a study given the limited number of patients available for orphan or ultra-orphan indications; |
| · | the number of patients who participate, the rate of enrollment, and the ratio of randomized to evaluable patients in each clinical study; |
| · | the number and location of sites and the rate of site initiation in each study; |
| · | the duration of patient treatment and follow-up; |
| · | the potential for additional safety monitoring or other post-marketing studies that may be requested by regulatory agencies; |
| · | the time and cost to manufacture clinical trial material and commercial product, including process development and scale-up activities, and to conduct stability studies, which can last several years; |
| · | the degree of difficulty and cost involved in securing alternate manufacturers or suppliers of drug product, components or delivery devices, as necessary to meet FDA requirements and/or commercial demand; |
| · | the costs, requirements, timing of, and the ability to, secure regulatory approvals; |
| · | the extent to which Innovate increases its workforce and the costs involved in recruiting, training and incentivizing new employees; |
| · | the costs related to developing, acquiring and/or contracting for sales, marketing and distribution capabilities, supply chain management capabilities, and regulatory compliance capabilities, if Innovate obtains regulatory approval for a product candidate and commercializes it without a partner; |
| 44 |
| · | the costs involved in evaluating competing technologies and market developments or the loss in sales in case of such competition; and |
| · | the costs involved in establishing, enforcing or defending patent claims and other proprietary rights. |
Additional capital may not be available when Innovate needs it, on terms that are acceptable to it or at all. If adequate funds are not available to Innovate on a timely basis, it will be required to delay, limit, reduce or terminate manufacturing, development activities or other activities that may be necessary to commercialize its product candidates, conduct preclinical or clinical studies, distribution capabilities, its establishment of sales and marketing, or other development activities.
If Innovate raises additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, it may have to relinquish certain valuable rights to its product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable. If Innovate raises additional capital through public or private equity offerings, the ownership interest of its stockholders will be diluted and the terms of any new equity securities may have preferential rights over its common stock. If Innovate raises additional capital through debt financing, it may be subject to covenants limiting or restricting its ability to take specific actions, such as incurring additional debt or making capital expenditures, or subject to specified financial ratios, any of which could restrict its ability to develop and commercialize its product candidates or operate as a business.
Innovate has not generated any revenue from product sales and may never be profitable.
Innovate has no products approved for commercialization and has never generated any revenue from product sales. Innovate’s ability to generate revenue and achieve profitability depends on its ability, alone or with strategic collaboration partners, to successfully complete the development of, and obtain the requisite regulatory approvals necessary to commercialize, one or more of its product candidates.
The recently passed comprehensive tax reform bill could adversely affect Innovate’s business and financial condition.
On December 22, 2017, President Trump signed into law new legislation that significantly revises the Code. The newly enacted federal income tax law, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses), limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, elimination of U.S. tax on foreign earnings (subject to certain important exceptions), immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new federal tax law is uncertain and Innovate’s business and financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the newly enacted federal tax law. The impact of this tax reform on holders of Innovate’s common stock is also uncertain and could be adverse. We urge our stockholders to consult with their legal and tax advisors with respect to this legislation and the potential tax consequences of investing in or holding our common stock.
Risks Related to Innovate’s Business Strategy and Operations
Innovate does not have any products that are approved for commercial sale.
Innovate currently does not have any therapeutic products approved for commercial sale. Innovate has not received, and may not receive within the next several years, if at all, any revenues from the commercialization of its product candidates if approved.
| 45 |
Innovate is substantially dependent upon the clinical, regulatory and commercial success of its three product candidates, INN-202, INN-108 and INN-329. Clinical drug development involves a lengthy and expensive process with an uncertain outcome, results of earlier studies and trials may not be predictive of future trial results, and Innovate’s clinical trials may fail to adequately demonstrate to the satisfaction of regulatory authorities the safety and efficacy of its three product candidates.
The success of Innovate’s business is dependent on its ability to advance the clinical development of INN-202 for the treatment of celiac disease, INN-108 for the treatment of mild to moderate ulcerative colitis, and INN-329 for MRCP. INN-202 has had successful completion of Phase 2 trials and Phase 3 pivotal studies and long-term safety studies remain to be conducted. INN-108 will be entering into Phase 2 efficacy trials for mild to moderate ulcerative colitis. INN-329 requires some additional studies to be performed for completion of Phase 3 trials.
Clinical testing is expensive and can take many years to complete. The outcome of this testing is inherently uncertain. A failure of one or more of Innovate’s clinical trials can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of Innovate’s product candidates may not necessarily be predictive of the results of later-stage clinical trials. There is a high failure rate for drugs proceeding through clinical trials, and product candidates in later stages of clinical trials may fail to show the required safety and efficacy despite having progressed through preclinical studies and initial clinical trials. Many companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier clinical trials, and Innovate cannot be certain that it will not face similar setbacks. Even if Innovate’s clinical trials are completed, the results may not be sufficient to obtain regulatory approval for its product candidates.
Because of the developmental nature of Innovate’s product candidates, Innovate is subject to risks associated with initiating, completing and achieving positive outcomes from its current and future clinical trials, including:
| · | inability to enroll enough patients in the clinical trials; |
| · | slow implementation, enrollment and completion of the clinical trials; |
| · | low patient compliance and adherence to dosing and reporting requirements, such as incomplete reporting of patient reported outcomes in the clinical trials or missed doses; |
| · | lack of safety and efficacy in the clinical trials; |
| · | delays in the manufacture of supplies for drug components due to delays in formulation, process development, or manufacturing activities; |
| · | requirements for additional nonclinical or clinical studies based on changes to formulation and/or changes to regulatory requirements; |
| · | requirements for additional clinical studies based on inconclusive clinical results or changes in market, standard of care, and/or regulatory requirements; |
| 46 |
If Innovate successfully completes the necessary clinical trials for its product candidates, its success will be subject to the risks associated with obtaining regulatory approvals, product launch, and commercialization, including:
| · | delays during regulatory review and/or requirements for additional CMC, nonclinical, or clinical studies, resulting in increased costs and/or delays in marketing approval and subsequent commercialization of the product candidates in the United States and other markets; |
| · | FDA rejection of Innovate’s New Drug Application (“NDA”) submissions for its product candidates; |
| · | regulatory rejection in the EU, Japan, and other markets; |
| · | inability to consistently manufacture commercial supplies of drug and delivery devices resulting in slowed market development and lower revenue; |
| · | poor commercial sales due to: |
| o | the ability of Innovate’s future sales organization or its potential commercialization partners to effectively sell the product candidates; |
| o | Innovate’s lack of success in educating physicians and patients about the benefits, administration, and use of its product candidates; |
| o | low patient demand for the product candidates; |
| o | the availability, perceived advantages, relative cost, relative safety and relative efficacy of other products or treatments for the targeted indications of the product candidates; |
| o | poor prescription coverage and inadequate reimbursement for its product candidates; |
| · | Innovate’s inability to enforce its intellectual property rights in and to its product candidates; and |
| · | reduction in the safety profile of its product candidates following approval. |
Many of these clinical, regulatory and commercial matters are beyond Innovate’s control and are subject to other risks described elsewhere in this “Risk Factors” annex. Accordingly, Innovate cannot assure that it will be able to advance its product candidates further through final clinical development, or obtain regulatory approval of, commercialize or generate significant revenue from them. If Innovate cannot do so, or is significantly delayed in doing so, its business will be materially harmed.
If Innovate fails to attract and retain senior management and key scientific personnel, it may be unable to successfully develop and commercialize its product candidates.
Innovate has historically operated with a limited number of employees. As of December 31, 2017, Innovate had four full-time employees, including one employee engaged part-time in research and development. Therefore, institutional knowledge is concentrated within a small number of employees. Innovate’s success depends in part on its continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. Innovate’s future success is highly dependent upon the contributions of its senior management team. The loss of services of any of these individuals could delay or prevent the successful development of its product pipeline, completion of its planned clinical trials or the commercialization of its product candidates.
| 47 |
There may be intense competition from other companies and organizations for qualified personnel. Other companies and organizations with which Innovate competes for personnel may have greater financial and other resources and different risk profiles than Innovate, and a history of successful development and commercialization of its product candidates. Replacing key employees may be difficult and costly; and Innovate may not have other personnel with the capacity to assume all the responsibilities of a key employee upon his/her departure. If Innovate cannot attract and retain skilled personnel, as needed, Innovate may not achieve its development and other goals.
In addition, the success of Innovate’s business will depend on its ability to develop and maintain relationships with respected service providers and industry-leading consultants and advisers. If Innovate cannot develop and maintain such relationships, as needed, the rate and success at which Innovate can develop and commercialize product candidates may be limited. In addition, its outsourcing strategy, which has included engaging consultants to manage key functional areas, may subject Innovate to scrutiny under labor laws and regulations, which may divert management time and attention and have an adverse effect on its business and financial condition.
Innovate has identified a material weakness in its internal control over financial reporting and may identify additional material weaknesses in the future or otherwise fail to maintain an effective system of internal control, which may impair its ability to produce accurate financial statements or prevent fraud.
Currently, Innovate has limited resources to address its internal controls and procedures and relies on part-time consultants to assist Innovate with its financial accounting and compliance obligations. In connection with the preparation of Innovate’s audited financial statements for the year ended December 31, 2016, Innovate’s independent auditors advised management that a material weakness existed in internal controls over financial reporting due to Innovate’s inability to adequately segregate duties as a result of Innovate’s limited number of accounting personnel. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the subject company’s annual or interim financial statements will not be prevented or detected on a timely basis. Although Innovate is committed to continuing to improve its internal control processes and intends to implement a plan to remediate this material weakness, Innovate cannot be certain of the effectiveness of such plan or that, in the future, additional material weaknesses or significant deficiencies will not exist or otherwise be discovered. If Innovate is unable to maintain proper and effective internal controls, it may not be able to produce timely and accurate financial statements and prevent fraud. In addition, if Innovate is unable to successfully remediate the material weaknesses in our internal controls or if Innovate is unable to produce accurate and timely financial statements, Innovate’s stock price may be adversely affected and Innovate may be unable to maintain compliance with applicable stock exchange listing requirements.
Innovate’s employees, independent contractors and consultants, principal investigators, CROs, CMOs and other vendors, and any future commercial partners may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for Innovate and harm its reputation.
Innovate is exposed to the risk that its employees, independent contractors and consultants, principal investigators, clinical research organizations (CROs), contract manufacturing organizations (CMOs) and other vendors, and any future commercial partners may engage in fraudulent conduct or other misconduct. This type of misconduct may include intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, to provide accurate information to the FDA or comparable foreign regulatory authorities, to comply with manufacturing standards required by cGMP or Innovate standards, to comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, and to report financial information or data accurately or disclose unauthorized activities to them. The misconduct of its employees and other Innovate service providers could involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to its reputation. Innovate intends to adopt a code of business ethics and conduct, but it is not always possible to identify and deter such misconduct, and the precautions Innovate takes to detect and prevent this activity, such as the implementation of a quality system which entails vendor audits by quality experts, may not be effective in controlling unknown or unmanaged risks or losses or in protecting Innovate from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against them, and Innovate is not successful in defending itself or asserting its rights, those actions could have a significant impact on its business and results of operations, including the imposition of significant fines or other sanctions.
| 48 |
Innovate does not have, and does not have plans to establish manufacturing facilities. Innovate completely relies on third parties for the manufacture and supply of its clinical trial drug and delivery device supplies and, if approved, commercial product materials. The loss of any of these vendors or a vendor’s failure to provide Innovate with an adequate supply of clinical trial or commercial product material in a timely manner and on commercially acceptable terms, or at all, could harm its business.
Innovate outsources the manufacture of its product candidates and does not plan to establish its own manufacturing facilities. To manufacture Innovate’s product candidates, Innovate has made numerous custom modifications at CMOs, making Innovate highly dependent on these CMOs. For clinical and commercial supplies, if approved, Innovate has supply agreements with third party CMOs for drug substance and finished drug product. While Innovate has secured long-term commercial supply agreements with many of the third party CMOs, Innovate would need to negotiate agreements for commercial supply with several important CMOs, and Innovate may not be able to reach agreement on acceptable terms. In addition, Innovate relies on these third parties to conduct or assist Innovate in key manufacturing development activities, including qualification of equipment, developing and validating methods, defining critical process parameters, releasing component materials and conducting stability testing, among other things. If these third parties are unable to perform their tasks successfully in a timely manner, whether for technical, financial or other reasons, Innovate may be unable to secure clinical trial material, or commercial supply material if approved, which likely would delay the initiation, conduct or completion of its clinical studies or prevent Innovate from having enough commercial supply material for sale, which would have a material and adverse effect on its business.
Currently, Innovate does not have alternative vendors to back up its primary vendors of clinical trial material or, if approved, commercial supply material. Identification of and discussions with other vendors may be protracted and/or unsuccessful, or these new vendors may be unsuccessful in producing the same results as the current primary vendors producing the material. Therefore, if its primary vendors become unable or unwilling to perform their required activities, Innovate could experience protracted delays or interruptions in the supply of clinical trial material and, ultimately, product for commercial sale, which would materially and adversely affect its development programs, commercial activities, operating results and financial condition. In addition, the FDA or regulatory authorities outside of the United States may require Innovate to have an alternate manufacturer of a drug product before approving it for marketing and sale in the United States or abroad and securing such alternate manufacturer before approval of an NDA could result in considerable additional time and cost prior to NDA approval.
Any new manufacturer or supplier of finished drug product or its component materials, including drug substance and delivery devices, would be required to qualify under applicable regulatory requirements and would need to have sufficient rights under applicable intellectual property laws to the method of manufacturing of such product or ingredients required by Innovate. The FDA or foreign regulatory agency may require Innovate to conduct additional clinical studies, collect stability data and provide additional information concerning any new supplier, or change in a validated manufacturing process, including scaling-up production, before Innovate could distribute products from that manufacturer or supplier or revised process. For example, if Innovate were to engage a third party other than its current CMOs to supply the drug substance or drug product for future clinical trial, or commercial product, the FDA or regulatory authorities outside of the United States may require Innovate to conduct additional clinical and nonclinical studies to ensure comparability of the drug substance or drug product manufactured by its current CMOs to that manufactured by the new supplier.
| 49 |
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling-up initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, and shortages of qualified personnel. Innovate’s product candidates have not been manufactured at the scale Innovate believes will be necessary to maximize its commercial value and, accordingly, Innovate may encounter difficulties in attempting to scale-up production and may not succeed in that effort on a timely basis or at all. In addition, the FDA or other regulatory authorities may impose additional requirements as Innovate scales-up initial production capabilities, which may delay its scale-up activities and/or add expense.
All manufacturers of Innovate’s clinical trial material and, if approved, commercial product, including drug substance manufacturers, must comply with cGMP requirements enforced by the FDA through its facilities inspection program and applicable requirements of foreign regulatory authorities. These requirements include quality control, quality assurance and the maintenance of records and documentation. Manufacturers of Innovate’s clinical trial material may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. While Innovate or its representatives generally monitor and audit its manufacturers’ systems, Innovate does not have full control over their ongoing compliance with these regulations. And while the responsibility to maintain cGMP compliance is shared between Innovate and the third-party manufacturer, Innovate bears ultimate responsibility for its supply chain and compliance with regulatory standards. Failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay or failure to obtain product approval, product seizure or recall, or withdrawal of product approval.
If Innovate’s manufacturers encounter any of the aforementioned difficulties or otherwise fail to comply with their contractual obligations or there are delays entering commercial supply agreements due to capital constraints, Innovate may have insufficient quantities of material to support ongoing and/or planned clinical studies or to meet commercial demand, if approved. In addition, any delay or interruption in the supply of materials necessary or useful to manufacture its product candidates could delay the completion of its clinical studies, increase the costs associated with its development programs and, depending upon the period of delay, require Innovate to commence new clinical studies at significant additional expense or terminate the studies completely. Delays or interruptions in the supply of commercial product could result in increased cost of goods sold and lost sales. Innovate cannot provide assurance that manufacturing or quality control problems will not arise in connection with the manufacture of its clinical trial material or commercial product, if approved, or that third-party manufacturers will be able to maintain the necessary governmental licenses and approvals to continue manufacturing such clinical trial material or commercial product, as applicable. In addition, if Innovate products are manufactured entirely or partially outside the United States, Innovate may experience interruptions in supply due to shipping or customs difficulties or regional instability. Furthermore, changes in currency fluctuations, shipping costs, or import tariffs could adversely affect cost of goods sold. Any of the above factors could cause Innovate to delay or suspend anticipated or ongoing trials, regulatory submissions or commercialization of its product candidates, entail higher costs or result in Innovate being unable to effectively commercialize its products. Innovate’s dependence upon third parties for the manufacture of its clinical trial material may adversely affect its future costs and its ability to develop and commercialize its product candidates on a timely and competitive basis.
| 50 |
Innovate currently relies significantly on third parties to conduct its nonclinical testing and clinical studies and other aspects of its development programs and if those third parties do not satisfactorily perform their contractual obligations or meet anticipated deadlines, the development of its product candidates could be adversely affected.
Innovate does not currently employ personnel or possess the facilities necessary to conduct many of the activities associated with its programs. Innovate engages consultants, advisors, clinical research organizations (CROs), and others to assist in the design and conduct of nonclinical and clinical studies of its product candidates, with interpretation of the results of those studies and with regulatory activities, and Innovate expects to continue to outsource all or a significant amount of such activities. As a result, many important aspects of its development programs are and will continue to be outside its direct control, and its third-party service providers may not perform their activities as required or expected including the maintenance of GCP, GLP and GMP compliance, which are ultimately Innovate’s responsibility to ensure. Further, such third parties may not be as committed to the success of Innovate’s programs as Innovate’s own employees and, therefore, may not devote the same time, thoughtfulness or creativity to completing projects or problem-solving as Innovate’s own employees would. To the extent Innovate is unable to successfully manage the performance of third-party service providers, its business may be adversely affected.
The CROs that Innovate may engage to execute its clinical studies play a significant role in the conduct of the studies, including the collection and analysis of study data, and Innovate likely will depend on CROs and clinical investigators to conduct future clinical studies and to assist in analyzing data from completed studies and developing regulatory strategies for its product candidates. Individuals working at the CROs with which it will contract, as well as investigators at the sites at which its studies are conducted, are not Innovate’s employees, and Innovate has limited control over the amount or timing of resources that they devote to their programs. If Innovate’s CROs, study investigators, and/or third-party sponsors fail to devote sufficient time and resources to studies of its product candidates, if Innovate and/or its CROs do not comply with all GLP and GCP regulatory and contractual requirements, or if their performance is substandard, it may delay commencement and/or completion of these studies, submission of applications for regulatory approval, regulatory approval, and commercialization of its product candidates. Failure of CROs to meet their obligations to Innovate could adversely affect development of its product candidates.
In addition, CROs Innovate engages may have relationships with other commercial entities, some of which may compete with Innovate. Through intentional or unintentional means, Innovate’s competitors may benefit from lessons learned on the Innovate project that could ultimately harm Innovate’s competitive position. Moreover, if a CRO fails to properly, or at all, perform its activities during a clinical study, Innovate may not be able to enter into arrangements with alternative CROs on acceptable terms or in a timely manner, or at all. Switching CROs may increase costs and divert management time and attention. In addition, there likely would be a transition period before a new CRO commences work. These challenges could result in delays in the commencement or completion of Innovate’s clinical studies, which could materially impact its ability to meet its desired and/or announced development timelines and have a material adverse impact on its business and financial condition.
| 51 |
Innovate may not achieve its projected development goals within the time frames that Innovate has announced.
Innovate has set goals for accomplishing certain objectives material to the successful development of its product candidates. The actual timing of these events may vary due to many factors, including delays or failures in its nonclinical testing, clinical studies and manufacturing and regulatory activities and the uncertainties inherent in the regulatory approval process. From time to time, Innovate creates estimates for the completion of enrollment of or announcement of data from clinical studies of its product candidates. However, predicting the rate of enrollment or the time from completion of enrollment to announcement of data for any clinical study requires Innovate to make significant assumptions that may prove to be incorrect. As discussed in other risk factors above, its estimated enrollment rates and the actual rates may differ materially and the time required to complete enrollment of any clinical study may be considerably longer than Innovate estimates. Such delays may adversely affect its financial condition and results of operations.
Even if Innovate completes a clinical study with successful results, Innovate may not achieve its projected development goals within the periods Innovate initially anticipates or announces. If a development plan for a product candidate becomes more extensive and costly than anticipated, Innovate may determine that the associated time and cost are not financially justifiable and, as a result, may discontinue development in a particular indication or of the product candidate as a whole. In addition, even if a study did complete with successful results, changes may occur in regulatory requirements or policy during the period of product development and/or regulatory review of an NDA that relate to the data required to be included in NDAs which may require additional studies that may be costly and time consuming. Any of these actions may be viewed negatively, which could adversely impact its financial condition.
Further, throughout development, Innovate must provide adequate assurance to the FDA and other regulatory authorities that Innovate can consistently develop and produce its product candidates in conformance with GLP, GCP, cGMP, and other regulatory standards. As discussed above, Innovate relies on CMOs for the manufacture of clinical, and future commercial, quantities of its product candidates. If future FDA or other regulatory authority inspections identify cGMP compliance deficiencies at these third-party facilities, production of its clinical trial material or, in the future, commercial product, could be disrupted, causing potentially substantial delay in or failure of development or commercialization of its product candidates.
Innovate currently has limited marketing capabilities and no sales organization. If Innovate is unable to establish sales and marketing capabilities on its own or through third parties, it will be unable to successfully commercialize its products, if approved, or generate product revenue.
To commercialize Innovate’s products, if approved, in the United States and other jurisdictions it seeks to enter, Innovate must build its marketing, sales, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and it may not be successful in doing so. If Innovate’s products receive regulatory approval, it expects to market such products in the United States through a focused, specialized sales force, which will be costly and time consuming. Innovate has no prior experience in the marketing and sale of pharmaceutical products and there are significant risks involved in building and managing a sales organization, including its ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively manage a geographically dispersed sales and marketing team. Outside of the United States, Innovate may consider collaboration arrangements. If Innovate is unable to enter into such arrangements on acceptable terms or at all, it may not be able to successfully commercialize its products in certain markets. Any failure or delay in the development of its internal sales, marketing and distribution capabilities would adversely impact the commercialization of its products. If Innovate is not successful in commercializing its products, either on its own or through collaborations with one or more third parties, its future product revenue will suffer and it would incur significant additional losses.
| 52 |
To establish a sales and marketing infrastructure and expand its manufacturing capabilities, Innovate will need to increase the size of its organization, and Innovate may experience difficulties in managing this growth.
As of December 31, 2017, Innovate had four full-time employees, including one employee engaged part-time in research and development. As Innovate advances its product candidates through the development process and to commercialization, it will need to continue to expand its development, regulatory, quality, managerial, sales and marketing, operational, finance and other resources to manage its operations and clinical trials, continue its development activities and commercialize its product candidates, if approved. As its operations expand, Innovate expects that it will need to manage additional relationships with various manufacturers and collaborative partners, suppliers and other organizations.
Due to Innovate’s limited financial resources and its limited experience in managing a company with such anticipated growth, Innovate may not be able to effectively manage the expansion of its operations or recruit and train additional qualified personnel. In addition, the physical expansion of its operations may lead to significant costs and may divert its management and resources. Any inability to manage growth could delay the execution of its development and strategic objectives, or disrupt its operations, which could materially impact its business, revenue and operating results.
Innovate’s product candidates may cause undesirable side effects or adverse events, or have other properties that could delay or prevent its clinical development, regulatory approval or commercialization.
As with many pharmaceutical products, undesirable side effects or adverse events caused by Innovate’s product candidates could interrupt, delay or halt clinical studies and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all indications, and in turn prevent Innovate from commercializing its product candidates. A significant challenge in clinical development is that the patient population in early studies, where small numbers of patients are required, is different from the patient population observed in later stage studies, where larger groups of patients are required. For example, patients in earlier stage studies may be sicker, more compliant, or otherwise motivated than patients in larger studies.
If undesirable side effects occur, they could possibly prevent approval, which would have a material and adverse effect on its business.
If any of its product candidates receive marketing approval and Innovate or others later identify undesirable side effects caused by the product:
| · | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| · | Innovate may be required to change the way the product is administered, conduct additional clinical studies or change the labeling of the product; and |
| · | regulatory authorities may withdraw approval of the product; |
| · | its reputation may suffer. |
| 53 |
Any of these events could prevent Innovate from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent Innovate from generating significant revenue from its sale.
Innovate’s business and operations would suffer in the event of third-party computer system failures, cyber-attacks on third-party systems or deficiency in its cyber security.
Innovate relies on information technology systems, including third-party “cloud based” service providers, to keep financial records, maintain laboratory data, clinical data and corporate records, to communicate with staff and external parties, and to operate other critical functions. This includes critical systems such as email, other communication tools, electronic document repositories, and archives. If any of these third-party information technology (IT) providers are compromised due to computer viruses, unauthorized access, malware, natural disasters, fire, terrorism, war and telecommunication failures, electrical failures, cyber-attacks or cyber-intrusions over the internet, then sensitive emails or documents could be exposed or deleted. Similarly, Innovate could incur business disruption if its access to the internet is compromised and Innovate is unable to connect with third-party IT providers. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. In addition, Innovate relies on those third parties to safeguard important confidential personal data regarding its employees and patients enrolled in its clinical trials. If a disruption event were to occur and cause interruptions in a third-party IT provider’s operations, it could result in a disruption of its drug development programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in its regulatory approval efforts and significantly increase its costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to its data or applications, or inappropriate disclosure of confidential or proprietary information, Innovate could incur liability and development of its product candidates could be delayed, or could fail.
Risks Related to Drug Development and Commercialization
Innovate depends on the successful completion of clinical studies of its product candidates, and any positive results in prior clinical studies do not ensure that ongoing or future clinical studies will be successful.
Pharmaceutical products are subject to stringent regulatory requirements covering quality, safety, and efficacy. The burden of proof is on the manufacturer, such as Innovate, to show with substantial clinical data that the risk/benefit profile for any new drug is favorable. Only after successfully completing extensive pharmaceutical development, nonclinical testing, and clinical studies may a product be considered for regulatory approval.
If Innovate licenses rights to develop its product candidates to independent third parties or otherwise permit such third parties to evaluate its product candidates in clinical studies, Innovate may have limited control over those clinical studies. Any safety or efficacy concern identified in a third-party sponsored study could adversely affect its or another licensee’s development of its product candidate and prospects for its regulatory approval, even if the data from that study are subject to varying interpretations and analyses.
There is significant risk that ongoing and future clinical studies of its product candidates are unsuccessful. Negative or inconclusive results could cause the FDA and other regulatory authorities to require Innovate to repeat or conduct additional clinical studies, which could significantly increase the time and expense associated with development of that product candidate or cause Innovate to elect to discontinue one or more clinical programs. Failure to complete a clinical study of a product candidate or an unsuccessful result of a clinical study could have a material adverse effect on its business.
| 54 |
Clinical drug development involves a lengthy and expensive process, with an uncertain outcome. Innovate may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of its drug candidates.
Clinical studies are expensive, difficult to design and implement, may take many years to complete, and outcomes are inherently uncertain. A drug product may fail to demonstrate positive results at any stage of testing despite having progressed satisfactorily through nonclinical testing and initial clinical studies. There is significant risk in clinical development where later stage clinical studies are designed and powered based on the analysis of data from earlier studies, with these earlier studies involving a smaller number of patients, and the results of the earlier studies being driven primarily by a subset of responsive patients. In addition, interim results of a clinical study do not necessarily predict final results. Further, clinical study data frequently are susceptible to varying interpretations. Medical professionals and/or regulatory authorities may analyze or weigh study data differently than the sponsor company, resulting in delay or failure to obtain marketing approval for a product candidate. Additionally, the possible lack of standardization across multiple investigative sites may induce variability in the results, which can interfere with the evaluation of treatment effects.
Delays in commencement and completion of clinical studies are common and have many causes. Delays in clinical studies of Innovate’s product candidates could increase overall development costs and jeopardize its ability to obtain regulatory approval and successfully commercialize any approved products.
Clinical studies may not commence on time or be completed on schedule, if at all. The commencement and completion of clinical studies can be delayed for a variety of reasons, including:
| · | inability to raise sufficient funding to initiate or to continue a clinical study; |
| · | delays in obtaining regulatory approval to commence a clinical study; |
| · | delays in identifying and reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical study sites and investigators, which agreements can be subject to extensive negotiation and may vary significantly among study sites; |
| · | delays in obtaining regulatory approval in a prospective country; |
| · | delays in obtaining ethic committee approval to conduct a clinical study at a prospective site; |
| · | delays in reaching agreements on acceptable terms with prospective contract manufacturing organizations, or CMOs, or other vendors for the production and supply of clinical trial material and, if necessary, drug administration devices, which agreements can be subject to extensive negotiation; |
| · | delays in the production or delivery of sufficient quantities of clinical trial material from its CMOs and other vendors to initiate or continue a clinical study; |
| · | delays due to product candidate recalls as a result of stability failure, excessive product complaints or other failures of the product candidate during its use or testing; |
| 55 |
| · | invalidation of clinical data caused by premature unblinding or integrity issues; |
| · | invalidation of clinical data caused by mixing up of the active drug and placebo through randomization or manufacturing errors; |
| · | delays on the part of its CROs, CMOs, and other third-party contractors in developing procedures and protocols or otherwise conducting activities in accordance with applicable policies and procedures and in accordance with agreed upon timelines; |
| · | delays in identifying and hiring or engaging, as applicable, additional employees or consultants to assist in managing clinical study-related activities; |
| · | delays in recruiting and enrolling individuals to participate in a clinical study, which historically can be challenging in orphan diseases; |
| · | delays caused by patients dropping out of a clinical study due to side effects, concurrent disorders, difficulties in adhering to the study protocol, unknown issues related to different patient profiles than in previous studies, or otherwise; |
| · | delays in having patients complete participation in a clinical study, including returning for post-treatment follow-up; |
| · | delays resulting from study sites dropping out of a trial, providing inadequate staff support for the study, problems with shipment of study supplies to clinical sites, or focusing its staff’s efforts on enrolling studies that compete for the same patient population; |
| · | suspension of enrollment at a study site or the imposition of a clinical hold by the FDA or other regulatory authority following an inspection of clinical study operations at study sites or finding of a drug-related serious adverse event; and |
| · | delays in quality control/quality assurance procedures necessary for study database lock and analysis of unblinded data. |
Innovate may experience difficulties in the enrollment of patients in its clinical trials, which may delay or prevent Innovate from obtaining regulatory approval.
Innovate may not be able to continue clinical trials for its product candidates if Innovate is unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States. In particular, because Innovate is focused on diseases in genomically defined patient populations, our ability to enroll eligible patients may be limited or may result in slower enrollment than we anticipate. In addition, some of our competitors have ongoing clinical trials for drug candidates that treat the same indications as our drug candidates, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ drug candidates.
Patient enrollment, a critical component to successful completion of a clinical study, is affected by many factors, including:
| · | the size of the target patient population; |
| · | other ongoing studies competing for the same patient population; |
| 56 |
| · | the eligibility criteria for the clinical trial; |
| · | the design of the clinical study; |
| · | the perceived risks and benefits of the product candidate under study; |
| · | the efforts to facilitate timely enrollment in clinical trials; |
| · | the proximity and availability of clinical trial sites for prospective patients; and |
| · | the ability to monitor patients adequately during and after treatment. |
Clinical studies may not begin on time or be completed in the time frames Innovate anticipates. The length of time necessary to successfully complete clinical studies varies significantly and is difficult to predict accurately. Innovate may make statements regarding anticipated timing for completion of enrollment in and/or availability of results from its clinical studies, but such predictions are subject to a number of significant assumptions and actual timing may differ materially for a variety of reasons, including patient enrollment rates, length of time needed to prepare raw study data for analysis and then to review and analyze it, and other factors described above. If Innovate experiences delays in the completion of a clinical study, if a clinical study is terminated, or if failure to conduct a study in accordance with regulatory requirements or the study’s protocol leads to deficient safety and/or efficacy data, the regulatory approval and/or commercial prospects for its product candidates may be harmed and its ability to generate product revenue will be delayed. In addition, any delays in completing its clinical studies likely will increase its development costs. Further, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical studies may ultimately lead to the denial of regulatory approval of a product candidate. Even if Innovate ultimately commercializes its product candidates, the standard of care may have changed or other therapies for the same indications may have been introduced to the market in the interim and may establish a competitive threat to Innovate or may diminish the need for Innovate’s products.
Clinical studies are very expensive, difficult to design and implement, often take many years to complete, and the outcome is inherently uncertain.
Clinical development of pharmaceutical products for humans is generally very expensive, takes many years to complete and failures can occur at any stage of clinical testing. Innovate estimates that clinical development of its product candidates will take several additional years to complete, but because of the variety of factors that can affect the design, timing, and outcome of clinical studies, Innovate is unable to estimate the exact funds required to complete research and development, to obtain regulatory approval and to commercialize all of its product candidates. Innovate will need significant additional capital to continue to advance its products as per current business plans.
Failure at any stage of clinical testing is not uncommon and Innovate may encounter problems that would require additional, unplanned studies or cause Innovate to abandon a clinical development program.
In addition, a clinical study may be suspended or terminated by Innovate, an IRB, a data safety monitoring board, the FDA or other regulatory authorities due to a number of factors, including:
| · | lack of adequate funding to continue the study; |
| · | failure to conduct the study in accordance with regulatory requirements or the study’s protocol; |
| 57 |
| · | inspection of clinical study operations or sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| · | unforeseen safety issues, including adverse side effects; or |
| · | changes in governmental regulations or administrative actions. |
Changes in governmental regulations and guidance relating to clinical studies may occur and Innovate may need to amend study protocols to reflect these changes, or Innovate may amend study protocols for other reasons. Amendments may require Innovate to resubmit protocols to IRBs for reexamination and approval or renegotiate terms with CROs, study sites and investigators, all of which may adversely impact the costs or timing of or its ability to successfully complete a trial.
There is significant uncertainty regarding the regulatory approval process for any investigational new drug, substantial further testing and validation of its product candidates and related manufacturing processes may be required, and regulatory approval may be conditioned, delayed or denied, any of which could delay or prevent Innovate from successfully marketing its product candidates and substantially harm its business.
Pharmaceutical products generally are subject to rigorous nonclinical testing and clinical studies and other approval procedures mandated by the FDA and foreign regulatory authorities. Various federal and foreign statutes and regulations also govern or materially influence the manufacturing, safety, labeling, storage, record keeping and marketing of pharmaceutical products. The process of obtaining these approvals and the subsequent compliance with appropriate U.S. and foreign statutes and regulations is time-consuming and requires the expenditure of substantial resources.
Innovate is preparing INN-202, larazotide acetate, for Phase 3 clinical trials, the success of which will be needed for FDA approval to market INN-202 in the United States to treat celiac disease in patients with persistent symptoms while adhering to a gluten free diet. While significant communication with the FDA on the Phase 3 study design has occurred, even if the Phase 3 clinical study meets all of its statistical goals and protocol end points, the FDA may not view the results as robust and convincing. They may require additional clinical studies and/or other costly studies, which could require Innovate to expend substantial additional resources and could significantly extend the timeline for clinical development prior to market approval. Additionally, Innovate is required by the FDA to conduct a long-term safety study. The results of this study will not be known until a short time prior to potential submission of an NDA for INN-202. If the safety study cannot be completed for technical or other reasons, or provides results that the FDA determines to be concerning, this may cause a delay or failure in obtaining approval for INN-202.
INN-108 plans to initiate Phase 2 clinical trials for mild-to-moderate ulcerative colitis. Concurrently, Innovate plans to make formulation changes to INN-108 that would simplify the dosing in pediatric patients. While this change is expected by Innovate to reduce studies and/or other documentation requirements, the regulatory agencies may require additional clinical or nonclinical studies prior to approval, even if current clinical studies are deemed successful, which could require Innovate to expend substantial additional resources and significantly extend the timeline for clinical development of INN-108.
| 58 |
Innovate is preparing INN-329, secretin, for additional testing in its Phase 3 clinical trial, the success of which will be needed for FDA approval to market INN-329 in the United States for MRCP procedures. While significant communication with the FDA on the Phase 3 study design has occurred in the past, Innovate will be required to initiate communication with the FDA to finalize the study design and to seek their approval for the additional Phase 3 trial design. Even if the Phase 3 clinical study meets all of its statistical goals and protocol end points, the FDA may not view the results as robust and convincing. The FDA may require additional clinical studies and/or other costly studies, which could require Innovate to expend substantial additional resources and could significantly extend the timeline for clinical development prior to market approval. Additionally, Innovate is required by the FDA to conduct a long term safety study. The results of this study will not be known until a short time prior to potential submission of an NDA for INN-329. If the safety study cannot be completed for technical or other reasons, or provides results that the FDA determines to be concerning, this may cause a delay or failure in obtaining approval for INN-329.
Significant uncertainty exists with respect to the regulatory approval process for any investigational new drug, including INN-202, INN-108 and INN-329. Regardless of any guidance the FDA or foreign regulatory agencies may provide a drug’s sponsor during its development, the FDA or foreign regulatory agencies retain complete discretion in deciding whether to accept an NDA or the equivalent foreign regulatory approval submission for filing or, if accepted, approve an NDA. There are many components to an NDA or marketing authorization application submission in addition to clinical study data. For example, the FDA or foreign regulatory agencies will review the sponsor’s internal systems and processes, as well as those of its CROs, CMOs and other vendors, related to development of its product candidates, including those pertaining to its clinical studies and manufacturing processes. Before accepting an NDA for review or before approving the NDA, the FDA or foreign regulatory agencies may request that Innovate provide additional information that may require significant resources and time to generate and there is no guarantee that its product candidates will be approved for any indication for which Innovate may apply. The FDA or foreign regulatory agencies may choose not to approve an NDA for any of a variety of reasons, including a decision related to the safety or efficacy data, manufacturing controls or systems, or for any other issues that the agency may identify related to the development of its product candidates. Even if one or more Phase 3 clinical studies are successful in providing statistically significant evidence of the efficacy and safety of the investigational drug, the FDA or foreign regulatory agencies may not consider efficacy and safety data from the submitted studies adequate scientific support for a conclusion of effectiveness and/or safety and may require one or more additional Phase 3 or other studies prior to granting marketing approval. If this were to occur, the overall development cost for the product candidate would be substantially greater and its competitors may bring products to market before Innovate, which could impair its ability to generate revenues from the product candidates, or even seek approval, if blocked by a competitor’s Orphan Drug exclusivity, which would have a material adverse effect on Innovate’s business, financial condition and results of operations.
Further, development of Innovate’s product candidates and/or regulatory approval may be delayed for reasons beyond its control. For example, U.S. federal government shut-down or budget sequestration, such as ones that occurred during 2013 and 2018, may result in significant reductions to the FDA’s budget, employees and operations, which may lead to slower response times and longer review periods, potentially affecting Innovate’s ability to progress development of its product candidates or obtain regulatory approval for its product candidates.
| 59 |
Even if the FDA or foreign regulatory agencies grant approvals for Innovate’s product candidates, the conditions or scope of the approval(s) may limit successful commercialization of the product candidates and impair Innovate’s ability to generate substantial sales revenue. The FDA or foreign regulatory agencies may also only grant marketing approval contingent on the performance of costly post-approval nonclinical or clinical studies, or subject to warnings or contraindications that limit commercialization. Additionally, even after granting approval, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for its products will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, and continued compliance with current good manufacturing processes, or cGMP, good clinical practices, international conference on harmonization regulations and good laboratory practices, which are regulations and guidelines that are enforced by the FDA or foreign regulatory agencies for all of its clinical development and for any clinical studies that Innovate conducts post-approval. The FDA or foreign regulatory agencies may decide to withdraw approval, add warnings or narrow the approved indications in the product label, or establish risk management programs that could restrict distribution of its products. These actions could result from, among other things, safety concerns, including unexpected side effects or drug-drug interaction problems, or concerns over misuse of a product. If any of these actions were to occur following approval, Innovate may have to discontinue commercialization of the product, limit its sales and marketing efforts, implement risk minimization procedures, and/or conduct post-approval studies, which in turn could result in significant expense and delay or limit its ability to generate sales revenues.
Regulations may be changed prior to submission of an NDA that require higher hurdles than currently anticipated. These may occur as a result of drug scandals, recalls, or a political environment unrelated to Innovate’s products.
Even if Innovate receives regulatory approval for a product candidate, Innovate may face regulatory difficulties that could materially and adversely affect its business, financial condition and results of operations.
Even if initial regulatory approval is obtained, as a condition to the initial approval the FDA or a foreign regulatory agency may impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or marketing surveillance programs, any of which would limit the commercial potential of the product. Its product candidates also will be subject to ongoing FDA requirements related to the manufacturing processes, labeling, packaging, storage, distribution, advertising, promotion, record-keeping and submission of safety and other post-market information regarding the product. For instance, the FDA may require changes to approved drug labels, require post-approval clinical studies and impose distribution and use restrictions on certain drug products. In addition, approved products, manufacturers and manufacturers’ facilities are subject to continuing regulatory review and periodic inspections. If previously unknown problems with a product are discovered, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, the FDA may impose restrictions on that product or Innovate, including requiring withdrawal of the product from the market. If Innovate or a CMO of Innovate’s fails to comply with applicable regulatory requirements, a regulatory agency may:
| · | issue warning letters or untitled letters; |
| · | impose civil or criminal penalties; |
| · | suspend or terminate any ongoing clinical studies; |
| · | close the facilities of a CMO; |
| · | refuse to approve pending applications or supplements to approved applications; |
| · | suspend or withdraw regulatory approval; |
| · | exclude its product from reimbursement under government healthcare programs, including Medicaid or Medicare; |
| 60 |
| · | impose restrictions or affirmative obligations on Innovate’s or its CMO’s operations, including costly new manufacturing requirements; or |
| · | seize or detain products or require a product recall. |
If any of Innovate’s product candidates for which Innovate receives regulatory approval fails to achieve significant market acceptance among the medical community, patients or third-party payers, the revenue Innovate generates from its sales will be limited and its business may not be profitable.
Innovate’s success will depend in substantial part on the extent to which its product candidates, if approved, are accepted by the medical community and patients and reimbursed by third-party payers, including government payers. Innovate cannot predict with reasonable accuracy whether physicians, patients, healthcare insurers or health maintenance organizations, or the medical community in general, will accept or utilize any of its products, if approved. If its product candidates are approved but do not achieve an adequate level of acceptance by these parties, Innovate may not generate sufficient revenue to become or to remain profitable. In addition, its efforts to educate the medical community and third-party payers regarding benefits of its products may require significant resources and may never be successful.
The degree of market acceptance with respect to each of its approved products, if any, will depend upon a number of factors, including:
| · | the safety and efficacy of its products as demonstrated in clinical studies; |
| · | acceptance in the medical and patient communities of its products as a safe and effective treatment; |
| · | the perceived advantages of its product over alternative treatments, including with respect to the incidence and severity of any adverse side effects and the cost of treatment; |
| · | the indications for which its product is approved; |
| · | claims or other information (including limitations or warnings) in its product’s approved labeling; |
| · | reimbursement and coverage policies of government and other third-party payers; |
| · | smaller than expected market size due to lack of disease awareness of a rare disease, or the patient population with a specific rare disease being smaller than anticipated; |
| · | availability of alternative treatments; |
| · | pricing and cost-effectiveness of its product relative to alternative treatments; |
| · | inappropriate diagnostic efforts due to limited knowledge and/or resources among clinicians; |
| · | the prevalence of off-label substitution of chemically equivalent products or alternative treatments; and |
| · | the resources Innovate devotes to marketing its product and restrictions on promotional claims Innovate can make with respect to the product. |
| 61 |
If Innovate determines that a product candidate may not achieve adequate market acceptance or that the potential market size does not justify additional expenditure on the program, Innovate may reduce its expenditures on the development and/or the process of seeking regulatory approval of the product candidate while Innovate evaluates whether and on what timeline to move the program forward.
Even if Innovate receives regulatory approval to market one or more of its product candidates in the United States, Innovate may never receive approval or commercialize its products outside of the United States, which would limit its ability to realize the full commercial potential of its product candidates.
In order to market products outside of the United States, Innovate must establish and comply with the numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. The time required to obtain approval in other countries generally differs from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States, as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the United States. As described above, such effects include the risks that its product candidates may not be approved for all indications requested, which could limit the uses of its product candidates and have an adverse effect on product sales, and that such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
Conversely, if the product candidates do receive approval outside the US in the future, Innovate may not meet the FDA requirements in the United States for approval.
Innovate must comply with the U.S. Foreign Corrupt Practices Act and similar foreign anti-corruption laws.
The U.S. Foreign Corrupt Practices Act, to which Innovate is subject, prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, to offer to pay or to authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. Other countries, such as the U.K., have similar laws with which Innovate must comply. Innovate faces the risk that an employee or agent could be accused of violating one or more of these laws, particularly in geographies where significant overlap exists between local government and healthcare industries. Such an accusation, even if unwarranted, could prove disruptive to Innovate’s developmental and commercialization efforts.
Risks Related to Innovate’s Intellectual Property
Innovate’s success will depend in part on obtaining and maintaining effective patent and other intellectual property protection for its product candidates and proprietary technology.
Innovate relies on patents and other intellectual property to maintain exclusivity for its product candidates. INN-202 and INN-108 are covered by several issued patents in the U.S. as well as patents outside the U.S., with patent applications pending in several jurisdictions. INN-329 is not protected by patents. Further, the INN-202 primary end point is a proprietary Patient Report Outcome measure (CeD PRO) that is protected by copyright. Intellectual property relating to the INN-202 program is exclusively licensed from Alba Therapeutics Corp. Intellectual property relating to INN-108 program is exclusively licensed from Seachaid Pharmaceuticals Inc. Innovate’s success will depend in part on its ability to:
| 62 |
| · | obtain and maintain patents and other exclusivity with respect to its products; |
| · | prevent third parties from infringing upon its proprietary rights; |
| · | maintain proprietary know-how and trade secrets; |
| · | operate without infringing upon the patents and proprietary rights of others; and |
| · | obtain and maintain appropriate licenses to patents or proprietary rights held by third parties if infringement would otherwise occur or if necessary to secure exclusive rights to them, both in the United States and in foreign countries. |
The patent and intellectual property positions of biopharmaceutical companies generally are highly uncertain, involve complex legal and factual questions, and have been and continue to be the subject of much litigation. There is no guarantee that Innovate has or will develop or obtain the rights to products or processes that are patentable, that patents will issue from any pending applications or that claims issued will be sufficient to protect the technology Innovate develops or has developed or that is used by Innovate, its CMOs or its other service providers. In addition, any patents that are issued and/or licensed to Innovate may be limited in scope or challenged, invalidated, infringed or circumvented, including by its competitors, and any rights Innovate has under issued and/or licensed patents may not provide competitive advantages to Innovate. If competitors can develop and commercialize technology and products similar to Innovate’s, its ability to successfully commercialize its technology and products may be impaired.
Patent applications in the United States are confidential for a period of time until they are published, and publication of discoveries in scientific or patent literature typically lags actual discoveries by several months. As a result, Innovate cannot be certain that the inventors listed in any patent or patent application owned or licensed by Innovate were the first to conceive of the inventions covered by such patents and patent applications (for U.S. patent applications filed before March 16, 2013), or that such inventors were the first to file patent applications for such inventions outside the United States and, after March 15, 2013, in the United States. In addition, changes in or different interpretations of patent laws in the United States and foreign countries may affect Innovate’s patent rights and limit the patents Innovate can obtain, which could permit others to use its discoveries or to develop and to commercialize Innovate’s technology and products without any compensation to Innovate.
Innovate also relies on unpatented know-how and trade secrets and continuing technological innovation to develop and maintain its competitive position, which Innovate seeks to protect, in part, through confidentiality agreements with employees, consultants, collaborators and others. Innovate also has invention or patent assignment agreements with its employees and certain consultants. The steps Innovate has taken to protect its proprietary rights, however, may not be adequate to preclude misappropriation of or otherwise protect its proprietary information or prevent infringement of its intellectual property rights, and Innovate may not have adequate remedies for any such misappropriation or infringement. In addition, it is possible that inventions relevant to Innovate’s business could be developed by a person not bound by an invention assignment agreement with Innovate or independently discovered by a competitor.
| 63 |
Innovate also intends to rely on regulatory exclusivity for protection of its product candidates, if approved for commercial sale. Implementation and enforcement of regulatory exclusivity, which may consist of regulatory data protection and market protection, varies widely from country to country. Failure to qualify for regulatory exclusivity, or failure to obtain or to maintain the extent or duration of such protections that Innovate expects for its product candidates, if approved, could affect its decision on whether to market the products in a particular country or countries or could otherwise have an adverse impact on its revenue or results of operations.
Innovate may rely on trademarks, trade names and brand names to distinguish its products, if approved for commercial sale, from the products of its competitors. However, Innovate’s trademark applications may not be approved. Third parties may also oppose Innovate’s trademark applications or otherwise challenge its use of the trademarks in which case Innovate may expend substantial resources to defend its proposed or approved trademarks and may enter into agreements with third parties that may limit Innovate’s use of its trademarks. In the event that Innovate’s trademarks are successfully challenged, Innovate could be forced to rebrand its products, which could result in loss of brand recognition and could require Innovate to devote significant resources to advertising and marketing these new brands. Further, Innovate’s competitors may infringe its trademarks or Innovate may not have adequate resources to enforce its trademarks.
Innovate’s success depends on its ability to prevent competitors from duplicating or developing and commercializing equivalent versions of its product candidates, and intellectual property protection may not be sufficient or effective to exclude this competition.
Innovate has patent protection in the United States and other countries to cover the composition of matter, formulation and method of use for INN-202 and INN-108. However, these patents may not provide Innovate with significant competitive advantages, because the validity, scope, term, or enforceability of the patents may be challenged and, if instituted, one or more of the challenges may be successful. Patents may be challenged in the United States under post-grant review proceedings, inter partes reexamination, ex parte re-examination, or challenged in district court. Any patents issued in foreign jurisdictions may be subjected to comparable proceedings lodged in various foreign patent offices, or courts. These proceedings could result in either loss of the patent or loss or reduction in the scope of one or more of the claims of the patent. Even if a patent issues, and is held valid and enforceable, competitors may be able to design around Innovate’s patent rights, such as by using pre-existing or newly developed technology, in which case competitors may not infringe Innovate’s issued claims and may be able to market and sell products that compete directly with Innovate’s before and after its patents expire.
Further, the INN-202 primary end point is a proprietary Patient Report Outcome measure (CeD PRO) that is protected by copyright. However, copyright protection may not be sufficient to exclude others from developing products that compete with INN-202.
The patent prosecution process is expensive and time-consuming. Innovate and any future licensors and licensees may not apply for or prosecute patents on certain aspects of its product candidates at a reasonable cost, in a timely fashion, or at all. Innovate may not have the right to control the preparation, filing and prosecution of some patent applications related to its product candidates or technologies. As a result, these patents and patent applications may not be prosecuted and enforced in a manner consistent with the best interests of Innovate. It is also possible that Innovate or any future or present licensors or licensees will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Further, it is possible that defects of form in the preparation or filing of Innovate’s patent applications may exist, or may arise in the future, such as with respect to proper priority claims, inventorship, assignment, term or claim scope. If there are material defects in the form or preparation of its patents or patent applications, such patents or applications may be invalid or unenforceable. In addition, one or more parties may independently develop similar technologies or methods, duplicate its technologies or methods, or design around the patented aspects of its products, technologies or methods. Any of these circumstances could impair Innovate’s ability to protect its products, if approved, in ways which may have an adverse impact on Innovate’s business, financial condition and operating results.
| 64 |
Furthermore, the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and Innovate’s owned and licensed patents may be challenged in the courts or patent offices in and outside of the United States. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit Innovate’s ability to use its patents to stop others from using or commercializing similar or identical products or technology, or to limit the duration of the patent protection of its technology and drugs. Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, Innovate’s owned and licensed patent portfolio may not provide Innovate with sufficient rights to exclude others from commercializing drugs similar to or identical to those of Innovate.
Enforcement of intellectual property rights in certain countries outside the United States, including China in particular, has been limited or non-existent. Future enforcement of patents and proprietary rights in many other countries will likely be problematic or unpredictable. Moreover, the issuance of a patent in one country does not assure the issuance of a similar patent in another country. Claim interpretation and infringement laws vary by nation, so the extent of any patent protection is uncertain and may vary in different jurisdictions.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and Innovate’s patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the United States Patent and Trademark Office, or USPTO, and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in decreased patent term or in abandonment or lapse of the patent or patent application, leading to partial or complete loss of patent rights in the relevant jurisdiction.
Third parties may claim that Innovate’s products, if approved, infringe on their proprietary rights and may challenge the approved use or uses of a product or its patent rights through litigation or administrative proceedings, and defending such actions may be costly and time consuming, divert management attention away from Innovate’s business, and result in an unfavorable outcome that could have an adverse effect on Innovate’s business.
Innovate’s commercial success depends on its ability and the ability of its CMOs and component suppliers to develop, manufacture, market and sell its products and product candidates and use its proprietary technologies without infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which Innovate is or may be developing products. Because patent applications can take many years to publish and issue, there currently may be pending applications, unknown to Innovate, that may later result in issued patents that its products, product candidates or technologies infringe, or that the process of manufacturing its products or any of its respective component materials, or the component materials themselves, infringe, or that the use of its products, product candidates or technologies infringe.
| 65 |
Innovate or its CMOs or component material suppliers may be exposed to, or threatened with, litigation by third parties alleging that Innovate’s products, product candidates and/or technologies infringe its patents and/or other intellectual property rights, or that one or more of the processes for manufacturing its products or any of its respective component materials, or the component materials themselves, or the use of its products, product candidates or technologies, infringe its patents and/or other intellectual property rights. If a third-party patent or other intellectual property right is found to cover its products, product candidates, technologies or its uses, or any of the underlying manufacturing processes or components, Innovate could be required to pay damages and could be unable to commercialize its products or to use its technologies or methods unless Innovate is able to obtain a license to the patent or intellectual property right. A license may not be available to Innovate in a timely manner or on acceptable terms, or at all. In addition, during litigation, the third-party alleging infringement could obtain a preliminary injunction or other equitable remedy that could prohibit Innovate from making, using, selling or importing its products, technologies or methods.
There generally is a substantial amount of litigation involving patent and other intellectual property rights in the industries in which Innovate operates and the cost of such litigation may be considerable. Innovate can provide no assurance that its product candidates or technologies will not infringe patents or rights owned by others, licenses to which might not be available to Innovate in a timely manner or on acceptable terms, or at all. If a third party claims that Innovate or its CMOs or component material suppliers infringe its intellectual property rights, Innovate may face a number of issues, including, but not limited to:
| · | infringement and other intellectual property claims which, with or without merit, may be expensive and time consuming to litigate and may divert management’s time and attention from Innovate’s core business; |
| · | substantial damages for infringement, including the potential for treble damages and attorneys’ fees, which Innovate may have to pay if it is determined that the product and/or its use at issue infringes or violates the third party’s rights; |
| · | a court prohibiting Innovate from selling or licensing the product unless the third-party licenses its intellectual property rights to Innovate, which it may not be required to do; |
| · | if a license is available from the third party, Innovate may have to pay substantial royalties, fees and/or grant cross-licenses to the third party; and |
| · | redesigning Innovate’s products or processes so they do not infringe, which may not be possible or may require substantial expense and time. |
No assurance can be given that patents do not exist, have not been filed, or could not be filed or issued, which contain claims covering Innovate’s products, product candidates or technology or those of its CMOs or component material suppliers or the use of its products, product candidates or technologies. Because of the large number of patents issued and patent applications filed in the industries in which Innovate operates, there is a risk that third parties may allege they have patent rights encompassing Innovate’s products, product candidates or technologies, or those of its CMOs or component material suppliers, or uses of its products, product candidates or technologies.
| 66 |
In the future, it may be necessary for Innovate to enforce its proprietary rights, or to determine the scope, validity and unenforceability of other parties’ proprietary rights, through litigation or other dispute proceedings, which may be costly, and to the extent Innovate is unsuccessful, adversely affect its rights. In these proceedings, a court or administrative body could determine that its claims, including those related to enforcing patent rights, are not valid or that an alleged infringer has not infringed its rights. The uncertainty resulting from the mere institution and continuation of any patent- or other proprietary rights-related litigation or interference proceeding could have a material and adverse effect on its business prospects, operating results and financial condition.
Risks Related to Innovate’s Industry
Innovate is subject to uncertainty relating to healthcare reform measures and reimbursement policies that, if not favorable to its products, could hinder or prevent its products’ commercial successes, if any of its product candidates are approved.
The unavailability or inadequacy of third-party payer coverage and reimbursement could negatively affect the market acceptance of its product candidates and the future revenues Innovate may expect to receive from those products. The commercial success of its product candidates, if approved, will depend in part on the extent to which the costs of such products will be covered by third-party payers, such as government health programs, commercial insurance and other organizations. Third-party payers are increasingly challenging the prices and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. If these third-party payers do not consider its products to be cost-effective compared to other therapies, Innovate may not obtain coverage for its products after approval as a benefit under the third-party payers’ plans or, even if Innovate does, the level of coverage or payment may not be sufficient to allow Innovate to sell its products on a profitable basis.
Significant uncertainty exists as to the reimbursement status for newly approved drug products, including coding, coverage and payment. There is no uniform policy requirement for coverage and reimbursement for drug products among third-party payers in the United States, therefore coverage and reimbursement for drug products can differ significantly from payer to payer. The coverage determination process is often a time-consuming and costly process that will require Innovate to provide scientific and clinical support for the use of its products to each payer separately, with no assurance that coverage and adequate payment will be applied consistently or obtained. The process for determining whether a payer will cover and how much it will reimburse a product may be separate from the process of seeking approval of the product or for setting the price of the product. Even if reimbursement is provided, market acceptance of its products may be adversely affected if the amount of payment for its products proves to be unprofitable for healthcare providers or less profitable than alternative treatments or if administrative burdens make its products less desirable to use. Third-party payer reimbursement to providers of its products, if approved, may be subject to a bundled payment that also includes the procedure of administering its products or third-party payers may require providers to perform additional patient testing to justify the use of its products. To the extent there is no separate payment for its product(s), there may be further uncertainty as to the adequacy of reimbursement amounts.
The continuing efforts of governments, private insurance companies, and other organizations to contain or to reduce costs of healthcare may adversely affect:
| · | Innovate’s ability to set an appropriate price for its products; |
| · | the rate and scope of adoption of its products by healthcare providers; |
| 67 |
| · | its ability to generate revenue or achieve or maintain profitability; |
| · | the future revenue and profitability of its potential customers, suppliers and collaborators; and |
| · | its access to additional capital. |
Innovate’s ability to successfully commercialize its products will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish what Innovate believes are appropriate coverage and reimbursement for its products. The containment of healthcare costs has become a priority of federal and state governments worldwide and the prices of drug products have been a focus in this effort. For example, there have been several recent U.S. Congressional inquiries and proposed bills designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs, and the new U.S. President has stated that reducing drug pricing is a priority for his administration. Innovate expects that federal, state and local governments in the United States, as well as in other countries, will continue to consider legislation directed at lowering the total cost of healthcare. In addition, in certain foreign markets, the pricing of drug products is subject to government control and reimbursement may in some cases be unavailable or insufficient. It is uncertain whether and how future legislation, whether domestic or abroad, could affect prospects for its product candidates or what actions federal, state, or private payers for healthcare treatment and services may take in response to any such healthcare reform proposals or legislation. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures reforms may prevent or limit its ability to generate revenue, attain profitability or commercialize its product candidates, especially in light of Innovate’s plans to price its product candidates at a high level.
Furthermore, Innovate expects that Congress will again attempt to pass reform measures that may be adopted in the future, including the possible repeal and replacement of the Affordable Care Act, which the Trump administration has stated is a priority. These potential courses of action are unpredictable; and the potential impact of new legislation on Innovate’s operations and financial position is uncertain, but may result in more rigorous coverage criteria, lower reimbursement, and additional downward pressure on the price Innovate may receive for an approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent Innovate from being able to generate revenue, attain profitability or commercialize its products, if approved.
Innovate expects competition in the marketplace for its product candidates, should any of them receive regulatory approval.
Larazotide acetate has issued patents for composition of matter, method of use and its formulation in the United States, Innovate’s primary market. INN-202 has either been issued patents or is prosecuting patent applications in numerous countries outside the United States. The barrier to entry for any company developing larazotide acetate for celiac disease is very high. Innovate believes that INN-202 is the first drug entering into Phase 3 clinical trials for celiac disease. Additionally, if larazotide acetate is the first drug granted FDA approval for celiac disease, competitors may need to license or to seek approval from Innovate for the usage of its CeD-PRO as an endpoint in subsequent celiac disease trials.
| 68 |
Innovate has received for Orphan Drug Designation from the FDA for INN-108 for pediatric ulcerative colitis. Orphan Drug Designation will provide market exclusivity in the U.S. for seven years, but only if (1) INN-108 receives market approval before a competitor using the same active compound for the same indication, (2) Innovate is able to produce sufficient supply to meet demand in the marketplace, and (3) another product with the same active ingredient(s) is not deemed clinically superior.
INN-329, secretin, has received Orphan Drug Designation from the FDA. Orphan Drug Designation will provide market exclusivity in the U.S. for seven years, but only if (1) INN-329 receives market approval before a competitor using a similar peptide for the same indication, (2) Innovate is able to produce sufficient supply to meet demand in the marketplace, and (3) another product with the same active ingredient is not deemed clinically superior.
The industries in which Innovate operates (biopharmaceutical, specialty pharmaceutical, biotechnology and pharmaceutical) are highly competitive and subject to rapid and significant changes. Developments by others may render potential application of any of its product candidates in a particular indication obsolete or noncompetitive, even prior to completion of its development and approval for that indication.
If successfully developed and approved, Innovate expects its product candidates will face competition. Innovate may not be able to compete successfully against organizations with competitive products, particularly large pharmaceutical companies. Many of its potential competitors have significantly greater financial, technical and human resources than Innovate, and may be better equipped to develop, manufacture, market and distribute products. Many of these companies operate large, well-funded research, development and commercialization programs, have extensive experience in nonclinical and clinical studies, obtaining FDA and other regulatory approvals and manufacturing and marketing products, and have multiple products that have been approved or are in late-stage development. These advantages may enable them to receive approval from the FDA or any foreign regulatory agency before Innovate and prevent Innovate from competing due to their orphan drug protections. Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and biotechnology companies. Furthermore, heightened awareness on the part of academic institutions, government agencies and other public and private research organizations of the potential commercial value of their inventions have led them to actively seek to commercialize the technologies they develop, which increases competition for investment in Innovate’s programs. Competitive products may be more effective, easier to dose, or more effectively marketed and sold, than theirs, which would have a material adverse effect on Innovate’s ability to generate revenue.
Innovate faces potential product liability exposure and, if successful claims are brought against it, Innovate may incur substantial liability for a product or product candidate and may have to limit its commercialization. In the future, Innovate anticipates that it will need to obtain additional or increased product liability insurance coverage and it is uncertain whether such increased or additional insurance coverage can be obtained on commercially reasonable terms, if at all.
Innovate’s business (in particular, the use of its product candidates in clinical studies and the sale of any products for which it obtains marketing approval) will expose Innovate to product liability risks. Product liability claims might be brought against Innovate by patients, healthcare providers, pharmaceutical companies or others selling or involved in the use of its products. If Innovate cannot successfully defend itself against any such claims, Innovate will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| · | significant costs of related litigation; |
| · | decreased demand for its products and loss of revenue; |
| 69 |
| · | impairment of its business reputation; |
| · | a “clinical hold,” suspension or termination of a clinical study or amendments to a study design; |
| · | delays in enrolling patients to participate in its clinical studies; |
| · | withdrawal of clinical study participants; |
| · | substantial monetary awards to patients or other claimants; and |
| · | the inability to commercialize its products and product candidates. |
Innovate maintains limited product liability insurance for its clinical studies, but its insurance coverage may not reimburse Innovate or may not be sufficient to reimburse Innovate for all expenses or losses it may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, Innovate may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect itself against losses.
Innovate expects that it will expand its insurance coverage to include the sale of commercial products if it obtains marketing approval for any of its product candidates, but Innovate may be unable to obtain product liability insurance on commercially acceptable terms or may not be able to maintain such insurance at a reasonable cost or in sufficient amounts to protect Innovate against potential losses. Large judgments have been awarded in class action lawsuits based on drug products that had unanticipated side effects. A successful product liability claim or series of claims brought against Innovate, if judgments exceed its insurance coverage, could decrease its cash and adversely affect its business.
Risks Related to Our Common Stock
The market price of the Company’s common stock is likely to be volatile, and the stock price may drop following the Merger.
The stock market in general and the market for pharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. A certain degree of market price volatility may also occur as a result of the completion of the Merger and listing of the shares of the combined company. The market price of the Company’s common stock following the closing of the Merger may be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control, including:
| · | regulatory or legal developments in the United States and foreign countries; |
| · | results from or delays in clinical trials of our product candidates; |
| · | announcements of regulatory approval or disapproval of INN-202 (for celiac disease), INN-108 (for ulcerative colitis), INN-329 (for magnetic resonance cholangiopancreatography or MRCP) or any future product candidates; |
| · | commercialization of our product candidates; |
| · | FDA or other U.S. or foreign regulatory actions affecting us or our industry; |
| 70 |
| · | introductions and announcements of new products by us, any commercialization partners or our competitors, and the timing of these introductions and announcements; |
| · | variations in our financial results or those of companies that are perceived to be similar to us; |
| · | changes in the structure of healthcare payment systems; |
| · | announcements by us or our competitors of significant acquisitions, licenses, strategic partnerships, joint ventures or capital commitments; |
| · | market conditions in the pharmaceutical and biopharmaceutical sectors and issuance of securities analysts’ reports or recommendations; |
| · | actual or anticipated quarterly variations in our results of operations or those of our future competitors; |
| · | changes in financial estimates or guidance, including our ability to meet our future revenue and operating profit or loss estimates or guidance; |
| · | sales of substantial amounts of our stock by insiders and large stockholders, or the expectation that such sales might occur; |
| · | general economic, industry and market conditions; |
| · | additions or departures of key personnel; |
| · | intellectual property, product liability or other litigation against us; |
| · | expiration or termination of our potential relationships with strategic partners; |
| · | negative reactions from investors to the prospects of the combined organization’s business and prospects from the Merger; |
| · | the effect of the Merger on the combined organization’s business and prospects is not consistent with the expectations of financial or industry analysts; |
| · | the combined organization does not achieve the perceived benefits of the Merger as rapidly or to the extent anticipated by financial or industry analysts; and |
| · | the other factors described in this section entitled “Risk Factors.” |
If securities or industry analysts do not publish research or publish unfavorable research about the Company’s business, the Company’s common stock price and trading volume could decline.
Equity research analysts do not currently provide research coverage of the Company’s common stock. In particular, as a smaller company, it may be difficult for us to attract the interest of equity research analysts. A lack of research coverage may adversely affect the liquidity of and market price of the Company’s common stock. To the extent we obtain equity research analyst coverage, we will not have any control of the analysts or the content and opinions included in their reports. The market price of the stock could decline if one or more equity research analysts downgrade the common stock or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of the Company, or fails to publish reports on us regularly, demand for the common stock could decrease, which in turn could cause the market price of the common stock or trading volume to decline.
| 71 |
Sales of substantial amounts of the Company’s common stock in the public markets, or the perception that such sales might occur, could cause the market price of the Company’s common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of the Company’s common stock in the public market could occur at any time. If our stockholders sell, or the market perceives that our stockholders intend to sell, substantial amounts of the Monster common stock in the public market following the closing of the Merger, the market price of the Company’s common stock could decline significantly.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful third-party claims against us and may reduce the amount of money available to us.
Our certificate of incorporation and restated bylaws following as they will be in effect following the Merger provide that we will indemnify our directors and officers, in each case to the fullest extent permitted by Delaware law.
To the extent that a claim for indemnification is brought by any of our directors or officers, it would reduce the amount of funds available for use in our business.
If the Company sells shares of its common stock in the future, stockholders may experience immediate dilution and, as a result, the market price of the Company’s common stock may decline.
The Company may from time to time issue additional shares of its common stock at a discount from the then-current trading price. As a result, the Company’s stockholders would experience immediate dilution upon the purchase of any shares of such common stock sold at such discount. In addition, as opportunities present themselves, the Company may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred stock or common stock. If the Company issues common stock or securities convertible into common stock, its common stockholders would experience additional dilution and, as a result, the market price of the Company’s common stock may decline.
Concentration of ownership of our common stock among our existing principal stockholders may effectively limit the voting power of other stockholders.
Our executive officers, directors and current beneficial owners of 5% or more of our common stock, in aggregate, beneficially own approximately 42% of our outstanding common stock. Accordingly, these stockholders, acting together, will continue to be able to significantly influence all matters requiring stockholder approval, including the election and removal of directors and any merger or other significant corporate transactions. These stockholders may therefore delay or prevent a change of control, even if such a change of control would benefit the other stockholders. The significant concentration of stock ownership may adversely affect the market price of the Company’s common stock due to investors’ perception that conflicts of interest may exist or arise.
| 72 |
Anti-takeover provisions in our corporate charter documents and under Delaware law could make an acquisition of us more difficult, which could discourage takeover attempts and lead to management entrenchment, and the market price of our common stock may be lower as a result.
Certain provisions in our certificate of incorporation and bylaws may make it difficult for a third party to acquire, or attempt to acquire, control of our company, even if a change in control was considered favorable by the stockholders. For example, the Board will have the authority to issue up to 10,000,000 shares of preferred stock. The Board can fix the price, rights, preferences, privileges, and restrictions of the preferred stock without any further vote or action by our stockholders. The issuance of shares of preferred stock may delay or prevent a change in control transaction. As a result, the market price of our common stock and the voting and other rights of our stockholders may be adversely affected. An issuance of shares of preferred stock may result in the loss of voting control to other stockholders.
Our charter documents will also contain other provisions that could have an anti-takeover effect, including provisions that:
| · | establish that the Board is divided into three classes, Class I, Class II and Class III, with each class serving staggered three year terms; |
| · | provide that vacancies on the Board may be filled only by a majority of directors then in office, even though less than a quorum; |
| · | provide that our directors may only be removed for cause; |
| · | eliminate cumulative voting in the election of directors; |
| · | authorize the Board to issue shares of preferred stock and determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval; |
| · | provide the Board with the exclusive right to elect a director to fill a vacancy or newly created directorship; |
| · | permit stockholders to only take actions at a duly called annual or special meeting and not by written consent; |
| · | prohibit stockholders from calling a special meeting of stockholders; |
| · | require that stockholders give advance notice to nominate directors or submit proposals for consideration at stockholder meetings; |
| · | authorize the Board, by a majority vote, to amend the bylaws; and |
| · | require the affirmative vote of at least 66 2/3% or more of the outstanding common stock to amend many of the provisions described above. |
In addition, we are subject to the anti-takeover provisions of Section 203 of the Delaware General Corporation Law, which limits the ability of stockholders owning in excess of 15% of our outstanding voting stock to merge or combine with us. Finally, our amended and restated certificate of incorporation will also provide that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders. These provisions could discourage potential acquisition proposals and could delay or prevent a change in control transaction. They could also have the effect of discouraging others from making tender offers for our common stock, including transactions that may be in your best interests. These provisions may also prevent changes in our management or limit the price that certain investors are willing to pay for our stock.
| 73 |
We may be subject to securities litigation, which is expensive and could divert management attention.
The market price of our common stock may be volatile, and in the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
Innovate has not paid cash dividends in the past and does not expect to pay dividends in the future. Any return on investment may be limited to the value of its common stock.
Innovate has never paid cash dividends on its common stock and does not anticipate paying cash dividends in the near future. The payment of dividends on its common stock will depend on earnings, financial condition and other business and economic factors affecting Innovate at such time as the board of directors may consider relevant. If Innovate does not pay dividends, its common stock may be less valuable because a return on investment will only occur if its stock price appreciates.
Our ability to use our net operating loss carryforwards and certain other tax attributes to offset future taxable income may be subject to certain limitations.
Innovate has U.S. federal net operating loss carryforwards, or NOLs, which expire in various years if not utilized. In addition, we have federal research and development credit carryforwards. The federal research and development credit carryforwards expire in various years if not utilized. Under Sections 382 and 383 of Internal Revenue Code of 1986, as amended, or the Code, if a corporation undergoes an “ownership change,” the corporation’s ability to use its pre-change NOLs and other pre-change tax attributes, such as research tax credits, to offset its future post-change income and taxes may be limited. In general, an “ownership change” occurs if there is a cumulative change in our ownership by “5% shareholders” that exceeds 50 percentage points over a rolling three-year period. Similar rules may apply under state tax laws. We have not performed a formal study to determine whether any of our NOLs are subject to these limitations. We have recorded deferred tax assets for our NOLs and research and development credits and have recorded a full valuation allowance against these deferred tax assets. In the event that it is determined that we have in the past experienced additional ownership changes, or if we experience one or more ownership changes as a result of future transactions in our stock, then we may be further limited in our ability to use our NOLs and other tax assets to reduce taxes owed on the net taxable income that we earn in the event that we attain profitability. Any such limitations on the ability to use our NOLs and other tax assets could adversely impact our business, financial condition and operating results in the event that we attain profitability.
We will incur costs as a result of operating as a public company and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices, including maintaining an effective system of internal control over financial reporting.
As a public company listed in the United States, and increasingly after we are no longer an “emerging growth company,” we will incur significant additional legal, accounting and other expenses that Innovate did not incur as a private company. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act and regulations implemented by the SEC and Nasdaq, may increase legal and financial compliance costs and make some activities more time consuming. These laws, regulations and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If notwithstanding our efforts to comply with new laws, regulations and standards, we fail to comply, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
| 74 |
As a public company in the United States, we will be required, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. We will need to disclose any material weaknesses identified by our management in our internal control over financial reporting, and, when we are no longer an “emerging growth company”, we will need to provide a statement that our independent registered public accounting firm has issued an opinion on our internal control over financial reporting.
The controls and other procedures are designed to ensure that information required to be disclosed by us in the reports that we file with the Securities and Exchange Commission, or SEC, is disclosed accurately and is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms. We are in the early stages of conforming our internal control procedures to the requirements of Section 404 and we may not be able to complete our evaluation, testing and any required remediation needed to comply with Section 404 in a timely fashion. Our independent registered public accounting firm was not engaged to perform an audit of our internal control over financial reporting for the year ended December 31, 2017, or for any other period. Accordingly, no such opinion will be expressed.
Even after we develop these new procedures, these new controls may become inadequate because of changes in conditions or the degree of compliance with these policies or procedures may deteriorate and material weaknesses in our internal control over financial reporting may be discovered. We may err in the design or operation of our controls, and all internal control systems, no matter how well designed and operated, can provide only reasonable assurance that the objectives of the control system are met. Because there are inherent limitations in all control systems, there can be no absolute assurance that all control issues have been or will be detected. If we are unable, or are perceived as unable, to produce reliable financial reports due to internal control deficiencies, investors could lose confidence in our reported financial information and operating results, which could result in a negative market reaction.
To fully comply with Section 404, we will need to retain additional employees to supplement our current finance staff, and we may not be able to do so in a timely manner, or at all. In addition, in the process of evaluating our internal control over financial reporting, we expect that certain of our internal control practices will need to be updated to comply with the requirements of Section 404 and the regulations promulgated thereunder, and we may not be able to do so on a timely basis, or at all. In the event that we are not able to demonstrate compliance with Section 404 in a timely manner, or are unable to produce timely or accurate financial statements, we may be subject to sanctions or investigations by regulatory authorities, such as the SEC or the stock exchange on which our stock is listed, and investors may lose confidence in our operating results and the price of our common stock could decline. Furthermore, if we are unable to certify that our internal control over financial reporting is effective and in compliance with Section 404, we may be subject to sanctions or investigations by regulatory authorities, such as the SEC or stock exchanges, and we could lose investor confidence in the accuracy and completeness of our financial reports, which could hurt our business, the price of our common stock and our ability to access the capital markets.
We also expect that being a public company will make it more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on committees of our board of directors or as members of senior management.
| 75 |
We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups, or JOBS, Act enacted in April 2012, and may remain an “emerging growth company” for up to five years following the completion of our initial public offering, although, if we have more than $1.0 billion in annual revenue, the market value of our common stock that is held by non-affiliates exceeds $700 million as of June 30 of any year, or we issue more than $1.0 billion of non-convertible debt over a three-year period before the end of that five-year period, we would cease to be an “emerging growth company” as of the following December 31. For as long as we remain an “emerging growth company,” we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not “emerging growth companies.” These exemptions include:
| · | being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s discussion and analysis of financial condition and results of operations” disclosure; |
| · | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
| · | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| · | reduced disclosure obligations regarding executive compensation; and |
| · | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. |
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards, delaying the adoption of these accounting standards until they would apply to private companies. We have irrevocably elected not to avail ourselves of this exemption and, as a result, our financial statements may not be comparable to the financial statements of issuers who are required to comply with the effective dates for new or revised accounting standards that are applicable to public companies. We cannot predict whether investors will find our common stock less attractive as a result of our reliance on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and the market price of our common stock may be reduced or more volatile.
Risks Related to the Company Following the Merger
Monster and Innovate stockholders may not realize a benefit from the Merger commensurate with the ownership dilution they will experience in connection with the Merger.
If the combined organization is unable to realize the full strategic and financial benefits currently anticipated from the Merger, Monster and Innovate stockholders will have experienced substantial dilution of their ownership interests in their respective companies without receiving any commensurate benefit, or only receiving part of the commensurate benefit to the extent the combined organization is able to realize only part of the strategic and financial benefits currently anticipated from the Merger.
| 76 |
Because the lack of a public market for Innovate shares makes it difficult to evaluate the fairness of the Merger, the stockholders of Innovate may receive consideration in the Merger that is less than the fair market value of the Innovate shares.
Prior to the Merger, the outstanding capital stock of Innovate was privately held and was not traded in any public market. The lack of a public market makes it extremely difficult to determine the fair market value of Innovate. Because the percentage of Monster equity issued to Innovate stockholders was determined based on negotiations between the parties, it is possible that the value of Monster common stock to be received by Innovate stockholders in the Merger is less than the fair market value of Innovate.
| 77 |
INNOVATE MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of Innovate’s financial condition and results of operations together with the section entitled “Selected Financial Data” and Innovate’s financial statements and related notes included elsewhere in this Current Report on Form 8-K. This discussion and other parts of this Current Report on Form 8-K contain forward-looking statements that involve risks and uncertainties, such as its plans, objectives, expectations, intentions and beliefs. Innovate’s actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those identified below and those discussed in the section entitled “Risk Factors” included elsewhere in this Current Report on Form 8-K.
Company Overview
Innovate is a clinical-stage biopharmaceutical company developing novel therapies for autoimmune and inflammatory disorders. Innovate’s lead product candidate, larazotide acetate (INN-202), is an orally administered 8-amino acid synthetic peptide that has the potential to be the first-to-market therapeutic for celiac disease (CeD), and is currently entering phase 3 registration trials. Innovate’s second product candidate, INN-108, is an oral tablet that uses an azo-bonded pro-drug approach linking mesalamine to 4-APAA and is in development for the treatment of mild to moderate ulcerative colitis (UC). INN-108 is entering a proof-of-concept Phase 2 trial in 2018, after having successfully completed a Phase 1 trial.
Since its inception in January 2012, Innovate has focused its efforts and resources on identifying and developing its programs. Innovate has not had any products approved for commercial sale and has incurred operating losses in each year since inception. Substantially all of its operating losses resulted from expenses incurred in connection with its research and development programs and from general and administrative costs associated with its operations.
Innovate expects to continue to incur significant expenses and increasing operating losses for the foreseeable future, which may fluctuate significantly between periods. Innovate anticipates that its expenses will increase substantially as it:
| • | continues research and development, including preclinical and clinical development of its existing product candidates; |
| • | potentially seeks regulatory approval for its product candidates; |
| • | commercializes any product candidates for which it obtains regulatory approval; |
| • | maintains and protects its intellectual property rights; |
| • | adds operational, financial and management information systems and personnel; and |
| • | incurs additional legal, accounting and other expenses in operating as a public company. |
Recent Developments
On January 29, 2018, Monster Digital, Inc., a Delaware corporation now known as Innovate Biopharmaceuticals, Inc. (the “Company”) completed its merger with privately-held Innovate Biopharmaceuticals Inc. (“IB Pharmaceuticals Inc.”) in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated July 3, 2017, whereby Monster Merger Sub., Inc. (“Merger Sub”), a wholly owned subsidiary of the Company merged with and into IB Pharmaceuticals Inc., with IB Pharmaceuticals Inc. surviving as the Company’s wholly owned subsidiary (the “Merger”). In connection with the Merger, the Company changed its name from Monster Digital, Inc. to Innovate Biopharmaceuticals, Inc. All references to the “Company” refer to Innovate Biopharmaceuticals, Inc. as of and following the closing of the Merger on January 29, 2018 (the “Closing Date”) and all references to “Monster” refer to Monster Digital, Inc. prior to the closing of the Merger on the Closing Date.
Immediately prior to the closing of the Merger, accredited investors purchased shares of common stock of IB Pharmaceuticals Inc. in a private placement for gross proceeds of approximately $18.13 million (the “Equity Issuance”). IB Pharmaceuticals Inc. issued five-year warrants to each purchaser of common stock with a price per exercise price of $1.2011 (subject to adjustment in connection with the Merger). Concurrently with the Equity Issuance, convertible promissory notes issued by IB Pharmaceuticals Inc. in the aggregate principal amount of approximately $8.65 million were converted into shares of IB Pharmaceuticals Inc. common stock at a price per share of $0.7206 (the “Conversion”).
| 78 |
H.C. Wainwright & Co., LLC (“HCW”) and GP Nurmenkari Inc. (“GPN”) were retained as the placement agents for the Equity Issuance. HCW was paid a flat fee of $250,000.00, a cash fee of $285,000.06 (equal to 10% of the gross proceeds of the Equity Issuance up to a certain cap), a cash fee of $9,018.15 (equal to 3.5% of the gross proceeds in excess of a certain cap), and non-accountable expense allowance of $50,000. GPN was paid a cash fee of $891,266.11 (equal to 10% of the gross proceeds of certain investors in the Equity Issuance) and non-accountable expense allowance of $50,000. IB Pharmaceuticals Inc. issued to affiliates of HCW five-year warrants to purchase 557,097 shares of common stock with an exercise price per share equal to $1.2011(subject to adjustment in connection with the Merger). IB Pharmaceuticals Inc. issued to GPN five-year warrants to purchase 927,529 shares of common stock with an exercise price per share equal to $1.2011(subject to adjustment in connection with the Merger); provided that a small number of warrants (representing 318,776 shares of underlying IB Pharmaceuticals Inc. common stock) were issued to affiliates of GPN with an exercise price per share equal to $0.9609 (subject to adjustment in connection with the Merger). Upon the closing of the Merger, the outstanding shares of IB Pharmaceuticals Inc.’s common stock were exchanged for shares of common stock of Monster at an exchange ratio of one share of IB Pharmaceuticals Inc. common stock to 0.37813802 shares of Monster common stock (the “Exchange Ratio”). Immediately following the closing of the Merger, after giving effect to the Equity Issuance and applying the Exchange Ratio, Monster’s securityholders owned approximately 5.8% of the outstanding common stock of the Company on a fully-diluted basis and IB Pharmaceuticals Inc.’s securityholders owned approximately 94.2% of the outstanding common stock of the Company.
On January 29, 2018, the Company entered into a Note Purchase Agreement and Senior Note Payable (“Note”) with a lender. The principal amount of the Note is $4,800,000 (“Principal”). The Note was issued at a discount of $1,800,000 and net of $20,000 for financing costs, for total proceeds of $2,980,000. The Note matures on September 30, 2018 (“Maturity Date”), however, the Maturity Date may be extended at the option of the lender under certain circumstances as outlined in the Note. Interest on the Note accrues starting on January 29, 2018 at a rate of 12.5% per annum and payments of interest only are due beginning on March 30, 2018 and compound quarterly. Upon the Maturity Date of the Note, the Company is required to pay the lender an amount representing 105% of all outstanding Principal, accrued and unpaid interest, and any unpaid late charges, if applicable (“Outstanding Amount”). The Note contains redemption features and certain non-financial covenants and penalties to the Company in the case of certain events of default, as defined in the Note.
Innovate announced on September 6, 2017, that it received U.S. Food and Drug Administration orphan drug designation for INN-108 as an oral therapy proposed for the treatment of pediatric ulceration colitis.
Critical Accounting Policies
Use of Estimates The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and disclosures made in the accompanying notes to the financial statements. Areas of the financial statements where estimates may have the most significant effect include accrued expenses, share-based compensation, deferred compensation, valuation allowance for income tax assets and management’s assessment of Innovate’s ability to continue as a going concern. Changes in the facts or circumstances underlying these estimates could result in material changes and actual results could differ from these estimates.
Accrued Expenses Innovate incurs periodic expenses such as research and development, salaries, taxes, and professional fees. An adjusting entry to accrue expenses is necessary when expenses have been incurred by Innovate prior to them being paid. When a vendor’s invoice is not received, Innovate is required to estimate its accrued expenses. This process involves reviewing quotations and contracts, identifying services that have been performed on Innovate’s behalf and estimating the level of service performed and the associated cost incurred for the service when Innovate has not yet been invoiced or otherwise notified of the actual cost. The majority of Innovate’s service providers invoice monthly in arrears for services performed or when contractual milestones are met. Innovate estimates accrued expenses as of each balance sheet date based on facts and circumstances known at that time. Innovate periodically confirms the accuracy of its estimates with the service providers and makes adjustments if necessary.
Research and Development Innovate expenses the cost of research and development as incurred. Research and development expenses comprise costs incurred from contracted services and other external costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity is performed or when the goods have been received, rather than when payment is made, in accordance with Accounting Standards Codification (“ASC”) 730, Research and Development.
Share-Based Compensation Innovate accounts for share-based compensation using the fair value method of accounting which requires all such compensation to employees, including the grant of employee stock options, to be recognized in the Statements of Operations based on its fair value at the grant date. The expense associated with share-based compensation is recognized on a straight-line basis over the requisite service period of each award; however, the amount of compensation expense recognized at any date must at least equal the portion of the grant-date value of the award that is vested at that date. For share-based compensation granted to non-employees, the measurement date is generally considered to be the date when all services have been rendered or the date that options are fully vested.
| 79 |
Liquidity and Going Concern The accompanying financial statements have been prepared on a basis which assumes that Innovate will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. There is substantial doubt that Innovate will continue as a going concern for at least 12 months following the date these financial statements are issued, without additional financing based on Innovate’s limited operating history and recurring operating losses. Management’s plans with regard to these matters include seeking additional debt or equity financing arrangements, including one or more bridge financings prior to closing of the Merger. There is no assurance such financing will be available to Innovate when required or that such financing will be available under favorable terms, or that Innovate can achieve its developmental milestones. The accompanying financial statements do not include any adjustments that might be necessary should Innovate be unable to continue as a going concern.
Results of Operations
Comparison of the Three Months Ended September 30, 2017 and 2016
The following table sets forth the key components of Innovate’s results of operations for the three months ended September 30, 2017 and 2016:
| Three Months Ended September 30, | ||||||||||||
| 2017 | 2016 | Change | ||||||||||
| Operating expenses: | ||||||||||||
| Research and development expenses | $ | 367,551 | $ | 239,297 | $ | 128,254 | ||||||
| General and administrative expenses | 1,973,256 | 1,733,246 | 240,010 | |||||||||
| Total operating expenses | 2,340,807 | 1,972,543 | 368,264 | |||||||||
| Other income (expense): | ||||||||||||
| Interest income | - | - | - | |||||||||
| Interest expense | (110,508 | ) | (57,164 | ) | (53,344 | ) | ||||||
| Total other expense, net | (110,508 | ) | (57,164 | ) | (53,344 | ) | ||||||
| Net loss | $ | (2,451,315 | ) | $ | (2,029,707 | ) | $ | (421,608 | ) | |||
Research and Development Expense
Research and development expense for the three months ended September 30, 2017, increased by approximately $128,000, or 53.6% as compared to the three months ended September 30, 2016. The increase was primarily due to a $314,000 increase in share-based compensation expense for stock options granted to consultants working on Innovate’s development programs plus an approximate $9,000 increase in clinical and regulatory costs for the development of Innovate’s INN-202 and INN-108 programs, partially offset by an approximate $193,000 decrease in manufacturing related expense.
General and Administrative Expense
General and administrative expense for the three months ended September 30, 2017, increased by approximately $240,000, or 13.8%, as compared to the three months ended September 30, 2016. The increase was primarily due to an approximate aggregate increase of $402,000 in accounting, legal, and transaction advisory services related to the Company’s anticipated Merger plus an approximate aggregate increase of $182,000 in business development expenses in connection with the Company’s INN-202 and INN-108 programs, partially offset by an approximate $374,000 net decrease in employee compensation and share-based compensation expense.
Interest Expense
Interest expense for the three months ended September 30, 2017, increased by approximately $53,000, or 93.3%, as compared to the three months ended September 30, 2016. The increase was due to the increase in the principal amount of convertible debt outstanding from $2.7 million at September 30, 2016, to $7.7 million at September 30, 2017.
| 80 |
Comparison of the Nine Months Ended September 30, 2017 and 2016
The following table sets forth the key components of Innovate’s results of operations for the nine months ended September 30, 2017 and 2016:
| Nine Months Ended September 30, | ||||||||||||
| 2017 | 2016 | Change | ||||||||||
| Operating expenses: | ||||||||||||
| Research and development expenses | $ | 2,832,787 | $ | 885,449 | $ | 1,947,338 | ||||||
| General and administrative expenses | 6,115,088 | 2,705,976 | 3,409,112 | |||||||||
| Total operating expenses | 8,947,875 | 3,591,425 | 5,356,450 | |||||||||
| Other income (expense): | ||||||||||||
| Interest income | - | 94 | (94 | ) | ||||||||
| Interest expense | (281,638 | ) | (150,101 | ) | (131,537 | ) | ||||||
| Total other expense, net | (281,638 | ) | (150,007 | ) | (131,631 | ) | ||||||
| Net loss | $ | (9,229,513 | ) | $ | (3,741,432 | ) | $ | (5,488,081 | ) | |||
Research and Development Expense
Research and development expense for the nine months ended September 30, 2017 increased approximately $1.9 million, or 220% as compared to the nine months ended September 30, 2016. The increase was primarily due to an increase of $1.6 million in share-based compensation expense for stock options granted to consultants working on Innovate’s development programs and to an approximate $845,000 net increase in manufacturing, clinical and regulatory costs for the development of Innovate’s INN-202 and INN-108 programs, partially offset by the absence of $525,000 in license fees.
General and Administrative Expense
General and administrative expense for the nine months ended September 30, 2017 increased approximately $3.4 million, or 126% as compared to the nine months ended September 30, 2016. The increase was primarily comprised of a $2.1 million increase in share-based compensation expense, an approximate $885,000 aggregate increase in legal, accounting, and transaction advisory services primarily related to the Company’s preparation for its anticipated Merger, plus an approximate aggregate increase of $384,000 in business development and patent support expenses primarily in connection with the Company’s INN-202 and INN-108 programs.
Interest Expense
Interest expense for the nine months ended September 30, 2017 increased by approximately $132,000, or 87.6%, as compared to the nine months ended September 30, 2016. The increase was due to the increase in the principal amount of convertible debt outstanding from $2.7 million at September 30, 2016 to $7.7 million at September 30, 2017.
| 81 |
Liquidity and Capital Resources
Sources of Liquidity
To date, Innovate has funded its operations primarily through the issuance of approximately $8.6 million of convertible promissory notes. As of September 30, 2017, Innovate had cash of approximately $1.5 million, compared to approximately $361,000 as of December 31, 2016. Innovate expects to incur substantial expenditures in the foreseeable future for the development and clinical trials of its INN-202 and INN-108 product candidates. Innovate will continue to require additional financing to develop its product candidates and fund operations for the foreseeable future. Innovate will continue to seek funds through debt or equity financings, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements, or other sources of financing. If Innovate is unable to raise additional funds when needed, its ability to develop its product candidates may be impaired. Innovate may also be required to delay, reduce, or terminate some or all of its development programs and clinical trials.
Cash Flows
The following table sets forth the primary sources and uses of cash for the nine months ended September 30, 2017 and 2016:
| Nine Months Ended September 30, | ||||||||
| 2017 | 2016 | |||||||
| Cash provided by (used in): | ||||||||
| Operating activities | $ | (3,165,404 | ) | $ | (1,533,549 | ) | ||
| Investing activities | (1,600 | ) | (80,731 | ) | ||||
| Financing activities | 4,285,024 | 1,853,000 | ||||||
| Net increase in cash | $ | 1,118,020 | $ | 238,720 | ||||
Operating Activities
For the nine months ended September 30, 2017, Innovate’s net cash used in operating activities of approximately $3.2 million primarily consisted of a net loss of $9.2 million, offset by adjustments for share-based compensation of $4.7 million, accrued interest of approximately $252,000, and an increase in accounts payable of $1.1 million.
For the nine months ended September 30, 2016, Innovate’s net cash used in operating activities of approximately $1.5 million primarily consisted of a net loss of $3.7 million, offset by adjustments for share-based compensation of $1.0 million and accrued interest of approximately $123,000, and increases of approximately $782,000, $409,000, and $128,000 in accrued expenses, accounts payable, and prepaid expenses and other current assets, respectively.
Investing Activities
Net cash used in investing activities for 2017 represented the purchase of computer equipment. Net cash used for investing activities in 2016 represented net loans to a related party and approximately $6,000 for the purchase of computer equipment.
Financing Activities
Net cash provided by financing activities for all periods presented primarily consisted of proceeds from convertible promissory notes.
Future Funding Requirements
Innovate has not generated any revenue from product sales or any other activities. Innovate does not expect to generate significant revenue unless and until it obtains regulatory approval of and commercializes, or out licenses, any of its product candidates and does not know when, or if, these will occur. In addition, Innovate expects its expenses to significantly increase in connection with its ongoing development activities, particularly as it continues the research, development and clinical trials of, and seeks regulatory approval for, its product candidates. In addition, subject to obtaining regulatory approval of its product candidates, Innovate expects to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. Innovate anticipates that it will need substantial additional funding in connection with its continuing operations, including increased costs associated with becoming a public company since its consummation of the Merger.
| 82 |
Contractual Obligations and Commitments
As of September 30, 2017, Innovate did not have any lease obligations for its operating facilities in Raleigh, North Carolina. The lease expired in April 2017, and Innovate was under a month-to-month lease at a cost of approximately $1,600 per month.
In October 2017, the Company entered into a new three-year lease for office space that expires on September 30, 2020. Base annual rent is $60,000, or $5,000 per month. The first two months of rent were paid in advance upon lease signing and the next ten months of rent were paid in advance on November 30, 2017. Beginning with month thirteen, monthly payments of $5,000 will be paid in advance of the first day of each month of the remaining term. A security deposit of $5,000 was paid in October 2017. The lease contains a two-year renewal option.
The Company has employment agreements with certain executives of the Company (the “Executive Agreements”). Under the terms of the Executive Agreements, the Company has agreed to pay the executives certain payments upon the achievement of financial milestone events. These milestone events were based on total debt or equity funding received by the Company. During the nine months ended September 30, 2017, the initial funding milestone was reached and the executives were paid $145,000 in accordance with the terms of the Executive Agreements. The executives are eligible to receive up to $1,595,000 in additional milestone payments upon the achievement of a financing event with gross proceeds of at least $45,000,000 by March 15, 2018. As of September 30, 2017, these deferred compensation payments were included in accrued expenses.
The following table summarizes Innovate’s contractual obligations as of September 30, 2017:
| Less than 1 Year | 1 to 3 years | 4 to 5 Years | After 5 Years | Total | ||||||||||||||||
| Principal payments of convertible debt | $ | 7,732,335 | - | - | - | $ | 7,732,335 | |||||||||||||
| Accrued interest on convertible debt | 416,075 | - | - | - | 416,075 | |||||||||||||||
| Deferred compensation | 1,595,000 | - | - | - | 1,595,000 | |||||||||||||||
| Total contractual obligations | $ | 9,743,410 | - | - | - | $ | 9,743,410 | |||||||||||||
During the period from October through January 24, 2018, the Company issued additional convertible debt in an aggregate amount of $845,000 to third-party investors.
Innovate is obligated to make future payments to third parties under in-license agreements, including sublicense fees, royalties, and payments that become due and payable on the achievement of certain development and commercialization milestones. As the amount and timing of sublicense fees and the achievement and timing of these milestones are not probable and estimable, such commitments have not been included on Innovate’s balance sheet or in the contractual obligations table above.
On January 29, 2018, the Company entered into a Note Purchase Agreement and Senior Note Payable (“Note”) with a lender. The principal amount of the Note is $4,800,000 (“Principal”). The Note was issued at a discount of $1,800,000 and net of $20,000 for financing costs, for total proceeds of $2,980,000. The Note matures on September 30, 2018 (“Maturity Date”), however, the Maturity Date may be extended at the option of the lender under certain circumstances as outlined in the Note. Interest on the Note accrues starting on January 29, 2018 at a rate of 12.5% per annum and payments of interest only are due beginning on March 30, 2018 and compound quarterly. Upon the Maturity Date of the Note, the Company is required to pay the lender an amount representing 105% of all outstanding Principal, accrued and unpaid interest, and any unpaid late charges, if applicable (“Outstanding Amount”). The Company has the right to redeem the Note at any time prior to the Maturity Date provided an event of default has not occurred. The redemption amount consists of the Outstanding Amount prior to the redemption date plus an additional amount of interest that would have accrued from the redemption date through January 29, 2019. The Note contains certain non-financial covenants and penalties to the Company in the case of certain events of default, as defined in the Note.
Off-Balance Sheet Arrangements
As of September 30, 2017, Innovate had no off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K as promulgated by the SEC.
| 83 |
Security Ownership of Certain Beneficial Owners and Management
The following table and the related notes present information on the beneficial ownership of shares of the Company’s capital stock as of January 29, 2018 by:
| · | each of the Company’s directors as of the closing of the Merger; |
| · | each of the Company’s executive officers as of the closing of the merger and each of the Company’s named executive officers for the year ended December 31, 2017; |
| · | all of the Company’s current directors and executive officers as a group; and |
| · | each stockholder known by the Company to beneficially own more than five percent of its common stock on an as converted basis. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Shares of common stock that may be acquired by an individual or group within 60 days of January 29, 2018, pursuant to the exercise of options or warrants, are deemed to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table.
Except as indicated in footnotes to this table, the Company believes that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them, based on information provided to the Company by such stockholders. Unless otherwise indicated, the address for each stockholder listed is: c/o Innovate Biopharmaceuticals, Inc., 8480 Honeycutt Road, Suite 200, Raleigh, NC 27615.
| Name and Address of Beneficial Owner | Shares Beneficially Owned | Percent of Outstanding | ||||||
| Principal Stockholders: | ||||||||
| BrynMawr Technology Holdings | 1,891,104 | 7.34 | % | |||||
| Moonstar Family Group | 2,697,290 | 10.47 | % | |||||
| The Sea Island Partnership | 2,902,060 | 11.26 | % | |||||
| Triangle Healthcare Partners | 1,726,260 | 6.70 | % | |||||
| Directors and Named Executive Officers: | ||||||||
| Jay Madan (1) | 1,087,117 | 4.21 | % | |||||
| Sandeep Laumas, M.D. (2) | 718,966 | 2.78 | % | |||||
| Christopher Prior, Ph.D. (3) | 1,990,686 | 7.17 | % | |||||
| Lorin K. Johnson, Ph.D. (4) | 234,445 | * | ||||||
| Anthony E. Maida III, Ph.D. (5) | 45,376 | * | ||||||
| Anna Kazanchyan, M.D. | - | - | ||||||
| Roy Proujanksy, M.D. | - | - | ||||||
| David H. Clarke (6) | 126,372 | * | ||||||
| Jonathan Clark | 28,500 | * | ||||||
| Stephen R. Brownsell | 13,500 | * | ||||||
| David Olert (7) | 8,684 | * | ||||||
| All directors and executive officers as a group (7 persons) (8) | 4,076,674 | 14.47 | % | |||||
* Represents beneficial ownership of less than 1% of the shares of common stock
| 84 |
| (1) | Includes 130,031 shares held by Madan Global, Inc., 122,516 shares held by OM Healthcare Partners LLC, 122,516 shares held by OM Healthcare Partners II LLC, and 122,516 shares held by OM Healthcare Partners III LLC, and 65,546 shares issuable upon the exercise of an option held by Mr. Madan that are exercisable within 60 days of January 29, 2018. |
| (2) | Includes 651,322 shares held by Bearing Circle Capital LLC and 67,644 shares issuable upon the exercise of an option held by Dr. Laumas that are exercisable within 60 days of January 29, 2018. Dr. Laumas is affiliated with Bearing Circle Capital LLC and has voting and investment power over the shares held by Bearing Circle Capital LLC. |
| (3) | Consists of 1,990,686 shares issuable upon the exercise of options held by Dr. Prior that are exercisable within 60 days of January 29, 2018. |
| (4) | Consists of 234,445 shares issuable upon the exercise of options held by Dr. Johnson that are exercisable within 60 days of January 29, 2018. |
| (5) | Consists of 45,376 shares issuable upon the exercise of an option held by Dr. Maida that is exercisable within 60 days of January 29, 2018. |
| (6) | Includes 56,720 shares held by Mr. Clarke, 1,807 shares held by Leslie Clarke, Mr. Clarke’s wife, and 52,835 shares held by GBS Holdings, Inc., an entity which may be deemed controlled by Mr. Clarke but which is owned by Leslie Clarke and the children of Mr. Clarke. Also includes warrants to purchase (i) 548 shares of common stock held by Mr. Clarke, (ii) 539 shares of common stock held by Leslie Clarke, and (iii) 13,923 shares of common stock held by GBS Holdings, Inc. Mr. Clarke may be deemed the indirect beneficial owner of these securities since he has shared sale, voting and investment control over the securities with his wife. The address of GSB Holdings, Inc. and Mr. Clarke is 14179 Laurel Trail, Wellington, Florida 33414. |
| (7) | Includes 1,684 shares issuable upon the exercise of options held by Mr. Olert that are exercisable within 60 days of January 29, 2018. |
| (8) | Includes 2,403,694 shares issuable upon the exercise of options held by the Company’s current directors and executive that are exercisable within 60 days of January 29, 2018. |
| 85 |
Executive Officers and Directors
At the effective time of the merger, each of Sandeep Laumas, Christopher Prior, Jay Madan, Lorin Johnson, Anna Kazanchyan, Anthony Maida, and Roy Proujansky was appointed to the Board and such individuals constitute our Board as of the date of this report. Additionally, pursuant to the Merger Agreement, our executive management team changed at the effective time of the merger by the resignation of the then-serving executive officers of Monster Digital, Inc. and the appointment of Sandeep Laumas as our Executive Chairman, Christopher Prior as our Chief Executive Officer, and Jay Madan as our President.
The following table sets forth the names, ages and positions of each of our directors and executive officers as of the date of this report:
| Name | Age | Position(s) | ||
| Executive Officers | ||||
| Sandeep Laumas, M.D. | 49 | Executive Chairman | ||
| Christopher Prior, Ph.D. | 65 | Chief Executive Officer and Director | ||
| Jay Madan, M.S. | 52 | President and Director | ||
| Lorin K. Johnson, Ph.D. | 65 | Director | ||
| Anna Kazanchyan, M.D. | 49 | Director | ||
| Anthony E. Maida, Ph.D., M.A., M.B.A | 65 | Director | ||
| Roy Proujansky, M.D. | 61 | Director |
Executive Officers
Sandeep Laumas, M.D. Dr. Laumas joined Innovate in 2014 as its Executive Chairman. In August 2007 Dr. Laumas founded Bearing Circle Capital, LP and has served as its Managing Director since such time. Dr. Laumas began his career at Goldman Sachs & Co. in 1996 as an equity analyst in the healthcare investment banking division working on mergers, acquisitions, and corporate finance transactions before transitioning to the healthcare equity research division. After leaving Goldman Sachs in 2000, Dr. Laumas moved to the buy side as an analyst at Balyasny Asset Management from 2001 to 2003. Dr. Laumas was a Managing Director of North Sound Capital from 2003 to 2007, where he was responsible for the global healthcare investment portfolio. From February 2011 to 2012 he was a member of the board of directors of Super Religare Laboratories Limited, Southeast Asia’s largest clinical laboratory service company. Dr. Laumas also served as a Director of Parkway Holdings Ltd. (acquired by IHH Healthcare for $3 Billion: Singapore: IHH) from May through August 2010. Dr. Laumas received his A.B. (Chemistry) from Cornell University in 1990, M.D. from Albany Medical College in 1995 with a research gap year at the Dana-Farber Cancer Institute and completed his medical internship in 1996 from the Yale University School of Medicine.
The Company believes that Dr. Laumas’ prior board service and years of experience investing in the healthcare industry qualifies Dr. Laumas to serve on the Board.
Christopher P. Prior, Ph.D. Dr. Prior joined Innovate as its Chief Executive Officer in 2015. From April 2008 to October 2014, he served as the Chief Executive Officer of Phasebio Pharmaceuticals, Inc., a clinical stage biopharmaceutical company. Prior to that, he founded Principia Pharmaceutical Corporation, a company that develops biopharmaceutical products for chronic diseases, where he served as President, and BioRexis Pharmaceuticals Corporation, a biopharmaceutical company developing diabetes candidates and novel therapeutic agents, where he served as the President and Chief Scientific Officer. During the course of his 30-year career, he has generated more than 25 INDs and achieved four product approvals from the FDA. Dr. Prior received his Bachelor of Science, with honors, in Chemistry from the University of London, and received a Ph.D. in Biochemistry from Columbia University. Dr. Prior also completed a research fellowship at The Rockefeller Medical Institute in New York. Dr. Prior is a member of the New York Academy of Sciences and is the author of numerous publications and patents focused on the development of therapeutics.
| 86 |
The Company believes that Dr. Prior’s role as chief executive officer of Innovate and extensive experience as an executive in the biopharmaceutical industry qualifies him to serve on the board of directors.
Jay P. Madan, M.S. Mr. Madan founded Innovate in 2012 and has served as its President and as a member of the board of directors since such time. Prior to that, Mr. Madan was an independent contractor advising multiple life sciences companies, including Reliance Life Sciences, Millipore, Baxter, Dade Behring, and Goodwin. This experience in working across multiple teams led him to develop a global network of healthcare professionals. From July 2007 to November 2008, Mr. Madan served as the VP of Business Development at Reliance Biopharmaceuticals Pvt. Ltd., a part of Reliance Industries Ltd., India’s largest conglomerate. While at Reliance and Goodwin, Mr. Madan was focused on the development of their contract manufacturing businesses. Mr. Madan holds a Bachelor of Science degree in Chemical Engineering from University of Mumbai and an M.S. in Chemical Engineering from Washington State University.
The Company believes that Mr. Madan’s role as a co-founder of Innovate and extensive experience in the life sciences and biotech industries qualifies him to serve on the Board.
Non-Employee Directors
Lorin K. Johnson, Ph.D. Dr. Johnson is the founder and Chief Scientist of Glycyx PharmaVentures Ltd., a biopharma investment and development company. In 1989, he co-founded Salix Pharmaceuticals, Inc. (NASDAQ: SLXP), a specialty pharmaceutical company, and held senior leadership positions prior to its $15.8 billion acquisition by Valeant Pharmaceuticals International, Inc. (NYSEA: VRX) in April 2015. Prior to Salix, Dr. Johnson served as Director of Scientific Operations and Chief Scientist at Scios, Inc. (formerly, California Biotechnology, Inc). He is a board member of Sigmoid Pharma, a GI specialty drug delivery company based in Dublin, Ireland. In addition to his career in industry, Dr. Johnson has served as an Assistant Professor of Pathology at Stanford University Medical Center and held academic positions at Stanford University School of Medicine and the University of California, San Francisco. He is the co-author of 75 journal articles and book chapters and is the co-inventor on 18 issued patents. Dr. Johnson holds a PhD from the University of Southern California and was a Postdoctoral Fellow at the University of California, San Francisco.
The Company believes that Dr. Johnson’s extensive experience in the pharmaceutical and life science industries, both as an executive and investor, qualifies him to serve on the Board.
Anna Kazanchyan, M.D. Dr. Kazanchyan founded Saghmos Therapeutics, a company focused on the prevention of contrast-induced acute kidney injury, in September 2016 and serves as its CEO and Chairwoman. Dr. Kazanchyan has served as a member of the board of directors of Foamix Pharmaceuticals (NASDAQ: FOMX) since December 2014 and currently serves on its compensation committee. She is also the founder and Managing Partner since April 2004 of Primary i-Research, LLC, where she provides due diligence to leading healthcare investment funds and evaluates investment prospects of biopharmaceutical companies based on the scientific, clinical, regulatory, and commercial outlook for their products. In addition, she has been a strategic advisor to CEOs of biopharmaceutical companies (start-ups to global companies) and has advised companies on matters related to business development, regulatory strategy, marketing, and commercial/competitive landscape. Previously, Dr. Kazanchyan was Senior Biotechnology Analyst at Wachovia Securities, and was a member of the #1 and #2 Institutional-Investor ranked Biotechnology Equity Research teams at Goldman Sachs and Citigroup, respectively. She received an M.D. from Harvard Medical School and a B.A. in Biology, summa cum laude, from Clark University.
The Company believes that Dr. Kazanchyan’s 20 years of experience leading and advising companies in the biopharmaceutical and therapeutics industries qualifies her to serve on the Board.
| 87 |
Anthony E. Maida III, Ph.D., M.A., M.B.A. Dr. Maida has wide experience in the biotechnology industry for more than two decades serving as a CEO, member of the board of directors and working with biotechnology investors. From 1997 through 2010, Dr. Maida served as Chairman, Founder and Director of BioConsul Drug Development Corporation and Principal of Anthony Maida Consulting International, servicing pharmaceutical and investment firms, in the clinical development of therapeutic products and product/company acquisitions. From June 2009 through June 2010, Dr. Maida served as Vice President of Clinical Research and General Manager, Oncology, Worldwide for PharmaNet, Inc., a clinical research organization. Since June 2010, Dr. Maida has served as Senior Vice President, Clinical Research for Northwest Biotherapeutics, Inc., a cancer vaccine company focused on therapy for patients with glioblastoma multiforme and prostate cancer. From 1992 to September of 1999, Dr. Maida was President and Chief Executive Officer of Jenner Biotherapies, Inc., an immunotherapy company. Dr. Maida has served in a number of executive roles including President and CEO of Replicon NeuroTherapeutics, Inc. Dr. Maida is currently a member of the Board of Directors and Audit Chair of Spectrum Pharmaceuticals, Inc (NASDAQ GS: SPPI)., Vitality Biopharma, Inc. (OTCQB: VBIO) and OncoSec Medical Inc. (OTCQB: ONCS). Dr. Maida holds a B.A. in Biology and History, an M.B.A., an M.A. in Toxicology and a Ph.D. in Immunology. He is a member of the American Society of Clinical Oncology (ASCO), the American Association for Cancer Research (AACR), the Society of Neuro-Oncology, the International Society for Biological Therapy of Cancer and the American Chemical Society (ACS).
The Company believes that Dr. Maida’s extensive experience as an executive at various biotechnology and biopharmaceutical companies as well as his service on private and public company boards qualifies him to serve on the Board.
Roy Proujansky, M.D. Dr. Proujansky is a pediatric gastroenterologist who since July 2013 has served as the Executive Vice President and Chief Executive of Delaware Valley Operations (DuPont Hospital for Children) for the Nemours Children’s Health System, a non-profit children’s health organization. Before his current position, Dr. Proujansky served as Executive Vice President for Patient Operations and Chief Operating Officer of Nemours from 2006 to July 2013. From 2000 to 2006, Dr. Proujansky was the Robert L. Brent Professor and Chairman of Pediatrics and Associate Dean for Jefferson Medical College at Thomas Jefferson University. Additionally, from 1998 to 2015, Dr. Proujansky was the co-director or direct supervisor of Nemours Research Programs and has authored forty-seven original publications and book chapters in the field of pediatric gastroenterology. Dr. Proujansky received an M.D. from Northwestern University, an MBA from the University of Massachusetts at Amherst and a B.S. in Medical Science from Northwestern University.
The Company believes Dr. Proujansky’s extensive knowledge and experience in the field of pediatric gastroenterology qualifies him to serve on the Board.
| 88 |
Composition of the Board of Directors
The Board consists of seven directors, and each director’s term expires upon the election and qualification of successor directors at the annual meeting of the stockholders to be held in 2018.
There are no family relationships among any of the directors and executive officers.
Director Independence
The Board has determined that a majority of its directors are independent as defined under NASDAQ listing standards. The Board has also determined that each current member of each of the Nominating and Corporate Governance Committee and Compensation Committee is independent as defined under NASDAQ listing standards and that each current member of the Audit Committee and Compensation Committee is independent as defined under NASDAQ listing standards and applicable SEC rules. In making this determination, the Board found that none of these directors had a material or other disqualifying relationship with the Company.
Committees of the Board of Directors
The Board has an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee.
Audit Committee
The Audit Committee of the Board consists of Anthony Maida, Lorin Johnson and Anna Kazanchyan, with Dr. Maida acting as the chair. The primary functions of the Audit Committee include, among other things:
| • | reviewing and approving the engagement of the independent registered public accounting firm to perform audit services and any permissible non-audit services; |
| • | evaluating the performance of the independent registered public accounting firm and deciding whether to retain their services; |
| • | monitoring the rotation of partners on the engagement team of the independent registered public accounting firm; |
| 89 |
| • | reviewing annual and quarterly financial statements and reports and discussing the statements and reports with the Company’s independent registered public accounting firm and management, including a review of disclosures under “Management’s Discussion and Analysis of Financial Condition and Results of Operations;” |
| • | considering and approving or disapproving all related party transactions; |
| • | reviewing, with the Company’s independent registered public accounting firm and management, significant issues that may arise regarding accounting principles and financial statement presentation, as well as matters concerning the scope, adequacy and effectiveness of financial controls; |
| • | conducting an annual assessment of the performance of the Audit Committee and its members, and the adequacy of its charter; and |
| • | establishing procedures for the receipt, retention and treatment of complaints received by the Company regarding financial controls, accounting or auditing matters. |
Each member of the Audit Committee satisfies the independence requirements under NASDAQ listing standards and Rule 10A-3(b)(1) of the Exchange Act and is a person who the Board has determined has the requisite financial expertise required under the applicable requirements of NASDAQ. In arriving at this determination, the Board examined each Audit Committee member’s scope of experience and the nature of their employment in the corporate finance sector. The Board has also determined that Dr. Maida qualifies as an “audit committee financial expert,” as defined in applicable SEC rules.
Compensation Committee
The Compensation Committee of the Board consists of Anna Kazanchyan, Lorin Johnson and Anthony Maida, with Dr. Kazanchyan acting as the chair. The functions of the Compensation Committee include, among other things:
| • | determining the compensation and other terms of employment of the chief executive officer and its other executive officers and reviewing and approving corporate performance goals and objectives relevant to such compensation; |
| • | reviewing and recommending to the full Board the compensation of the Monster directors; |
| • | evaluating and administering the equity incentive plans, compensation plans and similar programs advisable for the Company, as well as reviewing and recommending to the Board the adoption, modification or termination of plans and programs; |
| • | establishing policies with respect to equity compensation arrangements; |
| • | if required, reviewing with management the Company’s disclosures under the caption “Compensation Discussion and Analysis” and recommending to the full the Board its inclusion in the Company’s periodic reports to be filed with the SEC; and |
| • | reviewing and evaluating, at least annually, the performance of the Compensation Committee and the adequacy of its charter. |
| 90 |
The Board has determined that each current member of the Compensation Committee is independent under NASDAQ listing standards, a “non-employee director” as defined in Rule 16b-3 promulgated under the Exchange Act and an “outside director” as that term is defined in Section 162(m) of the Code.
Nominating and Corporate Governance Committee
The Nominating and Governance Committee of the Board currently consists of Lorin Johnson, Anna Kazanchyan and Anthony Maida, with Dr. Johnson acting as the chair. The functions of the Nominating and Corporate Governance Committee include, among other things, the following:
| • | reviewing periodically and evaluating director performance on the Board and its applicable committees, and recommending to the Board and management areas for improvement; |
| • | interviewing, evaluating, nominating and recommending individuals for membership on the Board; |
| • | reviewing and recommending to our board of directors any amendments to the Company’s corporate governance policies; and |
| • | reviewing and assessing, at least annually, the performance of the Nominating and Corporate Governance committee and the adequacy of its charter. |
The Board has determined that each member of the Nominating and Corporate Governance Committee is independent under Nasdaq listing standards.
The Board may from time to time establish other committees.
2016 Innovate Director Compensation
Innovate did not have any directors in the year ended December 31, 2016, who were not employed by Innovate.
Compensation Committee Interlocks and Insider Participation
Each member of the Compensation Committee is an “outside” director as that term is defined in Section 162(m) of the Internal Revenue Code, a “non-employee” director within the meaning of Rule 16b-3 of the rules promulgated under the Exchange Act and independent within the meaning of the independent director guidelines of the NasdaqCM. None of the executive officers serve as a member of the board of directors or compensation committee of any entity that has one or more executive officers who serves on the Board or Compensation Committee.
| 91 |
Executive Compensation
This section discusses the material components of the executive compensation program offered to Innovate’s named executive officers identified below.
2016 Summary Compensation Table
The following table provides information regarding Innovate’s named executive officers during the fiscal year ended December 31, 2016. These individuals are referred to elsewhere in this current report as the “named executive officers” of Innovate.
| Name and Principal Position | Year | Salary | Bonus | Option Awards(1) | Total | |||||||||||||||
| Sandeep Laumas, M.D. Executive Chairman | 2016 | $ | 18,000 | (2) | $ | 2,100 | $ | — | $ | 20,100 | ||||||||||
| Christopher Prior, Ph.D. Chief Executive Officer | 2016 | $ | 18,000 | $ | 2,100 | $ | 1,250,392 | $ | 1,270,492 | |||||||||||
| Jay P. Madan President, Corporate Development | 2016 | $ | 30,000 | (3) | $ | 4,500 | $ | — | $ | 34,500 | ||||||||||
| (1) | The amounts in the “Option Awards” column reflect the aggregate grant date fair value of stock options granted during the calendar year computed in accordance with the provisions of Accounting Standards Codification (ASC) 718, Compensation — Stock Compensation. The assumptions that Innovate used to calculate these amounts are discussed in the notes to the December 31, 2016 and 2015 audited financial statements of Innovate included elsewhere in this proxy statement/information statement. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the stock options, the exercise of the stock options, or the sale of the common stock underlying such stock options. |
| (2) | As described below under the heading “Employment and Severance Agreements,” under the terms of Dr. Laumas’ employment agreement, a portion of the amount of the 2016 base salary set forth in the agreement was deferred and would be paid if Innovate reached a specified financial milestone prior to March 15, 2017. The milestone was not reached by that date, and the amount in the table reflects the amounts paid in 2016. |
| (3) | As described below under the heading “Employment and Severance Agreements,” under the terms of Mr. Madan’s employment agreement, a portion of the amount of the 2016 base salary set forth in the agreement was deferred and would be paid if Innovate reached a specified financial milestone prior to March 15, 2017. The milestone was not reached by that date, and the amount in the table reflects the amounts paid in 2016. |
Narrative Disclosure to Summary Compensation Table
The primary elements of compensation for Innovate’s named executive officers are base salary, bonus and equity-based compensation awards. The named executive officers also participate in employee benefit plans and programs that Innovate offers to its other full-time employees on the same basis.
Base Salary
The base salary payable to Innovate’s named executive officers is intended to provide a fixed component of compensation that reflects the executive’s skill set, experience, role and responsibilities.
Bonus
Although Innovate does not have a written bonus plan, the Innovate Board may, in its discretion, award bonuses to its executive officers on a case-by-case basis. These awards are structured to reward named executive officers for the successful performance of Innovate as a whole and of each participating named executive officer as an individual. The bonus amounts awarded in 2016 were on an entirely discretionary basis. In addition, as described under the heading “Employment and Severance Agreements,” each of the named executive officers is eligible under the terms of their respective employment agreements to receive set bonus amounts based on Innovate’s achievement of certain financial milestones.
| 92 |
Health, Welfare and Additional Benefits
Each of Innovate’s named executive officers is eligible to participate in Innovate’s employee benefit plans and programs, including medical, dental and vision benefits, to the same extent as its other full-time employees, subject to the terms and eligibility requirements of those plans.
Although Innovate does not have a formal policy with respect to the grant of equity incentive awards to its executive officers or any formal equity ownership guidelines applicable to them, Innovate believes that equity grants provide its executives with a strong link to Innovate’s long-term performance, create an ownership culture and help to align the interests of Innovate’s executives and its stockholders. In addition, Innovate believes that equity grants with a time-based vesting feature promote executive retention because this feature incentivizes executive officers to remain in Innovate’s employment during the vesting period. In 2016, Innovate granted Dr. Prior options to purchase up to an aggregate of 5,400,000 shares (as adjusted for a one-for-three stock split in 2016). The options have an exercise price of $0.1133, and vested as to 4,050,000 shares on November 2, 2015, with 37,500 vesting each month over a period of three years thereafter.
2016 Outstanding Equity Awards at Year-End
The following table presents the outstanding equity awards held by Innovate’s named executive officers as of December 31, 2016.
| Option Awards | ||||||||||||||
| Name | Number of Securities Underlying Unexercised Options Exercisable | Number of Securities Underlying Unexercised Options Unexercisable | Option Exercise price | Option Expiration date | ||||||||||
| Christopher Prior, Ph.D. | 4,537,500 | 862,500 | $ | 0.1133 | June 30, 2026 | |||||||||
Employment and Severance Agreements
Innovate has entered into employment agreements with each of its named executive officers described below, and standard confidential information and/or inventions assignment agreements, under which each of its named executive officers has agreed not to disclose Innovate’s confidential information. Each agreement is for an initial term of three years from the Minimum Financial Milestone Event, defined as the sale by Innovate of its equity securities in a bona fide equity financing in which Innovate receives gross proceeds of not less than $5,000,000, and is thereafter automatically renewed until the employment agreement is terminated or either party provides notice of non-renewal.
| 93 |
Sandeep Laumas, M.D.
Innovate entered into an executive employment agreement with Dr. Laumas in October 2015, which was subsequently amended in February 2016, March 2017 and August 2017.
The agreement provided for an initial base salary of $75,000, which was increased to $111,000 effective July 1, 2016. The agreement provides that the base salary was to be deferred until the time of the Minimum Financial Milestone Event; however, if such Minimum Financial Milestone Event did not occur on or before March 15, 2017, Dr. Laumas agreed to forfeit such base salary for the period of January 1, 2016, through December 31, 2016. The Minimum Financial Milestone Event occurred after March 15, 2017.
Commencing January 1, 2017, Dr. Laumas’ annual base salary exceeding $75,000 was subjected to deferral, with such deferral and salary accrual commencing January 1, 2017 and continuing until the Minimum Financial Milestone Event occurred, so long as the Minimum Financial Milestone Event occurred on or prior to March 15, 2018. If the Minimum Financial Milestone Event does not occur on or before March 15, 2018, Dr. Laumas agreed to forfeit such 2017 deferred salary for the period of January 1, 2017, through December 31, 2017.
From and after the occurrence of the Minimum Financial Milestone Event, Dr. Laumas’ annual base salary shall be $150,000 and shall not be subject to deferral. Upon the occurrence of the Second Financial Milestone Event, Dr. Laumas’ annual base salary increases to $160,000. Upon the occurrence of the Third Financial Milestone Event, Dr. Laumas’ annual base salary increases to $175,000. Upon the occurrence of the Fourth Financial Milestone Event, Dr. Laumas’ annual base salary would increase to $300,000.
The agreement also provides that Dr. Laumas will be eligible to receive a one-time lump sum cash bonus in the amount of $25,000 upon the occurrence of the Minimum Financial Milestone Event, a one-time lump sum cash bonus in the amount of $110,000 upon the occurrence of the Second Financial Milestone Event, a one-time lump sum cash bonus in the appoint of $175,000 upon the occurrence of the Minimum Third Milestone Event, and a one-time lump sum cash bonus in the amount of $175,000 upon the occurrence of the Minimum Fourth Milestone Event.
For the months of July, August and September 2016, Dr. Laumas was eligible for a discretionary monthly bonus in the amount of $700 per month. If a Minimum Financial Milestone Event has not occurred by March 15, 2017, Dr. Laumas was eligible for a discretionary bonus of $75,000, awarded in Innovate’s discretion upon the achievement of certain corporate objectives on or before December 31, 2017.
Dr. Laumas is also eligible to receive periodic stock or option awards in the discretion of Innovate.
Christopher P. Prior, Ph.D.
Innovate entered into an executive employment agreement with Dr. Prior in November 2015, which was subsequently amended in February 2016, twice in March 2017, and in August 2017.
Upon the occurrence of the Minimum Financial Milestone Event, Dr. Prior was entitled to an annual base salary of $240,000. Upon the occurrence of the Second Financial Milestone Event, defined as the sale by Innovate of its equity securities in a bona fide equity financing or the sale of assets or entry into out-licensing and/or partnering agreements in which Innovate receives gross proceeds of not less than $10,000,000 (including proceeds from the Minimum Financial Milestone Event), Dr. Prior’s annual base salary increases to $260,000. Upon the occurrence of the Third Financial Milestone Event, defined as the sale by Innovate of its equity securities in a bona fide equity financing or the sale of assets or entry into out-licensing and/or partnering agreements in which Innovate receives gross proceeds of not less than $25,000,000 (including proceeds from the Minimum Financial Milestone Event and the Second Financial Milestone Event), Dr. Prior’s annual base salary increases to $300,000. Upon the occurrence of the Fourth Financial Milestone Event, defined as the sale by Innovate of its equity securities in a bona finde equity financing or the sale of assets or entry into out-licensing and/or partnering agreements in which Innovate receives gross proceeds of not less than $45,000,000 (including proceeds from the Minimum Financial Milestone Event, the Second Milestone Financial Event and the Third Milestone Financial Event), Dr. Prior’s annual base salary increases to $425,000.
| 94 |
The agreement also provides that Dr. Prior will be eligible to receive a one-time lump sum cash bonus in the amount of $60,000 upon the occurrence of the Minimum Financial Milestone Event, a one-time lump sum cash bonus in the amount of $125,000 upon the occurrence of the Second Financial Milestone Event, a one-time lump sum cash bonus in the appoint of $175,000 upon the occurrence of the Minimum Third Milestone Event, and a one-time lump sum cash bonus in the appoint of $175,000 upon the occurrence of the Minimum Fourth Milestone Event.
Following the completion of the Minimum Financial Milestone Event, Dr. Prior became eligible for an annual grant of restricted stock for each year of service subject to the completion of certain milestones and the approval of the Innovate Board. Such grants would vest with respect to 25% of the restricted stock on the one year anniversary of the date of grant and thereafter with respect to 75% of the stock over the following three years. Upon a change of control, 100% of the unvested shares of restricted stock would vest.
Jay P. Madan, M.S.
Innovate entered into an executive employment agreement with Mr. Madan in October 2015, which was subsequently amended in February 2016, March 2017 and August 2017.
The agreement provided for an initial base salary of $90,000, which was increased to $150,000 effective July 1, 2016. The agreement provides that the 2016 base salary was to be deferred until the time of the Minimum Financial Milestone Event; however, if such Minimum Financial Milestone Event did not occur on or before March 15, 2017, Mr. Madan agreed to forfeit such base salary for the period of January 1, 2016, through December 31, 2016. The Minimum Financial Milestone Event occurred after March 15, 2017.
Commencing January 1, 2017, Mr. Madan’s annual base salary exceeding $90,000 was subjected to deferral, with such deferral and salary accrual continuing until the Minimum Financial Milestone Event occurred; however, if the Minimum Financial Milestone Event does not occur on or before March 15, 2018, Mr. Madan agreed to forfeit the 2017 deferred salary.
From and after the occurrence of the Minimum Financial Milestone Event, the agreement provides Mr. Madan’s annual base salary shall be $180,000 and shall not be subject to deferral. Upon the occurrence of the Second Financial Milestone Event, Mr. Madan’s annual base salary increases to $210,000. Upon the occurrence of the Third Financial Milestone Event, Mr. Madan’s annual base salary increases to $250,000. Upon the occurrence of the Fourth Financial Milestone Event, Mr. Madan’s annual base salary increases to $350,000.
The agreement also provides that Mr. Madan will be eligible to receive a one-time lump sum cash bonus in the amount of $30,000 upon the occurrence of the Minimum Financial Milestone Event, a one-time lump sum cash bonus in the amount of $115,000 upon the occurrence of the Second Financial Milestone Event, a one-time lump sum cash bonus in the appoint of $150,000 upon the occurrence of the Minimum Third Milestone Event, and a one-time lump sum cash bonus in the amount of $125,000 upon the occurrence of the Minimum Fourth Milestone Event.
For the months of July, August and September 2016, Mr. Madan was eligible for a discretionary monthly bonus in the amount of $1,500 per month. If a Minimum Financial Milestone Event has not occurred by March 15, 2017, Mr. Madan was eligible for a discretionary bonus of $90,000, awarded in Innovate’s discretion upon the achievement of certain corporate objectives on or before December 31, 2017.
Mr. Madan is also eligible to receive periodic stock or option awards in the discretion of Innovate.
| 95 |
Potential Payments Upon Termination of Employment or Change in Control
Pursuant to the terms of the executive employment agreement with Dr. Prior, upon termination of the agreement if Innovate does not renew the agreement for a reason unrelated to “cause” (as defined in the agreement), by Dr. Prior for “good reason” (as defined in the agreement”), or by Innovate for reasons other than “cause” (as defined in the agreements), death, or disability, liquidation or dissolution of Innovate, then, subject to Dr. Prior timely signing and not revoking a separation agreement and release of claims agreement, Dr. Prior would be entitled to receive:
| • | a lump sum payment equal to (i) twelve months of his then-current base salary; and |
| • | reimbursements for payments he makes for continued healthcare coverage pursuant to COBRA until the earlier of (i) the date twelve months from the termination date or (ii) the date on which he obtains reasonably comparable coverage. |
Pursuant to the terms of the executive employment agreement with each of Dr. Laumas and Mr. Madan, upon termination of the agreement if Innovate does not renew the agreement for a reason unrelated to “cause” (as defined in the agreements), by the executive for “good reason” (as defined in the agreements), or by Innovate for reasons other than “cause” (as defined in the agreements), death, or disability, liquidation or dissolution of Innovate, then, subject to the executive’s timely signing and not revoking a separation agreement and release of claims agreement, the executive would be entitled to receive:
| • | a lump sum payment equal to (i) six months of his then-current base salary; and |
| • | reimbursements for payments he makes for continued healthcare coverage pursuant to COBRA until the earlier of (i) the date six months from the termination date or (ii) the date on which he obtains reasonably comparable coverage. |
Additionally, upon termination of the agreement with each of the above-named executives, pursuant to expiration of the term based on a non-renewal notice or by the executive for “good reason” or for other than “good reason” upon 30 days’ notice, Innovate may elect to pay an amount equal to the executive’s then current base salary for all or any portion of the applicable notice periods required pursuant to the agreements in lieu of all or any portion of such notice period.
| 96 |
Indemnification of Officers and Directors
Innovate has entered into agreements to indemnify its directors, executive officers and other employees as determined by the board of directors. With specified exceptions, these agreements provide for indemnification for related expenses including, among other things, attorneys’ fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding. Innovate believes that the provisions in its Bylaws and indemnification agreements described above are necessary to attract and retain talented and experienced officers and directors.
| 97 |
Described below are transactions occurring since January 1, 2015, and any currently proposed transactions to which Innovate was a party and in which:
| • | The amounts involved exceeded or will exceed $120,000; and |
| • | A director, executive officer, holder of more than 5% of the outstanding capital stock of Innovate, or any member of such person’s immediate family had or will have a direct or indirect material interest, other than compensation, termination and change of control arrangements that are described under the section titled “Executive Compensation” in this proxy statement/information statement. |
In 2016, Innovate made a loan to Jay Madan and his affiliates of $135,000. Mr. Madan repaid $60,000 of the borrowed amount in 2016 and $75,000 remains outstanding.
| 98 |
Indemnification of Directors and Officers
Section 145 of the Delaware General Corporation Law authorizes a court to award, or a corporation’s board of directors to grant, indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities, including reimbursement for expenses incurred, arising under the Securities Act of 1933, as amended. The Company’s amended and restated certificate of incorporation provides for indemnification of our directors, officers, employees and other agents to the maximum extent permitted by the Delaware General Corporation Law, and our amended and restated bylaws provide for indemnification of our directors, officers, employees and other agents to the maximum extent permitted by the Delaware General Corporation Law.
We have entered into indemnification agreements with our directors and executive officers, whereby we have agreed to indemnify our directors and executive officers to the fullest extent permitted by law, including indemnification against expenses and liabilities incurred in legal proceedings to which the director or executive officer was, or is threatened to be made, a party by reason of the fact that such director or executive officer is or was our director, officer, employee or agent, provided that such director or executive officer acted in good faith and in a manner that the director or executive officer reasonably believed to be in, or not opposed to, the our best interest. At present, there is no pending litigation or proceeding involving any of our directors or executive officers regarding which indemnification is sought, nor are we aware of any threatened litigation that may result in claims for indemnification.
We maintain insurance policies that indemnify our directors and officers against various liabilities arising under the Securities Act of 1933, as amended, and the Securities Exchange Act of 1934, as amended, that might be incurred by any director or officer in his capacity as such.
Reference is made to the financial statements and pro forma financial information relating to Innovate contained in item 9.01 of this Current Report on form 8-K, which is incorporated herein by reference.
| 99 |
Item 9.01. Financial Statements and Exhibits.
| (a) | Financial Statements of Business Acquired. |
Innovate
Biopharmaceuticals, Inc.
Condensed Financial Statements
For the Three and Nine-Months Ended September 30, 2017 and 2016
| 100 |
Innovate Biopharmaceuticals, Inc.
Table of Contents
Condensed Financial Statements
| 101 |
INNOVATE BIOPHARMACEUTICALS, INC.
Condensed Balance Sheets
| September 30, | December 31, | |||||||
| 2017 | 2016 | |||||||
| (Unaudited) | (Note 1) | |||||||
| Assets | ||||||||
| Cash | $ | 1,478,831 | $ | 360,811 | ||||
| Due from related party | 75,000 | 75,000 | ||||||
| Prepaid expenses and other current assets | 52,528 | 12,085 | ||||||
| Total current assets | 1,606,359 | 447,896 | ||||||
| Computer equipment, net | 6,845 | 7,767 | ||||||
| Total assets | $ | 1,613,204 | $ | 455,663 | ||||
| Liabilities | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 2,819,292 | $ | 1,752,045 | ||||
| Accrued expenses | 1,726,536 | 1,723,225 | ||||||
| Convertible promissory notes, net | 7,475,444 | - | ||||||
| Convertible promissory notes, related party, net | 243,065 | - | ||||||
| Accrued interest | 416,075 | - | ||||||
| Total current liabilities | 12,680,412 | 3,475,270 | ||||||
| Convertible promissory notes, net | - | 3,166,137 | ||||||
| Convertible promissory notes, related party, net | - | 238,199 | ||||||
| Accrued interest | - | 163,611 | ||||||
| Total liabilities | 12,680,412 | 7,043,217 | ||||||
| Commitments and contingencies (Note 8) | ||||||||
| Stockholders’ deficit | ||||||||
| Common stock; $0.001 par value, 250,000,000 shares authorized, 31,545,000 shares issued and outstanding | $ | 31,545 | $ | 31,545 | ||||
| Additional paid-in-capital | 5,878,634 | 1,128,800 | ||||||
| Stock subscription receivable | - | (25 | ) | |||||
| Accumulated deficit | (16,977,387 | ) | (7,747,874 | ) | ||||
| Total stockholders’ deficit | (11,067,208 | ) | (6,587,554 | ) | ||||
| Total liabilities and stockholders’ deficit | $ | 1,613,204 | $ | 455,663 | ||||
The accompanying notes are an integral part of these financial statements.
| F-2 |
INNOVATE BIOPHARMACEUTICALS, INC.
Condensed Statements of Operations and Comprehensive Loss
(Unaudited)
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development expenses | $ | 367,551 | $ | 239,297 | $ | 2,832,787 | $ | 885,449 | ||||||||
| General and administrative expenses | 1,973,256 | 1,733,246 | 6,115,088 | 2,705,976 | ||||||||||||
| Total operating expenses | 2,340,807 | 1,972,543 | 8,947,875 | 3,591,425 | ||||||||||||
| Other income (expense): | ||||||||||||||||
| Interest income | - | - | - | 94 | ||||||||||||
| Interest expense | (110,508 | ) | (57,164 | ) | (281,638 | ) | (150,101 | ) | ||||||||
| Total other expense, net | (110,508 | ) | (57,164 | ) | (281,638 | ) | (150,007 | ) | ||||||||
| Net loss | $ | (2,451,315 | ) | $ | (2,029,707 | ) | $ | (9,229,513 | ) | $ | (3,741,432 | ) | ||||
| Comprehensive loss | $ | (2,451,315 | ) | $ | (2,029,707 | ) | $ | (9,229,513 | ) | $ | (3,741,432 | ) | ||||
| Net loss per common share - basic and diluted | $ | (0.08 | ) | $ | (0.06 | ) | $ | (0.29 | ) | $ | (0.12 | ) | ||||
| Weighted-average common shares outstanding - basic and diluted | 31,545,000 | 31,545,000 | 31,545,000 | 31,545,000 | ||||||||||||
The accompanying notes are an integral part of these financial statements.
| F-3 |
INNOVATE BIOPHARMACEUTICALS, INC.
Condensed Statements of Cash Flows
(Unaudited)
| Nine Months Ended September 30, | ||||||||
| 2017 | 2016 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (9,229,513 | ) | $ | (3,741,432 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Share-based compensation | 4,749,834 | 1,023,000 | ||||||
| Accrued interest on convertible promissory notes | 252,464 | 123,021 | ||||||
| Amortization of debt discount | 29,174 | 27,080 | ||||||
| Depreciation | 2,522 | 235 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other assets | (40,443 | ) | (128,355 | ) | ||||
| Accounts payable | 1,067,247 | 408,719 | ||||||
| Accrued expense | 3,311 | 782,348 | ||||||
| Due to related party | - | (28,165 | ) | |||||
| Net cash used by operating activities | (3,165,404 | ) | (1,533,549 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of computer equipment | (1,600 | ) | (5,731 | ) | ||||
| Advances to related party | - | (135,000 | ) | |||||
| Payments from related party | - | 60,000 | ||||||
| Net cash used by investing activities | (1,600 | ) | (80,731 | ) | ||||
| Cash flows from financing activities | ||||||||
| Borrowings from convertible promissory notes | 4,284,999 | 1,853,000 | ||||||
| Payment of stock subscription receivable | 25 | - | ||||||
| Net cash provided by financing activities | 4,285,024 | 1,853,000 | ||||||
| Net increase (decrease) in cash | 1,118,020 | 238,720 | ||||||
| Cash as of beginning of period | 360,811 | 4,207 | ||||||
| Cash as of end of period | $ | 1,478,831 | $ | 242,927 | ||||
| Supplemental schedule of noncash financing activities | ||||||||
| Conversion of due to related party to convertible promissory notes | $ | - | $ | 35,737 | ||||
| Conversion of accrued interest to convertible promissory notes | $ | - | $ | 28,574 | ||||
| Debt conversion feature | $ | - | $ | 80,000 | ||||
The accompanying notes are an integral part of these financial statements.
| F-4 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 1: | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Business Description
Innovate Biopharmaceuticals, Inc. (the “Company”) was originally incorporated in the state of North Carolina on January 12, 2012, as GI Therapeutics, Inc. The Company is developing clinical stage products for celiac disease, pancreatic imaging and ulcerative colitis/inflammatory bowel diseases.
On April 10, 2014, the Company changed its name from GI Therapeutics, Inc. to Innovate Biopharmaceuticals, Inc. and reincorporated in Delaware on June 23, 2014.
Business Risks
The Company faces risks associated with companies whose products are in the early stage of development. These risks include, among others, the Company’s need for additional financing to achieve key development milestones, the need to defend intellectual property rights, and the dependence on key members of management.
Basis of Presentation
The unaudited interim condensed financial statements as of and for the nine months ended September 30, 2017, have been prepared by the Company pursuant to the rules and regulations of the Securities and Exchange Commission (the “SEC”) for interim financial reporting. These financial statements are unaudited and, in the opinion of management, include all adjustments (consisting of normal recurring adjustments and accruals) necessary for a fair statement of the balance sheets, operating results, and cash flows for the periods presented in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). Operating results for the three and nine months ended September 30, 2017, are not necessarily indicative of the results that may be expected for the fiscal year ending December 31, 2017. Certain information and footnote disclosures normally included in annual financial statements prepared in accordance with U.S. GAAP have been omitted in accordance with the SEC’s rules and regulations for interim reporting.
The accompanying unaudited financial statements and related notes should be read in conjunction with the Company’s audited financial statements for the years ended December 31, 2016 and 2015.
| F-5 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 1: | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
Significant Accounting Policies
There have been no material changes to the Company’s significant accounting policies during the nine months ended September 30, 2017 and 2016, as compared to the significant accounting policies disclosed in Note 1 of the financial statements for the years ended December 31, 2016 and 2015. However, the following accounting policies are the most critical in fully understanding the Company’s financial condition and results of operations.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and disclosures made in the accompanying notes to the financial statements. Areas of the financial statements where estimates may have the most significant effect include accrued expenses, share-based compensation, deferred compensation, valuation allowance for income tax assets and management’s assessment of the Company’s ability to continue as a going concern. Changes in the facts or circumstances underlying these estimates could result in material changes and actual results could differ from these estimates.
Accrued Expenses
The Company incurs periodic expenses such as research and development, salaries, taxes, and professional fees. An adjusting entry to accrue expenses is necessary when expenses have been incurred by the Company prior to them being invoiced. When a vendor’s invoice is not received, the Company is required to estimate its accrued expenses. This process involves reviewing quotations and contracts, identifying services that have been performed on the Company’s behalf and estimating the level of service performed and the associated cost incurred for the service when the Company has not yet been invoiced or otherwise notified of the actual cost. The majority of the Company’s service providers invoice monthly in arrears for services performed or when contractual milestones are met. The Company estimates accrued expenses as of each balance sheet date based on facts and circumstances known at that time. The Company periodically confirms the accuracy of its estimates with the service providers and makes adjustments if necessary.
| F-6 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
NOTE 1: SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Accrued expenses consisted of the following as of September 30, 2017 and December 31, 2016:
| 2017 | 2016 | |||||||
| Compensation and benefits | $ | 1,617,776 | $ | 1,682,900 | ||||
| Research and development | 27,760 | 15,833 | ||||||
| Professional fees | 81,000 | 24,492 | ||||||
| Total | $ | 1,726,536 | $ | 1,723,225 | ||||
Research and Development
The Company expenses the cost of research and development as incurred. Research and development expenses comprise costs incurred from contracted services and other external costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity is performed or when the goods have been received, rather than when payment is made, in accordance with Accounting Standards Codification (“ASC”) 730, Research and Development.
Share-Based Compensation
The Company accounts for share-based compensation using the fair value method of accounting which requires all such compensation to employees, including the grant of employee stock options, to be recognized in the Statements of Operations and Comprehensive Loss based on its fair value at the grant date. The expense associated with share-based compensation is recognized on a straight-line basis over the requisite service period of each award; however, the amount of compensation expense recognized at any date must at least equal the portion of the grant-date value of the award that is vested at that date. For share-based compensation granted to non-employees, the measurement date is generally considered to be the date when all services have been rendered or the date that options are fully vested.
| F-7 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 1: | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
Net Loss per Share
The Company calculates net loss per share as a measurement of the Company’s performance while giving effect to all dilutive potential common shares that were outstanding during the reporting period. The Company had a net loss for all periods presented; accordingly, the inclusion of common stock options or other similar instruments would be anti-dilutive. Therefore, the weighted average shares used to calculate both basic and diluted earnings per share are the same.
For the three months ended September 30, 2017 and 2016, 4,755,727 and 4,320,773 potentially dilutive securities related to stock options issued and outstanding have been excluded from the computation of diluted weighted shares outstanding because the effect would be anti-dilutive. For the nine months ended September 30, 2017 and 2016, 4,551,142 and 635,000 potentially dilutive securities related to stock options issued and outstanding have been excluded from the computation of diluted weighted shares outstanding because the effect would be anti-dilutive.
Comprehensive Loss
Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. The Company is required to record all components of comprehensive loss in the financial statements in the period in which they are recognized. Net loss and other comprehensive loss, including foreign currency translation adjustments and unrealized gains and losses on investments are reported, net of their related tax effect, to arrive at a comprehensive loss. For the nine months ended September 30, 2017 and 2016, comprehensive loss was equal to the net loss.
| F-8 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 1: | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued) |
Recent Accounting Pronouncements
In May 2017, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2017-09, Compensation-Stock Compensation (Topic 718): Scope of Modification Accounting. The FASB issued ASU 2017-09 to clarify and reduce both (i) diversity in practice and (ii) cost and complexity when applying the guidance in Topic 718, to a change to the terms and conditions of a share-based payment award. This guidance is effective for the Company for the year-ending December 31, 2018. Early adoption is permitted. The amendments in this ASU should be applied prospectively to an award modified on or after the adoption date. The Company is currently evaluating the effect of this guidance on its financial statements.
Reclassification of Prior Year Presentation
Certain prior year amounts have been reclassified for consistency with the current period presentation. On the condensed balance sheet, certain amounts have been reclassified from “Convertible promissory notes, net” to “Convertible promissory notes, related party, net.” These reclassifications had no effect on the reported results of operations or on the reported amount of cash flows from financing activities for the prior year.
| NOTE 2: | LIQUIDITY AND GOING CONCERN |
The accompanying financial statements have been prepared on a basis which assumes that the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. There is substantial doubt that the Company will continue as a going concern for at least 12 months following the date these financial statements are issued, without additional financing based on the Company’s limited operating history and recurring operating losses. Management’s plans with regard to these matters include seeking additional debt or equity financing arrangements (Note 9). There is no assurance such financing will be available to the Company when required or that such financing will be available under favorable terms, or that the Company can achieve its developmental milestones. The accompanying financial statements do not include any adjustments that might be necessary should the Company be unable to continue as a going concern.
| F-9 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 3: | RELATED PARTY TRANSACTIONS |
Certain owners of the Company were hired as executive employees (“Executives”) of the Company during 2016. In prior years, these Executives paid certain expenses on behalf of the Company and provided certain consulting services through companies owned by the Executives (“Executive Companies”). During 2016, the Company advanced payments to Executive Companies under short-term note receivable arrangements. As of September 30, 2017, and December 31, 2016, there was $75,000 included in due from related party for a note receivable owed to the Company with a due date of March 31, 2018.
During the three and nine months ended September 30, 2017 and 2016, certain Executives and the CEO of the Company provided services to the Company in accordance with the terms of their executive agreements (Note 8). The Company recorded compensation expense for services performed of approximately $166,000 and $470,000, for the three months ended September 30, 2017 and 2016, respectively, and $871,000 and $936,000 for the nine months ended September 30, 2017 and 2016, respectively. Included in accrued expenses as of September 30, 2017 and December 31, 2016, was approximately $1,618,000 and $1,683,000 of compensation expense, respectively.
As of September 30, 2017, and December 31, 2016, there was approximately $195,000 of convertible promissory notes and approximately $21,000 and $12,000 of accrued interest, respectively, owed to certain Executives, including entities where these Executives were deemed to have beneficial ownership. In addition, as of September 30, 2017 and December 31, 2016, the Company owes certain relatives of the CEO approximately $50,000 for convertible promissory notes and approximately $6,000 and $3,000 for accrued interest, respectively.
The Company obtains legal services from a law firm that owns a minority portion of the Company’s common stock. The Company incurred expenses with this law firm of approximately $24,000 and $13,000 during the three months ended September 30, 2017 and 2016, respectively, and $68,000 and $79,000, during the nine months ended September 30, 2017 and 2016, respectively. As of September 30, 2017 and December 31, 2016, approximately $119,000 and $113,000 was recorded in accounts payable to this law firm, respectively.
| F-10 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 4: | CONVERTIBLE PROMISSORY NOTES |
During 2013, the Company entered into a convertible note purchase agreement (the “2013 Notes”) and issued $120,025 in convertible notes which were to convert at 80% of the price paid per share in the next equity financing of $2,500,000 or more. The 2013 Notes bore interest at 5% annually and the outstanding principal and accrued interest were due and payable on December 31, 2015.
During 2015, the Company entered into convertible note purchase agreements (the “2015 Notes”) and issued $650,000 in convertible notes which were to convert at 80.00% of the price paid per share in the next equity financing of $2,500,000 or more. The 2015 Notes bore interest at 5% annually and the outstanding principal and accrued interest were due and payable on December 31, 2015, for certain notes issued and December 31, 2017, for other notes issued, provided no conversion had occurred on or prior to such date.
The 2013 Notes and 2015 Notes included a redemption provision that required the Company, unless the note is converted, if there is a liquidity event, as defined in the agreement, to redeem the note in the amount equal to 150% of the principal balance, plus accrued and unpaid interest. The Company evaluated this redemption feature under the provisions of ASC 405, Accounting for Contingencies, and determined that the likelihood of the Company being required to redeem the note at 150% of its principal balance was not probable.
During January 2016, the Company issued an additional $150,000 of 2015 Notes. On January 22, 2016, the 2013 Notes and 2015 Notes and accrued interest totaling approximately $949,000 were exchanged for new convertible promissory notes (the “2016 Notes”) bearing an interest rate of 7% annually with a maturity date for the outstanding principal and accrued interest of January 22, 2018. The 2016 Notes will convert at 75% of the price paid per share in the next equity financing of $7,500,000 or more. The redemption provision from the 2015 Notes and 2013 Notes was eliminated upon the issuance of the 2016 Notes.
| F-11 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 4: | CONVERTIBLE PROMISSORY NOTES (continued) |
The Company examined the terms of the exchange of the 2015 Notes and 2013 Notes and determined that the exchange did not result in a debt extinguishment under the guidance of ASC 470-50, Debt Modifications and Extinguishments. There was a change in the fair value of the embedded conversion option from changing the conversion rate from 80% to 75% immediately before and after the modification, which resulted in an increase to the fair value of the embedded conversion feature. In accordance with ASC 470-50, the carrying amount of the debt instrument must be adjusted for an increase in the fair value of the embedded conversion option resulting from the modification.
The estimated fair value of the change in the embedded conversion option approximated $80,000 and was recorded as a debt discount and additional paid-in-capital at the modification date of January 22, 2016. The estimated fair value of the embedded conversion option was calculated as the difference in the conversion amount of the original conversion option of 80% versus the new conversion option of 75% at the exchange date, certain future dates and the maturity date of the 2016 Notes based on a probability-weighted scenario, discounted using the effective interest rate of the 2013 and 2015 Notes. Debt discount amortized to interest expense using the effective interest method was approximately $9,000 and $10,000 during the three months ended September 30, 2017 and 2016 and approximately $29,000 and $27,000 during the nine months ended September 30, 2017 and 2016.
After the conversion of the 2015 Notes and 2013 Notes, the Company issued approximately $2,499,000 of additional 2016 Notes in 2016, and approximately $1,885,000 of additional 2016 Notes during the nine months ended September 30, 2017.
In April 2017 the Company issued new convertible promissory notes (the “April 2017 Notes”) bearing an interest rate of 7% annually with a maturity date for the outstanding principal and accrued interest of June 30, 2018. The April 2017 Notes will convert at 75% of the price paid per share in the next equity financing of $20,000,000 or more. For the nine months ended September 30, 2017, the Company issued $1,000,000 in April 2017 Notes.
In September 2017, the Company issued new convertible promissory notes (the “September 2017 Notes”) bearing an interest rate of 7% annually with a maturity date for the outstanding principal and accrued interest of June 30, 2018. The September 2017 Notes will convert at 75% of the price paid per share in the next equity financing of $7,500,000 or more. For the nine months ended September 30, 2017 the Company issued $1,400,000 in September 2017 Notes (Note 9).
The 2016 Notes, April 2017 Notes and September 2017 Notes are secured by all assets of the Company.
The conversion discount embedded in the 2016 Notes, April 2017 Notes and September 2017 Notes creates a contingent beneficial conversion feature which will be recorded as a charge to interest expense when the contingency occurs.
| F-12 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 4: | CONVERTIBLE PROMISSORY NOTES (continued) |
The convertible promissory notes consist of the following as of September 30, 2017 and December 31, 2016:
| 2017 | 2016 | |||||||
| Convertible promissory notes | $ | 7,486,936 | $ | 3,201,937 | ||||
| Less debt discount | (11,492 | ) | (35,800 | ) | ||||
| Total | $ | 7,475,444 | $ | 3,166,137 | ||||
The convertible promissory notes, related party consist of the following as of September 30, 2017 and December 31, 2016:
| 2017 | 2016 | |||||||
| Convertible promissory notes, related party | $ | 245,399 | $ | 245,399 | ||||
| Less debt discount | (2,334 | ) | (7,200 | ) | ||||
| Total | $ | 243,065 | $ | 238,199 | ||||
| NOTE 5: | LICENSE AGREEMENTS |
During 2015, the Company entered into an Option Agreement (the “Alba Option”) with Alba Therapeutics Corporation (“Alba”). The Alba Option provided the Company with a period of time to evaluate Alba’s intellectual property and enter into a license agreement with Alba. During 2015, the Company paid $225,000 to Alba for the Alba Option, which was recorded as research and development expense in the Company’s Statements of Operations. In January 2016, the Company paid the remaining $25,000 option fee and exercised its rights under the Alba Option and in February 2016, entered into another agreement with Alba (the “Alba License”) to obtain the rights to certain intellectual property relating to larazotide acetate and related compounds. The Company’s initial area of focus for these assets relates to the treatment of celiac disease.
Upon execution of the Alba License, the Company paid Alba a non-refundable license fee of $500,000. In addition, the Company is required to make milestone payments to Alba upon the achievement of certain clinical and regulatory milestones totaling up to $1,500,000 and payments upon regulatory approval and commercial sales of a licensed product totaling up to $150,000,000, which is based on sales ranging from $100,000,000 to $1,500,000,000. There was no research and development expense related to the Alba License for the nine months ended September 30, 2017. The Company recorded $525,000 in research and development expenses from the Alba License for the three and nine months ended September 30, 2016.
| F-13 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 5: | LICENSE AGREEMENTS (continued) |
Upon the Company paying Alba $2,500,000 for the first commercial sale of a licensed product, the Alba License becomes perpetual and irrevocable. Upon the achievement of net sales in a year exceeding $1,500,000,000, the Alba License also becomes milestone fee free. The Alba License provides Alba with certain termination rights; including failure of the Company to use Commercially Reasonable Efforts to develop the licensed products.
During 2013, the Company entered into an exclusive license agreement with Seachaid Pharmaceuticals, Inc. (the “Seachaid Agreement”) to further develop and commercialize the licensed product. The agreement shall continue in effect on a country-by-country basis, unless terminated sooner in accordance with the termination provisions of the agreement, until the expiration of the royalty term for such product and such country. The royalty term for each such product and such country shall continue until the earlier of the expiration of certain patent rights (as defined in the agreement) or the date that the sales for one or more generic equivalents makes up a certain percentage of sales in an applicable country during a calendar year. There was no expense recorded from the Seachaid Agreement for the nine months ended September 30, 2017 and 2016.
The agreement also calls for milestone payments totaling up to $6,000,000 to be paid when certain clinical and regulatory milestones are met. There are also commercialization milestone payments ranging from $1,000,000 to $2,500,000 depending on net sales of the products in a single calendar year, followed by royalty payments to be made based on net product sales.
During 2014, the Company entered into an Asset Purchase Agreement with Repligen Corporation (“Repligen”) to acquire Repligen’s RG-1068 program for the development of secretin for the Pancreatic Imaging Market and Magnetic Resonance Cholangiopancreatography. This program is now referred to as INN-329 by the Company. Upon commercialization of a product, royalty payments are to be made based on a percentage of net product sales.
| F-14 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 6: | STOCKHOLDERS’ DEFICIT |
Pursuant to the Articles of Incorporation and Bylaws approved in January 2012, the Company was authorized to issue 1,000,000 shares of common stock having no par value. The Company filed Amended and Restated Articles of Incorporation and Amended and Restated Bylaws in February 2014, which included a 1-to-4 stock split, and increased the authorized shares to 5,000,000 shares of common stock having no par value. During September 2015, the Company amended its Articles of Incorporation and Amended and Restated Bylaws further to increase the authorized shares of stock to 100,000,000 shares of common stock at a par value of $.001 including a 1-to-3 stock split. During August 2016, the Company amended its Articles of Incorporation and Amended and Restated Bylaws further to increase the authorized shares of stock to 250,000,000 shares of common stock at a par value of $0.001 including a 1-to-3 stock split. As a result, the Company recorded a reclassification to increase the amount recorded for common stock to equal its par value as of December 31, 2016. This reclassification had no effect on the results of operations or the total amount of stockholders’ deficit. Share amounts for all periods presented are shown at post-split amounts.
| NOTE 7: | SHARE-BASED COMPENSATION |
The Company has reserved 20,000,000 shares of common stock for issuance to officers, directors, employees and consultants of the Company in accordance with the terms of its 2015 Stock Incentive Plan (the “Stock Plan”).
As of September 30, 2017, there were 1,841,425 shares available for future stock option grants under the Stock Plan. The terms of the agreements are determined by the Company’s board of directors. The Company’s awards vest based on the terms in the agreements with some awards vesting immediately and others over a period of nine months to four years with a term of ten years.
The following summarizes share-based compensation expense recognized in the Company’s financial statements for the three and nine months ended September 30, 2017 and 2016:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Research and development expenses | $ | 314,206 | $ | - | $ | 1,627,606 | $ | - | ||||||||
| General and administrative expenses | 962,728 | 1,023,000 | 3,122,228 | 1,023,000 | ||||||||||||
| Total | $ | 1,276,934 | $ | 1,023,000 | $ | 4,749,834 | $ | 1,023,000 | ||||||||
| F-15 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 7: | SHARE-BASED COMPENSATION (continued) |
The Company utilizes the Black-Scholes option pricing model to value awards under its Stock Plan. Key valuation assumptions include:
| § | Expected dividend yield. The expected dividend is assumed to be zero as the Company has never paid dividends and has no current plans to pay any dividends on the Company’s common stock. |
| § | Expected stock-price volatility. As the Company’s common stock is not publicly traded, the expected volatility is derived from the average historical volatilities of publicly traded companies within the Company’s industry that the Company considers to be comparable to the Company’s business over a period approximately equal to the expected term. |
| § | Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant for zero coupon U.S. Treasury notes with maturities approximately equal to the expected term. |
| § | Expected term. The expected term represents the period that the stock-based awards are expected to be outstanding. The Company’s historical share option exercise experience does not provide a reasonable basis upon which to estimate an expected term because of a lack of sufficient data. Therefore, for employee option, the Company estimates the expected term by using the simplified method provided by the Securities and Exchange Commission. The simplified method calculates the expected term as the average of the time-to-vesting and the contractual life of the options. The expected term for awards to non-employees is the contractual term of the options. |
The fair value of stock options was estimated using the following assumptions for the three and nine months ended September 30, 2017 and September 30, 2016:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Expected volatility | 62.06 | % | 70.60 | % | 73.00 | % | 70.60 | % | ||||||||
| Risk free rate | 1.56 | % | 1.00 | % | 2.20 | % | 1.00 | % | ||||||||
| Dividend yield | 0 | % | 0 | % | 0 | % | 0 | % | ||||||||
| Expected term in years (weighted average) | 6.5 | 5.3 | 8.7 | 5.3 | ||||||||||||
| F-16 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 7: | SHARE-BASED COMPENSATION (continued) |
The following table summarizes stock option activity under the Stock Plan:
| Number of Shares |
Weighted- Average Exercise Price |
Aggregate Intrinsic Value |
Weighted- Average Remaining Contractual Life (In Years) |
|||||||||||||
| Outstanding at September 30, 2016 | 5,400,000 | $ | 0.11 | $ | 1,004,400 | 9.76 | ||||||||||
| Options granted | - | - | - | - | ||||||||||||
| Options forfeited | - | - | - | - | ||||||||||||
| Options exercised | - | - | - | - | ||||||||||||
| Outstanding at December 31, 2016 | 5,400,000 | 0.11 | 3,618,000 | 8.84 | ||||||||||||
| Options granted | 12,758,575 | 0.79 | - | - | ||||||||||||
| Options forfeited | - | - | - | - | ||||||||||||
| Options exercised | - | - | - | - | ||||||||||||
| Outstanding at September 30, 2017 | 18,158,575 | 0.59 | 5,255,540 | 9.29 | ||||||||||||
| Exercisable at September 30, 2017 | 11,876,520 | 0.51 | 4,378,896 | 9.19 | ||||||||||||
| Vested and expected to vest at September 30, 2017 |
17,785,738 | $ | 0.59 | $ | 5,214,800 | 9.28 | ||||||||||
The weighted average grant date fair value of options granted was $0.55 and $0.23 during the three months ended September 30, 2017 and 2016, respectively, and $0.60 and $0.23 for the nine months ended September 30, 2017 and 2016, respectively.
As of September 30, 2017, there was approximately $3,674,000 of total unrecognized compensation cost related to un-vested stock-based compensation arrangements. This cost is expected to be recognized over a weighted average period of 2.5 years.
The Stock Plan provides for accelerated vesting under certain change-of-control transactions.
| F-17 |
Innovate Biopharmaceuticals, Inc.
Notes to Condensed Financial Statements
| NOTE 8: | COMMITMENTS AND CONTINGENCIES |
The Company has employment agreements with certain executives of the Company (the “Executive Agreements”). Under the terms of the Executive Agreements, the Company has agreed to pay the executives certain payments upon the achievement of financial milestone events. These milestone events were based on total debt or equity funding received by the Company. During the nine months ended September 30, 2017, the initial funding milestone was reached and the executives were paid $145,000 in accordance with the terms of the Executive Agreements. The executives are eligible to receive up to $1,595,000 in additional milestone payments upon the achievement of a financing event with gross proceeds of at least $45,000,000 by March 15, 2018. As of September 30, 2017, these deferred compensation payments were included in accrued expenses.
| NOTE 9: | SUBSEQUENT EVENTS |
During July 2017, the Company entered into a definitive Merger Agreement with Monster Digital, Inc. (“Monster”), a publicly held company listed on the NASDAQ Exchange, under which the shareholders of the Company would become the majority owners of Monster. The Company expects to close this Merger in 2017.
In late September 2017, the Company issued additional September 2017 Notes in the aggregate amount of $250,000 to third-party investors. (Note 4). The proceeds from these notes were received in early October 2017.
In November 2017 through January 2018, the Company issued additional 2016 Notes in the aggregate amount of $865,000 to third-party investors (Note 4).
On January 29, 2018, Monster Digital, Inc., a Delaware corporation now known as Innovate Biopharmaceuticals, Inc. (the “Company”) completed its merger with privately-held Innovate Biopharmaceuticals Inc. (“IB Pharmaceuticals Inc.”) in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated July 3, 2017, whereby Monster Merger Sub., Inc. (“Merger Sub”), a wholly owned subsidiary of the Company merged with and into IB Pharmaceuticals Inc., with IB Pharmaceuticals Inc. surviving as the Company’s wholly owned subsidiary (the “Merger”). In connection with the Merger, the Company changed its name from Monster Digital, Inc. to Innovate Biopharmaceuticals, Inc. All references to the “Company” refer to Innovate Biopharmaceuticals, Inc. as of and following the closing of the Merger on January 29, 2018 (the “Closing Date”) and all references to “Monster” refer to Monster Digital, Inc. prior to the closing of the Merger on the Closing Date.
Immediately prior to the closing of the Merger, accredited investors purchased shares of common stock of IB Pharmaceuticals Inc. in a private placement for gross proceeds of approximately $18.13 million (the “Equity Issuance”). IB Pharmaceuticals Inc. issued five-year warrants to each purchaser of common stock with a price per exercise price of $1.2011 (subject to adjustment in connection with the Merger). Concurrently with the Equity Issuance, convertible promissory notes issued by IB Pharmaceuticals Inc. in the aggregate principal amount of approximately $8.65 million were converted into shares of IB Pharmaceuticals Inc. common stock at a price per share of $0.7206 (the “Conversion”).
H.C. Wainwright & Co., LLC (“HCW”) and GP Nurmenkari Inc. (“GPN”) were retained as the placement agents for the Equity Issuance. HCW was paid a flat fee of $250,000.00, a cash fee of $285,000.06 (equal to 10% of the gross proceeds of the Equity Issuance up to a certain cap), a cash fee of $9,018.15 (equal to 3.5% of the gross proceeds in excess of a certain cap), and non-accountable expense allowance of $50,000. GPN was paid a cash fee of $891,266.11 (equal to 10% of the gross proceeds of certain investors in the Equity Issuance) and non-accountable expense allowance of $50,000. IB Pharmaceuticals Inc. issued to affiliates of HCW five-year warrants to purchase 557,097 shares of common stock with an exercise price per share equal to $1.2011(subject to adjustment in connection with the Merger). IB Pharmaceuticals Inc. issued to GPN five-year warrants to purchase 927,529 shares of common stock with an exercise price per share equal to $1.2011(subject to adjustment in connection with the Merger); provided that a small number of warrants (representing 318,776 shares of underlying IB Pharmaceuticals Inc. common stock) were issued to affiliates of GPN with an exercise price per share equal to $0.9609 (subject to adjustment in connection with the Merger). Upon the closing of the Merger, the outstanding shares of IB Pharmaceuticals Inc.’s common stock were exchanged for shares of common stock of Monster at an exchange ratio of one share of IB Pharmaceuticals Inc. common stock to 0.37813802 shares of Monster common stock (the “Exchange Ratio”). Immediately following the closing of the Merger, after giving effect to the Equity Issuance and applying the Exchange Ratio, Monster’s securityholders owned approximately 5.8% of the outstanding common stock of the Company on a fully-diluted basis and IB Pharmaceuticals Inc.’s securityholders owned approximately 94.2% of the outstanding common stock of the Company.
On January 29, 2018, the Company entered into a Note Purchase Agreement and Senior Note Payable (“Note”) with a lender. The principal amount of the Note is $4,800,000 (“Principal”). The Note was issued at a discount of $1,800,000 and net of $20,000 for financing costs, for total proceeds of $2,980,000. The Note matures on September 30, 2018 (“Maturity Date”), however, the Maturity Date may be extended at the option of the lender under certain circumstances as outlined in the Note. Interest on the Note accrues starting on January 29, 2018 at a rate of 12.5% per annum and payments of interest only are due beginning on March 30, 2018 and compound quarterly. Upon the Maturity Date of the Note, the Company is required to pay the lender an amount representing 105% of all outstanding Principal, accrued and unpaid interest, and any unpaid late charges, if applicable (“Outstanding Amount”). The Note contains redemption features and certain non-financial covenants and penalties to the Company in the case of certain events of default, as defined in the Note.
In October 2017, the Company entered into a new three-year lease for office space that expires on September 30, 2020. Base annual rent is $60,000, or $5,000 per month. The first two months of rent were paid in advance upon lease signing and the next ten months of rent will be paid in advance on November 30, 2017. Beginning with month thirteen, monthly payments of $5,000 will be paid in advance of the first day of each month of the remaining term. A security deposit of $5,000 was paid in October 2017. The lease contains a two-year renewal option.
Subsequent events have been evaluated through February 2, 2018, the date at which the financial statements were available to be issued.
| F-18 |
| (b) | Pro Forma Financial Information. |
UNAUDITED PRO FORMA CONDENSED CONSOLIDATED
FINANCIAL STATEMENTS
UNAUDITED PRO FORMA CONDENSED CONSOLIDATED FINANCIAL DATA
The following unaudited pro forma condensed consolidated financial data presents the pro forma financial position and results of operations of (1) Monster based on the historical consolidated financial statements of Monster, after giving effect to the spin-off of all of the business, assets and certain liabilities of Monster; and (2) the consolidated business based on the historical consolidated financial statements of Monster and Innovate, after giving effect to the Monster spin-off and Merger.
The unaudited pro forma consolidated financial information, including the notes thereto, should be read in conjunction with the separate historical financial statements of Monster and Innovate and the sections of this Form 8-K statement entitled “Innovate Management’s Discussion and Analysis of Financial Condition and Results of Operations.” Monster’s historical unaudited consolidated financial statements as of and for the nine months ended September 30, 2017 are included in its Quarterly Report on Form 10-Q filed on November 8, 2017. Monster’s audited consolidated financial statements for the year ended December 31, 2016 are included in its Annual Report on Form 10-K for the year ended December 31, 2016 filed with the SEC on March 31, 2017. Innovate’s historical unaudited financial statements for the nine months ended September 30, 2017 and 2016, and audited financial statements for the year ended December 31, 2016, which are included elsewhere in this Form 8-K.
Unaudited Pro Forma Financial Information For Spin-Co Adjustment
The following selected unaudited pro forma financial data presents the pro forma financial position and results of operations of Monster based on the historical consolidated financial statements of Monster, after giving effect to the spin-off transaction whereby, all of the business, assets and certain of the liabilities of Monster not assumed by Innovate have been acquired by Holdco (“Spin-Co”).
The unaudited pro forma consolidated balance sheet data as of September 30, 2017 gives effect to the Spin-Co transaction as if it took place on September 30, 2017. The unaudited pro forma consolidated statement of operations data for the nine months ended September 30, 2017 gives effect to the Spin-Co transaction as if it took place on January 1, 2017. The unaudited pro forma consolidated statement of operations data for the year ended December 31, 2016 gives effect to the Spin-Co transaction as if it took place on January 1, 2016.
Because the unaudited pro forma consolidated balance sheet data reflects the financial information of Monster as of September 30, 2017, it does not reflect any changes to the current assets which have occurred since September 30, 2017 or which may occur following the date of this Form 8-K.
Unaudited Pro Forma Condensed Consolidated Balance Sheet
| September 30, 2017 (in thousands, except share data and per share data) |
Historical Monster |
SpinCo Adjustments (Note 3) |
Monster Merger Sub | Pro Forma Adjustments | Note 4 | Adjusted Historical Monster |
||||||||||||||||||
| ASSETS | ||||||||||||||||||||||||
| Current assets: | ||||||||||||||||||||||||
| Cash | $ | 174 | $ | (174 | ) | — | (a)(c) | $ | — | |||||||||||||||
| Accounts receivable | 127 | (127 | ) | — | — | — | ||||||||||||||||||
| Inventory | 498 | (498 | ) | — | — | — | ||||||||||||||||||
| Prepaids & other | 257 | (257 | ) | — | — | — | ||||||||||||||||||
| Total current assets | 1,056 | (1,056 | ) | — | — | — | ||||||||||||||||||
| Deposits | 14 | (14 | ) | — | — | |||||||||||||||||||
| Trademark | 2,319 | (2,319 | ) | — | — | — | ||||||||||||||||||
| Total | $ | 3,389 | $ | (3,389 | ) | — | — | $ | — | |||||||||||||||
| LIABILITIES AND STOCKHOLDERS’ (DEFICIT) EQUITY | ||||||||||||||||||||||||
| Current liabilities: | ||||||||||||||||||||||||
| Line of credit | $ | (107 | ) | $ | (107 | ) | — | $ | — | |||||||||||||||
| Accounts payable | 624 | (351 | ) | 273 | 273 | |||||||||||||||||||
| Accrued expenses | 1,506 | (763 | ) | 743 | — | 743 | ||||||||||||||||||
| Customer deposits | 1,336 | (736 | ) | 600 | (600 | ) | (b) | — | ||||||||||||||||
| Due to related parties | 34 | (34 | ) | — | — | — | ||||||||||||||||||
| Notes payable | 1,270 | (38 | ) | 1,232 | (1,232 | ) | (d) | — | ||||||||||||||||
| Total current liabilities | 4,877 | (2,029 | ) | 2,848 | (1,832 | ) | — | 1,016 | ||||||||||||||||
| Stockholders’ (deficit) equity: | ||||||||||||||||||||||||
| Common stock | 1 | — | 1 | (1) | — | |||||||||||||||||||
| Additional paid-in capital | 35,986 | — | 35,986 | (35,986 | ) | (a)(b)(c)(d)(e)(f) | — | |||||||||||||||||
| Accumulated deficit | (37,475 | ) | (1,360 | ) | (38,835 | ) | 37,819 | (a)(c)(e)(f) | (1,016) | |||||||||||||||
| Total stockholders’ (deficit) equity | (1,488 | ) | (1,360 | ) | (2,848 | ) | 1,832 | (1,016) | ||||||||||||||||
| Total | $ | 3,389 | $ | (3,389 | ) | — | — | $ | — | |||||||||||||||
The accompanying notes are an integral part of these unaudited pro forma financial statements.
Unaudited Pro Forma Condensed Consolidated Statement of Operations
| For the Nine Months Ended September 30, 2017 (in thousands, except per share data) | Historical Monster | SpinCo Adjustments (Note 3) | Monster Merger Sub, Inc. | Pro Forma Adjustments | Note 4 | Adjusted Historical Monster | ||||||||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||||||||||
| Revenue | $ | 1,277 | $ | (1,277 | ) | — | — | $ | — | |||||||||||||
| Cost of sales | (1,432 | ) | 1,432 | — | — | — | ||||||||||||||||
| Gross profit | (155 | ) | 155 | — | — | — | ||||||||||||||||
| Operating expenses: | ||||||||||||||||||||||
| Research and development | 170 | (170 | ) | — | — | — | ||||||||||||||||
| Sales and marketing | 1,286 | (1,286 | ) | — | — | — | ||||||||||||||||
| General and administrative | 3,807 | (889 | ) | 2,918 | 1,585 | (aa) | 4,503 | |||||||||||||||
| Total operating expenses | 5,263 | (2,345 | ) | 2,918 | 1,585 | 4,503 | ||||||||||||||||
| Operating Income (loss) | (5,418 | ) | 2,500 | (2,918 | ) | |||||||||||||||||
| Other (income) expense: | ||||||||||||||||||||||
| Interest expense | 37 | (1 | ) | 36 | (1,585 | ) | 36 | |||||||||||||||
| Gain on settlement of debt | (68 | ) | 68 | — | — | — | ||||||||||||||||
| Income tax | — | — | — | — | — | |||||||||||||||||
| Total other (income) expense, net | (67 | ) | 67 | — | ||||||||||||||||||
| Net income (loss) | $ | (5,387 | ) | $ | 2,433 | (2,954 | ) | (1,585 | ) | $ | (4,539 | ) | ||||||||||
| Net loss per share | ||||||||||||||||||||||
| Basic and diluted | $ | (0.62 | ) | |||||||||||||||||||
| Weighted-average common shares outstanding: | ||||||||||||||||||||||
| Basic and diluted | 8,684 | |||||||||||||||||||||
The accompanying notes are an integral part of these unaudited pro forma financial statements.
Unaudited Pro Forma Condensed Consolidated Statement of Operations
| For the Year Ended December 31, 2016 (in thousands, except per share data) | Historical Monster | SpinCo Adjustments (Note 3) | Adjusted Historical Monster | |||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||
| Revenue | $ | 4,065 | $ | (4,065 | ) | a | $ | |||||||
| Cost of sales | (3,329 | ) | (3,329 | ) | a | — | ||||||||
| Gross profit | 736 | (736 | ) | — | ||||||||||
| Operating expenses: | ||||||||||||||
| Research and development | 270 | (270 | ) | a | — | |||||||||
| Sales and marketing | 2,425 | (2,425 | ) | a | — | |||||||||
| General and administrative | 3,984 | (526 | ) | a | 3,458 | |||||||||
| Total operating expenses | 6,679 | (3,221 | ) | 3,458 | ||||||||||
| Other (income) expense: | ||||||||||||||
| Interest expense | 825 | (71 | ) | a | 754 | |||||||||
| Gain on settlement of debt | (557 | ) | — | (557 | ) | |||||||||
| Income tax | 2 | (1 | ) | a | 1 | |||||||||
| Total other (income) expense, net | 270 | (72 | ) | 198 | ||||||||||
| Net income (loss) | $ | (6,213 | ) | $ | 2,557 | $ | (3,656 | ) | ||||||
| Net loss per share | ||||||||||||||
| Basic and diluted | $ | (3.33 | ) | $ | (1.96 | ) | ||||||||
| Weighted-average common shares outstanding: | ||||||||||||||
| Basic and diluted | 1,863 | 1,863 | ||||||||||||
The accompanying notes are an integral part of these unaudited pro forma financial statements.
NOTES TO UNAUDITED PRO FORMA
CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
| 1) | Basis of Presentation |
The unaudited pro forma condensed consolidated financial information was prepared in accordance with U.S. GAAP and pursuant to the rules and regulations of SEC Regulation S-X.
In the unaudited pro forma condensed consolidated financial data, the Merger has been accounted for as a capital transaction rather than as a business combination as the business of Monster was spun off prior to the Merger. In accordance with U.S. GAAP, the Merger will be accounted for as a reverse recapitalization, equivalent to the issuance of common shares by Innovate for the net monetary assets of Monster accompanied by a re-capitalization. The accounting is similar to that resulting from a reverse acquisition, except that no goodwill or other intangible assets are recorded. Monster will be the legal acquirer but, for accounting purposes, Innovate will be treated as the accounting acquirer. Innovate will record Monster’s liabilities assumed upon the consummation of the Merger at fair value. Effective with the consummation of the Merger, the historical financial statements of Innovate became the historical financial statements of the consolidated company.
The unaudited pro forma consolidated financial data is based on the audited financial statements of Monster and Innovate as of December 31, 2016 and the unaudited financial statements of Monster and of Innovate as of September 30, 2017. As such, the financial data set forth below is not a prediction or estimate of the amounts that would be reflected in Monster’s balance sheet as of the day of closing of the transactions. Other than as disclosed in the footnotes thereto, the unaudited pro forma consolidated financial data does not reflect any additional liabilities, off-balance sheet commitments or other obligations that may become payable after the date of such financial data.
The unaudited pro forma consolidated financial information has been prepared for illustrative purposes only and is not necessarily indicative of the financial position or results of operations in future periods or the results that actually would have been realized had Monster and Innovate been a consolidated company during the specified periods.
| 2) | Description of Transaction |
On January 29, 2018, Monster Digital, Inc., a Delaware corporation now known as Innovate Biopharmaceuticals Inc. (the “Company”) completed its merger with privately-held Innovate Biopharmaceuticals Inc. (“IB Pharmaceuticals Inc.”) in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated July 3, 2017, whereby Monster Merger Sub., Inc. (“Merger Sub”), a wholly owned subsidiary of the Company merged with and into IB Pharmaceuticals Inc., with IB Pharmaceuticals Inc. surviving as the Company’s wholly owned subsidiary (the “Merger”). In connection with the Merger, the Company changed its name from Monster Digital, Inc. to Innovate Biopharmaceuticals, Inc. All references to the “Company” refer to Innovate Biopharmaceuticals, Inc. as of and following the closing of the Merger on January 29, 2018 (the “Closing Date”) and all references to “Monster” refer to Monster Digital, Inc. prior to the closing of the Merger on the Closing Date.
Immediately prior to the closing of the Merger, accredited investors purchased shares of common stock of IB Pharmaceuticals Inc. in a private placement for gross proceeds of approximately $18.1 million (the “Equity Issuance”). IB Pharmaceuticals Inc. issued five-year warrants to each purchaser of common stock with a price per exercise price of $1.2011 (subject to adjustment in connection with the Merger). Concurrently with the Equity Issuance, convertible promissory notes issued by IB Pharmaceuticals Inc. in the aggregate principal amount of approximately $8.65 million were converted into shares of IB Pharmaceuticals Inc. common stock at a price per share of $0.7206 (the “Conversion”).
H.C. Wainwright & Co., LLC (“HCW”) and GP Nurmenkari Inc. (“GPN”) were retained as the placement agents for the Equity Issuance. HCW was paid a flat fee of $250,000, a cash fee of $285,000 (equal to 10% of the gross proceeds of the Equity Issuance up to a certain cap), a cash fee of $9,018 (equal to 3.5% of the gross proceeds in excess of a certain cap), and non-accountable expense allowance of $50,000. GPN was paid a cash fee of $891,266 (equal to 10% of the gross proceeds of certain investors in the Equity Issuance) and non-accountable expense allowance of $50,000. IB Pharmaceuticals Inc. issued to affiliates of HCW five-year warrants to purchase up to 557,097 shares of common stock with an exercise price per share equal to $1.2011(subject to adjustment in connection with the Merger). IB Pharmaceuticals Inc. issued to affiliates of GPN five-year warrants to purchase up to 927,529 shares of common stock with an exercise price per share equal to $1.2011(subject to adjustment in connection with the Merger); provided that a small number of warrants (representing 318,776 shares of underlying IB Pharmaceuticals Inc. common stock) were issued to affiliates of GPN with an exercise price per share equal to $0.9609 (subject to adjustment in connection with the Merger).
Upon the closing of the Merger, the outstanding shares of IB Pharmaceuticals Inc.’s common stock were exchanged for shares of common stock of Monster at an exchange ratio of one share of IB Pharmaceuticals Inc. common stock to 0.37813802 shares of Monster common stock (the “Exchange Ratio”). Immediately following the closing of the Merger, after giving effect to the Equity Issuance and applying the Exchange Ratio, Monster’s securityholders owned approximately 5.8% of the outstanding common stock of the Company on a fully-diluted basis and IB Pharmaceuticals Inc.’s securityholders owned approximately 94.2% of the outstanding common stock of the Company on a fully-diluted basis.
| 3) | Spin-Co Adjustments |
Spin-Co is the action sports camera business operated by Monster Digital, Inc. In regards to the September 30, 2017 pro forma balance sheet presentation, other than certain liabilities of approximately $1.0 million that were assumed by Innovate, all assets and liabilities of Spin-Co are eliminated as Spin-Co adjustments with net assets distributed to the stockholders of Monster Digital, Inc. In regards to the December 31, 2016 and September 30, 2017 statements of operations, the revenue and cost of sales related to the camera business and the expenses associated with the generation of those revenues are eliminated as Spin-Co adjustments.
| (aa) | These adjustments reflect the spin-out transaction on the effective date of the Merger, whereby all of the business, assets and certain of the liabilities of Monster not assumed by Innovate further to the Merger were acquired by Spin-Co. The remaining general and administrative expenses shown in the Adjusted Historical Monster column consist of public company operating expenses including legal fees, insurance, executive salaries and stock compensation associated with operating a public company. |
Unaudited Pro Forma Financial Information for Merger
The following selected unaudited pro forma consolidated financial data presents the pro forma financial position and results of operations of the consolidated business based on the historical consolidated financial statements of Monster and Innovate, after giving effect to the Spin-Co adjustment and the Merger.
The unaudited pro forma consolidated balance sheet data as of September 30, 2017 gives effect to the Monster Spin-Co adjustment and the Merger as if each took place on September 30, 2017. The unaudited pro forma consolidated statement of operations data for the six months ended September 30, 2017 gives effect to the Spin-Co adjustment and the Merger as if each took place on January 1, 2017. The unaudited pro forma consolidated statement of operations data for the year ended December 31, 2016 gives effect to the Spin-Co adjustment and the Merger as if each took place on January 1, 2016.
The pro forma condensed consolidated balance sheet and condensed consolidated statement of operations information gives effect to the Equity Issuance and the Conversion at a $60.0 million pre-Merger valuation amount (the “Valuation”).
Unaudited Pro Forma Condensed Consolidated Balance Sheet
| Historical | Adjusted | Adjusted | ||||||||||||||||||
| September 30, 2017 | Innovate | Proforma | Historical | Proforma | ||||||||||||||||
| (in thousands, except share data and per share data) | (Unaudited) | Adjustments | Monster | Total | ||||||||||||||||
| Assets | ||||||||||||||||||||
| Cash | $ | 1,479 | $ | 16,532 | (g) | $ | - | $ | 20,765 | |||||||||||
| 2,980 | (g) | |||||||||||||||||||
| - | (226 | ) | (h) | - | - | |||||||||||||||
| Prepaid expenses and other | 128 | 20 | (g) | - | 148 | |||||||||||||||
| Total current assets | 1,607 | 19,306 | - | 20,913 | ||||||||||||||||
| Equipment, net | 7 | - | - | 7 | ||||||||||||||||
| Total assets | $ | 1,614 | $ | 19,306 | $ | - | $ | 20,920 | ||||||||||||
| Liabilities | ||||||||||||||||||||
| Current liabilities: | ||||||||||||||||||||
| Accounts payable | $ | 2,819 | $ | (1,116 | ) | (h) | $ | 273 | (l) | $ | 1,976 | |||||||||
| Accrued expenses including interest | 2,143 | (416 | ) | (h) | 743 | (l) | 4,088 | |||||||||||||
| - | 1,618 | (j) | - | - | ||||||||||||||||
| Debt | - | 4,800 | (g) | - | 4,800 | |||||||||||||||
| Debt Discount | - | (1,800 | ) | (g) | - | (1,800 | ) | |||||||||||||
| Convertible promissory notes, net | 7,490 | (7,490 | ) | (h) | - | - | ||||||||||||||
| Convertible promissory notes, related party, net | 228 | (228 | ) | (h) | - | - | ||||||||||||||
| Total current liabilities | $ | 12,680 | $ | (4,632 | ) | $ | 1,016 | $ | 9,064 | |||||||||||
| Total liabilities | $ | 12,680 | $ | (4,632 | ) | $ | 1,016 | $ | 9,064 | |||||||||||
| Stockholders’ deficit | ||||||||||||||||||||
| Common stock | $ | 32 | (29 | ) | (k) | - | $ | 3 | ||||||||||||
| Additional paid-in-capital | 5,879 | 16,532 | (g) | - | 35,249 | |||||||||||||||
| - | 9,230 | (h) | - | - | ||||||||||||||||
| - | 3,579 | (i) | - | - | ||||||||||||||||
| - | 29 | (k) | - | - | ||||||||||||||||
| Accumulated deficit | (16,977 | ) | (206 | ) | (h) | (1,016 | )(l) | (23,396 | ) | |||||||||||
| - | (3,579 | ) | (i) | - | - | |||||||||||||||
| - | (1,618 | ) | (j) | - | - | |||||||||||||||
| Total stockholders’ (deficit) equity | $ | (11,066 | ) | $ | 23,938 | $ | (1,016 | ) | $ | 11,856 | ||||||||||
| Total liabilities and stockholders’ (deficit) equity | $ | 1,614 | $ | 19,306 | $ | - | $ | 20,920 | ||||||||||||
| (1) | See the Spin-Co adjustments in the “Unaudited Pro Forma Financial Information for Spin-Co Adjustment” for the Adjusted Historical Monster adjustments. |
The accompanying notes are an integral part of these unaudited pro forma financial statements.
Unaudited Pro Forma Condensed Consolidated Statement of Operations
| For the Nine Months Ended September 30, 2017 (in thousands, except per share data) |
Historical Innovate |
Adjusted Historical Monster(1) |
Pro Forma Adjustments |
Pro Forma Consolidated |
||||||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||||||
| Revenue | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Cost of sales | — | — | — | — | ||||||||||||||
| Gross profit | — | — | — | — | ||||||||||||||
| Operating expenses: | ||||||||||||||||||
| Research and development | 2,833 | — | — | 2,833 | ||||||||||||||
| Sales and marketing | — | — | — | — | ||||||||||||||
| General and administrative | 6,115 | 4,503 | (258 | ) | a | 10,360 | ||||||||||||
| Total operating expenses | 8,948 | 4,503 | (258 | ) | 13,193 | |||||||||||||
| Other (income) expense: | ||||||||||||||||||
| Interest expense | 282 | 36 | — | 318 | ||||||||||||||
| Gain on settlement of debt | — | — | — | — | ||||||||||||||
| Income tax | — | — | — | — | ||||||||||||||
| Total other expense, net | 282 | 36 | — | 318 | ||||||||||||||
| Net income (loss) | $ | (9,230 | ) | $ | (4,539 | ) | $ | 258 | $ | (13,511 | ) | |||||||
| Net loss per share | ||||||||||||||||||
| Basic and diluted | $ | (0.47 | ) | $ | (0.44 | ) | $ | $ | (0.52 | ) | ||||||||
| Weighted-average common shares outstanding: | ||||||||||||||||||
| Basic and diluted | 11,928 | 1,863 | 11,980 | m | 25,771 | |||||||||||||
| (1) | See the Spin-Co adjustments in the “Unaudited Pro Forma Financial Information for Spin-Co Adjustment” for the Adjusted Historical Monster adjustments. |
The accompanying notes are an integral part of these unaudited pro forma financial statements.
Unaudited Pro Forma Condensed Consolidated Statement of Operations
| For the Year Ended December 31, 2016 (in thousands, except per share data) | Historical Innovate | Adjusted Historical Monster(1) | Pro Forma Adjustments | Pro Forma Consolidated | ||||||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||||||
| Revenue | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Cost of sales | — | — | — | — | ||||||||||||||
| Gross profit | — | — | — | — | ||||||||||||||
| Operating expenses: | ||||||||||||||||||
| Research and development | 1,946 | — | — | 1,946 | ||||||||||||||
| Sales and marketing | — | — | — | — | ||||||||||||||
| General and administrative | 3,470 | 3,458 | — | 6,928 | ||||||||||||||
| Total operating expenses | 5,416 | 3,458 | — | 8,874 | ||||||||||||||
| Other (income) expense: | ||||||||||||||||||
| Interest expense | 204 | 754 | — | 958 | ||||||||||||||
| Gain on settlement of debt | — | (557 | ) | — | (557 | ) | ||||||||||||
| Income tax | — | 1 | — | 1 | ||||||||||||||
| Total other expense, net | 204 | 198 | — | 402 | ||||||||||||||
| Net loss | $ | (5,620 | ) | $ | (3,656 | ) | $ | — | $ | (9,276 | ) | |||||||
| Net loss per share | ||||||||||||||||||
| Basic and diluted | $ | (0.18 | ) | $ | (0.66 | ) | $ | $ | (0.04 | ) | ||||||||
| Weighted-average common shares outstanding: | ||||||||||||||||||
| Basic and diluted | 31,545 | 5,524 | k | 212,612 | ||||||||||||||
| (1) | See the Spin-Co adjustments in the “Unaudited Pro Forma Financial Information for Spin-Co Adjustment” for the Adjusted Historical Monster adjustments. |
The accompanying notes are an integral part of these unaudited pro forma financial statements.
Notes to the Unaudited Pro Forma Condensed Consolidated Financial Information
Description of Transaction and Basis of Presentation
The unaudited pro forma condensed consolidated financial information was prepared in accordance with U.S. GAAP and pursuant to the rules and regulations of SEC Regulation S-X, and present the pro forma financial position and results of operations of the consolidated companies based upon the historical data of Monster and Innovate. The pro forma condensed consolidated financial information assume that the Merger occurred on September 30, 2017, and do not provide a reasonable estimate of the assets of the consolidated company on or following the date of the closing. In particular, the pro forma financials do not reflect the reduction in either Monster or Innovate’s cash, resulting from the operations of such entities since September 30, 2017 or since the date of this proxy statement.
| 2) | Pro Forma Adjustments |
Pro forma adjustments are necessary to reflect the acquisition consideration exchanged and to adjust amounts related to the tangible assets and liabilities of Monster to reflect the preliminary estimate of their fair values, and to reflect the impact on the statements of operations of the Merger as if the companies had been consolidated during the periods presented therein. The unaudited pro forma condensed consolidated financial information includes pro forma adjustments that are (i) directly attributable to the transaction, (ii) factually supportable, and (iii) with respect to the unaudited pro forma condensed consolidated statements of operations, expected to have a continuing impact on the results of operations of the consolidated company. Such adjustments do not contemplate the consumption of cash resources to fund continuing operating costs of Monster for the period subsequent to September 30, 2017, which are expected to be material and will therefore affect the exchange ratio calculation. The pro forma adjustments included in the unaudited pro forma condensed consolidated financial statements are as follows:
| a) | Represents the exercise of warrants that occurred in November 2017. The proceeds of the warrant exercise were used to fund the operations of Monster. |
| b) | Represents a conversion of debt to equity that occurred in November 2017 |
| c) | Represents shares of Monster common stock issued in January 2018. The proceeds of the exercise funded the operations of Monster. |
| d) | Represents the full conversion of convertible debt at the consummation of the reverse merger |
| e) | Represents the elimination of Monster’s historical accumulated deficit of $38,835. |
| f) | Represents $384 of unamortized, non-cash, stock-based compensation related to the issuance of restricted common stock and options of Monster. |
| (g) | Represents the net proceeds of $16,532 from the sale of $18,133 of Innovate common stock (the Equity Issuance), net of $1,580 of offering costs, plus the $2,980 in proceeds from a $4,800 debt financing, net of a $1,800 debt discount and $20 in debt legal costs. |
| (h) | Represents convertible promissory notes of $8,647 and related accrued interest through January 29, 2018 of $582 that converted concurrent with the consummation of the Merger, at a discount to equity securities issued by Innovate pursuant to the Equity Issuance. This amount excludes $200 in convertible promissory notes which matured on January 22 for which the holders chose to redeem their notes and accrued interest for cash instead of converting. Promissory notes entered subsequent to September 30, 2017 are being used for general operating purposes. |
| (i) | The conversion described in (b) creates a beneficial conversion feature, which is recorded as additional interest expense and additional paid-in capital. This pro forma adjustment is not reflected in the unaudited pro forma condensed combined statements of operations as this amount is not expected to have a continuing effect on the operating results of the Company. |
| (j) | Represents accrued investment banker and legal fees directly related to the merger. This pro forma adjustment is not reflected in the unaudited pro forma condensed combined statements of operations as this amount is not expected to have a continuing effect on the operating results of the Company. |
| (k) | Adjusts outstanding common shares to their par value. |
| (l) | Represents accrued expenses that are directly attributable to the closing of the transaction, including approximately $402 for tail insurance coverage to be purchased by Monster, for its directors and officers, and estimated transaction costs to complete the transaction of approximately $600. |
| m) | The basic and diluted shares outstanding on a pro forma consolidated basis were calculated based on the shares issued for the Conversion and Equity Issuance of Innovate upon Merger close. The pro forma consolidated shares outstanding is calculated as follows: |
| Description | ($000s) Pro Forma | |||
| Innovate common shares | 11,928 | |||
| Innovate convertible debt and accrued interest | 3,775 | |||
| Innovate Equity Issuance | 8,205 | |||
| Innovate warrants and options | 8,855 | |||
| Monster common shares | 1,863 | |||
| Monster convertible debt and accrued interest | - | |||
| Monster warrants and options | 154 | |||
| Total pro forma shares outstanding | 34,780 | |||
(d) Exhibits.
| # | Indicates management contract or compensatory plan |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| Date: February 2, 2018 | INNOVATE BIOPHARMACEUTICALS, INC. a Delaware corporation | |||
| By: | /s/ Jay P. Madan | |||
| Jay P. Madan | ||||
| President | ||||