Attached files
| file | filename |
|---|---|
| EX-31.1 - VBI Vaccines Inc/BC | ex31-1.htm |
| EX-32.2 - VBI Vaccines Inc/BC | ex32-2.htm |
| EX-32.1 - VBI Vaccines Inc/BC | ex32-1.htm |
| EX-31.2 - VBI Vaccines Inc/BC | ex31-2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-Q
(Mark One)
| [X] | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended September 30, 2017
OR
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-37769
VBI VACCINES INC.
(Exact name of registrant as specified in its charter)
| British Columbia, Canada | N/A | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) |
222 Third Street, Suite 2241 Cambridge, Massachusetts |
02142 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: 617-830-3031
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [X] No [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer [ ] | Accelerated filer [ ] |
| Non-accelerated filer [ ] (Do not check if a smaller reporting company) | Smaller reporting company [X] |
| Emerging growth company [X] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [X]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date.
| Common Shares, no par value per share | 63,804,781 |
| (Class) | Outstanding at November [9], 2017 |
VBI VACCINES INC.
FORM 10-Q FOR THE QUARTERLY PERIOD ENDED SEPTEMBER 30, 2017
TABLE OF CONTENTS
| 2 |
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS AND OTHER INFORMATION
CONTAINED IN THIS REPORT
This quarterly report on Form 10-Q (this “Form 10-Q”) contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 and the provisions of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements give our current expectations or forecasts of future events. You can identify these statements by the fact that they do not relate strictly to historical or current facts. You can find many (but not all) of these statements by looking for words such as “approximates,” “believes,” “hopes,” “expects,” “anticipates,” “estimates,” “projects,” “intends,” “plans,” “would,” “should,” “could,” “may,” or other similar expressions in this Form 10-Q. In particular, these include statements relating to future actions; prospective products, applications, customers and technologies; future performance or results of anticipated products; anticipated expenses; and projected financial results. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this Quarterly Report on Form 10-Q and are subject to a number of risks, uncertainties and assumptions that could cause actual results to differ materially from our historical experience and our present expectations or projections described under the sections in this Quarterly Report on Form 10-Q entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in the sections entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our 2016 annual report on the Form 10-K filed with the Securities and Exchange Commission on March 20, 2017. Factors that could cause actual results to differ from those discussed in the forward-looking statements include, but are not limited to:
| ● | the timing of, and our ability to, obtain and maintain regulatory approvals for our clinical trials, products and product candidates; | |
| ● | the timing and results of our ongoing and planned clinical trials for products and product candidates; | |
| ● | the amount of funds we require for our immuno-oncology and infectious disease vaccine candidate pipeline; | |
| ● | the potential benefits of strategic partnership agreements and our ability to enter into strategic partnership arrangements; | |
| ● | our ability to effectively execute and deliver our plans related to commercialization, marketing and manufacturing capabilities and strategy; | |
| ● | our ability to license our intellectual property; | |
| ● | our ability to maintain a good relationship with our employees; | |
| ● | the suitability and adequacy of our office, manufacturing and research facilities and our ability to secure term extensions or expansions of leased space; | |
| ● | our ability to manufacture, or to have manufactured, any products we develop to the standards and requirements of regulatory agencies; | |
| ● | the ability of our vendors to manufacture and deliver materials that meet regulatory agency and our standards and requirements in order to meet planned timelines and milestones; |
| 3 |
| ● | our compliance with all laws, rules and regulations applicable to our business and products; | |
| ● | our ability to continue as a going concern; | |
| ● | our history of losses; | |
| ● | our ability to generate revenues and achieve profitability; | |
| ● | emerging competition and rapidly advancing technology in our industry that may outpace our technology; | |
| ● | customer demand for our products and product candidates; | |
| ● | the impact of competitive or alternative products, technologies and pricing; | |
| ● | general economic conditions and events and the impact they may have on us and our potential customers; | |
| ● | our ability to obtain adequate financing in the future on reasonable terms, as and when we need it; | |
| ● | our ability to implement network systems and controls that are effective at preventing cyber-attacks, malware intrusions, malicious viruses and ransomware threats; | |
| ● | our ability to secure and maintain protection over our intellectual property; | |
| ● | our ability to maintain our existing licenses for intellectual property; and | |
| ● | our success at managing the risks involved in the foregoing items. |
Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for us to predict all risk factors and uncertainties. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
Unless otherwise stated or the context otherwise requires, the terms “VBI,” “we,” “us,” “our” and the “Company” refer to VBI Vaccines Inc. and its subsidiaries.
Unless indicated otherwise, all references to the U.S. Dollar, Dollar or $ are to the United States Dollar, the legal currency of the United States of America and all references to € mean Euros, the legal currency of the European Union. We may also refer to NIS, which is the New Israeli Shekel, the legal currency of Israel, and the Canadian Dollar or CAD, which is the legal currency of Canada.
Except for per share amounts or as otherwise specified to be in millions, amounts presented are stated in thousands.
| 4 |
| Item 1. | Condensed Consolidated Financial Statements |
VBI Vaccines Inc. and Subsidiaries
Condensed Consolidated Balance Sheets
(in thousands, except number of shares)
| September 30, 2017 | December 31, 2016 | |||||||
| (Unaudited) | ||||||||
| CURRENT ASSETS | ||||||||
| Cash | $ | 10,054 | $ | 32,282 | ||||
| Accounts receivable, net | 203 | 10 | ||||||
| Inventory, net | 701 | 830 | ||||||
| Other current assets | 1,993 | 1,236 | ||||||
| Total current assets | 12,951 | 34,358 | ||||||
| NON-CURRENT ASSETS | ||||||||
| Long-term deposits | 172 | 167 | ||||||
| Other long term assets | 514 | 487 | ||||||
| Property and equipment, net | 2,153 | 1,850 | ||||||
| Intangible assets, net | 63,669 | 59,507 | ||||||
| Goodwill | 9,021 | 8,385 | ||||||
| Total non-current assets | 75,529 | 70,396 | ||||||
| TOTAL ASSETS | $ | 88,480 | $ | 104,754 | ||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable | $ | 3,446 | $ | 2,018 | ||||
| Other current liabilities | 8,138 | 5,562 | ||||||
| Deferred revenues | - | 34 | ||||||
| Current portion of long-term debt | 1,000 | - | ||||||
| Total current liabilities | 12,584 | 7,614 | ||||||
| NON-CURRENT LIABILITIES | ||||||||
| Long-term debt, net of debt discount of $2,460 and $3,344, as of September 30, 2017 and December 31, 2016, respectively and current portion | 11,840 | 11,956 | ||||||
| Long-term deferred tax liability | - | 428 | ||||||
| Liabilities for severance pay | 414 | 356 | ||||||
| Deferred revenues, net of current portion | 669 | 669 | ||||||
| Total non-current liabilities | 12,923 | 13,409 | ||||||
| COMMITMENTS AND CONTINGENCIES (NOTE 10) | - | - | ||||||
| STOCKHOLDERS’ EQUITY | ||||||||
| Common shares (unlimited authorized; no par value) (40,229,371 and 40,018,495 shares issued and outstanding at September 30, 2017 and December 31, 2016, respectively) | 134,046 | 133,312 | ||||||
| Additional paid-in capital | 59,721 | 58,595 | ||||||
| Accumulated other comprehensive income (loss) | 1,638 | (3,196 | ) | |||||
| Accumulated deficit | (132,432 | ) | (104,980 | ) | ||||
| Total stockholders’ equity | 62,973 | 83,731 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 88,480 | $ | 104,754 | ||||
See accompanying Notes to Condensed Consolidated Financial Statements
| 5 |
VBI Vaccines Inc. and Subsidiaries
Condensed Consolidated Statements of Operations and Comprehensive Loss
(Unaudited)
(in thousands, except per share data)
| Three
months ended September 30 | Nine
months ended September 30 | |||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Revenues | $ | 193 | $ | 281 | $ | 664 | $ | 411 | ||||||||
| Operating expenses: | ||||||||||||||||
| Cost of revenue | 1,334 | 1,501 | 3,966 | 2,581 | ||||||||||||
| Research and development | 5,205 | 3,371 | 14,387 | 5,748 | ||||||||||||
| General and administration | 2,795 | 3,149 | 8,611 | 8,267 | ||||||||||||
| Total operating expenses | (9,334 | ) | (8,021 | ) | (26,964 | ) | (16,596 | ) | ||||||||
| Loss from operations | (9,141 | ) | (7,740 | ) | (26,300 | ) | (16,185 | ) | ||||||||
| Interest expense, net | 742 | 62 | 2,188 | 90 | ||||||||||||
| Foreign exchange gain | (81 | ) | (503 | ) | (605 | ) | (1,481 | ) | ||||||||
| Loss before incomes taxes | (9,802 | ) | (7,299 | ) | (27,883 | ) | (14,794 | ) | ||||||||
| Income tax benefit | - | - | 431 | - | ||||||||||||
| NET LOSS | $ | (9,802 | ) | $ | (7,299 | ) | $ | (27,452 | ) | $ | (14,794 | ) | ||||
| Net loss per share of common shares, basic and diluted | $ | (0.24 | ) | $ | (0.20 | ) | $ | (0.68 | ) | $ | (0.54 | ) | ||||
| Weighted-average number of common shares outstanding, basic and diluted | 40,229,371 | 36,150,936 | 40,115,689 | 27,592,940 | ||||||||||||
| Other comprehensive income (loss): | ||||||||||||||||
| Foreign currency translation adjustments | 2,818 | (506 | ) | 4,834 | (816 | ) | ||||||||||
| COMPREHENSIVE LOSS | $ | (6,984 | ) | $ | (7,805 | ) | $ | (22,618 | ) | $ | (15,610 | ) | ||||
See accompanying Notes to Condensed Consolidated Financial Statements
| 6 |
VBI Vaccines Inc. and Subsidiaries
Condensed Consolidated Statement of Stockholders’ Equity
(Unaudited)
(in thousands, except number of shares)
| Number of Common Shares | Share Capital | Additional Paid-in Capital | Accumulated Other Comprehensive Income (Loss) - Foreign Currency Translation Adjustments | Accumulated Deficit | Total Stockholders’ Equity | |||||||||||||||||||
| BALANCE AS OF DECEMBER 31, 2016 | 40,018,495 | $ | 133,312 | $ | 58,595 | $ | (3,196 | ) | $ | (104,980 | ) | $ | 83,731 | |||||||||||
| Stock-based compensation | 179,499 | 633 | 1,126 | 1,759 | ||||||||||||||||||||
| Common shares issued for services | 25,000 | 85 | 85 | |||||||||||||||||||||
| Common shares issued on exercise of stock options | 6,377 | 16 | 16 | |||||||||||||||||||||
| Net loss | (27,452 | ) | (27,452 | ) | ||||||||||||||||||||
| Foreign currency translation adjustments | 4,834 | 4,834 | ||||||||||||||||||||||
| BALANCE AS OF SEPTEMBER 30, 2017 | 40,229,371 | $ | 134,046 | $ | 59,721 | $ | 1,638 | $ | (132,432 | ) | $ | 62,973 | ||||||||||||
See accompanying Notes to Condensed Consolidated Financial Statements
| 7 |
VBI Vaccines Inc. and Subsidiaries
Condensed Consolidated Statements of Cash Flows
(Unaudited)
(in thousands)
| For
the Nine months Ended September 30 |
||||||||
| 2017 | 2016 | |||||||
| CASH FLOWS FROM: | ||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
| Net loss | $ | (27,452 | ) | $ | (14,794 | ) | ||
| Adjustments to reconcile net loss to cash used in operating activities: | ||||||||
| Depreciation and amortization | 539 | 440 | ||||||
| Deferred taxes | (431 | ) | - | |||||
| Stock-based compensation | 1,844 | 1,808 | ||||||
| Amortization of debt discount | 884 | 9 | ||||||
| Impairment charge of intangible assets | 300 | - | ||||||
| Net change in operating working capital items, net of business acquisitions: | ||||||||
| (Increase) in accounts receivable | (187 | ) | (35 | ) | ||||
| Decrease in inventory | 198 | 272 | ||||||
| (Increase) decrease in other current assets | (686 | ) | 639 | |||||
| Decrease in other long-term assets | 26 | 15 | ||||||
| Increase in trade account payable and other current liabilities | 3,169 | 1,835 | ||||||
| (Decrease) in deferred revenues | (94 | ) | (758 | ) | ||||
| Net cash flows used in operating activities | (21,890 | ) | (10,569 | ) | ||||
| INVESTING ACTIVITIES | ||||||||
| Cash acquired in business combination | - | 2,126 | ||||||
| Changes in long-term deposits | - | (215 | ) | |||||
| Purchase of property and equipment | (526 | ) | (311 | ) | ||||
| Net cash flows (used in)/provided by investing activities | (526 | ) | 1,600 | |||||
| FINANCING ACTIVITIES | ||||||||
| Proceeds from issuance of common shares for cash | - | 13,610 | ||||||
| Proceeds from exercise of stock options | 16 | - | ||||||
| Repayment of long-term debt | - | (375 | ) | |||||
| Net cash flows provided by financing activities | 16 | 13,235 | ||||||
| Effect of exchange rates on cash | 172 | (27 | ) | |||||
| CHANGE IN CASH FOR THE PERIOD | (22,228 | ) | 4,266 | |||||
| CASH, BEGINNING OF PERIOD | 32,282 | 12,476 | ||||||
| CASH, END OF PERIOD | $ | 10,054 | $ | 16,715 | ||||
| Supplementary disclosure of cash flow information: | ||||||||
| Cash paid during the period for interest | $ | 1,375 | $ | 111 | ||||
| Non-cash investing and financing activities: | ||||||||
| Common shares, options and warrants issued on acquisition of VBI | - | 67,494 | ||||||
| Capital expenditures included in other current liabilities | 145 | 31 | ||||||
See accompanying Notes to Condensed Consolidated Financial Statements
| 8 |
VBI Vaccines Inc. and Subsidiaries
Notes to Condensed Consolidated Financial Statements
(Unaudited)
(in thousands, except share and per share amounts)
| 1. | NATURE OF BUSINESS AND CONTINUATION OF BUSINESS |
Corporate Overview
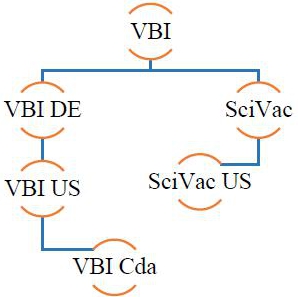
VBI Vaccines Inc. (formerly SciVac Therapeutics, Inc.) was incorporated under the laws of British Columbia, Canada on April 9, 1965 and has the following wholly-owned subsidiaries: VBI Vaccines (Delaware) Inc., a Delaware corporation (“VBI DE”); VBI DE’s wholly-owned subsidiary, Variation Biotechnologies (US), Inc., a Delaware corporation (“VBI US”); Variation Biotechnologies, Inc. a Canadian company (“VBI Cda”) and the wholly-owned subsidiary of VBI US; SciVac Ltd. an Israeli company (“SciVac”); and SciVac USA, LLC, a Florida limited liability company (“SciVac US”) and the wholly owned subsidiary of SciVac. VBI Vaccines Inc. and its subsidiaries are collectively referred to as the “Company,” “we,” “us,” “our” or “VBI”.
The Company’s principal office is located at 222 Third Street, Suite 2241, Cambridge, Massachusetts 02142. In addition, the Company has manufacturing facilities located in Rehovot, Israel and research facilities located in Ottawa, Ontario, Canada.
The Company operates in one segment and therefore segment information is not presented.
Principal Operations
We are a commercial stage biopharmaceutical company developing next generation vaccines to address unmet needs in infectious disease and immuno-oncology. VBI’s first marketed product is Sci-B-Vac®, a hepatitis B (“HBV”) vaccine that mimics all three viral surface antigens of the hepatitis B virus. Sci-B-Vac is approved for use in Israel and 14 other countries. Our wholly-owned subsidiary, SciVac Ltd., manufactures Sci-B-Vac in Rehovot, Israel. VBI is also planning to continue advancing Sci-B-Vac®, in a Phase III clinical study to be conducted in the United States, Europe and Canada.
Following our merger with VBI DE on May 6, 2016 (the “VBI-SciVac Merger”), we are also advancing our two platform technologies – our Enveloped Virus-Like Particle (“eVLP”) platform technology and our Lipid Particle Vaccine (“LPV”) technology. We are advancing a pipeline of eVLP vaccines, with lead programs in both infectious disease, with our congenital cytomegalovirus (“CMV”) vaccine, and in immuno-oncology, with our therapeutic glioblastoma multiforme (“GBM” or “glioblastoma”) vaccine candidate.
| 9 |
Liquidity and Going Concern
The Company has a limited operating history and faces a number of risks, including but not limited to, uncertainties regarding the success of its development activities, demand and market acceptance of the Company’s products and reliance on major customers. The Company anticipates that it will continue to incur significant operating costs and losses in connection with the development of its products.
The Company has an accumulated deficit of $132,432 as of September 30, 2017, and cash outflows from operating activities of $21,890 for the nine months ended September 30, 2017.
The Company will require significant additional funds to conduct clinical and non-clinical studies, achieve regulatory approvals, and, subject to such approvals, commercially launch its products. The Company plans to finance future operations with a combination of existing cash reserves, proceeds from the issuance of equity securities, the issuance of additional debt, and revenues from potential collaborations, if any. There is no assurance the Company will manage to obtain these sources of financing. The above conditions raise substantial doubt about the Company’s ability to continue as a going concern. The report of our independent registered public accounting firm on our consolidated financial statements for the year ended December 31, 2016 contains an explanatory paragraph regarding our ability to continue as a going concern. The condensed consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result should the Company be unable to continue as a going concern.
On May 15, 2017, the Company entered into an equity distribution agreement (the “Distribution Agreement”) with a registered broker-dealer, as sales agent (the “Sales Agent”), pursuant to which the Company may offer and sell, from time to time, through the Sales Agent its common shares having an aggregate offering price of up to $30 million. The Company is not obligated to sell any common shares under the Distribution Agreement. Subject to the terms and conditions of the Distribution Agreement, the Sales Agent will use commercially reasonable efforts consistent with its normal trading and sales practices, applicable state and federal law, rules and regulations, and the rules of the NASDAQ Capital Market to sell shares from time to time based upon the Company’s instructions, including any price, time or size limits specified by the Company. The Company will pay the Sales Agent a commission of 3.0% of the aggregate gross proceeds from each sale of common shares occurring pursuant to the Distribution Agreement, if any. The Distribution Agreement may be terminated by the Sales Agent or the Company at any time upon ten days’ notice to the other party, or by the Sales Agent at any time in certain circumstances. To-date no amounts have been raised under this Distribution Agreement.
See Note 13, Subsequent Events, for a description of financings completed in October 2017.
| 2. | SIGNIFICANT ACCOUNTING POLICIES |
Basis of Presentation and Consolidation
The Company’s fiscal year ends on December 31 of each calendar year. The accompanying unaudited condensed consolidated financial statements have been prepared in U.S. dollars (“USD”) and pursuant to the rules and regulations of the United States Securities and Exchange Commission (“SEC”), for interim reporting. Accordingly, certain information and footnote disclosures normally included in the financial statements prepared in accordance with United States of America generally accepted accounting principles (“U.S. GAAP”), have been condensed or omitted pursuant to such rules and regulations. The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the condensed consolidated financial statements and the reported amounts of revenue and expenses during the reporting periods. Actual results could differ from these estimates and are not necessarily indicative of the results to be expected for any future period or the entire fiscal year. The December 31, 2016 consolidated balance sheet in this document was derived from the audited consolidated financial statements and does not include all of the disclosures required by U.S. GAAP. The condensed consolidated financial statements and notes included in this quarterly report on Form 10-Q (this “Form 10-Q”) should be read in conjunction with the financial statements and notes included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2016 (the “2016 10-K”), as filed with the SEC on March 20, 2017.
| 10 |
The condensed consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries: SciVac, SciVac USA, and from May 6, 2016 the accounts of VBI DE, VBI US and VBI Cda. Intercompany balances and transactions between the Company and its subsidiaries are eliminated in the condensed consolidated financial statements.
In the opinion of management, these condensed consolidated financial statements include all adjustments and accruals of a normal and recurring nature necessary to fairly state the financial statements for the dates and the periods presented. The results for the periods presented are not necessarily indicative of results to be expected for the full year or for any future periods.
Significant Accounting Policies
The significant accounting policies used in the preparation of these condensed consolidated financial statements are disclosed in the 2016 10-K, and there have been no changes to the Company’s significant accounting policies during the nine months ended September 30, 2017.
Foreign currency
The functional and reporting currency of the Company is the USD. Each of the Company’s subsidiaries determines its own respective functional currency, and this currency is used to separately measure each entity’s financial position and operating results.
Assets and liabilities of foreign operations with a different functional currency from that of the Company are translated at the closing rate at the end of each reporting period. Profit or loss items are translated at average exchange rates for all the relevant periods. All resulting translation differences are recognized as a component of accumulated other comprehensive income (loss).
Foreign exchange gains and losses arising from transactions denominated in a currency other than the functional currency of the entity involved, are included in the condensed consolidated statements of operations.
Use of Estimates
Preparation of the condensed consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the condensed consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual amounts could differ from the estimates made and changes in estimates may occur. We continually evaluate estimates used in the preparation of the condensed consolidated financial statements for reasonableness. Appropriate adjustments, if any, to the estimates used are made prospectively based upon such periodic evaluation. The significant areas of estimation include determining the deferred tax valuation allowance, the estimated lives of property and equipment and intangible assets, the inputs in determining the fair value of equity based awards and warrants issued as well as the values ascribed to assets acquired and liabilities assumed in a business combination. Actual results may differ from those estimates.
| 11 |
Goodwill and In-Process Research and Development
The Company’s intangibles including in-process research and development (“IPR&D”) and goodwill, are tested for impairment annually, or more frequently if events or circumstances indicate that the assets might be impaired.
Goodwill represents the excess of the purchase price over the fair value of the net tangible and identifiable intangible assets acquired in a business combination. Goodwill can become impaired in certain circumstances, including but not limited to: (1) a significant adverse change in legal factors or in business climate, (2) unanticipated competition, or (3) an adverse action or assessment by a regulator. When evaluating goodwill for impairment, we may first perform an assessment qualitatively whether it is more likely than not that a reporting unit’s carrying amount exceeds its fair value, referred to as a “step zero” approach. The Company adopted Accounting Standards Update (“ASU”) 2017-04, “Intangibles – Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment” effective August 31, 2017 which has eliminated Step 2 from the goodwill impairment test. Under this update, an entity should perform its goodwill impairment test by comparing the fair value of a reporting unit with its carrying amount. The Company has established August 31st as the date for its annual impairment test of goodwill. There was no goodwill impairment determined as a result of the Company’s annual testing on August 31, 2017.
The costs of rights to IPR&D projects acquired in an asset acquisition are expensed in the consolidated statements of operations unless the project has an alternative future use. These costs include initial payments incurred prior to regulatory approval in connection with research and development agreements that provide rights to develop, manufacture, market and/or sell pharmaceutical products.
IPR&D acquired in a business combination is capitalized as an intangible asset and tested for impairment at least annually until commercialization, after which time the IPR&D is amortized over its estimated useful life. The impairment test compares the carrying amount of the IPR&D asset to its fair value. If the carrying amount exceeds the fair value of the asset, such excess is recorded as an impairment loss. The Company performed its annual impairment test on its IPR&D assets on August 31, 2017 and recorded an impairment of $300 related to certain IPR&D assets. The fair value of the IRR&D assets included in the impairment test on August 31, 2017 was determined using the income approach method.
Fair value measurements of financial instruments
Accounting guidance defines fair value as the price that would be received to sell an asset or paid to transfer a liability (the exit price) in an orderly transaction between market participants at the measurement date. The accounting guidance outlines a valuation framework and creates a fair value hierarchy in order to increase the consistency and comparability of fair value measurements and the related disclosures. In determining fair value, the Company uses quoted prices and observable inputs. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from independent sources.
The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
Level 1 — Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities.
Level 2 — Valuations based on observable inputs and quoted prices in active markets for similar assets and liabilities.
Level 3 — Valuations based on inputs that are unobservable and models that are significant to the overall fair value measurement.
Financial instruments recognized in the condensed consolidated balance sheet consist of cash, accounts receivable and other current assets, accounts payable and other current liabilities. The Company believes that the carrying value of the aforementioned financial instruments approximates their fair values due to the short-term nature of these instruments. The Company does not hold any derivative financial instruments.
The carrying amounts of the Company’s deposits and other long-term assets approximate their respective fair values.
At September 30, 2017 and December 31, 2016, the fair value of the Company’s outstanding debt is estimated to be approximately $15,512 and $15,012, respectively.
| 12 |
In determining the fair value of the long-term debt as of September 30, 2017 and December 31, 2016 the Company used the following assumptions:
| September 30, 2017 | December 31, 2016 | |||||||
| Long-term debt: | ||||||||
| Interest rate | 12.2 | % | 12.0 | % | ||||
| Discount rate | 11.0 | % | 13.5 | % | ||||
| Expected time to payment in months | 26 | 35 | ||||||
| 3. | NEW ACCOUNTING PRONOUNCEMENTS |
Recently Adopted Accounting Pronouncements
Stock Compensation
In March 2016, the Financial Accounting Standards Board (the “FASB”) issued ASU No. 2016-09, “Compensation - Stock Compensation (Topic 718),” which simplifies several aspects of the accounting for share-based payment award transactions, including the income tax consequences, classification of awards as either equity or liabilities, classification on the statement of cash flows and accounting for forfeitures. ASU No. 2016-09 is effective for fiscal years beginning after December 15, 2016, including periods within those fiscal years. Our adoption of this ASU in the first quarter of 2017 did not have a material impact on our condensed consolidated financial statements.
Cash Flow Classification
The FASB issued ASU 2016-15, an accounting standard that affects the classification of certain cash receipts and cash payments on the statement of cash flows. The standard provides guidance on eight issues: debt prepayment or extinguishment costs, settlement of zero-coupon bonds or bonds issued at a discount with insignificant cash coupon, contingent consideration payments made after a business combination, proceeds from the settlement of insurance claims, proceeds from the settlement of corporate-owned life insurance policies, distributions received from equity method investees, beneficial interests in securitization transactions, separately identifiable cash flows and applying the predominance principle. The standard is effective for public business entities for fiscal years beginning after December 15, 2017 including periods within those fiscal years.
The FASB issued ASU 2016-18, an accounting standard that requires companies to include cash and cash equivalents that have restrictions on withdrawal or use in total cash and cash equivalents on the statement of cash flows. The standard does not define restricted cash or restricted cash equivalents, but companies will need to disclose the nature of the restrictions. The standard is effective for public business entities for fiscal years beginning after December 15, 2017 including interim periods within those fiscal years.
Our adoption of these ASUs in the first quarter of 2017 did not have an impact on our condensed consolidated financial statements.
Recently Issued Accounting Standards, not yet Adopted
Revenue from Contracts with Customers
In May 2014, the FASB issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”). ASU 2014-09 outlines a single comprehensive model to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. ASU 2014-09 also requires entities to disclose sufficient information, both quantitative and qualitative, to enable users of financial statements to understand the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers. An entity should apply the amendments in this ASU using one of the following two methods: (1) retrospectively to each prior reporting period presented with a possibility to elect certain practical expedients, or, (2) on a modified retrospective basis with the cumulative effect of initially applying ASU 2014-09 recognized at the date of initial application. If an entity elects the latter transition method, it also should provide certain additional disclosures. For a public entity, the ASU as amended is effective for annual periods beginning after December 15, 2017, including interim reporting periods within that reporting period. Given the Company’s current level of revenue, we do not expect a significant impact from the adoption of this new accounting guidance on our financial statements and footnote disclosures.
| 13 |
Leases
In February 2016 the FASB issued ASU 2016-02: Leases. The ASU introduces a lessee model that results in most leases impacting the balance sheet by requiring reporting entities to recognize lease assets and lease liabilities for substantially all lease arrangements. The update is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. The Company is currently evaluating the impact this new guidance will have on its financial statements and related disclosures.
Accounting for Income Taxes on Intercompany Transfers
The FASB recently issued ASU 2016-16, an accounting standard that requires the seller and buyer to recognize at the transaction date the current and deferred income tax consequences of intercompany asset transfers. The FASB expects the new standard to cause volatility in companies’ effective tax rates, particularly for those that transfer intangible assets to subsidiaries. The standard is effective for public business entities for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. While the Company continues to assess the potential impact of this standard, the adoption of this standard is not expected to have a material impact on our financial statements.
Recognition and Measurement of Financial Assets and Financial Liabilities
In January 2016, the FASB issued ASU 2016-01, “Financial Instruments – Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities”. This update will change the income statement impact of equity investments held by an entity; disclosures related to fair value of financial instruments and presentation of financial assets and liabilities. ASU 2016-01 is effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. Entities must apply the standard using a cumulative-effect adjustment to the balance sheet as of the beginning of the fiscal year of adoption. Except for certain early application guidance, early adoption is not permitted. The Company is currently assessing the impact that adopting this new ASU will have on our financial statements and footnote disclosures.
Clarifying the Scope of Asset Derecognition Guidance and Accounting for Partial Sales of Nonfinancial Assets
In February 2017, The FASB issued ASU 2017-05, “Other Income – Gains and Losses from the Derecognition of Nonfinancial Assets (Subtopic 610-12): Clarifying the Scope of Assets Derecognition Guidance and Accounting for Partial Sales of Nonfinancial Assets”. This amendment in this update clarifies the guidance on accounting for derecognition of a nonfinancial asset and an in-substance nonfinancial asset and applies only when the asset (or asset group) does not meet the definition of a business; defines in-substance nonfinancial assets; and provides guidance for partial sales of nonfinancial assets. ASU 2017-05 is effective for annual reporting periods beginning after December 15, 2017, including interim periods within those fiscal years. While the Company continues to assess the potential impact of this standard, the adoption of this standard is not expected to have a material impact on its financial statements.
| 14 |
| 4. | INTANGIBLES |
| September 30, 2017 | ||||||||||||||||||||
| Gross
Carrying amount | Accumulated
Amortization | Impairment Charge | Cumulative Currency Translation | Net
Book Value | ||||||||||||||||
| Patents | $ | 669 | $ | (381 | ) | $ | - | $ | 27 | $ | 315 | |||||||||
| IPR&D assets | 61,500 | - | (300 | ) | 2,154 | 63,354 | ||||||||||||||
| $ | 62,169 | $ | (381 | ) | $ | (300 | ) | $ | 2,181 | $ | 63,669 | |||||||||
| December 31, 2016 | ||||||||||||||||
| Gross
Carrying amount | Accumulated
Amortization | Cumulative Currency Translation | Net
Book Value | |||||||||||||
| Patents | $ | 669 | $ | (334 | ) | $ | (4 | ) | $ | 331 | ||||||
| IPR&D assets | 61,500 | - | (2,324 | ) | 59,176 | |||||||||||
| $ | 62,169 | $ | (334 | ) | $ | (2,328 | ) | $ | 59,507 | |||||||
The Company amortizes intangible assets with finite lives on a straight-line basis over their estimated useful lives. The amortization expense for the three and nine months ended September 30, 2017 was $16 and $46, respectively compared to the three and nine months ended in 2016 of $15 and $46, respectively.
The IPR&D assets relate to the May 6, 2016 VBI-SciVac Merger and will not begin amortizing until the Company commercializes its products. Future costs incurred to extend the life of the patents will be expensed. At August 31, 2017, the Company prepared its annual impairment test. As a result of that test, there was an impairment charge of $300 related to certain IPR&D assets. Such amount is included in research and development expenses in the accompanying condensed consolidated statements of operations and comprehensive loss.
| 5. | LOSS PER SHARE OF COMMON SHARES |
Basic loss per share is computed by dividing net loss applicable to common stockholders by the weighted average number of common shares outstanding during each period. Diluted loss per share includes the effect, if any, from the potential exercise or conversion of securities, such as warrants, and stock options, which would result in the issuance of incremental common shares unless such effect is anti-dilutive. In computing the basic and diluted net loss per share applicable to common stockholders, the weighted average number of shares remains the same for both calculations due to the fact that when a net loss exists, dilutive shares are not included in the calculation as their effect would be anti-dilutive. These potentially dilutive securities are more fully described in Note 8, Stockholders’ Equity and Additional Paid-in Capital.
The following potentially dilutive securities outstanding at September 30, 2017 and 2016 have been excluded from the computation of diluted weighted average shares outstanding, as they would be antidilutive:
| As at September 30, | ||||||||
| 2017 | 2016 | |||||||
| Warrants | 2,069 | 364 | ||||||
| Stock options and equity awards | 2,871 | 2,766 | ||||||
| 4,940 | 3,130 | |||||||
| 15 |
| 6. | LONG-TERM DEBT |
As at September 30, 2017 and the December 31, 2016, the outstanding debt is as follows:
| September 30, 2017 | December 31, 2016 | |||||||
| Long-term debt, net of deferred financing costs and unamortized debt discount based on an imputed interest rate of 20.5% of $ 2,460 and $3,344 at September 30, 2017 and December 31, 2016, respectively | $ | 12,840 | $ | 11,956 | ||||
| Less: current portion | 1,000 | - | ||||||
| $ | 11,840 | $ | 11,956 | |||||
As a result of the VBI-SciVac Merger, the Company through VBI DE assumed a term loan facility with Perceptive Credit Holdings, LP (the “Lender”) in the amount of $6,000 (the “Facility”), with an initial advance of $3,000 drawn down on prior to the VBI-SciVac Merger. As of the date of the VBI-SciVac Merger, the Company assumed an amount of $2,361 in the Facility. On December 6, 2016, the Company amended the Facility (the “Amended Facility”) and raised an additional $13,200 which was combined with the remaining balance from the Facility of $1,800. The total principal outstanding at September 30, 2017, including the $300 exit fee discussed below, is $15,300 before the net deferred financing costs and unamortized debt discount of $2,460. The principal on the Amended Facility accrues interest at an annual rate equal to the greater of (a) one-month LIBOR (subject to a 5.00% cap) or (b) 1.00%, plus the applicable margin. The applicable margin will be 11.00%. The first eighteen months are interest only. The interest rate as of September 30, 2017 was 12.25%. Upon the occurrence of an event of default, and during the continuance, of an event of default, the applicable margin will be increased by 4.00% per annum. The Company begins monthly principal repayments of $200 in May 2018 for 19 months upon which the remaining balance is due on maturity. The Amended Facility matures December 6, 2019 and includes both financial and non-financial covenants, including a minimum cash balance requirement of $2,500. The Company was in compliance with these covenants as of September 30, 2017. Pursuant to the Amended Facility, the Company agreed to appoint a representative of the Lender to the Board who is also a portfolio manager of the Company’s largest shareholder.
The Company’s obligations under the Amended Facility are secured on a senior basis by a lien on substantially all of the assets of the Company, including the Company’s interest in its subsidiaries, and its U.S. and Canadian subsidiaries and guaranteed by the Company and its subsidiaries. The Amended Facility also contains customary events of default.
In connection with the Amended Facility, on December 6, 2016, the Company issued to the Lender two tranches of warrants. The first tranche was a warrant to purchase 363,771 common shares at an exercise price of $4.13 per share and the second tranche was a warrant to purchase 1,341,282 common shares at an exercise price of $3.355 per share. The total proceeds from the Amended Facility attributed to the warrants was $2,793, based on the relative fair value of the warrants as compared to the sum of the fair values of the warrants and debt. This attribution resulted in the debt being issued at a discount. The Company incurred $360 of debt issuance costs and is required to pay an exit fee of $300 upon full repayment of the debt resulting in additional debt discount. The total debt discount of $3,453 is being charged to interest expense using the effective interest method over the term of the debt. As of September 30, 2017, the unamortized debt discount is $2,460. The Company recorded $884 of interest expense related to the amortization of the debt discount during the nine months ended September 30, 2017.
The following table summarizes the future principal payments that the Company expects to make for long-term debt:
| Period
ending September 30, | Principal
payments on Amended Facility and exit fee | |||
| 2017 | $ | - | ||
| 2018 | 1,000 | |||
| 2019 | 14,300 | |||
| $ | 15,300 | |||
| 16 |
| 7. | DEFERRED REVENUE AND RELATED PARTY TRANSACTIONS |
Prior to the VBI-SciVac Merger, one of the Company’s directors was also the chairman of the board of Kevelt AS (“Kevelt”), a wholly owned subsidiary of OAO Pharmsynthez (“Pharmsynthez”), a shareholder of the Company and was also the chairman of the board of Pharmsynthez. Following the VBI-SciVac Merger, in accordance with the merger agreement, this director resigned.
On April 26, 2013, SciVac entered into a Development and Manufacturing Agreement (“DMA”) with Kevelt, pursuant to which SciVac agreed to develop the manufacturing process for the production of clinical and commercial quantities of certain materials in drug substance form. On July 30, 2016, the Company received a letter of termination from Kevelt, in part containing a request for refund of $2.5 million it had previously transferred to the Company. Such amount is included in other current liabilities in the accompanying condensed consolidated financial statements. See Note 13, Subsequent Events for a description on the Kevelt settlement agreement.
SciVac entered into a services agreement with OPKO Biologics Ltd. (“OPKO Bio”), a wholly-owned subsidiary of OPKO Health, Inc., a related party shareholder of the Company, dated as of March 15, 2015, as amended on January 25, 2016, pursuant to which SciVac agreed to provide certain aseptic process filling services to OPKO Bio. The terms of the services agreement are based on market rates and comparable to other non-related party service agreements.
See Note 6, Long-term Debt, for the Facility from a lender that is also affiliated with the Company’s largest shareholder.
Nine months ended September 30 | ||||||||
| 2017 | 2016 | |||||||
| Services revenues from related parties: | ||||||||
| OPKO Bio | $ | 4 | $ | 12 | ||||
Subsequent to the VBI-SciVac Merger on May 6, 2016, Kevelt and Pharmsynthez are no longer considered related parties due to the common shareholder no longer having significant influence.
| 8. | STOCKHOLDERS’ EQUITY AND ADDITIONAL PAID-IN CAPITAL |
Stock option plans
The Company’s stock option plans are approved by and administered by the Company’s board of directors (the “Board”) and its Compensation Committee. The Board designates, in connection with recommendations from the Compensation Committee, eligible participants to be included under the plan, and designates the number of options, exercise price and vesting period of the new options.
2006 VBI US Stock Option Plan
No further options will be issued under the 2006 VBI US Stock Option Plan (the “2006 Plan”). As at September 30, 2017, there were 1,290,153 options outstanding under the 2006 Plan.
2013 Stock Incentive Plan
No further options will be issued under the 2013 Equity Incentive Plan (the “2013 Plan”). As at September 30, 2017, there were 4,613 options outstanding under the 2013 Plan.
2014 Equity Incentive Plan
No further options will be issued under the 2014 Equity Incentive Plan (the “2014 Plan”). As at September 30, 2017, there were 713,716 options outstanding under the 2014 Plan.
| 17 |
2016 VBI Equity Incentive Plan
The 2016 VBI Equity Incentive Plan (the “2016 Plan”) is a rolling incentive plan that sets the number of common shares issuable under the 2016 Plan, together with any other security-based compensation arrangement of the Company, at a maximum of 10% of the aggregate common shares issued and outstanding on a non-diluted basis at the time of any grant under the 2016 Plan. The 10% maximum is inclusive of options granted under all equity incentive plans. The 2016 Plan is an omnibus equity incentive plan pursuant to which the Company may grant equity and equity-linked awards to eligible participants in order to promote the success of the Company following the VBI-SciVac Merger by providing a means to offer incentives and to attract, motivate, retain and reward persons eligible to participate in the 2016 Plan. Grants under the 2016 Plan include a grant or right consisting of one or more options, stock appreciation rights (“SARs”), restricted share units (“RSUs”), performance share units (“PSUs”), shares of restricted stock or other such award as may be permitted under the 2016 Plan. As at September 30, 2017, there were 412,250 options and 450,250 stock awards outstanding under the 2016 Plan.
The aggregate number of common shares remaining available for issuance for awards under this plan was 648,702 at September 30, 2017.
Activity related to stock options is as follows (in thousands, except for weighted average exercise price):
| Number of Stock Options | Weighted
Average Exercise Price | |||||||
| Balance outstanding at December 31, 2016 | 2,168 | $ | 4.45 | |||||
| Granted | 304 | $ | 3.60 | |||||
| Exercised | (6 | ) | $ | 2.50 | ||||
| Forfeited | (45 | ) | $ | 4.41 | ||||
| Balance outstanding at September 30, 2017 | 2,421 | $ | 4.43 | |||||
| Exercisable at September 30, 2017 | 1,634 | $ | 4.47 | |||||
Information relating to restricted stock units is as follow (in thousands, except for weighted average fair value at grant date):
| Number of Stock Awards | Weighted Average Fair Value at Grant Date |
|||||||
| Unvested shares outstanding at December 31, 2016 | 639 | $ | 3.88 | |||||
| Granted | 57 | $ | 4.72 | |||||
| Vested | (212) | $ | 4.23 | |||||
| Forfeited | (33) | $ | 3.90 | |||||
| Unvested shares outstanding at September 30, 2017 | 451 | $ | 3.96 | |||||
| 18 |
The fair value of the options and restricted stock units will be recognized as an expense on a straight-line basis over the vesting period. The total stock-based compensation expense recorded in the three and nine months ended September 30, 2017 and 2016 was as follows:
| Three
months ended September 30 |
Nine
months ended September 30 |
|||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Research and development | $ | 166 | $ | 334 | $ | 553 | $ | 616 | ||||||||
| General and administrative | 410 | 368 | 1,241 | 1,128 | ||||||||||||
| Cost of revenues | 17 | - | 50 | - | ||||||||||||
| Total stock based compensation | $ | 593 | $ | 702 | $ | 1,844 | $ | 1,744 | ||||||||
| 9. | INCOME TAXES |
The Company operates in U.S., Israel and Canadian tax jurisdictions. Its income is subject to varying rates of tax, and losses incurred in one jurisdiction cannot be used to offset income taxes payable in another.
The Company’s effective tax rate on loss before tax for the three and nine months ended September 30, 2017 of 0% and 1.5%, respectively (0% - 2016) differs from the Canadian statutory rate of 26% primarily due to the recognition of a valuation allowance on the Canadian deferred tax assets as well as on the other deferred tax assets in all other jurisdictions.
The Company maintains a valuation allowance on its net deferred tax assets. A valuation allowance is required when, based upon an assessment of various factors, including recent operating loss history, anticipated future earnings, and prudent and reasonable tax planning strategies, it is more likely than not that some portion of the deferred tax assets will not be realized.
| 10. | COMMITMENTS AND CONTINGENCIES |
Licensing
| (a) | The Company’s manufactured and marketed product, Sci-B-Vac™, is a recombinant third generation hepatitis B vaccine whose sales and territories are governed by the Ferring License Agreement (“License Agreement”). Under the License Agreement, the Company is committed to pay Ferring royalties equal to 7% of net sales (as defined therein). Royalty payments have been de minimis for the three and nine months ended September 30, 2017 and 2016, respectively. |
| (b) | Under an Assignment and Assumption Agreement, the Company is required to pay royalties to SciGen Singapore equal to 5% of net sales of Sci-B-Vac. Royalty payments have been de minimis for the three and nine months ended September 30, 2017 and 2016. |
Legal Proceedings
From time to time, the Company may be involved in certain claims and litigation arising out of the ordinary course of business. Management assesses such claims and, if it considers that it is probable that an asset had been impaired or a liability had been incurred and the amount of loss can be reasonably estimated, provisions for loss are made based on management’s assessment of the most likely outcome. The Company believes that it maintains adequate insurance coverage for any such litigation matters arising in the normal course of business.
| 19 |
Operating Leases
The Company has entered into various non-cancelable lease agreements for its office, lab and manufacturing facilities. These arrangements expire at various times through 2022. Rent expense for the three months ended September 30, 2017 and 2016 was $233 and $184, respectively and for the nine months ended September 30, 2017 and 2016 was $689 and $401, respectively.
The future annual minimum payments under these leases is as follows:
| Year ending December 31 | ||||
| Remaining 2017 | $ | 228 | ||
| 2018 | 738 | |||
| 2019 | 685 | |||
| 2020 | 448 | |||
| 2021 | 448 | |||
| Thereafter | 37 | |||
| Total | $ | 2,584 | ||
| 11. | REVENUE BY GEOGRAPHIC REGION |
| Three
months ended September 30 | Nine
months ended September 30 | |||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Israel | $ | 153 | $ | 116 | $ | 400 | $ | 206 | ||||||||
| Asia | 40 | - | 101 | 4 | ||||||||||||
| North America | - | - | 150 | - | ||||||||||||
| South America | - | - | 2 | - | ||||||||||||
| Europe | - | 165 | 11 | 201 | ||||||||||||
| Total | $ | 193 | $ | 281 | $ | 664 | $ | 411 | ||||||||
| 12. | PROPERTY AND EQUIPMENT, NET BY GEOGRAPHIC REGION |
| September 30, 2017 | December 31, 2016 | |||||||
| Property and equipment in Israel | $ | 2,015 | $ | 1,850 | ||||
| Property and equipment in North America | 138 | - | ||||||
| Total | $ | 2,153 | $ | 1,850 | ||||
| 13. | SUBSEQUENT EVENTS |
On October 30, 2017, the Company closed an underwritten public offering and a concurrent registered direct offering of an aggregate of 23,575,410 common shares at a price of $3.05 per share. In addition, in connection with the registered direct offering, the Company issued four-year warrants to purchase 550,000 common shares at an exercise price of $3.34 per share. The aggregate estimate net proceeds from the offerings to the Company were approximately $67.4 million.
On November 8, 2017, SciVac Ltd.entered into a settlement agreement with Kevelt, whereby SciVac Ltd. agreed to pay Kevelt $1,000,000 in cash by November 10, 2017, and issue 274,000 common shares of the Company by no later than December 18, 2017, to settle certain ongoing disputes arising out of SciVac Ltd.’s development and manufacturing agreement with Kevelt. As part of the settlement, the development and manufacturing agreement was terminated and Kevelt and SciVac Ltd. entered into mutual release agreements, whereby each of them released the other from all claims and liabilities arising under the development and manufacturing agreement.
| 20 |
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion of our financial condition and results of operations in conjunction with the condensed consolidated financial statements and the related notes included elsewhere in this Form 10-Q and with our audited consolidated financial statements included in our annual report on Form 10-K for the year ended December 31, 2016, as filed with the U.S. Securities and Exchange Commission (the “SEC”).
Overview
VBI- SciVac Merger
On May 6, 2016, the Company completed its acquisition of VBI DE, pursuant to which Seniccav Acquisition Corporation, a Delaware corporation and a wholly owned subsidiary of the Company, merged with and into VBI DE, with VBI DE continuing as the surviving corporation and as a wholly-owned subsidiary of the Company (the “VBI-SciVac Merger”). Upon completion of the VBI-SciVac Merger, the Company changed its name to “VBI Vaccines Inc.” and its common shares commenced trading on The NASDAQ Capital Market. The common shares began trading on The NASDAQ Capital Market under the ticker symbol “VBIV.” Prior to the VBI-SciVac Merger, the Company’s common shares were only listed on the Toronto Stock Exchange (the “TSX”) under the symbol “VAC.” Following the effective time of the VBI-SciVac Merger, the common shares began to trade on the TSX under the new symbol “VBV.”
Principal Operations
We are a commercial stage biopharmaceutical company developing next generation vaccines to address unmet needs in infectious disease and immuno-oncology. Our first marketed product is Sci-B-Vac®, a hepatitis B virus (“HBV”) vaccine that mimics all three viral surface antigens of the hepatitis B virus. Sci-B-Vac is approved for use in Israel and 14 other countries. Our wholly-owned subsidiary, SciVac Ltd., manufactures Sci-B-Vac in Rehovot, Israel.
We are also advancing our two platform technologies – our “enveloped” virus-like particle (“eVLP”) platform technology and our thermostable lipid particle vaccine (“LPV”) technology.
| - | Our eVLP platform technology enables the development of enveloped virus-like particle vaccines that closely mimic the target virus to elicit a potent immune response. We are advancing a pipeline of eVLP vaccines, with lead programs in both infectious disease, with our congenital cytomegalovirus (“CMV”) vaccine, and in immuno-oncology, with our therapeutic glioblastoma multiforme (“GBM”) vaccine candidate. | |
| - | Our LPV thermostability technology is a proprietary formulation of lipids and process that allows vaccines and biologics to preserve stability, potency, and safety at temperatures outside of the most common cold chain storage requirements of 2oC to 8 oC. |
Sci-B-Vac Clinical Program
On November 14, 2016, we announced results from an interim analysis of a post-marketing Phase IV clinical study in Israel for Sci-B-Vac to confirm a new in-house reference standard for regulatory and quality control purposes. Sci-B-Vac has not yet been approved by the U.S. Food and Drug Administration (the “FDA”), the European Medicines Agency (the “EMA”) or Health Canada (“HC”). The post-marketing Phase IV clinical study was completed in June 2017. On July 11, 2017, we announced plans for a global Phase III clinical program for Sci-B-Vac following discussions with the FDA, the EMA, and HC. The Phase III program is expected to be a global 15-month program, consisting of two concurrent Phase III studies – a safety and immunogenicity study (“PROTECT”) and a lot-to-lot consistency study (“CONSTANT”), enrolling approximately 4,800 subjects. The Phase III program is expected to be conducted at approximately 40 sites across the U.S., Europe, and Canada.
| 21 |
PROTECT – Safety and Immunogenicity Study
| ● | PROTECT will be a double-blind, two-arm, randomized, controlled study. Approximately 1,600 adult subjects, age 18 years and older, will be randomized in a 1:1 ratio to receive either a three-dose course of Sci-B-Vac 10μg or a three-dose course of the control vaccine, Engerix-B® 20μg. Enrollment will be stratified by age group. |
The co-primary objectives of the study will be:
| ● | To demonstrate non-inferiority of the seroprotection rate induced by Sci-B-Vac as compared to Engerix-B four weeks after the third vaccination in adults age 18 and older. | |
| ● | To demonstrate superiority of the seroprotection rate induced by Sci-B-Vac as compared to Engerix-B four weeks after the third vaccination in adults older than 45 years of age. |
The study will also include multiple secondary objectives to evaluate the speed to seroprotection and the overall safety and tolerability of Sci-B-Vac as compared to Engerix-B.
CONSTANT – Lot-to-Lot Consistency Study
| ● | CONSTANT will be a double-blind, four-arm, randomized, controlled study. Approximately 3,200 adult subjects, age 18-45 years, will be randomized in a 1:1:1:1 ratio to receive one of four three-dose courses: Lot A of Sci-B-Vac 10μg, Lot B of Sci-B-Vac 10μg, Lot C of Sci-B-Vac 10μg, or the control vaccine Engerix-B® 20μg. |
The primary objective of this study will be to demonstrate lot-to-lot consistency for immune response as measured by geometric mean concentration (GMC) of antibodies across three independent, consecutive lots of Sci-B-Vac four weeks after the third vaccination.
The secondary objective will be to evaluate safety and efficacy of Sci-B-Vac as compared to Engerix-B.
On August 25, 2017, the FDA accepted our investigational new drug application (“IND”) for the Sci-B-Vac Phase III clinical program. Previously, on June 14, 2017, HC provided us with a No Objection Letter for both Sci-B-Vac Phase III clinical studies, which authorized us to proceed with the Sci-B-Vac Phase III clinical studies as described in our clinical trial application. Acceptance of the IND and the receipt of the No Objection Letter enable us to initiate the the Sci-B-Vac Phase III clinical study in both the United States and Canada.
Congenital CMV Vaccine Candidate (eVLP)
Our second lead program is a vaccine for congenital CMV, a leading cause of birth defects, that is in Phase I development. Our congenital CMV vaccine candidate uses our eVLP platform and is adjuvanted with aluminum phosphate (alum), an adjuvant used in FDA-approved products.
In July 2017, we announced interim data read-out of safety data through day 84 of the study and initial immunogenicity signals in participant samples collected one month after the second of three planned vaccine doses as follows:
| ● | Safety: The vaccine was well tolerated at all doses, with no safety signals. | |
| ● | Immunogenicity: |
| ● | The vaccine induced antibody responses against the CMV glycoprotein B (gB) antigen with clear evidence of dose-dependent boosting after the second vaccination. | |
| ● | Immunization with the highest dose of the vaccine induced seroconversion in 100% of subjects after just two vaccinations. | |
| ● | After two of the three planned vaccinations, neutralizing antibodies against epithelial cell infection were demonstrated in 17% of subjects who received the highest dose of VBI-1501A. | |
| ● | The highest dose of VBI-1501A (2.0μg of gB-G content with alum) has approximately 10-fold less antigen content than that used in several other VLP-based vaccines or in past CMV vaccine candidates. | |
| ● | Formulation of the vaccine with alum enhanced antibody titers. |
| 22 |
The final data read-out of safety and immunogenicity, following a third vaccination is expected in the first half of 2018.
Therapeutic GBM Vaccine (eVLP)
We also have a therapeutic glioblastoma brain cancer (GBM) vaccine program, VBI-1901, with respect to which an IND was accepted by the FDA on August 11, 2017. We expect to initiate enrollment in a multi-center Phase I/IIA clinical study evaluating VBI-1901 in patients with recurrent GBM in the fourth quarter of 2017. We expect results from an ongoing biomarker analysis in the first half of 2018, and initial efficacy indications in the second half of 2018. On October 3, 2017, the U.S. Patent and Trademark Office granted a patent on VBI-1901.
At present, our operations are focused on:
| ● | manufacturing in Rehovot, Israel and sale of Sci-B-Vac in territories where it is currently registered; | |
| ● | preparing for the Sci-B-Vac Phase III clinical program to support various marketing authorization applications in the U.S., Europe, Canada; | |
| ● | completing the Phase I clinical study with our CMV vaccine candidate, VBI-1501; | |
| ● | preparing for the planned Phase I/IIA clinical study of our GBM vaccine candidate, VBI-1901; | |
| ● | scaling-up Sci-B-Vac manufacturing capabilities to further commercialize this product in additional markets where we may obtain regulatory approval; | |
| ● | continuing the research and development of our product candidates, including the exploration and development of new product candidates; | |
| ● | adding operational, financial and management information systems and human resources support, including additional personnel to support our vaccine development and commercialization activities; and | |
| ● | maintaining, expanding and protecting our intellectual property portfolio. |
R&D Services
Pursuant to an agreement with the Office of the Chief Scientist in Israel, we are required to make services available for the biotechnology industry in Israel. These services include relevant activities for development and manufacturing of therapeutic proteins according to international standards and cGMP quality level suitable for toxicological studies in animals and clinical studies (Phase I & II) in humans. Service activities include analytics/bio analytics methods for development and process development of therapeutic proteins starting with a lead candidate clone through the upstream, purification, formulation and filling processes and manufacturing for Phase I & II clinical studies.
These R&D services are primarily marketed to the Israeli research community in academia and Israeli biotechnology companies in the life sciences lacking the infrastructure or experience in the development and production of therapeutic proteins and the standards and quality required for clinical studies for human use. In 2016 and 2015, we provided services to more than 10 biotech companies including analytical development, upstream development process, protein purification and formulation and filling for Phase I clinical studies.
| 23 |
VBI Cda also provides R&D services pursuant to a research agreement and certain governmental research and development grants.
Financial Overview
Overall Performance
We had net losses of approximately $9,802 and $7,299 for the three months ended September 30, 2017 and 2016, respectively and $27,452 and $14,794 for the nine months ended September 30, 2017 and 2016, respectively. At September 30, 2017, we had an accumulated deficit of $132,432, cash on hand of $10,054 and net working capital of approximately $367.
R&D Expenses
R&D expenses consist primarily of costs incurred for the development of our CMV, GBM and Sci-B-Vac vaccines, which include:
| ● | the cost of acquiring, developing and manufacturing clinical study materials and other consumables and lab supplies used in our pre-clinical studies; | |
| ● | expenses incurred under agreements with contractors or contract manufacturing organizations to advance the vaccines into clinical studies; and | |
| ● | employee-related expenses, including salaries, benefits, travel and stock-based compensation expense. |
We expense R&D costs when we incur them.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for executive and other administrative personnel and consultants, including stock-based compensation and travel expenses. Other general and administrative expenses include professional fees for legal, patent protection, consulting and accounting services, travel and conference fees, including board and scientific advisory board meeting costs, rent, maintenance of facilities, depreciation, office supplies and expenses, insurance and other general expenses.
We expect that our general and administrative expenses will increase in the future as a result of adding employees and scaling our operations commensurate with advancing clinical candidates and continuing to support a public company infrastructure. These increases will likely include increased costs for insurance, hiring of additional personnel, board committees, outside consultants, investor relations, lawyers and accountants, among other expenses.
Interest Income
Interest income consists principally of interest income earned on cash balances and on R&D tax refunds.
Interest Expense
Interest expense is associated with our previously outstanding convertible notes and the credit facility entered into on July 25, 2014 and subsequently amended on December 6, 2016.
| 24 |
Results of Operations
Three and Nine months Ended September 30, 2017 Compared to the Three and Nine months Ended September 30, 2016
All amounts stated below are in thousands, unless otherwise indicated.
Revenues
Revenue by Geographic Region
| Three
months ended September 30, 2017 | Three
months ended September 30, 2016 | $ Change | % Change | |||||||||||||
| Revenue in Israel | $ | 153 | $ | 116 | $ | 37 | 32 | % | ||||||||
| Revenue in Asia | 40 | - | 40 | N/A | % | |||||||||||
| Revenue in Europe | - | 165 | (165 | ) | N/A | % | ||||||||||
| Total Revenue | $ | 193 | $ | 281 | $ | (88 | ) | (32 | )% | |||||||
| Nine
months ended September 30, 2017 | Nine
months ended September 30, 2016 | $ Change | % Change | |||||||||||||
| Revenue in Israel | $ | 400 | $ | 206 | $ | 194 | 94 | % | ||||||||
| Revenue in Asia | 101 | 4 | 97 | 2,425 | % | |||||||||||
| Revenue in North America | 150 | - | 150 | N/A | % | |||||||||||
| Revenue in South America | 2 | - | 2 | N/A | % | |||||||||||
| Revenue in Europe | 11 | 201 | (190 | ) | (95 | )% | ||||||||||
| Total Revenue | $ | 664 | $ | 411 | $ | 253 | 62 | % | ||||||||
Revenue earned in Israel for the three months ended September 30, 2017 was $153 as compared to $116 for the three months ended September 30, 2016. The revenue earned in Israel increased by $37, or 32%, primarily as a result of completion of a fee for service project during the third quarter of 2017 compared to 2016. In Asia, we completed a contract with one customer in the three and nine month periods ended September 30, 2017 compared to negligible sales in 2016 in that region. For the nine month period ended September 30, 2017, the revenue earned in Israel increased by 94% due to several new contracts in 2017 while there was a factory shutdown in 2016.
Revenue earned in North America for the nine months ended September 30, 2017 was a result of a services agreement that was completed, whereas there were no similar agreements in North America in the comparative period in 2016 that generated revenue.
Cost of Revenues
Cost of revenues for the three months ended September 30, 2017 was $1,334 as compared to $1,501 for the three months ended September 30, 2016. The decrease in the cost of revenues of $167, or 11%, was a result of lower sales for the three months ended September 30, 2017 compared to the three months ended September 30, 2016.
Cost of revenues for the nine months ended September 30, 2017 was $3,966 as compared to $2,581 for the nine months ended September 30, 2016. The increase in the cost of revenues of $1,385, or 54%, was a result of a returning to regular vaccine manufacturing production, as noted above.
| 25 |
Research and Development
Research and Development (“R&D”) expenses for the three months ended September 30, 2017 were $5,205, as compared to $3,371 for the three months ended September 30, 2016. The increase of $1,834 was mainly a result of the costs associated with various research projects that are going on in Q3 2017 and us having a lower level of activity in the three months ended September 30, 2016. In addition, in the three months ended September 30, 2017, the Company recorded a $300 impairment related to certain IPR&D assets.
R&D expenses for the nine months ended September 30, 2017 were $14,387, as compared to $5,748 for the nine months ended September 30, 2016. The increase of $8,639 was mainly a result of a full nine months of expenses in 2017 compared to five months post acquisition expenses for the nine months ended September 30, 2016, as well as a higher level of activity associated with research projects and trials during the nine months ended September 30, 2017.
General and Administrative
General and administrative (“G&A”) expenses for the three months ended September 30, 2017 were $2,795 compared to the three months ended September 30, 2016 of $3,149. The decrease of $354 was due to additional costs associated with post merger activities incurred during the three month ended September 30, 2016 compared to the period ended September 30, 2017.
G&A expenses for the nine months ended September 30, 2017 were $8,611 compared to the nine months ended September 30, 2016 of $8,267. G&A expenses for the nine months ended September 30, 2017 were $8,611 compared to the nine months ended September 30, 2016 of $8,267. G&A costs for the nine months ended September 30, 2016 included G&A costs for five months of activity incurred for the combined Company following the VBI-SciVac Merger as compared to G&A costs for nine months of activity incurred for the combined Company during the nine months in the period ended September 30, 2017. Additionally, during the nine months ended September 30, 2016, the Company incurred certain acquisition costs that were not incurred in the nine months ended September 30, 2017.
Loss from Operations
Losses from operations were $9,141 and $26,300 for the three and nine months ended September 30, 2017, compared to $7,740 and $16,185 for the three and nine months ended September 30, 2016. The losses for all periods presented are a result of the items noted above.
Interest expense, net
Interest expense, net is $742 and $2,188 for the three and nine months ended September 30, 2017. This includes the interest on our outstanding long term-debt plus the amortization of the debt discount. In 2016, these amounts were minimal as a majority of the long term debt was drawn in December 2016.
Foreign exchange gain
The foreign exchange gains of $81 and $605 for the three and nine months ended September 30, 2017 and the foreign exchange gains of $503 and $1,481 for the three and nine month periods ended September 30, 2016 are a result of the change in the foreign currencies in which the foreign currency transactions were denominated for each of those periods.
Income tax benefit
The income tax benefit for the nine month period ended September 30, 2017 was a result of the losses incurred by VBI Cda up to the amount of deferred tax asset that is more likely than not to be realized.
Net loss
Losses of $9,802 and $27,452 for the three and nine months ended September 30, 2017 compared to $7,299 and $14,794 for the three and nine months ended September 30, 2016 are a result of the items discussed above.
| 26 |
Liquidity and Capital Resources
| September 30, 2017 | December 31, 2016 | $ Change | % Change | |||||||||||||
| Cash | $ | 10,054 | $ | 32,282 | $ | (22,228 | ) | (69 | )% | |||||||
| Current Assets | 12,951 | 34,358 | (21,507 | ) | (63 | )% | ||||||||||
| Current Liabilities | 12,584 | 7,614 | 4,870 | 64 | % | |||||||||||
| Working Capital | 367 | 26,744 | (26,377 | ) | (99 | )% | ||||||||||
| Accumulated Deficit | (132,432 | ) | (104,980 | ) | (27,452 | ) | 26 | % | ||||||||
As at September 30, 2017, we had cash of $10,054 as compared to $32,282 as at December 31, 2016. As at September 30, 2017, we had working capital of $367 as compared to working capital of $26,744 at December 31, 2016. Working capital is calculated by subtracting current liabilities from current assets.
On June 20, 2016, we closed an equity private placement, pursuant to which we sold an aggregate of 3,269,688 common shares at a price of approximately $4.16 per share for total gross proceeds of approximately $13.6 million.
On December 6, 2016, we raised $10.6 million in an equity financing transaction with Perceptive Life Sciences Master Fund Ltd. and Titan-Perc Ltd., pursuant to which we sold an aggregate of 3,475,000 common shares at a price of $3.05 per share, for total gross proceeds of approximately $10.6 million. In a concurrent debt financing transaction with Perceptive Credit Holdings, LP (“Perceptive Credit”), we raised an additional $12.8 million, net of $360 in deferring financing charges. In conjunction with the additional debt funding, we issued a 5-year warrant to Perceptive Credit for the purchase of an aggregate of 1,705,053 common shares. Up to 363,771 common shares underlying the warrant may be exercised at a price of $4.13 per share and up to 1,341,282 common shares underlying the warrant may be exercised at a price of $3.355 per share.
On October 30, 2017, we closed an underwritten public offering and a concurrent registered direct offering of an aggregate of 23,575,410 common shares at a price of $3.05 per share. In addition, in connection with the registered direct offering, we issued four-year warrants to purchase 550,000 common shares at an exercise price of $3.34 per share. The aggregate estimate net proceeds from the offerings to the Company were approximately $67.4 million.
Liquidity Outlook
VBI’s income generating activities to-date have been from sales of its Sci-B-Vac product in markets that have generated a limited number of sales to-date, as well as fees from research and development (“R&D”) services. VBI has incurred significant net losses and negative operating cash flows since inception. As of September 30, 2017, VBI had an accumulated deficit of approximately $132,432. Our ability to maintain our status as an operating company is dependent upon obtaining adequate cash to finance our clinical development, our administrative overhead and our research and development activities. We plan to finance future operations with a combination of existing cash reserves, proceeds from the issuance of equity securities, the issuance of additional debt, and revenues from potential collaborations, if any. There is no assurance the Company will manage to obtain these sources of financing on reasonable terms or at all. These factors raise substantial doubt about our ability to continue as a going concern. The accompanying financial statements have been prepared assuming that we will continue as a going concern. The financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result should we be unable to continue as a going concern.
We have incurred operating losses since inception, have not generated significant product sales revenue and have not achieved profitable operations. We incurred a net loss of $9,802 and $27,452 for the three and nine months ended September 30, 2017 and we expect to continue to incur substantial losses in future periods. We anticipate that our operating expenses will increase substantially as we continue the clinical development of our Sci-B-Vac product and CMV vaccine candidates, as well as advance our pre-clinical-stage product candidate, GBM. These include expenses related to:
| 27 |
| ● | completing the CMV Phase I clinical study, a planned GBM Phase I/IIA clinical study and preparation to start a Sci-B-Vac Phase III program; | |
| ● | continuing the research and development of our product candidates; | |
| ● | scaling-up manufacturing capabilities through sub-contractors to commercialize products and dose forms for which we may obtain regulatory approval; | |
| ● | maintaining, expanding and protecting our intellectual property portfolio; | |
| ● | hiring additional clinical, manufacturing, and scientific personnel or contractors; and | |
| ● | adding operational, financial and management information systems and human resources support, including additional personnel, to support our vaccine development and commercialization activities. |
Our actual future capital requirements will depend on many factors, including the progress and results of our clinical studies, the duration and cost of discovery and preclinical development, laboratory testing and clinical studies for our products, the timing and outcome of regulatory review of our products, product sales outside of Israel, the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights, the number and development requirements of other product candidates that we pursue and the costs of commercialization activities, including product marketing, sales and distribution.
As stated above, we will require significant additional funds to conduct clinical and non-clinical studies, achieve regulatory approvals, and, subject to such approvals, commercially launch our products.
Net cash used in Operating Activities
We incurred net losses of $27,452 and $14,794 in the nine months ended September 30, 2017 and 2016, respectively. We used $21,890 and $10,569 in cash for operating activities during the nine months ended September 30, 2017 and 2016, respectively. The increase in cash outflows is largely as a result of increased professional fees and additional operating costs related to increased R&D expenses related to the advancement of the CMV, GBM and Sci-B-Vac vaccines.
Net cash used in and provided by Investing Activities
Cash flows used in investing activities decreased by $2,126 from $1,600 cash provided by investing activities for the nine months ended September 30, 2016 to $526 of cash used in investing activities for the nine months ended September 30, 2017 largely related to the purchase of additional property and equipment in SciVac in 2017. Going forward, we will be required to refresh some information technology equipment and to purchase additional R&D equipment.
Net cash used in and provided by Financing Activities
Cash flows related to financing activities were not significant in nine months ended September 30, 2017. For the nine month period ended September 30, 2016, the cash received from financing activities reflects the financing obtained.
Off-Balance Sheet Arrangements
As of September 30, 2017, we had no off-balance sheet arrangements.
| 28 |
Commitment and Contingencies
Leases
The remaining minimum annual lease commitments relating to office, lab and manufacturing facilities during the next five years are as follows. See Note 10, Commitments and Contingencies to the condensed consolidated financial statements for further discussion.
The future annual minimum payments under these leases is as follows:
| Year ending December 31 | ||||
| Remaining 2017 | $ | 228 | ||
| 2018 | 738 | |||
| 2019 | 685 | |||
| 2020 | 448 | |||
| 2021 | 448 | |||
| Thereafter | 37 | |||
| Total | $ | 2,584 | ||
Critical Accounting Policies and Estimates
There have been no changes to our critical accounting policies during the three months ended September 30, 2017. Critical accounting policies and the significant accounting estimates made in accordance with such policies are regularly discussed with the Audit Committee of the Company’s board of directors. Those policies are discussed under “Critical Accounting Policies” in our “Management’s Discussion and Analysis of the Financial Condition and Results of Operations” included in Item 7, as well as in our condensed consolidated financial statements and the footnotes thereto, included in our 2016 annual report on the Form 10-K filed with the Securities and Exchange Commission on March 20, 2017.
Trends, Events and Uncertainties
As with other companies that are in the process of commercializing novel vaccines, we will need to successfully manage normal business and scientific risks. R&D of new technologies is, by its nature, unpredictable. We cannot assure you that our technology will be adopted, that we will ever earn revenues sufficient to support our operations, or that we will ever be profitable. Furthermore, other than as discussed in this Form 10-Q, we have no committed source of financing and may not be able to raise money as and when we need it to continue our operations. If we cannot raise funds as and when we need them, we may be required to severely curtail, or even to cease, our operations.
Other than as discussed above and elsewhere in this Form 10-Q, we are not aware of any trends, events or uncertainties that are likely to have a material effect on our financial condition.
Recent Accounting Pronouncements
See Note 3, New Accounting Pronouncements, of Notes to the Condensed Consolidated Financial Statements.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
Not applicable.
Item 4. Controls and Procedures
Disclosure Controls and Procedures
Our management has evaluated, under the supervision and with the participation of our Chief Executive Officer (our principal executive officer) and our Senior Vice-President, Finance (our principal financial officer), the effectiveness of our disclosure controls and procedures as defined in Rule 13a-15(e) or Rule 15d-15(e) under the Exchange Act as of the end of the period covered by this Form 10-Q. Based on that evaluation, our Chief Executive Officer and our Senior Vice-President, Finance have concluded that, as of the end of the period covered by this Form 10-Q, our disclosure controls and procedures are effective in ensuring that information required to be disclosed in our Exchange Act reports is (1) recorded, processed, summarized and reported in a timely manner, and (2) accumulated and communicated to our management, including our Chief Executive Officer and our Senior Vice-President, Finance, as appropriate, to allow timely decisions regarding required disclosure.
| 29 |
Changes in Internal Control Over Financial Reporting
There has been no significant changes in our internal control over financial reporting that occurred during our last fiscal quarter ended September 30, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
From time to time, we may be involved in certain claims and litigation arising out of the ordinary course and conduct of business. Management assesses such claims and, if it considers that it is probable that an asset had been impaired or a liability had been incurred and the amount of loss can be reasonably estimated, provisions for loss are made based on management’s assessment of the most likely outcome.
The following description of risk factors includes any material changes to, and supersedes the description of, risk factors associated with our business, financial condition and results of operations previously disclosed in “Item 1A. Risk Factors” of our annual report on Form 10-K for the fiscal year ended December 31, 2016, as filed with the SEC on March 20, 2017, and our Quarterly Report on Form 10-Q for the fiscal quarter ended June 30, 2017, as filed with the SEC on July 31, 2017. Our business, financial condition and operating results can be affected by a number of factors, whether currently known or unknown, including but not limited to those described below, any one or more of which could, directly or indirectly, cause our actual financial condition and operating results to vary materially from past, or from anticipated future, financial condition and operating results. Any of these factors, in whole or in part, could materially and adversely affect our business, financial condition, operating results and stock price.
The following discussion of risk factors contains forward-looking statements. These risk factors may be important to understanding other statements in this Form 10-Q. The following information should be read in conjunction with the condensed consolidated financial statements and related notes in Part I, Item 1, “Financial Statements” and Part I, Item 2, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this Form 10-Q.
Product Development Risks
Because our vaccine product development efforts depend on new and rapidly evolving technologies, we cannot be certain that our efforts will be successful.
Our vaccine development efforts depend on new, rapidly evolving technologies and on the marketability and profitability of our products. Commercialization of our vaccines could fail for a variety of reasons, and include the possibility that:
| ● | Sci-B-Vac may not be approved for sale in the United States, Europe or Canada; | |
| ● | our eVLP vaccine technologies, any or all of the products based on such technologies or our manufacturing process will be ineffective or unsafe, or otherwise fail to receive necessary regulatory clearances or achieve commercial viability; | |
| ● | our lipid particle vaccine technologies, any or all of the products produced using such technologies will be ineffective or unsafe, or otherwise fail to receive necessary regulatory clearances or achieve commercial viability; | |
| ● | we may be unable to develop a scale-up method for our manufacturing protocols in a cost-effective manner; | |
| ● | the products, if safe and effective, will be difficult to manufacture on a large-scale or may be uneconomical to market; | |
| ● | our subcontracted third party manufacturing facility may fail to continue to pass regulatory inspections; | |
| ● | proprietary rights of third parties will prevent us or our collaborators from exploiting technologies, and manufacturing or marketing products; and | |
| ● | third-party competitors will gain greater market share due to superior products or marketing capabilities. |
The FDA and corresponding foreign regulatory agencies may require more clinical trials for our Sci-B-Vac than we currently expect or are conducting before granting regulatory approval, if regulatory approval is granted at all.
Our registration and commercial timelines for Sci-B-Vac depend on further discussions with the FDA and corresponding foreign regulatory agencies and requirements and requests they may make for additional data or completion of additional clinical trials. Any such requirements or requests could:
| o | adversely affect our ability to timely and successfully commercialize or market Sci-B-Vac in the United States and other jurisdictions where Sci-B-Vac is not currently approved; | |
| o | result in significant additional costs; | |
| o | potentially diminish any competitive advantages for Sci-B-Vac; | |
| o | potentially limit the markets for Sci-B-Vac; | |
| o | adversely affect our ability to enter into collaborations or receive milestone payments or royalties from potential collaborators; | |
| o | cause us to abandon the further development of the affected product candidate to comply with requests by the FDA or other jurisdictions where it is not currently approved; or | |
| o | limit our ability to obtain additional financing on acceptable terms, if at all. |
| 30 |
Pre-clinical and clinical trials will be lengthy and expensive. Delays in clinical trials are common for many reasons and any such delays could result in increased costs to us and jeopardize or delay our ability to obtain regulatory approval and commence product sales as currently contemplated.
As part of the regulatory process, we must conduct clinical trials for each vaccine candidate to demonstrate safety and efficacy to the satisfaction of the regulatory authorities, including the FDA for the United States, the EMA for the European Union and HC for Canada. Clinical trials are subject to rigorous regulatory requirements and are expensive and time-consuming to design and implement. We may experience delays in clinical trials for any of our vaccine candidates, and the projected timelines for continued development of the technologies and related vaccine candidates by us may otherwise be subject to delay or suspension. Our planned clinical trials might not begin on time; may be interrupted, delayed, suspended, or terminated once commenced; might need to be redesigned; might not enroll a sufficient number of patients; or might not be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including the following:
| ● | delays in obtaining regulatory approval to commence a trial; | |
| ● | imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or other regulatory authorities; | |
| ● | imposition of a clinical hold because of safety or efficacy concerns by the FDA, a data safety monitoring board or committee, a clinical trial site’s Institutional Review Board (“IRB”), or us; | |
| ● | delays in reaching agreement on acceptable terms with prospective contract research organizations (“CROs”) and clinical trial sites; | |
| ● | delays in obtaining required IRB approval at each site for clinical trial protocols; | |
| ● | delays in identifying, recruiting and training suitable clinical investigators; | |
| ● | delays in recruiting suitable patients to participate in a trial; | |
| ● | delays in having patients complete participation in a trial or return for post-treatment follow-up; | |
| ● | clinical sites dropping out of a trial to the detriment of enrollment; | |
| ● | time required to add new sites; | |
| ● | delays in obtaining sufficient supplies of clinical trial materials, including comparator drugs; | |
| ● | delays resulting from negative or equivocal findings of a data safety monitoring board for a trial; or | |
| ● | adverse or inconclusive results from pre-clinical testing or clinical trials. |
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials, and clinicians’ and patients’ perceptions as to the potential advantages of the biologic being studied in relation to other available therapies, including any new biologics that may be approved for the indications we are investigating. Any of these delays in completing our clinical trials could increase costs, slow down the product development and approval process, and jeopardize our ability to commence product sales and generate revenue.
| 31 |
Development of sufficient and appropriate clinical protocols to demonstrate safety and efficacy are required, and we may not adequately develop such protocols to support approval.
In addition to FDA requirements and those of other regulatory authorities, an independent IRB or an independent ethics committee at each medical institution proposing to participate in the conduct of the clinical trial generally must review and approve the clinical trial design and patient informed consent form before commencement of the study at the respective medical institution. The IRBs approve the clinical trial protocols, which describe the type of people who may participate in the clinical trial, the schedule of tests and procedures, the medications and dosages to be studied, the length of the study, the study’s objectives, and other details. In general, the IRB will consider, among other matters, ethical factors, the safety of human subjects and the possibility of liability of the institution conducting the trial. Our preclinical studies may not be adequate proof of safety and efficacy, and as a result, we may not be successful in developing clinical trial protocols necessary to support IRB approval. Any delay or failure to obtain IRB approval to conduct a clinical trial at a prospective site could materially impact the costs, timing or successful completion of a clinical trial.
We rely on CROs, third party investigators, and independent sites to conduct our clinical trials. If these third parties do not fulfill their contractual obligations or meet expected deadlines, our planned clinical trials may be extended, delayed, modified, or terminated and we may fail to obtain the regulatory approvals necessary to commercialize our product candidates.
We rely on third party CROs to conduct our clinical trials, including the Sci-B-Vac Phase III clinical studies. If these CROs do not perform their obligations or meet expected deadlines, our planned clinical trials may be extended, delayed, modified or terminated. We rely on the processes of our CROs to ensure that accurate records are maintained to support the results of the clinical trials. While we or our CROs conduct regular monitoring of clinical sites, we are dependent on the processes and quality control efforts of our third party contractors to ensure that detailed, quality records are maintained to support the results of the clinical trials that they are conducting on our behalf. Any extension, delay, modification or termination of our clinical trials or failure to ensure adequate documentation and the quality of the results in the clinical trials could delay or otherwise adversely affect our ability to commercialize our products and product candidates and could have a material adverse effect on our business and operations.
We may rely upon independent sites and investigators, such as universities and medical institutions and their faculty or staff, to conduct our clinical trials. These sites and investigators are not our employees and we cannot control the amount or timing of resources that they devote to our programs. If these investigators or collaborators fail to devote sufficient time and resources to our product development programs, or if their performance is substandard, the approval of our regulatory submissions and our introductions of new products will be delayed or prevented.
Our potential collaborators may also have relationships with other commercial entities, some of which may compete with us. If outside collaborators assist our competitors to our detriment, the approval of our regulatory submissions will be delayed and the sales from our products, if and when commercialized, will be less than expected. Even if clinical trials are completed as planned, their results may not support expectations or intended marketing claims. The clinical trials process may fail to demonstrate that our product candidates are safe and effective for indicated uses. Such failure could cause us to abandon a product candidate and could delay development of other product candidates.
Additional delays to the completion of clinical studies may result from modifications being made to the protocol during the clinical trial, if such modifications are warranted and/or required by the occurrences in the given trial.
Each modification to a protocol for a clinical trial must be submitted to the FDA or foreign regulatory authorities and the IRBs. This submission could result in the delay or suspension of a clinical trial while the modification is evaluated. In addition, depending on the magnitude and nature of the changes made, the FDA and other regulatory authorities could take the position that the data generated by the clinical trial prior to the protocol modification cannot be pooled with the data collected after the modification because the same protocol was not used throughout the trial. This prohibition might require the enrollment of additional subjects, which could result in the extension of the clinical trial and the FDA and other regulatory authorities delaying approval of a product candidate.
| 32 |
We may be required to suspend or discontinue clinical trials because of adverse side effects or other safety risks that could preclude approval of our biologic candidates.
Our clinical trials may be suspended or terminated at any time for a number of reasons. A clinical trial may be suspended or terminated by us, our collaborators, the FDA, or other regulatory authorities because of a failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, presentation of unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using the investigational biologic, changes in governmental regulations or administrative actions, lack of adequate funding to continue the clinical trial, or negative or equivocal findings of the data safety monitoring board or the IRB for a clinical trial. An IRB may also suspend or terminate our clinical trials for failure to protect patient safety or patient rights. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants. If we elect or are forced to suspend or terminate any clinical trial of any proposed product that we develop, the commercial prospects of such proposed product will be harmed and our ability to generate product revenue from such proposed product will be delayed or eliminated. Any of these occurrences may harm our business, financial condition, results of operations and prospects significantly.
The future results of our current or future clinical trials may not support our product candidate claims or may result in the discovery of unexpected adverse side effects.
Even if our clinical trials are completed as planned, we cannot be certain that the results will support our product candidate claims or that the FDA or foreign regulatory authorities will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses. If the FDA or foreign regulatory authorities conclude that the clinical trials for any of our product candidates for which we might seek approval have failed to demonstrate safety and effectiveness, we would not receive regulatory approval to market that product in the United States or in other jurisdictions for the indications sought. In addition, such an outcome could cause us to abandon the product candidate and might delay development of others. Any delay or termination of our clinical trials will delay the filing of any product submissions with the FDA or foreign regulatory authorities and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile. Adverse clinical trial results, such as death or injury due to side effects, could jeopardize regulatory approval, and if approval is granted, such results may also lead to marketing restrictions or prohibitions. In addition, the clinical trials performed until now involve a relatively small patient population. Because of the small sample size, their results may not be indicative of future results.
International commercialization of Sci-B-Vac and our vaccine candidates faces significant obstacles, including obtaining regulatory approvals. Failure to obtain regulatory approval in foreign jurisdictions will prevent us from marketing or selling our products in such jurisdictions.
Sci-B-Vac is approved for sale in Israel and 14 other countries. In countries where we do not currently have the required approvals (including the United States, the European Union and Canada), we will need to obtain separate approvals from the relevant regulatory, pricing and reimbursement authorities to market or sell Sci-B-Vac or any of our vaccine candidates. Pursuing regulatory approvals will be time-consuming and expensive, and we may not obtain foreign regulatory approvals on a timely basis, if at all. The regulations vary among countries, and regulatory authorities in one market may require different or additional clinical trials than those required to obtain approval for our vaccine candidates in another market, and the time required to obtain approval may differ in one market from that required to obtain approval for our vaccine candidates in another market. Obtaining approval in one country does not ensure approval by regulatory authorities in other countries.
In addition, we have limited foreign regulatory, clinical and commercial resources. We currently market or sell Sci-B-Vac through collaborative relationships with foreign partners and may plan to do so with other vaccine candidates in the future, and, as such, current and future partners are critical to our international success. We may not be able to maintain current, or enter into future, collaboration agreements with appropriate partners for important foreign markets on acceptable terms, if at all. Current and future collaborations with foreign partners may not be effective or profitable.
| 33 |
Future legislation, regulations and policies adopted by the FDA or other regulatory authorities may increase the time and costs required for us to conduct and complete clinical trials for our vaccine candidates.
The FDA has established regulations, guidelines and policies to govern the pharmaceutical development and approval process, as have foreign regulatory authorities. We expect there will continue to be federal and state laws and/or regulations, proposed and implemented, that could impact our operations and business. Any change in regulatory requirements resulting from the adoption of new legislation, regulations or policies may require us to amend existing clinical trial protocols or add new clinical trials to comply with these changes. Such amendments to existing protocols or clinical trial applications or the need for new ones, may significantly and adversely affect the cost, timing and completion of the clinical trials for our candidates.
In addition, the FDA’s policies and those of other regulatory authorities may change and additional government regulations may be issued that could prevent, limit or delay regulatory approval of our product candidates, or impose more stringent product labeling and post-marketing testing and other requirements.
Developments by competitors may establish standards of care that affect our ability to conduct our clinical trials as planned.
Changes in standards related to clinical trial design could affect our ability to design and conduct clinical trials as planned. For example, regulatory authorities may not allow us to compare one or more of our product candidates to a placebo in a particular clinical indication where approved products are available. In that case, both the cost and the amount of time required to conduct a clinical trial could increase.
We face product liability exposure, which, if not covered by insurance, could result in significant financial liability.
The risk of product liability is inherent in the research, development, manufacturing, marketing and use of pharmaceutical products. Sci-B-Vac, our product candidates and products that we may commercially market in the future may cause, or may appear to have caused, injury or dangerous drug reactions, and expose us to product liability claims. These claims might be made by patients who use the product, healthcare providers, pharmaceutical companies, our corporate collaborators or others selling such products. If our current products or any of our product candidates during clinical trials were to cause adverse side effects, we may be exposed to substantial liabilities. Regardless of the merits or eventual outcome, product liability claims or other claims related to our products or product candidates may result in:
| ● | decreased demand for our products due to negative public perception; | |
| ● | injury to our reputation; | |
| ● | withdrawal of clinical trial participants or difficulties in recruiting new trial participants; | |
| ● | initiation of investigations by regulators; | |
| ● | costs to defend or settle the related litigation; | |
| ● | a diversion of management’s time and our resources; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
| ● | loss of revenues from product sales; and | |
| ● | the inability to commercialize any of our product candidates, if approved. |
| 34 |
We currently maintain product liability insurance, and we generally obtain clinical trial insurance once a clinical trial is initiated. However, the insurance coverage may not be sufficient to reimburse us for any expenses or losses we may suffer. Insurance coverage is becoming increasingly expensive, and, in the future, we, or any of our collaborators, may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts or at all to protect us against losses due to liability. Even if our agreements with any current or future collaborators entitle us to indemnification against product liability losses, such indemnification may not be available or adequate should any claim arise. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against product liability claims could prevent or inhibit the commercialization of our product candidates. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may not be sufficient to cover such claims and our business operations could be impaired.
Should any of the events described above occur, this could have a material adverse effect on our business, financial condition and results of operations.
Even if we obtain regulatory approval for one or more of our product candidates, we will still face extensive, ongoing regulatory requirements and review and our products may face future development and regulatory difficulties.
Even if we obtain regulatory approval for one or more of our product candidates in the United States, which we cannot guarantee, the FDA may still impose significant restrictions on a product’s indicated uses or marketing or impose conditions for approval, or impose ongoing requirements for potentially costly post-approval studies, including Phase IV clinical trials or post-market surveillance. As a condition to granting marketing approval of a product, the FDA may require us to conduct additional clinical trials. The results generated in these post-approval clinical trials could result in loss of marketing approval, changes in product labeling, or new or increased concerns about side effects or efficacy of a product. For example, the labeling for our product candidates, if approved, may include restrictions on use or warnings. The Food and Drug Administration Amendments Act of 2007 gives the FDA enhanced post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information and compliance with FDA-approved Risk Evaluation and Mitigation Strategies (“REMS programs”). If approved, our vaccine candidates will also be subject to ongoing FDA requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, record keeping and reporting of safety and other post-market information. The FDA’s exercise of its authority could result in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved products. Foreign regulatory agencies often have similar authority and may impose comparable costs. Post-marketing studies, whether conducted by us or by others and whether mandated by regulatory agencies or voluntary, and other emerging data about marketed products, such as adverse event reports, may also adversely affect sales of our product candidates once approved, and potentially our other marketed products. Further, the discovery of significant problems with a product similar to one of our products that implicate (or are perceived to implicate) an entire class of products could have an adverse effect on sales of our approved products. Accordingly, new data about our products could negatively affect demand because of real or perceived side effects or uncertainty regarding efficacy and, in some cases, could result in product withdrawal or recall. Furthermore, new data and information, including information about product misuse, may lead government agencies, professional societies and practice management groups or organizations involved with various diseases to publish guidelines or recommendations related to the use of our products or the use of related therapies or place restrictions on sales. Such guidelines or recommendations may lead to lower sales of our products.
The holder of an approved biologics license application (“BLA”) also is subject to obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the BLA. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Application holders must also submit advertising and other promotional material to the FDA. Advertising and promotional materials must comply with FDA rules in addition to other potentially applicable federal and state laws, including, by way of example, the Federal Trade Commission Act. Any sales and promotional activities are also potentially subject to federal and state consumer protection and unfair competition laws. The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA, or such other regulatory agencies as reflected in the product’s approved labeling. In particular, any labeling approved by such regulatory agencies for our product candidates may also include restrictions on use. Such regulatory agencies may impose further requirements or restrictions on the distribution or use of our product candidates as part of a mandatory plan, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. If we receive marketing approval for one or more of our product candidates, physicians may nevertheless prescribe such products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. In particular, the U.S. federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
| 35 |
Depending on the circumstances, failure to meet post-approval requirements by us or our third-party collaborators can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, FDA issuance of Form 483, untitled letters, and/or warning letters, suspension or termination of any ongoing clinical trials, or refusal to allow us to enter into supply contracts, including government contracts. Any government investigation of alleged violations of law could require us to expend significant amounts of time and resources in response, and could generate negative publicity and significantly inhibit our ability to bring to market or continue to market our products and generate revenue.
We may not succeed at in-licensing product candidates or technologies to expand our product pipeline.
We may not successfully in-license product candidates or technologies to expand our product pipeline. The number of such candidates and technologies is limited. Competition among large pharmaceutical companies and biopharmaceutical companies for promising product candidates and technologies is intense because such companies generally desire to expand their product pipelines through in-licensing. If we fail to carry out such in-licensing and expand our product pipeline, our potential future revenues may suffer especially if our current products or product candidates fail to generate material revenue.
The failure by us or our current or future manufacturers to obtain FDA or other regulatory agencies’ approval for their manufacturing facilities could have a material adverse impact on our business, results of operations, financial condition and prospects.
The facilities of any of our current and future manufacturers, whether the facilities are ours or third-party manufacturer facilities, must be approved by the FDA after we submit our BLA and before approval or by the regulators in other jurisdictions for our product candidate to be manufactured for commercial production. In the event that we are approved to market a drug product in the United States, we or our third-party manufacturers must register the manufacturing facilities with the FDA and are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with the FDA’s current Good Manufacturing Practices (“cGMP”) regulations. Similar rules apply in the event we are approved to market a medicinal product in the European Union. Other than Sci-B-Vac, which is currently manufactured by us, we are completely dependent on these third-party manufacturers for compliance with the requirements of U.S. and non-U.S. regulators for the manufacture of our finished products. If we or our third-party manufacturers cannot successfully produce material that conforms to our specifications and current good manufacturing practice requirements of any regulatory agency, we will not be able to secure approval for our manufacturing facilities. If the FDA or another regulatory agency does not approve these facilities for commercial production, we will need to find alternative suppliers, which would result in significant delays in obtaining required regulatory approvals. In addition, if we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility, or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing, requiring new warnings or other labeling changes to limit use of the drug, requiring that we conduct additional clinical trials, imposing new monitoring requirements or requiring that we establish a REMS program. These challenges may have a material adverse impact on our business, results of operations, financial condition and prospects.
We manufacture clinical and commercial supplies of Sci-B-Vac at a single location. Any disruption in the operations of our manufacturing facility could adversely affect our business and results of operations.
We rely on our manufacturing facility in Rehovot, Israel, for the manufacture of all clinical and commercial supplies of Sci-B-Vac. Our current manufacturing facility contains highly specialized equipment and utilizes complicated production processes developed over a number of years, which would be difficult, time-consuming and costly to duplicate or, though a remote risk, may be impossible to duplicate. If our facility were damaged or destroyed, or otherwise subject to disruption, including contamination, it would require substantial lead-time to replace our manufacturing capabilities and could cause costly delays. In such event, we would be forced to identify and rely entirely on third-party contract manufacturers for an indefinite period of time, which we may not be able to do in a timely manner and would further increase our production costs. Any disruptions or delays at our facility or its failure to meet regulatory compliance would significantly impair our ability to manufacture Sci-B-Vac for sale in the jurisdictions where it is approved for sale and for our proposed clinical studies in jurisdictions where we are seeking regulatory approval, which would result in increased costs and losses and adversely affect our business and results of operations.
| 36 |
If the supplier of our raw materials and certain reagents, fails to provide sufficient quantities to us, we may not be able to obtain an alternative supply on a timely or acceptable basis.
We rely on a single source for our supply of raw materials and certain reagents required for the manufacture of Sci-B-Vac. We do not have a written or oral agreement with this source of supply, as all orders are handled through individual purchase orders or an order-by-order basis. Alternative sources from which we can obtain our supply of most of these materials exist. However, we may not be able to find alternative suppliers in a timely manner that would provide supplies of these raw materials or reagents at acceptable quantities and prices, if at all. Any interruption in the supply of these materials would disrupt our ability to manufacture Sci-B-Vac for further development, current and future clinical trials, and commercial manufacturing, and could have a material adverse effect on our business, commercialization of Sci-B-Vac and future profit margins, if any.
We do not manufacture any of our raw materials nor do we plan to develop any capacity to do so. Instead, we rely on multiple sources to supply our raw materials so that we can manufacture sufficient quantities of Sci-B-Vac at our manufacturing facility. Some of the countries for our raw materials are not the same as our drug manufacturing location. Any disruption in supply of raw materials from a qualified supplier could result in significant delays with our manufacturing, clinical trials, BLA filing, BLA approval or commercial sale of the finished product due to contract delays, the need to manufacture new raw materials, out of specification raw materials, the need for import and export permits, and the failure of the newly sourced raw materials to perform to the standards of the previously sourced raw materials. These delays could have a material adverse effect on our business and future profit margins, if any.
We expect the healthcare industry to face increased limitations on reimbursement, rebates and other payments as a result of continued healthcare reform changes, which could adversely affect third-party coverage of our products and how much or under what circumstances healthcare providers will prescribe or administer our products.
In both the United States and other countries, sales of our products will depend in part upon the availability of reimbursement from third-party payers, which include governmental authorities, managed care organizations and other private health insurers. Third-party payers are increasingly challenging the price and examining the cost effectiveness of medical products and services.
Increasing expenditures for healthcare have been the subject of considerable public attention in the United States. Both private and government entities are seeking ways to reduce or contain healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system have been introduced or proposed in Congress and in some state legislatures, including reducing reimbursement for prescription products and reducing the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products.
Although we cannot predict the full effect on our business of the implementation of existing legislation or the enactment of additional legislation pursuant to healthcare and other legislative reform, we believe that legislation or regulations that would reduce reimbursement for, or restrict coverage of, our products could adversely affect how much or under what circumstances healthcare providers will prescribe or administer our products. As such, this legislation or regulations could materially and adversely affect our business by reducing our ability to generate revenue, raise capital, obtain additional collaborators and market our products. In addition, we believe the increasing emphasis on managed care in the United States has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact product sales.
Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues.
In some countries, particularly the countries of the European Union, the pricing and/or reimbursement of prescription pharmaceuticals is subject to governmental control. In Canada, the prices of patented medicines are subject to price controls. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our products to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be harmed, possibly materially.
| 37 |
We face intense competition and rapid technological change, which may make it more difficult to achieve significant market penetration. If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our product candidates is characterized by intense competition and rapid technological advances. For example, if it is approved in the future, Sci-B-Vac will compete in the United States with established HBV vaccines marketed by Merck & Co. and GlaxoSmithKline plc and outside the United States with vaccines from those companies and several additional established pharmaceutical companies. If competitors’ existing products or new products are more effective than or considered superior to our current or future products, the commercial opportunity for our products will be reduced or eliminated. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. We face competition from fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of our competitors have products or product candidates already approved or in development. In addition, many of these competitors, either alone or together with their collaborative partners, are larger than us and have substantially greater financial, technical, research, marketing, sales, distribution and other resources. Existing and potential competitors may develop or market products that are more effective or commercially attractive than any that we are developing or marketing. Competitors may obtain regulatory approvals and introduce and commercialize products before we do. These developments could have a significant negative effect on our financial condition. Even if we are able to compete successfully, we may not be able to do so in a profitable manner.
We may be exposed to liability claims associated with the use of hazardous materials and chemicals.
Our research and development activities involve the controlled use of hazardous materials and chemicals. Although we believe that our safety procedures for using, storing, handling and disposing of these materials comply with federal, state, provincial and local laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for any resulting damages and any liability could materially adversely affect our business, financial condition and results of operations. In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business and financial condition.
Our vaccine candidates may never achieve market acceptance, even if we obtain regulatory approvals.
Even if we receive regulatory approvals for the commercial sale of our vaccine candidates, the commercial success of these vaccine candidates will depend on, among other things, their acceptance by physicians, patients, third-party payers such as health insurance companies and other members of the medical community as a vaccine and a cost-effective alternative to competing products. If our vaccine candidates fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business. Market acceptance of, and demand for, any product that we may develop and commercialize will depend on many factors, including:
| ● | our ability to provide acceptable evidence of safety and efficacy; | |
| ● | the prevalence and severity of adverse side effects; | |
| ● | whether our vaccines are differentiated from other vaccines based on immunogenicity; | |
| ● | availability, relative cost and relative efficacy of alternative and competing treatments; | |
| ● | the effectiveness of our marketing and distribution strategy; | |
| ● | publicity concerning our products or competing products and treatments; and | |
| ● | our ability to obtain sufficient third-party insurance coverage or reimbursement. |
| 38 |
In particular, there are significant challenges to obtaining regulatory approval for CMV vaccines developed for the target market (pre-pregnant women) due to the relatively low tolerance for risk to these populations. The risk-benefit analysis undertaken by the FDA and other regulators in deciding whether or not to approve this product candidate will be high relative to other vaccines and biologic products that target less sensitive populations.
If our vaccine candidates do not become widely accepted by physicians, patients, third-party payers and other members of the medical community, our business, financial condition and results of operations would be materially and adversely affected.
If we are unable to manufacture our vaccines in sufficient quantities, at sufficient yields or are unable to obtain regulatory approvals for a manufacturing facility for our vaccines, we may experience delays in product development, clinical trials, regulatory approval and commercial distribution.
Completion of our clinical trials and commercialization of our eVLP vaccine candidates require access to, or development of, facilities to manufacture our eVLP vaccine candidates at sufficient yields and at commercial-scale. We have limited experience manufacturing any of our eVLP vaccine candidates in the volumes that will be necessary to support large-scale clinical trials or commercial sales. Efforts to establish these capabilities may not meet initial expectations as to scheduling, scale-up, reproducibility, yield, purity, cost, potency or quality.
If we are unable to manufacture our eVLP vaccine candidates in clinical or commercial quantities, as the case may be, in sufficient yields, with sufficient purity, potency, quality, and identity, then we must find, qualify, and rely on third parties. Any new third-party manufacturers must also receive FDA approval before we may use product manufactured by them as our commercial products and product candidates. Our vaccines may be in competition with other products for access to these facilities and may be subject to delays in manufacture if our third party manufacturers give other products greater priority. Although we regularly evaluate potential manufacturers, we have a third party manufacturing agreement in place with Paragon and have reserved resource capability for the manufacture of our Phase I and Phase I/IIa clinical trial materials. We have conducted technology transfer related to our proprietary product. Despite progress achieved to date, any delays experienced by Paragon, whether directly by Paragon or by its raw material suppliers in relation to our project, may result in delays.
As a result, any delay or interruption could have a material adverse effect on our business, financial condition, results of operations and cash flows.
Our Sci-B-Vac vaccine is manufactured in the manufacturing facility of our subsidiary in Rehovot, Israel.
In light of our current resources and limited experience, we may need to establish successful third-party relationships to successfully commercialize our product candidates.
The near and long-term viability of our vaccine candidates may depend, in part, on our ability to successfully establish new strategic collaborations with pharmaceutical and biotechnology companies, non-profit organizations and government agencies. Establishing strategic collaborations and obtaining government funding is difficult and time-consuming. Potential collaborators may reject collaborations based upon their assessment of our financial, regulatory or intellectual property position or based on their internal pipeline; government agencies may reject contract or grant applications based on their assessment of public need, the public interest, the ability of our products to address these areas, or other reasons beyond our expectations or control. If we fail to establish a sufficient number of collaborations or government relationships on acceptable terms, we may not be able to commercialize our vaccine candidates or generate sufficient revenue to fund further research and development efforts.
Even if we establish new collaborations or obtain government funding, these relationships may never result in the successful development or commercialization of any vaccine candidates for several reasons, including the fact that:
| ● | we may not have the ability to control the activities of our partners and cannot provide assurance that they will fulfill their obligations to us, including with respect to the license, development and commercialization of vaccine candidates, in a timely manner or at all; | |
| ● | such partners may not devote sufficient resources to our vaccine candidates or properly maintain or defend our intellectual property rights; | |
| ● | relationships with our collaborators could also be subject to certain fraud and abuse laws if not structured properly to comply with such laws; | |
| ● | any failure on the part of our partners to perform or satisfy their obligations to us could lead to delays in the development or commercialization of our vaccine candidates and affect our ability to realize product revenue; and | |
| ● | disagreements, including disputes over the ownership of technology developed with such collaborators, could result in litigation, which would be time-consuming and expensive, and may delay or terminate research and development efforts, regulatory approvals and commercialization activities. |
| 39 |
If we or our collaborators fail to maintain our existing agreements or in the event we fail to establish agreements as necessary, we could be required to undertake research, development, manufacturing and commercialization activities solely at our own expense. These activities would significantly increase our capital requirements and, given our lack of sales, marketing and distribution capabilities, significantly delay the commercialization of our vaccine candidates.
Risks Related to Our Capital Requirements and Financings
We will need additional financing to continue our operations. If we are unable to obtain additional financing on acceptable terms, we may have to curtail or cease our development plans and operations.
Since inception, we and our subsidiaries collectively have raised approximately $124.7 million in total equity and debt financing to support clinical and research development and general business operations. Our revenue generating activities include product sales and research and development services pursuant to fee for service agreements, research collaboration agreements and certain governmental research and development grants. However, our revenues have not been significant to date. Our long-term success and ability to continue as a going concern is dependent upon obtaining sufficient capital to fund the research and development of our products, to bring about their successful commercial release, if approved, to generate revenue and, ultimately, to attain profitable operations or alternatively advance the products and technology to such a point that an acquirer would find attractive. We face substantial demand on our cash resources to fund operations and our growth plans in the future.
To date, we have been able to obtain financing; however, there is no assurance that financing will be available in the future, or if it is, that it will be available at terms acceptable to us. Additional financings may be effected through debt financing and/or the issuance of equity securities, there being no assurance that any type of financing on terms acceptable to us will be available or otherwise occur. Debt financing must be repaid regardless of whether we generate revenues or cash flows from operations and may be secured by substantially all of our assets. Any equity financing or debt financing that requires the issuance of equity securities or securities convertible into equity securities would cause the percentage ownership of our shareholders to be diluted, which dilution may be substantial. Also, any additional equity securities issued may have rights, preferences or privileges senior to those of existing shareholders. Furthermore, if we issue additional securities, whether equity or debt, or if investors believe we may issue additional securities, the market price of our common shares could decline. If such financing is not available when required or is not available on acceptable terms, we may be required to reduce or eliminate certain product candidates and development activities, and it may ultimately require us to suspend or cease operations, which could cause investors to lose the entire amount of their investment.
We have incurred significant losses since inception and anticipate that we will incur continued losses for the foreseeable future.
We have incurred significant net losses and negative operating cash flows since inception. We incurred net losses of approximately $23.2 million in 2016 and $26.2 million in 2015. We incurred net losses of approximately $27.5 million and $14.8 million during the nine months ended September 30, 2017 and 2016, respectively. As of September 30, 2017, we had an accumulated deficit of $132.4 million. Our income generating activities have been from sales of our Sci-B-Vac product in markets that have generated a limited number of sales to-date and fees from research and development services. We expect to incur significant and increasing operating losses for the next several years as we continue to advance Phase III clinical program for Sci-B-Vac and support regulatory submissions, expand our research and development, advance other vaccine candidates into and through clinical development, including CMV and GBM vaccine candidates, complete clinical trials and seek regulatory approval. Because of the numerous risks and uncertainties associated with developing and commercializing pharmaceutical products, as well as those related to our expectations for Sci-B-Vac Phase III clinical program, we are unable to predict the extent of any future losses or guarantee when, or if, our company will become profitable or cash flow positive. If we never achieve profitability or positive cash flows, or achieve either later than we anticipate, you may lose some or all of your investment in us.
| 40 |
The report of our independent registered public accounting firm for the year ended December 31, 2016, contains an explanatory paragraph as to our ability to continue as a going concern, and our financial statements for the three and nine months ended September 30, 2017, state that certain factors raise substantial doubt as to our ability to continue as a going concern, which could prevent us from obtaining new financing on reasonable terms or at all.
Because we have had recurring losses and negative cash flows from operating activities and have significant future commitments, substantial doubt exists regarding our ability to continue as a going concern. Accordingly, the report of EisnerAmper LLP, our independent registered public accounting firm, with respect to our financial statements for the year ended December 31, 2016, includes an explanatory paragraph as to our potential inability to continue as a going concern, and our financial statements for the three and nine months ended September 30, 2017, state that certain factors raise substantial doubt about our ability to continue as a going concern. Such doubts regarding our ability to continue as a going concern may adversely affect our ability to obtain new financing on reasonable terms or at all.
Risks Related to Our Business
Our future results will suffer if we do not effectively manage our expanded operations.
As a result of our acquisition of VBI Vaccines (Delaware) Inc. on May 6, 2016 (the “VBI-SciVac Merger”), we became a larger company than either we or VBI Vaccines (Delaware) Inc. was, on a stand-alone basis, prior to the VBI-SciVac Merger, and our business became more complex. There can be no assurance that we will effectively manage this increased complexity without experiencing operating inefficiencies or control deficiencies. Our failure to successfully manage the increased complexity could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We have international operations, which subject us to risks inherent with operations outside of the United States.
We have international operations and we may seek to obtain market approvals in foreign markets that we deem to generate significant opportunities. However, even with the cooperation of a commercialization partner, conducting drug development in foreign countries involves inherent risks, including, but not limited to: difficulties in staffing, funding and managing foreign operations; different and unexpected changes in regulatory requirements; export restrictions; tariffs and other trade barriers; different reimbursement systems; economic weaknesses or political instability in particular foreign economies and markets; compliance with tax, employment, immigration and labor laws for employees living or travelling abroad; supply chain and raw materials management; difficulties in protecting, acquiring, enforcing and litigating intellectual property rights; fluctuations in currency exchange rates; and potentially adverse tax consequences.
If we were to experience any of the difficulties listed above, or any other difficulties, our international development activities and our overall financial condition may suffer and cause us to reduce or discontinue our international development and market approval efforts.
We may not be successful in hiring and retaining key employees, in which case our business may be harmed.
Our business is highly dependent upon the continued services of our senior management and key scientific and technical personnel. As such, our future success depends on our ability to identify, attract, hire or engage, retain and motivate well-qualified managerial, technical, clinical and regulatory personnel. Our operations require qualified personnel with expertise in nonclinical pharmacology and toxicology, pharmaceutical development, clinical research, regulatory affairs, manufacturing, sales and marketing. We must compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and, when the need arises, we may not be able to hire the personnel necessary to support our efforts. There can be no assurance that these professionals will be available in the market, or that we will be able to retain existing professionals or to meet or to continue to meet their compensation requirements. Furthermore, the cost base in relation to such compensation, which may include equity compensation, may increase significantly, which could have a material adverse effect on us. Failure to establish and maintain an effective management team and work force could adversely affect our ability to operate, grow and manage our business.
| 41 |
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to:
| ● | comply with FDA regulations or similar regulations of comparable foreign regulatory authorities; | |
| ● | provide accurate information to the FDA or comparable foreign regulatory authorities; | |
| ● | comply with manufacturing standards that we have established; | |
| ● | comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities; | |
| ● | properly protect patient information which is subject to federal and state privacy and security laws or similar laws in foreign countries; | |
| ● | report financial information or data accurately; or | |
| ● | disclose unauthorized activities to us. |
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commissions, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions that we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us and we are not successful in defending or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
We are subject to federal, provincial and state laws and regulations relating to our business and our failure to comply with those laws could have a material adverse effect on our results of operations and financial conditions.
We are subject to healthcare regulation and enforcement by the U.S. federal government and the states and other jurisdictions in which we conduct our business. The laws that may affect our ability to operate include the following:
| ● | the federal healthcare program Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under government healthcare programs such as the Medicare and Medicaid programs; | |
| ● | the federal Ethics in Patient Referrals Act of 1989, commonly known as the Stark Law, which prohibits a physician from referring a patient for certain items or services covered by Medicare or Medicaid to an entity in which the physician or a family has a financial interest; | |
| ● | the federal False Claims Act and related laws that prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other government healthcare programs that are false or fraudulent; |
| 42 |
| ● | the so-called qui tam provisions of the federal and state false claims acts which permit whistleblowers to sue in the name the federal or state governments’ healthcare providers and others for alleged violations of those laws and which permit whistleblowers to obtain a reward for bringing the case. These qui tam cases have been on the rise in recent years; | |
| ● | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; | |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payer, including commercial insurers; | |
| ● | the Patient Protection and Affordable Care Act (the “Affordable Care Act”) which imposes reporting requirements on device and pharmaceutical manufacturers to make annual public disclosures of payments to healthcare providers and ownership of their stock by healthcare providers. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value, or ownership or investment interests that are not reported; | |
| ● | The Prescription Drug Marketing Act, as amended, which governs the distribution of prescription drug samples to healthcare practitioners; | |
| ● | The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; and | |
| ● | State law equivalents of HIPAA related to the privacy and security of patient information. |
Further, the Affordable Care Act, among other things, amends the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes. A person or entity can now be found guilty of fraud or false claims under the Affordable Care Act without actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Possible sanctions for violation of these anti-kickback laws include monetary fines, civil and criminal penalties, exclusion from Medicare, Medicaid and other government programs and forfeiture of amounts collected in violation of such prohibitions. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against such claims, could result in a material adverse effect on our reputation, business, results of operations and financial condition.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians for marketing. Some states, such as California, Massachusetts and Vermont, mandate implementation of corporate compliance programs, along with the tracking and reporting of gifts, compensation and other remuneration to physicians.
The scope and enforcement of these laws is uncertain and subject to change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. We are not able to predict the impact on our business of any changes in these laws. Federal or state regulatory authorities may challenge our future activities under these laws. Any such challenge could have a material adverse effect on our reputation, business, results of operations and financial condition. Any state or federal regulatory review of the Company, regardless of the outcome, would be costly and time-consuming.
In addition, we expect that the current presidential administration and U.S. Congress will seek to modify, repeal, or otherwise invalidate all, or certain provisions of, the Affordable Care Act. Since taking office, President Trump has continued to support the repeal of all or portions of the Affordable Care Act. In January 2017, the House and Senate passed a budget resolution that authorizes congressional committees to draft legislation to repeal all or portions of the Affordable Care Act and permits such legislation to pass with a majority vote in the Senate. President Trump has also recently issued an executive order in which he stated that it is his administration’s policy to seek the prompt repeal of the Affordable Care Act and directed executive departments and federal agencies to waive, defer, grant exemptions from, or delay the implementation of the provisions of the Affordable Care Act to the maximum extent permitted by law. There is still uncertainty with respect to the impact President Trump’s administration and the U.S. Congress may have, if any, and any changes will likely take time to unfold, and could have an impact on coverage and reimbursement for healthcare items and services covered by plans that were authorized by the Affordable Care Act. However, we cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us.
| 43 |
In the context of securing and protecting any patient information that our clinical sites may obtain which may be subject to HIPAA or state law protections, we intend to include in the written agreements we will enter into with such clinical sites provisions requiring such clinical sites to have appropriate policies, procedures and systems in place to satisfy the privacy and security requirements. Our efforts, however, cannot protect against every potential threat to such patient information. For example, cyber attacks or improper actions of an employee could result in a breach of our systems, resulting in immediate costs to address and correct the breach and notify any impacted parties, as well as potential litigation or governmental proceedings which could result in monetary fines and/or criminal sanctions. A breach of protected information could result in material adverse effects on our reputation, business operations and financial condition.
We may expand our business through the acquisition of rights to new product candidates that could disrupt our business and harm our financial condition.
We may expand our product offerings, and we may seek acquisitions of product candidates or technologies to do so. We may also seek to expand our business through the acquisition of businesses or companies having rights to new product candidates. Acquisitions involve numerous risks, including substantial cash expenditures; potentially dilutive issuances of equity securities; incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of the acquisition; difficulties in assimilating the acquired technologies or the operations of the acquired companies; diversion of management’s attention away from other business concerns; risks of entering markets in which we have limited or no direct experience; and the potential loss of key employees or key employees of the acquired companies.
There can be no assurance that any acquisition by us will result in short-term or long-term benefits to us. We may incorrectly judge the value or worth of an acquired product, company or business. In addition, future success of the combined company will depend in part on our ability to manage the rapid growth associated with some of these acquisitions. There can be no assurance that we will be able to make the combination of our business with that of any acquired products, businesses or companies work or be successful. Furthermore, the development or expansion of our business or any acquired products, businesses or companies may require a substantial capital investment by us. We may not have these necessary funds or such funds might not be available on acceptable terms or at all. We may also seek to raise funds by selling capital stock or instruments convertible into or exercisable for capital stock, which could dilute each shareholder’s ownership interest.
Business interruptions could limit our ability to operate our business.
Our operations, as well as those of any collaborators on which we depend, are vulnerable to damage or interruption from computer viruses, human error, natural disasters, extreme weather, electrical and telecommunication failures, international acts of terror and similar events. Our formal disaster recovery plan and back-up operations and business interruption insurance may not be adequate to compensate us for losses we may suffer. A significant business interruption could result in losses or damages incurred by us and require us to cease or curtail our operations.
Our business and operations would suffer in the event of computer system failures, cyber-attacks or deficiencies in our cyber-security.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, research data, our proprietary business information and that of our suppliers, technical information about our products, clinical trial plans and employee records. Similarly, our third-party providers possess certain of our sensitive data and confidential information. The secure maintenance of this information is critical to our operations and business strategy. Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, ransomware, cyber fraud, natural disasters, terrorism, war, telecommunication and electrical failures, cyber-attacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, encrypted, lost or stolen. Any such access, inappropriate disclosure of confidential or proprietary information or other loss of information, including our data being breached at third-party providers, could result in legal claims or proceedings, liability or financial loss under laws that protect the privacy of personal information, disruption of our operations or our product development programs and damage to our reputation, which could adversely affect our business. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data.
| 44 |
Under current U.S., Canadian and Israeli law, we may not be able to enforce covenants not to compete or to prevent the breach of confidentiality agreements, and therefore, may be unable to prevent our competitors from benefiting from the expertise of some of our former employees.
We generally enter into non-competition agreements with our employees and certain key consultants. These agreements prohibit our employees and certain key consultants, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period of time. However, under current U.S., Canadian and Israeli law, we may be unable to enforce these agreements, in whole or in part, and therefore, we cannot be sure that these employees and key consultants will not compete with us. For example, in the past, Israeli courts have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the secrecy of a company’s confidential commercial information or the protection of its intellectual property. If we are unable to demonstrate that harm would be caused to us or otherwise enforce these non-competition agreements, in whole or in part, we may be unable to prevent our competitors from benefitting from the expertise our former employees or consultants developed while working for us and our ability to remain competitive may be diminished.
We rely on confidential information that we seek to protect through confidentiality agreements with our employees and other parties. If these agreements are breached, competitors may obtain and use our confidential information to gain a competitive advantage over us or could substantially delay product development or harm our commercialization activities. We may not have any remedies against our competitors and any remedies that may be available to us may not be adequate to protect our business or compensate us for the damaging disclosure. In addition, we may have to expend resources to protect our interests from possible infringement by others, which may divert our available funds away from our business activities.
We have significant operations located in Israel and, therefore, our results may be adversely affected by political, economic and military instability in Israel.
Our subsidiary’s operations are located in Rehovot, Israel. Accordingly, political, economic and military conditions in Israel may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its neighboring countries. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect our business and results of operations.
Any armed conflicts, terrorist activities or political instability in the region could adversely affect business conditions and could harm our results of operations and could make it more difficult for us to raise capital. Parties with whom we do business have sometimes declined to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary in order to meet our business partners face to face. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements.
Since the Gaza Strip’s 2007 coup, by which the terrorist organization Hamas seized control, there have been a number of armed conflicts between Hamas and Israel – in December-January 2008-9, November 2012 and as recently as July-August 2014 – in all of which conflicts rockets were fired from Gaza into Israeli civilian population centers. During the summer of 2006, Israel was engaged in an armed conflict with Hezbollah, a Lebanese Islamist Shiite militia group and political party backed by Iran and controlling large swathes of Lebanon. These conflicts involved missile strikes against civilian targets in various parts of Israel, including areas in which our Rehovot facilities, employees and some of our consultants are located, and negatively affected business conditions in Israel. Since February 2011, Egypt has experienced political turbulence and an increase in terrorist activity in the Sinai Peninsula following the resignation of Hosni Mubarak as president. This included protests throughout Egypt, and the appointment of a military regime in his stead, followed by the elections to parliament which brought groups affiliated with the Muslim Brotherhood (which had been previously outlawed by Egypt), and the subsequent overthrow of this elected government by a military regime instead. Such political turbulence and violence could affect the region as a whole. Similar civil unrest and political turbulence has occurred in other countries in the region, including Syria which shares a common border with Israel, and is affecting the political stability of those countries. Since April 2011, internal conflict in Syria has escalated, and evidence indicates that chemical weapons have been used in the region. Intervention may be contemplated by outside parties in order to prevent further chemical weapon use. The extreme Sunni jihadist group ISIS has taken over large parts of Syria and its neighbor to the east, Iraq, and committed widespread massacres against the local civilian populations in those areas, all the while continuing in its efforts to conquer further territories. Syria and Iraq are now widely viewed as failed states on the verge of disintegration into tribal fiefdoms. This instability and any intervention may lead to additional conflicts in the region. In addition, Iran has threatened to attack Israel and is widely believed to be developing nuclear weapons. Iran also has a strong influence among extremist groups in the region, such as Hamas in Gaza, Hezbollah in Lebanon, and both the Allawite regime and various rebel militia groups in Syria. These situations may potentially escalate in the future to more violent events which may affect Israel and us. Any armed conflicts, terrorist activities or political instability in the region could adversely affect business conditions and could harm our results of operations and could make it more difficult for us to raise capital.
| 45 |
Commercial insurance does not cover losses that may occur as a result of an event associated with the security situation in the Middle East. Although the Israeli government is currently committed to covering the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure that this government coverage will be maintained, or if maintained, will be sufficient to compensate us fully for damages incurred. Any losses or damages incurred by us could have a material adverse effect on our business. Any armed conflicts or political instability in the region would likely negatively affect business conditions generally and could harm our results of operations.
Political relations could limit our ability to sell or buy internationally.
We could be adversely affected by the interruption or reduction of trade between Israel and its trading partners. To date the State of Israel and Israeli companies have been repeatedly subjected to economic boycotts. Several countries, companies and organizations continue to participate in a boycott of Israeli firms and others doing business with Israel or with Israeli companies. Also, over the past several years there have been calls in Europe and elsewhere to reduce trade with Israel. These restrictive laws and policies may have an adverse impact on our operating results, financial condition or the expansion of our business
The operations of our subsidiary in Israel may be disrupted as a result of the obligation of Israeli citizens to perform military service.
Many Israeli citizens are obligated to perform several days, and in some cases more, of annual military reserve duty until they reach the age of 40 (or older, for reservists who are officers or who have certain special training) and, in the event of a military conflict, may be called to active duty. In response to increases in terrorist activity and recent armed conflicts, there have been periods of significant call-ups of military reservists. It is possible that there will be military reserve duty call-ups in the future. The operations of our subsidiary in Israel could be disrupted by such call-ups, which may include the call-up of our employees or the employees of our Israeli business partners. Such disruption could materially adversely affect our business, financial condition and results of operations.
Exchange rate fluctuations between the U.S. dollar, Canadian dollar and the New Israeli Shekel currencies may negatively affect our earnings cash flows.
Our functional currency is the U.S. dollar. We incur expenses in New Israeli Shekel, which we refer to as NIS, Canadian Dollars and U.S. dollars. As a result, we are exposed to the risks that the U.S. dollar may devalue relative to the Canadian Dollar or NIS, or, if the U.S. dollar appreciates relative to the Canadian Dollar or NIS, that the inflation rate in the United States may exceed such rate of devaluation of the U.S. dollar, or that the timing of such devaluation may lag behind inflation in the United States. The average exchange rate for the year ended December 31, 2016, was US$1.00 = NIS 3.8376 and US$ 1.00 = Canadian Dollar 1.3134. We cannot predict any future trends in the rate of inflation in the United States or the rate of devaluation, if any, of the U.S. dollar against the Canadian Dollar or NIS.
Risks Related to Our Intellectual Property
Our success depends on our ability to maintain the proprietary nature of our technology. We may become subject to third parties’ claims alleging infringement of patents and proprietary rights or seeking to invalidate our patents or proprietary rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development of our current or future product candidates or commercialization of our products.
Our success in large part depends on our ability to maintain the proprietary nature of our technology. To do so, we must, at significant cost, prosecute and maintain existing patents, obtain new patents and pursue trade secret and other intellectual property protection. We also must operate without infringing the proprietary rights of third parties or allowing third parties to infringe our rights. We currently have rights to over 120 fully owned or exclusively licensed allowed patents and patent applications. However, patent issues relating to pharmaceuticals and biologics involve complex legal, scientific and factual questions.
| 46 |
To date, no consistent policy has emerged regarding the breadth of biotechnology patent claims that are granted by the U.S. Patent and Trademark Office or enforced by the federal courts. Therefore, we do not know whether our patent applications will result in the issuance of patents, or that any patents issued to us will provide us with any competitive advantage. We also cannot be sure that we will develop additional proprietary products that are patentable. Furthermore, there is a risk that others will independently develop similar technology or products or circumvent the patents issued to us.
Even if we are issued patents for our technologies, there is always a risk that third parties will initiate post grant review proceedings to challenge the validity of one or more of our patents. These proceedings can result in the loss of patent rights. Even if we are successful in defending our patents during post grant review proceedings, these procedures are time consuming and expensive and may have a negative impact on our results.
There is also a risk that third parties may challenge our existing patents in court or claim that we are infringing their patents or proprietary rights. We cannot assure you that any of our products or current or future product candidates will not infringe existing or future patents. Because we have not conducted a formal freedom to operate analysis for patents related to our products or product candidates, we may not be aware of patents that have already been issued that a third party might assert are infringed by one of our products or current or future product candidates. Because patent applications can take many years to issue and may be confidential for eighteen months or more after filing, there also may be applications now pending of which we are unaware and which may later result in issued patents that we may infringe by commercializing any of our products or current or future product candidates. We could incur substantial costs in defending patent infringement suits or in filing suits against others to have their patents declared invalid or claim infringement. It is also possible that we may be required to obtain licenses from third parties to avoid infringing third-party patents or other proprietary rights. We cannot be sure that such third-party licenses would be available to us on acceptable terms, if at all. If we are unable to obtain required third-party licenses, we may be delayed in or prohibited from developing, manufacturing or selling products requiring such licenses.
Although our patent filings include claims covering various features of our vaccine candidates, including composition, methods of manufacture and use, our patents do not provide us with complete protection against the development of competing products. Some of our know-how and technology is not patentable. To protect our proprietary rights in unpatentable intellectual property and trade secrets, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. These agreements may not provide meaningful protection for our trade secrets, know-how or other proprietary information.
Sci-B-Vac is not currently protected by any pending patent application nor any unexpired patent. Accordingly, Sci-B-Vac may be subject to competition from the sale of generic products that could adversely affect our business and operations.
Sci-B-Vac has no patent production, and therefore, we will seek to rely on non-patent data exclusivity in the United States Biologics Price Competition and Innovation Act (the “BPCI Act”), which is described further under “—We may not be able to obtain marketing exclusivity in the United States under the BPCI Act or equivalent regulatory data exclusivity protection in other jurisdictions for our products.”
Sci-B-Vac is the only product we currently market. Failure to obtain and retain marketing exclusivity or expiration of the market exclusivity could seriously adversely affect the revenue potential for Sci-B-Vac in the jurisdictions where it is approved for sale.
Our ability to protect and enforce our patents does not guarantee that we will secure the right to commercialize the patents.
A patent is a limited monopoly right conferred upon an inventor, and any successors in title, in return for the making and disclosing of a useful, new, and non-obvious invention. This monopoly is of limited duration but, while in force, allows the patent holder to prevent others from making and/or using his invention. While a patent gives the holder this right to exclude others, it is not a license to commercialize the invention, where other permissions may be required for permissible commercialization to occur. For example, a drug cannot be marketed without the appropriate authorization from the FDA, regardless of the existence of a patent covering the product. Further, the invention, even if patented itself, cannot be commercialized if it infringes the valid patent rights of another party.
| 47 |
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. Patent and Trademark Office and various foreign governmental patent offices require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case, which could result in a material adverse effect on our business or results of operations.
We are dependent on technologies we have licensed and we may need to license in the future, and if we fail to obtain licenses we need, or fail to comply with our payment obligations in the agreements under which we in-license intellectual property and other rights from third parties, we could lose our ability to develop our product candidates.
We currently are dependent on licenses from third parties for certain of our key technologies relating to eVLP technology, including the licenses from the L’Universite Pierre et Marie Curie (“UPMC”). Under our license agreement with UPMC and other licensors, we are granted an exclusive license to a family of patents and patent applications that is expected to expire in the United States in 2022 and 2021 in other countries. Under this agreement, we are required to pay UPMC between 0.75% to 1.75% of net sales and certain lump-sum milestone payments. In addition, we expect we will need to license intellectual property from other third parties in the future and that these licenses will be material to our business. No assurance can be given that we will generate sufficient revenue or raise additional financing to meet our payment obligations in the license agreements with UPMC or other license agreements we enter into with third parties in the future. Any failure to make the payments required by the license agreements may permit the licensor to terminate the license. If we were to lose or otherwise be unable to maintain these licenses for any reason, it would halt our ability to develop our product candidates. Furthermore, such loss of these licenses may enable development of new products based on the eVLP platform that may compete with our product candidates, and our competitors may gain proprietary position. Any of the foregoing could result in a material adverse effect on our business or results of operations.
In addition, we do not own the patents or patent applications that we license, and as such, we may need to rely upon our licensors to properly prosecute and maintain those patent applications and prevent infringement of those patents. If our licensors are unable to adequately protect their proprietary intellectual property we license from legal challenges, infringement or alternative technologies, we will not be able to compete effectively in the drug discovery and development business.
If patent laws or the interpretation of patent laws change, our competitors may be able to develop and commercialize our discoveries.
Important legal issues remain to be resolved as to the extent and scope of available patent protection for biopharmaceutical products and processes in the United States and other important markets outside the United States, such as Europe, China and Japan. As such, litigation or administrative proceedings may be necessary to determine the validity, scope and ownership of certain of our and others’ proprietary rights. Any such litigation or proceeding may result in a significant commitment of resources in the future and could force us to do one or more of the following: cease selling or using any of our products that incorporate the challenged intellectual property, which would adversely affect our revenue; obtain a license or other rights from the holder of the intellectual property right alleged to have been infringed or otherwise violated, which license may not be available on reasonable terms, if at all; and redesign our products to avoid infringing or violating the intellectual property rights of third parties, which may be time-consuming or impossible to do. In addition, changes in patent laws in the United States and other countries may result in allowing others to use our discoveries or develop and commercialize our products. We cannot provide assurance that the patents we obtain or the unpatented technology we hold will afford us significant commercial protection.
| 48 |
We may not be able to enforce our intellectual property rights throughout the world. This risk is exacerbated for us because it expects that one or more of its product candidates will be manufactured and used in a number of foreign countries.
The laws of foreign countries may not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. This risk is exacerbated for us because we currently have one product manufactured, and we expect that one or more of our product candidates will be manufactured, and used in a number of foreign countries.
The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to life sciences. This could make it difficult for us to stop the infringement or other misappropriation of our intellectual property rights. For example, several foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents and trade secrets may provide limited or no benefit.
Most jurisdictions in which we have applied for, intend to apply for or have been issued patents have patent protection laws similar to those of the United States, but some of them do not. For example, we may do business in China, Indonesia and India in the future and the countries in these regions may not provide the same or similar protection as that provided in the United States. Additionally, due to uncertainty in patent protection law, we have not filed applications in many countries where significant markets exist.
Proceedings to enforce patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of our intellectual property.
We may not be able to monetize intangible assets including In Process Research and Development (“IPR&D”) which may result in the need to record an impairment charge.
Our balance sheet contains significant amounts of intangible assets. For IPR&D assets, the risk of failure is significant, and there can be no certainty that these assets ultimately will yield successful products. The nature of our business is high-risk and requires that we invest in a large number of projects in an effort to achieve a successful portfolio of approved products. Our ability to realize value on these significant investments is often contingent upon, among other things, regulatory approvals and market acceptance while we currently expect to be able to monetize our intangible assets, these IPR&D assets may become impaired and be written off at some time in the future. An example of an event that is indicative of impairment is a projection or forecast that indicates losses or reduced profits associated with an asset. For IPR&D projects, this could result from, among other things, a change in outlook based on clinical trial data, a delay in the projected launch date or additional expenditures to commercialize the product.
While all intangible assets other than goodwill can face events and circumstances that can lead to impairment, in general, intangible assets other than goodwill that are most at risk of impairment include IPR&D assets. IPR&D assets are high-risk, as research and development is an inherently risky activity.
We may not be able to obtain marketing exclusivity in the United States under the BPCI Act or equivalent regulatory data exclusivity protection in other jurisdictions for our products.
The BPCI Act, which is included in the Affordable Care Act, creates an approval pathway for biosimilar and interchangeable biological products and provides the manufacturer of innovator biologic to seek a twelve-year period of marketing exclusivity. Similar data exclusivity regimes exist in the European Union and in Canada, although the term of market exclusivity is shorter than in the United States. We intend to seek the maximum period of market exclusivity for our Sci-B-Vac product and our other vaccine candidate products in each jurisdiction, but there is no guarantee that any of our products will receive any marketing exclusivity under the BPCI Act, or under analogous legislation in other jurisdictions. Furthermore, changes in legislation could alter the period of market exclusivity or limit its availability. Our failure to obtain exclusivity for any product that is ultimately approved by the FDA, the EMA or HC may expose us to substantial competition, which could have significant adverse financial consequences.
| 49 |
Risks Related to Our Indebtedness
Our obligations under our credit facility are secured by substantially all of our assets, so if we default on those obligations, the lender could foreclose on our assets. As a result of these security interests, such assets would only be available to satisfy claims of our general creditors or to holders of our equity securities if we were to become insolvent at a time when the value of such assets exceeded the amount of our indebtedness and other obligations.
Perceptive Credit, the lender under our credit facility has a security interest in all of our assets other than excluded and future projects. As a result, if we default under our obligations to the lender, the lender could foreclose on its security interests and liquidate some or all of these assets, which would harm our business, financial condition and results of operations. The principal amount of the term loan as of September 30, 2017, was $15 million ($15.3 million including the exit fee).
In the event of a default in connection with our bankruptcy, insolvency, liquidation, or reorganization, the lender would have a prior right to substantially all of our assets to the exclusion of our general creditors. In that event, our assets would first be used to repay in full all indebtedness and other obligations secured by the lender, resulting in all or a portion of our assets being unavailable to satisfy the claims of any unsecured indebtedness. Only after satisfying the claims of any unsecured creditors would any amount be available for our equity holders. These events of default include, among other things, our failure to pay any amounts due under the Amended Credit Agreement or any of the other loan documents, a breach of covenants under the Amended Credit Agreement, our insolvency, a material adverse effect occurring, the occurrence of certain defaults under certain other indebtedness or certain final judgments against us.
The pledge of these assets and other restrictions may limit our flexibility in raising capital for other purposes. Because substantially all of our assets are pledged under the term loan, our ability to incur additional secured indebtedness or to sell or dispose of assets to raise capital may be impaired, which could have an adverse effect on our financial flexibility.
If we are unable to comply with certain financial and operating restrictions in our existing credit facility, we may be limited in our business activities and access to credit or may default under our credit facility.
Provisions in the Amended Credit Agreement impose restrictions or require prior approval on our ability, and the ability of certain of our subsidiaries to, among other things:
| ● | incur additional debt; | |
| ● | pay cash dividends and make distributions; | |
| ● | make certain investments and acquisitions; | |
| ● | guarantee the indebtedness of others or our subsidiaries; | |
| ● | redeem or repurchase capital shares; | |
| ● | create liens or encumbrances; | |
| ● | enter into transactions with affiliates; | |
| ● | engage in new lines of business; | |
| ● | sell, lease or transfer certain parts of our business or property; | |
| ● | incur obligations for capital expenditures; | |
| ● | issue additional capital shares; and | |
| ● | acquire new companies and merge or consolidate. |
| 50 |
The Amended Credit Agreement also contains other customary covenants, including covenants that require us to meet specified financial ratios and financial tests and maintain a minimum cash balance of $2.5 million. We may not be able to comply with these covenants in the future. Our failure to comply with these covenants may result in the declaration of an event of default, which, if not cured or waived, may result in the acceleration of the maturity of indebtedness outstanding under this agreement and would require us to pay all amounts outstanding. If the maturity of our indebtedness is accelerated, we may not have sufficient funds available for repayment or we may not have the ability to borrow or obtain sufficient funds to replace the accelerated indebtedness on terms acceptable to us or at all. Our failure to repay our indebtedness would result in our lender foreclosing on all or a portion of our assets and force us to curtail or cease our operations.
Our outstanding term loan obligations may adversely affect our cash flow and our ability to operate our business.
Pursuant to the terms of Amended Credit Agreement, the lender made a term loan to us in aggregate amount of $15.0 million. We are required to make average monthly payments of interest in the amount of approximately $140,000 (based on the one-month London Interbank Offered Rate being 1% or less until April 2018) and monthly payments of interest and principal in the amount of approximately $200,000 per month from May 2018 until the loan matures. The principal amount of the term loan as of September 30, 2017, was $15 million ($15.3 million including the exit fee). The term loan under the Amended Credit Agreement matures on December 6, 2019.
The terms of our term loan could have negative consequences to us, such as:
| ● | we may be unable to obtain additional financing to fund working capital, operating losses, capital expenditures or acquisitions on terms acceptable to us, or at all; | |
| ● | the amount of our interest expense may increase because our term loan has a variable rate of interest at any time dependent on one-month London Interbank Offered Rate greater than 1%; and | |
| ● | we may be more vulnerable to economic downturns and adverse developments in our industry or the economy in general. |
Our ability to meet our expenses and debt obligations will depend on our future performance, which will be affected by financial, business, economic, regulatory and other factors. We will be unable to control many of these factors, such as economic conditions. We cannot be certain that we will continue to have sufficient capital to allow us to pay the principal and interest on our debt and meet any other obligations. If we do not have enough money to service our debt, we may be required, but unable to refinance all or part of our existing debt, sell assets, borrow money or raise equity on terms acceptable to us, if at all, and the lender could foreclose on its security interests and liquidate some or all of our assets.
Risks Related to Our Common Shares
The price of our common shares has been, and may continue to be, volatile. This may affect the ability of our investors to sell their shares, and the value of an investment in our common shares may decline.
During the 12-month period ended October 25, 2017, our common shares traded as high as $6.60 per share and as low as $2.75 per share. Due to the volatility of the market for our common shares, the market price for our shares may be significantly affected by factors such as variations in quarterly and yearly operating results or changes in state, provincial or federal regulations affecting us and our industry. Furthermore, in recent years the stock market has experienced extreme price and volume fluctuations that are unrelated or disproportionate to the operating performance of the affected companies. Such broad market fluctuations may adversely affect the market price of our common shares.
We have no immediate plans to pay dividends.
We plan to reinvest all of our earnings, to the extent we have earnings, in order to market our products and to cover operating costs and to otherwise become and remain competitive. We do not plan to pay any cash dividends with respect to our securities in the foreseeable future. We cannot assure you that we would, at any time, generate sufficient surplus cash that would be available for distribution to the holders of our common shares as a dividend. In addition, our Amended Credit Agreement with Perceptive Credit prohibits us from declaring or paying cash dividends or making distributions on any class of our capital stock. We currently intend to retain earnings, if any, for reinvestment in our business. Therefore, holders of our common shares should not expect to receive cash dividends on our common shares.
| 51 |
Common shares eligible for future sale may cause the price of our common shares to decline.
From time to time, certain of our shareholders may be eligible to sell all or some of their common shares by means of ordinary brokerage transactions in the open market pursuant to Rule 144, promulgated under the Securities Act of 1933, as amended, subject to certain limitations. In general, pursuant to Rule 144, non-affiliate shareholders may sell freely after six months, subject only to the current public information requirement (which disappears after one year). Of the 40,229,371 common shares outstanding as of September 30, 2017, approximately 21,493,485 common shares are held by “non-affiliates,” all of which are freely tradable without restriction pursuant to Rule 144.
Any substantial sale of our common shares pursuant to Rule 144 or pursuant to any resale prospectus may have a material adverse effect on the market price of our common shares.
In addition, as of September 30, 2017, we had outstanding options and warrants for the purchase of 4,939,806 common shares. Of this amount, options and awards for the purchase of 588,345 common shares are held by non-affiliates, who may sell these shares in the public markets from time to time, without limitations on the timing, amount or method of sale. If our share price rises, the holders may exercise their options and sell a large number of shares. This could cause the market price of our common shares to decline.
As compared to previous years, we are required to comply with the domestic reporting regime under the Securities Exchange Act of 1934, as amended, and will incur significant legal, accounting and other expenses and resources, and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices.
Prior to December 31, 2016, we were a foreign private issuer and therefore were not required to comply with all of the periodic disclosure and current reporting requirements of the Securities Exchange Act of 1934, as amended, applicable to U.S. domestic issuers. As we are no longer a foreign private issuer as of January 1, 2017, we are required to comply with all of the periodic disclosure and current reporting requirements of the Securities Exchange Act of 1934, as amended, applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. In addition, our officers, directors and principal shareholders are no longer exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act and are no longer exempt from the requirements of Regulation FD promulgated by the Securities and Exchange Commission under the Securities Exchange Act of 1934, as amended. We are also no longer permitted to follow our home country rules in lieu of the corporate governance obligations imposed by The NASDAQ Stock Market LLC, and may be required to comply with the governance practices required of U.S. domestic issuers. The regulatory and compliance costs associated with the reporting and governance requirements applicable to U.S. domestic issuers may be significantly higher than the costs we previously incurred as a foreign private issuer. As a result, we expect that the loss of foreign private issuer status will increase our legal and financial compliance costs and will make some activities highly time consuming and costly. In addition, we need to develop our reporting and compliance infrastructure and may face challenges in complying with the new requirements applicable to us.
There are inherent limitations in all control systems, and misstatements due to error or fraud may occur and not be detected.
The ongoing internal control provisions of Section 404 of the Sarbanes-Oxley Act require us to identify material weaknesses in internal control over financial reporting, which is a process to provide reasonable assurance regarding the reliability of financial reporting for external purposes in accordance with accounting principles generally accepted in the United States. Our management, including our chief executive officer and principal financial officer, does not expect that our internal controls and disclosure controls will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. In addition, the design of a control system must reflect the fact that there are resource constraints and the benefit of controls must be relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, in our company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple errors or mistakes. Further, controls can be circumvented by individual acts of some persons, by collusion of two or more persons, or by management override of the controls. The design of any system of controls is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving our stated goals under all potential future conditions. Over time, a control may be inadequate because of changes in conditions, such as growth of the company or increased transaction volume, or the degree of compliance with the policies or procedures may deteriorate. Because of inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
| 52 |
In addition, discovery and disclosure of a material weakness, by definition, could have a material adverse impact on our financial statements. Such an occurrence could discourage certain customers or suppliers from doing business with us, cause downgrades in our future debt ratings leading to higher borrowing costs and affect how our common share trades. This could, in turn, negatively affect our ability to access public debt or equity markets for capital.
We are an “emerging growth company” and may elect to comply with reduced public company reporting requirements, which could make our common shares less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act. For as long as we continue to be an “emerging growth company”, we may take advantage of exemptions from various reporting requirements that are applicable to other public reporting companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in our periodic reports. We could be an “emerging growth company” up until December 31, 2021, although circumstances could cause us to lose that status earlier if our annual revenues exceed $1.07 billion, if we issue more than $1.0 billion in non-convertible debt in any three-year period or if the market value of our common shares held by non-affiliates exceeds $700.0 million as of any June 30th, in which case we would no longer be an “emerging growth company” as of the following December 31st. We cannot predict if investors will find our securities less attractive because we may rely on these exemptions. If some investors find our securities less attractive as a result, there may be a less active trading market for our securities and the price of our securities may be more volatile.
Even after we no longer qualify as an “emerging growth company”, we may still qualify as a “smaller reporting company,” which would allow us to take advantage of many of the same exemptions from disclosure requirements including exemption from compliance with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We cannot predict if investors will find our common shares less attractive because we may rely on these exemptions. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and the price for our common shares may be more volatile.
U.S. civil liabilities may not be enforceable against us or certain of our officers.
We are governed by the Business Corporations Act (British Columbia) (“BCBCA”) and a substantial portion of our assets, including our manufacturing facility in Rehovot, Israel, and our research facility in Ottawa, Canada, are located outside the United States. As a result, it may be difficult for investors to effect service of process within the United States upon us or to enforce judgments obtained against us in U.S. courts, in any action, including actions predicated upon the civil liability provisions of U.S. federal securities laws or any other laws of the United States. Additionally, rights predicated solely upon civil liability provisions of U.S. federal securities laws or any other laws of the United States may not be enforceable in original actions, or actions to enforce judgments obtained in U.S. courts, brought in Canadian or Israeli courts. In addition, two of our officers reside outside of the United States, and all or a substantial portion of their assets may be located outside the United States, which may make effecting service of process within the United States or enforcing judgments obtained against such persons in U.S. courts difficult.
We are governed by the corporate laws of British Columbia which in some cases have a different effect on shareholders than the corporate laws of Delaware, United States.
We are governed by the BCBCA and other relevant laws, which may affect the rights of shareholders differently than those of a company governed by the laws of a U.S. jurisdiction, and may, together with our charter documents, including the advance notice provisions in our Articles for the nomination of directors, have the effect of delaying, deferring or discouraging another party from acquiring control of our company by means of a tender offer, a proxy contest or otherwise, or may affect the price an acquiring party would be willing to offer in such an instance. The material differences between the BCBCA and Delaware General Corporation Law, or DGCL, that may have the greatest such effect include, but are not limited to, the following: (i) for material corporate transactions (such as mergers and amalgamations, other extraordinary corporate transactions or amendments to our articles) the BCBCA generally requires a two-thirds majority vote by shareholders, whereas DGCL generally only requires a majority vote; and (ii) under the BCBCA a holder of 5% or more of our common shares can requisition a special meeting of shareholders, whereas such right does not exist under the DGCL.
| 53 |
We may be subject to securities litigation, which is expensive and could divert management attention.
In the past, companies that have experienced volatility in the market price of their securities have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Litigation of this type could result in substantial costs and diversion of management’s attention and resources, which could seriously hurt our business. Any adverse determination in litigation could also subject us to significant liabilities.
The concentration of the capital stock ownership with our insiders will likely limit the ability of other shareholders to influence corporate matters.
As of October 23, 2017, approximately 46.6% of our outstanding common shares was controlled by our officers, directors, beneficial owners of 10% or more of our securities and their respective affiliates. As a result, these shareholders, if they acted together, may be able to determine or influence matters that require approval by our shareholders, including the election of directors and approval of significant corporate transactions. Corporate actions might be taken even if other shareholders oppose them. This concentration of ownership might also have the effect of delaying or preventing a corporate transaction that other shareholders may view as beneficial.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, the price of our common shares and trading volume could decline.
The trading market for our common shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. Although we currently have research coverage by securities and industry analysts, you should not invest in our common shares in anticipation that we will increase such coverage. If one or more of the analysts who covers us at any given time downgrades our common shares or publishes inaccurate or unfavorable research about our business, the price of our common shares would likely decline. If one or more of these analysts ceases coverage of us or fails to publish reports on us regularly, demand for our common shares could decrease, which could cause the price of our common shares and trading volume to decline.
| 54 |
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
a) Sales of Unregistered Securities
There have been no unregistered sales of securities during the period covered by this Form 10-Q that have not been previously reported in a current report on Form 8-K.
c) Issuer Purchases of Equity Securities
None.
Item 3. Defaults Upon Senior Securities
Not applicable.
Item 4. Mine Safety Disclosure
Not applicable.
None.
See the Exhibit Index following the signature page to this Form 10-Q for a list of exhibits filed or furnished with this Form 10-Q, which Exhibit Index is incorporated herein by reference.
| 55 |
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| Date: November 13, 2017 | VBI VACCINES INC. | |
| By: | /s/ Jeff Baxter | |
Jeff Baxter President & Chief Executive Officer (Principal Executive Officer) | ||
| By: | /s/ Athena Kartsaklis | |
| Athena Kartsaklis | ||
Senior Vice President, Finance (Principal Financial Officer) | ||
| 56 |
EXHIBIT INDEX
* Filed herewith.
** Furnished herewith.
+ Indicates a management contract or compensatory plan.
| 57 |