Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10 - K
[X]
ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
ACT OF 1934
For the
fiscal year ended June 30, 2017
or
[
]
TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
ACT OF 1934
For the
transition period from ___________ to __________
Commission
file number: 001-15543
PALATIN TECHNOLOGIES, INC.
(Exact
name of registrant as specified in its charter)
|
Delaware
|
|
95-4078884
|
|
(State
or other jurisdiction of
incorporation
or organization)
|
|
(I.R.S.
Employer Identification No.)
|
|
|
|
|
|
4B Cedar Brook Drive
Cranbury, New Jersey
|
|
08512
|
|
(Address
of principal executive offices)
|
|
(Zip
Code)
|
(609) 495-2200
(Registrant’s
telephone number, including area code)
Securities
registered pursuant to Section 12(b) of the Act:
|
Title of Each Class
|
|
Name of Each Exchange
on Which Registered
|
|
Common
Stock, par value $.01 per share
|
|
NYSE
MKT
|
Securities
registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as
defined in Rule 405 of the Securities Act. Yes [ ] No
[X]
Indicate
by check mark if the registrant is not required to file reports
pursuant to Section 13 or Section 15(d) of the Act. Yes [ ] No
[X]
Indicate
by check mark whether the registrant (1) has filed all reports
required to be filed by Section 13 or 15(d) of the Securities
Exchange Act of 1934 during the preceding 12 months (or for such
shorter period that the registrant was required to file such
reports), and (2) has been subject to such filing requirements for
the past 90 days.
Yes [X]
No [ ]
Indicate
by check mark whether the registrant has submitted electronically
and posted on its corporate Web site, if any, every Interactive
Data File required to be submitted and posted pursuant to Rule 405
of Regulation S-T during the preceding 12 months (or for such
shorter period that the registrant was required to submit and post
such files). Yes [X] No [
]
Indicate
by check mark if disclosure of delinquent filers pursuant to Item
405 of Regulation S-K is not contained herein, and will not be
contained, to the best of the registrant’s knowledge, in
definitive proxy or information statements incorporated by
reference in Part III of this Form 10-K or any amendment to this
Form 10-K. [ ]
Indicate
by check mark whether the registrant is a large accelerated filer,
an accelerated filer, a non-accelerated filer, a smaller reporting
company, or an emerging growth company. See the definitions of
“large accelerated filer,” “accelerated
filer,” “smaller reporting company,” and
“emerging growth company” in Rule 12b-2 of the Exchange
Act. (Check one):
|
Large accelerated filer
|
☐
|
Accelerated filer
|
☐
|
|
Non-accelerated
filer
|
☐ (Do not check if a smaller reporting
company)
|
Smaller reporting company
|
☒
|
|
|
|
Emerging
growth company
|
☐
|
If an
emerging growth company, indicate by check mark if the registrant
has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided
pursuant to Section 13(a) of the Exchange
Act. ☐
Indicate
by check mark whether the registrant is a shell company (as defined
in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
State
the aggregate market value of the voting and non-voting common
equity held by non-affiliates, computed by reference to the price
at which the common equity was last sold, or the average bid and
asked price of such common equity, as of the last business day of
the registrant’s most recently completed second fiscal
quarter (December 31, 2016): $67,224,210.
Indicate
the number of shares outstanding of each of the registrant’s
classes of common stock, as of the latest practicable date
(September 21, 2017): 179,045,453.
PALATIN TECHNOLOGIES, INC.
Table of Contents
|
|
Page
|
|
|
PART
I
|
||
|
Item
1.
|
Business
|
1
|
|
Item
1A.
|
Risk
Factors
|
16
|
|
Item
1B.
|
Unresolved Staff
Comments
|
36
|
|
Item
2.
|
Properties
|
37
|
|
Item
3.
|
Legal
Proceedings
|
37
|
|
Item
4.
|
Mine
Safety Disclosures
|
37
|
|
PART
II
|
||
|
Item
5.
|
Market
for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities
|
38
|
|
Item
6.
|
Selected Financial
Data
|
38
|
|
Item
7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
38
|
|
Item
7A.
|
Quantitative and
Qualitative Disclosures About Market Risk
|
43
|
|
Item
8.
|
Financial
Statements and Supplementary Data
|
44
|
|
Item
9.
|
Changes
in and Disagreements with Accountants on Accounting and Financial
Disclosure
|
70
|
|
Item
9A.
|
Controls and
Procedures
|
70
|
|
Item
9B.
|
Other
Information
|
70
|
|
PART
III
|
||
|
Item
10.
|
Directors,
Executive Officers and Corporate Governance
|
71
|
|
Item
11.
|
Executive
Compensation
|
76
|
|
Item
12.
|
Security Ownership
of Certain Beneficial Owners and Management and Related Stockholder
Matters
|
82
|
|
Item
13.
|
Certain
Relationships and Related Transactions, and Director
Independence
|
86
|
|
Item
14.
|
Principal
Accountant Fees and Services
|
86
|
|
PART
IV
|
||
|
Item
15.
|
Exhibits, Financial
Statement Schedules
|
87
|
|
Item
16.
|
Form
10-K Summary
|
88
|
Forward-Looking Statements
Statements
in this Annual Report on Form 10-K (this “Annual
Report”), as well as oral statements that may be made by us
or by our officers, directors, or employees acting on our behalf,
that are not historical facts constitute “forward-looking
statements,” which are made pursuant to the safe harbor
provisions of Section 21E of the Securities Exchange Act of 1934
(the “Exchange Act”). The forward-looking statements in
this Annual Report do not constitute guarantees of future
performance. Investors are cautioned that statements that are not
strictly historical facts contained in this Annual Report,
including, without limitation, those relating to our current or
future financial performance, management’s plans and
objectives for future operations, ability to raise capital or repay
debt, if required, clinical trials and results, uncertainties
associated with product research and development, product plans and
performance, management’s assessment of market factors, as
well as statements regarding our strategy and plans and those of
our strategic partners, constitute forward-looking statements. In
some cases, you can identify forward-looking statements by
terminology such as “believe,” will,”
“may,” “estimate,” “continue,”
“anticipated,” “intend,”
“should,” “plan,” “expect,”
“predict,” “could,”
“potentially,” or the negative of these terms or other
similar expressions. Such forward-looking statements involve
substantial risks, uncertainties and other factors that could cause
our actual results to be materially different from our historical
results or from any results expressed or implied by such
forward-looking statements. Our future operating results are
subject to risks and uncertainties and are dependent upon many
factors, including, without limitation, the risks identified under
the caption “Risk Factors” and elsewhere in this Annual
Report, and any of those made in our other reports filed with the
U.S. Securities and Exchange Commission (the “SEC”).
Except as required by law, we do not intend, and undertake no
obligation, to publicly update forward-looking statements to
reflect events or circumstances after the date of this document or
to reflect the occurrence of unanticipated events.
In this
Annual Report, references to “we,” “our,”
“us,” the “Company” or
“Palatin” means Palatin Technologies, Inc. and its
subsidiary.
PART I
Item 1. Business.
Overview
We are
a biopharmaceutical company developing targeted, receptor-specific
peptide therapeutics for the treatment of diseases with significant
unmet medical need and commercial potential. Our programs are based
on molecules that modulate the activity of the melanocortin and
natriuretic peptide receptor systems. Our lead product in clinical
development is bremelanotide for the treatment of premenopausal
women with hypoactive sexual desire disorder (“HSDD”),
which is a type of female sexual dysfunction (“FSD”),
defined as low desire with associated distress. In addition, we
have drug candidates and development programs for cardiovascular
diseases and inflammatory diseases.
The
following drug development programs are actively under
development:
●
Bremelanotide, an
as-needed subcutaneous injectable product for the treatment of HSDD
in premenopausal women. Bremelanotide is a synthetic peptide analog
of the naturally occurring hormone alpha-MSH
(melanocyte-stimulating hormone). In two pivotal Phase 3 clinical
studies of bremelanotide for HSDD in premenopausal women,
bremelanotide met the pre-specified co-primary efficacy endpoints
of improvement in desire and decrease in distress associated with
low sexual desire as measured using validated patient-reported
outcome instruments. We have licensed North American rights to
bremelanotide to AMAG Pharmaceuticals, Inc. (“AMAG”),
and rights in China, Taiwan, Hong Kong and Macau to Shanghai Fosun
Pharmaceutical Industrial Development Co., Ltd.
(“Fosun”);
●
Melanocortin
peptide system program, focused on development of treatments for a
variety of inflammatory disease indications. PL-8177 is a selective
melanocortin receptor 1 (“MC1r”) agonist peptide we
have designated as our lead clinical development candidate for
inflammatory bowel diseases. We are scheduled to file an IND
application this year, and may thereafter initiate a Phase 1
clinical safety study. A dual melanocortin receptor 1 and 5 peptide
we developed, PL-8331, is a preclinical development candidate for
treating ocular inflammation. We anticipate completing preclinical
IND enabling activities on PL-8331 this calendar year;
and
●
Natriuretic peptide
system program, including PL3994, a natriuretic peptide
receptor-A (“NPR-A”) agonist, for treatment of
cardiovascular indications. PL3994, a synthetic mimetic of
the neuropeptide hormone atrial natriuretic peptide
(“ANP”), is in development for treatment of heart
failure, and is scheduled to start Phase 2A clinical trials later
this calendar year. A dual natriuretic peptide receptor A and C
agonist we developed, PL-5028, is in preclinical development for
cardiovascular diseases, including reducing cardiac hypertrophy and
fibrosis. We may file an Investigational New Drug
(“IND”), application in the first half of calendar year
2018, and thereafter initiate a Phase 1 clinical safety
study.
The
following chart illustrates the status of our drug development
programs.
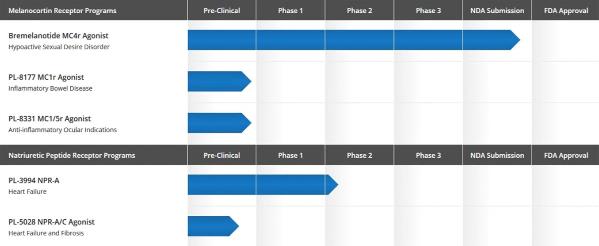
1
Our Strategy
Key
elements of our business strategy include:
●
Using our
technology and expertise to develop and commercialize products in
our active drug development programs;
●
Entering into
strategic alliances and partnerships with pharmaceutical companies
to facilitate the development, manufacture, marketing, sale and
distribution of our product candidates;
●
Partially funding
our product development programs with the cash flow generated from
existing license agreements, as well as any potential future
research, collaboration or license agreements with third parties;
and
●
Completing
development and seeking regulatory approval of certain of our
product candidates.
Our Melanocortin Receptor-Specific Programs
The
melanocortin system is involved in a large and diverse number of
physiologic functions. Therapeutic agents modulating this system
may have the potential to treat a variety of conditions and
diseases, including sexual dysfunction, obesity and related
disorders, pigmentation disorders and inflammation-related
diseases.
Bremelanotide for HSDD. We are developing subcutaneously
administered bremelanotide for the treatment of HSDD in
premenopausal women. HSDD is characterized by both a decrease in
sexual desire and significant personal distress or interpersonal
difficulty as a result of the lack of desire. Bremelanotide is a
melanocortin agonist with a mechanism of action which we believe
involves activation of endogenous neuronal pathways in the brain
regulating sexual arousal and desire responses.
We
completed last patient visits in the efficacy parts of our two
pivotal Phase 3 clinical studies of bremelanotide for the treatment
of HSDD in premenopausal women in the third quarter of calendar
year 2016. We announced topline efficacy results in the fourth
quarter of calendar year 2016. The open-label safety extension
portions of our pivotal Phase 3 clinical studies were completed in
the second quarter of calendar year 2017.
Our
Phase 3 clinical study program consisted of two randomized,
double-blinded, placebo-controlled Phase 3 studies, Studies 301 and
302, comparing the efficacy and safety of bremelanotide versus
placebo in premenopausal women diagnosed with HSDD. The primary
efficacy analysis population was the modified intent-to-treat
patient population, consisting of 1,202 women with HSDD in the
United States and Canada. Patients self-administered
either 1.75 mg of bremelanotide or placebo as needed in
anticipation of sexual activity. The efficacy portion of each study
consisted of a 24-week treatment evaluation period.
Based
on discussions with the U.S. Food and Drug Administration
(“FDA”), it was decided that the co-primary endpoints
for the Phase 3 clinical trials were the Female Sexual Function
Index: Desire Domain (“FSFI-D”) and Female Sexual
Distress Scale-Desires/Arousal/Orgasm (“FSDS-DAO”) Item
13. The FSFI-D is a validated patient reported outcome measurement
tool of sexual desire in the context of overall sexual function.
The FSDS-DAO Item 13 is a validated patient reported outcome
measurement tool of distress related to sexual dysfunction,
measuring personal distress associated with low sexual desire. Both
Phase 3 Studies 301 and 302 with bremelanotide for HSDD in
premenopausal women met the pre-specified co-primary efficacy
endpoints.
The
FSFI-D showed a statistically significant increase for
bremelanotide compared to placebo in both trials in the modified
intent-to-treat patient population:
Study
301: Mean change of 0.54 vs. 0.24, median change of 0.60 vs. 0.00,
p=0.0002; and,
Study
302: Mean change of 0.63 vs. 0.21, median change of 0.60 vs. 0.00,
p<0.0001.
The
FSDS-DAO Item 13 showed a statistically significant reduction in
distress related to low sexual desire for bremelanotide compared to
placebo in both trials in the modified intent-to-treat patient
population:
Study
301: Mean change of -0.73 vs. -0.36, median change of -1.0 vs. 0.0,
p<0.0001; and,
Study
302: Mean change of -0.71 vs. -0.42, median change of -1.0 vs. 0.0,
p=0.0053.
The
changes seen in both co-primary endpoints were clinically
significant. An independent committee evaluated the clinical
significance of co-primary endpoint study results using multiple
assessments of patient benefit, and was based on discussions with
the FDA and FDA guidance documents.
In the
safety population (1,247 patients), bremelanotide appeared to be
well tolerated. The most frequent adverse event was nausea, which
was generally mild in nature. The safety profile of bremelanotide
was consistent with prior clinical experience.
2
In the
Phase 3 clinical study program patients self-administered
bremelanotide with a single-use autoinjector pen. The bremelanotide
single-use autoinjector pen, intended to be the commercial drug
product, does not have a visible needle, is stored at room
temperature and is easy to use. Women administer bremelanotide by
pressing the autoinjector pen collar against either their thigh or
abdomen, and the autoinjector pen automatically introduces the
needle, administers the dose of bremelanotide under the skin and
audibly signals when the drug had been delivered and the needle has
been retracted.
Ongoing Studies and New Drug Application. We are conducting
multiple pharmacokinetic and safety pharmacology studies, including
an abuse-liability study and drug-to-drug interaction studies, as
well as certain chemistry, manufacturing and controls activities,
including a drug product process validation study. We anticipate
that the required human clinical studies will be completed this
calendar year. We currently expect that we will, with AMAG, our
North American licensee of bremelanotide, submit a New Drug
Application (“NDA”) to FDA for bremelanotide for the
treatment of HSDD in early calendar year 2018 following completion
of ongoing studies. We cannot assure you that a complete review of
the Phase 3 efficacy data and the pharmacokinetic and safety
pharmacology studies will support approval of bremelanotide for
HSDD or that the FDA will approve an NDA for
bremelanotide.
Medical Need — HSDD. HSDD, either with or without
arousal difficulties, is the largest single category of FSD. FSD is
a multifactorial condition that has anatomical, physiological,
medical, psychological and social components, and is defined as
persistent or recurring problems during one or more of the stages
of sexual response with associated distress. HSDD has a significant
impact on a patient’s self-image, relationships and general
well-being. The 2006 PRESIDE (Prevalence of Female Sexual Problems
Associated with Distress and Determinants of Treatment Seeking)
study, a cross-sectional, population-based survey of 31,581 female
adult respondents in the United States published in 2008 in the
journal Obstetrics &
Gynecology, found that approximately 22% of women reported a
sexual problem and 11% were women with HSDD. Based on the number of
premenopausal women in the United States according to the U.S.
Census, the presenting market size of premenopausal women with
primary HSDD is at least 5.8 million women.
Subcutaneous Bremelanotide. Bremelanotide, which is believed
to act through activation of melanocortin receptors in the central
nervous system, is a first-in-class pharmaceutical agent in
development as a treatment of HSDD. Bremelanotide is intended for
as needed use and is self-administered by the patient, using a
simple and patient-friendly single-use autoinjector pen, thirty
minutes to one hour prior to anticipated sexual
activity.
Partnering. In January 2017, we entered into a license
agreement with AMAG, pursuant to which we granted AMAG an exclusive
license in all countries of North America, with the right to grant
sublicenses, to research, develop and commercialize products
containing bremelanotide. AMAG also has a non-exclusive license,
with the right to grant sublicenses, to manufacture products
containing bremelanotide in North America, and to research, develop
and manufacture, but not commercialize, products containing
bremelanotide in countries outside North America. Upon the license
agreement becoming effective on February 2, 2017, AMAG paid us $60
million as a one-time initial payment, and is required to pay us up
to $25 million to reimburse us for direct out-of-pocket expenses
incurred in development and regulatory activities necessary to file
an NDA. In addition, we may receive up to $80 million in specified
regulatory payments upon achievement of certain regulatory
milestones, and up to $300 million in sales milestone payments
based on achievement of certain annual net sales amounts of
products containing bremelanotide. AMAG will also pay tiered
royalties on annual net sales of products containing bremelanotide
at rates ranging from the high single-digits to the low
double-digits.
In
early September 2017, we entered into a license agreement with
Fosun for exclusive rights to commercialize bremelanotide in the
territories of mainland China, Taiwan, Hong Kong S.A.R. and Macau
S.A.R. We will receive an upfront payment of $5.0 million and, when
regulatory approval for a bremelanotide product is obtained in
China, a $7.5 million milestone payment. We may receive up to $92.5
million in sales related milestones, and will receive high
single-digit to low double-digit royalties on net sales in the
licensed territories.
We
retain worldwide rights for bremelanotide for FSD, HSDD and all
other indications outside North America and the territories
licensed to Fosun. We are in active discussions with potential
partners for marketing and commercialization rights for
bremelanotide in other jurisdictions, including Europe. We may not
be able to enter into suitable agreements with potential partners
on acceptable terms, if at all.
Prior Clinical Trials. We have completed several Phase 1
clinical studies in which various safety parameters, including
blood pressure effects of subcutaneously administered
bremelanotide, were studied. Based in part on these studies, our
Phase 2B clinical trial assessed the magnitude and duration of
blood pressure effect, and determined that subcutaneous
administration of selected doses of bremelanotide for treatment of
HSDD in premenopausal women provides acceptable control of blood
pressure effects.
3
MC1r Peptide Agonists. We have conducted preclinical animal
studies with MC1r peptide drug candidates for a number of
inflammatory disease and autoimmune indications. The MC1r is
upregulated in a number of diseases, including inflammatory bowel
disease, nephritis, which is inflammation of the kidneys, and
rheumatoid arthritis, and in ocular indications such as uveitis and
dry eye. We believe that MC1r peptides have broad anti-inflammatory
effects and appear to utilize mechanisms engaged by the endogenous
melanocortin system in regulation of the immune system and
resolution of pro-inflammatory responses.
Our
MC1r peptide drug candidates are highly specific, with
substantially greater binding and activity at MC1r than at other
melanocortin receptors. In vitro safety studies have shown that our
MC1r peptide drug candidates have no activity in a wide range of
receptors, ion channels and kinases. We have selected one of our
MC1r peptide drug candidates, designated PL-8177, as a clinical
trial candidate. PL-8177 is a selective MC1r agonist peptide we
have designated as our lead clinical development candidate for
inflammatory bowel diseases. We have completed preclinical
toxicology testing on PL-8177 and chemistry, controls and
manufacturing activities to support Phase 1 studies, and anticipate
filing an IND application on PL-8177 this calendar year, and may
thereafter to initiate Phase 1 clinical safety
studies.
We are
also developing a peptide which is a dual melanocortin receptor 1
and 5 agonist, PL-8331, which is a preclinical development
candidate for treating ocular inflammatory diseases. We anticipate
completing preclinical IND enabling activities with PL-8331 by the
first half of calendar year 2018.
Next Generation Melanocortin Receptor 4 (“MC4r”)
Peptide and Small Molecule Agonists. We have developed a
series of highly selective MC4r peptides and orally active small
molecules. In developing these compounds, we examined effectiveness
in animal models of sexual response, obesity and related metabolic
signals, and also determined cardiovascular effects, primarily
looking at changes in blood pressure. Results of these studies
suggest that certain of these compounds may have significant
medical and commercial potential for treatment of conditions
responsive to MC4r activation, including HSDD, FSD, ED, obesity and
diabetes. We are seeking collaboration and development partners for
these compounds for obesity and related clinical indications, but
may not be able to enter into suitable agreements on acceptable
terms with potential partners, if at all.
Our Natriuretic Peptide Receptor-Specific Programs
The
natriuretic peptide receptor system has numerous cardiovascular
functions, and therapeutic agents modulating this system may be
useful in treatment of heart failure, acute asthma, other pulmonary
diseases and hypertension. While the therapeutic potential of
modulating this system is well appreciated, development of
therapeutic agents has been difficult due, in part, to the short
biological half-life of native peptide agonists.
We have
designed and are developing potential candidate drugs that are
selective for different natriuretic peptide receptors, including
NPR-A, natriuretic peptide receptor B (“NPR-B”),
natriuretic peptide receptor C, and both NPR-A and
NPR-B.
PL-3994. PL-3994 is our lead natriuretic peptide receptor
product candidate, and is a synthetic mimetic of the neuropeptide
hormone ANP and an NPR-A agonist. PL-3994 is in development for
treatment of heart failure (with preserved or reduced ejection
fraction) and may be suitable for replacement therapy in patients
with prohormone processing deficiencies. PL-3994 activates NPR-A, a
receptor known to play a role in cardiovascular homeostasis.
Consistent with being an NPR-A agonist, PL-3994 increases plasma
cyclic guanosine monophosphate (“cGMP”), levels, a
pharmacological response consistent with the effects of endogenous
(naturally produced) natriuretic peptides on cardiovascular
function and smooth muscle relaxation. PL-3994 also decreases
activity of the renin-angiotensin-aldosterone system
(“RAAS”), a hormone system that regulates blood
pressure and fluid balance. The RAAS system is frequently
over-activated in heart failure patients, leading to worsening of
cardiovascular function.
PL-3994,
our lead product development candidate which is ready for Phase 2
safety and efficacy studies, is one of a number of natriuretic
peptide receptor agonist compounds we have developed. In
conjunction with clinicians at a major research institution,
PL-3994 is scheduled to enter Phase 2A clinical trials later in
calendar year 2017. PL-3994 is a synthetic molecule incorporating a
novel and proprietary amino acid mimetic structure, and has an
extended circulation half-life and metabolic stability compared to
endogenous ANP. Based on the half-life and pharmacokinetics, we
believe that PL-3994 is amenable to once daily chronic use
subcutaneous administration.
Prior Clinical Studies with PL-3994. Human clinical studies
of PL-3994 commenced with a Phase 1 trial, which concluded in 2008.
This was a randomized, double-blind, placebo-controlled study in 26
healthy volunteers who received either PL-3994 or a placebo
subcutaneously. Dosing concluded with the successful achievement of
the primary endpoint of the study, a pre-specified reduction in
systemic blood pressure. No volunteer experienced a serious or
severe adverse event. Elevations in plasma cGMP levels, increased
diuresis and increased natriuresis were all observed for several
hours after single subcutaneous doses. Later in 2008, we conducted
a trial in volunteers with controlled hypertension who were
receiving one or more conventional antihypertensive medications. No
volunteer experienced a serious or severe adverse event. Elevations
in plasma cGMP levels were observed for several hours after single
subcutaneous doses. Based on the studies to date, PL-3994 is ready
for Phase 2 safety and efficacy studies.
4
PL-5028. We are in preclinical development with PL-5028, a
dual natriuretic peptide receptor A and C agonist we developed, for
cardiovascular disease indications, including reducing cardiac
hypertrophy and fibrosis. We may file an IND application in the
first half of calendar year 2018, and thereafter initiate a Phase 1
clinical safety study.
Administration of PL-3994 and PL-5028. For heart failure and
other cardiovascular disease indications we believe that
subcutaneous administration may be employed. In studies to date,
PL-3994 is well absorbed through the subcutaneous route of
administration. In human studies with PL-3994, the pharmacokinetic
and pharmacodynamic half-lives were on the order of hours,
significantly longer than the comparable half-lives of endogenous
natriuretic peptides. We believe that subcutaneous PL-3994 or
PL-5028, if successful, will be appropriate for self-administration
by patients, similar to insulin and other self-administered
drugs.
Heart Failure. Heart failure is an illness in which the
heart is unable to pump blood efficiently, and includes acutely
decompensated heart failure with dyspnea (shortness of breath) at
rest or with minimal activity. Endogenous natriuretic peptides have
a number of beneficial effects, including vasodilation (relaxation
of blood vessels), natriuresis (excretion of sodium) and diuresis
(excretion of fluids).
Patients
who have been admitted to the hospital with an episode of worsening
heart failure have an increased risk of either death or hospital
readmission in the three months following discharge. Up to 15% of
patients die in this period and as many as 30% need to be
readmitted to the hospital. We believe that decreasing mortality
and hospital readmission in patients discharged following
hospitalization for worsening heart failure is a large unmet
medical need for which PL-3994 may be effective. PL-3994 could
potentially be utilized as an adjunct to existing heart failure
medications, and may, if successfully developed, be
self-administered by patients as a subcutaneous injection following
hospital discharge. We believe that our natriuretic peptide
products under development may, if successful, reduce cardiac
hypertrophy (increase in heart size due to disease), which is an
independent risk factor for cardiovascular morbidity and
mortality.
According
to a report from the American Heart Association published in 2014
in the journal Circulation,
an estimated 5.7 million Americans suffer from heart failure, with
870,000 new cases of heart failure diagnosed each year, with
disease incidence expected to increase with the aging of the
American population. Heart failure has tremendous human and
financial costs. The same report estimated that the 2012 total
costs in the United States for heart failure were $30.7 billion,
with heart failure constituting the leading cause of
hospitalization in people over 65 years of age and with over 1
million hospital discharges for heart failure in 2010. Heart
failure is a high mortality disease, with approximately one-half of
heart failure patients dying within five years of initial
diagnosis.
Patient
populations have been identified which have reduced levels of
endogenous active natriuretic peptides, including endogenous active
ANP. The reduced levels have a variety of causes, including
mutations in endogenous natriuretic peptides and in enzymes
necessary to convert natriuretic peptide sequences to their active
form. Patients with reduced levels of endogenous active natriuretic
peptides are reported to have a poor response to current drug
therapies and to have increased rates of cardiac remodeling and
cardiac events.
We
believe that PL-3994 has the potential to treat heart failure with
preserved ejection fraction (“HFpEF”), which is a high
unmet medical need with no approved treatment options, heart
failure with reduced ejection fraction (“HFrEF”), and
patients with reduced levels of endogenous active natriuretic
peptides, such as corin deficiencies, which is a high unmet medical
need in patients with a poor response to current therapies, with
the objective to restore normal natriuretic peptide
function.
Technologies We Use
We used
a rational drug design approach to discover and develop proprietary
peptide, peptide mimetic and small molecule agonist compounds,
focusing on melanocortin and natriuretic peptide receptor systems.
Computer-aided drug design models of receptors are optimized based
on experimental results obtained with peptides and small molecules
that we develop. With our approach, we believe we are developing an
advanced understanding of the factors which drive
agonism.
We have
developed a series of proprietary technologies used in our drug
development programs. One technology employs novel amino acid
mimetics in place of selected amino acids. These mimetics provide
the receptor-binding functions of conventional amino acids while
providing structural, functional and physiochemical advantages. The
amino acid mimetic technology is employed in PL-3994, our compound
in development for treatment of heart failure.
Some
compound series have been derived using our proprietary and
patented platform technology, called MIDAS™, or Metal Ion-induced Distinctive Array of Structures. This technology employs
metal ions to fix the three-dimensional configuration of peptides,
forming conformationally rigid molecules that remain folded
specifically in their active state. These MIDAS molecules are
generally simple to synthesize, are chemically and proteolytically
stable, and have the potential to be orally bioavailable. In
addition, MIDAS molecules are information-rich and provide data on
structure-activity relationships that may be used to design small
molecule, non-peptide drugs.
5
Amount Spent on Research and Development Activities
Research
and development expenses were approximately $45.7 million for the
fiscal year ended June 30, 2017 (“ fiscal 2017”), $43.1
million for the fiscal year ended June 30, 2016 (“fiscal
2016”), and $24.6 million for the fiscal year ended June 30,
2015 (“fiscal 2015”).
Competition
General. Our products under development will compete on the
basis of quality, performance, cost effectiveness and application
suitability with numerous established products and technologies. We
have many competitors, including pharmaceutical, biopharmaceutical
and biotechnology companies. Furthermore, there are several
well-established products in our target markets that we will have
to compete against. Products using new technologies which may be
competitive with our proposed products may also be introduced by
others. Most of the companies selling or developing competitive
products have financial, technological, manufacturing and
distribution resources significantly greater than ours and may
represent significant competition for us. In addition, if any of
our product candidates are approved by FDA, they will eventually
face competition from generic versions that will sell at
significantly reduced prices, be preferred by managed care and
health insurance payers, and be eligible for automatic pharmacy
substitution even when a prescriber writes a prescription for our
product. The timing and extent of future generic competition is
dependent upon both our intellectual property rights and the FDA
regulatory process, but cannot be accurately
predicted.
The
pharmaceutical and biotechnology industries are characterized by
extensive research efforts and rapid technological change. Many
biopharmaceutical companies have developed or are working to
develop products similar to ours or that address the same markets.
Such companies may succeed in developing technologies and products
that are more effective or less costly than any of those that we
may develop. Such companies may be more successful than us in
developing, manufacturing and marketing products.
We
cannot guarantee that we will be able to compete successfully in
the future or that developments by others will not render our
proposed products under development or any future product
candidates obsolete or noncompetitive or that our collaborators or
customers will not choose to use competing technologies or
products.
Bremelanotide for Treatment of HSDD. There is competition
and financial incentive to develop, market and sell drugs for the
treatment of HSDD and other forms of FSD. Flibanserin, sold under
the trade name Addyi®, is the only drug currently approved in
the United States for treatment of HSDD. Flibanserin, a
non-hormonal oral serotonin 5-HT1A agonist, 5-HT2A antagonist,
which requires chronic dosing, was approved by the FDA on August
18, 2015 for treatment of premenopausal women with HSDD. The FDA
approval included a risk evaluation and mitigation strategy
(“REMS”) because of the increased risk of severe
hypotension and syncope due to the interaction between flibanserin
and alcohol, and a Boxed Warning to highlight the risks of severe
hypotension and syncope in patients who drink alcohol during
treatment with flibanserin, in those who also use moderate or
strong CYP3A4 inhibitors, and in those who have liver impairment.
We are aware of several other drugs at various stages of
development, most of which are taken on a chronic, typically
once-daily, basis. There are other companies reported to be
developing new drugs for FSD indications, some of which may be in
clinical trials in the United States or elsewhere. We are not aware
of any company actively developing a melanocortin receptor agonist
drug for HSDD.
PL-3994 and PL-5028 for Heart Failure Indications.
Nesiritide (sold under the trade name Natrecor®), a
recombinant human B-type natriuretic peptide drug, is marketed in
the United States by Scios Inc., a Johnson & Johnson company.
Nesiritide is approved for treatment of acutely decompensated
congestive heart failure patients who have dyspnea at rest or with
minimal activity. Other peptide drugs, including carperitide, a
recombinant human ANP drug, and ularitide, a synthetic form of
urodilatin, a naturally occurring human natriuretic peptide related
to ANP, have been investigated for treatment of congestive heart
failure, but we are not aware of any active development in the
United States. We are aware of other companies developing
intravenously administered natriuretic peptide drugs, with at least
one reported to have completed Phase 2 clinical trials for acute
heart failure. A combination drug comprised of sacubitril and
valsartan developed by Novartis AG, sold under the trade name
Entresto®, inhibits both the angiotensin II receptor and
neprilysin (an enzyme which inactivates endogenous active
natriuretic peptides). This combination drug, which was approved by
the FDA in July 2015, results in increases of endogenous active ANP
levels, and thus has a mechanism of action with similarities to
PL-3994 and PL-5028. In a Phase 3 trial, the combination drug was
compared to an angiotensin-converting-enzyme inhibitor, enalapril,
in heart failure patients with reduced ejection fraction. It
significantly improved the rate of death from cardiovascular
causes, significantly reduced hospitalization for heart failure and
significantly improved heart failure symptoms. This combination
drug demonstrated that upregulation of the natriuretic peptide
system in combination with angiotensin-converting-enzyme inhibition
is superior to angiotensin-converting-enzyme inhibition alone, and
thus provides validation of the natriuretic peptide system as a
target for improving outcomes in treating heart failure patients.
In addition, there are a number of approved drugs and drugs in
development for treatment of heart failure through mechanisms or
pathways other than agonism of NPR-A.
6
MC1r Peptides for Inflammatory Disease-Related Indications.
Many inflammatory disease-related indications are treated using
systemic steroids or other immunosuppressant drugs, all of which
have side effects which can be dose limiting. There are a large
number of approved biological drugs and biological drugs under
development for treatment of inflammatory disease-related
indications. For inflammatory bowel diseases, FDA-approved drugs
include mesalazine and immunosuppressive drugs such as prednisone
and other steroids, tumor necrosis factor inhibitors such as
infliximab and adalimumab, and immune system suppressants such as
azathioprine, mercaptopurine and methotrexate.
Obesity. There are a number of FDA-approved drugs and
medical devices for the treatment of obesity, and a large number of
products in clinical development by other companies, including
products which target melanocortin receptors. At least one Phase 2
study has been reported on use of an MC4r agonist for obesity
indications.
Patents and Proprietary Information
Patent Protection. Our success will depend in substantial
part on our ability to obtain, defend and enforce patents, maintain
trade secrets and operate without infringing upon the proprietary
rights of others, both in the United States and abroad. We own a
number of issued United States patents and have pending United
States patent applications, many with issued or pending counterpart
patents in selected foreign countries. We seek patent protection
for our technologies and products in the United States and those
foreign countries where we believe patent protection is
commercially important.
We own
two issued United States patents claiming the bremelanotide
substance and an issued patent claiming the bremelanotide substance
in each of Australia, Austria, Belgium, Brazil, Canada, Cyprus,
Denmark, Finland, France, Germany, Greece, Hong Kong, Ireland,
Italy, Japan, Korea, Luxembourg, Mexico, Monaco, Netherlands, New
Zealand, Portugal, Spain, Sweden, Switzerland, and the United
Kingdom. The issued United States patents have a term until 2020,
which term may be subject to extension for a maximum period of up
to five years as compensation for patent term lost during drug
development and the FDA regulatory review process, pursuant to the
Drug Price Competition and Patent Term Restoration Act of 1984, or
the Hatch-Waxman Amendments. Whether we will be able to obtain
patent term extensions under the Hatch-Waxman Amendments and the
length of any such extension cannot be determined until the FDA
approves for marketing, if ever, a product in which bremelanotide
is the active ingredient. In addition, the claims of issued patents
covering bremelanotide may not provide meaningful protection.
Further, third parties may challenge the validity or scope of any
issued patent, and under the Hatch-Waxman Amendments, potentially
receive approval of a competing generic version of our product or
products even before a court rules on the validity or infringement
of our patents.
We own
two issued United States patents and pending patent applications in
the United States for methods of treating FSD with bremelanotide,
and related patent applications are pending in Australia, Brazil,
Canada, China, Georgia, Hong Kong, India, Indonesia, Israel, Japan,
Korea, Malaysia, Mexico, New Zealand, Philippines, South Africa,
Ukraine, Vietnam and before the European and Eurasian patent
offices. The issued United States patent has a term until 2033.
Whether we will be able to obtain a patent term extension in the
United States under the Hatch-Waxman Amendments, assuming that a
relevant patent issues in the United States, and the length of any
such extension, cannot be determined until the FDA approves for
marketing, if ever, a product utilizing bremelanotide by methods
claimed in the patent. Issued patents and pending applications in
the United States and elsewhere in the world have a presumptive
term, if a patent is issued, until 2033.
We have
patents and patent applications on an alternative class of
melanocortin receptor-specific peptides for treatment of sexual
dysfunction and other indications, including obesity, consisting of
two issued patents in the United States, an issued patent in each
of Australia, Canada, China, France, Germany, Ireland, Israel,
Japan, Korea, Mexico, New Zealand, Russia, Switzerland and the
United Kingdom, and pending patent applications on the same class
in Brazil, India, and South Africa. The presumptive term of the
issued patents and pending patent applications is until 2029. We
also have patents and pending patent applications for a second
class of alternative melanocortin receptor-specific peptides for
treatment of sexual dysfunction and other indications, including
obesity, consisting of issued patents in the United States,
Australia, China, Japan, Israel, Korea, New Zealand, Russia, and
South Africa and pending patent applications on the same class in
Brazil, Canada, China, India, Mexico, and before the European
patent office. The presumptive term of the issued patents and
pending patent applications is until 2030. Until one or more
product candidates covered by a claim of one of these patents and
patent applications are developed for commercialization, which may
never occur, we cannot evaluate the duration of any potential
patent term extension under the Hatch-Waxman
Amendments.
We own
issued patents in the United States, Mexico, New Zealand, South
Africa and Russia claiming highly selective MC1r agonist peptides
for treatment of inflammation-related diseases and disorders and
related indications, and pending patent applications on two broad
classes of highly selective MC1r agonist peptides in the United
States, Australia, Brazil, Canada, China, India, Israel, Japan,
Korea, and Mexico and before the European patent office. The
presumptive term of the issued patents and pending patent
applications is until 2030. Until one or more product candidates
covered by a claim of one of these patent applications are
developed for commercialization, which may never occur, we cannot
evaluate the duration of any potential patent term extension under
the Hatch-Waxman Amendments.
7
We own
two issued United States patents claiming the PL-3994 substance and
other natriuretic peptide receptor agonist compounds that we have
developed and an issued United States patent claiming a precursor
molecule to the PL-3994 substance, both of which expire in 2027.
Corresponding patents on the PL-3994 substance and other
natriuretic peptide receptor agonist compounds were issued in
Australia, Austria, Belgium, China, Colombia, Denmark, Finland,
France, Germany, Hong Kong, Hungary, India, Ireland, Israel, Italy,
Japan, Korea, Mexico, Netherlands, Philippines, Russia, South
Africa, Spain, Sweden, Switzerland, and the United Kingdom, with
terms until 2027. Patent applications on the PL-3994 substance and
other natriuretic peptide receptor agonist compounds are pending in
Brazil and Canada, with presumptive terms until 2027. Applications
claiming precursor molecules for the PL-3994 substance and other
compounds have issued in the United States, Australia, China,
France, Germany, Hong Kong, India, Ireland, Israel, Japan, Mexico,
Netherlands, Philippines, Korea, South Africa, Sweden, Switzerland
and the United Kingdom, and expire in 2027. Patent applications on
the precursor molecules are pending in Brazil, Canada, and before
the Eurasian Patent Office, with presumptive terms until 2027. We
also own an issued United States patent claiming use of the PL-3994
substance for treatment of acute asthma and chronic obstructive
pulmonary disease, which expires in 2031. We do not know the full
scope of patent coverage we will obtain, or whether any patents
will issue other than the patents already issued. Until one or more
product candidates covered by a claim of the issued patents or one
of these patent applications are developed for commercialization,
which may never occur, we cannot evaluate the duration of any
potential patent term extension under the Hatch-Waxman
Amendments.
We
additionally have 31 issued United States patents on melanocortin
receptor specific peptides and small molecules, and five issued
United States patents on natriuretic peptide receptor agonist
compounds, but we are not actively developing any product candidate
covered by a claim of any of these patents.
In the
event that a third party has also filed a patent application
relating to an invention we claimed in a patent application, we may
be required to participate in an interference proceeding
adjudicated by the United States Patent and Trademark Office
(“USPTO”) to determine priority of invention. The
possibility of an interference proceeding could result in
substantial uncertainties and cost, even if the eventual outcome is
favorable to us. An adverse outcome could result in the loss of
patent protection for the subject of the interference, subjecting
us to significant liabilities to third parties, the need to obtain
licenses from third parties at undetermined cost, or requiring us
to cease using the technology.
Future Patent Infringement. We do not know for certain that
our commercial activities will not infringe upon patents or patent
applications of third parties, some of which may not even have been
issued. Although we are not aware of any valid United States
patents which are infringed by bremelanotide or PL-3994, we cannot
exclude the possibility that such patents might exist or arise in
the future. We may be unable to avoid infringement of any such
patents and may have to seek a license, defend an infringement
action, or challenge the validity of such patents in court. Patent
litigation is costly and time consuming. If such patents are valid
and we do not obtain a license under any such patents, or we are
found liable for infringement, we may be liable for significant
monetary damages, may encounter significant delays in bringing
products to market, or may be precluded from participating in the
manufacture, use or sale of products or methods of treatment
covered by such patents.
Proprietary Information. We rely on proprietary information,
such as trade secrets and know-how, which is not patented. We have
taken steps to protect our unpatented trade secrets and know-how,
in part through the use of confidentiality and intellectual
property agreements with our employees, consultants and certain
contractors. If our employees, scientific consultants,
collaborators or licensees develop inventions or processes
independently that may be applicable to our product candidates,
disputes may arise about the ownership of proprietary rights to
those inventions and processes. Such inventions and processes will
not necessarily become our property, but may remain the property of
those persons or their employers. Protracted and costly litigation
could be necessary to enforce and determine the scope of our
proprietary rights.
If
trade secrets are breached, our recourse will be solely against the
person who caused the secrecy breach. This might not be an adequate
remedy to us because third parties other than the person who causes
the breach will be free to use the information without
accountability to us. This is an inherent limitation of the law of
trade secret protection.
8
U.S. Governmental Regulation of Pharmaceutical
Products
General
Regulation
by governmental authorities in the United States and other
countries will continue to significantly impact our research,
product development, manufacturing and marketing of any
pharmaceutical products. The nature and the extent to which
regulations apply to us will vary depending on the nature of any
such products. Our potential pharmaceutical products will require
regulatory approval by governmental agencies prior to
commercialization. The products we are developing are subject to
federal regulation in the United States, principally by the FDA
under the Federal Food, Drug, and Cosmetic Act
(“FFDCA”), and by state and local governments, as well
as regulatory and other authorities in foreign governments that
include rigorous preclinical and clinical testing and other
approval procedures. Such regulations govern or influence, among
other things, the research, development, testing, manufacture,
safety and efficacy requirements, labeling, storage, recordkeeping,
licensing, advertising, promotion, distribution and export of
products, manufacturing and the manufacturing process. In many
foreign countries, such regulations also govern the prices charged
for products under their respective national social security
systems and availability to consumers.
All
drugs intended for human use are subject to rigorous regulation by
the FDA in the United States and similar regulatory bodies in other
countries. The steps ordinarily required by the FDA before an
innovative new drug product may be marketed in the United States
are similar to steps required in most other countries and include,
but are not limited to:
●
completion of
preclinical laboratory tests, preclinical animal testing and
formulation studies;
●
submission to the
FDA of an IND, which must be in effect before clinical trials may
commence;
●
submission to the
FDA of an NDA that includes preclinical data, clinical trial data
and manufacturing information;
●
payment of
substantial user fees for filing the NDA and other recurring user
fees;
●
FDA review of the
NDA;
●
satisfactory
completion of an FDA pre-approval inspection of the manufacturing
facilities; and
●
FDA approval of the
NDA, including approval of all product labeling.
For
combination products deemed to have a
‘‘drug’’ primary mode of action, primary
review of the product will be conducted by the appropriate division
within the Center for Drug Evaluation and Research
(“CDER”), but CDER will consult with the Center for
Devices and Radiological Health to ensure that the device
components of the product meet all applicable device
requirements.
The
research, development and approval process requires substantial
time, effort and financial resources, and approvals may not be
granted on a timely or commercially viable basis, if at
all.
Preclinical
testing includes laboratory evaluations to characterize the
product’s composition, impurities, stability, and mechanism
of its pharmacologic effect, as well as animal studies to assess
the potential safety and efficacy of each product. Preclinical
safety tests must be conducted by laboratories that comply with FDA
regulations regarding Good Laboratory Practices and the U.S.
Department of Agriculture’s Animal Welfare Act. Violations of
these laws and regulations can, in some cases, lead to invalidation
of the tests, requiring such tests to be repeated and delaying
approval of the NDA. The results of the preclinical tests, together
with manufacturing information and analytical data, are submitted
to the FDA as part of an IND and are reviewed by the FDA before the
commencement of human clinical trials. Unless the FDA objects to an
IND by placing the study on clinical hold, the IND will go into
effect 30 days following its receipt by the FDA. The FDA may
authorize trials only on specified terms and may suspend ongoing
clinical trials at any time on various grounds, including a finding
that patients are being exposed to unacceptable health risks. If
the FDA places a study on clinical hold, the sponsor must resolve
all of the FDA’s concerns before the study may begin or
continue. The IND application process may become extremely costly
and substantially delay development of products. Similar
restrictive requirements also apply in other countries.
Additionally, positive results of preclinical tests will not
necessarily indicate positive results in clinical
trials.
Clinical
trials involve the administration of the investigational product to
humans under the supervision of qualified principal investigators.
Our clinical trials must be conducted in accordance with Good
Clinical Practice regulations under protocols submitted to the FDA
as part of an IND. In addition, each clinical trial is approved and
conducted under the auspices of an institutional review board
(“IRB”), and requires the patients’ informed
consent. An IRB considers, among other things, ethical factors, the
safety of human subjects, and the possibility of liability of the
institutions conducting the trial. The IRB at each institution at
which a clinical trial is being performed may suspend a clinical
trial at any time for a variety of reasons, including a belief that
the test subjects are being exposed to an unacceptable health risk.
As the sponsor, we can also suspend or terminate a clinical trial
at any time.
9
Clinical
trials are typically conducted in three sequential phases, Phases
1, 2, and 3, involving an increasing number of human subjects.
These phases may sometimes overlap or be combined. Phase 1 trials
are performed in a small number of healthy human subjects or
subjects with the targeted condition, and involve testing for
safety, dosage tolerance, absorption, distribution, metabolism and
excretion. Phase 2 studies, which may involve up to hundreds of
subjects, seek to identify possible adverse effects and safety
risks, preliminary information related to the efficacy of the
product for specific targeted diseases, dosage tolerance, and
optimal dosage. Finally, Phase 3 trials may involve up to thousands
of individuals often at geographically dispersed clinical trial
sites, and are intended to provide the documentation of
effectiveness and important additional safety data required for
approval. Prior to commencing Phase 3 clinical trials many sponsors
elect to meet with FDA officials to discuss the conduct and design
of the proposed trial or trials.
In
addition, federal law requires the listing, on a publicly-available
website, of detailed information on clinical trials for
investigational drugs. Some states have similar or supplemental
clinical trial reporting laws.
Success
in early-stage animal studies and clinical trials does not
necessarily assure success in later-stage clinical trials. Data
obtained from animal studies and clinical activities are not always
conclusive and may be subject to alternative interpretations that
could delay, limit or even prevent regulatory
approval.
All
data obtained from the preclinical studies and clinical trials, in
addition to detailed information on the manufacture and composition
of the product, would be submitted in an NDA to the FDA for review
and approval for the manufacture, marketing and commercial
shipments of any of our products. FDA approval of the NDA is
required before commercial marketing or non-investigational
interstate shipment may begin in the United States. The FDA may
also conduct an audit of the clinical trial data used to support
the NDA.
The FDA
may deny or delay approval of an NDA that does not meet applicable
regulatory criteria. For example the FDA may determine that the
preclinical or clinical data or the manufacturing information does
not adequately establish the safety and efficacy of the drug. The
FDA has substantial discretion in the approval process and may
disagree with an applicant’s interpretation of the data
submitted in its NDA. The FDA can request additional information,
seek clarification regarding information already provided in the
submission or ask that new additional clinical trials be conducted,
all of which can delay approval. Similar types of regulatory
processes will be encountered as efforts are made to market any
drug internationally. We will be required to assure product
performance and manufacturing processes from one country to
another.
Even if
the FDA approves a product, it may limit the approved uses for the
product as described in the product labeling, require that
contraindications, warning statements or precautions be included in
the product labeling, require that additional studies be conducted
following approval as a condition of the approval, impose
restrictions and conditions on product distribution, prescribing or
dispensing in the form of a REMS, or otherwise limit the scope of
any approval or limit labeling. Once it approves an NDA, the FDA
may revoke or suspend the product approval if compliance with
post-market regulatory standards is not maintained or if problems
occur after the product reaches the marketplace. In addition, the
FDA may require post-marketing studies to monitor the effect of
approved products, and may limit further marketing of the product
based on the results of these post-market studies. The FDA and
other government agencies have broad post-market regulatory and
enforcement powers, including the ability to levy civil and
criminal penalties, suspend or delay issuance of approvals, seize
or recall products and revoke approvals.
Pharmaceutical
manufacturers, distributors and their subcontractors are required
to register their facilities with the FDA and state agencies.
Manufacturers are required to list their marketed drugs with the
FDA, are subject to periodic inspection by the FDA and other
authorities, where applicable, and must comply with the FDA’s
current Good Manufacturing Practices (“GMP”)
regulations, and the product specifications set forth in the
approved NDA. The GMP requirements for pharmaceutical products are
extensive and compliance with them requires considerable time,
resources and ongoing investment. The regulations require
manufacturers and suppliers of raw materials and components to
establish validated systems and to employ and train qualified
employees to ensure that products meet high standards of safety,
efficacy, stability, sterility (where applicable), purity, and
potency. The requirements apply to all stages of the manufacturing
process, including the synthesis, processing, sterilization,
packaging, labeling, storage and shipment of the drug product. For
all drug products, the regulations require investigation and
correction of any deviations from GMP requirements and impose
documentation requirements upon us and any third-party
manufacturers that we may decide to use. Manufacturing
establishments are subject to mandatory user fees, and to periodic
unannounced inspections by the FDA and state agencies for
compliance with all GMP requirements. The FDA is authorized to
inspect manufacturing facilities without a warrant at reasonable
times and in a reasonable manner.
10
We or
our present or future suppliers may not be able to comply with GMP
and other FDA regulatory requirements. Failure to comply with the
statutory and regulatory requirements subjects the manufacturer
and/or the NDA sponsor or distributor to possible legal or
regulatory action, such as a delay or refusal to approve an NDA,
suspension of manufacturing, seizure or recall of a product, or
civil or criminal prosecution of the company or individual officers
or employees.
Post-Marketing Regulation
Any
drug products manufactured or distributed by us pursuant to FDA
approvals, as well as the materials and components used in our
products, are subject to pervasive and continuing regulation by the
FDA, including:
●
recordkeeping
requirements;
●
periodic reporting
requirements;
●
GMP requirements
related to all stages of manufacturing, testing, storage,
packaging, labeling and distribution of finished dosage forms of
the product;
●
monitoring and
reporting of adverse experiences with the product; and
●
advertising and
promotional reporting requirements and restrictions.
Adverse
experiences with the product must be reported to the FDA and could
result in the imposition of market restriction through labeling
changes or product removal. Product approvals may be revoked if
compliance with regulatory requirements is not maintained or if
problems concerning safety or effectiveness of the product occur
following approval. The FDA is developing a national electronic
drug safety tracking system known as SENTINEL that may impose
additional safety monitoring burdens, and enhanced FDA enforcement
authority, beyond the extensive requirements already in effect. As
a condition of NDA approval, the FDA may require post-approval
testing and surveillance to monitor a product’s safety or
efficacy. The FDA also may impose other conditions, including
labeling restrictions which can materially impact the potential
market and profitability of a product.
With
respect to post-market product advertising and promotion, the FDA
and other government agencies including the Department of Health
and Human Services and the Department of Justice, and individual
States, impose a number of complex regulations on entities that
advertise and promote pharmaceuticals, including, among others,
standards and restrictions on direct-to-consumer advertising,
off-label promotion, industry-sponsored scientific and educational
activities and promotional activities involving the Internet. The
FDA has very broad enforcement authority under the FFDCA, and
failure to abide by these regulations can result in administrative
and judicial enforcement actions, including the issuance of a
Warning Letter directing correction of deviations from FDA
standards, a requirement that future advertising and promotional
materials be pre-cleared by the FDA, False Claims Act prosecution
based on alleged off-label marketing seeking monetary and other
penalties, including potential exclusion of the drug and/or the
company from participation in government health care programs, and
state and federal civil and criminal investigations and
prosecutions. Foreign regulatory bodies also strictly enforce these
and other regulatory requirements and drug marketing may be
prohibited in whole or in part in other countries.
We, our
collaborators or our third-party contract manufacturers may not be
able to comply with the applicable regulations. After regulatory
approvals are obtained, the subsequent discovery of previously
unknown problems, or the failure to maintain compliance with
existing or new regulatory requirements, may result
in:
●
restrictions on the
marketing or manufacturing of a product;
●
Warning Letters or
Untitled Letters from the FDA asking us, our collaborators or
third-party contractors to take or refrain from taking certain
actions;
●
withdrawal of the
product from the market;
●
the FDA’s
refusal to approve pending applications or supplements to approved
applications;
●
voluntary or
mandatory product recall;
●
fines or
disgorgement of profits or revenue;
●
suspension or
withdrawal of regulatory approvals;
●
refusals to permit
the import or export of products;
●
product seizure;
and
●
injunctions or the
imposition of civil or criminal penalties.
11
We may
also be subject to healthcare laws, regulations and enforcement and
our failure to comply with any such laws, regulations or
enforcement could adversely affect our business, operations and
financial condition. Certain federal and state healthcare laws and
regulations pertaining to fraud and abuse and patients’
rights are and will be applicable to our business. We are subject
to regulation by both the federal government and the states in
which we or our partners conduct our business. The laws and
regulations that may affect our ability to operate
include:
●
the federal
Anti-Kickback Statute, which prohibits, among other things, any
person or entity from knowingly and willfully offering, soliciting,
receiving or providing any remuneration (including any kickback,
bribe or rebate), directly or indirectly, overtly or covertly, in
cash or in kind, to induce either the referral of an individual or
in return for the purchase, lease, or order of any good, facility
item or service, for which payment may be made, in whole or in
part, under federal healthcare programs such as the Medicare and
Medicaid programs;
●
federal civil and
criminal false claims laws and civil monetary penalty laws,
including, for example, the federal civil False Claims Act, which
impose criminal and civil penalties, including civil whistleblower
or qui tam actions, against individuals or entities for, among
other things, knowingly presenting, or causing to be presented, to
the federal government, including the Medicare and Medicaid
programs, claims for payment that are false or fraudulent or making
a false statement to avoid, decrease or conceal an obligation to
pay money to the federal government;
●
the federal Health
Insurance Portability and Accountability Act of 1996
(“HIPAA”), which created new federal criminal statutes
that prohibit knowingly and willfully executing, or attempting to
execute, a scheme to defraud any healthcare benefit program or
obtain, by means of false or fraudulent pretenses, representations
or promises, any of the money or property owned by, or under the
custody or control of, any healthcare benefit program, regardless
of the payer (e.g., public or private), knowingly and willfully
embezzling or stealing from a health care benefit program,
willfully obstructing a criminal investigation of a health care
offense and knowingly and willfully falsifying, concealing or
covering up by any trick or device a material fact or making any
materially false statements in connection with the delivery of, or
payment for, healthcare benefits, items or services relating to
healthcare matters;
●
HIPAA, as amended
by the Health Information Technology for Economic and Clinical
Health Act, and their implementing regulations, which impose
obligations on covered entities, including healthcare providers,
health plans, and healthcare clearinghouses, as well as their
respective business associates that create, receive, maintain or
transmit individually identifiable health information for or on
behalf of a covered entity, with respect to safeguarding the
privacy, security and transmission of individually identifiable
health information;
●
the federal
physician sunshine requirements under the Patient Protection and
Affordable Care Act ( “Affordable Care Act”), which
require manufacturers of drugs, devices, biologics and medical
supplies to report annually to the Centers for Medicare &
Medicaid Services information related to payments and other
transfers of value provided to physicians and teaching hospitals,
and ownership and investment interests held by physicians and their
immediate family members; and
●
state law
equivalents of each of the above federal laws, such as
anti-kickback and false claims laws, which may apply to items or
services reimbursed by any third-party payer, including commercial
insurers; state laws that require pharmaceutical companies to
comply with the pharmaceutical industry’s voluntary
compliance guidelines and the applicable compliance guidance
promulgated by the federal government, or otherwise restrict
payments that may be provided to healthcare providers and other
potential referral sources; state laws that require drug
manufacturers to report information related to payments and other
transfers of value to healthcare providers or marketing
expenditures; and state laws governing the privacy and security of
health information in certain circumstances, many of which differ
from each other in significant ways and may not have the same
effect, thus complicating compliance efforts.
Because
of the breadth of these laws and the narrowness of the statutory
exceptions and safe harbors available, it is possible that some of
our business activities could be subject to challenge under one or
more of such laws. In addition, recent health care reform
legislation has strengthened these laws. For example, the
Affordable Care Act, among other things, amended the intent
requirement of the federal Anti-Kickback Statute and certain
criminal healthcare fraud statutes. A person or entity no longer
needs to have actual knowledge of the statute or specific intent to
violate it. In addition, the Affordable Care Act provided that the
government may assert that a claim including items or services
resulting from a violation of the federal Anti-Kickback Statute
constitutes a false or fraudulent claim for purposes of the federal
civil False Claims Act.
12
Achieving
and sustaining compliance with these laws may prove costly. In
addition, any action against us for violation of these laws, even
if we successfully defend against it, could cause us to incur
significant legal expenses and divert our management’s
attention from the operation of our business. If our operations are
found to be in violation of any of the laws described above or any
other governmental laws or regulations that apply to us, we may be
subject to penalties, including administrative, civil and criminal
penalties, damages, fines, disgorgement, the exclusion from
participation in federal and state healthcare programs, individual
imprisonment or the curtailment or restructuring of our operations,
any of which could adversely affect our ability to operate our
business and our financial results.
Generic Competition
Orange Book Listing. In seeking approval for a drug through
an NDA, applicants are required to list with the FDA each patent
whose claims cover the applicant’s product. Upon approval of
a drug, the applicant identifies all patents that claim the
approved product’s active ingredient(s), the drug
product’s approved formulation, or an approved method of use
of the drug. Each of the identified patents are then published in
the FDA’s Approved Drug Products with Therapeutic Equivalence
Evaluations, commonly known as the Orange Book. Drugs listed in the
Orange Book can, in turn, be cited by potential generic competitors
in support of approval of an abbreviated new drug application
(“ANDA”). An ANDA provides for marketing of a drug
product that has the same active ingredients in the same strengths
and dosage form as the listed drug and has been shown through
bioequivalence testing, unless such testing is waived by the FDA,
as is the case with some injectable drug products, to be
therapeutically equivalent to the listed drug. Other than
bioequivalence testing, ANDA applicants are not required to
conduct, or submit results of, preclinical or clinical tests to
prove the safety or effectiveness of their drug product. Drugs
approved in this way are commonly referred to as
‘‘generic equivalents’’ to the listed drug,
and can usually be substituted by pharmacists under prescriptions
written for the original listed drug.
The
ANDA applicant is required to certify to the FDA concerning any
patents listed for the approved product in the FDA’s Orange
Book. Specifically, the applicant must certify either that: (1) the
required patent information has not been filed (a Paragraph I
Certification); (2) the listed patent has expired (a Paragraph II
Certification); (3) the listed patent has not expired, but will
expire on a particular date and the generic approval is being
sought only after patent expiration (a Paragraph III
Certification); or (4) the listed patent is invalid, unenforceable,
or will not be infringed by the proposed generic product (a
Paragraph IV Certification). In certain circumstances, the ANDA
applicant may also elect to submit a ‘‘section
(viii)’’ statement instead of a Paragraph IV
Certification, certifying that its proposed ANDA label does not
contain (or carves out) any language regarding the patented
method-of-use rather than certify to a listed method-of-use patent.
If the application contains only Paragraph I or Paragraph II
Certifications, the ANDA may be approved as soon as FDA completes
its review and concludes that all approval requirements have been
met. If the ANDA contains one or more Paragraph III Certifications,
the ANDA cannot not be approved until each listed patent for which
a Paragraph III Certification was filed have expired.
If the
ANDA applicant has provided a Paragraph IV certification to the
FDA, the applicant must also send notice of the Paragraph IV
certification to the NDA holder and patent owner once the ANDA has
been accepted for filing by the FDA. The patent owner or NDA holder
may then commence a patent infringement lawsuit in response to the
notice of the Paragraph IV certification. The filing of a patent
infringement lawsuit within 45 days of the receipt of a Paragraph
IV certification automatically prevents the FDA from approving the
ANDA until the earlier of 30 months (the ‘‘30-month
stay’’), expiration of the patent, settlement of the
lawsuit in which the patent owner admits that the patent is invalid
or not infringed by the ANDA product, or a decision in the
infringement case that holds the patent to be invalid or not
infringed, or an order by the court shortening the 30-month stay
due to actions by the patent holder to delay the litigation. In
most circumstances, NDA holder is only eligible for one 30-month
stay against an ANDA.
If a
patent infringement action is filed against an ANDA applicant, any
settlement of the litigation must be submitted to the Federal Trade
Commission (“FTC”). If the FTC believes the terms or
effects of the settlement are anticompetitive, FTC may bring an
antitrust enforcement action against the parties. Private parties
may also bring antitrust lawsuits against drug companies based on
such patent litigation settlements.
The
ANDA also will not be approved until any applicable non-patent
regulatory exclusivity listed in the Orange Book for the referenced
product has expired.
13
Regulatory Exclusivity. Upon NDA approval of a new chemical
entity (“NCE”), which is a drug that contains no active
moiety that has been approved by the FDA in any other NDA, that
drug receives five years of marketing exclusivity during which the
FDA cannot receive for review any ANDA seeking approval of a
generic version of that drug. An ANDA containing a Paragraph IV
Certification may be received by FDA 4 years after the NCE
drug’s approval, but any 30-month stay that ensues would be
extended so that it expires seven and one half years after the NCE
approval date, subject to early termination by reason of a court
decision or settlement as described above.
Certain
changes to an NDA drug, such as the addition of a new indication to
the package insert, for which new clinical trials, conducted or
sponsored by the applicant are deemed by FDA to be essential to the
approval of the change, can be eligible for a three-year period of
exclusivity during which the FDA cannot approve an ANDA for a
generic drug that includes the change. An ANDA that contains a
section (viii) statement to a method of use patent may be approved
with labeling that omits the patented use before the use patent
expires. Generic drugs approved with such a labeling carve out may
be substituted by pharmacists for the original branded drug before
the method of use patent expires.
Section 505(b)(2) NDAs. Most drug products obtain FDA
marketing approval pursuant to an NDA or an ANDA. A third
alternative is a special type of NDA, commonly referred to as a
505(b)(2) NDA, which enables the applicant to rely, in part, on the
FDA’s previous approval of a similar product, or published
literature, in support of its application.
505(b)(2)
NDAs often provide an alternate path to FDA approval for new or
improved formulations or new uses of previously approved products.
A 505(b)(2) NDA may be used where at least some of the information
required for approval comes from studies not conducted by, or for,
the applicant and for which the applicant has not obtained a right
of reference. If the 505(b)(2) applicant can establish that
reliance on the FDA’s previous approval is scientifically
appropriate, it may eliminate the need to conduct certain
preclinical or clinical studies of the new product. The FDA may
also require companies to perform additional studies or
measurements to support the change from the approved product. The
FDA may then approve the new product candidate for all, or some, of
the label indications for which the referenced product has been
approved, as well as for any new indication or conditions of use
sought by the Section 505(b)(2) applicant.
To the
extent that the Section 505(b)(2) applicant is relying on studies
conducted for an already approved product, the applicant is
required to certify to the FDA concerning any patents listed for
the approved product in the Orange Book to the same extent that an
ANDA applicant would. As a result, approval of a 505(b)(2) NDA can
be stalled until all the listed patents claiming the referenced
product have expired, until any non-patent exclusivity, such as
exclusivity for obtaining approval of a new chemical entity, listed
in the Orange Book for the referenced product has expired, and, in
the case of a Paragraph IV certification and subsequent patent
infringement suit, until the expiration of any 30-month stay,
subject to early termination of the stay as described
above.
Changing Legal and Regulatory Landscape
Periodically,
legislation is introduced in the U.S. Congress that could change
the statutory and regulatory provisions governing the approval,
manufacturing and marketing of our drugs. In addition, the FDCA,
FDA regulations and guidance are often revised or reinterpreted by
the FDA or the courts in ways that may significantly affect our
business and products. We cannot predict whether or when
legislation or court decisions impacting our business will be
enacted or issued, what FDA regulations, guidance or
interpretations may change, or what the impact of such changes, if
any, may be in the future.
Third-Party Reimbursements
Successful
sales of our proposed products in the United States and other
countries depend, in large part, on the availability of adequate
reimbursement from third-party payers such as governmental
entities, managed care organizations, health maintenance
organizations (“HMOs”), and private insurance plans.
Reimbursement by a third-party payer depends on a number of
factors, including the payer’s determination that the product
has been approved by the FDA for the indication for which the claim
is being made, that it is neither experimental nor investigational,
and that the use of the product is safe and efficacious, medically
necessary, appropriate for the specific patient and cost
effective.
Since
reimbursement by one payer does not guarantee reimbursement by
another, we or our licensees may be required to seek approval from
each payer individually. Seeking such approvals is a time-consuming
and costly process. Third-party payers routinely limit the products
that they will cover and the amount of money that they will pay
and, in many instances, are exerting significant pressure on
medical suppliers to lower their prices.
14
Payers
frequently employ a tiered system in reimbursing end users for
pharmaceutical products, with tier designation affecting copay or
deductible amounts. The only approved product for treating HSDD is
flibanserin, sold under the trade name Addyi®. There is
significant uncertainty concerning the extent and scope of
third-party reimbursement for products treating HSDD. Based on
third-party reimbursement for approved products treating ED, we
believe bremelanotide will be classified as a Tier 3 drug, so that
reimbursement will be limited for bremelanotide for treatment of
premenopausal women with HSDD, assuming the product is approved by
the FDA. Less than full reimbursement by governmental and other
third-party payers may adversely affect the market acceptance of
bremelanotide. Further, healthcare reimbursement systems vary from
country to country, and third-party reimbursement might not be made
available for bremelanotide for HSDD under any other reimbursement
system.
Manufacturing and Marketing
To be
successful, our proposed products will need to be manufactured in
commercial quantities under GMP prescribed by the FDA and at
acceptable costs. We do not have the facilities to manufacture any
of our proposed products under GMP. We intend to rely on
collaborators, licensees or contract manufacturers for the
commercial manufacture of our proposed products.
Our
bremelanotide product candidate is a synthetic peptide. While the
production process involves well-established technology, there are
few manufacturers capable of scaling up to commercial quantities
under GMP at acceptable costs. We identified one third-party
manufacturer for the production of bremelanotide, Lonza Ltd., and
have validated manufacturing of the bremelanotide drug substance
under GMP with that manufacturer. AMAG, as the exclusive licensee
for North America, has assumed responsibility for manufacturing
bremelanotide drug substance.
Our
bremelanotide product candidate will be a combination product,
incorporating both the bremelanotide drug substance and a delivery
device. AMAG, as the exclusive licensee for North America, has
assumed responsibility for manufacturing the bremelanotide
combination product. We relied on a third-party manufacturer,
Ypsomed AG, to make the selected autoinjector pen delivery device.
A third-party contract manufacturer, Catalent Belgium S.A.,
performs fill, finish and packaging of our bremelanotide product
candidate. We negotiated a long-term commercial supply agreement
with Catalent Belgium S.A., and have assigned this agreement to
AMAG.
Our
PL-3994 product candidate is a peptide mimetic molecule,
incorporating a proprietary amino acid mimetic structure and amino
acids. We identified a manufacturer that made the product in
quantities sufficient for Phase 1 and Phase 2, and are evaluating
commercial-scale manufacturers. Scaling up to commercial quantities
may involve production, purification, formulation and other
problems not present in the scale of manufacturing done to
date.
Our
MC1r and MC4r agonist product candidates are synthetic peptides,
which we have manufactured only at laboratory scale. We have not
contracted with a third-party manufacturer to produce these
synthetic peptides for either clinical trials or commercial
purposes. While the production process involves well-established
technology, there are few manufacturers capable of scaling up to
commercial quantities under GMP at acceptable costs. Additionally,
scaling up to commercial quantities may involve production,
purification, formulation and other problems not present in the
scale of manufacturing done to date.
The
failure of any manufacturer or supplier to comply with FDA
regulations, including GMP or medical device quality systems
regulations (“QSR”), or to supply the device component
or drug substance and services as agreed, would force us or our
licensees to seek alternative sources of supply and could interfere
with our and our licensees’ ability to deliver product on a
timely and cost effective basis or at all. Establishing
relationships with new manufacturers or suppliers, any of whom must
be FDA-approved, is a time-consuming and costly
process.
Product Liability and Insurance
Our
business may be affected by potential product liability risks that
are inherent in the testing, manufacturing, marketing and use of
our proposed products. We have liability insurance providing $10
million coverage in the aggregate as to certain clinical trial
risks.
Employees
As of
September 21, 2017, we employed 22 people full time, of whom 16 are
engaged in research and development activities and 6 are engaged in
administration and management, and did not have any part-time
employees. While we have been successful in attracting skilled and
experienced scientific personnel, competition for personnel in our
industry is intense. None of our employees are covered by a
collective bargaining agreement. All of our employees have executed
confidentiality and intellectual property agreements. We consider
relations with our employees to be good.
15
We rely
on contractors and scientific consultants to work on specific
research and development programs. We also rely on independent
organizations, advisors and consultants to provide services,
including aspects of manufacturing, testing, preclinical
evaluation, clinical management, regulatory strategy and market
research. Our independent advisors, contractors and consultants
sign agreements that provide for confidentiality of our proprietary
information and that we have the rights to any intellectual
property developed while working for us.
Corporate Information
We were
incorporated under the laws of the State of Delaware on November
21, 1986 and commenced operations in the biopharmaceutical area in
1996. Our corporate offices are located at 4B Cedar Brook Drive,
Cranbury, New Jersey 08512 and our telephone number is (609)
495-2200. We maintain an Internet site at www.palatin.com, where among other
things, we make available free of charge on and through this
website our Forms 3, 4 and 5, annual reports on Form 10-K,
quarterly reports on Form 10-Q, current reports on Form 8-K, and
amendments to those reports filed or furnished pursuant to Section
13(a) or 15(d) and Section 16 of the Exchange Act as soon as
reasonably practicable after we electronically file such material
with, or furnish it to, the SEC. Our website and the information
contained in it or connected to it are not incorporated into this
Annual Report. The reference to our website is an inactive textual
reference only.
Item 1A. Risk Factors.
Risks Related to Our Financial Results and Need for
Financing
We have a history of substantial net losses, and expect to continue
to incur substantial net losses over the next few years, and we may
never achieve or maintain profitability.
As of
June 30, 2017, we had an accumulated deficit of $356. 7 million and
incurred a net loss for the year ended June 30, 2017 of $13.3
million. We may not achieve or sustain profitability in future
years, which is dependent on numerous factors, including whether
and when development and sales milestones are met, regulatory
actions by the FDA and other regulatory bodies, the performance of
our licensees, and market acceptance of our products. If we attain
sustained profitability, it will not be until the fiscal year
ending June 30, 2019 at the earliest.
We
expect to incur additional losses as we continue our development of
natriuretic peptide and MC1r products. These losses, among other
things, have had and will continue to have an adverse effect on our
stockholders’ equity, total assets and working
capital.
Since
2005 we have not had any products available for commercial sale and
have not received any revenues from the sale of our product
candidates. For the foreseeable future, we will have to fund all of
our operations and capital expenditures from license and contract
revenue under collaborative development agreements, existing cash
balances and outside sources of financing, which may not be
available on acceptable terms, if at all. Unless and until we
receive approval from the FDA or other equivalent regulatory
authorities outside the United States, we cannot sell our products
and will not have product revenues from them. We have devoted
substantially all of our efforts to research and development,
including preclinical and clinical trials. Because of the numerous
risks associated with developing drugs, we are unable to predict
the extent of future losses, whether or when any of our product
candidates will become commercially available, or when we will
become profitable, if at all.
We will need additional funding, including funding to complete
clinical trials for our product candidates other than
bremelanotide, which may not be available on acceptable terms, if
at all.
Under
the license agreement with AMAG, we are contractually required to
complete development and regulatory activities necessary to file an
NDA for bremelanotide for HSDD in the United States. AMAG will
reimburse us for up to an aggregate amount of $25 million for all
reasonable, documented, direct out-of-pocket expenses we incur in
completing these development and regulatory activities. To the
extent that our expenses exceed this amount, we will be responsible
for the required additional funding.
In
addition to our responsibilities under the license agreement with
AMAG, we intend to focus efforts on our other product candidates,
including our natriuretic peptide and MC1r programs. As of June 30,
2017, we had cash, cash equivalents, accounts receivable and
investments of $55.6 million, with current liabilities of $19.9
million, net of deferred revenue of $35.1 million. After giving
effect to the proceeds from our license agreements with AMAG and
Fosun and the proceeds from the financing transactions on August 4,
2016 and December 6, 2016, we believe we currently have sufficient
existing capital resources to fund our planned operations through
at least the 2018 calendar year. We will need additional funding to
complete development activities and required clinical trials for
our other product candidates and development programs and, if those
clinical trials are successful (which we cannot predict), to
complete submission of required regulatory applications to the
FDA.
16
Until
the FDA approves bremelanotide for HSDD and marketing commences, if
at all, we will not have any recurring revenue. Even if
bremelanotide is approved and marketing commences, we cannot
predict product sales or our resulting royalties, so we may not
have any source of significant recurring revenue and may need to
depend on financing or partnering to sustain our operations. We may
raise additional funds through public or private equity or debt
financings, collaborative arrangements on our product candidates,
or other sources. However, such financing arrangements may not be
available on acceptable terms, or at all. To obtain additional
funding, we may need to enter into arrangements that require us to
develop only certain of our product candidates or relinquish rights
to certain technologies, product candidates and/or potential
markets.
If we
are unable to raise sufficient additional funds when needed, we may
be required to curtail operations significantly, cease clinical
trials and decrease staffing levels. We may seek to license, sell
or otherwise dispose of our product candidates, technologies and
contractual rights on the best possible terms available. Even if we
are able to license, sell or otherwise dispose of our product
candidates, technologies and contractual rights, it is likely to be
on unfavorable terms and for less value than if we had the
financial resources to develop or otherwise advance our product
candidates, technologies and contractual rights
ourselves.
Our
future capital requirements depend on many factors,
including:
●
our ability to
enter into one or more licensing or similar agreements for
bremelanotide outside of North America and China;
●
the timing of, and
the costs involved in, obtaining regulatory approvals for
bremelanotide for HSDD and our other product
candidates;
●
the number and
characteristics of any additional product candidates we develop or
acquire;
●
the scope,
progress, results and costs of researching and developing our
future product candidates, and conducting preclinical and clinical
trials;
●
the cost of
commercialization activities if any future product candidates are
approved for sale, including marketing, sales and distribution
costs;
●
the cost of
manufacturing any future product candidates and any products we
successfully commercialize;
●
our ability to
establish and maintain strategic collaborations, licensing or other
arrangements and the terms and timing of such
arrangements;
●
the degree and rate
of market acceptance of any future approved products;
●
the emergence,
approval, availability, perceived advantages, relative cost,
relative safety and relative efficacy of alternative and competing
products or treatments;
●
any product
liability or other lawsuits related to our products;
●
the expenses needed
to attract and retain skilled personnel;
●
the costs involved
in preparing, filing, prosecuting, maintaining, defending and
enforcing patent claims, including litigation costs and the outcome
of such litigation; and
●
the timing, receipt
and amount of sales of, or royalties on, future approved products,
if any.
We have a limited operating history upon which to base an
investment decision.
Our
operations are primarily focused on acquiring, developing and
securing our proprietary technology, conducting preclinical and
clinical studies and formulating and manufacturing on a small-scale
basis our principal product candidates. These operations provide a
limited basis for stockholders to assess our ability to
commercialize our product candidates.
We have
not yet demonstrated our ability to perform the functions necessary
for the successful commercialization of any of our current product
candidates. The successful commercialization of our product
candidates will require us to perform a variety of functions,
including:
●
continuing to
conduct preclinical development and clinical trials;
●
participating in
regulatory approval processes;
●
formulating and
manufacturing products, or having third parties formulate and
manufacture products;
●
post-approval
monitoring and surveillance of our products;
●
conducting sales
and marketing activities, either alone or with a partner;
and
●
obtaining
additional capital.
17
If we
are unable to obtain regulatory approval of any of our product
candidates, to successfully commercialize any products for which we
receive regulatory approval or to obtain additional capital, we may
not be able to recover our investment in our development
efforts.
The
clinical and commercial success of our product candidates will
depend on a number of factors, including the
following:
●
the ability to
raise additional capital on acceptable terms, or at
all;
●
timely completion
of our clinical trials, which may be significantly slower or cost
more than we currently anticipate and will depend substantially
upon the performance of third-party contractors;
●
whether we are
required by the FDA or similar foreign regulatory agencies to
conduct additional clinical trials beyond those planned to support
the approval and commercialization of our product candidates or any
future product candidates;
●
acceptance of our
proposed indications and primary endpoint assessments relating to
the proposed indications of our product candidates by the FDA and
similar foreign regulatory authorities;
●
our ability to
demonstrate to the satisfaction of the FDA and similar foreign
regulatory authorities, the safety and efficacy of our product
candidates or any future product candidates;
●
the prevalence,
duration and severity of potential side effects experienced with
our product candidates or future approved products, if
any;
●
the timely receipt
of necessary marketing approvals from the FDA and similar foreign
regulatory authorities;
●
achieving and
maintaining, and, where applicable, ensuring that our third-party
contractors achieve and maintain, compliance with our contractual
obligations and with all regulatory requirements applicable to our
product candidates or any future product candidates or approved
products, if any;
●
the ability of
third parties with whom we contract to manufacture clinical trial
and commercial supplies of our product candidates or any future
product candidates, remain in good standing with regulatory
agencies and develop, validate and maintain commercially viable
manufacturing processes that are compliant with GMP;
●
a continued
acceptable safety profile and efficacy during clinical development
and following approval of our product candidates or any future
product candidates;
●
our ability to
successfully commercialize our product candidates or any future
product candidates in the United States and internationally, if
approved for marketing, sale and distribution in such countries and
territories, whether alone or in collaboration with
others;
●
acceptance by
physicians and patients of the benefits, safety and efficacy of our
product candidates or any future product candidates, if approved,
including relative to alternative and competing
treatments;
●
our and our
partners’ ability to establish and enforce intellectual
property rights in and to our product candidates or any future
product candidates;
●
our and our
partners’ ability to avoid third-party patent interference or
intellectual property infringement claims; and
●
our ability to
in-license or acquire additional product candidates or
commercial-stage products that we believe can be successfully
developed and commercialized.
If we
do not achieve one or more of these factors, many of which are
beyond our control, in a timely manner or at all, we could
experience significant delays or an inability to obtain regulatory
approvals or commercialize our product candidates. Even if
regulatory approvals are obtained, we may never be able to
successfully commercialize any of our product candidates.
Accordingly, we cannot assure you that we will be able to generate
sufficient revenue through the sale of our product candidates or
any future product candidates to continue our
business.
Raising additional capital may cause dilution to existing
shareholders, restrict our operations or require us to relinquish
rights.
We may
seek the additional capital necessary to fund our operations
through public or private equity offerings, collaboration
agreements, debt financings or licensing arrangements. To the
extent that we raise additional capital through the sale of equity
or convertible debt securities, existing shareholders’
ownership interests will be diluted and the terms may include
liquidation or other preferences that adversely affect their rights
as a shareholder. Debt financing, if available, may involve
agreements that include covenants limiting or restricting our
ability to take specific actions such as incurring additional debt,
making capital expenditures or declaring dividends. If we raise
additional funds through collaborations and licensing arrangements
with third parties, we may have to relinquish valuable rights to
our technologies or product candidates, or grant licenses on terms
that are not favorable to us.
18
Risks Related to Our Business, Strategy and Industry
We are substantially dependent on the clinical and commercial
success of our product candidates, primarily our lead product
candidate, bremelanotide for HSDD, but we and our licensees may
never obtain regulatory approval for or successfully commercialize
bremelanotide for HSDD or any of our product
candidates.
To
date, we have invested most of our efforts and financial resources
in the research and development of bremelanotide for HSDD, which is
currently our lead product candidate. We licensed to AMAG all
rights to bremelanotide in North America, but are contractually
obligated to complete development and regulatory activities
necessary to file an NDA for bremelanotide for HSDD in the United
States, with AMAG reimbursing us for up to $25 million for
reasonable, documented, out-of-pocket expenses we incur. We
licensed to Fosun rights to bremelanotide for China, but depending
on the regulatory approval pathway utilized in China, approval in
China may be contingent on approval of an NDA for bremelanotide for
HSDD in the United States.
Our
near-term prospects, including our ability to finance our company
and generate revenue, will depend heavily on the successful
development, regulatory approval and commercialization of
bremelanotide for HSDD, as well as any future product candidates.
The clinical and commercial success of our product candidates will
depend on a number of factors, including the
following:
●
timely completion
of, or need to conduct additional clinical trials and studies,
including for bremelanotide for HSDD, which may be significantly
slower or cost more than we currently anticipate and will depend
substantially upon the accurate and satisfactory performance of
third-party contractors;
●
the ability to
demonstrate to the satisfaction of the FDA the safety and efficacy
of bremelanotide for HSDD or any future product candidates through
clinical trials;
●
whether we or our
licensees are required by the FDA or other similar foreign
regulatory agencies to conduct additional clinical trials to
support the approval of bremelanotide for HSDD or any future
product candidates;
●
the acceptance of
parameters for regulatory approval, including our proposed
indication, primary endpoint assessment and primary endpoint
measurement, relating to our lead indications of bremelanotide for
HSDD;
●
the success of our
licensees in educating physicians and patients about the benefits,
administration and use of bremelanotide for HSDD, if
approved;
●
the prevalence and
severity of adverse events experienced with bremelanotide for HSDD
or any future product candidates or approved products;
●
the adequacy and
regulatory compliance of the autoinjector device, supplied by an
unaffiliated third party, to be used as part of the bremelanotide
combination product;
●
the timely receipt
of necessary marketing approvals from the FDA and similar foreign
regulatory authorities;
●
our ability to
raise additional capital on acceptable terms to achieve our
goals;
●
achieving and
maintaining compliance with all regulatory requirements applicable
to bremelanotide for HSDD or any future product candidates or
approved products;
●
the availability,
perceived advantages, relative cost, relative safety and relative
efficacy of alternative and competing treatments;
●
the effectiveness
of our own or our future potential strategic collaborators’
marketing, sales and distribution strategy and
operations;
●
the ability to
manufacture clinical trial supplies of bremelanotide for HSDD or
any future product candidates and to develop, validate and maintain
a commercially viable manufacturing process that is compliant with
current GMP;
●
the ability of AMAG
to successfully commercialize bremelanotide for HSDD;
●
our ability to
successfully commercialize any future product candidates, if
approved for marketing and sale, whether alone or in collaboration
with others;
●
our ability to
enforce our intellectual property rights in and to bremelanotide
for HSDD or any future product candidates;
●
our ability to
avoid third-party patent interference or intellectual property
infringement claims;
●
acceptance of
bremelanotide for HSDD or any future product candidates, if
approved, as safe and effective by patients and the medical
community; and
19
●
a continued
acceptable safety profile and efficacy of bremelanotide for HSDD or
any future product candidates following approval.
If we
fail to satisfy any one of these prerequisites to our commercial
success, many of which are beyond our control, in a timely manner
or at all, we could experience significant delays or an inability
to successfully commercialize our product candidates. Accordingly,
we cannot assure you that we will be able to generate sufficient
revenue through the sale of bremelanotide for HSDD by AMAG and
Fosun or through the sale of any future product candidate to
continue our business. In addition to preventing us from executing
our current business plan, any delays in our clinical trials, or
inability to successfully commercialize our products could impair
our reputation in the industry and the investment community, and
could hinder our ability to fulfill our existing contractual
commitments. As a result, our share price would likely decline
significantly, and we would have difficulty raising necessary
capital for future projects.
We do not control the development or commercialization of
bremelanotide in North America, which is licensed to AMAG, and as a
result we may not realize a significant portion of the potential
value of the license arrangement with AMAG.
Although
we will conduct all development work to support an NDA for
bremelanotide in HSDD, the license agreement with AMAG for
bremelanotide in North America limits our control over development
activities, including regulatory approvals, and we do not have any
direct control over commercialization efforts. AMAG may abandon
further development of bremelanotide in its licensed territory,
including terminating the agreement, for any reason, including a
change of priorities within AMAG or lack of success in ancillary
clinical trials necessary to obtain regulatory approvals. Because
the potential value of the license arrangement with AMAG is
contingent upon the successful development and commercialization of
bremelanotide in the United States and other countries in the
licensed territory, the ultimate value of this license will depend
on the efforts of AMAG. If AMAG does not succeed in obtaining
regulatory approval of bremelanotide in the United States for any
reason, does not succeed in securing market acceptance of
bremelanotide in the United States, or elects for any reason to
discontinue development of bremelanotide, we will be unable to
realize the potential value of this arrangement and would
experience significant delays or an inability to successfully
commercialize bremelanotide.
Production and supply of bremelanotide depend on contract
manufacturers over whom neither we nor AMAG have any control, and
we may not have adequate supplies of bremelanotide.
We do
not have the facilities to manufacture the bremelanotide active
drug ingredient or the autoinjector pen component of the
bremelanotide combination product, or to fill, assemble and package
the bremelanotide combination product. AMAG, our exclusive licensee
for North America for bremelanotide, has assumed responsibility for
contract manufacturing. The contract manufacturers must perform
these manufacturing activities in a manner that complies with FDA
regulations. AMAG’s ability to control third-party compliance
with FDA requirements is limited to contractual remedies and rights
of inspection. The manufacturers of approved products and their
manufacturing facilities will be subject to ongoing review and
periodic inspections by the FDA and other authorities where
applicable, and must comply with regulatory requirements, including
FDA regulations concerning GMP. Failure of third-party
manufacturers to comply with GMP, medical device quality system
regulations, or other FDA requirements may result in enforcement
action by the FDA. Failure to conduct their activities in
compliance with FDA regulations could delay bremelanotide
development programs or negatively impact AMAG’s ability to
receive FDA approval of the bremelanotide potential products or
continue marketing bremelanotide products if they are approved.
Establishing relationships with new suppliers, who must be
FDA-approved, is a time-consuming and costly process. If AMAG is
not able to obtain adequate supplies of bremelanotide, it will be
difficult for AMAG to develop bremelanotide and compete
effectively.
Most of our product candidates are still in the early stages of
development, and all of our product candidates remain subject to
clinical testing and regulatory approval. If we are unable to
successfully develop and test our product candidates, we will not
be successful.
Our
product candidates are at various stages of research and
development, will require regulatory approval, and may never be
successfully developed or commercialized. Our product candidates
will require significant further research, development and testing
before we can seek regulatory approval to market and sell them. We
must demonstrate that our product candidates are safe and effective
for use in patients in order to receive regulatory approval for
commercial sale. Preclinical studies in animals, using various
doses and formulations, must be performed before we can begin human
clinical trials. Even if we obtain favorable results in the
preclinical studies, the results in humans may be different.
Numerous small-scale human clinical trials may be necessary to
obtain initial data on a product candidate’s safety and
efficacy in humans before advancing to large scale human clinical
trials. We face the risk that the results of our trials in later
phases of clinical trials may be inconsistent with those obtained
in earlier phases. Adverse or inconclusive results could delay the
progress of our development programs and may prevent us from filing
for regulatory approval of our product candidates. Additional
factors that could inhibit the successful development of our
product candidates include:
20
●
lack of
effectiveness of any product candidate during clinical trials or
the failure of our product candidates to meet specified
endpoints;
●
failure to design
appropriate clinical trial protocols;
●
uncertainty
regarding proper dosing;
●
inability to
develop or obtain a supplier for an autoinjector device that meets
the FDA’s medical device requirements;
●
insufficient data
to support regulatory approval;
●
inability or
unwillingness of medical investigators to follow our clinical
protocols;
●
inability to add a
sufficient number of clinical trial sites; or
●
the availability of
sufficient capital to sustain operations and clinical
trials.
You
should evaluate us in light of these uncertainties, difficulties
and expenses commonly experienced by early stage biopharmaceutical
companies, as well as unanticipated problems and additional costs
relating to:
●
product approval or
clearance;
●
regulatory
compliance;
●
good manufacturing
practices;
●
intellectual
property rights;
●
product
introduction; and
●
marketing and
competition.
If clinical trials for our product candidates are prolonged or
delayed, we may be unable to commercialize our product candidates
on a timely basis, which would require us to incur additional costs
and delay our receipt of any revenue from potential product
sales.
We may
be unable to commercialize our product candidates on a timely basis
due to unexpected delays in our human clinical trials. Potential
delaying events include:
●
discovery of
serious or unexpected toxicities or side effects experienced by
study participants or other safety issues;
●
slower than
expected rates of subject recruitment and enrollment rates in
clinical trials resulting from numerous factors, including the
prevalence of other companies’ clinical trials for their
product candidates for the same indication, or clinical trials for
indications for which patients do not as commonly seek
treatment;
●
difficulty in
retaining subjects who have initiated a clinical trial but may
withdraw at any time due to adverse side effects from the therapy,
insufficient efficacy, fatigue with the clinical trial process or
for any other reason;
●
difficulty in
obtaining IRB approval for studies to be conducted at each
site;
●
delays in
manufacturing or obtaining, or inability to manufacture or obtain,
sufficient quantities of materials for use in clinical
trials;
●
inadequacy of or
changes in our manufacturing process or the product formulation or
method of delivery;
●
changes in
applicable laws, regulations and regulatory policies;
●
delays or failure
in reaching agreement on acceptable terms in clinical trial
contracts or protocols with prospective contract research
organizations (“CROs”), clinical trial sites and other
third-party contractors;
●
failure of our CROs
or other third-party contractors to comply with contractual and
regulatory requirements or to perform their services in a timely or
acceptable manner;
●
failure by us, our
employees, our CROs or their employees or any partner with which we
may collaborate or their employees to comply with applicable FDA or
other regulatory requirements relating to the conduct of clinical
trials or the handling, storage, security and recordkeeping for
drug, medical device and biologic products;
●
delays in the
scheduling and performance by the FDA of required inspections of
us, our CROs, our suppliers, or our clinical trial sites, and
violations of law or regulations by discovered in the course of FDA
inspections;
●
scheduling
conflicts with participating clinicians and clinical institutions;
or
●
difficulty in
maintaining contact with subjects during or after treatment, which
may result in incomplete data.
21
Any of
these events or other delaying events, individually or in the
aggregate, could delay the commercialization of our product
candidates and have a material adverse effect on our business,
results of operations and financial condition.
We may not be able to secure and maintain research institutions to
conduct our clinical trials.
We rely
on research institutions to conduct our clinical trials, and we
therefore have limited control over the timing and cost of clinical
trials and our ability to recruit subjects. If we are unable to
reach agreements with suitable research institutions on acceptable
terms, or if any such agreement is terminated, we may be unable to
quickly replace the research institution with another qualified
institution on acceptable terms. We may not be able to secure and
maintain suitable research institutions to conduct our clinical
trials.
Even if our product candidates receive regulatory approval, they
may never achieve market acceptance, in which case our business,
financial condition and results of operation will be materially
adversely affected.
Regulatory
approval for the marketing and sale of any of our product
candidates does not assure the product’s commercial success.
Any approved product will compete with other products manufactured
and marketed by major pharmaceutical and other biotechnology
companies. If any of our product candidates are approved by the FDA
and do not achieve adequate market acceptance, our business,
financial condition and results of operations will be materially
adversely affected. The degree of market acceptance of any such
product will depend on a number of factors, including:
●
perceptions by
members of the healthcare community, including physicians, about
the safety and effectiveness of any such product;
●
cost-effectiveness
relative to competing products and technologies;
●
availability of
reimbursement for our products from third-party payers such as
health insurers, HMOs and government programs such as Medicare and
Medicaid; and
●
advantages over
alternative treatment methods.
There
is one FDA approved product for treatment of HSDD, flibanserin,
which is sold under the trade name Addyi®, and started
marketing in October 2015. The actual market size and market
dynamics for HSDD are unknown, and there is significant uncertainty
concerning the extent and scope of third-party reimbursement for
products treating HSDD. While we believe that an on-demand drug for
HSDD has competitive advantages compared to chronic or daily use
hormones and other drugs, we may not be able to realize this
perceived advantage in the market. Bremelanotide is administered by
subcutaneous injection. While the single-use, disposable
autoinjector pen format is designed to maximize market
acceptability, bremelanotide as a subcutaneous injectable drug for
HSDD may never achieve significant market acceptance. In addition,
we believe reimbursement of bremelanotide from third-party payers
such as health insurers, HMOs or other third-party payers of
healthcare costs will be similar to reimbursement for ED drugs, and
that the ultimate user may pay a substantial part of the cost of
bremelanotide for HSDD. If the market opportunity for bremelanotide
is smaller than we anticipate, it may also be difficult for us to
find marketing partners and, as a result, we may be unable to
generate revenue and business from bremelanotide. If bremelanotide
for HSDD does not achieve adequate market acceptance at an
acceptable price point, our business, financial condition and
results of operations will be materially adversely
affected.
Even if our product candidates receive regulatory approval in the
United States, we may never receive approval or commercialize our
products outside of the United States.
In
order to market any products outside of the United States, we must
establish and comply with numerous and varying regulatory
requirements of other countries regarding safety and efficacy.
Approval procedures vary among countries and can involve additional
product testing and additional administrative review periods. The
time required to obtain approval in other countries might differ
from that required to obtain FDA approval. The regulatory approval
process in other countries may include all of the risks detailed
above regarding FDA approval in the United States as well as other
risks. Regulatory approval in one country does not ensure
regulatory approval in another, but a failure or delay in obtaining
regulatory approval in one country may have a negative effect on
the regulatory process in others. Failure to obtain regulatory
approval in other countries or any delay or setbacks in obtaining
such approval would impair our ability to develop foreign markets
for our product candidates and may have a material adverse effect
on our results of operations and financial condition.
22
If side effects emerge that can be linked to our product candidates
(either while they are in development or after they are approved
and on the market), we may be required to perform lengthy
additional clinical trials, change the labeling of any such
products, or withdraw such products from the market, any of which
would hinder or preclude our ability to generate
revenues.
If we
identify side effects or other problems occur in future clinical
trials, we may be required to terminate or delay clinical
development of the product candidate. Furthermore, even if any of
our product candidates receive marketing approval, as greater
numbers of patients use a drug following its approval, if the
incidence of side effects increases or if other problems are
observed after approval that were not seen or anticipated during
pre-approval clinical trials, a number of potentially significant
negative consequences could result, including:
●
regulatory
authorities may withdraw their approval of the
product;
●
we may be required
to reformulate such products or change the way the product is
manufactured;
●
we may become the
target of lawsuits, including class action suits; and
●
our reputation in
the market place may suffer resulting in a significant drop in the
sales of such products.
Any of
these events could substantially increase the costs and expenses of
developing, commercializing and marketing any such product
candidates or could harm or prevent sales of any approved
products.
The number of subjects in our study pools in our clinical trials
may be deemed by regulators to be too small, which could cause
unanticipated delays or higher than anticipated costs.
Our
clinical trials have been conducted on a pool of subjects that is
structured for such research. Nevertheless, there is the
possibility that for statistical reasons, the pool of subjects may
be determined by the FDA or another regulatory body to be too small
to verify statistical significance. In such a case, the conclusions
from the previous trials will need to be established with at least
another set of clinical trials testing the relevant issue. Due to
the need to find new subjects for any additional clinical trials
and the limited pool from which such subjects can be selected, any
such determination by the FDA could result in a delay in obtaining
FDA approval or require additional financial
expenditures.
We may not be able to keep up with the rapid technological change
in the biotechnology and pharmaceutical industries, which could
make any future approved products obsolete and reduce our
revenue.
Biotechnology
and related pharmaceutical technologies have undergone and continue
to be subject to rapid and significant change. Our future will
depend in large part on our ability to maintain a competitive
position with respect to these technologies. Our competitors may
render our technologies obsolete by advances in existing
technological approaches or the development of new or different
approaches, potentially eliminating the advantages in our drug
discovery process that we believe we derive from our research
approach and proprietary technologies. In addition, any future
products that we develop, including our clinical product
candidates, may become obsolete before we recover expenses incurred
in developing those products, which may require that we raise
additional funds to continue our operations.
Competing products and technologies may make our proposed products
noncompetitive.
Flibanserin,
a daily-use oral drug sold under the trade name Addyi®, has
been approved by the FDA for HSDD in premenopausal women. There are
other products being developed for HSDD and other FSD indications,
including a number of oral combination drugs, some of which
incorporate testosterone, antidepressants or PDE-5 inhibitors.
There is competition to develop drugs for treatment of HSDD and FSD
in both premenopausal and postmenopausal patients. Our
bremelanotide drug product is intended to be administered by
subcutaneous injection, and an as needed drug product for the same
indication which utilizes another route of administration, such as
a conventional oral drug product, may make subcutaneous
bremelanotide noncompetitive.
There
are several products approved for use in treatment of obesity and
related indications, and a number of other products being developed
for treatment of obesity, including products in clinical trials.
There is intense competition to develop drugs for treatment of
obesity and related indications.
There
are a number of products approved for use in treating inflammatory
diseases and indications, and other products are being developed,
including products in clinical trials.
We are
aware of one recombinant natriuretic peptide product for acutely
decompensated congestive heart failure approved and marketed in the
United States, and another recombinant natriuretic peptide product
approved and marketed in Japan. Clinical trials on other
natriuretic peptide products are being conducted in the United
States. In addition, other products for treatment of heart failure
are either currently being marketed or in development, including a
combination drug which increases active levels of ANP.
23
As
discussed above, the biopharmaceutical industry is highly
competitive. We are likely to encounter significant competition
with respect to bremelanotide, other melanocortin receptor agonist
compounds and PL-3994. Most of our competitors have substantially
greater financial and technological resources than we do. Many of
them also have significantly greater experience in research and
development, marketing, distribution and sales than we do.
Accordingly, our competitors may succeed in developing, marketing,
distributing and selling products and underlying technologies more
rapidly than we can. These competitive products or technologies may
be more effective and useful or less costly than bremelanotide,
other melanocortin receptor agonist compounds or PL-3994. In
addition, academic institutions, hospitals, governmental agencies
and other public and private research organizations are also
conducting research and may develop competing products or
technologies on their own or through strategic alliances or
collaborative arrangements.
We rely on third parties over whom we have no control to conduct
preclinical studies, clinical trials and other research for our
product candidates and their failure to timely perform their
obligations could significantly harm our product
development.
We have
limited research and development staff and do not have dedicated
research or development facilities. We rely on third parties and
independent contractors, such as researchers at CROs and
universities, in certain areas that are particularly relevant to
our research and product development plans. We engage such
researchers to conduct our preclinical studies, clinical trials and
associated tests. These outside contractors are not our employees
and may terminate their engagements with us at any time. In
addition, we have limited control over the resources that these
contractors devote to our programs and they may not assign as great
a priority to our programs or pursue them as diligently as we would
if we were undertaking such programs ourselves. There is also
competition for these relationships, and we may not be able to
maintain our relationships with our contractors on acceptable
terms. If our third-party contractors do not carry out their duties
under their agreements with us, fail to meet expected deadlines or
fail to comply with appropriate standards for preclinical or
clinical research, our ability to develop our product candidates
and obtain regulatory approval on a timely basis, if at all, may be
materially adversely affected.
Production and supply of our product candidates depend on contract
manufacturers over whom we have no control, with the risk that we
may not have adequate supplies of our product candidates or
products.
We do
not have the facilities to manufacture our early stage potential
products such as PL-3994, PL-8177 and other melanocortin receptor
agonist compounds for use in preclinical studies and clinical
trials. Contract manufacturers must perform these manufacturing
activities in a manner that complies with FDA regulations. Our
ability to control third-party compliance with FDA requirements is
limited to contractual remedies and rights of inspection. The
manufacturers of our potential products and their manufacturing
facilities will be subject to continual review and periodic
inspections by the FDA and other authorities where applicable, and
must comply with ongoing regulatory requirements, including FDA
regulations concerning GMP. Failure of third-party manufacturers to
comply with GMP, medical device QSR, or other FDA requirements may
result in enforcement action by the FDA. Failure to conduct their
activities in compliance with FDA regulations could delay our
development programs or negatively impact our ability to receive
FDA approval of our potential products. Establishing relationships
with new suppliers, who must be FDA-approved, is a time-consuming
and costly process.
If we are unable to establish sales and marketing capabilities
within our organization or enter into and maintain agreements with
third parties to market and sell our product candidates, we may be
unable to generate product revenue.
We do
not currently have any experience in sales, marketing and
distribution of pharmaceutical products. We will need to establish
sales and marketing capabilities within our organization or
establish and maintain agreements with third parties to market and
sell our product candidates. We do not have marketing partners for
any of our products, including bremelanotide and PL-3994. If any of
these products are approved by the FDA or other regulatory
authorities, we must either develop marketing, distribution and
selling capacity and expertise, which will be costly and time
consuming, or enter into agreements with other companies to provide
these capabilities. We may not be able to enter into suitable
agreements on acceptable terms, if at all. Engaging a third party
to perform these services could delay the commercialization of any
of our product candidates, if approved for commercial sale. If we
are unable to establish adequate sales, marketing and distribution
capabilities, whether independently or with third parties, we may
not be able to generate product revenue and our business would
suffer. In addition, if we enter into arrangements with third
parties to perform sales, marketing and distribution services, our
product revenues are likely to be lower than if we could market and
sell any products that we develop ourselves.
We will need to hire additional employees in order to commercialize
our product candidates in the future. Any inability to manage
future growth could harm our ability to commercialize our product
candidates, increase our costs and adversely impact our ability to
compete effectively.
In
order to commercialize our product candidates, we will need to hire
experienced sales and marketing personnel to sell and market those
product candidates we decide to commercialize, and we will need to
expand the number of our managerial, operational, financial and
other employees to support commercialization. Competition exists
for qualified personnel in the biopharmaceutical
field.
24
Future
growth will impose significant added responsibilities on members of
management, including the need to identify, recruit, maintain and
integrate additional employees. Our future financial performance
and our ability to commercialize our product candidates and to
compete effectively will depend, in part, on our ability to manage
any future growth effectively.
Our ability to achieve revenues from the sale of our products in
development will depend, in part, on our ability to obtain adequate
reimbursement from Medicare, Medicaid, private insurers and other
healthcare payers.
Our
ability to successfully commercialize our products in development
will depend, in significant part, on the extent to which we or our
marketing partners can obtain reimbursement for our products and
also reimbursement at appropriate levels for the cost of our
products. Obtaining reimbursement from governmental payers,
insurance companies, HMOs and other third-party payers of
healthcare costs is a time-consuming and expensive process. There
is no guarantee that our products will ultimately be reimbursed.
There is significant uncertainty concerning third-party
reimbursement for the use of any pharmaceutical product
incorporating new technology and third-party reimbursement might
not be available for our proposed products once approved, or if
obtained, might not be adequate.
There
is significant uncertainty concerning the extent and scope of
third-party reimbursement for products treating HSDD and other
forms of FSD. Based on third-party reimbursement for approved
products treating ED, we believe bremelanotide for HSDD will be
classified as a Tier 3 drug, so that reimbursement will be limited
for bremelanotide for treatment of premenopausal women with HSDD,
assuming the product is approved by the FDA. If we are able to
obtain reimbursement, continuing efforts by governmental and
third-party payers to contain or reduce costs of healthcare may
adversely affect our future revenues and ability to achieve
profitability. Third-party payers are increasingly challenging the
prices charged for diagnostic and therapeutic products and related
services. Reimbursement from governmental payers is subject to
statutory and regulatory changes, retroactive rate adjustments,
administrative rulings and other policy changes, all of which could
materially decrease the range of products for which we are
reimbursed or the rates of reimbursement by government payers. In
addition, recent legislation reforming the healthcare system may
result in lower prices or the actual inability of prospective
customers to purchase our products in development. The cost
containment measures that healthcare payers and providers are
instituting and the effect of any healthcare reform could
materially and adversely affect our ability to operate profitably.
Furthermore, even if reimbursement is available, it may not be
available at price levels sufficient for us to realize a positive
return on our investment, which would have a material adverse
effect on our business, financial condition and results of
operations.
Even if we receive regulatory approval for our products in Europe,
we may not be able to secure adequate pricing and reimbursement in
Europe for us or any strategic partner to achieve
profitability.
Even if
one or more of our products are approved in Europe, we may be
unable to obtain appropriate pricing and reimbursement for such
products. In most European markets, demand levels for healthcare in
general and for pharmaceuticals in particular are principally
regulated by national governments. Therefore, pricing and
reimbursement for our products will have to be negotiated on a
“Member State by Member State” basis according to
national rules, as there does not exist a centralized European
process. As each Member State has its own national rules governing
pricing control and reimbursement policy for pharmaceuticals, there
are likely to be uncertainties attaching to the review process, and
the level of reimbursement that national governments are prepared
to accept. In the current economic environment, governments and
private payers or insurers are increasingly looking to contain
healthcare costs, including costs on drug therapies. If we are
unable to obtain adequate pricing and reimbursement for our
products in Europe, we or a potential strategic partner or
collaborator may not be able to cover the costs necessary to
manufacture, market and sell the product, limiting or preventing
our ability to achieve profitability.
We may incur substantial liabilities and may be required to limit
commercialization of our products in response to product liability
lawsuits.
The
testing and marketing of medical products entails an inherent risk
of product liability. If we cannot successfully defend ourselves
against product liability claims, we may incur substantial
liabilities or be required to limit commercialization of our
products or cease clinical trials. Our inability to obtain
sufficient product liability insurance at an acceptable cost to
protect against potential product liability claims could prevent or
inhibit the commercialization of pharmaceutical products we
develop, alone or with corporate collaborators. We currently carry
$10 million liability insurance in the aggregate as to certain
clinical trial risks, but we do not have and have not obtained
quotations for commercial product liability insurance. We, or any
corporate collaborators, may not in the future be able to obtain
insurance at a reasonable cost or in sufficient amounts, if at all.
Even if our agreements with any future corporate collaborators
entitle us to indemnification against losses, such indemnification
may not be available or adequate should any claim
arise.
25
Our internal computer systems, or those of our third-party
contractors or consultants, may fail or suffer security breaches,
which could result in a material disruption of our product
development programs.
Despite
the implementation of security measures, our internal computer
systems and those of our third-party contractors and consultants
are vulnerable to damage from computer viruses, unauthorized
access, natural disasters, terrorism, war and telecommunication and
electrical failures. We rely on industry accepted measures and
technology to secure confidential and proprietary information
maintained on our computer systems. However, these measures and
technology may not adequately prevent security breaches. While we
do not believe that we have experienced any such system failure,
accident, or security breach to date, if such an event were to
occur and cause interruptions in our operations, it could result in
a loss of clinical trial data for our product candidates which
could result in delays in our regulatory approval efforts and
significantly increase our costs to recover or reproduce the data.
To the extent that any disruption or security breach results in a
loss of or damage to our data or applications or other data or
applications relating to our technology, intellectual property,
research and development or product candidates, or inappropriate
disclosure of confidential or proprietary information, we could
incur liabilities and the further development of our product
candidates could be delayed.
We may be subject, directly or indirectly, to federal and state
healthcare fraud and abuse laws, false claims laws, and health
information privacy and security laws. If we are unable to comply,
or have not fully complied, with such laws, we could face
substantial penalties.
If we
begin commercializing any of our products in the United States, our
operations may be directly, or indirectly through our customers,
subject to various federal and state fraud and abuse laws,
including, without limitation, the federal Anti-Kickback Statute,
the federal False Claims Act, and physician sunshine laws and
regulations. These laws may impact, among other things, our
proposed sales, marketing, and education programs. In addition, we
may be subject to patient privacy regulation by both the federal
government and the states in which we conduct our business. The
laws that may affect our ability to operate include:
●
the federal
Anti-Kickback Statute, which prohibits, among other things, persons
or entities from soliciting, receiving, offering or providing
remuneration, directly or indirectly, in return for or to induce
either the referral of an individual for, or the purchase order or
recommendation of, any item or services for which payment may be
made under a federal health care program such as the Medicare and
Medicaid programs;
●
federal civil and
criminal false claims laws and civil monetary penalty laws, which
prohibit, among other things, individuals or entities from
knowingly presenting, or causing to be presented, claims for
payment from Medicare, Medicaid, or other third-party payors that
are false or fraudulent;
●
HIPAA, which
created new federal criminal statutes that prohibit executing a
scheme to defraud any healthcare benefit program and making false
statements relating to healthcare matters;
●
HIPPA, as amended
by the Health Information Technology and Clinical Health Act, and
its implementing regulations, which imposes certain requirements
relating to the privacy, security, and transmission of individually
identifiable health information;
●
The federal
physician sunshine requirements under the Affordable Care Act,
which require manufacturers of drugs, devices, biologics, and
medical supplies to report annually to the U.S. Department of
Health and Human Services information related to payments and other
transfers of value to physicians, other healthcare providers, and
teaching hospitals, and ownership and investment interests held by
physicians and other healthcare providers and their immediate
family members and applicable group purchasing organizations;
and
●
state law
equivalents of each of the above federal laws, such as
anti-kickback and false claims laws that may apply to items or
services reimbursed by any third-party payor, including commercial
insurers, state laws that require pharmaceutical companies to
comply with the pharmaceutical industry's voluntary compliance
guidelines and the relevant compliance guidance promulgated by the
federal government, or otherwise restrict payments that may be made
to healthcare providers and other potential referral sources; state
laws that require drug manufacturers to report information related
to payments and other transfers of value to physicians and other
healthcare providers or marketing expenditures; and state laws
governing the privacy and security of health information in certain
circumstances, many of which differ from each other in significant
ways and may not have the same effect, thus complicating compliance
efforts.
Because
of the breadth of these laws and the narrowness of the statutory
exceptions and safe harbors available, it is possible that some of
our business activities could be subject to challenge under one or
more of such laws. In addition, recent health care reform
legislation has strengthened these laws. For example, the
Affordable Care Act, among other things, amends the intent
requirement of the federal anti-kickback and criminal healthcare
fraud statutes. A person or entity no longer needs to have actual
knowledge of this statute or specific intent to violate it.
Moreover, the Affordable Care Act provides that the government may
assert that a claim including items or services resulting from a
violation of the federal anti-kickback statute constitutes a false
or fraudulent claim for purposes of the False Claims
Act.
26
If our
operations are found to be in violation of any of the laws
described above or any other governmental regulations that apply to
us, we may be subject to penalties, including civil and criminal
penalties, damages, fines, exclusion from participation in
government health care programs, such as Medicare and Medicaid,
imprisonment, and the curtailment or restructuring of our
operations, any of which could adversely affect our ability to
operate our business and our results of operations
We are highly dependent on our management team, senior staff
professionals and third-party contractors and consultants, and the
loss of their services could materially adversely affect our
business.
We rely
on our relatively small management team and staff as well as
various contractors and consultants to provide critical services.
Our ability to execute our clinical programs for bremelanotide and
PL-3994 and our preclinical programs for MC1r and MC4r peptide drug
candidates depends on our continued retention and motivation of our
management and senior staff professionals, including executive
officers and senior members of product development and management,
including commercialization, who possess significant technical
expertise and experience and oversee our development and
commercialization programs. If we lose the services of existing key
personnel, our development programs could be adversely affected if
suitable replacement personnel are not recruited quickly. Our
success also depends on our ability to develop and maintain
relationships with contractors, consultants and scientific
advisors.
There
is competition for qualified personnel, contractors and consultants
in the pharmaceutical industry, which makes it difficult to attract
and retain the qualified personnel, contractors and consultants
necessary for the development and growth of our business. Our
failure to attract and retain such personnel, contractors and
consultants could have a material adverse effect on our business,
results of operations and financial condition.
Because we expect bremelanotide for the treatment of HSDD to be
classified as a Tier 3 drug with reimbursement by third-party
payers similar to approved products for treating ED, demand for
this product will be tied to discretionary spending levels of our
targeted patient population and particularly affected by
unfavorable economic conditions.
The
market for HSDD may be particularly vulnerable to unfavorable
economic conditions. We expect bremelanotide for the treatment of
HSDD to have significant copay or deductible requirements and to be
only partially reimbursed by third-party payers and, as a result,
demand for this product may be tied to discretionary spending
levels of our targeted patient population. The recent global
financial crisis caused extreme volatility and disruptions in the
capital and credit markets. A severe or prolonged economic downturn
could result in a variety of risks to our business, including
weakened demand for bremelanotide for HSDD or any future product
candidates, if approved, and our ability to raise additional
capital when needed on acceptable terms, if at all. This is
particularly true in Europe, which is undergoing a continued severe
economic crisis. A weak or declining economy could also strain our
suppliers, possibly resulting in supply disruption, or cause our
customers to delay making payments for our services. Any of the
foregoing could harm our business and we cannot anticipate all of
the ways in which future economic climates and financial market
conditions could adversely impact our business.
Risks Related to Government Regulation
Both before and after marketing approval, our product candidates
are subject to ongoing regulatory requirements and, if we fail to
comply with these continuing requirements, we could be subject to a
variety of sanctions and the sale of any approved commercial
products could be suspended.
Both
before and after regulatory approval to market a particular product
candidate, the manufacturing, labeling, packaging, adverse event
reporting, storage, advertising and promotion and record keeping
related to the product candidates are subject to extensive
regulatory requirements. If we fail to comply with the regulatory
requirements of the FDA and other applicable U.S. and foreign
regulatory authorities, we could be subject to administrative or
judicially imposed sanctions, including:
●
restrictions on the
products or manufacturing process;
●
warning
letters;
●
civil or criminal
penalties;
●
fines;
●
injunctions;
●
imposition of a
Corporate Integrity Agreement requiring heightened monitoring of
our compliance functions, overseen by outside monitors, and
enhanced reporting requirements to, and oversight by, the FDA and
other government agencies;
●
product seizures or
detentions and related publicity requirements;
●
suspension or
withdrawal of regulatory approvals;
27
●
regulators or IRBs
may not authorize us or any potential future collaborators to
commence a clinical trial or conduct a clinical trial at a
prospective trial site;
●
total or partial
suspension of production; and
●
refusal to approve
pending applications for marketing approval of new product
candidates.
Changes
in the regulatory approval policy during the development period,
changes in or the enactment of additional regulations or statutes,
or changes in the regulatory review for each submitted product
application may cause delays in the approval or rejection of an
application. Even if the FDA approves a product candidate, the
approval may impose significant restrictions on the indicated uses,
conditions for use, labeling, advertising, promotion, marketing
and/or production of such product, and may impose ongoing
requirements for post-approval studies, including additional
research and development and clinical trials. The approval may also
impose REMS on a product if the FDA believes there is a reason to
monitor the safety of the drug in the marketplace. REMS may include
requirements for additional training for health care professionals,
safety communication efforts and limits on channels of
distribution, among other things. The sponsor would be required to
evaluate and monitor the various REMS activities and adjust them if
need be. The FDA also may impose various civil or criminal
sanctions for failure to comply with regulatory requirements,
including withdrawal of product approval.
Furthermore,
the approval procedure and the time required to obtain approval
varies among countries and can involve additional testing beyond
that required by the FDA. Approval by one regulatory authority does
not ensure approval by regulatory authorities in other
jurisdictions. The FDA has substantial discretion in the approval
process and may refuse to accept any application or may decide that
our data are insufficient for approval and require additional
preclinical, clinical or other studies.
In
addition, varying interpretations of the data obtained for
preclinical and clinical testing could delay, limit or prevent
regulatory approval of a product candidate. Even if we submit an
application to the FDA for marketing approval of a product
candidate, it may not result in marketing approval from the
FDA.
We do
not expect to receive regulatory approval for the commercial sale
of any of our product candidates that are in development in the
near future, if at all. The inability to obtain FDA approval or
approval from comparable authorities in other countries for our
product candidates would prevent us or any potential future
collaborators from commercializing these product candidates in the
United States or other countries.
We may not be able to obtain regulatory approval of bremelanotide
for HSDD even if the product is effective in treating
HSDD.
Clinical
drug development programs for our product candidates are very
expensive, time-consuming, difficult to design and implement and
their outcome is inherently uncertain. Approval of bremelanotide
for treatment of HSDD in premenopausal women requires a
determination by the FDA that the product is both safe and
effective. Our Phase 3 clinical trials for HSDD demonstrated what
we believe to be an acceptable safety profile and statistically
significant efficacy. However, the FDA may ultimately disagree with
our safety analysis, definition of efficacy in HSDD, our clinical
trial designs, or our interpretation of our clinical trial results.
It is not possible to predict, with any assurance, whether the FDA
will approve bremelanotide for any indications. The FDA may deny or
delay approval of any application for bremelanotide if the FDA
determines that the clinical data do not adequately establish the
safety of the drug even if efficacy is established. If FDA approves
bremelanotide, the approved labeling of the product may be limited
or restricted in such ways as to inhibit or prevent the successful
market acceptance and profitability of the product. Bremelanotide
could take a significantly longer time to obtain approval than we
expect and it may never gain approval. If regulatory approval of
bremelanotide is delayed, limited or never obtained, our business,
financial condition and results of operations would be materially
adversely affected.
The regulatory approval process is lengthy, expensive and
uncertain, and may prevent us from obtaining the approvals that we
require.
Government
authorities in the United States and other countries extensively
regulate the advertising, labeling, storage, record-keeping,
safety, efficacy, research, development, testing, manufacture,
promotion, marketing and distribution of drug products. Drugs are
subject to rigorous regulation in the United States by the FDA and
similar regulatory bodies in other countries. The steps ordinarily
required by the FDA before a new drug may be marketed in the United
States include:
●
completion of
non-clinical tests including preclinical laboratory and formulation
studies and animal testing and toxicology;
●
submission to the
FDA of an IND application, which must become effective before
clinical trials may begin, and which may be placed on
“clinical hold” by the FDA, meaning the trial may not
commence, or must be suspended or terminated prior to
completion;
28
●
performance of
adequate and well-controlled Phase 1, 2 and 3 human clinical trials
to establish the safety and efficacy of the drug for each proposed
indication, and potentially post-approval or Phase 4 studies to
further define the drug’s efficacy and safety, generally or
in specific patient populations;
●
submission to the
FDA of an NDA that must be accompanied by a substantial “user
fee” payment;
●
FDA review and
approval of the NDA before any commercial marketing or sale;
and
●
compliance with
post-approval commitments and requirements.
Satisfaction
of FDA pre-market approval requirements for new drugs typically
takes a number of years and the actual time required for approval
may vary substantially based upon the type, complexity and novelty
of the product or disease to be treated by the drug. The results of
product development, preclinical studies and clinical trials are
submitted to the FDA as part of an NDA. The NDA also must contain
extensive manufacturing information, demonstrating compliance with
applicable GMP requirements. Once the submission has been accepted
for filing, the FDA generally has twelve months to review the
application and respond to the applicant. Such response may be an
approval, or may be a “complete response letter”
outlining additional data or steps that must be completed prior to
further FDA review of the NDA. The review process is often
significantly extended by FDA requests for additional information
or clarification. Success in early stage clinical trials does not
assure success in later stage clinical trials. Data obtained from
clinical trials is not always conclusive and may be susceptible to
varying interpretations that could delay, limit or prevent
regulatory approval. The FDA may refer the NDA to an advisory
committee for review, evaluation and recommendation as to whether
the application should be approved, but the FDA is not bound by the
recommendation of the advisory committee. The FDA may deny or delay
approval of applications that do not meet applicable regulatory
criteria or if the FDA determines that the clinical data do not
adequately establish the safety and efficacy of the drug.
Therefore, our proposed products could take a significantly longer
time than we expect or may never gain approval. If regulatory
approval is delayed or never obtained, our business, financial
condition and results of operations would be materially adversely
affected.
Some of
our products or product candidates, including bremelanotide, may be
used in combination with a drug delivery device, such as an
injector or other delivery system. Medical products containing a
combination of new drugs, biological products or medical devices
are regulated as “combination products” in the United
States. A combination product generally is defined as a product
comprised of components from two or more regulatory categories
(e.g., drug/device, device/biologic, drug/biologic). Each component
of a combination product is subject to the requirements established
by the FDA for that type of component, whether a new drug, biologic
or device. In order to facilitate pre-market review of combination
products, the FDA designates one of its centers to have primary
jurisdiction for the pre-market review and regulation of the
overall product based upon a determination by the FDA of the
primary mode of action of the combination product. The
determination whether a product is a combination product or two
separate products is made by the FDA on a case-by-case basis. Our
product candidates intended for use with such devices, or expanded
indications that we may seek for our products used with such
devices, may not be approved or may be substantially delayed in
receiving approval if the devices do not gain and/or maintain their
own regulatory approvals or clearances. Where approval of the drug
product and device is sought under a single application, the
increased complexity of the review process may delay approval. In
addition, because these drug delivery devices are provided by
single source unaffiliated third-party companies, we are dependent
on the sustained cooperation and effort of those third-party
companies both to supply the devices, maintain their own regulatory
compliance, and, in some cases, to conduct the studies required for
approval or other regulatory clearance of the devices. We are also
dependent on those third-party companies continuing to maintain
such approvals or clearances once they have been received. Failure
of third-party companies to supply the devices, to successfully
complete studies on the devices in a timely manner, or to obtain or
maintain required approvals or clearances of the devices, and
maintain compliance with all regulatory requirements, could result
in increased development costs, delays in or failure to obtain
regulatory approval and delays in product candidates reaching the
market or in gaining approval or clearance for expanded labels for
new indications.
Upon
approval, a product candidate may be marketed only in those dosage
forms and for those indications approved by the FDA. Once approved,
the FDA may withdraw the product approval if compliance with
regulatory requirements is not maintained or if problems occur
after the product reaches the marketplace. In addition, the FDA may
require post-marketing studies, referred to as Phase 4 studies, to
monitor the approved products in a specific subset of patients or a
larger number of patients than were required for product approval
and may limit further marketing of the product based on the results
of these post-market studies. The FDA has broad post-market
regulatory and enforcement powers, including the ability to seek
injunctions, levy fines and civil penalties, criminal prosecution,
withdraw approvals and seize products or request
recalls.
29
If
regulatory approval of any of our product candidates is granted, it
will be limited to certain disease states or conditions, patient
populations, duration or frequency of use, and will be subject to
other conditions as set forth in the FDA-approved labeling. Adverse
experiences with the product must be reported to the FDA and could
result in the imposition of market restriction through labeling
changes or in product removal. Product approvals may be withdrawn
if compliance with regulatory requirements is not maintained or if
problems concerning safety or efficacy of the product occur
following approval.
Outside
the United States, our ability to market our product candidates
will also depend on receiving marketing authorizations from the
appropriate regulatory authorities. The foreign regulatory approval
process generally includes all of the risks associated with FDA
approval described above. The requirements governing the conduct of
clinical trials and marketing authorization vary widely from
country to country. At present, foreign marketing authorizations
are applied for at a national level, although within the European
Community (“EC”), registration procedures are available
to companies wishing to market a product to more than one EC member
state. If the regulatory authority is satisfied that adequate
evidence of safety, quality and efficiency has been presented, a
marketing authorization will be granted. If we do not obtain, or
experience difficulties in obtaining, such marketing
authorizations, our business, financial condition and results of
operations may be materially adversely affected.
Legislative or regulatory healthcare reforms in the United States
may make it more difficult and costly for us to obtain regulatory
clearance or approval of bremelanotide for HSDD or any future
product candidates and to produce, market and distribute our
products after clearance or approval is obtained.
From
time to time, legislation is drafted and introduced in Congress,
and court decisions are issued, that could significantly change the
statutory provisions governing the regulatory clearance or
approval, manufacture and marketing of regulated products or the
reimbursement thereof. In addition, FDA regulations and guidance
are often revised or reinterpreted by the FDA in ways that may
significantly affect our business and our products. Any new
regulations or revisions or reinterpretations of existing
regulations may impose additional costs or lengthen review times of
bremelanotide for HSDD or any future product candidates. We cannot
determine what effect changes in regulations, statutes, court
decisions, legal interpretation or policies, when and if
promulgated, enacted, issued or adopted may have on our business in
the future. Such changes could, among other things:
●
require changes to
manufacturing methods;
●
require recall,
replacement or discontinuance of one or more of our
products;
●
require additional
recordkeeping;
●
limit or restrict
our ability to engage in certain types of marketing or promotional
activities;
●
alter or eliminate
the scope or terms of any currently available regulatory
exclusivities; and
●
restrict or
eliminate our ability to settle any patent litigation we may bring
against potential generic competitors.
Each of
these would likely entail substantial time and cost and could
materially harm our business and our financial results. In
addition, delays in receipt of or failure to receive regulatory
clearances or approvals for any future products would harm our
business, financial condition and results of
operations.
Changes in healthcare policy could adversely affect our
business.
U.S.
and foreign governments continue to propose and pass legislation
designed to reduce the cost of healthcare. For example, the
Medicare Prescription Drug Improvement and Modernization Act of
2003 (“MMA”), expanded Medicare coverage for drugs
purchased by Medicare beneficiaries and introduced new
reimbursement methodologies. In addition, this law provided
authority for limiting the number of drugs that will be covered in
any therapeutic class. We do not know what impact the MMA and
similar laws will have on the availability of coverage for and the
price that we receive for any approved products. Moreover, while
the MMA applies only to drug benefits for Medicare beneficiaries,
private payers often follow Medicare policies in setting their own
reimbursement policies, and any reduction in reimbursement that
results from the MMA may result in similar reductions by private
payers.
The
Affordable Care Act, as amended by the Health Care and Education
Affordability Reconciliation Act (together the “ACA”),
was adopted in 2010. This law has resulted in an increase in the
number of people who are covered by both public and private
insurance and has changed the way health care is financed by both
government health program and private insurers, with significant
impacts on the pharmaceutical industry. The ACA contains a number
of provisions that may impact our business and operations in ways
that may negatively affect our potential revenues in the future.
For example, the ACA imposes a non-deductible excise tax on
pharmaceutical manufacturers or importers that sell branded
prescription drugs to U.S. government programs that we believe will
increase the cost of any products that we develop. In addition, as
part of the ACA’s provisions closing a funding gap that
currently exists in the Medicare Part D prescription drug program
(commonly known as the “donut hole”), we will be
required to provide a 50% discount on any branded prescription
drugs that we develop sold to beneficiaries who fall within the
donut hole. We cannot predict all of the specific effects the ACA
or any future healthcare reform legislation will have on our
business, but they could have a material adverse effect on our
business and financial condition.
30
Efforts
have been made to repeal, or repeal and replace, the ACA. It is not
known whether the ACA will be repealed, amended, replaced or
otherwise modified within the next several years. It is possible
that any repeal, amendment or replacement of the ACA will decrease
the number of people who are covered by both public and private
insurance and change the way health care is financed by both
government health programs and private insurers, which could
significantly impact the pharmaceutical industry.
The
availability of government reimbursement for prescription drugs
will be impacted by the Budget Control Act of 2011, which was
signed into law on August 2, 2011. This law is expected to result
in federal spending cuts totaling between $1.2 trillion and $1.5
trillion over the next decade, over half of which will include cuts
in Medicare and other health-related spending.
Risks Related to Our Intellectual Property
If we fail to adequately protect or enforce our intellectual
property rights or secure rights to patents of others, the value of
our intellectual property rights would diminish.
Our
success, competitive position and future revenues will depend in
part on our ability and the abilities of our licensors to obtain
and maintain patent protection for our products, methods, processes
and other technologies, to preserve our trade secrets, to prevent
third parties from infringing on our proprietary rights and to
operate without infringing the proprietary rights of third parties.
We cannot predict:
●
the degree and
range of protection any patents will afford us against competitors,
including whether third parties will find ways to invalidate or
otherwise circumvent our patents;
●
if and when patents
will be issued;
●
whether or not
others will obtain patents claiming aspects similar to those
covered by our patents and patent applications; and
●
whether we will
need to initiate litigation or administrative proceedings, which
may be costly whether we win or lose.
If our
products, methods, processes and other technologies infringe the
proprietary rights of other parties we could incur substantial
costs and we may have to:
●
obtain licenses,
which may not be available on commercially reasonable terms, if at
all;
●
redesign our
products or processes to avoid infringement;
●
stop using the
subject matter claimed in the patents held by others;
●
pay damages;
or
●
defend litigation
or administrative proceedings, which may be costly whether we win
or lose, and which could result in a substantial diversion of our
management resources.
We may become involved in lawsuits to protect or enforce our
patents or other intellectual property or the patents of our
licensors, which could be expensive and time
consuming.
Competitors
may infringe our intellectual property, including our patents or
the patents of our licensors. As a result, we may be required to
file infringement claims to stop third-party infringement or
unauthorized use. This can be expensive, particularly for a company
of our size, and time-consuming. In addition, in an infringement
proceeding, a court may decide that a patent of ours is not valid
or is unenforceable, or may refuse to stop the other party from
using the technology at issue on the grounds that our patent claims
do not cover its technology or that the factors necessary to grant
an injunction against an infringer are not satisfied.
An
adverse determination of any litigation or other proceedings could
put one or more of our patents at risk of being invalidated or
interpreted narrowly and could put our patent applications at risk
of not issuing.
Interference,
derivation or other proceedings brought at the USPTO may be
necessary to determine the priority or patentability of inventions
with respect to our patent applications or those of our licensors
or collaborators. Litigation or USPTO proceedings brought by us may
fail or may be invoked against us by third parties. Even if we are
successful, domestic or foreign litigation or USPTO or foreign
patent office proceedings may result in substantial costs and
distraction to our management. We may not be able, alone or with
our licensors or collaborators, to prevent misappropriation of our
proprietary rights, particularly in countries where the laws may
not protect such rights as fully as in the United
States.
Furthermore,
because of the substantial amount of discovery required in
connection with intellectual property litigation or other
proceedings, there is a risk that some of our confidential
information could be compromised by disclosure during this type of
litigation or proceedings. In addition, during the course of this
kind of litigation or proceedings, there could be public
announcements of the results of hearings, motions or other interim
proceedings or developments or public access to related documents.
If investors perceive these results to be negative, the market
price for our common stock could be significantly
harmed.
31
If we infringe or are alleged to infringe intellectual property
rights of third parties, our business could be harmed.
Our
research, development and commercialization activities may infringe
or otherwise violate or be claimed to infringe or otherwise violate
patents owned or controlled by other parties. There may also be
patent applications that have been filed but not published that,
when issued as patents, could be asserted against us. These third
parties could bring claims against us that would cause us to incur
substantial expenses and, if successful against us, could cause us
to pay substantial damages. Further, if a patent infringement suit
were brought against us, we could be forced to stop or delay
research, development, manufacturing or sales of the product or
product candidate that is the subject of the suit.
As a
result of patent infringement claims, or to avoid potential claims,
we may choose or be required to seek licenses from third parties.
These licenses may not be available on acceptable terms, or at all.
Even if we are able to obtain a license, the license would likely
obligate us to pay license fees or royalties or both, and the
rights granted to us might be nonexclusive, which could result in
our competitors gaining access to the same intellectual property.
Ultimately, we could be prevented from commercializing a product,
or be forced to cease some aspect of our business operations, if,
as a result of actual or threatened patent infringement claims, we
are unable to enter into licenses on acceptable terms, if at
all.
There
has been substantial litigation and other proceedings regarding
patent and other intellectual property rights in the pharmaceutical
industry. In addition to infringement claims against us, we may
become a party to other patent litigation and other proceedings,
including interference, derivation or post-grant proceedings
declared or granted by the USPTO and similar proceedings in foreign
countries, regarding intellectual property rights with respect to
our current or future products. The cost to us of any patent
litigation or other proceeding, even if resolved in our favor,
could be substantial. Some of our competitors may be able to
sustain the costs of such litigation or proceedings more
effectively than we can because of their substantially greater
financial resources. Patent litigation and other proceedings may
also absorb significant management time. Uncertainties resulting
from the initiation and continuation of patent litigation or other
proceedings could impair our ability to compete in the marketplace.
The occurrence of any of the foregoing could have a material
adverse effect on our business, financial condition or results of
operations.
We may not be able to protect our intellectual property rights
throughout the world.
Filing,
prosecuting and defending patents on product candidates in all
countries throughout the world would be prohibitively expensive,
and our intellectual property rights in some countries outside the
United States can be less extensive than those in the United
States. In addition, the laws of some foreign countries do not
protect intellectual property rights to the same extent as federal
and state laws in the United States and in some cases may even
force us to grant a compulsory license to competitors or other
third parties. Consequently, we may not be able to prevent third
parties from practicing our inventions in all countries outside the
United States, or from selling or importing products made using our
inventions in and into the United States or other jurisdictions.
Competitors may use our technologies in jurisdictions where we have
not obtained patent protection to develop their own products and
further, may export otherwise infringing products to territories
where we have patent protection, but enforcement is not as strong
as that in the United States. These products may compete with our
products and our patents or other intellectual property rights may
not be effective or sufficient to prevent them from
competing.
Many
companies have encountered significant problems in protecting and
defending intellectual property rights in foreign jurisdictions.
The legal systems of certain countries, particularly certain
developing countries, do not favor the enforcement of patents and
other intellectual property protection, particularly those relating
to biopharmaceuticals, which could make it difficult for us to stop
the infringement of our patents or marketing of competing products
in violation of our proprietary rights generally. Proceedings to
enforce our patent rights in foreign jurisdictions could result in
substantial costs and divert our efforts and attention from other
aspects of our business, could put our patents at risk of being
invalidated or interpreted narrowly and our patent applications at
risk of not issuing and could provoke third parties to assert
claims against us. We may not prevail in any lawsuits that we
initiate and the damages or other remedies awarded, if any, may not
be commercially meaningful. Accordingly, our efforts to enforce our
intellectual property rights around the world may be inadequate to
obtain a significant commercial advantage from the intellectual
property that we develop or license.
In
addition, our ability to protect and enforce our intellectual
property rights may be adversely affected by unforeseen changes in
domestic and foreign intellectual property laws.
32
If we are unable to keep our trade secrets confidential, our
technologies and other proprietary information may be used by
others to compete against us.
In
addition to our reliance on patents, we attempt to protect our
proprietary technologies and processes by relying on trade secret
laws and agreements with our employees and other persons who have
access to our proprietary information. These agreements and
arrangements may not provide meaningful protection for our
proprietary technologies and processes in the event of unauthorized
use or disclosure of such information, and may not provide an
adequate remedy in the event of unauthorized disclosure of
confidential information. In addition, our competitors may
independently develop substantially equivalent technologies and
processes or gain access to our trade secrets or technology, either
of which could materially and adversely affect our competitive
position.
Risks Related to Obligations in Our 2014 and 2015 Private
Placements
Under agreements relating to our 2014 and 2015 private placements,
we are required to allow purchasers in the 2014 and 2015 private
placements to participate in certain future equity and debt
financings, which may restrict our ability to raise funds on
acceptable terms, or at all.
For
four years after our 2014 and 2015 private placements, unless the
FDA earlier approves bremelanotide for HSDD, the purchasers have
the right of first negotiation on any subsequent equity or debt
financing. If we do not agree to terms of a financing with the
purchasers, depending on pricing of the financing, the purchasers
have the right to purchase between 83.5% and all of the financing.
We will require significant additional resources and capital for
our product development programs. The right of first negotiation
and right of participation granted to the purchasers in our 2014
and 2015 private placements may make it more difficult to raise
additional funding through public or private equity or debt
financings or other sources. If we are able to obtain additional
funding, such funding may not be available on acceptable
terms
Under agreements relating to our 2014 and 2015 private placements,
so long as any Series C 2014 and Series E 2015 warrants are
outstanding, we are required to redeem such warrants at the option
of the holders in the event of any takeover, change of control or
other fundamental transaction which we permit.
Under
the purchase agreements and forms of our Series C and Series E
warrants for our 2014 and 2015 private placements, if we permit,
make or allow a takeover, change of control or other fundamental
transaction, including any transfer of all or substantially all of
our properties or assets, then so long as any warrants remain
outstanding we are required, as elected by such warrant holders, to
pay such holders a warrant early termination price tied to the
greater of the then market price of our common stock or the amount
per share paid to any other person. The application of these
provisions could adversely affect our financial position and have
the effect of delaying or preventing a change of control or other
fundamental transaction, which could adversely affect the market
price of our common stock, and could make any potential acquisition
or change of control more costly.
Under agreements relating to our 2014 and 2015 private placements,
so long as any Series C 2014 and Series E 2015 warrants are
outstanding, we are required to oppose any takeover or change of
control that does not provide specified rights to holders of such
warrants.
Under
the purchase agreements and forms of our Series C and Series E
warrants for our 2014 and 2015 private placements, so long as any
such warrants remain outstanding we are required to (i) not permit,
(ii) take necessary action to prevent both the occurrence or
consummation of, and (iii) not be a party to any fundamental
transaction, change of control or similar event unless
contractually-specified rights are provided with respect to payment
of any such warrant early termination price tied to the greater of
the then market price of our common stock or the amount per share
paid to any other person.
We are
also required, subject to the exercise by our board of its
fiduciary duties, to take all reasonable efforts to adopt a poison
pill or any other anti-takeover provision or method necessary to
prevent the fundamental transaction, change of control or similar
event. The application of these provisions could have the effect of
delaying or preventing a change of control or other fundamental
transaction, which could adversely affect the market price of our
common stock, and could make any potential acquisition or change of
control more costly.
Risks Related to the Ownership of Our Common Stock
Our stock price is volatile and may fluctuate in a way that is
disproportionate to our operating performance and we expect it to
remain volatile, which could limit investors’ ability to sell
stock at a profit.
The
volatile price of our stock makes it difficult for investors to
predict the value of their investment, to sell shares at a profit
at any given time or to plan purchases and sales in advance. A
variety of factors may affect the market price of our common stock.
These include, but are not limited to:
●
publicity regarding
actual or potential clinical results relating to products under
development by our competitors or us;
33
●
delay or failure in
initiating, completing or analyzing preclinical or clinical trials
or unsatisfactory designs or results of these trials;
●
interim decisions
by regulatory agencies, including the FDA, as to clinical trial
designs, acceptable safety profiles and the benefit/risk ratio of
products under development;
●
achievement or
rejection of regulatory approvals by our competitors or by
us;
●
announcements of
technological innovations or new commercial products by our
competitors or by us;
●
developments
concerning proprietary rights, including patents;
●
developments
concerning our collaborations;
●
regulatory
developments in the United States and foreign
countries;
●
economic or other
crises and other external factors;
●
period-to-period
fluctuations in our revenue and other results of
operations;
●
changes in the
structure of healthcare payment systems or other actions that
affect the effective reimbursement rates for treatment regimens
containing our products;
●
changes in
financial estimates and recommendations by securities analysts
following our business or our industry;
●
sales of our common
stock (or the perception that such sales could occur);
and
●
the other factors
described in this “Risk Factors” section.
We will
not be able to control many of these factors, and we believe that
period-to-period comparisons of our financial results will not
necessarily be indicative of our future performance. If our
revenues, if any, in any particular period do not meet
expectations, we may not be able to adjust our expenditures in that
period, which could cause our operating results to suffer further.
If our operating results in any future period fall below the
expectations of securities analysts or investors, our stock price
may fall by a significant amount.
For the
12-month period ended June 30, 2017, the price of our stock has
been volatile, ranging from a high of $0.90 per share to a low of
$0.29 per share. In addition, the stock market in general, and the
market for biotechnology companies in particular, has experienced
extreme price and volume fluctuations that may have been unrelated
or disproportionate to the operating performance of individual
companies. These broad market and industry factors may seriously
harm the market price of our common stock, regardless of our
operating performance.
As a public company in the United States, we are subject to the
Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley”). We can
provide no assurance that we will, at all times, in the future be
able to report that our internal controls over financial reporting
are effective.
Companies
that file reports with the SEC, including us, are subject to the
requirements of Section 404 of Sarbanes-Oxley. Section 404 requires
management to establish and maintain a system of internal control
over financial reporting, and annual reports on Form 10-K filed
under the Exchange Act, must contain a report from management
assessing the effectiveness of a company’s internal control
over financial reporting. Ensuring that we have adequate internal
financial and accounting controls and procedures in place to
produce accurate financial statements on a timely basis will be a
costly and time-consuming effort that needs to be re-evaluated
frequently. Failure on our part to have effective internal
financial and accounting controls would cause our financial
reporting to be unreliable, could have a material adverse effect on
our business, operating results, and financial condition, and could
cause the trading price of our common stock to fall
dramatically.
If securities or industry analysts do not publish research or
publish unfavorable research about our business, our stock price
and trading volume could decline.
As a
smaller company, it may be difficult for us to attract or retain
the interest of equity research analysts. A lack of research
coverage may adversely affect the liquidity of and market price of
our common stock. We do not have any control of the equity research
analysts or the content and opinions included in their reports. The
price of our stock could decline if one or more equity research
analysts downgrade our stock or issue other unfavorable commentary
or research. If one or more equity research analysts ceases
coverage of our company, or fails to publish reports on us
regularly, demand for our stock could decrease, which in turn could
cause our stock price or trading volume to decline.
34
Holders of our preferred stock may have interests different from
our common stockholders.
We are
permitted under our certificate of incorporation to issue up to
10,000,000 shares of preferred stock. We can issue shares of our
preferred stock in one or more series and can set the terms of the
preferred stock without seeking any further approval from our
common stockholders. 4,030 shares of our Series A Preferred Stock
remain outstanding as of September 21, 2017. Each share of Series A
Preferred Stock is convertible at any time, at the option of the
holder, and such conversion could dilute the value of our common
stock to current stockholders and could adversely affect the market
price of our common stock. The conversion price decreases if we
sell common stock (or equivalents) for a price per share less than
the conversion price or less than the market price of the common
stock and is also subject to adjustment upon the occurrence of a
merger, reorganization, consolidation, reclassification, stock
dividend or stock split which results in an increase or decrease in
the number of shares of common stock outstanding. Upon (i)
liquidation, dissolution or winding up of the Company, whether
voluntary or involuntary, (ii) sale or other disposition of all or
substantially all of the assets of the Company, or (iii) any
consolidation, merger, combination, reorganization or other
transaction in which the Company is not the surviving entity or in
which the shares of common stock constituting in excess of 50% of
the voting power of the Company are exchanged for or changed into
other stock or securities, cash and/or any other property, after
payment or provision for payment of the debts and other liabilities
of the Company, the holders of Series A Preferred Stock will be
entitled to receive, pro rata and in preference to the holders of
any other capital stock, an amount per share equal to $100 plus
accrued but unpaid dividends, if any. Any preferred stock that we
issue may rank ahead of our common stock in terms of dividend
priority or liquidation premiums and may have greater voting rights
than our common stock.
Because we do not anticipate paying any cash dividends on our
common stock in the foreseeable future, capital appreciation, if
any, will be your sole source of gains.
We do
not anticipate paying any cash dividends in the foreseeable future
and intend to retain future earnings, if any, for the development
and expansion of our business. Our outstanding Series A Preferred
Stock, consisting of 4,030 shares on September 21, 2017, provides
that we may not pay a dividend or make any distribution to holders
of any class of stock unless we first pay a special dividend or
distribution of $100 per share to the holders of the Series A
Preferred Stock. In addition, the terms of existing or future
agreements may limit our ability to pay dividends. As a result,
capital appreciation, if any, of our common stock will be your sole
source of gain for the foreseeable future.
Anti-takeover provisions of Delaware law and our charter documents
may make potential acquisitions more difficult and could result in
the entrenchment of management.
We are
incorporated in Delaware. Anti-takeover provisions of Delaware law
and our charter documents may make a change in control or efforts
to remove management more difficult. Also, under Delaware law, our
board of directors may adopt additional anti-takeover measures.
Under Section 203 of the Delaware General Corporation Law, a
corporation may not engage in a business combination with an
“interested stockholder” for a period of three years
after the date of the transaction in which the person first becomes
an “interested stockholder,” unless the business
combination is approved in a prescribed manner.
We are
authorized to issue up to 300,000,000 shares of common stock. To
the extent that we sell or otherwise issue authorized but currently
unissued shares, this could have the effect of making it more
difficult for a third party to acquire a majority of our
outstanding voting stock.
Our
charter authorizes us to issue up to 10,000,000 shares of preferred
stock and to determine the terms of those shares of stock without
any further action by our stockholders. If we exercise this right,
it could be more difficult for a third party to acquire a majority
of our outstanding voting stock.
In
addition, our equity incentive plans generally permit us to
accelerate the vesting of options and other stock rights granted
under these plans in the event of a change of control. If we
accelerate the vesting of options or other stock rights, this
action could make an acquisition more costly.
The
application of these provisions could have the effect of delaying
or preventing a change of control, which could adversely affect the
market price of our common stock.
We are a smaller reporting company and the reduced disclosure
requirements applicable to smaller reporting companies may make our
Common Stock less attractive to investors.
We are
currently a “smaller reporting company” as defined in
the Exchange Act. Smaller reporting companies are able to provide
simplified executive compensation disclosures in their filings, are
exempt from the provisions of Section 404(b) of the Sarbanes-Oxley
Act requiring that an independent registered public accounting firm
provide an attestation report on the effectiveness of internal
control over financial reporting, and have certain other decreased
disclosure obligations in their SEC filings. We cannot predict
whether investors will find our common stock less attractive
because of our reliance on any of these exemptions. If some
investors find our common stock less attractive as a result, there
may be a less active trading market for our common stock and our
stock price may be more volatile.
35
As of September 21, 2017 there were 57,174,292 shares of common
stock underlying outstanding convertible preferred stock, options,
restricted stock units and warrants. Stockholders may experience
dilution from the conversion of preferred stock, exercise of
outstanding options and warrants and vesting of restricted stock
units.
As of
September 21, 2017, holders of our outstanding dilutive securities
had the right to acquire the following amounts of underlying common
stock:
●
60,592 shares
issuable on the conversion of immediately convertible Series A
Convertible preferred stock, subject to adjustment, for no further
consideration;
●
8,977,812 shares
issuable on the exercise of stock options, at exercise prices
ranging from $0.37 to $6.60 per share;
●
5,263,617 shares
issuable under restricted stock units which vest on dates between
December 8, 2017 and September 7, 2021, subject to the fulfillment
of service conditions; and
●
42,872,271 shares
issuable on the exercise of warrants at exercise prices ranging
from $0.01 to $0.91 per share, of which 16,472,418 are prefunded
warrants with an exercise price of $0.01.
If the
holders convert, exercise or receive these securities, or similar
dilutive securities we may issue in the future, stockholders may
experience dilution in the net book value of their common stock. In
addition, the sale or availability for sale of the underlying
shares in the marketplace could depress our stock price. We have
registered or agreed to register for resale substantially all of
the underlying shares listed above. Holders of registered
underlying shares could resell the shares immediately upon
issuance, which could result in significant downward pressure on
our stock price.
Our failure to meet the continued listing requirements of the NYSE
MKT could result in a de-listing of our common stock.
Our
common shares are listed on the NYSE MKT, a national securities
exchange, under the symbol “PTN”. Although we currently
meet the NYSE MKT’s listing standards, which generally
mandate that we meet certain requirements relating to
stockholders’ equity, market capitalization, aggregate market
value of publicly held shares and distribution requirements, we
cannot assure you that we will be able to continue to meet the NYSE
MKT’s listing requirements. If we fail to satisfy the
continued listing requirements of the NYSE MKT, such as the
corporate governance requirements or the minimum closing bid price
requirement, the NYSE MKT may take steps to de-list our common
stock. If the NYSE MKT delists our securities for trading on its
exchange, we could face significant material adverse consequences,
including:
●
a limited
availability of market quotations for our securities;
●
reduced liquidity
with respect to our securities;
●
a determination
that our shares of common stock are “penny stock” which
will require brokers trading in our shares of common stock to
adhere to more stringent rules, possibly resulting in a reduced
level of trading activity in the secondary trading market for our
shares of common stock;
●
a limited amount of
news and analyst coverage for our company; and
●
a decreased ability
to issue additional securities or obtain additional financing in
the future.
Such a
de-listing would likely have a negative effect on the price of our
common stock and would impair your ability to sell or purchase our
common stock when you wish to do so. In the event of a de-listing,
we may take actions to restore our compliance with the NYSE
MKT’s listing requirements, but we can provide no assurance
that any such action taken by us would allow our common stock to
become listed again, stabilize the market price or improve the
liquidity of our common stock, prevent our common stock from
dropping below the NYSE MKT minimum bid price requirement or
prevent future non-compliance with the NYSE MKT’s listing
requirements.
The
National Securities Markets Improvement Act of 1996, which is a
federal statute, prevents or preempts the states from regulating
the sale of certain securities, which are referred to as
“covered securities.” Our common shares are considered
to be covered securities because they are listed on the NYSE MKT.
Although the states are preempted from regulating the sale of our
securities, the federal statute does allow the states to
investigate companies if there is a suspicion of fraud, and, if
there is a finding of fraudulent activity, then the states can
regulate or bar the sale of covered securities in a particular
case. Further, if we were no longer listed on the NYSE MKT, our
common stock would not be covered securities and we would be
subject to regulation in each state in which we offer our
securities.
Item 1B. Unresolved Staff Comments
None.
36
Item
2. Properties
Our
corporate offices are located at 4B Cedar Brook Drive, Cedar Brook
Corporate Center, Cranbury, NJ 08512, where we lease approximately
10,000 square feet of office space under a lease that expires in
June 2020. We also lease approximately 1,700 square feet of
laboratory space in the Township of South Brunswick, NJ, under a
lease that expires in August 2018. We believe our present
facilities are adequate for our current needs. We do not own any
real property.
Item
3. Legal Proceedings
We are
involved, from time to time, in various claims and legal
proceedings arising in the ordinary course of our business. We are
not currently a party to any claim or legal
proceeding.
Item
4. Mine Safety Disclosures
Not
applicable.
37
PART II
Item
5. Market for Registrant’s Common Equity, Related Stockholder
Matters and Issuer Purchases of Equity
Securities.
The
table below provides, for the fiscal quarters indicated, the
reported high and low sales prices for our common stock on the NYSE
MKT since July 1, 2016.
|
FISCAL YEAR
ENDED JUNE 30, 2017
|
HIGH
|
LOW
|
|
Fourth
Quarter
|
$0.50
|
$0.29
|
|
Third
Quarter
|
0.62
|
0.32
|
|
Second
Quarter
|
0.90
|
0.45
|
|
First
Quarter
|
0.86
|
0.45
|
|
|
|
|
|
FISCAL YEAR
ENDED JUNE 30, 2016
|
HIGH
|
LOW
|
|
Fourth
Quarter
|
$0.74
|
$0.40
|
|
Third
Quarter
|
0.69
|
0.36
|
|
Second
Quarter
|
0.90
|
0.62
|
|
First
Quarter
|
1.18
|
0.80
|
|
|
|
|
Our
common stock has been listed on NYSE MKT under the symbol
“PTN” since December 21, 1999. It previously traded on
The Nasdaq SmallCap Market under the symbol
“PLTN.”
On
September 21, 2017, we had approximately 151 record holders of common stock
and the closing sales price of our common stock as reported on the
NYSE MKT was $0.60 per share. The aggregate market
value of the common and non-voting common equity held by
non-affiliates on such date, computed by reference to the closing
sales price of our common stock on that date, was
$106,223,409.
Issuer purchases of equity securities. We have not and do
not currently intend to retire or repurchase any of our capital
securities other than providing our employees with the option to
withhold shares to satisfy tax withholding amounts due from
employees upon the vesting of restricted stock units in connection
with our 2011 Stock Incentive Plan. The following 67,723 shares
were withheld during the quarter ended June 30, 2017 at the
direction of the employees as permitted under the 2011 Stock
Incentive Plan in order to pay the minimum amount of tax liability
owed by the employee from the vesting of those units:
|
Period
|
Total Number of Shares Purchased (1)
|
Weighted Average Price Paid per Share
|
Total Number of Shares Purchased as Part of Publicly Announced
Plans or Programs
|
Maximum
Number of Shares that May Yet be Purchased Under Announced Plans or
Programs
|
|
April
1-30, 2017
|
-
|
$-
|
-
|
-
|
|
May
1-31, 2017
|
-
|
-
|
-
|
-
|
|
June
1-30, 2017
|
67,723
|
0.36
|
-
|
-
|
|
Total
|
67,723
|
$0.36
|
-
|
-
|
(1)
Consists solely of 67,723 shares that were withheld to satisfy tax
withholding amounts due from employees upon the vesting of
previously issued restricted stock units.
Dividends and dividend policy. We have never declared or
paid any dividends. We currently intend to retain earnings, if any,
for use in our business. We do not anticipate paying dividends in
the foreseeable future.
Dividend restrictions. Our outstanding Series A Preferred
Stock, consisting of 4,030 shares on September 21, 2017, provides
that we may not pay a dividend or make any distribution to holders
of any class of stock unless we first pay a special dividend or
distribution of $100 per share to the holders of the Series A
Preferred Stock.
Equity Compensation Plan Information. Reference is
made to the information contained in the Equity Compensation Plan
table contained in Item 12 of this Annual Report.
Item 6. Selected Financial Data.
Not
Applicable.
Item
7. Management’s Discussion and Analysis of Financial
Condition and Results of Operations.
The
following discussion and analysis should be read in conjunction
with the consolidated financial statements and notes to the
consolidated financial statements filed as part of this Annual
Report.
38
Forward-Looking Statements
The
following discussion and analysis contains forward-looking
statements within the meaning of the federal securities laws. You
are urged to carefully review our description and examples of
forward-looking statements included earlier in this Annual Report
on Form 10-K immediately prior to Part I, under the heading
“Forward-Looking Statements.” Forward-looking
statements are subject to risks and uncertainties that could cause
actual results to differ materially from those expressed in the
forward-looking statements. You are urged to carefully review the
disclosures we make concerning risks and other factors that may
affect our business and operating results, including those made in
Part I, Item 1A of this Annual Report on Form 10-K, and any of
those made in our other reports filed with the SEC. You are
cautioned not to place undue reliance on the forward-looking
statements included herein, which speak only as of the date of this
document. We do not intend, and undertake no obligation, to publish
revised forward-looking statements to reflect events or
circumstances after the date of this document or to reflect the
occurrence of unanticipated events.
Critical Accounting Policies and Estimates
Our
significant accounting policies are described in Note 2 to the
consolidated financial statements included in this Annual Report.
We believe that our accounting policies and estimates relating to
revenue recognition, accrued expenses and stock-based compensation
charges are the most critical.
Revenue Recognition.
Revenue
is recognized in accordance with Financial Accounting Standards
Board (“FASB”) Accounting Standards Codification
(“ASC”) Topic 605-25, Revenue Recognition for Arrangements with
Multiple Elements, which addresses the determination of
whether an arrangement involving multiple deliverables contains
more than one unit of accounting. A delivered item within an
arrangement is considered a separate unit of accounting only if
both of the following criteria are met:
●
the delivered item
has value to the customer on a stand-alone basis; and
●
if the arrangement
includes a general right of return relative to the delivered item,
delivery or performance of the undelivered item is considered
probable and substantially in control of the vendor.
Under
FASB ASC Topic 605-25, if both of the criteria above are not met,
then separate accounting for the individual deliverables is not
appropriate.
Revenue
resulting from the achievement of development milestones is
recorded in accordance with the accounting guidance for the
milestone method of revenue recognition.
Amounts
received prior to satisfying the revenue recognition criteria are
recorded as deferred revenue on the Company’s consolidated
balance sheet. Amounts expected to be recognized as revenue in the
next 12 months following the balance sheet date are classified as
current liabilities.
Accrued Expenses.
Third
parties perform a significant portion of our development
activities. We review the activities performed under significant
contracts each quarter and accrue expenses and the amount of any
reimbursement to be received from our collaborators based upon the
estimated amount of work completed. Estimating the value or stage
of completion of certain services requires judgment based on
available information. If we do not identify services performed for
us but not billed by the service-provider, or if we underestimate
or overestimate the value of services performed as of a given date,
reported expenses will be understated or overstated.
Stock-based Compensation.
The
fair value of stock options granted has been calculated using the
Black-Scholes option pricing model, which requires us to make
estimates of expected volatility and expected option lives. We
estimate these factors at the time of grant based on our own prior
experience, public sources of information and information for
comparable companies. The amount of recorded compensation related
to an option grant is not adjusted for subsequent changes in these
estimates or for actual experience. The amount of our recorded
compensation is also dependent on our estimates of future option
forfeitures. If we initially over-estimate future forfeitures, our
reported expenses will be understated until such time as we adjust
our estimate. Changes in estimated forfeitures will affect our
reported expenses in the period of change and future periods. In
addition, awards containing a market condition are valued using a
multifactor Monte Carlo simulation.
The
amount and timing of compensation expense to be recorded in future
periods related to grants of restricted stock units may be affected
by employment terminations. As a result, stock-based compensation
charges may vary significantly from period to period.
See
Note 3 to the consolidated financial statements included in this
Annual Report for a description of recent accounting pronouncements
that affect us.
39
Results of Operations
Year Ended June 30, 2017 Compared to the Year Ended June 30,
2016:
Revenue – For the fiscal year ended June 30, 2017
(“fiscal 2017”), we recognized $44,723,827 in revenue
pursuant to our license agreement with AMAG. For the fiscal year
ended June 30, 2016 (“fiscal 2016”), we did not
recognize any revenue.
On
January 8, 2017, we entered into the license agreement with AMAG
which provided for $60,000,000 as a one-time initial payment, which
was received on the effective date of February 2, 2017. Pursuant to
the terms of and subject to the conditions in the license
agreement, AMAG is required to pay us up to $25,000,000 in
reimbursement for reasonable, documented, direct out-of-pocket
expenses we incur following the effective date of the license
agreement in connection with the development and regulatory
activities necessary to file an NDA for bremelanotide for HSDD in
the United States. As of June 30, 2017, $4,657,577 was received and
$15,116,822 was included in accounts receivable relating to
reimbursable expenses.
Research and Development – Research and development
expenses were $45,683,174 for fiscal 2017 compared to $43,071,051
for fiscal 2016. These costs primarily relate to our bremelanotide
Phase 3 clinical trial program.
Research
and development expenses related to our bremelanotide, PL-3994,
MC1r, MC4r and other preclinical programs were $41,146,970 and
$39,371,908 in fiscal years 2017 and 2016, respectively. Spending
to date has been primarily related to our bremelanotide for the
treatment of HSDD program. The increase in research and development
expenses is mainly attributable to the continued progress of Phase
3 clinical trial and development of bremelanotide for HSDD. The
amount of such spending and the nature of future development
activities are dependent on a number of factors, including
primarily the availability of funds to support future development
activities, success of our clinical trials and preclinical and
discovery programs, and our ability to progress compounds in
addition to bremelanotide and PL-3994 into human clinical
trials.
The
amounts of project spending above exclude general research and
development spending, which was $4,536,204 and $3,699,143 in fiscal
years 2017 and 2016, respectively. The increase in general research
and development spending is primarily attributable to additional
staffing and secondarily to the recognition of stock-based
compensation.
Cumulative
spending from inception to June 30, 2017 was approximately
$279,000,000 on our bremelanotide program and approximately
$125,400,000 on all our other programs (which include PL-3994,
PL-8177, other melanocortin receptor agonists, other discovery
programs and terminated programs). Due to various risk factors
described herein under “Risk Factors,” including the
difficulty in currently estimating the costs and timing of future
Phase 1 clinical trials and larger-scale Phase 2 and Phase 3
clinical trials for any product under development, we cannot
predict with reasonable certainty when, if ever, a program will
advance to the next stage of development or be successfully
completed, or when, if ever, related net cash inflows will be
generated.
General and Administrative – General and
administrative expenses, which consist mainly of compensation and
related costs, were $9,610,147 for fiscal 2017 compared to
$6,179,084 for fiscal 2016. The increase in general and
administrative expenses is primarily attributable to payment for
professional services of Greenhill & Co. LLC relating to
entering into our license agreement with AMAG and secondarily
attributable to employee related expenses recognized in the
year.
Other Income (Expense) – Total other expense, net was
$(2,262,039) and $(2,462,801) for fiscal 2017 and fiscal 2016,
respectively. For fiscal 2017, we recognized $26,270 of investment
income offset by $(2,288,309) of interest expense primarily related
to our venture debt. For fiscal 2016, we recognized $50,226 of
investment income offset by $(2,513,027) of interest expense
primarily related to our venture debt.
Income Taxes – Income tax expense was $500,000 in
fiscal 2017 compared to no income tax expense or benefit in fiscal
2016. The fiscal year 2017 income tax expense relates to
alternative minimum tax ("AMT") expense based on federal
alternative minimum taxable income attributable to the $60,000,000
initial payment from AMAG.
Year Ended June 30, 2016 Compared to the Year Ended June 30,
2015:
Revenue – For the fiscal year ended June 30, 2016
(fiscal 2016), we did not recognize any revenue. For the fiscal
year ended June 30, 2015 (fiscal 2015), we recognized $12,951,730
in revenue pursuant to our license, co-development and
commercialization agreement with Gedeon Richter.
In
August 2014, we entered into a license, co-development and
commercialization agreement with Gedeon Richter, which provided for
$9,763,347 in upfront payments. The non-refundable portion of the
upfront payment, $4,932,315, was recorded as revenue in the three
months ended September 30, 2014 and the remaining balance was
recorded as revenue in the three months ended December 31, 2014,
which became non-refundable upon initiation of our Phase 3 clinical
trial program in the United States. We also recognized $3,188,383
in the three months ended December 31, 2014 relating to the
milestone payment due upon initiation of our Phase 3 clinical trial
program in the United States, which was initiated in December 2014.
On September 16, 2015, we entered into a termination agreement
pursuant to which we and Gedeon Richter agreed to mutually and
amicably terminate the license agreement.
40
Research and Development – Research and development
expenses were $43,071,051 for fiscal 2016 compared to $24,560,233
for fiscal 2015. These costs primarily relate to our bremelanotide
Phase 3 clinical trial program.
Research
and development expenses related to our bremelanotide, PL-3994,
MC1r, MC4r and other preclinical programs were $39,371,908 and
$21,879,136 in fiscal years 2016 and 2015, respectively. Spending
to date has been primarily related to our bremelanotide for the
treatment of HSDD program. The increase in research and development
expenses is mainly attributable to the continued progress of Phase
3 clinical trial and development of bremelanotide for HSDD. The
amount of such spending and the nature of future development
activities are dependent on a number of factors, including
primarily the availability of funds to support future development
activities, success of our clinical trials and preclinical and
discovery programs, and our ability to progress compounds in
addition to bremelanotide and PL-3994 into human clinical
trials.
The
amounts of project spending above exclude general research and
development spending, which were $3,699,143 and $2,681,097 in
fiscal years 2016 and 2015, respectively. The increase in general
research and development spending is primarily attributable to
additional staffing and secondarily to the recognition of
stock-based compensation primarily related to the restricted stock
units granted in December 2015.
Cumulative
spending from inception to June 30, 2016 is approximately
$234,800,000 on our bremelanotide program and approximately
$123,900,000 on all our other programs (which include PL-3994,
PL-8177, other melanocortin receptor agonists, other discovery
programs and terminated programs). Due to various risk factors
described herein under “Risk Factors,” including the
difficulty in currently estimating the costs and timing of future
Phase 1 clinical trials and larger-scale Phase 2 and Phase 3
clinical trials for any product under development, we cannot
predict with reasonable certainty when, if ever, a program will
advance to the next stage of development or be successfully
completed, or when, if ever, related net cash inflows will be
generated.
General and Administrative – General and
administrative expenses, which consist mainly of compensation and
related costs, were $6,179,084 for fiscal 2016 compared to
$5,677,654 for fiscal 2015. The increase in general and
administrative expenses is primarily attributable to the
recognition of stock-based compensation primarily related to the
restricted stock units granted in December 2015.
Other Income (Expense) – Total other income expense,
net was $(2,462,801) and $(910,914) for fiscal 2016 and fiscal
2015, respectively. For fiscal 2016, we recognized $50,226 of
investment income offset by $(2,513,027) of interest expense
primarily related to our venture debt. For fiscal 2015, we
recognized $35,439 of investment income offset by a $(284,656)
foreign exchange transaction loss and $(661,697) of interest
expense primarily related to our venture debt.
Income Tax Benefit – For fiscal 2016 the Company had
no income tax expense or a tax benefit from the sale of New Jersey
state net operating loss carryforwards on account that it has
reached the state limits on sale of New Jersey state net operating
loss carryforwards. For fiscal 2015, income tax benefits recorded
of $531,508 related to the sale of New Jersey state net operating
loss carryforwards and tax credits. The amount of such losses and
tax credits that we were able to sell depended on annual pools and
allocations established by the state of New Jersey. This program
enables approved, unprofitable biotechnology businesses to sell
their unused net operating loss carryovers and unused research and
development tax credits to unaffiliated, profitable corporate
taxpayers in the state of New Jersey.
Effects of Inflation
We do
not believe that inflation has had a material impact on our
business, revenues or operating results during the periods
presented.
Liquidity and Capital Resources
Since
inception, we have incurred net operating losses, primarily related
to spending on our research and development programs. We have
financed our net operating losses primarily through debt and equity
financings and amounts received under collaborative
agreements.
Our
product candidates are at various stages of development and will
require significant further research, development and testing and
some may never be successfully developed or commercialized. We may
experience uncertainties, delays, difficulties and expenses
commonly experienced by early stage biopharmaceutical companies,
which may include unanticipated problems and additional costs
relating to:
●
the development and
testing of products in animals and humans;
●
product approval or
clearance;
●
regulatory
compliance;
●
GMP
compliance;
●
intellectual
property rights;
41
●
product
introduction;
●
marketing, sales
and competition; and
●
obtaining
sufficient capital.
Failure
to enter into or successfully perform under collaboration
agreements and obtain timely regulatory approval for our product
candidates and indications would impact our ability to increase
revenues and could make it more difficult to attract investment
capital for funding our operations. Any of these possibilities
could materially and adversely affect our operations and require us
to curtail or cease certain programs.
During
fiscal 2017, net cash provided by operating activities was
$12,881,527, compared to cash used in operating activities of
$47,363,814 in fiscal 2016 and $13,358,042 in fiscal 2015. The
difference of cash provided by and cash used in operations in
fiscal 2017 compared to fiscal 2016 was primarily the result of the
receipt of the initial payment of $60,000,000 relating to the
license agreement with AMAG. Higher net cash outflows from
operations in fiscal 2016 compared to fiscal 2015 were primarily
the result of spending on our bremelanotide for the treatment of
HSDD program. Our periodic prepaid expenses, accounts payable and
accrued expenses balances will continue to be highly dependent on
the timing of our operating costs.
During
fiscal 2017, net cash provided by investing activities was
$991,596, which consisted of $1,124,999 of proceeds from the
maturity of investments offset by $133,403 used for the acquisition
of equipment. During fiscal 2016, net cash used in investing
activities was $1,404,717 consisting primarily of the purchase of
investments. We did not engage in any investing activities in
fiscal 2015.
During
fiscal 2017, net cash provided by financing activities was
$18,324,533, which consisted of net proceeds from our underwritten
offerings of our units in August and December 2016 of $23,856,973
and proceeds from the exercise of warrants of $164,358, offset by
$5,696,798 for the payments on notes payable, capital lease
payments and the payment of withholding taxes related to restricted
stock units. During fiscal 2016, net cash provided by financing
activities was $29,471,931, which consisted of a private placement
with net proceeds of $19,834,278, a loan of $9,853,885, net of
related debt issuance costs offset by $216,232 for the payment of
withholding taxes related to restricted stock units and capital
lease payments. During fiscal 2015, net cash provided by financing
activities was $28,472,705, which consisted of a private placement
with net proceeds of $18,556,111, a loan of $9,790,634, net of
related issuance costs and $254,148 of proceeds from the exercise
of common stock warrants offset by $128,188 for the payment of
withholding taxes related restricted stock units and capital lease
payments.
We have
incurred cumulative negative cash flows from operations since our
inception, and have expended, and expect to continue to expend in
the future, substantial funds to complete our planned product
development efforts. Continued operations are dependent upon our
ability to complete equity or debt financing activities and enter
into licensing or collaboration arrangements. As of June 30, 2017,
our cash, cash equivalents, accounts receivable and investments
were $55,566,983 and our current liabilities were $19,911,724, net
of deferred revenue of $35,050,572.
We
intend to utilize existing capital resources for general corporate
purposes and working capital, including required ancillary studies
of bremelanotide for HSDD and preparing and filing an NDA on
bremelanotide, preclinical and clinical development of our MC1r and
MC4r peptide programs and PL3994 natriuretic peptide, and
development of other portfolio products.
We
believe that our existing capital resources, along with the
additional proceeds from our September 2017 license agreement with
Fosun (Note 16), will be adequate to fund our planned operations
through at least the 2018 calendar year. We will need additional
funding to complete required clinical trials for our other product
candidates and development programs and, if those clinical trials
are successful (which we cannot predict), to complete submission of
required regulatory applications to the FDA.
We
anticipate incurring additional losses over at least the next few
years. To achieve or maintain profitability, if ever, we, alone or
with others, must successfully develop and commercialize our
technologies and proposed products, conduct preclinical studies and
clinical trials, obtain required regulatory approvals and
successfully manufacture and market our technologies and proposed
products. The time required to reach profitability is highly
uncertain, and we do not know whether we will be able to achieve
profitability on a sustained basis, if at all.
42
Off-Balance Sheet Arrangements
None.
Contractual Obligations
We have
entered into various contractual obligations and commercial
commitments. The following table summarizes our most significant
contractual obligations as of June 30, 2017:
|
|
Payments due by Period
|
||||
|
|
Total
|
Less than 1 Year
|
1 - 3 Years
|
3 - 5 Years
|
More than 5 Years
|
|
|
|
|
|
|
|
|
Facility
operating leases
|
$734,267
|
$280,923
|
$453,344
|
$-
|
$-
|
|
Capital
lease obligations
|
14,568
|
14,568
|
-
|
-
|
-
|
|
Notes
payable, including interest
|
16,593,250
|
8,974,750
|
7,618,500
|
-
|
-
|
|
|
|
|
|
|
|
|
Total
contractual obligations
|
$17,342,085
|
$9,270,241
|
$8,071,844
|
$-
|
$-
|
Item 7A. Quantitative and Qualitative Disclosures About Market
Risk.
Not
applicable.
43
Item 8. Financial Statements and Supplementary Data.
Table of Contents
Consolidated Financial Statements
The
following consolidated financial statements are filed as part of
this Annual Report:
|
|
Page
|
|
|
|
|
Report
of Independent Registered Public Accounting Firm
|
45
|
|
|
|
|
Consolidated
Balance Sheets
|
46
|
|
|
|
|
Consolidated
Statements of Operations
|
47
|
|
|
|
|
Consolidated
Statements of Comprehensive Loss
|
48
|
|
|
|
|
Consolidated
Statements of Stockholders’ (Deficiency) Equity
|
49
|
|
|
|
|
Consolidated
Statements of Cash Flows
|
50
|
|
|
|
|
Notes
to Consolidated Financial Statements
|
51
|
44
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
The
Board of Directors and Stockholders
Palatin
Technologies, Inc.:
We have
audited the accompanying consolidated balance sheets of Palatin
Technologies, Inc. and subsidiary as of June 30, 2017 and 2016, and
the related consolidated statements of operations, comprehensive
loss, stockholders’ (deficiency) equity, and cash flows for
each of the years in the three-year period ended June 30, 2017.
These consolidated financial statements are the responsibility of
the Company’s management. Our responsibility is to express an
opinion on these consolidated financial statements based on our
audits.
We
conducted our audits in accordance with the standards of the Public
Company Accounting Oversight Board (United States). Those standards
require that we plan and perform the audit to obtain reasonable
assurance about whether the financial statements are free of
material misstatement. An audit includes examining, on a test
basis, evidence supporting the amounts and disclosures in the
financial statements. An audit also includes assessing the
accounting principles used and significant estimates made by
management, as well as evaluating the overall financial statement
presentation. We believe that our audits provide a reasonable basis
for our opinion.
In our
opinion, the consolidated financial statements referred to above
present fairly, in all material respects, the financial position of
Palatin Technologies, Inc. and subsidiary as of June 30, 2017 and
2016, and the results of their operations and their cash flows for
each of the years in the three-year period ended June 30, 2017, in
conformity with U.S. generally accepted accounting
principles.
/s/
KPMG LLP
Philadelphia,
Pennsylvania
September
25, 2017
45
PALATIN TECHNOLOGIES,
INC .
and Subsidiary
Consolidated Balance Sheets
|
|
June 30,
2017
|
June 30,
2016
|
|
ASSETS
|
|
|
|
Current
assets:
|
|
|
|
Cash
and cash equivalents
|
$40,200,324
|
$8,002,668
|
|
Available-for-sale
investments
|
249,837
|
1,380,556
|
|
Accounts
receivable
|
15,116,822
|
-
|
|
Prepaid
expenses and other current assets
|
1,011,221
|
1,313,841
|
|
Total
current assets
|
56,578,204
|
10,697,065
|
|
|
|
|
|
Property
and equipment, net
|
198,153
|
97,801
|
|
Other
assets
|
56,916
|
63,213
|
|
Total
assets
|
$56,833,273
|
$10,858,079
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ DEFICIENCY
|
|
|
|
Current
liabilities:
|
|
|
|
Accounts
payable
|
$1,551,367
|
$713,890
|
|
Accrued
expenses
|
10,521,098
|
7,767,733
|
|
Notes
payable, net of discount
|
7,824,935
|
5,374,951
|
|
Capital
lease obligations
|
14,324
|
27,424
|
|
Deferred
revenue
|
35,050,572
|
-
|
|
Total
current liabilities
|
54,962,296
|
13,883,998
|
|
|
|
|
|
Notes
payable, net of discount
|
6,281,660
|
14,106,594
|
|
Capital
lease obligations
|
-
|
14,324
|
|
Other
non-current liabilities
|
753,961
|
439,130
|
|
Total
liabilities
|
61,997,917
|
28,444,046
|
|
|
|
|
|
Commitments
and contingencies (Note 12)
|
|
|
|
|
|
|
|
Stockholders’
deficiency:
|
|
|
|
Preferred stock of $0.01 par value – authorized 10,000,000
shares:
|
|
|
|
Series
A Convertible: issued and outstanding 4,030 shares as of June 30,
2017 and June 30, 2016
|
40
|
40
|
|
Common stock of $0.01 par value – authorized 300,000,000
shares;
|
|
|
|
issued
and outstanding 160,515,361 shares as of June 30, 2017 and
68,568,055 as of June 30, 2016, respectively
|
1,605,153
|
685,680
|
|
Additional
paid-in capital
|
349,974,538
|
325,142,509
|
|
Accumulated
other comprehensive loss
|
(590)
|
(1,944)
|
|
Accumulated
deficit
|
(356,743,785)
|
(343,412,252)
|
|
Total
stockholders’ deficiency
|
(5,164,644)
|
(17,585,967)
|
|
Total
liabilities and stockholders’ deficiency
|
$56,833,273
|
$10,858,079
|
The
accompanying notes are an integral part of these consolidated
financial statements
46
PALATIN
TECHNOLOGIES, INC.
and
Subsidiary
|
Consolidated Statements of Operations
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended June 30,
|
||
|
|
2017
|
2016
|
2015
|
|
|
|
|
|
|
REVENUES
|
|
|
|
|
License
and contract
|
$44,723,827
|
$-
|
$12,951,730
|
|
|
|
|
|
|
OPERATING
EXPENSES
|
|
|
|
|
Research
and development
|
45,683,174
|
43,071,051
|
24,560,233
|
|
General
and administrative
|
9,610,147
|
6,179,084
|
5,677,654
|
|
Total
operating expenses
|
55,293,321
|
49,250,135
|
30,237,887
|
|
|
|
|
|
|
Loss
from operations
|
(10,569,494)
|
(49,250,135)
|
(17,286,157)
|
|
|
|
|
|
|
OTHER
INCOME (EXPENSE)
|
|
|
|
|
Investment
income
|
26,270
|
50,226
|
35,439
|
|
Interest
expense
|
(2,288,309)
|
(2,513,027)
|
(661,697)
|
|
Foreign
exchange transaction loss
|
-
|
-
|
(284,656)
|
|
Total
other expense, net
|
(2,262,039)
|
(2,462,801)
|
(910,914)
|
|
|
|
|
|
|
Loss
before income taxes
|
(12,831,533)
|
(51,712,936)
|
(18,197,071)
|
|
Income
tax (expense) benefit
|
(500,000)
|
-
|
531,508
|
|
|
|
|
|
|
NET
LOSS
|
$(13,331,533)
|
$(51,712,936)
|
$(17,665,563)
|
|
|
|
|
|
|
Basic
and diluted net loss per common share
|
$(0.07)
|
$(0.33)
|
$(0.15)
|
|
|
|
|
|
|
Weighted
average number of common shares outstanding used in computing basic
and diluted net loss per common share
|
184,087,719
|
156,553,534
|
121,014,506
|
The
accompanying notes are an integral part of these consolidated
financial statements
47
PALATIN TECHNOLOGIES, INC.
and
Subsidiary
|
Consolidated Statements of Comprehensive Loss
|
|||
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended June 30,
|
||
|
|
2017
|
2016
|
2015
|
|
|
|
|
|
|
Net
loss
|
$(13,331,533)
|
$(51,712,936)
|
$(17,665,563)
|
|
|
|
|
|
|
Other
comprehensive loss:
|
|
|
|
|
Unrealized
gain (loss) on available-for-sale investments
|
1,354
|
(1,944)
|
-
|
|
|
|
|
|
|
Total
comprehensive loss
|
$(13,330,179)
|
$(51,714,880)
|
$(17,665,563)
|
|
The
accompanying notes are an integral part of these consolidated
financial statements
|
48
|
PALATIN TECHNOLOGIES, INC.
|
||||||||
|
and Subsidiary
|
||||||||
|
Consolidated Statements of Stockholders’ (Deficiency)
Equity
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
Acculumated Other
|
|
|
|
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Comprehensive
|
Accumulated
|
|
||
|
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Loss
|
Deficit
|
Total
|
|
Balance,
June 30, 2014
|
4,697
|
47
|
39,416,595
|
394,166
|
283,428,356
|
-
|
(274,033,753)
|
9,788,816
|
|
Stock-based
compensation
|
-
|
-
|
705,833
|
7,058
|
1,160,221
|
-
|
-
|
1,167,279
|
|
Sale
of common stock units, net of costs
|
-
|
-
|
2,050,000
|
20,500
|
18,535,611
|
-
|
-
|
18,556,111
|
|
Issuance
of warrants on debt
|
-
|
-
|
-
|
-
|
267,820
|
-
|
-
|
267,820
|
|
Withholding
taxes related to restricted stock units
|
-
|
-
|
(174,568)
|
(1,746)
|
(162,390)
|
-
|
-
|
(164,136)
|
|
Warrant
exercises
|
-
|
-
|
15,130,573
|
151,306
|
102,842
|
-
|
-
|
254,148
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(17,665,563)
|
(17,665,563)
|
|
Balance,
June 30, 2015
|
4,697
|
47
|
57,128,433
|
571,284
|
303,332,460
|
-
|
(291,699,316)
|
12,204,475
|
|
Stock-based
compensation
|
-
|
-
|
662,186
|
6,622
|
1,836,743
|
-
|
-
|
1,843,365
|
|
Sale
of common stock units, net of costs
|
-
|
-
|
-
|
-
|
19,834,278
|
-
|
-
|
19,834,278
|
|
Issuance
of warrants on debt
|
-
|
-
|
-
|
-
|
305,196
|
-
|
-
|
305,196
|
|
Withholding
taxes related to restricted stock units
|
-
|
-
|
(123,483)
|
(1,235)
|
(57,166)
|
-
|
-
|
(58,401)
|
|
Warrant
exercises
|
|
|
10,890,889
|
108,909
|
(108,909)
|
-
|
-
|
-
|
|
Series
A Conversion
|
(667)
|
(7)
|
10,030
|
100
|
(93)
|
-
|
-
|
-
|
|
Unrealized
loss on investments
|
-
|
-
|
-
|
-
|
-
|
(1,944)
|
-
|
(1,944)
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(51,712,936)
|
(51,712,936)
|
|
Balance,
June 30, 2016
|
4,030
|
40
|
68,568,055
|
685,680
|
325,142,509
|
(1,944)
|
(343,412,252)
|
(17,585,967)
|
|
Stock-based
compensation
|
-
|
-
|
579,400
|
5,794
|
1,751,465
|
-
|
-
|
1,757,259
|
|
Sale
of common stock units, net of costs
|
-
|
-
|
36,866,097
|
368,661
|
23,488,312
|
-
|
-
|
23,856,973
|
|
Withholding
taxes related to restricted stock units
|
-
|
-
|
(75,993)
|
(760)
|
(26,328)
|
-
|
-
|
(27,088)
|
|
Warrant
exercises
|
|
|
54,577,802
|
545,778
|
(381,420)
|
-
|
-
|
164,358
|
|
Unrealized
gains on investments
|
-
|
-
|
-
|
-
|
-
|
1,354
|
-
|
1,354
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(13,331,533)
|
(13,331,533)
|
|
Balance,
June 30, 2017
|
4,030
|
$40
|
160,515,361
|
$1,605,153
|
$349,974,538
|
$(590)
|
$(356,743,785)
|
$(5,164,644)
|
|
The
accompanying notes are an integral part of these consolidated
financial statements
|
49
|
PALATIN TECHNOLOGIES, INC.
|
|||
|
and Subsidiary
|
|||
|
Consolidated Statements of Cash Flows
|
|||
|
|
|
|
|
|
|
Year Ended June 30,
|
||
|
|
2017
|
2016
|
2015
|
|
CASH
FLOWS FROM OPERATING ACTIVITIES:
|
|
|
|
|
Net loss
|
$(13,331,533)
|
$(51,712,936)
|
$(17,665,563)
|
|
Adjustments to reconcile net loss to net cash provided
by
|
|
|
|
|
(used) in operating activities:
|
|
|
|
|
Depreciation
and amortization
|
33,051
|
43,052
|
117,590
|
|
Non-cash
interest expense
|
298,790
|
327,479
|
87,087
|
|
Stock-based
compensation
|
1,757,259
|
1,843,365
|
1,167,279
|
|
Changes
in operating assets and liabilities:
|
|
|
|
|
Accounts
receivable
|
(15,116,822)
|
-
|
-
|
|
Prepaid
expenses and other assets
|
308,917
|
503,785
|
(1,667,139)
|
|
Accounts
payable
|
837,477
|
(392,594)
|
845,204
|
|
Accrued
expenses
|
2,728,985
|
1,676,209
|
4,666,196
|
|
Deferred
revenue
|
35,050,572
|
-
|
(1,000,000)
|
|
Other
non-current liabilities
|
314,831
|
347,826
|
91,304
|
|
Net
cash provided by (used in) operating activities
|
12,881,527
|
(47,363,814)
|
(13,358,042)
|
|
|
|
|
|
|
CASH
FLOWS FROM INVESTING ACTIVITIES:
|
|
|
|
|
Proceeds
from sale/maturity of investments
|
1,124,999
|
-
|
-
|
|
Purchases
of investments
|
-
|
(1,387,022)
|
-
|
|
Purchases
of property and equipment
|
(133,403)
|
(17,695)
|
-
|
|
Net
cash provided by (used in) investing activities
|
991,596
|
(1,404,717)
|
-
|
|
|
|
|
|
|
CASH
FLOWS FROM FINANCING ACTIVITIES:
|
|
|
|
|
Payments
on capital lease obligations
|
(27,424)
|
(25,872)
|
(12,380)
|
|
Payment
of withholding taxes related to restricted
|
|
|
|
|
stock units
|
(2,708)
|
(190,360)
|
(115,808)
|
|
Payment
on debt obligations
|
(5,666,666)
|
-
|
-
|
|
Proceeds
from exercise of common stock warrants
|
164,358
|
-
|
254,148
|
|
Proceeds
from the sale of common stock and warrants, net
|
|
|
|
|
of costs
|
23,856,973
|
19,834,278
|
18,556,111
|
|
Proceeds
from the issuance of notes payable and warrants
|
-
|
10,000,000
|
10,000,000
|
|
Payment
of debt issuance costs
|
-
|
(146,115)
|
(209,366)
|
|
Net
cash provided by financing activities
|
18,324,533
|
29,471,931
|
28,472,705
|
|
|
|
|
|
|
NET
INCREASE (DECREASE) IN CASH
|
|
|
|
|
AND CASH EQUIVALENTS
|
32,197,656
|
(19,296,600)
|
15,114,663
|
|
|
|
|
|
|
CASH
AND CASH EQUIVALENTS, beginning of year
|
8,002,668
|
27,299,268
|
12,184,605
|
|
|
|
|
|
|
CASH
AND CASH EQUIVALENTS, end of year
|
$40,200,324
|
$8,002,668
|
$27,299,268
|
|
|
|
|
|
|
SUPPLEMENTAL
CASH FLOW INFORMATION:
|
|
|
|
|
Cash
paid for interest
|
$1,676,954
|
$1,836,743
|
$483,306
|
|
Equipment
acquired under capital lease
|
-
|
-
|
80,000
|
|
Issuance
of warrants in connection with debt financing
|
-
|
305,196
|
267,820
|
The accompanying notes are an integral part of these consolidated
financial statements
50
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
(1)
ORGANIZATION
Nature of Business – Palatin Technologies, Inc.
(“Palatin” or “the Company”) is a
biopharmaceutical company developing targeted, receptor-specific
peptide therapeutics for the treatment of diseases with significant
unmet medical need and commercial potential. Palatin’s
programs are based on molecules that modulate the activity of the
melanocortin and natriuretic peptide receptor systems. The
melanocortin system is involved in a large and diverse number of
physiologic functions, and therapeutic agents modulating this
system may have the potential to treat a variety of conditions and
diseases, including sexual dysfunction and inflammation-related
diseases. The natriuretic peptide receptor system has numerous
cardiovascular functions, and therapeutic agents modulating this
system may be useful in treatment of heart failure and other
cardiovascular diseases.
The
Company’s primary product in development is bremelanotide for
the treatment of hypoactive sexual desire disorder
(“HSDD”), which is a type of female sexual dysfunction
(“FSD”). The Company also has drug candidates or
development programs for cardiovascular diseases, including heart
failure and fibrosis, and inflammatory diseases, including
inflammatory bowel disease and ocular indications.
Key
elements of the Company’s business strategy include using its
technology and expertise to develop and commercialize therapeutic
products; entering into alliances and partnerships with
pharmaceutical companies to facilitate the development,
manufacture, marketing, sale and distribution of product candidates
that the Company is developing; and partially funding its product
candidate development programs with the cash flow generated from
third parties.
Business Risk and Liquidity – Since inception, the
Company has incurred negative cash flows from operations, and has
expended, and expects to continue to expend, substantial funds to
complete its planned product development efforts. As shown in the
accompanying consolidated financial statements, the Company had an
accumulated deficit as of June 30, 2017 of $356,743,785 and
incurred a net loss for fiscal 2017 of $13,331,533. The Company
anticipates incurring additional losses in the future as a result
of spending on its development programs and will require
substantial additional financing to continue to fund its planned
developmental activities. To achieve profitability, if ever, the
Company, alone or with others, must successfully develop and
commercialize its technologies and proposed products, conduct
successful preclinical studies and clinical trials, obtain required
regulatory approvals and successfully manufacture and market such
technologies and proposed products. The time required to reach
profitability is highly uncertain, and the Company may never be
able to achieve profitability on a sustained basis, if at
all.
On
January 8, 2017, the Company entered into an exclusive license
agreement (“License Agreement”) with AMAG for
bremelanotide for North America (Note 5). The License Agreement
became effective on February 2, 2017, and the Company received an
upfront payment of $60,000,000 pursuant to the License Agreement on
that date.
As of
June 30, 2017, the Company’s cash, cash equivalents, accounts
receivable and investments were $55,566,983 and current liabilities
were $19,911,724, net of deferred revenue of $35,050,572. The
Company intends to utilize existing capital resources for general
corporate purposes and working capital, including required
ancillary studies with bremelanotide for HSDD preparatory to filing
a New Drug Application (“NDA”) with the U.S. Food and
Drug Administration (“FDA”), and preclinical and
clinical development of our other product candidates and programs,
including natriuretic peptide receptor and melanocortin receptor
programs.
On
September 6, 2017, the Company entered into a collaboration and
license agreement with Shanghai Fosun Pharmaceutical Industrial
Development Co., Ltd., a subsidiary of Shanghai Fosun
Pharmaceutical (Group) Co., Ltd. (“Fosun”), for
exclusive rights to develop and commercialize bremelanotide for FSD
indications in the territories of mainland China, Taiwan, Hong Kong
S.A.R. and Macau S.A.R. (Note 16).
Management
believes that its existing capital resources, including the
proceeds from the License Agreement with Fosun, will be adequate to
fund its planned operations through at least the 2018 calendar
year. The Company will need additional funding to complete required
clinical trials for its other product candidates and, assuming
those clinical trials are successful, as to which there can be no
assurance, to complete submission of required applications to the
FDA. If the Company is unable to obtain approval or otherwise
advance in the FDA approval process, the Company’s ability to
sustain its operations would be materially adversely
affected.
51
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
The
Company may seek the additional capital necessary to fund its
operations through public or private equity offerings,
collaboration agreements, debt financings or licensing
arrangements. Additional capital that is required by the Company
may not be available on reasonable terms, or at all.
Concentrations – Concentrations in the Company’s
assets and operations subject it to certain related risks.
Financial instruments that subject the Company to concentrations of
credit risk primarily consist of cash and cash equivalents and
available-for-sale investments. The Company’s cash and cash
equivalents are primarily invested in one money market account
sponsored by a large financial institution. For the year ended June
30, 2017, 100% of revenues were from AMAG. For the year ended June
30, 2016, the Company had no revenues reported, and for the year
ended June 30, 2015, 100% of revenues were from Gedeon Richter
(Note 4).
(2)
SUMMARY
OF SIGNIFICANT ACCOUNTING POLICIES
Principles of Consolidation – The consolidated
financial statements include the accounts of Palatin and its
wholly-owned inactive subsidiary. All intercompany accounts and
transactions have been eliminated in consolidation.
Use of Estimates – The preparation of consolidated
financial statements in conformity with accounting principles
generally accepted in the United States of America (“U.S.
GAAP”) requires management to make estimates and assumptions
that affect the reported amounts of assets and liabilities and
disclosure of contingent assets and liabilities at the date of the
consolidated financial statements and the reported amounts of
revenues and expenses during the reporting period. Actual results
could differ from those estimates.
Cash and Cash Equivalents – Cash and cash equivalents
include cash on hand, cash in banks and all highly liquid
investments with a purchased maturity of less than three months.
Cash equivalents consist of $40,019,336 and $7,782,243 in a money
market account at June 30, 2017 and 2016,
respectively.
Investments – The
Company determines the appropriate classification of its
investments in debt and equity securities at the time of purchase
and reevaluates such determinations at each balance sheet date.
Debt securities are classified as held-to-maturity when the Company
has the intent and ability to hold the securities to maturity. Debt
securities for which the Company does not have the intent or
ability to hold to maturity are classified as available-for-sale.
Held-to-maturity securities are recorded as either short-term or
long-term on the balance sheet, based on the contractual maturity
date and are stated at amortized cost. Marketable securities that
are bought and held principally for the purpose of selling them in
the near term are classified as trading securities and are reported
at fair value, with unrealized gains and losses recognized in
earnings. Debt and marketable equity securities not classified as
held-to-maturity or as trading are classified as available-for-sale
and are carried at fair market value, with the unrealized gains and
losses, net of tax, included in the determination of other
comprehensive (loss) income.
The fair value of substantially all securities is determined by
quoted market prices. The estimated fair value of securities for
which there are no quoted market prices is based on similar types
of securities that are traded in the market.
Fair Value of Financial Instruments – The
Company’s financial instruments consist primarily of cash
equivalents, accounts receivable, accounts payable and notes
payable. Management believes that the carrying values of cash
equivalents, available-for-sale investments, accounts
receivable and accounts payable are representative of their
respective fair values based on the short-term nature of these
instruments. Management believes that the carrying amount of its
notes payable approximates fair value based on terms of the
notes.
Credit Risk – Financial instruments which potentially
subject the Company to concentrations of credit risk consist
principally of cash and cash equivalents. Total cash and cash
equivalent balances have exceeded insured balances by the Federal
Depository Insurance Company (“FDIC”).
Property and Equipment – Property and equipment
consists of office and laboratory equipment, office furniture and
leasehold improvements and includes assets acquired under capital
leases. Property and equipment are recorded at cost. Depreciation
is recognized using the straight-line method over the estimated
useful lives of the related assets, generally five years for
laboratory and computer equipment, seven years for office furniture
and equipment and the lesser of the term of the lease or the useful
life for leasehold improvements. Amortization of assets acquired
under capital leases is included in depreciation expense.
Maintenance and repairs are expensed as incurred while expenditures
that extend the useful life of an asset are
capitalized.
52
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
Impairment of Long-Lived Assets – The Company reviews
its long-lived assets for impairment whenever events or changes in
circumstances indicate that the carrying amount of the assets may
not be fully recoverable. To determine recoverability of a
long-lived asset, management evaluates whether the estimated future
undiscounted net cash flows from the asset are less than its
carrying amount. If impairment is indicated, the long-lived asset
would be written down to fair value. Fair value is determined by an
evaluation of available price information at which assets could be
bought or sold, including quoted market prices, if available, or
the present value of the estimated future cash flows based on
reasonable and supportable assumptions.
Revenue Recognition – The Company has generated
revenue solely through license and collaboration agreements. The
Company recognizes revenue in accordance with Financial Accounting
Standards Board (“FASB”) Accounting Standards
Codification (“ASC”) Topic 605-25, Revenue Recognition for Arrangements with
Multiple Elements, which addresses the determination of
whether an arrangement involving multiple deliverables contains
more than one unit of accounting. A delivered item within an
arrangement is considered a separate unit of accounting only if
both of the following criteria are met:
●
the delivered item
has value to the customer on a stand-alone basis; and
●
if the arrangement
includes a general right of return relative to the delivered item,
delivery or performance of the undelivered item is considered
probable and substantially in control of the vendor.
Under
FASB ASC Topic 605-25, if both of the criteria above are not met,
then separate accounting for the individual deliverables is not
appropriate.
The
Company has determined that it is appropriate to recognize such
revenue using the input-based proportional method during the period
of Palatin’s development obligations as defined in the AMAG
License Agreement. Refer to Note 5 for additional
information.
Revenue
resulting from the achievement of development milestones is
recorded in accordance with the accounting guidance for the
milestone method of revenue recognition.
Amounts
received prior to satisfying the revenue recognition criteria are
recorded as deferred revenue on the Company’s consolidated
balance sheet. Amounts expected to be recognized as revenue in the
next 12 months following the balance sheet date are classified as
current liabilities.
Research and Development Costs – The costs of research
and development activities are charged to expense as incurred,
including the cost of equipment for which there is no alternative
future use.
Accrued Expenses – Third parties perform a significant
portion of our development activities. We review the activities
performed under all contracts each quarter and accrue expenses and
the amount of any reimbursement to be received from our
collaborators based upon the estimated amount of work completed.
Estimating the value or stage of completion of certain services
requires judgment based on available information. If we do not
identify services performed for us but not billed by the
service-provider, or if we underestimate or overestimate the value
of services performed as of a given date, reported expenses will be
understated or overstated.
Stock-Based Compensation – The Company charges to
expense the fair value of stock options and other equity awards
granted. The Company determines the value of stock options
utilizing the Black-Scholes option pricing model. Compensation
costs for share-based awards with pro-rata vesting are determined
using the quoted market price of the Company’s common stock
on the date of grant and allocated to periods on a straight-line
basis, while awards containing a market condition are valued using
multifactor Monte Carlo simulations.
Income Taxes – The Company and its subsidiary file
consolidated federal and separate-company state income tax returns.
Income taxes are accounted for under the asset and liability
method. Deferred tax assets and liabilities are recognized for the
future tax consequences attributable to differences between the
financial statement carrying amounts of assets and liabilities and
their respective tax basis and operating loss and tax credit
carryforwards. Deferred tax assets and liabilities are measured
using enacted tax rates expected to apply to taxable income in the
years in which those temporary differences or operating loss and
tax credit carryforwards are expected to be recovered or settled.
The effect on deferred tax assets and liabilities of a change in
tax rates is recognized in the period that includes the enactment
date. The Company has recorded a valuation allowance against its
deferred tax assets based on the history of losses
incurred.
53
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
Net Loss per Common Share – Basic and diluted earnings
per common share (“EPS”) are calculated in accordance
with the provisions of FASB ASC Topic 260, “Earnings per
Share,” which includes guidance pertaining to the warrants,
issued in connection with the July 3, 2012, December 23, 2014, and
July 2, 2015 private placement offerings and the August 4, 2016
underwritten offering, that are exercisable for nominal
consideration and, therefore, are to be considered in the
computation of basic and diluted net loss per common share. The
Series A 2012 warrants issued on July 3, 2012 to purchase up to
31,988,151 shares of common stock are included in the weighted
average number of common shares outstanding used in computing basic
and diluted net loss per common share for all periods presented in
the consolidated statements of operations.
The
Series B 2012 warrants issued on July 3, 2012 to purchase up to
35,488,380 shares of common stock are included in the weighted
average number of common shares outstanding used in computing basic
and diluted net loss per common share for all periods presented in
the consolidated statements of operations.
The
Series C 2014 warrants to purchase up to 24,949,325 shares of
common stock were exercisable starting at December 23, 2014 and,
therefore are included in the weighted average number of common
shares outstanding used in computing basic and diluted net loss per
common share starting on December 23, 2014.
The
Series E 2015 warrants to purchase up to 21,917,808 shares of
common stock were exercisable starting at July 2, 2015 and,
therefore are included in the weighted average number of common
shares outstanding used in computing basic and diluted net loss per
common share starting on July 2, 2015.
The
Series I 2016 warrants to purchase up to 2,218,045 shares of common
stock were exercisable starting at August 4, 2016 and, therefore
are included in the weighted average number of common shares
outstanding used in computing basic and diluted net loss per common
share starting on August 4, 2016 (Note 13).
As of
June 30, 2017, 2016 and 2015, there were 40,597,194, 32,167,737,
and 30,212,446 common shares issuable upon conversion of Series A
Convertible Preferred Stock, the exercise of outstanding options
and warrants (excluding the Series A 2012, Series B 2012, Series C
2014, and Series E 2015 warrants issued in connection with the July
3, 2012, December 23, 2014, and July 2, 2015 private placement
offerings), and the vesting of restricted stock units,
respectively. These share amounts have been excluded from the
calculation of net loss per share as the impact would be
anti-dilutive.
(3)
NEW
AND RECENTLY ADOPTED ACCOUNTING PRONOUNCEMENTS
In May
2017, the FASB issued ASU No. 2017-09, Compensation-Stock Compensation (Topic 718):
Scope of Modification Accounting, which clarifies when to
account for a change to the terms or conditions of a share-based
payment award as a modification. Under the new guidance,
modification accounting is required only if the fair value, the
vesting conditions, or the classification of the award (as equity
or liability) changes as a result of the change in terms or
conditions. It is effective prospectively for the annual period
ending June 30, 2019 and interim periods within that annual period.
Early adoption is permitted. The Company is currently evaluating
the effect that ASU No. 2017-09 will have on its consolidated
financial statements and related disclosures.
In June
2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses:
Measurement of Credit Losses on Financial Instruments which
requires measurement and recognition of expected credit losses for
financial assets held at the reporting date based on historical
experience, current conditions, and reasonable and supportable
forecasts. This is different from the current guidance as this will
require immediate recognition of estimated credit losses expected
to occur over the remaining life of many financial assets. The new
guidance will be effective for the Company on July 1, 2020. Early
adoption will be available on July 1, 2019. The Company is
currently evaluating the effect that ASU No. 2016-13 will have on
its consolidated financial statements and related
disclosures.
54
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
In
March 2016, the FASB issued ASU No. 2016-09, Compensation – Improvement to Employee
Share-Based Payment Accounting, which amends the current
guidance related to stock compensation. The updated guidance
changes how companies account for certain aspects of share-based
payment awards to employees, including the accounting for income
taxes, forfeitures, and statutory tax withholding requirements, as
well as classification in the statement of cash flows. The update
to the standard is effective for the Company on July 1, 2017. The
Company is currently evaluating the effect that ASU No. 2016-09
will have on its consolidated financial statements and related
disclosures.
In
February 2016, the FASB issued ASU No. 2016-02, Leases, related to the recognition of
lease assets and lease liabilities. The new guidance requires
lessees to recognize almost all leases on their balance sheet as a
right-of-use asset and a lease liability, other than leases that
meet the definition of a short- term lease, and requires expanded
disclosures about leasing arrangements. The recognition,
measurement, and presentation of expenses and cash flows arising
from a lease by a lessee have not significantly changed from the
current guidance. Lessor accounting is similar to the current
guidance, but updated to align with certain changes to the lessee
model and the new revenue recognition standard. The new guidance is
effective for the Company on July 1, 2019, with early adoption
permitted. The Company is currently evaluating the impact that ASU
No. 2016-02 will have on its consolidated financial statements and
related disclosures.
In
January 2016, the FASB issued ASU No. 2016-01, Financial Instruments: Recognition and
Measurement of Financial Assets and Financial Liabilities.
The new guidance relates to the recognition and measurement of
financial assets and liabilities. The new guidance makes targeted
improvements to GAAP impacting equity investments (other than those
accounted for under the equity method or consolidated), financial
liabilities accounted for under the fair value election, and
presentation and disclosure requirements for financial instruments,
among other changes. The new guidance is effective for the Company
on July 1, 2018, with early adoption prohibited other than for
certain provisions. The Company is currently evaluating the impact
that ASU No. 2016-01 will have on its consolidated financial
statements and related disclosures.
In
November 2015, the FASB issued ASU No. 2015-17, Income Taxes: Balance Sheet Classification
of Deferred Taxes which simplifies the balance sheet
classification of deferred taxes. The new guidance requires that
deferred tax liabilities and assets be classified as noncurrent in
a classified statement of financial position. The current
requirement that deferred tax liabilities and assets of a
tax-paying component of an entity be offset and presented as a
single amount is not affected by the new guidance. The new guidance
is effective for the Company on July 1, 2017. The new guidance may
be applied either prospectively to all deferred tax liabilities and
assets or retrospectively to all periods presented. The Company is
currently evaluating the impact that ASU No. 2015-17 will have on
its consolidated financial statements and related disclosures;
however, at the present time the Company has recorded a valuation
allowance against its deferred tax assets based on the history of
losses incurred.
In
April 2015, the FASB issued ASU No. 2015-03, Simplifying the Presentation of Debt Issuance
Costs, which requires debt issuance costs related to a
recognized debt liability to be presented on the balance sheet as a
direct deduction from the debt liability, similar to the
presentation of debt discounts. In August 2015, the FASB issued a
clarification that debt issuance costs related to line-of-credit
arrangements were not within the scope of the new guidance and
therefore should continue to be accounted for as deferred assets in
the balance sheet, consistent with existing GAAP. The Company
adopted the retrospective guidance as of July 1, 2016. As a result
of the adoption of ASU No. 2015 03, we made the following
adjustments to the June 30, 2016 consolidated balance sheet: a
$110,441 decrease to prepaid expenses and other current assets, a
$83,215 decrease to other assets, a $110,441 decrease to the
current portion of notes payable, net of discounts and debt
issuance costs, and a $83,215 decrease to the long-term portion of
notes payable, net of discounts and debt issuance
costs.
In
August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements-Going
Concern: Disclosures of Uncertainties about an Entity’s
Ability to Continue as a Going Concern. The amendments in
this update provide guidance in U.S. GAAP about management's
responsibility to evaluate whether there is substantial doubt about
an entity's ability to continue as a going concern and to provide
related footnote disclosures. In doing so, the amendments should
reduce diversity in the timing and content of footnote disclosures.
The new standard was effective for the Company for its fiscal year
ending June 30, 2017. The Company has adopted ASU No. 2014-15 as of
June 30, 2017 without material impact on its consolidated financial
statements or disclosures.
55
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with
Customers, which requires an
entity to recognize the amount of revenue to which it expects to be
entitled for the transfer of promised goods or services to
customers. The ASU will replace most existing revenue recognition
guidance in U.S. GAAP when it becomes effective. In July 2015, the
FASB voted to defer the effective date of the new standard until
fiscal years beginning after December 15, 2017 with early
application permitted for fiscal years beginning after December 15,
2016. With the deferral, the new standard is effective for the
Company on July 1, 2018, with early adoption permitted one year
prior. The standard permits the use of either the retrospective or
cumulative effect transition method. The Company is currently
evaluating the effect that ASU 2014-09 will have on its
consolidated financial statements and related disclosures. The
Company has not yet selected a transition method nor has it
determined the effect of the standard on its ongoing financial
reporting.
(4)
AGREEMENT
WITH GEDEON RICHTER
In
August 2014, the Company entered into a license, co-development and
commercialization agreement with Gedeon Richter on bremelanotide
for FSD in Europe and selected countries. On September 16, 2015,
the Company and Gedeon Richter mutually and amicably agreed to
terminate the license, co-development and commercialization
agreement. In connection with the termination of the license
agreement, all rights and licenses to co-develop and commercialize
bremelanotide for FSD indications granted by the Company under the
license agreement to Gedeon Richter terminated and reverted to the
Company, and neither party has any future material obligations
under the license agreement. Neither the Company nor Gedeon Richter
incurred any early termination penalties or other payment or
reimbursement obligations as a result of the termination of the
license agreement.
The
Company viewed the delivery of the license for bremelanotide as a
revenue generating activity that is part of its ongoing and central
operations. The other elements of the agreement with Gedeon Richter
were considered non-revenue activities associated with the
collaborative arrangement. The Company believes the license had
standalone value from the other elements of the collaborative
arrangement because it conveyed all of the rights necessary to
develop and commercialize bremelanotide in the licensed
territory.
In
August 2013, the Company received an initial payment of $1,000,000
from Gedeon Richter as a non-refundable option fee on the license,
co-development and commercialization agreement, and in September
2014, the Company received €6,700,000 ($8,763,347) on
execution of the definitive agreement. During the year ended June
30, 2015, the upfront payment of €7,500,000 ($9,763,347) was
recorded as license revenue in the consolidated statements of
operations. During the year ended June 30, 2015, the Company
recorded revenue related to a milestone payment of €2,500,000
($3,188,383) upon the initiation of the Company’s Phase 3
clinical trial program in the United States.
As a
result of fluctuations in the conversion rates between the Euro and
the U.S. Dollar between the transaction dates and the settlement
dates, the Company recorded a foreign exchange loss of $284,656 for
the year ended June 30, 2015.
(5)
AGREEMENT
WITH AMAG
On
January 8, 2017, the Company entered into the License Agreement
with AMAG. Under the terms of the License Agreement, the Company
granted to AMAG (i) an exclusive license in all countries of North
America (“the Territory”), with the right to grant
sub-licenses, to research, develop and commercialize products
containing bremelanotide (each a Product, and collectively,
Products), (ii) a non-exclusive license in the Territory, with the
right to grant sub-licenses, to manufacture Products, and (iii) a
non-exclusive license in all countries outside the Territory, with
the right to grant sub-licenses, to research, develop and
manufacture (but not commercialize) the Products.
Following
the satisfaction of certain conditions to closing, the License
Agreement became effective on February 2, 2017. On that date, AMAG
paid the Company $60,000,000 as a one-time initial payment.
Pursuant to the terms of and subject to the conditions in the
License Agreement, AMAG is required to reimburse the Company up to
an aggregate amount of $25,000,000 for reasonable, documented,
direct out-of-pocket expenses incurred by the Company following
February 2, 2017, in connection with the development and regulatory
activities necessary to file an NDA for bremelanotide for HSDD in
the United States related to Palatin’s development
obligations.
56
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
The
Company has determined there is no stand-alone value for the
license, and that the license and the reimbursable direct
out-of-pocket expenses, pursuant to the terms of the License
Agreement, represent a combined unit of accounting which totals
$85,000,000. The Company is recognizing revenue of the combined
unit of accounting over the arrangement using the input-based
proportional method as the Company completes its development
obligations. For year ended June 30, 2017, the Company recognized
$44,723,827 as license and contract revenue related to this
transaction. As of June 30, 2017, $4,657,577 was received and
$15,116,822 was included in accounts receivable relating to
reimbursable expenses. As of June 30, 2017, there is $35,050,572 of
current deferred revenue on the consolidated balance sheet related
to this transaction.
In
addition, pursuant to the terms of and subject to the conditions in
the License Agreement, the Company will be eligible to receive from
AMAG (i) up to $80,000,000 in specified regulatory milestone
payments upon achievement of certain regulatory milestones, and
(ii) up to $300,000,000 in sales milestone payments based on
achievement of certain annual net sales milestones for all Products
in the Territory.
AMAG is
also obligated to pay the Company tiered royalties on annual net
sales of Products, on a product-by-product basis, in the Territory
ranging from the high single-digits to the low double-digits. The
royalties will expire on a product-by-product and
country-by-country basis until the latest to occur of (i) the
earliest date on which there are no valid claims of the
Company’s patent rights covering such Product in such
country, (ii) the expiration of the regulatory exclusivity period
for such Product in such country and (iii) ten years following the
first commercial sale of such Product in such country. Such
royalties are subject to reductions in the event that:
(a) AMAG must license additional third party intellectual
property in order to develop, manufacture or commercialize a
Product, or (b) generic competition occurs with respect to a
Product in a given country, subject to an aggregate cap on such
deductions of royalties otherwise payable to the Company. After the
expiration of the applicable royalties for any Product in a given
country, the license for such Product in such country will become a
fully paid-up, royalty-free, perpetual and irrevocable
license.
The
Company engaged Greenhill & Co. LLC (“Greenhill”)
as the Company’s sole financial advisor in connection with a
potential transaction with respect to bremelanotide. Under the
engagement agreement with Greenhill, the Company was obligated to
pay Greenhill a fee equal to 2% of all proceeds and consideration
paid to the Company by AMAG in connection with the License
Agreement, subject to a minimum fee of $2,500,000. The minimum fee
of $2,500,000, less a credit of $50,000 for an advisory fee
previously paid by the Company, was paid to Greenhill upon the
closing of the licensing transaction. This amount will be credited
toward amounts that become due to Greenhill in the future, provided
that the aggregate fee payable to Greenhill will not be less than
2% of all proceeds and consideration paid to the Company by AMAG in
connection with the License Agreement. The Company will pay
Greenhill an aggregate total of 2% of all proceeds and
consideration paid to the Company by AMAG in connection with the
License Agreement, including future milestone and royalty payments,
after crediting the $2,500,000 that was paid to Greenhill upon
entering into the License Agreement with AMAG. The Company also
reimbursed Greenhill $7,263 for certain expenses incurred in
connection with its advisory services.
Pursuant
to the License Agreement, the Company has assigned to AMAG the
Company’s manufacturing and supply agreements with Catalent
Belgium S.A. to perform fill, finish and packaging of
bremelanotide.
(6)
PREPAID
EXPENSES AND OTHER CURRENT ASSETS
Prepaid
expenses and other
current assets consist of the following:
|
|
June 30,
|
June 30,
|
|
|
2017
|
2016
|
|
Clinical
study costs
|
$657,069
|
$1,146,975
|
|
Insurance
premiums
|
182,966
|
23,010
|
|
Other
|
171,186
|
143,856
|
|
|
$1,011,221
|
$1,313,841
|
57
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
(7)
INVESTMENTS
The
following summarizes the carrying value of our available-for-sale
investments, which consist of corporate debt
securities:
|
|
June 30,
|
June 30,
|
|
|
2017
|
2016
|
|
Cost
|
$1,387,022
|
$1,387,022
|
|
Matured
investments
|
(1,124,999)
|
-
|
|
Amortization
of premium
|
(11,596)
|
(4,522)
|
|
Gross
unrealized loss
|
(590)
|
(1,944)
|
|
Fair
value
|
$249,837
|
$1,380,556
|
(8)
FAIR
VALUE MEASUREMENTS
The
fair value of cash equivalents is classified using a hierarchy
prioritized based on inputs. Level 1 inputs are quoted prices
(unadjusted) in active markets for identical assets or liabilities.
Level 2 inputs are quoted prices for similar assets and liabilities
in active markets or inputs that are observable for the asset or
liability, either directly or indirectly through market
corroboration, for substantially the full term of the financial
instrument. Level 3 inputs are unobservable inputs based on
management’s own assumptions used to measure assets and
liabilities at fair value. A financial asset or liability’s
classification within the hierarchy is determined based on the
lowest level input that is significant to the fair value
measurement.
The
following table provides the assets carried at fair
value:
|
|
Carrying Value
|
Quoted prices in
active markets
(Level 1)
|
Other quoted/observable inputs
(Level 2)
|
Significant unobservable inputs
(Level 3)
|
|
June
30, 2017:
|
|
|
|
|
|
Money
Market Account
|
$40,019,336
|
$40,019,336
|
$-
|
$-
|
|
June
30, 2016:
|
|
|
|
|
|
Money
Market Account
|
$7,782,243
|
$7,782,243
|
$-
|
$-
|
(9)
PROPERTY
AND EQUIPMENT, NET
Property
and equipment, net, consists of the following:
|
|
June 30,
|
June 30,
|
|
|
2017
|
2016
|
|
Office
equipment
|
$1,180,210
|
$1,180,210
|
|
Laboratory
equipment
|
548,706
|
415,303
|
|
Leasehold
improvements
|
751,226
|
751,226
|
|
|
2,480,142
|
2,346,739
|
|
Less:
Accumulated depreciation and amortization
|
(2,281,989)
|
(2,248,938)
|
|
|
$198,153
|
$97,801
|
58
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
The
aggregate cost of assets acquired under capital leases was $146,115
as of June 30, 2017 and 2016. Accumulated amortization associated
with assets acquired under capital leases was $106,115 as of June
30, 2017 and $90,115 as of June 30, 2016.
(10)
ACCRUED
EXPENSES
Accrued
expenses consist of the
following:
|
|
June 30,
|
June 30,
|
|
|
2017
|
2016
|
|
Clinical
study costs
|
$9,138,827
|
$6,983,581
|
|
Other
research related expenses
|
217,307
|
69,609
|
|
Professional
services
|
434,768
|
231,482
|
|
Other
|
730,196
|
483,061
|
|
|
$10,521,098
|
$7,767,733
|
(11)
NOTES PAYABLE:
Notes
payable consist of the following:
|
|
June 30,
|
June 30,
|
|
|
2017
|
2016
|
|
Notes
payable under venture loan
|
$14,333,334
|
$20,000,000
|
|
Unamortized
related debt discount
|
(143,524)
|
(324,799)
|
|
Unamortized
debt issuance costs
|
(83,215)
|
(193,656)
|
|
Notes
payable
|
14,106,595
|
19,481,545
|
|
|
|
|
|
Less:
current portion
|
7,824,935
|
5,374,951
|
|
|
|
|
|
Long-term
portion
|
$6,281,660
|
$14,106,594
|
On
December 23, 2014, the Company closed on a $10,000,000 venture loan
which was led by Horizon Technology Finance Corporation
(“Horizon”). The debt facility is a four year senior
secured term loan that bears interest at a floating coupon rate of
one-month LIBOR (floor of 0.50%) plus 8.50%, and provides for
interest-only payments for the first eighteen months followed by
monthly payments of principal of $333,333 plus accrued interest
through January 1, 2019. The lenders also received five-year
immediately exercisable Series D 2014 warrants to purchase 666,666
shares of common stock exercisable at an exercise price of $0.75
per share. The Company recorded a debt discount of $267,820 equal
to the fair value of these warrants at issuance, which is being
amortized to interest expense over the term of the related debt.
This debt discount is offset against the note payable balance and
included in additional paid-in capital on the Company’s
balance sheet at June 30, 2017 and June 30, 2016. In addition, a
final incremental payment of $500,000 is due on January 1, 2019, or
upon early repayment of the loan. This final incremental payment is
being accreted to interest expense over the term of the related
debt. The Company incurred $209,367 of costs in connection with the
loan. These costs were capitalized as deferred financing costs and
are offset against the note payable balance. These debt issuance
costs are being amortized to interest expense over the term of the
related debt. In addition, if the Company repays all or a portion
of the loan prior to the applicable maturity date, it will pay the
lenders a prepayment penalty fee, based on a percentage of the then
outstanding principal balance, equal to 3% if the prepayment occurs
on or before 18 months after the funding date thereof or 1% if the
prepayment occurs more than 18 months after, but on or before 30
months after, the funding date.
59
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
On July
2, 2015, the Company closed on a $10,000,000 venture loan led by
Horizon. The debt facility is a four-year senior secured term loan
that bears interest at a floating coupon rate of one-month LIBOR
(floor of 0.50%) plus 8.50% and provides for interest-only payments
for the first eighteen months followed by monthly payments of
principal of $333,333 plus accrued interest through August 1, 2019.
The lenders also received five-year immediately exercisable Series
G warrants to purchase 549,450 shares of the Company’s common
stock exercisable at an exercise price of $0.91 per share. The
Company has recorded a debt discount of $305,196 equal to the fair
value of these warrants at issuance, which is being amortized to
interest expense over the term of the related debt. This debt
discount is offset against the note payable balance and is included
in additional paid-in capital on the Company’s balance sheet
at June 30, 2017 and June 30, 2016. In addition, a final
incremental payment of $500,000 is due on August 1, 2019, or upon
early repayment of the loan. This final incremental payment is
being accreted to interest expense over the term of the related
debt. The Company incurred $146,115 of costs in connection with the
loan agreement. These costs were capitalized as deferred financing
costs and are offset against the note payable balance. These debt
issuance costs are being amortized to interest expense over the
term of the related debt. In addition, if the Company repays all or
a portion of the loan prior to the applicable maturity date, it
will pay the lenders a prepayment penalty fee, based on a
percentage of the then outstanding principal balance, equal to 3%
if the prepayment occurs on or before 18 months after the funding
date thereof or 1% if the prepayment occurs more than 18 months
after, but on or before 30 months after, the funding
date.
The
Company’s obligations under the 2015 amended and restated
loan agreement, which includes both the 2014 venture loan and the
2015 venture loan, are secured by a first priority security
interest in substantially all of its assets other than its
intellectual property. The Company also has agreed to specified
limitations on pledging or otherwise encumbering its intellectual
property assets. The 2015 amended and restated loan agreement
includes customary affirmative and restrictive covenants, but does
not include any covenants to attain or maintain specified financial
metrics. The loan agreement includes customary events of default,
including payment defaults, breaches of covenants, change of
control and a material adverse change default. Upon the occurrence
of an event of default and following any applicable cure periods, a
default interest rate of an additional 5% may be applied to the
outstanding loan balances, and the lenders may declare all
outstanding obligations immediately due and payable and take such
other actions as set forth in the loan agreement. As of June 30,
2017, the Company was in compliance with all of its loan covenants.
Scheduled future principal payments related to notes payable as of
June 30, 2017 are as follows:
|
Year Ending June 30,
|
|
|
2018
|
$8,000,000
|
|
2019
|
6,000,000
|
|
2020
|
333,334
|
|
|
14,333,334
|
|
Less:
Unamortized debt discount and issuance costs
|
(226,739)
|
|
Net
|
$14,106,595
|
(12)
COMMITMENTS
AND CONTINGENCIES
Operating Leases – The Company currently leases
facilities under two non-cancelable operating leases. The lease on
our corporate offices was renewed effective July 1, 2015 and
expires on June 30, 2020 and in June 2016 we entered into a lease
for approximately 1,700 square feet of laboratory space which
expires in August 2018. Future minimum lease payments under these
leases are as follows:
|
Year Ending June 30,
|
|
|
2018
|
280,923
|
|
2019
|
232,000
|
|
2020
|
221,344
|
|
|
$734,267
|
60
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
For the
years ended June 30, 2017, 2016 and 2015, rent expense was
$261,580, $256,642, and $222,215, respectively.
Capital Leases – The Company has acquired certain of
its equipment under leases classified as capital leases. Scheduled
future payments related to capital leases as of June 30, 2017 are
as follows:
|
Year Ending June 30,
|
|
|
2018
|
$14,568
|
|
Amount
representing interest
|
(244)
|
|
Net
|
$14,324
|
Employment Agreements – The Company has employment
agreements with two executive officers which provide a stated
annual compensation amount, subject to annual increases, and annual
bonus compensation in an amount to be approved by the
Company’s Board of Directors. Each agreement allows the
Company or the employee to terminate the agreement in certain
circumstances. In some circumstances, early termination by the
Company may result in severance pay to the employee for a period of
18 to 24 months at the salary then in effect, continuation of
health insurance premiums over the severance period and immediate
vesting of all stock options and restricted stock units.
Termination following a change in control will result in a lump sum
payment of one and one-half to two times the salary then in effect
and immediate vesting of all stock options and restricted stock
units.
Employee Retirement Savings Plan – The Company
maintains a defined contribution 401(k) plan for the benefit of its
employees. The Company currently matches a portion of employee
contributions to the plan. For the years ended June 30, 2017, 2016
and 2015, Company contributions were $199,264, $138,184, and
$147,203, respectively.
Contingencies – The Company accounts for litigation
losses in accordance with ASC 450-20, “Loss
Contingencies.” Under ASC 450-20, loss contingency provisions
are recorded for probable losses when management is able to
reasonably estimate the loss. Any outcome upon settlement that
deviates from the Company’s best estimate may result in
additional expense or in a reduction in expense in a future
accounting period. The Company records legal expenses associated
with such contingencies as incurred.
The
Company is involved, from time to time, in various claims and legal
proceedings arising in the ordinary course of its business. The
Company is not currently a party to any such claims or proceedings
that, if decided adversely to it, would either individually or in
the aggregate have a material adverse effect on its business,
financial condition or results of operations.
(13)
STOCKHOLDERS’
DEFICIENCY
Series A Convertible Preferred Stock – As of June
30, 2017, 4,030 shares of Series A Convertible Preferred Stock were
outstanding. Each share of Series A Convertible Preferred Stock is
convertible at any time, at the option of the holder, into the
number of shares of common stock equal to $100 divided by the
Series A Conversion Price. As of June 30, 2017, the Series A
Conversion Price was $6.65, so each share of Series A Convertible
Preferred Stock is currently convertible into approximately 15.0
shares of common stock. The Series A Conversion Price is subject to
adjustment, under certain circumstances, upon the sale or issuance
of common stock for consideration per share less than either (i)
the Series A Conversion Price in effect on the date of such sale or
issuance, or (ii) the market price of the common stock as of the
date of such sale or issuance. The Series A Conversion Price is
also subject to adjustment upon the occurrence of a merger,
reorganization, consolidation, reclassification, stock dividend or
stock split which will result in an increase or decrease in the
number of shares of common stock outstanding. Shares of Series A
Convertible Preferred Stock have a preference in liquidation,
including certain merger transactions, of $100 per share, or
$403,000 in the aggregate as of June 30, 2017. Additionally, the
Company may not pay a dividend or make any distribution to holders
of any class of stock unless the Company first pays a special
dividend or distribution of $100 per share to holders of the Series
A Convertible Preferred Stock.
61
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
Financing Transactions – On December 6, 2016, the
Company closed on an underwritten public offering of units, with
each unit consisting of a share of common stock and a Series J
warrant to purchase 0.50 of a share of common stock. Gross proceeds
of the offering were $16,500,000, with net proceeds to the Company,
after deducting underwriting discounts and commissions and offering
expenses, of $15,386,076. The Company issued 25,384,616 shares of
common stock and Series J warrants to purchase 12,692,310 shares of
common stock at an initial exercise price of $0.80 per share, which
warrants are exercisable immediately upon issuance and expire on
the fifth anniversary of the date of issuance. The Series J
warrants are subject to a limitation on their exercise if the
holder and its affiliates would beneficially own more than 9.99%,
or 4.99% for certain holders, of the total number of the
Company’s shares of common stock following such
exercise.
On
August 4, 2016, the Company closed on an underwritten offering of
units, with each unit consisting of a share of common stock and a
Series H warrant to purchase 0.75 of a share of common stock.
Investors whose purchase of units in the offering would result in
them beneficially owning more than 9.99% of the Company’s
outstanding common stock following the completion of the offering
had the opportunity to acquire units with Series I prefunded
warrants substituted for any common stock they would have otherwise
acquired. Gross proceeds of the offering were $9,225,000, with net
proceeds to the Company, after deducting offering expenses, of
$8,470,897. The Company issued 11,481,481 shares of common stock
and ten-year prefunded Series I warrants to purchase 2,218,045
shares of common stock at an exercise price of $0.01, together with
Series H warrants to purchase 10,274,646 shares of common stock at
an exercise price of $0.70 per share.
The
Series I warrants were exercisable immediately upon issuance and
were exercised during the year ended June 30, 2017. The Series H
warrants are exercisable at an initial exercise price of $0.70 per
share, are exercisable commencing six months following the date of
issuance and expire on the fifth anniversary of the date of
issuance. The Series H warrants are subject to a limitation on
their exercise if the holder and its affiliates would beneficially
own more than 9.99% of the total number of the Company's shares of
common stock following such exercise.
On July
2, 2015, the Company closed on a private placement of Series E
warrants to purchase 21,917,808 shares of common stock and Series F
warrants to purchase 2,191,781 shares of common stock. Certain
funds managed by QVT Financial LP (“QVT”) invested
$5,000,000 and another accredited investment fund invested
$15,000,000. The funds paid $0.90 for each Series E warrant and
$0.125 for each Series F warrant, resulting in gross proceeds to
the Company of $20,000,000, with net proceeds, after deducting
estimated offering expenses, of approximately
$19,834,278.
The
Series E warrants, which may be exercised on a cashless basis, are
exercisable immediately upon issuance at an initial exercise price
of $0.01 per share and expire on the tenth anniversary of the date
of issuance. The Series E warrants are subject to a limitation
on their exercise if QVT and its affiliates would beneficially own
more than 9.99% (4.99% for the other accredited investment fund
holder) of the total number of Palatin's shares of common stock
following such exercise. The Series F warrants are exercisable at
an initial exercise price of $0.91 per share, exercisable
immediately upon issuance and expire on the fifth anniversary of
the date of issuance. The Series F warrants are subject to the same
beneficial ownership limitation as the Series E
warrants.
The
purchase agreement for the private placement provides that the
purchasers have certain rights until the earlier of approval of
bremelanotide for FSD by the U.S. Food and Drug Administration and
July 3, 2018, including rights of first refusal and participation
in any subsequent equity or debt financing. The purchase agreement
also contains certain restrictive covenants so long as the funds
continue to hold specified amounts of warrants or beneficially own
specified amounts of the outstanding shares of common
stock.
62
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
Outstanding Stock Purchase Warrants – As of June 30,
2017, the Company had outstanding warrants exercisable for shares
of common stock as follows:
|
Shares of Common
|
|
Exercise Price per
|
|
Latest Termination
|
|
Stock
|
|
Share
|
|
Date
|
|
6,732,307
|
|
0.01
|
|
September 27, 2022
|
|
666,666
|
|
0.75
|
|
December 23, 2019
|
|
11,513,514
|
|
0.01
|
|
December 23, 2024
|
|
2,191,781
|
|
0.91
|
|
July 2, 2020
|
|
549,450
|
|
0.91
|
|
July 2, 2020
|
|
16,917,808
|
|
0.01
|
|
July 2, 2025
|
|
10,274,646
|
|
0.70
|
|
August 4, 2021
|
|
25,000
|
|
0.70
|
|
August 4, 2021
|
|
12,692,310
|
|
0.80
|
|
December 6, 2021
|
|
61,563,482
|
|
|
|
|
During
the year ended June 30, 2017, the Company issued 38,141,991 shares
of common stock pursuant to the cashless exercise provisions of
warrants at an exercise price of $0.01 per share, and during the
year ended June 30, 2017, the Company received $164,358 and issued
16,435,811 shares of common stock pursuant to the exercise of
warrants at an exercise price of $0.01 per share. As of June 30,
2017, there were 35,163,629 warrants outstanding at an exercise
price of $0.01 per share.
During
the year ended June 30, 2016, the Company issued 10,890,889 shares
of common stock pursuant to the cashless exercise provisions of
warrants at an exercise price of $0.01. During the year ended June
30, 2015, the Company received $254,148 and issued 15,130,573
shares of common stock pursuant to the exercise of warrants at
exercise prices of $0.01 and $1.00, including warrants exercised
pursuant to cashless exercise provisions.
On
October 31, 2016, in connection with a contract for financial
advisory services, we issued to each of PSL Business Development
Consulting and SARL Avisius, or their permitted designees, as
partial consideration for services, a warrant to purchase up to
12,500 shares of the Company’s common stock at an exercise
price of $0.70 per share. The warrants are exercisable at any time,
and expire on August 4, 2021. The Company recorded stock-based
compensation related to these stock warrants of $6,885 for the year
ended June 30, 2017.
Stock Plan – The Company’s 2011 Stock Incentive
Plan was approved by the Company’s stockholders at the annual
meeting of stockholders held in May 2011 and amended at the annual
meeting of stockholders held on June 8, 2017. The 2011 Stock
Incentive Plan provides for incentive and nonqualified stock option
grants and other stock-based awards to employees, non-employee
directors and consultants for up to 22,500,000 shares of common
stock. The 2011 Stock Incentive Plan is administered under the
direction of the Board of Directors, which may specify grant terms
and recipients. Options granted by the Company generally expire ten
years from the date of grant and generally vest over three to four
years. The 2005 Stock Plan was terminated and replaced by the 2011
Stock Incentive Plan, and shares of common stock that were
available for grant under the 2005 Stock Plan became available for
grant under the 2011 Stock Incentive Plan. No new awards can be
granted under the 2005 Stock Plan, but awards granted under the
2005 Stock Plan remain outstanding in accordance with their terms.
As of June 30, 2017, 6,525,778 shares were available for grant
under the 2011 Stock Incentive Plan.
The
Company also has outstanding options that were granted under the
2005 Stock Plan. The Company expects to settle option exercises
under any of its plans with authorized but currently unissued
shares.
63
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
The
following table summarizes option activity and related information
for the years ended June 30, 2017, 2016 and 2015:
|
|
Number of Shares
|
Weighted Average Exercise Price
|
Weighted Average Remaining Term in Years
|
Aggregate Intrinsic Value
|
|
|
|
|
|
|
|
Outstanding
- July 1, 2014
|
4,241,973
|
$1.63
|
7.7
|
|
|
|
|
|
|
|
|
Granted
|
975,800
|
1.06
|
|
|
|
Forfeited
|
(78,810)
|
1.92
|
|
|
|
Expired
|
(8,733)
|
36.47
|
|
|
|
|
|
|
|
|
|
Outstanding
- June 30, 2015
|
5,130,230
|
1.46
|
7.3
|
|
|
|
|
|
|
|
|
Granted
|
355,000
|
0.54
|
|
|
|
Forfeited
|
(170,550)
|
0.80
|
|
|
|
Expired
|
(52,940)
|
22.53
|
|
|
|
|
|
|
|
|
|
Outstanding
- June 30, 2016
|
5,261,740
|
1.21
|
6.2
|
|
|
|
|
|
|
|
|
Granted
|
4,119,000
|
0.46
|
|
|
|
Forfeited
|
(410,388)
|
1.12
|
|
|
|
Expired
|
(43,220)
|
22.59
|
|
|
|
|
|
|
|
|
|
Outstanding
- June 30, 2017
|
8,927,132
|
$0.76
|
7.5
|
$-
|
|
|
|
|
|
|
|
Exercisable
at June 30, 2017
|
4,297,394
|
$1.02
|
5.3
|
$-
|
|
|
|
|
|
|
|
Expected
to vest at June 30, 2017
|
3,446,101
|
$0.54
|
9.5
|
$-
|
|
|
|
|
|
|
For the
years ended June 30, 2017, 2016 and 2015, the fair value of option
grants was estimated at the grant date using the Black-Scholes
model. The Company’s weighted average assumptions for the
years ended June 30, 2017, 2016 and 2015 were as
follows:
|
|
2017
|
2016
|
2015
|
|
|
|
|
|
|
Risk-free
interest rate
|
1.7%
|
1.4%
|
1.9%
|
|
Volatility
factor
|
75.0%
|
73.5%
|
83.4%
|
|
Dividend
yield
|
0%
|
0%
|
0%
|
|
Expected
option life (years)
|
6.2
|
5.7
|
6.1
|
|
Weighted
average grant date fair value per share
|
$0.27
|
$0.35
|
$0.76
|
Expected
volatilities are based on the Company’s historical
volatility. The expected term of options is based upon the
simplified method, which represents the average of the vesting term
and the contractual term. The risk-free interest rate is based on
U.S. Treasury yields for securities with terms approximating the
expected term of the option.
For the
years ended June 30, 2017, 2016 and 2015 the Company recorded
stock-based compensation related to stock options of $547,953,
$529,454 and $572,609, respectively. As of June 30, 2017, there was
$1,017,672 of unrecognized compensation cost related to unvested
options, which is expected to be recognized over a weighted-average
period of 2.86 years.
64
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
In June
2017, the Company granted 1,797,000 options to its executive
officers, 780,000 options to its employees and 378,000 options to
its non-employee directors under the Company’s 2011 Stock
Incentive Plan. The Company is amortizing the fair value of these
options of $445,533, $194,689 and $89,220, respectively, over the
vesting period.
In
September 2016, the Company granted 828,000 options to its
executive officers and 336,000 options to its employees under the
Company’s 2011 Stock Incentive Plan. The Company is
amortizing the fair value of the options vesting over a 48 month
period, consisting of 595,000 options granted to its executive
officers and all options granted to its employees, of $188,245 and
$106,303, respectively, over the vesting period. The remaining
233,000 options granted to its executive officers vest 12 months
from the date of grant, and the Company is amortizing the fair
value of these options of $67,160 over this vesting
period.
In June
2016, the Company granted 262,500 options to its non-employee
directors under the Company’s 2011 Stock Incentive Plan. The
Company amortized the fair value of these options of $81,435, over
the vesting period.
During
the year ended June 30, 2016, the Company granted an aggregate
92,500 options to certain employees under the Company’s 2011
Stock Incentive Plan. The Company is amortizing the fair value of
these options of $41,470, over the vesting period.
In June
2015, the Company granted 570,000 options to its executive
officers, 185,800 options to its employees and 160,000 options to
its non-employee directors under the Company’s 2011 Stock
Incentive Plan. The Company is amortizing the fair value of these
options of $446,748, $145,439 and $111,876, respectively, over the
vesting period.
Unless
otherwise stated, stock options granted to the Company’s
executive officers and employees vest over a 48 month period, while
stock options granted to its non-employee directors vest over a 12
month period.
Restricted Stock Units – The following table
summarizes restricted stock award activity for the years ended June
30, 2017, 2016 and 2015:
|
|
2017
|
2016
|
2015
|
|
Outstanding
at beginning of year
|
2,665,768
|
1,028,017
|
957,150
|
|
Granted
|
3,192,000
|
2,302,500
|
785,800
|
|
Forfeited
|
(68,751)
|
(2,563)
|
(9,100)
|
|
Vested
|
(579,400)
|
(662,186)
|
(705,833)
|
|
Outstanding
at end of year
|
5,209,617
|
2,665,768
|
1,028,017
|
For the
years ended June 30, 2017, 2016 and 2015 the Company recorded
stock-based compensation related to restricted stock units of
$1,202,421, $1,297,724 and $594,670, respectively.
In June
2017, the Company granted 1,140,000 restricted stock units to its
executive officers, 780,000 restricted stock units to its employees
and 378,000 restricted stock units to its non-employee directors
under the Company’s 2011 Stock Incentive Plan. The Company is
amortizing the fair value of these restricted stock units of
$421,800, $288,600, and $139,860, respectively, over the vesting
period.
In
September 2016, the Company granted 558,000 restricted stock units
to its executive officers, 415,000 of which vest over 24 months and
143,000 of which vest at 12 months, and 336,000 restricted stock
units to its employees under the Company’s 2011 Stock
Incentive Plan. The Company is amortizing the fair value of the
restricted stock units of $284,580, and $171,360, respectively,
over the vesting periods.
In June
2016, the Company granted 262,500 restricted stock units to its
non-employee directors under the Company’s 2011 Stock
Incentive Plan. The Company amortized the fair value of these
options of $131,250, over the vesting period.
65
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
In
December 2015, the Company granted 625,000 performance-based
restricted stock units to its executive officers and 200,000
performance-based restricted stock units to its employees under the
Company’s 2011 Stock Incentive Plan, which vest during the
performance period, ending December 31, 2017, if and upon the
earlier of: i) achievement of a closing price for the
Company’s common stock equal to or greater than $1.20 per
share for 20 consecutive trading days, which is considered a market
condition, or ii) entering into a collaboration agreement (U.S. or
global) of bremelanotide for FSD, which is considered a performance
condition. This performance condition was deemed met as of February
2, 2017, the effective date of the License Agreement on
bremelanotide with AMAG. Prior to meeting the performance
condition, the Company determined that it was not probable of
achievement on the date of grant since meeting the condition was
outside the control of the Company. The fair value of these awards,
as calculated under a multifactor Monte Carlo simulation, was
$338,250 and was recognized over the derived service period which
was through December 2016. Upon the achievement of the performance
condition, which occurred in the three month period ended March 31,
2017 the grant date fair value was utilized and an incremental
$222,075 was recognized as stock-based compensation expense during
the three months ended March 31, 2017. Also, in December 2015, the
Company granted 625,000 restricted stock units to its executive
officers, 340,000 restricted stock units to its non-employee
directors and 200,000 restricted stock units to its employees under
the Company’s 2011 Stock Incentive Plan. For executive
officers and employees, the restricted stock units vest 25% on the
date of grant and 25% on the first, second and third anniversary
dates from the date of grant. For non-employee directors, the
restricted stock units vest 50% on the first and second anniversary
dates from the date of grant. The Company is amortizing the fair
value of these restricted stock units of $425,000, $231,200 and
$136,000, respectively, over the vesting period.
In June
2015, the Company granted 400,000 restricted stock units to its
executive officers, 185,800 restricted stock units to its employees
and 160,000 restricted stock units to its non-employee directors
under the Company’s 2011 Stock Incentive Plan. The Company is
amortizing the fair value of these restricted stock units of
$432,000, $200,664, and $172,800, respectively, over the vesting
period. In addition, in June 2015, the Company granted 20,000
restricted stock units to former non-employee directors in
recognition of their prior services. These restricted stock units
vested upon issuance and the Company recognized the fair value of
these restricted stock units of $21,600 as stock-based compensation
expense which was included in general and administrative expense at
June 30, 2015.
In
January 2015, the Company granted 20,000 restricted stock units to
an employee and is amortizing the fair value of these restricted
stock units of $16,000 over a 24 month vesting period.
Unless
otherwise stated, restricted stock units granted to the
Company’s executive officers, employees and non-employee
directors in 2015 and 2014 vest over 24 months, 48 months and 12
months, respectively.
In
connection with the vesting of restricted share units during the
years ended June 30, 2017, 2016 and 2015, the Company withheld
75,993, 123,483 and 174,568 shares with aggregate values of
$27,088, $58,401 and $164,136, respectively, in satisfaction of
minimum tax withholding obligations.
(14)
INCOME
TAXES
For
fiscal 2017, the Company incurred $500,000 of federal AMT expense
based on federal alternative minimum taxable income attributable to
the $60,000,000 initial payment from AMAG. For fiscal 2016 the
Company had no income tax expense because of operating losses or a
tax benefit from the sale of New Jersey state net operating loss
carryforwards on account that it reached the state limits on the
sale of New Jersey state net operating loss carryforwards and tax
credits. In fiscal 2015 the Company recorded an income tax benefit
of $531,508 for amounts recognized for the sale of New Jersey state
net operating loss carryforwards and tax credits.
Deferred
tax assets and liabilities are determined based on the estimated
future tax effect of differences between the financial statement
and tax reporting basis of assets and liabilities, as well as for
AMT credit carryforwards, net operating loss carryforwards and
research and development credit carryforwards, given the provisions
of existing tax laws.
As of
June 30, 2017, the Company had state net operating loss
carryforwards of approximately $111,000,000, which will expire, if
not utilized, between 2017 and 2037, federal net operating loss
carryforwards of approximately $278,000,000, federal research and
development credits of approximately $11,900,000, which expire, if
not utilized, between 2020 and 2037, and AMT credits of $500,000,
which can be carried forward indefinitely if not
utilized.
66
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
The Tax
Reform Act of 1986 (the “Act”) provides for limitation
on the use of these net operating loss and research and development
tax credit carryforwards following certain ownership changes (as
defined by the Act) that could limit the Company’s ability to
utilize these carryforwards. Since its inception, the Company has
completed several financings and sales of common stock which has
resulted in multiple ownership changes defined by Section 382 of
the Act. Accordingly, the Company’s ability to utilize the
aforementioned carryforwards are subject to limitation under
Section 382. The Company does
have adequate levels of available net operating loss carryforwards
that are not subject to limitation under Section 382 to offset
taxable income during the tax year ended June 30, 2017. If the
Company undergoes a future ownership change or as it completes its
Section 382 limitation assessment, any unutilized carryforwards
that were not previously subject to a Section 382 limitation may
become subject to limitation which may result in a significant
limitation and loss of net operating loss carryforwards.
Additionally, U.S.
tax laws limit the time during which these carryforwards may be
applied against future taxes; therefore, the Company may not be
able to take full advantage of these carryforwards for federal
income tax purposes. Accordingly, a portion of the carryforwards
may expire unutilized.
The
Company’s net deferred tax assets are as
follows:
|
|
June 30,
|
June 30,
|
|
|
2017
|
2016
|
|
Net
operating loss carryforwards
|
$103,845,000
|
$114,081,000
|
|
Research
and development and AMT tax credits
|
12,360,000
|
9,965,000
|
|
Basis
difference in deferred revenue
|
14,318,000
|
-
|
|
Basis
differences in fixed assets and other
|
1,510,000
|
1,491,000
|
|
|
132,033,000
|
125,537,000
|
|
Valuation
allowance
|
(132,033,000)
|
(125,537,000)
|
|
Net
deferred tax assets
|
$-
|
$-
|
In
assessing the realizability of deferred tax assets, the Company
considers whether it is more likely than not that some portion or
all of the deferred tax assets will not be realized. The ultimate
realization of deferred tax assets is dependent upon the generation
of future taxable income and the application of loss limitation
provisions related to ownership changes. The Company assesses the
available positive and negative evidence to estimate if sufficient
future taxable income will be generated to use the existing
deferred tax assets. The Company also considers the scheduled
reversal of deferred tax liabilities (including the impact of
available carryback and carryforward periods), projected future
taxable income, and tax-planning strategies in making this
assessment. A significant piece of objective negative evidence
evaluated was the cumulative loss incurred over the three-year
period from July 1, 2014 through June 30, 2017. On the basis of
these considerations, the Company continued to recognize a full
valuation allowance against its net deferred tax assets as of June
30, 2017 and 2016.
The
deferred tax asset related to deferred revenue is attributable to
the AMAG arrangement at June 30, 2017 at a combined incremental tax
rate of approximately 40%.
The
Company recognizes interest expense and penalties on uncertain
income tax positions as a component of interest expense. No
interest expense or penalties were recorded for uncertain income
tax matters in fiscal 2017, 2016 or 2015. As of June 30, 2017 and
2016, the Company had no liabilities for uncertain income tax
matters.
(15)
CONSOLIDATED
QUARTERLY FINANCIAL DATA - UNAUDITED
The
following tables provide quarterly data for the years ended June
30, 2017 and 2016.
67
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
|
|
Three Months Ended
|
|||
|
|
June 30,
|
March 31,
|
December 31,
|
September 30,
|
|
|
2017
|
2017
|
2016
|
2016
|
|
|
(amounts
in thousands, except per share data)
|
|||
|
Revenues
|
$33,900
|
$10,824
|
$-
|
$-
|
|
Operating
expenses
|
19,581
|
13,836
|
9,441
|
12,435
|
|
Other
expense, net
|
(504)
|
(552)
|
(588)
|
(618)
|
|
Income
(loss) before income taxes
|
13,815
|
(3,564)
|
(10,029)
|
(13,053)
|
|
Income
taxes
|
(500)
|
-
|
-
|
-
|
|
Net
income (loss)
|
$13,315
|
$(3,564)
|
$(10,029)
|
$(13,053)
|
|
Basic
net income (loss) per common
share
|
$0.07
|
$(0.02)
|
$(0.06)
|
$(0.08)
|
|
Diluted
net income (loss) per common
share
|
$0.07
|
$(0.02)
|
$(0.06)
|
$(0.08)
|
|
Weighted average number of common
shares outstanding used
in computing basic net income
(loss) per common share
|
196,874,145
|
196,580,519
|
177,798,511
|
165,848,269
|
|
Weighted average number of common
shares outstanding used
in computing diluted net income
(loss) per common share
|
197,948,721
|
196,580,519
|
177,798,511
|
165,848,269
|
|
|
|
|
|
|
|
|
Three Months Ended
|
|||
|
|
June 30,
|
March 31,
|
December 31,
|
September 30,
|
|
|
2016
|
2016
|
2015
|
2015
|
|
|
(amounts
in thousands, except per share data)
|
|||
|
Revenues
|
$-
|
$-
|
$-
|
$-
|
|
Operating
expenses
|
12,738
|
12,086
|
12,628
|
11,798
|
|
Other
expense, net
|
(618)
|
(611)
|
(622)
|
(612)
|
|
Loss
before income taxes
|
(13,356)
|
(12,697)
|
(13,250)
|
(12,410)
|
|
Income
taxes
|
-
|
-
|
-
|
-
|
|
Net
loss
|
$(13,356)
|
$(12,697)
|
$(13,250)
|
$(12,410)
|
|
Basic
and diluted net loss per common
share
|
$(0.09)
|
$(0.08)
|
$(0.08)
|
$(0.08)
|
|
Weighted average number of common
shares outstanding used
in computing basic and diluted
net loss per common share
|
156,841,053
|
156,744,867
|
156,456,801
|
156,176,618
|
68
PALATIN TECHNOLOGIES, INC.
and Subsidiary
Notes to Consolidated Financial Statements
(16)
SUBSEQUENT
EVENTS
Licensing Agreement – On September 6, 2017, the
Company entered into a collaboration and license agreement with
Fosun, for exclusive rights to commercialize bremelanotide in the
territories of mainland China, Taiwan, Hong Kong S.A.R. and Macau
S.A.R.
Under
the terms of the agreement, Palatin will receive an upfront payment
of $5.0 million and a $7.5 million milestone payment when
regulatory approval in China is obtained. Palatin has the potential
to receive up to $92.5 million in sales related milestones payments
and high single-digit to low double-digit royalties on net sales in
the licensed territory. All development, regulatory, sales,
marketing, and commercial activities and associated costs in the
licensed territory will be the sole responsibility of
Fosun.
Outstanding Common Stock – Between July 1, 2017 and
September 21, 2017, the Company issued 7,091,736 shares of common
stock pursuant to the cashless exercise provisions of warrants at
an exercise price of $0.01 and issued 11,438,356 shares of common
stock pursuant to the exercise of warrants at an exercise price of
$0.01 per share. As of September 21, 2017, warrants with an
exercise price of $0.01 per share to purchase 16,472,412 shares of
common stock are outstanding, all of which include cashless
exercise provisions.
69
Item 9. Changes in and Disagreements with Accountants on Accounting
and Financial Disclosure
None.
Item 9A. Controls and Procedures.
Our
management carried out an evaluation, with the participation of our
Chief Executive Officer and our Chief Financial Officer, of the
effectiveness of our disclosure controls and procedures (as defined
in Rules 13a-15(e) or 15d-15(e) of the Exchange Act) as of the end
of the period covered by this report. Based upon this evaluation,
our Chief Executive Officer and our Chief Financial Officer
concluded that, as of June 30, 2017, our disclosure controls and
procedures were effective.
A
control system, no matter how well designed and operated, cannot
provide absolute assurance that the objectives of the control
system are met, and no evaluation of controls can provide absolute
assurance that all control issues and instances of fraud, if any,
within a company have been detected.
Management’s Report on Internal Control Over Financial
Reporting
Management
is responsible for establishing and maintaining adequate internal
control over financial reporting as defined in Rule 13a-15(f) or
15d-15(f) of the Exchange Act. Our internal control system was
designed to provide reasonable assurance to management and the
board of directors regarding the preparation and fair presentation
of published financial statements.
All
internal control systems, no matter how well designed, have
inherent limitations. Therefore, even those systems determined to
be effective can provide only reasonable assurance with respect to
financial statement preparation and presentation.
There
was no change in our internal control over financial reporting
during the fourth quarter of the period covered by this Annual
Report that has materially affected, or is reasonably likely to
materially affect, our internal control over financial
reporting.
Management
assessed the effectiveness of our internal control over financial
reporting as of June 30, 2017. In making this assessment, it used
the criteria set forth by the Committee of Sponsoring Organizations
of the Treadway Commission (COSO) in Internal Control-Integrated Framework as
adopted in 2013. Based on its assessment, management
believes that, as of June 30, 2017, our internal control over
financial reporting is effective based on those
criteria.
Item 9B. Other Information.
None.
70
PART III
Item 10. Directors, Executive Officers and Corporate
Governance.
Identification of Directors
The
following table sets forth the names, ages, positions and committee
memberships of our current directors. All directors hold office
until the next annual meeting of stockholders or until their
successors have been elected and qualified. All current directors
were elected at our annual stockholders’ meeting on June 8,
2017, except for Anthony M. Manning, Ph.D., who was appointed to
the board of directors on September 7, 2017.
|
Name
|
|
Age
|
|
Position with
Palatin
|
|
Carl
Spana, Ph.D.
|
|
55
|
|
Chief
Executive Officer, President and a Director
|
|
John
K.A. Prendergast, Ph.D. (3)
|
|
63
|
|
Director, Chairman
of the Board of Directors
|
|
Robert
K. deVeer, Jr. (1) (2)
|
|
71
|
|
Director
|
|
J.
Stanley Hull (1) (2)
|
|
65
|
|
Director
|
|
Alan W.
Dunton, M.D. (1) (2)
|
|
63
|
|
Director
|
|
Angela
Rossetti (1) (3)
|
|
64
|
|
Director
|
|
Arlene
M. Morris (2) (3)
|
|
65
|
|
Director
|
|
Anthony
M. Manning
|
|
55
|
|
Director
|
(1)
Member of the audit committee.
(2)
Member of the compensation committee.
(3)
Member of the nominating and corporate governance
committee.
CARL
SPANA, Ph.D., co-founder of Palatin, has been our Chief Executive
Officer and President since June 14, 2000. He has been a director
of Palatin since June 1996 and has been a director of our
wholly-owned subsidiary, RhoMed Incorporated, since July 1995. From
June 1996 through June 14, 2000, Dr. Spana served as an executive
vice president and our chief technical officer. From June 1993 to
June 1996, Dr. Spana was vice president of Paramount Capital
Investments, LLC, a biotechnology and biopharmaceutical merchant
banking firm, and of The Castle Group Ltd., a medical venture
capital firm. Through his work at Paramount Capital Investments and
The Castle Group, Dr. Spana co-founded and acquired several private
biotechnology firms. From July 1991 to June 1993, Dr. Spana was a
Research Associate at Bristol-Myers Squibb, a publicly-held
pharmaceutical company, where he was involved in scientific
research in the field of immunology. He was previously a member of
the board of the life science company AVAX Technologies, Inc. Dr.
Spana received his Ph.D. in molecular biology from The Johns
Hopkins University and his B.S. in biochemistry from Rutgers
University.
Dr.
Spana’s qualifications for our board include his leadership
experience, business judgment and industry experience. As a senior
executive of Palatin for over nineteen years, he provides in-depth
knowledge of our company, our drug products under development and
the competitive and corporate partnering landscape.
JOHN
K.A. PRENDERGAST, Ph.D., co-founder of Palatin, has served as the
non-executive Chairman of the board since June 14, 2000, and as a
director since August 1996. While Mr. Prendergast has served as a
member of the board, he does not, and has not, served in a
management or operational role with the company. Dr. Prendergast
has been president and sole stockholder of Summercloud Bay, Inc.,
an independent consulting firm providing services to the
biotechnology industry, since 1993. Dr. Prendergast is a director
and executive chairman of the board of directors of Antyra, Inc., a
privately held biopharmaceutical firm, and a director of Heat
Biologics, Inc., a publicly traded clinical stage immunotherapy
company. He was previously a member of the board of the life
science companies AVAX Technologies, Inc., Avigen, Inc. and
MediciNova, Inc. From October 1991 through December 1997, Dr.
Prendergast was a managing director of The Castle Group Ltd., a
medical venture capital firm. Dr. Prendergast received his M.Sc.
and Ph.D. from the University of New South Wales, Sydney, Australia
and a C.S.S. in administration and management from Harvard
University.
71
Dr.
Prendergast brings a historical perspective to our board coupled
with extensive industry experience in corporate development and
finance in the life sciences field. His prior service on other
publicly traded company boards provides experience relevant to good
corporate governance practices.
ROBERT
K. deVEER, Jr. has been a director of Palatin since November 1998.
Since January 1997, Mr. deVeer has been the president of deVeer
Capital LLC, a private investment company. He was a director of
Solutia Inc., a publicly-held chemical-based materials company,
until its merger with Eastman Chemical Company in July 2012. From
1995 until his retirement in 1996, Mr. deVeer served as Managing
Director, Head of Industrial Group, at New York-based Lehman
Brothers. From 1973 to 1995, he held increasingly responsible
positions at New York-based CS First Boston, including Head of
Project Finance, Head of Industrials and Head of Natural Resources.
He was a managing director, member of the investment banking
committee and a trustee of the First Boston Foundation. He received
a B.A. in economics from Yale University and an M.B.A. in finance
from Stanford Graduate School of Business.
Mr.
deVeer has extensive experience in investment banking and corporate
finance, including the financing of life sciences companies, and
serves as the audit committee’s financial
expert.
J.
STANLEY HULL has been a director of Palatin since September 2005.
Mr. Hull has over three decades of experience in the field of sales
and marketing. Mr. Hull joined GlaxoSmithKline, a research-based
pharmaceutical company, in October 1987 and retired as Senior Vice
President, Pharmaceuticals in May 2010, having previously served in
the R&D organization of Glaxo Wellcome as Vice President and
Worldwide Director of Therapeutic Development and Product Strategy
– Neurology and Psychiatry. Prior to that, he was Vice
President of Marketing – Infectious Diseases and
Gastroenterology for Glaxo Wellcome. Mr. Hull started his career in
the pharmaceutical industry with SmithKline and French Laboratories
in 1978. Mr. Hull received his B.S. in business administration from
the University of North Carolina at Greensboro.
Mr.
Hull has extensive experience in commercial operations, development
and marketing of pharmaceutical drugs and corporate alliances
between pharmaceutical companies and biotechnology
companies.
ALAN W.
DUNTON, M.D., has been a director of Palatin since June 2011. Since
November 2015, he has been senior vice president of research and
development for Purdue Pharma L.P., with responsibilities for
overall research strategy and development programs. He founded
Danerius, LLC, a biotechnology consulting company, in 2006. From
January 2007 to March 2009, Dr. Dunton served as president and
chief executive officer of Panacos Pharmaceuticals Inc. and he
served as a managing director of Panacos from March 2009 to January
2011. Dr. Dunton is currently a member of the board of directors of
the publicly traded company Oragenics, Inc. He previously served on
the board of directors of the publicly traded companies Targacept,
Inc., EpiCept Corporation (as Non-Executive Chairman), Adams
Respiratory Therapeutics, Inc. (acquired by Reckitt Benckiser Group
plc), MediciNova, Inc. and Panacos Pharmaceuticals, Inc. Dr. Dunton
has served as a director or executive officer of various
pharmaceutical companies, and from 1994 to 2001, Dr. Dunton was a
senior executive in various capacities in the Pharmaceuticals Group
of Johnson & Johnson. Dr. Dunton received his M.D. degree from
New York University School of Medicine, where he completed his
residency in internal medicine. He also was a Fellow in Clinical
Pharmacology at the New York Hospital/Cornell University Medical
Center.
Dr.
Dunton has extensive drug development and clinical research
experience, having played a key role in the development of more
than 20 products to regulatory approval, and also has extensive
experience as an executive and officer for both large
pharmaceutical companies and smaller biotechnology and
biopharmaceutical companies.
ANGELA
ROSSETTI has been a director of Palatin since June 2013. From June
2015 through July 2017, she served as Executive Vice President of
Cell Machines, Inc., an early stage biopharmaceutical company
developing and novel protein therapies. Previously, Ms. Rossetti
served in pharmaceutical marketing, communications and financial
roles, including as Vice President at Pfizer Inc., where she led a
global commercial medicine team for smoking cessation, and as an
Assistant Vice President at Wyeth, managing a global hemophilia
business. Previously, she was President of Ogilvy Healthworld, an
advertising business in the pharmaceutical and biotechnology
sectors, and served on the Biotech and Pharmaceutical Advisory
Board of Danske Capital for six years. Ms. Rossetti also serves as
chair of the board of directors of Viramal Limited, a privately
held London-based emerging pharmaceutical company. Ms. Rossetti
graduated from a joint program of the Albert Einstein College of
Medicine and Benjamin N. Cardozo School of Law with an M.S.in
Bioethics in 2014, has an M.B.A. in Finance from Columbia
University Graduate School of Business and a B.A. in Biology from
the University of Pennsylvania, and is an adjunct Assistant
Professor at New York Medical College.
72
Ms.
Rossetti has extensive experience in worldwide development and
marketing of specialty pharmaceuticals, including prefilled syringe
products, in communications and development of commercialization
plans and in pharmaceutical and biotechnology finance.
ARLENE
M. MORRIS has been a director of Palatin since June 2015. Since May
2015 she has served as the chief executive officer of Willow
Advisors, LLC. From April 2012 until May 2015 she was President and
Chief Executive Officer of Syndax Pharmaceuticals, Inc., a
privately held biopharmaceutical company focused on the development
and commercialization of an epigenetic therapy for
treatment-resistant cancers, and was a member of the board of
directors from May 2011 until May 2015. From 2003 to January 2011,
Ms. Morris served as the President, Chief Executive Officer and a
member of the board of directors of Affymax, Inc., a publicly
traded biotechnology company. Ms. Morris has also held various
management and executive positions at Clearview Projects, Inc., a
corporate advisory firm, Coulter Pharmaceutical, Inc., a publicly
traded pharmaceutical company, Scios Inc., a publicly traded
biopharmaceutical company, and Johnson & Johnson, a publicly
traded healthcare company. She is currently a member of the board
of directors of Viveve Medical, Inc., a publicly traded female
healthcare medical device company, Dimension Therapeutics, Inc., a
publicly traded gene therapy company, and Neovacs, SA, a French
publicly traded biotechnology company, and was a director of Biodel
Inc., a publicly traded specialty pharmaceutical company, from 2015
until its merger with Albireo Limited in 2016. Ms. Morris received
a B.A. in Biology and Chemistry from Carlow College.
Ms.
Morris has extensive experience in the biotechnology industry,
including prior leadership positions, current senior management and
board service, and experience as chief executive officer of
companies with product candidates in phase 3 clinical
trials.
ANTHONY
M. MANNING, Ph.D., has been a director of Palatin since September
2017. Since 2013, he has been senior vice president of research at
Momenta Pharmaceuticals, Inc., a publicly traded biopharmaceutical
company developing therapeutics for autoimmune indications,
biosimilars and generic versions of complex drugs. From 2011 to
2013, he was senior vice president of research and development at
Aileron Therapeutics, Inc., a publicly traded biopharmaceutical
company developing stapled peptide therapeutics for cancers and
other diseases. From 2007 to 2011, he was vice president and head
of inflammation and autoimmune diseases research at Biogen, Inc., a
publicly traded biopharmaceutical company developing medicines for
neurological and neurodegenerative conditions. From 2002 to 2007,
he was vice president and global therapy area head for Roche
Pharmaceuticals, the pharmaceutical division of Roche Holding AG,
and from 2000 to 2002 he was vice president of Pharmacia, a global
pharmaceutical company acquired by Pfizer in 2002. Dr. Manning
received his Ph.D., M.Sc. and B.Sc. from the University of Otago,
Dunedin, New Zealand.
Dr.
Manning has extensive experience in translational research and
development of new pharmaceutical products, and in pharmaceutical
and biotechnology research, development and business
strategy.
The Board and Its Committees
Committees and meetings. The board has an audit committee, a
compensation committee and a nominating and corporate governance
committee. During fiscal 2017, the board met six times, the audit
committee met four times, the compensation committee met three
times and the nominating and corporate governance committee met two
times. Each director attended at least 75% of the total number of
meetings of the board and committees of the board on which he
served. The independent directors meet in executive sessions at
least annually, following the annual board meeting. We do not have
a policy requiring our directors to attend stockholder meetings.
With the exception of Drs. Prendergast and Spana, the directors did
not attend the annual meeting of stockholders held on June 8,
2017.
Audit committee. The audit committee reviews the engagement
of the independent registered public accounting firm and reviews
the independence of the independent registered public accounting
firm. The audit committee also reviews the audit and non-audit fees
of the independent registered public accounting firm and the
adequacy of our internal control procedures. The audit committee is
currently composed of four independent directors, Mr. deVeer
(chair), and Dr. Dunton, Ms. Rossetti and Mr. Hull. The board has
determined that the members of the audit committee are independent,
as defined in the listing standards of the NYSE MKT, and satisfy
the requirements of the NYSE MKT as to financial literacy and
expertise. The board has determined that at least one member of the
committee, Mr. deVeer, is the audit committee financial expert as
defined by Item 407 of Regulation S-K. The responsibilities of the
audit committee are set forth in a written charter adopted by the
board and updated as of October 1, 2013, a copy of which is
available on our web site at www.palatin.com.
73
Compensation committee. The compensation committee reviews
and recommends to the board on an annual basis employment
agreements and compensation for our officers, directors and some
employees, and administers our 2011 Plan and the options still
outstanding which were granted under previous stock option plans.
The compensation committee is composed of Dr. Dunton (chair), Ms.
Morris and Messrs. deVeer and Hull. The board has determined that
the members of the compensation committee are independent, as
defined in the listing standards of the NYSE MKT. Our Chief
Executive Officer aids the compensation committee by providing
annual recommendations regarding the compensation of all executive
officers, other than himself. Our Chief Financial Officer supports
the committee in its work by gathering, analyzing and presenting
data on our compensation arrangements and compensation in the
marketplace.
The
responsibilities of the compensation committee are set forth in a
written charter adopted by the board effective October 1, 2013, a
copy of which is available on our web site at www.palatin.com. The
committee administers our 2011 Plan, under which it has delegated
to an officer its authority to grant stock options to employees and
to a single-member committee of the board its authority to grant
restricted stock units to officers and to grant options and
restricted stock units to our consultants, but in either instance
not to grant options or restricted stock units to themselves, any
member of the board or officer, or any person subject to Section 16
of the Exchange Act.
Nominating and corporate governance committee. The
nominating and corporate governance committee assists the board in
recommending nominees for directors, and in determining the
composition of committees. It also reviews, assesses and makes
recommendations to the board concerning policies and guidelines for
corporate governance, including relationships of the board, the
stockholders and management in determining our direction and
performance. The responsibilities of the nominating and corporate
governance committee are set forth in a written charter adopted by
the board and updated as of October 1, 2013, a copy of which is
available on our web site at www.palatin.com. The nominating and
corporate governance committee is composed of Dr. Prendergast
(chair) and Mss. Rossetti and Morris, each of whom meets the
independence requirements established by the NYSE MKT.
Duration of Office. Unless a director resigns, all directors
hold office until the next annual meeting of stockholders or until
their successors have been elected and qualified. Directors serve
as members of committees as the board determines from time to
time.
Communicating With Directors
Generally,
stockholders or other interested parties who have questions or
concerns should contact Stephen T. Wills, Secretary, Palatin
Technologies, Inc., 4B Cedar Brook Drive, Cranbury, NJ 08512.
However, any stockholder or other interest party who wishes to
address questions regarding our business directly to the board of
directors, or any individual director, can direct questions to the
board members or a director by regular mail to the Secretary at the
address above or by e-mail at boardofdirectors@palatin.com.
Stockholders or other interested parties may also submit their
concerns anonymously or confidentially by postal mail.
Communications
are distributed to the board, or to any individual directors as
appropriate, depending on the facts and circumstances outlined in
the communication, unless the Secretary determines that the
communication is unrelated to the duties and responsibilities of
the board, such as product inquiries, resumes, advertisements or
other promotional material. Communications that are unduly hostile,
threatening, illegal or similarly unsuitable will also not be
distributed to the board or any director. All communications
excluded from distribution will be retained and made available to
any non-management director upon request.
Board Role in Risk Oversight
Our
board, as part of its overall responsibility to oversee the
management of our business, considers risks generally when
reviewing our strategic plan, financial results, business
development activities, legal and regulatory matters. The board
satisfies this responsibility through regular reports directly from
our officers responsible for oversight of particular risks. The
board’s risk management oversight also includes full and open
communications with management to review the adequacy and
functionality of the risk management processes used by management.
The board’s role in risk oversight has no effect on the
board’s leadership structure. In addition, committees of the
board assist in its risk oversight responsibility,
including:
●
The audit committee
assists the board in its oversight of the integrity of the
financial reporting and our compliance with applicable legal and
regulatory requirements. It also oversees our internal controls and
compliance activities, and meets privately with representatives
from our independent registered public accounting
firm.
74
●
The compensation
committee assists the board in its oversight of risk relating to
compensation policies and practices. The compensation committee
annually reviews our compensation policies, programs and
procedures, including the incentives they create and mitigating
factors that may reduce the likelihood of excessive risk taking, to
determine whether they present a significant risk to our
company.
Board Leadership Structure
Since
2000, the roles of chairman of the board and chief executive
officer have been held by separate persons. John K.A. Prendergast,
Ph.D., a non-employee director, has served as Chairman of the board
since June 2000. Carl Spana, Ph.D., has been our Chief Executive
Officer and President since June 2000. Generally, the chairman is
responsible for advising the chief executive officer, assisting in
long-term strategic planning, and presiding over meetings of the
board, and the chief executive officer is responsible for leading
our day-to-day performance. While we do not have a written policy
with respect to separation of the roles of chairman of the board
and chief executive officer, the board believes that the existing
leadership structure, with the separation of these roles, provides
several important advantages, including: enhancing the
accountability of the chief executive officer to the board;
strengthening the board’s independence from management;
assisting the board in reaching consensus on particular strategies
and policies; and facilitating robust director, board, and
executive officer evaluation processes.
Code of Corporate Conduct and Ethics
We have
adopted a code of corporate conduct and ethics, updated as of March
11, 2016, that applies to all of our directors, officers and
employees, including our Chief Executive Officer and Chief
Financial Officer. You can view the code of corporate conduct and
ethics at our website, www.palatin.com. We will disclose any
amendments to, or waivers from, provisions of the code of corporate
conduct and ethics that apply to our directors, principal executive
and financial officers in a current report on Form 8-K, unless the
rules of the NYSE MKT permit website posting of any such amendments
or waivers.
Executive Officers
Executive
officers are appointed by the board and serve at the discretion of
the board. Each officer holds his position until his successor is
appointed and qualified. The current executive officers hold office
under employment agreements.
|
Name
|
|
Age
|
|
Position with
Palatin
|
|
Carl
Spana, Ph.D.
|
|
55
|
|
Chief
Executive Officer, President and Director
|
|
Stephen
T. Wills, MST, CPA
|
|
60
|
|
Chief
Financial Officer, Chief Operating Officer, Executive Vice
President, Secretary and Treasurer
|
Additional
information about Dr. Spana is included above under the heading
“Identification of Directors.”
STEPHEN
T. WILLS, CPA, MST, has been Executive Vice President, Secretary,
Treasurer and Chief Financial Officer since 1997 and was Executive
Vice President of Operations from 2005 until June 2011, when he was
appointed Chief Operating Officer and Executive Vice President. Mr.
Wills is currently a member of the board of directors of MediWound
Ltd., an Israeli biopharmaceutical company publicity traded on
Nasdaq. Mr. Wills served as executive chairman and interim
principal executive officer of Derma Sciences, Inc.
(“Derma”), a publicly-held company providing advanced
wound care products, from December 2015 until February 2017 when
Derma was acquired by Integra LifeSciences Holding Corporation. Mr.
Wills also served as the lead director of Derma until December 2015
and as Derma’s chief financial officer from 1997 to 2000. Mr.
Wills serves on the board of trustees and executive committee of
The Hun School of Princeton, and from 1991 to 2000 he was the
president and chief operating officer of Golomb, Wills &
Company, P.C., a public accounting firm. Mr. Wills, a certified
public accountant, received his B.S. in accounting from West
Chester University, and an M.S. in taxation from Temple
University.
Section 16(A) Beneficial Ownership Reporting
Compliance
The
rules of the SEC require us to disclose failures to file or late
filings of reports of stock ownership and changes in stock
ownership required to be filed by our directors, officers and
holders of more than 10% of our common stock. To the best of our
knowledge, all of the filings for our directors, officers and
holders of more than 10% of our common stock were made on a timely
basis in fiscal 2017.
75
Item 11. Executive Compensation.
Fiscal 2017 Summary Compensation Table
The
following table summarizes the compensation earned by or paid to
our principal executive officer and our principal financial
officer, who constitute all of our executive officers, for our
fiscal years ended June 30, 2017 and 2016. We have no defined
benefit or actuarial pension plan, and no deferred compensation
plan.
|
Name and
Principal Position
|
|
Fiscal
Year
|
Salary
($)
|
Stock
awards (1)
($)
|
Option
awards (1)
($)
|
Nonequity
incentive plan compensation (2)($)
|
All
other
compensation
(3)($)
|
Total
($)
|
|
Carl Spana,
Ph.D.,
|
|
2017
|
476,400
|
368,050
|
367,368
|
458,000
|
13,250
|
1,683,068
|
|
Chief Executive
Officer
|
|
2016
|
476,400
|
354,250
|
-
|
101,000(4)
|
13,598
|
945,248
|
|
and
President
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stephen T.
Wills,
|
|
2017
|
435,200
|
338,330
|
336,570
|
438,000
|
13,250
|
1,561,350
|
|
MST, CPA, Chief
Financial Officer,
|
|
2016
|
435,200
|
327,000
|
-
|
92,000(4)
|
13,568
|
867,768
|
|
Chief Operating
Officer and
|
|
|
|
|
|
|
|
|
|
Executive Vice
President
|
|
|
|
|
|
|
|
|
(1)
Amounts in these
columns represent the aggregate grant date fair value for stock
awards and option awards computed using either the Black-Scholes
model or a multifactor Monte Carlo simulation. For a description of
the assumptions we used to calculate these amounts, see Note 13 to
the consolidated financial statements included in this Annual
Report.
(2)
Bonus
amounts.
(3)
Consists of
matching contributions to 401(k) plan.
(4)
Bonus amount for
fiscal year 2016 paid after fiscal year end but accrued as of June
30, 2016.
Employment Agreements
Effective
July 1, 2016, we entered into employment agreements with Dr. Spana
and Mr. Wills which continue through June 30, 2019 unless
terminated earlier. Under these agreements, which replaced
substantially similar agreements that expired on June 30, 2016, Dr.
Spana is serving as Chief Executive Officer and President at a base
salary of $476,400 per year and Mr. Wills is serving as Chief
Financial Officer and Chief Operating Officer at a base salary of
$435,200 per year. Each agreement also provides for:
●
annual
discretionary bonus compensation, in an amount to be decided by the
compensation committee and approved by the board, based on
achievement of yearly performance objectives; and
●
participation in
all benefit programs that we establish, to the extent the
executive’s position, tenure, salary, age, health and other
qualifications make him eligible to participate.
Each
agreement allows us or the executive to terminate the agreement
upon written notice, and contains other provisions for termination
by us for “cause,” or by the employee for “good
reason” or due to a “change in control” (as these
terms are defined in the employment agreements and set forth
below). Early termination may, in some circumstances, result in
severance pay at the salary then in effect, plus continuation of
medical and dental benefits then in effect for a period of two
years (Dr. Spana) or 18 months (Mr. Wills). In addition, the
agreements provide that options and restricted stock units granted
to these officers accelerate upon termination of employment except
for voluntary resignation by the officer or termination for cause.
In the event of retirement, termination by the officer for good
reason, or termination by us other than for “cause”,
options may be exercised until the earlier of twenty-four months
following termination or expiration of the option term.
Arrangements with our named executive officers in connection with a
termination following a change in control are described below. Each
agreement includes non-competition, non-solicitation and
confidentiality covenants.
76
The
compensation committee awarded performance-based bonuses to our
named executive officers for fiscal 2017 and fiscal 2016, which
were paid in July 2017 and September 2016, respectively, based on
results of operations, including clinical trial operations and our
financial condition.
Stock Option and Restricted Stock Unit Grants
On
December 8, 2015, we granted 325,000 performance-based restricted
stock units and 325,000 non-performance-based restricted stock
units to Dr. Spana and 300,000 performance-based restricted stock
units and 300,000 restricted stock units to Mr. Wills. The
performance-based restricted stock units vest during the
performance period, ending December 31, 2017, if and upon the
earlier of: (i) achievement of a closing price for the
Company’s common stock equal to or greater than $1.20 per
share for 20 consecutive trading days, which is considered a market
condition, or (ii) entering into a collaboration agreement (U.S. or
global) of bremelanotide for FSD, which is considered a performance
condition. The performance-based restricted stock units were deemed
vested as of February 2, 2017, the effective date of our license
agreement with AMAG Pharmaceuticals, Inc. for North American rights
to develop and commercialize bremelanotide for FSD. The
non-performance-based restricted stock units vest 25% on the date
of grant and 25% on the first, second and third anniversary dates
from the date of grant.
On June
20, 2017, we granted 595,000 restricted stock units to Dr. Spana
and 545,000 restricted stock units to Mr. Wills, which vest as
to 50% on each anniversary of the grant date. We also granted
938,000 stock options to Dr. Spana and 859,000 stock options
to Mr. Wills, which vest as to 25% on each anniversary of the grant
date. These options have an exercise price of $0.37, the fair
market value of the common stock on the date of grant, and they
expire on June 20, 2027.
On
September 7, 2016, we granted 290,000 restricted stock units to Dr.
Spana and 268,000 restricted stock units to Mr. Wills, which vest
as to 182,500 restricted stock units for Dr. Spana and 168,000
restricted stock units for Mr. Wills on September 7, 2017 and
the remaining restricted stock units of 107,500 and 100,000 for Dr.
Spana and Mr. Wills, respectively on September 7, 2018. We
also granted 432,000 options to Dr. Spana and 396,000 options to
Mr. Wills, which vest as to 199,500 options for Dr. Spana and
182,250 options to Mr. Wills on September 7, 2017 and the remaining
options of 232,500 and 213,750 for Dr. Spana and Mr. Wills,
respectively, as to 33% on September 7, 2018, 2019 and 2010. These
options have an exercise price of $0.68 and expire on September 7,
2026.
77
Outstanding Equity Awards at 2017 Fiscal Year-End
The
following table summarizes all of the outstanding equity-based
awards granted to our named executive officers as of June 30, 2017,
the end of our fiscal year.
|
|
|
|
Option awards
(1)
|
Stock awards
(2)
|
||||
|
Name
|
|
Option or
stock
award
grant
date
|
Number
of
securities
underlying
unexercised
options
(#)
exercisable
|
Number
of
securities
underlying
unexercised
options
(#)
unexercisable
|
Option
exercise
price
($)
|
Option
expiration
date
|
Number of shares
or units of stock that have not vested
(#)
|
Market value of
shares or units of stock that have not vested
($)
(3)
|
|
Carl
Spana
|
|
03/26/08
|
28,125
|
-
|
2.80
|
03/26/18
|
|
|
|
|
|
03/26/08
|
4,687
|
-
|
5.00
|
03/26/18
|
|
|
|
|
|
03/26/08
|
4,688
|
-
|
6.60
|
03/26/18
|
|
|
|
|
|
07/01/08
|
25,000
|
-
|
1.80
|
07/01/18
|
|
|
|
|
|
07/01/09
|
25,000
|
-
|
2.80
|
07/01/19
|
|
|
|
|
|
06/22/11
|
300,000
|
-
|
1.00
|
06/22/21
|
|
|
|
|
|
07/17/12
|
150,000
|
-
|
0.72
|
07/17/22
|
|
|
|
|
|
06/27/13
|
275,000
|
-
|
0.62
|
06/27/23
|
|
|
|
|
|
06/25/14
|
131,250
|
43,750
|
1.02
|
06/25/24
|
|
|
|
|
|
06/11/15
|
150,000
|
150,000
|
1.08
|
06/11/25
|
|
|
|
|
|
09/07/16
|
-
|
432,000
|
0.68
|
09/07/26
|
|
|
|
|
|
06/20/17
|
-
|
938,000
|
0.37
|
06/20/27
|
|
|
|
|
|
12/08/15
|
|
|
|
|
162,500
|
69,875
|
|
|
|
09/07/16
|
|
|
|
|
290,000
|
124,700
|
|
|
|
06/20/17
|
|
|
|
|
595,000
|
255,850
|
|
|
|
Total Stock Awards
|
|
|
1,047,500
|
$450,425
|
||
|
|
|
|
|
|
|
|
|
|
|
Stephen T.
Wills
|
|
03/26/08
|
22,500
|
-
|
2.80
|
03/26/18
|
|
|
|
|
|
03/26/08
|
3,750
|
-
|
5.00
|
03/26/18
|
|
|
|
|
|
03/26/08
|
3,750
|
-
|
6.60
|
03/26/18
|
|
|
|
|
|
07/01/08
|
20,000
|
-
|
1.80
|
07/01/18
|
|
|
|
|
|
07/01/09
|
20,000
|
-
|
2.80
|
07/01/19
|
|
|
|
|
|
06/22/11
|
250,000
|
-
|
1.00
|
06/22/21
|
|
|
|
|
|
07/17/12
|
135,000
|
-
|
0.72
|
07/17/22
|
|
|
|
|
|
06/27/13
|
250,000
|
-
|
0.62
|
06/27/23
|
|
|
|
|
|
06/25/14
|
112,750
|
37,500
|
1.02
|
06/25/24
|
|
|
|
|
|
06/11/15
|
135,000
|
135,000
|
1.08
|
06/11/25
|
|
|
|
|
|
09/07/16
|
-
|
396,000
|
0.68
|
09/07/26
|
|
|
|
|
|
06/20/17
|
-
|
859,000
|
0.37
|
06/20/27
|
|
|
|
|
|
12/08/15
|
|
|
|
|
150,000
|
64,500
|
|
|
|
09/07/16
|
|
|
|
|
268,000
|
115,240
|
|
|
|
06/20/17
|
|
|
|
|
545,000
|
234,350
|
|
|
|
Total Stock Awards
|
|
|
963,000
|
$414,090
|
||
78
(1)
Stock option
vesting schedules: all options granted on or before June 27, 2013
have fully vested. Options granted after June 27, 2013 vest over
four years with 1/4 of the shares vesting per year starting on the
first anniversary of the grant date, provided that the named
executive officer remains an employee. See “Termination and
Change-In-Control Arrangements” below.
(2)
Time-based stock
award vesting schedule: stock awards consist of restricted stock
units granted December 8, 2015, as to 162,500 shares for Dr. Spana
and 150,000 shares for Mr. Wills, which will vest in equal amounts
over a two year period, provided that the named executive officer
remains an employee; restricted stock units granted on September 7,
2016, as to 182,500 shares for Dr. Spana and 168,000 shares for
Mr. Wills vested on September 7, 2017, the remaining shares of
107,500 and 100,000 for Dr. Spana and Mr. Wills, respectively,
vest on September 7, 2018, provided that the named executive
officer remains an employee; restricted stock units granted on June
20, 2017, as to 595,000 shares for Dr. Spana and 545,000 shares for
Mr. Wills, which will vest in equal amounts over a two year period,
provided that the named executive officer remains an employee. See
“Stock Options and Restricted Stock Unit Awards” above
and “Termination and Change-In-Control Arrangements”
below.
(3)
Calculated by
multiplying the number of restricted stock units by $0.43, the
closing market price of our common stock on June 30, 2017, the last
trading day of our most recently completed fiscal
year.
Termination
and Change-In-Control Arrangements
The
employment agreements, stock option agreements and restricted stock
unit agreements with Dr. Spana and Mr. Wills contain the following
provisions concerning severance compensation and the vesting of
stock options and restricted stock units upon termination of
employment or upon a change in control. The executive’s
entitlement to severance, payment of health benefits and
accelerated vesting of options is contingent on the executive
executing a general release of claims against us.
Termination Without Severance Compensation. Regardless of
whether there has been a change in control, if we terminate
employment for cause or the executive terminates employment without
good reason (as those terms are defined in the employment agreement
and set forth below), then the executive will receive only his
accrued salary and vacation benefits through the date of
termination. He may also elect to receive medical and dental
benefits pursuant to COBRA for up to two years (Dr. Spana) or 18
months (Mr. Wills), but must remit the cost of coverage to us.
Under the terms of our outstanding options and restricted stock
units, all unvested options and restricted stock units would
terminate immediately, and vested options would be exercisable for
three months after termination.
Severance Compensation After Death or Disability. In the
event of the executive’s death or disability, we will provide
lump sum severance pay equal to 24 months (for Dr. Spana) or 18
months (for Mr. Wills) of base pay, as well as the opportunity for
COBRA benefits as described above under “Termination Without
Severance Compensation.”
Severance Compensation Without a Change in Control. If we
terminate or fail to extend the employment agreement without cause,
or the executive terminates employment with good reason, then the
executive will receive as severance pay his salary then in effect,
paid in a lump sum, plus medical and dental benefits at our
expense, for a period of two years (Dr. Spana) or 18 months (Mr.
Wills) after the termination date. In addition, upon such event all
unvested options would immediately vest and be exercisable for two
years after the termination date or, if earlier, the expiration of
the option term, and all unvested restricted stock units would
accelerate and become fully vested.
Severance Compensation After a Change in Control. If, within
one year after a change in control, we terminate employment or the
executive terminates employment with good reason, then the
executive will receive as severance pay 200% (Dr. Spana) or 150%
(Mr. Wills) of his salary then in effect, paid in a lump sum, plus
medical and dental benefits at our expense, for a period of two
years (Dr. Spana) or 18 months (Mr. Wills) after the termination
date. We would also reimburse the executive for up to $25,000 in
fees and expenses during the six months following termination, for
locating employment. We would also reimburse the executive for any
excise tax he might incur on “excess parachute
payments” (as defined in Section 280G(b) of the Internal
Revenue Code). All unvested options would immediately vest and be
exercisable for two years after the termination date or, if
earlier, the expiration of the option term. All unvested restricted
stock units would vest upon a change in control, without regard to
whether the executive’s employment is
terminated.
Option and Restricted Stock Unit Vesting Upon a Change in
Control. Options and restricted stock units granted under
the 2011 Stock Incentive Plan vest upon a change in control. If any
options granted under the 2005 Stock Plan are to be terminated in
connection with a change in control, those options will vest in
full immediately before the change in control.
79
Definitions. Under the employment agreements, a
“change in control,” “cause” and
“good reason” are defined as follows:
A
“change in control” occurs when:
(a)
any person or
entity acquires more than 50% of the voting power of our
outstanding securities;
(b)
the individuals
who, during any twelve month period, constitute our board of
directors cease to constitute at least a majority of the board of
directors;
(c)
the consummation of
a merger or consolidation; or
(d)
we sell
substantially all our assets.
The
term “cause” means:
(a)
the occurrence of
(i) the executive’s material breach of, or habitual neglect
or failure to perform the material duties which he is required to
perform under, the terms of his employment agreement; (ii) the
executive’s material failure to follow the reasonable
directives or policies established by or at the direction of our
board of directors; or (iii) the executive’s engaging in
conduct that is materially detrimental to our interests such that
we sustain a material loss or injury as a result thereof, provided
that the breach or failure of performance is not cured, to the
extent cure is possible, within ten days of the delivery to the
executive of written notice thereof;
(b)
the willful breach
by the executive of his obligations to us with respect to
confidentiality, invention and non-disclosure, non-competition or
non-solicitation; or
(c)
the conviction of
the executive of, or the entry of a pleading of guilty or nolo
contendere by the executive to, any crime involving moral turpitude
or any felony.
The
term “good reason” means the occurrence of any of the
following, with our failure to cure such circumstances within 30
days of the delivery to us of written notice by the executive of
such circumstances:
(a)
any material
adverse change in the executive’s duties, authority or
responsibilities, which causes the executive’s position with
us to become of significantly less responsibility, or assignment of
duties and responsibilities inconsistent with the executive’s
position;
(b)
a material
reduction in the executive’s salary;
(c)
our failure to
continue in effect any material compensation or benefit plan in
which the executive participates, unless an equitable arrangement
has been made with respect to such plan, or our failure to continue
the executive’s participation therein (or in a substitute or
alternative plan) on a basis not materially less favorable, both in
terms of the amount of benefits provided and the level of the
executive’s participation relative to other
participants;
(d)
our failure to
continue to provide the executive with benefits substantially
similar to those enjoyed by the executive under any of our health
and welfare insurance, retirement and other fringe-benefit plans,
the taking of any action by us which would directly or indirectly
materially reduce any of such benefits, or our failure to provide
the executive with the number of paid vacation days to which he is
entitled; or
(e)
the relocation of
the executive to a location which is a material distance from
Cranbury, New Jersey.
80
Director Compensation
The
following table sets forth the compensation we paid to all
directors during fiscal 2017, except for Dr. Spana, whose
compensation is set forth above in the Summary Compensation Table
and related disclosure. Dr. Spana did not receive any separate
compensation for his services as a director.
|
Name
|
Fees earned or
paid in cash ($)
|
Stock awards ($)
(2)
|
Option awards
($) (1) (2)
|
Total
($)
|
|
John K.A.
Prendergast, Ph.D.
|
95,000
|
39,960
|
25,491
|
160,451
|
|
Robert K. deVeer,
Jr.
|
58,750
|
19,980
|
12,746
|
91,476
|
|
J. Stanley
Hull
|
52,500
|
19,980
|
12,746
|
85,226
|
|
Alan W. Dunton,
M.D.
|
58,750
|
19,980
|
12,746
|
91,476
|
|
Angela
Rossetti
|
50,000
|
19,980
|
12,746
|
82,726
|
|
Arlene
Morris
|
50,000
|
19,980
|
12,746
|
82,726
|
(1)
The aggregate
number of shares underlying option awards and stock awards
outstanding at June 30, 2017 for each director was:
|
|
Option
awards
|
Stock
awards
|
|
Dr.
Prendergast
|
492,750
|
208,000
|
|
Mr.
deVeer
|
276,500
|
104,000
|
|
Mr.
Hull
|
273,000
|
104,000
|
|
Dr.
Dunton
|
204,000
|
94,000
|
|
Ms.
Rossetti
|
156,500
|
84,000
|
|
Ms.
Morris
|
115,500
|
74,000
|
(2)
Amounts in these
columns represent the aggregate grant date fair value for stock
awards and option awards computed using the Black-Scholes model.
For a description of the assumptions we used to calculate these
amounts, see Note 13 to the consolidated financial statements
included in this Annual Report. Amounts in this column include
options granted on June 20, 2017 for our current fiscal year ending
June 30, 2018.
Non-Employee Directors’ Equity Grants. Our
non-employee directors receive an annual equity grant at the board
meeting closest to the beginning of each fiscal year, or such other
date as may be determined by the board.
On June
20, 2017, as the annual equity grant for our current fiscal year
ending June 30, 2018, the Chairman of the board received 108,000
restricted stock units which vest on June 20, 2018, and an option
to purchase 108,000 shares of common stock, and each other serving
non-employee director received 54,000 restricted stock units which
vest on June 20, 2018, and an option to purchase 54,000 shares of
common stock. All of the options have an exercise price of $0.37
per share, the closing price of our common stock on the date of
grant, vest in twelve monthly installments beginning on July 31,
2017, expire ten years from the date of grant and provide for
accelerated vesting in the event of involuntarily termination as a
director following a change in control, with exercise permitted
following accelerated vesting for up to the earlier of one year
after termination or the expiration date of the
option.
On
December 8, 2015, the compensation committee determined that
additional equity grants were necessary in order to reward,
motivate and retain the non-employee directors, and we granted the
Chairman of the board 100,000 restricted stock units and the other
serving non-employee directors a total of 240,000 restricted stock
units, consisting of 50,000 to each of Mr. deVeer, Mr. Hull and Dr.
Horovitz, 40,000 to Dr. Dunton, 30,000 to Ms. Rossetti and 20,000
to Ms. Morris. The restricted stock units vest as to 50% on the
first and second anniversary of the grant date, conditioned on
continued service as a director through the applicable vesting
dates.
On June
9, 2016, as the annual equity grant for our current fiscal year
ending June 30, 2017, the Chairman of the board received 75,000
restricted stock units which vested on June 9, 2017, and an option
to purchase 75,000 shares of common stock, and each other serving
non-employee director received 37,500 restricted stock units which
vest on June 9, 2017, and an option to purchase 37,500 shares of
common stock. All of the options have an exercise price of $0.50
per share, the closing price of our common stock on the date of
grant, vest in twelve monthly installments beginning on July 31,
2016, expire ten years from the date of grant and provide for
accelerated vesting in the event of involuntarily termination as a
director following a change in control, with exercise permitted
following accelerated vesting for up to the earlier of one year
after termination or the expiration date of the
option.
81
Non-Employee Directors’ Cash Compensation. Dr.
Prendergast serves as Chairman of the board and for our fiscal year
ended June 30, 2017 received an annual retainer of $87,500, payable
quarterly. Other non-employee directors received an annual base
retainer of $40,000, payable on a quarterly basis. The chairperson
of the audit committee received an additional annual retainer of
$12,500, the chairperson of the compensation committee received an
additional annual retainer of $12,500 and the chairperson of the
corporate governance committee received an additional annual
retainer of $7,500. Members of the foregoing committees, other than
the non-employee Chairman, received an additional retainer of
one-half the retainer payable to the committee chairperson. For the
fiscal year ending June 30, 2018, Dr. Prendergast serves as
Chairman of the board and will received an annual retainer of
$87,500, payable quarterly. Other non-employee directors will
receive an annual base retainer of $40,000, payable on a quarterly
basis. The chairperson of the audit committee will receive an
additional annual retainer of $15,000, the chairperson of the
compensation committee will receive an additional annual retainer
of $15,000 and the chairperson of the corporate governance
committee will receive an additional annual retainer of $10,000.
Members of the foregoing committees, other than the non-employee
Chairman, receive an additional retainer of one-half the retainer
payable to the committee chairperson.
Non-Employee Directors’ Expenses. Non-employee
directors are reimbursed for expenses incurred in performing their
duties as directors, including attending all meetings of the board
and any committees on which they serve.
Employee Directors. Employee directors are not separately
compensated for services as directors, but are reimbursed for
expenses incurred in performing their duties as directors,
including attending all meetings of the board and any committees on
which they serve.
Item 12. Security Ownership of Certain Beneficial Owners and
Management and Related Stockholder Matters.
Securities Authorized for Issuance Under Equity Compensation
Plans. The table below provides information on our equity
compensation plans as of June 30, 2017:
Equity Compensation Plan Information
as of June 30, 2017
|
Plan
category
|
Number of
securities to be issued upon exercise of outstanding options,
warrants and rights
|
Weighted-average
exercise price of outstanding options, warrants and
rights
|
Number of
securities remaining available for future issuance under equity
compensation plans (excluding securities reflected in column
(a))
|
|
|
(a)
|
(b)
|
(c)
|
|
Equity compensation
plans approved by security holders
|
14,136,749(1)
|
$0.76(2)
|
6,525,778
|
|
Equity compensation
plans not approved by security holders
|
25,000(3)
|
$0.70
|
-
|
|
Total
|
14,161,749
|
|
6,525,778
|
(1)
Consists of
8,592,162 options and 5,209,617 restricted stock units granted
under our 2011 Stock Incentive Plan and 334,970 options granted
under our 2005 Stock Plan. Our 2005 Stock Plan has terminated, but
termination does not affect awards that are currently outstanding
under this plan. The shares subject to outstanding awards under the
2005 Stock Plan, if forfeited prior to exercise, will become
available for issuance under the 2011 Stock Incentive
Plan.
(2)
The amount in
column (a) for equity compensation plans approved by security
holders includes 5,209,617 shares reserved for issuance on vesting
of outstanding restricted stock units, granted under our 2011 Stock
Incentive Plan, which vest on various dates through June 20, 2021,
subject to the fulfillment of service conditions. Because no
exercise price is required for issuance of shares on vesting of the
restricted stock units, the weighted-average exercise price in
column (b) does not take the restricted stock units into
account.
82
(3)
Consists of two
warrants to purchase 12,500 shares at $0.70 per share issued in
connection with a contract for financial advisory
services.
Beneficial Ownership Tables. The tables below show the
beneficial stock ownership and voting power, as of September 21,
2017, of:
●
each director, each
of the named executive officers, and all current directors and
officers as a group; and
●
all persons who, to
our knowledge, beneficially own more than five percent of the
common stock or Series A preferred stock.
“Beneficial
ownership” here means direct or indirect voting or investment
power over outstanding stock and stock which a person has the right
to acquire now or within 60 days after September 21, 2017. See the
footnotes for more detailed explanations of the holdings. Except as
noted, to our knowledge, the persons named in the tables
beneficially own and have sole voting and investment power over all
shares listed.
The
common stock has one vote per share and the Series A preferred
stock has approximately 15 votes per share of Series A preferred
stock. Voting power is calculated on the basis of the aggregate of
common stock and Series A preferred stock outstanding as of
September 21, 2017, on which date 179,045,453 shares of
common stock and 4,030 shares of Series A preferred stock,
convertible into 60,592 shares of common stock, were
outstanding.
The
address for all members of our management and directors is c/o
Palatin Technologies, Inc., 4B Cedar Brook Drive, Cranbury, NJ
08512. Addresses of other beneficial owners are in the
table.
MANAGEMENT:
|
Class
|
|
Name of
beneficial owner
|
Amount and
nature of beneficial ownership
|
Percent of
class
|
Percent of total
voting power
|
|
Common
|
|
Carl Spana,
Ph.D.
|
2,704,852(1)
|
1.5%
|
*
|
|
Common
|
|
Stephen T.
Wills
|
2,477,239(2)
|
1.4%
|
*
|
|
Common
|
|
John K.A.
Prendergast, Ph.D.
|
613,517(3)
|
*
|
*
|
|
Common
|
|
Robert K. deVeer,
Jr.
|
368,060(4)
|
*
|
*
|
|
Common
|
|
J. Stanley
Hull
|
339,000(5)
|
*
|
*
|
|
Common
|
|
Alan W. Dunton,
M.D.
|
268,020(6)
|
*
|
*
|
|
Common
|
|
Angela
Rossetti
|
208,000(7)
|
*
|
*
|
|
Common
|
|
Arlene M.
Morris
|
143,000(8)
|
*
|
*
|
|
Common
|
|
Anthony M. Manning,
Ph.D.
|
0
|
*
|
*
|
|
|
|
|
|
|
|
|
|
|
All current
directors and executive officers as a group (nine
persons)
|
7,121,688(9)
|
3.9%
|
1.1%
|
*Less
than one percent.
83
(1)
Includes 1,293,250
shares of common stock underlying outstanding options and 670,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(2)
Includes 1,134,750
shares of common stock underlying outstanding options and 618,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(3)
Includes 416,750
shares of common stock underlying outstanding options and 50,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(4)
Includes 238,500
shares of common stock underlying outstanding options and 25,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(5)
Includes 235,000
shares of common stock underlying outstanding options and 25,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(6)
Includes 168,000
shares of common stock underlying outstanding options and 20,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(7)
Includes 120,500
shares of common stock underlying outstanding options and 15,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(8)
Consists of 75,500
shares of common stock underlying outstanding options and 10,000
shares of common stock underlying restricted stock units, all of
which shares of common stock underlying restricted stock units have
vested but not been delivered under deferred delivery provisions
provision for delivery after the grantee’s separation from
service or a defined changed in control, but does not include
shares of common stock underlying outstanding options or restricted
stock unit awards that have not vested and will not vest within 60
days.
(9)
Includes 5,115,250
shares of common stock underlying outstanding options and
restricted stock units.
84
MORE THAN 5% BENEFICIAL OWNERS:
|
Class
|
|
Name and address
of beneficial owner
|
Amount and
nature of beneficial ownership (1)
|
Percent
of
class
|
Percent of total
voting power
|
|
|
|
|
|
|
|
|
Common
|
|
QVT Financial
LP
1177 Avenue of the
Americas, 9th Floor
New York, New York
10036
|
19,394,939(2)
|
9.9%
|
2.4%
|
|
Series A
Preferred
|
|
Steven N.
Ostrovsky
43 Nikki
Ct.
Morganville, NJ
07751
|
500
|
12.4%
|
*
|
|
Series A
Preferred
|
|
Thomas L. Cassidy
IRA Rollover
38 Canaan
Close
New Canaan, CT
06840
|
500
|
12.4%
|
*
|
|
Series A
Preferred
|
|
Jonathan E.
Rothschild
300 Mercer St.,
#28F
New York, NY
10003
|
500
|
12.4%
|
*
|
|
Series A
Preferred
|
|
Arthur J.
Nagle
19 Garden
Avenue
Bronxville, NY
10708
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Thomas P. and Mary
E. Heiser, JTWROS
10 Ridge
Road
Hopkinton, MA
01748
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Carl F.
Schwartz
31 West 87th
St.
New York, NY
10016
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Michael J.
Wrubel
3650 N. 36 Avenue,
#39
Hollywood, FL
33021
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Myron M.
Teitelbaum, M.D.
175 Burton
Lane
Lawrence, NY
11559
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Laura Gold
Galleries Ltd. Profit Sharing Trust Park South Gallery at Carnegie
Hall
154 West 57th
Street, Suite 114
New York, NY
10019
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Laura
Gold
180 W. 58th
Street
New York, NY
10019
|
250
|
6.2%
|
*
|
|
Series A
Preferred
|
|
Nadji T.
Richmond
20 E. Wedgewood
Glen
The Woodlands, TX
77381
|
230
|
5.7%
|
*
|
*Less
than one percent.
(1)
Unless otherwise indicated by footnote, all share amounts represent
outstanding shares of the class indicated, and all beneficial
owners listed have, to our knowledge, sole voting and dispositive
power over the shares listed.
85
(2) QVT
Financial LP (“QVT Financial”) is the investment
manager for QVT Fund V LP (Fund V) and other private investment
funds (collectively, the “Funds”). The information
contained herein with respect to the Funds is based on information
included in filings by QVT Financial and the Funds with the SEC.
Based on a Form 13F filed by QVT Financial on August 14, 2017 for
the period ending June 30, 2017, the Funds beneficially own in the
aggregate 19,394,939 shares of common stock, including 15,098,092
shares of common stock underlying outstanding warrants. QVT
Financial has the power to direct the vote and disposition of the
common stock held by the Funds, and accordingly QVT Financial may
be deemed to be the beneficial owner of an aggregate amount of
19,394,939 shares of common stock, consisting of the shares
beneficially owned by the Funds.
QVT
Financial GP LLC, as General Partner of QVT Financial, may be
deemed to beneficially own the same number of shares of common
stock reported by QVT Financial. QVT Associates GP LLC, as General
Partner of the Funds may be deemed to beneficially own the
aggregate number of shares of common stock beneficially owned by
the Funds, and accordingly, QVT Associates GP LLC may be deemed to
be the beneficial owner of an aggregate amount of 19,394,939 shares
of common stock.
Exercise
of warrants held by the Funds is restricted if, as a result of an
exercise, the beneficial ownership of the holder and its affiliates
and any other party or person that could be deemed to be a group
would exceed 9.99% of the outstanding common stock. Beneficial
ownership as listed in the table above excludes warrants which are
not exercisable because of that restriction, but includes warrants
which are exercisable based on the common shares
outstanding.
Item 13. Certain Relationships and Related Transactions, and
Director Independence.
The
board of directors has determined that all of the directors except
for Dr. Spana (our Chief Executive Officer and President) are
independent directors, as defined in the listing standards of the
NYSE MKT.
As a
condition of employment, we require all employees to disclose in
writing actual or potential conflicts of interest, including
related party transactions. Our code of corporate conduct and
ethics, which applies to employees, officers and directors,
requires that the audit committee review and approve related party
transactions. Since July 1, 2015, there have been no
transactions or proposed transactions in which we were or are to be
a participant, in which any related person had or will have a
direct or indirect material interest.
Item 14. Principal Accountant Fees and Services.
KPMG
LLP (“KPMG”), served as our independent registered
public accounting firm for fiscal 2017 and fiscal
2016.
Audit Fees. For fiscal 2017, KPMG billed us a total of
$382,600 for professional services rendered for the audit of our
annual consolidated financial statements, review of our
consolidated financial statements in our Forms 10-Q and services
provided in connection with regulatory filings. For fiscal 2016,
the total billed for the same services was $235,500.
Audit-Related Fees. For fiscal 2017 and 2016, KPMG did not
perform or bill us for any audit-related services.
Tax Fees. For fiscal 2017, KPMG billed us a total of
$208,651 for professional services rendered for tax compliance and
IRC Section 382 services. For fiscal 2016, KPMG billed us $16,000
for professional services rendered for tax compliance.
All Other Fees. KPMG did not perform or bill us for any
services other than those described above for fiscal 2017 and
2016.
Policy on Audit Committee Pre-Approval of Audit and Permissible
Non-Audit Services of Independent Auditors. Consistent with
SEC policies regarding auditor independence, the audit committee
has responsibility for appointing, setting compensation for and
overseeing the work of the independent registered public accounting
firm. In recognition of this responsibility, the audit committee
has established a policy to pre-approve all audit and permissible
non-audit services provided by the independent registered public
accounting firm.
The
audit committee pre-approves fees for each category of service. The
fees are budgeted and the audit committee requires the independent
registered public accounting firm and management to report actual
fees versus the budget periodically throughout the year by category
of service. During the year, circumstances may arise when it may
become necessary to engage the independent registered public
accounting firm for additional services not contemplated in the
original pre-approval. In those instances, the audit committee
requires specific pre-approval before engaging the independent
registered public accounting firm.
86
The
audit committee may delegate pre-approval authority to one or more
of its members. The member to whom such authority is delegated must
report, for informational purposes only, any pre-approval decisions
to the audit committee at its next scheduled meeting.
PART IV
Item 15. Exhibits, Financial Statement Schedules.
(a) Documents filed as part of the
report:
1.
Financial
statements: The following consolidated financial statements are
filed as a part of this report under Item 8 – Financial
Statements and Supplementary Data:
—
Report of Independent Registered Public Accounting
Firm
—
Consolidated Balance Sheets
—
Consolidated Statements of Operations
—
Consolidated Statements of Comprehensive Loss
—
Consolidated Statements of Stockholders’ (Deficiency)
Equity
—
Consolidated Statements of Cash Flows
—
Notes to Consolidated Financial Statements
2.
Financial statement
schedules: None.
87
3.
List of
Exhibits
The
following exhibits are incorporated by reference or filed as part
of this report:
|
Exhibit Number
|
|
Description
|
|
Filed Herewith
|
|
Form
|
|
Filing Date
|
|
SEC File No.
|
|
|
Restated Certificate of Incorporation of Palatin Technologies,
Inc., as amended.
|
|
|
|
10-K
|
|
September 27, 2013
|
|
001-15543
|
|
|
|
Bylaws of Palatin Technologies, Inc.
|
|
|
|
10-Q
|
|
February 8, 2008
|
|
001-15543
|
|
|
|
Form of Series A 2012 Warrant.
|
|
|
|
8-K
|
|
July 6, 2012
|
|
001-15543
|
|
|
|
Form of Series B 2012 Warrant.
|
|
|
|
8-K
|
|
July
6, 2012
|
|
001-15543
|
|
|
|
Form of Series C 2014 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
December 30, 2014
|
|
001-15543
|
|
|
|
Form of Series D 2014 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
December
30, 2014
|
|
001-15543
|
|
|
|
Form of Series E 2015 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
July 7, 2015
|
|
001-15543
|
|
|
|
Form of Series F 2015 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
July 7, 2015
|
|
001-15543
|
|
|
|
Form of Series G 2015 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
July 7, 2015
|
|
001-15543
|
|
|
|
Form of Series H 2016 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
August 2, 2016
|
|
001-15543
|
|
|
|
Form of Series I 2016 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
August 2, 2016
|
|
001-15543
|
|
|
|
Form of Series J 2016 Common Stock Purchase Warrant.
|
|
|
|
8-K
|
|
December 1, 2016
|
|
001-15543
|
|
|
|
Form of warrant issued to PSL Business Development Consulting and
SARL Avisius in connection with a contract for financial advisory
services.
|
|
|
|
10-Q
|
|
February 10, 2017
|
|
001-15543
|
|
|
10.1†
|
|
1996 Stock Option Plan, as amended.
|
|
|
|
10-K
|
|
September 28, 2009
|
|
001-15543
|
|
10.2†
|
|
Form of Option Certificate (Incentive Option) Under the 2005 Stock
Plan.
|
|
|
|
8-K
|
|
September 21, 2005
|
|
001-15543
|
|
10.3†
|
|
Form of Incentive Stock Option Under the 2005 Stock
Plan.
|
|
|
|
8-K
|
|
September 21, 2005
|
|
001-15543
|
|
10.4†
|
|
Form of Opinion Certificate (Non-Qualified
Opinion) Under the 2005 Stock Plan.
|
|
|
|
8-K
|
|
September 21, 2005
|
|
001-15543
|
|
10.5†
|
|
Form of Non-Qualified Stock Option Agreement Under the 2005 Stock
Plan.
|
|
|
|
8-K
|
|
September 21, 2005
|
|
001-15543
|
|
10.6†
|
|
2007 Change in Control Severance Plan.
|
|
|
|
10-Q
|
|
February 8, 2008
|
|
001-15543
|
|
10.7†
|
|
2005 Stock Plan, as amended.
|
|
|
|
10-Q
|
|
May 15, 2009
|
|
001-15543
|
|
10.8†
|
|
Form of Executive Officer Option Certificate.
|
|
|
|
10-Q
|
|
May 14, 2008
|
|
001-15543
|
|
10.9†
|
|
Form of Amended Restricted Stock Unit Agreement.
|
|
|
|
10-Q
|
|
May 14, 2008
|
|
001-15543
|
|
|
Form of Amended Option Certificate (Incentive Option) Under the
2005 Stock Plan.
|
|
|
|
10-Q
|
|
May 14, 2008 |
|
001-15543
|
|
|
|
Employment Agreement, effective as of July 1, 2016, between Carl
Spana and Palatin Technologies, Inc.
|
|
|
|
8-K
|
|
June 13, 2016
|
|
001-15543
|
|
|
|
Employment Agreement, effective as of July 1, 2016, between Stephen
T. Wills and Palatin Technologies, Inc.
|
|
|
|
8-K
|
|
June 13, 2016
|
|
001-15543
|
|
|
|
Underwriting Agreement, dated February
24, 2011, by and between Roth Capital Partners, LLC and Palatin
Technologies, Inc.
|
|
|
|
8-K
|
|
February 24, 2011
|
|
001-15543
|
|
|
|
2011 Stock Incentive Plan, as amended.
|
|
|
|
8-K
|
|
June 8, 2017
|
|
001-15543
|
|
|
|
Form of Restricted Share Unit Agreement Under the 2011 Stock
Incentive Plan.
|
|
|
|
10-Q
|
|
May 13, 2011
|
|
001-15543
|
|
|
|
Form of Nonqualified Stock Option Agreement under the 2011 Stock
Incentive Plan.
|
|
|
|
10-Q
|
|
May 13, 2011
|
|
001-15543
|
|
|
|
Form of Incentive Stock Option Agreement under the 2011 Stock
Incentive Plan.
|
|
|
|
10-Q
|
|
May 13, 2011
|
|
001-15543
|
|
|
|
Form of Restricted Share Unit Agreement under the 2011 Stock
Incentive Plan.
|
|
|
|
8-K
|
|
December 11, 2015
|
|
001-15543
|
|
|
|
Form of Performance-Based Restricted Share Unit Agreement under the
2011 Stock Incentive Plan.
|
|
|
|
8-K
|
|
December 11, 2015
|
|
001-15543
|
|
|
|
Form of Restricted Share Unit Agreement for Non-Employee Directors
under the 2011 Stock Incentive Plan.
|
|
|
|
8-K
|
|
December 11, 2015
|
|
001-15543
|
|
|
|
Amended form of Restricted Share Unit Agreement under the 2011
Stock Incentive Plan.
|
|
|
|
10-Q
|
|
February 12, 2016
|
|
001-15543
|
|
|
|
Amended form of Performance-Based Restricted Share Unit Agreement
under the 2011 Stock Incentive Plan.
|
|
|
|
10-Q
|
|
February 12, 2016
|
|
001-15543
|
|
|
|
Amended form of Restricted Share Unit Agreement for Non-Employee
Directors under the 2011 Stock Incentive Plan.
|
|
|
|
10-Q
|
|
February 12, 2016
|
|
001-15543
|
|
|
|
Form of Indenture.
|
|
|
|
S-3
|
|
August 3, 2015
|
|
333-206047
|
|
|
|
Letter Agreement, dated October 7, 2011, between Biotechnology
Value Fund, L.P. and Palatin Technologies, Inc.
|
|
|
|
8-K
|
|
October 7, 2011
|
|
001-15543
|
|
|
|
Securities Purchase Agreement, dated December 23, 2014, by and
between Palatin Technologies, Inc. and the investors named
therein.
|
|
|
|
8-K
|
|
December 30, 2014
|
|
001-15543
|
|
|
|
Registration Rights Agreement, dated December 23, 2014, by and
between Palatin Technologies, Inc. and the investors named
therein.
|
|
|
|
8-K
|
|
December 30, 2014
|
|
001-15543
|
|
|
|
Placement Engagement Letter, dated December 22, 2014, by and
between Palatin Technologies, Inc. and Piper Jaffray &
Co.
|
|
|
|
8-K
|
|
December 30, 2014
|
|
001-15543
|
|
|
|
Amended and Restated Venture Loan and Security Agreement, dated
July 2, 2015, by and between Palatin Technologies, Inc. and Horizon
Technology Finance Corporation, Fortress Credit Co LLC, Horizon
Credit II LLC and Fortress Credit Opportunities V CLO
Limited.
|
|
|
|
8-K
|
|
July 7, 2015
|
|
001-15543
|
|
|
|
Securities Purchase Agreement, dated July 2, 2015, by and between
Palatin Technologies, Inc. and the investors named
therein.
|
|
|
|
8-K
|
|
July 7, 2015
|
|
001-15543
|
|
|
|
Registration Rights Agreement, dated July 2, 2015, by and between
Palatin Technologies, Inc. and the investors named
therein.
|
|
|
|
8-K
|
|
July 7, 2015
|
|
001-15543
|
|
|
|
Underwriting Agreement, dated August 1, 2016, by and between
Palatin Technologies, Inc. and Canaccord Genuity Inc., on behalf of
itself and as representative of the underwriters named
therein.
|
|
|
|
8-K
|
|
August 2, 2016
|
|
001-15543
|
|
|
10.33††
|
|
License, Co-Development and Commercialization Agreement, dated
August 29, 2014, by and between Chemical Works of Gedeon Richter
Plc. and Palatin Technologies, Inc.
|
|
|
|
10-K/A
|
|
October 9, 2014
|
|
001-15543
|
|
|
Termination Agreement, dated September 16, 2015, by and
between Chemical Works of
Gedeon Richter Plc. and Palatin Technologies,
Inc.
|
|
|
|
10-K
|
|
September 18, 2015
|
|
001-15543
|
|
|
10.35††
|
|
Commercial Supply Agreement dated June 20, 2016, by and between
Catalent Belgium S.A. and Palatin Technologies, Inc.
|
|
|
|
10-K
|
|
September 19, 2016
|
|
001-15543
|
|
10.36††
|
|
Manufacturing Preparation and Services Agreement dated June 20,
2016, by and between Catalent Belgium S.A. and Palatin
Technologies, Inc.
|
|
|
|
10-K
|
|
September 19, 2016
|
|
001-15543
|
|
|
Underwriting Agreement, dated December 1, 2016, by and between
Palatin Technologies, Inc. and Canaccord Genuity Inc., on behalf of
itself and as representative of the underwriters named
therein.
|
|
|
|
8-K
|
|
December 1, 2016
|
|
001-15543
|
|
|
10.38††
|
|
License Agreement, dated January 8, 2017, by and between AMAG
Pharmaceuticals, Inc. and Palatin Technologies, Inc.
|
|
|
|
10-Q
|
|
February 10, 2017
|
|
001-15543
|
|
21
|
|
Subsidiaries of Palatin Technologies, Inc.
|
|
X
|
|
|
|
|
|
|
|
|
Consent of KPMG LLP.
|
|
X
|
|
|
|
|
|
|
|
|
|
Certification of Chief Executive Officer.
|
|
X
|
|
|
|
|
|
|
|
|
|
Certification of Chief Financial Officer.
|
|
X
|
|
|
|
|
|
|
|
|
|
Certification of principal executive officer pursuant to U.S.C.
Section 1350, as adopted pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
|
X
|
|
|
|
|
|
|
|
|
|
Certification of principal financial officer pursuant to U.S.C.
Section 1350, as adopted pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002.
|
|
X
|
|
|
|
|
|
|
|
|
101.INS
|
|
XBRL Instance Document.
|
|
X
|
|
|
|
|
|
|
|
101.SCH
|
|
XBRL Taxonomy Extension Schema Document.
|
|
X
|
|
|
|
|
|
|
|
101.CAL
|
|
XBRL Taxonomy Extension Calculation Linkbase Document.
|
|
X
|
|
|
|
|
|
|
|
101.LAB
|
|
XBRL Taxonomy Extension Label Linkbase Document.
|
|
X
|
|
|
|
|
|
|
|
101.PRE
|
|
XBRL Taxonomy Extension Presentation Linkbase
Document.
|
|
X
|
|
|
|
|
|
|
|
101.DEF
|
|
XBRL Taxonomy Extension Definition Linkbase Document.
|
|
X
|
|
|
|
|
|
|
† Management contract or compensatory plan or
arrangement.
††
Confidential treatment granted as to certain portions, which
portions are omitted and filed separately with the
SEC.
Item
16. Form 10-K Summary.
None.
88
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities
Exchange Act of 1934, the registrant has duly caused this report to
be signed on its behalf by the undersigned, thereunto duly
authorized.
|
|
PALATIN
TECHNOLOGIES, INC.
|
|
|
|
|
|
|
|
|
|
By:
|
/s/
Carl
Spana
|
|
|
|
|
Carl Spana, Ph.D. |
|
|
|
|
President and Chief
Executive Officer
(principal
executive officer)
|
|
Date:
September 25, 2017
Pursuant
to the requirements of the Securities Exchange Act of 1934, this
report has been signed below by the following persons on behalf of
the registrant and in the capacities and on the dates
indicated.
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
/s/
Carl Spana
|
|
President,
Chief Executive Officer and Director
|
|
September
25, 2017
|
|
Carl
Spana
|
|
(principal
executive officer)
|
|
|
|
|
|
|
|
|
|
/s/
Stephen T. Wills
|
|
Executive
Vice President, Chief Financial Officer
|
|
September
25, 2017
|
|
Stephen
T. Wills
|
|
and
Chief Operating Officer (principal financial and accounting
officer)
|
|
|
|
|
|
|
|
|
|
/s/
John K. A. Prendergast
|
|
Chairman
and Director
|
|
September
25, 2017
|
|
John K.
A. Prendergast
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Robert K. deVeer, Jr,
|
|
Director
|
|
September
25, 2017
|
|
Robert
K. deVeer, Jr.
|
|
|
|
|
|
|
|
|
|
|
|
/s/ J.
Stanley Hull
|
|
Director
|
|
September
25, 2017
|
|
J.
Stanley Hull
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Alan W. Dunton
|
|
Director
|
|
September
25, 2017
|
|
Alan W.
Dunton
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Angela Rossetti
|
|
Director
|
|
September
25, 2017
|
|
Angela
Rossetti
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Arlene M. Morris
|
|
Director
|
|
September
25, 2017
|
|
Arlene
M. Morris
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Anthony M. Manning
|
|
Director
|
|
September
25, 2017
|
|
Anthony
M. Manning
|
|
|
|
|
89