Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Teligent, Inc. | tlgt10-k12312016exhibit321.htm |
| EX-31.2 - EXHIBIT 31.2 - Teligent, Inc. | tlgt10-k12312016exhibit312.htm |
| EX-31.1 - EXHIBIT 31.1 - Teligent, Inc. | tlgt10-k12312016exhibit311.htm |
| EX-23.1 - EXHIBIT 23.1 - Teligent, Inc. | tlgt10-k12312016exhibit231.htm |
| EX-10.32 - EXHIBIT 10.32 - Teligent, Inc. | tlgt10-k12312016exhibit1032.htm |
| EX-10.31 - EXHIBIT 10.31 - Teligent, Inc. | tlgt10-k12312016exhibit1031.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
OR
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _________ to _________
Commission file number: 001-08568
Teligent, Inc.
(Formerly IGI Laboratories, Inc.)
(Exact name of registrant as specified in its charter)
Delaware | 01-0355758 | |
(State or other jurisdiction | (I.R.S. Employer Identification No.) | |
of incorporation or organization) | ||
105 Lincoln Ave., Buena, NJ | 08310 | |
(Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code (856) 697-1441
Securities registered pursuant to Section 12(b) of the Exchange Act:
Title of each class | Name of each exchange on which registered | |
Common Stock, $0.01 Par Value Per Share | The NASDAQ Stock Market | |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer¨ Accelerated filer ý
Non-accelerated filer¨ [Do not check if a smaller reporting company]
Smaller reporting company¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No ý
The aggregate market value of the registrant’s voting and non-voting common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) computed by reference to the price at which the common stock was last sold, as of the last business day of the registrant’s most recently completed second fiscal quarter was $306.1 million.
As of March 6, 2017, the registrant had 53,226,382 shares of common stock outstanding.
APPLICABLE ONLY TO REGISTRANTS INVOLVED IN BANKRUPTCY
PROCEEDINGS DURING THE PRECEDING FIVE YEARS:
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Sections 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court. Yes ¨ No ¨
DOCUMENTS INCORPORATED BY REFERENCE
The following documents (or parts thereof) are incorporated by reference into the following parts of this Form 10-K: Certain information required in Part III of this Annual Report on Form 10-K is incorporated from the Registrant’s Proxy Statement for the Annual Meeting of Stockholders to be held on May 18, 2017.
PART I
Item 1. BUSINESS
Our Company
Strategic Overview
Teligent, Inc., is a specialty generic pharmaceutical company. All references to "Teligent," the "Company," "we," "us," and "our" refer to Teligent, Inc. Our mission is to become a leader in the specialty generic pharmaceutical market. Under our own label, we currently market and sell generic topical and branded generic and generic injectable pharmaceutical products in the United States and Canada. In the United States we are currently marketing 16 generic topical pharmaceutical products and four branded generic pharmaceutical products. Through the completion of an acquisition, we now sell a total of 30 generic and branded generic injectable products and medical devices in Canada. Generic pharmaceutical products are bioequivalent to their brand name counterparts. We also provide development, formulation, and manufacturing services to the pharmaceutical, over-the-counter, or OTC, and cosmetic markets. We operate our business under one segment. Effective October 23, 2015, we changed our name from IGI Laboratories, Inc. to Teligent, Inc. On October 26, 2015, our common stock, which was previously listed on the NYSE MKT, began trading on the NASDAQ Global Select Market, under the trading symbol “TLGT.” Our principal executive office, laboratories and manufacturing facilities are located at 105 Lincoln Avenue, Buena, New Jersey. We have additional offices located in Iselin, New Jersey, Toronto, Canada, and Tallinn, Estonia.
Currently, we have two platforms for growth:
• | Developing, manufacturing and marketing a portfolio of generic pharmaceutical products in our own label in topical, injectable, complex and ophthalmic dosage forms; and |
• | Managing our current contract manufacturing and formulation services business. |
We have been in the contract manufacturing and development of topical products business since the early 1990s, but our strategy since 2010 has been focused on the growth of our own generic pharmaceutical business. Since 2010, we have focused on transitioning our business to include more customers in the topical pharmaceutical industry. In 2014, we broadened our target product focus from topical pharmaceuticals to include a wider specialty pharmaceutical approach. We believe that expanding our development and commercial base beyond topical generics, historically the cornerstone of our expertise, to include injectable generics, complex generics and ophthalmic generics (what we call our “TICO strategy”), will leverage our existing expertise and capabilities, and broaden our platform for more diversified strategic growth.
As of the date of this report, we have acquired 25 drug products that have been previously approved by the United States Food and Drug Administration, or FDA. Our pipeline includes 34 Abbreviated New Drug Applications, or ANDAs, on file with the FDA, for additional pharmaceutical products. In addition, we have five abbreviated new drug submissions, or ANDSs, on file with Health Canada. We have an additional 34 product candidates at various stages of our development pipeline, ten of which are in stability testing. In December 2015, we announced the approval by the FDA of Cefotan® (Cefotan for Injection). This was our first product approved from the portfolio of discontinued and withdrawn new drug applications, or NDAs, and ANDAs that we purchased from AstraZeneca Pharmaceuticals LP, or AstraZeneca, on September 25, 2014. We have also experienced an increased rate of review by the FDA of applications filed in Generic Drug User Fee Amendments, or GDUFA, Year 3 and Year 4, which began October 1, 2014, and October 1, 2015, respectively. We submitted 12 topical ANDAs in 2016. We expect to continue to expand our presence in the generic topical pharmaceutical market through the filing of additional ANDAs with the FDA and the subsequent launch of products as these applications are approved. We received nine approvals from our internally developed pipeline of topical generic products in 2016. We intend to continue to submit further ANDAs to the FDA and ANDSs to Health Canada in 2017. We will also seek to license or acquire further products, intellectual property, or pending applications to expand our portfolio.
In addition, we may continue to explore ways to accelerate our growth through the creation of unique opportunities from the acquisition of additional intellectual property, and the expansion of the use of our existing intellectual property.
Teligent Canada. On November 13, 2015, we acquired all of the rights, title and interest in the development, production, marketing, import and distribution of all products of Alveda Pharmaceuticals Inc., or Alveda, pursuant to two asset purchase agreements, one relating to the acquisition of all of the intellectual property-related assets of Alveda and the other relating to the acquisition of all other assets of Alveda.
2
In connection with the closing of the acquisition, we formed three subsidiaries: Teligent Luxembourg S.à.r.l., or LuxCo, a private limited company incorporated under the laws of the Grand Duchy of Luxembourg and wholly-owned by the Company; Teligent OÜ, a private limited company incorporated under the laws of the Republic of Estonia that is wholly-owned by LuxCo; and Teligent Canada Inc., a company incorporated under the laws of the Province of British Columbia that is wholly-owned by LuxCo.
Teligent Canada currently has nine employees, including a general manager of Teligent Canada, located in our offices in Toronto, Canada. Teligent Canada acquired all of the Alveda working capital, including accounts receivable, inventory, accounts payable, and capital assets. In addition, Teligent Canada acquired Alveda’s existing customer relations, all contracts necessary to execute the Canadian distribution activities, operational permits, and all intellectual property required to operate the marketing and distribution of products in Canada. Teligent Canada also transitioned a majority of the existing workforce as part of the acquisition. Teligent Canada currently markets and distributes 30 products. Teligent continues to transition these products to distribute them under a Teligent Canada label.
Teligent OÜ. Teligent OÜ currently has five employees, including a general manager of Teligent OÜ. Teligent OÜ is responsible for the development, enhancement, maintenance, protection and exploitation functions related to the intellectual property-related assets acquired from Alveda. In addition, Teligent OÜ is responsible for the management of the supply chain function and procurement of products for sale to Teligent Canada. In 2017, we intend to hire additional employees in Teligent OÜ. We secured a quality control laboratory space to support our Teligent US and Teligent Canada supply chain management and technical services teams.
Teligent Jersey Limited. On October 5, 2015, we, together with our wholly-owned subsidiary incorporated under the laws of Jersey, or Teligent Jersey, entered into and closed an Asset Purchase Agreement and certain other ancillary agreements with Concordia Pharmaceuticals Inc., S.à.r.l., Barbados Branch, or Concordia, pursuant to which we acquired all rights, title and interests of Concordia in the existing inventory and certain contracts associated with three currently marketed injectable pharmaceutical products (Fortaz®, Zinacef ™ , and Zantac® Injection), and Teligent Jersey acquired all rights, title and interests of Concordia in, among other things, certain other contracts, product registrations and books and records associated with those products. In consideration for the purchase of those assets, we paid Concordia an aggregate of $10,100,000 in cash. The transaction is accounted for as a purchase of the product and product rights. In addition, we purchased approximately $1.2 million of inventory related to the three products acquired.
Facility Expansion. We completed the first phase of our facility expansion in July 2016, with the complete interior renovation of our building at 101 Lincoln Avenue in Buena, New Jersey. This building now houses our new product development laboratory for work on topical and sterile pharmaceuticals. This laboratory integrates our formulation and analytical chemistry teams into one lab, which we expect will increase productivity across the drug development process. This building renovation also houses our regulatory affairs, supply chain and corporate service teams.
We are also progressing with the significant expansion and utilities upgrade of our manufacturing facility at 105 Lincoln Avenue in Buena, New Jersey. The expanded facility will increase our manufacturing capacity for topical products, and will also enable the production of sterile injectable products in both vial and ampule presentations. We are using this facility expansion as an opportunity to upgrade and improve the degree of automation and capacity in our existing topical production suite. The sterile production area is designed around isolator-based technology. The facility will include a versatile vial and ampule filling line capable of four million units per year, with space and critical utilities included in the build-out for a potential future higher-speed filling line. The current plans consider a total capital outlay for 105 Lincoln Avenue of approximately $55 million. We have been partnering with contract manufacturing organizations, or CMOs, for the development, registration and manufacture of some of our sterile injectable and ophthalmic products. Upon completion of the site expansion, we may transfer the manufacture of some of these injectable products to this facility. We will also use the new sterile production capability to support our internal R&D pipeline of sterile injectable products in vial and ampule presentations.
Our Generic Pharmaceutical Business
In September 2010, we leveraged our existing formulation and manufacturing capabilities to begin the Company’s transformation from being solely a contract manufacturing and development company into a generic pharmaceutical company with our own portfolio of products, as recognized by our first ANDA submission to the FDA. ANDAs are submitted to the FDA for generic drug products that have the same active ingredient, strength, dosage form, and route of administration as brand name innovator drug products to which they are bioequivalent, meaning that there is no significant difference between the drugs in their rate and extent of absorption in the body. In the United States, approved ANDA generic drugs are usually interchangeable with the innovator drug. This means that the generic version may generally be substituted for the branded product by either a physician or pharmacist when dispensing a prescription. Our commercialization of each of these product candidates requires approval of the respective ANDA by the FDA.
3
In December 2012, we launched our first generic topical pharmaceutical products under our own label. In March 2014, we received our first approval from the FDA for an ANDA for the generic equivalent of lidocaine hydrochloride USP 4% topical solution. We also have a number of additional product candidates in various stages of development.
Based on Quintiles IMS data, the addressable market, as of January 2017, for the 34 products we have pending at the FDA totals approximately $2.0 billion in annual sales. We expect to continue to expand our presence in the generic topical pharmaceutical market through the submission of additional ANDAs to the FDA and the subsequent launch of products if and when these applications are approved by the FDA. We also plan to file further ANDSs with Health Canada in 2017.
As part of our growth strategy, we also seek opportunities to acquire additional products and ANDAs or ANDSs. On February 1, 2013, we acquired assets and intellectual property, including an approved ANDA, for econazole nitrate cream 1%, which we launched under our label in September 2013. On September 24, 2014, we acquired from AstraZeneca previously approved ANDAs and NDAs associated with 18 products, 17 of which are injectable products and one non-injectable product for pain management. On September 30, 2014, we acquired previously marketed and approved ANDAs associated with two ophthalmic products from Valeant Pharmaceuticals LLC and Valeant Pharmaceuticals Luxembourg SARL, or Valeant, in addition to the exclusive right to acquire three additional previously marketed and approved injectable products from Valeant. In November 2014, we completed the purchase of one of those three optioned injectable products and its related NDA from Valeant. In March 2015, we completed the purchase of the final two optioned injectable products and their related NDAs from Valeant.
Our Contract Manufacturing and Development Business
We also develop, manufacture, fill and package topical semi-solid and liquid products for branded and generic pharmaceutical customers, as well as the OTC and cosmetic industries. These products are used in a wide range of applications, from purely cosmetic to the prescription treatment of conditions like dermatitis, psoriasis and eczema.
Our contract manufacturing and development business includes two services: contract formulation and contract manufacturing. These services are offered to pharmaceutical, OTC and cosmetic customers. For our pharmaceutical contract services customers, we formulate, test and/or manufacture prescription drugs and medical devices. The products include cosmetics sold by retail stores directly to the public, as well as prescription drug products promoted directly to physicians. All contract manufacturing products are produced under our customers’ labels. We do not expect to record significant revenues from our contract formulation services in 2017 and beyond.
We have filed several 510(k) submissions with the FDA to obtain clearance on behalf of our customers for the marketing and distribution of certain medical devices. In addition, we have two additional ANDAs pending approval at the FDA that we submitted under joint development and commercialization agreements with our partners. In December 2012, after completion of the required formulation and regulatory requirements, we submitted two ANDAs on behalf of one of our pharmaceutical partners, both of which were approved in 2016. In December 2013, we submitted another of the ANDAs associated with a generic topical pharmaceutical drug product, which, once approved, will be licensed, marketed and distributed by one of our large multi-national pharmaceutical partners, West-Ward Pharmaceuticals Corp. In June 2014, we submitted an ANDA under a joint development and commercialization agreement with Impax Laboratories, Inc.
We believe that our quality contract manufacturing and development business provides a consistent and reliable source of products and services to our customers. We offer flexibility in batch sizing and package design, which gives our customers the opportunity to select the appropriate presentation for each product. Our high-speed packaging lines can accommodate a variety of tubes, bottles, pumps and jars. As a result of the rollout of our TICO strategy and the increased focus and commitment of R&D and technical resources toward internal projects, we anticipate that revenue from our contract services business will decrease over time.
Our Financings
On December 22, 2014, we consummated the sale of an aggregate of $143.75 million in principal of our notes, or the Notes, to Deutsche Bank Securities Inc. and J.P. Morgan Securities LLC, as the initial purchasers, including the initial purchasers’ exercise of their option to purchase an $18.75 million in principal of Notes. The Notes were sold in a private placement to qualified institutional buyers pursuant to Rule 144A under the Securities Act of 1933, as amended. In connection with the sale of the Notes, we entered into an indenture with Wilmington Trust, National Association, as trustee. The Notes bear interest at a rate of 3.75% per year, payable semi-annually in arrears on June 15 and December 15 of each year, commencing June 15, 2015. The Notes will mature on December 15, 2019, unless earlier repurchased or redeemed by the Company or converted by holders, pursuant to the terms therein. Additionally, subject to certain conditions, we may redeem for cash any or all outstanding Notes on or after December 19, 2017 in an amount equal to the outstanding principal amount of such Notes, plus accrued and unpaid interest. No sinking fund is provided for the Notes. The Notes are the Company’s senior unsecured obligations and will not be guaranteed by any of our existing or future
4
subsidiaries. Aggregate net proceeds from the transaction were approximately $139 million, after deducting underwriter commissions and other expenses paid by us.
Corporate Information
We were incorporated in Delaware in 1977, and on May 7, 2008, our stockholders approved our name change from IGI, Inc. to IGI Laboratories, Inc. Effective October 23, 2015, we changed our name to Teligent Inc. Our principal offices are located at 105 Lincoln Avenue, Buena, New Jersey 08310. Our telephone number is (856) 697-1441. We maintain a website at www.teligent.com. We make available on or through our website our periodic reports that we file with the Securities and Exchange Commission, or the SEC. This information is available on our website free of charge as soon as reasonably practicable after we electronically file the information with or furnish it to the SEC. The contents of our website are not incorporated by reference into this document and shall not be deemed “filed” under the Securities Exchange Act of 1934, as amended, or the Exchange Act.
Our Competitive Strategy
Our goal is to become a leader in the specialty generic pharmaceutical market. Under our own label, we currently market and sell generic topical and branded generic injectable pharmaceutical products in the United States and Canada. We also provide development, formulation, and manufacturing services to the pharmaceutical, OTC, and cosmetic industries. We have been in the contract manufacturing and development of topical products business since the early 1990s, but our strategy since 2010 has been focused on the growth of our own generic pharmaceutical business. In 2014, we started the transformation of our business from working toward being a leader in the topical generic pharmaceutical industry to becoming a leader in the specialty pharmaceutical markets. We believe that expanding our development and commercial base beyond topical generics, the cornerstone of our expertise, to injectable generics, complex generics and ophthalmic generics (what we call our TICO strategy), will leverage existing expertise and capabilities, diversify our commercial opportunities and broaden our platform for long-term strategic growth.
Our TICO Strategy
Our TICO strategy originated from our opportunity to leverage the industry value chain, which we have developed and strengthened through our topical portfolio. This value chain includes our internal expertise in product and molecule selection and development, manufacturing, sales, logistics and distribution, as well as our relationships with our customers and consumers. With the notable exception of manufacturing capabilities, we see the potential to effectively leverage our existing infrastructure across this value chain and to further expand our strategic reach to the injectable, complex and ophthalmic generic pharmaceutical markets.
Topical (T) - Our focus on the topical market has been the foundation for our growth. While we have manufactured topical products since the early 1990s, we began to focus our strategy on the topical generic market in 2010. In December 2012, we launched our first generic topical pharmaceutical products under our own label. Currently, we market 16 topical products under our own label. We have received FDA approvals for nine topical generic products from our internally developed pipeline in 2016. In our topical pipeline, we have 34 ANDAs submitted to the FDA that are awaiting approval. We intend to continue to develop topical generic products and utilize our expertise in drug formulation and manufacture to expand our own generic topical prescription drug portfolio. We are targeting to develop and file further regulatory submissions with the FDA in 2017. Upon regulatory approval, we would market these products under the Teligent label to national chain drug stores and drug wholesalers through our internal sales efforts. Based on Quintiles IMS data, the addressable market, as of January 2017, for the 34 products we have pending at the FDA totals approximately $2.0 billion in annual sales.
In our topical contract services business, we have developed strong customer relationships that we believe provide us with both recurring revenue streams from those customers and opportunities to selectively increase our product offerings to our customers. We intend to continue to capitalize on our strong customer relationships to maintain some contract manufacturing and development revenues.
We have an FDA-registered, cGMP-compliant facility that is equipped for manufacturing topical, semi-solid and liquid products. The design and configuration of our manufacturing facility provides flexibility in manufacturing batch sizes from 250 kg up to 4,000 kg. We intend to leverage this flexibility and capacity to support our growth in the topical prescription markets. We are progressing with the significant expansion and utilities upgrade in this facility which will increase our manufacturing capacity for topical products to accommodate the expected growth created by the eventual commercial launch of the 34 topical generic pharmaceutical products in our pipeline.
Injectable (I) - As part of the injectable phase of our TICO strategy, on September 24, 2014, we acquired from AstraZeneca previously approved ANDAs and NDAs associated with 18 products, 17 of which are injectable products and one of which is a non-injectable product for pain management. Of the products we acquired, two of the products are currently on the FDA drug
5
shortage list. We have received FDA approval for our first product in this portfolio, Cefotan® (Cefotetan for Injection), which we launched in the first quarter of 2016.
On September 30, 2014, we acquired previously marketed and approved ANDAs associated with two ophthalmic products from Valeant, in addition to the exclusive right to acquire three additional previously marketed and approved injectable products from Valeant. In November 2014, we completed the purchase of one of those three optioned injectable products and its related NDA from Valeant. In March 2015, we completed the purchase of the final two optioned injectable products and their related NDAs from Valeant.
On October 5, 2015, we acquired three currently marketed injectable pharmaceutical products (Fortaz®, Zinacef ™ and Zantac® Injection) from Concordia Pharmaceuticals Inc., S.à.r.l., Barbados Branch.
On November 13, 2015, we formed Teligent Canada, and completed the acquisition of Alveda. Teligent Canada currently has nine employees, including a general manager located in our offices in Toronto, Canada. Teligent Canada acquired all of the Alveda working capital, including accounts receivable, inventory, accounts payable, and capital assets. In addition, Teligent Canada acquired Alveda’s existing customer relations, all contracts necessary to execute the Canadian distribution activities, operational permits, and all intellectual property required to operate the marketing and distribution of Alveda’s products in Canada. Teligent Canada also transitioned a majority of the existing workforce as part of the acquisition. Teligent Canada currently markets and distributes 30 injectable products.
We intend to leverage our existing topical value chain as we build our injectable generic portfolio. We have entered into partnerships with contract manufacturing organizations, or CMOs, for the manufacture of some of our products in our portfolio of sterile products. Longer term, we expect to bring much of this production capability in-house.
The facility expansion, which began construction activities in the beginning of 2016, will also enable the production of sterile injectable products in both vial and ampule presentations. The sterile production area is designed around forward-thinking isolator-based technology. We have been partnering with CMOs for the development, registration and manufacture of some of our sterile injectable and ophthalmic products. Upon completion of the site expansion, we may transfer the manufacture of some of these products to our Buena, New Jersey facility. We will also use the new sterile production capability to support our internal R&D pipeline of sterile injectable products in vial and ampule presentations.
We plan to continue to review business development opportunities to expand our injectable portfolio.
Complex (C) - We have begun three projects that we consider to be part of the complex portfolio of our TICO strategy. We consider our focus on complex products or markets to be broadly defined to include potential complexity in one of the critical areas of our industry value chain. As part of our complex program, we are researching two 505(b)(2) projects. A 505(b)(2) submission is an NDA that contains full safety and effectiveness reports, but permits some of the information required for approval to come from studies not conducted by or for the applicant, thereby avoiding unnecessary duplication of studies already performed on a product. In addition, we are currently working with a contract research organization, or CRO, to develop a generic equivalent of a pharmaceutical drug product designated for a chronic rare disease. The intent of this opportunity is to provide patients with a lower cost alternative of an approved orphan drug. The Orphan Drug Designation program at the FDA provides orphan status to drugs and biologics which are defined as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases/disorders that affect fewer than 200,000 people in the U.S., or that affect more than 200,000 persons, but are not expected to recover the costs of developing and marketing a treatment drug. We will continue to seek opportunities relevant to building our complex portfolio of products.
Ophthalmic (O) - As part of the ophthalmic portfolio of our TICO strategy, on September 30, 2014, we acquired previously marketed and approved ANDAs associated with two ophthalmic products from Valeant. Similar to our injectable portfolio, we are forming partnerships with CMOs for commercial production. We plan to continue to review business development opportunities to expand our ophthalmic portfolio.
6
Our Customers
Generic Pharmaceutical Business. The manufacturing and commercialization of generic specialty pharmaceutical markets is competitive, and there are established manufacturers, suppliers and distributors actively engaged in all phases of our business. We currently manufacture and sell topical generic pharmaceutical products under our own label. In October 2015, we acquired and began to sell our first generic injectable products. We currently market 30 products in Canada. As we continue to execute our TICO strategy, we will compete in other markets, including the injectable and ophthalmic generic pharmaceutical markets, and expect to face other competitors.
For the years ended December 31, 2016, and 2015, 41% and 43% of our total product sales, net, respectively, were to the three large wholesale drug distributors: AmerisourceBergen Corporation, or ABC; Cardinal Health, Inc., or Cardinal; and McKesson Drug Company, or McKesson. As of December 31, 2016, Cardinal accounted for 56% of our accounts receivable, McKesson accounted for 20% of our accounts receivable, and ABC accounted for approximately 11% of our accounts receivable. As of December 31, 2015, ABC represented 28% of our accounts receivable.
ABC, Cardinal and McKesson are key distributors of our products, as well as a broad range of health care products for many other companies. None of these distributors is an end user of our products. Generally, if sales to any one of these distributors were to diminish or cease, we believe that the end users of our products would likely find little difficulty obtaining our products either directly from us or from another distributor. However, the loss of one or more of these distributors, together with a delay or inability to secure an alternative distribution source for end users, could have a material adverse effect on our revenue, business, financial condition and results of operations. Furthermore, ABC, Cardinal and McKesson have entered into strategic alliances with Walgreens, CVS Caremark and Rite-Aid, respectively. Since Walgreens, CVS Caremark and Rite-Aid are customers for several of our products, the loss of our distributor relationship with any of the three large wholesalers could result in a reduction to our revenues.
We consider our business relationships with ABC, Cardinal and McKesson to be in good standing and have fee for services contracts with each of them. However, a change in purchasing patterns, a decrease in inventory levels, an increase in returns of our products, delays in purchasing products and delays in payment for products by one or more of these distributors could have a material adverse effect on our revenue, business, financial condition and results of operations. We continue to analyze the market for other opportunities to expand our current relationships with other customers, while we continue to seek to diversify our existing portfolio of specialty generic drug products through internal research and development. In addition, we continue to explore business development opportunities to add additional products and /or capabilities to our existing portfolio.
Contract Manufacturing and Development Business. Our customers in the contract manufacturing business generally consist of pharmaceutical companies, as well as cosmetic and OTC product marketers, who require product development/manufacturing support. For the year ended December 31, 2016, approximately 90% of our contract services revenue was derived from pharmaceutical customers, as compared to 86% of total contract services revenue for the year ended December 31, 2015. One contract manufacturing customer represented 10% of total revenue for the year ended December 31, 2016, and one of our contract manufacturing services customers represented 11% of total revenue for the year ended December 31, 2015. We do not expect any contract manufacturing or formulation services customers to exceed 10% of revenue for 2017 and beyond.
Concentration of credit risk. In 2016, we had sales to three customers which individually accounted for more than 10% of our total revenue. These customers had sales of $13.5 million, $8.6 million and $6.8 million, respectively, and represented 43% of total revenues in the aggregate. Accounts receivable related to these major customers comprised 81% of all accounts receivable as of December 31, 2016.
In 2015, we had sales to three customers which individually accounted for more than 10% of our total revenue. These customers had sales of $12.3 million, $5.8 million and $5.0 million, respectively, and represented 52% of total revenues in the aggregate. Accounts receivable related to these major customers comprised 83% of all accounts receivable as of December 31, 2015.
In 2014, we had sales to two customers which individually accounted for more than 10% of our total revenue. These customers had sales of $10.5 million and $4.4 million, respectively, and represented 44% of total revenues in the aggregate. Accounts receivable related to these major customers comprised 42% of all accounts receivable as of December 31, 2014.
Expansion into foreign operations in the fourth quarter of 2015 has generated net revenues greater than 10% outside of the United States. For the year ended December 31, 2016, domestic net revenues were $56.1 million and foreign net revenues were $10.8 million. As of December 31, 2016, domestic net assets were $120.0 million and foreign assets were $63.2 million.
7
Our Products
We recorded net revenue from one product, econazole nitrate cream 1%, which accounted for 8% and 45% of total revenues in 2016 and 2015, respectively. We had net revenue from lidocaine ointment 5%, which accounted for 23% of total revenues in 2016, which we launched at the end of the first quarter of 2016.
Teligent United States Topical Pharmaceutical Products
Product | Formulation | Presentations | Brand equivalent | Therapeutic Classification |
Desoximetasone 0.25% | Ointment | 15g, 60g, 100g | Topicort® | Topical Corticosteroid |
Diclofenac Sodium 1.5% | Topical Solution | 150mL | Pennsaid® | Topical Anti-inflammatory |
Fluocinolone Acetonide 0.01% | Topical Solution | 60mL | Synalar® | Topical Corticosteroid |
Fluocinolone Acetonide 0.025% | Ointment | 15g, 60g | Synalar® | Topical Corticosteroid |
Fluocinolone Acetonide 0.025% | Cream | 15g, 60g | Synalar® | Topical Corticosteroid |
Fluocinolone Acetonide 0.01% | Cream | 15g, 60g | Synalar® | Topical Corticosteroid |
Econazole Nitrate 1% | Cream | 15g, 30g, 85g | Spectazole® | Topical Anti-fungal |
Lidocaine USP 5% | Ointment | 35.44g | Xylocaine® | Topical Anesthetic |
Lidocaine 4% | Topical Solution | 50mL | Xylocaine® | Topical Anesthetic |
Triamcinolone Acetonide USP 0.1% | Ointment | 15g, 80g, 1lb jar | Kenalog® | Topical Corticosteroid |
Triamcinolone Acetonide USP 0.025% | Lotion | 60ml | Kenalog® | Topical Corticosteroid |
Triamcinolone Acetonide USP 0.1% | Lotion | 60mL | Kenalog® | Topical Corticosteroid |
Clobetasol Propionate 0.05% | Lotion | 2oz, 4oz | Clobex® | Topical Corticosteroid |
Flurandrenolide USP 0.05% | Ointment | 15g, 30g, 60g | Cordran® | Topical Corticosteroid |
Clindamycin Phosphate 1% | Topical Solution | 30mL, 60mL | Cleocin® | Topical Anti-infective |
Nystatin and Triamcinolone Acetonide USP | Ointment | 15g, 30g, 60g | Mycolog® | Topical Anti-fungal and Corticosteroid |
Triamcinolone Acetonide USP, 0.5% (1) | Ointment | 15g | Kenalog® | Topical Corticosteroid |
Clobetasol Propionate Gel 0.05% (2) | Gel | 15g, 30g, 60g | Temovate® | Topical Corticosteroid |
(1) ANDA approved by the FDA on March 3, 2017. We expect to launch the product in the second quarter of 2017.
(2) ANDA approved by the FDA on March 7, 2017. We expect to launch the product in the second quarter of 2017.
8
Teligent United States Injectable Products
Product | Strength | Formulation | Presentations | Dossier type held by Teligent | Therapeutic Classification |
Cefotan (Cefotetan) ® | 1g, 2g | Injectable | Vial | NDA | Antibacterial for systemic use |
Fortaz (Ceftazidime) ® | 500mg, 1g, 2g, 6g | Injectable | Vial, Twist Vial, Frozen Bag | NDA | Antibacterial for systemic use |
Zantac (Ranitidine) ® | 50mg, 150mg, 1g | Injectable | Vials | NDA | Drugs for Peptic Ulcer and gastro-oesophageal related disorders (GORD) |
Zinacef (Cefuroxime) ™ | 750mg, 1.5g, 7.5g | Injectable | Vial, Twist Vial, Frozen Bag | NDA | Antibacterial for systemic use |
Teligent Canada Products (1)
Product | Strength | Formulation | Presentations | Brand equivalent | Dossier type held by Teligent | Therapeutic Classification |
Acetylcysteine | 2 g, 6 g | Injectable | 10ml and 30 ml vials | Mucomyst® Parvolex® | ANDS | Antidote |
Atropine Injection BP | 0.4 mg | Injectable | 1 ml ampoules | N/A | DIN | Antimuscarnic, antispasmodic |
Atropine Injection BP | 0.6 mg | Injectable | 1 ml ampoules | N/A | DIN | Antimuscarnic, antispasmodic |
Baclofen Injection | 0.05 mg | Injectable | 1mL ampoules | Lioresal® | ANDS | Muscle Relaxant |
Baclofen Injection | 10 mg | Injectable | 5mL, 20mL ampoules | Lioresal® | ANDS | Muscle Relaxant |
Baclofen Injection | 40 mg | Injectable | 20mL ampoules | Lioresal® | ANDS | Muscle Relaxant |
Ibuprofen for Intravenous Infusion | 800 mg | Injectable | 8 ml vials | Caldolor® | NDS | Nonsteroidal Antiinflammatory Agent |
Diazepam Injection USP | 10 mg | Injectable | 2mL ampoules | Valium® | ANDS | Anxiolytic |
Dimenhydrinate Injection USP | 50 mg | Injectable | 1 ml ampoule | Gravol® | DIN | Antihistamine |
Dimenhydrinate Injection USP with preservative | 250 mg | Injectable | 5 ml vial | Gravol® | DIN | Antihistamine |
Epinephrine Injection | 1 mg | Injectable | 1 ml ampoule | Adrenalin® | DIN | Cardiac Stimulant |
Ergonovine Maleate Injection | 0.25 mg | Injectable | 1 ml ampoule | N/A | ANDS | Oxytocic |
Fentanyl Citrate Injection USP | 100 mcg | Injectable | 2mL ampoule | Sublimaze® | ANDS | Opiate Anesthetic |
Furosemide Injection USP | 20 mg | Injectable | 2 ml ampoule | Lasix® | ANDS | Diuretic |
Gentamicin Injection USP | 80 mg | Injectable | 2mL ampoule | Garamycin® | NDS | Antibiotic |
Irinotecan Hydrochloride | 40 mg, 100 mg, 500 mg | Injectable | 2 ml, 5 ml, 25 ml vials | Camptosar® | ANDS | Antineoplastic agent |
Lidocaine Hydrochloride Injection (1% Preservative Free) | 50 mg, 100 mg | Injectable | 5 ml and 10 ml polyampoule | Xylocaine® | DIN | Local Anesthetic |
9
Lidocaine Hydrochloride Injection with Preservative (1%) | 200 mg, 500 mg | Injectable | 20 ml and 50 ml vials | Xylocaine® | DIN | Local Anesthetic |
Lidocaine Hydrochloride Injection (2% Preservative Free) | 100 mg, 200 mg | Injectable | 5 ml and 10 ml polyampoule | Xylocaine® | DIN | Local Anesthetic |
Lidocaine Hydrochloride Injection with Preservative (2%) | 400 mg, 1 g | Injectable | 20 ml and 50 ml vials | Xylocaine® | DIN | Local Anesthetic |
Lidocaine 2% and Epinephrine 1:100,000 Injection | 400 mg, 1 g | Injectable | 20 ml and 50 ml vials | Xylocaine® | DIN | Local Anesthetic |
Lidocaine Hydrochloride Topical Solution USP 4% | 2000 mg | Topical Solution | 50mL | Xylocaine® | DIN | Topical Anesthetic |
Lidocaine Ointment USP 5% | 1750 mg | Ointment | 35g | Xylocaine® | DIN | Topical Anesthetic |
Methylene Blue Injection USP | 50 mg | Injectable | 5mL ampoule | N/A | DIN | Antidote |
Naloxone Hydrochloride Injection USP | 0.4 mg | Injectable | 1mL ampoule | Narcan® | ANDS | Opiate Antagonist |
Piperacillin and Tazobactam for Injection | 2 g, 0.25 g, 3 g, 0.375 g, 4 g, 0.5 g | Injectable | 2.25 g, 3.375 g, 4.5 g vials | Tazocin® | ANDS | Antibacterial for systemic use |
Sodium Chloride Injection USP 0.9% | 90 mg | Injectable | 10 ml vials | N/A | DIN | Diluent |
Sterile Water for Injection USP | 100% | Injectable | 10 ml polyampoule | N/A | DIN | Diluent |
Succinylcholine Chloride Injection USP | 200 mg, 400 mg | Injectable | 10 ml and 20 ml vials | Quelicin® | DIN | Muscle Relaxant |
(1) Table does not include Euflexxa®, which is not owned by Teligent Canada but is distributed and sold by Teligent Canada.
10
Teligent United States Other Products
Below is a listing of the previously marketed products that were purchased from AstraZeneca and Valeant, along with a description of each respective formulation, presentation, brand equivalent, dossier and indication
11
Product | Strength | Formulation | Presentations | Brand equivalent | Dossier type held by Teligent | Therapeutic Classification |
Ciprofloxacin | 0.3% | Ophthalmic Solution | 2.5ml, 5ml, 10ml bottles | Ciloxan ® | ANDA | Antibacterial for systemic use |
Betaxolol | 0.5% | Ophthalmic Solution | 5ml, 7.5ml, 15ml bottles | Betopic ® | ANDA | Beta Blocking Agent |
Phytonadione | 10mg, 1mg | Injectable | 0.5ml, 1ml ampoules; 3cc, 6cc vials | AquaMephyton ® | NDA | Hemostatic |
Amikacin Sulfate | 50mg/ml, 250mg/ml | Injectable | 2ml, 4ml vials | Amikacin Sulfate | ANDA | Antibacterial for systemic use |
Calcitonin Salmon | 200IU/ml | Injectable | 2ml vials | Miacalcin ® | ANDA | Anti-parathyroid Agent |
Cefotetan Disodium | 20mg/ml | Injectable (bag) | 50ml bags | Cefotan ® | NDA | Antibacterial for systemic use |
Cefotetan | 10mg/ml | Injectable | 2ml vials | Cefotan ® | NDA | Antibacterial for systemic use |
Clindamycin Phosphate | 150mg/ml | Injectable | 2ml, 4ml, 6ml, 60ml vials | Cleocin ® | ANDA | Antibacterial for systemic use |
Dobutamine HCl | 12.5mg/ml | Injectable | 20ml, 40ml vials | Dobutamine HCl | ANDA | Cardiac Stimulant |
Dopamine HCl | 40mg/ml | Injectable | 5ml, 10ml (vials and syringes) | Dopamine HCl | NDA / ANDA | Cardiac Stimulant |
Dopamine HCl | 80mg/ml | Injectable | 5ml, 10ml (vials, ampoules, and syringes) | Dopamine HCI | NDA / ANDA | Cardiac Stimulant |
Dopamine HCl | 160mg/ml | Injectable | 5ml (vials and ampoules) | Dopamine HCl | NDA / ANDA | Cardiac Stimulant |
Droperidol | 2.5mg/ml | Injectable | 10ml vials, 2ml and 5ml ampoules, and 2ml syringes | Inapsine ® | ANDA | Anti-Psychotic |
Furosemide | 10mg/ml | Injectable | 2ml, 4ml, 8ml, and 10ml vials, 4ml and 10ml syringes | Furosemide | ANDA | Diuretic |
Mannitol | USP 25% | Injectable | 50ml (vials and syringes) | Mannitol | ANDA | Diuretic |
Meperidine HCl | 25mg/ml, 50mg/ml, 75mg/ml, 100mg/ml | Injectable | 1ml and 30ml vials, 1ml and 1.5ml ampoules, and 1ml syringes | Demerol ® | ANDA | Systemic analgesic |
Midazolam HCl | 5mg/ml | Injectable | 2ml syringe | Midazolam | ANDA | Sedative |
Orphenadrine | 30 mg/mL | Injectable | 2 mL ampule | Orphenadrine Citrate | NDA | Muscle Relaxant |
Edrophonium | 10 mg/mL | Injectable | 1 mL ampule and 10 mL vial | Enlon® | NDA | Acetylcholinesterase inhibitor |
MVI-12 | N/A | Injectable | 10 mL ampules and 5 mL vials | N/A | NDA | Systemic multivitamin |
Naloxone HCl | 0.4 mg/mL, 1 mg/mL | Injectable | 1 mL 5 mLand 10 mL vials | N/A | ANDA | Opiate Antagonist |
Naloxone HCl (preservative free) | 0.4 mg/mL | Injectable | 1 mL vials | N/A | ANDA | Opiate Antagonist |
Pancuronium | 1 mg/ml, 2mg/ml | Injectable | 10ml vials, 2mg/ml (vials, ampoules, and syringes) | N/A | ANDA | General anesthesia |
Tobramycin Sulfate | 10 mg/mL, 40 mg/mL | Injectable | 2 mLand 35 mL vials | N/A | ANDA | Antibacterial for systemic use |
Nalbuphine | 10 mg/mL and 20 mg/mL | Injectable | 1 mL and 10 mL vials | Nubain® | ANDA | Systemic analgesic |
12
Our Suppliers
We require a supply of quality raw materials and components to manufacture and package pharmaceutical products for ourselves and third parties with which we have contracted. The principal components of our products are active and inactive pharmaceutical ingredients and certain packaging materials. The APIs and other materials and supplies used in our pharmaceutical manufacturing operations are generally available and purchased from many different U.S. and non-U.S. suppliers. However, in some cases, the raw materials used to manufacture pharmaceutical products are available only from a single supplier. Even when more than one supplier exists, we may choose, and in some cases have chosen, only to list one supplier in our applications submitted to the FDA. Any change in a supplier not previously approved must then be submitted through a formal approval process with the FDA. No raw materials or components suppliers represented 10% or more of our purchases in 2016, 2015 or 2014.
Research and Development
Our R&D activities are integral to our business and are conducted at our facility in Buena, New Jersey. Our R&D department is led by our Chief Scientific Officer, Stephen Richardson, who joined the Teligent team in October 2015. The R&D team consists of 33 full-time employees and their responsibilities include: formulation, reverse engineering, methods development, analytical and microbiologic testing and scale up, and regulatory expertise. Our employees have specific expertise in developing injectable products and topical products in a wide range of dosage forms, including simple solutions through complex creams. All ANDA topical development is conducted in-house except for bioequivalence testing, which is performed by a contract research organization.
We have been steadily increasing our investment in R&D as we believe that R&D is the future of the Company. We incurred $17.1, $13.2, and $6.9 million in R&D expenses in 2016, 2015, and 2014, respectively. We expect to increase our R&D spending in 2017 to approximately 24% to 27% of revenue in 2017 in order to expand our ANDA submissions and pipeline. As the business continues to grow over the next three to five years, we expect research and development costs as a percentage of revenue to decline.
Product Development and Government Regulation
United States
Prescription pharmaceutical products in the U.S. are generally marketed as either brand or generic drugs. Brand products are usually marketed under brand names through marketing programs that are designed to generate physician and consumer loyalty. Brand products generally are patent protected, which provides a period of market exclusivity during which time they are sold with little or no competition for the compound, although there typically are other participants in the therapeutic area. Additionally, brand products may benefit from other periods of non-patent market exclusivity. Exclusivity normally provides brand products with the ability to maintain their profitability for relatively long periods of time and brand products typically continue to play a significant role in the market due to physician and consumer loyalties after the end of patent protection or other market exclusivities.
Generic pharmaceutical products are the pharmaceutical and therapeutic equivalents of the brand product, also known as the reference listed drug, or RLD. A reference listed brand drug is an approved drug product listed in the FDA publication entitled Approved Drug Products with Therapeutic Equivalence Evaluations, popularly known as the Orange Book. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, provides that generic drugs may enter the market after the approval of an ANDA. An ANDA approval requires that bioequivalence to the reference listed drug be demonstrated and also requires that any patents on the corresponding reference listed drug be expired, invalidated, non-infringed and/or any other relevant market exclusivity periods related to the reference listed drug be expired as well. Generic drugs are bioequivalent to their reference brand name counterparts. Accordingly, generic products provide a safe, effective and cost-efficient alternative to users of these reference brand products. Branded generic pharmaceutical products are generic products in that they are approved for marketing under an ANDA, but they may be more responsive to promotion efforts generally used to promote branded pharmaceutical products. Growth in the generic pharmaceutical industry has been, and will continue to be, driven by the increased market acceptance of generic drugs, as well as the number of brand drugs for which patent terms and/or other market exclusivities have expired.
We obtain new generic products primarily through internal product development. Additionally, we license or co-develop products through arrangements with other companies. All applications for FDA approval must contain information relating to product formulation, raw material suppliers, stability, manufacturing processes, packaging, labeling and quality control. Information to support the bioequivalence of generic drug products or the safety and effectiveness of new drug products for their intended use is also required to be submitted. There are generally two types of applications used for obtaining FDA approval of new products:
• | New Drug Application — An NDA is filed when approval is sought to market a newly developed branded product and, in certain instances, for a new dosage form, a new delivery system or a new indication for a previously approved drug. |
13
• | Abbreviated New Drug Application — An ANDA is filed when approval is sought to market a generic equivalent of a drug product previously approved under an NDA and listed in the FDA’s Orange Book or for a new dosage strength for a drug previously approved under an ANDA. |
The ANDA development process is generally less time-consuming and complex than the NDA development process. It typically does not require new preclinical and clinical studies, because it relies on the studies establishing safety and efficacy conducted for the RLD previously approved through the NDA process. The ANDA process, however, does typically require one or more bioequivalence studies to show that the ANDA drug is bioequivalent to the previously approved reference listed brand drug. Bioequivalence studies compare the bioavailability of the proposed drug product with that of the RLD product containing the same active ingredient. Bioavailability is a measure of the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. Thus, a demonstration of bioequivalence confirms the absence of a significant difference between the proposed product and the reference listed brand drug in terms of the rate and extent to which the active ingredient or active moiety becomes available at the site of drug action when administered at the same molar dose under similar conditions.
Generic products are generally introduced to the marketplace at the expiration of patent protection for the brand product or at the end of a period of non-patent market exclusivity. However, if an ANDA applicant files an ANDA containing a certification of invalidity, non-infringement or unenforceability related to a patent listed in the Orange Book with respect to a reference drug product, the applicant may be able to market the generic equivalent prior to the expiration of patent protection for the brand product. Such patent certification is commonly referred to as a Paragraph IV certification. If the holder of the NDA sues, claiming infringement or invalidation, within 45 days of notification by the applicant, the FDA may not approve the ANDA application until the earlier of the rendering of a court decision favorable to the ANDA applicant or the expiration of 30 months. An ANDA applicant that is first to file a Paragraph IV certification is eligible for a period of generic marketing exclusivity. This exclusivity, which under certain circumstances may be required to be shared with other ANDA sponsors that have made Paragraph IV certifications, lasts for 180 days, during which the FDA cannot grant final approval to other ANDA applications for a generic equivalent to the same reference drug.
In addition to patent exclusivity, the holder of the NDA for the listed drug may be entitled to a period of non-patent market exclusivity, during which the FDA cannot approve an application for a generic version product. If the reference drug is a new chemical entity, the FDA may not accept an ANDA for a generic product for up to five years following approval of the NDA for the new chemical entity. If it is not a new chemical entity, but the holder of the NDA conducted clinical trials essential to approval of the NDA or a supplement thereto, the FDA may not approve an ANDA for a reference NDA product before the expiration of three years. Certain other periods of exclusivity may be available if the RLD is indicated for treatment of a rare disease or the sponsor conducts pediatric studies in accordance with FDA requirements.
Supplemental ANDAs are required for approval of various types of changes to an approved application and these supplements may be under review for six months or more. In addition, certain types of changes may only be approved once new bioequivalence studies are conducted or other requirements are satisfied.
An additional requirement for FDA approval of NDAs and ANDAs is that our manufacturing procedures and operations conform to FDA requirements and guidelines, generally referred to as current Good Manufacturing Practices, or cGMPs. The requirements for FDA approval encompass all aspects of the production process, including validation and recordkeeping, the standards around which are continuously changing and evolving.
Facilities, procedures, operations and/or testing of products are subject to periodic inspection by the FDA, the U.S. Drug Enforcement Administration, or DEA, and other authorities. In addition, the FDA conducts pre-approval and post-approval reviews and plant inspections to determine whether our systems and processes are in compliance with cGMP and other FDA regulations. Our suppliers are subject to similar regulations and periodic inspections.
In 2012, the U.S. Food and Drug Administration Safety and Innovation Act, or the FDASIA, was enacted into law. FDASIA is intended to enhance the safety and security of the U.S. drug supply chain by holding all drug manufacturers supplying products to the U.S. to the same FDA inspection standards and schedules. Specifically, prior to the passage of FDASIA, U.S. law required U.S. based manufacturers to be inspected by the FDA every two years but remained silent with respect to foreign manufacturers, causing some foreign manufacturers to go as many as nine years without a routine FDA cGMP inspection, according to the Government Accountability Office.
FDASIA also included GDUFA, a novel user fee program to provide FDA with approximately $1.5 billion in total user fees through 2018 focused on three key aims:
14
• | Safety – Ensure that industry participants, foreign or domestic, are held to consistent quality standards and are inspected with parity using a risk-based approach. |
• | Access – Expedite the availability of generic drugs by bringing greater predictability to the review times for abbreviated new drug applications, amendments and supplements and improving timeliness in the review process. |
• | Transparency – Enhance FDA’s visibility into the complex global supply environment by requiring the identification of facilities involved in the manufacture of generic drugs and associated APIs, and improve FDA’s communications and feedback with industry. |
Under GDUFA, 70% of the total fees are being derived from facility fees paid by Finished Dosage Form manufacturers and API facilities listed in pending or approved generic drug applications. The remaining 30% of the total fees are being derived from application fees, including generic drug application fees, prior approval supplement fees and fees for certain types of Drug Master Files, or DMFs.
Canada
In Canada, the registration process for approval of all generic pharmaceuticals has two tracks that proceed in parallel. The first track of the process involves an examination of the proposed generic product by Health Canada, the federal department responsible for national public health, to ensure that the quality, safety and efficacy of the proposed generic product meets Canadian standards and bioequivalence requirements. The second track concerns patent rights of the brand drug owner. Companies may submit an application called an abbreviated new drug submission, or ANDS, to Health Canada that compares the proposed generic drug to another drug marketed in Canada under a Notice of Compliance, or NOC, issued to a first person. When Health Canada is satisfied that the generic pharmaceutical product described in the ANDS satisfies the statutory requirements, it issues an NOC for that product for the uses specified in the ANDS, subject to any court order that may be made in the second track of the approval process.
The second track of the approval process is governed by the Patented Medicines NOC Regulations, or the Regulations. The owner or exclusive licensee of patents relating to the brand drug for which it has an NOC may have established a list of patents administered by Health Canada that enumerates all the patents claiming the medicinal ingredient, formulation, dosage form or the use of the medicinal ingredient. It is possible that even though the patent for the API may have expired, the originator may have other patents on the list which relate to new forms of the API, a formulation or additional uses. Most brand name drugs have an associated patent list containing one or more unexpired patents claiming the medicinal ingredient itself or a use of the medicinal ingredient (a claim for the use of the medicinal ingredient for the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state or its symptoms). In its ANDS, a generic applicant must make at least one of the statutory allegations with respect to each patent on the patent list, for example, alleging that the patent is invalid or would not be infringed and explaining the basis for that allegation. In conjunction with filing its ANDS, the generic applicant is required to serve the originator a Notice of Allegation, or NOA, which gives a detailed statement of the factual and legal basis for its allegations in the ANDS. The originator may commence a court application within 45 days after it has been served with the NOA, if it takes the position that the allegations are not justified. When the application is filed in court and served on Health Canada, Health Canada may not issue an NOC until the earlier of the determination by the court after a hearing or the expiration of 24 months from the commencement of the generic drug application. The period may be shortened or lengthened by the court in certain circumstances. An NOC can be obtained for a generic product only if the generic respondent is successful in dismissing the application under the Regulations in court. The legal costs incurred in connection with the application could be substantial.
Section C.08.004.1 of the Canadian Food and Drug Regulations is the so-called data protection provision, and the current version of this section applies in respect of all drugs for which an NOC was issued on or after June 17, 2006. A subsequent applicant for approval to market a drug for which an NOC has already been issued does not need to perform duplicate clinical trials similar to those conducted by the first NOC holder, but is permitted to demonstrate safety and efficacy by submitting data demonstrating that its formulation is bioequivalent to the formulation that was issued for the first NOC. The first party to obtain an NOC for a drug will have an eight-year period of exclusivity starting from the date it received its NOC based on those clinical data. A subsequent applicant for approval that seeks to establish safety and efficacy by comparing its product to the product that received the first NOC will not be able to file its own application until six years after the issuance of the first NOC. The Minister of Health will not be permitted to issue an NOC to that applicant until eight years after the issuance of the first NOC — this additional two-year period will correspond in most cases to the 24-month automatic stay under the Regulations. If the first person provides the Minister with the description and results of clinical trials relating to the use of the drug in pediatric populations, it will be entitled to an extra six months of data protection. A drug is only entitled to data protection so long as it is being marketed in Canada.
Facilities, procedures, operations and/or testing of products are subject to periodic inspection by Health Canada. In addition, Health Canada conducts pre-approval and post-approval reviews and plant inspections to determine whether our systems are in compliance
15
with the Good Manufacturing Practices in Canada, Drug Establishment Licensing requirements and other provisions of the Regulations. Competitors are subject to similar regulations and inspections.
The federal government, provinces and territories in Canada operate drug benefit programs through which eligible recipients receive drugs through public funding; these drugs are listed on provincial or territorial Drug Benefit Formularies (each, a “Formulary”). Eligible recipients include First Nations and Inuit clients, seniors, persons on social assistance, low-income earners, and those with certain specified conditions or diseases. Formulary listings are also used by private payors to reimburse generic products. To be listed in a Formulary, drug products must have received an NOC from Health Canada and must comply with each jurisdiction’s individual review process.
The primary regulatory approval for pharmaceutical manufacturers, distributors and importers selling pharmaceuticals to be marketed in Canada is the issuance of an establishment license, or EL. An EL is issued to a Canadian facility once Health Canada has approved the facilities in which the pharmaceuticals are manufactured, distributed or imported. A key requirement for EL-issuance is compliance with the Good Manufacturing Practices as set out by Health Canada. For pharmaceuticals that are imported into Canada, the license for the Canadian importing facility must list all foreign sites at which imported pharmaceuticals, and their active ingredients, are manufactured and tested. To be listed on our EL, all our foreign sites must demonstrate compliance with relevant Good Manufacturing Practices recognized by Health Canada.
Sales and Marketing
We manufacture, sell, distribute and market our prescription drug products to national chain drug stores and drug wholesalers and distributors and group purchasing organizations, or GPOs, in the United States and Canada. This commercialization infrastructure includes satisfying our state, provincial, territorial, or national licensing requirements, implementing procedures with our third-party logistics partners, and maintaining appropriate sales order to cash administrative processes and a manager of national accounts to manage our sales.
Competition
In our generic topical prescription drug business, we face competition from other generic drug manufacturers and brand-name pharmaceutical companies through authorized generics. Although there are a significant number of competitors in the generic drug market, there are fewer competitors in the topical generic drug market. The five dominant companies in the topical generic drug market are: Sandoz (the generic pharmaceutical division of Novartis AG), Taro Pharmaceutical Industries, Ltd., Perrigo Company, Teva Pharmaceutical Industries, Ltd. and Akorn, Inc. Collectively, these five competitors control approximately 58% of the generic topical market by value based on Quintiles IMS data from December 2016. We believe the concentrated nature of the topical generic drug market creates an opportunity for us to be able to compete based on a variety of factors, including our focus on niche opportunities within the market segment and our dedication to quality in every area of our business.
In our generic injectable prescription drug business, we also face competition from other generic drug manufacturers and brand-name pharmaceutical companies through authorized generics. Although there are a significant number of competitors in the generic drug market, there are fewer dominant competitors in the injectable generic drug market. The 7 dominant companies in the injectable generic drug market in the United States consist of Hospira, Inc. (a subsidiary of Pfizer, Inc.), Fresenius Kabi USA, Sandoz (the generic pharmaceutical division of Novartis AG), Grifols USA, LLC, West-Ward (a subsidiary of Hikma Pharmaceuticals PLC), Mylan, Inc., and Winthrop (a subsidiary of Sanofi-Aventis U.S. LLC.). Collectively, these seven competitors control approximately 63% of the generic injectable market by value based on Quintiles IMS data from December 2016. In Canada, we face competition from largely the same firms as in the United States as well as certain Canada-only firms. The Canadian generic injectable market is dominated by Sandoz (the generic pharmaceutical division of Novartis AG), Pfizer Injectables and Fresenius Kabi Canada.
Our generic injectable strategy is focused on injectable products with limited competition, and products that have a history of lack of supply, or instability in the supply chain, where we can add value and leverage on our ability to be a reliable supplier to the marketplace. We believe the concentrated nature of some molecules within the injectable generic drug market, and history of lack of supply of certain molecules in the marketplace, create opportunities for us that we believe will enable us to compete based on a variety of factors, including our focus on niche opportunities within the market segment and our dedication to quality in every area of our business.
The contract manufacturing services market is highly competitive and includes larger organizations with substantially greater resources than us. Many of our competitors are companies that commercialize and/or manufacture their required products at their own facilities. These competitors include major pharmaceutical companies, generic drug manufacturers and consumer health product companies that generally have substantially greater manufacturing, R&D, marketing and financial resources than us and, in some cases, have more geographically diversified international operations. We compete specifically with a number of different privately-
16
held contract manufacturing companies, including DPT Laboratories, Ltd. Although this market is competitive, the competition is limited due to the need for specific expertise in topical formulations and cGMP facilities. We believe that we have the expertise required and we will continue to service our existing customers in this market by providing high quality, customer-oriented service, complemented by our contract development expertise in topical formulations.
Environmental Matters
Our operations are subject to a variety of environmental, health and safety laws and regulations, including those of the United States Environmental Protection Agency and equivalent state and local regulatory agencies. These laws and regulations govern, among other things, air emissions, wastewater discharges, the use, handling and disposal of hazardous substances and wastes, soil and groundwater contamination and employee health and safety. Our manufacturing facility uses, in varying degrees, hazardous substances in its processes. Contamination at our facility can result and has resulted in liability to us, for which we have recorded appropriate reserves as needed. For example, two of the Company’s facilities have undergone remediation of environmental contamination. See Note 14 to the Company’s Consolidated Financial Statements included elsewhere in this Annual Report.
Intellectual Property
To compete effectively, we need to develop and maintain a proprietary position with regard to our own technology, product candidates and business. Our goal is to safeguard our trade secrets and know-how, attain, maintain and enforce patent protection for our product candidates, formulations, processes, methods and other proprietary technologies, and operate without infringing on the proprietary rights of others. We seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information and proprietary technology. We seek to achieve this protection through a combination of contractual arrangements and patents.
We depend upon the skills, knowledge, experience and know-how of our management and R&D personnel, as well as that of our consultants, advisors and collaborators. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently rely, and will continue to rely in the future, on confidentiality agreements to protect our interests. We require our employees, consultants, advisors and collaborators to enter into confidentiality agreements that prohibit the disclosure of confidential information to any other parties. We also require our employees and consultants to disclose and assign to us their ideas, developments, discoveries and inventions. We understand that these agreements may not provide us with adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure.
We also seek to obtain patent protection when necessary, and we understand that this may not provide us with complete protection against competitors who may attempt to circumvent our patents.
Facility and Operations
The Company’s executive administrative offices are located in Buena, New Jersey, in two facilities of approximately 33,000 square feet built on 8.44 acres of land in 1995, which we own. This facility is used for production, product development, marketing and warehousing for our pharmaceutical, cosmeceutical and cosmetic products. We are in the process of expanding our facility to total approximately 103,000 square feet. Our manufacturing capabilities encompass a full suite of competencies, including regulatory, quality assurance and in-house validation.
The facility is equipped to manufacture semi-solids, ointments, gels and liquids in solution form. The facility is also configured to provide flexibility in manufacturing. Pilot batches typically range from 30 kg to 250 kg, while commercial batches may range from 250 kg to 4,000 kg.
We operate our facility in accordance with cGMP, utilizing the same high standards as our pharmaceutical customers. Our facility is registered with the FDA. We believe that our facility and equipment are in good condition, are well-maintained and are able to operate at present levels. Our manufacturing operations are focused on regulatory compliance, continuous improvement, process standardization and excellence in quality and execution across the organization.
We also lease an additional 11,000 square feet of warehouse space in Vineland, New Jersey.
The Company also leases approximately 7,500 square feet of corporate office space in Iselin, New Jersey, approximately 4,000 square feet of office space in Toronto, Canada and approximately 2,605 square feet of office and laboratory space in Tallinn, Estonia.
Employees
17
On December 31, 2016, we had a total of 153 full-time employees, including nine full-time employees in Canada and five full-time employees in Estonia. In addition, as the need arises, we occasionally utilize short-term, part-time employees who are paid on an hourly basis. We also utilize temporary employees provided by a third-party on a regular basis, primarily in our production department. We do not have a collective bargaining agreement with our employees and we believe that our employee relations are good.
Item 1A. RISK FACTORS
Our current business and future results may be affected by a number of risks and uncertainties, including those described below. The risks and uncertainties described below are not the only risks and uncertainties we face. Additional risks and uncertainties not currently known to us or that we currently deem immaterial also may impair our business operations. If any of the following risks actually occur, our business, results of operations and financial condition could suffer. The risks discussed below also include forward-looking statements and our actual results may differ substantially from those discussed in these forward-looking statements.
Risks Related to Our Business
We have a history of losses and cannot assure you that we will become profitable. As a result, we may have to cease operations and liquidate our business.
With the exception of 2015, our expenses exceeded our revenue in each of the last 12 years, and no net income has been available to common stockholders during each of these years. As of December 31, 2016, our stockholders’ equity was $56.7 million and we had an accumulated deficit of $44.9 million. Our future profitability depends on revenue exceeding expenses, but we cannot assure you that this will occur. If we do not become profitable or continue to raise external financing, we could be forced to curtail operations and sell or liquidate our business, and you could lose some or all of your investment.
We rely on a limited number of customers for a large portion of our revenues.
We depend on a limited number of customers for a large portion of our revenue. Three of our customers accounted for 43% of our revenue for the year ended December 31, 2016, and three of our customers accounted for 52% of our revenue for the year ended December 31, 2015. The loss of one or more of these customers could have a significant impact on our revenues and harm our business and results of operations.
Due to our dependence on a limited number of products, our business will be materially adversely affected if these products do not perform as well as expected.
We expect to generate a significant portion of our total revenues and gross margin from the sale of a limited number of products. While we continue to diversify our product portfolio, one of our products accounted for 8% and 38% of our revenue for the years ended December 31, 2016 and 2015, respectively. Any material adverse developments, including increased competition, loss of customers, pricing pressures and supply shortages, with respect to the sale or use of our products and prospective products, or our failure to successfully introduce such products, could have a material adverse effect on our revenues and gross margin.
The pharmaceutical industry in which we operate is intensely competitive. We are particularly subject to the risks of competition. For example, the competition we encounter may have a negative impact upon the prices we may charge for our products, the market share of our products and our revenue and profitability.
The pharmaceutical industry in which we operate is intensely competitive. The competition that we encounter has an effect on our product prices, market share, revenue and profitability. Depending upon how we respond to this competition, its effect may be materially adverse to us.
We compete with:
• | the original manufacturers of the brand-name equivalents of our generic products; and |
• | other generic drug manufacturers. |
Most of the products that we are developing are either generic drugs or products without patent protection. These drugs and products do not benefit from patent protection and are therefore more subject to the risk of competition than patented products. In addition, because many of our competitors have substantially greater financial, production and research and development
18
resources, substantially larger sales and marketing organizations, and substantially greater name recognition than we have, we are particularly subject to the risks inherent in competing with them. For example, many of our competitors may be able to develop products and processes competitive with, or superior to, our own. Furthermore, we may not be able to successfully develop or introduce new products that are less costly than those of our competitors or offer purchasers of our products payment and other commercial terms as favorable as those offered by our competitors.
Furthermore, in the current political climate in which drug prices are a focus of the current administration, Congress, government and private payors, and the public more broadly, we cannot predict whether new legislative, regulatory, or other measures related to drug pricing may be enacted. If enacted, such drug pricing measures could have an impact on our drug prices or our gross margins from product sales, which could significantly and adversely impact our financial condition and cash flows.
As our competitors introduce their own generic equivalents of our generic pharmaceutical products, our revenues and gross margin from such products may decline, potentially rapidly.
Revenues and gross margin derived from generic pharmaceutical products often follow a pattern based on regulatory and competitive factors that we believe are unique to the generic pharmaceutical industry. As the patent(s) for a brand name product and the statutory marketing exclusivity period (if any) expires, the first generic manufacturer to receive regulatory approval for a generic equivalent of the product often is able to capture a substantial share of the market. However, as other generic manufacturers receive regulatory approvals for identical competing products, that market share, and the price of that product, may decline depending on several factors, including the number of competitors, the price of the brand product and the pricing strategy of the new competitors. In addition, the FDA has continued to shorten the review and response time to certain ANDAs, as a result of their guidelines established under GDUFA. If this trend continues, and the FDA is successful in reducing the current backlog of unapproved ANDAs, currently pending approval at the FDA, competitors could potentially enter the markets in which we compete more quickly. We cannot provide assurance that we will be able to continue to develop such products or that the number of competitors with such products will not increase to such an extent that we may stop marketing a product for which we previously obtained approval, which may have a material adverse impact on our revenues and gross margin.
Our strategy depends on our ability to successfully develop and launch new pharmaceutical products ahead of our competitors.
Our continued growth is dependent upon our ability to develop and commercialize products in a timely manner. We may encounter delays in testing and manufacturing new pharmaceutical products, submitting applications for regulatory approval, receiving approval from the relevant authorities and commercializing new products. This process is costly and time-consuming. Delays at any stage could prevent us from successfully launching new products ahead of our competitors and could have a material adverse effect on our business, financial condition and results of operations.
If pharmaceutical companies are successful in limiting the use of generics through their legislative, regulatory and other efforts, sales of our generic products may be adversely impacted.
Many pharmaceutical companies increasingly have used state and federal legislative and regulatory means to delay generic competition. These efforts have included:
• | pursuing new patents for existing products that may be granted just before the expiration of earlier patents, which could extend patent protection for additional years or otherwise delay the launch of generics; |
• | selling the brand product as an “authorized generic,” either by the brand company directly, through an affiliate or by a marketing partner; |
• | using the Citizen Petition process to request amendments to FDA standards or otherwise delay generic drug approvals; |
• | seeking changes to the U.S. Pharmacopeia, an FDA- and industry-recognized compendia of drug standards; |
• | attaching patent extension amendments to non-related federal legislation; |
• | engaging in state-by-state initiatives to enact legislation that restricts the substitution of some generic drugs, which could have an impact on products that we are developing; and |
19
• | seeking patents on methods of manufacturing certain active pharmaceutical ingredients. |
If pharmaceutical companies or other third parties are successful in limiting the use of generic products through these or other means, our sales of our generic products may decline. If we experience a material decline in generic product sales, our results of operations, financial condition and cash flows may be significantly and adversely impacted.
Our generics business also faces increasing competition from brand-name manufacturers that do not face any significant regulatory approval or other barriers to enter into the generics market.
Our generics business also faces increasing competition from brand-name manufacturers that do not face any significant regulatory approval or other barriers to enter into the generics market. These brand-name companies sell “authorized generic” versions of their products to the market directly, acquire or form strategic alliances with our competitor generic pharmaceutical companies, or grant them rights to sell “authorized generics.” Moreover, brand-name companies continually seek new ways to delay the introduction of generic products and decrease the impact of generic competition, such as filing new patents on drugs whose original patent protection is about to expire, developing patented controlled-release products, changing product claims and product labeling, or developing and marketing as over-the-counter products those branded products that are about to face generic competition, when feasible (given that significant new clinical data must be provided to FDA in a “switch” application and some drug products are not safe enough to be sold over-the-counter). Our competitors, which include major multinational corporations, are consolidating in both the branded and generics industries, and the strength of the combined companies could affect our competitive position in all of our business areas. Furthermore, if one of our competitors or its customers acquires any of our customers or suppliers, we may lose business from the customer or lose a supplier of a critical raw material.
We may need to raise additional capital that will be required to operate and grow our business, and we may not be able to raise capital on terms acceptable to us or at all.
Operating our business and maintaining our growth efforts will require additional cash outlays and capital expenditures. If cash on hand and cash generated from operations are not sufficient to meet our cash requirements, we will need to seek additional capital, potentially through debt or equity financings, to fund our growth. We cannot assure you that we will be able to raise needed cash on terms acceptable to the Company, our significant stockholders, or at all. Financings may be on terms that are dilutive or potentially dilutive to our stockholders, and the prices at which new investors would be willing to purchase our securities may be lower than the current price per share of our common stock. The holders of new securities may also have rights, preferences or privileges which are senior to those of existing holders of common stock. If new sources of financing are required, but are insufficient or unavailable, we will be required to modify our growth and operating plans based on available funding, if any, which would harm our ability to grow our business or even stay in business.
Our business and operations have experienced rapid growth, and if we do not appropriately manage any future growth, our business will be adversely affected.
We have experienced, and are continuing to experience, rapid growth over the last several years, and additional growth through acquisitions is possible in the future. Such growth has put significant demands on our management and infrastructure. Our success will depend in part upon our ability to manage this growth effectively. As we continue to grow, we must improve our operational, financial and management controls and our reporting systems and procedures. We must ensure that our policies and procedures evolve to reflect our current operations. We must also continue to effectively manage existing employees and to hire, train and manage new employees as needed. Any failure to expand these areas and implement appropriate procedures and controls in an efficient manner and at a pace consistent with our business objectives could have a material adverse effect on our business, financial condition and results of operations.
Sales of our products may continue to be adversely affected by the continuing consolidation of our distribution network and the concentration of our customer base. The result of such developments could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
Our principal customers are wholesale drug distributors and major retail drug store chains. These customers comprise a significant part of the distribution network for pharmaceutical products in the U.S. This distribution network is continuing to undergo significant consolidation marked by mergers and acquisitions, alliances and partnerships among wholesale distributors and the growth of large retail drug store chains. As a result, a small number of large wholesale distributors control a significant share of the market, and the number of independent drug stores and small drug store chains has decreased. We expect that consolidation of drug wholesalers and retailers will increase pricing and other competitive pressures on drug manufacturers. In addition, the Company generally does not enter into long-term supply agreements with its customers that would require them to purchase our products.
20
The result of these developments may have a material adverse impact on our business, financial position and results of operations, and could cause the market value of our common stock to decline.
We face intense competition in the consumer products business.
Our business competes with large, well-financed cosmetic, pharmaceutical and consumer products companies with development and marketing groups that are experienced in the industry and possess far greater resources than those available to us. There is no assurance that we can compete successfully against our competitors or that we can develop and market products that will be favorably received in the marketplace.
Lack of availability, issues with quality or significant increases in the cost of raw materials used in manufacturing our products could adversely impact our profit margins and operating results.
Affordable, high quality raw materials and packaging components are essential to our business due to the nature of the products we manufacture. Raw materials and packaging components are generally available from multiple suppliers. Supplies of certain raw materials, and finished goods purchased by us are limited, or are available from one or only a few suppliers that have been pre-approved by FDA for use in the manufacture of our products. In this type of limited-supplier situation, increased prices, rationing and/or shortages can occur. In response to the situation, we try to identify alternative materials or suppliers for such raw materials and finished goods like containers and closures. However, FDA requirements for products approved through the ANDA or NDA process could substantially lengthen the time for approval of an alternate material source. Certain material shortages and approval of alternate sources could adversely affect our financial results. The rapid increase in cost of many raw materials from inflationary forces, such as increased energy costs, and our ability or inability to pass on these increases to our customers, could have a material impact on our financial results.
In addition, raw materials purchased from third parties, including those from foreign countries, may contain counterfeit ingredients or other adulterants. We maintain a strict program of verification and product testing throughout the ingredient sourcing and manufacturing process to identify potential counterfeit ingredients, adulterants and toxic substances. Nevertheless, discovery of previously unknown problems with the raw materials or product manufacturing processes or new data suggesting an unacceptable safety risk associated therewith, could result in a voluntary or mandatory withdrawal of a potentially contaminated product from the marketplace, either temporarily or permanently. In addition, because regulatory authorities must generally approve raw material sources for pharmaceutical products, changes in raw material suppliers or the quality of their products may result in production delays or higher raw material costs. Also, any future recall or removal would result in additional costs to us, and may give rise to product liability or other litigation, either of which could have a material adverse effect on our operating results.
Our products, and the raw materials used to make those products, generally have limited shelf lives. Our inventory levels are based, in part, on expectations regarding future sales. We may experience build-ups in inventory if sales slow. Any significant shortfall in sales may result in higher inventory levels of raw materials and finished products, thereby increasing the risk of inventory spoilage and corresponding inventory write-downs and write-offs, which may materially and adversely affect our results of operations. Additionally, labeling changes required for regulatory compliance could render packaging inventories obsolete. Cargo thefts and/or diversions and economically or maliciously motivated product tampering in store shelves may be experienced from time to time, causing unexpected shortages.
We depend on a limited number of suppliers for API. Generally, only a single source of API is qualified for use in each product due to the costs and time required to validate a second source of supply. Changes in API suppliers must usually be approved through a Prior Approval Supplement by the FDA.
We maintain several single-source supplier relationships, either because alternative sources are not available or because the relationship is advantageous due to regulatory, performance, quality, support, or price considerations. Unavailability or delivery delays of single-source components or products could adversely affect our ability to ship the relevant product in a timely manner. The effect of unavailability or delivery delays would be more severe if associated with our higher volume or more profitable products. Even where alternative sources of supply are available, qualifying the alternate suppliers and establishing reliable supplies could cost more or could result in delays and a loss of revenues. As a result, the loss of a single-source supplier could have a material adverse effect on our results of operations.
Incidents related to hazardous materials could materially adversely affect our reputation, business, financial condition, operating results and cash flows.
There are portions of our operations that require the controlled use of hazardous materials. Although we are diligent in designing and implementing safety procedures to comply with the standards prescribed by federal, state, and local regulations, the risk of
21
accidental contamination of property or injury to individuals from these materials cannot be completely eliminated. In the event of such an incident, we could be liable for any damages that result, which could materially adversely affect our reputation, business, financial condition, operating results and cash flows.
We are subject to stringent regulatory requirements. Failure to adhere to such requirements could harm our business and results of operations.
In the United States, we and our suppliers of raw materials are also subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other current and potential future federal, state or local regulations. Failure to adhere to such regulations, by either us or our suppliers, could harm our business and results of operations. In addition, our analytical department uses certain hazardous materials and chemicals in limited and controlled quantities. We have implemented safety procedures for handling and disposing of such materials, however, such procedures may not comply with the standards prescribed by federal, state and local regulations. Even if we follow such safety procedures for handling and disposing of hazardous materials and chemicals and such procedures comply with applicable law, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages and any such liability could exceed our resources.
Our operations and properties are also subject to a wide variety of increasingly complex and stringent federal, state and local environmental laws and regulations, including those governing the remediation of contaminated soil and groundwater. Such environmental laws may apply to conditions at properties and facilities presently or formerly owned or operated by us, as well as to conditions at properties at which wastes or other contamination attributable to us have been sent or otherwise come to be located. One of our facilities has undergone remediation of environmental contamination, and one of our facilities is currently undergoing remediation of environmental contamination. The total estimated costs for the clean-up and remediation is $0.9 million as of December 31, 2016, and remaining costs accrued at December 31, 2016 totaled $0.1 million. Based on information provided to us from our environmental consultants and what is known to date, we believe the reserves are sufficient for the remaining remediation of the environmental contamination. There is a possibility, however, that the remediation costs may exceed our estimates. In addition, we can give no assurance that the future cost of compliance with existing environmental laws will not give rise to additional significant expenditures or liabilities that would be material to us. Future events, such as new information, changes in existing environmental laws or their interpretation, and more vigorous enforcement policies of federal, state or local regulatory agencies, may have a material adverse effect on our business, financial condition and results of operations.
In Canada, we and our suppliers of raw materials are also subject to regulation under Hazardous Products Act, Controlled Products Regulations, Consumer Product Safety Act. Canadian Environmental Protection Act and other current and potential future federal, provincial/territorial or local regulations. Failure to adhere to such regulations, by either us or our suppliers, could harm our business and results of operations. In addition, our analytical department uses certain hazardous materials and chemicals in limited and controlled quantities. We have implemented safety procedures for handling and disposing of such materials, however, such procedures may not comply with the standards prescribed by federal, provincial/territorial and local regulations. Even if we follow such safety procedures for handling and disposing of hazardous materials and chemicals and such procedures comply with applicable law, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages and any such liability could exceed our resources.
Future events, such as new information, changes in existing environmental laws or their interpretation, and more vigorous enforcement policies of federal, provincial/territorial or local regulatory agencies, may have a material adverse effect on our business, financial condition and results of operations.
We are subject to extensive government regulation by the FDA and other federal, state and local regulatory authorities that increases our costs and could prevent us from marketing or selling our products.
The manufacturing, processing, formulation, packaging, labeling, testing, storing, distributing, marketing, advertising and sale of our products, among other things, are subject to extensive regulation by one or more U.S. agencies, including the FDA, the Federal Trade Commission and the Consumer Products Safety Commission, as well as by several state and local agencies in localities where our products are stored, distributed or sold. In addition, we manufacture and market certain of our products in accordance with standards set by organizations, such as the United States Pharmacopeial Convention, or USP, a scientific nonprofit organization that sets standards for the identity, strength, quality, and purity of medicines, food ingredients, and dietary supplements manufactured, distributed and consumed worldwide. USP’s drug standards are enforceable in the United States by the FDA.
The FDA regulates the testing, manufacture, labeling, marketing and sale of pharmaceutical products. Approval by the FDA is required before any new drug, including any new generic drug, may be marketed or sold in the United States. In order to receive approval from the FDA for our product candidates that are generic versions of brand-name drugs, we intend to use the Abbreviated
22
New Drug Application, or ANDA, route, which requires us to demonstrate to the FDA that each generic product candidate has the same active ingredient, strength, dosage form, route of administration and intended use as a corresponding approved drug product and is bioequivalent to the branded drug product (approved under a New Drug Application, or NDA), meaning that there is no significant difference between the drugs in their rate and extent of absorption in the body. However, if the FDA determines that an ANDA for a generic drug product is not adequate to support approval, it could deny our application or request additional data or information, which could delay approval of the product and impair our ability to compete with the brand-name drug product and/or other generic versions of the product.
If our product candidates receive FDA approval through the ANDA process, the labeling claims and marketing statements that we can make for our generic drugs are generally limited to the claims approved by the FDA for use in the brand-name product’s label. In addition, following regulatory approval, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the product will be subject to extensive and ongoing regulatory requirements.
As a manufacturer of pharmaceutical products, we must also comply with cGMPs, or current Good Manufacturing Practices, which include requirements related to production processes, quality control and assurance and recordkeeping. Our manufacturing facilities and procedures and those of our suppliers are subject to periodic inspection by the FDA and foreign regulatory agencies. Any material deviations from pharmaceutical cGMPs or other applicable requirements identified during such inspections may result in recalls or other enforcement actions, including warning letters, a delay or suspension in manufacturing operations, consent decrees or civil or criminal penalties. Further, discovery of previously unknown problems with a product or manufacturer may result in restrictions or sanctions, including suspension or withdrawal of marketing approvals, seizures or recalls of products from the market, or civil or criminal fines or penalties, any of which could significantly and adversely affect supplies of our products.
We are subject to extensive government regulation by Health Canada and other federal, state provincial/territorial and local regulatory authorities that increases our costs and could prevent us from marketing or selling our products.
The manufacturing, processing, formulation, packaging, labeling, testing, storing, distributing, marketing, advertising and sale of our products, among other things, are subject to extensive regulation by one or more Canadian agencies, including Health Canada, as well as by several state and local agencies in localities where our products are stored, distributed or sold. In addition, we market certain of our products in accordance with standards set by organizations, such as the United States Pharmacopeial Convention, or USP, and the British Pharmacopeia, or BP, scientific nonprofit organizations that sets standards for the identity, strength, quality, and purity of medicines, food ingredients, and dietary supplements manufactured, distributed and consumed worldwide. Adherence to USP and BP published drug standards are prescribed by the Food and Drug Regulations.
Health Canada regulates the testing, manufacture, labeling, marketing and sale of pharmaceutical products. Approval by Health Canada is required before any new drug, including any new generic drug, may be marketed or sold in Canada. In order to receive approval from Health Canada for our product candidates that are generic versions of brand-name drugs, we intend to use the ANDS, or Drug Identification Number Application, or DINA, routes, which requires us to demonstrate to Health Canada that each generic product candidate has the same active ingredient, strength, dosage form, route of administration and intended use as a corresponding approved drug product and is bioequivalent to the branded drug product (approved under a New Drug Submission or NDS or Drug Identification Number Application, or DINA), meaning that there is no significant difference between the drugs in their rate and extent of absorption in the body. However, if Health Canada determines that an ANDS or DINA for a generic drug product is not adequate to support approval, it could deny our application or request additional data or information, which could delay approval of the product and impair our ability to compete with the brand-name drug product and/or other generic versions of the product.
If our product candidates receive Health Canada approval through the ANDS or DINA process, the labeling claims and marketing statements that we can make for our generic drugs are generally limited to the claims approved by Health Canada for use in the brand-name product’s label. In addition, following regulatory approval, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the product will be subject to extensive and ongoing regulatory requirements.
As an importer and distributor of pharmaceutical products, we must also comply with cGMPs, or current Good Manufacturing Practices, which include requirements related to production processes, quality control and assurance and recordkeeping. Our facilities and procedures and those of our suppliers are subject to periodic inspection by Health Canada and foreign regulatory agencies. Any material deviations from pharmaceutical cGMPs or other applicable requirements identified during such inspections may result in recalls or other enforcement actions, including non-compliance ratings, a delay or suspension in manufacturing operations. Further, discovery of previously unknown problems with a product or manufacturer may result in restrictions or sanctions, including suspension or withdrawal of marketing approvals, seizures or recalls of products from the market, and revoking of licenses, any of which could significantly and adversely affect supplies of our products.
23
Our global operations expose us to certain risks, including challenges associated with political and economic instability, major hostilities and acts of terrorism.
We are a global company with operations outside of the United States. We face numerous risks inherent in conducting business internationally, including terrorist acts, acts of war, political unrest, public health concerns, labor disputes and national disasters. Such events may lead to economic and political uncertainties and contribute to global economic instability. We may not be successful in developing and implementing policies and strategies to address the foregoing events in a timely and effective manner. Consequently, the occurrence of one or more of the foregoing events could have a material adverse impact on our business, operating results and financial condition, including loss of sales or customers.
Violations of cGMP and other government regulations could have a material adverse effect on our reputation, business, financial condition and results of operations.
All facilities and manufacturing techniques used to manufacture pharmaceutical products for clinical use or for commercial sale in the United States and other Teligent markets must be operated in conformity with cGMP regulations as required by the FDA and other regulatory bodies. Our suppliers’ facilities are subject to scheduled periodic regulatory and customer inspections to ensure compliance with cGMP and other requirements applicable to such products. A finding that we or one or more of our suppliers had materially violated these requirements could result in one or more regulatory sanctions, loss of a customer contract, disqualification of data for client submissions to regulatory authorities and a mandated closing of our suppliers’ facilities, which in turn could have a material adverse effect on our reputation, business, financial condition, operating results and cash flows.
During our efforts to expand our existing manufacturing facility, as well as potentially select and build out an additional manufacturing facility, we could experience business interruptions, as well as incur significant capital expenditures to complete the expansions, which may have a material adverse effect on our business, financial position and results of operations.
We manufacture drug products at one domestic manufacturing facility. This facility may be forced to shut down or may be unable to operate at full capacity as a result of potential expansion plans. A significant disruption at this facility, even on a short-term basis, could impair our ability to produce and ship drug products to the market on a timely basis, which may have a material adverse effect on our business, financial position and results of operations.
We could experience business interruptions at our manufacturing facility, which may have a material adverse effect on our business, financial position and results of operations.
We manufacture drug products at one domestic manufacturing facility. This facility may be forced to shut down or may be unable to operate at full capacity as a result of hurricanes, tornadoes, earthquakes, storms and other extreme weather events as well as strikes, war, violent upheavals, terrorist acts and other force majeure events. A significant disruption at this facility, even on a short-term basis, could impair our ability to produce and ship drug products to the market on a timely basis, which may have a material adverse effect on our business, financial position and results of operations.
We are currently in the process of expanding our manufacturing facilities. Any delays in the expansion process or in the receipt of certain regulatory approvals in connection therewith could have a material adverse effect on our business and results of operations.
We are in the process of expanding and upgrading our existing manufacturing facilities in Buena, New Jersey. Upon the completion of this expansion, we intend to transfer the manufacture of certain sterile injectable, for which we currently rely on CMOs, to this facility. Any delays in the expansion process could increase the overall cost of the expansion and could force us to postpone the planned transfer of our manufacturing to this facility. In addition, any delays or denials of the regulatory approvals needed to begin manufacturing products at this facility could have a material adverse effect on our business.
Our reporting and payment obligations related to our participation in federal health care programs, including Medicare and Medicaid, are complex and often involve subjective decisions that could change. Any failure to comply with those obligations could subject us to investigation, penalties, and sanctions.
Federal laws regarding reporting and payment obligations with respect to a pharmaceutical company’s participation in federal health care programs, including Medicare and Medicaid, are complex. These programs generally require us to pay rebates or provide discounts to government payors in connection with our products that are dispensed to beneficiaries of these programs. In
24
some cases, such as with the Medicaid Drug Rebate Program, the rebates are based on pricing and rebate calculations that we report on a monthly and quarterly basis to the government agencies that administer the programs. Because our processes for calculating applicable government prices and the judgments involved in making these calculations involve subjective decisions and complex methodologies, these calculations are subject to risk of errors and differing interpretations. In addition, they are subject to review and challenge by the applicable governmental agencies, and it is possible that such reviews could result in changes that may have material adverse legal, regulatory, or economic consequences. Responding to current and future changes may increase our costs and the complexity of compliance will be time-consuming, and could have a material adverse effect on our results of operations.
In addition, the Office of Inspector General has recently increased its focus on the methodologies used by manufacturers to calculate the average manufacturer price, or AMP, and best price, or BP, to assess manufacturer compliance with reporting requirements under the Medicaid Drug Rebate Program. We are liable for errors associated with our submission of pricing data and for overcharging government payors. For example, failure to submit monthly/quarterly AMP and BP data on a timely basis could result in a civil monetary penalty of $10,000 per day for each day the submission is late beyond the due date. Failure to make necessary disclosures and/or to identify overpayments could result in allegations against us under the Federal False Claims Act and other laws and regulations.
Our policies regarding returns, allowances and chargebacks, failure to supply penalties and marketing programs adopted by wholesalers may reduce revenues in future fiscal periods.
We, like other generic drug manufacturers, have agreements with customers allowing chargebacks, product returns, administrative fees, failure to supply penalties and other rebates. Under many of these arrangements, we may match lower prices offered to customers by competitors. If we choose to lower our prices, we generally give the customer a credit on the products that the customer is holding in inventory, which could reduce sales revenue and gross margin for the period the credit is provided. Under many of these arrangements, we may have failure to supply penalties, which in the event we are unable to supply a certain product and are unable to meet the needs of our customers, we may incur failure to supply penalties which may be significant. Like our competitors, we also give credits for chargebacks to wholesalers with whom we have contracts for their sales to hospitals, group purchasing organizations, pharmacies or other customers. A chargeback is the difference between the price at which we invoice the wholesaler and the price that the wholesaler’s end-customer pays for a product. Although we establish reserves based on prior experience and our best estimates of the impact that these policies may have in subsequent periods, we cannot ensure that our reserves are adequate or that actual product returns, allowances, and chargebacks will not exceed our estimates. As we continue to experience the consolidation of our customers, which may result in changes to previous patterns of ordering and/or pricing of our products, this could disrupt our established methodologies for calculating our provisions for chargebacks and other accruals.
We are subject to federal and state healthcare fraud and abuse and false claims laws and may be subject to related litigation brought by the government or private individuals.
We are subject to state and federal healthcare laws pertaining to fraud and abuse, physician payment transparency and laws that govern the submission of claims for reimbursement. These laws include the following:
• | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs, such as Medicare and Medicaid. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
• | the federal False Claims Act, or FCA, which imposes civil liability and criminal fines on individuals or entities that knowingly submit, or cause to be submitted, false or fraudulent claims for payment to the government. The FCA also allows private individuals to bring a suit on behalf of the government against an individual or entity for violations of the FCA. These suits, also known as qui tam actions, may be brought by, with only a few exceptions, any private citizen who believes that he has material information of a false claim that has not yet been previously disclosed. These suits have increased significantly in recent years because the FCA allows an individual to share in any amounts paid to the federal government in fines or settlement as a result of a successful qui tam action; |
• | federal criminal laws that prohibit executing a scheme to defraud any federal healthcare benefit program or making false statements relating to healthcare matters; |
25
• | the federal Physician Payment Sunshine Act, which requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the government information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and applicable manufacturers and group purchasing organizations to report annually ownership and investment interests held by physicians (as defined above) and their immediate family members and payments or other “transfers of value” to such physician owners and their immediate family members; |
• | the Federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; and |
• | analogous state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
If our past or present operations are found to be in violation of any of such laws or any other governmental regulations that may apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from federal health care programs, and/or the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment, or restructuring of our operations could adversely affect our ability to operate our business and our financial results, action against us for violation of these laws, even if we successfully defend against them, it could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the Affordable Care Act, was passed, which substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressures.
Even after our products receive regulatory approval, such products may not achieve expected levels of market acceptance.
Even if we are able to obtain regulatory approvals for our generic pharmaceutical products the success of those products is dependent upon market acceptance. Levels of market acceptance for our products could be impacted by several factors, including but not limited to:
• | the availability of alternative products from our competitors; |
• | the price of our products relative to that of our competitors; |
• | the timing of our market entry; |
• | the ability to market our products effectively to the different levels in the distribution chain; |
• | other competitor actions; and |
• | the continued acceptance of and/or reimbursement for our products by government and private formularies and/or third party payors. |
26
Additionally, studies of the proper utilization, safety, and efficacy of pharmaceutical products are being conducted by the industry, government agencies, and others. Such studies, which increasingly employ sophisticated methods and techniques, including methods to investigate the comparative effectiveness of different products used for similar indications, can call into question the utilization, safety, and efficacy of previously marketed as well as future products. In some cases, studies have resulted, and may in the future result, in the discontinuance of product marketing or other risk management programs, such as the need for a patient registry, as well as delays in approvals. The occurrence of any of the above risks could adversely affect our profitability, business, financial position, results of operations and/or cash flow, and could cause the market value of our common stock to decline.
Product recalls could harm our business.
Product recalls or product field alerts may be issued at our discretion or required by the FDA and Health Canada, other governmental agencies or other companies having regulatory authority for pharmaceutical product sales. From time to time, we may recall products for various reasons, including failure of our products to maintain their stability through their expiration dates or other quality issues. Any recall or product field alert has the potential of damaging our reputation or the reputation of the product. Any significant recalls could materially affect our sales. In these cases, our business, financial condition, results of operations and cash flows could be materially adversely affected.
We are susceptible to product liability claims that may not be covered by insurance and could require us to pay substantial sums.
We face the risk of loss resulting from, and adverse publicity and reputational harm associated with, product liability lawsuits, whether or not such claims are valid. We may not be able to avoid such claims. In addition, our product liability insurance may not be adequate to cover such claims and we may not be able to obtain adequate insurance coverage in the future at acceptable costs. A successful product liability claim that exceeds our policy limits could require us to pay substantial sums. In addition, product liability coverage for pharmaceutical companies is becoming more expensive and increasingly difficult to obtain and, as a result, we may not be able to obtain the type and amount of coverage we desire or to maintain our current coverage.
The manufacture and storage of pharmaceutical and other products are subject to inherent risk.
Because chemical ingredients are used in the manufacture of our products and due to the nature of the manufacturing process itself, there is a risk of incurring liability for damages caused by or during the storage or manufacture of both the chemical ingredients and the finished products. Although we have never incurred any material liability for damages of that nature, we may be subject to liability in the future. In addition, while we believe our insurance coverage is adequate, it is possible that a successful claim would exceed our coverage, requiring us to pay a substantial sum.
The testing required for the regulatory approval of our products is conducted by independent third parties. Any failure by any of these third parties to perform this testing properly and in a timely manner may have an adverse effect upon our ability to obtain regulatory approvals.
Our applications for the regulatory approval of our products incorporate the results of testing and other information that is conducted or gathered by independent third parties (including, for example, manufacturers of raw materials, testing laboratories, CROs or independent research facilities). Our ability to obtain regulatory approval of the products being tested is dependent upon the quality of the work performed by these third parties, the quality of the third parties’ facilities, and the accuracy of the information provided to us by third parties. We have little or no control over any of these factors. If this testing is not performed properly, our ability to obtain regulatory approvals could be restricted or delayed. In addition, if third party fraud or other recordkeeping problems are discovered after our products are approved for marketing, any government investigations or findings could result in any products that incorporated those fraudulent results having their regulatory approvals withdrawn.
The failure to obtain, maintain or protect patents, trade secrets, know-how and other intellectual property could impact our ability to compete effectively.
To compete effectively, we need to develop and maintain a proprietary position with regard to our own technology, products and business. We rely on a combination of patents, trade secrets, proprietary know-how and other intellectual property to protect our proprietary technology and rights. We also maintain a number of trade secrets, know-how and other intellectual property.
The risks and uncertainties that we face with respect to patents and other proprietary rights include the following:
27
• | the pending patent applications we have filed or may file, or to which we have exclusive rights, may not result in issued patents, or may take longer than we expect to result in issued patents; |
• | changes in U.S. patent laws may adversely affect our ability to obtain or maintain our patent protection; |
• | we may be subject to interference proceedings; |
• | the claims of any patents that are issued may not provide meaningful protection; |
• | we may not be able to develop additional proprietary technologies that are patentable; |
• | the patents licensed or issued to us or our collaborators may not provide a competitive advantage; |
• | other companies may challenge patents licensed or issued to us or our collaborators; |
• | other companies may independently develop similar or alternative technologies, or duplicate our technology; |
• | other companies may design around technologies we have licensed or developed; and |
• | enforcement of patents is complex, uncertain and expensive. |
The trademark applications we have filed or may file may not result in trademark registrations, which would result in lesser protections for our brands.
Our product offerings and our customers’ products may infringe on the intellectual property rights of third parties.
From time to time, third parties have asserted intellectual property infringement claims against us and our customers and there can be no assurance that third parties will not assert infringement claims against either us or our customers in the future. While we believe that our product offerings do not infringe in any material respect upon proprietary rights of other parties and/or that meritorious defenses would exist with respect to any assertions to the contrary, there can be no assurance that we would not be found to infringe on the proprietary rights of others.
Patent applications in the U.S. and some foreign countries are generally not publicly disclosed until they are published or the patent is issued, and we may not be aware of currently filed patent applications that relate to our offerings or processes. If patents later issue on these applications, we may be found liable for subsequent infringement. There has been substantial litigation in the pharmaceutical and biotechnology industries with respect to the manufacture, use and sale of products and processes that are the subject of conflicting patent rights.
Any claims that our product offerings or processes infringe these rights, regardless of their merit or resolutions, could be costly and may divert the efforts and attention of our management and technical personnel. We may not prevail in such proceedings given the complex technical issues and inherent uncertainties in intellectual property litigation. If such proceedings result in an adverse outcome, we could, among other things, be required to:
• | pay damages in the form of lost profits and/or a reasonable royalty for any infringement; |
• | pay substantial damages (potentially treble damages in the U.S. if any such infringement is found to be willful); |
• | pay attorney fees of a prevailing party, if the case is found to be exceptional; |
• | cease the manufacture, use or sale of the infringing offerings or processes; |
• | discontinue the use of the infringing technology; |
• | expend significant resources to design around patented technology and develop non-infringing technology; and |
28
• | license patented technology from the third party claiming infringement, which license may not be available on commercially reasonable terms, or may not be available at all. |
In addition, our customers’ products may be subject to claims of intellectual property infringement and such claims could materially affect our business if their products cease to be manufactured and they have to discontinue the use of the infringing technology which we may provide. Further, depending on the particular circumstances of any given claim, it may be the case that we may be responsible for indemnifying our customers for a claim of intellectual property infringement.
If we were to assert any of our own intellectual property against third parties and the third parties were found not to infringe our intellectual property or our intellectual property was found to be invalid, and/or unenforceable, we would lose the opportunity to leverage our own intellectual property, for example, through licensing of our technology to others, collection of damages and/or royalty payments based upon successful assertion of our intellectual property rights via enjoining others from practicing the technology at issue.
Any of the foregoing could affect our ability to compete or have a material adverse effect on our business, financial condition and results of operations.
Significant balances of intangible assets, including goodwill, are subject to impairment testing and may result in impairment charges, which may materially and adversely affect our results of operations and financial condition.
A significant amount of our total assets is related to goodwill and intangible assets. As of December 31, 2016 the value of our goodwill and intangible assets net of accumulated amortization was $52.9 million. Goodwill and other intangible assets are tested for impairment annually when events occur or circumstances change that could potentially reduce the fair value of the reporting unit or intangible asset. Impairment testing compares the fair value of the reporting unit or intangible asset to its carrying amount. Any future goodwill or other intangible asset impairment, if any, would be recorded in operating income and could have a material adverse effect on our results of operations and financial condition.
We may not be able to fully realize the expected benefits from the acquisition of certain products and/or companies.
Our recent acquisition of certain products and a company subjects us to additional operational and financial risks, including the following:
• | additional costs that we may need to incur in order to return the products to the market and to comply with regulatory requirements; |
• | difficulties in coordinating research and development activities; |
• | uncertainties in the business relationships with our customers and suppliers; and |
• | lack of previous experiences in manufacturing, commercializing, and distributing products in therapeutic areas outside of the topical generic pharmaceutical market and in markets outside of the United States. |
Our approved products may not achieve commercialization at levels of market acceptance that allow us to achieve profitability, which could have a material adverse effect on our business, financial position and results of operations.
We seek to develop, license or acquire products that we can commercialize at levels of market acceptance that would allow us to recoup the costs of development and commercialization, grow market share, and achieve profitability. Even if we are able to obtain regulatory approvals for certain pharmaceutical products, if we fail to accurately predict demand for such products, our business, financial position, and results of operations could be adversely impacted. Levels of market acceptance for products could be impacted by several factors, including but not limited to:
• | the availability of alternative products from our competitors; |
• | the price of our products relative to that of our competitors; |
• | the effectiveness of our marketing relative to that of our competitors; |
• | the timing of our market entry; |
29
• | the ability to market our products effectively to the retail level; and |
• | the acceptance of our products by government and private formularies. |
Some of these factors are not within our control and, if any arises, our profitability, business, financial position and results of operations could be materially adversely affected.
Future acquisitions and investments could disrupt our business and harm our financial condition and operating results.
Our growth will depend, in part, on our continued ability to develop, commercialize and expand our drug products, including in response to changing regulatory and competitive pressures. In some circumstances, we accelerate our growth through the acquisition of complementary products and technologies rather than through internal development. The identification of suitable products to be acquired can be difficult, time-consuming and costly, and we may not be able to successfully complete or successfully execute strategies for identified acquisitions. The risks faced in connection with acquisitions include:
• | diversion of management time and focus from operating our business to addressing acquisition and/or product integration challenges; |
• | coordination of research and development and sales and marketing functions; |
• | retention of key employees from the acquired company; |
• | integration of the acquired company’s accounting, management information, human resources and other administrative systems; |
• | the need to implement or improve controls, procedures, and policies at a business that prior to the acquisition may have lacked effective controls, procedures and policies; |
• | liability for activities of the acquired company and/or products before the acquisition, including patent infringement claims, violations of laws, commercial disputes, tax liabilities and other known and unknown liabilities; |
• | unanticipated write-offs or charges; and |
• | litigation or other claims in connection with the acquired company or product, including claims from product users, former stockholders or other third parties. |
In any acquisition that we may undertake, our failure to address these risks or other problems encountered in connection with any acquisitions and investments could cause us to fail to realize the anticipated benefits of these acquisitions or investments, cause us to incur unanticipated liabilities, and harm our business generally.
We may become involved in legal proceedings from time to time which may result in losses, damage to our business and reputation and place a strain on our internal resources.
In the ordinary course of our business, we may be involved in legal proceedings with both private parties and certain government agencies, including FDA. Enforcement actions and litigation may result in verdicts against us, which may include significant monetary awards, judgments that certain of our intellectual property rights are invalid or unenforceable and injunctions preventing the manufacture, marketing and sale of our products. If disputes are resolved unfavorably, our business, financial condition and results of operations may be adversely affected.
Any government enforcement action or litigation, whether or not successful, may damage our reputation. Furthermore, we are likely to incur substantial expense in defending these actions and lawsuits, and the time demands of such enforcement actions and lawsuits could divert management’s attention from ongoing business concerns and interfere with our normal operations.
In the normal course of business, we periodically enter into employment agreements, legal settlements, and other agreements which incorporate indemnification provisions. We maintain insurance coverage which we believe will effectively mitigate our obligations under these indemnification provisions. However, should our obligation under an indemnification provision exceed our coverage or should coverage be denied, it could have a material adverse effect on our business, financial position and results of operations.
30
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our product development programs. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability and the further development of our product candidates may be delayed.
In addition, we rely on complex information technology systems, including Internet-based systems, to support our supply chain processes as well as internal and external communications. The size and complexity of our systems make them potentially vulnerable to breakdown or interruption, whether due to computer viruses or other causes that may result in the loss of key information or the impairment of production and other supply chain processes. Such disruptions and breaches of security could adversely affect our business.
Compliance with ongoing post-marketing obligations for our approved ANDAs, NDAs, NDSs, and ANDSs may uncover new safety information that could give rise to a product recall, updated warnings, or other regulatory actions that could have an adverse impact on our business.
After the FDA or Health Canada approves a drug for marketing under an NDA, ANDA, NDS, or ANDS, the product’s sponsor must comply with several post-marketing obligations that continue until the product is discontinued. These post-marking obligations include the prompt reporting of serious adverse events to the agency, the submission of product-specific annual reports that include changes in the distribution, manufacturing, and labeling information, and notification when a drug product is found to have significant deviations from its approved manufacturing specifications (among others). Our ongoing compliance with these types of mandatory reporting requirements could result in additional requests for information from the FDA or Health Canada and, depending on the scope of a potential product issue that the FDA or Health Canada may decide to pursue, potentially also result in a request from the agency to conduct a product recall or to strengthen warnings and/or revise other label information about the product. Any of these post-marketing regulatory actions could materially affect our sales and, therefore, they have the potential to adversely affect our business, financial condition, results of operations and cash flows.
Economic conditions could severely impact us.
Current economic conditions may cause a decline in business and consumer spending which could adversely affect our business and financial performance. Our operating results are impacted by the health of the North American economies. Our business and financial performance, including collection of our accounts receivable, realization of inventory, recoverability of assets including investments, may be adversely affected by current and future economic conditions, such as a reduction in the availability of credit, financial market volatility and recession.
Adverse conditions in the economy and disruption of financial markets could negatively impact our customers and therefore our results of operations.
An economic downturn in the businesses or geographic areas in which we sell our products could reduce demand for these products and result in a decrease in sales volume that could have a negative impact on our results of operations. Volatility and disruption of financial markets could limit our customers’ ability to obtain adequate financing or credit to purchase and pay for our products in a timely manner, or to maintain operations, and result in a decrease in sales volume that could have a negative impact on our results of operations. Additionally, economic conditions and market turbulence may also impact our suppliers causing them to be unable to supply in a timely manner sufficient quantities of product components, thereby impairing our ability to manufacture on schedule and at commercially reasonable costs.
If the U.S. economy rapidly contracts or expands, we may have difficulty quickly scaling our operations in response, which may negatively impact our business and financial position.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
We will need to hire additional qualified personnel with expertise in nonclinical testing, government regulation, formulation and manufacturing, sales and marketing and finance. We compete for qualified individuals with numerous pharmaceutical and consumer products companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be
31
certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
If we are unable to satisfy regulatory requirements relating to internal controls, our stock price could suffer.
Section 404 of the Sarbanes-Oxley Act of 2002 requires companies to conduct a comprehensive evaluation of the effectiveness of their internal control over financial reporting. At the end of each fiscal year, we must perform an evaluation of our internal control over financial reporting, include in our annual report the results of the evaluation and have our external auditors also publicly attest to the effectiveness of our internal control over financial reporting. If material weaknesses are found in our internal controls in the future, if we fail to complete future evaluations on time or if our external auditors cannot attest to the effectiveness of our internal control over financial reporting, we could fail to meet our regulatory reporting requirements and be subject to regulatory scrutiny and a loss of public confidence in our internal controls, which could have an adverse effect on our stock price.
Currency fluctuations and changes in exchange rates could adversely affect our business, financial condition, results of operations, cash flows, and/or common stock price.
Although we report our financial results in U.S. Dollars, a portion of our revenues and other liabilities and our costs are denominated in non-U.S. currencies, including the Euro and Canadian Dollar. Our results of operations and, in some cases, cash flows, have in the past been and may in the future be adversely affected by certain movements in currency exchange rates. The occurrence of any of the above risks could cause a material adverse effect on our business, financial condition, results of operations, cash flows, and/or share price.
The Company is exposed to market risk from fluctuations in currency exchange rates.
The Company operates in multiple jurisdictions denominated in currencies of the local jurisdiction. Additionally, the Company
may enter into acquisition, licensing, borrowing or other financial transactions that may give rise to currency exposure. Since
the Company cannot, with certainty, foresee and mitigate against such adverse fluctuations, fluctuations in currency exchange
rates could negatively affect the Company’s results of operations, financial position and cash flows.
Our ability to use our net operating loss carry forwards and certain other tax attributes may be limited.
As of December 31, 2016, we had federal net operating loss carry forwards, or NOLs, of approximately $34.6 million which expire from 2020 through 2035. Our ability to utilize our NOLs may be limited under Section 382 of the Internal Revenue Code. The limitations apply if an ownership change, as defined by Section 382, occurs. Generally, an ownership change occurs when certain shareholders increase their aggregate ownership by more than 50 percentage points over their lowest ownership percentage in a testing period (typically three years). Our ability to use net operating loss carry forwards is subject to substantial limitation in future periods under certain provisions of Section 382 of the Internal Revenue Code, which limit the utilization of net operating losses upon a more than 50% change in ownership of our stock that is held by 5% or greater stockholders. We examined the application of Section 382 with respect to an ownership change that took place during 2010, as well as the limitation on the application of net operating loss carry forwards. We believe that operating losses subsequent to the change date in 2010 (aggregating $15.3 million) are not subject to Section 382 limitations. We have estimated that the annual limitation starting in 2010 aggregates from $1.0 million to $2.3 million per year including the effect of amortization of built in gains.
We are currently involved in antitrust litigation related to our pricing practices.
Complaints have been filed against us in each of the U.S. District Court for the District of New Jersey and the U.S. District Court for the Eastern District of Pennsylvania alleging violations of various provisions of federal and state antitrust laws in connection with the sale of our antifungal skin cream Econazole Nitrate 1% product. While we intend to vigorously defend our position in connection with both lawsuits, the outcome of the litigation could result in serious fines being levied on us, along with harm to our reputation. Any negative outcome from this or any other investigation related to our pricing could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Common Stock
Shares of our common stock can be relatively illiquid which may affect the trading price of our common stock.
For the year ended December 31, 2016, the average daily trading volume of our common stock on the NASDAQ Global Select Market was approximately 299,310 shares. As a result of our relatively small public float, our common stock may be less liquid
32
than the stock of companies with broader public ownership. Among other things, trading of a relatively small volume of our common stock may have a greater impact on the trading price for our shares than would be the case if our public float were larger.
We have not paid dividends to our common stockholders in the past nor do we expect to pay dividends in the foreseeable future, and any return on investment may be limited to potential future appreciation on the value of our common stock.
We currently intend to retain any future earnings to support the development and expansion of our business and do not anticipate paying cash dividends in the foreseeable future. Our payment of any future dividends will be at the discretion of our Board of Directors after taking into account various factors, including without limitation, our financial condition, operating results, cash needs, growth plans and the terms of any credit agreements that we may be a party to at the time. To the extent we do not pay dividends, our stock may be less valuable because a return on investment will only occur if and to the extent our stock price appreciates, which may never occur. In addition, investors must rely on sales of their common stock after price appreciation as the only way to realize their investment, and if the price of our stock does not appreciate, then there will be no return on investment. Investors seeking cash dividends should not purchase our common stock.
If we fail to comply with the reporting obligations of the Exchange Act and Section 404 of the Sarbanes-Oxley Act of 2002, or if we fail to achieve and maintain adequate disclosure controls and procedures and internal control over financial reporting, our business results of operations and financial condition, and investors’ confidence in us, could be materially adversely affected.
As a public company, we are required to comply with the periodic reporting obligations of the Exchange Act including preparing annual reports, quarterly reports and current reports. Our failure to prepare and disclose this information in a timely manner could subject us to penalties under federal securities laws, expose us to lawsuits and restrict our ability to access financing. In addition, we are required under applicable law and regulations to integrate our systems of disclosure controls and procedures and internal control over financial reporting. Our management assessed our existing disclosure controls and procedures as of December 31, 2016 and December 31, 2015, and our management concluded that our disclosure controls and procedures were effective as of such times. The year ended December 31, 2014 was the first year in which we were required to have our external auditors issue an attestation report on the effectiveness of internal controls over financial reporting.
If we fail to achieve and maintain the adequacy of our disclosure controls and procedures and internal control over financial reporting, we may not be able to ensure that we can conclude that we have effective disclosure controls and procedures and internal control over financial reporting in accordance with the Sarbanes-Oxley Act of 2002. Moreover, effective disclosure controls and procedures and internal control over financial reporting are necessary for us to produce reliable financial reports and are important to help prevent fraud. As a result, our failure to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 on a timely basis could result in the loss of investor confidence in the reliability of our financial statements, which in turn could harm our business and negatively impact the trading price of our common stock.
Our principal stockholders, directors and executive officers own a significant percentage of our stock and will be able to exercise significant influence over our affairs.
Our current principal stockholders, directors and executive officers own in the aggregate a significant portion of the voting power of our capital stock. As a result, these stockholders, if acting together, would be able to influence or control matters requiring approval by our stockholders, including the election of directors and the approval of mergers, acquisitions or other extraordinary transactions. They may also have interests that differ from yours and may vote in a way with which you disagree and which may be adverse to your interests. This concentration of ownership may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company and might ultimately affect the market price of our common stock. If such stockholders sold a significant amount of stock it could have an adverse effect on the price of the stock.
Due to the concentration of common stock owned by significant stockholders, the sale of such stock might adversely affect the price of our common stock.
Our largest stockholders own shares of common stock that have been registered for resale under the Securities Act. The sale of such stock, depending on the interplay of numerous factors, including, without limitation, the method and timing of the sales, could substantially depress the value of our common stock.
Our stock price is, and we expect it to remain, volatile and subject to wide fluctuations, which may make it difficult for stockholders to sell shares of common stock at or above the price for which they were acquired.
33
Our stock price is, and we expect it to remain, volatile, which could limit investors’ ability to sell stock at a profit. During the last two fiscal years, our stock price has closed at a low of $4.46 in the first quarter of 2016 and a high of $12.05 in the first quarter of 2015. The volatile price of our stock makes it difficult for investors to predict the value of their investment, to sell shares at a profit at any given time, or to plan purchases and sales in advance. A variety of factors may affect the market price of our common stock. These include, but are not limited to:
• | publicity regarding actual or potential clinical results relating to products under development by our competitors or us; |
• | delay or failure in initiating, completing or analyzing nonclinical or clinical trials or the unsatisfactory design or results of these trials; |
• | achievement or rejection of regulatory approvals by our competitors or us; |
• | announcements of technological innovations or new commercial products by our competitors or us; |
• | developments concerning proprietary rights, including patents; |
• | developments concerning our collaborations; |
• | regulatory developments in the U.S. and foreign countries; |
• | economic or other crises, especially given the recent financial deterioration in the markets in which we compete, and other external factors; |
• | stock market price and volume fluctuations of other publicly traded companies and, in particular, those that are in the cosmetic, pharmaceutical and consumer products industry; |
• | actual or anticipated sales of our common stock, including sales by our directors, officers or significant stockholders; |
• | period-to-period fluctuations in our revenues and other results of operations; and |
• | speculation about our business in the press or the investment community. |
In the past, securities class action litigation has often been instituted against companies following periods of volatility in their stock price. This type of litigation, even if it does not result in liability for us, could result in substantial costs to us and divert management’s attention and resources.
If we fail to meet the continued listing standards of the NASDAQ Global Select Market, our common stock could be delisted and our liquidity and stock price could suffer.
Our common stock is listed on the NASDAQ Global Select Market, a national securities exchange, which imposes continued listing requirements with respect to listed shares. If we fail to meet the continued listing standards of the NASDAQ Global Select Market, our common stock could be delisted and our stock price could suffer. A delisting of our shares of common stock could negatively impact us by further reducing the liquidity and market price of our shares of common stock and the number of investors willing to hold or acquire our shares of common stock, which could negatively impact our ability to raise equity financing.
Risks Related to the Notes
We may not have the ability to raise the funds necessary to settle conversions of the Notes, purchase the Notes as required pursuant to the terms of the indenture governing the Notes or pay the redemption price for any Notes we redeem, and our future debt may contain limitations on our ability to pay cash upon conversion or repurchase of the Notes.
On December 16, 2014, we completed the sale of $125 million aggregate principal amount of our 3.75% Convertible Senior Notes due 2019, or the Notes, to Deutsche Bank Securities Inc. and J.P. Morgan Securities LLC as the initial purchasers and on December 22, 2014, we issued to the initial purchasers an additional $18.75 million aggregate principal amount of the Notes. Pursuant to the terms of the indenture governing the Notes, following certain events, holders of Notes will have the right to require us to purchase their Notes for cash. Such event may also constitute an event of default or prepayment under, and result in the acceleration of the
34
maturity of, our then-existing indebtedness. We cannot assure you that we will have sufficient financial resources, or will be able to arrange financing, to pay the purchase price in cash with respect to any Notes surrendered by holders for purchase at that time, make cash payments upon conversions or pay the redemption price for any Notes we redeem. In addition, restrictions in our then existing credit facilities or other indebtedness, if any, may not allow us to purchase the Notes (even if required pursuant to the terms of the indenture), make cash payments upon conversions of the Notes or pay the redemption price for any Notes we redeem would result in an event of default with respect to the Notes which could, in turn, constitute a default under the terms of our other indebtedness, if any. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and purchase the Notes, make cash payments upon conversions thereof or pay the redemption price for any Notes we redeem.
Servicing our debt requires a significant amount of cash, and we may not have sufficient cash flow from our business to pay our debt.
Our ability to make scheduled payments of the principal of, to pay interest on, to pay any cash due upon conversion of or to refinance our indebtedness, including the Notes, depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. Our business may not continue to generate cash flow from operations in the future sufficient to service our debt and make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
To the extent we issue shares of our common stock to satisfy all or a portion of our conversion obligation, conversions of the Notes will dilute the ownership interest of our existing stockholders, including holders who had previously converted their Notes.
The holders of our Notes can require us, under certain circumstances, to convert their Notes. We have the option to satisfy this conversion obligation with cash, shares of our common stock or a combination of cash and shares of our common stock at our election. To the extent we issue shares of our common stock to satisfy all or a portion of our conversion obligation, the conversion of some or all of the Notes will dilute the ownership interests of our existing stockholders. Any sales in the public market of our common stock issuable upon such conversion could adversely affect prevailing market prices of our common stock. In addition, the existence of the Notes may encourage short selling by market participants because the conversion of the Notes could depress the price of our common stock.
Our substantial indebtedness could materially adversely affect our business, financial condition or results of operations and prevent us from fulfilling our obligations under the Notes.
After giving effect to the issuance of the Notes, we will have a substantial amount of indebtedness. As of December 31, 2016, our total consolidated indebtedness was $143.75 million. Our substantial level of indebtedness increases the possibility that we may be unable to generate cash sufficient to pay, when due, the principal of, interest on, or other amounts due in respect of our indebtedness. Our substantial indebtedness, combined with our other financial obligations and contractual commitments, may have a material adverse impact on us. For example, it could
• | make it difficult for us to satisfy our obligations with respect to our outstanding and other future debt obligations; |
• | increase our vulnerability to general adverse economic conditions or a downturn in the industries in which we operate; |
• | impair our ability to obtain additional financing in the future for working capital, investments, acquisitions and other general corporate purposes; |
• | require us to dedicate a substantial portion of our cash flows to the payment to our financing sources, thereby reducing the availability of our cash flows to fund working capital, investments, acquisitions and other general corporate purposes; and |
• | place us at a disadvantage compared to our competitors. |
35
We will continue to have the ability to incur debt; if we incur substantial additional debt, these higher levels of debt may affect our ability to pay the principal of and interest on the Notes.
We and our subsidiaries may be able to incur substantial additional debt in the future, subject to the restrictions contained in our debt instruments, some of which may be secured debt. The indenture governing the Notes does not restrict our ability to incur additional indebtedness or require us to maintain financial ratios or specified levels of net worth or liquidity. If we incur substantial additional indebtedness in the future, these higher levels of indebtedness may affect our ability to pay the principal of and interest on the Notes, or any fundamental change purchase price or any cash due upon conversion, and our creditworthiness generally.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
Item 2. PROPERTIES
The Company’s executive administrative offices are located in Buena, New Jersey, in two facilities totaling approximately 33,000 square feet built on 8.44 acres of land in 1995, which we own. One of those facilities is used for production, product development, marketing and warehousing for our own generic prescription pharmaceutical products and pharmaceutical, cosmeceutical and cosmetic products. In July 2016, the Company completed the first phase of the facility expansion in the Buena, New Jersey location. The facility now houses our new product development laboratory for work on topical and sterile pharmaceuticals. The other facility is currently being expanded to increase our manufacturing capacity for topical products, and will also enable the production of sterile injectable products in both vial and ampule presentations. We lease an additional 11,000 square feet of warehouse space in Vineland, New Jersey, lease approximately 7,500 square feet of corporate office space in Iselin, New Jersey, and lease approximately 4,000 square feet of office space in Toronto, Canada. The Company also leases approximately 2,605 square feet of office and laboratory space in Tallinn, Estonia.
36
Item 3. LEGAL PROCEEDINGS
On March 2, 2001, the Company became aware of environmental contamination resulting from an unknown heating oil leak at its former manufacturing facility. The Company immediately notified the New Jersey Department of Environmental Protection, or NJ DEP, and the local authorities, and hired a contractor to assess the exposure and required clean up. The total estimated costs for the clean-up and remediation was $889,000, of which approximately $123,000 remains accrued as of December 31, 2016. Based on information provided to the Company from its environmental consultant and what is known to date, the Company believes the reserve is sufficient for the remaining remediation of the environmental contamination. There is a possibility, however, that the remediation costs may exceed the Company’s estimates.
The restricted cash, included in other assets on the Consolidated Balance Sheet of $122,000 as of December 31, 2016 and $124,000 as of December 31, 2015, represents a restricted escrow account set up on the requirement of the NJ DEP for the soil remediation work. These funds will be released to the Company upon the NJ DEP approval when the remediation is completed.
On December 19, 2013, we filed a complaint in the United States District Court for the District of Delaware against Mallinckrodt LLC, Mallinckrodt, Inc. and Nuvo Research Inc., which we collectively refer to as Mallinckrodt, seeking a declaration of non-infringement of United States Patent Nos. 8,217,078 and 8,546,450 so that we can bring our generic diclofenac sodium topical solution 1.5% to market at the earliest possible date under applicable statutory and FDA regulatory provisions. On January 10, 2014, Mallinckrodt filed an answer and counterclaim alleging that we infringed the patents at issue. On June 26, 2014, we entered into a settlement agreement with Mallinckrodt, pursuant to which Mallinckrodt granted us a non-exclusive license to launch our diclofenac sodium topical solution 1.5% product on March 28, 2015. There was no material impact on our financial statements as a result of the settlement. We received approval to sell our diclofenac sodium topical solution 1.5% from the FDA in July 2015.
On May 21, 2015, Horizon Pharma Ireland Limited, HZNP Limited and Horizon Pharma USA, Inc., which we collectively refer to as Horizon, filed a complaint in the United States District Court for the District of New Jersey against the Company alleging infringement of certain United States patents based upon our submission to the FDA of an Abbreviated New Drug Application, or ANDA seeking FDA approval to market diclofenac topical solution 2% w/w before the expiration of the patents asserted in the complaint. On June 30, 2015, August 11, 2015, September 17, 2015, October 27, 2015 and February 5, 2016, Horizon filed additional complaints in the United States District Court for the District of New Jersey against the Company alleging infringement of other of its United States patents in relation to the Company’s submission of the same ANDA. On July 21, 2015, September 11, 2015, October 6, 2015, October 21, 2015, December 17, 2015 and March 17, 2016, the Company filed answers, affirmative defenses and counterclaims with respect to the complaints filed by Horizon. In those filings, the Company asserted that the patents alleged to be infringed in the complaints filed by Horizon are invalid and not infringed by us. On April 27, 2016, Horizon and the Company filed a stipulation of dismissal to dismiss the cases. The court entered an order dismissing the cases on May 2, 2016. On May 9, 2016, Horizon and the Company entered into a settlement agreement. Under the settlement agreement, the Company obtained a license to market diclofenac topical solution 2% no later than January 10, 2029 or earlier in certain circumstances, including the resolution by settlement or court decision of other third party litigation involving diclofenac topical solution 2% or the market entry by other third party generic versions of diclofenac topical solution 2%. At this time, the Company cannot estimate if or when any of those earlier events might occur. No consideration was exchanged as part of the settlement. The Company has not recorded accruals related to this case and has not yet received final approval.
On December 4, 2015, Galderma Laboratories, L.P. and Galderma S.A., which we collectively refer to as Galderma, filed a complaint in the United States District Court for the Northern District of Texas against us alleging infringement of United States Patent No. 6,106,848 based upon our submission to the FDA of an ANDA seeking FDA approval to market clobetasol propionate lotion 0.05% before the expiration patent asserted in the complaint. On January 5, 2016, Galderma and the Company entered into a Settlement and License Agreement, the terms of which are confidential. On January 22, 2016, the case was dismissed with prejudice.
From December 20, 2016 to January 26, 2017, eight putative class action antitrust lawsuits were filed against the Company, along with co-defendants including Taro Pharmaceuticals U.S.A., Inc., Perrigo New York Inc., Fougera Pharmaceuticals Inc., and Sandoz, Inc. The actions are currently pending in the District of New Jersey and Eastern District of Pennsylvania, and a consolidation motion is currently pending before the Judicial Panel on Multidistrict Litigation.
The class plaintiffs seek to represent nationwide or state classes consisting of persons who directly purchased, indirectly purchased or reimbursed patients for the purchase of generic econazole from any of the defendants from June 1, 2014 (or later in some complaints) until the time the defendants’ allegedly unlawful conduct ceased or will cease.
The plaintiffs allege a conspiracy to fix prices for generic econazole, in violation of federal antitrust laws or state antitrust, consumer protection, and other laws. Plaintiffs seek treble damages for alleged price overcharges for generic econazole during the alleged
37
period of conspiracy, and the indirect purchaser class plaintiffs seek injunctive relief against Teligent.
All of these cases are in their initial stages. The parties are negotiating briefing schedules for motions to dismiss, which will be filed after the Judicial Panel on Multidistrict Litigation determines an appropriate forum. Due to the early stage of these cases, we are unable to form a judgment at this time as to whether an unfavorable outcome is either probable or remote or to provide an estimate of the amount or range of potential loss. We intend to vigorously defend against these claims.
Item 4. MINE SAFETY DISCLOSURES
Not applicable.
38
PART II
Item 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
We transferred the listing of our common stock from the NYSE MKT to the NASDAQ Global Select Market. Our common stock ceased trading on the NYSE MKT under the symbol “IG” at the close of business on October 23, 2015 and began trading on the NASDAQ Global Select Market under the symbol “TLGT” on October 26, 2015.
The following table sets forth, for the periods indicated, the high and low sales prices for our common stock, as reported by the NYSE MKT and the NASDAQ Global Select Market, as applicable.
Common Stock | ||||||
High | Low | |||||
2016 | ||||||
First Quarter | 8.88 | 4.46 | ||||
Second Quarter | 7.39 | 4.79 | ||||
Third Quarter | 8.66 | 6.96 | ||||
Fourth Quarter | 7.99 | 5.75 | ||||
2015 | ||||||
First Quarter | 12.05 | 7.75 | ||||
Second Quarter | 9.40 | 4.75 | ||||
Third Quarter | 8.98 | 6.13 | ||||
Fourth Quarter | 9.18 | 5.90 | ||||
Stockholders
As of March 6, 2017, there were approximately 378 stockholders of record of our 53,226,382 outstanding shares of common stock.
Dividends
We have not paid cash dividends to our stockholders since inception and we do not plan to pay cash dividends in the foreseeable future. We currently intend to retain earnings, if any, to finance the growth of the Company.
Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report.
39
Performance Graph
Comparison of Cumulative Total Return among Teligent, Inc.,
the S&P 500 Index and the Dow Jones - US Health Care Index
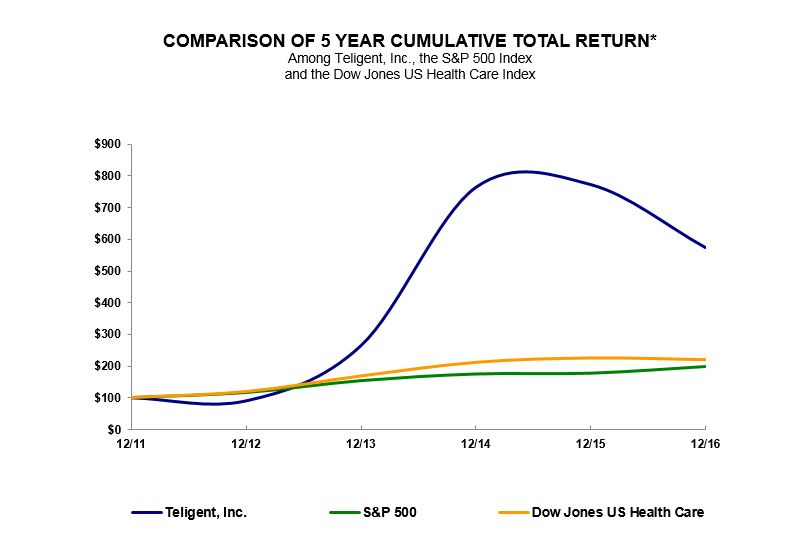
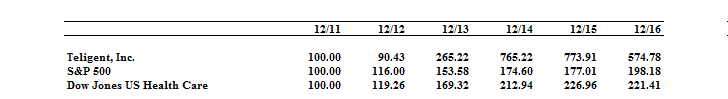
Unregistered Sales of Securities
None.
Issuer Purchases of Equity Securities
None.
40
Item 6. SELECTED FINANCIAL DATA
The following table sets forth consolidated financial data with respect to the Company for each of the five-year periods ended December 31. The selected financial data for each of the five-year periods ended December 31 have been derived from the consolidated financial statements of the Company, which financial statements have been audited by EisnerAmper LLP, independent registered public accounting firm. The foregoing consolidated financial statements and the report thereon are included elsewhere in this Annual Report on Form 10-K. The information below should be read in conjunction with the consolidated financial statements (and notes thereon) and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” included in Item 7.
As of and For the Years Ended December 31, | ||||||||||||||||||||
2016 | 2015 | 2014 | 2013 | 2012 | ||||||||||||||||
(In thousands, except per share data) | ||||||||||||||||||||
Revenues | $ | 66,881 | $ | 44,250 | $ | 33,740 | $ | 18,224 | $ | 8,563 | ||||||||||
Gross profit | 34,687 | 21,315 | 16,972 | 6,145 | 2,776 | |||||||||||||||
Operating income (loss) | 2,542 | (3,192 | ) | 3,906 | (82 | ) | (3,136 | ) | ||||||||||||
Interest and other non-operating income (expense) | (14,240 | ) | 9,895 | 1,518 | (199 | ) | (975 | ) | ||||||||||||
Pretax income (loss) | (11,698 | ) | 6,703 | 5,424 | (281 | ) | (4,111 | ) | ||||||||||||
Income tax provision (benefit) | 287 | 35 | 173 | (197 | ) | (184 | ) | |||||||||||||
Net income (loss) | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | $ | (84 | ) | $ | (3,927 | ) | |||||||
Preferred stock dividend | — | — | — | (1,308 | ) | — | ||||||||||||||
Net income (loss) attributable to common stockholders | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | $ | (1,392 | ) | (3,927 | ) | ||||||||
Weighted average shares outstanding: | ||||||||||||||||||||
Basic | 53,078 | 52,873 | 49,818 | 43,518 | 39,786 | |||||||||||||||
Diluted | 53,078 | 67,112 | 64,207 | 43,518 | 39,786 | |||||||||||||||
PER SHARE: | ||||||||||||||||||||
Net income (loss): | ||||||||||||||||||||
Basic | (0.23 | ) | 0.13 | 0.11 | (0.03 | ) | (0.10 | ) | ||||||||||||
Diluted | (0.23 | ) | (0.07 | ) | 0.09 | (0.03 | ) | (0.10 | ) | |||||||||||
Share Price: High | 8.88 | 12.05 | 11.28 | 3.39 | 1.48 | |||||||||||||||
Low | 4.46 | 4.75 | 2.93 | 1.00 | 0.94 | |||||||||||||||
BALANCE SHEET DATA: | ||||||||||||||||||||
Current assets | $ | 103,296 | $ | 116,801 | $ | 177,218 | $ | 10,558 | $ | 6,139 | ||||||||||
Net property, plant & equipment | 26,215 | 8,706 | 3,262 | 2,623 | 2,691 | |||||||||||||||
Total assets | 183,226 | 184,762 | 197,078 | 15,427 | 9,427 | |||||||||||||||
Current liabilities | 14,963 | 10,768 | 13,002 | 5,221 | 1,976 | |||||||||||||||
Long-term obligations, less current installments | 111,596 | 107,235 | 144,942 | 3,015 | 1,024 | |||||||||||||||
Shareholders’ equity | 56,667 | 66,759 | 39,134 | 7,191 | 6,427 | |||||||||||||||
CASH FLOW DATA: | ||||||||||||||||||||
Cash provided by (used in) operating activities | $ | 1,098 | $ | (15,513 | ) | $ | (3,891 | ) | $ | (618 | ) | $ | (2,373 | ) | ||||||
Cash used in investing activities | (21,972 | ) | (53,068 | ) | (3,792 | ) | (2,113 | ) | (342 | ) | ||||||||||
Cash provided by (used in) financing activities | (10 | ) | (3,111 | ) | 164,465 | 2,296 | 2,337 | |||||||||||||
Increase/(Decrease) in cash and cash equivalents | (20,884 | ) | (71,692 | ) | 156,782 | (435 | ) | (378 | ) | |||||||||||
41
Item 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Forward-Looking Statements
This “Management’s Discussion and Analysis of Financial Condition and Results of Operation” section and other sections of this Annual Report on Form 10-K contain forward-looking statements that are based on current expectations, estimates, forecasts and projections about the industry and markets in which the Company operates and on management’s beliefs and assumptions. In addition, other written or oral statements, which constitute forward-looking statements, may be made by or on behalf of the Company. Words such as “expects,” “anticipates,” “intends,” “plans,” “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements. These statements are not guarantees of future performance, and involve certain risks, uncertainties and assumptions, which are difficult to predict. See “Item 1A: Risk Factors” above. Therefore, actual outcomes and results may differ materially from what is expressed or forecasted in such forward-looking statements. The Company undertakes no obligation to update publicly any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
Company Overview
Strategic Overview
Teligent, Inc. is a specialty generic pharmaceutical company. All references to "Teligent," the "Company," "we," "us," and "our" refer to Teligent, Inc. Our mission is to become a leader in the specialty generic pharmaceutical market. Under our own label, we currently market and sell generic topical and branded generic injectable pharmaceutical products in the United States and Canada. In the United States we are currently marketing 16 generic topical pharmaceutical products and four branded generic pharmaceutical products. Through the completion of an acquisition, we now sell a total of 30 generic and branded generic injectable products and medical devices in Canada. Generic pharmaceutical products are bioequivalent to their brand name counterparts. We also provide development, formulation, and manufacturing services to the pharmaceutical, over-the-counter, or OTC, and cosmetic markets. We operate our business under one segment. Effective October 23, 2015, we changed our name from IGI Laboratories, Inc. to Teligent, Inc. On October 26, 2015, our common stock, which was previously listed on the NYSE MKT, began trading on the NASDAQ Global Select Market under the trading symbol “TLGT.” Our principal executive office, laboratories and manufacturing facilities are located at 105 Lincoln Avenue, Buena, New Jersey. We have additional offices located in Iselin, New Jersey, Toronto, Canada, and Tallinn, Estonia.
Currently, we have two platforms for growth:
• | Developing, manufacturing and marketing a portfolio of generic pharmaceutical products in our own label in topical, injectable, complex and ophthalmic dosage forms; and |
• | Managing our current contract manufacturing and formulation services business. |
We have been in the contract manufacturing and development of topical products business since the early 1990s, but our strategy since 2010 has been focused on the growth of our own generic pharmaceutical business. Since 2010, we have focused on transitioning our business to include more customers in the topical pharmaceutical industry. In 2014, we broadened our target product focus from topical pharmaceuticals to include a wider specialty pharmaceutical approach. We believe that expanding our development and commercial base beyond topical generics, historically the cornerstone of our expertise, to include injectable generics, complex generics and ophthalmic generics (what we call our “TICO strategy”), will leverage our existing expertise and capabilities, and broaden our platform for more diversified strategic growth.
As of the date of this report, we have acquired 25 drug products that have been previously approved by the United States Food and Drug Administration, or FDA. Our pipeline includes 34 Abbreviated New Drug Applications, or ANDAs filed with the FDA, for additional pharmaceutical products. In addition, we have five abbreviated new drug submissions, or ANDSs, on file with Health Canada. We have an additional 34 product candidates at various stages of our development pipeline, ten of which are in stability testing. In December 2015, we announced the approval by the FDA of Cefotan® (Cefotan for Injection). This was our first product approved from the portfolio of discontinued and withdrawn new drug applications, or NDAs, and ANDAs that we purchased from AstraZeneca on September 25, 2014. We have also experienced an increased rate of review by the FDA of applications filed in Generic Drug User Fee Amendments, or GDUFA, Year 3 and Year 4, which began October 1, 2014, and October 1, 2015, respectively. We submitted 12 topical ANDAs in 2016. We expect to continue to expand our presence in the generic topical
42
pharmaceutical market through the filing of additional ANDAs with the FDA and the subsequent launch of products as these applications are approved. We received nine approvals from our internally developed pipeline of topical general products in 2016. We intend to continue to submit further ANDAs to the FDA and ANDSs to Health Canada in 2017. We will also seek to license or acquire further products, intellectual property, or pending applications to expand our portfolio.
Effective October 26, 2015, we transferred the listing of our common stock from the NYSE MKT to the NASDAQ Global Select Market. Our common stock ceased trading on the NYSE MKT under the symbol “IG” at the close of business on October 23, 2015 and began trading on the NASDAQ Global Select Market under the symbol “TLGT” on October 26, 2015.
On November 13, 2015, we acquired all of the rights, title and interest in the development, production, marketing, import and distribution of all pharmaceutical products of Alveda Pharmaceuticals Inc., or Alveda, pursuant to two asset purchase agreements, one relating to the acquisition of all of the intellectual property-related assets of Alveda and the other relating to the acquisition of all other assets of Alveda.
We also develop, manufacture, fill, and package topical semi-solid and liquid products for branded and generic pharmaceutical customers, as well as the OTC and cosmetic markets. These products are used in a wide range of applications from cosmetics and cosmeceuticals to the prescription treatment of conditions like dermatitis, psoriasis, and eczema.
Results of Operations
Fiscal Year 2016 Compared to Fiscal Year 2015
We had a net loss of $12.0 million, or $0.23 per share, in 2016 compared to net income of $6.7 million, or $0.13 per share, in 2015.
Revenues (in thousands):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
Components of Revenue: | 2016 | 2015 | $ | % | |||||||||||
Product sales, net | $ | 65,904 | $ | 43,497 | $ | 22,407 | 52 | % | |||||||
Research and development services and other income | 977 | 753 | 224 | 30 | % | ||||||||||
Total Revenues | $ | 66,881 | $ | 44,250 | $ | 22,631 | 51 | % | |||||||
The increase in product sales for the year ended December 31, 2016 as compared to the same period in 2015 was primarily due to the increased revenue from our own generic pharmaceutical product line and our entry into the specialty generic injectable market in the U.S. and Canada. In addition, our contract manufacturing revenues increased over the same period in the prior year, primarily due to our acquisition of two new customers, one in the fourth quarter of 2015 and one in the first six months of 2016, for which we manufactured one of our generic topical products in a private label, offset by a slight decline in purchase orders. We do not expect revenue from these two contract manufacturing customers in 2017 and beyond.
Research and development services and other income will not be consistent and will vary, from period to period, depending on the required timeline of each development project and/or agreement.
Costs and expenses (in thousands):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
2016 | 2015 | $ | % | ||||||||||||
Cost of revenues | 32,194 | $ | 22,935 | $ | 9,259 | 40 | % | ||||||||
Selling, general and administrative | 15,005 | 11,336 | 3,669 | 32 | % | ||||||||||
Product development and research | 17,140 | 13,171 | 3,969 | 30 | % | ||||||||||
Totals costs and expenditures | $ | 64,339 | $ | 47,442 | $ | 16,897 | 36 | % | |||||||
43
Cost of revenues increased for the year ended December 31, 2016 as compared to the same period in 2015 as a result of the increase in total revenue. Cost of revenues decreased as a percentage of total revenue to 48% for the year ended December 31, 2016 as compared to 52% for the same period in 2015. The decrease in cost of revenue as a percentage of sales was primarily due to the increased revenue from our own generic pharmaceutical product line driven by new product launches and our entry in to the specialty generic injectable market. Sales related to our own label products generally have lower cost of revenues percentages than our contract manufacturing product revenues; however, sales to one new contract manufacturing customer, where we sold more of our generic products in a private label, did reduce cost of revenues as a percentage of sales. In addition, our costs of revenue include the provision for the write-down of inventory of $1.4 million. This write-down includes the write-down of the inventory step up in basis in the amount of $0.5 million. The inventory step-up was initially recorded in connection with our acquisition of Alveda Pharmaceuticals, Inc. in November 2015. Our research and development income results primarily from services rendered under contractual agreements and, therefore, cost of revenues as a percentage of our research and development income is relatively low. Consistent with our strategy, we expect cost of revenues as a percentage of total revenue to decline over time.
Selling, general and administrative expenses for the year ended December 31, 2016 increased by $3.7 million as compared to the same period in 2015. In 2016, there were increases of $2.3 million in amortization expense related to assets acquired in the fourth quarter of 2015, $1.3 million in expenses related to our Canadian operations, $0.8 million in salaries and related costs, $0.5 million in expenses related to our Estonia operations, $0.4 million in recruiting fees, other corporate expenses of $0.3 million, bad debt expense of $0.3 million, $0.1 million from the issuance of stock-based compensation related to options and restricted stock, $0.1 million in conferences and seminars and $0.1 million in board of directors fees, offset by a decrease of $2.5 million in professional fees.
Product development and research expenses for the year ended December 31, 2016 increased by $4.0 million as compared to the same period in 2015. Consistent with our strategy to expand our portfolio of generic prescription pharmaceutical products, we increased headcount, which resulted in an increase of $1.8 million in salaries and related costs, $1.4 million in exhibit and pilot batch costs, $0.7 million in clinical studies, $0.5 million in expenses related to Canadian operations, $0.4 million in stock based compensation related to options and restricted stock and $0.3 million in overhead costs. These were partially offset by decreases in consulting fees of $0.9 million, $0.1 million in fees related to Generic Drug User Fee Act, or GDUFA, and the associated filing of our applications with the FDA and technology license fees of $0.1 million.
Interest and Other Expense, net (in thousands):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
2016 | 2015 | $ | % | ||||||||||||
Interest and other expense, net | $ | (13,304 | ) | $ | (13,358 | ) | $ | 54 | — | % | |||||
Foreign exchange (loss) / gain | $ | (936 | ) | $ | 109 | $ | (1,045 | ) | 100 | % | |||||
Change in the fair value of derivative liability | $ | — | $ | 23,144 | $ | (23,144 | ) | (100 | )% | ||||||
Interest expense increased for the year ended December 31, 2016 as compared to the same period in 2015. The increase is related to the interest expense, amortization of debt discount and amortization of debt issuance costs of the Notes (see Note 6), partially offset by capitalized interest related to our facility expansion. Foreign exchange loss of $0.9 million was recorded for the year ended December 31, 2016, primarily related to the foreign currency translation of our intercompany loans denominated in U.S. dollars to our foreign subsidiaries. These loans are to be repaid in November 2022. Depending on the changes in foreign currency exchange rates, we will continue to record a non-cash gain or loss on translation for the remainder of the term of these loans. Due to the nature of this transaction, there is no economic benefit to the Company to hedge this transaction. During the year ended December 31, 2015, we recorded a $23.1 million change in the fair value of the derivative liability as a result of the change in the fair value of our derivative liability, caused primarily by the decrease in the price of our common stock. Due to the approval of the sufficient shares at the Company's annual shareholder meeting, the liability for the embedded derivative was reclassified to equity on May 20, 2015, and as such there is no change in the fair value of the derivative liability recorded for the year ended December 31, 2016.
Net (loss) income attributable to common stockholders (in thousands, except per share numbers):
44
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
2016 | 2015 | $ | % | ||||||||||||
Net (loss) income attributable to common stockholders | $ | (11,985 | ) | $ | 6,668 | $ | (18,653 | ) | (280 | )% | |||||
Basic (loss) income per share | $ | (0.23 | ) | $ | 0.13 | $ | (0.36 | ) | (277 | )% | |||||
Diluted loss per share | $ | (0.23 | ) | $ | (0.07 | ) | $ | (0.16 | ) | 229 | % | ||||
Net loss for the year ended December 31, 2016 was $12.0 million as compared to net income of $6.7 million for the year ended December 31, 2015. The decrease is due to increases in costs and expenses in 2016, foreign currency exchange loss of $0.9 million, and the absence of change in the fair value of the derivative liability that occurred in 2015 in the amount of $23.1 million, partially offset by increases in revenues in 2016.
Fiscal Year 2015 Compared to Fiscal Year 2014
We had net income of $6.7 million in 2015 compared to net income of $5.3 million in 2014. Net income attributable to common stockholders was $6.7 million, or $0.13 per share in 2015, and net income applicable to common stockholders was $5.3 million, or $0.11 per share, in 2014:
Revenues (in thousands):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
Components of Revenue: | 2015 | 2014 | $ | % | |||||||||||
Product sales, net | $ | 43,497 | $ | 32,104 | $ | 11,393 | 35 | % | |||||||
Research and development services and other income | 753 | 1,636 | (883 | ) | (54 | )% | |||||||||
Total Revenues | $ | 44,250 | $ | 33,740 | $ | 10,510 | 31 | % | |||||||
The increase in product sales for the year ended December 31, 2015 as compared to the same period in 2014 was primarily due to the increased revenue from our own generic pharmaceutical product line that was launched in the first quarter of 2013, the launch of an additional Company label product in July 2015, the purchase of three commercialized injectable products in October 2015 and the launch of two additional Company label products in June 2014. There was a decrease in product sales in our contract services business to three of our pharmaceutical customers and one cosmetic customer, which was only partially offset by increased sales to three of our pharmaceutical customers. Research and development income will not be consistent and will vary, from period to period, depending on the required timeline of each development project. Licensing, royalty and other revenue decreased slightly due to a decrease in other revenue, while licensing and royalty revenue remained the same.
Costs and expenses (in thousands):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
2015 | 2014 | $ | % | ||||||||||||
Cost of revenues | $ | 22,935 | $ | 16,948 | $ | 5,987 | 35 | % | |||||||
Selling, general and administrative | 11,336 | 5,976 | 5,360 | 90 | % | ||||||||||
Product development and research | 13,171 | 6,910 | 6,261 | 91 | % | ||||||||||
Totals costs and expenditures | $ | 47,442 | $ | 29,834 | $ | 17,608 | 59 | % | |||||||
Cost of revenues increased for the year ended December 31, 2015 as compared to the same period in 2014 as a result of the increase in total revenue. Cost of revenues as a percentage of total revenue was 52% for the year ended December 31, 2015 as compared to 50% for 2014. During 2015, approximately 86% of our revenue from contract and formulation services came from pharmaceutical customers as compared to 79% in 2014. Our research and development income results primarily from services rendered under contractual agreements and, therefore, cost of revenues as a percentage of our research and development income is relatively low. Consistent with our strategy, we expect cost of revenues as a percentage of total revenue to decline over time.
45
Selling, general and administrative expenses for the year ended December 31, 2015 increased by $5.4 million as compared to the same period in 2014. During the fourth quarter of 2015, the Company recorded acquisition related costs in the amount of $2.3 million. In addition, in 2015 there were increases of $1.1 million from the issuance of stock-based compensation related to options and restricted stock, other corporate expenses of $0.5 million, professional fees of $0.4 million, amortization of product acquired in 2015 of $0.4 million, expenses related to Canadian operations of $0.2 million, travel-related costs of $0.2 million, website expenses of $0.1 million, overhead costs of $0.1 million, recruiting and human resources expenses of $0.1 million, contributions of $0.1 million and stockholder relations expense of $44,000 during the year ended December 31, 2016 as compared to the same period in 2015. These increases were partially offset by a decrease of $47,000 in salaries and related costs.
Product development and research expenses for the year ended December 31, 2015 increased by $6.3 million as compared to the same period in 2014. Consistent with our strategy to expand our portfolio of generic prescription pharmaceutical products, we increased headcount, including hiring our Chief Scientific Officer in October of 2015, which resulted in an increase of $0.8 million in salaries and related costs; we increased spending on clinical studies by $2.7 million, costs related to our exhibit batches by $1.0 million, contract research by $0.9 million, professional fees by $0.3 million, $0.2 million in expenses related to Canadian operations, stock based compensation related to options and restricted stock of $0.2 million, consulting fees by $0.1 million and overhead costs by $0.1 million. In addition, fees related to GDUFA, and the associated filing of our applications with the FDA, increased by $0.1 million.
Interest and Other Expense, net (in thousands):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
2015 | 2014 | $ | % | ||||||||||||
Interest and other expense, net | $ | (13,358 | ) | $ | (782 | ) | $ | (12,576 | ) | 1,608 | % | ||||
Foreign exchange gain | $ | 109 | $ | — | $ | 109 | 100 | % | |||||||
Change in the fair value of derivative liability | $ | 23,144 | $ | 2,300 | $ | 20,844 | 906 | % | |||||||
Interest expense increased for the year ended December 31, 2015 as compared to the same period in 2014, primarily due to the inclusion in 2015 of approximately $12.8 million of interest expense; amortization of debt discount and amortization of debt issuance costs related to the Convertible 3.75% Senior Notes (see Note 6 to the Company’s Consolidated Financial Statements). Gain on foreign exchange in 2015 resulted from the change in exchange rates applied to funds due from Teligent Canada at December 31, 2015. We also recorded a $23.1 million change in the fair value of the derivative liability as a result in the change in the fair value of our derivative liability, caused primarily by the decrease in the price of our common stock in 2015.
Net income attributable to common stockholders (in thousands, except per share numbers):
Year Ended December 31, | Increase/(Decrease) | ||||||||||||||
2015 | 2014 | $ | % | ||||||||||||
Net income attributable to common stockholders | $ | 6,668 | $ | 5,251 | $ | 1,417 | 27 | % | |||||||
Basic income per share | $ | 0.13 | $ | 0.11 | $ | 0.02 | 18 | % | |||||||
Diluted (loss) income per share | $ | (0.07 | ) | $ | 0.09 | $ | (0.16 | ) | (178 | )% | |||||
Net income for the year ended December 31, 2015 increased as compared to the year ended December 31, 2014 due to the change in the fair value of the derivative liability, partially offset by the increase in revenues and the increase in costs and expenses noted above.
Liquidity and Capital Resources
Our principal sources of liquidity were cash and cash equivalents of approximately $66.0 million at December 31, 2016 and cash from operations. The Company terminated its $10 million credit facility with General Electric Capital Corporation, as agent, and GE Capital Bank and certain other institutions, as lenders, in February 2016. We had working capital of $88.3 million at December 31, 2016. We may require additional funding and this funding will depend, in part, on the timing and structure of potential business arrangements. If necessary, we may continue to seek to raise additional capital through the sale of our equity
46
or through a strategic alliance with a third party. There may also be additional acquisition and growth opportunities that may require external financing. There can be no assurance that such financing will be available on terms acceptable to us, or at all. We believe that our existing capital resources will be sufficient to support our current business plan beyond March 2018.
On December 10, 2014, we entered into a purchase agreement, pursuant to which we agreed to sell our 3.75% Convertible Senior Notes due 2019, or the Notes, to Deutsche Bank Securities Inc. and J.P. Morgan Securities LLC, as the initial purchasers. We received net proceeds of approximately $139 million, after expenses of $4.8 million, upon completion of the transaction. The sale was completed on December 16, 2014. See Note 6.
On June 27, 2014, we announced the pricing of our underwritten public offering of 4,650,000 shares of our common stock at a price to the public of $5.00 per share. The offering closed on July 2, 2014, and, after giving effect to the underwriters’ exercise of the over-allotment option in full, we sold an aggregate of 5,347,500 shares of common stock. The net proceeds of the offering were approximately $24.9 million, after deducting the underwriters’ commission and offering expenses.
Our operating activities provided $1.1 million of cash during the year ended December 31, 2016 compared to $15.5 million and $3.9 million of cash used during the years ended December 31, 2015 and 2014, respectively. The cash provided for the year ended December 31, 2016 was mostly due to the collection of the Canadian goods and services tax, or GST, and the harmonized sales tax, or HST, of $5.2 million, in addition to other changes in operating assets and liabilities, offset by $7.6 million of interest expense related to our Notes. The use of cash for the year ended December 31, 2015 was a result of $5.2 million paid related to Canadian GST and HST and interest expense in the amount of $6.7 million related to our Notes. In connection with the acquisition of Alveda, we paid $2.2 million in acquisition costs. The remaining use of cash was primarily a result of the $3.8 million in changes in operating assets and liabilities, which included a $6.0 million payment related to the AstraZeneca assets acquired in September of 2015, offset by the net income for the year. The use of cash for the year ended December 31, 2014 was substantially a result of the changes in operating assets and liabilities offset by the net income for the year.
Our investing activities used $22.0 million during the year ended December 31, 2016 compared to $53.1 million of cash used in the year ended December 31, 2015 and $3.8 million of cash used in the year ended December 31, 2014. The funds used for the year ended December 31, 2016 included $18.6 million in capital expenditures, for which the majority were for the facility expansion in Buena, as well as expenditures for the Estonian lab, and $3.4 million in product acquisition costs including Sebela and the buyout of the royalty stream related to AstraZeneca (See Note 16). The funds used for the year ended December 31, 2015 included $35.4 million in cash paid to acquire the assets of Alveda in November 2015. We completed the acquisition of five products, which used $11.7 million in cash in 2015. We also used $6.0 million for the purchase of capital expenditures related to additional scientific and manufacturing equipment and costs related to the planning phase of our expansion. The funds used during the year ended December 31, 2014 were for the purchase of products (see Note 7 to the Company’s Condensed Consolidated Financial Statements) and capital expenditures related to additional computer equipment and scientific equipment and improvements incurred to expand our R&D.
Our financing activities used $10,000 of cash during the year ended December 31, 2016 compared to $3.1 million of cash used in financing activities in the year ended December 31, 2015 and $164.5 million of cash provided by financing activities in the year ended December 31, 2014. The cash used during the year ended December 31, 2016 was mainly $70,000 of principal payments on capital lease obligations offset by $96,000 in proceeds from the exercise of common stock warrants and options. The cash used during the year ended December 31, 2015 in the amount of $3.1 million was used to pay down debt. The cash provided for the year ended December 31, 2014 was mainly $139 million net proceeds from the Notes, $24.9 million net proceeds from the public offering of common stock and $0.8 million net proceeds from the exercise of common stock warrants and options.
Off-Balance Sheet Arrangements
We have no significant off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to our shareholders.
Contractual Obligations
As more fully described under Item 2, Properties, we lease a warehouse in Vineland, New Jersey, office space in Iselin, New Jersey, office space in Toronto, Canada and office and laboratory space in Tallinn, Estonia. Our remaining obligations under these leases are summarized in the table below.
As of December 31, 2016, our principal outstanding debt obligation related to our Notes is a total of $143.75 million and are due in December of 2019.
47
Payments Due by Period (in thousands) | ||||||||||||||||||||
Contractual Obligations | Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | |||||||||||||||
Convertible Senior Notes | $ | 143,750 | $ | — | $ | 143,750 | $ | — | $ | — | ||||||||||
Capital Lease | — | — | — | — | — | |||||||||||||||
Operating Lease | 2,987 | 548 | 879 | 818 | 742 | |||||||||||||||
Total | $ | 146,737 | $ | 548 | $ | 144,629 | $ | 818 | $ | 742 | ||||||||||
Critical Accounting Policies and Estimates
Our consolidated financial statements were prepared in accordance with U.S. generally accepted accounting principles, which require us to make subjective decisions, assessments and estimates about the effect of matters that are inherently uncertain. As the number of variables and assumptions affecting the judgment increases, such judgments become even more subjective. While we believe our assumptions are reasonable and appropriate, actual results may be materially different than estimated.
Fair Value of Financial Instruments
The carrying amounts of cash and cash equivalents, trade receivables, restricted cash, notes payable, accounts payable, capital leases and other accrued liabilities at December 31, 2016 approximate their fair value for all periods presented. The Company measures fair value in accordance with ASC 820-10, Fair Value Measurements and Disclosures (formerly SFAS 157, Fair Value Measurements). ASC 820-10 clarifies that fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or a liability. As a basis for considering such assumptions, ASC 820-10 establishes a three-tier value hierarchy, which prioritizes the inputs used in the valuation methodologies in measuring fair value:
Level 1 Inputs: Unadjusted quoted prices in active markets for identical assets or liabilities accessible to the reporting entity at the measurement date.
Level 2 Inputs: Other than quoted prices included in Level 1 inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the asset or liability.
Level 3 Inputs: Unobservable inputs for the asset or liability used to measure fair value to the extent that observable inputs are not available, thereby allowing for situations in which there is little, if any, market activity for the asset or liability at measurement date. The fair value hierarchy also requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
The Company measures its derivative liability at fair value. The derivative convertible option related to the aggregate of $143.75 million in principal notes issued on December 16, 2014, to Deutsche Bank Securities Inc. and J.P. Morgan Securities LLC, as the initial purchasers, or the Notes, was valued using the “with” and “without” analysis. A “with” and “without” analysis is a standard valuation technique for valuing embedded derivatives by first considering the value of the Notes with the option and then considering the value of the Notes without the option. The difference is the fair value of the embedded derivatives. The embedded derivative is classified within Level 3 because it is valued using the “with” and “without” method, which does utilize inputs that are unobservable in the market.
On May 20, 2015, the Company received approval to increase its authorized shares sufficiently to allow for the conversion of the Notes into equity at the annual shareholders meeting. Therefore, the derivative liability of $18.3 million was reclassified into stockholders equity. The Company recorded a change in the fair value of the derivative liability through May 20, 2015 of $23.1 million for the year ended December 31, 2015. On May 20, 2015, the Company reclassified the fair value of the derivative liability into stockholders equity due to the approval of sufficient shares. Based on the closing price of the Company’s common stock as of December 31, 2016, the net carrying value of the Notes was approximately $111.4 million compared to their face value of $143.75 million as of December 31, 2016. However, this variance is due to the conversion feature in the Notes rather than to changes in market interest rates. The Notes carry a fixed interest rate and therefore do not subject the Company to interest rate risk.
48
Allowance for Doubtful Accounts
The Company extends credit to its contract services customers, based upon credit evaluations, in the normal course of business, primarily with 30-day terms. The Company does not require collateral from its customers. Bad debt provisions are provided for on the allowance method based on historical experience and management’s evaluation of outstanding accounts receivable. The Company reviews the allowance for doubtful accounts regularly, and past due balances are reviewed individually for collectability. The Company charges off uncollectible receivables against the allowance when the likelihood of collection is remote.
The Company extends credit to wholesaler and distributor customers and national retail chain customers, based upon credit evaluations, in the normal course of business, primarily with 90-day terms. The Company maintains customer-related accruals and allowances that consist primarily of chargebacks, rebates, sales returns, shelf stock allowances, administrative fees and other incentive programs. Some of these adjustments relate specifically to the generic prescription pharmaceutical business. Typically, the aggregate gross-to-net adjustments related to these customers can exceed 50% of the gross sales through this distribution channel. Certain of these accruals and allowances are recorded in the balance sheet as current liabilities and others are recorded as a reduction to accounts receivable.
Revenue Recognition
The Company considers revenue realized or realizable and earned when it has persuasive evidence of an arrangement, delivery has occurred or contractual services rendered, the sales price is fixed or determinable, and collection is reasonably assured in conformity with ASC 605, Revenue Recognition.
The Company derives its revenues from three basic types of transactions: sales of its own pharmaceutical products, sales of manufactured product for its customers included in product sales, and research and product development services and other services performed for third parties. Due to differences in the substance of these transaction types, the transactions require, and the Company utilizes, different revenue recognition policies for each.
Product Sales: Product Sales, net, include Company Product Sales and Contract Manufacturing Sales.
Company Product Sales: The Company records revenue from Company product sales when title and risk of ownership have been transferred to the customer, which is typically upon delivery of products to the customer.
Revenue and Provision for Sales Returns and Allowances
As is customary in the pharmaceutical industry, the Company’s gross product sales from Company label products are subject to a variety of deductions in arriving at reported net product sales. When the Company recognizes revenue from the sale of products, an estimate of sales returns and allowances, or SRA, is recorded, which reduces product sales. Accounts receivable and/or accrued expenses are also reduced and/or increased by the SRA amount. These adjustments include estimates for chargebacks, rebates, cash discounts and returns and other allowances. These provisions are estimates based on historical payment experience, historical relationship to revenues, estimated customer inventory levels and current contract sales terms with direct and indirect customers. The estimation process used to determine our SRA provision has been applied on a consistent basis and no material adjustments have been necessary to increase or decrease our reserves for SRA as a result of a significant change in underlying estimates. The Company will use a variety of methods to assess the adequacy of our SRA reserves to ensure that our financial statements are fairly stated. These will include periodic reviews of customer inventory data, customer contract programs, subsequent actual payment experience and product pricing trends to analyze and validate the SRA reserves.
The provision for chargebacks is our most significant sales allowance. A chargeback represents an amount payable in the future to a wholesaler for the difference between the invoice price paid to the Company by our wholesale customer for a particular product and the negotiated contract price that the wholesaler’s customer pays for that product. The Company’s chargeback provision and related reserve varies with changes in product mix, changes in customer pricing and changes to estimated wholesaler inventories. The provision for chargebacks also takes into account an estimate of the expected wholesaler sell-through levels to indirect customers at contract prices. The Company will validate the chargeback accrual quarterly through a review of the inventory reports obtained from our largest wholesale customers. This customer inventory information is used to verify the estimated liability for future chargeback claims based on historical chargeback and contract rates. These large wholesalers represent 90% - 95% of the Company’s chargeback payments. The Company continually monitors current pricing trends and wholesaler inventory levels to ensure the liability for future chargebacks is fairly stated.
49
Net revenues and accounts receivable balances in the Company’s consolidated financial statements are presented net of SRA estimates. Certain SRA balances are included in accounts payable and accrued expenses.
Contract Manufacturing Sales: The Company recognizes revenue when title transfers to its customers, which is generally upon shipment of products. These shipments are made in accordance with sales commitments and related sales orders entered into with customers either verbally or in written form. The revenues associated with these transactions, net of appropriate cash discounts, product returns and sales reserves, are recorded upon shipment of the products included in product sales, net in the Company's Condensed Consolidated Statement of Operations.
Research and Development Services and Other Income: The Company establishes agreed upon product development agreements with its customers to perform product development services. Product development revenues are recognized in accordance with the product development agreement upon the completion of the phases of development and when the Company has no future performance obligations relating to that phase of development. Revenue recognition requires the Company to assess progress against contracted obligations to assure completion of each stage. These payments are generally non-refundable and are reported as deferred until they are recognizable as revenue. If no such arrangement exists, product development fees are recognized ratably over the entire period during which the services are performed. Other types of revenue include royalty or licensing revenue, and would be recognized based upon the contractual agreement upon completion of the earnings process.
In making such assessments, judgments are required to evaluate contingencies such as potential variances in schedule and the costs, the impact of change orders, liability claims, contract disputes and achievement of contractual performance standards. Changes in total estimated contract cost and losses, if any, are recognized in the period they are determined. Billings on research and development contracts are typically based upon terms agreed upon by the Company and customer and are stated in the contracts themselves and do not always align with the revenues recognized by the Company.
Derivatives
The Company accounts for its derivative instruments in accordance with ASC 815-10, Derivatives and Hedging, or ASC 815-10. ASC 815-10 establishes accounting and reporting standards requiring that derivative instruments, including derivative instruments embedded in other contracts, be recorded on the balance sheet as either an asset or liability measured at its fair value. ASC 815-10 also requires that changes in the fair value of derivative instruments be recognized currently in results of operations unless specific hedge accounting criteria are met. The Company has not entered into hedging activities to date. The Company's derivative liability was the embedded convertible option of its Notes issued December 16, 2014 (see Note 6), which has been recorded as a liability at fair value until May 20, 2015, and was revalued at each reporting date, with changes in the fair value of the instruments included in the consolidated statements of operations as non-operating income (expense). Due to the approval of the sufficient shares at the Company’s annual shareholder meeting, the liability for the embedded derivative was reclassified to equity on May 20, 2015. The Company has no derivatives at December 31, 2016 and December 31, 2015.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include derivatives, SRA allowances, allowances for excess and obsolete inventories, allowances for doubtful accounts, provisions for income taxes and related deferred tax asset valuation allowances, stock based compensation and accruals for environmental cleanup and remediation costs. Actual results could differ from those estimates.
Recent Accounting Pronouncements
In December 2016, the FASB issued ASU 2016-20, Technical Corrections and Improvements to Topic 606, Revenue from Contracts with Customers". The update provides users with classification guidance on thirteen specific areas of correction or improvement topics as follows: 1) Loan Guarantee Fees, 2) Contract Costs - Impairment Testing, 3) Contract Costs - Interaction of Impairment Testing with Guidance in Other Topics, 4) Provisions for Losses on Construction-Type and Production-Type Contracts, 5) Scope of Topic 606, 6) Disclosure of Remaining Performance Obligations, 7) Disclosure of Prior-Period Performance Obligations, 8) Contract Modifications Example, 9) Contract Asset versus Receivable, 10) Refund Liability, 11) Advertising Costs, 12) Fixed-Odds Wagering Contracts in the Casino Industry and 13) Cost Capitalization for Advisors to Private Funds and Public Funds. The amendments affect the guidance in update 2014-09, which is effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years.
50
For us, the amendments are effective January 1, 2018. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In December 2016, the FASB issued ASU 2016-19, Technical Corrections and Improvements. The update is to address suggestions received from stakeholders on the Accounting Standards Codification and to make other incremental improvements to GAAP. It contains amendments that affect a wide variety of Topics in the Accounting Standards Codification and applies to all reporting entities within the scope of the affected accounting guidance. Most of the amendments are effective upon issuance of the update. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In November 2016, the FASB issued ASU 2016-18, Statement of Cash Flows (Topic 230): "Restricted Cash (a consensus of the FASB Emerging Issues Task Force)". The update addresses the diversity in the industry with respect to classification and presentation of changes in restricted cash on the statement of cash flows. These amendments require that a statement of cash flows explain the restricted cash change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. It affects those reporting entities that are required to evaluate whether they should consolidate a VIE. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For us, the amendments are effective January 1, 2018. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In October 2016, the FASB issued ASU 2016-17, Consolidation (Topic 810): "Interests Held through Related Parties That Are Under Common Control". The update was issued to amend the consolidation guidance on how a reporting entity that is a single decision maker of a variable interest entity ("VIE") should treat indirect interests in the entity held through related parties that are under common control with the reporting entity when determining whether it is the primary beneficiary of that VIE. It affects those reporting entities that are required to evaluate whether they should consolidate a VIE. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For us, the amendments are effective January 1, 2018. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In October 2016, the FASB issued ASU 2016-16, Income Taxes (Topic 740): "Intra-Entity Transfers of Assets Other Than Inventory". The update addresses income tax consequences of intra-entity transfers of assets other than inventory. It seeks to clarify the authoritative guidance about the prohibition of the recognition of current and deferred income taxes for intra-entity asset transfers until the asset has been sold to an outside party. Instead, this update now eliminates that prohibition and states that an entity should recognize the income tax consequences when the transfer occurs. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For us, the amendments are effective January 1, 2018. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230): “Classification of Certain Cash Receipts and Cash Payments (a Consensus of the Emerging Issues Task Force)”. The update provides users with classification guidance on eight specific cash flow topics as follows: 1) Debt Prepayment or Debt Extinguishment Costs, 2) Settlement of Zero-Coupon Debt Instruments or Other Debt Instruments with Coupon Interest Rates That Are Insignificant in Relation to the Effective Interest Rate of the Borrowing, 3) Contingent Consideration Payments Made after a Business Combination, 4) Proceeds from the Settlement of Insurance Claims, 5) Proceeds from the Settlement of Corporate-Owned Life Insurance Policies, including Bank-Owned Life Insurance Policies, 6) Distributions Received from Equity Method Investees, 7) Beneficial Interests in Securitization Transactions and 8) Separately Identifiable Cash Flows and Application of the Predominance Principle. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For us, the amendments are effective January 1, 2018. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In June 2016, the FASB issued ASU 2016-13, Financial Instruments - Credit Losses (Topic 326): “Measurement of Credit Losses on Financial Instruments”. The update provides users with more useful information for decision making regarding expected credit losses on financial instruments/commitments to extend credit held by a reporting entity at each reporting date. The amendments affect loans, debt securities, trade receivables, net investments in leases, off-balance-sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. Credit quality of the entity’s assets now plays a key role in this update. The amendments in this update are effective for fiscal years beginning after December 15, 2019 for public business entities, including interim periods within those fiscal years. For us, the amendments are effective January 1, 2020. We are currently evaluating the impact of this ASU on our consolidated financial statements.
51
In May 2016, the FASB issued ASU 2016-12, Revenue from Contracts with Customers (Topic 606): “Narrow-Scope Improvements and Practical Expedients”. The update addresses issues identified by the FASB-IASB Joint Transition Resource Group (TRG), a group formed in June, 2014 in order to inform the Boards about potential implementation issues that could arise as a result of organizations implementing the May, 2014 revenue guidance. It affects entities that enter into contracts with customers to transfer goods or services within an entity’s ordinary activities in exchange for consideration. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In May 2016, the FASB issued ASU 2016-11, Revenue Recognition (Topic 605) and Derivatives and Hedging (Topic 815): “Rescission of SEC Guidance Because of Accounting Standards Updates 2014-09 and 2014-16 Pursuant to Staff Announcements at the March 3, 2016 EITF Meeting (SEC Update)”. The update is a result of adoption of Topic 606, Revenue from Contracts with Customers. SEC Staff Observer comments found in Topic 605 are therefore not recommended to be relied upon and have been superseded. The comments are found in the following topics: 1) Revenue and Expense Recognition for Freight Services in Process, 2) Accounting for Shipping and Handling Fees and Costs, 3) Accounting for Consideration Given by a Vendor to a Customer (including Reseller of the Vendor’s Products), and 4) Accounting for Gas-Balancing Arrangements. As these amendments require changes to the U.S. GAAP Financial Reporting Taxonomy, they will be incorporated into the proposed 2017 Taxonomy and finalized as part of the annual release process. We are currently evaluating the impact of this ASU on our consolidated financial statements.
In March 2016, the FASB issued ASU No. 2016-09, Compensation-Stock Compensation (Topic 718): “Improvements to Employee Share-Based Payment Accounting”. The update includes multiple provisions intended to simplify various aspects of the accounting for share-based payments, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. The amendments in this update are effective for public companies for annual periods beginning after December 15, 2016, and interim periods within those annual periods. Early adoption is permitted for any interim or annual period. We have evaluated the impact of this ASU on our consolidated financial statements and as a result, will adjust retained earnings in 2017 for the amounts previously recognized as windfall tax benefits in additional paid in capital.
In February 2016, the FASB issued Accounting Standards Update (“ASU”) 2016-02, Leases (Topic 842): “Recognition and Measurement of Financial Assets and Financial Liabilities”. The update supersedes Topic 840, Leases and requires the recognition of lease assets and lease liabilities by lessees for those leases classified as operating leases under previous GAAP. Topic 842 retains a distinction between finance leases and operating leases, with cash payments from operating leases classified within operating activities in the statement of cash flows. The amendments in this update are effective for fiscal years beginning after December 15, 2018 for public business entities, which for us means January 1, 2019. We are currently evaluating the impact of this ASU on our consolidated financial statements
In September 2015, the FASB issued ASU 2015-16, Business Combinations (Topic 805): “Simplifying the Accounting for Measurement-Period Adjustments”. The update eliminates the requirement to retrospectively adjust the provisional amounts recognized at the acquisition date with a corresponding adjustment to goodwill during the measurement period when new information is obtained about the facts and circumstances that existed as of the acquisition date, that if known, would have affected the measurement of the amounts initially recognized or would have resulted in the recognition of additional assets or liabilities. The amendments in this update are effective for fiscal years beginning after December 15, 2015, which for the Company means January 1, 2016, and should be applied prospectively to adjustments to provisional amounts that occur after the effective date of this update. Early application is permitted for financial statements that have not been issued. We have concluded that the adoption of this ASU will not have any significant impact on our consolidated financial statements.
In July 2015, the FASB issued ASU 2015-11, Inventory (Topic 330): “Simplifying the Measurement of Inventory”. ASU 2015-11 requires inventory measured using any method other than last-in, first out (“LIFO”) or the retail inventory method to be subsequently measured at the lower of cost or net realizable value, rather than at the lower of cost or market. Under this ASU, subsequent measurement of inventory using the LIFO and retail inventory method is unchanged. ASU 2015-11 is effective prospectively for fiscal years, and for interim periods within those years, beginning after December 15, 2016. Early application is permitted. We do not expect the adoption of this ASU will have any significant impact on its consolidated financial statements.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As of December 31, 2016, our principal debt obligation was related to our Notes. Interest accrues at a fixed rate of 3.75% on the outstanding principal amount of the Notes and is paid semi-annually every June 15 and December 15 until the Notes mature on December 15, 2019. Since the interest rate is fixed, we have no market risk related to the Notes.
52
We had a revolving Credit and Security Agreement with General Electric Capital Corporation that called for interest to accrue based on a premium above either the current prime rate or current LIBOR rates. We terminated this credit facility in February 2016.
Our financial instruments include cash and cash equivalents, accounts receivable, accounts payable and the Notes. The fair values of cash and cash equivalents, accounts receivable and accounts payable approximate book value because of the short maturity of these instruments. Based on the closing price of our common stock as of December 31, 2016, the fair value of our Notes was approximately $111.4 million compared to their face value of $143.75 million as of December 31, 2016. However, this variance is due to the conversion feature in the Notes rather than to changes in market interest rates. As noted above, the Notes carry a fixed interest rate and therefore do not subject us to interest rate risk. As a result in the change in fair value, we recorded a $23.1 million change in the fair value of the derivative liability on our consolidated statements of operations in 2015.
At December 31, 2016, the bulk of our cash and cash equivalents was invested in overnight instruments, the interest rates of which may change daily. Accordingly, these overnight investments are subject to market risk.
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
See Index to Financial Statements on page F-1.
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON AUDITING AND FINANCIAL DISCLOSURE
None.
Item 9a. CONTROLS AND PROCEDURES
(a) Evaluation of Disclosure Controls and Procedures. Our principal executive officer and principal financial officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) as of the end of the period covered by this Annual Report on Form 10-K, have concluded that, based on such evaluation, our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and is accumulated and communicated to our management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
(b) Changes in Internal Controls. There were no changes in our internal control over financial reporting identified in connection with the evaluation of such internal control that occurred during the fourth quarter of our last fiscal year that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. The Company’s internal control over financial reporting includes those policies and procedures that:
• | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; |
• | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and |
• | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
53
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2016. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control-Integrated Framework (2013).
Based on our assessment, management believes that, as of December 31, 2016, the Company’s internal control over financial reporting is effective based on those criteria.
EisnerAmper LLP, the independent registered public accounting firm that audited the consolidated financial statements in this Annual Report on Form 10-K for the year ended December 31, 2016, has issued a report concerning the effectiveness of our internal control over financial reporting for that year, which is included in Part II, Item 8 of this Annual Report on Form 10-K.
Item 9B. OTHER INFORMATION
In the interest of maintaining consistency with the Company's 2016 Equity Incentive Plan, on March 13, 2017, the Company entered into (i) an amendment to the option agreements governing each option grant currently outstanding under the Company's 2009 Equity Incentive Plan, and (ii) an amendment to the restricted stock unit, or RSU, agreements governing each RSU grant currently outstanding under the 2009 Plan. The amendments provide for the automatic vesting upon a change of control of the Company of each option grant and RSU grant, as applicable, outstanding under the 2009 Plan. The forms of amendment are attached hereto as Exhibits 10.31 and 10.32, respectively, and are incorporated by reference herein.
PART III
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Management and Corporate Governance Matters,” “Section 16(a) Beneficial Ownership Reporting Compliance,” and “Code of Conduct and Ethics” in the Company’s Proxy Statement for the 2017 Annual Meeting of Stockholders.
Item 11. EXECUTIVE COMPENSATION
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Executive Officer and Director Compensation,” “Compensation Discussion and Analysis,” “Management and Corporate Governance Matters,” “Compensation Committee Report” and “Compensation Discussion and Analysis” in the Company’s Proxy Statement for the 2017 Annual Meeting of Stockholders.
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in the Company’s Proxy Statement for the 2017 Annual Meeting of Stockholders.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Certain Relationships and Related Person Transactions” and “Management and Corporate Governance” in the Company’s Proxy Statement for the 2017 Annual Meeting of Stockholders.
Item 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The response to this item is incorporated by reference from the discussion responsive thereto under the caption “Independent Registered Public Accounting Firm” in the Company’s Proxy Statement for the 2017 Annual Meeting of Stockholders.
PART IV
Item 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
54
(a) | The following documents are filed as part of this Annual Report on Form 10-K: |
(a)(1) | See “Index to Consolidated Financial Statements and Financial Statement Schedules” at Item 8 to this Annual Report on Form 10-K. |
(a)(2) | Other financial statement schedules have not been included because they are not applicable or the information is included in the financial statements or notes thereto. |
(a)(3) | The following is a list of exhibits filed as part of this Annual Report on Form 10-K. |
Exhibits | |
(3.1) | Amended and Restated Certificate of Incorporation of Teligent, Inc., dated October 23, 2015 (incorporated by reference to Exhibit 3.1 to the Company’s Report on Form 8-K, filed October 23, 2015). |
(3.2) | Amended and Restated Bylaws of IGI Laboratories, Inc., effective May 7, 2008 (incorporated by reference to Exhibit 3.2 to the Company’s Report on Form 8-K, filed May 12, 2008). |
(4.1) | Specimen stock certificate for shares of Common Stock, par value $.01 per share (incorporated by reference to Exhibit 4 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2000, filed March 28, 2001 (“the 2000 Form 10-K”)). |
(4.2) | Indenture dated as of December 16, 2014, by and between IGI Laboratories, Inc. and Wilmington Trust, National Association (incorporated by reference to Exhibit 4.1 to the Company’s Report on Form 8-K, filed December 17, 2014). |
(10.1)# | IGI, Inc. 1998 Directors Stock Plan, as amended (incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form S-8 (Registration No. 333-160342), filed June 30, 2009). |
(10.2)# | IGI, Inc. 1999 Director Stock Option Plan, as amended (incorporated by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-8 (Registration No. 333-160342, filed June 30, 2009). |
(10.3)# | IGI, Inc. 1999 Stock Incentive Plan, as amended (incorporated by reference to Exhibit 4.3 to the Company’s Registration Statement on Form S-8 (Registration No. 333-160342), filed June 30, 2009). |
(10.4)# | IGI Laboratories, Inc. 2009 Equity Incentive Plan, as amended and restated (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed June 4, 2014). |
(10.5)# | Form of Non-Qualified Stock Option Agreement under the IGI Laboratories, Inc. 2009 Equity Incentive Plan (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed July 2, 2009). |
(10.6)# | Form of Stock Option Award Agreement under the IGI Laboratories, Inc. 2009 Equity Incentive Plan (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed July 20, 2011). |
(10.7)# | Form of Award Agreement for Restricted Shares under the IGI Laboratories, Inc. 2009 Equity Incentive Plan (incorporated by reference to Exhibit 10.3 to the Company’s Report on Form 8-K, filed July 2, 2009). |
(10.8)# | Form of Indemnification Agreement for Certain Directors (incorporated by reference to Exhibit 10.11 to the March 2009 8-K). |
55
(10.9)# | Employment Agreement dated July 14, 2011 between IGI Laboratories, Inc. and Jenniffer Collins (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed July 20, 2011). |
(10.10)# | Employment Agreement dated July 30, 2012 between IGI Laboratories, Inc. and Jason Grenfell-Gardner (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed July 30, 2012). |
(10.11)+ | Purchase and Sale Agreement between the Company and Prasco, LLC for the purchase of econazole nitrate cream 1%, dated February 1, 2013, (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed August 9, 2013). |
(10.12) | Asset Purchase Agreement dated as of September 30, 2014, by and between IGI Laboratories, Inc. and Valeant Pharmaceuticals North America, LLC and Valeant Pharmaceuticals Luxembourg SARL (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed October 1, 2014). |
(10.13) | Asset Purchase Agreement dated as of September 30, 2014, by and between IGI Laboratories, Inc. and Valeant Pharmaceuticals North America, LLC and Valeant Pharmaceuticals Luxembourg SARL (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed October 1, 2014). |
(10.14)+ | Asset Purchase Agreement dated as of September 24, 2014, by and between IGI Laboratories, Inc. and AstraZeneca Pharmaceuticals LP (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 10-Q, filed November 13, 2014). |
(10.15) | Credit Agreement dated as of November 18, 2014, by and among IGI Laboratories, Inc., Igen, Inc., and IGI Labs, Inc. as Borrowers, the other Persons party thereto that are designated as Credit Parties, General Electric Capital Corporation as Agent for all Lenders, GE Capital Bank as a Lender, and the other financial institutions party thereto as Lenders (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed November 24, 2014). |
(10.16) | Guaranty and Security Agreement dated as of November 18, 2014, by and among IGI Laboratories, Inc., Igen, Inc., and IGI Labs, Inc. as Borrowers and each other Grantor from time to time party thereto in favor of General Electric Capital Corporation as Agent (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed November 24, 2014). |
(10.17) | Purchase Agreement dated December 10, 2014, by and between IGI Laboratories, Inc. and the initial purchasers set forth on Schedule 1 thereto (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed December 17, 2014). |
(10.18) | Second Amendment to Credit Agreement, dated as of August 14, 2015, by and among Teligent, Inc., Igen, Inc. and Teligent Pharma, Inc. as Borrowers, General Electric Capital Corporation as Agent, and the Lenders signatory thereto (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 10-Q, filed November 9, 2015). |
(10.19) | Third Amendment to Credit Agreement, dated as of September 16, 2015, by and among Teligent, Inc., Igen, Inc. and Teligent Pharma, Inc. as Borrowers, General Electric Capital Corporation as Agent, and the Lenders signatory thereto (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 10-Q, filed November 9, 2015). |
(10.20)+ | Asset Purchase Agreement, dated as of October 5, 2015, by between Concordia Pharmaceuticals Inc., S.à.r.l., Barbados Branch, on the one hand, and Teligent, Inc. and Teligent Jersey Limited, on the other hand (incorporated by reference to Exhibit 10.3 to the Company’s Report on Form 10-Q, filed November 9, 2015). |
(10.21) | Asset Purchase Agreement, dated October 12, 2015, between IGI Laboratories, Inc. and Alveda Pharmaceuticals, Inc. (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed October 13, 2015). |
(10.22) | Asset Purchase Agreement, dated October 12, 2015, between IGI Laboratories, Inc. and Alveda Pharmaceuticals, Inc. (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed October 13, 2015). |
56
(10.23) | Contribution Agreement, by and between the Teligent, Inc. and Teligent Luxembourg S.à.r.l., dated as of November 13, 2015 (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed November 16, 2015). |
(10.24) | Loan Agreement, by and between Teligent, Inc. and Teligent Luxembourg S.à.r.l., dated as of November 13, 2015 (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed November 16, 2015). |
(10.25) | Loan Agreement, by and between Teligent, Inc. and Teligent Canada Inc., dated as of November 13, 2015 (incorporated by reference to Exhibit 10.3 to the Company’s Report on Form 8-K, filed November 16, 2015). |
(10.26) | Distribution Agreement, by and between Teligent OÜ and Teligent Canada Inc., dated as of November 13, 2015 (incorporated by reference to Exhibit 10.4 to the Company’s Report on Form 8-K, filed November 16, 2015). |
(10.27) | First Amendment to Asset Purchase Agreement, by and between Teligent, Inc. and AstraZeneca Pharmaceuticals, LP, dated as of November 30, 2015 (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed December 4, 2015). |
(10.28) | First Amendment to Asset Purchase Agreement, dated December 10, 2015, by and between Concordia Pharmaceuticals Inc., S.à.r.l., Barbados Branch, on the one hand, and Teligent, Inc. and Teligent Jersey Limited, on the other hand (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed December 15, 2015). |
(10.29) | Trademark Assignment Agreement, dated December 10, 2015, by and between Concordia Pharmaceuticals Inc., S.à.r.l., Barbados Branch, on the one hand, and Teligent Jersey Limited, on the other hand (incorporated by reference to Exhibit 10.2 to the Company’s Report on Form 8-K, filed December 15, 2015). |
(10.30)# | Teligent, Inc. 2016 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 8-K, filed May 27, 2016). |
(10.31)#* | Form of Amendment to Outstanding Option Agreements under the Company’s 2009 Equity Incentive Plan. |
(10.32)#* | Form of Amendment to Outstanding RSU Agreements under the Company’s 2009 Equity Incentive Plan. |
(21) | List of Subsidiaries (incorporated by reference to Exhibit 10.1 to the Company’s Report on Form 10-K, filed March 15, 2016). |
(23.1)* | Consent of EisnerAmper LLP. |
(31.1)* | Certification of the President and Chief Executive Officer Pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
(31.2)* | Certification of the Chief Financial Officer Pursuant to Rule 13a-14(a) under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
(32.1)* | Certification of the President and Chief Executive Officer and of the Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
(101)* | The following financial information from this Annual Report on Form 10-K for the year ended December 31, 2016, formatted in XBRL (Extensible Business Reporting Language) and furnished electronically herewith: (i) the Consolidated Statements of Operations; (ii) the Consolidated Balance Sheets; (iii) the Consolidated Statements of Cash Flows; and (iv) the Notes to Consolidated Financial Statements, tagged as blocks of text. |
*Filed herewith.
57
#Indicates management contract or compensatory plan.
+Portions of this Exhibit were omitted and filed separately with the Secretary of the SEC pursuant to a request for confidential treatment that has been granted by the SEC.
58
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Teligent, Inc. | ||
By: | /s/ Jason Grenfell-Gardner | |
Jason Grenfell-Gardner | ||
President and Chief Executive Officer | ||
Date: March 15, 2017
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated below and on the dates indicated.
Signature | Title | Date | ||
/s/ Jason Grenfell-Gardner | Director, President and Chief Executive Officer | March 15, 2017 | ||
Jason Grenfell-Gardner | (Principal Executive Officer) | |||
/s/ Jenniffer Collins | Chief Financial Officer | March 15, 2017 | ||
Jenniffer Collins | (Principal Financial Officer) | |||
/s/ Steven Koehler | Director | March 15, 2017 | ||
Steven Koehler | ||||
/s/ James Gale | Director | March 15, 2017 | ||
James Gale | ||||
/s/ Narendra Borkar | Director | March 15, 2017 | ||
Narendra Borkar | ||||
/s/ Bhaskar Chaudhuri | Director | March 15, 2017 | ||
Bhaskar Chaudhuri | ||||
/s/ John Celentano | Director | March 15, 2017 | ||
John Celentano | ||||
/s/ Carole Ben-Maimon | Director | March 15, 2017 | ||
Carole Ben-Maimon | ||||
59
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Teligent, Inc. and subsidiaries (formerly known as IGI Laboratories, Inc.)
We have audited the accompanying consolidated balance sheets of Teligent, Inc. and subsidiaries (the "Company") as of December 31, 2016 and 2015, and the related consolidated statements of operations, comprehensive income (loss), stockholders' equity, and cash flows for each of the years in the three-year period ended December 31, 2016. In connection with our audits of the consolidated financial statements, we have also audited financial statement schedule "Schedule II - Valuation and Qualifying Accounts" for each of the years in the three-year period ended December 31, 2016. The financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Teligent, Inc. and Subsidiaries as of December 31, 2016 and 2015, and the consolidated results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2016 in conformity with accounting principles generally accepted in the United States of America. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly, in all material respects, the information stated therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Teligent, Inc. and Subsidiaries' internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"), and our report dated March 15, 2017 expressed an unqualified opinion thereon.
/s/ EISNERAMPER LLP
Iselin, New Jersey
March 15, 2017
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Teligent, Inc. and subsidiaries (formerly known as IGI Laboratories, Inc.)
We have audited Teligent, Inc. and Subsidiaries' (the "Company") internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
An entity’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. An entity’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the entity; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the entity are being made only in accordance with authorizations of management and directors of the entity; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the entity’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Teligent, Inc. and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on criteria established in the 2013 Internal Control - Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Teligent, Inc. and Subsidiaries as of December 31, 2016 and 2015, and the related consolidated statements of operations, comprehensive income, stockholders' equity, and cash flows for each of the years in the three-year period ended December 31, 2016, and our report dated, March 15, 2017, expressed an unqualified opinion thereon.
/s/ EISNERAMPER LLP
Iselin, New Jersey
March 15, 2017
F-3
TELIGENT, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share information)
December 31, 2016 | December 31, 2015 | |||||||
ASSETS | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 66,006 | $ | 87,191 | ||||
Accounts receivable, net | 21,735 | 14,028 | ||||||
Inventories | 12,708 | 8,985 | ||||||
Prepaid expenses and other receivables | 2,847 | 6,597 | ||||||
Total current assets | 103,296 | 116,801 | ||||||
Property, plant and equipment, net | 26,215 | 8,706 | ||||||
Intangible assets, net | 52,465 | 54,320 | ||||||
Goodwill | 446 | 426 | ||||||
Other | 804 | 482 | ||||||
Total assets | $ | 183,226 | $ | 180,735 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 4,614 | $ | 3,955 | ||||
Accrued expenses | 10,349 | 6,267 | ||||||
Deferred income, current | — | 476 | ||||||
Capital lease obligation, current | — | 70 | ||||||
Total current liabilities | 14,963 | 10,768 | ||||||
Convertible 3.75% senior notes, net of debt discount and debt issuance costs (face of $143,750) | 111,391 | 102,964 | ||||||
Deferred tax liability | 205 | 244 | ||||||
Total liabilities | 126,559 | 113,976 | ||||||
Stockholders’ equity: | ||||||||
Series A Convertible Preferred stock, $0.01 par value, 100 shares authorized; 0 shares issued and outstanding as of December 31, 2016 and 2015, respectively | — | — | ||||||
Series C Convertible Preferred stock, $0.01 par value, 1,550 shares authorized; 0 shares issued and outstanding as of December 31, 2016 and 2015, respectively | — | — | ||||||
Common stock, $0.01 par value, 100,000,000 shares authorized; 53,148,441 and 53,000,689 shares issued and outstanding as of December 31, 2016 and December 31, 2015, respectively | 551 | 549 | ||||||
Additional paid-in capital | 102,624 | 99,258 | ||||||
Accumulated deficit | (44,903 | ) | (32,918 | ) | ||||
Accumulated other comprehensive loss, net of taxes | (1,605 | ) | (130 | ) | ||||
Total stockholders’ equity | 56,667 | 66,759 | ||||||
Total liabilities and stockholders’ equity | $ | 183,226 | $ | 180,735 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
F-4
TELIGENT, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
For the years ended December 31, 2016, 2015 and 2014
(in thousands, except shares and per share information)
2016 | 2015 | 2014 | ||||||||||
Revenues: | ||||||||||||
Product sales, net | $ | 65,904 | $ | 43,497 | $ | 32,104 | ||||||
Research and development services and other income | 977 | 753 | 1,636 | |||||||||
Total revenues | 66,881 | 44,250 | 33,740 | |||||||||
Costs and Expenses: | ||||||||||||
Cost of revenues | 32,194 | 22,935 | 16,948 | |||||||||
Selling, general and administrative expenses | 15,005 | 11,336 | 5,976 | |||||||||
Product development and research expenses | 17,140 | 13,171 | 6,910 | |||||||||
Total costs and expenses | 64,339 | 47,442 | 29,834 | |||||||||
Operating income (loss) | 2,542 | (3,192 | ) | 3,906 | ||||||||
Other Income (Expense): | ||||||||||||
Change in the fair value of derivative liability | — | 23,144 | 2,300 | |||||||||
Foreign currency exchange gain (loss) | (936 | ) | 109 | — | ||||||||
Interest and other expense, net | (13,304 | ) | (13,358 | ) | (782 | ) | ||||||
Income (loss) before income tax expense (benefit) | (11,698 | ) | 6,703 | 5,424 | ||||||||
Income tax expense | 287 | 35 | 173 | |||||||||
Net income (loss) income attributable to common stockholders | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | |||||
Basic earnings (loss) per share | $ | (0.23 | ) | $ | 0.13 | $ | 0.11 | |||||
Diluted earnings (loss) per share | $ | (0.23 | ) | $ | (0.07 | ) | $ | 0.09 | ||||
Weighted average shares of common stock outstanding: | ||||||||||||
Basic | 53,078,158 | 52,872,814 | 49,817,721 | |||||||||
Diluted | 53,078,158 | 67,111,995 | 64,207,190 | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-5
TELIGENT, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
For the years ended December 31, 2016, 2015 and 2014
(in thousands)
2016 | 2015 | 2014 | ||||||||||
Net income (loss) | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | |||||
Other comprehensive loss, net of tax | ||||||||||||
Foreign currency translation adjustment | (1,475 | ) | (130 | ) | — | |||||||
Other comprehensive loss | (1,475 | ) | (130 | ) | — | |||||||
Comprehensive income (loss) | $ | (13,460 | ) | $ | 6,538 | $ | 5,251 | |||||
The accompanying notes are an integral part of the consolidated financial statements.
F-6
TELIGENT, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended December 31, 2016, 2015 and 2014
2016 | 2015 | 2014 | ||||||||||
Cash flows from operating activities: | ||||||||||||
Net income (loss) | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | |||||
Reconciliation of net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||
Depreciation and amortization of fixed assets | 946 | 560 | 415 | |||||||||
Amortization of license fee | — | 100 | 100 | |||||||||
Provision for write down of inventory | 1,400 | 50 | 228 | |||||||||
Issuance of stock to consultant | 189 | — | 80 | |||||||||
Stock based compensation | 3,090 | 2,273 | 823 | |||||||||
Amortization of debt issuance costs | 828 | 1,132 | 107 | |||||||||
Amortization of intangibles | 2,833 | 514 | 120 | |||||||||
Foreign currency exchange loss (gain) | 936 | (109 | ) | — | ||||||||
Amortization of debt discount on convertible 3.75% senior notes | 7,599 | 6,680 | 261 | |||||||||
Change in the fair value of derivative liability | — | (23,144 | ) | (2,300 | ) | |||||||
Loss on disposal of property | 16 | — | — | |||||||||
Changes in operating assets and liabilities: | ||||||||||||
Accounts receivable | (7,681 | ) | 1,250 | (9,419 | ) | |||||||
Inventories | (5,042 | ) | (3,578 | ) | (143 | ) | ||||||
Prepaid expenses and other current receivables | 3,427 | (5,408 | ) | (1,075 | ) | |||||||
Other assets | 316 | (14 | ) | — | ||||||||
Accounts payable and accrued expenses | 4,702 | (2,849 | ) | 2,346 | ||||||||
Deferred income | (476 | ) | 362 | (685 | ) | |||||||
Net cash provided by (used in) operating activities | 1,098 | (15,513 | ) | (3,891 | ) | |||||||
Cash flows from investing activities: | ||||||||||||
Capital expenditures | (18,551 | ) | (5,998 | ) | (834 | ) | ||||||
Acquisition of product rights and other related assets | — | (35,418 | ) | — | ||||||||
Product acquisition costs, net | (3,421 | ) | (11,652 | ) | (2,958 | ) | ||||||
Net cash used in investing activities | (21,972 | ) | (53,068 | ) | (3,792 | ) | ||||||
Cash flows from financing activities: | ||||||||||||
Proceeds from convertible 3.75% senior notes, net of $4,765 | — | — | 138,985 | |||||||||
Proceeds from issuance of stock, net | — | (3 | ) | 24,858 | ||||||||
Proceeds from note payable, net of debt issuance costs | — | — | 2,755 | |||||||||
Principal payments on note payable, bank | — | (3,160 | ) | (3,000 | ) | |||||||
Proceeds from exercise of common stock options and warrants | 96 | 165 | 837 | |||||||||
Principal payments on capital lease obligations | (70 | ) | (132 | ) | (64 | ) | ||||||
Recovery from stockholder, net | (36 | ) | 19 | — | ||||||||
Excess tax benefits from stock compensation | — | — | 94 | |||||||||
Net cash provided by (used in) financing activities | (10 | ) | (3,111 | ) | 164,465 | |||||||
Effect of exchange rate on cash and cash equivalents | (301 | ) | — | — | ||||||||
Net increase (decrease) in cash and cash equivalents | (20,884 | ) | (71,692 | ) | 156,782 | |||||||
Cash and cash equivalents at beginning of year | 87,191 | 158,883 | 2,101 | |||||||||
Cash and cash equivalents at end of year | $ | 66,006 | $ | 87,191 | $ | 158,883 | ||||||
Supplemental Cash flow information: | ||||||||||||
Cash payments for interest | $ | 5,393 | $ | 5,517 | $ | 178 | ||||||
Cash payments for income taxes | 113 | 123 | 23 | |||||||||
Non cash investing and financing transactions: | ||||||||||||
Reclassification of derivative liability to equity | — | 18,256 | — | |||||||||
Issuance of stock to consultant | — | 31 | — | |||||||||
Issuance of restricted stock | — | 347 | — | |||||||||
Bifurcation of derivative from convertible 3.75% senior notes | — | — | 41,400 | |||||||||
Payable related to product acquisition costs | — | — | 6,000 | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-7
TELIGENT, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
For the years ended December 31, 2016, 2015 and 2014
(in thousands, except share information)
Series A Convertible | Series C Convertible | Additional | Accumulated Other | Total | |||||||||||||||||||||||||||||||||||||
Preferred Stock | Preferred Stock | Common Stock | Paid-In | Comprehensive | Accumulated | Treasury | Stockholders’ | ||||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Shares | Amount | Capital | Loss | Deficit | Stock | Equity | |||||||||||||||||||||||||||||||
Balance, December 31, 2013 | — | $ | — | — | $ | — | 46,748,575 | $ | 487 | $ | 51,541 | $ | — | $ | (44,837 | ) | $ | — | $ | 7,191 | |||||||||||||||||||||
Issuance of stock pursuant to a public offering, net of associated fees of $1,868 | 5,347,500 | 53 | 24,805 | 24,858 | |||||||||||||||||||||||||||||||||||||
Issuance of stock to consultant | 10,000 | 80 | 80 | ||||||||||||||||||||||||||||||||||||||
Stock based compensation expense | 823 | 823 | |||||||||||||||||||||||||||||||||||||||
Stock warrants exercised | 270,546 | 3 | 325 | 328 | |||||||||||||||||||||||||||||||||||||
Stock options exercised | 443,166 | 5 | 504 | 509 | |||||||||||||||||||||||||||||||||||||
Excess tax benefits from stock compensation | 94 | 94 | |||||||||||||||||||||||||||||||||||||||
Net income | — | — | — | — | — | — | — | — | 5,251 | — | 5,251 | ||||||||||||||||||||||||||||||
Balance, December 31, 2014 | — | $ | — | — | $ | — | 52,819,787 | $ | 548 | $ | 78,172 | $ | — | $ | (39,586 | ) | $ | — | $ | 39,134 | |||||||||||||||||||||
Issuance of stock to consultant | 5,000 | 31 | 31 | ||||||||||||||||||||||||||||||||||||||
Stock based compensation expense | 2,273 | 2,273 | |||||||||||||||||||||||||||||||||||||||
Stock warrants exercised | 67,636 | 82 | 82 | ||||||||||||||||||||||||||||||||||||||
Stock options exercised | 75,766 | 1 | 82 | 83 | |||||||||||||||||||||||||||||||||||||
Issuance of restricted stock | 32,500 | 346 | 346 | ||||||||||||||||||||||||||||||||||||||
Reclassification of derivative liability to equity | 18,256 | 18,256 | |||||||||||||||||||||||||||||||||||||||
Recovery from stockholder, net | 19 | 19 | |||||||||||||||||||||||||||||||||||||||
Costs related to stock issuance | (3 | ) | (3 | ) | |||||||||||||||||||||||||||||||||||||
Cumulative translation adjustment | (130 | ) | (130 | ) | |||||||||||||||||||||||||||||||||||||
Net income | — | — | — | — | — | — | — | — | 6,668 | — | 6,668 | ||||||||||||||||||||||||||||||
Balance, December 31, 2015 | — | $ | — | — | $ | — | 53,000,689 | $ | 549 | $ | 99,258 | $ | (130 | ) | $ | (32,918 | ) | $ | — | $ | 66,759 | ||||||||||||||||||||
Issuance of stock to consultant | 25,000 | 189 | 189 | ||||||||||||||||||||||||||||||||||||||
Stock based compensation expense | 3,090 | 3,090 | |||||||||||||||||||||||||||||||||||||||
Stock options exercised | 61,834 | 1 | 95 | 96 | |||||||||||||||||||||||||||||||||||||
Issuance of stock for vested restricted stock units | 60,918 | 1 | 1 | ||||||||||||||||||||||||||||||||||||||
Recovery from stockholder, net | (36 | ) | (36 | ) | |||||||||||||||||||||||||||||||||||||
Tax benefit related to stock options | 28 | 28 | |||||||||||||||||||||||||||||||||||||||
Cumulative translation adjustment | (1,475 | ) | (1,475 | ) | |||||||||||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | — | (11,985 | ) | — | (11,985 | ) | ||||||||||||||||||||||||||||
Balance, December 31, 2016 | — | $ | — | — | $ | — | 53,148,441 | $ | 551 | $ | 102,624 | $ | (1,605 | ) | $ | (44,903 | ) | $ | — | $ | 56,667 | ||||||||||||||||||||
The accompanying notes are an integral part of the condensed consolidated financial statements.
F-8
TELIGENT, INC. AND SUBSIDIARIES
(FORMERLY IGI LABORATORIES, INC. AND SUBSIDIARIES)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Summary of Significant Accounting Policies
Nature of the Business
Teligent, Inc. is a Delaware corporation incorporated in 1977. On October 22, 2015, the Company announced the change of its name from IGI Laboratories, Inc. to Teligent, Inc. effective as of October 23, 2015. The Company’s office, research and development facilities and manufacturing facilities are located at 105 Lincoln Avenue, Buena, New Jersey. The Company is a specialty generic pharmaceutical company. Under our own label, the Company currently markets and sells generic topical and branded generic and generic injectable pharmaceutical products in the United States and Canada. In the US, we are currently marketing 16 generic topical pharmaceutical products and four branded generic pharmaceutical products. Through the completion of an acquisition, we now sell a total of 30 generic and branded generic injectable products and medical devices in Canada. Generic pharmaceutical products are bioequivalent to their brand name counterparts. We also provide development, formulation, and manufacturing services to the pharmaceutical, over-the-counter, or OTC, and cosmetic markets. The Company also provides development, formulation and manufacturing services to the pharmaceutical, over-the-counter (“OTC”) and cosmetic markets. We operate our business under one segment.
As of the date of this report, the Company has 34 Abbreviated New Drug Applications, or ANDAs, on file with the United States Food and Drug Administration, or FDA, for additional pharmaceutical products.
On October 14, 2015, the Company provided written notice to the NYSE MKT LLC (the “NYSE MKT”) that the Company intended to transfer the listing of the Company’s common stock from the NYSE MKT to the NASDAQ Global Select Market, and withdraw the listing and registration of the common stock from the NYSE MKT. The Company’s common stock ceased trading on the NYSE MKT at the close of business on October 23, 2015, and began trading on the NASDAQ Global Select Market on October 26, 2015.
As discussed in Note 7, on November 13, 2015, the Company acquired all of the rights, title and interest in the development, production, marketing, import and distribution of all pharmaceutical products of Alveda Pharmaceuticals Inc. (“Alveda”) pursuant to two asset purchase agreements, one relating to the acquisition of all of the intellectual property-related assets of Alveda (the “IP-Related APA”) and the other relating to the acquisition of all other assets of Alveda (the “Non-IP Related APA,” and, together with the IP-Related APA, the “APAs”).
Teligent also develops, manufactures, fills, and packages topical semi-solid and liquid products for branded and generic pharmaceutical customers, as well as the OTC and cosmetic markets. These products are used in a wide range of applications from cosmetics and cosmeceuticals to the prescription treatment of conditions like dermatitis, psoriasis, and eczema. Teligent has also started selling injectable products as of the fourth quarter of 2015, consistent with the Company's TICO strategy. Teligent has continued to make progress on its facility expansion in Buena, New Jersey, to support the increased capacity demand expected from future product approvals from the FDA. As the Company continues to execute its TICO strategy, it will compete in other markets, including the ophthalmic generic pharmaceutical market, and expects to face other competitors.
Principles of Consolidation
The consolidated financial statements include the accounts of Teligent, Inc. and its wholly-owned and majority-owned subsidiaries. All inter-company accounts and transactions have been eliminated. The Company consolidated the following entities: Igen, Inc., Teligent Pharma. Inc., Teligent Luxembourg S.à.r.l., Teligent OÜ, Teligent Canada Inc., and Teligent Jersey Limited., in addition to the following inactive entities: Microburst Energy, Inc., Blood Cells, Inc. and Flavorsome, Ltd.
Cash Equivalents
Cash equivalents consist of short-term investments, which have original maturities of 90 days or less. These include direct obligations of the U.S. Treasury, including bills, notes and bonds, as well as obligations issued or guaranteed by agencies or instrumentalities of the U.S. government including government-sponsored enterprises, or GSEs.
Fair Value of Financial Instruments
F-9
The carrying amounts of cash and cash equivalents, trade receivables, restricted cash, notes payable, accounts payable, capital leases and other accrued liabilities at December 31, 2016 approximate their fair value for all periods presented.
The Company measures fair value in accordance with ASC 820-10, “Fair Value Measurements and Disclosures”. ASC 820-10 clarifies that fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or a liability. As a basis for considering such assumptions, ASC 820-10 establishes a three-tier value hierarchy, which prioritizes the inputs used in the valuation methodologies in measuring fair value:
Level 1 Inputs: Unadjusted quoted prices in active markets for identical assets or liabilities accessible to the reporting entity at the measurement date.
Level 2 Inputs: Other than quoted prices included in Level 1 inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the asset or liability.
Level 3 Inputs: Unobservable inputs for the asset or liability used to measure fair value to the extent that observable inputs are not available, thereby allowing for situations in which there is little, if any, market activity for the asset or liability at measurement date. The fair value hierarchy also requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
The Company measures its derivative liability at fair value. The derivative convertible option related to the Notes issued December 16, 2014 was valued using the “with” and “without” analysis. A “with” and “without” analysis is a standard valuation technique for valuing embedded derivatives by first considering the value of the Notes with the option and then considering the value of the Notes without the option. The difference is the fair value of the embedded derivatives. The convertible note derivative is classified within Level 3 because it is valued using the “with” and “without” method, which does utilize inputs that are unobservable in the market.
On May 20, 2015, the Company received approval to increase its authorized shares sufficient to allow for the conversion of the entire note into equity at the annual shareholders meeting. Therefore, the derivative liability of $18.3 million was reclassified into stockholders equity. As of December 31, 2016, the net carrying value of the Notes was approximately $111.4 million compared to their face value of $143.75 million. However, this variance is due to the conversion feature in the Notes rather than to changes in market interest rates. The Notes carry a fixed interest rate and therefore do not subject the Company to interest rate risk. The Company recorded a change in the fair value of the derivative liability through May 20, 2015 of $23.1 million for the year ended December 31, 2015. On May 20, 2015, the Company recorded the final change in fair value and subsequently reclassified the value of the derivative liability into stockholders equity due to the approval of sufficient shares.
Accounts Receivable and Allowance for Doubtful Accounts
The Company extends credit to its contract services customers based upon credit evaluations in the normal course of business, primarily with 30-day terms. The Company does not require collateral from its customers. Bad debt provisions are provided for on the allowance method based on historical experience and management’s evaluation of outstanding accounts receivable. The Company reviews the allowance for doubtful accounts regularly, and past due balances are reviewed individually for collectability. The Company charges off uncollectible receivables against the allowance when the likelihood of collection is remote.
The Company extends credit to wholesaler and distributor customers and national retail chain customers, based upon credit evaluations, in the normal course of business, primarily with 60-day terms. The Company maintains customer-related accruals and allowances that consist primarily of chargebacks, rebates, sales returns, shelf stock allowances, administrative fees and other incentive programs. Some of these adjustments relate specifically to the generic prescription pharmaceutical business. Typically, the aggregate gross-to-net adjustments related to these customers can exceed 50% of the gross sales through this distribution channel. Certain of these accruals and allowances are recorded in the balance sheet as current liabilities and others are recorded as a reduction to accounts receivable.
Concentration of Credit Risk
Financial instruments, which subject the Company to concentrations of credit risk, consist primarily of cash equivalents and trade receivables. These include direct obligations of the U.S. Treasury, including bills, notes and bonds, as well as obligations issued or guaranteed by agencies or instrumentalities of the U.S. government including GSEs, which are not federally insured.
F-10
The Company maintains its cash in accounts with quality financial institutions. Although the Company currently believes that the financial institutions with which the Company does business will be able to fulfill their commitments to us, there is no assurance that those institutions will be able to continue to do so.
In 2016, the Company had sales to three customers which individually accounted for more than 10% of the Company’s total revenue. These customers had sales of $13.5 million, $8.6 million and $6.8 million, respectively, and represented 43% of total revenues in the aggregate. Accounts receivable related to the Company’s major customers comprised 81% of all accounts receivable as of December 31, 2016.
In 2015, the Company had sales to three customers which individually accounted for more than 10% of the Company’s total revenue. These customers had sales of $12.3 million, $5.8 million and $5.0 million, respectively, and represented 52% of total revenues in the aggregate. Accounts receivable related to the Company’s major customers comprised 83% of all accounts receivable as of December 31, 2015.
In 2014, the Company had sales to two customers which individually accounted for more than 10% of the Company’s total revenue. These customers had sales of $10.5 million and $4.4 million, respectively, and represented 44% of total revenues in the aggregate. Accounts receivable related to the Company’s major customers comprised 42% of all accounts receivable as of December 31, 2014.
The Company had net revenue from one product, econazole nitrate cream, which accounted for 8%, 45% and 38% of total revenues in 2016, 2015 and 2014, respectively. The Company had net revenue in 2016 from lidocaine ointment, which accounted for 23% of total revenues, which we launched at the end of the first quarter of 2016.
Inventories
Inventories are valued at the lower of cost, using the first-in, first-out (“FIFO”) method, or market. The Company records an inventory reserve for losses associated with dated and expired raw materials. This reserve is based on management’s current knowledge with respect to inventory levels, planned production, and extension capabilities of materials on hand. Management does not believe the Company’s inventory is subject to significant risk of obsolescence in the near term. Reserve for obsolescence included in inventory at December 31, 2016 and 2015 were $0.3 million and $0.1 million respectively.
Property, Plant and Equipment
Depreciation and amortization of property, plant and equipment is provided for under the straight-line method over the assets’ estimated useful lives as follows:
Useful Lives | ||
Buildings and improvements | 10 - 30 years | |
Machinery and equipment | 3 - 15 years | |
Repair and maintenance costs are charged to operations as incurred while major improvements are capitalized. When assets are retired or disposed, the related cost and accumulated depreciation thereon are removed and any gains or losses are included in operating results.
Intangible Assets
Definite-lived intangible assets are stated at cost less accumulated amortization. Amortization of definite-lived intangible assets is computed on a straight-line basis over the assets’ estimated useful lives, generally for periods ranging from 10 to 15 years. The Company continually evaluates the reasonableness of the useful lives of these assets. Indefinite-lived intangible assets are not amortized, but instead are tested at least annually for impairment. Costs to renew or extend the term of a recognized intangible asset are expensed as incurred.
In-Process Research and Development
Amounts allocated to in-process research and development (“IPR&D”) in connection with a business combination are recorded at fair value and are considered indefinite-lived intangible assets subject to the impairment testing in accordance with the Company’s
F-11
impairment testing policy for indefinite-lived intangible assets. As products in development are approved for sale, amounts will be allocated to product rights and will be amortized over their estimated useful lives. These valuations reflect, among other things, the impact of changes to the development programs, the projected development and regulatory time frames and the current competitive environment. Changes in any of the Company’s assumptions may result in a reduction to the estimated fair value of the IPR&D asset and could result in future impairment charges.
Goodwill
Goodwill, which represents the excess of purchase price over the fair value of net assets acquired, is carried at cost. Goodwill is tested for impairment on an annual basis during the fourth quarter of each fiscal year or more frequently if events or changes in circumstances indicate that the asset might be impaired. The Company first performs a qualitative assessment to determine if the quantitative impairment test is required. If changes in circumstances indicate an asset may be impaired, the Company performs the quantitative impairment test. In accordance with accounting standards, a two-step quantitative method is used for determining goodwill impairment. In the first step, the Company determines the fair value. If the net book value exceeds its fair value, the second step of the impairment test which requires allocation of the fair value to all of its assets and liabilities using the acquisition method prescribed under authoritative guidance for business combinations would then be performed. Any residual fair value is allocated to goodwill. An impairment charge is recognized only if the implied fair value of our reporting unit’s goodwill is less than its carrying amount.
The carrying value of goodwill at December 31, 2016 was $0.4 million. We believe it is unlikely that there will be a material change in the future estimates or assumptions used to test for impairment losses on goodwill. However, if actual results were not consistent with our estimates or assumptions, we could be exposed to an impairment charge.
Acquisitions
The Company accounts for acquired businesses using the acquisition method of accounting, which requires with limited exceptions, that assets acquired and liabilities assumed be recognized at their estimated fair values as of the acquisition date. Transaction costs are expensed as incurred. Any excess of the consideration transferred over the assigned values of the net assets acquired is recorded as goodwill. When net assets that do not constitute a business are acquired, no goodwill is recognized.
Contingent consideration, if any, is included as part of the acquisition cost and is recognized at fair value as of the acquisition date. Any liability resulting from contingent consideration is remeasured to fair value at each reporting date until the contingency is resolved. These changes in fair value are recognized in earnings.
Long-Lived Assets
In accordance with the provisions of ASC 360-10-55, the Company reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. In performing such review for recoverability, the Company compares expected future cash flows of assets to the carrying value of the long-lived assets and related identifiable intangibles. If the expected future cash flows (undiscounted) are less than the carrying amount of such assets, the Company recognizes an impairment loss for the difference between the carrying value of the assets and their estimated fair value, with fair values being determined using projected discounted cash flows at the lowest level of cash flows identifiable in relation to the assets being reviewed. As of December 31, 2016, no impairments existed.
Foreign Currency Translation
The net assets of international subsidiaries where the local currencies have been determined to be the functional currencies are translated into U.S. dollars using current exchange rates. The U.S. dollar effects that arise from translating the net assets of these subsidiaries at changing rates are recorded in the foreign currency translation account, which is included in Accumulated other comprehensive income (loss) (AOCI) and reflected as a separate component of equity. For those subsidiaries where the U.S. dollar has been determined to be the functional currency, non-monetary foreign currency assets and liabilities are translated using historical rates, while monetary assets and liabilities are translated at current rates, with the U.S. dollar effects of rate changes included in Other (income) expense, net.
Accrued Expenses
Accrued expenses represent various obligations of the Company including certain operating expenses and taxes payable.
F-12
For the fiscal year ended December 31, 2016, and 2015, the largest components of accrued expenses were:
2016 | 2015 | |||||||
(in thousands) | ||||||||
Wholesaler fees | $ | 3,505 | $ | 2,523 | ||||
Capital expenditures | 2,475 | 482 | ||||||
Payroll | 1,706 | 1,167 | ||||||
Royalties | 843 | 744 | ||||||
Consulting fees | 608 | 432 | ||||||
Interest expense | 240 | 240 | ||||||
Other | 972 | 679 | ||||||
$ | 10,349 | $ | 6,267 | |||||
License Fee
License fee is amortized on a straight-line basis over the life of the agreement (10 years).
Accounting for Environmental Costs
Accruals for environmental remediation are recorded when it is probable a liability has been incurred and costs are reasonably estimable. The estimated liabilities are recorded at undiscounted amounts. Environmental insurance recoveries are included in the statement of operations in the year in which the issue is resolved through settlement or other appropriate legal process.
Income Taxes
The Company records income taxes in accordance with ASC 740-10, “Accounting for Income Taxes,” under the asset and liability method of accounting for income taxes. Under the asset and liability method, deferred income taxes are recognized for the tax consequences of temporary differences by applying enacted statutory tax rates applicable to future years to operating loss and tax credit carry forwards and differences between the financial statement carrying amounts and the tax bases of existing assets and liabilities. The effect on deferred taxes of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is recorded based on a determination of the ultimate realizability of future deferred tax assets. A valuation allowance equal to 100% of the net deferred tax assets has been recognized due to uncertainty regarding the future realization of these assets.
The Company complies with the provisions of ASC 740-10-25 that clarifies the accounting for uncertainty in income taxes recognized in an entity’s financial statements in accordance with ASC 740-10, “Accounting for Income Taxes,” and prescribes a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities. Additionally, ASC 740-10 provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. There were no unrecognized tax benefits as of the date of adoption. As such, there are no unrecognized tax benefits included in the balance sheet that would, if recognized, affect the effective tax rate.
Revenue Recognition
The Company considers revenue realized or realizable and earned when it has persuasive evidence of an arrangement, delivery has occurred or contractual services rendered, the sales price is fixed or determinable, and collection is reasonably assured in conformity with ASC 605, Revenue Recognition.
The Company derives its revenues from three basic types of transactions: sales of its own generic pharmaceutical topical products, sales of manufactured product for its customers included in product sales, and research and product development services and other services performed for third parties. Due to differences in the substance of these transaction types, the transactions require, and the Company utilizes, different revenue recognition policies for each.
Product Sales: Product Sales, net, include Company Product Sales and Contract Manufacturing Sales.
F-13
Company Product Sales: The Company records revenue from Company product sales when title and risk of ownership have been transferred to the customer, which is typically upon delivery of products to the customer.
Revenue and Provision for Sales Returns and Allowances
As is customary in the pharmaceutical industry, the Company’s gross product sales from Company label products are subject to a variety of deductions in arriving at reported net product sales. When the Company recognizes revenue from the sale of products, an estimate of sales returns and allowances (“SRA”) is recorded, which reduces product sales. Accounts receivable and/or accrued expenses are also reduced and/or increased by the SRA amount. These adjustments include estimates for chargebacks, rebates, cash discounts and returns and other allowances. These provisions are estimates based on historical payment experience, historical relationship to revenues, estimated customer inventory levels and current contract sales terms with direct and indirect customers. The estimation process used to determine our SRA provision has been applied on a consistent basis and no material adjustments have been necessary to increase or decrease our reserves for SRA as a result of a significant change in underlying estimates. The Company will use a variety of methods to assess the adequacy of our SRA reserves to ensure that our financial statements are fairly stated. These will include periodic reviews of customer inventory data, customer contract programs, subsequent actual payment experience, and product pricing trends to analyze and validate the SRA reserves.
The provision for chargebacks is our most significant sales allowance. A chargeback represents an amount payable in the future to a wholesaler for the difference between the invoice price paid to the Company by our wholesale customer for a particular product and the negotiated contract price that the wholesaler’s customer pays for that product. The Company’s chargeback provision and related reserve varies with changes in product mix, changes in customer pricing and changes to estimated wholesaler inventories. The provision for chargebacks also takes into account an estimate of the expected wholesaler sell-through levels to indirect customers at contract prices. The Company will validate the chargeback accrual quarterly through a review of the inventory reports obtained from our largest wholesale customers. This customer inventory information is used to verify the estimated liability for future chargeback claims based on historical chargeback and contract rates. These large wholesalers represent 90% - 95% of the Company’s chargeback payments. The Company continually monitors current pricing trends and wholesaler inventory levels to ensure the liability for future chargebacks is fairly stated.
Net revenues and accounts receivable balances in the Company’s consolidated financial statements are presented net of SRA estimates. Certain SRA balances are included in accounts payable and accrued expenses.
Gross-To-Net Sales Deductions
Years ended December 31, | ||||||||||||
2016 | 2015 | 2014 | ||||||||||
Gross Company product sales | $ | 217,633 | $ | 99,721 | $ | 51,136 | ||||||
Reduction to gross product sales: | ||||||||||||
Chargebacks and billbacks | 141,343 | 50,127 | 26,940 | |||||||||
Sales discounts and other allowances | 27,419 | 17,974 | 4,366 | |||||||||
Total reduction to gross product sales | $ | 168,762 | $ | 68,101 | $ | 31,306 | ||||||
Company product sales, net | $ | 48,871 | $ | 31,620 | $ | 19,830 | ||||||
F-14
The annual activity in the Company's allowance for customer deductions and doubtful accounts for the three years ended December 31, 2016 is as follows (in thousands):
Returns | Chargebacks & Rebates | Discounts | Doubtful Accounts | TOTAL | ||||||||||||||||
Balance at December 31, 2013 | $ | 56 | $ | 1,746 | $ | 142 | $ | 16 | $ | 1,960 | ||||||||||
Provision | 767 | 31,040 | 1,060 | — | 32,867 | |||||||||||||||
Charges processed | (149 | ) | (28,234 | ) | (857 | ) | — | (29,240 | ) | |||||||||||
Balance at December 31, 2014 | $ | 674 | $ | 4,552 | $ | 345 | $ | 16 | $ | 5,587 | ||||||||||
Provision | 1,724 | 65,713 | 2,201 | 74 | 69,712 | |||||||||||||||
Charges processed | (1,464 | ) | (57,815 | ) | (1,754 | ) | — | (61,033 | ) | |||||||||||
Balance at December 31, 2015 | $ | 934 | $ | 12,450 | $ | 792 | $ | 90 | $ | 14,266 | ||||||||||
Provision | 3,568 | 160,556 | 4,667 | 347 | 169,138 | |||||||||||||||
Charges processed | (2,192 | ) | (137,125 | ) | (2,156 | ) | (20 | ) | (141,493 | ) | ||||||||||
Balance at December 31, 2016 | $ | 2,310 | $ | 35,881 | $ | 3,303 | $ | 417 | $ | 41,911 | ||||||||||
Accounts receivable are presented net of SRA balances of $41.5 million and $14.2 million at December 31, 2016 and 2015, respectively. Accounts payable and accrued expenses include $3.5 million and $2.5 million at December 31, 2016 and 2015, respectively, for certain fees related to services provided by the wholesalers. Wholesale fees of $3.7 million , $6.3 million and $3.1 million for the years ended December 31, 2016, 2015 and 2014, respectively, were included in cost of goods sold.
In addition, in connection with four of the 16 products the Company currently manufactures, markets and distributes in its own label in the U.S., in accordance with an agreement entered into in December of 2011, the Company is required to pay a royalty calculated based on net sales to one of its pharmaceutical partners. The royalty is calculated based on contracted terms of 40% of net sales for the four products, which is to be paid quarterly to the pharmaceutical partner that the agreement is with. In accordance with the agreement, net sales exclude fees related to services provided by the wholesalers. Accounts payable and accrued expenses include $0.8 million and $0.7 million at December 31, 2016 and 2015, respectively, related to these royalties. Royalty expense of $3.0 million, $3.6 million and $3.6 million was included in cost of goods sold for the years ended December 31, 2016, 2015, and 2014 respectively. The Company includes significant estimates to arrive at net product sales arising from wholesaler chargebacks, Medicaid and Medicare rebates, allowances and other pricing and promotional programs.
Contract Manufacturing Sales: The Company recognizes revenue when title transfers to its customers, which is generally upon shipment of products. These shipments are made in accordance with sales commitments and related sales orders entered into with customers either verbally or in written form. The revenues associated with these transactions, net of appropriate cash discounts, product returns and sales reserves, are recorded upon shipment of the products and are included in product sales, net on the Company's Consolidated Statement of Operations.
Research and Development Income: The Company establishes agreed upon product development agreements with its customers to perform product development services. Product development revenues are recognized in accordance with the product development agreement upon the completion of the phases of development and when the Company has no future performance obligations relating to that phase of development. Revenue recognition requires the Company to assess progress against contracted obligations to assure completion of each stage. These payments are generally non-refundable and are reported as deferred until they are recognizable as revenue. If no such arrangement exists, product development fees are recognized ratably over the entire period during which the services are performed. Other types of revenue include royalty or licensing revenue, and would be recognized based upon the contractual agreement upon completion of the earnings process.
In making such assessments, judgments are required to evaluate contingencies such as potential variances in schedule and the costs, the impact of change orders, liability claims, contract disputes and achievement of contractual performance standards. Changes in total estimated contract cost and losses, if any, are recognized in the period they are determined. Billings on research and development contracts are typically based upon terms agreed upon by the Company and customer and are stated in the contracts themselves and do not always align with the revenues recognized by the Company.
Licensing and Royalty Income: Revenues earned under licensing or sublicensing contracts are recognized as earned in accordance with the terms of the agreements. The Company recognizes royalty revenue based on royalty reports received from the licensee. The Company does not have current plans to have meaningful revenue from licensing and royalty agreements in 2017.
F-15
Stock-Based Compensation
ASC 718-10 defines the fair-value-based method of accounting for stock-based employee compensation plans and transactions used by the Company to account for its issuances of equity instruments to record compensation cost for stock-based employee compensation plans at fair value as well as to acquire goods or services from non-employees. Transactions in which the Company issues stock-based compensation to employees, directors and advisors and for goods or services received from non-employees are accounted for based on the fair value of the equity instruments issued. The Company utilizes pricing models in determining the fair values of options and warrants issued as stock-based compensation. These pricing models utilize the market price of the Company’s common stock and the exercise price of the option or warrant, as well as time value and volatility factors underlying the positions. Stock-based compensation expense is recognized over the vesting period of the grant.
Debt Issuance Costs
Expenses related to debt financing activities are capitalized and amortized on an effective interest method, over the term of the loan. See detailed amounts per year in Notes 5 and 8.
ASU 2015-3 specifies that debt issuance costs are to be netted against the carrying value of the financial liability. Under prior guidance, debt issuance costs were recognized as a deferred charge and reported as a separate asset on the balance sheet. The updated guidance aligns the treatment of debt issuance costs and debt discounts in that both reduce the carrying value of the liability. Amortization of debt issuance costs is to be recorded as interest expense on the income statement.
Product Development and Research
The Company’s research and development costs are expensed as incurred.
Shipping and Handling Costs
Costs related to shipping and handling is comprised of outbound freight and the associated labor. These costs are recorded in costs of sales.
Earnings (Loss) per Common Share
Basic earnings (loss) per share of common stock is computed based on the weighted average number of shares of common stock outstanding during the period. Diluted earnings (loss) per share of common stock is computed using the weighted average number of shares of common stock and potential dilutive common stock equivalents outstanding during the period. Potential dilutive common stock equivalents include shares issuable upon the conversion of the notes and the exercise of options and warrants. Due to the net loss for the year ended December 31, 2016, the effect of the Company’s potential dilutive common stock equivalents was anti-dilutive; as a result, the basic and diluted weighted average number of common shares outstanding and net loss per common share are the same. As of December 31, 2016, the shares of common stock issuable in connection with stock options and warrants have been excluded from the diluted earnings (loss) per share, as their effect would have been anti-dilutive.
F-16
For the years ended December 31, 2016, 2015 and 2014
(in thousands except shares and per share data)
2016 | 2015 | 2014 | ||||||||||
Basic earnings (loss) per share computation: | ||||||||||||
Net income (loss) attributable to common stockholders —basic | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | |||||
Weighted average common shares —basic | 53,078,158 | 52,872,814 | 49,817,721 | |||||||||
Basic earnings (loss) per share | $ | (0.23 | ) | $ | 0.13 | $ | 0.11 | |||||
Dilutive earnings (loss) per share computation: | ||||||||||||
Net income (loss) attributable to common stockholders —basic | $ | (11,985 | ) | $ | 6,668 | $ | 5,251 | |||||
Interest expense related to convertible 3.75% senior notes | — | 5,391 | 224 | |||||||||
Amortization of discount related to convertible 3.75% senior notes | — | $ | 6,680 | $ | — | |||||||
Change in the fair value of derivative | — | $ | (23,144 | ) | $ | — | ||||||
Net income (loss) attributable to common stockholders —diluted | $ | (11,985 | ) | $ | (4,405 | ) | $ | 5,475 | ||||
Share Computation: | ||||||||||||
Weighted average common shares —basic | 53,078,158 | 52,872,814 | 49,817,721 | |||||||||
Effect of convertible 3.75% senior notes | — | 12,732,168 | 12,732,168 | |||||||||
Effect of dilutive stock options and warrants | — | 1,507,013 | 1,657,301 | |||||||||
Weighted average common shares outstanding —diluted | 53,078,158 | 67,111,995 | 64,207,190 | |||||||||
Diluted net earnings (loss) per share | $ | (0.23 | ) | $ | (0.07 | ) | $ | 0.09 | ||||
Derivatives
The Company accounts for its derivative instruments in accordance with ASC 815-10, “Derivatives and Hedging” (“ASC 815-10”). ASC 815-10 establishes accounting and reporting standards requiring that derivative instruments, including derivative instruments embedded in other contracts, be recorded on the balance sheet as either an asset or liability measured at its fair value. ASC 815-10 also requires that changes in the fair value of derivative instruments be recognized currently in results of operations unless specific hedge accounting criteria are met. The Company has not entered into hedging activities to date. The Company’s derivative liability is the embedded convertible option of its Convertible Notes issued December 16, 2014 (as defined in Note 6), which has been recorded as a liability at fair value until May 20, 2015, and was revalued at each reporting date, with changes in the fair value of the instruments included in the consolidated statements of operations as non-operating income (expense). Due to the approval of the sufficient shares at the Company’s annual shareholder meeting, the liability for the embedded derivative was reclassified to equity on May 20, 2015. The Company has no derivatives at December 31, 2016.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include valuation of the derivative liability, SRA allowances, allowances for excess and obsolete inventories, allowances for doubtful accounts, provisions for income taxes and related deferred tax asset valuation allowances, stock based compensation, the impairment of long-lived assets (including intangibles and goodwill) and accruals for environmental cleanup and remediation costs. Actual results could differ from those estimates.
Recent Accounting Pronouncements
In December 2016, the FASB issued ASU 2016-20, Technical Corrections and Improvements to Topic 606, Revenue from Contracts with Customers". The update provides users with classification guidance on thirteen specific areas of correction or improvement topics as follows: 1) Loan Guarantee Fees, 2) Contract Costs - Impairment Testing, 3) Contract Costs - Interaction of Impairment Testing with Guidance in Other Topics, 4) Provisions for Losses on Construction-Type and Production-Type Contracts, 5) Scope of Topic 606, 6) Disclosure of Remaining Performance Obligations, 7) Disclosure of Prior-Period Performance Obligations, 8) Contract Modifications Example, 9) Contract Asset versus Receivable, 10) Refund Liability, 11) Advertising Costs, 12) Fixed-Odds Wagering Contracts in the Casino Industry and 13) Cost Capitalization for Advisors to Private Funds and Public Funds. The amendments affect the guidance in update 2014-09, which is effective for fiscal years beginning after December 15, 2017 for
F-17
public business entities, including interim periods within those fiscal years. For the Company, the amendments are effective January 1, 2018. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In December 2016, the FASB issued ASU 2016-19, Technical Corrections and Improvements. The update is to address suggestions received from stakeholders on the Accounting Standards Codification and to make other incremental improvements to GAAP. It contains amendments that affect a wide variety of Topics in the Accounting Standards Codification and applies to all reporting entities within the scope of the affected accounting guidance. Most of the amendments are effective upon issuance of the update. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In November 2016, the FASB issued ASU 2016-18, Statement of Cash Flows (Topic 230): "Restricted Cash (a consensus of the FASB Emerging Issues Task Force)". The update addresses the diversity in the industry with respect to classification and presentation of changes in restricted cash on the statement of cash flows. These amendments require that a statement of cash flows explain the restricted cash change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. It affects those reporting entities that are required to evaluate whether they should consolidate a variable interest entity "VIE". The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For the Company, the amendments are effective January 1, 2018. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In October 2016, the FASB issued ASU 2016-17, Consolidation (Topic 810): "Interests Held through Related Parties That Are Under Common Control". The update was issued to amend the consolidation guidance on how a reporting entity that is a single decision maker of a VIE should treat indirect interests in the entity held through related parties that are under common control with the reporting entity when determining whether it is the primary beneficiary of that VIE. It affects those reporting entities that are required to evaluate whether they should consolidate a VIE. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For the Company, the amendments are effective January 1, 2018. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In October 2016, the FASB issued ASU 2016-16, Income Taxes (Topic 740): "Intra-Entity Transfers of Assets Other Than Inventory". The update addresses income tax consequences of intra-entity transfers of assets other than inventory. It seeks to clarify the authoritative guidance about the prohibition of the recognition of current and deferred income taxes for intra-entity asset transfers until the asset has been sold to an outside party. Instead, this update now eliminates that prohibition and states that an entity should recognize the income tax consequences when the transfer occurs. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For the Company, the amendments are effective January 1, 2018. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230): “Classification of Certain Cash Receipts and Cash Payments (a Consensus of the Emerging Issues Task Force)”. The update provides users with classification guidance on eight specific cash flow topics as follows: 1) Debt Prepayment or Debt Extinguishment Costs, 2) Settlement of Zero-Coupon Debt Instruments or Other Debt Instruments with Coupon Interest Rates That Are Insignificant in Relation to the Effective Interest Rate of the Borrowing, 3) Contingent Consideration Payments Made after a Business Combination, 4) Proceeds from the Settlement of Insurance Claims, 5) Proceeds from the Settlement of Corporate-Owned Life Insurance Policies, including Bank-Owned Life Insurance Policies, 6) Distributions Received from Equity Method Investees, 7) Beneficial Interests in Securitization Transactions and 8) Separately Identifiable Cash Flows and Application of the Predominance Principle. The amendments in this update are effective for fiscal years beginning after December 15, 2017 for public business entities, including interim periods within those fiscal years. For the Company, the amendments are effective January 1, 2018. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In June 2016, the FASB issued ASU 2016-13, Financial Instruments - Credit Losses (Topic 326): “Measurement of Credit Losses on Financial Instruments”. The update provides users with more useful information for decision making regarding expected credit losses on financial instruments/commitments to extend credit held by a reporting entity at each reporting date. The amendments affect loans, debt securities, trade receivables, net investments in leases, off-balance-sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. Credit quality of the entity’s assets now plays a key role in this update. The amendments in this update are effective for fiscal years beginning after December 15, 2019 for public business entities, including interim periods within those fiscal years. For the Company, the amendments are effective January 1, 2020. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
F-18
In May 2016, the FASB issued ASU 2016-12, Revenue from Contracts with Customers (Topic 606): “Narrow-Scope Improvements and Practical Expedients”. The update addresses issues identified by the FASB-IASB Joint Transition Resource Group (TRG), a group formed in June 2014 in order to inform the Boards about potential implementation issues that could arise as a result of organizations implementing the May 2014 revenue guidance. It affects entities that enter into contracts with customers to transfer goods or services within an entity’s ordinary activities in exchange for consideration. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In May 2016, the FASB issued ASU 2016-11, Revenue Recognition (Topic 605) and Derivatives and Hedging (Topic 815): “Rescission of SEC Guidance Because of Accounting Standards Updates 2014-09 and 2014-16 Pursuant to Staff Announcements at the March 3, 2016 EITF Meeting (SEC Update)”. The update is a result of adoption of Topic 606, Revenue from Contracts with Customers. SEC Staff Observer comments found in Topic 605 are therefore not recommended to be relied upon and have been superseded. The comments are found in the following topics: 1) Revenue and Expense Recognition for Freight Services in Process, 2) Accounting for Shipping and Handling Fees and Costs, 3) Accounting for Consideration Given by a Vendor to a Customer (including Reseller of the Vendor’s Products), and 4) Accounting for Gas-Balancing Arrangements. As these amendments require changes to the U.S. GAAP Financial Reporting Taxonomy, they will be incorporated into the proposed 2017 Taxonomy and finalized as part of the annual release process. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In March 2016, the FASB issued ASU No. 2016-09, Compensation-Stock Compensation (Topic 718): “Improvements to Employee Share-Based Payment Accounting”. The update includes multiple provisions intended to simplify various aspects of the accounting for share-based payments, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. The amendments in this update are effective for public companies for annual periods beginning after December 15, 2016, and interim periods within those annual periods. Early adoption is permitted for any interim or annual period. The Company has evaluated the impact of this ASU on its consolidated financial statements and as a result, will adjust retained earnings in 2017 for the amounts previously recognized as windfall tax benefits in additional paid in capital.
In February 2016, the FASB issued Accounting Standards Update (“ASU”) 2016-02, Leases (Topic 842): “Recognition and Measurement of Financial Assets and Financial Liabilities”. The update supersedes Topic 840, Leases and requires the recognition of lease assets and lease liabilities by lessees for those leases classified as operating leases under previous GAAP. Topic 842 retains a distinction between finance leases and operating leases, with cash payments from operating leases classified within operating activities in the statement of cash flows. The amendments in this update are effective for fiscal years beginning after December 15, 2018 for public business entities, which for the Company means January 1, 2019. The Company is currently evaluating the impact of this ASU on its consolidated financial statements.
In September 2015, the FASB issued ASU 2015-16, Business Combinations (Topic 805): “Simplifying the Accounting for Measurement-Period Adjustments”. The update eliminates the requirement to retrospectively adjust the provisional amounts recognized at the acquisition date with a corresponding adjustment to goodwill during the measurement period when new information is obtained about the facts and circumstances that existed as of the acquisition date, that if known, would have affected the measurement of the amounts initially recognized or would have resulted in the recognition of additional assets or liabilities. The amendments in this update are effective for fiscal years beginning after December 15, 2015, which for the Company means January 1, 2016, and should be applied prospectively to adjustments to provisional amounts that occur after the effective date of this update. Early application is permitted for financial statements that have not been issued. The Company has concluded the adoption of this ASU will not have any significant impact on its consolidated financial statements.
In July 2015, the FASB issued ASU 2015-11, Inventory (Topic 330): “Simplifying the Measurement of Inventory”. ASU 2015-11 requires inventory measured using any method other than last-in, first out (“LIFO”) or the retail inventory method to be subsequently measured at the lower of cost or net realizable value, rather than at the lower of cost or market. Under this ASU, subsequent measurement of inventory using the LIFO and retail inventory method is unchanged. ASU 2015-11 is effective prospectively for fiscal years, and for interim periods within those years, beginning after December 15, 2016. Early application is permitted. The Company does not expect the adoption of this ASU will have any significant impact on its consolidated financial statements.
2. Liquidity
The Company’s principal sources of liquidity are cash and cash equivalents of approximately $66 million at December 31, 2016 and cash from operations. The Company terminated its $10 million credit facility with General Electric Capital Corporation, as agent, and GE Capital Bank and certain other institutions, as lenders, in February 2016. See Note 8.
F-19
The Company may require additional funding and this funding will depend, in part, on the timing and structure of potential business arrangements. If necessary, the Company may continue to seek to raise additional capital through the sale of its equity or through a strategic alliance with a third party. There may also be additional acquisition and growth opportunities that may require external financing. There can be no assurance that such financing will be available on terms acceptable to the Company, or at all. The Company also has the ability to defer certain product development and other programs, if necessary. The Company believes that our existing capital resources will be sufficient to support its current business plan and operations beyond March 2018.
3. Inventories
Inventories as of December 31, 2016 and 2015 consisted of:
2016 | 2015 | |||||||
(in thousands) | ||||||||
Raw materials | $ | 6,546 | $ | 4,833 | ||||
Work in progress | — | 128 | ||||||
Finished goods | 6,162 | 4,024 | ||||||
$ | 12,708 | $ | 8,985 | |||||
4. Property, Plant and Equipment
Property, plant and equipment, at cost, as of December 31, 2016 and 2015, consisted of:
2016 | 2015 | |||||||
(in thousands) | ||||||||
Land | $ | 257 | $ | 257 | ||||
Building and improvements | 8,515 | 5,296 | ||||||
Machinery and equipment | 8,583 | 5,270 | ||||||
Construction in progress | 15,496 | 3,594 | ||||||
32,851 | 14,417 | |||||||
Less accumulated depreciation and amortization | (6,636 | ) | (5,711 | ) | ||||
Property, plant and equipment, net | $ | 26,215 | $ | 8,706 | ||||
The Company recorded depreciation and amortization expense of $946,000, $560,000 and $415,000 in 2016, 2015 and 2014, respectively.
5. Convertible 3.75% Senior Notes
On December 16, 2014, the Company issued $125 million aggregate principal amount of 3.75% Convertible Senior Notes due 2019, or the Notes. On December 22, 2014, the Company announced the closing of the initial purchasers’ exercise in full of their option to purchase an additional $18.75 million aggregate principal amount. The Notes were offered and sold only to qualified institutional buyers pursuant to Rule 144A under the Securities Act of 1933, as amended (the “Securities Act”). The net proceeds from the sale of the Notes were approximately $139 million, after deducting underwriting fees and other related expenses of approximately $4.8 million. Accrued interest in the amount of $0.2 million related to the Notes was included in accrued expenses.
The Notes bear interest at a fixed rate of 3.75% per year, payable semiannually in arrears on June 15 and December 15 of each year, beginning on June 15, 2015 and mature on December 15, 2019, unless earlier repurchased, redeemed or converted. The Notes are convertible into shares of the Company’s common stock, cash or a combination thereof.
The Notes are convertible at an initial conversion price of approximately $11.29 per share, which is equivalent to an initial conversion rate of 88.5716 shares per $1,000 principal amount of Notes, subject to adjustment in certain events, such as distributions of dividends or stock splits. Holders may convert their Notes at their option prior to September 15, 2019, when or if certain conditions have been met or circumstances have occurred, such as the Company’s stock price exceeds 130% of the conversion
F-20
price under the Notes for a designated period of time, or the trading price of the Notes is, for a designated period of time, less than 98% of the closing sale price of the Company’s common stock multiplied by the then-current conversion rate of the Notes, or the Company calls Notes for redemption, or certain specified corporate events occur. Holders may also convert their Notes at their option at any time on or after September 15, 2019 and prior to the close of business on the business day immediately preceding the stated maturity date. In addition, following the occurrence of certain changes of control of the Company described in the Indenture governing the Notes or termination of trading of the Company’s common stock or other securities into which the Notes are convertible (a “make-whole fundamental change”) or the delivery by the Company of a notice of redemption, the conversion rate for a holder who elects to convert its Notes in connection with such make-whole fundamental change or such notice of redemption will increase in certain circumstances. Additionally, subject to certain conditions, the Company may redeem for cash any or all outstanding Notes on or after December 19, 2017 in an amount equal to the outstanding principal amount of such Notes, plus accrued and unpaid interest.
The Notes and any common stock issuable upon conversion of the Notes have not been registered under the Securities Act, applicable state securities laws or the securities laws of any other jurisdiction, and may not be offered or sold in the United States without registration or an applicable exemption from registration requirements. The Company does not intend to file a registration statement for the resale of the Notes or any common stock issuable upon conversion of the Notes, nor shall there be any sale of these securities in any jurisdiction in which such offer, solicitation, or sale would be unlawful.
Since the Company did not have sufficient authorized shares available to share-settle the conversion option in full prior to May 20, 2015, the embedded conversion option did not qualify for equity classification and instead was separately valued and accounted for as a derivative liability. On December 16, 2014, the initial value allocated to the derivative liability was $43.7 million of the $143.75 million principal amount of the Notes, which represents a discount to the debt to be amortized through interest expense using the effective interest method through the maturity of the Notes. Accordingly, the effective interest rate used to amortize the debt discount on the Notes is 12.94%. During each reporting period through May 20, 2015, the derivative liability was marked to fair value with the change in fair value recorded in the consolidated statement of operations. This resulted in a change in the fair value of the derivative liability of $23.1 million for the year ended December 31, 2015.
On May 20, 2015, the Company received shareholder approval for the increase in the number of shares of common stock authorized and available for issuance upon conversion of the Notes. As a result, the conversion option can now be share-settled in full, and now qualifies for equity classification, and the bifurcated derivative liability no longer needs to be accounted for as a separate derivative on a prospective basis as of May 20, 2015. The remaining unamortized debt discount that arose at the date of debt issuance from the original bifurcation will continue to be amortized using the effective interest method through interest expense. After adjusting the derivative liability to market value on May 20, 2015, the Company reclassified the remaining $18.3 million value of the derivative liability to stockholders equity.
The remaining unamortized discount and unamortized debt financing costs will be amortized over the remaining term of the debt of 3.0 years. At December 31, 2016 and December 31, 2015, the net carrying amount of the liability component and the remaining unamortized debt discount were as follows:
December 31, 2016 | December 31, 2015 | |||||
(in thousands) | (in thousands) | |||||
Face amount of the Notes | $ | 143,750 | $ | 143,750 | ||
Unamortized discount | 29,160 | 36,759 | ||||
Debt issuance costs | $ | 3,199 | $ | 4,027 | ||
Carrying amount of the Notes | $ | 111,391 | $ | 102,964 | ||
Debt issuance costs associated with the Notes, include fees of $3.2 million at December 31, 2016 and $4.0 million at December 31, 2015. The assumptions used in connection with the valuation of the convertible option of the Notes issued December 16, 2014 utilizing the “with” and “without” method, discussed in Note 2 was as follows:
F-21
Initial Measurement December 16, 2014 | Measurement December 31, 2014 | Measurement May 20, 2015 | ||||||||||
Issue date | 12/17/2014 | 12/17/2014 | 12/17/2014 | |||||||||
Maturity date | 12/15/2019 | 12/15/2019 | 12/15/2019 | |||||||||
Term | 4.99 | 4.92 | 4.57 | |||||||||
Principal (millions) | 143.75 | 143.75 | 143.75 | |||||||||
Coupon | 3.75 | % | 3.75 | % | 3.75 | % | ||||||
Seniority | Senior unsecured | Senior unsecured | Senior unsecured | |||||||||
Conversion shares | 88.572 | 88.572 | 88.572 | |||||||||
Conversion price | $ | 11.29 | $ | 11.29 | $ | 11.29 | ||||||
Stock price | $ | 9.45 | $ | 8.80 | $ | 5.73 | ||||||
Risk free rate | 1.61 | % | 1.64 | % | 1.44 | % | ||||||
Volatility (rounded) | 40.00 | % | 40.00 | % | 46.00 | % | ||||||
The table below provides a reconciliation of beginning and ending balances for the liability measured at fair value using significant observable and unobservable inputs (Level 3). The table reflects the gains associated with the decrease in fair value and the reclassification of the balance of the derivative liability.
Initial Measurement December 16, 2014 | Decrease in Fair Value | December 31, 2014 | Decrease in Fair Value January 1, 2015 to May 20, 2015 | Reclassification of derivative liability to equity on May 20, 2015 | December 31, 2015 | |||||||||||||||||||
Fair value of convertible feature of 3.75% senior notes | $ | 43,700 | $ | 2,300 | $ | 41,400 | $ | 23,144 | $ | 18,256 | $ | — | ||||||||||||
For the year ended December 31, 2016 and December 31, 2015, the Company recorded the following expenses in relation to the Notes:
December 31, 2016 | December 31, 2015 | December 31, 2014 | ||||||||
(in thousands) | (in thousands) | (in thousands) | ||||||||
Interest Expense at 3.75% coupon rate | $ | 5,391 | $ | 5,391 | $ | 225 | ||||
Debt discount amortization | 7,599 | 6,680 | 261 | |||||||
Amortization of deferred financing costs | 828 | 728 | 28 | |||||||
Total interest expense (1) | $ | 13,818 | $ | 12,799 | $ | 514 | ||||
(1) Included within “Interest and other expense, net” on the Consolidated Statements of Operations
F-22
6. Acquisitions
On November 13, 2015, the Company completed its acquisition of all of the rights, title and interest in the development, production, marketing, import and distribution of all pharmaceutical products of Alveda.
In connection with the closing of the Acquisition, the Company formed three subsidiaries: Teligent Luxembourg S.à.r.l., a private limited company incorporated under the laws of the Grand Duchy of Luxembourg and wholly-owned by the Company ("LuxCo"); Teligent OÜ, a private limited company incorporated under the laws of the Republic of Estonia that is wholly-owned by LuxCo ("EstoniaCo"); and Teligent Canada Inc., a company incorporated under the laws of the Province of British Columbia that is wholly-owned by LuxCo ("CanadaCo"). Effective immediately prior to the closing, the Company assigned its rights and obligations under the IP-Related APA to EstoniaCo and assigned its rights and obligations under the Non-IP Related APA to CanadaCo, The Company capitalized these subsidiaries and funded the Acquisition as follows: the Company funded LuxCo by way of an equity contribution in the amount of $3,374,549 in accordance with the terms and conditions of the Contribution Agreement, by and between the Company and LuxCo, dated as of November 13, 2015 (the "Contribution Agreement"), and extended a loan in a principal amount of $28,185,847 in accordance with the terms and provisions of a loan agreement, by and between the Company and LuxCo dated as of November 13, 2015 (the "LuxCo Loan Agreement"). The LuxCo Loan Agreement has a maturity date of November 4, 2022. The initial interest rate under the LuxCo Loan Agreement is 0.49% per annum, which shall reset annually to be equal to the short-term Applicable Federal Rate published by the Internal Revenue Service (the "AFR"). LuxCo, in turn, extended a loan to EstoniaCo in the same principal amount on the same terms (except that the interest rate is increased by 25 basis points). In addition, the Company funded CanadaCo by extending a loan in a principal amount of $3,746,094 in accordance with the terms and provisions of a loan agreement, by and between the Company and CanadaCo dated as of November 13, 2015 (the "CanadaCo Loan Agreement"). The initial interest rate under the CanadaCo Loan Agreement is 0.49% per annum, which shall reset annually to be equal to the short-term AFR.
Also in connection with the closing of the Acquisition, CanadaCo and EstoniaCo entered into a distribution agreement pursuant to which CanadaCo has agreed to purchase from EstoniaCo certain products and act as the exclusive distributor of such products in Canada (the "Distribution Agreement"). In consideration for the supply of the products, CanadaCo agrees to pay an established per-unit amount for each packaging configuration unit of the product, with such price to be negotiated by the parties from time to time throughout the effective period. As a result, EstoniaCo shall manage all contract manufacturing arrangements with third parties and the sale of all such manufactured products to CanadaCo, which, in turn, shall manage the sale to third parties of all such finished products in Canada. The Distribution Agreement shall have an initial term of two years from the effective date, with automatic one-year renewals, unless terminated earlier by either party.
The Company has the right to use licenses and product registrations, access to intellectual property rights (“IPR”) (including know-how, and patents) to use, import, have imported, offer for sale, sell, manufacture and commercialize the devices in Canada.
The Alveda Acquisition has been accounted for using the acquisition method of accounting. This method requires that assets acquired and liabilities assumed in a business combination be recognized at their fair values as of the acquisition date. Acquisition related costs were expensed as incurred.
The Company utilized the Multi-Period Excess Earnings Method (MPEEM) income approach to value intangible assets. Key assumptions utilized in the analysis of the intangibles were the revenue base, cost of goods sold, operating expenses, income tax rate and discount rate.
The following table summarizes the consideration paid for Alveda, the total acquisition related costs incurred by the Company during 2015 in connection with the acquisition, and the fair values of the assets acquired and liabilities assumed (amounts in thousands):
F-23
Consideration: | |||
Fair value of total consideration transferred | $ | 35,418 | |
Acquisition-related costs* : | $ | 2,256 | |
Estimated fair value of identifiable assets acquired and liabilities assumed: | |||
Accounts receivable | $ | 911 | |
Inventories | 2,673 | ||
Prepaid expenses and other current assets | 4 | ||
Property and equipment | 6 | ||
Goodwill, deductible | 440 | ||
Developed Technology | 24,858 | ||
In-process research and development | 3,816 | ||
Customer relationships | 3,615 | ||
Accounts payable and other assumed liabilities | (661 | ) | |
Deferred tax liability | (244 | ) | |
*At closing, the Company also paid $5.2 million related to Canadian goods and services tax (GST) and the harmonized sales tax (HST), which is not included in the above table as consideration or acquisition related costs, and all amounts have been refunded in 2016.
The following sets forth the major categories of the Company’s intangible assets acquired from Alveda and the weighted-average remaining amortization period as of December 31, 2015 for those assets that are not already fully amortized (dollar amounts in thousands):
Gross Carrying Amount at 12/31/15 | Accumulated Amortization at 12/31/15 | Net Carrying Amount at 12/31/15 | Weighted Average Remaining Amortization Period | ||||||||
Technology | 25,243 | (210 | ) | 25,033 | 14.9 years | ||||||
In-process research and development ("IPR&D") | 3,875 | — | 3,875 | N/A - Indefinite lived | |||||||
Customer relationships | 3,460 | (43 | ) | 3,417 | 9.9 years | ||||||
Total | 32,578 | (253 | ) | 32,325 | |||||||
As of December 31, 2016, $10.8 million of revenues and $1.6 million net income from CanadaCo are included in the Company’s earnings.
Pro Forma Information (unaudited): The following pro forma information presents the results of operations for the years ended December 31, 2015, and December 31, 2014, as if the Alveda acquisition occurred on January 1, 2014:
F-24
(amounts in thousands, except for per share amounts) | ||||||||
For the Years Ended | ||||||||
December 31, 2015 | December 31, 2014 | |||||||
Total Revenue | $ | 55,767 | $ | 47,284 | ||||
Net income | $ | 8,443 | $ | 6,525 | ||||
Basic earnings per share | $ | 0.16 | $ | 0.13 | ||||
Diluted earnings (loss) per share | $ | (0.04 | ) | $ | 0.11 | |||
The above pro forma results are presented for illustrative purposes and are not intended to represent or be indicative of the actual results of operations of the merged companies that would have been achieved had the acquisition occurred at January 1, 2014, nor are they intended to represent or be indicative of future results of operations. These pro forma results require significant estimates and judgments.
7. Goodwill and Intangible Assets
Goodwill
The Company acquired the assets of Canadian pharmaceutical company Alveda Pharmaceuticals, Inc., in November 2015. As a result of the acquisition, we recorded goodwill of $0.4 million. We assess the recoverability of the carrying value of goodwill in the fourth quarter of each year, and whenever events occur or circumstances change that would, more likely than not, reduce the fair value of our reporting unit below its carrying value. There have been no events or changes in circumstances that would have reduced the fair value of our reporting unit below its carrying value. No impairment losses were recognized during the year ended December 31, 2016.
Changes in goodwill during the two years ended December 31, 2016 were as follows (in thousands):
Goodwill | |||
December 31, 2014 | $ | — | |
Acquisition | 440 | ||
Impairments | — | ||
Foreign currency translation | (14 | ) | |
December 31, 2015 | 426 | ||
Acquisition | — | ||
Impairments | — | ||
Foreign currency translation | 20 | ||
December 31, 2016 | $ | 446 | |
Intangible Assets
The following sets forth the major categories of the Company’s intangible assets and the weighted-average remaining amortization period as of December 31, 2016 and December 31, 2015 for those assets that are not already fully amortized (dollar amounts in thousands):
F-25
December 31, 2016 | ||||||||||||||
Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Weighted Average Remaining Amortization Period | |||||||||||
Trademarks and Technology | $ | 35,403 | $ | (3,123 | ) | $ | 32,280 | 13.8 | ||||||
In-process research and development (“IPR&D”) | 17,024 | — | 17,024 | N/A - Indefinite lived | ||||||||||
Customer relationships | 3,565 | (404 | ) | 3,161 | 9.1 | |||||||||
Total | $ | 55,992 | $ | (3,527 | ) | $ | 52,465 | |||||||
December 31, 2015 | ||||||||||||||
Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Weighted Average Remaining Amortization Period | |||||||||||
Trademarks and Technology | $ | 37,203 | $ | (651 | ) | $ | 36,552 | 14.8 | ||||||
In-process research and development (“IPR&D”) | 14,351 | — | 14,351 | N/A - Indefinite lived | ||||||||||
Customer relationships | 3,460 | (43 | ) | 3,417 | 9.9 | |||||||||
Total | $ | 55,014 | $ | (694 | ) | $ | 54,320 | |||||||
Changes in intangibles during the two years ended December 31, 2016 were as follows (in thousands):
Trademarks and Technology | IPR&D | Customer Relationships | ||||||||||
December 31, 2014 | $ | 1,646 | $ | — | $ | — | ||||||
Acquisition | 34,992 | 14,292 | 3,615 | |||||||||
Amortization | (471 | ) | — | (43 | ) | |||||||
Foreign currency translation | 385 | 59 | (155 | ) | ||||||||
December 31, 2015 | 36,552 | 14,351 | 3,417 | |||||||||
Acquisition | 661 | 2,811 | — | |||||||||
Amortization | (2,472 | ) | — | (361 | ) | |||||||
Foreign currency translation | (2,461 | ) | (138 | ) | 105 | |||||||
December 31, 2016 | $ | 32,280 | $ | 17,024 | $ | 3,161 | ||||||
Assuming no additions, disposals or adjustments are made to the carrying values and/or useful lives of the intangible assets, annual amortization expense on product rights and other related intangibles for each of the following five years is estimated to be as follows (in thousands):
F-26
Year ending | Amortization | |
December 31, | Expense * | |
2017 | 2,712 | |
2018 | 2,712 | |
2019 | 2,712 | |
2020 | 2,712 | |
2021 | 2,712 | |
Thereafter | 22,017 | |
*IPR&D amounts will be amortized once products become saleable, and are not included in the table
The useful lives of the Company’s intangibles is as follows:
Intangibles Category | Amortizable Life | |
Trademarks and Technology | 15 years | |
Customer Relationships | 10 years | |
8. Note Payable - General Electric Capital Corporation
On November 18, 2014, the Company entered into an asset-based revolving senior secured credit facility (the “Credit Agreement”) with General Electric Capital Corporation, as agent (the “Agent”), and GE Capital Bank and the other financial institutions party thereto, as lenders (the “Lenders”), pursuant to which the Lenders agreed to extend credit facilities to the Company (the “Financing”).
To secure payment of the amounts financed under the Credit Agreement, the Company and the Agent entered into a Guaranty and Security Agreement (the “Guaranty and Security Agreement”). Under the terms of the Guaranty and Security Agreement, the Company granted to the Agent, for the benefit of the Lenders and other secured parties, a continuing security interest in and against substantially all of its tangible and intangible assets, except intellectual property, and each of the Company’s direct and indirect future subsidiaries shall guarantee the Company’s’ obligations under the Credit Agreement.
Under the Credit Agreement, the Company could request revolving loan advances up to an aggregate total amount of $10 million which may be increased to $15 million at the request of the Company if certain conditions are met. The Company may also request an incremental facility for revolving loan commitments of up to $10 million. Borrowings under the Credit Agreement may be made as prime rate loans with an applicable margin of 3.0% per annum or 1, 2, 3 or 6 month LIBOR loans with an applicable margin of 4.0% per annum. At December 31, 2016, the interest rate in effect was 4.2%. Availability under the Credit Agreement is calculated as 85% of the book value of eligible accounts at such time multiplied by a liquidity factor, less any reserves established by the Agent. We had $3.2 million outstanding loans under our GE Capital Credit Agreement at December 31, 2015. The Company paid the balance of $3.2 million on August 24, 2015. In accordance with the Credit Agreement, the Company is required to provide the Lenders information related to working capital by which the Lenders will calculate the available line of credit, defined in the agreement as the Borrowing Base Certificate.
The Credit Agreement was amended on September 16, 2015 to reduce the unused line fee from 0.5% to 0.375%.
The term of the Credit Agreement is up to five years from November 18, 2014. The Credit Agreement contains customary representations and warranties and customary affirmative and negative covenants, including, among others, covenants that limit or restrict the Company’s ability to incur indebtedness, grant liens, merge or consolidate, dispose of assets, make investments, make acquisitions, enter into certain transactions with affiliates, pay dividends or make distributions, or repurchase stock, in each case, subject to customary exceptions for a loan facility of this size and type. In addition, the Credit Agreement contains customary events of default (subject to customary cure periods for certain events of default), including, among others, non-payment, inaccuracy of representations and warranties, covenant defaults, cross-default to material agreements, cross-default to material indebtedness, bankruptcy and insolvency and material judgment defaults. The Company must meet certain financial reporting and audit requirements, as defined by the credit agreement. In December 2015, the Company notified the bank of the termination of the line of credit. On February 10, 2016, the Company formally terminated the line of credit. The Company was in compliance with all covenants of the Credit Agreement for the borrowing period up to February 10, 2016.
F-27
Debt issuance costs of $432,000 were being amortized over the life of the Credit Agreement. The Company included the unamortized financing costs at December 31, 2015 in the amount of $351,000 in interest expense, net in 2015.
9. Stock-Based Compensation
The 1999 Director Stock Option Plan, as amended (the “Director Plan”), provides for the grant of stock options to non-employee directors of the Company at an exercise price equal to the fair market value per share on the date of the grant. An aggregate of 1,975,000 shares have been approved and authorized for issuance pursuant to the Director Plan. A total of 2,634,798 options have been granted to non-employee directors through December 31, 2016 and 807,782 of those have been forfeited through December 31, 2016 and returned to the option pool for future issuance. The options granted under the Director Plan vest in full one year after their respective grant dates and have a maximum term of ten years. As of December 31, 2016, there were 650,000 shares of common stock options outstanding. As of December 31, 2016, the 147,984 options available were transferred to a plan that has superseded the Director Plan, as discussed further in this section.
The 1999 Stock Incentive Plan, as amended (“1999 Plan”), replaced all previously authorized employee stock option plans, and no additional options may be granted under those previous plans. Under the 1999 Plan, options or stock awards may be granted to all of the Company’s employees, officers, directors, consultants and advisors to purchase a maximum of 3,200,000 shares of common stock. However, pursuant to the terms of the 1999 Plan, no awards may be granted after March 16, 2009. A total of 2,892,500 options, having a maximum term of ten years, have been granted at 100% of the fair market value of the Company’s common stock at the date of grant. Options outstanding under the 1999 Plan are generally exercisable in cumulative increments over four years commencing one year from date of grant.
On June 26, 2009, the Board of Directors adopted, and the Company’s stockholders subsequently approved by partial written consent, the IGI Laboratories, Inc. 2009 Equity Incentive Plan (the “2009 Plan”). The 2009 Plan became effective on July 29, 2009. The 2009 Plan allows the Company to continue to grant options and restricted stock, as under the 1999 Plan, but also authorizes the Board of Directors to grant a broad range of other equity-based awards, including stock appreciation rights, restricted stock units ("RSUs") and performance awards. The 2009 Plan has been created, pursuant to and consistent with the Company’s current compensation philosophy, to assist the Company in attracting, retaining and rewarding designated employees, directors, consultants and other service providers of the Company and its subsidiaries and affiliates, in a manner that will be cost efficient to the Company from both an economic and financial accounting perspective. On April 12, 2010, the Board of Directors adopted, and the Company’s stockholders subsequently approved, an amendment and restatement of the 2009 Plan to increase the number of shares of Common Stock available for grant under such plan by adding 2,000,000 shares of Common Stock. The 2009 Plan, as amended on May 29, 2010, authorizes up to 5,000,000 shares of the Company’s common stock for issuance pursuant to the terms of the 2009 Plan. The maximum number of shares that may be subject to awards made to any individual in any single calendar year under the 2009 Plan is 1,000,000 shares. As of December 31, 2016, there were 179,900 RSUs outstanding, 1,341,746 shares of stock outstanding, and 3,216,369 shares of common stock options outstanding. As of December 31, 2016, the 92,883 options available were transferred to a plan that has superseded the 2009 Plan, as discussed further in this section.
On May 25, 2016, the Board of Directors approved the Company's 2016 Equity Incentive Plan (the "2016 Plan"). The 2016 Plan provides for the issuance of awards of up to 2,000,000 shares of the Company's common stock, plus any shares of common stock that are represented by awards granted under our Director Plan and 2009 Plan that are forfeited, expire or are canceled without delivery of shares of common stock or which result in the forfeiture of shares of common stock back to the Company on or after May 25, 2016. Generally, shares of common stock reserved for awards under the 2016 Plan that lapse or are canceled, will be added back to the share reserve available for future awards. However, shares of common stock tendered in payment for an award or shares of common stock withheld for taxes will not be available again for grant. The 2016 Plan provides that no participant may receive awards for more than 1,000,000 shares of common stock in any fiscal year. As the 2016 Plan supersedes both the Director Plan and the 2009 Plan, any available shares from both are now incorporated into the 2016 Plan. As of December 31, 2016, there were 20,000 shares of common stock outstanding and options to purchase 239,000 shares of common stock outstanding. As of December 31, 2016, there were a total of 1,981,867 shares of common stock available under the 2016 Plan.
As of December 31, 2016, there were options to purchase 4,105,369 shares of common stock outstanding collectively in the Director Plan, 2009 Plan, and the 2016 Plan.
In the interest of maintaining consistency with the Company's 2016 Equity Incentive Plan, on March 13, 2017, the Company entered into (i) an amendment to the option agreements governing each option grant currently outstanding under the Company's 2009 Equity Incentive Plan, and (ii) an amendment to the restricted stock unit, or RSU, agreements governing each RSU grant currently outstanding under the 2009 Plan. The amendments provide for the automatic vesting upon a change of control of the
F-28
Company of each option grant and RSU grant, as applicable, outstanding under the 2009 Plan. The forms of amendment are attached hereto as Exhibits 10.31 and 10.32, respectively, and are incorporated by reference herein.
Stock Options
The fair value of each option award is estimated on the date of grant using the Black-Scholes option-pricing formula that uses assumptions noted in the following table. Expected volatilities and risk-free interest rates are based upon the expected life of the grant.
Assumptions | 2016 | 2015 | 2014 | ||||||
Expected dividends | 0 | % | 0 | % | 0 | % | |||
Risk free rate | 1.14 | % | 1.11 | % | 0.74% – 1.2% | ||||
Expected volatility | 68.0% - 71.3% | 52.7% - 68.3% | 44.0% - 53.0% | ||||||
Expected term (in years) | 3.1 – 3.3 years | 3.2 – 3.3 years | 3.2 - 3.3 years | ||||||
Estimated volatility was calculated using the historical volatility of the Company’s stock over the expected life of the options. The expected life of the options was estimated based on the Company’s historical data. The forfeiture rates are estimated based on historical employment/directorship termination experience. The risk free interest rate is based on U.S. Treasury yields for securities with terms approximating the terms of the grants. The assumptions used in the Black-Scholes option valuation model are highly subjective, and can materially affect the resulting valuation.
Stock option transactions in each of the past three years under the aforementioned plans in total were:
Shares | Exercise Price Per Share | Weighted Average Exercise Price | ||||||||
January 1, 2014 shares issuable under options | 2,643,500 | $ .55 - $3.03 | $ | 1.12 | ||||||
Granted | 397,500 | 2.96 - 10.55 | 5.89 | |||||||
Exercised | (443,166 | ) | .55 - 1.95 | 1.15 | ||||||
Expired | — | — | — | |||||||
Forfeited | (161,000 | ) | 1.10 - 5.65 | 2.71 | ||||||
December 31, 2014 shares issuable under options | 2,436,834 | .76 - 10.55 | 1.79 | |||||||
Granted | 1,357,000 | 5.55 - 10.67 | 9.20 | |||||||
Exercised | (75,766 | ) | .76 - 3.62 | 1.10 | ||||||
Expired | — | — | — | |||||||
Forfeited | (125,334 | ) | 1.40 – 10.67 | 8.99 | ||||||
December 31, 2015 shares issuable under options | 3,592,734 | .79 - 10.67 | 4.36 | |||||||
Granted | 739,135 | 4.72 - 8.81 | 7.26 | |||||||
Exercised | (61,834 | ) | 1.10 - 6.51 | 1.54 | ||||||
Expired | — | — | — | |||||||
Forfeited | (164,666 | ) | 4.55 - 10.67 | 8.37 | ||||||
December 31, 2016 shares issuable under options | 4,105,369 | $ .79 - $10.67 | 4.76 | |||||||
The following table summarizes information concerning outstanding and exercisable options as of December 31, 2016:
F-29
Options Outstanding | Options Exercisable | |||||||||||||||
Range of Exercise Price | Number of Options | Weighted Average Remaining Life (Years ) | Weighted Average Exercise Price | Number of Options | Weighted Average Exercise Price | |||||||||||
$0.79 to $1.00 | 50,000 | 3.01 | $ | 0.79 | 50,000 | $ | 0.79 | |||||||||
1.01 to 1.50 | 1,808,400 | 5.11 | 1.07 | 1,808,400 | 1.07 | |||||||||||
1.51 to 10.67 | 2,246,969 | 8.35 | 7.82 | 805,803 | 7.15 | |||||||||||
$0.79 to $10.67 | 4,105,369 | 6.86 | $ | 4.76 | 2,664,203 | $ | 2.90 | |||||||||
The following table summarizes information concerning outstanding and exercisable options as of December 31, 2015:
Options Outstanding | Options Exercisable | |||||||||||||||
Range of Exercise Price | Number of Options | Weighted Average Remaining Life (Years ) | Weighted Average Exercise Price | Number of Options | Weighted Average Exercise Price | |||||||||||
$0.79 to $1.00 | 50,000 | 4.01 | $ | 0.79 | 50,000 | $ | 0.79 | |||||||||
1.01 to 1.50 | 1,862,400 | 6.14 | 1.07 | 1,851,400 | 1.07 | |||||||||||
1.51 to 10.67 | 1,680,334 | 8.96 | 8.10 | 289,997 | 4.02 | |||||||||||
$0.79 to $10.67 | 3,592,734 | 7.43 | $ | 4.36 | 2,191,397 | $ | 1.45 | |||||||||
The following table summarizes information concerning outstanding and exercisable options as of December 31, 2014:
Options Outstanding | Options Exercisable | |||||||||||||||
Range of Exercise Price | Number of Options | Weighted Average Remaining Life (Years ) | Weighted Average Exercise Price | Number of Options | Weighted Average Exercise Price | |||||||||||
$0.76 to $1.00 | 97,000 | 3.04 | $ | 0.78 | 97,000 | $ | 0.78 | |||||||||
1.01 to 1.50 | 1,887,500 | 7.14 | 1.07 | 1,448,664 | 1.08 | |||||||||||
1.51 to 10.55 | 452,334 | 8.68 | 5.02 | 110,333 | 1.78 | |||||||||||
$0.76 to $10.55 | 2,436,834 | 7.26 | $ | 1.79 | 1,655,997 | $ | 1.11 | |||||||||
The Company has recorded an aggregate of $2.3 million, $1.7 million and $0.3 million related to its stock option based expenses in cost of sales, product development and research expenses, and selling, general and administrative expenses on the accompanying Consolidated Statements of Operations for the years ended December 31, 2016, 2015 and 2014, respectively.
The aggregate intrinsic value of options outstanding was $11.5 million at December 31, 2016, $17.4 million at December 31, 2015 and $17.1 million at December 31, 2014. The aggregate intrinsic value of the options exercisable was $11.3 million at December 31, 2016, $16.3 million at December 31, 2015 and $12.7 million at December 31, 2014. The total intrinsic value of the options exercised during 2016, 2015 and 2014 was $0.3 million, $0.6 million and $3.4 million, respectively.
A summary of non-vested options at December 31, 2016 and changes during the year ended December 31, 2016 is presented below:
F-30
Options | Weighted Average Grant Date Fair Value | ||||||
Non-vested options at January 1, 2016 | 1,401,337 | $ | 3.60 | ||||
Granted | 739,135 | 3.45 | |||||
Vested | (574,469 | ) | 3.46 | ||||
Forfeited | (124,837 | ) | 3.42 | ||||
Non-vested options at December 31, 2016 | 1,441,166 | $ | 3.58 | ||||
As of December 31, 2016, there was $3.4 million of total unrecognized compensation cost related to non-vested share-based compensation arrangements under the Plan. The costs will be recognized through December 2018.
Restricted Stock and RSUs
The Company periodically grants restricted stock and RSU awards to certain officers and other employees that typically vest one to three years from their grant date. On December 30, 2013, in accordance with the terms of the employment agreement between Jason Grenfell-Gardner, President and CEO, and the Company executed on July 30, 2012, a restricted stock award in the amount of 325,000 shares was granted to Jason Grenfell-Gardner, with one third of the shares of restricted stock vested on December 31, 2013, and the remaining two thirds of the shares of restricted stock vesting in equal amounts on July 30, 2014 and July 30, 2015. The Company recognized $0.8 million, $0.6 million and $0.5 million, respectively, of compensation expense during the years ended December 31, 2016, 2015 and 2014 related to restricted stock awards and RSUs. Stock compensation expense is recognized over the vesting period of the restricted stock and RSUs. At December 31, 2016, the Company had approximately $1.0 million of total unrecognized compensation cost related to non-vested restricted stock and RSUs, all of which will be recognized through September 2018.
A summary of non-vested shares of restricted stock and changes during each of the past three years is as follows:
Number of Restricted Stock | Weighted Average Issuance Price | ||||||
Non-vested balance at January 1, 2014 | 246,001 | $ | 2.64 | ||||
Changes during the period: | |||||||
Shares granted | — | — | |||||
Shares vested | (137,667 | ) | 2.46 | ||||
Shares forfeited | — | ||||||
Non-vested balance at January 1, 2015 | 108,334 | $ | 2.86 | ||||
Changes during the period: | |||||||
Shares granted | 32,500 | 10.67 | |||||
Shares vested | (140,834 | ) | 4.66 | ||||
Shares forfeited | — | ||||||
Non-vested balance at December 31, 2015 | — | $ | — | ||||
There has been no activity in the year ended 2016. A summary of non-vested RSUs and changes during each of the past three years is as follows:
F-31
Number of RSUs | Weighted Average Issuance Price | ||||||
Non-vested balance at January 1, 2015 | — | $ | — | ||||
Changes during the period: | |||||||
Shares granted | 230,250 | 10.32 | |||||
Shares vested | (32,500 | ) | 10.67 | ||||
Shares forfeited | (15,000 | ) | 10.67 | ||||
Non-vested balance at December 31, 2015 | 182,750 | $ | 10.23 | ||||
Changes during the period: | |||||||
Shares granted | 58,068 | 7.50 | |||||
Shares vested | (60,918 | ) | 10.13 | ||||
Shares forfeited | — | — | |||||
Non-vested balance at December 31, 2016 | 179,900 | $ | 9.35 | ||||
There was no RSU activity for the year ended December 31, 2014.
10. Stock Warrants
Stock warrants as of December 31, 2016, 2015 and 2014 consisted of:
2016 | 2015 | 2014 | |||||||||||||||||||
Warrants | Weighted Average Exercise Price | Warrants | Weighted Average Exercise Price | Warrants | Weighted Average Exercise Price | ||||||||||||||||
Beginning balance | — | $ | — | 84,000 | $ | 1.21 | 354,546 | $ | 1.21 | ||||||||||||
Stock warrants granted | — | — | — | — | — | — | |||||||||||||||
Stock warrants expired | — | — | (16,364 | ) | 1.21 | — | — | ||||||||||||||
Stock warrants exercised | — | — | (67,636 | ) | 1.21 | (270,546 | ) | 1.21 | |||||||||||||
Ending balance | — | $ | — | — | $ | — | 84,000 | $ | 1.21 | ||||||||||||
In connection with the private placement of the Company’s Common Stock on December 8, 2010, the Company granted common stock warrants to purchase up to 338,182 and 16,364 shares, respectively, to each of its two placement agents for $1.21 per share which expired on December 8, 2015. There were no stock warrant activities during 2016 and no outstanding warrants as of December 31, 2016.
11. Income Taxes
During the year ended December 31, 2016, the Company began significant operations in certain foreign countries and is, accordingly, subject to tax in those foreign jurisdictions.
Income (Loss) before income tax for the years ended December 31, 2016, 2015 and 2014 consisted of the following (in thousands):
F-32
2016 | 2015 | 2014 | ||||||||||
(in thousands) | ||||||||||||
U.S. operations | (9,514 | ) | 6,911 | 5,424 | ||||||||
Foreign operations | (2,184 | ) | (208 | ) | — | |||||||
Global Total | $ | (11,698 | ) | $ | 6,703 | $ | 5,424 | |||||
The Company’s current tax expense was $287,000, $35,000 and $173,000 for the years ended December 31, 2016, 2015 and 2014, respectively. The provision for income taxes attributable to continuing operations before income taxes for the years ended December 31, 2016, 2015 and 2014 is as follows (in thousands):
2016 | 2015 | 2014 | ||||||||||
Current tax expense (benefit): | ||||||||||||
Federal | $ | 26 | $ | — | $ | 97 | ||||||
State and local | 35 | 19 | 76 | |||||||||
Foreign | 272 | 28 | ||||||||||
Total current tax expense | 333 | 47 | 173 | |||||||||
Deferred tax expense: | ||||||||||||
Federal | — | — | — | |||||||||
State and local | — | — | — | |||||||||
Foreign | (46 | ) | (12 | ) | ||||||||
Total deferred tax expense | (46 | ) | (12 | ) | — | |||||||
Total income tax expense | $ | 287 | $ | 35 | $ | 173 | ||||||
The provision for (benefit from) income taxes differed from the amount of income taxes determined by applying the applicable federal tax rate (34%) to pretax income (loss) from continuing operations as a result of the following (in thousands):
2016 | 2015 | 2014 | ||||||||||
Expected Statutory expense (benefit) | $ | (3,977 | ) | $ | 2,244 | $ | 1,844 | |||||
Change in the fair values of derivative and amortization of debt discount | 2,584 | (5,597 | ) | (693 | ) | |||||||
Other non-deductible expenses | 63 | 7 | 3 | |||||||||
Change in valuation allowance | 590 | 3,254 | (1,031 | ) | ||||||||
Rate differential - foreign vs. US | 822 | 114 | — | |||||||||
State income taxes, net of federal benefit | 23 | 13 | 50 | |||||||||
Federal tax impact of state tax benefit, net | 154 | — | — | |||||||||
Exchange gain | $ | 28 | $ | — | $ | — | ||||||
$ | 287 | $ | 35 | $ | 173 | |||||||
Deferred tax assets included in the Consolidated Balance Sheets as of December 31, 2016 and 2015 consisted of the following:
F-33
2016 | 2015 | |||||||
(in thousands) | ||||||||
Current Assets: | ||||||||
Allowance for doubtful accounts | $ | 118 | $ | 6 | ||||
Inventory reserve | 467 | 157 | ||||||
Accrued expenses | 831 | 1,005 | ||||||
Total current assets | 1,416 | 1,168 | ||||||
Long Term Assets (Liabilities): | ||||||||
Property, plant and equipment | 317 | 348 | ||||||
Intangible assets | (205 | ) | (256 | ) | ||||
Tax operating loss carry forwards | 10,962 | 11,283 | ||||||
Tax credit carry forwards | 254 | 291 | ||||||
Non-employee stock options | 2,302 | 1,238 | ||||||
Other | (1 | ) | (7 | ) | ||||
Total Long Term Assets (Liabilities) | 13,629 | 12,897 | ||||||
Gross Deferred Tax Asset (Liability) | 15,045 | 14,065 | ||||||
Less Valuation Allowance | (15,250 | ) | (14,309 | ) | ||||
Deferred taxes, net | $ | (205 | ) | $ | (244 | ) | ||
The Company evaluates the recoverability of its net deferred tax assets based on its history of operating results, its expectations for the future, and the expiration dates of the net operating loss carry forwards. Based on the preponderance of the evidence, the Company has concluded that it is more likely than not that it will be unable to realize the net deferred tax assets in the immediate future and has established a valuation allowance for all such net deferred tax assets. Accordingly, the Company has provided a valuation allowance of $15.3 million and $14.3 million for the years ended December 31, 2016 and 2015, respectively, on its net deferred tax assets. The valuation allowance increased during the year 2015 by $1.0 million related to changes in deferred tax assets for the year ended December 31, 2016.
Operating loss and tax credit carry forwards as of December 31, 2016 were as follows:
2016 | 2015 | |||||||
(in thousands) | ||||||||
Federal: | ||||||||
Net operating losses (expiring through 2035) | $ | 31,336 | $ | 32,870 | ||||
Research tax credits (expiring through 2025) | 168 | 168 | ||||||
Alternative minimum tax credits (available without expiration) | 86 | 70 | ||||||
State: | ||||||||
Net Operating Losses: | ||||||||
Tennessee (expiring in 2030) | 529 | 568 | ||||||
New Jersey (expiring in 2035) | 1,764 | 822 | ||||||
Illinois (expiring in 2035) | 222 | 255 | ||||||
Foreign | ||||||||
Net operating losses (no expiration) | 232 | 10 | ||||||
At December 31, 2016, the Company’s U.S. federal net operating loss carryforwards will expire as follows:
F-34
Year | Net Operating Loss (in thousands) | ||
2017 - 2021 | $ | 7,187 | |
2022 - 2026 | 2,268 | ||
2027 - 2031 | 11,373 | ||
2032 and thereafter | 10,508 | ||
Total | $ | 31,336 | |
The above excludes net operating losses of $3.3 million which, if realized would be accounted for as additional paid-in capital.
The Company’s ability to use net operating loss carry forwards is subject to substantial limitation in future periods under certain provisions of Section 382 of the Internal Revenue Code of 1986, as amended, which limit the utilization of net operating losses upon a more than 50% change in ownership of the Company’s stock that is held by 5% or greater stockholders. The Company examined the application of Section 382 with respect to an ownership change that took place during 2010, as well as the limitation on the application of net operating loss carry forwards. The Company believes that operating losses subsequent to the change date in 2010 (aggregating $15.3 million) are not subject to Section 382 limitations. The Company has estimated that the annual limitation starting in 2010 aggregates from $1.0 million to $2.3 million per year including the effect of amortization of built in gains. The Company's loss carryforwards may be further limited in the future if additional ownership changes occur.
ASU 2016-09 “Improvements to Employee Share-Based Payment Accounting” issued by FASB, allows recognition of windfall profits as a tax benefit, either as a reduction of current income tax expense or (as in the case of the Company) a deferred tax asset resulting from an increase in net operating loss carryforwards. The effective date for implementation of this change is for years beginning after December 15, 2016 with a cumulative adjustment to retained earnings as of the beginning of the year of adoption. The Company does not anticipate any adjustment to retained earnings related to the increase in net operating losses since deferred tax assets related to net operating losses are fully reserved. The Company had previously recognized $122,000 of windfall tax benefits in additional paid in capital and will adjust retained earnings by that amount during the first quarter of 2017.
The Company is subject to the provisions of ASC 740-10-25, Income Taxes (ASC 740). ASC 740 prescribes a more likely-than-not threshold for the financial statement recognition of uncertain tax positions. ASC 740 clarifies the accounting for income taxes by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. On a quarterly basis, the Company undergoes a process to evaluate whether income tax accruals are in accordance with ASC 740 guidance on uncertain tax positions. For federal purposes, post 1998 tax years remain open to examination as a result of net operating loss carryforwards. The Company is currently open to audit by the appropriate state income taxing authorities for tax years 2012 through 2015. The Company has not recorded any liability for uncertain tax positions at December 31, 2016 or December 31, 2015.
12. Lease Commitments
The Company’s commitments and contingencies consisted of operating leases for warehouse and office space and equipment. Future minimum lease payments under non-cancelable operating leases are as follows:
Commitments | |||
2017 | $ | 548 | |
2018 | 458 | ||
2019 | 421 | ||
2020 | 426 | ||
2021 | 392 | ||
Thereafter | 742 | ||
$ | 2,987 | ||
Rent expense was $934,400, $360,000 and $216,200 for the years ended December 31, 2016, 2015 and 2014, respectively.
F-35
13. Legal and U.S. Regulatory Proceedings
On March 2, 2001, the Company became aware of environmental contamination resulting from an unknown heating oil leak at its former manufacturing facility. The Company immediately notified the New Jersey Department of Environmental Protection (“NJ DEP”) and the local authorities, and hired a contractor to assess the exposure and required clean up. The total estimated costs for the clean-up and remediation is $889,000, of which approximately $122,000 remains accrued as of December 31, 2016. Based on information provided to the Company from its environmental consultant and what is known to date, the Company believes the reserve is sufficient for the remaining remediation of the environmental contamination. There is a possibility, however, that the remediation costs may exceed the Company’s estimates.
The restricted cash, included in other assets on the Consolidated Balance Sheet of $122,000 as of December 31, 2016 and $124,000 as of December 31, 2015 represents a restricted escrow account set up on the requirement of the NJ DEP for the soil remediation work. These funds will be released to the Company upon the NJ DEP’s approval when the remediation is completed.
On December 19, 2013, the Company filed a complaint in the United States District Court for the District of Delaware against Mallinckrodt LLC, Mallinckrodt, Inc. and Nuvo Research Inc., collectively refer to as Mallinckrodt, seeking a declaration of non-infringement of United States Patent Nos. 8,217,078 and 8,546,450 so that the Company can bring our generic diclofenac sodium topical solution 1.5% to market at the earliest possible date under applicable statutory and FDA regulatory provisions. On January 10, 2014, Mallinckrodt filed an answer and counterclaim alleging that the Company infringed the patents at issue. On June 26, 2014, the Company entered into a settlement agreement with Mallinckrodt, pursuant to which Mallinckrodt granted us a non-exclusive license to launch the Company’s diclofenac sodium topical solution 1.5% product on March 28, 2015. There was no material impact on the Company’s financial statements as a result of the settlement. We received approval to sell the diclofenac sodium topical solution 1.5% from the FDA in July 2015.
On May 21, 2015, Horizon Pharma Ireland Limited, HZNP Limited and Horizon Pharma USA, Inc. (collectively, “Horizon”) filed a complaint in the United States District Court for the District of New Jersey against the Company alleging infringement of certain United States patents based upon our submission to the FDA of an Abbreviated New Drug Application (“ANDA”) seeking FDA approval to market diclofenac topical solution 2% w/w before the expiration of the patents asserted in the complaint. On June 30, 2015, August 11, 2015, September 17, 2015, October 27, 2015 and February 5, 2016, Horizon filed additional complaints in the United States District Court for the District of New Jersey against the Company alleging infringement of other of its United States patents in relation to the Company’s submission of the same ANDA. On July 21, 2015, September 11, 2015, October 6, 2015, October 21, 2015 and December 17, 2015, and March 17, 2016 the Company filed answers, affirmative defenses and counterclaims with respect to the complaints filed by Horizon. In those filings, the Company asserted that the patents alleged to be infringed in the complaints filed by Horizon are invalid and not infringed by us. On April 27, 2016, Horizon and the Company filed a stipulation of dismissal to dismiss the cases. The court entered an order dismissing the cases on May 2, 2016. On May 9, 2016, Horizon and the Company entered into a settlement agreement. Under the settlement agreement, the Company obtained a license to market diclofenac topical solution 2% no later than January 10, 2029 or earlier in certain circumstances, including the resolution by settlement or court decision of other third party litigation involving diclofenac topical solution 2% or the market entry by other third party generic versions of diclofenac topical solution 2%. At this time, the Company cannot estimate if or when any of those earlier events might occur. No consideration was exchanged as part of the settlement. The Company has not recorded accruals related to this case and has not yet received final approval.
On December 4, 2015, Galderma Laboratories, L.P. and Galderma S.A. (collectively, “Galderma”) filed a complaint in the United States District Court for the Northern District of Texas against the Company alleging infringement of United States Patent No. 6,106,848 based upon the Company’s submission to the FDA of an ANDA seeking FDA approval to market clobetasol propionate lotion 0.05% before the expiration patent asserted in the complaint. On January 5, 2016, Galderma and the Company entered into a Settlement and License Agreement the terms of which are confidential. On January 22, 2016, the case was dismissed with prejudice.
From December 20, 2016 to January 26, 2017, eight putative class action antitrust lawsuits have been filed against Teligent Inc. along with co-defendants including Taro Pharmaceuticals U.S.A., Inc., Perrigo New York Inc., Fougera Pharmaceuticals Inc., and Sandoz, Inc. The actions are currently pending in the District of New Jersey and Eastern District of Pennsylvania, and a consolidation motion is currently pending before the Judicial Panel on Multidistrict Litigation.
The class plaintiffs seek to represent nationwide or state classes consisting of persons who directly purchased, indirectly purchased or reimbursed patients for the purchase of generic econazole from any of the defendants from June 1, 2014 (or later in some complaints) until the time the defendants’ allegedly unlawful conduct ceased or will cease.
The plaintiffs allege a conspiracy to fix prices for generic econazole, in violation of federal antitrust laws or state antitrust, consumer
F-36
protection, and other laws. Plaintiffs seek treble damages for alleged price overcharges for generic econazole during the alleged period of conspiracy, and the indirect purchaser class plaintiffs seek injunctive relief against Teligent.
All of these cases are in their initial stages. The parties are negotiating briefing schedules for motions to dismiss, which will be filed after the Judicial Panel on Multidistrict Litigation determines an appropriate forum. Due to the early stage of these cases, we are unable to form a judgment at this time as to whether an unfavorable outcome is either probable or remote or to provide an estimate of the amount or range of potential loss. We believe these cases are without merit, and we intend to vigorously defend against these claims.
14. Employee Benefits
The Company has a 401(k) contribution plan, pursuant to which employees may elect to contribute to the plan, in whole percentages, up to 100% of compensation. Employees’ contributions are subject to a minimum contribution by participants of 1% of compensation and a maximum contribution of $18,000 for 2016, $18,000 for 2015 and $17,500 for 2014, plus a catch-up contribution of up to $6,000 for 2016, $6,000 for 2015 and $5,500 for 2014, if a participant qualifies. The Company matches 100% of the first 3% of compensation contributed by participants and 50% of the next 2% of compensation contributed by participants. The Company contribution is in the form of cash, which is vested immediately. The Company has recorded charges to expense related to this plan of approximately $228,619, $172,965 and $126,600 in 2016, 2015 and 2014, respectively.
F-37
15. Asset Purchase Agreements
Sebela
On March 31, 2016, the Company entered into an Asset Purchase Agreement and certain other ancillary agreements with Sebela International Limited, an Irish company resident in Bermuda ("Sebela"). The Company acquired all rights, title and interests of Sebela in its existing inventory and certain of its contracts associated with two medical devices, which the Company had previously developed.
The transaction is accounted for as a purchase of the product and product rights, and as such the initial payment and related costs to acquire the assets (excluding inventory) are included as part of product acquisition costs totaling $330,000. The Company will amortize the costs over fifteen years, the useful life of the acquired products and product rights. In addition, the Company purchased approximately $69,000 of inventory related to the products acquired.
Concordia
On October 5, 2015, the Company, together with a wholly-owned subsidiary of the Company incorporated under the laws of Jersey (the “Company Subsidiary”, and together with the Company, the “Purchasers”), entered into an Asset Purchase Agreement (the “Purchase Agreement”) and certain other ancillary agreements with Concordia Pharmaceuticals Inc., S.à.r.l., Barbados Branch (“Concordia”), pursuant to which the Company acquired all rights, title and interests of Concordia in the existing inventory and certain contracts associated with three currently marketed injectable pharmaceutical products (Fortaz®, Zinacef ™ , and Zantac® Injection) (the “Inventory”), and the Company Subsidiary acquired all rights, title and interests of Concordia in, among other things, certain other contracts, product registrations and books and records associated with those products (together with the Inventory and other assets acquired by the Company, the “Purchased Assets”). The transaction was also completed on October 5, 2015.
The transaction is accounted for as a purchase of the product and product rights, and as such the initial payment and related costs to acquire the assets (excluding inventory) are included as part of product acquisition costs totaling $10,100,000. The Company will amortize the costs over fifteen years, the useful life of the acquired products and products rights. In addition, Teligent Pharma Inc. purchased approximately $1,200,000 of inventory related to the three products acquired.
On December 10, 2015, the Company, together with Teligent Jersey Limited, a wholly-owned subsidiary of the Company incorporated under the laws of Jersey (the “Company Subsidiary,” and together with the Company, the “Purchasers”) , entered into a First Amendment to the Asset Purchase Agreement (the “First Amendment”) with Concordia, which amends the Asset Purchase Agreement, dated October 5, 2015, by and between the Purchasers and the Seller (the “Purchase Agreement”). The First Amendment amends the Purchase Agreement to include certain additional intellectual property rights in the Purchased Assets relating to the “TWISTVIAL” trademark. In connection with the First Amendment, on December 10, 2015, the Company Subsidiary also entered into a Trademark Assignment Agreement by and between the Company Subsidiary and the Seller, pursuant to which the Seller sold, conveyed, assigned, transferred and delivered the “TWISTVIAL” trademark associated with certain of the Purchased Assets.
AstraZeneca
On September 24, 2014, the Company entered into an Asset Purchase Agreement with AstraZeneca, in which the Company acquired all rights, titles and interests of AstraZeneca and its affiliates in Abbreviated New Drug Applications and New Drug Applications associated with eighteen products (collectively the “Purchased Regulatory Approvals”) and certain documents relating thereto (together with the Purchased Regulatory Approvals, the “Purchased Assets”).
The transaction was accounted for as a purchase of the product and product rights, and as such the initial payment, related costs to acquire the assets, and a milestone payment were all included as part of product acquisition costs totaling $6.9 million. In addition, the Company agreed to pay, for each product manufactured by the Company pursuant to a Purchased Regulatory Approval, a royalty on future gross profits from product sales. Notwithstanding the foregoing, as amended in the First Amendment to the Asset Purchase Agreement by and between the Company and AstraZeneca, dated November 13, 2015, the Company at any time prior to June 30, 2016, could satisfy in full its royalty obligations with a single payment of $3.0 million. The Company elected to make the single payment as of June 30, 2016, bringing the total costs related to the asset purchase to $9.9 million and relieving the Company from any future royalties to AstraZeneca. The Company will amortize the costs over fifteen years, the useful life of the acquired products and product rights, commencing when the product can be sold.
F-38
Valeant
On September 30, 2014, the Company entered into two Asset Purchase Agreements (each, a “Valeant Purchase Agreement” and together, the “Valeant Asset Purchase Agreements”) one with Valeant Pharmaceuticals North America LLC and one with Valeant Pharmaceuticals Luxembourg SARL (together, “Valeant”), pursuant to which the Company acquired all rights, titles and interests of Valeant and their respective affiliates in Abbreviated New Drug Applications and New Drug Applications associated with two products (collectively, the “Valeant Purchased Regulatory Approvals”) and certain documents relating thereto (together with the Valeant Purchased Regulatory Approvals, the “Valeant Purchased Assets”). Pursuant to the terms of the Valeant Asset Purchase Agreements, the Company also acquired the option (each, an “Option” and, collectively, the “Options”) to purchase Abbreviated New Drug Applications and New Drug Applications associated with three additional products (the “Additional Assets”).
The transaction is accounted for as a purchase of the product and product rights, and as such the initial payment and related costs to acquire the asset are included as part of product acquisition costs totaling $3,565,000. The Company will amortize the costs over fifteen years, the useful life of the acquired products and products rights, commencing when the product can be sold. In consideration for the purchase of the Valeant Purchased Assets, the Company paid Valeant an aggregate of $1,500,000 in cash. In consideration for the purchase of the Additional Assets, the Company may exercise any Option, in its sole discretion, and pay $750,000 for each of two additional products and $500,000 for one additional product, for a total aggregate of $2,000,000 if all Options are exercised. The Company exercised its Option and purchased the one additional product for $0.5 million on November 18, 2014. On March 27, 2015, the Company exercised its Option and purchased the two additional products for a total of $1.5 million in cash. The transaction is accounted for as a purchase of the product and product rights and, as such, the initial payment and related costs to acquire the Valeant Purchased Assets are included as part of product acquisition costs totaling $3.5 million. The Company will amortize the costs over fifteen years, the useful life of the acquired product and product rights, commencing when the product can be sold.
F-39
16. Quarterly Results (Unaudited)
The following is a summary of certain quarterly financial information for the fiscal years 2016 and 2015:
First Quarter | Second Quarter | Third Quarter | Fourth Quarter | Total | ||||||||||||||||
(in thousands, except per share data) | ||||||||||||||||||||
Year Ended December 31, 2016 | ||||||||||||||||||||
Total revenues, net | $ | 15,657 | $ | 17,138 | $ | 16,151 | $ | 17,935 | $ | 66,881 | ||||||||||
Gross profit | 7,955 | 9,556 | 8,014 | 9,162 | 34,687 | |||||||||||||||
Operating income (loss) | 837 | 1,076 | 303 | 326 | 2,542 | |||||||||||||||
Net loss | (950 | ) | (2,901 | ) | (2,703 | ) | (5,431 | ) | (11,985 | ) | ||||||||||
Net loss attributable to common stockholders | (950 | ) | (2,901 | ) | (2,703 | ) | (5,431 | ) | (11,985 | ) | ||||||||||
Basic loss per share | $ | (0.02 | ) | $ | (0.05 | ) | $ | (0.05 | ) | $ | (0.11 | ) | $ | (0.23 | ) | |||||
Diluted loss per share | $ | (0.02 | ) | $ | (0.05 | ) | $ | (0.05 | ) | $ | (0.11 | ) | $ | (0.23 | ) | |||||
Year Ended December 31, 2015 | ||||||||||||||||||||
Total revenues, net | $ | 10,671 | $ | 8,893 | $ | 11,615 | $ | 13,071 | $ | 44,250 | ||||||||||
Gross profit | 5,628 | 3,666 | 6,077 | 5,944 | 21,315 | |||||||||||||||
Operating income (loss) | 1,098 | (1,911 | ) | 391 | (2,769 | ) | (3,192 | ) | ||||||||||||
Net income (loss) | 6,555 | 9,376 | (2,888 | ) | (6,375 | ) | 6,668 | |||||||||||||
Net income (loss) attributable to common stockholders | 6,555 | 9,376 | (2,888 | ) | (6,375 | ) | 6,668 | |||||||||||||
Basic income (loss) per share | $ | 0.12 | $ | 0.18 | $ | (0.05 | ) | $ | (0.12 | ) | $ | 0.13 | ||||||||
Diluted income (loss) per share | $ | 0.01 | $ | (0.03 | ) | $ | (0.05 | ) | $ | (0.12 | ) | $ | (0.07 | ) | ||||||
F-40
TELIGENT, INC.
SCHEDULE II-VALUATION AND QUALIFYING ACCOUNTS
(in thousands)
Additions | |||||||||||||||||
Balance at Beginning of Year | Charged to Costs and Expenses | Charged other Accounts | Deductions | Balance at End of Year | |||||||||||||
Year Ended December 31, 2014 | |||||||||||||||||
Change in Tax Valuation Allowance | $ | 12,062 | — | — | 1,092 | $ | 10,970 | ||||||||||
Allowance for Doubtful Accounts | $ | 16 | — | — | — | $ | 16 | ||||||||||
Reserve for Inventory Obsolescence | $ | 166 | 228 | (12 | ) | 170 | $ | 212 | |||||||||
Year Ended December 31, 2015 | |||||||||||||||||
Change in Tax Valuation Allowance | $ | 10,970 | — | 3,339 | — | $ | 14,309 | ||||||||||
Allowance for Doubtful Accounts | $ | 16 | 74 | — | — | $ | 90 | ||||||||||
Reserve for Inventory Obsolescence | $ | 212 | 51 | (8 | ) | 134 | $ | 121 | |||||||||
Year Ended December 31, 2016 | |||||||||||||||||
Change in Tax Valuation Allowance | $ | 14,309 | — | 941 | — | $ | 15,250 | ||||||||||
Allowance for Doubtful Accounts | $ | 90 | 347 | 20 | $ | 417 | |||||||||||
Reserve for Inventory Obsolescence | $ | 121 | 777 | — | 583 | $ | 315 | ||||||||||
F-41