Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - PRA Health Sciences, Inc. | prah-20161231ex322f2e48b.htm |
| EX-32.1 - EX-32.1 - PRA Health Sciences, Inc. | prah-20161231ex321d7131d.htm |
| EX-31.2 - EX-31.2 - PRA Health Sciences, Inc. | prah-20161231ex312d7112c.htm |
| EX-31.1 - EX-31.1 - PRA Health Sciences, Inc. | prah-20161231ex311d6191f.htm |
| EX-23.1 - EX-23.1 - PRA Health Sciences, Inc. | prah-20161231ex231e0327b.htm |
| EX-21.1 - EX-21.1 - PRA Health Sciences, Inc. | prah-20161231ex2114c9692.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10‑K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission file number: 001‑36732
PRA Health Sciences, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware |
46‑3640387 |
|
(State or other jurisdiction of |
(I.R.S. Employer |
|
incorporation or organization) |
Identification No.) |
4130 ParkLake Avenue, Suite 400, Raleigh, NC 27612
(Address of principal executive offices) (Zip Code)
(919) 786‑8200
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
|
Name of each exchange on which registered |
|
|
Common Stock, par value $0.01 per share |
|
Nasdaq Global Select Market |
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well‑known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S‑T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S‑K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10‑K or any amendment to this Form 10‑K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non‑accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b‑2 of the Exchange Act.
|
Large accelerated filer ☒ |
Accelerated filer ☐ |
Non-accelerated filer ☐ |
Smaler reporting company ☐ |
|
|
|
(Do not check if a |
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b‑2 of the Act). Yes ☐ No ☒
The aggregate market value of the voting and non‑voting common equity held by non‑affiliates of the registrant, based upon the closing sale price as reported on the Nasdaq Global Select Market on June 30, 2016, the last business day of the registrant’s most recently completed second fiscal quarter, was approximately $1.3 billion. For purposes of this computation, shares of the registrant’s common stock held by each executive officer, director, and each person known to the registrant to own 10% or more of the outstanding voting power have been excluded in that such persons are affiliates.
Indicate the number of shares outstanding of each of the issuer’s classes of Common Stock, as of the latest practicable date.
|
Class |
|
Number of Shares Outstanding |
|
Common Stock $0.01 par value |
61,655,141 shares outstanding as of February 17, 2017 |
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission relating to the 2017 Annual Meeting of Stockholders are incorporated herein by reference into Part III of this Annual Report on Form 10‑K to the extent stated herein. Such Proxy Statement will be filed with the Securities and Exchange Commission within 120 days after the end of the fiscal year to which this report relates.
PRA HEALTH SCIENCES, INC.
ANNUAL REPORT ON FORM 10‑K
FOR FISCAL YEAR ENDED DECEMBER 31, 2016
|
Item |
|
|
|
Number |
|
Page No. |
|
|
|
|
|
|
|
|
| 2 | ||
| 17 | ||
| 37 | ||
| 37 | ||
| 37 | ||
| 37 | ||
|
|
|
|
|
|
|
|
| 38 | ||
| 39 | ||
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
41 | |
| 61 | ||
| 63 | ||
|
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
108 | |
| 108 | ||
| 108 | ||
|
|
|
|
|
|
|
|
| 109 | ||
| 109 | ||
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
109 | |
|
Certain Relationships and Related Transactions, and Director Independence |
109 | |
| 109 | ||
|
|
|
|
|
|
|
|
| 109 | ||
| 109 | ||
|
|
110 | |
|
|
111 |
i
FORWARD‑LOOKING STATEMENTS
This Annual Report on Form 10‑K contains forward‑looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Such forward‑looking statements reflect, among other things, our current expectations and anticipated results of operations, all of which are subject to known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements, market trends, or industry results to differ materially from those expressed or implied by such forward‑looking statements. Therefore, any statements contained herein that are not statements of historical fact may be forward‑looking statements and should be evaluated as such. Without limiting the foregoing, the words “anticipates,” “believes,” “estimates,” “expects,” “intends,” “may,” “plans,” “projects,” “should,” “targets,” “will” and the negative thereof and similar words and expressions are intended to identify forward‑looking statements. These forward‑looking statements are subject to a number of risks, uncertainties and assumptions, including those described in “Risk Factors” in Part I, Item 1A of this report. Unless legally required, we assume no obligation to update any such forward‑looking information to reflect actual results or changes in the factors affecting such forward‑looking information.
Website and Social Media Disclosure
We use our website (www.prahs.com) and our corporate Twitter account (@PRAHSciences) as channels of distribution of company information. The information we post through these channels may be deemed material. Accordingly, investors should monitor these channels, in addition to following our press releases, Securities and Exchange Commission, or SEC, filings and public conference calls and webcasts. The contents of our website and social media channels are not, however, a part of this report.
1
Overview
We are one of the world’s leading global contract research organizations, or CROs, by revenue, providing outsourced clinical development services to the biotechnology and pharmaceutical industries. We believe we are one of a select group of CROs with the expertise and capability to conduct clinical trials across all major therapeutic areas on a global basis. We have therapeutic expertise in areas that are among the largest in pharmaceutical development, including oncology, central nervous system, inflammation and infectious diseases. We believe we provide our clients with one of the most flexible clinical development service offerings, which includes both traditional, project‑based Phase I through Phase IV services as well as embedded and functional outsourcing services. We believe we further differentiate ourselves from our competitors through our investments in medical informatics and clinical technologies designed to enhance efficiencies, improve study predictability and provide better transparency for our clients throughout their clinical development processes.
We are one of the largest CROs in the world by revenue, focused on executing clinical trials on a global basis. Our global clinical development platform includes approximately 70 offices across North America, Europe, Asia, Latin America, South Africa, Australia and the Middle East and over 13,000 employees worldwide. Since 2000, we have participated in approximately 3,500 clinical trials worldwide, we have worked on marketed drugs across several therapeutic areas and conducted the pivotal or supportive trials that led to U.S. Food and Drug Administration, or FDA, or international regulatory approval of more than 70 drugs.
We believe we are a leader in the transformation of the CRO engagement model via our flexible clinical development service offerings, which include embedded and functional outsourcing services in addition to traditional, project‑based clinical trial services. In September 2013, we completed the acquisition of ReSearch Pharmaceutical Services, or RPS, a global CRO providing clinical development services primarily to large pharmaceutical companies, which provides a highly complementary fit with our historical focus on biotechnology and small‑ to mid‑sized pharmaceutical companies. RPS, now known as our Strategic Solutions offerings, provides Embedded Solutions™ and functional outsourcing services in which our teams are fully integrated within the client’s internal clinical development operations and are responsible for managing functions across the entire breadth of the client’s drug development pipeline. We believe that our Strategic Solutions offerings represent an innovative alternative to the traditional, project‑based approach and allow our clients to maintain greater control over their clinical development processes. Our flexible clinical development service offerings expand our addressable market beyond the traditional outsourced clinical development market to include the clinical development spending that biopharmaceutical companies historically have retained in‑house.
Over the past 30 years, we have developed strong client relationships and have performed services for more than 300 biotechnology and pharmaceutical clients. In the year ended December 31, 2016, we derived 14% of our service revenue from small‑ to mid‑sized pharmaceutical companies, 19% of our service revenue from large biotechnology companies and 15% of our service revenue from all other biotechnology companies. We believe that we have built a reputation as a strategic partner of choice for biotechnology and small‑ to mid‑sized pharmaceutical companies as a result of our competitively differentiated platform and our long‑term track record of serving these companies. We expect to benefit from growth in clinical development investment from these customers given the favorable capital raising environment in recent years. Our acquisition of RPS significantly expanded our relationships with large pharmaceutical companies, which represented 52% of our service revenue for the year ended December 31, 2016 and includes all of the top 15 largest pharmaceutical companies. We believe we are well positioned to broaden our relationships and pursue strategic alliances with these large pharmaceutical companies due to our global presence, broad therapeutic expertise and flexible clinical development service offerings.
CRO Industry
CROs provide drug development services, regulatory and scientific support, and infrastructure and staffing support to provide their clients with the flexibility to supplement their in‑house capabilities or to provide a fully outsourced solution. The CRO industry has grown from providing limited clinical trial services in the 1970s to a full service industry characterized by broad relationships with clients and by service offerings that encompass the entire drug
2
development process. Today, CROs provide a comprehensive range of clinical services, including protocol design and management and monitoring of Phase I through Phase IV clinical trials, data management, laboratory testing, medical and safety reviews and statistical analysis. In addition, CROs provide services that generate high quality and timely data in support of applications for regulatory approval of new drugs or reformulations of existing drugs as well as new and existing marketing claims. CROs leverage selected information technologies and procedures to efficiently capture, manage and analyze the large streams of data generated during a clinical trial.
Drug development processes
Discovering and developing new drugs is an expensive and time‑consuming process and is highly regulated and monitored through approval processes that vary by region. Before a new prescription drug reaches commercialization, it must undergo extensive pre‑clinical and clinical testing and regulatory review, to verify that the drug is safe and effective.
A drug is first tested in pre‑clinical studies, which can take several years to complete. When a new molecule is synthesized or discovered, it is tested for therapeutic value using various animal and tissue models. If the drug warrants further development, additional studies are completed and an investigational new drug application, or IND, is submitted to the FDA. Once the IND becomes effective, the drug may proceed to the human clinical trial phase which generally consists of the following interrelated phases, which may overlap:
Stages of Clinical Development
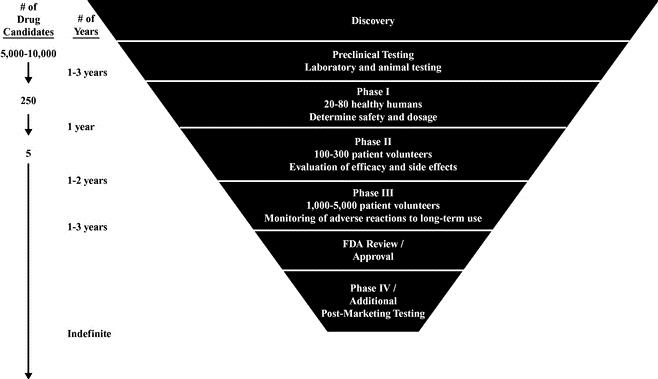
Market trends
Industry Standard Research, or ISR, a market research firm, estimated in its “2016 CRO Market Size Projections 2015-2020” report, or ISR 2016 Market Report, that the size of the worldwide CRO market was approximately $28 billion in 2015 and will grow at a 7% CAGR to $38 billion in 2020. This growth will be driven by an increase in the amount of research and development expenditure and levels of clinical development outsourcing by biopharmaceutical companies.
Increased R&D spending
ISR estimates in the ISR 2016 Market Report that R&D expenditures by biopharmaceutical companies were approximately $263 billion in 2015 and will grow approximately 3% per year through 2020. Of this amount, approximately $108 billion was spent on development, including $77 billion on Phase I through IV clinical development. Growth drivers of R&D spending among biopharmaceutical companies include the need to replenish lost
3
revenues resulting from the patent expirations of a large number of high‑profile drugs in recent years and, a robust capital raising environment among biotechnology companies.
|
· |
Patent Expirations—Since 2012 a significant bolus of branded drugs have lost patent protection which represents in aggregate an estimated $84 billion in revenue. This surge of patent expirations has resulted in the need for biopharmaceutical companies to increase their R&D expenditures to eventually fill this revenue void with new drug approvals. |
|
· |
Biotechnology Capital Raising—According to BioWorld, over $23.8 billion has been raised by biotechnology companies for the year to date period ending on September 18, 2014. We believe these biotechnology companies primarily use the capital to fund clinical trials, and due to the general lack of existing infrastructure, these trials are often contracted to CROs. We expect the favorable capital raising environment will continue to be a source of strong growth for R&D spending. |
The expected increase in R&D expenditures is supported by the recent increase in IND submissions, which will lead to higher clinical development spending as these compounds move through the drug development process. In 2013, the FDA received approximately 7,000 IND submissions, a 17% increase from the approximately 6,000 IND submissions in 2007.
Higher outsourcing penetration
ISR estimates in the ISR 2016 Market Report that approximately 38% of Phase I through IV of clinical development spend is outsourced to CROs, and the levels of penetration are expected to increase to approximately 44% by 2020. We believe this increase in outsourcing is due to several factors, including the need to maximize R&D productively, the increasing burden of clinical trial complexity, the desire to pursue simultaneous registration in multiple countries, and strong growth in Phase II through Phase IV trials.
|
· |
Maximizing Productivity and Reducing Cost—Productivity within the biopharmaceutical industry has declined over the past several years and the cost of developing a new drug, which is now estimated to be $1.4 billion per drug, has significantly increased. The combined impact of declining R&D productivity and increased development costs has translated into significant pressure on margins and short‑term earnings for biopharmaceutical companies. We believe that the need for these companies to maximize productivity and lower costs will lead them to increasingly partner with CROs that can improve efficiency, and increase flexibility and speed across their clinical operations. |
|
· |
Increasing Clinical Trial Complexity—Over the last decade, the burden of clinical trial complexity has been increasingly difficult to manage due to requirements from regulatory authorities worldwide for greater amounts of clinical trial and safety data to support the approval of new drugs, and requirements for adherence to increasingly complex and diverse regulations and guidelines. In an effort to minimize potential risks, these regulatory agencies also typically require a greater amount of post‑approval information and monitoring of drugs on the market. To balance the conflicting demands of a growing market with the need to control R&D expenses, biopharmaceutical companies partner with CROs that can provide services designed to generate high quality and timely data in support of regulatory approvals of new drugs or the reformulations of existing drugs as well as support of post‑approval regulatory requirements. |
|
· |
Simultaneous Multi‑Country Registration—Given their desire to maximize efficiency and global market penetration to achieve higher potential returns on their R&D expenditures, biopharmaceutical companies are increasingly pursuing simultaneous, rather than sequential, regulatory new drug submissions and approvals in multiple countries. However, most biotechnology and small‑ to mid‑sized pharmaceutical companies do not possess the capability or capacity to simultaneously conduct large‑scale clinical trials in more than one country. In addition, establishing and maintaining internal global infrastructure to pursue multiple regulatory approvals in different therapeutic categories and jurisdictions can be costly. |
|
· |
Growth in Phase II through Phase IV Trials—Biopharmaceutical companies are also devoting an increasing amount of resources to Phase II through IV trials. According to clinicaltrials.gov, there were approximately 8,300 Phase II through IV trials submitted in 2016, an increase of 11% when compared with the |
4
approximately 7,500 that were submitted in 2011. Complex late‑stage trials, especially those in which sponsors seek to recruit patients with specific conditions on a global basis, are ideally suited for outsourcing to the select group of global CROs with expertise to execute these studies and access to industry leading investigators and trial sites globally. We believe the increase in the quantity and complexity of clinical trials exceeds the capacity and expertise of many biopharmaceutical companies, and is causing them to increasingly seek outsourced solutions. |
Our History and Corporate Information
PRA Health Sciences, Inc. was incorporated in Delaware in June 2013 under the name Pinnacle Holdco Parent, Inc. On December 19, 2013, Pinnacle Holdco Parent, Inc. changed its name to PRA Global Holdings, Inc. and on July 10, 2014, PRA Global Holdings, Inc. changed its name to PRA Health Sciences, Inc. Our wholly‑owned subsidiary, PRA Holdings, Inc., or PRA Holdings, was incorporated in Delaware in July 2007 and its predecessors date back to 1982. Our qualified and experienced clinical and scientific staff has been delivering clinical drug development services to our clients for more than 30 years and our service offerings now encompass the spectrum of the clinical drug development process.
We are a subsidiary of KKR PRA Investors L.P., a Delaware limited partnership controlled by KKR, or KKR PRA Investors.
Our Competitive Strengths
Global CRO platform
We are one of the largest CROs in the world by revenue focused on executing clinical trials on a global basis. Our global clinical development platform includes approximately 70 offices across North America, Europe, Asia, Latin America, South Africa, Australia and the Middle East and over 13,000 employees worldwide. We are dedicated to the seamless execution of integrated clinical trials on multiple continents concurrently. We believe our global presence and scale are important differentiators as biopharmaceutical companies are increasingly focused on greater patient access for increasingly complex clinical trials and gaining regulatory approval for new products in multiple jurisdictions simultaneously.
Broad and flexible service offering
We believe that we are one of a select group of CROs capable of providing both traditional, project‑based CRO services as well as embedded and functional outsourcing services. Our broad and flexible service offering allows us to meet the clinical research needs of a wide range of clients, from small biotechnology companies to large pharmaceutical companies. Through more than 30 years of experience, we have developed significant expertise executing complex drug development projects that span Phase I through Phase IV clinical trials. Our Product Registration offerings consist primarily of traditional, project‑based CRO services, where we have gained the reputation as a strategic partner of choice to biotechnology and pharmaceutical companies. Our Strategic Solutions offerings primarily cater to the needs of large pharmaceutical companies that seek to maintain greater control over their clinical trial processes.
Therapeutic expertise in large segments of drug development
Our therapeutic expertise encompasses areas that are among the largest in pharmaceutical development, including oncology, central nervous system, inflammation and infectious diseases. We have participated in more than 2,100 clinical trials in these key areas since 2005, accounting for a substantial majority of our total clinical trials during this period. We employ drug development experts with extensive experience across numerous therapeutic areas in preparing development plans, establishing study and protocol designs, identifying investigative sites and patients and submitting regulatory filings. Our staff is highly experienced and includes approximately 620 Ph.Ds, 600 medical doctors and 250 doctors of pharmacy worldwide.
Innovative approach to clinical trials using medical informatics
We are committed to being an industry leader in developing global, scalable and sustainable solutions for our clients. We aim to continuously improve our systems and processes by investing in medical informatics, technology,
5
analytics and IT infrastructure. Our information delivery system enables rapid, web‑based delivery of clinical trial data to clients and project teams. We believe our proprietary analysis and application of this data are key differentiators and allow us to identify more productive investigative sites and speed up overall patient enrollment, thereby decreasing drug development timelines. We have invested in and acquired large databases of aggregated patient medical data, which we refer to as medical informatics, to better understand patient distribution and location. Specifically, we have acquired data sources that give us significant amounts of information about patient populations within the United States to enhance enrollment, including medical claims data, hospital master charge data, pharmacy data, laboratory data and payor data. Capitalizing on our investments in medical informatics, we have the capability to identify potential patient populations by location, diagnostic code, treating physician, medications, date diagnosed, last treatment and other relevant metrics. Our medical informatics suite includes physician, hospital and pharmacy databases that cover more than 280 million patient lives and approximately 10 billion patient and pharmacy claims in the United States.
Diversified and attractive client base
Over the past 30 years, we have performed services for more than 300 biotechnology and pharmaceutical clients. We believe we are one of a select group of global, large scale CROs with a long‑term track record serving biotechnology and small‑ to mid‑sized pharmaceutical companies, and we believe that these companies represent an attractive growth opportunity. In the year ended December 31, 2016, we derived 14% of our service revenue from small‑ to mid‑sized pharmaceutical companies, 19% of our service revenue from large biotechnology companies and 15% of our service revenue from all other biotechnology companies. Going forward, we believe that we will benefit from growth in clinical development investment from these customers that has resulted from the active capital raising environment over the past several years. In addition, our acquisition of RPS significantly expanded our relationships and positioned us to pursue strategic alliances with large pharmaceutical companies, which currently include all of the top 15 largest pharmaceutical companies. Our client relationships are also broad and diversified, and in the year ended December 31, 2016 our top 10 clients represented 66% of service revenue, with our largest client representing approximately 11% of service revenue and our largest single study accounting for approximately 4% of our service revenue.
Innovative management team
We are led by a dedicated and experienced executive management team that has an average of 20 years of experience across the global clinical research, pharmaceutical and life sciences industries. This team has been responsible for building our global platform, successfully integrating our acquisitions, developing our advanced IT‑enabled infrastructure and realizing our significant growth in revenue and earnings over the past five years.
Our Growth Strategy
Leverage our strong market position within the biotechnology and small‑ to mid‑sized pharmaceutical market
We believe our long‑term track record serving biotechnology and small‑ to mid‑sized pharmaceutical companies has resulted in our earning a reputation as a strategic partner of choice for these companies. We believe that biotechnology and small‑ to mid‑sized pharmaceutical companies rely on full service CROs to deliver fast, effective and thorough support throughout the clinical development and regulatory processes, as these companies generally lack a global clinical development infrastructure. We intend to leverage our strong relationships with biotechnology and small‑ to mid‑sized pharmaceutical companies to capture additional business from these companies. In particular we believe the CRO strategic alliances that have become prevalent with large pharmaceutical companies over the past several years will increasingly be utilized by biotechnology and small‑ to mid‑sized pharmaceutical companies. We believe we are well positioned to take advantage of these opportunities given the depth of our relationships and our proven track record serving these customers.
Build deeper and broader relationships with large pharmaceutical companies
Large pharmaceutical companies have increasingly focused on partnering with multi‑national CROs that offer a wide array of global therapeutic and service capabilities. We have invested significantly in our global scale and infrastructure over the past several years to enhance our status as a service provider for these companies. Our acquisition of RPS significantly increased the depth of our relationships with large pharmaceutical companies. We intend to expand
6
our relationships beyond the Embedded Solutions provided through our Strategic Solutions offering to include traditional, project‑based clinical trial services.
Expand our leading therapeutic expertise in existing and new areas
We believe that our therapeutic expertise in all clinical phases of drug development is critical to the proper design and management of clinical trials and we intend to continue to capitalize on our strong market positions in several large therapeutic categories. We have established, and will continue to refine, our scientific and therapeutic business development initiatives, which link our organization to key clinical opinion leaders and medical informatics data to more effectively leverage therapeutic expertise throughout our client engagement. Specifically, we believe that oncology, central nervous system, inflammation and infectious diseases, which together represent the majority of all drug candidates currently in clinical development by biotechnology and pharmaceutical companies, will be significant drivers of our growth. In the area of oncology, we believe that the growth of targeted therapies, companion diagnostics and personalized medicine will continue to drive drug development. With the aging demographics we believe we will see significant growth in the area of dementia and Alzheimer’s research and drug development, which is complemented by our specialty and focus in neurology. Additionally, we believe that development of niche therapeutic drugs (orphan drugs) will see considerable growth moving forward and we have a dedicated staff focused on the design and conduct of trials for these drugs.
Continue to realize financial synergies and strategic benefits from recent acquisitions
We believe we will continue to realize financial synergies and strategic benefits from the acquisitions we have completed over the past three years, resulting in additional revenue growth and margin improvements. We have substantially completed the operational integration of these acquisitions, and are in the process of executing our strategy to eliminate redundancies in corporate and overhead functions and achieve cost efficiencies resulting from the scale of the combined business. We believe that our strategic acquisitions are complementary to our customer base and expect to generate incremental revenue growth by cross‑selling our full set of services to our existing and new customers, thereby expanding the scope of our customer relationships and generating additional revenue.
Pursue selective and complementary acquisition strategy
We are a selectively acquisitive company, focused on growing our core service offerings, therapeutic capabilities and geographic reach into areas of high market growth. We have acquired 18 companies since 1997 and have established programs to help us identify acquisition targets and integrate them successfully. Our acquisition strategy is driven by our comprehensive commitment to serve client needs and we are continuously assessing the market for potential opportunities.
Service Offerings
We perform a broad array of services across the spectrum of clinical development programs, from the filing of INDs and similar regulatory applications to conducting all phases of clinical trials. Our core service offerings include:
|
· |
Product Registration, which includes Phase IIb through III product registration trials and Phase IV trials, inclusive of post‑marketing commitments and registries; |
|
· |
Strategic Solutions, which provides Embedded Solutions and functional outsourcing services, in which our teams are fully integrated within the client’s internal clinical development operations and responsible for managing functions across the entire breadth of the client’s drug development pipeline; and |
|
· |
Early Development Services, which includes Phase I through Phase IIa clinical trials and bioanalytical laboratory services. |
We provide many back office services to clients as well, including processing the payments to investigators and volunteers. We also collaborate with third‑party vendors for services such as imaging, central lab and patient recruitment services.
7
Product Registration
Product Registration encompasses the design, management and implementation of study protocols for Phase II through Phase III clinical trials, which are the critical building blocks of product development programs, as well as Phase IV, or post‑approval, clinical trials. We have extensive resources and expertise to design and conduct studies on a global basis, develop integrated global product databases, collect and analyze trial data and prepare and submit regulatory submissions in the United States, Europe and other jurisdictions. A typical full‑scale program or project may involve the following components:
|
· |
clinical program development, review and consultation and lifecycle management planning; |
|
· |
design of the clinical protocol and electronic case report forms, or CRFs; |
|
· |
feasibility studies for investigator interest and patient access and availability; |
|
· |
patient recruitment and retention services; |
|
· |
project management; |
|
· |
investigator and site analysis for selection and qualification; |
|
· |
investigator handbook and meetings; |
|
· |
investigational site support and clinical monitoring; |
|
· |
data management; |
|
· |
patient medical and safety management; |
|
· |
analysis and reporting; |
|
· |
medical and scientific publications; and |
|
· |
preparation of regulatory filings. |
As described below, we offer a suite of product registration service offerings to our clients to address the several components involved in conducting a full‑scale program or project.
Clinical Trial Management—Our clinical trial management services, used by biotechnology and pharmaceutical clients, may be performed exclusively by us or in collaboration with the client’s internal staff or other CROs. With our broad clinical trial management capabilities, we conduct single site studies, multi‑site U.S. and international studies and global studies on multiple continents. Through our electronic trial master file, we can create, collect, store, edit and retrieve any electronic document in any of our office locations worldwide, enabling our global project teams to work together efficiently regardless of where they are physically located and allowing seamless transfer of work to a more efficient locale.
Project Management—Our project management group manages the development process, setting specific targets and utilizing various metrics to ensure that a project moves forward in the right trajectory, resources are used optimally and client satisfaction is met. This group also oversees the implementation of a work breakdown structure, communication plan, and a risk and contingency program for each study. We believe that the management structure of our service delivery model sets us apart in the industry. Each individual project is assigned a director of project delivery and key strategic accounts are also assigned a general partner. As a member of the senior management team, the general partner works with the director of project delivery, the project management group and client representatives to ensure the highest level of client satisfaction. With more than 330 project directors and project managers, we match our project management personnel to projects based on experience and study specific parameters.
8
Regulatory Affairs—Our team of global regulatory professionals has extensive experience working with biotechnology and pharmaceutical companies and regulatory authorities worldwide. Our regulatory affairs group is comprised of an internal network of local regulatory experts who are native speakers in countries across North America, Latin America, Western and Eastern Europe, Africa and Asia Pacific. Regulatory team members and local regulatory experts act as clients’ representatives for submissions and direct communications with regulatory authorities in all regions. The group’s regulatory expertise enables rapid study start‑up and facilitates competitive product development plans and effective submission strategies.
Therapeutic Expertise—Our therapeutic expertise group provides scientific and medical expertise and patient access and retention services worldwide across a broad range of therapeutic areas. Our broad experience throughout various therapeutic areas allows us to offer a more complete global service offering to our clients. Our diverse therapeutic expertise group leverages best‑in‑class data assets to assist our clients with the design and implementation of entire clinical development programs and our current and potential clients increasingly seek partners who can provide these capabilities. We provide clients with therapeutic expertise in the design and implementation of high‑quality product development programs and help them achieve key development milestones in a cost and time effective manner. Our therapeutic expertise is used by both emerging biotechnology companies that lack clinical development infrastructure and pharmaceutical companies that have limited internal medical resources or are exploring new therapeutic areas.
Clinical Operations—Our clinical operations group provides clients with a full set of study site management and monitoring services in over 90 countries worldwide, through our highly experienced team of clinical research associates and specialists. This experience includes knowledge of local regulations, medical practices, safety and individual therapeutic areas. We provide our clients with fully trained and locally based clinical teams led by experienced clinical team managers that initiate site start‑up, monitor activities and review data. Based in the Americas, Europe, Asia Pacific and Africa, these teams work from a strategic foundation that combines reliance on proven, consistent processes with the flexibility to adapt innovative ideas and technologies. Given our expertise executing clinical trials around the world we are positioned to meet our clients’ diverse needs and expectations. Our study start‑up services group, a unit within clinical operations, manages the key components of rapid site activation and investigational site set‑up for clinical trials by utilizing our global and region specific expertise.
Data and Programming Services—Our global data and programming services group offers an innovative suite of technologies that gather and organize clinical trial data. We employ industry leading electronic data capture technologies and innovative delivery systems to produce high quality and standardized data and reports. We focus on evaluating a client’s needs, presenting optimal solutions for each trial and implementing the chosen solution effectively during project execution. To support these goals, we have built a group of technological experts in drug research that has a strong foundation in data management fundamentals and core programming abilities.
Safety and Risk Management—Our dedicated safety and risk management group helps clients design, implement and operationalize the proper safety procedures from development through to post‑marketing, allowing for clear assessment and the communication of patient safety profiles. We have centralized drug safety centers in Mannheim, Germany; Swansea, United Kingdom; Charlottesville, Virginia, United States (with a satellite center in Lenexa, Kansas); Sao Paulo, Brazil; and Singapore. Centers are staffed with experienced drug safety associates. These associates are responsible for integrating an effective risk minimization strategy for a drug product and generating useable information through ongoing risk evaluation. Our safety and risk management team provides risk mitigation strategies for our clients at all stages of the drug development cycle along with core signal detection capabilities.
Biostatistics and Medical Writing—Our global biostatistics and medical writing operations integrate our biostatistics, medical writing, pharmacokinetics and regulatory publishing groups. With a staff of industry experienced and therapeutically trained biostatisticians and medical writers, we offer clients expertise in statistical analysis, data pooling and regulatory reporting. This global team provides specialist consulting expertise and support to clients from the first stage of protocol design through post‑marketing surveillance and Phase IV studies. For publishing, we use a specialized electronic system that enables us to seamlessly assemble, manage and publish complex documents in compliance with applicable regulatory guidelines.
Quality Assurance Services—Our global quality assurance group is staffed by a team of experienced professionals in the Americas, Europe and Asia Pacific. Our quality assurance department is entirely separate from and independent of the personnel engaged in the direction and conduct of clinical trials. The objective of the quality
9
assurance group is the global promotion of ongoing quality awareness and continuous improvement of our processes. This group serves these efforts by performing audits on the processes and systems used in the management of clinical trials to ensure compliance with study protocol and applicable regulatory requirements. This group has performed audits for a wide range of medical indications and in all phases of clinical trials across the globe.
Late Phase Services—Our global late phase services group supports global and regional post‑approval trials with management locations centralized in Pennsylvania, Germany and Singapore. Our experienced late‑phase services team assists clients with the post‑marketing process by helping identify trends and signals in large populations as well as planning and conducting safety surveillance studies, large‑sample trials, registries, restricted access programs, risk management programs, diagnostic trials and biomarker research. The team consists of industry leading strategic experts, operational specialists and epidemiologists who work with clients to identify post‑marketing research objectives and goals and translate them into comprehensive study designs.
Strategic Solutions
Our Strategic Solutions offerings enable biotechnology and pharmaceutical companies to execute their internally‑managed development portfolio with greater flexibility and to leverage their existing infrastructure to minimize redundancy. These offerings provide a broad spectrum of solutions that allow for the efficient management and execution of critical clinical development functions for pharmaceutical clients. These services are embedded or integrated within the client’s internal clinical development operations to support the entire breadth of the client’s drug development pipeline. By embedding our employees within our clients’ infrastructure, we create a strategic and interdependent relationship that allows us to anticipate our clients’ clinical trial demands and efficiently deploy our skilled clinical professionals to meet our clients’ needs. Clinical functions supported by this service offering include study start‑up activities, site monitoring, study management, data management, biostatistics, regulatory and product safety. We focus our solutions primarily on our clients’ Phase II through Phase IV development programs. While traditional, project‑based CRO offerings target the outsourced component of biopharmaceutical industry spending, our Strategic Solutions offerings address the total Phase II through IV development market. We pioneered the embedded services model described below, and have extensive experience helping customers re‑align their operating model to more efficiently manage their development portfolio with greater flexibility and control.
Our Strategic Solutions offerings include:
Embedded Solutions—We believe we are the only company in the industry to offer a strategically scalable, fully‑embedded clinical development solution. Our Embedded Solutions model is designed to merge clinical operations expertise, management, infrastructure and support to create a flexible and integrated operating model. The goal of our Embedded Solutions model is to enable our client’s internally‑managed development processes to be executed with greater flexibility. These solutions can be further enhanced by leveraging our systems and technology as required. In our Embedded Solutions model, we typically work with our partners to assist in redesigning existing systems and processes to drive greater efficiency, speed and quality and to implement innovative approaches and enhanced technology. We employ a strong joint governance structure and robust metrics to measure and ensure strong quality, cycle time, productivity and service‑level performance.
Functional Services Provider Solutions—Our functional services provider offering provides dedicated capacity management within a single operating platform and within one function or across multiple functions and geographies. While the customer provides direction and functional management, we provide resources and line management, training and support. We also utilize business level metrics to help ensure that staff are deployed with the relevant experience and are producing consistent, repeatable results.
Staff Augmentation Solutions—Our staff augmentation solutions offering provides customers with the ability to address their dynamic staffing needs by supplying access to resources qualified to meet their clinical development needs. This allows clients to maintain flexibility while also reducing fixed costs. In order to rapidly attract and recruit qualified employees for these situations, we have assembled what we believe is the largest team in the industry focused on personnel recruitment. These individual professionals are hired as our employees and managed by our teams, minimizing co‑employment related issues. The customer has the ability to define the resources required according to the therapeutic‑ and disease‑specific experience required. These resources can be on site at the customer’s facility, at our offices, or regionally based.
10
Custom‑Built Development Solutions—Our custom‑built development solutions are designed to offer people, process, systems and development expertise that enable the efficient internal development of a company’s product portfolio with greater control and flexibility, accelerated development timelines and substantially reduced costs. With the client’s core leadership in control, we help to build the development team our clients need, while enabling them to maintain the flexibility to be nimble during the development lifecycle.
Commercialization Services—Through our commercialization services offering, we assist our clients in addressing the challenge of commercializing products. We do this by deploying professionals who are knowledgeable in launch preparation and product lifecycle management. We assist customers in managing the product lifecycle by working with them to create concise messaging, engage thought leadership and health care providers, generate consumer enthusiasm for the product, and prepare for post‑marketing commitments. Our commercialization services offering utilizes our flexible service model and, as such, can be delivered as an Embedded Solution, through our functional service provider model, or through staff augmentation.
Early Development Services
Our Early Development Services business unit, or EDS, offers a full range of services for Phase I and Phase IIa studies as well as bioanalytical analysis. We have conducted studies for major pharmaceutical companies in Europe, the United States and Japan, as well as for many smaller and emerging biotechnology companies. We have also built direct relationships with a large base of available subjects, including healthy volunteers and patient populations with specific medical conditions.
Our December 2013 acquisition of CRI Holding Company, LLC, or CRI Lifetree, significantly expanded our Phase I to Phase II services. CRI Lifetree is a specialized CRO focused on the conduct and design of early stage patient population studies, and is therapeutically focused in human abuse liability, or HAL, addiction, pain, psychiatric, neurological, pediatric and infectious disease services. CRI Lifetree is one of the largest providers of patient population for Phase I and confined Phase II to Phase III services in the United States, and is one of only a few CROs in the world which has the ability to design and conduct HAL studies, a regulatory‑required study for central nervous system compounds. We believe this acquisition enables us to provide our clients with a full range of Phase I to Phase II clinical research services in specialized patient populations for both inpatient and outpatient settings.
EDS also supports a variety of additional services, ranging from protocol development to data management and pharmacy services, including manufacturing of investigational medicinal products. Our state‑of‑the‑art laboratories provide pharmacokinetics, the branch of pharmacology concerned with the movement of drugs within the body, and pharmacodynamics, the branch of pharmacology concerned with the effects of drugs and the mechanism of their action analyses, including biomarkers, as needed. Our safety laboratory supports our own clinics and also acts as a central lab for medium sized Phase II trials. We also provide clinical study reports, statistical analysis, medical writing and regulatory support.
We focus on high‑end Phase I studies and specialize in more complex types of studies in which safety, intelligent design, and a wide range of pharmacodynamics assessments are critical factors. We believe our Phase I team is a leader in new developments such as microdosing studies, pain models, HAL studies and multi‑purpose protocols with adaptive designs. We have developed extensive methodologies enabling us to conduct studies with pharmacokinetics and/or pharmacodynamics objectives.
We have more than 1,000 early development specialists working in seven clinical pharmacology units located across four different countries, including the United States, the Netherlands and countries in Central and Eastern Europe. We are equipped with the technologies and infrastructure for high‑quality, efficient studies on a wide range of drugs and indications. Over the past five years we have conducted more than 700 high‑level, complex early development clinical trials and more than 200 bioanalytical studies per year over the previous five years.
Phase I through IIa Studies—For in‑house Phase I studies, we offer approximately 450 beds worldwide and accommodate volunteers in our state‑of‑the‑art clinical pharmacology units, some of which are hospital based. At these centers, volunteers are under constant medical supervision by a team of highly experienced medical professionals. We have an active pool of more than 100,000 study participants (both healthy volunteers and various specific patient populations).
11
In addition to in‑house studies, we use an innovative “unit‑on‑demand” business model that brings a Phase I center to patients. This model establishes a Phase I study environment in central medical facilities that specialize in the treatment of the target patient population. Physicians can recruit high volumes of patients using extensive networks of referring specialists and general practitioners. The studies occur in single center and multi‑national settings. We have also built an extensive patient network and database in areas including depression, schizophrenia, diabetes and hepatitis C. In addition to conducting Phase I and IIa studies in subjects, these sites act as investigative sites in Phase IIb and III trials.
We also offer full pharmacy capabilities and we operate a manufacturing site that complies with applicable current Good Manufacturing Practice regulations and is designed for fast and flexible manufacturing of small batches of investigational medicinal product for studies. In addition, dedicated data management professionals who can process clinical data into specific deliverables are integrated in each clinical pharmacology unit.
Since a large proportion of drug compounds do not succeed in Phase I, we utilize IND trials that include “microdose” or “low‑dose” studies to screen multiple candidates at an early stage and minimize the number of failing clinical product candidates. We have been closely involved in the field of microdose studies over the past ten years and have conducted more than 30 microdose studies.
Bioanalytical Laboratory—We offer clients two state‑of‑the‑art bioanalytical laboratories located in Assen, the Netherlands, and Lenexa, Kansas, United States. These bioanalytical laboratories have been harmonized with respect to standard operating procedures, work instructions and equipment. This provides a high level of consistency, continuity and efficiency. It also provides our clients with the ability to run studies in either laboratory, depending on the requirements of the study, and ensures that they will receive the same high level of service. Both bioanalytical laboratories are located within close proximity to their respective Phase I clinical pharmacology unit, ensuring rapid sample processing for critical dose escalation decision making involving pharmacokinetic assays. Both facilities include laboratories for mass spectrometry and ultra‑ performance liquid chromatography, typically applied to small molecule analysis. For large molecules, such as biologicals and biomarkers, our laboratories operate a wide variety of specialized assays, including ligand binding assays with a variety of detection methodologies and immunogenicity. In our fully licensed isotope laboratory, bioanalytical support is provided for mass balance and microdosing studies. The laboratories, combined with expert and highly educated staff, provide a full range of analytical services throughout the development process.
Clients and Suppliers
We serve a wide range of client types, including biotechnology and pharmaceutical companies. We have developed numerous strategic relationships in the last five years. In the year ended December 31, 2016, we derived 52% of our service revenue from large pharmaceutical companies, 14% of our service revenue from small‑ to mid‑sized pharmaceutical companies, 19% of our service revenue from large biotechnology companies and 15% of our service revenue from all other biotechnology companies. In 2016 and 2015, our top five clients represented approximately 45% and 41% of service revenue, respectively; this revenue was derived from a combination of fixed‑fee contracts, fee‑for‑service contracts and time and materials contracts. Two of our clients accounted for 11.0% and 10.4% of service revenue during the year ended December 31, 2016, respectively. One client accounted for 10.7% of service revenue during the year ended December 31, 2015. No individual project accounted for 10% or more of service revenue for the years ended December 31, 2016 and 2015.
We utilize a number of suppliers in our business, including central laboratory services, drug storage and shipping, foreign language translation services and information technology. In 2016, our largest individual supplier was paid $11.9 million. In addition, our top 10 suppliers together received payments during 2016 of approximately $72.4 million. We believe that we will continue to be able to meet our current and future supply needs.
Sales and Marketing
We have a proven sales team with the ability to build relationships with new clients and to grow within existing clients. Critical to our sales process is the involvement of our operations and global scientific and medical affairs teams who contribute their knowledge to project implementation strategies presented in client proposals. These teams also work closely with the sales team to build long‑term relationships with biotechnology and pharmaceutical companies. Our therapeutic expertise team supports the sales effort by developing robust service offerings in its core therapeutic
12
areas, which link our organization to key clinical opinion leaders, global investigator networks and best‑in‑class vendors. We rely heavily on our past project performance, qualified teams, medical informatics data and therapeutic expertise in winning new business.
Our approach to proposal development, led by seasoned proposal developers in conjunction with insight from our drug development experts, allows us to submit proposals that address client requirements in a creative and tailored manner. Proposal teams conduct research on competing drugs and conduct feasibility studies among potential investigators to assess their interest and patient availability for proposals and presentations. Our proprietary, automated estimation system allows for rapid and accurate creation of project budgets, which forms the initial basis for business management of budgets subsequent to award of the study.
Refer to Note 20 to our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K for further details regarding our foreign and domestic operations in 2016, 2015 and 2014. For a discussion of risks associated with our foreign operations, see “Item 1A. Risk Factors.”
Competition
We compete primarily with other full‑service CROs and in‑house research and development departments of pharmaceutical and established biotech companies. Our principal traditional CRO competitors are ICON plc, INC Research Holdings, Inc., inVentiv Health Inc., Laboratory Corporation of America Holdings, PAREXEL International Corporation, Pharmaceutical Product Development LLC, and Quintiles IMS Holdings Inc.
CROs compete on the basis of a number of factors, including reliability, past performance, expertise and experience in specific therapeutic areas, scope of service offerings, strengths in various geographic markets, technological capabilities, ability to manage large scale global clinical trials, and price.
The CRO industry remains highly fragmented, with several hundred smaller, limited service providers and a small number of full‑service companies with global capabilities. We believe there are significant barriers to becoming a global provider offering a broad range of services and products. These barriers include:
|
· |
the cost and experience necessary to develop broad therapeutic expertise; |
|
· |
the ability to manage large, complex international clinical programs; |
|
· |
the ability to deliver high‑quality services consistently for large drug development projects; |
|
· |
the experience to prepare regulatory submissions on a global basis; and |
|
· |
the infrastructure and knowledge to respond to the global needs of clients. |
Backlog
Our studies and projects are performed over varying durations, ranging from several months to several years. Backlog represents anticipated service revenue from contracted new business awards that either have not started or are in process but have not been completed. Cancelled contracts and scope reductions are removed from backlog as they occur. Our backlog at December 31, 2016, 2015 and 2014 was approximately $2.9 billion, $2.4 billion and $2.1 billion, respectively. Cancellations totaled $290.6 million, $231.0 million and $251.7 million for the years ended December 31, 2016, 2015 and 2014, respectively.
We believe our backlog as of any date is not necessarily a meaningful indicator of our future results for a variety of reasons. First, studies vary in duration. For instance, some studies that are included in our backlog may be completed in 2017, while others may be completed in later years. Second, the scope of studies may change, which may either increase or decrease the amount of backlog. Third, studies may be terminated or delayed at any time by the client or regulatory authorities. Delayed contracts remain in our backlog until a determination of whether to continue, modify or cancel the study is made.
13
We had $2,076.5 million, $1,696.6 million, and $1,493.7 million in net new business awards in the years ended December 31, 2016, 2015, and 2014, respectively. Net new business represents gross new business awards less cancellations for the period.
For more details regarding risks related to our backlog, see “Risk Factors—Our backlog may not convert to service revenue at the historical conversion rate.”
Intellectual Property
We have a pending patent application for our “Early Warning System” a solution for automated identification and qualification of risk to the deliverables in complex projects such as clinical trials. We also maintain and protect trade secrets, know‑how and other proprietary information regarding many of our business processes and related systems. We also hold various federal trademark registrations and pending applications, including PRA Health Sciences (design) PRA® (including a design), PRA International® and Predictivv (including design).
Government Regulation
In the United States, the FDA governs the conduct of clinical trials of drug products in human subjects, the form and content of regulatory applications, including, but not limited to, IND applications for human clinical testing and the development, approval, manufacture, safety, labeling, storage, record keeping, and marketing of drug products. The FDA has similar authority and similar requirements with respect to the clinical testing of biological products and medical devices. In the European Union, or EU, similar laws and regulations apply which may vary slightly from one member state to another and are enforced by the European Medicines Agency or respective national member states’ authorities, depending on the case.
Governmental regulation directly affects our business. Increased regulation leads to more complex clinical trials and an increase in potential business for us. Conversely, a relaxation in the scope of regulatory requirements, such as the introduction of simplified marketing applications for pharmaceutical and biological products, could decrease the business opportunities available to us.
We must perform our clinical drug and biologic services in compliance with applicable laws, rules and regulations, including “Good Clinical Practices,” or GCP, which govern, among other things, the design, conduct, performance, monitoring, auditing, recording, analysis, and reporting of clinical trials. Before a human clinical trial may begin, the manufacturer or sponsor of the clinical product candidate must file an IND with the FDA, which contains, among other things, the results of preclinical tests, manufacturer information, and other analytical data. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Each clinical trial must be conducted in accordance with an effective IND. In addition, under GCP, each human clinical trial we conduct is subject to the oversight of an independent institutional review board, or IRB, which is an independent committee that has the regulatory authority to review, approve and monitor a clinical trial for which the IRB has responsibility. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the study subjects are being exposed to an unacceptable health risk. In the EU, we must perform our clinical drug services in compliance with essentially similar laws and regulations.
In order to comply with GCP and other regulations, we must, among other things:
|
· |
comply with specific requirements governing the selection of qualified investigators; |
|
· |
obtain specific written commitments from the investigators; |
|
· |
obtain IRB review and approval of the clinical trial; |
|
· |
verify that appropriate patient informed consent is obtained before the patient participates in a clinical trial; |
|
· |
ensure adverse drug reactions resulting from the administration of a drug or biologic during a clinical trial are medically evaluated and reported in a timely manner; |
|
· |
monitor the validity and accuracy of data; |
14
|
· |
verify drug or biologic accountability; |
|
· |
instruct investigators and study staff to maintain records and reports; and |
|
· |
permit appropriate governmental authorities access to data for review. |
We must also maintain reports in compliance with applicable regulatory requirements for each study for auditing by the client and the FDA or similar regulatory authorities.
A failure to comply with applicable regulations relating to the conduct of clinical trials or the preparation of marketing applications could lead to a variety of sanctions. For example, violations of GCP could result, depending on the nature of the violation and the type of product involved, in the issuance of a warning letter, suspension or termination of a clinical study, refusal of the FDA to approve clinical trial or marketing applications or withdrawal of such applications, injunction, seizure of investigational products, civil penalties, criminal prosecutions, or debarment from assisting in the submission of new drug applications.
We monitor our clinical trials to test for compliance with applicable laws and regulations in the United States and the non‑U.S. jurisdictions in which we operate. We have adopted standard operating procedures that are designed to satisfy regulatory requirements and serve as a mechanism for controlling and enhancing the quality of our clinical trials. In the United States, our procedures were developed to ensure compliance with GCP and associated guidelines. Within Europe, all work is carried out in accordance with the European Community Note for Guidance (CPMP/ICH/135/95). In order to facilitate global clinical trials, we have implemented common standard operating procedures across our regions to assure consistency whenever feasible.
The Standards for Privacy of Individually Identifiable Health Information, or the Privacy Rule, and the Security Rule, issued under the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health, or HITECH, Act of 2009, collectively HIPAA, as well as applicable state privacy and security laws and regulations restrict the use and disclosure of certain protected health information, or PHI, and establishes national standards to protect individuals’ electronic PHI that is created, received, used or maintained by certain entities. Under the Privacy Rule, “covered entities” may not use or disclose PHI without the authorization of the individual who is the subject of the PHI, unless such use or disclosure is specifically permitted by the Privacy Rule or required by law.
We are not a covered entity under HIPAA. However, in connection with our clinical development activities, we do receive PHI from covered entities subject to HIPAA. In order for those covered entities to disclose PHI to us, the covered entity must obtain an authorization from the research subject that meets the Privacy Rule requirements, or make such disclosure pursuant to an exception to the Privacy Rule’s authorization requirement. We are both directly and indirectly affected by the privacy provisions surrounding individual authorizations because many investigators with whom we are involved in clinical trials are directly subject to them as a HIPAA “covered entity” and because we obtain identifiable health information from third parties that are subject to such regulations. Because of recent amendments to the HIPAA data security and privacy rules that were promulgated on January 25, 2013, some of which went into effect on March 26, 2013, there are some instances where we may be a HIPAA “business associate” of a “covered entity,” meaning that we may be directly liable for any breaches in protected health information and other HIPAA violations. As part of our research activities, we require covered entities that perform research activities on our behalf to comply with HIPAA, including the Privacy Rule’s authorization requirement, and applicable state privacy and security laws and regulations.
In Europe, EC Directive 95/46, or the Directive, is intended to protect the personal data of individuals by, among other things, imposing restrictions on the manner in which personal data can be collected, transferred, processed, and disclosed and the purposes for which personal data can be used. National laws and regulations implementing the Directive or dealing with personal data include provisions which, in certain EU Member States, are more stringent than the Directive’s mandates and/or cover areas that do not fall within the scope of the Directive. While we strive to comply with all privacy laws potentially applicable to our operations in Europe, we cannot guarantee that our business complies with all of these laws, which vary in scope and complexity in the multiple jurisdictions in which we operate.
15
We maintain a registration with the Drug Enforcement Administration, or DEA, that enables us to use controlled substances in connection with our research services. Controlled substances are those drugs and drug products that appear on one of five schedules promulgated and administered by DEA under the Controlled Substances Act. This act governs, among other things, the distribution, recordkeeping, handling, security, and disposal of controlled substances. Our DEA registration authorizes us to receive, conduct testing on, and distribute controlled substances in Schedules II through V. A failure to comply with the DEA’s regulations governing these activities could lead to a variety of sanctions, including the revocation or the denial of a renewal of our DEA registration, injunctions, or civil or criminal penalties.
Environmental Regulation and Liability
We are subject to various laws and regulations relating to the protection of the environment and human health and safety in the countries in which we do business, including laws and regulations governing the management and disposal of hazardous substances and wastes, the cleanup of contaminated sites and the maintenance of a safe workplace. Our operations include the use, generation, and disposal of hazardous materials and medical wastes. We may, in the future, incur liability under environmental statutes and regulations for contamination of sites we own or operate (including contamination caused by prior owners or operators of such sites), the off‑site disposal of hazardous substances and for personal injuries or property damage arising from exposure to hazardous materials from our operations. We believe that we have been and are in substantial compliance with all applicable environmental laws and regulations and that we currently have no liabilities under such environmental requirements that could reasonably be expected to materially harm our business, results of operations or financial condition.
Liability and Insurance
We may be liable to our clients for any failure to conduct their studies properly according to the agreed‑upon protocol and contract. If we fail to conduct a study properly in accordance with the agreed‑upon procedures, we may have to repeat a study or a particular portion of the services at our expense, reimburse the client for the cost of the services and/or pay additional damages.
At our clinical pharmacology units we study the effects of drugs on healthy volunteers. In addition, in our clinical business we, on behalf of our clients, contract with physicians who render professional services, including the administration of the substance being tested, to participants in clinical trials, many of whom are seriously ill and are at great risk of further illness or death as a result of factors other than their participation in a trial. As a result, we could be held liable for bodily injury, death, pain and suffering, loss of consortium, or other personal injury claims and medical expenses arising from a clinical trial. In addition, we sometimes engage the services of vendors necessary for the conduct of a clinical trial, such as laboratories or medical diagnostic specialists. Because these vendors are engaged as subcontractors, we are responsible for their performance and may be held liable for damages if the subcontractors fail to perform in the manner specified in their contract.
To reduce our potential liability, and as a requirement of the GCP regulations, informed consent is required from each volunteer and patient. In addition, our clients provide us with contractual indemnification for all of our service related contracts. These indemnities generally do not, however, protect us against certain of our own actions such as those involving negligence or misconduct. Our business, financial condition and operating results could be harmed if we were required to pay damages or incur defense costs in connection with a claim that is not indemnified, that is outside the scope of an indemnity or where the indemnity, although applicable, is not honored in accordance with its terms.
We maintain errors, omissions, and professional liability insurance in amounts we believe to be appropriate. This insurance provides coverage for vicarious liability due to negligence of the investigators who contract with us, as well as claims by our clients that a clinical trial was compromised due to an error or omission by us. If our insurance coverage is not adequate, or if insurance coverage does not continue to be available on terms acceptable to us, our business, financial condition, and operating results could be materially harmed.
Employees
As of December 31, 2016, we had over 13,000 employees, of which approximately 46% were in the United States, approximately 32% were in Europe, approximately 3% were in Canada, and approximately 19% were in Africa, Latin America, and Asia Pacific. Some of our employees located outside of the United States are represented by workers
16
council or labor unions. We believe that our employee relations are satisfactory. Approximately 40% of employees hold a Master’s level degree or higher. We have approximately 1,600 employees that hold a Ph.D, M.D. or other doctorate level degrees.
Available Information
We are subject to the informational requirements of the Exchange Act and, in accordance therewith, file reports, including annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission, or the SEC. Copies of our annual reports on Form 10‑K, quarterly reports on Form 10‑Q, current reports on Form 8‑K and our Proxy Statements for our annual meetings of stockholders, and any amendments to those reports, as well as Section 16 reports filed by our insiders, are available free of charge on our website as soon as reasonably practicable after we file the reports with, or furnish the reports to the SEC. Our website address is http://www.prahs.com, and our investor relations website is located at investor.prahs.com. Information on our website is not incorporated by reference herein. Our SEC filings are also available for reading and copying at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. Information on the operation of the Public Reference Room may be obtained by calling the SEC at 1‑800‑SEC‑0330. In addition, the SEC maintains an Internet site (http://www.sec.gov) containing reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
You should consider carefully the risks and uncertainties described below together with the other information included in this Annual Report on Form 10‑K, including our consolidated financial statements and related notes thereto. The occurrence of any of the following risks may materially and adversely affect our business, financial condition, results of operations and future prospects, which could in turn materially affect the price of our common stock.
The potential loss, delay or non‑renewal of our contracts, or the non‑payment by our clients for services that we have performed, could adversely affect our results.
We routinely experience termination, cancellation and non‑renewals of contracts by our clients in the ordinary course of business, and the number of cancellations can vary significantly from year to year.
Most of our clients for traditional, project‑based clinical trial services can terminate our contracts without cause upon 30 to 60 days’ notice. For example, our cancellation percentage for traditional, project‑based Phase I through IV trials was 18% for the years ended December 31, 2016 and 2015. Our traditional, project‑based clients may delay, terminate or reduce the scope of our contracts for a variety of reasons beyond our control, including but not limited to:
|
· |
decisions to forego or terminate a particular clinical trial; |
|
· |
lack of available financing, budgetary limits or changing priorities; |
|
· |
actions by regulatory authorities; |
|
· |
production problems resulting in shortages of the drug being tested; |
|
· |
failure of the drug being tested to satisfy safety requirements or efficacy criteria; |
|
· |
unexpected or undesired clinical results; |
|
· |
insufficient patient enrollment in a trial; |
|
· |
insufficient investigator recruitment; |
|
· |
decisions to downsize product development portfolios; |
|
· |
dissatisfaction with our performance, including the quality of data provided and our ability to meet agreed upon schedules; |
17
|
· |
shift of business to another CRO or internal resources; |
|
· |
product withdrawal following market launch; or |
|
· |
shut down of our clients’ manufacturing facilities. |
In addition, our clients for our Strategic Solutions offerings may elect not to renew our contracts for a variety of reasons beyond our control, including in the event that we are unable to provide staff sufficient in number or experience as required for a project.
In the event of termination, our contracts often provide for fees for winding down the study, but these fees may not be sufficient for us to maintain our profit margins, and termination or non‑renewal may result in lower resource utilization rates, including with respect to personnel who we are not able to place on another client engagement.
Clinical trials can be costly and a material portion of our revenue is derived from emerging biotechnology and small to mid‑sized pharmaceutical companies, which may have limited access to capital. In addition, we provide services to such companies before they pay us for some of our services. There is a risk that we may initiate a clinical trial for a client, and the client subsequently becomes unwilling or unable to fund the completion of the trial. In such a situation, notwithstanding the client’s ability or willingness to pay for or otherwise facilitate the completion of the trial, we may be legally or ethically bound to complete or wind down the trial at our own expense.
Because the contracts included in our backlog can generally be terminated without cause, we do not believe that our backlog as of any date is necessarily a meaningful predictor of future results. In addition, we may not realize the full benefits of our backlog of contractually committed services if our clients cancel, delay or reduce their commitments under our contracts with them. Thus, the loss or delay of a large contract or the loss or delay of multiple contracts could adversely affect our service revenue and profitability. In addition, the terminability of our contracts puts increased pressure on our quality control efforts, since not only can our contracts be terminated by clients as a result of poor performance, but any such termination may also affect our ability to obtain future contracts from the client involved and others. We believe the risk of loss or delay of multiple contracts is even greater in those cases where we are party to broader partnering arrangements with global biopharmaceutical companies.
We bear financial risk if we underprice our fixed‑fee contracts or overrun cost estimates, and our financial results can also be adversely affected by failure to receive approval for change orders or delays in documenting change orders.
Most of our traditional, project‑based Phase I through IV contracts are fixed‑fee contracts. We bear the financial risk if we initially underprice our contracts or otherwise overrun our cost estimates. In addition, contracts with our clients are subject to change orders, which we commonly experience and which occur when the scope of work we perform needs to be modified from that originally contemplated by our contract with the client. Modifications can occur, for example, when there is a change in a key trial assumption or parameter, a significant change in timing or a change in staffing needs. Furthermore, if we are not successful in converting out‑of‑scope work into change orders under our current contracts, we bear the cost of the additional work. Such underpricing, significant cost overruns or delay in documentation of change orders could have a material adverse effect on our business, results of operations, financial condition or cash flows.
Our backlog may not convert to service revenue at the historical conversion rate.
Backlog represents anticipated service revenue from contracted new business awards that either have not started or are in process but have not been completed and was $2.9 billion, $2.4 billion, and $2.1 billion at December 31, 2016, 2015, and 2014, respectively. Our revenue conversion rate is based on a financial and operational analysis performed by our project management teams and represents the level of effort expected to be expended at a specific point in time. Once work begins on a project, revenue is recognized over the duration of the project. Projects may be terminated or delayed by the client or delayed by regulatory authorities for reasons beyond our control. To the extent projects are delayed, the timing of our revenue could be affected. In the event that a client cancels a contract, we generally would be entitled to receive payment for all services performed up to the cancellation date and subsequent client authorized services related to terminating the canceled project. Generally, however, we have no contractual right to the full amount of the revenue reflected in our backlog in the event of a contract cancellation. The duration of the projects included in
18
our backlog, and the related revenue recognition, range from a few months to many years. Our backlog may not be indicative of our future results, and we may not realize all the anticipated future revenue reflected in our backlog. A number of factors may affect the realization of our revenue from backlog, including:
|
· |
the size, complexity and duration of the projects; |
|
· |
the cancellation or delay of projects; and |
|
· |
change in the scope of work during the course of a project. |
Fluctuations in our reported backlog levels also result from the fact that we may receive a small number of relatively large orders in any given reporting period that may be included in our backlog. Because of these large orders, our backlog in that reporting period may reach levels that may not be sustained in subsequent reporting periods.
As we increasingly compete for and enter into large contracts that are more global in nature, there can be no assurance about the rate at which our backlog will convert into revenue. A decrease in this conversion rate would mean that the rate of revenue recognized on contracts may be slower than what we have experienced in the past, which could impact our service revenue and results of operations on a quarterly and annual basis. The revenue recognition on larger, more global projects could be slower than on smaller, less global projects for a variety of reasons, including but not limited to, an extended period of negotiation between the time the project is awarded to us and the actual execution of the contract, as well as an increased timeframe for obtaining the necessary regulatory approvals. Additionally, delayed projects will remain in backlog and will not generate revenue at the rate originally expected. Thus, the relationship of backlog to realized revenues is indirect and may vary over time.
Our operating margins and profitability will be adversely affected if we are unable to either achieve efficiencies in our operating expenses or grow revenues at a rate faster than expenses.
We operate in a highly competitive environment and experience competitive pricing pressure. To achieve our operating margins over the last three years, we have implemented initiatives to control the rate of growth of our operating expenses. We will continue to utilize these initiatives in the future with a view to offsetting these pricing pressures; however, we cannot be certain that we will be able to achieve the efficiency gains necessary to maintain or grow our operating margins or that the magnitude of our growth in service revenue will be faster than the growth in our operating costs. If we are unable to grow our service revenue at a faster rate than our operating costs, our operating margins will be adversely affected. Our initiatives and any future cost initiatives may also adversely affect us, as they may decrease employee morale or make it more difficult for us to meet operational requirements.
If we are unable to attract suitable investigators and patients for our clinical trials, our clinical development business may suffer.
The recruitment of investigators and patients for clinical trials is essential to our business. Patients typically include people from the communities in which the clinical trials are conducted. Our clinical development business could be adversely affected if we are unable to attract suitable and willing investigators or patients for clinical trials on a consistent basis. For example, if we are unable to engage investigators to conduct clinical trials as planned or enroll sufficient patients in clinical trials, we may need to expend additional funds to obtain access to resources or else be compelled to delay or modify the clinical trial plans, which may result in additional costs to us. These considerations might result in our being unable to successfully achieve our projected development timelines, or potentially even lead us to consider the termination of ongoing clinical trials or development of a product.
Our embedded and functional outsourcing solutions could subject us to significant employment liability.
With our embedded and functional outsourcing services, we place employees at the physical workplaces of our clients. The risks of this activity include claims of errors and omissions, misuse or misappropriation of client proprietary information, theft of client property and torts or other claims under employment liability, co‑employment liability or joint employment liability. We have policies and guidelines in place to reduce our exposure to such risks, but if we fail to follow these policies and guidelines we may suffer reputational damage, loss of client relationships and business, and monetary damages.
19
If we lose the services of key personnel or are unable to recruit experienced personnel, our business could be adversely affected.
Our success substantially depends on the collective performance, contributions and expertise of our senior management team and other key personnel including qualified management, professional, scientific and technical operating staff and qualified sales representatives for our contract sales services. There is significant competition for qualified personnel in the biopharmaceutical services industry, particularly those with higher educational degrees, such as a medical degree, a Ph.D or an equivalent degree. The departure of any key executive, the payment of increased compensation to attract and retain qualified personnel, or our inability to continue to identify, attract and retain qualified personnel or replace any departed personnel in a timely fashion, may impact our ability to grow our business and compete effectively in our industry and may negatively affect our ability to meet financial and operational goals.
Our effective income tax rate may fluctuate, which may adversely affect our operations, earnings and earnings per share.
Our effective income tax rate is influenced by our projected profitability in the various taxing jurisdictions in which we operate. The global nature of our business increases our tax risks. In addition, as a result of increased funding needs by governments resulting from fiscal stimulus measures, revenue authorities in many of the jurisdictions in which we operate are known to have become more active in their tax collection activities. Changes in the distribution of profits and losses among taxing jurisdictions may have a significant impact on our effective income tax rate, which in turn could have an adverse effect on our net income and earnings per share. The application of tax laws in various taxing jurisdictions, including the United States, is subject to interpretation, and tax authorities in various jurisdictions may have diverging and sometimes conflicting interpretations of the application of tax laws. Changes in tax laws or tax rulings, such as tax reform proposals currently under consideration in the United States or other tax jurisdictions in which we operate, could materially impact our effective tax rate.
Factors that may affect our effective income tax rate include, but are not limited to:
|
· |
the requirement to exclude from our quarterly worldwide effective income tax calculations losses in jurisdictions where no income tax benefit can be recognized; |
|
· |
actual and projected full year pre‑tax income, including differences between actual and anticipated income before taxes in various jurisdictions; |
|
· |
changes in tax laws, or in the interpretation or application of tax laws, in various taxing jurisdictions; |
|
· |
audits or other challenges by taxing authorities; and |
|
· |
the establishment of valuation allowances against a portion or all of certain deferred income tax assets if we determined that it is more likely than not that future income tax benefits will not be realized. |
These changes may cause fluctuations in our effective income tax rate that could adversely affect our results of operations and cause fluctuations in our earnings and earnings per share.
Our business depends on the continued effectiveness and availability of our information systems, including the information systems we use to provide our services to our clients, and failures of these systems may materially limit our operations.
Due to the global nature of our business and our reliance on information systems to provide our services, we intend to increase our use of web‑enabled and other integrated information systems in delivering our services. We also provide access to similar information systems to certain of our clients in connection with the services we provide them. As the breadth and complexity of our information systems continue to grow, we will increasingly be exposed to the risks inherent in the development, integration and ongoing operation of evolving information systems, including:
|
· |
disruption, impairment or failure of data centers, telecommunications facilities or other key infrastructure platforms; |
20
|
· |
security breaches of, cyberattacks on and other failures or malfunctions in our critical application systems or their associated hardware; and |
|
· |
excessive costs, excessive delays or other deficiencies in systems development and deployment. |
The materialization of any of these risks may impede the processing of data, the delivery of databases and services, and the day‑to‑day management of our business and could result in the corruption, loss or unauthorized disclosure of proprietary, confidential or other data. While we have disaster recovery plans in place, they might not adequately protect us in the event of a system failure. Despite any precautions we take, damage from fire, floods, hurricanes, power loss, telecommunications failures, computer viruses, information system security breaches and similar events at our various computer facilities could result in interruptions in the flow of data to our servers and from our servers to our clients. Corruption or loss of data may result in the need to repeat a trial at no cost to the client, but at significant cost to us, or result in the termination of a contract or damage to our reputation. Additionally, significant delays in system enhancements or inadequate performance of new or upgraded systems once completed could damage our reputation and harm our business. Finally, long‑term disruptions in the infrastructure caused by events such as natural disasters, the outbreak of war, the escalation of hostilities and acts of terrorism, particularly involving cities in which we have offices, could adversely affect our business. Although we carry property and business interruption insurance, our coverage might not be adequate to compensate us for all losses that may occur.
Unauthorized disclosure of sensitive or confidential data, whether through system failure or employee negligence, fraud or misappropriation, could damage our reputation and cause us to lose clients. Similarly, unauthorized access to or through our information systems or those we develop for our clients, whether by our employees or third parties, including a cyber‑attack by computer programmers and hackers who may develop and deploy viruses, worms or other malicious software programs could result in negative publicity, significant remediation costs, legal liability and damage to our reputation and could have a material adverse effect on our results of operations. In addition, our liability insurance might not be sufficient in type or amount to adequately cover us against claims related to security breaches, cyber‑attacks and other related breaches. To date, cyber security attacks directed at us have not had a material impact on our financial results. Due to the evolving nature of security threats, however, the impact of any future incidents cannot be predicted.
Upgrading the information systems that support our operating processes and evolving the technology platform for our services pose risks to our business.
Continued efficient operation of our business requires that we implement standardized global business processes and evolve our information systems to enable this implementation. We have continued to undertake significant programs to optimize business processes with respect to our services. Our inability to effectively manage the implementation and adapt to new processes designed into these new or upgraded systems in a timely and cost‑effective manner may result in disruption to our business and negatively affect our operations.
We have entered into agreements with certain vendors to provide systems development and integration services that develop or license to us the IT platform for programs to optimize our business processes. If such vendors fail to perform as required or if there are substantial delays in developing, implementing and updating the IT platform, our client delivery may be impaired, and we may have to make substantial further investments, internally or with third parties, to achieve our objectives. Additionally, our progress may be limited by parties with existing or claimed patents who seek to enjoin us from using preferred technology or seek license payments from us.
Meeting our objectives is dependent on a number of factors which may not take place as we anticipate, including obtaining adequate technology enabled services, creating IT‑enabled services that our clients will find desirable and implementing our business model with respect to these services. Also, increased IT‑related expenditures may negatively impact our profitability.
Our operations might be affected by the occurrence of a natural disaster or other catastrophic event.
We depend on our clients, investigators, laboratories and other facilities for the continued operation of our business. Although we have contingency plans in place for natural disasters or other catastrophic events, these events, including terrorist attacks, pandemic flu, hurricanes and ice storms, could nevertheless disrupt our operations or those of our clients, investigators and collaboration partners, which could also affect us. In particular, our headquarters are in
21
Raleigh, North Carolina where hurricanes might occur. Even though we carry business interruption insurance policies and typically have provisions in our contracts that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our insurance policies or for which we do not have coverage. Any natural disaster or catastrophic event affecting us or our clients, investigators or collaboration partners could have a significant negative impact on our operations and financial performance.
We may be adversely affected by client concentration or concentration in therapeutic classes in which we conduct clinical trials.
We derive the majority of our revenues from a limited number of large clients. In 2016 and 2015, we derived 45% and 41%, respectively, of our service revenue from our top five clients. In addition, almost 46% of our backlog, as of December 31, 2016, is concentrated among five clients. If any large client decreases or terminates its relationship with us, our business, results of operations or financial condition could be materially adversely affected.
Additionally, we conduct multiple clinical trials for different clients in single therapeutic classes, particularly in the areas of oncology and central nervous system. Conducting multiple clinical trials for different clients in a single therapeutic class involving drugs with the same or similar chemical action has in the past, and may in the future, adversely affect our business if some or all of the trials are canceled because of new scientific information or regulatory judgments that affect the drugs as a class or if industry consolidation results in the rationalization of drug development pipelines.
Our business is subject to international economic, political and other risks that could negatively affect our results of operations and financial condition.
We have significant operations in non‑U.S. countries that may require complex arrangements to deliver services on global contracts for our clients. Additionally, we have established operations in locations remote from our most developed business centers. As a result, we are subject to heightened risks inherent in conducting business internationally, including the following:
|
· |
conducting a single trial across multiple countries is complex, and issues in one country, such as a failure to comply with local regulations or restrictions, may affect the progress of the trial in the other countries, for example, by limiting the amount of data necessary for a trial to proceed, resulting in delays or potential cancellation of contracts, which in turn may result in loss of revenue; |
|
· |
non‑U.S. countries could enact legislation or impose regulations or other restrictions, including unfavorable labor regulations or tax policies, which could have an adverse effect on our ability to conduct business in or expatriate profits from those countries; |
|
· |
tax rates in certain non‑U.S. countries may exceed those in the United States and non‑U.S. earnings may be subject to withholding requirements or the imposition of tariffs, exchange controls or other restrictions, including restrictions on repatriation; |
|
· |
certain non‑U.S. countries are expanding or may expand their regulatory framework with respect to patient informed consent, protection and compensation in clinical trials, which could delay or inhibit our ability to conduct trials in such jurisdictions or which could materially increase the risks associated with performing trials in such jurisdictions; |
|
· |
the regulatory or judicial authorities of non‑U.S. countries may not enforce legal rights and recognize business procedures in a manner to which we are accustomed or would reasonably expect; |
|
· |
we may have difficulty complying with a variety of laws and regulations in non‑U.S. countries, some of which may conflict with laws in the United States; |
|
· |
changes in political and economic conditions may lead to changes in the business environment in which we operate, as well as changes in non‑U.S. currency exchange rates; |
22
|
· |
clients in non‑U.S. jurisdictions may have longer payment cycles, and it may be more difficult to collect receivables in non‑U.S. jurisdictions; and |
|
· |
natural disasters, pandemics or international conflict, including terrorist acts, could interrupt our services, endanger our personnel or cause project delays or loss of trial materials or results. |
These risks and uncertainties could negatively impact our ability to, among other things, perform large, global projects for our clients. Furthermore, our ability to deal with these issues could be affected by applicable U.S. laws and the need to protect our assets. In addition, we may be more susceptible to these risks as we enter and continue to target growth in emerging countries and regions, including India, China, Eastern Europe and Latin America, which may be subject to a relatively higher risk of political instability, economic volatility, crime, corruption and social and ethnic unrest, all of which are exacerbated in many cases by a lack of an independent and experienced judiciary and uncertainties in how local law is applied and enforced. The materialization of any such risks could have an adverse impact on our financial condition and results of operations.
Due to the global nature of our business, we may be exposed to liabilities under the Foreign Corrupt Practices Act and various non‑U.S. anti‑corruption laws, and any allegation or determination that we violated these laws could have a material adverse effect on our business.
We are required to comply with the U.S. Foreign Corrupt Practices Act, or the FCPA, and other U.S. and non‑U.S. anti‑corruption laws, which prohibit companies from engaging in bribery, including corruptly or improperly offering, promising, or providing money or anything else of value to non‑U.S. officials and certain other recipients. In addition, the FCPA imposes certain books, records, and accounting control obligations on public companies and other issuers. We operate in parts of the world in which corruption can be common and compliance with anti‑bribery laws may conflict with local customs and practices. Our global operations face the risk of unauthorized payments or offers being made by employees, consultants, sales agents, and other business partners outside of our control or without our authorization. It is our policy to implement safeguards to prohibit these practices by our employees and business partners with respect to our operations. However, irrespective of these safeguards, or as a result of monitoring compliance with such safeguards, it is possible that we or certain other parties may discover or receive information at some point that certain employees, consultants, sales agents, or other business partners may have engaged in corrupt conduct for which we might be held responsible. Violations of the FCPA or other non‑U.S. anti‑corruption laws may result in restatements of, or irregularities in, our financial statements as well as severe criminal or civil sanctions, and we may be subject to other liabilities, which could negatively affect our business, operating results and financial condition. In some cases, companies that violate the FCPA may be debarred by the U.S. government and/or lose their U.S. export privileges. Changes in anti‑corruption laws or enforcement priorities could also result in increased compliance requirements and related costs which could adversely affect our business, financial condition and results of operations. In addition, the U.S. or other governments may seek to hold us liable for successor liability FCPA violations or violations of other anti‑corruption laws committed by companies in which we invest or that we acquired or will acquire.
If we are unable to successfully develop and market new services or enter new markets, our growth, results of operations or financial condition could be adversely affected.
A key element of our growth strategy is the successful development and marketing of new services and entering new markets that complement or expand our existing business. As we develop new services or enter new markets, including services targeted at participants in the broader healthcare industry, we may not have or adequately build the competencies necessary to perform such services satisfactorily, may not receive market acceptance for such services or may face increased competition. If we are unable to succeed in developing new services, entering new markets or attracting a client base for our new services or in new markets, we will be unable to implement this element of our growth strategy, and our future business, reputation, results of operations and financial condition could be adversely affected.
If we fail to perform our services in accordance with contractual requirements, government regulations and ethical considerations, we could be subject to significant costs or liability and our reputation could be adversely affected.
We contract with biotechnology and pharmaceutical companies to perform a wide range of services to assist them in bringing new drugs to market. Our services include monitoring clinical trials, data and laboratory analysis, electronic data capture, patient recruitment and other related services. Such services are complex and subject to
23
contractual requirements, government regulations, and ethical considerations. For example, we are subject to regulation by the FDA and comparable non‑U.S. regulatory authorities relating to our activities in conducting pre‑clinical and clinical trials. The clinical trial process must be conducted in accordance with regulations promulgated by the FDA under the Federal Food, Drug and Cosmetic Act, which requires the drug to be tested and studied in certain ways. In the United States, before human clinical testing may begin, a manufacturer must file an IND with the FDA. Further, an IRB for each medical center proposing to participate in the clinical trial must review and approve the protocol for the clinical trial before the medical center’s investigators participate. Once initiated, clinical trials must be conducted pursuant to and in accordance with the applicable IND, the requirements of the relevant IRBs, and GCP regulations. Similarly, before clinical trials begin, a drug is tested in pre‑clinical studies that are expected to comply with Good Laboratory Practice requirements. We are also subject to regulation by the DEA which regulates the distribution, recordkeeping, handling, security, and disposal of controlled substances. If we fail to perform our services in accordance with these requirements, regulatory authorities may take action against us. Such actions may include injunctions or failure to grant marketing approval of products, imposition of clinical holds or delays, suspension or withdrawal of approvals, rejection of data collected in our studies, license revocation, product seizures or recalls, operational restrictions, civil or criminal penalties or prosecutions, damages or fines. Clients may also bring claims against us for breach of our contractual obligations and patients in the clinical trials and patients taking drugs approved on the basis of those trials may bring personal injury claims against us. Any such action could have a material adverse effect on our results of operations, financial condition and reputation.
Such consequences could arise if, among other things, the following occur:
Improper performance of our services. The performance of clinical development services is complex and time‑consuming. For example, we may make mistakes in conducting a clinical trial that could negatively impact or obviate the usefulness of the trial or cause the results of the trial to be reported improperly. If the trial results are compromised, we could be subject to significant costs or liability, which could have an adverse impact on our ability to perform our services and our reputation would be harmed. As examples:
|
· |
non‑compliance generally could result in the termination of ongoing clinical trials or the disqualification of data for submission to regulatory authorities; |
|
· |
compromise of data from a particular trial, such as failure to verify that adequate informed consent was obtained from patients, could require us to repeat the trial under the terms of our contract at no further cost to our client, but at a potentially substantial cost to us; and |
|
· |
breach of a contractual term could result in liability for damages or termination of the contract. |
Large clinical trials can cost tens of millions of dollars, and while we endeavor to contractually limit our exposure to such risks, improper performance of our services could have a material adverse effect on our financial condition, damage our reputation and result in the cancellation of current contracts by the affected client or other current clients or failure to obtain future contracts from the affected client or other current or potential clients.
Investigation of clients. From time to time, one or more of our clients are investigated by regulatory authorities or enforcement agencies with respect to regulatory compliance of their clinical trials, programs or the marketing and sale of their drugs. In these situations, we have often provided services to our clients with respect to the clinical trials, programs or activities being investigated, and we are called upon to respond to requests for information by the authorities and agencies. There is a risk that either our clients or regulatory authorities could claim that we performed our services improperly or that we are responsible for clinical trial or program compliance. If our clients or regulatory authorities make such claims against us and prove them, we could be subject to damages, fines or penalties. In addition, negative publicity regarding regulatory compliance of our clients’ clinical trials, programs or drugs could have an adverse effect on our business and reputation.
If we fail to comply with federal, state, and non‑U.S. healthcare laws, including fraud and abuse laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
Even though we do not order healthcare services or bill directly to Medicare, Medicaid or other third‑party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse are and will be applicable
24
to our business. We could be subject to healthcare fraud and abuse laws of both the federal government and the states in which we conduct our business. Because of the breadth of these laws and the narrowness of available statutory and regulatory exceptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, imprisonment and the curtailment or restructuring of our operations, any of which could materially adversely affect our ability to operate our business and our financial results.
Our services could subject us to potential liability that may adversely affect our results of operations and financial condition.
Our business involves the testing of new drugs on patients in clinical trials. Our involvement in the clinical trial and development process creates a risk of liability for personal injury to or death of patients, particularly those with life‑threatening illnesses, resulting from adverse reactions to the drugs administered during testing or after regulatory approval. For example, we may be sued in the future by individuals alleging personal injury due to their participation in clinical trials and seeking damages from us under a variety of legal theories. If we are required to pay damages or incur defense costs in connection with any personal injury claim that is outside the scope of indemnification agreements we have with our clients, if any indemnification agreement is not performed in accordance with its terms or if our liability exceeds the amount of any applicable indemnification limits or available insurance coverage, our financial condition, results of operations and reputation could be materially and adversely affected. We might also not be able to obtain adequate insurance or indemnification for these types of risks at reasonable rates in the future.
We also contract with physicians to serve as investigators in conducting clinical trials. Investigators are typically located at hospitals, clinics or other sites and supervise the administration of the investigational drug to patients during the course of a clinical trial. If the investigators commit errors or make omissions during a clinical trial that result in harm to trial patients or after a clinical trial to a patient using the drug after it has received regulatory approval, claims for personal injury or products liability damages may result. Additionally, if the investigators engage in fraudulent or negligent behavior, trial data may be compromised, which may require us to repeat the clinical trial or subject us to liability or regulatory action. We do not believe we are legally responsible for the medical care rendered by such third‑party investigators, and we would vigorously defend any claims brought against us. However, it is possible we could be found liable for claims with respect to the actions of third‑party investigators.
Some of our services involve direct interaction with clinical trial patients and operation of Phase I and IIa clinical facilities, which could create potential liability that may adversely affect our results of operations and financial condition.
We operate facilities where Phase I to IIa clinical trials are conducted, which ordinarily involve testing an investigational drug on a limited number of individuals to evaluate its safety, determine a safe dosage range and identify side effects. Failure to operate such a facility in accordance with applicable regulations could result in disruptions to our operations. Additionally, we face risks associated with adverse events resulting from the administration of such drugs and the professional malpractice of medical care providers. We also directly employ nurses and other trained employees who assist in implementing the testing involved in our clinical trials, such as drawing blood from subjects. Any professional malpractice or negligence by such investigators, nurses or other employees could potentially result in liability to us in the event of personal injury to or death of a subject in clinical trials. This liability, particularly if it were to exceed the limits of any indemnification agreements and insurance coverage we may have, may adversely affect our financial condition, results of operations and reputation.
Our insurance may not cover all of our indemnification obligations and other liabilities associated with our operations.
We maintain insurance designed to provide coverage for ordinary risks associated with our operations and our ordinary indemnification obligations. The coverage provided by such insurance may not be adequate for all claims we may make or may be contested by our insurance carriers. If our insurance is not adequate or available to pay liabilities associated with our operations, or if we are unable to purchase adequate insurance at reasonable rates in the future, our profitability may be adversely impacted.
25
We do not currently maintain key person life insurance policies on any of our employees. If any of our key employees were to join a competitor or to form a competing company, some of our clients might choose to use the services of that competitor or new company instead of our own. Furthermore, clients or other companies seeking to develop in‑house capabilities may hire some of our senior management or key employees. We cannot assure you that a court would enforce the non‑competition provisions in our employment agreements.
Exchange rate fluctuations may affect our results of operations and financial condition.
During 2016, approximately 17% of our service revenue and 37% of our expenses were denominated in currencies other than the U.S. dollar, particularly the Euro and the Pound Sterling. Because a portion of our service revenue and expenses are denominated in currencies other than the U.S. dollar and our financial statements are reported in U.S. dollars, changes in non‑U.S. currency exchange rates could significantly affect our results of operations and financial condition.
The revenue and expenses of our non‑U.S. operations are generally denominated in local currencies and translated into U.S. dollars for financial reporting purposes. Accordingly, exchange rate fluctuations will affect the translation of non‑U.S. results into U.S. dollars for purposes of reporting our consolidated results.
We are subject to non‑U.S. currency transaction risk for fluctuations in exchange rates during the period of time between the consummation and cash settlement of a transaction. We earn revenue from our service contracts over a period of several months and, in some cases, over several years. Accordingly, exchange rate fluctuations during this period may affect our profitability with respect to such contracts.
We may limit these risks through exchange rate fluctuation provisions stated in our service contracts, or we may hedge our transaction risk with non‑U.S. currency exchange contracts or options. We have not, however, hedged any of our non‑U.S. currency transaction risk, and we may experience fluctuations in financial results from our operations outside the United States and non‑U.S. currency transaction risk associated with our service contracts.
If we do not keep pace with rapid technological changes, our services may become less competitive or obsolete.
The biopharmaceutical industry generally, and drug development and clinical research more specifically, are subject to rapid technological changes. Our current competitors or other businesses might develop technologies or services that are more effective or commercially attractive than, or render obsolete, our current or future technologies and services. If our competitors introduce superior technologies or services and if we cannot make enhancements to remain competitive, our competitive position would be harmed. If we are unable to compete successfully, we may lose clients or be unable to attract new clients, which could lead to a decrease in our revenue and financial condition.
Our relationships with existing or potential clients who are in competition with each other may adversely impact the degree to which other clients or potential clients use our services, which may adversely affect our results of operations.
The biopharmaceutical industry is highly competitive, with companies each seeking to persuade payors, providers and patients that their drug therapies are more cost‑effective than competing therapies marketed or being developed by competing firms. In addition to the adverse competitive interests that biopharmaceutical companies have with each other, these companies also have adverse interests with respect to drug selection and reimbursement with other participants in the healthcare industry, including payors and providers. Biopharmaceutical companies also compete to be first to the market with new drug therapies. We regularly provide services to biopharmaceutical companies who compete with each other, and we sometimes provide services to such clients regarding competing drugs in development. Our existing or future relationships with our biopharmaceutical clients have in the past and may continue to deter other biopharmaceutical clients from using our services or in certain instances has resulted in our clients seeking to place limits on our ability to serve their competitors and other industry participants. In addition, our further expansion into the broader healthcare market may adversely impact our relationships with biopharmaceutical clients, and such clients may elect not to use our services, reduce the scope of services that we provide to them or seek to place restrictions on our ability to serve clients in the broader healthcare market with interests that are adverse to theirs. Any loss of clients or reductions in the level of revenues from a client could have a material adverse effect on our results of operations, business and prospects.
26
If we are unable to manage our joint ventures and identify, acquire and integrate future acquisitions and joint ventures with our existing business, services and technologies, our business, results of operations and financial condition could be adversely impacted.
We have historically grown our business both organically and through acquisitions, and we anticipate that a portion of our future growth may come from acquiring existing businesses, services or technologies and entering into strategic alliances and joint ventures. The success of any acquisition will depend upon, among other things, our ability to effectively integrate acquired personnel, operations, products and technologies into our business, to obtain regulatory approvals, and to retain the key personnel and clients of our acquired businesses. Failure to successfully integrate any acquired business may result in reduced levels of revenue, earnings or operating efficiency than might have been achieved if we had not acquired such businesses. In addition, any future acquisitions could result in the incurrence of additional debt and related interest expense, contingent liabilities and amortization expenses related to intangible assets, which could have a material adverse effect on our business, financial condition, operating results and cash flow.
The success of any joint venture will involve, among other things, learning about new markets and regulations, ensuring quality controls are adequate and not inadvertently creating competitors. In addition, we may be unable to identify suitable acquisition opportunities, properly evaluate the price of such acquisitions or obtain any necessary financing on commercially acceptable terms.
We may also spend time and money investigating and negotiating with potential acquisition targets and strategic alliance partners but not complete the transaction. Acquisitions involve other risks, including, among others, the assumption of additional liabilities and expenses, difficulties and expenses in connection with integrating the acquired companies and achieving the expected benefits, issuances of potentially dilutive securities or debt, loss of key employees of the acquired companies, transaction costs, diversion of management’s attention from other business concerns and, with respect to the acquisition of non‑U.S. companies, the inability to overcome differences in non‑U.S. business practices, language and customs. Our failure to identify potential acquisitions, complete targeted acquisitions and integrate completed acquisitions or identify and manage strategic alliances or joint ventures could have a material adverse effect on our business, financial condition and results of operations.
We have a significant amount of goodwill and intangible assets on our balance sheet, and our results of operations may be adversely affected if we fail to realize the full value of our goodwill and intangible assets.
Our balance sheet reflects goodwill and intangibles assets of $972.0 million and $474.0 million, respectively, as of December 31, 2016. Collectively, goodwill and intangibles assets represented 66% of our total assets as of December 31, 2016. In accordance with generally accepted accounting principles, or GAAP, goodwill and indefinite-lived intangible assets are not amortized, but are subject to a periodic impairment evaluation. We assess the realizability of our indefinite-lived intangible assets and goodwill annually and conduct an interim evaluation whenever events or changes in circumstances, such as operating losses or a significant decline in earnings associated with the acquired business or asset, indicate that these assets may be impaired. In addition, we review long‑lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets might not be recoverable. If indicators of impairment are present, we evaluate the carrying value in relation to estimates of future undiscounted cash flows. Our ability to realize the value of the goodwill and intangible assets will depend on the future cash flows of the businesses we have acquired, which in turn depend in part on how well we have integrated these businesses into our own business. The carrying amount of the goodwill could be impaired if there is a downturn in our business or our industry or other factors that affect the fair value of our business, in which case a charge to earnings would become necessary. If we are not able to realize the value of the goodwill and intangible assets, we may be required to incur material charges relating to the impairment of those assets. Such impairment charges could materially and adversely affect our operating results and financial condition. See Note 2 to our consolidated financial statements included elsewhere in this Annual Report on Form 10‑K for a further discussion of our goodwill and intangible asset impairment testing.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
Under Sections 382 and 383 of the U.S. Internal Revenue Code, if a corporation undergoes an “ownership change” (generally defined as a greater than 50 percentage point change, by value, in the aggregate stock ownership of certain stockholders over a three‑year period), the corporation’s ability to use its pre‑change net operating loss carryforwards to offset its future taxable income and other pre‑change tax attributes may be limited. We have experienced at least one ownership change in the past. We may experience additional ownership changes in the future. In
27
addition, future changes in our stock ownership (including future sales by KKR) could result in additional ownership changes. Any such ownership changes could limit our ability to use our net operating loss carryforwards to offset any future taxable income and other tax attributes. State and non‑U.S. tax laws may also impose limitations on our ability to utilize net operating loss carryforwards and other tax attributes.
Our business could be harmed if we are unable to manage our growth effectively.
We believe that sustained growth places a strain on operational, human and financial resources. To manage our growth, we must continue to improve our operating and administrative systems and to attract and retain qualified management, professional, scientific and technical operating personnel. We believe that maintaining and enhancing both our systems and personnel at reasonable cost are instrumental to our success. We cannot assure you that we will be able to enhance our current technology or obtain new technology that will enable our systems to keep pace with developments and the sophisticated needs of our clients. The nature and pace of our growth introduces risks associated with quality control and client dissatisfaction due to delays in performance or other problems. In addition, non‑U.S. operations involve the additional risks of assimilating differences in non‑U.S. business practices, hiring and retaining qualified personnel and overcoming language barriers. Failure to manage growth effectively could have an adverse effect on our business.
Our operations involve the use and disposal of hazardous substances and waste which can give rise to liability that could adversely impact our financial condition.
We conduct activities that have involved, and may continue to involve, the controlled use of hazardous materials and the creation of hazardous substances, including medical waste and other highly regulated substances. Although we believe that our safety procedures for handling the disposal of such materials generally comply with the standards prescribed by non‑U.S., state and federal laws and regulations, our operations nevertheless pose the risk of accidental contamination or injury caused by the release of these materials and/or the creation of hazardous substances, including medical waste and other highly regulated substances. In the event of such an accident, we could be held liable for damages and cleanup costs which, to the extent not covered by existing insurance or indemnification, could harm our business. In addition, other adverse effects could result from such liability, including reputational damage resulting in the loss of additional business from certain clients.
We rely on third parties for important products and services.
We depend on certain third parties to provide us with products and services critical to our business. Such services include, among others, suppliers of drugs for patients participating in trials, suppliers of kits for use in our laboratories, suppliers of reagents for use in our testing equipment and providers of maintenance services for our equipment. The failure of any of these third parties to adequately provide the required products or services, or to do so in compliance with applicable regulatory requirements, could have a material adverse effect on our business.
We have only a limited ability to protect our intellectual property rights, and these rights are important to our success.
Our success depends, in part, upon our ability to develop, use and protect our proprietary methodologies, analytics, systems, technologies and other intellectual property. Existing laws of the various countries in which we provide services or solutions offer only limited protection of our intellectual property rights, and the protection in some countries may be very limited. We rely upon a combination of trade secrets, confidentiality policies, nondisclosure, invention assignment and other contractual arrangements, and copyright, trademark and trade secret laws, to protect our intellectual property rights. These laws are subject to change at any time and certain agreements may not be fully enforceable, which could further restrict our ability to protect our innovations. Our intellectual property rights may not prevent competitors from independently developing services similar to or duplicative of ours. Further, the steps we take in this regard might not be adequate to prevent or deter infringement or other misappropriation of our intellectual property by competitors, former employees or other third parties, and we might not be able to detect unauthorized use of, or take appropriate and timely steps to enforce, our intellectual property rights. Enforcing our rights might also require considerable time, money and oversight, and we may not be successful in enforcing our rights.
Depending on the circumstances, we might need to grant a specific client greater rights in intellectual property developed in connection with a contract than we otherwise generally do. In certain situations, we might forego all rights to the use of intellectual property we create, which would limit our ability to reuse that intellectual property for other
28
clients. Any limitation on our ability to provide a service or solution could cause us to lose revenue generating opportunities and require us to incur additional expenses to develop or license new or modified solutions for future projects.
Our business has experienced substantial expansion and contraction in the past and we might not properly manage any expansion or contraction in the future.
Rapid expansion or contraction, both of which we have experienced, could strain our operational, human and financial resources and facilities. If we fail to properly manage any changes, our expenses might grow more than revenue and our results of operations and financial condition might be negatively affected. In order to manage expansion or contraction, we must, among other things, do the following:
|
· |
continue to improve our operating, administrative and information systems; |
|
· |
accurately predict our future personnel, resource and facility needs to meet our commitments; |
|
· |
track the progress of ongoing projects; and |
|
· |
attract and retain qualified management, sales, professional, scientific and technical operating personnel. |
In addition, we have numerous business groups, subsidiaries and divisions. If we cannot properly manage these groups, subsidiaries or divisions, it will disrupt our operations. We also face additional risks in expanding our non‑U.S. operations. Specifically, we might find it difficult to:
|
· |
assimilate differences in non‑U.S. business practices and regulations; |
|
· |
properly integrate systems and operating procedures; |
|
· |
hire and retain qualified personnel; and |
|
· |
overcome language and cultural barriers. |
The biopharmaceutical services industry is fragmented and highly competitive.
The biopharmaceutical services industry is fragmented and highly competitive and if we do not compete successfully, our business will suffer. We often compete for business with other biopharmaceutical services companies, universities, niche providers and discovery and development departments within our clients, some of which are large biopharmaceutical services companies in their own right with greater resources than ours. As part of our business model, we have formed preferred vendor relationships. These relationships generally are not contractual and are subject to change at any time. As a result of these relationships, we may find reduced access to certain potential clients due to preferred vendor arrangements with other competitors. There are few barriers to entry for smaller specialized companies considering entering the industry. Because of their size and focus, these companies might compete effectively against larger companies like us, which could have a material adverse impact on our business. Additionally, the industry is highly fragmented, with numerous smaller specialized companies and a handful of full‑service companies with global capabilities similar to ours. Increased competition has led to price and other forms of competition, such as acceptance of less favorable contract terms that could adversely affect our operating results. As a result of competitive pressures, in recent years our industry has experienced consolidation. This trend is likely to produce more competition from the resulting larger companies for both clients and acquisition candidates.
Outsourcing trends in the biopharmaceutical industry and changes in aggregate spending and R&D budgets could adversely affect our operating results and growth rate.
We provide services to the biopharmaceutical industry and our revenues depend on the outsourcing trends and R&D expenditures in the industry. Economic factors and industry trends that affect biopharmaceutical companies affect our business. Biopharmaceutical companies continue to seek long‑term strategic collaborations with global CROs with favorable pricing terms. Competition for these collaborations is intense and we may decide to forego an opportunity or we may not be selected, in which case a competitor may enter into the collaboration and our business with the client, if
29
any, may be limited. In addition, if the biopharmaceutical industry reduces its outsourcing of clinical trials or such outsourcing fails to grow at projected rates, our operations and financial condition could be materially and adversely affected. We may also be negatively affected by consolidation and other factors in the biopharmaceutical industry, which may slow decision making by our clients or result in the delay or cancellation of clinical trials. All of these events could adversely affect our business, results of operations or financial condition.
Consolidation in the biopharmaceutical industry could lead to a reduction in our revenues.
Several large biopharmaceutical companies have completed mergers and acquisitions that will consolidate the outsourcing trends and R&D expenditures into fewer companies. As a result of the RPS Acquisition and the expansion of our relationship with large pharmaceutical companies, pharmaceutical companies have become an increasing portion of our customer base. The pharmaceutical industry is currently undergoing a period of increased merger activity. As a result of this and future consolidations, our client diversity may decrease and our business may be adversely affected.
We may be affected by healthcare reform and potential additional reforms.
Numerous government bodies are considering or have adopted various healthcare reforms and may undertake, or are in the process of undertaking, efforts to control growing healthcare costs through legislation, regulation and voluntary agreements with medical care providers and biopharmaceutical companies. By way of example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, was signed into law, which, among other things, expanded, over time, health insurance coverage, imposed health industry cost containment measures, enhanced remedies against healthcare fraud and abuse, added new transparency requirements for healthcare and health insurance industries, imposed new taxes and fees on pharmaceutical and medical device manufacturers and imposed additional health policy reforms, any of which may significantly impact the biopharmaceutical industry, including many of our customers. We are uncertain as to the effects of these recent reforms on our business and are unable to predict what legislative proposals, if any, will be adopted in the future. If regulatory cost containment efforts limit the profitability of new drugs, our clients may reduce their R&D spending, which could reduce the business they outsource to us. Similarly, if regulatory requirements are relaxed or simplified drug approval procedures are adopted, the demand for our services could decrease.
Government bodies may also adopt healthcare legislation or regulations that are more burdensome than existing regulations. For example, product safety concerns and recommendations by the Drug Safety Oversight Board could change the regulatory environment for drug products, and new or heightened regulatory requirements may increase our expenses or limit our ability to offer some of our services. Additionally, new or heightened regulatory requirements may have a negative impact on the ability of our clients to conduct industry sponsored clinical trials, which could reduce the need for our services. Furthermore, a relaxation of the scope of regulatory requirements, such as the introduction of simplified marketing applications for pharmaceuticals and biologics, could decrease the business opportunities available to us.
Actions by regulatory authorities or clients to limit the scope of or withdraw an approved drug from the market could result in a loss of revenue.
Government regulators have the authority, after approving a drug, to limit its indication for use by requiring additional labeled warnings or to withdraw the drug’s approval for its approved indication based on safety concerns. Similarly, clients may act to voluntarily limit the availability of approved drugs or withdraw them from the market after we begin our work. If we are providing services to clients for drugs that are limited or withdrawn, we may be required to narrow the scope of or terminate our services with respect to such drugs, which would prevent us from earning the full amount of service revenue anticipated under the related service contracts.
Current and proposed laws and regulations regarding the protection of personal data could result in increased risks of liability or increased cost to us or could limit our service offerings.
The confidentiality, collection, use and disclosure of personal data, including clinical trial patient‑specific information, are subject to governmental regulation generally in the country in which the personal data was collected or used. For example, U.S. federal regulations under HIPAA generally require individuals’ written authorization, in addition to any required informed consent, before protected health information may be used for research and such regulations specify standards for de‑identifications and for limited data sets. We may also be subject to applicable state
30
privacy and security laws and regulations in states in which we operate. We are both directly and indirectly affected by the privacy provisions surrounding individual authorizations because many investigators with whom we are involved in clinical trials are directly subject to them as a HIPAA “covered entity” and because we obtain identifiable health information from third parties that are subject to such regulations. Because of recent amendments to the HIPAA data security and privacy rules that were promulgated on January 25, 2013, some of which went into effect on March 26, 2013, there are some instances where we may be a HIPAA “business associate” of a “covered entity,” meaning that we may be directly liable for any breaches in protected health information and other HIPAA violations. These amendments may subject us to HIPAA’s enforcement scheme, which, as amended, can result in up to $1.5 million in annual civil penalties for each HIPAA violation.
In the EU and other jurisdictions, personal data includes any information that relates to an identified or identifiable natural person with health information carrying additional obligations, including obtaining the explicit consent from the individual for collection, use or disclosure of the information. In addition, we are subject to laws and regulations with respect to cross‑border transfers of such data out of certain jurisdictions in which we operate, including the EU. If we are unable to transfer data between and among countries and regions in which we operate, it could affect the manner in which we provide our services or adversely affect our financial results. The United States, the EU and its member states, and other countries where we have operations, such as Japan, continue to issue new privacy and data protection rules and regulations that relate to personal data and health information. Failure to comply with these data protection and privacy regulations and rules in various jurisdictions, or to resolve any serious privacy or security complaints, could subject us to regulatory sanctions, criminal prosecution or civil liability. Federal, state and non‑U.S. governments may propose or have adopted additional legislation governing the collection, possession, use or dissemination of personal data, such as personal health information, and personal financial data as well as security breach notification rules for loss or theft of such data. Additional legislation or regulation of this type might, among other things, require us to implement new security measures and processes or bring within the legislation or regulation de‑identified health or other personal data, each of which may require substantial expenditures or limit our ability to offer some of our services. Additionally, if we violate applicable laws, regulations or duties relating to the use, privacy or security of personal data, we could be subject to civil liability or criminal prosecution, be forced to alter our business practices and suffer reputational harm. In the next few years, the European data protection framework may be revised as a generally applicable data regulation. The text has not yet been finalized, but it contains new provisions specifically directed at the processing of health information, sanctions of up to 2% of worldwide gross revenue and extra‑territoriality measures intended to bring non‑EU companies under the proposed regulation.
The biopharmaceutical industry has a history of patent and other intellectual property litigation, and we might be involved in costly intellectual property lawsuits.
The biopharmaceutical industry has a history of intellectual property litigation, and these lawsuits will likely continue in the future. Accordingly, we may face patent infringement suits by companies that have patents for similar business processes or other suits alleging infringement of their intellectual property rights. Legal proceedings relating to intellectual property could be expensive, take significant time and divert management’s attention from other business concerns, regardless of the outcome of the litigation. If we do not prevail in an infringement lawsuit brought against us, we might have to pay substantial damages, and we could be required to stop the infringing activity or obtain a license to use technology on unfavorable terms.
Circumstances beyond our control could cause the CRO industry to suffer reputational or other harm that could result in an industry‑wide reduction in demand for CRO services, which could harm our business.
Demand for our services may be affected by perceptions of our clients regarding the CRO industry as a whole. For example, other CROs could engage in conduct that could render our clients less willing to do business with us or any CRO. Although to date no event has occurred causing material industry‑wide reputational harm, one or more CROs could engage in or fail to detect malfeasance, such as inadequately monitoring sites, producing inaccurate databases or analysis, falsifying patient records, and performing incomplete lab work, or take other actions that would reduce the confidence of our clients in the CRO industry. As a result, the willingness of biopharmaceutical companies to outsource R&D services to CROs could diminish and our business could thus be harmed materially by events outside our control.
31
Our substantial indebtedness could adversely affect our financial condition and prevent us from fulfilling our debt obligations and may otherwise restrict our activities.
As of December 31, 2016, we had total indebtedness of $836.4 million, including $91.4 million principal amount of 9.5% senior notes due 2023, or Senior Notes, $120.0 million principal amount of variable rate accounts receivable financing agreement due 2019 and $625.0 million principal amount of variable rate first lien term loan due 2021, or 2016 First Lien Term Loan. We had no outstanding borrowings under our revolving line of credit, or the 2016 Revolver. The 2016 First Lien Term Loan and 2016 Revolver are collectively known as the 2016 Credit Facilities.
Specifically, our high level of debt could have important consequences to the holders of the Senior Notes, including:
|
· |
making it more difficult for us to satisfy our obligations with respect to the Senior Notes and our other debt; |
|
· |
limiting our ability to obtain additional financing to fund future working capital, capital expenditures, investments or acquisitions or other general corporate requirements; |
|
· |
requiring a substantial portion of our cash flows to be dedicated to debt service payments instead of other purposes, thereby reducing the amount of cash flow available for working capital, capital expenditures, investments or acquisitions and other general corporate purposes; |
|
· |
increasing our vulnerability to adverse changes in general economic, industry and competitive conditions; |
|
· |
exposing us to the risk of increased interest rates as certain of our borrowings, including borrowings under the 2016 Credit Facilities and accounts receivable financing agreement, are at variable rates of interest; |
|
· |
limiting our flexibility in planning for and reacting to changes in the industry in which we compete; |
|
· |
placing us at a disadvantage compared to other, less leveraged competitors; and |
|
· |
increasing our cost of borrowing. |
Despite our level of indebtedness, we may incur more debt and undertake additional obligations. Incurring such debt or undertaking such additional obligations could further exacerbate the risks to our financial condition.
Although the credit agreement governing the 2016 Credit Facilities and the indenture governing the Senior Notes contain restrictions on the incurrence of additional indebtedness, these restrictions are subject to a number of qualifications and exceptions and the indebtedness incurred in compliance with these restrictions could increase. To the extent new debt is added to our current debt levels, the risks to our financial condition would increase.
While the credit agreement governing the 2016 Credit Facilities and the indenture governing the Senior Notes also contain restrictions on our ability to make loans and investments, these restrictions are subject to a number of qualifications and exceptions, and the investments incurred in compliance with these restrictions could be substantial.
If we do not comply with the covenants in our financing agreements, we may not have the funds necessary to pay all of our indebtedness that could become due.
The credit agreement governing the 2016 Credit Facilities, the accounts receivable financing agreement, and the indenture governing the Senior Notes require us to comply with certain covenants. In particular, our credit agreement and indenture prohibit us from incurring any additional indebtedness, except in specified circumstances, or amending the terms of agreements relating to certain existing junior indebtedness, if any, in a manner materially adverse to the lenders under our credit agreement and holders of our Senior Notes, without their respective approval. Further, our credit agreement, the accounts receivable financing agreement and indenture contain customary covenants, including covenants that restrict our ability to acquire and dispose of assets, engage in mergers or reorganizations, pay dividends or make investments. A violation of any of these covenants could cause an event of default under our financing agreements.
32
If we default on our financing agreements as a result of our failure to pay principal or interest when due, our material breach of any representation, warranty or covenant, or any other reason, all outstanding amounts could become immediately due and payable. In such case, we may not have sufficient funds to repay all the outstanding amounts. In addition, or in the alternative, the lenders under our financing agreements could exercise their rights under the security documents entered into in connection with these agreements. If any of the holders of our indebtedness accelerate the repayment of such indebtedness, there can be no assurance that we will have sufficient assets to repay our indebtedness. If we were unable to repay those amounts, the holders of our secured indebtedness could proceed against the collateral granted to them to secure that indebtedness. Any acceleration of amounts due or the substantial exercise by the lenders of their rights under the security documents would likely have a material adverse effect on us.
We may not be able to generate sufficient cash to service all of our indebtedness, and may be forced to take other actions to satisfy our obligations under our indebtedness that may not be successful.
Our ability to satisfy our debt obligations will depend upon, among other things:
|
· |
our future financial and operating performance, which will be affected by prevailing economic conditions and financial, business, regulatory and other factors, many of which are beyond our control; and |
|
· |
the future availability of borrowings under our 2016 Credit Facilities, which depends on, among other things, our complying with the covenants in those facilities. |
It cannot be assured that our business will generate sufficient cash flow from operations, or that future borrowings will be available to us under our 2016 Credit Facilities or otherwise, in an amount sufficient to fund our liquidity needs.
If our cash flows and capital resources are insufficient to service our indebtedness, we may be forced to reduce or delay capital expenditures, sell assets, seek additional capital or restructure or refinance our indebtedness. These alternative measures may not be successful and may not permit us to meet our scheduled debt service obligations. Our ability to restructure or refinance our debt will depend on the condition of the capital markets and our financial condition at such time. Any refinancing of our debt could be at higher interest rates and may require us to comply with more onerous covenants, which could further restrict our business operations. In addition, the terms of existing or future debt agreements, may restrict us from adopting some of these alternatives. In the absence of such operating results and resources, we could face substantial liquidity problems and might be required to dispose of material assets or operations to meet our debt service and other obligations. We may not be able to consummate those dispositions for fair market value or at all and any proceeds that we could realize from any such dispositions may not be adequate to meet our debt service obligations then due.
Interest rate fluctuations may affect our results of operations and financial condition.
Because a portion of our debt is variable‑rate debt, fluctuations in interest rates could have a material effect on our business. We currently utilize derivative financial instruments such as interest rate swaps to hedge our exposure to interest rate fluctuations, but such instruments may not be effective in reducing our exposure to interest fluctuations, and we may discontinuing utilizing them at any time. As a result, we may incur higher interest costs if interest rates increase. These higher interest costs could have a material adverse impact on our financial condition and the levels of cash we maintain for working capital.
The parties to the Stockholders Agreement have significant influence over us, including control over decisions that require the approval of our stockholders, which could limit your ability to influence the outcome of matters submitted to stockholders for a vote.
KKR owned approximately 37% of our outstanding common stock as of December 31, 2016. As a result, it has the ability to exert a significant amount of influence over our management and over corporate actions requiring stockholder approval, irrespective of how our other stockholders may vote, including:
|
· |
the election and removal of directors and the size of our board of directors; |
|
· |
any amendment of our articles of incorporation or bylaws; or |
33
|
· |
the approval of mergers and other significant corporate transactions, including a sale of substantially all of our assets. |
Although we are no longer a "controlled company" within the meaning of the NASDAQ rules, we are relying on exemptions from certain corporate governance requirements during transition periods of up to one year.
We are no longer a "controlled company" within the meaning of the NASDAQ listing rules. Consequently, as of May 6, 2016, the NASDAQ requires that:
|
· |
we appoint at least a majority of independent directors to our compensation committee within 90 days and have a fully independent compensation committee within one year; and |
|
· |
we appoint a majority of independent directors to our Board within one year. |
We intend to utilize the transition periods described above to achieve full compliance with these NASDAQ requirements. As a result, at this time we have a majority of independent directors, but our compensation committee does not consist entirely of independent directors. Accordingly, our stockholders do not, and during these transition periods will not, have the same protections afforded to stockholders of companies that are subject to all of the corporate governance requirements of the NASDAQ. In addition, if we are unable to comply with the heightened corporate governance requirements prior to the prescribed NASDAQ deadlines, we may incur penalties or our shares could be delisted.
Provisions of our corporate governance documents and Delaware law could make any change in our board of directors or in control of our company more difficult.
Our amended and restated certificate of incorporation and our amended and restated bylaws and Delaware law contain provisions, such as provisions authorizing, without a vote of stockholders, the issuance of one or more series of preferred stock, that could make it difficult or expensive for a third party to pursue a tender offer, change in control or takeover attempt that is opposed by our management and board of directors even if such a transaction would be beneficial to our stockholders. We also have a staggered board of directors that could make it more difficult for stockholders to change the composition of our board of directors in any one year. These anti‑takeover provisions could substantially impede the ability of public stockholders to change our management or board of directors.
Our operating results and share price may be volatile, which could cause the fair value of our stockholders’ investments to decline.
Securities markets worldwide have experienced, and are likely to continue to experience, significant price and volume fluctuations. This market volatility, as well as general economic, market or political conditions, could subject the market price of our shares to wide price fluctuations regardless of our operating performance. Our operating results and the trading price of our shares may fluctuate in response to various factors, including:
|
· |
market conditions in the broader stock market; |
|
· |
actual or anticipated fluctuations in our quarterly financial and operating results; |
|
· |
introduction of new products or services by us or our competitors; |
|
· |
the public’s reaction to our press releases, our other public announcements and our filings with the SEC; |
|
· |
changes in, or failure to meet, earnings estimates or recommendations by research analysts who track our common stock or the stock of other companies in our industries; |
|
· |
strategic actions by us, our customers or our competitors, such as acquisitions or restructurings; |
|
· |
changes in accounting standards, policies, guidance, interpretations or principles; |
34
|
· |
issuance of new or changed securities analysts’ reports or recommendations or termination of coverage of our common stock by securities analysts; |
|
· |
sales, or anticipated sales, of large blocks of our stock; |
|
· |
the granting or exercise of employee stock options; |
|
· |
volume of trading in our common stock; |
|
· |
additions or departures of key personnel; |
|
· |
regulatory or political developments; |
|
· |
litigation and governmental investigations; |
|
· |
changing economic conditions; |
|
· |
defaults on our indebtedness; and |
|
· |
exchange rate fluctuations. |
These and other factors, many of which are beyond our control, may cause our operating results and the market price and demand for our shares to fluctuate substantially. While we believe that operating results for any particular quarter are not necessarily a meaningful indication of future results, fluctuations in our quarterly operating results could limit or prevent investors from readily selling their shares and may otherwise negatively affect the market price and liquidity of our shares. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management from our business, which could significantly harm our profitability and reputation.
A significant portion of our total outstanding shares may be sold into the market in the near future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock into the public market could occur at any time. We have 61,597,705 outstanding shares of common stock as of December 31, 2016. Certain of our stockholders have demand registration rights and “piggyback” registration rights with respect to future registered offerings of our common stock. KKR and other stockholders, who collectively own 36.9% of our common stock, may sell shares of our common stock. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. We also registered all shares of common stock that we may issue under our equity compensation plans and they can be freely sold in the public market upon issuance, subject to restrictions on transfer contained in management stockholders agreements entered into between certain recipients of equity compensation and KKR. As restrictions on transfer end, the market price of our stock could decline if the holders of currently restricted shares sell them or are perceived by the market as intending to sell them.
Because we have no current plans to pay regular cash dividends on our common stock, you may not receive any return on investment unless you sell your common stock for a price greater than that which you paid for it.
Although we have previously declared dividends to our stockholders, we do not anticipate paying any regular cash dividends on our common stock. Any decision to declare and pay dividends in the future will be made at the discretion of our board of directors and will depend on, among other things, our results of operations, financial condition, cash requirements, contractual restrictions and other factors that our board of directors may deem relevant. In addition, our ability to pay dividends is, and may be, limited by covenants of existing and any future outstanding indebtedness we or our subsidiaries incur, including under our existing credit facilities. Therefore, any return on investment in our common stock is solely dependent upon the appreciation of the price of our common stock on the open market, which may not occur. See Part II, Item 5. “Market for Registrants Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities—Dividend Policy” for more detail.
35
Failure to maintain effective internal control over financial reporting in accordance with Section 404 of the Sarbanes Oxley Act could have a material adverse effect on our business and share price.
The Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. Under Section 404 of the Sarbanes-Oxley Act, we are required to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment must include disclosure of any material weaknesses identified by management in our internal control over financial reporting. A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting.
Our compliance with Section 404 requires that we compile the system and process documentation necessary to perform an appropriate evaluation. During the evaluation and testing process, if we identify one or more material weaknesses in its internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that its internal control over financial reporting is effective, or if our independent registered public accounting firm determines that we have a material weakness or significant deficiency in our internal control over financial reporting once that firm begin its reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by NASDAQ, the Securities and Exchange Commission or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
We are incurring significant costs as a result of operating as a public company, and our management is devoting substantial time to new compliance initiatives.
As a publicly traded company, we are incurring significant legal, accounting and other expenses to comply with laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd‑Frank Wall Street Reform and Consumer Protection Act and the rules and regulations promulgated and to be promulgated thereunder, as well as under the Sarbanes-Oxley Act, and the rules and regulations of the SEC, and the NASDAQ. In addition these compliance requirements are making some activities more difficult, time‑consuming or costly. For example, the Exchange Act requires us, among other things, to file annual, quarterly and current reports with respect to our business and operating results, and to provide an annual assessment of the effectiveness of our internal control over financial reporting. In order to maintain and improve the effectiveness of our disclosure controls and procedures and internal controls over financial reporting as required by the Exchange Act, significant resources and management oversight are required.
Being a public company and being subject to new rules and regulations makes it more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors may therefore strain our resources, divert management’s attention and affect our ability to attract and retain qualified members of our board of directors.
Furthermore, the need to maintain the corporate infrastructure demanded of a public company may divert management’s attention from implementing our growth strategy, which could prevent us from improving our business, results of operations and financial condition. We have made, and will continue to make, changes to our internal controls and procedures for financial reporting and accounting systems to meet our reporting obligations as a publicly traded company. If we are not able to implement or maintain the necessary procedures and processes, we may be unable to report our financial information on a timely basis and thereby could subject us to adverse regulatory consequences, including sanctions by the SEC or violations of applicable stock exchange listing rules, and could result in a breach of covenants under the agreements governing any of our financing agreements. There could also be a negative reaction in the financial markets due to a loss of investor confidence in us and the reliability of our financial statements.
36
Item 1B. Unresolved Staff Comments
None.
We lease a facility for our corporate headquarters in Raleigh, North Carolina. We also lease other offices in North America, Europe, Africa, Latin America, Australia and Asia. In 2016, our total rental expense for our facilities and offices was approximately $31.9 million. We do not own any real estate. We believe that our properties, taken as a whole, are in good operating condition and are suitable for our business operations.
We are currently involved, as we are from time to time, in legal proceedings that arise in the ordinary course of our business. We believe that we have adequately accrued for these liabilities and that there is no other litigation pending that could materially harm our results of operations and financial condition. See Note 13 to our consolidated financial statements included elsewhere in this Annual Report on Form 10‑K for a further discussion of our current legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
37
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information for Common Stock
Our common stock trades on the NASDAQ under the symbol “PRAH.” The following table sets forth the high and low sales prices per share of our common stock as reported by the NASDAQ for the periods indicated.
|
|
|
High |
|
Low |
|
||
|
Fiscal Year 2016 |
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
60.96 |
|
$ |
50.87 |
|
|
Third Quarter |
|
$ |
56.77 |
|
$ |
39.25 |
|
|
Second Quarter |
|
$ |
51.35 |
|
$ |
39.52 |
|
|
First Quarter |
|
$ |
47.84 |
|
$ |
35.60 |
|
|
|
|
High |
|
Low |
|
||
|
Fiscal Year 2015 |
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
50.25 |
|
$ |
33.00 |
|
|
Third Quarter |
|
$ |
46.35 |
|
$ |
33.74 |
|
|
Second Quarter |
|
$ |
37.41 |
|
$ |
26.91 |
|
|
First Quarter |
|
$ |
31.99 |
|
$ |
22.40 |
|
Holders of Record
On February 17, 2017, we had approximately 44 common stockholders of record. This number does not include beneficial owners for whom shares are held by nominees in street name.
Dividend Policy
We have not paid any cash dividends during the two most recent fiscal years. We also have no current plans to pay any cash dividends on our common stock for the foreseeable future and instead intend to retain earnings, if any, for future operations, expansion and debt repayment. Any decision to declare and pay dividends in the future will be made at the discretion of our board of directors and will depend on, among other things, our results of operations, cash requirements, financial condition, contractual restrictions and other factors that our board of directors may deem relevant. In addition, our ability to pay dividends is limited by covenants in the credit agreement governing our 2016 Credit Facilities and in the indenture governing our Senior Notes. See Part II, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operation—Liquidity and Capital Resources—2016 Credit Facilities” and “—Senior Notes” for restrictions on our ability to pay dividends.
Recent Sales of Unregistered Securities
There were no unregistered sales of equity securities in 2016.
Purchases of Equity Securities by the Issuer
None.
Stock Performance Graph
This performance graph shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or incorporated by reference into any filing of PRA Health Sciences, Inc.
The following graph shows a comparison from November 13, 2014 (the date our common stock commenced trading on the NASDAQ) through December 31, 2016 of the cumulative total return for our common stock, the Nasdaq Composite Index and the Nasdaq Health Care Index.
The graph assumes that $100 was invested at the market close on November 13, 2014 in the common stock of PRA Health Sciences, Inc., the Nasdaq Composite Index and the Nasdaq Health Care Index, and assumes reinvestments
38
of dividends, if any. The stock price performance of the following graph is not necessarily indicative of future stock price performance.
Item 6. Selected Financial Data
The following tables set forth, for the periods and at the dates indicated, our selected historical consolidated financial data. We have derived the selected consolidated financial data for the years ended December 31, 2014, 2015 and 2016, and as of December 31, 2015 and 2016, from our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 10‑K. We have derived the selected consolidated financial data for the year ended December 31, 2012, the period from January 1 through September 22, 2013, the period from September 23 through December 31, 2013, and as of December 31, 2012, 2013 and 2014 from our consolidated financial statements not appearing elsewhere in this Annual Report on Form 10‑K. Our historical results are not necessarily indicative of the results we may achieve in any future period.
The accompanying consolidated statements of operations, cash flows and stockholders’ equity are presented for two periods: Predecessor and Successor, which relate to the period preceding the Company’s acquisition by KKR, or KKR Transaction, and the period succeeding the KKR Transaction, respectively. The Company refers to the operations of PRA Health Sciences, Inc. and subsidiaries for both the Predecessor and Successor periods.
Historical results are not necessarily indicative of the results to be expected in the future.
You should read the following information together with the more detailed information contained in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated
financial statements and the accompanying notes appearing elsewhere in this Annual Report on Form 10‑K.
39
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Predecessor |
|
|
Successor |
||||||||||||||
|
|
|
December 31, |
|
January 1, 2013 - |
|
|
September 23, 2013 - |
|
December 31, |
|
December 31, |
|
December 31, |
||||||
|
(in thousands, except per share data) |
|
2012 |
|
September 22, 2013 |
|
|
December 31, 2013 |
|
2014 |
|
2015 |
|
2016 |
||||||
|
Consolidated statement of operations data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Service revenue |
|
$ |
597,072 |
|
$ |
508,539 |
|
|
$ |
324,362 |
|
$ |
1,266,596 |
|
$ |
1,375,847 |
|
$ |
1,580,023 |
|
Reimbursement revenue |
|
|
102,664 |
|
|
103,531 |
|
|
|
54,854 |
|
|
192,990 |
|
|
238,036 |
|
|
231,688 |
|
Total revenue |
|
|
699,736 |
|
|
612,070 |
|
|
|
379,216 |
|
|
1,459,586 |
|
|
1,613,883 |
|
|
1,811,711 |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Direct costs |
|
|
358,572 |
|
|
304,102 |
|
|
|
222,776 |
|
|
859,218 |
|
|
886,528 |
|
|
1,032,688 |
|
Reimbursable out-of-pocket costs |
|
|
102,664 |
|
|
103,531 |
|
|
|
54,854 |
|
|
192,990 |
|
|
238,036 |
|
|
231,688 |
|
Selling, general and administrative |
|
|
160,643 |
|
|
142,880 |
|
|
|
69,730 |
|
|
253,970 |
|
|
246,417 |
|
|
269,893 |
|
Transaction-related costs |
|
|
— |
|
|
47,486 |
|
|
|
29,180 |
|
|
— |
|
|
— |
|
|
44,834 |
|
Depreciation and amortization |
|
|
30,687 |
|
|
25,144 |
|
|
|
25,333 |
|
|
96,564 |
|
|
77,952 |
|
|
69,506 |
|
Loss on disposal of fixed assets |
|
|
1,560 |
|
|
225 |
|
|
|
— |
|
|
5 |
|
|
652 |
|
|
753 |
|
Income (loss) from operations |
|
|
45,610 |
|
|
(11,298) |
|
|
|
(22,657) |
|
|
56,839 |
|
|
164,298 |
|
|
162,349 |
|
Interest expense, net |
|
|
(32,823) |
|
|
(32,719) |
|
|
|
(23,703) |
|
|
(81,939) |
|
|
(61,747) |
|
|
(54,913) |
|
Loss on modification or extinguishment of debt |
|
|
(9,683) |
|
|
(21,678) |
|
|
|
(7,211) |
|
|
(25,036) |
|
|
— |
|
|
(38,178) |
|
Foreign currency (losses) gains, net |
|
|
(7,841) |
|
|
(3,641) |
|
|
|
(4,117) |
|
|
10,538 |
|
|
14,048 |
|
|
24,029 |
|
Other income (expense), net |
|
|
183 |
|
|
(530) |
|
|
|
1,180 |
|
|
(2,254) |
|
|
(1,434) |
|
|
607 |
|
(Loss) income before income taxes and equity in (losses) gains of unconsolidated joint ventures |
|
|
(4,554) |
|
|
(69,866) |
|
|
|
(56,508) |
|
|
(41,852) |
|
|
115,165 |
|
|
93,894 |
|
(Benefit from) provision for income taxes |
|
|
(1,847) |
|
|
(22,079) |
|
|
|
(17,186) |
|
|
(8,154) |
|
|
30,004 |
|
|
28,494 |
|
(Loss) income before equity in (losses) gains of unconsolidated joint ventures |
|
|
(2,707) |
|
|
(47,787) |
|
|
|
(39,322) |
|
|
(33,698) |
|
|
85,161 |
|
|
65,400 |
|
Equity in (losses) gains of unconsolidated joint ventures, net of tax |
|
|
— |
|
|
(603) |
|
|
|
(621) |
|
|
(2,044) |
|
|
(3,396) |
|
|
2,775 |
|
Net (loss) income |
|
$ |
(2,707) |
|
$ |
(48,390) |
|
|
$ |
(39,943) |
|
$ |
(35,742) |
|
$ |
81,765 |
|
$ |
68,175 |
|
Net (loss) income per share (1): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
(0.07) |
|
$ |
(1.22) |
|
|
$ |
(1.02) |
|
$ |
(0.83) |
|
$ |
1.36 |
|
$ |
1.12 |
|
Diluted |
|
$ |
(0.07) |
|
$ |
(1.22) |
|
|
$ |
(1.02) |
|
$ |
(0.83) |
|
$ |
1.29 |
|
$ |
1.06 |
|
Cash dividends declared per common stockholders: |
|
$ |
2.31 |
|
$ |
2.83 |
|
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
Weighted average common shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
39,641 |
|
|
39,643 |
|
|
|
39,337 |
|
|
42,897 |
|
|
59,965 |
|
|
60,759 |
|
Diluted |
|
|
39,641 |
|
|
39,643 |
|
|
|
39,337 |
|
|
42,897 |
|
|
63,207 |
|
|
64,452 |
|
Cash flow data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) operating activities (5) |
|
$ |
99,259 |
|
$ |
54,044 |
|
|
$ |
(25,666) |
|
$ |
34,034 |
|
$ |
152,428 |
|
$ |
160,047 |
|
Net cash used in investing activities (5) |
|
|
(18,058) |
|
|
(54,753) |
|
|
|
(1,008,419) |
|
|
(11,472) |
|
|
(71,686) |
|
|
(34,614) |
|
Net cash (used in) provided by financing activities (5) |
|
|
(42,157) |
|
|
(42,065) |
|
|
|
1,115,041 |
|
|
(5,956) |
|
|
(42,444) |
|
|
(101,595) |
|
Other financial data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Backlog (at period end) (2) |
|
$ |
1,382,826 |
|
$ |
— |
|
|
$ |
1,939,666 |
|
$ |
2,141,112 |
|
$ |
2,440,123 |
|
|
2,934,823 |
|
Net new business (3) |
|
|
653,529 |
|
|
462,046 |
|
|
|
312,298 |
|
|
1,493,652 |
|
|
1,696,635 |
|
|
2,076,484 |
|
|
|
Predecessor |
|
|
|
|
|
Successor |
|||||||||||
|
|
|
As of |
|
|
|
|
|
|
|
|
As of |
|
As of |
|
As of |
||||
|
|
|
December 31, |
|
|
|
|
|
As of December 31, |
|
December 31, |
|
December 31, |
|
December 31, |
|||||
|
|
|
2012 |
|
|
|
|
|
2013 |
|
2014 |
|
2015 |
|
2016 |
|||||
|
Consolidated balance sheet data |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
109,211 |
|
|
|
|
|
$ |
72,155 |
|
$ |
85,192 |
|
$ |
121,065 |
|
$ |
144,623 |
|
Accounts receivable and unbilled services, net |
|
|
184,891 |
|
|
|
|
|
|
294,984 |
|
|
338,781 |
|
|
415,077 |
|
|
439,053 |
|
Working capital |
|
|
18,317 |
|
|
|
|
|
|
(11,270) |
|
|
22,367 |
|
|
43,796 |
|
|
60,538 |
|
Total assets (4) |
|
|
982,525 |
|
|
|
|
|
|
2,357,673 |
|
|
2,214,484 |
|
|
2,228,743 |
|
|
2,190,391 |
|
Total long-term debt, net (4) |
|
|
451,076 |
|
|
|
|
|
|
1,208,751 |
|
|
924,444 |
|
|
889,514 |
|
|
797,052 |
|
Total liabilities (4) |
|
|
806,568 |
|
|
|
|
|
|
1,890,338 |
|
|
1,537,669 |
|
|
1,526,021 |
|
|
1,461,139 |
|
Total stockholders' equity |
|
|
175,957 |
|
|
|
|
|
|
467,335 |
|
|
676,815 |
|
|
702,722 |
|
|
729,252 |
|
Total liabilities and stockholders' equity (4) |
|
|
982,525 |
|
|
|
|
|
|
2,357,673 |
|
|
2,214,484 |
|
|
2,228,743 |
|
|
2,190,391 |
|
(1) |
Because of the KKR Transaction, our capital structure for periods before and after the KKR Transaction are not comparable and therefore we are adjusting the number of shares to reflect the stock split only for the successor periods. See Note 1 to our consolidated financial statements included elsewhere in this Annual Report on Form 10‑K for a further discussion of our reverse stock split. |
|
(2) |
Our backlog consists of anticipated service revenue from new business awards that either have not started or are but have not been completed. Backlog varies from period to period depending upon new business awards and contract increases, cancellations and the amount of service revenue recognized under existing contracts. |
|
(3) |
For our Strategic Solutions offering, the value of new business awards is the anticipated service revenue to be recognized in the corresponding quarter of the next fiscal year. For the remainder of our business, net new business is the value of services awarded during the period from projects under signed contracts, letters of intent and, in some cases, pre‑contract commitments that are supported by written communications, adjusted for contracts that were modified or canceled during the period. For the fiscal year 2013, net new business excludes the RPS Acquisition. |
|
(4) |
In 2015 we early adopted ASU No. 2015-03 and ASU No. 2015-15. The balance sheet data for 2014, 2013, and 2012 has been restated to reflect the presentation of debt issuance costs as a reduction of long-term debt. |
|
(5) |
In 2016, we early adopted ASU No. 2016-15 and ASU No. 2016-18. The consolidated statement of cash flows data for the years ended December 31, 2015 and 2014, and the period from January 1 through September 22, 2013, and the period from September 23 through December 31, 2013 has been restated to reflect the adoption of the new standards. |
40
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our “Selected Financial Data” and the consolidated financial statements and the related notes included elsewhere in “Financial Statements and Supplementary Data.” Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report, including information with respect to our plans and strategy for our business, includes forward‑looking statements that involve risks and uncertainties. You should read the “Risk Factors” section of this Annual Report on Form 10‑K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward‑looking statements contained in the following discussion and analysis.
Overview
We are one of the world’s leading global CROs, by revenue, providing outsourced clinical development services to the biotechnology and pharmaceutical industries. We believe we are one of a select group of CROs with the expertise and capability to conduct clinical trials across major therapeutic areas on a global basis. Our therapeutic expertise includes areas that are among the largest in pharmaceutical development, and we focus in particular on oncology, central nervous system inflammation, respiratory, cardiometabolic and infectious diseases. We believe that we further differentiate ourselves from our competitors through our investments in medical informatics and clinical technologies designed to enhance efficiencies, improve study predictability and provide better transparency for our clients throughout their clinical development processes.
Contracts define the relationships with our clients and establish the way we earn revenue. Three types of relationships are most common: a fixed‑price contract, a time and materials contract and fee‑for‑service arrangements. In cases where the contracts are fixed price, we may bear the cost of overruns for the contracted scope, or we may benefit if the costs are lower than we anticipated for the contracted scope. In cases where our contracts are fee‑for‑service, the contracts contain an overall budget for contracted resources. If actual resources used are lower than anticipated, the client generally keeps the savings and we may be responsible for covering the cost of the unused resource if we are unable to redeploy the resource. For time and material contracts, we bill the client only for the actual hours we spend to complete the contracted scope based upon stated hourly rates by position. The duration of our contracts range from a few months to several years. Revenue for services is recognized only after persuasive evidence of an arrangement exists, the sales price is determinable, services have been rendered, and collectability is reasonably assured. Once these criteria have been met, we recognize revenue for the services provided on fixed‑fee contracts based on the proportional performance methodology, which determines the proportion of outputs or performance obligations which have been completed or delivered compared to the total contractual outputs for performance obligations. To measure performance, we compare the contract costs incurred to estimated total contract costs through completion. As part of the client proposal and contract negotiation process, we develop a detailed project budget for the direct costs based on the scope of the work, the complexity of the study, the geographical location involved and our historical experience. We then establish the individual contract pricing based on our internal pricing guidelines, discount agreements, if any, and negotiations with the client. The estimated total contract costs are reviewed and revised periodically throughout the lives of the contracts, with adjustments to revenue resulting from such revisions being recorded on a cumulative basis in the period in which the revisions are first identified. Our costs consist of expenses necessary to carry out the clinical development project undertaken by us on behalf of the client. These costs primarily include the expense of obtaining appropriately qualified labor to administer the project, which we refer to as direct cost headcount. Other costs we incur are attributable to the expense of operating our business generally, such as leases and maintenance of information technology and equipment. Revenue from time and materials contracts is recognized as hours are incurred. Revenues and the related costs of fee‑for‑service contracts are recognized in the period in which services are performed.
How We Assess the Performance of Our Business
In addition to our GAAP financial measures, we review various financial and operational metrics, including, new business awards, cancellations, and backlog. Many of our current contracts include clinical trials covering multiple geographic locations. We utilize the same management systems and reporting tools to monitor and manage these activities on the same basis worldwide. For this reason, we consider our operations to be a single business segment, and we present our results of operations as a single reportable segment.
41
Our gross new business awards for the years ended December 31, 2016, 2015 and 2014 were $2,367.1 million, $1,927.6 million and $1,745.4 million, respectively. New business awards arise when a client selects us to execute its trial and is documented by written or electronic correspondence or for our Strategic Solutions offering when the amount of revenue expected to be recognized is measurable. The number of new business awards can vary significantly from year to year, and awards can have terms ranging from several months to several years. For our Strategic Solutions offering, the value of a new business award is the anticipated service revenue to be recognized in the corresponding quarter of the next fiscal year. For the remainder of our business, the value of a new award is the anticipated service revenue over the life of the contract, which does not include reimbursement activity or investigator fees.
In the normal course of business, we experience contract cancellations, which are reflected as cancellations when the client provides us with written or electronic correspondence that the work should cease. During the years ended December 31, 2016, 2015 and 2014 we had $290.6 million, $231.0 million, and $251.7 million, respectively, of cancellations for which we received correspondence from the client. The number of cancellations can vary significantly from year to year. The value of the cancellation is the remaining amount of unrecognized service revenue, less the estimated effort to transition the work back to the client.
Our backlog consists of anticipated service revenue from new business awards that either have not started or are in process but have not been completed. Backlog varies from period to period depending upon new business awards and contract modifications, cancellations, and the amount of service revenue recognized under existing contracts. Our backlog at December 31, 2016, 2015 and 2014 was $2.9 billion, $2.4 billion, and $2.1 billion, respectively.
Industry Trends
ISR estimated in its ISR 2016 Market Report that the size of the worldwide CRO market was approximately $28 billion in 2015 and will grow at a 7% CAGR to $38 billion over the next five years. This growth will be driven by an increase in the amount of research and development expenditures and higher levels of clinical development outsourcing by biopharmaceutical companies.
Acquisition of PRA by Kohlberg Kravis Roberts & Co. L.P.
On September 23, 2013, we were acquired by affiliates of KKR for $1.4 billion pursuant to a plan of merger by and among the Company, merger sub and Genstar, or Merger. Upon completion of the KKR Transaction, merger sub was merged with and into PRA Holdings, Inc., Predecessor Company, which became a subsidiary of Pinnacle Holdco Parent, Inc., or Parent. On December 19, 2013, Pinnacle Holdco Parent, Inc. changed its name to PRA Global Holdings, Inc. and on July 10, 2014, PRA Global Holdings, Inc. changed its name to PRA Health Sciences, Inc.
Business Combinations
We have completed a number of acquisitions during 2015 and 2016 to enhance our capabilities and service offerings in certain areas.
On June 8, 2015, we purchased the assets of Value Health Solutions Inc., or VHS, a software development firm, for $0.5 million in cash and 47,598 unregistered shares of our common stock with a fair market value of $1.6 million; an additional $0.4 million of common stock will be issued in June 2017, less amounts reimbursable to us for any indemnification obligations of the seller. The asset purchase agreement also includes contingent consideration in the form of potential earn-out payments of up to $16.0 million.
On March 18, 2016, we acquired all of the outstanding shares of Nextrials, Inc., or Nextrials, a developer of web-based software which integrates electronic health records with clinical trials, for $4.8 million in cash and contingent consideration in the form of potential earn-out payments of up to $3.0 million.
On May 6, 2016, as part of the dissolution of the WuXiPRA Clinical Research (Shanghai) Co., Ltd. joint venture, or WuXiPRA, we acquired WuXiPRA’s Hong Kong operations for $0.3 million.
The results of operations of acquired businesses have been included since the date of acquisition.
42
See Note 4 to our audited consolidated financial statements found elsewhere in this Annual Report on Form 10-K for additional information with respect to the acquisitions.
Joint Ventures
On May 6, 2016, we and WuXi AppTec (Shanghai) Co., Ltd., or WuXi, finalized an agreement to dissolve the WuXiPRA joint venture. Under the agreement, we sold our 49% portion of the joint venture located in mainland China for $4.0 million, which subsequently became a wholly owned subsidiary of WuXi. The portion of the joint venture located in Hong Kong became our wholly owned subsidiary and was acquired for $0.3 million. As a result of the transaction, we recognized a $3.3 million gain on the sale, which is recorded in the equity in gains (losses) of unconsolidated joint ventures in the accompanying consolidated statement of operations.
During April 2015, prior to the dissolution of the WuXiPRA joint venture, we made a $3.0 million contribution to WuXiPRA, along with WuXi, to fund the joint venture’s working capital needs. Our interest in WuXiPRA remained at 49% after the capital contribution. We recorded reductions to the investment balance of $0.7 million (excluding the gain on the sale), $2.9 million, and $2.1 million during the years ended December 31, 2016, 2015, and 2014, respectively, for our equity in the venture’s net loss for the periods, which are recorded in the equity in gains (losses) of unconsolidated joint ventures, net of tax in our consolidated statement of operations. The investment was adjusted for our equity in the venture’s net income (loss), cash contributions, distributions, and other adjustments required by the equity method of accounting. Our investment in WuXiPRA totaled $1.1 million as of December 31, 2015.
We entered into a joint venture agreement with A2 Healthcare Corporation (formerly part of Asklep, Inc.). The joint venture provides research and development outsourcing solutions in Japan to the biopharmaceutical and medical device industries. This joint venture is based in Tokyo, Japan and is owned by us (49%) and Asklep (51%). On October 17, 2014, the joint venture changed its name from RPS Asklep, Inc. to A2PRA Corporation, or A2PRA. We recorded a $0.1 million increase to the investment balance during the year ended December 31, 2016. There was no change in the investment balance for the year ended December 31, 2015 and we recorded a $0.1 million reduction to the investment balance during the year ended December 31, 2014, for our equity in the venture’s net income (loss) for the period, which is recorded in the equity in gains (losses) of unconsolidated joint venture, net of tax in our consolidated statement of operations. The investment will be adjusted for our equity in the venture’s net income (loss), cash contributions, distributions, and other adjustments required by the equity method of accounting. Our investment in A2PRA totaled $0.3 million and $0.2 million at December 31, 2016 and 2015, respectively.
In August 2015, we entered into a joint venture, along with an affiliate of KKR. The joint venture was dissolved in December 2015. The purpose of the joint venture included, among other things, the evaluation of investments or acquisitions to enhance our strategic objectives. The joint venture was jointly owned by us (11%) and KKR (89%). We contributed $20.0 million to the joint venture in August 2015 and received $19.5 million in December 2015 when the joint venture was dissolved. We recorded the $0.5 million reduction to the investment balance in equity in gains (losses) of unconsolidated joint ventures, net of tax in the consolidated statement of operations. The investment in the joint venture was adjusted for our equity in the venture’s net income (loss), cash contributions, distributions, and other adjustments required by the equity method of accounting.
Sources of Revenue
Total revenues are comprised of service revenue and reimbursement revenue, each of which is described below.
Service Revenue
We generally enter into contracts with customers to provide services with payments based on either fixed‑fee, time and materials, or fee‑for‑service arrangements. Revenue for services is recognized only after persuasive evidence of an arrangement exists, the sales price is determinable, services have been rendered, and collectability is reasonably assured.
Once these criteria have been met, we recognize revenue for the services provided on fixed‑fee contracts based on the proportional performance methodology, which determines the proportion of outputs or performance obligations which have been completed or delivered compared to the total contractual outputs for performance obligations. To measure performance, we compare the contract costs incurred to estimated total contract costs through completion. As
43
part of the client proposal and contract negotiation process, we develop a detailed project budget for the direct costs based on the scope of the work, the complexity of the study, the geographical location involved and our historical experience. We then establish the individual contract pricing based on our internal pricing guidelines, discount agreements, if any, and negotiations with the client. The estimated total contract costs are reviewed and revised periodically throughout the lives of the contracts, with adjustments to revenue resulting from such revisions being recorded on a cumulative basis in the period in which the revisions are first identified. Revenue from time and materials contracts is recognized as hours are incurred. Billable hours typically fluctuate during the terms of individual contracts, as services we provide generally increase at the beginning of a study and decrease toward the end of a study. Revenues and the related costs of fee‑for‑service contracts are recognized in the period in which services are performed.
A majority of our contracts undergo modifications over the contract period and our contracts provide for these modifications. During the modification process, we recognize revenue to the extent we incur costs, provided client acceptance and payment is deemed reasonably assured.
We often offer volume discounts to our large customers based on annual volume thresholds. We record an estimate of the annual volume rebate as a reduction of revenue throughout the period based on the estimated total rebate to be earned for the period.
Most of our contracts can be terminated by the client either immediately or after a specified period, typically 30 to 60 days, following notice. In the case of early termination, these typically contracts require payment to us of fees earned to date, the fees, and in some cases, a termination fee or some portion of the fees or profit that we could have earned under the contract if it had not been terminated early. Based on ethical, regulatory, and health considerations, this wind‑down activity may continue for several quarters or years. Therefore, revenue recognized prior to cancellation generally does not require a significant adjustment upon cancellation.
Increases in the estimated total direct costs to complete a contract without a corresponding proportional increase to the total contract price result in a cumulative adjustment to the amount of revenue recognized in the period the change in estimate is determined.
Our service revenue was $1,580.0 million, $1,375.8 million, and $1,266.6 million for the years ended December 31, 2016, 2015 and 2014, respectively. Changes in service revenue from period to period are driven primarily by changes in backlog at the beginning of a period, as well as new business awards during such period. Additionally, service revenue and billable hours will generally be impacted by the mix of studies that are active during a period, as different studies have different staffing requirements, as well as the life cycles of projects that are active during a period.
Our service revenues are derived from a wide range of client types. During the year ended December 31, 2016, we derived 52% of our service revenue from large pharmaceutical companies, 14% of our service revenue from small‑ to mid‑sized pharmaceutical companies, 19% of our service revenue from large biotechnology companies and 15% of our service revenue from all other biotechnology companies. For the years ended December 31, 2016, 2015, and 2014, our top five clients represented approximately 45%, 41%, and 38%, respectively, of service revenue; this revenue was derived from a combination of fixed‑fee contracts, fee‑for‑service contracts and time and materials contracts. Two of our clients accounted for 11.0% and 10.4% of service revenue during the year ended December 31, 2016, respectively. One client accounted for 10.7% of service revenue during the year ended December 31, 2015. No client accounted for 10% or more of service revenue for the year ended December 31, 2014. No individual project accounted for 10% or more of service revenue for the years ended December 31, 2016, 2015 and 2014.
Reimbursement Revenue and Reimbursable Out‑of‑Pocket Costs
We incur out‑of‑pocket costs, which are reimbursable by our customers. We include these out‑of‑pocket costs as reimbursement revenue and reimbursable out‑of‑pocket expenses in our consolidated statement of operations.
As is customary in our industry, we also routinely enter into separate agreements on behalf of our clients with independent physician investigators in connection with clinical trials. We also receive funds from our clients for investigator fees, which are netted against the related costs, since such fees are the obligation of our clients, without risk or reward to us. We are not obligated either to perform the service or to pay the investigator in the event of default by the client. In addition, we do not pay the independent physician investigator until funds are received from the client. Accordingly, unlike reimbursable out‑of‑pocket costs, we do not recognize these investigator fees in revenue.
44
Reimbursement costs and investigator fees are not included in our backlog because they are pass‑through costs to our clients.
We believe that the fluctuations in reimbursement costs and reimbursement revenue from period to period are not meaningful to our underlying performance.
Costs and Expenses
Our costs and expenses are comprised primarily of our direct costs, selling, general and administrative costs, depreciation and amortization and income taxes. In addition, we incur reimbursable out‑of‑pocket expenses; however, as noted above, our reimbursable out‑of‑pocket expenses are directly offset by our reimbursement revenue. Since reimbursement revenue is offset by our out‑of‑pocket reimbursable expenses, we monitor and measure costs as a percentage of service revenue rather than total revenue as we believe this is a more meaningful comparison and better reflects the operations of our business.
Direct Costs
Our direct costs consist primarily of labor‑related charges. They include elements such as salaries, benefits and incentive compensation for our employees. In addition, we utilize staffing agencies to procure primarily part time individuals to perform work on our contracts. Labor-related charges as a percentage of our total direct costs were 96.6%, 95.7%, and 94.8% for the years ended December 31, 2016, 2015 and 2014, respectively. The cost of labor procured through staffing agencies is included in these percentages and represents 5.1%, 4.1%, and 5.1% of total direct costs for the years ended December 31, 2016, 2015 and 2014, respectively. Our remaining direct costs are items such as travel, meals, postage and freight, patient costs, medical waste and supplies. The total of all these items as a percentage of total direct cost was 3.4%, 4.3%, and 5.2% for the year ended December 31, 2016, 2015 and 2014, respectively.
Historically, direct costs have increased with an increase in service revenues. The future relationship between direct costs and service revenues may vary from historical relationships. Direct costs as a percentage of service revenues were 65.4%, 64.4%, and 67.8% during the years ended December 31, 2016, 2015, and 2014, respectively. Several factors will cause direct costs to decrease as a percentage of service revenues. Deployment of our billable staff in an optimally efficient manner has the most impact on our ratio of direct cost to service revenue. The most effective deployment of our staff is when they are fully engaged in billable work and are accomplishing contract related activities at a rate that meets or exceeds budgeted targets. We also seek to optimize our efficiency by performing work using the employee with the lowest cost. Generally, the following factors may cause direct costs to increase as a percentage of service revenues: our staff are not fully deployed, as is the case when there are unforeseen cancellations or delays, or when our staff are accomplishing tasks at levels of effort that exceed budget, such as rework; as well as pricing pressure from increased competition.
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist of administration payroll and benefits, marketing expenditures, and overhead costs such as information technology and facilities costs. These expenses also include central overhead costs that are not directly attributable to our operating business and include certain costs related to insurance, professional fees and property.
Loss on Modification or Extinguishment of Debt
Loss on extinguishment of debt for the year ended December 31, 2016 was associated with our cash tender offer on our 9.5% senior notes due 2023, or Senior Notes, and the refinancing of our variable rate first lien term loan due 2020, or 2013 First Lien Term Loan, and revolving line of credit, or 2013 Revolver, collectively known as the 2013 Credit Facilities. Loss on extinguishment of debt for the year ended December 31, 2014 consists of previously capitalized unamortized debt financing costs that were expensed as a result of the repricing and the debt repayment in conjunction with our initial public offering, or IPO.
45
Transaction‑Related Costs
Transaction-related costs consist of expenses incurred with our secondary offerings, the closing of our accounts receivable financing agreement, transaction-related stock-based compensation awards and our refinancing of the 2013 Credit Facilities.
Depreciation and Amortization
Depreciation represents the depreciation charged on our fixed assets. The charge is recorded on a straight‑line method, based on estimated useful lives of three to seven years for computer hardware and software and five to seven years for furniture and equipment. Leasehold improvements are depreciated over the lesser of the life of the lease term or the useful life of the improvements. Amortization expense consists of amortization recorded on acquisition‑related intangible assets. Customer relationships, backlog and finite‑lived trade names are amortized on an accelerated basis, which coincides with the period of economic benefit we expect to receive. All other finite‑lived intangibles are amortized on a straight‑line basis. In accordance with GAAP, we do not amortize goodwill and indefinite‑lived intangible assets.
Income Taxes
Because we conduct operations on a global basis, our effective tax rate has and will continue to depend upon the geographic distribution of our pre‑tax earnings among several different taxing jurisdictions. Our effective tax rate can also vary based on changes in the tax rates of the different jurisdictions. Our effective tax rate is also impacted by tax credits and the establishment or release of deferred tax asset valuation allowances and tax reserves, as well as significant non‑deductible items such as portions of transaction‑related costs.
Foreign subsidiaries are taxed separately in their respective jurisdictions. We have foreign net operating loss carryforwards in some jurisdictions. The carryforward periods for these losses vary from five years to an indefinite carryforward period depending on the jurisdiction. Our ability to offset future taxable income with the net operating loss carryforwards may be limited in certain instances, including changes in ownership.
Exchange Rate Fluctuations
The majority of our foreign operations transact in the Euro or British Pound. As a result, our revenue and expenses are subject to exchange rate fluctuations with respect to these currencies. We have translated these currencies into U.S. dollars using the following average exchange rates:
|
|
|
Years Ended December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
U.S. Dollars per: |
|
|
|
|
|
|
|
|
Euro |
|
1.11 |
|
1.11 |
|
1.33 |
|
|
British Pound |
|
1.35 |
|
1.53 |
|
1.65 |
|
46
Results of Operations
Year Ended December 31, 2016 Compared to the Year Ended December 31, 2015
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31, |
|
||||
|
(in thousands) |
|
2016 |
|
2015 |
|
||
|
Revenue |
|
|
|
|
|
|
|
|
Service revenue |
|
$ |
1,580,023 |
|
$ |
1,375,847 |
|
|
Reimbursement revenue |
|
|
231,688 |
|
|
238,036 |
|
|
Total revenue |
|
|
1,811,711 |
|
|
1,613,883 |
|
|
Operating expenses |
|
|
|
|
|
|
|
|
Direct costs |
|
|
1,032,688 |
|
|
886,528 |
|
|
Reimbursable out-of-pocket costs |
|
|
231,688 |
|
|
238,036 |
|
|
Selling, general and administrative |
|
|
269,893 |
|
|
246,417 |
|
|
Transaction-related costs |
|
|
44,834 |
|
|
— |
|
|
Depreciation and amortization |
|
|
69,506 |
|
|
77,952 |
|
|
Loss on disposal of fixed assets |
|
|
753 |
|
|
652 |
|
|
Income from operations |
|
|
162,349 |
|
|
164,298 |
|
|
Interest expense, net |
|
|
(54,913) |
|
|
(61,747) |
|
|
Loss on modification or extinguishment of debt |
|
|
(38,178) |
|
|
— |
|
|
Foreign currency gains, net |
|
|
24,029 |
|
|
14,048 |
|
|
Other income (expense), net |
|
|
607 |
|
|
(1,434) |
|
|
Income before income taxes and equity in gains (losses) of unconsolidated joint ventures |
|
|
93,894 |
|
|
115,165 |
|
|
Provision for income taxes |
|
|
28,494 |
|
|
30,004 |
|
|
Income before equity in gains (losses) of unconsolidated joint ventures |
|
|
65,400 |
|
|
85,161 |
|
|
Equity in gains (losses) of unconsolidated joint ventures, net of tax |
|
|
2,775 |
|
|
(3,396) |
|
|
Net income |
|
$ |
68,175 |
|
$ |
81,765 |
|
Service revenue increased by $204.2 million, or 14.8%, from $1,375.8 million during the year ended December 31, 2015 to $1,580.0 million during the year ended December 31, 2016. Service revenue for the year ended December 31, 2016 benefited from an increase in billable hours and an increase in the effective rate of the hours billed on our studies, offset by an unfavorable impact of $5.3 million from foreign currency exchange rate fluctuations. The growth in service revenue and the increase in billable hours were due largely to the increase in our backlog as we entered the year, the type of services we are providing on our active studies, which was driven by the life cycles of projects that were active during the period, the growth in new business awards as a result of higher demand for our services across the industries we serve, and more effective sales efforts and the growth in the overall CRO market. New business awards arise when a client selects us to execute its trial. The number of awards can vary significantly from period to period and our studies have terms ranging from several months to several years. The increase in our effective rate of the hours billed on our studies is attributable to the contract pricing terms on our current mix of active studies and the mix of clients and the services that we provide to those clients.
Direct costs increased by $146.2 million, or 16.5%, from $886.5 million during the year ended December 31, 2015 to $1,032.7 million during the year ended December 31, 2016. The increase in direct costs was primarily due to an increase in labor-related costs of $170.6 million, as we continued to hire billable staff to support our current projects and as we hired additional staff in anticipation of our growing portfolio of studies, offset by a favorable impact of $21.3 million from foreign currency exchange rate fluctuations. Direct costs as a percentage of service revenue increased from 64.4% during the year ended December 31, 2015 to 65.4% during the year ended December 31, 2016. This increase in direct costs as a percentage of service revenue is primarily due to the $8.3 million impact of research and development credits, or R&D Credits, recorded during the year ended December 31, 2015 that related to prior years. The R&D Credits are the result of a comprehensive analysis we have been performing across the organization to determine whether expenditures incurred qualify as research and development as defined by the respective jurisdiction.
Selling, general and administrative expenses increased by $23.5 million, or 9.5%, from $246.4 million during the year ended December 31, 2015 to $269.9 million during the year ended December 31, 2016. Selling, general and administrative expenses as a percentage of service revenue decreased from 17.9% during the year ended December 31, 2015 to 17.1% during the year ended December 31, 2016. This decrease in selling, general and
47
administrative expenses as a percentage of service revenue is primarily related to our continued efforts to effectively manage our selling and administrative functions as we continue to grow.
During the year ended December 31, 2016, we incurred transaction-related expenses of $44.8 million. The costs consist of $10.1 million of stock-based compensation expense associated with the release of the transfer restrictions on a portion of shares issuable upon exercise of vested service-based options in connection with the announcement of our March, May, and November 2016 secondary offerings. The costs also include $32.0 million of stock-based compensation expense related to the vesting and release of the transfer restrictions of certain performance-based stock options. In addition, we incurred $2.7 million of third-party fees associated with the secondary offerings and the closing of our accounts receivable financing agreement. There were no transaction-related expenses incurred for the year ended December 31, 2015.
Depreciation and amortization expense decreased by $8.4 million, or 10.8%, from $78.0 million during the year ended December 31, 2015 to $69.5 million during the year ended December 31, 2016. Depreciation and amortization expense as a percentage of service revenue was 5.7% during the year ended December 31, 2015 and 4.4% during the year ended December 31, 2016. The decrease in depreciation and amortization expense as a percentage of service revenue is primarily due the continued decline in amortization of our acquired intangibles, which are amortized on an accelerated basis.
Interest expense, net decreased by $6.8 million from $61.7 million during the year ended December 31, 2015 to $54.9 million during the year ended December 31, 2016. The cash tender on our Senior Notes during 2016, as well as a 0.8% decrease in the weighted average interest rate on our outstanding debt as compared to the year ended December 31, 2015, resulted in a $9.7 million reduction in interest expense. Additionally, interest expense decreased $1.6 million due to lower amortization of debt issuance costs, which was offset by an increase of $4.7 million related to the amortization of our terminated interest rate swaps and interest expense on our current interest rate swap.
Losses on modification or extinguishment of debt were $38.2 million during the year ended December 31, 2016 and there were no losses on modification of debt during the year ended December 31, 2015. The $38.2 million loss on extinguishment of debt incurred during the year ended December 31, 2016 was associated with our cash tender offer on our Senior Notes and our refinancing of the 2013 Credit Facilities. The loss of $21.5 million due to our cash tender offer consisted of a $17.4 million early tender premium, a $3.7 million write-off of unamortized debt issuance cost and $0.4 million of fees associated with the transaction. The refinancing of our 2013 Credit Facilities resulted in a $16.7 million loss on extinguishment of debt, which consisted of the write-off of $15.8 million write-off of unamortized debt issuance costs and $0.9 million of fees associated with the transaction.
Foreign currency gains, net increased by $10.0 million from $14.0 million during the year ended December 31, 2015 to $24.0 million during the year ended December 31, 2016. The foreign currency gains and losses are due to fluctuations in the U.S. dollar, gains or losses that arise in connection with the revaluation of short-term inter-company balances between our domestic and international subsidiaries, and gains or losses from foreign currency transactions, such as those resulting from the settlement of third-party accounts receivables and payables denominated in a currency other than the local currency of the entity making the payment. During the year ended December 31, 2016, the foreign currency gains were primarily a result of the weakening of the British Pound against the U.S. dollar by 16.7% following the decision by voters in the United Kingdom, to approve a referendum to exit the European Union, commonly referred to as Brexit, in June 2016. During the year ended December 31, 2015, foreign currency gains were primarily due to the devaluation of the Canadian dollar, Euro and British Pound against the U.S. dollar by 16.1%, 10.1% and 4.6%, respectively.
Provision for income taxes decreased by $1.5 million from $30.0 million during the year ended December 31, 2015 to $28.5 million during the year ended December 31, 2016. Our effective tax rate was 30.3% and 26.1% during the years ended December 31, 2016 and December 31, 2015, respectively. The change in the effective tax rate was primarily attributable to a decrease in global pre-tax income related to an increase in the overall U.S. loss, the impact of that loss on the valuation allowance and the increase of foreign earnings that are taxed currently in the U.S.
48
Year Ended December 31, 2015 Compared to the Year Ended December 31, 2014
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31, |
|
||||
|
(in thousands) |
|
2015 |
|
2014 |
|
||
|
Revenue |
|
|
|
|
|
|
|
|
Service revenue |
|
$ |
1,375,847 |
|
$ |
1,266,596 |
|
|
Reimbursement revenue |
|
|
238,036 |
|
|
192,990 |
|
|
Total revenue |
|
|
1,613,883 |
|
|
1,459,586 |
|
|
Operating expenses |
|
|
|
|
|
|
|
|
Direct costs |
|
|
886,528 |
|
|
859,218 |
|
|
Reimbursable out-of-pocket costs |
|
|
238,036 |
|
|
192,990 |
|
|
Selling, general and administrative |
|
|
246,417 |
|
|
253,970 |
|
|
Depreciation and amortization |
|
|
77,952 |
|
|
96,564 |
|
|
Loss on disposal of fixed assets |
|
|
652 |
|
|
5 |
|
|
Income from operations |
|
|
164,298 |
|
|
56,839 |
|
|
Interest expense, net |
|
|
(61,747) |
|
|
(81,939) |
|
|
Loss on modification or extinguishment of debt |
|
|
— |
|
|
(25,036) |
|
|
Foreign currency gains, net |
|
|
14,048 |
|
|
10,538 |
|
|
Other expense, net |
|
|
(1,434) |
|
|
(2,254) |
|
|
Income (loss) before income taxes and equity in losses of unconsolidated joint ventures |
|
|
115,165 |
|
|
(41,852) |
|
|
Provision for (benefit from) income taxes |
|
|
30,004 |
|
|
(8,154) |
|
|
Income (loss) before equity in losses of unconsolidated joint ventures |
|
|
85,161 |
|
|
(33,698) |
|
|
Equity in losses of unconsolidated joint ventures, net of tax |
|
|
(3,396) |
|
|
(2,044) |
|
|
Net income (loss) |
|
$ |
81,765 |
|
$ |
(35,742) |
|
Service revenue increased by $109.3 million, or 8.6%, from $1,266.6 million during the year ended December 31, 2014 to $1,375.8 million during the year ended December 31, 2015. Service revenue for the year ended December 31, 2015 benefited from an increase in billable hours and the effective rate of the hours billed on our studies, offset by an unfavorable impact of $45.4 million from foreign currency exchange rate fluctuations. The growth in service revenue and the increase in billable hours were due largely to the increase in our backlog as we entered the year, the type of services we are providing on our active studies, which was driven by the life cycles of projects that were active during the period, the growth in new business awards as a result of higher demand for our services across the industries we serve, and more effective sales efforts and the growth in the overall CRO market. New business awards arise when a client selects us to execute its trial. The number of awards can vary significantly from period to period and our studies have terms ranging from several months to several years. The increase in our effective rate of the hours billed on our studies is attributable to the contract pricing terms on our current mix of active studies and the mix of clients and the services that we provide to those clients.
Direct costs increased by $27.3 million, or 3.2%, from $859.2 million during the year ended December 31, 2014 to $886.5 million during the year ended December 31, 2015. The increase in direct costs was primarily due to an increase in salaries and related benefits of $104.3 million, as we continued to hire billable staff to support our current projects and as we hired additional staff in anticipation of our growing portfolio of studies, offset by an $8.3 million benefit due to the favorable impact of the R&D Credits recorded in the current period that relate to prior tax years and a favorable impact of $64.2 million from foreign currency exchange rate fluctuations. The R&D Credits are the result of a comprehensive analysis we have been performing across the organization to determine whether expenditures incurred qualify as research and development as defined by the respective jurisdiction. Direct costs as a percentage of service revenue decreased from 67.8% during the year ended December 31, 2014 to 64.4% during the year ended December 31, 2015. This decrease in direct costs as a percentage of service revenue is primarily due to the favorable impact from foreign currency exchange rate fluctuations and the impact of the R&D Credits.
Selling, general and administrative expenses decreased by $7.6 million, or 3.0%, from $254.0 million during the year ended December 31, 2014 to $246.4 million during the year ended December 31, 2015. Selling, general and administrative expenses as a percentage of service revenue decreased from 20.1% during the year ended December 31, 2014 to 17.9% during the year ended December 31, 2015. This decrease in selling, general and administrative expenses as a percentage of service revenue is primarily related to our continued ability to effectively manage our selling and
49
administrative functions and a termination fee of $11.9 million we paid KKR in connection with the completion of the IPO during the year ended December 31, 2014.
Depreciation and amortization expense decreased by $18.6 million, or 19.3%, from $96.6 million during the year ended December 31, 2014 to $78.0 million during the year ended December 31, 2015. Depreciation and amortization expense as a percentage of service revenue was 7.6% during the year ended December 31, 2014 and 5.7% during the year ended December 31, 2015. The decrease in depreciation and amortization expense as a percentage of service revenue is primarily due the continued decline in amortization of our acquired intangibles, which are amortized on an accelerated basis.
Interest expense, net decreased by $20.2 million from $81.9 million during the year ended December 31, 2014 to $61.7 million during the year ended December 31, 2015. This decrease in interest expense is related to the paydowns to our 2013 First Lien Term Loan and Senior Notes made using proceeds from our IPO in November 2014 and repayments made to our 2013 First Lien Term Loan during 2015.
Losses on modification or extinguishment of debt were $25.0 million during the year ended December 31, 2014 and there were no losses on modification of debt during the year ended December 31, 2015. The $25.0 million loss on modification or extinguishment of debt incurred in the year ended December 31, 2014 was due to a $1.4 million loss on modification debt as a result of our repricing transaction which took place on March 24, 2014, and a $23.7 million loss on modification debt due to a $14.3 million prepayment penalty and $9.4 million of unamortized debt costs being written off as a result of our repayment which took place on November 18, 2014 in conjunction with our IPO.
Foreign currency gains, net increased by $3.5 million from $10.5 million during the year ended December 31, 2014 to $14.0 million during the year ended December 31, 2015. The foreign currency gains and losses are due to fluctuations in the U.S. dollar, gains or losses that arise in connection with the revaluation of short-term inter-company balances between our domestic and international subsidiaries, and gains or losses from foreign currency transactions, such as those resulting from the settlement of third-party accounts receivables and payables denominated in a currency other than the local currency of the entity making the payment. During the year ended December 31, 2015, foreign currency gains were primarily due to the devaluation of the Canadian dollar, Euro and British Pound against the U.S. dollar by 16.1%, 10.1% and 4.6%, respectively. During the year ended December 31, 2014 foreign currency gains were primarily due to the devaluation of the Euro, Canadian dollar and British Pound against the U.S. dollar by 11.8%, 8.1% and 5.7%, respectively.
Provisions for (benefit from) income taxes increased by $38.2 million from a benefit of $8.2 million during the year ended December 31, 2014 to an income tax provision of $30.0 million during the year ended December 31, 2015. Our effective tax rate was 26.1% during the year ended December 31, 2015 and was a benefit rate of 19.5% for the year ended December 31, 2014. The change in the effective tax rate was primarily attributable to fact that the Company was in an overall pre-tax income position for the year ended December 31, 2015 as compared to an overall pre-tax loss for the year-ended December 31, 2014.
Seasonality
Although our business is not generally seasonal, we typically experience a slight decrease in our revenue growth rate during the fourth quarter due to holiday vacations and a similar decrease in new business awards in the first quarter due to our clients’ budgetary cycles and vacations during the year‑end holiday period.
Liquidity and Capital Resources
We assess our liquidity in terms of our ability to generate cash to fund our operating, investing and financing activities. Our principal source of liquidity is operating cash flows. As of December 31, 2016, we had approximately $144.6 million of cash and cash equivalents of which $54.3 million was held by our foreign subsidiaries. Our expected primary cash needs on both a short and long‑term basis are for capital expenditures, expansion of services, geographic expansion, debt repayments, and other general corporate purposes. We have historically funded our operations and growth, including acquisitions, with cash flow from operations, borrowings, and issuances of equity securities. We expect to continue expanding our operations through internal growth and strategic acquisitions and investments. We expect these activities will be funded from existing cash, cash flow from operations and, if necessary or appropriate, borrowings under our existing or future credit facilities. Our sources of liquidity could be affected by our dependence on
50
a small number of industries and clients, compliance with regulations, international risks, and personal injury, environmental or other material litigation claims.
Cash Collections
Cash collections from accounts receivable were $2,074.1 million during the year ended December 31, 2016, including $248.2 million of funds received from customers to pay independent physician investigators, or investigators, as compared to $1,831.1 million during the year ended December 31, 2015, including $231.4 million of funds received from customers to pay investigators, $1,654.2 million during the year ended December 31, 2014, including $248.2 million of funds received from customers to pay investigators. The increase in cash collections is related to our increase in revenue, driven by an increase in new business awards and backlog.
Discussion of Cash Flows
Cash Flow from Operating Activities
Cash provided by operating activities increased by $7.6 million during the year ended December 31, 2016 as compared to 2015. The increase in operating cash flow reflects increased cash flows from our operating performance and a reduction in interest payments, which was partially offset by an increase in cash outflows primarily from working capital. The changes in working capital were driven by a $2.1 million decrease in accounts payable and accrued expenses during the year ended December 31, 2016 as compared to $21.3 million increase during the year ended December 31, 2015 and is attributable to the timing and payment of invoices. This is partially offset by a $9.1 million improvement in cash outflows from our accounts receivable, unbilled services, and advanced billings accounts, driven by a slower rate of increase in our days sales outstanding during the year ended December 31, 2016.
Cash provided by operating activities increased by $118.4 million during the year ended December 31, 2015 as compared to 2014. The increase in operating cash flow reflects an increase in net income, a reduction in interest payments, as well as reduction in cash outflows from working capital. Interest payments decreased by $26.1 million, primarily due to the debt payments made in conjunction with the IPO during November 2014. Additionally, net income for the year ended December 31, 2014 included a $11.9 million fee paid to terminate our monitoring agreement with KKR. DSO contributed to a $31.3 million improvement in cash flow from operations and reflects a slower rate of increase in our DSO in 2015 as compared to 2014. DSO can shift significantly at each reporting period depending on the timing of cash receipts under contractual payment terms relative to the recognition of revenue over a project lifecycle.
Cash Flow from Investing Activities
Net cash used in investing activities decreased by $37.1 million during the year ended December 31, 2016 as compared to 2015. The decrease in the cash outflows was primarily due to the $32.9 million payment for the termination of our interest rate swaps during 2015 and a $7.2 million change in cash flows related to our unconsolidated joint ventures. During the year ended December 31, 2015, we made $23.0 million in capital contributions to our unconsolidated joint ventures and received $19.5 million from the dissolution of our joint venture with KKR, and during the year ended December 31, 2016, we received $3.7 million from the sale of our ownership stake in our WuXiPRA joint venture.
Net cash used in investing activities increased by $60.2 million during the year ended December 31, 2015 as compared to 2014. The increase in the cash outflows was due, in part, to a $32.9 million payment for the termination of our interest rate swaps during 2015. Cash flows relating to working capital adjustments associated with our recent acquisitions contributed to an additional $17.5 million of the change. A $5.5 million increase in fixed asset purchases during 2015, primarily related to our new bioanalytical laboratory in the Netherlands and the capitalized software costs for our Predictivv platform, also contributed to the change.
Cash Flow from Financing Activities
Net cash used in financing activities during the year ended December 31, 2016 was $101.6 million compared to $42.4 million for the same period of 2015. During the year ended December 31, 2016, we entered into an accounts receivable financing agreement and received proceeds of $120.0 million which was used to repay $133.6 million aggregate principal on our Senior Notes, as part of a cash tender offer. In addition, we voluntarily repaid $689.0 million
51
in principal balance on the 2013 First Lien Term Loan and received $625.0 million in proceeds from the 2016 First Lien Term Loan. During 2016, we also paid $25.5 million in debt extinguishment and debt issuance costs. For the year ended December 31, 2015, we voluntarily repaid $40.0 million on our 2013 First Lien Term Loan.
Net cash used in financing activities during the year ended December 31, 2015 was $42.4 million compared to $6.0 million of net cash used in financing activities for the same period of 2014. During 2015, we voluntarily repaid $40.0 million on our 2013 First Lien Term Loan. During 2014, we received $328.0 million of net proceeds from the issuance of 19,523,255 shares of common stock in connection with our IPO, we repaid $308.8 million of previously existing bank and subordinated debt and paid $14.3 million in debt prepayment and debt extinguishment costs.
Indebtedness
On March 17, 2016, we repaid $133.6 million aggregate principal on our Senior Notes, as part of a cash tender offer. In accordance with the guidance in ASC 470-50 the debt repayment was accounted for as a partial debt extinguishment. The repayment resulted in a $17.4 million early tender premium, a $3.7 million write-off of unamortized debt issuance costs and $0.4 million of fees associated with the transaction, which are included in loss on modification or extinguishment of debt in the consolidated statement of operations.
On March 22, 2016, we entered into a $140.0 million accounts receivable financing agreement, of which $120.0 million was outstanding as of December 31, 2016. The borrowings were used to repay amounts outstanding on the Company’s revolving credit facility that were used to fund the cash tender offer for the Senior Notes.
On December 6, 2016, we refinanced our 2013 Credit Facilities to obtain a lower interest rate. We entered into a new credit agreement with a syndicate of banks for an aggregate principal amount of $625.0 million of first lien term debt, or 2016 First Lien Term Debt, and a $125.0 million revolving line of credit, or 2016 Revolver, or collectively known as the 2016 Credit Facilities. The proceeds from the 2016 Credit Facilities were used to repay the 2013 First Lien Term Loan principal balance of $614.0 million and approximately $8.0 million in related fees and expenses.
2016 Credit Facilities
Wells Fargo Bank, National Association acted as the lead arranger and bookrunner for the 2016 Credit Facilities.
The 2016 Credit Facilities provide senior secured financing of up to $750.0 million, consisting of:
|
· |
the 2016 First Lien Term Loan in an aggregate principal amount of up to $625.0 million; and |
|
· |
the 2016 Revolver in an aggregate principal amount of up to $125.0 million. |
The borrower of the 2016 First Lien Term Loan and the 2016 Revolver is Pharmaceutical Research Associates, Inc., a wholly-owned subsidiary of PRA Health Sciences, Inc. The 2016 Revolver includes borrowing capacity available for letters of credit up to $25.0 million and for up to $20.0 million of borrowings on same‑day notice, referred to as swingline loans.
The 2016 Credit Facilities provides that we have the right at any time to request incremental term loans and/or revolving commitments in an aggregate principal amount of up to (a) $275.0 million, plus (b) all voluntary prepayments and corresponding voluntary commitment reductions of the Senior Secured Credit Facilities, other than from proceeds of long-term indebtedness, prior to the date of any such incurrence, plus (c) an additional amount which, after giving effect to the incurrence of such amount, we would not exceed a consolidated net first lien secured leverage to consolidated EBITDA ratio of 3.0 to 1.0 pro forma for such incremental facilities, minus (d) the sum of (i) the aggregate principal amount of new term loan commitments and new revolving credit commitments incurred and (ii) the aggregate principal amount of certain other indebtedness incurred. The lenders under these facilities are not under any obligation to provide any such incremental commitments or loans, and any such addition of or increase in commitments or loans is subject to certain customary conditions precedent.
52
Interest Rate and Fees
Borrowings under the 2016 First Lien Term Loan and the 2016 Revolver bear interest at a rate equal to, at our option, either (a) London Interbank Offered Rate, or LIBOR, for the relevant interest period, plus an applicable margin; provided that solely with respect to the 2016 First Lien Term Loan LIBOR shall be deemed to be no less than 0.00% per annum or (b) an adjusted base rate, or the ABR rate, plus an applicable margin.
The applicable margin on our 2016 First Lien Term Loan is based on our ratio of total debt to EBITDA ratio per the table below:
|
Pricing |
|
Total indebtedness |
|
Letter of |
|
ABR Margin |
|
Adjusted LIBOR |
|
Commitment |
|
I |
|
> 3.75x |
|
2.25% |
|
1.25% |
|
2.25% |
|
0.40% |
|
II |
|
< 3.75x but > 3.00x |
|
2.00% |
|
1.00% |
|
2.00% |
|
0.35% |
|
III |
|
< 3.00x but > 2.25x |
|
1.75% |
|
0.75% |
|
1.75% |
|
0.30% |
|
IV |
|
< 2.25x but > 1.50x |
|
1.50% |
|
0.50% |
|
1.50% |
|
0.25% |
|
V |
|
< 1.50x |
|
1.25% |
|
0.25% |
|
1.25% |
|
0.20% |
In addition to paying interest on outstanding principal under the 2016 Credit Revolver, the Company is required to pay a commitment fee to the lenders under the 2016 Revolver in respect of the unutilized commitments thereunder. The commitment fee rate will be based on the ratio of total indebtedness to EBITDA on a given date. We are also required to pay customary letter of credit fees.
Prepayments
The 2016 Credit Facilities require us to prepay outstanding term loans, subject to certain exceptions, with:
|
· |
100% of the net cash proceeds of the incurrence or issuance of certain debt; and |
|
· |
100% of the net cash proceeds of $5.0 million of certain non-ordinary course asset sales and casualty and condemnation events, subject to reinvestment rights and certain other exceptions. |
The foregoing mandatory prepayments will be applied first to accrued interest and fees and second, to the scheduled installments of principal of the 2016 Credit Facilities in direct order of maturity.
We may voluntarily repay outstanding loans under the 2016 Credit Facilities at any time without premium or penalty, subject to reimbursements of the lenders’ redeployment costs actually incurred in the case of a prepayment of LIBOR borrowings other than on the last day of the relevant interest period.
Amortization and Final Maturity
The 2016 First Lien Term Loan has scheduled fixed quarterly principal payments as follows:
|
· |
1.25% by quarterly term loan amortization payments, or $7.8 million per quarter, to be made commencing March 31, 2017 and made on or prior to December 31, 2017; |
|
· |
1.88% by quarterly term loan amortization payments, or $11.7 million per quarter, to be made on or after March 31, 2018, but on or prior to December 31, 2019; |
|
· |
2.50% by quarterly term loan amortization payments, or $15.6 million per quarter, to be made on or after March 31, 2020, but on or prior to December 31, 2020; |
|
· |
3.13% by quarterly term loan amortization payments, or $19.5 million per quarter, to be made on or after March 31, 2021, but prior to September 30, 2021; and |
|
· |
60.63% (or if less, the remaining principal amount of the term loan) on December 06, 2021. |
Principal amounts outstanding under the 2016 Revolver are due and payable in full at maturity, on or about December 6, 2021.
53
Guarantee and Security
All obligations of the borrower under the 2016 Credit Facilities are unconditionally guaranteed by us and all our material, wholly‑owned U.S. restricted subsidiaries, with customary exceptions including where providing such guarantees is not permitted by law, regulation or contract or would result in material adverse tax consequences.
All obligations of the borrower under the 2016 Credit Facilities, and the guarantees of such obligations, are secured, subject to permitted liens and other exceptions, by substantially all of the assets of the borrower and each guarantor, including but not limited to: (i) a perfected pledge of all of the capital stock issued by the borrower and each guarantor and (ii) perfected security interests in substantially all other tangible and intangible assets of the borrower and the guarantors (subject to certain exceptions and exclusions).
Certain Covenants and Events of Default
The 2016 Credit Facilities contain a number of covenants that, among other things, restrict, subject to certain exceptions, our ability to:
|
· |
create any liens; |
|
· |
make investments and acquisitions; |
|
· |
incur or guarantee additional indebtedness; |
|
· |
enter into mergers or consolidations and other fundamental changes; |
|
· |
conduct sales and other dispositions of property or assets; |
|
· |
enter into sale-leaseback transactions or hedge agreements; |
|
· |
prepay subordinated debt; |
|
· |
pay dividends or make other payments in respect of capital stock; |
|
· |
change the line of business; |
|
· |
enter into transactions with affiliates; |
|
· |
enter into burdensome agreements with negative pledge clauses and clauses restriction; and |
|
· |
subsidiary distributions. |
Our 2016 Credit Facilities contain customary events of default (subject to exceptions, thresholds and grace periods), including, without limitation: (i) nonpayment of principal or interest; (ii) failure to perform or observe covenants; (iii) inaccuracy or breaches of representations and warranties; (iv) cross‑defaults with certain other indebtedness; (v) certain bankruptcy related events; (vi) impairment of certain security interests in collateral, guarantees or invalidity or unenforceability of certain 2016 Credit Facilities documents; (vii) monetary judgment defaults; (viii) certain ERISA matters; and (ix) certain change of control events.
In addition, the 2016 Revolver requires us to maintain a consolidated total debt to consolidated EBITDA ratio of 4.25 to 1.0 and consolidated EBITDA to fixed charges no less than 3.0 to 1.0 for any four consecutive fiscal quarters for which financial statements have been provided to the administrative agent as required by the Senior Secured Credit Agreement.
The 2016 Credit Facilities also contain certain customary affirmative covenants and events of default, including a change of control.
54
Senior Notes
PRA Holdings has $91.4 million aggregate principal amount of 9.5% senior notes outstanding, which mature on October 1, 2023, pursuant to the indenture. Interest on the notes is payable on April 1 and October 1 of each year.
Redemption
On or prior to October 1, 2018, we may redeem the Senior Notes, in whole or in part, at a redemption price equal to 100% of the principal amount plus accrued and unpaid interest to the redemption date plus a make‑whole premium as set forth in the indenture governing the Senior Notes.
After October 1, 2018, we may redeem the Senior Notes, in whole or in part, at redemption prices specified in the indenture governing the Senior Notes.
See Note 9 to our consolidated financial statements included elsewhere in this Annual Report on Form 10‑K for further information on the range of prepayments premiums.
Change of Control
Upon the occurrence of a change of control, which is defined in the indenture, each holder of the notes has the right to require PRA Holdings to repurchase some or all of such holder’s notes at a purchase price in cash equal to 101% of the principal amount thereof, plus accrued and unpaid interest, if any, to the repurchase date.
Covenants
The indenture contains covenants limiting, among other things, PRA Health Sciences’ ability and the ability of its restricted subsidiaries to (subject to certain exceptions):
|
· |
incur additional debt or issue certain preferred shares; |
|
· |
pay dividends on or make other distributions in respect of capital stock, purchase or redeem equity interests of the issuer, prepay or repurchase subordinated indebtedness, make certain investments; |
|
· |
sell or transfer certain assets; |
|
· |
create liens on certain assets to secure debt; |
|
· |
consolidate, merge, sell or otherwise dispose of all or substantially all of our assets; |
|
· |
enter into certain transactions with affiliates; and |
|
· |
designate subsidiaries as unrestricted subsidiaries. |
Events of Default
The indenture also provides for events of default which, if any of them occurs, would permit or require the principal of and accrued interest on the notes to become or to be declared due and payable, including without limitation: (i) nonpayment of principal or interest; (ii) failure to perform or observe covenants; (iii) cross‑defaults with certain other indebtedness; (iv) certain bankruptcy related events; (v) impairment of certain security interests in collateral, guarantees or invalidity or unenforceability of certain Senior Secured Credit Facility documents; and (vi) monetary judgment defaults.
Accounts Receivable Financing Agreement
We entered into an accounts receivable financing agreement with PNC Bank, National Association, as administrative agent and lender on March 22, 2016.
55
We may borrow up to $140.0 million from PNC, secured by liens on our accounts receivables and other assets. We are liable for customary representations, warranties, covenants and indemnities. In addition, we have guaranteed the performance of the obligations and will guarantee the obligations of any additional servicer that may become party to the accounts receiveable financing agreement. As of December 31, 2016, the outstanding balance was $120.0 million.
The accounts receivable financing agreement terminates on March 22, 2019, unless terminated earlier pursuant to its term.
Interest Rate and Fees
Loans under the accounts receivable financing agreement will accrue interest at either a reserve-adjusted LIBOR or a base rate, plus 1.60%. As of December 31, 2016, the weighted average interest rate on the accounts receivable financing agreement was 2.31%. We may prepay loans upon one business day prior notice and may terminate the accounts receivable financing agreement with 15 days’ prior notice.
Covenants and Events of Default
The accounts receivable financing agreement contains various customary representations and warranties and covenants, and default provisions which provide for the termination and acceleration of the commitments and loans under the accounts receivable financing agreement in circumstances including, but not limited to, failure to make payments when due, breach of representations, warranties or covenants, certain insolvency events or failure to maintain the security interest in the trade receivables, and defaults under other material indebtedness.
Contractual Obligations and Commercial Commitments
The following table summarizes our future minimum payments for all contractual obligations and commercial commitments for years subsequent to the year ended December 31, 2016:
|
|
|
Payments Due by Period |
|
|||||||||||||
|
|
|
Less than 1 |
|
|
|
|
|
|
|
More than 5 |
|
|
|
|
||
|
|
|
year |
|
1 - 3 years |
|
3 - 5 years |
|
years |
|
Total |
|
|||||
|
|
|
(in thousands) |
|
|||||||||||||
|
Principal payments on long-term debt (1) |
|
$ |
31,250 |
|
$ |
213,750 |
|
$ |
500,000 |
|
$ |
91,441 |
|
$ |
836,441 |
|
|
Interest payments on long-term debt (1) |
|
|
30,257 |
|
|
63,539 |
|
|
53,289 |
|
|
15,203 |
|
|
162,288 |
|
|
Service purchase commitments (2) |
|
|
13,848 |
|
|
10,108 |
|
|
2,486 |
|
|
6 |
|
|
26,448 |
|
|
Operating leases |
|
|
37,975 |
|
|
61,064 |
|
|
51,915 |
|
|
117,083 |
|
|
268,037 |
|
|
Less: sublease income |
|
|
(539) |
|
|
(593) |
|
|
(540) |
|
|
(308) |
|
|
(1,980) |
|
|
Uncertain income tax positions (3) |
|
|
3,725 |
|
|
— |
|
|
— |
|
|
— |
|
|
3,725 |
|
|
Customer dispute settlement (4) |
|
|
1,500 |
|
|
— |
|
|
— |
|
|
— |
|
|
1,500 |
|
|
Total |
|
$ |
118,016 |
|
$ |
347,868 |
|
$ |
607,150 |
|
$ |
223,425 |
|
$ |
1,296,459 |
|
|
(1) |
Principal payments are based on the terms contained in our credit agreements. Interest payments are based on the interest rate in effect on December 31, 2016. |
|
(2) |
Service purchase commitments are defined as agreements to purchase goods or services that are enforceable and legally binding and that specify all significant terms, including fixed or minimum quantities to be purchased. |
|
(3) |
As of December 31, 2016, our liability related to uncertain income tax positions was approximately $12.4 million, of which $8.7 million has not been included in the above table as we are unable to predict when these liabilities will be paid due to the uncertainties in timing of the settlement of the income tax positions. |
|
(4) |
On May 5, 2014, we reached a settlement in the amount of $9.0 million related to a customer dispute. We paid $4.5 million, $1.5 million and $1.5 million related to this dispute in 2014, 2015 and 2016, respectively. The remaining $1.5 million is payable on December 31, 2017. |
56
Off‑Balance Sheet Arrangements
We have no off‑balance sheet arrangements. The term “off‑balance sheet arrangement” generally means any transaction, agreement or other contractual arrangement to which an entity unconsolidated with us is a party, under which we have any obligation arising under a guarantee contract, derivative instrument or variable interest or a retained or contingent interest in assets transferred to such entity or similar arrangement that serves as credit, liquidity or market risk support for such assets.
Recent Accounting Pronouncements
For information on new accounting pronouncements and the impact, if any, on our financial position or results of operations, see Note 2 to our audited consolidated financial statements found elsewhere in this Annual Report on Form 10-K.
Critical Accounting Policies and Estimates
In preparing our financial statements in conformity with GAAP, management is required to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Our actual results could differ from those estimates. We believe that the following are some of the more critical judgment areas in the application of our accounting policies that affect our financial condition and results of operations. We have discussed the application of these critical accounting policies with our board of directors.
Revenue Recognition
The majority of our service revenue is recorded regionally on a proportional performance basis. Revenue for service is recognized only after persuasive evidence of an arrangement exists, the sales price is determinable and collectability is reasonably assured. To measure performance, we compare contract costs incurred to estimated total contract costs through completion. We believe this is the best indicator of the performance of the contract obligations because the costs relate to the amount of labor hours incurred to perform the service. Direct costs are primarily comprised of labor‑related charges associated with the delivery of services. Each month we accumulate costs on each project and compare them to the total current estimated costs to determine the proportional performance. We then multiply the proportion completed by the contract value to determine the amount of revenue that can be recognized. Each month we review the total current estimated costs on each project to determine if these estimates are still accurate and, if necessary, we adjust the total estimated costs for each project. During our monthly contract review process, we review each contract’s performance to date, current cost trends, and circumstances specific to each study. The original or current cost estimates are reviewed and if necessary the estimates are adjusted and refined to reflect any changes in the anticipated performance under the study. In the normal course of business, we conduct this review each month in all service delivery locations. As the work progresses, original estimates might be deemed incorrect due to, among other things, revisions in the scope of work or patient enrollment rate, and a contract modification might be negotiated with the customer to cover additional costs. If not, we bear the risk of costs exceeding our original estimates. Management assumes that actual costs incurred to date under the contract are a valid basis for estimating future costs. Should management’s assumption of future cost trends fluctuate significantly, future margins could be reduced. In the past, we have had to commit unanticipated resources to complete projects, resulting in lower margins on those projects. Should our actual costs exceed our estimates on fixed price contracts, future margins could be reduced, absent our ability to negotiate a contract modification. We accumulate information on each project to refine our bidding process. Historically, the majority of our estimates and assumptions have been materially correct, but these estimates might not continue to be accurate in the future.
Allowance for Doubtful Accounts
Included in “Accounts receivable and unbilled services, net” on our consolidated balance sheets is an allowance for doubtful accounts. Generally, before we do business with a new client, we perform a credit check, as our allowance for doubtful accounts requires that we make an accurate assessment of our customers’ creditworthiness. Approximately 15% of our client base is small- to mid-sized biotech companies, creating a heightened risk related to the creditworthiness for a portion of our client base. We manage and assess our exposure to bad debt on each of our contracts. We age our billed accounts receivable and assess exposure by client type, by aged category, and by specific
57
identification. After all attempts to collect a receivable have failed, the receivable is written off against the allowance. Historically, we have not had any write‑offs in excess of our allowance. If, at December 31, 2016, our aged accounts receivable balance greater than 90 days were to increase by 10% (for the U.S. operations), no material adjustments to bad debt expense would be required.
Income Taxes
Changes in judgment as to recognition or measurement of tax positions can materially affect the estimate of our effective tax rate and, consequently, our operating results. We consider many factors when evaluating and estimating our tax positions and tax benefits, which may require periodic adjustments and may not accurately anticipate actual outcomes.
We have to use estimates and judgments in calculating certain tax liabilities and determining the recoverability of certain deferred tax assets, which arise from net operating losses, tax credit carry forwards and temporary differences between the tax and financial statement recognition of revenue and expense. We are also required to reduce our deferred tax assets by a valuation allowance if, based on the weight of available evidence, it is more likely than not that some portion or all of the recorded deferred tax assets will not be realized in future periods.
In evaluating our ability to recover our deferred tax assets, in full or in part, we consider all available positive and negative evidence, including our past operating results, the existence of cumulative losses in the most recent fiscal years and our forecast of future taxable income on a jurisdiction‑by‑jurisdiction basis. In determining future taxable income, assumptions include the amount of state, federal and international pretax operating income, international transfer pricing policies, the reversal of temporary differences and the implementation of feasible and prudent tax planning strategies. These assumptions require significant judgment about the forecasts of future taxable income and are consistent with the plans and estimates we use to manage the underlying businesses. Based on our analysis of the above factors, we determined that a valuation allowance of $21.7 million was required as of December 31, 2016 relating to the U.S. net deferred tax asset, state net operating loss carryforwards, foreign net operating loss carryforwards and state tax credit carryforwards. Changes in our assumptions could result in an adjustment to the valuation allowance, up or down, in the future.
In addition, the calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax regulations in a multitude of jurisdictions. We determine our liability for uncertain tax positions globally under the provisions in Financial Accounting Standards Board’s, or FASB, Accounting Standards Codification, or ASC, 740, Income Taxes. As of December 31, 2016, we had recorded a liability for uncertain tax positions of $12.4 million. If events occur such that payment of these amounts ultimately proves to be unnecessary, the reversal of liabilities would result in tax benefits being recognized in the period when we determine the liabilities are no longer necessary. If our calculation of liability related to uncertain tax positions proves to be more or less than the ultimate assessment, a tax expense or benefit to expense, respectively, would result. The total liability reversal that could affect the tax rate is $7.6 million.
Stock‑Based Compensation
In accordance with the ASC 718, Stock Compensation, as modified and supplemented, we estimate the value of employee stock options on the date of grant using either the Black‑Scholes model for all options with a service condition or a lattice model for options with market and performance conditions. The determination of fair value of stock‑based payment awards on the date of grant using an option‑pricing model is affected by the stock price of similar entities as well as assumptions regarding a number of highly complex and subjective variables. These variables include the expected stock price volatility over the term of the awards, and actual and projected employee stock option exercise behaviors. The Black‑Scholes and lattice models require extensive actual employee exercise behavior data and the use of a number of complex assumptions including expected volatility, risk‑free interest rate, expected dividends, and expected life. In developing our assumption, we take into account the following:
|
· |
We use the historical volatilities of a selected peer group as we do not have sufficient history to estimate the volatility of our common share price. We calculate expected volatility based on reported data for selected reasonably similar publicly traded companies for which the historical information is available. For the purpose of identifying peer companies, we consider characteristics such as industry, length of trading history, similar vesting terms and in‑the‑money option status. We plan to continue to use the guideline peer |
58
group volatility information until the historical volatility of our common shares is relevant to measure expected volatility for future award grants. |
|
· |
The risk‑free interest rate assumption is based upon observed interest rates appropriate for the term of our employee stock options. |
|
· |
The dividend yield assumption is based on the history and expectation of dividend payouts. |
|
· |
For those options valued using the Black-Scholes model, the expected life is based upon the guidance provided by the FASB. For those options with a market condition valued under the lattice model, the expected life varies depending on the target stock price that triggers vesting. |
|
· |
As stock‑based compensation expense recognized in the consolidated statement of operations is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. We estimate forfeitures based on our company experience. Current accounting guidance requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. |
Due to the absence of an active market for our common stock prior to our IPO, the fair value of our common stock on the date of the grant was determined in good faith by our Board of Directors with the assistance of management, based on a number of factors consistent with the methodologies outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. Subsequent to the IPO, the fair value of our common stock is based upon the market price of our common stock on the date of the grant as listed on the NASDAQ.
Long‑Lived Assets, Goodwill and Indefinite‑Lived Intangible Assets
As a result of our acquisitions we have recorded goodwill and other identifiable finite and indefinite‑lived acquired intangibles. The identification and valuation of these intangible assets at the time of acquisition require significant management judgment and estimates.
We review long‑lived asset groups for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset group might not be recoverable. If indicators of impairment are present, we evaluate the carrying value of property and equipment in relation to estimates of future undiscounted cash flows. As a result of our acquisitions we have recorded goodwill and other identifiable finite and indefinite‑lived acquired intangibles. The identification and valuation of these intangible assets at the time of acquisition require significant management judgment and estimates. In connection with the acquisition of PRA by KKR on September 23, 2013, the purchases of ClinStar on February 28, 2013, RPS on September 23, 2013, CRI Lifetree on December 2, 2013, VHS on June 8, 2015, and Nextrials on March 18, 2016, valuations were completed, and value was assigned to identifiable finite‑lived and indefinite‑lived intangible assets and goodwill, based on the purchase price of the transactions.
We test goodwill for impairment on at least an annual basis by comparing the carrying value to the estimated fair value of our reporting units. On October 1, 2016, we reviewed goodwill for impairment and our analysis indicated that the fair value of goodwill exceeded the carrying value and, therefore, no impairment exists. When evaluating for impairment, we may first perform a qualitative assessment to determine whether it is more likely than not that a reporting unit or indefinite-lived intangible asset is impaired. If we do not perform a qualitative assessment, or if it determines that it is not more likely than not that the fair value of the reporting unit or indefinite-lived intangible asset exceeds its carrying amount, we will calculate the estimated fair value of the reporting unit’s or indefinite-lived intangible asset. Our decision to perform a qualitative impairment assessment for an individual reporting unit in a given year is influenced by a number of factors, inclusive of the size of the reporting unit's goodwill, the significance of the excess of the reporting unit's estimated fair value over carrying value at the last quantitative assessment date, the amount of time in between quantitative fair value assessments and the date of acquisition. During 2016, as part of our annual impairment analysis, we performed the qualitative assessment for approximately $850.0 million, or 87.5%, of our total goodwill balance of $972.0 million, which resides in our PR and SS reporting units, and for our indefinite-lived trade name intangible asset.
59
If we do not perform a qualitative assessment, goodwill impairment is determined by comparing the fair value of each reporting unit, determined using various valuation techniques, with the primary technique being a discounted cash flow analysis, to its carrying value. This process is inherently subjective and dependent upon the estimates and assumptions we make. In determining the expected future cash flows of our company, we assume that we will continue to enter into new contracts, execute the work on these contracts profitably, collect receivables from customers, and thus generate positive cash flows. In addition, our analysis could be impacted by future adverse change such as future declines in our operating results, a further significant slowdown in the worldwide economy or pharmaceutical and biotechnology industry or failure to meet the performance projections included in our forecast.
The estimated fair value of the EDS reporting unit closely approximated its carrying value when we performed its annual goodwill impairment test during the fourth quarter of 2014. We made operational improvements during 2015 and 2016 in order to improve the profitability of the EDS reporting unit. As a result of these changes, EDS saw growth in both backlog and new business awards that contributed to its improved financial performance during the year and led to us updating its forecast for future periods. We considered all of these factors when we performed our most recent goodwill impairment test during the fourth quarter of 2016 and it was concluded that the estimated fair value of the EDS reporting unit exceeded its carrying value by approximately $70.0 million or 33%. Any negative changes in assumptions on revenue, new business awards, cancellations, or our ability to improve operations while maintaining a competitive cost structure could adversely affect the fair value of EDS and result in significant goodwill impairment charges in 2017 or later.
Fair Value Measurements
We record certain assets and liabilities at fair value. Fair value is defined as a price that would be received to sell an asset or paid to transfer a liability in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants at the measurement date. A three‑level hierarchy that prioritizes the inputs used to measure fair value is further described in Note 2 to our audited consolidated financial statements included elsewhere in this Annual Report on Form 10‑K.
Fair Value Measurements on a Recurring Basis
At December 31, 2016 and 2015, we used Level 3 inputs to measure liabilities totaling $2.8 million and $1.0 million, respectively. The liability at December 31, 2016 relates to contingent consideration issued in connection with our acquisition of VHS and Nextrials. The liability at December 31, 2015 related to contingent consideration issued in connection with our acquisition of VHS.
All derivatives are measured at fair value and recognized as either assets or liabilities on the consolidated balance sheets. The fair value of our interest rate swaps, measured using Level 2 inputs, was a liability of $0.6 million and an asset of $0.1 million at December 31, 2016 and 2015, respectively. No other liabilities or assets are remeasured at fair value.
Inflation
Our long‑term contracts, those in excess of one year, generally include an inflation or cost of living adjustment for the portion of the services to be performed beyond one year from the contract date. As a result, we expect that inflation generally will not have a material adverse effect on our operations or financial condition. Historically our projection of inflation contained within our contracts has not significantly impacted our operating income. Should inflation be in excess of the estimates within our contracts our operating margins would be negatively impacted if we were unable to negotiate contract modifications with our clients.
Potential Liability and Insurance
Our clients provide us with contractual indemnification for all of our service related contracts. In addition, we attempt to manage our risk of liability for personal injury or death to patients from administration of products under study through measures such as stringent operating procedures and insurance. We monitor our clinical trials in a manner designed to ensure compliance with government regulations and guidelines. We have adopted global standard operating procedures intended to satisfy regulatory requirements in the United States and in many foreign countries that serve as a tool for controlling and enhancing the quality of our clinical trials. We currently maintain professional liability insurance
60
coverage with limits we believe are adequate and appropriate. If our insurance coverage is not adequate to cover actual claims, or if insurance coverage does not continue to be available on terms acceptable to us, our business, financial condition, and operating results could be materially harmed. Historically we have experienced infrequent and immaterial claims. Should a material claim arise that exceeds our insurance coverage levels, there would be a dollar for dollar impact to operating income for the amount in excess of our insurance coverage.
Dividend History
We have not declared or paid dividends during 2016, 2015 and 2014.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Market risk is the potential loss arising from adverse changes in market rates and prices, such as foreign currency exchange rates, interest rates and other relevant market rate or price changes. In the ordinary course of business, we are exposed to various market risks, including changes in foreign currency exchange rates and interest rates, and we regularly evaluate our exposure to such changes. Our overall risk management strategy seeks to balance the magnitude of the exposure and the cost and availability of appropriate financial instruments.
Interest Rate Risk
We are subject to market risk associated with changes in interest rates. Our 2016 Credit Facilities is subject to interest rates based on LIBOR, subject to a 0.00% LIBOR floor, plus an applicable margin ranging from 1.25% to 2.25%, or ABR rates, ranging from 0.25% to 1.25%. At December 31, 2016, we had $625.0 million outstanding under our 2016 First Lien Term Loan and no outstanding balance under our 2016 Revolver subject to these variable interest rates. In conjunction with the closing of the 2016 Credit Facilities in December 2016, our 2015 Swap was amended to modify the rate, repricing dates and embedded floor, or the Modified 2015 Swap. The Company re-designated the modify swap against refinanced debt under the 2016 Credit Facilities. The Modified 2015 Swap will mature on September 6, 2018. The interest rate swap is used to hedge the Company’s variable rate on our 2016 Credit Facilities. Our accounts receivable financing agreement is subject to interest rates based on LIBOR, or a base rate, plus 1.60%. At December 31, 2016, we had $120.0 million outstanding under our accounts receiveable financing agreement. Once the interest rate swap is effective, each quarter percentage point increase or decrease in the variable rate would result in our interest expense changing by approximately $1.2 million per year under our unhedged variable rate debt.
Foreign Exchange Risk
Since we operate on a global basis, we are exposed to various foreign currency risks. First, our consolidated financial statements are denominated in U.S. dollars, but a significant portion of our revenue is generated in the local currency of our foreign subsidiaries. Accordingly, changes in exchange rates between the applicable foreign currency and the U.S. dollar will affect the translation of each foreign subsidiary’s financial results into U.S. dollars for purposes of reporting consolidated financial results. A hypothetical change of 10% in average exchange rates used to translate all foreign currencies to U.S. dollars would have impacted income (loss) before income taxes and equity in gains (losses) of unconsolidated joint ventures by approximately $9.0 million for the year ended December 31, 2016. The process by which each foreign subsidiary’s financial results are translated into U.S. dollars is as follows: income statement accounts are translated at average exchange rates for the period; balance sheet asset and liability accounts are translated at end of period exchange rates; and equity accounts are translated at historical exchange rates. Translation of the balance sheet in this manner affects the stockholders’ equity account, referred to as the cumulative translation adjustment account. This account exists only in the foreign subsidiary’s U.S. dollar balance sheet and is necessary to keep the foreign balance sheet stated in U.S. dollars in balance. Accumulated currency translation adjustments recorded as a separate component of stockholders’ equity were $(201.1) million and $(106.1) million at December 31, 2016 and 2015, respectively. We do not have significant operations in countries in which the economy is considered to be highly‑inflationary.
In addition, two specific risks arise from the nature of the contracts we enter into with our clients, which from time to time are denominated in currencies different than the particular subsidiary’s local currency. These risks are generally applicable only to a portion of the contracts executed by our foreign subsidiaries providing clinical services. The first risk occurs as revenue recognized for services rendered is denominated in a currency different from the currency in which the subsidiary’s expenses are incurred. As a result, the subsidiary’s earnings can be affected by fluctuations in exchange rates.
61
The second risk results from the passage of time between the invoicing of clients under these contracts and the ultimate collection of client payments against such invoices. Because the contract is denominated in a currency other than the subsidiary’s local currency, we recognize a receivable at the time of invoicing for the local currency equivalent of the foreign currency invoice amount. Changes in exchange rates from the time the invoice is prepared until payment from the client is received will result in our receiving either more or less in local currency than the local currency equivalent of the invoice amount at the time the invoice was prepared and the receivable established. This difference is recognized by us as a foreign currency transaction gain or loss, as applicable, and is reported in other expense or income in our consolidated statements of operations. Historically, fluctuations in exchange rates from those in effect at the time contracts were executed have not had a material effect on our consolidated financial results.
62
Item 8. Financial Statements and Supplementary Data
Management’s Report on Internal Control Over Financial Reporting
Management of PRA Health Sciences, Inc. (the “Company”) is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our consolidated financial statements for external reporting purposes in accordance with accounting principles generally accepted in the United States of America. Internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company, (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors, and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements in the consolidated financial statements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2016. In making these assessments, management used the framework established by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control — Integrated Framework (2013). Based on management’s assessment and the criteria in the COSO framework, management has concluded that the Company’s internal control over financial reporting as of December 31, 2016 was effective.
The Company’s independent registered public accounting firm has issued a report on the Company’s internal control over financial reporting. This report appears on page 65 in this Annual Report on Form 10-K.
|
/s/ Colin Shannon |
|
/s/ Linda Baddour |
|
|
|
|
|
Colin Shannon |
|
Linda Baddour |
|
President, Chief Executive Officer and Chairman of the Board of Directors |
|
Executive Vice President and Chief Financial Officer |
|
(Principal Executive Officer) |
|
(Principal Financial Officer) |
63
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
PRA Health Sciences, Inc.
Raleigh, North Carolina
We have audited the accompanying consolidated balance sheets of PRA Health Sciences, Inc. and subsidiaries (the "Company") as of December 31, 2016 and 2015, and the related consolidated statements of operations, comprehensive (loss) income, changes in stockholders' equity, and cash flows for each of the three years in the period ended December 31, 2016. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of PRA Health Sciences, Inc. and subsidiaries as of December 31, 2016 and 2015, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2016, in conformity with accounting principles generally accepted in the United States of America.
As discussed in Note 2 to the financial statements, the Company retrospectively adopted ASU No. 2016-15, “Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments” and ASU No. 2016-18, “Statement of Cash Flows (Topic 230): Restricted Cash.”
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company's internal control over financial reporting as of December 31, 2016, based on the criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 23, 2017 expressed an unqualified opinion on the Company's internal control over financial reporting.
/s/ Deloitte & Touche LLP
Raleigh, North Carolina
February 23, 2017
64
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
PRA Health Sciences, Inc.
Raleigh, North Carolina
We have audited the internal control over financial reporting of PRA Health Sciences, Inc. and subsidiaries (the "Company") as of December 31, 2016, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company's management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying “Management's Report on Internal Control Over Financial Reporting.” Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed by, or under the supervision of, the company's principal executive and principal financial officers, or persons performing similar functions, and effected by the company's board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis.
Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on the criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements as of and for the year ended December 31, 2016 of the Company, and our report dated February 23, 2017 expressed an unqualified opinion on those financial statements and included an explanatory paragraph regarding the Company’s retrospective adoption of ASU No. 2016-15, “Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments” and ASU No. 2016-18, “Statement of Cash Flows (Topic 230): Restricted Cash.”
/s/ Deloitte & Touche LLP
Raleigh, North Carolina
February 23, 2017
65
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
(in thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
144,623 |
|
$ |
121,065 |
|
|
Restricted cash |
|
|
4,715 |
|
|
5,060 |
|
|
Accounts receivable and unbilled services, net |
|
|
439,053 |
|
|
415,077 |
|
|
Prepaid expenses and other current assets |
|
|
35,367 |
|
|
30,175 |
|
|
Income taxes receivable |
|
|
979 |
|
|
2,399 |
|
|
Total current assets |
|
|
624,737 |
|
|
573,776 |
|
|
Fixed assets, net |
|
|
87,577 |
|
|
80,691 |
|
|
Goodwill |
|
|
971,980 |
|
|
1,014,798 |
|
|
Intangible assets, net |
|
|
473,976 |
|
|
533,938 |
|
|
Deferred tax assets |
|
|
6,568 |
|
|
3,069 |
|
|
Investment in unconsolidated joint ventures |
|
|
284 |
|
|
1,288 |
|
|
Deferred financing fees |
|
|
1,762 |
|
|
2,490 |
|
|
Other assets |
|
|
23,507 |
|
|
18,693 |
|
|
Total assets |
|
$ |
2,190,391 |
|
$ |
2,228,743 |
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Current portion of long-term debt |
|
$ |
31,250 |
|
$ |
— |
|
|
Accounts payable |
|
|
51,335 |
|
|
57,096 |
|
|
Accrued expenses and other current liabilities |
|
|
123,589 |
|
|
119,893 |
|
|
Income taxes payable |
|
|
25,524 |
|
|
19,262 |
|
|
Advanced billings |
|
|
332,501 |
|
|
333,729 |
|
|
Total current liabilities |
|
|
564,199 |
|
|
529,980 |
|
|
Deferred tax liabilities |
|
|
73,703 |
|
|
81,691 |
|
|
Long-term debt, net |
|
|
797,052 |
|
|
889,514 |
|
|
Other long-term liabilities |
|
|
26,185 |
|
|
24,836 |
|
|
Total liabilities |
|
|
1,461,139 |
|
|
1,526,021 |
|
|
Commitments and contingencies (Note 13) |
|
|
|
|
|
|
|
|
Stockholders' equity: |
|
|
|
|
|
|
|
|
Preferred stock, $0.01 par value; 100,000,000 shares authorized, 0 shares issued and outstanding at December 31, 2016 and 2015, respectively |
|
|
— |
|
|
— |
|
|
Common stock, $0.01 par value, 1,000,000,000 authorized shares at December 31, 2016 and December 31, 2015; 61,597,705 and 60,245,009 issued and outstanding at December 31, 2016 and December 31, 2015, respectively |
|
|
616 |
|
|
602 |
|
|
Additional paid-in capital |
|
|
879,067 |
|
|
828,347 |
|
|
Accumulated other comprehensive loss |
|
|
(224,686) |
|
|
(132,307) |
|
|
Retained earnings |
|
|
74,255 |
|
|
6,080 |
|
|
Total stockholders' equity |
|
|
729,252 |
|
|
702,722 |
|
|
Total liabilities and stockholders' equity |
|
$ |
2,190,391 |
|
$ |
2,228,743 |
|
The accompanying notes are an integral part of the consolidated financial statements.
66
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Revenue: |
|
|
|
|
|
|
|
|
|
|
|
Service revenue |
|
$ |
1,580,023 |
|
$ |
1,375,847 |
|
$ |
1,266,596 |
|
|
Reimbursement revenue |
|
|
231,688 |
|
|
238,036 |
|
|
192,990 |
|
|
Total revenue |
|
|
1,811,711 |
|
|
1,613,883 |
|
|
1,459,586 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
Direct costs |
|
|
1,032,688 |
|
|
886,528 |
|
|
859,218 |
|
|
Reimbursable out-of-pocket costs |
|
|
231,688 |
|
|
238,036 |
|
|
192,990 |
|
|
Selling, general and administrative |
|
|
269,893 |
|
|
246,417 |
|
|
253,970 |
|
|
Transaction-related costs |
|
|
44,834 |
|
|
— |
|
|
— |
|
|
Depreciation and amortization |
|
|
69,506 |
|
|
77,952 |
|
|
96,564 |
|
|
Loss on disposal of fixed assets |
|
|
753 |
|
|
652 |
|
|
5 |
|
|
Income from operations |
|
|
162,349 |
|
|
164,298 |
|
|
56,839 |
|
|
Interest expense, net |
|
|
(54,913) |
|
|
(61,747) |
|
|
(81,939) |
|
|
Loss on modification or extinguishment of debt |
|
|
(38,178) |
|
|
— |
|
|
(25,036) |
|
|
Foreign currency gains, net |
|
|
24,029 |
|
|
14,048 |
|
|
10,538 |
|
|
Other income (expense), net |
|
|
607 |
|
|
(1,434) |
|
|
(2,254) |
|
|
Income (loss) before income taxes and equity in gains (losses) of unconsolidated joint ventures |
|
|
93,894 |
|
|
115,165 |
|
|
(41,852) |
|
|
Provision for (benefit from) income taxes |
|
|
28,494 |
|
|
30,004 |
|
|
(8,154) |
|
|
Income (loss) before equity in gains (losses) of unconsolidated joint ventures |
|
|
65,400 |
|
|
85,161 |
|
|
(33,698) |
|
|
Equity in gains (losses) of unconsolidated joint ventures, net of tax |
|
|
2,775 |
|
|
(3,396) |
|
|
(2,044) |
|
|
Net income (loss) |
|
$ |
68,175 |
|
$ |
81,765 |
|
$ |
(35,742) |
|
|
Net income (loss) per share attributable to common stockholders: |
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
$ |
1.12 |
|
$ |
1.36 |
|
$ |
(0.83) |
|
|
Diluted |
|
$ |
1.06 |
|
$ |
1.29 |
|
$ |
(0.83) |
|
|
Weighted average common shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
Basic |
|
|
60,759 |
|
|
59,965 |
|
|
42,897 |
|
|
Diluted |
|
|
64,452 |
|
|
63,207 |
|
|
42,897 |
|
The accompanying notes are an integral part of the consolidated financial statements.
67
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE (LOSS) INCOME
(in thousands)
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Net income (loss) |
|
$ |
68,175 |
|
$ |
81,765 |
|
$ |
(35,742) |
|
|
Other comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
Foreign currency translation adjustments |
|
|
(95,019) |
|
|
(52,433) |
|
|
(68,700) |
|
|
Unrealized losses on derivative instruments, net of income taxes of $(622), $(578), and $(3,298) |
|
|
(978) |
|
|
(11,273) |
|
|
(17,681) |
|
|
Reclassification adjustments: |
|
|
|
|
|
|
|
|
|
|
|
Losses on derivatives included in net income (loss), net of income taxes, $2,303, $0, and $0 |
|
|
3,618 |
|
|
908 |
|
|
3 |
|
|
Comprehensive (loss) income |
|
$ |
(24,204) |
|
$ |
18,967 |
|
$ |
(122,120) |
|
The accompanying notes are an integral part of the consolidated financial statements.
68
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other |
|
Retained |
|
|
|
|
||
|
|
|
|
|
|
|
|
Additional |
|
Comprehensive |
|
Earnings |
|
|
|
|
|||
|
|
|
Common Stock |
|
Paid-in |
|
(Loss) Income |
|
(Accumulated |
|
|
|
|
||||||
|
|
|
Shares |
|
Amount |
|
Capital |
|
(Note 16) |
|
Deficit) |
|
Total |
|
|||||
|
Balance at December 31, 2013 |
|
40,268 |
|
$ |
403 |
|
$ |
490,006 |
|
$ |
16,869 |
|
$ |
(39,943) |
|
$ |
467,335 |
|
|
Exercise of common stock options |
|
11 |
|
|
— |
|
|
33 |
|
|
— |
|
|
— |
|
|
33 |
|
|
Issuance of common stock |
|
19,535 |
|
|
195 |
|
|
351,326 |
|
|
— |
|
|
— |
|
|
351,521 |
|
|
Common stock issuance costs |
|
— |
|
|
— |
|
|
(23,421) |
|
|
— |
|
|
— |
|
|
(23,421) |
|
|
Stock-based compensation |
|
— |
|
|
— |
|
|
3,467 |
|
|
— |
|
|
— |
|
|
3,467 |
|
|
Net loss |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(35,742) |
|
|
(35,742) |
|
|
Other comprehensive loss, net of tax |
|
— |
|
|
— |
|
|
— |
|
|
(86,378) |
|
|
— |
|
|
(86,378) |
|
|
Balance at December 31, 2014 |
|
59,814 |
|
|
598 |
|
|
821,411 |
|
|
(69,509) |
|
|
(75,685) |
|
|
676,815 |
|
|
Exercise of common stock options |
|
257 |
|
|
3 |
|
|
78 |
|
|
— |
|
|
— |
|
|
81 |
|
|
Issuance of common stock |
|
174 |
|
|
1 |
|
|
1,582 |
|
|
— |
|
|
— |
|
|
1,583 |
|
|
Stock-based compensation |
|
— |
|
|
— |
|
|
5,276 |
|
|
— |
|
|
— |
|
|
5,276 |
|
|
Net income |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
81,765 |
|
|
81,765 |
|
|
Other comprehensive loss, net of tax |
|
— |
|
|
— |
|
|
— |
|
|
(62,798) |
|
|
— |
|
|
(62,798) |
|
|
Balance at December 31, 2015 |
|
60,245 |
|
|
602 |
|
|
828,347 |
|
|
(132,307) |
|
|
6,080 |
|
|
702,722 |
|
|
Exercise of common stock options |
|
1,303 |
|
|
13 |
|
|
642 |
|
|
— |
|
|
— |
|
|
655 |
|
|
Stock-based compensation |
|
50 |
|
|
1 |
|
|
49,232 |
|
|
— |
|
|
— |
|
|
49,233 |
|
|
Income tax benefit from stock-based award activities |
|
— |
|
|
— |
|
|
846 |
|
|
— |
|
|
— |
|
|
846 |
|
|
Net income |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
68,175 |
|
|
68,175 |
|
|
Other comprehensive loss, net of tax |
|
— |
|
|
— |
|
|
— |
|
|
(92,379) |
|
|
— |
|
|
(92,379) |
|
|
Balance at December 31, 2016 |
|
61,598 |
|
$ |
616 |
|
$ |
879,067 |
|
$ |
(224,686) |
|
$ |
74,255 |
|
$ |
729,252 |
|
The accompanying notes are an integral part of the consolidated financial statements.
69
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) |
|
$ |
68,175 |
|
$ |
81,765 |
|
$ |
(35,742) |
|
|
Adjustments to reconcile net income (loss) to net cash provided by operating activities: |
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
69,506 |
|
|
77,952 |
|
|
96,564 |
|
|
Amortization of debt issuance costs and discount |
|
|
4,433 |
|
|
5,983 |
|
|
5,737 |
|
|
Amortization of terminated interest rate swaps |
|
|
4,961 |
|
|
731 |
|
|
— |
|
|
Stock-based compensation expense |
|
|
7,067 |
|
|
5,276 |
|
|
3,467 |
|
|
Non-cash transaction related costs |
|
|
42,166 |
|
|
— |
|
|
— |
|
|
Unrealized foreign currency gains |
|
|
(24,499) |
|
|
(16,464) |
|
|
(12,222) |
|
|
Loss on modification or extinguishment of debt |
|
|
38,178 |
|
|
— |
|
|
25,036 |
|
|
Loss on disposal of fixed assets |
|
|
753 |
|
|
652 |
|
|
5 |
|
|
Change in acquisition-related contingent consideration |
|
|
(527) |
|
|
89 |
|
|
504 |
|
|
Equity in (gains) losses of unconsolidated joint ventures |
|
|
(2,775) |
|
|
3,396 |
|
|
2,044 |
|
|
Unrealized loss on derivatives |
|
|
47 |
|
|
1,787 |
|
|
1,731 |
|
|
Other reconciling items |
|
|
(652) |
|
|
443 |
|
|
978 |
|
|
Excess tax benefit from stock-based compensation |
|
|
(846) |
|
|
— |
|
|
— |
|
|
Deferred income taxes |
|
|
(10,469) |
|
|
(3,219) |
|
|
(31,968) |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable and unbilled services |
|
|
(31,313) |
|
|
(83,211) |
|
|
(32,781) |
|
|
Prepaid expenses and other assets |
|
|
(10,071) |
|
|
(11,675) |
|
|
(10,944) |
|
|
Accounts payable and other liabilities |
|
|
(1,474) |
|
|
36,135 |
|
|
19,727 |
|
|
Income taxes |
|
|
7,308 |
|
|
9,958 |
|
|
15,634 |
|
|
Advanced billings |
|
|
79 |
|
|
42,830 |
|
|
(13,736) |
|
|
Net cash provided by operating activities |
|
|
160,047 |
|
|
152,428 |
|
|
34,034 |
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
Purchase of fixed assets |
|
|
(33,143) |
|
|
(32,814) |
|
|
(27,323) |
|
|
Cash paid for interest on interest rate swap |
|
|
(913) |
|
|
(302) |
|
|
— |
|
|
Cash paid to terminate interest rate swaps |
|
|
— |
|
|
(32,907) |
|
|
— |
|
|
Acquisition of Nextrials, Inc., net of cash acquired |
|
|
(4,268) |
|
|
— |
|
|
— |
|
|
Acquisition of Value Health Solutions, Inc., net of cash acquired |
|
|
— |
|
|
(543) |
|
|
— |
|
|
Proceeds from RPS Parent Holding Corp. working capital settlement |
|
|
— |
|
|
— |
|
|
15,000 |
|
|
Proceeds from CRI Holding Company, LLC working capital settlement |
|
|
— |
|
|
— |
|
|
851 |
|
|
Payment of ClinStar, LLC working capital settlement |
|
|
— |
|
|
(1,693) |
|
|
— |
|
|
Distributions from unconsolidated joint ventures |
|
|
3,700 |
|
|
19,529 |
|
|
|
|
|
Contributions to unconsolidated joint ventures |
|
|
— |
|
|
(23,000) |
|
|
— |
|
|
Proceeds from the sale of fixed assets |
|
|
10 |
|
|
44 |
|
|
— |
|
|
Net cash used in investing activities |
|
|
(34,614) |
|
|
(71,686) |
|
|
(11,472) |
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of long-term debt |
|
|
625,000 |
|
|
— |
|
|
— |
|
|
Proceeds from accounts receivable financing agreement |
|
|
120,000 |
|
|
— |
|
|
— |
|
|
Repayment of long-term debt |
|
|
(822,559) |
|
|
(40,000) |
|
|
(308,775) |
|
|
Borrowings on line of credit |
|
|
110,000 |
|
|
90,000 |
|
|
105,000 |
|
|
Repayments of line of credit |
|
|
(110,000) |
|
|
(90,000) |
|
|
(115,000) |
|
|
Payment of debt prepayment and debt extinguishment costs |
|
|
(17,824) |
|
|
— |
|
|
(14,250) |
|
|
Payment for debt issuance costs |
|
|
(7,713) |
|
|
— |
|
|
— |
|
|
Proceeds from common stock issued, net of underwriters discount |
|
|
— |
|
|
— |
|
|
333,950 |
|
|
Payment of common stock issuance costs |
|
|
— |
|
|
(525) |
|
|
(5,325) |
|
|
Excess tax benefit from stock-based compensation |
|
|
846 |
|
|
— |
|
|
— |
|
|
Proceeds from stock option exercises |
|
|
655 |
|
|
81 |
|
|
33 |
|
|
Payment of acquisition-related contingent consideration |
|
|
— |
|
|
(2,000) |
|
|
(1,589) |
|
|
Net cash used in financing activities |
|
|
(101,595) |
|
|
(42,444) |
|
|
(5,956) |
|
|
Effects of foreign exchange changes on cash, cash equivalents, and restricted cash |
|
|
(625) |
|
|
(3,702) |
|
|
(5,992) |
|
|
Change in cash, cash equivalents, and restricted cash |
|
|
23,213 |
|
|
34,596 |
|
|
10,614 |
|
|
Cash, cash equivalents, and restricted cash, beginning of period |
|
|
126,125 |
|
|
91,529 |
|
|
80,915 |
|
|
Cash, cash equivalents, and restricted cash, end of period |
|
$ |
149,338 |
|
$ |
126,125 |
|
$ |
91,529 |
|
The accompanying notes are an integral part of the consolidated financial statements.
70
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2016
(1) Basis of Presentation
Description of Business
PRA Health Sciences, Inc. and its subsidiaries (collectively, the Company) is a full‑service global contract research organization providing a broad range of product development services for pharmaceutical and biotechnology companies around the world. The Company’s integrated services include data management, statistical analysis, clinical trial management, and regulatory and drug development consulting.
Organization and Initial Public Offering, or IPO
On November 13, 2014, the Company’s common stock began trading on the NASDAQ Global Select Market under the symbol “PRAH,” at a price to the public of $18.00 per share. The Company issued and sold 19,523,255 shares of common stock, including 2,546,511 common shares issued pursuant to the full exercise of the underwriters’ option to purchase additional shares. The offering raised net proceeds of approximately $328.0 million after deducting underwriting discounts and commissions and offering expenses.
On September 23, 2013, all of the outstanding stock of PRA Holdings, Inc., or the Predecessor Company, was acquired by affiliates of Kohlberg Kravis Roberts & Co. L.P., or KKR, pursuant to a plan of merger by and among Pinnacle Holdco Parent, Inc., or Parent, Pinnacle Merger Sub, Inc., or merger sub, and Genstar Capital Partners V, L.P., or Genstar. Upon completion of the merger, or the Merger, the merger sub was folded into the Predecessor Company, which became a subsidiary of the Parent. On December 19, 2013, Pinnacle Holdco Parent, Inc. changed its name to PRA Global Holdings, Inc. and on July 10, 2014, PRA Global Holdings, Inc. changed its name to PRA Health Sciences, Inc.
Basis of Presentation
Our consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States, or GAAP, and include our accounts and the accounts of our subsidiaries.
Reverse Stock Split
On September 29, 2014, the Board of Directors of the Company approved, and made legally effective, a 2.34539 to 1 reverse stock split of the Company’s common stock. All shares, stock options and per share information presented in the consolidated financial statements have been adjusted to reflect the reverse stock split on a retroactive basis for all Successor periods presented. There was no change in number of authorized shares or the par value of the Company’s common stock.
Secondary Offerings
During 2016, KKR and certain executive officers of the Company sold a total of 17,500,000 shares of the Company’s common stock as part of three seperate secondary offerings, or the Secondary Offerings. The Company incurred professional fees in connection with the Secondary Offerings of $1.3 million during year ended December 31, 2016. The fees are included in transaction-related costs in the accompanying consolidated statement of operations. As of December 31, 2016, KKR owned 36.9% of the Company’s outstanding common stock.
(2) Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements include the accounts and results of operations of the Company. All intercompany balances and transactions have been eliminated.
71
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Variable Interest Entities
Financial Accounting Standards Board’s, or FASB, accounting guidance concerning variable interest entities, or VIE, addresses the consolidation of business enterprise to which the usual condition of consolidation (ownership of a majority voting interest) does not apply. This guidance focuses on controlling financial interests that may be achieved through arrangements that do not involve voting interests. The guidance requires an assessment of who the primary beneficiary is and whether the primary beneficiary should consolidate the VIE. The primary beneficiary is identified as the variable interest holder that has both the power to direct the activities of the variable interest entity that most significantly impacts the entity’s economic performance and the obligation to absorb losses or the right to receive benefits from the entity that could potentially be significant to the variable interest entity. Application of the VIE consolidation requirements may require the exercise of significant judgment by management.
Accounts Receiveable Financing Agreement
On March 22, 2016, the Company entered into a three-year accounts receivable financing agreement and related arrangements to securitize certain of its accounts receivable. Under the accounts receivable financing agreement, certain of the Company’s U.S. accounts receivable and unbilled services balances are sold by certain of its consolidated subsidiaries to another of its consolidated subsidiaries, a wholly-owned bankruptcy-remote special purpose entity, or SPE. The SPE in turn may borrow up to $140.0 million from a third party lender, secured by liens on the receivables and other assets of the SPE.
The Company retains the servicing of the securitized accounts receivable portfolio and has a variable interest in the SPE by holding the residual equity. The Company determined that the SPE is a VIE and it is the primary beneficiary because (i) the Company’s servicing responsibilities for the securitized portfolio gives it the power to direct the activities that most significantly impact the performance of the VIE and (ii) its variable interest in the VIE gives it the obligation to absorb losses and the right to receive residual returns that could potentially be significant. As a result, the Company has consolidated the VIE within its financial statements.
Refer to Note 9, Current Borrowings and Long-Term Debt, for additional information regarding the accounts receivable financing agreement.
CNS Research Institute, Inc.
In order to comply with laws in New Jersey and Pennsylvania prohibiting the corporate practice of medicine, the Company has management contracts for medical services with a professional corporation, CNS Research Institute, Inc., or CNS, for physician investigators working in New Jersey and Pennsylvania. CNS is owned by an employee of the Company. The management contracts expire on January 30, 2025.
The Company pays CNS a management fee equal to the salary, bonus and benefits for the physicians. The Company manages all aspects of CNS’ operations including providing administrative support and making all significant operational decisions. Additionally, CNS cannot provide services to any other party without the prior written approval of the Company.
After evaluating all of the factors noted above, it was concluded that CNS should be included in the Company’s consolidated financial statements as it is a variable interest entity and the Company is the primary beneficiary. The Company paid CNS $0.8 million, $0.7 million and $0.9 million during the years ended December 31, 2016, 2015 and 2014, respectively, as compensation under the management contract, which was eliminated in consolidation. CNS had no net income for the years ended December 31, 2016, 2015 and 2014.
72
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Risks and Other Factors
The Company’s revenues are dependent on research and development expenditures of the pharmaceutical and biotechnology industries. Any significant reduction in research and development expenditures by the pharmaceutical and biotechnology industries could have a material adverse effect on the Company and its results of operations.
Clients of the Company generally may terminate contracts without cause upon 30 to 60 days’ notice. While the Company generally negotiates deposit payments and early termination fees up front, such terminations could significantly impact the future level of staff utilization and have a material adverse effect on the Company and the results of future operations.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. In particular, the Company’s primary method of revenue recognition requires estimates of costs to be incurred to fulfill existing long‑term contract obligations. Actual results could differ from those estimates. Estimates are also used when accounting for certain items such as allowance for doubtful accounts, depreciation and amortization, asset impairment, certain acquisition‑related assets and liabilities, income taxes, fair market value determinations, and contingencies.
Reportable Segments
The Company is solely focused on the execution of clinical trials on a global basis. The Company has considered whether the delivery of the different types of capabilities in various stages of clinical development constitute separate products or lines of service in accordance with ASC 280, “Segment Reporting,” or ASC 280, and has concluded that there are substantial similarities and overlaps in the capabilities delivered at each stage of clinical development, with the primary differences between the Early Development Services, or EDS, compared to the Product Registration, or PR, and Strategic Solutions, or SS, relating to the points during the life cycle of a clinical trial at which such capabilities are delivered. After review and analysis of the operating characteristics of each service offering and using the aggregation characteristics under ASC 280, the Company has concluded that the services provided are similar across most characteristics.
The Company's operations consist of one reportable segment, which represents management's view of the Company's operations based on its management and internal reporting structure. The Company considered the guidance in ASC 350, “Intangibles—Goodwill and Other,” which notes that a reporting unit is an operating segment or one level below an operating segment. PR, EDS, and SS are the business units that are one level below the Company’s sole operating segment and the Company determined that they meet the definition of “components,” as discrete financial information exists and this information is regularly reviewed by segment management.
Business Combinations
Business combinations are accounted for using the acquisition method and, accordingly, the identifiable assets acquired, the liabilities assumed, and any non‑controlling interest in the acquiree are recorded at their estimated fair values on the date of the acquisition. Goodwill represents the excess of the purchase price over the estimated fair value of the net assets acquired, including the amount assigned to identifiable intangible assets.
Contingent Liabilities
The Company provides for contingent liabilities when (1) it is probable that an asset has been impaired or a liability has been incurred at the date of the consolidated financial statements and (2) the amount of the loss can be
73
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
reasonably estimated. Disclosure in the notes to the consolidated financial statements is required for loss contingencies that do not meet both these conditions if there is a reasonable possibility that a loss may have been incurred. The Company expenses as incurred the costs of defending legal claims against the Company.
Cash Equivalents
The Company considers all highly‑liquid investments purchased with an original maturity of three months or less to be cash equivalents. As of December 31, 2016 and 2015, substantially all of the Company’s cash and cash equivalents were held in or invested with large financial institutions. Certain bank deposits may at times be in excess of the Federal Deposit Insurance Corporation insurance limits.
Restricted cash
The Company receives cash advances from its customers to be used for the payment of investigator costs and other pass‑through expenses. The terms of certain customer contracts require that such advances be maintained in separate escrow accounts; these accounts are not commingled with the Company’s cash and cash equivalents and are presented separately in the consolidated balance sheets as restricted cash.
Additionally, as part of the acquisition of Nextrials, Inc., or Nextrials, the Company was required to transfer $0.5 million to an escrow account held by a subsidiary. As of December 31, 2016, the balance of the cash held in escrow was $0.5 million. These funds are expected to be distributed in 2017.
Also, as part of the acquisition of ClinStar, LLC, or ClinStar, the Company was required to transfer $1.0 million to an escrow account held by a subsidiary. The funds were used to pay deferred compensation to certain former ClinStar employees. During the year ended December 31, 2014, the Company distributed all of the remaining funds held in the escrow account.
The following table provides a reconciliation of cash, cash equivalents, and restricted cash reported within the consolidated balance sheets that sum to the total of the same amounts shown in the consolidated statements of cash flows:
|
|
|
December 31, |
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Cash and cash equivalents |
|
$ |
144,623 |
|
$ |
121,065 |
|
$ |
85,192 |
|
Restricted cash |
|
|
4,715 |
|
|
5,060 |
|
|
6,337 |
|
Total cash, cash equivalents, and restricted cash |
|
$ |
149,338 |
|
$ |
126,125 |
|
$ |
91,529 |
Accounts Receivable and Unbilled Services
Accounts receivable represent amounts for which invoices have been sent to clients based upon contract terms. Unbilled services represent amounts earned for services that have been rendered but for which clients have not been billed and include reimbursement revenue. Unbilled services are generally billable upon submission of appropriate billing information, achievement of contract milestones or contract completion.
Allowances for Doubtful Accounts
The Company maintains an allowance for estimated losses resulting from the inability of its customers to make required payments. The Company performs credit reviews of each customer, monitors collections and payments from our customers, and determines the allowance based upon historical experience and specific customer collection issues. The Company ages billed accounts receivable and assesses exposure by customer type, by aged category, and by specific identification. After all attempts to collect a receivable have failed, the receivable is written off against the allowance or, to the extent unreserved, to bad debt expense.
74
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Advanced Billings
Advanced billings represent amounts associated with services, reimbursement revenue and investigator fees that have been received but have not yet been earned or paid.
Fixed Assets
Fixed assets and software purchased or developed for internal use are recorded at cost and are depreciated on a straight‑line basis over the following estimated useful lives:
|
Furniture, fixtures and equipment |
|
|
5-7 years |
|
|
Computer hardware and software |
|
|
3-7 years |
|
|
Leasehold improvements |
|
|
Lesser of the life of the lease or useful life of the improvements |
|
Internal Use Software
The Company accounts for internal use software in accordance with the provisions of accounting standards, which require certain direct costs and interest costs incurred during the application stage of development to be capitalized and amortized over the useful life of the software.
Derivative Financial Instruments
All derivatives are measured at fair value and recognized as either assets or liabilities on the consolidated balance sheets. Derivatives that are not determined to be effective hedges are adjusted to fair value with a corresponding effect on earnings. Changes in the fair value of derivatives that are designated and determined to be effective as part of a hedge transaction have no immediate effect on earnings and depending on the type of hedge, are recorded either as part of other comprehensive loss and will be included in earnings in the period in which earnings are affected by the hedged item, or are included in earnings as an offset to the earnings impact of the hedged item. Any ineffective portion of hedges is reported in earnings as it occurs. The Company utilizes interest rate swap and cap agreements, or interest rate contracts, to manage changes in market conditions related to debt obligations. Amounts previously recorded in accumulated other comprehensive loss related to these interest rate swaps will be reclassified into earnings over the term of the previously hedged borrowing using the swaplet method. The Company has elected the accounting policy that cash flows associated with interest rate derivative contracts are classified as cash flows from investing activities.
Fair Value Measurements
The Company records certain assets and liabilities at fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants at the measurement date. A three‑level fair value hierarchy that prioritizes the inputs used to measure fair value is described below. This hierarchy requires entities to maximize the use of observable inputs and minimize the use of unobservable inputs. The three levels of inputs used to measure fair value are as follows:
|
· |
Level 1—Quoted prices in active markets for identical assets or liabilities. |
|
· |
Level 2—Observable inputs other than quoted prices included in Level 1, such as quoted prices for similar assets and liabilities in active markets; quoted prices for identical or similar assets and liabilities in markets that are not active; or other inputs that are observable or can be corroborated by observable market data. |
75
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
|
· |
Level 3—Unobservable inputs that are supported by little or no market activity. This includes certain pricing models, discounted cash flow methodologies and similar techniques that use significant unobservable inputs. |
The carrying amount of financial instruments, including cash and cash equivalents, accounts receivable, unbilled services, accounts payable and advanced billings, approximate fair value due to the short maturities of these instruments.
Recurring Fair Value Measurements
The following table summarizes the fair value of the Company’s financial assets and liabilities that are measured on a recurring basis as of December 31, 2016 (in thousands):
|
|
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest rate swap |
|
$ |
— |
|
$ |
590 |
|
$ |
— |
|
$ |
590 |
|
|
Contingent consideration |
|
|
— |
|
|
— |
|
|
2,754 |
|
|
2,754 |
|
|
Total |
|
$ |
— |
|
$ |
590 |
|
$ |
2,754 |
|
$ |
3,344 |
|
The following table summarizes the fair value of the Company’s financial assets and liabilities that are measured on a recurring basis as of December 31, 2015 (in thousands):
|
|
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Assets: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest rate swap |
|
$ |
— |
|
$ |
28 |
|
$ |
— |
|
$ |
28 |
|
|
Total |
|
$ |
— |
|
$ |
28 |
|
$ |
— |
|
$ |
28 |
|
|
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contingent consideration |
|
$ |
— |
|
$ |
— |
|
$ |
999 |
|
$ |
999 |
|
|
Total |
|
$ |
— |
|
$ |
— |
|
$ |
999 |
|
$ |
999 |
|
The Company values contingent consideration, related to business combinations, using a weighted probability of potential payment scenarios discounted at rates reflective of the weighted average cost of capital for the businesses acquired. Key assumptions used to estimate the fair value of contingent consideration include revenue and operating forecasts and the probability of achieving the specific targets. Interest rate swaps and caps are measured at fair value using a market approach valuation technique. The valuation is based on an estimate of net present value of the expected cash flows using relevant mid‑market observable data inputs and based on the assumption of no unusual market conditions or forced liquidation.
76
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The following table summarizes the changes in Level 3 financial liabilities measured on a recurring basis (in thousands):
|
|
|
Contingent Consideration |
|
|
|
|
|
- Accrued expenses and |
|
|
|
|
|
Other long-term liabilities |
|
|
|
Balance at December 31, 2014 |
|
$ |
1,911 |
|
|
Revaluations included in earnings |
|
|
89 |
|
|
Payments on ClinStar contingent consideration |
|
|
(2,000) |
|
|
Initial estimate of VHS contingent consideration |
|
|
999 |
|
|
Balance at December 31, 2015 |
|
$ |
999 |
|
|
Initial estimate of Nextrials contingent consideration |
|
|
2,282 |
|
|
Revaluations included in earnings |
|
|
(527) |
|
|
Balance at December 31, 2016 |
|
$ |
2,754 |
|
Non‑recurring Fair Value Measurements
Certain assets and liabilities are carried on the accompanying consolidated balance sheets at cost and are not remeasured to fair value on a recurring basis. These assets include finite‑lived intangible assets which are tested when a triggering event occurs and goodwill and identifiable indefinite‑lived intangible assets which are tested for impairment annually on October 1 or when a triggering event occurs.
As of December 31, 2016, assets carried on the balance sheet and not remeasured to fair value on a recurring basis totaling approximately $1,446.0 million were identified as Level 3. These assets are comprised of goodwill of $972.0 million and identifiable intangible assets, net of $474.0 million.
Refer to Note 9, Current Borrowings and Long-Term Debt, for additional information regarding the fair value of long-term debt balances.
Impairment of Long‑Lived Assets
The Company reviews the recoverability of its long‑lived asset groups, including furniture and equipment, computer hardware and software, leasehold improvements, and other finite‑lived intangibles, when events or changes in circumstances occur that indicate the carrying value of the asset group may not be recoverable. The assessment of possible impairment is based on the Company’s ability to recover the carrying value of the asset group from the expected future pre‑tax cash flows (undiscounted and without interest charges) of the related operations. If these cash flows are less than the carrying value of such asset, an impairment loss is recognized for the difference between estimated fair value and carrying value. The Company’s primary measure of fair value is based on discounted cash flows. The measurement of impairment requires the Company to make estimates of these cash flows related to long‑lived assets, as well as other fair value determinations.
Goodwill and Other Intangibles
Goodwill and indefinite‑lived intangible assets are tested for impairment annually or more frequently if an event or circumstance indicates that an impairment loss may have been incurred. Separate intangible assets that have finite useful lives are amortized over their estimated useful lives or over the period in which economic benefit is received. The Company’s primary finite lived intangibles are customer relationships and customer backlog, which are amortized on an accelerated basis, which coincides with the period of economic benefit received by the Company.
The Company reviews the carrying value of goodwill to determine whether impairment may exist on an annual basis or whenever it has reason to believe goodwill may not be recoverable. The annual impairment test of goodwill is
77
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
performed during the fourth quarter of each fiscal year. The Company did not have an impairment for any of the years presented.
When evaluating for impairment, the Company may first perform a qualitative assessment to determine whether it is more likely than not that a reporting unit or indefinite-lived intangible asset is impaired. If the Company does not perform a qualitative assessment, or if it determines that it is not more likely than not that the fair value of the reporting unit or indefinite-lived intangible asset exceeds its carrying amount, the Company will calculate the estimated fair value of the reporting unit or indefinite-lived intangible asset. The Company’s decision to perform a qualitative impairment assessment for an individual reporting unit in a given year is influenced by a number of factors, inclusive of the size of the reporting unit's goodwill, the significance of the excess of the reporting unit's estimated fair value over carrying value at the last quantitative assessment date, the amount of time in between quantitative fair value assessments and the date of acquisition. During 2016, as part of the Company’s annual impairment analysis, the Company performed the qualitative assessment for approximately $850.0 million, or 87.5%, of its total goodwill balance of $972.0 million, which resides in its PR and SS reporting units, and for its indefinite-lived trade name intangible asset.
If the Company does not perform a qualitative assessment, goodwill impairment is determined by the Company using a two‑step process. The first step of the goodwill impairment test is used to identify potential impairment by comparing the fair value of each reporting unit, determined using various valuation techniques, with the primary technique being a discounted cash flow analysis, to its carrying value. If the fair value of a reporting unit exceeds its carrying amount, goodwill of the reporting unit is considered not impaired and the second step of the impairment test is unnecessary. If the carrying amount of a reporting unit exceeds its fair value, the second step of the goodwill impairment test is performed to measure the amount of impairment loss, if any. The second step of the goodwill impairment test compares the implied fair value of the reporting unit’s goodwill with the carrying amount of that goodwill. If the carrying amount of the reporting unit’s goodwill exceeds the implied fair value of that goodwill, an impairment loss is recognized.
The estimated fair value of the EDS reporting unit closely approximated its carrying value when the Company performed its annual goodwill impairment test during the fourth quarter of 2014. The Company made operational improvements during 2015 and 2016 in order to improve the profitability of the EDS reporting unit. As a result of these changes, EDS saw growth in both backlog and new business awards that contributed to its improved financial performance during the year and led to the Company to update its forecast for future periods. The Company considered all of these factors when it performed its most recent goodwill impairment test during the fourth quarter of 2016 and it was concluded that the estimated fair value of the EDS reporting unit exceeded its carrying value by approximately $70.0 million or 33%. Any negative changes in assumptions on revenue, new business awards, cancellations, or the Company’s ability to improve operations while maintaining a competitive cost structure could adversely affect the fair value of EDS and result in significant goodwill impairment charges in 2017 or later.
Revenue Recognition
The Company generally enters into contracts with customers to provide services with payments based on either fixed‑fee, time and materials, or fee‑for‑service arrangements. Revenue for services is recognized only after persuasive evidence of an arrangement exists, the sales price is determinable, services have been rendered, and collectability is reasonably assured.
Once these criteria have been met, the Company recognizes revenue for the services provided on fixed‑fee contracts based on the proportional performance methodology, which determines the proportion of outputs or performance obligations which have been completed or delivered compared to the total contractual outputs for performance obligations. To measure performance, the Company compares the contract costs incurred to estimated total contract costs through completion. As part of the client proposal and contract negotiation process, the Company develops a detailed project budget for the direct costs based on the scope of the work, the complexity of the study, the geographical location involved and the Company’s historical experience. The Company then establishes the individual contract pricing based on the Company’s internal pricing guidelines, discount agreements, if any, and negotiations with
78
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
the client. The estimated total contract costs are reviewed and revised periodically throughout the lives of the contracts, with adjustments to revenue resulting from such revisions being recorded on a cumulative basis in the period in which the revisions are first identified. Contract costs consist primarily of direct labor and other project‑related costs. Revenue from time and materials contracts is recognized as hours are incurred. Revenues and the related costs of fee‑for‑service contracts are recognized in the period in which services are performed.
A majority of the Company’s contracts undergo modifications over the contract period and the Company’s contracts provide for these modifications. During the modification process, the Company recognizes revenue to the extent it incurs costs, provided client acceptance and payment is deemed reasonably assured.
The Company often offers volume discounts to certain of its large customers based on annual volume thresholds. The Company records an estimate of the annual volume rebate as a reduction of revenue throughout the period based on the estimated total rebate to be earned for the period.
Most of the Company’s contracts can be terminated by the client either immediately or after a specified period following notice. These contracts require the client to pay the Company the fees earned through the termination date, the fees and expenses to wind down the study, and, in some cases, a termination fee or some portion of the fees or profit that the Company could have earned under the contract if it had not been terminated early. Therefore, revenue recognized prior to cancellation generally does not require a significant adjustment upon cancellation.
Reimbursement Revenue and Reimbursable Out‑of‑Pocket Costs
The Company incurs out‑of‑pocket costs, in excess of contract amounts, which are reimbursable by its customers. The Company includes out‑of‑pocket costs both as reimbursement revenue and as reimbursable out‑of‑pocket costs in the consolidated statements of operations.
As is customary in the industry, the Company routinely enters into separate agreements on behalf of its clients with independent physician investigators in connection with clinical trials. The funds received for investigator fees are netted against the related cost because such fees are the obligation of the Company’s clients, without risk or reward to the Company. The Company is not obligated either to perform the service or to pay the investigator in the event of default by the client. In addition, the Company does not pay the independent physician investigator until funds are received from the client. Total payments to investigators were $249.6 million, $208.0 million, and $211.5 million for the years ended December 31, 2016, 2015, and 2014, respectively.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to credit risk consist of cash and cash equivalents, accounts receivable, and unbilled services. As of December 31, 2016, substantially all of the Company’s cash and cash equivalents were held in or invested with large financial institutions. Accounts receivable include amounts due from pharmaceutical and biotechnology companies. The Company establishes an allowance for potentially uncollectible receivables. In management’s opinion, there is no additional material credit risk beyond amounts provided for such losses.
Service revenue from individual customers greater than 10% of consolidated service revenue in the respective periods was as follows:
|
|
|
Years Ended December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
Customer A |
|
11.0% |
|
— |
|
— |
|
|
Customer B |
|
10.4% |
|
10.7% |
|
— |
|
79
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Accounts receivable and unbilled receivables from individual customers that were equal to or greater than 10% of consolidated accounts receivable and unbilled receivables at the respective dates were as follows:
|
|
|
December 31, |
|
||
|
|
|
2016 |
|
2015 |
|
|
Customer A |
|
12.0% |
|
— |
|
|
Customer B |
|
— |
|
13.4% |
|
|
Customer C |
|
— |
|
10.1% |
|
Foreign Currency
The assets and liabilities of the Company’s foreign subsidiaries are translated into U.S. dollars at exchange rates in effect as of the end of the period. Equity activities are translated at the spot rate effective at the date of the transaction. Revenue and expense accounts and cash flows of these operations are translated at average exchange rates prevailing during the period the transactions occurred. Translation gains and losses are included as an adjustment to the accumulated other comprehensive loss account in stockholders’ equity.
Translation gains and losses from foreign currency transactions, such as those resulting from the settlement and revaluation of foreign receivables and payables, are included in the determination of net income (loss). These amounts are included in foreign currency gains, net in the consolidated statement of operations. In addition, gains or losses related to the Company’s intercompany loans payable and receivable denominated in a foreign currency other than the subsidiary’s functional currency that are deemed to be of a long‑term investment nature are remeasured to cumulative translation and recorded in accumulated other comprehensive loss in the consolidated balance sheets.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets are recognized for future deductible temporary differences, along with net operating loss carryforwards and credit carryforwards, if it is more likely than not that the tax benefits will be realized. To the extent a deferred tax asset cannot be recognized under the preceding criteria, a valuation allowance is established to reduce the deferred tax asset to the amount that is more likely than not to be realized. Deferred tax liabilities are recognized for future taxable temporary differences. Deferred tax assets and liabilities are measured using enacted tax rates in effect for the year in which those temporary differences are expected to be recovered or settled.
There are inherent uncertainties related to the interpretation of tax regulations in the jurisdictions in which the Company transacts business. The judgments and estimates made at a point in time may change based on the outcome of tax audits, as well as changes to, or further interpretations of, regulations. Income tax expense is adjusted in the period in which these events occur, and these adjustments are included in the Company’s consolidated statement of operations. If such changes take place, there is a risk that the Company’s effective tax rate may increase or decrease in any period. A company must recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the financial statements from such a position are measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate resolution.
Stock‑Based Compensation
The primary type of stock‑based compensation utilized by the Company is stock options. Stock options are awards which allow the employee to purchase shares of the Company’s stock at a fixed price. The Company measures compensation cost at the grant date, based on fair value of the award, and recognizes it as expense over the employees’ requisite service period.
80
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The fair value of each option issued during these periods was estimated on the date of grant using the Black‑Scholes option pricing model for service condition awards and a lattice model for market and performance condition awards with the following weighted average assumptions:
|
|
|
Years Ended December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
Risk-free interest rate |
|
1.5% |
|
1.7% |
|
2.0% |
|
|
Expected life, in years |
|
6.3 |
|
6.3 |
|
6.5 |
|
|
Dividend yield |
|
N/A |
|
N/A |
|
N/A |
|
|
Volatility |
|
31.2% |
|
34.4% |
|
39.5% |
|
The risk‑free interest rate is based on the United States Treasury yield curve in effect at the time of the grant. The expected life represents the period of time the grants are expected to be outstanding. The Company uses the historical volatilities of a selected peer group as it does not have sufficient history to estimate the volatility of its common share price. The Company calculates expected volatility based on reported data for selected reasonably similar publicly traded companies for which the historical information is available. For the purpose of identifying peer companies, the Company considers characteristics such as industry, length of trading history, similar vesting terms and in‑the‑money option status.
Due to the absence of an active market for the Company’s common shares prior to the Company’s IPO, the fair value of our common shares for purposes of determining the exercise price for award grants was determined in good faith by the Company’s Board of Directors, with the assistance and upon the recommendation of management based on a number of market factors, including: the common shares underlying the award involved illiquid securities in a private company; results of operations and financial position; and the market performance of publicly traded companies compared to the Company.
The Company accounts for its stock‑based compensation for restricted share awards and restricted share units, or collectively, RSAs/RSUs, based on the closing market price of the Company’s common stock on the trading day immediately prior to the grant date.
Net Income (Loss) Per Share
The calculation of net income (loss) per share, or EPS, is based on the weighted average number of common shares or common stock equivalents outstanding during the applicable period. The dilutive effect of common stock equivalents is excluded from basic earnings per share and is included in the calculation of diluted earnings per share, unless the effect of inclusion would be anti‑dilutive.
Debt Issuance Costs
Debt issuance costs relating to the Company’s long‑term debt are recorded as a direct reduction of long-term debt; these costs are deferred and amortized to interest expense using the effective interest method, over the respective terms of the related debt. Debt issuance costs relating to the Company’s revolving credit facilities are recorded as an asset; these costs are deferred and amortized to interest expense using the straight‑line method.
Compensated Absences
The Company accrues for the costs of compensated absences to the extent that the employee’s right to receive payment relates to service already rendered, the obligation vests or accumulates, payment is probable and the amount can be reasonably estimated. The Company’s policies related to compensated absences vary by jurisdiction and obligations are recorded net of estimated forfeiture due to turnover when reasonably predictable.
81
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Operating Leases
The Company records rent expense for operating leases, some of which have escalating rent over the term of the lease, on a straight‑line basis over the initial effective lease term. The Company begins depreciation on the date of initial possession, which is generally when the Company enters the space and begins to make improvements in preparation for its intended use. Some of the Company’s facility leases provide for concessions by the landlords, including payments for leasehold improvements considered tenant assets, free rent periods, and other lease inducements. The Company reflects these concessions as deferred rent in the accompanying consolidated financial statements. The Company accounts for the difference between rent expense and rent paid as deferred rent. For tenant allowances for improvements considered to be tenant assets, rent holidays and other lease incentives, the Company records a deferred rent liability at the inception of the lease term and amortizes the deferred rent over the term of the lease as a reduction to rent expense. For tenant allowances considered to be property owner assets, the payment is treated as a reimbursement for the cost of the lessor asset.
Recently Implemented Accounting Standards
In August 2016, the FASB issued Accounting Standard Update, or ASU, No. 2016-15, “Statement of Cash Flows Classification of Certain Cash Receipts and Cash Payments,” which clarifies existing guidance related to accounting for cash receipts and cash payments and classification on the statement of cash flows. In November 2016, the FASB issued ASU No. 2016-18, “Statement of Cash Flows (Topic 230): Restricted Cash,” which requires restricted cash be included with cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2017, with early adoption permitted. The guidance for both standards requires application using a retrospective transition method.
The Company early adopted both ASUs in the accompanying consolidated financial statements. As a result of the retrospective application of ASU 2016-15, $14.3 million of payments of debt prepayment and debt extinguishment costs originally recorded as operating cash outflows were reclassified to financing outflows in the consolidated statement of cash flows for the year ended December 31, 2014. The retrospective application of ASU 2016-18 resulted in restricted cash being reclassified as a component of cash, cash equivalents, and restricted cash in the consolidated statement of cash flows for all periods presented.
In August 2014, the FASB issued ASU No. 2014‑15, “Presentation of Financial Statements—Going Concern.” This ASU clarifies management’s responsibility to evaluate whether there is a substantial doubt about the entity’s ability to continue as a going concern and provides guidance for related footnote disclosures. This ASU became effective beginning in 2016. The adoption of this ASU did not impact the Company’s consolidated financial statements.
Recently Issued Accounting Pronouncements
In May 2014, the FASB issued ASU No. 2014-09, "Revenue from Contracts with Customers.” The new revenue standard establishes a single revenue recognition model for recognizing contracts from customers. Under the new standard, revenue is recognized when a customer obtains control of promised goods or services and is recognized in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. In addition, the standard requires disclosure of the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers. The FASB has recently issued several amendments to the standard, including clarification on principal versus agent considerations, identifying performance obligations, and accounting for licenses of intellectual property.
The new standard is effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period. The standard permits the use of either the retrospective or modified retrospective (cumulative effect) transition method.
82
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The Company plans to adopt the new revenue guidance as of January 1, 2018 and is currently evaluating the transition methods and the potential impact to the Company’s consolidated financial statements. The Company has established an implementation team that consists of both internal resources and external advisors to assist with the adoption of the new standard. The evaluation and implementation process is ongoing and is expected to continue through 2017 as the Company performs an analysis on the contract portfolio to identify potential differences from its current accounting policies, and as it reviews the business processes, systems and controls required to support recognition and disclosure under the new standard.
In February 2016, the FASB issued ASU No. 2016-02, “Leases,” which revises the accounting related to lessee accounting. Under the new guidance, lessees will be required to recognize a lease liability and a right-of-use asset for all leases with terms greater than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The provisions of ASU No. 2016-02 are effective for fiscal years beginning after December 15, 2018 and should be applied through a modified retrospective transition approach for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. Early adoption is permitted. The Company is currently assessing the potential impact of ASU No. 2016-02 on the Company’s consolidated financial statements.
In March 2016, the FASB issued ASU No. 2016-09, “Compensation-Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting.” This update includes provisions intended to simplify various aspects of accounting for share-based compensation. In addition, ASU No. 2016-09 will take effect for public companies for the annual periods beginning after December 15, 2016. The Company will adopt this ASU beginning with the first quarter of 2017. The adoption of this ASU will have the following effects on the consolidated financial statements:
Income taxes - The new guidance requires excess tax benefits and tax deficiencies to be recorded as income tax benefit or expense in the statement of operations. The Company will apply the modified retrospective adoption approach beginning in 2017 and expects to record a cumulative-effect adjustment to retained earnings and reduce its deferred tax liabilities by $12.6 million with an offsetting increase to the valuation allowance of $12.6 million. As such, it is expected that the net impact to retained earnings will be zero. The Company continuously evaluates its need for a valuation allowance on its net deferred tax assets based upon the weight of available evidence. If the Company is able to support the recognition of certain net deferred tax assets in the future, it is noted that an additional tax benefit from the release of this additional valuation could occur in the future. This adjustment relates to tax assets that had previously arisen from tax deductions for equity compensation expenses that were greater than the compensation recognized for financial reporting.
Forfeitures – The standard provides an accounting policy election to account for forfeitures as they occur. The Company plans to make this accounting policy election and does not expect the modified retrospective adoption for this component of the standard to have a material impact on its financial statements.
Statements of Cash Flows - Cash flows related to excess tax benefits will no longer be separately classified as a financing activity apart from other income tax cash flows. The Company will adopt this component of the standard on a prospective basis.
Earnings Per Share - The Company uses the treasury stock method to compute diluted earnings per share, unless the effect would be anti-dilutive. Under this method, the Company will no longer be required to estimate the tax rate and apply it to the dilutive share calculation for determining the dilutive earnings per share.
(3) Joint Ventures
On May 6, 2016, the Company and WuXi AppTec (Shanghai) Co., Ltd., or WuXi, finalized an agreement to dissolve the WuXiPRA joint venture. Under the agreement, the Company sold its 49% portion of the joint venture located in mainland China for $4.0 million, which subsequently became a wholly owned subsidiary of WuXi. The
83
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
portion of the joint venture located in Hong Kong became a wholly owned subsidiary of the Company and was acquired for $0.3 million. As a result of the transaction, the Company recognized a $3.3 million gain on the sale, which is recorded in the equity in gains (losses) of unconsolidated joint ventures in the accompanying consolidated statement of operations.
During April 2015, prior to the dissolution of the WuXiPRA joint venture, both the Company and WuXi made a $3.0 million contribution to WuXiPRA to fund the joint venture’s working capital needs. The Company’s interest in WuXiPRA remained at 49% after the capital contribution. The Company recorded reductions to the investment balance of $0.7 million (excluding the gain on the sale), $2.9 million, and $2.1 million during the years ended December 31, 2016, 2015, and 2014, respectively, for our equity in the venture’s net loss for the period, which is recorded in the equity in gains (losses) of unconsolidated joint ventures, net of tax in our consolidated statement of operations. The investment was adjusted for our equity in the venture’s net income (loss), cash contributions, distributions, and other adjustments required by the equity method of accounting. The investment in WuXiPRA totaled $1.1 million as of December 31, 2015.
The Company entered into a joint venture agreement with A2 Healthcare Corporation (formerly part of Asklep, Inc.). The joint venture provides research and development outsourcing solutions in Japan to the biopharmaceutical and medical device industries. This joint venture is based in Tokyo, Japan and is owned by the Company (49%) and Asklep (51%). On October 17, 2014, the joint venture changed its name from RPS Asklep, Inc. to A2PRA Corporation, or A2PRA. The Company recorded changes to the investment balance totaling $0.1 million, $0.0 million, and $(0.1) million during the years ended December 31, 2016, 2015, and 2014, respectively, for the Company’s equity in the venture’s net income (loss) for the period, which is recorded in the equity in gains (losses) of unconsolidated joint venture, net of tax in our consolidated statement of operations. The investment will be adjusted for the Company’s equity in the venture’s net income (loss), cash contributions, distributions, and other adjustments required by the equity method of accounting. The investment in A2PRA totaled $0.3 million and $0.2 million at December 31, 2016 and 2015, respectively.
In August 2015, the Company and an affiliate of KKR entered into a joint venture. The joint venture was dissolved in December 2015. The purpose of the joint venture included, among other things, the evaluation of investments or acquisitions to enhance the strategic objectives of the Company. The joint venture was jointly owned by the Company (11%) and KKR (89%). The Company contributed $20.0 million to the joint venture in August 2015 and received $19.5 million in December 2015 when the joint venture was dissolved. The Company recorded the $0.5 million reduction to the investment balance in equity in gains (losses) of unconsolidated joint ventures, net of tax in the consolidated statement of operations. The investment in the joint venture was adjusted for the Company’s equity in the venture’s net income (loss), cash contributions, distributions, and other adjustments required by the equity method of accounting.
(4) Business Combinations
Acquisition of VHS
On June 8, 2015, the Company purchased the assets of Value Health Solutions Inc., or VHS, a software development firm, for $0.5 million in cash and 47,598 unregistered shares of the Company’s common stock with a fair market value of $1.6 million; an additional $0.4 million of common stock will be issued in June 2017, less amounts reimbursable to the Company for any indemnification obligations of the seller. The asset purchase agreement also includes contingent consideration in the form of potential earn-out payments of up to $16.0 million. Earn-out payments totaling $1.0 million and $15.0 million are contingent upon the achievement of project milestones and certain external software sales targets, respectively, during the 48-month period following closing. The Company has recognized a liability of approximately $1.0 million as the estimated acquisition date fair value of the earn-out; this amount is included in the accrued expenses and other current liabilities in the consolidated balance sheet. The fair value of the contingent consideration was based on significant inputs not observed in the market and thus represented a Level 3 measurement. Any change in the fair value of the contingent consideration subsequent to the acquisition date will be
84
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
recognized in earnings in the period of the change. With this acquisition, the Company expects to enhance its ability to serve customers throughout the clinical research process with technologies that include improved efficiencies by reducing study durations and costs through integrated operational management.
The acquisition of VHS was accounted for as a business combination and, accordingly, the assets acquired and the liabilities assumed have been recorded at their respective fair values as of the acquisition date. In connection with the acquisition, the Company recorded approximately $1.0 million of goodwill, which is deductible for income tax purposes. The goodwill is attributable to the workforce of the acquired business and expected synergies with the Company’s existing information technology operations.
The Company’s purchase price allocation is as follows (in thousands):
|
|
|
Purchase |
|
Weighted |
|
|
|
|
|
Price |
|
Amortization |
|
|
|
|
|
Allocation |
|
Period |
|
|
|
Software intangible |
|
$ |
2,500 |
|
5 years |
|
|
Property, plant and equipment |
|
|
43 |
|
|
|
|
Estimated fair value of net assets acquired |
|
|
2,543 |
|
|
|
|
Purchase price, including contingent consideration |
|
|
3,499 |
|
|
|
|
Total goodwill |
|
$ |
956 |
|
|
|
Pro forma information is not provided as the acquisition did not have a material effect on the Company’s consolidated results.
Acquisition of Nextrials
On March 18, 2016, the Company acquired all of the outstanding shares of Nextrials, Inc., or Nextrials, a developer of web-based software which integrates electronic health records with clinical trials, for $4.8 million in cash and contingent consideration in the form of potential earn-out payments of up to $3.0 million. Earn-out payments totaling $2.0 million and $1.0 million are contingent upon the achievement of project milestones and certain external software sales targets, respectively, during the 30-month period following closing. The Company recognized a liability of approximately $2.3 million as the estimated acquisition date fair value of the earn-out; the fair value was based on significant inputs not observed in the market and thus represented a Level 3 measurement. Changes in the fair value of the earn-out subsequent to the acquisition date were recognized in earnings in the period of the change. The fair value of the contingent consideration decreased by $0.5 million during the year ended December 31, 2016. As of December 31, 2016, the earn-out liability totaled $1.8 million; $0.7 million of the balance is included in accrued expenses and other current liabilities and the remaining $1.1 million is included in other long-term liabilities in the consolidated balance sheet. With this acquisition, the Company expects to enhance its ability to serve customers throughout the clinical research process with technologies that include improved efficiencies by reducing study durations and costs through integrated operational management.
The acquisition of Nextrials was accounted for as a business combination and, accordingly, the assets acquired and the liabilities assumed have been recorded at their respective fair values as of the acquisition date. In connection with the acquisition, the Company recorded approximately $4.3 million of goodwill, which is not deductible for income tax purposes. The goodwill is attributable to the workforce of the acquired business and expected synergies with the Company’s existing information technology operations.
85
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The Company’s purchase price allocation is as follows (in thousands):
|
|
|
Purchase |
|
Weighted |
|
|
|
|
|
Price |
|
Amortization |
|
|
|
|
|
Allocation |
|
Period |
|
|
|
Cash and cash equivalents |
|
$ |
94 |
|
|
|
|
Accounts receivable |
|
|
211 |
|
|
|
|
Other current assets |
|
|
96 |
|
|
|
|
Property, plant and equipment |
|
|
111 |
|
|
|
|
Software intangible |
|
|
5,574 |
|
5 years |
|
|
Accounts payable and accrued expenses |
|
|
(1,585) |
|
|
|
|
Other long-term liabilities |
|
|
(1,663) |
|
|
|
|
Estimated fair value of net assets acquired |
|
|
2,838 |
|
|
|
|
Purchase price, including contingent consideration |
|
|
7,145 |
|
|
|
|
Total goodwill |
|
$ |
4,307 |
|
|
|
Since the acquisition date, goodwill increased by $2.0 million, primarily as a result of adjustments to the acquired income tax balances. Pro forma information is not provided as the acquisition did not have a material effect on the Company’s consolidated results.
Acquisition of WuXiPRA’s Hong Kong Operations
As noted in Note 3, the Company acquired WuXiPRA’s Hong Kong operations for $0.3 million when the joint venture was dissolved on May 6, 2016. The acquisition was accounted for as a business combination and, accordingly, the assets acquired and the liabilities assumed have been recorded at their respective fair values as of the acquisition date. In connection with the acquisition, the Company recorded approximately $0.6 million of goodwill, which is attributable to the workforce of the acquired business. Pro forma information is not provided as the acquisition did not have a material effect on the Company’s consolidated results.
(5) Accounts Receivable and Unbilled Services
Accounts receivable and unbilled services include service revenue, reimbursement revenue, and amounts associated with work performed by investigators. Accounts receivable and unbilled services were (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Accounts receivable |
|
$ |
284,647 |
|
$ |
290,963 |
|
|
Unbilled services |
|
|
155,609 |
|
|
126,755 |
|
|
|
|
|
440,256 |
|
|
417,718 |
|
|
Less allowance for doubtful accounts |
|
|
(1,203) |
|
|
(2,641) |
|
|
Total accounts receivable and unbilled services, net |
|
$ |
439,053 |
|
$ |
415,077 |
|
A rollforward of the allowance for doubtful accounts is as follows (in thousands):
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Beginning balance |
|
$ |
2,641 |
|
$ |
1,819 |
|
$ |
129 |
|
|
Charged (credited) to income from operations |
|
|
(652) |
|
|
443 |
|
|
976 |
|
|
Write-offs, recoveries and the effects of foreign currency exchange |
|
|
(786) |
|
|
379 |
|
|
714 |
|
|
Ending balance |
|
$ |
1,203 |
|
$ |
2,641 |
|
$ |
1,819 |
|
86
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
(6) Fixed Assets
The carrying amount of fixed assets is as follows (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Leasehold improvements |
|
$ |
25,083 |
|
$ |
20,606 |
|
|
Computer hardware and software |
|
|
92,095 |
|
|
79,853 |
|
|
Furniture and equipment |
|
|
33,751 |
|
|
27,208 |
|
|
|
|
|
150,929 |
|
|
127,667 |
|
|
Accumulated depreciation |
|
|
(63,352) |
|
|
(46,976) |
|
|
Total fixed assets, net |
|
$ |
87,577 |
|
$ |
80,691 |
|
All fixed assets are included as collateral for the payment and performance in full of the term loans pledged by the Company and its subsidiaries.
Depreciation expense was $24.1 million, $21.2 million, and $22.2 million for the years ended December 31, 2016, 2015 and 2014, respectively.
(7) Goodwill and Intangible Assets
Goodwill
The changes in the carrying amount of goodwill are as follows (in thousands):
|
Balance December 31, 2014 |
|
$ |
1,033,999 |
|
|
Acquisition of VHS |
|
|
956 |
|
|
Currency translation |
|
|
(20,157) |
|
|
Balance December 31, 2015 |
|
$ |
1,014,798 |
|
|
Acquisition of Nextrials |
|
|
4,307 |
|
|
Acquisition of the WuXiPRA joint venture’s Hong Kong operations |
|
|
570 |
|
|
Currency translation |
|
|
(47,695) |
|
|
Balance December 31, 2016 |
|
$ |
971,980 |
|
There are no accumulated impairment charges as of December 31, 2016 and 2015.
87
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Intangible Assets
Intangible assets consist of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Customer relationships |
|
$ |
360,328 |
|
$ |
380,721 |
|
|
Customer backlog |
|
|
119,223 |
|
|
127,871 |
|
|
Trade names (finite-lived) |
|
|
25,740 |
|
|
25,693 |
|
|
Patient list and other intangibles |
|
|
28,974 |
|
|
23,400 |
|
|
Non-competition agreements |
|
|
2,737 |
|
|
2,657 |
|
|
Total finite-lived intangible assets, gross |
|
|
537,002 |
|
|
560,342 |
|
|
Accumulated amortization |
|
|
(181,036) |
|
|
(144,414) |
|
|
Total finite-lived intangible assets, net |
|
|
355,966 |
|
|
415,928 |
|
|
Trade names (indefinite-lived) |
|
|
118,010 |
|
|
118,010 |
|
|
Total intangible assets, net |
|
$ |
473,976 |
|
$ |
533,938 |
|
The Company conducts its annual impairment test of indefinite‑lived intangibles during the fourth quarter of the fiscal year. For the periods ended December 31, 2016, 2015, and 2014, the Company concluded that the fair value of indefinite‑lived intangibles exceeded the carrying value and, therefore, no impairment exists. Amortization expense was $45.4 million, $56.7 million and $74.4 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Estimated amortization expense related to finite‑lived intangible assets for the next five years and thereafter is as follows (in thousands):
|
2017 |
|
|
|
|
$ |
35,221 |
|
|
2018 |
|
|
|
|
|
31,118 |
|
|
2019 |
|
|
|
|
|
25,859 |
|
|
2020 |
|
|
|
|
|
24,612 |
|
|
2021 |
|
|
|
|
|
22,507 |
|
|
2022 and thereafter |
|
|
|
|
|
216,649 |
|
|
Total |
|
|
|
|
$ |
355,966 |
|
(8) Accrued Expenses and Other Current Liabilities
Accrued expenses consisted of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Compensation, including bonuses, fringe benefits and payroll taxes |
|
$ |
86,160 |
|
$ |
77,650 |
|
|
Other |
|
|
33,813 |
|
|
36,093 |
|
|
Interest |
|
|
3,616 |
|
|
6,150 |
|
|
Total accrued expenses and other liabilities |
|
$ |
123,589 |
|
$ |
119,893 |
|
88
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
(9) Current Borrowings and Long‑Term Debt
Long‑term debt consists of the following (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Term loans, first lien |
|
$ |
625,000 |
|
$ |
689,000 |
|
|
Senior notes |
|
|
91,441 |
|
|
225,000 |
|
|
Accounts receivable financing agreement |
|
|
120,000 |
|
|
— |
|
|
|
|
|
836,441 |
|
|
914,000 |
|
|
Less debt issuance costs and discount |
|
|
(8,139) |
|
|
(24,486) |
|
|
|
|
|
828,302 |
|
|
889,514 |
|
|
Less current portion |
|
|
(31,250) |
|
|
— |
|
|
Total long-term debt, net |
|
$ |
797,052 |
|
$ |
889,514 |
|
Principal payments on long‑term debt are due as follows (in thousands):
|
Current maturities of long-term debt: |
|
|
|
|
|
|
|
|
2017 |
|
|
|
|
$ |
31,250 |
|
|
2018 |
|
|
|
|
|
46,875 |
|
|
2019 |
|
|
|
|
|
166,875 |
|
|
2020 |
|
|
|
|
|
62,500 |
|
|
2021 |
|
|
|
|
|
437,500 |
|
|
2022 and thereafter |
|
|
|
|
|
91,441 |
|
|
Total |
|
|
|
|
$ |
836,441 |
|
2016 Credit Facilities
On December 6, 2016, the Company through its wholly-owned subsidiary, Pharmaceutical Research Associates, Inc., entered into new senior secured credit facilities, or the 2016 Credit Facilities, totaling $750.0 million. The 2016 Credit Facilities are comprised of a $625.0 million first lien term loan due 2021, or 2016 First Lien Term Loan, and a five-year $125.0 million revolving line of credit, or 2016 Revolver.
The proceeds from the 2016 Credit Facilities were used to repay the then outstanding 2013 First Lien Term Loan (defined below). In accordance with the guidance in ASC 470‑50, “Debt—Modifications and Extinguishments,” the debt repayment was accounted for as a partial debt extinguishment. The repayment resulted in a $16.7 million loss on extinguishment of debt, consisting of $15.8 million write-off of unamortized debt issuance costs and $0.9 million of fees associated with the transaction, which is included in loss on modification or extinguishment of debt in the consolidated statement of operations for the year ended December 31, 2016.
As collateral for borrowings under the 2016 Credit Facilities, the Company granted a pledge on primarily all of its assets, and the stock of wholly‑owned U.S. restricted subsidiaries. The Company was subject to certain financial covenants, which require the Company to maintain certain debt‑to‑EBITDA and interest expense-to-EBITDA ratios. The 2016 Credit Facilities also contain covenants that, among other things, restrict the Company’s ability to incur create any liens, make investments and acquisitions, incur or guarantee additional indebtness, enter into mergers or consolidations and other fundamental changes, conduct sales and other dispositions of property or assets, enter into sale-leaseback transactions or hedge agreements, prepay subordinated debt, pay dividends or make other payments in respect of capital stock, change the line of business, enter into transactions with affiliates, enter into burdensome agreements with negative pledge clauses and clauses restriction, and make subsidiary distributions. After giving effect to the applicable restrictions on the payment of dividends under the 2016 Credit Facilities, subject to compliance with applicable law, as of December 31, 2016, all amounts in retained earnings were free of restriction and were available for the payment of dividends. The Company does not expect to pay dividends in the foreseeable future. The Company does not expect these covenants to
89
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
restrict its liquidity, financial condition or access to capital resources in the foreseeable future. The 2016 Credit Facilities also contains customary representations, warranties, affirmative covenants, and events of default.
2016 First Lien Term Loan
The 2016 First Lien Term Loan is a floating rate term loan with scheduled, fixed quarterly principal payments as follows:
|
· |
1.25% by quarterly term loan amortization payments, or $7.8 million per quarter, to be made commencing March 31, 2017 and made on or prior to December 31, 2017; |
|
· |
1.88% by quarterly term loan amortization payments, or $11.7 million per quarter, to be made on or after March 31, 2018, but on or prior to December 31, 2019; |
|
· |
2.50% by quarterly term loan amortization payments, or $15.6 million per quarter, to be made on or after March 31, 2020, but on or prior to December 31, 2020; |
|
· |
3.13% by quarterly term loan amortization payments, or $19.5 million per quarter, to be made on or after March 31, 2021, but prior to September 30, 2021; and |
|
· |
60.63% (or if less, the remaining principal amount of the term loan) on December 06, 2021. |
The variable interest rate is a rate equal to the London Interbank Offered Rate, or LIBOR, or the adjusted base rate, or ABR rate, at the election of the Company, plus a margin based on the ratio of total indebtedness to EBITDA and ranges from 1.25% to 2.25%, in the case of LIBOR rate loans, and 0.25% to 1.25%, in the case of ABR rate loans. The Company has the option of 1, 2, 3 or 6 month base interest rates. As of December 31, 2016, the weighted average interest rate on the first lien term loan was 2.70%. There are no prepayment penalties.
2016 Revolver
The Company’s 2016 Revolver provides for $125.0 million of potential borrowings and expires on December 6, 2021. The interest rate on the 2016 Revolver is based on the LIBOR with a 0% LIBOR floor or ABR rate, at the election of the Company, plus an applicable margin, based on the leverage ratio of the Company. The Company, at its discretion, may elect interest periods of 1, 2, 3 or 6 months. In addition, the Company was required to pay to the lenders a commitment fee of 0.3% quarterly for unused commitments on the revolver from December 6, 2016 to December 31, 2016. Following December 31, 2016, the commitment fee will range from 0.2% to 0.4% based on the Company’s debt-to-EBITDA ratio. At December 31, 2016, the Company had no outstanding borrowings under the 2016 Revolver. In addition, at December 31, 2016, the Company had $7.0 million in letters of credit outstanding, which are secured by the 2016 Revolver.
2013 Credit Facilities
In September 2013, the Company entered into a senior secured credit facilities, or the 2013 Credit Facilities, for an aggregate principal amount of $825.0 million of first lien term loan, or 2013 First Lien Term Loan, and a $125.0 million revolving line of credit, or 2013 Revolver. In September 2013, the Company also issued $375.0 million in senior notes, or Senior Notes. The proceeds from the 2013 Credit Facilities and the Senior Notes issuances were used in conjunction with the acquisition by KKR, to fund the acquisition of RPS, repay existing debt, and pay for fees and expenses related to the aforementioned events. The Company paid an $8.3 million debt discount in connection with the 2013 First Lien Term Loan.
On December 2, 2013, the Company borrowed $65.0 million under the first lien term loan facility of the 2013 Credit Facilities, or the Incremental Term Loan Borrowing. The proceeds were used to fund the acquisition of CRI Lifetree. In accordance with the guidance in ASC 470‑50, “Debt—Modifications and Extinguishments,” the Incremental Term Loan Borrowing was accounted for as a debt modification.
90
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
On March 24, 2014, the Company completed a repricing transaction, or the Repricing, associated with the 2013 First Lien Term Loan that reduced the applicable margin from 4.0% to 3.5%. As part of the repricing, eight previous lenders did not consent to the repricing terms; therefore the non‑consenting lenders were replaced by new lenders. In accordance with the guidance in ASC 470‑50, “Debt—Modifications and Extinguishments,” the Repricing was accounted for as a partial debt extinguishment based on non‑consenting lenders no longer having a holding interest. As a result of the partial debt extinguishment, the Company recognized a loss of extinguishment of debt totaling $1.3 million, which was recorded during the year ended December 31, 2014. The Company incurred $0.1 million in expenses for the repricing transaction, which were expensed during the year ended December 31, 2014.
As collateral for borrowings under the 2013 Credit Facilities, the Company granted a pledge on primarily all of its assets, and the stock of designated subsidiaries. The Company was subject to certain financial covenants, which require the Company to maintain certain debt‑to‑EBITDA ratios. The 2013 Credit Facilities also contains covenants that, among other things, restrict the Company’s ability to incur additional indebtedness, grant liens, make investments, loans, guarantees or advances, make restricted junior payments, including dividends, redemptions of capital stock and voluntary prepayments or repurchase of certain other indebtedness, engage in mergers, acquisitions or sales of assets, enter into sale and leaseback transactions or engage in certain transactions with affiliates and otherwise restrict certain corporate activities. After giving effect to the applicable restrictions on the payment of dividends under the 2013 Credit Facilities, subject to compliance with applicable law, as of December 31, 2015, there was approximately $3.0 million free of restriction, which was available for the payment of dividends. The 2013 Credit Facilities also contained customary representations, warranties, affirmative covenants, and events of default.
2013 First Lien Term Loan
The 2013 First Lien Term Loan was a floating rate term loan with scheduled, fixed quarterly principal payments of 0.25% of the original principal balance through September 2020. The voluntary prepayments made during 2014, using proceeds from the IPO, fully satisfied all required quarterly principal payments through maturity. The variable interest rate was based on the LIBOR, with a 1.0% LIBOR floor, plus an applicable margin of 3.5%. The applicable margin was dependent upon the Company’s debt to consolidated EBITDA ratio as defined in the 2013 Credit Facilities. The 2013 Credit Facilities required us to prepay outstanding term loans, subject to certain exceptions, with 50% of our annual Excess Cash Flow, which percentage would be reduced to 25% if PRA achieves a debt‑to‑EBITDA ratio of less than or equal to 3.75 to 1.0, but greater than 3.25 to 1.0 on the date of prepayment for the most recent test period and no prepayment would be required if PRA achieves a debt‑to‑EBITDA ratio of less than or equal to 3.25 to 1.0 on the date of prepayment for the most recent test period, commencing in 2014.
The Company had the option of 1, 2, 3 or 6 month base interest rates. As of December 31, 2015, the weighted average interest rate on the first lien term loan was 4.5%. There were no prepayment penalties.
On November 18, 2014, the Company repaid $152.1 million in principal using proceeds from the Company’s IPO. In accordance with the guidance in ASC 470‑50, “Debt—Modifications and Extinguishments,” the debt repayment was accounted for as a partial debt extinguishment. The repayment resulted in the write‑off of $4.8 million in unamortized debt issuance costs which is included in loss on modification or extinguishment of debt in the consolidated statement of operations during the year ended December 31, 2014.
2013 Revolver
The Company’s 2013 Revolver provided for $125.0 million of potential borrowings and would have expired on September 23, 2018. The interest rate on the 2013 Revolver was based on the LIBOR plus an applicable rate, based on the leverage ratio of the Company. The Company, at its discretion, may have chosen interest periods of 1, 2, 3 or 6 months. In addition, the Company was required to pay to the lenders a commitment fee of 0.5% quarterly for unused commitments on the revolver, subject to a step‑down to 0.375% based upon achievement of a certain leverage ratio. At December 31, 2015, the Company had no outstanding borrowings under the 2013 Revolver. In addition, at December 31, 2015, the Company had $4.4 million in letters of credit outstanding, which were secured by the 2013 Revolver.
91
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Senior Notes
In September 2013, the Company issued $375.0 million of Senior Notes. The Senior Notes do not require principal payments and mature on October 1, 2023. The Senior Notes bear interest at a rate of 9.50% per year payable on April 1, and October 1 of each year, beginning April 1, 2014.
The Company may redeem the Senior Notes, in whole or in part, at any time prior to October 1, 2018 subject to a prepayment premium calculated in accordance with the Senior Notes indenture. From October 1, 2018 through October 1, 2019, the prepayment premium is 4.75% declining ratably to 0% beginning on October 1, 2021. In the event of a change in control, the Company may be required to offer to repurchase the Senior Notes at a price equal to the outstanding principal balance and a 1% prepayment premium plus accrued and unpaid interest.
The Senior Notes include covenants which place limitations on incurring additional indebtedness, selling certain assets, and making certain distributions.
The Senior Notes agreement contains certain provisions that restrict the payment of dividends from the Company’s subsidiaries to the parent company. As a result, there are no material balances present within the parent company that are available for the payment of dividends as the parent company did not have any net income during 2016 that was free of restrictions. The Company does not expect to pay dividends in the foreseeable future.
On November 18, 2014, the Company repaid $150.0 million in principal and a $14.3 million prepayment penalty using proceeds from the Company’s IPO. In accordance with the guidance in ASC 470‑50, the debt repayment was accounted for as a partial debt extinguishment. The repayment also resulted in the write‑off of $4.6 million in unamortized debt issuance costs, which is included in loss on modification or extinguishment of debt in the consolidated statement of operations during the year ended December 31, 2014.
On March 17, 2016, the Company repurchased $133.6 million aggregate principal amount of its Senior Notes as part of a cash tender offer. In accordance with the guidance in ASC 470-50, the debt repurchase was accounted for as a partial debt extinguishment. The repurchase resulted in a $21.5 million loss on extinguishment of debt, which consists of a $17.4 million early tender premium, a $3.7 million write-off of unamortized debt issuance cost and $0.4 million of fees associated with the transaction which is included in loss on modification or extinguishment of debt in the consolidated statement of operations during the year ended December 31, 2016.
Accounts Receivable Financing Agreement
In March 2016, the Company entered into a $140.0 million accounts receivable financing agreement, of which $120.0 million was outstanding as of December 31, 2016. The borrowings were used to repay amounts outstanding on the Company’s revolving credit facility that were used to fund the cash tender offer for the Senior Notes.
Loans under the accounts receivable financing agreement accrue interest at either a reserve-adjusted LIBOR or a base rate, plus 1.6%. The Company may prepay loans upon one business day prior notice and may terminate the accounts receivable financing agreement with 15 days’ prior notice. As of December 31, 2016, the weighted average interest rate on the accounts receivable financing agreement was 2.31%.
The accounts receivable financing agreement contains various customary representations and warranties and covenants, and default provisions which provide for the termination and acceleration of the commitments and loans under the agreement in circumstances including, but not limited to, failure to make payments when due, breach of representations, warranties or covenants, certain insolvency events or failure to maintain the security interest in the trade receivables, and defaults under other material indebtedness.
92
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The accounts receivable financing agreement terminates on March 22, 2019, unless terminated earlier pursuant to its terms. At December 31, 2016, there was $20.0 million of remaining capacity available under the accounts receivable financing agreement.
Fair Value of Debt
The estimated fair value of the Company’s debt was $844.2 million and $924.9 million at December 31, 2016 and 2015, respectively. The fair value of the Senior Notes, which totaled $99.2 million and $246.2 million at December 31, 2016 and 2015, respectively, was determined based on Level 2 inputs using the market approach, which is primarily based on rates at which the debt is traded among financial institutions. The fair value of the term loans, borrowings under credit facilities, and accounts receivable financing agreement which totaled $745.0 million and $678.7 million at December 31, 2016 and 2015, respectively, was determined based on Level 3 inputs, which is primarily based on rates at which the debt is traded among financial institutions adjusted for the Company’s credit standing.
(10) Stockholders’ Equity
Authorized Shares
The Company is authorized to issue up to one billion shares of common stock, with a par value of $0.01. The Company is authorized to issue up to one hundred million shares of preferred stock, with a par value of $0.01.
(11) Stock‑Based Compensation
On September 23, 2013 and in connection with the acquisition of the Company by KKR, the Board of Directors approved the formation of the 2013 Stock Incentive Plan for Key Employees of Pinnacle Holdco Parent, Inc. and its subsidiaries, or the Plan. The Plan allowed for the issuance of stock options and other stock‑based awards as permitted by applicable laws. The number of shares available for grant under the Plan is 12.5% of the outstanding shares at closing on a fully diluted basis. The Company rolled over 2,052,909 stock options under the Plan; this amount is comprised of 2,016,581 and 36,328 options rolled over by employees of the Predecessor Company and RPS, respectively. The fair value of the options that were rolled over equaled the fair value of the options in the Predecessor Company and, therefore, there was no additional stock‑based compensation expense recorded.
All stock options granted under the Plan are subject to transfer restrictions of the stock option’s underlying shares once vested and exercised. This lack of marketability was included as a discount, calculated using the Finnerty Model, when determining the grant date value of these options. In conjunction with the Secondary Offerings, the transfer restrictions on a portion of such shares issuable upon exercise of vested options granted under the Plan were released. The release of the transfer restrictions is considered a modification under ASC 718, “Stock Compensation.” As a result of these modifications, the Company incurred approximately $10.1 million of incremental compensation expense associated with service-based options during the year ended December 31, 2016, which is included in transaction-related costs in the accompanying consolidated statement of operations.
On November 23, 2014 and in connection with the Company’s IPO, the Board of Directors approved the formation of the 2014 Omnibus Plan for Key Employees, or the 2014 Omnibus Plan. The 2014 Omnibus Plan allows for the issuance of stock options, stock appreciation rights, restricted shares and restricted stock units, other stock‑based awards, and performance compensation awards as permitted by applicable laws. The number of shares available for grant under the 2014 Omnibus Plan is 3,200,000.
Generally, the Company grants stock options with exercise prices greater than or equal to the fair market value of the Company’s common stock on the date of grant. The stock option compensation cost calculated under the fair value approach is recognized on a pro‑rata basis over the vesting period of the stock options (usually five years under the Plan and four years under the 2014 Omnibus Plan). Most stock option grants are subject to graded vesting as services are rendered and have a contractual life of ten years.
93
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
In December 2013, the Company granted certain employees market-based options under the Plan that vest only upon the achievement of a specified internal rate of return from a liquidity event (“2.0x Options” and “2.5x Options”). At the time of grant, no compensation expense was recorded as the 2.0x Options and 2.5x Options vest upon a liquidity event, which is not considered probable until the date it occurs. On January 20, 2016, the Compensation Committee of the Board of Directors adopted a resolution to adjust the vesting criteria for all 2.0x Options granted and still outstanding on such date. Under the revised vesting criteria, the 2.0x Options vest upon the announcement of a secondary offering. The Company did not record compensation expense on the January 20, 2016 modification date as the Company determined the modification resulted in Type IV Improbable-to-Improbable modification as the secondary offering was deemed improbable since the event was outside of the Company’s control and could not be considered probable until the date it occured. On March 2, 2016, the Company announced a secondary offering of shares by KKR and certain management stockholders, and it became probable that the 2.0x Options would vest. Due to the modification of the terms of the 2.0x Options, the Company calculated the fair value of these options using the Black‑Scholes option pricing model with the following assumptions: expected life of 2.92 years; risk-free rate of 1.04%; volatility of 45%; dividend yield of 0%; and a Finnerty discount of approximately 16%. In total, 835,551 2.0x Options held by current employees were modified. As a result of this modification, and the modifications associated with the transfer restrictions releases noted above, the Company incurred approximately $25.7 million of incremental compensation expense associated with the 2.0x Options during the year ended December 31, 2016, which is included in transaction-related costs in the accompanying consolidated statement of operations.
On November 16, 2016, the 2.5x Options vested upon the achievement of a specified internal rate of return and multiple on invested capital in connection with the closing of a secondary offering of shares by KKR. In total, 809,755 2.5x Options held by current employees vested. The Company incurred approximately $6.4 million of incremental compensation expense associated with the vesting and transfer restriction release of the 2.5x Options during the year ended December 31, 2016, which is included in transaction-related costs in the accompanying consolidated statement of operations.
As of December 31, 2016, there was $16.2 million of unrecognized compensation cost related to unvested stock-based compensation, which is expected to be recognized over a weighted average period of 2 years. The total fair value of options vested during the years ended December 31, 2016, 2015 and 2014 was $27.3 million, $3.1 million and $2.8 million, respectively.
Aggregated information regarding the Company’s option plans is summarized below:
|
|
|
|
|
|
|
|
Wtd. Average |
|
|
|
|
|
|
|
|
|
Wtd. Average |
|
Remaining |
|
Intrinsic Value |
|
||
|
|
|
Options |
|
Exercise Price |
|
Contractual Life |
|
(in millions) |
|
||
|
Outstanding at December 31, 2015 |
|
6,839,129 |
|
$ |
11.39 |
|
6.8 |
|
$ |
231.7 |
|
|
Granted |
|
489,000 |
|
|
46.10 |
|
|
|
|
|
|
|
Exercised |
|
(1,492,797) |
|
|
6.77 |
|
|
|
|
|
|
|
Expired/forfeited |
|
(327,985) |
|
|
17.23 |
|
|
|
|
|
|
|
Outstanding at December 31, 2016 |
|
5,507,347 |
|
$ |
15.38 |
|
6.7 |
|
$ |
218.9 |
|
|
Exercisable at December 31, 2016 |
|
3,622,518 |
|
$ |
10.40 |
|
6.0 |
|
$ |
162.0 |
|
The weighted‑average fair value of service‑based options granted was $15.57, $10.87 and $6.71 during the years ended December 31, 2016, 2015 and 2014, respectively.
94
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Selected information regarding the Company’s stock options as of December 31, 2016 is as follows:
|
Options Outstanding |
|
Options Exercisable |
|
|||||||||||||
|
|
|
|
|
|
Wtd Average |
|
|
|
|
|
|
Wtd. Average |
|
|
|
|
|
|
|
|
Number of |
|
Remaining Life |
|
Wtd. Average |
|
Number of |
|
Remaining Life |
|
Wtd. Average |
|
||
|
Exercise Price |
|
Options |
|
(in Years) |
|
Exercise Price |
|
Options |
|
(in Years) |
|
Exercise Price |
|
|||
|
$ |
2.94 |
|
871,026 |
|
2.8 |
|
$ |
2.94 |
|
871,026 |
|
2.8 |
|
$ |
2.94 |
|
|
$ |
11.73 |
|
3,420,136 |
|
7.0 |
|
$ |
11.73 |
|
2,541,911 |
|
7.0 |
|
$ |
11.73 |
|
|
$ |
16.42 |
|
144,060 |
|
7.5 |
|
$ |
16.42 |
|
73,706 |
|
7.3 |
|
$ |
16.42 |
|
|
$ |
25.35 - 54.69 |
|
1,072,125 |
|
8.7 |
|
$ |
36.97 |
|
135,875 |
|
7.6 |
|
$ |
30.01 |
|
Restricted Stock Awards and Units
The Company’s RSAs/RSUs will settle in shares of the Company’s common stock on the applicable vesting date. RSAs/RSUs granted to employees vest 100% on the third anniversary of the date of grant. RSAs/RSUs granted to our non-employee directors vest 50% on the first anniversary of the date of grant and 50% on the second anniversary of the date of grant.
Activity related to the Company’s RSAs/RSUs in 2016 is as follows:
|
|
|
|
|
Wtd. Average |
|
Intrinsic |
|
||
|
|
|
|
|
Grant-Date |
|
Value |
|
||
|
|
|
Awards |
|
Fair Value |
|
(millions) |
|
||
|
Unvested at December 31, 2015 |
|
143,984 |
|
$ |
28.42 |
|
$ |
6.5 |
|
|
Granted |
|
50,487 |
|
|
43.85 |
|
|
|
|
|
Vested |
|
(5,881) |
|
|
25.51 |
|
|
|
|
|
Unvested at December 31, 2016 |
|
188,590 |
|
$ |
32.63 |
|
$ |
10.4 |
|
Stock‑based compensation expense related to employee stock options and RSAs/RSUs is summarized below (in thousands):
|
|
|
Years Ended December 31, |
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Direct costs |
|
$ |
1,813 |
|
$ |
1,218 |
|
$ |
752 |
|
Selling, general and administrative |
|
|
5,254 |
|
|
4,058 |
|
|
2,715 |
|
Transaction-related costs |
|
|
42,166 |
|
|
— |
|
|
— |
|
Total stock-based compensation expense |
|
$ |
49,233 |
|
$ |
5,276 |
|
$ |
3,467 |
(12) Income Taxes
The components of income (loss) before income taxes and equity in gains (losses) of unconsolidated joint ventures are as follows (in thousands):
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Domestic |
|
$ |
(61,226) |
|
$ |
(23,400) |
|
$ |
(92,040) |
|
|
Foreign |
|
|
155,120 |
|
|
138,565 |
|
|
50,188 |
|
|
|
|
$ |
93,894 |
|
$ |
115,165 |
|
$ |
(41,852) |
|
95
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The components of the provision for (benefit from) income taxes were as follows (in thousands):
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Current: |
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
$ |
151 |
|
$ |
1,132 |
|
$ |
— |
|
|
State |
|
|
1,842 |
|
|
1,507 |
|
|
(617) |
|
|
Foreign |
|
|
36,970 |
|
|
30,584 |
|
|
24,431 |
|
|
Total current income tax expense |
|
|
38,963 |
|
|
33,223 |
|
|
23,814 |
|
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
(2,230) |
|
|
(1,349) |
|
|
(22,002) |
|
|
State |
|
|
(451) |
|
|
1,564 |
|
|
(1,867) |
|
|
Foreign |
|
|
(7,788) |
|
|
(3,434) |
|
|
(8,099) |
|
|
Total deferred income tax benefit |
|
|
(10,469) |
|
|
(3,219) |
|
|
(31,968) |
|
|
Total income tax expense (benefit) |
|
$ |
28,494 |
|
$ |
30,004 |
|
$ |
(8,154) |
|
Income taxes computed at the statutory U.S. federal income tax rate of 35.0% are reconciled to the benefit from income taxes as follows:
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Statutory federal income tax rate |
|
|
35.0% |
|
|
35.0% |
|
|
35.0% |
|
|
State income taxes, net of federal benefit |
|
|
0.3% |
|
|
2.0% |
|
|
5.5% |
|
|
Tax on foreign earnings: |
|
|
|
|
|
|
|
|
|
|
|
Foreign rate differential |
|
|
(17.7%) |
|
|
(13.6%) |
|
|
4.3% |
|
|
Foreign earnings taxed in the U.S. |
|
|
17.5% |
|
|
7.3% |
|
|
(10.9%) |
|
|
Non-U.S. research and development credits |
|
|
(3.9%) |
|
|
(4.4%) |
|
|
3.8% |
|
|
Stock-based compensation |
|
|
1.9% |
|
|
0.2% |
|
|
(0.8%) |
|
|
Change in liability for uncertain tax positions |
|
|
— |
|
|
(0.6%) |
|
|
(14.6%) |
|
|
Nondeductible expenses |
|
|
0.1% |
|
|
0.3% |
|
|
(2.0%) |
|
|
Other |
|
|
(2.9%) |
|
|
(0.1%) |
|
|
(0.8%) |
|
|
Effective income tax rate |
|
|
30.3% |
|
|
26.1% |
|
|
19.5% |
|
96
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Components of the deferred tax assets and liabilities were as follows (in thousands):
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Net operating loss carryforwards |
|
$ |
29,470 |
|
$ |
48,689 |
|
|
Accruals and reserves |
|
|
12,986 |
|
|
13,002 |
|
|
Equity based compensation |
|
|
17,392 |
|
|
8,418 |
|
|
Prepaid expenses and other |
|
|
25,232 |
|
|
18,311 |
|
|
Deferred and unbilled revenue |
|
|
25,718 |
|
|
29,560 |
|
|
Tax credits |
|
|
5,295 |
|
|
3,680 |
|
|
|
|
|
116,093 |
|
|
121,660 |
|
|
Valuation allowance |
|
|
(21,689) |
|
|
(23,205) |
|
|
Deferred tax assets |
|
|
94,404 |
|
|
98,455 |
|
|
Identified intangibles |
|
|
(148,576) |
|
|
(164,645) |
|
|
Depreciable, amortizable and other property |
|
|
(12,963) |
|
|
(12,432) |
|
|
Deferred tax liabilities |
|
|
(161,539) |
|
|
(177,077) |
|
|
Total deferred tax liability |
|
$ |
(67,135) |
|
$ |
(78,622) |
|
|
Long-term deferred tax asset |
|
$ |
6,568 |
|
$ |
3,069 |
|
|
Long-term deferred tax liability |
|
$ |
(73,703) |
|
$ |
(81,691) |
|
The Company’s foreign subsidiaries are taxed separately in their respective jurisdictions. As of December 31, 2016, the Company has cumulative foreign net operating loss carryforwards of approximately $9.7 million. In addition, the Company has federal net operating loss carryforwards of approximately $70.9 million and state net operating loss carryforwards of approximately $279.4 million.
The carryforward periods for the Company’s net operating losses vary from five years to an indefinite number of years depending on the jurisdiction. The Company’s ability to offset future taxable income with net operating loss carryforwards may be limited in certain instances, including changes in ownership.
The Company also has federal and state income tax credit carryforwards available to potentially offset future federal and state income tax of $3.1 million and $1.6 million, respectively. The federal credits are indefinitely-lived. The state credits begin expiring in 2022. The Company has provided a partial valuation allowance against the benefits of these credits.
In determining the extent to which a valuation allowance for deferred tax assets is required, the Company evaluates all available evidence including projections of future taxable income, carry back opportunities, reversal of certain deferred tax liabilities, and other tax‑planning strategies. The valuation allowance at December 31, 2016 relates to the U.S. net federal deferred tax asset (including the federal net operating loss), certain foreign net operating losses, state net operating losses and state tax credit carryforwards. Based upon the available evidence, the Company has concluded that it is not more likely than not that a certain portion of these net deferred tax assets will be realized as of December 31, 2016. If the Company determines at some point in the future that utilization of these deferred tax assets becomes more likely than not, the Company will reduce the valuation allowance accordingly at that time.
97
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
A reconciliation of the beginning and ending amount of gross unrecognized income tax benefits is presented below (in thousands):
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Beginning balance |
|
$ |
11,729 |
|
$ |
16,207 |
|
$ |
11,284 |
|
|
Additions based on tax positions related to current year |
|
|
1,196 |
|
|
1,333 |
|
|
5,221 |
|
|
Additions for income tax positions of prior years |
|
|
542 |
|
|
95 |
|
|
1,559 |
|
|
Impact of changes in exchange rates |
|
|
(127) |
|
|
(594) |
|
|
(1,005) |
|
|
Settlements with tax authorities |
|
|
(559) |
|
|
— |
|
|
— |
|
|
Reductions for income tax positions for prior years |
|
|
(349) |
|
|
(4,308) |
|
|
(452) |
|
|
Reductions due to lapse of applicable statute of limitations |
|
|
— |
|
|
(1,004) |
|
|
(400) |
|
|
Ending balance |
|
$ |
12,432 |
|
$ |
11,729 |
|
$ |
16,207 |
|
As of December 31, 2016, 2015, and 2014, the total gross unrecognized tax benefits were $12.4 million, $11.7 million, and $16.2 million, respectively. As of December 31, 2016, the total amount of gross unrecognized tax benefits which, if recognized, would impact the Company’s effective tax rate is $7.6 million. The Company anticipates changes in total unrecognized tax benefits due to the expiration of statute of limitations within the next 12 months and an income tax audit resolution. Specifically, adjustments related to transfer pricing and foreign tax exposures are expected to be resolved in various jurisdictions. A reasonable estimate of the change in the total gross unrecognized tax benefit expected to be recognized as a result is $3.7 million as of the balance sheet date.
The Company’s policy for recording interest and penalties associated with uncertain tax positions is to record such items as a component of income tax expense. The Company recorded an increase of $0.1 million, a decrease of $0.1 million, and an increase of $0.1 million during the years ended December 31, 2016, 2015 and 2014, respectively. As of December 31, 2016, the Company has a total of $2.4 million recognized on uncertain tax positions. To the extent interest and penalties are not incurred with respect to uncertain tax positions, amounts accrued will be reduced and reflected as a reduction in income tax expense.
The Company has analyzed filing positions in all of the significant federal, state and foreign jurisdictions where the Company is required to file income tax returns. The only periods subject to examination by the major tax jurisdictions where the Company does business are the 2008 through 2015 tax years.
The Company has concluded that a portion of the undistributed earnings of its foreign subsidiaries related to certain previously taxed income is not indefinitely reinvested. With respect to the previously taxed income, as of December 31, 2016 and December 31, 2015, there is no liability recorded for the effect of repatriating those foreign earnings due to the movement in foreign exchange rates which would cause a foreign exchange loss if the previously taxed income were distributed. As of December 31, 2015 the Company recorded a deferred tax liability in the amount of $0.3 million on Russian earnings of $3.4 for which there was not an indefinite reinvestment assertion. The Company has since concluded that this portion of Russian earnings is indefinitely reinvested and has removed the prior year deferred tax liability in the amount of $0.3 million as of December 31, 2016. For the remaining undistributed earnings of $256.6 million, $236.7 million, and $186.1 million as of December 31, 2016, 2015, and 2014, respectively, the Company has concluded that these earnings would be permanently reinvested in the local jurisdictions and not repatriated to the United States. Accordingly, the Company has not provided for U.S. federal and foreign withholding taxes on those undistributed earnings of its foreign subsidiaries. It is not practicable to estimate the amount that might be payable if some or all of such earnings were to be remitted. The amount of the tax liability that would result from a repatriation is impracticable to calculate given the uncertainty as to which repatriation structure would be used should the Company change its assertion and repatriate foreign earnings. Furthermore, given the uncertainty as to the repatriation structure, the Company cannot analyze the availability and amount of foreign tax credits that might be available. These earnings will provide the Company with the opportunity to continue to expand the Company’s global footprint and fund the working capital needs of the Company’s foreign locations for future growth.
98
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
A rollforward of the deferred tax asset valuation allowance accounts is as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31, |
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|||
|
Beginning balance |
|
$ |
23,205 |
|
$ |
16,142 |
|
$ |
6,120 |
|
Additions - purchase accounting |
|
|
— |
|
|
— |
|
|
1,069 |
|
Additions - other comprehensive income |
|
|
— |
|
|
3,892 |
|
|
5,165 |
|
Additions - charged to expense |
|
|
3,421 |
|
|
3,770 |
|
|
8,476 |
|
Deductions - charged to expense (including translation adjustments) |
|
|
(4,937) |
|
|
(599) |
|
|
(4,688) |
|
Ending balance |
|
$ |
21,689 |
|
$ |
23,205 |
|
$ |
16,142 |
The valuation allowance is primarily related to U.S. federal loss carryforwards, state loss carryforwards, state credit carryforwards, and loss carryforwards in various foreign jurisdictions.
(13) Commitments and Contingencies
Operating Leases
The Company leases office space under operating lease agreements expiring at various times through 2030. The Company has sublease agreements for certain facilities to reduce rent expense and accommodate expansion needs. The subleases expire at various times through 2022. The Company also leases certain office equipment under the terms of operating leases expiring at various times through 2021.
Rent expense under operating leases, net of sublease rental income, was $31.9 million, $30.1 million and $33.4 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Future minimum lease commitments on non‑cancelable operating leases are as follows (in thousands):
|
Year Ending December 31, |
|
Leases |
|
Sublease |
|
|
Net Total |
|
||
|
2017 |
|
$ |
37,975 |
|
$ |
(539) |
|
$ |
37,436 |
|
|
2018 |
|
|
32,450 |
|
|
(323) |
|
|
32,127 |
|
|
2019 |
|
|
28,614 |
|
|
(270) |
|
|
28,344 |
|
|
2020 |
|
|
26,845 |
|
|
(270) |
|
|
26,575 |
|
|
2021 |
|
|
25,070 |
|
|
(270) |
|
|
24,800 |
|
|
2022 and thereafter |
|
|
117,083 |
|
|
(308) |
|
|
116,775 |
|
|
Total |
|
$ |
268,037 |
|
$ |
(1,980) |
|
$ |
266,057 |
|
Employment Agreements
The Company has entered into employment and non‑compete agreements with certain management employees. In the event of termination of employment for certain instances, employees will receive severance payments for base salary and benefits for a specified period (six months for vice presidents, nine months for senior vice presidents and twelve months for executive vice presidents, the president and chief executive officer). Each employment agreement also contains provisions that restrict the employee’s ability to compete directly with the Company for a comparable period after employment terminates. In addition, stock option grant agreements for these employees provide the Company with the right to repurchase from the employee, or the employee with the right to sell to the Company, stock owned by the employee in certain limited instances of termination.
99
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
Legal Proceedings
The Company is involved in legal proceedings from time to time in the ordinary course of its business, including employment claims and claims related to other business transactions. Although the outcome of such claims is uncertain, management believes that these legal proceedings will not have a material adverse effect on the financial condition or results of future operations of the Company.
The Company is currently a party to litigation with the City of Sao Paulo, Brazil. The dispute relates to whether the export of services provided by the Company is subject to a local tax on services. The Company has not recorded a liability associated with the claim, which totaled $4.9 million at December 31, 2016, given that it is not deemed probable the Company will incur a loss related to this case. However, a deposit totaling $4.9 million has been made to the Brazilian court in order to annul the potential tax obligation and to avoid the accrual of additional interest and penalties. This balance is recorded in other assets on the consolidated balance sheet. During June 2015, the Judiciary Court of Justice of the State of Sao Paulo ruled in the favor of the Company, however, the judgment was appealed by the City of Sao Paulo. The Company expects to recover the full amount of the deposit when the case is settled.
Insurance
The Company currently maintains insurance for risks associated with the operation of its business, provision of professional services, and ownership of property. These policies provide coverage for a variety of potential losses, including, without limitation, loss or damage to property, bodily injury, general commercial liability, professional errors and omissions, and medical malpractice.
The Company’s retentions and deductibles associated with these insurance policies range up to a maximum of $0.5 million.
Employee Health Insurance
The Company is self‑insured for health insurance for employees within the United States. The Company maintains stop‑loss insurance on a “claims made” basis for expenses in excess of $0.3 million per member per year. As of December 31, 2016 and 2015, the Company maintained a reserve of approximately $4.1 million and $3.6 million, respectively, included in accrued expense and other current liabilities on the consolidated balance sheets, to cover open claims and estimated claims incurred but not reported.
(14) Employee Benefit Plans
Defined contribution or profit sharing style plans are offered in Australia, Belgium, Germany, Hong Kong, India, Israel, the Netherlands, New Zealand, the Philippines, South Africa, Spain, Sweden, Thailand, and the United Kingdom. In some cases these plans are required by local laws or regulations.
The Company maintains a 401(k) Plan in the United States, which covers substantially all employees of its U.S. subsidiaries. The Company matches 50% of each participant's voluntary contributions after six months of employment, subject to a maximum contribution of 6% of the participant's compensation. The employer contributions to the 401(k) Plan were approximately $9.9 million, $6.6 million and $4.4 million for the years ended December 31, 2016, 2015 and 2014, respectively.
(15) Derivatives
The Company is exposed to certain risks relating to our ongoing business operations. The primary risk that the Company seeks to manage by using derivative instruments is interest rate risk. Accordingly, the Company has instituted interest rate hedging programs that are accounted for in accordance with ASC 815, “Derivatives and Hedging.” The interest rate hedging program is a cash flow hedge program designed to minimize interest rate volatility. The Company
100
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
swaps the difference between fixed and variable interest amounts calculated by reference to an agreed‑upon notional principal amount, at specified intervals. The Company also employed an interest rate cap that would have compensated the Company if variable interest rates had risen above a pre‑determined rate. The Company’s interest rate contracts are designated as hedging instruments.
On October 2, 2013, the Company entered into interest rate swap agreements with an aggregate notional principal amount of $620.0 million, or the 2013 Swaps. The interest rate swaps were set to begin on September 23, 2015. The interest rate swaps were to be used to hedge the Company’s variable rate debt. The interest rate swaps had maturity dates ranging from one to five years. During the third quarter of 2015, the Company paid $32.9 million to terminate the 2013 Swaps. Amounts previously recorded in accumulated other comprehensive loss related to these interest rate swaps, totaling $29.6 million, are being reclassified into earnings over the term of the previously hedged borrowing using the swaplet method. For the terminated swaps, the Company reclassified $4.7 million and $0.7 million previously recorded in accumulated other comprehensive loss into interest expense during the years ended December 31, 2016 and 2015, respectively.
In addition, on October 2, 2013 the Company also entered into an interest rate cap with an aggregate notional principal amount of $800.0 million. The interest rate cap began on September 23, 2014. The interest rate cap was used to hedge the variable rate of the Company’s 2013 First Lien Term Loan to the extent that the LIBOR exceeded 4.00%. During the third quarter of 2015, the Company’s interest rate cap with a notional principal amount of $800.0 million expired.
Subsequent to the termination of all existing interest rate swaps, the Company entered into a new interest rate swap agreement with a notional principal amount of $250.0 million and a fixed three month LIBOR rate of 1.48%, or the 2015 Swap. The interest rate swap began on September 23, 2015 and will mature on September 23, 2018. The interest rate swap is being used to hedge the Company’s variable rate debt.
In conjunction with the closing of the 2016 Credit Facilities in December 2016, the 2015 Swap was amended to modify the fixed rate, repricing dates and embedded floor, or the Modified 2015 Swap. The Company re-designated the Modified 2015 Swap against the refinanced debt under the 2016 Credit Facilities. As a result of the re-designation, all amounts previously recorded in accumulated other comprehensive loss related to the 2015 Swap, totaling $0.8 million, were frozen and will be amortized into earnings over the term of the previously hedged borrowing using the swaplet method. The closing of the 2016 Credit Facilities did not impact the amortization of the losses frozen in accumulated other comprehensive loss associated with the 2013 Swaps.
The following table presents the notional amounts and fair values (determined using level 2 inputs) of the Company’s derivatives as of December 31, 2016 and 2015 (in thousands):
|
|
|
Balance Sheet |
|
December 31, 2016 |
|
December 31, 2015 |
|
||||||
|
|
|
Classification |
|
Notional amount |
|
Asset/(Liability) |
|
Notional amount |
|
Asset/(Liability) |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Derivatives in an asset position: |
|
Other assets |
$ |
— |
$ |
— |
|
$ |
250,000 |
|
$ |
28 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Derivatives in a liability position: |
|
Other long-term liabilities |
|
250,000 |
|
(590) |
|
|
— |
|
|
— |
|
The Company records the effective portion of any change in the fair value of derivatives designated as hedging instruments under ASC 815 to other accumulated comprehensive loss in the consolidated balance sheets, net of deferred taxes, and will later reclassify into earnings when the hedged item affects earnings or is no longer expected to occur. Gains and losses from the ineffective portion of any hedge are recognized in earnings immediately. For other derivative contracts that do not qualify or no longer qualify for hedge accounting, changes in the fair value of the derivatives are recognized in earnings each period.
101
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
During 2014, due to the debt repayments made in conjunction with the Company’s IPO and related changes to forecasted voluntary debt repayments in future periods, the Company determined interest rate swaps with a notional principal amount of $47.5 million no longer qualified for hedge accounting and interest rate swaps with a notional principal amount of $297.5 million experienced ineffectiveness. During 2015, the Company further revised its forecasted voluntary debt repayments in future periods; as a result of the change in expected future cash flows, an interest rate swap with a notional principal amount of $17.5 million experienced ineffectiveness. This also caused interest rate swaps with a notional principal amount of $40.0 million, which experienced ineffectiveness during 2014, to no longer qualify for hedge accounting. All of abovementioned interest rate swaps were terminated during the third quarter of 2015.
The table below presents the effect of our derivatives on the consolidated statements of operations and comprehensive (loss) income (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31, |
|
|||||||
|
Derivatives in Cash Flow Hedging Relationships (Interest Rate Contracts) |
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Amount of pretax loss recognized in other comprehensive income (loss) on derivatives |
|
$ |
(1,600) |
|
$ |
(11,851) |
|
$ |
(20,976) |
|
|
Amount of loss recognized in other income (expense), net on derivatives (ineffective portion) |
|
|
1 |
|
|
(444) |
|
|
(1,275) |
|
|
Amount of loss recognized in other income (expense), net on derivatives (no longer qualify for hedge accounting) |
|
|
— |
|
|
(1,137) |
|
|
(453) |
|
|
Amount of loss reclassified from accumulated other comprehensive loss into interest expense, net on derivatives |
|
|
(5,921) |
|
|
(908) |
|
|
(3) |
|
The Company expects that $6.9 million of unrealized losses will be reclassified out of accumulated other comprehensive loss and into interest expense, net over the next 12 months.
102
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
(16) Accumulated Other Comprehensive (Loss) Income
Below is a summary of the components of accumulated other comprehensive (loss) income (in thousand):
|
|
|
Foreign |
|
|
|
|
|
|||
|
|
|
Currency |
|
Derivative |
|
|
|
|||
|
|
|
Translation |
|
Instruments |
|
Total |
|
|||
|
Balance at December 31, 2013 |
|
$ |
15,061 |
|
$ |
1,808 |
|
$ |
16,869 |
|
|
Other comprehensive loss before reclassifications, net of tax |
|
|
(68,700) |
|
|
(17,681) |
|
|
(86,381) |
|
|
Reclassification adjustments, net of tax |
|
|
— |
|
|
3 |
|
|
3 |
|
|
Balance at December 31, 2014 |
|
|
(53,639) |
|
|
(15,870) |
|
|
(69,509) |
|
|
Other comprehensive loss before reclassifications, net of tax |
|
|
(52,433) |
|
|
(11,273) |
|
|
(63,706) |
|
|
Reclassification adjustments, net of tax |
|
|
— |
|
|
908 |
|
|
908 |
|
|
Balance at December 31, 2015 |
|
|
(106,072) |
|
|
(26,235) |
|
|
(132,307) |
|
|
Other comprehensive loss before reclassifications, net of tax |
|
|
(95,019) |
|
|
(978) |
|
|
(95,997) |
|
|
Reclassification adjustments, net of tax |
|
|
— |
|
|
3,618 |
|
|
3,618 |
|
|
Balance at December 31, 2016 |
|
$ |
(201,091) |
|
$ |
(23,595) |
|
$ |
(224,686) |
|
Foreign Currency Translation
The change in the foreign currency translation adjustment during the year ended December 31, 2016 was primarily due to the movements in the British Pound, or GBP, Euro, or EUR, Canadian dollar, or CAD, and Russian Ruble, or RUB, exchange rates against the U.S. dollar, or USD. The USD strengthened by 16.7% and 3.6% versus the GBP and EUR, respectively, during the year ended December 31, 2016, and the USD depreciated by 3.1% and 19.5% against the CAD and RUB, respectively, during the same period. The movement in the GBP and EUR represented $90.2 million and $8.4 million, respectively, of the $95.0 million loss recorded to accumulated other comprehensive loss during the year ended December 31, 2016. The overall change was partially offset by gains in the CAD and RUB, representing $1.1 million and $4.0 million of the adjustment, respectively.
The change in the foreign currency translation adjustment during the year ended December 31, 2015 was primarily due to the movements in the GBP, EUR, and CAD exchange rates against the USD. The USD strengthened by 4.6%, 10.1%, and 16.1% against the GBP, EUR, and CAD, respectively. The movement of the GBP, EUR, and CAD represented $25.8 million, $16.4 million, and $7.1 million, respectively, of the $52.4 million loss recorded to accumulated other comprehensive loss during the year ended December 31, 2015.
The change in the foreign currency translation adjustment during the year ended December 31, 2014 was primarily due to the movements in the GBP and EUR exchange rates against the USD. The USD strengthened by 5.7% and 11.8% against the GBP and EUR, respectively. The movement of the GBP and EUR represented $32.3 million and $20.6 million, respectively, of the $68.7 million loss recorded to accumulated other comprehensive loss during the year ended December 31, 2014.
Derivative Instruments
See Note 15 for further information on changes to accumulated other comprehensive income related to the derivative instruments.
(17) Net Income (Loss) Per Share
Basic net income (loss) per share is calculated by dividing net income (loss) by the weighted average number of common shares outstanding for the applicable period. Diluted net income (loss) per share is calculated after adjusting the denominator of the basic net income (loss) per share calculation for the effect of all potentially dilutive common shares, which in the Company’s case, includes shares issuable under the stock option and incentive award plan.
103
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
The following table reconciles the basic to diluted weighted average shares outstanding (in thousands):
|
|
|
Years Ended December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|
Basic weighted average common shares outstanding |
|
60,759 |
|
59,965 |
|
42,897 |
|
|
Effect of dilutive stock options and RSAs |
|
3,693 |
|
3,242 |
|
— |
|
|
Diluted weighted average common shares outstanding |
|
64,452 |
|
63,207 |
|
42,897 |
|
|
Anti-dilutive shares |
|
305 |
|
115 |
|
1,223 |
|
The anti‑dilutive shares disclosed above were calculated using the treasury stock method. The treasury stock method calculates dilution assuming the exercise of all in-the-money options and vesting of RSAs/RSUs, reduced by the repurchase of shares with the proceeds from the assumed exercises, and unrecognized compensation expense for outstanding awards. As the Company was in a net loss position during the year ended December 31, 2014, all options and RSAs outstanding (as disclosed in Note 11) would be anti‑dilutive.
(18) Related Party Transactions
KKR, a significant stockholder of the Company, was a participant in the syndicate of lenders that provided financing under the 2013 Credit Facilities. KKR contributed $28.0 million of the $887.8 million of 2013 First Lien Term Loan issued under the 2013 Credit Facilities, which makes up approximately 3% of the 2013 First Lien Term Loan. Based on the limited contribution of KKR in the 2013 Credit Facilities, the Company represents that the 2013 Credit Facilities was arranged at an arm’s length basis. KKR and UBS were the underwriters of the Incremental Term Loan Borrowing for $32.5 million each. At December 31, 2015, KKR held $14.7 million in 2013 First Lien Term Loan. The 2013 Credit Facilities was extinguished in December 2016. For further discussion of the 2013 Credit Facilities transaction and extinguishment, see Note 9.
In connection with the Merger, the Company entered into a monitoring agreement with KKR pursuant to which KKR provided management services to the Company and its affiliates. The monitoring agreement provided a termination fee based on the net present value of future payment obligations under the monitoring agreement, under certain circumstances in which the monitoring agreement was terminated by us. In connection with the IPO, the Company paid a termination fee of $11.9 million during the year ended December 31, 2014, and therefore, no management fees to KKR were incurred subsequent to the IPO. Prior to the termination of the monitoring agreement, the Company paid management fees of $1.6 million for the year ended December 31, 2014. Also, in connection with the IPO, the Company paid an underwriting discount and commission of $4.0 million to affiliates of KKR.
The Company also entered into a joint venture with an affiliate of KKR during 2015. The joint venture was dissolved during the same year. For further discussion on the related party transaction, refer to Note 3.
104
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
(19) Supplemental Cash Flow Information
The following table presents the Company’s supplemental cash flow information (in thousands):
|
|
Years Ended December 31, |
|
|||||||
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Cash paid during the period for: |
|
|
|
|
|
|
|
|
|
|
Income taxes, net of refunds |
$ |
27,644 |
|
$ |
17,148 |
|
$ |
6,778 |
|
|
Interest |
|
48,156 |
|
|
54,632 |
|
|
80,699 |
|
|
Non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|
Issuance of common stock for the acquisition of Value Health Solutions, Inc. |
|
— |
|
|
1,582 |
|
|
— |
|
|
IPO cost incurred but not paid |
|
— |
|
|
— |
|
|
525 |
|
|
Accrued fixed assets purchases |
|
2,644 |
|
|
2,733 |
|
|
1,644 |
|
|
Cashless exercises of stock options |
|
9,456 |
|
|
1,672 |
|
|
— |
|
(20) Operations by Geographic Area
The table below presents certain enterprise‑wide information about the Company’s operations by geographic area for the years ended December 31, 2016, 2015 and 2014. The Company attributes revenues to geographical locations based upon where the services are performed.
The Company’s operations within each geographical region are further broken down to show each country which accounts for 10% or more of the totals (in thousands):
|
|
|
Years Ended December 31, |
|
|||||||
|
|
|
2016 |
|
2015 |
|
2014 |
|
|||
|
Service revenue: |
|
|
|
|
|
|
|
|
|
|
|
Americas: |
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
1,063,625 |
|
$ |
898,637 |
|
$ |
822,220 |
|
|
Other |
|
|
33,320 |
|
|
32,802 |
|
|
28,925 |
|
|
Americas |
|
|
1,096,945 |
|
|
931,439 |
|
|
851,145 |
|
|
Europe, Africa, and Asia-Pacific |
|
|
|
|
|
|
|
|
|
|
|
United Kingdom |
|
|
394,363 |
|
|
364,476 |
|
|
313,535 |
|
|
Netherlands |
|
|
68,118 |
|
|
57,739 |
|
|
67,208 |
|
|
Other |
|
|
20,597 |
|
|
22,193 |
|
|
34,708 |
|
|
Europe, Africa, and Asia-Pacific |
|
|
483,078 |
|
|
444,408 |
|
|
415,451 |
|
|
Total service revenue |
|
|
1,580,023 |
|
|
1,375,847 |
|
|
1,266,596 |
|
|
Reimbursement revenues |
|
|
231,688 |
|
|
238,036 |
|
|
192,990 |
|
|
Total revenue |
|
$ |
1,811,711 |
|
$ |
1,613,883 |
|
$ |
1,459,586 |
|
105
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
|
|
|
December 31, |
|
||||
|
|
|
2016 |
|
2015 |
|
||
|
Long-lived assets: |
|
|
|
|
|
|
|
|
Americas: |
|
|
|
|
|
|
|
|
United States |
|
$ |
60,462 |
|
$ |
54,058 |
|
|
Other |
|
|
802 |
|
|
889 |
|
|
Americas |
|
|
61,264 |
|
|
54,947 |
|
|
Europe, Africa, and Asia-Pacific |
|
|
|
|
|
|
|
|
United Kingdom |
|
|
3,569 |
|
|
4,773 |
|
|
Netherlands |
|
|
13,313 |
|
|
10,850 |
|
|
Other |
|
|
9,431 |
|
|
10,121 |
|
|
Europe, Africa, and Asia-Pacific |
|
|
26,313 |
|
|
25,744 |
|
|
Total long-lived assets |
|
$ |
87,577 |
|
$ |
80,691 |
|
(21) Quarterly Financial Data (unaudited)
The following table summarizes the Company’s unaudited quarterly results of operations (in thousands, except per share data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2016 |
|
||||||||||
|
|
|
First Quarter |
|
Second Quarter |
|
Third Quarter |
|
Fourth Quarter |
|
||||
|
Service revenue |
|
$ |
372,320 |
|
$ |
394,249 |
|
$ |
399,841 |
|
$ |
413,613 |
|
|
Reimbursement revenue |
|
|
57,903 |
|
|
61,598 |
|
|
53,414 |
|
|
58,773 |
|
|
Total revenue |
|
|
430,223 |
|
|
455,847 |
|
|
453,255 |
|
|
472,386 |
|
|
Income from operations (1) |
|
|
18,946 |
|
|
50,348 |
|
|
54,814 |
|
|
38,241 |
|
|
(Benefit from) provision for income taxes |
|
|
(5,264) |
|
|
12,312 |
|
|
10,821 |
|
|
10,625 |
|
|
(Losses) income before equity in (losses) gains of unconsolidated joint ventures (2) |
|
|
(15,431) |
|
|
35,423 |
|
|
31,416 |
|
|
13,992 |
|
|
Equity in (losses) gains of unconsolidated joint ventures |
|
|
(538) |
|
|
3,247 |
|
|
33 |
|
|
33 |
|
|
Net (loss) income |
|
|
(15,969) |
|
|
38,670 |
|
|
31,449 |
|
|
14,025 |
|
|
Comprehensive (loss) income |
|
$ |
(22,251) |
|
$ |
567 |
|
$ |
21,982 |
|
$ |
(24,502) |
|
|
Basic (losses) earnings per share (3) |
|
$ |
(0.27) |
|
$ |
0.64 |
|
$ |
0.52 |
|
$ |
0.23 |
|
|
Diluted (losses) earnings per share (3) |
|
$ |
(0.27) |
|
$ |
0.60 |
|
$ |
0.49 |
|
$ |
0.22 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2015 |
|
||||||||||
|
|
|
First Quarter |
|
Second Quarter |
|
Third Quarter |
|
Fourth Quarter |
|
||||
|
Service revenue |
|
$ |
331,968 |
|
$ |
336,518 |
|
$ |
345,096 |
|
$ |
362,265 |
|
|
Reimbursement revenue |
|
|
56,610 |
|
|
56,330 |
|
|
58,414 |
|
|
66,682 |
|
|
Total revenue |
|
|
388,578 |
|
|
392,848 |
|
|
403,510 |
|
|
428,947 |
|
|
Income from operations |
|
|
32,937 |
|
|
38,321 |
|
|
49,179 |
|
|
43,861 |
|
|
Provision for income taxes |
|
|
8,022 |
|
|
5,623 |
|
|
10,696 |
|
|
5,663 |
|
|
Income before equity in (losses) gains of unconsolidated joint ventures |
|
|
18,124 |
|
|
13,220 |
|
|
25,978 |
|
|
27,839 |
|
|
Equity in (losses) gains of unconsolidated joint ventures |
|
|
(937) |
|
|
(805) |
|
|
(2,319) |
|
|
665 |
|
|
Net income |
|
|
17,187 |
|
|
12,415 |
|
|
23,659 |
|
|
28,504 |
|
|
Comprehensive (loss) income |
|
$ |
(29,391) |
|
$ |
44,470 |
|
$ |
(4,086) |
|
$ |
7,974 |
|
|
Basic earnings per share (3) |
|
$ |
0.29 |
|
$ |
0.21 |
|
$ |
0.39 |
|
$ |
0.47 |
|
|
Diluted earnings per share (3) |
|
$ |
0.27 |
|
$ |
0.20 |
|
$ |
0.37 |
|
$ |
0.45 |
|
106
PRA HEALTH SCIENCES, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
DECEMBER 31, 2016
|
(1) |
Transaction-related costs for the three months ended March 31, 2016, June 30, 2016 and December 31, 2016 were $28.9 million, $2.9 million and $13.0 million, respectively. There were no transaction-related costs for the three months ended September 30, 2016. Transaction-related costs primarily relate to costs incurred in connection with the March, May and November 2016 secondary offerings and receivables financing agreement. These costs include $42.1 million of non-cash stock-based compensation expense and $2.7 million of third-party fees. |
|
(2) |
During the three months ended March 31, 2016 and December 31, 2016, the Company recorded a loss on extinguishment of debt of $21.5 million and $16.7 million, respectively. The loss on extinguishment of debt recorded during the three months ended March 31, 2016 related to the cash tender offer on the Company’s Senior Notes. The loss on extinguishment of debt recorded during the three months ended December 31, 2016 related to the refinancing of the Company’s 2013 Credit Facilities. Refer to Note 9, Current Borrowings and Long-Term Debt, for additional information regarding the cash tender on the Senior Notes and the 2013 Credit Facilities refinancing. |
|
(3) |
The sum of the quarterly per share amounts may not equal per share amounts reported for year‑to‑date periods. This is due to changes in the number of weighted average shares outstanding and the effects of rounding for each period. |
107
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
As of December 31, 2016, we carried out an evaluation under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures. Regulations under the Exchange Act require public companies, including us, to maintain “disclosure controls and procedures,” which are defined in Rule 13a‑15(e) and Rule 15d‑15(e) of the Exchange Act to mean a company’s controls and other procedures that provide reasonable assurance that information required to be disclosed in reports that it files or submits under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to management, including our principal executive officer and principal financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosures. There are inherent limitations to the effectiveness of any system of disclosure controls and procedures, including the possibility of human error and the circumvention or overriding of the controls and procedures. Accordingly, even effective disclosure controls and procedures can only provide reasonable assurance of achieving their control objectives. Based upon our evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this report to accomplish their objectives at a reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Our management’s report on internal control over financial reporting is set forth in Part II, Item 8 of this Annual Report on Form 10-K and is incorporated herein by reference.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting during the quarter ended December 31, 2016 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Annual Meeting Date
The Board of Directors of the Company has fixed the date of the 2017 Annual Meeting of Stockholders for June 1, 2017.
108
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item will be included in our definitive proxy statement (or the “2017 Proxy Statement”) to be filed with the SEC within 120 days of the end of our fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 11. Executive Compensation
The information required by this item will be included in our 2017 Proxy Statement to be filed with the SEC within 120 days of the end of our fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item will be included in our 2017 Proxy Statement to be filed with the SEC within 120 days of the end of our fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item will be included in our 2017 Proxy Statement to be filed with the SEC within 120 days of the end of our fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services
The information required by this item will be included in our 2017 Proxy Statement to be filed with the SEC within 120 days of the end of our fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 15. Exhibits, Financial Statement Schedules
(1)Financial Statements
The following financial statements and supplementary data are included in Item 8 of this annual report:
|
|
Page |
|
64 |
|
|
66 |
|
|
67 |
|
|
68 |
|
|
69 |
|
|
70 |
|
|
71 |
(2)Financial Statement Schedules
The information required to be submitted in the Financial Statement Schedules for PRA Health Sciences, Inc. and subsidiaries has either been shown in the financial statements or notes, or is not applicable or required under Regulation S‑X; therefore, those schedules have been omitted.
(3)Exhibits
The exhibits listed in the accompanying Exhibit Index following the signature page are filed or furnished as a part of this report and are incorporated herein by reference.
None.
109
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf on February 23, 2017 by the undersigned, thereunto duly authorized.
|
|
PRA Health Sciences, Inc. |
||
|
|
By: |
/s/ Linda Baddour |
|
|
|
|
Name: |
Linda Baddour |
|
|
|
Title: |
Executive Vice President and Chief Financial Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities indicated on February 23, 2017.
|
Signature |
|
Capacity |
|
|
|
|
|
|
|
/s/ Colin Shannon Colin Shannon |
|
President, Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ Linda Baddour Linda Baddour |
|
Executive Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
/s/ Jeffrey T. Barber Jeffrey T. Barber |
|
Director |
|
|
|
|
|
|
|
/s/ Max C. Lin Max C. Lin |
|
Director |
|
|
|
|
|
|
|
/s/ James C. Momtazee James C. Momtazee |
|
Director |
|
|
|
|
|
|
|
/s/ Ali J. Satvat Ali J. Satvat |
|
Director |
|
|
|
|
|
|
|
/s/ Matthew P. Young Matthew P. Young |
|
Director |
|
|
|
|
|
|
|
/s/ LINDA S GRAIS Linda S. Grais |
|
Director |
|
110
|
Exhibit |
|
|
|
Number |
|
Description of Exhibit |
| 3.1 |
|
Amended and Restated Certificate of Incorporation of PRA Health Sciences, Inc. (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8‑K filed on November 18, 2014 (No. 001‑36732)) |
| 3.2 |
|
Amended and Restated Bylaws of PRA Health Sciences, Inc. (incorporated by reference to Exhibit 3.2 to the Registrant’s Current Report on Form 8‑K filed on November 18, 2014 (No. 001‑36732)) |
| 4.1 |
|
Stockholders Agreement, dated as of November 18, 2014, among PRA Health Sciences, Inc. and the other parties named thereto (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8‑K filed on November 18, 2014 (No. 001‑36732)) |
| 4.2 |
|
Sale Participation Agreement of KKR PRA Investors L.P., dated September 23, 2013 (incorporated by reference to Exhibit 4.3 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.1** |
|
2014 Omnibus Incentive Plan (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8‑K filed on November 18, 2014 (No. 001‑36732)) |
|
10.2** |
|
PRA Global Holdings, Inc. 2013 Equity Incentive Plan (incorporated by reference to Exhibit 10.2 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.3** |
|
PRA Holdings, Inc. 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.3 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.4** |
|
PRA International 2004 Incentive Award Plan (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.5** |
|
PRA Holdings, Inc. 2001 Stock Option Plan (incorporated by reference to Exhibit 10.5 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.6** |
|
Form of Stock Option Agreement (incorporated by reference to Exhibit 10.6 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.7** |
|
Form of Rollover Option Agreement (incorporated by reference to Exhibit 10.7 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.8** |
|
Amended and Restated Employment Agreement, effective as of January 1, 2010, by and between PRA International and Colin Shannon, as amended (incorporated by reference to Exhibit 10.8 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.9** |
|
Employment Agreement, effective July 1, 2014, between PRA Global Holdings, Inc., PRA International and Colin Shannon (incorporated by reference to Exhibit 10.9 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.10** |
|
Executive Employment Agreement, effective July 1, 2015, between PRA Health Sciences, Inc. and Linda Baddour (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on July 6, 2015 (No. 001‑36732)) |
|
10.11** |
|
Employment and Non‑Competition Agreement, effective as of March 1, 2009, by and between Pharmaceutical Research Associates, Inc. and David W. Dockhorn (incorporated by reference to Exhibit 10.11 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.12 |
|
Senior Secured Credit Agreement, dated as of September 23, 2013, by and among PRA Holdings, Inc., UBS AG, Stamford Branch, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.12 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.13 |
|
Amendment No. 1 to the Senior Secured Credit Agreement, dated as of March 14, 2014, by and among PRA Holdings, Inc., UBS AG, Stamford Branch, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.13 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.14 |
|
Security Agreement, dated as of September 23, 2013, by and among PRA Holdings, Inc., UBS AG, Stamford Branch, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.14 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.15 |
|
Guarantee Agreement, dated as of September 23, 2013, by and among PRA Holdings, Inc., UBS AG, Stamford Branch, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.15 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.16 |
|
Indenture, dated as of September 23, 2013, among Pinnacle Merger Sub, Inc., as Issuer, the Guarantors named therein and Wells Fargo Bank, National Association, as Trustee (incorporated by reference to Exhibit 10.16 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.17 |
|
Registration Rights Agreement among KKR PRA Investors L.P., KKR PRA Investors GP LLC and PRA Health Sciences, Inc. (f/k/a Pinnacle Holdco Parent, Inc.) (incorporated by reference to Exhibit 10.17 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
111
| 10.18 |
|
Monitoring Agreement of PRA Health Sciences, Inc. (f/k/a) Pinnacle Holdco Parent, Inc., dated September 23, 2013 (incorporated by reference to Exhibit 10.18 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.19 |
|
Indemnification Agreement among KKR PRA Investors L.P., KKR PRA Investors GP LLC, PRA Health Sciences, Inc. (f/k/a Pinnacle Holdco Parent, Inc.), PRA Holdings, Inc. and Kohlberg Kravis Roberts & Co. L.P. dated September 23, 2013 (incorporated by reference to Exhibit 10.19 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.20 |
|
Transaction Fee Agreement between PRA Health Sciences, Inc. (f/k/a Pinnacle Holdco Parent, Inc.) and Kohlberg Kravis Roberts & Co. L.P. dated September 23, 2013 (incorporated by reference to Exhibit 10.20 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.21 |
|
Stockholders Agreement, dated as of November 18, 2014, among PRA Health Sciences, Inc. and the other parties named thereto (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8‑K (No. 001‑36732)) |
|
10.22** |
|
Form of Non‑Qualified Stock Option Agreement under the PRA Holdings, Inc. 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.22 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.23** |
|
Form of Non‑Qualified Stock Option Agreement (Time‑Based Vesting) under the PRA Holdings, Inc. 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.23 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.24** |
|
Form of Non‑Qualified Stock Option Agreement (Performance‑Based Vesting) under the PRA Holdings, Inc. 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.24 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.25** |
|
Form of Option Agreement of PRA International (incorporated by reference to Exhibit 10.25 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.26** |
|
Amendment to Employment Agreement effective July 1, 2014, dated as of September 22, 2014, between PRA Health Sciences, Inc., PRA International, and Colin Shannon (incorporated by reference to Exhibit 10.26 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
|
10.27** |
|
Form of Restricted Stock Grant Notice under the PRA Health Sciences, Inc. 2014 Omnibus Incentive Plan (incorporated by reference to Exhibit 10.27 to the Registrant’s Registration Statement on Form S‑1 (No. 333‑198644)) |
| 10.28 |
|
Receivables Financing Agreement, dated as of March 22, 2016, by and among PRA Holdings, Inc., PNC Bank, National Association, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8‑K filed on March 25, 2016 (No. 001‑36732)) |
| 10.29 |
|
Purchase and Sale Agreement, dated as of March 22, 2016, by and among PRA Holdings, Inc., PNC Bank, National Association, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8‑K filed on March 25, 2016 (No. 001‑36732)) |
| 10.30 |
|
Credit Agreement, dated as of December 6, 2016, by and among Pharmaceutical Research Associates, Inc., Wells Fargo Bank, National Association, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8‑K filed on December 7, 2016 (No. 001‑36732)) |
| 10.31 |
|
Security Agreement, dated as of December 6, 2016, by and among Pharmaceutical Research Associates, Inc., Wells Fargo Bank, National Association, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8‑K filed on December 7, 2016 (No. 001‑36732)) |
| 10.32 |
|
Guarantee Agreement, dated as of December 6, 2016, by and among Pharmaceutical Research Associates, Inc., Wells Fargo Bank, National Association, as administrative agent, and other agents and lenders party thereto (incorporated by reference to Exhibit 10.3 to the Registrant’s Current Report on Form 8‑K filed on December 7, 2016 (No. 001‑36732)) |
|
21.1* |
|
Subsidiaries of the Registrant |
|
23.1* |
|
Consent of Deloitte & Touche LLP |
|
31.1* |
|
Certification of the Chief Executive Officer pursuant to Rule 13a‑14(a) or 15d‑14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes‑Oxley Act of 2002 |
|
31.2* |
|
Certification of the Chief Financial Officer pursuant to Rule 13a‑14(a) or 15d‑14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes‑Oxley Act of 2002 |
112
|
32.1* |
|
Certification of the Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes‑Oxley Act of 2002 |
|
32.2* |
|
Certification of the Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes‑Oxley Act of 2002 |
|
101* |
|
The following financial information from PRA Health Sciences, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2016 formatted in XBRL: (i) Consolidated Balance Sheets as of December 31, 2016 and December 31, 2015, (ii) Consolidated Statements of Operations for the years ended December 31, 2016, 2015 and 2014, (iii) Consolidated Statements of Comprehensive Income (Loss) for the years ended December 31, 2016, 2015 and 2014, (iv) Consolidated Statements of Cash Flows for the years ended December 31, 2016, 2015 and 2014, and (v) Notes to Consolidated Financial Statements |
* Filed herewith.
** This document has been identified as a management contract or compensatory plan or arrangement.
113