Attached files
| file | filename |
|---|---|
| EX-23 - EXHIBIT 23 - ANGIODYNAMICS INC | ex23angio2015consent531161.htm |
| EX-32.2 - EXHIBIT 32.2 - ANGIODYNAMICS INC | ex3225311610-k.htm |
| EX-32.1 - EXHIBIT 32.1 - ANGIODYNAMICS INC | ex3215311610-k.htm |
| EX-31.2 - EXHIBIT 31.2 - ANGIODYNAMICS INC | ex3125311610-k.htm |
| EX-31.1 - EXHIBIT 31.1 - ANGIODYNAMICS INC | ex3115311610-k.htm |
| EX-21 - EXHIBIT 21 - ANGIODYNAMICS INC | ex2153116.htm |
| EX-10.4.3 - EXHIBIT 10.4.3 - ANGIODYNAMICS INC | ex104353116.htm |
| EX-10.1.5 - EXHIBIT 10.1.5 - ANGIODYNAMICS INC | ex101553116.htm |
| EX-3.1.2 - EXHIBIT 3.1.2 - ANGIODYNAMICS INC | ex31253116.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended May 31, 2016 | |
OR
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to | |
Commission file number 0-50761
AngioDynamics, Inc.
(Exact name of registrant as specified in its charter)
Delaware | 11-3146460 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
14 Plaza Drive Latham, New York | 12110 | |
(Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code (518) 795-1400
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered | |
Common Stock, par value $.01 per share | NASDAQ Global Select Market | |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨ | Accelerated filer x | ||
Non-accelerated filer ¨ | Smaller reporting company ¨ | ||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of November 30, 2015, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the registrant’s common stock held by non-affiliates was approximately $431,798,128 computed by reference to the last sale price of the common stock on that date as reported by The NASDAQ Global Select Market.
As of July 22, 2016, there were 36,422,398 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The information required for Part III of this annual report on Form 10-K is incorporated by reference to the registrant’s Proxy Statement for its 2016 Annual Meeting of Stockholders to be filed within 120 days of the registrant's fiscal year ended May 31, 2016.
AngioDynamics, Inc. and Subsidiaries
INDEX
Page | ||
Part I: | ||
Item 1. | ||
Item 1A. | ||
Item 1B. | ||
Item 2. | ||
Item 3. | ||
Item 4. | ||
Part II: | ||
Item 5. | ||
Item 6. | ||
Item 7. | ||
Item 7A. | ||
Item 8. | ||
Item 9. | ||
Item 9A. | ||
Item 9B. | ||
Part III: | ||
Item 10. | ||
Item 11. | ||
Item 12. | ||
Item 13. | ||
Item 14. | ||
Part IV: | ||
Item 15. | ||
1
Part I
Item 1. | Business. |
Overview
AngioDynamics, Inc. (together with its subsidiaries, "AngioDynamics," the "Company," "we," "our" or "us") designs, manufactures and sells a wide range of medical, surgical and diagnostic devices used by professional healthcare providers for vascular access, for the treatment of peripheral vascular disease and for use in oncology and surgical settings. Our devices are generally used in minimally invasive, image-guided procedures.
History
AngioDynamics was founded in 1988 and we completed our initial public offering in 2004, raising net proceeds of approximately $21.7 million at an offering price of $11.00 per share. In 2006 we completed a follow-on offering, raising net proceeds of approximately $61.9 million at a public offering price of $24.07 per share.
Products
Our product offerings fall within three product groupings: Peripheral Vascular, Vascular Access and Oncology/Surgery. All products discussed below have been cleared for sale in the United States by the FDA. International regulatory clearances vary by product and jurisdiction.
Peripheral Vascular Products
Our Peripheral Vascular products include Fluid Management, Venous, Thrombus Management, as well as other core products.
Fluid Management Products
Our Fluid Management product offering includes the NAMIC® Fluid Management portfolio. Since 1969, the NAMIC product line has been a leader in providing clinicians high quality, dependable devices that help in the diagnosis and treatment of cardiovascular and peripheral vascular disease. The NAMIC product line includes an extensive offering of manifolds, contrast management systems, closed fluid systems, guidewires, disposable transducers and interventional accessories. These devices are utilized together and allow clinicians to aspirate or inject contrast, saline, remove waste and monitor invasive blood pressures throughout the procedure.
Venous Products
Our venous products focus on the treatment of varicose veins and consist of our VenaCure EVLT® laser system, Asclera® (polidocanol) injection and Sotradecol®.
Our VenaCure EVLT laser system products are used in endovascular laser procedures to treat superficial venous disease (varicose veins). Superficial venous disease is a malfunction of one or more valves in the leg veins whereby blood refluxes or does not return to the heart. Venous laser treatment is an outpatient procedure that generally allows the patient to quickly return to normal activities with minimal post-operative pain.
With our VenaCure EVLT laser system, laser energy is used to stop the reflux by ablating, or collapsing and destroying, the affected vein. The body subsequently re-routes the blood to other healthy veins. Our products are sold as a system that includes diode laser hardware with our family of disposable laser fiber components, training and marketing materials. The disposable components in the system include a laser fiber system featuring our NeverTouch® gold-tip technology, an access sheath, access wires and needles. The procedure kits come in a variety of lengths and configurations to accommodate varied patient anatomies. Our VenaCure EVLT 1470 nanometer wavelength laser allows physicians to more efficiently heat the vein wall using lower power settings thereby reducing the risk of collateral damage.
Asclera® (polidocanol) injection is an FDA approved sclerosing drug that we distribute through a global agreement with the manufacturer and their distributor. Asclera is used for sclerotherapy to treat uncomplicated spider veins and uncomplicated reticular veins in the lower extremity. In a clinical study, it was proven to be 95% successful and patients were more satisfied with Asclera than alternative sclerosants.
2
Sotradecol® (sodium tetradecyl sulfate injection) is an FDA approved sclerosing drug that we distribute through a global agreement with the manufacturer. Sotradecol® has been shown to be an effective non-surgical treatment of small, uncomplicated varicose veins of the lower extremities that show simple dilation with competent valves.
Thrombus Management
Our Thrombus Management product offerings include our AngioVac and thrombolytic products.
AngioVac
In fiscal 2013, we released our AngioVac venous drainage system which includes a Venous Drainage Cannula and Cardiopulmonary Bypass Circuit. The AngioVac devices are for use with other manufacturer’s off-the-shelf pump, filter, and reinfusion cannula, to facilitate venous drainage as part of an extracorporeal bypass procedure. The cannula is indicated for use as a venous drainage cannula and for removal of fresh, soft thrombi or emboli during extracorporeal bypass. The cardiopulmonary bypass circuit is indicated for use in procedures requiring extracorporeal circulatory support for periods of up to six hours.
The AngioVac venous drainage cannula is a 22F coil-reinforced cannula designed with a balloon actuated, expandable funnel shaped distal tip. The proprietary funnel shaped tip enhances venous drainage flow when the balloon is inflated, prevents clogging of the cannula with commonly encountered undesirable intravascular material, and facilitates en bloc removal of such extraneous material.
Thrombolytic Products
Thrombolytic catheters are used to deliver thrombolytic agents, which are drugs that dissolve blood clots in hemodialysis access grafts, arteries, veins and surgical bypass grafts.
Core Products
Our other core peripheral vascular products include Angiographic products and accessories, drainage, micro access and other products.
Angiographic Products and Accessories
Angiographic products and accessories are used during peripheral vascular interventional procedures. These products permit interventional physicians to reach targeted locations within the vascular system to deliver contrast media for visualization purposes and therapeutic agents and devices, such as percutaneous transluminal angioplasty (PTA) balloons. Angiographic products consist primarily of angiographic catheters, but also include entry needles and guidewires specifically designed for peripheral interventions and fluid management products. We manufacture angiographic catheters and guidewires that are available in more than 500 tip configurations and lengths.
Drainage Products
Drainage products percutaneously drain abscesses and other fluid pockets. An abscess is a tender inflamed mass that typically must be drained by a physician.
Our line of drainage products, The Total Abscession® Family of Drainage Catheters, consists of our Total Abscession General, Biliary, and Nephrostomy drainage catheters. These products feature our proprietary soft shaft with Blue Silk™ finish for a more comfortable patient fit. The kink-resistant shaft recovers rapidly, even if severely bent, knotted, or twisted. This is particularly beneficial when patients roll over and risk a potential kinking of the catheter during sleep. The thermal molded tip allows for less buckling and kinking upon insertion. Also important is that the shaft diameter equals the inner diameter of the catheter hub to maximize flow. Our Total Abscession drainage catheters feature a tamper-resistant locking mechanism called the Vault® which securely fixes the pigtail and prevents tampering or accidental removal. This locking mechanism helps to prevent the drain from becoming unlocked during routine use, thus reducing a physician’s time by avoiding a possible “redo” case, and increasing patient satisfaction by not having to repeat the procedure. The Total Abscession catheter permits aspiration in the locked or unlocked position thus allowing more accurate placement and greater versatility for draining complex situations.
3
Micro Access Kits
Our Micro Access sets provide interventional physicians a smaller introducer system for minimally-invasive procedures. Our Micro Access product line provides physicians with the means to build a custom set from the wide selection of configurations available, including four wires in two different lengths, seven needle options and three sheath dilator options.
Vascular Access
Image-guided vascular access, or IGVA, involves the use of advanced imaging equipment to guide the placement of catheters that deliver primarily short-term drug therapies, such as chemotherapeutic agents and antibiotics, into the central venous system. Delivery to the circulatory system allows drugs to mix with a large volume of blood as compared to intravenous drug delivery into a superficial vessel. IGVA procedures include the placement of PICC lines, implantable ports and central venous catheters, or CVCs.
BioFlo®
Our BioFlo products incorporate Endexo Technology into the manufacturing and design of our Vascular Access products. Endexo is a fluorine based additive that creates a non-eluting (permanent), non-heparin based catheter material that is designed to reduce thrombus accumulation and platelet adhesion to all surfaces of the catheter. BioFlo’s long-term durability and efficacy is intended to provide clinicians a high degree of safety and confidence in providing better patient care and improved patient outcomes.
PICC Products
A peripherally inserted central catheter, or PICC, is a long thin catheter that is inserted into a peripheral vein, typically in the upper arm, and advanced until the catheter tip terminates in a large vein in the chest near the heart to obtain intravenous access. PICCs can typically be used for prolonged periods of time and provide an alternative to central venous catheters. Our PICC product offerings include:
• | BioFlo® PICC: Our BioFlo line is the only power injectable PICC available that incorporates Endexo Technology into the manufacturing and design of the catheter. Advanced features such as large lumen diameters allow the BioFlo® PICC to deliver the power injection flow rates required for contrast-enhanced computed tomography (CT) scans compatible with up to 325 psi CT injections. |
• | Xcela PICC: The Xcela® PICC line is designed to provide a high degree of safety, ease and confidence in patient care. Advanced features such as large lumen diameters allow the Xcela® PICC to deliver the power injection flow rates required for contrast-enhanced CTs compatible with up to 325 psi CT injections. |
• | PASV® Valve Technology: The PASV® Valve Technology is available in both BioFlo and Xcela lines and is designed to automatically resist backflow and reduce blood reflux that could lead to catheter-related complications. |
Port Products
Ports are implantable devices utilized for the central venous administration of a variety of medical therapies and for blood sampling and diagnostic purposes. Central venous access facilitates a more systemic delivery of treatment agents, while mitigating certain harsh side effects of certain treatment protocols and eliminating the need for repeated access to peripheral veins. Depending upon needle gauge size and the port size, a port can be utilized for up to approximately 2,000 accesses once implanted in the body. Our ports are used primarily in systemic or regional short and long-term cancer treatment protocols that require frequent infusions of highly concentrated or toxic medications (such as chemotherapy agents, antibiotics or analgesics) and frequent blood samplings.
Our port products and accessories include:
• | BioFlo® Port: Our BioFlo line is the only port available that incorporates Endexo Technology into the manufacturing and design of the catheter. Advanced features include multiple profile and catheter options, a large septum area for ease of access and the ability to administer contrast through a CT (Computed Tomography) injection for purposes of imaging. |
• | SmartPort®: The Smart Port power-injectable port with Vortex technology offers the ability for a clinician to access a vein for both the delivery of medications or fluids and for administering power-injected contrast to perform a Computed Tomography (CT) scan. The ability to access a port for power-injected contrast studies |
4
eliminates the need for additional needle sticks in the patient’s arm and wrist veins. Once implanted, repeated access to the bloodstream can be accomplished with greater ease and less discomfort. Our Smart Port is available in mini and low-profiles to accommodate more patient anatomies.
• | Vortex®: Our Vortex port technology line of ports is a clear-flow port technology that, we believe, revolutionized port design. With its rounded chamber, the Vortex port is designed to have no sludge-harboring corners or dead spaces. This product line consists of titanium, plastic and dual-lumen offerings. |
• | PASV® Valve Technology: The PASV® Valve Technology is designed to automatically resist backflow and reduce blood reflux that could lead to catheter-related complications. |
• | LifeGuard®: The LifeGuard Safety Infusion Set and The LifeGuard Vision are used to infuse our ports and complement our port and vascular access catheters. The needles’ low profile design is intended to allow clinicians to easily dress the site. |
Dialysis Products
We market a complete line of dialysis products that provide short and long-term vascular access for dialysis patients. Dialysis, or cleaning of the blood, is necessary in conditions such as acute renal failure, chronic renal failure and end-stage renal disease (ESRD).
We currently offer a wide variety of dialysis catheters, including:
• | BioFlo®: Our BioFlo line is the only dialysis catheter available that incorporates Endexo Technology into the manufacturing and design of the catheter. Advanced features include large inner diameter lumens designed for long term patency, a proprietary guidewire lumen to facilitate catheter exchanges and Curved Tip Technology that allows the catheter to self-center in the SVC (Superior Vena Cava). |
• | DuraMax®. The DuraMax catheter is a stepped-tip catheter designed to improve ease of use, dialysis efficiency and overall patient outcomes. |
Oncology / Surgery Products
Our Oncology/Surgery product offerings include our Microwave Ablation products, our Radiofrequency Ablation (RFA) and our NanoKnife product lines.
Microwave Ablation Products
The Acculis Microwave Tissue Ablation (MTA) System complements the full range of ablative technologies we offer. When configured for use with the Accu2i pMTA Applicators, it includes the Sulis VpMTA Generator, optional MTA Temperature Probes, Acculis Local Control Station (LCS) and Accu2i pMTA Applicators. Designed for physicians trained in image-guided ablation procedures, intraoperative ultrasound and/or CT guided needle placement, the system is used for thermal coagulation of soft tissue. By utilizing 2.45 GHz of microwave energy, the Acculis MTA System can complete ablations up to 5 cm in six minutes with a single applicator. Applicators are available in 14 cm, 19 cm and 29 cm lengths, offering flexibility in selecting the appropriate length for the procedure. Additionally, an antenna transmits energy directly to the targeted tissue, eliminating the need for electrosurgical grounding pads, while the single, simple to place insertion applicator eliminates the need to deploy an active array.
Radiofrequency Ablation Products
Radiofrequency Ablation (RFA) products use radiofrequency energy to provide a minimally invasive approach to ablating solid cancerous or benign tumors. Our system delivers radiofrequency energy to raise the temperature of cells above 45-50°C, causing cellular death.
The physician inserts the disposable needle electrode device into the targeted body tissue, typically under ultrasound, computed tomography or magnetic resonance imaging guidance. Once the device is inserted, pushing on the handle of the device causes a group of curved wires to be deployed from the tip of the electrode. When the power is turned on, these wires deliver radiofrequency energy throughout the tumor. In addition, temperature sensors on the tips of the wires measure tissue temperature throughout the procedure.
During the procedure, our system automatically adjusts the amount of energy delivered in order to maintain the temperature necessary to ablate the targeted tissue. For a typical 5cm ablation using our StarBurst ® Xli-enhanced disposable
5
device, the ablation process takes approximately ten minutes. When the ablation is complete, pulling back on the handle of the device causes the curved wire array to be retracted into the device so it can be removed from the body.
The RFA system consists of a radiofrequency generator and a family of disposable devices. We also market the Habib ® 4X ® resection device under a distribution agreement with EMcision Limited. In addition to the intra-operative (open surgery) device Habib 4X, AngioDynamics markets a minimally-invasive version of the Habib 4X device, a Laparoscopic 4X unit, which is used in minimally invasive laparoscopic surgery (MILS) procedures in surgical specialties such as: Hepato-Biliary, GI, Surgical Oncology, Transplant Surgery and Urology (Partial Nephrectomy Resections). It is clinically indicated to assist in coagulation of tissue during intraoperative and laparoscopic procedures.
NanoKnife® Ablation System Products
The NanoKnife® Ablation System is for the surgical ablation of soft tissue. The NanoKnife Ablation System utilizes low energy direct current electrical pulses to permanently open pores in target cell membranes. These permanent pores or nano-scale defects in the cell membranes result in cell death. The treated tissue is then removed by the body’s natural processes in a matter of weeks, mimicking natural cell death. Unlike other ablation technologies, NanoKnife Ablation System does not achieve tissue ablation using thermal energy.
The NanoKnife Ablation System consists of two major components: a Low Energy Direct Current, or LEDC Generator and needle-like electrode probes. Up to six (6) electrode probes can be placed into or around the targeted soft tissue. Once the probes are in place, the user enters the appropriate parameters for voltage, number of pulses, interval between pulses, and the pulse length into the generator user interface. The generator then delivers a series of short electric pulses between each electrode probe. The energy delivery is hyperechoic and can be monitored under real-time ultrasound.
Research & Development
Our growth depends in large part on the continuous introduction of new and innovative products, together with ongoing enhancements to our existing products, through internal product development, technology licensing and strategic alliances. We recognize the importance of, and intend to continue to make investments in, research and development.
Our R&D development teams work closely with our sales force to incorporate customer feedback into our development and design process. We believe that we have a reputation among interventional physicians as a strong partner for product development because of our tradition of close physician collaboration, dedicated market focus, responsiveness and execution capabilities for product development and commercialization.
Competition
We encounter significant competition across our product lines and in each market in which our products are sold. These markets are characterized by rapid change resulting from technological advances and scientific discoveries. We face competitors ranging from large manufacturers with multiple business lines to small manufacturers that offer a limited selection of products.
Our primary device competitors include: Boston Scientific Corporation; Cook Medical; C.R. Bard; Medical Components, Inc. (Medcomp); TeleFlex Medical; Smiths Medical, a subsidiary of Smiths Group plc; Vascular Solutions; Medtronic; Merit Medical; Terumo Medical Corporation; Johnson and Johnson and Total Vein Systems.
Many of our competitors have substantially greater financial, technological, research and development, regulatory, marketing, sales and personnel resources than we do. Competitors may also have greater experience in developing products, obtaining regulatory approvals, and manufacturing and marketing such products. Additionally, competitors may obtain patent protection or regulatory approval or clearance, or achieve product commercialization before us, any of which could materially adversely affect us.
We believe that our products compete primarily on the basis of their quality, clinical outcomes, ease of use, reliability, physician familiarity and cost-effectiveness. In the current environment of managed care, which is characterized by economically motivated buyers, consolidation among health care providers, increased competition and declining reimbursement rates, we have been increasingly required to compete on the basis of price. We believe that our continued competitive success will depend upon our ability to develop or acquire scientifically advanced technology, apply our technology cost-effectively across product lines and markets, develop or acquire proprietary products, attract and retain skilled development personnel, obtain patent or other protection for our products, obtain required regulatory and reimbursement
6
approvals, manufacture and successfully market our products either directly or through outside parties and maintain sufficient inventory to meet customer demand.
Sales and Marketing
We sell our broad line of quality devices in the United States primarily through a direct sales force and internationally through a combination of direct sales and distributor relationships. We support our customers and sales organization with a marketing staff that includes product managers, customer service representatives and other marketing specialists.
We focus our sales and marketing efforts on interventional radiologists, interventional cardiologists, vascular surgeons, urologists and interventional and surgical oncologists.
Backlog
Historically, we ship the majority of products within 24-48 hours of receipt of the orders, and accordingly our backlog is not significant.
Manufacturing
We manufacture certain proprietary components and products and assemble, inspect, test and package our finished products. By designing and manufacturing many of our products from raw materials, and assembling and testing our subassemblies and products, we believe that we are able to maintain better quality control, ensure compliance with applicable regulatory standards and our internal specifications, and limit outside access to our proprietary technology. We have custom-designed proprietary manufacturing and processing equipment and have developed proprietary enhancements for existing production machinery.
We own or lease four primary manufacturing properties providing capabilities which include manufacturing, service, engineering and research, distribution warehouses and offices. These facilities are registered with the FDA and have been certified to ISO 13485 standards, as well as the CMD/CAS Canadian Medical Device Regulations. ISO 13485 is a quality system standard that satisfies European Union regulatory requirements, thus allowing us to market and sell our products in European Union countries. Our manufacturing facilities are subject to periodic inspections by regulatory authorities to ensure compliance with domestic and non-U.S. regulatory requirements. See “Government Regulation” section of this report for additional information. See Item 2 "Properties" for details on each manufacturing location.
Intellectual Property
Patents, trademarks and other proprietary rights are very important to our business. We also rely upon trade secrets, manufacturing know-how, technological innovations and licensing opportunities to maintain and improve our competitive position. We regularly monitor and review third-party proprietary rights, including patents and patent applications, as available, to aid in the development of our intellectual property strategy, avoid infringement of third-party proprietary rights, and identify licensing opportunities.
The company owns an extensive portfolio of patents and patent applications in the United States and in certain foreign countries. The portfolio also includes exclusive licenses to third party patents and applications.
Most of our products are sold under the AngioDynamics trade name or trademark. Additionally, many are also sold under product trademarks and/or registered product trademarks owned by AngioDynamics, Inc., or an affiliate or subsidiary. Some products contain trademarks of companies other than AngioDynamics.
See Part I. Item 3 of this report for additional details on litigation regarding proprietary technology.
Litigation
We operate in an industry characterized by extensive patent litigation. Patent litigation can result in significant damage awards and injunctions that could prevent the manufacture and sale of affected products or result in significant royalty payments in order to continue selling the products. While it is not possible to predict the outcome of patent litigation incidents to our business, we believe the costs associated with this type of litigation could have a material adverse impact on our consolidated results of operations, financial position, or cash flows. The medical device industry is also susceptible to significant product liability claims. These claims may be brought by individuals seeking relief on their own behalf or
7
purporting to represent a class. In addition, product liability claims may be asserted against us in the future based on events we are not aware of at the present time. At any given time, we are involved in a number of product liability actions. For additional information, see both Part I. Item 3 of this report and Note O to the consolidated financial statements in this annual report on Form 10-K.
Government Regulation
The products we manufacture and market are subject to regulation by the FDA under the Federal Food, Drug, and Cosmetic Act, or FDCA, and international regulations to our specific target markets.
United States FDA Regulation - Before a new medical device can be introduced into the market, a manufacturer generally must obtain marketing clearance or approval from the FDA through either a 510(k) submission (a premarket notification) or a premarket approval application (PMA).
The 510(k) procedure is available only in particular circumstances. The 510(k) clearance procedure is available only if a manufacturer can establish that its device is “substantially equivalent” in intended use and in safety and effectiveness to a “predicate device,” which is a legally marketed device with 510(k) clearance in class I or II or preamendment status based upon products commercially distributed on or before May 28, 1976. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance. The 510(k) clearance procedure including questions and responses, may take up to 12 months. In some cases, supporting clinical data may be required. The FDA may determine that a new or modified device is not substantially equivalent to a predicate device or may require that additional information, including clinical data, be submitted before a determination is made, either of which could significantly delay the introduction of new or modified device products. If a device cannot demonstrate substantial equivalence it may be subject to either a de novo submission or a PMA.
The PMA application procedure is more comprehensive than the 510(k) procedure and typically takes several years to complete. The PMA application must be supported by scientific evidence providing pre-clinical and clinical data relating to the safety and efficacy of the device and must include other information about the device and its components, design, manufacturing and labeling. The FDA will approve a PMA application only if a reasonable assurance that the device is safe and effective for its intended use can be provided. As part of the PMA application review, the FDA will inspect the manufacturer’s facilities for compliance with its Quality System Regulation, or QSR. As part of the PMA approval the FDA may place restrictions on the device, such as requiring additional patient follow-up for an indefinite period of time. If the FDA’s evaluation of the PMA application or the manufacturing facility is not favorable, the FDA may deny approval of the PMA application or issue a “not approvable” letter. The FDA may also require additional clinical trials, which can delay the PMA approval process by several years. After the PMA is approved, if significant changes are made to a device, its manufacturing or labeling, a PMA supplement containing additional information must be filed for prior FDA approval.
Historically, our products have been introduced into the market using the 510(k) procedure and we have never had to use the PMA procedure.
The process of FDA submissions requires extensive and expensive validations and testing. The financial outlay for this is large and requires a significant time period. Recent changes in both regulations and FDA perspectives have increased both time and testing requirements, this has caused significant delays and increased costs for approvals. The parameters for increased testing have and will continue to cause severe delays. The increased focus by the FDA on such issues as chemical identification of all colorants, non-acceptance of certain colorants (certain forms of carbon black) continue to cause problems and delays. In addition changes to existing products call into question previously approved devices and result in additional costs for testing and material analysis.
After a product is placed on the market, the product and its manufacturer are subject to pervasive and continuing regulation by the FDA. The FDA enforces these requirements by inspection and market surveillance. Our suppliers also may be subject to FDA inspection, this has resulted in several suppliers altering price structures for medical device companies. The additional costs due to testing and potential for lawsuits due to material contamination or unforeseen chemical/allergenic reactions has led to some manufacturers actively refusing to supply to medical device companies. The financial expenditure needed to maintain compliance to the requirements of the FDA are extensive and ever increasing. Specific systems are needed to maintain compliance to baseline requirements. In addition complex systems are needed to ensure that specific violations such as ‘off label promotion’ are avoided. The FDA has specific requirements for labeling and marketing materials. These need extensive policing and evaluation in order to avoid falling foul of the vague FDA constraints. Penalties for breach of off label promotion can result in fines of several million dollars.
8
The devices manufactured by us also are subject to the QSR, which imposes elaborate testing, control, documentation and other quality assurance procedures. Every phase of production, including raw materials, components and subassembly, manufacturing, testing, quality control, labeling, tracing of consignees after distribution and follow-up and reporting of complaint information is governed by the FDA’s QSR. Device manufacturers are required to register their facilities and list their products with the FDA and certain state agencies. The FDA periodically inspects manufacturing facilities and, if there are alleged violations, the operator of a facility must correct them or satisfactorily demonstrate the absence of the violations or face regulatory action. Penalties for failure to maintain compliance to the QSR include warning letters and potentially consent decrees. AngioDynamics has recently removed three warning letters, and the failure to maintain the QSR appropriately could result in the development of further warning letters. In addition non-compliance with applicable FDA requirements can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the FDA to grant marketing approvals, withdrawal of marketing approvals, a recommendation by the FDA to disallow us to enter into government contracts, and criminal prosecutions. The FDA also has the authority to request repair, replacement or refund of the cost of any device manufactured or distributed by us.
Other - We and our products are also subject to a variety of state and local laws in those jurisdictions where our products are or will be marketed, and federal, state and local laws relating to matters such as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. In addition, we are subject to various federal and state laws governing our relationships with the physicians and others who purchase or make referrals for our products. For instance, federal law prohibits payments of any form that are intended to induce a referral for any item payable under Medicare, Medicaid or any other federal healthcare program. Many states have similar laws. There can be no assurance that we will not be required to incur significant costs to comply with such laws and regulations now or in the future or that such laws or regulations will not have a material adverse effect upon our ability to do business.
International Regulation - Internationally, all of our current products are considered medical devices under applicable regulatory regimes, and we anticipate that this will be true for all of our future products. Sales of medical devices are subject to regulatory requirements in many countries. The regulatory review process may vary greatly from country to country. For example, the European Union has a dedicated set of regulations regarding medical devices, specifically regulating their design, manufacturing, clinical trials, labeling and adverse event reporting. Devices that comply with those requirements are entitled to bear a Conformité Européenne, or CE Mark, indicating that the device conforms to the essential requirements of the applicable directives and can be commercially distributed in countries that are members of the European Union. Similar regulations are in place for Canada, Japan, China and most other countries.
In some cases, we rely on our international distributors to obtain regulatory approvals, complete product registrations, comply with clinical trial requirements and complete those steps that are customarily taken in the applicable jurisdictions.
International sales of medical devices manufactured in the United States that are not approved or cleared by the FDA for use in the United States, or are banned or deviate from lawful performance standards, are subject to FDA export requirements. Before exporting such products to a foreign country, we must first comply with the FDA’s regulatory procedures for exporting unapproved devices.
The process of obtaining approval to distribute medical products is costly and time-consuming in virtually all of the major markets where we sell medical devices. We cannot assure that any new medical devices we develop will be approved in a timely or cost-effective manner or approved at all. There can be no assurance that new laws or regulations regarding the release or sale of medical devices will not delay or prevent sale of our current or future products.
Third-Party Reimbursement and Anti-Fraud and Corrupt Practices Regulation
United States - The delivery of our devices is subject to regulation by the Department of Health and Human Services (HHS) and comparable state and non-U.S. agencies responsible for reimbursement and regulation of health care items and services. U.S. laws and regulations are imposed primarily in connection with the Medicare and Medicaid programs, as well as the government’s interest in regulating the quality and cost of health care. Foreign governments also impose regulations in connection with their health care reimbursement programs and the delivery of health care items and services.
U.S. federal health care laws apply when we or customers submit claims for items or services that are reimbursed under Medicare, Medicaid, or other federally-funded health care programs. The principal U.S. federal laws include: (1) the Anti-kickback Statute which prohibits offers to pay or receive remuneration of any kind for the purpose of including or rewarding referrals of items or services reimbursable by a federal health care program; (2) the False Claims Act which prohibits the submission of false or otherwise improper claims for payment to a federally-funded health care program, including claims resulting from a violation
9
of the Anti-kickback Statute; (3) the Stark law which prohibits physicians from referring Medicare or Medicaid patients to a provider that bills these programs for the provision of certain designated health services if the physician (or a member of the physician’s immediate family) has a financial relationship with that provider; and (4) health care fraud statutes that prohibit false statements and improper claims to any third-party payer. There are often similar state false claims, anti-kickback, and anti-self-referral and insurance laws that apply to state-funded Medicaid and other health care programs and private third-party payers. In addition, the U.S. Foreign Corrupt Practices Act (FCPA) can be used to prosecute companies in the U.S. for arrangements with physicians or other parties outside the U.S. if the physician or party is a government official of another country and the arrangement violates the law of that country.
International - Our success in international markets will depend largely upon the availability of reimbursement from the third-party payors through which healthcare providers are paid in those markets. Reimbursement and healthcare payment systems vary significantly by country. The main types of healthcare payment systems are government sponsored healthcare and private insurance. Reimbursement approval must be obtained individually in each country in which our products are marketed. Outside the United States, we generally rely on our distributors to obtain reimbursement approval in the countries in which they will sell our products. There can be no assurance that reimbursement approvals will be received.
Insurance
Our product liability insurance coverage is limited to a maximum of $10,000,000 per product liability claim and an annual aggregate policy limit of $10,000,000, subject to a self-insured retention of $500,000 per occurrence and $2,000,000 in the aggregate. The policy covers, subject to policy conditions and exclusions, claims of bodily injury and property damage from any product sold or manufactured by us.
There is no assurance that this level of coverage is adequate. We may not be able to sustain or maintain this level of coverage and cannot assure you that adequate insurance coverage will continue to be available on commercially reasonable terms, or at all. A successful product liability claim or other claim with respect to uninsured or underinsured liabilities could have a material adverse effect on our business.
Environmental
We are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain hazardous and potentially hazardous substances used in connection with our operations. Although we believe that we have complied with these laws and regulations in all material respects and, to date, have not been required to take any action to correct any noncompliance, there can be no assurance that we will not be required to incur significant costs to comply with environmental regulations in the future.
Employees
As of May 31, 2016, we had approximately 1,300 full time employees. None of our employees are represented by a labor union and we have never experienced a work stoppage.
Executive Officers of the Company
The following table sets forth certain information with respect to our executive officers.
Name | Age | Position | ||
James C. Clemmer | 51 | President and Chief Executive Officer | ||
Peter J. Kish | 61 | Interim Chief Financial Officer | ||
Mark Stephens | 47 | Senior Vice President Administration | ||
Stephen A. Trowbridge | 42 | Senior Vice President and General Counsel | ||
Barbara Kucharczyk | 42 | Senior Vice President Global Operations | ||
Gary Barrett | 47 | Senior Vice President Quality & Regulatory Affairs | ||
Ben Davis | 51 | Senior Vice President Business Development | ||
10
James C. Clemmer became our President and Chief Executive Officer in April 2016. Prior to joining AngioDynamics, Mr. Clemmer served as President of the Medical Supplies segment at Covidien plc from September 2006 to January 2015. In this role, Mr. Clemmer directed the strategic and day-to-day operations for global business divisions that collectively manufactured 23 different product categories. In addition, he managed global manufacturing, research and development, operational excellence, business development and all other functions associated with the Medical Supplies business. Prior to his role at Covidien, Mr. Clemmer served as Group President at Kendall Healthcare from July 2004 to September 2006, where he managed the US business across five divisions and built the strategic plan for the Medical Supplies segment before it was spun off from Tyco. Mr. Clemmer served as interim president at the Massachusetts College of Liberal Arts from August 2015 until March 1, 2016. Mr. Clemmer is a graduate of the Massachusetts College of Liberal Arts.
Peter J. Kish has served as a consultant to the Company since May 16, 2016 and was designated as the principal financial officer and principal accounting officer by the Board of Directors of the company on July 27, 2016. Currently, Mr. Kish is a CFO Engagement Partner with Tatum, a Randstad Company. Prior to that he held Executive Finance positions with various Private Equity Portfolio Companies. Mr. Kish also spent 15 years at Kulicke & Soffa Industries, Inc. a global semiconductor manufacturing company listed on the Nasdaq and served as Vice President and Corporate Controller. He also worked in a management capacity for Bristol Myers Squibb and MacMillan Publishing. Mr. Kish earned both his Bachelor of Science in Accounting and Masters in Business Administration from Rutgers, the State University of New Jersey.
Mark Stephens joined AngioDynamics in January 2013 as Senior Vice President, Administration. Prior to joining AngioDynamics, Mr. Stephens most recently led the global human resources organization for Smith and Nephew Orthopedics. Before joining Smith and Nephew, Mr. Stephens held the position of Vice-President, Human Resources, at Ingersoll Rand Corporation and served as Director of talent management with the Robert Bosch Corporation. He holds a MBA in Human Resources from Murray State University and a BS in Business Administration with a concentration in Economics and Finance from the University of Tennessee.
Stephen A. Trowbridge joined AngioDynamics as corporate counsel in June 2008, becoming our Vice President and General Counsel in June 2010 and Senior Vice President and General Counsel in August 2013. Prior to joining AngioDynamics, Mr. Trowbridge was corporate counsel for Philips Healthcare from November 2006 through June 2008, and corporate counsel for Intermagnetics General Corporation from April 2006 until its acquisition by Philips Healthcare in November 2006. Mr. Trowbridge began his career at Cadwalader, Wickersham & Taft LLP in New York City in September 2000. Mr. Trowbridge holds a BS in Science and Technology Studies from Rensselaer Polytechnic Institute, a Juris Doctor from the University of Pennsylvania Law School and an MBA from Duke University’s Fuqua School of Business.
Barbara Kucharczyk joined AngioDynamics in June 2012 and was promoted to Senior Vice President Global Operations in June 2015. Prior to joining AngioDynamics, Mrs. Kucharczyk most recently was the Focus Factory Manager for Vascular Therapy at Covidien. Before joining Covidien, Mrs. Kucharczyk was the Plant Manager for the Forest Products Group at Hexion Specialty Chemicals, Inc. She holds an MBA from Rensselaer Polytechnic Institute, a BS in Chemical Engineering from the State University of New York Center at Buffalo and a BS in Chemistry from the State University of New York College at Fredonia.
Gary Barrett joined AngioDynamics in May 2014 and was promoted to Senior Vice President, Quality and Regulatory Affairs in June 2015. Prior to joining AngioDynamics, Mr. Barrett most recently was the Head of RA Development/Business Development at DEKRA. Before joining DEKRA, Mr. Barrett was the Vice President Regulatory Affairs for Merit Medical Systems Inc. He holds a PhD in Biotechnology from the Cranfield Biotechnology Center, a MBA from the Cranfield School of Management, a Masters in Environmental Diagnostics and Bachelors in Zoology.
Ben Davis joined AngioDynamics as Senior Vice President, Business Development in March 2015. Prior to joining AngioDynamics, Mr. Davis most recently was the Vice President Business Integration at C.R. Bard, Inc. where he was previously the Divisional Head of Business Development from 2004 -2013. Before joining C.R. Bard, Inc. Mr. Davis held the position of Chief Financial Officer, Treasurer at Axya Medical Inc. He holds an MBA in Business Administration - Finance from Bentley College Graduate School and BS in Business Administration from Bryant College.
Available Information
Our corporate headquarters is located at 14 Plaza Drive, Latham, New York 12110. Our phone number is (518) 795-1400. Our website is www.angiodynamics.com.
We make available, free-of-charge through our website, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after we electronically file or furnish such materials to
11
the Securities and Exchange Commission, or SEC. In addition, our website includes, among other things, charters of the various committees of our Board of Directors and our code of business conduct and ethics applicable to all employees, officers and directors. Any stockholder also may obtain copies of these documents, free of charge, by sending a request in writing to our investor relations firm: FTI Consulting, 88 Pine Street, 32nd Floor New York, NY 10005, Attention: Evan Smith. Information on our website or connected to our website is not incorporated by reference into this annual report on Form 10-K.
Item 1A. | Risk Factors. |
In addition to the other information contained in this annual report on Form 10-K, the following risk factors should be considered carefully in evaluating the Company's business. Our financial and operating results are subject to a number of factors, many of which are not within our control. These factors include those set forth below. Our business, financial condition or results of operations could be materially and adversely affected by any of these risks. Additional risks not presently known to us or that we currently deem immaterial may also adversely affect our business, financial condition or results of operations.
If we are unable to manage our growth profitably, our business, financial results and stock price could suffer.
Our future financial results will depend in part on our ability to profitably manage our growth. Management will need to maintain existing customers and attract new customers, recruit, retain and effectively manage employees, as well as expand operations and integrate customer support and financial control systems. If integration-related expenses and capital expenditure requirements are greater than anticipated or if we are unable to manage our growth profitably, our financial results and the market price of our common stock may decline.
In recent years we have begun to implement our operational excellence initiatives which include a number of restructuring, realignment and cost reduction initiatives. While we have realized some efficiencies from these actions, we may not realize the benefits of these initiatives to the extent we anticipated. Further, such benefits may be realized later than expected, and the ongoing difficulties in implementing these measures may be greater than anticipated, which could cause us to incur additional costs or result in business disruptions. In addition, if these measures are not successful or sustainable, we may undertake additional realignment and cost reduction efforts, which could result in significant additional charges. Moreover, if our restructuring and realignment efforts prove ineffective, our ability to achieve our other strategic goals and business plans may be adversely affected
We are dependent on single and limited source suppliers which subjects our business and results of operations to risks of supplier business interruptions.
We currently purchase significant amounts of several key products and product components from single and limited source suppliers and anticipate that we will do so for future products as well. Any delays in delivery of or shortages in those or other products and components could interrupt and delay manufacturing of our products and result in the cancellation of orders for our products. Any or all of these suppliers could discontinue the manufacture or supply of these products and components at any time. Due to FDA and other business considerations, we may not be able to identify and integrate alternative sources of supply in a timely fashion or at all. Any transition to alternate suppliers may result in production delays and increased costs and may limit our ability to deliver products to our customers. Furthermore, if we are unable to identify alternative sources of supply, we would have to modify our products to use substitute components, which may cause delays in shipments, increased design and manufacturing costs and increased prices for our products.
In addition, we purchase certain products as a distributor for the manufacturer of those products, including Asclera, Sotradecol and our Celerity tip location system. Operational, quality or regulatory issues of the manufacturers of the products we distribute could constrain or interrupt the availability of those products or services. Any constraint or interruption in the supply of finished products that we distribute could have a material adverse effect on our ability to sell products, our financial condition and our results of operations.
Changes in reimbursement levels by governmental or other third-party payors for procedures using our products may cause our revenues to decline.
Our products are purchased principally by hospitals or physicians which typically bill various third-party payors, such as governmental programs (e.g. Medicare, Medicaid and comparable foreign programs), private insurance plans and managed care plans, for the healthcare services provided to their patients. The ability of our customers to obtain appropriate reimbursement for products and services from third-party payors is critical to the success of medical device companies because it affects which products customers purchase and the prices they are willing to pay. Reimbursement varies by country and can significantly impact the acceptance of new technology. Implementation of healthcare reforms in the United States and in other countries may limit, reduce or eliminate reimbursement for our products and adversely affect both our pricing flexibility and the demand for
12
our products. Even when we develop a promising new product, we may find limited demand for the product unless reimbursement approval is obtained from private and governmental third party payors.
Third-party payors have adopted, and are continuing to adopt, a number of healthcare policies intended to curb rising healthcare costs. These policies include:
• | controls on government-funded reimbursement for healthcare services and price controls on medical products and services providers; |
• | challenges to the pricing of medical procedures or limits or prohibitions on reimbursement for specific devices and therapies through other means; and |
• | the introduction of managed care systems in which healthcare providers contract to provide comprehensive healthcare for a fixed cost per person. |
We are unable to predict whether federal, state or local healthcare reform legislation or regulation affecting our business may be proposed or enacted in the future, or what effect any such legislation or regulation would have on our business. Changes in healthcare systems in the United States or elsewhere in a manner that significantly reduces reimbursement for procedures using our medical devices or denies coverage for these procedures, or adverse decisions relating to our products by administrators of these systems in coverage or reimbursement issues, would have an adverse impact on the acceptance of our products and the prices which our customers are willing to pay for them.
Failure to secure adequate reimbursement for our products could materially impair our ability to grow revenue and drive profitability.
Our products are used in medical procedures generally covered by government or private health plans.
In general, a third-party payor only covers a medical product or procedure when the plan administrator is satisfied that the product or procedure improves health outcomes, including quality of life or functional ability, in a safe and cost-effective manner. Even if a device has received clearance or approval for marketing by the FDA, there is no assurance that third-party payors will cover the cost of the device and related procedures.
In many instances, third-party payors use price schedules that do not vary to reflect the cost of the products and equipment used in performing those procedures. In other instances, payment or reimbursement is separately available for the products and equipment used, in addition to payment or reimbursement for the procedure itself. Even if coverage is available, third-party payors may place restrictions on the circumstances where they provide coverage or may offer reimbursement that is not sufficient to cover the cost of our products.
Third-party payors who cover the cost of medical products or equipment, in addition to allowing a general charge for the procedure, often maintain lists of exclusive suppliers or approved lists of products deemed to be cost-effective. Authorization from those third-party payors is required prior to using products that are not on these lists as a condition of reimbursement. If our products are not on the approved lists, healthcare providers must determine if the additional cost and effort required in obtaining prior authorization, and the uncertainty of actually obtaining coverage, is justified by any perceived clinical benefits from using our products.
Finally, the advent of contracted fixed rates per procedure has made it difficult to receive reimbursement for disposable products, even if the use of these products improves clinical outcomes. In addition, many third-party payors are moving to managed care systems in which providers contract to provide comprehensive healthcare for a fixed cost per person. Managed care providers often attempt to control the cost of healthcare by authorizing fewer elective surgical procedures. Under current prospective payment systems, such as the diagnosis related group system and the hospital out-patient prospective payment system, both of which are used by Medicare and in many managed care systems used by private third-party payors, the cost of our products will be incorporated into the overall cost of a procedure and not be separately reimbursed. As a result, we cannot be certain that hospital administrators and physicians will purchase our products, despite the clinical benefits and opportunity for cost savings that we believe can be derived from their use. If hospitals and physicians cannot obtain adequate reimbursement for our products or the procedures in which they are used, our business, financial condition, results of operations, and cash flows could suffer a material adverse impact.
Our success in international markets will depend largely upon the availability of reimbursement from the third-party payors through which healthcare providers are paid in those markets. Reimbursement and healthcare payment systems vary significantly by country. The main types of healthcare payment systems are government sponsored healthcare and private insurance. Reimbursement approval must be obtained individually in each country in which our products are marketed. Outside
13
the United States, we generally rely on our distributors to obtain reimbursement approval in the countries in which they will sell our products. There can be no assurance that reimbursement approvals will be received. The failure to secure reimbursement approvals in international markets could materially impact our financial position and results of operations.
Cost-containment efforts of group purchasing organizations could adversely affect our selling prices, financial position and results of operations.
Many of our existing and potential customers have become members of group purchasing organizations, or GPOs, and integrated delivery network, or IDNs, in an effort to reduce costs. GPOs and IDNs negotiate pricing arrangements with healthcare product manufacturers and distributors and offer the negotiated prices to affiliated hospitals and other members. GPOs and IDNs typically award contracts on a category-by-category basis through a competitive bidding process. Bids are generally solicited from multiple manufacturers with the intention of driving down pricing. Due to the highly competitive nature of the GPO and IDN contracting processes, we may not be able to obtain market prices for our products or obtain or maintain contract positions with major GPOs and IDNs, which could adversely impact our profitability. Also, sales through a GPO or IDN can be significant to our business and if we are unable to retain contracts with our customers, or acquire additional contracts, our financial results may be negatively impacted.
If we are unable to convince customers that our products can improve the cost structure of their business, our revenue growth and profitability may be materially adversely impacted.
Worldwide initiatives to contain healthcare costs have led government and the private sector to enact cost containment efforts as a means of managing the growth of health care utilization. Common techniques include policies on price regulation, competitive pricing, bidding and tender mechanics, coverage and payment, comparative effectiveness of therapies, technology assessments, and managed-care arrangements. These changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective medical devices and therapies. Government programs, including Medicare and Medicaid, private health care insurance, and managed-care plans have attempted to control costs by limiting the amount of reimbursement they will pay for particular procedures or treatments, tying reimbursement to outcomes, shifting to population health management, and other mechanisms designed to constrain utilization and contain costs. Simultaneously, hospitals are redefining their role in health care delivery as many assume much more risk and control of the total cost of patient care. To successfully make this transformation health systems are consolidating, purchasing or partnering with physicians, post-acute care providers, while also narrowing networks thus allowing greater control over outcomes. Today, many systems are becoming ‘mini’ payer/provider organizations. These newly redesigned health systems are creating mechanisms such as value analysis and centralized purchasing functions that set pricing and in some cases limit the number of vendors that can participate in the purchasing program. Hospitals are also aligning interests with physicians through employment and other arrangements, such as gainsharing, where a hospital agrees with physicians to share any realized cost savings resulting from the physicians’ collective change in practice patterns such as standardization of devices where medically appropriate. This has created an increasing level of price sensitivity among customers for our products. Some third-party payers must also approve coverage and set reimbursement levels for new or innovative devices or therapies before they will reimburse health care providers who use the medical devices or therapies. Even though a new medical device may have been cleared for commercial distribution, we may find limited demand for the device until coverage and sufficient reimbursement levels have been obtained from governmental and private third-party payers. In addition, some private third-party payers require that certain procedures or that the use of certain products be authorized in advance as a condition of reimbursement. International examples of cost containment initiatives and health care reforms advancing clinical outcomes as the key to market access are emerging in France, Germany, the Netherlands and the UK. This new criteria can severely restrict coverage, reduce reimbursement and delay access to key markets with requirements for incremental clinical benefit and coverage with evidence development.
If we fail to develop or market new products and enhance existing products, we could lose market share to our competitors and our results of operations could suffer.
The market for interventional devices is characterized by rapid technological change, new product introductions, technological improvements, changes in physician requirements and evolving industry standards. To be successful, we must continue to develop and commercialize new products and to enhance versions of our existing products. Our products are technologically complex and require significant research, planning, design, development and testing before they may be marketed. This process generally takes at least 12 to 18 months from initial concept and may take up to several years. In addition, product life cycles are relatively short because medical device manufacturers continually develop smaller, more effective and less expensive versions of existing devices in response to physician demand.
Our success in developing and commercializing new and enhanced versions of our products is affected by our ability to:
• | recruit engineers; |
14
• | timely and accurately identify new market trends; |
• | accurately assess customer needs; |
• | minimize the time and costs required to obtain regulatory clearance or approval; |
• | adopt competitive pricing; |
• | timely manufacture and deliver products; |
• | accurately predict and control costs associated with the development, manufacturing and support of our products; and |
• | anticipate and compete effectively with our competitors’ efforts. |
Market acceptance of our products depends in part on our ability to demonstrate that our products are cost-effective and easier to use, as well as offer technological advantages. Additionally, we may experience design, manufacturing, marketing or other difficulties that could delay or prevent our development, introduction or marketing of new products or new versions of our existing products. As a result of such difficulties and delays, our development expenses may increase and, as a consequence, our results of operations could suffer.
Development and sales of our products are dependent on a number of factors beyond our control, and our inability to successfully complete our research and development, design, marketing strategy and regulatory clearance with respect to the respective products may adversely affect our business, financial condition and results of operations.
A significant aspect of our growth strategy is the continued market development of products including NanoKnife, AngioVac and BioFlo products.
There can be no guarantee that we will be able to develop and manufacture additional next generation or updated products on commercially favorable terms, or at all. NanoKnife and AngioVac are developing technologies and the inability of either of them to achieve clinical acceptance, as well as our inability to generate meaningful clinical data to convince providers of the clinical and economic benefits of our BioFlo platform, could severely limit our ability to drive revenue growth.
We currently have FDA 510(k) clearance to market NanoKnife products for soft tissue ablation. If we are not able to secure FDA approval to conduct investigational device exemption (IDE) trials or marketing approval for additional or more specific indications, through 510(k) clearance, pre-market approval or otherwise, our ability to market our NanoKnife products will be restricted which may have an adverse effect on our business, financial condition and results of operations.
We face intense competition in the medical device industry. We may be unable to compete effectively with respect to technological innovation and price which may have an adverse effect on our revenues, financial condition or results of operations.
The markets for our products are highly competitive, and we expect competition to continue to intensify. We may not be able to compete effectively, and we may lose market share to our competitors. Our primary device competitors include: Boston Scientific Corporation; Cook Medical; C.R. Bard; Medical Components, Inc. (Medcomp); TeleFlex Medical; Smiths Medical, a subsidiary of Smiths Group plc; Vascular Solutions; Medtronic; Merit Medical; Terumo Medical Corporation; Vascular Solutions; Johnson and Johnson and Total Vein Systems. Many of our competitors have substantially greater:
• | financial and other resources to devote to product acquisitions, research and development, marketing and manufacturing; |
• | variety of products; |
• | technical capabilities; |
• | history of developing and introducing new products; |
• | patent portfolios that may present an obstacle to our conduct of business; |
• | name recognition; and |
• | distribution networks and in-house sales forces. |
Our competitors may succeed in developing technologies and products earlier, in obtaining patent protection or regulatory clearance earlier, or in commercializing new products or technologies more rapidly than us. Our competitors may also develop products and technologies that are superior to those we are developing or that otherwise could render our products obsolete or noncompetitive. In addition, we may face competition from providers of other medical therapies, such as pharmaceutical companies, that may offer non-surgical therapies for conditions that are currently, or in the future, may be treated using our products. Our products are generally sold at higher prices than those of our competitors. However, in the current environment of managed care, which is characterized by economically motivated buyers, consolidation among healthcare providers,
15
increased competition and declining reimbursement rates, we are increasingly being required to compete on the basis of price. If we are not able to compete effectively, our market share and revenues may decline.
If we fail to adequately protect our intellectual property rights, we may not be able to generate revenues from new or existing products and our business may suffer.
Our success depends in part on obtaining, maintaining and enforcing our patents, trademarks and other proprietary rights, and our ability to avoid infringing the proprietary rights of others. We take precautionary steps to protect our technological advantages and intellectual property. We rely upon patent, trade secret, copyright, know-how and trademark laws, as well as license agreements and contractual provisions, to establish our intellectual property rights and protect our products. However, no assurances can be made that any pending or future patent applications will result in the issuance of patents, that any current or future patents issued to, or licensed by, us will not be challenged or circumvented by our competitors, or that our patents will not be found invalid.
Patent positions of medical device companies, including our company, are uncertain and involve complex and evolving legal and factual questions. The coverage sought in a patent application can be denied or significantly reduced either before or after the patent is issued. Consequently, there can be no assurance that any of our pending patent applications will result in an issued patent. There is also no assurance that any existing or future patent will provide significant protection or commercial advantage, or whether any existing or future patent will be circumvented by a more basic patent, thus requiring us to obtain a license to produce and sell the product. Generally, patent applications can be maintained in secrecy for at least 18 months after their earliest priority date. In addition, publication of discoveries in the scientific or patent literature often lags behind actual discoveries. Therefore, we cannot be certain that we were the first to invent the subject matter covered by each of our pending U.S. patent applications or that we were the first to file non-U.S. patent applications for such subject matter.
Additionally, we rely on trade secret protection for certain unpatented aspects of our proprietary technology. There can be no assurance that others will not independently develop or otherwise acquire substantially equivalent proprietary information or techniques, that others will not gain access to our proprietary technology or disclose such technology, or that we can meaningfully protect our trade secrets. We have a policy of requiring key employees and consultants to execute confidentiality agreements upon the commencement of an employment or consulting relationship with us. Our confidentiality agreements also require our employees to assign to us all rights to any inventions made or conceived during their employment with us. We also generally require our consultants to assign to us any inventions made during the course of their engagement by us. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for us in the event of unauthorized use, transfer or disclosure of confidential information or inventions.
If we are not able to adequately protect our intellectual property, our market share, financial condition and results of operations may suffer.
If third parties claim that our products infringe their intellectual property rights, we may be forced to expend significant financial resources and management time defending against such actions and our financial condition and our results of operations could suffer.
Third parties may claim that our products infringe their patents and other intellectual property rights. Identifying third-party patent rights can be particularly difficult because, in general, patent applications can be maintained in secrecy for at least 18 months after their earliest priority date. Some companies in the medical device industry have used intellectual property infringement litigation to gain a competitive advantage. If a competitor were to challenge our patents, licenses or other intellectual property rights, or assert that our products infringe its patent or other intellectual property rights, we could incur substantial litigation costs, be forced to make expensive changes to our product design, pay royalties or other fees to license rights in order to continue manufacturing and selling our products, or pay substantial damages. Third-party infringement claims, regardless of their outcome, would not only consume our financial resources but also divert our management’s time and effort. Such claims could also cause our customers or potential customers to purchase competitors’ products or defer or limit their purchase or use of our affected products until resolution of the claim.
If we cannot obtain and maintain marketing clearance or approval from governmental agencies, we will not be able to sell our products.
Our products are medical devices that are subject to extensive regulation in the United States and in the foreign countries in which they are sold. Unless an exemption applies, each medical device that we wish to market in the United States must receive either 510(k) clearance or premarket approval (PMA) from the FDA before the product can be sold. Either process can be lengthy and expensive. The FDA’s 510(k) clearance procedure, also known as “premarket notification,” is the process we
16
have used for our current products. This process usually takes from four to 12 months from the date the premarket notification is submitted to the FDA, but may take significantly longer. Although we have obtained 510(k) clearances for our current products, our clearances may be revoked by the FDA if safety or effectiveness problems develop with the devices. The PMA process is much more costly, lengthy and uncertain. It generally takes from one to three years from the date the application is submitted to, and filed with, the FDA, and may take even longer. Regulatory regimes in other countries similarly require approval or clearance prior to our marketing or selling products in those countries. We rely on our distributors to obtain regulatory clearances or approvals of our products outside of the United States. If we are unable to obtain additional clearances or approvals needed to market existing or new products in the United States or elsewhere or obtain these clearances or approvals in a timely fashion or at all, or if our existing clearances are revoked, our revenues and profitability may decline.
If we or some of our suppliers fail to comply with the FDA’s Quality System Regulation, or QSR, and other applicable postmarket requirements, our manufacturing operations could be disrupted, our product sales and profitability could suffer, and we may be subject to a wide variety of FDA enforcement actions.
After a device is placed on the market, numerous regulatory requirements apply. We are subject to inspection and marketing surveillance by the FDA to determine our compliance with all regulatory requirements. Our failure to comply with applicable regulatory requirements could result in the FDA or a court instituting a wide variety of enforcement actions against us, including a public "Warning Letter"; an order to shut down some or all manufacturing operations; a recall of products; fines or civil penalties; seizure or detention of our products; refusing our requests for 510(k) clearance or a PMA of new or modified products; withdrawing 510(k) clearance or PMA approvals already granted to us; and criminal prosecution.
Our manufacturing processes and those of some of our suppliers must comply with the FDA’s Quality System Regulation, or QSR, which governs the methods used in, and the facilities and controls used for, the design, testing, manufacture, control, quality assurance, installation, servicing, labeling, packaging, storage and shipping of medical devices. The FDA enforces the QSR through unannounced inspections. If we, or one of our suppliers, fail a QSR inspection, or if a corrective action plan adopted by us or one of our suppliers is not sufficient, the FDA may bring an enforcement action, and our operations could be disrupted and our manufacturing delayed. We are also subject to the FDA’s general prohibition against promoting our products for unapproved or “off-label” uses, the FDA’s adverse event reporting requirements and the FDA’s reporting requirements for field correction or product removals. The FDA has recently placed increased emphasis on its scrutiny of compliance with the QSR and these other postmarket requirements.
If we, or one of our suppliers, violate the FDA’s requirements or fail to take adequate corrective action in response to any significant compliance issue raised by the FDA, the FDA can take various enforcement actions which could cause our product sales and profitability to suffer.
In addition, most other countries require us and our suppliers to comply with manufacturing and quality assurance standards for medical devices that are similar to those in force in the United States before marketing and selling our products in those countries. If we, or our suppliers, should fail to do so, we would lose our ability to market and sell our products in those countries.
Even after receiving regulatory clearance or approval, our products may be subject to product recalls, which may harm our reputation and divert managerial and financial resources.
The FDA and similar governmental authorities in other countries have the authority to order mandatory recall of our products or order their removal from the market if there are material deficiencies or defects in design, manufacture, installation, servicing or labeling of the device, or if the governmental entity finds that our products would cause serious adverse health consequences. A government mandated voluntary recall or field action by us could occur as a result of component failures, manufacturing errors or design defects, including labeling defects. Any recall of our products may harm our reputation with customers and divert managerial and financial resources.
We may be subject to fines, penalties, injunctions or costly investigations if we are determined to be promoting the use of our products for unapproved or “off-label” uses.
If we are incorrect in our belief that our promotional materials and training methods regarding physicians are conducted in compliance with regulations of the FDA and other applicable regulations, and the FDA determines that our promotional materials or training constitutes promotion of an unapproved use, the FDA could request that we modify our training or promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider promotional or training materials to constitute promotion of an unapproved use, which could result
17
in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. Any of these results could have a material adverse effect on our financial position or results of operations.
In June 2014 we received a subpoena from the U.S. Department of Justice (the “DOJ”) requesting documents in relation to a criminal and civil investigation the DOJ is conducting regarding BTG International, Inc.’s LC Bead product beginning in 2003. RITA Medical Systems and AngioDynamics, Inc., after our acquisition of RITA, was the exclusive distributor of LC Beads in the United States from 2006 through December 31, 2011. We are cooperating fully with this investigation and at this time are unable to predict its scope, duration or outcome. In April 2015 we received a subpoena from the DOJ requesting documents in relation to a criminal and civil investigation the DOJ is conducting regarding purported promotion of certain of our VenaCure EVLT products for un-cleared indications. We are cooperating fully with this investigation and at this time are unable to predict its scope, duration or outcome.
We may be exposed to risks associated with acquisitions, including integration risks and risks associated with methods of financing and the impact of accounting treatment. Accordingly, completed acquisitions may not enhance our financial position or results of operations as they are based projections and assumptions which are uncertain and subject to change.
Part of our growth strategy is to acquire businesses and technologies that are complementary to ours. There is no assurance that acquisition opportunities will be available on acceptable terms, or at all, or that we will be able to obtain necessary financing or regulatory approvals. Any acquisitions that we do undertake would be accompanied by the risks commonly encountered in acquisitions, including the:
• | potential disruption of our business while we evaluate opportunities, complete acquisitions and develop and implement new business strategies to take advantage of these opportunities; |
• | inability of our management to maximize our financial and strategic position by incorporating an acquired technology or business into our existing offerings; |
• | our inability to achieve the cost savings and operating synergies anticipated in the acquisition, which would prevent us from achieving the positive earnings gains expected as a result of the acquisition; |
• | diversion of management attention from ongoing business concerns to integration matters; |
• | difficulty of maintaining uniform standards, controls, procedures and policies; |
• | challenges in demonstrating to our customers that the acquisition will not result in adverse changes in customer service standards or business focus; |
• | possible cash flow interruption or loss of revenue as a result of change of ownership transitional matters; |
• | difficulty of assimilating the operations and personnel of acquired businesses; |
• | potential loss of key employees of acquired businesses, and the impairment of relationships with employees and customers as a result of changes in management; and |
• | uncertainty as to the long-term success of any acquisitions we may make including the impact on contingent liabilities. |
There is no assurance that any completed acquisition will be accretive to our margins or profits in the short term or in the long term. If we proceed with one or more significant acquisitions in which the consideration consists of cash, a substantial portion of our available cash could be used to consummate the acquisitions. If we consummate one or more acquisitions in which the consideration consists of capital stock, our stockholders could suffer significant dilution of their interest in us. In addition, we could incur or assume significant amounts of indebtedness in connection with acquisitions. Further, acquisitions could also result in significant goodwill and/or amortization charges for acquired businesses or technologies.
We have incurred significant indebtedness which imposes operating and financial restrictions on us which, together with our debt service obligations, could significantly limit our ability to execute our business strategy and increase the risk of default under our debt obligations.
We borrowed an aggregate of approximately $121 million as of May 31, 2016. The terms of our credit facilities require us to comply with certain financial maintenance covenants. In addition, the terms of our indebtedness also include certain covenants restricting or limiting our ability to take certain actions.
These covenants may adversely affect our ability to finance future operations or limit our ability to pursue certain business opportunities or take certain corporate actions. The covenants may also restrict our flexibility in planning for changes in our business and the industry and make us more vulnerable to economic downturns and adverse developments.
18
Our ability to meet our cash requirements, including our debt service obligations, will be dependent upon our operating performance, which will be subject to general economic and competitive conditions and to financial, business and other factors affecting our operations, many of which are or may be beyond our control. We cannot provide assurance that our business operations will generate sufficient cash flows from operations to fund these cash requirements and debt service obligations. If our operating results, cash flow or capital resources prove inadequate, we could face substantial liquidity problems and might be required to dispose of material assets or operations to meet our debt and other obligations. If we are unable to service our debt, we could be forced to reduce or delay planned expansions and capital expenditures, sell assets, restructure or refinance our debt or seek additional equity capital, and we may be unable to take any of these actions on satisfactory terms or in a timely manner. Further, any of these actions may not be sufficient to allow us to service our debt obligations or may have an adverse impact on our business. Our debt agreements limit our ability to take certain of these actions. Our failure to generate sufficient operating cash flow to pay our debts or to successfully undertake any of these actions could have a material adverse effect on us.
In addition, the degree to which we are leveraged as a result of the indebtedness incurred in connection with an acquisition or otherwise could materially and adversely affect our ability to obtain additional financing for working capital, capital expenditures, acquisitions, debt service requirements or other purposes, could make us more vulnerable to general adverse economic, regulatory and industry conditions, could limit our flexibility in planning for, or reacting to, changes and opportunities in the markets in which we compete, could place us at a competitive disadvantage compared to our competitors that have less debt or could require us to dedicate a substantial portion of our cash flow to service our debt.
The presence of a significant stockholder who may sell our common stock, could cause our stock price to decline or affect the ability of a third party to acquire control of us.
The former Navilyst stockholders, including investment funds affiliated with Avista Capital Partners, beneficially own approximately 26% of our outstanding common stock. The sale of a substantial number of our shares by such parties or our other stockholders within a short period of time could cause our stock price to decline, make it more difficult for us to raise funds through future offerings of our common stock or acquire other businesses using our common stock as consideration.
Certain of the former Navilyst stockholders entered into a stockholders agreement at the closing of the acquisition that permits investment funds affiliated with Avista Capital Partners to appoint two directors to our Board of Directors until such time as, with respect to the first director, certain of the former Navilyst stockholders’ beneficial ownership in us has been reduced below 20% of the then outstanding voting shares and, with respect to the second director, certain of the former Navilyst stockholders’ beneficial ownership in us has been reduced below 10% of the then outstanding voting shares. Although these directors will not constitute a majority of the Board of Directors, they may exercise influence over the decisions of the board. David Burgstahler and Sriram Venkataraman were appointed to our Board of Directors on May 22, 2012.
Having certain of the former Navilyst stockholders as significant stockholders of us may have the effect of making it more difficult for a third party to acquire, or of discouraging a third party from seeking to acquire, a majority of our outstanding common stock or control of our Board of Directors through a proxy solicitation. In that regard, these stockholders and their controlled affiliates are obligated pursuant to the stockholders agreement, in certain circumstances, not to transfer their shares of our common stock, in whole or in part, pursuant to any recapitalization, reclassification, consolidation, merger, share exchange or other business combination transaction involving us or pursuant to any tender, exchange or other similar offer for our common stock unless, in each case, the Board of Directors recommends such transaction or offer or fails to recommend that our stockholders reject such transaction or offer.
For the period from the date that is one year from the date of the stockholders agreement until the first date that certain of the former Navilyst stockholders no longer beneficially own at least ten percent (10%) of the voting securities outstanding at such time, the applicable former Navilyst stockholders agree to vote all voting securities then owned by them either, in the sole discretion of each stockholder, (1) in accordance with the recommendation of our Board or (2) in proportion to the votes cast with respect to the voting securities not owned by the applicable former Navilyst stockholders with respect to any business or proposal on which our stockholders are entitled to vote. If at any time following one (1) year from the date of the stockholders agreement, certain of the former Navilyst stockholders beneficially own less than fifteen percent (15%) of the voting securities then outstanding and there is no stockholder designee then serving on our Board pursuant to the stockholders agreement, the applicable former Navilyst stockholders may vote all voting securities then owned by them in their own discretion.
International and national economic and industry conditions constantly change, and could materially and adversely affect our business, financial condition and results of operations.
Our business, financial condition and results of operation are affected by many changing economic, industry and other conditions beyond our control. Actual or potential changes in international, national, regional and local economic, business and
19
financial conditions, including recession, inflation and trade protection measures, may negatively affect consumer preferences, perceptions, spending patterns or demographic trends, any of which could adversely affect our business, financial condition or results of operations. Our customers may experience financial difficulties or be unable to borrow money to fund their operations, which may adversely impact their ability or decision to purchase or pay for our products. Disruptions in the credit markets have previously resulted, and could again result, in volatility, decreased liquidity, widening of credit spreads, and reduced availability of financing. There can be no assurance that future financing will be available to us on acceptable terms, if at all. An inability to obtain necessary additional financing on acceptable terms may have an adverse impact on us and on our ability to execute on our business plan.
Our industry is experiencing greater scrutiny and regulation by governmental authorities, which has led to certain costs and business distractions as we respond to inquiries and comply with new regulations, and may lead to greater governmental regulation in the future.
Our medical devices and our business activities are subject to rigorous regulation by the FDA and numerous other federal, state and foreign governmental authorities. These authorities and members of Congress have been increasing their scrutiny of our industry. In addition, certain states, including Massachusetts, have recently passed or are considering legislation restricting our interactions with health care providers and requiring disclosure of many payments to them. The federal government has recently introduced similar legislation, which may or may not preempt state laws. Recent Supreme Court case law has clarified that the FDA’s authority over medical devices preempts state tort laws, but legislation has been introduced at the federal level to allow state intervention, which could lead to increased and inconsistent regulation at the state level. We anticipate that the government will continue to scrutinize our industry closely, and that additional regulation by governmental authorities may increase compliance costs, exposure to litigation and other adverse effects to our operations.
Our international sales and operations are subject to risks and uncertainties that vary by country and which could have a material adverse effect on our business and/or results of operations.
Sales outside the United States accounted for approximately 19% of our net sales during our fiscal year ended May 31, 2016. We anticipate that sales from international operations will continue to represent a significant portion of our total sales, and we intend to continue our expansion into emerging and/or faster-growing markets outside the United States. Our sales and profitability from our international operations are subject to risks and uncertainties that can vary by country, and include those related to political and economic conditions, foreign currency exchange rate fluctuations, changes in tax laws, regulatory and reimbursement programs and policies, and the protection of intellectual property rights. These risks and uncertainties could have a material adverse effect on our business and/or results of operations.
Foreign currency exchange rate may adversely affect our business, financial condition and results of operations.
We are exposed to a variety of market risks, including the effects of changes in foreign currency exchange rates. Products manufactured in, and sold into, foreign markets represent a significant portion of our operations. When the United States dollar strengthens or weakens in relation to the foreign currencies of the countries in which we sell or manufacture our products, such as the euro, our United States dollar-reported revenue and income will fluctuate. As a result of the June 23, 2016 referendum by British voters to exit the European Union, global markets and foreign currencies have been adversely impacted and the value of the Pound Sterling has sharply declined as compared to the U.S. Dollar and other currencies. This volatility in foreign currencies is expected to continue as the U.K. negotiates and executes its exit from the European Union but it is uncertain over what time period this will occur. The effects of currency rate fluctuations and changes in the relative values of currencies may, in some instances, have a significant effect on our business, financial condition, results of operations and cash flows.
Consolidation in the healthcare industry could have an adverse effect on our revenues and results of operations.
Many healthcare industry companies, including medical device companies, are consolidating to create new companies with greater market power. As the healthcare industry consolidates, competition to provide goods and services to industry participants will become more intense. These industry participants may try to use their market power to negotiate price concessions or reductions for medical devices that incorporate components produced by us. If we are forced to reduce our prices because of consolidation in the healthcare industry, our revenues would decrease and our consolidated earnings, financial condition, or cash flow would suffer.
20
We may be limited in our ability to utilize, or may not be able to utilize, net operating loss carryforwards to reduce our future tax liability.
IRC Section 382 and related provisions contain rules that limit for U.S. federal income tax purposes the ability of a company that undergoes an “ownership change” to utilize its net operating loss carryforwards and certain other tax attributes existing as of the date of such ownership change. Our Federal net operating loss carryforwards as of May 31, 2016 after considering IRC Section 382 limitations are $151.7 million. The expiration of the Federal net operating loss carryforwards is as follows: $29.8 million between 2017 and 2023 and $121.9 million between 2027 and 2036. Our state net operating loss carryforwards as of May 31, 2016 after considering remaining IRC Section 382 limitations are $30.8 million which expire in various years from 2016 to 2036. Future ownership changes within the meaning of IRC Section 382 may also subject our tax loss carryforwards to annual limitations which would restrict our ability to use them to offset our taxable income in periods following the ownership changes.
See Note I to our consolidated financial statements included in our Annual Report on Form 10-K for the fiscal year ended May 31, 2016 for a further discussion of our tax loss carryovers.
Fluctuations in our effective tax rate and changes to tax laws may adversely affect us.
As an international company, we are subject to taxation in numerous countries, states and other jurisdictions. Our effective tax rate is derived from a combination of applicable tax rates in the various countries, states and other jurisdictions in which we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of these jurisdictions. Our effective tax rate may, however, differ from the estimated amount due to numerous factors, including a change in the mix of our profitability from country to country and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations, which could have an adverse effect on our business, financial condition and results of operations and cash flows.
If we do not maintain our reputation with interventional physicians, our growth will be limited and our business could be harmed.
Physicians typically influence the medical device purchasing decisions of the hospitals and other healthcare institutions in which they practice. Consequently, our reputation with interventional physicians is critical to our continued growth. We believe that we have built a positive reputation based on the quality of our products, our physician-driven product development efforts, our marketing and training efforts and our presence at medical society meetings. Any actual or perceived diminution in the quality of our products, or our failure or inability to maintain these other efforts, could damage our reputation with interventional physicians and cause our growth to be limited and our business to be harmed.
Our business could be harmed if we lose the services of our key personnel.
Our business depends upon our ability to attract and retain highly qualified personnel, including managerial, sales and technical personnel. We compete for key personnel with other companies, healthcare institutions, academic institutions, government entities and other organizations. We do not have written employment agreements with our executive officers, other than the CEO. Our ability to maintain and expand our business may be impaired if we are unable to retain our current key personnel or hire or retain other qualified personnel in the future. In addition, our sales force is highly talented and there is high competition in the sales industry which could have an adverse effect on our business if there is significant turnover.
Undetected defects may increase our costs and impair the market acceptance of our products.
Our products have occasionally contained, and may in the future contain, undetected defects. When these problems occur, we must divert the attention of our engineering personnel to address them. There is no assurance that we will not incur warranty or repair costs, be subject to liability claims for damages related to product defects, or experience manufacturing, shipping or other delays or interruptions as a result of these defects in the future. Our insurance policies may not provide sufficient protection should a claim be asserted. In addition, the occurrence of defects may result in significant customer relations problems and injury to our reputation, and may impair market acceptance of our products.
If a product liability claim is brought against us or our product liability insurance coverage is inadequate, our business could be harmed.
The design, manufacture and marketing of the types of medical devices we sell entail an inherent risk of product liability. Our products are used by physicians to treat seriously ill patients. We are periodically subject to product liability claims, and patients or customers may in the future bring claims against us in a number of circumstances and for a number of reasons,
21
including if our products were misused, if a component of our product fails, if their manufacture or design was flawed, if they produced unsatisfactory results or if the instructions for use and operating manuals and disclosure of product related risks for our products were found to be inadequate. In addition, individuals or groups seeking to represent a class may file suit against us. The outcome of litigation, particularly class action lawsuits, is difficult to assess or quantify. Plaintiffs in these types of lawsuits often seek recovery of very large or indeterminate amounts, including not only actual damages, but also punitive damages. The magnitude of the potential losses relating to these lawsuits may remain unknown for substantial periods of time.
We carry a product liability policy with a limit of $10,000,000 per product liability claim and an aggregate policy limit of $10,000,000, subject to a self-insured retention of $500,000 per occurrence and $2,000,000 in the aggregate. We believe, based on claims made against us in the past, our existing product liability insurance coverage is reasonably adequate to protect us from any liabilities we might incur. However, there is no assurance that this coverage will be sufficient to satisfy any claim made against us. In addition, we may not be able to continue to maintain adequate coverage at a reasonable cost and on reasonable terms, if at all. Any product liability claim brought against us, with or without merit, could increase our product liability insurance rates or prevent us from securing any coverage in the future. Additionally, if one or more product liability claims is brought against us for uninsured liabilities or is in excess of our insurance coverage, our financial condition and results of operations could be negatively impacted. Further, such claims may require us to recall some of our products, which could result in significant costs to us and could divert management’s attention from our business.
Modifications to our current products may require new marketing clearances or approvals or require us to cease marketing or recall the modified products until such clearances or approvals are obtained.
Any modification to an FDA-cleared medical device that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, requires a new FDA 510(k) clearance or, possibly, a premarket approval. The FDA requires every manufacturer to make its own determination as to whether a modification requires a new 510(k) clearance or premarket approval, but the FDA may review and disagree with any decision reached by the manufacturer. We have modified aspects of some of our devices since receiving regulatory clearance. We believed that some of these modifications did not require new 510(k) clearance or premarket approval and, therefore, we did not seek new 510(k) clearances or premarket approvals. In the future, we may make additional modifications to our products after they have received FDA clearance or approval and, in appropriate circumstances, determine that new clearance or approval is unnecessary. Regulations in other countries in which we market or sell, or propose to market or sell, our products may also require that we make judgments about changes to our products and whether or not those changes are such that regulatory approval or clearance should be obtained. In the United States and elsewhere, regulatory authorities may disagree with our past or future decisions not to seek new clearance or approval and may require us to obtain clearance or approval for modifications to our products. If that were to occur for a previously cleared or approved product, we may be required to cease marketing or recall the modified device until we obtain the necessary clearance or approval. Under these circumstances, we may also be subject to significant regulatory fines or other penalties. If any of the foregoing were to occur, our financial condition and results of operations could be negatively impacted.
We are subject to healthcare fraud and abuse regulations that could result in significant liability, require us to change our business practices and restrict our operations in the future.
We are subject to various federal, state and local laws targeting fraud and abuse in the healthcare industry, including anti-kickback and false claims laws. Violations of these laws are punishable by criminal or civil sanctions, including substantial fines, imprisonment and exclusion from participation in healthcare programs such as Medicare and Medicaid and health programs outside the United States. These laws and regulations are wide ranging and subject to changing interpretation and application, which could restrict our sales or marketing practices. Furthermore, since many of our customers rely on reimbursement from Medicare, Medicaid and other governmental programs to cover a substantial portion of their expenditures, our exclusion from such programs as a result of a violation of these laws could have a material adverse effect on our business, results of operations, financial condition and cash flow.
If our employees or agents violate the U.S. Foreign Corrupt Practices Act or anti-bribery laws in other jurisdictions, we may incur fines or penalties, or experience other adverse consequences.
We are subject to the U.S. Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery laws in international jurisdictions, including the UK Anti-Bribery Act, which generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. Because of the predominance of government-sponsored healthcare systems around the world, many of our customer relationships outside of the United States are with governmental entities and are therefore subject to such anti-bribery laws. Our sales to customers and distributors outside of the United States have been increasing and we expect them to continue to increase in the future. If our employees or
22
agents violate the provisions of the FCPA or other anti-bribery laws, we may incur fines or penalties, we may be unable to market our products in other countries or we may experience other adverse consequences which could have a material adverse effect on our operating results or financial condition.
Any disaster at our manufacturing facilities could disrupt our ability to manufacture our products for a substantial amount of time, which could cause our revenues to decrease.
We conduct our manufacturing and assembly at facilities in Queensbury, New York, Glens Falls, New York, Manchester, Georgia, and Denmead, England. It would be difficult, expensive and time-consuming to transfer resources from one facility to the other, replace, or repair these facilities and our manufacturing equipment if they were significantly affected by a disaster. Additionally, we might be forced to rely on third-party manufacturers or to delay production of our products. Insurance for damage to our properties and the disruption of our business from disasters may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all. In addition, if one of our principal suppliers were to experience a similar disaster, uninsured loss or under-insured loss, we might not be able to obtain adequate alternative sources of supplies or products or could face significant delays and incur substantial expense in doing so. Any significant uninsured loss, prolonged or repeated disruption, or inability to operate experienced by us or any of our principal suppliers could cause significant harm to our business, financial condition and results of operations.
Our future operating results are difficult to predict and may vary significantly from quarter to quarter, which may adversely affect the price of our common stock.
The ongoing introduction of new products and services that affect our overall product mix make the prediction of future operating results difficult. You should not rely on our past results as any indication of future operating results. The price of our common stock will likely fall in the event that our operating results do not meet the expectations of analysts and investors. Comparisons of our quarterly operating results are an unreliable indication of our future performance because they are likely to vary significantly based on many factors, including:
• | the level of sales of our products and services in our markets; |
• | our ability to introduce new products or services and enhancements in a timely manner; |
• | the demand for and acceptance of our products and services; |
• | the success of our competition and the introduction of alternative products or services; |
• | our ability to command favorable pricing for our products and services; |
• | the growth of the market for our devices and services; |
• | the expansion and rate of success of our direct sales force in the United States and internationally and our independent distributors internationally; |
• | actions relating to ongoing FDA compliance; |
• | the effect of intellectual property disputes; |
• | the size and timing of orders from independent distributors or customers; |
• | the attraction and retention of key personnel, particularly in sales and marketing, regulatory, manufacturing and research and development; |
• | unanticipated delays or an inability to control costs; |
• | general economic conditions as well as those specific to our customers and markets; and |
• | seasonal fluctuations in revenue due to the elective nature of some procedures. |
Our stock price may be volatile, which may cause the value of our stock to decline or subject us to a securities class action litigation.
The trading price of our common stock price may be volatile and could be subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including:
• | general economic, industry and market conditions; |
• | actions by institutional or other large stockholders; |
• | the depth and liquidity of the market for our common stock; |
• | volume and timing of orders for our products; |
• | developments generally affecting medical device companies; |
• | the announcement of new products or product enhancements by us or our competitors; |
• | changes in earnings estimates or recommendations by securities analysts; |
• | investor perceptions of us and our business, including changes in market valuations of medical device companies; and |
• | our results of operations and financial performance. |
23
In addition, the stock market in general, and the NASDAQ Stock Market and the market for medical devices in particular, have experienced substantial price and volume volatility that is often seemingly unrelated to the operating performance of particular companies. These broad market fluctuations may cause the trading price of our common stock to decline. In the past, securities class action litigation has often been brought against a company after a period of volatility in the market price of its common stock. We may become involved in this type of litigation in the future. Any securities litigation claims brought against us could result in substantial expense and the diversion of management’s attention from our business.
Anti-takeover provisions in our organizational documents and Delaware law may discourage or prevent a change of control, even if an acquisition would be beneficial to our stockholders, which could cause our stock price to decline and prevent attempts by our stockholders to replace or remove our current management.
Our amended and restated certificate of incorporation and our amended and restated bylaws contain provisions that may enable our management to resist a change in control. These provisions may discourage, delay or prevent a change in the ownership of our company or a change in our management. In addition, these provisions could limit the price that investors would be willing to pay in the future for shares of our common stock. Such provisions include:
• | our board of directors is authorized, without prior stockholder approval, to create and issue “blank check” preferred stock, with rights senior to those of our common stock; |
• | our board of directors is classified so that not all members of our board of directors are elected at one time, which may make it more difficult for a person who acquires control of a majority of our outstanding voting stock to replace our directors; |
• | advance notice requirements for stockholders to nominate individuals to serve on our board of directors or for stockholders to submit proposals that can be acted upon at stockholder meetings; |
• | stockholder action by written consent is prohibited; and |
• | stockholders are not permitted to cumulatively vote for the election of directors. |
We are also subject to the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock.
These and other provisions in our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law could make it more difficult for stockholders or potential acquirers to obtain control of our board of directors or initiate actions that are opposed by our then-current board of directors, including delaying or impeding a merger, tender offer or proxy contest involving our company. Any delay or prevention of a change of control transaction or changes in our board of directors could cause the market price of our common stock to decline.
Our goodwill, intangible assets and fixed assets are subject to potential impairment.
A significant portion of our assets consists of goodwill, intangible assets and fixed assets, the carrying value of which may be reduced if we determine that those assets are impaired.
Most of our intangible and fixed assets have finite useful lives and are amortized or depreciated over their useful lives on either a straight-line basis or over the expected period of benefit or as revenues are earned from the sales of the related products. The underlying assumptions regarding the estimated useful lives of these intangible assets are reviewed annually and more often if an event or circumstance occurs making it likely that the carrying value of the assets may not be recoverable and are adjusted through accelerated amortization if necessary. Whenever events or changes in circumstances indicate that the carrying value of the assets is not recoverable we test intangible assets for impairment based on estimates of future cash flows. If an intangible asset is considered to be impaired, the amount of the impairment will equal the excess of the carrying value over the fair value of the asset.
We review our single reporting unit for potential goodwill impairment in the third fiscal quarter of each year as part of our annual goodwill impairment testing, and more often if an event or circumstance occurs making it likely that impairment exists. We conduct impairment testing based on our current business strategy in light of present industry and economic conditions, as well as future expectations. The annual goodwill impairment review performed in December 2015 indicated no goodwill impairments.
If actual results differ from the assumptions and estimates used in the goodwill and intangible asset calculations, we could incur future impairment or amortization charges, which could negatively impact our results of operations.
24
Failure to attract additional capital which we may require to expand our business could curtail our growth.
We may require additional capital to expand our business. If cash generated internally is insufficient to fund capital requirements, we will require additional debt or equity financing. In addition, we may require financing to fund any significant acquisitions we may seek to make. Needed financing may not be available or, if available, may not be available on terms satisfactory to us and may result in significant stockholder dilution. Covenants in our existing financing agreements may also restrict our ability to obtain additional debt financing. If we fail to obtain sufficient additional capital in the future, we could be forced to curtail our growth strategy by reducing or delaying capital expenditures and acquisitions, selling assets, restructuring our operations or refinancing our indebtedness.
We rely on the proper function, availability and security of information technology systems to operate our business and a cyber-attack or other breach of these systems could have a material adverse effect on our business, financial condition or results of operations.
We rely on information technology systems to process, transmit, and store electronic information in our day-to-day operations. Similar to other large multi-national companies, the size and complexity of our information technology systems makes them vulnerable to a cyber-attack, malicious intrusion, breakdown, destruction, loss of data privacy, or other significant disruption. Our information systems require an ongoing commitment of significant resources to maintain, protect, and enhance existing systems and develop new systems to keep pace with continuing changes in information processing technology, evolving systems and regulatory standards, the increasing need to protect patient and customer information, and changing customer patterns. In addition, third parties may attempt to hack into our products to obtain data relating to patients with our products or our proprietary information. Any failure by us to maintain or protect our information technology systems and data integrity, including from cyber-attacks, intrusions or other breaches, could result in the unauthorized access to patient data and personally identifiable information, theft of intellectual property or other misappropriation of assets, or otherwise compromise our confidential or proprietary information and disrupt our operations. Any of these events, in turn, may cause us to lose existing customers, have difficulty preventing, detecting, and controlling fraud, have disputes with customers, physicians, and other health care professionals, be subject to legal claims and liability, have regulatory sanctions or penalties imposed, have increases in operating expenses, incur expenses or lose revenues as a result of a data privacy breach or theft of intellectual property, or suffer other adverse consequences, any of which could have a material adverse effect on our business, financial condition or results of operations.
We, our competitors or other third parties, may engage in clinical trials with respect to our products. The results of these trials may be unfavorable, or perceived as unfavorable by the market, and could have a material adverse effect on our business, financial condition or results of operations.
Our products may be the subject of clinical trials conducted by us, our competitors or third parties for the purposes of obtaining regulatory clearances or to gather market data. Unfavorable or inconsistent clinical data from existing or future clinical trials conducted by us, by our competitors or by third parties, or the FDA's or the market's perception of this clinical data, may adversely impact our ability to obtain product approvals, our position in, and share of, the markets in which we participate and our business, financial condition, results of operations or future prospects
Item 1B. | Unresolved Staff Comments. |
None.
Item 2. | Properties. |
During the year ended May 31, 2016, we operated in the following locations:
Location | Purpose | Approx. Sq. Ft. | Property Type | ||||
Latham, NY | Corporate headquarters | 55,000 | Leased | ||||
Glens Falls, NY | Manufacturing and distribution | 189,000 | Owned | ||||
Queensbury, NY | Manufacturing and distribution | 129,000 | Owned | ||||
Manchester, GA | Manufacturing and distribution | 60,000 | Leased | ||||
Marlborough, MA | Research & Development | 31,000 | Leased | ||||
Denmead, U.K. | Manufacturing | 7,500 | Leased | ||||
Amsterdam, NL | Selling, Marketing & Administrative | 10,100 | Leased | ||||
In addition, we lease sales offices in various other jurisdictions.
25
Item 3. | Legal Proceedings. |
AngioDynamics v. biolitec
On January 2, 2008, we commenced an action in the United States District Court for the Northern District of New York entitled AngioDynamics, Inc. v. biolitec, Inc. In this action, we sought judgment against biolitec for defense and indemnification in two lawsuits which we previously settled. Our claims arise out of a Supply and Distribution Agreement (“SDA”) entered into with biolitec on April 1, 2002. On September 27, 2011, the U.S. District Court granted key portions of our motion for summary judgment in our legal case against biolitec. The Court also dismissed biolitec’s counterclaims against us. The court denied one portion of our summary judgment motion, which sought to recover additional costs from biolitec, leaving this for adjudication at trial. On November 8, 2012, the Court granted partial judgment to us in the amount of $23.2 million. Biolitec appealed this judgment. On August 23, 2013, the U.S. Court of Appeals for the Second Circuit dismissed biolitec’s appeal.
In October 2009, we commenced an action in the United States District Court for the District of Massachusetts entitled AngioDynamics, Inc. v. biolitec AG and Wolfgang Neuberger. The Complaint in this action was amended in March 2010. This action seeks to recover against biolitec, Inc.’s parent entities and CEO for tortiously interfering with biolitec, Inc.’s contractual obligation to defend and indemnify us, and also seeks to pierce the corporate veil of biolitec, Inc. and to invalidate certain alleged fraudulent transfers in order to hold biolitec, Inc.’s parent entities jointly and severally liable for the alleged breach of the SDA. On September 13, 2012, the Massachusetts Court granted our request for a preliminary injunction prohibiting the downstream merger of biolitec AG with its Austrian subsidiary. On April 1, 2013, the U.S. Court of Appeals for the First Circuit affirmed the preliminary injunction. On January 14, 2014, the District Court entered judgment in our favor as to liability. On March 18, 2014, the District Court entered judgment in our favor against Biolitec AG, Biomed Technology Holdings, Ltd., and Wolfgang Neuberger, jointly and severally, in the amount of $74.9 million. On March 11, 2015, the U.S. Court of Appeals for the First Circuit affirmed the judgment. The defendants petitioned to the U.S. Supreme Court for a writ of certiorari. The Supreme Court denied the petition on November 30, 2015. The defendants have also filed an appeal with the U.S. Court of Appeals for the First Circuit regarding civil contempt sanctions imposed by the Massachusetts District Court as a result of defendants’ completion of the downstream merger in violation of the Court’s injunction. On May 6, 2016, the First Circuit issued an opinion rejecting this latest appeal. On February 18, 2016, the Massachusetts District Court issued an order compelling the Massachusetts defendants to provide post-judgment discovery intended to aid us in potentially collecting our judgment. On March 21, 2016, the Massachusetts defendants noticed an appeal from this order. On June 27, 2016, we filed a motion asking the Massachusetts District Court to impose sanctions on the Massachusetts defendants for their failure to comply with the post-judgment discovery order.
On November 13, 2014, the U.S. District Court for the District of Massachusetts issued summonses to four Biolitec entities - Biolitec U.S., Inc., Biolitec Holding U.S., Inc., Biolitec Medical Devices, Inc., and CeramOptec Industries, Inc. - pursuant to Massachusetts trustee process. We sought to use this process to attach the assets of these entities in order to satisfy our judgment. The trustee process was automatically stayed when the four Biolitec entities filed Chapter 7 petitions in the U.S. Bankruptcy Court for the District of Delaware. However, on November 3, 2015, the Delaware Bankruptcy Court granted our request to modify the automatic stay to allow us to seek a default against the four Biolitec entities pursuant to trustee process. On January 21, 2016, the four Chapter 7 cases were transferred at our request to the U.S. Bankruptcy Court for the District of New Jersey.
On August 29, 2013, we became co-plaintiffs in an adversary proceeding in the United States Bankruptcy Court for the District of New Jersey entitled Cyganowski, Trustee, et al. v. Biolitec U.S., Inc., et al. In this action, we assert claims of conversion, unjust enrichment, tortious interference, and unfair competition against various biolitec entities for alleged violation of Bankruptcy Court settlement and sale orders under which we acquired certain assets of Biolitec, Inc. On September 3, 2013, we, along with our co-plaintiff, obtained a temporary restraining order against the defendants in this action. On January 22, 2015, the Bankruptcy Court entered a permanent injunction on our behalf for an additional two years.
C.R. Bard, Inc. v. AngioDynamics, Inc.
On January 11, 2012, C.R. Bard, Inc. (“Bard”) filed a suit in the United States District Court of Utah claiming certain of our implantable port products infringe on three U.S. patents held by Bard (the "Utah Action"). Bard is seeking unspecified damages and other relief. The Court denied Bard’s motion for pre-trial consolidation with separate actions it filed on the same day against Medical Components, Inc. and Smiths Medical ASD, Inc., but had asked for supplemental briefing on the issue of whether to conduct a common Markman hearing. Meanwhile, we filed petitions for reexamination in the US Patent and Trademark Office ("PTO") which seek to invalidate all three patents asserted by Bard in the litigation. Our petitions were granted and 40 of Bard's 41 patent claims were rejected and, following further proceedings, the Patent Office issued a Final
26
Rejection of all 40 claims subject to reexamination. Thereafter, Bard filed appeals to the PTO Board of Appeals and Interferences for all three reexams. The parties completed briefing on the appeals and oral argument was held on June 18, 2015. The Patent Office has issued decisions in the three appeals. In one (issued on March 11, 2016 for US Patent No. 7,785,302), the rejections of six of the ten claims under reexamination were affirmed, but were reversed on four of the ten claims. In the second (issued on March 24, 2016 for U.S. Patent No. 7,959,615), the rejections of eight of the ten claims under reexamination were affirmed but the rejections of the other two of the ten claims were reversed. In the third (issued on March 29 for U.S. Patent No. 7,947.022) the rejections of all twenty claims under reexamination were affirmed. Bard has since filed Requests for Rehearing in all three reexamination appeals and the Company filed Requests for Rehearing in two of the reexamination appeals (the ‘302 and ‘615 patent reexaminations). Each party has filed comments in Opposition to the other party’s Rehearing Requests, and we are awaiting the PTO determinations in all three reexaminations. The Utah Action has been stayed pending final resolution of the PTO process. We believe these claims are without merit and intend to defend them vigorously. We have not recorded an expense related to the outcome of this litigation because it is not yet possible to determine if a potential loss is probable nor reasonably estimable.
On March 10, 2015, C.R. Bard, Inc. and Bard Peripheral Vascular, Inc. (“Bard”) filed suit in the United States District Court for the District of Delaware claiming certain of our implantable port products infringe on three U.S. patents held by Bard (the “Delaware Action). Bard is seeking unspecified damages and other relief. The patents asserted in the Delaware Action are different than those asserted in the Utah Action. On June 1, 2015, we filed two motions in response to Bard’s Complaint - one sought transfer to the District of Utah where the Utah Action is currently pending, and the other sought dismissal of the entire complaint on grounds that none of the claims in the asserted patents is directed to patent eligible subject matter under Section 101 of the Patent Statute and in light of recent authority from the U. S. Supreme Court. On January 12, 2016, the court issued a decision denying both motions. We have since served an Answer and Counterclaim to which Bard has served a Reply. On March 10, 2016, the Court held a case management conference, and, on March 14, 2016, the court entered a Scheduling Order which set, inter alia, a Markman hearing for March 10, 2017, a summary judgment hearing for December 8, 2017 and trial for March 12, 2018. The parties have since served various discovery requests on each other; on May 27, 2016 Bard served its Infringement Contentions which identified all the port products accused of infringement; and, on June 24, 2016, we served Invalidity Contentions which detail various grounds for invalidating the three asserted patents. We believe these claims are without merit and intend to defend them vigorously. We have not recorded an expense related to the outcome of this litigation because it is not yet possible to determine if a potential loss is probable nor reasonably estimable.
Governmental Investigations
LC Beads
In June 2014 we received a subpoena from the U.S. Department of Justice (the “DOJ”) requesting documents in relation to a criminal and civil investigation the DOJ is conducting regarding BTG International, Inc.’s LC Bead® product beginning in 2003. RITA Medical Systems and AngioDynamics, Inc., after its acquisition of RITA, was the exclusive distributor of LC Beads in the United States from 2006 through December 31, 2011. We are cooperating fully with this investigation and at this time are unable to predict its scope, duration or outcome. We are unable at this time to reasonably estimate the amount or range of any loss, although it is possible that the amount of such loss could be material. In accordance with ASC 450, "Contingencies," or "ASC 450," no amount in respect of any potential liability in this matter, including for penalties, fines or other sanctions, has been accrued in the consolidated financial statements.
EVLT
In April 2015 we received a subpoena from the DOJ requesting documents in relation to a criminal and civil investigation the DOJ is conducting regarding purported promotion of certain of AngioDynamics’ VenaCure EVLT products for un-cleared indications. We are cooperating fully with this investigation and at this time are unable to predict its scope, duration or outcome. We are unable at this time to reasonably estimate the amount or range of any loss, although it is possible that the amount of such loss could be material. In accordance with ASC 450, "Contingencies," or "ASC 450," no amount in respect of any potential liability in this matter, including for penalties, fines or other sanctions, has been accrued in the consolidated financial statements.
Item 4. | Mine Safety Disclosures. |
Not applicable.
Part II
27
Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities. |
Our common stock is traded on The Global Select Market tier of The NASDAQ Stock Market LLC (formerly the Nasdaq National Market), under the symbol “ANGO.”
The following table sets forth, for the fiscal quarters indicated, the high and low sale prices for our common stock as reported by The NASDAQ Stock Market.
Sale Price | |||||||
High | Low | ||||||
Year ended May 31, 2016 | |||||||
Fourth Quarter | $ | 12.72 | $ | 10.76 | |||
Third Quarter | $ | 12.70 | $ | 10.02 | |||
Second Quarter | $ | 14.87 | $ | 11.24 | |||
First Quarter | $ | 16.80 | $ | 14.31 | |||
Sale Price | |||||||
High | Low | ||||||
Year ended May 31, 2015 | |||||||
Fourth Quarter | $ | 18.89 | $ | 15.54 | |||
Third Quarter | $ | 19.72 | $ | 17.29 | |||
Second Quarter | $ | 17.85 | $ | 13.29 | |||
First Quarter | $ | 16.60 | $ | 13.80 | |||
As of July 22, 2016, there were 202 holders of record of our common stock.
Dividends
We did not declare any cash dividends on our common stock during our last three fiscal years. We do not anticipate paying any cash dividends on our common stock for the foreseeable future.
Performance Graph
The graph below matches AngioDynamics, Inc.’s cumulative 5-year total shareholder return on common stock with the cumulative total returns of the NASDAQ Composite index, the RDG SmallCap Medical Devices index, and the NASDAQ Medical Equipment index. The graph tracks the performance of a $100 investment in our common stock and in each index (with the reinvestment of all dividends) from May 31, 2011 to May 31, 2016. The stock price performance included in this graph is not necessarily indicative of future stock price performance.
28
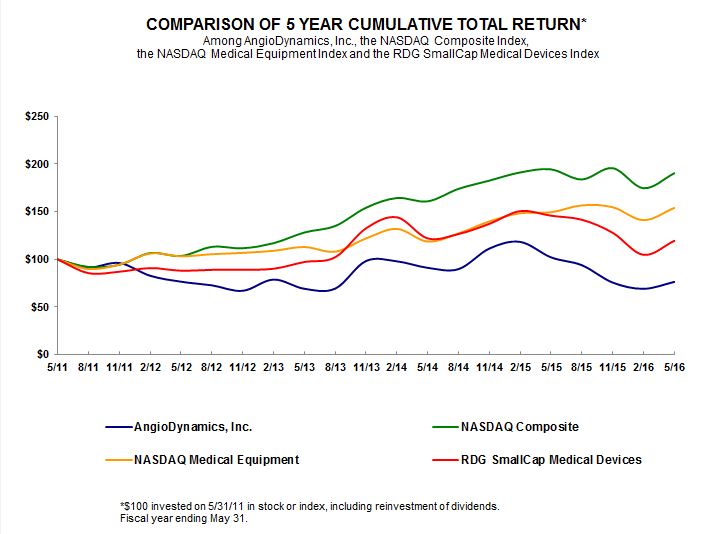
29
Item 6. | Selected Financial Data. |
You should read the following selected financial data in conjunction with our consolidated financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this annual report on Form 10-K.
The consolidated statements of operations data for the fiscal years ended May 31, 2016, May 31, 2015, and May 31, 2014, and the consolidated balance sheet data as of May 31, 2016 and May 31, 2015, are derived from the consolidated financial statements that are included elsewhere in this annual report on Form 10-K. The consolidated statements of operations data for the fiscal years ended May 31, 2013 and May 31, 2012, and the consolidated balance sheet data as of May 31, 2014, May 31, 2013 and May 31, 2012, are derived from our audited consolidated financial statements not included in this annual report on Form 10-K. Historical results are not necessarily indicative of the results of operations to be expected for future periods. See Note N of “Notes to Consolidated Financial Statements” for a description of the method that we used to compute our historical basic and diluted net income per share attributable to common stockholders.
We made adjustments to correct immaterial errors within this selected financial data. For a detailed explanation of these adjustments, please refer to Note R, "Immaterial Error Corrections".
Year ended | |||||||||||||||||||
(Amounts in thousands, except per share information) | |||||||||||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | May 31, 2013 | May 31, 2012 | |||||||||||||||
Consolidated Statements of Operations Data: | |||||||||||||||||||
Net sales | $ | 353,890 | $ | 356,534 | $ | 354,425 | $ | 341,916 | $ | 221,917 | |||||||||
Gross profit | 174,316 | 175,796 | 180,174 | 168,514 | 125,309 | ||||||||||||||
Operating expenses | |||||||||||||||||||
Research and development | 25,053 | 26,594 | 28,124 | 26,091 | 20,511 | ||||||||||||||
Sales and marketing | 84,723 | 83,220 | 85,305 | 77,564 | 65,757 | ||||||||||||||
General and administrative | 29,603 | 29,162 | 26,902 | 26,035 | 19,033 | ||||||||||||||
Amortization of intangibles | 17,964 | 17,966 | 16,562 | 16,599 | 9,393 | ||||||||||||||
Change in fair value of contingent consideration | 948 | (8,096 | ) | (1,908 | ) | 1,583 | — | ||||||||||||
Acquisition, restructuring and other items, net (a) | 12,591 | 26,257 | 10,873 | 13,800 | 15,859 | ||||||||||||||
Medical device excise tax | 2,416 | 4,142 | 3,829 | 1,600 | — | ||||||||||||||
Total operating expenses | 173,298 | 179,245 | 169,687 | 163,272 | 130,553 | ||||||||||||||
Operating income (loss) | 1,018 | (3,449 | ) | 10,487 | 5,242 | (5,244 | ) | ||||||||||||
Total other (expenses) income, net | (4,271 | ) | (4,682 | ) | (5,301 | ) | (6,579 | ) | (409 | ) | |||||||||
Net income (loss) | $ | (43,590 | ) | $ | (3,388 | ) | $ | 2,347 | $ | (1,051 | ) | $ | (5,331 | ) | |||||
Earnings (loss) per share | |||||||||||||||||||
Basic | $ | (1.21 | ) | $ | (0.09 | ) | $ | 0.07 | $ | (0.03 | ) | $ | (0.21 | ) | |||||
Diluted | $ | (1.21 | ) | $ | (0.09 | ) | $ | 0.07 | $ | (0.03 | ) | $ | (0.21 | ) | |||||
(a) | Acquisition, restructuring and other items, net consists of fixed and long-term asset impairments, intangible impairments, cost associated with litigation, recalls, the operational excellence program and other miscellaneous items. |
30
As of | |||||||||||||||||||
(Amounts in thousands) | |||||||||||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | May 31, 2013 | May 31, 2012 | |||||||||||||||
Consolidated Balance Sheet Data: | |||||||||||||||||||
Cash, cash equivalents and marketable securities | $ | 33,986 | $ | 20,080 | $ | 17,914 | $ | 23,955 | $ | 40,309 | |||||||||
Working capital | 79,527 | 90,283 | 81,071 | 71,643 | 99,068 | ||||||||||||||
Total assets | 727,063 | 773,058 | 798,576 | 790,561 | 719,903 | ||||||||||||||
Total debt | 121,410 | 137,660 | 142,660 | 142,500 | 150,000 | ||||||||||||||
Contingent consideration | 38,275 | 47,384 | 67,231 | 75,049 | — | ||||||||||||||
Total long-term liabilities | 153,108 | 167,444 | 195,750 | 201,317 | 142,827 | ||||||||||||||
Total stockholders’ equity | 507,228 | 545,099 | 536,885 | 526,324 | 523,306 | ||||||||||||||
Item 7. | Management’s Discussion and Analysis of Financial Conditions and Results of Operations. |
The following information should be read together with the audited consolidated financial statements and the notes thereto and other information included elsewhere in this annual report on Form 10-K.
Forward-Looking Statements
This annual report on Form 10-K, including the sections entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. All statements regarding AngioDynamics’ expected future financial position, results of operations, cash flows, business strategy, budgets, projected costs, capital expenditures, products, competitive positions, growth opportunities, acquisitions, plans and objectives of management for future operations, as well as statements that include the words such as “expects,” “reaffirms,” “intends,” “anticipates,” “plans,” “believes,” “seeks,” “estimates,” or variations of such words and similar expressions, are forward-looking statements. These forward looking statements are not guarantees of future performance and are subject to risks and uncertainties. Investors are cautioned that actual events or results may differ from our expectations. Factors that may affect the actual results include, without limitation, our ability to develop our existing and new products, future actions by the FDA or other regulatory agencies, results of pending or future clinical trials, the results of ongoing litigation, overall economic conditions, general market conditions, market acceptance, foreign currency exchange rate fluctuations, the effects on pricing from group purchasing organizations and competition, the loss of any of our key customers or reduction in the purchase of our products by any such customers, and our ability to integrate acquired businesses as well as the risk factors listed in Part I, Item 1A of this annual report on Form 10-K.
Although we believe that the assumptions underlying the forward-looking statements contained herein are reasonable, any of the assumptions could be inaccurate and, therefore, there can be no assurance that the forward-looking statements included in this annual report on Form 10-K will prove to be accurate. In light of the significant uncertainties inherent in the forward-looking statements included herein, the inclusion of such information should not be regarded as a representation by us or any other person that our objectives and plans will be achieved. Any forward-looking statements are made pursuant to the Private Securities Litigation Reform Act of 1995 and, as such, speak only as of the date made. We disclaim any obligation to update the forward-looking statements. Investors are cautioned not to place undue reliance on these forward-looking statements which speak only as of the date stated, or if no date is stated, as of the date of this document.
Overview
We design, manufacture and sell a wide range of medical, surgical and diagnostic devices used by professional healthcare providers for vascular access, for the treatment of peripheral vascular disease and for use in oncology and surgical settings. Our devices are generally used in minimally invasive, image-guided procedures. Most of our products are intended to be used once and then discarded, or they may be temporarily implanted for short- or long-term use.
Our sales and profitability growth depends, in part, on the introduction of new and innovative products, together with ongoing enhancements to our existing products. Expansions to our product offerings are created through internal product development, technology licensing and strategic alliances. In recent years we have acquired or developed, and launched several new products, including the AngioVac cannula and circuit, the BioFlo family of products, NanoKnife and the Acculis
31
microwave system, which are all expected to be growth drivers of our business. We recognize the importance of, and intend to continue to make investments in, research and development activities and business development opportunities.
We sell our products in the United States through a direct sales force and outside the U.S. through a combination of direct sales and distributor relationships. We expect our international business to grow in both sales and profit through geographic expansion, market penetration, and increasing our direct presence.
Our ability to further increase our profitability will depend in part on improving gross profit and operating margins. A portion of improved gross margin we expect to deliver through the acquisition, development and sale of innovative products, such as those mentioned above. Additionally, we have an active company-wide operational excellence program designed to create manufacturing efficiencies and drive improved business performance. Further, we anticipate being able to manage increases in our operating expenses at a rate slower than our sales growth to provide further operating margin expansion.
Recent Developments
On March 31, 2016, Joseph DeVivo, former President and Chief Executive Officer, decided to pursue other interests outside of the Company and on April 4, 2016, James C. Clemmer was appointed as the new President and Chief Executive Officer. As part of the separation agreement with Joseph DeVivo, his stock options, restricted stock units and performance shares will continue to vest for one year.
On November 3, 2015, Mark Frost resigned as Executive Vice President and Chief Financial Officer (CFO). Michael Trimarchi, Vice President and Global Controller, assumed the responsibilities as principal accounting officer of the Company and interim CFO until his resignation on May 13, 2016. On July 22, 2016, Michael Greiner was appointed Executive Vice President and Chief Financial Officer of the Company, effective August 16, 2016. On July 27, 2016, Peter J. Kish was designated as the principal financial officer and principal accounting officer of the Company by the Board of Directors of the Company. Mr. Kish will serve in this role until Mr. Greiner begins his service as Chief Financial Officer on August 16, 2016.
On December 18, 2015, President Obama signed into law H.R. 2029, the “Consolidated Appropriations Act, 2016”, which includes a two-year moratorium on the medical device excise tax, effective January 1, 2016. The 2.3 percent tax on sales of medical devices (except certain devices sold at retail) was enacted as part of the Affordable Care Act in 2010 and applied to device sales beginning on January 1, 2013. Absent further legislative action, the tax will be automatically reinstated for medical device sales starting on January 1, 2018. As presented on our Consolidated Condensed Statement of Operations we have incurred $12.0 million cumulatively since the enactment of the tax on January 1, 2013 through the May 31, 2016. In the absence of this tax, the Company will seek opportunities to further invest in growth drivers to create long-term shareholder value.
On November 17, 2015, the Company received a letter from the FDA closing out the warning letter the Company received from FDA in January 2011 regarding certain promotional activities related to the NanoKnife System. On November 25, 2015, the Company received letters from the FDA closing out the warning letters the Company received from FDA in May 2011 related to the Company’s Queensbury facility and in November 2014 related to the Company’s Glens Falls facility. These close out letters resolved all outstanding warning letters against the Company.
During the quarter ended May 31, 2016, we made the decision to discontinue the Celerity tip location and navigation product line. The discontinuance of the product line was the result of performance and quality issues with the product and a strategic shift to focus on other product within the Vascular Access business. We recorded a write-off of approximately $5.8 million of inventory and $0.1 million of hardware assets during the fourth quarter.
During the quarter ended May 31, 2016 we entered into an agreement with Merz North America where we became the exclusive sub-distributor of ASCLERA in the vein market in the United States and received the Merz customer list for the designated market territory. As part of the agreement we receive a personal, non-exclusive, non-transferable, non-assignable, non-sub licensable, license to use the trademarks, service marks and trades names from Merz. As a result of this agreement we recorded $3.3 million of intangible assets for the exclusive distribution rights and customer lists obtained. The Asclera product is the replacement for Sotradecol.
Management's Use of Non-GAAP Measures
Net sales “on a constant currency basis” is a non-GAAP measure. The company analyzes net sales on a constant currency basis to better measure the comparability of results between periods. Because changes in foreign currency exchange rates have a non-operating impact on net sales, the company believes that evaluating growth in net sales on a constant currency basis
32
provides an additional and meaningful assessment of net sales to both management and the company’s investors. Constant currency growth rates are calculated by translating the current period's local currency sales by the prior period’s exchange rate.
Constant currency growth rates are not indicative of changes in corresponding cash flows. The limitation of these non-GAAP measures is that they do not reflect results on a standardized reporting basis. Non-GAAP measures are intended to supplement the applicable GAAP disclosures and should not be viewed as replacements of GAAP results.
Critical Accounting Policies and Use of Estimates
Our significant accounting policies are summarized in Note A to Notes to Consolidated Financial Statements included elsewhere in this annual report on Form 10-K. While all these significant accounting policies affect the reporting of our financial condition and results of operations, we view certain of these policies as critical. Policies determined to be critical are those policies that have the most significant impact on our financial statements and require us to use a greater degree of judgment and/or estimates. Actual results may differ from those estimates. The accounting policies identified as critical are as follows:
Revenue Recognition
We recognize revenue in accordance with generally accepted accounting principles as outlined in the SEC’s authoritative guidance on revenue recognition which requires that four criteria be met before revenue can be recognized: (i) persuasive evidence that an arrangement exists; (ii) the price is fixed or determinable; (iii) collectability is reasonably assured; and (iv) product delivery has occurred or services have been rendered. Decisions relative to criterion (iii) regarding collectability are based upon our judgments, as discussed under “Accounts Receivable” in Note A, and should conditions change in the future and cause us to determine this criterion is not met; our results of operations may be affected. We recognize revenue, net of sales taxes assessed by any governmental authority, as products are shipped, based on F.O.B. shipping point terms when title and risk of loss passes to customers. We negotiate shipping and credit terms on a customer-by-customer basis and products are shipped at an agreed upon price. All product returns must be pre-approved by us and customers may be subject to a 20% restocking charge. To be accepted, a returned product must be unadulterated, undamaged and have at least 12 months remaining prior to its expiration date. Charges for discounts, returns, rebates and other allowances are recognized as a deduction from revenue on an accrual basis in the period in which the revenue is recorded. The accrual for product returns, discounts and other allowances is based on the company’s history.
Income Taxes
In preparing our financial statements, we calculate income tax expense for each jurisdiction in which we operate. This involves estimating actual current taxes due plus assessing temporary differences arising from differing treatment for tax and accounting purposes that are recorded as deferred tax assets and liabilities. We periodically evaluate deferred tax assets, capital loss carryforwards and tax credit carryforwards to determine their recoverability based primarily on our ability to generate future taxable income and capital gains. Where it is more-likely-than-not these will not be recovered, we estimate a valuation allowance and record a corresponding additional tax expense in our statement of operations.
We file income tax returns in the U.S. federal jurisdiction and various state and foreign jurisdictions. In the normal course of business we are subject to examination by taxing authorities throughout the world. Fiscal years 2012 through 2016 remain open to examination by the various tax authorities. New York State is currently auditing AngioDynamic’s franchise tax filings for 2011 through 2013, and we do not anticipate any material adjustments will result. We analyzed filing positions in all of the Federal and state jurisdictions where we are required to file income taxes, as well as all open tax years in these jurisdictions and believe that our income tax filing positions and deductions will be sustained on audit and we do not anticipate any adjustments will result in a material adverse effect on our financial condition, results of operations or cash flows.
Acquisitions and Contingent Consideration
In a business combination, the acquisition method of accounting requires that the identifiable assets acquired and liabilities assumed be measured at their fair value, with goodwill being the excess value of consideration paid over the fair value of the net identifiable assets acquired. IP R&D is capitalized and recorded as an indefinite-lived intangible asset at the acquisition date, contingent consideration is recorded at fair value at the acquisition date, and transaction costs are expensed as incurred. When the company acquires net assets that are not accounted for as a business combination, no goodwill is recognized.
33
The fair value of the liability for contingent consideration recorded on the acquisition date is based on probability weighted estimated cash flow streams, discounted back to present value using a discount rate determined in accordance with accepted valuation methods. The liability for contingent consideration is remeasured to fair value at each reporting period with changes recorded in earnings until the contingency is resolved.
Goodwill and Intangible Assets
Intangible assets other than goodwill, indefinite lived intangible assets and IP R&D are amortized over their estimated useful lives, which range between three and twenty years, on either a straight-line basis over the expected period of benefit or as revenues are earned from the sales of the related products. We periodically review the estimated useful lives of our intangible assets and review such assets for impairment whenever events or changes in circumstances indicate that the carrying value of the assets may not be recoverable. Our determination of impairment is based on estimates of future cash flows. If an intangible asset is considered to be impaired, the amount of the impairment will equal the excess of the carrying value over the fair value of the asset.
Acquired IP R&D is not amortized until completion and development of the project, at which time the IP R&D becomes an amortizable asset with an appropriate useful life and an amortization method is determined. If the related project is not completed in a timely manner or the project is terminated or abandoned, we may have an impairment related to the IP R&D, calculated as the excess of the asset’s carrying value over its fair value.
Our policy defines IP R&D as the value assigned to those projects for which the related products have not received regulatory approval and have no alternative future use. Determining the portion of the purchase price allocated to IP R&D requires us to make significant estimates. The amount of the purchase price allocated to IP R&D is determined by estimating the future cash flows of each project or technology and discounting the net cash flows back to their present values. The discount rate used is determined at the time of measurement in accordance with accepted valuation methods. These methodologies include consideration of the risk of the project not achieving commercial feasibility.
Goodwill and other intangible assets that have indefinite useful lives are not amortized, but rather, are tested for impairment annually or more frequently if impairment indicators arise. Goodwill represents the excess of the purchase price over the fair value of the net tangible and identifiable intangible assets acquired in each business combination. Goodwill and intangible assets have been recorded at either incurred or allocated cost. Allocated costs were based on respective fair market values at the date of acquisition.
For goodwill, the impairment test requires a comparison of the estimated fair value of the reporting unit to which the goodwill is assigned to the sum of the carrying value of the assets and liabilities of that unit. If the sum of the carrying value of the assets and liabilities of a reporting unit exceeds the fair value of the reporting unit, the carrying value of the reporting unit’s goodwill is reduced to its implied fair value through an adjustment to the goodwill balance, resulting in an impairment charge. Our determination of impairment is based on estimates of future cash flows. We consider our business to be a single operating segment entity – the development, manufacture and sale on a global basis of medical devices for vascular access, surgery, peripheral vascular disease and oncology.
Inventories
Inventories are stated at the lower of cost (using the first-in, first-out method) or market. Appropriate consideration is given to deterioration, obsolescence, expiring and other factors in evaluating net realizable value.
Results of Operations for the years ended May 31, 2016 and 2015
For the fiscal year ended May 31, 2016, we reported a net loss of $(43.6) million, or $(1.21) loss per diluted share, on net sales of $353.9 million compared to a fiscal 2015 net loss of $(3.4) million, or $(0.09) loss per diluted share, on net sales of $356.5 million.
Net Sales
Net sales - Net sales are derived from the sale of our products and related freight charges, less discounts and returns.
Net sales for the year ended May 31, 2016 and 2015 were:
34
For the years ended May 31, | |||||||||||||
2016 | 2015 | % Growth | Currency Impact (Pos) Neg | Constant Currency Growth | |||||||||
Net Sales by Product Category | |||||||||||||
Peripheral Vascular | $ | 202,780 | $ | 192,713 | 5% | ||||||||
Vascular Access | 99,375 | 107,754 | -8% | ||||||||||
Oncology/Surgery | 48,895 | 51,890 | -6% | ||||||||||
Total Excluding Supply Agreement | 351,050 | 352,357 | 0% | 0% | 0% | ||||||||
Supply Agreement | 2,840 | 4,177 | -32% | 0% | -32% | ||||||||
Total | $ | 353,890 | $ | 356,534 | -1% | 1% | 0% | ||||||
Net Sales by Geography | |||||||||||||
United States | $ | 283,519 | $ | 280,611 | 1% | 0% | 1% | ||||||
International | 67,531 | 71,746 | -6% | 4% | -2% | ||||||||
Supply Agreement | 2,840 | 4,177 | -32% | 0% | -32% | ||||||||
Total | $ | 353,890 | $ | 356,534 | -1% | 1% | 0% | ||||||
For year ended May 31, 2016, net sales decreased $(2.6) million to $353.9 million compared to the year ended May 31, 2015. As shown in the table above, while consolidated net sales decreased by 1%, excluding the planned reduction in sales under our supply agreement and the negative impact from fluctuations in currency exchange rates, our sales were flat year over year. The decline in net sales from vascular access and oncology surgery was partially offset by 5% year over year growth in our peripheral vascular franchise. Our international sales were significantly impacted by unfavorable movement in currency exchange rates, particularly the Euro, British pound and Canadian dollar.
Peripheral Vascular sales increased $10.1 million primarily attributable to increased sales of AngioVac, Core and Venus products. While Vascular Access sales decreased $8.4 million primarily in our non-BioFlo businesses, our BioFlo line of products continued to gain traction in the marketplace. Oncology/Surgery sales decreased $3.0 million primarily due to fewer capital sales across all product lines compared to prior year. This was partially offset by increases in the sales of disposables in our Microwave and NanoKnife product lines.
U.S. sales increased $2.9 million due to growth in the Peripheral Vascular products, offset by a reduction in Vascular Access and Oncology/Surgery sales. While total US Vascular Access sales declined by $6.2 million overall, we saw growth in our U.S. BioFlo product lines of 18% year over year. U.S. Oncology/Surgery declined by $1.6 million, driven primarily through lower capital sales offset by growth in disposables. International sales decreased 2% on a constant-currency basis, due to a decline in Thermal Ablation and in the Vascular Access product lines.
Our supply agreement arrangement, which we do not include in either the U.S. or International geographic sales, declined by $1.3 million as we continue to wind down that relationship.
35
Gross Profit, Operating expenses, and Other income (expense)
For the year ended May 31, | |||||||||||
2016 | 2015 | % Change | |||||||||
Gross profit | $ | 174.3 | $ | 175.8 | -0.9 | % | |||||
Gross profit % of sales | 49.3 | % | 49.3 | % | |||||||
Research and development | $ | 25.1 | $ | 26.6 | -5.6 | % | |||||
% of sales | 7.1 | % | 7.5 | % | |||||||
Selling and marketing | $ | 84.7 | $ | 83.2 | 1.8 | % | |||||
% of sales | 23.9 | % | 23.3 | % | |||||||
General and administrative | $ | 29.6 | $ | 29.2 | 1.4 | % | |||||
% of sales | 8.4 | % | 8.2 | % | |||||||
Medical device excise tax | $ | 2.4 | $ | 4.1 | -41.5 | % | |||||
% of sales | 0.7 | % | 1.2 | % | |||||||
Gross profit - Gross profit consists of net sales less the cost of goods sold, which includes the costs of materials, products purchased from third parties and sold by us, manufacturing personnel, royalties, freight, business insurance, depreciation of property and equipment and other manufacturing overhead. The $(1.5) million decrease compared to 2015 is primarily attributable to a $5.9 million charge related to the write-off of Celerity inventory on hand and hardware assets after the business decision to no longer pursue the Celerity Navigation project. The prior year gross profit included a $4.8 million charge related to the voluntary Morpheus recall.
Research and development expenses - Research and development (“R&D”) expenses include internal and external costs to develop new products, enhance existing products, validate new and enhanced products, manage clinical, regulatory and medical affairs. The decrease in R&D costs for the year ended May 31, 2016 is due to reductions in project spend and restructuring. As a percentage of net sales, R&D expenses were 7.1% for fiscal 2016, compared to 7.5% for fiscal 2015.
Sales and marketing expenses - Sales and marketing (“S&M”) expenses consist primarily of salaries, commissions, travel and related business expenses, attendance at medical society meetings, product promotions and marketing activities. Increases in S&M expense for the year ended May 31, 2016 is the result of investments made in the US sales force focused around retention and improved sales performance along with an increase in credit card fees. As a percentage of net sales, S&M expenses were 23.9% for fiscal 2016 compared to 23.3% for fiscal 2015.
General and administrative expenses - General and administrative (“G&A”) expenses include executive management, finance, information technology, human resources, business development, legal, and the administrative and professional costs associated with those activities. Increases in G&A expenses for the year ended May 31, 2016 are primarily the result of increased legal and audit fees.
Medical device excise tax - Medical device excise tax is assessed on our U.S. product sales subject to exclusions and adjustments. The decrease as compared to the prior year is attributable to the suspension of the medical device excise tax as of January 1, 2016.
For the year ended May 31, | ||||||||||||
2016 | 2015 | $ Change | ||||||||||
Amortization of intangibles | $ | 18.0 | $ | 18.0 | $ | — | ||||||
Change in fair value of contingent consideration | $ | 0.9 | $ | (8.1 | ) | $ | 9.0 | |||||
Acquisition, restructuring and other items, net | $ | 12.6 | $ | 26.3 | $ | (13.7 | ) | |||||
Other expense | $ | (4.3 | ) | $ | (4.7 | ) | $ | 0.4 | ||||
Amortization of intangibles - Amortization of intangibles for the year ended May 31, 2016 increased primarily related to intangible asset amortization associated with the Merz intangibles acquired as part of the agreement that was entered into in Q4 2016.
36
Change in fair value of contingent consideration - Represents changes in contingent consideration driven by changes to estimated future payments on earn-out liabilities created through acquisitions and amortization of present value discounts on long-term contingent consideration. The decrease from the prior year is due to a $10.5 million gain recognized as a result of reducing the estimated present value of future payments due on earn-outs in the prior year compared to $1 million in gains in 2016. These gains were partially offset in each period by amortization of the present value discount on the contingent liabilities.
Acquisition, restructuring and other items, net - Expense for fiscal 2016 consists of $7.5 million of litigation expense, $2.5 million of M&A related expenses, $1.9 million of severance, $0.7 million of a gain related to the modification of stock based compensation awards for the former CEO and $1.0 million of accelerated depreciation associated with our operational excellence program, and other miscellaneous items.
Other expenses - Other expenses include interest expense, foreign currency impacts, bank fees, and amortization of deferred financing costs. The increase in other expenses was primarily related to foreign currency losses.
For year ended May 31, | ||||||||
2016 | 2015 | |||||||
Income tax expense (benefit) | $ | 40.3 | $ | (4.7 | ) | |||
Effective tax rate including discrete items | (1,240 | )% | 58 | % | ||||
Income tax provision (benefit) - Our effective tax rate was (1,240)% for fiscal 2016 compared with 58% for the prior year. The current year rate reflects expense of $40.4 million related to a full valuation allowance on our US net deferred tax assets. The prior year rate reflects the benefit of $9.2 million nontaxable adjustment to the contingent liabilities related to Vortex Medical and Clinical Devices, and a seven month benefit from the R&D tax credit that expired on December 31, 2014, offset by non-deductible interest expense related to contingent payments, true-ups of our fiscal year 2014 US income tax returns and the impact of the elimination of the ASC 718 APIC pool.
At May 31, 2016, we had a net deferred tax liability of $21.7 million, after recording a valuation allowance of $42.2 million. The increase in the valuation allowance was $40.4 million.
While the net deferred tax asset at May 31, 2016 before the valuation allowance was $19.9 million, the Company was required to record a valuation allowance of $40.4 million due to deferred tax liabilities related to intangibles of $20.5 million that have an indefinite reversal period and can not be used to support the deferred tax asset.
A valuation allowance is provided if based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. The Company’s analysis of the need for a valuation allowance considered that the Company has incurred a cumulative loss in the U.S. over the three year period ending May 31, 2016. A majority of the cumulative loss has been caused by the charges associated with the product recall and discontinuance and the impairment of fixed and intangible assets recorded in the quarter end February 28, 2015, From the time when the Company initially incurred a three year cumulative loss in the quarter ended February 28, 2015, and in each subsequent quarter through the quarter ended February 28, 2016, the Company could still demonstrate a recent history of core earnings , and had anticipated a return to profitability for the full fiscal year 2016. However, in the quarter ended May 31, 2016 the Company did not return to profitability for the full fiscal year and could no longer demonstrate a recent history of core earnings. Consequently after careful consideration and weighing of all the available positive and negative evidence, the weight given to the three year cumulative loss and lack of a recent history of core earnings was difficult to overcome and a full valuation allowance related to the U.S. deferred tax assets was established. Management will continue to reevaluate the positive and negative evidence at each reporting period and if future results as projected in the U.S. and our tax planning strategies are favorable, the valuation allowance may be removed, which could have a favorable material impact on our results of operations in the period in which it is recorded.
Results of Operations for the years ended May 31, 2015 and 2014
For the fiscal year ended May 31, 2015, we reported net loss of $(3.4) million, or $(0.09) loss per basic and diluted common share, on net sales of $356.5 million compared to a fiscal 2014 net income of $2.3 million, or $0.07 per basic and diluted common share, on net sales of $354.4 million.
37
Net Sales
Net sales - Net sales are derived from the sale of our products and related freight charges, less discounts and returns.
Net sales for the year ended May 31, 2015 and 2014 were:
For the years ended May 31, | |||||||||||||
2015 | 2014 | % Growth | Currency Impact (Pos) Neg | Constant Currency Growth | |||||||||
Net Sales by Product Category | |||||||||||||
Peripheral Vascular | $ | 192,713 | $ | 192,626 | 0% | ||||||||
Vascular Access | 107,754 | 106,394 | 1% | ||||||||||
Oncology/Surgery | 51,890 | 49,360 | 5% | ||||||||||
Total Excluding Supply Agreement | 352,357 | 348,380 | 1% | 1% | 2% | ||||||||
Supply Agreement | 4,177 | 6,045 | -31% | 0% | -31% | ||||||||
Total | $ | 356,534 | $ | 354,425 | 1% | 0% | 1% | ||||||
Net Sales by Geography | |||||||||||||
United States | $ | 280,611 | $ | 280,161 | 0% | 0% | 0% | ||||||
International | 71,746 | 68,219 | 5% | 4% | 9% | ||||||||
Supply Agreement | 4,177 | 6,045 | -31% | 0% | -31% | ||||||||
Total | $ | 356,534 | $ | 354,425 | 1% | 0% | 1% | ||||||
For year ended May 31, 2015, net sales increased $2.1 million to $356.5 million compared to the year ended May 31, 2014. As shown in the table above, while consolidated net sales increased by 1%, excluding the planned reduction in sales under our supply agreement and a negative impact from fluctuations in currency exchange rates, our sales increased 2% year over year. Growth was driven by our international business, which grew 9% excluding the negative impact of currency. Sales were significantly impacted by unfavorable movement in currency exchange rates, particularly the Euro, British pound and Canadian dollar, with the remainder of the changes as compared to the prior year driven by volume.
From a product line perspective, Peripheral Vascular sales increased $0.1 million primarily attributable to increased sales of AngioVac and Fluid Management products, offset by declines in our venous business and a negative impact of currency rate movements noted above. Vascular access sales increased $1.4 million as our BioFlo line of products continued to gain traction in the marketplace, offset by the voluntary recall and discontinuance of our Morpheus product line in the third fiscal quarter. Oncology/Surgery sales increased $2.5 million primarily due to the performance of NanoKnife products, particularly in the international markets.
From a geographic perspective, U.S. sales increased $0.5 million due to growth in the Peripheral Vascular and Vascular Access products, offset by a reduction in Oncology/Surgery sales. While total U.S. Oncology/Surgery declined by $0.9 million, our U.S. NanoKnife sales growth exceeded 10%. International sales increased 9% on a constant-currency basis, with the largest driver being NanoKnife.
Our supply agreement arrangement, which we do not include in either the U.S. or International geographic sales, declined by $1.9 million as we continue to wind down that relationship.
38
Gross Profit, Operating expenses, and Other income (expense)
For the year ended May 31, | |||||||||||
2015 | 2014 | % Change | |||||||||
Gross profit | $ | 175.8 | $ | 180.2 | -2.4 | % | |||||
Gross profit % of sales | 49.3 | % | 50.8 | % | |||||||
Research and development | $ | 26.6 | $ | 28.1 | -5.3 | % | |||||
% of sales | 7.5 | % | 7.9 | % | |||||||
Selling and marketing | $ | 83.2 | $ | 85.3 | -2.5 | % | |||||
% of sales | 23.3 | % | 24.1 | % | |||||||
General and administrative | $ | 29.2 | $ | 26.9 | 8.6 | % | |||||
% of sales | 8.2 | % | 7.6 | % | |||||||
Medical device excise tax | $ | 4.1 | $ | 3.8 | 7.9 | % | |||||
% of sales | 1.2 | % | 1.1 | % | |||||||
Gross profit - Gross profit consists of net sales less the cost of goods sold, which includes the costs of materials, products purchased from third parties and sold by us, manufacturing personnel, royalties, freight, business insurance, depreciation of property and equipment and other manufacturing overhead. The $4.4 million decrease compared to 2014 is mostly attributable to a $4.8 million charge related to Morpheus PICC inventory on hand at the time of the product discontinuance. Further decreases were the result of currency exchange fluctuations which negatively impacted our sales with minimal reduction to our cost of sales. These headwinds were partially offset by product cost reductions generated by our active Operational Excellence program and favorable shifts in product mix.
Research and development expenses - Research and development (“R&D”) expenses include internal and external costs to develop new products, enhance existing products, validate new and enhanced products, manage clinical, regulatory and medical affairs. The decrease in R&D costs for the year ended May 31, 2015 relates to savings generated by restructuring activities in fiscal 2015.
Sales and marketing expenses - Sales and marketing (“S&M”) expenses consist primarily of salaries, commissions, travel and related business expenses, attendance at medical society meetings, product promotions and marketing activities. Decreases in S&M expense for the year ended May 31, 2015 is the result of a reorganization of our international sales organization, combined with the impact of attrition in our U.S. sales force. While the U.S. sales force attrition benefited our operating expenses in fiscal 2015, retaining our sales employees is important to our long-term revenue growth and operating results.
General and administrative expenses - General and administrative (“G&A”) expenses include executive management, finance, information technology, human resources, business development, legal, and the administrative and professional costs associated with those activities. Increases in G&A expenses for the year ended May 31, 2015 are primarily the result of higher depreciation and maintenance expenses particularly as a result of the ERP implementation in the prior year and increased incentive and stock-based compensation costs.
Medical device excise tax - Medical device excess tax is assessed on our U.S. product sales subject to exclusions and adjustments. The slight increase as compared to the prior year is attributable to the mix of taxable products within the U.S. market.
For the year ended May 31, | ||||||||||||
2015 | 2014 | $ Change | ||||||||||
Amortization of intangibles | $ | 18.0 | $ | 16.6 | $ | 1.4 | ||||||
Change in fair value of contingent consideration | $ | (8.1 | ) | $ | (1.9 | ) | $ | (6.2 | ) | |||
Acquisition, restructuring and other items, net | $ | 26.3 | $ | 10.9 | $ | 15.4 | ||||||
Other expense | $ | (4.7 | ) | $ | (5.3 | ) | $ | 0.6 | ||||
39
Amortization of intangibles - Amortization of intangibles for the year ended May 31, 2015 increased primarily related to intangible asset amortization associated with our AngioVac technologies.
Change in fair value of contingent consideration - represents changes in contingent consideration driven by changes to estimated future payments on earn-out liabilities created through acquisitions and amortization of present value discounts on long-term contingent consideration. Fiscal 2015 included $10.5 million in gains recognized as a result of reducing the estimated present value of future payments due on earn-outs. These gains were partially offset in each period by amortization of the present value discount on the contingent liabilities.
Acquisition, restructuring and other items, net - Expense for fiscal 2015 consists of $9.1 million of fixed and long-term asset impairments, $6.4 million of impairment on the NAMIC trademark, other costs associated with litigation, the recall of Morpheus, our operational excellence program, and other miscellaneous items. The impairment charges were primarily driven by a change in strategy within our fluid management product development pipeline, as we moved away from our planned design of an Automated Power Injector.
Other expenses - Other expenses include interest expense, foreign currency impacts, bank fees, and amortization of deferred financing costs. Expenses were consistent year over year in amount and composition.
For year ended May 31, | ||||||||
2015 | 2014 | |||||||
Income tax expense (benefit) | $ | (4.7 | ) | $ | 2.8 | |||
Effective tax rate including discrete items | 58 | % | 54 | % | ||||
Income tax provision (benefit) - Our effective tax rate was 58% for fiscal 2015 compared with 54% for the prior year. The current year rate reflects the benefit of the $9.2 million nontaxable adjustment to the contingent liabilities related to Vortex Medical and Clinical Devices, and a seven month benefit from the R&D tax credit that expired on December 31, 2014, offset by non-deductible interest expense related to contingent payments, true-ups of our fiscal year 2014 US income tax returns and the impact of the elimination of the ASC 718 APIC pool. The prior year rate reflects the benefit of the $5.0 million nontaxable adjustment to the contingent liability related to Vortex Medical, Inc., offset by the impact of a New York State tax law change that resulted in a $1.2 million net write off of tax assets, non-deductible interest expense related to contingent payments, a seven month benefit from the R&D tax credit that expired on December 31, 2013, true ups of our fiscal year 2013 US income tax returns and the impact of the elimination of the ASC 718 APIC pool.
Liquidity and Capital Resources
Our cash and cash equivalents totaled $32.3 million as of May 31, 2016, compared with $18.4 million as of May 31, 2015. Marketable securities totaled $1.7 million and $1.7 million as of years ended May 31, 2016 and May 31, 2015, respectively, and consist of auction rate securities. As of May 31, 2016, total debt was $121.4 million comprised of a term loan and revolving credit facility. The fair value of contingent consideration payments as of May 31, 2016 was $38.3 million.
The table below summarizes our cash flows for the years ended May 31, 2016, 2015 and 2014:
For the year ended May 31, | |||||||||||
2016 | 2015 | 2014 | |||||||||
Cash provided by (used in): | |||||||||||
Operating activities | $ | 45,216 | $ | 25,685 | $ | 24,681 | |||||
Investing activities | (7,569 | ) | (12,736 | ) | (16,448 | ) | |||||
Financing activities | (23,663 | ) | (10,465 | ) | (14,016 | ) | |||||
Effect of exchange rate changes on cash and cash equivalents | (42 | ) | (198 | ) | 86 | ||||||
Net change in cash and cash equivalents | $ | 13,942 | $ | 2,286 | $ | (5,697 | ) | ||||
Cash provided by operating activities during the twelve months ended May 31, 2016 and 2015 was primarily the result of net loss adjusted for non-cash items offset by favorable shifts in working capital in 2016 compared to unfavorable shifts in working capital in 2015. In the current year, DSO improvement and inventory management, coupled with reductions in payables and accrued expenses contributed to $12.9 million improvement in operating activities. The $12.9 million improvement includes approximately $4.8 million in non-cash changes to inventory reserves.
40
The net cash used in investing activities for the current year consisted of $2.3 million in fixed asset additions as a result of the agreement with EmboMedics, $2.0 million in warrant additions and $3.3 million in intangible asset additions related to the Merz Distribution Agreement. The prior year use of cash consisted primarily of $11.4 million in fixed asset additions, a large portion of which is associated with facility investments, and $1.4 million in intangible asset additions.
The net cash used in financing activities is the result of $9.9 million in payments on contingent liabilities and $16.3 million of payments on our credit facility, partially offset by $2.4 million of stock option and ESPP activity proceeds.
We believe that our current cash and investment balances, together with cash generated from operations and our remaining revolving credit facility capacity of $63.6 million as of May 31, 2016, will provide sufficient liquidity to meet our anticipated needs for capital for at least the next 12 months. If we seek to make significant acquisitions of other businesses or technologies in the future for cash, we may require external financing.
Our contractual obligations as of May 31, 2016 are set forth in the table below (in thousands). We have no variable interest entities or other off-balance sheet obligations.
Cash Payments Due By Period as of May 31, 2016 | |||||||||||||||||||
Total | Less than One Year | 1-3 Years | 3-5 Years | After 5 Years | |||||||||||||||
Contractual Obligations: | |||||||||||||||||||
Long term debt and interest | $ | 125,428 | $ | 18,369 | $ | 107,059 | $ | — | $ | — | |||||||||
Operating leases(1) | 8,884 | 2,183 | 5,766 | 935 | — | ||||||||||||||
Purchase obligations(1) | 50,949 | 13,805 | 28,435 | 8,709 | — | ||||||||||||||
Acquisition-related future obligations (2) | 40,844 | 13,359 | 10,958 | 7,737 | 8,790 | ||||||||||||||
Other | 1,000 | 167 | 501 | 332 | — | ||||||||||||||
$ | 227,105 | $ | 47,883 | $ | 152,719 | $ | 17,713 | $ | 8,790 | ||||||||||
(1) | The non-cancelable operating leases and inventory purchase obligations are not reflected on our consolidated balance sheets under accounting principles generally accepted in the United States of America. |
(2) | Acquisition-related future obligations include scheduled minimum payments and contingent payments based upon achievement of performance measures or milestones such as sales or profitability targets, the achievement of research and development objectives or the receipt of regulatory approvals. The amount represents the undiscounted value of contingent liabilities recorded on the balance sheet. Timing of payments are as contractually scheduled, or where contingent, the Company's best estimate of payment timing. |
Recent Accounting Pronouncements
Refer to Note A for Recently issued Accounting Pronouncements.
Item 7A. | Quantitative and Qualitative Disclosures about Market Risk. |
We are exposed to market risk from changes in currency exchange rates, as well as interest rate fluctuations on our credit facility and investments that could impact our results of operations and financial position.
We transact sales in currencies other than the U.S. Dollar, particularly the Euro, British pound and Canadian dollar. Approximately 7% of our sales in fiscal 2016 were denominated in foreign currencies. We do not have expenses denominated in foreign currencies at the level of our sales and as a result, our profitability is exposed to currency fluctuations. When the U.S. Dollar strengthens, our sales and gross profit will be negatively impacted. In addition, we have assets and liabilities denominated in non-functional currencies which are remeasured at each reporting period, with the offset to changes presented as a component of Other Income (Expenses). Significant non-functional balances include a Euro-denominated contingent liability and accounts receivable due from a sub-section of our international customers.
In June 2012, we entered in an interest rate swap agreement, with an initial notional amount of $100 million, to limit the effect of variability due to interest rates on our debt. The swap agreement, which qualifies for hedge accounting under authoritative guidance, is a contract to exchange floating interest rate payments for fixed interest rate payments of 3.26% of the
41
outstanding balance of loan over the life of the swap agreement without the exchange of the underlying notional amounts. The Swap matured in May 2016. We do not currently engage in any other hedging or market risk management tools.
On September 19, 2013, we entered into a Credit Agreement (the "Credit Agreement") which provides for a $100 million senior secured term loan facility (“ Term Loan") and a $100 million senior secured revolving credit facility (the “Revolving Facility", and together with the Term Loan, the "Facilities"). Interest on both the Term Loan and Revolver is based on a base rate or Eurodollar rate plus an applicable margin which increases as our total leverage ratio increases, with the base rate and Eurodollar rate having ranges of 0.50% to 1.25% and 1.50% to 2.25% respectively. In the event of default, the interest rate may be increased by 2.0%. Changes in the interest rate would not be material.
Our excess cash is invested in highly liquid, short-term, investment grade securities with maturities primarily of less than two years. These investments are not held for speculative or trading purposes. Changes in interest rates may affect the investment income we earn on cash, cash equivalents and marketable securities and therefore affect our cash flows and results of operations. We hold investments in auction rate securities (“ARS”) in order to generate higher than typical money market investments. ARS typically are high credit quality, generally issued with municipal bond insurance. Credit risks are eased by the historical track record of bond insurers, which back a majority of this market. Sell orders for any security traded through an auction process could exceed bids. Such instances are usually the result of a drastic deterioration of issuer credit quality. Should there be a failed auction, we may be unable to liquidate our position in the securities in the near term. We have $1.7 million in investments in two auction rate securities issued by New York state and local government authorities that have failed auctions. The authorities are current in their interest payments on the securities.
Item 8. | Financial Statements and Supplementary Data. |
Financial statements and supplementary data required by Part II, Item 8 are included in Part IV of this report as indexed as Item 15 (a) (1) and (2) of this report, and are incorporated by reference into this Item 8.
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. |
None.
Item 9A. | Controls and Procedures. |
Evaluation of disclosure controls and procedures
As of the end of the period covered by this report, our management, under the supervision and with the participation of our Chief Executive Officer and our Interim Chief Financial Officer, evaluated the effectiveness of the design and operation of our disclosure controls and procedures pursuant to Rule 13a-15(b) of the Securities Exchange Act of 1934, as amended. Based on that evaluation, the Chief Executive Officer and the Interim Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within the time periods specified in the SEC rules and forms and is accumulated and communicated to management, including our Chief Executive Officer and Interim Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting for our company. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Securities Exchange Act of 1934, as amended, as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States and includes those policies and procedures that:
• | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
• | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States, and that our receipts and |
42
expenditures are being made only in accordance with authorizations of our management and members of our board of directors; and
• | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management has assessed the effectiveness of our internal control over financial reporting as of May 31, 2016. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on this evaluation, management concluded that our internal control over financial reporting was effective as of May 31, 2016.
The effectiveness of our internal control over financial reporting as of May 31, 2016 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears herein.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting for the fiscal quarter ended May 31, 2016 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. | Other Information. |
None.
43
Part III
Certain information required by Part III is omitted from this annual report on Form 10-K because we will file a definitive proxy statement within 120 days after the end of our fiscal year end pursuant to Regulation 14A (the “Proxy Statement”) for our annual meeting of Stockholders, currently scheduled for October 2016. The information included in the Proxy Statement under the respective headings noted below is incorporated herein by reference.
Item 10. | Directors, Executive Officers and Corporate Governance. |
Information required in this annual report on Form 10-K with respect to Executive Officers is contained in the discussion titled “Executive Officers of the Company” in Part I of this annual report on Form 10-K. The balance of the information required by Item 10 is incorporated herein by reference to our Proxy Statement under the heading “Election of Directors”.
Item 11. | Executive Compensation. |
The information required by Item 11 is incorporated herein by reference to our Proxy Statement under the heading “Executive Compensation”.
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The information required by this caption is incorporated herein by reference to our Proxy Statement under the heading “Ownership of Securities”.
Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
The information required by this caption is incorporated herein by reference to our Proxy Statement under the heading “Certain Relationships and Related Transactions”.
Item 14. | Principal Accounting Fees and Services. |
The information required by this caption is incorporated herein by reference to our Proxy Statement under the headings “Audit Matters—Principal Accounting Fees and Services and—Policy on Audit Committee Pre-approval of Audit and Permissible Non-Audit Services of Independent Registered Public Accounting Firm”.
44
Part IV
Item 15. | Exhibits, Financial Statement Schedules. |
(a)(1) Financial Statements
The following consolidated financial statements and supplementary data of Registrant and its subsidiaries required by Part II, Item 8, are included in Part IV of this report:
47 | ||
48 | ||
49 | ||
50 | ||
51 | ||
52 | ||
54 | ||
(2) Financial Statement Schedules
The following consolidated financial statement schedule is included in Part IV of this report:
All other schedules are omitted because they are not applicable, or not required, or because the required information is included in the consolidated financial statements or notes thereto.
45
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of AngioDynamics, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, of comprehensive income (loss), of stockholders’ equity, and of cash flows present fairly, in all material respects, the financial position of AngioDynamics, Inc. and its subsidiaries (the Company) at May 31, 2016 and May 31, 2015, and the results of their operations and their cash flows for each of the three years in the period ended May 31, 2016 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the index appearing under Item 15(a)(2) presents fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also, in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of May 31, 2016, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements, on the financial statement schedule, and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
As discussed in Note A to the consolidated financial statements, the Company changed the manner in which it accounts for the classification of deferred taxes in the consolidated balance sheets due to the adoption of ASU 2015-17, Balance Sheet Classification of Deferred Taxes.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
August 1, 2016
46
AngioDynamics, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
Years ended | |||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | |||||||||
Net sales | $ | 353,890 | $ | 356,534 | $ | 354,425 | |||||
Cost of sales | 179,574 | 180,738 | 174,251 | ||||||||
Gross profit | 174,316 | 175,796 | 180,174 | ||||||||
Operating expenses | |||||||||||
Research and development | 25,053 | 26,594 | 28,124 | ||||||||
Sales and marketing | 84,723 | 83,220 | 85,305 | ||||||||
General and administrative | 29,603 | 29,162 | 26,902 | ||||||||
Amortization of intangibles | 17,964 | 17,966 | 16,562 | ||||||||
Change in fair value of contingent consideration | 948 | (8,096 | ) | (1,908 | ) | ||||||
Acquisition, restructuring and other items, net | 12,591 | 26,257 | 10,873 | ||||||||
Medical device excise tax | 2,416 | 4,142 | 3,829 | ||||||||
Total operating expenses | 173,298 | 179,245 | 169,687 | ||||||||
Operating income (loss) | 1,018 | (3,449 | ) | 10,487 | |||||||
Other (expenses) income | |||||||||||
Interest income | 11 | 4 | — | ||||||||
Interest expense | (3,396 | ) | (3,197 | ) | (3,656 | ) | |||||
Other expense | (886 | ) | (1,489 | ) | (1,645 | ) | |||||
Total other expenses, net | (4,271 | ) | (4,682 | ) | (5,301 | ) | |||||
Income (loss) before income tax expense (benefit) | (3,253 | ) | (8,131 | ) | 5,186 | ||||||
Income tax expense (benefit) | 40,337 | (4,743 | ) | 2,839 | |||||||
Net income (loss) | $ | (43,590 | ) | $ | (3,388 | ) | $ | 2,347 | |||
Earnings (loss) per share | |||||||||||
Basic | $ | (1.21 | ) | $ | (0.09 | ) | $ | 0.07 | |||
Diluted | $ | (1.21 | ) | $ | (0.09 | ) | $ | 0.07 | |||
Weighted average shares outstanding | |||||||||||
Basic | 36,161 | 35,683 | 35,136 | ||||||||
Diluted | 36,161 | 35,683 | 35,440 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
47
AngioDynamics, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(in thousands)
Years ended | |||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | |||||||||
Net income (loss) | $ | (43,590 | ) | $ | (3,388 | ) | $ | 2,347 | |||
Other comprehensive income (loss), before tax: | |||||||||||
Unrealized gain (loss) on marketable securities | (11 | ) | (120 | ) | (16 | ) | |||||
Unrealized gain (loss) on interest rate swap | 257 | 296 | (32 | ) | |||||||
Foreign currency translation gain (loss) | (112 | ) | (264 | ) | 442 | ||||||
Other comprehensive income (loss), before tax | 134 | (88 | ) | 394 | |||||||
Income tax benefit (expense) related to items of other comprehensive income (loss) | (92 | ) | (64 | ) | 18 | ||||||
Other comprehensive income (loss), net of tax | 42 | (152 | ) | 412 | |||||||
Total comprehensive income (loss), net of tax | $ | (43,548 | ) | $ | (3,540 | ) | $ | 2,759 | |||
The accompanying notes are an integral part of these consolidated financial statements.
48
AngioDynamics, Inc. and Subsidiaries
CONSOLIDATED BALANCE SHEETS
(in thousands, except share data)
May 31, 2016 | May 31, 2015 | ||||||
ASSETS | |||||||
Current Assets | |||||||
Cash and cash equivalents | $ | 32,333 | $ | 18,391 | |||
Marketable securities, at fair value | 1,653 | 1,689 | |||||
Accounts receivable, net of allowances of $4,372 and $3,043, respectively | 52,867 | 58,428 | |||||
Inventories | 55,370 | 67,388 | |||||
Prepaid income taxes | 788 | 770 | |||||
Prepaid expenses and other | 3,243 | 4,132 | |||||
Total current assets | 146,254 | 150,798 | |||||
Property, Plant and Equipment, net | 48,284 | 54,450 | |||||
Other Assets | 4,696 | 5,398 | |||||
Intangible Assets, net | 166,577 | 181,652 | |||||
Goodwill | 361,252 | 361,252 | |||||
Deferred Income Taxes, long term | — | 19,508 | |||||
Total Assets | $ | 727,063 | $ | 773,058 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current Liabilities | |||||||
Accounts payable | $ | 15,616 | $ | 23,048 | |||
Accrued liabilities | 21,896 | 18,109 | |||||
Income taxes payable | 46 | 439 | |||||
Current portion of long-term debt | 16,250 | 8,750 | |||||
Current portion of contingent consideration | 12,919 | 9,969 | |||||
Other current liabilities | — | 200 | |||||
Total current liabilities | 66,727 | 60,515 | |||||
Long-term Debt, net of current portion | 105,160 | 128,910 | |||||
Deferred Income Taxes, long term | 21,684 | 1,119 | |||||
Contingent Consideration, net of current portion | 25,356 | 37,415 | |||||
Other Long Term Liabilities | 908 | — | |||||
Total Liabilities | 219,835 | 227,959 | |||||
Commitments and Contingencies (Note O) | |||||||
Stockholders’ Equity | |||||||
Preferred stock, par value $.01 per share, 5,000,000 shares authorized; no shares issued and outstanding | — | — | |||||
Common stock, par value $.01 per share, 75,000,000 shares authorized; 36,420,403 and 36,043,725 shares issued and 36,278,098 and 35,901,420 shares outstanding at May 31, 2016 and May 31, 2015, respectively | 363 | 360 | |||||
Additional paid-in capital | 525,775 | 520,101 | |||||
Retained earnings | (16,015 | ) | 27,575 | ||||
Treasury stock, 142,305 shares, at cost | (2,104 | ) | (2,104 | ) | |||
Accumulated other comprehensive loss | (791 | ) | (833 | ) | |||
Total Stockholders' Equity | 507,228 | 545,099 | |||||
Total Liabilities and Stockholders' Equity | $ | 727,063 | $ | 773,058 | |||
The accompanying notes are an integral part of these consolidated financial statements.
49
AngioDynamics, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
Years ended May 31, 2016, May 31, 2015 and May 31, 2014
(in thousands, except share data)
Common Stock | Additional paid in capital | Retained earnings | Accumulated other comprehensive income (loss) | Treasury Stock | Total | ||||||||||||||||||||||||
Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||
Balance at May 31, 2013 | 35,060,351 | $ | 351 | $ | 500,554 | $ | 28,616 | $ | (1,093 | ) | (142,305 | ) | $ | (2,104 | ) | $ | 526,324 | ||||||||||||
Net loss | 2,347 | 2,347 | |||||||||||||||||||||||||||
Exercise of stock options | 105,676 | 1,085 | 1,085 | ||||||||||||||||||||||||||
Tax effect of exercise of stock options | (146 | ) | (146 | ) | |||||||||||||||||||||||||
Issuance of restricted shares, net | 129,702 | 1 | (1,358 | ) | (1,357 | ) | |||||||||||||||||||||||
Purchase of common stock under Employee Stock Purchase Plan | 146,275 | 1 | 2,717 | 2,718 | |||||||||||||||||||||||||
Stock-based compensation | 5,502 | 5,502 | |||||||||||||||||||||||||||
Other comprehensive income (loss), net of tax | 412 | 412 | |||||||||||||||||||||||||||
Balance at May 31, 2014 | 35,442,004 | $ | 353 | $ | 508,354 | $ | 30,963 | $ | (681 | ) | (142,305 | ) | $ | (2,104 | ) | $ | 536,885 | ||||||||||||
Net income | (3,388 | ) | (3,388 | ) | |||||||||||||||||||||||||
Exercise of stock options | 341,446 | 3 | 4,335 | 4,338 | |||||||||||||||||||||||||
Tax effect of exercise of stock options | — | ||||||||||||||||||||||||||||
Issuance of restricted shares, net | 141,274 | 2 | — | 2 | |||||||||||||||||||||||||
Purchase of common stock under Employee Stock Purchase Plan | 119,001 | 2 | 1,414 | 1,416 | |||||||||||||||||||||||||
Stock-based compensation | 5,998 | 5,998 | |||||||||||||||||||||||||||
Other comprehensive income (loss), net of tax | (152 | ) | (152 | ) | |||||||||||||||||||||||||
Balance at May 31, 2015 | 36,043,725 | $ | 360 | $ | 520,101 | $ | 27,575 | $ | (833 | ) | (142,305 | ) | $ | (2,104 | ) | $ | 545,099 | ||||||||||||
Net loss | (43,590 | ) | (43,590 | ) | |||||||||||||||||||||||||
Exercise of stock options | 101,040 | 1 | 1,296 | 1,297 | |||||||||||||||||||||||||
Tax effect of exercise of stock options | — | ||||||||||||||||||||||||||||
Issuance of restricted shares, net | 137,681 | 1 | (332 | ) | (331 | ) | |||||||||||||||||||||||
Purchase of common stock under Employee Stock Purchase Plan | 137,957 | 1 | 1,470 | 1,471 | |||||||||||||||||||||||||
Stock-based compensation | 3,240 | 3,240 | |||||||||||||||||||||||||||
Other comprehensive income (loss), net of tax | 42 | 42 | |||||||||||||||||||||||||||
Balance at May 31, 2016 | 36,420,403 | $ | 363 | $ | 525,775 | $ | (16,015 | ) | $ | (791 | ) | (142,305 | ) | $ | (2,104 | ) | $ | 507,228 | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
50
AngioDynamics, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
Years ended | |||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | |||||||||
Cash flows from operating activities: | |||||||||||
Net income (loss) | $ | (43,590 | ) | $ | (3,388 | ) | $ | 2,347 | |||
Adjustments to reconcile net income (loss) to net cash provided by operating activities: | |||||||||||
Depreciation and amortization | 28,115 | 29,861 | 28,329 | ||||||||
Amortization of acquired inventory basis step-up | — | — | 150 | ||||||||
Tax effect of exercise of stock options and issuance of performance shares | — | — | (146 | ) | |||||||
Deferred income tax provision | 39,983 | (5,123 | ) | 2,716 | |||||||
Stock based compensation | 3,240 | 5,998 | 5,502 | ||||||||
Changes in accounts receivable allowances | 2,377 | 1,448 | 465 | ||||||||
Change in fair value of contingent consideration | 948 | (8,096 | ) | (1,908 | ) | ||||||
Loss on impairment/disposal of long-term assets | 806 | 9,381 | — | ||||||||
Loss on impairment of intangible assets | 384 | 6,400 | — | ||||||||
Other | 90 | 181 | 130 | ||||||||
Changes in operating assets and liabilities, net of effects of acquisitions: | |||||||||||
Accounts receivable | 3,131 | 2,095 | (14,786 | ) | |||||||
Inventories | 11,976 | (5,648 | ) | (6,114 | ) | ||||||
Prepaid expenses and other | 712 | (1,170 | ) | 1,208 | |||||||
Accounts payable and accrued liabilities | (2,956 | ) | (6,254 | ) | 6,788 | ||||||
Net cash provided by operating activities | 45,216 | 25,685 | 24,681 | ||||||||
Cash flows from investing activities: | |||||||||||
Additions to property, plant and equipment | (2,326 | ) | (11,383 | ) | (11,172 | ) | |||||
Acquisition of businesses, net of cash acquired | — | — | (4,169 | ) | |||||||
Acquisition of intangible assets | (3,268 | ) | (1,353 | ) | (1,435 | ) | |||||
Acquisition of warrants | (2,000 | ) | — | — | |||||||
Purchases of marketable securities | — | — | (25 | ) | |||||||
Proceeds from sale or maturity of marketable securities | 25 | — | 353 | ||||||||
Net cash used in investing activities | (7,569 | ) | (12,736 | ) | (16,448 | ) | |||||
Cash flows from financing activities: | |||||||||||
Repayment of long-term debt | (16,250 | ) | (20,000 | ) | (146,250 | ) | |||||
Proceeds from issuance of and borrowings on long-term debt | — | 15,000 | 146,410 | ||||||||
Proceeds from exercise of stock options and ESPP | 2,437 | 5,757 | 2,444 | ||||||||
Payment of acquisition related contingent consideration | (9,850 | ) | (11,222 | ) | (15,943 | ) | |||||
Deferred financing costs on long-term debt | — | — | (677 | ) | |||||||
Net cash used in financing activities | (23,663 | ) | (10,465 | ) | (14,016 | ) | |||||
Effect of exchange rate changes on cash and cash equivalents | (42 | ) | (198 | ) | 86 | ||||||
Increase (decrease) in cash and cash equivalents | 13,942 | 2,286 | (5,697 | ) | |||||||
Cash and cash equivalents at beginning of year | 18,391 | 16,105 | 21,802 | ||||||||
Cash and cash equivalents at end of year | $ | 32,333 | $ | 18,391 | $ | 16,105 | |||||
The accompanying notes are an integral part of these consolidated financial statements.
51
AngioDynamics, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF CASH FLOWS—(Continued)
(in thousands)
Years ended | |||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | |||||||||
Supplemental disclosures of cash flow information: | |||||||||||
Supplemental disclosure of non-cash operating, investing and financing activities: | |||||||||||
Contractual obligations for acquisition of fixed assets | $ | 75 | $ | 140 | $ | 4,970 | |||||
Contractual obligations for acquisition of intangibles and business | — | — | 2,249 | ||||||||
Contractual obligations for tax basis adjustment | — | 779 | — | ||||||||
Cash paid during the period for: | |||||||||||
Interest | $ | 3,063 | $ | 3,151 | $ | 3,591 | |||||
Income taxes | 332 | 699 | 182 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
52
AngioDynamics, Inc. and Subsidiaries
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE A—BASIS OF PRESENTATION, BUSINESS DESCRIPTION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
1. Basis of Presentation and Description of Business
The consolidated financial statements include the accounts of AngioDynamics, Inc. and its wholly owned subsidiaries, (collectively, the “Company”). We design, manufacture and sell a wide range of medical, surgical and diagnostic devices used by professional healthcare providers for vascular access, for the treatment of peripheral vascular disease and in oncology and surgical settings. Our devices are generally used in minimally invasive, image-guided procedures. Most of our products are intended to be used once and then discarded, or they may be temporarily implanted for short- or long-term use.
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements. Estimates also affect reported amounts of sales and expenses during the reporting period. Actual results could differ from those estimates.
Assets and liabilities of non-U.S. dollar functional currency entities are translated to U.S. dollars at period-end exchange rates, and the resulting gains and losses arising from the translation of those net assets are recorded as a cumulative translation adjustment, a component of accumulated other comprehensive loss on the consolidated balance sheets.
All intercompany balances and transactions have been eliminated.
2. Cash and Cash Equivalents
We consider all unrestricted highly liquid investments purchased with an initial maturity of less than three months to be cash equivalents. We maintain cash and cash equivalent balances with financial institutions in the United States in excess of amounts insured by the Federal Deposit Insurance Corporation.
3. Marketable Securities
Marketable securities, which include auction rate investments, are classified as “available-for-sale securities” and are reported at fair value, with unrealized gains and losses excluded from operations and reported as a component of accumulated other comprehensive income (loss), net of the related tax effects, in stockholders’ equity. Cost is determined using the specific identification method. We hold investments in auction rate securities in order to generate higher than typical money market rate investment returns. Auction rate securities typically are high credit quality, generally achieved with municipal bond insurance. Credit risks are eased by the historical track record of bond insurers, which back a majority of this market. Sell orders for any security traded through an auction process could exceed bids and, in such cases, the auction fails and we may be unable to liquidate our position in the securities in the near term. As of May 31, 2016 and 2015, we had $1.7 million and $1.7 million, respectively, in investments in two auction rate securities issued by New York state and local government authorities that failed auctions. The authorities are current in their interest payments on the securities.
4. Accounts Receivable
Accounts receivable, principally trade, are generally due within 30 to 90 days and are stated at amounts due from customers, net of an allowance for sales returns and doubtful accounts. We perform ongoing credit evaluations of our customers and adjust credit limits based upon payment history and the customer’s current creditworthiness, as determined by a review of their current credit information. We continuously monitor aging reports, collections and payments from customers, and a provision for estimated credit losses is maintained based upon our historical experience and any specific customer collection issues that have been identified. While such credit losses have historically been within our expectations and the provisions established, we cannot guarantee that the same credit loss rates will be experienced in the future. We write off accounts receivable when they are determined to be uncollectible.
5. Inventories
Inventories are stated at the lower of cost (using the first-in, first-out method) or market. Appropriate consideration is given to deterioration, obsolescence, expiring and other factors in evaluating net realizable value.
53
6. Property, Plant and Equipment
Property, plant and equipment are stated at cost, less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the assets. Refer below for useful lives by category:
Estimated useful lives | ||
Building and building improvements | 39 years | |
Machinery and equipment | 3 to 8 years | |
Computer software and equipment | 3 to 10 years | |
We evaluate these assets for impairment periodically or as changes in circumstances or the occurrence of events suggest the remaining value is not recoverable. Expenditures for repairs and maintenance are charged to expense as incurred. Renewals and betterments are capitalized.
7. Goodwill and Intangible Assets
Intangible assets other than goodwill and acquired IP R&D are amortized over their estimated useful lives, which range between two and eighteen years, on either a straight-line basis over the expected period of benefit or as revenues are earned from the sales of the related products. We periodically review the estimated useful lives of our intangible assets and review such assets for impairment whenever events or changes in circumstances indicate that the carrying value of the assets is not recoverable. Our determination of impairment is based on estimates of future cash flows. If an intangible asset is considered to be impaired, the amount of the impairment will equal the excess of the carrying value over the fair value of the asset.
Acquired IP R&D has an indefinite life and is not amortized until completion of the development of the project, at which time the IP R&D becomes an amortizable asset. If the related project is not completed in a timely manner or the project is terminated or abandoned, we may have an impairment related to the IP R&D, calculated as the excess of the asset’s carrying value over its fair value.
Our policy defines IP R&D as the value assigned to those projects for which the related products have not received regulatory approval and have no alternative future use. Determining the portion of the purchase price allocated to IP R&D requires us to make significant estimates. The amount of the purchase price allocated to IP R&D is determined by estimating the future cash flows of each project or technology and discounting the net cash flows back to their present values. The discount rate used is determined at the time of measurement in accordance with accepted valuation methods. These methodologies include consideration of the risk of the project not achieving commercial feasibility.
Goodwill and other intangible assets that have indefinite useful lives are not amortized, but rather, are tested for impairment annually or more frequently if impairment indicators arise. Goodwill represents the excess of the purchase price over the fair value of the net tangible and identifiable intangible assets acquired in each business combination. Goodwill and intangible assets have been recorded at either incurred or allocated cost. Allocated costs were based on respective fair market values at the date of acquisition.
For goodwill, the impairment test requires a comparison of the estimated fair value of the reporting unit to which the goodwill is assigned to the sum of the carrying value of the assets and liabilities of that unit. If the sum of the carrying value of the assets and liabilities of a reporting unit exceeds the fair value of the reporting unit, the carrying value of the reporting unit’s goodwill is reduced to its implied fair value through an adjustment to the goodwill balance, resulting in an impairment charge. Our determination of impairment is based on estimates of future cash flows.
8. Contingent Consideration
The fair value of the liability for contingent consideration recorded on the acquisition date for a business combination is based on probability weighted estimated cash flow streams, discounted back to present value using a discount rate determined in accordance with accepted valuation methods. The liability for contingent consideration is remeasured to fair value at each reporting period with changes recorded in earnings until the contingency is resolved.
9. Revenue Recognition
We recognize revenue when the following four criteria has been met: (i) persuasive evidence that an arrangement exists; (ii) the price is fixed or determinable; (iii) collectability is reasonably assured; and (iv) product delivery has occurred or services have been rendered. We recognize revenue, net of sales taxes assessed by any governmental authority, as products are shipped, based on shipping terms, and when title and risk of loss passes to customers. We negotiate shipping and credit terms
54
on a customer-by-customer basis and products are shipped at an agreed upon price. All product returns must be pre-approved by us and customers may be subject to a 20% restocking charge. To be accepted, a returned product must be unadulterated, undamaged and have at least twelve months remaining prior to its expiration date. Charges for discounts, returns, rebates and other allowances are recognized as a deduction from revenue on an accrual basis in the period in which the revenue is recorded. The accrual for product returns, discounts and other allowances is based on the company’s history.
Shipping and handling costs, associated with the distribution of finished products to customers, are recorded in costs of goods sold and are recognized when the related finished product is shipped to the customer. Amounts charged to customers for shipping are recorded in net sales.
10. Research and Development
Research and development costs, including salaries, consulting fees, building costs, utilities and administrative expenses that are related to developing new products, enhancing existing products, validating new and enhanced products, managing clinical, regulatory and medical affairs are expensed as incurred.
11. Income Taxes
In preparing our financial statements, we calculate income tax expense for each jurisdiction in which we operate. This involves estimating actual current taxes due plus assessing temporary differences arising from differing treatment for tax and accounting purposes that are recorded as deferred tax assets and liabilities. We periodically evaluate deferred tax assets, capital loss carryforwards and tax credit carryforwards to determine their recoverability based primarily on our ability to generate future taxable income and capital gains. Where it is more-likely-than-not these will not be recovered, we estimate a valuation allowance and record a corresponding additional tax expense in our statement of operations.
We file income tax returns in the U.S. federal jurisdiction and various state and foreign jurisdictions. In the normal course of business we are subject to examination by taxing authorities throughout the world. Fiscal years 2012 through 2016 remain open to examination by the various tax authorities. New York State is currently auditing AngioDynamic’s franchise tax filings for 2011 through 2013, and we do not anticipate any material adjustments will result. We analyzed filing positions in all of the Federal and state jurisdictions where we are required to file income taxes, as well as all open tax years in these jurisdictions and believe that our income tax filing positions and deductions will be sustained on audit and we do not anticipate any adjustments will result in a material adverse effect on our financial condition, results of operations or cash flows.
12. Derivative Financial Instruments
We are exposed to market risks, including changes in interest rates. We periodically enter into certain derivative financial instruments to hedge the underlying economic exposure. The derivative instruments used are floating-to-fixed rate interest rate swaps, which are subject to hedge accounting treatment.
Derivative instruments are presented in the consolidated financial statements at their fair value. Changes in the fair value of derivative financial instruments are either recognized periodically in income or in stockholders’ equity as a component of accumulated other comprehensive income (loss) depending on whether the derivative financial instrument qualifies for hedge accounting and, if so, whether it qualifies as a fair value or cash flow hedge. Generally, the changes in the fair value of derivatives accounted for as fair value hedges are recorded in income along with the portions of the changes in the fair value of hedged items that relate to the hedged risks. Changes in the fair value of derivatives accounted for as cash flow hedges, to the extent they are effective as hedges, are recorded in accumulated other comprehensive income (loss).
13. Supplier Concentrations
We are dependent upon the ability of our suppliers to provide products on a timely basis and on favorable pricing terms. The loss of our principal suppliers or a significant reduction in product availability from these suppliers could have a material adverse effect on us. We believe that our relationships with these suppliers are satisfactory.
14. Recent Events
On March 31, 2016, Joseph DeVivo, former President and Chief Executive Officer, decided to pursue other interests outside of the Company and on April 4, 2016, James C. Clemmer was appointed as the new President and Chief Executive Officer. As part of the separation agreement with Joseph DeVivo, his stock options, restricted stock units and performance shares will continue to vest for one year.
55
On November 3, 2015, Mark Frost resigned as Executive Vice President and Chief Financial Officer (CFO). Michael Trimarchi, Vice President and Global Controller, assumed the responsibilities as principal accounting officer of the Company and interim CFO until his resignation on May 13, 2016. On July 22, 2016, Michael Greiner was appointed Executive Vice President and Chief Financial Officer of the Company, effective August 16, 2016. On July 27, 2016, Peter J. Kish was designated as the principal financial officer and principal accounting officer of the Company by the Board of Directors of the Company. Mr. Kish will serve in this role until Mr. Greiner begins his service as Chief Financial Officer on August 16, 2016.
On December 18, 2015, President Obama signed into law H.R. 2029, the “Consolidated Appropriations Act, 2016”, which includes a two-year moratorium on the medical device excise tax, effective January 1, 2016. The 2.3 percent tax on sales of medical devices (except certain devices sold at retail) was enacted as part of the Affordable Care Act in 2010 and applied to device sales beginning on January 1, 2013. Absent further legislative action, the tax will be automatically reinstated for medical device sales starting on January 1, 2018. As presented on our Consolidated Condensed Statement of Operations we have incurred $12.0 million cumulatively since the enactment of the tax on January 1, 2013 through the May 31, 2016. In the absence of this tax, the company will seek opportunities to further invest in growth drivers to create long-term shareholder value.
On November 17, 2015, the Company received a letter from the FDA closing out the warning letter the Company received from FDA in January 2011 regarding certain promotional activities related to the NanoKnife System. On November 25, 2015, the Company received letters from the FDA closing out the warning letters the Company received from FDA in May 2011 related to the Company’s Queensbury facility and in November 2014 related to the Company’s Glens Falls facility. These close out letters resolved all outstanding warning letters against the Company.
During the quarter ended May 31, 2016, we made the decision to discontinue the Celerity tip location and navigation product line. The discontinuance of the product line was the result of performance and quality issues with the product and a strategic shift to focus on other product within the Vascular Access business. We recorded a write-off of approximately $5.8 million of inventory and $0.1 million of hardware assets during the fourth quarter.
During the quarter ended May 31, 2016 we entered into an agreement with Merz North America where we became the exclusive sub-distributor of ASCLERA in the vein market in the United States and received the Merz customer list for the designated market territory. As part of the agreement we receive a personal, non-exclusive, non-transferable, non-assignable, non-sub licensable, license to use the trademarks, service marks and trades names from Merz. As a result of this agreement we recorded $3.3 million of intangible assets for the exclusive distribution rights and the customer lists obtained. The Asclera product is the replacement for Sotradecol.
Regulatory Matters
On May 27, 2011, we received a Warning Letter from the U.S. Food and Drug Administration ("FDA") in connection with its inspection of our Queensbury, NY manufacturing facility. In the Warning Letter, FDA cited deficiencies in the response letter we provided FDA pertaining to the inspection that occurred from January 4 to January 13, 2011. The deficiencies related to our internal procedures for medical device reporting, corrections and removals and complaint handling. We responded to the Warning Letter and completed corrective and preventive actions to address the observations noted.
In December 2011, we initiated a comprehensive Quality Call to Action Program to review and augment our Quality Management Systems at our Queensbury facility. To accelerate implementation of the program, we engaged a team of external regulatory and quality experts and reallocated a significant number of engineering and product development resources to support this corporate initiative. From inception of the Quality Call to Action Program through fiscal 2014, we have incurred $3.2 million in direct costs associated with the program.
On February 10, 2012, we received from FDA a Form 483, List of Investigational Observations, in connection with its inspection of our Queensbury facility from November 14, 2011 to February 10, 2012. The Form 483 contained 12 observations related to, among other things, our CAPA (Corrective and Preventive Action) system, MDR (Medical Device Reporting), complaint investigation, corrections and removals, acceptance criteria and training. Some of the observations contained in the Form 483 were repeat observations from the May 27, 2011 Warning Letter described above.
On February 13, 2012, we received from FDA a Form 483 in connection with its inspection of our Fremont facility from January 12, 2012 to February 13, 2012. The Form 483 contained six observations related to, among other things, our CAPA system, design controls, risk management and training. We provided responses to FDA within 15 business days of our receipt of the Form 483s.
56
On September 24, 2012, we received from FDA a Form 483 in connection with its subsequent inspection of our Queensbury, NY facility from September 6 to September 14, and September 19 to September 24. This re-inspection followed our response to the original Form 483 issued by FDA on February 13, 2012. The Form 483 contained 5 observations related to 510(k) decisions, complaint investigations, acceptance criteria, corrective and preventive actions and training. All but one of the observations in the Form 483 related to events that occurred before the date that we had indicated to FDA in our previous responses that our corrective and remediation activities related to our Quality Call to Action would be completed. We provided responses to FDA within 15 business days of our receipt of the Form 483.
On February 4, 2014, FDA completed a comprehensive follow-up inspection of our Queensbury facility. The inspection began on January 14, 2014 and resulted in FDA issuing a Form 483 containing one observation. The observation related to the inconsistency of certain complaint investigation elements in certain devices that have hardware and disposable components. The Form 483 observation was annotated to reflect that during the inspection we had corrected the issue, and this correction was verified by the inspector. In addition, we provided a response to FDA within 15 business days of our receipt of the Form 483. We believe that the results of this inspection validate that all of the Quality System and current Good Manufacturing Practice issues raised in the 483s described above have been fully addressed.
On March 31, 2014, FDA completed an inspection of our Glens Falls, NY facility. The inspection began on March 17, 2014 and resulted in FDA issuing a form 483 containing 3 observations. The observations were related to 1) inconsistency of a manufacturing product test process used among similar products, 2) a particular verification test of a product, and 3) non-conforming product control procedure. We responded to the FDA within 15 business days of the receipt of the Form 483.
During the fourth quarter of our fiscal year ended May 31, 2014, we received Certificate to Foreign Governments ("CFGs") from the FDA covering all Vascular Access and Peripheral Vascular products manufactured in our Queensbury facility. During the first quarter of our fiscal year ended May 31, 2016, we received CFGs for our NanoKnife product.
On November 5, 2014, we received a Warning Letter from the FDA relating to observations noted during FDA’s inspection of our Navilyst Medical facilities located in Marlborough, Massachusetts and Glens Falls, New York in 2014. The matters raised in the Warning Letter and observations focused on design control processes related to packaging validations and accelerated and real time aging testing in connection with our fluid management and PICC families of products, inconsistency of a manufacturing product test process used among similar valved PICC products, a particular verification test of valved PICC products and non-conforming product control procedures.
On November 17, 2015, we received a letter from the FDA closing out the warning letter we received from FDA in January 2011 regarding certain promotional activities related to the NanoKnife System. On November 25, 2015, we received letters from the FDA closing out the warning letters we received from FDA in May 2011 related to the Company’s Queensbury facility and in November 2014 related to the Company’s Glens Falls facility. These close out letters resolved all outstanding warning letters against the Company.
Recently Issued Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board ("FASB") issued ASU No. 2014-09, "Revenue from Contracts with Customers" ("ASU 2014-09"). ASU 2014-09 provides a single, comprehensive accounting model for revenues arising from contracts with customers that supersedes most of the existing revenue recognition guidance, including industry-specific guidance. Under this model, revenue is recognized at an amount that an entity expects to be entitled to upon transferring control of goods or services to a customer, as opposed to when risks and rewards transfer to a customer under existing revenue recognition guidance. ASU 2014-09 is effective for the Company beginning in its fiscal year 2018, and may be applied retrospectively to all prior periods presented or through a cumulative adjustment to the opening retained earnings balance in the year of adoption. The Company is currently in the process of evaluating the impact of ASU 2014-09 on its consolidated financial statements.
In June 2014, the FASB issued ASU 2014-12, Accounting for Share-Based Payments When the Terms of the Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period, that clarified that entities should treat performance targets that can be met after the requisite service period of a share-based payment award as performance conditions that affect vesting. Therefore, an entity would not record compensation expense related to an award for which transfer to the employee is contingent on the entity’s satisfaction of a performance target until it becomes probable that the performance target is met. This ASU is effective for the Company in its first quarter beginning after January 1, 2016 and did not have a material impact on the Company’s consolidated financial statements.
57
In April 2015, the FASB issued ASC Update No. 2015-03, Interest-Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs. Update No. 2015-03 requires debt issuance costs related to a recognized debt liability to be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. Update No. 2015-03 is effective for annual reporting periods beginning after December 15, 2015 and interim periods within those reporting periods. Early adoption is permitted for financial statements that have not been previously issued. This update is not expected to impact the results of our operations.
In July 2015, the FASB issued ASC Update No. 2015-11, Inventory (Topic 330): Simplifying the Measurement of Inventory. Update No. 2015-11 more closely aligns the measurement of inventory in U.S. GAAP with the measurement of inventory in International Financial Reporting Standards by requiring companies using the first-in, first-out and average costs methods to measure inventory using the lower of cost and net realizable value, where net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Update No. 2015-11 is effective for annual reporting periods beginning after December 15, 2016 and interim periods within those fiscal years. Update No. 2015-11 should be applied prospectively with earlier application permitted as of the beginning of an interim or annual reporting period. The adoption of Update No. 2015-11 is not expected to have a material impact on our financial position or results of operations.
In November 2015, the FASB issued ASC Update No. 2015-17, “Balance Sheet Classification of Deferred Taxes” as part of its simplification initiatives. This update requires deferred tax liabilities and assets to be classified as non-current on the consolidated condensed balance sheet for fiscal years beginning after December 15, 2016, and interim periods within those annual periods. Early application is permitted. An entity can elect to adopt prospectively or retrospectively to all periods presented. This update was applied retrospectively as of November 30, 2015. The current deferred tax asset balance of $4.4 million was classified as a non-current deferred tax asset for the period ended May 31, 2015 in the consolidated condensed balance sheet.
In January 2016, the FASB issued ASU 2016-01, Financial Instruments - Overall (Subtopic 825-10). Update No. 2016-01 addresses certain aspects of recognition, measurement, presentation and disclosure of financial instruments. Update No. 2016-01 is effective for annual reporting periods beginning after December 15, 2017 and interim periods within those fiscal years and early application is permitted. The adoption of Update No. 2016-01 is not expected to have a material impact on our financial position or results of operations.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842). ASU 2016-02 increases transparency and comparability among organizations by recognizing lease assets and liabilities on the balance sheet and disclosing key information about leasing arrangements. For leases with a term or twelve months or less, a lessee is permitted to make an accounting policy election by class of underlying asset not to recognize lease assets and liabilities. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018 and early application is permitted. The Company is currently in the process of evaluating the impact of ASU 2016-02 on its consolidated financial statements.
In March 2016, the FASB issued ASU 2016-09, Compensation - Stock Based Compensation (Topic 718: Improvements to Employee Share-Based Payment Accounting. ASU 2016-09 simplifies and improves various aspects of ASC 718 for share-based payments, including income tax items and the classification of these items on the statement of cash flows. ASU 2016-09 is effective for annual periods beginning after December 31, 2016 and early application is permitted. The Company is currently in the process of evaluating the impact of ASU 2016-09 on its consolidated financial statements.
58
NOTE B—OTHER ASSETS
On March 2, 2015, the Company filed an 8-K stating that it executed a non-binding letter of intent to enter into a strategic relationship with privately-held EmboMedics Inc., which develops injectable and resorbable embolic microspheres. On April 9, 2015, the Company entered into a License, Distribution, Manufacturing and Purchase Option Agreement with EmboMedics Inc, subject to certain approvals by EmboMedics shareholders. Under the terms of the agreement, AngioDynamics received an exclusive worldwide license to market and sell, upon regulatory clearances, EmboMedics’ microsphere technology. AngioDynamics has the ability to determine the manufacturing of the products.
On December 7, 2015, AngioDynamics made an initial $2.0 million purchase of non-transferable warrants in a subsidiary of EmboMedics which become exercisable upon a change of control of EmboMedics. The Company does not have significant influence, or control of the subsidiary. This initial investment is recorded at cost and the Company will review for impairment at each balance sheet date. The warrants are not exercisable at the original issue date or the balance sheet date as they only become exercisable upon a change of control, termination of the agreement or delivery of an offer notice. Based on the achievement of certain development activities, the Company will make an additional $5.0 million purchase of non-transferable warrants and an additional $4.0 million in milestone payments based on regulatory approvals. In the future, AngioDynamics could execute an exclusive option to acquire this subsidiary of EmboMedics.
59
NOTE C—FAIR VALUE OF FINANCIAL INSTRUMENTS
Our financial instruments include cash and cash equivalents, accounts receivable, marketable securities, accounts payable, interest rate swap agreement and contingent earn outs. The carrying amount of cash and cash equivalents, accounts receivable, and accounts payable approximates fair value due to the immediate or short-term maturities. The marketable securities and interest rate swap agreement has been recorded at its fair value based on a valuation received from an independent third party. The contingent earn out has been recorded at fair value using the income approach.
Fair value is the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. This policy establishes a fair value hierarchy which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. The policy describes three levels of inputs that may be used to measure fair value which are provided in the table below.
Level 1 | Quoted prices in active markets for identical assets or liabilities. Level 1 assets include money market funds that are traded in an active exchange market. |
Level 2 | Observable inputs other than Level 1 prices such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. When quoted market prices are unobservable, we obtain pricing information from an independent pricing vendor. The pricing vendor uses various pricing models for each asset class that are consistent with what other market participants would use. The inputs and assumptions to the model of the pricing vendor are derived from market observable sources including: benchmark yields, reported trades, broker/dealer quotes, issuer spreads, benchmark securities, bids, offers, and other market-related data. The pricing vendor considers all available market observable inputs in determining the evaluation for a security. Thus, certain securities may not be priced using quoted prices, but rather determined from market observable information. Included in Level 2 assets is our interest rate swap agreement which is valued using a mid-market valuation model. |
Level 3 | Unobservable inputs that are supported by little or no market activity and are significant to the fair value of the assets or liabilities. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires significant management judgment or estimation. This category includes the auction rate securities where independent pricing information was not able to be obtained and the contingent consideration related to the acquisitions of Vortex, Microsulis and Clinical Devices. Our investments in auction-rate securities were classified as Level 3 as quoted prices were unavailable since these auction rate securities issued by New York state and local government authorities failed auction. Due to limited market information, we utilized a discounted cash flow (“DCF”) model to derive an estimate of fair value for contingent considerations for all periods presented. The assumptions used in preparing the DCF model included estimates with respect to the discount rate, amount and timing of future interest and principal payments and forward projections. Assumptions associated with the auction rate securities include the interest rate benchmarks, the probability of full repayment of the principal considering the credit quality and guarantees in place, and the rate of return required by investors to own such securities given the current liquidity risk. |
60
The following tables provide information by level for assets and liabilities that are measured at fair value (in thousands):
Fair Value Measurements using inputs considered as: | |||||||||||||||
Level 1 | Level 2 | Level 3 | Fair Value at May 31, 2016 | ||||||||||||
Financial Assets | |||||||||||||||
Marketable securities | |||||||||||||||
New York State government agency obligations | $ | — | $ | — | $ | 1,653 | $ | 1,653 | |||||||
Total | — | — | 1,653 | 1,653 | |||||||||||
Total Financial Assets | $ | — | $ | — | $ | 1,653 | $ | 1,653 | |||||||
Financial Liabilities | |||||||||||||||
Contingent liability for acquisition earn out | — | — | 38,275 | 38,275 | |||||||||||
Total Financial Liabilities | $ | — | $ | — | $ | 38,275 | $ | 38,275 | |||||||
Fair Value Measurements using inputs considered as: | |||||||||||||||
Level 1 | Level 2 | Level 3 | Fair Value at May 31, 2015 | ||||||||||||
Financial Assets | |||||||||||||||
Marketable securities | |||||||||||||||
New York State government agency obligations | $ | — | $ | — | $ | 1,689 | $ | 1,689 | |||||||
Total | — | — | 1,689 | 1,689 | |||||||||||
Total Financial Assets | $ | — | $ | — | $ | 1,689 | $ | 1,689 | |||||||
Financial Liabilities | |||||||||||||||
Interest rate swap agreements | $ | — | $ | 257 | $ | — | $ | 257 | |||||||
Contingent liability for acquisition earn out | — | — | 47,384 | 47,384 | |||||||||||
Total Financial Liabilities | $ | — | $ | 257 | $ | 47,384 | $ | 47,641 | |||||||
There were no transfers in and out of Level 1, 2 and 3 measurements for the years ended May 31, 2016 and 2015.
The components of Level 3 fair value instruments as of May 31, 2016 are shown below (in thousands):
Financial Assets | Financial Liabilities | ||||||
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | Fair Value Measurements Using Significant Unobservable Inputs | ||||||
Balance at May 31, 2015 | $ | 1,689 | $ | 47,384 | |||
Change in fair value of contingent consideration (1) | — | 948 | |||||
Currency (gain) loss from remeasurement | — | 43 | |||||
Fair market value adjustments | (36 | ) | — | ||||
Contingent consideration payments | — | (10,100 | ) | ||||
Balance at May 31, 2016 | $ | 1,653 | $ | 38,275 | |||
The components of Level 3 fair value instruments as of May 31, 2015 are shown below (in thousands):
61
Financial Assets | Financial Liabilities | ||||||
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) | Fair Value Measurements Using Significant Unobservable Inputs | ||||||
Balance at May 31, 2014 | $ | 1,809 | $ | 67,331 | |||
Change in fair value of contingent consideration (1) | — | (8,196 | ) | ||||
Currency (gain) loss from remeasurement | — | (529 | ) | ||||
Included in other comprehensive income (loss) | (120 | ) | — | ||||
Contingent consideration payments | — | (11,222 | ) | ||||
Balance at May 31, 2015 | $ | 1,689 | $ | 47,384 | |||
(1) Change in the fair value of contingent consideration is included in earnings and comprised of changes in estimated earn out payments based on projections of company performance and amortization of the present value discount.
Contingent Liability for Acquisition Earn Outs
Certain of our business combinations involve the potential for the payment of future contingent consideration upon the achievement of certain product development milestones and/or various other favorable operating conditions. Payment of the additional consideration is generally contingent on the acquired company reaching certain performance milestones, including attaining specified revenue levels or achieving product development targets. Contingent consideration is recorded at the estimated fair value of the contingent milestone payments on the acquisition date. The fair value of the contingent milestone consideration is remeasured at the estimated fair value at each reporting period with the change in fair value recognized as income or expense within change in fair value of contingent consideration in the consolidated statement of operations. We measure the initial liability and remeasure the liability on a recurring basis using Level 3 inputs as defined under authoritative guidance for fair value measurements.
Contingent consideration liabilities will be remeasured to fair value each reporting period using projected net sales, discount rates, probabilities of payment and projected payment dates. Projected contingent payment amounts are discounted back to the current period using a discounted cash flow model. Projected net sales are based on our internal projections and extensive analysis of the target market and the sales potential. Increases in projected net sales and probabilities of payment may result in higher fair value measurements in the future. Increases in discount rates and the projected time to payment may result in lower fair value measurements in the future. Increases or decreases in any valuation inputs in isolation may result in a significantly lower or higher fair value measurement in the future.
The recurring Level 3 fair value measurements of the contingent consideration liabilities include the following significant unobservable inputs as of May 31, 2016 (in thousands of dollars):
Fair value at May 31, 2016 | Valuation Technique | Unobservable Input | Range | ||||||
Revenue based payments | $ | 35,325 | Discounted cash flow | Discount rate Probability of payment Projected fiscal year of payment | 4% 75% - 100% 2017 - 2022 | ||||
Milestone based payments | 2,950 | Discounted cash flow | Discount rate Probability of payment Projected fiscal year of payment | 16% 75% - 100% 2017 | |||||
$ | 38,275 | ||||||||
At May 31, 2016, the estimated potential amount of undiscounted future contingent consideration that we expect to pay as a result of all completed acquisitions is approximately $40.8 million. The milestones associated with the contingent consideration must be reached in future periods ranging from fiscal years 2017 to 2022 in order for the consideration to be paid.
62
NOTE D—MARKETABLE SECURITIES AND INVESTMENTS
As of May 31, 2016, marketable securities consisted of the following:
Amortized cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||||||||||
(in thousands) | |||||||||||||||
Available-for-sales securities | |||||||||||||||
New York State government agency obligations | $ | 1,800 | $ | — | $ | (147 | ) | $ | 1,653 | ||||||
$ | 1,800 | $ | — | $ | (147 | ) | $ | 1,653 | |||||||
As of May 31, 2015, marketable securities consisted of the following:
Amortized cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||||||||||
(in thousands) | |||||||||||||||
Available-for-sales securities | |||||||||||||||
New York State government agency obligations | $ | 1,825 | $ | — | $ | (136 | ) | $ | 1,689 | ||||||
$ | 1,825 | $ | — | $ | (136 | ) | $ | 1,689 | |||||||
The amortized cost and fair value of marketable securities as of May 31, 2016, by contractual maturity, are shown below. Expected maturities may differ from contractual maturities because borrowers may have the right to call or prepay obligations with or without call or prepayment penalties.
Amortized cost | Fair Value | ||||||
(in thousands) | |||||||
As of May 31, 2016: | |||||||
Due in one year or less | $ | — | $ | — | |||
Due after one through five years | — | — | |||||
Due after five through twenty years | 1,800 | 1,653 | |||||
$ | 1,800 | $ | 1,653 | ||||
63
NOTE E—INVENTORIES
As of May 31, 2016 and 2015, inventories consisted of the following:
May 31, 2016 | May 31, 2015 | ||||||
(in thousands) | |||||||
Raw materials | $ | 21,669 | $ | 28,040 | |||
Work in process | 10,700 | 11,910 | |||||
Finished goods | 23,001 | 27,438 | |||||
Total | $ | 55,370 | $ | 67,388 | |||
Total inventory is reduced to net realizable value by $12.6 million compared to $7.8 million in the prior year. Of the $12.6 million in the current year, $5.8 million relates to the write-off of Celerity.
NOTE F—PREPAID EXPENSES AND OTHER
As of May 31, 2016 and 2015, prepaid expenses and other consisted of the following:
May 31, 2016 | May 31, 2015 | ||||||
(in thousands) | |||||||
Deposits | $ | 190 | $ | 2,011 | |||
Other prepaid taxes | 160 | 165 | |||||
License fees | 108 | 121 | |||||
Software licenses | 282 | 667 | |||||
Trade shows | 278 | 279 | |||||
Rent | 127 | 77 | |||||
Other | 2,098 | 812 | |||||
Total | $ | 3,243 | $ | 4,132 | |||
64
NOTE G—PROPERTY, PLANT AND EQUIPMENT, AT COST
As of May 31, 2016 and 2015, property, plant and equipment are summarized as follows:
May 31, 2016 | May 31, 2015 | ||||||
(in thousands) | |||||||
Building and building improvements | $ | 39,585 | $ | 33,853 | |||
Machinery and equipment | 24,078 | 23,626 | |||||
Computer software and equipment | 24,691 | 24,431 | |||||
Construction in progress | 1,743 | 7,033 | |||||
90,097 | 88,943 | ||||||
Less accumulated depreciation and amortization | (43,536 | ) | (36,197 | ) | |||
46,561 | 52,746 | ||||||
Land and land improvements | 1,723 | 1,704 | |||||
$ | 48,284 | $ | 54,450 | ||||
Depreciation expense for fiscal 2016, 2015 and 2014 was $8.2 million, $9.8 million and $8.4 million, respectively. In conjunction with the Operational Excellence Program that was announced in December 2013, we updated the estimated useful lives on certain manufacturing equipment. As a result of the change in the useful life, we recorded additional depreciation of $1.0 million, $1.5 million and $0.8 million for the years ended May 31, 2016, 2015 and 2014, respectively.
65
NOTE H—GOODWILL AND INTANGIBLE ASSETS
Intangible assets other than goodwill and indefinite lived intangible assets are amortized over their estimated useful lives, which range between two and eighteen years, on either a straight-line basis or proportionately to the benefit being realized. We periodically review the estimated useful lives of our intangible assets and review such assets for impairment, based on estimated future cash flows, whenever events or changes in circumstances indicate that the carrying value of the assets may not be recoverable. If an intangible asset is considered to be impaired, the amount of the impairment will equal the excess of the carrying value over the fair value of the asset.
We consider our business to be a single operating segment entity, and a single reporting unit engaged in the development, manufacture and sale on a global basis of medical devices for vascular access, peripheral vascular disease, oncology and surgery.
Goodwill and other intangible assets that have indefinite useful lives are not amortized, but rather, are tested for impairment annually or more frequently if impairment indicators arise. Goodwill represents the excess of the purchase price over the fair value of the net tangible and identifiable intangible assets acquired in each business combination. Goodwill and intangible assets have been recorded at either incurred or allocated costs based on respective fair market values at the date of acquisition.
For goodwill, the impairment test requires a comparison of the estimated fair value of the reporting unit to which the goodwill is assigned to the sum of the carrying value of the assets and liabilities of that unit. If the sum of the carrying value of the assets and liabilities of a reporting unit exceeds the fair value of the reporting unit, the carrying value of the reporting unit’s goodwill is reduced to its implied fair value through an adjustment to the goodwill balance, resulting in an impairment charge.
To determine fair value of our single reporting unit we utilized a market-based approach and an income approach. We determined the discounted cash flow as the best indicator to determine fair value and therefore assigned a weight of 75% with the remaining 25% assigned to the market approach.
Under the market-based approach, we utilized information of our Company as well as publicly available information of certain peer companies within our industry to determine earnings multiples (EBITDA).
Under the income approach, we determined fair value based on estimated future cash flows of the reporting unit, discounted by our estimated weighted-average cost of capital, which reflects the overall level of inherent risk of a reporting unit and the rate of return an outside investor would expect to earn. We used a discount rate of 12.5% to calculate the fair value of our reporting unit.
Use of the income approach in determining the fair value of a reporting unit requires the use of significant estimates and assumptions, including revenue growth rates, operating margins, discount rates and future market conditions, among others. These assumptions are highly sensitive and changes in these estimates could result in impairment. We applied gross margin assumptions, showing improvement over historical trends, improvements over historical trends of revenue of certain product lines and used a capitalization rate of 8.5% to calculate the terminal value of our reporting unit.
We completed our annual goodwill impairment test of our single reporting unit as of December 31, 2015. Our assessment of goodwill impairment indicated that the fair value of our reporting unit exceeded its carrying value and therefore goodwill was not impaired. The fair value of our reporting unit exceeded its carrying value by 9.2%.
At times our stock market capitalization has been lower than our shareholders’ equity or book value. However, our reporting unit has continued to generate significant cash flows from operations, and we expect to continue to do so in fiscal 2016 and beyond. Furthermore, we believe that a reasonable potential buyer would offer a control premium for our business that would adequately cover the difference between our stock market capitalization and our book value. The implied control premium in our annual goodwill impairment test as of December 31, 2015 is comparable to premiums identified in recent acquisitions of companies of similar size and in similar industries.
Even though we determined that there was no goodwill impairment as of December 31, 2015, the future occurrence of a potential indicator of impairment, such as a significant adverse change in legal, regulatory, business or economic conditions or a more-likely-than-not expectation that the reporting unit or a significant portion of the reporting unit will be sold or disposed of, would require an interim assessment for the reporting unit prior to the next required annual assessment as of December 31, 2016. We continued to assess impairment through May 31, 2016 and noted no events that would be considered a triggering event.
66
It is not possible at this time to determine if any such future impairment charge would result or, if it does, whether such charge would be material. Events that could, in the future, result in impairment include, but are not limited to, declining sales for a significant product or in a significant geographic region.
As of May 31, 2016 and 2015, intangible assets consisted of the following:
May 31, 2016 | |||||||||||||
Gross carrying value | Accumulated amortization | Net carrying value | Weighted average useful life | ||||||||||
(in thousands) | (years) | ||||||||||||
Product technologies | $ | 148,387 | $ | (51,313 | ) | $ | 97,074 | 10.2 | |||||
Customer relationships | 88,389 | (47,133 | ) | 41,256 | 11.9 | ||||||||
Trademarks | 28,470 | (6,242 | ) | 22,228 | 10.7 | ||||||||
In process R&D | 3,600 | — | 3,600 | Indefinite | |||||||||
Licenses | 7,931 | (6,716 | ) | 1,215 | 7.6 | ||||||||
Distributor relationships | 2,150 | (946 | ) | 1,204 | 5.2 | ||||||||
$ | 278,927 | $ | (112,350 | ) | $ | 166,577 | |||||||
May 31, 2015 | |||||||||||||
Gross carrying value | Accumulated amortization | Net carrying value | Weighted avg useful life | ||||||||||
(in thousands) | (years) | ||||||||||||
Product technologies | $ | 148,776 | $ | (41,447 | ) | $ | 107,329 | 10.2 | |||||
Customer relationships | 86,371 | (42,813 | ) | 43,558 | 12.0 | ||||||||
Trademarks | 28,545 | (3,383 | ) | 25,162 | 10.7 | ||||||||
In process R&D | 3,600 | — | 3,600 | Indefinite | |||||||||
Licenses | 7,913 | (5,910 | ) | 2,003 | 8.3 | ||||||||
Distributor relationships | 900 | (900 | ) | — | 3.0 | ||||||||
$ | 276,105 | $ | (94,453 | ) | $ | 181,652 | |||||||
Amortization expense was $18.0 million, $18.0 million and $16.6 million for fiscal 2016, 2015 and 2014, respectively.
Annual amortization of these intangible assets is expected to approximate the following amounts for each of the next five fiscal years (in thousands):
2017 | $ | 19,049 | |
2018 | $ | 19,903 | |
2019 | $ | 22,062 | |
2020 | $ | 23,329 | |
2021 | $ | 24,370 | |
Adjustments to goodwill for the fiscal year ended May 31, 2016 and May 31, 2015 are as follows (in thousands):
Total | |||
Balance, May 31, 2014 | $ | 360,473 | |
Tax basis adjustment | 779 | ||
Balance, May 31, 2015 | $ | 361,252 | |
— | |||
Balance, May 31, 2016 | $ | 361,252 | |
67
During fiscal 2015, goodwill and deferred tax liabilities were adjusted by $0.8 million due to the receipt of a New York State tax grant that was not correctly accounted for as part of purchase accounting in fiscal 2012. The Company has determined that this adjustment was not material to any current or prior annual or interim periods.
NOTE I-INCOME TAXES
The components of income (loss) before income tax provision for the years ended May 31 are as follows:
2016 | 2015 | 2014 | |||||||||
(in thousands) | |||||||||||
(Loss) income before tax provision: | |||||||||||
US | $ | (4,444 | ) | $ | (8,965 | ) | $ | 4,645 | |||
Non-US | 1,191 | 834 | 541 | ||||||||
$ | (3,253 | ) | $ | (8,131 | ) | $ | 5,186 | ||||
Income tax (benefit) provision analyzed by category and by statement of operations classification for the years ended May 31 is summarized as follows:
2016 | 2015 | 2014 | |||||||||
(in thousands) | |||||||||||
Current | |||||||||||
Federal | $ | 34 | $ | (242 | ) | $ | (133 | ) | |||
State and local | 103 | 205 | 99 | ||||||||
Non U.S. | 217 | 417 | 157 | ||||||||
354 | 380 | 123 | |||||||||
Deferred | 39,983 | (5,123 | ) | 2,716 | |||||||
Income tax (benefit) provision | $ | 40,337 | $ | (4,743 | ) | $ | 2,839 | ||||
The significant components of deferred income tax (benefit) expense from operations for the years ended May 31 consist of the following:
2016 | 2015 | 2014 | |||||||||
(in thousands) | |||||||||||
Net effect of temporary differences | $ | 133 | $ | (1,916 | ) | $ | 1,543 | ||||
Adjustments for beginning-of-the-year valuation allowance balance for changes in circumstances | 40,418 | — | — | ||||||||
Impact of NYS tax reform legislation | — | — | 1,173 | ||||||||
Net operating loss carryforward | (568 | ) | (3,207 | ) | — | ||||||
$ | 39,983 | $ | (5,123 | ) | $ | 2,716 | |||||
68
Temporary differences that give rise to deferred tax assets and liabilities are summarized as follows:
May 31, 2016 | May 31, 2015 | ||||||
(in thousands) | |||||||
Deferred tax assets | |||||||
Net operating loss carryforward | $ | 52,593 | $ | 52,025 | |||
Stock-based compensation | 4,135 | 4,468 | |||||
Federal and state R&D tax credit carryforward | 2,145 | 1,646 | |||||
Inventories | 4,535 | 2,808 | |||||
Expenses incurred not currently deductible | 3,018 | 2,107 | |||||
Accrued liabilities | 339 | 114 | |||||
Gross deferred tax asset | 66,765 | 63,168 | |||||
Deferred tax liabilities | |||||||
Excess tax over book depreciation and amortization | 46,240 | 42,988 | |||||
46,240 | 42,988 | ||||||
Valuation Allowance | (42,209 | ) | (1,791 | ) | |||
Net deferred tax asset (liability) | $ | (21,684 | ) | $ | 18,389 | ||
Our Federal net operating loss carryforwards as of May 31, 2016 after considering IRC Section 382 limitations are $151.7 million. The expiration of the Federal net operating loss carryforwards are as follows: $29.8 million between 2017 and 2023 and $121.9 million between 2027 and 2036.
Our state net operating loss carryforwards as of May 31, 2016 after considering remaining IRC Section 382 limitations are $30.8 million which expire in various years from 2016 to 2036.
As a result of certain realization requirements of ASC 718, the table of deferred tax assets and liabilities shown above does not include certain deferred tax assets as of May 31, 2016 and 2015 that arose directly from tax deductions related to equity compensation greater than compensation recognized for financial reporting. Equity will be increased by $0.4 million if and when such excess tax benefits are ultimately realized.
At May 31, 2016, we had a net deferred income tax liability of $21.7 million, after recording a valuation allowance of $42.2 million. The valuation allowance increased by $40.4 million in 2016.
While the net deferred tax asset at May 31, 2016 before the valuation allowance was $19.9 million, the Company was required to record a valuation allowance of $40.4 million due to deferred tax liabilities related to intangibles of $20.5 million that have an indefinite reversal period and can not be used to support the deferred tax asset.
A valuation allowance is provided if based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. The Company’s analysis of the need for a valuation allowance considered that the Company has incurred a cumulative loss in the U.S. over the three year period ending May 31, 2016. A majority of the cumulative loss has been caused by the charges associated with the product recall and discontinuance and the impairment of fixed and intangible assets recorded in the quarter end February 28, 2015, From the time when the Company initially incurred a three year cumulative loss in the quarter ended February 28, 2015, and in each subsequent quarter through the quarter ended February 28, 2016, the Company could still demonstrate a recent history of core earnings , and had anticipated a return to profitability for the full fiscal year 2016. However, in the quarter ended May 31, 2016 the Company did not return to profitability for the full fiscal year and could no longer demonstrate a recent history of core earnings. Consequently after careful consideration and weighing of all the available positive and negative evidence, the weight given to the three year cumulative loss and lack of a recent history of core earnings was difficult to overcome and a full valuation allowance related to the U.S. deferred tax assets was established. Management will continue to reevaluate the positive and negative evidence at each reporting period and if future results as projected in the U.S. and our tax planning strategies are favorable, the
69
valuation allowance may be removed, which could have a favorable material impact on our results of operations in the period in which it is recorded.
Our consolidated income tax provision has differed from the amount that would be provided by applying the U.S. Federal statutory income tax rate to our income before income taxes for the following reasons:
2016 | 2015 | 2014 | |||||||||
(in thousands) | |||||||||||
Income tax (benefit) provision at statutory tax rate of 35% | $ | (1,139 | ) | $ | (2,845 | ) | $ | 1,814 | |||
Effect of graduated tax rates | 33 | 81 | (51 | ) | |||||||
State income taxes, net of Federal tax benefit | (215 | ) | (21 | ) | 111 | ||||||
Impact of Non US operations | (162 | ) | 133 | (27 | ) | ||||||
Research and development tax credit | (499 | ) | (604 | ) | (236 | ) | |||||
Meals and entertainment | 329 | — | — | ||||||||
Nondeductible interest on contingent payments | 262 | 265 | 540 | ||||||||
Nontaxable gain on revaluation of contingent consideration liability | (170 | ) | (3,102 | ) | (1,734 | ) | |||||
Tax law changes | — | (454 | ) | 1,173 | |||||||
Adjustment to beginning of year valuation allowance | 40,685 | — | — | ||||||||
Effect of elimination of stock compensation APIC pool | 739 | 1,253 | 440 | ||||||||
Nondeductible stock-based compensation | — | — | 176 | ||||||||
Other nondeductible expenses | 207 | 498 | 384 | ||||||||
Over (under) accrual of prior year Federal and state taxes | 356 | 38 | 249 | ||||||||
Other | (89 | ) | 15 | — | |||||||
Income tax (benefit) expense | $ | 40,337 | $ | (4,743 | ) | $ | 2,839 | ||||
The following table provides a reconciliation of the beginning and ending amount of unrecognized tax benefits, all of which, if recognized, would impact the effective tax rate.
2016 | 2015 | 2014 | |||||||||
(in thousands) | |||||||||||
Unrecognized tax benefits balance at June 1 | $ | — | $ | — | $ | — | |||||
Increase in gross amounts of tax positions related to prior years | 899 | — | — | ||||||||
Decrease in gross amounts of tax positions related to prior years | — | — | — | ||||||||
Increase in gross amounts of tax positions related to current year | — | — | — | ||||||||
Decrease due to settlements with tax authorities | — | — | — | ||||||||
Decrease due to lapse in statute of limitations | — | — | — | ||||||||
Unrecognized tax benefits balance at May 31 | $ | 899 | $ | — | $ | — | |||||
The table above includes unrecognized tax benefits associated with the calculation of limitations placed on the utilization of tax attributes related to an acquired company. If recognized, $0.1 million of the balance of unrecognized tax benefits as of May 31, 2016 would affect the effective tax rate and the balance of $0.8 million would result in adjustments to other tax accounts.
70
We recognize interest and penalties related to unrecognized tax benefits within its global operations as a component of income tax expense. There are no accrued interest and penalties recognized in the consolidated balance sheet as of May 31, 2016 and May 31, 2015.
We file income tax returns in the U.S. federal jurisdiction and various state and foreign jurisdictions. In the normal course of business we are subject to examination by taxing authorities throughout the world.
Fiscal years 2012 through 2016 remain open to examination by the various tax authorities. New York State is currently auditing AngioDynamic's franchise tax filings for 2011 through 2013. We do not anticipate any adjustments will result in a material adverse effect on our financial condition, results of operations or cash flows.
We do not anticipate that the amount of unrecognized tax benefits will significantly change in the next twelve months.
The accumulated undistributed earnings of the Company’s foreign operations were approximately $4.6 million, and are intended to remain indefinitely invested in foreign operations. Accordingly, no taxes have been provided on these earnings at May 31, 2016. If these earnings were distributed, the Company would be subject to both foreign withholding taxes and U.S. income taxes that may not be fully offset by foreign tax credits. A reasonable estimate of the deferred tax liability on these earnings is not practicable at this time.
NOTE J—ACCRUED LIABILITIES
As of May 31, 2016 and 2015, accrued liabilities consist of the following:
May 31, 2016 | May 31, 2015 | ||||||
(in thousands) | |||||||
Payroll and related expenses | $ | 9,414 | $ | 10,297 | |||
Royalties | 2,489 | 2,237 | |||||
Accrued severance | 1,524 | 158 | |||||
Sales and franchise taxes | 565 | 489 | |||||
Interest rate swap liability | — | 257 | |||||
Outside services | 2,063 | 1,522 | |||||
Deferred Rent | 35 | 808 | |||||
Other | 5,806 | 2,341 | |||||
Total | $ | 21,896 | $ | 18,109 | |||
NOTE K—LONG-TERM DEBT
On September 19, 2013, we entered into a Credit Agreement (the "Credit Agreement") with the lenders party thereto, JPMorgan Chase Bank, N.A., as administrative agent, Bank of America, N.A. and Keybank National Association as co-syndication agents, and J.P. Morgan Securities LLC, Merrill Lynch, Pierce, Fenner & Smith Incorporated and Keybank National Association as joint bookrunners and joint lead arrangers.
The Credit Agreement provides for a $100 million senior secured term loan facility (the "Term Loan") and a $100 million senior secured revolving credit facility, which includes up to a $20 million sublimit for letters of credit and a $5 million sublimit for swingline loans (the "Revolving Facility", and together with the Term Loan, the "Facilities").
The proceeds of the Revolving Facility may be used for general corporate purposes of the Company and its subsidiaries. The Facilities have a five years maturity. The Term Loan has a quarterly repayment schedule equal to 5%, 5%, 10%, 15% and 65% of its principal amount in years one through five. Interest on both the Term Loan and Revolving Facility will be based on a base rate or Eurodollar rate plus an applicable margin which increases as our total leverage ratio increases, with the base rate and Eurodollar rate having ranges of 0.5% to 1.25% and 1.5% to 2.25% respectively. After default, the interest rate may be increased by 2.0%. The Revolving Facility will also carry a commitment fee of 0.2% to 0.35% per annum on the unused portion.
71
Our obligations under the Facilities are unconditionally guaranteed, jointly and severally, by our material direct and indirect domestic subsidiaries (the "Guarantors"). All obligations of the Company and the Guarantors under the Facilities are collateralized by first priority security interests in substantially all of the assets of the Company and the Guarantors.
We have entered in an interest rate swap agreement, (the "Swap Agreement"), with an initial notional amount of $100 million, to limit the effect of rising of interest rates. The Swap Agreement, which qualified for hedge accounting, was a contract to exchange floating interest rate payments for fixed interest rate payments of 3.26% on the outstanding balance of the loan over the life of the agreement without the exchange of the underlying notional amounts. The Swap Agreement provides for a fixed rate of 0.74% above the applicable rate provided for in the Credit Agreement. The Swap matured in May 2016.
On September 19, 2013, we borrowed $100 million under the Term Facility and approximately $41.4 million under the Revolving Facility to repay the prior credit agreement. As of May 31, 2016, $85.0 million and $36.4 million were outstanding under the Term Facility and Revolving Facility, respectively. As of May 31, 2016 and 2015 the carrying value of long-term debt approximates its fair market value. The Credit Agreement includes customary representations, warranties and covenants, and acceleration, indemnity and events of default provisions, including, among other things, two financial covenants. The first financial covenant requires us to maintain, as of the end of each of our fiscal quarters, a ratio of (i) consolidated EBITDA minus consolidated capital expenditures to (ii) consolidated interest expense paid or payable in cash plus scheduled principal payments in respect of indebtedness under the Credit Agreement of not less than 1.35 to 1.00. The second financial covenant requires us to maintain, as of the end of each of our fiscal quarters, a ratio of consolidated total indebtedness to consolidated EBITDA of not greater than 3.75 to 1.00. We were in compliance with both financial covenants as of May 31, 2016.
As of May 31, 2016, future minimum principal payments on long-term debt were as follows (in thousands):
2017 | $ | 16,250 | |
2018 | 26,250 | ||
2019 | 78,910 | ||
$ | 121,410 | ||
NOTE L—RETIREMENT PLANS
We have a 401(k) plan under which eligible employees can defer a portion of their compensation, part of which is matched by us. Matching contributions were $3.7 million, $3.7 million and $2.8 million in 2016, 2015 and 2014, respectively. There are also various immaterial foreign retirement plans.
NOTE M—STOCKHOLDERS’ EQUITY
1. Capitalization
On October 29, 2014, our Board of Directors approved our Amended and Restated Certificate of Incorporation (the “Amended Certificate”). Under the Amended Certificate, the authorized capital stock is 80,000,000 shares, consisting of 75,000,000 shares of common stock, par value $.01 per share and 5,000,000 shares of preferred stock, par value $.01 per share.
The holders of common stock are entitled to one vote for each share held. Subject to preferences applicable to any outstanding shares of preferred stock, the holders of common stock are entitled to receive ratably dividends, if any, as may be declared by the Board of Directors out of funds legally available for dividend payments. If we liquidate, dissolve, or wind up, the holders of common stock are entitled to share ratably in all assets remaining after payment of liabilities and liquidation preferences of any outstanding shares of preferred stock. Holders of common stock have no pre-emptive rights or rights to convert their common stock into any other securities. There are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences and privileges of the holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate in the future.
Our board of directors has the authority to (i) issue the undesignated preferred stock in one or more series, (ii) determine the powers, preferences and rights and the qualifications, limitations or restrictions granted to or imposed upon any wholly un-
72
issued series of undesignated preferred stock and (iii) fix the number of shares constituting any series and the designation of the series, without any further vote or action by our stockholders.
2. Stock Options
1997 Stock Option Plan
In 1997, we adopted a Stock Option Plan (the “1997 Plan”). The 1997 Plan provided for the grant to key employees of both nonqualified stock options and incentive stock options and to members of the Board of Directors and consultants of nonqualified stock options. A total of 1,497,674 shares of our common stock were available to be issued under the 1997 Plan pursuant to the exercise of options. All stock options were to have an exercise price of not less than the fair market value of the shares on the date of grant. Options are exercisable over a period of time to be designated by the administrators of the 1997 Plan (but not more than 10 years from the date of grant) and are subject to such other terms and conditions as the administrators may determine. The vesting schedule is subject to the discretion of our Board of Directors. Options are exercisable immediately upon vesting. In addition, all options, whether vested or not, become exercisable in full immediately upon a change of control, as defined under the 1997 Plan. The 1997 Plan terminated in March 2007 and as such, no further options will be granted under this plan.
2004 Stock and Incentive Award Plan
The 2004 Stock and Incentive Award Plan (the “2004 Plan”) provides for the grant of incentive options to our employees and for the grant of non-statutory stock options, restricted stock, stock appreciation rights, performance units, performance shares and other incentive awards to our employees, directors and other service providers. A total of 6,750,000 shares of our common stock have been reserved for issuance under the 2004 Plan, of which up to 800,000 shares may be issued upon the exercise of incentive stock options. The compensation committee of the Board of Directors administers the 2004 Plan. The committee determines vesting terms and the exercise price of options granted under the 2004 Plan, but for all incentive stock options the exercise price must at least be equal to the fair market value of our common stock on the date of grant. The term of an incentive stock option may not exceed ten years.
On October 5, 2011, we amended the 2004 Stock and Incentive Award Plan to increase the maximum number of shares of our common stock with respect to which stock options may be granted during any calendar year to one employee from 200,000 shares to 500,000 shares.
The following table summarizes information about stock options activity for the fiscal year ended May 31, 2016.
2016 | ||||||||||||
Shares | Weighted- average exercise price | Weighted average remaining contractual life | Aggregate intrinsic value (in thousands) | |||||||||
Outstanding at beginning of year | 2,299,313 | $ | 14.86 | |||||||||
Granted | 564,380 | $ | 14.22 | |||||||||
Exercised | (145,260 | ) | $ | 13.73 | ||||||||
Forfeited | (304,563 | ) | $ | 13.85 | ||||||||
Expired | (132,252 | ) | $ | 22.75 | ||||||||
Outstanding at end of year | 2,281,618 | $ | 14.45 | 3.79 | $ | 164 | ||||||
Options exercisable at year-end | 1,313,698 | $ | 14.25 | 2.34 | $ | 130 | ||||||
Options expected to vest in future periods | 854,189 | $ | 14.72 | 5.76 | $ | 30 | ||||||
As of May 31, 2016, there remained approximately 2.2 million shares available for granting of options under the 2004 Plan. Options are exercisable into common stock.
73
The following table summarizes information about stock options outstanding at May 31, 2016.
Range of exercise prices | Number outstanding | Weighted- average remaining life in years | Weighted- average exercise price | Number Exercisable | Weighted- average exercise price | ||||||||||
$10.25 - $14.24 | 1,398,385 | 3.46 | $ | 12.93 | 957,571 | $ | 13.14 | ||||||||
$14.25 - $18.24 | 684,427 | 4.75 | 16.04 | 228,571 | 15.84 | ||||||||||
$18.25 - $22.24 | 183,695 | 3.01 | 19.30 | 112,445 | 19.23 | ||||||||||
$22.25 - $26.24 | 14,250 | 0.48 | 23.62 | 14,251 | 23.62 | ||||||||||
$26.25 - $30.24 | 861 | 0.60 | 26.94 | 860 | 26.94 | ||||||||||
2,281,618 | 3.79 | $ | 14.45 | 1,313,698 | $ | 14.25 | |||||||||
Stock options are granted at exercise prices equal to the quoted market price of our common stock at the date of the grant. Options vest 25% per year over four years for employees and 100% after one year for consultants. Grants to directors vest 33.33% per year over three years. Stock options granted prior to May 1, 2007 expire on the tenth anniversary of the grant date. Stock options granted on or after May 1, 2007, expire on the seventh anniversary of the grant date.
We measure the fair value of each stock option grant at the date of grant using a Black-Scholes option pricing model. The weighted average grant-date fair value of options granted during the years ended May 31, 2016, 2015 and 2014 was $4.16, $4.74, and $4.10, respectively. The following assumptions were used in arriving at the fair value of options granted during 2016, 2015 and 2014, respectively: risk-free interest rates of 1.48%, 1.54% and 1.44%; expected volatility of 31%, 31%, and 34%; and expected lives of 4.81 years, 4.76 years, and 4.74 years. We do not declare dividends therefore a dividend yield of zero was used for the years ended 2016, 2015 and 2014. Risk-free interest rates reflect the yield on zero-coupon U.S. Treasury bonds whose maturity period equals the expected term of the option. Expected volatilities are based on the historical volatility of our stock. The expected option lives are based on our historical experience of employee exercise behavior.
The total intrinsic value of options exercised during the years ended 2016, 2015 and 2014 amounted to $0.1 million, $1.6 million, and $1.0 million, respectively. As of May 31, 2016, there was $3.3 million of total unrecognized compensation cost related to non-vested options. The cost is expected to be recognized over a weighted average period of 3 years.
Cash received from option exercises during 2016, 2015 and 2014 was $1.3 million, $4.3 million and $1.3 million, respectively. The tax benefit realized from stock options exercised during the years ended 2016, 2015 and 2014 were $0.1 million, $0.5 million and $0.1 million, respectively.
3. Performance Share and Restricted Stock Unit Awards
We grant restricted stock units to certain employees under the 2004 Plan which give the recipients the right to receive shares of our stock upon vesting. The restricted stock unit awards vest in equal annual installments over the term of the grants. Unvested restricted stock unit awards will be forfeited if the recipient ceases to be employed by us, competes with our business or otherwise engages in activities detrimental to our business before such date.
The following table summarizes information about restricted stock unit activity for the year ended May 31, 2016.
Non-Vested Stock Award Units | Weighted Average Grant-Date Fair Value | |||||
Non-vested at beginning of year | 563,101 | $ | 13.73 | |||
Granted | 259,917 | $ | 15.21 | |||
Vested | (160,845 | ) | $ | 15.39 | ||
Canceled | (112,396 | ) | $ | 13.81 | ||
Non-vested at end of year | 549,777 | $ | 14.62 | |||
Awards expected to vest in future periods | 482,594 | $ | 14.62 | |||
The fair value of each restricted stock unit is the market price of our stock on the date of grant. The weighted average grant date fair value of restricted stock units granted during the years ended May 31, 2016, 2015 and 2014 was $15.21, $14.75
74
and $13.23, respectively. The total intrinsic value of restricted stock units vesting during the years ended 2016, 2015 and 2014 was $2.5 million, $2.4 million, and $1.8 million, respectively. As of May 31, 2016, there was $5.4 million of total unrecognized compensation cost related to non-vested restricted stock awards. The cost is expected to be recognized over a weighted average period of 2 years.
We grant performance share awards to certain employees under the 2004 Plan which gives the recipients the right to receive shares of our stock if certain criteria is met. The performance criteria is established by the compensation committee for vesting of the performance share awards and may include factors such as the achievement of relative total shareholder return ("TSR"), certain sales, operating income and earnings per share (“EPS”) goals. Performance share awards are subject to additional conditions, including the recipient’s continued employment with us.
Performance share units are valued using a Monte Carlo simulation model on the date of grant. As of May 31, 2016, 2015 and 2014, the weighted average grant date fair market value for new grants was $18.07, $19.83 and $25.56, respectively. Compensation cost is recognized over the performance period which is typically three years. As of May 31, 2016, 0.4 million performance share units with a weighted average remaining contractual term of 3 years and $4.6 million of unrecognized compensation cost were outstanding. As of May 31, 2015, 0.2 million performance share units with a weighted average remaining contractual term of 3 years and $2.2 million of unrecognized compensation cost were outstanding.
4. Employee Stock Purchase Plan
The Employee Stock Purchase Plan (the “Stock Purchase Plan”) provides a means by which our employees (the “participants”) are given an opportunity to purchase our common stock through payroll deductions. A total of 2,000,000 shares of our common stock have been reserved for issuance under the Stock Purchase Plan. Shares are offered through two purchase periods, each with duration of approximately 6 months, commencing on the first business day of the first and third fiscal quarters. An employee is eligible to participate in an offering period if, on the first day of an offering period, he or she has been employed in a full-time capacity for at least six months, with a customary working schedule of 20 or more hours per week and more than five months in a calendar year. Employees who own stock possessing 5% or more of the total combined voting power or value of all classes of our stock are not eligible to participate in the Stock Purchase Plan. The purchase price of the shares of common stock acquired on each purchase date will be the lower of (i) 85% of the fair market value of a share of common stock on the first day of the offering period or (ii) 85% of the fair market value of a share of common stock on the last day of the purchase period, subject to adjustments made by the Board of Directors. The Stock Purchase Plan is intended to qualify as an “employee stock purchase plan” within the meaning of Section 423 of the Internal Revenue Code. During the year ended May 31, 2015, an additional 800,000 shares of our common stock have been reserved for issuance under the Stock Purchase Plan.
We use the Black-Scholes option-pricing model to calculate the purchase date fair value of the shares issued under the Stock Purchase Plan and recognize expense related to shares purchased ratably over the offering period. During the years ended 2016, 2015 and 2014, 137,957, 119,001 and 146,275 shares, respectively, were issued at an average price of $10.67, $11.89 and $9.30, respectively, under the Stock Purchase Plan. As of May 31, 2016, 1.0 million shares remained available for future purchases under the Stock Purchase Plan.
5. Other items
Share-based compensation expense recognized in the consolidated statement of operations during the years ended 2016, 2015 and 2014 amounted to $3.2 million, $6.0 million and $5.5 million, respectively. The income tax benefit recognized in earnings on the compensation expense recognized for all share-based compensation arrangements amounted to $1.0 million, $2.0 million and $1.8 million, respectively. Income tax expense of $0.8 million, $1.3 million and $0.5 million was recorded in continuing operations for the excess of cumulative book deductions over actual tax deductions for the years ended 2016, 2015 and 2014, respectively. The income tax benefit on all share based compensation expense in 2016 as well as the income tax expense on the excess book deductions over actual tax deductions in 2016 are negated by the full valuation allowance established at May 31, 2016. Additional income tax expense of $0.1 million for 2014 was recorded in shareholder’s equity for the excess book deductions to the extent prior tax deductions exceeded prior book deductions.
As part of his employment agreement, the Company awarded James C. Clemmer, the Company’s President and CEO, 250,000 performance shares, 200,000 options and 50,000 restricted stock units. The award was unanimously approved by the Company's independent compensation committee. The performance shares have a three years term with payouts to be made in
75
shares of the Company's common stock at the end of the term ranging between 0 and 200 percent of the grant amount depending on the Company's total shareholder return relative to a peer group of companies. The options will vest in four equal installments beginning on the first anniversary of the grant date, have a strike price equal to the closing price of the company's common stock as of April 4, 2016 and expire, if not exercised, on April 4, 2023. The restricted stock units will vest in four equal installments beginning on the first anniversary of the grant date.
NOTE N—EARNINGS PER SHARE
Basic earnings per share are based on the weighted average number of common shares outstanding without consideration of potential common stock. In addition, diluted earnings per share include the dilutive effect of potential common stock consisting of stock options, restricted stock units and performance stock units, provided that the inclusion of such securities is not anti-dilutive. In periods with a net loss, stock options and restricted stock units are not included in the computation of basic loss per share as the impact would be anti-dilutive.
The following table reconciles basic to diluted weighted average shares outstanding for the years ended May 31, 2016, 2015 and 2014:
2016 | 2015 | 2014 | ||||||
Basic | 36,161,383 | 35,683,139 | 35,135,689 | |||||
Effect of dilutive securities | — | — | 304,161 | |||||
Diluted | 36,161,383 | 35,683,139 | 35,439,850 | |||||
Securities excluded as their inclusion would be anti-dilutive | 3,277,037 | 2,862,414 | 2,347,426 | |||||
NOTE O—COMMITMENTS AND CONTINGENCIES
Leases
We are committed under non-cancelable operating leases for facilities and equipment. During fiscal 2016, 2015 and 2014, aggregate rental costs under all operating leases were approximately $2.5 million, $3.4 million and $2.0 million, respectively. Future annual payments under non-cancelable operating leases in the aggregate, of which one includes an escalation clause, with initial remaining terms of more than one year at May 31, 2016, are summarized as follows (in thousands):
2017 | $ | 2,183 | |
2018 | 2,013 | ||
2019 | 1,981 | ||
2020 | 1,772 | ||
2021 | 935 | ||
$ | 8,884 | ||
Litigation Matters
AngioDynamics v. biolitec
On January 2, 2008, we commenced an action in the United States District Court for the Northern District of New York entitled AngioDynamics, Inc. v. biolitec, Inc. In this action, we sought judgment against biolitec for defense and indemnification in two lawsuits which we previously settled. Our claims arise out of a Supply and Distribution Agreement (“SDA”) entered into with biolitec on April 1, 2002. On September 27, 2011, the U.S. District Court granted key portions of our motion for summary judgment in our legal case against biolitec. The Court also dismissed biolitec’s counterclaims against us. The court denied one portion of our summary judgment motion, which sought to recover additional costs from biolitec, leaving this for adjudication at trial. On November 8, 2012, the Court granted partial judgment to us in the amount of $23.2 million. Biolitec appealed this judgment. On August 23, 2013, the U.S. Court of Appeals for the Second Circuit dismissed biolitec’s appeal.
76
In October 2009, we commenced an action in the United States District Court for the District of Massachusetts entitled AngioDynamics, Inc. v. biolitec AG and Wolfgang Neuberger. The Complaint in this action was amended in March 2010. This action seeks to recover against biolitec, Inc.’s parent entities and CEO for tortiously interfering with biolitec, Inc.’s contractual obligation to defend and indemnify us, and also seeks to pierce the corporate veil of biolitec, Inc. and to invalidate certain alleged fraudulent transfers in order to hold biolitec, Inc.’s parent entities jointly and severally liable for the alleged breach of the SDA. On September 13, 2012, the Massachusetts Court granted our request for a preliminary injunction prohibiting the downstream merger of biolitec AG with its Austrian subsidiary. On April 1, 2013, the U.S. Court of Appeals for the First Circuit affirmed the preliminary injunction. On January 14, 2014, the District Court entered judgment in our favor as to liability. On March 18, 2014, the District Court entered judgment in our favor against Biolitec AG, Biomed Technology Holdings, Ltd., and Wolfgang Neuberger, jointly and severally, in the amount of $74.9 million. On March 11, 2015, the U.S. Court of Appeals for the First Circuit affirmed the judgment. The defendants petitioned to the U.S. Supreme Court for a writ of certiorari. The Supreme Court denied the petition on November 30, 2015. The defendants have also filed an appeal with the U.S. Court of Appeals for the First Circuit regarding civil contempt sanctions imposed by the Massachusetts District Court as a result of defendants’ completion of the downstream merger in violation of the Court’s injunction. On May 6, 2016, the First Circuit issued an opinion rejecting this latest appeal. On February 18, 2016, the Massachusetts District Court issued an order compelling the Massachusetts defendants to provide post-judgment discovery intended to aid us in potentially collecting our judgment. On March 21, 2016, the Massachusetts defendants noticed an appeal from this order. On June 27, 2016, we filed a motion asking the Massachusetts District Court to impose sanctions on the Massachusetts defendants for their failure to comply with the post-judgment discovery order.
On November 13, 2014, the U.S. District Court for the District of Massachusetts issued summonses to four Biolitec entities - Biolitec U.S., Inc., Biolitec Holding U.S., Inc., Biolitec Medical Devices, Inc., and CeramOptec Industries, Inc. - pursuant to Massachusetts trustee process. We sought to use this process to attach the assets of these entities in order to satisfy our judgment. The trustee process was automatically stayed when the four Biolitec entities filed Chapter 7 petitions in the U.S. Bankruptcy Court for the District of Delaware. However, on November 3, 2015, the Delaware Bankruptcy Court granted our request to modify the automatic stay to allow us to seek a default against the four Biolitec entities pursuant to trustee process. On January 21, 2016, the four Chapter 7 cases were transferred at our request to the U.S. Bankruptcy Court for the District of New Jersey.
On August 29, 2013, we became co-plaintiffs in an adversary proceeding in the United States Bankruptcy Court for the District of New Jersey entitled Cyganowski, Trustee, et al. v. Biolitec U.S., Inc., et al. In this action, we assert claims of conversion, unjust enrichment, tortious interference, and unfair competition against various biolitec entities for alleged violation of Bankruptcy Court settlement and sale orders under which we acquired certain assets of Biolitec, Inc. On September 3, 2013, we, along with our co-plaintiff, obtained a temporary restraining order against the defendants in this action. On January 22, 2015, the Bankruptcy Court entered a permanent injunction on our behalf for an additional two years.
C.R. Bard, Inc. v. AngioDynamics, Inc.
On January 11, 2012, C.R. Bard, Inc. (“Bard”) filed a suit in the United States District Court of Utah claiming certain of our implantable port products infringe on three U.S. patents held by Bard (the "Utah Action"). Bard is seeking unspecified damages and other relief. The Court denied Bard’s motion for pre-trial consolidation with separate actions it filed on the same day against Medical Components, Inc. and Smiths Medical ASD, Inc., but had asked for supplemental briefing on the issue of whether to conduct a common Markman hearing. Meanwhile, we filed petitions for reexamination in the US Patent and Trademark Office ("PTO") which seek to invalidate all 3 patents asserted by Bard in the litigation. Our petitions were granted and 40 of 41 patent claims were rejected and, following further proceedings, the Patent Office issued a Final Rejection of all 40 claims subject to reexamination. Thereafter, Bard filed appeals to the PTO Board of Appeals and Interferences for all three reexams. The parties completed briefing on the appeals and oral argument was held on June 18, 2015. The Patent Office has issued decisions in the three appeals. In one (issued on March 11, 2016 for US Patent No. 7,785,302), the rejections of six of the ten claims under reexamination were affirmed, but were reversed on four of the ten claims. In the second (issued on March 24, 2016 for U.S. Patent No. 7,959,615), the rejections of eight of the ten claims under reexamination were affirmed but the rejections of the other two of the ten claims were reversed. In the third (issued on March 29 for U.S. Patent No. 7,947.022) the rejections of all twenty claims under reexamination were affirmed. Bard has since filed Requests for Rehearing in all three reexamination appeals and the Company filed Requests for Rehearing in two of the reexamination appeals (the ‘302 and ‘615 patent reexaminations). Each party has filed comments in Opposition to the other party’s Rehearing Requests, and we are awaiting the PTO determinations in all three reexaminations. The Utah Action has been stayed pending final resolution of the
77
PTO process. We believe these claims are without merit and intend to defend them vigorously. We have not recorded an expense related to the outcome of this litigation because it is not yet possible to determine if a potential loss is probable nor reasonably estimable.
On March 10, 2015, C.R. Bard, Inc. and Bard Peripheral Vascular, Inc. (“Bard”) filed suit in the United States District Court for the District of Delaware claiming certain of our implantable port products infringe on three U.S. patents held by Bard (the “Delaware Action). Bard is seeking unspecified damages and other relief. The patents asserted in the Delaware Action are different than those asserted in the Utah Action. On June 1, 2015, we filed two motions in response to Bard’s Complaint - one sought transfer to the District of Utah where the Utah Action is currently pending, and the other sought dismissal of the entire complaint on grounds that none of the claims in the asserted patents is directed to patent eligible subject matter under Section 101 of the Patent Statute and in light of recent authority from the U. S. Supreme Court. On January 12, 2016, the court issued a decision denying both motions. We have since served an Answer and Counterclaim to which Bard has served a Reply. On March 10, 2016, the Court held a case management conference, and, on March 14, 2016, the court entered a Scheduling Order which set, inter alia, a Markman hearing for March 10, 2017, a summary judgment hearing for December 8, 2017 and trial for March 12, 2018. The parties have since served various discovery requests on each other; on May 27, 2016 Bard served its Infringement Contentions which identified all the port products accused of infringement; and, on June 24, 2016, we served Invalidity Contentions which detail various grounds for invalidating the three asserted patents. We believe these claims are without merit and intend to defend them vigorously. We have not recorded an expense related to the outcome of this litigation because it is not yet possible to determine if a potential loss is probable nor reasonably estimable.
Governmental Investigations
LC Beads
In June 2014 we received a subpoena from the U.S. Department of Justice (the “DOJ”) requesting documents in relation to a criminal and civil investigation the DOJ is conducting regarding BTG International, Inc.’s LC Bead® product beginning in 2003. RITA Medical Systems and AngioDynamics, Inc., after its acquisition of RITA, was the exclusive distributor of LC Beads in the United States from 2006 through December 31, 2011. We are cooperating fully with this investigation and at this time are unable to predict its scope, duration or outcome. We are unable at this time to reasonably estimate the amount or range of any loss, although it is possible that the amount of such loss could be material. In accordance with ASC 450, "Contingencies," or "ASC 450," no amount in respect of any potential liability in this matter, including for penalties, fines or other sanctions, has been accrued in the consolidated financial statements.
EVLT
In April 2015 we received a subpoena from the DOJ requesting documents in relation to a criminal and civil investigation the DOJ is conducting regarding purported promotion of certain of AngioDynamics’ VenaCure EVLT products for un-cleared indications. We are cooperating fully with this investigation and at this time are unable to predict its scope, duration or outcome. We are unable at this time to reasonably estimate the amount or range of any loss, although it is possible that the amount of such loss could be material. In accordance with ASC 450, "Contingencies," or "ASC 450," no amount in respect of any potential liability in this matter, including for penalties, fines or other sanctions, has been accrued in the consolidated financial statements.
Future Purchase Obligations
We have entered into commitments for future minimum inventory purchases related to several core products. Total future non-cancelable purchase obligations through fiscal 2021 amount to $8.9 million. There are no such obligations thereafter.
NOTE P—SEGMENTS AND GEOGRAPHIC INFORMATION
Segment information
We consider our business to be a single segment entity related to the development, manufacture and sale on a global basis of medical devices for vascular access, surgery, peripheral vascular disease and oncology. Our chief operating decision maker (CEO) evaluates the various global product portfolios on a net sales basis. Executives reporting in to the CEO include those responsible for operations and supply chain management, research and development, sales, franchise marketing and certain
78
corporate functions. The CEO evaluates profitability, investment and cash flow metrics on a consolidated worldwide basis due to shared infrastructure and resources.
Total sales by product category are summarized below (in thousands):
Year Ended | |||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | |||||||||
Net sales by Product Category | |||||||||||
Peripheral Vascular | $ | 202,780 | $ | 192,713 | $ | 192,626 | |||||
Vascular Access | 99,375 | 107,754 | 106,394 | ||||||||
Oncology/Surgery | 48,895 | 51,890 | 49,360 | ||||||||
Supply Agreement | 2,840 | 4,177 | 6,045 | ||||||||
Total | $ | 353,890 | $ | 356,534 | $ | 354,425 | |||||
Geographic information
Total sales for geographic areas are summarized below (in thousands):
Year Ended | |||||||||||
May 31, 2016 | May 31, 2015 | May 31, 2014 | |||||||||
Net sales by Geography | |||||||||||
United States | $ | 283,519 | $ | 280,611 | $ | 280,161 | |||||
International | 67,531 | 71,746 | 68,219 | ||||||||
Supply Agreement | 2,840 | 4,177 | 6,045 | ||||||||
Total | $ | 353,890 | $ | 356,534 | $ | 354,425 | |||||
For fiscal years 2016, 2015 and 2014, International sales as a percentage of total net sales were 19%, 20% and 19%, respectively. Sales to any one country outside the U.S., as determined by shipment destination, did not comprise a material portion of our net sales in any of the last three fiscal years. 99% of our total assets are located within the United States.
NOTE Q—RESTRUCTURING
On December 5, 2013, we announced a company-wide operational excellence program designed to save between $15 and $18 million during the course of the next three years. The initiative is expected to create greater efficiencies and drive business performance improvements by focusing on several key elements, including product rationalization, lean manufacturing initiatives, supply chain optimization and enterprise resource planning (ERP) implementation. The plan also incorporates the consolidation of our New York plants to establish a single manufacturing center in Glens Falls and a distribution center in Queensbury. During the course of the three years program, we reduced our New York employee base by approximately 80-100 positions as a result of this plant consolidation and reorganization. We have invested $5.4 million in facility improvements to date. In addition, total restructuring charges are estimated to be $4.9 million. During the year ended May 31, 2016, the cost incurred was $1.5 million, consisting of $1.0 million of accelerated depreciation and $0.5 million of other related costs. During the year ended May 31, 2015, the cost incurred was $2.0 million, consisting of $0.5 million of severance and related costs and $1.5 million of accelerated depreciation. These costs are included in “Acquisition, restructuring and other items, net” in the statements of income.
During the year ended May 31, 2015, we initiated a restructuring of finance, R&D and S&M organizations to improve our profitability. As part of the restructuring, we recorded $1.9 million and $0.8 million, for the years ended May 31, 2016 and 2015, respectively, in severance expense which is included in "Acquisition, restructuring and other items, net" in the statement of operations. In addition, we recorded a gain of $0.7 million related to the modification of the stock based compensation for the former CEO.
79
NOTE R—IMMATERIAL ERROR CORRECTIONS
During the financial closing process for the fourth quarter of fiscal year 2016, the Company determined that certain consolidated financial statement amounts were not accounted for correctly in prior periods. The Company has evaluated these errors together with errors identified in prior periods and concluded that they were not material individually or in the aggregate to any of its previously issued annual and interim financial statements. However, the financial statements included herein have been revised to correct for the impact of these items.
The Company has corrected the relevant financial information from previous reporting periods contained in these financial statements. The immaterial error corrections identified were primarily related to a misclassification of credit card fees from Other Expense to Sales and Marketing (refer to the tables below for the impact in each period), intercompany foreign exchange losses (cumulative impact of approximately $0.7 million), research and development expenses (cumulative impact of approximately $0.4 million), lease expense (cumulative impact of approximately $0.3 million) and other individually immaterial items. The impacts of these revisions are shown in the tables below:
Year ended May 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 356,974 | $ | (440 | ) | $ | 356,534 | ||||
Cost of sales | 180,085 | 653 | 180,738 | ||||||||
Gross profit | 176,889 | (1,093 | ) | 175,796 | |||||||
Research and development | 26,931 | (337 | ) | 26,594 | |||||||
Sales and marketing | 80,623 | 2,597 | 83,220 | ||||||||
General and administrative | 29,871 | (709 | ) | 29,162 | |||||||
Amortization of intangibles | 17,912 | 54 | 17,966 | ||||||||
Change in fair value of contingent consideration | (8,196 | ) | 100 | (8,096 | ) | ||||||
Acquisition, restructuring and other items, net | 26,600 | (343 | ) | 26,257 | |||||||
Total operating expenses | 177,883 | 1,362 | 179,245 | ||||||||
Operating income | (994 | ) | (2,455 | ) | (3,449 | ) | |||||
Other expense | (3,812 | ) | 2,323 | (1,489 | ) | ||||||
Total other income (expense) | (7,005 | ) | 2,323 | (4,682 | ) | ||||||
Income (loss) before taxes | (7,999 | ) | (132 | ) | (8,131 | ) | |||||
Income tax (benefit) expense | (4,731 | ) | (12 | ) | (4,743 | ) | |||||
Net income (loss) | (3,268 | ) | (120 | ) | (3,388 | ) | |||||
Year ended May 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | (3,268 | ) | $ | (120 | ) | $ | (3,388 | ) | ||
Foreign currency translation | (411 | ) | 147 | (264 | ) | ||||||
Other comprehensive income (loss), before tax | (235 | ) | 147 | (88 | ) | ||||||
Other comprehensive income (loss), net of tax | (299 | ) | 147 | (152 | ) | ||||||
Total comprehensive income (loss), net of tax | (3,567 | ) | 27 | (3,540 | ) | ||||||
80
As of May 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Prepaid expenses and other | $ | 4,783 | $ | (651 | ) | $ | 4,132 | ||||
Total current assets | 151,449 | (651 | ) | 150,798 | |||||||
Property, Plant and Equipment, net | 54,560 | (110 | ) | 54,450 | |||||||
Other Assets | 5,288 | 110 | 5,398 | ||||||||
Intangible Assets, net | 181,806 | (154 | ) | 181,652 | |||||||
Deferred Income Taxes, long term | 19,268 | 240 | 19,508 | ||||||||
Total Assets | 773,623 | (565 | ) | 773,058 | |||||||
Accounts payable | 23,668 | (620 | ) | 23,048 | |||||||
Accrued liabilities | 18,331 | (222 | ) | 18,109 | |||||||
Other current liabilities | — | 200 | 200 | ||||||||
Total current liabilities | 61,157 | (642 | ) | 60,515 | |||||||
Total Liabilities | 228,601 | (642 | ) | 227,959 | |||||||
Retained earnings | 28,233 | (658 | ) | 27,575 | |||||||
Accumulated other comprehensive loss | (1,568 | ) | 735 | (833 | ) | ||||||
Total Stockholders' Equity | 545,022 | 77 | 545,099 | ||||||||
Total Liabilities and Stockholders' Equity | 773,623 | (565 | ) | 773,058 | |||||||
Year ended May 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 26,242 | $ | (557 | ) | $ | 25,685 | ||||
Net cash provided by (used in) investing activities | (13,293 | ) | 557 | (12,736 | ) | ||||||
Net cash provided by (used in) financing activities | (10,465 | ) | — | (10,465 | ) | ||||||
81
Year ended May 31, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Cost of sales | $ | 174,757 | $ | (506 | ) | $ | 174,251 | ||||
Gross profit | 179,668 | 506 | 180,174 | ||||||||
Research and development | 27,486 | 638 | 28,124 | ||||||||
Sales and marketing | 83,200 | 2,105 | 85,305 | ||||||||
General and administrative | 26,639 | 263 | 26,902 | ||||||||
Amortization of intangibles | 16,622 | (60 | ) | 16,562 | |||||||
Change in fair value of contingent consideration | (1,808 | ) | (100 | ) | (1,908 | ) | |||||
Acquisition, restructuring and other items, net | 10,760 | 113 | 10,873 | ||||||||
Total operating expenses | 166,728 | 2,959 | 169,687 | ||||||||
Operating income | 12,940 | (2,453 | ) | 10,487 | |||||||
Other expense | (3,544 | ) | 1,899 | (1,645 | ) | ||||||
Total other income (expense) | (7,200 | ) | 1,899 | (5,301 | ) | ||||||
Income (loss) before taxes | 5,740 | (554 | ) | 5,186 | |||||||
Income tax (benefit) expense | 3,074 | (235 | ) | 2,839 | |||||||
Net income (loss) | 2,666 | (319 | ) | 2,347 | |||||||
Year ended May 31, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | 2,666 | $ | (319 | ) | $ | 2,347 | ||||
Foreign currency translation | 295 | 147 | 442 | ||||||||
Other comprehensive income (loss), before tax | 247 | 147 | 394 | ||||||||
Other comprehensive income (loss), net of tax | 265 | 147 | 412 | ||||||||
Total comprehensive income (loss), net of tax | 2,931 | (172 | ) | 2,759 | |||||||
82
As of May 31, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Inventories | $ | 61,234 | $ | 506 | $ | 61,740 | |||||
Prepaid expenses and other | 5,471 | (591 | ) | 4,880 | |||||||
Total current assets | 147,097 | (85 | ) | 147,012 | |||||||
Property, Plant and Equipment, net | 66,590 | (135 | ) | 66,455 | |||||||
Other Assets | 3,926 | (120 | ) | 3,806 | |||||||
Intangible Assets, net | 205,256 | (203 | ) | 205,053 | |||||||
Deferred Income Taxes, long term | 15,028 | 228 | 15,256 | ||||||||
Total Assets | 798,891 | (315 | ) | 798,576 | |||||||
Accrued liabilities | 16,652 | (712 | ) | 15,940 | |||||||
Current portion of contingent consideration | 10,918 | (100 | ) | 10,818 | |||||||
Other current liabilities | 599 | — | 599 | ||||||||
Total current liabilities | 66,753 | (812 | ) | 65,941 | |||||||
Other Long Term Liabilities | 84 | 447 | 531 | ||||||||
Total Liabilities | 262,056 | (365 | ) | 261,691 | |||||||
Retained earnings | 31,501 | (538 | ) | 30,963 | |||||||
Accumulated other comprehensive loss | (1,269 | ) | 588 | (681 | ) | ||||||
Total Stockholders' Equity | 536,835 | 50 | 536,885 | ||||||||
Total Liabilities and Stockholders' Equity | 798,891 | (315 | ) | 798,576 | |||||||
Year ended May 31, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 24,681 | $ | — | $ | 24,681 | |||||
Net cash provided by (used in) investing activities | (16,448 | ) | — | (16,448 | ) | ||||||
Net cash provided by (used in) financing activities | (14,016 | ) | — | (14,016 | ) | ||||||
Year ended May 31, 2013 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Research and development | $ | 26,319 | $ | (228 | ) | $ | 26,091 | ||||
Sales and marketing | 76,121 | 1,443 | 77,564 | ||||||||
General and administrative | 26,186 | (151 | ) | 26,035 | |||||||
Amortization of intangibles | 16,617 | (18 | ) | 16,599 | |||||||
Total operating expenses | 162,226 | 1,046 | 163,272 | ||||||||
Operating income | 6,288 | (1,046 | ) | 5,242 | |||||||
Other expense | (2,707 | ) | 1,296 | (1,411 | ) | ||||||
Total other income (expense) | (7,875 | ) | 1,296 | (6,579 | ) | ||||||
Income (loss) before taxes | (1,587 | ) | 250 | (1,337 | ) | ||||||
Income tax (benefit) expense | (376 | ) | 90 | (286 | ) | ||||||
Net income (loss) | (1,211 | ) | 160 | (1,051 | ) | ||||||
83
As of May 31, 2013 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Property, Plant and Equipment, net | $ | 62,391 | $ | (16 | ) | $ | 62,375 | ||||
Intangible Assets, net | 214,673 | (150 | ) | 214,523 | |||||||
Deferred Income Taxes, long term | 18,016 | (7 | ) | 18,009 | |||||||
Total Assets | 790,734 | (173 | ) | 790,561 | |||||||
Accrued liabilities | 16,356 | (395 | ) | 15,961 | |||||||
Total current liabilities | 63,315 | (395 | ) | 62,920 | |||||||
Total Liabilities | 264,632 | (395 | ) | 264,237 | |||||||
Retained earnings | 28,835 | (219 | ) | 28,616 | |||||||
Accumulated other comprehensive loss | (1,534 | ) | 441 | (1,093 | ) | ||||||
Total Stockholders' Equity | 526,102 | 222 | 526,324 | ||||||||
Total Liabilities and Stockholders' Equity | 790,734 | (173 | ) | 790,561 | |||||||
Year ended May 31, 2012 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Sales and marketing | $ | 64,505 | $ | 1,252 | $ | 65,757 | |||||
Amortization of intangibles | 9,309 | 84 | 9,393 | ||||||||
Total operating expenses | 129,217 | 1,336 | 130,553 | ||||||||
Operating income | (3,908 | ) | (1,336 | ) | (5,244 | ) | |||||
Other expense | (2,096 | ) | 1,105 | (991 | ) | ||||||
Total other income (expense) | (1,514 | ) | 1,105 | (409 | ) | ||||||
Income (loss) before taxes | (5,422 | ) | (231 | ) | (5,653 | ) | |||||
Income tax (benefit) expense | (239 | ) | (83 | ) | (322 | ) | |||||
Net income (loss) | (5,183 | ) | (148 | ) | (5,331 | ) | |||||
As of May 31, 2012 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Intangible Assets, net | $ | 147,363 | $ | (168 | ) | $ | 147,195 | ||||
Deferred Income Taxes, long term | 44,194 | 83 | 44,277 | ||||||||
Total Assets | 719,988 | (85 | ) | 719,903 | |||||||
Retained earnings | 30,046 | (379 | ) | 29,667 | |||||||
Accumulated other comprehensive loss | (1,274 | ) | 294 | (980 | ) | ||||||
Total Stockholders' Equity | 523,391 | (85 | ) | 523,306 | |||||||
Total Liabilities and Stockholders' Equity | 719,988 | (85 | ) | 719,903 | |||||||
84
NOTE S—QUARTERLY INFORMATION (unaudited)
Quarterly results of operations during the fiscal years ended May 31, 2016 and 2015 are as follows:
2016 | |||||||||||||||
First quarter | Second quarter | Third quarter | Fourth quarter | ||||||||||||
(in thousands, except per share data) | |||||||||||||||
Net sales | $ | 83,753 | $ | 89,284 | $ | 87,434 | $ | 93,419 | |||||||
Gross profit | 43,371 | 45,884 | 43,534 | 41,527 | |||||||||||
Net income (loss) | (775 | ) | (334 | ) | 594 | (43,075 | ) | ||||||||
Earnings (loss) per common share | |||||||||||||||
Basic | (0.02 | ) | (0.01 | ) | 0.02 | (1.19 | ) | ||||||||
Diluted | (0.02 | ) | (0.01 | ) | 0.02 | (1.19 | ) | ||||||||
2015 | |||||||||||||||
First quarter | Second quarter | Third quarter | Fourth quarter | ||||||||||||
(in thousands, except per share data) | |||||||||||||||
Net sales | $ | 87,091 | $ | 92,149 | $ | 86,597 | $ | 90,697 | |||||||
Gross profit | 45,964 | 46,828 | 37,673 | 45,331 | |||||||||||
Net income (loss) | 284 | 1,069 | (4,153 | ) | (588 | ) | |||||||||
Earnings (loss) per common share | |||||||||||||||
Basic | 0.01 | 0.03 | (0.12 | ) | (0.02 | ) | |||||||||
Diluted | 0.01 | 0.03 | (0.12 | ) | (0.02 | ) | |||||||||
The data in the schedules above has been intentionally rounded to the nearest thousand and therefore the quarterly amounts may not sum to the full year amounts. We made adjustments to correct immaterial errors within this selected financial data. For a detailed explanation of these adjustments, please refer to Note R, "Immaterial Error Corrections".
Three months ended February 29, 2016 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 87,384 | $ | 50 | $ | 87,434 | |||||
Gross profit | 43,484 | 50 | 43,534 | ||||||||
Sales and marketing | 20,301 | 654 | 20,955 | ||||||||
General and administrative | 6,784 | 117 | 6,901 | ||||||||
Total operating expenses | 40,797 | 771 | 41,568 | ||||||||
Operating income | 2,687 | (721 | ) | 1,966 | |||||||
Other expense | (868 | ) | 654 | (214 | ) | ||||||
Total other income (expense) | (1,675 | ) | 654 | (1,021 | ) | ||||||
Income (loss) before taxes | 1,012 | (67 | ) | 945 | |||||||
Income tax (benefit) expense | 382 | (31 | ) | 351 | |||||||
Net income (loss) | 630 | (36 | ) | 594 | |||||||
85
Nine months ended February 29, 2016 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 260,321 | $ | 150 | $ | 260,471 | |||||
Cost of sales | 127,829 | (147 | ) | 127,682 | |||||||
Gross profit | 132,492 | 297 | 132,789 | ||||||||
Research and development | 18,189 | (73 | ) | 18,116 | |||||||
Sales and marketing | 61,429 | 2,105 | 63,534 | ||||||||
General and administrative | 22,300 | 597 | 22,897 | ||||||||
Total operating expenses | 127,418 | 2,629 | 130,047 | ||||||||
Other expense | (2,861 | ) | 2,291 | (570 | ) | ||||||
Total other income (expense) | (5,464 | ) | 2,291 | (3,173 | ) | ||||||
Income (loss) before taxes | (390 | ) | (41 | ) | (431 | ) | |||||
Income tax (benefit) expense | 99 | (15 | ) | 84 | |||||||
Net income (loss) | (489 | ) | (26 | ) | (515 | ) | |||||
As of February 29, 2016 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Intangible Assets, net | $ | 168,080 | $ | (154 | ) | $ | 167,926 | ||||
Deferred Income Taxes, long term | 19,563 | 255 | 19,818 | ||||||||
Total Assets | 753,513 | 101 | 753,614 | ||||||||
Other current liabilities | — | 50 | 50 | ||||||||
Total current liabilities | 61,579 | 50 | 61,629 | ||||||||
Total Liabilities | 202,768 | 50 | 202,818 | ||||||||
Retained earnings | 27,744 | (684 | ) | 27,060 | |||||||
Accumulated other comprehensive loss | (1,790 | ) | 735 | (1,055 | ) | ||||||
Total Stockholders' Equity | 550,745 | 51 | 550,796 | ||||||||
Total Liabilities and Stockholders' Equity | 753,513 | 101 | 753,614 | ||||||||
Nine months ended February 29, 2016 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 26,672 | $ | — | $ | 26,672 | |||||
Net cash provided by (used in) investing activities | (3,888 | ) | — | (3,888 | ) | ||||||
Net cash provided by (used in) financing activities | (19,167 | ) | — | (19,167 | ) | ||||||
86
Three months ended November 30, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 89,234 | $ | 50 | $ | 89,284 | |||||
Gross profit | 45,834 | 50 | 45,884 | ||||||||
Sales and marketing | 20,569 | 809 | 21,378 | ||||||||
General and administrative | 8,089 | (7 | ) | 8,082 | |||||||
Total operating expenses | 44,517 | 802 | 45,319 | ||||||||
Operating income | 1,317 | (752 | ) | 565 | |||||||
Other expense | (1,048 | ) | 809 | (239 | ) | ||||||
Total other income (expense) | (2,045 | ) | 809 | (1,236 | ) | ||||||
Income (loss) before taxes | (728 | ) | 57 | (671 | ) | ||||||
Income tax (benefit) expense | (366 | ) | 29 | (337 | ) | ||||||
Net income (loss) | (362 | ) | 28 | (334 | ) | ||||||
Six months ended November 30, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 172,937 | $ | 100 | $ | 173,037 | |||||
Cost of sales | 83,929 | (147 | ) | 83,782 | |||||||
Gross profit | 89,008 | 247 | 89,255 | ||||||||
Research and development | 12,381 | (73 | ) | 12,308 | |||||||
Sales and marketing | 41,128 | 1,450 | 42,578 | ||||||||
General and administrative | 15,516 | 480 | 15,996 | ||||||||
Total operating expenses | 86,621 | 1,857 | 88,478 | ||||||||
Operating income | 2,387 | (1,610 | ) | 777 | |||||||
Other expense | (1,993 | ) | 1,636 | (357 | ) | ||||||
Total other income (expense) | (3,789 | ) | 1,636 | (2,153 | ) | ||||||
Income (loss) before taxes | (1,402 | ) | 26 | (1,376 | ) | ||||||
Income tax (benefit) expense | (283 | ) | 16 | (267 | ) | ||||||
Net income (loss) | (1,119 | ) | 10 | (1,109 | ) | ||||||
As of November 30, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Intangible Assets, net | $ | 172,511 | $ | (154 | ) | $ | 172,357 | ||||
Deferred Income Taxes, long term | 19,826 | 224 | 20,050 | ||||||||
Total Assets | 760,371 | 70 | 760,441 | ||||||||
Accrued liabilities | 16,975 | (117 | ) | 16,858 | |||||||
Other current liabilities | — | 100 | 100 | ||||||||
Total current liabilities | 60,104 | (17 | ) | 60,087 | |||||||
Total Liabilities | 212,732 | (17 | ) | 212,715 | |||||||
Retained earnings | 27,114 | (648 | ) | 26,466 | |||||||
Accumulated other comprehensive loss | (1,922 | ) | 735 | (1,187 | ) | ||||||
Total Stockholders' Equity | 547,639 | 87 | 547,726 | ||||||||
Total Liabilities and Stockholders' Equity | 760,371 | 70 | 760,441 | ||||||||
87
Six months ended November 30, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 14,278 | $ | — | $ | 14,278 | |||||
Net cash provided by (used in) investing activities | (1,143 | ) | — | (1,143 | ) | ||||||
Net cash provided by (used in) financing activities | (12,370 | ) | — | (12,370 | ) | ||||||
Three months ended August 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 83,703 | $ | 50 | $ | 83,753 | |||||
Cost of sales | 40,529 | (147 | ) | 40,382 | |||||||
Gross profit | 43,174 | 197 | 43,371 | ||||||||
Research and development | 6,202 | (73 | ) | 6,129 | |||||||
Sales and marketing | 20,559 | 641 | 21,200 | ||||||||
General and administrative | 7,427 | 487 | 7,914 | ||||||||
Total operating expenses | 42,104 | 1,055 | 43,159 | ||||||||
Operating income | 1,070 | (858 | ) | 212 | |||||||
Other expense | (945 | ) | 827 | (118 | ) | ||||||
Total other income (expense) | (1,744 | ) | 827 | (917 | ) | ||||||
Income (loss) before taxes | (674 | ) | (31 | ) | (705 | ) | |||||
Income tax (benefit) expense | 83 | (13 | ) | 70 | |||||||
Net income (loss) | (757 | ) | (18 | ) | (775 | ) | |||||
As of August 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Other Assets | $ | 4,818 | $ | 110 | $ | 4,928 | |||||
Intangible Assets, net | 177,399 | (154 | ) | 177,245 | |||||||
Deferred Income Taxes, long term | 19,436 | 253 | 19,689 | ||||||||
Total Assets | 771,789 | 209 | 771,998 | ||||||||
Other current liabilities | — | 150 | 150 | ||||||||
Total current liabilities | 60,651 | 150 | 60,801 | ||||||||
Total Liabilities | 224,666 | 150 | 224,816 | ||||||||
Retained earnings | 27,476 | (676 | ) | 26,800 | |||||||
Accumulated other comprehensive loss | (1,615 | ) | 735 | (880 | ) | ||||||
Total Stockholders' Equity | 547,123 | 59 | 547,182 | ||||||||
Total Liabilities and Stockholders' Equity | 771,789 | 209 | 771,998 | ||||||||
Three months ended August 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 4,699 | $ | — | $ | 4,699 | |||||
Net cash provided by (used in) investing activities | (743 | ) | — | (743 | ) | ||||||
Net cash provided by (used in) financing activities | (2,071 | ) | — | (2,071 | ) | ||||||
88
Three months ended May 31, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 90,897 | $ | (200 | ) | $ | 90,697 | ||||
Cost of sales | 45,340 | 26 | 45,366 | ||||||||
Gross profit | 45,557 | (226 | ) | 45,331 | |||||||
Research and development | 7,289 | (738 | ) | 6,551 | |||||||
Sales and marketing | 20,218 | 933 | 21,151 | ||||||||
General and administrative | 7,658 | (210 | ) | 7,448 | |||||||
Total operating expenses | 44,217 | (15 | ) | 44,202 | |||||||
Operating income | 1,340 | (211 | ) | 1,129 | |||||||
Other expense | (860 | ) | 630 | (230 | ) | ||||||
Total other income (expense) | (1,607 | ) | 630 | (977 | ) | ||||||
Income (loss) before taxes | (267 | ) | 419 | 152 | |||||||
Income tax (benefit) expense | 547 | 193 | 740 | ||||||||
Net income (loss) | (814 | ) | 226 | (588 | ) | ||||||
Three months ended February 28, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Cost of sales | $ | 48,746 | $ | 178 | $ | 48,924 | |||||
Gross profit | 37,851 | (178 | ) | 37,673 | |||||||
Research and development | 6,855 | (277 | ) | 6,578 | |||||||
Sales and marketing | 19,355 | 444 | 19,799 | ||||||||
General and administrative | 6,917 | (304 | ) | 6,613 | |||||||
Amortization of intangibles | 5,106 | 82 | 5,188 | ||||||||
Change in fair value of contingent consideration | (10,044 | ) | 250 | (9,794 | ) | ||||||
Acquisition, restructuring and other items, net | 18,779 | (103 | ) | 18,676 | |||||||
Total operating expenses | 48,002 | 92 | 48,094 | ||||||||
Operating income | (10,151 | ) | (270 | ) | (10,421 | ) | |||||
Other expense | (971 | ) | 497 | (474 | ) | ||||||
Total other income (expense) | (1,828 | ) | 497 | (1,331 | ) | ||||||
Income (loss) before taxes | (11,979 | ) | 227 | (11,752 | ) | ||||||
Income tax (benefit) expense | (7,717 | ) | 118 | (7,599 | ) | ||||||
Net income (loss) | (4,262 | ) | 109 | (4,153 | ) | ||||||
89
Nine months ended February 28, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 266,077 | $ | (240 | ) | $ | 265,837 | ||||
Cost of sales | 134,745 | 627 | 135,372 | ||||||||
Gross profit | 131,332 | (867 | ) | 130,465 | |||||||
Research and development | 19,642 | 401 | 20,043 | ||||||||
Sales and marketing | 60,405 | 1,664 | 62,069 | ||||||||
General and administrative | 22,213 | (499 | ) | 21,714 | |||||||
Amortization of intangibles | 13,182 | 54 | 13,236 | ||||||||
Change in fair value of contingent consideration | (8,626 | ) | 100 | (8,526 | ) | ||||||
Acquisition, restructuring and other items, net | 23,745 | (343 | ) | 23,402 | |||||||
Total operating expenses | 133,666 | 1,377 | 135,043 | ||||||||
Operating income | (2,334 | ) | (2,244 | ) | (4,578 | ) | |||||
Other expense | (2,950 | ) | 1,693 | (1,257 | ) | ||||||
Total other income (expense) | (5,398 | ) | 1,693 | (3,705 | ) | ||||||
Income (loss) before taxes | (7,732 | ) | (551 | ) | (8,283 | ) | |||||
Income tax (benefit) expense | (5,278 | ) | (205 | ) | (5,483 | ) | |||||
Net income (loss) | (2,454 | ) | (346 | ) | (2,800 | ) | |||||
Three months ended February 28, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | (4,262 | ) | $ | 109 | $ | (4,153 | ) | |||
Foreign currency translation | (624 | ) | 37 | (587 | ) | ||||||
Other comprehensive income (loss), before tax | (508 | ) | 37 | (471 | ) | ||||||
Other comprehensive income (loss), net of tax | (551 | ) | 37 | (514 | ) | ||||||
Total comprehensive income (loss), net of tax | (4,813 | ) | 146 | (4,667 | ) | ||||||
Nine months ended February 28, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | (2,454 | ) | $ | (346 | ) | $ | (2,800 | ) | ||
Foreign currency translation | (728 | ) | 110 | (618 | ) | ||||||
Other comprehensive income (loss), before tax | (568 | ) | 110 | (458 | ) | ||||||
Other comprehensive income (loss), net of tax | (627 | ) | 110 | (517 | ) | ||||||
Total comprehensive income (loss), net of tax | (3,081 | ) | (236 | ) | (3,317 | ) | |||||
90
As of February 28, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Inventories | $ | 68,710 | $ | 57 | $ | 68,767 | |||||
Prepaid expenses and other | 4,859 | (221 | ) | 4,638 | |||||||
Total current assets | 154,654 | (164 | ) | 154,490 | |||||||
Property, Plant and Equipment, net | 58,295 | (80 | ) | 58,215 | |||||||
Other Assets | 4,060 | 224 | 4,284 | ||||||||
Intangible Assets, net | 186,547 | (154 | ) | 186,393 | |||||||
Deferred Income Taxes, long term | 19,107 | 433 | 19,540 | ||||||||
Total Assets | 783,136 | 259 | 783,395 | ||||||||
Accounts payable | 21,696 | 278 | 21,974 | ||||||||
Accrued liabilities | 19,946 | (461 | ) | 19,485 | |||||||
Total current liabilities | 59,687 | (183 | ) | 59,504 | |||||||
Other Long Term Liabilities | — | 628 | 628 | ||||||||
Total Liabilities | 239,380 | 445 | 239,825 | ||||||||
Retained earnings | 29,047 | (884 | ) | 28,163 | |||||||
Accumulated other comprehensive loss | (1,896 | ) | 698 | (1,198 | ) | ||||||
Total Stockholders' Equity | 543,756 | (186 | ) | 543,570 | |||||||
Total Liabilities and Stockholders' Equity | 783,136 | 259 | 783,395 | ||||||||
Nine months ended February 28, 2015 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 15,436 | $ | (557 | ) | $ | 14,879 | ||||
Net cash provided by (used in) investing activities | (12,042 | ) | 557 | (11,485 | ) | ||||||
Net cash provided by (used in) financing activities | 641 | — | 641 | ||||||||
Three months ended November 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Cost of sales | $ | 44,493 | $ | 828 | $ | 45,321 | |||||
Gross profit | 47,656 | (828 | ) | 46,828 | |||||||
Research and development | 6,069 | 207 | 6,276 | ||||||||
Sales and marketing | 20,983 | 553 | 21,536 | ||||||||
General and administrative | 7,973 | (187 | ) | 7,786 | |||||||
Amortization of intangibles | 4,061 | (50 | ) | 4,011 | |||||||
Change in fair value of contingent consideration | 617 | (75 | ) | 542 | |||||||
Acquisition, restructuring and other items, net | 2,302 | (240 | ) | 2,062 | |||||||
Total operating expenses | 43,081 | 208 | 43,289 | ||||||||
Operating income | 4,575 | (1,036 | ) | 3,539 | |||||||
Other expense | (954 | ) | 580 | (374 | ) | ||||||
Total other income (expense) | (1,746 | ) | 580 | (1,166 | ) | ||||||
Income (loss) before taxes | 2,829 | (456 | ) | 2,373 | |||||||
Income tax (benefit) expense | 1,491 | (187 | ) | 1,304 | |||||||
Net income (loss) | 1,338 | (269 | ) | 1,069 | |||||||
91
Six months ended November 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 179,480 | $ | (240 | ) | $ | 179,240 | ||||
Cost of sales | 85,999 | 449 | 86,448 | ||||||||
Gross profit | 93,481 | (689 | ) | 92,792 | |||||||
Research and development | 12,787 | 678 | 13,465 | ||||||||
Sales and marketing | 41,050 | 1,221 | 42,271 | ||||||||
General and administrative | 15,296 | (195 | ) | 15,101 | |||||||
Amortization of intangibles | 8,076 | (28 | ) | 8,048 | |||||||
Change in fair value of contingent consideration | 1,418 | (150 | ) | 1,268 | |||||||
Acquisition, restructuring and other items, net | 4,966 | (240 | ) | 4,726 | |||||||
Total operating expenses | 85,664 | 1,285 | 86,949 | ||||||||
Operating income | 7,817 | (1,974 | ) | 5,843 | |||||||
Other expense | (1,979 | ) | 1,196 | (783 | ) | ||||||
Total other income (expense) | (3,570 | ) | 1,196 | (2,374 | ) | ||||||
Income (loss) before taxes | 4,247 | (778 | ) | 3,469 | |||||||
Income tax (benefit) expense | 2,439 | (323 | ) | 2,116 | |||||||
Net income (loss) | 1,808 | (455 | ) | 1,353 | |||||||
Three months ended November 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | 1,338 | $ | (269 | ) | $ | 1,069 | ||||
Foreign currency translation | (104 | ) | 37 | (67 | ) | ||||||
Other comprehensive income (loss), before tax | (100 | ) | 37 | (63 | ) | ||||||
Other comprehensive income (loss), net of tax | (101 | ) | 37 | (64 | ) | ||||||
Total comprehensive income (loss), net of tax | 1,237 | (232 | ) | 1,005 | |||||||
Six months ended November 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | 1,808 | $ | (455 | ) | $ | 1,353 | ||||
Foreign currency translation | (104 | ) | 74 | (30 | ) | ||||||
Other comprehensive income (loss), before tax | (60 | ) | 74 | 14 | |||||||
Other comprehensive income (loss), net of tax | (76 | ) | 74 | (2 | ) | ||||||
Total comprehensive income (loss), net of tax | 1,732 | (381 | ) | 1,351 | |||||||
92
As of November 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Inventories | $ | 75,315 | $ | 57 | $ | 75,372 | |||||
Prepaid expenses and other | 6,753 | (191 | ) | 6,562 | |||||||
Total current assets | 159,355 | (134 | ) | 159,221 | |||||||
Property, Plant and Equipment, net | 67,552 | (829 | ) | 66,723 | |||||||
Other Assets | 2,741 | (15 | ) | 2,726 | |||||||
Intangible Assets, net | 197,362 | (175 | ) | 197,187 | |||||||
Deferred Income Taxes, long term | 11,327 | 551 | 11,878 | ||||||||
Total Assets | 799,331 | (602 | ) | 798,729 | |||||||
Accrued liabilities | 19,177 | (588 | ) | 18,589 | |||||||
Current portion of contingent consideration | 9,795 | (250 | ) | 9,545 | |||||||
Total current liabilities | 57,937 | (838 | ) | 57,099 | |||||||
Other Long Term Liabilities | 124 | 568 | 692 | ||||||||
Total Liabilities | 255,760 | (270 | ) | 255,490 | |||||||
Retained earnings | 33,309 | (993 | ) | 32,316 | |||||||
Accumulated other comprehensive loss | (1,345 | ) | 662 | (683 | ) | ||||||
Total Stockholders' Equity | 543,571 | (332 | ) | 543,239 | |||||||
Total Liabilities and Stockholders' Equity | 799,331 | (602 | ) | 798,729 | |||||||
Six months ended November 30, 2014 | ||||||||||
As previously reported | Adjustments | As revised | ||||||||
Net cash provided by (used in) operating activities | $ | 3,205 | $ | (557 | ) | 2,648 | ||||
Net cash provided by (used in) investing activities | (7,773 | ) | 557 | (7,216 | ) | |||||
Net cash provided by (used in) financing activities | 3,381 | — | 3,381 | |||||||
Three months ended August 31, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net sales | $ | 87,331 | $ | (240 | ) | $ | 87,091 | ||||
Cost of sales | 41,506 | (379 | ) | 41,127 | |||||||
Gross profit | 45,825 | 139 | 45,964 | ||||||||
Research and development | 6,718 | 470 | 7,188 | ||||||||
Sales and marketing | 20,067 | 668 | 20,735 | ||||||||
General and administrative | 7,323 | (8 | ) | 7,315 | |||||||
Amortization of intangibles | 4,015 | 22 | 4,037 | ||||||||
Change in fair value of contingent consideration | 801 | (75 | ) | 726 | |||||||
Total operating expenses | 42,583 | 1,077 | 43,660 | ||||||||
Operating income | 3,242 | (938 | ) | 2,304 | |||||||
Other expense | (1,025 | ) | 616 | (409 | ) | ||||||
Total other income (expense) | (1,824 | ) | 616 | (1,208 | ) | ||||||
Income (loss) before taxes | 1,418 | (322 | ) | 1,096 | |||||||
Income tax (benefit) expense | 948 | (136 | ) | 812 | |||||||
Net income (loss) | 470 | (186 | ) | 284 | |||||||
93
Three months ended August 31, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net income (loss) | $ | 470 | $ | (186 | ) | $ | 284 | ||||
Foreign currency translation | — | 37 | 37 | ||||||||
Other comprehensive income (loss), before tax | 41 | 37 | 78 | ||||||||
Other comprehensive income (loss), net of tax | 26 | 37 | 63 | ||||||||
Total comprehensive income (loss), net of tax | 496 | (149 | ) | 347 | |||||||
As of August 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Inventories | $ | 70,421 | $ | 885 | $ | 71,306 | |||||
Prepaid expenses and other | 6,777 | (311 | ) | 6,466 | |||||||
Total current assets | 150,638 | 574 | 151,212 | ||||||||
Property, Plant and Equipment, net | 66,794 | (605 | ) | 66,189 | |||||||
Other Assets | 3,345 | (53 | ) | 3,292 | |||||||
Intangible Assets, net | 201,440 | (225 | ) | 201,215 | |||||||
Deferred Income Taxes, long term | 12,903 | 364 | 13,267 | ||||||||
Total Assets | 796,114 | 55 | 796,169 | ||||||||
Accrued liabilities | 17,700 | (178 | ) | 17,522 | |||||||
Current portion of contingent consideration | 10,897 | (175 | ) | 10,722 | |||||||
Total current liabilities | 63,707 | (353 | ) | 63,354 | |||||||
Other Long Term Liabilities | 32 | 507 | 539 | ||||||||
Total Liabilities | 256,430 | 154 | 256,584 | ||||||||
Retained earnings | 31,971 | (724 | ) | 31,247 | |||||||
Accumulated other comprehensive loss | (1,243 | ) | 625 | (618 | ) | ||||||
Total Stockholders' Equity | 539,684 | (99 | ) | 539,585 | |||||||
Total Liabilities and Stockholders' Equity | 796,114 | 55 | 796,169 | ||||||||
Three months ended August 30, 2014 | |||||||||||
As previously reported | Adjustments | As revised | |||||||||
Net cash provided by (used in) operating activities | $ | 5,352 | $ | (410 | ) | $ | 4,942 | ||||
Net cash provided by (used in) investing activities | (5,258 | ) | 410 | (4,848 | ) | ||||||
Net cash provided by (used in) financing activities | (2,391 | ) | — | (2,391 | ) | ||||||
94
AngioDynamics, Inc. and Subsidiaries
SCHEDULE II -VALUATION AND QUALIFYING ACCOUNTS | |||||||||||||||
(in thousands) | |||||||||||||||
Column A | Column B | Column C | Column D | Column E | |||||||||||
Description | Balance at Beginning of Year | Additions - Charged to costs and expenses | Deductions | Balance at End of Period | |||||||||||
Year Ended May 31, 2014 | |||||||||||||||
Allowance for deferred tax asset (a) | 712 | 819 | — | 1,531 | |||||||||||
Allowance for sales returns and doubtful accounts (b) | 1,272 | 7,342 | (6,878 | ) | 1,736 | ||||||||||
Totals | $ | 1,984 | $ | 8,161 | $ | (6,878 | ) | $ | 3,267 | ||||||
Year Ended May 31, 2015 | |||||||||||||||
Allowance for deferred tax asset (a) | 1,531 | 467 | (207 | ) | 1,791 | ||||||||||
Allowance for sales returns and doubtful accounts (b) | 1,736 | 1,846 | (539 | ) | 3,043 | ||||||||||
Totals | $ | 3,267 | $ | 2,313 | $ | (746 | ) | $ | 4,834 | ||||||
Year Ended May 31, 2016 | |||||||||||||||
Allowance for deferred tax asset (a) | 1,791 | 40,685 | (267 | ) | 42,209 | ||||||||||
Allowance for sales returns and doubtful accounts (b) | 3,043 | 3,748 | (2,419 | ) | 4,372 | ||||||||||
Totals | $ | 4,834 | $ | 44,433 | $ | (2,686 | ) | $ | 46,581 | ||||||
(a) | Fully reserved deferred tax assets and expiration of fully reserved state net operating loss. |
We made adjustments to correct immaterial errors within this selected financial data. For a detailed explanation of these adjustments, please refer to Note R, "Immaterial Error Corrections".
(b) | Previously reserved sales returns and accounts written off as uncollectible. |
95
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ANGIODYNAMICS, INC. | |||||
Date: | August 1, 2016 | By: | /S/ HOWARD W. DONNELLY | ||
Howard W. Donnelly, Chairman of the Board, Director | |||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Date: | August 1, 2016 | /S/ HOWARD W. DONNELLY | |
Howard W. Donnelly, | |||
Chairman of the Board, Director | |||
Date: | August 1, 2016 | /S/ JAMES C. CLEMMER | |
James C. Clemmer, | |||
Chief Executive Officer (Principal Executive Officer) | |||
Date: | August 1, 2016 | /S/ PETER KISH | |
Peter Kish, | |||
Interim—Chief Financial Officer, (Principal Financial and Chief Accounting Officer) | |||
Date: | August 1, 2016 | /S/ WESLEY E. JOHNSON, JR. | |
Wesley E. Johnson, Jr., | |||
Director | |||
Date: | August 1, 2016 | /S/ JEFFREY G. GOLD | |
Jeffrey G. Gold, | |||
Director | |||
Date: | August 1, 2016 | /S/ DENNIS S. METENY | |
Dennis S. Meteny, | |||
Director | |||
Date: | August 1, 2016 | /S/ STEVEN R. LAPORTE | |
Steven R. LaPorte, | |||
Director | |||
Date: | August 1, 2016 | /S/ KEVIN J. GOULD | |
Kevin J. Gould, | |||
Director | |||
Date: | August 1, 2016 | /S/ DAVID BURGSTAHLER | |
David Burgstahler, | |||
Director | |||
Date: | August 1, 2016 | /S/ SRIRAM VENKATARAMAN | |
Sriram Venkataraman, | |||
Director | |||
96
EXHIBITS
(b) | Exhibits |
2.1 | Stockholders Agreement, dated as of May 22, 2012, among AngioDynamics, Inc. and the stockholders set forth on the signature pages thereto (incorporated by reference to Exhibit 2.2 of the Company’s current report on Form 8-K filed with the Commission on May 25, 2012). |
2.2 | Stock Purchase Agreement, dated as of October 8, 2012, by and among AngioDynamics, Inc., Vortex Medical, Inc. (“Vortex”), the stockholders of Vortex set forth on the signature pages thereto, the option holders of Vortex set forth on the signature pages thereto and CHTP Management Services, Inc., as sellers’ representative (incorporated by reference to Exhibit 2.1 of the Company’s current report on Form 8-K, filed with the Commission on October 12, 2012). |
3.1.1 | Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 of the Company’s quarterly report on Form 10-Q, filed with the Commission on October 7, 2005). |
3.1.2 | Certificate of Amendment to the Amended and Restated Certificate of Incorporation of AngioDynamics, Inc. (incorporated by reference to Exhibit 3.1.2 of the Company's annual report on Form 10-K, filed with the Commission on August 10, 2015). |
3.2 | Second Amended and Restated By-Laws, effective October 16, 2015 (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K, filed with the Commission on October 21, 2015). |
10.1 | Credit Agreement, dated as of September 19, 2013, by and among AngioDynamics, Inc., the lenders party thereto, JPMorgan Chase Bank, N.A., as administrative agent, Bank of America, N.A. and Keybank National Association as co-syndication agents, and J.P. Morgan Securities LLC, Merrill Lynch, Pierce, Fenner & Smith Incorporated and Keybank National Association as joint bookrunners and joint lead arrangers (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K filed with the Commission on September 24, 2013). |
10.1.1 | AngioDynamics, Inc. 1997 Stock Option Plan, as amended by the Board and Shareholders on February 27, 2004 (incorporated by reference to Exhibit 10.2 of the Company’s registration statement on Form S-1, filed on March 5, 2004). |
10.1.2 | AngioDynamics, Inc. 2004 Stock and Incentive Award Plan (as amended) (incorporated by reference to the Company’s Definitive Proxy Statement on Schedule 14A filed with the Commission on September 17, 2014). |
10.1.3 | AngioDynamics 2013 Total Shareholder Return Performance Unit Agreement Program (incorporated by reference to Exhibit 10.2 of the Company's current report on Form 8-K filed with the Commission on November 5, 2013). |
10.1.4 | AngioDynamics 2014 Total Shareholder Return Performance Unit Agreement Program (incorporated by reference to Exhibit 10.1.4 of the Company's annual report on Form 10-K filed with the Commission on January 12, 2015). |
10.1.5 | AngioDynamics 2015 Total Shareholder Return Performance Unit Agreement Program (incorporated by reference to Exhibit 10.1.5 of the Company’s annual report on Form 10-K filed with the Commission on August 10, 2015). |
10.2 | AngioDynamics, Inc. Employee Stock Purchase Plan (as amended) (incorporated by reference to the Company’s Definitive Proxy Statement on Schedule 14A filed with the Commission on September 17, 2014). |
10.3 | Form of Non-Statutory Stock Option Agreement pursuant to the AngioDynamics, Inc. Stock and Incentive Award Plan (incorporated by reference to Exhibit 10.1 of the Company’s quarterly report on Form 10-Q, filed with the Commission on October 12, 2004). |
10.4.1 | Form of 2013 Performance Share Award Agreement pursuant to the AngioDynamics, Inc. 2004 Stock and Incentive Award Plan (incorporated by reference to Exhibit 10.2 of the Company’s current report on Form 8-K, filed with the Commission on May 12, 2005). |
10.4.2 | Form of 2014 Performance Share Award Agreement pursuant to the AngioDynamics, Inc. 2004 Stock and Incentive Award Plan (incorporated by reference to Exhibit 10.4.2 of the Company's annual report on Form 10-K filed with the Commission on January 12, 2015). |
10.4.3 | Form of 2015 Performance Share Award Agreement pursuant to the AngioDyanmics, Inc. 2004 Stock and Incentive Award Plan (incorporated by reference to Exhibit 10.4.3 of the Company’s annual report on Form 10-K filed with the Commission on August 10, 2015). |
10.5 | Form of Restricted Stock Award Agreement pursuant to the AngioDynamics, Inc. 2004 Stock and Incentive Award Plan (incorporated by reference to the Company’s current report on Form 8-K, filed with the Commission on May 12, 2005). |
10.6 | Rita Medical Systems, Inc. 1994 Incentive Stock Plan (incorporated by reference to Exhibit 10.2 of Rita Medical Systems registration statement on Form S-1, filed with the Commission on May 3, 2000) |
97
10.7 | Horizon Medical Products, Inc. 1998 Stock Incentive Plan (incorporated by reference to Exhibit 10.11 of Horizon Medical Products’ registration statement on Form S-1, filed with the Commission on February 13, 1998. |
10.8 | Rita Medical Systems, Inc. 2000 Stock Plan (incorporated by reference to Exhibit 10.3 of Rita Medical Systems registration statement on Form S-1/A, filed with the Commission on June 14, 2000). |
10.9 | Rita Medical Systems, Inc. 2000 Directors’ Stock Plan, as amended on June 8, 2005 (incorporated by reference to Exhibit 99.2 of Rita Medical System’s registration statement on Form S-8, filed with the Commission on July 8, 2005). |
10.10 | Rita Medical Systems, Inc. 2005 Stock and Incentive Plan (incorporated by reference to Exhibit 99.1 of Rita Medical System’s registration statement on Form S-8, filed with the Commission on July 8, 2005). |
10.11 | Form of Indemnification Agreement of AngioDynamics, Inc. (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K, filed with the Commission on May 12, 2006). |
10.11.1 | Employment Agreement, dated April 1, 2016, between AngioDynamics, Inc. and James C. Clemmer (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K, filed with the Commission on April 6, 2016). |
10.12 | Change in Control Agreement, dated April 1, 2016, between AngioDynamics, Inc. and James C. Clemmer (incorporated by reference to Exhibit 10.2 of the Company’s current report on Form 8-K, filed with the Commission on April 6, 2016). |
10.12.1 | Form of Severance Agreement of AngioDynamics, Inc. (incorporated by reference to Exhibit 10.1 of the Company’s current report on form 8-K, filed with the Commission on October 31, 2007). |
10.13 | Form of Change in Control Agreement (incorporated by reference to Exhibit 10.13 of the Company's annual report on Form 10-K filed with the Commission on January 12, 2015). |
10.13.1 | Performance Share Award Agreement, with a grant date of April 4, 2016, between AngioDynamics, Inc. and James C. Clemmer (incorporated by reference to Exhibit 10.3 of the Company’s current report on Form 8-K, filed with the Commission on April 6, 2016). |
10.14 | AngioDynamics, Inc. Total Shareholder Return Performance Share Award Program - Performance Period Ending July 2019 (incorporated by reference to Exhibit 10.4 of the Company’s current report on Form 8-K, filed with the Commission on April 6, 2016). |
10.15 | Stock Option Award Agreement, with a grant date of April 4, 2016, between AngioDynamics, Inc. and James C. Clemmer (incorporated by reference to Exhibit 10.5 of the Company’s current report on Form 8-K, filed with the Commission on April 6, 2016). |
10.16 | Restricted Stock Unit Award Agreement, with a grant date of April 4, 2016, between AngioDynamics, Inc. and James C. Clemmer (incorporated by reference to Exhibit 10.6 of the Company’s current report on Form 8-K, filed with the Commission on April 6, 2016). |
10.17 | Separation Agreement and General Release, dated April 22, 2016, between AngioDynamics, Inc. and Joseph M. DeVivo (incorporated by reference to Exhibit 10.1 of the Company’s current report on Form 8-K, filed with the Commission on April 27, 2016). |
10.19 | AngioDynamics, Inc. Fiscal Year 2012 Senior Executive Equity Incentive Program (incorporated by reference to Exhibit 10.30 of the Company’s annual report on Form 10-K, filed with the commission on August 12, 2011). |
14 | Code of Ethics (incorporated by reference to Exhibit 14 of the Company’s current report on Form 8-K, filed with the Commission on May 12, 2006). |
98
21 | Subsidiaries |
23 | Consent of PricewaterhouseCoopers LLP, an independent registered public accounting firm. |
31.1 | Certification by the Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
31.2 | Certification by the Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
32.1 | Certification by the Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
32.2 | Certification by the Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
101.INS | XBRL Instance Document |
101.SCH | XBRL Schema Document |
101.CAL | XBRL Calculation Linkbase Documents |
101.DEF | XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB | XBRL Labels Linkbase Documents |
101.PRE | XBRL Presentation Linkbase Documents |
99